WO2013039988A1 - Azaindazoles - Google Patents
Azaindazoles Download PDFInfo
- Publication number
- WO2013039988A1 WO2013039988A1 PCT/US2012/054785 US2012054785W WO2013039988A1 WO 2013039988 A1 WO2013039988 A1 WO 2013039988A1 US 2012054785 W US2012054785 W US 2012054785W WO 2013039988 A1 WO2013039988 A1 WO 2013039988A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- pyridinyl
- oxo
- pyridine
- pyrazolo
- Prior art date
Links
- KXXLCLVJGUVBTJ-UHFFFAOYSA-N CC(C)[n](c1nc(-c(cc2)cc(C3)c2NC3=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c(cc2)cc(C3)c2NC3=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O KXXLCLVJGUVBTJ-UHFFFAOYSA-N 0.000 description 1
- KLLPIDYLSWTUIN-UHFFFAOYSA-N CC(C)[n](c1nc(-c(cc2)ccc2C(NC)=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c(cc2)ccc2C(NC)=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O KLLPIDYLSWTUIN-UHFFFAOYSA-N 0.000 description 1
- AKBZTJSWWZBIFH-UHFFFAOYSA-N CC(C)[n](c1nc(-c(cc2)ccc2NS(C)(=O)=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c(cc2)ccc2NS(C)(=O)=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O AKBZTJSWWZBIFH-UHFFFAOYSA-N 0.000 description 1
- BSNAZGHGZOMFTG-UHFFFAOYSA-N CC(C)[n](c1nc(-c(cc2)ccc2S(C)(=O)=O)c2)ncc1c2C(O)=O Chemical compound CC(C)[n](c1nc(-c(cc2)ccc2S(C)(=O)=O)c2)ncc1c2C(O)=O BSNAZGHGZOMFTG-UHFFFAOYSA-N 0.000 description 1
- SXFSERCDFHFHJK-UHFFFAOYSA-N CC(C)[n](c1nc(-c(cc2)ccc2S(N(C)C)(=O)=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c(cc2)ccc2S(N(C)C)(=O)=O)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O SXFSERCDFHFHJK-UHFFFAOYSA-N 0.000 description 1
- OBASFAPNSSLYMR-UHFFFAOYSA-N CC(C)[n](c1nc(-c(cc2)cnc2N2CCOCC2)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c(cc2)cnc2N2CCOCC2)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O OBASFAPNSSLYMR-UHFFFAOYSA-N 0.000 description 1
- CPVQOGUURKEJJM-UHFFFAOYSA-N CC(C)[n](c1nc(-c2c[nH]nc2)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c2c[nH]nc2)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O CPVQOGUURKEJJM-UHFFFAOYSA-N 0.000 description 1
- WGUMQTCFJOVEEG-UHFFFAOYSA-N CC(C)[n](c1nc(-c2c[s]cc2)c2)ncc1c2C(O)=O Chemical compound CC(C)[n](c1nc(-c2c[s]cc2)c2)ncc1c2C(O)=O WGUMQTCFJOVEEG-UHFFFAOYSA-N 0.000 description 1
- ROLJNRNHZVGSDA-UHFFFAOYSA-N CC(C)[n](c1nc(-c2cc(NS(c3ccccc3)(=O)=O)cnc2)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c2cc(NS(c3ccccc3)(=O)=O)cnc2)c2)ncc1c2C(NCC1=C(C)C=C(C)NC1=O)=O ROLJNRNHZVGSDA-UHFFFAOYSA-N 0.000 description 1
- OTXZCSZGICIBRE-UHFFFAOYSA-N CC(C)[n](c1nc(-c2ccncc2)c2)ncc1c2C(NCC1=C(C2CC2)C=C(C)NC1=O)=O Chemical compound CC(C)[n](c1nc(-c2ccncc2)c2)ncc1c2C(NCC1=C(C2CC2)C=C(C)NC1=O)=O OTXZCSZGICIBRE-UHFFFAOYSA-N 0.000 description 1
- HCNLJCWUXKHIRQ-UHFFFAOYSA-N CC(C)[n](c1nc(Cl)c2)nc(C)c1c2C(O)=O Chemical compound CC(C)[n](c1nc(Cl)c2)nc(C)c1c2C(O)=O HCNLJCWUXKHIRQ-UHFFFAOYSA-N 0.000 description 1
- CKQCYGJHRBQQHV-UHFFFAOYSA-N CC(C=C(C)N1)=C(CNC(c2c(cn[n]3CCN)c3nc(C3CC3)c2)=O)C1=O Chemical compound CC(C=C(C)N1)=C(CNC(c2c(cn[n]3CCN)c3nc(C3CC3)c2)=O)C1=O CKQCYGJHRBQQHV-UHFFFAOYSA-N 0.000 description 1
- ZPVOFDSZWZKZJS-UHFFFAOYSA-N CC(C=C(C)N1)=C(CNC(c2c(cn[n]3N)c3nc(C3CC3)c2)=O)C1=O Chemical compound CC(C=C(C)N1)=C(CNC(c2c(cn[n]3N)c3nc(C3CC3)c2)=O)C1=O ZPVOFDSZWZKZJS-UHFFFAOYSA-N 0.000 description 1
- AWQYPAVOIYFYRY-UHFFFAOYSA-N CC(C=C(C1CC1)N1)=C(CN)C1=O Chemical compound CC(C=C(C1CC1)N1)=C(CN)C1=O AWQYPAVOIYFYRY-UHFFFAOYSA-N 0.000 description 1
- JHWYUCIVJQZDEF-UHFFFAOYSA-N CCOC(C(CC(c(cc1)ccc1OC)=O)=O)=O Chemical compound CCOC(C(CC(c(cc1)ccc1OC)=O)=O)=O JHWYUCIVJQZDEF-UHFFFAOYSA-N 0.000 description 1
- VEXJCJWHJAUSQZ-UHFFFAOYSA-N CCOC(c1c(c(C)n[n]2C3CCCCC3)c2nc(C2CC2)c1)=O Chemical compound CCOC(c1c(c(C)n[n]2C3CCCCC3)c2nc(C2CC2)c1)=O VEXJCJWHJAUSQZ-UHFFFAOYSA-N 0.000 description 1
- KQOHEMACLCRIKG-UHFFFAOYSA-N Cc1n[n](C2CC2)c2nc(C3CC3)cc(C(NCC3=C(C)C=C(C)NC3=O)=O)c12 Chemical compound Cc1n[n](C2CC2)c2nc(C3CC3)cc(C(NCC3=C(C)C=C(C)NC3=O)=O)c12 KQOHEMACLCRIKG-UHFFFAOYSA-N 0.000 description 1
- UOUDFDCOISMLQB-UHFFFAOYSA-N Cc1n[n](C2CC2)c2nc(C3CC3)cc(C(O)=O)c12 Chemical compound Cc1n[n](C2CC2)c2nc(C3CC3)cc(C(O)=O)c12 UOUDFDCOISMLQB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- This invention relates to substituted azaindazoles which inhibit EZH2 and thus are useful for inhibiting the proliferation of and/or inducing apoptosis in cancer cells.
- Epigenetic modifications play an important role in the regulation of many cellular processes including cell proliferation, differentiation, and cell survival.
- Global epigenetic modifications are common in cancer, and include global changes in DNA and/or histone methylation, dysregulation of non-coding RNAs and nucleosome remodeling leading to aberrant activation or inactivation of oncogenes, tumor suppressors and signaling pathways.
- these epigenetic changes can be reversed through selective inhibition of the enzymes involved.
- Several methylases involved in histone or DNA methylation are known to be dysregulated in cancer. Thus, selective inhibitors of particular methylases will be useful in the treatment of proliferative diseases such as cancer.
- EZH2 (enhancer of zeste homolog 2; human EZH2 gene: Cardoso, C, et al;
- Histone H3 is one of the five main histone proteins involved in the structure of chromatin in eukaryotic cells.
- Histones are involved with the structure of the nucleosomes, a 'beads on a string' structure. Histone proteins are highly post-translationally modified however Histone H3 is the most extensively modified of the five histones. The term "Histone H3" alone is purposely ambiguous in that it does not distinguish between sequence variants or modification state. Histone H3 is an important protein in the emerging field of epigenetics, where its sequence variants and variable modification states are thought to play a role in the dynamic and long term regulation of genes.
- Increased EZH2 expression has been observed in numerous solid tumors including those of the prostate, breast, skin, bladder, liver, pancreas, head and neck and correlates with cancer aggressiveness, metastasis and poor outcome (Varambally et al, 2002; Kleer et al, 2003; Breuer et al, 2004; Bachmann et al, 2005; Weikert et al, 2005; Sudo et al, 2005; Bachmann et al., 2006).
- this invention relates to compounds of formula (I)
- X and Z are selected independently from the group consisting of hydrogen, (Ci- C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, unsubstituted or substituted (C3-Cg)cycloalkyl, unsubstituted or substituted (C 3 -Cg)cycloalkyl-(Ci-Cg)alkyl or -(C 2 -C 8 )alkenyl, unsubstituted or substituted (C 5 -C 8 )cycloalkenyl, unsubstituted or substituted (C 5 - Cg)cycloalkenyl-(Ci-Cg)alkyl or -(C 2 -Cg)alkenyl, (C 6 -Cio)bicycloalkyl, unsubstituted or substituted heterocycloalkyl, unsubstituted or substituted heterocycloalkyl-(Ci
- Y is H or halo
- R 1 is (Ci-Cg)alkyl, (C 2 -Cg)alkenyl, (C 2 -Cg)alkynyl, unsubstituted or substituted (C 3 -C 8 )cycloalkyl, unsubstituted or substituted (C 3 -C 8 )cycloalkyl-(Ci-Cg)alkyl or - (C 2 -C 8 )alkenyl, unsubstituted or substituted (Cs-Cg)cycloalkenyl, unsubstituted or substituted (C 5 -Cg)cycloalkenyl-(Ci-Cg)alkyl or -(C 2 -Cg)alkenyl, unsubstituted or substituted (C 6 -Cio)bicycloalkyl, unsubstituted or substituted heterocycloalkyl or - (C 2 -C 8 )alkenyl, unsubstituted or substitute
- R 3 is hydrogen, (Ci-C 8 )alkyl, cyano, trifluoromethyl, -NR a R b , or halo;
- R 6 is selected from the group consisting of hydrogen, halo, (Ci-Cg)alkyl,
- cycloalkenyl, bicycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by 1 , 2 or 3 groups independently selected from the group consisting of
- any aryl or heteroaryl moiety of said aryl, heteroaryl, aryl(Ci-C 4 )alkyl, or heteroaryl(Ci-C 4 )alkyl is optionally substituted by 1, 2 or 3 groups independently selected from the group consisting of halo,
- R a and R b are each independently hydrogen, (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl,
- heterocycloalkyl aryl, heteroaryl, wherein said (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 - Cs)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C 4 )alkoxy, amino, (Ci-C 4 )alkylamino, ((Ci-C 4 )alkyl)((Ci-C 4 )alkyl)amino, - C0 2 H, -C0 2 (Ci-C 4 )alkyl, -CONH 2 ,-CONH(Ci-C 4 )alkyl, - CON((Ci-C 4 )alkyl), -S0 2
- R a and R b taken together with the nitrogen to which they are attached represent a 5-8 membered saturated or unsaturated ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C 4 )alkyl, (Ci-
- C 4 )haloalkyl amino, (Ci-C 4 )alkylamino, ((Ci-C 4 )alkyl)((Ci-C 4 )alkyl)amino, hydroxyl, oxo, (Ci-C 4 )alkoxy, and (Ci-C 4 )alkoxy(Ci-C 4 )alkyl, wherein said ring is optionally fused to a (C 3 -Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- R a and R b taken together with the nitrogen to which they are attached represent a 6- to 10-membered bridged bicyclic ring system optionally fused to a (C 3 -Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- each R c is independently (Ci-C 4 )alkylamino, -NR a S0 2 R b , -SOR a , -S0 2 R a , - NR a C(0)OR a , -NR a R b , or -C0 2 R a ;
- this invention relates to a method of treating cancer.
- compositions comprising compounds of formula (I) and pharmaceutically acceptable excipients.
- a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof in the preparation of a medicament for use in the treatment of a disorder mediated by inhibiting EZH2, such as inducing apoptosis in cancer cells.
- substituted means substituted by one or more defined groups.
- groups may be selected from a number of alternative groups the selected groups may be the same or different.
- an "effective amount” means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal or human that is being sought, for instance, by a researcher or clinician. Furthermore, the term
- terapéuticaally effective amount means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder.
- the term also includes within its scope amounts effective to enhance normal physiological function.
- alkyl refers to a straight- or branched-chain hydrocarbon radical having the specified number of carbon atoms, so for example, as used herein, the terms “Ci-Csalkyl” refers to an alkyl group having at least 1 and up to 8 carbon atoms respectively.
- Examples of such branched or straight-chained alkyl groups useful in the present invention include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, isobutyl, n-butyl, t-butyl, n-pentyl, isopentyl, n-hexyl, n-heptyl, and n-octyl and branched analogs of the latter 5 normal alkanes.
- alkoxy as used herein means -0(Ci_Cgalkyl) including -OCH3, -
- alkylthio as used herein is meant -S(Ci_C 8 alkyl) including -SCH3, -
- acyloxy means -OC(0)Ci_Cgalkyl and the like per the definition of alkyl above.
- Acylamino means-N(H)C(0)Ci_C 8 alkyl and the like per the definition of alkyl above.
- Aryloxy means -O(aryl), -0(substituted aryl), -O(heteroaryl) or -0(substituted heteroaryl).
- Arylamino means -NH(aryl), -NH(substituted aryl), -NH(heteroaryl) or - NH(substituted heteroaryl), and the like.
- alkenyl refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms and at least 1 and up to 5 carbon-carbon double bonds. Examples include ethenyl (or ethenylene) and propenyl (or propenylene).
- alkynyl refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms and at least 1 and up to 5 carbon-carbon triple bonds. Examples include ethynyl (or ethynylene) and propynyl (or propynylene).
- Haloalkyl refers to an alkyl group group that is substituted with one or more halo substituents, suitably from 1 to 6 substituents. Haloalkyl includes trifluoromethyl.
- cycloalkyl refers to a non-aromatic, saturated, cyclic
- Cs-Cscycloalkyl refers to a non-aromatic cyclic hydrocarbon ring having from three to eight carbon atoms.
- Exemplary "C3-C 8 cycloalkyl” groups useful in the present invention include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- Cs-Cscycloalkenyl refers to a non-aromatic monocyclic carboxycyclic ring having the specified number of carbon atoms and up to 3 carbon-carbon double bonds.
- Cycloalkenyl includes by way of example cyclopentenyl and cyclohexenyl.
- Cs-Csheterocycloalkyl means a non-aromatic heterocyclic ring containing the specified number of ring atoms being, saturated or having one or more degrees of unsaturation and containing one or more heteroatom substitutions
- Such a ring may be optionally fused to one or more other "heterocyclic" ring(s) or cycloalkyl ring(s). Examples are given herein below.
- Aryl refers to optionally substituted monocyclic or polycarbocyclic unfused or fused groups having 6 to 14 carbon atoms and having at least one aromatic ring that complies with Huckel's Rule.
- aryl groups are phenyl, biphenyl, naphthyl, anthracenyl, phenanthrenyl, and the like, as further illustrated below.
- Heteroaryl means an optionally substituted aromatic monocyclic ring or polycarbocyclic fused ring system wherein at least one ring complies with Huckel's Rule, has the specified number of ring atoms, and that ring contains at least one heteratom independently selected from N, O and S. Examples of “heteroaryl” groups are given herein below.
- event(s) may or may not occur, and includes both event(s), which occur, and events that do not occur.
- pharmaceutically-acceptable salts refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically-acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
- X and Z are selected from the group consisting of (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NR a R b , and -OR a ;
- Y is H or F
- R 1 is selected from the group consisting of (Ci-Cg)alkyl, (C 3 -C 8 )cycloalkyl, heterocycloalkyl, aryl, and heteroaryl;
- R 3 is selected from the group consisting of hydrogen, (Ci-Cg)alkyl, cyano, trifluoromethyl, -NR a R b , and halo;
- R 6 is selected from the group consisting of hydrogen, halo, cyano, trifluoromethyl, amino, (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl;, aryl, heteroaryl, acylamino; (C 2 -Cg)alkynyl, arylalkynyl, heteroarylalkynyl; -S0 2 R a ; -S0 2 NR a R b , and -NR a S0 2 R b ;
- any (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, (C 2 -Cg)alkynyl, arylalkynyl, heteroarylalkynyl group is optionally substituted by 1 , 2 or 3 groups independently selected from -0(Ci-C 6 )alkyl(R c )i_ 2 , -S(Ci-C 6 )alkyl(R c )i_ 2 , -(Ci-C 6 )alkyl(R c )i_ 2 ,
- each R c is independently (Ci-C 4 )alkylamino, -NR a S0 2 R b , -SOR a , -S0 2 R a , - NR a C(0)OR a , -NR a R b , or -C0 2 R a ;
- R a and R b are each independently hydrogen, (Ci-Cg)alkyl, (C 2 -C8)alkenyl, (C 2 -Cg)alkynyl, (C3-Cg)cycloalkyl, (C5-Cg)cycloalkenyl, (C6-Cio)bicycloalkyl,
- heterocycloalkyl aryl, heteroaryl, wherein said (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 - Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1 , 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C0 2 H, -C0 2 (Ci-C 4 )alkyl, -CONH 2 , -CONH(Ci-C 4 )alkyl, -
- An aryl or heteroaryl group in this particular subgroup A is selected independently from the group consisting of furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, oxadiazole, thiadiazole, triazole, tetrazole, benzofuran, benzothiophene, benzoxazole, benzothiazole, phenyl, pyridine, pyridazine, pyrimidine, pyrazine, triazine, tetrazine, quinoline, cinnoline, quinazoline, quinoxaline, and na hthyridine or another aryl or heteroaryl group as follows:
- A is O, NH, or S; B is CH or N, and C is hydrogen or Ci-Cg alkyl; or
- D is N or C optionally substituted by hydrogen or Ci-C 8 alkyl
- E is NH or CH 2 ;
- F is O or CO; and
- G is NH or CH 2 ; or
- J is O, S or CO;
- Q is CH or N
- M is CH or N
- L/(5) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, -COR a , - C0 2 R a , -CONR a R b , -CONR a NR a R b , -S0 2 R a , -S0 2 NR a R b , -NR a R b , -NR a C(0)R b - NR a S0 2 R b , -NR a S0 2 NR a R b , -NR a S0 2 NR a R b , -NR a NR a R b , -NR a NR a C(0)R b , -NR a NR a C(0)NR a R b , -OR a , wherein any (Ci-Cg)alkyl, (C 3 -Cg
- L/ 6 is NH or CH 2 ;
- M/(7) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, heterocycloalkyl, -COR a , -C0 2 R a , -CONR a R b , -CONR a NR a R b S0 2 R a , - S0 2 NR a R b , -NR a R b , -NR a C(0)R b ,-NR a S0 2 R b , -NR a S0 2 R b , -NR a S0 2 NR a R b , -NR a S0 2 NR a R b , -NR a NR a R b , -NR a NR a C(0)R b , -NR a NR a C(0)NR b , -NR a NR a C(0)NR a R
- any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl group is optionally substituted by 1 , 2 or 3 groups independently selected from
- P is CH 2 , NH, O, or S;
- Q/(8) is CH or N; and
- n is 0-2; or
- S/(9) and T(9) is C, or S/(9) is C and T(9) is N, or S/(9) is N and T/(9) is C;
- R is hydrogen, amino, methyl, trifluoromethyl, halo;
- U is hydrogen, halo, amino, cyano, nitro, trifluoromethyl, (Ci-Cg)alkyl, (C 3 - C 8 )cycloalkyl, -COR a , -C0 2 R a , -CONR a R b , -S0 2 R a , -S0 2 NR a R b , -NR a R b , - NR a C(0)R b ,-NR a S0 2 R b , -NR a S0 2 NR b , -NR a S0 2 NR a R b , -NR a S0 2 NR a R b , -NR a S0 2 NR a R b , -NR a NR a R b , -NR a NR a C(0)R b ,
- any (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, group is optionally
- X and Z are selected independently from the group consisting of (Ci-Cg)alkyl, (C 3 - Cg)cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NR a R b , and -OR a ;
- Y is H
- R 1 is (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, or heterocycloalkyl;
- R 3 is hydrogen, (Ci-Cg)alkyl or halo
- R 6 is hydrogen, halo, cyano, trifluoromethyl, amino, (Ci-Cg)alkyl, (C 3 - Cg)cycloalkyl;, aryl, heteroaryl, acylamino; (C 2 -Cg)alkynyl, arylalkynyl, heteroarylalkynyl; -S0 2 R a ; -S0 2 NR a R b , or -NR a S0 2 R b ;
- any (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, (C 2 -Cg)alkynyl, arylalkynyl, heteroarylalkynyl group is optionally substituted by 1 , 2 or 3 groups independently selected from halo, (Ci-C 6 )alkyl, (C 3 -Cg)cycloalkyl, (Cs-Cg)cycloalkenyl,
- (Ci-C 6 )haloalkyl cyano, -COR a , -C0 2 R a , -CONR a R b , -SR a , -SOR a , -S0 2 R a , - S0 2 NR a R b , nitro, -NR a R b , -NR a C(0)R b , -NR a C(0)NR a R b , -NR a C(0)OR a , - NR a S0 2 R b , -NR a S0 2 NR a R b , -OR a , -OC(0)R a , -OC(0)NR a R b , heterocycloalkyl, aryl, heteroaryl, aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
- R a and R b are each independently hydrogen, (Ci-Cg)alkyl, (C 2 -Cg)alkenyl,
- heterocycloalkyl aryl, heteroaryl, wherein said (Ci-Cg)alkyl, (C 2 -C 8 )alkenyl, (C 2 - Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C0 2 H, -C0 2 (Ci-C 4 )alkyl, -CONH 2 ,-CONH(Ci-C 4 )alkyl, - CON((Ci-C 4 )alkyl)((Ci-C 4 )alkyl), -S0 2 (Ci-C 4 )al
- 5- 8 membered saturated or unsaturated ring optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C 4 )alkyl, (Ci- C 4 )haloalkyl, amino, (Ci-C 4 )alkylamino, ((Ci-C 4 )alkyl)((Ci-C 4 )alkyl)amino, hydroxyl, oxo, (Ci-C )alkoxy, and (Ci-C )alkoxy(Ci-C )alkyl, wherein said ring is optionally fused to a (C 3 -Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
- Aryl and heteroaryl in this definition are selected from the group consisting of furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, oxadiazole, thiadiazole, triazole, tetrazole, benzofuran,
- benzothiophene benzoxazole, benzothiazole, phenyl, pyridine, pyridazine, pyrimidine, pyrazine, triazine, tetrazine, quinoline, cinnoline, quinazoline, quinoxaline, and na hthyridine as or a compound of or another aryl or heteroaryl group as follows:
- A is O, NH, or S; B is CH or N, and C is hydrogen or Ci-Cg alkyl; or
- D is N or C optionally substituted by hydrogen or Ci-Cg alkyl
- E is NH or CH 2 ;
- F is O or CO; and
- G is NH or CH 2 ; or
- J is O, S or CO;
- Q is CH or N
- M is CH or N
- L/(5) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, -COR'
- any (Ci-Cg)alkyl, (C 3 -C 8 )cycloalkyl, group is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C 6 )alkyl,
- R a and R b are defined as above;
- L/ 6 is NH or CH 2 ;
- M/(7) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, heterocycloalkyl, -COR a , -C0 2 R a , -CONR a R b , -CONR a NR a R b S0 2 R a , - S0 2 NR a R b , -NR a R b , -NR a C(0)R b ,-NR a S0 2 R b , -NR a S0 2 R b , -NR a S0 2 NR a R b , -NR a S0 2 NR a R b , -NR a NR a R b , -NR a NR a C(0)R b , -NR a NR a C(0)NR b , -NR a NR a C(0)NR a R
- any (Ci-Cg)alkyl, (C 3 -Cg)cycloalkyl, heterocycloalkyl group is optionally substituted by 1 , 2 or 3 groups independently selected from
- P is CH 2 , NH, O, or S;
- Q/(8) is CH or N; and
- n is 0-2; or
- S/(9) and T(9) is C, or S/(9) is C and T(9) is N, or S/(9) is N and T/(9) is C;
- R is hydrogen, amino, methyl, trifluoromethyl, halo
- U is hydrogen, halo, amino, cyano, nitro, trifluoromethyl, (Ci-Cg)alkyl, (C 3 - C 8 )cycloalkyl, -COR a , -C0 2 R a , -CONR a R b , -S0 2 R a , -S0 2 NR a R b , -NR a R b , - NR a C(0)R b ,-NR a S0 2 R b , -NR a S0 2 NR b , -NR a S0 2 NR a R b , -NR a S0 2 NR a R b , -NR a NR a R b , -NR a NR a C(0)R b , , -OR a , 4-(lH- pyrazol-4-yl),
- any (Ci-Cg)alkyl, (C 3 -C8)cycloalkyl, group is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C 6 )alkyl,
- X is methyl, ethyl, n-propyl, isopropyl , cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, trifluoromethyl, tetrahydropyran, hydroxymethyl, methoxymethyl, or benzyl;
- Y is H
- Z is methyl, ethyl, n-propyl, isopropyl, trifluoromethyl, or benzyl;
- R 1 is isopropyl, tert-butyl, cyclobutyl, cyclopentyl, cyclohexyl, (1- methylethyl)cyclopropyl, 1 , 1 -dioxo-tetrahydrothiophene-3-yl, 1 -Me-piperidin-4-yl, tetrahydrofuran-3-yl, tetrahydropyran-4-yl, N,N-dimethyl-l-propanaminyl, benzyl, or 4- pyridyl;
- R 3 is H, methyl, or Br
- R 6 is methyl, bis(l,l-dimethylethyl), bis(l-methylethyl), cyclopropyl, propyl, dimethylamino, ethylamino, (2-hydroxyethyl)amino, 2-propen-l-ylamino, 1-piperazinyl,
- this invention also relates to the exemplified compounds.
- this invention also relates to the following compounds:
- co-administering and derivatives thereof as used herein is meant either simultaneous administration or any manner of separate sequential administration of one or more additional pharmaceutically active compounds, whether for treating cancer, the side effects of cancer or cancer therapy, or some other disease.
- the administration is not simultaneous, the compounds are administered in a close time proximity to each other.
- the compounds are administered in a close time proximity to each other.
- one compound may be administered topically and another compound may be administered orally.
- compounds according to Formula I may contain an acidic functional group, one acidic enough to form salts.
- Representative salts include pharmaceutically-acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a pharmaceutically- acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically-acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, triethylamine, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
- pharmaceutically-acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts
- carbonates and bicarbonates of a pharmaceutically- acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium
- compounds according to Formula (I) may contain a basic functional group and are therefore capable of forming pharmaceutically-acceptable acid addition salts by treatment with a suitable acid.
- suitable acids include pharmaceutically- acceptable inorganic acids and pharmaceutically-acceptable organic acids.
- Representative pharmaceutically-acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphate. !
- ethanesulfonate (esylate), 2-hydroxyethanesulfonate, benzenesulfonate (besylate), p- aminobenzenesulfonate, /?-toluenesulfonate (tosylate) and napthalene-2-sulfonate.
- the compounds of formula (I) may be prepared in crystalline or non-crystalline form, and, if crystalline, may optionally be solvated, e.g. as the hydrate.
- This invention includes within its scope stoichiometric solvates (e.g. hydrates) as well as compounds containing variable amounts of solvent (e.g. water).
- Certain of the compounds described herein may contain one or more chiral atoms, or may otherwise be capable of existing as two enantiomers.
- the compounds claimed below include mixtures of enantiomers as well as purified enantiomers or enantiomerically enriched mixtures.
- Also included within the scope of the invention are the individual isomers of the compounds represented by formula (I), or claimed below, as well as any wholly or partially equilibrated mixtures thereof.
- the present invention also covers the individual isomers of the claimed compounds as mixtures with isomers thereof in which one or more chiral centers are inverted. Where there are different isomeric forms they may be separated or resolved one from the other by conventional methods, or any given isomer may be obtained by conventional synthetic methods or by stereospecific or asymmetric syntheses.
- compositions which includes a compound of formula (I) and salts, solvates and the like, and one or more pharmaceutically acceptable carriers, diluents, or excipients.
- the compounds of formula (I) and salts, solvates, etc, are as described above.
- the carrier(s), diluent(s) or excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- a process for the preparation of a pharmaceutical formulation including admixing a compound of the formula (I), or salts, solvates etc, with one or more pharmaceutically acceptable carriers, diluents or excipients.
- prodrugs for compounds of the invention include: esters, carbonate esters, hemi-esters, phosphate esters, nitro esters, sulfate esters, sulfoxides, amides, carbamates, azo-compounds, phosphamides, glycosides, ethers, acetals and ketals.
- the compounds and compositions of the invention are used to treat cellular proliferation diseases. Disease states which can be treated by the methods and
- compositions provided herein include, but are not limited to, cancer (further discussed below), autoimmune disease, fungal disorders, arthritis, graft rejection, inflammatory bowel disease, proliferation induced after medical procedures, including, but not limited to, surgery, angioplasty, and the like. It is appreciated that in some cases the cells may not be in a hyper or hypo proliferation state (abnormal state) and still requires treatment. For example, during wound healing, the cells may be proliferating "normally", but
- the invention herein includes application to cells or individuals afflicted or impending affliction with any one of these disorders or states.
- compositions and methods provided herein are particularly deemed useful for the treatment of cancer including tumors such as prostate, breast, brain, skin, cervical carcinomas, testicular carcinomas, etc. They are particularly useful in treating metastatic or malignant tumors. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to tumor types such as astrocytic, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck,
- these compounds can be used to treat: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell,
- bronchiolar carcinoma bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma;
- nephroblastoma lymphoma, leukemia
- bladder and urethra squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver: hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma,
- angiosarcoma hepatocellular adenoma, hemangioma
- Biliary tract gall bladder carcinoma, ampullary carcinoma, cholangiocarcinoma
- Bone osteogenic sarcoma
- nerveous system skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans), meninges (meningioma, meningiosarcoma, gliomatosis), brain (astrocytoma, meduUoblastoma, glioma, ependymoma, germinoma (pinealoma), glioblasto
- cancer carcinoma
- Hematologic blood (myeloid leukemia (acute and chronic), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplasia syndrome), Hodgkin's disease, non-Hodgkin's lymphoma (malignant lymphoma); Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Karposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma.
- the term "cancerous cell” as provided herein includes a cell afflicted by any one or related of the above identified conditions.
- Combination therapies according to the invention comprise the administration of at least one compound of the invention and the use of at least one other treatment method.
- combination therapies according to the invention comprise the administration of at least one compound of the invention and surgical therapy.
- combination therapies according to the invention comprise the administration of at least one compound of the invention and radiotherapy.
- combination therapies according to the invention comprise the administration of at least one compound of the invention and at least one supportive care agent (e.g., at least one anti-emetic agent).
- combination therapies according to the present invention comprise the administration of at least one compound of the invention and at least one other chemotherapeutic agent.
- the invention comprises the administration of at least one compound of the invention and at least one anti-neoplastic agent.
- the invention comprises a therapeutic regimen where the EZH2 inhibitors of this disclosure are not in and of themselves active or significantly active, but when combined with another therapy, which may or may not be active as a standalone therapy, the combination provides a useful therapeutic outcome.
- co-administering and derivatives thereof as used herein is meant either simultaneous administration or any manner of separate sequential administration of an EZH2 inhibiting compound, as described herein, and a further active ingredient or ingredients, known to be useful in the treatment of cancer, including chemotherapy and radiation treatment.
- further active ingredient or ingredients includes any compound or therapeutic agent known to or that demonstrates advantageous properties when administered to a patient in need of treatment for cancer.
- the compounds are administered in a close time proximity to each other.
- one compound may be administered topically and another compound may be administered orally.
- any anti-neoplastic agent that has activity versus a susceptible tumor being treated may be co-administered in the treatment of specified cancers in the present invention.
- examples of such agents can be found in Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6 th edition (February 15, 2001),
- anti-neoplastic agents useful in the present invention include, but are not limited to, anti-microtubule agents such as diterpenoids and vinca alkaloids; platinum coordination complexes; alkylating agents such as nitrogen mustards, oxazaphosphorines, alkylsulfonates, nitrosoureas, and triazenes; antibiotic agents such as anthracyclins, actinomycins and bleomycins; topoisomerase II inhibitors such as epipodophyllotoxins; antimetabolites such as purine and pyrimidine analogues and anti-folate compounds; topoisomerase I inhibitors such as camptothecins; hormones and hormonal analogues; DNA methyltransferase inhibitors such as azacitidine and decitabine; signal transduction pathway
- any chemotherapeutic agent that has activity against a susceptible neoplasm being treated may be utilized in combination with the compounds the invention, provided that the particular agent is clinically compatible with therapy employing a compound of the invention.
- Typical anti-neoplastic agents useful in the present invention include, but are not limited to: alkylating agents, anti-metabolites, antitumor antibiotics, antimitotic agents, nucleoside analogues, topoisomerase I and II inhibitors, hormones and hormonal analogues; retinoids, histone deacetylase inhibitors; signal transduction pathway inhibitors including inhibitors of cell growth or growth factor function, angiogenesis inhibitors, and serine/threonine or other kinase inhibitors; cyclin dependent kinase inhibitors; antisense therapies and immunotherapeutic agents, including monoclonals, vaccines or other biological agents.
- Nucleoside analogues are those compounds which are converted to
- HDAC Histone deacetylase
- Signal transduction pathway inhibitors are those inhibitors which block or inhibit a chemical process which evokes an intracellular change. As used herein this change is cell proliferation or differentiation or survival.
- Signal transduction pathway inhibitors useful in the present invention include, but are not limited to, inhibitors of receptor tyrosine kinases, non-receptor tyrosine kinases, SH2/SH3 domain blockers, serine/threonine kinases, phosphatidyl inositol-3-OH kinases, myoinositol signaling, and Ras oncogenes. Signal transduction pathway inhibitors may be employed in combination with the compounds of the invention in the compositions and methods described above.
- Receptor kinase angiogenesis inhibitors may also find use in the present invention.
- Inhibitors of angiogenesis related to VEGFR and TIE-2 are discussed above in regard to signal transduction inhibitors (both are receptor tyrosine kinases).
- Other inhibitors may be used in combination with the compounds of the invention.
- anti-VEGF antibodies which do not recognize VEGFR (the receptor tyrosine kinase), but bind to the ligand; small molecule inhibitors of integrin (alpha v beta 3 ) that inhibit angiogenesis;
- endostatin and angiostatin may also prove useful in combination with the compounds of the invention.
- VEGFR antibody is bevacizumab
- Trastuzumab (Herceptin ® ) is an example of an anti-erbB2 antibody inhibitor of growth factor function.
- One example of an anti-erbBl antibody inhibitor of growth factor function is cetuximab (ErbituxTM, C225).
- Bevacizumab (Avastin ® ) is an example of a monoclonal antibody directed against VEGFR.
- small molecule inhibitors of epidermal growth factor receptors include but are not limited to lapatinib (TykerbTM) and erlotinib (TARCEVA ® ).
- Imatinib mesylate is one example of a PDGFR inhibitor.
- VEGFR inhibitors include pazopanib, ZD6474, AZD2171, PTK787, sunitinib and sorafenib.
- Anti-microtubule or anti-mitotic agents are phase specific agents active against the microtubules of tumor cells during M or the mitosis phase of the cell cycle.
- anti-microtubule agents include, but are not limited to, diterpenoids and vinca alkaloids.
- Diterpenoids which are derived from natural sources, are phase specific anti - cancer agents that operate at the G 2 /M phases of the cell cycle. It is believed that the diterpenoids stabilize the ⁇ -tubulin subunit of the microtubules, by binding with this protein. Disassembly of the protein appears then to be inhibited with mitosis being arrested and cell death following. Examples of diterpenoids include, but are not limited to, paclitaxel and its analog docetaxel.
- Paclitaxel 5 ,20-epoxy-l,2a,4,7 ,10 ,13a-hexa-hydroxytax-l l-en-9-one 4,10- diacetate 2-benzoate 13-ester with (2R,3S)-N-benzoyl-3-phenylisoserine; is a natural diterpene product isolated from the Pacific yew tree Taxus brevifolia and is commercially available as an injectable solution TAXOL ® . It is a member of the taxane family of terpenes. It was first isolated in 1971 by Wani et al. J. Am. Chem, Soc, 93:2325.
- Paclitaxel has been approved for clinical use in the treatment of refractory ovarian cancer in the United States (Markman et al., Yale Journal of Biology and Medicine, 64:583, 1991; McGuire et al, Ann. Intern, Med., 111 :273,1989) and for the treatment of breast cancer (Holmes et al., J. Nat. Cancer Inst., 83: 1797,1991.) It is a potential candidate for treatment of neoplasms in the skin (Einzig et. al., Proc. Am. Soc. Clin. Oncol., 20:46) and head and neck carcinomas (Forastire et. al, Sem. Oncol., 20:56, 1990).
- the compound also shows potential for the treatment of polycystic kidney disease (Woo et. al, Nature, 368:750. 1994), lung cancer and malaria.
- Treatment of patients with paclitaxel results in bone marrow suppression (multiple cell lineages, Ignoff, R.J. et. al, Cancer Chemotherapy Pocket Guidei 1998) related to the duration of dosing above a threshold concentration (50nM) (Kearns, CM. et. al, Seminars in Oncology, 3(6) p.16-23, 1995).
- Docetaxel (2R,3S)- N-carboxy-3-phenylisoserine,N-tert-butyl ester, 13-ester with 5 -20-epoxy-l,2a,4,7 ,10 ,13a-hexahydroxytax-l l-en-9-one 4-acetate 2-benzoate, trihydrate; is commercially available as an injectable solution as TAXOTERE ® .
- Docetaxel is indicated for the treatment of breast cancer.
- Docetaxel is a semisynthetic derivative of paclitaxel q.v., prepared using a natural precursor, 10-deacetyl-baccatin III, extracted from the needle of the European Yew tree. The dose limiting toxicity of docetaxel is neutropenia.
- Vinca alkaloids are phase specific anti-neoplastic agents derived from the periwinkle plant. Vinca alkaloids act at the M phase (mitosis) of the cell cycle by binding specifically to tubulin. Consequently, the bound tubulin molecule is unable to polymerize into microtubules. Mitosis is believed to be arrested in metaphase with cell death following. Examples of vinca alkaloids include, but are not limited to, vinblastine, vincristine, and vinorelbine.
- Vinblastine vincaleukoblastine sulfate
- VELBAN ® an injectable solution
- Myelosuppression is the dose limiting side effect of vinblastine.
- Vincristine vincaleukoblastine, 22-oxo-, sulfate
- ONCOVIN ® an injectable solution.
- Vincristine is indicated for the treatment of acute leukemias and has also found use in treatment regimens for Hodgkin's and non-Hodgkin's malignant lymphomas.
- Alopecia and neurologic effects are the most common side effect of vincristine and to a lesser extent myelosupression and gastrointestinal mucositis effects occur.
- Vinorelbine 3',4'-didehydro -4'-deoxy-C'-norvincaleukoblastine [R-(R*,R*)-2,3- dihydroxybutanedioate (l :2)(salt)], commercially available as an injectable solution of vinorelbine tartrate (NAVELBINE ® ), is a semisynthetic vinca alkaloid.
- Vinorelbine is indicated as a single agent or in combination with other chemotherapeutic agents, such as cisplatin, in the treatment of various solid tumors, particularly non-small cell lung, advanced breast, and hormone refractory prostate cancers. Myelosuppression is the most common dose limiting side effect of vinorelbine.
- Platinum coordination complexes are non-phase specific anti-cancer agents, which are interactive with DNA.
- the platinum complexes enter tumor cells, undergo, aquation and form intra- and interstrand crosslinks with DNA causing adverse biological effects to the tumor.
- Examples of platinum coordination complexes include, but are not limited to, cisplatin and carboplatin.
- Cisplatin cis-diamminedichloroplatinum, is commercially available as
- Cisplatin is primarily indicated in the treatment of metastatic testicular and ovarian cancer and advanced bladder cancer.
- the primary dose limiting side effects of cisplatin are nephrotoxicity, which may be controlled by hydration and diuresis, and ototoxicity.
- Carboplatin, platinum, diammine [l,l-cyclobutane-dicarboxylate(2-)-0,0'], is commercially available as PARAPLATIN ® as an injectable solution.
- Carboplatin is primarily indicated in the first and second line treatment of advanced ovarian carcinoma. Bone marrow suppression is the dose limiting toxicity of carboplatin.
- Alkylating agents are non-phase anti-cancer specific agents and strong
- alkylating agents form covalent linkages, by alkylation, to DNA through nucleophilic moieties of the DNA molecule such as phosphate, amino, sulfhydryl, hydroxyl, carboxyl, and imidazole groups. Such alkylation disrupts nucleic acid function leading to cell death.
- alkylating agents include, but are not limited to, nitrogen mustards such as cyclophosphamide, melphalan, and chlorambucil; alkyl sulfonates such as busulfan; nitrosoureas such as carmustine; and triazenes such as dacarbazine.
- Cyclophosphamide 2-[bis(2-chloroethyl)amino]tetrahydro-2H-l,3,2- oxazaphosphorine 2-oxide monohydrate, is commercially available as an injectable solution or tablets as CYTOXAN ® . Cyclophosphamide is indicated as a single agent or in combination with other chemotherapeutic agents, in the treatment of malignant lymphomas, multiple myeloma, and leukemias. Alopecia, nausea, vomiting and leukopenia are the most common dose limiting side effects of cyclophosphamide.
- Melphalan 4-[bis(2-chloroethyl)amino]-L-phenylalanine, is commercially available as an injectable solution or tablets as ALKERAN ® .
- Melphalan is indicated for the palliative treatment of multiple myeloma and non-resectable epithelial carcinoma of the ovary. Bone marrow suppression is the most common dose limiting side effect of melphalan.
- Chlorambucil 4-[bis(2-chloroethyl)amino]benzenebutanoic acid, is commercially available as LEUKERAN ® tablets. Chlorambucil is indicated for the palliative treatment of chronic lymphatic leukemia, and malignant lymphomas such as lymphosarcoma, giant follicular lymphoma, and Hodgkin's disease. Bone marrow suppression is the most common dose limiting side effect of chlorambucil.
- Busulfan 1 ,4-butanediol dimethanesulfonate, is commercially available as MYLERAN ® TABLETS. Busulfan is indicated for the palliative treatment of chronic myelogenous leukemia. Bone marrow suppression is the most common dose limiting side effects of busulfan. Carmustine, l,3-[bis(2-chloroethyl)-l-nitrosourea, is commercially available as single vials of lyophilized material as BiCNU ® .
- Carmustine is indicated for the palliative treatment as a single agent or in combination with other agents for brain tumors, multiple myeloma, Hodgkin's disease, and non-Hodgkin's lymphomas. Delayed myelosuppression is the most common dose limiting side effects of carmustine.
- DTIC-Dome ® Commercially available as single vials of material as DTIC-Dome ® .
- dacarbazine is indicated for the treatment of metastatic malignant melanoma and in combination with other agents for the second line treatment of Hodgkin's Disease. Nausea, vomiting, and anorexia are the most common dose limiting side effects of dacarbazine.
- Antibiotic anti-neoplastics are non-phase specific agents, which bind or intercalate with DNA. Typically, such action results in stable DNA complexes or strand breakage, which disrupts ordinary function of the nucleic acids leading to cell death.
- antibiotic anti-neoplastic agents include, but are not limited to, actinomycins such as dactinomycin, anthrocyclins such as daunorubicin and doxorubicin; and bleomycins.
- Dactinomycin also know as Actinomycin D, is commercially available in injectable form as COSMEGEN ® . Dactinomycin is indicated for the treatment of Wilm's tumor and rhabdomyosarcoma. Nausea, vomiting, and anorexia are the most common dose limiting side effects of dactinomycin.
- Daunorubicin (8S-cis-)-8-acetyl-10-[(3-amino-2,3,6-trideoxy-a-L-lyxo- hexopyranosyl)oxy]-7,8,9, 10-tetrahydro-6,8, 11 -trihydroxy- 1 -methoxy-5, 12
- naphthacenedione hydrochloride is commercially available as a liposomal injectable form as DAUNOXOME ® or as an injectable as CERUBIDINE ® .
- Daunorubicin is indicated for remission induction in the treatment of acute nonlymphocytic leukemia and advanced HIV associated Kaposi's sarcoma. Myelosuppression is the most common dose limiting side effect of daunorubicin.
- Doxorubicin (8S, 10S)-10-[(3-amino-2,3,6-trideoxy-a-L-lyxo- hexopyranosyl)oxy]-8-glycoloyl, 7,8,9, 10-tetrahydro-6,8, 11 -trihydroxy- 1 -methoxy-5, 12 naphthacenedione hydrochloride, is commercially available as an injectable form as RUBEX ® or ADRIAMYCIN RDF ® .
- Doxorubicin is primarily indicated for the treatment of acute lymphoblastic leukemia and acute myeloblastic leukemia, but is also a useful component in the treatment of some solid tumors and lymphomas. Myelosuppression is the most common dose limiting side effect of doxorubicin.
- Bleomycin a mixture of cytotoxic glycopeptide antibiotics isolated from a strain of Streptomyces verticillus, is commercially available as BLENOXANE ® .
- Bleomycin is indicated as a palliative treatment, as a single agent or in combination with other agents, of squamous cell carcinoma, lymphomas, and testicular carcinomas. Pulmonary and cutaneous toxicities are the most common dose limiting side effects of bleomycin.
- Topoisomerase II inhibitors include, but are not limited to, epipodophyllotoxins.
- Epipodophyllotoxins are phase specific anti-neoplastic agents derived from the mandrake plant. Epipodophyllotoxins typically affect cells in the S and G 2 phases of the cell cycle by forming a ternary complex with topoisomerase II and DNA causing DNA strand breaks. The strand breaks accumulate and cell death follows.
- Examples of epipodophyllotoxins include, but are not limited to, etoposide and teniposide.
- Etoposide, 4'-demethyl-epipodophyllotoxin 9[4,6-0-(R )-ethylidene- -D- glucopyranoside] is commercially available as an injectable solution or capsules as
- VePESID ® and is commonly known as VP-16.
- Etoposide is indicated as a single agent or in combination with other chemotherapy agents in the treatment of testicular and non- small cell lung cancers. Myelosuppression is the most common side effect of etoposide. The incidence of leucopenia tends to be more severe than thrombocytopenia.
- Teniposide 4'-demethyl-epipodophyllotoxin 9[4,6-0-(R )-thenylidene- -D- glucopyranoside], is commercially available as an injectable solution as VUMON ® and is commonly known as VM-26. Teniposide is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia in children.
- Teniposide can induce both leucopenia and thrombocytopenia.
- Antimetabolite neoplastic agents are phase specific anti-neoplastic agents that act at S phase (DNA synthesis) of the cell cycle by inhibiting DNA synthesis or by inhibiting purine or pyrimidine base synthesis and thereby limiting DNA synthesis. Consequently, S phase does not proceed and cell death follows.
- Examples of antimetabolite anti-neoplastic agents include, but are not limited to, fluorouracil, methotrexate, cytarabine,
- 5-fluorouracil 5-fluoro-2,4- (1H,3H) pyrimidinedione
- fluorouracil is commercially available as fluorouracil.
- Administration of 5-fluorouracil leads to inhibition of thymidylate synthesis and is also incorporated into both RNA and DNA. The result typically is cell death.
- 5-fluorouracil is indicated as a single agent or in combination with other chemotherapy agents in the treatment of carcinomas of the breast, colon, rectum, stomach and pancreas. Myelosuppression and mucositis are dose limiting side effects of 5- fluorouracil.
- Other fluoropyrimidine analogs include 5-fluoro deoxyuridine (floxuridine) and 5-f uorodeoxyuridine monophosphate.
- Cytarabine 4-amino-l-P-D-arabinofuranosyl-2 (lH)-pyrimidinone, is
- CYTOSAR-U commercially available as CYTOSAR-U ® and is commonly known as Ara-C. It is believed that cytarabine exhibits cell phase specificity at S-phase by inhibiting DNA chain elongation by terminal incorporation of cytarabine into the growing DNA chain.
- Cytarabine is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia.
- Other cytidine analogs include 5-azacytidine and 2', 2 '-difluorodeoxy cytidine (gemcitabine). Cytarabine induces leucopenia, thrombocytopenia, and mucositis.
- Mercaptopurine l,7-dihydro-6H-purine-6-thione monohydrate
- PURINETHOL ® is commercially available as PURINETHOL ® .
- Mercaptopurine exhibits cell phase specificity at S-phase by inhibiting DNA synthesis by an as of yet unspecified mechanism.
- Mercaptopurine is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia. Myelosuppression and gastrointestinal mucositis are expected side effects of mercaptopurine at high doses.
- a useful mercaptopurine analog is azathioprine.
- Thioguanine 2-amino-l,7-dihydro-6H-purine-6-thione, is commercially available as TABLOID ® .
- Thioguanine exhibits cell phase specificity at S-phase by inhibiting DNA synthesis by an as of yet unspecified mechanism.
- Thioguanine is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia.
- Myelosuppression including leucopenia, thrombocytopenia, and anemia, is the most common dose limiting side effect of thioguanine administration. However, gastrointestinal side effects occur and can be dose limiting.
- Other purine analogs include pentostatin, erythrohydroxynonyladenine, fludarabine phosphate, and cladribine.
- Gemcitabine 2'-deoxy-2', 2'-difluorocytidine monohydrochloride ( ⁇ -isomer), is commercially available as GEMZAR ® .
- Gemcitabine exhibits cell phase specificity at S- phase and by blocking progression of cells through the Gl/S boundary.
- Gemcitabine is indicated in combination with cisplatin in the treatment of locally advanced non-small cell lung cancer and alone in the treatment of locally advanced pancreatic cancer.
- Myelosuppression including leucopenia, thrombocytopenia, and anemia, is the most common dose limiting side effect of gemcitabine administration.
- Methotrexate N-[4[[(2,4-diamino-6-pteridinyl) methyljmethylamino] benzoyl]-L- glutamic acid, is commercially available as methotrexate sodium. Methotrexate exhibits cell phase effects specifically at S-phase by inhibiting DNA synthesis, repair and/or replication through the inhibition of dyhydrofolic acid reductase which is required for synthesis of purine nucleotides and thymidylate. Methotrexate is indicated as a single agent or in combination with other chemotherapy agents in the treatment of
- choriocarcinoma meningeal leukemia, non-Hodgkin's lymphoma, and carcinomas of the breast, head, neck, ovary and bladder.
- Myelosuppression leucopenia, thrombocytopenia, and anemia
- mucositis are expected side effect of methotrexate administration.
- Camptothecins including, camptothecin and camptothecin derivatives are available or under development as Topoisomerase I inhibitors. Camptothecins cytotoxic activity is believed to be related to its Topoisomerase I inhibitory activity. Examples of camptothecins include, but are not limited to irinotecan, topotecan, and the various optical forms of 7-(4-methylpiperazino-methylene)-10,l l-ethylenedioxy-20-camptothecin described below.
- Irinotecan is a derivative of camptothecin which binds, along with its active metabolite SN-38, to the topoisomerase I - DNA complex. It is believed that cytotoxicity occurs as a result of irreparable double strand breaks caused by interaction of the topoisomerase I : DNA : irintecan or SN-38 ternary complex with replication enzymes. Irinotecan is indicated for treatment of metastatic cancer of the colon or rectum.
- the dose limiting side effects of irinotecan HC1 are myelosuppression, including neutropenia, and GI effects, including diarrhea.
- Topotecan HC1 (S)- 10-[(dimethylamino)methyl]-4-ethyl-4,9-dihydroxy- 1 H- pyrano[3 ' ,4 ' ,6,7]indolizino[ 1 ,2-b]quinoline-3 , 14-(4H, 12H)-dione monohydrochloride, is commercially available as the injectable solution HYCAMTIN ® .
- Topotecan is a derivative of camptothecin which binds to the topoisomerase I - DNA complex and prevents religation of singles strand breaks caused by Topoisomerase I in response to torsional strain of the DNA molecule.
- Topotecan is indicated for second line treatment of metastatic carcinoma of the ovary and small cell lung cancer.
- the dose limiting side effect of topotecan HC1 is myelosuppression, primarily neutropenia.
- compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose.
- a unit may contain, for example, 0.5 mg to 1 g, preferably 1 mg to 700 mg, more preferably 5 mg to 100 mg of a compound of the formula (I), depending on the condition being treated, the route of administration and the age, weight and condition of the patient, or pharmaceutical compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose.
- Preferred unit dosage compositions are those containing a daily dose or sub-dose, as herein above recited, or an appropriate fraction thereof, of an active ingredient.
- such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
- compositions may be adapted for administration by any combination of ingredients.
- compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association a compound of formal (I) with the carrier(s) or excipient(s).
- compositions adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths.
- Glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation.
- a disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
- Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate,
- Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
- Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like. Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets.
- a powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate.
- the powder mixture can be granulated by tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil. The lubricated mixture is then compressed into tablets.
- the compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps.
- a clear or opaque protective coating consisting of a sealing coat of shellac, a coating of sugar or polymeric material and a polish coating of wax can be provided.
- Dyestuffs can be added to these coatings to distinguish different unit dosages.
- Oral fluids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of a compound of formula (I).
- Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
- dosage unit pharmaceutical compositions for oral can also be added.
- administration can be microencapsulated.
- the formulation can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
- compositions adapted for rectal administration may be presented as suppositories or as enemas.
- compositions adapted for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
- compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the composition isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the pharmaceutical compositions may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- compositions may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
- a therapeutically effective amount of a compound of the present invention will depend upon a number of factors including, for example, the age and weight of the intended recipient, the precise condition requiring treatment and its severity, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant prescribing the medication.
- an effective amount of a compound of formula (I) for the treatment of anemia will generally be in the range of 0.001 to 100 mg/kg body weight of recipient per day, suitably in the range of .01 to 10 mg/kg body weight per day.
- the actual amount per day would suitably be from 7 to 700 mg and this amount may be given in a single dose per day or in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same.
- An effective amount of a salt or solvate, etc. may be determined as a proportion of the effective amount of the compound of formula (I) per se. It is envisaged that similar dosages would be appropriate for treatment of the other conditions referred to above.
- the compounds of this invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention as prepared are given in the examples.
- the present invention includes both possible stereoisomers and includes not only racemic compounds but the individual enantiomers as well.
- a compound When a compound is desired as a single enantiomer, it may be obtained by stereospecific synthesis or by resolution of the final product or any convenient intermediate. Resolution of the final product, an intermediate, or a starting material may be effected by any suitable method known in the art. See, for example, Stereochemistry of Organic Compounds by E. L. Eliel, S. H. Wilen, and L. N. Mander (Wiley-Interscience, 1994).
- Celite® registered trademark of Celite Corp. brand of diatomaceous earth DBU l,8-diazabicyclo[5.4.0]undeca-7-ene
- Preparative normal phase silica gel chromatography was carried out using either a Teledyne ISCO CombiFlash Companion instrument with RediSep or ISCO Gold silica gel cartridges (4 g-330 g), or an Analogix IF280 instrument with SF25 silica gel cartridges (4 g - 3-00g), or a Biotage SP1 instrument with HP silica gel cartridges (lOg - 100 g).
- a PE Sciex API 150 single quadrupole mass spectrometer (PE Sciex, Thornhill, Ontario, Canada) was operated using electrospray ionization in the positive ion detection mode.
- the nebulizing gas was generated from a zero air generator (Balston Inc.,
- Method A LCMS Method A LCMS. Samples were introduced into the mass spectrometer using a CTC PAL autosampler (LEAP Technologies, Carrboro, NC) equipped with a hamilton 10 uL syringe which performed the injection into a Valco 10-port injection valve.
- the HPLC pump was a Shimadzu LC-lOADvp (Shimadzu Scientific Instruments, Columbia, MD) operated at 0.3 mL/min and a linear gradient 4.5% A to 90% B in 3.2 min. with a 0.4 min. hold.
- the mobile phase was composed of 100% (H 2 0 0.02%> TFA) in vessel A and 100%) (CH 3 CN 0.018% TFA) in vessel B.
- the stationary phase is Aquasil (CI 8) and the column dimensions were 1 mm x 40 mm. Detection was by UV at 214 nm, evaporative light- scattering (ELSD) and MS.
- Method B LCMS.
- an Agilent 1100 analytical HPLC system with an LC/MS was used and operated at 1 mL/min and a linear gradient 5% A to 100% B in 2.2 min with a 0.4 min hold.
- the mobile phase was composed of 100% (H 2 0 0.02% TFA) in vessel A and 100% (CH 3 CN 0.018% TFA) in vessel B.
- the stationary phase was Zobax (C8) with a 3.5 um partical size and the column dimensions were 2.1 mm x 50 mm.
- Detection was by UV at 214 nm, evaporative light-scattering (ELSD) and MS. Method C, LCMS.
- an MDSSCIEX API 2000 equipped with a capillary column of (50 x 4.6 mm, 5 ⁇ ) was used.
- HPLC was done on Agilent-1200 series UPLC system equipped with column Zorbax SB-C18 (50 x 4.6 mm, 1.8 ⁇ ) eluting with CH 3 CN: ammonium acetate buffer. The reactions were performed in the microwave (CEM, Discover).
- Analytical HPLC Products were analyzed by Agilent 1100 Analytical Chromatography system, with 4.5 x 75 mm Zorbax XDB-C18 column (3.5 um) at 2 mL/min with a 4 min gradient from 5% CH 3 CN (0.1% formic acid) to 95% CH 3 CN (0.1% formic acid) in H 2 0 (0.1% formic acid) and a 1 min hold.
- Scheme 1 illustrates two methods to synthesize a compound of formula (VII).
- Substituted aminopyrazoles of formula (I) are heated with diethyl oxobutanedione in benzene or toluene at 62 °C overnight.
- Treatment of the putative intermediate with acetic acid and typically heating at reflux furnishes azaindazole compounds of formula (II).
- Compounds of formula (II) are converted to compounds of formula (III) by base-catalyzed hydrolysis of the ethyl ester and then chlorination of the putative carboxylic acid intermediate with POCl 3 under standard conditions to afford compounds of formula (IV).
- compounds of formula (II) are converted to the corresponding triflate (Ila) using standard methods.
- Compounds of formula (Ila) are then substituted at the 6-pos. using standard palladium mediated cross-coupling conditions, followed by base-catalyzed hydrolysis of the ethyl ester group to afford compounds of formula (IVa).
- Treatment of compounds of formula (IVa) with substituted aminomethyl pyridones of formula (V) using EDC, HO AT, N- methylmorpholine, and DMSO at room temperature for a period of no less than 12 h stirring at room temperature affords compounds of formula (VII).
- Scheme 4 illustrates the method to synthesize a compound of formula (V).
- 6-Cyclopropyl-4-methyl-2-oxo-l,2-dihydro-3-pyridinecarbonitrile (0.35 g, 2.01 mmol) was added to methanol (20 mL) and the stirring contents cooled to -10 °C.
- di-tert-butyloxycarbonyl (0.933 mL, 4.02 mmol) and the suspension was stirred for 15 min.
- N1CI 2 -6H 2 O 0.055 g, 0.201 mmol
- NaBH 4 0.532 g, 14.06 mmol
- the crude product was purified by silica gel chromatography (eluent: 10% Methanol in Dichloromethane). The collected product was dried under hi-vacuum for 1 h, and then treated with ether and filtered. After drying in vacuum oven at 45 °C for 2 h, the product was collected as 0.28 g (50%).
- 6-Chloro-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (0.12g, 0.501 mmol), l-hydroxy-7-azabenzotriazole (0.102 g, 0.751 mmol), EDC (0.144 g, 0.751 mmol), and 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (0.123 g, 0.651 mmol) were dissolved in dimethyl sulfoxide (3.0 mL) and stirred at room temperature. Added next to the stirring contents was N-methylmorpholine (0.220 mL, 2.003 mmol) via syringe at once.
- the title compound was prepared in the same manner as described in example 7 from 6-cyclopropyl-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (250 mg, 1.019 mmol), 3-(aminomethyl)-6-methyl-4-(l-methylethyl)-2(lH)-pyridinone » TFA (300 mg, 1.019 mmol), HO AT (208 mg, 1.529 mmol), EDC (293 mg, 1.529 mmol), N- methylmorpholine (0.448 mL, 4.08 mmol), and DMF(6 mL), wherein the reaction time was 48 h.
- the stirring contents were allowed to cool (15 min.) then fitted with a balloon of H 2 and allowed to stir at room temperature overnight. The contents were then purged with N 2 . The mixture was then diluted with 10% MeOH/DCM (20mL), stirred for 10 min., and filtered through Celite. The filtrate was concentrated in vacuo and the crude solid was triturated with ethanol. After filtration and washing the collected solid with additional ethanol (cold), the solid was air-dried for 15 min, then in vacuum oven at 40 °C overnight. The final product was collected as 185 mg (80%>).
- the title compound was prepared in the same manner as described in example 19 from 6-cyclopropyl-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (0.12 g, 0.489 mmol), l-hydroxy-7-azabenzotriazole (0.100 g, 0.734 mmol), 3-(aminomethyl)-6- methyl-4-(trifluoromethyl)-2(lH)-pyridinone (0.154 g, 0.636 mmol, DMSO (3.0 mL), N- methylmorpholine (0.215 mL, 1.957 mmol), and EDC (0.141 g, 0.734 mmol)
- the crude solid was purified by silica gel chromatography (eluent: gradient 5-100% of 10%> 2M NH 3 (in MeOH/DCM) and DCM) and the collected product dried in vacuum oven for 5h.
- reaction mixture was diluted with water (40 mL), adjusted to pH 6-7, and stirred for 15 min. The contents were filtered and washed with water. The product was dried in vacuum oven at 50 °C for 5 hr. The product was obtained as 57mg (79%).
- the stirring suspension was degassed with nitrogen for 5 min., wherein an emulsion had formed.
- Added next were phenylacetylene (0.110 mL, 1.003 mmol) and Pd(Ph 3 P) 4 (0.039 g, 0.033 mmol).
- the sealed reaction mixture was placed onto a heat block, stirred at 90 °C for 3 hr, and then allowed to cool to room temperature overnight.
- the contents were poured onto water and 20% THF/EtOAc, stirred, and the layers separted.
- the organic layer was washed with brine, dried over MgS0 4 , filtered, and concentrated in vacuo.
- the filter pad was washed with additional EtOAc.
- the combined filtrates were concentrated in vacuo to a yellow/orange residue that was dried on hi-vac pump.
- the crude solid was then pre-adsorbed onto silica gel and purified by silica gel chromatography (dry loaded, eluent: 5-80 % gradient of DCM and chloroform containing 10% 2M Ammonia (in methanol)).
- the isolated product was obtained as a yellow solid which was then further purified by reverse phase HPLC (mobile phase: 20-90% ACN in H 2 0, 0.1% TFA, Gradient time: 8min).
- the isolated solid was dissolved in 10% MeOH/ CH 2 C1 2 and treated with 0.6 g of Silicycle carbonate resin for 30 min.
- the contents were transferred to a heat block and heated at 135 °C for 16 hr., and then at 145 °C for an additional 12 h. After cooling to room temperature, the contents were diluted with CH 2 CI 2 and pre-absorbed onto silica gel.
- the crude product was purified by silica gel chromatography (dry loaded, eluent; gradient of 5-80% DCM and chloroform containing 10% 2M Ammonia (in methanol)). The isolated solid was triturated with MTBE, filtered, and washed with additional MTBE. The collected solid was dried in vacuum oven at 45 °C overnight to afford the final product as 0.067 g (55%).
- LCMS E-S (M+H) 445.3.
- N-methylmorpholine (0.11 ml, 0.96 mmol) and EDC (79 mg, 0.41 mmol) were added, the vessel was sealed, and the light brown mixture was stirred at room temperature for 2 days. Next added 2 mL of water and the mixture was stirred for 10 min. Solids that precipitated were sonicated, and allowed to stand at room temperature for 10 min. The contents were filtered, washed with water, and dried to afford the title compound (68 mg, 68%) as a light pink solid.
- LCMS E-S (M+H) 354.3.
- N-methylmorpholine (0.1 ml, 0.87 mmol) and EDC (72 mg, 0.37 mmol) were added, the vessel was sealed, and the bright yellow mixture was stirred at room temperature for 2 days. Next added 2 mL of water, and the contents were stirred for 10 min. Solids that precipitated were sonicated, and allowed to stand at room temperature for 10 min. The reaction contents were filtered and washed with water. The solid was treated with 2 mL of EtOH, sonicated and heated, and then allowed to cool to room temperature. The contents were filtered, washed with water and dried to afford the title compound (74 mg, 70%) as a white solid.
- LCMS E-S (M+H) 416.3.
- PdCl 2 (dppf)- CH 2 C1 2 adduct (8.7 mg, 0.01 1 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min.
- Sodium bicarbonate (53.9 mg, 0.64 mmol) was added, the vessel was sealed, and the insoluble green mixture was irradiated (microwave) at 150 °C for 20 min. After cooling, 2 mL of water was added to the dark green mixture and solids that precipitated were filtered. EtOAc was added, the mixture was heated and some hexanes were added. Solids were filtered, dissolved in DCM/MeOH (1 : 1), and filtered through a pad of silica gel and the filtrate was evaporated.
- PdCl 2 (dppf)-CH 2 Cl 2 adduct (8.2 mg, 0.01 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min.
- Sodium bicarbonate (50.6 mg, 0.6 mmol) was added , the vessel was sealed, and the insoluble mixture was heated in a microwave at 150 °C for 30 min. After cooling, 2 mL of water was added to the mixture and solids that precipitated were filtered.
- DCM/MeOH (1 : 1) was added and the solution was filtered through a pad of silica gel and the filtrate was evaporated.
- the reaction mixture was degassed with nitrogen for 5 min.
- Sodium bicarbonate (67.4 mg, 0.802 mmol) was added and the contents sealed and irradiated (microwave) at 140 °C.
- the reaction mixture was cooled to room temperature and poured on a silica column (through Na 2 S0 4 ) and purified by silica gel chromatography (eluent : 5% MeOH/CH 2 Cl 2 ) which provided the desired product as an off-white solid after preciptation from EtOAc/MeOH.
- the final product was collected as 0.090 g (76%).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Herein are disclosed azaindazoles of formula (I), (I), where the various groups are defined herein, and which are useful for treating cancer.
Description
AZAINDAZOLES
FIELD OF THE INVENTION
This invention relates to substituted azaindazoles which inhibit EZH2 and thus are useful for inhibiting the proliferation of and/or inducing apoptosis in cancer cells.
BACKGROUND OF THE INVENTION
Epigenetic modifications play an important role in the regulation of many cellular processes including cell proliferation, differentiation, and cell survival. Global epigenetic modifications are common in cancer, and include global changes in DNA and/or histone methylation, dysregulation of non-coding RNAs and nucleosome remodeling leading to aberrant activation or inactivation of oncogenes, tumor suppressors and signaling pathways. However, unlike genetic mutations which arise in cancer, these epigenetic changes can be reversed through selective inhibition of the enzymes involved. Several methylases involved in histone or DNA methylation are known to be dysregulated in cancer. Thus, selective inhibitors of particular methylases will be useful in the treatment of proliferative diseases such as cancer.
EZH2 (enhancer of zeste homolog 2; human EZH2 gene: Cardoso, C, et al;
European J of Human Genetics, Vol. 8, No. 3 Pages 174-180, 2000) is the catalytic subunit of the Polycomb Repressor Complex 2 (PRC2) which functions to silence target genes by tri-methylating lysine 27 of histone H3 (H3K27me3). Histone H3 is one of the five main histone proteins involved in the structure of chromatin in eukaryotic cells.
Featuring a main globular domain and a long -terminal tail, Histones are involved with the structure of the nucleosomes, a 'beads on a string' structure. Histone proteins are highly post-translationally modified however Histone H3 is the most extensively modified of the five histones. The term "Histone H3" alone is purposely ambiguous in that it does not distinguish between sequence variants or modification state. Histone H3 is an important protein in the emerging field of epigenetics, where its sequence variants and variable modification states are thought to play a role in the dynamic and long term regulation of genes.
Increased EZH2 expression has been observed in numerous solid tumors including those of the prostate, breast, skin, bladder, liver, pancreas, head and neck and correlates with cancer aggressiveness, metastasis and poor outcome (Varambally et al, 2002; Kleer
et al, 2003; Breuer et al, 2004; Bachmann et al, 2005; Weikert et al, 2005; Sudo et al, 2005; Bachmann et al., 2006). For instance, there is a greater risk of recurrence after prostatectomy in tumors expressing high levels of EZH2, increased metastasis, shorter disease-free survival and increased death in breast cancer patients with high EZH2 levels (Varambally et al., 2002; Kleer et al., 2003). More recently, inactivating mutations in UTX (ubiquitously transcribed tetratricopeptixe repeats X), a H3K27 demethylase which functions in opposition to EZH2, have been identified in multiple solid and hematological tumor types (including renal, glioblastoma, esophageal, breast, colon, non-small cell lung, small cell lung, bladder, multiple myeloma, and chronic myeloid leukemia tumors), and low UTX levels correlate with poor survival in breast cancer suggesting that loss of UTX function leads to increased H3K27me3 and repression of target genes (Wang et al, 2010). Together, these data suggest that increased H3K27me3 levels contribute to cancer aggressiveness in many tumor types and that inhibition of EZH2 activity may provide therapeutic benefit.
Numerous studies have reported that direct knockdown of EZH2 via siRNA or shRNA or indirect loss of EZH2 via treatment with the SAH hydrolase inhibitor 3- deazaneplanocin A (DZNep) decreases cancer cell line proliferation and invasion in vitro and tumor growth in vivo (Gonzalez et al, 2008, GBM 2009). While the precise mechanism by which aberrant EZH2 activity leads to cancer progression is not known, many EZH2 target genes are tumor suppressors suggesting that loss of tumor suppressor function is a key mechanism (refs). In addition, EZH2 overexpression in immortalized or primary epithelial cells promotes anchorage independent growth and invasion and requires EZH2 catalytic activity. (Kleer et al, 2003; Cao et al, 2008).
Thus, there is strong evidence to suggest that inhibition of EZH2 activity decreases cellular proliferation and invasion. Accordingly, compounds that inhibit EZH2 activity would be useful for the treatment of cancer. The azaindazoles of this invention provide such treatment.
SUMMARY OF THE INVENTION
In a first instance, this invention relates to compounds of formula (I)

