WO2013046136A1 - 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh - Google Patents
3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh Download PDFInfo
- Publication number
- WO2013046136A1 WO2013046136A1 PCT/IB2012/055133 IB2012055133W WO2013046136A1 WO 2013046136 A1 WO2013046136 A1 WO 2013046136A1 IB 2012055133 W IB2012055133 W IB 2012055133W WO 2013046136 A1 WO2013046136 A1 WO 2013046136A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- pyrimidin
- phenyl
- alkyl
- group
- Prior art date
Links
- 239000003112 inhibitor Substances 0.000 title description 10
- DPXAMRSETFKEGX-UHFFFAOYSA-N O=C1OCCN1C1=CC=NC=N1 Chemical class O=C1OCCN1C1=CC=NC=N1 DPXAMRSETFKEGX-UHFFFAOYSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 176
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 81
- 150000003839 salts Chemical class 0.000 claims abstract description 76
- 230000003538 neomorphic effect Effects 0.000 claims abstract description 62
- 201000010099 disease Diseases 0.000 claims abstract description 44
- 208000035475 disorder Diseases 0.000 claims abstract description 37
- 102100037845 Isocitrate dehydrogenase [NADP], mitochondrial Human genes 0.000 claims abstract description 21
- 125000000217 alkyl group Chemical group 0.000 claims description 134
- -1 C3_g cycloalkyi Chemical group 0.000 claims description 112
- 229910052739 hydrogen Inorganic materials 0.000 claims description 105
- 239000001257 hydrogen Substances 0.000 claims description 105
- 125000001424 substituent group Chemical group 0.000 claims description 100
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 92
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 71
- 125000005843 halogen group Chemical group 0.000 claims description 69
- 238000000034 method Methods 0.000 claims description 64
- 150000002431 hydrogen Chemical group 0.000 claims description 61
- 125000001072 heteroaryl group Chemical group 0.000 claims description 54
- 125000003545 alkoxy group Chemical group 0.000 claims description 49
- 229910052805 deuterium Inorganic materials 0.000 claims description 46
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 44
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 44
- 125000000623 heterocyclic group Chemical group 0.000 claims description 43
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 40
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 38
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 36
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 35
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 32
- 239000003814 drug Substances 0.000 claims description 30
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 30
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 27
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 27
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 24
- 125000006569 (C5-C6) heterocyclic group Chemical group 0.000 claims description 23
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 22
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical group [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 claims description 22
- 125000004043 oxo group Chemical group O=* 0.000 claims description 22
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 22
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 claims description 21
- 125000001188 haloalkyl group Chemical group 0.000 claims description 20
- 125000003118 aryl group Chemical group 0.000 claims description 19
- 229940124597 therapeutic agent Drugs 0.000 claims description 19
- 125000001153 fluoro group Chemical group F* 0.000 claims description 18
- 239000008194 pharmaceutical composition Substances 0.000 claims description 17
- 125000004076 pyridyl group Chemical group 0.000 claims description 17
- CZZYITDELCSZES-UHFFFAOYSA-N diphenylmethane Chemical compound C=1C=CC=CC=1CC1=CC=CC=C1 CZZYITDELCSZES-UHFFFAOYSA-N 0.000 claims description 14
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 14
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims description 14
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 claims description 13
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 12
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 12
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 11
- GOEGYHBXKPIDJE-UHFFFAOYSA-N 3-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)N1CCOC1=O GOEGYHBXKPIDJE-UHFFFAOYSA-N 0.000 claims description 10
- 239000003937 drug carrier Substances 0.000 claims description 10
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 10
- 125000003386 piperidinyl group Chemical group 0.000 claims description 9
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 claims description 8
- 125000004193 piperazinyl group Chemical group 0.000 claims description 8
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 8
- 125000003107 substituted aryl group Chemical group 0.000 claims description 8
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 claims description 7
- 229910017711 NHRa Inorganic materials 0.000 claims description 7
- BUGOPWGPQGYYGR-UHFFFAOYSA-N thiane 1,1-dioxide Chemical class O=S1(=O)CCCCC1 BUGOPWGPQGYYGR-UHFFFAOYSA-N 0.000 claims description 7
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 claims description 6
- 229910003827 NRaRb Inorganic materials 0.000 claims description 6
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 claims description 5
- 108010081348 HRT1 protein Hairy Proteins 0.000 claims description 5
- 102100021881 Hairy/enhancer-of-split related with YRPW motif protein 1 Human genes 0.000 claims description 5
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims description 5
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 claims description 5
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 claims description 5
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 5
- 125000002757 morpholinyl group Chemical group 0.000 claims description 5
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 5
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 5
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 5
- 125000000335 thiazolyl group Chemical group 0.000 claims description 5
- 125000001425 triazolyl group Chemical group 0.000 claims description 5
- WCYWZMWISLQXQU-FIBGUPNXSA-N trideuteriomethane Chemical compound [2H][C]([2H])[2H] WCYWZMWISLQXQU-FIBGUPNXSA-N 0.000 claims description 5
- PEEWKHRIJDGILX-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-[3-(4-chlorophenyl)-1,2,4-oxadiazol-5-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2ON=C(N=2)C=2C=CC(Cl)=CC=2)=N1 PEEWKHRIJDGILX-SWLSCSKDSA-N 0.000 claims description 4
- VNRQSKACLLUOAJ-HNAYVOBHSA-N (4s)-3-[5-fluoro-2-[[(1s)-1-[4-(2-methylpropoxy)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=CC(OCC(C)C)=CC=C1[C@H](C)NC1=NC=C(F)C(N2C(OC[C@@H]2C(C)C)=O)=N1 VNRQSKACLLUOAJ-HNAYVOBHSA-N 0.000 claims description 4
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 claims description 4
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 claims description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 4
- FCSBPEDVQHJOBB-QFBILLFUSA-N (4s)-3-[2-[[(1s)-1-[4-(2-methylpropoxy)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=CC(OCC(C)C)=CC=C1[C@H](C)NC1=NC=CC(N2C(OC[C@@H]2C(C)C)=O)=N1 FCSBPEDVQHJOBB-QFBILLFUSA-N 0.000 claims description 3
- 125000005960 1,4-diazepanyl group Chemical group 0.000 claims description 3
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 claims description 3
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 3
- 125000001246 bromo group Chemical group Br* 0.000 claims description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 claims description 3
- ZPANBBAEJLXOHF-LAUBAEHRSA-N n-[4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]phenyl]cyclohexanecarboxamide Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(NC(=O)C3CCCCC3)=CC=2)=N1 ZPANBBAEJLXOHF-LAUBAEHRSA-N 0.000 claims description 3
- 125000002971 oxazolyl group Chemical group 0.000 claims description 3
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 claims description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 3
- 125000001544 thienyl group Chemical group 0.000 claims description 3
- NXLIAJSDZZVZGW-KBXCAEBGSA-N (4s)-3-[2-[[(1s)-1-(2-fluoro-4-propan-2-ylphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C(=CC(=CC=2)C(C)C)F)=N1 NXLIAJSDZZVZGW-KBXCAEBGSA-N 0.000 claims description 2
- DOKKAMWYERTTEW-DZGCQCFKSA-N (4s)-3-[2-[[(1s)-1-(5-phenyl-1,3,4-thiadiazol-2-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2SC(=NN=2)C=2C=CC=CC=2)=N1 DOKKAMWYERTTEW-DZGCQCFKSA-N 0.000 claims description 2
- PVCIBEUBAOJSPW-OXJNMPFZSA-N (4s)-3-[2-[[(1s)-1-(6-phenylpyridin-3-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=NC(=CC=2)C=2C=CC=CC=2)=N1 PVCIBEUBAOJSPW-OXJNMPFZSA-N 0.000 claims description 2
- PQMAWDDODIHUII-HNAYVOBHSA-N (4s)-3-[2-[[(1s)-1-[1-(3-methoxyphenyl)pyrazol-4-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound COC1=CC=CC(N2N=CC(=C2)[C@H](C)NC=2N=C(C=CN=2)N2C(OC[C@@H]2C(C)C)=O)=C1 PQMAWDDODIHUII-HNAYVOBHSA-N 0.000 claims description 2
- IMYNKVGASYVFPQ-KBXCAEBGSA-N (4s)-3-[2-[[(1s)-1-[2-fluoro-4-(1-methylcyclopropyl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C(=CC(=CC=2)C2(C)CC2)F)=N1 IMYNKVGASYVFPQ-KBXCAEBGSA-N 0.000 claims description 2
- XNWMMXIFOLWPGU-XHDPSFHLSA-N (4s)-3-[2-[[(1s)-1-[2-fluoro-4-(trifluoromethyl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C(=CC(=CC=2)C(F)(F)F)F)=N1 XNWMMXIFOLWPGU-XHDPSFHLSA-N 0.000 claims description 2
- GOBJPRCTHVAYDE-GOEBONIOSA-N (4s)-3-[2-[[(1s)-1-[3-(3-methylphenyl)-1,2,4-oxadiazol-5-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2ON=C(N=2)C=2C=C(C)C=CC=2)=N1 GOBJPRCTHVAYDE-GOEBONIOSA-N 0.000 claims description 2
- VXBCBCASQHFSAS-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-[3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2ON=C(N=2)C=2C=CC(F)=CC=2)=N1 VXBCBCASQHFSAS-SWLSCSKDSA-N 0.000 claims description 2
- VNTDYDNOVYQSFZ-PGRDOPGGSA-N (4s)-3-[2-[[(1s)-1-[3-fluoro-4-[(3,3,4-trimethylpiperazin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C(CN3CC(C)(C)N(C)CC3)=CC=2)=N1 VNTDYDNOVYQSFZ-PGRDOPGGSA-N 0.000 claims description 2
- SWZRRQKQLVMABW-LAUBAEHRSA-N (4s)-3-[2-[[(1s)-1-[3-fluoro-4-[(4-methylpiperazin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C(CN3CCN(C)CC3)=CC=2)=N1 SWZRRQKQLVMABW-LAUBAEHRSA-N 0.000 claims description 2
- OKXLHEMRMSXABA-PKOBYXMFSA-N (4s)-3-[2-[[(1s)-1-[3-methyl-4-(2-methylpropoxy)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=C(C)C(OCC(C)C)=CC=C1[C@H](C)NC1=NC=CC(N2C(OC[C@@H]2C(C)C)=O)=N1 OKXLHEMRMSXABA-PKOBYXMFSA-N 0.000 claims description 2
- AGSQVFXRDDNSQD-HNAYVOBHSA-N (4s)-3-[2-[[(1s)-1-[4-(1-methylpyrazol-4-yl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(=CC=2)C2=CN(C)N=C2)=N1 AGSQVFXRDDNSQD-HNAYVOBHSA-N 0.000 claims description 2
- IPBMCQWLGDMECF-HRAATJIYSA-N (4s)-3-[2-[[(1s)-1-[4-(4-fluorophenoxy)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(OC=3C=CC(F)=CC=3)=CC=2)=N1 IPBMCQWLGDMECF-HRAATJIYSA-N 0.000 claims description 2
- NWEKVDPZORGKLH-GHTZIAJQSA-N (4s)-3-[2-[[(1s)-1-[4-[(4-methylpiperazin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCN(C)CC3)=CC=2)=N1 NWEKVDPZORGKLH-GHTZIAJQSA-N 0.000 claims description 2
- VNCZODINPSDRIC-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-[5-(4-chlorophenyl)-1,2,4-oxadiazol-3-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N=C(ON=2)C=2C=CC(Cl)=CC=2)=N1 VNCZODINPSDRIC-SWLSCSKDSA-N 0.000 claims description 2
- HBHNBVQOMXUYKF-HRAATJIYSA-N (4s)-3-[2-[[(1s)-1-[5-(4-fluoro-3-methylphenyl)pyridin-2-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N=CC(=CC=2)C=2C=C(C)C(F)=CC=2)=N1 HBHNBVQOMXUYKF-HRAATJIYSA-N 0.000 claims description 2
- UCHDSSAFUHZSQS-KBXCAEBGSA-N (4s)-3-[2-[[(1s)-1-[5-(4-fluorophenoxy)pyrazin-2-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N=CC(OC=3C=CC(F)=CC=3)=NC=2)=N1 UCHDSSAFUHZSQS-KBXCAEBGSA-N 0.000 claims description 2
- AUBGVCAXOQPLIS-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-[5-(4-fluorophenyl)-1,3,4-oxadiazol-2-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2OC(=NN=2)C=2C=CC(F)=CC=2)=N1 AUBGVCAXOQPLIS-SWLSCSKDSA-N 0.000 claims description 2
- SXBNORYEQFOLBM-MGPUTAFESA-N (4s)-3-[5-fluoro-2-[[(1s)-1-[3-fluoro-4-(piperidine-1-carbonyl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=NC(N[C@@H](C)C=2C=C(F)C(C(=O)N3CCCCC3)=CC=2)=NC=C1F SXBNORYEQFOLBM-MGPUTAFESA-N 0.000 claims description 2
- ZXJPIOXEBIYSLQ-UQBPGWFLSA-N (4s)-4-methyl-3-[2-[[(1s)-1-[4-[(4-methylpiperazin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@@]1(C)COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCN(C)CC3)=CC=2)=N1 ZXJPIOXEBIYSLQ-UQBPGWFLSA-N 0.000 claims description 2
- ZUSUQVVEAKBKTO-SIKLNZKXSA-N (4s)-4-propan-2-yl-3-[2-[[(1s)-1-[4-[(3,3,4-trimethylpiperazin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CC(C)(C)N(C)CC3)=CC=2)=N1 ZUSUQVVEAKBKTO-SIKLNZKXSA-N 0.000 claims description 2
- GPTQTESJYSBSOC-KBXCAEBGSA-N (4s)-4-propan-2-yl-3-[2-[[(1s)-1-[5-[3-(trifluoromethyl)phenyl]pyrimidin-2-yl]ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N=CC(=CN=2)C=2C=C(C=CC=2)C(F)(F)F)=N1 GPTQTESJYSBSOC-KBXCAEBGSA-N 0.000 claims description 2
- QVKJMYIIQYXWEG-MGPUTAFESA-N 2-chloro-n-cyclopentyl-4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]benzamide Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(Cl)C(C(=O)NC3CCCC3)=CC=2)=N1 QVKJMYIIQYXWEG-MGPUTAFESA-N 0.000 claims description 2
- ONXIZYRKUYBMHT-OTYBAMDLSA-N 2-fluoro-n-(4-hydroxy-4-methylcyclohexyl)-4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]benzamide Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C(C(=O)NC3CCC(C)(O)CC3)=CC=2)=N1 ONXIZYRKUYBMHT-OTYBAMDLSA-N 0.000 claims description 2
- WRVQRGXMUQLSAU-JTQLQIEISA-N 3-[2-[[(1s)-1-[3-(4-chlorophenyl)-1,2,4-oxadiazol-5-yl]ethyl]amino]-5-fluoropyrimidin-4-yl]-4,4-dimethyl-1,3-oxazolidin-2-one Chemical compound N([C@@H](C)C=1ON=C(N=1)C=1C=CC(Cl)=CC=1)C(N=1)=NC=C(F)C=1N1C(=O)OCC1(C)C WRVQRGXMUQLSAU-JTQLQIEISA-N 0.000 claims description 2
- NUKCLTBGGMSGTG-UHFFFAOYSA-N 3-[5-fluoro-2-[1-[5-(4-fluoro-3-methylphenyl)pyridin-2-yl]ethylamino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C=1C=C(C=2C=C(C)C(F)=CC=2)C=NC=1C(C)NC(N=1)=NC=C(F)C=1N1CCOC1=O NUKCLTBGGMSGTG-UHFFFAOYSA-N 0.000 claims description 2
- NNDMNJIAZMBOPZ-AWEZNQCLSA-N 3-[5-fluoro-2-[[(1s)-1-(4-phenoxyphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound N([C@@H](C)C=1C=CC(OC=2C=CC=CC=2)=CC=1)C(N=1)=NC=C(F)C=1N1CCOC1=O NNDMNJIAZMBOPZ-AWEZNQCLSA-N 0.000 claims description 2
- FWKRVPIRNNHJBL-VIFPVBQESA-N 3-[6-chloro-2-[[(1s)-1-[3-(4-chlorophenyl)-1,2,4-oxadiazol-5-yl]ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound N([C@@H](C)C=1ON=C(N=1)C=1C=CC(Cl)=CC=1)C(N=1)=NC(Cl)=CC=1N1CCOC1=O FWKRVPIRNNHJBL-VIFPVBQESA-N 0.000 claims description 2
- YBUPWRYTXGAWJX-UHFFFAOYSA-N 4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)C1COC(=O)N1 YBUPWRYTXGAWJX-UHFFFAOYSA-N 0.000 claims description 2
- SVKZPLRWJXWQQI-QUJKESNLSA-N CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C(C(=O)N[C@@H]3CC[C@@H](O)CC3)=CC=2)=N1 Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C(C(=O)N[C@@H]3CC[C@@H](O)CC3)=CC=2)=N1 SVKZPLRWJXWQQI-QUJKESNLSA-N 0.000 claims description 2
- 239000004305 biphenyl Substances 0.000 claims description 2
- 235000010290 biphenyl Nutrition 0.000 claims description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 2
- QACQWPWIMWZTHR-HRAATJIYSA-N n-cyclohexyl-2-fluoro-4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]benzamide Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C(C(=O)NC3CCCCC3)=CC=2)=N1 QACQWPWIMWZTHR-HRAATJIYSA-N 0.000 claims description 2
- OCZCXJFDMAWAIM-FXAWDEMLSA-N (4s)-3-[2-[[(1s)-1-[4-[(4,4-difluoropiperidin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCC(F)(F)CC3)=CC=2)=N1 OCZCXJFDMAWAIM-FXAWDEMLSA-N 0.000 claims 1
- CYBTVJWHWQFLKH-GHTZIAJQSA-N (4s)-3-[2-[[(1s)-1-[4-[(4-amino-4-methylpiperidin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCC(C)(N)CC3)=CC=2)=N1 CYBTVJWHWQFLKH-GHTZIAJQSA-N 0.000 claims 1
- OELBJGXZLKHRER-LMVRJCEZSA-N (4s)-3-[2-[[(1s)-1-[4-[[(2r,6s)-2,6-dimethylmorpholin-4-yl]methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3C[C@@H](C)O[C@@H](C)C3)=CC=2)=N1 OELBJGXZLKHRER-LMVRJCEZSA-N 0.000 claims 1
- OXPZKAYILDTYSK-WMZHIEFXSA-N (4s)-3-[2-[[(1s)-1-[4-[[4-(dimethylamino)piperidin-1-yl]methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCC(CC3)N(C)C)=CC=2)=N1 OXPZKAYILDTYSK-WMZHIEFXSA-N 0.000 claims 1
- JDCYIMQAIKEACU-HNAYVOBHSA-N (4s)-3-[2-[[(1s)-1-[5-(4-fluoro-3-methylphenyl)pyrimidin-2-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N=CC(=CN=2)C=2C=C(C)C(F)=CC=2)=N1 JDCYIMQAIKEACU-HNAYVOBHSA-N 0.000 claims 1
- QZSFGXVFAIYNNF-KBXCAEBGSA-N (4s)-3-[2-[[(1s)-1-[5-(4-fluorophenoxy)pyrimidin-2-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N=CC(OC=3C=CC(F)=CC=3)=CN=2)=N1 QZSFGXVFAIYNNF-KBXCAEBGSA-N 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 106
- 206010028980 Neoplasm Diseases 0.000 abstract description 49
- 201000011510 cancer Diseases 0.000 abstract description 42
- 208000035269 cancer or benign tumor Diseases 0.000 abstract description 10
- 230000005764 inhibitory process Effects 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 390
- 235000019439 ethyl acetate Nutrition 0.000 description 186
- 239000000543 intermediate Substances 0.000 description 174
- 239000000243 solution Substances 0.000 description 161
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 140
- 239000011541 reaction mixture Substances 0.000 description 124
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 124
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 118
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 116
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 88
- 238000004896 high resolution mass spectrometry Methods 0.000 description 81
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 78
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 75
- 238000005481 NMR spectroscopy Methods 0.000 description 67
- 238000006243 chemical reaction Methods 0.000 description 65
- 239000007787 solid Substances 0.000 description 63
- 239000000047 product Substances 0.000 description 61
- 238000005160 1H NMR spectroscopy Methods 0.000 description 60
- 238000002360 preparation method Methods 0.000 description 60
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 55
- 239000012267 brine Substances 0.000 description 54
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 50
- 238000010898 silica gel chromatography Methods 0.000 description 46
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 44
- 238000000926 separation method Methods 0.000 description 43
- 108010075869 Isocitrate Dehydrogenase Proteins 0.000 description 38
- 102000012011 Isocitrate Dehydrogenase Human genes 0.000 description 38
- 230000002829 reductive effect Effects 0.000 description 36
- 239000003921 oil Substances 0.000 description 35
- 235000019198 oils Nutrition 0.000 description 35
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- 239000010410 layer Substances 0.000 description 32
- HWXBTNAVRSUOJR-UHFFFAOYSA-N 2-hydroxyglutaric acid Chemical compound OC(=O)C(O)CCC(O)=O HWXBTNAVRSUOJR-UHFFFAOYSA-N 0.000 description 31
- 238000004007 reversed phase HPLC Methods 0.000 description 31
- 239000008346 aqueous phase Substances 0.000 description 30
- 238000003756 stirring Methods 0.000 description 29
- 239000000126 substance Substances 0.000 description 29
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 28
- 238000007792 addition Methods 0.000 description 27
- 239000012044 organic layer Substances 0.000 description 27
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 26
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 26
- 239000000741 silica gel Substances 0.000 description 26
- 229910002027 silica gel Inorganic materials 0.000 description 26
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 24
- 239000003480 eluent Substances 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 238000000746 purification Methods 0.000 description 22
- 229920006395 saturated elastomer Polymers 0.000 description 22
- 239000002904 solvent Substances 0.000 description 20
- 239000000725 suspension Substances 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 19
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 19
- 229940093499 ethyl acetate Drugs 0.000 description 18
- 230000035772 mutation Effects 0.000 description 18
- 101000599886 Homo sapiens Isocitrate dehydrogenase [NADP], mitochondrial Proteins 0.000 description 17
- 239000012071 phase Substances 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 16
- 238000004440 column chromatography Methods 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- 101001042041 Bos taurus Isocitrate dehydrogenase [NAD] subunit beta, mitochondrial Proteins 0.000 description 15
- 208000003174 Brain Neoplasms Diseases 0.000 description 15
- 101000960234 Homo sapiens Isocitrate dehydrogenase [NADP] cytoplasmic Proteins 0.000 description 15
- 102100039905 Isocitrate dehydrogenase [NADP] cytoplasmic Human genes 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N hexane Substances CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- CMYCIRFZJKISNU-SSDOTTSWSA-N (4s)-3-(2-chloropyrimidin-4-yl)-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(Cl)=N1 CMYCIRFZJKISNU-SSDOTTSWSA-N 0.000 description 14
- 239000012043 crude product Substances 0.000 description 14
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 13
- 229960004132 diethyl ether Drugs 0.000 description 13
- 239000000706 filtrate Substances 0.000 description 13
- 239000012074 organic phase Substances 0.000 description 13
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 12
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- 206010018338 Glioma Diseases 0.000 description 12
- 208000032839 leukemia Diseases 0.000 description 12
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 12
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 12
- 206010025323 Lymphomas Diseases 0.000 description 11
- 125000004429 atom Chemical group 0.000 description 11
- 229910002092 carbon dioxide Inorganic materials 0.000 description 11
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 11
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 11
- 239000012047 saturated solution Substances 0.000 description 11
- 239000011734 sodium Substances 0.000 description 11
- 238000002560 therapeutic procedure Methods 0.000 description 11
- 208000032612 Glial tumor Diseases 0.000 description 10
- 208000017604 Hodgkin disease Diseases 0.000 description 10
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 10
- 206010039491 Sarcoma Diseases 0.000 description 10
- 208000005017 glioblastoma Diseases 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 10
- 206010003571 Astrocytoma Diseases 0.000 description 9
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 9
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 9
- 239000013058 crude material Substances 0.000 description 9
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 9
- 125000005842 heteroatom Chemical group 0.000 description 9
- 201000001441 melanoma Diseases 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 9
- 201000000849 skin cancer Diseases 0.000 description 9
- 239000012453 solvate Substances 0.000 description 9
- 206010009944 Colon cancer Diseases 0.000 description 8
- 201000006762 D-2-hydroxyglutaric aciduria Diseases 0.000 description 8
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 8
- SHGAZHPCJJPHSC-NUEINMDLSA-N Isotretinoin Chemical compound OC(=O)C=C(C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-NUEINMDLSA-N 0.000 description 8
- 208000000453 Skin Neoplasms Diseases 0.000 description 8
- 208000024770 Thyroid neoplasm Diseases 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 238000003818 flash chromatography Methods 0.000 description 8
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 8
- 238000004128 high performance liquid chromatography Methods 0.000 description 8
- 230000035935 pregnancy Effects 0.000 description 8
- 201000002510 thyroid cancer Diseases 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 7
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 7
- 206010060862 Prostate cancer Diseases 0.000 description 7
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 7
- 230000001154 acute effect Effects 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 208000029742 colonic neoplasm Diseases 0.000 description 7
- 125000000392 cycloalkenyl group Chemical group 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 201000005202 lung cancer Diseases 0.000 description 7
- 208000020816 lung neoplasm Diseases 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 6
- CESUXLKAADQNTB-SSDOTTSWSA-N 2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@](N)=O CESUXLKAADQNTB-SSDOTTSWSA-N 0.000 description 6
- KPGXRSRHYNQIFN-UHFFFAOYSA-L 2-oxoglutarate(2-) Chemical compound [O-]C(=O)CCC(=O)C([O-])=O KPGXRSRHYNQIFN-UHFFFAOYSA-L 0.000 description 6
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 6
- 208000005243 Chondrosarcoma Diseases 0.000 description 6
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 6
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 6
- 208000009277 Neuroectodermal Tumors Diseases 0.000 description 6
- 206010061332 Paraganglion neoplasm Diseases 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 229960000473 altretamine Drugs 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- 208000006990 cholangiocarcinoma Diseases 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 208000007312 paraganglioma Diseases 0.000 description 6
- 235000018102 proteins Nutrition 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 102200093149 rs77938727 Human genes 0.000 description 6
- 229910052938 sodium sulfate Inorganic materials 0.000 description 6
- 235000011152 sodium sulphate Nutrition 0.000 description 6
- 238000004808 supercritical fluid chromatography Methods 0.000 description 6
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 6
- PWCCTIRBZBAJSA-XJKSGUPXSA-N 4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]benzaldehyde Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(C=O)=CC=2)=N1 PWCCTIRBZBAJSA-XJKSGUPXSA-N 0.000 description 5
- 108010024976 Asparaginase Proteins 0.000 description 5
- 206010006187 Breast cancer Diseases 0.000 description 5
- 208000026310 Breast neoplasm Diseases 0.000 description 5
- 201000005262 Chondroma Diseases 0.000 description 5
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 5
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 5
- 206010014967 Ependymoma Diseases 0.000 description 5
- 201000008808 Fibrosarcoma Diseases 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 description 5
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 5
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 5
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 5
- 239000007789 gas Substances 0.000 description 5
- 230000002496 gastric effect Effects 0.000 description 5
- 201000005263 juxtacortical chondroma Diseases 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 5
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- VBIIOCJWMOULOM-WNWIJWBNSA-N n-[(1s)-1-(4-bromo-2-fluorophenyl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C1=CC=C(Br)C=C1F VBIIOCJWMOULOM-WNWIJWBNSA-N 0.000 description 5
- 238000007911 parenteral administration Methods 0.000 description 5
- 208000024975 periosteal chondroma Diseases 0.000 description 5
- 238000010791 quenching Methods 0.000 description 5
- 125000006413 ring segment Chemical group 0.000 description 5
- 102200069690 rs121913500 Human genes 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 208000011580 syndromic disease Diseases 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- NWHBYLZBPGSARH-JTQLQIEISA-N (4r)-3-(2-chloropyrimidin-4-yl)-4-phenyl-1,3-oxazolidin-2-one Chemical compound ClC1=NC=CC(N2C(OC[C@H]2C=2C=CC=CC=2)=O)=N1 NWHBYLZBPGSARH-JTQLQIEISA-N 0.000 description 4
- ZRHYDOZKIROIIX-CQSZACIVSA-N (4s)-4-(4-phenylphenyl)-1,3-oxazolidin-2-one Chemical compound C1OC(=O)N[C@H]1C1=CC=C(C=2C=CC=CC=2)C=C1 ZRHYDOZKIROIIX-CQSZACIVSA-N 0.000 description 4
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 4
- TVFLYOWMGULWTE-UHFFFAOYSA-N 1-bromo-4-(difluoromethyl)-2-fluorobenzene Chemical compound FC(F)C1=CC=C(Br)C(F)=C1 TVFLYOWMGULWTE-UHFFFAOYSA-N 0.000 description 4
- BQFZXBFMRXHADS-UHFFFAOYSA-N 4-phenyl-1,8-dioxa-3-azaspiro[4.5]decan-2-one Chemical compound C1COCCC21OC(=O)NC2C1=CC=CC=C1 BQFZXBFMRXHADS-UHFFFAOYSA-N 0.000 description 4
- 201000009030 Carcinoma Diseases 0.000 description 4
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 4
- 208000021309 Germ cell tumor Diseases 0.000 description 4
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 4
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 4
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 4
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 4
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 4
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 4
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 4
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 4
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 4
- 229940024606 amino acid Drugs 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 235000019441 ethanol Nutrition 0.000 description 4
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 4
- 239000012634 fragment Substances 0.000 description 4
- 150000004677 hydrates Chemical class 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 230000002267 hypothalamic effect Effects 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 230000000155 isotopic effect Effects 0.000 description 4
- 201000007270 liver cancer Diseases 0.000 description 4
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- XWCCTMBMQUCLSI-UHFFFAOYSA-N n-ethyl-n-propylpropan-1-amine Chemical compound CCCN(CC)CCC XWCCTMBMQUCLSI-UHFFFAOYSA-N 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 210000002784 stomach Anatomy 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- BVXQMBGFIRABTR-VIFPVBQESA-N tert-butyl n-[(1s)-1-(3-fluoro-4-formylphenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=C(C=O)C(F)=C1 BVXQMBGFIRABTR-VIFPVBQESA-N 0.000 description 4
- WOFCTRSNCBCJCV-VIFPVBQESA-N tert-butyl n-[(1s)-1-(3-hydroxyphenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=CC(O)=C1 WOFCTRSNCBCJCV-VIFPVBQESA-N 0.000 description 4
- OHOZMZZOCYTORQ-MAUKXSAKSA-N tert-butyl n-[(1s)-1-(4-bromophenyl)ethyl]-n-[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]carbamate Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N([C@@H](C)C=2C=CC(Br)=CC=2)C(=O)OC(C)(C)C)=N1 OHOZMZZOCYTORQ-MAUKXSAKSA-N 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 4
- QJXZUGFDCFVWNP-VIFPVBQESA-N (1s)-1-(4-pyrimidin-5-yloxyphenyl)ethanamine Chemical compound C1=CC([C@@H](N)C)=CC=C1OC1=CN=CN=C1 QJXZUGFDCFVWNP-VIFPVBQESA-N 0.000 description 3
