WO2013028471A1 - Methods and intermediates for preparing macrolactams - Google Patents
Methods and intermediates for preparing macrolactams Download PDFInfo
- Publication number
- WO2013028471A1 WO2013028471A1 PCT/US2012/051182 US2012051182W WO2013028471A1 WO 2013028471 A1 WO2013028471 A1 WO 2013028471A1 US 2012051182 W US2012051182 W US 2012051182W WO 2013028471 A1 WO2013028471 A1 WO 2013028471A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- solution
- compounds
- added
- hcv
- Prior art date
Links
- 0 CC(C)(C)[C@@](C(O)=O)NC(O[C@]1[C@@](CCCC#Cc2nc(ccc(OC)c3)c3nc2O[C@](C2)CN(*)[C@@]2C(OC)=O)C1)=O Chemical compound CC(C)(C)[C@@](C(O)=O)NC(O[C@]1[C@@](CCCC#Cc2nc(ccc(OC)c3)c3nc2O[C@](C2)CN(*)[C@@]2C(OC)=O)C1)=O 0.000 description 4
- ZLNLGHIMCOKISX-IJLUTSLNSA-N CC(C)(C)[C@@H](C(O)=O)NC(O[C@H]1[C@H](CCCC#C)C1)=O Chemical compound CC(C)(C)[C@@H](C(O)=O)NC(O[C@H]1[C@H](CCCC#C)C1)=O ZLNLGHIMCOKISX-IJLUTSLNSA-N 0.000 description 3
- ZYJYPPYJVAFFOX-DOMZBBRYSA-N CC(C)(C)OC(N(C[C@@H](C1)Oc2nc3cc(OC)ccc3nc2Cl)[C@@H]1C(OC)=O)=O Chemical compound CC(C)(C)OC(N(C[C@@H](C1)Oc2nc3cc(OC)ccc3nc2Cl)[C@@H]1C(OC)=O)=O ZYJYPPYJVAFFOX-DOMZBBRYSA-N 0.000 description 1
- NPDBDJFLKKQMCM-SCSAIBSYSA-N CC(C)(C)[C@@H](C(O)=O)N Chemical compound CC(C)(C)[C@@H](C(O)=O)N NPDBDJFLKKQMCM-SCSAIBSYSA-N 0.000 description 1
- BXBDLUGDQJEYEN-HSAIGVDASA-N CC(C)(C)[C@@H](C(O)=O)NC(O[C@H]1[C@H](CCCC#Cc2nc(ccc(OC)c3)c3nc2O[C@H](C[C@H]2C(OC)=O)CN2C(OC(C)(C)C)=O)C1)=O Chemical compound CC(C)(C)[C@@H](C(O)=O)NC(O[C@H]1[C@H](CCCC#Cc2nc(ccc(OC)c3)c3nc2O[C@H](C[C@H]2C(OC)=O)CN2C(OC(C)(C)C)=O)C1)=O BXBDLUGDQJEYEN-HSAIGVDASA-N 0.000 description 1
- WEPJLNOQGOQHBW-QVDQXJPCSA-N CC1(C)OB([C@H]2C(CCCCl)C2)OC1(C)C Chemical compound CC1(C)OB([C@H]2C(CCCCl)C2)OC1(C)C WEPJLNOQGOQHBW-QVDQXJPCSA-N 0.000 description 1
- AGAHETWGCFCMDK-UHFFFAOYSA-N COc(cc1N)ccc1N Chemical compound COc(cc1N)ccc1N AGAHETWGCFCMDK-UHFFFAOYSA-N 0.000 description 1
- CHTYMWBYHAIEOF-UHFFFAOYSA-N COc(ccc1n2)cc1nc(O)c2O Chemical compound COc(ccc1n2)cc1nc(O)c2O CHTYMWBYHAIEOF-UHFFFAOYSA-N 0.000 description 1
- LDUUPJQVWFGFFF-UHFFFAOYSA-N N#CCCCC(C1)=C1O Chemical compound N#CCCCC(C1)=C1O LDUUPJQVWFGFFF-UHFFFAOYSA-N 0.000 description 1
- LWDSJSSAQHMGHA-UHFFFAOYSA-N OC1C(CCCCl)C1 Chemical compound OC1C(CCCCl)C1 LWDSJSSAQHMGHA-UHFFFAOYSA-N 0.000 description 1
- LWDSJSSAQHMGHA-PHDIDXHHSA-N O[C@H]1[C@H](CCCCl)C1 Chemical compound O[C@H]1[C@H](CCCCl)C1 LWDSJSSAQHMGHA-PHDIDXHHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C35/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C35/02—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring monocyclic
- C07C35/04—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring monocyclic containing a three or four-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/04—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups from amines with formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/32—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C271/34—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of rings other than six-membered aromatic rings with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/14—Preparation of carboxylic acid esters from carboxylic acid halides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/013—Esters of alcohols having the esterified hydroxy group bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/02—Esters of acyclic saturated monocarboxylic acids having the carboxyl group bound to an acyclic carbon atom or to hydrogen
- C07C69/12—Acetic acid esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/16—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0808—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms, e.g. Val, Ile, Leu
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/12—Cyclic peptides with only normal peptide bonds in the ring
- C07K5/126—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- the present invention relates to process and intermediates that can be used for preparing macrolactams.
- One use of the methods and intermediates described herein is the production of macrolactam compounds able to inhibit HCV NS3 protease activity.
- HCV NS3 inhibitory compounds have therapeutic and research applications. BACKGROUND OF THE INVENTION
- HCV infection is a major health problem. HCV infection leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals.
- NS3 metalloprotease
- NS3 serine protease
- NS3 a helicase
- NS5B RNA- dependent RNA polymerase
- the NS3 protease is located in the N-terminal domain of the NS3 protein.
- NS4A provides a cofactor for NS3 activity.
- HCV protease activity include: Harper et al, WO2010011566; Liverton et al, WO2009134624;
- the present invention includes methods and intermediates for preparing macrolactams.
- One use of the methods and intermediates described herein is in the production of macrolactam compounds able to inhibit HCV NS3 protease activity.
- HCV NS3 inhibitory compounds have therapeutic and research applications.
- HCV inhibitory compound that can be produced using the method and intermediates described herein is Compound A, or a pharmaceutically salt thereof:
- a first aspect of the present invention describes a compound selected from group consisting of: , or C(0)NHCH(X)COOH; provided that X is a C 2 -C 6 alkyl, or a C3-C8 cycloalkyl and R is a Ci_ 6 alkyl or aryl,
- Compounds 6 are 7 are provided in a trans configuration.
- Additional embodiments include methods for producing Compound 6.
- Another aspect of the present invention describes a method of making a compound of: wherein R is C(0)NHCH(X)COOH, comprising the step of:
- Compound 8 wherein X is a C 2 -C6 alkyl, or a C3-C8 cycloalkyl. Additional embodiments include methods of making Compound 7.
- Another aspect of the present invention describes a method of producing Compound 13, comprising the step of:
- Additional embodiments include methods for producing Compound 11.
- PG refers to a protecting group.
- Another aspect of the present invention describes a method for making mpound 14 comprising the step of coupling Compounds 13 and 8:
- Additional embodiments include forming a macrolactam, adding functional groups; and producing Compound 13 and/or Compound 8 used in this aspect, by using methods described herein.
- a preferred macrolactam compound produced using methods described herein is Compound A.
- Figure 1 illustrates an X-ray diffraction pattern of a crystalline Compound 17.
- Figure 2 illustrates an X-ray diffraction pattern of a crystalline Compound 18.
- Compound A has the following structure:
- Functional groups that can be modified include a different heterocycle group, a different alkyl in place of the t-butyl group, and alteration of the cyclopropylsulfonyl functional group (e.g., with an ethyl group replacing the ethylene and/or a methylcyclopropyl group replacing the cyclopropyl group).
- Harper et al, WO2010011566 describes an alternative method for making Compound A. Harper et a/.,WO2010011566 et al, also includes data illustrating the ability of Compound A to inhibit HCV replicon activity and NS3/4A.
- Macrolactam compounds able to inhibit HCV activity have different uses including inhibiting HCV activity in vivo, inhibiting HCV activity in vitro, and inhibiting HCV NS3 enzymatic activity. In vivo inhibition of HCV activity can be used for therapeutic applications. Inhibiting HCV activity in vitro has different applications including being used to obtain HCV resistant mutants, further characterizing the ability of a functional group to inhibit HCV replicon or enzymatic activity, and studying HCV replication or protease activity. Cyclopropyl Linker Synthesis
- Scheme A illustrates an overall scheme for producing a cyclopropyl linker and different intermediates. Individual steps in Scheme A provide for additional embodiments. Further embodiments include steps upstream and downstream from a particular step.
- LG refers to leaving group
- Advantages of performing the different steps illustrated in Scheme A compared with a method of producing an alternative cyclopropyl linker having an ethylene group, described in Harper et al, WO 2010/011566, include: acetylide addition to the alkyl chloride having cyclopropane function to avoid forming double cyclopropanation, selective production of chiral intermediate (Compound 7), direct carbamate formation (which does not require protection), very high trans selectivity, avoiding the use of unstable enol silyl ether, and improved yield.
- Compound 8 can be used, for example, as illustrated in Scheme C infra., to produce a macrolactam.
- An as ect of the present invention is directed to a compound of Formula I: or salt thereof; wherein R is either H, C(0)R A , or C(0)NHCH(X)COOH; provided that X is a C 2 -C 6 alkyl, or a C3-C8 cycloalkyl and R A is a Ci_ 6 alkyl or aryl. Variations in the X group can be used to provide modifications to Compound A.
- R is H; R is acetyl; R is C(0)NHCH(X)COOH and X is t-butyl; R is C(0)NHCH(X)COOH and X is cyclohexyl; or R is C(0)NHCH(X)COOH and X is cyclopentyl.
- a compound of Formula I is present in a composition or mixture substantially free of its stereoisomers.
- the percent of the ee form of Formula I present, with respect to other stereoisomers is at least 90% ee, at least 95%> ee, at least 90% to about 98% ee, or at least 95% to about 98% ee.
- R is H and the ee form of the compound, with respect to other stereoisomers, is at least 90% ee, at least 95% ee, at least 90% to about 98% ee, or at least 95% to about 98% ee; R is
- C(0)NHCH(X)COOH and the ee form of the compound, with respect to other stereoisomers is at least 90%> ee, at least 95%> ee, at least 90%> to about 98%> ee, or at least 95%> to about 98%> ee; and R is C(0)NHCH(X)COOH and X is either t-butyl, cyclopentyl or cyclohexyl, preferably t- butyl, and the ee form of the compound, with respect to other stereoisomers, is at least 90%> ee, at least 95% ee, at least 90% to about 98% ee, or at least 95% to about 98% ee.
- Another aspect is directed to a method of producing a Formula I compound involving the enz matic resolution of Compound 6 to produce Compound 7:
- Suitable enzymes are lipases or proteases, examples of which include Candida cylindraceae lipase 2, Pseudomonas cepacia lipase 2, Mucor miehei lipase, papain, Amano protease S (Bacillus stearothermophilus protease), Amano protease A (Aspergillus niger),
- Protomax Novo, Enzyme Development Corp protese S20059, and Novozymes 435 (immobilized Candida antarctica lipase B).
- the protease from Bacillus stearothermophilus and Novozymes 435 are preferred, and Novozymes 435 is most preferred.
- the particular reaction conditions will vary depending upon the selected enzyme.
- the enzyme is Novozyme 435 added to a buffer saturated organic solvent solution of Compound 6 at a temperature between 0 and 50°C .
- Preferred reaction conditions are when the solvent is MTBE saturated with a 0.1 M K 2 HP0 4 solution and the reaction temperature is 10°C.
- X is either t-butyl, cyclopentyl or cyclohexyl.
- the reaction can be carried using a condensation reagent, optionally in the presence of base.
- condensation reagents including CDI, phosgene, diphosgene, triphogene, and chlorocarbonates.
- CDI is a preferred reagent.
- the reaction is carried out in the presence of base.
- suitable bases include typical bases and organic bases such as TEA, DIPEA, DABCO, and DBU.
- TEA and DIPEA are preferred reagents for this reaction.
- Compound 6 is produced from Compound 5 using an acetylating agent:
- acetylating agents include acetyl chloride, acetyl bromide and acetic anhydride.
- Suitable reaction conditions include the use of an aprotic organic solvent at a temperature between 0 and 50 °C followed by the addition of neutral aqueous solution and separation of the organic layer to obtain Compound 6.
- Compound 5 is produced by mixing Compound 4 with a metal acetylide
- the reaction can be carried out, for example, by mixing Compound 4 in with an organometallic reagent, in an aprotic organic solvent at a temperature between -78 and 30 °C. Mixing this solution with a polar aprotic solvent and a metallated acetylene at a temperature from 0-60°C followed by addition of an acidic aqueous solution and separation of the organic layer to obtain Compound 5.
- organometallic reagents include alkyl lithiums, alkyl magnesium halides, sodium and potassium hydride.
- polar aprotic solvents include, DMSO, DMF, N,N-dimethylacetamide, N-methyl pyrrolidinone, hexamethylphosphoramide and DMPU.
- metallated acetylene such as lithium acetylide-ethylene diamine complex or potassium acetylide is used, at a temperature from 0-60 °C followed by addition of an acidic aqueous solution and separation of the organic layer to obtain Compound 5.
- Compound 4 is produced by oxidizing Compound 3.
- the reaction can be carried out by mixing Compound 3 with an oxidant in a protic or aprotic organic solvent at a temperature between 0 and 100 °C.
- oxidants include hydrogen peroxide, alkyl hydrogen peroxides, sodium perborate, and sodium and potassium persulfate.
- Compound 3 is produced from Compound 2:
- the reaction can be performed, for example, by mixing Compound 2 with an alkyl zinc reagent, an acid, and a dihalomethane in a halogenated solvent at a temperature between 0 and 40 °C followed by addition of an acidic aqueous solution and separation of the organic layer to obtain Compound 3.
- suitable acids include alkyl and aryl carboxylic acids, sulfonic acids or phosphoric acids.
- Compound 2 can be prepared as described by Shirakawa et al. Synthesis 77: 1814-
- Scheme B illustrates a scheme for producing a quinoloxine and joining it to a hydroxyproline. Individual steps in Scheme B provide for additional embodiments. Further embodiments include steps upstream and downstream from a particular step. Alternative heterocycles can be joined to hydroxyproline using the provided procedures.
- 2010/011566 include eliminating a poor selective chloration intermediate, and eliminating four steps by allowing for the use of natural hydroxyproline.
- Another aspect of the present invention is directed to production of Compound
- the reaction can be carried out by SnAr replacement of Compound 11 with
- a general temperature for the reaction is 20-100 °C, with a preferred temperature being -50 °C.
- a wide range of solvents can be using including aprotic polar solvents, DMF, DM Ac, NMP, DMSO, and DMPU.
- a preferred solvent is DM Ac.
- Different bases can be used including CS 2 CO 3 , DBU, K 2 CO 3 , K 3 PO 4 , and KOtBu.
- a preferred base is DBU.
- Compound 11 is produced by:
- Com ound 10 is produced by:
- Preferred reaction conditions providing advantages compared with an alternative method described in Harper et ah, WO 2010/011566 include: use of the highly effective Sonogashira/Macrolactamization method in forming Compound 14, from Compounds 8 and 13; a one-pot procedure going from Compound 14 to 15 to 16 to 17; and producing Compound A from Compounds 18 and 19 using EDC instead of HATU.
- Compounds 18 and 19 can be carried out using pyridine or a pyridine derivatives, where HOBt is either not present or is present in very small amounts.
- HOBt is either not present or is present in very small amounts.
- the use of pyridine or a pyridine derivative instead of HOBt for coupling offers several advantages including higher yield and less emerization on the proline a-center.
- HOBt is shock sensitive in a dry state.
- An aspect of the present invention is directed to a compound selected from the consisting of:
- Another aspect of the present invention is directed to the production of
- a first embodiment involves Sonogashira cross coupling of Compounds 13 and 8 in the presence of tri tert-butylphosphine tetrafluoroborate salt in a solvent system.
- a preferred general temperature range is -50-100 °C, more preferable the temperature is about 80 °C.
- suitable solvent systems include CPME and MeCN, THF, 2-MeTHF, toluene CPME alone and MeCN alone.
- a preferred solvent system is CPME and MeCN.
- suitable catalysts include Pd and copper.
- a preferred catalyst is Pd(OAc) 2 .
- salts of Compound 8 are directly employed in the Sonogashira cross coupling reaction with Compound 13 to give Compound 14.
- This reaction can be carried out in the presence of tri fert-butylphosphine tetrafluoroborate salt in a solvent.
- the preferred solvent for this transformation is DMF.
- a preferred general temperature range is ⁇ 40- 80 °C, more preferable the temperature is about 65 °C, and the preferred catalyst is Pd(OAc) 2 .
- Examples of salt that can employed include dibenzylamine and t-butylamine.
- Suitable conditions include the use of a palladium catalyst and solvent.
- solvents include EtOAc, MeOH, and IPAc/MeOH mixture.
- IPAc/MeOH mixture is a preferred solvent.
- a general temperature range is 0 to 35 °C, preferably 15-20 °C.
- any of Compounds 15 and 16 can be provided as salts.
- Suitable conditions are a range of acids and solvents. Examples of acids are methanesulfonic acid, benzenesulfonic acid, p- toluenesulfonic acid, trifluoroacetic acid, hydrochloric acid, phosphoric acid, and
- solvents examples include THF, iPrOAc, MeCN, and CH 2 C1 2 .
- the second and third embodiments are performed in a one- pot procedure for the protection/macrolactamization/isolation to provide a crystal of Compound 17.
- Compound A is produced by coupling Compound 18 with
- General coupling (condensation) reagents can be employed.