wherein
X and Z are selected independently from the group consisting of hydrogen, (Ci- C8)alkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, unsubstituted or substituted (C3-Cg)cycloalkyl, unsubstituted or substituted (C3-Cg)cycloalkyl-(Ci-Cg)alkyl or -(C2-C8)alkenyl, unsubstituted or substituted (C5-C8)cycloalkenyl, unsubstituted or substituted (C5- Cg)cycloalkenyl-(Ci-Cg)alkyl or -(C2-Cg)alkenyl, (C6-Cio)bicycloalkyl, unsubstituted or substituted heterocycloalkyl, unsubstituted or substituted heterocycloalkyl-(Ci-Cg)alkyl or -(C2-C8)alkenyl, unsubstituted or substituted aryl, unsubstituted or substituted aryl-(Ci- Cg)alkyl or -(C2-Cg)alkenyl, unsubstituted or substituted heteroaryl, unsubstituted or substituted heteroaryl-(Ci-Cg)alkyl or -(C2-Cg)alkenyl, halo, cyano, -CORa, -C02Ra, - CONRaRb, -CONRaNRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -NRaNRaC(0)ORa, -ORa, - OC(0)Ra, and -OC(0)NRaRb;
Y is H or halo;
R1 is (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-Cg)alkynyl, unsubstituted or substituted (C3-C8)cycloalkyl, unsubstituted or substituted (C3-C8)cycloalkyl-(Ci-Cg)alkyl or - (C2-C8)alkenyl, unsubstituted or substituted (Cs-Cg)cycloalkenyl, unsubstituted or substituted (C5-Cg)cycloalkenyl-(Ci-Cg)alkyl or -(C2-Cg)alkenyl, unsubstituted or substituted (C6-Cio)bicycloalkyl, unsubstituted or substituted heterocycloalkyl or - (C2-C8)alkenyl, unsubstituted or substituted heterocycloalkyl-(Ci-C8)alkyl, unsubstituted or substituted aryl, unsubstituted or substituted aryl-(Ci-C8)alkyl or -(C2-Cg)alkenyl, unsubstituted or substituted heteroaryl, unsubstituted or substituted heteroaryl-(Ci- C8)alkyl or -(C2-C8)alkenyl, -CORa, -C02Ra, -CONRaRb, -CONRaNRaRb;
R3 is hydrogen, (Ci-C8)alkyl, cyano, trifluoromethyl, -NRaRb, or halo;
R6 is selected from the group consisting of hydrogen, halo, (Ci-Cg)alkyl,
(C2-C8)alkenyl, -B(OH)2, substituted or unsubstituted (C2-Cg)alkynyl, unsubstituted or substituted (C3-Cg)cycloalkyl, unsubstituted or substituted (C3-Cg)cycloalkyl-(Ci-Cg)alkyl, unsubstituted or substituted (C5-C8)cycloalkenyl, unsubstituted or substituted (C5- C8)cycloalkenyl-(Ci-C8)alkyl, (C6-Cio)bicycloalkyl, unsubstituted or substituted heterocycloalkyl, unsubstituted or substituted heterocycloalkyl-(Ci-C8)alkyl, unsubstituted or substituted aryl, unsubstituted or substituted aryl-(Ci-C8)alkyl, unsubstituted or substituted heteroaryl, unsubstituted or substituted heteroaryl-(Ci-C8)alkyl, cyano, -CORa, -C02Ra, -CONRaRb, -CONRaNRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -NRaNRaC(0)ORa, -ORa, - OC(0)Ra, -OC(0)NRaRb;
wherein any (Ci-Cg)alkyl, (C2-C8)alkenyl, (C2-Cg)alkynyl, cycloalkyl,
cycloalkenyl, bicycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by 1 , 2 or 3 groups independently selected from the group consisting of
-0(C1-C6)alkyl(Rc)1_2, -S(C1-C6)alkyl(Rc)1_2, -(C1-C6)alkyl(Rc)1_2, (C1-C8)alkyl- heterocycloalkyl, (C3-C8)cycloalkyl-heterocycloalkyl, halo, (Ci-C6)alkyl,
(C3-Cg)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, -CORa, - C02Ra,-CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -
ORa, -OC(0)Ra, -OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl,
aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
wherein any aryl or heteroaryl moiety of said aryl, heteroaryl, aryl(Ci-C4)alkyl, or heteroaryl(Ci-C4)alkyl is optionally substituted by 1, 2 or 3 groups independently selected from the group consisting of halo,
(Ci-C6)alkyl, (C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, -CONRaRb,
-SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb,
-NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)Ra, and -OC(0)NRaRb;
Ra and Rb are each independently hydrogen, (Ci-C8)alkyl, (C2-C8)alkenyl,
(C2-C8)alkynyl, (C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (C6-Cio)bicycloalkyl,
heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-C8)alkyl, (C2-C8)alkenyl, (C2-
Cs)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2,-CONH(Ci-C4)alkyl, - CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2,-S02NH(Ci-C4)alkyl, or - S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
or Ra and Rb taken together with the nitrogen to which they are attached represent a 5-8 membered saturated or unsaturated ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C4)alkyl, (Ci-
C4)haloalkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, hydroxyl, oxo, (Ci-C4)alkoxy, and (Ci-C4)alkoxy(Ci-C4)alkyl, wherein said ring is optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
or Ra and Rb taken together with the nitrogen to which they are attached represent a 6- to 10-membered bridged bicyclic ring system optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
each Rc is independently (Ci-C4)alkylamino, -NRaS02Rb, -SORa, -S02Ra, - NRaC(0)ORa, -NRaRb, or -C02Ra;
or a salt thereof.
In a further iteration of this invention it relates to a method of treating cancer.
Another aspect of the invention are pharmaceutical preparations comprising compounds of formula (I) and pharmaceutically acceptable excipients.
In a fourth aspect, there is provided the use of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, in the preparation of a medicament for use in the treatment of a disorder mediated by inhibiting EZH2, such as inducing apoptosis in cancer cells.
In a fifth aspect there is provided methods of co-administering the presently invented compounds of formula (I) with another active ingredients.
DETAILED DESCRIPTION OF THE INVENTION
For the avoidance of doubt, unless otherwise indicated, the term "substituted" means substituted by one or more defined groups. In the case where groups may be selected from a number of alternative groups the selected groups may be the same or different.
The term "independently" means that where more than one substituent is selected from a number of possible substituents, those substituents may be the same or different.
An "effective amount" means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal or human that is being sought, for instance, by a researcher or clinician. Furthermore, the term
"therapeutically effective amount" means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder. The term also includes within its scope amounts effective to enhance normal physiological function.
As used herein the term "alkyl" refers to a straight- or branched-chain hydrocarbon radical having the specified number of carbon atoms, so for example, as used herein, the terms "Ci-Csalkyl" refers to an alkyl group having at least 1 and up to 8 carbon atoms respectively. Examples of such branched or straight-chained alkyl groups useful in the present invention include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, isobutyl, n-butyl, t-butyl, n-pentyl, isopentyl, n-hexyl, n-heptyl, and n-octyl and branched analogs of the latter 5 normal alkanes.
The term "alkoxy" as used herein means -0(Ci_Cgalkyl) including -OCH3, -
OCH2CH3 and -OC(CH3)3 and the like per the definition of alkyl above.
The term "alkylthio" as used herein is meant -S(Ci_C8alkyl) including -SCH3, -
SCH2CH3 and the like per the definition of alkyl above.
The term "acyloxy" means -OC(0)Ci_Cgalkyl and the like per the definition of alkyl above.
"Acylamino" means-N(H)C(0)Ci_C8alkyl and the like per the definition of alkyl above.
"Aryloxy" means -O(aryl), -0(substituted aryl), -O(heteroaryl) or -0(substituted heteroaryl).
"Arylamino" means -NH(aryl), -NH(substituted aryl), -NH(heteroaryl) or - NH(substituted heteroaryl), and the like.
When the term "alkenyl" (or "alkenylene") is used it refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms and at least 1 and up to 5 carbon-carbon double bonds. Examples include ethenyl (or ethenylene) and propenyl (or propenylene).
When the term "alkynyl" (or "alkynylene") is used it refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms and at least 1 and up to 5 carbon-carbon triple bonds. Examples include ethynyl (or ethynylene) and propynyl (or propynylene).
"Haloalkyl" refers to an alkyl group group that is substituted with one or more halo substituents, suitably from 1 to 6 substituents. Haloalkyl includes trifluoromethyl.
When "cycloalkyl" is used it refers to a non-aromatic, saturated, cyclic
hydrocarbon ring containing the specified number of carbon atoms. So, for example, the term "Cs-Cscycloalkyl" refers to a non-aromatic cyclic hydrocarbon ring having from three to eight carbon atoms. Exemplary "C3-C8cycloalkyl" groups useful in the present invention include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
The term "Cs-Cscycloalkenyl" refers to a non-aromatic monocyclic carboxycyclic ring having the specified number of carbon atoms and up to 3 carbon-carbon double bonds. "Cycloalkenyl" includes by way of example cyclopentenyl and cyclohexenyl.
Where "Cs-Csheterocycloalkyl" is used, it means a non-aromatic heterocyclic ring containing the specified number of ring atoms being, saturated or having one or more degrees of unsaturation and containing one or more heteroatom substitutions
independently selected from O, S and N. Such a ring may be optionally fused to one or more other "heterocyclic" ring(s) or cycloalkyl ring(s). Examples are given herein below.
"Aryl" refers to optionally substituted monocyclic or polycarbocyclic unfused or fused groups having 6 to 14 carbon atoms and having at least one aromatic ring that complies with Huckel's Rule. Examples of aryl groups are phenyl, biphenyl, naphthyl, anthracenyl, phenanthrenyl, and the like, as further illustrated below.
"Heteroaryl" means an optionally substituted aromatic monocyclic ring or polycarbocyclic fused ring system wherein at least one ring complies with Huckel's Rule, has the specified number of ring atoms, and that ring contains at least one heteratom
independently selected from N, O and S. Examples of "heteroaryl" groups are given herein below.
The term "optionally" means that the subsequently described event(s) may or may not occur, and includes both event(s), which occur, and events that do not occur.
Herein, the term "pharmaceutically-acceptable salts" refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically-acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively.
While the compounds encompassed by the general structure of formula (I) as defined herein are believed to be useful for inducing apoptosis in cancer cells, some of these compounds are more active that others. In that vein, the following subgroups delineate certain compounds believed to have greater potency or other properties which suggest they may be a better choice for use in therapy, versus other. Those subgroups are represented as follows:
Subgroup A
X and Z are selected from the group consisting of (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NRaRb, and -ORa;
Y is H or F;
R1 is selected from the group consisting of (Ci-Cg)alkyl, (C3-C8)cycloalkyl, heterocycloalkyl, aryl, and heteroaryl;
R3 is selected from the group consisting of hydrogen, (Ci-Cg)alkyl, cyano, trifluoromethyl, -NRaRb, and halo;
R6 is selected from the group consisting of hydrogen, halo, cyano, trifluoromethyl, amino, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl;, aryl, heteroaryl, acylamino; (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl; -S02Ra; -S02NRaRb , and -NRaS02Rb ;
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl group is optionally substituted by 1 , 2 or 3 groups independently selected from -0(Ci-C6)alkyl(Rc)i_2, -S(Ci-C6)alkyl(Rc)i_2, -(Ci-C6)alkyl(Rc)i_2,
(Ci-Cg)alkyl-heterocycloalkyl, (C3-Cg)cycloalkyl-heterocycloalkyl, halo, (Ci-C6)alkyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, - CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -
NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl,
aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
each Rc is independently (Ci-C4)alkylamino, -NRaS02Rb, -SORa, -S02Ra, - NRaC(0)ORa, -NRaRb, or -C02Ra;
Ra and Rb are each independently hydrogen, (Ci-Cg)alkyl, (C2-C8)alkenyl, (C2-Cg)alkynyl, (C3-Cg)cycloalkyl, (C5-Cg)cycloalkenyl, (C6-Cio)bicycloalkyl,
heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-C8)alkyl, (C2-C8)alkenyl, (C2- Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1 , 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2, -CONH(Ci-C4)alkyl, -
CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2,-S02NH(Ci-C4)alkyl, and
S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
or Ra and Rb taken together with the nitrogen to which they are attached represent a
5-8 membered saturated or unsaturated ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C4)alkyl, (Ci- C4)haloalkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, hydroxyl, oxo, (Ci-C4)alkoxy, and (Ci-C4)alkoxy(Ci-C4)alkyl, wherein said ring is optionally fused to a (C3-C8)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
or Ra and Rb taken together with the nitrogen to which they are attached represent a
6- to 10-membered bridged bicyclic ring system optionally fused to a (C3-C8)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring. An aryl or heteroaryl group in this particular subgroup A is selected independently from the group consisting of furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, oxadiazole, thiadiazole, triazole, tetrazole, benzofuran, benzothiophene, benzoxazole, benzothiazole, phenyl, pyridine, pyridazine, pyrimidine, pyrazine, triazine, tetrazine, quinoline, cinnoline, quinazoline, quinoxaline, and na hthyridine or another aryl or heteroaryl group as follows:

wherein in (1),
A is O, NH, or S; B is CH or N, and C is hydrogen or Ci-Cg alkyl; or

wherein in (2),
D is N or C optionally substituted by hydrogen or Ci-C8 alkyl; or

wherein in (3),
E is NH or CH2; F is O or CO; and G is NH or CH2; or

wherein in (4),
J is O, S or CO; or

wherein in (5),
Q is CH or N;
M is CH or N; and
L/(5) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, -CORa, - C02Ra, -CONRaRb, -CONRaNRaRb, -S02Ra, -S02NRaRb, -NRaRb, -NRaC(0)Rb - NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa, wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, group is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, - NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)Ra, -OC(0)NRaRb; wherein RaandRb are defined as above; or

wherein in 6,
L/ 6) is NH or CH2; or

wherein in 7,
M/(7) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl, -CORa, -C02Ra, -CONRaRb, -CONRaNRaRb S02Ra, - S02NRaRb, -NRaRb, -NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, - NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa,
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl group is optionally substituted by 1 , 2 or 3 groups independently selected from
(Ci-C6)alkyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano,
CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02NRaRb, nitro, -NRaRb,
NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, ORa -OC(0)Ra, -OC(0)NRaRb; wherein RaandRb are defined as above; or

wherein in (8),
P is CH2, NH, O, or S; Q/(8) is CH or N; and n is 0-2; or

wherein in (9),
S/(9) and T(9) is C, or S/(9) is C and T(9) is N, or S/(9) is N and T/(9) is C;
R is hydrogen, amino, methyl, trifluoromethyl, halo;
U is hydrogen, halo, amino, cyano, nitro, trifluoromethyl, (Ci-Cg)alkyl, (C3- C8)cycloalkyl, -CORa, -C02Ra, -CONRaRb, -S02Ra, -S02NRaRb, -NRaRb, - NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, -ORa, 4-(lH- pyrazol-4-yl),
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, group is optionally
substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, -CORa, - C02Ra,-CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb; wherein Ra andRb are defined as above.
Subgroup B
X and Z are selected independently from the group consisting of (Ci-Cg)alkyl, (C3- Cg)cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NRaRb, and -ORa;
Y is H;
R1 is (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, or heterocycloalkyl;
R3 is hydrogen, (Ci-Cg)alkyl or halo;
R6 is hydrogen, halo, cyano, trifluoromethyl, amino, (Ci-Cg)alkyl, (C3- Cg)cycloalkyl;, aryl, heteroaryl, acylamino; (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl; -S02Ra; -S02NRaRb, or -NRaS02Rb;
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl group is optionally substituted by 1 , 2 or 3 groups independently selected from halo, (Ci-C6)alkyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl,
(Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, - S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, - NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)Ra, -OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl, aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
Ra and Rb are each independently hydrogen, (Ci-Cg)alkyl, (C2-Cg)alkenyl,
(C2-Cg)alkynyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (C6-Cio)bicycloalkyl,
heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-Cg)alkyl, (C2-C8)alkenyl, (C2- Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2,-CONH(Ci-C4)alkyl, -
CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2, -S02NH(Ci-C4)alkyl, and -S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
or Ra and Rb taken together with the nitrogen to which they are attached represent a
5- 8 membered saturated or unsaturated ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C4)alkyl, (Ci- C4)haloalkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, hydroxyl, oxo, (Ci-C )alkoxy, and (Ci-C )alkoxy(Ci-C )alkyl, wherein said ring is optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
or Ra and Rb taken together with the nitrogen to which they are attached represent
6- to 10-membered bridged bicyclic ring system optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring. Aryl and heteroaryl in this definition are selected from the group consisting of furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, oxadiazole, thiadiazole, triazole, tetrazole, benzofuran,
benzothiophene, benzoxazole, benzothiazole, phenyl, pyridine, pyridazine, pyrimidine, pyrazine, triazine, tetrazine, quinoline, cinnoline, quinazoline, quinoxaline, and na hthyridine as or a compound of or another aryl or heteroaryl group as follows:

wherein in (1),
A is O, NH, or S; B is CH or N, and C is hydrogen or Ci-Cg alkyl; or

wherein in (2),
D is N or C optionally substituted by hydrogen or Ci-Cg alkyl; or

wherein in (3),
E is NH or CH2; F is O or CO; and G is NH or CH2; or

wherein in (4),
J is O, S or CO; or

wherein in (5),
Q is CH or N;
M is CH or N; and
L/(5) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, -COR'
C02Ra, -CONRaRb, -CONRaNRaRb, -S02Ra, -S02NRaRb, -NRaRb, -NRaC(0)Rb,
NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa wherein any (Ci-Cg)alkyl, (C3-C8)cycloalkyl, group is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, -C02Ra -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, - NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)Ra, -OC(0)NRaRb,
wherein Ra and Rb are defined as above; or

wherein in 6,
L/ 6) is NH or CH2; or

wherein in 7,
M/(7) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl, -CORa, -C02Ra, -CONRaRb, -CONRaNRaRb S02Ra, -
S02NRaRb, -NRaRb, -NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, - NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa,
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl group is optionally substituted by 1 , 2 or 3 groups independently selected from
(Ci-C6)alkyl, (C3-Cg)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, - CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa -OC(0)Ra, -OC(0)NRaRb; wherein RaandRb are defined as above; or

wherein in (8),
P is CH2, NH, O, or S; Q/(8) is CH or N; and n is 0-2; or
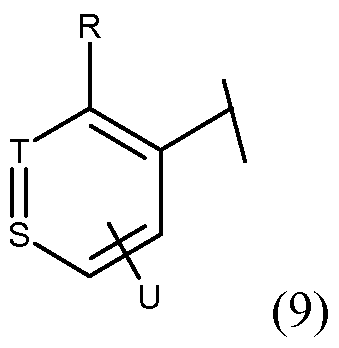
wherein in (9),
S/(9) and T(9) is C, or S/(9) is C and T(9) is N, or S/(9) is N and T/(9) is C;
R is hydrogen, amino, methyl, trifluoromethyl, halo;
U is hydrogen, halo, amino, cyano, nitro, trifluoromethyl, (Ci-Cg)alkyl, (C3- C8)cycloalkyl, -CORa, -C02Ra, -CONRaRb, -S02Ra, -S02NRaRb, -NRaRb, - NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, , -ORa, 4-(lH- pyrazol-4-yl),
wherein any (Ci-Cg)alkyl, (C3-C8)cycloalkyl, group is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-Cg)cycloalkyl, (C5-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, -CORa, - C02Ra,-CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb, wherein Ra and Rb are defined as above.
Subgroup C
X is methyl, ethyl, n-propyl, isopropyl , cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, trifluoromethyl, tetrahydropyran, hydroxymethyl, methoxymethyl, or benzyl;
Y is H;
Z is methyl, ethyl, n-propyl, isopropyl, trifluoromethyl, or benzyl;
R1 is isopropyl, tert-butyl, cyclobutyl, cyclopentyl, cyclohexyl, (1- methylethyl)cyclopropyl, 1 , 1 -dioxo-tetrahydrothiophene-3-yl, 1 -Me-piperidin-4-yl, tetrahydrofuran-3-yl, tetrahydropyran-4-yl, N,N-dimethyl-l-propanaminyl, benzyl, or 4- pyridyl;
R3 is H, methyl, or Br; and
R6 is methyl, bis(l,l-dimethylethyl), bis(l-methylethyl), cyclopropyl, propyl, dimethylamino, ethylamino, (2-hydroxyethyl)amino, 2-propen-l-ylamino, 1-piperazinyl,
1- piperidinyl, 4-morpholinyl, 4-piperidinylamino, tetrahydro-2H-pyran-4-ylamino, phenylamino, (phenylmethyl)amino, (4-pyridinylmethyl)amino, [2-(2- pyridinylamino)ethyl] amino, 2-(dimethylamino)ethyl]amino, 4-pyridinylamino , 4- (aminocarbonyl)phenyl]amino, 3 -hydroxy-3 -methyl- 1 -butyn- 1 -yl, 4-pyridinylethynyl, phenylethynyl, 2-furanyl, 3-thienyl; lH-pyrazol-4-yl, lH-indazol-5-yl, lH-indazol-6-yl, 3- methyl-lH-indazol-5-yl, lH-1 ,2,3-benzotriazol-5-yl, 2-oxo-2,3-dihydro-lH-benzimidazol- 5-yl, 2-oxo-2,3-dihydro-lH-indol-5-yl, 2-oxo-2,3-dihydro-lH-indol-6-yl, 2,1,3- benzoxadiazol-5-yl, 2-amino-6-quinazolinyl, 2,4-dioxo-l,2,3,4-tetrahydro-5-pyrimidinyl,
2- amino-5-pyrimidinyl, 7-oxo-l,5,6,7-tetrahydro-l,8-naphthyridin-3-yl, phenyl, 2- methylphenyl, 2-nitrophenyl, 2-phenylethyl, 3-aminophenyl, 4-aminophenyl, 4- chlorophenyl, 4-fluorophenyl, 4-(methyloxy)phenyl, 3-(acetylamino)phenyl, 4- (acetylamino)phenyl, 4-(aminocarbonyl)phenyl, 4-(lH-pyrazol-4-yl)phenyl, 4- (aminosulfonyl)phenyl, 4-(methylsulfonyl)phenyl, 4-[(dimethylamino)sulfonyl]phenyl, 4- [(methylamino)carbonyl]phenyl, 4- [(methylamino)sulfonyl]phenyl, 4- [(methylsulfonyl)amino]phenyl, 3-pyridinyl, 4-pyridinyl, 2-(4-morpholinyl)-4-pyridinyl, 2-amino-4-pyridinyl, 5-(methyloxy)-3-pyridinyl, 5-(methylsulfonyl)-3-pyridinyl, 5- [(cyclopropylsulfonyl)amino]-6-(methyloxy)-3-pyridinyl, 5-[(phenylsulfonyl)amino]-3- pyridinyl, 6-(4-methyl-l-piperazinyl)-3-pyridinyl, 6-(4-morpholinyl)-3-pyridinyl, 6- (acetylamino)-3-pyridinyl, 6-(dimethylamino)-3-pyridinyl, 6-(methyloxy)-3-pyridinyl, 6-
[(methylamino)carbonyl] -3 -pyridinyl, 6- [(methylamino)sulfonyl] -3 -pyridinyl, 6-methyl-3 - pyridinyl, 4-pyridinyloxy.
In another aspect, this invention also relates to the exemplified compounds.
In another aspect, this invention also relates to the following compounds:
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- (pyridin-3-yl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- phenyl-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)-6-(4- ((dimethylamino)methyl)phenyl)- 1 -isopropyl-3 -methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide;
N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)-6-(4-fluorophenyl)- 1 - isopropyl-3 -methyl- 1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
1 -isopropyl-N-((6-methyl-2-oxo-4-propyl- 1 ,2-dihydropyridin-3-yl)methyl)-6- phenyl-1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-(2-methyl-5-pyrimidinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-[4-(methyloxy)phenyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(41 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-3-methyl-6-[6-(4-methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4- ¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(41 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl-6- [6-(4-morpholinyl)-3 -pyridinyl] - IH-pyrazolo [3 ,4- ¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 , 1 -dimethylethy l)-3 -methyl-N- [(6-methyl-2-oxo-4-propyl- 1,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; l,6-dicyclopropyl-3-methyl-N-[(6-methyl-2-oxo-4-propyl-l,2-dihydro-3- pyridinyl)methyl] - IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide; l,6-dicyclopropyl-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3- methyl-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(cyclopropylamino)-N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)- 1 - isopropyl-3 -methyl- 1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-((4-benzyl-6-methyl-2-oxo- 1 ,2-dihydropyridin-3 -y l)methyl)- 1 -isopropyl- 1 H- pyrazolo[3,4-b]pyridine-4-carboxamide; N-((4-benzyl-6-methyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)- 1 -isopropyl-3- methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4-cyclopropyl-6-methyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- 1 -( 1 -methy lethyl)- lH-pyrazolo [3 ,4-¾]pyridine-4- carboxamide;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N- { [6-methyl-4-( 1 -methylethyl)-2-oxo- l ,2-dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide ; 6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N-[(6-methyl-2-oxo-4-propyl- 1 ,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide ;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N- { [6-methyl-2-oxo-4-(phenylmethyl)- l ,2-dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; or a pharmaceutically acceptable salt thereof.
Individual compounds can be found in the Examples set out below.
By the term "co-administering" and derivatives thereof as used herein is meant either simultaneous administration or any manner of separate sequential administration of one or more additional pharmaceutically active compounds, whether for treating cancer, the side effects of cancer or cancer therapy, or some other disease. Preferably, if the administration is not simultaneous, the compounds are administered in a close time proximity to each other. Furthermore, it does not matter if the compounds are
administered in the same dosage form, e.g. one compound may be administered topically and another compound may be administered orally.
In certain embodiments, compounds according to Formula I may contain an acidic functional group, one acidic enough to form salts. Representative salts include pharmaceutically-acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a pharmaceutically- acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically-acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, triethylamine, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
In certain embodiments, compounds according to Formula (I) may contain a basic functional group and are therefore capable of forming pharmaceutically-acceptable acid addition salts by treatment with a suitable acid. Suitable acids include pharmaceutically- acceptable inorganic acids and pharmaceutically-acceptable organic acids. Representative pharmaceutically-acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphate.! acetate, hydroxy acetate, phenylacetate, propionate, butyrate, isobutyrate, valerate, maleate, hydroxymaleate, acrylate, fumarate, malate, tartrate, citrate, salicylate, / aminosalicyclate, glycollate, lactate, heptanoate, phthalate, oxalate, succinate, benzoate, o-acetoxybenzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, mandelate, tannate, formate, stearate, ascorbate, palmitate, oleate, pyruvate, pamoate, malonate, laurate, glutarate, glutamate, estolate, methanesulfonate (mesylate),
ethanesulfonate (esylate), 2-hydroxyethanesulfonate, benzenesulfonate (besylate), p- aminobenzenesulfonate, /?-toluenesulfonate (tosylate) and napthalene-2-sulfonate.
All tautomeric forms of the compounds described herein, including mixtures thereof, are intended to be encompassed within the scope of the invention. Generally, the compounds exemplified herein have been assigned names based on the structure of the tautomer of formaula (IA). It should be understood that any reference to named compounds of this invention is intended to encompass all tautomers of the named compounds and any mixtures of tautomers of the named compounds.
The compounds of formula (I) may be prepared in crystalline or non-crystalline form, and, if crystalline, may optionally be solvated, e.g. as the hydrate. This invention includes within its scope stoichiometric solvates (e.g. hydrates) as well as compounds containing variable amounts of solvent (e.g. water).
Certain of the compounds described herein may contain one or more chiral atoms, or may otherwise be capable of existing as two enantiomers. The compounds claimed below include mixtures of enantiomers as well as purified enantiomers or enantiomerically enriched mixtures. Also included within the scope of the invention are the individual isomers of the compounds represented by formula (I), or claimed below, as well as any wholly or partially equilibrated mixtures thereof. The present invention also covers the individual isomers of the claimed compounds as mixtures with isomers thereof in which one or more chiral centers are inverted.
Where there are different isomeric forms they may be separated or resolved one from the other by conventional methods, or any given isomer may be obtained by conventional synthetic methods or by stereospecific or asymmetric syntheses.
While it is possible that, for use in therapy, a compound of formula (I), as well as salts, solvates and the like, may be administered as a neat preparation, i.e. no additional carrier, the more usual practice is to present the active ingredient confected with a carrier or diluent. Accordingly, the invention further provides pharmaceutical compositions, which includes a compound of formula (I) and salts, solvates and the like, and one or more pharmaceutically acceptable carriers, diluents, or excipients. The compounds of formula (I) and salts, solvates, etc, are as described above. The carrier(s), diluent(s) or excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. In accordance with another aspect of the invention there is also provided a process for the preparation of a pharmaceutical formulation including admixing a compound of the formula (I), or salts, solvates etc, with one or more pharmaceutically acceptable carriers, diluents or excipients.
It will be appreciated by those skilled in the art that certain protected derivatives of compounds of formula (I), which may be made prior to a final deprotection stage, may not possess pharmacological activity as such, but may, in certain instances, be administered orally or parenterally and thereafter metabolised in the body to form compounds of the invention which are pharmacologically active. Such derivatives may therefore be described as "prodrugs". Further, certain compounds of the invention may act as prodrugs of other compounds of the invention. All protected derivatives and prodrugs of compounds of the invention are included within the scope of the invention. It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as "pro- moieties" may be placed on appropriate functionalities when such functionalities are present within compounds of the invention. Preferred prodrugs for compounds of the invention include: esters, carbonate esters, hemi-esters, phosphate esters, nitro esters, sulfate esters, sulfoxides, amides, carbamates, azo-compounds, phosphamides, glycosides, ethers, acetals and ketals.
Treatments
The compounds and compositions of the invention are used to treat cellular proliferation diseases. Disease states which can be treated by the methods and
compositions provided herein include, but are not limited to, cancer (further discussed
below), autoimmune disease, fungal disorders, arthritis, graft rejection, inflammatory bowel disease, proliferation induced after medical procedures, including, but not limited to, surgery, angioplasty, and the like. It is appreciated that in some cases the cells may not be in a hyper or hypo proliferation state (abnormal state) and still requires treatment. For example, during wound healing, the cells may be proliferating "normally", but
proliferation enhancement may be desired. Thus, in one embodiment, the invention herein includes application to cells or individuals afflicted or impending affliction with any one of these disorders or states.
The compositions and methods provided herein are particularly deemed useful for the treatment of cancer including tumors such as prostate, breast, brain, skin, cervical carcinomas, testicular carcinomas, etc. They are particularly useful in treating metastatic or malignant tumors. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to tumor types such as astrocytic, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck,
hepatocellular, laryngeal, lung, oral, ovarian, prostate and thyroid carcinomas and sarcomas. More specifically, these compounds can be used to treat: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell,
undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar
(bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Karposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm's tumor
(nephroblastoma), lymphoma, leukemia), bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma), prostate (adenocarcinoma, sarcoma), testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma); Liver: hepatoma (hepatocellular carcinoma), cholangiocarcinoma, hepatoblastoma,
angiosarcoma, hepatocellular adenoma, hemangioma; Biliary tract: gall bladder
carcinoma, ampullary carcinoma, cholangiocarcinoma; Bone: osteogenic sarcoma
(osteosarcoma), fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, malignant giant cell tumor chordoma, osteochronfroma (osteocartilaginous exostoses), benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma and giant cell tumors; Nervous system: skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans), meninges (meningioma, meningiosarcoma, gliomatosis), brain (astrocytoma, meduUoblastoma, glioma, ependymoma, germinoma (pinealoma), glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors), spinal cord neurofibroma, meningioma, glioma, sarcoma); Gynecological: uterus (endometrial carcinoma), cervix (cervical carcinoma, pre -tumor cervical dysplasia), ovaries (ovarian carcinoma (serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma), granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma), vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma), vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma), fallopian tubes
(carcinoma); Hematologic: blood (myeloid leukemia (acute and chronic), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplasia syndrome), Hodgkin's disease, non-Hodgkin's lymphoma (malignant lymphoma); Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Karposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term "cancerous cell" as provided herein, includes a cell afflicted by any one or related of the above identified conditions.
The instant compounds can be combined with or co-administered with other therapeutic agents, particularly agents that may enhance the activity or time of disposition of the compounds. Combination therapies according to the invention comprise the administration of at least one compound of the invention and the use of at least one other treatment method. In one embodiment, combination therapies according to the invention comprise the administration of at least one compound of the invention and surgical therapy. In one embodiment, combination therapies according to the invention comprise the administration of at least one compound of the invention and radiotherapy. In one embodiment, combination therapies according to the invention comprise the
administration of at least one compound of the invention and at least one supportive care agent (e.g., at least one anti-emetic agent). In one embodiment, combination therapies according to the present invention comprise the administration of at least one compound of the invention and at least one other chemotherapeutic agent. In one particular
embodiment, the invention comprises the administration of at least one compound of the invention and at least one anti-neoplastic agent. In yet another embodiment, the invention comprises a therapeutic regimen where the EZH2 inhibitors of this disclosure are not in and of themselves active or significantly active, but when combined with another therapy, which may or may not be active as a standalone therapy, the combination provides a useful therapeutic outcome.
By the term "co-administering" and derivatives thereof as used herein is meant either simultaneous administration or any manner of separate sequential administration of an EZH2 inhibiting compound, as described herein, and a further active ingredient or ingredients, known to be useful in the treatment of cancer, including chemotherapy and radiation treatment. The term further active ingredient or ingredients, as used herein, includes any compound or therapeutic agent known to or that demonstrates advantageous properties when administered to a patient in need of treatment for cancer. Preferably, if the administration is not simultaneous, the compounds are administered in a close time proximity to each other. Furthermore, it does not matter if the compounds are
administered in the same dosage form, e.g. one compound may be administered topically and another compound may be administered orally.
Typically, any anti-neoplastic agent that has activity versus a susceptible tumor being treated may be co-administered in the treatment of specified cancers in the present invention. Examples of such agents can be found in Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6th edition (February 15, 2001),
Lippincott Williams & Wilkins Publishers. A person of ordinary skill in the art would be able to discern which combinations of agents would be useful based on the particular characteristics of the drugs and the cancer involved. Typical anti-neoplastic agents useful in the present invention include, but are not limited to, anti-microtubule agents such as diterpenoids and vinca alkaloids; platinum coordination complexes; alkylating agents such as nitrogen mustards, oxazaphosphorines, alkylsulfonates, nitrosoureas, and triazenes; antibiotic agents such as anthracyclins, actinomycins and bleomycins; topoisomerase II inhibitors such as epipodophyllotoxins; antimetabolites such as purine and pyrimidine
analogues and anti-folate compounds; topoisomerase I inhibitors such as camptothecins; hormones and hormonal analogues; DNA methyltransferase inhibitors such as azacitidine and decitabine; signal transduction pathway inhibitors; non-receptor tyrosine kinase angiogenesis inhibitors; immunotherapeutic agents; proapoptotic agents; and cell cycle signaling inhibitors.
Typically, any chemotherapeutic agent that has activity against a susceptible neoplasm being treated may be utilized in combination with the compounds the invention, provided that the particular agent is clinically compatible with therapy employing a compound of the invention. Typical anti-neoplastic agents useful in the present invention include, but are not limited to: alkylating agents, anti-metabolites, antitumor antibiotics, antimitotic agents, nucleoside analogues, topoisomerase I and II inhibitors, hormones and hormonal analogues; retinoids, histone deacetylase inhibitors; signal transduction pathway inhibitors including inhibitors of cell growth or growth factor function, angiogenesis inhibitors, and serine/threonine or other kinase inhibitors; cyclin dependent kinase inhibitors; antisense therapies and immunotherapeutic agents, including monoclonals, vaccines or other biological agents.
Nucleoside analogues are those compounds which are converted to
deoxynucleotide triphosphates and incorporated into replicating DNA in place of cytosine. DNA methyltransferases become covalently bound to the modified bases resulting in an inactive enzyme and reduced DNA methylation. Examples of nucleoside analogues include azacitidine and decitabine which are used for the treatment of myelodysplasia disorder.Histone deacetylase (HDAC) inhibitors include vorinostat, for the treatment of cutaneous T-cell lymphoma. HDACs modify chromatin through the deactylation of histones. In addition, they have a variety of substrates including numerous transcription factors and signaling molecules. Other HDAC inhibitors are in development.
Signal transduction pathway inhibitors are those inhibitors which block or inhibit a chemical process which evokes an intracellular change. As used herein this change is cell proliferation or differentiation or survival. Signal transduction pathway inhibitors useful in the present invention include, but are not limited to, inhibitors of receptor tyrosine kinases, non-receptor tyrosine kinases, SH2/SH3 domain blockers, serine/threonine kinases, phosphatidyl inositol-3-OH kinases, myoinositol signaling, and Ras oncogenes. Signal transduction pathway inhibitors may be employed in combination with the compounds of the invention in the compositions and methods described above.
Receptor kinase angiogenesis inhibitors may also find use in the present invention. Inhibitors of angiogenesis related to VEGFR and TIE-2 are discussed above in regard to signal transduction inhibitors (both are receptor tyrosine kinases). Other inhibitors may be used in combination with the compounds of the invention. For example, anti-VEGF antibodies, which do not recognize VEGFR (the receptor tyrosine kinase), but bind to the ligand; small molecule inhibitors of integrin (alphav beta3) that inhibit angiogenesis;
endostatin and angiostatin (non-RTK) may also prove useful in combination with the compounds of the invention. One example of a VEGFR antibody is bevacizumab
(AVASTIN®).
Several inhibitors of growth factor receptors are under development and include ligand antagonists, antibodies, tyrosine kinase inhibitors, anti-sense oligonucleotides and aptamers. Any of these growth factor receptor inhibitors may be employed in combination with the compounds of the invention in any of the compositions and methods/uses described herein. Trastuzumab (Herceptin®) is an example of an anti-erbB2 antibody inhibitor of growth factor function. One example of an anti-erbBl antibody inhibitor of growth factor function is cetuximab (Erbitux™, C225). Bevacizumab (Avastin®) is an example of a monoclonal antibody directed against VEGFR. Examples of small molecule inhibitors of epidermal growth factor receptors include but are not limited to lapatinib (Tykerb™) and erlotinib (TARCEVA®). Imatinib mesylate (GLEEVEC®) is one example of a PDGFR inhibitor. Examples of VEGFR inhibitors include pazopanib, ZD6474, AZD2171, PTK787, sunitinib and sorafenib.
Anti-microtubule or anti-mitotic agents are phase specific agents active against the microtubules of tumor cells during M or the mitosis phase of the cell cycle. Examples of anti-microtubule agents include, but are not limited to, diterpenoids and vinca alkaloids.
Diterpenoids, which are derived from natural sources, are phase specific anti - cancer agents that operate at the G2/M phases of the cell cycle. It is believed that the diterpenoids stabilize the β-tubulin subunit of the microtubules, by binding with this protein. Disassembly of the protein appears then to be inhibited with mitosis being arrested and cell death following. Examples of diterpenoids include, but are not limited to, paclitaxel and its analog docetaxel.
Paclitaxel, 5 ,20-epoxy-l,2a,4,7 ,10 ,13a-hexa-hydroxytax-l l-en-9-one 4,10- diacetate 2-benzoate 13-ester with (2R,3S)-N-benzoyl-3-phenylisoserine; is a natural
diterpene product isolated from the Pacific yew tree Taxus brevifolia and is commercially available as an injectable solution TAXOL®. It is a member of the taxane family of terpenes. It was first isolated in 1971 by Wani et al. J. Am. Chem, Soc, 93:2325. 1971), who characterized its structure by chemical and X-ray crystallographic methods. One mechanism for its activity relates to paclitaxel's capacity to bind tubulin, thereby inhibiting cancer cell growth. Schiff et al, Proc. Natl, Acad, Sci. USA, 77: 1561-1565 (1980);
Schiff et al, Nature, 277:665-667 (1979); Kumar, J. Biol, Chem, 256: 10435-10441 (1981). For a review of synthesis and anticancer activity of some paclitaxel derivatives see: D. G. I. Kingston et al., Studies in Organic Chemistry vol. 26, entitled "New trends in Natural Products Chemistry 1986", Attaur-Rahman, P.W. Le Quesne, Eds. (Elsevier, Amsterdam, 1986) pp 219-235.
Paclitaxel has been approved for clinical use in the treatment of refractory ovarian cancer in the United States (Markman et al., Yale Journal of Biology and Medicine, 64:583, 1991; McGuire et al, Ann. Intern, Med., 111 :273,1989) and for the treatment of breast cancer (Holmes et al., J. Nat. Cancer Inst., 83: 1797,1991.) It is a potential candidate for treatment of neoplasms in the skin (Einzig et. al., Proc. Am. Soc. Clin. Oncol., 20:46) and head and neck carcinomas (Forastire et. al, Sem. Oncol., 20:56, 1990). The compound also shows potential for the treatment of polycystic kidney disease (Woo et. al, Nature, 368:750. 1994), lung cancer and malaria. Treatment of patients with paclitaxel results in bone marrow suppression (multiple cell lineages, Ignoff, R.J. et. al, Cancer Chemotherapy Pocket Guidei 1998) related to the duration of dosing above a threshold concentration (50nM) (Kearns, CM. et. al, Seminars in Oncology, 3(6) p.16-23, 1995).
Docetaxel, (2R,3S)- N-carboxy-3-phenylisoserine,N-tert-butyl ester, 13-ester with 5 -20-epoxy-l,2a,4,7 ,10 ,13a-hexahydroxytax-l l-en-9-one 4-acetate 2-benzoate, trihydrate; is commercially available as an injectable solution as TAXOTERE®. Docetaxel is indicated for the treatment of breast cancer. Docetaxel is a semisynthetic derivative of paclitaxel q.v., prepared using a natural precursor, 10-deacetyl-baccatin III, extracted from the needle of the European Yew tree. The dose limiting toxicity of docetaxel is neutropenia.
Vinca alkaloids are phase specific anti-neoplastic agents derived from the periwinkle plant. Vinca alkaloids act at the M phase (mitosis) of the cell cycle by binding specifically to tubulin. Consequently, the bound tubulin molecule is unable to polymerize
into microtubules. Mitosis is believed to be arrested in metaphase with cell death following. Examples of vinca alkaloids include, but are not limited to, vinblastine, vincristine, and vinorelbine.
Vinblastine, vincaleukoblastine sulfate, is commercially available as VELBAN® as an injectable solution. Although, it has possible indication as a second line therapy of various solid tumors, it is primarily indicated in the treatment of testicular cancer and various lymphomas including Hodgkin's Disease; and lymphocytic and histiocytic lymphomas. Myelosuppression is the dose limiting side effect of vinblastine.
Vincristine, vincaleukoblastine, 22-oxo-, sulfate, is commercially available as ONCOVIN® as an injectable solution. Vincristine is indicated for the treatment of acute leukemias and has also found use in treatment regimens for Hodgkin's and non-Hodgkin's malignant lymphomas. Alopecia and neurologic effects are the most common side effect of vincristine and to a lesser extent myelosupression and gastrointestinal mucositis effects occur.
Vinorelbine, 3',4'-didehydro -4'-deoxy-C'-norvincaleukoblastine [R-(R*,R*)-2,3- dihydroxybutanedioate (l :2)(salt)], commercially available as an injectable solution of vinorelbine tartrate (NAVELBINE®), is a semisynthetic vinca alkaloid. Vinorelbine is indicated as a single agent or in combination with other chemotherapeutic agents, such as cisplatin, in the treatment of various solid tumors, particularly non-small cell lung, advanced breast, and hormone refractory prostate cancers. Myelosuppression is the most common dose limiting side effect of vinorelbine.
Platinum coordination complexes are non-phase specific anti-cancer agents, which are interactive with DNA. The platinum complexes enter tumor cells, undergo, aquation and form intra- and interstrand crosslinks with DNA causing adverse biological effects to the tumor. Examples of platinum coordination complexes include, but are not limited to, cisplatin and carboplatin.
Cisplatin, cis-diamminedichloroplatinum, is commercially available as
PLATINOL® as an injectable solution. Cisplatin is primarily indicated in the treatment of metastatic testicular and ovarian cancer and advanced bladder cancer. The primary dose limiting side effects of cisplatin are nephrotoxicity, which may be controlled by hydration and diuresis, and ototoxicity.
Carboplatin, platinum, diammine [l,l-cyclobutane-dicarboxylate(2-)-0,0'], is commercially available as PARAPLATIN® as an injectable solution. Carboplatin is primarily indicated in the first and second line treatment of advanced ovarian carcinoma. Bone marrow suppression is the dose limiting toxicity of carboplatin.
Alkylating agents are non-phase anti-cancer specific agents and strong
electrophiles. Typically, alkylating agents form covalent linkages, by alkylation, to DNA through nucleophilic moieties of the DNA molecule such as phosphate, amino, sulfhydryl, hydroxyl, carboxyl, and imidazole groups. Such alkylation disrupts nucleic acid function leading to cell death. Examples of alkylating agents include, but are not limited to, nitrogen mustards such as cyclophosphamide, melphalan, and chlorambucil; alkyl sulfonates such as busulfan; nitrosoureas such as carmustine; and triazenes such as dacarbazine.
Cyclophosphamide, 2-[bis(2-chloroethyl)amino]tetrahydro-2H-l,3,2- oxazaphosphorine 2-oxide monohydrate, is commercially available as an injectable solution or tablets as CYTOXAN®. Cyclophosphamide is indicated as a single agent or in combination with other chemotherapeutic agents, in the treatment of malignant lymphomas, multiple myeloma, and leukemias. Alopecia, nausea, vomiting and leukopenia are the most common dose limiting side effects of cyclophosphamide.
Melphalan, 4-[bis(2-chloroethyl)amino]-L-phenylalanine, is commercially available as an injectable solution or tablets as ALKERAN®. Melphalan is indicated for the palliative treatment of multiple myeloma and non-resectable epithelial carcinoma of the ovary. Bone marrow suppression is the most common dose limiting side effect of melphalan.
Chlorambucil, 4-[bis(2-chloroethyl)amino]benzenebutanoic acid, is commercially available as LEUKERAN® tablets. Chlorambucil is indicated for the palliative treatment of chronic lymphatic leukemia, and malignant lymphomas such as lymphosarcoma, giant follicular lymphoma, and Hodgkin's disease. Bone marrow suppression is the most common dose limiting side effect of chlorambucil.
Busulfan, 1 ,4-butanediol dimethanesulfonate, is commercially available as MYLERAN® TABLETS. Busulfan is indicated for the palliative treatment of chronic myelogenous leukemia. Bone marrow suppression is the most common dose limiting side effects of busulfan.
Carmustine, l,3-[bis(2-chloroethyl)-l-nitrosourea, is commercially available as single vials of lyophilized material as BiCNU®. Carmustine is indicated for the palliative treatment as a single agent or in combination with other agents for brain tumors, multiple myeloma, Hodgkin's disease, and non-Hodgkin's lymphomas. Delayed myelosuppression is the most common dose limiting side effects of carmustine.
Dacarbazine, 5-(3,3-dimethyl-l-triazeno)-imidazole-4-carboxamide, is
commercially available as single vials of material as DTIC-Dome®. Dacarbazine is indicated for the treatment of metastatic malignant melanoma and in combination with other agents for the second line treatment of Hodgkin's Disease. Nausea, vomiting, and anorexia are the most common dose limiting side effects of dacarbazine.
Antibiotic anti-neoplastics are non-phase specific agents, which bind or intercalate with DNA. Typically, such action results in stable DNA complexes or strand breakage, which disrupts ordinary function of the nucleic acids leading to cell death. Examples of antibiotic anti-neoplastic agents include, but are not limited to, actinomycins such as dactinomycin, anthrocyclins such as daunorubicin and doxorubicin; and bleomycins.
Dactinomycin, also know as Actinomycin D, is commercially available in injectable form as COSMEGEN®. Dactinomycin is indicated for the treatment of Wilm's tumor and rhabdomyosarcoma. Nausea, vomiting, and anorexia are the most common dose limiting side effects of dactinomycin.
Daunorubicin, (8S-cis-)-8-acetyl-10-[(3-amino-2,3,6-trideoxy-a-L-lyxo- hexopyranosyl)oxy]-7,8,9, 10-tetrahydro-6,8, 11 -trihydroxy- 1 -methoxy-5, 12
naphthacenedione hydrochloride, is commercially available as a liposomal injectable form as DAUNOXOME® or as an injectable as CERUBIDINE®. Daunorubicin is indicated for remission induction in the treatment of acute nonlymphocytic leukemia and advanced HIV associated Kaposi's sarcoma. Myelosuppression is the most common dose limiting side effect of daunorubicin.
Doxorubicin, (8S, 10S)-10-[(3-amino-2,3,6-trideoxy-a-L-lyxo- hexopyranosyl)oxy]-8-glycoloyl, 7,8,9, 10-tetrahydro-6,8, 11 -trihydroxy- 1 -methoxy-5, 12 naphthacenedione hydrochloride, is commercially available as an injectable form as RUBEX® or ADRIAMYCIN RDF®. Doxorubicin is primarily indicated for the treatment of acute lymphoblastic leukemia and acute myeloblastic leukemia, but is also a useful
component in the treatment of some solid tumors and lymphomas. Myelosuppression is the most common dose limiting side effect of doxorubicin.
Bleomycin, a mixture of cytotoxic glycopeptide antibiotics isolated from a strain of Streptomyces verticillus, is commercially available as BLENOXANE®. Bleomycin is indicated as a palliative treatment, as a single agent or in combination with other agents, of squamous cell carcinoma, lymphomas, and testicular carcinomas. Pulmonary and cutaneous toxicities are the most common dose limiting side effects of bleomycin.
Topoisomerase II inhibitors include, but are not limited to, epipodophyllotoxins. Epipodophyllotoxins are phase specific anti-neoplastic agents derived from the mandrake plant. Epipodophyllotoxins typically affect cells in the S and G2 phases of the cell cycle by forming a ternary complex with topoisomerase II and DNA causing DNA strand breaks. The strand breaks accumulate and cell death follows. Examples of epipodophyllotoxins include, but are not limited to, etoposide and teniposide.
Etoposide, 4'-demethyl-epipodophyllotoxin 9[4,6-0-(R )-ethylidene- -D- glucopyranoside], is commercially available as an injectable solution or capsules as
VePESID® and is commonly known as VP-16. Etoposide is indicated as a single agent or in combination with other chemotherapy agents in the treatment of testicular and non- small cell lung cancers. Myelosuppression is the most common side effect of etoposide. The incidence of leucopenia tends to be more severe than thrombocytopenia.
Teniposide, 4'-demethyl-epipodophyllotoxin 9[4,6-0-(R )-thenylidene- -D- glucopyranoside], is commercially available as an injectable solution as VUMON® and is commonly known as VM-26. Teniposide is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia in children.
Myelosuppression is the most common dose limiting side effect of teniposide. Teniposide can induce both leucopenia and thrombocytopenia.
Antimetabolite neoplastic agents are phase specific anti-neoplastic agents that act at S phase (DNA synthesis) of the cell cycle by inhibiting DNA synthesis or by inhibiting purine or pyrimidine base synthesis and thereby limiting DNA synthesis. Consequently, S phase does not proceed and cell death follows. Examples of antimetabolite anti-neoplastic agents include, but are not limited to, fluorouracil, methotrexate, cytarabine,
mecaptopurine, thioguanine, and gemcitabine.
5-fluorouracil, 5-fluoro-2,4- (1H,3H) pyrimidinedione, is commercially available as fluorouracil. Administration of 5-fluorouracil leads to inhibition of thymidylate synthesis and is also incorporated into both RNA and DNA. The result typically is cell death. 5-fluorouracil is indicated as a single agent or in combination with other chemotherapy agents in the treatment of carcinomas of the breast, colon, rectum, stomach and pancreas. Myelosuppression and mucositis are dose limiting side effects of 5- fluorouracil. Other fluoropyrimidine analogs include 5-fluoro deoxyuridine (floxuridine) and 5-f uorodeoxyuridine monophosphate.
Cytarabine, 4-amino-l-P-D-arabinofuranosyl-2 (lH)-pyrimidinone, is
commercially available as CYTOSAR-U® and is commonly known as Ara-C. It is believed that cytarabine exhibits cell phase specificity at S-phase by inhibiting DNA chain elongation by terminal incorporation of cytarabine into the growing DNA chain.
Cytarabine is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia. Other cytidine analogs include 5-azacytidine and 2', 2 '-difluorodeoxy cytidine (gemcitabine). Cytarabine induces leucopenia, thrombocytopenia, and mucositis.
Mercaptopurine, l,7-dihydro-6H-purine-6-thione monohydrate, is commercially available as PURINETHOL®. Mercaptopurine exhibits cell phase specificity at S-phase by inhibiting DNA synthesis by an as of yet unspecified mechanism. Mercaptopurine is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia. Myelosuppression and gastrointestinal mucositis are expected side effects of mercaptopurine at high doses. A useful mercaptopurine analog is azathioprine.
Thioguanine, 2-amino-l,7-dihydro-6H-purine-6-thione, is commercially available as TABLOID®. Thioguanine exhibits cell phase specificity at S-phase by inhibiting DNA synthesis by an as of yet unspecified mechanism. Thioguanine is indicated as a single agent or in combination with other chemotherapy agents in the treatment of acute leukemia. Myelosuppression, including leucopenia, thrombocytopenia, and anemia, is the most common dose limiting side effect of thioguanine administration. However, gastrointestinal side effects occur and can be dose limiting. Other purine analogs include pentostatin, erythrohydroxynonyladenine, fludarabine phosphate, and cladribine.
Gemcitabine, 2'-deoxy-2', 2'-difluorocytidine monohydrochloride (β-isomer), is commercially available as GEMZAR®. Gemcitabine exhibits cell phase specificity at S- phase and by blocking progression of cells through the Gl/S boundary. Gemcitabine is indicated in combination with cisplatin in the treatment of locally advanced non-small cell lung cancer and alone in the treatment of locally advanced pancreatic cancer.
Myelosuppression, including leucopenia, thrombocytopenia, and anemia, is the most common dose limiting side effect of gemcitabine administration.
Methotrexate, N-[4[[(2,4-diamino-6-pteridinyl) methyljmethylamino] benzoyl]-L- glutamic acid, is commercially available as methotrexate sodium. Methotrexate exhibits cell phase effects specifically at S-phase by inhibiting DNA synthesis, repair and/or replication through the inhibition of dyhydrofolic acid reductase which is required for synthesis of purine nucleotides and thymidylate. Methotrexate is indicated as a single agent or in combination with other chemotherapy agents in the treatment of
choriocarcinoma, meningeal leukemia, non-Hodgkin's lymphoma, and carcinomas of the breast, head, neck, ovary and bladder. Myelosuppression (leucopenia, thrombocytopenia, and anemia) and mucositis are expected side effect of methotrexate administration.
Camptothecins, including, camptothecin and camptothecin derivatives are available or under development as Topoisomerase I inhibitors. Camptothecins cytotoxic activity is believed to be related to its Topoisomerase I inhibitory activity. Examples of camptothecins include, but are not limited to irinotecan, topotecan, and the various optical forms of 7-(4-methylpiperazino-methylene)-10,l l-ethylenedioxy-20-camptothecin described below.
Irinotecan HC1, (4S)-4,1 l-diethyl-4-hydroxy-9-[(4-piperidinopiperidino) carbonyloxy]-lH-pyrano[3 ',4',6,7]indolizino[l ,2-b]quinoline-3, 14(4H, 12H)-dione hydrochloride, is commercially available as the injectable solution CAMPTOSAR®.
Irinotecan is a derivative of camptothecin which binds, along with its active metabolite SN-38, to the topoisomerase I - DNA complex. It is believed that cytotoxicity occurs as a result of irreparable double strand breaks caused by interaction of the topoisomerase I : DNA : irintecan or SN-38 ternary complex with replication enzymes. Irinotecan is indicated for treatment of metastatic cancer of the colon or rectum. The dose limiting side effects of irinotecan HC1 are myelosuppression, including neutropenia, and GI effects, including diarrhea.
Topotecan HC1, (S)- 10-[(dimethylamino)methyl]-4-ethyl-4,9-dihydroxy- 1 H- pyrano[3 ' ,4 ' ,6,7]indolizino[ 1 ,2-b]quinoline-3 , 14-(4H, 12H)-dione monohydrochloride, is commercially available as the injectable solution HYCAMTIN®. Topotecan is a derivative of camptothecin which binds to the topoisomerase I - DNA complex and prevents religation of singles strand breaks caused by Topoisomerase I in response to torsional strain of the DNA molecule. Topotecan is indicated for second line treatment of metastatic carcinoma of the ovary and small cell lung cancer. The dose limiting side effect of topotecan HC1 is myelosuppression, primarily neutropenia.
Pharmaceutical compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose. Such a unit may contain, for example, 0.5 mg to 1 g, preferably 1 mg to 700 mg, more preferably 5 mg to 100 mg of a compound of the formula (I), depending on the condition being treated, the route of administration and the age, weight and condition of the patient, or pharmaceutical compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose. Preferred unit dosage compositions are those containing a daily dose or sub-dose, as herein above recited, or an appropriate fraction thereof, of an active ingredient. Furthermore, such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
Pharmaceutical compositions may be adapted for administration by any
appropriate route, for example by the oral (including buccal or sublingual), rectal, nasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal) route. Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association a compound of formal (I) with the carrier(s) or excipient(s).
Pharmaceutical compositions adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths. Glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation. A disintegrating or solubilizing agent such as agar-agar,
calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
Moreover, when desired or necessary, suitable binders, lubricants, disintegrating agents and coloring agents can also be incorporated into the mixture. Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate,
carboxymethylcellulose, polyethylene glycol, waxes and the like. Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like. Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like. Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets. A powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate. The powder mixture can be granulated by tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil. The lubricated mixture is then compressed into tablets. The compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps. A clear or opaque protective coating consisting of a sealing coat of shellac, a coating of sugar or polymeric material and a polish coating of wax can be provided. Dyestuffs can be added to these coatings to distinguish different unit dosages.
Oral fluids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of a compound of formula (I). Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
Where appropriate, dosage unit pharmaceutical compositions for oral
administration can be microencapsulated. The formulation can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
Pharmaceutical compositions adapted for rectal administration may be presented as suppositories or as enemas.
Pharmaceutical compositions adapted for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
Pharmaceutical formulations adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the composition isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. The pharmaceutical compositions may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
It should be understood that in addition to the ingredients particularly mentioned above, the pharmaceutical compositions may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
A therapeutically effective amount of a compound of the present invention will depend upon a number of factors including, for example, the age and weight of the intended recipient, the precise condition requiring treatment and its severity, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant prescribing the medication. However, an effective amount of a compound of formula (I) for the treatment of anemia will generally be in the range of 0.001 to 100 mg/kg body weight of recipient per day, suitably in the range of .01 to 10 mg/kg body weight per day. For a 70kg adult mammal, the actual amount per day would suitably be from 7 to 700 mg and this amount may be given in a single dose per day or in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same. An effective amount of a salt or solvate, etc., may be determined as a proportion
of the effective amount of the compound of formula (I) per se. It is envisaged that similar dosages would be appropriate for treatment of the other conditions referred to above.
Chemical Background
The compounds of this invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention as prepared are given in the examples.
Compounds of general formula (I) may be prepared by methods known in the art of organic synthesis as set forth in part by the following synthesis schemes. In all of the schemes described below, it is well understood that protecting groups for sensitive or reactive groups are employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Green and P. G. M. Wuts (1991) Protecting Groups in Organic Synthesis, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection of processes as well as the reaction conditions and order of their execution shall be consistent with the preparation of compounds of formula (I). Those skilled in the art will recognize if a stereocenter exists in compounds of formula (I). Accordingly, the present invention includes both possible stereoisomers and includes not only racemic compounds but the individual enantiomers as well. When a compound is desired as a single enantiomer, it may be obtained by stereospecific synthesis or by resolution of the final product or any convenient intermediate. Resolution of the final product, an intermediate, or a starting material may be effected by any suitable method known in the art. See, for example, Stereochemistry of Organic Compounds by E. L. Eliel, S. H. Wilen, and L. N. Mander (Wiley-Interscience, 1994).
EXAMPLES
General Experimental Methods
The following abbreviations are used throughout the experimental and have the following meaning:
aq aqueous
BINAP 2,2'-bis(diphenylphosphino) -Ι, -binapthyl
ca. circa
CDCl3-<i chloroform-<i
Cs2C03 cesium carbonate
CHC13 chloroform
ACN acetonitrile
CH3CN acetonitrile
Celite® registered trademark of Celite Corp. brand of diatomaceous earth DBU l,8-diazabicyclo[5.4.0]undeca-7-ene
DCE dichloroethane
DCM methylene chloride
DME 1 ,2 dimethoxyethane
DMF Ν,Ν-dimethyl formamide
DIEA diisopropyl ethylamine
DMSO-< 6 dimethylsulfoxide-<i6
EtOAc ethyl acetate
EDC 1 -(3 -dimethylaminopropy l)-3 -ethylcarbodimmide hydrochloride h hour(s)
1H NMR proton nuclear magnetic resonance
HC1 hydrochloric acid
HO AT l-hydroxy-7-azabenzotriazole
HPLC high performance liquid chromatography
IPA 2-propanol
K2C03 potassium carbonate
KOH potassium hydroxide
LC/MS liquid chromatography/mass spectroscopy
MgS04 magnesium sulfate
MeOH methanol
min minute(s)
MTBE methyl tert-butyl ether
MS mass spectrometry
NaOH sodium hydroxide
Na2S04 sodium sulfate
NH4OH ammonium hydroxide
NMM 4-methylmorpholine
NMP N-methyl-2-pyrrolidone
Pd/C palladium (10% by wt) on carbon
PdCl2(dppf)-CH2Cl2 1 , 1 '-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex
Pd(Ph3P)4 tetrakis(triphenylphosphine)palladium(0)
SOCl2 thionyl chloride
SPhos 2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl
TFA trifluoroacetic acd
THF tetrahydrofuran
TLC thin layer chromatography
The following guidelines apply to all experimental procedures described herein. All reactions were conducted under a positive pressure of nitrogen using oven-dried glassware, unless otherwise indicated. Temperatures designated are external (i.e. bath temperatures), and are approximate. Air and moisture-sensitive liquids were transferred via syringe. Reagents were used as received. Solvents utilized were those listed as "anhydrous" by vendors. Molarities listed for reagents in solutions are approximate, and were used without prior titration against a corresponding standard. All reactions were agitated by stir bar, unless otherwise indicated. Heating was conducted using heating baths containing silicon oil, unless otherwise indicated. Reactions conducted by microwave irradiation (0 - 400 W at 2.45 GHz) were done so using a Biotage Initiator™ 2.0 instrument with Biotage microwave EXP vials (0.2 - 20 mL) and septa and caps. Irradiation levels utilized (i.e. high, normal, low) based on solvent and ionic charge were based on vendor specifications. Cooling to temperatures below -70 °C was conducted using dry ice/acetone or dry ice/2-propanol. Magnesium sulfate and sodium sulfate used as drying agents were of anhydrous grade, and were used interchangeably. Solvents
described as being removed "in vacuo " or "under reduced pressure" were done so by rotary evaporation.
Preparative normal phase silica gel chromatography was carried out using either a Teledyne ISCO CombiFlash Companion instrument with RediSep or ISCO Gold silica gel cartridges (4 g-330 g), or an Analogix IF280 instrument with SF25 silica gel cartridges (4 g - 3-00g), or a Biotage SP1 instrument with HP silica gel cartridges (lOg - 100 g).
Purification by reverse phase HPLC was conducted using a YMC-pack column (ODS-A 75x30mm) as solid phase, unless otherwise noted. A mobile phase of 25mL/min A (acetonitrile-0.1%TFA) : B (water-0.1% TFA), 10-80% gradient A (10 min) was utilized, with UV detection at 214 nM, unless otherwise noted.
A PE Sciex API 150 single quadrupole mass spectrometer (PE Sciex, Thornhill, Ontario, Canada) was operated using electrospray ionization in the positive ion detection mode. The nebulizing gas was generated from a zero air generator (Balston Inc.,
Haverhill, MA, USA) and delivered at 65 psi and the curtain gas was high purity nitrogen delivered from a Dewar liquid nitrogen vessel at 50 psi. The voltage applied to the electrospray needle was 4.8 kV. The orifice was set at 25 V and mass spectrometer was scanned at a rate of 0.5 scan/sec using a step mass of 0.2 amu and collecting profile data.
Method A LCMS. Samples were introduced into the mass spectrometer using a CTC PAL autosampler (LEAP Technologies, Carrboro, NC) equipped with a hamilton 10 uL syringe which performed the injection into a Valco 10-port injection valve. The HPLC pump was a Shimadzu LC-lOADvp (Shimadzu Scientific Instruments, Columbia, MD) operated at 0.3 mL/min and a linear gradient 4.5% A to 90% B in 3.2 min. with a 0.4 min. hold. The mobile phase was composed of 100% (H20 0.02%> TFA) in vessel A and 100%) (CH3CN 0.018% TFA) in vessel B. The stationary phase is Aquasil (CI 8) and the column dimensions were 1 mm x 40 mm. Detection was by UV at 214 nm, evaporative light- scattering (ELSD) and MS.
Method B, LCMS. Alternatively, an Agilent 1100 analytical HPLC system with an LC/MS was used and operated at 1 mL/min and a linear gradient 5% A to 100% B in 2.2 min with a 0.4 min hold. The mobile phase was composed of 100% (H20 0.02% TFA) in vessel A and 100% (CH3CN 0.018% TFA) in vessel B. The stationary phase was Zobax (C8) with a 3.5 um partical size and the column dimensions were 2.1 mm x 50 mm.
Detection was by UV at 214 nm, evaporative light-scattering (ELSD) and MS.
Method C, LCMS. Alternatively, an MDSSCIEX API 2000 equipped with a capillary column of (50 x 4.6 mm, 5 μτα) was used. HPLC was done on Agilent-1200 series UPLC system equipped with column Zorbax SB-C18 (50 x 4.6 mm, 1.8 μτα) eluting with CH3CN: ammonium acetate buffer. The reactions were performed in the microwave (CEM, Discover).
1H-NMR spectra were taken in deuterated DMSO (unless otherwise noted) and recorded at 400 MHz using a Bruker AVANCE 400 MHz instrument, with ACD Spect manager v. 10 used for reprocessing. Multiplicities indicated are: s=singlet, d=doublet, t=triplet, q=quartet, quint= quintet, sxt= sextet, m=multiplet, dd = doublet of doublets, dt=doublet of triplets etc. and br indicates a broad signal.
Analytical HPLC: Products were analyzed by Agilent 1100 Analytical Chromatography system, with 4.5 x 75 mm Zorbax XDB-C18 column (3.5 um) at 2 mL/min with a 4 min gradient from 5% CH3CN (0.1% formic acid) to 95% CH3CN (0.1% formic acid) in H20 (0.1% formic acid) and a 1 min hold.
The compounds of the present invention were prepared according to the following schemes 1-4 described in detail below. The groups and substituents shown in the schemes 1-4, such as X, Y, Z and the various R groups have the same definition in what follows as they have herein above. The solvents and conditions referred to are illustrative and are not intended to be limiting.
Scheme 1

