WO2013016081A1 - Notch pathway signaling inhibitor compound - Google Patents
Notch pathway signaling inhibitor compound Download PDFInfo
- Publication number
- WO2013016081A1 WO2013016081A1 PCT/US2012/047100 US2012047100W WO2013016081A1 WO 2013016081 A1 WO2013016081 A1 WO 2013016081A1 US 2012047100 W US2012047100 W US 2012047100W WO 2013016081 A1 WO2013016081 A1 WO 2013016081A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- compound
- pharmaceutically acceptable
- hydrate
- acceptable salt
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 71
- 108010070047 Notch Receptors Proteins 0.000 title claims abstract description 33
- 102000005650 Notch Receptors Human genes 0.000 title claims abstract description 33
- 230000011664 signaling Effects 0.000 title claims abstract description 20
- 230000037361 pathway Effects 0.000 title abstract description 10
- 239000003112 inhibitor Substances 0.000 title abstract description 8
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 66
- 201000011510 cancer Diseases 0.000 claims abstract description 40
- 150000003839 salts Chemical class 0.000 claims abstract description 35
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 8
- 229940125904 compound 1 Drugs 0.000 claims description 28
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 22
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 22
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 19
- 238000000034 method Methods 0.000 claims description 17
- 206010009944 Colon cancer Diseases 0.000 claims description 16
- 208000005017 glioblastoma Diseases 0.000 claims description 16
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 15
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 15
- 201000001441 melanoma Diseases 0.000 claims description 15
- 206010006187 Breast cancer Diseases 0.000 claims description 14
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 14
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 14
- 206010033128 Ovarian cancer Diseases 0.000 claims description 14
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 14
- 208000026310 Breast neoplasm Diseases 0.000 claims description 13
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 13
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 13
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 13
- 201000002528 pancreatic cancer Diseases 0.000 claims description 13
- 239000003814 drug Substances 0.000 claims description 12
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 claims description 11
- 230000002401 inhibitory effect Effects 0.000 claims description 8
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 7
- 208000031637 Erythroblastic Acute Leukemia Diseases 0.000 claims description 7
- 208000036566 Erythroleukaemia Diseases 0.000 claims description 7
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 7
- 208000000172 Medulloblastoma Diseases 0.000 claims description 7
- 206010060862 Prostate cancer Diseases 0.000 claims description 7
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 7
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 7
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 7
- 208000021841 acute erythroid leukemia Diseases 0.000 claims description 7
- 201000010881 cervical cancer Diseases 0.000 claims description 7
- 201000010536 head and neck cancer Diseases 0.000 claims description 7
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 7
- 201000007270 liver cancer Diseases 0.000 claims description 7
- 208000014018 liver neoplasm Diseases 0.000 claims description 7
- 201000005202 lung cancer Diseases 0.000 claims description 7
- 208000020816 lung neoplasm Diseases 0.000 claims description 7
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 7
- 201000000849 skin cancer Diseases 0.000 claims description 7
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 7
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 5
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- 229910016523 CuKa Inorganic materials 0.000 claims description 3
- 230000001154 acute effect Effects 0.000 claims description 3
- 230000005855 radiation Effects 0.000 claims description 2
- 238000002560 therapeutic procedure Methods 0.000 claims description 2
- 208000032839 leukemia Diseases 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 90
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 75
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 48
- 210000004027 cell Anatomy 0.000 description 44
- 238000003756 stirring Methods 0.000 description 44
- 239000000243 solution Substances 0.000 description 43
- 239000012141 concentrate Substances 0.000 description 36
- 239000000203 mixture Substances 0.000 description 36
- 239000007787 solid Substances 0.000 description 36
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 34
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 239000011541 reaction mixture Substances 0.000 description 18
- 239000010410 layer Substances 0.000 description 17
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 15
- 108090000623 proteins and genes Proteins 0.000 description 15
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- 230000005764 inhibitory process Effects 0.000 description 14
- 238000002360 preparation method Methods 0.000 description 14
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 13
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- XZOWIJDBQIHMFC-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O.CCCC(N)=O XZOWIJDBQIHMFC-UHFFFAOYSA-N 0.000 description 11
- 239000012091 fetal bovine serum Substances 0.000 description 11
- 229910052938 sodium sulfate Inorganic materials 0.000 description 11
- 235000011152 sodium sulphate Nutrition 0.000 description 11
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 10
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 239000012065 filter cake Substances 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 230000004060 metabolic process Effects 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000002207 metabolite Substances 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 8
- 241000699670 Mus sp. Species 0.000 description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 8
- 108700037638 Neurogenic locus notch homolog protein 1 Proteins 0.000 description 8
- 239000013078 crystal Substances 0.000 description 8
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 239000002953 phosphate buffered saline Substances 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 102000002659 Amyloid Precursor Protein Secretases Human genes 0.000 description 7
- 108010043324 Amyloid Precursor Protein Secretases Proteins 0.000 description 7
- 238000002965 ELISA Methods 0.000 description 7
- 230000001594 aberrant effect Effects 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 238000003818 flash chromatography Methods 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 6
- 229930182816 L-glutamine Natural products 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 238000003776 cleavage reaction Methods 0.000 description 6
- 230000029142 excretion Effects 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 239000008103 glucose Substances 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- 230000007017 scission Effects 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 208000031404 Chromosome Aberrations Diseases 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 101800001628 Notch 1 intracellular domain Proteins 0.000 description 5
- 102400000552 Notch 1 intracellular domain Human genes 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 231100000005 chromosome aberration Toxicity 0.000 description 5
- LGIBDQRYOFBMTC-UHFFFAOYSA-N dnc010031 Chemical compound C1=CC(O)=CC=C1C1C(=O)NC2=CC=CC=C2C2=C3C1=CNC3=NC=C2 LGIBDQRYOFBMTC-UHFFFAOYSA-N 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 230000036210 malignancy Effects 0.000 description 5
- 238000002483 medication Methods 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- GDZSEAOCYLAVOG-UHFFFAOYSA-N 5,7-dihydropyrido[2,3-d][3]benzazepin-6-one Chemical compound N1C(=O)CC2=CC=CC=C2C2=CC=CN=C21 GDZSEAOCYLAVOG-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 239000007995 HEPES buffer Substances 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 4
- 239000012980 RPMI-1640 medium Substances 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 229960001484 edetic acid Drugs 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 210000003141 lower extremity Anatomy 0.000 description 4
- 239000006166 lysate Substances 0.000 description 4
- 239000012139 lysis buffer Substances 0.000 description 4
- 108010082117 matrigel Proteins 0.000 description 4
- AMVCFIFDMKEIRE-UHFFFAOYSA-N methyl 2-(2-bromophenyl)acetate Chemical compound COC(=O)CC1=CC=CC=C1Br AMVCFIFDMKEIRE-UHFFFAOYSA-N 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 230000002018 overexpression Effects 0.000 description 4
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 238000010254 subcutaneous injection Methods 0.000 description 4
- 239000007929 subcutaneous injection Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 230000004614 tumor growth Effects 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- ANLRCVLRDIIGKK-BYPYZUCNSA-N (2s)-2-(4,4,4-trifluorobutanoylamino)propanoic acid Chemical compound OC(=O)[C@H](C)NC(=O)CCC(F)(F)F ANLRCVLRDIIGKK-BYPYZUCNSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- WRDABNWSWOHGMS-UHFFFAOYSA-N AEBSF hydrochloride Chemical compound Cl.NCCC1=CC=C(S(F)(=O)=O)C=C1 WRDABNWSWOHGMS-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 238000003109 Karl Fischer titration Methods 0.000 description 3
- 239000005909 Kieselgur Substances 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 229920004890 Triton X-100 Polymers 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 230000003466 anti-cipated effect Effects 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 210000000349 chromosome Anatomy 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000002559 cytogenic effect Effects 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 208000003747 lymphoid leukemia Diseases 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 210000004940 nucleus Anatomy 0.000 description 3
- 231100000590 oncogenic Toxicity 0.000 description 3
- 230000002246 oncogenic effect Effects 0.000 description 3
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 3
- 230000003285 pharmacodynamic effect Effects 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 238000000371 solid-state nuclear magnetic resonance spectroscopy Methods 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 2
- DWXSYDKEWORWBT-UHFFFAOYSA-N 2-(2-bromophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC=C1Br DWXSYDKEWORWBT-UHFFFAOYSA-N 0.000 description 2
- 125000006016 2-bromoethoxy group Chemical group 0.000 description 2
- XZKIHKMTEMTJQX-UHFFFAOYSA-N 4-Nitrophenyl Phosphate Chemical compound OP(O)(=O)OC1=CC=C([N+]([O-])=O)C=C1 XZKIHKMTEMTJQX-UHFFFAOYSA-N 0.000 description 2
- BTKKMGSOUTXABL-UHFFFAOYSA-N 7-amino-5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]-7h-pyrido[2,3-d][3]benzazepin-6-one Chemical compound NC1C(=O)N(CCO[Si](C)(C)C(C)(C)C)C2=NC=CC=C2C2=CC=CC=C21 BTKKMGSOUTXABL-UHFFFAOYSA-N 0.000 description 2
- VNIDFFLZHAANMV-UHFFFAOYSA-N 7-azido-5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]-7h-pyrido[2,3-d][3]benzazepin-6-one Chemical compound [N-]=[N+]=NC1C(=O)N(CCO[Si](C)(C)C(C)(C)C)C2=NC=CC=C2C2=CC=CC=C21 VNIDFFLZHAANMV-UHFFFAOYSA-N 0.000 description 2
- 206010069754 Acquired gene mutation Diseases 0.000 description 2