- the reaction can be done under standard coupling conditions with typically carbodiimide type of reagents, such as DCC, EDC, etc and in the presence or absence of HOBt, HOPO, or pyridine derivative etc.
- An example of suitable conditions includes using a carbodiimide, an activator and a tertiary organic base in a polar aprotic solvent at a temperature between 0 and 40°C.
- activators include HOBt and HO AT.
- about 1.2 equiv EDC about 2.4 equiv HOBt- 3 ⁇ 40, and about 2.4 equiv of DIPEA in about 5 volumes DMF are employed.
- Compound A is produced by coupling Compound 18 with Compound 19 using pyridine or a pyridine derivative.
- the reaction can be carried out using a coupling reagent, an aprotic organic solvent and pyridine or a pyridine derivative.
- a general temperature is 0 °C to 50 °C (preferably room temperature).
- Examples of coupling reagents include dicyclohexylcarbodiimide (DCC), ⁇ , ⁇ '- diisopropylcarbodiimide (DIC), and l-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC).
- Examples of aprotic organic solvents include acetonitrile, THF, and toluene. In an embodiment EDC is used. In an embodiment, at least 10 equivalents of pyridine are used with acetonitrile.
- Preferred pyridine derivatives have electron donating or neutral groups at the 3 and 4 position. Exam les of general structures covering pyridine and derivatives include:
- R 5 is either hydrogen, aryl, halogen, Cl-6 alkyl, O-Cl-6 alkyl or C3-C8 cycloalkyl.
- Preferred reagents are pyridine, 4-phenylpyridine, 4-alkylpyridine, methylpyridine,
- alkyl refers to a monovalent straight or branched chain, saturated aliphatic hydrocarbon radical having a number of carbon atoms in the specified range.
- Cl-6 alkyl refers to any of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and iso- propyl, ethyl, and methyl.
- Cl-4 alkyl refers to n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl, and methyl.
- cycloalkyl refers to any monocyclic ring of an alkane having a number of carbon atoms in the specified range.
- C3-8 cycloalkyl refers to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- halogen refers to fluorine, chlorine, bromine and iodine (alternatively referred to as fluoro, chloro, bromo, and iodo).
- haloalkyl refers to an alkyl group as defined above in which one or more of the hydrogen atoms have been replaced with a halogen (i.e., F, CI, Br and/or I).
- a halogen i.e., F, CI, Br and/or I.
- Cl-6 haloalkyl or “C1-C6 haloalkyl” refers to a Cl to C6 linear or branched alkyl group as defined above with one or more halogen substituents.
- fluoroalkyl has an analogous meaning except the halogen substituents are restricted to fluoro. Suitable fluoroalkyls include the series (CH2)0-4CF3 (i.e., trifluoromethyl, 2,2,2-trifluoroethyl, 3,3,3- trifluoro-n-propyl, etc.).
- PG indicates a protecting group. In different embodiments described throughout the application where a protecting group is employed: PG is either BOC or Fmoc; or PG is BOC.
- aryl is either phenyl, substituted phenyl, naphthyl, substituted naphthyl, heteroaryl, or substituted heteroaryl, provided that substituted phenyl, substituted naphthyl, and substituted heteroaryl, each have 1 to 5 substituents independently selected from the group consisting of:
- Cl-6 alkyl (2) C 1 -6 alkyl substituted with OH, O-C 1 -6 alkyl, O-C 1 -6 haloalkyl, CN, N02, N(RC)RD C(0)N(RC)RD C(0)RC, C02R C , SRC, S(0)RC, SO2RC, S02N(RC)RD ? N(RC)C(0)RD, N(RC)C02R°, N(RC)S02R D , N(RC)S02N(RC)RD OC(0)N(RC)RD ? N(RC)C(0)N(RC)RD ? or
- R ⁇ and R" are each independently H or Ci_ 6 alkyl.
- heteroaryl is a (i) a 5- or 6-membered heteroaromatic ring containing from 1 to 4 heteroatoms independently selected from N, O and S or (ii) a 9- or 10-membered bicyclic, fused ring system containing from 1 to 4 heteroatoms independently selected from N, O and S.
- the atoms in a compound described herein may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature.
- the present invention is meant to include all suitable isotopic variations of the compounds of described herein.
- different isotopic forms of hydrogen (H) include protium (IK) and deuterium (3 ⁇ 4).
- Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
- Isotopically-enriched compounds described herein can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples provided herein using appropriate isotopically-enriched reagents and/or intermediates.
- CDI 1 , 1 '-Carbonyldiimidazole
- DIPEA Diisopropylethylamine
- HATU 0-(7-azabenzotriazol- 1 -yl)-N,N,N',N'-tetramethyluronium hexafluorophoshate
- IP Ac Isopropyl acetate
- compositions for treating patients can be used with compounds for treating patients.
- Non-pharmaceutical salts may, however, be useful in the preparation of intermediate compounds.
- Pharmaceutically acceptable salts are suitable for administration to a patient, preferably, a human.
- Suitable salts include acid addition salts which may, for example, be formed by mixing a solution of a compound with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid.
- Compounds carrying an acidic moiety can be mixed with suitable pharmaceutically acceptable salts to provide, for example, alkali metal salts (e.g., sodium or potassium salts), alkaline earth metal salts (e.g., calcium or magnesium salts), and salts formed with suitable organic ligands such as quaternary ammonium salts.
- suitable organic ligands such as quaternary ammonium salts.
- pharmaceutically acceptable esters can be employed to modify the solubility or hydrolysis characteristics of the compound.
- compositions described herein having therapeutic applications can be administered to a patient infected with HCV.
- administration and variants thereof (e.g., “administering” a compound) means providing the compound to the individual in need of treatment.
- administration and its variants are each understood to include concurrent and sequential provision of the compound or salt and other agents.
- composition is intended to encompass a product comprising the specified ingredients, as well as any product which results, directly or indirectly, from combining the specified ingredients.
- pharmaceutically acceptable is meant the ingredients of the pharmaceutical composition must be compatible with each other and are suitable to the recipient thereof.
- subject refers to an animal, preferably a mammal, most preferably a human, who is the object of treatment, observation or experiment.
- an effective amount indicates a sufficient amount to exert a therapeutic or prophylactic effect.
- an effective amount is sufficient to achieve one or more of the following effects: reduce the ability of HCV to replicate, reduce HCV load, and increase viral clearance.
- an effective amount is sufficient to achieve one or more of the following: a reduced susceptibility to HCV infection, and a reduced ability of the infecting virus to establish persistent infection for chronic disease.
- the compounds can be administered by means that produces contact of the active agent with the agent's site of action. They can be administered by conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are
- a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- Compounds can, for example, be administered by one or more of the following routes: orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation (such as in a spray form), or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and pharmaceutically-acceptable carriers (e.g., a carrier suitable for administration to a human patient), adjuvants and vehicles.
- pharmaceutically-acceptable carriers e.g., a carrier suitable for administration to a human patient
- Liquid preparations suitable for oral administration e.g., suspensions, syrups, elixirs and the like
- media such as water, glycols, oils, alcohols and the like.
- Solid preparations suitable for oral administration can be prepared according to techniques known in the art and can employ solid excipients as such starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like.
- Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as solubility aids.
- injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose.
- Therapeutic compounds can be administered orally in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day in a single dose or in divided doses.
- mammal e.g., human
- One dosage range is 0.01 to 500 mg/kg body weight per day orally in a single dose or in divided doses.
- Another dosage range is 0.1 to 100 mg/kg body weight per day orally in single or divided doses.
- the compositions can be provided in the form of tablets or capsules containing 1.0 to 500 mg of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, and 750 mg of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
- HCV NS3 activity HCV replicon activity
- HCV replication activity can be evaluated using techniques well-known in the art. (See, for example, Carroll et al., J. Biol. Chem. 275: 11979-11984, 2003.)
- TRF time-resolved fluorescence
- a NS3 protease assay can be performed, for example, in a final volume of 100 ⁇ assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15 % glycerol, 0.15 % TRITON X-100, 10 mM DTT, and 0.1 % PEG 8000.
- NS3 and NS4A are pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM).
- NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 ⁇ of 500 mM MES, pH 5.5.
- Product fluorescence is detected using either a VICTOR V2 or FUSION fluorophotometer (Perkin Elmer Life and Analytical Sciences) with excitation at 340 nm and emission at 615 nm with a 400 delay. Testing concentrations of different enzyme forms are selected to result in a signal to background ratio (S/B) of 10-30.
- S/B signal to background ratio
- IC 50 values are derived using a standard four- parameter fit to the data. K; values are derived from IC 50 values using the following formula,
- reaction mixture was diluted with 200 mL of water.
- the biphasic mixture was transferred to a separatory funnel and the aqueous layer removed.
- the organic layer was washed with 200 mL of 2 N HCl and then with 300 mL of sat. NaHC0 3 prior to drying over MgSC ⁇ .
- the solvent was removed in vacuo to give 41.8 g of rac-6 (>99% yield).
- the mixture was aged for 30 min to dissolve all the solids and then transferred to a 100 L cylindrical extractor.
- the aqueous layer was then washed with 12 L of MTBE.
- the bottom aqueous layer was drained and the top MTBE layer was discarded.
- the aqueous layer was washed with 8 L of MTBE.
- the bottom aqueous layer was drained and the top MTBE layer was discarded.
- the MTBE layer was then transferred via vacuum into a 50 L round bottom flask equipped with a mechanical stirrer, thermocouple, and batch concentrator and the solvent was removed under reduced pressure keeping the internal temperature of the batch ⁇ 20 °C during the distillation.
- the solvent was then switched to cyclopentyl methyl ether (CPME) by flushing with ⁇ 5 L of CPME and then diluted to a final volume of ⁇ 20 L. This material was used in the next reaction without further purification.
- CPME cyclopentyl methyl ether
- the resulting grey slurry was then cooled to an internal temperature of 20 °C overnight
- the slurry was filtered, water (1.0-1.5 L/Kg) was used to help with the transfer.
- the light grey solids were washed with 2 cake volumes water (5.0-5.5 L/Kg).
- the solids were dried under vacuum/N 2 sweep for 24 hours, at which time the solids were still very wet.
- thermocouple and condenser was added to Compound 10 (3.8 kg), and charged slowly at room temperature with POCI 3 (5.92 L @ 99%). There was no initial temperature change. The grey slurry was heated to 98 °C for 20 hours. After 2-3 hours the slurry turned from grey to green, then to yellow and finally turned homogeneous red. As the slurry became homogenous in POCI 3 , significant amounts of HC1 off-gassing were produced. The reaction was monitored by HPLC. The dark red, homogenous solution was allowed to cool slowly to below 80 °C.
- the reaction mixture was concentrated and azotroped by MTBE until the KF of the solution was ⁇ 300 ppm, and adjusted to a total volume (18 L).
- the solution was seeded, and stirred at room temperature for 5 hours.
- Heptane (21.5 L) was slowly added over 2 hours.
- the resulting slurry was stirred at room temperature overnight, and at 5-10 °C for 2 hours.
- the crystalline solid was collected by filtration, washed with cold heptane/MTBE (3 : 1 , 16 L), heptane (10 L), and dried under vacuum with nitrogen sweep to afford desired product Compound 13 (5.68 kg, 96.7 A%, 95.5 wt%, 71% isolated yield after correction), m.p. 98.5-99 °C.
- Retention time for regioisomer 4.927 min.
- the reaction mixture was filtered through Solka floe eluting with IP Ac. The filtrate was then concentrated under reduced pressure in a 100 mL round bottom flask equipped with a thermocouple, mechanical stirrer, and batch concentrator. The solvent was switched to MeCN and a final volume of ⁇ 50 mL. Final assay of the combined batches was 7.25 g (89%) of the product Compound 15. The crude product was used without further purification in the next step.
- HPLC Conditions Zorbax Eclipse Plus C18 50 x 4.6 mm, 1.8 um, 1.5 mL/min, 230 nm, 25 °C, Eluents: Water 0.1% H 3 P0 4 (A), Acetonitrile (B). 90% A 0 min, 5% A 5 min, 5% A 6 min.
- the ethyl acetate/ THF solution was concentrated to 4 volumes (14 L) and then flushed with 18 L ethyl acetate keeping the volume constant at 14 L. To the slurry was added 38.5 L hexanes over 4 hours. The slurry was aged overnight at ambient temperature.
- the Boc-sulfonamide was charged in a 75 L 4-neck round bottomed flask fitted with overhead stirrer, temperature probe, and N 2 inlet. Ethyl acetate (22.6 L) was added followed by the TsOH. The mixture was aged at ambient temperature for 23.5 hours The mixture is homogeneous at the start, but forms precipitates during the overnight age giving a free flowing white slurry.
- Compound A was dissolved in acetone 4L at RT, filtered and transferred to a 12 L RBF with overhead stirring, rinsed with extra acetone 1L, heated to 50 °C, water 0.9L was added, seeded lOg, aged 15minutes, then added water 0.8L over 2.5 hours, extra water 3.3v over 2.5 hours was added, stopped heating, cooled to RT, aged at RT overnight, filtered, washed with water/acetone (1 : 1 v/v) 4L, and dried in air under vacuum.
- Compound A Hydrate III, 670 g was obtained as an off-white solid.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Virology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Communicable Diseases (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Cephalosporin Compounds (AREA)
Abstract
The present invention includes compounds useful as intermediates in the preparation of macrolactams, methods for preparing the intermediates, and methods for preparing macrolactams. One use of the methods and intermediates described herein is in the production of macrolactam compounds able to inhibit HCV NS3 protease activity. HCV NS3 inhibitory compounds have therapeutic and research applica
Description
TITLE OF THE APPLICATION
METHODS AND INTERMEDIATES FOR PREPARING MACROLACTAMS
FIELD OF THE INVENTION
The present invention relates to process and intermediates that can be used for preparing macrolactams. One use of the methods and intermediates described herein is the production of macrolactam compounds able to inhibit HCV NS3 protease activity. HCV NS3 inhibitory compounds have therapeutic and research applications. BACKGROUND OF THE INVENTION
Hepatitis C virus (HCV) infection is a major health problem. HCV infection leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals.
Several virally-encoded enzymes are putative targets for therapeutic intervention, including a metalloprotease (NS2-3), a serine protease (NS3), a helicase (NS3), and an RNA- dependent RNA polymerase (NS5B). The NS3 protease is located in the N-terminal domain of the NS3 protein. NS4A provides a cofactor for NS3 activity.
Potential treatments for HCV infection are discussed in different references including Balsano, Mini Rev. Med. Chem. S(¾):307-318, 2008, Ronn et al, Current Topics in Medicinal Chemistry 5:533-562, 2008, Sheldon et al, Expert Opin. Investig. Drugs 16(8): \ \1\-
1181, 2007, and De Francesco et al, Antiviral Research 55: 1-16, 2003.
Examples of publications describing macrolactam compounds able to inhibit
HCV protease activity include: Harper et al, WO2010011566; Liverton et al, WO2009134624;
McCauley et al, WO2009108507; Liverton et a/.,WO2009010804; Liverton et
a/.,WO2008057209; Liverton et al, WO2008051477; Liverton et a/.,WO2008051514; Liverton et al, WO2008057208; Crescenzi et al, WO2007148135; Di Francesco et al, WO2007131966;
Holloway et al, WO2007015855; Holloway et a/.,WO2007015787; Holloway et al,
WO2007016441; Holloway et al, WO2006119061; Liverton et al, J. Am. Chem. Soc,
130:4607-4609, 2008; and Liverton et al, Antimicrobial Agents and Chemotherapy 54:305-311, 2010.
SUMMARY OF THE INVENTION
The present invention includes methods and intermediates for preparing macrolactams. One use of the methods and intermediates described herein is in the production of macrolactam compounds able to inhibit HCV NS3 protease activity. HCV NS3 inhibitory compounds have therapeutic and research applications.
An example of a HCV inhibitory compound that can be produced using the method and intermediates described herein is Compound A, or a pharmaceutically salt thereof:

Compound A
A first aspect of the present invention describes a compound selected from group consisting of:
 , or C(0)NHCH(X)COOH; provided that X is a C2-C6 alkyl, or a C3-C8 cycloalkyl and R is a Ci_6 alkyl or aryl,
, or C(0)NHCH(X)COOH; provided that X is a C2-C6 alkyl, or a C3-C8 cycloalkyl and R is a Ci_6 alkyl or aryl,

(Compound 14) (Compound and salts thereof, wherein Y is either a protecting group or hydrogen.
aspect of the present invention is directed to a method of making

(Compound 7)
comprising the step of:
Enzyme
(Compound 6) (Compound 7)
by enzymatic resolution of Compound 6. Compounds 6 are 7 are provided in a trans configuration.
Additional embodiments include methods for producing Compound 6.
Another aspect of the present invention describes a method of making a compound of:
 wherein R is C(0)NHCH(X)COOH, comprising the step of:
wherein R is C(0)NHCH(X)COOH, comprising the step of:

(Compound 8) wherein X is a C2-C6 alkyl, or a C3-C8 cycloalkyl. Additional embodiments include methods of making Compound 7.
Another aspect of the present invention describes a method of producing Compound 13, comprising the step of:

(Compound 12)
(Compound 13)
Additional embodiments include methods for producing Compound 11. "PG" refers to a protecting group.
Another aspect of the present invention describes a method for making mpound 14 comprising the step of coupling Compounds 13 and 8:

Additional embodiments include forming a macrolactam, adding functional groups; and producing Compound 13 and/or Compound 8 used in this aspect, by using methods described herein. A preferred macrolactam compound produced using methods described herein is Compound A.
Other embodiments, aspects and features of the present invention are either further described herein or will be apparent from the ensuing description, examples and appended claims.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 illustrates an X-ray diffraction pattern of a crystalline Compound 17. Figure 2 illustrates an X-ray diffraction pattern of a crystalline Compound 18.
DETAILED DESCRIPTION OF THE INVENTION
The methods and intermediates described herein can be used to synthesize macrolactams such as Compound A and compounds varying from Compound A by one or more functional group. Compound A has the following structure:

Compound A
Functional groups that can be modified include a different heterocycle group, a different alkyl in place of the t-butyl group, and alteration of the cyclopropylsulfonyl functional group (e.g., with an ethyl group replacing the ethylene and/or a methylcyclopropyl group replacing the cyclopropyl group).
Different intermediates and synthesis protocols are illustrated herein where Compound A was ultimately obtained. However, it is understood that based on the guidance provided herein other macrolactams can be produced using appropriate intermediates and by adding or modifying different functional groups. Examples of different macrolactams having different functional groups are provided in McCauley et al, WO2011014487; Harper et al,
WO2010011566; Liverton et al., WO2009134624; McCauley et al., WO2009108507; Liverton et a/.,WO2009010804; Liverton et a/.,WO2008057209; Liverton et al, WO2008051477;
Liverton et a/.,WO2008051514; Liverton et al, WO2008057208; Crescenzi et al,
WO2007148135; Di Francesco et al, WO2007131966; Holloway et al, WO2007015855;
Holloway et a/.,WO2007015787; Holloway et al, WO2007016441; Holloway et al,
WO2006119061; Liverton et al, J. Am. Chem. Soc, 130:4607-4609, 2008; and Liverton et al, Antimicrobial Agents and Chemotherapy 54:305-311, 2010.
Harper et al, WO2010011566 describes an alternative method for making Compound A. Harper et a/.,WO2010011566 et al, also includes data illustrating the ability of Compound A to inhibit HCV replicon activity and NS3/4A.
Intermediates and procedures that can be used to produce macrolactams can be illustrated by taking into account: (1) cyclopropyl linker synthesis: (2) heterocycle synthesis; and (3) forming a macrolactam using the cyclopropyl linker and heterocycle group, and optionally adding or modifying different functional groups. The optionally added functional groups can be
used to provide for, or enhance, the ability of a compound to inhibit HCV NS3 activity and/or HCV replication.
Macrolactam compounds able to inhibit HCV activity have different uses including inhibiting HCV activity in vivo, inhibiting HCV activity in vitro, and inhibiting HCV NS3 enzymatic activity. In vivo inhibition of HCV activity can be used for therapeutic applications. Inhibiting HCV activity in vitro has different applications including being used to obtain HCV resistant mutants, further characterizing the ability of a functional group to inhibit HCV replicon or enzymatic activity, and studying HCV replication or protease activity. Cyclopropyl Linker Synthesis
Scheme A illustrates an overall scheme for producing a cyclopropyl linker and different intermediates. Individual steps in Scheme A provide for additional embodiments. Further embodiments include steps upstream and downstream from a particular step.
Scheme A

Enzyme


"LG" refers to leaving group.
The compounds illustrated in Scheme A are in the neutral form. It should be understood that different embodiment described throughout the application include appropriate acid or base forms of the different compounds.
Advantages of performing the different steps illustrated in Scheme A, compared with a method of producing an alternative cyclopropyl linker having an ethylene group, described in Harper et al, WO 2010/011566, include: acetylide addition to the alkyl chloride having cyclopropane function to avoid forming double cyclopropanation, selective production of chiral intermediate (Compound 7), direct carbamate formation (which does not require protection), very high trans selectivity, avoiding the use of unstable enol silyl ether, and improved yield.
Important compounds illustrated in Scheme A include Compound 7 and
Compound 8. Compound 8 can be used, for example, as illustrated in Scheme C infra., to produce a macrolactam.
An as ect of the present invention is directed to a compound of Formula I:
 or salt thereof;
wherein R is either H, C(0)RA, or C(0)NHCH(X)COOH; provided that X is a C2-C6 alkyl, or a C3-C8 cycloalkyl and RA is a Ci_6 alkyl or aryl. Variations in the X group can be used to provide modifications to Compound A.
or salt thereof;
wherein R is either H, C(0)RA, or C(0)NHCH(X)COOH; provided that X is a C2-C6 alkyl, or a C3-C8 cycloalkyl and RA is a Ci_6 alkyl or aryl. Variations in the X group can be used to provide modifications to Compound A.
In a first series of embodiments, R is H; R is acetyl; R is C(0)NHCH(X)COOH and X is t-butyl; R is C(0)NHCH(X)COOH and X is cyclohexyl; or R is C(0)NHCH(X)COOH and X is cyclopentyl.
In another series of embodiments, a compound of Formula I is present in a composition or mixture substantially free of its stereoisomers. Preferably, the percent of the ee form of Formula I present, with respect to other stereoisomers, is at least 90% ee, at least 95%> ee, at least 90% to about 98% ee, or at least 95% to about 98% ee. In other embodiments R is H and the ee form of the compound, with respect to other stereoisomers, is at least 90% ee, at least 95% ee, at least 90% to about 98% ee, or at least 95% to about 98% ee; R is
C(0)NHCH(X)COOH and the ee form of the compound, with respect to other stereoisomers, is at least 90%> ee, at least 95%> ee, at least 90%> to about 98%> ee, or at least 95%> to about 98%> ee; and R is C(0)NHCH(X)COOH and X is either t-butyl, cyclopentyl or cyclohexyl, preferably t- butyl, and the ee form of the compound, with respect to other stereoisomers, is at least 90%> ee, at least 95% ee, at least 90% to about 98% ee, or at least 95% to about 98% ee.
Another aspect is directed to a method of producing a Formula I compound involving the enz matic resolution of Compound 6 to produce Compound 7:

Suitable enzymes are lipases or proteases, examples of which include Candida cylindraceae lipase 2, Pseudomonas cepacia lipase 2, Mucor miehei lipase, papain, Amano protease S (Bacillus stearothermophilus protease), Amano protease A (Aspergillus niger),
Protomax Novo, Enzyme Development Corp protese S20059, and Novozymes 435 (immobilized Candida antarctica lipase B). The protease from Bacillus stearothermophilus and Novozymes 435 are preferred, and Novozymes 435 is most preferred. The particular reaction conditions will vary depending upon the selected enzyme. In an embodiment, the enzyme is Novozyme 435 added to a buffer saturated organic solvent solution of Compound 6 at a temperature between 0 and 50°C . Preferred reaction conditions are when the solvent is MTBE saturated with a 0.1 M K2HP04 solution and the reaction temperature is 10°C. Peferably, after the reaction is complete, the MTBE is removed by distillation and the resulting mixture of desired alcohol and undesired ester separated on a chromatography system.
In an embodiment Compound 7 is used to produce a Formula I compound wherein R is C(0)NHCH(X)COOH; and X either C2-C6 alkyl or C3-C8 cycloalkyl:
i-

Preferably, X is either t-butyl, cyclopentyl or cyclohexyl.
The reaction can be carried using a condensation reagent, optionally in the presence of base. Examples of condensation reagents including CDI, phosgene, diphosgene, triphogene, and chlorocarbonates. CDI is a preferred reagent. In an embodiment, the reaction is carried out in the presence of base. Examples of suitable bases include typical bases and organic bases such as TEA, DIPEA, DABCO, and DBU. TEA and DIPEA are preferred reagents for this reaction.
In another embodiment Compound 6 is produced from Compound 5 using an acetylating agent:

5 Acetylating agent 6
Examples of acetylating agents include acetyl chloride, acetyl bromide and acetic anhydride. Suitable reaction conditions include the use of an aprotic organic solvent at a temperature between 0 and 50 °C followed by the addition of neutral aqueous solution and separation of the organic layer to obtain Compound 6.
In another embodiment, Compound 5 is produced by mixing Compound 4 with a metal acetylide

The reaction can be carried out, for example, by mixing Compound 4 in with an organometallic reagent, in an aprotic organic solvent at a temperature between -78 and 30 °C. Mixing this solution with a polar aprotic solvent and a metallated acetylene at a temperature from 0-60°C followed by addition of an acidic aqueous solution and separation of the organic layer to obtain Compound 5. Examples of organometallic reagents include alkyl lithiums, alkyl magnesium halides, sodium and potassium hydride. Examples of polar aprotic solvents include,
DMSO, DMF, N,N-dimethylacetamide, N-methyl pyrrolidinone, hexamethylphosphoramide and DMPU. In an embodiment, metallated acetylene, such as lithium acetylide-ethylene diamine complex or potassium acetylide is used, at a temperature from 0-60 °C followed by addition of an acidic aqueous solution and separation of the organic layer to obtain Compound 5.
In another embodiment Compound 4 is produced by oxidizing Compound 3.