- KJTZUOPRFPEYPX-YFKPBYRVSA-N (1s)-1-[4-(difluoromethyl)-2-fluorophenyl]ethanamine Chemical compound C[C@H](N)C1=CC=C(C(F)F)C=C1F KJTZUOPRFPEYPX-YFKPBYRVSA-N 0.000 description 3
- DRGRSSFCCDTWRF-CQSZACIVSA-N (2s)-2-amino-2-(4-phenylphenyl)ethanol Chemical compound C1=CC([C@@H](CO)N)=CC=C1C1=CC=CC=C1 DRGRSSFCCDTWRF-CQSZACIVSA-N 0.000 description 3
- XFHTWOCOWZUAOD-DPJUBJNGSA-N (ne)-2-methyl-n-[(4-pyrimidin-5-yloxyphenyl)methylidene]propane-2-sulfinamide Chemical compound C1=CC(/C=N/[S@](=O)C(C)(C)C)=CC=C1OC1=CN=CN=C1 XFHTWOCOWZUAOD-DPJUBJNGSA-N 0.000 description 3
- LHZWIUXIEXFXIL-ROBMAMTRSA-N (ne)-n-[[4-(difluoromethyl)-2-fluorophenyl]methylidene]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)\N=C\C1=CC=C(C(F)F)C=C1F LHZWIUXIEXFXIL-ROBMAMTRSA-N 0.000 description 3
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 3
- FNVZJXZTKQKXSN-UHFFFAOYSA-N 1-[(2-chloro-5-fluoropyrimidin-4-yl)amino]-2-methylpropan-2-ol Chemical compound CC(C)(O)CNC1=NC(Cl)=NC=C1F FNVZJXZTKQKXSN-UHFFFAOYSA-N 0.000 description 3
- KLSXATQRBXFQTO-UHFFFAOYSA-N 1-[4-(4-fluorophenoxy)pyrimidin-2-yl]ethanol Chemical compound CC(O)C1=NC=CC(OC=2C=CC(F)=CC=2)=N1 KLSXATQRBXFQTO-UHFFFAOYSA-N 0.000 description 3
- HMFQTHBOWURKDL-UHFFFAOYSA-N 1-[4-(4-fluorophenoxy)pyrimidin-2-yl]ethanone Chemical compound CC(=O)C1=NC=CC(OC=2C=CC(F)=CC=2)=N1 HMFQTHBOWURKDL-UHFFFAOYSA-N 0.000 description 3
- CGJTZXSYVQSSSF-UHFFFAOYSA-N 1-[5-[3-(trifluoromethyl)phenyl]pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1C1=CC=CC(C(F)(F)F)=C1 CGJTZXSYVQSSSF-UHFFFAOYSA-N 0.000 description 3
- XIRPMPKSZHNMST-UHFFFAOYSA-N 1-ethenyl-2-phenylbenzene Chemical group C=CC1=CC=CC=C1C1=CC=CC=C1 XIRPMPKSZHNMST-UHFFFAOYSA-N 0.000 description 3
- 229940044613 1-propanol Drugs 0.000 description 3
- BTTNYQZNBZNDOR-UHFFFAOYSA-N 2,4-dichloropyrimidine Chemical compound ClC1=CC=NC(Cl)=N1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 3
- KVACJTVQJOBLIB-UHFFFAOYSA-N 2-(1-azidoethyl)-4-(4-fluorophenoxy)pyrimidine Chemical compound [N-]=[N+]=NC(C)C1=NC=CC(OC=2C=CC(F)=CC=2)=N1 KVACJTVQJOBLIB-UHFFFAOYSA-N 0.000 description 3
- SMJJOVOLSHRMFK-BJDAYTSDSA-N 2-methyl-n-[(1s)-1-(4-pyrimidin-5-yloxyphenyl)ethyl]propane-2-sulfinamide Chemical compound C1=CC([C@@H](NS(=O)C(C)(C)C)C)=CC=C1OC1=CN=CN=C1 SMJJOVOLSHRMFK-BJDAYTSDSA-N 0.000 description 3
- KWACGKAMCAMFCX-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-4-phenyl-1,8-dioxa-3-azaspiro[4.5]decan-2-one Chemical compound ClC1=NC=CC(N2C(OC3(CCOCC3)C2C=2C=CC=CC=2)=O)=N1 KWACGKAMCAMFCX-UHFFFAOYSA-N 0.000 description 3
- BMBBBYKHOYWKSV-UHFFFAOYSA-N 4-(4-fluorophenoxy)pyrimidine-2-carbonitrile Chemical compound C1=CC(F)=CC=C1OC1=CC=NC(C#N)=N1 BMBBBYKHOYWKSV-UHFFFAOYSA-N 0.000 description 3
- UHIIEVZNTMPDKA-UHFFFAOYSA-N 4-(difluoromethyl)-2-fluorobenzaldehyde Chemical compound FC(F)C1=CC=C(C=O)C(F)=C1 UHIIEVZNTMPDKA-UHFFFAOYSA-N 0.000 description 3
- HTBWYNUHIPFGDA-UHFFFAOYSA-N 4-[amino(phenyl)methyl]oxan-4-ol Chemical compound C1COCCC1(O)C(N)C1=CC=CC=C1 HTBWYNUHIPFGDA-UHFFFAOYSA-N 0.000 description 3
- UFMGPLUKYBMTMX-UHFFFAOYSA-N 4-pyrimidin-5-yloxybenzaldehyde Chemical compound C1=CC(C=O)=CC=C1OC1=CN=CN=C1 UFMGPLUKYBMTMX-UHFFFAOYSA-N 0.000 description 3
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 3
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 3
- 102000015790 Asparaginase Human genes 0.000 description 3
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- 108010092160 Dactinomycin Proteins 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 3
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 3
- 239000012448 Lithium borohydride Substances 0.000 description 3
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 3
- 208000000172 Medulloblastoma Diseases 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- 201000010133 Oligodendroglioma Diseases 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 3
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000005587 bubbling Effects 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 3
- 208000002458 carcinoid tumor Diseases 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- PJGJQVRXEUVAFT-UHFFFAOYSA-N chloroiodomethane Chemical compound ClCI PJGJQVRXEUVAFT-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000007884 disintegrant Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- WGQSPCCCBKQNEV-UHFFFAOYSA-N ethyl 5,6-dichloropyridine-3-carboxylate Chemical compound CCOC(=O)C1=CN=C(Cl)C(Cl)=C1 WGQSPCCCBKQNEV-UHFFFAOYSA-N 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 210000004153 islets of langerhan Anatomy 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 201000003866 lung sarcoma Diseases 0.000 description 3
- 208000003747 lymphoid leukemia Diseases 0.000 description 3
- 208000030883 malignant astrocytoma Diseases 0.000 description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 229960004961 mechlorethamine Drugs 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- WZDFQSLQBLLQOG-VHSXEESVSA-N methyl (2s)-2-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]propanoate Chemical compound COC(=O)[C@H](C)NC1=NC=CC(N2C(OC[C@@H]2C(C)C)=O)=N1 WZDFQSLQBLLQOG-VHSXEESVSA-N 0.000 description 3
- LFNXGVQOIUBOOG-JTQLQIEISA-N methyl 4-[(1s)-1-[(2-methylpropan-2-yl)oxycarbonylamino]ethyl]benzoate Chemical compound COC(=O)C1=CC=C([C@H](C)NC(=O)OC(C)(C)C)C=C1 LFNXGVQOIUBOOG-JTQLQIEISA-N 0.000 description 3
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 3
- 201000005962 mycosis fungoides Diseases 0.000 description 3
- WXTQZYIVTDKTKN-WPCRTTGESA-N n-[(1s)-1-[4-(difluoromethyl)-2-fluorophenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C1=CC=C(C(F)F)C=C1F WXTQZYIVTDKTKN-WPCRTTGESA-N 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 3
- 239000008184 oral solid dosage form Substances 0.000 description 3
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 210000002307 prostate Anatomy 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 201000008205 supratentorial primitive neuroectodermal tumor Diseases 0.000 description 3
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 3
- ZSPHESMDSSMZSN-XUZZJYLKSA-N tert-butyl 2,2-dimethyl-4-[[4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]phenyl]methyl]piperazine-1-carboxylate Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CC(C)(C)N(C(=O)OC(C)(C)C)CC3)=CC=2)=N1 ZSPHESMDSSMZSN-XUZZJYLKSA-N 0.000 description 3
- NVZKZNRMBATQPU-UHFFFAOYSA-N tert-butyl n-(3-hydroxy-2-methylbutan-2-yl)carbamate Chemical compound CC(O)C(C)(C)NC(=O)OC(C)(C)C NVZKZNRMBATQPU-UHFFFAOYSA-N 0.000 description 3
- LOCVHXYTJHRXDL-JTQLQIEISA-N tert-butyl n-[(1s)-1-(5-phenyl-1,3,4-thiadiazol-2-yl)ethyl]carbamate Chemical compound S1C([C@@H](NC(=O)OC(C)(C)C)C)=NN=C1C1=CC=CC=C1 LOCVHXYTJHRXDL-JTQLQIEISA-N 0.000 description 3
- GRGLNDFPDWQFED-JTQLQIEISA-N tert-butyl n-[(1s)-1-[3-fluoro-4-[methoxy(methyl)carbamoyl]phenyl]ethyl]carbamate Chemical compound CON(C)C(=O)C1=CC=C([C@H](C)NC(=O)OC(C)(C)C)C=C1F GRGLNDFPDWQFED-JTQLQIEISA-N 0.000 description 3
- WNIOYQBDDWKYPN-JTQLQIEISA-N tert-butyl n-[(1s)-1-[4-(chloromethyl)phenyl]ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=C(CCl)C=C1 WNIOYQBDDWKYPN-JTQLQIEISA-N 0.000 description 3
- IKDSQAZPZFBIIK-JTQLQIEISA-N tert-butyl n-[(1s)-1-[4-(hydroxymethyl)phenyl]ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=C(CO)C=C1 IKDSQAZPZFBIIK-JTQLQIEISA-N 0.000 description 3
- BCGANEVUMGTAKD-QGZVFWFLSA-N tert-butyl n-[(1s)-2-hydroxy-1-(4-phenylphenyl)ethyl]carbamate Chemical compound C1=CC([C@@H](CO)NC(=O)OC(C)(C)C)=CC=C1C1=CC=CC=C1 BCGANEVUMGTAKD-QGZVFWFLSA-N 0.000 description 3
- 210000001685 thyroid gland Anatomy 0.000 description 3
- 229960001727 tretinoin Drugs 0.000 description 3
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 3
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 210000000239 visual pathway Anatomy 0.000 description 3
- 230000004400 visual pathway Effects 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- KETZXWRRCDXDFT-YFKPBYRVSA-N (1s)-1-(2,3-difluorophenyl)ethanamine Chemical compound C[C@H](N)C1=CC=CC(F)=C1F KETZXWRRCDXDFT-YFKPBYRVSA-N 0.000 description 2
- KRCZPHITDAGPPK-NSHDSACASA-N (1s)-1-(3-phenoxyphenyl)ethanamine Chemical compound C[C@H](N)C1=CC=CC(OC=2C=CC=CC=2)=C1 KRCZPHITDAGPPK-NSHDSACASA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 2
- QQOAHLJDKWZJPD-ZCFIWIBFSA-N (4r)-4-(2-methylpropyl)-1,3-oxazolidin-2-one Chemical compound CC(C)C[C@@H]1COC(=O)N1 QQOAHLJDKWZJPD-ZCFIWIBFSA-N 0.000 description 2
- ZGSKFDUQVOXAOF-MRXNPFEDSA-N (4s)-3-(2-chloropyrimidin-4-yl)-4-(4-phenylphenyl)-1,3-oxazolidin-2-one Chemical compound ClC1=NC=CC(N2C(OC[C@@H]2C=2C=CC(=CC=2)C=2C=CC=CC=2)=O)=N1 ZGSKFDUQVOXAOF-MRXNPFEDSA-N 0.000 description 2
- KESKTKDGWWKBBO-SSDOTTSWSA-N (4s)-3-(2-fluoropyrimidin-4-yl)-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(F)=N1 KESKTKDGWWKBBO-SSDOTTSWSA-N 0.000 description 2
- NEKGXIIMUAXULQ-UUSAFJCLSA-N (4s)-3-[2-[1-(4-piperidin-1-ylphenyl)ethylamino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(NC(C)C=2C=CC(=CC=2)N2CCCCC2)=N1 NEKGXIIMUAXULQ-UUSAFJCLSA-N 0.000 description 2
- NKJZELHWGLSGFP-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-(4-bromophenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(Br)=CC=2)=N1 NKJZELHWGLSGFP-SWLSCSKDSA-N 0.000 description 2
- OABBAWSEQBHGDN-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-(4-hydroxyphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(O)=CC=2)=N1 OABBAWSEQBHGDN-SWLSCSKDSA-N 0.000 description 2
- UXYGHYPCCFQPOI-XJKSGUPXSA-N (4s)-3-[2-[[(1s)-1-[4-(hydroxymethyl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CO)=CC=2)=N1 UXYGHYPCCFQPOI-XJKSGUPXSA-N 0.000 description 2
- RJPUQNWYWITUJJ-AFYYWNPRSA-N (4s)-4-propan-2-yl-3-[2-[(2,2,2-trifluoro-1-phenylethyl)amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(NC(C=2C=CC=CC=2)C(F)(F)F)=N1 RJPUQNWYWITUJJ-AFYYWNPRSA-N 0.000 description 2
- VNTHYLVDGVBPOU-QQYBVWGSSA-N (7s,9s)-9-acetyl-7-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 VNTHYLVDGVBPOU-QQYBVWGSSA-N 0.000 description 2
- SGMRADNZPJGFBY-JBNJFFQFSA-N (NE,R)-2-methyl-N-[(5-methyl-1H-pyrazol-4-yl)methylidene]propane-2-sulfinamide Chemical compound CC1=NNC=C1\C=N\[S@](=O)C(C)(C)C SGMRADNZPJGFBY-JBNJFFQFSA-N 0.000 description 2
- CEPGPYNLLCJZGY-SNQWNFELSA-N (ne)-n-[(2,3-difluorophenyl)methylidene]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)\N=C\C1=CC=CC(F)=C1F CEPGPYNLLCJZGY-SNQWNFELSA-N 0.000 description 2
- OFQWCKSOOWFGBM-SNQWNFELSA-N (ne)-n-[(4-bromo-2-fluorophenyl)methylidene]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)\N=C\C1=CC=C(Br)C=C1F OFQWCKSOOWFGBM-SNQWNFELSA-N 0.000 description 2
- UJUFOUVXOUYYRG-UHFFFAOYSA-N 1-(3,4-dichlorophenyl)ethanamine Chemical compound CC(N)C1=CC=C(Cl)C(Cl)=C1 UJUFOUVXOUYYRG-UHFFFAOYSA-N 0.000 description 2
- KATCFWMBDRASHY-UHFFFAOYSA-N 1-(4-phenylphenyl)ethanamine;hydrochloride Chemical compound Cl.C1=CC(C(N)C)=CC=C1C1=CC=CC=C1 KATCFWMBDRASHY-UHFFFAOYSA-N 0.000 description 2
- RZYYUYOKHGRGLE-UHFFFAOYSA-N 1-[4-(4-fluorophenoxy)pyrimidin-2-yl]ethanamine Chemical compound CC(N)C1=NC=CC(OC=2C=CC(F)=CC=2)=N1 RZYYUYOKHGRGLE-UHFFFAOYSA-N 0.000 description 2
- HJXYKQDGYQLTQQ-UHFFFAOYSA-N 1-[5-(4-fluorophenoxy)pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1OC1=CC=C(F)C=C1 HJXYKQDGYQLTQQ-UHFFFAOYSA-N 0.000 description 2
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 2
- UCNGGGYMLHAMJG-UHFFFAOYSA-N 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound C1=NN(C)C=C1B1OC(C)(C)C(C)(C)O1 UCNGGGYMLHAMJG-UHFFFAOYSA-N 0.000 description 2
- ZEMZPXWZVTUONV-UHFFFAOYSA-N 2-(2-dicyclohexylphosphanylphenyl)-n,n-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 ZEMZPXWZVTUONV-UHFFFAOYSA-N 0.000 description 2
- HUHGPYXAVBJSJV-UHFFFAOYSA-N 2-[3,5-bis(2-hydroxyethyl)-1,3,5-triazinan-1-yl]ethanol Chemical compound OCCN1CN(CCO)CN(CCO)C1 HUHGPYXAVBJSJV-UHFFFAOYSA-N 0.000 description 2
- WJLYSQLBESVSSC-QMMMGPOBSA-N 2-chloro-4-[(1s)-1-[(2-methylpropan-2-yl)oxycarbonylamino]ethyl]benzoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=C(C(O)=O)C(Cl)=C1 WJLYSQLBESVSSC-QMMMGPOBSA-N 0.000 description 2
- BKUKCRBAZPIOTJ-QMMMGPOBSA-N 2-fluoro-4-[(1s)-1-[(2-methylpropan-2-yl)oxycarbonylamino]ethyl]benzoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=C(C(O)=O)C(F)=C1 BKUKCRBAZPIOTJ-QMMMGPOBSA-N 0.000 description 2
- MFNXWZGIFWJHMI-UHFFFAOYSA-N 2-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)NC(C)(C)C(O)=O MFNXWZGIFWJHMI-UHFFFAOYSA-N 0.000 description 2
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 2
- AKSMSHPCIZQROJ-VXJOIVPMSA-N 2-methyl-n-[(1s)-1-(5-methyl-1h-pyrazol-4-yl)ethyl]propane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C=1C=NNC=1C AKSMSHPCIZQROJ-VXJOIVPMSA-N 0.000 description 2
- FJKBYGGOTYJXKF-UHFFFAOYSA-N 3-(2-chloro-5-fluoropyrimidin-4-yl)-5,5-dimethyl-1,3-oxazolidin-2-one Chemical compound O=C1OC(C)(C)CN1C1=NC(Cl)=NC=C1F FJKBYGGOTYJXKF-UHFFFAOYSA-N 0.000 description 2
- QULBHIBEAZTUQA-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3-oxazolidin-2-one Chemical compound CC1(C)NC(=O)OC1(C)C QULBHIBEAZTUQA-UHFFFAOYSA-N 0.000 description 2
- JPYDCXGSUMJQQX-QMMMGPOBSA-N 4,6-difluoro-n-[(1s)-1-phenylethyl]pyrimidin-2-amine Chemical compound N([C@@H](C)C=1C=CC=CC=1)C1=NC(F)=CC(F)=N1 JPYDCXGSUMJQQX-QMMMGPOBSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- RHMPLDJJXGPMEX-UHFFFAOYSA-N 4-fluorophenol Chemical compound OC1=CC=C(F)C=C1 RHMPLDJJXGPMEX-UHFFFAOYSA-N 0.000 description 2
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 2
- MLTPFSSREOWLQN-UHFFFAOYSA-N 5-chloro-6-(1,1-difluoroethyl)pyridine-3-carbaldehyde Chemical compound CC(F)(F)C1=NC=C(C=O)C=C1Cl MLTPFSSREOWLQN-UHFFFAOYSA-N 0.000 description 2
- UYLUGCQAIWKVHI-UHFFFAOYSA-N 5-chloro-6-(2,2,2-trifluoroethoxy)pyridine-3-carbaldehyde Chemical compound FC(F)(F)COC1=NC=C(C=O)C=C1Cl UYLUGCQAIWKVHI-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 206010060971 Astrocytoma malignant Diseases 0.000 description 2
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- 206010004593 Bile duct cancer Diseases 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 206010006143 Brain stem glioma Diseases 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 206010007275 Carcinoid tumour Diseases 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- 101000960235 Dictyostelium discoideum Isocitrate dehydrogenase [NADP] cytoplasmic Proteins 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 2
- 208000012468 Ewing sarcoma/peripheral primitive neuroectodermal tumor Diseases 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 206010061252 Intraocular melanoma Diseases 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 2
- 206010023825 Laryngeal cancer Diseases 0.000 description 2
- 206010025557 Malignant fibrous histiocytoma of bone Diseases 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- 229920000881 Modified starch Polymers 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 2
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 2
- SHGAZHPCJJPHSC-UHFFFAOYSA-N Panrexin Chemical compound OC(=O)C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-UHFFFAOYSA-N 0.000 description 2
- 229910002666 PdCl2 Inorganic materials 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 description 2
- 206010061934 Salivary gland cancer Diseases 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 2
- 240000006394 Sorghum bicolor Species 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 2
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 201000009365 Thymic carcinoma Diseases 0.000 description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 2
- 201000005969 Uveal melanoma Diseases 0.000 description 2
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 2
- JMKXEKPBEDYIFU-UHFFFAOYSA-N [5-chloro-6-(1,1-difluoroethyl)pyridin-3-yl]methanol Chemical compound CC(F)(F)C1=NC=C(CO)C=C1Cl JMKXEKPBEDYIFU-UHFFFAOYSA-N 0.000 description 2
- IQJKMTYNYLCJHN-UHFFFAOYSA-N [5-chloro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl]methanol Chemical compound OCC1=CN=C(OCC(F)(F)F)C(Cl)=C1 IQJKMTYNYLCJHN-UHFFFAOYSA-N 0.000 description 2
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 2
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- VPEPQDBAIMZCGV-UHFFFAOYSA-N boron;5-ethyl-2-methylpyridine Chemical compound [B].CCC1=CC=C(C)N=C1 VPEPQDBAIMZCGV-UHFFFAOYSA-N 0.000 description 2
- LTEJRLHKIYCEOX-OCCSQVGLSA-N brivanib alaninate Chemical compound C1=C2NC(C)=CC2=C(F)C(OC2=NC=NN3C=C(C(=C32)C)OC[C@@H](C)OC(=O)[C@H](C)N)=C1 LTEJRLHKIYCEOX-OCCSQVGLSA-N 0.000 description 2
- 229950005993 brivanib alaninate Drugs 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- HGXJOXHYPGNVNK-UHFFFAOYSA-N butane;ethenoxyethane;tin Chemical compound CCCC[Sn](CCCC)(CCCC)C(=C)OCC HGXJOXHYPGNVNK-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- STNNHWPJRRODGI-UHFFFAOYSA-N carbonic acid;n,n-diethylethanamine Chemical compound [O-]C([O-])=O.CC[NH+](CC)CC.CC[NH+](CC)CC STNNHWPJRRODGI-UHFFFAOYSA-N 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 201000007335 cerebellar astrocytoma Diseases 0.000 description 2
- 208000030239 cerebral astrocytoma Diseases 0.000 description 2
- 230000002490 cerebral effect Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 229960002436 cladribine Drugs 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 229910000365 copper sulfate Inorganic materials 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 239000002254 cytotoxic agent Substances 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 150000001975 deuterium Chemical group 0.000 description 2
- HQWPLXHWEZZGKY-UHFFFAOYSA-N diethylzinc Chemical compound CC[Zn]CC HQWPLXHWEZZGKY-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 201000004101 esophageal cancer Diseases 0.000 description 2
- JJJHITSREACPMI-UHFFFAOYSA-N ethyl 5-chloro-6-(1,1-difluoroethyl)pyridine-3-carboxylate Chemical compound CCOC(=O)C1=CN=C(C(C)(F)F)C(Cl)=C1 JJJHITSREACPMI-UHFFFAOYSA-N 0.000 description 2
- TVRPISOHRPXBRP-UHFFFAOYSA-N ethyl 5-chloro-6-(2,2,2-trifluoroethoxy)pyridine-3-carboxylate Chemical compound CCOC(=O)C1=CN=C(OCC(F)(F)F)C(Cl)=C1 TVRPISOHRPXBRP-UHFFFAOYSA-N 0.000 description 2
- FKONNDVONXFZOL-UHFFFAOYSA-N ethyl 6-acetyl-5-chloropyridine-3-carboxylate Chemical compound CCOC(=O)C1=CN=C(C(C)=O)C(Cl)=C1 FKONNDVONXFZOL-UHFFFAOYSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 2
- 229960000752 etoposide phosphate Drugs 0.000 description 2
- 208000024519 eye neoplasm Diseases 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 150000004795 grignard reagents Chemical class 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 229940003183 hexalen Drugs 0.000 description 2
- 208000029824 high grade glioma Diseases 0.000 description 2
- WJRBRSLFGCUECM-UHFFFAOYSA-N hydantoin Chemical compound O=C1CNC(=O)N1 WJRBRSLFGCUECM-UHFFFAOYSA-N 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 229960005280 isotretinoin Drugs 0.000 description 2
- 210000000244 kidney pelvis Anatomy 0.000 description 2
- 206010023841 laryngeal neoplasm Diseases 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 201000011614 malignant glioma Diseases 0.000 description 2
- 208000006178 malignant mesothelioma Diseases 0.000 description 2
- 229960002510 mandelic acid Drugs 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 229960001924 melphalan Drugs 0.000 description 2
- 206010027191 meningioma Diseases 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- OXGSYLWMSHXMLT-UHFFFAOYSA-N methyl 2-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound COC(=O)C(C)(C)NC(=O)OC(C)(C)C OXGSYLWMSHXMLT-UHFFFAOYSA-N 0.000 description 2
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 208000025113 myeloid leukemia Diseases 0.000 description 2
- CUBNTJXLXXSMMG-RCDICMHDSA-N n-[(1s)-1-(1-benzyl-3-methylpyrazol-4-yl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound N1=C(C)C([C@@H](N[S@](=O)C(C)(C)C)C)=CN1CC1=CC=CC=C1 CUBNTJXLXXSMMG-RCDICMHDSA-N 0.000 description 2
- MTRYWPCUDNJHSD-WNWIJWBNSA-N n-[(1s)-1-(2,3-difluorophenyl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C1=CC=CC(F)=C1F MTRYWPCUDNJHSD-WNWIJWBNSA-N 0.000 description 2
- ZKZJICAAYKEGIV-JEOXALJRSA-N n-[(1s)-1-(2-fluoro-4-prop-1-en-2-ylphenyl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C1=CC=C(C(C)=C)C=C1F ZKZJICAAYKEGIV-JEOXALJRSA-N 0.000 description 2
- RZOYZXHYRYEKHP-JEOXALJRSA-N n-[(1s)-1-(2-fluoro-4-propan-2-ylphenyl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)C1=CC=C([C@H](C)N[S@](=O)C(C)(C)C)C(F)=C1 RZOYZXHYRYEKHP-JEOXALJRSA-N 0.000 description 2
- NMCCHQSXZISLHB-APBUJDDRSA-N n-[(1s)-1-(4-cyclopropyl-2-fluorophenyl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound C1=C(F)C([C@@H](N[S@](=O)C(C)(C)C)C)=CC=C1C1CC1 NMCCHQSXZISLHB-APBUJDDRSA-N 0.000 description 2
- BLEGNILWSMOBMQ-XTZNXHDOSA-N n-[(1s)-1-(6-cyclopropylpyridin-3-yl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound N1=CC([C@@H](N[S@](=O)C(C)(C)C)C)=CC=C1C1CC1 BLEGNILWSMOBMQ-XTZNXHDOSA-N 0.000 description 2
- SHUNSRMORFMTKU-JEOXALJRSA-N n-[(1s)-1-(6-tert-butylpyridin-3-yl)ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C1=CC=C(C(C)(C)C)N=C1 SHUNSRMORFMTKU-JEOXALJRSA-N 0.000 description 2
- GHDRPMCEOLUCNL-PRWKNARSSA-N n-[(1s)-1-[2-fluoro-4-(1-methylcyclopropyl)phenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound C1=C(F)C([C@@H](N[S@](=O)C(C)(C)C)C)=CC=C1C1(C)CC1 GHDRPMCEOLUCNL-PRWKNARSSA-N 0.000 description 2
- LSRLPJUERSLJKX-KPWVOAKYSA-N n-[(1s)-1-[2-fluoro-4-(1-methylpyrazol-4-yl)phenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound C1=C(F)C([C@@H](N[S@](=O)C(C)(C)C)C)=CC=C1C1=CN(C)N=C1 LSRLPJUERSLJKX-KPWVOAKYSA-N 0.000 description 2
- KECUKBMYAYRDNG-FFVOIRBGSA-N n-[(1s)-1-[2-fluoro-4-(trifluoromethyl)phenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C1=CC=C(C(F)(F)F)C=C1F KECUKBMYAYRDNG-FFVOIRBGSA-N 0.000 description 2
- IGFHADHVSVZEQN-WIUDPPPLSA-N n-[(1s)-1-[4-(1-cyanocyclopropyl)-2-fluorophenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound C1=C(F)C([C@@H](N[S@](=O)C(C)(C)C)C)=CC=C1C1(C#N)CC1 IGFHADHVSVZEQN-WIUDPPPLSA-N 0.000 description 2
- ZCZWWLUXKFOKQV-AMXDTQDGSA-N n-[(1s)-1-[4-(1-ethoxycyclopropyl)-2-fluorophenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound C=1C=C([C@H](C)N[S@](=O)C(C)(C)C)C(F)=CC=1C1(OCC)CC1 ZCZWWLUXKFOKQV-AMXDTQDGSA-N 0.000 description 2
- HYUKXASYENDFPT-WIUDPPPLSA-N n-[(1s)-1-[4-(1-ethoxyethenyl)-2-fluorophenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound CCOC(=C)C1=CC=C([C@H](C)N[S@](=O)C(C)(C)C)C(F)=C1 HYUKXASYENDFPT-WIUDPPPLSA-N 0.000 description 2
- 208000018795 nasal cavity and paranasal sinus carcinoma Diseases 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 201000008106 ocular cancer Diseases 0.000 description 2
- 201000002575 ocular melanoma Diseases 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- 230000002611 ovarian Effects 0.000 description 2
- 125000001715 oxadiazolyl group Chemical group 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- WBXPDJSOTKVWSJ-ZDUSSCGKSA-N pemetrexed Chemical compound C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 WBXPDJSOTKVWSJ-ZDUSSCGKSA-N 0.000 description 2
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 2
- 210000002381 plasma Anatomy 0.000 description 2
- 208000010626 plasma cell neoplasm Diseases 0.000 description 2
- 229940063179 platinol Drugs 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- 239000011698 potassium fluoride Substances 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 2
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 2
- 108091006082 receptor inhibitors Proteins 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 210000004872 soft tissue Anatomy 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 229940032147 starch Drugs 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 229960004964 temozolomide Drugs 0.000 description 2
- DILZCRKRYCQDAC-UHFFFAOYSA-N tert-butyl n-(2-methyl-3-oxobutan-2-yl)carbamate Chemical compound CC(=O)C(C)(C)NC(=O)OC(C)(C)C DILZCRKRYCQDAC-UHFFFAOYSA-N 0.000 description 2
- AKRFQGJUPWHOOT-UHFFFAOYSA-N tert-butyl n-(3-hydroxy-2,3-dimethylbutan-2-yl)carbamate Chemical compound CC(C)(C)OC(=O)NC(C)(C)C(C)(C)O AKRFQGJUPWHOOT-UHFFFAOYSA-N 0.000 description 2
- HWXIAJBLXAISMQ-AWEZNQCLSA-N tert-butyl n-[(1s)-1-(3-cyclohexyloxyphenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=CC(OC2CCCCC2)=C1 HWXIAJBLXAISMQ-AWEZNQCLSA-N 0.000 description 2
- AUYOTVVWLYJVEW-ZDUSSCGKSA-N tert-butyl n-[(1s)-1-(3-cyclopentyloxyphenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=CC(OC2CCCC2)=C1 AUYOTVVWLYJVEW-ZDUSSCGKSA-N 0.000 description 2
- KXJPXAVUCDVRKS-LBPRGKRZSA-N tert-butyl n-[(1s)-1-[3-chloro-4-(cyclopentylcarbamoyl)phenyl]ethyl]carbamate Chemical compound ClC1=CC([C@@H](NC(=O)OC(C)(C)C)C)=CC=C1C(=O)NC1CCCC1 KXJPXAVUCDVRKS-LBPRGKRZSA-N 0.000 description 2
- INCYWHYASZKZBO-HNNXBMFYSA-N tert-butyl n-[(1s)-1-[3-fluoro-4-[(3,3,4-trimethylpiperazin-1-yl)methyl]phenyl]ethyl]carbamate Chemical compound FC1=CC([C@@H](NC(=O)OC(C)(C)C)C)=CC=C1CN1CC(C)(C)N(C)CC1 INCYWHYASZKZBO-HNNXBMFYSA-N 0.000 description 2
- ITMAASUVPAQYAK-PGRDOPGGSA-N tert-butyl n-[(1s)-1-[4-(1-methylpyrazol-4-yl)phenyl]ethyl]-n-[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]carbamate Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N([C@@H](C)C=2C=CC(=CC=2)C2=CN(C)N=C2)C(=O)OC(C)(C)C)=N1 ITMAASUVPAQYAK-PGRDOPGGSA-N 0.000 description 2
- GCTCFRAUQWFBTQ-AWEZNQCLSA-N tert-butyl n-[(1s)-1-[4-(6,8-dihydro-5h-[1,2,4]triazolo[4,3-a]pyrazin-7-ylmethyl)phenyl]ethyl]carbamate Chemical compound C1=CC([C@@H](NC(=O)OC(C)(C)C)C)=CC=C1CN1CC2=NN=CN2CC1 GCTCFRAUQWFBTQ-AWEZNQCLSA-N 0.000 description 2
- CHKKXQNAVZACJY-GBXCKJPGSA-N tert-butyl n-[(1s)-1-[4-(cyclohexanecarbonylamino)phenyl]ethyl]-n-[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]carbamate Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N([C@@H](C)C=2C=CC(NC(=O)C3CCCCC3)=CC=2)C(=O)OC(C)(C)C)=N1 CHKKXQNAVZACJY-GBXCKJPGSA-N 0.000 description 2
- FRRCSAZNGYCTLR-MRVPVSSYSA-N tert-butyl n-[(1s)-1-cyclopropyl-2-hydroxyethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@H](CO)C1CC1 FRRCSAZNGYCTLR-MRVPVSSYSA-N 0.000 description 2
- CNCQQHJVACAXNZ-JTQLQIEISA-N tert-butyl n-[(2s)-1-(2-benzoylhydrazinyl)-1-oxopropan-2-yl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C(=O)NNC(=O)C1=CC=CC=C1 CNCQQHJVACAXNZ-JTQLQIEISA-N 0.000 description 2
- FRNDCHOMCRLCJX-YFKPBYRVSA-N tert-butyl n-[(2s)-1-hydrazinyl-1-oxopropan-2-yl]carbamate Chemical compound NNC(=O)[C@H](C)NC(=O)OC(C)(C)C FRNDCHOMCRLCJX-YFKPBYRVSA-N 0.000 description 2
- RMDGQPFDHSRKFU-VIFPVBQESA-N tert-butyl n-[(3s)-2-methyl-4-oxopentan-3-yl]carbamate Chemical compound CC(C)[C@@H](C(C)=O)NC(=O)OC(C)(C)C RMDGQPFDHSRKFU-VIFPVBQESA-N 0.000 description 2
- XRIPRJRSUDUZQF-UHFFFAOYSA-N tert-butyl n-[1-[methoxy(methyl)amino]-2-methyl-1-oxopropan-2-yl]carbamate Chemical compound CON(C)C(=O)C(C)(C)NC(=O)OC(C)(C)C XRIPRJRSUDUZQF-UHFFFAOYSA-N 0.000 description 2
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- 206010044412 transitional cell carcinoma Diseases 0.000 description 2
- 208000018417 undifferentiated high grade pleomorphic sarcoma of bone Diseases 0.000 description 2
- 210000000626 ureter Anatomy 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- RQEUFEKYXDPUSK-ZETCQYMHSA-N (1S)-1-phenylethanamine Chemical compound C[C@H](N)C1=CC=CC=C1 RQEUFEKYXDPUSK-ZETCQYMHSA-N 0.000 description 1
- LHXIMNAJBPVMKU-PPHPATTJSA-N (1s)-1-(1-benzyl-3-methylpyrazol-4-yl)ethanamine;hydrochloride Chemical compound Cl.N1=C(C)C([C@@H](N)C)=CN1CC1=CC=CC=C1 LHXIMNAJBPVMKU-PPHPATTJSA-N 0.000 description 1
- KDAJNNPXBWHWAY-NSHDSACASA-N (1s)-1-(3-cyclohexyloxyphenyl)ethanamine Chemical compound C[C@H](N)C1=CC=CC(OC2CCCCC2)=C1 KDAJNNPXBWHWAY-NSHDSACASA-N 0.000 description 1
- KEPXHYJZNKVHME-MERQFXBCSA-N (1s)-1-(3-cyclohexyloxyphenyl)ethanamine;hydrochloride Chemical compound Cl.C[C@H](N)C1=CC=CC(OC2CCCCC2)=C1 KEPXHYJZNKVHME-MERQFXBCSA-N 0.000 description 1
- RYPHNGPMFRLDAP-JTQLQIEISA-N (1s)-1-(3-cyclopentyloxyphenyl)ethanamine Chemical compound C[C@H](N)C1=CC=CC(OC2CCCC2)=C1 RYPHNGPMFRLDAP-JTQLQIEISA-N 0.000 description 1
- CYVBLVQCGGRCBA-PPHPATTJSA-N (1s)-1-(3-cyclopentyloxyphenyl)ethanamine;hydrochloride Chemical compound Cl.C[C@H](N)C1=CC=CC(OC2CCCC2)=C1 CYVBLVQCGGRCBA-PPHPATTJSA-N 0.000 description 1
- SOZMSEPDYJGBEK-LURJTMIESA-N (1s)-1-(4-bromophenyl)ethanamine Chemical compound C[C@H](N)C1=CC=C(Br)C=C1 SOZMSEPDYJGBEK-LURJTMIESA-N 0.000 description 1
- PCMZVOJIMQGBCW-MERQFXBCSA-N (1s)-1-(4-phenoxyphenyl)ethanamine;hydrochloride Chemical compound Cl.C1=CC([C@@H](N)C)=CC=C1OC1=CC=CC=C1 PCMZVOJIMQGBCW-MERQFXBCSA-N 0.000 description 1
- LDHQMPZBBRWDJH-ZETCQYMHSA-N (1s)-1-(5-phenyl-1,3,4-thiadiazol-2-yl)ethanamine Chemical compound S1C([C@@H](N)C)=NN=C1C1=CC=CC=C1 LDHQMPZBBRWDJH-ZETCQYMHSA-N 0.000 description 1
- VPSSPAXIFBTOHY-ZCFIWIBFSA-N (2r)-2-amino-4-methylpentan-1-ol Chemical compound CC(C)C[C@@H](N)CO VPSSPAXIFBTOHY-ZCFIWIBFSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- HIHKCTSALSNXCH-DTWKUNHWSA-N (2s)-2-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]propanehydrazide Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C(=O)NN)=N1 HIHKCTSALSNXCH-DTWKUNHWSA-N 0.000 description 1
- QFVJNEASAAJIDF-ZETCQYMHSA-N (2s)-2-cyclopropyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)C1CC1 QFVJNEASAAJIDF-ZETCQYMHSA-N 0.000 description 1
- SZXBQTSZISFIAO-ZETCQYMHSA-N (2s)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoic acid Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)OC(C)(C)C SZXBQTSZISFIAO-ZETCQYMHSA-N 0.000 description 1
- WSADMGUUGLMCER-MGPUTAFESA-N (4S)-3-[2-[[(1S)-1-[4-(4-oxo-3H-pyridin-2-yl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C(C)(C)[C@@H]1N(C(OC1)=O)C1=NC(=NC=C1)N[C@@H](C)C1=CC=C(C=C1)C1=NC=CC(C1)=O WSADMGUUGLMCER-MGPUTAFESA-N 0.000 description 1
- QOGOAGWFCKVFDC-UGSOOPFHSA-N (4r)-3-[2-[[(1s)-1-(3,4-dichlorophenyl)ethyl]amino]pyrimidin-4-yl]-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1([C@H]2N(C(OC2)=O)C=2C=CN=C(N=2)N[C@@H](C)C=2C=C(Cl)C(Cl)=CC=2)=CC=CC=C1 QOGOAGWFCKVFDC-UGSOOPFHSA-N 0.000 description 1
- LVPITHJLZOMCTJ-HRAATJIYSA-N (4r)-4-(4-methoxyphenyl)-5,5-dimethyl-3-[2-[[(1s)-1-phenylethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1=CC(OC)=CC=C1[C@@H]1C(C)(C)OC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC=CC=2)=N1 LVPITHJLZOMCTJ-HRAATJIYSA-N 0.000 description 1
- QDMNNMIOWVJVLY-QMMMGPOBSA-N (4r)-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1OC(=O)N[C@@H]1C1=CC=CC=C1 QDMNNMIOWVJVLY-QMMMGPOBSA-N 0.000 description 1
- SAPIHOMJNKGPFR-DVECYGJZSA-N (4r)-4-phenyl-3-[2-[[(1r)-1-(4-phenylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1([C@H]2N(C(OC2)=O)C=2C=CN=C(N=2)N[C@H](C)C=2C=CC(=CC=2)C=2C=CC=CC=2)=CC=CC=C1 SAPIHOMJNKGPFR-DVECYGJZSA-N 0.000 description 1
- SAPIHOMJNKGPFR-CYFREDJKSA-N (4r)-4-phenyl-3-[2-[[(1s)-1-(4-phenylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1([C@H]2N(C(OC2)=O)C=2C=CN=C(N=2)N[C@@H](C)C=2C=CC(=CC=2)C=2C=CC=CC=2)=CC=CC=C1 SAPIHOMJNKGPFR-CYFREDJKSA-N 0.000 description 1
- NHFLDELZXRJYMK-LBPRGKRZSA-N (4s)-3-(2-chloropyrimidin-4-yl)-5,5-dimethyl-4-phenyl-1,3-oxazolidin-2-one Chemical compound C1([C@@H]2N(C(=O)OC2(C)C)C=2N=C(Cl)N=CC=2)=CC=CC=C1 NHFLDELZXRJYMK-LBPRGKRZSA-N 0.000 description 1