Scheme 1 illustrates two methods to synthesize a compound of formula (VII). Substituted aminopyrazoles of formula (I) are heated with diethyl oxobutanedione in benzene or toluene at 62 °C overnight. Treatment of the putative intermediate with acetic acid and typically heating at reflux furnishes azaindazole compounds of formula (II). Compounds of formula (II) are converted to compounds of formula (III) by base-catalyzed hydrolysis of the ethyl ester and then chlorination of the putative carboxylic acid intermediate with POCl3 under standard conditions to afford compounds of formula (IV). Treatment of compounds of formula (IV) with substituted aminomethyl pyridones of formula (V) using EDC, HO AT, N-methylmorpholine, and DMSO for a period of no less than 12 h stirring typically at room temperature (in some instances, heating at 40 °C may be required), affords compounds of formula (VI). Compounds of formula (VI) are
substituted at the 6-position using standard methods known to those skilled in the art (i.e. nucleophillic substitution, palladium mediated cross couplings), to afford compounds of formula (VII). Alternatively, compounds of formula (VII) can be obtained from a compound of formula (II) via compounds (II a) and (IVa). In this route, compounds of formula (II) are converted to the corresponding triflate (Ila) using standard methods. Compounds of formula (Ila) are then substituted at the 6-pos. using standard palladium mediated cross-coupling conditions, followed by base-catalyzed hydrolysis of the ethyl ester group to afford compounds of formula (IVa). Treatment of compounds of formula (IVa) with substituted aminomethyl pyridones of formula (V) using EDC, HO AT, N- methylmorpholine, and DMSO at room temperature for a period of no less than 12 h stirring at room temperature affords compounds of formula (VII).
Scheme 2

Compounds of formula (VII) are also prepared as depicted in scheme 2. In this embodiment, substituted oxobutanediones of formula (VIII) (prepared by Claisen condensation between an appropriately substitutued ketone and diethyloxalate) are heated with substituted aminopyrazoles of formula (I) (as described for scheme 1), to afford compounds of formula (IX). Compounds of formula (IX) are converted to compounds of formula (X) by base-catalyzed hydrolysis. Substitutions respectively at either the R3-
position, or on the R6 substituted group of compounds of formulas (IX) and (X) are done so using methods known to those skilled in the art (e.g. bromination, nitration). Treatment of compounds of formula (X) with substituted aminomethyl pyridones of formula (V) using EDC, HO AT, N-methylmorpholine, and DMSO at room temperature for a period no less than 12 h stirring at room temperature, affords compounds of formula (VII).
Scheme 3

Compounds of formula (VII) are also prepared as depicted in scheme 3. In this embodiment, substituted aminopyrazoles of formula (I) are heated with a keto-ester of formula (XI) in benzene containing catalytic acetic acid at 62 °C overnight. Exposure of the putative intermediate to refluxing Dowtherm A overnight affords a compound of formula (XII). Compounds of formula (XI) that are not commercially available are prepared using standard methods known to those skilled in the art, and are described herein. Compounds of formula (XII) are converted to compounds of formula (XIII) by bromination with refluxing POBr3 in toluene/DMF for lh. Heating of compounds of formula (XIII) with dicyano zinc, tris(dibenzylideneacetone)dipalladium(0), and SPhos in DMFand water at 120° Cfor 2 hours furnishes compounds of formula (XIV). The compounds of formula (XIV) are hydrolyzed to the compounds of formula (X) using standard base-catalyzed hydrolysis conditions.
Compounds of formula (X) are treated with substituted aminomethyl pyridones of formula (V) using EDC, HO AT, N-methylmorpholine, and DMSO at room temperature for a period no less than 12 h stirring at room temperature, to afford compounds of formula (VII).
Scheme 4

Scheme 4 illustrates the method to synthesize a compound of formula (V).
Heating of compounds of formula (XV) with cyanoacetamide in ethanol at reflux containing catalytic piperidine for typically 30 min, affords compounds of formula (XVII). Alternatively, treatment of compounds of formula (XVI) with cyanoacetamide in DMSO at room temperature with an excess of potassium tert-butoxide under an atmosphere of oxygen for ca. 90 min. also affords compounds of formula (XVII). Regioisomeric mixtures, are separable and the individual compounds regiochemical assignments were confirmed by 2D HNMR techniques. Compounds of formula (XV) and (XVI) which are not commercially available are prepared using standard methods known to those skilled in the art, and are described herein. Compounds of formula (XVII) can be converted to compounds of formula (V) either by hydrogenation using sodium acetate, palladium on carbon, and platinum oxide, or reduction conditions using NaBH4 with either iodine or NiCl2-6H20.
Examples
Intermediate 1
6-Hydroxy-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

l-(l-methylethyl)-lH-pyrazol-5-amine

To a solution of ethyl (2Z)-2-cyano-3-(ethyloxy)-2-propenoate (114.2 g, 0.67 mol) in ethanol (250 mL) was slowly added isopropylhydrazine (55 g, 0.74 mol) in a dropwise manner. The mixture was heated at reflux for 4 h, and then cooled to room temperature. The mixture was concentrated in vacuo. Approximately half of the crude 5 -amino- 1- isopropyl-lH-pyrazole-4-carboxylic acid ethyl ester (50 g) was suspended in an aqueous solution of sodium hydroxide (4M, 130 mL), and stirred with heating at reflux for 2 h. The reaction mixture was then cooled to room temperature and adjusted to pH=3.5 with concentrated HCl, wherein precipitate formation ensued. The solid was collected by filtration and dried in vacuum oven overnight to afford crude 5-amino-l-isopropyl-lH- pyrazole-4-carboxylic acid (30 g). The solid was suspended in diphenyl ether (120 mL) and stirred with heating at 160-165 °C for 2 h. The solution was then cooled to room temperature and the solvent removed in vacuo. The crude product was purified by silica gel chromatography (eluent: petroleum ether/EtOAc=l : l) to afford the product as 12 g. 1H NMR (400 MHz, DMSO-<¾) δ ppm 1.24 (d, 6H, J= 6.4 Hz), 4.32 (m, 1H), 5.06 (s, 2H), 5.21 (s, 1H), 7.00 (s, 1H).
Step 2:
To a solution of diethyl 2-oxobutanedione (96 g) in toluene (500 mL) was added 1-
(l-methylethyl)-lH-pyrazol-5-amine (30 g, 0.24 mol) and the mixture was stirred at 60 °C overnight. The mixture was concentrated in vacuo, the crude residue dissolved into acetic acid (500 mL), and then heated at reflux for 2 h. The mixture was then cooled to room temperature and concentrated in vacuo to give a residue, which was recrystallized from DCM to afford the product as a yellow colored solid, collected as 40 g. This solid was suspended in in ethanol (700 mL) and THF (100 mL), followed by addition of 3 M NaOH (150 mL). The reaction mixture was stirred at 40 °C for 40 min. The mixture was concentrated in vacuo to remove the volatiles, and the aqueous layer then acidified using 1M HCl. The resulting precipitate was collected by filtration and dried under high vacuum to give the title compound, 6-hydroxy-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-
carboxylic acid, as 27 g. LCMS E-S (M+H) = 222.0. 1H NMR (400 MHz, DMSO-d6) δ ppm l .48 (d, J=6.82 Hz, 6 H), 4.89 - 4.96 (m, 1H), 6.81 (s, 1H), 8.13 (s, 1H).
Intermediate 2
3-Methyl-l-(l-methylethyl)-6-oxo-6,7-dihydro-lH-pyrazolo[3,4-6]pyridine-4- carboxylic acid

Step 1:
3-Methyl-l-(l-methylethyl)-lH-pyrazol-5-amine

3-Amino-2-butenenitrile (33.3 g, 0.41 mol) and ethanol (170 mL) were combined and stirred at room temperature for 30 min., after which time isopropylhydrazine (50 g, 0.67 mol) was added at once. After stirring at room temperature for 5 min., the contents were then heated at reflux for 10 h. After cooling to room temperature, the mixture was concentrated in vacuo to give the desired product (85 g) which was used in the next step directly. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.21 (d, 6 H, J =6.4 Hz), 1.94 (s, 3H), 4.21 (m, 1H), 4.93 (s, 2H), 5.02 (s, 1H).
Step 2:
To a solution of diethyl 2-oxobutanedione (176 g, 0.94 mol) in toluene (2 L) was added 3 -methyl- l-(l-methylethyl)-lH-pyrazol-5 -amine (82.5 g, 0.59 mol), and the mixture stirred at 62 °C, overnight. After cooling to room temperature, the mixture was concentrated in vacuo and the crude residue dissolved into acetic acid (1.5 L). The mixture was heated at reflux for 2 h. After cooling to room temperature, the mixture was concentrated in vacuo to afford a solid residue, which was recrystallized from DCM to afford the desired product as a yellow colored solid. The collected solid was suspended in
ethanol (1510 niL) and THF (216 mL) followed by addition of 3N NaOH (334 mL) and the reaction mixture was stirred at 40 °C for 40 min. The mixture was concentrated in vacuo to remove the volatiles and the aqueous phase acidified using IN HC1. The resulting precipitate was collected by filtration and dried under high vacuum to give the title compound, 3-methyl-l-(l-methylethyl)-6-oxo-6,7-dihydro-lH-pyrazolo[3,4-¾]pyridine-4- carboxylic acid, as 51.38 g. LCMS E-S (Μ+Η) = 236.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.35 (d, J=6.8 Hz, 6 H), 2.41 (s, 3H), 4.84-4.91 (m, 1H), 6.64 (s, 1H). Carboxylic acid proton not observed.
Intermediate 3
Ethyl l-(l,l-dimethylethyl)-6-hydroxy-3-methyl-lH-pyrazolo[3,4-6]pyridine-4- carboxylate

l-(l,l-Dimethylethyl)-3 -methyl- lH-pyrazol-5 -amine (5 g, 32.6 mmol), diethyl 2- oxobutanedione (6.14 g, 32.6 mmol) and toluene (100 mL) were heated at 70 °C for 16 hours. The solvent was removed in vacuo, the crude residue dissolved in acetic acid (100 mL), and heated at reflux for 4 hours. The solvent was removed in vacuo, and the crude product purified via silica gel chromatography (eluent: gradient of 0 to 10%
EtOAc/Hexanes). The product was collected as a solid, 6.32 g (70%). LCMS E-S (M+H) = 278.4. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.39 (s, 1H), 6.82 (s, 1H), 4.36 (q, 2H, j = 7.2 Hz), 2.45 (s, 3H), 1.69 (s, 9H), 1.32 (t, 3H, j = 7.2 Hz).
Intermediate 4
l-(l,l-Dimethylethyl)-6-hydroxy-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

Sodium hydroxide (52.6 mL, 52.6 mmol) was added to an EtOH solution (100 mL) of ethyl l-(l,l-dimethylethyl)-3-methyl-6-oxo-6,7-dihydro-lH-pyrazolo[3,4-b]pyridine-4- carboxylate (7.3 g, 26.3 mmol) and stirred at room temperature for 16 h. The solvent was removed in vacuo. The crude residue was suspended in water, and extracted with EtOAc. The combined organic layers were washed with water, dried over MgSC^, filtered, and concentrated in vacuo. The solid product obtained was set aside. The aqueous phase was concentrated in vacuo and the crude product purified by reverse phase HPLC (mobile phase : 20 -50% ACN/H20, 0.1%TFA) to afford additional product. The combined products were collected as a solid, 5.76 g (88%). LCMS E-S (M+H) = 250.3. 1H NMR (400 MHz, DMSO- 6) 6 ppm l3.5 - 13.9 (br s, 1H), 11.2 - 11.5 (br s, 1H), 6.78 (s, 1H), 2.46 (s, 3H), 1.70 (s, 9H).
Intermediate 5
6-Chloro-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

To a 75 mL pressure vessel was added l-(l-methylethyl)-6-oxo-6,7-dihydro-lH- pyrazolo[3,4-b]pyridine-4-carboxylic acid (4.12 g, 18.62 mmol), followed by phosphorous oxychloride (26.0 ml, 279 mmol). The flask was sealed and the stirring mixture heated at ca. 105 °C for ca. 18 h. After cooling to room temperature, the contents were concentrated in vacuo to remove most of volatiles. The residual contents were poured into a mixture of
ice and 3M NaOH (60 mL), followed by additional 3M NaOH to ensure the pH stayed basic. The mixture was stirred for 30 min., and then cooled in an ice bath. The heterogenous mixture was slowly acidified to pH = 3-4 with 6M HC1. The resulting suspension was extracted with EtOAc (3x). The combined organic layers were dried over MgS04, filtered, and concentrated in vacuo to afford a yellow solid which was dried in a hi-vac oven overnight. The title compound was collected as 4.11 g (90%), and used without further purification. LCMS E-S (M+H) = 240.2/242.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.51 (d, J=6.57 Hz, 6 H) 5.17 (quin, J=6.63 Hz, 1 H) 7.65 (s, 1 H) 8.41 (s, 1 H) 14.25 (br. s., 1 H).
Intermediate 6
6-Chloro-3-methyl-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

3 -Methyl- 1 -(1 -methylethyl)-6-oxo-6,7-dihydro- lH-pyrazolo[3,4-b]pyridine-4- carboxylic acid (1.5 g, 6.38 mmol) was added to a solution of THF (15 mL) followed by addition of phosphorous oxychloride (8.9 mL, 96 mmol), and the contents heated at 105 °C overnight. After cooling to room temperature, the contents were concentrated in vacuo to remove most of the volatiles. The residual contents were slowly poured into a solution of iced water and IN NaOH (10 mL), and the contents were stirred at room temperature for 24 h, during which time solid precipitation ensued. Additional IN NaOH was added, upon which the solids went into solution. After stirring at room temperature for an additional 30 min., the contents were cooled in an ice bath and the mixture slowly acidified to pH = 3-4 by slow addition of 6N HC1, to afford a heterogenous mixture. The mixture was filtered and a white solid set aside. The aq. layer was further extracted with EtOAc and DCM. The combined organic layers dried were dried over MgS0 , filtered and concentrated in vacuo. The resultant light brown solid was triturated in EtOAc/EtOH (1 : 1) and filtered to afford a first crop of white solid product, which was set aside. The filtrate was again concentrated in vacuo. The residue was diluted with EtOAc, sonicated, treated with hexanes, and filtered. The process was repeated. The isolated solid product
crops were dried under vacuum (3 h) and collected as 1.33 g (80%). LCMS E-S (M+H) :
254.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.46 (d, 6 H), 2.60 (s, 3 H), 5.10 (quin,
J=6.63 Hz, 1 H), 7.50 (s, 1 H), 14.17 (br. s., 1 H).
Intermediate 7
6-Chloro-l-(l,l-dimethylethyl)-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic
acid

l-(l,l-Dimethylethyl)-3-methyl-6-oxo-6,7-dihydro-lH-pyrazolo[3,4-b]pyridine-4- carboxylic acid (5.07 g, 20.34 mmol) and phosphorus oxychloride (28.4 ml, 305 mmol) were heated at 100 °C for 16 hours. The contents were concentrated in vacuo. The residue was added to ice water followed by IN NaOH until basic (pH > 10). After stirring for 15 minutes, the mixture was adjusted to pH 3-4 by addition of IN HCl. The contents were extracted with EtOAc, then washed with water, brine and concentrated in vacuo. The crude product was purified by reverse phase HPLC (40 - 70% ACN/H20, 0.1 %TFA). The product was collected as a solid, 0.50 g (9%). LCMS E-S (M+H) =268.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.73 (s, 9 H), 2.56 (s, 3 H), 7.48 (s, 1 H), 14.17 (br. s., 1 H).
Intermediate 8
3-(Aminomethyl)-4,6-dim pyridinone hydrochloride

Palladium on carbon (10%) (3.24 g) was charged into a 2L dry Parr bottle and a small amount of acetic acid was added. Next added 4,6-dimethyl-2-oxo-l,2-dihydro- pyridine- 3-carbonitrile (30 g , 202.7 mmol), sodium acetate (30.75 g, 375.0 mmol), platinum oxide (0.218 g), and acetic acid (1 L).. The bottle was capped, placed on Parr apparatus, and shaken under an atmosphere of H2 (100 psi) for 2 days. The reaction mixture was filtered.
The solvent was removed to give a residue, which was treated with 150 mL of cone. HCl,
and the formed solids were filtered. The yellow filtrate was concentrated . To the crude compound was added 30 mL of cone. HCl and 150 mL EtOH, the contents cooled to 0 °C, and stirred at 0 °C for 2h. The formed solids were filtered, washed with cold EtOH, ether, and dried. The product was collected as 36 g. This batch was combined with other batches prepared on smaller scales and triturated with ether to give 51 g of pure compound.. 1H NMR (400 MHz, DMSO- 6) δ ppm 11.85 (br s,l H) 8.13 (br s, 3 H) 5.93 - 6.01 (m, 1 H) 3.72 - 3.80 (m, 2 H) 2.22 (s, 3 H) 2.16 (s, 3 H).
Intermediate 9
3-(Aminomethyl)-6-meth -4-(trifluoromethyl)-2(lH)-pyridinone

To a dried 500 mL Parr bottle equipped with nitrogen inlet were placed sodium acetate (1.502 g, 18.30 mmol), 10% palladium on carbon (1.579 g, 0.742 mmol), platinum(IV) oxide (0.011 g, 0.049 mmol) and a small amount of acetic acid to wet the catalysts under nitrogen stream. Next added 2-hydroxy-6-methyl-4-(trifluoromethyl)-3- pyridinecarbonitrile (2.0g, 9.89 mmol) followed by acetic acid (175 mL) while under nitrogen atmosphere. The contents were sealed, placed on a Parr shaker, and reacted at 40 psi of H2 for ca. 6 hr., keeping the H2 psi between 20 and 40 psi (vessel was refilled twice). The vessel was purged with nitrogen and the reaction mixture filtered through Celite, and the filter pad was further washed with a small amount of acetic acid. The volatiles were removed in vacuo to afford a residue, which was dried under hi-vacuum for 45 min. The solid was suspended in cone. HCl (12 mL), stirred, and filtered (removed NaCl). The clear filtrate was concentrated in vacuo and the residue dried under hi- vacuum. The collected solid was suspended in cone. HCl (2 mL) and diluted with EtOH (13 mL). The contents were agitated (i.e. spatula) and stored at ca. 0 °C (i.e. freezer) for 30 min to give a white solid. The solid was filtered and washed with cold ethanol (5 mL). The solid was filtered and dried in vacuum oven for 1 h. The final product was collected as 0.95 g (40%). LCMS E-S (M+H) = 206.9. 1H NMR (400 MHz, DMSO- 6) δ ppm 2.31 (s, 3 H), 3.87 (d, J=5.05 Hz, 2 H), 6.41 (s, 1H), 8.12 - 8.37 (m, 3 H).
Intermediate 10
3-(Aminomethyl)-4-cyclohexyl-6-methyl-2(lH)-pyridinone

Step 1
To a stirred suspension of CrCl2 (58 g, 472.8 mmol in THF (1500 mL) was added a
THF solution (500 mL) of l,l-dichloro-2-propanone ( 10 g, 78.8 mmol) and
cyclohexanecarbaldehyde (8.84 g, 78.8 mmol). The reaction mixture was heated at reflux for 2 h, and then quenched by the addition of 1.0 M HCl. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo. The crude residue ( 10 g) was added to a solution of DMSO (150 mL) containing t-BuOK (7.5 g, 65.7 mmol), and cyanoacetamide (6.1 g, 72.3 mmol) and stirred at room temperature for 30 min.
Additional t-BuOK (22.5 g, 197.1 mmol) was added and the reaction mixture was stirred under an atmosphere of oxygen for an additional lh. The contents were purged with argon, diluted with 4 volumes of H20, and then 5 volumes of 4 N HCl, which were added slowly. The reaction mixture was filtered, washed with water and dried to give 4- cyclohexyl-6-methyl-2-oxo-l,2-dihydro-3-pyridinecarbonitrile as 4.5 g (32%). 1H NMR (400 MHz, DMSO-de) δ ppm 6.25 (s, 1H), 2.61-2.65 (m, 1H), 2.22 (s, 3H), 1.66-1.79 (m, 4H), 1.24-1.46 (m, 6H). Step 2
To an ice-bath cooled THF (100 mL) solution of the product from step 1 (2 g, 9.26 mmol) were added NaBH4 (0.81 g, 21.3mmol), and I2 (2.3 g, 9.26 mmol), and the mixture stirred for 30 min. The reaction mixture was then heated at reflux for 3h, and then allowed to cool to room temperature. After cooling to 0 °C, the reaction mixture was acidified by slow addition of 3N HCl (ImL). The reaction mixture was concentrated in vacuo and the crude product purified by reverse phase HPLC to give the title compound as a solid (TFA salt), 0.5 g (25%). LCMS E-S (M+H) = 221.1. 1H NMR (400 MHz, DMSO-d6) δ ppm
1 1.8 - 1 1.9 (br s, 1H), 7.80-7.93 (br s, 3H), 6.07 (s, 1H), 3.69 (s, 2H), 2.67-2.75 (m, 1H), 2.17 (s, 3H), 1.58-1.72 (m, 5H), 1.19 - 1.41 (m, 5H).
Intermediate 11
3-(Aminomethyl)-4-cyclopropyl-6-methyl-2(lH)-pyridinone hydrochloride

The title compound was prepared in the same manner as described for intermediate 10 (step 2) from 4-cyclopropyl-6-methyl-2-oxo-l ,2-dihydro-3-pyridinecarbonitrile (5 g, 28.7 mmol). The product was collected as a TFA salt, 0.50 g. LCMS E-S (Μ+Η) = 179.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1 1.76 - 1 1.78 (br s, 1H), 7.82 - 7.92 (br s, 3H), 5.61 (s, 1H), 3.94 - 3.99 (m, 2H), 2.1 1 (s, 3H), 1.98 - 2.05 (m, 1H), 0.95 - 1.01 (m, 2H), 0.74 - 0.79 (m, 2H).
Intermediate 12
3-(Aminomethyl)-6-methyl-4-propyl-2(lH)-pyridinone

Step 1
To a solution of DMSO (300 mL) containing t-BuOK (20 g, 178 mmol) and cyanoacetamide (16.5 g, 196 mmol) was added (3E)-3-hepten-2-one (20 g, 178 mmol), and contents stirred at room temperature for 30 min. Additional t-BuOK (60 g, 534 mmol) was added and the reaction mixture was under an atmosphere of oxygen for an additional 1 h. The reaction mixture was purged with argon, diluted with 4 volumes of H20, and then 5 volumes of 4 N HCl, which were added slowly. The reaction mixture was filtered, washed with water, and dried to give the product as 10 g (32%). 1H NMR (400 MHz, DMSO-d6) δ ppm 12.25 - 12.40 (br s, 1H), 6.18 (s, 1H), 2.53 (t, 2H), 2.22 (s, 3H), 1.57 - 1.64 (m, 2H), 0.84 (t, 3H).
Step 2
The title compound was prepared in the same manner as described for intermediate 10 (step 2) from the product of step 1 (2 g, 1 1.2 mmol). The product was collected as 1.2 g (60%). LCMS E-S (M+H) = 181.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.85 - 7.95 (br s, 3H), 5.99 (s, 1H), 3.80 - 3.85 (m, 2H), 2.42 (t, 2H), 2.14 (s, 3H), 1.43 - 1.49 (m, 2H), 0.86 (t, 3H).
Intermediate 13
3-(Aminomethyl)-6-methyl-4-phenyl-2(lH)-pyridinone

The title compound was prepared in the same manner as described for intermediate 12 (steps 1 and 2) from (3E)-4-phenyl-3-buten-2-one (20 g, 137 mmol). The crude nitrile intermediate was obtained as 10 g (35%), of which 4 g of this putative intermediate was converted to the title compound 1.2 g as a TFA salt. LCMS E-S (Μ+Η) = 215.0. 1H NMR (400 MHz, DMSO-de) δ ppm 12.2 - 12.3 (br s, 1H), 7.88 - 8.00 (br s, 3H), 7.43 - 7.51 (m, 3H), 7.29 - 7.38 (m, 2H), 6.08 (s, 1H), 3.67 - 3.70 (m, 2H), 2.23 (s, 3H).
Intermediate 14
3-(Aminomethyl)-6-methyl-4-(l-methylethyl)-2(lH)-pyridinone

The title compound was prepared in the same manner as described for intermediate 12 (steps 1 and 2) from (3E)-5-methyl-3-hexen-2-one (20 g, 137 mmol). The crude nitrile intermediate was obtained as 7 g (22 %), of which 3 g of this putative intermediate was converted to the title compound 1.3 g as a TFA salt. LCMS E-S (Μ+Η) = 181.1. 1H
NMR (400 MHz, DMSO-d6) δ ppm 11.8 - 11.9 (br s, 1H), 7.86 - 7.96 (br s, 3H), 6,.10 (s, 1H), 3.82 - 3.86 (m, 2H), 3.02 - 3.09 (m, 1H), 2.17 (s, 3H), 1.08 (d, 6H).
Intermediate 15
3-(Aminomethyl)-4-methyl-6-propyl-2(lH)-pyridinone

Step 1
To a solution of NaNH2 (32.5 g ,862 mmol) in anhydrous ether (500 mL) at 30 °C was added dropwise a mixture of butyric acid ethyl ester (50 g ,431 mmol) and acetone (37.5 g 646.5 mol). After addition, the reaction mixture was stirred for 4 h. The reaction mixture was poured onto ice water with stirring. Additional ether was added, and the layers were separated. The aqueous layer was acidified to pH 5.0 with 2 N HC1 and then to pH 7.5 with Na2CC"3. The aq. layer was then extracted with ether. The combined organic layers were dried over Na2S04, filtered, and concentrated in vacuo. The crude product (20 g, 156 mmol) and 2-cyanoacetamide (13.12 g , 156 mmol) were suspended in EtOH (160 mL) at 75 °C, followed by addition of piperidine (13.2 g,156 mmol ). The contents were stirred and heated at reflux for lh. The mixture was cooled to room temperature, and filtered. The collected solid was suspended in water and stirred for 1 h. The mixture was filtered and dried to give 4-methyl-2-oxo-6-propyl-l,2-dihydro-3- pyridinecarbonitrile (1 1 g, 40%). LCMS E-S (M+H) = 181.1. 1H NMR (400 MHz,
DMSO-de) δ ppm 12.3 - 12.4 (br s, 1H), 6.25 (s, 1H), 3.64 (s, 3H), 2.50 (t, 2H), 1.63 (m, 2H), 0.94 (t, 3H).
Step 2
Sodium acetate (3.5 g, 42.6 mmol), palladium on carbon (0.81 g) and platinum oxide (0.1 g) were placed in a dried Parr bottle flushed with nitrogen, followed by addition of a small amount of acetic acid (to wet the catalysts). A solution of 4-methyl-2-oxo-6- propyl-l,2-dihydro-pyridine-3-carbonitrile (5 g, 28 mmol) in acetic acid was added to the Parr bottle followed by additional acetic acid (200 mL). The vessel was capped, placed on Parr apparatus and hydrogenated at 45 psi for 12 h. The reaction mixture was filtered and the filtrate concentrated in vacuo. The crude product was purified by preparative HPLC to
afford the title compound (TFA salt) as 4.1 g. LCMS E-S (M+H)) = 181.1. 1H NMR (400 MHz, DMSO-de) δ ppm 11.8 - 11.9 (br s, 1H), 7.83 - 7.88 (br s, 3H), 5.99 (s, 1H), 3.77 - 3.81 (m, 2H), 2.37 (t, 2H), 1.53 (m, 2H), 0.83 (t, 3H).
Intermediate 16
3-(Aminomethyl)-6-c lopropyl-4-methyl-2(lH)-pyridinone

Step 1
l-Cyclopropyl-l,3-butanedione
To a a stirring solution of THF (100 mL) was suspended potassium tert-butoxide (5.60 g, 49.5 mmol), followed by a mixture of cyclopropyl methyl ketone (3.27mL, 33 mmol) and ethyl acetate (9.69 mL, 99 mmol) in 30 mL THF at 35 °C, via addition funnel over a 25 min period. The contents were heated and stirred at 60 °C. After 3 h, the contents were removed from heating, and allowed to stir with cooling to room
temperature. The reaction mixture was carefully diluted with 30 mL 2N HCl and stirred for 10 min. The mixture was extracted with diethyl ether (3 x 50 mL), and the combined organic layers washed with brine (1 x 50 mL). The organic layer was dried over MgSC^, filtered, and concentrated in vacuo. The crude oil was chromatographed on silica gel (eluent: 0 to 15% EtOAc in hexanes) with good separation to afford the desired product as a light yellow colored oil, 3.9 g in -75% purity (residual solvent), for an overall yield of 70%. 1H NMR (400 MHz, CHLOROFORM- ) δ ppm 0.89 - 0.96 (m, 2 H), 1.09-1.15 (m, 2 H), 1.59-1.69 (m, 1H), 2.04 (s, 3H), 5.63 (s, 1 H), 15.5 - 16.0 (br s, 1H). Step 2
6-Cyclopropyl-4-methyl-2-o -l,2-dihydro-3-pyridinecarbonitrile
 To a stirring solution of ethanol (5 mL) were suspended l-cyclopropyl-1,3- butanedione (505 mg, 3.00 mmol) and cyanoacetamide (252 mg, 3.00 mmol), and the heterogenous contents heated until homogenous (ca. 75 °C). Next added piperidine (0.395 mL, 4.00 mmol), and the mixture was stirred with warming at reflux for 30 min. The reaction mixture was allowed to cool to room temperature, wherein precipitation ensued. The solid precipitate was filtered and set aside. The filtrate was concentrated in vacuo, and the oily residue treated with minimal EtOAc and then 10 mL hexanes to afford a 2nd crop of solid. The solid product crops were combined, suspended in water (7 mL), vigorously stirred, and vacuum filtered to afford a nearly white solid as 380 mg (73%). LCMS E-S (M+H) = 175.1. 1H NMR (400 MHz, CHLOROFORM-;/) δ ppm 1.01 - 1.09 (m, 2 H), 1.28 (dd, J=8.59, 2.27 Hz, 2 H), 1.95-2.01 (m, 1H), 2.43 (s, 3H), 5.82 (s, 1 H).
To a stirring solution of ethanol (5 mL) were suspended l-cyclopropyl-1,3- butanedione (505 mg, 3.00 mmol) and cyanoacetamide (252 mg, 3.00 mmol), and the heterogenous contents heated until homogenous (ca. 75 °C). Next added piperidine (0.395 mL, 4.00 mmol), and the mixture was stirred with warming at reflux for 30 min. The reaction mixture was allowed to cool to room temperature, wherein precipitation ensued. The solid precipitate was filtered and set aside. The filtrate was concentrated in vacuo, and the oily residue treated with minimal EtOAc and then 10 mL hexanes to afford a 2nd crop of solid. The solid product crops were combined, suspended in water (7 mL), vigorously stirred, and vacuum filtered to afford a nearly white solid as 380 mg (73%). LCMS E-S (M+H) = 175.1. 1H NMR (400 MHz, CHLOROFORM-;/) δ ppm 1.01 - 1.09 (m, 2 H), 1.28 (dd, J=8.59, 2.27 Hz, 2 H), 1.95-2.01 (m, 1H), 2.43 (s, 3H), 5.82 (s, 1 H).
Step 3
1 , 1-Dimethylethyl [(6-cyclopropyl-4-methyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] carbamate
6-Cyclopropyl-4-methyl-2-oxo-l,2-dihydro-3-pyridinecarbonitrile (0.35 g, 2.01 mmol) was added to methanol (20 mL) and the stirring contents cooled to -10 °C. Next added di-tert-butyloxycarbonyl (0.933 mL, 4.02 mmol) and the suspension was stirred for 15 min. Next added in N1CI2-6H2O (0.055 g, 0.201 mmol) as a solid and stirred for 5 min. Next added NaBH4 (0.532 g, 14.06 mmol) in 6 portions with 5 min. increments between each portion. After completed addition (ca. 30min), the ice bath was removed and the contents were stirred with warming to room temperature overnight. The reaction mixture was returned to - 10 °C, followed by addition of 3 more portions of NaBH4 (0.532 g, 14.06 mmol). The ice bath was removed and the mixture stirred at room temperature for 1 h. The contents were quenched by addition of diethylethylene amine (0.218 mL, 2.01 mmol) and stirred for 45 min. at room temperature. The volatiles were removed in vacuo and the residue suspended in EtOAc and sat. NaHC03. The organic layer was washed with additional NaHC03. The layers were separated, and the organic layer dried over
MgSC"4, filtered, and concentrated in vacuo. The crude product was purified by silica gel chromatography (eluent: 10% Methanol in Dichloromethane). The collected product was dried under hi-vacuum for 1 h, and then treated with ether and filtered. After drying in vacuum oven at 45 °C for 2 h, the product was collected as 0.28 g (50%). 1H NMR (400 MHz, DMSO-de) δ ppm 0.73 - 0.80 (m, 2 H), 0.88 - 0.96 (m, 2 H), 1.36 (s, 9 H), 1.70 - 1.82 (m, 1 H), 2.11 (s, 3 H), 3.95 (d, J=5.31 Hz, 2 H), 5.66 (s, 1 H), 6.51 (t, J=4.80 Hz, 1 H) ,11.50 (br. s., 1 H).
Step 4
3-(Aminomethyl)-6-cyclopropyl-4-methyl-2(lH)-pyridinone hydrochloride
1 , 1 -Dimethylethyl [(6-cyclopropyl-4-methyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]carbamate (0.28g, 1.006 mmol) was added to EtOAc (9 mL) and methanol (1.OmL). The suspension was stirred at room temperature for 5 min., followed by addition of 4M HC1 in dioxane (5.03 mL, 20.12 mmol), and the contents were stirred at room temperature overnight. The volatiles were then removed in vacuo to afford a solid. The solid was triturated with ether, filtered, and dried in a vacuum oven at 45°C for 4 h. The title compound was collected as 0.22 g (100% yield). 1H NMR (400 MHz, DMSO- d6) δ ppm 0.78 - 0.86 (m, 2 H), 0.95 - 1.03 (m, 2 H), 1.83 (tt, J=8.46, 5.05 Hz, 1 H), 2.16 - 2.22 (m, 3 H), 3.75 (q, J=5.47 Hz, 2 H), 5.79 (s, 1 H), 8.02 (br. s., 3 H), 11.92 (br. s., 1 H).
Example 1
6-Chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)- lH-pyrazolo[3,4-b]pyridine-4-carboxamide

6-Chloro-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (0.12g, 0.501 mmol), l-hydroxy-7-azabenzotriazole (0.102 g, 0.751 mmol), EDC (0.144 g, 0.751 mmol), and 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (0.123 g, 0.651 mmol) were dissolved in dimethyl sulfoxide (3.0 mL) and stirred at room temperature. Added next to the stirring contents was N-methylmorpholine (0.220 mL, 2.003 mmol) via syringe at
once. After stirring at room temperature overnight, the reaction mixture was slowly added to water (75 mL) and stirred for 10 min. After sitting for 10 min. at room temperature, the contents were filtered to afford a tan solid which was washed with water and then cold 50% aq EtOH. The contents were filtered, air-dried for 10 min., and then dried under vacuum for lhr. The collected solid was then further dried in a vacuum oven at 45 °C for
4 hr. The title compound was collected as 0.105 g (55%). LCMS E-S (M+H) = 373.9. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.49 (d, J=6.82 Hz, 6 H), 2.13 (s, 3 H), 2.20 (s, 3 H), 4.35 (d, J=4.80 Hz, 2 H), 5.07 - 5.20 (m, 1 H), 5.89 (s, 1H), 7.66 (s, 1 H), 8.39 (s, 1 H), 8.91 (t, J=4.80 Hz, 1 H), 11.55 (s, 1 H).
Example 2
6-Chloro- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l- methylethyl)-lH-pyraz -6]pyridine-4-carboxamide

6-chloro-3 -methyl- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid ( 1324 mg, 5.22 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone. HC1 (1329 mg, 7.05 mmol) and l-hydroxy-7-azabenzotriazole (1066 mg, 7.83 mmol) were stirred in 10 mL of DMSO for 10 min under nitrogen. N-methylmorpholine (2.3 mL, 20.88 mmol) was added along with EDC (1501 mg, 7.83 mmol) and the mixture became dark yellow. After a few hours solids precipitated and the contents became very thick, so 10 mL DMSO was added to facilitate stirring, The contents were stirred at RT overnight. Next added ice-water and then 10% K2CO3 (pH ~ 8-9). The contents were stirred at RT for 30 min and then allowed to stand at RT for another 30 min. The contents were filtered, washed with water, and dried in vacuo. The title compound was collected as 1.67g (81 >) and used without further purification. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.44 (d, 6 H), 2.12 (s, 3 H), 2.22 (s, 3 H), 2.40 (s, 3 H), 4.34 (d, J=5.05 Hz, 2 H), 5.05 (quin, J=6.63 Hz, 1 H), 5.88 (s, 1 H), 7.16 (s, 1 H), 8.78 (t, J=4.93 Hz, 1 H), 11.53 (br. s., 1 H). LCMS E-S (M+H) = 388.1.
Example 3
6-Chloro-l-(l,l-dimethylethyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-3-methyl- -pyrazolo[3,4-6]pyridine-4-carboxamide

To a solution of 6-chloro-l -(1,1 -dimethylethyl)-3 -methyl- lH-pyrazolo [3,4- b]pyridine-4-carboxylic acid (320 mg, 1.195 mmol) in DMSO(7 mL) were added 3- (aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (338 mg, 1.793 mmol), N- methylmorpholine (0.526 mL, 4.78 mmol), l-hydroxy-7-azabenzotriazole (325 mg, 2.391 mmol) and EDC (458 mg, 2.391 mmol), and the reaction mixture was stirred overnight. The reaction mixture was quenched with water (20 mL) and stirred for 10 min. The precipitate was collected by filtration and further dried under high vacuum to give the product as a solid, 450 mg (94%). LCMS E-S (M+H) = 402.1. 1H NMR (400 MHz, DMSO-d6) 5 ppm 1.71 (s, 9 H), 2.12 (s, 3 H), 2.22 (s, 3 H), 2.37 (s, 3 H), 4.33 (d, J= 4.80 Hz, 2 H), 5.88 (s, 1 H), 7.09 - 7.21 (m, 1 H), 8.77 (t, J= 4.93 Hz, 1 H), 11.53 (s, 1 H).
Example 4
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- pyridinyloxy)- -pyrazolo[3,4-6]pyridine-4-carboxamide

6-Chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (0.15 g, 0.401 mmol), 4- pyridinol (0.057 g, 0.602 mmol), cesium carbonate (0.261 g, 0.802 mmol), and l-(2- pyridinyl)-2-propanone (10.85 mg, 0.080 mmol) were sequentially dissolved in DMSO
(4.0 mL). Next added copper(I) bromide (5.76 mg, 0.040 mmol) and the suspension was
stirred under nitrogen (degassed) for 1 min. The sealed reaction mixture was stirred with heating at 110 °C (heating block) for 20 h, and then allowed to cool to room temperature overnight. The reaction mixture was diluted with EtOAc (25 mL) and water. The contents were vigorously stirred and then fitlered through Celite, washing the filter pad with 20%THF/EtOAc. The layers were separated and the aq. layer extracted with EtOAc. The combined organic layers were dried over MgSC^, filtered, and concentrated in vacuo to a dark residue that was dried under hi-vacuum overnight. The crude product was purified by silica gel chromatography (eluent: gradient of 5-95 % Dichloromethane/ Chloroform containing 2M Ammonia (in methanol). The collected product was washed with MTBE, filtered, and dried in a vacuum oven at 45 °C for 5 hr. The final product was collected as 0.075 g (42%). LCMS E-S (M+H) = 433.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1H NMR (400 MHz, DMSO-d6) δ ppm 1.52 (d, J=6.57 Hz, 6 H), 2.13 (s, 3 H), 2.22 (s, 3 H), 4.41 (d, J=5.05 Hz, 2 H), 5.18 - 5.30 (m, 1 H), 5.91 (s, 1H), 6.30 - 6.40 (m, 2 H), 7.91 (s, 1 H), 8.41 (s, 1 H), 8.57 - 8.65 (m, 2 H), 8.92 (t, J=4.93 Hz, 1 H), 11.58 (s, 1 H).
Example 5
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2- propen-l-ylamin -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

6-Chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (0.25 g, 0.669 mmol) was suspended in ethanol (4 mL) followed by addition of allylamine (0.753 mL, 10.03 mmol). The sealed contents were heated (heat block) at 140 °C for ca. 60 h, and then allowed to cool to room temperature. The reaction mixture was diluted with water (100 mL) and then filtered. The collected solid was washed with additional water and then dried in vacuum oven at 45 °C for 18h. The final product was isolated as an off-white solid, 0.225 g (83%). LCMS E-S (M+H) = 394.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.42 (d, J=6.82 Hz, 6 H), 2.12 (s, 3 H), 2.21 (s, 3 H), 4.00 (t, J=5.43 Hz, 2 H), 4.32 (d, J=5.05 Hz, 2 H), 4.94 (quin, J=6.69 Hz, 1 H), 5.09 (dd, J=10.36, 1.77 Hz, 1 H), 5.18 - 5.29 (m, 1 H), 5.85 - 6.01
(m, 2 H), 6.68 (s, 1 H), 7.37 (t, J=5.56 Hz, 1 H), 7.84 (s, 1 H), 8.45 (t, J=5.05 Hz, 1 H), 11.55 (br. s., 1 H).
Example 6
6-Amino- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)- lH-pyrazolo[3,4-6]pyridine-4-carboxamide

A mixture of 10% Pd/C (0.200 g, 0.188 mmol) and N-[(4,6-dimethyl-2-oxo-l,2- dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)-6-(2-propen- 1 -ylamino)- 1 H-pyrazolo [3 ,4- b]pyridine-4-carboxamide (0.200g, 0.507 mmol) was suspended in ethanol (10 mL) and stirred. Added next was methanesulfonic acid (0.033 mL, 0.507 mmol), and the stirring contents were heated at reflux for 2 h. After cooling to room temperature, the reaction mixture was diluted with DCM and filtered through Celite. The filter pad was washed with 10% MeOH/DCM. The filtrate was pre-adsorped onto silica gel, and the crude product purified by silica gel chromatography (dry loaded, eluent: gradient of 5-80%> Dichloromethane/Chloroform containing 10% 2M Ammonia (in methanol)). The collected product was dried in vacuum oven at 45 °C overnight. The final product was collected as 0.065 g (36%). LCMS E-S (M+H) = 355.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.40 (d, J=6.82 Hz, 6 H), 2.12 (s, 3 H), 2.21 (s, 3 H), 4.31 (d, J=5.05 Hz, 2 H), 4.85 - 5.00 (m, 1 H), 5.89 (s, 1H), 6.49 - 6.68 (m, 3 H), 7.85 (s, 1 H), 8.46 (t, J=5.05 Hz, 1 H), 11.54 (br. s., 1 H).
Example 7
6-Cyclopropyl-l-(l-methylethyl)- V-[(4-methyl-2-oxo-6-propyl-l,2-dihydro-3- pyridinyl)methy -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

6-Cyclopropyl- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid
(167 mg, 0.680 mmol), 3-(aminomethyl)-4-methyl-6-propyl-2(lH)-pyridinone
trifluoroacetate (200 mg, 0.680 mmol), ΗΟΑΤ (139 mg, 1.019 mmol), EDC (195 mg, 1.019 mmol), and N-methylmorpholine (0.299 mL, 2.72 mmol) were dissolved in DMF(6 mL) and stirred at 40 °C for 24 h. The reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated to an orange oil. The residue was dissolved in DMSO, and purified by reverse phase HPLC (mobile phase: 40-60% ACN in H20, 0.1% TFA). The isolated product was dried in a vacuum oven overnight and furnished the TFA salt of the title compound as a white solid, 0.113 g (32%). LCMS E-S (M+H) = 408.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.80 - 0.98 (m, 3 H), 1.06 (d, J= 7.07 Hz, 4 H), 1.46 (d, J= 6.82 Hz, 6 H), 1.52 - 1.67 (m, 2 H), 2.17 - 2.31 (m, 4 H), 2.37 (t, J= 7.58 Hz, 2 H), 4.36 (d, J= 4.80 Hz, 2 H), 5.02 - 5.27 (m, 1 H), 5.91 (s, 1 H), 7.43 (s, 1 H), 8.21 (s, 1 H), 8.62 - 8.87 (m, 1 H), 11.54 (br. s., 1 H).
Example 8
6-Cyclopropyl-l-(l-methylethyl)- V-{[6-methyl-4-(l-methylethyl)-2-oxo-l,2-dihydro- 3-pyridinyl]methyl}-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

Example 9
l-(l-Methylethyl)- V-[(4-methyl-2-oxo-6-propyl-l,2-dihydro-3-pyridinyl)methyl]-6-(4- pyridinyl)-l -pyrazolo[3,4-6]pyridine-4-carboxamide

l-(l-Methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (192 mg, 0.680 mmol), 3-(aminomethyl)-4-methyl-6-propyl-2(lH)-pyridinone»TFA (200 mg, 0.680 mmol), HO AT (139 mg, 1.019 mmol), EDC (195 mg, 1.019 mmol), and N- methylmorpholine (0.299 mL, 2.72 mmol) were sequentially added to DMF(6 mL), and the mixture stirred at 40 °C overnight. The reaction mixture was then filtered. The collected solid was washed with ethanol, and dried, affording the final product as a white solid, 0.160 g (53%). LCMS E-S (M+H) = 445.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.88 (t, 3 H), 1.46 - 1.65 (m, 8 H), 2.24 (s, 3 H), 2.38 (t, J= 7.58 Hz, 2 H), 4.43 (d, J = 4.80 Hz, 2 H), 5.37 (s, 1 H), 5.93 (s, 1 H), 8.23 (d, J= 5.81 Hz, 2 H), 8.30 (s, 1 H), 8.45 (s, 1 H), 8.78 (d, J= 5.31 Hz, 2 H), 9.00 (br. s., 1 H), 11.56 (s, 1 H).
Example 10
V-[(6-Ethyl-4-methyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- pyridinyl)-l -pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 9 from 1 -( 1 -methy lethyl)-6-(4-pyridinyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid (403 mg, 1.427 mmol), 3-(aminomethyl)-6-ethyl-4-methyl-2(lH)-pyridinone»TFA (400 mg, 1.427 mmol), HOAT (291 mg, 2.141 mmol), EDC (1094 mg, 5.71 mmol), N- methylmorpholine (0.628 mL, 5.71 mmol), and DMF(6 mL). The final product was collected as a white solid, 232 mg (38%). LCMS E-S (M+H) = 432.4. 1H NMR (400
MHz, DMSO-d6) δ ppm 1.14 (t, 3 H), 1.56 (d, J = 6.57 Hz, 6 H), 2.25 (s, 3 H), 2.42 (q, J = 7.41 Hz, 2 H), 4.43 (d, J = 4.55 Hz, 2 H), 5.29 - 5.45 (m, 1 H), 5.93 (s, 1 H), 8.22 (d, J = 6.06 Hz, 2 H), 8.30 (s, 1 H), 8.45 (s, 1 H), 8.78 (d, J= 5.81 Hz, 2 H), 9.01 (t, J = 4.55 Hz, 1 H), 1 1.58 (s, 1 H).
Example 11
l-(l-Methylethyl)- V-[(6-methyl-2-oxo-4-propyl-l,2-dihydro-3-pyridinyl)methyl]-6-(4- pyridinyl)-l -pyrazolo[3,4-6]pyridine-4-carboxamide

l-(l-Methylethyl)-6-(4-pyridinyl)-l H-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (150 mg, 0.531 mmol), 3-(aminomethyl)-6-methyl-4-propyl-2(lH)-pyridinone (1 15 mg, 0.531 mmol), EDC (122 mg, 0.638 mmol), HOAt (72.3 mg, 0.531 mmol), and N- methylmorpholine (0.233 mL, 2.12 mmol) were suspended in DMF (5 mL) and stirred at room temperature overnight, water was added to the reaction mixture, and the contents
were filtered. The filter cake was washed with addional water (2x). The crude solid was purified by reverse phase HPLC (mobile phase: 10-30% ACN in H20, 0.1% TFA). The isolated solid was then neutralized with saturated NaHC03, and washed with EtOAc (5 x 15 mL). The combined organic layers were dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as an off-white solid, 0.060 g (24%). LCMS E-S (M+H) = 445.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.84 - 0.92 (m, 3 H), 1.47 - 1.54 (m, 2 H), 1.57 (d, J=6.82 Hz, 6 H), 2.14 (s, 3 H), 2.48 - 2.52 (m, 2 H), 4.42 - 4.49 (m, 2 H), 5.33 - 5.43 (m, 1 H), 5.90 - 5.96 (m, 1 H), 8.28 - 8.37 (m, 3 H), 8.44 - 8.48 (m, 1 H), 8.82 - 8.87 (m, 2 H), 8.99 - 9.04 (m, 1 H), 11.58 (s, 1 H).
Example 12
l-(l-Methylethyl)- V-{[6-methyl-4-(l-methylethyl)-2-oxo-l,2-dihydro-3- pyridinyl]methyl}-6-(4-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

A DMF solution (6 mL) of l-(l-methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4- b]pyridine-4-carboxylic acid (288 mg, 1.019 mmol), 3-(aminomethyl)-6-methyl-4-(l- methylethyl)-2(lH)-pyridinone'TFA (300 mg, 1.019 mmol), HO AT (208 mg, 1.529 mmol), EDC (782 mg, 4.08 mmol), and N-methylmorpholine (0.448 mL, 4.08 mmol) was stirred overnight at 40 °C. After cooling to room temperature, the reaction mixture was poured into H20 (50 mL) and EtOAc (50 mL). The aqueous layer was extracted with EtOAC (2 x 50 mL). The organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was dissolved in DMSO and purified by reverse phase (15-40%) ACN in H20, 0.1% TFA). The product fractions were poured into sodium bicarbonate and extracted with ethyl acetate. The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo to afford the title compound as a white solid (123 mg, 27% yield). LCMS E-S (M+H) = 445.4. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.10 (m, 6 H), 1.56 (m, 6 H), 2.17 (s, 3 H), 3.17 - 3.28 (m, 1 H), 4.45 - 4.56 (m, 2 H), 5.29
- 5.43 (m, 1 H), 5.99 - 6.12 (m, 1 H), 8.16 - 8.23 (m, 2 H), 8.24 - 8.29 (m, 1 H), 8.40 - 8.46 (m, 1 H), 8.71 - 8.82 (m, 2 H), 8.96 - 9.04 (m, 1 H), 1 1.53 - 1 1.63 (m, 1 H).
Example 13
6-Cyclopropyl-l-(l-methylethyl)- V-[(6-methyl-2-oxo-4-propyl-l,2-dihydro-3- pyridinyl)methy -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 1 1 from 1 -( 1 -methylethyl)-6-(cyclopropyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid (150 mg, 0.612 mmol). The product was collected as a white solid, 0.043 g (16%).
LCMS E-S (M+H) = 408.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.89 (t, J=7.20 Hz, 3 H), 1.06 (d, J=2.78 Hz, 3 H), 1.08 (br. s., 1 H), 1.47 (d, J=6.57 Hz, 8 H), 1.49 - 1.57 (m, 2 H), 2.14 (s, 3 H), 2.19 - 2.30 (m, 1 H), 4.38 (d, J=4.80 Hz, 2 H), 5.03 - 5.18 (m, 1 H), 5.92 (s, 1 H), 7.40 (s, 1 H), 8.21 (s, 1 H), 8.69 - 8.76 (m, 1 H), 1 1.55 (s, 1 H).
Example 14
l-(l-Methylethyl)- V-[(6-methyl-2-oxo-4-phenyl-l,2-dihydro-3-pyridinyl)methyl]-6-(4- pyridinyl)-l -pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 1 1 from 1 -( 1 -methy lethyl)-6-(4-pyridinyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid (150 mg, 0.531 mmol) and 3-(aminomethyl)-6-methyl-4-phenyl-2(lH)-pyridinone (133 mg, 0.531 mmol). The product was collected as a white solid, 0.041 g (15%). LCMS E-S (M+H) = 479.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.57 (d, J=6.82 Hz, 6 H) 2.23 (s, 3 H) 4.23 - 4.27 (m, 2 H) 5.33 - 5.42 (m, 1 H) 6.02 - 6.04 (m, 1 H) 7.35 - 7.42 (m, 1 H)
7.43 (s, 4 H) 8.23 - 8.26 (m, 1 H) 8.34 - 8.39 (m, 2 H) 8.41 - 8.42 (m, 1 H) 8.84 - 8.88 (m, 2 H) 8.91 - 8.96 (m, 1 H) 11.91 (s, 1 H).
Example 15
V-[(4-Cyclohexyl-6-methyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-
6-(4-pyridinyl)- -pyrazolo[3,4-6]pyridine-4-carboxamide

A mixture of l-(l-methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-b]pyridine-4- carboxylic acid (169 mg, 0.6 mmol), 3-(aminomethyl)-4-cyclohexyl-6-methyl-2(lH)- pyridinone trifiuoroacetate (221 mg, 0.660 mmol), EDC (150 mg, 0.780 mmol), HOAT (106 mg, 0.780 mmol), and N-methylmorpholine (0.264 mL, 2.400 mmol) in DMF(3 mL) were stirred at room temperature for 6 days, water (15 mL) was added to the slurry, and it was stirred for an hour. The precipitate was then collected by vacuum filtration and rinsed with EtOH (4 mL), and the solid was dried in the vacuum oven overnight to give the title compounds as an off-white solid (137 mg, 45%). LCMS E-S (M+H) = 485.2. 1H NMR
(400 MHz, DMSO-de) δ ppm 1.09 - 1.44 (m, 5 H), 1.56 (d, J = 6.57 Hz, 6 H), 1.55 - 1.72 (m, 5 H), 2.15 (s, 3 H), 2.80 - 2.92 (m, 1 H), 4.53 (d, J= 4.55 Hz, 2 H), 5.36 (spt, J= 6.53 Hz, 1 H), 6.03 (s, 1 H), 8.18 - 8.23 (m, 2 H), 8.26 (s, 1 H), 8.45 (s, 1 H), 8.75 - 8.82 (m, 2 H), 9.01 (t, J= 4.55 Hz, 1 H), 11.59 (s, 1 H).
Example 16
TV- [(4-Cyclohexyl-6-methyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl] -6-cyclopropyl- 1- (l-methylethyl)- -pyrazolo[3,4-6]pyridine-4-carboxamide
The title compound was prepared in the same manner as described in example 15 from 6-cyclopropyl- l-(l-methylethyl)-l H-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (147 mg, 0.60 mmol), 3-(aminomethyl)-4-cyclohexyl-6-methyl-2(lH)-pyridinone
trifluoroacetate (221 mg, 0.660 mmol), EDC (150 mg, 0.780 mmol), HO AT (106 mg, 0.780 mmol), N-methylmorpholine (0.264 mL, 2.400 mmol) and DMF(3 mL), wherein the stir time was 3 d and the final product was not treated with EtOH. The final product was collected as 193 mg (68%). LCMS E-S (M+H) = 448.4. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.01 - 1.11 (m, 4 H), 1.15 - 1.43 (m, 5 H), 1.47 (d, J= 6.57 Hz, 6 H), 1.55 - 1.76 (m, 5 H), 2.15 (s, 3 H), 2.19 - 2.28 (m, 1 H), 2.83 (t, J= 11.12 Hz, 1 H), 4.45 (d, J= 5.05 Hz, 2 H), 5.12 (spt, J= 6.69 Hz, 1 H), 6.02 (s, 1 H), 7.37 (s, 1 H), 8.21 (s, 1 H), 8.74 (t, J = 4.80 Hz, 1 H), 11.56 (s, 1 H).
Example 17
6-Cyclopropyl-l-(l-methylethyl)-iV-[(6-methyl-2-oxo-4-phenyl-l,2-dihydro-3- pyridinyl)methy -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 11 from 1 -( 1 -methylethyl)-6-(cyclopropyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid
(150 mg, 0.612 mmol) and 3-(aminomethyl)-6-methyl-4-phenyl-2(lH)-pyridinone (153 mg, 0.612 mmol). The product was collected as a white solid, 0.067 g (24%). LCMS E-S (Μ+Η) = 442.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.04 - 1.11 (m, 3 H), 1.47 (d, J=6.57 Hz, 5 H), 2.22 (s, 3 H), 2.24 - 2.29 (m, 1 H), 3.17 (d, J=5.31 Hz, 2 H), 4.07 - 4.15 (m, 1 H), 4.19 (d, J=4.29 Hz, 2 H), 5.06 - 5.18 (m, 1 H), 6.01 (s, 1 H), 7.36 (s, 1 H), 7.38 - 7.49 (m, 5 H), 8.18 (s, 1 H), 8.70 (s, 1 H), 11.88 (s, 1 H).
Example 18
V-[(4-Cyclopropyl-6-methyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-6-(4-pyridinyl)- -pyrazolo[3,4-6]pyridine-4-carboxamide

l-(l-Methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (100 mg, 0.354 mmol), 3-(aminomethyl)-4-cyclopropyl-6-methyl-2(lH)-pyridinone (104 mg, 0.354 mmol), EDC (81 mg, 0.425 mmol), HOAt (48 mg, 0.354 mmol), and N- methylmorpholine (0.156 mL, 1.42 mmol) were suspended in DMF(5 mL) and stirred at room temperature overnight. The contents were filtered, washed with ethanol, and then concentrated in vacuo. The crude solid was purified by reverse phase HPLC (mobile phase : 20-40%, ACN in H20, 0.1% TFA). The isolated solid was then neutralized with saturated NaHC03, and washed with EtOAc (3x). The combined organic layers were dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as an off- white solid, 0.042 g (26%). LCMS E-S (M+H) = 443.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.71 - 0.78 (m, 2 H), 0.94 (dd, J=8.34, 2.27 Hz, 2 H), 1.57 (d, J=6.57 Hz, 6 H), 2.12 (s, 3 H), 2.13 - 2.18 (m, 1 H), 4.62 (d, J=4.80 Hz, 2 H), 5.38 (s, 1 H), 5.54 (s, 1 H), 8.37 (s, 1 H), 8.44 - 8.54 (m, 3 H), 8.91 (d, J=6.06 Hz, 2 H), 9.04 (s, 1 H), 11.51 (br. s., 1 H).
Example 19
6-Cyclopropyl- V-[(6-cyclopropyl-4-methyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l- (l-methylethyl)-lH-pyrazolo 3,4-6]pyridine-4-carboxamide

