WO2012149171A1 - Designing padlock probes for targeted genomic sequencing - Google Patents
Designing padlock probes for targeted genomic sequencing Download PDFInfo
- Publication number
- WO2012149171A1 WO2012149171A1 PCT/US2012/035227 US2012035227W WO2012149171A1 WO 2012149171 A1 WO2012149171 A1 WO 2012149171A1 US 2012035227 W US2012035227 W US 2012035227W WO 2012149171 A1 WO2012149171 A1 WO 2012149171A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- target
- probe
- ccaac
- dinucleotides
- sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6811—Selection methods for production or design of target specific oligonucleotides or binding molecules
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
Definitions
- the subject matter described herein relates generally to the fields of molecular biology and bioinformatics. More specifically, the subject matter described herein relates to systems, methods, and computer programs for designing probes for use in nucleic acid sequencing, particularly padlock probes useful in carrying out targeted genomic and methylation sequencing.
- Padlock Probe (PP) technology is a multiplex genomic enrichment method allowing for accurate targeted high-throughput sequencing.
- PP technology has been used to perform highly multiplexed genotyping, digital allele quantification, targeted bisulfite sequencing, and exome sequencing. See Hardenbol, P. et al., (2005) Genome Res. 15: 269-275; Wang, Y. et al., (2005) Nucleic Acids Res., 2005, 33(21) el 83: 1 -14; Porreca, G.J. et al., (2007) Nat. Methods 4(1 1): 931 -936; Zhang, K. et al., (2009) Nat. Methods 6: 613-618; Turner, E.H.
- Padlock probe technology may utilize a linear oligonucleotide molecule with two binding sequences at each end joined by a common linker sequence.
- the probe's binding arms may be hybridized several base pairs apart surrounding a target single-stranded genomic DNA region.
- a DNA polymerase (with each of the four standard dNTP molecules) may be used to fill in the gap between the two binding arms, and a DNA ligase may be used to circularize the resulting molecule.
- a mixture of exonucleases may then used to digest all arm; the resulting circular molecules can be amplified using rolling circle amplification or polymerase chain reaction to generate a DNA library compatible with modem high- throughput sequencers.
- Padlock probes are generally designed to have binding arms with
- each binding arm is generally designed to have a specific DNA melting temperature.
- padlock probe efficacy from molecule to molecule has been found to vary greatly, ranging across several orders of magnitude.
- Previous work identified a complex nonlinear relationship between padlock probe efficiency and many probe characteristics. (Deng, J. et al., (2009) Nat. Biotechnol. 27: 353-360; Li, J.B. et al., (2009) Genome Res. 19(9): 1606- 1615).
- the bias inherent in padlock probe design has been demonstrated to show a complex nonlinear relationship with many probe characteristics, confounding attempts to generate efficient probes. Because of this, padlock probe results are highly biased towards certain genomic regions. Certain genomic regions are therefore extremely difficult to target using padlock probes due to the lack of knowledge of probe capturing efficiency.
- the subject matter described herein relates to methods, systems, and computer program products that can be used to design oligonucleotide primers and probes, particularly padlock probes for high-density multiplex genome sequencing.
- the subject matter disclosed herein is particularly useful for detecting methylation status and/or single nucleotide variants in a target nucleic acid sequence of interest.
- the subject matter described herein provides a method of designing probes or primers for sequencing a target nucleic acid molecule, comprising the steps of selecting one or more inputs associated with efficiency of the probe or primer;
- the method further comprises synthesizing the probe or primer.
- the probe is a padlock probe.
- the one or more inputs may comprise target length, target folding energy, target GC content, extension arm A%, extension arm G%, target A%, target T%, target G%, number of "GG” dinucleotides in ligation arm, number of "AT” dinucleotides in extension arm, number of "GG” dinucleotides in extension arm, number of "AA” dinucleotides in target, number of "AT” dinucleotides in target, number of "TA” dinucleotides in target, number of "GT” dinucleotides in target, number of "GA” dinucleotides in target, ligation arm terminal dinucleotide, extension arm terminal dinucleotide, target 5' terminal dinucleotide, ligation arm melting temperature, extension arm melting temperature, ligation arm length, extension arm length, local single- stranded folding energy of the target, and dinucleotides present at the
- the target nucleic acid sequence is derived from a human.
- the target-capturing sequence may include a ligation arm and an extension arm.
- the target-capturing sequence contains one or more CpG dinucleotides.
- the target-capturing sequences in the first library may also contain all possible methylation state combinations of the one or more CpG dinucleotides.
- the extension arm comprises one or more priming sites for amplification of the target nucleic acid sequence and may be universal priming sites.
- the target capturing sequence may also include one or more restriction sites.
- the methods disclosed herein involve an algorithm that may comprise one or more neural networks.
- the one or more neural networks may comprise the one or more inputs, e.g., seven or more inputs.
- the method further comprises, after the extracting step, pooling the non-extracted probe or primer sequences and repeating certain steps defined herein.
- the linker sequence is a sequence that is common to each probe or primer sequence in the second library.
- an apparatus comprising at least one processor and at least one memory including code which when executed by the at least one processor provides operations comprising: selecting one or more inputs associated with efficiency of the probe or primer; selecting a target nucleic acid sequence; generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence; determining the efficiency of each probe or primer sequence in the first library by using an algorithm comprising the one or more selected inputs; ranking the probe or primer sequences in the first library by efficiency; extracting the probe or primer sequences having the highest efficiency to generate a second library; and adding a linker sequence to each of the probe or primer sequences in the second library.
- a computer-readable storage medium including code which when executed by at least one processor provides operations comprising: selecting one or more inputs associated with efficiency of the probe or primer; selecting a target nucleic acid sequence; generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence; determining the efficiency of each probe or primer sequence in the first library by using an algorithm comprising the one or more selected inputs; ranking the probe or primer sequences in the first library by efficiency; extracting the probe or primer sequences having the highest efficiency to generate a second library; and adding a linker sequence to each of the probe or primer sequences in the second library.
- Figure 1 is a block diagram of a single padlock probe capture experiment, demonstrating an example of a workflow for targeted genomic resequencing consistent with some of the exemplary embodiments described herein;
- Figure 2 is a flowchart of a process for designing padlock probes from input files consistent with some of the exemplary embodiments described herein;
- Figure 3 is a block diagram of a back propagation neural network used to derive the probe efficiency scoring equation consistent with some of the exemplary embodiments described herein;
- Figure 4 is a depiction of two examples of customized linker sequences and the function provided thereby, each of which is consistent with some of the exemplary embodiments described herein.
- Figure 4 discloses SEQ ID NOS 420-424, respectively, in order of appearance;
- FIG. 5 ⁇ -5 ⁇ depicts the design of padlock probes for targeted bisulfite sequencing.
- A is a diagram showing that each padlock probe has a common linker sequence flanked by two target-specific capturing arms (HI and H2). H I and H2 are melting temperature normalized, and a spacer sequence is included to normalize probe lengths. The linker sequence contains priming sites (API and AP2) for universal primers, two Mmel sites and a central Alul recognition site.
- (B) is a diagram depicting a CpG island (or other target region) covered by multiple padlock probes targeting partially overlapped regions on alternating strands.
- (C) is a diagram showing a library of padlock probes annealed to bisulfite-converted genomic DNA.
- (D) is a flow chart showing the generation of a shotgun sequencing library.
- (E) is a picture showing gel electrophoresis analysis of the padlock-captured products from two independent capturing reactions (1 and 2) and a no-template control (NTC).
- Figures 6A-6G are graphs summarizing an analysis of the effect of probe
- Figures 7A-7D depict experimental normalization of padlock-capturing efficiency.
- A shows the "subsetting" strategy.
- B depicts the 'suppressor oligo' strategy.
- C shows the distribution of normalized abundance for all captured targets with one 30,000-probe set and with four probe sets.
- the x-axis is the normalized abundance of each captured target, which is calculated by dividing the counts of the target by the average counts of all targets.
- the y-axis is the fraction of probes with the coverage equal to or greater than the normalized coverage.
- D Comparison of relative abundance for each target before and after
- the vertical dash lines indicate the clear separation of four subsets of targets, as well as the fifth set normalized with the suppressor oligos.
- FIGS. 8A-8C show the results of a validation experiment demonstrating the digital methylation assay.
- A Comparison of methylation measurements from both strands for the same CpG sites. The methylation levels of the forward strand were plotted against the levels of the reverse strand on 2697 CpG sites that were covered by on both strands by different probes.
- B Methylation levels of 182 randomly selected CpG sites in the B J fibroblast lines were measured by the conventional bisulfite Sanger sequencing.
- C Comparison of the methylation levels of 25,665 CpG sites (at least 50x sequencing reads per site) between two biological replicates on the IMR90 fibroblasts.
- FIG. 9 is a schematic for the probe design software (ppDesigner).
- Figure 10 are graphs comparing probe capture efficiencies between the DMR220K, LC4K probe sets and the CGI30K set. The first three plots were generated from data without subsetting or suppressor oligos to allow for a direct comparison of probe design.
- Figure 1 1 is a scatter plot of number of characterized CpG sites versus mappable sequencing data for the DMR330K probe set. Variability in sequencing quality of individual sequencing runs is responsible for the different number of CpG sites characterized with similar sequencing effort.
- Figure 12 is a graph showing the number of CpG sites called per sample as a function of sequencing effort.
- the horizontal dash line represents 4Gbps of sequences per library.
- Figures 13A-13B are graphs showing captured CpG sites that were tested for potential regulatory interactions with genes by GREAT.
- A Most CpG sites were interacting with 1-2 genes.
- B Distance of CpG sites to the transcriptional start sites (TSS) of the predicted regulating genes.
- Figures 14A-14E are graphs showing the accuracy of digital quantification by BSPP.
- A, B show a comparison of the methylation levels obtained at lOx depth from multiple capture reactions of the same sample (PGP HPS) within batches and between batches.
- C, D, E Within sample comparison of methylation levels obtained from different probes capturing the same CpG site on different strands at 1 Ox depth within one capture reaction.
- Figure 15 is a graph showing the comparison between BSPP and whole genome bisulfite sequencing (WGBS). Two HI ESC datasets were compared, using sites with at least lOx read depth in each.
- Figure 16 is a graph depicting the variation in amount of sequencing data obtained per sample in a multiplexed BSPP capture experiment. Forty-eight whole blood samples were captured and sequenced in one batch using the library-free BSPP method.
- Figure 17 depicts exemplary padlock probes ordered from (A) Agilent's
- oligonucleotide synthesis service (SEQ ID NOS 425-427, respectively, in order of appearance) and (B) LC Sciences' oligonucleotide synthesis service (SEQ ID NOS 428-430, respectively, in order of appearance).
- Figure 18 is a diagram depicting the addition of a second neural network specifically for bisulfite-con verted DNA.
- This network contains two hidden layers with 10 and 12 nodes, respectively, and accepts 25 pieces of information as input.
- read refers to a nucleic acid sequence of sufficient length (e.g. , at least about 30 bp) that can be used to identify a larger sequence or region, e.g. that can be aligned and specifically assigned to a chromosome or genomic region or gene.
- aligning refers to one or more sequences that are identified as a match in terms of the order of their nucleic acid molecules to a known sequence from a reference genome. Such alignment can be done manually or by a computer algorithm. Examples include, without limitation, the Efficient Local Alignment of Nucleotide Data (ELAND) computer program distributed as part of the Illumina Genomics Analysis, Bowtie, BWA, and SOAP2Align.
- ELAND Efficient Local Alignment of Nucleotide Data
- the matching of a sequence read in aligning can be a 100% sequence match or less than 100% (non-perfect match).
- Amplification or amplifying methods include but are not limited to the polymerase chain reaction (PCR), the ligase chain reaction (LCR) (e.g., Wu, D.Y. and Wallace, R.B. (1989) Genomics 4: 560-569; Landegren, U. et al., (1998) Science 241 (4869): 1077-1080; and Barringer, K.J. et al. (1990) Gene 89(1): 1 17-122), transcription
- PCR polymerase chain reaction
- LCR ligase chain reaction
- nucleic acid based sequence amplification (NABSA; U.S. Patent Nos. 5,409,818, 5,554,517, and 6,063,603 each of which is incorporated herein by reference).
- NABSA nucleic acid based sequence amplification
- Other amplification methods that may be used are described in U.S. Patent Nos. 5,242,794, 5,494,810, 4,988,617.
- a "nucleotide” is a monomer that includes a base, such as a pyrimidine, purine, or synthetic analogs thereof, linked to a sugar and one or more phosphate groups. Nucleotides include adenine (A) residues, guanine (G) residues, cytosine (C) residues, thymine (T) residues, and uracil (U) residues.
- the major nucleotides of DNA are deoxyadenosine 5'- triphosphate (dATP or A), deoxyguanosine 5'-triphosphate (dGTP or G), deoxycytidine 5'- triphosphate (dCTP or C) and deoxythymidine 5'-triphosphate (dTTP or T).
- the major nucleotides of RNA are adenosine '-triphosphate (ATP or A), guanosine 5 '-triphosphate (GTP or G), cytidine 5 '-triphosphate (CTP or C) and uridine 5'-triphosphate (UTP or U).
- Nucleotides also include chemical entities containing modified bases, modified sugar moieties and modified phosphate backbones, for example as described in U.S. Patent No. 5,866,336. Such modifications however, can allow for incorporation of the nucleotide into a growing nucleic acid chain or for binding of the nucleotide to the complementary nucleic acid chain.
- Nucleotides can be modified at any position on their structures. Examples include, but are not limited to, the modified nucleotides 5-fluorouracil, 5-bromouracil, 5-chlorouracil, 5-iodouracil, hypoxanthine, xanthine, acetylcytosine, 5-(carboxyhydroxylmethyl) uracil, 5- carboxymethylaminomethyl-2-thiouridine, 5-carboxymethylaminomethyluracil,
- dihydrouracil beta-D-galactosylqueosine, inosine, N-6-sopentenyl adenine, 1 -methylguanine, 1 -methylinosine, 2,2-dimethylguanine, 2-methyladenine, 2-methylguanine, 3-methylcytosine, 5-methylcytosine, N6-adenine, 7-methylguanine, 5-methylaminomethyluracil,
- methoxyarninomethyl-2-thiouracil beta-D-mannosylqueosine, 5'- methoxycarboxymethyluracil, 5-methoxyuracil, 2-methylthio-N6-isopentenyladenine, uracil- 5-oxyacetic acid, pseudouracil, queosine, 2-thiocytosine, 5-methyl-2-thiouracil, 2-thiouracil, 4-thiouracil, 5-methyluracil, uracil-5-oxyacetic acid methylester, uracil-S-oxyacetic acid, 5- methyl-2-thiouracil, 3-(3-amino-3-N-2-carboxypropyl) uracil, and 2,6-diaminopurinc.
- modified sugar moieties which can be used to modify nucleotides at any position on their structures include, but are not limited to: arabinose, 2-fluoroarabinose, xylose, and hexose, or a modified component of the phosphate backbone, such as phosphorothioate, a phosphorodithioate, a phosphoramidothioate, a pliosphoramidate, a phosphordiamidate, a methylphosphonate, an alkyl phosphotriester, or a formacetal or analog thereof.
- nucleic acid As used herein the terms “nucleic acid”, “nucleic acid molecule”, “polynucleotide” and “oligonucleotide” are used interchangeably and refers to the polymeric form of nucleotides, cither ribonucleotides and/or deoxyribonucleotides or a modified form of either type of nucleotide.
- Nucleic acids include, without limitation, cDNA, niRNA, genomic DNA, and synthetic (such as chemically synthesized) DNA or RNA, plasmids, amplicons, cosmids, and fragments thereof.
- the nucleic acid can be double stranded (ds) or single stranded (ss).
- the nucleic acid molecule can be the sense strand or the antisense strand.
- Nucleic acids can include natural nucleotides (such as adenine, thymine/uracil, cytosine, and guanine) and can also include analogs of natural nucleotides.
- a set of bases linked to a peptide backbone, as in a peptide nucleic acid (PNA), can be used as a substitute for a nucleic acid molecule.
- Nucleic acids can be modified by means of a fluorophore that is directly or indirectly excitable.
- Fluorescent DNA dye as used herein may refer to a composition, for example SYBR Green I or SYBR Gold that becomes fluorescently excitable when it associates with double-stranded DNA.
- fluorescent DNA dyes include, e.g., 5 -carboxy fluorescein, 2'7'-dimethoxy-4'5 , -dichloro-6- carboxyfluorescein, fluorescein (FL); N,N,N',N'-tetramethyl-6-carboxyrhodamine; 6- carboxy-X-rhodamine; CY3; CY5; tetrachloro-fluorescein; and hexachloro-fluorescein; NED; 6-FAM; VIC; PET; LIZ, SID, TED, and TAZ.
- a "target" nucleic acid molecule is a nucleic acid to be sequenced, identified, or detected, and can be obtained or isolated in purified form, by any method known to those skilled in the art (for example, as described in U.S. Patent No. 5,674,743), but need not be in purified form.
- Various other biomolecules can also be present with the target nucleic acid molecule.
- the target nucleic acid molecule can be present in a cell or a biological sample (which can include other nucleic acid molecules and proteins).
- the target nucleic acid molecule may be a whole genome or a portion of a genome, such as
- the target nucleic acid molecule can be derived from any species, including but not limited to, vertebrates such as humans, cows, dogs, cats, mice, rats, sheep, horse, goat, invertebrates, bacteria, viruses, fungi, and the like.
- the target nucleic acid may be derived from any source, including tissues, primary cells, cultured cells, cell lines, tumor specimens, bodily fluids, and the like.
- the target nucleic acid may also be wholly or partly synthetic.
- a "complementary" nucleic acid molecule is complementary to the target nucleic acid molecule and is the nucleic acid strand that is elongated when sequencing the target nucleic acid molecule.
- oligonucleotide refers to a linear nucleic acid molecule (such as DNA or R A) sequence of at least 6 nucleotides, for example at least 9, at least 15, at least 18, at least 24, at least 30, at least 50, at least 100, at least 200 or even at least 500 nucleotides long. However shorter or longer oligonucleotides may be used. Oligonucleotides may be designed to have different length. In some embodiments, the sequence of the nucleic acid molecule may be divided up into a plurality of shorter sequences that can be synthesized in parallel and assembled into a single or a plurality of desired nucleic acid molecules using the methods described herein.
- the oligonucleotides are designed to provide the full sense and antisense strands of the nucleic acid molecule. After hybridization of the plus and minus strand oligonucleotides, two double stranded oligonucleotides are subjected to ligation or polymerization in order to form a first subassembly product. Subassembly products are then subjected to ligation or polymerization to form a larger DNA or the full DNA sequence.
- a "primer” is a short nucleic acid molecule, for example sequences of at least 9 nucleotides, which can be annealed to a complementary target nucleic acid molecule by nucleic acid hybridization to form a hybrid between the primer and the target nucleic acid strand.
- a primer can be extended along the target nucleic acid molecule by a polymerase enzyme. Therefore, individual primers can be used for nucleic acid sequencing, wherein the sequence of the primer is specific for the target nucleic acid molecule, for example so that the primer will hybridize to the target nucleic acid molecule under stringent hybridization conditions.
- a primer is at least 10 nucleotides in length, such as at least 10 contiguous nucleotides complementary to a target nucleic acid molecule to be sequenced.
- longer primers can be employed, such as primers having at least 12, at least 15, at least 20, or at least 30 contiguous nucleotides
- a "probe” refers to an oligonucleotide sequence that may or may not be extended in the amplification reaction by a DNA polymerase. Probes that are very specific for a perfectly complementary target sequence and strongly reject closely related sequences having one or a few mismatched bases are known in the art as "allele
- Probes that hybridize under at least one applicable detection condition not only to perfectly complementary sequences, but also to partially complementary sequences having one or more mismatched bases, are "mismatch tolerant" probes.
- oligonucleotide set refers to a collection of primers or primers and probes for performing amplification or sequencing reactions.
- oligonucleotide library refers to a collection of primers or primers and probes for performing amplification or sequencing reactions.
- probe library refers to a collection of primers or primers and probes for performing amplification or sequencing reactions.
- methods of assembling libraries containing nucleic acids, primers, and probes having predetermined sequence variations are provided herein.
- libraries of nucleic acids are libraries of sequence variants. Sequence variants may be variants of a single naturally-occurring sequence. However, in some embodiments, sequence variants may be variants of a plurality of different sequences.
- a high-density nucleic acid library may include more than 100 different sequence variants (e.g.
- the libraries may contain information obtained from sequencing reactions, such as target nucleic acid sequences, sequence reads, sequence alignments, bisulfite-converted sequences, methylation frequencies, allele frequencies, single nucleotide variants or polymorphisms, among others. Libraries may be stored or kept on high-density arrays, microarrays, microchips, computer-readable media, or by any method or apparatus known in the art. ⁇ -
- the oligonucleotides, primers, and probes may comprise universal (common to all oligonucleotides), semi-universal (common to at least of portion of the oligonucleotides) or individual or unique primer (specific to each oligonucleotide) binding sites (also referred to herein as "priming sites") on either the 5' end or the 3' end or both.
- the term "universal" primer or primer binding site means that a sequence used to amplify the oligonucleotide is common to all oligonucleotides such that all such oligonucleotides can be amplified using a single set of universal primers.
- an oligonucleotide contains a unique primer binding site.
- unique primer binding site refers to a set of primer recognition sequences that selectively amplifies a subset of oligonucleotides.
- an oligonucleotide contains both universal and unique amplification sequences, which can optionally be used sequentially.
- a “linker” or “linker sequence” is a structure, which can be a unique nucleic acid sequence, that joins one molecule to another, such as attachment of a probe as described herein to another molecule or a substrate, wherein one portion of the linker is operably linked to a substrate, and wherein another portion of the linker is operably linked to the probe.
- a linker or linker sequence may refer to a common nucleic acid sequence that is preferably non-complementary to a target nucleic acid sequence.
- oligonucleotides, probes or primers as described herein may each contain a single common linker sequence.
- a "barcode” or “barcode sequence” as used herein refers to a short stretch of nucleotides in a particular order, and different barcodes are different combinations of nucleotides.
- a barcode may be of any length, but is preferably between 4 to 15, more preferably between 4 to 10, and most preferably between 4 and 8, such as 4, 5, 6, 7 or 8 nucleotides long. Ideally, the barcodes are designed such that they can be unambiguously called post-sequencing or post-amplification.
- the sequence of the barcode may be identical or different for each nucleic acid molecule in a particular sequencing run, or according to a number of different parameters, such as target nucleic acid origin, sequence reads derived from the 5' strand or the 3 ' strand, sequence length or molecular weight of a sequence read.
- Barcode sequences may be derived computationally or by hand. In some aspects, barcode sequences can be randomly generated using an iterative script program, such as Perl.
- Oligonucleotides, primers, and probes may also contain one or more sites ("restriction sites”) for cleavage by restriction endonucleases (also known as “restriction enzymes").
- Type 1 enzymes cut DNA at random far from their recognition sequences.
- Type II enzymes cut DNA at defined positions close to or within their recognition sequences.
- Type III enzymes are also large combination restriction-and-modification enzymes. They cleave outside of their recognition sequences and require two such sequences in opposite orientations within the same DNA molecule to accomplish cleavage.
- Type IV enzymes recognize modified, typically methylated DNA. Restriction endonucleascs are commercially available and restriction site sequences are well known in the art (New England Biolabs, Beverly, MA). Any restriction site sequence may be included in oligonucleotides, primers, and probes as described herein.
- Oligonucleotides, primers, and probes may be isolated from natural sources or purchased from commercial sources. Oligonucleotides, primers, and probes described herein may also be synthesized by any suitable method, e.g., standard phosphoramidite methods such as those described by Beaucage, S.L. and Caruthers, M.H. (1981 ) Tetrahedron Lett. 22: 1859-1862 or the triester method according to Matteucci, M.D. and Caruthers, M.H. (1981) J Am. Chem. Soc.
- oligonucleotide synthesizer or high-throughput, high-density array methods known in the art (see U.S. Patent Nos. 5,602,244, 5,574, 146, 5,554,744, 5,428, 148, 5,264,566, 5,141 ,813, 5,959,463, 4,861 ,571 and 4,659,774, incorporated herein by reference in its entirety for all purposes).
- Pre-synthesized oligonucleotides may also be obtained commercially from a variety of vendors. Oligonucleotides, primers, and probes may be prepared using a variety of microarray technologies known in the art.
- Pre-synthesized oligonucleotide and/or polynucleotide sequences may be attached to a support or synthesized in situ using light-directed methods, flow channel and spotting methods, inkjet methods, pin- based methods and bead-based methods set forth in the following references: McGall et al. (1996) Proc. Natl. Acad. Sci. U.S.A. 93(24): 13555-13560; Synthetic DNA Arrays In Genetic Engineering, Vol. 20: 1 1 1, Plenum Press (1998); Duggan, D.J. et al., (1999) Nat. Genet.
- T m melting temperatures
- the T m of a primer is a calculated value using any method knows in the art, particularly the "% GC” method (Wetmar, J.G. (1991) Crit. Rev. Diochem. Mol. Biol. 26: 227-259), the "2(A+T) plus 4(G+C)” method at a standard condition of primer and salt concentration, or methods described in Santa Lucia, J. (1998) Proc. Natl. Acad. Sci. 95(4): 1460-1465 and von Ahsen, N. et al., (2001) Clin. Chem. 47: 1956-1961.
- methylation denotes the type of chemical modification of nucleic acids that involves the addition of a methyl group, for example to the C5 carbon atom of the cytosine pyrimidine ring or to the N6 nitrogen atom of the adenosine purine ring, with the first option being particularly preferred.
- This modification can be inherited and subsequently removed without changing the original nucleic acid sequence. As such, it is part of the epigenetic code and the most well characterized epigenetic mechanism.
- Methylation is reversible: methyl -transferases catalyze the transfer of a methyl group from S- adenosyl-L-methionine to cytosine or adenosine residues. Polymerases such as DNA polymerases do not copy the methylated status during replication (reviewed, e.g. , in
- CpG dinucleotide sites refers to regions of DNA where a cytosine nucleotide is located immediately adjacent to a guanine nucleotide in the linear sequence.
- CpG refers to cytosine and guanine separated by a phosphate (i.e., - -C--phosphate— G— ).
- the "CpG” notation is used to distinguish a cytosine followed by guanine from a cytosine base paired to a guanine. Regions of the DNA that have a higher frequency or concentration of CpG sites are known as "CpG islands”.
- CpG islands may also define a contiguous region of genomic DNA that satisfies the criteria of (1) having a frequency of CpG dinucleotides corresponding to an "observed/expected ratio" greater than 0.6; (2) having a "GC content” greater than 0.5; and (3) having a length of at least 0.2 kb (as described in Gardiner-Garden et al., (1987) J. Mol. Biol , 196: 262-282), with the exception that repeat regions matching these criteria are excluded (or masked).
- CpG island fragment or “CGI fragment” arc used interchangeably to refer to a nucleic acid molecule fragment mapping to and containing at least part of a CpG island.
- CpG target sequence refers to a stretch of bases targeted to selectively enrich for CpG island fragments and/or other methylation informative GC-rich fragments.
- Many genes in mammalian genomes have CpG islands associated with the transcriptional start site (including the promoter) of the gene, which play a pivotal role in controlling gene expression.
- Methylation at the C5 of cytosine has been found in bacteria, fungi, plant and mammalian genomes. Approximately 60-90% of CpG dinucleotides are methylated in most mammalian cell types. The CpG dinucleotides are not uniformly distributed in mammalian genomes. For example, sequence analysis of the human genome has estimated nearly 30,000 CpG islands, which accounts for about 0.7% of the genome. CpG dinucleotides in the remaining 99.3% of the genome are sparsely distributed. Because of the high cytosine- guanine frequency of CpG islands, it is possible to identify them without knowledge of the methylation pattern of the DNA.
- CpG islands are often unmethylated but a subset of islands becomes methylated during oncogenesis, cellular development, and various disease states.
- Hypermethylation i.e. an increased level of methylation
- CpG sites within the promoters of genes can lead to their silencing, a feature found, e.g. , in a number of human cancers (for example the silencing of tumor suppressor genes).
- hypomethylation i.e. a reduced level of methylation
- oncogenes within cancer cells (reviewed, e.g. , in Robertson, K.D. and Wolffe, A.P. (2000) Nat. Rev. Genet. 1(1 ): 1 1 -19; Li, E. (2002) Nat. Rev. Genet. 3: 662-673; Bird, A.P.
- methylation assay refers to any assay for determining the methylation status of one or more CpG dinucleotide sequences within one or more nucleic acid sequences.
- Methodylation frequency refers to a measure of methylation status of one or more CpG dinucleotide sequences within one or more nucleic acid sequences.
- Methodylation frequency refers to a measure of methylation status of one or more CpG dinucleotide sequences within one or more nucleic acid sequences.
- the methylation status of a particular nucleic acid fragment or sequence can indicate the methylation state of every base in the sequence or can indicate the methylation state of a subset of the base pairs (e.g. , whether the base is cytosine or 5-methylcytosine) within the sequence.
- Methylation states at one or more particular CpG methylation sites (each having two CpG dinucleotide sequences) within a nucleic acid sequence may include "unmethylated,” “fully-methylated” and "hemi-methylated” sites.
- Methylation status can also indicate information regarding regional methylation density within the sequence without specifying the exact location at the single nucleotide position level.
- a "methylation profile” refers to a set of data representing the methylation states or methylation frequencies of one or more loci within a molecule of DNA from e.g. , the genome of an individual or cells or tissues from an individual.
- the profile can indicate the methylation state of every base in an individual, can have information regarding a subset of the base pairs (e.g. , the methylation state of specific promoters or quantity of promoters) in a genome, or can have information regarding regional methylation density or methylation frequency of one or more loci with or without specifying the exact location at the single nucleotide position level.
- “Differential methylation” denotes a condition in which a particular candidate genomic locus is (at one or more nucleic acid sites comprised in its sequence) methylated in at least one target sample but unmethylated in at least one reference sample, or vice versa, in which a particular candidate genomic locus is (at one or more nucleic acid sites comprised in its sequence) unmethylated in the reference sample but methylated in the target sample.
- the determination of the differential methylation pattern or frequency of the one or more candidate genes/loci already includes the identification of the exact nucleic acid sites (i.e. sequence elements, genetic loci) comprised in the one or more candidate genes.
- the nucleic acid sites comprised in the one or more candidate genes/loci that are differentially methylated are CpG dinucleotide sites.
- methylation is determined by means of one or more methods selected from reverse-phase HPLC, thin-layer chromatography, Sssl
- methyltransferases with incorporation of labeled methyl groups the chloracetaldehyde reaction, differentially sensitive restriction enzymes, hydrazine or permanganate treatment (m5C is cleaved by permanganate treatment but not by hydrazine treatment), bisulfite sequencing, combined bisulfite-restriction analysis, pyrosequencing, methylation-sensitive single-strand conformation analysis (MS-SSCA), high resolution melting analysis (HRM), methylation-sensitive single nucleotide primer extension (MS-SnuPE), base-specific cleavage/MALDI-TOF, methylation-specific PCR (MSP), microarray-based methods, and Mspl cleavage (reviewed, e.g., in Rein, T.
- MS-SSCA methylation-sensitive single-strand conformation analysis
- HRM high resolution melting analysis
- MS-SnuPE methylation-sensitive single nucleotide primer extension
- MSP methylation-specific
- Methods for identifying methylation may be based on differential cleavage by restriction enzymes are used. Methylation-sensitive restriction analysis followed by PCR amplification or Southern analysis have been disclosed, for example, in Huang, T.H. et al. (1997) Cancer Res. 57: 1030-1034; Zuccotti, M. et al, (1993) Meth. Enzymol. 225: 557-567; Carrel, L. et al. (1996) Am. J. Med. Genet. 64: 27-30; and Chang et al. (1992) Plant Mol. Biol. Rep. 10: 362-366.
- enzymes that include at least one CpG dinucleotide in the recognition site may be used.
- Enzymes with a recognition site that includes the sequence CCGG include, for example, Mspl, Hpa l, Age], Xmal, Smal, NgoMW, Nael, and BspEl.
- Enzymes with a recognition site that includes the sequence CGCG include, for example, BstUl (CGCG, MSRE), Mlul (ACGCGT, MSRE), Sacll (CCGCGG, MSRE), AwHlI (GCGCGC, MSRE) and Nrwl (TCGCGA, MSRE).
- Enzymes with a recognition site that includes the sequence GCGC include, for example, HinP , Hhal, Afe , Kasl, Nar , Sfol, Bbel, and Fspl.
- Enzymes with a recognition site that includes the sequence TCGA include, for example, Taql, Clal (MSRE), BspOl (MSRE), PaeWll, Tlil, Xltol, Sail, and BstB .
- restriction enzymes that contain CpG in the recognition sequence and for information about the enzyme's sensitivity to methylation, see, for example, the New England Biolabs catalog and web site.
- two restriction enzymes may have a different recognition sequence but generate identical overhangs or compatible cohesive ends.
- the overhangs generated by cleavage with Hpall or Mspl can be ligated to the overhang generated by cleavage with Taql.
