WO2012151561A1 - Compounds for inhibiting cell proliferation in egfr-driven cancers - Google Patents
Compounds for inhibiting cell proliferation in egfr-driven cancers Download PDFInfo
- Publication number
- WO2012151561A1 WO2012151561A1 PCT/US2012/036683 US2012036683W WO2012151561A1 WO 2012151561 A1 WO2012151561 A1 WO 2012151561A1 US 2012036683 W US2012036683 W US 2012036683W WO 2012151561 A1 WO2012151561 A1 WO 2012151561A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- alkenyl
- heteroalkyl
- alkynyl
- Prior art date
Links
- 0 C*(*)c(c(C)c1OCCCN(*)c1c1)c1Nc1ncc(*)c(Nc2ccc(*)c(*)c2*)n1 Chemical compound C*(*)c(c(C)c1OCCCN(*)c1c1)c1Nc1ncc(*)c(Nc2ccc(*)c(*)c2*)n1 0.000 description 10
- WVHGIUIVYMKAAS-UHFFFAOYSA-N COc(c(Nc(nc1Nc2ccccc2P(C)(C)=O)ncc1Cl)c1)cnc1NC(C=C)=O Chemical compound COc(c(Nc(nc1Nc2ccccc2P(C)(C)=O)ncc1Cl)c1)cnc1NC(C=C)=O WVHGIUIVYMKAAS-UHFFFAOYSA-N 0.000 description 1
- GTOBYFTWGYBBQY-RMKNXTFCSA-N COc(ccc(NC(/C=C/CN1CCOCC1)=O)c1)c1Nc(nc1)nc(Nc2ccccc2P(C)(C)=O)c1Cl Chemical compound COc(ccc(NC(/C=C/CN1CCOCC1)=O)c1)c1Nc(nc1)nc(Nc2ccccc2P(C)(C)=O)c1Cl GTOBYFTWGYBBQY-RMKNXTFCSA-N 0.000 description 1
- VVZBBMFNXAVCAR-UHFFFAOYSA-N COc(ccc(NC(C=C)=O)c1)c1Nc(nc1Nc(cccc2OC)c2P(C)(C)=O)ncc1Cl Chemical compound COc(ccc(NC(C=C)=O)c1)c1Nc(nc1Nc(cccc2OC)c2P(C)(C)=O)ncc1Cl VVZBBMFNXAVCAR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/4025—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil not condensed and containing further heterocyclic rings, e.g. cromakalim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/538—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with carbocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- This invention relates to pharmaceutical compositions and methods for inhibiting the proliferation of cells.
- kinase inhibitors In human clinical studies with non-small cell lung cancer (NSCLC) patients, the kinase inhibitors, erlotinib and gefitinib have been found to be effective, but in only a subset of patients. It was later determined that the responsive patients had certain mutations in the gene for epidermal growth factor receptor (EGFR).
- EGFR epidermal growth factor receptor
- the mutant forms of EGFR are enzymatically active without the need for ligand stimulation. They are also particularly sensitive to kinase inhibitors like erlotinib and gefitinib, which competitively bind to the ATP binding site of the EGFR kinase domain. Those mutations have been cataloged and described at length in the scientific literature.
- mutations in the EGFR gene e.g., the T790M mutation, produces mutant EGFR proteins to which drugs like erlotinib and gefitinib bind less well. Those mutations are associated with resistance to the drugs and to relapse in cancer patients bearing such mutation.
- New diagnostic methods and therapies are needed for the treatment of EGFR-driven cancers in which mutations confer resistance to front line tyrosine kinase inhibitor ("TKI") therapies.
- TKI front line tyrosine kinase inhibitor
- new therapies for inhibiting cells expressing such gefitinib-resistant or erlotinib-resistant EGFR genes could be of profound benefit.
- the invention features a class of active inhibitors of EGFR-driven cancers, including cancers driven by EGFR mutants (e.g., mutants harboring the T790M mutation or any other mutation which is associated with resistance to erlotinib and gefitinib).
- EGFR mutants e.g., mutants harboring the T790M mutation or any other mutation which is associated with resistance to erlotinib and gefitinib.
- the inhibitors have the structure of formula (I), below:
- the invention features a compound of formula (I):
- U 1 and U 2 are both N and U 3 is C-R e ; or U 3 is N, one of U' and U 2 is N, and the other is C-R d ; or U 3 is C-R e , one of U 1 and U 2 is N, and the other is C-R d ; V 1 is O or NH; V 1 is O, S, NR V ,
- R is H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, or aryl;
- e d e is H, CF 3 , CN, C 1.4 alkenyl, C, A alkyl, C alkoxy, or halo; and R is H or NH 2 ; or R and R , together with the pyrimidine ring atoms to which they are attached, form a 5- or 6-membered ring containing one, two or three heteroatoms, independently selected from N, S and O, wherein the 5- or 6- membered ring is substituted by R h ; R h is H, C M alkyl, or halo; R g is H, -P(0)(R 3A )(R 3B ), - S(0)N(R 3C )(R 3D ),-S(0) 2 R 3E , -C(0)N(R 3F )(R 3G ), -OC(0)N(R 3F )(R 3G ), -NR 3H C(0)OR 31 , a 5 or 6 member heterocyclic ring including 1 , 2, 3
- R g2 and R 8 together with the atoms to which they are attached form a 5- to 7-member heterocyclic ring including 1 - 3 hetero atoms independently selected from P, N , O and S, the heterocyclic ring being unsubstituted or substituted;
- R gl is H, F, -P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ), -S(0) 2 R 3E , -C(0)N(R 3F )(R 3G ), -OC(0)N(R 3F )(R 3G ), -NR 3H C(0)OR 31 , or a 5 or 6 member heterocyclic ring including 1 or 2 N atoms, the heterocyclic ring being unsubstituted or substituted;
- Ring A is selected from:
- R is H, F, or is a 5 or 6 member heterocyclic ring containing 1 , 2 or 3 N or O atoms, the heterocyclic ring being unsubstituted or substituted;
- R M is H, F, W 1 , C : . 6 alkoxy, C 3 .6 alkenyloxy, or C 3 .
- R 5A , R 5B , R 5C , and R 5D is, independently, selected from H, alkyl, alkenyl, alkynyl, and heteroalkyl, or R 5A and R 5B , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted;
- R A L combines with R M to form a 6 member heterocyclic ring, or
- R A2 is H, C
- . 6 alkyl, C : . 6 alkoxy, C 3 hopefully 6 alkenyloxy, or C 3 . 6 cycloalkyloxy; each Y is, independently, a bond, -0-, -S- or -NR 1 -; each occurrence of R 1 and R 2 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl; each of X : and X 2 is, independently, selected from CH and N; R 4 is selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl; W 1 is a moiety selected from -NR 7 C(0)CH CH 2 , -NR
- R 7 is H, alkyl, or heteroalkyl
- R 8 is C M alkyl
- each occurrence of R 9 and R 10 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl
- R 1 1 is -C(0)-OR 12 , -CH 2 N(CH 3 ) 2 , H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl
- R 12 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl
- R 13 is H or C al
- U 1 and U 2 are both N and U 3 is C-R E ; or U 3 is N, one of U 1 and U 2 is N, and the other is C-R D ; or U 3 is C-R E , one of U 1 and U 2 is N, and the other is C-R D ;
- V 1 is O, S, NR V , CO, CH 2 , or CF 2 ;
- R V is H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, or aryl;
- R is H, CF 3 , CN, CM alkenyl, C alkyl, CM alkoxy, or halo; and R is H or NH 2 ; or R and R E , together with the pyrimidine ring atoms to which they are attached, form a 5- or 6-membered ring containing one, two or three heteroatoms, independently selected from N, S and O, wherein the 5- or 6-membered ring is substituted by R H ;
- R H is H, C alkyl, or halo;
- R 6 is H, -P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ),-S(0) 2 R 3E , -C(0)N(R 3F )(R 3G ), -OC(0)N(R 3F )(R 3G ), -NR 3H C(0)OR 31 , a 5 or 6 member heterocyclic ring comprising 1 , 2, 3 or 4 N atoms, or combined with R s2 forms a 5- to 7-member heterocyclic ring, wherein each of R 3A , R 3B , R 3C , R 3D , R 3E , R 3F , R 3G , R 3H , and R 31 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and heteroalkyl
- R g2 is H, F, W', -P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ),-S(0) 2 R 3E ,
- R 8 ' is H, F, -P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ) ,-S(0) 2 R 3E , -C(0)N(R 3F )(R 3G ),
- Ring A is selected from:
- R M is H, F, or is a 5 or 6 member heterocyclic ring containing 1 , 2 or 3 N or O atoms, the heterocyclic ring being unsubstituted or substituted;
- R M is H, F, W 1 , C,.6 alkoxy, C 3 . 6 alkenyloxy, or C 3 . 6 cycloalkyloxy, -OC(0)N(R 5A )(R 5B ), - NR 5C C(0)OR 5D ; a 5 or 6 member heterocyclic ring comprising 1 , 2 or 3 N or O atoms, the heterocyclic ring being unsubstituted or substituted, or, R M and R al together with the atoms to which they are attached form a 6 member heterocyclic ring comprising 1 , 2 or 3 N or O atoms which is unsubstituted or substituted;
- each of R 5A , R 5B , R 5C , and R 5D is, independently, selected from H, alkyl, alkenyl, alkynyl, and heteroalkyl, or R 5A and R 5B , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted;
- R AL combines with R M to form a 6 member heterocyclic ring, or is H, halo, W 1
- R is H, Ci_6 alkyl, Ci. 6 alkoxy, C 3 . 6 alkenyloxy, or C 3 . 6 cycloalkyloxy;
- each Y is, independently, a bond, -0-, -S- or -NR 1 -;
- each occurrence of R 1 and R 2 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl;
- each of i and X 2 is, independently, selected from CH and N;
- R 4 is selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl;
- R 7 is H, alkyl, or heteroalkyl
- R 8 is C,.4 alkyl
- each occurrence of R 9 and R 10 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl;
- R" is -C(0)-OR 12 , -CH 2 N(CH 3 ) 2 , H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl;
- R 1 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl;
- R 13 is H or C M alkyl, or a pharmaceutically acceptable salt thereof, wherein one of R l , R g2 , and R M comprises W 1 , and wherein said compound comprises at least one -P(0)(R 6A )(R 6B ) wherein each of R 6A and R 6B is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and heteroalkyl, or R 6A and R 6B , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted.
- the compound can further be described by formula (la), formula (lb), formula (Ic), formula (Id), or formula (Ie):
- Ring A, R e , R d , R 8 , R sl , and R 82 are as defined in formula (I).
- the compound can further be described by formula (Ila), formula (lib), or formula (lie):
- V 1 is O, S, NR V , CO, CH 2 , or CF 2 ;
- R V is H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, or aryl;
- R is H, CF 3 , CN, C,. 4 alkenyl, C alkyl, C alkoxy, or halo; and e d e
- R is H or NH 2 ; or R and R , together with the pyrimidine ring atoms to which they are attached, form a 5- or 6-membered ring containing one, two or three heteroatoms, independently selected from N, S and O, wherein the 5- or 6-membered ring is substituted by R H ;
- R H is H, C alkyl, or halo;
- R" 2 is H, C, . 6 alkyl, C,. 6 alkoxy, C 3 . 6 alkenyloxy, or C 3 . 6 cycloalkyloxy;
- R 6 is -P(0)(R 3A )(R 3B ),
- R 3A , R 3 B , R 3C , R 3D , R 3E , R 3 F , R 3G , R 3H , and R 31 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and heteroalkyl, or R 3A and R 3B , or R 3C and R 3D , or R 3 F and R 3G , together with the atoms to which they are
- R G2 and R S together with the atoms to which they are attached form a 5- to 7-member heterocyclic ring including 1 - 3 hetero atoms independently selected from P, N , O and S, the heterocyclic ring being unsubstituted or substituted;
- R G L is H, F, ⁇ P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ) ,-S(0) 2 R 3E ,-C(0)N(R 3F )(R 3G ), -OC(0)N(R 3F )(R 3G ), - NR J H C(0)OR ;LI , or a 5 or 6 member heterocyclic ring including 1 or 2 N atoms, the heterocyclic ring being unsubstituted or substituted;
- R B2 is H, F, or is a 5 or 6 member heterocyclic ring containing 1 , 2 or 3 N or O atoms
- R 7 is H, alkyl, or heteroalkyl
- R 8 is C 1-4 alkyl
- each occurrence of R 9 and R 10 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl
- R" is -C(0)-OR 12 , -CH 2 N(CH 3 ) 2 , H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl
- R 12 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl
- R 13 is H or C al
- R 6A P(0)(R 6A )(R 6B ), -S(0)N(R 6C )(R 6D ),-S(0) 2 R 6b , wherein each of R , R , R 6 , R 6U , and R 6b is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and heteroalkyl, or R and R , or R 6C and R , or R and R , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted.
- the present disclosure provides a compound described by formula (Ila), formula (lib), or formula (lie):
- V' is O, S, NR V , CO, CH?, or CF 2 ;
- R v is H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, or aryl;
- R is H, CF 3 , CN, C M alkenyl, C M alkyl, C alkoxy, or halo; and R is H or NH 2 ; or R and e
- R together with the pyrimidine ring atoms to which they are attached, form a 5- or 6-membered ring containing one, two or three heteroatoms, independently selected from N, S and O, wherein the 5- or 6-membered ring is substituted by R h ;
- R h is H, C M alkyl, or halo
- R 32 is H, Cj.6 alkyl, C]. 6 alkoxy, C 3 .6 alkenyloxy, or C 3 . 6 cycloalkyloxy;
- R 8 is -P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ) -S(0) 2 R 3E , -C(0)N(R 3F )(R 3G ),
- R jA , R 3B , R jC , R 3D , R 3E , R 3F , R 3G , R 3H , and R 31 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and heteroalkyl, or R 3A and R 3B , or R 3C and R 3D , or R 3F and R 3G , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted;
- R 82 is H, F, W 1 , -P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ) ,-S(0) 2 R 3E ,
- R 8 ' is H, F, -P(0)(R 3A )(R 3B ), -S(0)N(R 3C )(R 3D ) ,-S(0) 2 R 3E , -C(0)N(R 3F )(R 3G ),
- R B2 is H, F, or is a 5 or 6 member heterocyclic ring containing 1 , 2 or 3 N or O atoms, the heterocyclic ring being unsubstituted or substituted;
- R M is H, F, W 1 , C
- a 5 or 6 member heterocyclic ring comprising 1 , 2 or 3 N or O atoms, the heterocyclic ring being unsubstituted or substituted, or, R M and R A L together with the atoms to which they are attached form a 6 member heterocyclic ring comprising 1 , 2 or 3 N or O atoms which is unsubstituted or substituted;
- each of R 5A , R 5B , R 5C , and R 5D is, independently, selected from H, alkyl, alkenyl, alkynyl, and heteroalkyl, or R 5A and R 5B , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted;
- R A L combines with R M to form a 6 member heterocyclic ring, or is H, halo, W 1 ,
- each Y is, independently, a bond, -0-, -S- or -NR 1 -;
- each occurrence of R 1 and R 2 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl;
- each of Xi and X 2 is, independently, selected from CH and N;
- R 4 is selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl;
- R 7 is H, alkyl, or heteroalkyl
- R is C,_4 alkyl
- each occurrence of R 9 and R 10 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl;
- R" is -C(0)-OR 12 , -CH 2 N(CH 3 ) 2 , H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl;
- R 12 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl;
- R 13 is H or C, .4 alkyl
- R 6A and R 6B are, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and heteroalkyl, or R 6A and R 6B , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted.
- the compound is further described by any of formulas (Illa)-(IIIe), or a pharmac
- the compound is further described by any of formulas (IVa)-(IVe), or a pharmaceutically acceptable salt thereof:
- R a2 ; R b2 ; R M ; R 8 ; R gl ; R 82 ; R d ; and R h are as defined in formula (I).
- the compound is further described by any of formulas (Va)-(Ve), or a pharmaceutically acceptable salt thereof:
- the compound can further be described by any of formulas (Vla)-(VIe), or a pharmaceutically acceptable salt thereof:
- the compound can further be described by any of formulas (Vlla)-(VIIe), or a
- the compound can further be described by any of formulas (VUIa)-(VIIIe), or a pharmaceutically acceptable salt thereof:
- each Y is, independently, a bond, -0-, -S- or -NR 1 -;
- W 1 is as defined in formula (I); each occurrence of R 1 and R 2 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl; each of X, and X 2 is, independently, selected from CH and N; and R 4 is selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl.
- the compound can further be described by any of formulas (IXa)-(IXe), or a
- each Y is, independently, a bond, -0-, -S- or -NR 1 -;
- W' is as defined in formula (I); each occurrence of R' and R 2 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl; each of X, and X 2 is, independently, selected from CH and N; and R 4 is selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl.
- the compound can further be described by any of formulas (Xa)-(Xe), or a pharmaceutically acceptable salt thereof:
- each Y is, independently, a bond, -0-, -S- or -NR 1 -;
- W 1 is as defined in formula (I); each occurrence of R 1 and R 2 is, independently, selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl; each of X] and X 2 is, independently, selected from CH and N; and R 4 is selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic and heteroaryl.
- R" 2 is H, methoxy, ethoxy, methyl, or ethyl.
- R D is H, CI, F, Br, I, CN, CH 3 , CF 3 , or cyclopropyl.
- R B2 is H.
- R 8 ' is H and R 2 is H, F, Ci_ 6 alkyl, or C alkoxy.
- R 8 is -P(0)(R jA )(R 3B ) or
- R 3A ; R 3B ; and R 3E are as defined in formula (I).
- R 8 can be selected from -P(0)(CH 3 ) 2 and -S(0) 2 (CH(CH 3 ) 2 ).
- R al is a 5 or 6 member heterocyclic ring including 1 or 2 N or O atoms which is unsubstituted or substituted with an alkyl group.
- R al can be selected from any of the following groups:
- R 32 is methoxy; R d is CI, F, Br, I, or
- R 8 is -P(0)(CH 3 ) 2 or -S(0) 2 (CH(CH 3 ) 2 ).
- U 1 is N
- U 2 is C-R d
- U 3 is C-R e
- U' is C-R d
- U 2 is N
- U 3 is C-R e
- U 1 is N
- U 2 is N
- U 3 is C-R e
- U 3 is N, one of U 1 and U 2 is N, and the other is C-R d .
- R a2 is OCH 3 ;
- R 8 or R gl is -P(0)(R 3A )(R 3B ); each of R 3A and R 3B is, independently, selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, and heteroalkyl, or R jA and R 3B , together with the atoms to which they are attached, combine to form a 5- or 6-membered heterocyclic ring which is unsubstituted or substituted;
- R 1 1 is -C(0)
- R" is -C(0)-OR 12 , -CH 2 N(CH 3 ) 2 , H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl;
- R 12 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroalkyl, heterocyclic, or heteroaryl;
- R d is CI; and
- R E or R gl is selected from -P(0)(CH 3 ) 2 and
- the invention features a method for treating an EGFR-driven cancer in a subject by administering to the subject a therapeutically effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof.
- the invention features a method for treating an EGFR-driven cancer in a subject, the method including (a) providing a subject having an EGFR-driven cancer characterized by the presence of a mutation in epidermal growth factor receptor kinase (EGFR), and (b) administering to the subject a therapeutically effective amount of a compound of the invention, or a
- EGFR epidermal growth factor receptor kinase
- EGFR-driven cancer is characterized by the presence of one or more mutations selected from: (i) L858R, (ii) T790M, (iii) both L858R and T790M, (iv) delE746_A750, and (v) both delE746_A750 and T790M.
- the EGFR-driven cancer can be a non-small cell lung cancer (NSCLS); glioblastoma; pancreatic cancer; head and neck cancer (e.g., squamous cell carcinoma); breast cancer; colorectal cancer; epithelial cancer; ovarian cancer; prostate cancer; an adenocarcinoma, or any EGFR-driven cancer described herein.
- NSCMS non-small cell lung cancer
- glioblastoma pancreatic cancer
- head and neck cancer e.g., squamous cell carcinoma
- breast cancer colorectal cancer
- epithelial cancer ovarian cancer
- prostate cancer an adenocarcinoma, or any EGFR-driven cancer described herein.
- the method further includes administering to the subject a first kinase inhibitor selected from erlotinib, gefitinib, and pharmaceutically acceptable salts thereof, within 6 days (e.g., within 2 weeks, 1 week, 6 days, 5 days, 4 days, 3 days, 2 days, 1 day, or simultaneously) of administering the compound of the invention (e.g., a compound of any of formulas (I), (Ia)-(Ic), (Ila)-(IIc), (Illa)-(IIIe), (IVa)-(IVe), (Va)-(Ve), (Vla)-(VIe), (Vllla)-(VIIIe), (IXa)-(IXe), and (Xa)-(Xe)), wherein each of the compound of the invention and the first kinase inhibitor are administered in an amount that together is sufficient to treat the EGFR-driven cancer.