To a 4 mL solution of DMSO were sequentially added 6-cyclopropyl-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (0.11 g, 0.448 mmol), 3- (aminomethyl)-6-cyclopropyl-4-methyl-2(lH)-pyridinone hydrochloride (0.106 g, 0.493 mmol), l-hydroxy-7-azabenzotriazole (0.073 g, 0.538 mmol), EDC (0.103 g, 0.538 mmol), and then N-methylmorpholine (0.197 mL, 1.79 mmol) via syringe. After stirring overnight at room temperature, the suspension was diluted with 50 mL of water and stirred for 15 min. After standing at room temperature for 15 min., the reaction mixture was filtered and the collected solid washed with additional water. The solid was then dried in a vacuum oven at 45 °C for 4 h. The product was obtained as 0.165 g (89%). LCMS E-S (M+H) = 406.5. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.76 - 0.82 (m, 2 H), 0.91 - 0.98 (m, 2 H), 1.03 - 1.12 (m, 4 H), 1.46 (d, J=6.82 Hz, 6 H), 1.74 - 1.84 (m, 1 H), 2.19 (s, 3 H), 2.21 - 2.31 (m, 1 H), 4.34 (d, J=5.05 Hz, 2 H), 5.05 - 5.18 (m, 1 H), 5.73 (s, 1 H), 7.42 (s, 1 H), 8.21 (s, 1 H), 8.72 (t, J=4.93 Hz, 1 H), 11.61 (br. s., 1 H).
Example 20
V-[(5-Fluoro-4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)- 6-(4-pyridinyl)- -pyrazolo[3,4-6]pyridine-4-carboxamide

Example 21
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(ethylamino)-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

To a 10 mL microwave vial equipped with stir bar, septum cap and nitrogen inlet were added 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (23 mg, 0.062 mmol) and ethanol (1 mL). Added next to the stirring suspension was ethylamine (0.100 mL, 1.230 mmol) via syringe at once. The sealed reaction mixture was irradiated (microwave) at 110 °C for 40 min, followed by addition of 0.1 mL ethylamine and further irradiation for 7 hr at 130 °C. After cooling to room temperature, about 50% of the volatiles were removed by a stream of nitrogen. The reaction mixture was diluted with water (20 mL) and stirred for 10 min. The mixture was then filtered and the collected solid washed with water. The solid was dried in vacuum oven at 50 °C for 18h, to afford the title compound as 0.17 g (71 %). LCMS E-S (M+H) = 383.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.17 (t, j=7.07 Hz, 3 H), 1.43 (d, J=6.57 Hz, 6 H), 2.12 (s, 3 H), 2.21 (s, 3 H), 3.35 - 3.39 (m, 2 H), 4.31 (d,J=5.05 Hz, 2 H), 4.89 - 5.01 (m, 1 H), 5.89 (s, 1 H), 6.61 (s, 1 H), 7.18 (t, j=5.31 Hz, 1 H), 7.83 (s, 1 H), 8.44 (t, j=5.05 Hz, 1 H), 11.55 (br. s., 1 H).
Example 22
N- [(4,6-Dimethyl-2-oxo- 1 ,2-dihydro-3-py ridinyl)methyl] - l-(l-methylethyl)- 1H- pyrazolo [3, -b] pyridine-4-carboxamide

To a 50 mL round bottom flask fitted with a 3 -way inlet was placed 10% Pd/C (0.071 g, 0.033 mmol) followed by degassing with N2, and then addition of 2 mL ethanol. Added next to the stirring slurry was 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (.25 g, 0.669 mmol). Additional ethanol (8mL) and THF (lOmL) were added with slight warming to facilitate dissolution. The stirring contents were allowed to cool (15 min.) then fitted with a balloon of H2 and allowed to stir at room temperature overnight. The contents were then purged with N2. The mixture was then diluted with 10% MeOH/DCM (20mL), stirred for 10 min., and filtered through Celite. The filtrate was concentrated in vacuo and the crude solid was triturated with ethanol. After filtration and washing the collected solid with additional ethanol (cold), the solid was air-dried for 15 min, then in vacuum oven at 40 °C overnight. The final product was collected as 185 mg (80%>).
LCMS E-S (M+H) = 340.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.50 (d, J=6.82 Hz, 6 H), 2.13 (s, 3 H), 2.22 (s, 3 H), 4.37 (d, J=4.80 Hz, 2 H), 5.18 - 5.31 (m, 1 H), 5.89 (s, 1H), 7.54 (d, J=4.80 Hz, 1 H), 8.36 (s, 1 H), 8.63 (d, J=4.55 Hz, 1 H), 8.82 (t, J=4.93 Hz, 1 H), 11.56 (s, 1 H).
Example 23
6-Cyclopropyl- l-(l-methylethyl)-N- { [6-methyl-2-oxo-4-(trifluor omethyl)- 1 ,2- dihydro-3-pyridinyl]meth l}-lH-pyrazolo[3,4-b]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 19 from 6-cyclopropyl-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (0.12 g, 0.489 mmol), l-hydroxy-7-azabenzotriazole (0.100 g, 0.734 mmol), 3-(aminomethyl)-6- methyl-4-(trifluoromethyl)-2(lH)-pyridinone (0.154 g, 0.636 mmol, DMSO (3.0 mL), N- methylmorpholine (0.215 mL, 1.957 mmol), and EDC (0.141 g, 0.734 mmol) The crude solid was purified by silica gel chromatography (eluent: gradient 5-100% of 10%> 2M NH3 (in MeOH/DCM) and DCM) and the collected product dried in vacuum oven for 5h. The final product was collected as 0.1 12g (50%). LCMS E-S (M+H) = 434.3. 1H NMR (400 MHz, DMSO-de) δ ppm 1.03 - 1.12 (m, 4 H), 1.47 (d, J=6.82 Hz, 6 H), 2.20 - 2.33 (m, 4 H), 4.34 - 4.54 (m, 2 H), 5.12 (quin, J=6.63Hz, 1 H), 6.33 (s, 1 H), 7.39 (s, 1 H), 8.18 (s, 1 H), 8.70 (t, J=4.04 Hz, 1 H), 12.43 (s, 1 H).
Example 24
6-(Dimethylamino)-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH- razolo[3,4-b]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 21 from 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -
methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (23 mg, 0.062 mmol), ethanol (0.7 mL), and dimethylamine (0.461 mL, 0.923 mmol). The product was dried in vacuum oven at 50 °C for 5 hr and collected as 0.016 g (67%). LCMS E-S (M+H) = 383.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.44 (d, J=6.57 Hz, 6 H), 2.05 - 2.25 (m, 6 H), 3.14 (s, 6 H), 4.34 (d, J=4.80 Hz, 2 H), 4.99 (dt, J=13.39, 6.69 Hz, 1 H), 5.89 (s, 1 H), 6.94 (s, 1 H), 7.96 (s, 1 H), 8.64 (t, J=4.93 Hz, 1 H), 11.56 (br. s., 1 H).
Example 25
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(l- piperidinyl)-lH-p razolo[3,4-b]pyridine-4-carboxamide

To a 10 mL reaction vial containing stir bar were added 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (.060g, 0.160 mmol), ethanol (1.5 mL), and then piperidine (0.318 mL, 3.21 mmol) via syringe at once. The contents were capped, placed into a heat block, and heated at 120 °C for 18 h. After cooling to room temperature, the reaction mixture was diluted with water (40 mL), adjusted to pH 6-7, and stirred for 15 min. The contents were filtered and washed with water. The product was dried in vacuum oven at 50 °C for 5 hr. The product was obtained as 57 mg (82%). LCMS E-S (M+H) = 422.8. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.43 (d, J=6.82 Hz, 6 H), 1.53 - 1.67 (m, 6 H), 2.12 (s, 3 H), 2.20 (s, 3 H), 3.61 - 3.73 (m, 4 H), 4.34 (d, J=5.05Hz, 2 H), 4.91 - 5.04 (m, 1 H), 5.89 (s, 1 H), 7.10 (s, 1 H), 7.97 (s, 1 H), 8.65 (t, J=4.93 Hz, 1 H), 11.51 (br. s., 1 H).
Example 26
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- morpholinyl)-lH- razolo[3,4-b]pyridine-4-carboxamide

To a 10 mL reaction vial containing stir bar were added 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (.060 g, 0.160 mmol), ethanol (1.5 mL), and then morpholine (0.280 mL, 3.21 mmol) via syringe at once. The contents were capped, placed into a heat block, and heated at 120 °C for 18 h and then at 135 °C for 2 hr. After cooling to room temperature, the reaction mixture was diluted with water (40 mL), adjusted to pH 6-7, and stirred for 15 min. The contents were filtered and washed with water. The product was dried in vacuum oven at 50 °C for 5 hr. The product was obtained as 57mg (79%).
LCMS E-S (M+H) = 425.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.44 (d, J=6.57 Hz, 6 H), 2.12 (s, 3 H), 2.19 (s, 3 H), 3.56 - 3.66 (m, 4 H), 3.69 - 3.78 (m, 4 H), 4.35 (d, J=4.80 Hz, 2 H), 5.00 (quin, J=6.63 Hz, 1 H), 5.89 (s, 1 H), 7.13 (s, 1 H), 8.03 (s, 1 H), 8.65 (t, J=4.93 Hz, 1 H), 11.56 (br. s., 1 H).
Example 27
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l-methylethyl)- 6- [6-(4-methyl- l-piperazinyl)-3-pyridinyl] - lH-pyrazolo [3,4-6] pyridine-4- carboxamide

Example 28
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(l- piperazinyl)-lH-p razolo [3,4-b] pyridine-4-carboxamide

To a 10 mL microwave vial equipped with stir bar were added 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridiny l)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (.060g, 0.160 mmol), ethanol (1.5 mL), and 1,1-dimethylethyl 1-piperazinecarboxylate (0.299 g, 1.605 mmol). The stirring suspension was placed onto heat block and heated at 120 °C for 18 h, and then irradiated (microwave) at 160 °C for 2 hr. The contents were cooled to room temperature, diluted into water (40 mL), and then adjusted to pH 6-7 and stirred for 15 min. The contents were filtered, washed with water,
and then dried in hi-vac oven at 50 °C for 5 hr. The collected solid (52 mg) was dissolved in CH2CI2/TFA (3 mL, 2: 1) and stirred at room temperature for 2 hr. The volatiles were removed in vacuo to afford a residue which was then dissolved in CH2CI2 and 10% 2M Ammonia (in methanol) in Chloroform. The crude product was purified by silica gel chromatography (eluent : gradient of 10% 2M Ammonia (in Methanol/Chloroform) and Dichloromethane). The isolated solid was dried in vacuum oven at 45 °C for 18 hr. to afford the final product as 38 mg (55 % yield). LCMS E-S (M+H) = 423.9. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.43 (d, J=6.82 Hz, 6 H), 2.08 - 2.24 (m, 6 H), 2.75 - 2.89 (m, 4 H), 3.53 - 3.64 (m, 4 H), 4.34 (d, J=4.80 Hz, 2H), 4.98 (quin, J=6.69 Hz, 1 H), 5.89 (s, 1 H), 7.10 (s, 1 H), 8.00 (s, 1 H), 8.65 (t, J=4.93 Hz, 1 H), 11.55 (br. s., 1 H).
Example 29
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(3-hydroxy-3-methyl-l- butyn-l-yl)-l-(l-methylethyl -lH-pyrazolo[3,4-b]pyridine-4-carboxamide

To a 10 mL microwave vial were sequentially added 6-chloro-N-[(4,6-dimethyl-2- oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethy 1)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (.15 g, 0.401 mmol), sodium iodide (0.012 g, 0.080 mmol), zinc (5.25 mg, 0.080 mmol), DMSO(2.5 mL), triethylamine (0.112 mL, 0.802 mmol), and DBU (0.121 mL, 0.802 mmol). The suspension was stirred and degassed with nitrogen for 5 min, forming an emulsion. Next added 2-methyl-3-butyn-2-ol (0.194 mL, 2.006 mmol) and
Pd(Ph3P)4 (0.046 g, 0.040 mmol). The stirring contents were heated at 90 °C for 3 hr, and then allowed to cool to room temperature. The reaction mixture was poured into a solution of water and 20% THF/EtOAc, and stirred. The layers were separated, and the organic layer washed with brine. The organic layer was dried over MgS04, and then filtered through Celite, washing the filter pad with additional EtOAc. The filtrate was concentrated in vacuo and the crude residue dried overnight on a hi-vac pump. The crude product was purified by silica gel chromatography (eluent: gradient: 5-80%> of DCM and Chloroform containing 10% 2M Ammonia (in methanol)). The collected product was dried in vacuum oven at 45 °C overnight, to afford the final product as 0.116 g (67%
yield). LCMS E-S (M+H) = 421.9. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.44 - 1.55 (m, 12 H), 2.13 (s, 3 H), 2.20 (s, 3 H), 4.35 (d, J=4.80 Hz, 2 H), 5.15 - 5.26 (m, 1 H), 5.70 (s, 1 H), 5.89 (s, 1 H), 7.65 (s, 1 H), 8.38 (s, 1 H), 8.93 (t, J=4.80 Hz, 1 H), 11.55 (s, 1 H).
Example 30
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(3- methyl-lH-indazol-5-yl)-lH- razolo[3,4-b]pyridine-4-carboxamide
To a 10 mL microwave vial were sequentially added 6-chloro-N-[(4,6-dimethyl-2- oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethy 1)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (0.12 g, 0.321 mmol), 1 ,1-dimethylethyl 3-methyl-5-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)-lH-indazole-l-carboxylate (0.138 g, 0.385 mmol), potassium phosphate (tribasic) (0.204 g, 0.963 mmol), 1,4-dioxane (3 mL), and water (0.75 mL). The stirring suspension was degassed with nitrogen for 10 min., wherein an emulsion had formed. Next added PdCl2(dppf)-CH2Cl2 adduct (0.039 g, 0.048 mmol) and the contents were placed onto a heat block and stirred at 105 °C overnight. After cooling to room temperature, the reaction mixture was diluted with EtOAc and filtered through Celite. The filter pad was washed with 50% THF/EtOAc. Silica gel was added to the combined filtrates and the mixture concentrated in vacuo to a solid. The contents were purified by silica gel chromatography (dry loaded, eluent: 5 - 80% gradient of DCM and chloroform containing 10% 2M Ammonia (in methanol)). The isolated solid was dried in vacuum oven at 45 °C overnight to afford the final product as 0.126 g (81%). LCMS E-S (M+H) = 470.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.56 (d, J=6.57 Hz, 6 H), 2.13 (s, 3 H), 2.25 (s, 3 H), 2.60 (s, 3 H), 4.43 (d, J=5.05 Hz, 2 H), 5.31 - 5.43 (m, 1H), 5.91 (s, 1 H), 7.60 (d, J=8.84 Hz, 1 H), 8.22 (s, 1 H), 8.29 - 8.37 (m, 2 H), 8.60 (s, 1 H), 8.97 (t, J=5.05 Hz, 1 H), 11.58 (br. s., 1 H), 12.84 (s, 1 H).
Example 31
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6- (phenylethynyl)-lH- razolo [3,4-b] pyridine-4-carboxamide

To a 10 mL microwave vial were sequentially added 6-chloro-N-[(4,6-dimethyl-2- oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethy 1)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (0.125 g, 0.334 mmol), sodium iodide (10.02 mg, 0.067 mmol) and zinc (4.37 mg, 0.067 mmol), DMSO(2.5 mL), triethylamine (0.093 mL, 0.669 mmol) and DBU (0.101 mL, 0.669 mmol). The stirring suspension was degassed with nitrogen for 5 min., wherein an emulsion had formed. Added next were phenylacetylene (0.110 mL, 1.003 mmol) and Pd(Ph3P)4 (0.039 g, 0.033 mmol). The sealed reaction mixture was placed onto a heat block, stirred at 90 °C for 3 hr, and then allowed to cool to room temperature overnight. The contents were poured onto water and 20% THF/EtOAc, stirred, and the layers separted. The organic layer was washed with brine, dried over MgS04, filtered, and concentrated in vacuo. The filter pad was washed with additional EtOAc. The combined filtrates were concentrated in vacuo to a yellow/orange residue that was dried on hi-vac pump. The crude solid was then pre-adsorbed onto silica gel and purified by silica gel chromatography (dry loaded, eluent: 5-80 % gradient of DCM and chloroform containing 10% 2M Ammonia (in methanol)). The isolated product was obtained as a yellow solid which was then further purified by reverse phase HPLC (mobile phase: 20-90% ACN in H20, 0.1% TFA, Gradient time: 8min). The isolated solid was dissolved in 10% MeOH/ CH2C12 and treated with 0.6 g of Silicycle carbonate resin for 30 min. The contents were filtered through Celite and the filter pad washed with additional 10% MeOH/CH2Cl2. The combined filtrates were concentrated in vacuo to afford a solid which was dried in vacuum oven for 18 hr. The final product was collected as 0.045 g (30%). LCMS E-S (M+H) = 440.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.51 (d, J=6.82 Hz, 6 H), 2.13 (s, 3 H), 2.21 (s, 3 H), 4.37 (d, J=4.80 Hz, 2 H), 5.18 - 5.33 (m, 1 H), 5.90 (s, 1H), 7.45 - 7.56 (m, 3 H), 7.64 - 7.73 (m, 2 H), 7.86 (s, 1 H), 8.42 (s, 1 H), 8.92 (t, J=4.80 Hz, 1 H), 11.55 (br. s., 1
Example 32
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2- phenylethyl)- -pyrazolo[3,4-6]pyridine-4-carboxamide

To a suspension of palladium on carbon (0.063 g, 0.059 mmol) in ethanol (1 mL) under nitrogen was added N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-6-(4-pyridinylethynyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (0.13 g, 0.295 mmol) and then ethanol (5 mL) and tetrahydrofuran (THF) (1.5 mL). The suspension was stirred under an atmosphere of hydrogen (ca. 1 atm, balloon) overnight. The reaction mixture was then evacuated with nitrogen, and diluted with 10%
MeOH/DCM. Celite was added and the contents stirred for 15 min., and then filtered through Celite (analytical grade) and washed with 10%MeOH/DCM. The filtrate was concentrated in vacuo and purified by silica gel chromatography (eluent: gradient of 5-95 %. Dichloromethane/Chloroform containing 10% 2M ammonia (in methanol). The collected solid was dried in vacuum oven at 45 °C for 18 h. The final product was collected as 0.112 g (84%). LCMS E-S (M+H) = 445.3. 1H NMR (400 MHz, DMSO- 6) δ ppm 1.46 (d, J=6.57 Hz, 6 H), 2.13 (s, 3 H), 2.21 (s, 3 H), 3.08 - 3.18 (m, 2 H), 3.22 - 3.29 (m, 2 H), 4.36 (d, J=5.05Hz, 2 H), 5.18 (quin, J=6.69 Hz, 1 H), 5.90 (s, 1 H), 7.28 (d, J=6.06 Hz, 2 H), 7.53 (s, 1 H), 8.26 (s, 1 H), 8.38 - 8.47 (m, 2 H), 8.72 (t, J=5.05 Hz, 1 H),11.56 (s, 1 H).
Example 33
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- pyridinylethynyl)-lH- razolo [3,4-b] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 31 from 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (.15 g, 0.401 mmol), sodium iodide (0.012 g, 0.080 mmol), zinc (5.25 mg, 0.080 mmol), DMSO (4.0 mL),
triethylamine (0.168 mL, 1.204 mmol), DBU (0.121 mL, 0.802), 4-ethynylpyridine (0.112 g, 0.802 mmol), and Pd(Ph3P)4 (0.046 g, 0.040 mmol). The crude product was purified by silica gel chromatography (eluent: Gradient of 5-100% of DCM and chloroform containing 10% 2M Ammonia (in methanol)). The isolated product was dried in vacuum oven for 18 hr. to afford the final product as 0.032 g (18%). LCMS E-S (M+H) = 441.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.51 (d, J=6.57 Hz, 6 H), 2.13 (s, 3 H), 2.21 (s, 3 H), 4.37 (d, J=4.80 Hz, 2 H), 5.20 - 5.32 (m, 1 H), 5.90 (s, 1 H), 7.62 - 7.69 (m, 2 H), 7.93 (s, 1 H), 8.45 (s, 1 H), 8.67 - 8.75 (m, 2 H), 8.93 (t, J=4.80 Hz, 1 H), 11.55 (s, 1 H).
Example 34
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6- (phenylamino)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide

To a 10 mL microwave vial were sequentially added 6-chloro-N-[(4,6-dimethyl-2- oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethy 1)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (0.100 g, 0.267 mmol), cesium carbonate (0.305 g, 0.936 mmol), 1,4-
Dioxane (2.5 mL) and aniline (0.049mL, 0.535 mmol). The stirring suspension was
degassed with nitrogen for 10 min. Added next were BINAP (0.033 g, 0.053 mmol) and palladium(II) acetate (6.01 mg, 0.027 mmol). The sealed mixture was stirred at 105 °C overnight. After cooling to room temperature, the contents were poured onto EtOAc and filtered through Celite. The filter pad was washed with additional 50% THF/EtOAc. The combined filtrates were treated with silica gel and concentrated in vacuo. The crude product was purified by silica gel chromatography (dry loaded, eluent: Gradient 5-80%> of DCM and chloroform containing 10% 2M Ammonia (in methanol))). The isolated solid was dried in a vacuum oven overnight and the final product collected as 0.061 g (52%). LCMS E-S (M+H) = 431.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.50 (d, J=6.57 Hz, 6 H), 2.13 (s, 3 H), 2.23 (s, 3 H), 4.35 (d, J=5.05 Hz, 2 H), 4.98 - 5.11 (m, 1 H), 5.90 (s, 1H), 6.93 - 7.00 (m, 2 H), 7.34 (t, J=7.96 Hz, 2 H), 7.86 (d, J=7.58 Hz, 2 H), 7.97 (s, 1 H), 8.59 (t, J=5.05 Hz, 1 H), 9.62 (s, 1 H), 11.58 (br. s., 1 H).
Example 35
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6- [(phenylmethyl)amino] -lH- razolo [3,4-b] pyridine-4-carboxamide

To a 10 mL microwave vial were sequentially added 6-chloro-N-[(4,6-dimethyl-2- oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethy 1)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (0.100 g, 0.267 mmol), ethanol (2.0mL) and then benzylamine (0.350 mL, 3.21 mmol) via syringe at once. The sealed contents were irradiated at 140 °C for 3 hr. The contents were transferred to a heat block and heated at 135 °C for 16 hr., and then at 145 °C for an additional 12 h. After cooling to room temperature, the contents were diluted with CH2CI2 and pre-absorbed onto silica gel. The crude product was purified by silica gel chromatography (dry loaded, eluent; gradient of 5-80% DCM and chloroform containing 10% 2M Ammonia (in methanol)). The isolated solid was triturated with MTBE, filtered, and washed with additional MTBE. The collected solid was dried in vacuum oven at 45 °C overnight to afford the final product as 0.067 g (55%). LCMS E-S (M+H) = 445.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.38 (d, J=6.57 Hz, 6 H), 2.12 (s, 3 H), 2.20 (s, 3 H), 4.31 (d, J=5.05 Hz, 2 H), 4.56 (d, J=6.06 Hz, 2 H), 4.92 (quin, J=6.69
Hz, 1 H), 5.88 (s, 1 H), 6.69 (s, 1 H), 7.18 - 7.25 (m, 1 H), 7.26 - 7.35 (m, 2 H), 7.35 - 7.43 (m, 2 H), 7.77 (t, J=5.94 Hz, 1 H), 7.83 (s, 1H), 8.47 (t, J=5.05 Hz, 1 H), 11.55 (br. s., 1 H).
Example 36
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-methyl-l-(l-methylethyl)- lH-pyrazolo -6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-methyl-l-(l- methylethyl)-lH-pyrazolo [3, 4-b] pyridine-4-carboxylic acid (60 mg, 0.27 mmol) and 3- (aminomethyl)-4,6-dimethyl-2(lH)-pyridinone.HCl (62 mg, 0.33 mmol) in DMSO (3 mL). l-hydroxy-7-azabenzotriazole (56 mg, 0.41 mmol) was added and the resulting mixture was degassed with nitrogen for 10 minutes. N-methylmorpholine (0.11 ml, 0.96 mmol) and EDC (79 mg, 0.41 mmol) were added, the vessel was sealed, and the light brown mixture was stirred at room temperature for 2 days. Next added 2 mL of water and the mixture was stirred for 10 min. Solids that precipitated were sonicated, and allowed to stand at room temperature for 10 min. The contents were filtered, washed with water, and dried to afford the title compound (68 mg, 68%) as a light pink solid. LCMS E-S (M+H) = 354.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.54 (s, 1 H), 8.69 (t, J=4.80 Hz, 1 H), 8.26 (s, 1 H), 7.46 (s, 1 H), 5.89 (s, 1 H), 5.20 (quin, J=6.69 Hz, 1 H), 4.36 (d, J=5.05 Hz, 2 H), 2.63 (s, 3 H), 2.21 (s, 3 H), 2.13 (s, 3 H), 1.48 (d, J=6.57 Hz, 6 H).
Example 37
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l,6-bis(l-methylethyl)-lH- pyrazolo [ -6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 52 using l,6-bis(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (70 mg, 0.28 mmol) to afford an off-white solid (95 mg, 86%). LCMS E-S (M+H) = 382.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.56 (br. s., 1 H, 8.76 (t, J=4.93 Hz, 1 H), 8.26 (s, 1 H), 7.50 (s, 1 H), 5.90 (s, 1 H), 5.15 - 5.25 (m, 1 H), 4.37 (d, J=5.05 Hz, 2 H), 3.13 - 3.23 (m, 1 H), 2.22 (s, 3 H), 2.13 (s, 3 H), 1.50 (d, J=6.57 Hz, 6 H), 1.32 (d, J=6.82 Hz, 6 H).
Example 38
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-phenyl- lH-pyrazo -6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined l-(l-methylethyl)-6- phenyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (70 mg, 0.25 mmol) and 3- (aminomethyl)-4,6-dimethyl-2(lH)-pyridinone.HCl (56.3 mg, 0.3 mmol) in DMSO (3 mL). l-hydroxy-7-azabenzotriazole (51 mg, 0.37 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. N-methylmorpholine (0.1 ml, 0.87 mmol) and EDC (72 mg, 0.37 mmol) were added, the vessel was sealed, and the bright yellow mixture was stirred at room temperature for 2 days. Next added 2 mL of water, and the contents were stirred for 10 min. Solids that precipitated were sonicated, and allowed to
stand at room temperature for 10 min. The reaction contents were filtered and washed with water. The solid was treated with 2 mL of EtOH, sonicated and heated, and then allowed to cool to room temperature. The contents were filtered, washed with water and dried to afford the title compound (74 mg, 70%) as a white solid. LCMS E-S (M+H) = 416.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.58 (s, 1 H), 8.98 (t, J=4.80 Hz, 1 H), 8.38 (s, 1 H), 8.25 - 8.30 (m, 2 H), 8.17 (s, 1 H), 7.49 - 7.59 (m, 3 H), 5.91 (s, 1 H), 5.30 - 5.38 (m, 1 H), 4.42 (d, J=4.80 Hz, 2 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.55 (d, J=6.57 Hz, 6 H).
Example 39
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(4-fluorophenyl)-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (90 mg, 0.24 mmol), (4-fluorophenyl)boronic acid (33.7 mg, 0.24 mmol) in 1,4-dioxane (3 mL). PdCl2(dppf)-CH2Cl2 adduct (19.7 mg, 0.024 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium carbonate (77 mg, 0.72 mmol) was added, the vessel was sealed, and the mixture was heated at 85 °C for 2 hrs and then at 100 °C overnight. Added an additional 2 eq of boronic acid and 0.2 eq of PdCl2(dppf)-CH2Cl2 adduct. The contents were heated at 120 °C for 4 hr. and then irradiated (microwave) first at 160 °C for 90 min, and then at 190 °C for 2 hrs. After cooling to room temperature, the solids were filtered, washed with DMSO and the filtrate was evaporated. The crude product was purified first by reverse-phase HPLC (CI 8, 5% to 80% CH3CN in water with 0.1% TFA, 18 minute gradient) and then silica gel chromatography (eluent: gradient 0 to 90: 10: 1 DCM/MeOH/NH4OH). The collected solid was dried to afford the title compound (13 mg, 12%>) as a white solid. LCMS E-S (M+H) = 434.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.58 (br. s., 1 H), 8.96 (t, J=4.67 Hz, 1 H), 8.38 (s, 1 H), 8.33 (dd, J=8.84, 5.56 Hz, 2 H), 8.16 (s, 1 H), 7.40
(t, J=8.84 Hz, 2 H), 5.91 (s, 1 H), 5.33 (dt, J=13.26, 6.76 Hz, 1 H), 4.41 (d, J=4.55 Hz, 2 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.55 (d, J=6.57 Hz, 6 H).
Example 40
6-{4-[(Dimethylamino)sulfonyl]phenyl}- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (85 mg, 0.23 mmol) and {4- [(dimethylamino)sulfonyl]phenyl}boronic acid (104 mg, 0.46 mmol) in DME/water (3 ml: l ml). PdCl2(dppf)-CH2Cl2 adduct (9.3 mg, 0.011 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium bicarbonate (57.3 mg, 0.68 mmol) was added and the insoluble light brown mixture was heated in an oil bath at 110 °C for 3 hrs. After cooling, 2 mL of water was added to the black mixture and solids that precipitated were filtered. DMF was added along with a few drops of water and solids were filtered. DCM/MeOH (1 : 1) was added to the grey solids and they were filtered and dried to afford the title compound (69 mg, 57%) as a grayish solid. LCMS E-S (M+H) = 523.2 . 1H NMR (400 MHz, DMSO-d6) δ ppm 11.59 (br. s., 1 H), 9.01 (br. s., 1 H), 8.51 (m, J=8.34 Hz, 2 H), 8.44 (s, 1 H), 8.27 (s, 1 H), 7.93 (m, J=8.34 Hz, 2 H), 5.91 (s, 1 H), 5.36 (dt, J=13.14, 6.57 Hz, 1 H), 4.43 (d, J=4.55 Hz, 2 H), 2.66 (s, 6 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.56 (d, J=6.57 Hz, 6 H).
Example 41
6-[6-(Dimethylamino)-3-pyridinyl]- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and [6-(dimethylamino)-3- pyridinyljboronic acid (53.3 mg, 0.32 mmol) in DME/water (3 mL : 1 mL). PdCl2 (dppf)- CH2C12 adduct (8.7 mg, 0.01 1 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium bicarbonate (53.9 mg, 0.64 mmol) was added, the vessel was sealed, and the insoluble green mixture was irradiated (microwave) at 150 °C for 20 min. After cooling, 2 mL of water was added to the dark green mixture and solids that precipitated were filtered. EtOAc was added, the mixture was heated and some hexanes were added. Solids were filtered, dissolved in DCM/MeOH (1 : 1), and filtered through a pad of silica gel and the filtrate was evaporated. The residue was dissolved in DCM and purified by Si02 chromatography (eluent: gradient of 0 to 90: 10 DCM/MeOH) to afford a residual oil that was triturated with EtOH/EtOAc (1 : 1). The resultant solid was filtered and dried to afford the title compound (46 mg, 45%) as a yellow solid. LCMS E-S (M+H) = 460.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1 1.58 (s, 1 H), 9.02 (d, J=2.27 Hz, 1 H), 8.87 (t, J=4.67 Hz, 1 H), 8.36 (dd, J=8.84, 2.53 Hz, 1 H), 8.31 (s, 1 H), 8.08 (s, 1 H), 6.79 (d, J=8.84 Hz, 1 H), 5.91 (s, 1 H), 5.25 - 5.34 (m, 1 H), 4.40 (d, J=4.80 Hz, 2 H), 3.12 (s, 6 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.53 (d, J=6.57 Hz, 6 H).
Example 42
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-{4- [(methylamino)sulfonyl]phenyl}-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4- carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (75 mg, 0.2 mmol), {4-[(methylamino)sulfonyl]phenyl}boronic acid (69 mg, 0.32 mmol) in DME/water (3 ml: l ml). PdCl2(dppf)-CH2Cl2 adduct (8.2 mg, 0.01 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium bicarbonate (50.6 mg, 0.6 mmol) was added , the vessel was sealed, and the insoluble mixture was heated in a microwave at 150 °C for 30 min. After cooling, 2 mL of water was added to the mixture and solids that precipitated were filtered. DCM/MeOH (1 : 1) was added and the solution was filtered through a pad of silica gel and the filtrate was evaporated. The residue was purified by silica gel chromatography (eluent: gradient of 0 to 90: 10 DCM/MeOH) and the isolated solid triturated in EtOAc. The solids were filtered and dried to afford the title compound (29 mg, 28%) as an off-white solid. LCMS E-S (M+H) = 509.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.58 (br. s., 1 H), 8.99 (t, J=4.93 Hz, 1 H), 8.47 (m, J=8.59 Hz, 2 H), 8.43 (s, 1 H), 8.25 (s, 1 H), 7.95 (m, J=8.59 Hz, 2 H), 7.60 (br. s., 1 H), 5.91 (s, 1 H), 5.36 (quin, J=6.69 Hz, 1 H), 4.42 (d, J=4.80 Hz, 2 H), 2.46 (s, 3 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.56 (d, J=6.82 Hz, 6 H).
Example 43
6-(4-Aminophenyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-l -pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and (4-aminophenyl)boronic acid (44 mg, 0.32 mmol) in DME/water (3 ml: l ml). PdCl2(dppf)-CH2Cl2 adduct (8.74 mg, 0.011 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min.
Sodium bicarbonate (53.9 mg, 0.64 mmol) was added, the vessel was sealed, and the insoluble light pink mixture was irradiated (microwave) at 150 °C for 30 min. After cooling, DCM/MeOH (1 : 1) was added, it was pre-absorbed on silica gel and purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1 DCM/MeOH/NH4OH). The isolated solid was treated with MeOH. The solids that precipitated were filtered and dried to afford the title compound (63 mg, 66%) as a yellow solid. LCMS E-S (M+H) = 431.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.56 (s, 1 H), 8.88 (t, J=4.93 Hz, 1 H), 8.27 (s, 1 H), 8.01 (s, 1 H), 7.99 (d, J=2.02 Hz, 2 H), 6.69 (s, 1 H), 6.67 (s, 1 H), 5.90 (s, 1 H), 5.61 (s, 2 H), 5.24 - 5.32 (m, 1 H), 4.40 (d, J=4.80 Hz, 2 H), 2.22 (s, 3 H), 2.13 (s, 3 H), 1.53 (s, 3 H), 1.51 (s, 3 H).
Example 44
!tylamino)phenyl] -TV- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl] - 1- (l-methylethyl)- -pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and [4-(acetylamino)phenyl]boronic acid (57.5 mg, 0.32 mmol) in DME/water (3 ml: l ml). PdCl2(dppf)-CH2Ci2 adduct (8.7 mg, 0.010 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium bicarbonate (53.9 mg, 0.64 mmol) was added, the vessel was sealed, and the insoluble light brown mixture was heated at 110 °C for 2.5 h. After cooling, 2 mL of water was added to the dark grey mixture and solids that precipitated were filtered. EtOAc was added, the mixture was heated and some hexanes were added. The solids were filtered, dissolved in DCM/MeOH (1 : 1) and 1 mL of DMF. The contents were pre-absorbed onto silica gel and purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1
DCM/MeOH/NH4OH). The isolated product was treated with MeOH, and the solids that precipitated were filtered, washed with hexanes and dried to afford the title compound (55 mg, 52%) as a white solid. LCMS E-S (M+H) = 473.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.57 (s, 1 H), 10.18 (s, 1 H), 8.93 (t, J=4.93 Hz, 1 H), 8.35 (s, 1 H), 8.22 (m, J=8.84 Hz, 2 H), 8.13 (s, 1 H), 7.76 (m, J=8.84 Hz, 2 H), 5.91 (s, 1 H), 5.28 - 5.37 (m, 1 H), 4.41 (d, J=4.80 Hz, 2 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 2.09 (s, 3 H), 1.54 (d, J=6.82 Hz, 6 H).
Example 45
-aminophenyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and (3-aminophenyl)boronic acid (46.9 mg, 0.34 mmol) in DME/water (3 ml: l ml). PdCl2(dppf)-CH2Cl2 adduct (8.74 mg, 0.01 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min.
Sodium bicarbonate (53.9 mg, 0.64 mmol) was added, the vessel was sealed, and the reaction mixture was irradiated (microwave) at 150 °C for 25 min. After cooling, 2 mL of water was added to the black mixture and solids that precipitated were filtered.
DCM/MeOH (1 : 1) was added, the mixture was pre-absorbed on silica gel and purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1 DCM/MeOH/NH4OH). The isolated product was treated with EtOH/EtOAc/hexanes (1 : 1 :1). The solids that precipitated were filtered and dried to afford the title compound (40 mg, 42%) as a light grey solid. LCMS E-S (M+H) = 431.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.56 (br. s., 1 H), 8.96 (t, J=4.93 Hz, 1 H), 8.34 (s, 1 H), 8.02 (s, 1 H), 7.46 (t, J=1.89 Hz, 1 H), 7.36 (d, J=7.58 Hz, 1 H), 7.18 (t, J=7.71 Hz, 1 H), 6.69 (dd, J=7.83, 1.52 Hz, 1 H), 5.90 (s, 1 H), 5.25 - 5.37 (m, 3 H), 4.40 (d, J=4.80 Hz, 2 H), 2.22 (s, 3 H), 2.13 (s, 3 H), 1.55 (s, 3 H), 1.54 (s, 3 H).
Example 46
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-{4- [(methylamino)carbonyl]phenyl}-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4- carboxamide

The title compound was prepared in the same manner as described in example 61 using (4-[(methylamino) carbonyl] phenyl} boronic acid (57.5 mg, 0.32 mmol) to afford a light grey solid (70 mg, 68%). LCMS E-S (M+H) = 473.1. 1H NMR (400 MHz, DMSO- d6) δ ppm 11.58 (s, 1 H), 9.00 (t, J=4.80 Hz, 1 H), 8.59 (q, J=4.38 Hz, 1 H), 8.41 (s, 1 H), 8.35 (m, J=8.59 Hz, 2 H), 8.24 (s, 1 H) 8.01 (m, J=8.59 Hz, 2 H), 5.91 (s, 1 H), 5.33 - 5.41 (m, 1 H), 4.42 (d, J=4.80 Hz, 2 H), 2.83 (d, J=4.55 Hz, 3 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.56 (s, 3 H), 1.55 (s, 3 H).
Example 47
6- [3-(Acetylamino)phenyl] -TV- [(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl] - l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and [3-(acetylamino)phenyl]boronic acid (57.5 mg, 0.32 mmol) in DME/water (3 ml: l ml). PdCl2(dppf)-CH2Ci2 adduct (8.7 mg, 0.010 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min.
Sodium bicarbonate (53.9 mg, 0.64 mmol) was added, the vessel was sealed and the reaction mixture was irradiated (microwave) at 150 °C for 30 min. After cooling, 2 mL of water was added and solids that precipitated were filtered. DCM/MeOH (1 : 1) was added, the mixture was pre-absorbed on silica gel and purified by silica gel chromatography (eluent: gradient 0 to 90 : 10 : 1 DCM/MeOH/NH4OH). The isolated product was treated with EtOH and DCM, filtered, and dried to afford the title compound (65 mg, 63%) as a white solid. LCMS E-S (M+H) = 473.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.58 (s, 1 H), 9.02 (t, J=4.80 Hz, 1 H), 8.61 - 8.68 (m, 2 H), 8.37 - 8.43 (m, 2 H), 8.21 (s, 1 H), 7.94 (d, J=7.83 Hz, 1 H), 7.65 (t, J=7.83 Hz, 1 H), 5.91 (s, 1 H), 5.37 (quin, J=6.63 Hz, 1 H), 4.42 (d, J=4.80 Hz, 2 H), 2.84 (d, J=4.29 Hz, 3 H), 2.24 (s, 3 H), 2.13 (s, 3 H), 1.57 (s, 3 H), 1.55 (s, 3 H).
Example 48
TV- [(4,6-Dimethyl-2-oxo- 1 ,2-dihydro-3-py ridinyl)methyl] -6-(2,4-dioxo- 1 ,2,3,4- tetrahydro-5-pyrimidinyl)-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4- carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and (2,4-dioxo-l,2,3,4-tetrahydro-5- pyrimidinyl)boronic acid (50 mg, 0.32 mmol) in DME/water (3 ml: l ml). PdCi2(dppf)-
CH2CI2 adduct (8.74 mg, 0.011 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium bicarbonate (53.9 mg, 0.64 mmol) was added, the vessel was sealed, and reaction mixture was heated in a microwave at 150 °C for 30 min. After cooling, 2 mL of water was added to the black mixture and solids that precipitated were filtered. DCM/MeOH (1 : 1) was added, the mixture was pre-absorbed on silica gel and purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1
DCM/MeOH/NH4OH). The isolated product was treated with EtOH/EtOAc (1 : 1) and sonicated. The solids that precipitated were filtered, washed with EtOH and DCM, and
dried to afford the title compound (41 mg, 42%) as a light grey solid. LCMS E-S (M+H) 449.9. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.58 (s, 1 H), 11.52 (br. s., 1 H), 11.47 (s 1 H), 8.75 (t, J=5.05 Hz, 1 H), 8.36 (s, 1 H), 8.31 (s, 1 H), 8.23 (s, 1 H), 5.90 (s, 1 H), 5.27 (quin, J=6.69 Hz, 1 H), 4.37 (d, J=5.05 Hz, 2 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.52 (s 3 H), 1.50 (s, 3 H).
Example 49
6-(2-Amino-4-pyridinyl)- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihy dro-3-pyridinyl)methyl] - 1
(l-methylethyl)-l -pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as example 48 using 4-
(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2-pyridinamine (65.9 mg, 0.3 mmol) to afford an off-white solid (60 mg, 73%). LCMS E-S (M+H) = 432.3. 1H NMR (400 MHz, DMSO-de) δ ppm 11.57 (s, 1 H), 8.99 (t, J=4.80 Hz, 1 H), 8.41 (s, 1 H), 8.05 - 8.13 (m, 2 H), 7.24 - 7.32 (m, 2 H), 6.14 (s, 2 H), 5.91 (s, 1 H), 5.33 (quin, J=6.69 Hz, 1 H), 4.41 (d, J=4.80 Hz, 2 H), 2.22 (s, 3 H), 2.13 (s, 3 H), 1.55 (d, J=6.57 Hz, 6 H).
Example 50
6-[4-(Aminosulfonyl)phenyl]- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as example 48 using [4- (aminosulfonyl) phenyl] boronic acid (64.5 mg, 0.32 mmol) to afford a light grey solid (76
mg, 74 %). LCMS E-S (M+H) = 495.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.58 (br. s., 1 H), 8.99 (t, J=4.80 Hz, 1 H), 8.41 - 8.48 (m, 3 H), 8.25 (s, 1 H), 8.00 (d, J=8.59 Hz, 2 H), 7.50 (s, 2 H), 5.91 (s, 1 H), 5.37 (quin, J=6.69 Hz, 1 H), 4.43 (d, J=4.80 Hz, 2 H), 2.23 (s, 3H), 2.14 (s, 3 H), 1.56 (d, J=6.57 Hz, 6 H).
Example 51
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-{4- [(methylsulfonyl)amino]phenyl}-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (75 mg, 0.2 mmol) and {4-
[(methylsulfonyl)amino]phenyl}boronic acid (43.1 mg, 0.2 mmol) in DME/water (3 ml: l ml). PdCl2(dppf)-CH2Cl2 adduct (8.2 mg, 0.01 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium bicarbonate (50.6 mg, 0.6 mmol) was added, the vessel was sealed, and the reaction mixture was irradiated (microwave) at 150 °C for 25 min. After cooling, 2 mL of water was added to the black mixture and solids that precipitated were filtered. DCM/MeOH (1 : 1) was added, the mixture was pre- absorbed on silica gel and purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1 DCM/MeOH/NH4OH). The isolated product was suspended in EtOAc heated, and sonicated. Some hexanes was added and the contents allowed to cool to room temperature. Solids that precipitated were filtered. The solid was then suspended in EtOH, heated, and sonicated. After cooling to room temperature, the solids were filtered and dried to afford the title compound (38 mg, 36%) as a beige solid. LCMS E-S (M+H) = 509.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.57 (s, 1 H), 10.05 (br. s., 1 H), 8.93 (t, J=4.93 Hz, 1 H), 8.35 (s, 1 H), 8.24 (m, J=8.59 Hz, 2 H), 8.11 (s, 1 H), 7.37 (m, J=8.84 Hz, 2 H), 5.91 (s, 1 H), 5.32 (quin, J=6.69 Hz, 1 H), 4.41 (d, J=4.80 Hz, 2 H), 3.08 (s, 3 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.55 (s, 3 H), 1.54 (s, 3 H).
Example 52
6-{4-[(Dimethylamino)sulfonyl]phenyl}- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-3-methyl-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4- carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] -3 -methyl- 1 -( 1 -methylethyl)- 1 H- pyrazolo[3,4-b]pyridine-4-carboxamide (75 mg, 0.19 mmol) and N,N-dimethyl-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)benzenesulfonamide (90 mg, 0.29 mmol) in
DME/water (3 ml: 1 ml). PdCl2 (dppf)-CH2Cl2 adduct (7.9 mg, 0.009 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium bicarbonate (48.7 mg, 0.58 mmol) was added, the vessel was sealed, and the reaction mixture was irradiated (microwave) at 150 °C for 30 min. After cooling, 2 mL of water was added and solids that precipitated were filtered. DCM/MeOH (1 :1) was added, the mixture was pre- absorbed on silica gel and purified by silica gel chromatography (eluent: gradient 0 to
90: 10: 1 DCM/MeOH/NH4OH). The isolated product was treated with EtOH/EtOAc (1 : 1), the solids filtered and washed with EtOAc and DCM, and then dried to afford the title compound (83 mg, 78%) as a white solid. LCMS E-S (M+H) = 537.1. 1H NMR (400 MHz, DMSO-de) δ ppm 11.54 (s, 1 H) 8.79 (t, J=4.93 Hz, 1 H) 8.47 (m, J=8.59 Hz, 2 H) 7.90 (m, J=8.59 Hz, 2 H) 7.80 (s, 1 H) 5.89 (s, 1 H) 5.28 (quin, J=6.69 Hz, 1 H) 4.40 (d, J=5.05 Hz, 2 H) 2.66 (s, 6 H) 2.47 (s, 3 H) 2.25 (s, 3 H) 2.12 (s, 3 H) 1.53 (s, 3 H) 1.51 (s, 3 H).
Example 53
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- piperidinylamino)-l -pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4- b]pyridine-4-carboxamide ( 60 mg, 0.16 mmol) and 4-piperidinamine (2 mL) in EtOH (3 mL). The vessel was sealed and the reaction mixture was irradiated (microwave) at 125 °C for 5 hr and then at 160 °C for 90 min. The mixture was evaporated under vacuum and the resulting residue was partitioned between EtOAc and water. Organics were washed with water (2x) and brine, dried over MgSC^, filtered and evaporated. The residue was purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1 DCM/MeOH/NH4OH). The isolated product was treated with EtOAc/ether (1 : 1) and allowed to stand at room temperature overnight. The solids that had precipitated were triturated, filtered, and dried to afford the title compound as a pale yellow solid 25 mg (35%). LCMS E-S (M+H) = 438.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.66 (t, J=4.67 Hz, 1 H) 7.98 (s, 1 H) 7.12 (s, 1 H) 5.89 (s, 1 H) 4.98 (dt, J=13.33, 6.60 Hz, 1 H) 4.30 - 4.41 (m, 4 H) 3.01 (t, J=11.62 Hz, 2 H) 2.85 (br. s., 1 H) 2.20 (s, 3 H) 2.12 (s, 3 H) 1.80 (d, J=10.36 Hz, 2 H) 1.44 (s, 3 H) 1.43 (s, 3 H) 1.18 - 1.30 (m, 3 H) 0.79 - 0.91 (m, 1 H)
Example 54
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6- (tetrahydro-2H-pyran-4-ylamino)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (60 mg, 0.16 mmol) and tetrahydro-2H-pyran-4-amine (2 ml, 19 mmol) in EtOH (3 mL). The vessel was sealed and the reaction mixture was irradiated (microwave) at 125 °C for 2 hr. and then at 170 °C for 1 hr. The mixture was concentrated to ca. 20% volume and NMP (2 mL) added to the reaction mixture. The contents were again irradiated (microwave) first at 180 °C for 2.5 h and then at 190 °C for 75 min. The mixture was evaporated under vacuum, and the resulting residue was purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1 DCM/MeOH/NH4OH). The isolated product was treated with EtOAc/hexanes (1 : 1), sonicated, and heated. After cooling, solids that precipitated were filtered and dried to afford the title compound (46 mg, 64%) as an off-white solid. LCMS E-S (M+H) = 439.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.55 (s, 1 H) 8.44 (t, J=5.05 Hz, 1 H) 7.83 (s, 1 H) 7.20 (d, J=7.33 Hz, 1 H) 6.63 (s, 1 H) 5.89 (s, 1 H) 4.93 (quin, J=6.69 Hz, 1 H) 4.32 (d, J=5.05 Hz, 2 H) 3.99 - 4.06 (m, 1 H) 3.88 (dt, J=11.49, 3.47 Hz, 2 H) 3.45 (td, J=11.37, 2.02 Hz, 2 H) 2.21 (s, 3 H) 2.13 (s, 3 H) 1.95 (d, J=10.61 Hz, 2 H) 1.41 - 1.51 (m, 8 H).
Example 55
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[(4- pyridinylmethyl)ami -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (60 mg, 0.16 mmol) and (4-pyridinylmethyl)amine (0.065 ml, 0.64 mmol) in EtOH (2 mL). The vessel was sealed and the reaction mixture was heated at 90 °C overnight. An additional 1 mL of (4-pyridinylmethyl) amine was added and the mixture was heated in a microwave at 160 °C for 90 min. The EtOH solvent was removed under reduced pressure, 2 mL of NMP was added, and the mixture was then irradiated (microwave) at 180 °C for 90 min. The mixture was evaporated under vacuum, and the resulting residue was partitioned between EtOAc and water. The crude residue was extracted with EtOAc, DCM and DCM/isopropanol (70:30). The combined organics were washed with water (2x) and brine, dried over MgS0 , filtered and evaporated. The residue was purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1
DCM/MeOH/NH40H). The isolated yellow oil was treated with EtOAc and sonicated.
The filtered solid was washed with hexanes and dried to afford the title compound (26 mg, 36%) as a white solid. LCMS E-S (M+H) = 446.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.56 (s, 1 H) 8.50 (t, J=5.18 Hz, 1 H) 8.43 - 8.48 (m, 2 H) 7.89 (t, J=5.94 Hz, 1 H) 7.84 (s, 1 H) 7.34 (d, J=6.06 Hz, 2 H) 6.73 (s, 1 H) 5.89 (s, 1 H) 4.83 (quin, J=6.69 Hz, 1 H) 4.58 (d, J=5.81 Hz, 2 H) 4.32 (d, J=5.05 Hz, 2 H) 2.21 (s, 3 H) 2.13 (s, 3 H) 1.32 (d, J=6.82 Hz, 6 H).
Example 56
6- { [2-(Dimethylamino)ethyl] amino}- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and (2-aminoethyl)dimethylamine (37.7 mg, 0.43 mmol) in NMP (2 mL). The vessel was sealed and the reaction mixture was irradiated (microwave) at 150 °C for lh. The mixture was evaporated under vacuum, and the resulting residue was partitioned between EtOAc and water. The contents were extracted with EtOAc, DCM and DCM/isopropanol (70:30). The combined organics were washed with water (2x) and brine, dried over MgSC^, filtered and evaporated. The residue was purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1
DCM/MeOH/NH40H). The isolated yellow oil was treated with EtOAc and sonicated. Solids that precipitated were filtered, washed with hexanes, and dried to afford the title compound (60 mg, 65%) as a white solid. LCMS E-S (M+H) = 425.9. 1H NMR (400 MHz, DMSO-de) δ ppm 11.55 (br. s., 1 H) 8.43 (t, J=5.05 Hz, 1 H) 7.84 (s, 1 H) 7.08 (t, J=5.05 Hz, 1 H) 6.67 (s, 1 H) 5.89 (s, 1 H) 4.94 (quin, J=6.63 Hz, 1 H) 4.32 (d, J=5.05 Hz, 2 H) 3.44 (q, J=6.48 Hz, 2 H) 2.44 (t, J=6.69 Hz, 2 H) 2.17 - 2.22 (m, 9 H) 2.12 (s, 3 H) 1.43 (d, J=6.57 Hz, 6 H).
Example 57
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-[(2-hydroxyethyl)amino] l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6- dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3,4- b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and 2-aminoethanol (1 ml) in NMP (2 mL). The vessel was sealed and the reaction mixture was irradiated (microwave) at 130 °C for 1 h and then at 140 °C for 90 min. The mixture was evaporated under vacuum and the residue was purified by silica gel chromatography (eluent: gradient 0 to 90: 10: 1
DCM/MeOH/NH^OH). The isolated yellow oil was treated with EtOAc, sonicated, and treated with hexanes. The solids were filtered, dissolved in DMF (1 mL), and a few drops of water were added. The solution was allowed to stand at room temperature overnight, and then sonicated. The filtered solid was washed with Hexanes/EtOAc (1 : 1), and then dried to afford the title compound (55 mg, 63%) as a light yellow solid. LCMS E-S (M+H) = 399.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.56 (br. s., 1 H) 8.44 (br. s., 1 H) 7.84 (s, 1 H) 7.20 (br. s., 1 H) 6.68 (s, 1 H) 5.89 (s, 1 H) 4.95 (dt, J=13.01, 6.38 Hz, 1 H) 4.72 (t, J=4.93 Hz, 1 H) 4.32 (d, J=4.80 Hz, 2 H) 3.58 (d, J=5.56 Hz, 2 H) 3.38 - 3.46 (m, 2 H) 2.21 (s, 3 H) 2.12 (s, 3 H) 1.43 (d, J=6.57 Hz, 6 H)
Example 58
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[5- (methyloxy)-3-pyridinyl -lH-pyrazolo [3,4-b] pyridine-4-carboxamide

To a 20 mL microwave vial were sequentially added 6-chloro-N-[(4,6-dimethyl-2- oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethy 1)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (100 mg, 0.267 mmol, 3-(methyloxy)-5-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)pyridine (69.2 mg, 0.294 mmol), PdCl2(dppf)-CH2Ci2 adduct (21.84 mg, 0.027 mmol), (DME) (5 mL), and water (2 mL). The reaction mixture was degassed with nitrogen for 5 min. Sodium bicarbonate (67.4 mg, 0.802 mmol) was added and the contents sealed and irradiated (microwave) at 140 °C. The reaction mixture was cooled to room temperature and poured on a silica column (through Na2S04) and purified by silica gel chromatography (eluent : 5% MeOH/CH2Cl2) which provided the desired product as an off-white solid after preciptation from EtOAc/MeOH. The final product was collected as 0.090 g (76%). LCMS E-S (M+H) 447.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.57 (s., 1 H), 9.06 (d, J=1.52 Hz, 1 H), 8.92 - 9.00 (m, 1 H), 8.39 - 8.48 (m, 2 H), 8.24 (s, 1 H), 8.14 (d, J=1.77 Hz, 1 H), 5.91 (s, 1 H), 5.30 - 5.45 (m, 1 H), 4.42 (d, J=4.80 Hz, 2 H), 3.97 (s, 3 H), 2.24 (s, 3 H), 2.13 (s, 3 H), 1.55 (d, J=6.82 Hz, 6 H).
Example 59
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[2- (methyloxy)-4-pyridinyl] - lH-pyrazolo [3,4-6] pyridine-4-carboxamide

Example 60
6-(6-Amino-3-pyridinyl)- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihy dro-3-pyridinyl)methyl] - 1-
(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using 6-aminopyridine-3-boronic acid pinacol ester (64.8 mg, 0.294 mmol) to give the desired product as a grey solid after evaporation and preciptation from EtOAc/MeOH.
The final product was collected as 0.060 g (49%). LCMS E-S (M+H) = 432.2. 1H NMR (400 MHz, DMSO-de) δ ppm 11.57 (br. s., 1 H) 8.85 - 8.92 (m, 2 H) 8.31 (s, 1 H) 8.26 (dd, J=8.59, 2.53 Hz, 1 H) 8.04 (s, 1 H) 6.57 (d, J=8.59 Hz, 1 H) 6.46 (s, 2 H) 5.90 (s, 1 H) 5.25 - 5.33 (m, 1 H) 4.40 (d, J=4.80 Hz, 2 H) 2.22 (s, 3 H) 2.13 (s, 3 H) 1.53 (d, 6 H).
Example 61
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[5- (methylsulfony -3-pyridinyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using [5-(methylsulfonyl)-3-pyridinyl]boronic acid (59.1 mg, 0.294 mmol) to give the desired product as an off-white solid after evaporation and preciptation from
EtOAc/MeOH. The final product was collected as 0.079 g (59%). LCMS E-S (M+H) = 495.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.59 (br. s., 1 H), 9.76 (d, J=2.27 Hz, 1 H), 9.21 (d, J=2.02 Hz, 1 H), 9.00 - 9.06 (m, 2 H), 8.46 (s, 1 H), 8.37 (s, 1 H), 5.91 (s, 1 H), 5.36 - 5.44 (m, 1 H), 4.43 (d, J=4.80 Hz, 2 H), 3.45 (s, 3 H), 2.24 (s, 3 H), 2.13 (s, 3 H), 1.56 (d, 6 H).
Example 62
TV- [(4,6-Dimethyl-2-oxo- 1 ,2-dihydr o-3-pyridinyl)methyl] -6-(2-furanyl)- 1-(1- methylethyl)- -pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 2-(2-furanyl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (57.1 mg, 0.294 mmol) to give the desired product as an white solid after evaporation and preciptation from
EtOAc/MeOH. The final product was collected as 0.049 g (44%). LCMS E-S (M+H) 406.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.57 (s, 1 H) 8.93 (t, J=4.93 Hz, 1 H) 8.33
(s, 1 H) 7.97 (s, 1 H) 7.94 (d, J=1.01 Hz, 1 H) 7.34 (d, J=2.78 Hz, 1 H) 6.73 (dd, J=3.54, 1.77 Hz, 1 H) 5.90 (s, 1 H) 5.19 - 5.32 (m, 1 H) 4.39 (d, J=4.80 Hz, 2 H) 2.23 (s, 3 H) 2.13 (s, 3 H) 1.52 (d, 6 H).
Example 63
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-{6-
[(methylamino)carbonyl]-3-pyridinyl}-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine- -carboxamide
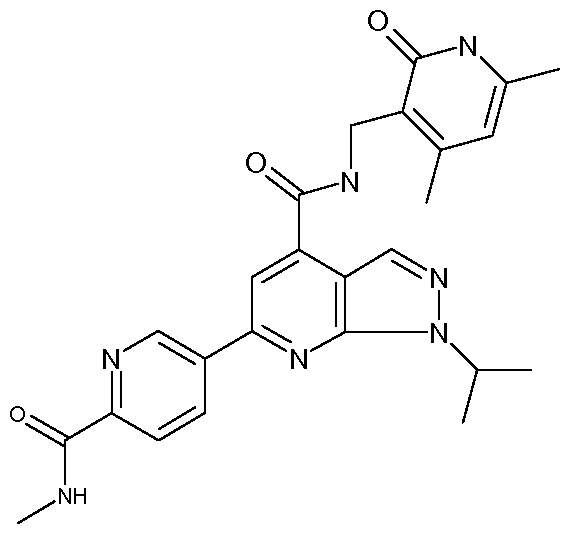
The title compound was prepared in the same manner as described in example 58 using 2-(N-methylamidocarboxy)-5-pyridineboronic acid pinacol ester (70.1 mg, 0.267 mmol) to give the desired product as an off-white solid after evaporation and preciptation from EtOAc/MeOH. The final product was collected as 0.080 g (60%). LCMS E-S (M+H) = 474.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.59 (br. s., 1 H), 9.46 (d, J=l .52 Hz, 1 H), 8.98 (t, J=4.55 Hz, 1 H), 8.85 (d, J=5.05 Hz, 1 H), 8.78 (dd, J=8.21, 2.15 Hz, 1 H), 8.44 (s, 1 H), 8.31 (s, 1 H), 8.20 (d, J=8.08 Hz, 1 H), 5.91 (s, 1 H), 5.30 - 5.42 (m, 1 H), 4.42 (d, J=4.55 Hz, 2 H), 2.87 (d, J=4.80 Hz, 3 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.56 (d, 6 H).
Example 64
6- [5- [(Cyclopropylsulfonyl)amino] -6-(methyloxy)-3-pyridinyl] -N- [(4,6-dimethyl-2- oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine- -carboxamide

Example 65
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-{5- [(phenylsulfonyl)amino]-3-pyridinyl}-lH-pyrazolo[3,4-b]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using N-[5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-3-pyridinyl]benzenesulfonamide (96 mg, 0.267 mmol) to give the desired product as an off-whitesolid after evaporation and preciptation from EtOAc/MeOH. The final product was collected as 0.050 g (33%). LCMS E-S (M+H) = 572.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.57 (s, 1 H), 10.83 (s, 1 H), 9.10 (d, J=1.77 Hz, 1 H), 8.95 (t, J=5.05 Hz, 1 H), 8.37 - 8.45 (m, 2 H), 8.32 (t, J=2.15 Hz, 1 H), 8.17 (s, 1 H), 7.82 - 7.91 (m, 2 H), 7.55 - 7.69 (m, 3 H), 5.90 (s, 1 H),
5.18 - 5.30 (m, 1 H), 4.41 (d, J=5.05 Hz, 2 H), 2.22 (s, 3 H), 2.13 (s, 3 H), 1.58 (d, J=6.82 Hz, 6 H).
Example 66
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[2-(4- morpholinyl)-4-pyridinyl] - -pyrazolo [3,4-b] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using 4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2-pyridinyl]morpholine (78 mg, 0.267 mmol) to give the desired product as an off-white solid after evaporation and preciptation from EtOAc/MeOH. The final product was collected as 0.110 g (58%). LCMS E-S (M+H) 502.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.57 (s, 1 H), 8.95 (t, J=5.05 Hz, 1 H), 8.40 (s, 1 H), 8.31 (d, J=5.30 Hz, 1 H), 8.18 (s, 1 H), 7.58 (s, 1 H), 7.51 (dd, J=5.31, 1.26 Hz, 1 H), 5.90 (s, 1 H), 5.39-5.30 (m, 1 H), 4.42 (d, J=4.80 Hz, 2 H), 3.72 - 3.80 (m, 4 H), 3.54 - 3.62 (m, 4 H), 2.24 (s, 3 H), 2.13 (s, 3 H), 1.55 (d, J=6.57 Hz, 6 H).
Example 67
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[6-(4- morpholinyl)-3-pyridiny -lH-pyrazolo [3,4-b] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using 6-morpholinopyridine-3-boronic acid (55.6 mg 0.267 mmol) to give the desired product as an off-white solid after evaporation and preciptation from EtOAc/MeOH. The final product was collected as 0.110 g (82%). LCMS E-S (M+H) 502.1. 1H NMR (400 MHz, DMSO-de) δ ppm 11.57 (s, 1 H), 9.06 (d, J=2.27 Hz, 1 H), 8.88 (t, J=4.93 Hz, 1 H), 8.41 (dd, J=8.97, 2.40 Hz, 1 H), 8.33 (s, 1 H), 8.11 (s, 1 H), 7.00 (d, J=9.09 Hz, 1 H), 5.91
(s, 1 H), 5.34-5.28 (m, 1 H), 4.40 (d, J=4.80 Hz, 2 H), 3.71 - 3.78 (m, 4 H), 3.55 - 3.62 (m, 4 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.54 (d, J=6.82 Hz, 6 H).
Example 68
N-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[6- (methyloxy)-3-pyridinyl] -lH-pyrazolo [3,4-b] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using [6-(methyloxy)-3-pyridinyl]boronic acid ( 40.9 mg 0.267 mmol) to give the desired product as an off-white solid after evaporation and preciptation from EtOAc/MeOH. The final product was collected as 0.060 g (50%). LCMS E-S (M+H) 447.2. 1H NMR (400
MHz, DMSO- g) δ ppm 11.58 (s, 1 H), 9.07 (d, J=2.02 Hz, 1 H), 8.90 (t, J=4.93 Hz, 1 H), 8.55 (dd, J=8.59, 2.53 Hz, 1 H), 8.37 (s, 1 H), 8.17 (s, 1 H), 7.01 (d, J=8.84 Hz, 1 H), 5.91 (s, 1 H), 5.36-5.28 (m, 1 H), 4.41 (d, J=5.05 Hz, 2 H), 3.95 (s, 3 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.55 (d, J=6.57 Hz, 6 H).
Example 69
6-[6-(Acetylamino)-3-pyridinyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using 2-acetamidopyridine-5-boronic acid pinacol ester (77 mg, 0.294 mmol) to give the desired product as an off-white solid after evaporation and preciptation from
EtOAc/MeOH. The final product was collected as 0.065 g (51%). LCMS E-S (M+H) =
474.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.57 (s, 1 H), 10.76 (s, 1 H), 9.19 (d, J=1.77 Hz, 1 H), 8.93 (t, J=4.80 Hz, 1 H), 8.61 (dd, J=8.84, 2.27 Hz, 1 H), 8.38 (s, 1 H), 8.25 (d, J=8.84 Hz, 1 H), 8.20 (s, 1 H), 5.91 (s, 1 H), 5.30 - 5.40 (m, 1 H), 4.42 (d, J=5.05 Hz, 2 H), 2.23 (s, 3 H), 2.14 (s, 3 H), 2.13 (s, 3 H), 1.54 (d, J=6.82 Hz, 6 H).
Example 70
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2- methylphenyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 58 using 2-methylphenyl boronic acid (40.0 mg, 0.294 mmol) to give the desired product as an off-white solid after evaporation and preciptation from MeOH. The final product was collected as 0.065 g (51%). LCMS E-S (M+H) = 430.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.54 (s, 1 H) 8.89 (t, J=4.93 Hz, 1 H) 8.39 (s, 1 H) 7.74 (s, 1 H) 7.56 (d, J=7.07 Hz, 1 H) 7.31 - 7.41 (m, 3 H) 5.89 (s, 1 H) 5.20 - 5.27 (m, 1 H) 4.38 (d, J=4.80 Hz, 2 H) 2.40 (s, 3 H) 2.21 (s, 3 H) 2.12 (s, 3 H) 1.53 (d, 6 H).
Example 71
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(lH- pyrazol-4-yl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