- Some restriction enzymes that include CpG in the recognition site are unable to cleave if the site is methylated; these are methylation sensitive restriction enzymes (MSRE).
- MIRE methylation insensitive restriction enzymes
- MDRE methylation dependent restriction enzymes
- Examples of MIREs that have a CpG in the recognition sequence include, for example, BsaVJl (WCCGGW), BsoBl, BssS , Mspl, and Taql.
- MSREs that include a CpG in the recognition site
- examples of MSREs include Aatl , Aci , AcH, Afel, Agel, Ascl, Aval, BmgBl, Bsa AI, BsaHl, BspDl, CM, Eagl, Fsel, Faul, Haelll, Hpall, H/nPlI, Mini, Narl, Noil, Nrul, Pvnl, Sacll, Sail, Smal and SnaBl.
- a pair of enzymes that have differential sensitivity to methylation and cleave at the same recognition sequence with one member of the pair being a MSRE and the other member being MIRE is used.
- Still other enzymes include BthQl, Glal, Hpal, HmPlI, Dpnl, Mbo , Chal and BstKTl.
- Bisulfite sequencing is a commonly used method in the art for generating methylation data at single-base resolution.
- the term “bisulfite conversion” refers to a biochemical process for converting unmethylated cytosine residue to uracil or thymine residues, whereby methylated cytosine residues are preserved.
- “Bisulfite conversion” may be carried out computationally from a nucleic acid sequence contained in a computer file (such as those in FASTA, FASTQ or any file format known in the art), wherein all cytosine residues in a sequence of interest are changed to thymine or uracil residues.
- Exemplary reagents for bisulfite conversion include sodium bisulfite and magnesium bisulfite.
- “Bisulfite reagent” refers to a reagent comprising bisulfite, disulfite, hydrogen sulfite or combinations thereof, useful as disclosed herein to distinguish between methylated and unmethylated CpG dinucleotide sequences.
- One way to obtain such melhylation data for the CGIs is to sequence the entire epigenome directly. Due to the difficulty in mapping bisulfite converted sequence reads and the methylation heterogeneity in a cell population, approximately 100 gigabases (Gb) of sequence data would be needed to generate a high-resolution human DNA methylation map (Lister, R. et al., (2009) Nature, 462(7271): 315-322).
- methylation profiling approaches include array capture (Hodges, E. et al., (2009) Genome Res. 19(9): 1593-1605), padlock probe capture (Deng, J. et al., (2009) Nat. Biotech. 27: 353-360; Ball, M.P. et al., (2009) Nat. Biotech. , 27(4): 361 -368) and reduced representation bisulfite sequencing (Gu et al., (2010) Nat. Methods 7(2): 133-136).
- bisulfite sequencing involves conversion of unmethylated cytosine to uracil or thymine through a three-step process during sodium bisulfite modification.
- the steps are sulfonation to convert cytosine to cytosine sulfonate, deamination to convert cytosine sulfonate to uracil sulfonate or thymine sulfonate and alkali desulfonation to convert uracil sulfonate to uracil or thymine sulfonate to thymine.
- Conversion of methylated cytosine is much slower and is not observed at significant levels in a 4-16 hour reaction (Clark, S.J.
- cytosine is methylated it will remain a cytosine. If the cytosine is unmethylated, it will be converted to uracil or thymine.
- a G will be incorporated in the inteiTOgation position (opposite the C being interrogated) if the C was methylated and an A will be incorporated in the interrogation position if the C was unmethylated.
- Kits for DNA bisulfite modification are commercially available from, for example, Human Genetic Signatures' Melhyleasy and Chemicon's CpGenome Modification Kit. See also, WO04096825, which describes bisulfite modification methods and Olek, A. et al.
- a catalyst such as diethylenetriamine may be used in conjunction with bisulfite treatment, see Komiyama, M. and Oshima, S., (1994) Tetrahedron Lett. 35(44): 8185-8188.
- Diethylenetriamine has been shown to catalyze bisulfite ion-induced deamination of 2'- deoxycytidine to 2'-deoxyuridine at pH 5 efficiently.
- Other catalysts include ammonia, ethylene-diamine, 3,3'-diaminodipropylamine, and spermine.
- deamination is performed using sodium bisulfite solutions of 3-5 M with an incubation period of 12-16 hours at about 50°C.
- a faster procedure has also been reported using 9-10 M bisulfite pH 5.4 for about 10 minutes at 90°C, see Hayatsu, H. et al., (2004) Proc. Jpn. Acad. Ser. B 80(4): 189-194.
- Bisulfite treatment allows the methylation status of cytosines to be detected by a variety of methods.
- any method that may be used to detect a single nucleotide polymorphism (SNP) may be used, for examples, see Syvanen, A.C. (2001) Nature Rev. Gen. 2(12): 930-942.
- bisulfite sequencing methods, systems, and computer program products described herein may provide information regarding not only methylation frequencies or methylation status of a sequence of interest at single base resolution, but also information regarding SNPs, preferably in the same sequencing run. Other methods such as single base extension (SBE) may be used or hybridization of sequence specific probes similar to allele specific hybridization methods.
- SBE single base extension
- “Variants” or “alleles” generally refer to one of a plurality of species each encoding a similar sequence composition, but with a degree of distinction from each other.
- the distinction may include any type of variation known to those of ordinary skill in the related art, that include, but are not limited to, polymorphisms such as SNPs, insertions or deletions (the combination of insertion/deletion events are also referred to as "indels"), differences in the number of repeated sequences (also referred to as tandem repeats), and structural variations. Detection of such variants or alleles is also within the ambit of the subject matter described herein.
- MIP molecular inversion probes
- a MIP may be designed for each cytosine to be interrogated.
- the MIP includes a locus specific region that hybridizes upstream and one that hybridizes downstream of an inteiTOgation site and can be extended through the interrogation site, incorporating a base that is complementary to the interrogation position.
- the interrogation position may be the cytosine of interest after bisulfite modification and amplification of the region and the detection can be similar to detection of a polymorphism. Separate reactions may be performed for each NTP so extension only takes place in the reaction containing the base corresponding to the interrogation base or the different products may be differentially labeled.
- PLP padlock probe
- PLPs refers to circularized nucleic acid molecules which may combine specific molecular recognition and universal amplification (or specific amplification and general recognition), thereby increasing sensitivity and multiplexing capabilities without limiting the range of potential target organisms.
- PLPs are long oligonucleotides of approximately 100 bases (but can be of any length), containing target complementary regions (referred to herein as “target-capturing sequences”) at both their 5' and 3' ends (See, for example, Figure 5). These regions recognize adjacent sequences on the target nucleic acid sequence (Nilsson, M., et al.
- binding arms which comprise “extension arms” having priming sites (e.g. , universal priming sites"), sites recognized by ligase enzymes, and unique sequence identifiers, sometimes referred to as a "ZipCode” or "barcode”.
- binding arms Upon hybridization, the ends of the probes are situated into adjacent position, and can be joined by enzymatic ligation at the ligation sites (also referred to herein as “ligation arms”) converting the probe into a circular molecule (also known in the art and referred to herein as an "amplicon”) that is threaded on the target strand.
- Non-circularized probes may be removed by exonuclease treatment, while the circularized entities may be amplified with universal primers, may or may not contain barcode or ZipCode sequences. This mechanism ensures reaction specificity, even in a complex nucleotide extract with a large number of padlock probes. Subsequent ly, the target- specific products are detected by a universal cZipCode microarray (Shoemaker, D.D., et al., (1996) Nat. Genet. 14: 450-456). PLPs have high specificity and multiplexing capabilities in genotyping assays (Hardenbol, P., et al., (2003) Nat. BiotechnoL, 21 : 673-678.).
- a “formula,” “algorithm,” or “model” is any mathematical equation, algorithmic, analytical or programmed process, or statistical technique that takes one or more continuous or categorical inputs (herein called “parameters”) and calculates an output value, sometimes referred to as an "index” or “index value.”
- Parameters continuous or categorical inputs
- Non-limiting examples of “algorithms” include sums, ratios, and regression operators, such as coefficients or exponents, value
- a "neural network” can be an Artificial Neural Network (ANN) and are information processing systems composed of varying numbers of simple, elements called neurons distributed into layers. Neurons are organized in an input layer, one or more hidden layers, and an output layer. The connections between elements determine network function just as in natural biological nervous systems.
- ANN is an intelligent technique that mimics the functioning of a human brain, and emulates human intuition of making decisions and drawing conclusions even when presented with complex, noisy, irrelevant and partial information.
- ANNs may have any number of hidden layers.
- the neurons are connected to each other by weighted links over which signals can pass.
- Each neuron receives multiple inputs from other neurons, except the neurons in the input layer, in proportion to their connection weights and then generates a single output in accordance with an activation function.
- An activation function can be linear or nonlinear depending on the application. Sigmoid or Hyperbolic Tangent activation function can be used to improve the performance of ANNs in power system applications.
- An ANN can be trained to perform a particular function by adjusting values of the interconnections called weights, and neuron thresholds. The process of adjusting
- Training an ANN consists of adjusting interconnection weights of neurons using a learning algorithm. Back propagation with momentum is the commonly used learning algorithm. Multilayer Feed Forward ANNs with Error Back Propagation learning algorithm are also commonly used. Feed Forward calculations, and propagating error from output layer to input layer and weight updating in hidden and output layers are major steps of training algorithm.
- the neural networks described herein comprise one or more inputs that are associated with efficiency of the probe or primer described herein.
- efficiency is meant the amount of target nucleic acid sequence represented by a particular probe or primer in a sequencing library. Standard methods can be used to calculate the efficiency by measuring or counting the amount of target nucleic acid(s) and the amount of unbound target nucleic acid(s) via sequencing.
- the efficiency of a probe or primer described herein is typically compared to the capture efficiency of a control probe or primer under the same incubation conditions (e.g. , using same buffer and temperature).
- Such inputs comprise, without limitation, target length, target folding energy, target GC content, extension arm A%, extension arm G%, target A%, target T%, target G%, number of "GG” dinucleotides in ligation arm, number of "AT” dinucleotides in extension arm, number of "GG” dinucleotides in extension arm, number of "AA” dinucleotides in target, number of "AT” dinucleotides in target, number of "TA” dinucleotides in target, number of "GT” dinucleotides in target, number of "GA” dinucleotides in target, ligation arm terminal dinucleotide, extension arm terminal dinucleotide, target 5' terminal dinucleotide, ligation arm melting temperature, extension arm melting temperature, ligation arm length, extension arm length, local single-stranded folding energy of the target, and the dinucleotides present at the extension site and ligation site during probe capture.
- the neural networks described herein may comprise one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, twenty-two, twenty-three, twenty-four, twenty-five or more inputs.
- the methods, computer program products, and systems described herein may comprise one, two, three, four, five or more neural networks.
- the probe designing algorithm used to predict efficiency of probes or primers as described herein utilizes two neural networks.
- a process may be implemented as a software application or computer program product (also known as software, programs, applications, components, or code) stored in memory and executed by one or more processors contained in a system.
- the software application or computer program product may accept a complete genome and a tabular list of specific regions of interest, along with several customizable probe properties (such as the inputs described herein, including, e.g., a desired target length, a desired binding arm length, and a desired DNA melting temperature).
- the software application or computer program product may use this information in an intelligent decision mechanism, such as for example a back propagation neural network-derived equation, to predict probe efficiency based on many probe characteristics.
- the software application or computer program product may output the optimal set of probes to obtain maximum coverage of a desired set of genomic regions in a high-throughput sequencing experiment.
- the software application or computer program product may also add a customized linker sequence to each probe molecule, allowing easy usage in modern high-throughput sequencers.
- the probe design software application may also perform an in silico bisulfite conversion prior to probe design in order to generate an optimal set of probes for targeted bisulfite sequencing.
- An additional feature may be implemented to allow the probe design software application or computer program product to output the most efficient padlock probe molecules for specific unique regions of a genome, allowing for efficient multiplex organism detection.
- the methods, systems, and computer program products described herein may exhibit features, such as an increased number of targetable genomic regions for padlock probe sequencing, more efficient and unbiased capture of genomic regions, and reduced cost of sequencing to obtain target coverage.
- the method, system, and computer program product described herein may extend to designing efficient padlock probes for SNP genotyping, mutation discovery, targeted genomic sequencing, quantification of allele specific gene expression, analysis of DNA methylation profiles or frequencies, and organism presence detection.
- Figure 2 depicts a process 200 which may be implemented by a system comprising one or more processors and at least one memory including code which when executed by a processor provide the one or more of the operations depicted at process 200.
- the process 200 may comprise a software application or computer program product implemented on at least one processor and at least one memory.
- the software application or computer program product may read three files presented by the user, such as for example, a job file, a target file, and/or a genome or chromosome file.
- the system may optionally comprise one or more databases containing libraries of information related to sequencing data, such as raw sequence reads, sequence alignments, methylation status or frequencies of a target nucleic acid sequence, and information regarding SNPs, allele frequencies, or other variations in nucleic acid sequences of interest.
- the system may also comprise one or more communication links connecting the one or more processors to the one or more databases.
- the job file may include several customizable parameters or inputs, such as for example a range for desired binding arm size, a range for target region size, and/or a flag for whether single stranded folding energy of each target should be calculated using an external module.
- the job file may also include links to one or more software modules to be used and the location on disk of the software, target file, and/or genome/chromosome file.
- the genome/chromosome file may include the genome or chromosome on which targeted sequencing is to be performed.
- the file may be configured in a FASTA or FASTQ format.
- FASTA an organism or chromosome name may be presented, beginning with the ">" character; example: ">chrl " may be the first line of a FASTA or FASTQ file.
- the subsequent lines may contain a string of DNA base characters (A, T, G, and C) or DNA ambiguity characters as determined by IUPAC (M, R, W, S, Y, , V, H, D, B, or N). Line break characters may be allowed to enable easier reading.
- Multi-chromosome organisms (such as, for example, humans) may still require one ">" character per file; multiple chromosomes may be split into separate files (for example "human_chrl .fa" or
- the target file may include a tab-delimited list of specific genomic regions or target nucleic acid sequences to be sequenced with padlock probes.
- the first column in the file may include a specific identifier chosen (e.g., by an experimenter) to describe the target nucleic acid molecule, for example the name of a specific gene or specific regulatory region of interest.
- the second column may list the chromosome or genome for the specific targeted region.
- the third and fourth columns may list the beginning and end of the specific target region, in linear bases; the distance spanned can be as short as a single base pair (for genotyping) or thousands of base pairs (for larger-scale sequencing).
- the fifth column (which may not be included in the file) may accept a strand designation; if one is provided, only probes targeting one specifically chosen strand of DNA (either forward or reverse) may be designed; otherwise, probes may be designed utilizing both strands in combination to provide optimal capture.
- the software application or computer program product implemented on one or more processors and at least one memory may read in the specified job file to obtain desired probe parameters or inputs.
- the software application or computer program product may then proceed sequentially through the target file, designing optimal probe sets for each target nucleic acid molecule.
- the software application or computer program product may begin by opening the first specified genome/chromosome and loading, for example, the entire sequence into random access memory for rapid access.
- the software application or computer program product may then extract the specific target region of interest or target capture sequence, along with flanking sequence both linearly upstream and downstream of the target, from the genome.
- the software application or computer program may design most, if not all, possible binding arms (which include extension arms and ligation arms as defined herein) for a given target nucleic acid molecule or region using the extracted sequence.
- the software application or computer program product may take into account the desired binding arm size from the job file, and remove from consideration any very low-complexity binding arms (i.e., more than six nucleotides of the same kind in a row).
- the software application or computer program product may next associate each binding arm with every possible partner binding arm, taking into account the desired target region length provided in the job file. These arm pairs would represent the two sequences surrounding the common linker.
- the software application or computer program product may designs arm pairs using both the forward and reverse strand of the provided genome unless the target file specifies a chosen strand; in this case, arm pairs will only be generated for the designated strand.
- the software application or computer program product may then obtains a portion of information or inputs (e.g., six to seven key pieces of information) about each binding arm pair, such as for example the target length, the target GC content, the melting temperature of each binding arm, the length of each binding ami, and/or optionally the local single-stranded folding energy of the target.
- the software application or computer program product may then use a previously developed equation to predict the probe efficiency. This equation may be developed by an intelligent decision mechanism, such as a neural network, pattern recognizer, and/or other numerical techniques.
- the equation to predict the probe efficiency may be developed by performing a multiplex genome sequencing reaction using hundreds of thousands of separate padlock probe molecules; the capturing efficiency of each molecule is measured in this sequencing experiment and modeled using one or more neural networks, including for example a back propagation neural network (with three layers containing 13, 8, and 5 nodes respectively, see Figure 3) to the aforementioned 6-7 pieces of information as input. Additional neural networks may be added (such as, e.g. , a network with two hidden layers having 10 and 12 nodes, see Figure 18) with at least 25 pieces of information as input.
- the software application or computer program product may then divide each probe into separate categories based on which regions of the target sequence are captured, and ranks each available probe by probe efficiency. The probes having the highest efficiency are extracted and included in the generation of a library or probe set. The non- extracted probes of lower efficiency can be pooled and resubmitted for additional rounds of probe design as defined by the methods described herein.
- the software application or computer program product may then generate candidate "probe sets” or "libraries” from the list of valid probes.
- the software application or computer program product may consider all possible sets of probe sets, and choose the set that provides maximum coverage of the sample and the highest aggregate probe scores, under the constraint that no binding arms in a probe set can bind to the same sequence (in order to prevent probe competition for hybridization).
- the software application or computer program product may also penalize sets that require many probes in order to control the cost of sequencing; this parameter may be adjustable.
- the software application or computer program product may then report all binding arm pairs for the optimal probe set to the user. If the user is satisfied, the software application may then attach a common linker sequence between the two binding arms and generate a single molecular sequence for each probe that can be generated using commercial DNA synthesis services (such as that provided by Integrated DNA Technologies) or via other high-throughput methods (see, e.g. , U.S. Patent Application Ser. No. 60/765,978).
- Each linker sequence may be customizable to include specific adaptor sequences for current high- throughput sequencers, allowing designed padlock probes to be converted into linear sequencing libraries in just one experimental step. Linker sequences can also be customized to include barcode sequences for even greater multiplex capability or restriction enzyme sites for easy linearization of the circular padlock probe modules (see, e.g. , Figure 4). The user may thus integrate the generated probes into many custom experimental environments.
- the software application or computer program product may output it and proceed to the next target listed in the target file.
- the software application or computer program product may then repeat the genomic loading and probe design process.
- the software application or computer program product may exclude probes having the highest efficiency and pool the remaining non-extracted probes and repeat the steps of generating a library or set of probe or primer sequences, determining efficiency of the probes in the generated library or set, and ranking the probe or primer sequences in the library or set by efficiency.
- the methods, systems, and computer program products described herein are related to characterizing the DNA methylation profile of a sample using bisulfite sequencing and include mapping a genomic sequence of interest to determine methylation status, methylation frequency, and detection of single nucleotide polymorphisms.
- bisulfite sequencing all cytosines present in the DNA molecule except those that are methylated are converted to thymines. Almost every methylated cytosine is present as a cytosine-guanine dinucleotide (CpG), though not all CpGs are methylated.
- CpG cytosine-guanine dinucleotide
- the software application or computer program product performs an in silico bisulfite conversion, computationally converting all cytosines except those present in a CpG to thymines.
- the application or program then designs probes as previously, but penalizes the inclusion of CpG sites in each binding arm.
- the software application or computer program product generates multiple probes for those arm pairs containing a CpG; one probe assumes a methylated state (and contains a CpG dinucleotide) while the other assumes an unmethylated state (and contains a TpG instead). This procedure allows for efficient targeted bisulfite sequencing of hundreds of thousands of CpG sites in parallel.
- the methods, systems, and computer program products described herein use the probes or primers to obtain sequence reads of a target genome or sequence of interest by bisulfite sequencing and loading it into memory.
- a software application or computer program product encodes the sequence reads by predicting the forward and reverse orientation of each of the sequence reads to generate at least one forward sequence read and at least one reverse sequence read.
- the forward and reverse sequence reads are then converted by the software application or computer program product by computationally changing all cytosine residues in the forward sequence reads to thymine residues in silico, and changing all guanine residues to adenine residues in the reverse sequence reads.
- the bisulfite-converted genome sequence and all forward and reverse sequence reads are then aligned computationally by an alignment software application or computer program (e.g. , ELAND, SOAP2 Align, Bowtie, BWA, BLAST or any other alignment program known in the art).
- the alignment application or program can be a standalone application or integrated into the system, software application or computer program product described herein.
- the aligned sequences are then combined to create a map of the target genomic sequence.
- the software application or computer program product analyzes and computes methylation frequencies or methylation status of the mapped sequences in entirety.
- the mapped sequences may also be analyzed by the software application or computer program product for the presence of single nucleotide polymorphisms. Because bisulfite sequencing provides sequence read information at single-base resolution, this technique (and modifications thereof described in the methods, systems, and computer program products described herein) is particularly advantageous for calculating methylation frequencies and detecting SNPs in a single sequencing reaction.
- the methods, systems and computer program products described herein are related to organism detection in a mixed sample. Many cellular samples are heterogeneous, and contain mixtures of organisms in unknown quantities;
- padlock probes can be used to detect which and how many organisms of each of a given type are present.
- the software application or computer program product may accept a fourth input file, known as a "homer file," which contains lists of preferred arm sequences in FASTA format (generally those found via genome annotation to be unique to a given genome or chromosome).
- the job file also contains at least one additional parameter: the number of probes to generate per genome or chromosome.
- the software application or computer program product then designs binding arm pairs as previously described, but favors binding arms containing a user-provided homer sequence.
- the software application or computer program instead of creating a "probe set" to maximize coverage of a target region, the software application or computer program instead returns the user-specified number of probes (in order of decreasing capturing efficiency) per genome. Probes designed for many separate genomes can be combined into a single padlock probe reaction, allowing detection of multiple organisms present at low frequency in a mixed population.
- control module may be realized in digital electronic circuitry, integrated circuitry, specially designed ASICs (application specific integrated circuits), computer hardware, firmware, software, and/or combinations thereof.
- ASICs application specific integrated circuits
- These various implementations may include implementation in one or more computer programs that are executable and/or interpretable on a programmable system including at least one programmable processor, which may be special or general purpose, coupled to receive data and instructions from, and to transmit data and instructions to, a storage system, at least one input device, and at least one output device.
- These software applications or computer program products include machine instructions for a programmable processor, and may be implemented in a high-level procedural and/or object-oriented programming language, and/or in assembly/machine language.
- computer-readable medium refers to any computer program product, apparatus and/or device (e.g. , magnetic discs, optical disks, memory, Programmable Logic Devices (PLDs)) used to provide machine instructions and/or data to a programmable processor, including a machine-readable medium.
- PLDs Programmable Logic Devices
- a computer may include any type of computer platform such as a workstation, a personal computer, a server, or any other present or future computer.
- Computers typically include known components such as a processor, an operating system, system memory, memory storage devices, input-output controllers, input-output devices, and display devices.
- a processor typically includes known components such as a processor, an operating system, system memory, memory storage devices, input-output controllers, input-output devices, and display devices.
- Many possible configurations and components of a computer exist in the art and may also include cache memory, a data backup unit, and other additional devices.
- Display devices may include display devices that provide visual information, this information typically may be logically and/or physically organized as an array of pixels.
- An interface controller may also be included that may comprise any of a variety of known or future software programs for providing input and output interfaces.
- interfaces may include "Graphical User Interfaces" (often referred to as GUI's) that provide one or more graphical representations to a user. Interfaces are typically enabled to accept user inputs using means of selection or input known to those o ordinary skill in the related art.
- applications on a computer may employ an interface that includes what are referred to as "command line interfaces" (often referred to as CLI's).
- CLPs typically provide a text based interaction between an application and a user.
- command line interfaces present output and receive input as lines of text through display devices.
- some implementations may include a "shell” such as Unix Shells known to those of ordinary skill in the related art, or Microsoft Windows Powershell that employs object-oriented type programming architectures such as the Microsoft .NET framework.
- Interfaces may include one or more GUI's, CLI's or a combination thereof.
- a processor may include a commercially available processor such as an Itanium® or Pentium® processor made by Intel Corporation, a SPARC® processor made by Sun Microsystems, an AthlonTM or OpteronTM processor made by AMD corporation, or it may be one of other processors that are or will become available.
- Some embodiments of a processor may also include Multi-core processors and/or employ parallel processing technology in a single or multi-core configuration.
- a multi-core architecture typically comprises two or more processor "execution cores". Each execution core may perform as an independent processor that enables parallel execution of multiple threads.
- a processor may be configured in what is generally referred to as 32 or 64 bit architectures, or other architectural configurations now known or that may be developed in the future.
- a processor typically executes an operating system, which may be, for example, a Windows®-type operating system (such as Windows® XP, Windows Vista®, Windows 7) from the Microsoft Corporation; the Mac OS X operating system from Apple Computer Corp. (such as 7.5 Mac OS X v l 0.4 "Tiger” or 7.6 Mac OS X vl 0.5 "Leopard” operating systems); a Unix® or Linux-type operating system available from many vendors or an open source; another or a future operating system; or some combination thereof.
- An operating system interfaces with firmware and hardware in a well-known manner, and facilitates the processor in coordinating and executing the functions of various computer programs that may be written in a variety of programming languages.
- An operating system typically in cooperation with a processor, coordinates and executes functions of the other components of a computer.
- An operating system also provides scheduling, input-output control, file and data management, memory management, and communication control and related services, all in accordance with known techniques.
- System memory may include any of a variety of known or future memory storage devices. Examples include any commonly available random access memory (RAM), magnetic medium such as a resident hard disk or tape, an optical medium such as a read and write compact disc, or other memory storage device.
- Memory storage devices may include any of a variety of known or future devices, including a compact disk drive, a tape drive, a removable hard disk drive, USB or flash drive, or a diskette drive.
- Such types of memory storage devices typically read from, and/or write to, a program storage medium such as, respectively, a compact disk, magnetic tape, removable hard disk, USB or flash drive, or floppy diskette. Any of these program storage media, or others now in use or that may later be developed, may be considered a computer program product.
- these program storage media typically store a computer software program and/or data.
- Computer software programs, also called computer control logic typically are stored in system memory and/or the program storage device used in conjunction with memory storage device.
- a computer program product comprising a computer usable medium having control logic (computer software program, including program code) stored therein.
- the control logic when executed by a processor, causes the processor to perform functions described herein.
- some functions are implemented primarily in hardware using, for example, a hardware state machine.
- Input-output controllers could include any of a variety of known devices for accepting and processing information from a user, whether a human or a machine, whether local or remote. Such devices include, for example, modem cards, wireless cards, network interface cards, sound cards, or other types of controllers for any of a variety of known input devices. Output controllers could include controllers for any of a variety of known display devices for presenting information to a user, whether a human or a machine, whether local or remote. As presently described herein, the functional elements of a computer may communicate with each other via a system bus. Some embodiments of a computer may communicate with some functional elements using network or other types of remote communications.
- an instrument control and/or a data processing application if implemented in software, may be loaded into and executed from system memory and/or a memory storage device. All or portions of the instrument contiol and/or data processing applications may also reside in a read-only memory or similar device of the memory storage device, such devices not requiring that the instrument control and/or data processing applications first be loaded through input-output controllers. It will be understood by those skilled in the relevant art that the instrument control and/or data processing applications, or portions of it, may be loaded by a processor in a known manner into system memory, or cache memory, or both, as advantageous for execution.
- a computer may include one or more library files, experiment data files, and an internet client stored in system memory.
- experiment data could include data related to one or more experiments or assays such as detected signal values, or other values associated with one or more sequencing experiments or processes.
- an internet client may include an application enabled to accesses a remote service on another computer using a network and may for instance comprise what are generally referred to as "Web Browsers".
- some commonly employed web browsers include Microsoft® Internet Explorer available from Microsoft Corporation, Mozilla Firefox® from the Mozilla Corporation, Safari from Apple Computer Corp., Google Chrome available from Google, Inc., or other type of web browser currently known in the art or to be developed in the future.
- An internet client may include, or could be an element of, specialized software applications enabled to access remote information via a network such as a data processing application for sequencing applications.
- a network may include one or more of the many various types of networks well known to those of ordinary skill in the art.
- a network may include a local or wide area network that employs what is commonly referred to as a TCP/IP protocol suite to communicate
- a network may include a network comprising a worldwide system of interconnected computer networks that is commonly referred to as the internet, or could also include various intranet architectures.
- Some users in networked environments may prefer to employ what are generally referred to as "firewalls" (also sometimes referred to as Packet Filters, or Border Protection Devices) to control information traffic to and from hardware and/or software systems.
- firewalls may comprise hardware or software elements or some combination thereof and are typically designed to enforce security policies put in place by users, such as for instance network administrators, etc.
- the subject matter described herein may also make use of additional computer program products and software for a variety of purposes, such as probe design, management of data, analysis, and instrument operation. See, U.S. Patent Nos. 5,593,839, 5,795,716, 5,733,729, 5,974,164, 6,066,454, 6,090,555, 6,185,561 , 6,188,783, 6,223,127, 6,229,91 1 and 6,308,170. Additionally, the subject matter described herein may also make use of methods for providing genetic information over networks such as the Internet as shown in U.S. Patent Application Ser. Nos. 10/197,621 , 10/063,559 (United States Publication No. 20020183936), 10/065,856, 10/065,868, 10/328,818, 10/328,872, 10/423,403, and 60/482,389.
- oligonucleotides, primers, and probes useful for general sequencing reactions and assays.
- Commercial sequencing by synthesis platforms are available, such as the Genome Sequencer from Roche/454 Life Sciences, the Genome Analyzer from Illumina/Solexa, the SOLiD system from Applied BioSystems, Pacific Biosystems and the Heliscope system from Helicos Biosciences.
- Exemplary sequencing platforms may have one or more of the following features: 1) four differently optically labeled nucleotides are utilized (e.g. , Genome
- Such sequencing reactions and assays include sequencing by ligation methods commercialized by Applied Biosystems (e.g. , SOLiD sequencing).
- double stranded fragment nucleic acid molecules can be prepared by the methods described herein, and then incorporated into a water-in-oil emulsion along with polystyrene beads and amplified, for example by PCR.
- alternative amplification methods can be employed in the water-in-oil emulsion such as any of the methods provided herein.
- the amplified product in each water microdroplet formed by the emulsion can interact, bind, or hybridize with the one or more beads present in that microdroplet leading to beads with a plurality of amplified products of substantially one sequence.
- the methods can include a step of rendering the nucleic acid bound to the beads single-stranded or partially single stranded.
- Sequencing primers are then added along with a mixture of four different fluorescently labeled oligonucleotide probes.
- the probes bind specifically to the two bases in the nucleic acid molecule to be sequenced immediately adjacent and 3' of the sequencing primer to determine which of the four bases are at those positions.
- a ligase is added.
- the ligase cleaves the oligonucleotide probe between the fifth and sixth bases, removing the fluorescent dye from the nucleic acid molecule to be sequenced. The whole process is repeated using a different sequence primer until all of the intervening positions in the sequence are imaged. The process allows the simultaneous reading of millions of DNA fragments in a "massively parallel” manner.
- This "sequence-by-ligation" technique uses probes that encode for two bases rather than just one, allowing error recognition by signal mismatching and leading to increased base determination accuracy.
- sequencing methods include sequencing by synthesis methods commercialized by 454/Roche Life Sciences including but not limited to the methods and apparatus described in Margulies et al., Nature (2005) 437:376-380 (2005); and U.S. Patent Nos. 7,244,559; 7,335,762; 7,211 ,390; 7,244,567; 7,264,929; and 7,323,305.
- double stranded fragment nucleic acid molecules can be prepared by the methods described herein, immobilized onto beads, and compartmentalized in a water-in-oil PCR emulsion.
- alternative amplification methods can be employed in the water-in-oil emulsion such as any of the methods provided herein.
- the methods can include a step of rendering the nucleic acid bound to the beads single stranded or partially single stranded.
- the beads can be enriched and loaded into wells of a fiber optic slide so that there is approximately 1 bead in each well.
- Nucleotides are flowed across and into the wells in a fixed order in the presence of polymerase, sulfhydrolase, and luciferase. Addition of nucleotides complementary to the target strand can result in a chemiluminescent signal that is recorded, such as by a camera. The combination of signal intensity and positional information generated across the plate allows software to determine the DNA sequence.
- double stranded fragment nucleic acid molecules can be isolated and purified, then immobilized onto a flow-cell surface.
- the methods can include a step of rendering the nucleic acid bound to the flow-cell surface stranded or partially single stranded.
- Polymerase and labeled nucleotides are then flowed over the immobilized DNA. After fluorescently labeled nucleotides are incorporated into the DNA strands by a DNA polymerase, the surface is illuminated with a laser, and an image is captured and processed to record single molecule incorporation events to produce sequence data.
- Other methods include sequencing by ligation methods commercialized by Dover Systems. Generally, oligonucleotides, primers, and probes can be prepared by the methods described herein. The nucleic acid molecules can then be amplified in an emulsion in the presence of magnetic beads. Any amplification methods can be employed in the water- in-oil emulsion.