- a first kinase inhibitor selected from erlotinib, gefitinib
- the invention features a method of inhibiting the proliferation of a cell expressing an EGFR mutant, the method including contacting the cell with a compound of the invention, or a pharmaceutically acceptable salt thereof, in an amount sufficient to inhibit the proliferation.
- the EGFR mutant can be characterized by the presence of one or more mutations in epidermal growth factor receptor kinase (EGFR) selected from: (i) L858R, (ii) T790M, (iii) both L858R and T790M, (iv) delE746_A750, (v) both delE746_A750 and T790M, and any other EGFR mutations described herein.
- EGFR epidermal growth factor receptor kinase
- the cell is a cancer cell (e.g., a cell from a non-small cell lung cancer (NSCLS); glioblastoma; pancreatic cancer; head and neck cancer (e.g., squamous cell carcinoma); breast cancer; colorectal cancer; epithelial cancer; ovarian cancer; prostate cancer; an adenocarcinoma, or any other EGFR expressing cancer described herein).
- NSC non-small cell lung cancer
- glioblastoma pancreatic cancer
- head and neck cancer e.g., squamous cell carcinoma
- breast cancer colorectal cancer
- epithelial cancer ovarian cancer
- prostate cancer an adenocarcinoma, or any other EGFR expressing cancer described herein.
- the invention further features a method of treating an EGFR-driven cancer refractory to a first kinase inhibitor selected from erlotinib, gefitinib, and pharmaceutically acceptable salts thereof, in a subject by administering to the subject a compound of the invention, or a pharmaceutically acceptable salt thereof, in an amount sufficient to treat the cancer.
- a first kinase inhibitor selected from erlotinib, gefitinib, and pharmaceutically acceptable salts thereof
- the compound can be either in its free base form, or a pharmaceutically acceptable salt.
- the response criteria for the methods of the invention can be graded according to the response evaluation criteria in solid tumors (RECIST) guidelines (see Eur. J. Cancer 45 :228 (2009)) that define when cancer patients improve ("respond"), stay the same (“stabilize”), or worsen ("progression") during treatments.
- a complete response is characterized by: (i) disappearance of all target lesions; and (ii) any pathological lymph nodes (whether target or non-target) must have reduction in short axis to ⁇ 10 mm.
- a partial response is characterized by: (i) at least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters.
- a progressive disease is characterized by (i) at least a 5%, 10%, or 20% increase in the sum of diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study); or (ii) the appearance of one or more new lesions.
- administering refers to a method of giving a dosage of a pharmaceutical composition to a mammal, where the method is, e.g., oral, intravenous,
- intraperitoneal, intraarterial, or intramuscular intraperitoneal, intraarterial, or intramuscular.
- the preferred method of administration can vary depending on various factors, e.g., the components of the pharmaceutical composition, site of the potential or actual disease and severity of disease.
- EGFR-driven cancer is meant a cancer characterized by inappropriately high expression of an EGFR gene or by a mutation in an EGFR gene that alters the biological activity of an EGFR nucleic acid molecule or polypeptide.
- EGFR-driven cancers can arise in any tissue, including brain, blood, connective tissue, liver, mouth, muscle, spleen, stomach, testis, and trachea.
- EGFR-driven cancers can include non-small cell lung cancer (NSCLS), including one or more of squamous cell carcinoma, adenocarcinoma, adenocarcinoma, bronchioloalveolar carcinoma (BAC), BAC with focal invasion, adenocarcinoma with BAC features, and large cell carcinoma; neural tumors, such as glioblastomas; pancreatic cancer; head and neck cancers (e.g., squamous cell carcinoma); breast cancer; colorectal cancer; epithelial cancer, including squamous cell carcinoma; ovarian cancer; prostate cancer; adenocarcinomas; and including cancers which are EGFR mediated.
- NSCS non-small cell lung cancer
- BAC bronchioloalveolar carcinoma
- BAC bronchioloalveolar carcinoma
- BAC BAC with focal invasion, adenocarcinoma with BAC features, and large cell carcinoma
- neural tumors such as glioblastomas
- pancreatic cancer head
- an "EGFR mutant” or “mutant” includes one or more deletions, substitutions, or additions in the amino acid or nucleotide sequences of EGFR protein, or EGFR coding sequence.
- the EGFR mutant can also include one or more deletions, substitutions, or additions, or a fragment thereof, as long as the mutant retains or increases tyrosine kinase activity, compared to wild type EGFR.
- kinase or phosphorylation activity can be increased (e.g., by at least 5%, 10%, 15%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or even 100%), as compared to wild type EGFR.
- Particular EGFR mutants are described herein, where mutations are provided relative to the position of an amino acid in human EGFR, as described in the sequence provided in NCBI GenBank Reference Sequence: NP_005219.2.
- the term "inhibiting the proliferation of a cell expressing an EGFR mutant” refers to measurably slowing, stopping, or reversing the growth rate of the EGFR-expressing cells in vitro or in vivo. Desirably, a slowing of the growth rate is by at least 10%, 20%, 30%, 50%, or even 70%), as determined using a suitable assay for determination of cell growth rates (e.g., a cell growth assay described herein).
- the EGFR mutant can be any EGFR mutant described herein.
- refractory refers to a cancer which is progressive in response to a given particular therapy.
- the cancer can be refractory either from the initial administration of the therapy; or become refractory over time in response to the therapy.
- sequence identity is meant the shared identity between two or more nucleic acid sequence, or two or more amino acid sequences, expressed in the terms of the identity between the sequences. Sequence identity can be measured in terms of percentage identity; the higher the percentage, the more identical the sequences are. Homologs or orthologs of nucleic acid or amino acid sequences possess a relatively high degree of sequence identity when aligned using standard methods. Methods of alignment of sequences for comparison are well known in the art. Various programs and alignment algorithms are described in: Smith and Watermann, Adv. Appl. Math. 2:482 ( 1981 ); Needleman and Wunsch, J. Mol. Biol. 48:443 (1970); Pearson and Lipman, Proc. Natl. Acad. Sci.
- NCBI National Center for Biological Information
- the option can be set as follows: -i is set to a file containing the first nucleic acid sequence to be compared; -j is set to a file containing the second nucleic acid sequence to be compared; -p is set to blastn; -o is set to any desired file name; -q is set to -1 ; -r is set to 2; and all other options are left at their default setting.
- the number of matches is determined by counting the number of positions where an identical nucleotide or amino acid residue is present in both sequences.
- the percent sequence identity is determined by dividing the number of matches either by the length of the sequence set forth in the identified sequence, or by an articulated length (such as 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, or 400 consecutive nucleotides or amino acid residues from a sequence set forth in an identified sequence), followed by multiplying the resulting value by 100.
- an articulated length such as 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200, 250, 300, 350, or 400 consecutive nucleotides or amino acid residues from a sequence set forth in an identified sequence
- Nucleic acid molecules that hybridize under stringent conditions to an EGFR gene sequence typically hybridize to a probe based on either an entire EGFR gene or selected portions of the gene (e.g., the kinase domain or a segment of the gene that contains the mutated codons described herein), under conditions described above.
- treating refers to administering a pharmaceutical composition for prophylactic and/or therapeutic purposes.
- To “prevent disease” refers to prophylactic treatment of a subject who is not yet ill, but who is susceptible to, or otherwise at risk of, a particular disease.
- To “treat disease” or use for “therapeutic treatment” refers to administering treatment to a subject already suffering from a disease to improve or stabilize the subject's condition.
- treating is the administration to a subject either for therapeutic or prophylactic purposes.
- alkyl refers to linear, branched, cyclic, and polycyclic non aromatic hydrocarbon groups, which may be substituted or unsubstituted. Unless otherwise specified, “alkyl” groups contain one to eight, and preferably one to six carbon atoms. Lower alkyl refers to alkyl groups containing 1 to 6 carbon atoms.
- alkyl examples include, without limitation, methyl, ethyl, n- propyl, isopropyl, cyclopropyl, butyl, isobutyl, sec-butyl, tert-butyl, cyclobutyl, pentyl, isopentyl tert- pentyl, cyclopentyl, hexyl, isohexyl, cyclohexyl, and n-heptyl, among others.
- exemplary substituted alkyl groups include, without limitation, haloalkyl groups (e.g., fluoromethyl, difluoromethyl, trifluoromethyl, 2-fluoroethyl, 3-fluoropropyl), hydroxy methyl, 2-hydroxyethyl,
- haloalkyl groups e.g., fluoromethyl, difluoromethyl, trifluoromethyl, 2-fluoroethyl, 3-fluoropropyl
- hydroxy methyl 2-hydroxyethyl
- alkoxy refers to a subset of alkyl in which an alkyl group as defined above with the indicated number of carbons attached through an oxygen bridge, -O-alkyl, wherein the alkyl group contains 1 to 8 carbons atoms and is substituted or unsubstituted.
- alkoxy groups include, without limitation, methoxy, ethoxy, n-propoxy,
- haloalkyl refers to a subset of alkyl in which an alkyl group as defined above having one or more hydrogen atoms of the alkyl substituted with a halogen atom.
- exemplary haloalkyl groups include, without limitation, trifluoromethyl, trichloromethyl, pentafluoroethyl and the like.
- alkenyl refers to a branched or unbranched hydrocarbon group containing one or more double bonds and having from 2 to 8 carbon atoms.
- An alkenyl may optionally include monocyclic or polycyclic rings, in which each ring desirably has from three to six members.
- the alkenyl group may be substituted or unsubstituted.
- Alkenyl groups include, without limitation, vinyl, allyl, 2-cyclopropyl-l -ethenyl,
- alkynyl refers to a branched or unbranched hydrocarbon group containing one or more triple bonds and having from 2 to 8 carbon atoms.
- the alkynyl group may be substituted or unsubstituted.
- Alkynyls include, without limitation, ethynyl, 1 -propynyl, 2-propynyl, 1 -butynyl, 2- butynyl, and 3-butynyl.
- cycloalkyl refers to cyclic or polycyclic hydrocarbon groups of from 3 to 13 carbon atoms, any of which is saturated. Cycloalkyl groups may be substituted or unsubstituted.
- cycloalkyl groups include, without limitation, cyclopropyl, norbornyl,
- cycloalkenyl refers to cyclic or polycyclic hydrocarbon groups of from 3 to 13 carbon atoms, preferably from 5 to 8 carbon atoms, containing one or more double bonds.
- Cycloalkenyl groups may be substituted or unsubstituted.
- Exemplary cycloalkenyl groups include, without limitation, cyclopentenyl, cyclohexenyl, and cyclooctenyl .
- cycloalkynyl refers to cyclic or polycyclic hydrocarbon groups of from 5 to 13 carbon atoms containing one or more triple bonds. Cycloalkynyl groups may be substituted or unsubstituted.
- heteroalkyl is meant a branched or unbranched alkyl, alkenyl, or alkynyl group having from 1 to 14 carbon atoms in addition to 1 , 2, 3 or 4 heteroatoms independently selected from the group consisting of N, O, S, and P.
- Heteroalkyls include, without limitation, tertiary amines, secondary amines, ethers, thioethers, amides, thioamides, carbamates, thiocarbamates, hydrazones, imines, phosphodiesters, phosphoramidates, sulfonamides, and disulfides.
- a heteroalkyl may optionally include monocyclic, bicyclic, or tricyclic rings, in which each ring desirably has three to six members.
- the heteroalkyl group may be substituted or unsubstituted.
- heteroalkyls include, without limitation, polyethers, such as methoxymethyl and ethoxyethyl.
- heterocyclic ring and “heterocyclyl” refer to non-aromatic ring systems having five to fourteen ring atoms in which one or more ring carbons, preferably one to four, are each replaced by a heteroatom such as N, O, S, or P, which may be used alone or as part of a larger moiety as in “heterocyclyl-alkyl” (a heterocyclyl-substituted C). 6 alkyl), “heterocyclyl-alkoxy” (a heterocyclyl-substituted C 1-6 alkoxy), or “heterocycloxy-alkyl” (a heterocycloxy-substituted C
- heterocyclic rings may be substituted or unsubstituted and may include one, two, or three fused or unfused ring systems.
- the heterocyclic ring is a 5- to 7-membered monocyclic or 7- to 14-membered bicyclic heterocyclic ring consisting of 2 to 6 carbon atoms and 1 , 2, 3, or 4 heteroatoms independently selected from N, O, and S and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- heterocyclic rings include, without limitation, 3- lH-benzimidazol-2-one, ( 1 -substituted)-2-oxo-benzimidazol-3-yl, 2-tetrahydrofuranyl, 3-tetrahydrofuranyl,
- a heterocyclyl group can include two or more of the ring systems listed above.
- Heterocyclic rings include those in which a non-aromatic heteroatom-containing ring is fused to one or more aromatic or non-aromatic rings, such as in an indolinyl, chromanyl, phenanthridinyl, or tetrahydroquinolinyl, where the radical or point of attachment is on the non-aromatic heteroatom- containing ring.
- aryl used alone or as part of a larger moiety as in “aralkyl” (an aryl-substituted C,.
- aralkoxy an aryl-substituted Ci. 6 alkoxy
- aryloxyalkyl an aryloxy-substituted Ci. 6 alkyl
- aromatic monocyclic or polycyclic ring groups having six to fourteen ring atoms, such as phenyl, 1 -naphthyl, 2-naphthyl, 1 -anthracyl, and 2-anthracyl and includes aralkyl, aralkoxy, and aryloxyalkyl groups.
- An "aryl” ring may be substituted or unsubstituted.
- aryl includes fused polycyclic aromatic ring systems in which an aromatic ring is fused to one or more rings.
- Non- limiting examples of aryl groups include phenyl, hydroxyphenyl, halophenyl, alkoxyphenyl, dialkoxyphenyl, trialkoxyphenyi, alkylenedioxyphenyl, naphthyl, phenanthryl, anthryl, phenanthro, 1 - naphthyl, 2-naphthyl, 1 -anthracyl, and 2-anthracyl.
- aryl is a group in which an aromatic ring is fused to one or more non-aromatic rings, such as in a indanyl, phenanthridinyl, or tetrahydronaphthyl, where the radical or point of attachment is on the aromatic ring.
- heteroaryl refers to stable heterocyclic, and polyheterocyclic aromatic moieties having 5 - 14 ring atoms. Heteroaryl groups may be substituted or unsubstituted and include both monocyclic and polycyclic ring systems.
- heteroaryl rings include 5-membered monocyclic rings, such as thienyl, pyrrolyl, imidazolyl, pyrazolyl, furyl, isothiazolyl, furazanyl, isoxazolyl, and thiazolyl; 6-membered monocyclic rings, such as pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, and triazinyl; and polycyclic heterocyclic rings, such as benzo[b]thienyl, naphtho[2,3-b]thienyl, thianthrenyl, isobenzofuranyl, chromenyl, xanthenyl, phenoxathienyl, indolizinyl, isoindolyl, indolyl, indazolyl, purinyl, isoquinolyl, quinolyl, phthalazinyl, naphthyridinyl, quinox
- heteroaryl rings include, without limitation, 2- furanyl, 3-furanyl, N-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl, 3-isoxazolyl, 4-isoxazoIyl, 5-isoxazolyl, 2-oxadiazolyl, 5-oxadiazolyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 1 -pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 5-pyrimidyl,
- Heteroaryl groups further include a group in which a heteroaromatic ring is fused to one or more aromatic or nonaromatic rings where the radical or point of attachment is on the heteroaromatic ring, such as tetrahydroquinoline, tetrahydroisoquinoline, and pyrido[3,4- d]pyrimidinyl, imidazo[l ,2-a]pyrimidyl, imidazo[l ,2-a]pyrazinyl, imidazo[l ,2-a]pyiridinyl, imidazo[l ,2-c]pyrimidyl, pyrazolo[ l ,5-a][l ,3,5]triazinyl, pyrazolo[l ,5-c]pyrimidyl, imidazo[l ,2- bjpyridazinyl, imidazo[l ,5-a]pyrimidyl, pyrazolo[l ,5-b]
- An aryl group or heteroaryl group may contain one or more substituents.
- substituents for aryl or heteroaryl group include halogen (F, CI, Br or I), alkyl, alkenyl, alkynyl, heteroalkyl, -NO2, -CN, -R A , -OR B , -S(O) R B , (wherein r is 0, 1 or 2), -S0 2 NR A R B ,— NR A R B , -O-
- R c is selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroaryl, and heterocyclyl.
- each of R A and R B is, independently, selected from hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroaryl, and heterocyclyl.
- R A , R B and R c optionally bears one or more substituents selected from amino, alkylamino, dialkylamino, aminocarbonyl, halogen, alkyl, aryl, heteroalkyl, heteroaryl, carbocycle, heterocycle, alkylaminocarbonyl, dialkylaminocarbonyl, alkylaminocarbonyloxy,
- dialkylaminocarbonyloxy, nitro, cyano, carboxy, alkoxycarbonyl, alkylcarbonyl, alkoxy, haloalkoxy groups, hydroxy, protected hydroxyl groups (e.g., -O-X, where X is acyl, phenyl, substituted phenyl, benzyl, substituted benzyl, phenethyl, or substituted phenethyl), -M -heteroaryl, -M-heterocycle, -M- aryl, -M-OR B , -M-SR B , -M-NR A R B , -M-OC(0)NR A R B -M-C( NR B )NR A R B ,
- R A , R B or R c group include haloalkyl and trihaloalkyl, alkoxyalkyl, halophenyl, chloromethyl,
- the invention features compounds which can be useful for treating patients who have an EGFR-driven cancer, including cancers which are, or have become, refractory to erlotinib or gefitinib, or cancers which bear an EGFR mutation identified herein, by administering a compound of formula (I) to the patient.
- the EGFR-driven cancers which can be treated using the compositions and method of the invention include, for example, EGFR mutants including one or more deletions, substitutions, or additions in the amino acid or nucleotide sequences of EGFR, or fragments thereof.
- Mutations in EGFR can occur in any part of the EGFR sequence.
- EGFR mutants arise from mutations in the kinase domain (i.e., exons 18-24 in the EGFR sequence) or in the extracellular domain (i.e., exons 2-16 in the EGFR sequence).
- mutations typically occur in the kinase domain, including one or more of a point mutation in exon 1 8 (e.g., L688P, V689M, P694L/S, N700D, L703V, E709K/Q/A/G/V, I715S, L718P, G719C/A/S/R, or S720P/F), a deletion in exon 19 that may or may not include an insertion (e.g., delG719, delE746_E749, delE746_A750, delE746_A750insRP, delE746_A750insQP, delE746_T751 , delE746_T751 insA/I/V,
- a point mutation in exon 1 8 e.g., L688P, V689M, P694L/S, N700D, L703V, E709K/Q/A/G/
- S768_D770dupSVD A767_V769dupASV, or H773dupH
- a point mutation in exon 20 e.g., D761 , A763V, V765A/M, S768I, V769L/M, S768I, P772R, N771T, H773R/Y/L, V774M, R776G/H/C, G779S/F, T783A, T784F, L792P, L798H/F, T790M, R803W, 806E, or L814P), or a point mutation in exon 21 (e.g., G810S, N826S, L833V, H835L, L838V, A839T, K846R, T847I, H850N, V851 I/A, I853T, L858M/R, A859T, L861 Q/R, G863
- activation mutants are typical, and 90% deletion of 746-750 (ELREA) and L858R result in sustained phosphorylation of EGFR without ligand stimulation.
- ELREA 746-750
- L858R L858R
- mutations typically, but not exclusively, occur in the extracellular domain, including EGFR variant I (EGFRvI) lacking the extracellular domain and resembling the v-erbB oncoprotein; EGFRvII lacking 83 amino acids from domain IV; and EGFRvIII lacking amino acids 30-297 from domains I and II, which is the most common amplification and is reported in 30-50%o of glioblastomas and 5% of squamous cell carcinoma.
- EGFRvI EGFR variant I
- EGFRvIII lacking 83 amino acids from domain IV
- EGFRvIII lacking amino acids 30-297 from domains I and II, which is the most common amplification and is reported in 30-50%o of glioblastomas and 5% of squamous cell carcinoma.
- Other mutations for glioblastoma include one or more of point mutations in exon 2 (e.g., D46N or G63R), exon 3 (e.g., R108 in domain I), exon 7 (e.g., T263P or A289D/T7V in domain II), exon 8 (e.g., R324L or E330K), exon 15 (e.g., P596L or G598V in domain IV), or exon 21 (L861 Q in the kinase domain).