- 108010039627 Aprotinin Proteins 0.000 description 2
- 238000000035 BCA protein assay Methods 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- MQUQNUAYKLCRME-INIZCTEOSA-N N-tosyl-L-phenylalanyl chloromethyl ketone Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N[C@H](C(=O)CCl)CC1=CC=CC=C1 MQUQNUAYKLCRME-INIZCTEOSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 102000001753 Notch4 Receptor Human genes 0.000 description 2
- 108010029741 Notch4 Receptor Proteins 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 208000009052 Precursor T-Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- XXAXKCWOTRABOW-UHFFFAOYSA-N [1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]azanium;(4-nitrophenyl) phosphate Chemical compound OCC(N)(CO)CO.OCC(N)(CO)CO.OP(O)(=O)OC1=CC=C([N+]([O-])=O)C=C1 XXAXKCWOTRABOW-UHFFFAOYSA-N 0.000 description 2
- 230000035508 accumulation Effects 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 201000011186 acute T cell leukemia Diseases 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 229960004405 aprotinin Drugs 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- PXXJHWLDUBFPOL-UHFFFAOYSA-N benzamidine Chemical compound NC(=N)C1=CC=CC=C1 PXXJHWLDUBFPOL-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 210000000013 bile duct Anatomy 0.000 description 2
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000010405 clearance mechanism Effects 0.000 description 2
- 201000010989 colorectal carcinoma Diseases 0.000 description 2
- 238000005388 cross polarization Methods 0.000 description 2
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 2
- 108010073357 cyanoginosin LR Proteins 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- AGCOMFFHXJMNLN-UHFFFAOYSA-N dichloromethane;dihydrochloride Chemical compound Cl.Cl.ClCCl AGCOMFFHXJMNLN-UHFFFAOYSA-N 0.000 description 2
- 238000002050 diffraction method Methods 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 2
- 235000011180 diphosphates Nutrition 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 208000037902 enteropathy Diseases 0.000 description 2
- 230000004049 epigenetic modification Effects 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 238000000265 homogenisation Methods 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 208000028774 intestinal disease Diseases 0.000 description 2
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 2
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 2
- 108010052968 leupeptin Proteins 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- ZYZCGGRZINLQBL-GWRQVWKTSA-N microcystin-LR Chemical compound C([C@H](OC)[C@@H](C)\C=C(/C)\C=C\[C@H]1[C@@H](C(=O)N[C@H](CCC(=O)N(C)C(=C)C(=O)N[C@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]([C@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1)C(O)=O)C(O)=O)C)C1=CC=CC=C1 ZYZCGGRZINLQBL-GWRQVWKTSA-N 0.000 description 2
- DIDLWIPCWUSYPF-UHFFFAOYSA-N microcystin-LR Natural products COC(Cc1ccccc1)C(C)C=C(/C)C=CC2NC(=O)C(NC(CCCNC(=N)N)C(=O)O)NC(=O)C(C)C(NC(=O)C(NC(CC(C)C)C(=O)O)NC(=O)C(C)NC(=O)C(=C)N(C)C(=O)CCC(NC(=O)C2C)C(=O)O)C(=O)O DIDLWIPCWUSYPF-UHFFFAOYSA-N 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- QNDVLZJODHBUFM-WFXQOWMNSA-N okadaic acid Chemical compound C([C@H](O1)[C@H](C)/C=C/[C@H]2CC[C@@]3(CC[C@H]4O[C@@H](C([C@@H](O)[C@@H]4O3)=C)[C@@H](O)C[C@H](C)[C@@H]3[C@@H](CC[C@@]4(OCCCC4)O3)C)O2)C(C)=C[C@]21O[C@H](C[C@@](C)(O)C(O)=O)CC[C@H]2O QNDVLZJODHBUFM-WFXQOWMNSA-N 0.000 description 2
- VEFJHAYOIAAXEU-UHFFFAOYSA-N okadaic acid Natural products CC(CC(O)C1OC2CCC3(CCC(O3)C=CC(C)C4CC(=CC5(OC(CC(C)(O)C(=O)O)CCC5O)O4)C)OC2C(O)C1C)C6OC7(CCCCO7)CCC6C VEFJHAYOIAAXEU-UHFFFAOYSA-N 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 238000005192 partition Methods 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 2
- 229910000105 potassium hydride Inorganic materials 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 2
- 239000003531 protein hydrolysate Substances 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 230000037439 somatic mutation Effects 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- LSGOVYNHVSXFFJ-UHFFFAOYSA-N vanadate(3-) Chemical compound [O-][V]([O-])([O-])=O LSGOVYNHVSXFFJ-UHFFFAOYSA-N 0.000 description 2
- CGRCVIZBNRUWLY-HNNXBMFYSA-N (2s)-2-[(4-methylphenyl)sulfonylamino]-3-phenylpropanoic acid Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 CGRCVIZBNRUWLY-HNNXBMFYSA-N 0.000 description 1
- QFELHROMWPLTIU-NHYWBVRUSA-N (2s)-2-amino-n-[(7s)-5-(2-hydroxyethyl)-6-oxo-7h-pyrido[2,3-d][3]benzazepin-7-yl]propanamide Chemical compound OCCN1C(=O)[C@@H](NC(=O)[C@@H](N)C)C2=CC=CC=C2C2=CC=CN=C21 QFELHROMWPLTIU-NHYWBVRUSA-N 0.000 description 1
- KFNRNFXZFIRNEO-NSHDSACASA-N (2s)-5-(diaminomethylideneamino)-2-[(4-methylphenyl)sulfonylamino]pentanoic acid Chemical compound CC1=CC=C(S(=O)(=O)N[C@@H](CCCN=C(N)N)C(O)=O)C=C1 KFNRNFXZFIRNEO-NSHDSACASA-N 0.000 description 1
- AEMWUHCKKDPRSK-UHFFFAOYSA-N (ne)-n-diazo-2,4,6-tri(propan-2-yl)benzenesulfonamide Chemical compound CC(C)C1=CC(C(C)C)=C(S(=O)(=O)N=[N+]=[N-])C(C(C)C)=C1 AEMWUHCKKDPRSK-UHFFFAOYSA-N 0.000 description 1
- PKDNWHHCOAPXJN-UHFFFAOYSA-N 1-amino-5-(2-hydroxyethyl)-7h-pyrido[3,2-a][3]benzazepin-6-one Chemical compound OCCN1C(=O)CC2=CC=CC=C2C2=C1N=CC=C2N PKDNWHHCOAPXJN-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- JBKINHFZTVLNEM-UHFFFAOYSA-N 2-bromoethoxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCCBr JBKINHFZTVLNEM-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- ZJSQZQMVXKZAGW-UHFFFAOYSA-N 2H-benzotriazol-4-ol hydrate Chemical compound O.OC1=CC=CC2=C1N=NN2 ZJSQZQMVXKZAGW-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- WTUCTMYLCMVYEX-UHFFFAOYSA-N 4,4,4-trifluorobutanoic acid Chemical compound OC(=O)CCC(F)(F)F WTUCTMYLCMVYEX-UHFFFAOYSA-N 0.000 description 1
- FPOOEWLDYMORLM-UHFFFAOYSA-N 5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]-7h-pyrido[2,3-d][3]benzazepin-6-one Chemical compound C1C(=O)N(CCO[Si](C)(C)C(C)(C)C)C2=NC=CC=C2C2=CC=CC=C21 FPOOEWLDYMORLM-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 description 1
- 241001136792 Alle Species 0.000 description 1
- 206010003497 Asphyxia Diseases 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 241001000171 Chira Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000016718 Chromosome Inversion Diseases 0.000 description 1
- 206010052360 Colorectal adenocarcinoma Diseases 0.000 description 1
- 230000007067 DNA methylation Effects 0.000 description 1
- 230000033616 DNA repair Effects 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- 208000037162 Ductal Breast Carcinoma Diseases 0.000 description 1
- 230000010665 Enzyme Interactions Effects 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 208000034951 Genetic Translocation Diseases 0.000 description 1
- 208000031448 Genomic Instability Diseases 0.000 description 1
- 108010033040 Histones Proteins 0.000 description 1
- 101000978766 Homo sapiens Neurogenic locus notch homolog protein 1 Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 238000005004 MAS NMR spectroscopy Methods 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 101000978776 Mus musculus Neurogenic locus notch homolog protein 1 Proteins 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 108700019961 Neoplasm Genes Proteins 0.000 description 1
- 102000048850 Neoplasm Genes Human genes 0.000 description 1
- 230000005913 Notch signaling pathway Effects 0.000 description 1
- 102000001760 Notch3 Receptor Human genes 0.000 description 1
- 108010029756 Notch3 Receptor Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 101710163806 Trypsin/chymotrypsin inhibitor Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 210000003484 anatomy Anatomy 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- 239000003855 balanced salt solution Substances 0.000 description 1
- RLMHWGDKMJIEHH-QRPNPIFTSA-N benzyl (2s)-2-aminopropanoate;hydrochloride Chemical compound Cl.C[C@H](N)C(=O)OCC1=CC=CC=C1 RLMHWGDKMJIEHH-QRPNPIFTSA-N 0.000 description 1
- DHCLVCXQIBBOPH-UHFFFAOYSA-N beta-glycerol phosphate Natural products OCC(CO)OP(O)(O)=O DHCLVCXQIBBOPH-UHFFFAOYSA-N 0.000 description 1
- GHRQXJHBXKYCLZ-UHFFFAOYSA-L beta-glycerolphosphate Chemical compound [Na+].[Na+].CC(CO)OOP([O-])([O-])=O GHRQXJHBXKYCLZ-UHFFFAOYSA-L 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 201000008274 breast adenocarcinoma Diseases 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- VDPVJUVIPQXKKF-UHFFFAOYSA-N butanamide hydrate Chemical compound O.CCCC(N)=O VDPVJUVIPQXKKF-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- JJWKPURADFRFRB-UHFFFAOYSA-N carbonyl sulfide Chemical compound O=C=S JJWKPURADFRFRB-UHFFFAOYSA-N 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 208000037516 chromosome inversion disease Diseases 0.000 description 1
- 210000001726 chromosome structure Anatomy 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000013256 coordination polymer Substances 0.000 description 1
- 238000005384 cross polarization magic-angle spinning Methods 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000834 fixative Substances 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 235000015203 fruit juice Nutrition 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000003540 gamma secretase inhibitor Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000003304 gavage Methods 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 235000008216 herbs Nutrition 0.000 description 1
- IZZWJPQHPPRVLP-UHFFFAOYSA-N hexane;2-methoxy-2-methylpropane Chemical class CCCCCC.COC(C)(C)C IZZWJPQHPPRVLP-UHFFFAOYSA-N 0.000 description 1
- 102000045609 human NOTCH1 Human genes 0.000 description 1
- 210000004293 human mammary gland Anatomy 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- GPGMRSSBVJNWRA-UHFFFAOYSA-N hydrochloride hydrofluoride Chemical compound F.Cl GPGMRSSBVJNWRA-UHFFFAOYSA-N 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 238000013394 immunophenotyping Methods 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000007154 intracellular accumulation Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 206010073095 invasive ductal breast carcinoma Diseases 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 210000001853 liver microsome Anatomy 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- AIICBFSEEBAMMB-UHFFFAOYSA-N methyl 2-[2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]acetate Chemical compound COC(=O)CC1=CC=CC=C1B1OC(C)(C)C(C)(C)O1 AIICBFSEEBAMMB-UHFFFAOYSA-N 0.000 description 1
- 210000001589 microsome Anatomy 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 1
- 230000006654 negative regulation of apoptotic process Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 238000004172 nitrogen cycle Methods 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 230000004783 oxidative metabolism Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000009131 signaling function Effects 0.000 description 1
- 231100000161 signs of toxicity Toxicity 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000030968 tissue homeostasis Effects 0.000 description 1
- 238000004481 total suppression of sideband Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- -1 tri-t-butylphosphonium tetrafluoroborate Chemical compound 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 230000036325 urinary excretion Effects 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06026—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atom, i.e. Gly or Ala
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- Notch signaling is an evolutionary conserved pathway that plays an integral role in development and tissue homeostasis in mammals.