- the resulting beads with immobilized clonal nucleic acid polonies are then purified by magnetic separation, capped, amine functionalized, and covalently immobilized in a series of flow cells.
- the methods can include a step of rendering the nucleic acid bound to the flow-cell surface stranded or partially single stranded.
- a series of anchor primers are flowed through the cell, where they hybridize to the synthetic oligonucleotide sequences at the 3' or 5' end of proximal or distal genomic DNA tags. Once an anchor primer is hybridized, a mixture of fully degenerate nonanucleotides ("nonamers”) and T4 DNA ligase is flowed into the cell.
- Each of the nonamer mixture's four components is labeled with one of four fluorophores, which correspond to the base type at the query position.
- the fluorophore- tagged nonamers selectively ligate onto the anchor primer, providing a fluorescent signal that identifies the corresponding base on the genomic DNA tag.
- the array is imaged in four colors. Each bead on the array will fluoresce in only one of the four images, indicating whether there is an A, C, G, or T at the position being queried.
- the array of annealed primer-fluorescent probe complex, as well as residual enzyme are chemically striped using guanidine HCl and sodium hydroxide.
- oligonucleotides, primers, and probes can be prepared by the methods described herein to produce amplified nucleic acid sequences tagged at one (e.g. , ( ⁇ )/( ⁇ ') or both ends (e.g., (A)I(A') and (C)/(C))-
- amplified nucleic acid sequences tagged at one or both ends is amplified by the methods described herein (e.g. , by SPIA or linear PCR).
- the resulting nucleic acid is then denatured and the single stranded amplified nucleic acid molecules are randomly attached to the inside surface of flow-cell channels. Unlabeled nucleotides are added to initiate solid-phase bridge amplification to produce dense clusters of double- stranded DNA.
- To initiate the first base sequencing cycle four labeled reversible terminators, primers, and DNA polymerase are added. After laser excitation, fluorescence from each cluster on the flow cell is imaged. The identity of the first base for each cluster is then recorded. Cycles of sequencing are performed to determine the fragment sequence one base at a time. For paired-end sequencing, such as for example, when the nucleic acid molecules are labeled at both ends by the methods described herein, sequencing templates can be regenerated in-situ so that the opposite end of the fragment can also be sequenced.
- Still other sequencing methods include those commercialized by Pacific
- oligonucleotides, primers and probes can be prepared by the methods described herein.
- Target nucleic acid molecules can then be immobilized in zero mode waveguide arrays.
- the methods may include a step of rendering the nucleic acid bound to the waveguide arrays single stranded or partially single stranded.
- Polymerase and labeled nucleotides are added in a reaction mixture, and nucleotide incorporations are visualized via fluorescent labels attached to the terminal phosphate groups of the nucleotides. The fluorescent labels are clipped off as part of the nucleotide incorporation.
- circular templates are utilized to enable multiple reads on a single molecule.
- a nanopore can be a small hole of the order of one nanometer in diameter. Immersion of a nanopore in a conducting fluid and application of a potential across it can result in a slight electrical current due to conduction of ions through the nanopore. The amount of current that flows is sensitive to the size of the nanopore. As a DNA molecule passes through a nanopore, each nucleotide on the DNA molecule obstructs the nanopore to a different degree. Thus, the change in the current passing through the nanopore as the DNA molecule passes through the nanopore can represent a reading of the DNA sequence.
- Ion Torrent e.g., using the Ion Personal Genome Machine (PGM)
- Ion Torrent technology can use a semiconductor chip with multiple layers, e.g. , a layer with micro-machined wells, an ion-sensitive layer, and an ion sensor layer.
- Nucleic acids can be introduced into the wells, e.g. , a clonal population of single nucleic can be attached to a single bead, and the bead can be introduced into a well.
- one type of deoxyribonucleotide e.g.
- dATP, dCTP, dGTP, or dTTP can be introduced into the wells.
- protons hydrogen ions
- the semiconductor chip can then be washed and the process can be repeated with a different deoxyribonucleotide.
- a plurality of nucleic acids can be sequenced in the wells of a semiconductor chip.
- the semiconductor chip can comprise chemical-sensitive field effect transistor (chemFET) arrays to sequence DNA (for example, as described in U.S. Patent Application Publication No. 20090026082). Incorporation of one or more triphosphates into a new nucleic acid strand at the 3' end of the sequencing primer can be detected by a change in current by a chemFET.
- An array can have multiple chemFET sensors.
- Example 1 Changes in DNA methylation detected by targeted bisulfite sequencing
- a method to specifically capture an arbitrary subset of genomic targets for single-molecule bisulfite sequencing and digital quantification of DNA methylation at single- nucleotide resolution is presented herein.
- a set of -30,000 padlock probes was designed to assess the methylation state of -66,000 CpG sites within 2,020 CpG islands on human chromosome 12, chromosome 20, and 34 selected regions.
- iPS Induced pluripotent stem
- Padlock probes have been previously used for exon capture and resequencing (Porreca, G.J. et al. (2007) Nat. Methods 4: 931 -936).
- This approach to targeted bisulfite sequencing involves the in situ synthesis of long ( ⁇ 150 nt) oligonucleotides on programmable microarrays, followed by their cleavage and enzymatic conversion into padlock probes.
- a library of padlock probes was annealed to the template DNA, circularized, and amplified by PCR before shotgun sequencing (Figure 5A-5C). There are, however, two major challenges in performing padlock capture for bisulfite sequencing.
- a probe design algorithm was developed to search for an optimal set of padlock probes covering an arbitrary set of non-repetitive genomic targets. This algorithm weights candidate probes based on several sequence features that were previously not considered in eMIP probe design, including the melting temperature, size and word statistics (distribution of 12-mers in the bisulfite-converted genome) of the capturing arms, and gap sizes.
- the capturing arms contain one or more CpG dinucleotides
- oligonucleotides (-150 nt) were synthesized by ink-jet printing on a programmable microarray and released (Agilent Technologies). The estimated total yield is 10 frnol per library.
- PCR amplification was performed in 32-96 reactions (100 ⁇ each) with 0.1 nM template oligonucleotides, 200 ⁇ dNTPs, 400 nM Apl V4IU primer, 400 nM Ap2V4 primer, 0.8x SybrGreen I, 36 units JumpStart Tag polymerase in l x
- JumpStart buffer (Sigma), at 94°C for 2 minutes, 22 cycles of 94°C for 30 seconds, 55°C for 2 minutes, 72°C for 45 seconds and, finally, 72°C for 5 minutes.
- the amplicons were purified by either column purification (Zymo DNA Concentrator- 100 columns) or ethanol precipitation.
- Genomic DNA was extracted from frozen pellets of fibroblast, iPS or hES cells using Qiagen DNeasy columns and bisulfite converted with the Zymo DNA Methylation Gold Kit (Zymo Research).
- Padlock probes (60 nM) and 200 ng of bisulfite-converted genomic DNA were mixed in 10 ⁇ 1 x Ampligase Buffer (Epicentre), denatured at 95°C for 10 minutes, then hybridized at 55°C for 18 hours, after which 1 ⁇ gap-filling mix (200 ⁇ dNTPs, 2 U AmpliTaq Stoffel Fragment (ABI) and 0.5 units Ampligase (Epicentre) in 1 * Ampligase buffer) was added to the reaction.
- the amplicons of the expected size range (344-394 bp) were purified with 6% PAGE (6% TBE gel; Invitrogen).
- the fragmented DNA was column purified, and end repaired at 25°C for 45 minutes in 25- ⁇ 1 reactions containing 2.5 ⁇ 10x buffer, 2.5 ⁇ dNTP mix (2.5 mM each), 2.5 ⁇ ATP (10 mM), 1 ⁇ end-repair enzyme mix (Epicentre), and 15 ⁇ DNA.
- 2.5 ⁇ 10x buffer 2.5 ⁇ dNTP mix (2.5 mM each)
- 2.5 ⁇ ATP 2.5 mM
- 1 ⁇ end-repair enzyme mix Epicentre
- NEB QuickLigase Buffer
- Ligation products of 150-175 bp in size were size selected with 6% PAGE, and amplified by PCR in 100 ⁇ reactions with 15 ⁇ template, 200 n Solexa PCR primers, 0.8 x SybrGreen I and 50 ⁇ iProof High-Fidelity Master Mix (Bio-Rad) at 98°C for 30 seconds, 12 cycles of 98°C for 10 seconds, 65°C for 20 seconds, 72°C for 20 seconds and 72°C for 3 minutes.
- the PCR amplicons were purified with Qiaquick PCR purification columns, and sequenced on Illumina Genome Analyzer. All primer sequences are listed in Table 1.
- Table 3 List of 26 selected genes and 8 ENCODE regions
- BJHues6-Hybridl A line of hybrid stem cells (BJHues6-Hybridl), which were reprogrammed by fusing the human fibroblasts (BJ) with hES cells (Hues6), as well as three hES cell lines (Hues 12, Hues42, Hues63) were also characterized.
- Bisulfite conversion, padlock capture and construction of shotgun sequencing libraries were performed on each DNA sample. Each library in one lane was sequenced in the flow cell of an Illumina Genome Analyzer, and yielding 2-3 million reads that were mapped to the targeted regions. The bisulfite conversion rates were >98.5%. To avoid stochastic sampling drift, CpG sites that were covered by ⁇ 10 reads were removed from the following analyses.
- CpG islands are not defined in a functional manner, the CpG islands were divided into three categories.
- the first comprises CpG islands in the regions from 2 kb upstream to 500 bp downstream of TSS. These "upstream regions” often include promoter regions.
- the second class (“gene body CpG islands”) comprises CpG islands in the regions from 500 bp downstream of TSS to the ends of the last exons.
- the final category comprises CpG islands outside of gene body and promoter regions. CpG islands in each category were further divided into three groups according to CpG density.
- Padlock capture is more efficient than full-genome bisulfite sequencing (Cokus, S.J. et al. (2008) Nature 452: 215-219; Lister, R. et al. (2008) Cell 133: 523-536) for quantifying DNA methylation, as it allows for focused sequencing on the most informative genomic regions. It also provides a much greater flexibility than reduced representation bisulfite sequencing (Meissner, A. et al. (2008) Nature 454: 766-770; Ball, M.P. et al. (2009) Nat. Biotechnol. 27(4): 361 -8) in the selection of genomic targets, because the latter method is limited to genomic regions closely adjacent to the recognition sites of restriction enzymes.
- the program ppDesigner was developed to aid in the design of efficient padlock probes for bisulfite analysis. It accepts as input the genome of any organism, a list of user-specified arbitrary targets and user-desired probe constraints matching requirements of the experimental protocol. It 'bisulfite-converts' the genome in silico (that is, it changes all cytosines to thymines) and outputs padlock probes to cover the chosen targets while avoiding CpGs on the capturing arms that could be methylated and not converted to be recognized as thymine. ppDesigner uses a back-propagation neural network to predict probe efficiency (Figure 9). This network was previously trained using data from probes for exomic targets (Gore, A.
- ppDesigner can explain ⁇ 50% of the variance in capturing efficiency for genomic DNA and ⁇ 20% of the variance in capturing efficiency for bisulfite-converted DNA; additional variation could be due to factors such as variability in oligonucleotide synthesis and sample DNA quality. ppDesigner is extremely flexible and has been used to design a variety of genomic and bisulfite probes for Homo sapiens (Liu, G.H. et al.
- Table 4 herein shows the number of enzymatic reactions, number of purifications, cost per sample, and mapping rates for first-generation padlock probes, second-generation library-free padlock probes, reduced representation bisulfite sequencing (RRBS), and whole genome bisulfite sequencing (WGBS).
- size selection is typically performed on 48-96 pooled libraries.
- the library preparation cost (including probes) with our protocol was comparable to that of the restricted-representation bisulfite sequencing and whole-genome bisulfite sequencing protocols, and the sequencing cost was much lower than that of whole-genome bisulfite sequencing owing to targeting of CpG sites of interest.
- Restricted-representation bisulfite sequencing is more cost-effective than BSPPs, but the former lacks BSPPs' flexibility in selecting specific sites or regions.
- bisReadMapper Another bottleneck in sequencing of bisulfite-converted DNA is a lack of computational tools to efficiently analyze sequencing data generated from hundreds of samples.
- an analysis pipeline for read mapping and methylation quantification was developed, called bisReadMapper.
- reads had been mapped only against target regions owing to the computational requirements of sequence alignment (Deng, J. et al. (2009) Nat. Biotechnol. 27: 353-360).
- bisReadMapper was designed to map to the full genome sequence, allowing processing of data from both targeted and whole-genome sequencing of bisulfite-con verted DNA.
- bisReadMapper also determines the origin strand of the read based on base composition and maps reads as if they were fully bisulfite-converted to a fully bisulfite-converted genome sequence, allowing mapping of both bi- and unidirectional bisulfite libraries in an unbiased manner. Another feature is the capability to call single-nucleotide polymorphisms (SNPs) from sequences of bisulfite-converted DNA; this feature not only allows for analysis of allele-specific methylation (Shoemaker, R. et al. (2010) Genome Res. 20: 883-889) but also allows accurate sample tracking in large-scale experiments. Finally, bisReadMapper can call methylation levels at both CpG and non-CpG sites.
- SNPs single-nucleotide polymorphisms
- oligonucleotides ( ⁇ 150 nt) were synthesized by ink-jet printing on programmable microarrays (Agilent Technologies) and released to form a combined library of 330,000 oligonucleotides.
- oligonucleotides were amplified by PCR in 96 reactions (100 ⁇ each) with 0.02 nM template oligonucleotide, 400 nM each of pAP l V61 U primer and AP2V6 primer (Table 6), and 50 ⁇ of APA SYBG fast Universal 2* qPCR Master Mix (Kapabiosystems) at 95°C for 30 seconds, 15-16 cycles of 95°C for 3 seconds; 55°C for 30 seconds; and 60°C for 20 seconds and 60°C for 2 minutes.
- Table 6 Primer sequences used for padlock probe production, padlock capture, sequencing library construction, and Ulumina sequencing
- the amplicons were purified by ethanol precipitation and repurified with Qiaquick PCR purification columns (Qiagen). Approximately 20 ⁇ of the purified amplicons were digested with 50 units of lambda exonuclease (5 U/ ⁇ ; New England Biolabs (NEB)) at 37°C for 1 hour in lambda exonuclease reaction buffer. The resulting single-strand amplicons were purified with Qiaquick PCR purification column. Approximately 5-8 ⁇ g of single strand amplicons were subsequently digested with 5 units USER ( 1 U ⁇ - ⁇ , NEB) at 37°C for 1 hour.
- the digested DNA was annealed to 5.88 ⁇ KE-Dp>iU-V6 guide oligo (Table 6) and denatured at 94°C for 2 minutes decreased the temperature to 37°C and incubated at 37 °C for 3 minutes.
- the mixture was digested with 50 U of Dpnll (10 U ⁇ - ⁇ , NEB) in NEBuffer DpnW at 37°C for 2 hours.
- the mixture was further digested with 5 U of USER at 37°C for 2 hours followed by enzyme inactivation at 75°C for 20 minutes.
- the USER and Z /wIi-digested DNA was purified with Qiaquick PCR purification column.
- the single-strand 102-nt probes were purified with 6% denaturing PAGE (6% TB-urea two dimensional (2D) gel; Invitrogen).
- the oligonucleotides (100 nt) were synthesized using a programmable microfluidic microarray platform (LC Sciences) and released to form a mix of 3,91 8 oligonucleotides.
- the oligonucleotides were amplified by two-step PCR in a 200 ⁇ reaction with 1 nM template oligonucleotides, 400 nM each of eMIP_CA l_F primer and
- eMIP_CAl_R primer (Table 6), and 100 ⁇ of KAPA SYBR fast Universal qPCR Master Mix at 95°C for 30 seconds, 5 cycles of 95°C for 5 seconds; 52°C for 1 minute; and 72°C for 30 seconds, 10-12 cycles of 95°C for 5 seconds; 60°C for 30 seconds; and 72°C for 30 seconds, and 72°C for 2 minutes.
- the resultant amplicons were purified with Qiaquick PCR purification columns and re-amplified in 32 PCRs (100 ⁇ each) with 0.02 nM first round amplicons, 400 nM each of eMIP CA l F primer and eMIP_CAl_R primer and 50 ⁇ of KAPA SYBR fast Universal qPCR master mix at 95°C for 30 seconds, 1 3-15 cycles of 95°C for 5 seconds; 60°C for 30 seconds; and 72°C for 30 seconds and 72°C for 2 minutes.
- the resultant amplicons were purified by ethanol precipitation and repurified with Qiaquick PGR purification columns as described above.
- Approximately 4 ng of the purified amplicons were digested with 100 U of Nt .AM (100 U/ ⁇ , NEB) at 37°C for 1 hour in NEBuffer 2. The enzyme was heat-inactivated at 80°C for 20 minutes. The digested amplicons were then incubated with 100 U of Nb.5riDI (10 U ⁇ - ⁇ , NEB) at 65°C for 1 hour. The nicked DNA was purified by Qiaquick PCR purification column. The probe molecules ( ⁇ 70 bases) were purified by 6% denaturing PAGE (6% TB-urea 2D gel).
- Genomic DNA was extracted using the AllPrep DNA/RNA Mini kit (Qiagen) and bisulfite-converted with the EZ-96 DNA methylation Gold kit (Zymoresearch) in a 96- well plate. Normalized amount of padlock probes, 200 ng of bisulfite converted gDNA and 4.2 nM oligo suppressor were mixed in 25 ⁇ 1 * Ampligase buffer (Epicentre) in 96-well plate, denatured at 95°C for 10 minutes, gradually lowered the temperature at 0.02°C/s to 55°C in a thermocycler and hybridized at 55°C for 20 hours.
- Ampligase buffer Epicentre
- exonuclease mix (10 U/ ⁇ exonuclease 1 and 100 U/ ⁇ exonuclease III, USB) was added to the reactions, and the reactions were incubated at 37°C for 2 hours and then inactivated at 94°C for 2 minutes.
- Indl 21 ACCAAT CAAGCAGAAGACGGCATACGAGATACCAATGCTAGGAACGATGAGCCT 156
- CTCCGT CAAGCAGAAGACGGCATACGAGATCTCCGTGCTAGGAACGATGAGCCT 267
- CAGCCA CAAGCAGAAGACGGCATACGAGATCAGCCAGCTAGGAACGATGAGCCT 365
- AAACCT CAAGCAGAAGACGGCATACGAGATAAACCTGCTAGGAACGATGAGCCT 376
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Methods, systems, and computer programs for designing probes or primers for nucleic acid sequencing, generating libraries of nucleic acid sequences, and mapping genomic sequences are provided herein,
Description
DESIGNING PADLOCK PROBES FOR TARGETED GENOMIC
SEQUENCING
SEQUENCE LISTING
[0001] The instant application contains a Sequence Listing which has been submitted in ASCII format via EFS-Web and is hereby incorporated by reference in its entirety. Said ASCII copy, created on April 26, 2012, is named 378665WO-l .txt and is 99, 190 bytes in size.
TECHNICAL FIELD
[0002] The subject matter described herein relates generally to the fields of molecular biology and bioinformatics. More specifically, the subject matter described herein relates to systems, methods, and computer programs for designing probes for use in nucleic acid sequencing, particularly padlock probes useful in carrying out targeted genomic and methylation sequencing.
BACKGROUND
[0003] Padlock Probe (PP) technology is a multiplex genomic enrichment method allowing for accurate targeted high-throughput sequencing. PP technology has been used to perform highly multiplexed genotyping, digital allele quantification, targeted bisulfite sequencing, and exome sequencing. See Hardenbol, P. et al., (2005) Genome Res. 15: 269-275; Wang, Y. et al., (2005) Nucleic Acids Res., 2005, 33(21) el 83: 1 -14; Porreca, G.J. et al., (2007) Nat. Methods 4(1 1): 931 -936; Zhang, K. et al., (2009) Nat. Methods 6: 613-618; Turner, E.H. et al., (2009) Nat. Methods 6: 315-316; Deng, J. et al., (2009) Nat. Biotechnol. 27: 353-360; Shoemaker, R. et al., (2010) Genome Res. 20: 883-889, and Gore, A. et al., (201 1) Nature 471 (7336): 63-67.
[0004] Padlock probe technology may utilize a linear oligonucleotide molecule with two binding sequences at each end joined by a common linker sequence. The probe's binding arms may be hybridized several base pairs apart surrounding a target single-stranded genomic DNA region. A DNA polymerase (with each of the four standard dNTP molecules) may be used to fill in the gap between the two binding arms, and a DNA ligase may be used to circularize the resulting molecule. A mixture of exonucleases may then used to digest all
arm; the resulting circular molecules can be amplified using rolling circle amplification or polymerase chain reaction to generate a DNA library compatible with modem high- throughput sequencers.
fOOOS] Previous padlock probe work has relied on relatively arbitrary methods for padlock probe design. Padlock probes are generally designed to have binding arms with
complementary sequence around a chosen target region of approximately 100-200 base pairs; each binding arm is generally designed to have a specific DNA melting temperature.
However, padlock probe efficacy from molecule to molecule has been found to vary greatly, ranging across several orders of magnitude. Previous work identified a complex nonlinear relationship between padlock probe efficiency and many probe characteristics. (Deng, J. et al., (2009) Nat. Biotechnol. 27: 353-360; Li, J.B. et al., (2009) Genome Res. 19(9): 1606- 1615). The bias inherent in padlock probe design has been demonstrated to show a complex nonlinear relationship with many probe characteristics, confounding attempts to generate efficient probes. Because of this, padlock probe results are highly biased towards certain genomic regions. Certain genomic regions are therefore extremely difficult to target using padlock probes due to the lack of knowledge of probe capturing efficiency.
[0006] There remains a need in the art for methods and algorithms that are useful for designing probes from a large set of probe characteristics to ensure optimal capture of DNA sequences, particularly genomic DNA.
SUMMARY
[0007] The subject matter described herein relates to methods, systems, and computer program products that can be used to design oligonucleotide primers and probes, particularly padlock probes for high-density multiplex genome sequencing. The subject matter disclosed herein is particularly useful for detecting methylation status and/or single nucleotide variants in a target nucleic acid sequence of interest.
[0008] Accordingly, in some aspects, the subject matter described herein provides a method of designing probes or primers for sequencing a target nucleic acid molecule, comprising the steps of selecting one or more inputs associated with efficiency of the probe or primer;
selecting a target nucleic acid sequence; generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence; determining the efficiency of each probe or primer sequence in the
first library by using an algorithm comprising the one or more selected inputs defined herein; ranking the probe or primer sequences in the first library by efficiency; extracting the probe or primer sequences having the highest efficiency to generate a second library; and adding a linker sequence to each of the probe or primer sequences in the second library. In some embodiments, the method further comprises synthesizing the probe or primer.
[0009] In some embodiments, the probe is a padlock probe. The one or more inputs may comprise target length, target folding energy, target GC content, extension arm A%, extension arm G%, target A%, target T%, target G%, number of "GG" dinucleotides in ligation arm, number of "AT" dinucleotides in extension arm, number of "GG" dinucleotides in extension arm, number of "AA" dinucleotides in target, number of "AT" dinucleotides in target, number of "TA" dinucleotides in target, number of "GT" dinucleotides in target, number of "GA" dinucleotides in target, ligation arm terminal dinucleotide, extension arm terminal dinucleotide, target 5' terminal dinucleotide, ligation arm melting temperature, extension arm melting temperature, ligation arm length, extension arm length, local single- stranded folding energy of the target, and dinucleotides present at the extension site and ligation site during probe capture.
[0010] In some embodiments, the target nucleic acid sequence is derived from a human.
[0011] The target-capturing sequence may include a ligation arm and an extension arm. In some embodiments, the target-capturing sequence contains one or more CpG dinucleotides. The target-capturing sequences in the first library may also contain all possible methylation state combinations of the one or more CpG dinucleotides. In some embodiments, the extension arm comprises one or more priming sites for amplification of the target nucleic acid sequence and may be universal priming sites. The target capturing sequence may also include one or more restriction sites.
[0012] The methods disclosed herein involve an algorithm that may comprise one or more neural networks. The one or more neural networks may comprise the one or more inputs, e.g., seven or more inputs.
[0013] In certain embodiments, the method further comprises, after the extracting step, pooling the non-extracted probe or primer sequences and repeating certain steps defined herein.
[0014] In some embodiments, the linker sequence is a sequence that is common to each probe or primer sequence in the second library.
[0015] In another aspect, an apparatus is provided, comprising at least one processor and at least one memory including code which when executed by the at least one processor provides operations comprising: selecting one or more inputs associated with efficiency of the probe or primer; selecting a target nucleic acid sequence; generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence; determining the efficiency of each probe or primer sequence in the first library by using an algorithm comprising the one or more selected inputs; ranking the probe or primer sequences in the first library by efficiency; extracting the probe or primer sequences having the highest efficiency to generate a second library; and adding a linker sequence to each of the probe or primer sequences in the second library.
[0016] In another aspect of the subject matter described herein, a computer-readable storage medium including code is provided, which when executed by at least one processor provides operations comprising: selecting one or more inputs associated with efficiency of the probe or primer; selecting a target nucleic acid sequence; generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence; determining the efficiency of each probe or primer sequence in the first library by using an algorithm comprising the one or more selected inputs; ranking the probe or primer sequences in the first library by efficiency; extracting the probe or primer sequences having the highest efficiency to generate a second library; and adding a linker sequence to each of the probe or primer sequences in the second library.
[0017] Other features and advantages of the subject matter described herein will be apparent from the following detailed description and claims.
BRIEF DESCRIPTION OF THE DRAWINGS
[0018] The following Detailed Description, given by way of example, but not intended to limit the subject matter described herein to specific embodiments described, may be understood in conjunction with the accompanying figures, incorporated herein by reference, in which:
[0019] Figure 1 is a block diagram of a single padlock probe capture experiment, demonstrating an example of a workflow for targeted genomic resequencing consistent with some of the exemplary embodiments described herein;
[0020| Figure 2 is a flowchart of a process for designing padlock probes from input files consistent with some of the exemplary embodiments described herein;
[0021] Figure 3 is a block diagram of a back propagation neural network used to derive the probe efficiency scoring equation consistent with some of the exemplary embodiments described herein;
[0022] Figure 4 is a depiction of two examples of customized linker sequences and the function provided thereby, each of which is consistent with some of the exemplary embodiments described herein. Figure 4 discloses SEQ ID NOS 420-424, respectively, in order of appearance;
[0023] Figures 5Λ-5Ε depicts the design of padlock probes for targeted bisulfite sequencing. (A) is a diagram showing that each padlock probe has a common linker sequence flanked by two target-specific capturing arms (HI and H2). H I and H2 are melting temperature normalized, and a spacer sequence is included to normalize probe lengths. The linker sequence contains priming sites (API and AP2) for universal primers, two Mmel sites and a central Alul recognition site. (B) is a diagram depicting a CpG island (or other target region) covered by multiple padlock probes targeting partially overlapped regions on alternating strands. (C) is a diagram showing a library of padlock probes annealed to bisulfite-converted genomic DNA. (D) is a flow chart showing the generation of a shotgun sequencing library. (E) is a picture showing gel electrophoresis analysis of the padlock-captured products from two independent capturing reactions (1 and 2) and a no-template control (NTC).
[0024] Figures 6A-6G are graphs summarizing an analysis of the effect of probe
characteristics on capturing efficiency. The statistical package R and its effects module were used for this analysis. A linear model was used, and each individual factor was assumed to be independent. The dashed lines represent a 95% confidence interval. (A) High GC content in the ligation arm was found to increase probe capturing efficiency. (B) Longer ligation arms were found to capture probes with higher efficiency than short ligation arms. (C) The frequency of 12-mers within the ligation arm in the human genome did hot have a statistically significant effect on probe efficiency. (D) GC content of the extension arm did not have a statistically significant effect on probe efficiency. (E) Extension arm length did not have a statistically significant effect on probe efficiency; the trend was that a longer extension arm would be better. (F) The frequency of 12-mers within the extension arm in the human
genome did not have a statistically significant effect on probe efficiency. (G) Shorter target sequences were captured with higher efficiency than long target sequences.
[0025] Figures 7A-7D depict experimental normalization of padlock-capturing efficiency. (A) shows the "subsetting" strategy. (B) depicts the 'suppressor oligo' strategy. (C) shows the distribution of normalized abundance for all captured targets with one 30,000-probe set and with four probe sets. The x-axis is the normalized abundance of each captured target, which is calculated by dividing the counts of the target by the average counts of all targets. The y-axis is the fraction of probes with the coverage equal to or greater than the normalized coverage. (D) Comparison of relative abundance for each target before and after
normalization. The vertical dash lines indicate the clear separation of four subsets of targets, as well as the fifth set normalized with the suppressor oligos.
[0026J Figures 8A-8C show the results of a validation experiment demonstrating the digital methylation assay. (A) Comparison of methylation measurements from both strands for the same CpG sites. The methylation levels of the forward strand were plotted against the levels of the reverse strand on 2697 CpG sites that were covered by on both strands by different probes. (B) Methylation levels of 182 randomly selected CpG sites in the B J fibroblast lines were measured by the conventional bisulfite Sanger sequencing. (C) Comparison of the methylation levels of 25,665 CpG sites (at least 50x sequencing reads per site) between two biological replicates on the IMR90 fibroblasts.
[0027J Figure 9 is a schematic for the probe design software (ppDesigner).
[0028] Figure 10 are graphs comparing probe capture efficiencies between the DMR220K, LC4K probe sets and the CGI30K set. The first three plots were generated from data without subsetting or suppressor oligos to allow for a direct comparison of probe design.
[0029] Figure 1 1 is a scatter plot of number of characterized CpG sites versus mappable sequencing data for the DMR330K probe set. Variability in sequencing quality of individual sequencing runs is responsible for the different number of CpG sites characterized with similar sequencing effort.
[0030] Figure 12 is a graph showing the number of CpG sites called per sample as a function of sequencing effort. The horizontal dash line represents 4Gbps of sequences per library.
[0031] Figures 13A-13B are graphs showing captured CpG sites that were tested for potential regulatory interactions with genes by GREAT. (A) Most CpG sites were interacting with 1-2
genes. (B) Distance of CpG sites to the transcriptional start sites (TSS) of the predicted regulating genes.
[0032] Figures 14A-14E are graphs showing the accuracy of digital quantification by BSPP. (A, B) show a comparison of the methylation levels obtained at lOx depth from multiple capture reactions of the same sample (PGP HPS) within batches and between batches. (C, D, E) Within sample comparison of methylation levels obtained from different probes capturing the same CpG site on different strands at 1 Ox depth within one capture reaction.
[0033] Figure 15 is a graph showing the comparison between BSPP and whole genome bisulfite sequencing (WGBS). Two HI ESC datasets were compared, using sites with at least lOx read depth in each.
[0034] Figure 16 is a graph depicting the variation in amount of sequencing data obtained per sample in a multiplexed BSPP capture experiment. Forty-eight whole blood samples were captured and sequenced in one batch using the library-free BSPP method.
[0035] Figure 17 depicts exemplary padlock probes ordered from (A) Agilent's
oligonucleotide synthesis service (SEQ ID NOS 425-427, respectively, in order of appearance) and (B) LC Sciences' oligonucleotide synthesis service (SEQ ID NOS 428-430, respectively, in order of appearance).
[0036] Figure 18 is a diagram depicting the addition of a second neural network specifically for bisulfite-con verted DNA. This network contains two hidden layers with 10 and 12 nodes, respectively, and accepts 25 pieces of information as input.
DETAILED DESCRIPTION
[0037] The features, structures, or characteristics described throughout this specification may be combined in any suitable manner in one or more embodiments. For example, the usage of the phrases "exemplary embodiments," "example embodiments," "some embodiments," or other similar language, throughout this specification refers to the fact that a particular feature, structure, or characteristic described in connection with an embodiment may be included in at least one embodiment described herein. Thus, appearances of the phrases "exemplary embodiments," "example embodiments," "in some embodiments," "in other embodiments," or other similar language, throughout this specification do not necessarily all refer to the same group of embodiments, and the described features, structures, or characteristics can be combined in any suitable manner in one or more embodiments.
[0038] To facilitate the understanding of this disclosure, a number of terms are defined below. Tenns defined herein have meanings as commonly understood by a person of ordinary skill in the areas relevant to the subject matter described herein. Terms such as "a", "an" and "the" are not intended to refer to only a singular entity, but include the general class of which a specific example may be used for illustration. The terminology herein is used to describe specific embodiments of the subject matter described herein, but their usage does not delimit the subject matter, except as outlined in the claims.
[0039] The term "read" refers to a nucleic acid sequence of sufficient length (e.g. , at least about 30 bp) that can be used to identify a larger sequence or region, e.g. that can be aligned and specifically assigned to a chromosome or genomic region or gene.