- exon 2 e.g., D46N or G63R
- exon 3 e.g., R108 in domain I
- exon 7 e.g., T263P or A289D/T7V in domain II
- exon 8 e.g., R324L or E330K
- exon 15 e.g., P596L or G598V in domain IV
- exon 21 L861 Q in the kinas
- EGFR mutants also include those with a combination of two or more mutations, as described herein.
- Exemplary combinations include S768I and G719A; S768I and V769L; H773R and
- W731 Stop R776G and L858R; R776H and L861 Q; T790M and L858R; T790M and delE746_A750; R803W and delE746_T751 insVA; delL747_E749 and A750P; delL747_S752 and E746V;
- T790M e.g., T790M and L858R or T790M and delE746_A750, with or without concomitant inhibition of single mutants L858R and delE746_A750.
- EGFR mutants can be either activation mutants or resistant mutants.
- Activation mutants include those with substitutions that increase drug sensitivity (e.g., G719C/S/A, delE746_A750, or
- Resistant mutants include those with substitutions that increase drug resistance (e.g., T790M or any combination including T790M).
- EGFR-driven cancers include those having any mutant described herein.
- EGFRvIII is commonly found in glioblastoma and has also been reported in breast, ovarian, prostate, and lung carcinomas.
- Exemplary EGFR-driven cancers glioblastoma, lung cancer (e.g., squamous cell carcinoma, non-small cell lung cancer, adenocarcinoma, bronchioloalveolar carcinoma (BAC), BAC with focal invasion, adenocarcinoma with BAC features, and large cell carcinoma), pancreatic cancer, head and neck cancers (e.g., squamous cell carcinoma), breast cancer, colorectal cancer, epithelial cancer (e.g., squamous cell carcinoma), ovarian cancer, and prostate cancer.
- lung cancer e.g., squamous cell carcinoma, non-small cell lung cancer, adenocarcinoma, bronchioloalveolar carcinoma (BAC), BAC with focal invasion, adenocar
- the invention described herein would benefit patient populations having higher risk for TKI-resistant mutations.
- About 8,000 to 16,000 new cases per year can be estimated based on: incidence of non-small cell lung cancer (about 160,000 new cases in the U.S.), the response to erlonitinib in the general population (about 10%, resulting in a sensitive population of 16,000), the presence of activation mutations (10-20% in white and 30-40% in Asian population, resulting in a sensitive population of 16,000-32,000), acquisition of secondary resistance (most if not all patients, resulting in a sensitive population of 16,000-32,000), and percentage of patients carrying the T790M point mutations (about 50%, resulting in a sensitive population of 8,000-16,000).
- Patients having TKI-resistant mutations include those patients having cancers resistant to one or more of erlotinib, gefitinib, CL-387,785, BIBW 2992 (CAS Reg. No. 439081 - 18-2), CI-1033, neratinib (HKI-272), MP- 412 (AV-412), PF-299804, AEE78, and XL64.
- the inventions relates to treatment of EGFR-driven cancers having the T790M point mutation.
- reversible inhibitors e.g., CI-1033, neratinib (HKI-272), and PF-299804
- CI-1033, neratinib (HKI-272), and PF-299804 are less potent in cell lines having the T790M mutation and do not inhibit T790M at clinically achievable concentrations. Since the ATP Km of T790M and WT are similar, concentrations that inhibit the mutant will inhibit the WT and result in gastrointestinal and cutaneous events.
- An EGFR mutant also includes other amino acid and nucleotide sequences of EGFR with one or more deletions, substitutions, or additions, such as point mutations, that retain or increase tyrosine kinase or phosphorylation activity.
- preferable substitutions are conservative substitutions, which are substitutions between amino acids similar in properties such as structural, electric, polar, or hydrophobic properties.
- the substitution can be conducted between basic amino acids (e.g., Lys, Arg, and His), or between acidic amino acids (e.g., Asp and Glu), or between amino acids having non-charged polar side chains (e.g., Gly, Asn,
- Gin, Ser, Thr, Tyr, and Cys amino acids having hydrophobic side chains (e.g., Ala, Val, Leu, He, Pro, Phe, and Met), or between amino acids having branched side chains (e.g., Thr, Val, Leu, and He), or between amino acids having aromatic side chains (e.g., Tyr, Trp, Phe, and His).
- amino acids having hydrophobic side chains e.g., Ala, Val, Leu, He, Pro, Phe, and Met
- amino acids having branched side chains e.g., Thr, Val, Leu, and He
- amino acids having aromatic side chains e.g., Tyr, Trp, Phe, and His
- the DNA encoding an EGFR mutant protein may comprise a nucleotide sequence capable of hybridizing to a complement sequence of the nucleotide sequence encoding an EGFR mutant, as defined herein, under stringent conditions.
- the stringent conditions include low, medium or high stringent conditions.
- An example of the stringent conditions includes hybridization at approximately 42-55°C in approximately 2-6 x SSC, followed by wash at approximately 50-65°C in approximately 0.1 - 1 x SSC containing approximately 0.1 -0.2% SDS, where 1 x SSC is a solution containing 0.15 M NaCl and 0.015 M Na citrate, pH 7.0. Wash can be performed once or more.
- stringent conditions may be set at a temperature approximately 5°C lower than a melting temperature (Tm) of a specific nucleotide sequence at defined ionic strength and pH.
- Tm melting temperature
- GenBank accession numbers for EGFR [Homo sapiens] include MIM131550, AAI28420, NM_005228, NP_005219.2, and GenelD: 1956.
- compositions and methods of the invention can be used to treat subjects having an EGFR-driven cancer (i.e., cancers characterized by EGFR mutant expression or overexpression).
- an EGFR-driven cancer i.e., cancers characterized by EGFR mutant expression or overexpression.
- EGFR mutant expression or overexpression can be determined in a diagnostic or prognostic assay by evaluating levels of EGFR mutants in biological sample, or secreted by the cell (e.g., via an immunohistochemistry assay using anti-EGFR antibodies or anti-p-EGFR antibodies; FACS analysis, etc.).
- FISH fluorescent in situ hybridization using a nucleic acid based probe corresponding to an EGFR mutant-encoding nucleic acid or the complement thereof
- FISH fluorescent in situ hybridization using a nucleic acid based probe corresponding to an EGFR mutant-encoding nucleic acid or the complement thereof
- PCR polymerase chain reaction
- RT-PCR real time quantitative PCR
- DNA of cells isolated by the methods of the invention can be sequenced, or certain sequence characteristics (e.g.,
- polymorphisms and chromosomal abnormalities can be identified using standard techniques, e.g., FISH or PCR.
- the chemical components of cells, and other analytes, may also be assayed after isolation.
- Cells may also be assayed without lysis, e.g., using extracellular or intracellular stains or by other observation, e.g., morphology or growth characteristics in various media.
- FISH fluorescent in situ hybridization
- FISH is a cytogenetic technique which can be used to detect and localize the presence or absence of specific DNA or RNA sequences on chromosomes.
- FISH incorporates the use of fluorescently labeled nucleic acid probes which bind only to those parts of the chromosome with which they show a high degree of sequence similarity. Fluorescence microscopy can be used to find out where the fluorescent probe bound to the chromosome. The basic steps of FISH are outlined below.
- Exemplary FISH probes include Vysis EGFR SpectrumOrange/ CEP SpectrumGreen Probe (Abbott, Downers Grove, IL), which hybridizes to band 7pl 2; and ZytoLight SPEC EGFR/CEN 7 Dual Color Probe (Zyto Vision), which hybridizes to the alpha-satellite sequences of the centromere of chromosome 7.
- Probes are generally labeled with fluorophores, with targets for antibodies, with biotin, or any combination thereof. This can be done in various ways, for example using random priming, nick translation, and PCR using tagged nucleotides.
- a sample or aliquot of a population of cells is used for FISH analysis.
- cells are trypsinized to disperse into single cells, cytospun onto glass slides, and then fixed with paraformaldehyde before storing in 70% ethanol.
- the chromosomes are firmly attached to a substrate, usually glass. After preparation, the probe is applied to the chromosome RNA and starts to hybridize. In several wash steps, all unhybridized or partially hybridized probes are washed away.
- An epifluorescence microscope can be used for observation of the hybridized sequences.
- the white light of the source lamp is filtered so that only the relevant wavelengths for excitation of the fluorescent molecules arrive onto the sample.
- Emission of the fluorochromes happens, in general, at larger wavelengths, which allows one to distinguish between excitation and emission light by mean of another optical filter. With a more sophisticated filter set, it is possible to distinguish between several excitation and emission bands, and thus between several fluorochromes, which allows observation of many different probes on the same strand.
- FISH can have resolution ranging from huge chromosomes or tiny ( ⁇ 100 kilobase) sequences.
- the probes can be quantified simply by counting dots or comparing color.
- Allele-specific quantitative real time-PCR may also be used to identify a nucleic acid encoding a mutant EGFR protein (see, for e.g., Diagnostic Innovations DxS BCR-ABL T3151 Mutation Test Kit, and Singer et al., Methods in Molec. Biol. 181 : 145 (2001 )).
- This technique utilizes Taq DNA polymerase, which is extremely effective at distinguishing between a match and a mismatch at the 3 '-end of the primer (when the 3 '-base is mismatched, no efficient amplification occurs).
- the 3 '-end of the primer may be designed to specifically hybridize to a nucleic acid sequence that corresponds to a codon that encodes a mutant amino acid in an EGFR mutant, as described herein.
- the specific mutated sequences can be selectively amplified in a patient sample.
- This technique further utilizes a Scorpion probe molecule, which is a bifunctional molecule containing a PCR primer, a fluorophore, and a quencher. The f uorophore in the probe interacts with a quencher, which reduces fluorescence.
- the Scorpion probe binds to the amplicon, the fluorophore and quencher in the Scorpion probe become separated, which leads to an increase in fluorescence from the reaction tube.
- Any of the primers described herein may be used in allele-specific quantitative real time PCR.
- a biological sample can be analyzed to detect a mutation in an EGFR gene, or expression levels of an EGFR gene, by methods that are known in the art. For example, methods such as direct nucleic acid sequencing, altered hybridization, aberrant electrophoretic gel migration, binding or cleavage mediated by mismatch binding proteins, single-strand conformational polymorphism (SSCP) analysis, or restriction fragment length polymorphism (RFLP) analysis of PCR products derived from a patient sample can be used to detect a mutation in an EGFR gene; ELISA can be used to measure levels of EGFR polypeptide; and PCR can be used to measure the level of an EGFR nucleic acid molecule.
- methods such as direct nucleic acid sequencing, altered hybridization, aberrant electrophoretic gel migration, binding or cleavage mediated by mismatch binding proteins, single-strand conformational polymorphism (SSCP) analysis, or restriction fragment length polymorphism (RFLP) analysis of PCR products derived from a patient sample can be used to detect
- Any of these techniques may be used to facilitate detection of a mutation in a candidate gene, and each is well known in the art; examples of particular techniques are described, without limitation, in Orita et al. (Proc. Natl. Acad. Sci. USA 86:2766 (1989)) and Sheffield et al. (Proc. Natl. Acad. Sci. USA 86:232 (1989)).
- expression of the candidate gene in a biological sample may be monitored by standard Northern blot analysis or may be aided by PCR (see, e.g., Ausubel et al., Current Protocols in Molecular Biology, John Wiley & Sons, New York, NY (1995); PCR Technology: Principles and Applications for DNA Amplification, H.A. Ehrlich, Ed., Stockton Press, NY; Yap et al., Nucl. Acids. Res. 19:4294 (1991 )).
- nucleic acid or protein sequence may identify in a nucleic acid or protein sequence a residue (e.g., amino acid or nucleotide) or codon that corresponds to a residue or codon in wild-type EGFR or EGFR mutants using a number of sequence alignment software programs (e.g., NCBI BLAST website). Such software programs may allow for gaps in the alignment of the compared sequences. Using such software, one skilled in the art may identify a nucleotide, amino acid, or amino acid that
- EGFR expression in a biological sample can be determined by using any of a number of standard techniques that are well known in the art or described herein.
- Exemplary biological samples include plasma, blood, sputum, pleural effusion, bronchoalveolar lavage, or biopsy, such as a lung biopsy and lymph node biopsy.
- EGFR expression in a biological sample e.g., a blood or tissue sample
- PCR Technology Principles and Applications for DNA Amplification, H.A. Ehrlich, Ed., Stockton Press, NY; Yap et al., Nucl. Acids. Res. 19:4294 ( 1991 )).
- compounds of formula (I) in which R e is H and R d is H, CI, CF 3 , or CH 3 can be synthesized from 2,4-dichloropyrimidine, 2,4,5-trichloropyrimidine, 2,4-dichloro-5- (trifluoromethyl)pyrimidine, or 2,4-dichloro-5-methylpyrimidine, respectively, as described in PCT Publication No. WO/2009/143389.
- Compounds of Formula I can be formulated into a pharmaceutical composition that comprises a compound of Formula I (as an active pharmaceutical ingredient) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable excipient.
- a pharmaceutical composition comprising a compound of Formula 1 or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable excipient.
- compositions containing a compound of Formula I suitable for administration may be formulated using conventional materials and methods, a wide variety of which are well known. Suitable dosage forms include those in solution, suspension or emulsion form, and solid oral dosage forms such as capsules, tablets, gel caps, caplets, etc.. Methods well known in the art for making formulations, including the foregoing unit dosage forms, are found, for example, in "Remington: The Science and Practice of Pharmacy” (20th ed., ed. A.R. Gennaro, 2000, Lippincott Williams & Wilkins).
- Compounds of formula (I) can be formulated for any route of administration (e.g., orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by transdermal patch, powders, ointments, or drops), sublingually, bucally, as an oral or nasal spray) effective for use in the methods of the invention.
- routes of administration e.g., orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by transdermal patch, powders, ointments, or drops), sublingually, bucally, as an oral or nasal spray
- routes of administration e.g., orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by transdermal patch, powders, ointments, or drops), sublingually, bucally, as an oral or nasal spray
- compounds of formula (I) are
- a compound of formula (I) can be formulated for as a capsule for oral administration containing nominally 10 mg, 50 mg, 100 mg, 150 mg, 250 mg, 500 mg, or any dosage amounts described herein as the free base or acid addition salt of the compound (e.g., the hydrochloride salt).
- the unit dosage forms of the invention can include a compound of the invention, or a salt thereof, formulated with excipients, fillers, flow enhancers, lubricants, and/or disintegrants as needed.
- a unit dosage form can include colloidal silicon dioxide (a flow enhancer), lactose anhydrous (a filler), magnesium stearate (a lubricant), microcrystalline cellulose (a filler), and/or sodium starch glycolate (a disintegrant).
- colloidal silicon dioxide a flow enhancer
- lactose anhydrous a filler
- magnesium stearate a lubricant
- microcrystalline cellulose a filler
- sodium starch glycolate a disintegrant
- Compounds of formula (I) can be useful for treating EGFR-driven cancers.
- the compounds can be useful for treating EGFR-driven cancers that express EGFR mutants and for treating EGFR-driven cancers that are refractory to TKI therapies (e.g., erlotinib or gefitinib).
- Such cancers can include, among others, non-small cell lung cancer (NSCLS), including one or more of squamous cell carcinoma, adenocarcinoma, adenocarcinoma, bronchioloalveolar carcinoma (BAC), BAC with focal invasion, adenocarcinoma with BAC features, and large cell carcinoma; neural tumors, such as glioblastomas; pancreatic cancer; head and neck cancers (e.g., squamous cell carcinoma); breast cancer; colorectal cancer; epithelial cancer, including squamous cell carcinoma; ovarian cancer; prostate cancer; adenocarcinomas; and including cancers which are EGFR mediated.
- NSCS non-small cell lung cancer
- BAC bronchioloalveolar carcinoma
- BAC bronchioloalveolar carcinoma
- BAC BAC with focal invasion, adenocarcinoma with BAC features, and large cell carcinoma
- neural tumors such as glioblastomas
- pancreatic cancer head
- the present invention is based upon the discovery that compounds of formula (I) can be used to treat EGFR-driven cancers, EGFR-driven cancers that express EGFR mutants, and for treating EGFR-driven cancers that are refractory to TKI therapy, such as erlotinib or gefitinib.
- Compounds of formula (I) can also be used in a maintenance role to prevent recurrence of cancer in patients in need of such a treatment.
- the effective systemic dose of a compound of formula (I) will typically be in the range of an average daily dose of from 10 mg to 2,000 mg of the compound per kg of patient body weight, administered in single or multiple doses.
- a compound of the invention may be administered to patients in need of such treatment in a daily dose range of about 50 to about 2,000 mg per patient. Administration may be once or multiple times daily, weekly (or at some other multiple- day interval) or on an intermittent schedule.
- the compound may be administered one or more times per day on a weekly basis (e.g. every Monday) indefinitely or for a period of weeks, e.g. 4 - 10 weeks.
- it may be administered daily for a period of days (e.g.
- a compound of the invention may be administered daily for 5 days, then discontinued for 9 days, then administered daily for another 5 day period, then discontinued for 9 days, and so on, repeating the cycle indefinitely, or for a total of 4 - 10 times.
- each component of the combination therapy may be administered at their monotherapy dosing levels and schedules.
- erlotinib has been administered orally for the treatment of NSCLC at 150 mg daily and of pancreatic cancer at 100 mg daily.
- gefitinib has been administered orally for the treatment of NSCLC at 250 mg daily.
- the effective systemic dose of a compound of the invention will typically be in the range of an average daily dose of from 10 mg to 2,000 mg of the compound per kg of patient body weight, administered in single or multiple doses.
- a compound of the invention may be administered to patients in need of such treatment in a daily dose range of about 50 to about 2,000 mg per patient. Administration may be once or multiple times daily, weekly (or at some other multiple- day interval) or on an intermittent schedule.
- the compound may be administered one or more times per day on a weekly basis (e.g. every Monday) indefinitely or for a period of weeks, e.g. 4 - 10 weeks.
- it may be administered daily for a period of days (e.g.
- a compound of the invention may be administered daily for 5 days, then discontinued for 9 days, then administered daily for another 5 day period, then discontinued for 9 days, and so on, repeating the cycle indefinitely, or for a total of 4 - 10 times.
- a TKI e.g., erlotinib or gefitinib
- a compound of formula (I) with a reduced dosing level in one or both components.
- Step 1 synthesis of compound 1
- the aniline (280 mg, 0.606 mmol) was dissolved in 8 mL of DCM and 0.3 mL of triethylamine was added. The mixture was cooled to -35 °C and acryloyl chloride (54.8 mg, 49 ⁇ , 0.606 mmol, 1.0 eq) was added in portions. The reaction was stirred around -30 °C for 15 min and quenched with saturated Na 2 C0 3 . The mixture was worked up with sat. Na 2 C0 3 /DCM and purified with preparation plates to give a light brown solid, 205 mg, in 66% yield.
- Step 1 Compound 31 was prepared according to the procedure described for the synthesis of compound 1 in Example 1 , using 2-iodo-3-methylaniline instead of 2-iodoaniline as the starting material.
- a suspension of 31 (0.53 mmol), 2,4,5-trichloropyrimidine (1.0 eq), potassium carbonate ( 1 .2 eq), and tetrabutylammonium hydrogensulfate (0.1 eq) in DMF was stirred at 65 °C for 1 8 hrs. Upon cooling the reaction mixture was filtered and the filtrate was concentrated. The residue was taken up into a mixture of EtOAc and water. After extraction with EtOAc (3 x), the combined organic phases were concentrated to give essentially pure material which was used directly in next step reaction.
- Step 2 A solution of 32 (0.82 mmol), 2-methoxy-5-nitroaniline 33 ( l eq) and TFA (3 eq) in 2-BuOH (3 mL) was heated at 100 °C for 18 hrs. Upon cooling EtOAc and aq. NaHC0 3 were added to the reaction mixture. Extraction (3 x) and concentration of combined extracts gave a solid which was purified on silica gel column (ISCO machine) with 10% MeOH in CH 2 CI 2 as the eluents, furnishing 34 as a brownish solid (55%).
- Step 3 To a suspension of 34 (0.46 mmol) and zinc powder (6 eq) in acetone (9 mL) and water ( 1 mL) was added ammonium chloride (10 eq) at 0 °C. After the mixture was stirred at room temperature for 30 min, HPLC indicated a complete conversion. Acetone was removed on rotavap and the residue was suspended in DCM and water. Filtration was carried out and the filtrate was extracted with DCM. Concentration of combined organic layers gave crude aniline 35, which was used in the next step without purification.
- Step 4 To a solution of aniline 35 (0.43 mmol) and N, N-diisopropylethylamine (1.1 eq) in DCM (2 mL) was added acryloyl chloride ( 1 .05 eq) at 0 °C. After the mixture was stirred at room temperature overnight, the volatile components were removed on rotavap. The residue was purified on silica gel column with 3% MeOH in DCM as eluents, furnishing amide 36 as beige solid (48 mg, 21 %).