- the Notch receptors and ligands contain single-pass transmembrane domains, are expressed on the cell surface and, for that reason, Notch signaling is particularly important in mediating communication between adjacent cells expressing the receptors and ligands.
- Notch 1 to Notch 4 There are four known Notch receptors found in rodents and humans, termed Notch 1 to Notch 4.
- the Notch receptors are heterodimeric proteins composed of extracellular and intracellular domains that are initially synthesized as a single polypeptide. Receptor-ligand interaction triggers a series of proteolytic cleavages of the Notch receptor polypeptide in which ⁇ -secretase activity is involved.
- Notch intracellular domain 3SIICD
- Notch intracellular domain 3SIICD
- Notch signaling functions include proliferation, differentiation, apoptosis, angiogenesis, migration and self-renewal. These diverse roles of Notch signaling during the development and maintenance of normal tissues are aberrantly activated in different forms of cancer.
- the oncogenic functions of Notch signaling include the inhibition of apoptosis and the promotion of cell proliferation.
- ⁇ -Secretase plays a pivotal role in the Notch activation cascade.
- inhibitors of ⁇ -secretase have been actively investigated for their potential to block Notch receptor activation.
- the compounds exemplified in WO 98/28268 such as 7C-203 are represe tative of such ⁇ -secretase inhibitors.
- no commercial Notch inhibitor chemotherapeutic agents have emerged.
- Figure 1 is a representative X-ray powder diffraction pattern for the compound of Example 2.
- One aspect of the invention is to provide a Notch signaling inhibitor compound of the structure:
- a second aspect of the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising 4,4,4-trifluoro-N-[(l S)-2-[[(7S)-5-(2 iydroxyethyl)-6-oxo-7H-pyrido[2,3- d] [3]benzazepm-7-yl]amino]- 1 -memyl-2-oxo-emyl]butanamide,
- the pharmaceutical composition comprises 4,4,4-trifluoro-N-[(l S)-2[[(7S)-5-(2-hydroxyetiiyl)-6-oxo-7H- pyrido[2,3-d] [3]benzoazepm-7-yl]amino]- 1 -methyl-2-oxo-ethyl]butanamide, or a pharmaceutically acceptable salt or hydrate thereof, together with a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- a third aspect of the present invention provides a method of inhibiting Notch signaling in a cancer patient in need, thereof, comprising administering a therapeutically effective amount of, 4,4,4-trifluoro-N-[(lS)-2-[[(7S)-5-(2-hydroxyethyl)-6-oxo-7H- pyrido[2,3-d][3]benzazepin-7-yl]amino]-l -meihyl-2-oxo-ethyl]butanamide, or a pharmaceutically acceptable salt or hydrate thereof, to said patient.
- a fourth aspect of the present invention provides a method of treating a cancer which is T-cell acute lymphoblastic leukemia, acute lymphoblastic leukemia, acute myelogenous leukemia, chronic myelogenous leukemia, erythroleukemia, breast cancer, ovarian cancer, melanoma, lung cancer, pancreatic cancer, glioblastoma, colorectal cancer, head and neck cancer, cervical cancer, prostate cancer, liver cancer, squamous cell carcinoma (oral), skin cancer or medulloblastoma in a patient comprising administering to a patient in need thereof a therapeutically effective amount of 4,4,4-trifluoro-N-[(l S)-2- [[(7S)-5-(2 ⁇ hydroxyethyl)-6-oxo-7H ⁇ pyrid.o[2,3-cl][3]benzazepin-7-yl]amino]-l-methyl- 2-oxo-ethyl]butanamide, or a pharmaceutically
- a fifth aspect of the present invention provides a method of treating a cancer which is T-cell acute lymphoblastic leukemia, acute lymphoblastic leukemia, chronic myelogenous leukemia, erythroieukemia, breast cancer, ovarian cancer, melanoma, pancreatic cancer, glioblastoma or colorectal cancer in a patient comprising administering to a patient in need thereof a therapeutically effective amount of 4,4,4-tri.fluoro-N-[(l S)- 2-[[(7S)-5-(2 iydroxyethyl)-6-oxo-7H ⁇ pyrid.o[2,3-d][3]beiizazepin-7-yl]amino]-l- methyl-2-oxo-ethyl]butanamide, or a pharmaceutically acceptable salt or hydrate thereof.
- a sixth aspect of the present invention provides a compound 4,4,4-trifluoro-N- [(lS)-2-[[(7S)-5-(2-hydroxyethyl)-6-oxo-7H-pyrido[2,3-d][3]benzazepin-7-yl]ammo]-l- methyl-2-oxo-ethyl]butanamide, or a pharmaceutically acceptable salt or hy drate thereof, for use in therapy.
- a seventh aspect of the present invention provides a compound 4,4,4-trifluoro-N- [( 1 S)-2- [ [(7S)-5-(2-hydroxyethyl)-6-oxo-7H-pyrido[2, 3 -d] [3 ] benzazepin-7-y 3] amino] ⁇ 1 ⁇ methyl-2-oxo-ethyl]butanamide, or a pharmaceutically acceptable salt or hydrate thereof, for use in the treatment of a cancer which is T-cell acute lymphoblastic leukemia, acute lymphoblastic leukemia, acute myelogenous leukemia, chronic myelogenous leukemia, erythroieukemia, breast cancer, ovarian cancer, melanoma, lung cancer, pancreatic cancer, glioblastoma, colorectal cancer, head and neck cancer, cervical cancer, prostate cancer, liver cancer, squamous cell carcinoma (oral), skin cancer or medulloblastoma.
- a cancer which is T
- An eighth aspect of the present invention provides a compound 4,4,4-trifluoro-N- [(lS)-2-[[(7S)-5-(2-hy ⁇ xyethyl)-6-oxo-7H-pyrido[2,3-d][3]benzazepin-7-yl]amino]-l- methyl-2-oxo-ethyl]butanamide, or a pharmaceutically acceptable salt or hydrate thereof, for use in the treatment of a cancer which is T-cell acute lymphoblastic leukemia, acute lymphoblastic leukemia, chronic myelogenous leukemia, erythroieukemia, breast cancer, ovarian cancer, melanoma, pancreatic cancer, glioblastoma or colorectal cancer.
- a ninth aspect of the present invention provides use of a compound. 4,4,4- trifluoro-N-[(l S)-2-[[(7S)-5-(2- ⁇
- yl]amino]-l-methyl-2-oxo-ethyl]butanarnide or a pharmaceutically acceptable salt or hydrate thereof, for the manufacture of a medicament for the treatment of a cancer which is T-celi acute lymphoblastic leukemia, acute lymphoblastic leukemia, acute
- myelogenous leukemia chronic myelogenous leukemia, eiythroleukem a, breast cancer, ovarian cancer, melanoma, lung cancer, pancreatic cancer, glioblastoma, colorectal cancer, head and neck cancer, cervical cancer, prostate cancer, liver cancer, squamous cell carcinoma (oral), skin cancer or medulloblastoma.
- a tenth aspect of the present invention provides use of a compound 4,4,4- trifluoro-N-[(l S)-2-[[(7S)-5-(2-hydroxye l)-6-oxo-7H-pyrido[2,3-d][3]benzazepin-7- yl]amino]- l-methyi-2-oxo-ethyl]butanamide, or a pharmaceutically acceptable salt or hydrate thereof, for the manufacture of a medicament for the treatment of a cancer which is T-cell acute lymphoblastic leukemia, acute lymphoblastic leukemia, chronic myelogenous leukemia, eiythroleukemia. breast cancer, ovarian cancer, melanoma, pancreatic cancer, glioblastoma or colorectal cancer.
- a cancer which is T-cell acute lymphoblastic leukemia, acute lymphoblastic leukemia, chronic myelogenous leukemia, eiythroleukemia.