[0040] As used herein, the terms "aligned", "alignment", or "aligning" refer to one or more sequences that are identified as a match in terms of the order of their nucleic acid molecules to a known sequence from a reference genome. Such alignment can be done manually or by a computer algorithm. Examples include, without limitation, the Efficient Local Alignment of Nucleotide Data (ELAND) computer program distributed as part of the Illumina Genomics Analysis, Bowtie, BWA, and SOAP2Align. The matching of a sequence read in aligning can be a 100% sequence match or less than 100% (non-perfect match).
[0041] "Amplification" or "amplifying" methods include but are not limited to the polymerase chain reaction (PCR), the ligase chain reaction (LCR) (e.g., Wu, D.Y. and Wallace, R.B. (1989) Genomics 4: 560-569; Landegren, U. et al., (1998) Science 241 (4869): 1077-1080; and Barringer, K.J. et al. (1990) Gene 89(1): 1 17-122), transcription
amplification (Kwoh, D.Y. et al., (1989) Proc. Natl Acad. Sci. 86(4): 1 173-1 177 and WO88/10315), self-sustained sequence replication (Guatelli, J.C. et al., (1990) Proc. Nat. Acad. Sci. 87(5): 1874-1878 and WO90/06995), selective amplification of target polynucleotide sequences (U.S. Patent No. 6,410,276), consensus sequence primed polymerase chain reaction (CP-PCR) (U.S. Patent No. 4,437,975), arbitrarily primed polymerase chain reaction (AP-PCR) (U.S. Patent Nos. 5,413,909, 5,861 ,245) and nucleic acid based sequence amplification (NABSA; U.S. Patent Nos. 5,409,818, 5,554,517, and 6,063,603 each of which is incorporated herein by reference). Other amplification methods that may be used are described in U.S. Patent Nos. 5,242,794, 5,494,810, 4,988,617.
Additional methods of sample preparation and techniques for reducing the complexity of a nucleic sample are described in Dong, S. et al., (2001) Genome Res. 1 1 (8): 1418-1424, in
U.S. Patent Nos. 6,361 ,947, 6,391 ,592 and U.S. Patent Application Ser. Nos. 09/916,135, 09/920,491 , 09/910,292, and 10/013,598.
[0042] A "nucleotide" is a monomer that includes a base, such as a pyrimidine, purine, or synthetic analogs thereof, linked to a sugar and one or more phosphate groups. Nucleotides include adenine (A) residues, guanine (G) residues, cytosine (C) residues, thymine (T) residues, and uracil (U) residues. The major nucleotides of DNA are deoxyadenosine 5'- triphosphate (dATP or A), deoxyguanosine 5'-triphosphate (dGTP or G), deoxycytidine 5'- triphosphate (dCTP or C) and deoxythymidine 5'-triphosphate (dTTP or T). The major nucleotides of RNA are adenosine '-triphosphate (ATP or A), guanosine 5 '-triphosphate (GTP or G), cytidine 5 '-triphosphate (CTP or C) and uridine 5'-triphosphate (UTP or U). Nucleotides also include chemical entities containing modified bases, modified sugar moieties and modified phosphate backbones, for example as described in U.S. Patent No. 5,866,336. Such modifications however, can allow for incorporation of the nucleotide into a growing nucleic acid chain or for binding of the nucleotide to the complementary nucleic acid chain.
[0043] Nucleotides can be modified at any position on their structures. Examples include, but are not limited to, the modified nucleotides 5-fluorouracil, 5-bromouracil, 5-chlorouracil, 5-iodouracil, hypoxanthine, xanthine, acetylcytosine, 5-(carboxyhydroxylmethyl) uracil, 5- carboxymethylaminomethyl-2-thiouridine, 5-carboxymethylaminomethyluracil,
dihydrouracil, beta-D-galactosylqueosine, inosine, N-6-sopentenyl adenine, 1 -methylguanine, 1 -methylinosine, 2,2-dimethylguanine, 2-methyladenine, 2-methylguanine, 3-methylcytosine, 5-methylcytosine, N6-adenine, 7-methylguanine, 5-methylaminomethyluracil,
methoxyarninomethyl-2-thiouracil, beta-D-mannosylqueosine, 5'- methoxycarboxymethyluracil, 5-methoxyuracil, 2-methylthio-N6-isopentenyladenine, uracil- 5-oxyacetic acid, pseudouracil, queosine, 2-thiocytosine, 5-methyl-2-thiouracil, 2-thiouracil, 4-thiouracil, 5-methyluracil, uracil-5-oxyacetic acid methylester, uracil-S-oxyacetic acid, 5- methyl-2-thiouracil, 3-(3-amino-3-N-2-carboxypropyl) uracil, and 2,6-diaminopurinc.
[0044] Examples of modified sugar moieties which can be used to modify nucleotides at any position on their structures include, but are not limited to: arabinose, 2-fluoroarabinose, xylose, and hexose, or a modified component of the phosphate backbone, such as phosphorothioate, a phosphorodithioate, a phosphoramidothioate, a pliosphoramidate, a
phosphordiamidate, a methylphosphonate, an alkyl phosphotriester, or a formacetal or analog thereof.
[0045] As used herein the terms "nucleic acid", "nucleic acid molecule", "polynucleotide" and "oligonucleotide" are used interchangeably and refers to the polymeric form of nucleotides, cither ribonucleotides and/or deoxyribonucleotides or a modified form of either type of nucleotide. Nucleic acids include, without limitation, cDNA, niRNA, genomic DNA, and synthetic (such as chemically synthesized) DNA or RNA, plasmids, amplicons, cosmids, and fragments thereof. The nucleic acid can be double stranded (ds) or single stranded (ss). Where single stranded, the nucleic acid molecule can be the sense strand or the antisense strand. Nucleic acids can include natural nucleotides (such as adenine, thymine/uracil, cytosine, and guanine) and can also include analogs of natural nucleotides. A set of bases linked to a peptide backbone, as in a peptide nucleic acid (PNA), can be used as a substitute for a nucleic acid molecule. Nucleic acids can be modified by means of a fluorophore that is directly or indirectly excitable. "Fluorescent DNA dye" as used herein may refer to a composition, for example SYBR Green I or SYBR Gold that becomes fluorescently excitable when it associates with double-stranded DNA. Other examples of fluorescent DNA dyes are known in the art and include, e.g., 5 -carboxy fluorescein, 2'7'-dimethoxy-4'5,-dichloro-6- carboxyfluorescein, fluorescein (FL); N,N,N',N'-tetramethyl-6-carboxyrhodamine; 6- carboxy-X-rhodamine; CY3; CY5; tetrachloro-fluorescein; and hexachloro-fluorescein; NED; 6-FAM; VIC; PET; LIZ, SID, TED, and TAZ.
[0046] A "target" nucleic acid molecule is a nucleic acid to be sequenced, identified, or detected, and can be obtained or isolated in purified form, by any method known to those skilled in the art (for example, as described in U.S. Patent No. 5,674,743), but need not be in purified form. Various other biomolecules can also be present with the target nucleic acid molecule. For example, the target nucleic acid molecule can be present in a cell or a biological sample (which can include other nucleic acid molecules and proteins). The target nucleic acid molecule may be a whole genome or a portion of a genome, such as
chromosomal sequences, or may be extrachromosomal sequences such as plasmids. The target nucleic acid molecule can be derived from any species, including but not limited to, vertebrates such as humans, cows, dogs, cats, mice, rats, sheep, horse, goat, invertebrates, bacteria, viruses, fungi, and the like. The target nucleic acid may be derived from any source, including tissues, primary cells, cultured cells, cell lines, tumor specimens, bodily fluids, and
the like. The target nucleic acid may also be wholly or partly synthetic. A "complementary" nucleic acid molecule is complementary to the target nucleic acid molecule and is the nucleic acid strand that is elongated when sequencing the target nucleic acid molecule.
[0047J An "oligonucleotide" refers to a linear nucleic acid molecule (such as DNA or R A) sequence of at least 6 nucleotides, for example at least 9, at least 15, at least 18, at least 24, at least 30, at least 50, at least 100, at least 200 or even at least 500 nucleotides long. However shorter or longer oligonucleotides may be used. Oligonucleotides may be designed to have different length. In some embodiments, the sequence of the nucleic acid molecule may be divided up into a plurality of shorter sequences that can be synthesized in parallel and assembled into a single or a plurality of desired nucleic acid molecules using the methods described herein. In certain embodiments, the oligonucleotides are designed to provide the full sense and antisense strands of the nucleic acid molecule. After hybridization of the plus and minus strand oligonucleotides, two double stranded oligonucleotides are subjected to ligation or polymerization in order to form a first subassembly product. Subassembly products are then subjected to ligation or polymerization to form a larger DNA or the full DNA sequence.
[0048] A "primer" is a short nucleic acid molecule, for example sequences of at least 9 nucleotides, which can be annealed to a complementary target nucleic acid molecule by nucleic acid hybridization to form a hybrid between the primer and the target nucleic acid strand. A primer can be extended along the target nucleic acid molecule by a polymerase enzyme. Therefore, individual primers can be used for nucleic acid sequencing, wherein the sequence of the primer is specific for the target nucleic acid molecule, for example so that the primer will hybridize to the target nucleic acid molecule under stringent hybridization conditions. In particular examples, a primer is at least 10 nucleotides in length, such as at least 10 contiguous nucleotides complementary to a target nucleic acid molecule to be sequenced. In order to enhance specificity, longer primers can be employed, such as primers having at least 12, at least 15, at least 20, or at least 30 contiguous nucleotides
complementary to a target nucleic acid molecule to be sequenced. Methods for preparing and using primers are described in, for example, Sambrook et al. (1989) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor, NY; Ausubel et al. (1987) Current Protocols in Molecular Biology, Greene Publ. Assoc. & Wiley-Intersciences.
[0049] As used herein a "probe" refers to an oligonucleotide sequence that may or may not be extended in the amplification reaction by a DNA polymerase. Probes that are very specific for a perfectly complementary target sequence and strongly reject closely related sequences having one or a few mismatched bases are known in the art as "allele
discriminating". Probes that hybridize under at least one applicable detection condition not only to perfectly complementary sequences, but also to partially complementary sequences having one or more mismatched bases, are "mismatch tolerant" probes.
[0050] As used herein an "oligonucleotide set", "primer set" or "probe set" refers to a collection of primers or primers and probes for performing amplification or sequencing reactions. Another analogous term is "oligonucleotide library", "primer library", or "probe library". In some embodiments, methods of assembling libraries containing nucleic acids, primers, and probes having predetermined sequence variations are provided herein.
Assembly strategies provided herein can be used to generate very large libraries
representative of many different nucleic acid probe sequences of interest. In some embodiments, libraries of nucleic acids are libraries of sequence variants. Sequence variants may be variants of a single naturally-occurring sequence. However, in some embodiments, sequence variants may be variants of a plurality of different sequences. A high-density nucleic acid library may include more than 100 different sequence variants (e.g. , about 102 to 103; about 103 to 104; about 104 to 105; about 10s to 106; about 106 to 107; about 107 to 108; about 108 to 109; about 109 to 1010; about 1010 to 10"; about 10" to 1012; about 1012 to 1013; about 1013 to 1014; about 1014 to 1015; or more different sequences) wherein a percentage of the different sequences are specified sequences as opposed to random sequences (e.g. , more than about 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 91 , 92, 93, 94, 95, 96, 97, 98, 99% of the sequences are predetermined sequences of interest). In some embodiments, the libraries may contain information obtained from sequencing reactions, such as target nucleic acid sequences, sequence reads, sequence alignments, bisulfite-converted sequences, methylation frequencies, allele frequencies, single nucleotide variants or polymorphisms, among others. Libraries may be stored or kept on high-density arrays, microarrays, microchips, computer-readable media, or by any method or apparatus known in the art. <-
[0051] In some embodiments, the oligonucleotides, primers, and probes may comprise universal (common to all oligonucleotides), semi-universal (common to at least of portion of
the oligonucleotides) or individual or unique primer (specific to each oligonucleotide) binding sites (also referred to herein as "priming sites") on either the 5' end or the 3' end or both. As used herein, the term "universal" primer or primer binding site means that a sequence used to amplify the oligonucleotide is common to all oligonucleotides such that all such oligonucleotides can be amplified using a single set of universal primers. In other circumstances, an oligonucleotide contains a unique primer binding site. As used herein, the term "unique primer binding site" refers to a set of primer recognition sequences that selectively amplifies a subset of oligonucleotides. In yet other circumstances, an oligonucleotide contains both universal and unique amplification sequences, which can optionally be used sequentially.
[0052] A "linker" or "linker sequence" is a structure, which can be a unique nucleic acid sequence, that joins one molecule to another, such as attachment of a probe as described herein to another molecule or a substrate, wherein one portion of the linker is operably linked to a substrate, and wherein another portion of the linker is operably linked to the probe. As used herein, a linker or linker sequence may refer to a common nucleic acid sequence that is preferably non-complementary to a target nucleic acid sequence. A library of
oligonucleotides, probes or primers as described herein may each contain a single common linker sequence.
[0053] A "barcode" or "barcode sequence" as used herein refers to a short stretch of nucleotides in a particular order, and different barcodes are different combinations of nucleotides. A barcode may be of any length, but is preferably between 4 to 15, more preferably between 4 to 10, and most preferably between 4 and 8, such as 4, 5, 6, 7 or 8 nucleotides long. Ideally, the barcodes are designed such that they can be unambiguously called post-sequencing or post-amplification. The sequence of the barcode may be identical or different for each nucleic acid molecule in a particular sequencing run, or according to a number of different parameters, such as target nucleic acid origin, sequence reads derived from the 5' strand or the 3 ' strand, sequence length or molecular weight of a sequence read. Barcode sequences may be derived computationally or by hand. In some aspects, barcode sequences can be randomly generated using an iterative script program, such as Perl.
[0054] Oligonucleotides, primers, and probes may also contain one or more sites ("restriction sites") for cleavage by restriction endonucleases (also known as "restriction enzymes"). Type 1 enzymes cut DNA at random far from their recognition sequences. Type II enzymes
cut DNA at defined positions close to or within their recognition sequences. Type III enzymes are also large combination restriction-and-modification enzymes. They cleave outside of their recognition sequences and require two such sequences in opposite orientations within the same DNA molecule to accomplish cleavage. Type IV enzymes recognize modified, typically methylated DNA. Restriction endonucleascs are commercially available and restriction site sequences are well known in the art (New England Biolabs, Beverly, MA). Any restriction site sequence may be included in oligonucleotides, primers, and probes as described herein.
[0055] Oligonucleotides, primers, and probes may be isolated from natural sources or purchased from commercial sources. Oligonucleotides, primers, and probes described herein may also be synthesized by any suitable method, e.g., standard phosphoramidite methods such as those described by Beaucage, S.L. and Caruthers, M.H. (1981 ) Tetrahedron Lett. 22: 1859-1862 or the triester method according to Matteucci, M.D. and Caruthers, M.H. (1981) J Am. Chem. Soc. 103(1 1): 3185-3191 , or by other chemical methods using either a commercial automated oligonucleotide synthesizer or high-throughput, high-density array methods known in the art (see U.S. Patent Nos. 5,602,244, 5,574, 146, 5,554,744, 5,428, 148, 5,264,566, 5,141 ,813, 5,959,463, 4,861 ,571 and 4,659,774, incorporated herein by reference in its entirety for all purposes). Pre-synthesized oligonucleotides may also be obtained commercially from a variety of vendors. Oligonucleotides, primers, and probes may be prepared using a variety of microarray technologies known in the art. Pre-synthesized oligonucleotide and/or polynucleotide sequences may be attached to a support or synthesized in situ using light-directed methods, flow channel and spotting methods, inkjet methods, pin- based methods and bead-based methods set forth in the following references: McGall et al. (1996) Proc. Natl. Acad. Sci. U.S.A. 93(24): 13555-13560; Synthetic DNA Arrays In Genetic Engineering, Vol. 20: 1 1 1, Plenum Press (1998); Duggan, D.J. et al., (1999) Nat. Genet. 21 (Suppl 1): 10-14; Microarrays: Making Them and Using Them In Microarray Bioinformatics, Cambridge University Press, 2003; U.S. Patent Application Publication Nos. 20030068633 and 20020081582; U.S. Patent Nos. 6,833,450, 6,830,890, 6,824,866, 6,800,439, 6,375,903 and 5,700,637; and PCT Application Nos. WO 04/031399, WO 04/031351 , WO 04/029586, WO 03/100012, WO 03/066212, WO 03/065038, WO 03/064699, WO 03/064027, WO 03/064026, WO 03/046223, WO 03/040410 and WO 02/24597.
[0056] In the context of the subject matter described herein, references are made to melting temperatures (Tm) of oligonucleotides, primers, and probes. Tm means the temperature at which half of the subject material exists in double-stranded form and the remainder is single stranded. Generally, the Tm of a primer is a calculated value using any method knows in the art, particularly the "% GC" method (Wetmar, J.G. (1991) Crit. Rev. Diochem. Mol. Biol. 26: 227-259), the "2(A+T) plus 4(G+C)" method at a standard condition of primer and salt concentration, or methods described in Santa Lucia, J. (1998) Proc. Natl. Acad. Sci. 95(4): 1460-1465 and von Ahsen, N. et al., (2001) Clin. Chem. 47: 1956-1961.
[0057] The term "methylation" as used herein, denotes the type of chemical modification of nucleic acids that involves the addition of a methyl group, for example to the C5 carbon atom of the cytosine pyrimidine ring or to the N6 nitrogen atom of the adenosine purine ring, with the first option being particularly preferred. This modification can be inherited and subsequently removed without changing the original nucleic acid sequence. As such, it is part of the epigenetic code and the most well characterized epigenetic mechanism.
Methylation is reversible: methyl -transferases catalyze the transfer of a methyl group from S- adenosyl-L-methionine to cytosine or adenosine residues. Polymerases such as DNA polymerases do not copy the methylated status during replication (reviewed, e.g. , in
Robertson, .D. and Wolffe, A.P. (2000) Nat. Rev. Genet. 1(1): 1 1 -19; Li, E. (2002) Nat. Rev. Genet. 3: 662-673; Bird, A.P. (2002) Genes Dev. 16: 6-21).
[0058] The term "CpG dinucleotide sites" (or "CpG sites"), as used herein, refers to regions of DNA where a cytosine nucleotide is located immediately adjacent to a guanine nucleotide in the linear sequence. "CpG" refers to cytosine and guanine separated by a phosphate (i.e., - -C--phosphate— G— ). The "CpG" notation is used to distinguish a cytosine followed by guanine from a cytosine base paired to a guanine. Regions of the DNA that have a higher frequency or concentration of CpG sites are known as "CpG islands". "CpG islands" may also define a contiguous region of genomic DNA that satisfies the criteria of (1) having a frequency of CpG dinucleotides corresponding to an "observed/expected ratio" greater than 0.6; (2) having a "GC content" greater than 0.5; and (3) having a length of at least 0.2 kb (as described in Gardiner-Garden et al., (1987) J. Mol. Biol , 196: 262-282), with the exception that repeat regions matching these criteria are excluded (or masked). "CpG island fragment" or "CGI fragment" arc used interchangeably to refer to a nucleic acid molecule fragment mapping to and containing at least part of a CpG island. The term "CpG target sequence"
refers to a stretch of bases targeted to selectively enrich for CpG island fragments and/or other methylation informative GC-rich fragments. Many genes in mammalian genomes have CpG islands associated with the transcriptional start site (including the promoter) of the gene, which play a pivotal role in controlling gene expression.
[0059] Methylation at the C5 of cytosine has been found in bacteria, fungi, plant and mammalian genomes. Approximately 60-90% of CpG dinucleotides are methylated in most mammalian cell types. The CpG dinucleotides are not uniformly distributed in mammalian genomes. For example, sequence analysis of the human genome has estimated nearly 30,000 CpG islands, which accounts for about 0.7% of the genome. CpG dinucleotides in the remaining 99.3% of the genome are sparsely distributed. Because of the high cytosine- guanine frequency of CpG islands, it is possible to identify them without knowledge of the methylation pattern of the DNA.
[0060] In normal tissue, CpG islands are often unmethylated but a subset of islands becomes methylated during oncogenesis, cellular development, and various disease states.
Hypermethylation (i.e. an increased level of methylation) of CpG sites within the promoters of genes can lead to their silencing, a feature found, e.g. , in a number of human cancers (for example the silencing of tumor suppressor genes). In contrast, the hypomethylation (i.e. a reduced level of methylation) of CpG sites has been associated with the over-expression of oncogenes within cancer cells (reviewed, e.g. , in Robertson, K.D. and Wolffe, A.P. (2000) Nat. Rev. Genet. 1(1 ): 1 1 -19; Li, E. (2002) Nat. Rev. Genet. 3: 662-673; Bird, A.P. (2002) Genes Dev. 16: 6-21 ; Klose, R.J. and Bird, A.P. (2006) Trends Biochem. Sci. 31 : 89-97). Accordingly, there is great interest in determining the methylation status or profiles of promoters and CpG islands (CGIs) in various tissues, especially with regard to methylation differences accounting for altered patterns of expression in normal development and in various disease states which would greatly improve understanding of these processes and provide potential diagnostic markers and therapeutic targets for diseases (Berman, B.P. et al., (2009) Nat. Biotech. , 27(4): 341-342).
[0061] The term "methylation assay" refers to any assay for determining the methylation status of one or more CpG dinucleotide sequences within one or more nucleic acid sequences. "Methylation frequency", "methylation state" or "methylation status" refers to a
determination of the presence or absence of 5-methylcytosine ("5-mCyt") or any other methylation modification at one or a plurality of CpG dinucleotides within a target nucleic
acid sequence by a methylation assay. The methylation status of a particular nucleic acid fragment or sequence can indicate the methylation state of every base in the sequence or can indicate the methylation state of a subset of the base pairs (e.g. , whether the base is cytosine or 5-methylcytosine) within the sequence. Methylation states at one or more particular CpG methylation sites (each having two CpG dinucleotide sequences) within a nucleic acid sequence may include "unmethylated," "fully-methylated" and "hemi-methylated" sites. Methylation status can also indicate information regarding regional methylation density within the sequence without specifying the exact location at the single nucleotide position level.
[0062] A "methylation profile" refers to a set of data representing the methylation states or methylation frequencies of one or more loci within a molecule of DNA from e.g. , the genome of an individual or cells or tissues from an individual. The profile can indicate the methylation state of every base in an individual, can have information regarding a subset of the base pairs (e.g. , the methylation state of specific promoters or quantity of promoters) in a genome, or can have information regarding regional methylation density or methylation frequency of one or more loci with or without specifying the exact location at the single nucleotide position level.
[0063] "Differential methylation" denotes a condition in which a particular candidate genomic locus is (at one or more nucleic acid sites comprised in its sequence) methylated in at least one target sample but unmethylated in at least one reference sample, or vice versa, in which a particular candidate genomic locus is (at one or more nucleic acid sites comprised in its sequence) unmethylated in the reference sample but methylated in the target sample. The determination of the differential methylation pattern or frequency of the one or more candidate genes/loci already includes the identification of the exact nucleic acid sites (i.e. sequence elements, genetic loci) comprised in the one or more candidate genes. Preferably, the nucleic acid sites comprised in the one or more candidate genes/loci that are differentially methylated are CpG dinucleotide sites.
[0064] Generally, the determination of the methylation pattern, methylation frequency, or methylation status of the one or more candidate genes/loci may be accomplished by any means known in the art. Preferably, methylation is determined by means of one or more methods selected from reverse-phase HPLC, thin-layer chromatography, Sssl
methyltransferases with incorporation of labeled methyl groups, the chloracetaldehyde
reaction, differentially sensitive restriction enzymes, hydrazine or permanganate treatment (m5C is cleaved by permanganate treatment but not by hydrazine treatment), bisulfite sequencing, combined bisulfite-restriction analysis, pyrosequencing, methylation-sensitive single-strand conformation analysis (MS-SSCA), high resolution melting analysis (HRM), methylation-sensitive single nucleotide primer extension (MS-SnuPE), base-specific cleavage/MALDI-TOF, methylation-specific PCR (MSP), microarray-based methods, and Mspl cleavage (reviewed, e.g., in Rein, T. et al. (1998) Nucl. Acids Res. 26: 2255-2264). Methods for detecting methylation status have been described in, for example U.S. Patent Nos. 6,214,556, 5,786,146, 6,017,704, 6,265,171 , 6,200,756, 6,251 ,594, 5,912,147,
6,331 ,393, 6,605,432, 5,786, 146, 6,143,504, 6,596,493, 6,884,586, 6,300,071 , and 7,195,870 and U.S. Patent Application Publication Nos. 20030148327, 20030148326, 20030143606, 20050009059, and 20060292564, each of which are incorporated herein by reference. Other array based methods of methylation analysis are disclosed in U.S. Patent Application Publication No. 20050196792. See also, Oakeley, E.J., (1999) Pharmacol. Ther. 84: 389- 400; Fraga et al., (2002) BioTechniques 33: 632-649; and Dahl et al., (2003) Biogerontology 4: 233-250.
[0065] Methods for identifying methylation may be based on differential cleavage by restriction enzymes are used. Methylation-sensitive restriction analysis followed by PCR amplification or Southern analysis have been disclosed, for example, in Huang, T.H. et al. (1997) Cancer Res. 57: 1030-1034; Zuccotti, M. et al, (1993) Meth. Enzymol. 225: 557-567; Carrel, L. et al. (1996) Am. J. Med. Genet. 64: 27-30; and Chang et al. (1992) Plant Mol. Biol. Rep. 10: 362-366.
[0066] In some aspects, enzymes that include at least one CpG dinucleotide in the recognition site may be used. Enzymes with a recognition site that includes the sequence CCGG include, for example, Mspl, Hpa l, Age], Xmal, Smal, NgoMW, Nael, and BspEl. Enzymes with a recognition site that includes the sequence CGCG include, for example, BstUl (CGCG, MSRE), Mlul (ACGCGT, MSRE), Sacll (CCGCGG, MSRE), AwHlI (GCGCGC, MSRE) and Nrwl (TCGCGA, MSRE). Noil, BstZl, Csp and Eagl have two CpGs in their recognition sites and cleavage is blocked by CpG methylation. Enzymes with a recognition site that includes the sequence GCGC include, for example, HinP , Hhal, Afe , Kasl, Nar , Sfol, Bbel, and Fspl. Enzymes with a recognition site that includes the sequence TCGA include, for example, Taql, Clal (MSRE), BspOl (MSRE), PaeWll, Tlil, Xltol, Sail,
and BstB . For additional enzymes that contain CpG in the recognition sequence and for information about the enzyme's sensitivity to methylation, see, for example, the New England Biolabs catalog and web site. In some aspects two restriction enzymes may have a different recognition sequence but generate identical overhangs or compatible cohesive ends. For example, the overhangs generated by cleavage with Hpall or Mspl can be ligated to the overhang generated by cleavage with Taql. Some restriction enzymes that include CpG in the recognition site are unable to cleave if the site is methylated; these are methylation sensitive restriction enzymes (MSRE). Other enzymes that contain CpG in their recognition site can cleave regardless of the presence of methylation; these are methylation insensitive restriction enzymes (MIRE). A third type of enzyme cleaves only when the recognition site is methylated, and are referred to herein as methylation dependent restriction enzymes (MDRE). Examples of MIREs that have a CpG in the recognition sequence include, for example, BsaVJl (WCCGGW), BsoBl, BssS , Mspl, and Taql. Examples of MSREs, that include a CpG in the recognition site, include Aatl , Aci , AcH, Afel, Agel, Ascl, Aval, BmgBl, Bsa AI, BsaHl, BspDl, CM, Eagl, Fsel, Faul, Haelll, Hpall, H/nPlI, Mini, Narl, Noil, Nrul, Pvnl, Sacll, Sail, Smal and SnaBl. In preferred aspects a pair of enzymes that have differential sensitivity to methylation and cleave at the same recognition sequence with one member of the pair being a MSRE and the other member being MIRE is used. Still other enzymes include BthQl, Glal, Hpal, HmPlI, Dpnl, Mbo , Chal and BstKTl.
[0067] Bisulfite sequencing is a commonly used method in the art for generating methylation data at single-base resolution. The term "bisulfite conversion" refers to a biochemical process for converting unmethylated cytosine residue to uracil or thymine residues, whereby methylated cytosine residues are preserved. "Bisulfite conversion" may be carried out computationally from a nucleic acid sequence contained in a computer file (such as those in FASTA, FASTQ or any file format known in the art), wherein all cytosine residues in a sequence of interest are changed to thymine or uracil residues. Exemplary reagents for bisulfite conversion include sodium bisulfite and magnesium bisulfite. "Bisulfite reagent" refers to a reagent comprising bisulfite, disulfite, hydrogen sulfite or combinations thereof, useful as disclosed herein to distinguish between methylated and unmethylated CpG dinucleotide sequences. One way to obtain such melhylation data for the CGIs is to sequence the entire epigenome directly. Due to the difficulty in mapping bisulfite converted sequence reads and the methylation heterogeneity in a cell population, approximately 100 gigabases
(Gb) of sequence data would be needed to generate a high-resolution human DNA methylation map (Lister, R. et al., (2009) Nature, 462(7271): 315-322). Other methylation profiling approaches include array capture (Hodges, E. et al., (2009) Genome Res. 19(9): 1593-1605), padlock probe capture (Deng, J. et al., (2009) Nat. Biotech. 27: 353-360; Ball, M.P. et al., (2009) Nat. Biotech. , 27(4): 361 -368) and reduced representation bisulfite sequencing (Gu et al., (2010) Nat. Methods 7(2): 133-136).
[0068] In particular, bisulfite sequencing involves conversion of unmethylated cytosine to uracil or thymine through a three-step process during sodium bisulfite modification. The steps are sulfonation to convert cytosine to cytosine sulfonate, deamination to convert cytosine sulfonate to uracil sulfonate or thymine sulfonate and alkali desulfonation to convert uracil sulfonate to uracil or thymine sulfonate to thymine. Conversion of methylated cytosine is much slower and is not observed at significant levels in a 4-16 hour reaction (Clark, S.J. et al, (1994) Nucleic Acids Res. , 22(15): 2990-7). If the cytosine is methylated it will remain a cytosine. If the cytosine is unmethylated, it will be converted to uracil or thymine. When the modified strand is copied, through, for example, extension of a locus specific primer, a random or degenerate primer or a primer to an adaptor, a G will be incorporated in the inteiTOgation position (opposite the C being interrogated) if the C was methylated and an A will be incorporated in the interrogation position if the C was unmethylated. When the double stranded extension product is amplified, those Cs that were converted to Us or Ts and resulted in incorporation of A in the extended primer will be replaced by Ts during amplification. Those Cs that were not modified and resulted in the incorporation of G will remain as C. Bisulfite treatment can degrade the DNA making it difficult to amplify. The sequence degeneracy resulting from the treatment also complicates primer design. The treatment may also result in incomplete desulfonation, depurination and other as yet uncharacterized DNA damage, making downstream processing more challenging. The treatment can also result in preferential amplification of unmethylated DNA relative to methylated DNA. This may be mitigated by increasing the PCR extension time.
[0069] Kits for DNA bisulfite modification are commercially available from, for example, Human Genetic Signatures' Melhyleasy and Chemicon's CpGenome Modification Kit. See also, WO04096825, which describes bisulfite modification methods and Olek, A. et al.
(1994) Nucl. Acids Res. 24(24): 5064-6, which discloses methods of performing bisulfite treatment and subsequent amplification on material embedded in agarose beads. In some
aspects a catalyst such as diethylenetriamine may be used in conjunction with bisulfite treatment, see Komiyama, M. and Oshima, S., (1994) Tetrahedron Lett. 35(44): 8185-8188. Diethylenetriamine has been shown to catalyze bisulfite ion-induced deamination of 2'- deoxycytidine to 2'-deoxyuridine at pH 5 efficiently. Other catalysts include ammonia, ethylene-diamine, 3,3'-diaminodipropylamine, and spermine. In some aspects, deamination is performed using sodium bisulfite solutions of 3-5 M with an incubation period of 12-16 hours at about 50°C. A faster procedure has also been reported using 9-10 M bisulfite pH 5.4 for about 10 minutes at 90°C, see Hayatsu, H. et al., (2004) Proc. Jpn. Acad. Ser. B 80(4): 189-194.
[0070] Bisulfite treatment allows the methylation status of cytosines to be detected by a variety of methods. For example, any method that may be used to detect a single nucleotide polymorphism (SNP) may be used, for examples, see Syvanen, A.C. (2001) Nature Rev. Gen. 2(12): 930-942. In a preferred aspect, bisulfite sequencing methods, systems, and computer program products described herein may provide information regarding not only methylation frequencies or methylation status of a sequence of interest at single base resolution, but also information regarding SNPs, preferably in the same sequencing run. Other methods such as single base extension (SBE) may be used or hybridization of sequence specific probes similar to allele specific hybridization methods. "Variants" or "alleles" generally refer to one of a plurality of species each encoding a similar sequence composition, but with a degree of distinction from each other. The distinction may include any type of variation known to those of ordinary skill in the related art, that include, but are not limited to, polymorphisms such as SNPs, insertions or deletions (the combination of insertion/deletion events are also referred to as "indels"), differences in the number of repeated sequences (also referred to as tandem repeats), and structural variations. Detection of such variants or alleles is also within the ambit of the subject matter described herein.