The reaction can be carried out by mixing Compound 3 with an oxidant in a protic or aprotic organic solvent at a temperature between 0 and 100 °C. Examples of oxidants include hydrogen peroxide, alkyl hydrogen peroxides, sodium perborate, and sodium and potassium persulfate.
In another embodiment, Compound 3 is produced from Compound 2:

The reaction can be performed, for example, by mixing Compound 2 with an alkyl zinc reagent, an acid, and a dihalomethane in a halogenated solvent at a temperature between 0 and 40 °C followed by addition of an acidic aqueous solution and separation of the organic layer to obtain Compound 3. Examples of suitable acids include alkyl and aryl carboxylic acids, sulfonic acids or phosphoric acids.
Compound 2 can be prepared as described by Shirakawa et al. Synthesis 77: 1814-
1820, 2004.
Heterocycle Synthesis
Scheme B illustrates a scheme for producing a quinoloxine and joining it to a hydroxyproline. Individual steps in Scheme B provide for additional embodiments. Further embodiments include steps upstream and downstream from a particular step. Alternative heterocycles can be joined to hydroxyproline using the provided procedures.
Scheme B

12
13
The compounds illustrated in Scheme B are in the neutral form. It should be understood that different embodiments described throughout the application include appropriate acid or base forms of the different compounds.
Advantages of the Scheme B heterocycle production and joining to hydroxylproline, compared to an alternative method described in Harper et al., WO
2010/011566 include eliminating a poor selective chloration intermediate, and eliminating four steps by allowing for the use of natural hydroxyproline.
Another aspect of the present invention is directed to production of Compound
13:

12
13
The reaction can be carried out by SnAr replacement of Compound 11 with
Compound 12 in the presence of a base. Compound 11 is described by Sarges et al, J. Med.
Chem. 33:2240-2254, 1990. A general temperature for the reaction is 20-100 °C, with a preferred temperature being -50 °C. A wide range of solvents can be using including aprotic
polar solvents, DMF, DM Ac, NMP, DMSO, and DMPU. A preferred solvent is DM Ac.
Different bases can be used including CS2CO3, DBU, K2CO3, K3PO4, and KOtBu. A preferred base is DBU. An advantage of the reaction was high regioselectivity and no detected
isomerization of the hydroxyproline.
In a first embodiment Compound 11 is produced by:

Suitable conditions are illustrated in the Examples infra.
In a second embodiment Com ound 10 is produced by:

10
Suitable conditions are illustrated in the Examples infra.
Macrolactam Production
Marcolactam formation using a cyclopropyl linker and a heterocycle joined to hydroxyproline, followed by side chain addition is illustrated in Scheme C. Individual steps in Scheme C provide for additional embodiments. Further embodiments include steps upstream and downstream from a particular step. Important intermediate compounds illustrated in Scheme C include Compound 14, Compound 15, and Compound 16.
Alternative macrolactams can be produced based on the guidance provided herein. Potential variations include using a different linker, heterocycle, and adding different functional groups.
Scheme C

The compounds illustrated in Scheme C are in the neutral form. It should be understood that different embodiments described throughout the application include appropriate acid or base forms of the different compounds.
Preferred reaction conditions, providing advantages compared with an alternative method described in Harper et ah, WO 2010/011566 include: use of the highly effective Sonogashira/Macrolactamization method in forming Compound 14, from Compounds 8 and 13; a one-pot procedure going from Compound 14 to 15 to 16 to 17; and producing Compound A from Compounds 18 and 19 using EDC instead of HATU. Preferably, EDC coupling of
Compounds 18 and 19 can be carried out using pyridine or a pyridine derivatives, where HOBt is either not present or is present in very small amounts. The use of pyridine or a pyridine derivative instead of HOBt for coupling offers several advantages including higher yield and less emerization on the proline a-center. In addition, HOBt is shock sensitive in a dry state.
An aspect of the present invention is directed to a compound selected from the consisting of:

Another aspect of the present invention is directed to the production of
Compound 14:

14
wherein any of Compounds 13, 8, and 14 (e.g., 1, 2 or all three) can be provided as salts.
A first embodiment involves Sonogashira cross coupling of Compounds 13 and 8 in the presence of tri tert-butylphosphine tetrafluoroborate salt in a solvent system. A preferred general temperature range is -50-100 °C, more preferable the temperature is about 80 °C.
Examples of suitable solvent systems include CPME and MeCN, THF, 2-MeTHF, toluene CPME alone and MeCN alone. A preferred solvent system is CPME and MeCN. Examples of suitable catalysts include Pd and copper. A preferred catalyst is Pd(OAc)2.
In another embodiment, salts of Compound 8 are directly employed in the Sonogashira cross coupling reaction with Compound 13 to give Compound 14. This reaction can be carried out in the presence of tri fert-butylphosphine tetrafluoroborate salt in a solvent. The preferred solvent for this transformation is DMF. A preferred general temperature range is ~40-
80 °C, more preferable the temperature is about 65 °C, and the preferred catalyst is Pd(OAc)2. Examples of salt that can employed include dibenzylamine and t-butylamine.
In a second embodiment, Compound 15 is produced by hydrogenation of
Compound 14:

wherein any of Compounds 14 and 15 can be provided as salts. Suitable conditions include the use of a palladium catalyst and solvent. Examples of solvents include EtOAc, MeOH, and IPAc/MeOH mixture. IPAc/MeOH mixture is a preferred solvent. A general temperature range is 0 to 35 °C, preferably 15-20 °C.
In a second embodiment Compound 16 is produced:

wherein any of Compounds 15 and 16 can be provided as salts. Suitable conditions are a range of acids and solvents. Examples of acids are methanesulfonic acid, benzenesulfonic acid, p- toluenesulfonic acid, trifluoroacetic acid, hydrochloric acid, phosphoric acid, and
tetrafluoroboric acid. Examples of solvents are THF, iPrOAc, MeCN, and CH2C12.
In a third embodiment, Compound 17 is produced:

In a sub-embodiment, the second and third embodiments are performed in a one- pot procedure for the protection/macrolactamization/isolation to provide a crystal of Compound 17.
In a fourth embodiment Compound 18 is produced:

wherein any of Compounds 17 and 18 can be provided as salts. Suitable conditions are illustrated in the Examples infra.
In a fifth embodiment Compound A is produced by coupling Compound 18 with
Compound 19:
Compound A

18
wherein any of Compounds 18, 19, or A can be provided as salts.
General coupling (condensation) reagents can be employed. The reaction can be done under standard coupling conditions with typically carbodiimide type of reagents, such as DCC, EDC, etc and in the presence or absence of HOBt, HOPO, or pyridine derivative etc. An example of suitable conditions includes using a carbodiimide, an activator and a tertiary organic base in a polar aprotic solvent at a temperature between 0 and 40°C. Examples of activators include HOBt and HO AT. In a sub-embodiment about 1.2 equiv EDC, about 2.4 equiv HOBt- ¾0, and about 2.4 equiv of DIPEA in about 5 volumes DMF are employed.
In another aspect, Compound A is produced by coupling Compound 18 with Compound 19 using pyridine or a pyridine derivative. Preferably, no detectable HOBt is present. The reaction can be carried out using a coupling reagent, an aprotic organic solvent and pyridine or a pyridine derivative. A general temperature is 0 °C to 50 °C (preferably room temperature). Examples of coupling reagents include dicyclohexylcarbodiimide (DCC), Ν,Ν'- diisopropylcarbodiimide (DIC), and l-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC). Examples of aprotic organic solvents include acetonitrile, THF, and toluene. In an embodiment EDC is used. In an embodiment, at least 10 equivalents of pyridine are used with acetonitrile.
Preferred pyridine derivatives have electron donating or neutral groups at the 3 and 4 position. Exam les of general structures covering pyridine and derivatives include:

wherein R5 is either hydrogen, aryl, halogen, Cl-6 alkyl, O-Cl-6 alkyl or C3-C8 cycloalkyl. Preferred reagents are pyridine, 4-phenylpyridine, 4-alkylpyridine, methylpyridine,
3- or 4- mono or dialkylpyridine, wherein the alkyl group can be a Cl-6 alkyl.
In additional embodiments: Compound 8 used to produce Compound 14 is produced using one or more embodiments of Scheme A, preferably all the steps in Scheme A; and/or Compound 13 used to produce Compound 14 is produced using one or more
embodiments of Scheme B, preferably all the steps provided in Scheme B.
Compounds
The term "alkyl" refers to a monovalent straight or branched chain, saturated aliphatic hydrocarbon radical having a number of carbon atoms in the specified range. Thus, for example, "Cl-6 alkyl" refers to any of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and iso- propyl, ethyl, and methyl. As another example, "Cl-4 alkyl" refers to n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl, and methyl.
The term "cycloalkyl" refers to any monocyclic ring of an alkane having a number of carbon atoms in the specified range. Thus, for example, "C3-8 cycloalkyl" (or "C3- C8 cycloalkyl") refers to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
The term "halogen" (or "halo") refers to fluorine, chlorine, bromine and iodine (alternatively referred to as fluoro, chloro, bromo, and iodo).
The term "haloalkyl" refers to an alkyl group as defined above in which one or more of the hydrogen atoms have been replaced with a halogen (i.e., F, CI, Br and/or I). Thus, for example, "Cl-6 haloalkyl" (or "C1-C6 haloalkyl") refers to a Cl to C6 linear or branched alkyl group as defined above with one or more halogen substituents. The term "fiuoroalkyl" has an analogous meaning except the halogen substituents are restricted to fluoro. Suitable fluoroalkyls include the series (CH2)0-4CF3 (i.e., trifluoromethyl, 2,2,2-trifluoroethyl, 3,3,3- trifluoro-n-propyl, etc.).
Reference to "PG" indicates a protecting group. In different embodiments described throughout the application where a protecting group is employed: PG is either BOC or Fmoc; or PG is BOC.
An "aryl" is either phenyl, substituted phenyl, naphthyl, substituted naphthyl, heteroaryl, or substituted heteroaryl, provided that substituted phenyl, substituted naphthyl, and substituted heteroaryl, each have 1 to 5 substituents independently selected from the group consisting of:
(1) Cl-6 alkyl,
(2) C 1 -6 alkyl substituted with OH, O-C 1 -6 alkyl, O-C 1 -6 haloalkyl, CN, N02, N(RC)RD C(0)N(RC)RD C(0)RC, C02RC, SRC, S(0)RC, SO2RC, S02N(RC)RD? N(RC)C(0)RD, N(RC)C02R°, N(RC)S02RD, N(RC)S02N(RC)RD OC(0)N(RC)RD? N(RC)C(0)N(RC)RD? or
N(RC)C(0)C(0)N(RC)RD,
3) O-C 1-6 alkyl,
4) C 1-6 haloalkyl,
5) O-C 1-6 haloalkyl,
6) OH,
(7) halogen,
8) CN,
9) NO2,
10) N(RC)RD
1 1) C(0)N(RC)RD,
(12) C(0)RC
13) C(0)-Cl-6 haloalkyl,
14) C(0)0RC
15) OC(0)N(RC)RD,
16) SRC
(17) S(0)RC,

(22) N(RC)C(0)RD,
23) N(RC)C(0)N(RC)RD,
24) N(RC)C(0)C(0)N(RC)RD, 0r
R^ and R" are each independently H or Ci_6 alkyl.
A "heteroaryl" is a (i) a 5- or 6-membered heteroaromatic ring containing from 1 to 4 heteroatoms independently selected from N, O and S or (ii) a 9- or 10-membered bicyclic, fused ring system containing from 1 to 4 heteroatoms independently selected from N, O and S.
The atoms in a compound described herein may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of described herein. For example, different
isotopic forms of hydrogen (H) include protium (IK) and deuterium (¾). Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
Isotopically-enriched compounds described herein can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples provided herein using appropriate isotopically-enriched reagents and/or intermediates. Abbreviations
BOC: t-Butyloxyxcarbomate
CDI : 1 , 1 '-Carbonyldiimidazole
CPMB: Cyclopentyl methyl ether
DABCO: 1,4-diazabicyclo [2.2.2.] octane
DBU: 1 ,8-Diazobicyclo[5 AO jundec-7-ene
DCC : N^-Dicyclohexylcarbodiimide
DIC : N,N'-diisopropylcarbodiimide
DIPEA: Diisopropylethylamine
DM Ac: AvV-Dimethylacetamide
DMF: Α/,/Υ-Dimethy!formamide
DMPU: N,N-dimethylpropyleneurea
DMSO: Dimethylsulfoxide
EDC: l-Ethyl-3-(3-dimethylaminopropyl) carbodiimide
EtOAc: Ethyl acetate
Fmoc: 9-Fluoreiiyimethyloxycarbonyl
HATU: 0-(7-azabenzotriazol- 1 -yl)-N,N,N',N'-tetramethyluronium hexafluorophoshate
HO AT: 1 -Hydroxy-7-azahenzotriazo f e
HOBt: 1 -Hydroxybenzotriazole
HOPO: 2-Hydroxypyridine-N-oxide
IP Ac: Isopropyl acetate
MTBE: t-butyl methyl ether
NMP: N-Methyipyrro!idine
TEA: Triethylamine
THF: Tetrahydrofuran
TsOH: p-Toluenesulfonic acid
Salts
Compounds described herein having appropriate functional groups can be provided as salts. Examples of such compounds are described herein by reference to possible salts. Such reference is for illustration purposes, and additional embodiments include salts of the any compounds described herein having suitable groups.
Pharmaceutically acceptable salts can be used with compounds for treating patients. Non-pharmaceutical salts may, however, be useful in the preparation of intermediate compounds.
Pharmaceutically acceptable salts are suitable for administration to a patient, preferably, a human. Suitable salts include acid addition salts which may, for example, be formed by mixing a solution of a compound with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid. Compounds carrying an acidic moiety can be mixed with suitable pharmaceutically acceptable salts to provide, for example, alkali metal salts (e.g., sodium or potassium salts), alkaline earth metal salts (e.g., calcium or magnesium salts), and salts formed with suitable organic ligands such as quaternary ammonium salts. Also, in the case of an acid (-COOH) or alcohol group being present, pharmaceutically acceptable esters can be employed to modify the solubility or hydrolysis characteristics of the compound.
Administration and Compositions
Compounds described herein having therapeutic applications, such as Compound A, can be administered to a patient infected with HCV. The term "administration" and variants thereof (e.g., "administering" a compound) means providing the compound to the individual in need of treatment. When a compound is provided in combination with one or more other active agents (e.g., antiviral agents useful for treating HCV infection), "administration" and its variants are each understood to include concurrent and sequential provision of the compound or salt and other agents.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients, as well as any product which results, directly or indirectly, from combining the specified ingredients.
By "pharmaceutically acceptable" is meant the ingredients of the pharmaceutical composition must be compatible with each other and are suitable to the recipient thereof.
The term "subject" (alternatively referred to herein as "patient") as used herein refers to an animal, preferably a mammal, most preferably a human, who is the object of treatment, observation or experiment.
The term "effective amount" indicates a sufficient amount to exert a therapeutic or prophylactic effect. For a patient infected with HCV, an effective amount is sufficient to achieve one or more of the following effects: reduce the ability of HCV to replicate, reduce HCV load, and increase viral clearance. For a patient not infected with HCV, an effective amount is sufficient to achieve one or more of the following: a reduced susceptibility to HCV infection, and a reduced ability of the infecting virus to establish persistent infection for chronic disease.
For the purpose of inhibiting HCV NS3 protease and treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection, the compounds, optionally in the form of a salt, can be administered by means that produces contact of the active agent with the agent's site of action. They can be administered by conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are
administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
Compounds can, for example, be administered by one or more of the following routes: orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation (such as in a spray form), or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and pharmaceutically-acceptable carriers (e.g., a carrier suitable for administration to a human patient), adjuvants and vehicles. Liquid preparations suitable for oral administration (e.g., suspensions, syrups, elixirs and the like) can be prepared according to techniques known in the art and can employ media such as water, glycols, oils, alcohols and the like. Solid preparations suitable for oral administration (e.g., powders, pills, capsules and tablets) can be prepared according to techniques known in the art and can employ solid excipients as such starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like. Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as solubility aids. Injectable
solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose.
Further guidance for methods suitable for use in preparing pharmaceutical compositions is provided in Remington: The Science and Practice of Pharmacy, 21th edition (Lippincott Williams & Wilkins, 2006).
Therapeutic compounds can be administered orally in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day in a single dose or in divided doses. One dosage range is 0.01 to 500 mg/kg body weight per day orally in a single dose or in divided doses. Another dosage range is 0.1 to 100 mg/kg body weight per day orally in single or divided doses. For oral administration, the compositions can be provided in the form of tablets or capsules containing 1.0 to 500 mg of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, and 750 mg of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated. The specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy. HCV Inhibitory Activity
The ability of a compound to inhibit HCV NS3 activity, HCV replicon activity, and HCV replication activity can be evaluated using techniques well-known in the art. (See, for example, Carroll et al., J. Biol. Chem. 275: 11979-11984, 2003.)
One such assay is a HCV NS3 protease time-resolved fluorescence (TRF) assay as described below and in Mao et al, Anal. Biochem. 573: 1-8, 2008 and Mao et al.,
WO 2006/102087. A NS3 protease assay can be performed, for example, in a final volume of 100 μΐ assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15 % glycerol, 0.15 % TRITON X-100, 10 mM DTT, and 0.1 % PEG 8000. NS3 and NS4A are pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM). NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 μΐ of 500 mM MES, pH 5.5. Product fluorescence is detected using either a VICTOR V2 or FUSION fluorophotometer (Perkin Elmer Life and Analytical Sciences) with excitation at 340 nm and emission at 615 nm
with a 400 delay. Testing concentrations of different enzyme forms are selected to result in a signal to background ratio (S/B) of 10-30. IC50 values are derived using a standard four- parameter fit to the data. K; values are derived from IC50 values using the following formula,
IC50 = K1 (1 + [S] / KM), Eqn (l),
where [S] is the concentration of substrate peptide in the reaction and KM is the Michaelis constant. See P. Gallinari et al., 38 BIOCHEM. 5620-32(1999); P. Gallinari et al., 72 J. Vl OL. 6758-69 (1998); M. Taliani et al., 240 ANAL. BIOCHEM. 60-67 (1996); and Mao et al., Analytical Biochemistry 373: 1 -8, (2008). Examples
The examples provided below are intended to illustrate the invention and its practice. Unless otherwise provided in the claims, the examples are not to be construed as limitations on the scope or spirit of the invention. Preparation of 2-r2-(3-Chloro-propyl)-cvclopropyl1-4,4,5,5-tetramethyl- n,3,21dioxaborolane (Compound 3)