- DLOGZKKGKKWDLN-MRVPVSSYSA-N (4s)-3-(2-fluoropyrimidin-4-yl)-4-propan-2-yl-1,3-oxazolidine Chemical compound CC(C)[C@H]1COCN1C1=CC=NC(F)=N1 DLOGZKKGKKWDLN-MRVPVSSYSA-N 0.000 description 1
- JBDRREUKUCSNKV-AWKYBWMHSA-N (4s)-3-[2-[1-(2,3-dihydro-1,4-benzodioxin-6-yl)ethylamino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(NC(C)C=2C=C3OCCOC3=CC=2)=N1 JBDRREUKUCSNKV-AWKYBWMHSA-N 0.000 description 1
- NIXWPEDNSULKIR-AWKYBWMHSA-N (4s)-3-[2-[1-(3,4-dimethoxyphenyl)ethylamino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=C(OC)C(OC)=CC=C1C(C)NC1=NC=CC(N2C(OC[C@@H]2C(C)C)=O)=N1 NIXWPEDNSULKIR-AWKYBWMHSA-N 0.000 description 1
- JBDRREUKUCSNKV-UKRRQHHQSA-N (4s)-3-[2-[[(1r)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@H](C)C=2C=C3OCCOC3=CC=2)=N1 JBDRREUKUCSNKV-UKRRQHHQSA-N 0.000 description 1
- NIXWPEDNSULKIR-UKRRQHHQSA-N (4s)-3-[2-[[(1r)-1-(3,4-dimethoxyphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=C(OC)C(OC)=CC=C1[C@@H](C)NC1=NC=CC(N2C(OC[C@@H]2C(C)C)=O)=N1 NIXWPEDNSULKIR-UKRRQHHQSA-N 0.000 description 1
- RAJZEQSIULHTBW-DYESRHJHSA-N (4s)-3-[2-[[(1r)-1-(4-phenylphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@H](C)C=2C=CC(=CC=2)C=2C=CC=CC=2)=N1 RAJZEQSIULHTBW-DYESRHJHSA-N 0.000 description 1
- QFATVDJWCRBEEE-RDTXWAMCSA-N (4s)-3-[2-[[(1r)-1-[1-(4-fluorophenyl)pyrazol-4-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@H](C)C2=CN(N=C2)C=2C=CC(F)=CC=2)=N1 QFATVDJWCRBEEE-RDTXWAMCSA-N 0.000 description 1
- LSXSGJKNEVEKER-DNVCBOLYSA-N (4s)-3-[2-[[(1r)-1-[1-(4-methoxyphenyl)pyrazol-4-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=CC(OC)=CC=C1N1N=CC([C@@H](C)NC=2N=C(C=CN=2)N2C(OC[C@@H]2C(C)C)=O)=C1 LSXSGJKNEVEKER-DNVCBOLYSA-N 0.000 description 1
- XRHQJGGGJFOMPH-MAUKXSAKSA-N (4s)-3-[2-[[(1s)-1-(1-phenylpyrazol-4-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C2=CN(N=C2)C=2C=CC=CC=2)=N1 XRHQJGGGJFOMPH-MAUKXSAKSA-N 0.000 description 1
- JBDRREUKUCSNKV-DZGCQCFKSA-N (4s)-3-[2-[[(1s)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C3OCCOC3=CC=2)=N1 JBDRREUKUCSNKV-DZGCQCFKSA-N 0.000 description 1
- HNBCEOIQRPXITK-WDEREUQCSA-N (4s)-3-[2-[[(1s)-1-(2-methyl-1,2,4-triazol-3-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N(N=CN=2)C)=N1 HNBCEOIQRPXITK-WDEREUQCSA-N 0.000 description 1
- QZEYGPXLALEWAY-XHDPSFHLSA-N (4s)-3-[2-[[(1s)-1-(3,4-dichlorophenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(Cl)C(Cl)=CC=2)=N1 QZEYGPXLALEWAY-XHDPSFHLSA-N 0.000 description 1
- NIXWPEDNSULKIR-DZGCQCFKSA-N (4s)-3-[2-[[(1s)-1-(3,4-dimethoxyphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=C(OC)C(OC)=CC=C1[C@H](C)NC1=NC=CC(N2C(OC[C@@H]2C(C)C)=O)=N1 NIXWPEDNSULKIR-DZGCQCFKSA-N 0.000 description 1
- GOXCOJSFYMWXQQ-HNAYVOBHSA-N (4s)-3-[2-[[(1s)-1-(3,5-dimethyl-1-phenylpyrazol-4-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C2=C(N(N=C2C)C=2C=CC=CC=2)C)=N1 GOXCOJSFYMWXQQ-HNAYVOBHSA-N 0.000 description 1
- WRHUJMBXYHYKGG-HNAYVOBHSA-N (4s)-3-[2-[[(1s)-1-(3-fluoro-4-pyridin-3-yloxyphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C(OC=3C=NC=CC=3)=CC=2)=N1 WRHUJMBXYHYKGG-HNAYVOBHSA-N 0.000 description 1
- GLVHYTGQNYTKRK-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-(3-fluorophenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(F)C=CC=2)=N1 GLVHYTGQNYTKRK-SWLSCSKDSA-N 0.000 description 1
- QNUAOGCTXZPJMR-LAUBAEHRSA-N (4s)-3-[2-[[(1s)-1-(4-benzoylphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(=CC=2)C(=O)C=2C=CC=CC=2)=N1 QNUAOGCTXZPJMR-LAUBAEHRSA-N 0.000 description 1
- JCWYQRYUDAIGKI-SWLSCSKDSA-N (4s)-3-[2-[[(1s)-1-(4-chlorophenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(Cl)=CC=2)=N1 JCWYQRYUDAIGKI-SWLSCSKDSA-N 0.000 description 1
- SMDJNCKIRDZUJW-MAUKXSAKSA-N (4s)-3-[2-[[(1s)-1-(4-imidazol-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(=CC=2)N2C=NC=C2)=N1 SMDJNCKIRDZUJW-MAUKXSAKSA-N 0.000 description 1
- RAJZEQSIULHTBW-LAUBAEHRSA-N (4s)-3-[2-[[(1s)-1-(4-phenylphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(=CC=2)C=2C=CC=CC=2)=N1 RAJZEQSIULHTBW-LAUBAEHRSA-N 0.000 description 1
- NEKGXIIMUAXULQ-FXAWDEMLSA-N (4s)-3-[2-[[(1s)-1-(4-piperidin-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(=CC=2)N2CCCCC2)=N1 NEKGXIIMUAXULQ-FXAWDEMLSA-N 0.000 description 1
- GYHCFZKUTSEBCZ-GXSJLCMTSA-N (4s)-3-[2-[[(1s)-1-(5-methyl-1,3,4-oxadiazol-2-yl)ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2OC(C)=NN=2)=N1 GYHCFZKUTSEBCZ-GXSJLCMTSA-N 0.000 description 1
- QFATVDJWCRBEEE-KBXCAEBGSA-N (4s)-3-[2-[[(1s)-1-[1-(4-fluorophenyl)pyrazol-4-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C2=CN(N=C2)C=2C=CC(F)=CC=2)=N1 QFATVDJWCRBEEE-KBXCAEBGSA-N 0.000 description 1
- LSXSGJKNEVEKER-HNAYVOBHSA-N (4s)-3-[2-[[(1s)-1-[1-(4-methoxyphenyl)pyrazol-4-yl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound C1=CC(OC)=CC=C1N1N=CC([C@H](C)NC=2N=C(C=CN=2)N2C(OC[C@@H]2C(C)C)=O)=C1 LSXSGJKNEVEKER-HNAYVOBHSA-N 0.000 description 1
- CNIVKZGESGWRFV-GHTZIAJQSA-N (4s)-3-[2-[[(1s)-1-[4-(4,7-diazaspiro[2.5]octan-4-ylmethyl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3C4(CC4)CNCC3)=CC=2)=N1 CNIVKZGESGWRFV-GHTZIAJQSA-N 0.000 description 1
- FJTTYDFAASXNGC-XJKSGUPXSA-N (4s)-3-[2-[[(1s)-1-[4-(chloromethyl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CCl)=CC=2)=N1 FJTTYDFAASXNGC-XJKSGUPXSA-N 0.000 description 1
- XQYMOEFQAQPZFV-FXAWDEMLSA-N (4s)-3-[2-[[(1s)-1-[4-(piperazin-1-ylmethyl)phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCNCC3)=CC=2)=N1 XQYMOEFQAQPZFV-FXAWDEMLSA-N 0.000 description 1
- CBTSWOIZJPAPMH-DVVHHAQGSA-N (4s)-3-[2-[[(1s)-1-[4-[(2-methyl-1,3,3a,4,6,6a-hexahydropyrrolo[3,4-c]pyrrol-5-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CC4CN(C)CC4C3)=CC=2)=N1 CBTSWOIZJPAPMH-DVVHHAQGSA-N 0.000 description 1
- CNLXSIGCRODFOP-FXAWDEMLSA-N (4s)-3-[2-[[(1s)-1-[4-[(3,3-difluoropiperidin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CC(F)(F)CCC3)=CC=2)=N1 CNLXSIGCRODFOP-FXAWDEMLSA-N 0.000 description 1
- IABDWAJMKBNPNA-GHTZIAJQSA-N (4s)-3-[2-[[(1s)-1-[4-[(3,3-dimethylpiperazin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CC(C)(C)NCC3)=CC=2)=N1 IABDWAJMKBNPNA-GHTZIAJQSA-N 0.000 description 1
- BEWLUEOYYPKPQL-PGRDOPGGSA-N (4s)-3-[2-[[(1s)-1-[4-[(4-acetylpiperazin-1-yl)methyl]phenyl]ethyl]amino]pyrimidin-4-yl]-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCN(CC3)C(C)=O)=CC=2)=N1 BEWLUEOYYPKPQL-PGRDOPGGSA-N 0.000 description 1
- LVPITHJLZOMCTJ-KKSFZXQISA-N (4s)-4-(4-methoxyphenyl)-5,5-dimethyl-3-[2-[[(1s)-1-phenylethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1=CC(OC)=CC=C1[C@H]1C(C)(C)OC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC=CC=2)=N1 LVPITHJLZOMCTJ-KKSFZXQISA-N 0.000 description 1
- YVODIRDFZUWKBG-MQBBKVSDSA-N (4s)-4-methyl-4-phenyl-3-[2-[1-(4-piperidin-1-ylphenyl)ethylamino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1([C@@]2(C)COC(=O)N2C=2C=CN=C(N=2)NC(C)C=2C=CC(=CC=2)N2CCCCC2)=CC=CC=C1 YVODIRDFZUWKBG-MQBBKVSDSA-N 0.000 description 1
- YVODIRDFZUWKBG-CCLHPLFOSA-N (4s)-4-methyl-4-phenyl-3-[2-[[(1s)-1-(4-piperidin-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1([C@@]2(C)COC(=O)N2C=2C=CN=C(N=2)N[C@@H](C)C=2C=CC(=CC=2)N2CCCCC2)=CC=CC=C1 YVODIRDFZUWKBG-CCLHPLFOSA-N 0.000 description 1
- YBUPWRYTXGAWJX-RXMQYKEDSA-N (4s)-4-propan-2-yl-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1 YBUPWRYTXGAWJX-RXMQYKEDSA-N 0.000 description 1
- JXQQNLDFVQDOOM-VQIMIIECSA-N (4s)-4-propan-2-yl-3-[2-[[(1r)-1-(3-pyrrol-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@H](C)C=2C=C(C=CC=2)N2C=CC=C2)=N1 JXQQNLDFVQDOOM-VQIMIIECSA-N 0.000 description 1
- QKEFSEHNRHABOP-VQIMIIECSA-N (4s)-4-propan-2-yl-3-[2-[[(1r)-1-(4-pyrrol-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@H](C)C=2C=CC(=CC=2)N2C=CC=C2)=N1 QKEFSEHNRHABOP-VQIMIIECSA-N 0.000 description 1
- JXQQNLDFVQDOOM-QFBILLFUSA-N (4s)-4-propan-2-yl-3-[2-[[(1s)-1-(3-pyrrol-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=C(C=CC=2)N2C=CC=C2)=N1 JXQQNLDFVQDOOM-QFBILLFUSA-N 0.000 description 1
- QKEFSEHNRHABOP-QFBILLFUSA-N (4s)-4-propan-2-yl-3-[2-[[(1s)-1-(4-pyrrol-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(=CC=2)N2C=CC=C2)=N1 QKEFSEHNRHABOP-QFBILLFUSA-N 0.000 description 1
- QTNNXCXGAWXJSN-JXFKEZNVSA-N (4s)-5,5-dimethyl-4-phenyl-3-[2-[[(1s)-1-phenylethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1([C@@H]2N(C(OC2(C)C)=O)C=2C=CN=C(N=2)N[C@@H](C)C=2C=CC=CC=2)=CC=CC=C1 QTNNXCXGAWXJSN-JXFKEZNVSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- DUAMQFWFRDHEGN-BKMCDNFVSA-N (ne)-n-[[2-fluoro-4-(trifluoromethyl)phenyl]methylidene]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)\N=C\C1=CC=C(C(F)(F)F)C=C1F DUAMQFWFRDHEGN-BKMCDNFVSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- AQVHNNSIODHDPL-UHFFFAOYSA-N 1,2,2-trimethylpiperazine;dihydrochloride Chemical compound Cl.Cl.CN1CCNCC1(C)C AQVHNNSIODHDPL-UHFFFAOYSA-N 0.000 description 1
- ARHYWWAJZDAYDJ-UHFFFAOYSA-N 1,2-dimethylpiperazine Chemical compound CC1CNCCN1C ARHYWWAJZDAYDJ-UHFFFAOYSA-N 0.000 description 1
- KEQGZUUPPQEDPF-UHFFFAOYSA-N 1,3-dichloro-5,5-dimethylimidazolidine-2,4-dione Chemical compound CC1(C)N(Cl)C(=O)N(Cl)C1=O KEQGZUUPPQEDPF-UHFFFAOYSA-N 0.000 description 1
- IGERFAHWSHDDHX-UHFFFAOYSA-N 1,3-dioxanyl Chemical group [CH]1OCCCO1 IGERFAHWSHDDHX-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- ILWJAOPQHOZXAN-UHFFFAOYSA-N 1,3-dithianyl Chemical group [CH]1SCCCS1 ILWJAOPQHOZXAN-UHFFFAOYSA-N 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- HKDFRDIIELOLTJ-UHFFFAOYSA-N 1,4-dithianyl Chemical group [CH]1CSCCS1 HKDFRDIIELOLTJ-UHFFFAOYSA-N 0.000 description 1
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 1
- IOAUCNVBBMEIQG-UHFFFAOYSA-N 1-(1-phenylpyrazol-4-yl)ethanamine Chemical compound C1=C(C(N)C)C=NN1C1=CC=CC=C1 IOAUCNVBBMEIQG-UHFFFAOYSA-N 0.000 description 1
- ASNVMKIDRJZXQZ-UHFFFAOYSA-N 1-(3-fluorophenyl)ethanamine Chemical compound CC(N)C1=CC=CC(F)=C1 ASNVMKIDRJZXQZ-UHFFFAOYSA-N 0.000 description 1
- MRPLWYVUULIYMY-UHFFFAOYSA-N 1-(3-pyrrol-1-ylphenyl)ethanamine Chemical compound CC(N)C1=CC=CC(N2C=CC=C2)=C1 MRPLWYVUULIYMY-UHFFFAOYSA-N 0.000 description 1
- PINPOEWMCLFRRB-UHFFFAOYSA-N 1-(4-chlorophenyl)ethanamine Chemical compound CC(N)C1=CC=C(Cl)C=C1 PINPOEWMCLFRRB-UHFFFAOYSA-N 0.000 description 1
- TZRGEQLNGUDZDO-UHFFFAOYSA-N 1-(4-piperidin-1-ylphenyl)ethanamine Chemical compound C1=CC(C(N)C)=CC=C1N1CCCCC1 TZRGEQLNGUDZDO-UHFFFAOYSA-N 0.000 description 1
- LMLXFODTRVVPKY-UHFFFAOYSA-N 1-(4-pyrrol-1-ylphenyl)ethanamine Chemical compound C1=CC(C(N)C)=CC=C1N1C=CC=C1 LMLXFODTRVVPKY-UHFFFAOYSA-N 0.000 description 1
- NZGSEUQFYKCKRU-UHFFFAOYSA-N 1-(5-bromopyrimidin-2-yl)ethanone Chemical compound CC(=O)C1=NC=C(Br)C=N1 NZGSEUQFYKCKRU-UHFFFAOYSA-N 0.000 description 1
- NNJJXGKCENQOJW-UHFFFAOYSA-N 1-(5-fluoropyrimidin-2-yl)ethanone Chemical compound CC(=O)C1=NC=C(F)C=N1 NNJJXGKCENQOJW-UHFFFAOYSA-N 0.000 description 1
- WZLIKWWAFFVTNX-UHFFFAOYSA-N 1-(5-pyridin-3-yloxypyrimidin-2-yl)ethanamine Chemical compound C1=NC(C(N)C)=NC=C1OC1=CC=CN=C1 WZLIKWWAFFVTNX-UHFFFAOYSA-N 0.000 description 1
- VEOMTIGCLUMAPX-UHFFFAOYSA-N 1-[1-(4-fluorophenyl)pyrazol-4-yl]ethanamine;hydrochloride Chemical compound Cl.C1=C(C(N)C)C=NN1C1=CC=C(F)C=C1 VEOMTIGCLUMAPX-UHFFFAOYSA-N 0.000 description 1
- FSDZABAZZCBYKG-UHFFFAOYSA-N 1-[1-(4-methoxyphenyl)pyrazol-4-yl]ethanamine;hydrochloride Chemical compound Cl.C1=CC(OC)=CC=C1N1N=CC(C(C)N)=C1 FSDZABAZZCBYKG-UHFFFAOYSA-N 0.000 description 1
- IHEOQTUFVVJBFK-UHFFFAOYSA-N 1-[2-(4-fluorophenoxy)pyrimidin-5-yl]ethanamine Chemical compound N1=CC(C(N)C)=CN=C1OC1=CC=C(F)C=C1 IHEOQTUFVVJBFK-UHFFFAOYSA-N 0.000 description 1
- HNDBFFBSXOAWPV-UHFFFAOYSA-N 1-[3-(4-chlorophenyl)-1,2,4-oxadiazol-5-yl]ethanamine Chemical compound O1C(C(N)C)=NC(C=2C=CC(Cl)=CC=2)=N1 HNDBFFBSXOAWPV-UHFFFAOYSA-N 0.000 description 1
- HRFMMVUOKYYHHU-UHFFFAOYSA-N 1-[5-(2,3-dichlorophenyl)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1C1=CC=CC(Cl)=C1Cl HRFMMVUOKYYHHU-UHFFFAOYSA-N 0.000 description 1
- HVFIKRNTTZAEPC-UHFFFAOYSA-N 1-[5-(2,3-dichlorophenyl)pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1C1=CC=CC(Cl)=C1Cl HVFIKRNTTZAEPC-UHFFFAOYSA-N 0.000 description 1
- BWCKCNXNAWNWQB-UHFFFAOYSA-N 1-[5-(2,4-difluorophenoxy)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1OC1=CC=C(F)C=C1F BWCKCNXNAWNWQB-UHFFFAOYSA-N 0.000 description 1
- GZQVGOACOWQMMP-UHFFFAOYSA-N 1-[5-(3,4-dichlorophenyl)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1C1=CC=C(Cl)C(Cl)=C1 GZQVGOACOWQMMP-UHFFFAOYSA-N 0.000 description 1
- VFUFFSZGCSMEDI-UHFFFAOYSA-N 1-[5-(3,4-dichlorophenyl)pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1C1=CC=C(Cl)C(Cl)=C1 VFUFFSZGCSMEDI-UHFFFAOYSA-N 0.000 description 1
- GIQOBBRNBUUCRP-UHFFFAOYSA-N 1-[5-(3-chloro-4-fluorophenoxy)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1OC1=CC=C(F)C(Cl)=C1 GIQOBBRNBUUCRP-UHFFFAOYSA-N 0.000 description 1
- WCQKXVBOHGKMLH-UHFFFAOYSA-N 1-[5-(3-fluoro-2-methylphenyl)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1C1=CC=CC(F)=C1C WCQKXVBOHGKMLH-UHFFFAOYSA-N 0.000 description 1
- WKGXBOOZHCMSJE-UHFFFAOYSA-N 1-[5-(3-fluoro-2-methylphenyl)pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1C1=CC=CC(F)=C1C WKGXBOOZHCMSJE-UHFFFAOYSA-N 0.000 description 1
- MGACTMZWOWBKHR-UHFFFAOYSA-N 1-[5-(4-fluoro-2-methylphenyl)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1C1=CC=C(F)C=C1C MGACTMZWOWBKHR-UHFFFAOYSA-N 0.000 description 1
- KNVBNFIBSAPEAE-UHFFFAOYSA-N 1-[5-(4-fluoro-2-methylphenyl)pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1C1=CC=C(F)C=C1C KNVBNFIBSAPEAE-UHFFFAOYSA-N 0.000 description 1
- ODANCJSBPJWMPY-UHFFFAOYSA-N 1-[5-(4-fluoro-3-methylphenyl)pyridin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=CC=C1C1=CC=C(F)C(C)=C1 ODANCJSBPJWMPY-UHFFFAOYSA-N 0.000 description 1
- COOBLEUJQLUQEX-UHFFFAOYSA-N 1-[5-(4-fluoro-3-methylphenyl)pyridin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=CC=C1C1=CC=C(F)C(C)=C1 COOBLEUJQLUQEX-UHFFFAOYSA-N 0.000 description 1
- GBITUYYBLXKEOT-UHFFFAOYSA-N 1-[5-(4-fluoro-3-methylphenyl)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1C1=CC=C(F)C(C)=C1 GBITUYYBLXKEOT-UHFFFAOYSA-N 0.000 description 1
- RCPVGTGTBAYXCZ-UHFFFAOYSA-N 1-[5-(4-fluoro-3-methylphenyl)pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1C1=CC=C(F)C(C)=C1 RCPVGTGTBAYXCZ-UHFFFAOYSA-N 0.000 description 1
- YAQJERLLGYMXSR-UHFFFAOYSA-N 1-[5-(4-fluorophenoxy)pyrazin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=CN=C1OC1=CC=C(F)C=C1 YAQJERLLGYMXSR-UHFFFAOYSA-N 0.000 description 1
- PZXXMEYQBAPDEQ-UHFFFAOYSA-N 1-[5-(4-fluorophenoxy)pyridin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=CC=C1OC1=CC=C(F)C=C1 PZXXMEYQBAPDEQ-UHFFFAOYSA-N 0.000 description 1
- QQYZEKPOFDMCPR-UHFFFAOYSA-N 1-[5-(4-fluorophenoxy)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1OC1=CC=C(F)C=C1 QQYZEKPOFDMCPR-UHFFFAOYSA-N 0.000 description 1
- VFIBYODBRLETTD-UHFFFAOYSA-N 1-[5-(5-bromopyridin-3-yl)oxypyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1OC1=CN=CC(Br)=C1 VFIBYODBRLETTD-UHFFFAOYSA-N 0.000 description 1
- BDAZNJBPZVCKMJ-UHFFFAOYSA-N 1-[5-(5-fluoro-2-methylphenyl)pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1C1=CC(F)=CC=C1C BDAZNJBPZVCKMJ-UHFFFAOYSA-N 0.000 description 1
- MFHHGGVBMKNHJU-UHFFFAOYSA-N 1-[5-(5-fluoro-2-methylphenyl)pyrimidin-2-yl]ethanone Chemical compound C1=NC(C(=O)C)=NC=C1C1=CC(F)=CC=C1C MFHHGGVBMKNHJU-UHFFFAOYSA-N 0.000 description 1
- USNZVQWXANJMLX-UHFFFAOYSA-N 1-[5-[3-(trifluoromethyl)phenyl]pyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1C1=CC=CC(C(F)(F)F)=C1 USNZVQWXANJMLX-UHFFFAOYSA-N 0.000 description 1
- RJYTXQIUTCNLOD-UHFFFAOYSA-N 1-[5-[5-(trifluoromethyl)pyridin-2-yl]oxypyrimidin-2-yl]ethanamine Chemical compound C1=NC(C(N)C)=NC=C1OC1=CC=C(C(F)(F)F)C=N1 RJYTXQIUTCNLOD-UHFFFAOYSA-N 0.000 description 1
- CXOSLOJKAMXJKJ-KRWDZBQOSA-N 1-[5-fluoro-2-[[(1s)-1-(4-phenoxyphenyl)ethyl]amino]pyrimidin-4-yl]-3-oxa-1-azaspiro[4.4]nonan-2-one Chemical compound N([C@@H](C)C=1C=CC(OC=2C=CC=CC=2)=CC=1)C(N=1)=NC=C(F)C=1N1C(=O)OCC11CCCC1 CXOSLOJKAMXJKJ-KRWDZBQOSA-N 0.000 description 1
- LXQMHOKEXZETKB-UHFFFAOYSA-N 1-amino-2-methylpropan-2-ol Chemical compound CC(C)(O)CN LXQMHOKEXZETKB-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- TZTXTIBZSSSFDI-UHFFFAOYSA-N 1-pyridin-2-ylpropan-2-one Chemical compound CC(=O)CC1=CC=CC=N1 TZTXTIBZSSSFDI-UHFFFAOYSA-N 0.000 description 1
- NMIZONYLXCOHEF-UHFFFAOYSA-N 1h-imidazole-2-carboxamide Chemical compound NC(=O)C1=NC=CN1 NMIZONYLXCOHEF-UHFFFAOYSA-N 0.000 description 1
- DNCYBUMDUBHIJZ-UHFFFAOYSA-N 1h-pyrimidin-6-one Chemical compound O=C1C=CN=CN1 DNCYBUMDUBHIJZ-UHFFFAOYSA-N 0.000 description 1
- DZCAUMADOBDJJH-UHFFFAOYSA-N 2,2,2-trifluoro-1-phenylethanamine Chemical compound FC(F)(F)C(N)C1=CC=CC=C1 DZCAUMADOBDJJH-UHFFFAOYSA-N 0.000 description 1
- WDBAXYQUOZDFOJ-UHFFFAOYSA-N 2,3-difluorobenzaldehyde Chemical compound FC1=CC=CC(C=O)=C1F WDBAXYQUOZDFOJ-UHFFFAOYSA-N 0.000 description 1
- DPVIABCMTHHTGB-UHFFFAOYSA-N 2,4,6-trichloropyrimidine Chemical compound ClC1=CC(Cl)=NC(Cl)=N1 DPVIABCMTHHTGB-UHFFFAOYSA-N 0.000 description 1
- NTSYSQNAPGMSIH-UHFFFAOYSA-N 2,4,6-trifluoropyrimidine Chemical compound FC1=CC(F)=NC(F)=N1 NTSYSQNAPGMSIH-UHFFFAOYSA-N 0.000 description 1
- WHPFEQUEHBULBW-UHFFFAOYSA-N 2,4-dichloro-5-fluoropyrimidine Chemical compound FC1=CN=C(Cl)N=C1Cl WHPFEQUEHBULBW-UHFFFAOYSA-N 0.000 description 1
- JZSYSXZDIQUOGP-UHFFFAOYSA-N 2,4-difluoropyrimidine Chemical compound FC1=CC=NC(F)=N1 JZSYSXZDIQUOGP-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- WXHLLJAMBQLULT-UHFFFAOYSA-N 2-[[6-[4-(2-hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4-yl]amino]-n-(2-methyl-6-sulfanylphenyl)-1,3-thiazole-5-carboxamide;hydrate Chemical compound O.C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1S WXHLLJAMBQLULT-UHFFFAOYSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- KFEHNXLFIGPWNB-UHFFFAOYSA-N 2-fluoro-4-(trifluoromethyl)benzaldehyde Chemical compound FC1=CC(C(F)(F)F)=CC=C1C=O KFEHNXLFIGPWNB-UHFFFAOYSA-N 0.000 description 1
- HSKFWUNQSAWFDR-UHFFFAOYSA-N 3,4-dimethyl-1,3-oxazolidin-2-one Chemical compound CC1COC(=O)N1C HSKFWUNQSAWFDR-UHFFFAOYSA-N 0.000 description 1
- UTQLXMBGKPYVFW-UHFFFAOYSA-N 3-(2,6-dichloropyrimidin-4-yl)-4,4-dimethyl-1,3-oxazolidin-2-one Chemical compound CC1(C)COC(=O)N1C1=CC(Cl)=NC(Cl)=N1 UTQLXMBGKPYVFW-UHFFFAOYSA-N 0.000 description 1
- TVHRDYCNZAAOHN-UHFFFAOYSA-N 3-(2-chloro-5-fluoropyrimidin-4-yl)-1,3-oxazolidin-2-one Chemical compound FC1=CN=C(Cl)N=C1N1C(=O)OCC1 TVHRDYCNZAAOHN-UHFFFAOYSA-N 0.000 description 1
- DMBVRSPBXIJEIB-UHFFFAOYSA-N 3-(2-fluoropyrimidin-4-yl)-1,3-oxazolidin-2-one Chemical compound FC1=NC=CC(N2C(OCC2)=O)=N1 DMBVRSPBXIJEIB-UHFFFAOYSA-N 0.000 description 1
- WFRNDUQAIZJRPZ-LURJTMIESA-N 3-[(1s)-1-aminoethyl]phenol Chemical compound C[C@H](N)C1=CC=CC(O)=C1 WFRNDUQAIZJRPZ-LURJTMIESA-N 0.000 description 1
- CVMZIXONAFYCLC-UHFFFAOYSA-N 3-[2-[1-[3-(4-chlorophenyl)-1,2,4-oxadiazol-5-yl]ethylamino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound N=1C(C=2C=CC(Cl)=CC=2)=NOC=1C(C)NC(N=1)=NC=CC=1N1CCOC1=O CVMZIXONAFYCLC-UHFFFAOYSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ROADCYAOHVSOLQ-UHFFFAOYSA-N 3-oxetanone Chemical compound O=C1COC1 ROADCYAOHVSOLQ-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- SVSUYEJKNSMKKW-UHFFFAOYSA-N 4,4,5,5-tetramethyl-2-prop-1-en-2-yl-1,3,2-dioxaborolane Chemical compound CC(=C)B1OC(C)(C)C(C)(C)O1 SVSUYEJKNSMKKW-UHFFFAOYSA-N 0.000 description 1
- SYARCRAQWWGZKY-UHFFFAOYSA-N 4,4-dimethyl-1,3-oxazolidin-2-one Chemical compound CC1(C)COC(=O)N1 SYARCRAQWWGZKY-UHFFFAOYSA-N 0.000 description 1
- YXTHNONOTJBSFP-MRXNPFEDSA-N 4,4-dimethyl-3-[2-[[(1r)-1-(4-piperidin-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound N([C@H](C)C=1C=CC(=CC=1)N1CCCCC1)C(N=1)=NC=CC=1N1C(=O)OCC1(C)C YXTHNONOTJBSFP-MRXNPFEDSA-N 0.000 description 1
- YXTHNONOTJBSFP-INIZCTEOSA-N 4,4-dimethyl-3-[2-[[(1s)-1-(4-piperidin-1-ylphenyl)ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound N([C@@H](C)C=1C=CC(=CC=1)N1CCCCC1)C(N=1)=NC=CC=1N1C(=O)OCC1(C)C YXTHNONOTJBSFP-INIZCTEOSA-N 0.000 description 1
- HORKHFJECNTMFZ-LURJTMIESA-N 4,6-difluoro-n-[(1s)-1-[2-fluoro-4-(trifluoromethyl)phenyl]ethyl]pyrimidin-2-amine Chemical compound N([C@@H](C)C=1C(=CC(=CC=1)C(F)(F)F)F)C1=NC(F)=CC(F)=N1 HORKHFJECNTMFZ-LURJTMIESA-N 0.000 description 1
- LXCICYRNWIGDQA-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2-oxazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CON=C1 LXCICYRNWIGDQA-UHFFFAOYSA-N 0.000 description 1
- LVPITHJLZOMCTJ-BJQOMGFOSA-N 4-(4-methoxyphenyl)-5,5-dimethyl-3-[2-[[(1s)-1-phenylethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C1=CC(OC)=CC=C1C1C(C)(C)OC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC=CC=2)=N1 LVPITHJLZOMCTJ-BJQOMGFOSA-N 0.000 description 1
- YGJRIDMDDKKESX-YFKPBYRVSA-N 4-[(1s)-1-aminoethyl]-2-fluorobenzoic acid Chemical compound C[C@H](N)C1=CC=C(C(O)=O)C(F)=C1 YGJRIDMDDKKESX-YFKPBYRVSA-N 0.000 description 1
- HNDFWZASJWHBCZ-RGMNGODLSA-N 4-[(1s)-1-aminoethyl]phenol;hydrochloride Chemical compound Cl.C[C@H](N)C1=CC=C(O)C=C1 HNDFWZASJWHBCZ-RGMNGODLSA-N 0.000 description 1
- YUCBLVFHJWOYDN-PPIALRKJSA-N 4-[(r)-[(2r,4s,5r)-5-ethyl-1-azabicyclo[2.2.2]octan-2-yl]-(6-methoxyquinolin-4-yl)methoxy]-1-[(r)-[(2r,4r,5s)-5-ethyl-1-azabicyclo[2.2.2]octan-2-yl]-(6-methoxyquinolin-4-yl)methoxy]phthalazine Chemical compound C1=C(OC)C=C2C([C@@H](OC=3C4=CC=CC=C4C(O[C@@H]([C@@H]4N5CC[C@@H]([C@@H](C5)CC)C4)C=4C5=CC(OC)=CC=C5N=CC=4)=NN=3)[C@H]3C[C@@H]4CCN3C[C@@H]4CC)=CC=NC2=C1 YUCBLVFHJWOYDN-PPIALRKJSA-N 0.000 description 1
- YFCIFWOJYYFDQP-PTWZRHHISA-N 4-[3-amino-6-[(1S,3S,4S)-3-fluoro-4-hydroxycyclohexyl]pyrazin-2-yl]-N-[(1S)-1-(3-bromo-5-fluorophenyl)-2-(methylamino)ethyl]-2-fluorobenzamide Chemical compound CNC[C@@H](NC(=O)c1ccc(cc1F)-c1nc(cnc1N)[C@H]1CC[C@H](O)[C@@H](F)C1)c1cc(F)cc(Br)c1 YFCIFWOJYYFDQP-PTWZRHHISA-N 0.000 description 1
- ZHSKUOZOLHMKEA-UHFFFAOYSA-N 4-[5-[bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid;hydron;chloride Chemical compound Cl.ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 ZHSKUOZOLHMKEA-UHFFFAOYSA-N 0.000 description 1
- UPCARQPLANFGQJ-UHFFFAOYSA-N 4-bromo-2-fluorobenzaldehyde Chemical compound FC1=CC(Br)=CC=C1C=O UPCARQPLANFGQJ-UHFFFAOYSA-N 0.000 description 1
- SWHUROFMIMHWKS-UHFFFAOYSA-N 4-bromo-3-fluorobenzaldehyde Chemical compound FC1=CC(C=O)=CC=C1Br SWHUROFMIMHWKS-UHFFFAOYSA-N 0.000 description 1
- AKONCVZBQYODEU-UHFFFAOYSA-N 4-chloropyrimidine-2-carbonitrile Chemical compound ClC1=CC=NC(C#N)=N1 AKONCVZBQYODEU-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- UOQXIWFBQSVDPP-UHFFFAOYSA-N 4-fluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C=C1 UOQXIWFBQSVDPP-UHFFFAOYSA-N 0.000 description 1
- ISDBWOPVZKNQDW-UHFFFAOYSA-N 4-phenylbenzaldehyde Chemical compound C1=CC(C=O)=CC=C1C1=CC=CC=C1 ISDBWOPVZKNQDW-UHFFFAOYSA-N 0.000 description 1
- UMEIYBJBGZKZOS-UHFFFAOYSA-N 5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine Chemical compound C1NCCN2C=NN=C21 UMEIYBJBGZKZOS-UHFFFAOYSA-N 0.000 description 1
- RNRLTTNKVLFZJS-UHFFFAOYSA-N 5,6-dichloropyridine-3-carboxylic acid Chemical compound OC(=O)C1=CN=C(Cl)C(Cl)=C1 RNRLTTNKVLFZJS-UHFFFAOYSA-N 0.000 description 1
- OUSVTWOQVSAYJW-UHFFFAOYSA-N 5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine Chemical compound C1=NCCN2C=NN=C21 OUSVTWOQVSAYJW-UHFFFAOYSA-N 0.000 description 1
- NWDMGTFNIOCVDU-UHFFFAOYSA-N 5-methyl-1h-pyrazole-4-carbaldehyde Chemical compound CC=1NN=CC=1C=O NWDMGTFNIOCVDU-UHFFFAOYSA-N 0.000 description 1
- YQURLNGUWNDBIR-UHFFFAOYSA-N 5-methyl-2,3,3a,4,6,6a-hexahydro-1h-pyrrolo[3,4-c]pyrrole Chemical compound C1NCC2CN(C)CC21 YQURLNGUWNDBIR-UHFFFAOYSA-N 0.000 description 1
- DXJKIFIEEXRIEZ-UHFFFAOYSA-N 7-(2-chloropyrimidin-4-yl)-8-phenyl-2,5-dioxa-7-azaspiro[3.4]octan-6-one Chemical compound ClC1=NC=CC(N2C(OC3(COC3)C2C=2C=CC=CC=2)=O)=N1 DXJKIFIEEXRIEZ-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- 208000002008 AIDS-Related Lymphoma Diseases 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 206010061424 Anal cancer Diseases 0.000 description 1
- 206010073127 Anaplastic meningioma Diseases 0.000 description 1
- 206010073128 Anaplastic oligodendroglioma Diseases 0.000 description 1
- 208000007860 Anus Neoplasms Diseases 0.000 description 1
- 101100452478 Arabidopsis thaliana DHAD gene Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010065869 Astrocytoma, low grade Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000032800 BCR-ABL1 positive blast phase chronic myelogenous leukemia Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 206010007279 Carcinoid tumour of the gastrointestinal tract Diseases 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241000202285 Claravis Species 0.000 description 1
- 206010073140 Clear cell sarcoma of soft tissue Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 102100021906 Cyclin-O Human genes 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 102000003915 DNA Topoisomerases Human genes 0.000 description 1
- 108090000323 DNA Topoisomerases Proteins 0.000 description 1
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- GUGHGUXZJWAIAS-QQYBVWGSSA-N Daunorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 GUGHGUXZJWAIAS-QQYBVWGSSA-N 0.000 description 1
- 208000021994 Diffuse astrocytoma Diseases 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 101100224482 Drosophila melanogaster PolE1 gene Proteins 0.000 description 1
- 239000001692 EU approved anti-caking agent Substances 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 206010014968 Ependymoma malignant Diseases 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000588698 Erwinia Species 0.000 description 1
- 208000017259 Extragonadal germ cell tumor Diseases 0.000 description 1
- GISRWBROCYNDME-PELMWDNLSA-N F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C Chemical compound F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C GISRWBROCYNDME-PELMWDNLSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical group FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000007818 Grignard reagent Substances 0.000 description 1
- 229920002907 Guar gum Polymers 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000897441 Homo sapiens Cyclin-O Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010021042 Hypopharyngeal cancer Diseases 0.000 description 1
- 206010056305 Hypopharyngeal neoplasm Diseases 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 108010022337 Leucine Enkephalin Proteins 0.000 description 1
- 206010062038 Lip neoplasm Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- 206010025312 Lymphoma AIDS related Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000004059 Male Breast Neoplasms Diseases 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 208000030070 Malignant epithelial tumor of ovary Diseases 0.000 description 1
- 208000032271 Malignant tumor of penis Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000002030 Merkel cell carcinoma Diseases 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 208000033833 Myelomonocytic Chronic Leukemia Diseases 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- BAWFJGJZGIEFAR-NNYOXOHSSA-O NAD(+) Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-O 0.000 description 1
- 229910017974 NH40H Inorganic materials 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 206010029266 Neuroendocrine carcinoma of the skin Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 208000012247 Oligodendroglial tumor Diseases 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 206010031096 Oropharyngeal cancer Diseases 0.000 description 1
- 206010057444 Oropharyngeal neoplasm Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000007571 Ovarian Epithelial Carcinoma Diseases 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061328 Ovarian epithelial cancer Diseases 0.000 description 1
- 206010033268 Ovarian low malignant potential tumour Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 206010034299 Penile cancer Diseases 0.000 description 1
- 208000027190 Peripheral T-cell lymphomas Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 201000007286 Pilocytic astrocytoma Diseases 0.000 description 1
- 201000005746 Pituitary adenoma Diseases 0.000 description 1
- 206010061538 Pituitary tumour benign Diseases 0.000 description 1
- 201000007288 Pleomorphic xanthoastrocytoma Diseases 0.000 description 1
- 201000008199 Pleuropulmonary blastoma Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 206010057846 Primitive neuroectodermal tumour Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 229910006074 SO2NH2 Inorganic materials 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 1
- 208000031672 T-Cell Peripheral Lymphoma Diseases 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 239000012317 TBTU Substances 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 206010044407 Transitional cell cancer of the renal pelvis and ureter Diseases 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 206010046431 Urethral cancer Diseases 0.000 description 1
- 206010046458 Urethral neoplasms Diseases 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 206010047741 Vulval cancer Diseases 0.000 description 1
- 208000004354 Vulvar Neoplasms Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- WOAORAPRPVIATR-UHFFFAOYSA-N [3-(trifluoromethyl)phenyl]boronic acid Chemical compound OB(O)C1=CC=CC(C(F)(F)F)=C1 WOAORAPRPVIATR-UHFFFAOYSA-N 0.000 description 1
- VAOYLMISGFLPQE-LURJTMIESA-N [4-[(1s)-1-aminoethyl]-3-fluorophenyl]methanol Chemical compound C[C@H](N)C1=CC=C(CO)C=C1F VAOYLMISGFLPQE-LURJTMIESA-N 0.000 description 1
- ZMYUHMGMBDTYJS-UHFFFAOYSA-N [Ar].CC[Zn]CC Chemical compound [Ar].CC[Zn]CC ZMYUHMGMBDTYJS-UHFFFAOYSA-N 0.000 description 1
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- DFDGRKNOFOJBAJ-UHFFFAOYSA-N acifran Chemical compound C=1C=CC=CC=1C1(C)OC(C(O)=O)=CC1=O DFDGRKNOFOJBAJ-UHFFFAOYSA-N 0.000 description 1
- 230000001780 adrenocortical effect Effects 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 229940064305 adrucil Drugs 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 208000030002 adult glioblastoma Diseases 0.000 description 1
- QNAYBMKLOCPYGJ-UHFFFAOYSA-M alaninate Chemical compound CC(N)C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-M 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 229940110282 alimta Drugs 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 229940098174 alkeran Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229940022824 amnesteem Drugs 0.000 description 1
- 206010002224 anaplastic astrocytoma Diseases 0.000 description 1
- 208000014534 anaplastic ependymoma Diseases 0.000 description 1
- 208000013938 anaplastic oligoastrocytoma Diseases 0.000 description 1
- 201000011165 anus cancer Diseases 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 239000008122 artificial sweetener Substances 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 229960003005 axitinib Drugs 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- HPMLGNIUXVXALD-UHFFFAOYSA-N benzoyl fluoride Chemical compound FC(=O)C1=CC=CC=C1 HPMLGNIUXVXALD-UHFFFAOYSA-N 0.000 description 1