[0071] In a preferred aspect, molecular inversion probes (MIP), described in Hardenbol, P. et al. Genome Res. 15:269-275 (2005) and in U.S. Patent No. 6,858,412, may be used to determine methylation status after methylation dependent modification. A MIP may be designed for each cytosine to be interrogated. In a preferred aspect the MIP includes a locus specific region that hybridizes upstream and one that hybridizes downstream of an inteiTOgation site and can be extended through the interrogation site, incorporating a base that is complementary to the interrogation position. The interrogation position may be the
cytosine of interest after bisulfite modification and amplification of the region and the detection can be similar to detection of a polymorphism. Separate reactions may be performed for each NTP so extension only takes place in the reaction containing the base corresponding to the interrogation base or the different products may be differentially labeled.
[0072] The term "padlock probe" (PLP) refers to circularized nucleic acid molecules which may combine specific molecular recognition and universal amplification (or specific amplification and general recognition), thereby increasing sensitivity and multiplexing capabilities without limiting the range of potential target organisms. PLPs are long oligonucleotides of approximately 100 bases (but can be of any length), containing target complementary regions (referred to herein as "target-capturing sequences") at both their 5' and 3' ends (See, for example, Figure 5). These regions recognize adjacent sequences on the target nucleic acid sequence (Nilsson, M., et al. (1994) Science 265: 2085-2088) and may also contain "binding arms" which comprise "extension arms" having priming sites (e.g. , universal priming sites"), sites recognized by ligase enzymes, and unique sequence identifiers, sometimes referred to as a "ZipCode" or "barcode". Upon hybridization, the ends of the probes are situated into adjacent position, and can be joined by enzymatic ligation at the ligation sites (also referred to herein as "ligation arms") converting the probe into a circular molecule (also known in the art and referred to herein as an "amplicon") that is threaded on the target strand. This ligation and the resulting circular molecule can only take place when both ligation arm and extension arm segments recognize their target sequences correctly. Non-circularized probes may be removed by exonuclease treatment, while the circularized entities may be amplified with universal primers,
 may or may not contain barcode or ZipCode sequences. This mechanism ensures reaction specificity, even in a complex nucleotide extract with a large number of padlock probes. Subsequent ly, the target- specific products are detected by a universal cZipCode microarray (Shoemaker, D.D., et al., (1996) Nat. Genet. 14: 450-456). PLPs have high specificity and multiplexing capabilities in genotyping assays (Hardenbol, P., et al., (2003) Nat. BiotechnoL, 21 : 673-678.).
may or may not contain barcode or ZipCode sequences. This mechanism ensures reaction specificity, even in a complex nucleotide extract with a large number of padlock probes. Subsequent ly, the target- specific products are detected by a universal cZipCode microarray (Shoemaker, D.D., et al., (1996) Nat. Genet. 14: 450-456). PLPs have high specificity and multiplexing capabilities in genotyping assays (Hardenbol, P., et al., (2003) Nat. BiotechnoL, 21 : 673-678.).
[0073] A "formula," "algorithm," or "model" is any mathematical equation, algorithmic, analytical or programmed process, or statistical technique that takes one or more continuous or categorical inputs (herein called "parameters") and calculates an output value, sometimes referred to as an "index" or "index value." Non-limiting examples of "algorithms" include
sums, ratios, and regression operators, such as coefficients or exponents, value
transformations and normalizations, rules and guidelines, statistical classification models, pattern recognition, linear and quadratic discrimination, support vector machines, principal component analysis, nearest neighbor search, na'ive Bayesian classifiers, and neural networks.
[0074] A "neural network" can be an Artificial Neural Network (ANN) and are information processing systems composed of varying numbers of simple, elements called neurons distributed into layers. Neurons are organized in an input layer, one or more hidden layers, and an output layer. The connections between elements determine network function just as in natural biological nervous systems. ANN is an intelligent technique that mimics the functioning of a human brain, and emulates human intuition of making decisions and drawing conclusions even when presented with complex, noisy, irrelevant and partial information. ANNs may have any number of hidden layers. The neurons are connected to each other by weighted links over which signals can pass. Each neuron receives multiple inputs from other neurons, except the neurons in the input layer, in proportion to their connection weights and then generates a single output in accordance with an activation function. An activation function can be linear or nonlinear depending on the application. Sigmoid or Hyperbolic Tangent activation function can be used to improve the performance of ANNs in power system applications.
[0075] An ANN can be trained to perform a particular function by adjusting values of the interconnections called weights, and neuron thresholds. The process of adjusting
interconnection weights and neuron thresholds to achieve output of the ANN the same as the target value or desired output for a given input is referred to as "training" of ANN. Training an ANN consists of adjusting interconnection weights of neurons using a learning algorithm. Back propagation with momentum is the commonly used learning algorithm. Multilayer Feed Forward ANNs with Error Back Propagation learning algorithm are also commonly used. Feed Forward calculations, and propagating error from output layer to input layer and weight updating in hidden and output layers are major steps of training algorithm.
[0076] By way of example, the neural networks described herein comprise one or more inputs that are associated with efficiency of the probe or primer described herein. By "efficiency" is meant the amount of target nucleic acid sequence represented by a particular probe or primer in a sequencing library. Standard methods can be used to calculate the efficiency by measuring or counting the amount of target nucleic acid(s) and the amount of
unbound target nucleic acid(s) via sequencing. The efficiency of a probe or primer described herein is typically compared to the capture efficiency of a control probe or primer under the same incubation conditions (e.g. , using same buffer and temperature).
[0077] Such inputs comprise, without limitation, target length, target folding energy, target GC content, extension arm A%, extension arm G%, target A%, target T%, target G%, number of "GG" dinucleotides in ligation arm, number of "AT" dinucleotides in extension arm, number of "GG" dinucleotides in extension arm, number of "AA" dinucleotides in target, number of "AT" dinucleotides in target, number of "TA" dinucleotides in target, number of "GT" dinucleotides in target, number of "GA" dinucleotides in target, ligation arm terminal dinucleotide, extension arm terminal dinucleotide, target 5' terminal dinucleotide, ligation arm melting temperature, extension arm melting temperature, ligation arm length, extension arm length, local single-stranded folding energy of the target, and the dinucleotides present at the extension site and ligation site during probe capture. The neural networks described herein may comprise one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, twenty-two, twenty-three, twenty-four, twenty-five or more inputs.
|0078] The methods, computer program products, and systems described herein may comprise one, two, three, four, five or more neural networks. In a preferred embodiment, the probe designing algorithm used to predict efficiency of probes or primers as described herein utilizes two neural networks.
[0079] In some exemplary embodiments, there is provided methods, systems, and computer program products for designing large numbers of efficient, low-bias padlock probe molecules using a computational algorithm for probe or primer efficiency prediction. For example, a process may be implemented as a software application or computer program product (also known as software, programs, applications, components, or code) stored in memory and executed by one or more processors contained in a system. The software application or computer program product may accept a complete genome and a tabular list of specific regions of interest, along with several customizable probe properties (such as the inputs described herein, including, e.g., a desired target length, a desired binding arm length, and a desired DNA melting temperature). The software application or computer program product may use this information in an intelligent decision mechanism, such as for example a back propagation neural network-derived equation, to predict probe efficiency based on many
probe characteristics. The software application or computer program product may output the optimal set of probes to obtain maximum coverage of a desired set of genomic regions in a high-throughput sequencing experiment. The software application or computer program product may also add a customized linker sequence to each probe molecule, allowing easy usage in modern high-throughput sequencers. Alternatively, the probe design software application may also perform an in silico bisulfite conversion prior to probe design in order to generate an optimal set of probes for targeted bisulfite sequencing. An additional feature may be implemented to allow the probe design software application or computer program product to output the most efficient padlock probe molecules for specific unique regions of a genome, allowing for efficient multiplex organism detection.
[0080] In some implementations, the methods, systems, and computer program products described herein may exhibit features, such as an increased number of targetable genomic regions for padlock probe sequencing, more efficient and unbiased capture of genomic regions, and reduced cost of sequencing to obtain target coverage. Moreover, the method, system, and computer program product described herein may extend to designing efficient padlock probes for SNP genotyping, mutation discovery, targeted genomic sequencing, quantification of allele specific gene expression, analysis of DNA methylation profiles or frequencies, and organism presence detection.
[0081] In some exemplary embodiments, the methods, systems, and computer program products described herein may be applied, in some implementations, with particular advantage to easily generate optimal, high-efficiency sets of padlock probes for targeted genomic sequencing experiments. Figure 2 depicts a process 200 which may be implemented by a system comprising one or more processors and at least one memory including code which when executed by a processor provide the one or more of the operations depicted at process 200. For example, the process 200 may comprise a software application or computer program product implemented on at least one processor and at least one memory. In this example, the software application or computer program product may read three files presented by the user, such as for example, a job file, a target file, and/or a genome or chromosome file. Parameters or inputs provided in these files are associated with probe or primer efficiency and can be used to design padlock probes. The system may optionally comprise one or more databases containing libraries of information related to sequencing data, such as raw sequence reads, sequence alignments, methylation status or frequencies of a
target nucleic acid sequence, and information regarding SNPs, allele frequencies, or other variations in nucleic acid sequences of interest. The system may also comprise one or more communication links connecting the one or more processors to the one or more databases.
[0082] The job file may include several customizable parameters or inputs, such as for example a range for desired binding arm size, a range for target region size, and/or a flag for whether single stranded folding energy of each target should be calculated using an external module. The job file may also include links to one or more software modules to be used and the location on disk of the software, target file, and/or genome/chromosome file.
[0083J The genome/chromosome file may include the genome or chromosome on which targeted sequencing is to be performed. The file may be configured in a FASTA or FASTQ format. In FASTA, an organism or chromosome name may be presented, beginning with the ">" character; example: ">chrl " may be the first line of a FASTA or FASTQ file. The subsequent lines may contain a string of DNA base characters (A, T, G, and C) or DNA ambiguity characters as determined by IUPAC (M, R, W, S, Y, , V, H, D, B, or N). Line break characters may be allowed to enable easier reading. Multi-chromosome organisms (such as, for example, humans) may still require one ">" character per file; multiple chromosomes may be split into separate files (for example "human_chrl .fa" or
"human_chr2.fa").
[0084] The target file may include a tab-delimited list of specific genomic regions or target nucleic acid sequences to be sequenced with padlock probes. The first column in the file may include a specific identifier chosen (e.g., by an experimenter) to describe the target nucleic acid molecule, for example the name of a specific gene or specific regulatory region of interest. The second column may list the chromosome or genome for the specific targeted region. The third and fourth columns may list the beginning and end of the specific target region, in linear bases; the distance spanned can be as short as a single base pair (for genotyping) or thousands of base pairs (for larger-scale sequencing). The fifth column (which may not be included in the file) may accept a strand designation; if one is provided, only probes targeting one specifically chosen strand of DNA (either forward or reverse) may be designed; otherwise, probes may be designed utilizing both strands in combination to provide optimal capture.
[0085] The software application or computer program product implemented on one or more processors and at least one memory may read in the specified job file to obtain desired probe
parameters or inputs. The software application or computer program product may then proceed sequentially through the target file, designing optimal probe sets for each target nucleic acid molecule. The software application or computer program product may begin by opening the first specified genome/chromosome and loading, for example, the entire sequence into random access memory for rapid access. The software application or computer program product may then extract the specific target region of interest or target capture sequence, along with flanking sequence both linearly upstream and downstream of the target, from the genome.
[0086] The software application or computer program may design most, if not all, possible binding arms (which include extension arms and ligation arms as defined herein) for a given target nucleic acid molecule or region using the extracted sequence. The software application or computer program product may take into account the desired binding arm size from the job file, and remove from consideration any very low-complexity binding arms (i.e., more than six nucleotides of the same kind in a row). The software application or computer program product may next associate each binding arm with every possible partner binding arm, taking into account the desired target region length provided in the job file. These arm pairs would represent the two sequences surrounding the common linker. The software application or computer program product may designs arm pairs using both the forward and reverse strand of the provided genome unless the target file specifies a chosen strand; in this case, arm pairs will only be generated for the designated strand.
[0087] The software application or computer program product may then obtains a portion of information or inputs (e.g., six to seven key pieces of information) about each binding arm pair, such as for example the target length, the target GC content, the melting temperature of each binding arm, the length of each binding ami, and/or optionally the local single-stranded folding energy of the target. The software application or computer program product may then use a previously developed equation to predict the probe efficiency. This equation may be developed by an intelligent decision mechanism, such as a neural network, pattern recognizer, and/or other numerical techniques. For example, the equation to predict the probe efficiency may be developed by performing a multiplex genome sequencing reaction using hundreds of thousands of separate padlock probe molecules; the capturing efficiency of each molecule is measured in this sequencing experiment and modeled using one or more neural networks, including for example a back propagation neural network (with three layers
containing 13, 8, and 5 nodes respectively, see Figure 3) to the aforementioned 6-7 pieces of information as input. Additional neural networks may be added (such as, e.g. , a network with two hidden layers having 10 and 12 nodes, see Figure 18) with at least 25 pieces of information as input. The software application or computer program product may then divide each probe into separate categories based on which regions of the target sequence are captured, and ranks each available probe by probe efficiency. The probes having the highest efficiency are extracted and included in the generation of a library or probe set. The non- extracted probes of lower efficiency can be pooled and resubmitted for additional rounds of probe design as defined by the methods described herein.
[0088] The software application or computer program product may then generate candidate "probe sets" or "libraries" from the list of valid probes. The software application or computer program product may consider all possible sets of probe sets, and choose the set that provides maximum coverage of the sample and the highest aggregate probe scores, under the constraint that no binding arms in a probe set can bind to the same sequence (in order to prevent probe competition for hybridization). The software application or computer program product may also penalize sets that require many probes in order to control the cost of sequencing; this parameter may be adjustable.
[0089] The software application or computer program product may then report all binding arm pairs for the optimal probe set to the user. If the user is satisfied, the software application may then attach a common linker sequence between the two binding arms and generate a single molecular sequence for each probe that can be generated using commercial DNA synthesis services (such as that provided by Integrated DNA Technologies) or via other high-throughput methods (see, e.g. , U.S. Patent Application Ser. No. 60/765,978). Each linker sequence may be customizable to include specific adaptor sequences for current high- throughput sequencers, allowing designed padlock probes to be converted into linear sequencing libraries in just one experimental step. Linker sequences can also be customized to include barcode sequences for even greater multiplex capability or restriction enzyme sites for easy linearization of the circular padlock probe modules (see, e.g. , Figure 4). The user may thus integrate the generated probes into many custom experimental environments.
[0090] Once an optimal probe set is designed, the software application or computer program product may output it and proceed to the next target listed in the target file. The software application or computer program product may then repeat the genomic loading and probe
design process. In some embodiments, the software application or computer program product may exclude probes having the highest efficiency and pool the remaining non-extracted probes and repeat the steps of generating a library or set of probe or primer sequences, determining efficiency of the probes in the generated library or set, and ranking the probe or primer sequences in the library or set by efficiency.
[0091] In some exemplary embodiments, the methods, systems, and computer program products described herein are related to characterizing the DNA methylation profile of a sample using bisulfite sequencing and include mapping a genomic sequence of interest to determine methylation status, methylation frequency, and detection of single nucleotide polymorphisms. During bisulfite sequencing, all cytosines present in the DNA molecule except those that are methylated are converted to thymines. Almost every methylated cytosine is present as a cytosine-guanine dinucleotide (CpG), though not all CpGs are methylated. In such embodiments, after the genome/chromosome is loaded into memory, the software application or computer program product performs an in silico bisulfite conversion, computationally converting all cytosines except those present in a CpG to thymines. The application or program then designs probes as previously, but penalizes the inclusion of CpG sites in each binding arm. During linker sequence insertion, the software application or computer program product generates multiple probes for those arm pairs containing a CpG; one probe assumes a methylated state (and contains a CpG dinucleotide) while the other assumes an unmethylated state (and contains a TpG instead). This procedure allows for efficient targeted bisulfite sequencing of hundreds of thousands of CpG sites in parallel.
[0092] In some exemplary embodiments, the methods, systems, and computer program products described herein use the probes or primers to obtain sequence reads of a target genome or sequence of interest by bisulfite sequencing and loading it into memory. A software application or computer program product encodes the sequence reads by predicting the forward and reverse orientation of each of the sequence reads to generate at least one forward sequence read and at least one reverse sequence read. The forward and reverse sequence reads are then converted by the software application or computer program product by computationally changing all cytosine residues in the forward sequence reads to thymine residues in silico, and changing all guanine residues to adenine residues in the reverse sequence reads. The bisulfite-converted genome sequence and all forward and reverse sequence reads are then aligned computationally by an alignment software application or
computer program (e.g. , ELAND, SOAP2 Align, Bowtie, BWA, BLAST or any other alignment program known in the art). The alignment application or program can be a standalone application or integrated into the system, software application or computer program product described herein. The aligned sequences are then combined to create a map of the target genomic sequence. The software application or computer program product then analyzes and computes methylation frequencies or methylation status of the mapped sequences in entirety. In preferred embodiments, the mapped sequences may also be analyzed by the software application or computer program product for the presence of single nucleotide polymorphisms. Because bisulfite sequencing provides sequence read information at single-base resolution, this technique (and modifications thereof described in the methods, systems, and computer program products described herein) is particularly advantageous for calculating methylation frequencies and detecting SNPs in a single sequencing reaction.
[0093] In some exemplary embodiments, the methods, systems and computer program products described herein are related to organism detection in a mixed sample. Many cellular samples are heterogeneous, and contain mixtures of organisms in unknown quantities;
padlock probes can be used to detect which and how many organisms of each of a given type are present. In this example, the software application or computer program product may accept a fourth input file, known as a "homer file," which contains lists of preferred arm sequences in FASTA format (generally those found via genome annotation to be unique to a given genome or chromosome). The job file also contains at least one additional parameter: the number of probes to generate per genome or chromosome. The software application or computer program product then designs binding arm pairs as previously described, but favors binding arms containing a user-provided homer sequence. Instead of creating a "probe set" to maximize coverage of a target region, the software application or computer program instead returns the user-specified number of probes (in order of decreasing capturing efficiency) per genome. Probes designed for many separate genomes can be combined into a single padlock probe reaction, allowing detection of multiple organisms present at low frequency in a mixed population.
[0094] The subject matter described herein may be embodied in systems, computer program products, and methods, depending on the desired configuration. For example, the control module may be realized in digital electronic circuitry, integrated circuitry, specially designed ASICs (application specific integrated circuits), computer hardware, firmware, software,
and/or combinations thereof. These various implementations may include implementation in one or more computer programs that are executable and/or interpretable on a programmable system including at least one programmable processor, which may be special or general purpose, coupled to receive data and instructions from, and to transmit data and instructions to, a storage system, at least one input device, and at least one output device.
[0095] These software applications or computer program products include machine instructions for a programmable processor, and may be implemented in a high-level procedural and/or object-oriented programming language, and/or in assembly/machine language. As used herein, the term "computer-readable medium" refers to any computer program product, apparatus and/or device (e.g. , magnetic discs, optical disks, memory, Programmable Logic Devices (PLDs)) used to provide machine instructions and/or data to a programmable processor, including a machine-readable medium.
[0096] A computer may include any type of computer platform such as a workstation, a personal computer, a server, or any other present or future computer. Computers typically include known components such as a processor, an operating system, system memory, memory storage devices, input-output controllers, input-output devices, and display devices. Many possible configurations and components of a computer exist in the art and may also include cache memory, a data backup unit, and other additional devices.
[0097] Display devices may include display devices that provide visual information, this information typically may be logically and/or physically organized as an array of pixels. An interface controller may also be included that may comprise any of a variety of known or future software programs for providing input and output interfaces. For example, interfaces may include "Graphical User Interfaces" (often referred to as GUI's) that provide one or more graphical representations to a user. Interfaces are typically enabled to accept user inputs using means of selection or input known to those o ordinary skill in the related art.
[0098] In the same or alternative embodiments, applications on a computer may employ an interface that includes what are referred to as "command line interfaces" (often referred to as CLI's). CLPs typically provide a text based interaction between an application and a user. Typically, command line interfaces present output and receive input as lines of text through display devices. For example, some implementations may include a "shell" such as Unix Shells known to those of ordinary skill in the related art, or Microsoft Windows Powershell
that employs object-oriented type programming architectures such as the Microsoft .NET framework. Interfaces may include one or more GUI's, CLI's or a combination thereof.
[0099] A processor may include a commercially available processor such as an Itanium® or Pentium® processor made by Intel Corporation, a SPARC® processor made by Sun Microsystems, an Athlon™ or Opteron™ processor made by AMD corporation, or it may be one of other processors that are or will become available. Some embodiments of a processor may also include Multi-core processors and/or employ parallel processing technology in a single or multi-core configuration. For example, a multi-core architecture typically comprises two or more processor "execution cores". Each execution core may perform as an independent processor that enables parallel execution of multiple threads. In addition, a processor may be configured in what is generally referred to as 32 or 64 bit architectures, or other architectural configurations now known or that may be developed in the future.
[00100] A processor typically executes an operating system, which may be, for example, a Windows®-type operating system (such as Windows® XP, Windows Vista®, Windows 7) from the Microsoft Corporation; the Mac OS X operating system from Apple Computer Corp. (such as 7.5 Mac OS X v l 0.4 "Tiger" or 7.6 Mac OS X vl 0.5 "Leopard" operating systems); a Unix® or Linux-type operating system available from many vendors or an open source; another or a future operating system; or some combination thereof. An operating system interfaces with firmware and hardware in a well-known manner, and facilitates the processor in coordinating and executing the functions of various computer programs that may be written in a variety of programming languages. An operating system, typically in cooperation with a processor, coordinates and executes functions of the other components of a computer. An operating system also provides scheduling, input-output control, file and data management, memory management, and communication control and related services, all in accordance with known techniques.
[00101 ] System memory may include any of a variety of known or future memory storage devices. Examples include any commonly available random access memory (RAM), magnetic medium such as a resident hard disk or tape, an optical medium such as a read and write compact disc, or other memory storage device. Memory storage devices may include any of a variety of known or future devices, including a compact disk drive, a tape drive, a removable hard disk drive, USB or flash drive, or a diskette drive. Such types of memory storage devices typically read from, and/or write to, a program storage medium such as,
respectively, a compact disk, magnetic tape, removable hard disk, USB or flash drive, or floppy diskette. Any of these program storage media, or others now in use or that may later be developed, may be considered a computer program product. As will be appreciated, these program storage media typically store a computer software program and/or data. Computer software programs, also called computer control logic, typically are stored in system memory and/or the program storage device used in conjunction with memory storage device.
[00102] In some embodiments, a computer program product is described comprising a computer usable medium having control logic (computer software program, including program code) stored therein. The control logic, when executed by a processor, causes the processor to perform functions described herein. In other embodiments, some functions are implemented primarily in hardware using, for example, a hardware state machine.
Implementation of the hardware state machine so as to perform the functions described herein will be apparent to those skilled in the relevant arts.
[00103] Input-output controllers could include any of a variety of known devices for accepting and processing information from a user, whether a human or a machine, whether local or remote. Such devices include, for example, modem cards, wireless cards, network interface cards, sound cards, or other types of controllers for any of a variety of known input devices. Output controllers could include controllers for any of a variety of known display devices for presenting information to a user, whether a human or a machine, whether local or remote. As presently described herein, the functional elements of a computer may communicate with each other via a system bus. Some embodiments of a computer may communicate with some functional elements using network or other types of remote communications.
[00104] As will be evident to those skilled in the relevant art, an instrument control and/or a data processing application, if implemented in software, may be loaded into and executed from system memory and/or a memory storage device. All or portions of the instrument contiol and/or data processing applications may also reside in a read-only memory or similar device of the memory storage device, such devices not requiring that the instrument control and/or data processing applications first be loaded through input-output controllers. It will be understood by those skilled in the relevant art that the instrument control and/or data processing applications, or portions of it, may be loaded by a processor in
a known manner into system memory, or cache memory, or both, as advantageous for execution.
[00105] Also a computer may include one or more library files, experiment data files, and an internet client stored in system memory. For example, experiment data could include data related to one or more experiments or assays such as detected signal values, or other values associated with one or more sequencing experiments or processes. Additionally, an internet client may include an application enabled to accesses a remote service on another computer using a network and may for instance comprise what are generally referred to as "Web Browsers". In the present example some commonly employed web browsers include Microsoft® Internet Explorer available from Microsoft Corporation, Mozilla Firefox® from the Mozilla Corporation, Safari from Apple Computer Corp., Google Chrome available from Google, Inc., or other type of web browser currently known in the art or to be developed in the future. An internet client may include, or could be an element of, specialized software applications enabled to access remote information via a network such as a data processing application for sequencing applications.
[00106] A network may include one or more of the many various types of networks well known to those of ordinary skill in the art. For example, a network may include a local or wide area network that employs what is commonly referred to as a TCP/IP protocol suite to communicate, A network may include a network comprising a worldwide system of interconnected computer networks that is commonly referred to as the internet, or could also include various intranet architectures. Some users in networked environments may prefer to employ what are generally referred to as "firewalls" (also sometimes referred to as Packet Filters, or Border Protection Devices) to control information traffic to and from hardware and/or software systems. For example, firewalls may comprise hardware or software elements or some combination thereof and are typically designed to enforce security policies put in place by users, such as for instance network administrators, etc.
[00107] The subject matter described herein may also make use of additional computer program products and software for a variety of purposes, such as probe design, management of data, analysis, and instrument operation. See, U.S. Patent Nos. 5,593,839, 5,795,716, 5,733,729, 5,974,164, 6,066,454, 6,090,555, 6,185,561 , 6,188,783, 6,223,127, 6,229,91 1 and 6,308,170. Additionally, the subject matter described herein may also make use of methods for providing genetic information over networks such as the Internet as shown in U.S. Patent
Application Ser. Nos. 10/197,621 , 10/063,559 (United States Publication No. 20020183936), 10/065,856, 10/065,868, 10/328,818, 10/328,872, 10/423,403, and 60/482,389.
[00108] The subject matter described herein also provides for design of
oligonucleotides, primers, and probes useful for general sequencing reactions and assays. Commercial sequencing by synthesis platforms are available, such as the Genome Sequencer from Roche/454 Life Sciences, the Genome Analyzer from Illumina/Solexa, the SOLiD system from Applied BioSystems, Pacific Biosystems and the Heliscope system from Helicos Biosciences. Exemplary sequencing platforms may have one or more of the following features: 1) four differently optically labeled nucleotides are utilized (e.g. , Genome
Analyzer); 2) sequencing-by-ligation is utilized (e.g. , SOLiD); 3) pyrosequencing is utilized (e.g. , Roche/454); and 4) four identically optically labeled nucleotides are utilized (e.g. , Helicos).
[00109] Such sequencing reactions and assays include sequencing by ligation methods commercialized by Applied Biosystems (e.g. , SOLiD sequencing). In general, double stranded fragment nucleic acid molecules can be prepared by the methods described herein, and then incorporated into a water-in-oil emulsion along with polystyrene beads and amplified, for example by PCR. In some cases, alternative amplification methods can be employed in the water-in-oil emulsion such as any of the methods provided herein. The amplified product in each water microdroplet formed by the emulsion can interact, bind, or hybridize with the one or more beads present in that microdroplet leading to beads with a plurality of amplified products of substantially one sequence. When the emulsion is broken, the beads float to the top of the sample and are placed onto an array. The methods can include a step of rendering the nucleic acid bound to the beads single-stranded or partially single stranded. Sequencing primers are then added along with a mixture of four different fluorescently labeled oligonucleotide probes. The probes bind specifically to the two bases in the nucleic acid molecule to be sequenced immediately adjacent and 3' of the sequencing primer to determine which of the four bases are at those positions. After washing and reading the fluorescence signal from the first incorporated probe, a ligase is added. The ligase cleaves the oligonucleotide probe between the fifth and sixth bases, removing the fluorescent dye from the nucleic acid molecule to be sequenced. The whole process is repeated using a different sequence primer until all of the intervening positions in the sequence are imaged. The process allows the simultaneous reading of millions of DNA fragments in a "massively
parallel" manner. This "sequence-by-ligation" technique uses probes that encode for two bases rather than just one, allowing error recognition by signal mismatching and leading to increased base determination accuracy.
[00110] Other sequencing methods include sequencing by synthesis methods commercialized by 454/Roche Life Sciences including but not limited to the methods and apparatus described in Margulies et al., Nature (2005) 437:376-380 (2005); and U.S. Patent Nos. 7,244,559; 7,335,762; 7,211 ,390; 7,244,567; 7,264,929; and 7,323,305. In general, double stranded fragment nucleic acid molecules can be prepared by the methods described herein, immobilized onto beads, and compartmentalized in a water-in-oil PCR emulsion. In some cases, alternative amplification methods can be employed in the water-in-oil emulsion such as any of the methods provided herein. When the emulsion is broken, amplified fragments remain bound to the beads. The methods can include a step of rendering the nucleic acid bound to the beads single stranded or partially single stranded. The beads can be enriched and loaded into wells of a fiber optic slide so that there is approximately 1 bead in each well. Nucleotides are flowed across and into the wells in a fixed order in the presence of polymerase, sulfhydrolase, and luciferase. Addition of nucleotides complementary to the target strand can result in a chemiluminescent signal that is recorded, such as by a camera. The combination of signal intensity and positional information generated across the plate allows software to determine the DNA sequence.
[00111] Other sequencing methods include those commercialized by Helicos
Biosciences Corporation (Cambridge, MA) as described in U.S. Patent Application Ser. No. 1 1/167,046, and U.S. Patent Nos. 7,501 ,245; 7,491 ,498; 7,276,720; and in U.S. Patent Application Publication Nos. 20090061439; 20080087826; 20060286566; 2006002471 1 ; 20060024678; 20080213770; and 20080103058. In general, double stranded fragment nucleic acid molecules can be isolated and purified, then immobilized onto a flow-cell surface. The methods can include a step of rendering the nucleic acid bound to the flow-cell surface stranded or partially single stranded. Polymerase and labeled nucleotides are then flowed over the immobilized DNA. After fluorescently labeled nucleotides are incorporated into the DNA strands by a DNA polymerase, the surface is illuminated with a laser, and an image is captured and processed to record single molecule incorporation events to produce sequence data.
[00112] Other methods include sequencing by ligation methods commercialized by Dover Systems. Generally, oligonucleotides, primers, and probes can be prepared by the methods described herein. The nucleic acid molecules can then be amplified in an emulsion in the presence of magnetic beads. Any amplification methods can be employed in the water- in-oil emulsion. The resulting beads with immobilized clonal nucleic acid polonies are then purified by magnetic separation, capped, amine functionalized, and covalently immobilized in a series of flow cells. The methods can include a step of rendering the nucleic acid bound to the flow-cell surface stranded or partially single stranded. A series of anchor primers are flowed through the cell, where they hybridize to the synthetic oligonucleotide sequences at the 3' or 5' end of proximal or distal genomic DNA tags. Once an anchor primer is hybridized, a mixture of fully degenerate nonanucleotides ("nonamers") and T4 DNA ligase is flowed into the cell. Each of the nonamer mixture's four components is labeled with one of four fluorophores, which correspond to the base type at the query position. The fluorophore- tagged nonamers selectively ligate onto the anchor primer, providing a fluorescent signal that identifies the corresponding base on the genomic DNA tag. Once the probes are ligated, fluorescently labeling the beads, the array is imaged in four colors. Each bead on the array will fluoresce in only one of the four images, indicating whether there is an A, C, G, or T at the position being queried. After imaging, the array of annealed primer-fluorescent probe complex, as well as residual enzyme, are chemically striped using guanidine HCl and sodium hydroxide. After each cycle of base reads at a given position have been completed, and the primer-fluorescent probe complex has been stripped, the anchor primer is replaced, and a new mixture of fluorescently tagged nonamers is introduced, for which the query position is shifted one base further into the genomic DNA tag. Seven bases are queried in this fashion, with the sequence performed from the 5' end of the proximal tag, followed by six base reads with a different anchor primer from the 3' end of the proximal tag, for a total of 13 base pair reads for this tag. This sequence is then repeated for the 5' and 3' ends of the distal tag, resulting in another 13 base pair reads. The ultimate result is a read length of 26 bases (thirteen from each of the paired tags). However, it is understood that this method is not limited to 26 base read lengths.
[00113] Other useful methods for sequencing include those commercialized by Illumina as described U.S. Patent Nos. 5,750,341 ; 6,306,597; and 5,969, 1 19. In general, oligonucleotides, primers, and probes can be prepared by the methods described herein to
produce amplified nucleic acid sequences tagged at one (e.g. , (Α)/(Α') or both ends (e.g., (A)I(A') and (C)/(C))- In some cases, single stranded nucleic acid tagged at one or both ends is amplified by the methods described herein (e.g. , by SPIA or linear PCR). The resulting nucleic acid is then denatured and the single stranded amplified nucleic acid molecules are randomly attached to the inside surface of flow-cell channels. Unlabeled nucleotides are added to initiate solid-phase bridge amplification to produce dense clusters of double- stranded DNA. To initiate the first base sequencing cycle, four labeled reversible terminators, primers, and DNA polymerase are added. After laser excitation, fluorescence from each cluster on the flow cell is imaged. The identity of the first base for each cluster is then recorded. Cycles of sequencing are performed to determine the fragment sequence one base at a time. For paired-end sequencing, such as for example, when the nucleic acid molecules are labeled at both ends by the methods described herein, sequencing templates can be regenerated in-situ so that the opposite end of the fragment can also be sequenced.