- patient means mammal and “mammal” includes, but is not limited to, a human.
- “Therapeutically effective amount” or “effective amount” means the dosage of the compound, or pharmaceutically acceptable salt or hydrate thereof, or pharmaceutical composition containing the compound, or pharmaceutically acceptable salt or hydrate thereof, necessary to inhibit Notch signaling in a cancer patient, and either destroy the target cancer cells or slow or arrest the progression of the cancer in a patient.
- Anticipated dosages of Compound 1 or a pharmaceutically acceptable salt or hydrate thereof are in the range of 0.1 to 200 mg/patient/day. Preferred dosages are anticipated to be in the range of 1 to 175 mg/patient/day. Most preferred dosages are anticipated to be in the range of 5 to 150 mg/patient/day.
- the exact dosage required to treat a patient and the length of treatment time will be determined by a physician in view of the stage and severity of the disease as well as the specific needs and. response of the individual patient. Although expressed as dosage on a per day basis, the dosing regimen may be adjusted to provide a more optimal therapeutic benefit to a patient and to manage and ameliorate mucoid enteropathy (hypersecretion and accumulation of mucus in the gastrointestinal tract).
- dosing every other day Q2D
- every other day over a five day- period followed by two days without dosing T.T.W.
- every third day Q3D
- a dosing regimen of every other day, l. l.W .. or every third day is preferred along with administration (pre-, concomitant, or post-administration of Compound 1 of dexameihasone to manage or ameliorate mucoid enteropathy.
- treatment are meant to include the full spectrum of intervention for the cancer from which the patient is suffering, such as administration of the active compound to alle viate to slow or reverse one or more of the symptoms and to delay progression of the cancer even if the cancer is not actually eliminated.
- the patient to be treated is a mammal, in particular a human being.
- the compound, of the present invention is preferably formulated as a
- compositions using a pharmaceutically acceptable carrier and administered by a variety of routes.
- such compositions are for oral administration.
- Such pharmaceutical compositions and processes for preparing them are well known in the art. See, e.g., REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (A.
- the pharmaceutical composition comprises 4,4,4-trifluoro-N-[(l S)-2-[[(7S)-5-(2- hydroxy ethyl) - 6 - oxo-7H-pyrido[2,3-d] [3]benzoazepin-7-yl]amino]- 1 -methyl-2-oxo- ethyl Jbutanamide, or a pharmaceutically acceptable salt or hydrate thereof, together with a pharmaceutically acceptable carrier and optionally other therapeutic ingredients particularly for treatment of cancer generally or a specific cancer type.
- the compound of the present invention is capable of reaction with a number of inorganic and organic acids to form pharmaceutically acceptable acid addition salts.
- pharmaceutically acceptable salts and common methodology for preparing them are well known in the art. See, e.g., P. Stahl, et ai, HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE, (VCHA/Wiley-VCH, 2002); S.M. Berge, et a!., "Pharmaceutical Salts, " Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977.
- Compound 1, or a pharmaceutically acceptable salt or hydrate thereof may be prepared by a variety of procedures known in the art, as well as those described below. The specific synthetic steps may be combined in different ways to prepare Compound 1 , or a pharmaceutically acceptable salt or hydrate thereof.
- Compound 1 is named: 4,4,4-trifluoro-N-[(l S)-2-[[(7S)-5-(2-hydroxyethyl)-6- oxo-7H-pyrido[2,3-d] [3]benzazepm-7-yl]ammo]- 1 -methyl ⁇ 2-oxo-ethyf]butanamid.e; and may also be named: -[(l S)-2 ⁇ [[(7S)-6,7-dihydro-5-(2-hydroxyethyl)-6-oxo-5H- pyrido[3,2-a] [3]benzazepin-7-yl]amino]-l -methyl-2-oxoethyl]-4,4,4-trifluorobutanamide; and other names may be used to unambiguously identify Compound 1.
- Atropisomer 2 exists as atropisomers, or specific conformers.
- atropisomer 2 is detected by i f NMR and LC-MS in equilibs "ium with atropisomer 1 (major atropisomer) at ambient temperature after 24 hours.
- atropisomer 2 is detected by ⁇ NMR and LC-MS in equilibrium with atropisomer 1 .
- atropisomer 2 is not isolable.
- the intermediates and Compound 1 are named using a SymaxDraw version 3.2 Drawing Program, from the structures, as the TUPAC name consistently applied.
- dichloromethane (430 mL) and separate the layers. Discard the organic layer and to the aqueous phase add dichloromethane (645 mL) and adjust the pH to approximately 10 using aqueous sodium hydroxide (20%. 252 mL). Separate the layers, extract the aqueous layer with additional dichloromethane (2 x 430 mL), and combine the organic phases. Concentrate the organic under reduced pressure below 45 "C to an approximate volume of 130- 150 mL, and add tetraliydrofuran (322 mL). Concentrate the solution under reduced pressure below 45 °C to an approximate volume of 200-220 mL, and add additional tetrahydrofuran (213 mL).
- the sample is scanned between 4 and 40° in 2 ⁇ , with a step size of 0.0087° in 2 ⁇ and a scan rate of 0.5 seconds/step, and with 0.6 mm divergence, 5,28mm fixed anti-scatter, and 9.5 mm detector slits.
- the dry powder is packed, on a quartz sample holder and a smooth surface is obtained using a glass slide.
- peak positions can shift due to a variation in the temperature or humidity at which a sample is analyzed, sample displacement, or the presence or absence of an internal standard.
- a peak position variability of ⁇ 0.2 in 2 ⁇ will take into account these potential variations without hindering the unequivocal identification of the indicated crystal form.
- Confirmation of a crystal form may be made based on any unique combination of distinguishing peaks (in units of ° 2 ⁇ ), typically the more prominent peaks.
- the crystal form diffraction pattern is collected at ambient temperature (19-25°C) and relative humidity (20-60%).
- a prepared sample of the compound of Example 2 is characterized by an XRPD pattern using CuKa radiation as having diffraction peaks (2-theta values) as described in Table 1 below.
- the form is crystalline and contains a peak at 22.97 degree in combination with one or more of the peaks selected from the group consisting of 11 .96, 18.81 , 20.78, and 21.07 degrees 2-theta, with a tolerance for the diffraction angles of 0.2 degrees.
- Example 2 Goniometer.
- the control standard is determined using Hydranol® as a water standard in duplicate. Run the sample in triplicate and record the average percentage of water to determine the amount of water in a sample. Karl Fischer Titration average result of Example 2 is 3.9% water. Theoretic percentage of one molar equivalent of water in Example 2 is 3.7%.
- Cancer is increasingly recognized as a heterogeneous collection of diseases whose initiation and progression are induced by the aberrant function of one or more genes that regulate DNA repair, genome stability, cell proliferation, cell death, adhesion, angiogenesis, invasion, and. metastasis in cell and tissue microenvirome ts.
- Variant or aberrant function of the "cancer" genes may result from naturally occurring DNA polymorphism, changes in genome copy number (through amplification, deletion, chromosome loss, or duplication), changes in gene and chromosome structure (through chromosomal translocation, inversion, or other rearrangement that leads to deregulated gene expression), and point mutations.
- Cancerous neoplasms may be induced by one aberrant gene function, and maintained by the same aberrant gene function, or maintenance and. progression exacerbated by additional aberrant gene functions.
- each of the cancers may also include epigenetic modifications of the genome including DNA methylation, genomic imprinting, and. histone modification by acetylation, methvlation, or phosphorylation.
- An epigenetic modification may play a role in the induction and/or maintenance of the malignancy.
- chromosome aberrations in suspected cases of cancer can be determined through cytogenetic analysis such as fluorescence in situ hybridization (FISH), karyotyping, spectral karyotyping (SKY), multiplex FISH (M- FISH), comparative genomic hybridization (CGH), single nucleotide polymorphism arrays (SNP Chips) and other diagnostic and analysis tests known and used by those skilled in the art.
- FISH fluorescence in situ hybridization
- SKY karyotyping
- M- FISH multiplex FISH
- CGH single nucleotide polymorphism arrays
- SNP Chips single nucleotide polymorphism arrays
- lymphoblastic leukemia acute myelogenous leukemia, chronic myelogenous leukemia and erythroleukemia.
- Aberrant constitutive Notch signaling due to mutation or over expression of ligands and/or receptors is also implicated in a number of solid tumor malignancies including breast cancer, ovarian cancer (Park et al. Cancer Research, 2006(66):6312-6318), melanoma (Gast et al. Genes, Chromosomes & Cancer,
- Notch 3 and Notch 4 receptors Assays evidencing Notch pathway signaling inhibitory activity and efficacy may be carried out substantially as follows or by similar assays affording similar data. Notch 1 N 1 ICD N ucl ear Aec mul ati on Cell lar Imagi g A say
- ⁇ 293 ⁇ 12 cells (HEK293 cells are engineered to stably express mouse Notch 1 cDNA coding for amino acid 1703-2183, NP 032740.3, with 23 amino acid signal peptide sequence, MPRL.LTPLLCLTLLPALAARGLR (SEQ ID NO: l), at its N- terminus) are plated at 5000 cells/well in 96 well plates, incubated in Dulbecco's Modified Eagle's Medium-high glucose with 5% fetal bovine serum at 37 °C, 5 % CO? for 24 hours.
- Cells are treated with test compound, dosing at 10 points of 1 :3 dilutions across the range of 1000 nM to 0.05 tiM, and with final dimethyl sulfoxide (DM SO) concentration at 0.2%.