- 2-(Dimethylaminomethyl)acrylic acid 39 was prepared according to a literature procedure ⁇ Synth. Comm. 1995, 25, 641 ). To a solution of 39 (65 mg, 0.5 mmol), coupling reagent TBTU ( 1 .2 eq) and N, N-diisopropylethylamine (3.0 eq) in DMF (5 mL) and DCM (20 mL) was added 5 ( 1 eq). After the mixture was stirred at room temperature overnight, the volatile components were removed on rotavap and the residue was purified by reverse phase prep-HPLC, furnishing the title compound as a tan solid (23 mg, 9%).
- Step 1 the starting material 41 was prepared from 3-fluoro-4-chIorophenoi via nitration and subsequent O-mefhylation, according to a published procedure (US Patent Publication No.
- Steps 2 and 3 A degassed suspension of 42 (0.96 g, 3.4 mmol), benzophenone imine (1.5 eq), palladium acetate (0.1 eq), xantphos (0.2 eq) and cesium carbonate (1.6 eq) in DMF (20 mL) was heated at 1 10 °C overnight. Upon cooling the reaction mixture was filtered and the filtrate was concentrated. The solid residue was dissolved in dioxane and 2M aq. HC1 ( 1 : 1 , 40 mL) and then heated at 70 °C for 2 hrs. Upon removing dioxane on rotavap, the water layer was washed with DCM and then basified with aq.
- Step 4 To a solution of 43 (0.38 g, 1.42 mmol) in THF ( 15 mL) was added NaH (2 eq.) under N 2 at 0 °C in multiple portions. After bubbles of H 2 were no longer observed, Boc 2 0 (4 eq.) was added. The resulting reaction mixture was heated at 50 °C and then refluxed for 2 hrs. The reaction was quenched with MeOH. Usual workup followed by silica gel column chromatograph (5% MeOH in DCM as eluents) furnished 44 (0.46 g, 88%).
- Step 5 With EtOAc as the solvent 44 (0.46 g) was hydrogenated under 50 psi to afford 45 (0.42 g, 99%). Upon removing the solvent, the crude material was used directly in the next step.
- Step 6 A degassed suspension of 45 (0.42 g, 3.4 mmol), 2 ( 1 .5 eq), palladium acetate (0.1 eq), xantphos (0.2 eq) and cesium carbonate ( 1.3 eq) in DMF ( 10 mL) was heated at 1 10 °C for 48 hrs. Usual workup followed by silica gel column chromatograph (5% MeOH in DCM as eluents) furnished 46 (0.49 g, 64%).
- Step 7 To a solution of 46 in DCM was added excessive TFA. After the mixture was stirred at room temperature for 2 hrs, the volatile components were removed on rotavap. The residue was dissolved in EtOAC and the solution was basified with aq. NaHC0 3 . Extraction and concentration gave 47 as tan solid.
- Step 8 Crude 47 (100 mg) was converted to 48 by using the procedure described in Example 14, step 4. The final product was purified by reverse phase prep-HPLC ( 10.4 mg, 9%).
- Step 2 To a degassed suspension of 49 (0.275 g, 1.0 mmol), Pd 2 (dba) 3 (0.1 eq), 2-(di-t- butylphosphino)-N, N-dimethylbiphenylamine (0.1 eq) and sodium t-butoxide (1.4 eq) in dioxane (10 mL) was added a solution of NH 3 in dioxane (0.5 M in a N 2 -sealed bottle, 10 mL). The resulting mixture was heated at 80 °C for 3 hrs. Usual workup followed by silica gel column chromatograph (15% MeOH in DCM as eluents) furnished 50 (0.15 g, 55%).
- Steps 3 to 7 The poly-substituted aniline 50 was converted to the title compound 51 according to the procedure described in Example 1 8 by substituting 50 for 43.
- 2,4-Dimethoxy-5-nitroaniline 53 was prepared from l ,5-difluoro-2,4-dinitrobenzene via double S N Ar substitution to generate 52 and subsequent mono-reduction of nitro groups, according to a published procedure ( J. Org. Chem. 2005, 70, 10660). This was converted to the title compound 54 as for Example 14 by substituting 53 for 33 and 2 for 32.
- Step 1 A degassed suspension of 2-nitro-4-bromoanisole (2.32 g, 10 mmol), N,N-dimethyl- 1 ,3-propanediamine (1.1 eq), Pd 2 (dba) 3 (0.02 eq), dppf (0.04 eq) and sodium
- Steps 2 to 6 The secondary amine 55 was converted to the title compound 56 as for Example 18 by substituting 55 for 43.
- Example 22
- Step 1 Acetic anhydride (7.8 mL, 82.6 mmol) was added dropwise with vigorously stirring to a suspension of 3-fluoro-4-aminophenol (10 g, 78.6 mmol) in water (20 mL). Insoluble amide started to precipitate as a white solid in a few minutes. After the reaction mixture was stirred for 10 more minutes, the white solid was collected via filtration and washed with cold water. Silica gel column chromatograph (5% MeOH in DCM as eluent) gave pure 57 (8.75 g, 66%).
- Step 2 To a suspension of 57 (8.75 g, 51 .73 mmol) and K 2 C0 3 (1.1 eq) in THF (30 rtiL) was added methyl iodide (1.2 eq). The mixture was heated in a sealed tube at 60 °C overnight. Filtration and concentration followed by silica gel column chromatograph (5% MeOH in DCM as eluent) furnished pure 58 (7.17 g, 89%).
- Step 3 Nitric acid (70%, 3.83 mL) was added dropwise to a solution of 58 (7 g) in DCM (70 mL) with vigorously stirring. After stirred at room temperature for 1 hr, the reaction mixture was refluxed for 3 hrs. DCM was removed on rotavap and the residue was washed with cold water and then subjected to a silica gel column chromatograph purification (5% MeOH in DCM as eluent) to afford 59 (3.26 g, 37%).
- Step 4 Poly-substituted nitrobenzene 59 was reduced to corresponding aniline 60 according to the procedure described in Example 14, step 3.
- Step 5 Poly-substituted aniline 60 (200 mg, 1 mmol) was coupled with precursor 2 ( 1 .5 eq) to afford 61 (320 mg, 67%) via the procedure described in Example 1 8, step 6.
- Step 6 N-arylacetamide 61 (320 mg) was heated at reflux in 6N HC1 for 30 min. After basification, extraction and concentration aryl amine 62 was obtained (290 mg, 98%).
- Step 7 Crude 62 ( 150 mg) was converted to 63 by using the procedure described in Example 14, step 4. The final product was purified by reverse phase prep-HPLC (41 mg, 24%).
- Step 1 2,4-Dchloro-7H-pyrrolo[2,3-d]pyrimidine (5.0 g, 27 mmol) was suspended in MeCN
- Step 2 A solution of 64 ( 1 .22 g, 6 mmol) in THF ( 10 mL) was slowly added to a suspension of NaH (1 eq) in THF ( 10 mL) at 0 °C. After the mixture was stirred for 10 min, a solution of tosyl chloride (1 eq) in THF (5 mL) was slowly added. Stirring was continued for 30 min at 0 °C and then at room temperature overnight. The reaction was shown to be complete via HPLC monitoring and was quenched via the addition of aq. NH 4 C1 ( 1 - 2 mL). The reaction mixture was then filtered through celite and the filtrate was evaporated. The crude product was purified via column chromatography (ISCO machine, EtO Ac/Heptane, EtOAc on a 0- 100% gradient) to give 65 ( 1.22 g, 56%).
- Step 3 In a microwave vessel (20 mL) were placed 65 (600 mg, 1.7 mmol), 1 (282 mg, 1.7 mmol) and isopropanol ( 10 mL). After HC1 (1.3 mL, 4 M in dioxane) was added, the resulting mixture was stirred in the microwave reactor at 150 °C for 2 hrs. The solvents were evaporated and the residue was purified via silica column chromatography (ISCO machine, EtOAc/Heptane, 0- 100% EtOAc to elute impurities and then MeOH DCM, 0-20% MeOH) to give pure 66 (900 mg, 54%).
- Step 4 Intermediate 66 (493 mg, 1 mmol), 2-methoxy-5-nitroaniline 33 ( 168 mg, 1 mmol), K 2 C0 3 (1.4 mmol), Pd 2 dba 3 (5 mol%) and X-Phos (10 mol%) were weighed into a 100 mL round bottom flask and placed under N 2 .
- the solvents toluene ( 10 mL) and tert-b t ol (2 mL) were added as a mixture and the stirring solution was evacuated and backfilled with N 2 three times. The resulting mixture was then stirred overnight at 1 10 °C. HPLC showed the reaction to be complete and the solvents were evaporated.
- the residue was purified by silica column chromatography on ISCO machine (5% MeOH in DCM as eluents) to give coupling product 67 (570 mg, 91 %).
- Step 5 Poly-substituted nitrobenzene 67 was reduced to corresponding aniline 68 according to the procedure described in Example 14, step 3.
- Step 6 Crude aniline 68 ( 123 mg) was converted to 69 by using the procedure described in Example 14, step 4. The product was purified by silica column chromatography on ISCO machine (5% MeOH in DCM as eluents). Yield: 69 mg, 51 %.
- Step 7 A solution of 69 (60 mg) and TBAF (1 M in THF, 0.3 mL) in THF ( 10 mL) was refluxed for 5 hrs. HPLC indicated a complete reaction. After the solvent was evaporated, the residue was purified by silica column chromatography on ISCO machine (5% MeOH in DCM as eluents). The product was co-eluted with TBAF; water wash furnished pure product ( 10 mg, 21 %).
- Step 1 To a suspension of 5-fluoro-2-nitroanisole (50 mmol, 8.5 g) and zinc powder (3.5 eq, 1 1 .4 g) in acetone (45 mL) and water (5 mL) was added ammonium chloride (1 1 eq, 29.3 g) at 0 °C in multiple portions. After the mixture was stirred at r.t. overnight, HPLC indicated a complete conversion. Acetone was removed on rotavap and the residue was suspended in DCM and water. Filtration was carried out and the filtrate was extracted with DCM. Concentration of combined organic layers gave crude aniline ( ⁇ 7.0 g), which was used in the next step without purification.
- Step 2 To a suspension of 4-fluoro-2-methoxyaniline (5.1 g, 36.1 mmol) in concentrated sulfuric acid (55mL) was added guanidine nitrate (4.38g, 36.1 mmol) in portion wise under ice cooling over 15 min. The mixture was stirred at the same temperature for additional 15 min. The reaction was then poured into a saturated cold NaHC0 3 solution and the precipitated solid were collected by filtration. The residue was taken up in EtOAc and dried over anhydrous Na 2 S0 4 . The solvent was stripped off to afford the B (4.72g). Step 3 : The above compound B (O.
- Step 4 NaH (0.039g, 0.96 mmol, 60% dispersion in oil) was taken up in a dry capped microwave vial. To this, l -(2-hydroxyethyl)-4-methylpiperazine (0.023g, 0.16mmol) dissolved in dry tetrahydrofuran ( 1 .6mL) was added dropwise. The mixture was stirred at rt for 20 min. Intermediate C ( 0.075g, 0.16mmol) was then added in one portion to this suspension and the mixture was heated to 67° C in the closed seal tube for 25 min. The mixture was allowed to reach at rt and quenched with a few drops of methanol. Solvent was removed under vacuum and the resultant crude was subjected to FCC eluting with DCM- MeOH (95/5) to furnish the desired product D ( 0.081 g).
- Step 5 Compound D (0.078g, 0.13mmol) was dissolved in a mixture of acetone ( 1.3mL) and water (0.3mL). To this, zinc nano powder (0.07g, 1 .3mmol) was added immediately followed by addition of NH 4 C1 (0.16g, 2.6mmol) in small portions. The mixture was vigorously stirred at r.t for 30 min. Anhydrous Na2S04 was then added to this stirring mixture and the resultant crude was filtered, solvents were evaporated and the residue were taken up in DCM and directly loaded on the silica gel cartridge and eluted with DCM- MeOH-NH 3 (90/ 10) to furnish the desired product E ( 0.044g).
- Step 6 To a solution of E (0.044g, 0.078mmol) in dry tetrahydrofuran (0.52mL) was added D1PEA (0.027mL, 0.156 mmol) at 0° C under stirring. This was followed by the addition of acryloyl chloride (0.007g, 0.078mmol). The reaction was stirred at that temperature for additional l h. Solvent was stripped off under vacuum and the crude was purified by FCC using DCM- MeOH-NH 3 (90/10) to furnish a gum which was further triturated with Et 2 0 to furnish a solid material F (0.02g).
- D1PEA 0.027mL, 0.156 mmol
- acryloyl chloride 0.007g, 0.078mmol
- Examples 32-34 were made in accordance with the methods shown in Scheme 26 by substituting secondary amines for (3-dimethylamino)pyrrolidine.
- Example 35 was made in accordance with the methods shown in Scheme 27.
- Example 36 was made in accordance with the methods shown in Scheme 28.
- Example 37 was made in accordance with the methods shown in Scheme 34.
- Step 1 To a solution of benzyl glycidyl ether (5.0 g) in MeOH (5.0 mL) was added NaOMe (5.0 mL, 25% in methanol). The mixture was warmed up to 50°C for 1 h and then heated to reflux for 5min. The mixture was treated with wet NaHC0 3 and filtered. Solvent was evaporated and the residue was dissolved in DCM (20 mL). The solution was dried and evaporated to give yellowish oil (4.8 g, yield 80%).
- Step 2 A solution of step 1 product (3.0 g) in DCM ( 100 mL) was treated with PDC (6.0 g) and molecular sieves (4.0 g). The mixture was stirred at room temperature for 4h and diluted with Et 2 0 (l OOmL). The mixture was filtered through a Celite pad and solvent evaporated to give yellow oil (1.7 g, yield 57%).
- Step 3 The compound was synthesized according to the following procedure: To a mixture of 2,2- dimethyl- l ,3-dioxan-5-one (2.6 g), dimethyl amine HC1 salt (1.8 g) in DCM (50 mL) was added NaHB(OAc) 3 (6.0 g) and Et 3 N (3.0 mL). The reaction mixture was stirred at room temperature overnight and then diluted with aq.NaHC03. The organic layer was dried and evaporated to give colorless oil (2.5 g, yield 79%).
- Step 4 A solution of step-3 product (1.5 g) in MeOH (10 mL) was charged with Pd-C (0.5 g, 10% wet) and hydrogenated under a hydrogen balloon at room temperature overnight. The catalyst was filtered off and the solvent was evaporated to give the product (0.64 g, yield 71%) as colorless oil.
- Step 5 The compound was synthesized according the general procedure from compound B and step- 4 product as a yellow solid.
- Example 37 was synthesized according to a similar procedure to the following: To a mixture of tert-butyl (4-(4-((5-chloro-4-((2-(dimethylphosphoryl)phenyl) amino)pyrimidin-2-yl)amino)-5- methoxy-2-nitrophenyl)-2-methylbut-3-yn-2-yl)carbamate (240 mg) in acetone (1.0 mL) and Zn dust (0.3 g), NH 4 C1 (0.15 g), was added drops of water. The mixture was stirring at room temperature for 15 min and then diluted with DCM (5 mL). After filtration, the organic solution was evaporated and the residue was used for next step.
- Example 38 was made in accordance with the methods shown in Scheme 35.
- Step 1 To a solution of N,N'-dimethylethylenediamine (300 mg) in DMF (2.0 mL) was added K 2 C0 3 ( 1.0 g) and compound B (466 mg). The mixture was heated at 80°C for 3h. Solvent was evaporated and the residue was extracted with DCM and then purified by a prep-TLC plate
- Step 2 A solution of step 1 product (l eq) in DMF (3.0 mL) was treated with NaHC0 3 (0.5 g) and, respective bromides (2.5eq) at 50°C for 5h. Solvent was evaporated and the products were purified by a prep-TLC plate (8%MeOH/DCM) to give product as yellow solids.
- Step 3 Example 38 was synthesized according to a similar procedure to that used in Step 6 of Example 37.
- Example 39 was made in accordance with the methods shown in Scheme 36.
- Step 1 A mixture of N-(2-methoxyethyl)methylamine ( 1.8 g) and ethyl bromoacetate (3.4 g) in acetonitrile (20 mL) was treated with K 2 C0 3 (4.0 g) and Nal (20 mmol). The mixture was refluxed overnight. Solvent was evaporated and the residue was extracted with DCM and then purified on Silica gel column (0-8% MeOH/DCM) to give the product as colorless oil (3.2 g, yield 93%).
- Step 2 To a solution of step- ] product (3.5 g) in THF (20 mL) was added LAH (800 mg) portion wise. The resulting mixture was stirred at room temperature overnight and then quenched with EtOAc and water. After filtration, the organic solution was evaporated to give colorless oil (2.0 g, yield 75%).
- Step 3 The step 3 product compound was synthesized according the general procedure from compound B (400 mg) and step-4 product to give the title compound as a yellow solid (200 mg, yield 40%).
- Step 4 Example 39 was synthesized according to a similar procedure to that used in Step 6 of Example 37.
- Example 40 was made in accordance with the methods shown in Scheme 39.
- Step 1 To a solution of 3-methoxy-4-nitrobenzyl alcohol (5g) in DCM ( 100 mL) was added PDC (1 .5 eq) and molecular sieves (6.0 g). The mixture was stirred at room temperature for 2h and diluted with Et 2 0 (100 mL). The mixture was filtered through a Celite pad and solvent was evaporated. The residue was washed with a small amount MeOH to give off white solid (3.7 g, yield 74%).
- Step 2 To a solution of step-1 product (0.91 g) in DCM ( 10 mL) was added
- Step 3 A solution of step-2 product (0.52 g) in MeOH ( 10 mL) was charged with Pd-C (0.5 g, 10% wet) and hydrogenated under a hydrogen balloon at room temperature overnight. The catalyst was filtered off and solvent was evaporated to give yellow oil (0.45 g, yield 97%).
- Step 4 A vial was charged with H 2 S0 4 (2.0 mL) and cooled to 0°C. Step-3 product (0.4 g) was carefully introduced. Guanidine nitrate (leq) was added. The mixture was stirred at 0°C for 2h and at room temperature for 1 h. The mixture was treated with excess wet NaHC03 and extracted with DCM (10 mL). The product was and purified by prep-TLC plates (8%MeOH/DCM) to give orange solid (0.34 g, yield 71 %).
- Step 5 A solution of compound C (320 mg), step-4 product (268 mg) and TFA (0.3 mL) in 2-BuOH (2 mL) was heated at 100°C for 18 hrs. Upon cooling EtOAc and aq. NaHC0 3 were added to the reaction mixture. Extraction (3 ⁇ ) and concentration of combined extracts gave a solid which purified by prep-TLC plates (15%MeOH/DCM) to give orange solid (410 mg, yield 71 %).
- Step 6 To a suspension of step-5 product (400 mg) in MeOH (2.0 mL) was added K 2 C0 3 ( 1.0 g) and water (0.5 mL). The reaction vial was capped and heated at 60°C for 15min.
- the mixture was cooled down to room temperature and the top layer was transferred to a new vial and diluted with water.
- the pH was adjusted to 5-6 by adding aq HCl (2N) and the product was collected by filtration as a yellow solid (310 mg, yield 86%).
- Step7 To a mixture of step-6 product (260 mg) and Et 2 NH ( 1.1 mmol) in DMF (2.0 mL) was added HBTU ( 1.3 mmol) and Et 3 N (0.14 mL). The mixture was stirred at room temperature for 2h and diluted with DCM (5.0 mL). The mixture was washed with aq. K 2 C0 3 and evaporated. The residue was purified by prep-TLC plates (15%MeOH/DCM) to give an orange solid (250 mg, yield 87%).
- Step 8 To a solution of step-7 product (250 mg) in THF (1.0 mL) was added BH 3 Me 2 S (4.0 mL, 2.0M solution in THF).
- Step 9 Example 40 was synthesized according to a similar procedure to that used in Step 6 of Example 37.
- Example 41 was made in accordance with the methods shown in Scheme 40.
- Step 1 N,N-diethyl-2-(3-methoxy-4-nitrophenyl)acetamide was made in accordance with the methods disclosed herein.
- Step 2 To a solution of N,N-diethyl-2-(3-methoxy-4-nitrophenyl)acetamide ( 1.0 g) in THF (5.0 mL) was added BH 3 Me 2 S (20. mL, 2.0M solution in THF). The mixture was stirred at 60C for 2h and solvent was evaporated. The residue was dissolved in MeOH ( 10 ml) and treated with wet 2 C0 3 in a capped vial at 70°C for lh. The organic solution was evaporated and the residue was purified on Silica gel column (5% MeOH/DCM) to give the product as an orange oil (0.62 g, yield 65%)
- Step 3 A solution of step-2 product (600 mg) in MeOH ( 10 mL) was charged with Pd-C (0.5 g) and hydrogenated under a hydrogen balloon at room temperature for 3h. The catalyst was filtered off and the solvent was evaporated to give a yellow oil (430 mg, yield 81 %)
- Step 4 The product of step 4 was synthesized according the procedure of step 4 of Scheme 39 as an orange oil.