- DVDWCHHLKXYBCX-WNCULLNHSA-N benzyl 4-[[4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]phenyl]methyl]piperazine-1-carboxylate Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCN(CC3)C(=O)OCC=3C=CC=CC=3)=CC=2)=N1 DVDWCHHLKXYBCX-WNCULLNHSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 229940108502 bicnu Drugs 0.000 description 1
- 208000026900 bile duct neoplasm Diseases 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- HTJWUNNIRKDDIV-UHFFFAOYSA-N bis(1-adamantyl)-butylphosphane Chemical compound C1C(C2)CC(C3)CC2CC13P(CCCC)C1(C2)CC(C3)CC2CC3C1 HTJWUNNIRKDDIV-UHFFFAOYSA-N 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 208000012172 borderline epithelial tumor of ovary Diseases 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 201000002143 bronchus adenoma Diseases 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 229940112133 busulfex Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- XAAHAAMILDNBPS-UHFFFAOYSA-L calcium hydrogenphosphate dihydrate Chemical compound O.O.[Ca+2].OP([O-])([O-])=O XAAHAAMILDNBPS-UHFFFAOYSA-L 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 208000011654 childhood malignant neoplasm Diseases 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 239000000460 chlorine Chemical group 0.000 description 1
- 229910052801 chlorine Chemical group 0.000 description 1
- 125000000068 chlorophenyl group Chemical group 0.000 description 1
- 208000020719 chondrogenic neoplasm Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 201000010902 chronic myelomonocytic leukemia Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229940031301 claravis Drugs 0.000 description 1
- 229940060799 clarus Drugs 0.000 description 1
- 201000000292 clear cell sarcoma Diseases 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000013066 combination product Substances 0.000 description 1
- 229940127555 combination product Drugs 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 229940088547 cosmegen Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 1
- 208000017763 cutaneous neuroendocrine carcinoma Diseases 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- PNZXMIKHJXIPEK-UHFFFAOYSA-N cyclohexanecarboxamide Chemical compound NC(=O)C1CCCCC1 PNZXMIKHJXIPEK-UHFFFAOYSA-N 0.000 description 1
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical compound NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 1
- XCIXKGXIYUWCLL-UHFFFAOYSA-N cyclopentanol Chemical compound OC1CCCC1 XCIXKGXIYUWCLL-UHFFFAOYSA-N 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229940059359 dacogen Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 229940052372 daunorubicin citrate liposome Drugs 0.000 description 1
- 229940041983 daunorubicin liposomal Drugs 0.000 description 1
- 229940107841 daunoxome Drugs 0.000 description 1
- 150000004691 decahydrates Chemical class 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 229940095079 dicalcium phosphate anhydrous Drugs 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 125000006001 difluoroethyl group Chemical group 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- DGODWNOPHMXOTR-UHFFFAOYSA-N dipotassium;dioxido(dioxo)osmium;dihydrate Chemical compound O.O.[K+].[K+].[O-][Os]([O-])(=O)=O DGODWNOPHMXOTR-UHFFFAOYSA-N 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229940073038 elspar Drugs 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethanedisulfonate group Chemical group C(CS(=O)(=O)[O-])S(=O)(=O)[O-] AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- 229940052303 ethers for general anesthesia Drugs 0.000 description 1
- OJSGTTFKLAKIFA-UHFFFAOYSA-N ethyl 5-chloro-6-(1-ethoxyethenyl)pyridine-3-carboxylate Chemical compound CCOC(=C)C1=NC=C(C(=O)OCC)C=C1Cl OJSGTTFKLAKIFA-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 201000008819 extrahepatic bile duct carcinoma Diseases 0.000 description 1
- 201000001169 fibrillary astrocytoma Diseases 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 239000011737 fluorine Chemical group 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 201000007116 gestational trophoblastic neoplasm Diseases 0.000 description 1
- 201000011610 giant cell glioblastoma Diseases 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 239000000665 guar gum Substances 0.000 description 1
- 235000010417 guar gum Nutrition 0.000 description 1
- 229960002154 guar gum Drugs 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 229940096120 hydrea Drugs 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical group [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 201000006866 hypopharynx cancer Diseases 0.000 description 1
- 229940099279 idamycin Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229940090411 ifex Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- ODBLHEXUDAPZAU-UHFFFAOYSA-N isocitric acid Chemical compound OC(=O)C(O)C(C(O)=O)CC(O)=O ODBLHEXUDAPZAU-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- DWKPPFQULDPWHX-VKHMYHEASA-N l-alanyl ester Chemical compound COC(=O)[C@H](C)N DWKPPFQULDPWHX-VKHMYHEASA-N 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 1
- URLZCHNOLZSCCA-UHFFFAOYSA-N leu-enkephalin Chemical compound C=1C=C(O)C=CC=1CC(N)C(=O)NCC(=O)NCC(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=CC=C1 URLZCHNOLZSCCA-UHFFFAOYSA-N 0.000 description 1
- 229940063725 leukeran Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 201000006721 lip cancer Diseases 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 230000000527 lymphocytic effect Effects 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 201000003175 male breast cancer Diseases 0.000 description 1
- 208000010907 male breast carcinoma Diseases 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 229940087732 matulane Drugs 0.000 description 1
- 208000020968 mature T-cell and NK-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 210000000716 merkel cell Anatomy 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 208000037970 metastatic squamous neck cancer Diseases 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- GJDICGOCZGRDFM-LURJTMIESA-N methyl (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound COC(=O)[C@H](C)NC(=O)OC(C)(C)C GJDICGOCZGRDFM-LURJTMIESA-N 0.000 description 1
- DKYLCANTGPRCHY-QMMMGPOBSA-N methyl (2s)-2-cyclopropyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetate Chemical compound CC(C)(C)OC(=O)N[C@H](C(=O)OC)C1CC1 DKYLCANTGPRCHY-QMMMGPOBSA-N 0.000 description 1
- JRDIMKBADQPZQD-RGMNGODLSA-N methyl 4-[(1s)-1-aminoethyl]-3-fluorobenzoate;hydrochloride Chemical compound Cl.COC(=O)C1=CC=C([C@H](C)N)C(F)=C1 JRDIMKBADQPZQD-RGMNGODLSA-N 0.000 description 1
- XSYGLHLLQZGWPT-ZETCQYMHSA-N methyl 4-[(1s)-1-aminoethyl]benzoate Chemical compound COC(=O)C1=CC=C([C@H](C)N)C=C1 XSYGLHLLQZGWPT-ZETCQYMHSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- LSEFCHWGJNHZNT-UHFFFAOYSA-M methyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(C)C1=CC=CC=C1 LSEFCHWGJNHZNT-UHFFFAOYSA-M 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- RAHBGWKEPAQNFF-UHFFFAOYSA-N motesanib Chemical compound C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 RAHBGWKEPAQNFF-UHFFFAOYSA-N 0.000 description 1
- 229950003968 motesanib Drugs 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 206010051747 multiple endocrine neoplasia Diseases 0.000 description 1
- 229940087004 mustargen Drugs 0.000 description 1
- 201000006462 myelodysplastic/myeloproliferative neoplasm Diseases 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 229940090009 myleran Drugs 0.000 description 1
- 201000004057 myxopapillary ependymoma Diseases 0.000 description 1
- KRKPYFLIYNGWTE-UHFFFAOYSA-N n,o-dimethylhydroxylamine Chemical compound CNOC KRKPYFLIYNGWTE-UHFFFAOYSA-N 0.000 description 1
- CHPYTKRPVYHZTJ-APBUJDDRSA-N n-[(1s)-1-[4-(cyanomethyl)-2-fluorophenyl]ethyl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H](C)C1=CC=C(CC#N)C=C1F CHPYTKRPVYHZTJ-APBUJDDRSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YUNHKYILTJWSII-YCRPNKLZSA-N n-cyclohexyl-2-fluoro-4-[(1s)-1-[[5-fluoro-4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]benzamide Chemical compound CC(C)[C@H]1COC(=O)N1C1=NC(N[C@@H](C)C=2C=C(F)C(C(=O)NC3CCCCC3)=CC=2)=NC=C1F YUNHKYILTJWSII-YCRPNKLZSA-N 0.000 description 1
- RIVIDPPYRINTTH-UHFFFAOYSA-N n-ethylpropan-2-amine Chemical compound CCNC(C)C RIVIDPPYRINTTH-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 208000007538 neurilemmoma Diseases 0.000 description 1
- 229940080607 nexavar Drugs 0.000 description 1
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229940109551 nipent Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000006501 nitrophenyl group Chemical group 0.000 description 1
- VWBWQOUWDOULQN-UHFFFAOYSA-N nmp n-methylpyrrolidone Chemical compound CN1CCCC1=O.CN1CCCC1=O VWBWQOUWDOULQN-UHFFFAOYSA-N 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-M octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC([O-])=O QIQXTHQIDYTFRH-UHFFFAOYSA-M 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 208000013937 oligoastrocytic tumor Diseases 0.000 description 1
- 206010073131 oligoastrocytoma Diseases 0.000 description 1
- 208000022982 optic pathway glioma Diseases 0.000 description 1
- 201000005443 oral cavity cancer Diseases 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 201000006958 oropharynx cancer Diseases 0.000 description 1
- 208000021284 ovarian germ cell tumor Diseases 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- UFSCUAXLTRFIDC-UHFFFAOYSA-N oxalosuccinic acid Chemical compound OC(=O)CC(C(O)=O)C(=O)C(O)=O UFSCUAXLTRFIDC-UHFFFAOYSA-N 0.000 description 1
- JMJRYTGVHCAYCT-UHFFFAOYSA-N oxan-4-one Chemical compound O=C1CCOCC1 JMJRYTGVHCAYCT-UHFFFAOYSA-N 0.000 description 1
- 125000005880 oxathiolanyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 238000005895 oxidative decarboxylation reaction Methods 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 201000002530 pancreatic endocrine carcinoma Diseases 0.000 description 1
- 229940096763 panretin Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- VMZMNAABQBOLAK-DBILLSOUSA-N pasireotide Chemical compound C([C@H]1C(=O)N2C[C@@H](C[C@H]2C(=O)N[C@H](C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](C(N[C@@H](CC=2C=CC(OCC=3C=CC=CC=3)=CC=2)C(=O)N1)=O)CCCCN)C=1C=CC=CC=1)OC(=O)NCCN)C1=CC=CC=C1 VMZMNAABQBOLAK-DBILLSOUSA-N 0.000 description 1
- 229960005415 pasireotide Drugs 0.000 description 1
- 108700017947 pasireotide Proteins 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- ANRQGKOBLBYXFM-UHFFFAOYSA-M phenylmagnesium bromide Chemical compound Br[Mg]C1=CC=CC=C1 ANRQGKOBLBYXFM-UHFFFAOYSA-M 0.000 description 1
- 208000028591 pheochromocytoma Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 201000003113 pineoblastoma Diseases 0.000 description 1
- 229960005141 piperazine Drugs 0.000 description 1
- 208000021310 pituitary gland adenoma Diseases 0.000 description 1
- 208000010916 pituitary tumor Diseases 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 238000000634 powder X-ray diffraction Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000036316 preload Effects 0.000 description 1
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 208000029340 primitive neuroectodermal tumor Diseases 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940117820 purinethol Drugs 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- QLULGIRFKAWHOJ-UHFFFAOYSA-N pyridin-4-ylboronic acid Chemical compound OB(O)C1=CC=NC=C1 QLULGIRFKAWHOJ-UHFFFAOYSA-N 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- LTXJLQINBYSQFU-UHFFFAOYSA-N pyrimidin-5-ol Chemical compound OC1=CN=CN=C1 LTXJLQINBYSQFU-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 description 1
- 208000030859 renal pelvis/ureter urothelial carcinoma Diseases 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229940061969 rheumatrex Drugs 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 206010039667 schwannoma Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 201000008261 skin carcinoma Diseases 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 201000002314 small intestine cancer Diseases 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- LYPGDCWPTHTUDO-UHFFFAOYSA-M sodium;methanesulfinate Chemical compound [Na+].CS([O-])=O LYPGDCWPTHTUDO-UHFFFAOYSA-M 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 229940034345 sotret Drugs 0.000 description 1
- 208000037969 squamous neck cancer Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- 201000004059 subependymal giant cell astrocytoma Diseases 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229940099419 targretin Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229940061353 temodar Drugs 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- DVMUNQAGXAMHOR-UHFFFAOYSA-N tert-butyl 2,2-dimethylpiperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1(C)C DVMUNQAGXAMHOR-UHFFFAOYSA-N 0.000 description 1
- ITVHTQLHEQGQNG-UHFFFAOYSA-N tert-butyl 4,7-diazabicyclo[4.2.0]octane-7-carboxylate Chemical compound C1CNCC2N(C(=O)OC(C)(C)C)CC21 ITVHTQLHEQGQNG-UHFFFAOYSA-N 0.000 description 1
- JGOYBCZJFUTGRS-SHSPXAOASA-N tert-butyl 4-[[4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]phenyl]methyl]-4,7-diazabicyclo[4.2.0]octane-7-carboxylate Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CC4N(CC4CC3)C(=O)OC(C)(C)C)=CC=2)=N1 JGOYBCZJFUTGRS-SHSPXAOASA-N 0.000 description 1
- PPBFNRSAZMHTJV-QMMMGPOBSA-N tert-butyl n-[(1s)-1-(4-bromo-3-fluorophenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=C(Br)C(F)=C1 PPBFNRSAZMHTJV-QMMMGPOBSA-N 0.000 description 1
- QJRGKGZVTLHEAI-JTQLQIEISA-N tert-butyl n-[(1s)-1-(4-formylphenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](C)C1=CC=C(C=O)C=C1 QJRGKGZVTLHEAI-JTQLQIEISA-N 0.000 description 1
- RRBFCGUIFHFYQK-VIFPVBQESA-N tert-butyl n-[(2s)-1-[methoxy(methyl)amino]-3-methyl-1-oxobutan-2-yl]carbamate Chemical compound CON(C)C(=O)[C@H](C(C)C)NC(=O)OC(C)(C)C RRBFCGUIFHFYQK-VIFPVBQESA-N 0.000 description 1
- VIKWWQXKCQEMIB-GKAPJAKFSA-N tert-butyl n-[(3s)-2-hydroxy-4-methylpentan-3-yl]carbamate Chemical compound CC(C)[C@@H](C(C)O)NC(=O)OC(C)(C)C VIKWWQXKCQEMIB-GKAPJAKFSA-N 0.000 description 1
- FQDMQDQEPYFXMK-XUZZJYLKSA-N tert-butyl n-[4-methyl-1-[[4-[(1s)-1-[[4-[(4s)-2-oxo-4-propan-2-yl-1,3-oxazolidin-3-yl]pyrimidin-2-yl]amino]ethyl]phenyl]methyl]piperidin-4-yl]carbamate Chemical compound CC(C)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2C=CC(CN3CCC(C)(CC3)NC(=O)OC(C)(C)C)=CC=2)=N1 FQDMQDQEPYFXMK-XUZZJYLKSA-N 0.000 description 1
- CKXZPVPIDOJLLM-UHFFFAOYSA-N tert-butyl n-piperidin-4-ylcarbamate Chemical compound CC(C)(C)OC(=O)NC1CCNCC1 CKXZPVPIDOJLLM-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- YMBCJWGVCUEGHA-UHFFFAOYSA-M tetraethylammonium chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC YMBCJWGVCUEGHA-UHFFFAOYSA-M 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000005247 tetrazinyl group Chemical group N1=NN=NC(=C1)* 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 208000008732 thymoma Diseases 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 229940035307 toposar Drugs 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 229940066958 treanda Drugs 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 208000029387 trophoblastic neoplasm Diseases 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 208000037965 uterine sarcoma Diseases 0.000 description 1
- 206010046885 vaginal cancer Diseases 0.000 description 1
- 208000013139 vaginal neoplasm Diseases 0.000 description 1
- 201000005102 vulva cancer Diseases 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 229940053890 zanosar Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention is directed to novel 3-pyrimidinyl-4-yl-oxazolidin-2-one compounds, compositions containing these compounds, the use of such compounds in the inhibition of mutant IDH proteins having a neomorphic activity and in the treatment of diseases or disorders associated with such mutant IDH proteins including, but not limited to, cell-proliferation disorders, such as cancer.
- Isocitrate dehydrogenase is a key family of enzymes found in cellular metabolism. They are NADP + / NAD + and metal dependent oxidoreductases of the enzyme class EC 1 .1 .1.42.
- the wild type proteins catalyze the oxidative decarboxylation of isocitrate to alpha-ketoglutarate generating carbon dioxide and NADPH / NADH in the process. They are also known to convert oxalosuccinate into alpha-ketoglutarate.
- IDH1 cytosolic
- IDH2 mitochondrial
- glioma glioblastoma multiforme
- paraganglioma supratentorial primordial neuroectodermal tumors
- acute myeloid leukemia (AML) prostate cancer
- thyroid cancer colon cancer
- chondrosarcoma cholangiocarcinoma
- peripheral T-cell lymphoma peripheral T-cell lymphoma
- melanoma See L. Deng et al., Trends Mol. Med., 2010, 16, 387; T. Shibata et al., Am. J. Pathol., 201 1 , 178(3), 1395; Gaal et al., J.
- this invention provides for a compound of formula (I)
- R ⁇ -R ⁇ are defined herein.
- this invention provides for a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- this invention provides for the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, as an inhibitor of a mutant IDH protein having a neomorphic activity such as reducing alpha-ketoglutarate to 2-hydroxyglutarate (2-HG neomorphic activity).
- this invention provides for the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, as an inhibitor of mutant IDH1 having a neomorphic activity, such as 2-HG neomorphic activity, and/or mutant IDH2 having a neomorphic activity, such as 2-HG neomorphic activity.
- This invention further provides for the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, as an inhibitor of IDH1 having a mutation at residue 97, 100 or 132, for example G97D, R100Q, R132H, R132C, R132S, R132G, R132L, and R132V; and/or an inhibitor of I DH2 having a mutation at residue 140 or 172, for example R172K, R172M, R172S, R172G, and R172W.
- a compound of formula (I), or a pharmaceutically acceptable salt thereof as an inhibitor of IDH1 having a mutation at residue 97, 100 or 132, for example G97D, R100Q, R132H, R132C, R132S, R132G, R132L, and R132V; and/or an inhibitor of I DH2 having a mutation at residue 140 or 172, for example R172K, R172M, R172S, R172G, and R172W.
- this invention provides for a method of treating a disease or disorder associated with a mutant I DH protein having a neomorphic activity comprising administration of an effective amount of a compound according to formula (I), or a pharmaceutically acceptable salt thereof, to a subject in need thereof.
- the disease or disorder is a cell proliferation disorder, such as cancer.
- the cancer is brain cancer, such as glioma, glioblastoma multiforme, paraganglioma, and supratentorial primordial neuroectodermal tumors (pNET); leukemia, such as acute myeloid leukemia (AML), myelodysplastic syndrome, and chronic myelogenous leukemia (CML); skin cancer, including melanoma; prostate cancer; thyroid cancer; colon cancer; lung cancer; sarcoma, including central chondrosarcoma, central and periosteal chondroma; and fibrosarcoma.
- the disease or disorder is D-2-hydroxyglutaric aciduria.
- the invention provides for a compound of formula (I), or a pharmaceutically acceptable salt thereof, in combination with another therapeutic agent.
- the present invention is directed to a compound of formula (I)
- R and R 2 are each independently hydrogen, deuterium, halo, hydroxyl, NH2, aryl, heteroaryl, or optionally substituted C-1.4 alkyl,
- C-1.4 alkyl is optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, and NH2;
- R ⁇ A is hydrogen, deuterium, C-
- R3b is hydrogen, deuterium, or C-
- R ⁇ A and R 3b are joined together forming an optionally substituted 3-7 membered cycloalkyi ring or an optionally substituted 4-7 membered heterocyclic ring,
- cycloalkyi and heterocyclic rings are each optionally substituted with one or two substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH2, and C-1.3 alkyl;
- R4a is hydrogen, C-
- phenyl, benzyl, and heteroaryl rings are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.4 alkoxy, C-1.3 haloalkyl, C-1.3 haloalkoxy, C-
- R ⁇ b js hydrogen, deuterium, or C-1.3 alkyl
- R 4A and R 4B are joined together forming an optionally substituted 3-7 membered cycloalkyi ring or an optionally substituted 4-7 membered heterocyclic ring,
- cycloalkyi and heterocyclic rings are optionally substituted with one or two substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH2, and C-1.3 alkyl,
- R5a js hydrogen or deuterium
- R5b js hydrogen, deuterium, methyl, ethyl, CD3, CF3, CH2F, or CHF2 and
- R6 is optionally substituted C-
- aryl, heteroaryl, heterocyclic and C3.-10 cycloalkyi are optionally substituted with one to three substituents each independently selected from the group consisting of: halo; hydroxyl; cyano; nitro; C-1.4 alkoxy; C-1.3 haloalkyi; C-1.3 haloalkoxy;
- _5 alkyl C3_g cycloalkyi optionally substituted with one to three substituents each independently selected from the group consisting of: hydroxyl, cyano, C-1.3 alkyl, C-1.3 alkoxy, and C-1.3 haloalkyi; phenyl optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-
- -NR b R b 5-6 membered heteroaryl optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, C-1.3 alkyl, C-1.3 alkoxy; 5-6 membered heterocyclic optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH 2 , and C-
- n 1 , 2, or 3 and
- said C3.7 cycloalkyi and group of formula (a) are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyi, C-1.3 haloalkoxy, C-
- phenyl and heteroaryl are optionally substituted with one to three substituents each independently selected from the group consisting of halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyi, C-1.3 haloalkoxy, and C-1.3 alkyl,
- heterocyclic is optionally substituted with one to three substituents each independently selected from the group consisting of halo, hydroxyl, oxo, C-1.3 alkoxy, C-1.3 haloalkyl, C-1.3 haloalkoxy, C-1.4 alkyl, C3.5 cycloalkyl, -C(0)R' 3 , and -NRbRb; and
- substituents each independently selected from the group consisting of halo, hydroxyl, oxo, C-
- Alkyl refers to a monovalent saturated hydrocarbon chain having the specified number of carbon atoms.
- .g alkyl refers to an alkyl group having from 1 to 6 carbon atoms.
- Alkyl groups may be optionally substituted with one or more substituents as defined in formula (I).
- Alkyl groups may be straight or branched. Representative branched alkyl groups have one, two, or three branches.
- alkyl groups include, but are not limited to, methyl, ethyl, propyl (n-propyl and isopropyl), butyl (n-butyl, isobutyl, sec-butyl, and t-butyl), pentyl (n-pentyl, isopentyl, and neopentyl), and hexyl.
- Alkoxy refers to any alkyl moiety attached through an oxygen bridge (i.e. a -O-
- _3 alkyl group wherein C-1.3 alkyl is as defined herein).
- examples of such groups include, but are not limited to, methoxy, ethoxy, and propoxy.
- Aryl refers to a hydrocarbon ring system having an aromatic ring. Aryl groups are monocyclic ring systems or bicyclic ring systems. Monocyclic aryl ring refers to phenyl. Bicyclic aryl rings refer to naphthyl and to rings wherein phenyl is fused to a C5.
- Aryl groups may be optionally substituted with one or more substituents as defined in formula (I).
- Cycloalkyl refers to a saturated hydrocarbon ring system having the specified number of carbon atoms. Cycloalkyl groups are monocyclic or bicyclic ring systems. For example, C .10 cycloalkyl refers to a cycloalkyl group having from 5 to 10 carbon atoms.
- Cycloalkyl groups may be optionally substituted with one or more substituents as defined in formula (I).
- Examples of cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and adamantanyl.
- Cycloalkenyl refers to an unsaturated hydrocarbon ring system having the specified number of carbon atoms and having a carbon-carbon double bond within the ring.
- C5.7 cycloalkenyl refers to a cycloalkenyl group having from 5 to 7 carbon atoms.
- cycloalkenyl groups have one carbon-carbon double bond within the ring.
- cycloalkeneyl groups have more than one carbon-carbon double bond within the ring.
- Cycloalkenyl rings are not aromatic. Cycloalkenyl groups may be optionally substituted with one or more substituents as defined in formula (I).
- Halo refers to the halogen radicals fluoro, chloro, bromo, and iodo.
- Haloalkyi refers to an alkyl group wherein at least one hydrogen atom attached to a carbon atom within the alkyl group is replaced with halo.
- the number of halo substituents includes, but is not limited to, 1 , 2, 3, 4, 5, or 6 substituents.
- Haloalkyi includes, but is not limited to, monofluoromethyl, difluoroethyl, and trifluoromethyl.
- Haloalkoxy refers to a haloalkyi moiety attached through an oxygen bridge (i.e. a -0-C-
- An example of a haloalkoxy group is trifluoromethoxy.
- Heteroaryl refers to an aromatic ring system containing from 1 to 5 heteroatoms. Heteroaryl groups containing more than one heteroatom may contain different heteroatoms. Heteroaryl groups may be optionally substituted with one or more substituents as defined in formula (I). Heteroaryl groups are monocyclic ring systems or are fused bicyclic ring systems. Monocyclic heteroaryl rings have from 5 to 6 ring atoms. Bicyclic heteroaryl rings have from 8 to 10 member atoms. Bicyclic heteroaryl rings include those ring systems wherein a heteroaryl ring is fused to a phenyl ring.
- Heteroaryl includes, but is not limited to, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, oxadiazolyl (including 1 ,3,4-oxadiazolyl and 1 ,2,4-oxadiazolyl), thiazolyl, isothiazolyl, thiadiazolyl, furanyl, furanzanyl, thienyl, triazolyl, pyridinyl (including 2-, 3-, and 4- pyridinyl), pyrimidinyl, pyridazinyl, pyrazinyl, trazinyl, tetrazinyl, tetrzolyl, indonyl, isoindolyl, indolizinyl, indazolyl, purinyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl, benzimidazolyl
- Heteroatom refers to a nitrogen, oxygen, or sulfur atom.
- Heterocyclic refers to a 3 to 1 1 membered saturated or unsaturated monocyclic or bicyclic ring containing from 1 to 4 heteroatoms. Heterocyclic ring systems are not aromatic. Heterocyclic groups containing more than one heteroatom may contain different heteroatoms. Heterocyclic includes ring systems wherein a sulfur atom is oxidized to form SO or S02- Heterocyclic groups may be optionally substituted with one or more substituents as defined in formula (I). Heterocyclic groups are monocyclic, spiro, or fused or bridged bicyclic ring systems. Monocyclic heterocyclic rings have 3 to 7 ring atoms.
- Examples of monocyclic heterocyclic groups include oxtanyl, tetrahydrofuranyl, dihydrofuranyl, 1 ,4-dioxanyl, morpholinyl, 1 ,4-dithianyl, piperazinyl, piperidinyl, 1 ,3- dioxolanyl, imidazolidinyl, imidazolinyl, pyrrolinyl, pyrrolidinyl, tetrahydropyranyl, dihydropyranyl, oxathiolanyl, dithiolanyl, 1 ,3-dioxanyl, 1 ,3-dithianyl, oxathianyl, thiomorpholinyl, tetrahydro-thiopyran 1 , 1 -dioxide, 1 ,4-diazepanyl, and the like.
- Fused heterocyclic ring systems have from 8 to 1 1 ring atoms and include groups wherein a heterocyclic ring is fused to a phenyl ring, a heteroaryl ring or another heterocyclic ring.
- fused heterocyclic rings include 2,3-dihydrobenzo[b][1 ,4]dioxinyl, octahydro- pyrrolo[1 ,2-a]pyrazinyl, octahydro-pyrido[1 ,2-a]pyrazinyl, octahydro-pyrrolo[3,4-c]pyrrolyl, 5,6,7,8-tetrahydro-[1 ,2,4]triazolo[4,3-a]pyrazinyl, 5,6,7,8-tetrahydro-imidazo[1 ,2- a]pyrazinyl and the like.
- bridged heterocyclic groups examples include 3,8-diaza- bicyclo[3.2.1]octanyl, 3,8-diaza-bicyclo[4.2.0]octanyl and the like.
- spiro heterocyclic groups examples include 4,7-diaza-spiro[2.5]octanyl and the like.
- 4-7 membered heterocyclic refers to a heterocyclic group as defined above, having from 4 to 7 ring atoms and containing from 1 to 4 heteroatoms.
- 5-6 membered heterocylic refers to a heterocyclic group as defined above, having 5 or 6 ring atoms and containing from 1 to 4 heteroatoms.
- Optionally substituted indicates that a group, such as an alkyl, cycloalkyl, heteroaryl, heterocyclic, phenyl, and benzyl may be unsubstitued or the group may be substituted with one or more substituents as defined in formula (I).
- “Pharmaceutically acceptable” means a compound which is suitable for pharmaceutical use.
- Salts and solvates (e.g. hydrates and hydrates of salts) of compounds of the invention which are suitable for use in medicine are those where in the counterion or associated solvent is pharmaceutically acceptable.
- salts and solvates having non-pharmaceutically acceptable counterions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of the invention and their pharmaceutically acceptable salts and solvates.
- Substituted in reference to a group such as alkyl, phenyl, benzyl, heteroaryl, and heterocyclic, indicates that one or more hydrogen atoms attached to an atom within the group is replaced with a substituent selected from the group of defined substituents. It should be understood that the term “substituted” includes the implicit provision that such substitution be in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound (i.e. one that does not spontaneously undergo transformation, for example, by hydrolysis, rearrangement, cyclization, or elimination and that is sufficiently robust to survive isolation from a reaction mixture).
- a group may contain one or more substituents, one or more (as appropriate) atoms within the group may be substituted.
- a single atom within the group may be substituted with more than one substituent as long as such substitution is accordance with the permitted valence of the atom.
- Suitable substituents are defined for each substituted or optionally substituted group.
- salts including pharmaceutically acceptable salts, of the compounds according to formula (I) may be prepared. These salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- the pharmaceutically acceptable salts of the present invention can be synthesized from a basic or acidic moiety, by conventional chemical methods.
- such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid.
- a stoichiometric amount of the appropriate base such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like
- Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two.
- use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable.
- Solvates, including pharmaceutically acceptable solvates, of the compounds of formula (I) may also be prepared.
- “Solvate” refers to a complex of variable stoichiometry formed by a solute and solvent. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute. Examples of suitable solvents include, but are not limited to, water, MeOH, EtOH, and AcOH. Solvates wherein water is the solvent molecule are typically referred to as hydrates. Hydrates include compositions containing stoichiometric amounts of water, as well as compositions containing variable amounts of water.
- the compounds of formula (I), including salts and solvates thereof, may exist in crystalline forms, non-crystalline forms, or mixtures thereof.