To a 5-mL microwave vial were added 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2- dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (70 mg, 0.187 mmol), 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-
pyrazole (47.2 mg, 0.243 mmol), DMSO(1.5 mL) and sodium carbonate (0.281 mL, 0.562 mmol), and the mixture was degassed with nitrogen for 5 min. Next added
bis(triphenylphosphine) palladium(II) chloride (10.51 mg, 0.015 mmol) and the vial was sealed. The mixture was irradiated (microwave) at 140 °C for 12h. The reaction mixture was filtered and the residue was washed with DMSO. The DMSO solution of crude
product was purified using reverse-phase HPLC. The TFA salt of the product obtained was neutralized with saturated NaHC03, washed with water, and dried under high vacuum to give 12 mg (16%) of product. LCMS: (M+H)+=406.2 1H NMR (400 MHz, DMSO-d6) 5 ppm 1.45 - 1.60 (m, J= 6.4 Hz, 6 H), 2.13 (s, 3 H), 2.23 (s, 3 H), 4.39 (d, J= 4.6 Hz, 2 H), 5.27 (quin, J= 6.7 Hz, 1 H), 5.90 (s, 1 H), 7.91 (s, 1 H), 8.22 - 8.31 (m, 1 H), 8.33 (m, 2 H), 8.77 (t, J= 4.7 Hz, 1 H).
Example 72
6-(2-Amino-5-pyrimidinyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

To a 5-mL microwave vial was added DMSO (2 mL) and it was degassed with nitrogen for 5 min. Next added 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl] - 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2-pyrimidinamine (49.7 mg, 0.225 mmol), sodium carbonate (59.5 mg, 0.562 mmol) and
bis(triphenylphosphine)palladium(II) chloride (9.20 mg, 0.013 mmol). The mixture was degassed for additional 5 min, sealed, and irradiated (microwave) at 135 °C for 15h. The mixture was filtered and the residue was washed with DMSO. The DMSO solution of the crude product was purified using reverse-phase HPLC. The TFA salt of the product was neutralized with saturated NaHC03 solution, filtered, washed with water, and dried under high vacuum to give 14 mg (17%) of product. LCMS E-S (M+H) = 433.2 1H NMR (400 MHz, DMSO-de) δ ppm 1.52 (d, J= 6.4 Hz, 6 H), 2.13 (s, 3 H), 2.22 (s, 3 H), 4.40 (d, J =
4.6 Hz, 2 H) 5.22 - 5.41 (m, 1 H), 5.90 (s, 1 H), 7.18 (m, 2 H), 8.08 (s, 1 H), 8.34 (s, 1 H), 8.86 (m, 1 H), 9.11 (s, 2 H), 11.58 (br. s., 1 H).
Example 73
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(3-pyridinyl)- lH-pyrazolo[3,4-6]pyridine-4-carboxamide

To a 5-mL microwave vial were added 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2- dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide (70 mg, 0.187 mmol), 3-pyridinylboronic acid (29.9 mg, 0.243 mmol),
DMSO (1.5 mL) and sodium carbonate (0.281 mL, 0.562 mmol), and the mixture was degassed with nitrogen for 10 min. Next was added bis(triphenylphosphine)palladium(II) chloride (10.51 mg, 0.015 mmol) and the vial was sealed. The reaction mixture was
irradiated (microwave) at 140 °C overnight. The reaction mixture was filtered and the residue was washed with DMSO. The crude product in DMSO was purified using
reverse-phase HPLC. The TFA salt of the product was neutralized using saturated
NaHC03, filtered, washed with water and dried under high vacuum to give 33 mg (42%) of product. LCMS E-S (M+H) = 417.2 1H NMR (400 MHz, DMSO- 6) 5 ppm l .56 (d, J
= 6.4 Hz , 6 H), 2.13 (s, 3 H), 2.23 (s, 3 H), 4.42 (d, J= 4.29 Hz, 2 H), 5.26 - 5.43 (m, 1
H), 5.91 (s, 1 H), 7.62 (dd, J= 7.3, 5.0 Hz, 1 H), 8.26 (s, 1 H), 8.43 (s, 1 H), 8.64 (d, J =
7.8 Hz, 1 H), 8.70 - 8.79 (m, 1 H), 8.96 (br. s., 1 H), 9.46 (br. s., 1 H), 11.59 (br. s., 1 H).
Example 74
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(lH-indazol-5-yl)-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

To a 5-mL microwave vial were added 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-indazole (59.4 mg, 0.243 mmol), DMSO(1.5 mL) and sodium carbonate (0.281 mL, 0.562 mmol), and the mixture was degassed with nitrogen for 10 min. Next was added bis(triphenylphosphine)palladium(II) chloride (10.51 mg, 0.015 mmol). The contents were sealed and irradiated (microwave) at 140 °C overnight. The mixture was filtered and the residue was washed with DMSO. The crude product in DMSO was purified using reverse-phase HPLC. The TFA salt of the product was neutralized using saturated NaHC03, filtered, washed with water and dried under high vacuum to give 41 mg (48%) of product. LCMS E-S (M+H) = 456.0 1H NMR (400 MHz, DMSO- 6) δ ppm 1.56 (d, J= 6.8 Hz , 6 H), 2.14 (s, 3 H), 2.23 (s, 3 H), 4.38 - 4.53 (m, 2 H), 5.27 - 5.49 (m, 1 H), 5.91 (s, 1 H), 7.59 - 7.75 (m, 1 H), 8.24 (s, 2 H), 8.37 (s, 2 H), 8.69 (s, 1 H), 8.95 - 9.09 (m, 1 H), 11.49 - 11.67 (m, 1 H), 13.21 - 13.35 (m, 1 H).
Example 75
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(lH-indazol-6-yl)-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

DMSO(1.5 mL), sodium carbonate (0.281 mL, 0.562 mmol), and
bis(triphenylphosphine)palladium(II) chloride (10.51 mg, 0.015 mmol). The final product was collected as 55 mg (65%). LCMS E-S (M+H) = 456.0 1H NMR (400 MHz, DMSO- d6) δ ppm 1.49 - 1.65 (m, J= 6.4 Hz, 6 H), 2.14 (s, 3 H), 2.23 (s, 3 H), 4.43 (d, J = 4.6
Hz, 2 H), 5.36 (quin, J= 6.7 Hz, 1 H), 5.87 - 5.97 (m, 1 H), 7.52 - 7.72 (m, 2 H) 7.92 (d, J= 8.6 Hz, 1 H), 8.08 (d, J= 8.6 Hz, 1 H), 8.16 (s, 1 H), 8.26 (s, 1 H) , 8.37 - 8.49 (m, 2
H), 8.96 - 9.14 (m, 1 H), 11.59 (br. s., 1 H).
Example 76
6-(lH-l,2,3-Benzotriazol-5-yl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 1H-1,2,3- benzotriazol-5-ylboronic acid (39.7 mg, 0.243 mmol), DMSO(2 mL), sodium carbonate
(0.281 mL, 0.562 mmol) and bis(triphenylphosphine)palladium(II) chloride (10.51 mg,
0.015 mmol). The final product was collected as 22 mg (26%). LCMS E-S (M+H) =
457.1 1H NMR (400 MHz, DMSO-d6) δ ppm 1.57 (d, 6 J = 6.4 Hz, H), 2.14 (s, 3 H), 2.24 (s, 3 H), 4.43 (d, J= 4.3 Hz, 2 H), 5.31 - 5.46 (m, 1 H), 5.91 (s, 1 H), 8.07 (br. s., 1 H),
8.34 (s, 1 H), 8.41 (m, 2 H), 8.85 (br. s., 1 H), 9.02 (br. s., 1 H), 11.59 (br. s., 1 H).
Example 77
V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l-methylethyl)-6-(2- oxo-2,3-dihydro-lH-benzimidazol-5- l)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 5- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l,3-dihydro-2H-benzimidazol-2-one (61.0 mg, 0.235 mmol), DMSO(1.5 mL) sodium carbonate (0.281 mL, 0.562 mmol), and
bis(triphenylphosphine)palladium(II) chloride (10.13 mg, 0.014 mmol). The final product was collected as 19 mg (21%). LCMS E-S (M+H) = 486.3 1H NMR (400 MHz, DMSO- d6) 5 ppm 1.50 (d, J= 6.8 Hz, 6 H), 2.12 (s, 3 H), 2.24 (s, 3 H), 2.43 (s, 3 H), 4.39 (d, J = 4.6 Hz, 2 H), 5.15 - 5.35 (m, 1 H), 5.89 (s, 1 H), 7.05 (d, J= 8.1 Hz, 1 H), 7.61 (s, 1 H), 7.77 - 7.93 (m, 2 H), 8.70 - 8.85 (m, 1 H), 10.79 (br. s., 1 H), 10.87 (br. s., 1 H), 11.54 (br. s., 1 H).
Example 78
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2-oxo- 2,3-dihydro-lH-indol-6-yl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 6-
(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l,3-dihydro-2H-indol-2-one (63.1 mg, 0.243 mmol), DMSO(1.5 mL), sodium carbonate (0.281 mL, 0.562 mmol), and
bis(triphenylphosphine)palladium(II) chloride (10.51 mg, 0.015 mmol). The final product was collected as 10 mg (11%). LCMS E-S (M+H) = 471.1 1H NMR (400 MHz, DMSO- d6) δ ppm 1.55 (d, J= 6.8 Hz, 6 H), 2.13 (s, 3 H), 2.22 (s, 3 H), 3.58 (s, 2 H), 4.41 (d, J = 4.8 Hz, 2 H), 5.31 (quin, J= 6.6 Hz, 1 H), 5.91 (s, 1 H), 7.38 (d, J= 7.6 Hz, 1 H), 7.73 (d, J= 1.0 Hz, 1 H), 7.87 (dd, J=7.8, 1.5 Hz, 1 H), 8.12 (s, 1 H), 8.37 (s, 1 H), 9.00 (t, J = 4.8 Hz, 1 H), 10.55 (s, 1 H), 11.58 (s, 1 H).
Example 79
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2-oxo- 2,3-dihydro-lH-indol-5-yl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 5-
(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l,3-dihydro-2H-indol-2-one (63.1 mg, 0.243 mmol), DMSO(1.5 mL), sodium carbonate (0.281 mL, 0.562 mmol), and
bis(triphenylphosphine)palladium(II) chloride (10.51 mg, 0.015 mmol). The final product was collected as 30 mg (34%). LCMS E-S (M+H) = 471.3 1H NMR (400 MHz, DMSO- d6) δ ppm 1.54 (d, J= 6.4 Hz, 6 H), 2.13 (s, 3 H), 2.22 (s, 3 H), 3.40 (br. s., 2 H), 4.41 (d, J= 4.6 Hz, 2 H), 5.25 - 5.41 (m, 1 H), 5.91 (s, 1 H), 6.97 (d, J= 8.1 Hz, 1 H), 8.08 - 8.21 (m, 3 H), 8.34 (s, 1 H), 8.96 (br. s., 1 H), 10.64 (s, 1 H), 11.58 (br. s., 1 H).
Example 80
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2-oxo-2,3- dihydro-lH-benzimidazol-5- l)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 5- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l,3-dihydro-2H-benzimidazol-2-one (63.3 mg, 0.243 mmol), DMSO(2 mL), sodium carbonate (0.281 mL, 0.562 mmol), and
bis(triphenylphosphine)palladium(II) chloride (10.51 mg, 0.015 mmol). The final product was collected as 35 mg (40%). LCMS E-S (M+H) = 472.4 1H NMR (400 MHz, DMSO- d6) δ ppm 1.55 (d, J= 6.4 Hz, 6 H), 2.13 (s, 3 H), 2.22 (s, 3 H), 4.41 (d, J= 4.8 Hz, 2 H), 5.30 (quin, J= 6.6 Hz, 1 H), 5.91 (s, 1 H), 7.07 (d, J= 8.3 Hz, 1 H), 7.86 (s, 1 H), 7.92
(dd, J= 8.3, 1.8 Hz, 1 H), 8.10 (s, 1 H), 8.30 - 8.41 (m, 1 H), 8.98 (t, J= 4.8 Hz, 1 H),
10.82 (s, 1 H), 10.89 (s, 1 H). 11.58 (s, 1 H).
Example 81
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[6-(4-methyl-l- piper azinyl)-3-py ridinyl] - lH-pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-
methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 1-methyl- 4-[5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2-pyridinyl]piperazine (73.8 mg, 0.243 mmol), DMSO(2 mL), sodium carbonate (0.281 mL, 0.562 mmol), and
bis(triphenylphosphine)palladium(II) chloride (10.51 mg, 0.015 mmol). The final product was collected as 12 mg (13%). LCMS E-S (M+H) = 515.1 1H NMR (400 MHz, DMSO- d6) δ ppm 1.53 (d, J = 6.4 Hz , 6 H), 2.13 (s, 3 H), 2.23 (m, 6 H), 2.37 - 2.46 (m, 4 H), 3.56 - 3.69 (m, 4 H), 4.40 (d, J= 4.6 Hz, 2 H), 5.30 (quin, J= 6.7 Hz, 1 H), 5.91 (s, 1 H), 6.99 (d, J= 9.1 Hz, 1 H), 8.10 (s, 1 H), 8.32 (s, 1 H), 8.38 (dd, J= 9.1, 2.5 Hz, 1 H), 8.91 (br. s., 1 H), 9.03 (d, J=2.3 Hz, 1 H).
Example 82
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[6-(4- methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 1-methyl- 4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]piperazine (73.6 mg, 0.243 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1 mL) sodium carbonate (0.281 mL, 0.562 mmol), and bis(triphenylphosphine)palladium(II) chloride (12.23 mg, 0.015 mmol), wherein the reaction time was 30 min. The crude product was purified by column chromatography (eluent: gradient of 0 to 15% (9 : 1 MeOH/NH4OH)/DCM). The product was dried under high vacuum and collected as 58 mg (59%). LCMS E-S (M+H) = 514.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.51 - 1.58 (m, 6 H), 2.13 (s, 3 H), 2.23 (s, 6 H), 2.24 (s, 3H), 2.47 (m, 4H), 3.24 - 3.30 (m, 4 H), 4.41 (d, J= 5.05 Hz, 2 H), 5.30 (quin, J = 6.63 Hz, 1 H), 5.90 (s, 1 H), 7.07 (d, J= 9.09 Hz, 2 H), 8.06 (s, 1 H), 8.15 (d, J= 8.84 Hz, 2H), 8.30 (s, 1 H), 8.92 (t, J= 4.80 Hz, 1 H), 11.55 (s, 1 H).
Example 83
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l-methylethyl)- 6- [6-(4-morpholinyl)-3-pyridinyl] - lH-pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.180 mmol), [6-(4- morpholinyl)-3-pyridinyl]boronic acid (48.8 mg, 0.235 mmol), DMSO(2.0 mL), sodium carbonate (0.271 mL, 0.541 mmol), and bis(triphenylphosphine)palladium(II) chloride (10.13 mg, 0.014 mmol), wherein the reaction time was 8 h. The mixture was filtered and the residue was washed with DMSO. The crude product in DMSO was purified by reverse-phase HPLC (mobile phase: 25 - 60 % CAN in H20, 0.1%TFA). The TFA product salt obtained was neutralized with saturated NaHCC , filtered, washed with water, and dried under high vacuum to give the product as 24 mg (25%). LCMS E-S (M+H) = 516.2 1H NMR (400 MHz, DMSO- e) δ ppm 1.50 (d, J= 6.57 Hz, 6 H), 2.12 (s, 3 H), 2.25 (s, 3 H), 2.42 (s, 3 H), 3.54 - 3.63 (m, 4 H), 3.73 (m, 4 H), 4.39 (d, J= 4.80 Hz, 2 H), 5.14 - 5.29 (m, 1 H), 5.89 (s, 1 H), 6.97 (d, J= 8.84 Hz, 1 H), 7.63 (s, 1 H), 8.37 (dd, J = 9.09, 2.53 Hz, 1 H), 8.68 (t, J = 4.93 Hz, 1 H), 8.98 (d, J= 2.27 Hz, 1 H), 11.51 (s, 1 H).
Example 84
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- pyridinylamino)-lH-pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (50 mg, 0.134 mmol), 4- pyridinamine (12.59 mg, 0.134 mmol), cesium carbonate (131 mg, 0.401 mmol), 1,4- dioxane (2 mL), palladium(II) acetate (1.501 mg, 6.69 μιηοΐ) and BINAP (8.33 mg, 0.013 mmol), wherein the reaction time was 2h. The final product was collected as 29 mg
(50%). LCMS E-S (M+H) = 432.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.45 - 1.59 (d, J= 6.8 Hz, 6 H), 2.14 (s, 3 H), 2.23 (s, 3 H), 4.36 (d, J= 5.0 Hz, 2 H), 5.16 (m, 1 H), 5.91 (s, 1 H), 7.09 (s, 1 H), 7.99 (d, J= 6.1 Hz, 2 H), 8.12 (s, 1 H) 8.49 (d, J= 6.6 Hz, 2 H), 8.74 (t, J= 5.1 Hz, 1 H), 10.58 (s, 1 H), 11.58 (s, 1 H).
Example 85
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[(2-oxo-2,; dihydro- lH-benzimidazol-5-yl)amino] - IH-pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.187 mmol), 5-amino- l,3-dihydro-2H-benzimidazol-2-one (33.5 mg, 0.225 mmol), cesium carbonate (92 mg,
0.281 mmol), Ν,Ν-dimethylacetamide (DMA) (2 mL), palladium(II) acetate (2.52 mg, 0.011 mmol) and Xantphos (10.83 mg, 0.019 mmol) wherein the reaction temperature was 150 °C and reaction time was 8h. The final product was collected as 25 mg (27%). LCMS E-S (M+H) = 487.2 1H NMR (400 MHz, DMSO-d6) δ ppm 1.50 - 1.61 (m, 6 H), 2.13 (s, 3 H), 2.23 (s, 3 H), 4.38 (d, J= 5.0 Hz, 2 H), 5.20 (quin, J= 6.6 Hz, 1 H), 5.90 (s, 1 H), 6.79 - 6.91 (m, 2 H), 8.04 (d, J= 8.3 Hz, 1 H), 8.26 - 8.33 (m, 2 H), 8.84 (t, J= 5.0 Hz, 1 H), 11.50 (s, 1 H).
Example 86
6-{[4-(Aminocarbonyl)phenyl]amino}- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.180 mmol), 4- aminobenzamide (29.5 mg, 0.217 mmol), cesium carbonate (88 mg, 0.271 mmol), N,N- dimethylacetamide (DMA) (1.5 mL), palladium(II) acetate (2.431 mg, 10.83 μιηοΐ) and Xantphos (10.44 mg, 0.018 mmol) wherein the reaction temperature was 150 °C and reaction time was lh. The final product was collected as 12 mg (14%). LCMS E-S (M+H) = 488.2 1H NMR (400 MHz, DMSO-d6) δ ppm 1.48 (d, J= 6.4 Hz, 6 H), 2.13 (s, 3 H), 2.23 (s, 3 H), 2.32 (s, 3 H), 4.35 (d, J= 4.8 Hz, 2 H), 4.99 (quin, J= 6.8 Hz, 1 H), 5.89 (s, 1 H), 6.65 (s, 1 H), 7.15 (br. s., 1 H), 7.75 - 7.82 (m, 1 H), 7.84 - 7.96 (m, 4 H), 8.56 (t, J= 4.9 Hz, 1 H), 9.77 (s, 1 H).
Example 87
l-(l,l-Dimethylethyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3- methyl-6-[6-(4-methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4-6]pyridine-4- carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro- 1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3- pyridinyl)methyl]-3-methyl-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.174 mmol), l-methyl-4-[5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2- pyridinyljpiperazine (68.7 mg, 0.226 mmol), DME (3 mL), water (1.00 mL), sodium
carbonate (0.261 mL, 0.523 mmol) and PdCl2(dppi>CH2Ci2 adduct (11.38 mg, 0.014
mmol), wherein the reaction time was 40 min. The crude product was purified by column chromatography (eluent: gradient of 0 to 15% (9: 1 MeOH/NH4OH)/DCM). The final product was collected as a solid, 41 mg (43%).. LCMS E-S (M+H) = 543.2 1H NMR
(400 MHz, DMSO-d6) δ ppm 1.78 (s, 9 H), 2.12 (s, 3 H), 2.24 (m, 7 H), 2.38 (s, 3 H),
2.39 - 2.49 (m, 4 H), 3.34 (s, 3 H), 4.38 (m, 2 H), 5.88 (s, 1 H), 6.98 (d, J= 9.09 Hz, 1 H), 7.59 (s, 1 H), 8.93 - 8.97 (m, 1 H), 11.49 - 11.57 (m, 1 H), 11.53 (s, 1 H), 11.53 (s, 1 H).
Example 88
l-(l,l-Dimethylethyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-6- (2-oxo-2,3-dihydro-lH-benzimidazol-5- l)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

(11.38 mg, 0.014 mmol) wherein the reaction time was 40 min. The crude product was purified by column chromatography (eluent: gradient of 0 to 15% (9:1 MeOH/NH4OH)/DCM). The final product was collected as a solid, 24 mg (27%). LCMS E-S (M+H) = 500.2 1H NMR (400 MHz, DMSO-de) δ ppm 1.77 (m, 9 H), 2.12 (s, 3 H), 2.24 (s, 3 H), 2.40 (s, 3 H), 4.38 (d, J= 4.55 Hz, 2 H), 5.89 (s, 1 H), 7.06 (d, J= 8.08 Hz, 1 H), 7.59 (s, 1 H), 7.72 - 7.86 (m, 2 H), 8.73 (br. s., 2 H), 10.74 - 10.92 (m, 1 H), 11.53 (br. s., 1 H).
Example 89
l-(l,l-Dimethylethyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-6- [6-(4-morpholin l)-3-pyridinyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6- chloro- 1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.174 mmol), [6-(4-morpholinyl)-3- pyridinyljboronic acid (47.1 mg, 0.226 mmol), DME (3 mL), water (1.00 mL), sodium carbonate (0.261 mL, 0.523 mmol) and PdCl2(dppf)-CH2Cl2 adduct (11.38 mg, 0.014 mmol) wherein the reaction time was 40 min. The crude product was purified by column chromatography (eluent: gradient of 0 to 15% (9: 1 MeOH/NH4OH)/DCM). The final product was collected as a solid, 61 mg (65%). LCMS E-S (M+H) = 530.0 1H NMR (400 MHz, DMSO-d6) δ ppm 1.78 (s, 9 H), 2.12 (s, 3 H), 2.24 (s, 3 H), 2.38 (s, 3 H), 3.52 - 3.64 (m, 4 H), 3.68 - 3.80 (m, 4 H), 4.38 (d, J = 5.05 Hz, 2 H), 5.88 (s, 1 H), 6.99 (d, J= 9.09 Hz, 1 H), 7.61 (s, 1 H), 8.34 (dd, J=8.84, 2.53 Hz, 1 H), 8.68 (t, J= 5.05 Hz, 1 H), 8.97 (d, J= 2.27 Hz, 1 H), 11.53 (s, 1H).
Example 90
6-(2, 1 ,3-Benzoxadiazol-5-yl)- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihy dr o-3-pyridinyl)methyl] -3- methyl-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.180 mmol), 5- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2,l,3-benzoxadiazole (57.7 mg, 0.235 mmol), DMSO(2 mL), sodium carbonate (0.271 mL, 0.541 mmol), and
bis(triphenylphosphine)palladium(II) chloride (12.67 mg, 0.018 mmol) wherein the
reaction time was 8h. The final product was collected as 17 mg (20%). LCMS LCMS E- S (M+H) = 472.4 1H NMR (400 MHz, DMSO- e) δ ppm 1.53 (d, J= 6.8 Hz ,6 H), 2.12 (s, 3 H), 2.27 (s, 3 H) 4.42 (d, J= 4.8 Hz, 2 H) 5.32 (quin, J= 6.6 Hz, 1 H), 5.90 (s, 1 H), 8.00 (s, 1 H), 8.23 (d, J= 9.4 Hz, 1 H), 8.54 - 8.64 (m, 1 H), 8.79 (t, J= 4.7 Hz, 1 H),
8.91 (s, 1 H), 11.54 (br. s., 1 H).
Example 91
6-(2- Amino-6-quinazolinyl)- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihy dr o-3-pyridinyl)methyl] -3- methyl-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 74 using 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (70 mg, 0.180 mmol), 5-
(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2,l,3-benzoxadiazole (57.7 mg, 0.235 mmol), DMSO(2 mL), sodium carbonate (0.271 mL, 0.541 mmol), and
bis(triphenylphosphine)palladium(II) chloride (12.67 mg, 0.018 mmol), wherein the reaction time was 8h. The final product was collected as 28 mg (31%). LCMS E-S (M+H) = 497.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.52 (d, J= 6.8 Hz , 6 H), 2.12 (s, 3 H), 2.26 (s, 3 H), 4.41 (d, J= 5.0 Hz, 2 H) 5.30 (quin, J= 6.6 Hz, 1 H), 5.89 (s, 1 H), 7.16 (br. s., 2 H), 7.56 (d, J= 9.1 Hz, 1 H), 7.78 (s, 1 H) 8.60 (dd, J= 8.8, 2.0 Hz, 1 H), 8.69 (d, J= 1.8 Hz, 1 H), 8.76 (t, J= 4.9 Hz, 1 H), 9.28 (s, 1 H), 11.53 (s, 1 H).
Example 92
TV- [(4,6-Dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl] -3-methyl-6-{4-
[(methylamino)sulfonyl]phenyl}-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4- carboxamide

6-Chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (80 mg, 0.21 mmol), {4- [(methylamino)sulfonyl]phenyl}boronic acid (66.5 mg, 0.31 mmol) and
bis(triphenylphosphine)palladium(II) chloride (8.4 mg, 0.01 mmol) were suspended in DME/water (4 mL, 3: 1) and stirred for 10 min under nitrogen at room temperature.
Sodium bicarbonate (52 mg, 0.62 mmol) was added and the heterogenous mixture was irradiated (microwave) at 150 °C for 30 min. After cooling to room temperature, water was added to the black mixture, and the contents were vacuum filtered. The crude product was dissolved in DCM/MeOH (1 : 1) and preabsorbed onto silica gel. The product was purified by silica gel chromatography (eluent: DCM/MeOH/NH^H, gradient of 0 to
90: 10: 1). The light beige solid that was collected was suspended in EtOH, sonicated, and filtered. The solid was then air-dried for 15 min, and then in vacuum oven overnight. The final product was collected as 77mg (70%). LCMS E-S (M+H) = 523.2 1H NMR (400
MHz, DMSO-de) δ ppm 11.54 (br. s., 1 H) 8.78 (t, J=5.05 Hz, 1 H) 8.43 (m, J=8.59 Hz, 2 H) 7.93 (m, J=8.59 Hz, 2 H) 7.78 (s, 1 H) 7.58 (br. s., 1 H) 5.89 (s, 1 H) 5.29 (quin, J=6.63 Hz, 1 H) 4.40 (d, J=4.80 Hz, 2 H) 2.46 (d, J=1.26 Hz, 6 H) 2.25 (s, 3 H) 2.12 (s, 3 H) 1.52 (s, 3 H) 1.51 (s, 3 H).
Example 93
6- [4-(Acetylamino)phenyl] -TV- [(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl] - 3-methyl-l-(l-methyleth l)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 93 using 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (80 mg, 0.21 mmol), [4- (acetylamino)phenyl]boronic acid (55.4 mg, 0.31 mmol),
bis(triphenylphosphine)palladium(II) chloride (8.4 mg, 0.01 mmol), DME/water (4 mL, 3: 1) and sodium bicarbonate (52 mg, 0.62 mmol). The crude product was dissolved in DCM/MeOH (1 : 1) and preabsorbed onto silica gel. The product was purified by silica gel chromatography (eluent: DCM/MeOH/NH4OH. gradient of 0 to 80:20:2). The collected solid was suspended in EtOH/EtOAc (1 : 1), sonicated, and filtered. After further washing with hexanes, the solid was then air-dried for 15 min, and then in vacuum oven overnight. The final product was collected as 70 mg (69%). LCMS E-S (M+H) = 487.3 1H NMR (400 MHz, DMSO-de) δ ppm 11.53 (br. s., 1 H) 10.16 (s, 1 H) 8.73 (t, J=5.05 Hz, 1 H) 8.17 (m, J=8.84 Hz, 2 H) 7.74 (m, J=8.84 Hz, 2 H) 7.64 (s, 1 H) 5.89 (s, 1 H) 5.25 (quin, J=6.69 Hz, 1 H) 4.39 (d, J=4.80 Hz, 2 H) 2.43 (s, 3 H) 2.25 (s, 3 H) 2.12 (s, 3 H) 2.09 (s, 3 H) 1.51 (s, 3 H) 1.49 (s, 3 H).
Example 94
6- [4-(Aminocarbonyl)phenyl] -TV- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-l-(l-methyleth l)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 93 using 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (80 mg, 0.21 mmol), [4- (aminocarbonyl)phenyl]boronic acid (52.9 mg, 0.31 mmol),
bis(triphenylphosphine)palladium(II) chloride (8.7 mg, 0.01 mmol), DME/water (4 mL, 3: 1) and sodium bicarbonate (54 mg, 0.62 mmol). The crude product was dissolved in DCM/MeOH (1 : 1) and preabsorbed onto silica gel. The product was purified by silica gel chromatography (eluent: DCM/MeOH/NH4OH (gradient 0 to 90: 10: 1). The collected solid was suspended in EtOH, sonicated, and filtered. After further washing with EtOH/DCM, the filtered solid was then air-dried for 15 min, and then in vacuum oven overnight. The final product was collected as 70 mg (69%). LCMS E-S (M+H) = 459.3
1H NMR (400 MHz, DMSO-d6) δ ppm 11.58 (s, 1 H) 9.00 (t, J=4.93 Hz, 1 H) 8.41 (s, 1 H) 8.36 (s, 1 H) 8.34 (s, 1 H) 8.24 (s, 1 H) 8.13 (s, 1 H) 8.06 (s, 1 H) 8.04 (s, 1 H) 7.51 (s, 1 H) 5.91 (s, 1 H) 5.37 (quin, J=6.69 Hz, 1 H) 4.42 (d, J=4.80 Hz, 2 H) 2.23 (s, 3 H) 2.13 (s, 3 H) 1.56 (s, 3 H) 1.55 (s, 3 H).
Example 95
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-{[2-(2- pyridinylamino)ethyl]amino -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

6-Chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (80 mg, 0.21 mmol) and (2- aminoethyl)2-pyridinylamine (88 mg, 0.64 mmol) were suspended in 2 mL of NMP and irradiated (microwave) as follows: 180 °C for 30 min, 200 °C for 30 min, 220°C for 30 min, 230°C for 30 min and then 240 °C for 1 h. After cooling to room temperature, some of the solvent was removed in vacuo and the residue was purified by silica gel
chromatography (eluent: DCM/MeOH/NH4OH, gradient 0 to 90: 10: 1). The collected solid was partitioned between water and EtO Ac/toluene (1 : 1) and then extracted twice with DCM/isopropanol. The combined organic layers were washed with water, dried over MgS04, filtered, and concentrated in vacuo to afford a dark brown oil. After drying under vacuum overnight, the residue was again purified by silica gel chromatography (eluent: DCM/MeOH/NH4OH (gradient 0 to 90: 10: 1). The collected solid was suspended in
EtOH, sonicated, and filtered. The solid was further washed with EtOH/DCM, filtered, and dried in vacuum-oven for 2 days. The final product was collected as 47 mg (45%). LCMS E-S (M+H) = 475.0 1H NMR (400 MHz, DMSO-d6) δ ppm 11.56 (br. s., 1 H) 8.47 (t, J=5.05 Hz, 1 H) 7.97 (dd, J=4.93, 1.14 Hz, 1 H) 7.85 (s, 1 H) 7.32 - 7.39 (m, 2 H) 6.65 (s, 1 H) 6.61 (t, J=5.43 Hz, 1 H) 6.46 - 6.51 (m, 2 H) 5.89 (s, 1 H) 4.97 (quin, J=6.69 Hz, 1 H) 4.32 (d, J=5.05 Hz, 2 H) 3.49 (dt, J=15.28, 5.62 Hz, 4 H) 2.21 (s, 3 H) 2.13 (s, 3 H) 1.44 (s, 3 H) 1.43 (s, 3 H).
Example 96
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(7-oxo- l,5,6,7-tetrahydro-l,8-naphth ridin-3-yl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

6-Bromo-3,4-dihydro-l,8-naphthyridin-2(lH)-one (200 mg, 0.881 mmol) (J. Med.
Chem. 2003; 46; 9; 1627-1635), bis(pinacolato)diboron (268 mg, 1.057 mmol), Pd(dppf) (35.7 mg, 0.044 mmol) and potassium acetate (259 mg, 2.64 mmol) were suspended in 1,4-Dioxane (8 mL), and stirred with heating at 100 °C for lh. After cooling to room temperature, 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (329 mg, 0.881 mmol), bis(triphenylphosphine)palladium(II) chloride (71.9 mg, 0.088 mmol) and sodium bicarbonate (222 mg, 2.64 mmol) were added, followed by DME (5 mL) and water (3 mL). The reaction mixture was irradiated (microwave) at 120 °C for 2 h. The reaction mxiture was cooled cooled to room temperature and filtered through Na2SC"4. The contents were purified directly by silica gel chromatography (eluent : 5% MeOH/CELCb) to furnish the desired product as a grey solid after evaporation and preciptation from warm EtOAc/MeOH (1 :9). The product was collected as 55 mg (13%). LCMS E-S (M+H) = 486.3 1H NMR (400 MHz, OMSO-de) δ ppm 11.57 (br. s., 1 H), 10.74 (s, 1 H), 8.99 (d, J=2.27 Hz, 1 H), 8.93 (t, J=4.93 Hz, 1 H), 8.44 (d, J=1.77 Hz, 1 H), 8.37 (s, 1 H), 8.16 (s, 1 H), 5.91 (s, 1 H), 5.29 - 5.40 (m, 1 H), 4.42 (d, J=5.05 Hz, 2 H), 3.04 (t, J=7.58 Hz, 2 H), 2.53 - 2.62 (m, 2 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.54 (d, 6 H).
Example 97
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-{6- [(methylamino)sulfonyl]-3-pyridinyl}-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-
4-carboxamide

5-Bromo-N-methyl-2-pyridinesulfonamide (225 mg, 0.896 mmol),
Bis(pinacolato)diboron (296 mg, 1.165 mmol), Pd(dppf) (35.7 mg, 0.044 mmol) and potassium acetate (264 mg, 2.69 mmol) were suspended in 1,4-Dioxane (8 mL), and stirred with heating at 100 °C for lh. After cooling to room temperature, 6-chloro-N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)- 1 H- pyrazolo[3,4-b]pyridine-4-carboxamide (368 mg, 0.986 mmol),
bis(triphenylphosphine)palladium(II) chloride (73.2 mg, 0.090 mmol) and sodium bicarbonate (226 mg, 2.69 mmol) were added, followed by DME (5 mL) and water (3 mL). The reaction mixture was irradiated (microwave) at 120 °C for 2h. The reaction mxiture was cooled cooled to room temperature and filtered through Na2SC"4. The contents were purified directly by silica gel chromatography (eluent : 15% MeOH/CH2Cl2) to furnish the desired product as a grey solid after evaporation and preciptation from warm EtOAc/MeOH (1 :9). The product was again purified by silica gel chromatography
(eluent : 5% MeOH/CH2Cl2) to afford the final product as a white solid, 83 mg (18%). LCMS E-S (M+H) = 510.0 1H NMR (400 MHz, DMSO-d6) δ ppm 11.59 (br. s., 1 H),
9.56 (d, J=1.52 Hz, 1 H), 8.96 (s, 1 H), 8.86 (dd, J=8.34, 2.27 Hz, 1 H), 8.46 (s, 1 H), 8.34 (s, 1 H), 8.12 (d, J=8.34 Hz, 1 H), 7.85 (br. s., 1 H), 5.91 (s, 1 H), 5.27 - 5.46 (m, 1 H), 4.43 (d, J=4.80 Hz, 2 H), 2.60 (s, 3 H), 2.23 (s, 3 H), 2.13 (s, 3 H), 1.56 (d, 6 H).
Intermediate 17
5-Bromo-N-meth l-2-pyridinesulfonamide

5-Bromo-2-pyridinesulfonyl chloride (500 mg, 1.949 mmol) was added to a 0 °C solution of pyridine (0.315 ml, 3.90 mmol), methylamine (0.975 ml, 1.949 mmol, 2M in THF) and CH2CI2 (2 mL). The reaction mixture was stirred at room temperature for 1 hr, then quenched with brine. The contents were extracted with DCM, dried, filtered, and concentrated in vacuo. The crude product was purified by silica gel chromatography (eluent: 50%, EtOAC/CH2Cl2). The product was collected as a clear oil, 225 mg (75%>). LCMS E-S (M+H) = 251.1. XH NMR (400 MHz, Chloroform-;/) δ ppm 8.77 (s, 1 H), 8.07 (dd, J=8.34, 2.27 Hz, 1 H), 7.92 (d, J=8.34 Hz, 1 H), 5.45 (d, J=5.05 Hz, 1 H), 2.76 (d, 3 H).
Intermediate 18
l-{[4-(Methyloxy)phen l]methyl}-lH-pyrazol-5-amine

Hydrazine hydrate (12.82 g, 400 mmol) was added dropwise to a cooled (< 20 °C) solution of 2-propenenitrile (21.76 g, 410 mmol) and ethanol (200 mL). After 16 h stirring the reaction mixture was cooled in an ice water bath and 4- (methyloxy)benzaldehyde (53.8 g, 395 mmol) was added dropwise. The mixture was stirred at room temperature for 16 h. The reaction mixture was concentrated to dryness. The residue was dissolved in n-butanol (200 mL), sodium hydroxide was added (1 g, 25.00 mmol), and the mixture heated at 120 °C for 6 h. The reaction mixture was concentrated to 50%> volume under reduced pressure, poured onto 300 mL of water, and then extracted with Et20 (2 x 200 mL). The combined ether phases were extracted with IN HC1 (3 x 100 mL). The combined HC1 extracts were combined and cooled in an ice/water bath. Added next was 6N NaOH until basic (pH > 12). The contents were extracted with Et20 (4 x 100 mL), washed with water, dried over MgS04, filtered, and
concentrated under reduced pressure. The residue was purified via silica gel
chromatography (eluent : 0 - 50% EtOAc:Hex). The final product was collected as 8.94 g (11%). 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 3.79 (s, 3 H), 5.14 (s, 2 H), 5.55 (d, J=1.77 Hz, 1 H), 6.81 - 6.94 (m, 2 H), 7.12 (d, J=8.84 Hz, 2 H), 7.31 (d, J=2.02 Hz, 1 H).
Intermediate 19
l-Cyclobut l-3-methyl-lH-pyrazol-5-amine

(2Z)-3-Amino-2 -butenenitrile (1.339 g, 16.31 mmol) and cyclobutylhydrazine HC1 (2 g, 16.31 mmol) were added to ethanol (20 mL) and heated at 75 °C for 16 h. After cooling to room temperature, the solvent was removed under reduced pressure. The crude residue was suspended in saturated NaHC03 (30 mL) and EtOAc (50 mL), and stirred for 10 min. The phases were separated and the aq. phase extracted with EtOAc (3 x 50 mL). The combined EtOAc extracts were washed with brine, dried over MgS04, filtered, and concentrated in vacuo. The crude residue was purified via silica gel chromatography (eluent: 0 to 5% EtOAc :DCM, then gradient to 100% EtOAc). The final product was collected as 0.4 g (16%). LCMS E-S (M+H) =: 152.0. 1H NMR (400 MHz,
CHLOROFORM-d) δ ppm 1.67 - 2.00 (m, 2 H) 2.21 (s, 3 H) 2.32 - 2.43 (m, 2 H) 2.69 (ddd, J=10.55, 9.41, 2.53 Hz, 2 H) 3.26 - 3.55 (br s, 2 H) 4.48 - 4.59 (m, 1 H) 5.36 (s, 1 H)
Intermediate 20
l-Cyclopentyl-3-meth l-lH-pyrazol-5-amine

The title compound was prepared in the same manner as described for intermediate 19 using cyclopentylhydrazine hydrochloride (2 g, 14.64 mmol), (2Z)-3-amino-2- butenenitrile (1.202 g, 14.64 mmol) and ethanol (20 mL). The final product was collected
as 0.57 g (24%). LCMS E-S (M+H) =166.0.1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.60 - 1.69 (m, 2 H), 1.86 - 1.95 (m, 2 H), 1.99 - 2.09 (m, 4 H), 2.19 (s, 3 H), 3.46 (br. s., 2 H), 4.38 (quin, J=7.89 Hz, 1 H), 5.36 (s, 1 H).
Intermediate 21
lylmeth l)- lH-pyrazol-5-amine
A solution of hydrazine hydrate (14.55 mL, 300 mmol) and ethanol (75 mL) was cooled in an ice water bath and then 2-propenenitrile (15.3 g, 288 mmol) was added dropwise. After stirring at room temperature for 2 h, benzaldehyde (31.8 g, 300 mmol) was added dropwise and the reaction mixture was allowed to stir at room temperature for 2 d. The solvent was removed under reduced pressure. The crude oil was cooled in an ice/water bath followed by dropwise addition of a n-BuONa solution (Na 6.9 g, (300 mmol) in n-Butanol (300 mL)). The reaction mixture was heated at reflux for 1 h, and then cooled to room temperature. The contents were poured onto 300 mL of water and then extracted with Et20 (2 x 200 mL). The ether phase was extracted with IN HC1 (3 x 100 mL). The combined HC1 extracts were combined and cooled in a ice/water bath, followed by addition of 6N NaOH until basic (pH > 12). The mixture was extracted with Et20 (4 x 100 mL), washed with water, dried over MgS04, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography (eluent: 0 to 50% EtOAc:Hex). The final product was collected as 6.96 g (14%). LCMS E-S (M+H) = 174.0. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 3.39 (br. s., 2 H), 5.23 (s, 2 H), 5.59 (d, J=1.77 Hz, 1 H), 7.17 (d, J=6.82 Hz, 2 H), 7.28 - 7.37 (m, 4 H)
Intermediate 22
1,1-Dimethylethyl 2-(tetrah dro-2H-pyran-4-yl)hydrazinecarboxylate

Tetrahydro-4H-pyran-4-one (9.69 g, 97 mmol) was added to a solution of 1,1- dimethylethyl hydrazinecarboxylate (14.07 g, 106 mmol) in methanol (100 mL) and stirred at room temperature for 3 h. The solvent was removed in vacuo, and the crude residue was suspended in acetic acid (140 mL). Next added sodium cyanoborohydride (6.69 g, 106 mmol) in portions over 3 minutes. The contents were stirred at room temperature for 60 h. The solvent was removed in vacuo, and the crude residue was suspended in DCM (100 mL). The reaction mixture was adjusted to pH 7 with 6N NaOH. The layers were separated, and the aq. layer extracted with DCM. The combined organic layers were washed with saturated NaHC03, and brine. The organic layer was dried over MgS04, filtered, and concentrated in vacuo to afford the product as a solid, 18.4 g (88%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.22 (m, 2 H), 1.39 (s, 9 H), 1.43 - 1.48 (m, 1 H), 1.59 - 1.69 (m, 2 H), 2.82 - 2.98 (m, 1 H), 3.20 - 3.32 (m, 2 H), 3.80 (d, J=l 1.62 Hz, 2 H), 4.36 (br. s., 1 H), 8.07 - 8.34 (m, 1 H).
Intermediate 23
Tetrahydro-2H-pyran-4-ylhydrazine

1,1-Dimethylethyl 2-(tetrahydro-2H-pyran-4-yl)hydrazinecarboxylate was added to 1,4-Dioxane (10 mL) followed by hydrochloric acid (4M in 1,4-Dioxane, 10 mL, 329 mmol). The reaction mixture was stirred at room temperature for 60 h. The reaction mixture was filtered to afford the product as a solid, 1.02 g. (72%). 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.45 (qd, J=l 1.79, 4.29 Hz, 2 H), 1.81 - 2.01 (m, 2 H), 3.00 - 3.19 (m, 1 H), 3.20 - 3.35 (m, 2 H), 3.78 - 3.97 (m, 2 H), 6.5-9.5(br m, 3 H).
Intermediate 24
3-Methyl-l-(tetrahydro-2H- ran-4-yl)-lH-pyrazol-5-amine

(2Z)-3-Amino-2-butenenitrile (0.538 g, 6.55 mmol), tetrahydro-2H-pyran-4- ylhydrazine (1 g, 6.55 mmol) and triethylamine (0.913 mL, 6.55 mmol) were added to ethanol (300 mL), and the reaction mixture was stirred at 75 °C for 16 hours. The solvent was removed in vacuo, and the crude material suspended in EtOAc. The organic phase was washed with water and then brine. The organic layer was then dried over MgSC^, filtered, and concentrated in vacuo. Half of the crude material was purified by reverse HPLC (mobile phase : 0 - 30% ACN/H20, 0.1%TFA) to afford 380 mg of the desired product. The other half of the crude material was purified via silica gel chromatography (eluent : 0% to 100% EtOAc:Hex then 0% to 20% MeOH:DCM). An additional 300 mg of the desired product was obtained (overall yield : 57%). LCMS E-S (M+H) = 182.0. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.78 (dd, 2 H), 1.94 (qd, J=12.13, 4.55 Hz, 2 H), 2.19 (s, 3 H), 3.30 - 3.46 (m, 2 H), 3.99 (dd, J=11.49, 4.17 Hz, 2 H), 4.49 (m, J=11.65, 11.65, 4.11, 3.92 Hz, 1 H), 5.53 (s, 1 H).
Intermediate 25
6-Methyl-l-(phenylmethyl)-lH-pyrazolo[3,4-6]pyridin-4-ol

A mixture of l-(phenylmethyl)-lH-pyrazol-4-amine (6.25 g, 36.1 mmol), ethyl 3- oxobutanoate (4.70 g, 36.1 mmol), acetic acid (.2 mL, 3.49 mmol) and benzene (50 mL) were refluxed for 16 h (Dean-Stark trap used). The solvent was removed under reduced pressure and the crude residue purified via silica gel chromatography (eluent; 0 to 60%> EtOAc:Hex). The collected product (7.35 g, 25.8 mmol) was then dissolved in 10 mL of
Dowtherm A and this solution added dropwise to 10 mL of refluxing Dowtherm A. After refluxing for an additional 20 minutes, the reaction mixture was cooled to room
temperature, and 20 mL of petroleum ether were added. After stirring at room
temperature for 16 hr, the solid product was filtered and dried on hi-vacuum. The final product was collected as 6.23 g (72%). LCMS E-S (M+H) = 240.0. 1H NMR (400 MHz, MeOD) ppm 4.89 (s, 3 H), 5.59 (s, 2 H), 6.17 (br. s., 1 H), 7.17 (d, J=6.82 Hz, 2 H), 7.25 - 7.37 (m, 3 H), 8.05 (s, 1 H).
Intermediate 26
4-Bromo-6-methyl-l-(phenylmethyl)-lH-pyrazolo[3,4-6]pyridine

Phosphorus oxybromide (5.31 g, 18.54 mmol) was added to a suspension of 6- methyl-l-(phenylmethyl)-lH-pyrazolo[3,4-b]pyridin-4-ol (3.08 g, 12.87 mmol) and toluene (25 mL). The contents were heated to reflux for 1 h, wherein 10 mL DMF were added during this process. The solvent was removed under reduced pressure. The crude residue was suspended in 30 mL water containing ice. The contents were adjusted to pH = 10 by addition of sat. NaHC03. The solid product was filtered and dried under hi-vacuum. The final product was collected as 3.55 g (91%). LCMS E-S (M+H) = 302.1/304.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.61 (s, 3 H), 5.65 (s, 2 H), 7.16 - 7.35 (m, 5 H), 7.48 (s, 1 H), 8.12 s, 1 H).
Intermediate 27
6-Methyl-l-(phenylmethyl)-lH- razolo[3,4-6]pyridine-4-carbonitrile

tris(dibenzylideneacetone)dipalladium(0) (0.523 g, 0.571 mmol), SPhos (0.562 g, 1.370 mmol), DMF(49 mL), and water (0.5 mL) was degassed with nitrogen for 10 minutes. The reaction mixture was heated at 120 °C for 2 hours, and then cooled to room temperature. The contents were concentrated to 50% volume and then 50 mL of IN NaOH and 50 mL of EtOAc were added. The solids were filtered off. The phases were separated and the aqueous phase extracted with EtOAc (3 x 50 mL). The combined EtOAc extracts were washed with water (3 x 50 mL), brine (50 mL), dried over MgS04, filtered, and concentrated in vacuo. The crude product containing SPhos reagent (-30- 40%) was collected as 3.69 g and used without further purification. LCMS E-S (M+H) = 249.1. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.77 (s, 3 H), 5.73 (s, 2 H), 7.08 - 7.44 (m, 6 H), 8.16 (s, 1 H)
Intermediate 28
6-Methyl-l-(phenylmethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

6-Methyl- 1 -(phenylmethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carbonitrile ( 1 g), sodium hydroxide (0.805 g, 20.14 mmol), ethanol (25 mL) and water (10 mL) were heated at reflux for 2 hours. After cooling to room temperature, the solvent was removed under reduced pressure. The crude product was suspended in water (50 mL) and acidified by dropwise addition of 6N HCl. The solid product was filtered off and dissolved in 100ml of EtOAc. The solution dried over MgS04, filtered, and concentrated in vacuo. The final product was obtained as 0.730 g. LCMS E-S (M+H) =267.9. 1H NMR (400 MHz, DMSO- d6) δ ppm 2.71 (s, 3 H), 5.70 (s, 2 H), 7.15 - 7.37 (m, 5 H), 7.63 (s, 1 H), 8.33 (s, 1 H), 13.86 (s, 1 H).
Intermediate 29
ethyl 4-cyclopropyl-2,4-dioxobutanoate

Sodium metal (2.411 g, 105 mmol) was dissolved in ethanol (50 mL). The solution was heated to reflux followed by addition of a mixture of 1-cyclopropylethanone (8.4 g, 100 mmol) and diethyl oxalate (14.59 g, 100 mmol) dropwise over 30 minutes. The reaction mixture was heated at reflux for an additional 2h, and then allowed to cool to room temperature over a 2 d period. The contents were diluted with water (200 mL) and acidified by dropwise addition of 6N HC1. The contents were extracted with EtOAc (3 x 75 mL), washed with water, brine, dried over MgS04, filtered, and concentrated in vacuo. The final product was collected as 14.3g (74%). LCMS E-S (M+H) =184.8 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.03 (d, J=7.83 Hz, 2 H) 1.13 - 1.19 (m, 2 H) 1.31 (t, J=7.07 Hz, 3 H) 1.81 - 1.90 (m, 1 H) 4.29 (q, J=7.16 Hz, 2 H) 6.43 (s, 1 H). Intermediate 30
Ethyl 6-cyclopropyl-l-{[4-(methyloxy)phenyl]methyl}-lH-pyrazolo[3,4-6]pyridine-4- carboxylate
A mixture of l-{[4-(methyloxy)phenyl]methyl}-lH-pyrazol-5-amine (3 g, 14.76 mmol), ethyl 4-cyclopropyl-2,4-dioxobutanoate (2.72 g, 14.76 mmol) and benzene (50 mL) were heated at 63 °C for 16 h. The solvent was removed under reduced pressure. The crude residue was purified via silica gel chromatography (eluent: 0 to 25%
EtOAc:Hex) to afford 2.56 g of the desired cyclized product and 1.71 g of the uncyclized adduct. The uncyclized adduct was dissolved in 25 mL of AcOH and heated to reflux for 16 hours. The solvent was removed under reduced pressure and the residue purified via
silica gel chromatography (eluent: 0 to 25% EtOAc:Hex) to afford an additional 1.15 g of the desired cyclized product (combined yield = 71%). LCMS E-S (M+H) = 352.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.07 - 1.13 (m, 4 H) 1.38 (t, J=7.07 Hz, 3 H) 2.39 (s, 1 H) 3.67 (s, 3 H) 4.41 (q, J=7.07 Hz, 2 H) 5.51 (s, 2 H) 6.84 (d, J=8.84 Hz, 2 H) 7.21 (d, J=8.59 Hz, 2 H) 7.64 (s, 1 H) 8.23 (s, 1 H).
Intermediate 31
Ethyl 6-cyclopropyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

To a 20 mL microwave vial were combined ethyl 6-cyclopropyl-l-{[4-
(methyloxy)phenyl]methyl}-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (2.05 g, 5.83 mmol), trifluoroacetic acid (6.74 ml, 88 mmol) and anisole (1.912 ml, 17.50 mmol). The reaction vessel was sealed and irradiated (microwave) at 100 °C for 5 minutes. After cooling to room temperature, the reaction mixture was concentrated in vacuo. The residue was diluted with water (20 mL) and then saturated NaHC03 until basic. The contents were extracted with DCM (4 x 20 mL). The combined organic layers were washed with water, brine, dried over MgS04, filtered and concentrated in vacuo. The crude product was purified via silica gel chromatography (eluent: 0 to 20% EtOAc:Hex). The final product was obtained as 1.06 g (79%). LCMS E-S (M+H) = 232.2. 1H NMR (400 MHz, DMSO-de) δ ppm 0.95 - 1.21 (m, 4 H) 1.41 (t, J=7.07 Hz, 3 H) 2.33 - 2.46 (m, 1 H) 4.44 (q, J=7.07 Hz, 2 H) 7.67 (s, 1 H) 8.26 (d, J=1.26 Hz, 1 H) 13.75 (s, 1 H).
Intermediate 32
Ethyl l-amino-6-cycloprop l-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

Ethyl 6-cyclopropyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (200 mg, 0.865 mmol) was dissolved in N-Methyl-2-pyrrolidone (NMP) (8 mL), followed by addition of potassium tert-butoxide (116 mg, 1.038 mmol). After 20 minutes stirring, 0-{[4- (methyloxy)phenyl]carbonyl}hydroxylamine l-[(aminooxy)carbonyl]-4- (methyloxy)benzene (289 mg, 1.730 mmol) was added and the mixture stirred at room temperature for 16 h. The contents were diluted with EtOAc, and then washed with brine, and saturated NaHC03. The organic phase was dried over MgSC^, filtered, and concentrated in vacuo. The crude product was purified via silica gel chromatography (eluent: 0 to 50% EtOAc). The final product as collected as 106 mg (50%). LCMS E-S (M+H) = 274.4. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.00 - 1.14 (m, 4 H), 1.41 (t, J=7.07 Hz, 3 H), 2.34 - 2.47 (m, 1 H), 4.44 (q, J=7.07 Hz, 2 H), 6.39 (s, 2 H), 7.64 (s, 1 H), 8.09 (s, 1 H).
Intermediate 33
Ethyl 6-cyclopropyl-3-methyl-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4- carbox late

A solution of ethyl 4-cyclopropyl-2,4-dioxobutanoate (700 mg, 3.76 mmol), 3- methyl-l-(l-methylethyl)-lH-pyrazol-5 -amine (523 mg, 3.76 mmol) and benzene (20 mL) were heated at 65 °C for 6 hr, and then allowed to cool to room temperature for 2 d. The solvent was removed under reduced pressure and the residue purified via silica gel chromatography (eluent: 0 to 25% EtOAc:Hex). The final product was collected as 0.77 g
(71%). LCMS E-S (M+H) = 288.0. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.04 - 1.11 (m, 2 H), 1.13 - 1.20 (m, 4 H), 1.47 (t, J=7.20 Hz, 6 H), 1.55 (d, J=6.82 Hz, 12 H), 2.16 - 2.25 (m, 2 H), 2.68 (s, 6 H), 4.49 (q, J=7.07 Hz, 4 H), 5.20 (spt, J=6.78 Hz, 2 H), 7.41 (s, 1 H).
Intermediate 34
Ethyl l-cyclobutyl-6-cyclopropyl-3-meth l-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

The title compound was prepared in the same manner as described for
intermediate 33 using l-cyclobutyl-3 -methyl- lH-pyrazol-5 -amine (400 mg, 2.65 mmol), ethyl 4-cyclopropyl-2,4-dioxobutanoate (487 mg, 2.65 mmol) and benzene (50 mL), wherein the reaction time was 4 h. The final product was collected as 0.556 g (70%). LCMS E-S (M+H) = 300.6 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.02 - 1.22 (m, 4 H) 1.47 (t, J=7.07 Hz, 3 H) 1.80 - 2.01 (m, 2 H) 2.15 - 2.28 (m, 1 H) 2.38 - 2.51 (m, 2 H) 2.70 (s, 3 H) 2.82 (td, J=9.85, 2.53 Hz, 2 H) 4.49 (q, J=7.07 Hz, 2 H) 5.32 - 5.57 (m, 1 H) 7.41 (s, 1 H).
Intermediate 35
ethyl l-cyclopentyl-6-cyclopropyl-3-methyl-lH-pyrazolo[3,4-6]pyridine-4- carbox late

The title compound was prepared in the same manner as described for
intermediate 33 using l-cyclopentyl-3 -methyl- lH-pyrazol-5 -amine (570 mg, 3.45 mmol),
ethyl 4-cyclopropyl-2,4-dioxobutanoate (635 mg, 3.45 mmol), and benzene (50 mL) wherein the reaction time was 4 h. The crude product was purified via silica gel chromatography (eluent: 0 to 10% EtOAc:Hex). The final product was collected as 0.740 g (68%). LCMS E-S (M+H) = 314.3 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.04 - 1.21 (m, 4 H) 1.47 (t, J=7.20 Hz, 3 H) 1.73 (br. s., 2 H) 2.00 (d, J=2.78 Hz, 2 H) 2.08 - 2.16 (m, 4 H) 2.21 (s, 1 H) 2.67 (s, 3 H) 4.49 (q, J=7.16 Hz, 2 H) 5.32 (t, J=7.83 Hz, 1 H) 7.40 (s, 1 H).
Intermediate 36
ethyl 6-cyclopropyl-l-(l,l-dimethylethyl)-3-methyl-lH-pyrazolo[3,4-6]pyridine-4- carbox late

The title compound was prepared in the same manner as described for
intermediate 33 using ethyl 4-cyclopropyl-2,4-dioxobutanoate (481 mg, 2.61 mmol), 1- (l,l-dimethylethyl)-3 -methyl- lH-pyrazol-5 -amine (400 mg, 2.61 mmol), and toluene (20 mL), wherein the reaction time was 3 h. The crude product was purified via silica gel chromatography (eluent: 0 to 10% EtOAc:Hex). The final product was collected as 0.24 g (30%). LCMS E-S (M+H) = 302.5. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.02 - 1.23 (m, 4 H) 1.47 (t, J=7.20 Hz, 3 H) 1.79 (s, 9 H) 2.19 (s, 1 H) 2.66 (s, 3 H) 4.49 (q, J=7.07 Hz, 2 H) 7.44 (s, 1 H).
Intermediate 37
Ethyl 6-cyclopropyl-l-(l-cyclopropylethyl)-3-methyl-lH-pyrazolo[3,4-6]pyridine-4- carbox late

l-(l-Cyclopropylethyl)-3 -methyl- lH-pyrazol-5 -amine (500 mg, 3.03 mmol) and ethyl 4-cyclopropyl-2,4-dioxobutanoate (557 mg, 3.03 mmol) were suspended in Toluene (10 mL) and heated at 70 °C for 16 h. The solvent was removed in vacuo and the crude residue was purified via silica gel chromatography (eluent: gradient of 0 to 10%
EtOAc:Hex). The final product was collected as a solid, 0.722 g (76%). LCMS E-S (M+H) = 314.3 1H NMR (400 MHz, DMSO-de) ppm 0.17 - 0.32 (m, 2 H), 0.39 (m, J=9.69, 4.82, 4.82, 4.67 Hz, 1 H), 0.51 - 0.62 (m, 1 H), 0.94 - 1.11 (m, 4 H), 1.28 - 1.41 (m, 4 H), 1.58 (d, J=6.82 Hz, 3 H), 2.30 - 2.38 (m, 1 H), 2.56 (s, 3 H), 4.19 (dq, J=9.44, 6.87 Hz, 1 H), 4.43 (q, J=7.07 Hz, 2 H), 7.48 (s, 1 H) Intermediate 38
Ethyl l-cyclohexyl-6-cyclopropyl-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 37 using l-(l-cyclohexyl)-3 -methyl- lH-pyrazol-5 -amine (500 mg, 2.79 mmol) and ethyl 4-cyclopropyl-2,4-dioxobutanoate (514 mg, 2.79 mmol). The final product was collected as a solid, 0.827 g (91%). LCMS E-S (M+H) = 328.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.01 - 1.10 (m, 3 H), 1.20 - 1.30 (m, 1 H), 1.37 (t, J=7.07 Hz, 3 H), 1.41 - 1.52 (m, 2
H), 1.70 (d, 1 H), 1.79 - 1.98 (m, 6 H), 2.30 - 2.38 (m, 1 H), 2.54 (s, 3 H), 4.42 (q, J=7.07 Hz, 2 H), 4.60 - 4.74 (m, 1 H), 7.44 (s, 1 H).
Intermediate 39
Ethyl 6-cyclopropyl-3-methyl-l-(l-methyl-4-piperidinyl)-lH-pyrazolo[3,4-6]pyridine-
4-carbox late

3 -Methyl- l-(l-methyl-4-piperidinyl)-lH-pyrazol-5 -amine (500 mg, 2.57 mmol) and ethyl 4-cyclopropyl-2,4-dioxobutanoate (474 mg, 2.57 mmol) were suspended in Toluene (10 mL) and heated at 70 °C for 5 h. The solvent was removed in vacuo and the crude residue was purified via silica gel chromatography (eluent: gradient of 0 to 10% MeOH:DCM). The final product was collected as a solid, 0.722 g (76%). LCMS E-S (M+H) = 343.1 1H NMR (400 MHz, DMSO-d6) δ ppm 1.03 - 1.11 (m, 4 H), 1.38 (t, J=7.07 Hz, 3 H), 1.83 (d, J=6.06 Hz, 2 H), 2.12 - 2.22 (m, 4 H), 2.27 (s, 3 H), 2.35 (m, J=7.83, 7.83, 5.05, 4.80 Hz, 1 H), 2.54 (s, 3 H), 2.94 (d, J=6.57 Hz, 2 H), 4.42 (q, J=7.16 Hz, 2 H), 4.57 - 4.73 (m, 1 H), 7.46 (s, 1 H).
Intermediate 40
Ethyl 6-cyclopropyl-3-methyl-l-(tetrahydro-2H-pyran-4-yl)-lH-pyrazolo[3,4- b] pyridine-4-carbox late
(43%). LCMS E-S (M+H) = 330.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.00 - 1.12 (m, 4 H), 1.37 (t, J=7.20 Hz, 3 H), 1.82 (dd, J=12.51, 2.40 Hz, 2 H), 2.16 (qd, J=12.21, 4.55 Hz, 2 H), 2.29 - 2.42 (m, 1 H), 2.54 (s, 3 H), 3.47 - 3.60 (m, 2 H), 3.99 (dd, J=11.37, 3.79 Hz, 2 H), 4.42 (q, J=7.24 Hz, 2 H), 4.92 (tt, J=l 1.59, 4.20 Hz, 1 H), 7.46 (s, 1 H).
Intermediate 41
Ethyl 6-cyclopropyl-l-[2-({[(l,l-dimethylethyl)oxy]carbonyl}amino)ethyl]-lH- pyrazolo [3,4-6] pyridine-4-carboxylate

Ethyl 6-cyclopropyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (125 mg, 0.541 mmol) was dissolved in DMF(10 mL), followed by addition of potassium tert-butoxide (79 mg, 0.703 mmol). After stirring for 15 minutes, 1,1-dimethylethyl (2- bromoethyl)carbamate (121 mg, 0.541 mmol) was added and the mixture was stirred at room temperature for 16 h. The reaction mixture was concentrated to dryness. The crude residue was diluted with water (50 mL) and acidified with acetic acid. The contents were extracted with DCM (4 x 50 mL). The combined organic layers were washed with water, brine, dried over MgS04, filtered, and concentrated in vacuo. The crude residue was purified via silica gel chromatography (eluent: 0 to 50% EtOAc:Hex). The final product was obtained as 0.114 g (56%). LCMS E-S (M+H) = 375.1. 1H NMR (400 MHz, DMSO-de) δ ppm 1.04 - 1.14 (m, 4 H), 1.15 - 1.20 (m, 2 H), 1.27 (s, 7 H), 1.41 (t, J=7.07
Hz, 3 H), 2.36 - 2.45 (m, 1 H), 3.31 - 3.41 (m, 3 H), 4.36 - 4.50 (m, 5 H), 6.84 (t, J=5.81 Hz, 1 H), 7.65 (s, 1 H), 8.24 (s, 3 H).
Intermediate 42
Ethyl 6-cyclopropyl-3-methyl-l-(4-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4- carbox late