- DM SO dimethyl sulfoxide
- cell plates are processed through following steps sequentially: fix ceils with ⁇ /welI PREFERTM fixative for 30 minutes at room temperature (RT); permeablize cells with ⁇ /well 0.1% TRITON® X 100 in phosphate buffered saline (PBS) for 20 min at RT; wash 3 times with 1 ⁇ /well PBS each; add 50 ⁇ rabbit anti-N lICD (Notch 1 Intracellular Domain) antibody, at 1 :2000 in PBS with 1 % bovine serum albumin and incubate 1.5 hours at 37 °C; wash 3 times with ⁇ /well PBS each; incubate with 50 ⁇ 1 ⁇ 13 goat anti-rabbit IgG Alexa 488 at 1 : 1000
- NIICD The antibody that recognizes cleaved Notch 1 or NIICD is raised, to a human peptide corresponding to the amino terminal cleavage site of human Notch 1 at Vail 744.
- NIICD generated from Notch 1 will translocate and accumulate in nucleus.
- Concentration response and the IC5.3 are determined by curve fitting to a four parameter logistic for the nuclear NIICD signal, while the %cell number is plotted in the same graph for cytotoxicity profiling. Performing the assay essentially as described above, the average IC5.3 for
- Compound 1 has affinity for Notch 1 and inhibits the intracellular accumulation of the Notch 1 intracellular domain cell signaling peptide.
- A2780 is a human ovarian cell line (Sigma- Aidrich, No. 93112519); MIA PaCa-2 is a human pancreas cell line (ATCC No. CRL- 1420); BxPC-3 is a human pancreas cell line (ATCC No. CRL-1687); SW480 is a human colorectal cell line (ATCC No. CCL-228); HCT 116 is a human colorectal cell line (ATCC No. CCL-247); DLD-1 is a human colorectal cell line (ATCC No.
- MD.A-MB-231 is a human mammary gland cell line (ATCC No. HTB-26); U-87 MG is a human glioblastoma cell line (ATCC No. HTB-14); A375 is a human malignant melanoma cell line (ATCC No. CRL-1619); CCRF-CEM is a human acute lymphoblastic leukemia (ALL) cell line (ATCC No. CCL-i 19); SUP-T1 is a human T-cell
- lymphoblastic leukemia cell line (ATCC No. CRL-1942); K-562 is a human chronic myelogenous leukemia (CML) cell line characterized by the presence of a fusion transcript comprised of the Bcr and Abll genes (ATCC No. CCL-243); Jurkat, Clone E6- 1 is a human acute T-cell leukemia cell line (ATCC No. TIB-152); MOLT-3 is a human acute lymphoblastic leukemia (ALL) cell line (ATCC No. CRL-1552); M.OLT-4 is a human acute lymphoblastic leukemia (ALL) cell line (ATCC No. CRL-1582); HEL 92.1 .7 is a human erythroleukemia cell line (ATCC No.
- Each of the cell lines are obtained from the American Type Culture Collection (ATCC) at the ATCC number stated,, except the A2780 cell line which is obtained from Sigma- Aldrich at the stated catalog number.
- ATCC American Type Culture Collection
- the cells are grown in their respective culture media at 37°C in 5% C0 2 with humidity in the atmosphere.
- Cell culture media for A2780 human ovarian carcinoma is RPMI-1640 (without phenol red) with. 2.05 mM L-glutamine, added 2 mM.
- HCT 1 16 human colorectal carcinoma is McCoy's 5A with 1.5 mM L-glutamine, 0.075% Na-bicarbonate and 10% FBS
- SW480 human colorectal carcinoma is RPMI- 1640 with 2.05 mM L- glutamine, 20 mM HEPES and 10% FBS
- U-87 MG human glioblastoma is Minimal Essential MediumEarl's Balanced Salt Solution with 2 mM L-glutamine, 0.1 mM Nonessential amino acids (NEA.A), 1 mM Na-pyruvate, and 10% FBS
- MIA PaCa-2 human pancreatic carcinoma is Dulbecco's Modified Eagles Medium (DMEM) without Na-pyruvate, with high glucose (4500 mg/ml), 4 mM L-glutamine, 2.5% horse serum and 10% FBS
- K-562 human CML is DMEM with Dulbecco's Modified Eagles Medium (DMEM) without Na-pyruvate
- DLD-1 human colorectal adenocarcinoma SUP-T1 human lymphoblastic leukemia, MOLT-3 human ALL, Molt-4 human ALL, CCRF-CEM human ALL, and HEL 92.1.7 human erythroleukemia is RPMI-1640 with 2.05 mM L-gmtamine, 2.5 g/L glucose, 20 mM HEPES, 1 mM Na- pyruvate, and 10% FBS.
- cell plates are processed essentially through the following steps sequentially: Cells are collected after rypsinization, washed once with ice-cold PBS, and lysed in 100 ⁇ ice cold XY lysis buffer (25 mM Tris pH 7.5, 10 ⁇ g/ml Tiypsin/Chymotrypsm inhibitor, 10 .ug/nil Aprotinin, 60 mM Beta-glycerol phosphate , 1 % Triton® X-100, 10 mM NaF, 2.5 mM pyrophosphate, 150 mM NaCl, 15 mM ethylene diamine tetra acetic acid.
- DMSO dimethyl sulfoxide
- EDTA ethylene g1yco1-bis(2-ammoethylether)-N,N,N',N'-ietra acetic acid
- EGTA ethylene g1yco1-bis(2-ammoethylether)-N,N,N',N'-ietra acetic acid
- 1 mM Na Vanadate 10 g/ml Leupeptin, 1 mM dithiothreitol, 1 ⁇ microcystin LR, 10 ⁇ g/mi N-p-tosyi-L-phenylalanine chloromethyl ketone (TPCK).
- TAME 2 mM N -p- tosyl-L-arginine methyl ester hydrochloride
- PNPP 4-nitrophenyl phosphate di(tris) salt
- AEBSF 4-(2-aminoethyl) benzenesulfoiiyl fluoride hydrochloride
- Table 1 evidences the potency of Compound 1 in its ability to inhibit NIICD signaling peptide generation by inhibiting ⁇ -secretase activity and as a result NIICD signaling peptide accumulation in specific human tumor ceil lines.
- A2780 (2 x 10 6 ), SW480 (6 x 1 Q 6 ), HCT 1 16 (6 x 1 Q 6 ), U-87 MG (6 x lO 6 ), and A-375 ( 10 x 10 6 ) cells in a 1 : 1 matrigel mix (0.2 mL volume) are implanted by subcutaneous injection in the hind leg of 6-8 weeks of age athymic nude female mice (Harlan Laboratories).
- K-562 (6 x 10 n ) ceils in a 1 : 1 matrigel mix (0.2 mL volume) are implanted by subcutaneous injection in the hind leg of 6-8 weeks of age CD1 ⁇ / ⁇ female mice (Charles River Laboratories).
- HEL 92.1.7 (7 x 10°) in a 1 : 1 matrigel mix (0.2 mL volume) are implanted, by subcutaneous injection in the hind leg of 6-8 weeks of age CB17 severely combined immune deficient female mice (Taconic Farms).
- Patient- derived tumors are minced into 1-2 mm pieces and mixed with matrigel ( 1 : 1 ) in 0.2 ml volume and implanted by subcutaneous injection in the hind leg of 6-8 weeks of age athymic nude female mice (Harlan Laboratories).
- Patients -derived tumor models include: human glioblastoma (EL2144), human triple negative invasive ductal breast carcinoma (EL I 997), and human colon carcinoma (EL1989, EL 1986, and EL 2056) with samples obtained after patient consent and hospital approval from IL ! Health, Cincinnati Hospital, Indiana, USA 46206. A total of 7 to 10 mice are used for each group.
- mice are irradiated (450 Total Body Irradiation). Mice are fed ad libitum on normal chow. Treatment is initiated, with oral administration (gavage) of compound or vehicle ( 1% Na-CMC in 0,25% Tween-80) in 0,2 niL volume when tumor size reached to 150 ⁇ 50 mm 3 . At designated time points following treatment, animals are sacrificed by C0 2 asphyxiation and cervical dislocation. Tumors are removed and used for PD response analysis. Tumor growth and. body weight are monitored, over time to evaluate efficacy and signs of toxicity.
- Tumor volume data are transformed, to a log scale to equalize variance across time and treatment groups.
- the log volume data are analyzed with a two-way repeated measures analysis of variance by time and treatment using the MIXEDTM procedures in SASTM software (version 8.2).
- the correlation model for the repeated, measures is spatial power.
- Treated groups are compared to the control group at each time point.
- the MIXEDTM procedure is also used separately for each treatment group to calculate adjusted means and standard errors at each time point.
- TGI tumor growth inhibition percentage
- N 1ICD levels in tumors approximately 75 mg is cut from the frozen tumor and minced prior to homogenization (actual mass recorded). Frozen tumor samples are transferred to Lysing Matrix-DTM tubes and re-suspended in ice-cold XY lysis buffer (25 mM Tris pH 7.5, 10 g/ml Trypsin/Chymotrypsin inhibitor, 10 g/ml Aprotinin, 60 mM Beta-giycerol phosphate , 1% Triton® X-100, 10 mM NaF, 2.5 mM
- pyrophosphate 150 niM ' NaCl, 15 mM ethylene diamine tetra acetic acid (EDTA) pH 8.0, 5 mM ethylene g3yco]-bis(2-am.inoethyiether)-N, ,N',N '-tetra acetic acid (EGTA) pH 8.0, 1 mM Na Vanadate, 10 .ug/ml Leupeptin, I mM dithiothreitol, 1 ⁇ microcystin LR, 10 pg/'nil N-p-tosyl-L-phenylalanine cliloromethyi ketone (TPCK), 2 mM Na-p-tosyl-L- arginine methyl ester hydrochloride (TAME), 15 mM 4-nitrophenyl phosphate di(tris) salt (PNPP), 0.1 mM 4 ⁇ (2-aminoethyl) benzenesul
- Tissues are homogenized in a Fast Prep FP120 homogenizer (Thermo Scientific, Rockford, IL) at a speed of 6.0 for 30 seconds at 4°C, followed by 15 minute incubation on ice. This is repeated for a total of 2-- 3 cycles until homogenization is complete. Lysates are spun in a 4°C eppendorf centrifuge at 30,000 rpm for 15 minutes to remove debris.