To a 5 L round bottomed flask equipped with a nitrogen inlet, mechanical stirrer, dropping funnel and thermocouple under N2 was added 800 mL dichloromethane and 800 mL of a 1M diethylzinc solution in heptane (0.8 mol, 1.07 equiv). The solution was cooled with an ice bath to an internal temperature of 3 °C. To the flask was then added from the dropping funnel a solution of 57.6 mL trifluoroacetic acid (0.748 mol, 1.0 equiv) in 200 mL dichloromethane over
1 hour, keeping the internal temperature below 10 °C. The resulting suspension was stirred for 30 min at 3 °C. To the flask was then added 72.4 mL diiodomethane (0.897 mol, 1.2 equiv) in a single portion. After stirring at 3 °C for 30 min, 172 mL of 2 (0.748 mol, 1.0 equiv) was added to the solution in a single portion. The flask was then allowed to warm to room temperature and a white precipitate began to form. After 3 hours, GC analysis indicated the reaction was at 90 % conversion. The suspension was aged for an additional 17 hour or until complete consumption of
2 is observed. At that point, 800 mL of 1M HCI (0.8 mol, 1.07 equiv) was added and a +5 C
exotherm was observed. The biphasic mixture was stirred for 30 min to dissolve the precipitated solids and the organic layer was separated. Extraction of the aqueous layer with 200 mL dichloromethane, washing of the combined organic layers with 500 mL brine and concentration in vacuo gave 194 g of 3 as a yellow oil (74 wt % in DCM, 79 % yield). 1H NMR (400 MHz, CDCls) δ 3.59 (t, 2H, J= 6.7 Hz), 1.90 (pent, 2H, J= 7.1 Hz), 1.49 (sext, 1H, J= 7.0 Hz), 1.36 (sext, 1H, J= 7.0 Hz), 1.23 (s, 12H), 0.93 (m, 1H), 0.71 (m, 1H), 0.44 (m, 1H), -0.35 (dt, 1H, J = 9.4, 5.7 Hz); 13C NMR (100 MHz, CDC13) δ 82.82, 44.74, 32.67, 32.22, 24.64, 17.22, 11.24, 0.5 (bs); GC: HP1 (30 m x 0.32 mm; 0.25 μιη), 25 psi, 200 °C front inlet. 5 min @ 50 °C, ramp 25 °C / min to 250 °C then hold for 4 min, tr(2)=9.78 min, tr(3)=10.08 min.
Preparation of 2-(3-Chloro-propyl)-cyclopropanol (Compound 4).

To a 3 L round bottomed flask equipped with a nitrogen inlet, mechanical stirrer, dropping funnel and thermocouple was added 143 g of 3 (0.585 mol, 1.0 equiv) in 1 L methanol. The solution was cooled with an acetone / water / dry-ice bath to an internal temperature of -8 °C. To the flask was then added from the dropping funnel 58.5 mL of 10M sodium hydroxide (0.585 mol, 1.0 equiv) over 30 min, keeping the internal temperature below 10 °C. After stirring for 30 min, 120 mL of 30 wt% hydrogen peroxide solution (1.17 mol, 2 equiv) was slowly added from the dropping funnel over lh, keeping the internal temperature below 10 °C. Upon completion of the addition, the cooling bath was removed and the resulting colorless slurry was stirred at RT (room temperature) for 30 min or until complete consumption of 3 is observed by GC. The suspension was then cooled in an ice bath to an internal temperature 2 °C and 375 mL 2 M HC1 is added from the dropping funnel over 30 min, keeping the internal temperature below 10 °C. To this clear yellow solution at 4 °C is then slowly added 500 mL of a 1 M solution of Na2S03 from the dropping funnel, keeping the internal temperature below 10 °C. The resulting suspension is then filtered and extracted 3 x 200 mL MTBE. Concentration followed by silica gel column chromatography (6:4 hexane:ethyl acetate), to remove pinacol, gave 60.6 g of product 4 as a clear oil (90 wt %, 69 % yield). 1H NMR (400 MHz, CDC13) δ 3.62 (t, 2H, J= 6.6 Hz), 3.27 (dt, 1H, J= 6.3, 2.6 Hz), 1.89 (pent, 2H, J= 6.8 Hz), 1.85 (bs, OH), 1.43 (sext, 1H, J =
7.0 Hz), 1.28 (sext, 1H, J= 7.0 Hz), 0.94 (m, 1H), 0.75 (m, 1H), 0.38 (q, lH, J= 6.0 Hz); 13C NMR (100 MHz, CDC13) δ 52.21, 44.69, 31.91, 28.69, 19.69, 14.15; GC: HP1 (30 m x 0.32 mm; 0.25 μιη), 25 psi, 200 °C front inlet. 5 min @ 50 °C, ramp 25 °C / min to 250 °C then hold for 4 min, tr(3)= 10.08 min, tr(4)=7.15 min.
Preparation of 2-Pent-4-vnyl-cvclopropanol (raoCompound 5)

DMPU, RT-50°C
To a 2-neck 15-mL round bottomed flask equipped with a temperature probe, N2 inlet, and septum was added 1 g of 4 (7.28 mmol, 1.0 equiv) and 3.0 mL THF. The solution was cooled to an internal temperature of 0 °C with an ice bath. To this solution was added 2.95 mL of 33 wt% n-Hexyllithium (7.28 mmol, 1.0 equiv) slowly via syringe pump over 1 hour. Internal temperature rose to 6.8 °C and solution became yellow. In a separate 3 -neck 100-mL round bottomed flask equipped with a temperature probe, N2 inlet, and septum 0.82 g of lithium acetylide-ethylenediamine complex (8.01 mmol, 1.1 equiv) was slurried in 5.0 mL of DMPU at room temperature. To this room temperature slurry, the cold solution of the deprotonated cyclopropanol was transferred via cannula over 5 min. After the addition, the brown mixture was heated to an internal temperature of 52 °C with a heating mantle for 3 hours or until greater than 98% conversion was observed by GC. The brown mixture was cooled with an ice bath to 3 °C and then the ice bath was removed to prevent freezing. To this was slowly added 17.5 mL of 0.5 N HC1 and an ice bath was applied to maintain an internal temperature below 21 °C. The mixture was then diluted with 10 mL MTBE and 5 mL of water before transfer to a separatory funnel and removal of the aqueous layer. The aqueous layer was extracted once with 15 mL MTBE and then the combined organic layers were washed with 20 mL water followed by 20 mL brine. The organic layer was then concentrated in vacuo to afford 1.27 g of rac-5 as a yellow oil (72 wt%, 98% yield). 1H NMR (400 MHz, CDC13) δ 3.24 (dt, 1H, J= 2.6, 5.3 Hz), 2.25 (dt, 2H, J= 2.6, 7.6 Hz), 1.96 (t, 1H, J= 2.6 Hz), 1.92 (s, 1H, OH), 1.64 (pent, 2H, J= 7.3 Hz), 1.38 (sext, 1H, J = 6.9 Hz), 1.24 (sext, 1H, J= 6.9 Hz), 0.93 (m, 1H), 0.72 (m, 1H), 0.35 (q, lH, J= 6.0 Hz); 13C NMR (100 MHz, CDC13) δ 84.49, 68.37, 52.45, 30.50, 27.74, 20.17, 18.01, 14.25; GC: HP1 (30
m x 0.32 mm; 0.25 μιη), 25 psi, 200 C front inlet. 5 min @ 50 C, ramp 25 C / min to 250 C then hold for 4 min, tr(4) = 7.15 min, tr(rac-5) = 6.72 min.
Preparation of Acetic Acid racemic tra/7s-2-pent-4-vnyl-cvclopropyl ester OaoCompound 6)

rac-5 rac-6
To a 5 L round bottomed flask equipped with a nitrogen inlet, mechanical stirrer, dropping funnel and thermocouple under N2 was added 31.2 g of rac-5 (251 mmol, 1.0 equiv), 350 mL of MTBE and 45.5 mL of triethylamine (327 mmol, 1.3 equiv) prior to cooling the solution in an acetone/ice bath to an internal temp of < 5 °C. To the solution was added from the dropping funnel 23.7 mL acetyl chloride (301 mmol, 1.1 equiv) over a 30 min period while maintaining the internal temp <10 °C. The resulting slurry was then warmed to room temperature and aged for 2 hours. At this point, the reaction mixture was diluted with 200 mL of water. The biphasic mixture was transferred to a separatory funnel and the aqueous layer removed. The organic layer was washed with 200 mL of 2 N HCl and then with 300 mL of sat. NaHC03 prior to drying over MgSC^. The solvent was removed in vacuo to give 41.8 g of rac-6 (>99% yield). 1H NMR (400 MHz, CDC13) δ 3.84 (dt, 1H, J= 6.7, 2.9 Hz), 2.25 (dt, 2H, J= 2.7, 7.0 Hz), 2.03 (s, 3H), 1.95 (t, 1H, J= 2.6 Hz), 1.67 (m, 2H), 1.39 (m, 2H), 1.01 (m, 1H), 0.89 (m, 1H), 0.57 (q, 1H, J= 6.5 Hz); 13C NMR (100 MHz, CDC13) δ 171.60, 84.15, 68.47, 54.20, 30.12, 27.40, 20.85, 17.92, 17.83, 11.81; GC: Restek RT-Bdex SA (30 m x 0.25mm x 0.25 μιη), 60 cm/s linear velocity, 20: 1 split, 120°C isothermal, tr(5) = 25.0, 29.6 min, tr(6) = 17.1, 17.5 min.
Preparation of (IR, 2i?)-2-Pent-4-vnyl-cvclopropanol (e/?t-Compound-5)
Novozym 435

rac-6 ent-5 ent-6
To a 1-L round bottom flask equipped with an overhead stirrer and temperature probe was added a 60 wt% solution of rac-6 in MTBE (44.8 g, 0.27 mol) and an additional 730 ml of MTBE that had been saturated with aqueous 0.1 M pH 7 phosphate buffer, giving a final solution concentration of rac-6 of 60 g/1. The flask was placed in an ice bath to maintain an
internal temperature of approximately 10 °C throughout the hydrolysis reaction, which was initiated by the addition of 730 mg Novozym 435. The reaction was aged at 10 °C for
approximately 4 hr until conversion had reached 41%, at which point the ee of ent-5 was 96%. The reaction mixture was then filtered through a 150-ml medium-pore glass filter funnel and the solid immobilized enzyme was washed three times with 80 ml MTBE. The resulting MTBE solution was then solvent switched to heptane. The mixture in heptane (39.2 kg, approximately 50 L) was applied to a Biotage Flash 400L cartridge (40 x 60 cm, 40 kg silica gel, 60 angstrom, 40-63 um) and eluted sequentially with 165 L of 2.5:97.5, 75 L of 10:90, and 330 L of 25:75 EtO Ac/heptane (v/v). After the mixture was applied to the column, 18 L fractions were taken. The rich cut fractions of the alcohol ent-5 were located by TLC (silica, 20% EtO Ac/heptane) and then analyzed by GC (HP-1, 30 m x 320 um x 0.25 um film, 9.14 psi constant He pressure, 15: 1 split, 50 °C for 5 min then 25 deg/min to 275 °C and hold 5 min, RT of alcohol 8.8 min).
Fractions 15-21 were concentrated to give 3.48 kg (80 wt%, 92 %>ee, 30%> yield from rac-6) of the desired ent-5.
GC: Restek RT-Bdex SA (30 m x 0.25mm x 0.25 μιη), 60 cm/s linear velocity, 20:1 split, 120°C isothermal, tr(5) = 25.0, 29.6 min, tr(6) = 17.1, 17.5 min.
Preparation of (S -3.3-dimethyl-2-(Y(Y 1 R,2R -2-(pent-4-yn- 1 - yl)cvclopropoxy)carbonyl)amino)butanoic acid (Compound 8)