The title compound was prepared in the same manner as described for intermediate 30 using 3 -methyl- l-(4-pyridinyl)-lH-pyrazol-5 -amine (310 mg, 1.780 mmol), ethyl 4- cyclopropyl-2,4-dioxobutanoate (328 mg, 1.780 mmol), benzene (50 mL), and acetic acid (25 mL). The final product was collected as 3.71 (71% overall). LCMS E-S (M+H) = 323.5 1H NMR (400 MHz, DMSO-d6) δ ppm 1.15 - 1.22 (m, 4 H), 1.41 (t, J=7.07 Hz, 3 H), 2.64 - 2.69 (m, 4 H), 4.47 (q, J=7.07 Hz, 2 H), 7.74 (s, 1 H), 8.27 - 8.36 (m, 2 H), 8.66 - 8.71 (m, 2 H).
Intermediate 43
6-Cyclopropyl-l-ethyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

To a solution of ethyl 6-cyclopropyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (200 mg, 0.865 mmol) in DMF(10 mL) was carefully added sodium hydride (29.1 mg, 1.211 mmol). After 15 minutes stirring, iodoethane (0.077 mL, 0.951 mmol) was added and the mixture stirred at room temperature for 2 hr. Sodium hydroxide (1 mL, 1.000 mmol) was added and the mixture allowed to stir at room temperature for 1 h. The contents were concentrated in vacuo. The crude residue was diluted with water (50 mL)
and acidified with acetic acid. The contents were then extracted with DCM (4 x 50 mL). The combined organic layers were washed with water, brine, dried over MgS04, filtered and concentrated in vacuo. The crude product was purified by silica gel chromatography (eluent: hexanes to 100% EtOAc, then DCM to 20% MeOH:DCM). The final product was collected as 90 mg (45%). LCMS E-S (M+H) = 232.1 1H NMR (400 MHz, DMSO-d6) δ ppm 1.06 - 1.17 (m, 4 H) 1.41 (t, J=7.33 Hz, 3 H) 2.35 - 2.45 (m, 1 H) 4.34 - 4.57 (m, 2 H) 7.62 (s, 1 H) 8.24 (s, 1 H) 13.83 (br. s., 1 H). Regiochemical assignment supported by 2D HNMR. Intermediate 44
6-Cyclopropyl-l-propyl-l -pyrazolo[3,4-6]pyridine-4-carboxylic acid
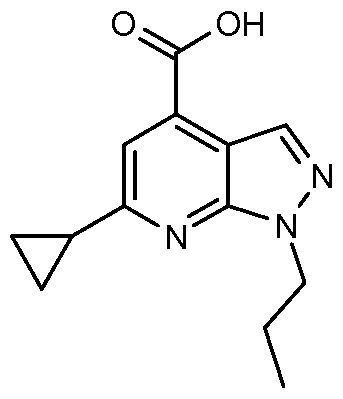
The title compound was prepared as in the same manner as described for intermediate 43 using ethyl 6-cyclopropyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (200 mg, 0.865 mmol), sodium hydride (29.1 mg, 1.211 mmol), DMF (10 mL), 1-iodopropane (162 mg, 0.951 mmol), and NaOH (1 mL). The crude product was purified by silica gel chromatography (eluent: 0 to 10% MeOH:DCM) to afford the final product as a solid, 90 mg (42%). Regiochemical assignment supported by 2D HNMR. LCMS E-S (M+H) = 246.0 1H NMR (400 MHz, DMSO-d6) δ ppm 13.83 (br s, 1H), 8.24 (s, 1H), 7.62 (s, 1H), 4.26 (t, 2H, j = 7.2 Hz), 2.37 - 2.40 (m, 2H), 1.83 - 1.89 (m, 2H), 1.07 - 1.11 (m, 4H), 0.77 (t, 3H, j = 7.2 Hz).
Intermediate 45
l-Amino-6-cyclopropyl-l -pyrazolo[3,4-6]pyridine-4-carboxylic acid

Intermediate 46
6-Cyclopropyl-3-methyl-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

To a solution of ethyl 6-cyclopropyl-3 -methyl- 1-(1 -methylethyl)-lH-pyrazolo [3,4- b]pyridine-4-carboxylate (200 mg, 0.696 mmol) in ethanol (5 mL) was added sodium hydroxide (2.088 mL, 2.088 mmol), and the mixture stirred at room temperature for 30 min. The reaction mixture was concentrated in vacuo. The residue was diluted with water (30 mL) and then acidified with acetic acid. The contents were extracted with DCM (3 x 30 mL). The combined organic layers were washed with water, brine, dried over MgSC^, filtered, and concentrated in vacuo. The final product was obtained as 0.17 g (94%).
LCMS E-S (M+H) = 260.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.97 - 1.12 (m, 4 H) 1.44 (d, J=6.57 Hz, 6 H) 2.27 - 2.38 (m, 1 H) 2.55 (s, 3 H) 5.07 (quin, J=6.69 Hz, 1 H) 7.43 (s, 1 H) 13.30 - 14.08 (m, 1 H).
Intermediate 47
6-Cyclopropyl- 1- [2-({ [(1 , l-dimethylethyl)oxy] carbonyl} amino)ethyl] - 1H- pyrazolo[3,4- ridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate
46 using ethyl 6-cyclopropyl-l-[2-({[(l,l-dimethylethyl)oxy]carbonyl}amino)ethyl]-lH- pyrazolo[3,4-b]pyridine-4-carboxylate (110 mg, 0.294 mmol), sodium hydroxide (1 mL, 0.294 mmol), and ethanol (10 mL) wherein the stir time was 16h. The final product was collected as 0.060 g (59%). LCMS E-S (M+H) = 347.1 1H NMR (400 MHz, DMSO-d6) δ ppm 1.06 - 1.13 (m, 4 H) 1.27 (s, 9 H) 2.33 - 2.43 (m, 1 H) 3.37 (q, J=5.81 Hz, 2 H) 4.44 (t, J=5.94 Hz, 2 H) 6.84 (t, J=5.81 Hz, 1 H) 7.62 (s, 5 H) 8.24 (s, 1 H).
Intermediate 48
l-Cyclobutyl-6-cyclopropyl-3-meth l-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 46 using ethyl l-cyclobutyl-6-cyclopropyl-3 -methyl- IH-pyrazolo [3, 4-b]pyridine-4- carboxylate (550 mg, 1.837 mmol), sodium hydroxide (3 ml, 3.00 mmol), and ethanol (30 mL) wherein the stir time was lh. The final product was collected as 0.490 g (98%). LCMS E-S (M+H) = 272.5 1H NMR (400 MHz, DMSO-d6) δ ppm 1.03 - 1.10 (m, 4 H), 1.80 - 1.91 (m, 2 H), 2.26 - 2.43 (m, 3 H), 2.58 (s, 3 H), 2.59 - 2.71 (m, 2 H), 5.35 (dq, J=8.59, 8.42 Hz, 1 H), 7.44 (s, 1 H), 13.73 (br. s., 1 H).
Intermediate 49
l-Cyclopentyl-6-cyclopropyl-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 46 using ethyl l-cyclopentyl-6-cyclopropyl-3 -methyl- IH-pyrazolo [3, 4-b]pyridine-4- carboxylate (740 mg, 2.361 mmol), sodium hydroxide (4ml, 4.00 mmol), and ethanol (30 mL), wherein the stir time was lh. The final product was collected as 0.530 g (79%). LCMS E-S (M+H) = 286.3 1H NMR (400 MHz, DMSO-d6) δ ppm 0.96 - 1.12 (m, 4 H) 1.52 - 1.74 (m, 2 H) 1.82 - 2.13 (m, 6 H) 2.26 - 2.37 (m, 1 H) 2.55 (s, 3 H) 5.17 - 5.30 (m, 1 H) 7.43 (s, 1 H) 13.70 (br. s., 1 H).
Intermediate 50
6-Cyclopropyl-l-(l,l-dimethylethyl)-3-methyl-lH-pyrazolo[3,4-6]pyridine-4- arboxylic acid

The title compound was prepared in the same manner as described for intermediate 46 using ethyl 6-cyclopropyl- 1 -( 1 , 1 -dimethylethyl)-3 -methyl- 1 H-pyrazolo [3 ,4- b]pyridine-4-carboxylate (240 mg, 0.796 mmol), sodium hydroxide (4 mL, 4.00 mmol), and ethanol (30 mL), wherein the stir time was lh. The final product was collected as 0.210 g (96%). LCMS E-S (M+H) = 274.4. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.05 (m, 4 H) 1.70 (s, 9 H) 2.33 (m, 1 H) 2.51 (s, 3 H) 7.46 (s, 1 H) 13.70 (br. s., 1 H).
Intermediate 51
6-Cyclopropyl-3-methyl-l-(4-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 46 using ethyl 6-cyclopropyl-l-(4-pyridinyl)-3 -methyl- lH-pyrazolo [3, 4-b]pyridine-4- carboxylate (240 mg, 0.796 mmol), sodium hydroxide (4 mL, 4.00 mmol), and ethanol (30 mL), wherein the stir time was 1 h. The final product was collected as 0.210 g (89%). LCMS E-S (M+H) = 295.3.
Intermediate 52
6-Cyclopropyl-l-(l-cyclopropylethyl)-3-methyl-lH-pyrazolo[3,4-6]pyridine-4- carboxylic acid

To an EtOH (10 mL) solution of ethyl 6-cyclopropyl-l-(l-cyclopropylethyl)-3- methyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (720 mg, 2.297 mmol) was added sodium hydroxide (6.89 mL, 6.89 mmol) and the mixture heated at 70 °C for 1 hour. The solvent was removed in vacuo and the residue was dissolved in 20 mL of water. The contents were acidifed with acetic acid, and extracted with EtOAc (4 x 30mL). The combined organic extracts were washed with water, brine, dried over MgSC^, filtered, and concentrated in vacuo. The final product was collected as a solid, 0.560 g (85%). LCMS
E-S (M+H) = 286.3 1H NMR (400 MHz, DMSO-d6) δ ppm 0.15 - 0.33 (m, 2 H), 0.38 (dq, J=9.57, 4.89 Hz, 1 H), 0.50 - 0.65 (m, 1 H), 0.90 - 1.10 (m, 4 H), 1.27 - 1.42 (m, 1 H),
1.57 (d, J=6.82 Hz, 3 H), 2.23 - 2.36 (m, 1 H), 2.57 (s, 3 H), 4.18 (dq, J=9.32, 6.83 Hz, 1 H), 7.44 (s, 1 H), 12.79 (br. s., 1H).
Intermediate 53
l-cyclohexyl-6-cyclopropyl-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 52 using ethyl 6-cyclopropyl-l-(l-cyclohexyl)-3 -methyl- lH-pyrazolo [3, 4-b]pyridine-4- carboxylate (820 mg, 2.5 mmol) and sodium hydroxide (7.51 mL, 7.51 mmol). The final product was collected as a solid, 0.660 g (88%). LCMS E-S (M+H) = 300.4 1H NMR (400 MHz, DMSO-de) δ ppm 1.01 - 1.11 (m, 4 H), 1.16 - 1.31 (m, 1 H), 1.36 - 1.55 (m, 2 H), 1.70 (d, J=12.38 Hz, 1 H), 1.80 - 1.98 (m, 6 H), 2.32 (m, J=7.80, 7.80, 5.05, 4.86 Hz, 1 H), 2.55 (s, 3 H), 4.60 - 4.73 (m, 1 H), 7.40 (s, 1 H), 13.71 (br. s., 1 H). Intermediate 54
6-Cyclopropyl-3-methyl-l-(l-methyl-4-piperidinyl)-lH-pyrazolo[3,4-6]pyridine-4- carboxylic acid

To an EtOH solution (10 mL) of ethyl 6-cyclopropyl-3 -methyl- 1-(1 -methyl-4- piperidinyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (620 mg, 1.811 mmol) was added sodium hydroxide (5.43 mL, 5.43 mmol) and the mixture heated at 70 °C for 1 hour. The solvent was removed in vacuo and the residue was dissolved in 20 mL of water. The contents were acidifed with acetic acid, and extracted with EtOAc (4 x 30mL). The combined organic extracts were washed with water, brine, dried over MgSC^, filtered, and
concentrated in vacuo to afford a minor amount of product. The aqueous phase was concentrated in vacuo and the crude residue purified by reverse phase HPLC purification (eluent : 0% ACN/H20, 0.1%TFA to 45% ACN/H20, 0.1% TFA). The isolated solid product was concentrated from toluene to afford the final product as 510 mg (90%>).
LCMS E-S (M+H) = 315.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.08 (d, J=6.57 Hz, 4 H), 2.10 (d, J=13.14 Hz, 2 H), 2.30 - 2.42 (m, 3 H), 2.55 (s, 3 H), 2.77 - 2.92 (m, 3 H), 3.30 (br. s., 2 H), 3.57 (d, J=12.13 Hz, 2 H), 4.98 (m, J=11.78, 11.78, 3.85, 3.66 Hz, 1 H), 7.46 (s, 1 H), 9.87 (br. s., 1 H).
Intermediate 55
6-Cyclopropyl-3-methyl-l-(tetrahydro-2H-pyran-4-yl)-lH-pyrazolo[3,4-6]pyridine-4- carboxylic acid

To an EtOH solution (30 mL) of ethyl 6-cyclopropyl-3 -methyl- l-(tetrahy dro-2H- pyran-4-yl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (300 mg, 0.911 mmol) was added sodium hydroxide (1.82 mL, 1.82 mmol) and the mixture stirred at room temperature for 16 h. The solvent was removed in vacuo and the residue was dissolved in 20 mL of water. The contents were acidifed with acetic acid, and extracted with EtOAc (4 x 30mL). The combined organic extracts were washed with water, brine, dried over MgSC^, filtered, and concentrated in vacuo to afford the final product as 265 mg (97%>). LCMS E-S (M+H)=
302.4 1H NMR (400 MHz, DMSO-de) δ ppm 1.02 - 1.09 (m, 4 H), 1.81 (dd, J=12.38, 2.27 Hz, 2 H), 2.15 (qd, J=12.25, 4.42 Hz, 2 H), 2.28 - 2.37 (m, 1 H), 2.54 (s, 3 H), 3.46 - 3.60 (m, 2 H), 3.89 - 4.02 (m, 2 H), 4.91 (m, J=11.56, 11.56, 4.17, 4.04 Hz, 1 H), 7.42 (s, 1 H), 13.73 (br. s., 1 H).
Intermediate 56
(4,6-dimethyl-2-oxo- 1 ,2-dihydr o-3-pyridinyl)methyl 6-cyclopropyl- 1- [2-({ [(1,1- dimethylethyl)oxy]carbonyl}amino)ethyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

To a solution of DMSO (10 mL) were sequentially added 6-cyclopropyl- 1 -[2-
({[(1,1 -dimethylethyl)oxy] carbonyl} amino)ethyl] - 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxylic acid (270 mg, 0.779 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (178 mg, 1.169 mmol), and l-hydroxy-7-azabenzotriazole (212 mg, 1.559 mmol), and the mixture stirred at room temperature for 10 min. Next was added EDC (299 mg, 1.559 mmol) and N-methylmorpholine (0.343 mL, 3.12 mmol), and reaction mixture was stirred at room temperature for 16 h.. The contents were diluted with water (25 mL) and stirred for 10 min. The solid product was filtered off, dried, and collected as 0.290 g (98%). LCMS E-S (M+H) = 381.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.00 - 1.15 (m, 4 H), 1.30 (s, 9H), 2.13 (s, 3 H), 2.18 - 2.32 (m, 4 H), 4.36 (d, J=4.80 Hz, 2 H), 4.42 (t, J=5.81 Hz, 2 H), 5.90 (s, 1 H), 6.85 (t, J=5.56 Hz, 1 H), 7.44 (s, 1 H), 8.22 (s, 2 H), 8.73 (t, J=4.67 Hz, 1 H), 11.57 (br. s., 1 H).
Example 98
l-(2- Aminoethyl)-6-cyclopropyl- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

To a solution of 1,1-dimethylethyl {2-[6-cyclopropyl-4-({[(4,6-dimethyl-2-oxo-
1 ,2-dihydro-3 -pyridinyl)methyl] amino } carbonyl)- 1 H-pyrazolo [3 ,4-b]pyridin- 1 - yl]ethyl} carbamate (260 mg, 0.541 mmol) in DCM (15 mL) was added trifluoroacetic acid (3 ml, 38.9 mmol) and the mixture stirred at room temperature for 2 h. The contents were concentrated in vacuo and the crude residue purified by reverse phase HPLC. (mobile phase : 10 to 70% ACN in H20, 0.1% NH4OH). The final product was collected as 0.170 g (89%). LCMS E-S (M+H) = 353.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.07 (d, J=6.32 Hz, 4 H), 2.13 (s, 3 H), 2.21 (s, 4 H), 3.00 (t, J=6.19 Hz, 2 H), 4.29 - 4.47 (m, 4 H), 5.90 (s, 1 H,) 7.43 (s, 1 H), 8.22 (s, 1 H), 8.75 (t, 1 H).
Example 99
6-Cyclopropyl-7V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl] - 1- {2-
[(methylsulfonyl)amino]ethyl}-lH-pyrazolo [3,4-6] pyridine-4-carboxamide

Example 100
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-methyl-l-(phenylmethyl)- lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate 56 using 6-methyl-l-(phenylmethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (200 mg, 0.748 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (142 mg, 0.935 mmol), l-hydroxy-7-azabenzotriazole (204 mg, 1.497 mmol) , DMSO (10 mL), EDC (287 mg, 1.497 mmol), and N-methylmorpholine (0.329 mL, 2.99 mmol). The final product was collected as 0.25 g (83%). LCMS E-S (M+H) = 402.2. 1H NMR (400 MHz, DMSO-de) δ ppm 0.93 - 1.12 (m, 4 H), 1.42 (d, J=6.82 Hz, 6 H), 2.12 (s, 3 H), 2.22 (s, 4 H), 2.36 (s, 3 H), 4.34 (d, J=4.80 Hz, 2 H), 4.90 - 5.11 (m, 1 H), 5.87 (s, 1 H), 6.99 (s, 1 H), 8.58 (s, 1 H), 11.51 (s, 1 H.
Example 101
6-Cyclopropyl-7V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl] - 1-ethyl- 1H- pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate
56 using 6-cyclopropyl-l -ethyl- IH-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (91 mg, 0.394 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (75 mg, 0.493 mmol), 1- hydroxy-7-azabenzotriazole (107 mg, 0.788 mmol), DMSO(10 mL), EDC (151 mg, 0.788 mmol), and N-methylmorpholine (0.173 mL, 1.577 mmol). The final product was collected as 0.090 g (63%). LCMS E-S (M+H) = 366.3 1H NMR (400 MHz, DMSO-d6) δ ppm 1.07 (d, J=6.32 Hz, 4 H), 1.39 (t, J=7.20 Hz, 3 H), 2.13 (s, 3 H), 2.19 - 2.32 (m, 4 H), 4.36 (d, J=4.80 Hz, 2 H), 4.42 (q, J=7.07 Hz, 2 H), 5.90 (s, 1 H), 7.43 (s, 1 H), 8.21 (s, 1 H), 8.75 (t, J=4.67 Hz, 1 H), 1 1.57 (br. s., 1 H).
Example 102
6-Cyclopropyl-7V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl] - 1-pr opyl- IH- pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate 56 using 6-cyclopropyl-l -propyl- IH-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (90 mg, 0.368 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (70 mg, 0.460 mmol), 1-
hydroxy-7-azabenzotriazole (100 mg, 0.736 mmol), DMSO(10 mL), EDC (141 mg, 0.736 mmol), and N-methylmorpholine (0.162 mL, 1.472 mmol). The final product was collected as 0.118 g (84%). LCMS E-S (M+H) = 380.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.78 (t, 3 H), 0.99 - 1.16 (m, 4 H), 1.85 (d, J=7.33 Hz, 2 H), 2.13 (s, 3 H), 2.22 (s, 4 H), 4.25 - 4.43 (m, 4 H), 5.90 (s, 1 H), 7.43 (s, 1 H), 8.21 (s, 1 H), 8.75 (s, 1 H), 11.57 (s, 1 H).
Example 103
6-Cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l- (l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate 56 using 6-cyclopropyl-l-(2-methylpropyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (80 mg, 0.309 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (70.4 mg, 0.463 mmol), l-hydroxy-7-azabenzotriazole (84 mg, 0.617 mmol), DMSO(10 mL), EDC (118 mg, 0.617 mmol), and N-methylmorpholine (0.136 mL, 1.234 mmol). The final product was collected as 0.123 g (100%). LCMS E-S (M+H) = 394.2 1H NMR (400 MHz, DMSO-de) δ ppm 0.93 - 1.12 (m, 4 H), 1.42 (d, J=6.82 Hz, 6 H), 2.12 (s, 3 H), 2.22 (s, 4 H), 2.36 (s, 3 H), 4.34 (d, J=4.80 Hz, 2 H), 4.90 - 5.11 (m, 1 H), 5.87 (s, 1 H), 6.99 (s, 1 H), 8.58 (s, 1 H), 11.51 (s, 1 H).
Example 104
l-Amino-6-cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-lH- pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate
56 using l-amino-6-cyclopropyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (73 mg, 0.335 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (76 mg, 0.502 mmol), 1- hydroxy-7-azabenzotriazole (91 mg, 0.669 mmol), DMSO(10 mL), EDC (128 mg, 0.669 mmol), and N-methylmorpholine (0.147 mL, 1.338 mmol). The aq. phase was extracted with EtOAc (5 x 30 mL). The combined EtOAc extracts were dried over MgS04, filtered, and concentrated in vacuo. The final product was collected as 0.068 g (58%). LCMS E-S (M+H) = 353.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.99 - 1.14 (m, 4 H), 2.13 (s, 3 H), 2.22 (s, 4 H), 4.35 (d, J=4.80 Hz, 2 H), 5.90 (s, 1 H), 6.31 (s, 2 H), 7.41 (s, 1 H), 8.04 (s, 1 H), 8.74 (s, 1 H), 11.58 (br. s., 1 H).
Example 105
l-Cyclobutyl-6-cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]- 3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate using l-cyclobutyl-6-cyclopropyl-3 -methyl- lH-pyrazolo [3, 4-b]pyridine-4-carboxylic
acid (164 mg, 0.604 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (129 mg, 0.846mmol), l-hydroxy-7-azabenzotriazole (165 mg, 1.209 mmol), EDC (232 mg, 1.209 mmol), N-methylmorpholine (0.266 mL, 2.418 mmol), and DMSO(10 mL). The final product was collected as 0.240 g (98%). LCMS E-S (M+H) = 406.3. 1H NMR (400 MHz, DMSO-de) δ ppm 1.03 (d, J=6.06 Hz, 4 H), 1.85 (dd, J=9.35, 5.05 Hz, 3 H), 2.12 (s, 4 H), 2.16 - 2.28 (m, 6 H), 2.30 - 2.44 (m, 7 H), 2.63 (d, J=19.96 Hz, 3 H), 4.34 (d, J=4.29 Hz, 2 H), 5.31 (t, J=8.34 Hz, 1 H), 5.87 (s, 1 H), 7.01 (s, 1 H), 8.59 (br. s., 1 H), 11.52 (br. s., 1 H).
Example 106
l-Cyclopentyl-6-cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-3-meth -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate 56 using l-cyclopentyl-6-cyclopropyl-3 -methyl- lH-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (160 mg, 0.561 mmol), 3- (aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (119 mg, 0.785 mmol), l-hydroxy-7-azabenzotriazole (153 mg, 1.121 mmol), EDC (215 mg, 1.121 mmol), N-methylmorpholine (0.247 mL, 2.243 mmol) and DMSO(10 mL). The final product was collected as 0.205 g (87%). LCMS E-S (M+H) = 420.0. 1H NMR (400 MHz, DMSO-de) δ ppm 1.03 (d, J=6.06 Hz, 4 H), 1.85 (dd, J=9.35, 5.05 Hz, 3 H), 2.12 (s, 4 H), 2.16 - 2.28 (m, 6 H), 2.30 - 2.44 (m, 7 H), 2.63 (d, J=19.96 Hz, 3 H), 4.34 (d, J=4.29 Hz, 2 H), 5.31 (t, J=8.34 Hz, 1 H), 5.87 (s, 1 H), 7.01 (s, 1 H), 8.59 (br. s., 1 H), 11.52 (br. s., 1 H).
Example 107
6-Cyclopropyl- 1-(1 , l-dimethylethyl)- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-3-meth -lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate
56 using 6-cyclopropyl- 1 -(1,1 -dimethylethyl)-3 -methyl- 1 H-pyrazolo [3, 4-b]pyridine -4- carboxylic acid (210 mg, 0.768 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (158 mg, 1.037 mmol), l-hydroxy-7-azabenzotriazole (209 mg, 1.537 mmol), EDC (295 mg, 1.537 mmol), N-methylmorpholine (0.338 mL, 3.07 mmol), and DMSO(10 mL). The final product was collected as 0.280 g (89%). LCMS E-S (M+H) = 408.2. 1H NMR (400 MHz, DMSO-de) δ ppm 1.02 (dddd, 4 H), 1.69 (m, 9 H), 2.12 (s, 3 H), 2.22 (m, 4 H), 2.33 (s, 3 H), 4.34 (d, J=4.80 Hz, 2 H), 5.87 (s, 1 H), 7.04 (s, 1 H), 8.55 (m, 1 H), 11.51 (s, 1 H).
Example 108
6-Cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(4- pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described for intermediate 56 using 6-cyclopropyl-3 -methyl- l-(4-pyridinyl)-l H-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (89 mg, 0.302 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (69.0 mg,
0.454 mmol), l-hydroxy-7-azabenzotriazole (82 mg, 0.605 mmol), DMSO(20 mL), EDC (116 mg, 0.605 mmol), and N-methylmorpholine (0.133 mL, 1.210 mmol). The final product was collected as 0.110 g (80%). LCMS E-S (M+H) = 429.1 1H NMR (400 MHz, DMSO-de) δ ppm 1.15 (br. s., 4 H), 2.13 (br. s., 3H), 2.25 (br. s., 4 H), 3.34 (br. s., 3 H), 4.37 (br. s., 2 H), 5.89 (br. s., 1 H), 7.30 (br. s., 1 H), 8.33 (br. s., 2 H), 8.67 (br. s., 3 H), 11.54 (br. s., 1 H).
Example 109
6-Cyclopropyl- l-(l-cyclopropylethyl)-7V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

6-Cyclopropyl- 1 -( 1 -cyclopropylethyl)-3 -methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxylic acid (186 mg, 0.652 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (160 mg, 0.847 mmol), l-hydroxy-7-azabenzotriazole (177 mg, 1.304 mmol), EDC (250 mg, 1.304 mmol) and N-methylmorpholine (0.287 mL, 2.61 mmol) were dissolved in DMSO (10 mL) and stirred at room temperature for 3 days. The reaction mixture was diluted with water (25 mL), stirred, and filtered. The product was dried and collected as a solid, 0.200 g (73%). LCMS E-S (M+H) = 420.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.14 - 0.30 (m, 2 H), 0.32 - 0.43 (m, 1 H), 0.48 - 0.64 (m, 1 H), 0.89 - 1.11 (m, 4 H), 1.22 - 1.43 (m, 1 H), 1.55 (d, J=6.82 Hz, 3 H), 2.11 (s, 3 H), 2.18 - 2.28 (m, 4 H), 2.37 (s, 3 H), 4.05 - 4.21 (m, 1 H), 4.34 (d, J=4.80 Hz, 2 H), 5.87 (s, 1 H), 6.99 (s, 1 H), 8.59 (t, J=4.67 Hz, 1 H), 11.51 (br. s., 1 H).
Example 110
l-Cyclohexyl-6-cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]- 3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using 6-cyclopropyl-l-(l -eye lohexyl)-3 -methyl- lH-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid (150 mg, 0.501 mmol), and 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (123 mg, 0.651 mmol). The product was collected as a solid, 0.190 g (87%). LCMS E-S (M+H) = 434.2. 1H NMR (400 MHz, DMSO-d ) δ ppm 0.95 - 1.07 (m, 4 H), 1.15 - 1.31 (m, 1 H), 1.36 - 1.52 (m, 2 H), 1.68 (br. s., 1 H), 1.77 - 1.99 (m, 6 H), 2.11 (s, 3 H), 2.18 - 2.29 (m, 4 H), 2.35 (s, 3 H), 4.33 (d, J=4.80 Hz, 2 H), 4.53 - 4.73 (m, 1 H), 5.87 (s, 1 H), 6.96 (s, 1 H), 8.58 (t, J=4.93 Hz, 1 H), 11.51 (s, 1 H).
Example 111
6-Cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l- (l-methyl-4-piperidinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

6-Cyclopropyl-3 -methyl- 1 -( 1 -methyl-4-piperidinyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxylic acid (150 mg, 0.477 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (117 mg, 0.620 mmol), l-hydroxy-7-azabenzotriazole (130 mg, 0.954 mmol), EDC (183 mg, 0.954 mmol) and N-methylmorpholine (0.210 mL, 1.909 mmol) were suspended in
DMSO(10 mL) and stirred at room temperature for 16 h. Next was added 25 mL of water, stirred for 10 minutes, and then extracted with EtOAc (5x). The organic layers were concentrated in vacuo to afford 20 mg product. The aqueous phase was concentrated in vacuo and the crude residue purified by reverse phase HPLC (mobile phase : 10 - 60% ACN in H20, 0. l%NH4OH) to afford additional product. The final product was collected as a solid, 0.100 g (47%). LCMS E-S (M+H) = 449.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.97 - 1.05 (m, 4 H), 1.79 (d, J=9.60 Hz, 2 H), 2.11 (s, 3 H), 2.15 (d, J=8.34 Hz, 3 H), 2.21 (s, 3 H), 2.23 - 2.27 (m, 3 H), 2.35 (s, 3 H), 2.91 (d, J=8.08 Hz, 2 H), 4.33 (d, J=4.80 Hz, 2 H), 4.49 - 4.72 (m, 1 H), 5.86 (s, 1 H), 6.97 (s, 1 H), 6.92 - 7.07 (m, 1 H), 8.58 (t, J=4.93 Hz, 1 H), 11.50 (s, 1 H).
Example 112
6-Cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l- (tetrahydro-2H-pyran-4-yl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using 6-cyclopropyl-3 -methyl- l-(tetrahydro-2H-pyran-4-yl)-lH-pyrazolo [3, 4-b]pyridine- 4-carboxylic acid (150 mg, 0.498 mmol), and 3-(aminomethyl)-4,6-dimethyl-2(lH)- pyridinone (98 mg, 0.647 mmol). The product was collected as a solid, 0.170 g (78%). LCMS E-S (M+H) = 436.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.95 - 1.14 (m, 4 H), 1.79 (dd, J=12.63, 2.27 Hz, 2 H), 2.05 - 2.18 (m, 5 H), 2.19 - 2.27 (m, 4 H), 2.36 (s, 3 H), 3.53 (t, J=11.12 Hz, 2 H), 3.98 (dd, J=11.49, 3.41 Hz, 2 H), 4.33 (d, J=4.80 Hz, 2 H), 4.87 (m, J=11.53, 11.53, 4.11, 3.92 Hz, 1 H), 5.87 (s, 1 H), 6.99 (s, 1 H), 8.59 (t, J=4.93 Hz, 1 H), 11.51 (s, 1 H).
Intermediate 57
Ethyl l-(l-methylethyl)-6-propyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

To a solution of ethyl 2,4-dioxoheptanoate (446 mg, 2.397 mmol) in benzene (5 mL) was added l-(l-methylethyl)-lH-pyrazol-5 -amine (300 mg, 2.397 mmol), and the mixture was stirred at 62 °C for 18 h. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography (Silica gel, gradient of 0 to 100%
EtOAc/hexanes) to give 370 mg (56%) of product. LCMS E-S (M+H) = 276.1. 1H NMR (400 MHz, CHLOROFORM- ) δ ppm 0.99 - 1.12 (m, 3 H), 1.46 - 1.54 (m, 3 H), 1.58 - 1.70 (m, 6 H), 1.82 - 1.96 (m, 2 H), 2.91 - 3.02 (m, 2 H), 4.51 (q, J= 7.2 Hz, 2 H), 5.28 - 5.44 (m, 1 H), 7.58 - 7.68 (s, 1 H), 8.27 - 8.40 (s, 1 H)
Intermediate 58
l-(l-Methylethyl)-6- ropyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

To a solution of ethyl l-(l-methylethyl)-6-propyl-lH-pyrazolo[3,4-b]pyridine-4- carboxylate (180 mg, 0.654 mmol) in ethanol (5 mL) and THF (1 mL) was added sodium hydroxide (1.090 mL, 3.27 mmol), and the mixture was stirred at 40 °C for lh. The volatiles were removed under reduced pressure. The aqueous phase was acidified to pH 3 with aq. IN HCl. The solid precipitate formed was collected by filtration and dried under high vacuum to give 144 mg (89%>) of product, which was used for next reaction without further purification. LCMS E-S (M+H) = 248.1 1H NMR (400 MHz, DMSO-d6) δ ppm 0.82 - 1.12 (m, 3 H), 1.50 (d, J = 6.6 Hz, 6 H), 1.78 (m, 2 H), 2.80 - 3.11 (m, 2 H), 5.09 - 5.38 (m, 1 H), 7.47 - 7.72 (s, 1 H), 8.18 - 8.46 (s, 1 H), 13.82 (s, 1 H).
Intermediate 60
Ethyl 4- [4-(methyloxy)phenyl] -2,4-dioxobutanoate

The title compound was prepared in the same manner as intermediate 29 using sodium metal (0.168 g, 7.32 mmol), l-[4-(methyloxy)phenyl]ethanone (lg, 6.66 mmol), and diethyl ethanedioate (0.903 mL, 6.66 mmol). The crude product was purified using column
chromatography (0 to 100 % EtOAc/hexanes) to give 0.95 g of product (57%). LCMS E-S (M+H) = 250.9. 1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.43 (m, 3 H), 3.91 (s, 3 H), 4.41 (q, J= 6.65 Hz, 2 H), 6.94 - 7.12 (m, 3 H), 8.01 (d, J= 8.59 Hz, 2 H).
Intermediate 61
Ethyl l-(l-methylethyl)-6-[4- methyloxy)phenyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

To a solution of ethyl 4-[4-(methyloxy)phenyl]-2,4-dioxobutanoate (600 mg, 2.397 mmol) in benzene (5 mL) was added l-(l-methylethyl)-lH-pyrazol-5 -amine (300 mg,
2.397 mmol), and the reaction mixture was stirred at 62 °C overnight. The mixture was concentrated in vacuo and the residue was dissolved into acetic acid (3 mL). The solution was stirred at reflux for lh and concentrated in vacuo. The residue was purified using
column chromatography (Silica gel, 0 to 100%) EtOAc/hexanes) to give 530 mg (65%>) of product. LCMS E-S (M+H) = 340.1 1H NMR (400 MHz, CHLOROFORM-^) δ ppm
1.47 - 1.57 (m, 3 H), 1.61 - 1.71 (m, 6 H), 3.88 - 3.96 (s, 3 H), 4.55 (q, J= 7.2 Hz, 2 H),
5.46 (m, 1 H), 7.03 - 7.12 (m, 2 H), 8.13 - 8.25 (m, 3 H), 8.36 - 8.43 (m, 1 H).
Intermediate 62
l-(l-Methylethyl)-6-[4-(methyloxy)phenyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

Example 113
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[4- (methyloxy)phenyl] - lH-pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using 1 -( 1 -methylethyl)-6-[4-(methyloxy)phenyl]- 1 H-pyrazolo[3 ,4-b]pyridine-4- carboxylic acid (70 mg, 0.225 mmol), DMSO(3 mL), 3-(aminomethyl)-4,6-dimethyl- 2(lH)-pyridinone (63.6 mg, 0.337 mmol) HC1 salt, N-methylmorpholine (0.099 mL, 0.899 mmol), l-hydroxy-7-azabenzotriazole (61.2 mg, 0.450 mmol), and EDC (86 mg, 0.450 mmol). The final product was collected as 89 mg (89%). LCMS E-S (M+H) = 446.5. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.54 (d, J= 6.8 Hz, 6 H), 2.13 (s, 3 H), 2.23 (s, 3 H), 3.85 (s, 3 H), 4.41 (d, J= 4.8 Hz, 2 H), 5.20 - 5.48 (m, 1 H), 5.91 (s, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 8.11 (s, 1 H), 8.24 (d, J= 8.8 Hz, 2 H), 8.34 (s, 1 H), 8.95 (m, 1 H), 11.58 (s, 1
Η).
Intermediate 63
Ethyl 2,4-dioxo-4-(4-pyridinyl)butanoate
 The title compound was prepared in the same manner as intermediate 29 using sodium metal (0.466 g, 20.26 mmol), l-(4-pyridinyl)ethanone (lg, 6.66 mmol), and diethyl ethanedioate (2.388 mL, 17.62 mmol). The crude product was purified using column chromatography (0 to 100 % EtOAc/hexanes) to give 0.95 g of product (24%). LCMS E-S (M+H) = 222.0. 1H NMR (400 MHz, CHLOROFORM-;/) δ ppm 1.37 - 1.55 (m, 3 H), 4.43 (q, J= 7.07 Hz, 2 H), 7.06 - 7.16 (m, 1 H), 7.72 - 7.92 (m, 2 H), 8.82 - 8.94 (m, 2 H).
The title compound was prepared in the same manner as intermediate 29 using sodium metal (0.466 g, 20.26 mmol), l-(4-pyridinyl)ethanone (lg, 6.66 mmol), and diethyl ethanedioate (2.388 mL, 17.62 mmol). The crude product was purified using column chromatography (0 to 100 % EtOAc/hexanes) to give 0.95 g of product (24%). LCMS E-S (M+H) = 222.0. 1H NMR (400 MHz, CHLOROFORM-;/) δ ppm 1.37 - 1.55 (m, 3 H), 4.43 (q, J= 7.07 Hz, 2 H), 7.06 - 7.16 (m, 1 H), 7.72 - 7.92 (m, 2 H), 8.82 - 8.94 (m, 2 H).
Intermediate 64
Ethyl l-(l-methylethyl)-6-(4-pyridinyl)-lH-pyrazolo [3,4-6] yridine-4-carboxylate

To a suspension of ethyl 2,4-dioxo-4-(4-pyridinyl)butanoate (442 mg, 1.997 mmol) in benzene (5 mL) was added l-(l-methylethyl)-lH-pyrazol-5 -amine (250 mg, 1.997 mmol), and the reaction mixture was stirred at 62 °C overnight. The reaction mixture was concentrated in vacuo and the residue was purified using column chromatography (Silica gel, 0 to 100% EtOAc/hexanes) to give 270 mg (43%) of product. LCMS E-S (M+H) = 311.3 1H NMR (400 MHz, CHLOROFORM- d) δ ppm 1.54 (t, J = 7.2 Hz, 3 H), 1.68 (d, J = 6.6 Hz, 6 H), 4.58 (q, J= 7.1 Hz, 2 H), 5.41 - 5.60 (m, 1 H), 8.04 - 8.19 (m, 2 H), 8.29 (s, 1 H), 8.44 - 8.55 (m, 2 H), 8.76 - 8.91 (m, 1 H).
Intermediate 65
l-(l-Methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 58 using ethyl l-(l-methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-b]pyridine-4- carboxylate (100 mg, 0.322 mmol), ethanol (4 mL), THF (0.8 mL), and sodium hydroxide (0.537 mL, 1.611 mmol). The final product was collected as 88 mg (97%). LCMS E-S
(M+H) = 283.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.58 (d, J = 6.4 Hz, 6 H), 5.40
(m, 1 H), 8.25 (d, J= 5.6 Hz, 2 H), 8.32 (s, 1 H), 8.44 (s, 1 H), 8.79 (d, J= 5.6 Hz, 2 H), 14.12 (br. s., 1 H).
Example 114
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4-pyridinyl)- lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using 1 -( 1 -methylethyl)-6-(4-pyridinyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid
(60 mg, 0.213 mmol), DMSO(2 mL), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone
(60.1 mg, 0.319 mmol) HC1 salt, N-methylmorpholine (0.093 mL, 0.850 mmol), 1- hydroxy-7-azabenzotriazole (57.9 mg, 0.425 mmol), and EDC (81 mg, 0.425 mmol). The final product was collected as 66 mg (74%). LCMS E-S (M+H) = 446.5. 1H NMR (400
MHz, DMSO- g) δ ppm 1.56 (d, J= 6.0 Hz, 6 H), 2.13 (br. s., 3 H), 2.23 (br. s., 3 H),
4.43 (m, 2 H), 5.37 (m, 1 H), 5.91 (br. s., 1 H), 8.15 - 8.34 (m, 3 H), 8.44 (s, 1 H), 8.78 (br.
s., 2 H), 9.00 (br. s., 1 H), 11.58 (br. s., 1 H).
Intermediate 66
Ethyl 4-(4-chlorophenyl)-2,4-dioxobutanoate

Sodium metal (0.143 g, 6.21 mmol) was dissolved into ethanol (3 mL) and the resulting solution was cooled with an ice bath. A mixture of l-(4-chlorophenyl)ethanone (0.756 mL, 5.65 mmol) and diethyl ethanedioate (0.766 mL, 5.65 mmol) were added
dropwise, and the reaction mixture was stirred for 16h. The reaction mixture was
quenched with ice water (5 mL) and acidified to ~pH 3 with IN HC1. The precipitate was
collected by filtration and dried under high vacuum to give 1.39 g (97%) of product.
LCMS E-S (M+H) = 255.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.29 (m, 3 H), 4.27 (d, J= 6.57 Hz, 2 H) ,7.60 (d, J= 7.58 Hz, 2 H), 8.02 (d, J= 6.57 Hz, 2 H).
Intermediate 67
Ethyl 6-(4-chlorophenyl)-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4- carboxylate

The title compound was prepared in the same manner as described for intermediate 61 using ethyl 4-(4-chlorophenyl)-2,4-dioxobutanoate (610 mg, 2.397 mmol), benzene (5 mL), l-(l-methylethyl)-lH-pyrazol-5 -amine (300 mg, 2.397 mmol), and acetic acid
(4mL). The crude product was purified by column chromatography (Silica gel, eluent: 0 to 100% EtOAc /hexanes) to give 621 mg (75%) of product. LCMS E-S (M+H) = 344.1. 1H NMR (400 MHz, CHLOROFORM-^) δ ppm 1.48 - 1.57 (m, 3 H), 1.64 - 1.74 (m, 6 H), 4.56 (q, J= 7.1 Hz, 2 H), 5.46 (m, 1 H), 7.49 - 7.55 (m, 2 H), 8.15 - 8.18 (m, 2 H), 8.21 (s, 1 H), 8.42 (s, 1 H).
Intermediate 68
6-(4-Chlorophenyl)-l-(l-meth lethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 58 using ethyl 6-(4-chlorophenyl)-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4- carboxylate (400 mg, 1.163 mmol), Ethanol (6 mL), THF (1 mL), and added sodium hydroxide (1.939 mL, 5.82 mmol) wherein the reaction time was 2h. The final product was collected as 330 mg (90%). LCMS E-S (M+H) = 315.8. 1H NMR (400 MHz,
DMSO-dg) δ ppm 1.56 (d, J= 6.8 Hz, 6 H), 5.26 - 5.47 (m, 1 H), 7.62 (d, J= 8.6 Hz, 2 H) ,8.20 (s, 1 H), 8.28 (d, J= 8.6 Hz , 2 H), 8.38 (s, 1 H), 14.03 (s, 1 H).
Example 115
6-(4-Chlorophenyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using 6-(4-chlorophenyl)-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (70 mg, 0.222 mmol), DMSO(2 mL), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone
HC1 salt (62.7 mg, 0.333 mmol), N-methylmorpholine (0.097 mL, 0.887 mmol), 1- hydroxy-7-azabenzotriazole (60.3 mg, 0.443 mmol) and EDC (85 mg, 0.443 mmol). The final product was collected as 77 mg (78%). LCMS E-S (M+H) = 450.3. 1H NMR (400
MHz, DMSO-dg) δ ppm 1.55 (d, J= 6.6 Hz, 6 H), 2.13 (s, 3 H), 2.22 (s, 3 H), 4.41 (d, J = 4.6 Hz, 2 H), 5.34 (quin, J= 6.7 Hz, 1 H), 5.91 (s, 1 H), 7.64 (d, J= 8.6 Hz, 2 H), 8.19 (s, 1 H), 8.31 (d, J= 8.6 Hz, 2 H), 8.39 (s, 1 H), 8.97 (t, J = 4.7 Hz, 1 H), 11.58 (s, 1 H).
Intermediate 69
Ethyl -dioxo-4-(2-thienyl)butanoate

The title compound was prepared in the same manner as for intermediate 66 using sodium metal (0.180 g, 7.85 mmol), l-(3-thienyl)ethanone (0.9 g, 7.13 mmol) and diethyl ethanedioate (0.967 mL, 7.13 mmol). The product was collected as 1.41 g (87%). LCMS E-S (M+H) = 226.9. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.30 (m, 3 H), 4.29 (q, J= 6.82 Hz, 2 H), 6.96 (br. s., 1 H), 7.60 - 7.81 (m, 2 H), 8.74 (br. s., 1 H).
Intermediate 70
Ethyl l-(l-methylethyl)-6-(3-thienyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 61 using ethyl 2,4-dioxo-4-(3-thienyl)butanoate (542 mg, 2.397 mmol), benzene (5 mL), l-(l-methylethyl)-lH-pyrazol-5 -amine (300 mg, 2.397 mmol), and acetic acid (5 ml). The crude product was purified using column chromatography (silica gel, eluent: 0 to 100% EtOAc/hexanes) to afford the product as 590 mg (78%). LCMS E-S (M+H) = 315.8. 1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.53 (t, J= 6.8 Hz, 3 H), 1.66 (d, J= 6.8 Hz, 6 H), 4.56 (q, J = 7.1 Hz, 2 H), 5.43 (spt, J= 6.7 Hz, 1 H), 7.47 (dd, J = 5.1, 3.0 Hz, 1 H), 7.84 - 7.95 (m, 1 H), 8.08 - 8.18 (m, 2 H), 8.32 - 8.43 (m, 1 H).
Intermediate 71
l-(l-Methylethyl)-6-(3-thienyl)-lH- razolo[3,4-6]pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate
58 using ethyl l-(l-methylethyl)-6-(3-thienyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (400 mg, 1.268 mmol), Ethanol (5 mL) THF (1 mL), and sodium hydroxide (2.114 mL, 6.34 mmol). The final product was collected as 370 mg (100%). LCMS E-S (M+H) = 287.9. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.55 (d, J= 6.8 Hz, 6 H), 5.34 (quin, J = 6.6 Hz, 1 H), 7.72 (dd, J= 5.0, 3.0 Hz, 1 H), 7.93 (dd, J= 5.0, 1.3 Hz, 1 H), 8.15 (s, 1 H), 8.34 (s, 1 H), 8.49 (dd, J= 3.0, 1.3 Hz, 1 H), 13.97 (s, 1 H).
Example 116
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(3- thienyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using l-(l-methylethyl)-6-(3-thienyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (70 mg, 0.244 mmol), DMSO(2 mL), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (68.9 mg, 0.365 mmol) HC1 salt, N-methylmorpholine (0.107 mL, 0.974 mmol), l-hydroxy-7- azabenzotriazole (33.2 mg, 0.244 mmol) and EDC (93 mg, 0.487 mmol). The final
product was collected as 75 mg (73%). LCMS E-S (M+H) = 422.2. 1H NMR (400 MHz,
DMSO-de) δ ppm 1.53 (d, J= 6.8 Hz, 6 H), 2.13 (s, 3 H), 2.23 (s, 3 H), 4.41 (d, J= 4.8
Hz, 2 H), 5.30 (quin, J= 6.6 Hz, 1 H), 5.91 (s, 1 H), 7.71 (dd, J= 5.0, 3.0 Hz, 1 H), 7.92
(d, J= 5.0 Hz, 1 H), 8.09 (s, 1 H), 8.34 (s, 1 H), 8.37 (d, J= 1.8 Hz, 1 H), 8.79 - 8.95 (m, 1 H), 11.58 (s, 1 H).
Intermediate 72
Ethyl 3-methyl-l-(l-methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 64 using ethyl 2,4-dioxo-4-(4-pyridinyl)butanoate (766 mg, 2.87 mmol), benzene (6 mL), and 3- methyl-l-(l-methylethyl)-lH-pyrazol-5 -amine (400 mg, 2.87 mmol). The crude product was purified using column chromatography (Silica gel ,eluent: 0 to 30%> MeOH/DCM) to give 95 mg (10%) of product. LCMS E-S (M+H) = 325.3. 1H NMR (400 MHz, CHLOROFORM-^) δ ppm
1.48 - 1.56 (m, 3 H), 1.61 - 1.73 (m, 6 H), 2.77 (s, 3 H), 4.56 (q, J =7.2 Hz, 2 H), 5.37 - 5.50 (m, 1 H), 8.02 - 8.16 (m, 3 H), 8.76 - 8.83 (m, 2 H).
Intermediate 73
3-Methyl-l-(l-methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic
acid

The title compound was prepared in the same manner as described for intermediate 58 using ethyl 3-methyl-l-(l-methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-b]pyridine-4- carboxylate (36 mg, 0.111 mmol), Ethanol (2 ml) and sodium hydroxide (0.185 mL, 0.555 mmol). The final product was collected as 26 mg 79%). LCMS E-S (M+H) = 297.2. 1H
NMR (400 MHz, DMSO-d6) δ ppm 1.54 (d, J= 6.4 Hz, 6 H), 2.65 (s, 3 H), 5.35 (m, 1 H), 8.17 (s, 1 H) 8.24 (m, 2 H), 8.78 (m, 2 H).
Example 117
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l-methylethyl)-6-(4- pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using 3 -methyl- 1 -( 1 -methylethyl)-6-(4-pyridinyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxylic acid (20 mg, 0.067 mmol), DMSO(l mL), 3-(aminomethyl)-4,6-dimethyl- 2(lH)-pyridinone (19.10 mg, 0.101 mmol), N-methylmorpholine (0.030 mL, 0.270 mmol), l-hydroxy-7-azabenzotriazole (18.37 mg, 0.135 mmol) and EDC (25.9 mg, 0.135 mmol).
The final product was collected as 29 mg (100%). LCMS E-S (M+H) = 431.2. 1H NMR
(400 MHz, DMSO-de) δ ppm 1.52 (d, J= 6.57 Hz, 6 H), 2.12 (s, 3 H), 2.25 (s, 3 H), 2.46
(s, 3H), 4.40 (d, J = 5.0 Hz, 2 H), 5.29 (quin, J= 6.7 Hz, 1 H), 5.89 (s, 1 H), 7.83 (s, 1 H), 8.19 (d, J = 6.1 Hz, 2 H), 8.64 - 8.89 (m, 3 H), 11.55 (br. s., 1 H).
Intermediate 74
Ethyl l,6-bis(l,l-dimethylethyl)-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 61 using ethyl 5,5-dimethyl-2,4-dioxohexanoate (392 mg, 1.958 mmol), benzene (10 mL), 1-(1,1- dimethylethyl)-3 -methyl- lH-pyrazol-5 -amine (300 mg, 1.958 mmol), and acetic acid (2 mL). The crude product was purified by column chromatography (Silica gel, eluent: 0 to 100% EtOAc /hexanes) to give 530 mg (85%) of product. LCMS E-S (M+H) = 318.3. 1H NMR (400 MHz, CHLOROFORM-;/) δ ppm 1.43 - 1.52 (m, 12 H), 1.84 (s, 9 H), 2.67 (s, 3 H), 4.50 (q, J= 7.07 Hz, 2 H), 7.59 (s, 1 H).
Intermediate 75
l,6-Bis(l,l-dimethylethyl)-3-meth l-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

To an EtOH solution (8 mL) of ethyl l,6-bis(l,l-dimethylethyl)-3-methyl-lH- pyrazolo[3,4-b]pyridine-4-carboxylate (528 mg, 1.663 mmol) was added sodium hydroxide (2.77 mL, 8.32 mmol), and the mixture was stirred for at room temperature for 2h. The volatiles were removed under reduced pressure and the aqueous phase was acidified using 1H HCl to ~pH 3. The precipitate was filtered, washed with water, and dried under high vacuum to give the product as 445 mg (92%). LCMS E-S (M+H) = 289.4. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.49 (s, 9 H), 1.76 (s, 9 H), 2.51 (s, 3 H), 7.51 (s, 1 H).
Example 118
1 ,6-Bis(l ,1 -dimethylethyl)- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihy dr o-3-pyridinyl)methyl] -3- methyl-lH- razolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using l,6-bis(l,l-dimethylethyl)-3-methyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (70 mg, 0.242 mmol), DMSO (2 mL), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (68.5 mg, 0.363 mmol), N-methylmorpholine (0.106 mL, 0.968 mmol), l-hydroxy-7-azabenzotriazole (65.9 mg, 0.484 mmol) and EDC (93 mg, 0.484 mmol). The final product was collected as 91 mg (87%). LCMS E-S (M+H) = 424.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 1H NMR (400 MHz, DMSO-d6) δ ppm 1.38 (s, 9 H), 1.74 (s, 9 H), 2.12 (s, 3 H), 2.23 (s, 3 H), 2.35 (s, 3 H), 4.35 (d, J= 4.80 Hz, 2 H), 5.88 (s, 1 H), 7.15 (s, 1 H), 8.61 (t, J= 4.93 Hz, 1 H), 11.52 (s, 1 H).
Intermediate 76
Ethyl 4- [4-(methylthio)phenyl] -2,4-dioxobutanoate

The title compound was prepared in the same manner as for intermediate 66 using sodium metal (0.160 g, 6.95 mmol), l-[4-(methylthio)phenyl]ethanone (1.05 g, 6.32 mmol) and diethyl ethanedioate (0.856 mL, 6.32 mmol). The product was collected as 1.58 g (59%). LCMS E-S (M+H) = 266.9. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.29 - 1.50 (m, 3 H), 2.41 - 2.62 (m, 3 H), 4.23 - 4.49 (m, 2 H), 7.15 - 7.38 (m, 2 H), 7.89 (dd, J= 8.72, 2.15 Hz, 2 H).
Intermediate 77
Ethyl l-(l-methylethyl)-6- [4-(methylthio)phenyl] -IH-pyrazolo [3,4-6] pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 61 using ethyl 4-[4-(methylthio)phenyl]-2,4-dioxobutanoate (851 mg, 3.20 mmol),
benzene (6 mL), l-(l-methylethyl)-lH-pyrazol-5 -amine (400 mg, 3.20 mmol), and acetic acid (3 mL). The crude product was purified by column chromatography (Silica gel,
eluent; 0 to 100% EtOAc/hexanes) to give 870 mg (63%) of product. LCMS E-S (M+H) = 433.2. 1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.47 - 1.57 (m, 3 H), 1.66 (d, J
= 6.8 Hz, 6 H), 2.58 (s, 3 H), 4.56 (q, J= 7.2 Hz, 2 H), 5.38 - 5.55 (m, 1 H), 7.36 - 7.46
(m, 2 H), 8.12 - 8.19 (m, 2 H), 8.22 (s, 1 H), 8.40 (s, 1 H).
Intermediate 78
Eth l l-(l-methylethyl)-6-[4-(methylsulfonyl)phenyl]-lH-pyrazolo[3,4-6]pyridine-4-
Oxone (1712 mg, 2.79 mmol) was added to a solution of ethyl l-(l-methylethyl)- 6-[4-(methylthio)phenyl]-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (330mg, 0.928 mmol) in acetone (6 mL) and water (2 mL), and the reaction mixture was stirred for 6 h. The
reaction mixture was quenched with water (10 mL) and then neutralized with NaHC03 solution. The mixture was extracted with EtOAc (3x) and the combined extracts dried over Na2S04, filtered and concentrated in vacuo. The residue was purified using column
chromatography (Silica gel, 0 to 100%) EtOAc/hexanes) to give 295 mg (82%) of product.
LCMS E-S (M+H) = 388.1. 1H NMR (400 MHz, CHLOROFORM-^/) δ ppm 1.54 (t, J =
7.2 Hz, 3 H), 1.67 - 1.69 (d, J= 6.8 Hz, 6 H), 3.14 (s, 3 H), 4.58 (q, J= 7.2 Hz, 2 H), 5.48 (spt, J= 6.7 Hz, 1 H), 8.06 - 8.18 (m, 2 H), 8.29 (s, 1 H), 8.39 - 8.44 (m, 2 H), 8.47 (s, 1 H).
Intermediate 79
l-(l-Methylethyl)-6-[4-(methylsulfonyl)phenyl]-lH-pyrazolo [3,4-6] pyridine-4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 58 using ethyl 1 -( 1 -methylethyl)-6- [4-(methylsulfonyl)phenyl] - 1 H-pyrazolo [3 ,4- b]pyridine-4-carboxylate (260 mg, 0.671 mmol), Ethanol (4 mL), THF (1 mL) and sodium hydroxide (2N, 1.118 mL, 3.36 mmol). The final product was collected as 231 mg (96%). LCMS E-S (M+H) = 360.1. 1H NMR (400 MHz, DMSO-</6) δ ppm 1.58 (d, J= 6.8 Hz , 6 H), 3.30 (s, 3H), 5.34 - 5.49 (m, 1 H), 8.11 (d, J= 8.6 Hz, 2 H), 8.30 (s, 1 H), 8.44 (s, 1 H), 8.51 (d, J= 8.6 Hz, 2 H).
Example 119
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[4- (methylsulfonyl)phenyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using 1 -( 1 -methylethyl)-6-[4-(methylsulfonyl)phenyl]- 1 H-pyrazolo[3 ,4-b]pyridine-4- carboxylic acid (90 mg, 0.250 mmol), DMSO(2 mL), 3-(aminomethyl)-4,6-dimethyl-
2(lH)-pyridinone (70.9 mg, 0.376 mmol), N-methylmorpholine (0.110 mL, 1.002 mmol), l-hydroxy-7-azabenzotriazole (68.2 mg, 0.501 mmol) and EDC (96 mg, 0.501 mmol).
The final product was collected as 106 mg (85%). LCMS E-S (M+H) = 494.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.56 (d, J= 5.6 Hz, 6 H), 2.13 (br. s., 3 H), 2.23 (br. s., 3
H), 4.42 (m, 2 H), 5.36 (br. s., 1 H), 5.91 (br. s., 1 H), 8.11 (d, J= 7.3 Hz, 2 H), 8.27 (br.
s., 1 H), 8.39 - 8.64 (m, 3 H), 9.01 (br. s., 1 H), 11.58 (br. s., 1 H).
Intermediate 80
Ethyl 4-(4-bromophenyl)-2,4-dioxobutanoate

The title compound was prepared in the same manner as for intermediate 66 using sodium metal (0.404 g, 17.58 mmol), l-(4-bromophenyl)ethanone (3.5 g, 17.58 mmol), and diethyl ethanedioate (2.384 mL, 17.58 mmol). The product was collected as 5.1 g (97%). LCMS E-S (M+H) = 299.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.22 - 1.37 (m, 3 H), 4.25 (d, J= 6.57 Hz, 2 H), 7.72 (m, 2 H), 7.92 (m, 2 H).
Intermediate 81
Ethyl 6-(4-bromophenyl)-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 61 using ethyl 4-(4-bromophenyl)-2,4-dioxobutanoate (956 mg, 3.20 mmol), benzene (10 mL), l-(l-methylethyl)-lH-pyrazol-5 -amine (400 mg, 3.20 mmol), and acetic acid (4 mL). The crude product was purified using column chromatography (Silica gel, eluent: 0 to
100% EtOAc/hexanes) to give 1.1 g (89%) of product. LCMS E-S (M+H) = 388.1. 1H
NMR (400 MHz, CHLOROFORM-^) δ ppm 1.49 - 1.57 (t, J= 67.2 Hz , 3 H), 1.63 - 1.74 (d, J= 6.8 Hz 6 H), 4.56 (q, J=7.2 Hz, 2 H), 5.46 (spt, J=6.7 Hz, 1 H), 7.65 - 7.71 (m, 2
H), 8.04 - 8.15 (m, 2 H), 8.21 (s, 1 H), 8.42 (s, 1 H).
Intermediate 82
l-(l-Methylethyl)-6- [4-(lH-pyrazol-4-yl)phenyl] -IH-pyrazolo [3,4-6] pyridine-4-carboxylic acid

To a 5-mL microwave vial were added ethyl 6-(4-bromophenyl)-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (80 mg, 0.206 mmol), 4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (56.0 mg, 0.288 mmol), 1,4-dioxane (2 mL) and sodium carbonate (0.206 mL, 0.412 mmol), and the mixture was degassed with nitrogen for 10 min. Next added PdP(Ph3)4 (19.05 mg, 0.016 mmol) and the vial was sealed. The reaction mixture was irradiated (microwave) at 120 °C overnight. The
reaction mixture was filtered and concentrated in vacuo. The residue was purified using reverse-phase HPLC to give 25 mg (35%) of product. LCMS E-S (M+H) = 348.1. 1H
NMR (400 MHz, DMSO-d6) δ ppm 1.58 (d, J= 6.8 Hz, 6 H), 5.38 (quin, J= 6.6 Hz, 1 H), 7.81 (d, J= 8.3 Hz, 2 H), 8.14 - 8.30 (m, 4H), 8.36 (s, 1 H).
Example 120
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[4-(lH- pyrazol-4-yl)phenyl] - IH-pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example using 1 -( 1 -methylethyl)-6- [4-( 1 H-pyrazol-4-yl)phenyl]- 1 H-pyrazolo [3 ,4-b]pyridine-4 carboxylic acid (20 mg, 0.058 mmol), DMSO(lmL), 3-(aminomethyl)-4,6-dimethyl-
2(lH)-pyridinone (16.29 mg, 0.086 mmol), N-methylmorpholine (0.025 mL, 0.230 mmol), l-hydroxy-7-azabenzotriazole (15.67 mg, 0.115 mmol) and EDC (22.07 mg, 0.115 mmol). The final product was collected as 12 mg (43%). LCMS E-S (M+H) = 482.0. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.56 (d, J= 6.4 Hz, 6 H), 2.14 (s, 3 H), 2.24 (s, 3 H), 4.42 (d, J= 4.8 Hz, 2 H) 5.27 - 5.47 (m, 1 H), 5.91 (s, 1 H), 7.79 (d, J= 8.3 Hz, 2 H), 8.04 (br. s., 1 H), 8.15 - 8.21 (m, 1 H), 8.27 (d, J= 8.3 Hz, 2 H), 8.35 - 8.42 (m, 1 H), 8.99 (t, J =
4.8 Hz, 1 H), 11.58 (br. s., 3 H).
Intermediate 83
Ethyl l-(l,l-dimethylethyl)-3-methyl-6-[4-(methyloxy)phenyl]-lH-pyrazolo[3,4- b] pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 61 using ethyl 4-[4-(methyloxy)phenyl]-2,4-dioxobutanoate (327 mg, 1.305 mmol), benzene (8 mL), l-(l,l-dimethylethyl)-3 -methyl- lH-pyrazol-5 -amine (200 mg, 1.305 mmol), and acetic acid (2 mL). The crude product was purified using column
chromatography (Silica gel, eluent: 0 to 100% EtOAc/hexanes) to give 0.41 g (85%>) of product. LCMS E-S (M+H) = 368.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.81 (s, 9 H), 2.55 (s, 3 H), 7.11 (d, J= 8.84 Hz, 2 H), 7.89 (s, 1 H), 8.16 (d, J = 8.84 Hz, 2 H).
Intermediate 84
l-(l,l-Dimethylethyl)-3-methyl-6-[4-(methyloxy)phenyl]-lH-pyrazolo[3,4-6]pyridine-
4-carboxylic acid

The title compound was prepared in the same manner as described for intermediate 75 using ethyl 1 -( 1 , 1 -dimethylethyl)-3 -methyl-6- [4-(methyloxy)phenyl] - 1 H-pyrazolo [3,4- b]pyridine-4-carboxylate (400 mg, 1.089 mmol), 3N sodium hydroxide (1.814 mL, 5.44 mmol)
and EtOH (6 mL). The product was collected as 347 mg (94%). LCMS E-S (M+H) = 340.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.81 (s, 9 H), 2.55 (s, 3 H), 7.1 1 (d, J= 8.84 Hz, 2 H), 7.89 (s, 1 H), 8.16 (d, J=8.84 Hz, 2 H). Example 121
l-(l,l-Dimethylethyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3- methyl-6- [4-(methyloxy)phenyl] - IH-pyrazolo [3,4-6] pyridine-4-carboxamide

To a solution of l-( 1 ,1 -dimethylethyl)-3 -methyl-6- [4-(methyloxy)phenyl]-lH- pyrazolo[3,4-b]pyridine-4-carboxylic acid (70 mg, 0.206 mmol) in DMSO (1 mL) were added 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (19.10 mg, 0.101 mmol), N- methylmorpholine (0.030 mL, 0.270 mmol), l-hydroxy-7-azabenzotriazole (18.37 mg, 0.135 mmol) and EDC (25.9 mg, 0.135 mmol), and the mixture was stirred overnight at room temperature. The reaction mixture was quenched with water (10 mL), stirred for 10 min.,and filtered. The contents were dried under high vacuum and collected as 71 mg
(71%). LCMS E-S (M+H) = 474.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 9 H), 2.12 (s, 3 H), 2.25 (s, 3 H), 2.39 (s, 3 H), 3.33 (s, 1 H), 4.38 (d, J = 4.80 Hz, 2 H), 5.88 (s, 1 H), 7.10 (d, J = 8.84 Hz, 2 H), 7.61 (s, 1 H), 8.16 (d, J = 8.84 Hz, 2 H), 8.70 (s, 1 H), 1 1.53 (s, 1 H).
Intermediate 85
Ethyl 1-(1 , l-dimethylethyl)-3-methyl-6- { [(trifluor omethyl)sulfonyl] oxy}- IH- pyrazolo [3,4-6] pyridine-4-carboxylate

Intermediate 86
Ethyl l-(l,l-dimethylethyl)-3-methyl-6-(3-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4- carbox late

Ethyl 1 -( 1 , 1 -dimethylethyl)-3 -methyl-6- { [(trifluoromethyl)sulfonyl]oxy } - 1 H- pyrazolo[3,4-b]pyridine-4-carboxylate (100 mg, 0.244 mmol), 3-pyridinylboronic acid (39.0 mg, 0.318 mmol), aq. saturated NaHC03 (1 mL) and 1,4-dioxane (3 mL) were degassed with nitrogen (10 min) followed by addition of PdCl2(dppf)-CH2Cl2 adduct (9.97 mg, 0.012 mmol). The sealed mixture was irradiated (microwave) at 120 °C for 40 min. The reaction mixture was diluted with EtOAc and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel chromatography (0 to 100% EtOAc/hexanes). The final product was collected as a solid, 72 mg (87%). LCMS E-S (M+H) = 339.3. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.46 - 1.57 (m, 3 H), 1.90 (s, 9 H), 2.72 (s, 3 H), 4.45 - 4.64 (m, 2 H), 7.47 (dd, J=8.08, 4.80 Hz, 1 H), 8.03 (s, 1 H), 8.46 (dt, J= 8.08, 2.02 Hz, 1 H), 8.72 (dd, J=4.80, 1.52 Hz, 1 H), 9.43 (d, J=1.52 Hz, 1 H).
Intermediate 87
l-(l,l-Dimethylethyl)-3-methyl-6-(3-pyridinyl)-lH-pyrazolo[3,4-6]pyridine-4- arboxylic acid

Ethyl 1 -( 1 , 1 -dimethylethy l)-3 -methyl-6-(3 -pyridinyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxylate (68 mg, 0.201 mmol) was suspended in ethanol (2 mL) followed by addition of sodium hydroxide (0.335 mL, 1.005 mmol). The mixture was stirred at room temperature for 2. 5h. The mixture was concentrated and the residue was diluted with water (1 mL) and acidified to pH 3 using IN HC1. The precipitate was collected by filtration and further dried under high vacuum to give the product as an HC1 salt, 60 mg (86%). LCMS E-S (M+H) = 311.5. 1H NMR (400 MHz, DMSO-de) δ ppm 1.82 (s, 9 H), 2.60 (s, 3 H), 7.60 (dd, J= 7.96, 4.67 Hz, 1 H), 8.08 (s, 1 H), 8.57 (dt, J = 8.34, 1.89 Hz, 1 H), 8.70 (dd, J= 4.80, 1.52 Hz, 1 H), 9.39 (d, J = 1.52 Hz, 1 H), 14.03 (br. s., 1 H).
Example 122
l-(l,l-Dimethylethyl)- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-6-
(3-pyridinyl)-lH- razolo[3,4-6]pyridine-4-carboxamide.