- N1ICD levels are determined using a custom N1ICD ELISA. Anaiyte is captured with a cleaved
- Notchl(Vall 744)-specific custom rabbit monoclonal antibody and detected with a C ⁇ terminal Notch.1 SULFO-TAGTM (Meso Scale Discovery, Gaithersburg, Maryland) polyclonal sheep antibody (R&D Systems, Minneapolis. M ). Lysates are diluted to 2 ⁇ &'' ⁇ ! in ice-cold ELISA iris lysis buffer (R60TX) (Meso Scale Discovery,
- the data in Table 2 evidences the tumor growth inhibition, and the inhibition of NTICD cleavage by Compound 1 in various xenograft models of human tumor.
- the data in Table 2 further provides an in vivo correlation to the functional activity cell data described in Table 1.
- CYP450 Cytochrome P450
- CYP450 metabolism leads to appreciable exposure of patients to active metabolites, efficacy and safety issues can arise from the greater variability associated with the contribution of multiple active species: therefore, generally a drug lacking active metabolites is preferred.
- Therapeutic agents evidencing low or no CYP450 metabolism and modulation are desirable and may have superior safety profiles in and for patients. Lynch et ai., Am. Fam. Physician, 76, 391 (2007).
- the potential for CYP450 enzyme interactions cannot be predicted based solely on the structure of an active pharmaceutical agent.
- Compound 1 is not an inhibitor or inducer of the major CYP450 enzymes and. was not metabolized to any appreciable extent by liver microsomes optimized for oxidative CYP450 metabolism. In rats and dogs in vivo, major oxidative metabolites were not observed in circulation or excreta. Therefore, Compound 1 has a low likelihood for CYP450-based interactions with other medications that could result in dose adjustments or a need to limit or stop additional medications in a patient being treated for cancer.
- Compound 1 was evaluated in vivo in bile-duct cannulated rats to study systemic pharmacokinetics (PK), excretion, and. metabolism. In vivo 51% of the IV dose was excreted unchanged, predominately in the urine, with low (2% of parent) systemic exposure to an active N-dealkylated metabolite, 4,4,4-trifIuoro-N-[(lS)-l-methyl-2-oxo- 2-[[(7S)-6-oxc 5,7-dihydropyrido[23-d][3]benzazepin-7-yf]amino]ethyl]butanamide,
- Profiling of rat plasma for metabolites indicated the absence of additional circulating metabolites. Further studies in bile-duct intact dogs also supported appreciable clearance by excretion of parent compound, as well as the absence of major circulating active metabolites.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description


















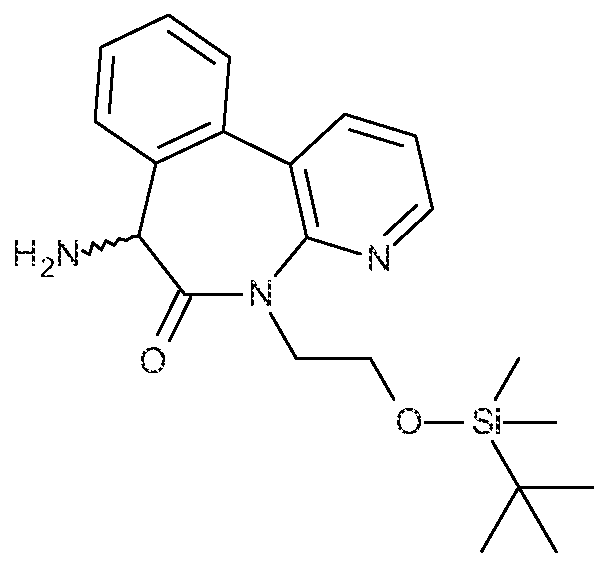






Claims


Priority Applications (24)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES12741192.4T ES2544937T3 (en) | 2011-07-27 | 2012-07-18 | Compound inhibitor of Notch path signaling |
| MYPI2014700183A MY184303A (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| PL12741192T PL2736920T3 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| JP2014522877A JP6027110B2 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compounds |
| UAA201314991A UA110834C2 (en) | 2011-07-27 | 2012-07-18 | Compound inhibitor of notch signaling pathway |
| CA2841178A CA2841178C (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| DK12741192.4T DK2736920T3 (en) | 2011-07-27 | 2012-07-18 | CONNECTION TO INHIBITION OF NOTCH SIGNAL ROAD |
| AP2014007362A AP4080A (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| MEP-2015-105A ME02171B (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| AU2012287251A AU2012287251B2 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| KR1020147001731A KR101578309B1 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| BR112014001600-3A BR112014001600B1 (en) | 2011-07-27 | 2012-07-18 | NOTCH SIGNALING PATH INHIBITOR COMPOUND, ITS USES, AND PHARMACEUTICAL COMPOSITION |
| MX2014001084A MX356536B (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound. |
| EP12741192.4A EP2736920B1 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| NZ618891A NZ618891B2 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| EA201490161A EA023044B1 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| CN201280037471.9A CN103732612B (en) | 2011-07-27 | 2012-07-18 | NOTCH pathway signal transduction inhibitor compound |
| RS20150496A RS54135B1 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| SI201230246T SI2736920T1 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
| TNP2013000515A TN2013000515A1 (en) | 2011-11-16 | 2013-12-12 | Notch pathway signaling inhibitor compound |
| IL229988A IL229988A (en) | 2011-07-27 | 2013-12-17 | 4,4,4-trifluoro-n-[(1s)-2[[(7s)-5-(2-hydroxyethyl)-6-oxo-7h-pyrido[2,3-d] [3]benzazepine-7-yl]amino]-1-methyl-2-oxo-ethyl]butanamide, its use in the manufacture of medicaments for treating cancer and crystalline hydrates thereof |
| MA36712A MA35611B1 (en) | 2011-07-27 | 2014-01-22 | Signal inhibitor compound by the notch route |
| HK14107480.6A HK1194086A1 (en) | 2011-07-27 | 2014-07-22 | Notch pathway signaling inhibitor compound notch |
| HRP20150771TT HRP20150771T1 (en) | 2011-07-27 | 2015-07-14 | Notch pathway signaling inhibitor compound |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161512016P | 2011-07-27 | 2011-07-27 | |
| US61/512,016 | 2011-07-27 | ||
| US201161560486P | 2011-11-16 | 2011-11-16 | |
| US61/560,486 | 2011-11-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013016081A1 true WO2013016081A1 (en) | 2013-01-31 |
Family
ID=46598983
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/047100 WO2013016081A1 (en) | 2011-07-27 | 2012-07-18 | Notch pathway signaling inhibitor compound |
Country Status (36)
| Country | Link |
|---|---|
| US (1) | US8569286B2 (en) |
| EP (1) | EP2736920B1 (en) |
| JP (1) | JP6027110B2 (en) |
| KR (1) | KR101578309B1 (en) |
| CN (1) | CN103732612B (en) |
| AP (1) | AP4080A (en) |
| AR (1) | AR087107A1 (en) |
| AU (1) | AU2012287251B2 (en) |
| BR (1) | BR112014001600B1 (en) |
| CA (1) | CA2841178C (en) |
| CL (1) | CL2014000175A1 (en) |
| CO (1) | CO6862159A2 (en) |
| CR (1) | CR20140036A (en) |
| CY (1) | CY1116645T1 (en) |
| DK (1) | DK2736920T3 (en) |
| DO (1) | DOP2014000011A (en) |
| EA (1) | EA023044B1 (en) |
| EC (1) | ECSP14013179A (en) |
| ES (1) | ES2544937T3 (en) |
| GT (1) | GT201400012A (en) |
| HK (1) | HK1194086A1 (en) |
| HR (1) | HRP20150771T1 (en) |
| HU (1) | HUE027534T2 (en) |
| IL (1) | IL229988A (en) |
| JO (1) | JO3148B1 (en) |
| MA (1) | MA35611B1 (en) |
| ME (1) | ME02171B (en) |
| MX (1) | MX356536B (en) |
| MY (1) | MY184303A (en) |
| PE (1) | PE20141061A1 (en) |
| PL (1) | PL2736920T3 (en) |
| PT (1) | PT2736920E (en) |
| RS (1) | RS54135B1 (en) |
| SI (1) | SI2736920T1 (en) |
| TW (1) | TWI568730B (en) |
| WO (1) | WO2013016081A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014110506A3 (en) * | 2013-01-14 | 2015-06-18 | The Trustees Of Columbia University In The City Of New York | Methods of treating, preventing and diagnosing leukemia and other blood diseases and disorders |
| WO2016106029A1 (en) | 2014-12-22 | 2016-06-30 | Eli Lilly And Company | Thieno[2,3-c]pyrrol-4-one derivatives as erk inhibitors |
| WO2016168014A1 (en) | 2015-04-14 | 2016-10-20 | Eli Lilly And Company | Targeted treatment of leiomyosarcoma |
| WO2017157825A1 (en) | 2016-03-15 | 2017-09-21 | F. Hoffmann-La Roche Ag | Combinations of lsd1 inhibitors for use in the treatment of solid tumors |
| WO2017180385A1 (en) | 2016-04-12 | 2017-10-19 | Eli Lilly And Company | Combination therapy with notch and pi3k/mtor inhibitors for use in treating cancer |
| WO2017180389A1 (en) | 2016-04-12 | 2017-10-19 | Eli Lilly And Company | Combination therapy with notch and cdk4/6 inhibitors for the treatment of cancer |
| WO2017200969A1 (en) | 2016-05-20 | 2017-11-23 | Eli Lilly And Company | Combination therapy with notch and pd-1 or pd-l1 inhibitors |
| WO2018044662A1 (en) | 2016-08-31 | 2018-03-08 | Eli Lilly And Company | Dosage regimen for treatment of solid tumors |