- the compound or salt or solvate thereof may also exhibit polymorphism, i.e. the capacity of occurring in different crystalline forms. These different crystalline forms are typically known as "polymorphs". Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, all of which may be used for identification.
- different polymorphs may be produced, for example, by changing or adjusting the conditions used in crystallizing/recrystallizing a compound of formula (I).
- the invention also includes various isomers of the compounds of formula (I).
- “Isomer” refers to compounds that have the same composition and molecular weight but differ in physical and/or chemical properties. The structural difference may be in constitution (geometric isomers) or in the ability to rotate the plane of polarized light (stereosiomers). With regard to stereoisomers, the compounds of formula (I) may have one or more asymmetric carbon atom and may occur as racemates, racemic mixtures and as individual enantiomers or diastereomers. All such isomeric forms are included within the present invention, including mixtures thereof. If the compound contains a double bond, the substituent may be in the E or Z configuration. If the compound contains a disubstituted cycloalkyl, the cycloalkyl substituent may have a cis- or transconfiguration. All tautomeric forms are also intended to be included.
- any asymmetric atom (e.g., carbon or the like) of a compound of formula (I) can be present in racemic or enantiomerically enriched, for example the (/?)-, (S)- or (Reconfiguration.
- each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 % enantiomeric excess, at least 80 % enantiomeric excess, at least 90 % enantiomeric excess, at least 95 % enantiomeric excess, or at least 99 % enantiomeric excess in the (R)- or (S)- configuration.
- Substituents at atoms with unsaturated double bonds may, if possible, be present in cis- (Z)- or trans- (£)- form.
- a compound of formula (I) can be in the form of one of the possible isomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as substantially pure geometric (cis or trans) isomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof.
- Any resulting mixtures of isomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
- any resulting racemates of final products or intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound.
- a basic moiety may thus be employed to resolve the compounds of the present invention into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-0,0'-p-toluoyl tartaric acid, mandelic acid, malic acid or camphor-10-sulfonic acid.
- Racemic products can also be resolved by chiral chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
- HPLC high pressure liquid chromatography
- the invention includes unlabeled forms as well as isotopically labeled forms of compounds of formula (I).
- Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 F 31 P, 32 P, 35 S, 36 CI, 125 l respectively.
- the invention includes various isotopically labeled compounds as defined herein, for example those into which radioactive isotopes, such as 3 H and 14 C, or those into which non-radioactive isotopes, such as 2 H and 13 C are present.
- isotopically labelled compounds are useful in metabolic studies (with 14 C), reaction kinetic studies (with, for example 2 H or 3 H), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients.
- PET positron emission tomography
- SPECT single-photon emission computed tomography
- an 18 F or labeled compound may be particularly desirable for PET or SPECT studies.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non- labeled reagent previously employed.
- D) may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index.
- deuterium in this context is regarded as a substituent of a compound of the formula (I).
- concentration of such a heavier isotope, specifically deuterium may be defined by the isotopic enrichment factor.
- isotopic enrichment factor as used herein means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
- One embodiment of the present invention is a compound according to formula (I) wherein:
- each R1 and R ⁇ is independently hydrogen, deuterium, halo, hydroxyl, NH2, aryl, heteroaryl, or optionally substituted C-1.4 alkyl,
- C-1.4 alkyl is optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, and NH2;
- R ⁇ a is hydrogen, deuterium, C-
- R3b is hydrogen, deuterium, or C-
- R ⁇ a and R 3b are joined together forming an optionally substituted 3-7 membered cycloalkyl ring or an optionally substituted 4-7 membered heterocyclic ring,
- cycloalkyl and heterocyclic rings are optionally substituted with one or two substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH2, and C1.3 alkyl;
- R4a is hydrogen, C-
- phenyl, benzyl, and heteroaryl rings are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyl, C-1.3 haloalkoxy, C-
- R4b is hydrogen, deuterium, or C-1.3 alkyl
- R ⁇ a and R 4b are joined together forming an optionally substituted 3-7 membered cycloalkyl ring or an optionally substituted 4-7 membered heterocyclic ring,
- cycloalkyl and heterocyclic rings are optionally substituted with one or two substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH2, and C-1.3 alkyl,
- R ⁇ a is hydrogen or deuterium
- R 5b is hydrogen, deuterium, methyl, ethyl, CD3, CF3, CH2F, or CHF2 and
- R6 is optionally substituted C-
- aryl, heteroaryl, heterocyclic and C .10 cycloalkyi are optionally substituted with one to three substituents each independently selected from the group consisting of: halo; hydroxyl; cyano; nitro; C-1.3 alkoxy; C-1.3 haloalkyi; C-1.3 haloalkoxy;
- NR B R B 5-6 membered heteroaryl; 5-6 membered heterocyclic optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH 2 , and C-
- R5t> and R ⁇ are joined together forming an optionally substituted C3.7 cycloalkyi group
- n 1 , 2, or 3 and
- said C3.7 cycloalkyi and group of formula (a) are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyi, C-1.3 haloalkoxy, C-
- R ⁇ is hydrogen, halo, or optionally substituted C-1.4 alkyl.
- R ⁇ is hydrogen, fluoro, chloro, or methyl.
- R ⁇ is hydrogen, fluoro or chloro.
- R ⁇ is hydrogen.
- R2 is hydrogen, halo or optionally substituted C-1.4 alkyl.
- R2 is hydrogen, fluoro, chloro, or methyl.
- R2 is hydrogen or fluoro.
- R2 is hydrogen.
- R ⁇ and R2 are both hydrogen.
- R ⁇ a is hydrogen, C ⁇
- R ⁇ a is hydrogen, methyl, or phenyl.
- R ⁇ a is hydrogen or methyl.
- R ⁇ a is hydrogen.
- R3b is hydrogen or methyl.
- R 3b is hydrogen.
- R ⁇ a and R ⁇ b are both hydrogen.
- R ⁇ a and R 3b are joined together forming oxetanyl or tetrahydro-2H-pyranyl.
- R ⁇ a is hydrogen, C-
- phenyl, benzyl, and heteroaryl rings are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyi, C-1.3 haloalkoxy, C-
- R ⁇ a j s hydrogen, C1.4 alkyl, optionally substituted phenyl, optionally substituted benzyl, optionally substituted heteroaryl, or methylene-dibenzene.
- R ⁇ a is hydrogen, C-1.4 alkyl, optionally substituted phenyl, optionally substituted benzyl, optionally substituted pyridinyl, or methylene-dibenzene. More suitably R ⁇ a is hydrogen, methyl, isopropyl, isobutyl, t- butyl, phenyl, 4-methoxyphenyl, 4-fluorophenyl, benzyl, or methylene-dibenzene.
- R ⁇ a j hydrogen, methyl, ethyl, isopropyl, phenyl, 4-fluorophenyl, 4- methoxyphenyl, biphenyl, benzyl, or pyridinyl.
- R ⁇ a js isopropyl.
- R 4b is hydrogen or methyl.
- R ⁇ a j s isopropyl and R ⁇ b js methyl. In another embodiment R ⁇ a j s isopropyl and R 4b is hydrogen.
- R ⁇ a and R 4b are joined together forming cyclopentyl.
- R ⁇ a j hydrogen. In another embodiment of the present invention R ⁇ a j s deuterium. In another embodiment of the present invention R5b is hydrogen, methyl, ethyl, or CF3. Suitably R 5b is methyl.
- R ⁇ is optionally substituted heteroaryl, optionally substituted heterocyclic or optionally substituted C .10 cycloalkyi.
- R ⁇ is methyl, C .10 cycloalkyi, optionally substituted phenyl, optionally substituted pyridinyl, optionally substituted pyrimidinyl, optionally substituted pyridazinyl, optionally substituted pyrazinyl, optionally substituted triazolyl, optionally substituted pyrazolyl, optionally substituted thiazolyl, optionally substitued 1 ,3,4-oxadiazolyl, optionally substituted 1 ,2,4-oxadiazolyl, optionally substitued isoxazolyl, thienyl, oxazolyl, quinolinyl, optionally substituted benzimidazolyl, benzthiazolyl, benzoxazolyl, tetrazolo[1 ,5-a]pyridinyl, imidazo[2, 1 -b][1 ,3,4]thiadiazolyl, optionally substituted piperidinyl, optionally
- R ⁇ is phenyl optionally substituted with one or two substituents.
- R ⁇ is optionally substituted 1 ,3,4-oxadiazolyl or 1 ,2,4- optionally substituted oxadiazolyl.
- R6 is pyrimidinyl optionally substituted with one substituent.
- R ⁇ is optionally substituted with one or two substituents each independently selected from the group consisting of: halo; hydroxy; nitro; C-1.4 alkoxy; C-1.3 haloalkyl; C-1.3 haloalkoxy; C-
- substituents each independently selected from the group consisting of: halo; hydroxy; nitro; C-1.4 alkoxy; C-1.3 haloalkyl; C-
- R 6 is substituted with one -CH2R a , -C(0)R a , -NHC(0)R a , -NHC(0)R b , -C(0)NHR a , -C(0)NHR b , -OR a , -NR a R b , -S0 2 NR b R b , -S02R a , or -SC>2R b group.
- R 6 is substituted with one -CH2R a , -C(0)R a , or -OR a group.
- R6 is phenyl substituted with one fluoro or chloro group and one -CH 2 R a , -C(0)R a , or -C(0)N HR a group wherein the -CH 2 R a , -C(0)R a , or
- R ⁇ is phenyl substituted with one fluoro group and one -CH2R a , -C(0)R a , or -C(0)NHR a group wherein the -CH2R a , -C(0)R a , or -C(0)NHR a group is in the para position of the phenyl ring.
- R ⁇ is phenyl substituted with one -CH2R a , -C(0)R a , or
- R ⁇ is phenyl substituted by -CH2R a in the para position.
- R a is phenyl optionally substituted with one or two substituents each independently selected from the group consisting of fluoro, chloro and bromo.
- R a is an optionally substituted 5-6 membered heteroaryl.
- R a is optionally substituted pyridinyl or optionally substituted pyrimidinyl.
- R a is pyridinyl or pyrimidinyl optionally substituted with one trifluoromethyl.
- R a is C5.7 cycloalkyl each of which is optionally substituted with one or two substituents each independently selected from the group consisting of fluoro, hydroxy, methyl, and C-1.3 haloalkoxy.
- R a is optionally substituted heterocyclic.
- R a is piperidinyl, piperazinyl, morpholinyl, tetrahydropyranyl, tetrahydro-thiopyran 1 , 1 -dioxide, 1 ,4-diazepanyl, 4,7-diaza-spiro[2.5]octanyl, 3,8-diaza-bicyclo[3.2.1 ]octanyl, 3,8-diaza- bicyclo[4.2.0]octanyl, octahydro-pyrrolo[1 ,2-a]pyrazinyl, octahydro-pyrido[1 ,2-a]pyrazinyl, octahydro-pyrrolo[3,4-c]pyrrolyl, and 5,6,7,8-tetrahydro-imidazo[1 ,2-a]pyrazinyl each of which is optionally substituted with one to
- R a is piperidinyl, piperazinyl, or morpholinyl each of which is optionally substitued with one to three substituents each independently selected from the group consisting of: hydroxy, fluoro, amino, dimethylamino, C-1.3 haloalkoxy, C-1.3 alkyl, and C3.5 cycloalkyi.
- R ⁇ b and R ⁇ are joined together forming an optionally substituted C3.7 cycloalkyi group or an optionally substituted group of formula (a).
- each Rb is independently hydrogen or methyl.
- R2 is fluoro and R ⁇ 3 , R ⁇ b, R4a anc
- R4b are each hydrogen.
- R 4b is hydrogen
- Selected compounds of the present invention include:
- Selected compounds of the present invention include:
- Selected compounds of the present invention include:
- Embodiment 1 A compound of formula (I)
- each R1 and R2 is independently hydrogen, deuterium, halo, hydroxyl, NH2, aryl, heteroaryl, or optionally substituted C-1.4 alkyl,
- C-1.4 alkyl is optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, and NH2;
- R3 3 is hydrogen, deuterium, C-
- R3b is hydrogen, deuterium, or C-
- R3a and R 3B are joined together forming an optionally substituted 3-7 membered cycloalkyl ring or an optionally substituted 4-7 membered heterocyclic ring,
- cycloalkyl and heterocyclic rings are each optionally substituted with one or two substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, N H2, and C-1.3 alkyl;
- R4a is hydrogen, C-
- phenyl, benzyl, and heteroaryl rings are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyi, C-1.3 haloalkoxy, C-
- R4b is hydrogen, deuterium, or C-1.3 alkyl
- R ⁇ a and R 4b are joined together forming an optionally substituted 3-7 membered cycloalkyl ring or an optionally substituted 4-7 membered heterocyclic ring,
- cycloalkyl and heterocyclic rings are optionally substituted with one or two substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH2, and C-1.3 alkyl, provided that only one of R ⁇ a and R 3b and R ⁇ a and R 4b are joined together forming a ring;
- R5a js hydrogen or deuterium
- R 5b is hydrogen, deuterium, methyl, ethyl, CD3, CF3, CH2F, or CHF2 and
- R6 is optionally substituted C-
- aryl, heteroaryl, heterocyclic and C5.-10 cycloalkyi are optionally substituted with one to three substituents each independently selected from the group consisting of: halo; hydroxyl; cyano; nitro; C-1.3 alkoxy; C-1.3 haloalkyi; C-1.3 haloalkoxy;
- NR b R b 5-6 membered heteroaryl; 5-6 membered heterocyclic optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, oxo, NH 2 , and C-
- R 5b and R6 are joined together forming an optionally substituted C3.7 cycloalkyi group
- n 1 , 2, or 3 and
- said C3.7 cycloalkyi and group of formula (a) are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyi, C-1.3 haloalkoxy, C-
- phenyl and heteroaryl are optionally substituted with one to three substituents each independently selected from the group consisting of halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyl, C-1.3 haloalkoxy, and C-1.3 alkyl,
- said 4-7 membered heterocyclic is optionally substituted with one to three substituents each independently selected from the group consisting of halo, hydroxyl, oxo, C-1.3 alkoxy, C-1.3 haloalkyl, C-1.3 haloalkoxy, and C-1.3 alkyl; and each R b is independently hydrogen or C-
- Embodiment 2 The compound according to embodiment 1 wherein R2 is hydrogen; or a pharmaceutically acceptable salt thereof.
- Embodiment 3 The compound according to embodiment 2 wherein is hydrogen, halo, or optionally substituted C-1.4 alkyl; or a pharmaceutically acceptable salt thereof.
- Embodiment 4 The compound according to embodiment 3 wherein is hydrogen, fluoro, chloro, or methyl; or a pharmaceutically acceptable salt thereof.
- Embodiment 5 The compound according to embodiment 4 wherein R ⁇ a is hydrogen, C-
- Embodiment 6 The compound according to embodiment 5 wherein R3 b is hydrogen or methyl; or a pharmaceutically acceptable salt thereof.
- Embodiment 7 The compound according to embodiment 6 wherein R ⁇ a is hydrogen, methyl, or phenyl; or a pharmaceutically acceptable salt thereof.
- Embodiment 8 The compound according to embodiment 7 wherein R ⁇ a is hydrogen, C-
- phenyl, benzyl, and heteroaryl rings are optionally substituted with one to three substituents each independently selected from the group consisting of: halo, hydroxyl, cyano, nitro, C-1.3 alkoxy, C-1.3 haloalkyl, C-1.3 haloalkoxy, C-
- Embodiment 9 The compound according to embodiment 8 wherein R ⁇ b j s hydrogen or methyl; or a pharmaceutically acceptable salt thereof.
- Embodiment 10 The compound according to embodiment 9 wherein R ⁇ a is hydrogen, C-
- Embodiment 1 1 .
- Embodiment 12 The compound according to embodiment 1 1 wherein R ⁇ a j s H; or a pharmaceutically acceptable salt thereof.
- Embodiment 13 The compound according to embodiment 12 wherein R ⁇ b j s hydrogen, methyl, ethyl, or CF3.
- Embodiment 14 The compound according to embodiment 13 wherein R ⁇ is isopropyl, optionally substituted aryl, optionally substituted pyrazolyl, optionally substituted pyridinyl, 2,3-dihydrobenzofuranyl, 2,3-dihydrobenzo[b][1 ,4]dioxinyl, or optionally substituted C .10 cycloalkyl; or a pharmaceutically acceptable salt thereof.
- Embodiment 15 A pharmaceutical composition comprising a compound according to embodiment 1 , or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- Embodiment 16 A method for the treatment of a disease or disorder associated with a mutant IDH protein having a neomorphic activity comprising administration of a therapeutically effective amount of a compound according to embodiment 1 , or a pharmaceutically acceptable salt thereof, to subject in need of thereof.
- Embodiment 17 A method for the treatment of a disease or disorder associated with a mutant IDH protein having a neomorphic activity comprising administration of a therapeutically effective amount of a compound according to embodiment 1 , or a pharmaceutically acceptable salt thereof, and another therapeutic agent to subject in need of thereof.
- the compounds of the present invention may be made by a variety of methods, including standard chemistry. Suitable synthetic routes are depicted in the Schemes given below.
- the compounds of formula (I) may be prepared by methods known in the art of organic synthesis as set forth in part by the following synthetic schemes. In the schemes described below, it is well understood that protecting groups for sensitive or reactive groups are employed where necessary in accordance with general principles or chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection processes, as well as the reaction conditions and order of their execution, shall be consistent with the preparation of compounds of formula (I).
- the present invention includes both possible stereoisomers and includes not only racemic compounds but the individual enantiomers and/or diastereomers as well.
- a compound When a compound is desired as a single enantiomer or diastereomer, it may be obtained by stereospecific synthesis or by resolution of the final product or any convenient intermediate. Resolution of the final product, an intermediate, or a starting material may be effected by any suitable method known in the art. See, for example, "Stereochemistry of Organic Compounds" by E. L. Eliel, S. H. Wilen, and L. N. Mander (Wiley-lnterscience, 1994).
- the compounds described herein may be made from commercially available starting materials or synthesized using known organic, inorganic, and/or enzymatic processes.
- Non-commercial aminoacids can be prepared following the procedures of Scheme 1 . Conversion of ketone 1 to the corresponding imidazolidine-2,4-dione 2 followed by hydrolysis provides aminoacid 3.
- aminoalcohol precursor of oxazolidinone
- R 3a R 3b
- protected aminoester 5 is treated with an appropriate Grignard reagent to give protected aminoalcohol 6 which goes through basic or acidic deprotection step.
- R 3a ⁇ R 3b protected aminoacid 8 is converted into Weinreb amide 9 which is treated with different Grignard reagents sequentially to provide protected aminoalcohol 10. Either basic or acidic deprotection of 10 gives 11.
- Oxazolidinone 12 is coupled with dihalogen-pyrimidine 13 in the presence of NaH and the resulting 14 is treated with primary amine 15 under several different reaction conditions as shown in Scheme 3 to provide 16.
- intermediate 14 can be prepared by coupling the amino alcohol 11 and dihalogen-pyrimidine 13 in the presence of a base such as diisopropylethyl amine resulting in intermediate 17 which can be treated with triphosgene in the presence of a base such as 2,6-lutidine resulting in intermediate 14.
- a base such as diisopropylethyl amine
- the compounds of the present invention are inhibitors of a mutant IDH protein having a neomorphic activity and are therefore useful in the treatment of diseases or disorders associated with such proteins including, but not limited to, cell proliferation disorders, such as cancer.
- mutant IDH protein having a neomorphic activity examples include mutant IDH1 and mutant IDH2.
- a neomorphic activity associated with mutant IDH1 and mutant IDH2 is the ability to produce 2-hydroxyglutarate (2-HG neomorphic activity), specifically R-2- HG (R-2-HG neomorphic activity).
- Mutations in IDH 1 associated with 2-HG neomorphic activity, specifically R-2-HG neomorphic activity include mutations at residues 97, 100, and 132, e.g. G97D, R100Q, R132H, R132C, R132S, R132G, R132L, and R132V.
- Mutations in IDH2 associated with 2-HG neoactivity, specifically R-2-HG neomorphic activity, include mutations at residues 140 and 172, e.g. R140Q, R140G, R172K, R172M, R172S, R172G, and R172W.
- Cell-proliferation disorders associated with a mutant IDH protein having a neomorphic activity include, but are not limited to, cancer.
- cancers include Acute Lymphoblastic Leukemia, Adult; Acute Lymphoblastic Leukemia, Childhood; Acute Myeloid Leukemia, Adult; Adrenocortical Carcinoma; Adrenocortical Carcinoma, Childhood; AIDS-Related Lymphoma; AIDS-Related Malignancies; Anal Cancer; Astrocytoma, Childhood Cerebellar; Astrocytoma, Childhood Cerebral; Bile Duct Cancer, Extrahepatic; Bladder Cancer; Bladder Cancer, Childhood; Bone Cancer, Osteosarcoma/Malignant Fibrous Histiocytoma; Brain Stem Glioma, Childhood; Brain Tumor, Adult; Brain Tumor, Brain Stem Glioma, Childhood; Brain Tumor, Cerebellar Astrocytoma, Childhood; Brain Tumor, Cerebral
- the cancer associated with a mutant IDH protein having a neomorphic acitvity is brain cancer, such as astrocytic tumor (e.g., pilocytic astrocytoma, subependymal giant-cell astrocytoma, diffuse astrocytoma, pleomorphic xanthoastrocytoma, anaplastic astrocytoma, astrocytoma, giant cell glioblastoma, glioblastoma, secondary glioblastoma, primary adult glioblastoma, and primary pediatric glioblastoma); oligodendroglial tumor (e.g., oligodendroglioma, and anaplastic oligodendroglioma); oligoastrocytic tumor (e.g., oligoastrocytoma, and anaplastic oligoastrocytoma); ependymoma (e.g., myxop
- the cancer associated with a mutant IDH protein having a neomorphic acitvity is leukemia, such as acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), chronic myelogenous leukemia (CML), myeloproliferative neoplasm (MPN), MDS.MPN including chronic myelomonocytic leukemia, post MDS AML, post MPN AML, post MDS/MPN AML, del(5q)-associated high risk MDS or AML, blast-phase chronic myelogenous leukemia, angioimmunoblastic lymphoma and acute lymphoblastic leukemia.
- AML acute myeloid leukemia
- MDS myelodysplastic syndrome
- CML chronic myelogenous leukemia
- MPN myeloproliferative neoplasm
- MDS.MPN including chronic myelomonocytic leukemia, post MDS AML, post
- the cancer associated with a mutant IDH protein having a neomorphic activity is skin cancer, including melanoma.
- the cancer associated with a mutant IDH protein having a neomorphic activity is prostate cancer, thyroid cancer, colon cancer, or lung cancer.
- the cancer associated with a mutant IDH protein having a neomorphic activity is sarcoma, including central chondrosarcoma, central and periosteal chondroma, and fibrosarcoma. In another embodiment the cancer associated with a mutant IDH protein having a neomorphic activity is cholangiocarcinoma.
- Another disease or disorder associated with a mutant IDH protein having R-2-HG neomorphic activity is D-2-hydroxyglutaric aciduria.
- Another disease or disorder associated with a mutant IDH protein having R-2-HG neomorphic activity is Diller disease and Mafucci syndrome.
- neomorphic activity refers to a gain of novel activity of a protein that the wild-type protein does not have or does not exhibit to a significant degree.
- a neomorphic activity associated with a mutant form of IDH 1 and IDH2 is the ability to reduce alpha-ketoglutarate to 2-hydroxyglutarate (i.e. 2-HG, specifically R-2-HG).
- the wild type form of IDH1 and IDH2 does not have the ability to reduce alpha-ketoglutarate to 2-hydroxyglutarate (i.e. 2-HG, specifically R-2-HG) or if it does have this ability, it does not produce significant (i.e. harmful or disease causing) amounts of 2-HG.
- the term "subject" refers to an animal. Typically the animal is a mammal. A subject also refers to for example, primates (e.g., humans, male or female), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice, fish, birds and the like. In certain embodiments, the subject is a primate. In yet other embodiments, the subject is a human.
- primates e.g., humans, male or female
- the subject is a primate.
- the subject is a human.
- the term "therapeutically effective amount" in reference to a compound of the invention means an amount of the compound sufficient to treat the subject's disease or condition, but low enough to avoid serious sides effects (at a reasonable benefit/risk ratio) within the scope of sound medical judgment.
- a therapeutically effective amount of a compound will vary with the particular compound chosen (e.g. consider the potency, efficacy, and half-life of the compound); the route of administration chosen; the condition being treated; the severity of the condition being treated; the age, size, weight, and physical condition of the subject being treated; the medical history of the subject being treated; the duration of the treatment; the nature of the concurrent therapy; the desired therapeutic effect; and like factors and can be routinely determined by the skilled artisan.
- the term “treat”, “treating” or “treatment” of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat”, “treating” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- “treat”, “treating” or “treatment” refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both.
- “treat”, “treating” or “treatment” refers to preventing or delaying the onset or development or progression of the disease or disorder.
- a subject is "in need of” a treatment if such subject would benefit biologically, medically or in quality of life from such treatment.
- the compounds of the present invention may be administered by any suitable route including oral and parenteral administration.
- Parenteral administration is typically by injection or infusion and includes intravenous, intramuscular, and subcontaneous injection or infusion.
- the compounds of the invention may be administered once or according to a dosing regimen wherein a number of doses are administered at varying intervals of time for a given period of time. For example, doses may be administered one, two, three, or four times per day. Doses may be administered until the desired therapeutic effect is achieved or indefinitely to maintain the desired therapeutic effect. Suitable dosing regimens for a compound of the invention depend on the pharmacokinetic properties of that compound, such as absorption, distribution and half life which can be determined by the skilled artisan.
- suitable dosing regimens including the duration such regimens are administered, for a compound of the invention depend on the disease or condition being treated, the severity of the disease or condition, the age and physical condition of the subject being treated, the medical history of the subject being treated, the nature of concurrent therapy, the desired therapeutic effect, and like factors within the knowledge and expertise of the skilled artisan. It will be further understood by such skilled artisans that suitable dosing regimens may require adjustment given an individual subject's response to the dosing regimen or over time as the individual subject needs change. Typical daily dosages may vary depending upon the particular route of administration chosen. Typical daily dosages for oral administration, to a human weighing approximately 70kg would range from about 5mg to about 500mg of a compound of formula (I).
- One embodiment of the present invention provides for a method of treating a disease or disorder associated with a mutant form of IDH having a neomorphic activity comprising administration of a therapeutically effective amount of a compound of formula (I) to a subject in need of treatment thereof.
- the disease or disorder associated with a mutant form of IDH having a neomorphic activity is a cell proliferation disorder.
- the cell proliferation disorder is cancer.
- the cancer is a cancer associated with mutant IDH1 having 2-HG neomorphic activity or mutant IDH2 having 2-HG neomorphic activity.
- the neomorphic activity is R-2-HG neomorphic activity.
- the cancer is associated with mutant IDH1 having 2-HG or R-2-HG neomorphic activity having a mutation at residues 97, 100, or 132, such as G97D, R100Q, R132H, R132C, R132S, R132G, R132L, and R132V.
- the cancer is associated with mutant IDH2 having 2-HG or R-2-HG neomorphic activity having a mutation at residues 140 or 172, e.g. R140Q, R140G, R172K, R172M, R172S, R172G, and R172W.
- the cancer is brain cancer, leukemia, skin cancer, prostate cancer, thyroid cancer, colon cancer, lung cancer or sarcoma.
- the cancer is glioma, glioblastoma multiforme, paraganglioma, suprantentorial primordial neuroectodermal tumors, acute myeloid leukemia, myelodysplastic syndrome, chronic myelogenous leukemia, melanoma, prostate, thyroid, colon, lung, central chondrosarcoma, central and periosteal chondroma tumors, fibrosarcoma, and cholangiocarcinoma.
- Another embodiment of the present invention provides for a method of treating a disease or disorder associated with a mutant form of IDH having R-2-HG neomorphic activity comprising administration of a therapeutically effective amount of a compound according to formula (I) to a subject in need thereof wherein the disease or disorder is D- 2-hydroxyglutaric aciduria, Oilier Disease, or Mafucci Syndrome.
- the therapy is a disease or disorder associated with a mutant form of IDH having a neomorphic activity.
- the therapy is a cell proliferation disorder associated with a mutant form of IDH having a neomorphic activity.
- the therapy is cancer.
- the therapy is a cancer associated with a mutant IDH protein having a neomorphic activity, such as mutant IDH1 having 2-HG neomorphic activity or mutant IDH2 having 2-HG neomorphic activity.
- the neomorphic activity is R-2-HG neomorphic activity.
- the cancer is associated with mutant IDH1 having 2-HG or R-2-HG neomorphic activity having a mutation at residues 97, 100, or 132, such as G97D, R100Q, R132H, R132C, R132S, R132G, R132L, and R132V.
- the cancer is associated with mutant IDH2 having 2-HG or R-2-HG neomorphic activity having a mutation at residue at residues R140 or 172, e.g. R140Q, R140G, R172K, R172M, R172S, R172G, and R172W.
- the cancer is brain cancer, leukemia, skin cancer, prostate cancer, thyroid cancer, colon cancer, lung cancer or sarcoma.
- the cancer is glioma, glioblastoma multiforme, paraganglioma, suprantentorial primordial neuroectodermal tumors, acute myeloid leukemia, myelodysplastic syndrome, chronic myelogenous leukemia, melanoma, prostate, thyroid, colon, lung, central chondrosarcoma, central and periosteal chondroma tumors, fibrosarcoma, and cholangiocarcinoma.
- Another embodiment of the present invention provides for the use of a compound of formula (I) in therapy wherein the therapy is D-2-hydroxyglutaric aciduria, Oilier Disease, or Mafucci Syndrome.
- Another embodiment of the present invention provides for the use of a compound according to formula (I) in the manufacture of a medicament for the treatment of disease or disorder associated with a mutant form of IDH having a neomorphic activity.
- the disease or disorder associated with a mutant form of IDH having a neomorphic activity is a cell proliferation disorder.
- the cell proliferation disorder is cancer.
- the cancer is a cancer associated with a mutant IDH protein having a neomorphic activity, such as mutant IDH1 having 2-HG neomorphic activity or mutant IDH2 having 2-HG neomorphic activity.
- the neomorphic activity is R-2-HG neomorphic activity.
- the cancer is associated with mutant IDH1 having 2-HG or R-2-HG neomorphic activity having a mutation at residues 97, 100, or 132, such as G97D, R100Q, R132H, R132C, R132S, R132G, R132L, and R132V.
- the cancer is associated with mutant IDH2 having 2-HG or R-2-HG neomorphic activity having a mutation at residue at residues 140 or 172, e.g. R140Q, R140G, R172K, R172M, R172S, R172G, and R172W.
- the cancer is brain cancer, leukemia, skin cancer, prostate cancer, thyroid cancer, colon cancer, lung cancer or sarcoma.
- the cancer is glioma, glioblastoma multiforme, paraganglioma, suprantentorial primordial neuroectodermal tumors, acute myeloid leukemia, myelodysplastic syndrome, chronic myelogenous leukemia, melanoma, prostate, thyroid, colon, lung, central chondrosarcoma, central and periosteal chondroma tumors, fibrosarcoma, and cholangiocarcinoma.
- Another embodiment of the present invention provides for the use of a compound according to formula (I) in the manufacture of a medicament for the treatment of disease or disorder associated with a mutant form of IDH having R-2-HG neomorphic activity wherein the disease or disorder is D-2-hydroxyglutaric aciduria, Oilier Disease, or Mafucci Syndrome.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) and a pharmaceutically acceptable carrier or excipient.
- the pharmaceutical compositions of the invention may be prepared and packaged in bulk form wherein a therapeutically effective amount of a compound of the invention can be extracted and then given to a subject, such as with powders or syrups.
- the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form wherein each physically discrete unit contains a therapeutically effective amount of a compound of the invention.
- the pharmaceutical compositions of the invention typically contain from about 5mg to 500mg of a compound of formula (I).
- pharmaceutically acceptable carrier or excipient means a pharmaceutically acceptable material, composition or vehicle that, for example, are involved in giving form or consistency to the pharmaceutical composition.
- Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a subject and interactions which would result in pharmaceutical compositions that are not pharmaceutically acceptable are avoided.
- each excipient must, of course, be of sufficiently high purity to render it pharmaceutically acceptable.
- the compound of the invention and the pharmaceutically acceptable carrier or excipient(s) will typically be formulated into a dosage form adapted for administration to the subject by the desired route of administration.
- dosage forms include those adapted for (1 ) oral administration such as tablets, capsules, caplets, pills, troches, powders, syrups, elixirs, suspensions, solutions, emulsions, sachets, and cachets; and (2) parenteral administration such as sterile solutions, suspensions, and powders for reconstitution.
- suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen.
- suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition.
- certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of the invention, once administered to the subject, from one organ or portion of the body to another organ or another portion of the body. Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
- Suitable pharmaceutically acceptable excipients include the following types of excipients: diluents, lubricants, binders, disintegrants, fillers, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweeteners, flavoring agents, flavor masking agents, coloring agents, anti-caking agents, hemectants, chelating agents, plasticizers, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents.
- Skilled artisans possess the knowledge and skill in the art to enable them to select suitable pharmaceutically acceptable carriers and excipients in appropriate amounts for the use in the invention.
- resources available to the skilled artisan which describe pharmaceutically acceptable carriers and excipients and may be useful in selecting suitable pharmaceutically acceptable carriers and excipients. Examples include Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
- compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
- the invention is directed to a solid oral dosage form such as a tablet or capsule comprising a therapeutically effective amount of a compound of the invention and a diluent or filler.
- Suitable diluents and fillers include lactose, sucrose, dextrose, mannitol, sorbitol, starch (e.g. corn starch, potato starch, and pre-gelatinized starch), cellulose and its derivatives, (e.g. microcrystalline cellulose), calcium sulfate, and dibasic calcium phosphate.
- the oral solid dosage form may further comprise a binder. Suitable binders include starch (e.g.
- the oral solid dosage form may further comprise a disintegrant. Suitable disintegrants include crospovidone, sodium starch glycolate, croscarmelose, alginic acid, and sodium carboxymethyl cellulose.
- the oral solid dosage form may further comprise a lubricant. Suitable lubricants include stearic acid, magnesium stearate, calcium stearate, and talc.
- dosage unit formulations for oral administration can be microencapsulated.
- the composition can also be prepared to prolong or sustain the release as, for example, by coating or embedding particulate material in polymers, wax, or the like.
- the compounds of the invention may also be coupled with soluble polymers as targetable drug carriers.
- soluble polymers can include polyvinylpyrrolidone, pyrancopolymer, polyhydroxypropylmethacrylamidephenol, polyhydroxyethylaspartamidephenol, or polyethyleneoxidepolylysine substituted with palmitoyl residues.
- the compounds of the invention may be coupled to a class of biodegradable polymers useful in achieving controlled release of a drug, for example polylactic acid, polepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- the invention is directed to a liquid oral dosage form.
- Oral liquids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of a compound of the invention.
- Syrups can be prepared by dissolving the compound of the invention in a suitably flavored aqueous solution; while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound of the invention in a non-toxic vehicle.
- Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additives such as peppermint oil or other natural sweeteners or saccharin or other artificial sweeteners and the like can also be added.
- the invention is directed to parenteral administration.
- compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the compositions may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- the compound of the present invention may be administered either simultaneously with, or before or after, one or more other therapeutic agent(s).
- the compound of the present invention may be administered separately, by the same or different route of administration, or together in the same pharmaceutical composition as the other agent(s).
- the invention provides a product comprising a compound of formula (I) and at least one other therapeutic agent as a combined preparation for simultaneous, separate or sequential use in therapy.
- the therapy is the treatment of a disease or disorder associated with a mutant form of IDH.
- Products provided as a combined preparation include a composition comprising the compound of formula (I) and the other therapeutic agent(s) together in the same pharmaceutical composition, or the compound of formula (I) and the other therapeutic agent(s) in separate form, e.g. in the form of a kit.
- the invention provides a pharmaceutical composition comprising a compound of formula (I) and another therapeutic agent(s).
- the pharmaceutical composition may comprise a pharmaceutically acceptable excipient, as described above.
- the invention provides a kit comprising two or more separate pharmaceutical compositions, at least one of which contains a compound of formula (I).
- the kit comprises means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- a container, divided bottle, or divided foil packet An example of such a kit is a blister pack, as typically used for the packaging of tablets, capsules and the like.
- the kit of the invention may be used for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit of the invention typically comprises directions for administration.
- the compound of the invention and the other therapeutic agent may be manufactured and/or formulated by the same or different manufacturers. Moreover, the compound of the invention and the other therapeutic agent may be brought together into a combination therapy: (i) prior to release of the combination product to physicians (e.g. in the case of a kit comprising the compound of the invention and the other therapeutic agent); (ii) by the physician themselves (or under the guidance of the physician) shortly before administration; (iii) in the patient themselves, e.g. during sequential administration of the compound of the invention and the other therapeutic agent.