The title compound was prepared in the same manner as described in example 121 using l-(l,l-dimethylethyl)-3-methyl-6-[3-pyridinyl]-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (58 mg, 0.167 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (47 mg, 0.251 mmol), N- methylmorpholine (0.092 mL, 0.836 mmol), l-hydroxy-7-azabenzotriazole (46 mg, 0.334 mmol), EDC (64 mg, 0.334 mmol), and DMSO (1 mL). The product was collected as 73 mg (96%). LCMS E-S (M+H) = 445.1. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.81 (s, 9 H), 2.12 (s, 3 H),
2.25 (s, 3 H), 2.42 (s, 3 H), 4.39 (d, J= 5.05 Hz, 2 H), 5.89 (s, 1 H), 7.59 (dd, J= 8.08, 4.80 Hz, 1 H), 7.78 (s, 1 H), 8.55 (dt, J= 8.15, 1.86 Hz, 1 H), 8.68 (dd, J= 4.80, 1.52 Hz, 1 H), 8.74 (t, J = 5.05 Hz, 1 H) 9.38 (d, J= 1.77 Hz, 1 H), 11.54 (s,l H).
Example 123
3-Bromo- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)- iyl-l -pyrazolo[3,4-6]pyridine-4-carboxamide

3-Bromo- 1 -(1 -methylethyl)-6-phenyl- lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (93 mg, 0.258 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone
hydrochloride (64 mg, 0.336 mmol), HOBT (60 mg, 0.387 mmol) and EDC (74 mg, 0.387 mmol) were suspended in DMSO (14 mL) and stirred at room temperature for 10 minutes, after which time DIEA (0.9 ml, 5.16 mmol) was added. After stirring for 2 h, 4- methylmorpholine (1 ml, 9.04 mmol) was added. The reaction mixture was stirred first at room temperature for 21h, and then at 80 °C (aluminum heating block) for 3 lh. The
reaction mixture was cooled to room temperature and then added dropwise to a cold,
slightly basic solution (pH ~ 8-10) of water (100 mL) and IN Na2C03 (8 mL). After
stirring for 20 min., the contents were extracted with EtOAc (2 x 100 mL). The combined organic layers are washed with brine, dried over Na2S04, filtered, and concentrated to a residue. The crude residue was dissolved in 10%MeOH/DCM and purified by silica gel chromatography (eluent: 10-95% gradient EtOAc/Hexanes and then 10%
(5%NH4OH/MeOH)/DCM and EtOAc, 10-90% gradient). The product was collected as an off-white solid. (35 mg, 27%). LCMS E-S (M+H) = 494.1 / 496.0. 1H NMR (400 MHz, CHLOROFORM-d) δ 11.58 (br. s., 1H), 8.11 (dd, J= 1.89, 7.71 Hz, 2H), 7.68 (br. s., 1H), 7.60 (br. s., 1H), 7.44 - 7.53 (m, 3H), 5.92 (s, 1H), 5.42 (quin, J= 6.69 Hz, 1H), 4.63 (br.
s., 1H), 2.41 (br. s., 3H), 2.11 (br. s., 3H), 1.62 (d, J= 6.57 Hz, 6H).
Intermediate 88
3-Bromo-l-(l-methylethyl)- -phenyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

Ethyl 3 -bromo- 1 -( 1 -methylethyl)-6-phenyl- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxylate (112 mg, 0.288 mmol) was suspended in THF (0.5 mL) and ethanol (1.5 mL), followed by addition of 3N NaOH (150 μΐ, 0.433 mmol). The reaction mixture was stirred at 55 °C for 3 hours, and then allowed to cool to room temperature. The reaction mixture was diluted with water (1.5 mL), cooled in an ice bath, and acidified with IN HC1 in a dropwise manner. The mixture was extracted with EtOAc. The organic layer was separated and concentrated in vacuo to afford a solid that dried on the high vacuum 3 hours. The product was collected as a pale yellow solid (93.6 mg, 90%). LCMS E-S (M+H) = 360.0 / 362.2. 1H NMR (400 MHz, MeOD) 58.22 (dd, J= 1.52, 8.08 Hz, 2H), 8.05 (s, 1H), 7.48 - 7.59 (m, 3H), 5.47 (quin, J= 6.76 Hz, 1H), 1.63 (d, J= 6.82 Hz, 6H)
Intermediate 89
Ethyl 3-bromo-l-(l-methyleth l)-6-phenyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

Ethyl 1 -( 1 -methylethyl)-6-phenyl- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylate (120 mg, 0.388 mmol), was suspended in acetic acid (2 mL) followed by addition of bromine (26 μΐ, 0.504 mmol). The reaction mixture was stirred with heating at 80 °C. After lh, a second portion of bromine was added (26 μΐ, 0.504 mmol) and the reaction mixture heated at 80 °C for an additional 2h. After cooling to room temperature, the solution was added to a saturated aqueous solution of sodium bicarbonate (6 mL) and extracted with
dichloromethane (2 x 10 mL) The combined organic layers were concentrated in vacuo. The crude product was purified by silica gel chromatography (eluent: EtOAc/Hexanes, 0- 50% gradient). The product was collected as a white powder (112 mg, 74%>). LCMS E-S
(M+H) = 388.0 / 390.2. 1H NMR (400 MHz, CHLOROFORM-d) δ 8.18 (dd, J= 1.52,
8.08 Hz, 2H), 8.00 (s, 1H), 7.48 - 7.58 (m, 3H), 5.47 (quin, J= 6.76 Hz, 1H), 4.58 (q, J = 7.16 Hz, 2H), 1.65 (d, J= 6.82 Hz, 6H), 1.52 (t, J= 7.20 Hz, 3H).
Intermediate 90
Ethyl l-(l-methylethyl)-6- henyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

The title compound was prepared in the same manner as described for intermediate 61 using ethyl 2,4-dioxo-4-phenylbutanoate (5 g, 22.7 mmol) , l-(l-methylethyl)-lH- pyrazol-5 -amine (2.84 g, 22.7 mmol) , benzene (70 mL), and acetic Acid (44 mL) The crude product was purified by silica gel chromatography (eluent: 0 to 100%
EtOAc/hexanes) to give 6.72 g (95%) as a pale yellow solid. LCMS E-S (M+H) = 310.5. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H), 8.17 - 8.28 (m, 3H), 7.49 - 7.64 (m, 3H), 5.38 (quin, J= 6.69 Hz, 1H), 4.49 (q, J= 7.07 Hz, 2H), 1.57 (d, J= 6.57 Hz, 6H), 1.44 (t, 3H).
Example 124
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2- nitrophenyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

l-(l-Methylethyl)-3-nitro-6-phenyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (24 mg, 0.074 mmol), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone hydrochloride (18 mg, 0.096 mmol), HO AT (17 mg, 0.110 mmol) and EDC (21 mg, 0.110 mmol) were suspended in DMSO (800 μί) and the reaction mixture stirred at room temperature. Next added 4-methylmorpholine (41 μΐ, 0.368 mmol), and the reaction was stirred at room temperature for 16 h. The reaction mixture was added dropwise to cold, slightly basic
solution of water (3 mL) and IN Na2CC"3 (0.5 mL). After stirring for 20 min., the precipitated solids were collected via vacuum filtration and washed with water. The solid was dried on the high vacuum (72 h). The product, was collected as an off- white powder (28 mg, 82%). LCMS E-S (M+H) = 461.1. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (br. s., 1H), 8.42 (s, 1H), 8.02 (ddd, J= 1.14, 4.99, 7.77 Hz, 2H), 7.99 (s, 1H), 7.86 (td, J= 1.26, 7.58 Hz, 1H), 7.72 - 7.77 (m, 1H), 5.90 (s, 1H), 5.04 (quin, J= 6.69 Hz, 1H), 4.41 (d, J = 4.55 Hz, 1H), 3.34 (s, 2H), 2.22 (s, 3H), 2.12 (s, 3H), 1.49 (d, J= 6.57 Hz, 6H).
Intermediate 91
l-(l-Methylethyl)-6-(2-nitrophenyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

Ethyl 1 -( 1 -methylethyl)-3 -nitro-6-phenyl- lH-pyrazolo [3 ,4-¾]pyridine-4- carboxylate was suspended in THF (0.5 ml) and ethanol (1 mL), followed by addition of 3N NaOH aq (50 μί). The reaction mixture was heated at 55 °C for lh, and then allowed to cool to room temperature. The volatiles were removed in vacuo and the residue was dissolved in water (1.5 mL). The reaction mixture was cooled in an ice bath and acidified by dropwise addition of IN HC1. After stirring for 20 minutes, the precipitated solids were collected by vacuum filtration dried under high vacuum overnight for 16 h. The product was collected as a white solid (24.5 mg, 76%). LCMS E-S (M+H) = 327.2. 1H NMR (400 MHz, DMSO-d6) δ 14.14 (br. s., 1H), 8.43 (s, 1H), 8.04 (dd, J= 1.14, 7.96 Hz, 1H), 7.98 - 8.02 (m, J= 1.26 Hz, 2H), 7.86 (td, J= 1.26, 7.58 Hz, 1H), 7.72 - 7.80 (m, 1H), 5.09 (qd, J= 6.57, 6.74 Hz, 1H), 1.51 (d, J= 6.57 Hz, 6H).
Intermediate 92
Ethyl l-(l-methylethyl)-6-(2-nitrophen l)-lH-pyrazolo[3,4-6]pyridine-4-carboxylate

stirring was diluted with water (3 mL) and then slowly with saturated Na2CC"3 (1 mL). The reaction mixture was extracted with EtOAc (2 x 6 mL). The combined organic layers were concentrated in vacuo and the crude product purified by silica gel chromatography (eluent: EtOAc/Hexanes, 0-70% gradient). The product, was collected as a white solid (35 mg,
26%). LCMS E-S (M+H) = 355.2. 1H NMR (400 MHz, CHLOROFORM-d) 8.46 (s, 1H), 7.99 (s, 1H), 7.94 (dd, J= 1.14, 7.96 Hz, 1H), 7.76 - 7.81 (m, 1H), 7.73 (td, J= 1.26, 7.58 Hz, 1H), 7.59 - 7.66 (m, 1H), 5.25 (ddd, J= 6.57, 6.69, 13.52 Hz, 1H), 4.51 - 4.59
(m, 2H), 1.61 (d, J= 6.82 Hz, 6H), 1.52 (t, J= 7.07 Hz, 3H).
Example 125
V-[(4,6-Dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-propyl-lH- pyrazolo [3,4-6] pyridine-4-carboxamide

The title compound was prepared in the same manner as described in example 109 using l-(l-methylethyl)-6-propyl-l H-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (70 mg, 0.283 mmol), DMSO(3 mL), 3-(aminomethyl)-4,6-dimethyl-2(lH)-pyridinone (80 mg, 0.425 mmol), N-methylmorpholine (0.124 mL, 1.132 mmol), l-hydroxy-7- azabenzotriazole (77 mg, 0.566 mmol), and EDC (109 mg, 0.566 mmol). The collected solid was washed with water and methanol and dried under high vacuum to give 73 mg
(68%) of product. LCMS E-S (M+H) = 382.3 1H NMR (400 MHz, DMSO-d6) δ ppm
0.82 - 1.10 (m, 3 H), 1.48 (d, J= 6.8 Hz, 6 H), 1.77 (m, 2 H), 2.13 (s, 3 H), 2.21 (s, 3 H), 2.85 (t, J= 7.6 Hz, 2 H), 4.36 (d, J= 4.8 Hz, 2 H), 5.20 (m, 1 H), 5.90 (s, 1 H), 7.38 - 7.58 (m, 1 H), 8.26 (s, 1 H), 8.73 (t, J= 4.7 Hz, 1 H), 11.57 (br. s., 1 H).
Intermediate 93
3-(Aminomethyl)-6-ethyl-4-methyl-2(lH)-pyridinone

Step 1
2,4-Hexanedione (2.283 g, 20 mmol) and cyanacetamide (1.682 g, 20.00 mmol) were added to ethanol (20 mL). The contents were initially heterogenous, but gradually dissolved upon reaching ca. 70 °C. Next added piperidine (1.976 mL, 20.00 mmol) and stirred with warming to reflux. After 30 min, the heterogenous contents were removed from heating and allowed to stir with cooling to room temperature. The contents were filtered in vacuo to afford a light yellowish colored solid and yellow filtrate. The collected solid filter cake was copiously washed with water which removed the yellow color. The final product was collected as fine white crystalline solid (1.66 g after drying). LCMS E-S (M+H) = 163.2. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.34 (t, J=7.58 Hz, 3H) 2.47 (s, 3H) 2.71 (q, J=7.58 Hz, 2H), 6.12 (s, 1 H).
Step 2
To a stirred solution of 6-ethyl-4-methyl-2-oxo-l,2-dihydro-3-pyridinecarbonitrile (1.462 g, 9.01 mmol) in Methanol (50 mL), cooled to 0 °C were added di- tertbutoxycarbonyl anhydride (4.19 mL, 18.03 mmol) and nickel(II) chloride•hexahydrate (0.214 g, 0.901 mmol). Sodium borohydride (2.387 g, 63.1 mmol) was added portionwise over 30 min, then the mixture was allowed to warm to room termperature and stirred overnight. Additional sodium borohydride (2.387 g, 63.1 mmol) was added and the reaction stirred overnight again. Diethylenetriamine (0.979 mL, 9.01 mmol) was added, and the reaction mixture was stirred for 30 min. at room temperature. The solvent was evaporated, and the residue was dissolved in ethyl acetate (20 mL) and extracted with sodium bicarbonate (2 x 10 mL). The organics were dried over sodium sulfate, and concentrated. The residue was dissolved in chloroform (25 mL) and TFA (3.47 mL, 45.1 mmol) was added. The reaction was heated to 50 °C for 3 h, then cooled to room temperature and concentrated. The reaction mixture was concentrated in vacuo and the crude product was purified by reverse phase HPLC (eluent; 0-30% gradient ACN in H20, 0.1% TFA) to afford the title compound (TFA salt, 1.37 g, 54.1% yield) as a white solid.
LC-MS(ES) m/z = 167 [M+H]+. 1H NMR (400 MHz, DMSO-d) δ ppm 1.12 (t, J=7.58 Hz, 3H), 2.20 (s, 3H), 2.44 (q, J=7.58 Hz, 2H), 3.78 - 3.83 (m, 2H), 6.01 (s, 1H), 7.75 - 7.9 (br s, 3H), 11.86 (s, 1H).
Intermediate 94
3-(Aminomethyl)-5-fluoro-4,6-dimethyl-2(lH)-pyridinone

Step 1
2,4-Pentanedione (12 g, 120 mmol) was suspended in deuterated MeCN (39 mL), to which was added Selectfluor (44.7 g, 120 mmol) in one portion. The resulting paste was then heated in an oil bath to 50 °C. After 5 min, an exotherm was observed, and the mixture was removed from the oil bath. The mixture was then heated at 50 °C for 1 h. The mixture was then aged at room temperature for 16 h, and then distilled under vacuum. The desired product distilled at 40-60 °C at 10-15 torr and was collected as a light yellowish liquid (1.8 mL, 1.95 g). A less pure fraction (ca. 80%) distilled at 25-35 °C at 15-20 torr, and was collected as a light yellowish liquid (3.33 g). 1H NMR (400 MHz, ACETONITRILE- 3) δ ppm 2.24 (s, 3 H), 2.26 (s, 3 H), 5.46 (d, J=48.0 Hz, 1 H).
Step 2
A mixture of 3-fluoro-2,4-pentanedione (247 mg, 2.10 mmol), cyanoacetamide (176 mg, 2.1 mmol, 1 equiv) and piperidine (0.21 mL, 2.1 mmol, 1 equiv) in 3 mL of EtOH was heated at 85 °C for 16 h, resulting in a clear but dark brown solution. The mixture was concentrated in vacuo. The residue was taken up in 2 mL of IN HCl to give a suspension, which was filtered. The tan solids collected were dried under vacuum to give the desired product as 80 mg. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.33 (s, 3H), 2.42 (s, 3H).
Step 3
5-Fluoro-4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinecarbonitrile (1.0 g, 6.02 mmol) was suspended in 50 mL of HO Ac at room temperature to give a dark brownish solution. NaOAc (1.0 g, 12.3 mmol, 2 equiv), Pt02 (20 mg) and 5% Pd/C (1.3 g) were
charged into a 500 mL Parr bottle, followed by wetting with some acetic acid under nitrogen. The substrate solution was then added, and rinsed with another 50 mL of acetic acid. The mixture was hydrogenated under a pressure of 40 psi. There was initial drop of hydrogen pressure and the vessel was refilled. The mixture was hydroganted for 6 h. The mixture was filtered through Celite, and concentrated in vacuo to give a solid residue, which was suspended in 6 mL of cone. HC1 to give a paste. The paste was filtered, and the cake was washed with 1 mL of cone HC1 (in duplicate). The filtrate was concentrated in vacuo. The solid residue dissolved in 0.4 mL of cone. HC1 and 6 mL of EtOH as a suspension, which was stored in the freezer for 2h, followed by filtration. The cake was washed with 2 mL of cold EtOH (in duplicate). The solids were dried under vacuum at room temperature for 18h to give the title compound as a cream-colored solid (1.03 g).
LC-MS (ES) m/z = 171 [M+H]+ and a dominant peak at 154. 1H NMR (400 MHz,
DMSO-de) δ ppm , 2.20 (d, J=3.0 Hz, 3 H), 2.22 (d, J=2.0 Hz, 3 H), 3.83 (q, J=5.2 Hz, 2 H), 7.99 (br. s., 3 H), 11.99 (br. s., 1 H).
Example 126
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- (pyridin-3-yl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide

In a 25 mL sealable tube under nitrogen were combined 6-chloro-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] -3 -methyl- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine- 4-carboxamide (90 mg, 0.23 mmol) and 3-pyridinylboronic acid (42.8 mg, 0.35 mmol) in dioxane/water (3: 1 mL). PdCi2(dppf)-CH2Ci2 adduct (9.47 mg, 0.012 mmol) was added and the resulting mixture was degassed with nitrogen for 10 min. Sodium carbonate (58.5 mg, 0.7 mmol) was added, the vessel was sealed, and the insoluble mixture was heated in a microwave to 110 °C for 20 min. Upon cooling, water was added and solids that precipitated were filtered, dissolved in DCM and purified by Si02 chromatography (eluent:
gradient 0 to 80:20:2 DCM/MeOH/NH4OH). The product containing fractions were evaporated and suspended in EtOH/EtOAc (1 : 1). The contents were sonicated, solids that precipitated were filtered, washed with hexanes and dried to afford the title compound as a white solid (78 mg, 77%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1 1.53 (br. s., 1 H) 9.39 (d, J=1.52 Hz, 1 H) 8.76 (t, J=5.05 Hz, 1 H) 8.69 (dd, J=4.67, 1.64 Hz, 1 H) 8.57 (dt, J=8.27, 1.80 Hz, 1 H) 7.78 (s, 1 H) 7.57 (dd, J=8.08, 5.56 Hz, 1 H) 5.89 (s, 1 H) 5.28 (quin, J=6.69 Hz, 1 H) 4.41 (s, 1 H) 4.39 (s, 1 H) 2.46 (s, 3 H) 2.25 (s, 3 H) 2.12 (s, 3 H) 1.52 (s, 3 H) 1.51 (s, 3 H); LC-MS (ES) m/z = 431.1 [M+H]+. Examples 127-129 were prepared from 6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl] -3 -methyl- 1 -( 1 -methy lethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide and the appropariate boronic acid reagent using the conditions described in the above example. The heating temperatures were between 100 -110 °C, and product triturations were performed using either EtOH/EtOAc or EtOAc/hexanes.
Example 127
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- phenyl-lH-pyrazolo [3,4-b] pyridine-4-carboxamide

1H NMR (400 MHz, DMSO-d6) δ ppm 11.53 (s, 1 H) 8.76 (t, J=4.93 Hz, 1 H) 8.22 (d, J=1.52 Hz, 1 H) 8.20 (s, 1 H) 7.69 (s, 1 H) 7.47 - 7.57 (m, 3 H) 5.89 (s, 1 H) 5.26 (quin, J=6.69 Hz, 1 H) 4.40 (s, 1 H) 4.39 (s, 1 H) 2.45 (s, 3 H) 2.25 (s, 3 H) 2.12 (s, 3 H) 1.52 (s, 3 H) 1.50 (s, 3H). LC-MS (ES) m/z = 429.9[M+H]+.
Example 128
N-((4,6-dimethyl-2-oxo- 1 ,2-dihydr opyridin-3-yl)methyl)-6-(4-
((dimethylamino)methyl)phenyl)-l-isopropyl-3-methyl-lH-pyrazolo[3,4-b]pyridine-
4-carboxamide

1H NMR (400 MHz, DMSO-d6) δ ppm 11.52 (s, 1 H) 8.74 (t, J=5.05 Hz, 1 H) 8.17 (s, 1 H) 8.15 (s, 1 H) 7.67 (s, 1 H) 7.46 (s, 1 H) 7.44 (s, 1 H) 5.89 (s, 1 H) 5.26 (quin, J=6.63 Hz, 1 H) 4.40 (br. s., 1 H) 4.39 (br. s., 1 H) 3.46 (s, 2 H) 2.45 (s, 3 H) 2.25 (s, 3 H) 2.18 (s, 6 H) 2.08 - 2.14 (m, 3 H) 1.51 (s, 3 H) 1.50 (s, 3 H). LC-MS (ES) m/z = 487.1 [M+H]+.
Example 129
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-6-(4-fluorophenyl)-l- isopropyl-3-methyl-lH-pyraz -b] pyridine-4-carboxamide

1H NMR (400 MHz, DMSO-d6) δ ppm 11.53 (s, 1 H) 8.74 (t, J=4.93 Hz, 1 H) 8.25 - 8.30 (m, 2 H) 7.69 (s, 1 H) 7.34 - 7.40 (m, 2 H) 5.89 (s, 1 H) 5.25 (quin, J=6.69 Hz, 1 H) 4.40 (s, 1 H) 4.39 (s, 1 H) 2.45 (s, 3 H) 2.25 (s, 3 H) 2.12 (s, 3 H) 1.51 (s, 3 H) 1.50 (s, 3 H). LC-MS (ES) m/z = 447.8 [M+H]+.
Example 130
l-isopropyl-N-((6-methyl-2-oxo-4-propyl-l,2-dihydropyridin-3-yl)methyl)-6-phenyl- lH-pyrazolo [3,4-b] pyridine-4-carboxamide

Ethyl l-(l-methylethyl)-6-phenyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylate (270 mg, 0.873 mmol) was suspended in THF (1913 μΐ) and Methanol (6378 μΐ). To the mixture was added 3N NaOH (436 μΐ, 1.309 mmol) and the contents heated on an aluminum block at 55 °C overnight. The solvent was removed in vacuo. The remaining orange residue was dissolved with water (10 mL) and cooled with stirring in an ice bath. The contents were acidifed by dropwise addition IN HCl until precipitation ceased. The white solid was collected via vacuum filtration and the filter cake was dried open to the air, under vacuum overnight. The white solid was added to a 20 mL vial followed by 3-(aminomethyl)-6- methyl-4-propyl-2(lH)-pyridinone (208 mg, 0.960 mmol), N-[3-(dimethylamino)propyl]- N'-ethylcarbodiimide hydrochloride (251 mg, 1.309 mmol), 3H-[l,2,3]triazolo[4,5- b]pyridin-3-ol hydrate (202 mg, 1.309 mmol) and DMSO (4 ml). To the same was added a magnetic stir bar and 4-methylmorpholine (618 mg, 6.11 mmol). The reaction was stirred at room temperature overnight. The reaction contents were slowly poured onto an aqueous solution of sat Na2C03 (6 mL) and water (20 mL) and stirred in an ice bath. The resulting suspension was stirred 20 minutes. The solid was collected by vacuum filtration and dried under high vacuum overnight. The title compound was obtained as an off- white solid (388 mg, 99%). 1H NMR (400 MHz, DMSO-56) δ 9.00 (t, J = 3.92 Hz, 1H), 8.38 (s, 1H), 8.23 - 8.29 (m, 2H), 8.16 (s, 1H), 7.46 - 7.62 (m, 3H), 5.93 (s, 1H), 5.34 (quin, J = 6.63 Hz, 1H), 4.45 (d, J = 4.55 Hz, 2H), 2.55 (s, 2H), 2.14 (s, 3H), 1.56 (d, J = 6.57 Hz, 6H), 1.45 - 1.54 (m, 2H), 0.88 (t, J = 7.20 Hz, 3H). LCMS(ES) [M+H]+ 444.3.
Example 131
l-cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-6-(2- methyl-5-pyrimidinyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide


Ethyl 1 -( 1 , 1 -dimethylethyl)-3 -methyl-6-hydroxy- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxylate (2 g, 7.21 mmol) and phosphorus oxychloride (10 ml, 107 mmol) were heated at 100 °C for 32 hours. Ice was added and the contents were extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSC^, filtered, and concentrated in vacuo. The crude residue was purified via silica gel chromatography (eluent: 0% to 50% gradient EtOAc:Hex). The product was collected as 310 mg. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.38 (t, J=7.20 Hz, 3 H), 2.58 (s, 3 H), 4.44 (q, J=7.16 Hz, 2 H), 7.53 (s, 1 H), 13.82 (br. s., 1 H). LCMS(ES) [M+H]+ 240.1 b) Methyl 6-chloro-l-cyclopropyl-3-methyl-lH-pyrazolo[3,4-6]pyridine-4- carboxylate

To a suspension of methyl 6-chloro-3 -methyl- 1 H-pyrazolo [3, 4-b]pyridine-4-carboxylate (510 mg, 2.260 mmol), cyclopropylboronic acid (388 mg, 4.52 mmol), sodium carbonate
(479 mg, 4.52 mmol) in 1 ,2-Dichloroethane (DCE) (150 mL) was added a suspension of copper(II) acetate (411 mg, 2.260 mmol), 2,2'-bipyridine (353 mg, 2.260 mmol) in hot 1,2- dichloroethane. The contents were heated at 70 °C for 16 hours. Added cyclopropylboronic acid (388 mg, 4.52 mmol), sodium carbonate (479 mg, 4.52 mmol) followed by copper(II) acetate (411 mg, 2.260 mmol), 2,2'-bipyridine (353 mg, 2.260 mmol) in hot 1 ,2-dichloroethane and heated at 70 °C for 16 hours. The contents were cooled to room temperature followed by addition of 100 mL of saturated NH4C1 solution and the phases were separated. The aqueous layer was extracted with DCM twice. The combined organic layers were washed with brine, dried over MgS04, filtered, and concentrated in vacuo. The crude residue was purified via silica gel chromatography (eluent: 0% to 20% gradient EtOAc:Hex). The product was collected as 310mg. 1H NMR (400 MHz, DMSO-de) δ ppm 0.95 - 1.26 (m, 4 H), 2.53 (s, 3 H), 3.75 - 3.91 (m, 1 H), 3.96 (s, 3 H), 7.55 (s, 1 H). LCMS(ES) [M+H]+266.0 c) 6-chloro-l-cyclopropyl-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

To a solution of methyl 6-chloro-l-cyclopropyl-3 -methyl- IH-pyrazolo [3, 4-b]pyridine-4- carboxylate (370 mg, 1.393 mmol) in ethanol (30 mL) was added IN sodium hydroxide (1.393 mL, 1.393 mmol) and heated at reflux for 2 hours. The solvent was removed in vacuo, the residue dissolved in 20 mL of water, and acidified with acetic acid. The contents were extracted with EtOAc (4x30 mL). The combined organic layers were washed with water, brine, dried over MgS04, filtered and concentrated in vacuo. The product was collected as 297mg. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.77 - 1.43 (m, 4 H), 2.55 (s, 3 H), 3.63 - 4.15 (m, 1 H), 7.52 (s, 1 H), 14.18 (br. s., 1 H). LCMS(ES) [M+H]+252.4
d) 6-chloro- l-cyclopropyl- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

A mixture of 6-chloro- l-cyclopropyl-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-3-methyl-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (85 mg, 0.220 mmol), 2-methyl-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyrimidine (58.2 mg,
0.264 mmol), sodium carbonate (0.330 mL, 0.661 mmol), 1 ,2-Dimethoxyethane (DME)
(3 mL) and Water (1 mL) were added to a microwave vial and degassed for 10 minutes.
Next added 1 , 1 '-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex (14.39 mg, 0.018 mmol). The contents were irradiated at 140
°C for 10 minutes. Water was added and insoluble material was filtered off. This insoluble material was dissolved in acetonitrile and purified by silica gel chromatography
(eluent: 0% to 20% (MeOH:NH40H/9: l):DCM). The product was collected as 23mg. 1H
NMR (400 MHz, DMSO-d6) δ ppm 1.00 - 1.34 (m, 4 H), 2.12 (s, 3 H), 2.25 (s, 3 H), 2.42 (s, 3 H), 2.72 (s, 3 H), 3.99 (dt, J=7.26, 3.57 Hz, 1 H), 4.39 (d, J=5.05 Hz, 2 H), 5.89 (s, 1
H), 7.86 (s, 1 H), 8.74 (s, 1 H), 9.47 (s, 1 H), 11.53 (br. s., 1 H). LCMS(ES) [M+H] 444.2
Examples 132-134 were prepared using the general procedures outlined for the above compound using 6-chloro- 1 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide and the appropriate boronic acid reagent. Example 132
l-cyclopropyl- V-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-6-[4- (methyloxy)phenyl] -lH-pyra -6] pyridine-4-carboxamide

Example 133
l-cyclopropyl- V- [(41-cyclopr opyl-TV- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3-methyl-6- [6-(4-methyl- l-piperazinyl)-3-pyridinyl] - 1H- pyrazolo [3,4-6] pyridine-4-carboxamide.

1H NMR (400 MHz, DMSO-d6) δ ppm 0.99 - 1.24 (m, 4 H), 2.12 (s, 3 H), 2.21 - 2.29 (m, 6 H), 2.38 (s, 3 H), 2.45 (br. s., 4 H), 3.63 (br. s., 4 H), 3.93 (m, J=7.26, 7.26, 3.79, 3.66 Hz, 1 H), 4.38 (d, J=5.05 Hz, 2 H), 5.88 (s, 1 H), 6.98 (d, J=9.09 Hz, 1 H), 7.64 (s, 1 H), 8.35 (dd, J=9.09, 2.53 Hz, 1 H), 8.70 (t, J=5.05 Hz, 1 H), 8.98 (d, J=2.27 Hz, 1 H), 11.52 (s, 1 H). LCMS(ES) [M+H]+ 527.1
Example 134
l-cyclopropyl- V- [(41-cyclopr opyl-TV- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3-methyl-6- [6-(4-morpholinyl)-3-pyridinyl] - IH-pyrazolo [3,4- b] pyridine-4-carboxamide.

1H NMR (400 MHz, DMSO-d6) δ ppm 1.03 - 1.32 (m, 4 H), 2.12 (s, 3 H), 2.24 (s, 3 H), 2.38 (s, 3 H), 3.49 - 3.64 (m, 4 H), 3.68 - 3.79 (m, 4 H), 3.93 (m, J=7.29, 7.29, 3.92, 3.73 Hz, 1 H), 4.38 (d, J=5.05 Hz, 2 H), 5.88 (s, 1 H), 6.98 (d, J=8.84 Hz, 1 H), 7.65 (s, 1 H),
8.38 (dd, J=8.97, 2.40 Hz, 1 H), 8.70 (t, J=4.93 Hz, 1 H), 9.00 (d, J=2.53 Hz, 1 H), 11.52 (s, 1 H). LCMS(ES) [M+H]+ 514.2
Example 135
6-cyclopropyl- 1-(1 , l-dimethylethyl)-3-methyl- V- [(6-methyl-2-oxo-4-propyl- 1 ,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide

a) Ethyl 6-cyclopropyl-l-(l,l-dimethylethyl)-3-methyl-lH-pyrazolo[3,4- b] pyridine-4-carboxylate

1 -(l,l-dimethylethyl)-3 -methyl- lH-pyrazol-5 -amine (166 mg, 1.086 mmol), ethyl (3Z)-4- cyclopropyl-4-hydroxy-2-oxo-3-butenoate (200 mg, 1.086 mmol) and acetic acid (10 mL) were heated to reflux for 2 hours. The solvent was removed in vacuo and the residue purified by silica gel chromatography (eluent: 0% to 7 % EtOAc:Hex). The product was collected as 278 mg. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.07 (dt, 2 H), 1.13 - 1.20 (m, 2 H), 1.46 (t, J=7.20 Hz, 3 H), 1.78 (s, 9 H), 2.18 (m, J=7.99, 7.99, 4.74, 4.55 Hz, 1 H), 2.65 (s, 3 H), 4.47 (q, J=7.07 Hz, 2 H), 7.43 (s, 1 H). LCMS(ES) [M+H]+ 302.2
a) 6-cyclopropyl- 1-(1 , l-dimethylethyl)-3-methyl- lH-py razolo [3,4-6] pyridine-4- carboxylic acid

To a solution of ethyl 6-cyclopropyl- 1-( 1,1 -dimethylethyl)-3 -methyl- lH-pyrazolo [3,4- b]pyridine-4-carboxylate (278 mg, 0.922 mmol) in ethanol (30 mL) was added sodium hydroxide (3.69 mL, 3.69 mmol) and the contents heated at reflux for 2 hours. The solvent was removed in vacuo, the residue dissolved in 20 mL of water, and acidifed by addition of acetic acid. The contents were extracted with EtOAc (4x30ml). The combined organic extracts were washed with water, brine, dried over MgSC^, filtered, and concentrated in vacuo. The product was collected as 233 mg. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.97 - 1.12 (m, 4 H), 1.69 (s, 9 H), 2.22 - 2.41 (m, 3 H), 2.52 (s, 3 H), 7.45 (s, 3 H). LCMS(ES) [M+H]+ 274.2 b) 6-cyclopropyl-l-(l,l-dimethylethyl)-3-methyl- V-[(6-methyl-2-oxo-4-propyl-l,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide
6-cyclopropyl- 1 -( 1 , 1 -dimethylethyl)-3 -methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxylic acid (53 mg, 0.194 mmol), 3-(aminomethyl)-6-methyl-4-propyl-2(lH)-pyridinone (71.3 mg, 0.242 mmol), l-hydroxy-7-azabenzotriazole (52.8 mg, 0.388 mmol), EDC (74.3 mg, 0.388 mmol) and N-methylmorpholine (0.085 mL, 0.776 mmol) were suspended in Dimethyl Sulfoxide (DMSO) (10 mL) and stirred at room temperature for 16 hours. Added 25 mL of water and stirred for 10 minutes. The contents were filtered and dried, and collected as 70 mg. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.93 (t, J=7.33 Hz, 3 H), 0.96 - 1.09 (m, 4 H), 1.54 (m, J=7.52, 7.52, 7.52, 7.52, 7.33 Hz, 2 H), 1.68 (s, 9 H), 2.12 (s, 2 H), 2.17 - 2.27 (m, 1 H), 2.33 (s, 3 H), 2.5(2H), 4.34 (d, J=5.05 Hz, 2 H), 5.89 (s, 1 H), 7.01 (s, 1 H), 8.54 (t, J=4.80 Hz, 1 H), 11.50 (s, 1 H). LCMS(ES) [M+H]+ 436.1.
Example 136
1 ,6-dicyclopropyl-3-methyl- V- [(6-methyl-2-oxo-4-propyl- 1 ,2-dihydro-3- pyridinyl)methyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide
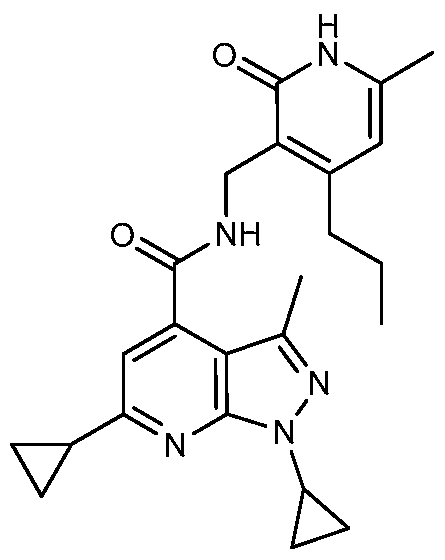

Nitrosonium tetrafluoroborate (2.415 g, 20.67 mmol) was added in several portions to a cooled (-30 °C) solution of 1,1-dimethylethyl cyclopropylcarbamate (2.5 g, 15.90 mmol) in anhydrous Pyridine (4 mL) and Acetonitrile (40 mL). The solution was stirred at -30 °C for 30 minutes then at 0 °C for 2 hours. Ice water and EtOAc were added. The organic phase was separated and washed with IN HC1, IN NaHC03, brine, dried over MgS04, filtered and concentrated in vacuo. The product was collected as 2.66 g. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 0.37 - 0.79 (m, 4 H), 1.64 (s, 9 H), 2.18 - 2.41 (m, 1 H). b) Cyclopropylhydrazine

1,1-dimethylethyl cyclopropyl(nitroso)carbamate (2.63 g, 14.12 mmol) was dissolved in methanol (130 mL) and cooled to -78 °C. Hydrochloric acid (12.48 mL, 150 mmol) was slowly added dropwise. Zinc was added in portions, and the contents were stirred at -78 °C for 6 hours. The contents were filtered through celite. The solvent was removed in vacuo. The residue was suspended in EtOH and concentrated in vacuo in triplicate (to
help azeotrope off water). The product was obtained as a sticky semi-solid which still had EtOH in sample (5 g). 1H NMR (400 MHz, DMSO-d6) δ ppm 0.34 - 0.79 (m, 5 H), 7.91 (d, 1 H), 8.87 (br. s., 2 H)
c) l-cyclopropyl-3-methyl-lH-pyrazol-5-amine

(2Z)-3-amino-2-butenenitrile (903 mg, 11.00 mmol), cyclopropylhydrazine (1194 mg, 11 mmol) and triethylamine (3.07 mL, 22.00 mmol) were suspended in ethanol (100 mL) and heated to 70 °C for 16 hours. The solvent was removed in vacuo. The residue was suspended in 50 mL of saturated NaHC03 and stirred for 10 minutes. The contents were extracted with DCM, dried over MgS04, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography (eluent: 0% to 100% gradient EtOAc:Hex). The product was collected as 310mg. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 0.95 - 1.13 (m, 4 H), 2.13 (s, 3 H), 3.06 (m, J=6.95, 6.95, 3.79, 3.54 Hz, 1 H), 3.76 - 3.88 (m, 2 H), 5.24 (s, 1 H). LCMS(ES) [M+H]+ 138.1 d ) Ethyl 1 ,6-dicyclopr opyl-3-methyl- lH-pyrazolo [3,4-6] pyridine-4-carboxylate

310mg. 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.05 - 1.13 (m, 4 H), 1.16 - 1.21 (m, 2 H), 1.25 - 1.30 (m, 2 H), 1.46 (t, J=7.20 Hz, 3 H), 2.16 - 2.31 (m, 1 H), 2.63 (s, 3 H),
3.78 (m, J=7.26, 7.26, 3.79, 3.66 Hz, 1 H), 4.47 (q, J=7.24 Hz, 2 H), 7.40 (s, 1 LCMS(ES) [M+H]+ 286.2 e) l,6-dicyclopropyl-3-methyl-lH-pyrazolo[3,4-6]pyridine-4-carboxylic acid

To a solution of ethyl l,6-dicyclopropyl-3-methyl-lH-pyrazolo[3,4-b]pyridine-4- carboxylate (310 mg, 1.086 mmol) in ethanol (30 mL) was added sodium hydroxide (4.35 mL, 4.35 mmol) and the mixture heated at reflux for 2 hours. The solvent was removed in vacuo, the residue suspended in water (20 mL), and acidified by addition of acetic acid. The contents wer extracted with EtOAc (4x30 mL). The combined EtOAc extracts were washed with water, brine, dried over MgSC^, filtered, and concentrated in vacuo. The product was collected as 250 mg. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.88 - 1.31 (m, 8 H), 2.21 - 2.42 (m, 1 H), 3.83 (m, J=7.39, 7.39, 3.79, 3.66 Hz, 1 H), 7.44 (s, 1 H), 13.72 (br. s., 1 H). LCMS(ES) [M+H]+ 258.2 f ) 1 ,6-dicyclopropyl-3-methyl- V- [(6-methyl-2-oxo-4-propyl- 1 ,2-dihydro-3- pyridinyl)methyl]-lH-pyrazolo[3,4-6]pyridine-4-carboxamide l,6-dicyclopropyl-3-methyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (125 mg, 0.486 mmol), 3-(aminomethyl)-6-methyl-4-propyl-2(lH)-pyridinone (126 mg, 0.583 mmol), 1- hydroxy-7-azabenzotriazole (132 mg, 0.972 mmol), EDC (186 mg, 0.972 mmol) and N- methylmorpholine (0.214 mL, 1.943 mmol) were suspended in Dimethyl Sulfoxide (DMSO) (10 mL) and stirred at room temperature for 16 hours. Added 25 mL of water and let stir for 10 minutes. The contents were filtered and dried. The product was collected as 200 mg. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.92 (t, J=7.07 Hz, 3 H), 1.04 (d, J=6.06 Hz, 8 H), 1.45 - 1.73 (m, 2 H), 2.12 (s, 3 H), 2.24 (br. s., 1 H), 2.33 (s, 3 H), 3.79 (br. s., 1 H), 4.34 (d, J=3.79 Hz, 2 H), 5.89 (br. s., 1 H), 6.99 (s, 1 H), 8.57 (br. s., 1 H), 11.51 (br. s., 1 H). MS(ES) [M+H]+ 420.2
Example 137
1 ,6-dicyclopropyl- V- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl] -3-methyl- lH-pyrazolo[3,4-6]pyridine-4-carboxamide.

The title compound was made using the same procedure as for example 11 (step f) from l,6-dicyclopropyl-3-methyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid and 3- (aminomethyl)-4,6-dimethyl-2(lH)-pyridinone. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.93 - 1.18 (m, 8 H), 2.11 (s, 3 H), 2.21 (s, 4 H), 2.32 (s, 3 H), 3.78 (m, J=7.33, 7.33, 3.92, 3.66 Hz, 1 H), 4.33 (d, J=5.05 Hz, 2 H), 5.87 (s, 1 H), 7.01 (s, 1 H), 8.58 (t, J=4.80 Hz, 1 H), 11.50 (s, 1 H). LCMS(ES) [M+H]+ 392.
Example 138
6-(cyclopropylamino)-N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l- isopropyl-3-methyl-lH-pyrazolo [3, -b] pyridine-4-carboxamide

6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide (lOOmg, 0.258 mmol) was dissolved in 1,4-dioxane (2.6 mL) followed by addition of cesium carbonate (336 mg, 1.031 mmol). After degassing with nitrogen for 5 min, Xantphos (44.8 mg, 0.077 mmol) was added. The mixture was degassed for 2 min, Pd2(dba)3 (35.4 mg, 0.039 mmol) was added, and then degassed for 1 min. Cyclopropanamine (0.090 mL, 1.289 mmol) was
added and the contents sealed. The suspension was stirred for 3h at 100 °C (heat block). The reaction mixture was poured onto water and extracted with EtOAc (2x). The combined organic layers were dried over MgSC^, filtered, and concentrated in vacuo to a solid residue. The aq. layer was extracted with 10%MeOH/DCM (2x). The combined organic layers were dried over MgS04, filtered, and concentrated in vacuo to afford a solid. The combined solid residues were dissolved in DMSO and acetonitrile spiked with TFA, and purified by reverse phase HPLC (Gradient B : 10-70%. A: Water + .1% TFA. B: CH3CN + .1% TFA) The title compound was collected as 11 mg (10%) ; 1H NMR (400 MHz, DMSO- 6) δ ppm 0.39 - 0.47 (m, 2 H), 0.68 - 0.76 (m, 2 H), 1.39 (d, J=6.57 Hz, 6 H), 2.11 (s, 3 H), 2.24 (s, 3 H), 2.20 (s, 3 H), 2.64 - 2.72 (m, 1 H), 4.30 (d, J=4.80 Hz, 2 H), 4.89 (quin, J=6.69 Hz, 1 H), 5.86 (s, 1 H), 6.34 (s, 1 H), 7.25 (d, J=3.03 Hz, 1 H), 8.41 (t, J=4.93 Hz, 1H), 11.49 (s, 1 H). LCMS(ES) [M+H]+ 409.1
Example 139
N-((4-benzyl-6-methyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-lH- pyrazolo [3,4-b] pyridine-4-carboxamide

a) l-isopropyl-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid

To a 100 mL round bottom flask was charged 10%> Pd/C(degussa) (0.213 g, 0.100 mmol), under N2 followed by addition of ca. 5 mL EtOH. The slurry was stirred followed by addition of 6-chloro-l-(l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (.48g, 2.003 mmol) and ethanol (22 mL). Next added Et3N (0.837 mL, 6.01 mmol) and stirred for 5 min under nitrogen. The contents were then stirred under an atmosphere of hydrogen (1 atm) for 6h at RT. The vessel was then flushed with nitrogen and the
contents diluted with DCM (10 mL) and a small amount of celite. The contents were stirred for 10 min., filtered through analytical grade celite and washed with 10% MeOH/DCM, EtOH, then DCM. The filtrate was concentrated in vacuo to a residue and dried on hi-vacuum overnight. The solid was treated with water and adjusted to pH 3 with 1M HC1. The contents were filtered, air-dried and then dried in hi-vacuum oven at 45 °C for 18h. The title compound was collected as 0.326g (78%). 1H NMR (400 MHz, DMSO- 6) δ ppm 1.52 (d, J=6.82 Hz, 6 H) 5.28 (quin, J=6.69 Hz, 1 H) 7.70 (d, J=4.55 Hz, 1 H) 8.39 (s, 1 H) 8.73 (d, J=4.55 Hz, 1 H) 13.91 (br. s., 1 H). LCMS(ES) [M+H]+ 205.9
b) N-((4-benzyl-6-methyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-lH- pyrazolo [3,4-b] pyridine-4-carboxamide
The title compound was prepared in the same manner as described for example 11 from 1- (l-methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (.060 g, 0.292 mmol) and 3(aminomethyl)-6-methyl-4-(phenylmethyl)-2(lH)-pyridinone (0.081 g, 0.307 mmol) wherein the product obtained was further purified by reverse phase HPLC (Gradient B: 5- 85%). A:Dichloromethane. B: 10% (2M Ammonia in Methanol) in Chloroform). The isolated product was concentrated from MTBE (2x) to afford a white solid (93 mg, 75%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.50 (d, J=6.57 Hz, 6 H), 2.10 (s, 3 H), 3.98 (s, 2 H), 4.45 (d, J=5.05 Hz, 2 H), 5.24 (quin, j=6.63 Hz, 1 H), 5.81 (s, 1 H), 7.12 - 7.30 (m, 5 H), 7.50 (d, J=4.55 Hz, 1 H), 8.33 (s, 1 H), 8.61 (d, J=4.55 Hz, 1 H), 8.88 (t, j=5.05 Hz, 1 H), 11.63 (s, 1 H). LCMS(ES) [M+H]+ 416.0
Example 140
N-((4-benzyl-6-methyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl- lH-pyrazolo [3,4-b] pyridine-4-carboxamide


The title compound was prepared in the same manner as described for example 139 (step a) from 10% Pd/C(degussa) (0.457 g, 0.215 mmol), and 6-chloro-3 -methyl- 1-(1 - methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxylic acid (1.09g, 4.30 mmol) wherein the stir time was 4h Upon acidification to pH 3 with 1M HCl, the contents were extracted with EtOAc (2x) and the combined organic layers dried over MgSC^, filtered, and concentrated in vacuo. The collected solid was dried in a vacuum oven at 45 °C for 3 h. The final product was collected as 0.826g (84%). 1H NMR (400 MHz, DMSO- 6) d ppm 1.48 (d, J=6.57 Hz, 6 H), 2.63 (s, 3 H), 5.21 (quin, J=6.69 Hz, 1 H), 7.52 (d, J=4.55 Hz, 1 H), 8.63 (d, J=4.55Hz, 1 H), 13.77 (br. s., 1 H). LCMS(ES) [M+H]+ 220.2
N-((4-benzyl-6-methyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl- lH-pyrazolo [3,4-b] pyridine-4-carboxamide
The title compound was prepared in the same manner as described for example 11 from 3- methyl- l-(l-methylethyl)-lH-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (.060g, 0.274 mmol) and 3-(aminomethyl)-6-methyl-4-(phenylmethyl)-2(lH)-pyridinone (0.076 g, 0.287 mmol). The product was collected as a white solid (92 mg, 77%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.46 (d, J=6.82 Hz, 6 H), 2.09 (s, 3 H), 2.41 (s, 3 H), 3.98 (s, 2 H), 4.43 (d, J=5.05 Hz, 2 H), 5.16 (quin, j=6.63 Hz, 1 H), 5.79 (s, 1 H), 7.04 (d, J=4.80 Hz, 1 H), 7.16 - 7.34 (m, 5 H), 8.49 (d, J=4.55 Hz, 1 H), 8.75 (t, j=5.05 Hz, 1 H), 11.59 (s, 1 H). LCMS(ES) [M+H]+ 429.9
Example 141
6-cyclopropyl- V- [(4-cyclopropyl-6-methyl-2-oxo- 1 ,2-dihydro-3-py ridinyl)methyl] -3- methyl-l-(l-methylethyl)-lH- razolo[3,4-6]pyridine-4-carboxamide

6-cyclopropyl-N- [(4-cyclopropyl-6-methyl-2-oxo- 1 ,2-dihydro-3-py ridinyl)methyl] -3- methyl-l-(l-methylethyl)-lH-pyrazolo[3,4-6]pyridine-4-carboxamide
6-cyclopropyl-l-isopropyl-3 -methyl- lH-pyrazolo [3, 4-b]pyridine-4-carboxylic acid (150 mg, 0.578 mmol), l-hydroxy-7-azabenzotriazole (118 mg, 0.868 mmol) and EDC (166 mg, 0.868 mmol) were suspended in Dimethyl Sulfoxide (DMSO) (10 mL). To the solution was added in one portion N-methylmorpholine (0.191 mL, 1.735 mmol), followed by 3-(aminomethyl)-4-cyclopropyl-6-methylpyridin-2(lH)-one (bis HCl salt) (189 mg, 0.752 mmol). and the reaction stirred at RT for 12 h. The reaction contents were poured onto ice water (200mL) and were stirred for 20 min. The contents were filtered and washed with water (10 mL) and then methanol/ice water (lOmL/10 mL). The product was dried to afford a tan solid which was collected as 180 mg (71%). 1H NMR (400 MHz, DMSO-de) d ppm 11.45 (s, 1 H) 8.64 (t, J=4.93 Hz, 1 H) 6.99 (s, 1 H) 5.49 (s, 1 H) 5.04 (quin, J=6.69 Hz, 1 H) 4.52 (d, J=5.05 Hz, 2 H) 2.37 (s, 3 H) 2.21 - 2.26 (m, 1 H) 2.12 - 2.17 (m, 1 H) 2.09 (s, 3 H) 1.42 (d, J=6.57 Hz, 6 H) 1.01 - 1.04 (m, 4 H) 0.95 - 1.00 (m, 2 H), 0.075-0.076 (m, 2H); LCMS: [M+H]+ = 420.3.
Examples 142-144 were prepared in the same manner as described for example 16 step c using 6-cyclopropyl-l-isopropyl-3 -methyl- lH-pyrazolo [3, 4-b]pyridine-4-carboxylic acid and the appropriately substituted 3-aminomethyl-pyridone intermediate.
Example 142
6-cyclopropyl-3-methyl-l-(l-methylethyl)- V-{[6-methyl-4-(l-methylethyl)-2- dihydro-3-pyridinyl]methyl}-lH- razolo[3,4-6]pyridine-4-carboxamide

Example 143
6-cyclopropyl-3-methyl-l-(l-methylethyl)- V-[(6-methyl-2-oxo-4-propyl-l,2-dihydro- 3-pyridinyl)methyl]-lH-pyrazolo[3,4-6] ridine-4-carboxamide

1H NMR (400 MHz, DMSO- 6) δ ppm 11.52 (br. s., 1 H) 8.57 (t, J=4.80 Hz, 1 H) 6.97 (s, 1 H) 5.90 (s, 1 H) 5.00-5.07 (m, 1 H) 4.35 (d, J=5.05 Hz, 2 H) 2.37 (s, 3 H) 2.20 - 2.26 (m, 1 H) 2.13 (s, 3 H) 1.51 - 1.59 (m, 2 H) 1.42 (d, J=6.57 Hz, 6 H) 0.99 - 1.06 (m, 5 H) 0.93 (t, J=7.33 Hz, 4 H); LCMS: [M+H]+ = 422.3
Example 144
6-cyclopropyl-3-methyl-l-(l-methylethyl)- V-{[6-methyl-2-oxo-4-(phenylmethyl)-l,2- dihydro-3-pyridinyl]methyl}-lH- razolo[3,4-6]pyridine-4-carboxamide

Intermediates 95 and 96
3-(aminomethyl)-6-methyl-4-(phenylmethyl)-2(lH)-pyridinone and 3-(aminomethyl)- 4-methyl-6-(phenylmethyl)-2(lH)-pyridinone


To a solution of NaNH2 (19.02 g, 480 mmol) in anhydrous ether (400 mL) under N2 at - 5 °C was added dropwise ethyl phenylacetate (19.2 g ,150 mmol) and then acetone (21.23 g, 370 mmol) with vigorous stirring. After addition, the reaction mixture was stirred at room temperature overnight. The mixture was then acidified to pH 4.0 - 5.0 with IN HCl. The organic layer was separated and concentrated in vacuo. The crude product was purified by silica gel chromatography to give l-phenyl-2,4-pentanedione (18.32 g, 44 %). 1H NMR
(400 MHz, CDC13-d3) δ 15.49 (br s, 1H), 7.33-7.45 (m, 5H), 5.53 (s, 1H), 3.66 (s, 2H), 2.10 (s, 3H). b) 6-methyl-2-oxo-4-(phenylmethyl)-l,2-dihydro-3-pyridinecarbonitrile and 4- methyl-2-oxo-6-(phenylmethyl)- 1 ,2-dihydro-3-pyridinecarbonitrile

1H NMR (400 MHz, DMSO-d6) (mixture of compounds) δ 7.21-7.31 (m, 10H), 6.06 (s,
2H), 3.89 (s, 2H), 3.79 (s, 2H), 2.24 (s, 3H), 2.15 (s, 3H).
a) 3-(aminomethyl)-6-methyl-4-(phenylmethyl)-2(lH)-pyridinone and 3- (aminomethyl)-4-methyl-6-(phenylmethyl)-2(lH)-pyridinone

Sodium acetate (6.14 g, 74.8 mmol), Pd/C (0.65 g, 1 mmol), and platinum (II) oxide (45 mg, 1 mmol) were placed in a dried Parr bottle equipped with nitrogen inlet. A small amount of acetic acid was added to wet the catalysts. A solution of 6-methyl-2-oxo-4- (phenylmethyl)- 1 ,2-dihydro-3-pyridinecarbonitrile and 4-methyl-2-oxo-6-(phenylmethyl)- l,2-dihydro-3-pyridinecarbonitrile (6 g, 26.7 mmol) in acetic acid (300 mL) was added to
the vessel. The contents were sealed and hydrogenated on Parr shaker at 45 psi for 12 h. The reaction mixture was filtered and washed with acetic acid. The filtrate was removed under reduced pressure. The residue was washed with methanol and filtered to afford a crude mixture of 3-(aminomethyl)-6-methyl-4-(phenylmethyl)-2(lH)-pyridinone and 3- (aminomethyl)-4-methyl-6-(phenylmethyl)-2(lH)-pyridinone. The reaction was run in duplicate to afford a total crude recovery of 14.5 g. To a solution of the above crude product mixture (4.0 g, 17.5 mmol) in THF (10 mL) and DMF (10 mL) was added di-tert- butoxycarbonyl anhydride (5.0 g, 23.4 mmoL) and triethylamine (5.2 g, 52.5 mmol) at 0 °C. The reaction mixture was stirred with warming to room temperature and then stirred for an additional 4 h. The contents were diluted with ice water and then filtered. The collected solid was dried and the products separated by HPLC to furnish 1.2 g of 1 , 1- dimethylethyl { [4-methyl-2-oxo-6-(phenylmethyl)- 1 ,2-dihydro-3 - pyridinyl]methyl} carbamate (1H NMR (400 MHz, DMSO-d6) 1 1.55-1.60 (br s, 1H), 7.20-7.29 (m, 5H), 5.85 (s, 1H), 3.92 (s, 2H), 3.90 (s, 2H), 2.10 (s, 3H), 1.32 (s, 9H) and 1.0 g of 1 , 1-dimethylethyl {[6-methyl-2-oxo-4-(phenylmethyl)-l ,2-dihydro-3- pyridinyl]methyl} carbamate (1H NMR (400 MHz, DMSO-d6) δ 11.50-1 1.55 (br s, 1H), 7.18-7.25 (m, 5H), 5.75 (s, 1H), 4.02 (s, 2H), 3.85 (s, 2H), 2.05 (s, 3H), 1.32 (s, 9H). d) 3-(aminomethyl)-4-methyl-6-(phenylmethyl)-2(lH)-pyridinone hydrochloride

A solution of 1 , 1-dimethylethyl {[4-methyl-2-oxo-6-(phenylmethyl)-l ,2-dihydro-3- pyridinyljmethyl} carbamate (1.2 g, 3.66 mmol) in 4N HCI (in 15 mL 1 ,4 dioxane) was heated to 60 °C for 1 h. The mixture was cooled to room temperature. The mixture was filtered and dried to give 3-(aminomethyl)-4-methyl-6-(phenylmethyl)-2(lH)-pyridinone as an HCI salt (0.725 g, 87%). LCMS MH+ = 229.1 1H NMR (400 MHz, DMSO-d6) δ 1 1.9-12.0 (br s, 1H), 7.99 (br s, 3H), 7.20 (s, 5H), 5.97 (s, 1H), 3.72-3.75 (m, 4H), 2.17 (s,
3H).
e) 3 -(aminomethyl)-6-methyl-4-(phenylmethyl)-2(lH)-pyridinone hydrochloride