[00114] Still other sequencing methods include those commercialized by Pacific
Biosciences as described in U.S. Patent Nos. 7,462,452; 7,476,504; 7,405,281 ; 7, 170,050; 7,462,468; 7,476,503; 7,315,019; 7,302,146; 7,3 13,308; and U.S. Patent Application
Publication Nos. US20090029385; US20090068655; US20090024331 ; and US20080206764. In general, oligonucleotides, primers and probes can be prepared by the methods described herein. Target nucleic acid molecules can then be immobilized in zero mode waveguide arrays. The methods may include a step of rendering the nucleic acid bound to the waveguide arrays single stranded or partially single stranded. Polymerase and labeled nucleotides are added in a reaction mixture, and nucleotide incorporations are visualized via fluorescent labels attached to the terminal phosphate groups of the nucleotides. The fluorescent labels are clipped off as part of the nucleotide incorporation. In some cases, circular templates are utilized to enable multiple reads on a single molecule.
[00115] Another example of a sequencing technique that can be used in the methods described herein is nanopore sequencing (see e.g. Soni, G.V. and Meller, A. (2007) Clin. Chem. 53: 1996-2001). A nanopore can be a small hole of the order of one nanometer in diameter. Immersion of a nanopore in a conducting fluid and application of a potential across it can result in a slight electrical current due to conduction of ions through the nanopore. The amount of current that flows is sensitive to the size of the nanopore. As a DNA molecule passes through a nanopore, each nucleotide on the DNA molecule obstructs the nanopore to a
different degree. Thus, the change in the current passing through the nanopore as the DNA molecule passes through the nanopore can represent a reading of the DNA sequence.
[00116] Another example of a sequencing technique that can be used is semiconductor sequencing provided by Ion Torrent (e.g., using the Ion Personal Genome Machine (PGM)). Ion Torrent technology can use a semiconductor chip with multiple layers, e.g. , a layer with micro-machined wells, an ion-sensitive layer, and an ion sensor layer. Nucleic acids can be introduced into the wells, e.g. , a clonal population of single nucleic can be attached to a single bead, and the bead can be introduced into a well. To initiate sequencing of the nucleic acids on the beads, one type of deoxyribonucleotide (e.g. , dATP, dCTP, dGTP, or dTTP) can be introduced into the wells. When one or more nucleotides are incorporated by DNA polymerase, protons (hydrogen ions) are released in the well, which can be detected by the ion sensor. The semiconductor chip can then be washed and the process can be repeated with a different deoxyribonucleotide. A plurality of nucleic acids can be sequenced in the wells of a semiconductor chip. The semiconductor chip can comprise chemical-sensitive field effect transistor (chemFET) arrays to sequence DNA (for example, as described in U.S. Patent Application Publication No. 20090026082). Incorporation of one or more triphosphates into a new nucleic acid strand at the 3' end of the sequencing primer can be detected by a change in current by a chemFET. An array can have multiple chemFET sensors.
[00117] Although a few variations have been described in detail above, other modifications or additions are possible. In particular, further features and/or variations may be provided in addition to those set forth herein. For example, the implementations described above may be directed to various combinations and subcombinations of the disclosed features and/or combinations and subcombinations of several further features disclosed above. In addition, the logic flow depicted in the accompanying figures and/or described herein does not require the particular order shown, or sequential order, to achieve desirable results. Other embodiments may be within the scope of the following claims.
EXAMPLES
Example 1 : Changes in DNA methylation detected by targeted bisulfite sequencing
[00118] A method to specifically capture an arbitrary subset of genomic targets for single-molecule bisulfite sequencing and digital quantification of DNA methylation at single- nucleotide resolution is presented herein. A set of -30,000 padlock probes was designed to
assess the methylation state of -66,000 CpG sites within 2,020 CpG islands on human chromosome 12, chromosome 20, and 34 selected regions. To investigate epigenetic differences associated with de-differentiation, methylation in three human fibroblast lines and eight human pluripotent stem cell lines was compared. Chromosome-wide methylation patterns were similar among all lines studied, but cytosine methylation was slightly more prevalent in the pluripotent cells than in the fibroblasts. Induced pluripotent stem (iPS) cells appeared to display more methylation than embryonic stem cells. In fibroblasts and pluripotent cells, 288 regions were methylated differently. This targeted approach is particularly useful for analyzing DNA methylation in large genomes.
[00119] Padlock probes have been previously used for exon capture and resequencing (Porreca, G.J. et al. (2007) Nat. Methods 4: 931 -936). This approach to targeted bisulfite sequencing involves the in situ synthesis of long (~150 nt) oligonucleotides on programmable microarrays, followed by their cleavage and enzymatic conversion into padlock probes. A library of padlock probes was annealed to the template DNA, circularized, and amplified by PCR before shotgun sequencing (Figure 5A-5C). There are, however, two major challenges in performing padlock capture for bisulfite sequencing. First, bisulfite treatment converts all unmethylated cytosines into uracils, resulting in marked reduction of sequence complexity. Achieving specific target capture on bisulfite-converted DNA is more difficult than on native genomic DNA. Second, low capturing sensitivity, high bias and random losses of alleles was initially observed with the "eMIP method" previously disclosed in the art (Porreca, G.J. et al. (2007) Nat. Methods 4: 931 -936). Obtaining accurate and efficient quantification of DNA methylation was not possible with the existing protocol, especially with the presence of allelic drop-outs.
Materials and Methods
[00120] Padlock probe design
[00121 ] A probe design algorithm was developed to search for an optimal set of padlock probes covering an arbitrary set of non-repetitive genomic targets. This algorithm weights candidate probes based on several sequence features that were previously not considered in eMIP probe design, including the melting temperature, size and word statistics (distribution of 12-mers in the bisulfite-converted genome) of the capturing arms, and gap sizes. When the capturing arms contain one or more CpG dinucleotides, we used multiple
probes to iterate all possible methylation state combinations of the CpGs contained within the arms. Chromosome positions of CpG islands were retrieved from the UCSC genome browser based on the hgl 8 annotation.
[00122] Padlock probe production
[00123] Libraries of long oligonucleotides (-150 nt) were synthesized by ink-jet printing on a programmable microarray and released (Agilent Technologies). The estimated total yield is 10 frnol per library. PCR amplification was performed in 32-96 reactions (100 μΐ each) with 0.1 nM template oligonucleotides, 200 μΜ dNTPs, 400 nM Apl V4IU primer, 400 nM Ap2V4 primer, 0.8x SybrGreen I, 36 units JumpStart Tag polymerase in l x
JumpStart buffer (Sigma), at 94°C for 2 minutes, 22 cycles of 94°C for 30 seconds, 55°C for 2 minutes, 72°C for 45 seconds and, finally, 72°C for 5 minutes. The amplicons were purified by either column purification (Zymo DNA Concentrator- 100 columns) or ethanol precipitation.
[00124] Approximately 40-60 μg of the purified PCR amplicons were digested with
40 units Lambda Exonuclease (5 U/μΙ; New England Biolabs (NEB)) in 1 * lambda exonuclease buffer (NEB) at 37°C for 2 hours, followed by denaturing at 90°C for 5 minutes, and purified with six Qiagen Qiaquick PCR purification columns. The resulting single- stranded DNA was subsequently digested with 6 units USER enzyme (1 U/μΙ; NEB) in l x DpnW buffer (NEB) at 37°C for 4 hours. Ten μΐ of 100 μ DpnW_ guide oligo was added into the reaction and denatured the mixture at 95°C for 5 minutes in a thermocycler, followed by a gradual decrease of temperature (0.1°C/s) to 60°C and a 20-minute incubation at 60°C. The mixture was digested with 100 U DpnW (50 U/μΙ) at 37°C for 2 hours. The single- stranded 102-nt probes were finally purified from the digestion with 6% denaturing PAGE (6% TB-Urea 2D gel; Invitrogen).
[00125] Multiplex capture on bisulfite-converted DNA
[00126] Genomic DNA was extracted from frozen pellets of fibroblast, iPS or hES cells using Qiagen DNeasy columns and bisulfite converted with the Zymo DNA Methylation Gold Kit (Zymo Research). Padlock probes (60 nM) and 200 ng of bisulfite-converted genomic DNA were mixed in 10 μΐ 1 x Ampligase Buffer (Epicentre), denatured at 95°C for 10 minutes, then hybridized at 55°C for 18 hours, after which 1 μΐ gap-filling mix (200 μΜ
dNTPs, 2 U AmpliTaq Stoffel Fragment (ABI) and 0.5 units Ampligase (Epicentre) in 1 * Ampligase buffer) was added to the reaction. For circularization, the reactions were incubated at 55°C for 4 hours, followed by five cycles of 95°C for 1 minute and 55°C for 4 hours. To digest linear DNA after circularization, 2 μΐ exonuclease mix containing 10 U/μΙ exonuclease I and 100 U/μΙ exonuclease III (USB) was added to the reaction, and the reactions were incubated at 37°C for 2 hours and then inactivated at 95°C for 5 minutes.
[00127] Capture circles amplification
[00128] Ten microliters of circularization products were amplified by PCR in 100 μΐ reactions with 200 nM AmpF6.2-SoL primer, 200 nM AmpR6.2-SoL primer, 0.4x
SybrGreen I and 50 μΐ iProof High-Fidelity Master Mix (Bio-Rad) at 98°C for 30 seconds, eight cycles of 98°C for 10 seconds, 58°C for 20 seconds, 72°C for 20 seconds, 14 cycles of 98°C for 10 seconds, 72°C for 20 seconds and 72°C for 3 minutes. The amplicons of the expected size range (344-394 bp) were purified with 6% PAGE (6% TBE gel; Invitrogen).
[00129] Shotgun sequencing library construction
[00130] Purified PCR products with the four probe sets on the same template DNA were pooled in equal molar ratio, and reamplified in 4x 100 μΐ reactions with 4-μ1 template (10- 15 ng/μΐ), 200μΜ dNTPs, 20 μΜ dUTP, 200 nM AmpF6.3 primer, 200 nM AmpR6.3 primer, 0.4x SybrGreen I and 200 μΐ 2χ Taq Master Mix (NEB) at 94°C for 3 minutes, 8 cycles of 94°C for 45 seconds, 55°C for 45 seconds, 72°C for 45 seconds and 72°C for 3 minutes. PCR amplicons were purified with Qiaquick columns, and digested with MmeV. -3.6 nmole purified PCR amplicons, 16 units of Mmel (2 U/μΙ; NEB), 100 μΜ SAM in 1 χ NEB Buffer 4 at 37°C for 1 hour. The digestions were again column purified, and digested with 3 U USER enzyme (1 U/μΙ) at 37°C for 2 hours, then with 10 units S I nuclease (10 U/μΙ; Invitrogen) in 1 χ S I nuclease buffer at 37°C for 10 minutes. The fragmented DNA was column purified, and end repaired at 25°C for 45 minutes in 25-μ1 reactions containing 2.5 μΐ 10x buffer, 2.5 μΐ dNTP mix (2.5 mM each), 2.5 μΐ ATP (10 mM), 1 μΐ end-repair enzyme mix (Epicentre), and 15 μΐ DNA. Approximately 100-500 ng of the end-repaired DNA was ligated with 60 μΜ Solexa sequencing adaptors in 30 μΐ of 1 QuickLigase Buffer (NEB) with 1 μΐ QuickLigase for 15 minutes at 25°C. Ligation products of 150-175 bp in size were size selected with 6% PAGE, and amplified by PCR in 100 μΐ reactions with 15 μΐ template,
200 n Solexa PCR primers, 0.8 x SybrGreen I and 50 μΐ iProof High-Fidelity Master Mix (Bio-Rad) at 98°C for 30 seconds, 12 cycles of 98°C for 10 seconds, 65°C for 20 seconds, 72°C for 20 seconds and 72°C for 3 minutes. The PCR amplicons were purified with Qiaquick PCR purification columns, and sequenced on Illumina Genome Analyzer. All primer sequences are listed in Table 1.
Table 1 : Sequences of primers used in padlock capture and sequencing library

"*" indicates a phosphorothioate bond
[00132] Read mapping and data analysis
[00133] Mapping of bisulfite sequencing reads was performed with SOAP (Li, R. et al.
(2008) Bioinformatics 24: 713-714) driven by a customized Perl script. An unbiased mapping strategy in which the mapping success rate is independent of the methylation status was developed. Sequences of the captured targets were extracted from the repeat-masked human genome (hgl 8), and both strands were "bisulfite converted" in silico assuming no methylation on all CpG dinucleotides. The raw sequencing reads were also converted to "unmcthylated reads". To do this, the C/T and A/G ratios for each read were first compared to determine whether the reads corresponding to the bisulfite-converted strand or the reverse- complementary strand. In the latter case, the raw sequence was reverse complemented. All Cs were replaced by Ts in the resulting sequences. The unmethylated reads were then aligned to the unmethylated template sequences using SOAP. The false mapping rate that was due to the use of captured targets instead of the full human genome sequence was 0.21 %. Finally, based on the mapping position, the methylation status of each CpG site was retrieved from the unconverted raw reads. Cluster analyses and statistical analyses were performed
with R, Cluster3 Per) module, and in-house Perl scripts. The UCSC Genome Browser and Multiexperiment Viewer were used for data visualization.
[00134] Discussion
[00135] Approximately 10,580 padlock probes were designed, each capturing a 175- to 225-bp region, including 9,350 probes covering 2,020 CpG islands (Table 2) on human chromosomes 12 and 20, 705 probes covering 237 promoters in eight ENCODE (the Encyclopedia of DNA Elements) regions, and 527 probes targeting 4-kb regions centered on the transcription start sites (TSS) of 26 genes related to development or pluripotency (Table 3).
[00136] Table 2: Summary statistics of padlock captured CGIs

*Calculated as the fraction of base-pairs within CGIs that were covered by the padlock probes. Some CGIs were not completely covered due to the presence of repetitive sequences, or extreme nucleotide composition such that no probe could be designed.
[00137] Table 3: List of 26 selected genes and 8 ENCODE regions

chr4:057466834-57470834 REST
chr4: 123965397-123969397 FGF2
chr5: 149524548-149528548 CDX
chr5: 153836005-153840005 HAND1
chr6:031244421-3 1248421 OCT4
chrX: 136474004-136478004 Z1C3
chr5: 153836005-153840005 HAND1
chr22:3033954-31833953 ENm004
chr21 :32668237-34364221 ENm005
chrX: 152767492- 15406308 1 ENm006
chrl 9:59023585-60024460 ENm007
chr7:26924046-27424045 . ENmOlO
chrl 1 : 1699992-2306039 ENmO l l
chrl2:38626477-39126476 ENrl 23
chr20:33304929-33804928 ENr333
[00138] The total size of captured fragments was 2.1 Mbp, representing 0.064% of the human genome. Because some probes contain CpG sites within the capturing arms, all possible C/T combinations were iterated on these CpG sites, and a total of 30,000 nondegenerate probes were synthesized. CpG islands were chosen to perform the proof-of- concept study primarily because they represent a relatively well-defined set of genomic features in the human genome annotation. To increase the sensitivity and reduce bias, the probe/target ratio was increased by more than tenfold, the reaction time was extended, and five additional cycles of circularization were added in comparison with the published protocol (Porreca, G.J. et al. (2007) Nat. Methods 4: 931-936). To integrate construction of sequencing libraries with padlock capture, a new method that uses a combination of uracil- specific excision reagent (USER) enzymes and SI nuclease to create fragments with random ends was developed (Figure 5D).
[00139] Validation of the targeted bisulfite sequencing method was achieved by capturing bisulfite-converted Jurkat cell genomic DNA with all 30,000 padlock probes in a single-tube reaction. PCR amplicons from the circularization reactions were template specific, and PAGE analysis showed the expected size distribution (Figure 5E). The specificity of the capturing reaction was estimated by ligating captured DNA fragments to a sequencing vector, cloning these into Escherichia colt, and sequencing 96 clones. Of 89 high-quality Sanger sequencing reads obtained, 80 were from the targeted regions, indicating a specificity of 90%. An Illumina Genome Analyzer was then used to sequence the ends of the captured fragments. Approximately 5.5 million reads were mapped to 10,364 of 10,582 targets, which translates to a sensitivity of 98%. These results indicate that padlock probes
can specifically extract a large set of genomic targets for single molecule bisulfite sequencing.
[00140] Although 98% of the targets were observed at least once in the end-sequencing analysis, the abundance of different captured fragments varied across a 10,000-fold range. Analysis of variance revealed that the bias resulted from a combination of factors, including GC content and length of the ligation arms, and the size of the targets to be captured (Figure 6). To normalize the relative abundance among different DNA fragments, a combination of two strategies was used: 'subsetting' and 'suppressor oligos' (Figure 7A-7D). All 30,000 padlock probes were ranked based on the capturing efficiency determined by end sequencing, and divided into four subsets, two containing 5,000, and two containing 10,000, oligos. The three less efficient subsets were resynthesized. For each DNA sample, four capturing reactions were performed separately using probes from the original set of 30,000 and the three resynthesized subsets. The PCR amplicons from the capturing reactions were pooled in equal molar ratios before constructing a shotgun sequencing library (Figure 7A). This subsetting strategy increased the relative abundance of less efficient targets by orders of magnitude. A very small number of probes were extremely efficient. For example, the top 48 (0.016%) most efficient probes account for 13.3% of mappable reads in the end- sequencing analysis.
|00141] Although the subsetting strategy allowed for adjusting the relative abundance among several relatively large subsets of probes, a method was also needed to specifically reduce the efficiency of a small number of probes in a library. For this, a set of 48 suppressor oligos was designed, which contained chimeric sequences: the 5' region was reverse- complementary to the extension arm H2, and the 3' region contained a short sequence unrelated to the ligation arm HI . When these suppressor oligos were mixed with padlock probes in a high molar ratio (100-fold molar excess of suppressor oligos), the 48 most efficient probes tended to anneal to the suppressor oligos, were extended from the 3' ends and yielded linear-extended sequences that were removed in the subsequent exonuclease digestion (Figure 7B). This normalization strategy was tested on the same bisulfite-converted Jurkat cell DNA, end sequencing on the captured DNA fragments was performed and 2.2 million mappable reads were obtained. The effect of normalization resulted in the fraction of probes with at least half of the average abundance increased from 31 % to 49%; the average efficiency for the 48 most abundant probes was reduced by fivefold (Figure 7C-7D).
[00142] To validate the measurement accuracy of this method, advantage was taken of the built-in redundancy in our probe design. Each CpG island was covered by multiple probes targeting partially overlapping DNA fragments on alternating strands (Figure 5B). The CpG sites in the overlapping regions were captured independently from two DNA strands with different probes. Because the sequencing reads were mapped in a strand-specific manner and CpG methylation is symmetric on the two DNA strands, the accuracy of the assay can be determined by comparing the methylation level of these CpG sites on the two strands. For 2,697 such CpG sites that were covered by >50 sequencing reads, the Pearson correlation coefficient (R) was 0.987 (Figure 8A). To confirm the measurement accuracy with an independent method, the methylation levels of 182 randomly selected CpG sites were quantified with conventional bisulfite Sanger sequencing. Correlation between the two assays was high (R = 0.975; Figure 8B). Finally, the methylation measurements from two batches of 1MR90 fibroblast cultures were compared. Correlation was observed between the biological replicates (R = 0.970; Figure 8C). Taken together, these three validation experiments indicate that this assay is highly robust.
[00143J To demonstrate the utility of targeted bisulfite sequencing, the changes of chromosome- wide methylation status during reprogramming of human fibroblasts to pluripotent cells was characterized. The methylation assay was performed on three sets of fibroblasts and iPS cells from three laboratories: IMR/IMR90-iPS 17 reprogrammed with four factors (Oct4, Sox2, Nanog and Lin28); hFib2/hFib2-iPS18 reprogrammed with a different set of factors (Oct4, Sox2, Klf4 and Myc); BJ/BJ-iPS l l/BJ-iPS 12 (Maherali, N. et al. (2008) Cell Stem Cell 3: 340-345) reprogrammed with the five factors (Oct4, Sox2, Nanog, Klf4 and Myc) controlled by an inducible promoter. A line of hybrid stem cells (BJHues6-Hybridl), which were reprogrammed by fusing the human fibroblasts (BJ) with hES cells (Hues6), as well as three hES cell lines (Hues 12, Hues42, Hues63) were also characterized. Bisulfite conversion, padlock capture and construction of shotgun sequencing libraries were performed on each DNA sample. Each library in one lane was sequenced in the flow cell of an Illumina Genome Analyzer, and yielding 2-3 million reads that were mapped to the targeted regions. The bisulfite conversion rates were >98.5%. To avoid stochastic sampling drift, CpG sites that were covered by <10 reads were removed from the following analyses.
[00144] The global methylation patterns in all 12 samples (1 1 cell lines plus a biological replicate on IMR90 fibroblasts) were visualized using the UCSC Genome
Browser. The chromosome-wide patterns of CpG island methylation were highly similar among all the cell lines. Globally, the methylation level of CpG dinucleotides followed similar bimodal distribution: 67% were weakly methylated (<20% methylation), 22% were highly methylated (>80% methylation) and the remaining 1 1 % had intermediate levels of methylation. To distinguish CpG islands with different methylation patterns, a histogram (bin size = 0.05) for the distribution of methylation on all CpG sites within a CpG island was generated. Treating such histograms as 20-component vectors, hierarchical clustering was performed to partition the CpG islands and divide all CpG islands into three clusters based on the similarity of distribution between pluripotent and fibroblast lines. In cluster 1 , the CpG islands (1 ,451 ; 77.3%) have similar distributions in the two cell types (R > 0.5); in cluster 2, CpG islands (252; 13.4%) have less similar distributions (0.5 > R > 0); in cluster 3, CpG islands (173; 9.2%) are anti-correlated (R < 0). Therefore, only a small fraction of CpG islands show cell-type-specific methylation.
[00145] Because CpG islands are not defined in a functional manner, the CpG islands were divided into three categories. The first comprises CpG islands in the regions from 2 kb upstream to 500 bp downstream of TSS. These "upstream regions" often include promoter regions. The second class ("gene body CpG islands") comprises CpG islands in the regions from 500 bp downstream of TSS to the ends of the last exons. The final category comprises CpG islands outside of gene body and promoter regions. CpG islands in each category were further divided into three groups according to CpG density. Consistent with previous findings, most (91.8%) CpG islands in promoter regions were weakly methylated (<20% methylation), 3.4% were highly methylated (>80% methylation) and the remaining 4.8% showed an intermediate level of methylation (20- 80% methylation). The distributions were quite similar among the three groups with different CpG densities. In contrast, only 45.2% of CpG islands in the gene body were weakly methylated, whereas roughly one-third of them (37.7%) were highly methylated. Methylated CpGs tended to locate in islands with low CpG density. In regions outside of gene body and promoter regions, more weakly methylated CpG islands (58.9%) than highly methylated CpG islands (26.6%) were found. Similarly, CpG islands with low CpG density were more methylated. There were 80 genes in the data set that contained both promoter-region CpG islands and gene-body CpG islands. Sixty-two of these genes were weakly methylated in promoter regions. Among these, 48.4% were highly methylated in the gene body and 29.0% displayed weak gene-body methylation.
[00146] In summary, these experiments demonstrated that padlock probes can specifically extract a large number of genomic regions in single-tube reactions for bisulfite sequencing analysis. The degree of multiplexity is at least four orders of magnitude greater than that possible with conventional PCR-based bisulfite sequencing. The high capturing specificity is contributed by the cooperative annealing of the two capturing arms on the target molecules in proper orientation and distance, the selectivity of DNA polymerase and ligase, and the removal of linear DNA with exonuclease.
[00147] Although padlock probes have been successfully applied to exon capturing (Porreca, G.J. et al. (2007) Nat. Methods 4: 93 1 -936) and SNP genotyping (Hardenbol, P. et al. (2003) Nat. Biotechnol. 21 : 673-678), these experiments demonstrate their use with bisulfite-converted DNA with highly skewed nucleotide composition and low sequence complexity. Recent studies showed that most methylation changes are restricted to a very small fraction of the genome outside of CpG islands (Meissner, A. et al. (2008) Nature 454: 766-770; Ball, M.P. et al. (2009) Nat. Biotechnol. 27(4): 361 -8; Irizarry, R.A. et al. (2009) Nat. Genet. 41 : 178- 186). Padlock capture is more efficient than full-genome bisulfite sequencing (Cokus, S.J. et al. (2008) Nature 452: 215-219; Lister, R. et al. (2008) Cell 133: 523-536) for quantifying DNA methylation, as it allows for focused sequencing on the most informative genomic regions. It also provides a much greater flexibility than reduced representation bisulfite sequencing (Meissner, A. et al. (2008) Nature 454: 766-770; Ball, M.P. et al. (2009) Nat. Biotechnol. 27(4): 361 -8) in the selection of genomic targets, because the latter method is limited to genomic regions closely adjacent to the recognition sites of restriction enzymes.
Example 2: Library-free methylation sequencing with bisulfite padlock probes
[00148] The program ppDesigner was developed to aid in the design of efficient padlock probes for bisulfite analysis. It accepts as input the genome of any organism, a list of user-specified arbitrary targets and user-desired probe constraints matching requirements of the experimental protocol. It 'bisulfite-converts' the genome in silico (that is, it changes all cytosines to thymines) and outputs padlock probes to cover the chosen targets while avoiding CpGs on the capturing arms that could be methylated and not converted to be recognized as thymine. ppDesigner uses a back-propagation neural network to predict probe efficiency (Figure 9). This network was previously trained using data from probes for exomic targets
(Gore, A. et al. (201 1 ) Nature 471 : 63-67) based on seven properties. Using bisulfite capture data from the first BSPPs (Deng, J. et al. (2009) Nat. Biotechnol. 27: 353-360), the network was refined with two additional factors. ppDesigner can explain ~50% of the variance in capturing efficiency for genomic DNA and ~20% of the variance in capturing efficiency for bisulfite-converted DNA; additional variation could be due to factors such as variability in oligonucleotide synthesis and sample DNA quality. ppDesigner is extremely flexible and has been used to design a variety of genomic and bisulfite probes for Homo sapiens (Liu, G.H. et al. (201 1) Nature 472: 221 -225; Liu, G.H. et al. (201 1 ) Cell Stem Cell 8: 688-694), Mus musculus (Xu, Y. et al. (2011 ) Mol. Cell 42: 451-464) and Drosophila melanogaster (Wang, H. et al. (2010) Genome Res. 29: 981-988).
100149] Key requirements for methylation analysis of large sample sizes include low cost, simple workflow and automation compatibility. As the cost of DNA sequencing has rapidly decreased, sample processing has become a bottleneck in terms of cost and throughput. A complicated workflow increases variability between samples and reduces power in large-scale studies. To address these issues, we extended a "library-free" protocol (Turner, E.H. et al. (2009) Nat. Methods 6: 315-316) to multiplexed BSPP capture. This method eliminates five steps from Illumina's library-construction protocol such that multiplexed libraries can be generated from DNA in only four steps (Table 4). Table 4 herein shows the number of enzymatic reactions, number of purifications, cost per sample, and mapping rates for first-generation padlock probes, second-generation library-free padlock probes, reduced representation bisulfite sequencing (RRBS), and whole genome bisulfite sequencing (WGBS).
[00150] Table 9: Comparison of bisulfite sequencing methods

includes the cost of ordering 400,000 synthesized probes from LC Sciences and reagents for preparing probes, bisulfite conversion, capture, and sequencing library preparation. Estimates assume that 10,000 samples will be processed.
'Estimated from: Gu et al., (2010) Nat. Methods 7(2): 133- 136.
"Adapted from: Beck et al., (2010) Nat. Biotechnol. 28: 1026-1028.
5Assumes sequencing using an lllumina HiSeq to generate 300 Gbps of sequencing data, with cost of $4920 for a flowcell, $6815 for sequencing reagents, and $2890 for service fee. ($48.75 per Gbps)
[00151] Using multiplexed primers with 6-base pair (bp) barcodes, libraries for 96 samples in 96-well plates were routinely generated and sequenced all at once in a single lllumina HiSeq flowcell. Additionally, barcodes to process 384 samples per batch were designed. As sample-specific barcodes were added, barcoded libraries can be pooled for size selection, which is the most time consuming, contamination-prone and error-prone step if performed individually. The protocol is compatible with the use of multichannel pipettes or liquid-handling devices. It dramatically reduced experimental cost and time, and improved reproducibility and read mapping rates (Tables 4 and 5).
[00152J Table 5: Representative cost per sample for oligonucleotide synthesis, sequencing library construction, and lllumina sequencing
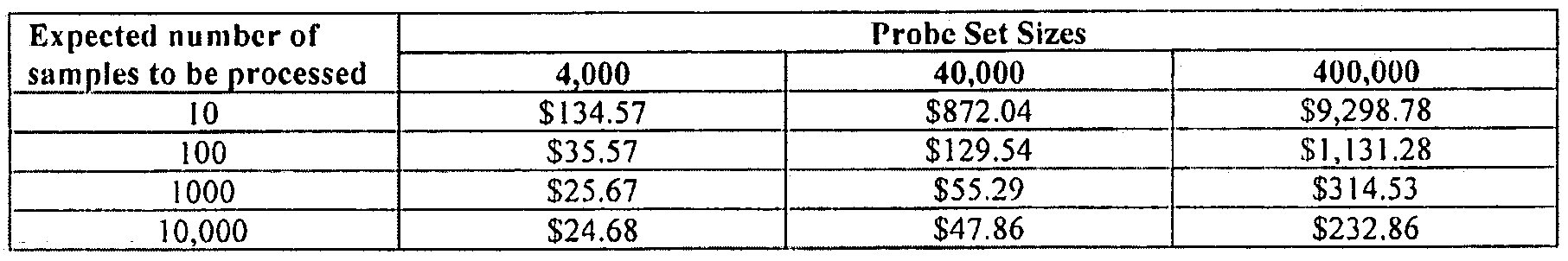
[00153] For large sample sizes, the library preparation cost (including probes) with our protocol was comparable to that of the restricted-representation bisulfite sequencing and whole-genome bisulfite sequencing protocols, and the sequencing cost was much lower than that of whole-genome bisulfite sequencing owing to targeting of CpG sites of interest.
Restricted-representation bisulfite sequencing is more cost-effective than BSPPs, but the former lacks BSPPs' flexibility in selecting specific sites or regions.
[00154] Another bottleneck in sequencing of bisulfite-converted DNA is a lack of computational tools to efficiently analyze sequencing data generated from hundreds of samples. To overcome this issue, an analysis pipeline for read mapping and methylation quantification was developed, called bisReadMapper. In previous padlock probe studies, reads had been mapped only against target regions owing to the computational requirements
of sequence alignment (Deng, J. et al. (2009) Nat. Biotechnol. 27: 353-360). In contrast, bisReadMapper was designed to map to the full genome sequence, allowing processing of data from both targeted and whole-genome sequencing of bisulfite-con verted DNA.
bisReadMapper also determines the origin strand of the read based on base composition and maps reads as if they were fully bisulfite-converted to a fully bisulfite-converted genome sequence, allowing mapping of both bi- and unidirectional bisulfite libraries in an unbiased manner. Another feature is the capability to call single-nucleotide polymorphisms (SNPs) from sequences of bisulfite-converted DNA; this feature not only allows for analysis of allele-specific methylation (Shoemaker, R. et al. (2010) Genome Res. 20: 883-889) but also allows accurate sample tracking in large-scale experiments. Finally, bisReadMapper can call methylation levels at both CpG and non-CpG sites.
[00155] To test this assay, a genome-scale probe set based on our previous results was generated, as well as new information about differential methylation (Deng, J. et al. (2009) Nat. Biotechnol. 27: 353-360). The new design was targeted for evaluation of methylation at genomic locations known to contain differentially methylated regions or differentially methylated sites (DMSs; Irizarry, R.A. et al. (2009) Nat. Genet. 41 : 178-186; Doi, A. et al. (2009) Nat. Genet. 41 : 1350-1353; Lister, R. et al. (2009) Nature 462: 315-322; Figueroa, M.E. et al. (2010) Cancer Cell 17: 13-27), transcriptional repressor CTCF binding sites and DNase 1 hypersensitive regions. All microRNA genes and all promoters for human U.S. National Center for Biotechnology Information reference sequence (RefSeq) genes were targeted. Using ppDesigner, -330,000 padlock probes that covered 140,749 non-overlapping regions with a total size of 34 megabases were designed. Capturing experiments and end- sequencing were performed, and found that these probes were slightly more specific (-96% on-target) and uniform than previous probes (Deng, J. et al. (2009) Nat. Biotechnol. 27: 353- 360) (Figure 10). To improve uniformity, the experimental capturing performance of these probes was normalized using subsetting and suppressor oligonucleotides as described previously (Deng, J. et al. (2009) Nat. Biotechnol. 27: 353-360). Roughly 500,000 CpG sites with ~4 gigabases of sequencing reads could be characterized, and additional sites became callable with deeper sequencing (Figures 1 1 and 12). A schematic for the padlock probes is illustrated in Figure 17.