- the invention provides the use of a compound of formula (I) for treating a disease or disorder associated with a mutant form of IDH, wherein the medicament is prepared for administration with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or disorder associated with a mutant form of IDH, wherein the medicament is administered with a compound of formula (I).
- the invention also provides a compound of formula (I) for use in a method of treating a disease or disorder associated with a mutant form of IDH, wherein the compound of formula (I) is prepared for administration with another therapeutic agent.
- the invention also provides another therapeutic agent for use in a method of treating a disease or disorder associated with a mutant form of IDH, wherein the other therapeutic agent is prepared for administration with a compound of formula (I).
- the invention also provides a compound of formula (I) for use in a method of treating a disease or disorder associated with a mutant form of IDH, wherein the compound of formula (I) is administered with another therapeutic agent.
- the invention also provides another therapeutic agent for use in a method of treating a disease or disorder associated with a mutant form of I DH, wherein the other therapeutic agent is administered with a compound of formula (I).
- the invention also provides the use of a compound of formula (I) for treating a disease or disorder associated with a mutant form of I DH, wherein the patient has previously (e.g. within 24 hours) been treated with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or disorder associated with a mutant form of I DH, wherein the patient has previously (e.g. within 24 hours) been treated with a compound of formula (I).
- the other therapeutic agent is selected from: vascular endothelial growth factor (VEGF) receptor inhibitors, topoisomerase II inhibitors, smoothen inhibitors, alkylating agents, anti-tumor antibiotics, anti-metabolites, retinoids, and other cytotoxic agents.
- VEGF vascular endothelial growth factor
- vascular endothelial growth factor (VEGF) receptor inhibitors include, but are not limited to, bevacizumab (sold under the trademark Avastin® by Genentech/Roche), axitinib, (A/-methyl-2-[[3-[(£)-2-pyridin-2-ylethenyl]-1 H-indazol-6- yl]sulfanyl]benzamide, also known as AG013736, and described in PCT Publication No.
- topoisomerase I I inhibitors include but are not limited to, etoposide (also known as VP-16 and Etoposide phosphate, sold under the tradenames Toposar®, VePesid® and Etopophos®), and teniposide (also known as VM-26, sold under the tradename Vumon®).
- etoposide also known as VP-16 and Etoposide phosphate, sold under the tradenames Toposar®, VePesid® and Etopophos®
- teniposide also known as VM-26, sold under the tradename Vumon®
- alkylating agents include but are not limited to, temozolomide (sold under the tradenames Temodar® and Temodal® by Schering-Plough/Merck), dactinomycin (also known as actinomycin-D and sold under the tradename Cosmegen®), melphalan (also known as L-PAM, L-sarcolysin, and phenylalanine mustard, sold under the tradename Alkeran®), altretamine (also known as hexamethylmelamine (HMM), sold under the tradename Hexalen®), carmustine (sold under the tradename BiCNU®), bendamustine (sold under the tradename Treanda®), busulfan (sold under the tradenames Busulfex® and Myleran®), carboplatin (sold under the tradename Paraplatin®), lomustine (also known as CCNU, sold under the tradename CeeNU®), cisplatin (also known as CDDP, sold under the tradenames P
- anti-tumor antibiotics include, but are not limited to, doxorubicin (sold under the tradenames Adriamycin® and Rubex®), bleomycin (sold under the tradename lenoxane®), daunorubicin (also known as dauorubicin hydrochloride, daunomycin, and rubidomycin hydrochloride, sold under the tradename Cerubidine®), daunorubicin liposomal (daunorubicin citrate liposome, sold under the tradename DaunoXome®), mitoxantrone (also known as DHAD, sold under the tradename Novantrone®), epirubicin (sold under the tradename EllenceTM), idarubicin (sold under the tradenames Idamycin®, Idamycin PFS®), and mitomycin C (sold under the tradename Mutamycin®).
- doxorubicin sold under the tradenames Adriamycin® and Rubex®
- bleomycin sold under the trade
- anti-metabolites include, but are not limited to, claribine (2- chlorodeoxyadenosine, sold under the tradename leustatin®), 5-fluorouracil (sold under the tradename Adrucil®), 6-thioguanine (sold under the tradename Purinethol®), pemetrexed (sold under the tradename Alimta®), cytarabine (also known as arabinosylcytosine (Ara-C), sold under the tradename Cytosar-U®), cytarabine liposomal (also known as Liposomal Ara-C, sold under the tradename DepoCytTM), decitabine (sold under the tradename Dacogen®), hydroxyurea (sold under the tradenames Hydrea®, DroxiaTM and MylocelTM), fludarabine (sold under the tradename Fludara®), floxuridine (sold under the tradename FUDR®), cladribine (also known as 2-ch
- retinoids examples include, but are not limited to, alitretinoin (sold under the tradename Panretin®), tretinoin (a ⁇ -trans retinoic acid, also known as ATRA, sold under the tradename Vesanoid®), Isotretinoin (13-c/s-retinoic acid, sold under the tradenames Accutane®, Amnesteem®, Claravis®, Clarus®, Decutan®, Isotane®, Izotech®, Oratane®, Isotret®, and Sotret®), and bexarotene (sold under the tradename Targretin®).
- Panretin® tretinoin (a ⁇ -trans retinoic acid, also known as ATRA, sold under the tradename Vesanoid®)
- Isotretinoin 13-c/s-retinoic acid
- cytotoxic agents include, but are not limited to, arsenic trioxide (sold under the tradename Trisenox®), asparaginase (also known as L-asparaginase, and Erwinia L-asparaginase, sold under the tradenames Elspar® and Kidrolase®).
- Trisenox® arsenic trioxide
- asparaginase also known as L-asparaginase, and Erwinia L-asparaginase, sold under the tradenames Elspar® and Kidrolase®.
- LCMS data (also reported herein as simply MS) were recorded using a Waters System (Acuity UPLC and a Micromass ZQ mass spectrometer; Column: Acuity HSS C18 1 .8- micron, 2.1 x 50 mm; gradient: 5-95 % acetonitrile in water with 0.05 % TFA over a 1.8 min period; flow rate 1.2 mL/min; molecular weight range 200-1500; cone Voltage 20 V; column temperature 50 °C). All masses reported are those of the protonated parent ions unless recorded otherwise.
- HRMS Method A ESI-MS data were recorded using a Synapt G2 HDMS (TOF mass spectrometer, Waters) with electrospray ionization source. The resolution of the MS system was approximately 15000.
- Leucine Enkephalin was used as lock mass (internal standards) infused from lockspary probe. The compound was infused into the mass spectrometer by UPLC (Acquity, Waters) from sample probe. The separation was performed on Acquity UPLC BEH C18 1x50 mm column at 0.2 mL/min flow rate with the gradient from 5% to 95% in 3 min.
- Solvent A was Water with 0.1 % Formic Acid and solvent B was Acetonitrile with 0.1 % Formic Acid. The mass accuracy of the system has been found to be ⁇ 5 ppm with lock mass.
- HRMS methods A and B are referred to throughout as HRMS(A) or HRMS(B), respectively.
- the reaction mixture was stirred at 4 °C for 15 min, warmed up to room temperature and stirred for an additional 1 h.
- the mixture was treated with saturated NH 4 CI (25 mL), followed by CH2CI2 (50 mL) and the resulting mixture was stirred for 20 min.
- the layers were separated and the organic layer was washed with water.
- the combined aqueous layers were extracted with CH2CI2 (50 mL).
- the combined organic layers were dried over Na2S04, filtered and concentrated to give (R)-4-isobutyloxazolidin-2-one (3.22 g) in 88% yield.
- the crude product was used for the next reaction without purification.
- Step 1 Preparation of 4-(amino(phenyl)methyl)tetrahydro-2H-pyran-4-ol
- the milky white mixture was extracted with DCM (1x), ethyl acetate/THF (1 :1 ; 1x) and ethyl acetate (2x).
- the organic layers (DCM and ethyl acetate solutions independently) were washed with saturated aqueous NaHC03 solution, dried over Na2S04, filtered off and concentrated under reduced pressure providing crude 4-(amino(phenyl)methyl)tetrahydro-2H-pyran-4-ol, which was directly used in the next reaction without further purification.
- Step 2 Preparation of 4-phenyl-1 ,8-dioxa-3-azaspiro[4.5]decan-2-one
- Step 2 Preparation of (S)-tert-butyl 1 -(biphenyl-4-yl)-2-hydroxyethylcarbamate
- Step 1 Preparation of Methyl 2-(tert-butoxycarbonylamino)-2-methylpropanoate
- 2-(tert-butoxycarbonylamino)-2-methylpropanoic acid 10.03 g, 49.4 mmol
- MeOH/DCM 60mL/140ml_
- Trimethylsilyl trimethylsilyl
- the reaction mixture was stirred for 30 minutes.
- Acetic acid was added drop wise to quench (trimethylsilyl)diazomethane.
- the reaction mixture was concentrated under reduced pressure to afford the desired product as a white solid (10.56 g).
- Step 2 Preparation of tert-butyl 3-hydroxy-2,3-dimethylbutan-2-ylcarbamate
- the capped tube was heated to 100°C for 16 h. After cooling the reaction mixture was diluted with EtOAc (10 mL) and washed with water (10 mL). After separation, the aqueous phase was extracted with EtOAc (3 x 10 mL). Combined organics were dried over Na2S04, filtered and concentrated. The crude material was purified through silica gel column chromatography (EtOAc in Heptane 12 to 100%) to give a white solid (50 mg, 49.3% yield).
- the vial was sealed, evacuated and purged with dry nitrogen three times before adding dioxane (1 .6 mL).
- the reaction mixture was heated to 100°C for 16 hours in an oil bath. After cooling the reaction was diluted with EtOAc (10 mL) and washed with water (10 mL). After separation, the aqueous phase was extracted with EtOAc (3 x 10 mL). Combined organics were dried over Na2S04, filtered and concentrated.
- the crude material was purified through silica gel column chromatography (EtOAc in Heptane 12 to 100%) to give a white solid (65 mg, 58.9% yield).
- DavePhos ligand [2-dicyclohexylphosphino-2'-(N,N-dimethylamino)biphenyl] (59 mg, 0.15 mmol), and Pd(OAc)2 (17 mg, 0.075 mmol) in 6 mL toluene was heated at 100°C for 1 h. The mixture was cooled to room temperature, and filtered through Celite. Filter cake was rinsed with 30 mL EtOAc. The filtrate was poured into 20 mL water. Layers were separated, and the aqueous was further extracted with EtOAc (20 mL).
- Step 1 A solution of 1 -(5-fluoropyrimidin-2-yl)ethanone (700 mg, 5.0 mmol) and 4- fluorophenol (616 mg, 5.50 mmol) in 6 mL DMF was treated with potassium carbonate (829 mg 6.0 mmol) and heated to 50°C for 3.5 h. The reaction mixture was poured into 20 mL water, and extracted with EtOAc (2 x 20 mL). Organics were washed with 20 mL each water, brine, and dried over Na2S04. Mixture was filtered and concentrated on silica gel.
- Step 2 1-(5-(4-fluorophenoxy)pyrimidin-2-yl)ethanone (290 mg, 1.25 mmol), NH40Ac (1.9 g, 24.6 mmol), and NaBH 3 CN (314 mg, 5.00 mmol) were taken up in 20 mL 200 proof EtOH, and heated at 130 C for 3 minutes in a microwave apparatus. The mixture was concentrated to remove the EtOH. Crude was taken up in 30 ml water + 25 mL EtOAc. 6N NaOH was added until aqueous pH was -10. Separated layers, and extracted aqueous with EtOAc (25 ml).
- Step 1 Preparation of tert-butyl 1 -(methoxy(methyl)amino)-2-methyl-1 -oxopropan- 2-yl carbamate
- Step 2 Preparation of (S)-tert-butyl 2-methyl-4-oxopentan-3-ylcarbamate
- THF 100 ml.
- methyl lithium 1.071 g, 48.7 mmol
- Cold bath was replaced with -40 °C bath (MeCN in dry ice) removed and the reaction was stirred for 4 hours.
- Saturated NH 4 CI solution (10ml_) was then added cautiously to quench the reaction.
- Step 1 Preparation of tert-butyl 1 -(methoxy(methyl)amino)-2-methyl-1 -oxopropan- 2-ylcarbamate
- Step 2 Preparation of (S)-tert-butyl (1 -cyclopropyl-2-hydroxyethyl)carbamate
- LiBH 4 LiBH 4
- methanol methanol
- Step 3 To a microwave vial with stir bar was added (R)-N-((S)-1-(4-bromo-2-fluorophenyl)ethyl)- 2-methylpropane-2- (1 g, 3.10 mmol), isopropenyl boronic acid pinacol ester (1.51 ml, 8.07 mmol), DME (8 ml), sodium carbonate (7.76 ml, 15.5 mmol) (2.0 M aq) and PdCI 2 (dppf). CH2CI2 adduct (0.127 g, 0.155 mmol). Vessel was capped and heated by microwave irradiation for 20 min at 100 °C. Reaction mixture was diluted with a saturated solution of NH 4 CI.
- Reaction mixture was cooled to 0 °C whereupon a second addition of diethylzinc (1.0M in hexanes) (13.1 mL, 13.1 mmol) took place followed by the addition of chloroiodomethane (0.95 mL, 13.1 mmol).
- Reaction mixture allowed to warm to room temperature and stirred 18 hours under argon.
- Reaction mixture was cooled to 0 °C in a ice bath and to the cold reaction mixture was slowly added a saturated solution of NH 4 CI.
- the aqueous mixture was extracted with EtOAc. Organic phases combined, washed with water, brine, dried (Na2S04), filtered and concentrated onto silica gel.
- 1 ,2-dibromoethane (0.1 1 ml, 1.22 mmol) and tetrabutylammonium bromide (19 mg, 0.06 mmol) were added and reaction mixture allowed to stir an additional 18 hours.
- a third addition of 1 ,2-dibromoethane (0.1 1 ml, 1 .22 mmol) was added and the reaction mixture heated to 50 °C for an additional 18 hours in a preheated aluminum tray.
- the reaction mixture was quenched with a saturated solution of NH 4 CI and the aqueous mixture extracted with EtOAc. Organics combined and washed twice with water, brine, dried (Na2S04), filtered and concentrated.
- Step 2 To a solution of (R,E)-N-(2-fluoro-4-(trifluoromethyl)benzylidene)-2-methylpropane-2- sulfinamide (7.3 g, 24.7 mmol) in CH2CI2 (247 mL) cooled to 0°C (water/ice bath) under nitrogen, was added 3M methyl magnesium bromide (33 mL, 99 mmol) in Et 2 0. Reaction mixture allowed to stir for 30 min at 0°C, then gradually allowed to warm to room temperature and stirred for 1 hour at room temperature. Reaction mixture was cooled to 0°C then quenched with the slow addition of a saturated solution of NH 4 CI. Aqueous mixture extracted with EtOAc.
- Step 1 To a round bottom flask with stir bar was added 4-((S)-1 aminoethyl-2-chlorobenzoic acid HCI salt (1.05 g, 4.45 mmol) followed by the addition of THF (40 ml_). To this solution was added DIEA (1 .86 ml, 10.7 mmol). The reaction mixture becomes cloudy white followed by the addition of di-tert-butyl dicarbonate (1 .07 g, 4.89 mmol). Resulting reaction mixture allowed to stir for 18 hours at room temperature. At which time the reaction mixture was then heated to 60 °C for 2 hours in a oil bath.
- Step 1 Mitsunobu A
- Step 1 Mitsunobu B
- Step 1 Preparation of 1 -bromo-4-(difluoromethyl)-2-fluorobenzene
- Step 3 Preparation of (R,E)-N-(4-(difluoromethyl)-2-fluorobenzylidene)-2- methylpropane-2-sulfinamide
- Step 2 Preparation of (R,E)-2-methyl-N-(4-(pyrimidin-5- yloxy)benzylidene)propane-2-sulfinamide
- Step 3 Preparation of 2-methyl-N-((S)-1 -(4-(pyrimidin-5- yloxy)phenyl)ethyl)propane-2-sulfinamide
- the suspension was stirred for -3 hour while slowly warming to -20 °C.
- the mixture was cooled to --40 °C, and additional methylmagnesium bromide (3M in diethylether; 0.4mL) was added.
- the suspension was stirred for 30 min and allowed to warm to -10 °C.
- the mixture was quenched slowly over 10 min with saturated aqueous NH 4 CI solution (10 mL).
- the mixture was diluted with saturated aqueous NH 4 CI solution (30 mL) and water (15 mL).
- the separated aqueous phase was extract with DCM (2x 75 mL).
- the combined organic layers were washed with brine (50 mL), dried over Na2S04, filtered off and concentrated under reduced pressure.
- Step 4 Preparation of (S)-1 -(4-(pyrimidin-5-yloxy)phenyl)ethanamine To 2-methyl-N-((S)-1-(4-(pyrimidin-5-yloxy)phenyl)ethyl)propane-2-sulfinamid (55 mg, 0.172 mmol) was added 4M HCI in dioxane (800 ⁇ _, 3.20 mmol) to give a white suspension. This resulting mixture was stirred at room temperature for -35 min and concentrated under reduced pressure to provide crude (S)-1-(4-(pyrimidin-5- yloxy)phenyl)ethanamine (44 mg) as its HCI salt, which was used without further purification. LCMS m/z 217.1 (M + H) + , Rt 0.37 min.
- Step 1 Preparation of ethyl 5,6-dichloronicotinate
- Step 3 Preparation of ethyl 5-chloro-6-(1 ,1 -difluoroethyl)nicotinate
- Step 4 Preparation of (5-chloro-6-(1 ,1 -difluoroethyl)pyridin-3-yl)methanol
- Step 5 Preparation of 5-chloro-6-(1 ,1 -difluoroethyl)nicotinaldehyde
- Step 1 Preparation of ethyl 5-chloro-6-(2,2,2-trifluoroethoxy)nicotinate
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Epidemiology (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description


























































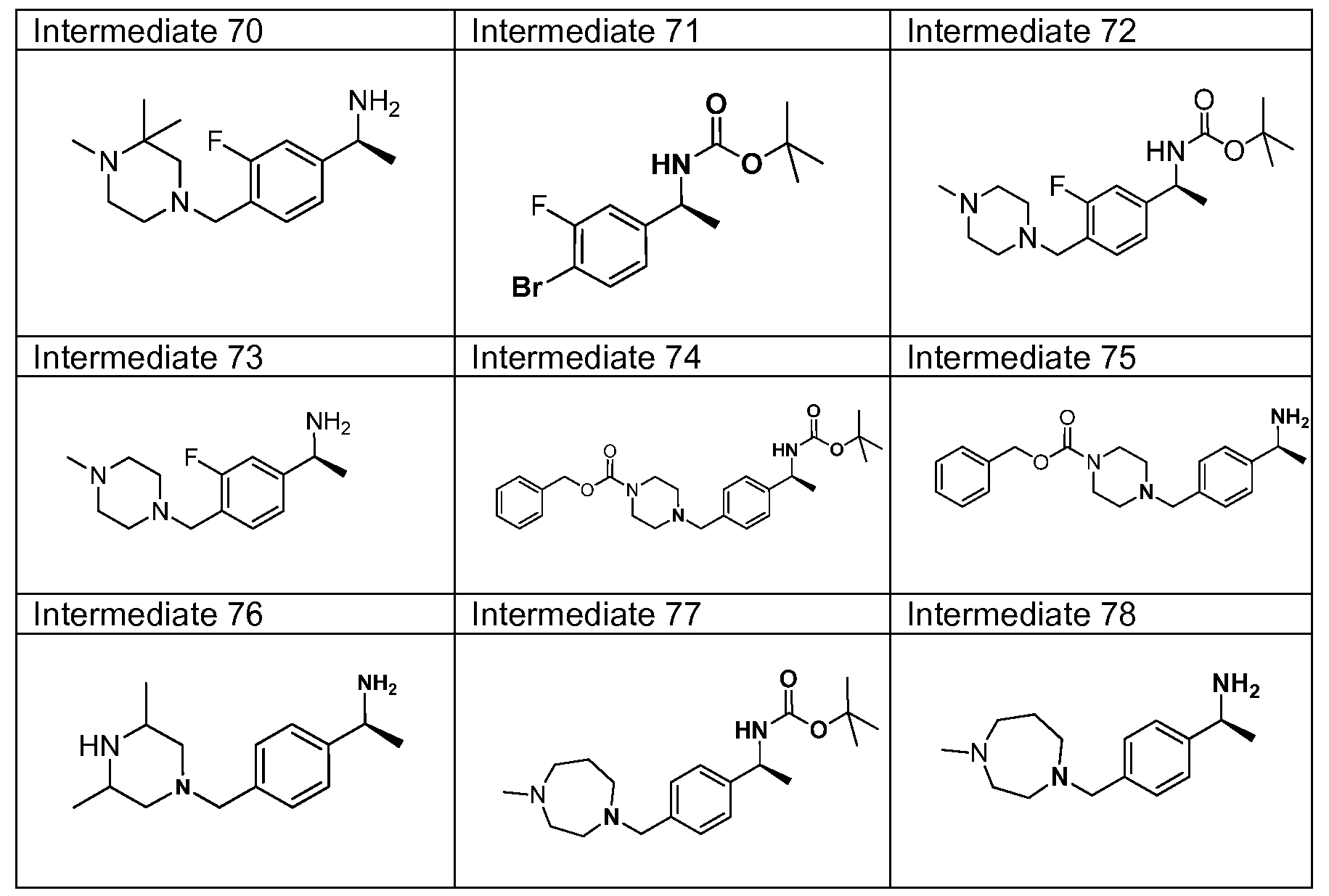












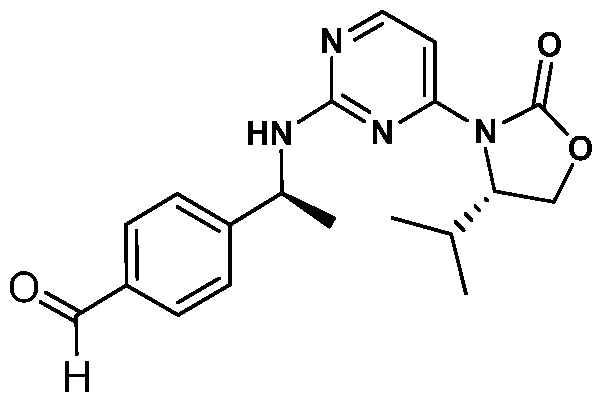











































































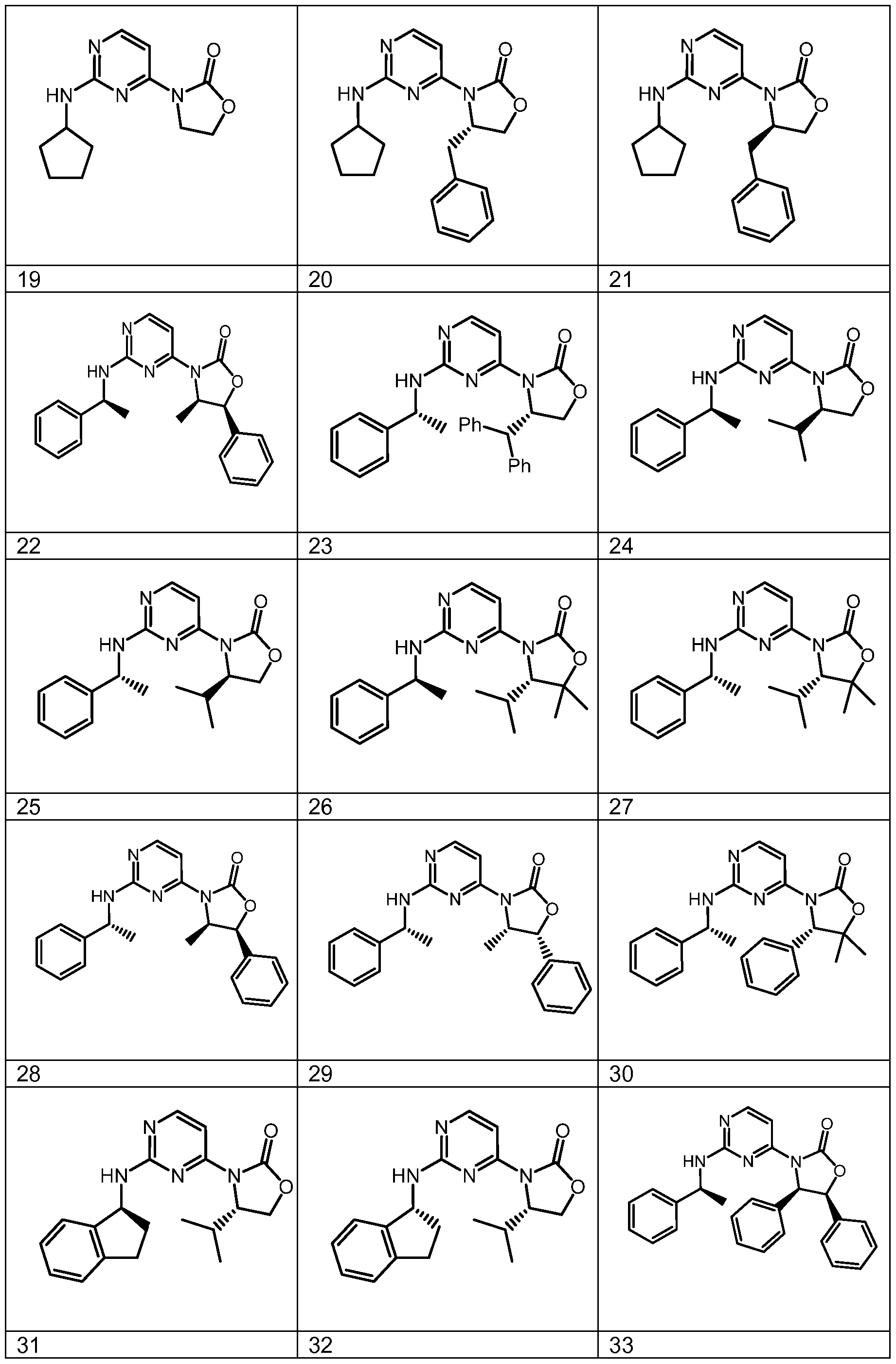








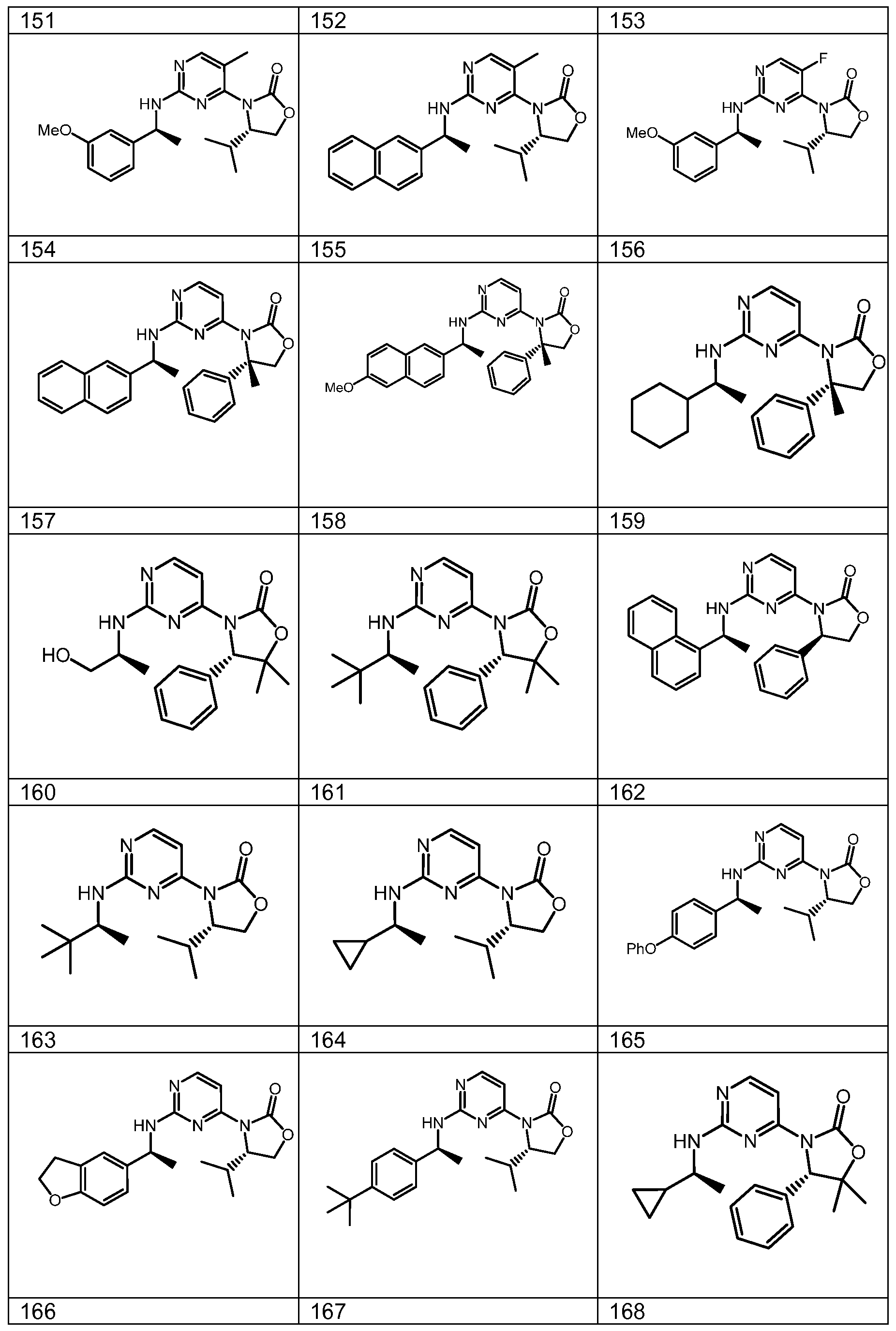









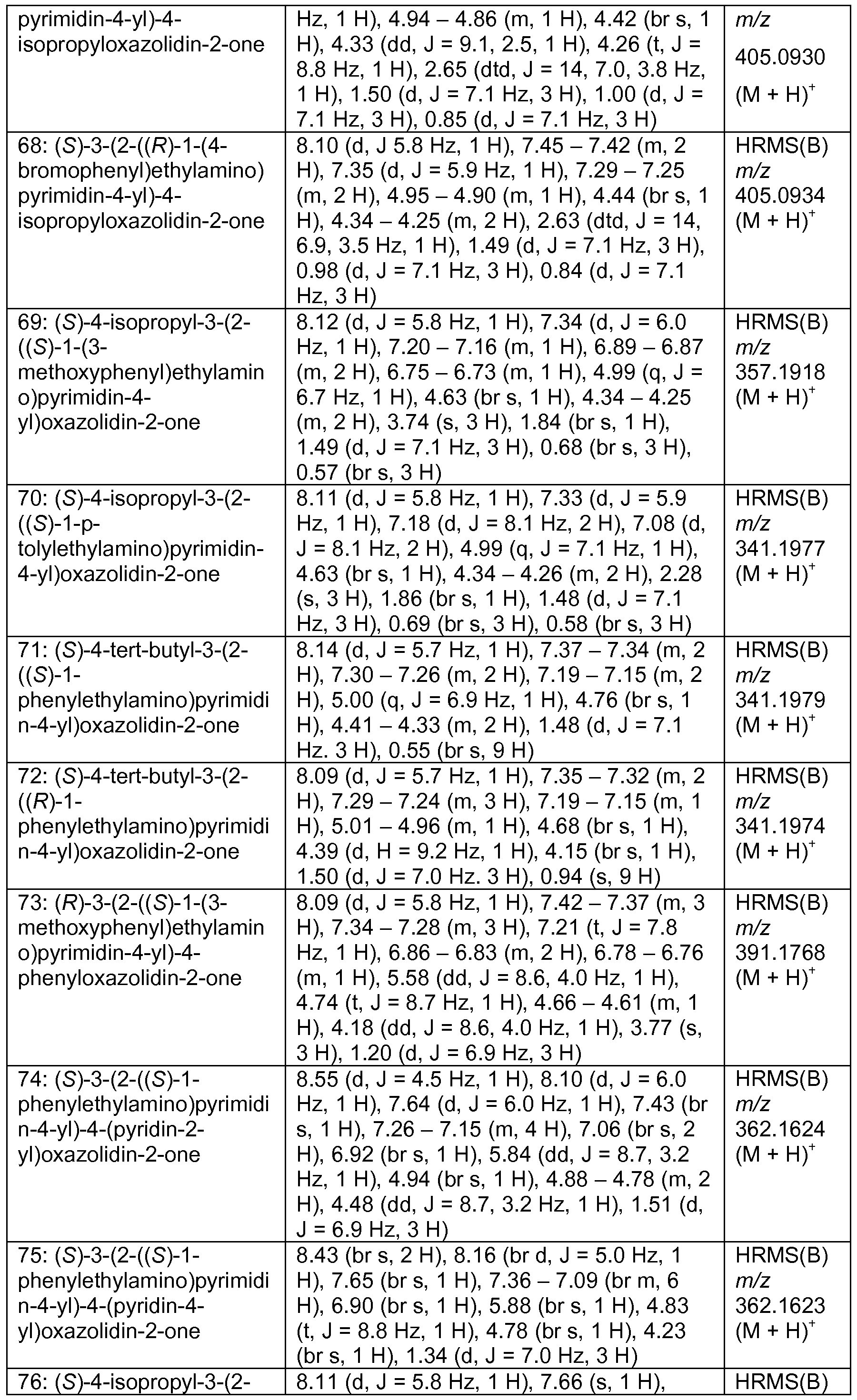



































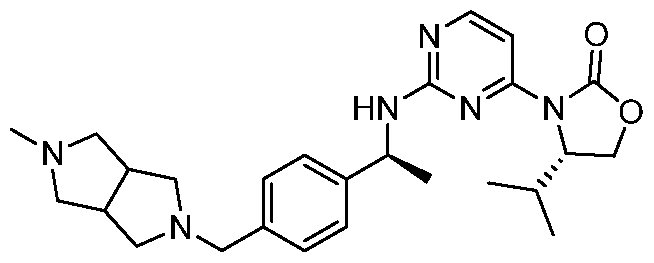



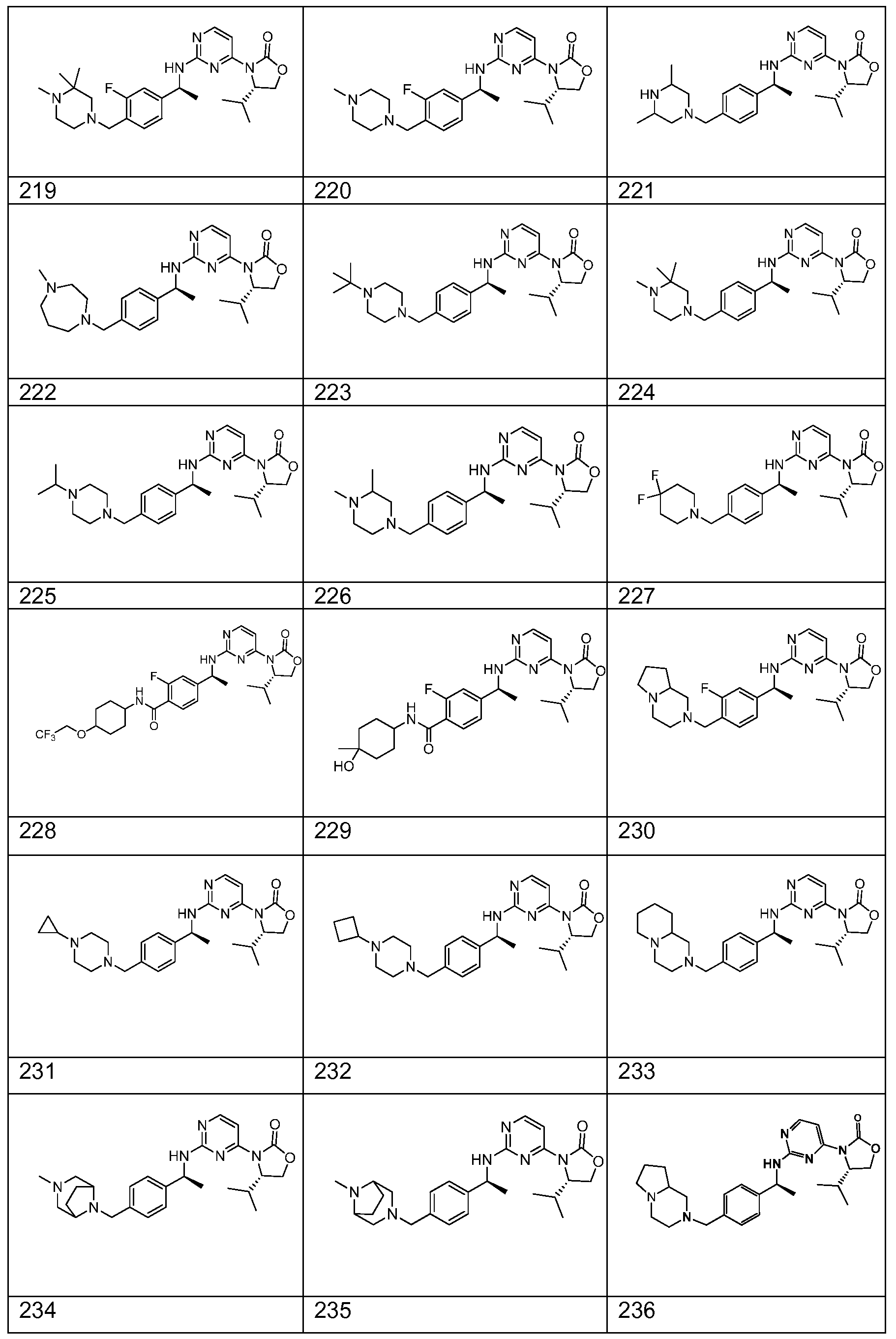
















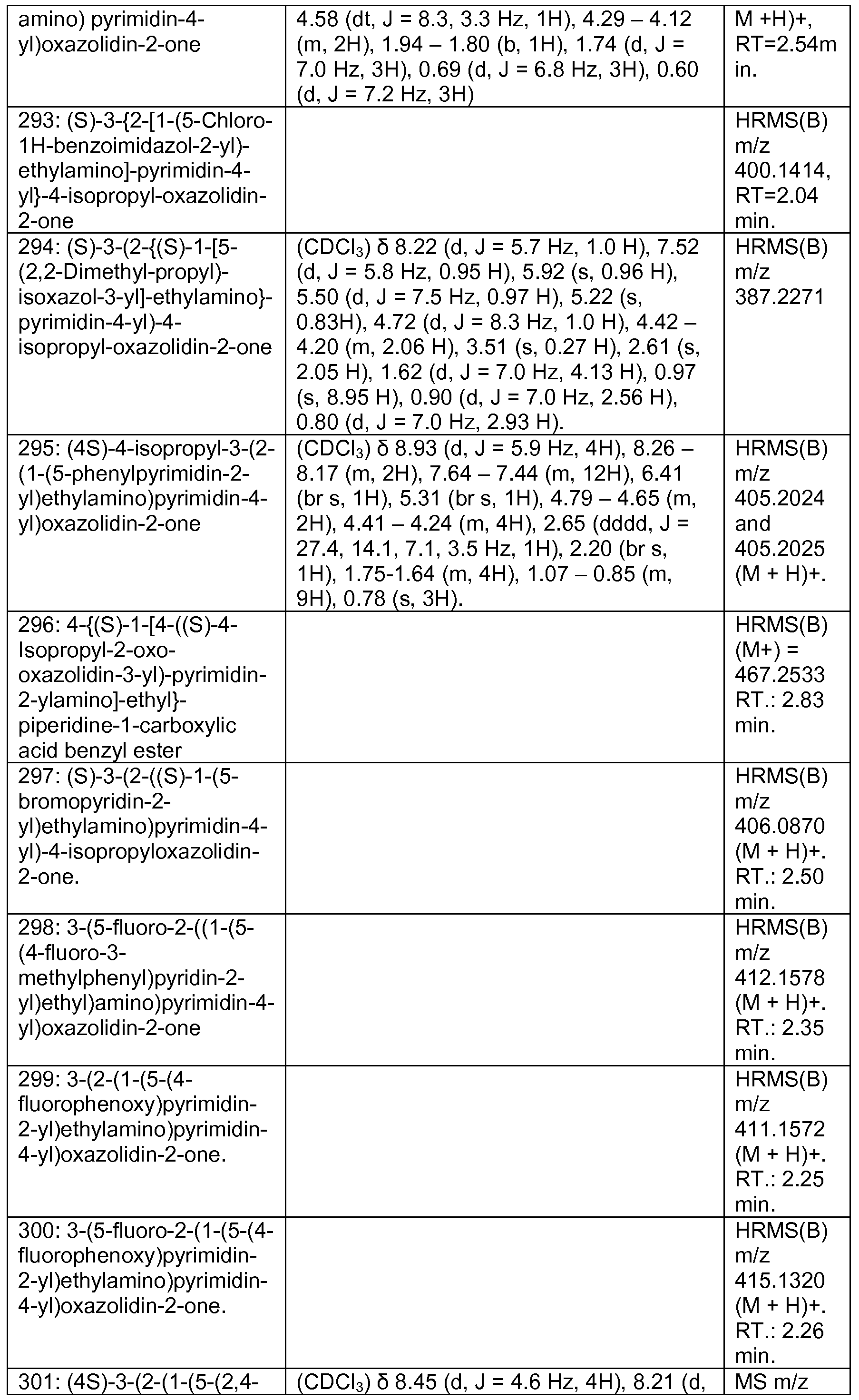
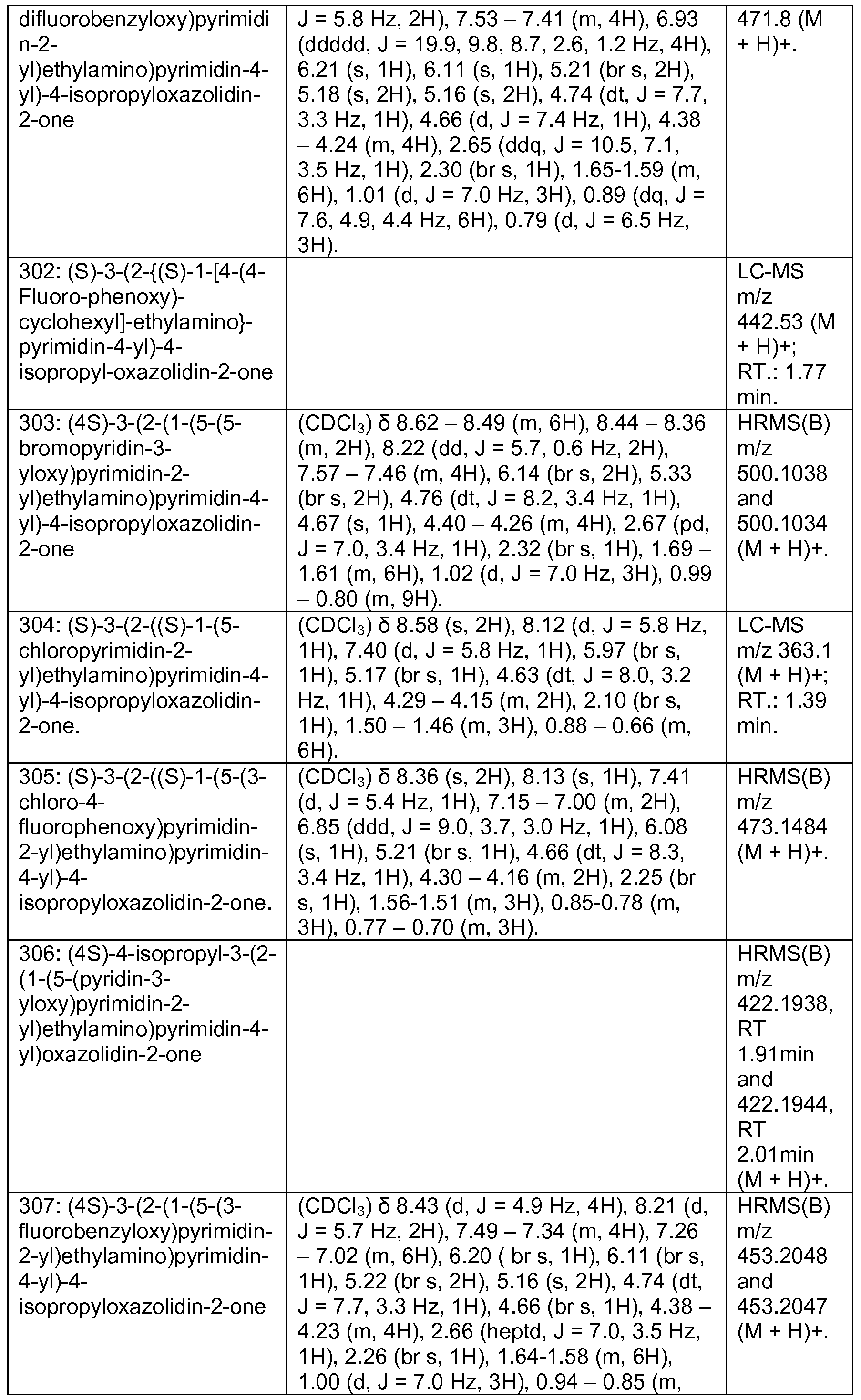

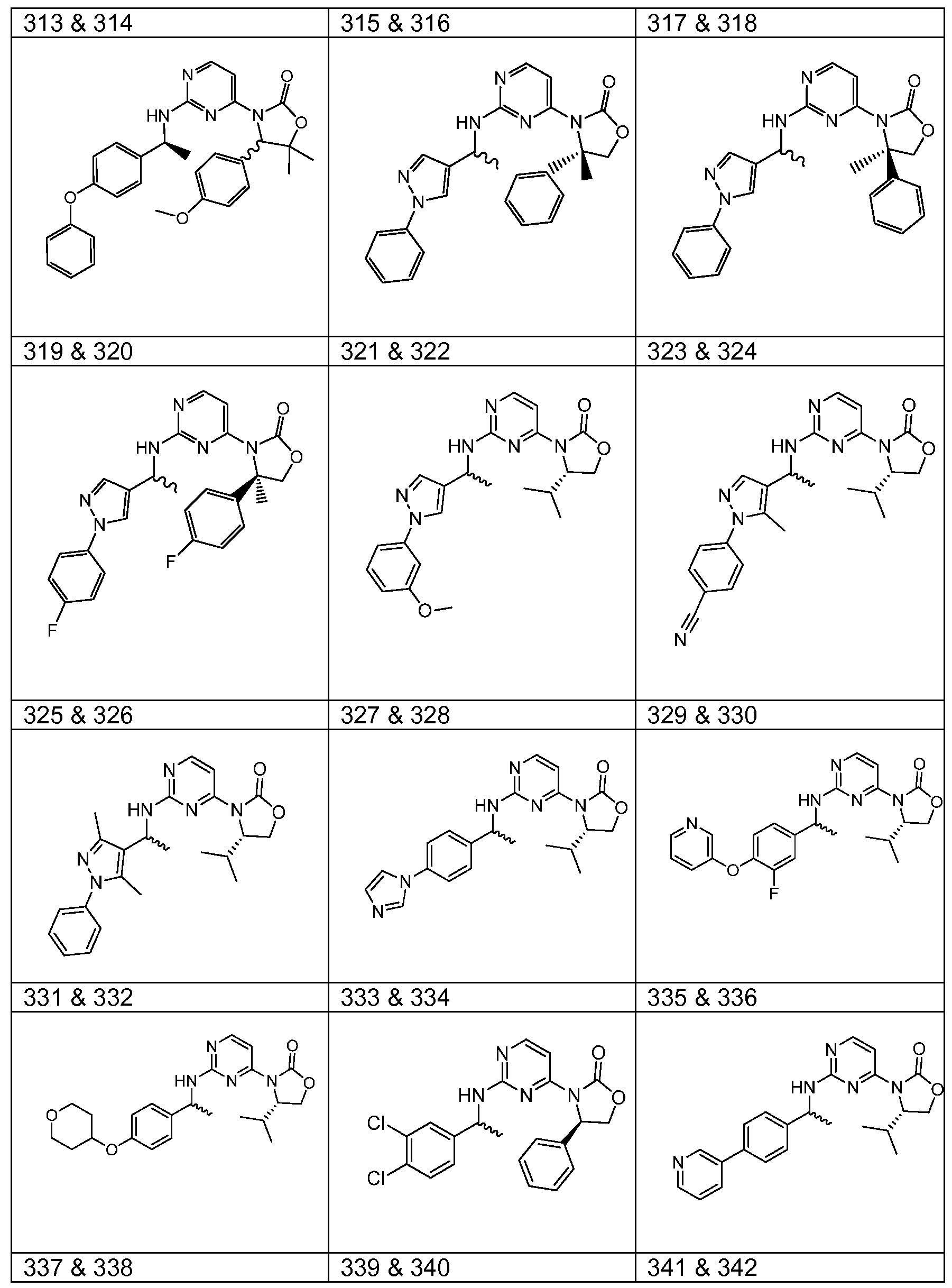

























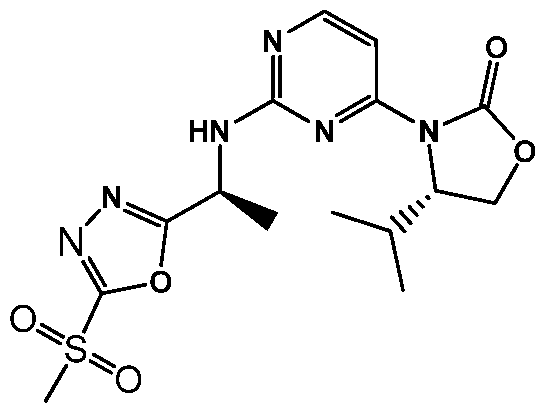




































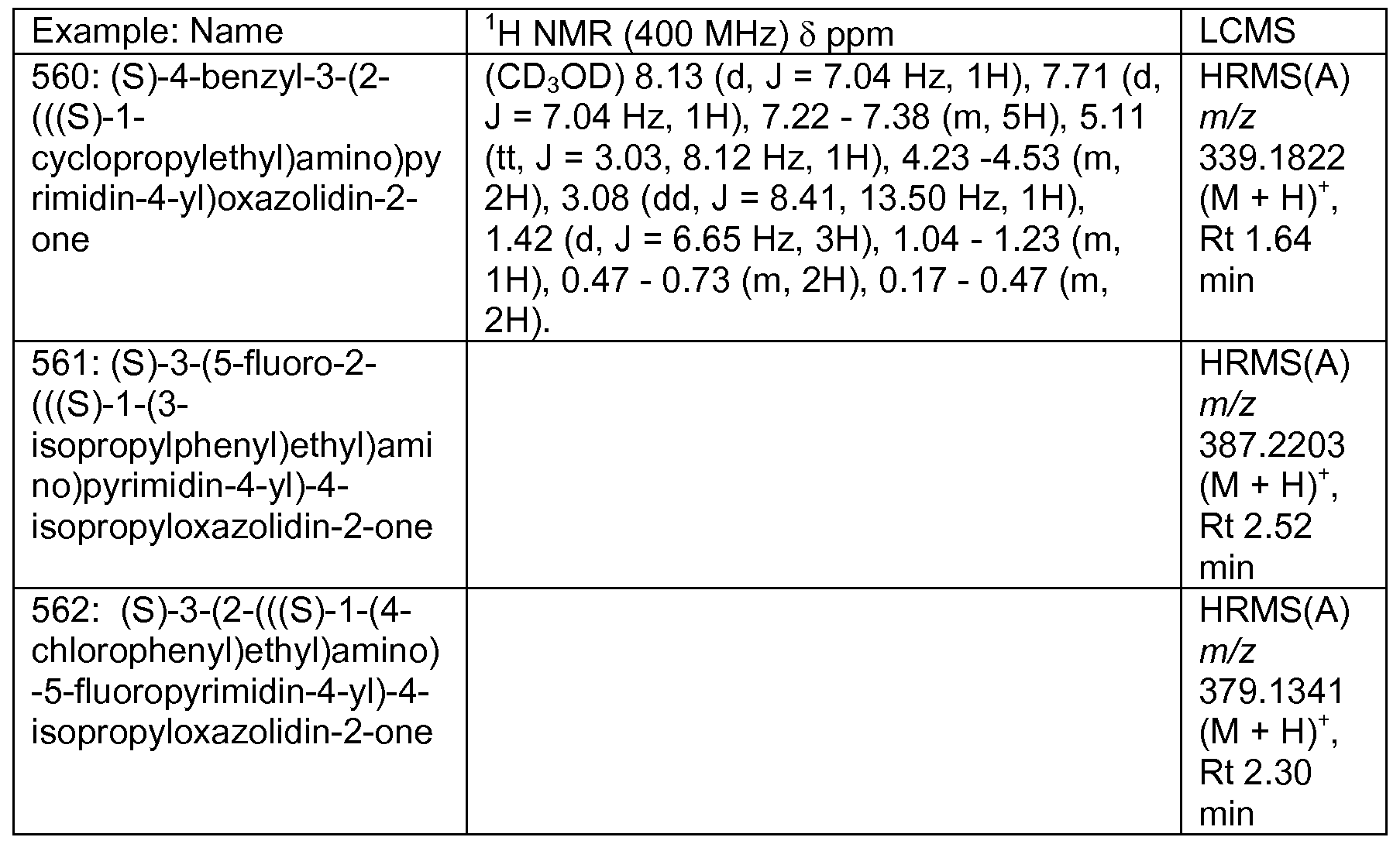
























Claims






Priority Applications (21)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2014003752A MX342326B (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh. |
| EP12784701.0A EP2771337B1 (en) | 2011-09-27 | 2012-09-26 | 3-(pyrimidin-4-yl)-oxazolidin-2-ones as inhibitors of mutant idh |
| AU2012313888A AU2012313888B2 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| KR1020147010827A KR20140069235A (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| UAA201403086A UA111503C2 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| EA201490696A EA025183B1 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| SG11201400989TA SG11201400989TA (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| CUP2014000036A CU24269B1 (en) | 2011-09-27 | 2012-09-26 | 3- PIRIMIDIN- 4-IL- OXAZOLIDIN- 2- INHIBITING WAVES OF THE MUTANT HDI |
| US14/347,481 US8957068B2 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| AP2014007602A AP3907A (en) | 2011-09-27 | 2012-09-26 | 3-Pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| ES12784701.0T ES2645968T3 (en) | 2011-09-27 | 2012-09-26 | 3- (pyrimidin-4-yl) -oxazolidin-2-ones as mutant HDI inhibitors |
| JP2014532533A JP6026544B2 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| BR112014007310A BR112014007310A2 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as mutant idh inhibitors |
| NZ624040A NZ624040B2 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| CA2849995A CA2849995A1 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| CN201280058052.3A CN103958506B (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| ZA2014/02144A ZA201402144B (en) | 2011-09-27 | 2014-03-24 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| MA36849A MA35452B1 (en) | 2011-09-27 | 2014-03-25 | 3-pyrimidin-4-yl-oxazolidin-2-ones as mutant idh inhibitors |
| TNP2014000128A TN2014000128A1 (en) | 2011-09-27 | 2014-03-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| CR20140143A CR20140143A (en) | 2011-09-27 | 2014-03-27 | 3-PIRMIDIN-4-IL-OXAZOLIDIN-2-ONAS AS INHIBITORS OF THE MUTANT HDI |
| IL231786A IL231786A (en) | 2011-09-27 | 2014-03-27 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161539553P | 2011-09-27 | 2011-09-27 | |
| US61/539,553 | 2011-09-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013046136A1 true WO2013046136A1 (en) | 2013-04-04 |
Family
ID=47178237
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2012/055133 WO2013046136A1 (en) | 2011-09-27 | 2012-09-26 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
Country Status (31)
| Country | Link |
|---|---|
| US (1) | US8957068B2 (en) |
| EP (1) | EP2771337B1 (en) |
| JP (1) | JP6026544B2 (en) |
| KR (1) | KR20140069235A (en) |
| CN (1) | CN103958506B (en) |
| AP (1) | AP3907A (en) |
| AR (1) | AR088175A1 (en) |
| AU (1) | AU2012313888B2 (en) |
| BR (1) | BR112014007310A2 (en) |
| CA (1) | CA2849995A1 (en) |
| CL (1) | CL2014000746A1 (en) |
| CO (1) | CO6920291A2 (en) |
| CR (1) | CR20140143A (en) |
| CU (1) | CU24269B1 (en) |
| DO (1) | DOP2014000058A (en) |
| EA (1) | EA025183B1 (en) |
| ES (1) | ES2645968T3 (en) |
| GE (1) | GEP20166432B (en) |
| GT (1) | GT201400057A (en) |
| IL (1) | IL231786A (en) |
| MA (1) | MA35452B1 (en) |
| MX (1) | MX342326B (en) |
| NI (1) | NI201400026A (en) |
| PE (1) | PE20141581A1 (en) |
| SG (1) | SG11201400989TA (en) |
| TN (1) | TN2014000128A1 (en) |
| TW (1) | TWI547493B (en) |
| UA (1) | UA111503C2 (en) |
| UY (1) | UY34352A (en) |
| WO (1) | WO2013046136A1 (en) |
| ZA (1) | ZA201402144B (en) |
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014141104A1 (en) * | 2013-03-14 | 2014-09-18 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| US8865894B2 (en) | 2012-02-24 | 2014-10-21 | Novartis Ag | Oxazolidin-2-one compounds and uses thereof |
| WO2015127172A1 (en) * | 2014-02-20 | 2015-08-27 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| CN105209460A (en) * | 2013-03-14 | 2015-12-30 | 诺华股份有限公司 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| US9296733B2 (en) | 2012-11-12 | 2016-03-29 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivative and use thereof for the treatment of conditions, diseases and disorders dependent upon PI3 kinases |