| WO2018071307A1 (en) | 2016-10-12 | 2018-04-19 | Eli Lilly And Company | Targeted treatment of mature t-cell lymphoma |
| WO2019090364A1 (en) | 2017-11-06 | 2019-05-09 | Juno Therapeutics, Inc. | Combination of a cell therapy and a gamma secretase inhibitor |
| WO2020072602A1 (en) | 2018-10-02 | 2020-04-09 | Frequency Therapeutics, Inc. | Pharmaceutical compositions comprising otic therapeutic agents and related methods |
| WO2020210388A1 (en) | 2019-04-08 | 2020-10-15 | Frequency Therapeutics, Inc. | Combination of chir99021 and valproic acid for treating hearing loss |
| US20210040050A1 (en) * | 2018-05-06 | 2021-02-11 | Ayala Pharmaceuticals Inc. | Combination compositions comprising bisfluoroalkyl-1,4- benzodiazepinone compounds and methods of use thereof |
| US11845803B2 (en) | 2017-02-17 | 2023-12-19 | Fred Hutchinson Cancer Center | Combination therapies for treatment of BCMA-related cancers and autoimmune disorders |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2938626A1 (en) | 2013-07-26 | 2015-01-29 | John Rothman | Compositions to improve the therapeutic benefit of bisantrene |
| TWI625332B (en) * | 2015-07-07 | 2018-06-01 | 美國禮來大藥廠 | Notch pathway signaling inhibitor compounds |
| CN109071504B (en) | 2016-02-05 | 2022-03-08 | 戴纳立制药公司 | Inhibitors of receptor interacting protein kinase 1 |
| CN110383066B (en) | 2016-12-09 | 2023-03-31 | 戴纳立制药公司 | Compounds, compositions and methods |
| TW201932482A (en) | 2017-11-01 | 2019-08-16 | 美商奇諾治療有限公司 | Chimeric antigen receptors specific for B-cell maturation antigen and encoding polynucleotides |
| AU2023206890A1 (en) | 2022-01-12 | 2024-08-22 | Denali Therapeutics Inc. | Crystalline forms of (s)-5-benzyl-n-(5-methyl-4-oxo-2, 3,4,5- tetrahydropyrido [3,2-b] [l,4]oxazepin-3-yl)-4h-l,2,4-triazole-3-carboxamide |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998028268A2 (en) | 1996-12-23 | 1998-07-02 | Elan Pharmaceuticals, Inc. | CYCLOALKYL, LACTAM, LACTONE AND RELATED COMPOUNDS AS β-AMYLOID PEPTIDE RELEASE INHIBITORS |
| WO2011060051A1 (en) * | 2009-11-12 | 2011-05-19 | University Of Massachusetts | Methods for treating glioblastoma |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU4707999A (en) | 1998-06-22 | 2000-01-10 | Elan Pharmaceuticals, Inc. | Compounds for inhibiting beta-amyloid peptide release and/or its synthesis |
| AU4710199A (en) * | 1998-06-22 | 2000-01-10 | Elan Pharmaceuticals, Inc. | Compounds for inhibiting beta-amyloid peptide release and/or its synthesis |
| BRPI0407262A (en) | 2003-02-04 | 2006-01-31 | Hoffmann La Roche | Malonamide derivatives as gamma secretase inhibitors |
| EP1711470B1 (en) | 2003-09-09 | 2009-04-15 | F. Hoffmann-La Roche Ag | Malonamide derivatives blocking the activtiy of gamma-secretase |
| KR100867035B1 (en) | 2003-10-06 | 2008-11-04 | 에프. 호프만-라 로슈 아게 | Substituted dibenzo-azepine and benzo-diazepine derivatives useful as gamma-secretase inhibitors |
| CA2645756C (en) | 2006-03-27 | 2013-12-31 | F. Hoffmann-La Roche Ag | Malonamide derivatives as gamma secretase inhibitors |
| MX2009007864A (en) * | 2007-02-02 | 2009-07-31 | Hoffmann La Roche | 6-oxo-6,7-dihydro-5h-dibenzo[b,d]azepin-7-yl derivatives. |
| AU2009203776A1 (en) | 2008-01-11 | 2009-07-16 | F. Hoffmann-La Roche Ag | Use of a gamma-secretase inhibitor for treating cancer |
-
2012
- 2012-07-10 JO JOP/2012/0190A patent/JO3148B1/en active
- 2012-07-10 AR ARP120102486A patent/AR087107A1/en not_active Application Discontinuation
- 2012-07-13 TW TW101125459A patent/TWI568730B/en active
- 2012-07-18 MY MYPI2014700183A patent/MY184303A/en unknown
- 2012-07-18 JP JP2014522877A patent/JP6027110B2/en active Active
- 2012-07-18 AU AU2012287251A patent/AU2012287251B2/en active Active
- 2012-07-18 KR KR1020147001731A patent/KR101578309B1/en active IP Right Grant
- 2012-07-18 ES ES12741192.4T patent/ES2544937T3/en active Active
- 2012-07-18 HU HUE12741192A patent/HUE027534T2/en unknown
- 2012-07-18 DK DK12741192.4T patent/DK2736920T3/en active
- 2012-07-18 ME MEP-2015-105A patent/ME02171B/en unknown
- 2012-07-18 BR BR112014001600-3A patent/BR112014001600B1/en active IP Right Grant
- 2012-07-18 EP EP12741192.4A patent/EP2736920B1/en active Active
- 2012-07-18 CA CA2841178A patent/CA2841178C/en active Active
- 2012-07-18 AP AP2014007362A patent/AP4080A/en active
- 2012-07-18 SI SI201230246T patent/SI2736920T1/en unknown
- 2012-07-18 PT PT127411924T patent/PT2736920E/en unknown
- 2012-07-18 US US13/551,681 patent/US8569286B2/en active Active
- 2012-07-18 PE PE2014000129A patent/PE20141061A1/en active IP Right Grant
- 2012-07-18 RS RS20150496A patent/RS54135B1/en unknown
- 2012-07-18 MX MX2014001084A patent/MX356536B/en active IP Right Grant
- 2012-07-18 WO PCT/US2012/047100 patent/WO2013016081A1/en active Application Filing
- 2012-07-18 CN CN201280037471.9A patent/CN103732612B/en active Active
- 2012-07-18 EA EA201490161A patent/EA023044B1/en not_active IP Right Cessation
- 2012-07-18 PL PL12741192T patent/PL2736920T3/en unknown
-
2013
- 2013-12-17 IL IL229988A patent/IL229988A/en active IP Right Grant
-
2014
- 2014-01-22 MA MA36712A patent/MA35611B1/en unknown
- 2014-01-22 DO DO2014000011A patent/DOP2014000011A/en unknown
- 2014-01-23 CL CL2014000175A patent/CL2014000175A1/en unknown
- 2014-01-24 GT GT201400012A patent/GT201400012A/en unknown
- 2014-01-24 CR CR20140036A patent/CR20140036A/en unknown
- 2014-01-24 CO CO14014585A patent/CO6862159A2/en active IP Right Grant
- 2014-01-27 EC ECSP14013179 patent/ECSP14013179A/en unknown
- 2014-07-22 HK HK14107480.6A patent/HK1194086A1/en unknown
-
2015
- 2015-07-14 HR HRP20150771TT patent/HRP20150771T1/en unknown
- 2015-08-25 CY CY20151100742T patent/CY1116645T1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998028268A2 (en) | 1996-12-23 | 1998-07-02 | Elan Pharmaceuticals, Inc. | CYCLOALKYL, LACTAM, LACTONE AND RELATED COMPOUNDS AS β-AMYLOID PEPTIDE RELEASE INHIBITORS |
| WO2011060051A1 (en) * | 2009-11-12 | 2011-05-19 | University Of Massachusetts | Methods for treating glioblastoma |
Non-Patent Citations (18)
| Title |
|---|
| "Characterization of Crystalline Solids by X-ray Powder Diffraction (XRPD", PHARMACOPIA 33 - NATIONAL FORMULARY, 1 October 2010 (2010-10-01) |
| "Compendium of Organic Synthetic Methods", vol. 1-10, WILEY INTERSCIENCE, pages: 1974 - 2002 |
| "Comprehensive Organic Transformations", 1989, VCH PUBLISHERS INC |
| "REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY", 1995, MACK PUBLISHING CO. |
| E. I. ELIEL; S. H. WILEN: "Stereochemistry of Organic Compounds", 1994, WILEY |
| FRANCIS A. CAREY; RICHARD J. SUNDBERG: "Advanced Organic Chemistry", 2000, KLUWER ACADEMIC / PLENUM PUBLISHERS |
| GAST ET AL., GENES, CHROMOSOMES & CANCER, no. 49, 2010, pages 733 - 745 |
| GRABHER ET AL., NATURE REVIEW CANCER, no. 6, 2006, pages 347 - 359 |
| J., JACQUES; A. COLLET; S. H. WILEN: "Enantiomers, Racemates, and Resolutions", 1991, WILEY |
| LYNCH, AM. FAM. PHYSICIAN, vol. 76, 2007, pages 391 |
| MICHAEL B. SMITH; JERRY MARCH: "Advanced Organic Chemistry, Reactions Mechanisms, and Structure", 2001, WILEY INTERSCIENCE |
| P. STAHL ET AL.: "HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE", 2002, VCHA/WILEY-VCH |
| PARK ET AL., CANCER RESEARCH, no. 66, 2006, pages 6312 - 6318 |
| RANGANATHAN ET AL., NATURE REVIEW CANCER, no. 11, 2011, pages 338 - 351 |
| S.M. BERGE ET AL.: "Pharmaceutical Salts", JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 66, no. 1, January 1977 (1977-01-01) |
| SHIH ET AL., CANCER RESEARCH, no. 67, 2007, pages 1879 - 1882 |
| WENG ET AL., SCIENCE, no. 306, 2004, pages 269 - 271 |
| WESTHOFF ET AL., PNAS, no. 106, 2009, pages 22293 - 22298 |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014110506A3 (en) * | 2013-01-14 | 2015-06-18 | The Trustees Of Columbia University In The City Of New York | Methods of treating, preventing and diagnosing leukemia and other blood diseases and disorders |
| US10350216B2 (en) | 2013-01-14 | 2019-07-16 | The Trustees Of Columbia University In The City Of New York | Methods of treating, preventing and diagnosing leukemia and other blood diseases and disorders |