- Step 5 The product of step 4 was synthesized according the procedure of step 5 of Scheme 13 to afford an orange solid.
- Step 6 Example 41 was synthesized according to a similar procedure to that used in Step 6 of Example 37.
- the compounds depicted below can be synthesized using methods analogous to those described herein and can be useful for treating EGFR-driven cancers.
- Kinase inhibitory activity of the compounds was measured against human EGFR (native sequence) and against EGFR bearing the L858R mution and the L858R/T790M double mutation (EGFR, L858R, and L858R/T790M, respectively in Table 1 ). Additional assays can be conducted with EGFR deletion mutants such as delE746 - A750 with or without the additional T790M mutation. Assay conditions included 10 pt curves with 3 ⁇ top concentration (singlicates) and 10 ⁇ ATP.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description










































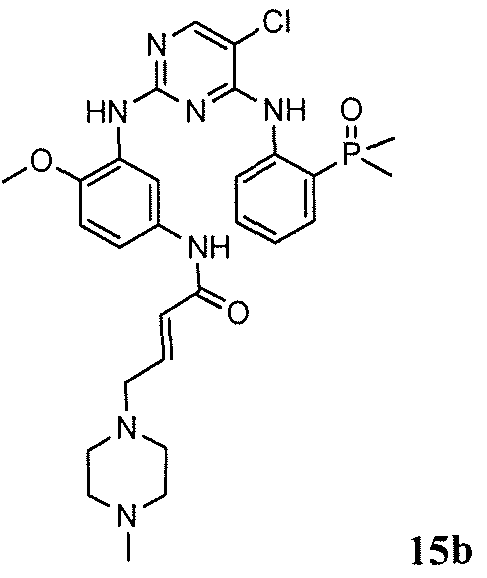























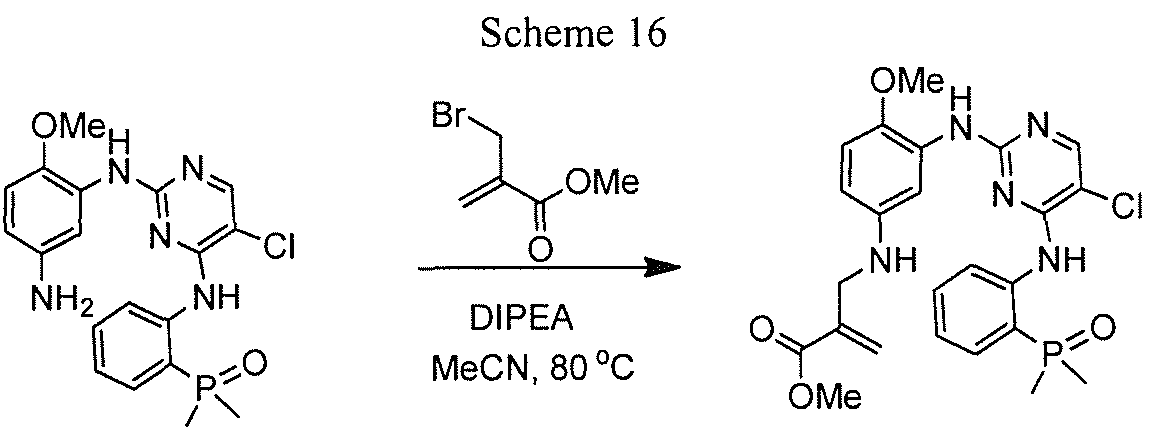
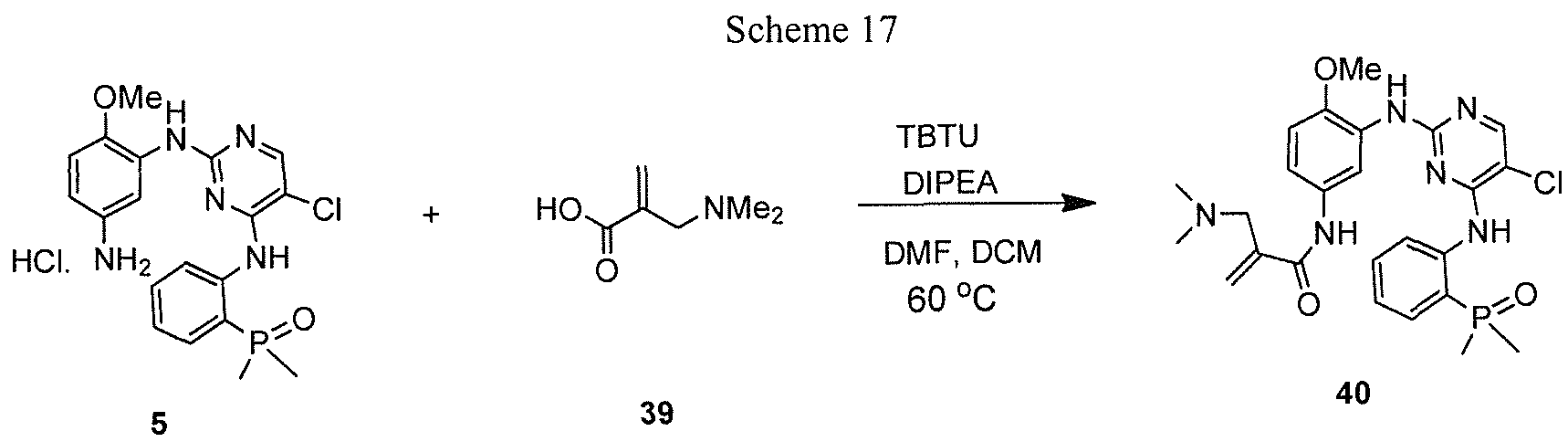



































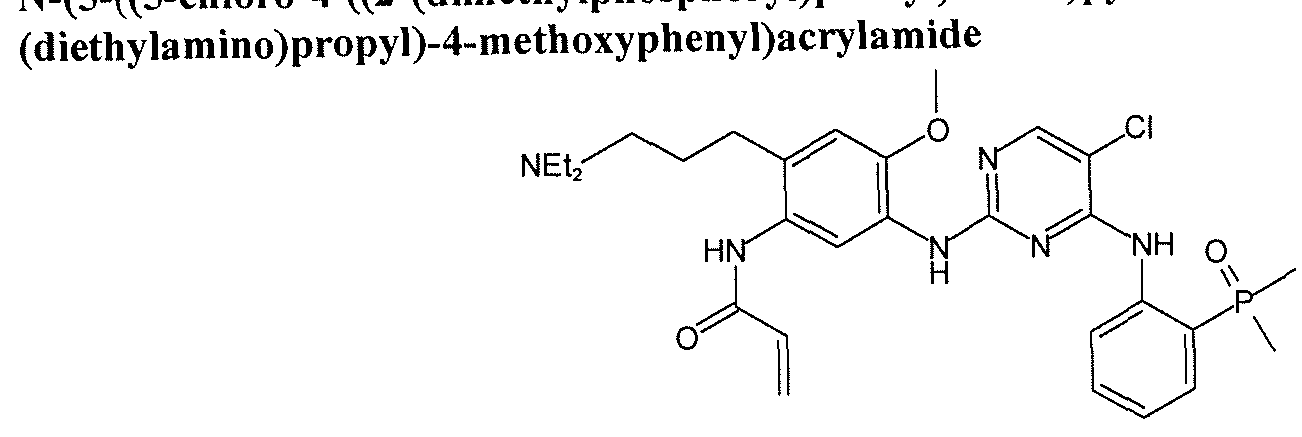
































Claims








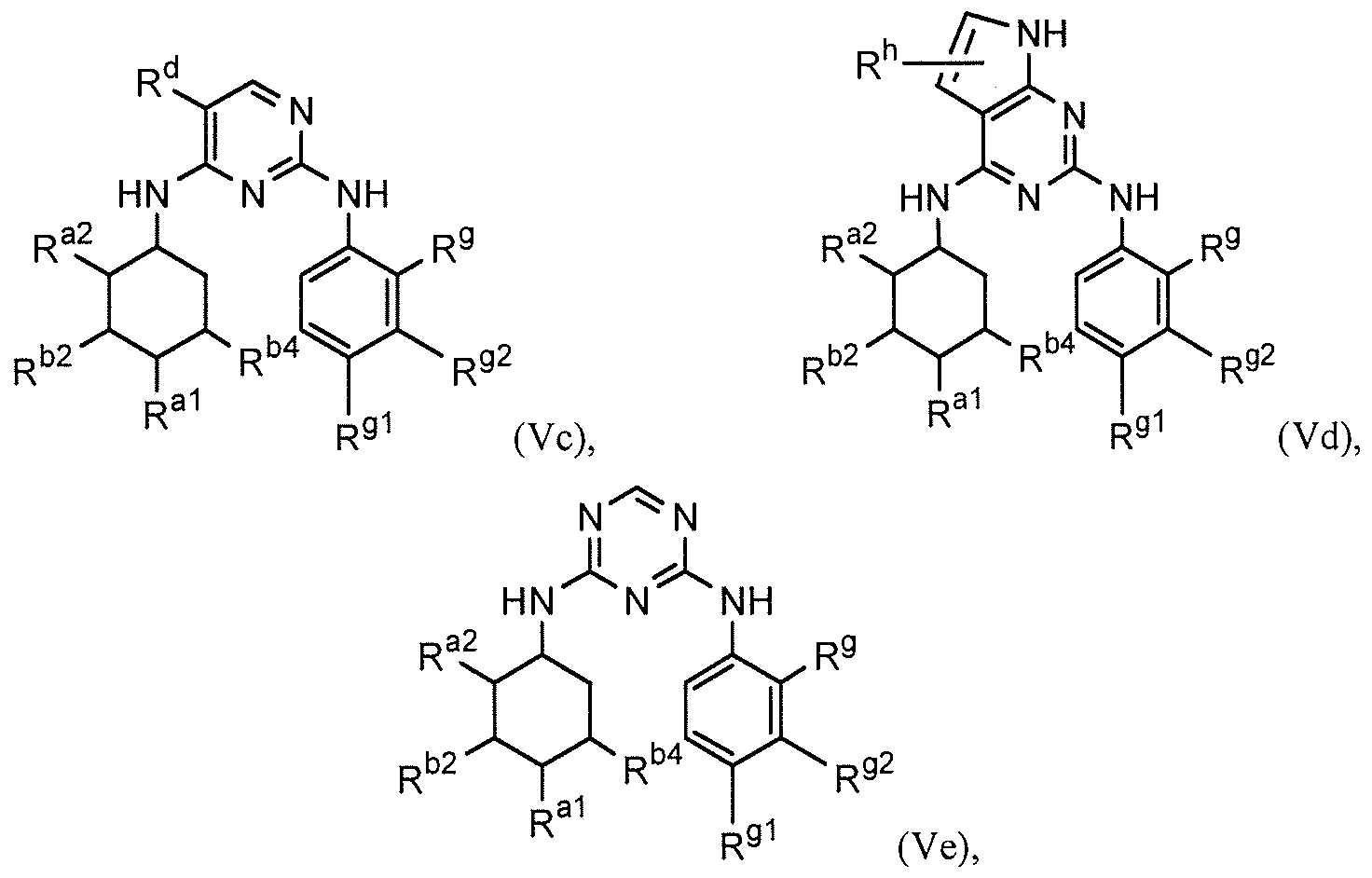









Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2832504A CA2832504C (en) | 2011-05-04 | 2012-05-04 | Compounds for inhibiting cell proliferation in egfr-driven cancers |
| KR1020137032214A KR101884010B1 (en) | 2011-05-04 | 2012-05-04 | Compounds for inhibiting cell proliferation in egfr-driven cancers |
| AU2012250517A AU2012250517B2 (en) | 2011-05-04 | 2012-05-04 | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| CN201280021702.7A CN103501612B (en) | 2011-05-04 | 2012-05-04 | The compound that cell is bred in cancer caused by suppression EGF-R ELISA |
| EP12779411.3A EP2704572B1 (en) | 2011-05-04 | 2012-05-04 | Compounds for inhibiting cell proliferation in egfr-driven cancers |
| MX2013012895A MX360404B (en) | 2011-05-04 | 2012-05-04 | Compounds for inhibiting cell proliferation in egfr-driven cancers. |
| JP2014509504A JP5999177B2 (en) | 2011-05-04 | 2012-05-04 | Compound for inhibiting cell proliferation of EGFR-activated cancer |
| BR112013027734A BR112013027734A2 (en) | 2011-05-04 | 2012-05-04 | compounds for inhibition of cell proliferation in egfr-driven cancers, method and pharmaceutical composition |
| EA201391626A EA201391626A1 (en) | 2011-05-04 | 2012-05-04 | COMPOUNDS FOR INHIBITING CELL PROLIFERATION IN EGFR-STIMULATED CANCER TYPES |
| IL228739A IL228739B (en) | 2011-05-04 | 2013-10-06 | Compounds for inhibiting cell proliferation in egfr driven cancers |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161482433P | 2011-05-04 | 2011-05-04 | |
| US61/482,433 | 2011-05-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012151561A1 true WO2012151561A1 (en) | 2012-11-08 |
Family
ID=47108074
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/036683 WO2012151561A1 (en) | 2011-05-04 | 2012-05-04 | Compounds for inhibiting cell proliferation in egfr-driven cancers |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9834518B2 (en) |
| EP (1) | EP2704572B1 (en) |
| JP (1) | JP5999177B2 (en) |
| KR (1) | KR101884010B1 (en) |
| CN (1) | CN103501612B (en) |
| AU (1) | AU2012250517B2 (en) |
| BR (1) | BR112013027734A2 (en) |
| CA (1) | CA2832504C (en) |
| EA (1) | EA201391626A1 (en) |
| IL (1) | IL228739B (en) |
| MX (1) | MX360404B (en) |
| WO (1) | WO2012151561A1 (en) |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013544273A (en) * | 2011-07-27 | 2013-12-12 | アストラゼネカ アクチボラグ | 2- (2,4,5-substituted-anilino) pyrimidine compounds |
| US9012462B2 (en) | 2008-05-21 | 2015-04-21 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
| US9145387B2 (en) | 2013-02-08 | 2015-09-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9273077B2 (en) | 2008-05-21 | 2016-03-01 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| AU2013204563B2 (en) * | 2012-05-05 | 2016-05-19 | Takeda Pharmaceutical Company Limited | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| JP2016528209A (en) * | 2013-07-11 | 2016-09-15 | エイシア バイオサイエンシーズ インコーポレイテッド | Pyrimidine derivatives as kinase inhibitors |
| JP2017506667A (en) * | 2014-02-25 | 2017-03-09 | シャンハイ ハイヤン ファーマシューティカル テクノロジー カンパニー リミテッドShanghai Haiyan Pharmaceutical Technology Co., Ltd. | 2,4-disubstituted benzene-1,5-diamine derivatives and uses thereof and pharmaceutical and medicinal compositions prepared therefrom |
| US9611283B1 (en) | 2013-04-10 | 2017-04-04 | Ariad Pharmaceuticals, Inc. | Methods for inhibiting cell proliferation in ALK-driven cancers |
| JP2017524703A (en) * | 2014-07-25 | 2017-08-31 | シャンハイ ハイヤン ファーマシューティカル テクノロジー カンパニー リミテッドShanghai Haiyan Pharmaceutical Technology Co., Ltd. | 2,4-disubstituted 7H-pyrrolo [2,3-d] pyrimidine derivatives, process for their preparation and use in medicine |
| US9834518B2 (en) | 2011-05-04 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| JP2017214390A (en) * | 2013-11-21 | 2017-12-07 | ファイザー・インク | 2,6-substituted purine derivatives and their use in treatment of proliferative disorders |
| US10005760B2 (en) | 2014-08-13 | 2018-06-26 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| US10449196B2 (en) | 2012-08-06 | 2019-10-22 | ACEA Therapeutics, Inc. | EGFR modulators and uses thereof |
| US10533011B2 (en) | 2015-10-09 | 2020-01-14 | ACEA Therapeutics, Inc. | Pharmaceutical salts, physical forms, and compositions of pyrrolopyrimidine kinase inhibitors, and methods of making same |
| US10596174B2 (en) | 2012-01-13 | 2020-03-24 | ACEA Therapeutics, Inc. | Pyrrolopyrimidine compounds as inhibitors of protein kinases |
| CN112055714A (en) * | 2018-04-20 | 2020-12-08 | 贵州伊诺其尼科技有限公司 | Dimethyloxyphosphonium compounds |
| EP3670500A4 (en) * | 2017-08-18 | 2020-12-16 | Beijing Hanmi Pharmaceutical Co., Ltd. | Chemical compound, pharmaceutical composition thereof, and use and application thereof |
| EP3675860A4 (en) * | 2017-08-28 | 2021-04-14 | Zhihong, Chen | Substituted pyrimidines, pharmaceutical compositions and therapeutic methods thereof |
| EP3656769A4 (en) * | 2017-07-19 | 2021-04-14 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Aryl-phosphorus-oxygen compound as egfr kinase inhibitor |
| WO2021073498A1 (en) * | 2019-10-17 | 2021-04-22 | 贝达药业股份有限公司 | Egfr inhibitor, composition, and method for preparation thereof |
| WO2021104441A1 (en) * | 2019-11-29 | 2021-06-03 | 江苏先声药业有限公司 | Polyaromatic compound as egfr kinase inhibitor |
| CN113677680A (en) * | 2019-04-04 | 2021-11-19 | 贝达药业股份有限公司 | EGFR inhibitor and composition and application thereof |
| EP3885344A3 (en) * | 2014-11-05 | 2021-12-08 | InventisBio Co., Ltd. | Pyrimidine compounds, preparation method therefor and pharmaceutical uses thereof |
| WO2022094354A1 (en) * | 2020-10-30 | 2022-05-05 | Lengo Therapeutics, Inc. | Pyrimidine compounds, compositions, and medicinal applications thereof |
| CN114728994A (en) * | 2020-07-03 | 2022-07-08 | 成都地奥九泓制药厂 | Aryl phosphorus oxygen compound and application thereof |
| WO2022199589A1 (en) * | 2021-03-23 | 2022-09-29 | 南京明德新药研发有限公司 | Pyrimidine derivatives |
| US11498922B2 (en) | 2017-04-07 | 2022-11-15 | ACEA Therapeutics, Inc. | Pharmaceutical composition comprising N-(3-((2-((3-fluoro-4-(4-methylpiperazin-1-yl phenyl)amino)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)phenylacrylamide |
| US11529350B2 (en) | 2019-07-03 | 2022-12-20 | Sumitomo Pharma Oncology, Inc. | Tyrosine kinase non-receptor 1 (TNK1) inhibitors and uses thereof |
| EP4129996A4 (en) * | 2020-03-23 | 2023-07-12 | Qilu Pharmaceutical Co., Ltd. | Novel aminopyrimidine egfr inhibitor |
| EP4137484A4 (en) * | 2020-04-14 | 2023-12-20 | Qilu Pharmaceutical Co., Ltd. | Tricyclic compounds as egfr inhibitors |
| US12084453B2 (en) | 2021-12-10 | 2024-09-10 | Incyte Corporation | Bicyclic amines as CDK12 inhibitors |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11351168B1 (en) | 2008-06-27 | 2022-06-07 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| EP3549934A1 (en) | 2008-06-27 | 2019-10-09 | Celgene CAR LLC | Heteroaryl compounds and uses thereof |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| TW201100441A (en) * | 2009-06-01 | 2011-01-01 | Osi Pharm Inc | Amino pyrimidine anticancer compounds |
| BR112013003388A2 (en) | 2010-08-10 | 2016-07-12 | Celgene Avilomics Res Inc | besylate salt of a btk inhibitor |
| WO2012061303A1 (en) | 2010-11-01 | 2012-05-10 | Avila Therapeutics, Inc. | Heteroaryl compounds and uses thereof |
| TWI632134B (en) | 2010-11-01 | 2018-08-11 | 阿維拉製藥公司 | Heterocyclic compounds and uses thereof |
| US8796255B2 (en) | 2010-11-10 | 2014-08-05 | Celgene Avilomics Research, Inc | Mutant-selective EGFR inhibitors and uses thereof |
| WO2012074951A1 (en) | 2010-11-29 | 2012-06-07 | OSI Pharmaceuticals, LLC | Macrocyclic kinase inhibitors |
| WO2013063401A1 (en) | 2011-10-28 | 2013-05-02 | Celgene Avilomics Research, Inc. | Methods of treating a bruton's tyrosine kinase disease or disorder |
| SI2825042T1 (en) | 2012-03-15 | 2019-01-31 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US9056839B2 (en) | 2012-03-15 | 2015-06-16 | Celgene Avilomics Research, Inc. | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| WO2014100748A1 (en) | 2012-12-21 | 2014-06-26 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US9492471B2 (en) | 2013-08-27 | 2016-11-15 | Celgene Avilomics Research, Inc. | Methods of treating a disease or disorder associated with Bruton'S Tyrosine Kinase |
| US9415049B2 (en) | 2013-12-20 | 2016-08-16 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| CN105315259B (en) * | 2014-07-29 | 2018-03-09 | 上海艾力斯医药科技有限公司 | Pyridine amine pyrimidine derivates, its preparation method and application |
| CN111170999B (en) * | 2014-10-11 | 2023-06-30 | 上海翰森生物医药科技有限公司 | EGFR inhibitor and preparation and application thereof |
| TWI618704B (en) * | 2014-12-11 | 2018-03-21 | 貝達醫藥公司 | Substituted 2-anilinopyrimidine derivatives as egfr modulators |
| WO2016187028A1 (en) * | 2015-05-15 | 2016-11-24 | Celgene Avilomics Research, Inc. | Heteroaryl compounds, synthesis thereof, and intermediates thereto |
| CN106187915A (en) * | 2015-05-27 | 2016-12-07 | 上海翰森生物医药科技有限公司 | There is inhibitor of ALK Yu EGFR double activity and its preparation method and application |
| CN106883213B (en) | 2015-12-15 | 2021-04-20 | 合肥中科普瑞昇生物医药科技有限公司 | Dual inhibitor of EGFR (epidermal growth factor receptor) and ALK (anaplastic lymphoma kinase) |
| CN108430972B (en) | 2015-12-24 | 2022-12-27 | 协和麒麟株式会社 | Alpha, beta-unsaturated amide compound |
| KR20190006991A (en) * | 2016-05-11 | 2019-01-21 | 베타 파마, 인크. | 2-anilinopyrimidine derivatives as therapeutics for the treatment of brain cancer |
| CN106083736A (en) * | 2016-06-21 | 2016-11-09 | 郑州泰基鸿诺医药股份有限公司 | A kind of pyrimidines, EGFR inhibitor and application thereof |
| WO2018108064A1 (en) * | 2016-12-13 | 2018-06-21 | 南京明德新药研发股份有限公司 | Spiro-aryl-phosphorus-oxygen compound as fourth generation of egfr kinase inhibitor |
| CN107522742B (en) * | 2017-10-20 | 2019-10-18 | 常州工程职业技术学院 | A kind of homogeneous " one kettle way " preparation method of Brigatinib key intermediate |
| US11254696B2 (en) | 2017-12-21 | 2022-02-22 | Shenzhen Targetrx, Inc. | Dianilinopyrimidine compound for inhibiting kinase activity |
| KR20190108080A (en) * | 2018-03-13 | 2019-09-23 | 보로노이바이오 주식회사 | 2,4,5-substituted pyrimidine derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating cancer or inflammatory disease in containing the same as an active ingredient |
| CN111718325A (en) * | 2019-03-22 | 2020-09-29 | 烟台药物研究所 | 2,4, 5-substituted pyrimidine compound and preparation method and application thereof |
| CN112538072B (en) * | 2019-09-21 | 2024-02-06 | 齐鲁制药有限公司 | Aminopyrimidine EGFR inhibitors |
| BR112022016045A2 (en) * | 2020-02-14 | 2022-10-04 | Betta Pharmaceuticals Co Ltd | PHOSPHINE QUINOLINE OXIDE COMPOUND, AND COMPOSITION AND APPLICATION THEREOF. |
| TW202214590A (en) * | 2020-06-08 | 2022-04-16 | 大陸商南京紅云生物科技有限公司 | Alkenyl pyrimidine compound, preparation method therefor, and application thereof |
| KR102685187B1 (en) * | 2021-10-15 | 2024-07-16 | 제이투에이치바이오텍 (주) | Compounds inhibiting ALK and/or EGFR mutation kinases and medical use thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009143389A1 (en) * | 2008-05-21 | 2009-11-26 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
Family Cites Families (278)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1129563A (en) | 1966-03-31 | 1968-10-09 | Ici Ltd | Phosphorus-containing pyrimidine derivatives and biologically active compositions containing them |
| ES2052556T3 (en) | 1986-04-17 | 1994-07-16 | Zeneca Ltd | FUNGICIDES. |
| US4966622A (en) | 1988-04-12 | 1990-10-30 | Ciba-Geigy Corporation | N-phenyl-N-pyrimidin-2-ylureas |
| GB8828222D0 (en) | 1988-12-02 | 1989-01-05 | Ici Plc | Reactive dyes |
| GB8903019D0 (en) | 1989-02-10 | 1989-03-30 | Ici Plc | Fungicides |
| GB9016584D0 (en) | 1990-07-27 | 1990-09-12 | Ici Plc | Fungicides |
| DE69129611T2 (en) | 1990-08-20 | 1998-12-17 | Eisai Co., Ltd., Tokio/Tokyo | Sulfonamide derivatives |
| GB9220964D0 (en) | 1991-11-15 | 1992-11-18 | Ici Plc | Anionic compounds |
| TW225528B (en) | 1992-04-03 | 1994-06-21 | Ciba Geigy Ag | |
| US5521184A (en) | 1992-04-03 | 1996-05-28 | Ciba-Geigy Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| AU693475B2 (en) | 1993-10-01 | 1998-07-02 | Novartis Ag | Pyrimidineamine derivatives and processes for the preparation thereof |
| CZ101496A3 (en) | 1993-10-12 | 1996-11-13 | Du Pont Merck Pharma | N-alkyl-n-aryl-pyrimidinamines and derivatives thereof |
| DE69630214T2 (en) | 1995-03-10 | 2004-07-15 | Berlex Laboratories, Inc., Richmond | BENZAMIDINE DERIVATIVES, THEIR PRODUCTION AND THEIR USE AS ANTI-COAGULANTS |
| US5728201A (en) | 1995-09-14 | 1998-03-17 | Canon Kabushiki Kaisha | Ink, and ink-jet recording method and instruments using the same |
| GB9519197D0 (en) | 1995-09-20 | 1995-11-22 | Affinity Chromatography Ltd | Novel affinity ligands and their use |
| GB9523675D0 (en) | 1995-11-20 | 1996-01-24 | Celltech Therapeutics Ltd | Chemical compounds |
| AU7692896A (en) | 1995-12-01 | 1997-06-27 | Novartis Ag | Quinazolin-2,4-diazirines as NPY receptor antagonist |
| GB9608771D0 (en) | 1996-04-27 | 1996-07-03 | Agrevo Uk Ltd | Pyrimethanil salts |
| JP3106966B2 (en) | 1996-07-17 | 2000-11-06 | 富士ゼロックス株式会社 | Ink jet recording ink and ink jet recording method |
| JPH1060338A (en) | 1996-08-21 | 1998-03-03 | Fuji Xerox Co Ltd | Ink for ink-jet recording and method of ink jet recording |