A solution of 1 , 1-dimethylethyl {[6-methyl-2-oxo-4-(phenylmethyl)-l ,2-dihydro-3- pyridinyljmethyl} carbamate (1.0 g, 3.0 mmol) in 4N HCI (in 15 mL 1 ,4 dioxane) was heated to 60 °C for 1 h. The mixture was cooled to room temperature. The mixture was filtered and dried to give 3-(aminomethyl)-6-methyl-4-(phenylmethyl)-2(lH)-pyridinone as an HCI salt (0.600 g, 86%). LCMS MH+ = 229.1 1H NMR (400 MHz, DMSO-d6) δ 1 1.9-12.0 (br s, 1H), 8.03 (br s, 3H), 7.16-7.30 (m, 5H), 5.84 (s, 1H), 3.91 (s, 2H), 3.81 (s, 2H), 2.10 (s, 3H).
Assay Protocol
Compounds contained herein were evaluated for their ability to inhibit the methyltransferase activity of EZH2 within the PRC2 complex. Human PRC2 complex was prepared by co-expressing each of the 5 member proteins (FLAG-EZH2, EED, SUZ12, RbAp48, AEBP2) in Sf9 cells followed by co-purification. Enzyme activity was measured in a scintillation proximity assay (SPA) where a tritiated methyl group is transferred from 3H-SAM to a lysine residue on Histone H3 of a mononucleosome, purified from HeLa cells. Mononucleosomes were captured on SPA beads and the resulting signal is read on a ViewLux plate reader.
Part A. Compound Preparation
1. Prepare 10 mM stock of compounds from solid in 100% DMSO.
2. Set up an 1 1-point serial dilution (1 :3 dilution, top concentration 10 mM) in 100% DMSO for each test compound in a 384 well plate leaving columns 6 and 18 for DMSO controls.
3. Dispense 100 nL of compound from the dilution plate into reaction plates (Grenier Bio-One, 384-well, Cat# 784075).
Part B. Reagent Preparation
Prepare the following solutions:
1. 50 mM Tris-HCl, pH 8: Per 1 L of base buffer, combine 1 M Tris-HCl, pH 8 (50 mL) and distilled water (950 mL).
2. lx Assay Buffer: Per 10 mL of lx Assay Buffer, combine 50 mM Tris-HCl, pH 8 (9958 uL), 1 M MgCl2 (20 uL), 2 M DTT (20 uL), and 10% Tween-20 (2 uL) to provide a final concentration of 50 mM Tris-HCl, pH 8, 2 mM MgCl2, 4 mM DTT, 0.002% Tween-20.
3. 2x Enzyme Solution: Per 10 mL of 2x Enzyme Solution, combine lx Assay Buffer and PRC2 complex to provide a final enzyme concentration of 10 nM.
4. SPA Bead Suspension: Per 1 mL of SPA Bead Suspension, combine PS-PEI
coated LEAD Seeker beads (40 mg) and ddH20 (1 mL) to provide a final concentration of 40 mg/mL.
5. 2x Substrate Solution: Per 10 mL of 2x Substrate Solution, combine lx Assay Buffer (9728.55 uL), 800 ug/mL mononucleosomes (125 uL), 1 mM cold SAM (4 uL), and 7.02 uM 3H-SAM (142.45 uL; 0.55 mCi/mL) to provide a final concentration of 5 ug/mL nucleosomes, 0.2 uM cold SAM, and 0.05 uM 3H-SAM.
6. 2.67x Quench/Bead Mixture: Per 10 mL of 2.67x Quench/Bead Mixture, combine ddH20 (9358 uL), 10 mM cold SAM (267 uL), 40 mg/mL Bead Suspension (375 uL) to provide a final concentration of 100 uM cold SAM and 0.5 mg/mL SPA beads.
Part C. Assay Reaction in 384-well Grenier Bio-One Plates
Compound Addition
1. Dispense 100 nL/well of lOOx Compound to test wells (as noted above).
2. Dispense 100 nL/well of 100% DMSO to columns 6 & 18 for high and low
controls, respectively.
Assay
1. Dispense 5 uL/well of lx Assay Buffer to column 18 (low control reactions).
2. Dispense 5 uL/well of 2x Enzyme Solution to columns 1-17, 19-24.
3. Spin assay plates for ~1 minute at 500 rpm.
4. Stack the assay plates, covering the top plate.
5. Incubate the compound/DMSO with the enzyme for 30 minutes at room
temperature.
6. Dispense 5 uL/well of 2x Substrate Solution to columns 1-24.
7. Spin assay plates for ~1 minute at 500 rpm.
8. Stack the assay plates, covering the top plate.
9. Incubate the assay plates at room temperature for 1 hour.
Quench/Bead Addition
1. Dispense 5 uL/well of the 3x Quench/Bead Mixture to columns 1-24.
2. Seal the top of each assay plate with adhesive TopSeal.
3. Spin assay plates for ~1 minute at 500 rpm.
4. Equilibrate the plates for > 20 min.
Read plates
1. Read the assay plates on the Viewlux Plate Reader utilizing the 613 nm emission filter with a 300 s read time.
Reagent addition can be done manually or with automated liquid handler.
*The final DMSO concentration in this assay is 1%.
*The positive control is in column 6; negative control is in column 18.
*Final starting concentration of compounds is 100 μΜ.
Part D. Data analysis
Percent inhibition was calculated relative to the DMSO control for each compound concentration and the resulting values were fit using standard IC50 fitting parameters within the ABASE data fitting software package.
Exemplified compounds of the present invention were generally tested according to the above or an analogous assay and were found to be inhibitors of EZH2. The IC50 values ranged from about 1 nM to about 10 μΜ; The IC50 values of the more active compounds range from about 1 nM to about 500 nM; The most active compounds are under 50 nM. As tested in the foregoing assay or an analogous assay, compounds of the various Examples gave the IC50 data (nM) in the paragraph below. Repeating the assay run(s) may result in a somewhat different.
Ex 1, 475; Ex 2, 806; Ex 4, 116; Ex 5, 705; Ex 6, 695; Ex 7, 1296; Ex 8, 167; Ex 9, 1309; Ex 10, 569; Ex 11, 18; Ex 12, 55; Ex 13, 55; Ex 14, 735; Ex 15, 179; Ex 16, 105; Ex 17, 2591; Ex 18, 40; Ex 19, 3372; Ex 20, 4647; Ex 21, 1040; Ex 22, 1362; Ex 23, 1428; Ex 24, 873; Ex 25, 685; Ex 26, 673; Ex 27, 24; Ex 28, 348; Ex 29, 234; Ex 30, 154; Ex 31, 232; Ex 32, 856; Ex 33, 70; Ex 35, 673; Ex 36, 924; Ex 37, 1095; Ex 38, 392; Ex 41, 86; Ex 42, 56; Ex 43, 204; Ex 44, 74; Ex 45, 248; Ex 46, 128; Ex 47, 88; Ex 48, 198; Ex 49, 115; Ex 50, 81; Ex 51, 161; Ex 53, 436; Ex 54, 514; Ex 55, 260; Ex 56, 2111; Ex 57, 784; Ex 58, 78; Ex 59, 155; Ex 60, 198; Ex 61, 112; Ex 62, 581; Ex 63, 96; Ex 64, 79; Ex 65, 55; Ex 66, 81; Ex 67, 58; Ex 68, 76; Ex 69, 25; Ex 70, 1893; Ex 71, 402; Ex 72, 171; Ex 73, 533; Ex 74, 151; Ex 75, 131; Ex 76, 82; Ex 77, 52; Ex 78, 43; Ex 79, 140; Ex 80, 71; Ex 81, 30; Ex 82, 108; Ex 83, 43; Ex 84, 99; Ex 85, 31; Ex 86, 142; Ex 87, 18; Ex 88, 52; Ex 89, 67; Ex 90, 173; Ex 92, 76; Ex 93, 83; Ex 94, 103; Ex 95, 489; Ex 96, 57; Ex 97, 55; Ex 99, 25044; Ex 100, 5747; Ex 103, 373; Ex 105, 315; Ex 106, 119; Ex 107, 75; Ex 109, 207; Ex 110, 231; Ex 111, 367; Ex 112, 693; Ex 113, 248; Ex 114, 199; Ex 117, 190; Ex 118, 273; Ex 119, 333; Ex 120, 270; Ex 121, 407; Ex 122, 153; Ex 123, 218; Ex 124, 1052; Ex 125, 2164.
The foregoing examples are provided to illustrate the invention and are not intended to limit it in any way. What is reserved to the inventors can be found by reference to the claims.
Claims
1. A compound of compounds of formula (I)

wherein
X and Z are selected independently from the group consisting of hydrogen, (Ci- C8)alkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, unsubstituted or substituted (C3-Cg)cycloalkyl, unsubstituted or substituted (C3-Cg)cycloalkyl-(Ci-Cg)alkyl or -(C2-Cg)alkenyl, unsubstituted or substituted (C5-C8)cycloalkenyl, unsubstituted or substituted (C5- C8)cycloalkenyl-(Ci-C8)alkyl or -(C2-C8)alkenyl, (C6-Cio)bicycloalkyl, unsubstituted or substituted heterocycloalkyl, unsubstituted or substituted heterocycloalkyl-(Ci-C8)alkyl or -(C2-C8)alkenyl, unsubstituted or substituted aryl, unsubstituted or substituted aryl-(Ci- C8)alkyl or -(C2-C8)alkenyl, unsubstituted or substituted heteroaryl, unsubstituted or substituted heteroaryl-(Ci-C8)alkyl or -(C2-C8)alkenyl, halo, cyano, -CORa, -C02Ra, - CONRaRb, -CONRaNRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -NRaNRaC(0)ORa, -ORa, - OC(0)Ra, and -OC(0)NRaRb;
Y is H or halo;
R1 is (Ci-Cg)alkyl, (C2-C8)alkenyl, (C2-Cg)alkynyl, unsubstituted or substituted
(C3-Cg)cycloalkyl, unsubstituted or substituted (C3-Cg)cycloalkyl-(Ci-Cg)alkyl or - (C2-C8)alkenyl, unsubstituted or substituted (C5-C8)cycloalkenyl, unsubstituted or substituted (C5-C8)cycloalkenyl-(Ci-C8)alkyl or -(C2-Cs)alkenyl, unsubstituted or substituted (C6-Cio)bicycloalkyl, unsubstituted or substituted heterocycloalkyl or - (C2-C8)alkenyl, unsubstituted or substituted heterocycloalkyl-(Ci-C8)alkyl, unsubstituted or substituted aryl, unsubstituted or substituted aryl-(Ci-C8)alkyl or -(C2-C8)alkenyl, unsubstituted or substituted heteroaryl, unsubstituted or substituted heteroaryl-(Ci- C8)alkyl or -(C2-C8)alkenyl, -CORa, -C02Ra, -CONRaRb, -CONRaNRaRb;
R3 is hydrogen, (Ci-Cg)alkyl, cyano, trifluoromethyl, -NRaRb, or halo;
R6 is selected from the group consisting of hydrogen, halo, (Ci-Cg)alkyl,
(C2-C8)alkenyl, -B(OH)2, substituted or unsubstituted (C2-Cg)alkynyl, unsubstituted or substituted (C3-Cg)cycloalkyl, unsubstituted or substituted (C3-Cg)cycloalkyl-(Ci-Cg)alkyl, unsubstituted or substituted (Cs-Cg)cycloalkenyl, unsubstituted or substituted (C5- Cg)cycloalkenyl-(Ci-C8)alkyl, (C6-Ci0)bicycloalkyl, unsubstituted or substituted heterocycloalkyl, unsubstituted or substituted heterocycloalkyl-(Ci-Cg)alkyl, unsubstituted or substituted aryl, unsubstituted or substituted aryl-(Ci-Cg)alkyl, unsubstituted or substituted heteroaryl, unsubstituted or substituted heteroaryl-(Ci-Cg)alkyl, cyano, -CORa, -C02Ra, -CONRaRb, -CONRaNRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -NRaNRaC(0)ORa, -ORa, - OC(0)Ra, -OC(0)NRaRb;
wherein any (Ci-Cg)alkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, cycloalkyl,
cycloalkenyl, bicycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by 1 , 2 or 3 groups independently selected from the group consisting of -0(Ci-C6)alkyl(Rc)1_2, -S(Ci-C6)alkyl(Rc)1_2, -(Ci-C6)alkyl(Rc)1_2, (Ci-C8)alkyl- heterocycloalkyl, (C3-C8)cycloalkyl-heterocycloalkyl, halo, (Ci-C6)alkyl,
(C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, - C02Ra,-CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl,
aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
wherein any aryl or heteroaryl moiety of said aryl, heteroaryl, aryl(Ci-C4)alkyl, or heteroaryl(Ci-C4)alkyl is optionally substituted by 1, 2 or 3 groups independently selected from the group consisting of halo, (Ci-C6)alkyl, (C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, -CONRaRb,
-SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb,
-NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -ORa,
-OC(0)Ra, and -OC(0)NRaRb; each Rc is independently (Ci-C4)alkylamino, -NRaS02Rb, -SORa, -S02Ra, - NRaC(0)ORa, -NRaRb, or -C02Ra;
Ra and Rb are each independently hydrogen, (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-C8)alkynyl, (C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (C6-Ci0)bicycloalkyl,
heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-
Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2,-CONH(Ci-C4)alkyl, - CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2,-S02NH(Ci-C4)alkyl, or - S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
or Ra and Rb taken together with the nitrogen to which they are attached represent a 5-8 membered saturated or unsaturated ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C4)alkyl, (Ci-
C4)haloalkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, hydroxyl, oxo, (Ci-C4)alkoxy, and (Ci-C4)alkoxy(Ci-C4)alkyl, wherein said ring is optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
or Ra and Rb taken together with the nitrogen to which they are attached represent a 6- to 10-membered bridged bicyclic ring system optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
or a salt thereof.
2. A compound of formula (I) according to claim 1 wherein:
X and Z are selected from the group consisting of (Ci-Cg)alkyl, (C3-C8)cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NRaRb, and -ORa;
Y is H or F;
R1 is selected from the group consisting of (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, and heteroaryl;
R3 is selected from the group consisting of hydrogen, (Ci-Cg)alkyl, cyano, trifluoromethyl, -NRaRb, and halo; R6 is selected from the group consisting of hydrogen, halo, cyano, trifluoromethyl, amino, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, aryl, heteroaryl, acylamino; (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl, -S02Ra, -S02NRaRb , and -NRaS02Rb ;
wherein any (Ci-Cg)alkyl, (C3-C8)cycloalkyl, (C2-C8)alkynyl, arylalkynyl, heteroarylalkynyl group is optionally substituted by 1 , 2 or 3 groups independently selected from -0(Ci-C6)alkyl(Rc)i_2, -S(d-C6)alkyl(Rc)i_2, -(Ci-C6)alkyl(Rc)i_2, (Ci-C8)alkyl-heterocycloalkyl, (C3-C8)cycloalkyl-heterocycloalkyl, halo,
(Ci-C6)alkyl, (C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, - CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl,
aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
each Rc is independently (Ci-C4)alkylamino, -NRaS02Rb, -SORa, -S02Ra, - NRaC(0)ORa, -NRaRb, or -C02Ra;
Ra and Rb are each independently hydrogen, (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-C8)alkynyl, (C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (C6-Ci0)bicycloalkyl,
heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2- Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2, -CONH(Ci-C4)alkyl, -
CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2,-S02NH(Ci-C4)alkyl, and -S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
or Ra and Rb taken together with the nitrogen to which they are attached represent a
5- 8 membered saturated or unsaturated ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C4)alkyl, (Ci- C4)haloalkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, hydroxyl, oxo, (Ci-C4)alkoxy, and (Ci-C4)alkoxy(Ci-C4)alkyl, wherein said ring is optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
or Ra and Rb taken together with the nitrogen to which they are attached represent a
6- to 10-membered bridged bicyclic ring system optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring. An aryl or heteroaryl group in this particular subgroup A is selected independently from the group consisting of furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, oxadiazole, thiadiazole, triazole, tetrazole, benzofuran, benzothiophene, benzoxazole, benzothiazole, phenyl, pyridine, pyridazine, pyrimidine, pyrazine, triazine, tetrazine, quinoline, cinnoline, quinazoline, quinoxaline, and na hthyridine or another aryl or heteroaryl group as follows: 
wherein in (1),
A is O, NH, or S; B is CH or N, and C is hydrogen or Ci-Cg alkyl; or 
wherein in (2),
D is N or C optionally substituted by hydrogen or Ci-Cg alkyl; or 
wherein in (3),
E is NH or CH2; F is O or CO; and G is NH or CH2; or 
wherein in (4),
J is O, S or CO; or 
wherein in (5),
Q is CH or N;
M is CH or N; and L/(5) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, -CORa, - C02Ra, -CONRaRb, -CONRaNRaRb, -S02Ra, -S02NRaRb, -NRaRb, -NRaC(0)Rb - NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa, wherein any (Ci-Cg)alkyl, (C3-C8)cycloalkyl, group is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, - NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)Ra, -OC(0)NRaRb; wherein RaandRb are defined as above; or

L/(6) is NH or CH2; or

wherein in 7,
M/(7) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl, -CORa, -C02Ra, -CONRaRb, -CONRaNRaRb, -S02Ra, - S02NRaRb, -NRaRb, -NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, - NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa,
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, heterocycloalkyl group is optionally substituted by 1 , 2 or 3 groups independently selected from
(Ci-C6)alkyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, - CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb; wherein RaandRb are defined as above; or 
wherein in (8),
P is CH2, NH, O, or S; Q/(8) is CH or N; and n is 0-2; or

wherein in (9),
S/(9) and T(9) is C, or S/(9) is C and T(9) is N, or S/(9) is N and T/(9) is C;
R is hydrogen, amino, methyl, trifluoromethyl, halo;
U is hydrogen, halo, amino, cyano, nitro, trifluoromethyl, (Ci-Cg)alkyl, (C3- C8)cycloalkyl, -CORa, -C02Ra, -CONRaRb, -S02Ra, -S02NRaRb, -NRaRb, - NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, , -ORa, 4-( 1 H- pyrazol-4-yl),
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, group is optionally
substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, -CORa, - C02Ra,-CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -
NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb; wherein Ra andRb are defined as above.
3. A compound of formula (I) according to claim 1 or 2 wherein:
X and Z are selected independently from the group consisting of (Ci-Cg)alkyl, (C3-
C8)cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NRaRb, and -ORa;
Y is H;
R1 is (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, or heterocycloalkyl;
R3 is hydrogen, (Ci-Cg)alkyl or halo;
R6 is hydrogen, halo, cyano, trifluoromethyl, amino, (Ci-Cg)alkyl, (C3- Cg)cycloalkyl;, aryl, heteroaryl, acylamino; (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl; -S02Ra; -S02NRaRb, or -NRaS02Rb; wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl group is optionally substituted by 1 , 2 or 3 groups independently selected from halo, (Ci-C6)alkyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl,
(Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, - S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -
NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)Ra, -OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl, aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
Ra and Rb are each independently hydrogen, (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-Cg)alkynyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (C6-Cio)bicycloalkyl,
heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-
Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2,-CONH(Ci-C4)alkyl, - CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2, -S02NH(Ci-C4)alkyl, and -S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
or Ra and Rb taken together with the nitrogen to which they are attached represent a 5-8 membered saturated or unsaturated ring, optionally containing an additional heteroatom selected from oxygen, nitrogen, and sulfur, wherein said ring is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C4)alkyl, (Ci-
C4)haloalkyl, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, hydroxyl, oxo, (Ci-C4)alkoxy, and (Ci-C4)alkoxy(Ci-C4)alkyl, wherein said ring is optionally fused to a (C3-Cg)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring;
or Ra and Rb taken together with the nitrogen to which they are attached represent a 6- to 10-membered bridged bicyclic ring system optionally fused to a (C3-C8)cycloalkyl, heterocycloalkyl, aryl, or heteroaryl ring. Aryl and heteroaryl in this definition are selected from the group consisting of furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, oxadiazole, thiadiazole, triazole, tetrazole, benzofuran,
benzothiophene, benzoxazole, benzothiazole, phenyl, pyridine, pyridazine, pyrimidine, pyrazine, triazine, tetrazine, quinoline, cinnoline, quinazoline, quinoxaline, and naphthyridine as or a compound of or another aryl or heteroaryl group as follows: 
wherein in (1),
A is O, NH, or S; B is CH or N, and C is hydrogen or Ci-Cg alkyl; or 
wherein in (2),
D is N or C optionally substituted by hydrogen or Ci-C8 alkyl; or 
wherein in (3),
E is NH or CH2; F is O or CO; and G is NH or CH2; or 
wherein in (4),
J is O, S or CO; or 
wherein in (5),
Q is CH or N;
M is CH or N; and
L/(5) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-C8)cycloalkyl, -COR'
C02Ra, -CONRaRb, -CONRaNRaRb, -S02R -S02NRaRb, -NRaRb, -NRaC(0)Rb,-
NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa, wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, group is optionally substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, - NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)R -OC(0)NRaRb,
wherein Ra and Rb are defined as above; or

wherein in (6),
L/ 6) is NH or CH2; or 
wherein in (7),
M/(7) is hydrogen, halo, amino, cyano, (Ci-Cg)alkyl, (C3-C8)cycloalkyl, heterocycloalkyl, -CORa, -C02Ra, -CONRaRb, -CONRaNR > aaRr> bD, -S02Ra, - S02NRaRb, -NRaRb, -NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, - NRaNRaC(0)Rb, -NRaNRaC(0)NRaRb, -ORa,
wherein any (Ci-Cg)alkyl, (C3-C8)cycloalkyl, heterocycloalkyl group is optionally substituted by 1 , 2 or 3 groups independently selected from
(Ci-C6)alkyl, (C3-Cg)cycloalkyl, (C5-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano,
CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02NRaRb, nitro, -NRaRb,
NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, ORa -OC(0)Ra, -OC(0)NRaRb; wherein RaandRb are defined as above; or

wherein in (8),
P is CH2, NH, O, or S; Q/(8) is CH or N; and n is 0-2; or

S/(9) and T(9) is C, or S/(9) is C and T(9) is N, or S/(9) is N and T/(9) is C;
R is hydrogen, amino, methyl, trifluoromethyl, halo;
U is hydrogen, halo, amino, cyano, nitro, trifluoromethyl, (Ci-Cg)alkyl, (C3- C8)cycloalkyl, -CORa, -C02Ra, -CONRaRb, -S02Ra, -S02NRaRb, -NRaRb, - NRaC(0)Rb,-NRaS02Rb, -NRaS02NRaRb, -NRaNRaRb, -NRaNRaC(0)Rb, , -ORa, 4-(lH- pyrazol-4-yl),
wherein any (Ci-Cg)alkyl, (C3-C8)cycloalkyl, group is optionally
substituted by 1, 2 or 3 groups independently selected from (Ci-C6)alkyl,
(C3-Cg)cycloalkyl,  (Ci-Ce)haloalkyl, cyano, -CORa, - C02Ra,-CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb, wherein Ra andRb are defined as above;
(Ci-Ce)haloalkyl, cyano, -CORa, - C02Ra,-CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb, wherein Ra andRb are defined as above;
or a salt thereof.
4. A compound according to any one of claims 1, 2 or 3 wherein:
X is methyl, ethyl, n-propyl, isopropyl , cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, trifluoromethyl, tetrahydropyran, hydroxymethyl, methoxymethyl, or benzyl;
Y is H;
Z is methyl, ethyl, n-propyl, isopropyl, trifluoromethyl, or benzyl;
R1 is isopropyl, tert-butyl, cyclobutyl, cyclopentyl, cyclohexyl, (1- methylethyl)cyclopropyl, 1 , 1 -dioxo-tetrahydrothiophene-3-yl, 1 -Me-piperidin-4-yl, tetrahydrofuran-3-yl, tetrahydropyran-4-yl, N,N-dimethyl-l-propanaminyl, benzyl, or 4- pyridyl;
R3 is H, methyl, or Br; and
R6 is methyl, bis(l,l-dimethylethyl), bis(l-methylethyl), cyclopropyl, propyl, dimethylamino, ethylamino, (2-hydroxyethyl)amino, 2-propen-l-ylamino, 1-piperazinyl, 1-piperidinyl, 4-morpholinyl, 4-piperidinylamino, tetrahydro-2H-pyran-4-ylamino, phenylamino, (phenylmethyl)amino, (4-pyridinylmethyl)amino, [2-(2- pyridinylamino)ethyl] amino, 2-(dimethylamino)ethyl]amino, 4-pyridinylamino , 4- (aminocarbonyl)phenyl]amino, 3 -hydroxy-3 -methyl- 1 -butyn- 1 -yl, 4-pyridinylethynyl, phenylethynyl, 2-furanyl, 3-thienyl; lH-pyrazol-4-yl, lH-indazol-5-yl, lH-indazol-6-yl, 3- methyl- lH-indazol-5-yl, lH-1 ,2,3-benzotriazol-5-yl, 2-oxo-2,3-dihydro-lH-benzimidazol- 5-yl, 2-oxo-2,3-dihydro-lH-indol-5-yl, 2-oxo-2,3-dihydro-lH-indol-6-yl, 2,1,3- benzoxadiazol-5-yl, 2-amino-6-quinazolinyl, 2,4-dioxo-l,2,3,4-tetrahydro-5-pyrimidinyl, 2-amino-5-pyrimidinyl, 7-oxo-l,5,6,7-tetrahydro-l,8-naphthyridin-3-yl, phenyl, 2- methylphenyl, 2-nitrophenyl, 2-phenylethyl, 3-aminophenyl, 4-aminophenyl, 4- chlorophenyl, 4-fluorophenyl, 4-(methyloxy)phenyl, 3-(acetylamino)phenyl, 4- (acetylamino)phenyl, 4-(aminocarbonyl)phenyl, 4-(lH-pyrazol-4-yl)phenyl, 4- (aminosulfonyl)phenyl, 4-(methylsulfonyl)phenyl, 4-[(dimethylamino)sulfonyl]phenyl, 4- [(methylamino)carbonyl]phenyl, 4- [(methylamino)sulfonyl]phenyl, 4- [(methylsulfonyl)amino]phenyl, 3-pyridinyl, 4-pyridinyl, 2-(4-morpholinyl)-4-pyridinyl, 2-amino-4-pyridinyl, 5-(methyloxy)-3-pyridinyl, 5-(methylsulfonyl)-3-pyridinyl, 5- [(cyclopropylsulfonyl)amino]-6-(methyloxy)-3-pyridinyl, 5-[(phenylsulfonyl)amino]-3- pyridinyl, 6-(4-methyl-l-piperazinyl)-3-pyridinyl, 6-(4-morpholinyl)-3-pyridinyl, 6- (acetylamino)-3-pyridinyl, 6-(dimethylamino)-3-pyridinyl, 6-(methyloxy)-3-pyridinyl, 6- [(methylamino)carbonyl] -3 -pyridinyl, 6- [(methylamino)sulfonyl] -3 -pyridinyl, 6-methyl-3 - pyridinyl, 4-pyridinyloxy;
or a salt thereof.
5. A compound of formula (I) according to claim 1 or 2 wherein:
X is (Ci-C8)alkyl or (C3-C8)cycloalkyl;
Y is H;
Z is (Ci-C3)alkyl;
R1 is (C3-Cg)alkyl, (C3-Cg)cycloalkyl, or heterocycloalkyl;
R3 is hydrogen, (Ci-C3)alkyl or halo;
R6 is selected from the group consisting of hydrogen, halo, cyano, trifluoromethyl, amino, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, aryl, heteroaryl, acylamino, (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl, -S02Ra; -S02NRaRb , and -NRaS02Rb ;
wherein any (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl group is optionally substituted by 1 , 2 or 3 groups independently selected from -0(Ci-C6)alkyl(Rc)i_2, -S(Ci-C6)alkyl(Rc)i_2, -(Ci-C6)alkyl(Rc)i_2, (Ci-Cg)alkyl-heterocycloalkyl, (C3-Cg)cycloalkyl-heterocycloalkyl, halo,
(Ci-C6)alkyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (Ci-Ce)haloalkyl, cyano, - CORa, -C02Ra, -CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, - NRaC(0)Rb, -NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, - ORa, -OC(0)Ra, -OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl,
aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl;
Ra and Rb are each independently hydrogen, (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-Cg)alkynyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (C6-Cio)bicycloalkyl, heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2- Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2, -CONH(Ci-C4)alkyl, -
CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2,-S02NH(Ci-C4)alkyl, and -S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
each Rc is independently (Ci-C4)alkylamino, -NRaS02Rb, -SORa, -S02Ra, - NRaC(0)ORa, -NRaRb, or -C02Ra;
or a pharmaceutically acceptable salt thereof.
6. A compound according to any preceeding claim wherein R6 is selected from the group consisting of: halo, cyano, trifiuoromethyl, amino, (Ci-Cg)alkyl, (C3-Cg)cycloalkyl, acylamino, (C2-Cg)alkynyl, arylalkynyl, heteroarylalkynyl, -S02Ra; -S02NRaRb , and - NRaS02Rb ; or a pharmaceutically acceptable salt thereof.
7. A compound according to any preceeding claim wherein R6 is aryl or heteroaryl, wherein said aryl or heteroaryl is optionally substituted by 1 , 2 or 3 groups independently selected from -0(Ci-C6)alkyl(Rc)i_2, -S(Ci-C6)alkyl(Rc)i_2, -(Ci-C6)alkyl(Rc)i_2, (d- C8)alkyl-heterocycloalkyl, (C3-Cg)cycloalkyl-heterocycloalkyl, halo, (Ci-C6)alkyl, (C3-C8)cycloalkyl, (C5-C8)cycloalkenyl, (Ci-C6)haloalkyl, cyano, -CORa, -C02Ra, - CONRaRb, -SRa, -SORa, -S02Ra, -S02NRaRb, nitro, -NRaRb, -NRaC(0)Rb, - NRaC(0)NRaRb, -NRaC(0)ORa, -NRaS02Rb, -NRaS02NRaRb, -ORa, -OC(0)Ra, - OC(0)NRaRb, heterocycloalkyl, aryl, heteroaryl, aryl(d-C4)alkyl, and
heteroaryl(Ci-C4)alkyl;
Ra and Rb are each independently hydrogen, (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-Cg)alkynyl, (C3-Cg)cycloalkyl, (Cs-Cg)cycloalkenyl, (C6-Cio)bicycloalkyl, heterocycloalkyl, aryl, heteroaryl, wherein said (Ci-Cg)alkyl, (C2-C8)alkenyl, (C2- Cg)alkynyl, cycloalkyl, cycloalkenyl, bicycloalkyl, heterocycloalkyl ,aryl or heteroaryl group is optionally substituted by 1, 2 or 3 groups independently selected from halo, hydroxyl, (Ci-C4)alkoxy, amino, (Ci-C4)alkylamino, ((Ci-C4)alkyl)((Ci-C4)alkyl)amino, - C02H, -C02(Ci-C4)alkyl, -CONH2, -CONH(Ci-C4)alkyl, -
CON((Ci-C4)alkyl)((Ci-C4)alkyl), -S02(Ci-C4)alkyl, -S02NH2,-S02NH(Ci-C4)alkyl, and -S02N((Ci-C4)alkyl)((Ci-C4)alkyl);
each Rc is independently (Ci-C4)alkylamino, -NRaS02Rb, -SORa, -S02Ra, - NRaC(0)ORa, -NRaRb, or -C02Ra;
or a pharmaceutically acceptable salt thereof.
8. A compound according to any preceeding claim which is:
6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
6-chloro-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l- methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-chloro- 1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- pyridinyloxy)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2- propen- 1 -ylamino)- lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
6-amino-N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 -methylethyl)-N-[(4-methyl-2-oxo-6-propyl- 1 ,2-dihydro-3 - pyridinyl)methyl] - lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 -methylethyl)-N- { [6-methyl-4-( 1 -methylethyl)-2-oxo- 1 ,2- dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -(1 -methylethyl)-N-[(4-methyl-2-oxo-6-propyl- 1 ,2-dihydro-3-pyridinyl)methyl]- 6-(4-pyridinyl)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide;
N- [(6-ethyl-4-methyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)-6- (4-pyridinyl)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide;
1 -(1 -methylethyl)-N-[(6-methyl-2-oxo-4-propyl- 1 ,2-dihydro-3-pyridinyl)methyl]- 6-(4-pyridinyl)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide; 1 -( 1 -methylethyl)-N- { [6-methyl-4-( 1 -methylethyl)-2-oxo- 1 ,2-dihydro-3 - pyridinyl]methyl}-6-(4-pyridinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 -methylethyl)-N-[(6-methyl-2-oxo-4-propyl- 1 ,2-dihydro-3 - pyridinyl)methyl] - lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
l-(l-methylethyl)-N-[(6-methyl-2-oxo-4-phenyl-l,2-dihydro-3-pyridinyl)meth 6-(4-pyridinyl)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide;
N-[(4-cyclohexyl-6-methyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4-cyclohexyl-6-methyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl] -6-cyclopropyl l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 -methylethyl)-N-[(6-methyl-2-oxo-4-phenyl- 1 ,2-dihy dro-3 - pyridinyl)methyl] - lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
N~ [(4-cyclopropyl-6-methyl-2-oxo- 1 ,2-dihy dro-3 -pyridinyl)methyl] - 1 -( 1 - methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(6-cyclopropyl-4-methyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N- [(5 -fluoro-4,6-dimethyl-2-oxo- 1 ,2-dihy dro-3 -pyridinyl)methyl] - 1 -( 1 - methylethyl)-6-(4-pyridinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N- [(4,6-dimethyl-2-oxo- 1 ,2-dihy dro-3 -pyridinyl)methyl] -6-(ethylamino)- 1 -( 1 - methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)- 1Η- pyrazolo[3,4-b]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 -methylethyl)-N- { [6-methyl-2-oxo-4-(trifluoromethyl)- 1,2- dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
6-(dimethylamino)-N- [(4,6-dimethyl-2-oxo- 1 ,2-dihy dro-3 -pyridinyl)methyl] - 1 -( 1 methylethyl)- lH-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-(l - piperidinyl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-(4- morpholinyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l- methylethyl)-6-[6-(4-methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4-¾]pyridine-4- carboxamide; N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-(l - piperazinyl)-l H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(3-hydroxy-3-methyl- 1 -butyn- 1 -yl)- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-(3- methyl- 1 H-indazol-5 -yl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6- (phenylethynyl)-l H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2- phenylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-(4- pyridinylethynyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6- (phenylamino)-l H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6- [(phenylmethyl)amino]-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-methyl-l-(l- methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l,6-bis(l-methylethyl)- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6- phenyl-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(4-fluorophenyl)-l-(l methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-{4-[(dimethylamino)sulfonyl]phenyl}-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-[6-(dimethylamino)-3-pyridinyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]- 1 -(1 -methylethyl)- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-6- {4- [(methylamino)sulfonyl]phenyl} - 1 -( 1 -methylethyl)- lH-pyrazolo[3 ,4-¾]pyridine-4- carboxamide;
6-(4-aminophenyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; 6-[4-(acetylamino)phenyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(3-aminophenyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-6- {4- [(methylamino)carbonyl]phenyl} - 1 -( 1 -methylethyl)- lH-pyrazolo[3 ,4-¾]pyridine-4- carboxamide;
6-[3-(acetylamino)phenyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(2,4-dioxo-l, 2,3,4- tetrahydro-5-pyrimidinyl)-l-(l -methylethyl)- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(2-amino-4-pyridinyl)-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]- l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-[4-(aminosulfonyl)phenyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6- {4- [(methylsulfonyl)amino]phenyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-{4-[(dimethylamino)sulfonyl]phenyl}-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl] -3 -methyl- 1 -( 1 -methylethyl)- lH-pyrazolo [3 ,4-¾]pyridine-4- carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- piperidinylamino)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6- (tetrahydro-2H-pyran-4-ylamino)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)-6- [(4- pyridinylmethyl)amino]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-{[2-(dimethylamino)ethyl]amino}-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-[(2- hydroxyethyl)amino]- 1 -( 1 -methylethyl)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-[5- (methyloxy)-3-pyridinyl]-lH-pyrazolo[3,4-b]pyridine-4-carboxamide; N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-[2- (methyloxy)-4-pyridinyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(6-amino-3-pyridinyl)-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-
1- (l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)-6- [5 - (methylsulfonyl)-3 -pyridinyl] - lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(2-furanyl)-l-(l- methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-6- {6- [(methylamino)carbonyl]-3-pyridinyl}-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4- carboxamide;
6- [5 - [(eye lopropylsulfonyl)amino] -6-(methyloxy)-3 -pyridinyl] -N-[(4,6-dimethyl-
2- oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l -methylethyl)- 1 H-pyrazolo [3, 4-b]pyridine-4- carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6- {5- [(phenylsulfonyl)amino]-3-pyridinyl}-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-[2- (4-morpholinyl)-4-pyridinyl]-l H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-[6- (4-morpholinyl)-3 -pyridinyl] -1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-[6- (methyloxy)-3 -pyridinyl] -1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
6-[6-(acetylamino)-3-pyridinyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]- 1 -( 1 -methylethyl)- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2- methylphenyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl]- 1 -( 1 -methylethyl)-6-( 1 H- pyrazol-4-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(2-amino-5-pyrimidinyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 - methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(3- pyridinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(lH-indazol-5-yl)-i
methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-(lH-indazol-6-yl)-l-(l- methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(lH-l,2,3-benzotriazol-5-yl)-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl] l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l-meth^
(2-oxo-2,3-dihydro-lH-benzimidazol-5-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(^ oxo-2,3-dihydro-lH-indol-6-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(^ oxo-2,3-dihydro-lH-indol-5-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(2-^ dihydro-lH-benzimidazol-5-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[6-(^ methyl- 1 -piperazinyl)-3 -pyridinyl] - 1 H-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-^
(4-methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l- methylethyl)-6-[6-(4-morpholinyl)-3-pyridinyl]-lH-pyrazolo[3,4-¾]pyridine-4- carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-^
pyridinylamino)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide;
N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)-6-[(2-oxo-2,3- dihydro-lH-benzimidazol-5-yl)amino]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-{[4-(aminocarbonyl)phenyl]amino}-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]- 1 -(1 -methylethyl)- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3- methyl-6-[6-(4-methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4-¾]pyridine-4- carboxamide;
1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-(2-oxo-2,3-dihydro-lH-benzimidazol-5-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; 1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-[6-(4-morpholinyl)-3-pyridinyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(2, 1 ,3-benzoxadiazol-5-yl)-N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-3- methyl- 1 -( 1 -methylethyl)- lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
6-(2-amino-6-quinazolinyl)-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3- methyl- 1 -( 1 -methylethyl)- lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-6-{4- [(methylamino)sulfonyl]phenyl} - 1 -( 1 -methylethyl)- lH-pyrazolo[3 ,4-¾]pyridine-4- carboxamide;
6-[4-(acetylamino)phenyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl] -3 -methyl- 1 -( 1 -methylethyl)- lH-pyrazolo [3 ,4-¾]pyridine-4- carboxamide;
6-[4-(aminocarbonyl)phenyl]-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl]- 1 -(1 -methylethyl)- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-{[2- (2-pyridinylamino)ethyl]amino}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(7- oxo-l,5,6,7-tetrahydro-l,8-naphthyridin-3-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-6- {6- [(methylamino)sulfonyl]-3-pyridinyl}-l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4- carboxamide;
l-(2-aminoethyl)-6-cyclopropyl-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3- pyridinyl)methyl] - lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-{2- [(methylsulfonyl)amino]ethyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-6-methyl-l- (phenylmethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -ethyl- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -propyl- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl- l-(l-methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; 1 -amino-6-cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -cyclobutyl-6-cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
1 -cyclopentyl-6-cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]- 1 -(4- pyridinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 -cyclopropylethyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
1 -cyclohexyl-6-cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl- l-(l-methyl-4-piperidinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl- l-(tetrahydro-2H-pyran-4-yl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[4- (methyloxy)phenyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(4- pyridinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(4-chlorophenyl)-N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l- methylethyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-(3- thienyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-3-methyl-l-(l-methylethyl)-6- (4-pyridinyl)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide;
1 ,6-bis(l , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridinyl)methyl]-l-(l-methylethyl)-6-[4- (methylsulfonyl)phenyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] - 1 -( 1 -methylethyl)-6-[4-(lH- pyrazol-4-yl)phenyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3- methyl-6- [4-(methyloxy)phenyl] - IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
1 -(1 , 1 -dimethylethyl)-N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-(3 -pyridinyl)- lH-pyrazolo[3 ,4-£]pyridine-4-carboxamide;
3 -bromo-N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] - 1 -( 1 - methylethyl)-6-phenyl-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l,2-dihydro-3-pyridiny
nitrophenyl)- lH-pyrazolo[3 ,4-¾]pyridine-4-carboxamide;
N-[(4,6-dimethyl-2-oxo-l ,2-dihydro-3-pyridinyl)methyl]- 1 -(1 -methylethyl)-6-propyl- 1H- pyrazolo[3,4-¾]pyridine-4-carboxamide;
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- (pyridin-3-yl)-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- phenyl-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)-6-(4- ((dimethylamino)methyl)phenyl)- 1 -isopropyl-3 -methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide;
N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)-6-(4-fluorophenyl)- 1 - isopropyl-3 -methyl- 1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
1 -isopropyl-N-((6-methyl-2-oxo-4-propyl- 1 ,2-dihydropyridin-3-yl)methyl)-6- phenyl-1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-(2-methyl-5-pyrimidinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-[4-(methyloxy)phenyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(41 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-3-methyl-6-[6-(4-methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4- ¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(41 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl-6- [6-(4-morpholinyl)-3 -pyridinyl] - IH-pyrazolo [3 ,4- ¾]pyridine-4-carboxamide; 6-cyclopropyl- 1 -( 1 , 1 -dimethylethy l)-3 -methyl-N- [(6-methyl-2-oxo-4-propyl- 1,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; l,6-dicyclopropyl-3-methyl-N-[(6-methyl-2-oxo-4-propyl-l,2-dihydro-3- pyridinyl)methyl] - lH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
1 ,6-dicyclopropyl-N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] -3 - methyl- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(cyclopropylamino)-N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)- 1 - isopropyl-3 -methyl- lH-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-((4-benzyl-6-methyl-2-oxo- 1 ,2-dihydropyridin-3 -y l)methyl)- 1 -isopropyl- 1 H- pyrazolo[3,4-b]pyridine-4-carboxamide;
N-((4-benzyl-6-methyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)- 1 -isopropyl-3 - methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4-cyclopropyl-6-methyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- 1 -( 1 -methy lethyl)- lH-pyrazolo [3 ,4-¾]pyridine-4- carboxamide;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N- { [6-methyl-4-( 1 -methylethyl)-2-oxo- l,2-dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide ;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N-[(6-methyl-2-oxo-4-propyl- 1 ,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide ;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N- { [6-methyl-2-oxo-4-(phenylmethyl)- l,2-dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; or a pharmaceutically acceptable salt thereof.
9. A compound according to any one of the proceeding claims, which is
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- (pyridin-3-yl)-l H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-l-isopropyl-3-methyl-6- phenyl-1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)-6-(4- ((dimethylamino)methyl)phenyl)- 1 -isopropyl-3 -methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4- carboxamide;
N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)-6-(4-fluorophenyl)- 1 - isopropyl-3 -methyl- 1 H-pyrazolo [3, 4-b]pyridine-4-carboxamide; 1 -isopropyl-N-((6-methyl-2-oxo-4-propyl- 1 ,2-dihydropyridin-3-yl)methyl)-6- phenyl-lH-pyrazolo[3,4-b]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-(2-methyl-5-pyrimidinyl)-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3-pyridinyl)methyl]-3-methyl- 6-[4-(methyloxy)phenyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(41 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl]-3-methyl-6-[6-(4-methyl-l-piperazinyl)-3-pyridinyl]-lH-pyrazolo[3,4- ¾]pyridine-4-carboxamide;
1 -cyclopropyl-N-[(41 -cyclopropyl-N-[(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl-6- [6-(4-morpholinyl)-3 -pyridinyl] - IH-pyrazolo [3 ,4- ¾]pyridine-4-carboxamide;
6-cyclopropyl- 1 -( 1 , 1 -dimethylethy l)-3 -methyl-N- [(6-methyl-2-oxo-4-propyl- 1,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; l,6-dicyclopropyl-3-methyl-N-[(6-methyl-2-oxo-4-propyl-l,2-dihydro-3- pyridinyl)methyl] - IH-pyrazolo [3 ,4-¾]pyridine-4-carboxamide;
1 ,6-dicyclopropyl-N- [(4,6-dimethyl-2-oxo- 1 ,2-dihydro-3 -pyridinyl)methyl] -3 - methyl- lH-pyrazolo[3,4-¾]pyridine-4-carboxamide;
6-(cyclopropylamino)-N-((4,6-dimethyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)- 1 isopropyl-3 -methyl- IH-pyrazolo [3, 4-b]pyridine-4-carboxamide;
N-((4-benzyl-6-methyl-2-oxo- 1 ,2-dihydropyridin-3 -y l)methyl)- 1 -isopropyl- 1 H- pyrazolo[3,4-b]pyridine-4-carboxamide;
N-((4-benzyl-6-methyl-2-oxo- 1 ,2-dihydropyridin-3-yl)methyl)- 1 -isopropyl-3 - methyl- 1 H-pyrazolo [3 ,4-b]pyridine-4-carboxamide;
6-cyclopropyl-N-[(4-cyclopropyl-6-methyl-2-oxo- 1 ,2-dihydro-3- pyridinyl)methyl] -3 -methyl- 1 -( 1 -methy lethyl)- IH-pyrazolo [3 ,4-¾]pyridine-4- carboxamide;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N- { [6-methyl-4-( 1 -methylethyl)-2-oxo l,2-dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide ;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N-[(6-methyl-2-oxo-4-propyl- 1 ,2- dihydro-3-pyridinyl)methyl]-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide ;
6-cyclopropyl-3 -methyl- 1 -( 1 -methylethyl)-N- { [6-methyl-2-oxo-4-(phenylmethyl) l,2-dihydro-3-pyridinyl]methyl}-lH-pyrazolo[3,4-¾]pyridine-4-carboxamide; or a pharmaceutically acceptable salt thereof.
10. A method of treating cancer comprising administering to a patient with cancer a therapeutically effective amount of a compound of formula (I) according to claim 1 or a pharmaceutically acceptable salt thereof, alone or admixed with a pharmaceutically acceptable carrier.
11. A pharmaceutically acceptable preparations comprising a compound of formula (I) according to claim 1 and pharmaceutically acceptable excipient.
12. A method of claim 10, wherein said cancer is selected from the group consisting of: brain (gliomas), glioblastomas, leukemias, lymphomas, Bannayan-Zonana syndrome, Cowden disease, Lhermitte-Duclos disease, breast, inflammatory breast cancer, Wilm's tumor, Ewing's sarcoma, Rhabdomyosarcoma, ependymoma, medulloblastoma, colon, gastric, bladder, head and neck, kidney, lung, liver, melanoma, renal, ovarian, pancreatic, prostate, sarcoma, osteosarcoma, giant cell tumor of bone and thyroid.
13. The use of a compound of formula (I) according to claim 1 or a pharmaceutically acceptable salt or solvate thereof, in the preparation of a medicament for use in the treatment of a disorder mediated by inhibiting EZH2.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/344,110 US9073924B2 (en) | 2011-09-13 | 2012-09-12 | Azaindazoles |
| EP12832131.2A EP2755962B1 (en) | 2011-09-13 | 2012-09-12 | Azaindazoles |
| ES12832131.2T ES2624986T3 (en) | 2011-09-13 | 2012-09-12 | Azaindazoles |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161534038P | 2011-09-13 | 2011-09-13 | |
| US61/534,038 | 2011-09-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013039988A1 true WO2013039988A1 (en) | 2013-03-21 |
Family
ID=47883669
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/054785 WO2013039988A1 (en) | 2011-09-13 | 2012-09-12 | Azaindazoles |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9073924B2 (en) |
| EP (1) | EP2755962B1 (en) |
| ES (1) | ES2624986T3 (en) |
| WO (1) | WO2013039988A1 (en) |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014062720A3 (en) * | 2012-10-15 | 2014-06-19 | Epizyme, Inc. | Methods of treating cancer |
| JP2015506985A (en) * | 2012-02-10 | 2015-03-05 | コンステレーション・ファーマシューティカルズ・インコーポレイテッドConstellation Pharmaceuticals,Inc. | Methyl group-modifying enzyme regulator, composition and use thereof |
| WO2015077193A1 (en) * | 2013-11-19 | 2015-05-28 | Bristol-Myers Squibb Company | Inhibitors of lysine methyl transferase |
| US9051269B2 (en) | 2011-11-18 | 2015-06-09 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US20150344459A1 (en) * | 2012-12-21 | 2015-12-03 | Epizyme, Inc. | 1,4-pyridone bicyclic heteroaryl compounds |
| WO2015153959A3 (en) * | 2014-04-04 | 2015-12-03 | The Regents Of The University Of Michigan | Small molecule inhibitors of mcl-1 and uses thereof |
| CN105503863A (en) * | 2015-12-11 | 2016-04-20 | 南京华威医药科技开发有限公司 | Novel anti-tumor compound |
| WO2016102493A1 (en) * | 2014-12-22 | 2016-06-30 | Bayer Pharma Aktiengesellschaft | Imidazopyridine ezh2 inhibitors |
| WO2016101956A2 (en) | 2014-12-23 | 2016-06-30 | University Of Copenhagen | Treatment of cancer by inhibiting ezh2 activity |
| US9527837B2 (en) | 2014-12-05 | 2016-12-27 | Eli Lilly And Company | Inhibitors of EZH2 |
| US9718838B2 (en) | 2015-08-27 | 2017-08-01 | Eli Lilly And Company | Inhibitors of EZH2 |
| CN107105651A (en) * | 2014-11-06 | 2017-08-29 | 达纳-法伯癌症研究所股份有限公司 | Ezh2 inhibitor and application thereof |
| US9745305B2 (en) | 2013-03-15 | 2017-08-29 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| CN107428742A (en) * | 2015-11-19 | 2017-12-01 | 江苏恒瑞医药股份有限公司 | Benzofuran derivative, its preparation method and its application in medicine |
| WO2017221100A1 (en) * | 2016-06-20 | 2017-12-28 | Novartis Ag | Imidazopyrimidine compounds useful for the treatment of cancer |
| US9944652B2 (en) | 2013-05-02 | 2018-04-17 | The Regents Of The University Of Michigan | Deuterated amlexanox |
| US9969716B2 (en) | 2013-08-15 | 2018-05-15 | Constellation Pharmaceuticals, Inc. | Indole derivatives as modulators of methyl modifying enzymes, compositions and uses thereof |
| EP3215160A4 (en) * | 2014-11-06 | 2018-08-08 | Dana-Farber Cancer Institute, Inc. | Use of compositions modulating chromatin structure for graft versus host disease (gvhd) |
| WO2018210302A1 (en) | 2017-05-18 | 2018-11-22 | 江苏恒瑞医药股份有限公司 | Crystal of benzofuran derivative free base and preparation method |
| US10214536B2 (en) | 2016-01-29 | 2019-02-26 | The Regents Of The University Of Michigan | Amlexanox analogs |
| US10245255B2 (en) | 2011-02-14 | 2019-04-02 | The Regents Of The University Of Michigan | Compositions and methods for the treatment of obesity and related disorders |
| WO2019091450A1 (en) | 2017-11-10 | 2019-05-16 | 江苏恒瑞医药股份有限公司 | Method for preparing benzofuran derivative |
| CN110072861A (en) * | 2016-09-07 | 2019-07-30 | Fgh生物科技公司 | For treating the disubstituted pyrazole class compound of disease |
| US10457640B2 (en) | 2016-10-19 | 2019-10-29 | Constellation Pharmaceuticals, Inc. | Synthesis of inhibitors of EZH2 |
| US10493076B2 (en) | 2015-08-24 | 2019-12-03 | Epizyme, Inc. | Method for treating cancer |
| WO2020011607A1 (en) | 2018-07-09 | 2020-01-16 | Fondation Asile Des Aveugles | Inhibition of prc2 subunits to treat eye disorders |
| US10577350B2 (en) | 2015-08-28 | 2020-03-03 | Constellation Pharmaceuticals, Inc. | Crystalline forms of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide |
| US10633371B2 (en) | 2016-04-22 | 2020-04-28 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| WO2020087170A1 (en) * | 2018-10-30 | 2020-05-07 | Repare Therapeutics Inc. | Compounds, pharmaceutical compositions, and methods of preparing compounds and of their use as atr kinase inhibitors |
| US10689378B2 (en) | 2016-06-20 | 2020-06-23 | Novartis Ag | Triazolopyridine compounds and uses thereof |
| WO2020259601A1 (en) * | 2019-06-28 | 2020-12-30 | Impact Therapeutics, Inc | Substituted fused heteroaromatic bicyclic compounds as kinase inhibitors and the use thereof |
| WO2021012049A1 (en) * | 2019-07-22 | 2021-01-28 | Repare Therapeutics Inc. | Substituted 2-morpholinopyridine derivatives as atr kinase inhibitors |
| US11091489B2 (en) | 2016-06-20 | 2021-08-17 | Novartis Ag | Crystalline forms of a triazolopyrimidine compound |
| RU2806857C2 (en) * | 2018-10-30 | 2023-11-08 | Рипэйр Терапьютикс Инк. | Compounds, pharmaceutical compositions, methods for preparing the compounds and their use as atr kinase inhibitors |
| WO2023244917A1 (en) | 2022-06-13 | 2023-12-21 | Treeline Biosciences, Inc. | 1,8-naphthyridin-2-one heterobifunctional bcl6 degraders |
| WO2023244918A1 (en) | 2022-06-13 | 2023-12-21 | Treeline Biosciences, Inc. | Quinolone bcl6 bifunctional degraders |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201318886D0 (en) * | 2013-10-25 | 2013-12-11 | Givaudan Sa | Improvements i or relating to organic compounds |
| GB201516396D0 (en) | 2015-09-16 | 2015-10-28 | Givaudan Sa | Improvements in or relating to organic compounds |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050059682A1 (en) * | 2003-09-12 | 2005-03-17 | Supergen, Inc., A Delaware Corporation | Compositions and methods for treatment of cancer |
| US20090012031A1 (en) | 2007-07-03 | 2009-01-08 | The Regents Of The University Of Michigan | EZH2 Cancer Markers |
| US20100113415A1 (en) * | 2008-05-29 | 2010-05-06 | Rajapakse Hemaka A | Epha4 rtk inhibitors for treatment of neurological and neurodegenerative disorders and cancer |
| WO2012005805A1 (en) * | 2010-05-07 | 2012-01-12 | Glaxosmithkline Llc | Azaindazoles |
| WO2012118812A2 (en) * | 2011-02-28 | 2012-09-07 | Epizyme, Inc. | Substituted 6,5-fused bicyclic heteroaryl compounds |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2889526B1 (en) | 2005-08-04 | 2012-02-17 | Aventis Pharma Sa | SUBSTITUTED 7-AZA-INDAZOLES, COMPOSITIONS CONTAINING SAME, PROCESS FOR PRODUCTION AND USE |
-
2012
- 2012-09-12 WO PCT/US2012/054785 patent/WO2013039988A1/en active Application Filing
- 2012-09-12 EP EP12832131.2A patent/EP2755962B1/en active Active
- 2012-09-12 ES ES12832131.2T patent/ES2624986T3/en active Active
- 2012-09-12 US US14/344,110 patent/US9073924B2/en not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050059682A1 (en) * | 2003-09-12 | 2005-03-17 | Supergen, Inc., A Delaware Corporation | Compositions and methods for treatment of cancer |
| US20090012031A1 (en) | 2007-07-03 | 2009-01-08 | The Regents Of The University Of Michigan | EZH2 Cancer Markers |
| US20100113415A1 (en) * | 2008-05-29 | 2010-05-06 | Rajapakse Hemaka A | Epha4 rtk inhibitors for treatment of neurological and neurodegenerative disorders and cancer |
| WO2012005805A1 (en) * | 2010-05-07 | 2012-01-12 | Glaxosmithkline Llc | Azaindazoles |
| WO2012118812A2 (en) * | 2011-02-28 | 2012-09-07 | Epizyme, Inc. | Substituted 6,5-fused bicyclic heteroaryl compounds |
Non-Patent Citations (19)
| Title |
|---|
| "Cancer Principles and Practice of Oncology", 15 February 2001, LIPPINCOTT WILLIAMS & WILKINS PUBLISHERS |
| CARDOSO, C ET AL., EUROPEAN J OF HUMAN GENETICS, vol. 8, no. 3, 2000, pages 174 - 180 |
| D. G. I. KINGSTON ET AL.: "Studies in Organic Chemistry", vol. 26, 1986, ELSEVIER, article "New trends in Natural Products Chemistry", pages: 219 - 235 |
| E. L. ELIEL; S. H. WILEN; L. N. MANDER: "Stereochemistry of Organic Compounds", 1994, WILEY-INTERSCIENCE |
| EINZIG, PROC. AM. SOC. CLIN. ONCOL., vol. 20, pages 46 |
| FORASTIRE, SEM. ONCOL., vol. 20, 1990, pages 56 |
| HOLMES ET AL., J. NAT. CANCER INST., vol. 83, 1991, pages 1797 |
| IGNOFF, R.J, CANCER CHEMOTHERAPY POCKET GUIDE, 1998 |
| KEARNS, C.M., SEMINARS IN ONCOLOGY, vol. 3, no. 6, 1995, pages 16 - 23 |
| KNUTSON ET AL.: "A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells.", NATURE CHEMICAL BIOLOGY, 30 September 2012 (2012-09-30), XP055078898 * |
| KUMAR, J. BIOL, CHEM, vol. 256, 1981, pages 10435 - 10441 |
| MARKMAN ET AL., YALE JOURNAL OF BIOLOGY AND MEDICINE, vol. 64, 1991, pages 583 |
| MCGUIRE ET AL., ANN. INTERN, MED., vol. 111, 1989, pages 273 |
| SCHIFF ET AL., NATURE, vol. 277, 1979, pages 665 - 667 |
| SCHIFF ET AL., PROC. NATL, ACAD, SCI. USA, vol. 77, 1980, pages 1561 - 1565 |
| See also references of EP2755962A4 * |
| T. W. GREEN; P. G. M. WUTS: "Protecting Groups in Organic Synthesis", 1991, JOHN WILEY & SONS |
| WANI ET AL., J. AM. CHEM, SOC., vol. 93, 1971, pages 2325 |
| WOO, NATURE, vol. 368, 1994, pages 750 |
Cited By (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10245255B2 (en) | 2011-02-14 | 2019-04-02 | The Regents Of The University Of Michigan | Compositions and methods for the treatment of obesity and related disorders |
| US9051269B2 (en) | 2011-11-18 | 2015-06-09 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US9409865B2 (en) | 2011-11-18 | 2016-08-09 | Constellation_Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US10016405B2 (en) | 2012-02-10 | 2018-07-10 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US9085583B2 (en) | 2012-02-10 | 2015-07-21 | Constellation—Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US9469646B2 (en) | 2012-02-10 | 2016-10-18 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US9980952B2 (en) | 2012-02-10 | 2018-05-29 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US9371331B2 (en) | 2012-02-10 | 2016-06-21 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| USRE47428E1 (en) | 2012-02-10 | 2019-06-11 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| JP2015506985A (en) * | 2012-02-10 | 2015-03-05 | コンステレーション・ファーマシューティカルズ・インコーポレイテッドConstellation Pharmaceuticals,Inc. | Methyl group-modifying enzyme regulator, composition and use thereof |
| WO2014062720A3 (en) * | 2012-10-15 | 2014-06-19 | Epizyme, Inc. | Methods of treating cancer |
| EP3659606A1 (en) * | 2012-10-15 | 2020-06-03 | Epizyme, Inc. | Methods of treating cancer |
| US9688665B2 (en) | 2012-10-15 | 2017-06-27 | Epizyme, Inc. | Methods of treating cancer |
| US9701666B2 (en) * | 2012-12-21 | 2017-07-11 | Epizyme, Inc. | 1,4-pyridone bicyclic heteroaryl compounds |
| US10150759B2 (en) | 2012-12-21 | 2018-12-11 | Epizyme, Inc. | 1,4-pyridone bicycic heteroaryl compounds |
| US20150344459A1 (en) * | 2012-12-21 | 2015-12-03 | Epizyme, Inc. | 1,4-pyridone bicyclic heteroaryl compounds |
| US9745305B2 (en) | 2013-03-15 | 2017-08-29 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US10590142B2 (en) | 2013-05-02 | 2020-03-17 | The Regents Of The University Of Michigan | Deuterated amlexanox |
| US9944652B2 (en) | 2013-05-02 | 2018-04-17 | The Regents Of The University Of Michigan | Deuterated amlexanox |
| US9969716B2 (en) | 2013-08-15 | 2018-05-15 | Constellation Pharmaceuticals, Inc. | Indole derivatives as modulators of methyl modifying enzymes, compositions and uses thereof |
| WO2015077193A1 (en) * | 2013-11-19 | 2015-05-28 | Bristol-Myers Squibb Company | Inhibitors of lysine methyl transferase |
| US9738630B2 (en) | 2013-11-19 | 2017-08-22 | Bristol-Myers Squibb Company | Inhibitors of lysine methyl transferase |
| US9394303B2 (en) | 2014-04-04 | 2016-07-19 | The Regents Of The University Of Michigan | Small molecule inhibitors of MCL-1 and uses thereof |
| WO2015153959A3 (en) * | 2014-04-04 | 2015-12-03 | The Regents Of The University Of Michigan | Small molecule inhibitors of mcl-1 and uses thereof |
| CN107105651A (en) * | 2014-11-06 | 2017-08-29 | 达纳-法伯癌症研究所股份有限公司 | Ezh2 inhibitor and application thereof |
| AU2015342774B2 (en) * | 2014-11-06 | 2020-01-30 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| EP3885343A1 (en) | 2014-11-06 | 2021-09-29 | Dana-Farber Cancer Institute, Inc. | Indole compounds as ezh2 inhibitors and uses thereof |
| EP3215160A4 (en) * | 2014-11-06 | 2018-08-08 | Dana-Farber Cancer Institute, Inc. | Use of compositions modulating chromatin structure for graft versus host disease (gvhd) |
| EP3214935A4 (en) * | 2014-11-06 | 2018-08-29 | Dana-Farber Cancer Institute, Inc. | Ezh2 inhibitors and uses thereof |
| AU2015342774C1 (en) * | 2014-11-06 | 2021-08-05 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| US11236082B2 (en) | 2014-11-06 | 2022-02-01 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| US20190000860A1 (en) * | 2014-11-06 | 2019-01-03 | Dana-Farber Cancer Institute, Inc. | Use of compositions modulating chromatin structure for graft versus host disease (gvhd) |
| US9527837B2 (en) | 2014-12-05 | 2016-12-27 | Eli Lilly And Company | Inhibitors of EZH2 |
| WO2016102493A1 (en) * | 2014-12-22 | 2016-06-30 | Bayer Pharma Aktiengesellschaft | Imidazopyridine ezh2 inhibitors |
| WO2016101956A2 (en) | 2014-12-23 | 2016-06-30 | University Of Copenhagen | Treatment of cancer by inhibiting ezh2 activity |
| US10898490B2 (en) | 2015-08-24 | 2021-01-26 | Epizyme, Inc. | Method for treating cancer |
| US10493076B2 (en) | 2015-08-24 | 2019-12-03 | Epizyme, Inc. | Method for treating cancer |
| US11642349B2 (en) | 2015-08-24 | 2023-05-09 | Epizyme, Inc. | Method for treating cancer |
| US9718838B2 (en) | 2015-08-27 | 2017-08-01 | Eli Lilly And Company | Inhibitors of EZH2 |
| US10577350B2 (en) | 2015-08-28 | 2020-03-03 | Constellation Pharmaceuticals, Inc. | Crystalline forms of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide |
| AU2016357900B2 (en) * | 2015-11-19 | 2020-09-17 | Jiangsu Hengrui Medicine Co., Ltd. | Benzofuran derivative, preparation method thereof and use thereof in medicine |
| EP3378859A4 (en) * | 2015-11-19 | 2018-09-26 | Jiangsu Hengrui Medicine Co. Ltd. | Benzofuran derivative, preparation method thereof and use thereof in medicine |
| CN107428742A (en) * | 2015-11-19 | 2017-12-01 | 江苏恒瑞医药股份有限公司 | Benzofuran derivative, its preparation method and its application in medicine |
| US11059811B2 (en) | 2015-11-19 | 2021-07-13 | Jiangsu Hengrui Medicine Co., Ltd. | Benzofuran derivative, preparation method thereof and use thereof in medicine |
| JP2018538257A (en) * | 2015-11-19 | 2018-12-27 | ジエンス ヘンルイ メデイシンカンパニー リミテッドJiangsu Hengrui Medicine Co.,Ltd. | Benzofuran derivatives, processes for their preparation and their use in medicine |
| US10759787B2 (en) | 2015-11-19 | 2020-09-01 | Jiangsu Hengrui Medicine Co., Ltd. | Benzofuran derivative, preparation method thereof and use thereof in medicine |
| CN107428742B (en) * | 2015-11-19 | 2020-05-08 | 江苏恒瑞医药股份有限公司 | Benzofuran derivative, preparation method and medical application thereof |
| CN105503863A (en) * | 2015-12-11 | 2016-04-20 | 南京华威医药科技开发有限公司 | Novel anti-tumor compound |
| US10214536B2 (en) | 2016-01-29 | 2019-02-26 | The Regents Of The University Of Michigan | Amlexanox analogs |
| US10633371B2 (en) | 2016-04-22 | 2020-04-28 | Dana-Farber Cancer Institute, Inc. | EZH2 inhibitors and uses thereof |
| US11548897B2 (en) | 2016-06-20 | 2023-01-10 | Novartis Ag | Crystalline forms of a triazolopyrimidine compound |
| US11091489B2 (en) | 2016-06-20 | 2021-08-17 | Novartis Ag | Crystalline forms of a triazolopyrimidine compound |
| US10689378B2 (en) | 2016-06-20 | 2020-06-23 | Novartis Ag | Triazolopyridine compounds and uses thereof |
| CN109790166A (en) * | 2016-06-20 | 2019-05-21 | 诺华股份有限公司 | Imidazopyridine is used for treating cancer |
| US10676479B2 (en) | 2016-06-20 | 2020-06-09 | Novartis Ag | Imidazolepyridine compounds and uses thereof |
| JP2019524872A (en) * | 2016-06-20 | 2019-09-05 | ノバルティス アーゲー | Imidazopyrimidine compounds useful for the treatment of cancer |
| WO2017221100A1 (en) * | 2016-06-20 | 2017-12-28 | Novartis Ag | Imidazopyrimidine compounds useful for the treatment of cancer |
| CN110072861A (en) * | 2016-09-07 | 2019-07-30 | Fgh生物科技公司 | For treating the disubstituted pyrazole class compound of disease |
| US11339142B2 (en) | 2016-09-07 | 2022-05-24 | Fgh Biotech, Inc. | Di-substituted pyrazole compounds for the treatment of diseases |
| CN110072861B (en) * | 2016-09-07 | 2022-11-11 | Fgh生物科技公司 | Disubstituted pyrazoles for the treatment of diseases |
| US10457640B2 (en) | 2016-10-19 | 2019-10-29 | Constellation Pharmaceuticals, Inc. | Synthesis of inhibitors of EZH2 |
| WO2018210302A1 (en) | 2017-05-18 | 2018-11-22 | 江苏恒瑞医药股份有限公司 | Crystal of benzofuran derivative free base and preparation method |
| US11155537B2 (en) | 2017-05-18 | 2021-10-26 | Jiangsu Hengrui Medicine Co., Ltd. | Crystal of benzofuran derivative free base and preparation method |
| WO2019091450A1 (en) | 2017-11-10 | 2019-05-16 | 江苏恒瑞医药股份有限公司 | Method for preparing benzofuran derivative |
| US11390614B2 (en) | 2017-11-10 | 2022-07-19 | Jiangsu Hengrui Medicine Co., Ltd. | Method for preparing benzofuran derivative |
| WO2020011607A1 (en) | 2018-07-09 | 2020-01-16 | Fondation Asile Des Aveugles | Inhibition of prc2 subunits to treat eye disorders |
| WO2020087170A1 (en) * | 2018-10-30 | 2020-05-07 | Repare Therapeutics Inc. | Compounds, pharmaceutical compositions, and methods of preparing compounds and of their use as atr kinase inhibitors |
| RU2806857C2 (en) * | 2018-10-30 | 2023-11-08 | Рипэйр Терапьютикс Инк. | Compounds, pharmaceutical compositions, methods for preparing the compounds and their use as atr kinase inhibitors |
| CN113454080A (en) * | 2018-10-30 | 2021-09-28 | 修复治疗公司 | Compounds, pharmaceutical compositions and methods of making compounds and methods of use thereof |
| WO2020259601A1 (en) * | 2019-06-28 | 2020-12-30 | Impact Therapeutics, Inc | Substituted fused heteroaromatic bicyclic compounds as kinase inhibitors and the use thereof |
| WO2021012049A1 (en) * | 2019-07-22 | 2021-01-28 | Repare Therapeutics Inc. | Substituted 2-morpholinopyridine derivatives as atr kinase inhibitors |
| CN114174292A (en) * | 2019-07-22 | 2022-03-11 | 修复治疗公司 | Substituted 2-morpholinopyridine derivatives as ATR kinase inhibitors |
| WO2023244917A1 (en) | 2022-06-13 | 2023-12-21 | Treeline Biosciences, Inc. | 1,8-naphthyridin-2-one heterobifunctional bcl6 degraders |
| WO2023244918A1 (en) | 2022-06-13 | 2023-12-21 | Treeline Biosciences, Inc. | Quinolone bcl6 bifunctional degraders |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140343056A1 (en) | 2014-11-20 |
| EP2755962A1 (en) | 2014-07-23 |
| ES2624986T3 (en) | 2017-07-18 |
| EP2755962B1 (en) | 2017-03-01 |
| US9073924B2 (en) | 2015-07-07 |
| EP2755962A4 (en) | 2015-04-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2566479B1 (en) | Azaindazoles | |
| US9073924B2 (en) | Azaindazoles | |
| US9649307B2 (en) | Indoles | |
| EP2566328B1 (en) | Indazoles | |
| EP2646020B1 (en) | Indoles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12832131 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14344110 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012832131 Country of ref document: EP |