Materials and Methods
[00156] Bisulfite padlock probe production (oligonucleotides from Agilent)
[00157J Libraries of oligonucleotides (~150 nt) were synthesized by ink-jet printing on programmable microarrays (Agilent Technologies) and released to form a combined library of 330,000 oligonucleotides. The oligonucleotides were amplified by PCR in 96 reactions (100 μΐ each) with 0.02 nM template oligonucleotide, 400 nM each of pAP l V61 U primer and AP2V6 primer (Table 6), and 50 μΐ of APA SYBG fast Universal 2* qPCR Master Mix (Kapabiosystems) at 95°C for 30 seconds, 15-16 cycles of 95°C for 3 seconds; 55°C for 30 seconds; and 60°C for 20 seconds and 60°C for 2 minutes.
[00158] Table 6: Primer sequences used for padlock probe production, padlock capture, sequencing library construction, and Ulumina sequencing

CP-2-SeqRead 1.x (Read 1 ) 5'-TACACGCCTATCGGGAAGCTGAAG-3 ' (SEQ ID NO: 32)
CP-2-lndSeq.x (IndexRead) 5'-ACACTACCGTCGGATGGCAGACCG-3 ' (SEQ ID NO: 33)
CP-2-SeqRead l .y (Read l ) 5'-TACACGCCTATCCGACGGTAGTGT-3' (SEQ ID NO: 34)
CP-2-IndSeq.y (IndexRead) 5'-CTTCAGCTTCCCGATGGCAGACCG-3' (SEQ ID NO: 35)
* Indicates a phosphorothioatc bond
[00159] The amplicons were purified by ethanol precipitation and repurified with Qiaquick PCR purification columns (Qiagen). Approximately 20 μ of the purified amplicons were digested with 50 units of lambda exonuclease (5 U/μΙ; New England Biolabs (NEB)) at 37°C for 1 hour in lambda exonuclease reaction buffer. The resulting single-strand amplicons were purified with Qiaquick PCR purification column. Approximately 5-8 μg of single strand amplicons were subsequently digested with 5 units USER ( 1 U μΐ-ΐ , NEB) at 37°C for 1 hour. The digested DNA was annealed to 5.88 μΜ KE-Dp>iU-V6 guide oligo (Table 6) and denatured at 94°C for 2 minutes decreased the temperature to 37°C and incubated at 37 °C for 3 minutes. The mixture was digested with 50 U of Dpnll (10 U μΐ-ΐ , NEB) in NEBuffer DpnW at 37°C for 2 hours. The mixture was further digested with 5 U of USER at 37°C for 2 hours followed by enzyme inactivation at 75°C for 20 minutes. The USER and Z /wIi-digested DNA was purified with Qiaquick PCR purification column. The single-strand 102-nt probes were purified with 6% denaturing PAGE (6% TB-urea two dimensional (2D) gel; Invitrogen).
[00160] Bisulfite padlock probe production (oligonucleotides from LC Sciences)
[00161 ] The oligonucleotides (100 nt) were synthesized using a programmable microfluidic microarray platform (LC Sciences) and released to form a mix of 3,91 8 oligonucleotides. The oligonucleotides were amplified by two-step PCR in a 200 μΐ reaction with 1 nM template oligonucleotides, 400 nM each of eMIP_CA l_F primer and
eMIP_CAl_R primer (Table 6), and 100 μΐ of KAPA SYBR fast Universal qPCR Master Mix at 95°C for 30 seconds, 5 cycles of 95°C for 5 seconds; 52°C for 1 minute; and 72°C for 30 seconds, 10-12 cycles of 95°C for 5 seconds; 60°C for 30 seconds; and 72°C for 30 seconds, and 72°C for 2 minutes. The resultant amplicons were purified with Qiaquick PCR purification columns and re-amplified in 32 PCRs (100 μΐ each) with 0.02 nM first round amplicons, 400 nM each of eMIP CA l F primer and eMIP_CAl_R primer and 50 μΐ of KAPA SYBR fast Universal qPCR master mix at 95°C for 30 seconds, 1 3-15 cycles of 95°C
for 5 seconds; 60°C for 30 seconds; and 72°C for 30 seconds and 72°C for 2 minutes. The resultant amplicons were purified by ethanol precipitation and repurified with Qiaquick PGR purification columns as described above. Approximately 4 ng of the purified amplicons were digested with 100 U of Nt .AM (100 U/μΙ, NEB) at 37°C for 1 hour in NEBuffer 2. The enzyme was heat-inactivated at 80°C for 20 minutes. The digested amplicons were then incubated with 100 U of Nb.5riDI (10 U μΐ-ΐ , NEB) at 65°C for 1 hour. The nicked DNA was purified by Qiaquick PCR purification column. The probe molecules (~70 bases) were purified by 6% denaturing PAGE (6% TB-urea 2D gel).
[00162] Sample preparation and capture
[00163] Genomic DNA was extracted using the AllPrep DNA/RNA Mini kit (Qiagen) and bisulfite-converted with the EZ-96 DNA methylation Gold kit (Zymoresearch) in a 96- well plate. Normalized amount of padlock probes, 200 ng of bisulfite converted gDNA and 4.2 nM oligo suppressor were mixed in 25 μΐ 1 * Ampligase buffer (Epicentre) in 96-well plate, denatured at 95°C for 10 minutes, gradually lowered the temperature at 0.02°C/s to 55°C in a thermocycler and hybridized at 55°C for 20 hours. 2.5 μΐ of SLN (Stoffel fragment, ligase, nucleotides) mix (100 μΜ dNTP, 2 U μΐ-ΐ AmpliTaq Stoffel Fragment (ABI) and 0.5 U/μΙ Ampligase (Epicentre) in 1 χ Ampligase buffer) was added to the reaction for gap-filling. For circularization, the reactions were incubated at 55°C for 20 hours, followed by enzyme inactivation at 94°C for 2 minutes. To digest linear DNA after circularization, 2 μΐ of exonuclease mix (10 U/μΙ exonuclease 1 and 100 U/μΙ exonuclease III, USB) was added to the reactions, and the reactions were incubated at 37°C for 2 hours and then inactivated at 94°C for 2 minutes.
[00164] Capture-circles amplification (library-free BSPP protocol, Agilent oligonucleotides)
[00165] Ten microliters of circularized DNA was amplified and barcoded in 100-μ1 reactions with 400 nM each of AmpF6.3Sol primer (Table 6) and AmpR6.3 indexing primer (Table 6), 0.4x SYBR Green I (Invitrogen) and 50 μΐ Phusion High-Fidelity 2 master mix (NEB) at 98°C for 30 seconds, 5 cycles of 98°C for 10 seconds; 58°C for 20 seconds; and 72°C for 20 seconds, 9-12 cycles of 98°C for 10 seconds; and 72°C for 20 seconds and 72°C for 3 minutes.
[00166) Capture-circles amplification (library-free BSPP protocol, LC Sciences oligonucleotides)
[00167] Ten microliters of circularized DNA was amplified in a 100-μ1 reaction with 200 nM each of CP-2-FA primer and CP-2-RA primer (Table 6) and 50 μΐ KAPA SYBR fast Universal qPCR Master Mix at 98°C for 30 seconds, 5 cycles of 98°C for 10 seconds; 52°C for 30 seconds; and 72°C for 30 seconds, 15 cycles of 98°C for 10 seconds; 60°C for 30 seconds; and 72°C for 30 seconds and 72°C for 3 minutes. The resultant amplicons with the corresponding expected size of -260 bp were purified by 6% PAGE (6% 5 -well gel, Invitrogen) and resuspended in 12 μΐ of TE buffer. Thirty percent of the gel-purified amplicons were reamplified and barcoded in a 100-μ1 reaction with 200 nM each of two different sets of primers to enable single-end sequencing of both ends of the amplicons (CP- 2-FA.IndSol primer and CP-2-RA.Sol primer or Switch. CP-2-FA and Switch.CP-2- RA.IndSol) and 50 μΐ KAPA SYBR fast Universal qPCR Master Mix at 98°C for 30 seconds, 4 cycles of 98°C for 10 seconds; 54°C for 30 seconds; and 72°C for 30 seconds and 72°C for 3 minutes.
[00168] Primer barcode design for multiplexing
[00169] An in-house Perl script was written to randomly generate 6-nt-long sequences.
A sequence was kept if it did not have more than two matching positions with another accepted barcode and if it had 2-4 guanines or cytosines. The script reiterated until the desired number of barcodes have been obtained. A total of 384 primers were designed (Table 7).
Table 7: Sequences of multiplexed barcoded primers used in the library-free

Ind5 CACTGT CAAGCAGAAGACGGCATACGAGATCACTGTGCTAGGAACGATGAGCCT 40
CCAAC
Ind6 ATTGGC CAAGCAGAAGACGGCATACGAGATATTGGCGCTAGGAACGATGAGCCT 41
CCAAC
Ind7 GATCTG CAAGCAGAAGACGGCATACGAGATGATCTGGCTAGGAACGATGAGCCT 42
CCAAC
Ind8 TCAAGT CAAGCAGAAGACGGCATACGAGATTCAAGTGCTAGGAACGATGAGCCT 43
CCAAC
lnd9 CTGATC CAAGCAGAAGACGGCATACGAGATCTGATCGCTAGGAACGATGAGCCT 44
CCAAC
Ind l O AAGCTA CAAGCAGAAGACGGCATACGAGATAAGCTAGCTAGGAACGATGAGCCT 45
CCAAC
Ind l l GTAGCC CAAGCAGAAGACGGCATACGAGATGTAGCCGCTAGGAACGATGAGCCT 46
CCAAC
lnd l 2 TACAAG CAAGCAGAAGACGGCATACGAGATTACAAGGCTAGGAACGATGAGCCT 47
CCAAC
Ind l 3 TCATGG CAAGCAGAAGACGGCATACGAGATTCATGGGCTAGGAACGATGAGCCT 48
CCAAC
Ind l 4 TGTCTT CAAGCAGAAGACGGCATACGAGATTGTCTTGCTAGGAACGATGAGCCT 49
CCAAC
lnd l 5 AGGAA CAAGCAGAAGACGGCATACGAGATAGGAAGGCTAGGAACGATGAGCC 50
G TCCAAC
Ind l 6 AACCCC CAAGCAGAAGACGGCATACGAGATAACCCCGCTAGGAACGATGAGCCT 5 1
CCAAC
lnd l 7 GATGAG CAAGCAGAAGACGGCATACGAGATGATGAGGCTAGGAACGATGAGCCT 52
CCAAC
Ind l 8 TGAACT CAAGCAGAAGACGGCATACGAGATTGAACTGCTAGGAACGATGAGCCT 53
CCAAC
Ind l 9 TGCGTC CAAGCAGAAGACGGCATACGAGATTGCGTCGCTAGGAACGATGAGCCT 5
CCAAC
Ind20 GACAGG CAAGCAGAAGACGGCATACGAGATGACAGGGCTAGGAACGATGAGCCT 55
. CCAAC
Ind2 1 GGGTTG CAAGCAGAAGACGGCATACGAGATGGGTTGGCTAGGAACGATGAGCCT 56
CCAAC
Ind22 TCCGAG CAAGCAGAAGACGGCATACGAGATTCCGAGGCTAGGAACGATGAGCCT 57
CCAAC
!nd23 TTTCGA CAAGCAGAAGACGGCATACGAGA'rrrTCGAGCTAGGAACGATGAGCCT 58
CCAAC
lnd24 GCGAAT CAAGCAGAAGACGGCATACGAGATGCGAATGCTAGGAACGATGAGCCT 59
CCAAC
Ind25 GCAGTA CAAGCAGAAGACGGCATACGAGATGCAGTAGCTAGGAACGATGAGCCT 60
CCAAC
Tnd26 TCACGA CAAGCAGAAGACGGCATACGAGATTCACGAGCTAGGAACGATGAGCCT 61
CCAAC
Ind27 CGCGTA CAAGCAGAAGACGGCATACGAGATCGCGTAGCTAGGAACGATGAGCCT 62
CCAAC
Ind28 GCACCT CAAGCAGAAGACGGCATACGAGATGCACCTGCTAGGAACGATGAGCCT 63
CCAAC
Ind29 GTTCGT CAAGCAGAAGACGGCATACGAGATG'ITCGTGCTAGGAACGATGAGCCT 64
CCAAC
Ind30 CACTAA CAAGCAGAAGACGGCATACGAGATCACTAAGCTAGGAACGATGAGCCT 65
CCAAC
Ind3 1 GTGGTG CAAGCAGAAGACGGCATACGAGATGTGGTGGCTAGGAACGATGAGCCT 66
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind32 CCTTGC CAAGCAGAAGACGGCATACGAGATCCTTGCGCTAGGAACGATGAGCCT 67
CCAAC
Ind33 GTTGCG CAAGCAGAAGACGGCATACGAGATGTTGCGGCTAGGAACGATGAGCCT 68
CCAAC
Ind34 TCAGTT CAAGCAGAAGACGGCATACGAGATTCAGTTGCTAGGAACGATGAGCCT 69
CCAAC
Ind35 CCCGAT CAAGCAGAAGACGGCATACGAGATCCCGATGCTAGGAACGATGAGCCT 70
CCAAC
Ind36 TGCTTG CAAGCAGAAGACGGCATACGAGATTGCTTGGCTAGGAACGATGAGCCT 71
CCAAC
lnd37 TGTAGC CAAGCAGAAGACGGCATACGAGATTGTAGCGCTAGGAACGATGAGCCT 72
CCAAC
Ind38 GCGTGA CAAGCAGAAGACGGCATACGAGATGCGTGAGCTAGGAACGATGAGCCT 73
CCAAC
!nd39 CTCTGG CAAGCAGAAGACGGCATACGAGATCTCTGGGCTAGGAACGATGAGCCT 74
CCAAC
Ind40 CCGTCA CAAGCAGAAGACGGCATACGAGATCCGTCAGCTAGGAACGATGAGCCT 75
CCAAC
Ind41 GTTCCC CAAGCAGAAGACGGCATACGAGATGTTCCCGCTAGGAACGATGAGCCT 76
CCAAC
Ind42 CTTTTC CAAGCAGAAGACGGCATACGAGATCTT]TCGCTAGGAACGATGAGCCT 77
CCAAC
Ind43 GGCACT CAAGCAGAAGACGGCATACGAGATGGCACTGCTAGGAACGATGAGCCT 78
CCAAC
Ind44 GGATGC CAAGCAGAAGACGGCATACGAGATGGATGCGCTAGGAACGATGAGCCT 79
CCAAC
Ind45 CGTAGT CAAGCAGAAGACGGCATACGAGATCGTAGTGCTAGGAACGATGAGCCT 80
CCAAC
Ind46 GAAATG CAAGCAGAAGACGGCATACGAGATGAAATGGCTAGGAACGATGAGCCT 81
CCAAC
Jnd47 GGAGA CAAGCAGAAGACGGCATACGAGATGGAGAGGCTAGGAACGATGAGCC 82
G TCCAAC
Ind48 TAACGT CAAGCAGAAGACGGCATACGAGATTAACGTGCTAGGAACGATGAGCCT 83
CCAAC
Ind49 ACACAG CAAGCAGAAGACGGCATACGAGATACACAGGCTAGGAACGATGAGCCT 84
CCAAC
lnd50 AAAGGT CAAGCAGAAGACGGCATACGAGATAAAGGTGCTAGGAACGATGAGCCT 85
CCAAC
lnd51 GCGA'l'A CAAGCAGAAGACGGCATACGAGATGCGATAGCTAGGAACGATGAGCCT 86
CCAAC
lndS2 CGTGTC CAAGCAGAAGACGGCATACGAGATCGTGTCGCTAGGAACGATGAGCCT 87
CCAAC
lnd53 GTAGAA CAAGCAGAAGACGGCATACGAGATGTAGAAGCTAGGAACGATGAGCCT 88
CCAAC
lnd54 GGACGT CAAGCAGAAGACGGCATACGAGATGGACGTGCTAGGAACGATGAGCCT 89
CCAAC
Ind55 AGTCGA CAAGCAGAAGACGGCATACGAGATAGTCGAGCTAGGAACGATGAGCCT 90
CCAAC
lnd56 GTCTGA CAAGCAGAAGACGGCATACGAGATGTCTGAGCTAGGAACGATGAGCCT 91
CCAAC
Ind57 GAAGG CAAGCAGAAGACGGCATACGAGATGAAGGAGCTAGGAACGATGAGCC 92
A TCCAAC
Ind58 ATGCTG CAAGCAGAAGACGGCATACGAGATATGCTGGCTAGGAACGATGAGCCT 93
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind59 TCTATC CAAGCAGAAGACGGCATACGAGATrCTATCGCTAGGAACGATGAGCCT 94
CCAAC
lnd60 ATCTGT CAAGCAGAAGACGGCATACGAGATATCTGTGCTAGGAACGATGAGCCT 95
CCAAC
Ind61 ATAGAG CAAGCAGAAGACGGCATACGAGATATAGAGGCTAGGAACGATGAGCCT 96
CCAAC
lnd62 GCTAAA CAAGCAGAAGACGGCATACGAGATGCTAAAGCTAGGAACGATGAGCCT 97
CCAAC
lnd63 ACCAGG CAAGCAGAAGACGGCATACGAGATACCAGGGCTAGGAACGATGAGCCT 98 '
CCAAC
Ind64 CCAACT CAAGCAGAAGACGGCATACGAGATCCAACTGCTAGGAACGATGAGCCT 99
CCAAC
Ind65 AAGGA CAAGCAGAAGACGGCATACGAGATAAGGAAGCTAGGAACGATGAGCC 100
A TCCAAC
Ind66 CCTCCA CAAGCAGAAGACGGCATACGAGATCCTCCAGCTAGGAACGATGAGCCT 101
CCAAC
Ind67 CACGTC CAAGCAGAAGACGGCATACGAGATCACGTCGCTAGGAACGATGAGCCT 102
CCAAC
Ind68 CATAAC CAAGCAGAAGACGGCATACGAGATCATAACGCTAGGAACGATGAGCCT 103
CCAAC
Ind69 CCATAT CAAGCAGAAGACGGCATACGAGATCCATATGCTAGGAACGATGAGCCT 104
CCAAC
Ind70 GAAGTC CAAGCAGAAGACGGCATACGAGATGAAGTCGCTAGGAACGATGAGCCT 105
CCAAC
Ind71 CAAAGA CAAGCAGAAGACGGCATACGAGATCAAAGAGCTAGGAACGATGAGCCT 106
CCAAC
Ind72 TGGCAG CAAGCAGAAGACGGCATACGAGATTGGCAGGCTAGGAACGATGAGCCT 107
CCAAC
Ind73 GAGTCC CAAGCAGAAGACGGCATACGAGATGAGTCCGCTAGGAACGATGAGCCT 108
CCAAC
Ind74 TCGCCA CAAGCAGAAGACGGCATACGAGATTCGCCAGCTAGGAACGATGAGCCT 109
CCAAC
Ind75 AAGTCG CAAGCAGAAGACGGCATACGAGATAAGTCGGCTAGGAACGATGAGCCT 1 10
CCAAC
Ind76 AATAGG CAAGCAGAAGACGGCATACGAGATAATAGGGCTAGGAACGATGAGCCT 1 1 1
CCAAC
lnd77 ACCCGT CAAGCAGAAGACGGCATACGAGATACCCGTGCTAGGAACGATGAGCCT 1 12
CCAAC
Ind78 AACACG CAAGCAGAAGACGGCATACGAGATAACACGGCTAGGAACGATGAGCCT 1 13
CCAAC
)nd79 GCTTGG CAAGCAGAAGACGGCATACGAGATGCTTGGGCTAGGAACGATGAGCCT 1 14
CCAAC
Ind80 TTACCA CAAGCAGAAGACGGCATACGAGATTTACCAGCTAGGAACGATGAGCCT 1 15
CCAAC
lnd8 l CCAGGT CAAGCAGAAGACGGCATACGAGATCCAGGTGCTAGGAACGATGAGCCT 1 16
CCAAC
Ind82 CGTTTG CAAGCAGAAGACGGCATACGAGATCGTTTGGCTAGGAACGATGAGCCT 1 17
CCAAC
lnd83 GACCAC CAAGCAGAAGACGGCATACGAGATGACCACGCTAGGAACGATGAGCCT 1 18
CCAAC
Ind84 ACAAGA CAAGCAGAAGACGGCATACGAGATACAAGAGCTAGGAACGATGAGCCT 1 19
CCAAC
Ind85 ACCGCA CAAGCAGAAGACGGCATACGAGATACCGCAGCTAGGAACGATGAGCCT 120
CCAAC
Barcode Barcode Primer SEQ ID ID NO: lnd86 TGGGTA CAAGCAGAAGACGGCATACGAGATTGGGTAGCTAGGAACGATGAGCCT 12 1
CCAAC
lnd87 ATTCCG CAAGCAGAAGACGGCATACGAGATATTCCGGCTAGGAACGATGAGCCT 122
CCAAC
lnd88 GAATGT CAAGCAGAAGACGGCATACGAGATGAATGTGCTAGGAACGATGAGCCT 123
CCAAC
Ind89 GCTGAT CAAGCAGAAGACGGCATACGAGATGCTGATGCTAGGAACGATGAGCCT 124
CCAAC
Ind90 AGTGCT CAAGCAGAAGACGGCATACGAGATAGTGCTGCTAGGAACGATGAGCCT 125
CCAAC
lnd91 CAGGGA CAAGCAGAAGACGGCATACGAGATCAGGGAGCTAGGAACGATGAGCCT 126
CCAAC
lnd92 CATGCG CAAGCAGAAGACGGCATACGAGATCATGCGGCTAGGAACGATGAGCCT 127
CCAAC
Ind93 TGCCTA CAAGCAGAAGACGGCATACGAGATTGCCTAGCTAGGAACGATGAGCCT 128
CCAAC
lnd94 CTATAC CAAGCAGAAGACGGCATACGAGATCTATACGCTAGGAACGATGAGCCT 129
CCAAC
lnd95 CCGAGT CAAGCAGAAGACGGCATACGAGATCCGAGTGCTAGGAACGATGAGCCT 130
CCAAC
Ind96 ACCTGC CAAGCAGAAGACGGCATACGAGATACCTGCGCTAGGAACGATGAGCCT 131
CCAAC
Ind97 CAGGAC CAAGCAGAAGACGGCATACGAGATCAGGACGCTAGGAACGATGAGCCT 132
CCAAC
lnd98 AAGATG CAAGCAGAAGACGGCATACGAGATAAGATGGCTAGGAACGATGAGCCT 133
CCAAC
Ind99 GGC'ITC CAAGCAGAAGACGGCATACGAGATGGCTTCGCTAGGAACGATGAGCCT 134
CCAAC
Ind ! OO GTGCTT CAAGCAGAAGACGGCATACGAGATGTGCTTGCTAGGAACGATGAGCCT 135
CCAAC
Ind l Ol TGCGCA CAAGCAGAAGACGGCATACGAGATTGCGCAGCTAGGAACGATGAGCCT 136
CCAAC
Indl 02 ACTAGC CAAGCAGAAGACGGCATACGAGATACTAGCGCTAGGAACGATGAGCCT 137
CCAAC
Indl03 TCGAAG CAAGCAGAAGACGGCATACGAGATTCGAAGGCTAGGAACGATGAGCCT 138.
CCAAC
Ind l 04 AGACTA CAAGCAGAAGACGGCATACGAGATAGACTAGCTAGGAACGATGAGCCT 139
CCAAC '
lnd ) 05 CGGGTT CAAGCAGAAGACGGCATACGAGATCGGGTTGCTAGGAACGATGAGCCT 140
CCAAC
Ind l 06 TGACTG CAAGCAGAAGACGGCATACGAGATTGACTGGCTAGGAACGATGAGCCT 14 1
CCAAC
Ind l 07 TTGTGT CAAGCAGAAGACGGCATACGAGATTTGTGTGCTAGGAACGATGAGCCT 142
CCAAC
Ind l 08 TCGCTG CAAGCAGAAGACGGCATACGAGATTCGCTGGCTAGGAACGATGAGCCT 143
CCAAC
Indl09 GATACA CAAGCAGAAGACGGCATACGAGATGATACAGCTAGGAACGATGAGCCT 144
CCAAC
Ind l l O TCCTTA CAAGCAGAAGACGGCATACGAGATTCCTTAGCTAGGAACGATGAGCCT 145
CCAAC
Ind l 1 1 CGATTT CAAGCAGAAGACGGCATACGAGATCGATrTGCTAGGAACGATGAGCCT 146
CCAAC
Ind l 12 TTACGG CAAGCAGAAGACGGCATACGAGATTTACGGGCTAGGAACGATGAGCCT 147
CCAAC
Barcode Barcode Primer SEQ ID TD NO:
Ind ) 13 TGTGAC CAAGCAGAAGACGGCATACGAGATTGTGACGCTAGGAACGATGAGCCT 148
CCAAC
Ind l 14 TTGGAA CAAGCAGAAGACGGCATACGAGATTTGGAAGCTAGGAACGATGAGCCT 149
CCAAC
Ind l 1 5 ACCATA CAAGCAGAAGACGGCATACGAGATACCATAGCTAGGAACGATGAGCCT 150
CCAAC
Ind l 16 GTCGAG CAAGCAGAAGACGGCATACGAGATGTCGAGGCTAGGAACGATGAGCCT 15 1 '
CCAAC
Indl 17 ACTGCC CAAGCAGAAGACGGCATACGAGATACTGCCGCTAGGAACGATGAGCCT 152
CCAAC
Indl 18 TCGGGT CAAGCAGAAGACGGCATACGAGATTCGGGTGCTAGGAACGATGAGCCT 153
CCAAC
Indl 19 GGCATG CAAGCAGAAGACGGCATACGAGATGGCATGGCTAGGAACGATGAGCCT 154 '
CCAAC
Indl20 GT'ITCT CAAGCAGAAGACGGCATACGAGATGTTTCTGCTAGGAACGATGAGCCT 155
CCAAC
Indl 21 ACCAAT CAAGCAGAAGACGGCATACGAGATACCAATGCTAGGAACGATGAGCCT 156
CCAAC
Ind l 22 ATGCAT CAAGCAGAAGACGGCATACGAGATATGCATGCTAGGAACGATGAGCCT 157
CCAAC
Ind l 23 TACGGC CAAGCAGAAGACGGCATACGAGATTACGGCGCTAGGAACGA TGAGCCT 158
CCAAC
Indl24 AGTCCC CAAGCAGAAGACGGCATACGAGATAGTCCCGCTAGGAACGATGAGCCT 159
CCAAC
Indl 25 CTGCAG CAAGCAGAAGACGGCATACGAGATCTGCAGGCTAGGAACGATGAGCCT 160
CCAAC
Indl 26 CTGTTG CAAGCAGAAGACGGCATACGAGATCTGTTGGCTAGGAACGATGAGCCT 161
CCAAC
Indl 27 CGGACA CAAGCAGAAGACGGCATACGAGATCGGACAGCTAGGAACGATGAGCCT 162
CCAAC
Ind l 28 TAAGCG CAAGCAGAAGACGGCATACGAGATTAAGCGGCTAGGAACGATGAGCCT 163
CCAAC
Ind l 29 GAGAGT CAAGCAGAAGACGGCATACGAGATGAGAGTGCTAGGAACGATGAGCCT 164
CCAAC
Ind l30 TACCCG CAAGCAGAAGACGGCATACGAGATTACCCGGCTAGGAACGATGAGCCT 165
CCAAC
Indl 31 'ITCGTA CAAGCAGAAGACGGCATACGAGATTTCGTAGCTAGGAACGATGAGCCT 166
CCAAC
Indl 32 AAAGTG CAAGCAGAAGACGGCATACGAGATAAAGTGGCTAGGAACGATGAGCCT 167
CCAAC
Indl 33 TTTGGT CAAGCAGAAGACGGCATACGAGATTTTGGTGCTAGGAACGATGAGCCT 168
CCAAC
Ind l 34 GTCCCT CAAGCAGAAGACGGCATACGAGATGTCCCTGCTAGGAACGATGAGCCT 169
CCAAC
IncJ 135 TAGCTT CAAGCAGAAGACGGCATACGAGATTAGCTTGCTAGGAACGATGAGCCT 170
CCAAC
Ind l 36 GCACTG CAAGCAGAAGACGGCATACGAGATGCACTGGCTAGGAACGATGAGCCT 171
CCAAC
Ind l 37 ACTATG CAAGCAGAAGACGGCATACGAGATACTATGGCTAGGAACGATGAGCCT 172
CCAAC
Ind l 38 GAACTT CAAGCAGAAGACGGCATACGAGATGAACTTGCTAGGAACGATGAGCCT 173
CCAAC
Ind l 39 TTTGAG CAAGCAGAAGACGGCATACGAGATTTTGAGGCTAGGAACGATGAGCCT 174
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind l40 AGGCCT CAAGCAGAAGACGGCATACGAGATAGGCCTGCTAGGAACGATGAGCCT 175
CCAAC
Ind l4 l ACACGC CAAGCAGAAGACGGCATACGAGATACACGCGCTAGGAACGATGAGCCT 176
CCAAC
lndl42 TTGTCG CAAGCAGAAGACGGCATACGAGATTTGTCGGCTAGGAACGATGAGCCT 177
CCAAC
Ind l43 TCTCAC CAAGCAGAAGACGGCATACGAGATTCTCACGCTAGGAACGATGAGCCT 178
CCAAC
lnd l 44 TAGACC CAAGCAGAAGACGGCATACGAGATTAGACCGCTAGGAACGATGAGCCT 179
CCAAC
Ind 145 CCTAAG CAAGCAGAAGACGGCATACGAGATCCTAAGGCTAGGAACGATGAGCCT 1 80
CCAAC
Ind l46 GTATCG CAAGCAGAAGACGGCATACGAGATGTATCGGCTAGGAACGATGAGCCT 181
CCAAC
Ind 147 TCCAGA CAAGCAGAAGACGGCATACGAGATTCCAGAGCTAGGAACGATGAGCCT 182
CCAAC
Ind l48 AGGTGA CAAGCAGAAGACGGCATACGAGATAGGTGAGCTAGGAACGATGAGCCT 183
CCAAC
Ind 149 CCCATC CAAGCAGAAGACGGCATACGAGATCCCATCGCTAGGAACGATGAGCCT 184
CCAAC
Indl 50 TTGCAC CAAGCAGAAGACGGCATACGAGATTTGCACGCTAGGAACGATGAGCCT 185
CCAAC
Ind 1 5 1 AACTCA CAAGCAGAAGACGGCA TACGAGATAACTCAGCTAGGAACGATGAGCCT 186
CCAAC
Ind 1 52 CGTATA CAAGCAGAAGACGGCATACGAGATCGTATAGCTAGGAACGATGAGCCT 187
CCAAC
Ind 153 AGCGAA CAAGCAGAAGACGGCATACGAGATAGCGAAGCTAGGAACGATGAGCCT 188
CCAAC
Ind 154 ACGGCT CAAGCAGAAGACGGCATACGAGATACGGCTGCTAGGAACGATGAGCCT 189
CCAAC
Ind 155 AGTGAG CAAGCAGAAGACGGCATACGAGATAGTGAGGCTAGGAACGATGAGCCT 190
CCAAC
Ind 1 56 TTTCTC CAAGCAGAAGACGGCATACGAGATTTTCTCGCTAGGAACGATGAGCCT 191
CCAAC
Ind 1 57 GCTCTA CAAGCAGAAGACGGCATACGAGATGCTCTAGCTAGGAACGATGAGCCT 192
CCAAC
Ind l 58 ACTTGA CAAGCAGAAGACGGCATACGAGATACTTGAGCTAGGAACGATGAGCCT 193
CCAAC
Ind 159 CGGTTC CAAGCAGAAGACGGCATACGAGATCGGTI'CGCTAGGAACGATGAGCCT 194
CCAAC
Ind 160 CATC AT CAAGCAGAAGACGGCATACGAGATCATCATGCTAGGAACGATGAGCCT 195
CCAAC
Indl61 CAAACG CAAGCAGAAGACGGCATACGAGATCAAACGGCTAGGAACGATGAGCCT 196
CCAAC
Ind 162 CTATGT CAAGCAGAAGACGGCATACGAGATCTATGTGCTAGGAACGATGAGCCT 197
CCAAC
Ind 163 AGCGTT CAAGCAGAAGACGGCA TACGAGATAGCGTTGCTAGGAACGATGAGCCT 198
CCAAC
lnd l 64 AAGACT CAAGCAGAAGACGGCATACGAGATAAGACTGCTAGGAACGATGAGCCT 199
CCAAC
lndl 65 CGATAA CAAGCAGAAGACGGCA TACGAGATCGATAAGCTAGGAACGATGAGCCT 200
CCAAC
Ind 166 CGGCTA CAAGCAGAAGACGGCATACGAGATCGGCTAGCTAGGAACGATGAGCCT 201
CCAAC
Barcode Barcode Primer SEQ ID ID NO: lnd l 67 TATACG CAAGCAGAAGACGGCATACGAGA'ITATACGGCTAGGAACGATGAGCCT 202
CCAAC
Ind l68 GAAACC CAAGCAGAAGACGGCATACGAGATGAAACCGCTAGGAACGATGAGCCT 203
CCAAC
Indl69 GAACCG CAAGCAGAAGACGGCATACGAGATGAACCGGCTAGGAACGATGAGCCT 204
CCAAC
Ind l70 TTAGGC CAAGCAGAAGACGGCATACGAGATTTAGGCGCTAGGAACGATGAGCCT 205
CCAAC
lndl71 GCCTTT CAAGCAGAAGACGGCATACGAGATGCCTTTGCTAGGAACGATGAGCCT 206
CCAAC
Ind l 72 GTTGGA CAAGCAGAAGACGGCATACGAGATGTTGGAGCTAGGAACGATGAGCCT 207
CCAAC
Ind l 73 GTACGC CAAGCAGAAGACGGCATACGAGATGTACGCGCTAGGAACGATGAGCCT 208
CCAAC
lnd l 74 TGGAAC CAAGCAGAAGACGGCATACGAGATTGGAACGCTAGGAACGATGAGCCT 209
CCAAC
Ind l75 AACAGA CAAGCAGAAGACGGCATACGAGATAACAGAGCTAGGAACGATGAGCCT 210
CCAAC
Ind l 76 AAGCAC CAAGCAGAAGACGGCATACGAGATAAGCACGCTAGGAACGATGAGCCT 21 1
CCAAC
lnd ! 77 ATGGTA CAAGCAGAAGACGGCATACGAGATATGGTAGCTAGGAACGATGAGCCT 212
CCAAC
Ind178 TCGTTC CAAGCAGAAGACGGCATACGAGATTCGTTCGCTAGGAACGATGAGCCT 213
CCAAC
lndl79 CTTCTA CAAGCAGAAGACGGCATACGAGATCTTCTAGCTAGGAACGATGAGCCT 214
CCAAC
Ind l SO TGGGAT CAAGCAGAAGACGGCATACGAGATTGGGATGCTAGGAACGATGAGCCT 215
CCAAC
Ind l S l ATCGCC CAAGCAGAAGACGGCATACGAGATATCGCCGCTAGGAACGATGAGCCT 216
CCAAC
lndl 82 AGTTGG CAAGCAGAAGACGGCATACGAGATAGTTGGGCTAGGAACGATGAGCCT 217
CCAAC
Ind l 83 AGCTCT CAAGCAGAAGACGGCATACGAGATAGCTCTGCTAGGAACGATGAGCCT 218
CCAAC
Ind l 84 GACGGT CAAGCAGAAGACGGCATACGAGATGACGGTGCTAGGAACGATGAGCCT 219
CCAAC
Ind l 85 CTCAGC CAAGCAGAAGACGGCATACGAGATCTCAGCGCTAGGAACGATGAGCCT 220
CCAAC
Ind i 86 CTTAGG CAAGCAGAAGACGGCATACGAGATCTTAGGGCTAGGAACGATGAGCCT 221
CCAAC
Ind l 87 CGACAG CAAGCAGAAGACGGCATACGAGATCGACAGGCTAGGAACGATGAGCCT 222
CCAAC
Ind l 88 ACATGT CAAGCAGAAGACGGCATACGAGATACATGTGCTAGGAACGATGAGCCT 223
CCAAC
Indl 89 ATACGA CAAGCAGAAGACGGCATACGAGATATACGAGCTAGGAACGATGAGCCT 224
CCAAC
Ind l 90 GAGCAT CAAGCAGAAGACGGCATACGAGATGAGCATGCTAGGAACGATGAGCCT 225
CCAAC
Ind I 91 ATCCTA CAAGCAGAAGACGGCATACGAGATATCCTAGCTAGGAACGATGAGCCT 226
CCAAC
Ind l 92 ACGTAA CAAGCAGAAGACGGCATACGAGATACGTAAGCTAGGAACGATGAGCCT 227
CCAAC
Ind 193 GGAAAC CAAGCAGAAGACGGCATACGAGATGGAAACGCTAGGAACGATGAGCCT 228
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Indl94 AGCAAC CAAGCAGAAGACGGCATACGAGATAGCAACGCTAGGAACGATGAGCCT 229
CCAAC
Ind l95 CTAGTG CAAGCAGAAGACGGCATACGAGATCTAGTGGCTAGGAACGATGAGCCT 230
CCAAC
Ind ) 96 CCGCTT CAAGCAGAAGACGGCATACGAGATCCGCTTGCTAGGAACGATGAGCCT 23 1
CCAAC
Ind l 97 CCAGCA CAAGCAGAAGACGGCATACGAGATCCAGCAGCTAGGAACGATGAGCCT 232
CCAAC
Indl98 ATACTC CAAGCAGAAGACGGCATACGAGATATACTCGCTAGGAACGATGAGCCT 233
CCAAC
Indl99 GGTGAA CAAGCAGAAGACGGCATACGAGATGGTGAAGCTAGGAACGATGAGCCT 234
CCAAC .