| US6008234A (en) | 1996-09-12 | 1999-12-28 | Berlex Laboratories, Inc. | Benzamidine derivatives substituted by cyclic amino acid and cyclic hydroxy acid derivatives and their use as anti-coagulants |
| NZ334264A (en) | 1996-09-12 | 2000-08-25 | Schering Ag | Benzamidine derivatives substituted by cyclic amino acid or cyclic hydroxy acid derivatives and their use as anti-coagulants |
| US6004985A (en) | 1996-10-09 | 1999-12-21 | Berlex Laboratories, Inc. | Thio acid derived monocylic N-heterocyclics as anticoagulants |
| CO4940418A1 (en) | 1997-07-18 | 2000-07-24 | Novartis Ag | MODIFICATION OF A CRYSTAL OF A DERIVATIVE OF N-PHENYL-2-PIRIMIDINAMINE, PROCESSES FOR ITS MANUFACTURE AND USE |
| US6573044B1 (en) | 1997-08-07 | 2003-06-03 | The Regents Of The University Of California | Methods of using chemical libraries to search for new kinase inhibitors |
| JPH1160573A (en) | 1997-08-22 | 1999-03-02 | Nippon Kayaku Co Ltd | Triazine derivative and telomerase inhibitor |
| AU1507199A (en) | 1997-12-15 | 1999-07-05 | Yamanouchi Pharmaceutical Co., Ltd. | Novel pyrimidine-5-carboxamide derivatives |
| EP1085807B1 (en) | 1998-06-10 | 2003-05-07 | Basf Aktiengesellschaft | Use of 2-(n-phenylamino)pyrimidines as fungicides, and novel 2-(n-phenylamino)pyrimidines |
| AU5648599A (en) | 1998-09-11 | 2000-04-03 | Kyorin Pharmaceutical Co. Ltd. | Phosphonic ester derivatives and process for producing the same |
| SK287269B6 (en) | 1998-11-10 | 2010-05-07 | Janssen Pharmaceutica N. V. | Pyrimidine derivative, its use, process for its preparation and pharmaceutical composition, combination and product containing thereof |
| BR9915401B1 (en) | 1998-11-17 | 2011-04-19 | pyrimidinylbenzimidazole or triazinylbenzimidazole derivatives, anilinotriazine and anilinopyrimidine, as well as agricultural / horticultural fungicide. | |
| US6262088B1 (en) | 1998-11-19 | 2001-07-17 | Berlex Laboratories, Inc. | Polyhydroxylated monocyclic N-heterocyclic derivatives as anti-coagulants |
| US6127376A (en) | 1998-12-04 | 2000-10-03 | Berlex Laboratories, Inc. | Aryl and heterocyclyl substituted pyrimidine derivatives as anti-coagulants |
| AU2871900A (en) | 1999-02-04 | 2000-08-25 | Millennium Pharmaceuticals, Inc. | G-protein coupled heptahelical receptor binding compounds and methods of use thereof |
| GB9910807D0 (en) | 1999-05-10 | 1999-07-07 | Prometic Biosciences Limited | Novel detoxification agents and their use |
| GB9914258D0 (en) | 1999-06-18 | 1999-08-18 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9918035D0 (en) | 1999-07-30 | 1999-09-29 | Novartis Ag | Organic compounds |
| ITMI992711A1 (en) | 1999-12-27 | 2001-06-27 | Novartis Ag | ORGANIC COMPOUNDS |
| KR20030024659A (en) * | 2000-02-17 | 2003-03-26 | 암겐 인코포레이티드 | Kinase Inhibitors |
| GB0004888D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| GB0004887D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| US7087608B2 (en) | 2000-03-03 | 2006-08-08 | Robert Charles Atkins | Use of PDGF receptor tyrosine kinase inhibitors for the treatment of diabetic nephropathy |
| US6525051B2 (en) | 2000-03-27 | 2003-02-25 | Schering Aktiengesellschaft | N-heterocyclic derivatives as NOS inhibitors |
| DK1282607T3 (en) | 2000-05-08 | 2016-02-01 | Janssen Pharmaceutica Nv | Prodrugs of HIV replication inhibiting pyrimidines |
| ES2542326T3 (en) | 2000-05-08 | 2015-08-04 | Janssen Pharmaceutica Nv | HIV replication inhibitors |
| AU2001294511A1 (en) | 2000-06-30 | 2002-01-08 | The Regents Of The University Of California | New strategy for leukemia therapy |
| GB0016877D0 (en) | 2000-07-11 | 2000-08-30 | Astrazeneca Ab | Chemical compounds |
| GB0022438D0 (en) | 2000-09-13 | 2000-11-01 | Novartis Ag | Organic Compounds |
| EP1341853B1 (en) | 2000-12-05 | 2005-08-24 | Clariant Finance (BVI) Limited | Trichromatic dyeing process |
| EP1343782B1 (en) | 2000-12-21 | 2009-05-06 | SmithKline Beecham Corporation | Pyrimidineamines as angiogenesis modulators |
| US6881737B2 (en) | 2001-04-11 | 2005-04-19 | Amgen Inc. | Substituted triazinyl acrylamide derivatives and methods of use |
| US6825190B2 (en) | 2001-06-15 | 2004-11-30 | Vertex Pharmaceuticals Incorporated | Protein kinase inhibitors and uses thereof |
| GB0115109D0 (en) | 2001-06-21 | 2001-08-15 | Aventis Pharma Ltd | Chemical compounds |
| US6960572B2 (en) | 2001-06-21 | 2005-11-01 | Ariad Pharmaceuticals, Inc. | Indolinones and uses thereof |
| EP1408978A4 (en) | 2001-06-21 | 2005-07-13 | Ariad Pharma Inc | Novel phenylamino-pyrimidines and uses thereof |
| WO2003023010A2 (en) | 2001-09-07 | 2003-03-20 | Irm Llc | Sensory neuron receptors |
| WO2003030909A1 (en) | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| US20060009642A1 (en) | 2001-10-12 | 2006-01-12 | Irm Llc, A Delaware Limited Liability Company | Methods for the synthesis of substituted purines |
| EP1578722A4 (en) | 2001-10-12 | 2006-09-06 | Irm Llc | Kinase inhibitor scaffolds and methods for their preparation |
| US6949644B2 (en) | 2001-10-12 | 2005-09-27 | Irm Llc | Methods for the synthesis of substituted purines |
| US6770652B2 (en) | 2001-10-18 | 2004-08-03 | Duquesne University Of The Holy Ghost | Multiple acting anti-angiogenic and cytotoxic compounds and methods for using the same |
| NZ531853A (en) | 2001-11-01 | 2006-02-24 | Janssen Pharmaceutica Nv | Heteroaryl amines as glycogen synthase kinase 3beta inhibitors (gsk3 inhibitors) |
| WO2003057165A2 (en) | 2002-01-04 | 2003-07-17 | The Rockefeller University | COMPOSITIONS AND METHODS FOR PREVENTION AND TREATMENT OF AMYLOID-β PEPTIDE-RELATED DISORDERS |
| EP1479693A4 (en) | 2002-02-05 | 2005-07-27 | Japan Science & Tech Agency | Egf/egfr complex |
| JP2005522438A (en) | 2002-02-08 | 2005-07-28 | スミスクライン ビーチャム コーポレーション | Pyrimidine compounds |
| GB0206215D0 (en) | 2002-03-15 | 2002-05-01 | Novartis Ag | Organic compounds |
| ATE367394T1 (en) | 2002-04-26 | 2007-08-15 | Gilead Sciences Inc | ENHANCEMENT IN THE CELL OF PHOSPHONATE ANALOGUES OF HIV PROTEASE INHIBITOR COMPOUNDS AND THE COMPOUNDS THEMSELVES |
| US20050239054A1 (en) | 2002-04-26 | 2005-10-27 | Arimilli Murty N | Method and compositions for identifying anti-HIV therapeutic compounds |
| US7704995B2 (en) | 2002-05-03 | 2010-04-27 | Exelixis, Inc. | Protein kinase modulators and methods of use |
| AU2003231231A1 (en) | 2002-05-06 | 2003-11-11 | Bayer Pharmaceuticals Corporation | Pyridinyl amino pyrimidine derivatives useful for treating hyper-proliferative disorders |
| AR039540A1 (en) | 2002-05-13 | 2005-02-23 | Tibotec Pharm Ltd | MICROBICIDE COMPOUNDS WITH PIRIMIDINE OR TRIAZINE CONTENT |
| US7019017B2 (en) | 2002-05-14 | 2006-03-28 | Baylor College Of Medicine | Small molecule inhibitors of HER2 expression |
| AU2003240714B2 (en) | 2002-05-27 | 2006-09-28 | Irm Llc | Bis-aromatic alkanols |
| GB0217431D0 (en) | 2002-07-27 | 2002-09-04 | Astrazeneca Ab | Novel compounds |
| AU2003265336B8 (en) | 2002-07-29 | 2009-04-23 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2004018435A1 (en) | 2002-08-24 | 2004-03-04 | Astrazeneca Ab | Pyrimidine derivatives as modulators of chemokine receptor activity |
| ATE454378T1 (en) | 2002-11-01 | 2010-01-15 | Vertex Pharma | COMPOUNDS ACTIVE AS INHIBITORS OF JAK AND OTHER PROTEIN KINASES |
| BRPI0407401A (en) | 2003-02-11 | 2006-02-21 | Irm Llc | bicyclic compounds and compositions |
| ATE433447T1 (en) | 2003-02-20 | 2009-06-15 | Smithkline Beecham Corp | PYRIMIIDINE COMPOUNDS |
| CL2004000306A1 (en) | 2003-02-20 | 2005-04-08 | Tibotec Pharm Ltd | COMPOUNDS DERIVED FROM PYRIMIDINE REPLACED WITH INDANO; PROCESS FOR PREPARATION; PHARMACEUTICAL COMPOSITION THAT UNDERSTANDS IT; PHARMACEUTICAL COMBINATION; AND ITS USE FOR THE TREATMENT OR PROFILAXIS OF AN INFECTIOUS DISEASE. |
| US7151096B2 (en) | 2003-03-05 | 2006-12-19 | Irm Llc | Cyclic compounds and compositions as protein kinase inhibitors |
| GB0305929D0 (en) | 2003-03-14 | 2003-04-23 | Novartis Ag | Organic compounds |
| TW200505902A (en) | 2003-03-20 | 2005-02-16 | Schering Corp | Cannabinoid receptor ligands |
| GB2400101A (en) | 2003-03-28 | 2004-10-06 | Biofocus Discovery Ltd | Compounds capable of binding to the active site of protein kinases |
| US20050014753A1 (en) | 2003-04-04 | 2005-01-20 | Irm Llc | Novel compounds and compositions as protein kinase inhibitors |
| WO2004093812A2 (en) | 2003-04-22 | 2004-11-04 | Irm Llc | Compounds that induce neuronal differentiation in embryonic stem cells |
| US7423031B2 (en) | 2003-05-01 | 2008-09-09 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| GB0311274D0 (en) | 2003-05-16 | 2003-06-18 | Astrazeneca Ab | Chemical compounds |
| US7317027B2 (en) | 2003-05-19 | 2008-01-08 | Sanofi-Aventis Deutschland Gmbh | Azaindole-derivatives as factor Xa inhibitors |
| MY150088A (en) | 2003-05-19 | 2013-11-29 | Irm Llc | Immunosuppressant compounds and compositions |
| WO2005011597A2 (en) | 2003-07-29 | 2005-02-10 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| PL1656372T3 (en) | 2003-07-30 | 2013-08-30 | Rigel Pharmaceuticals Inc | 2,4-pyrimidinediamine compounds for use in the treatment or prevention of autoimmune diseases |
| US20050113398A1 (en) | 2003-08-07 | 2005-05-26 | Ankush Argade | 2,4-pyrimidinediamine compounds and uses as anti-proliferative agents |
| EP1656378A4 (en) | 2003-08-15 | 2011-05-11 | Irm Llc | Compounds and compositions as inhibitors of receptor tyrosine kinase activity |
| BRPI0413616B8 (en) | 2003-08-15 | 2021-05-25 | Irm Llc | 2,4-pyrimidinediamines, their uses, and pharmaceutical composition |
| US7338957B2 (en) | 2003-08-28 | 2008-03-04 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| GB0321710D0 (en) | 2003-09-16 | 2003-10-15 | Novartis Ag | Organic compounds |
| JP2007505858A (en) | 2003-09-18 | 2007-03-15 | ノバルティス アクチエンゲゼルシャフト | 2,4-Di (phenylamino) pyrimidine useful for the treatment of proliferative disorders |
| US7189729B2 (en) | 2003-09-30 | 2007-03-13 | Irm Llc | Methods and compositions as protein kinase inhibitors |
| CN100455579C (en) | 2003-10-02 | 2009-01-28 | Irm责任有限公司 | Compounds and compositions as protein kinase inhibitors |
| JP4758349B2 (en) | 2003-10-08 | 2011-08-24 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as protein kinase inhibitors |
| DE10349587A1 (en) | 2003-10-24 | 2005-05-25 | Merck Patent Gmbh | Benzimidazolylderivate |
| JP2007508844A (en) | 2003-10-24 | 2007-04-12 | ギリアード サイエンシーズ, インコーポレイテッド | Methods and compositions for the identification of therapeutic compounds |
| MY141255A (en) | 2003-12-11 | 2010-03-31 | Memory Pharm Corp | Phosphodiesterase 4 inhibitors, including n-substituted diarylamine analogs |
| EP1694652A1 (en) | 2003-12-19 | 2006-08-30 | Rigel Pharmaceuticals, Inc. | Stereoisomers and stereoisomeric mixtures of 1-(2,4-pyrimidinediamino)-2-cyclopentanecarboxamide synthetic intermediates |
| EP1711467A2 (en) | 2004-01-16 | 2006-10-18 | Wyeth | Quinoline intermediates of receptor tyrosine kinase inhibitors and the synthesis thereof |
| EP1708571A4 (en) | 2004-01-16 | 2009-07-08 | Merck & Co Inc | Novel crystalline salts of a dipeptidyl peptidase-iv inhibitor |
| AU2005214352B2 (en) | 2004-02-14 | 2009-11-12 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| JP4812763B2 (en) | 2004-05-18 | 2011-11-09 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Cycloalkyl-substituted pyrimidinediamine compounds and uses thereof |
| EP1598343A1 (en) | 2004-05-19 | 2005-11-23 | Boehringer Ingelheim International GmbH | 2-Arylaminopyrimidine derivatives as PLK inhibitors |
| MXPA06014247A (en) | 2004-06-10 | 2007-03-12 | Irm Llc | Compounds and compositions as protein kinase inhibitors. |
| BRPI0512422A (en) | 2004-06-23 | 2008-03-04 | Irm Llc | compounds and compositions as protein kinase inhibitors |
| GB0512324D0 (en) | 2005-06-16 | 2005-07-27 | Novartis Ag | Organic compounds |
| US7521457B2 (en) | 2004-08-20 | 2009-04-21 | Boehringer Ingelheim International Gmbh | Pyrimidines as PLK inhibitors |
| GB0419161D0 (en) | 2004-08-27 | 2004-09-29 | Novartis Ag | Organic compounds |
| WO2006038594A1 (en) | 2004-10-04 | 2006-04-13 | Ono Pharmaceutical Co., Ltd. | N-type calcium channel inhibitor |
| CN101039919A (en) | 2004-10-13 | 2007-09-19 | 惠氏公司 | N-benzenesulfonyl substituted anilino-pyrimidine analogs |
| GT200500321A (en) | 2004-11-09 | 2006-09-04 | COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF PROTEIN KINASE. | |
| GB2420559B (en) | 2004-11-15 | 2008-08-06 | Rigel Pharmaceuticals Inc | Stereoisomerically enriched 3-aminocarbonyl bicycloheptene pyrimidinediamine compounds and their uses |
| JP5132319B2 (en) | 2004-12-21 | 2013-01-30 | スミスクライン ビーチャム コーポレーション | 2-pyrimidinylpyrazolopyridine ErbB kinase inhibitor |
| US8211929B2 (en) | 2004-12-30 | 2012-07-03 | Exelixis, Inc. | Pyrimidine derivatives as kinase modulators and method of use |
| MX2007007189A (en) | 2005-01-19 | 2010-09-28 | Rigel Pharmaceuticals Inc | Prodrugs of 2,4-pyrimidinediamine compounds and their uses. |
| RU2368602C2 (en) | 2005-01-26 | 2009-09-27 | Айрм Ллк | Compounds and compositions as protein kinase inhibitors |
| TW200639163A (en) | 2005-02-04 | 2006-11-16 | Genentech Inc | RAF inhibitor compounds and methods |
| BRPI0608513A2 (en) | 2005-03-15 | 2010-01-05 | Irm Llc | compounds and compositions as protein kinase inhibitors |
| JP2008533166A (en) | 2005-03-16 | 2008-08-21 | ターゲジェン インコーポレーティッド | Pyrimidine compounds and methods of use |
| DE102005016634A1 (en) | 2005-04-12 | 2006-10-19 | Merck Patent Gmbh | Novel aza heterocycles as kinase inhibitors |
| WO2006124874A2 (en) | 2005-05-12 | 2006-11-23 | Kalypsys, Inc. | Inhibitors of b-raf kinase |
| WO2006124462A2 (en) | 2005-05-13 | 2006-11-23 | Irm, Llc | Compounds and compositions as protein kinase inhibitors |
| ES2345629T3 (en) | 2005-05-16 | 2010-09-28 | Irm Llc | PIRROLOPIRIDINE DERIVATIVES AS PROTEIN KINASE INHIBITORS. |
| WO2006123165A2 (en) | 2005-05-19 | 2006-11-23 | Astex Therapeutics Limited | Pyrimidine derivatives as hsp90 inhibitors |
| WO2006128129A2 (en) | 2005-05-26 | 2006-11-30 | Synta Pharmaceuticals Corp. | Method for treating cancer |
| US20070032493A1 (en) | 2005-05-26 | 2007-02-08 | Synta Pharmaceuticals Corp. | Method for treating B cell regulated autoimmune disorders |
| WO2006129100A1 (en) | 2005-06-03 | 2006-12-07 | Glaxo Group Limited | Novel compounds |
| CN105348203B (en) | 2005-06-08 | 2018-09-18 | 里格尔药品股份有限公司 | Inhibit the composition and method of JAK approach |
| US20070203161A1 (en) | 2006-02-24 | 2007-08-30 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| US20070032514A1 (en) | 2005-07-01 | 2007-02-08 | Zahn Stephan K | 2,4-diamino-pyrimidines as aurora inhibitors |
| BRPI0613501A2 (en) | 2005-07-01 | 2011-01-11 | Irm Llc | compound, pharmaceutical composition and use thereof |