| WO2016052697A1 (en) * | 2014-10-01 | 2016-04-07 | 第一三共株式会社 | Isoxazole derivative as mutated isocitrate dehydrogenase 1 inhibitor |
| WO2016062770A1 (en) * | 2014-10-23 | 2016-04-28 | Bayer Pharma Aktiengesellschaft | 1-cyclohexyl-2-phenylaminobenzimidazoles as midh1 inhibitors for the treatment of tumors |
| WO2016089833A1 (en) | 2014-12-05 | 2016-06-09 | Merck Sharp & Dohme Corp. | Novel tricyclic compounds as inhibitors of mutant idh enzymes |
| JP2016523976A (en) * | 2013-07-11 | 2016-08-12 | アジオス ファーマシューティカルズ, インコーポレイテッド | 2,4- or 4,6-diaminopyrimidine compounds as IDH2 mutant inhibitors for the treatment of cancer |
| JP2016523935A (en) * | 2013-07-11 | 2016-08-12 | アジオス ファーマシューティカルズ, インコーポレイテッド | Therapeutically active compounds and methods of use thereof |
| JP2016525130A (en) * | 2013-07-25 | 2016-08-22 | アギオス ファーマシューティカルス,インコーポレーテッド | Therapeutically active compounds and uses thereof |
| JP2016526561A (en) * | 2013-07-11 | 2016-09-05 | アジオス ファーマシューティカルズ, インコーポレイテッド | Therapeutically active compounds and methods of their use |
| WO2017005674A1 (en) * | 2015-07-07 | 2017-01-12 | Bayer Pharma Aktiengesellschaft | 2-aryl- and 2-arylalkyl-benzimidazoles as midh1 inhibitors |
| WO2017019429A1 (en) * | 2015-07-27 | 2017-02-02 | Eli Lilly And Company | 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds and theit use as mutant idh1 inhibitors |
| WO2017016992A1 (en) * | 2015-07-27 | 2017-02-02 | Bayer Pharma Aktiengesellschaft | Inhibitor of the mutated isocitrate dehydrogenase idh1 r132h |
| JP2017518326A (en) * | 2014-06-17 | 2017-07-06 | キエージ・フアルマチエウテイチ・エツセ・ピ・ア | Indolizine derivatives as phosphoinositide 3-kinase inhibitors |
| WO2017140758A1 (en) | 2016-02-19 | 2017-08-24 | Debiopharm International S.A. | Derivatives of 2-amino-4-(2-oxazolidinon-3-yl)-pyrimidine fused with a five-membered heteroaromatic ring in 5,6-position which are useful for the treatment of various cancers |
| JP2017528492A (en) * | 2014-09-19 | 2017-09-28 | フォーマ セラピューティクス,インコーポレイテッド | Quinolinone pyrimidine compositions as mutant isocitrate dehydrogenase inhibitors |
| WO2017213910A1 (en) | 2016-06-06 | 2017-12-14 | Eli Lilly And Company | Mutant idh1 inhibitors |
| US9862703B2 (en) | 2014-09-22 | 2018-01-09 | National Health Research Institutes | Heterocyclic compounds and use thereof |
| WO2018111707A1 (en) * | 2016-12-16 | 2018-06-21 | Eli Lilly And Company | 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant idh1 and idh2 inhibitors |
| WO2018118793A1 (en) * | 2016-12-19 | 2018-06-28 | Isocure Biosciences Inc. | Inhibitors of mutant isocitrate dehydrogenases and compositions and methods thereof |
| US10047078B2 (en) | 2016-02-05 | 2018-08-14 | National Health Research Institutes | Aminothiazole compounds |
| US10703746B2 (en) | 2014-12-22 | 2020-07-07 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Mutant IDH1 inhibitors useful for treating cancer |
| US10807976B2 (en) | 2015-04-21 | 2020-10-20 | Forma Therapeutics, Inc. | Quinolinone five-membered heterocyclic compounds as mutant-isocitrate dehydrogenase inhibitors |
| WO2020216071A1 (en) | 2019-04-23 | 2020-10-29 | 上海仕谱生物科技有限公司 | Dimeric or polymeric form of mutant idh inhibitor |
| US10889567B2 (en) | 2014-09-19 | 2021-01-12 | Forma Therapeutics, Inc. | Pyridin-2(1H)-one quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| US11013734B2 (en) | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Treating patients harboring an isocitrate dehydrogenase-1 (IDH-1) mutation |
| US11013733B2 (en) | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mlDH-1) |
| US11247987B2 (en) | 2017-10-06 | 2022-02-15 | Forma Therapeutics, Inc. | Inhibiting ubiquitin specific peptidase 30 |
| US11311527B2 (en) | 2018-05-16 | 2022-04-26 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mIDH-1) |
| US11376246B2 (en) | 2018-05-16 | 2022-07-05 | Forma Therapeutics, Inc. | Inhibiting mutant IDH-1 |
| US11535618B2 (en) | 2018-10-05 | 2022-12-27 | Forma Therapeutics, Inc. | Fused pyrrolines which act as ubiquitin-specific protease 30 (USP30) inhibitors |
| EP3941908A4 (en) * | 2019-03-22 | 2023-05-17 | Yumanity Therapeutics, Inc. | Compounds and uses thereof |
| US11723905B2 (en) | 2018-05-16 | 2023-08-15 | Forma Therapeutics, Inc. | Solid forms of ((s)-5-((1-(6-chloro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl)amino)-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile |
| US11746115B2 (en) | 2021-08-13 | 2023-09-05 | Eli Lilly And Company | Solid forms of 7-[[(1S)-1-[4-[(1S)-2-cyclopropyl-1-(4-prop-2-enoylpiperazin-1-yl)ethyl]phenyl]ethyl]amino]-1-ethyl-4H-pyrimido[4,5-d][1,3]oxazin-2-one |
| US11873298B2 (en) | 2017-10-24 | 2024-01-16 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| US11970486B2 (en) | 2016-10-24 | 2024-04-30 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| US12049466B2 (en) | 2018-05-17 | 2024-07-30 | Forma Therapeutics, Inc. | Fused bicyclic compounds useful as ubiquitin-specific peptidase 30 inhibitors |
| US12098146B2 (en) | 2019-01-24 | 2024-09-24 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9474779B2 (en) | 2012-01-19 | 2016-10-25 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| EP2822944A1 (en) * | 2012-03-05 | 2015-01-14 | Amgen, Inc. | Oxazolidinone compounds and derivatives thereof |
| US20150031627A1 (en) | 2013-07-25 | 2015-01-29 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| SG11201601682RA (en) | 2013-09-06 | 2016-04-28 | Aurigene Discovery Tech Ltd | 1,2,4-oxadiazole derivatives as immunomodulators |
| CN106659765B (en) | 2014-04-04 | 2021-08-13 | 德玛医药 | Use of dianhydrogalactitol and analogs or derivatives thereof for treating non-small cell lung cancer and ovarian cancer |
| US10005734B2 (en) | 2014-09-19 | 2018-06-26 | Forma Therapeutics, Inc. | Pyridin-2(1H)-one quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| JP6751081B2 (en) | 2014-09-19 | 2020-09-02 | フォーマ セラピューティクス,インコーポレイテッド | Pyridinylquinolinone derivatives as mutant isocitrate dehydrogenase inhibitors |
| CN114213356A (en) | 2015-03-10 | 2022-03-22 | 奥瑞基尼探索技术有限公司 | 1,2, 4-oxadiazole and thiadiazole compounds as immunomodulators |
| MX2017011612A (en) * | 2015-03-10 | 2018-03-23 | Aurigene Discovery Tech Ltd | 3-substituted-1,2,4-oxadiazole and thiadiazole compounds as immunomodulators. |
| US10294206B2 (en) | 2015-04-21 | 2019-05-21 | Forma Tm2, Inc. | Fused-bicyclic aryl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| TWI681959B (en) * | 2015-06-09 | 2020-01-11 | 日商第一三共股份有限公司 | Isoxazole derivatives as inhibitors of variant isocitrate dehydrogenase 1 |
| CN107216312B (en) * | 2016-03-22 | 2023-08-01 | 上海海和药物研究开发股份有限公司 | Compound with mutant IDH inhibitory activity, preparation method and application thereof |
| WO2019061324A1 (en) | 2017-09-29 | 2019-04-04 | Curis Inc. | Crystal forms of immunomodulators |
| SG11202003081WA (en) | 2017-10-11 | 2020-05-28 | Aurigene Discovery Tech Ltd | Crystalline forms of 3-substituted 1,2,4-oxadiazole |
| WO2019087087A1 (en) | 2017-11-03 | 2019-05-09 | Aurigene Discovery Technologies Limited | Dual inhibitors of tim-3 and pd-1 pathways |
| EP3706798A1 (en) | 2017-11-06 | 2020-09-16 | Aurigene Discovery Technologies Limited | Conjoint therapies for immunomodulation |
| CN110407855A (en) * | 2018-04-26 | 2019-11-05 | 上海嗣新生物科技有限公司 | 2- aminopyrimidine piperidine derivatives and its preparation method and application |
| US10980788B2 (en) | 2018-06-08 | 2021-04-20 | Agios Pharmaceuticals, Inc. | Therapy for treating malignancies |
| KR102584855B1 (en) * | 2018-06-26 | 2023-10-05 | 케이피씨 파마슈티컬스 인코포레이티드 | Benzimidazole derivatives and their use as IDH1 inhibitors |
| US20220259203A1 (en) * | 2019-07-10 | 2022-08-18 | Aucentra Therapeutics Pty Ltd | Derivatives of 4-(imidazo[l,2-a]pyridin-3-yl)-n-(pyridinyl)pyrimidin- 2-amine as therapeutic agents |
| EP4319766A1 (en) * | 2021-04-08 | 2024-02-14 | Memorial Sloan Kettering Cancer Center | Nadk2 inhibition in cancer and fibrotic disorders |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001002369A2 (en) | 1999-07-02 | 2001-01-11 | Agouron Pharmaceuticals, Inc. | Indazole compounds and pharmaceutical compositions for inhibiting protein kinases, and methods for their use |
| WO2001060816A1 (en) * | 2000-02-17 | 2001-08-23 | Amgen Inc. | Kinase inhibitors |
| WO2002010192A2 (en) | 2000-08-01 | 2002-02-07 | Novartis Ag | Somatostatin analogues |
| WO2002066470A1 (en) | 2001-01-12 | 2002-08-29 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| WO2008080937A1 (en) * | 2006-12-28 | 2008-07-10 | Basf Se | 2-substituted pyrimidines i in therapy |
| WO2008081910A1 (en) | 2006-12-28 | 2008-07-10 | Banyu Pharmaceutical Co., Ltd. | Novel aminopyrimidine derivatives as plk1 inhibitors |
| WO2010090290A1 (en) * | 2009-02-06 | 2010-08-12 | 日本新薬株式会社 | Aminopyrazine derivative and medicine |
Family Cites Families (112)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB581334A (en) | 1943-09-29 | 1946-10-09 | Francis Henry Swinden Curd | New pyrimidine compounds |
| JPS4921148B1 (en) | 1970-12-28 | 1974-05-30 | ||
| JPS4921149A (en) | 1972-06-15 | 1974-02-25 | ||
| AT340933B (en) | 1973-08-20 | 1978-01-10 | Thomae Gmbh Dr K | PROCESS FOR THE PRODUCTION OF NEW PYRIMIDE DERIVATIVES AND THEIR ACID ADDITIONAL SALTS |
| DE2341925A1 (en) | 1973-08-20 | 1975-03-06 | Thomae Gmbh Dr K | 2,4, (opt.5-), 6-substd. pyriminidines as antithrombic agents - e.g. 6-methyl-5-nitro-2-piperazino-4-thiomorpolino-pyrimidine |
| US4994386A (en) | 1987-07-13 | 1991-02-19 | Pharmacia Diagnostics, Inc. | Production of HBLV virus in the HSB-2 cell line |
| US4929726A (en) | 1988-02-09 | 1990-05-29 | Georgia State University Foundation, Inc. | Novel diazines and their method of preparation |
| EP0330263A1 (en) | 1988-02-25 | 1989-08-30 | Merck & Co. Inc. | Piperazinylalkylpyrimidines as hypoglycemic agents |
| GB9012311D0 (en) | 1990-06-01 | 1990-07-18 | Wellcome Found | Pharmacologically active cns compounds |
| ATE139117T1 (en) | 1990-07-03 | 1996-06-15 | Mitsui Petrochemical Ind | PYRIMIDINE COMPOUND AND ITS PHARMACEUTICALLY ACCEPTABLE SALT |
| WO1996032384A1 (en) | 1995-04-13 | 1996-10-17 | Taiho Pharmaceutical Co., Ltd. | Novel 4,6-diarylpyrimidine derivatives and salts thereof |
| JP3734907B2 (en) | 1996-12-19 | 2006-01-11 | 富士写真フイルム株式会社 | Development processing method |
| KR100563514B1 (en) | 1997-07-24 | 2006-03-27 | 젠야쿠코교가부시키가이샤 | Heterocyclic compounds and antitumor agent containing the same as active ingredient |
| JPH11158073A (en) | 1997-09-26 | 1999-06-15 | Takeda Chem Ind Ltd | Adenosine a3 antagonist |
| US6440965B1 (en) | 1997-10-15 | 2002-08-27 | Krenitsky Pharmaceuticals, Inc. | Substituted pyrimidine derivatives, their preparation and their use in the treatment of neurodegenerative or neurological disorders of the central nervous system |
| US6150362A (en) | 1997-12-12 | 2000-11-21 | Henkin; Jack | Triazine angiogenesis inhibitors |
| SI0930302T1 (en) | 1998-01-16 | 2003-10-31 | F. Hoffmann-La Roche Ag | Benzosulfone derivatives |
| DE69919707T2 (en) | 1998-06-19 | 2005-09-01 | Chiron Corp., Emeryville | GLYCOGEN SYNTHASE KINASE 3 INHIBITORS |
| US7045519B2 (en) | 1998-06-19 | 2006-05-16 | Chiron Corporation | Inhibitors of glycogen synthase kinase 3 |
| US6495558B1 (en) | 1999-01-22 | 2002-12-17 | Amgen Inc. | Kinase inhibitors |
| AU768201B2 (en) | 1999-01-22 | 2003-12-04 | Amgen, Inc. | Kinase inhibitors |
| WO2001000214A1 (en) | 1999-06-30 | 2001-01-04 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| CA2376957A1 (en) | 1999-06-30 | 2001-01-04 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| AU5636900A (en) | 1999-06-30 | 2001-01-31 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| DE60005355T2 (en) | 1999-07-15 | 2004-07-01 | Pharmacopeia, Inc. | BRADIKININ B1 RECEPTOR ANTAGONISTS |
| JP2001089452A (en) | 1999-09-22 | 2001-04-03 | Sankyo Co Ltd | Pyrimidine derivative |
| AU2001242629B2 (en) | 2000-03-29 | 2005-08-11 | Cyclacel Limited | 2-substituted 4-heteroaryl-pyrimidines and their use in the treatment of proliferative disorders |
| DE60144322D1 (en) | 2000-04-27 | 2011-05-12 | Astellas Pharma Inc | CONDENSED HETEROARYL DERIVATIVES |
| EP1277741A4 (en) | 2000-04-28 | 2003-05-07 | Tanabe Seiyaku Co | Cyclic compounds |
| WO2002000647A1 (en) | 2000-06-23 | 2002-01-03 | Bristol-Myers Squibb Pharma Company | Heteroaryl-phenyl substituted factor xa inhibitors |
| CN100506801C (en) | 2000-09-06 | 2009-07-01 | 诺华疫苗和诊断公司 | Inhibitors of glycogen synthase kinase 3 |
| CN100355750C (en) | 2000-09-15 | 2007-12-19 | 沃泰克斯药物股份有限公司 | Pyrazole compounds useful as protein kinase inhibitors |
| SE0004053D0 (en) | 2000-11-06 | 2000-11-06 | Astrazeneca Ab | N-type calcium channel antagonists for the treatment of pain |
| ATE326462T1 (en) | 2000-12-21 | 2006-06-15 | Vertex Pharma | PYRAZOLE COMPOUNDS AS PROTEIN KINASE INHIBITORS |
| WO2002062766A2 (en) | 2001-02-07 | 2002-08-15 | Millennium Pharmaceuticals, Inc. | Melanocortin-4 receptor binding compounds and methods of use thereof |
| US20030130264A1 (en) | 2001-02-16 | 2003-07-10 | Tularik Inc. | Methods of using pyrimidine-based antiviral agents |
| JP2004531571A (en) | 2001-05-25 | 2004-10-14 | ベーリンガー インゲルハイム ファーマシューティカルズ インコーポレイテッド | Carbamate and oxamide compounds as inhibitors of cytokine production |
| HUP0402352A2 (en) | 2001-06-19 | 2005-02-28 | Bristol-Myers Squibb Co. | Pyrimidine derivatives for use as phosphodiesterase (pde)7 inhibitors and pharmaceutical compositions containing them |
| US6603000B2 (en) | 2001-07-11 | 2003-08-05 | Boehringer Ingelheim Pharmaceuticals, Inc. | Synthesis for heteroarylamine compounds |
| WO2003030909A1 (en) | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| AU2003218215A1 (en) | 2002-03-15 | 2003-09-29 | Vertex Pharmaceuticals, Inc. | Azolylaminoazines as inhibitors of protein kinases |
| ATE433973T1 (en) | 2002-03-15 | 2009-07-15 | Vertex Pharma | AZOLYLAMINOAZINES AS INHIBITORS OF PROTEIN KINASES |
| EP1485100B1 (en) | 2002-03-15 | 2010-05-05 | Vertex Pharmaceuticals Incorporated | Azinylaminoazoles as inhibitors of protein kinases |
| US6846928B2 (en) | 2002-03-15 | 2005-01-25 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| US20040082627A1 (en) | 2002-06-21 | 2004-04-29 | Darrow James W. | Certain aromatic monocycles as kinase modulators |
| AU2003275242B2 (en) | 2002-09-27 | 2010-03-04 | Merck Sharp & Dohme Corp. | Substituted pyrimidines |
| US20040171073A1 (en) | 2002-10-08 | 2004-09-02 | Massachusetts Institute Of Technology | Compounds for modulation of cholesterol transport |
| US7223870B2 (en) | 2002-11-01 | 2007-05-29 | Pfizer Inc. | Methods for preparing N-arylated oxazolidinones via a copper catalyzed cross coupling reaction |
| ES2375111T3 (en) | 2002-11-21 | 2012-02-24 | Novartis Ag | PYRIMIDINES 2,4,6-TRISUSTITUIDAS AS INHIBITORS OF FOSFOTIDILINOSITOL (PI) 3-QUINASA AND ITS USE IN THE TREATMENT OF C�? NCER. |
| CA2519677A1 (en) | 2003-03-24 | 2004-10-07 | Merck & Co., Inc. | Biaryl substituted 6-membered heterocycles as sodium channel blockers |
| US20050014753A1 (en) | 2003-04-04 | 2005-01-20 | Irm Llc | Novel compounds and compositions as protein kinase inhibitors |
| AU2004230928B2 (en) | 2003-04-09 | 2010-12-02 | Exelixis, Inc. | Tie-2 modulators and methods of use |
| CA2531490A1 (en) | 2003-07-15 | 2005-02-03 | Neurogen Corporation | Substituted pyrimidin-4-ylamina analogues as vanilloid receptor ligands |
| EP1644358A2 (en) | 2003-07-16 | 2006-04-12 | Neurogen Corporation | Biaryl piperazinyl-pyridine analogues |
| PE20050952A1 (en) | 2003-09-24 | 2005-12-19 | Novartis Ag | DERIVATIVES OF ISOQUINOLINE AS INHIBITORS OF B-RAF |
| EP1675830A4 (en) * | 2003-09-30 | 2008-08-20 | Scios Inc | Heterocyclic amides and sulfonamides |
| CA2561895A1 (en) | 2004-04-13 | 2005-10-27 | Icagen, Inc. | Polycyclic pyrimidines as potassium ion channel modulators |
| GB0415364D0 (en) | 2004-07-09 | 2004-08-11 | Astrazeneca Ab | Pyrimidine derivatives |
| GB0415365D0 (en) | 2004-07-09 | 2004-08-11 | Astrazeneca Ab | Pyrimidine derivatives |
| MY145822A (en) | 2004-08-13 | 2012-04-30 | Neurogen Corp | Substituted biaryl piperazinyl-pyridine analogues |
| CA2587642C (en) | 2004-11-30 | 2013-04-09 | Amgen Inc. | Substituted heterocycles and methods of use |
| AU2005317176A1 (en) | 2004-12-13 | 2006-06-22 | Neurogen Corporation | Piperazinyl-pyridine analogues |
| CN101080401A (en) | 2004-12-13 | 2007-11-28 | 神经能质公司 | Substituted biaryl analogues |
| KR101478933B1 (en) | 2004-12-28 | 2015-01-02 | 키넥스 파마슈티컬즈, 엘엘씨 | Compositions and methods of treating cell proliferation disorders |
| WO2006078992A2 (en) | 2005-01-19 | 2006-07-27 | Neurogen Corporation | Heteroaryl substituted piperazinyl-pyridine analogues |
| US20100130473A1 (en) | 2005-02-25 | 2010-05-27 | Marc Geoffrey Hummersone | Compounds |
| AR053712A1 (en) | 2005-04-18 | 2007-05-16 | Neurogen Corp | HETEROARILOS SUBSTITUTED, ANTAGONISTS OF CB1 (RECEIVER 1 CANABINOID) |
| GB2431156A (en) | 2005-10-11 | 2007-04-18 | Piramed Ltd | 1-cyclyl-3-substituted- -benzenes and -azines as inhibitors of phosphatidylinositol 3-kinase |
| AU2007204208A1 (en) | 2006-01-11 | 2007-07-19 | Astrazeneca Ab | Morpholino pyrimidine derivatives and their use in therapy |
| JO2660B1 (en) | 2006-01-20 | 2012-06-17 | نوفارتيس ايه جي | PI-3 Kinase inhibitors and methods of their use |
| CA2675558A1 (en) | 2007-02-06 | 2008-08-14 | Novartis Ag | Pi 3-kinase inhibitors and methods of their use |
| US7957951B2 (en) | 2007-03-16 | 2011-06-07 | Robert Bosch Gmbh | Address translation system for use in a simulation environment |
| EP2074118A2 (en) | 2007-07-09 | 2009-07-01 | AstraZeneca AB | Trisubstituted pyrimidine derivatives for the treatment of proliferative diseases |
| WO2009066084A1 (en) | 2007-11-21 | 2009-05-28 | F. Hoffmann-La Roche Ag | 2 -morpholinopyrimidines and their use as pi3 kinase inhibitors |
| AU2009221164B2 (en) | 2008-03-05 | 2012-07-26 | Novartis Ag | Use of pyrimidine derivatives for the treatment of EGFR dependent diseases or diseases that have acquired resistance to agents that target EGFR family members |
| EP2474323A3 (en) | 2008-03-26 | 2012-10-10 | Novartis AG | Imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| JP2011515462A (en) | 2008-03-27 | 2011-05-19 | アウククランド ウニセルビセス リミテッド | Substituted pyrimidines and triazines and their use in cancer therapy |
| JP5277256B2 (en) | 2008-04-09 | 2013-08-28 | 田辺三菱製薬株式会社 | Pyrimidine, pyridine and triazine derivatives as maxi-K channel openers |
| GB0815369D0 (en) | 2008-08-22 | 2008-10-01 | Summit Corp Plc | Compounds for treatment of duchenne muscular dystrophy |
| JP2012506898A (en) | 2008-10-31 | 2012-03-22 | ノバルティス アーゲー | Combination of phosphatidylinositol-3-kinase (PI3K) inhibitor and mTOR inhibitor |
| GB2465405A (en) | 2008-11-10 | 2010-05-19 | Univ Basel | Triazine, pyrimidine and pyridine analogues and their use in therapy |
| WO2010068863A2 (en) | 2008-12-12 | 2010-06-17 | Cystic Fibrosis Foundation Therapeutics, Inc. | Pyrimidine compounds and methods of making and using same |
| JP5747440B2 (en) | 2009-02-06 | 2015-07-15 | 住友化学株式会社 | Hydrazide compound and its use for pest control |
| WO2010105243A1 (en) | 2009-03-13 | 2010-09-16 | Agios Pharmaceuticals, Inc. | Methods and compositions for cell-proliferation-related disorders |
| WO2010120994A2 (en) | 2009-04-17 | 2010-10-21 | Wyeth Llc | Ureidoaryl-and carbamoylaryl-morpholino- pyrimidine compounds, their use as mtor kinase and pi3 kinase inhibitors, and their synthesis |
| ES2417355T3 (en) | 2009-05-19 | 2013-08-07 | Dow Agrosciences Llc | Compounds and methods for fungal control |
| AR080945A1 (en) | 2009-07-07 | 2012-05-23 | Pathway Therapeutics Inc | PIRIMIDINIL AND 1,3,5-TRIAZINIL BENZIMIDAZOLES AND ITS USE IN THERAPY AGAINST CANCER |
| US8609676B2 (en) | 2009-08-04 | 2013-12-17 | Merck Sharp & Dohme, Corp. | 4, 5, 6-trisubstituted pyrimidine derivatives as factor IXa inhibitors |
| AR077999A1 (en) | 2009-09-02 | 2011-10-05 | Vifor Int Ag | ANTIGONISTS OF PYRIMIDIN AND TRIAZIN-HEPCIDINE |
| CN102625708A (en) | 2009-09-09 | 2012-08-01 | 阿维拉制药公司 | Pi3 kinase inhibitors and uses thereof |
| JP5967827B2 (en) * | 2009-12-09 | 2016-08-10 | アジオス ファーマシューティカルズ, インコーポレイテッド | Therapeutically active compounds for the treatment of cancer characterized by having an IDH variant |
| GB201004200D0 (en) | 2010-03-15 | 2010-04-28 | Univ Basel | Spirocyclic compounds and their use as therapeutic agents and diagnostic probes |
| AR081626A1 (en) | 2010-04-23 | 2012-10-10 | Cytokinetics Inc | AMINO-PYRIDAZINIC COMPOUNDS, PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM AND USE OF THE SAME TO TREAT CARDIAC AND SKELETIC MUSCULAR DISORDERS |
| AR081331A1 (en) | 2010-04-23 | 2012-08-08 | Cytokinetics Inc | AMINO- PYRIMIDINES COMPOSITIONS OF THE SAME AND METHODS FOR THE USE OF THE SAME |
| WO2011133920A1 (en) | 2010-04-23 | 2011-10-27 | Cytokinetics, Inc. | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| WO2011143160A2 (en) | 2010-05-10 | 2011-11-17 | The Johns Hopkins University | Metabolic inhibitor against tumors having an idh mutation |
| JP6081354B2 (en) | 2010-07-16 | 2017-02-15 | アジオス ファーマシューティカルズ, インコーポレイテッド | Therapeutically active compositions and methods of their use |
| WO2012044727A2 (en) | 2010-10-01 | 2012-04-05 | Novartis Ag | Manufacturing process for pyrimidine derivatives |