Table 1
Materials MW Amount Mole Equiv
Cyclopropanol 7 124.18 2.816 kg 22.68 1.00
CDI 162.15 3.86 kg 23.81 1.05
Hunigs base 129.24 14.1 L
L-tert-leucine 131.17 3.57 kg 27.2 1.20
In a 50 L round bottom flask equipped with a mechanical stirrer, thermocouple and reflux condenser was added the starting Compound 7 (3.477 kg @ 81 wt%> by NMR, 92 %>
ee) and 14.1 L (5 L/kg) of Hunigs base. To the resulting homogeneous solution was added CDI portion wise as a solid while maintaining the internal temperature between 21-25 °C. The resulting slurry was aged at room temperature for 1 hour. To the slurry was added L-tert-leucine as a solid and the reaction mixture was heated to an internal temperature of 95 °C for 2.5 hours. The reaction mixture was cooled to room temperature with the aid of an ice-bath and diluted with 17 L of water. The mixture was aged for 30 min to dissolve all the solids and then transferred to a 100 L cylindrical extractor. The aqueous layer was then washed with 12 L of MTBE. The bottom aqueous layer was drained and the top MTBE layer was discarded. The aqueous layer was washed with 8 L of MTBE. The bottom aqueous layer was drained and the top MTBE layer was discarded.
The resulting aqueous layer was added back into the 100 L extractor and made acidic with concentrated HC1 to a final pH of 1.5-2.0. The biphasic mixture was extracted with MTBE (2 X 12 L) and the extracts were washed with 6 L of water and then 5 L of brine. NMR assay yield of the product Compound 8 at this point gave 4.49 kg (70.4 %, 20.7 kg wt of solution, 21.7 wt% product).
The MTBE layer was then transferred via vacuum into a 50 L round bottom flask equipped with a mechanical stirrer, thermocouple, and batch concentrator and the solvent was removed under reduced pressure keeping the internal temperature of the batch < 20 °C during the distillation. The solvent was then switched to cyclopentyl methyl ether (CPME) by flushing with ~ 5 L of CPME and then diluted to a final volume of ~ 20 L. This material was used in the next reaction without further purification.
An analytical sample was obtained by silica gel chromatography as a colorless oil: 1H NMR (CDC13, 400 MHz) δ 0.54 (q, 1H, J= 6.4 Hz), 0.83 (m, 1H), 0.99 (m, 1H), 1.01 (s, 9H), 1.40 (m, 2H), 1.67 (m, 2H), 1.94 (t, 1H, J= 2.6 Hz), 2.23 (m, 2H), 3.77 (br m, 1H), 4.20 (br m, 1H), 5.28 (br m, 1H), 9.40 (br s, 1H); 13C NMR (CDC13, 100 MHz) δ 11.8, 18.0, 26.5, 27.4, 30.1, 34.6, 55.0, 62.0, 68.4, 84.2, 156.7, 175.8.
Example 2: Heterocycle Production
Pre aration of 6-Methoxy-quinoxaline-2,3-diol (Compound 10)

9 10 Table 2
Materials MW Amount Mole Equiv
Diamine 9 21 1.09 2.597 kg 12.30 1.00
Oxalic acid 90.03 1.55 kg 17.22 1.40
3N HC1 17.8 L
In a 50 L round bottomed flask equipped with a mechanical stirrer, thermocouple and condenser was added 4-methoxy-l ,2-phenylenediamine dihydrochloride salt (Compound 9) (2.65 kg @ 98 wt%, 12.30 mol), oxalic acid (1.582 kg @ 98 wt.%, 17.22 mol) and 3N HC1 (aq) (17.8 L) under nitrogen. The grey heterogeneous slurry was heated to 90 °C with steam for 7.25 hours. The reaction was monitored by HPLC. The resulting grey slurry was then cooled to an internal temperature of 20 °C overnight The slurry was filtered, water (1.0-1.5 L/Kg) was used to help with the transfer. The light grey solids were washed with 2 cake volumes water (5.0-5.5 L/Kg). The solids were dried under vacuum/N2 sweep for 24 hours, at which time the solids were still very wet. The product was then slurry washed with methanol, and dried over 48 hours at 40-45 °C in a vacuum oven to give 2.304 kg of Compound 10 as an off-white product (97% yield) of 99.95% purity by HPLC assay. There was no methanol by NMR and the KF= 0.05 wt. %> water.
HPLC Conditions: Zorbax Eclipse Plus C18 50 x 4.6 mm, 1.8 um, 1.5 mL/min, 210 nm, 25 °C, Eluents: Water 0.1% H3P04 (A), Acetonitrile (B). 90% A 0 min, 5% A 5 min, 5% A 6 min
Compound 9 (diamine HC1 salt) 0.394 min
Compound 10 1.55 min (sometimes two peaks)
Preparation of 2,3-Dichloro-6-methoxyquinoxaline (Compound 1 1)

10 11
Chemical Formula: CgH8N203 Chemical Formula: CgH6Cl2N20
Molecular Weight: 192.17 Molecular Weight: 229.06
Table 3
Materials MW Amount Mole Equiv
Diol 10 192.17 3.80 kg 19.77 1.00
POCI3 153.33 5.92 L 63.5 3.20
MeCN 25 L
In a 22 L round bottomed flask equipped with a mechanical stirrer, thermocouple and condenser was added to Compound 10 (3.8 kg), and charged slowly at room temperature with POCI3 (5.92 L @ 99%). There was no initial temperature change. The grey slurry was heated to 98 °C for 20 hours. After 2-3 hours the slurry turned from grey to green, then to yellow and finally turned homogeneous red. As the slurry became homogenous in POCI3, significant amounts of HC1 off-gassing were produced. The reaction was monitored by HPLC. The dark red, homogenous solution was allowed to cool slowly to below 80 °C. At this point 19 L of acetonitrile (5.0 L/Kg) was charged which produced a dark brown slurry. The reaction was cooled to 10-15 °C in an ice bath and reverse quenched into 45.6 L of cold water (12.0 L/Kg) in a 100 L cylindrical vessel. This exothermic quench was kept below 27 °C. MeCN (~ 4L) was used to aide in slurry transfer. The brown slurry was filtered and 5 L of water was used to wash the flask. The solids were washed with 1 cake volume of water (~5 L). The pH of the filtrate was acidic. The solids were next displacement washed with 2 cake volumes of 5% sodium bicarbonate (-20.00 L). The pH was between 8-9. A slurry wash was performed with 2 cake volumes of water (20 L total). The pH did not change. The solids were dried for 72 hours under reduced pressure and nitrogen flow to give 4.464 kg of tan product Compound 1 1 (99% yield) of 99.5% purity by HPLC assay with KF= 0.5 wt. % water.
HPLC Conditions: Zorbax Eclipse Plus C18 50 x 4.6 mm, 1.8 um, 1.5 mL/min,
210 nm, 25 °C, Eluents: Water 0.1% H3PO4 (A), Acetonitrile (B). 90% A 0 min, 5% A 5 min, 5% A 6 min
Compound 10 1.55 min (sometimes two peaks)
Compound 1 1 4.55 min
Preparation of Ether (Compound 13)
Table 3
Materials MW Amount Moles Ratio
Compound 11 (93.5%) 229.06 4.286 kg 17.49 1
N-Boc-4-tra/75-hydroxy-L- 245.27 4.82 kg 19.24 1.1
Proline methyl ester (98%)
DBU (98%) 152.24 4.08 kg 26.2 1.5
DMAc 20 5 V
DMSO/water (l : l) 22 L
2.5 N HC1 10 L
MTBE 68 L
Heptane
16% NaCl 11 L
To a 100 L cylinder reactor, equipped with an overhead stirrer, thermocouple, and nitrogen inlet, was charged 2.3-dichloro-6-methoxyquinoxaline Compound 11 (4.286 kg, 93.5 wt%), N-Boc-4-trafts-hydroxy-L-Proline methyl ester Compound 12 (4.82 kg), and DMAc(20 L). The resulting reaction mixture was charged DBU (4.08 kg, or 4.04 L). The mixture was stirred at 50 °C for 20-30 h to afford products Compound 13 and a regioisomer (>98 LCAP%> conversion). The reaction mixture was cooled to room temperature. Water (20 L) and MTBE (34 L) were added. After phase separation, the aqueous layer was back to extract with MTBE (34 L). The combined organic layers were washed with DMSO/water (1 : 1, 2 x 11 L), 2.5 N HC1 (1 x 10 L), water (2 x 14 L) and 16% NaCl (11 L). Assay product Compound 12 in MTBE layer was about 6.72 kg (88%)
The reaction mixture was concentrated and azotroped by MTBE until the KF of the solution was <300 ppm, and adjusted to a total volume (18 L). The solution was seeded, and stirred at room temperature for 5 hours. Heptane (21.5 L) was slowly added over 2 hours. The resulting slurry was stirred at room temperature overnight, and at 5-10 °C for 2 hours. The
crystalline solid was collected by filtration, washed with cold heptane/MTBE (3 : 1 , 16 L), heptane (10 L), and dried under vacuum with nitrogen sweep to afford desired product Compound 13 (5.68 kg, 96.7 A%, 95.5 wt%, 71% isolated yield after correction), m.p. 98.5-99 °C.
HPLC Conditions: Zorbax Eclipse Plus C18 50 x 4.6 mm, 1.8 um, 1.5 mL/min,
230 nm, 25 °C, Eluents: Water 0.1% H3P04 (A), Acetonitrile (B). 90% A 0 min, 5% A 5 min, 5% A 6 min.
Retention time for Compound 1 1 : 4.486 min.
Retention time for Compound 12: 2.422 min.
Retention time for Compound 13 : 4.990 min.
Retention time for regioisomer: 4.927 min.
Example 3 : Macrolactam Production Preparation of (S -2-((((lR,2R -2-(5-(3-(((3R,5S -l-(tert-butoxycarbonvn-5-
(methoxycarbonyl)pyrrolidin-3 -yl)oxy)-6-methoxyquinoxalin-2-yl)pent-4-yn- 1 - yl)cyclopropoxy)carbonyl)amino)-3,3-dimethylbutanoic acid (Compound 14)

8
Table 4
Materials MW Amount mMole Equiv
Chloride 13 437.87 6.00 g 13.15 1.00
Acetylene 8 281.35 5.42 g 15.79 1.20
Pd(OAc)2 224.51 0.089 g 0.395 0.03
P(t-Bu)3HBF4 290.13 0.229 g 0.789 0.06
K2C03 138.21 4.55g 32.9 2.50
MeCN 16 L
1M H3P04 98.00 32.9 mL 65.8 5.00
The above solution containing 5.42 g of acetylene Compound 8 (82 Wt% solution in MTBE) was solvent switched to a final volume of ~ 32 mL of CPME. To the resulting solution was added as a solid 6.00 g (96 wt%) of Compound 13 and the sides of the reaction flask. The slurry was warmed to an internal temperature of ~ 28-32 °C while sparging with N2 and aged for 45 min to give a homogeneous solution.
In a separate 250 mL round bottom flask equipped with a mechanical stirrer, thermocouple, nitrogen inlet inserted in the flask, and reflux condenser was added sequentially as solids 89 mg of Pd(OAc)2, 229 mg of P(t-Bu)3HBF4, and 4.55 g of K2C03. The solids were diluted with 18 mL of MeCN and the slurry was sparged with N2 for 35 min at room
temperature. To the slurry was transferred via vacuum the above solution containing the acetylene and chloride starting materials and the final reaction mixture was sparged with N2 an additional 15 min. The reaction mixture was heated to an internal temperature of 80-85 °C and aged at this temperature for 2.0 hours. HPLC analysis at this point confirmed complete consumption of Compound 13 (conversion > 99%). The reaction mixture was cooled in an ice bath to bring the internal temperature to -25-30 °C.
In a separate 500 mL cylindrical extractor was placed 32.9 mL of a 1 M solution of H3P04. To the solution was added 100 mL of IPAc. To the biphasic mixture was transferred the above reaction mixture slowly over 30 min.
The pH of the aqueous layer was confirmed to be ~1 at this point. The layers were well mixed for 15 min and allowed to separate. The bottom aqueous layer was drained and discarded. The organic layer was washed with 100 mL of water and then with 100 mL of brine. HPLC of the organic layer gave 8.40 g (93.5%) of Compound 14.
An analytical sample was obtained by silica gel chromatography and as a colorless foam: 1H NMR (CDC13, 400 MHz) δ 0.57 (q, 1H, J= 6.3 Hz), 0.88 (m, 1H), 1.02 (s, 9H), 1.03 (m, 1H), 1.27 (m, 1H), 1.45 (br s, 11 H), 1.80 (m, 2H), 2.45 (m, 1H), 2.58-2.62 (m, 3H), 3.69-4.01 (m, 9H), 4.12 (m, 1H), 4.48 and 4.51 (t, due to rotamers, 1H, J= 7.8 Hz), 5.30 (br m, 1H), 5.71 (br m, 1H), 7.10 (s, 1H), 7.29 (d, 1H, J= 9.1 Hz), 7.84 (m, 1H, J= 9.1 Hz); 13C NMPv (CDCl3, 100 MHz) δ 11.9, 17.6, 18.3, 19.2, 26.2, 26.8, 27.2, 27.4, 28.0, 28.6, 29.1, 29.7, 30.2, 34.4, 35.6, 36.5, 52.0, 54.8, 55.1, 55.4, 55.6, 55.8, 57.8, 58.2, 62.1, 73.8, 74.1, 80.7, 97.4, 105.6, 106.1, 119.3, 119.8, 129.1, 129.6, 129.8, 134.3, 140.7, 153.9, 154.5, 156.4, 156.8, 161.4, 173.3, 174.4.
HPLC Conditions: Zorbax Eclipse Plus CI 8 50 X 4.6 mm, 1.8 um, 1.5 mL/min, 210 nm, 25 °C, Eluents: Water 0.1 % H3P04 (A); MeCN (B). 90% A to 50% A 2min, 27% A 5 min, 21% A 5.2 min, 5% A 6 min, 90% A 6.1 min
Compound 13 5.5 min
Compound 14 5.7 min
Preparation of (S -2-((((lR,2R -2-(5-(3-(((3R,5S -l-(tert-butoxycarbonvn-5-
(methoxycarbonyl)pyrrolidin-3-yl)oxy)-6-methoxyquinoxalin-2- yl)pentyl)cyclopropoxy)carbonyl)amino)-3,3-dimethylbutanoic acid (Compound 15)

Table 5
Materials MW Amount mMole Equiv
Acetylene SM 14 682.76 8.1 g 1 1.86 1.00
5% Pd/C 106.42 505 mg 0.237 0.02
To the crude acetylene 8.10 g in ~ 20 L of 2: 1 IPAc/MeOH was added 505 mg of 5% Pd/C. The reaction was placed under vacuum and purged with hydrogen (3X) and hydrogenated under balloon pressure of H2 for 2 hours at which point the reaction was complete by HPLC.
The reaction mixture was filtered through Solka floe eluting with IP Ac. The filtrate was then concentrated under reduced pressure in a 100 mL round bottom flask equipped with a thermocouple, mechanical stirrer, and batch concentrator. The solvent was switched to MeCN and a final volume of ~ 50 mL. Final assay of the combined batches was 7.25 g (89%) of the product Compound 15. The crude product was used without further purification in the next step.
An analytical sample was obtained by silica gel chromatography and as a colorless foam: 1H NMR (CDC13, 400 MHz) δ 0.50 (q, 1H, J = 6.3 Hz), 1.04 (br s, 1 1 H), 1.20 (br s, 3H), 1.45 (br s, 13 H), 1.72 (m, 2H), 2.40 (m, 1H), 2.63 (m, 1H), 2.93 9m, 2H), 3.68-3.94
(m, 9H), 4.15 (br m, 1H), 4.46 and 4.60 (t, due to rotamers, 1H, J= 7.8 Hz), 5.27 (br m, 1H), 5.78 (br m, 1H), 7.18 (m, 1H), 7.20 (m, 1H), 7.85 (m, 1H); 13C NMR (CDC13, 100 MHz) δ 1 1.9, 18.5, 26.6, 27.0, 28.1 , 28.3, 28.4, 29.1 , 30.9, 32.9, 34.1 , 35.7, 36.6, 49.4, 52.1 , 52.2, 52.4, 55.1 , 55.7, 57.7, 58.2, 62.3, 73.5, 74.1 , 80.7, 106.0, 1 18.8, 128.5, 133.7, 141.1 , 148.2, 153.9, 154.5, 155.3, 157.1 , 160.4, 173.2, 173.3, 174.4.
HPLC Conditions: Zorbax Eclipse Plus CI 8 50 X 4.6 mm, 1.8 um, 1.5 mL/min, 210 nm, 25 °C, Eluents: Water 0.1 % H3P04 (A); MeCN (B). 90% A to 50% A 2min, 27% A 5 min, 21% A 5.2 min, 5% A 6 min, 90% A 6.1 min
Compound 14 5.7 min
Compound 15 6.2 min
Preparation of Macrolactam Methyl Ester (Compound 17)