| MX2008000574A (en) | 2005-07-11 | 2008-03-14 | Sanofi Aventis | Novel 2,4-dianilinopyrimidine derivatives, the preparation thereof, their use as medicaments, pharmaceutical compositions and, in particular, as ikk inhibitors. |
| EP1937667B1 (en) | 2005-07-26 | 2010-10-13 | Vertex Pharmaceuticals Incorporated | Benzimidazoles useful as inhibitors of protein kinases |
| CA2615890A1 (en) | 2005-08-02 | 2007-02-08 | Irm Llc | 5-substituted thiazol-2-yl amino compounds and compositions as protein kinase inhibitors |
| AU2006279992A1 (en) | 2005-08-09 | 2007-02-22 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| US8071609B2 (en) | 2005-08-11 | 2011-12-06 | Ariad Pharmaceuticals, Inc. | Unsaturated heterocyclic derivatives |
| JP5016601B2 (en) | 2005-08-16 | 2012-09-05 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as protein kinase inhibitors |
| US7973060B2 (en) | 2005-10-13 | 2011-07-05 | Crystalgenomics, Inc. | Fab I inhibitor and process for preparing same |
| WO2007042299A1 (en) | 2005-10-13 | 2007-04-19 | Glaxo Group Limited | Pyrrolopyrimidine derivatives as syk inhibitors |
| US8372851B2 (en) | 2005-10-21 | 2013-02-12 | Exelixis, Inc. | Pyrazolo pyrimidines as casein kinase II (CK2) modulators |
| DE602006009539D1 (en) | 2005-10-28 | 2009-11-12 | Irm Llc | COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS |
| US8133900B2 (en) | 2005-11-01 | 2012-03-13 | Targegen, Inc. | Use of bi-aryl meta-pyrimidine inhibitors of kinases |
| US8604042B2 (en) | 2005-11-01 | 2013-12-10 | Targegen, Inc. | Bi-aryl meta-pyrimidine inhibitors of kinases |
| NZ567851A (en) | 2005-11-01 | 2011-09-30 | Targegen Inc | Bi-aryl meta-pyrimidine inhibitors of kinases |
| WO2007056151A2 (en) | 2005-11-03 | 2007-05-18 | Irm Llc | Protein kinase inhbitors |
| ES2412879T3 (en) | 2005-11-11 | 2013-07-12 | Boehringer Ingelheim International Gmbh | Combined cancer treatment comprising EGFR / HER2 inhibitors |
| ES2421450T3 (en) | 2005-12-05 | 2013-09-02 | Glaxosmithkline Llc | ErbB kinase inhibitor pyrimidinyl-pyrazolopyridines |
| US20080318989A1 (en) | 2005-12-19 | 2008-12-25 | Burdick Daniel J | Pyrimidine Kinase Inhibitors |
| DE102005062742A1 (en) | 2005-12-22 | 2007-06-28 | Bayer Schering Pharma Ag | New sulfoximine-substituted pyrimidines useful for treating e.g. diseases caused by inflammatory, allergic or proliferative processes, oncological diseases, cancer, eye, autoimmune and neurodegerative diseases |
| JO2660B1 (en) | 2006-01-20 | 2012-06-17 | نوفارتيس ايه جي | PI-3 Kinase inhibitors and methods of their use |
| TW200736232A (en) | 2006-01-26 | 2007-10-01 | Astrazeneca Ab | Pyrimidine derivatives |
| BRPI0706747A2 (en) | 2006-01-30 | 2011-04-05 | Exelixis Inc | 4-aryl-2-amino-pyrimidines or 4-aryl-2-aminoalkyl-pyrimidines as jak-2 modulators and pharmaceutical compositions containing them |
| US20090069327A1 (en) | 2006-02-06 | 2009-03-12 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| TW200804364A (en) | 2006-02-22 | 2008-01-16 | Boehringer Ingelheim Int | New compounds |
| US20070208164A1 (en) | 2006-02-27 | 2007-09-06 | Wyeth | Methods of synthesizing radiolabeled 3-cyano[14C]quinolines |
| JP5070278B2 (en) | 2006-03-30 | 2012-11-07 | テイボテク・フアーマシユーチカルズ | 5- (Hydroxymethylene and aminomethylene) substituted pyrimidines that inhibit HIV |
| AU2007233737B2 (en) | 2006-03-30 | 2012-11-29 | Janssen Sciences Ireland Uc | HIV inhibiting 5-amido substituted pyrimidines |
| GB0608386D0 (en) | 2006-04-27 | 2006-06-07 | Senexis Ltd | Compounds |
| US7601716B2 (en) | 2006-05-01 | 2009-10-13 | Cephalon, Inc. | Pyridopyrazines and derivatives thereof as ALK and c-Met inhibitors |
| BRPI0711628A2 (en) | 2006-05-15 | 2011-12-06 | Irm Llc | compound, pharmaceutical composition, use and process for preparing the compound |
| DE102006027156A1 (en) | 2006-06-08 | 2007-12-13 | Bayer Schering Pharma Ag | New sulfimide compounds are protein kinase inhibitors useful to treat e.g. cancer, Hodgkin's lymphoma, Kaposi's sarcoma, cardiovascular disease, Crohn's disease, endometriosis and hemangioma |
| AU2007269540B2 (en) | 2006-07-05 | 2013-06-27 | Exelixis, Inc. | Methods of using IGF1R and Abl kinase modulators |
| US20100010025A1 (en) | 2006-07-21 | 2010-01-14 | Norvartis Ag | Pyrimidine Derivatives |
| GB0614579D0 (en) | 2006-07-21 | 2006-08-30 | Black James Foundation | Pyrimidine derivatives |
| DE102006041382A1 (en) | 2006-08-29 | 2008-03-20 | Bayer Schering Pharma Ag | Carbamoyl sulfoximides as protein kinase inhibitors |
| WO2008039359A2 (en) | 2006-09-25 | 2008-04-03 | Janssen Pharmaceutica N.V. | Bicyclic pyrimidine kinase inhibitors |
| WO2008045978A1 (en) | 2006-10-10 | 2008-04-17 | Rigel Pharmaceuticals, Inc. | Pinane-substituted pyrimidinediamine derivatives useful as axl inhibitors |
| CA2598893C (en) | 2006-10-11 | 2012-04-10 | Astellas Pharma Inc. | Eml4-alk fusion gene |
| EP2090577B1 (en) | 2006-10-19 | 2017-04-05 | Signal Pharmaceuticals, LLC | Heteroaryl compounds, compositions thereof, and their use as protein kinase inhibitors |
| DE602007012363D1 (en) | 2006-10-19 | 2011-03-17 | Rigel Pharmaceuticals Inc | 2,4-pyridimediamine derivatives as inhibitors of JAK KINASES for the treatment of autoimmune diseases |
| CA2669111C (en) | 2006-10-23 | 2016-04-12 | Cephalon, Inc. | Fused bicyclic derivatives of 2,4-diaminopyrimidine as alk and c-met inhibitors |
| WO2008057280A1 (en) | 2006-10-27 | 2008-05-15 | Amgen Inc. | Multi-cyclic compounds and methods of use |
| JP5208123B2 (en) | 2006-12-08 | 2013-06-12 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as protein kinase inhibitors |
| GEP20156282B (en) | 2006-12-08 | 2015-05-11 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| US20100144732A1 (en) | 2006-12-19 | 2010-06-10 | Krueger Elaine B | Pyrimidine kinase inhibitors |
| EP1939185A1 (en) | 2006-12-20 | 2008-07-02 | Bayer Schering Pharma Aktiengesellschaft | New types of hetaryl-phenylendiamin-pyrimidines as protein kinase inhibitors for the treatment of cancer |
| KR20090103897A (en) | 2006-12-28 | 2009-10-01 | 다이쇼 세이야꾸 가부시끼가이샤 | Pyrazolopyrimidine compound |
| KR20090094073A (en) | 2006-12-29 | 2009-09-03 | 티보텍 파마슈티칼즈 리미티드 | Hiv inhibiting 6-substituted pyrimidines |
| EA020749B1 (en) | 2006-12-29 | 2015-01-30 | Тиботек Фармасьютикалз Лтд. | Hiv inhibiting 5,6-substituted pyrimidines |
| FR2911138B1 (en) | 2007-01-05 | 2009-02-20 | Sanofi Aventis Sa | NOVEL N, N'-2,4-DIANILINOPYRIMIDINE DERIVATIVES, THEIR PREPARATION AS MEDICAMENTS, PHARMACEUTICAL COMPOSITIONS AND IN PARTICULAR AS INHIBITORS OF IKK |
| AR065015A1 (en) | 2007-01-26 | 2009-05-13 | Smithkline Beecham Corp | ANTRANILAMIDE DERIVATIVES, PHARMACEUTICAL COMPOSITIONS CONTAINING THEM, AND USES FOR THE TREATMENT OF CANCER |
| WO2008094737A2 (en) | 2007-01-26 | 2008-08-07 | Irm Llc | Purine compounds and compositions as kinase inhibitors for the treatment of plasmodium related diseases |
| US8846704B2 (en) | 2007-01-31 | 2014-09-30 | YM Biosciences Austraila Pty Ltd | Thiopyrimidine-based compounds and uses thereof |
| DE102007010801A1 (en) | 2007-03-02 | 2008-09-04 | Bayer Cropscience Ag | Use of new and known 2,4-diaminopyrimidine derivatives as fungicides, especially for controlling phytopathogenic fungi |
| JP2010522186A (en) | 2007-03-20 | 2010-07-01 | スミスクライン ビーチャム コーポレーション | Compound |
| JP2010522188A (en) | 2007-03-20 | 2010-07-01 | スミスクライン ビーチャム コーポレーション | Compound |
| WO2008118822A1 (en) | 2007-03-23 | 2008-10-02 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| PT2142516E (en) | 2007-03-30 | 2013-03-07 | Sanofi Sa | Pyrimidine hydrazide compounds as pgds inhibitors |
| WO2008151183A1 (en) | 2007-06-04 | 2008-12-11 | Avila Therapeutics, Inc. | Heterocyclic compounds and uses thereof |
| EP2171090B1 (en) | 2007-06-08 | 2013-04-03 | Genentech, Inc. | Gene expression markers of tumor resistance to her2 inhibitor treatment |
| KR20100035635A (en) | 2007-06-21 | 2010-04-05 | 아이알엠 엘엘씨 | Protein kinase inhibitors and methods for using thereof |
| TWI389893B (en) | 2007-07-06 | 2013-03-21 | Astellas Pharma Inc | Di (arylamino) ary1 compound |
| KR20100049068A (en) | 2007-07-17 | 2010-05-11 | 리겔 파마슈티칼스, 인크. | Cyclic amine substituted pyrimidinediamines as pkc inhibitors |
| AR068054A1 (en) | 2007-08-08 | 2009-11-04 | Smithkline Beecham Corp | PIRROLOPIRIMIDINE COMPOUNDS |
| AU2008296479A1 (en) | 2007-08-28 | 2009-03-12 | Dana Farber Cancer Institute | Amino substituted pyrimidine, pyrollopyridine and pyrazolopyrimidine derivatives useful as kinase inhibitors and in treating proliferative disorders and diseases associated with angiogenesis |
| US8440681B2 (en) | 2007-08-28 | 2013-05-14 | Irm Llc | 2-biphenylamino-4-aminopyrimidine derivatives as kinase inhibitors |
| US7989465B2 (en) | 2007-10-19 | 2011-08-02 | Avila Therapeutics, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| TWI475996B (en) | 2007-10-19 | 2015-03-11 | Celgene Avilomics Res Inc | Heteroaryl compounds and uses thereof |
| EP3912973A3 (en) | 2007-11-28 | 2022-02-16 | Dana-Farber Cancer Institute, Inc. | Small molecule myristate inhibitors of bcr-abl and methods of use |
| CA2707653A1 (en) | 2007-12-03 | 2009-06-11 | Boehringer Ingelheim International Gmbh | Diaminopyridines for the treatment of diseases which are characterised by excessive or anomal cell proliferation |
| CA2710118A1 (en) | 2007-12-20 | 2009-07-02 | Cellzome Limited | Sulfamides as zap-70 inhibitors |
| CA2710144A1 (en) | 2007-12-21 | 2009-07-02 | Avila Therapeutics, Inc. | Hcv protease inhibitors and uses thereof |
| US8309685B2 (en) | 2007-12-21 | 2012-11-13 | Celgene Avilomics Research, Inc. | HCV protease inhibitors and uses thereof |
| US8293705B2 (en) | 2007-12-21 | 2012-10-23 | Avila Therapeutics, Inc. | HCV protease inhibitors and uses thereof |
| US9163061B2 (en) | 2007-12-21 | 2015-10-20 | Celgene Avilomics Research, Inc. | HCV protease inhibitors and uses thereof |
| AR070127A1 (en) | 2008-01-11 | 2010-03-17 | Novartis Ag | PIRROLO - PIRIMIDINAS AND PIRROLO -PIRIDINAS |
| WO2009091476A1 (en) | 2008-01-14 | 2009-07-23 | Irm Llc | Compositions and methods for treating cancers |
| US20110110923A1 (en) | 2008-02-12 | 2011-05-12 | The Brigham And Women's Hospital, Inc. | Fish assay for eml4 and alk fusion in lung cancer |
| ES2471452T3 (en) | 2008-03-05 | 2014-06-26 | Novartis Ag | Use of 5- (2,6-di-morpholin-4-yl-pyrimidin-4-yl) -4-trifluoromethyl-pyridin-2-ylamine for the treatment of non-small cell lung carcinoma with acquired resistance to modulators of the epidermal growth factor receptor (EGFR). |
| CA2717529A1 (en) | 2008-03-11 | 2009-09-17 | Cellzome Limited | Sulfonamides as zap-70 inhibitors |
| CL2009000600A1 (en) | 2008-03-20 | 2010-05-07 | Bayer Cropscience Ag | Use of diaminopyrimidine compounds as phytosanitary agents; diaminopyrimidine compounds; its preparation procedure; agent that contains them; procedure for the preparation of said agent; and method for combating pests of animals and / or harmful plant pathogenic fungi. |
| US8518931B2 (en) | 2008-04-07 | 2013-08-27 | Irm Llc | Compounds and compositions as kinase inhibitors |
| KR101238585B1 (en) | 2008-04-07 | 2013-02-28 | 노파르티스 아게 | Compounds and compositions as protein kinase inhibitors |
| WO2009127642A2 (en) | 2008-04-15 | 2009-10-22 | Cellzome Limited | Use of lrrk2 inhibitors for neurodegenerative diseases |
| EP2274280A2 (en) | 2008-04-16 | 2011-01-19 | Biolipox AB | Bis-aryl compounds for use as medicaments |
| CA2723185A1 (en) | 2008-04-22 | 2009-10-29 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| WO2009143018A2 (en) | 2008-05-19 | 2009-11-26 | Plexxikon, Inc. | Compounds and methods for kinase modulation, and indications therefor |
| US20130225527A1 (en) | 2008-05-21 | 2013-08-29 | Ariad Pharmaceuticals, Inc. | Phosphorus Derivatives as Kinase Inhibitors |
| US9273077B2 (en) | 2008-05-21 | 2016-03-01 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| US20130225528A1 (en) | 2008-05-21 | 2013-08-29 | Ariad Pharmaceuticals, Inc. | Phosphorus Derivatives as Kinase Inhibitors |
| CN102089278A (en) | 2008-06-11 | 2011-06-08 | Irm责任有限公司 | Compounds and compositions useful for the treatment of malaria |
| WO2010008739A2 (en) | 2008-06-20 | 2010-01-21 | Metabolex, Inc. | Aryl gpr119 agonists and uses thereof |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| EP3549934A1 (en) * | 2008-06-27 | 2019-10-09 | Celgene CAR LLC | Heteroaryl compounds and uses thereof |
| CN102203064A (en) | 2008-09-02 | 2011-09-28 | 贝林格尔.英格海姆国际有限公司 | Novel benzamides, production thereof, and use thereof as medicaments |
| US20110230478A1 (en) | 2008-09-03 | 2011-09-22 | Bayer ScienceCrop AG | 4-Alkyl-substituted diaminopyrimidines |
| JP2012501654A (en) | 2008-09-05 | 2012-01-26 | アビラ セラピューティクス, インコーポレイテッド | Algorithms for the design of irreversible inhibitors |
| EP2350053B1 (en) | 2008-10-17 | 2013-12-11 | Whitehead Institute For Biomedical Research | Modulators of MTOR Complexes |
| WO2010048149A2 (en) | 2008-10-20 | 2010-04-29 | Kalypsys, Inc. | Heterocyclic modulators of gpr119 for treatment of disease |
| US8110578B2 (en) | 2008-10-27 | 2012-02-07 | Signal Pharmaceuticals, Llc | Pyrazino[2,3-b]pyrazine mTOR kinase inhibitors for oncology indications and diseases associated with the mTOR/PI3K/Akt pathway |
| CN101723936B (en) | 2008-10-27 | 2014-01-15 | 上海睿星基因技术有限公司 | Kinase suppressor and pharmaceutical application thereof |
| WO2010056311A1 (en) | 2008-11-12 | 2010-05-20 | Ariad Pharmaceuticals, Inc. | Pyrazinopyrazines and derivatives as kinase inhibitors |
| JP2012511575A (en) | 2008-12-12 | 2012-05-24 | アリアド・ファーマシューティカルズ・インコーポレイテッド | Azaindole derivatives as kinase inhibitors |
| US8809343B2 (en) | 2008-12-26 | 2014-08-19 | Fudan University | Pyrimidine derivative, preparation method and use thereof |
| CA2748181C (en) | 2009-01-06 | 2019-07-16 | Nathanael S. Gray | Pyrimido-diazepinone kinase scaffold compounds and methods of treating disorders |
| US9364477B2 (en) | 2009-02-12 | 2016-06-14 | Cell Signaling Technology, Inc. | Mutant ROS expression in human cancer |
| WO2010098866A1 (en) | 2009-02-27 | 2010-09-02 | Supergen, Inc. | Cyclopentathiophene/cyclohexathiophene dna methyltransferase inhibitors |
| US20110071158A1 (en) | 2009-03-18 | 2011-03-24 | Boehringer Ingelheim International Gmbh | New compounds |
| WO2010111406A2 (en) | 2009-03-24 | 2010-09-30 | Myriad Pharmaceuticals, Inc. | Compounds and therapeutic uses thereof |
| US20120270237A9 (en) | 2009-04-03 | 2012-10-25 | Cellzone AG | Methods for the identification of kinase interacting molecules and for the purification of kinase proteins |
| WO2010129053A2 (en) | 2009-05-05 | 2010-11-11 | Dana Farber Cancer Institute | Egfr inhibitors and methods of treating disorders |