| WO2016106029A1 (en) | 2014-12-22 | 2016-06-30 | Eli Lilly And Company | Thieno[2,3-c]pyrrol-4-one derivatives as erk inhibitors |
| WO2016168014A1 (en) | 2015-04-14 | 2016-10-20 | Eli Lilly And Company | Targeted treatment of leiomyosarcoma |
| US10555951B2 (en) | 2015-04-14 | 2020-02-11 | Eli Lilly And Company | Targeted treatment of leiomyosarcoma |
| TWI609687B (en) * | 2015-04-14 | 2018-01-01 | 美國禮來大藥廠 | Targeted treatment of leiomyosarcoma |
| US10265279B2 (en) | 2016-03-15 | 2019-04-23 | Oryzon Genomics, S.A. | Combinations of LSD1 inhibitors for use in the treatment of solid tumors |
| WO2017157825A1 (en) | 2016-03-15 | 2017-09-21 | F. Hoffmann-La Roche Ag | Combinations of lsd1 inhibitors for use in the treatment of solid tumors |
| JP7288482B2 (en) | 2016-04-12 | 2023-06-07 | イーライ リリー アンド カンパニー | Combination therapy of Notch inhibitors and CDK4/6 inhibitors for the treatment of cancer |
| AU2017249078B2 (en) * | 2016-04-12 | 2022-10-20 | Eli Lilly And Company | Combination therapy with notch and CDK4/6 inhibitors for the treatment of cancer |
| KR20180129917A (en) * | 2016-04-12 | 2018-12-05 | 일라이 릴리 앤드 캄파니 | Combination therapy of Notch and PI3K / mTOR inhibitors for use in cancer therapy |
| KR20180129918A (en) * | 2016-04-12 | 2018-12-05 | 일라이 릴리 앤드 캄파니 | Combination therapy of Notch and CDK4 / 6 inhibitors for cancer treatment |
| US11564929B2 (en) | 2016-04-12 | 2023-01-31 | Eli Lilly And Company | Combination therapy with Notch and PI3K/mTOR inhibitors for use in treating cancer |
| JP7278331B2 (en) | 2016-04-12 | 2023-05-19 | イーライ リリー アンド カンパニー | Combination therapy with a Notch inhibitor and a PI3K/mTOR inhibitor for use in treating cancer |
| JP2019511529A (en) * | 2016-04-12 | 2019-04-25 | イーライ リリー アンド カンパニー | Combination therapy with a Notch inhibitor and a PI3K / mTOR inhibitor for use in the treatment of cancer |
| JP2019511526A (en) * | 2016-04-12 | 2019-04-25 | イーライ リリー アンド カンパニー | Combination therapy with Notch inhibitor and CDK4 / 6 inhibitor for the treatment of cancer |
| AU2017249988B2 (en) * | 2016-04-12 | 2022-10-27 | Eli Lilly And Company | Combination therapy with notch and PI3K/mTOR inhibitors for use in treating cancer |
| AU2017249988C1 (en) * | 2016-04-12 | 2023-02-16 | Eli Lilly And Company | Combination therapy with notch and PI3K/mTOR inhibitors for use in treating cancer |
| JP2021169474A (en) * | 2016-04-12 | 2021-10-28 | イーライ リリー アンド カンパニー | COMBINATION THERAPY WITH NOTCH AND PI3K/mTOR INHIBITORS FOR USE IN TREATING CANCER |
| WO2017180389A1 (en) | 2016-04-12 | 2017-10-19 | Eli Lilly And Company | Combination therapy with notch and cdk4/6 inhibitors for the treatment of cancer |
| KR102418766B1 (en) | 2016-04-12 | 2022-07-08 | 일라이 릴리 앤드 캄파니 | Combination Therapy of Notch and a PI3K/mTOR Inhibitor for Use in the Treatment of Cancer |
| KR102418765B1 (en) | 2016-04-12 | 2022-07-08 | 일라이 릴리 앤드 캄파니 | Combination Therapy of Notch and CDK4/6 Inhibitors for the Treatment of Cancer |
| WO2017180385A1 (en) | 2016-04-12 | 2017-10-19 | Eli Lilly And Company | Combination therapy with notch and pi3k/mtor inhibitors for use in treating cancer |
| US11298362B2 (en) | 2016-04-12 | 2022-04-12 | Eli Lilly And Company | Combination therapy with Notch and CDK4/6 inhibitors for the treatment of cancer |
| JP2021176858A (en) * | 2016-04-12 | 2021-11-11 | イーライ リリー アンド カンパニー | Combination therapy with notch inhibitor and cdk4/6 inhibitor for treatment of cancer |
| RU2747788C2 (en) * | 2016-04-12 | 2021-05-14 | Эли Лилли Энд Компани | Combination therapy with notch and cdk4/6 inhibitors for cancer treatment |
| RU2754452C2 (en) * | 2016-04-12 | 2021-09-02 | Эли Лилли Энд Компани | Combination therapy with notch and pi3k/mtor inhibitors for use in treatment of cancer |
| IL263110B (en) * | 2016-05-20 | 2022-07-01 | Lilly Co Eli | Combination therapy with notch and pd-1 or pd-l1 inhibitors |
| US11826317B2 (en) | 2016-05-20 | 2023-11-28 | Eli Lilly And Company | Combination therapy with notch and PD-1 or PD-L1 inhibitors |
| US10688104B2 (en) | 2016-05-20 | 2020-06-23 | Eli Lilly And Company | Combination therapy with Notch and PD-1 or PD-L1 inhibitors |
| WO2017200969A1 (en) | 2016-05-20 | 2017-11-23 | Eli Lilly And Company | Combination therapy with notch and pd-1 or pd-l1 inhibitors |
| US20190209581A1 (en) * | 2016-08-31 | 2019-07-11 | Eli Lilly And Company | Dosage regimen for treatment of solid tumors |
| WO2018044662A1 (en) | 2016-08-31 | 2018-03-08 | Eli Lilly And Company | Dosage regimen for treatment of solid tumors |
| CN109562114A (en) * | 2016-08-31 | 2019-04-02 | 伊莱利利公司 | For treating the dosage regimen of solid tumor |
| US11376259B2 (en) | 2016-10-12 | 2022-07-05 | Eli Lilly And Company | Targeted treatment of mature T-cell lymphoma |
| WO2018071307A1 (en) | 2016-10-12 | 2018-04-19 | Eli Lilly And Company | Targeted treatment of mature t-cell lymphoma |
| US11845803B2 (en) | 2017-02-17 | 2023-12-19 | Fred Hutchinson Cancer Center | Combination therapies for treatment of BCMA-related cancers and autoimmune disorders |
| WO2019090364A1 (en) | 2017-11-06 | 2019-05-09 | Juno Therapeutics, Inc. | Combination of a cell therapy and a gamma secretase inhibitor |
| US20210040050A1 (en) * | 2018-05-06 | 2021-02-11 | Ayala Pharmaceuticals Inc. | Combination compositions comprising bisfluoroalkyl-1,4- benzodiazepinone compounds and methods of use thereof |
| WO2020072602A1 (en) | 2018-10-02 | 2020-04-09 | Frequency Therapeutics, Inc. | Pharmaceutical compositions comprising otic therapeutic agents and related methods |
| WO2020072601A1 (en) | 2018-10-02 | 2020-04-09 | Frequency Therapeutics, Inc. | Pharmaceutical compositions comprising otic therapeutic agents and related methods |
| WO2020210388A1 (en) | 2019-04-08 | 2020-10-15 | Frequency Therapeutics, Inc. | Combination of chir99021 and valproic acid for treating hearing loss |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2736920B1 (en) | Notch pathway signaling inhibitor compound | |
| EP3989972B1 (en) | Glucagon-like peptide 1 receptor agonists | |
| EP2850082B1 (en) | 1-(3,3-dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido(2,3-d)pyrimidin-6-yl)phenyl)urea as Raf kinase inhibitor for the treatment of cancer | |
| EP3283079B1 (en) | Targeted treatment of leiomyosarcoma | |
| CA3144859A1 (en) | Quinazolin-4-one derivatives useful for the treatment of braf-associated diseases and disorders | |
| CN112771053A (en) | Biomarker-based therapeutic compositions | |
| TW202400608A (en) | Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compounds | |
| EP3640245B1 (en) | Poly(adp-ribose) polymerase inhibitor, preparation method and use | |
| KR20210135521A (en) | pyrrolopyrazole derivatives | |
| TWI546304B (en) | Protein tyrosine kinase inhibitors and their use | |
| NZ618891B2 (en) | Notch pathway signaling inhibitor compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12741192 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2841178 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20147001731 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014000175 Country of ref document: CL Ref document number: 201490161 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2014522877 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14014585 Country of ref document: CO Ref document number: CR2014-000036 Country of ref document: CR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 000129-2014 Country of ref document: PE Ref document number: MX/A/2014/001084 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2012287251 Country of ref document: AU Date of ref document: 20120718 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012741192 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201314991 Country of ref document: UA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014001600 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2015/0496 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: 112014001600 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140123 |