| SG10201708237SA (en) | 2010-10-18 | 2017-11-29 | Cerenis Therapeutics Holding Sa | Compounds, compositions and methods useful for cholesterol mobilisation |
| GB201106829D0 (en) | 2011-04-21 | 2011-06-01 | Proximagen Ltd | Heterocyclic compounds |
| JP2014505107A (en) | 2011-02-11 | 2014-02-27 | デイナ ファーバー キャンサー インスティチュート,インコーポレイテッド | Methods for inhibiting hamartoma tumor cells |
| CN102827073A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Therapeutically active compositions and application methods thereof |
| JP2014527542A (en) | 2011-09-01 | 2014-10-16 | ノバルティス アーゲー | PI3K inhibitor for use in the treatment of bone cancer or for preventing metastatic dissemination of primary cancer cells into bone |
| WO2013052395A1 (en) | 2011-10-06 | 2013-04-11 | Merck Sharp & Dohme Corp. | 1,3-substituted azetidine pde10 inhibitors |
| UY34632A (en) | 2012-02-24 | 2013-05-31 | Novartis Ag | OXAZOLIDIN- 2- ONA COMPOUNDS AND USES OF THE SAME |
| EP2849756A1 (en) | 2012-05-16 | 2015-03-25 | Novartis AG | Dosage regimen for a pi-3 kinase inhibitor |
| JP2015518888A (en) | 2012-06-06 | 2015-07-06 | ノバルティス アーゲー | Combination of a 17-alpha-hydroxylase (C17,20-lyase) inhibitor and a specific PI-3K inhibitor for the treatment of tumor diseases |
| KR20210049187A (en) | 2012-08-16 | 2021-05-04 | 노파르티스 아게 | Combination of pi3k inhibitor and c-met inhibitor |
| WO2014064058A1 (en) | 2012-10-23 | 2014-05-01 | Novartis Ag | Improved process for manufacturing 5-(2,6-di-4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine |
| US9296733B2 (en) | 2012-11-12 | 2016-03-29 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivative and use thereof for the treatment of conditions, diseases and disorders dependent upon PI3 kinases |
| CN103483345B (en) | 2013-09-25 | 2016-07-06 | 中山大学 | PI3K inhibitors of kinases, the pharmaceutical composition comprising it and application thereof |
| CN103694218B (en) | 2013-12-05 | 2016-04-27 | 中山大学 | Pyrimidine compound, PI3K inhibitor, the pharmaceutical composition comprising PI3K inhibitor and application |
-
2012
- 2012-09-26 EP EP12784701.0A patent/EP2771337B1/en active Active
- 2012-09-26 CU CUP2014000036A patent/CU24269B1/en unknown
- 2012-09-26 PE PE2014000427A patent/PE20141581A1/en not_active Application Discontinuation
- 2012-09-26 UA UAA201403086A patent/UA111503C2/en unknown
- 2012-09-26 CA CA2849995A patent/CA2849995A1/en active Pending
- 2012-09-26 US US14/347,481 patent/US8957068B2/en active Active
- 2012-09-26 KR KR1020147010827A patent/KR20140069235A/en not_active Application Discontinuation
- 2012-09-26 TW TW101135421A patent/TWI547493B/en active
- 2012-09-26 EA EA201490696A patent/EA025183B1/en not_active IP Right Cessation
- 2012-09-26 GE GEAP201213450A patent/GEP20166432B/en unknown
- 2012-09-26 ES ES12784701.0T patent/ES2645968T3/en active Active
- 2012-09-26 WO PCT/IB2012/055133 patent/WO2013046136A1/en active Application Filing
- 2012-09-26 AU AU2012313888A patent/AU2012313888B2/en not_active Ceased
- 2012-09-26 SG SG11201400989TA patent/SG11201400989TA/en unknown
- 2012-09-26 MX MX2014003752A patent/MX342326B/en active IP Right Grant
- 2012-09-26 AP AP2014007602A patent/AP3907A/en active
- 2012-09-26 JP JP2014532533A patent/JP6026544B2/en not_active Expired - Fee Related
- 2012-09-26 BR BR112014007310A patent/BR112014007310A2/en not_active IP Right Cessation
- 2012-09-26 CN CN201280058052.3A patent/CN103958506B/en not_active Expired - Fee Related
- 2012-09-27 AR ARP120103605A patent/AR088175A1/en unknown
- 2012-09-27 UY UY0001034352A patent/UY34352A/en not_active Application Discontinuation
-
2014
- 2014-03-24 ZA ZA2014/02144A patent/ZA201402144B/en unknown
- 2014-03-25 MA MA36849A patent/MA35452B1/en unknown
- 2014-03-26 NI NI201400026A patent/NI201400026A/en unknown
- 2014-03-26 TN TNP2014000128A patent/TN2014000128A1/en unknown
- 2014-03-26 CL CL2014000746A patent/CL2014000746A1/en unknown
- 2014-03-27 GT GT201400057A patent/GT201400057A/en unknown
- 2014-03-27 CR CR20140143A patent/CR20140143A/en unknown
- 2014-03-27 DO DO2014000058A patent/DOP2014000058A/en unknown
- 2014-03-27 IL IL231786A patent/IL231786A/en not_active IP Right Cessation
- 2014-03-27 CO CO14065647A patent/CO6920291A2/en active IP Right Grant
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001002369A2 (en) | 1999-07-02 | 2001-01-11 | Agouron Pharmaceuticals, Inc. | Indazole compounds and pharmaceutical compositions for inhibiting protein kinases, and methods for their use |
| WO2001060816A1 (en) * | 2000-02-17 | 2001-08-23 | Amgen Inc. | Kinase inhibitors |
| WO2002010192A2 (en) | 2000-08-01 | 2002-02-07 | Novartis Ag | Somatostatin analogues |
| WO2002066470A1 (en) | 2001-01-12 | 2002-08-29 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| WO2008080937A1 (en) * | 2006-12-28 | 2008-07-10 | Basf Se | 2-substituted pyrimidines i in therapy |
| WO2008081910A1 (en) | 2006-12-28 | 2008-07-10 | Banyu Pharmaceutical Co., Ltd. | Novel aminopyrimidine derivatives as plk1 inhibitors |
| WO2010090290A1 (en) * | 2009-02-06 | 2010-08-12 | 日本新薬株式会社 | Aminopyrazine derivative and medicine |
Non-Patent Citations (19)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 20th ed.,", 1985, MACK PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences", MACK PUBLISHING COMPANY |
| "The Handbook of Pharmaceutical Additives", GOWER PUBLISHING LIMITED |
| "The Handbook of Pharmaceutical Excipients", AMERICAN PHARMACEUTICAL ASSOCIATION AND THE PHARMACEUTICAL PRESS |
| AMARY ET AL., NATURE GENETICS, 2011 |
| BALSS, ACTA NEUROPATHOL., 2008 |
| DENG, TRENDS MOL. MED., vol. 16, 2010, pages 387 |
| E. L. ELIEL; S. H. WILEN; L. N. MANDER: "Stereochemistry of Organic Compounds", 1994, WILEY-INTERSCIENCE |
| GAAL ET AL., J. CLIN. ENDOCRINOL. METAB., 2010 |
| HAYDEN ET AL., CELL CYCLE, 2009 |
| KRANENDIJK, M., SCIENCE, vol. 330, 2010, pages 336 |
| L. DENG, NATURE, vol. 462, 2009, pages 739 |
| L. SELLNER, EUR. J. HAEMATOL., vol. 85, 2011, pages 457 |
| P.S. WARD, CANCER CELL, vol. 17, 2010, pages 225 |
| PANSURIYA ET AL., NATURE GENETICS, 2011 |
| S. GROSS, J. EXP. MED., vol. 207, no. 2, 2010, pages 339 |
| STAHL; WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| T. SHIBATA ET AL., AM. J. PATHOL., vol. 178, no. 3, 2011, pages 1395 |
| T. W. GREENE; P. G. M. WUTS: "Protective Groups in Organic Synthesis, Third edition,", 1999, WILEY |
Cited By (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8865894B2 (en) | 2012-02-24 | 2014-10-21 | Novartis Ag | Oxazolidin-2-one compounds and uses thereof |
| US9458177B2 (en) | 2012-02-24 | 2016-10-04 | Novartis Ag | Oxazolidin-2-one compounds and uses thereof |
| US9296733B2 (en) | 2012-11-12 | 2016-03-29 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivative and use thereof for the treatment of conditions, diseases and disorders dependent upon PI3 kinases |
| US10202371B2 (en) | 2012-11-12 | 2019-02-12 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivatives and the use thereof as phosphatidylinositol-3-kinase inhibitors |
| US9434719B2 (en) | 2013-03-14 | 2016-09-06 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| WO2014141104A1 (en) * | 2013-03-14 | 2014-09-18 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| CN105209460A (en) * | 2013-03-14 | 2015-12-30 | 诺华股份有限公司 | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| US20160039802A1 (en) * | 2013-03-14 | 2016-02-11 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| US10112931B2 (en) | 2013-03-14 | 2018-10-30 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| EA028033B1 (en) * | 2013-03-14 | 2017-09-29 | Новартис Аг | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| US9688672B2 (en) | 2013-03-14 | 2017-06-27 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH |
| JP2016523935A (en) * | 2013-07-11 | 2016-08-12 | アジオス ファーマシューティカルズ, インコーポレイテッド | Therapeutically active compounds and methods of use thereof |
| JP2016526561A (en) * | 2013-07-11 | 2016-09-05 | アジオス ファーマシューティカルズ, インコーポレイテッド | Therapeutically active compounds and methods of their use |
| JP2016523976A (en) * | 2013-07-11 | 2016-08-12 | アジオス ファーマシューティカルズ, インコーポレイテッド | 2,4- or 4,6-diaminopyrimidine compounds as IDH2 mutant inhibitors for the treatment of cancer |
| JP2016525130A (en) * | 2013-07-25 | 2016-08-22 | アギオス ファーマシューティカルス,インコーポレーテッド | Therapeutically active compounds and uses thereof |
| WO2015127172A1 (en) * | 2014-02-20 | 2015-08-27 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| JP2017518326A (en) * | 2014-06-17 | 2017-07-06 | キエージ・フアルマチエウテイチ・エツセ・ピ・ア | Indolizine derivatives as phosphoinositide 3-kinase inhibitors |
| US11498913B2 (en) | 2014-09-19 | 2022-11-15 | Forma Therapeutics, Inc. | Pyridin-2(1H)-one quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| JP2017528492A (en) * | 2014-09-19 | 2017-09-28 | フォーマ セラピューティクス,インコーポレイテッド | Quinolinone pyrimidine compositions as mutant isocitrate dehydrogenase inhibitors |
| US10889567B2 (en) | 2014-09-19 | 2021-01-12 | Forma Therapeutics, Inc. | Pyridin-2(1H)-one quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| US9862703B2 (en) | 2014-09-22 | 2018-01-09 | National Health Research Institutes | Heterocyclic compounds and use thereof |
| US9926298B2 (en) | 2014-09-22 | 2018-03-27 | National Health Research Institutes | Heterocyclic compounds and use thereof |
| CN106795146A (en) * | 2014-10-01 | 2017-05-31 | 第三共株式会社 | As the inhibitor isoxazole derivativeses of saltant type isocitric dehydrogenase 1 |
| WO2016052697A1 (en) * | 2014-10-01 | 2016-04-07 | 第一三共株式会社 | Isoxazole derivative as mutated isocitrate dehydrogenase 1 inhibitor |
| JPWO2016052697A1 (en) * | 2014-10-01 | 2017-04-27 | 第一三共株式会社 | Isoxazole derivatives as mutant isocitrate dehydrogenase 1 inhibitors |
| AU2015325279B2 (en) * | 2014-10-01 | 2020-02-27 | Daiichi Sankyo Company, Limited | Isoxazole derivative as mutant isocitrate dehydrogenase 1 inhibitor |
| JP6087033B2 (en) * | 2014-10-01 | 2017-03-01 | 第一三共株式会社 | Isoxazole derivatives as mutant isocitrate dehydrogenase 1 inhibitors |
| KR102440429B1 (en) | 2014-10-01 | 2022-09-05 | 다이이찌 산쿄 가부시키가이샤 | Isoxazole derivative as mutated isocitrate dehydrogenase 1 inhibitor |
| CN106795146B (en) * | 2014-10-01 | 2020-06-26 | 第一三共株式会社 | Isoxazole derivatives as inhibitors of mutant isocitrate dehydrogenase 1 |
| US10040791B2 (en) | 2014-10-01 | 2018-08-07 | Daiichi Sankyo Company, Limited | Isoxazole derivative as mutant isocitrate dehydrogenase 1 inhibitor |
| KR20170068501A (en) * | 2014-10-01 | 2017-06-19 | 다이이찌 산쿄 가부시키가이샤 | Isoxazole derivative as mutated isocitrate dehydrogenase 1 inhibitor |
| RU2692782C2 (en) * | 2014-10-01 | 2019-06-27 | Дайити Санкио Компани, Лимитед | Isoxazole derivative as inhibitor of mutant isocitrate dehydrogenase 1 |
| WO2016062770A1 (en) * | 2014-10-23 | 2016-04-28 | Bayer Pharma Aktiengesellschaft | 1-cyclohexyl-2-phenylaminobenzimidazoles as midh1 inhibitors for the treatment of tumors |
| US10137110B2 (en) | 2014-10-23 | 2018-11-27 | Bayer Pharma Aktiengesellschaft | 1-cyclohexyl-2-phenylaminobenzimidazoles as mIDH1 inhibitors for the treatment of tumors |
| CN107108554B (en) * | 2014-10-23 | 2020-11-06 | 德国癌症研究中心 | 1-cyclohexyl-2-phenylaminobenzimidazole as MIDH1 inhibitor for treating tumors |
| CN107108554A (en) * | 2014-10-23 | 2017-08-29 | 拜耳医药股份有限公司 | It is used for the phenyl amino benzimidazole of 1 cyclohexyl 2 for treating tumour as MIDH1 inhibitor |
| EP3226690A4 (en) * | 2014-12-05 | 2018-06-20 | Merck Sharp & Dohme Corp. | Novel tricyclic compounds as inhibitors of mutant idh enzymes |
| US10086000B2 (en) | 2014-12-05 | 2018-10-02 | Merck Sharp & Dohme Corp. | Tricyclic compounds as inhibitors of mutant IDH enzymes |
| WO2016089833A1 (en) | 2014-12-05 | 2016-06-09 | Merck Sharp & Dohme Corp. | Novel tricyclic compounds as inhibitors of mutant idh enzymes |
| US10703746B2 (en) | 2014-12-22 | 2020-07-07 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Mutant IDH1 inhibitors useful for treating cancer |
| US10807976B2 (en) | 2015-04-21 | 2020-10-20 | Forma Therapeutics, Inc. | Quinolinone five-membered heterocyclic compounds as mutant-isocitrate dehydrogenase inhibitors |
| US10179123B2 (en) | 2015-07-07 | 2019-01-15 | Bayer Pharma Aktiengesellschaft | 2-aryl- and 2-arylalkyl-benzimidazoles as mIDH1 inhibitors |
| WO2017005674A1 (en) * | 2015-07-07 | 2017-01-12 | Bayer Pharma Aktiengesellschaft | 2-aryl- and 2-arylalkyl-benzimidazoles as midh1 inhibitors |
| US10253041B2 (en) | 2015-07-27 | 2019-04-09 | Eli Lilly And Company | 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds and their use as mutant IDH1 inhibitors |
| CN107810181B (en) * | 2015-07-27 | 2021-06-22 | 德国癌症研究公共权益基金会 | Inhibitors of mutant isocitrate dehydrogenase IDH 1R 132H |
| AU2016300980B2 (en) * | 2015-07-27 | 2020-12-24 | Deutsches Krebsforschungszentrum, Stiftung Des Öffentlichen Rechts | Inhibitor of the mutated isocitrate dehydrogenase IDH1 R132H |
| US10344004B2 (en) | 2015-07-27 | 2019-07-09 | Bayer Pharma Aktiengesellschaft | Inhibitor of the mutated isocitrate dehydrogenase IDH1 R132H |
| WO2017019429A1 (en) * | 2015-07-27 | 2017-02-02 | Eli Lilly And Company | 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds and theit use as mutant idh1 inhibitors |
| CN107849059B (en) * | 2015-07-27 | 2020-04-28 | 伊莱利利公司 | 7-phenylethylamino-4H-pyrimido [4,5-D ] [1,3] oxazin-2-one compounds and their use as inhibitors of mutant IDH1 |
| WO2017016992A1 (en) * | 2015-07-27 | 2017-02-02 | Bayer Pharma Aktiengesellschaft | Inhibitor of the mutated isocitrate dehydrogenase idh1 r132h |
| CN107810181A (en) * | 2015-07-27 | 2018-03-16 | 拜耳制药股份公司 | The isocitric dehydrogenase IDH1 R132H of mutation inhibitor |
| CN107849059A (en) * | 2015-07-27 | 2018-03-27 | 伊莱利利公司 | 7 phenylethylcarbamate 4H pyrimidos [4,5 D] [the assimilation compound of 1,3] oxazines 2 and its purposes as mutant IDH1 inhibitor |
| US10047078B2 (en) | 2016-02-05 | 2018-08-14 | National Health Research Institutes | Aminothiazole compounds |
| WO2017140758A1 (en) | 2016-02-19 | 2017-08-24 | Debiopharm International S.A. | Derivatives of 2-amino-4-(2-oxazolidinon-3-yl)-pyrimidine fused with a five-membered heteroaromatic ring in 5,6-position which are useful for the treatment of various cancers |
| WO2017213910A1 (en) | 2016-06-06 | 2017-12-14 | Eli Lilly And Company | Mutant idh1 inhibitors |
| US10696665B2 (en) | 2016-06-06 | 2020-06-30 | Eli Lilly And Company | Mutant IDH1 inhibitors |
| US11970486B2 (en) | 2016-10-24 | 2024-04-30 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| AU2020260493B2 (en) * | 2016-12-16 | 2021-09-09 | Eli Lilly And Company | 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitors |
| EA036112B1 (en) * | 2016-12-16 | 2020-09-29 | Эли Лилли Энд Компани | 7-PHENYLETHYLAMINO-4H-PYRIMIDO[4,5-d][1,3]OXAZIN-2-ONE COMPOUNDS AS MUTANT IDH1 AND IDH2 INHIBITORS |
| WO2018111707A1 (en) * | 2016-12-16 | 2018-06-21 | Eli Lilly And Company | 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant idh1 and idh2 inhibitors |
| EP3763717A1 (en) * | 2016-12-16 | 2021-01-13 | Eli Lilly And Co. | Pyrido[4,3-d][1,3]oxazin-2-one and pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant idh1 and idh2 inhibitors |
| US11001596B2 (en) | 2016-12-16 | 2021-05-11 | Eli Lilly And Company | 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitors |
| US11649247B2 (en) | 2016-12-16 | 2023-05-16 | Eli Lilly And Company | 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitors |
| US11629156B2 (en) | 2016-12-16 | 2023-04-18 | Eli Lilly And Company | 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitors |
| AU2017378060B2 (en) * | 2016-12-16 | 2020-09-03 | Eli Lilly And Company | 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitors |
| KR102276022B1 (en) | 2016-12-16 | 2021-07-13 | 일라이 릴리 앤드 캄파니 | 7-phenylethylamino-4H-pyrimido[4,5-D][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitors |
| KR20190077539A (en) * | 2016-12-16 | 2019-07-03 | 일라이 릴리 앤드 캄파니 | 7-phenylethylamino-4H-pyrimido [4,5-D] [1,3] oxazin-2-one as a mutant IDH1 and IDH2 inhibitor |
| WO2018118793A1 (en) * | 2016-12-19 | 2018-06-28 | Isocure Biosciences Inc. | Inhibitors of mutant isocitrate dehydrogenases and compositions and methods thereof |
| US11149032B2 (en) | 2016-12-19 | 2021-10-19 | Isocure Biosciences Inc. | Inhibitors of mutant isocitrate dehydrogenases and compositions and methods thereof |
| EP3555079A4 (en) * | 2016-12-19 | 2020-08-19 | Isocure Biosciences Inc. | Inhibitors of mutant isocitrate dehydrogenases and compositions and methods thereof |
| AU2017379796B2 (en) * | 2016-12-19 | 2021-10-21 | Isocure Biosciences Inc. | Inhibitors of mutant isocitrate dehydrogenases and compositions and methods thereof |
| US11247987B2 (en) | 2017-10-06 | 2022-02-15 | Forma Therapeutics, Inc. | Inhibiting ubiquitin specific peptidase 30 |
| US11873298B2 (en) | 2017-10-24 | 2024-01-16 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| US12053463B2 (en) | 2018-05-16 | 2024-08-06 | Forma Therapeutics, Inc. | Solid forms of ((s)-5-((1-(6-chloro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl)amino)-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile |
| US11723905B2 (en) | 2018-05-16 | 2023-08-15 | Forma Therapeutics, Inc. | Solid forms of ((s)-5-((1-(6-chloro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl)amino)-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile |
| US11376246B2 (en) | 2018-05-16 | 2022-07-05 | Forma Therapeutics, Inc. | Inhibiting mutant IDH-1 |
| US11963956B2 (en) | 2018-05-16 | 2024-04-23 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mIDH-1) |
| US11311527B2 (en) | 2018-05-16 | 2022-04-26 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mIDH-1) |
| US11013733B2 (en) | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mlDH-1) |
| US11576906B2 (en) | 2018-05-16 | 2023-02-14 | Forma Therapeutics, Inc. | Inhibiting mutant IDH-1 |
| US11013734B2 (en) | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Treating patients harboring an isocitrate dehydrogenase-1 (IDH-1) mutation |
| US11738018B2 (en) | 2018-05-16 | 2023-08-29 | FORMA Therapeuetics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mIDH-1) |
| US12049466B2 (en) | 2018-05-17 | 2024-07-30 | Forma Therapeutics, Inc. | Fused bicyclic compounds useful as ubiquitin-specific peptidase 30 inhibitors |
| US11814386B2 (en) | 2018-10-05 | 2023-11-14 | Forma Therapeutics, Inc. | Fused pyrrolines which act as ubiquitin-specific protease 30 (USP30) inhibitors |
| US11535618B2 (en) | 2018-10-05 | 2022-12-27 | Forma Therapeutics, Inc. | Fused pyrrolines which act as ubiquitin-specific protease 30 (USP30) inhibitors |
| US12098146B2 (en) | 2019-01-24 | 2024-09-24 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| EP3941908A4 (en) * | 2019-03-22 | 2023-05-17 | Yumanity Therapeutics, Inc. | Compounds and uses thereof |
| EP3964508A4 (en) * | 2019-04-23 | 2022-12-21 | Epitas Biosciences (Shanghai) Co., Ltd. | Dimeric or polymeric form of mutant idh inhibitor |
| WO2020216071A1 (en) | 2019-04-23 | 2020-10-29 | 上海仕谱生物科技有限公司 | Dimeric or polymeric form of mutant idh inhibitor |
| US11746115B2 (en) | 2021-08-13 | 2023-09-05 | Eli Lilly And Company | Solid forms of 7-[[(1S)-1-[4-[(1S)-2-cyclopropyl-1-(4-prop-2-enoylpiperazin-1-yl)ethyl]phenyl]ethyl]amino]-1-ethyl-4H-pyrimido[4,5-d][1,3]oxazin-2-one |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2771337B1 (en) | 3-(pyrimidin-4-yl)-oxazolidin-2-ones as inhibitors of mutant idh | |
| US10112931B2 (en) | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant IDH | |
| EP2970242B1 (en) | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh | |
| US11591336B2 (en) | Substituted pyrazolo[3,4-b]pyrazines as SHP2 phosphatase inhibitors | |
| WO2014147586A1 (en) | 1-(2-(ethylamino)pyrimidin-4-yl)pyrrolidin-2-ones as inhibitors of mutant idh | |
| CA2828824A1 (en) | Thiazolopyrimidine compounds | |
| EP2970313A1 (en) | Substituted 7-azabicycles and their use as orexin receptor modulators | |
| NZ624040B2 (en) | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12784701 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2849995 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2014532533 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014000746 Country of ref document: CL Ref document number: 14347481 Country of ref document: US Ref document number: 12014500682 Country of ref document: PH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 231786 Country of ref document: IL Ref document number: 14065647 Country of ref document: CO Ref document number: 000427-2014 Country of ref document: PE Ref document number: MX/A/2014/003752 Country of ref document: MX Ref document number: CR2014-000143 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201403086 Country of ref document: UA |
|
| ENP | Entry into the national phase |
Ref document number: 20147010827 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012784701 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201490696 Country of ref document: EA Ref document number: 13450 Country of ref document: GE Ref document number: 2012784701 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2012313888 Country of ref document: AU Date of ref document: 20120926 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014007310 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014007310 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140327 |