| US8410126B2 (en) | 2009-05-29 | 2013-04-02 | Boehringer Ingelheim International Gmbh | Pyrimidine inhibitors of PKTK2 |
| US20120142667A1 (en) | 2009-06-10 | 2012-06-07 | Nigel Ramsden | Pyrimidine derivatives as zap-70 inhibitors |
| WO2010146132A1 (en) | 2009-06-18 | 2010-12-23 | Cellzome Limited | Sulfonamides and sulfamides as zap-70 inhibitors |
| EP2467142B1 (en) | 2009-08-17 | 2016-09-21 | Memorial Sloan-Kettering Cancer Center | 2-(Pyrimidin-5-yl)-thiopyrimidine derivatives as Hsp70 and Hsc70 modulators for the treatment of proliferative disorders |
| NZ598808A (en) | 2009-09-09 | 2014-07-25 | Celgene Avilomics Res Inc | Pi3 kinase inhibitors and uses thereof |
| SG179172A1 (en) | 2009-09-16 | 2012-04-27 | Avila Therapeutics Inc | Protein kinase conjugates and inhibitors |
| WO2011036566A1 (en) | 2009-09-25 | 2011-03-31 | Vertex Pharmaceuticals Incorporated | Methods for preparing pyrimidine derivatives useful as protein kinase inhibitors |
| MX341704B (en) | 2009-10-26 | 2016-08-31 | Signal Pharm Llc | Methods of synthesis and purification of heteroaryl compounds. |
| US20110207736A1 (en) | 2009-12-23 | 2011-08-25 | Gatekeeper Pharmaceuticals, Inc. | Compounds that modulate egfr activity and methods for treating or preventing conditions therewith |
| CN102812167A (en) | 2009-12-30 | 2012-12-05 | 阿维拉制药公司 | Ligand-directed Covalent Modification Of Protein |
| EP2536689A1 (en) | 2010-02-17 | 2012-12-26 | Amgen Inc. | Aryl carboxamide derivatives as sodium channel inhibitors for treatment of pain |
| US8383793B2 (en) | 2010-04-15 | 2013-02-26 | St. Jude Children's Research Hospital | Methods and compositions for the diagnosis and treatment of cancer resistant to anaplastic lymphoma kinase (ALK) kinase inhibitors |
| US20130137709A1 (en) | 2010-05-05 | 2013-05-30 | Nathanael S. Gray | Compounds that modulate EGFR activity and methods for treating or preventing conditions therewith |
| MX2012013127A (en) | 2010-05-13 | 2012-11-30 | Amgen Inc | Heteroaryloxyheterocyclyl compounds as pde10 inhibitors. |
| NZ627709A (en) * | 2010-06-23 | 2014-12-24 | Hanmi Science Co Ltd | Novel fused pyrimidine derivatives for inhibition of tyrosine kinase activity |
| IT1401326B1 (en) | 2010-07-29 | 2013-07-18 | Parlux S P A | SILENCER DEVICE FOR HAIRDRYER. |
| BR112013003388A2 (en) | 2010-08-10 | 2016-07-12 | Celgene Avilomics Res Inc | besylate salt of a btk inhibitor |
| WO2012022045A1 (en) | 2010-08-20 | 2012-02-23 | Hutchison Medipharma Limited | Pyrrolopyrimidine compounds and uses thereof |
| AU2011315831B2 (en) | 2010-10-14 | 2015-01-22 | Takeda Pharmaceutical Company Limited | Methods for inhibiting cell proliferation in EGFR-driven cancers |
| WO2012061303A1 (en) | 2010-11-01 | 2012-05-10 | Avila Therapeutics, Inc. | Heteroaryl compounds and uses thereof |
| TWI632134B (en) | 2010-11-01 | 2018-08-11 | 阿維拉製藥公司 | Heterocyclic compounds and uses thereof |
| US8796255B2 (en) | 2010-11-10 | 2014-08-05 | Celgene Avilomics Research, Inc | Mutant-selective EGFR inhibitors and uses thereof |
| EA201391626A1 (en) | 2011-05-04 | 2014-03-31 | Ариад Фармасьютикалз, Инк. | COMPOUNDS FOR INHIBITING CELL PROLIFERATION IN EGFR-STIMULATED CANCER TYPES |
| WO2013014448A1 (en) | 2011-07-27 | 2013-01-31 | Astrazeneca Ab | 2 - (2, 4, 5 - substituted -anilino) pyrimidine derivatives as egfr modulators useful for treating cancer |
| CA2847540C (en) | 2011-09-22 | 2016-05-17 | Pfizer Inc. | Pyrrolopyrimidine and purine derivatives |
| WO2013169401A1 (en) | 2012-05-05 | 2013-11-14 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in egfr-driven cancers |
| US20170166598A1 (en) | 2014-05-13 | 2017-06-15 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
| MA40240B1 (en) | 2014-06-19 | 2019-03-29 | Ariad Pharma Inc | Heteroaryl compounds of kinase inhibition |
| WO2016065028A1 (en) | 2014-10-21 | 2016-04-28 | Ariad Pharmaceuticals, Inc. | Crystalline forms of 5-chloro-n4-[-2-(dimethylphosphoryl) phenyl]-n2-{2-methoxy-4-[4-(4-methylpiperazin-1-yl) piperidin-1-yl] pyrimidine-2,4-diamine |
| US20210323976A1 (en) | 2015-05-13 | 2021-10-21 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
-
2012
- 2012-05-04 EA EA201391626A patent/EA201391626A1/en unknown
- 2012-05-04 CA CA2832504A patent/CA2832504C/en active Active
- 2012-05-04 KR KR1020137032214A patent/KR101884010B1/en active IP Right Grant
- 2012-05-04 WO PCT/US2012/036683 patent/WO2012151561A1/en active Application Filing
- 2012-05-04 MX MX2013012895A patent/MX360404B/en active IP Right Grant
- 2012-05-04 BR BR112013027734A patent/BR112013027734A2/en not_active IP Right Cessation
- 2012-05-04 CN CN201280021702.7A patent/CN103501612B/en active Active
- 2012-05-04 EP EP12779411.3A patent/EP2704572B1/en active Active
- 2012-05-04 JP JP2014509504A patent/JP5999177B2/en active Active
- 2012-05-04 US US13/464,921 patent/US9834518B2/en active Active
- 2012-05-04 AU AU2012250517A patent/AU2012250517B2/en active Active
-
2013
- 2013-10-06 IL IL228739A patent/IL228739B/en active IP Right Grant
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009143389A1 (en) * | 2008-05-21 | 2009-11-26 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| OGISO ET AL., CRYSTAL STRUCTURE OF THE COMPLEX OF HUMAN EPIDERMAL GROWTH FACTOR AND RECEPTOR EXTRACELLULAR DOMAINS IN CELL, vol. 110, no. 6, 2002, pages 775 - 787, XP002961395 * |
| See also references of EP2704572A4 * |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9012462B2 (en) | 2008-05-21 | 2015-04-21 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
| US9273077B2 (en) | 2008-05-21 | 2016-03-01 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| US9834518B2 (en) | 2011-05-04 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| JP2014094930A (en) * | 2011-07-27 | 2014-05-22 | Astrazeneca Ab | 2-(2,4,5-substituted-anilino)pyrimidine compound |
| JP2013544273A (en) * | 2011-07-27 | 2013-12-12 | アストラゼネカ アクチボラグ | 2- (2,4,5-substituted-anilino) pyrimidine compounds |
| US10596174B2 (en) | 2012-01-13 | 2020-03-24 | ACEA Therapeutics, Inc. | Pyrrolopyrimidine compounds as inhibitors of protein kinases |
| AU2013204563B2 (en) * | 2012-05-05 | 2016-05-19 | Takeda Pharmaceutical Company Limited | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US9834571B2 (en) | 2012-05-05 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US10449196B2 (en) | 2012-08-06 | 2019-10-22 | ACEA Therapeutics, Inc. | EGFR modulators and uses thereof |
| US11007197B2 (en) | 2012-08-06 | 2021-05-18 | ACEA Therapeutics, Inc. | EGFR modulators and uses thereof |
| US9145387B2 (en) | 2013-02-08 | 2015-09-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9980964B2 (en) | 2013-02-08 | 2018-05-29 | Celgene Car Llc | ERK inhibitors and uses thereof |
| US9796700B2 (en) | 2013-02-08 | 2017-10-24 | Celgene Car Llc | ERK inhibitors and uses thereof |
| US9561228B2 (en) | 2013-02-08 | 2017-02-07 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9504686B2 (en) | 2013-02-08 | 2016-11-29 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| US9611283B1 (en) | 2013-04-10 | 2017-04-04 | Ariad Pharmaceuticals, Inc. | Methods for inhibiting cell proliferation in ALK-driven cancers |
| JP2016528209A (en) * | 2013-07-11 | 2016-09-15 | エイシア バイオサイエンシーズ インコーポレイテッド | Pyrimidine derivatives as kinase inhibitors |
| US10562918B2 (en) | 2013-07-11 | 2020-02-18 | ACEA Therapeutics, Inc. | Heterocyclic compounds and uses thereof |
| JP2017214390A (en) * | 2013-11-21 | 2017-12-07 | ファイザー・インク | 2,6-substituted purine derivatives and their use in treatment of proliferative disorders |
| JP2017506667A (en) * | 2014-02-25 | 2017-03-09 | シャンハイ ハイヤン ファーマシューティカル テクノロジー カンパニー リミテッドShanghai Haiyan Pharmaceutical Technology Co., Ltd. | 2,4-disubstituted benzene-1,5-diamine derivatives and uses thereof and pharmaceutical and medicinal compositions prepared therefrom |
| JP2017524703A (en) * | 2014-07-25 | 2017-08-31 | シャンハイ ハイヤン ファーマシューティカル テクノロジー カンパニー リミテッドShanghai Haiyan Pharmaceutical Technology Co., Ltd. | 2,4-disubstituted 7H-pyrrolo [2,3-d] pyrimidine derivatives, process for their preparation and use in medicine |
| EP3173412A4 (en) * | 2014-07-25 | 2018-02-28 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | 2,4-disubstituted 7h-pyrrolo[2,3-d]pyrimidine derivative, preparation method and medicinal use thereof |
| US10202364B2 (en) | 2014-08-13 | 2019-02-12 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| US10005760B2 (en) | 2014-08-13 | 2018-06-26 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| US11498921B1 (en) | 2014-11-05 | 2022-11-15 | InventisBio Co., Ltd. | Pyrimidine or pyridine compounds, preparation method therefor and pharmaceutical uses thereof |
| EP3885344A3 (en) * | 2014-11-05 | 2021-12-08 | InventisBio Co., Ltd. | Pyrimidine compounds, preparation method therefor and pharmaceutical uses thereof |
| US10533011B2 (en) | 2015-10-09 | 2020-01-14 | ACEA Therapeutics, Inc. | Pharmaceutical salts, physical forms, and compositions of pyrrolopyrimidine kinase inhibitors, and methods of making same |
| US11498922B2 (en) | 2017-04-07 | 2022-11-15 | ACEA Therapeutics, Inc. | Pharmaceutical composition comprising N-(3-((2-((3-fluoro-4-(4-methylpiperazin-1-yl phenyl)amino)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)phenylacrylamide |
| EP3656769A4 (en) * | 2017-07-19 | 2021-04-14 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Aryl-phosphorus-oxygen compound as egfr kinase inhibitor |
| EP3670500A4 (en) * | 2017-08-18 | 2020-12-16 | Beijing Hanmi Pharmaceutical Co., Ltd. | Chemical compound, pharmaceutical composition thereof, and use and application thereof |
| EP3675860A4 (en) * | 2017-08-28 | 2021-04-14 | Zhihong, Chen | Substituted pyrimidines, pharmaceutical compositions and therapeutic methods thereof |
| CN112055714A (en) * | 2018-04-20 | 2020-12-08 | 贵州伊诺其尼科技有限公司 | Dimethyloxyphosphonium compounds |
| CN112055714B (en) * | 2018-04-20 | 2021-10-22 | 贵州伊诺其尼科技有限公司 | Dimethyloxyphosphonium compounds |
| CN113677680A (en) * | 2019-04-04 | 2021-11-19 | 贝达药业股份有限公司 | EGFR inhibitor and composition and application thereof |
| CN113677680B (en) * | 2019-04-04 | 2024-05-10 | 贝达药业股份有限公司 | EGFR inhibitor, composition and application thereof |
| US11529350B2 (en) | 2019-07-03 | 2022-12-20 | Sumitomo Pharma Oncology, Inc. | Tyrosine kinase non-receptor 1 (TNK1) inhibitors and uses thereof |
| WO2021073498A1 (en) * | 2019-10-17 | 2021-04-22 | 贝达药业股份有限公司 | Egfr inhibitor, composition, and method for preparation thereof |
| CN114430741A (en) * | 2019-10-17 | 2022-05-03 | 贝达药业股份有限公司 | EGFR inhibitor, composition and preparation method thereof |
| WO2021104441A1 (en) * | 2019-11-29 | 2021-06-03 | 江苏先声药业有限公司 | Polyaromatic compound as egfr kinase inhibitor |
| EP4129996A4 (en) * | 2020-03-23 | 2023-07-12 | Qilu Pharmaceutical Co., Ltd. | Novel aminopyrimidine egfr inhibitor |
| EP4137484A4 (en) * | 2020-04-14 | 2023-12-20 | Qilu Pharmaceutical Co., Ltd. | Tricyclic compounds as egfr inhibitors |
| CN114728994A (en) * | 2020-07-03 | 2022-07-08 | 成都地奥九泓制药厂 | Aryl phosphorus oxygen compound and application thereof |
| CN114728994B (en) * | 2020-07-03 | 2023-04-28 | 成都地奥九泓制药厂 | Aryl phosphorus oxide compound and application thereof |
| EP4177258A4 (en) * | 2020-07-03 | 2024-07-03 | Chengdu Diao Jiuhong Pharmaceutical Factory | Arylphosphine oxide compounds and use thereof |
| WO2022094354A1 (en) * | 2020-10-30 | 2022-05-05 | Lengo Therapeutics, Inc. | Pyrimidine compounds, compositions, and medicinal applications thereof |
| WO2022199589A1 (en) * | 2021-03-23 | 2022-09-29 | 南京明德新药研发有限公司 | Pyrimidine derivatives |
| US12084453B2 (en) | 2021-12-10 | 2024-09-10 | Incyte Corporation | Bicyclic amines as CDK12 inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112013027734A2 (en) | 2017-08-08 |
| US20120316135A1 (en) | 2012-12-13 |
| EP2704572A4 (en) | 2014-11-05 |
| US9834518B2 (en) | 2017-12-05 |
| CN103501612B (en) | 2017-03-29 |
| KR20140028057A (en) | 2014-03-07 |
| CN103501612A (en) | 2014-01-08 |
| MX360404B (en) | 2018-10-31 |
| IL228739A0 (en) | 2013-12-31 |
| CA2832504A1 (en) | 2012-11-08 |
| EP2704572B1 (en) | 2015-12-30 |
| AU2012250517B2 (en) | 2016-05-19 |
| JP2014514348A (en) | 2014-06-19 |
| MX2013012895A (en) | 2014-02-17 |
| AU2012250517A1 (en) | 2013-05-02 |
| EP2704572A1 (en) | 2014-03-12 |
| CA2832504C (en) | 2019-10-01 |
| KR101884010B1 (en) | 2018-07-31 |
| JP5999177B2 (en) | 2016-09-28 |
| EA201391626A1 (en) | 2014-03-31 |
| IL228739B (en) | 2018-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2704572B1 (en) | Compounds for inhibiting cell proliferation in egfr-driven cancers | |
| US9834571B2 (en) | Compounds for inhibiting cell proliferation in EGFR-driven cancers | |
| AU2011315831B2 (en) | Methods for inhibiting cell proliferation in EGFR-driven cancers | |
| KR102072869B1 (en) | Quinolines as fgfr kinase modulators | |
| EP2699551B1 (en) | Tetrahydroquinoline derivatives useful as bromodomain inhibitors | |
| WO2019101178A1 (en) | Uracil compound as c-met/axl inhibitor | |
| PT1294715E (en) | Quinazoline ditosylate salt compounds | |
| JP2010510974A (en) | Substituted dihydroimidazoles and their use in the treatment of tumors | |
| KR20180100441A (en) | A novel substituted cyanoindoline derivative as an NIK inhibitor | |
| CN111825658A (en) | Novel EGFR (epidermal growth factor receptor) triple-mutation inhibitor and application thereof | |
| WO2011132633A1 (en) | Substituted 5-hydroxypyrimidine-4-carboxamide compound | |
| WO2017088755A1 (en) | Aminopyrimidine heterocyclic compound with adenosine receptor antagonistic activity | |
| CN109867667B (en) | PARP and PI3K dual-target inhibitors containing pyridopyrimidine structure | |
| EP2844642B1 (en) | Compounds for inhibiting cell proliferation in egfr-driven cancers | |
| KR100843533B1 (en) | Novel diazine derivatives, their manufacture and use as pharmaceutical agents | |
| AU2013203904A1 (en) | Methods for inhibiting cell proliferation in EGFR-driven cancers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12779411 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012250517 Country of ref document: AU Date of ref document: 20120504 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2832504 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2014509504 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2013/012895 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012779411 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201391626 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20137032214 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013027734 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013027734 Country of ref document: BR Kind code of ref document: A2 Effective date: 20131029 |