Table 7
Materials MW Amount Moles Ratio
Compound 15 686.79 3.510 kg 5.1 1 1 1
PhS03H (98%) 158.18 2.062 kg 12.78 2.5
MeCN 28 L 8 V
Hunig's Base (98%)d=0.742 129.24 3.064 kg (4.09 L) 23.00 4.5
MeCN 24.6 L 7 V
HATU (97%) 380.23 2.70 kg 6.900 1.35
Cold MeCN 6 L
Heptane 8 L
To a 50 L cylinder reactor, equipped with an overhead stirrer, thermocouple, and nitrogen inlet, was charged Compound 15 (poly jug #1 : 9.9 kg x 19.1 wt%> = 1.89 kg; and poly jug #4: 9.0 kg x 18.0 wt%> = 1.62 kg) in acetonitrile solution. Benzenesulfonic acid (2.062 kg) in acetonitrile (10 L) was added over 10 min. The resulting solution was stirred at 50 °C for 3-5 hours (>99.5 A% conversion) to afford intermediate 16 A.
The reaction mixture was cooled to room temperature. Hunig's base (4.09 L) was slowly added at <30 °C. The resulting solution was stirred at room temperature for 0.5 hour. At this point, the pH of the solution should be about 8-9.
To a 100 L round bottomed flask, equipped with an overhead stirrer, thermocouple, and nitrogen inlet, was charged HATU [O-^-azabenzotriazol-l-y^-N^N'.N'- tetramethyluronium hexafluorophoshate] (2.70 kg) and acetonitrile (24.6 L) at room
temperature. To the resulting solution was slowly added intermediate Compound 16A in Hunig's base/acetonitrile solution over 8 hours with a rate about 80 mL/min. After the addition, product Compound 17 precipitated out. The resulting slurry was stirred at room temperature to allow the reaction to complete (100 A% conversion).
The slurry was concentrated to a total volume (18 L), and then aged at 5-10 °C for 3 hours. The crystalline solid was collected by filtration, washed with cold acetonitrile (6 L), and heptane (8 L), dried under vacuum with nitrogen sweep to afford desired product Compound 17 (1.90 kg, 65% isolated yield, 97.3 A% purity, assay 100 wt%), m.p. 230.3-231.3°C. The X-ray data of Compound 17 is shown in Figure 1.
HPLC Conditions: Zorbax Eclipse Plus C18 50 x 4.6 mm, 1.8 um, 1.5 mL/min, 230 nm, 25 °C, Eluents: Water 0.1% H3P04 (A), Acetonitrile (B). 90% A 0 min, 5% A 5 min, 5% A 6 min.
Retention time for Compound 15: 5.270 min.
Retention time for Compound 16A: 3.273 min.
Retention time for Compound 17: 5.423 min.
Saponification (Compound 18)

17 18
Table 8
Material MW Equiv Mass (kg) Mol vol m
Methyl Ester (17) 97.5% 568.66 1.0 3.5 6.0
LiOH 2 N 5.0 30.0 15.0
HCl 12 N 5.0 30.0 2.5
THF 17.5
Ethyl Acetate 53
Brine 10% 18
Hexanes 38.5
Hexanes/EtOAc 4: l 8.75
To a 100 L Buchi reactor vessel fitted with a N2 inlet and a temperature probe, was charged THF (17.5 L), methyl ester (Compound 17), and LiOH solution (15.0 L). The mixture was heated to 40 °C and aged for 1 hour. After cooling to 14 °C, concentrated HCl was added till a pH of 1.9 at 20 °C was obtained. To the mixture was added 53 L ethyl acetate. The layers were cut and the organic layer was washed once with 81 L 10% brine solution. The organic solution was stored overnight in poly jugs.
The ethyl acetate/ THF solution was concentrated to 4 volumes (14 L) and then flushed with 18 L ethyl acetate keeping the volume constant at 14 L. To the slurry was added 38.5 L hexanes over 4 hours. The slurry was aged overnight at ambient temperature.
The slurry was filtered, washed with 8.75 L of 4: 1 hexanes :EtO Ac, and dried under vacuum with a N2 sweep at ambient temperature for 60 hours to give product 18 (3.36 kg,
98% isolated yield, 98.7 LCAP%), m.p. 248.5-249.5°C. The X-ray data of Compound 18 is shown in Figure 2.
Analytical Data
Relative RT Area %
0.75 1.11
1.05 0.25
1.12 0.61
1.18 0.14 total imp 2.11
Acid 97.89
HPLC Conditions:
Column: Zorbax Eclipse Plus CI 8 50x4.6 nm 1.8 um
Gradient: A = ACN, C= Buffer 0.1% H3P04
10% A to 95% A over 6 min to 10% A over 0.1 min. hold till 8 min.
Flow = 1.5 mL/min
Temp 25 °C
Wavelength = 210 nm
Retention Times: Methyl ester 34 = 5.421 min, Carboxylic acid 17 = 4.827 min -protection of Side Chain (Compound 19)

19
Table 9
Material MW Equiv Mass (kg) Mol
Boc side chain 330.40 1 2.261 6.84
TsOH monohydrate 98.5% 190.22 1.8 2.34 12.12
Ethyl Acetate 33.6
The Boc-sulfonamide was charged in a 75 L 4-neck round bottomed flask fitted with overhead stirrer, temperature probe, and N2 inlet. Ethyl acetate (22.6 L) was added followed by the TsOH. The mixture was aged at ambient temperature for 23.5 hours The mixture is homogeneous at the start, but forms precipitates during the overnight age giving a free flowing white slurry.
The solids (Compound 19) were filtered, washed with 11 L ethyl acetate and dried under vacuum with a N2 sweep at ambient temperature for 48 hours.
1H NMR: Solids after drying = 0.5% w/w ethyl acetate.
Net Wt = 2.611 kg = 94.5% IY
Purity = 99.8 LCAP
Analytical Data
Relative RT Area %
0.41 0.10
1.31 0.25
total imp 0.35
side chain 99.65
HPLC Conditions:
Column: Zorbax Eclipse Plus C18 50x4.6 nm 1.8 um
Gradient: A = ACN, B= Buffer 0.1% HC104
5% A to 95% A over 6 min to 10% A over 0.1 min. hold till 8 min.
Flow = 0.5 mL/min
Temp 25 °C
Wavelength = 210 nm
Retention Times: Boc-protected side chain = 3.455 min; De-Boc Compound 19 = 0.646 min

Table 10
Materials MW Amount Moles Equiv carboxylic acid 18 554.63 2.70 kg 4.87 1.00 cyclopropyl amine 19 402.49 2.52 kg 5.84 1.20
HOBt-H20 153.14 1.79 kg 11.68 2.40
DIPEA 129.24 2.04 L 11.68 2.40
EDC 191.70 1.12 kg 5.84 1.20
DMF 15 L
MeCN 30 L
1M HC1 10.7 L 10.70 2.2
Water 33.5 L
In a 50 L jacketed cylindrical vessel equipped with a mechanical stirrer, thermocouple and nitrogen inlet containing 5 L DMF was added the carboxylic acid (Compound 18), the cyclopropyl amine (Compound 19) and HOBt-H20 with stirring. The walls of the flask were rinsed with 10 L DMF and to the resulting mixture was added the DIPEA while the flask was cooled at 10 °C. The resulting brown slurry was stirred for 30 min.
To the resulting solution was added portion wise the EDC. The resulting solution was then stirred at 18 °C for 18 hours. The brown solution was then transferred to a 100 L RBF containing 10 L MeCN and equipped with a mechanical stirrer, thermocouple, dropping funnel and nitrogen inlet. The 50 L jacketed cylindrical vessel was flushed with 5 L MeCN. To this brown solution was slowly added the 1M HC1 from the dropping funnel while cooling in an ice bath, maintaining an internal temperature below 22 °C.
To the resulting orange solution was then added slowly from the dropping funnel 20 L of water. The resulting slurry was stirred for 3 hours at ambient temperature and then filtered.
The wet cake was washed with 30 L of 1 :1 MeCN:H20 and then dried under
N2/vacuum sweep to give 3.66 kg of Compound A (94 wt%, 95.2 LCAP, 92 % yield).
HPLC Conditions: Zorbax Eclipse Plus C18 50 x 4.6 mm, 1.8 μιη, A=0.1% phosphoric acid, C=Acetonitrile: 10% to 95% C, 5 min; 95% C, 6 min; 10 %C, 6.1 min; 2 min post, 1.5 mL/min, 230 nm, 25 C.
Compound 18 4.85 min
Compound 19 0.79 min
Compound A 5.41 min
Example 4: Alternative Coupling Procedure for Producing Compound A

Compound A, hydrate form
Table 12
Materials MW Amount Moles
Mac-acid Penultimate (18) 554.6 1.06kg 1.91 (l .OOeq)
Amine-pTSA (19) 402.5 0.86kg 2.14 (1.12eq)
EDC-HC1 191.70 0.59kg 3.06 (1.60eq)
Pyridine 2.12L (13.7eq)
MeCN 7.42L
HCl 2N 11.1L 22.2 (11.6eq)
MeCN/water 8.0L
Water 6.0L
To a 50L round bottomed flask with overhead stirring was added Mac-acid 18 (1.06kg crude, l .OOeq), amine-pTSA (19, 862g crude, 1.12q) and MeCN 7.42L at 19 °C. The slurry was cooled in a water bath, pyridine (2.12L, 13.8eq) was added, aged 15 minutes, and then added EDC (586g, 1.60eq) in one portion, aged 1.5 hours while it turned into a clear
homogeneous solution.
The solution cooled in a water bath, then quenched with 2 N HCl ( 1.7 L), seeded (9.2 g), aged 15 minutes, and the rest of the aqueous HCl was added over 2.5 hours. A yellow slurry was formed. The slurry was aged overnight at RT, filtered, washed with MeCN/water (1 : 1 v/v) 8L, to obtain Compound A (Hydrate II).
Compound A was dissolved in acetone 4L at RT, filtered and transferred to a 12 L RBF with overhead stirring, rinsed with extra acetone 1L, heated to 50 °C, water 0.9L was
added, seeded lOg, aged 15minutes, then added water 0.8L over 2.5 hours, extra water 3.3v over 2.5 hours was added, stopped heating, cooled to RT, aged at RT overnight, filtered, washed with water/acetone (1 : 1 v/v) 4L, and dried in air under vacuum. Compound A Hydrate III, 670 g, was obtained as an off-white solid.
None of the references described throughout the present application are admitted to be prior art to the claimed invention.
Claims
1. A com ound selected from the group consisting of:  wherein R is either H, acetyl, or C(0)NHCH(X)COOH, and X alkyl, or a C3-C8 cycloalkyl,
wherein R is either H, acetyl, or C(0)NHCH(X)COOH, and X alkyl, or a C3-C8 cycloalkyl,

14 15 and salts thereof, wherein X is a protecting group
The compound of claim 1 , wherein said compound
3. A method of making Compound 6, comprising the step: 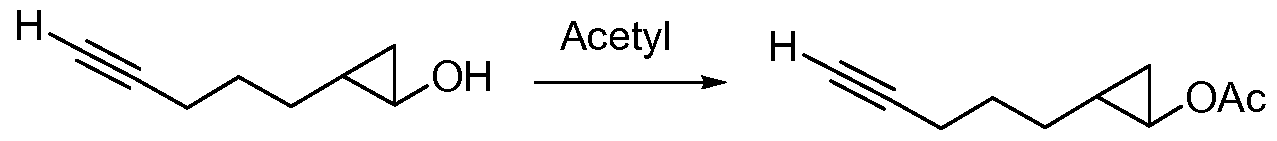
4. The method of claim 3, wherein Compound 5 is produced by: 
5. The method of claim 4, wherein Compound 4 is produced by oxidizing
Compound 3 : 
6. The method of claim 5, wherein Compound 3 is produced by: 
7. A method of making a compound of Formula I
H
C6 alkyl, or a C3-C8 cycloalkyl; or a salt thereof; comprising the step of: 
8
8. A method of producing Compound 5, comprising the step of: 
13 wherein PG is a protecting group.
9. The method of claim 8, comprising the following steps
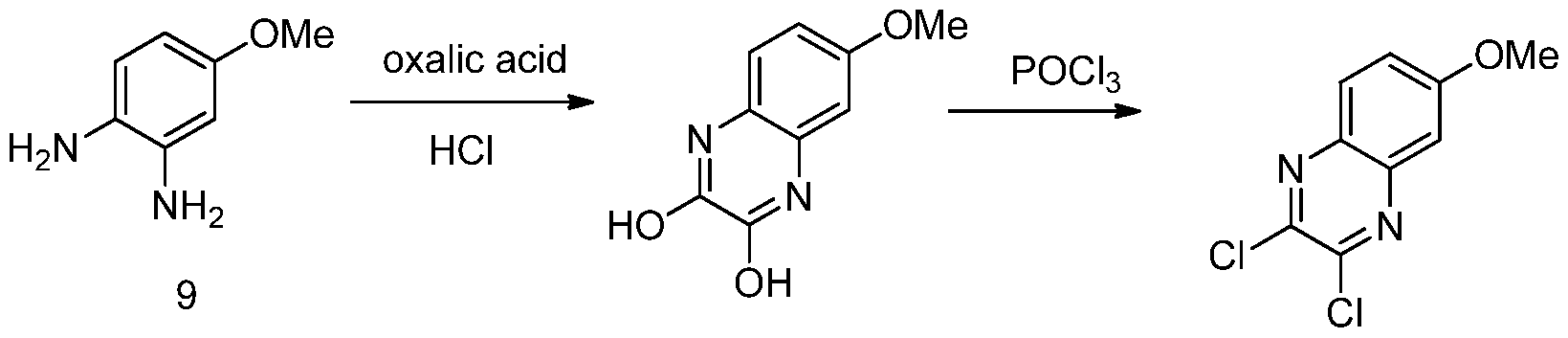
10
1 1 or salts thereof.
A method of producing a compound comprising the step of:

14 or salts thereof, wherein PG is a protecting group.
11. The method of claim 10, wherein the reaction uses tri tert-butylphosphine tetra fluoroborate salt in a solvent, and Compound 8 is provided as either a dibenzylamine salt or a t-butylamine salt.
12. The method of claim 10 or 11, further comprising the steps of: 
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12825726.8A EP2744331A4 (en) | 2011-08-19 | 2012-08-16 | Methods and intermediates for preparing macrolactams |
| US14/239,391 US9073825B2 (en) | 2011-08-19 | 2012-08-16 | Methods and intermediates for preparing macrolactams |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161525462P | 2011-08-19 | 2011-08-19 | |
| US61/525,462 | 2011-08-19 | ||
| US201161533439P | 2011-09-12 | 2011-09-12 | |
| US61/533,439 | 2011-09-12 | ||
| US201161533915P | 2011-09-13 | 2011-09-13 | |
| US61/533,915 | 2011-09-13 | ||
| US201161539540P | 2011-09-27 | 2011-09-27 | |
| US61/539,540 | 2011-09-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013028471A1 true WO2013028471A1 (en) | 2013-02-28 |
Family
ID=47746773
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/051168 WO2013028465A1 (en) | 2011-08-19 | 2012-08-16 | Crystal forms of a hcv protease inhibitor |
| PCT/US2012/051182 WO2013028471A1 (en) | 2011-08-19 | 2012-08-16 | Methods and intermediates for preparing macrolactams |
| PCT/US2012/051177 WO2013028470A1 (en) | 2011-08-19 | 2012-08-16 | Process and intermediates for preparing macrolactams |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/051168 WO2013028465A1 (en) | 2011-08-19 | 2012-08-16 | Crystal forms of a hcv protease inhibitor |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/051177 WO2013028470A1 (en) | 2011-08-19 | 2012-08-16 | Process and intermediates for preparing macrolactams |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US9073825B2 (en) |
| EP (3) | EP2744336B1 (en) |
| JP (2) | JP2014521750A (en) |
| KR (2) | KR20140059236A (en) |
| CN (2) | CN103874414A (en) |
| AU (2) | AU2012299223A1 (en) |
| BR (2) | BR112014003802A2 (en) |
| CA (2) | CA2844388A1 (en) |
| MX (2) | MX2014001944A (en) |
| RU (2) | RU2014110399A (en) |
| WO (3) | WO2013028465A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2015095437A1 (en) * | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Methods and intermediates for the preparation of macrolactams |
| US9073825B2 (en) | 2011-08-19 | 2015-07-07 | Merck Sharp & Dohme Limited | Methods and intermediates for preparing macrolactams |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9873707B2 (en) | 2013-10-18 | 2018-01-23 | Merck Sharp & Dohme Corp. | Methods and intermediates for preparing macrolactams |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA119315C2 (en) | 2012-07-03 | 2019-06-10 | Гіліад Фармассет Елелсі | Inhibitors of hepatitis c virus |
| EP2764866A1 (en) | 2013-02-07 | 2014-08-13 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| ES2735355T3 (en) | 2013-03-15 | 2019-12-18 | Gilead Sciences Inc | Hepatitis C virus macrocyclic and bicyclic inhibitors |
| RS65417B1 (en) * | 2014-01-24 | 2024-05-31 | Turning Point Therapeutics Inc | Diaryl macrocycles as modulators of protein kinases |
| MX2016016127A (en) * | 2014-06-06 | 2017-02-23 | Abbvie Inc | Crystal forms. |
| WO2017004342A1 (en) | 2015-07-02 | 2017-01-05 | Tp Therapeutics, Inc. | Chiral diaryl macrocycles as modulators of protein kinases |
| CN108026108B (en) | 2015-07-06 | 2021-06-22 | 特普医药公司 | Diaryl macrocyclic polymorphs |
| DK3325488T3 (en) * | 2015-07-21 | 2020-09-14 | Turning Point Therapeutics Inc | Chiral diaryl macrocycle and its use in the treatment of cancer |
| CN109562113A (en) | 2016-05-10 | 2019-04-02 | C4医药公司 | Loop coil degron body for target protein degradation |
| WO2017197055A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| CN109641874A (en) | 2016-05-10 | 2019-04-16 | C4医药公司 | C for target protein degradation3The glutarimide degron body of carbon connection |
| SG11201900163PA (en) | 2016-07-28 | 2019-02-27 | Tp Therapeutics Inc | Macrocycle kinase inhibitors |
| TWI808958B (en) | 2017-01-25 | 2023-07-21 | 美商特普醫葯公司 | Combination therapy involving diaryl macrocyclic compounds |
| IL271964B1 (en) | 2017-07-28 | 2024-08-01 | Turning Point Therapeutics Inc | Macrocyclic compounds and uses thereof |
| SI3728271T1 (en) | 2017-12-19 | 2023-01-31 | Turning Point Therapeutics, Inc. | Macrocyclic compounds for treating diseases |
| CN111057045A (en) * | 2019-12-18 | 2020-04-24 | 安徽红杉生物医药科技有限公司 | HCV NS3/4A protease inhibitor intermediate, and synthesis method and application thereof |
| CN112174982A (en) * | 2020-09-10 | 2021-01-05 | 上海希迈医药科技有限公司 | Lopitinib crystal form and preparation method thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4871868A (en) * | 1987-03-11 | 1989-10-03 | Takeda Chemical Industries, Ltd. | Production of substituted acetylenic compounds |
| WO2008057209A1 (en) * | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| US7507262B2 (en) * | 2006-03-07 | 2009-03-24 | The Procter & Gamble Company | Compositions for oxidatively dyeing keratin fibers and methods for using such compositions |
| WO2010011566A1 (en) * | 2008-07-22 | 2010-01-28 | Merck & Co., Inc. | Macrocyclic quinoxaline compounds as hcv ns3 protease inhibitors |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9207987D0 (en) * | 1992-04-10 | 1992-05-27 | Smithkline Beecham Plc | Novel container and closure |
| US5716960A (en) * | 1995-01-13 | 1998-02-10 | U.S. Bioscience Inc. And Individuals | Crystalline trimetrexate salts and the process for making the same |
| NO317155B1 (en) | 1997-02-04 | 2004-08-30 | Ono Pharmaceutical Co | <Omega> -cycloalkyl-prostaglandin-E <N> 2 </ N> derivatives |
| ZA98879B (en) * | 1997-02-04 | 1998-08-03 | Ono Pharmaceutical Co | Omega-cycloalkyl-prostaglandin e2 derivatives |
| CN102372704B (en) | 2005-02-18 | 2014-10-08 | 田边三菱制药株式会社 | Salt of proline derivative, solvate thereof, and production method thereof |
| US7834145B2 (en) | 2005-03-22 | 2010-11-16 | Merck Sharp & Dohme Corp. | HCV protease substrates |
| CA2606195C (en) | 2005-05-02 | 2015-03-31 | Merck And Co., Inc. | Hcv ns3 protease inhibitors |
| US7470664B2 (en) | 2005-07-20 | 2008-12-30 | Merck & Co., Inc. | HCV NS3 protease inhibitors |
| UA90909C2 (en) * | 2005-07-20 | 2010-06-10 | Мерк Шарп Энд Домэ Корп. | Hcv ns3 protease inhibitors |
| AU2006275605B2 (en) | 2005-08-01 | 2011-01-06 | Merck Sharp & Dohme Corp. | Macrocyclic peptides as HCV NS3 protease inhibitors |
| GB0609492D0 (en) | 2006-05-15 | 2006-06-21 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| GB0612423D0 (en) | 2006-06-23 | 2006-08-02 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| AU2007309546A1 (en) | 2006-10-24 | 2008-05-02 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | HCV NS3 protease inhibitors |
| JP5345941B2 (en) | 2006-10-24 | 2013-11-20 | メルク・シャープ・アンド・ドーム・コーポレーション | HCV NS3 protease inhibitor |
| EP2083844B1 (en) | 2006-10-27 | 2013-11-27 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| KR20100024920A (en) | 2007-05-03 | 2010-03-08 | 인터뮨, 인크. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| WO2009010804A1 (en) | 2007-07-19 | 2009-01-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic compounds as antiviral agents |
| WO2009108507A1 (en) | 2008-02-25 | 2009-09-03 | Merck & Co., Inc. | Therapeutic compounds |
| WO2009131196A1 (en) * | 2008-04-24 | 2009-10-29 | 武田薬品工業株式会社 | Substituted pyrrolidine derivative and use thereof |
| CA2722326A1 (en) | 2008-04-24 | 2009-10-29 | Incyte Corporation | Macrocyclic compounds and their use as kinase inhibitors |
| EP2271345B1 (en) | 2008-04-28 | 2015-05-20 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| CA2770338A1 (en) * | 2009-08-27 | 2011-03-03 | Merck Sharp & Dohme Corp. | Processes for preparing protease inhibitors of hepatitis c virus |
| CA2844388A1 (en) | 2011-08-19 | 2013-02-28 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
-
2012
- 2012-08-16 CA CA2844388A patent/CA2844388A1/en not_active Abandoned
- 2012-08-16 CA CA2844386A patent/CA2844386A1/en not_active Abandoned
- 2012-08-16 KR KR1020147006888A patent/KR20140059236A/en not_active Application Discontinuation
- 2012-08-16 WO PCT/US2012/051168 patent/WO2013028465A1/en active Application Filing
- 2012-08-16 RU RU2014110399/04A patent/RU2014110399A/en not_active Application Discontinuation
- 2012-08-16 US US14/239,391 patent/US9073825B2/en active Active
- 2012-08-16 JP JP2014526217A patent/JP2014521750A/en active Pending
- 2012-08-16 US US14/239,389 patent/US9242917B2/en active Active
- 2012-08-16 BR BR112014003802A patent/BR112014003802A2/en not_active IP Right Cessation
- 2012-08-16 EP EP12826404.1A patent/EP2744336B1/en not_active Not-in-force
- 2012-08-16 EP EP12825540.3A patent/EP2744507A4/en not_active Withdrawn
- 2012-08-16 AU AU2012299223A patent/AU2012299223A1/en not_active Abandoned
- 2012-08-16 US US14/239,393 patent/US9238604B2/en active Active
- 2012-08-16 WO PCT/US2012/051182 patent/WO2013028471A1/en active Application Filing
- 2012-08-16 MX MX2014001944A patent/MX2014001944A/en unknown
- 2012-08-16 CN CN201280050361.6A patent/CN103874414A/en active Pending
- 2012-08-16 MX MX2014001945A patent/MX2014001945A/en unknown
- 2012-08-16 WO PCT/US2012/051177 patent/WO2013028470A1/en active Application Filing
- 2012-08-16 EP EP12825726.8A patent/EP2744331A4/en not_active Withdrawn
- 2012-08-16 AU AU2012299218A patent/AU2012299218A1/en not_active Abandoned
- 2012-08-16 RU RU2014110400/04A patent/RU2014110400A/en not_active Application Discontinuation
- 2012-08-16 CN CN201280050382.8A patent/CN103889439A/en active Pending
- 2012-08-16 JP JP2014526215A patent/JP2014524442A/en active Pending
- 2012-08-16 KR KR1020147006858A patent/KR20140053330A/en not_active Application Discontinuation
- 2012-08-16 BR BR112014003798A patent/BR112014003798A2/en not_active IP Right Cessation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4871868A (en) * | 1987-03-11 | 1989-10-03 | Takeda Chemical Industries, Ltd. | Production of substituted acetylenic compounds |
| US7507262B2 (en) * | 2006-03-07 | 2009-03-24 | The Procter & Gamble Company | Compositions for oxidatively dyeing keratin fibers and methods for using such compositions |
| WO2008057209A1 (en) * | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2010011566A1 (en) * | 2008-07-22 | 2010-01-28 | Merck & Co., Inc. | Macrocyclic quinoxaline compounds as hcv ns3 protease inhibitors |
Non-Patent Citations (3)
| Title |
|---|
| BASSAN ET AL.: "Multikilogram-Scale Synthesis of a Chiral Cyclopropanol and an Investigation of the Safe Use of Lithium Acetylide Ethylene Diamine Complex", ORG PROCESS RES DEV, vol. 16, 7 December 2011 (2011-12-07), pages 87 - 95, XP055143025 * |
| See also references of EP2744331A4 * |
| SONG ET AL.: "Synthesis of Vaniprevir (MK-7009): Lactamization To Prepare a 22-Membered Macrocycle", J ORG CHEM, vol. 76, 12 August 2011 (2011-08-12), pages 7804 - 7815, XP055143024 * |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9527885B2 (en) | 2011-05-05 | 2016-12-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9073825B2 (en) | 2011-08-19 | 2015-07-07 | Merck Sharp & Dohme Limited | Methods and intermediates for preparing macrolactams |
| US9238604B2 (en) | 2011-08-19 | 2016-01-19 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| US9242917B2 (en) | 2011-08-19 | 2016-01-26 | Merck Sharp & Dohme Limited | Crystal forms of a HCV protease inhibitor |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9873707B2 (en) | 2013-10-18 | 2018-01-23 | Merck Sharp & Dohme Corp. | Methods and intermediates for preparing macrolactams |
| WO2015095437A1 (en) * | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Methods and intermediates for the preparation of macrolactams |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2014521750A (en) | 2014-08-28 |
| EP2744336B1 (en) | 2017-07-05 |
| KR20140053330A (en) | 2014-05-07 |
| CN103889439A (en) | 2014-06-25 |
| RU2014110400A (en) | 2015-09-27 |
| BR112014003802A2 (en) | 2017-06-13 |
| AU2012299223A1 (en) | 2014-02-27 |
| US9238604B2 (en) | 2016-01-19 |
| CA2844386A1 (en) | 2013-02-28 |
| US20140200343A1 (en) | 2014-07-17 |
| MX2014001945A (en) | 2014-03-27 |
| AU2012299218A1 (en) | 2014-02-20 |
| CA2844388A1 (en) | 2013-02-28 |
| EP2744336A1 (en) | 2014-06-25 |
| EP2744507A1 (en) | 2014-06-25 |
| WO2013028465A1 (en) | 2013-02-28 |
| US9073825B2 (en) | 2015-07-07 |
| US9242917B2 (en) | 2016-01-26 |
| JP2014524442A (en) | 2014-09-22 |
| BR112014003798A2 (en) | 2017-03-01 |
| EP2744331A1 (en) | 2014-06-25 |
| US20140206605A1 (en) | 2014-07-24 |
| CN103874414A (en) | 2014-06-18 |
| EP2744331A4 (en) | 2015-01-21 |
| EP2744507A4 (en) | 2015-01-28 |
| US20140243519A1 (en) | 2014-08-28 |
| MX2014001944A (en) | 2014-03-27 |
| WO2013028470A1 (en) | 2013-02-28 |
| EP2744336A4 (en) | 2014-12-31 |
| RU2014110399A (en) | 2015-09-27 |
| KR20140059236A (en) | 2014-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013028471A1 (en) | Methods and intermediates for preparing macrolactams | |
| JP6034802B2 (en) | Methods and intermediates for the preparation of macrocyclic lactams | |
| CN111343990B (en) | Benzodiazepine-2-one and benzodiazepine-2-one derivatives | |
| CN102140100B (en) | Polycyclic compound for inhibiting hepatitis C virus efficiently, preparation method thereof and application thereof | |
| TWI406660B (en) | Macrocyclic inhibitors of hepatitis c virus | |
| JP5021711B2 (en) | Hepatitis C inhibitor tripeptide | |
| WO2015095430A1 (en) | Methods and intermediates for the preparation of macrolactams | |
| RU2776703C2 (en) | Methods for separation of benzodiazepine-2-one and benzoazepine-2-one derivatives | |
| WO2015095437A1 (en) | Methods and intermediates for the preparation of macrolactams |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12825726 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14239391 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012825726 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012825726 Country of ref document: EP |