Ind200 CTCTAT CAAGCAGAAGACGGCATACGAGATCTCTATGCTAGGAACGATGAGCCT 235
CCAAC
Ind201 GACATA CAAGCAGAAGACGGCATACGAGATGACATAGCTAGGAACGATGAGCCT 236
CCAAC
lnd202 GGATCT CAAGCAGAAGACGGCATACGAGATGGATCTGCTAGGAACGATGAGCCT 237
CCAAC
Ind203 AACGAG CAAGCAGAAGACGGCATACGAGATAACGAGGCTAGGAACGATGAGCCT 238
CCAAC
Ind204 CACCTG CAAGCAGAAGACGGCATACGAGATCACCTGGCTAGGAACGATGAGCCT 239
CCAAC
Ind205 CATGAA CAAGCAGAAGACGGCATACGAGATCATGAAGCTAGGAACGATGAGCCT 240
CCAAC
Ind206 CGTGGA CAAGCAGAAGACGGCATACGAGATCGTGGAGCTAGGAACGATGAGCCT 241
CCAAC
Incl207 AGAAG CAAGCAGAAGACGGCATACGAGATAGAAGGGCTAGGAACGATGAGCC 242
G TCCAAC
lnd208 ATCCAG CAAGCAGAAGACGGCATACGAGATATCCAGGCTAGGAACGATGAGCCT 243
CCAAC
Ind209 TTCCTG CAAGCAGAAGACGGCATACGAGATTTCCTGGCTAGGAACGATGAGCCT 244
CCAAC
Ind210 CAACAA CAAGCAGAAGACGGCATACGAGATCAACAAGCTAGGAACGATGAGCCT 245
CCAAC
Ind21 1 CCTGTT CAAGCAGAAGACGGCATACGAGATCCTGTTGCTAGGAACGATGAGCCT 246
CCAAC
Ind212 CTCGTT CAAGCAGAAGACGGCATACGAGATCTCGTTGCTAGGAACGATGAGCCT 247
CCAAC
lnd213 CTGAGA CAAGCAGAAGACGGCATACGAGATCTGAGAGCTAGGAACGATGAGCCT 248
CCAAC
Ind2 ! 4 CGCTCA CAAGCAGAAGACGGCATACGAGATCGCTCAGCTAGGAACGATGAGCCT 249
CCAAC
lnd215 TACCAA CAAGCAGAAGACGGCATACGAGATTACCAAGCTAGGAACGATGAGCCT 250
CCAAC
lnd216 CGCAAG CAAGCAGAAGACGGCATACGAGATCGCAAGGCTAGGAACGATGAGCCT 251
CCAAC
Ind217 TACTGA CAAGCAGAAGACGGCATACGAGATTACTGAGCTAGGAACGATGAGCCT 252
CCAAC
lnd218 TCCACT CAAGCAGAAGACGGCATACGAGATTCCACTGCTAGGAACGATGAGCCT 253
CCAAC
Ind219 ATCGTG CAAGCAGAAGACGGCATACGAGATATCGTGGCTAGGAACGATGAGCCT 254
CCAAC
lnd220 TAGGCA CAAGCAGAAGACGGCATACGAGATTAGGCAGCTAGGAACGATGAGCCT 255
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind221 CAAGAG CAAGCAGAAGACGGCATACGAGATCAAGAGGCTAGGAACGATGAGCCT 256
CCAAC
lnd222 ACGACA CAAGCAGAAGACGGCATACGAGATACGACAGCTAGGAACGATGAGCCT 257
CCAAC
Ind223 GGTTCC CAAGCAGAAGACGGCATACGAGATGGTTCCGCTAGGAACGATGAGCCT 258
CCAAC
Ind224 TCTTAG CAAGCAGAAGACGGCATACGAGATTCTTAGGCTAGGAACGATGAGCCT 259
CCAAC
Ind225 AGAGG CAAGCAGAAGACGGCATACGAGATAGAGGAGCTAGGAACGATGAGCC 260
A .TCCAAC
Ind226 AAAGCA CAAGCAGAAGACGGCATACGAGATAAAGCAGC'I'AGGAACGATGAGCCT 261
CCAAC
Ind227 AGTCAT CAAGCAGAAGACGGCATACGAGATAGTCATGCTAGGAACGATGAGCCT 262
CCAAC
Ind228 TGCAGT CAAGCAGAAGACGGCATACGAGATTGCAGTGCTAGGAACGATGAGCCT 263
CCAAC
Ind229 TGTTCT CAAGCAGAAGACGGCATACGAGATTGTTCTGCTAGGAACGATGAGCCT 264
CCAAC
Ind230 TATCGC CAAGCAGAAGACGGCATACGAGATTATCGCGCTAGGAACGATGAGCCT 265
CCAAC
lnd231 GCATCA CAAGCAGAAGACGGCATACGAGATGCATCAGCTAGGAACGATGAGCCT 266
CCAAC
Ind232 CTCCGT CAAGCAGAAGACGGCATACGAGATCTCCGTGCTAGGAACGATGAGCCT 267
CCAAC
Ind233 TAAGAC CAAGCAGAAGACGGCATACGAGATTAAGACGCTAGGAACGATGAGCCT 268
CCAAC
Ind234 GAGGCT CAAGCAGAAGACGGCATACGAGATGAGGCTGCTAGGAACGATGAGCCT 269
CCAAC
lnd235 TGATTC CAAGCAGAAGACGGCATACGAGATTGATTCGCTAGGAACGATGAGCCT 270
CCAAC _
Ind236 GTCCAA CAAGCAGAAGACGGCATACGAGATGTCCAAGCTAGGAACGATGAGCCT 271
CCAAC
Ind237 ACTCTC CAAGCAGAAGACGGCATACGAGATACTCTCGCTAGGAACGATGAGCCT ' 272
CCAAC
Ind238 TCAACG CAAGCAGAAGACGGCATACGAGATTCAACGGCTAGGAACGATGAGCCT 1 273
CCAAC
Ind239 GGGTAC CAAGCAGAAGACGGCATACGAGATGGGTACGCTAGGAACGATGAGCCT 274
CCAAC
Ind240 ACGATT CAAGCAGAAGACGGCATACGAGATACGATTGCTAGGAACGATGAGCCT 275
CCAAC
Ind241 AACTTG CAAGCAGAAGACGGCATACGAGATAACTTGGCTAGGAACGATGAGCCT 276
CCAAC
Ind242 AACGTA CAAGCAGAAGACGGCATACGAGATAACGTAGCTAGGAACGATGAGCCT 277
CCAAC
Ind243 ACCCAC CAAGCAGAAGACGGCATACGAGATACCCACGCTAGGAACGATGAGCCT 278
CCAAC
Ind244 CGGTCT CAAGCAGAAGACGGCATACGAGATCGGTCTGCTAGGAACGATGAGCCT 279
CCAAC
]nd245 TTC AG CAAGCAGAAGACGGCATACGAGATTTCTAGGC'I'AGGAACGATGAGCCT 280
CCAAC
Incl246 GGTGTG CAAGCAGAAGACGGCATACGAGATGGTGTGGCTAGGAACGATGAGCCT 281
CCAAC
Ind247 ACCACC CAAGCAGAAGACGGCATACGAGATACCACCGCTAGGAACGATGAGCCT 282
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind248 GACTGC CAAGCAGAAGACGGCATACGAGATGACTGCGCTAGGAACGATGAGCCT 283
CCAAC
Ind249 CTACTT CAAGCAGAAGACGGCATACGAGATCTACTTGCTAGGAACGATGAGCCT 284
CCAAC
Ind250 TAGCGG CAAGCAGAAGACGGCATACGAGATTAGCGGGCTAGGAACGATGAGCCT 285
CCAAC
Ind251 TGTGCG CAAGCAGAAGACGGCATACGAGATTGTGCGGCTAGGAACGATGAGCCT 286
CCAAC
lnd252 CTGGCT CAAGCAGAAGACGGCATACGAGATCTGGCTGCTAGGAACGATGAGCCT 287
CCAAC
lnd253 CGAGAC CAAGCAGAAGACGGCATACGAGATCGAGACGCTAGGAACGATGAGCCT 288
CCAAC
lnd254 GGGATT CAAGCAGAAGACGGCATACGAGATGGGATTGCTAGGAACGATGAGCCT 289
CCAAC
Ind255 TACTCC CAAGCAGAAGACGGCATACGAGATTACTCCGCTAGGAACGATGAGCCT 290
CCAAC
lnd256 GTGGCA CAAGCAGAAGACGGCATACGAGATGTGGCAGCTAGGAACGATGAGCCT 291
CCAAC
Ind257 GTCGTC CAAGCAGAAGACGGCATACGAGATGTCGTCGCTAGGAACGATGAGCCT 292
CCAAC
Ind258 GCATTC CAAGCAGAAGACGGCATACGAGATGCATTCGCTAGGAACGATGAGCCT 293
CCAAC
Ind259 GAGCGA CAAGCAGAAGACGGCATACGAGATGAGCGAGCTAGGAACGATGAGCCT 294
CCAAC
Ind260 AGACAC CAAGCAGAAGACGGCATACGAGATAGACACGCTAGGAACGATGAGCCT 295
CCAAC
Ind261 TCTGGG CAAGCAGAAGACGGCATACGAGATTCTGGGGCTAGGAACGATGAGCCT 296
CCAAC
Ind262 GCCAGT CAAGCAGAAGACGGCATACGAGATGCCAGTGCTAGGAACGATGAGCCT 297
CCAAC
Ind263 CCAGTC CAAGCAGAAGACGGCATACGAGATCCAGTCGCTAGGAACGATGAGCCT 298
CCAAC
Ind264 TTGGCC CAAGCAGAAGACGGCATACGAGATTTGGCCGCTAGGAACGATGAGCCT 299
CCAAC
lnd265 GTAACA CAAGCAGAAGACGGCA TACGAGATGTAACAGCTAGGAACGATGAGCCT 300
CCAAC
lnd266 ATTACC CAAGCAGAAGACGGCA TACGAGATATTACCGCTAGGAACGATGAGCCT 301
CCAAC
lnd267 TCTCCT CAAGCAGAAGACGGCATACGAGAITCTCCTGCTAGGAACGATGAGCCT 302
CCAAC
Ind268 AGATCA CAAGCAGAAGACGGCATACGAGATAGATCAGCTAGGAACGATGAGCCT 303
CCAAC
Ind269 TCCTAT CAAGCAGAAGACGGCATACGAGATTCCTATGCTAGGAACGATGAGCCT 304
CCAAC
Ind270 ACTCGG CAAGCAGAAGACGGCATACGAGATACTCGGGCTAGGAACGATGAGCCT 305
CCAAC
lnd271 TGCCAT CAAGCAGAAGACGGCATACGAGATTGCCATGCTAGGAACGATGAGCCT 306
CCAAC
Ind272 CATCTC CAAGCAGAAGACGGCATACGAGATCATCTCGCTAGGAACGATGAGCCT 307
CCAAC
Ind273 CTTTGA CAAGCAGAAGACGGCATACGAGATCTITGAGCTAGGAACGATGAGCCT 308
CCAAC
Ind274 TCGCAT CAAGCAGAAGACGGCATACGAGATTCGCATGCTAGGAACGATGAGCCT 309
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind275 CACACC CAAGCAGAAGACGGCATACGAGATCACACCGCTAGGAACGATGAGCCT 310
CCAAC
lnd276 ACAGAA CAAGCAGAAGACGGCATACGAGATACAGAAGCTAGGAACGATGAGCCT 31 1
CCAAC
Ind277 ATGTCA CAAGCAGAAGACGGCATACGAGATATGTCAGCTAGGAACGATGAGCCT 312
CCAAC
lnd278 CTGTCC CAAGCAGAAGACGGCATACGAGATCTGTCCGCTAGGAACGATGAGCCT 3 13
CCAAC
Ind279 CGATCC CAAGCAGAAGACGGCATACGAGATCGATCCGCTAGGAACGATGAGCCT 3 14
CCAAC
Ind280 TGGAGG CAAGCAGAAGACGGCATACGAGATTGGAGGGCTAGGAACGATGAGCCT 3 15
CCAAC
Ind281 CGCCAA CAAGCAGAAGACGGCATACGAGATCGCCAAGCTAGGAACGATGAGCCT 3 16
CCAAC
Ind282 GAATAG CAAGCAGAAGACGGCATACGAGATGAATAGGCTAGGAACGATGAGCCT 317
CCAAC
Ind283 CAACGG CAAGCAGAAGACGGCATACGAGATCAACGGGCTAGGAACGATGAGCCT 318
CCAAC
Ind284 GCGGAA CAAGCAGAAGACGGCATACGAGATGCGGAAGCTAGGAACGATGAGCCT 319
CCAAC
Ind285 TCTGCA CAAGCAGAAGACGGCATACGAGATTCTGCAGCTAGGAACGATGAGCCT 320 .
CCAAC
Ind286 GATGGC CAAGCAGAAGACGGCATACGAGATGATGGCGCTAGGAACGATGAGCCT 321
CCAAC
Ind287 CCGATG CAAGCAGAAGACGGCATACGAGATCCGATGGCTAGGAACGATGAGCCT 322
CCAAC
Ind288 GATTTC ' CAAGCAGAAGACGGCATACGAGATGATTTCGCTAGGAACGATGAGCCT 323
CCAAC
Ind289 CCAAAC CAAGCAGAAGACGGCATACGAGATCCAAACGCTAGGAACGATGAGCCT 324
CCAAC
Ind290 AGGATC CAAGCAGAAGACGGCATACGAGATAGGATCGCTAGGAACGATGAGCCT 325
CCAAC
Ind291 CATTCA CAAGCAGAAGACGGCATACGAGATCATTCAGCTAGGAACGATGAGCCT 326
CCAAC
Ind292 AGATTG CAAGCAGAAGACGGCATACGAGATAGATTGGCTAGGAACGATGAGCCT 327
CCAAC
Ind293 CGAAGC CAAGCAGAAGACGGCATACGAGATCGAAGCGCTAGGAACGATGAGCCT 328
CCAAC
Ind294 GGAACG CAAGCAGAAGACGGCATACGAGATGGAACGGCTAGGAACGATGAGCCT 329
CCAAC
Ind295 CGACCA CAAGCAGAAGACGGCATACGAGATCGACCAGCTAGGAACGATGAGCCT 330
CCAAC
Ind296 AGCTTA CAAGCAGAAGACGGCATACGAGATAGCTTAGCTAGGAACGATGAGCCT 33 1
CCAAC
Ind297 TTCACG CAAGCAGAAGACGGCATACGAGA'ITTCACGGCTAGGAACGATGAGCCT 332
CCAAC
lnd298 CA1TAG CAAGCAGAAGACGGCATACGAGATCATTAGGCTAGGAACGATGAGCCT 333
CCAAC
Ind299 TAGGAG CAAGCAGAAGACGGCATACGAGATTAGGAGGCTAGGAACGATGAGCCT 334
CCAAC
lnd300 CTACCG CAAGCAGAAGACGGCATACGAGATCTACCGGCTAGGAACGATGAGCCT 335
CCAAC
Ind301 ATATCC CAAGCAGAAGACGGCATACGAGATATATCCGCTAGGAACGATGAGCCT 336
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind302 AATGGA CAAGCAGAAGACGGCATACGAGATAATGGAGCTAGGAACGATGAGCCT 337
CCAAC
Ind303 TTCGAC CAAGCAGAAGACGGCATACGAGATTTCGACGCTAGGAACGATGAGCCT 338
CCAAC
Ind304 AACCGG CAAGCAGAAGACGGCATACGAGATAACCGGGCTAGGAACGATGAGCCT 339
CCAAC
lnd305 AAGGCC CAAGCAGAAGACGGCATACGAGATAAGGCCGCTAGGAACGATGAGCCT 340
CCAAC
Ind306 CGTCCT CAAGCAGAAGACGGCATACGAGATCGTCCTGCTAGGAACGATGAGCCT 341
CCAAC
ind307 AAGAGC CAAGCAGAAGACGGCATACGAGATAAGAGCGCTAGGAACGATGAGCCT 342
CCAAC
Ind308 GTGAAA CAAGCAGAAGACGGCATACGAGATGTGAAAGCTAGGAACGATGAGCCT 343
CCAAC
Ind309 ACGAAC CAAGCAGAAGACGGCATACGAGATACGAACGCTAGGAACGATGAGCCT 344
CCAAC
Ind310 CCGAAA CAAGCAGAAGACGGCATACGAGATCCGAAAGCTAGGAACGATGAGCCT 345
CCAAC
Ind3 1 1 CAAGGC CAAGCAGAAGACGGCATACGAGATCAAGGCGCTAGGAACGATGAGCCT 346
CCAAC
Ind3 12 ATGTGC CAAGCAGAAGACGGCATACGAGATATGTGCGCTAGGAACGATGAGCCT 347
CCAAC
Ind3 13 CCCTTG CAAGCAGAAGACGGCATACGAGATCCC'ITGGCTAGGAACGATGAGCCT 348
CCAAC
Ind3 ! 4 GAGAA CAAGCAGAAGACGGCATACGAGATGAGAAGGCTAGGAACGATGAGCC 349
G TCCAAC
Ind315 GTAGTT CAAGCAGAAGACGGCATACGAGATGTAGTTGCTAGGAACGATGAGCCT 350
CCAAC
lnd3 16 TGGTGC CAAGCAGAAGACGGCATACGAGATTGGTGCGCTAGGAACGATGAGCCT 351
CCAAC
Ind3 17 GACTAT CAAGCAGAAGACGGCATACGAGATGACTATGCTAGGAACGATGAGCCT 352
CCAAC
Ind3 18 CTCGCA CAAGCAGAAGACGGCATACGAGATCTCGCAGCTAGGAACGATGAGCCT 353
CCAAC
Ind3 l 9 TCTTGT CAAGCAGAAGACGGCATACGAGATTCTTGTGCTAGGAACGATGAGCCT 354
CCAAC
lnd320 GCACAA CAAGCAGAAGACGGCATACGAGATGCACAAGCTAGGAACGATGAGCCT 355
CCAAC
lnd321 CCTTTA CAAGCAGAAGACGGCATACGAGATCCTTTAGCTAGGAACGATGAGCCT 356
CCAAC
Ind322 ACGTTG CAAGCAGAAGACGGCATACGAGATACGTTGGCTAGGAACGATGAGCCT 357
CCAAC
Ind323 AAGCGT CAAGCAGAAGACGGCATACGAGATAAGCGTGCTAGGAACGATGAGCCT 358
CCAAC
Ind324 AGGCAA CAAGCAGAAGACGGCATACGAGATAGGCAAGCTAGGAACGATGAGCCT 359
CCAAC
Ind325 TGAGCC CAAGCAGAAGACGGCATACGAGATTGAGCCGCTAGGAACGATGAGCCT 360
CCAAC
Ind326 TGAGAA CAAGCAGAAGACGGCATACGAGATTGAGAAGCTAGGAACGATGAGCCT 361
CCAAC
Ind327 GTACAG CAAGCAGAAGACGGCATACGAGATGTACAGGCTAGGAACGATGAGCCT 362
CCAAC
Ind328 ACGGAG CAAGCAGAAGACGGCATACGAGATACGGAGGCTAGGAACGATGAGCCT 363
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind329 ACATAC CAAGCAGAAGACGGCATACGAGATACATACGCTAGGAACGATGAGCCT 364
CCAAC
Ind330 CAGCCA CAAGCAGAAGACGGCATACGAGATCAGCCAGCTAGGAACGATGAGCCT 365
CCAAC
Ind33 1 TGTCAA CAAGCAGAAGACGGCATACGAGATTGTCAAGCTAGGAACGATGAGCCT 366
CCAAC
Ind332 TATTGG CAAGCAGAAGACGGCATACGAGATTATTGGGCTAGGAACGATGAGCCT 367
CCAAC
Jnd333 AGACCG CAAGCAGAAGACGGCATACGAGATAGACCGGCTAGGAACGATGAGCCT 368
CCAAC
Ind334 GCCAAC CAAGCAGAAGACGGCATACGAGATGCCAACGCTAGGAACGATGAGCCT 369
CCAAC
Ind335 ATGTAG CAAGCAGAAGACGGCATACGAGATATGTAGGCTAGGAACGATGAGCCT 370
CCAAC
lnd336 CGCTAC CAAGCAGAAGACGGCATACGAGATCGCTACGCTAGGAACGATGAGCCT 371
CCAAC
Ind337 CACTCG CAAGCAGAAGACGGCATACGAGATCACTCGGCTAGGAACGATGAGCCT 372
CCAAC
Ind338 GCTATT CAAGCAGAAGACGGCATACGAGATGCTATTGCTAGGAACGATGAGCCT 373
CCAAC
Ind339 CTCCAC CAAGCAGAAGACGGCATACGAGATCTCCACGCTAGGAACGATGAGCCT 374
CCAAC
lnd340 GTTAGC CAAGCAGAAGACGGCATACGAGATGTTAGCGCTAGGAACGATGAGCCT 375
CCAAC
Ind34 ! AAACCT CAAGCAGAAGACGGCATACGAGATAAACCTGCTAGGAACGATGAGCCT 376
CCAAC
Ind342 CTAGAT CAAGCAGAAGACGGCATACGAGATCTAGATGCTAGGAACGATGAGCCT 377
CCAAC
]nd343 ACCGTC CAAGCAGAAGACGGCATACGAGATACCGTCGC AGGAACGATGAGCCT 378
CCAAC
lnd344 TCACCC CAAGCAGAAGACGGCATACGAGATTCACCCGCTAGGAACGATGAGCCT 379
CCAAC
Ind345 GAAGAT CAAGCAGAAGACGGCATACGAGATGAAGATGCTAGGAACGATGAGCCT 380
CCAAC
lnd346 TGGCTC CAAGCAGAAGACGGCATACGAGATTGGCTCGCTAGGAACGATGAGCCT 381
CCAAC
lnd347 GGTTAT CAAGCAGAAGACGGCATACGAGATGGTTATGCTAGGAACGATGAGCCT 382
CCAAC
Ind348 ACACTT CAAGCAGAAGACGGCATACGAGATACACTTGCTAGGAACGATGAGCCT 383
CCAAC
Ind349 GGAAG CAAGCAGAAGACGGCA TACGAGATGGAAGAGCTAGGAACGATGAGCC 384
A TCCAAC
lnd350 TACGTG CAAGCAGAAGACGGCATACGAGATTACG TGGCTAGGAACGATGAGCCT 385
CCAAC
Ind351 CITGAC CAAGCAGAAGACGGCATACGAGATCTTGACGCTAGGAACGATGAGCCT 386
CCAAC
Ind352 ATGCCC CAAGCAGAAGACGGCATACGAGATATGCCCGCTAGGAACGATGAGCCT 387
CCAAC
]nd353 ATTCAC CAAGCAGAAGACGGCATACGAGATATTCACGCTAGGAACGATGAGCCT 388
CCAAC
Ind354 GCTTAC CAAGCAGAAGACGGCATACGAGATGCTTACGCTAGGAACGATGAGCCT 389
CCAAC
Ind355 TTCTGC CAAGCAGAAGACGGCATACGAGATTTCTGCGCTAGGAACGATGAGCCT 390
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind356 TGTATG CAAGCAGAAGACGGCATACGAGATTGTATGGCTAGGAACGATGAGCCT 391
CCAAC
Ind357 TGACGC CAAGCAGAAGACGGCATACGAGATTGACGCGCTAGGAACGATGAGCCT 392
CCAAC
Ind358 GTGTTA CAAGCAGAAGACGGCATACGAGATGTGTTAGCTAGGAACGATGAGCCT 393
CCAAC
lnd359 CATCGA CAAGCAGAAGACGGCATACGAGATCATCGAGCTAGGAACGATGAGCCT 394
CCAAC
Ind360 TGAGGT CAAGCAGAAGACGGCATACGAGATTGAGGTGCTAGGAACGATGAGCCT 395
CCAAC
Ind361 GGTCCA CAAGCAGAAGACGGCATACGAGATGGTCCAGCTAGGAACGATGAGCCT 396
CCAAC
Ind362 TATGCT CAAGCAGAAGACGGCATACGAGATTATGCTGCTAGGAACGATGAGCCT 397
CCAAC
Ind363 TTCATC CAAGCAGAAGACGGCATACGAGATTTCATCGCTAGGAACGATGAGCCT 398
CCAAC
Ind364 CCTCTG CAAGCAGAAGACGGCATACGAGATCCTCTGGCTAGGAACGATGAGCCT 399
CCAAC
Ind365 CCAAGG CAAGCAGAAGACGGCATACGAGATCCAAGGGCTAGGAACGATGAGCCT 400
CCAAC
Ind366 TAATGC CAAGCAGAAGACGGCATACGAGATTAATOCGCTAGGAACGATGAGCCT 401
CCAAC
lnd367 AGGGCA CAAGCAGAAGACGGCATACGAGATAGGGCAGCTAGGAACGATGAGCCT 402
CCAAC
Ind368 TAGAGA CAAGCAGAAGACGGCATACGAGATTAGAGAGCTAGGAACGATGAGCCT 403
CCAAC
Ind369 AGCACA CAAGCAGAAGACGGCATACGAGATAGCACAGCTAGGAACGATGAGCCT 404
CCAAC
lnd370 AGAGTC CAAGCAGAAGACGGCATACGAGATAGAGTCGCTAGGAACGATGAGCCT 405
CCAAC
Ind37 l ACTCAA CAAGCAGAAGACGGCATACGAGATACTCAAGCTAGGAACGATGAGCCT 406
CCAAC
lnd372 GTGACG CAAGCAGAAGACGGCATACGAGATGTGACGGCTAGGAACGATGAGCCT 407
CCAAC
lnd373 GGTAGG CAAGCAGAAGACGGCATACGAGATGGTAGGGCTAGGAACGATGAGCCT 408
CCAAC
Ind374 CTGAAT CAAGCAGAAGACGGCATACGAGATCTGAATGCTAGGAACGATGAGCCT 409
CCAAC
Ind37S GTCTAC CAAGCAGAAGACGGCATACGAGATGTCTACGCTAGGAACGATGAGCCT 410
CCAAC
Ind376 TCGGAC CAAGCAGAAGACGGCATACGAGATTCGGACGCTAGGAACGATGAGCCT 41 1
CCAAC
Ind377 A ACT AC CAAGCAGAAGACGGCATACGAGATAACTACGCTAGGAACGATGAGCCT 412
CCAAC
lnd378 AAGGTT CAAGCAGAAGACGGCATACGAGATAAGGTTGCTAGGAACGATGAGCCT 413
CCAAC
Ind379 AGGGAC CAAGCAGAAGACGGCATACGAGATAGGGACGCTAGGAACGATGAGCCT 414
CCAAC
Ind380 ACGCGA CAAGCAGAAGACGGCATACGAGATACGCGAGCTAGGAACGATGAGCCT 415
CCAAC
Ind381 'ITGCTA CAAGCAGAAGACGGCATACGAGATTTGCTAGCTAGGAACGATGAGCCT 416
CCAAC
Ind382 ATAACG CAAGCAGAAGACGGCATACGAGATATAACGGCTAGGAACGATGAGCCT 417
CCAAC
Barcode Barcode Primer SEQ ID ID NO:
Ind383 CCGGTA CAAGCAGAAGACGGCATACGAGATCCGGTAGCTAGGAACGATGAGCCT 418
CCA AC
Ind384 AGCTAG CAAGCAGAAGACGGCATACGAGATAGCTAGGCTAGGAACGATGAGCCT 419
CCAAC
[00171] Bisulfite read mapping and data analysis
[00172] The reference genome was computationally converted by changing all cytosincs to thymines on the two strands separately. Sequencing reads were encoded by (i) predicting the mapping orientation, (ii) converting all predicted forward mapping reads by changing all cytosincs to thymines and converting all predicted reverse complementary mapping reads by changing all guanines to adenines, the original reads are maintained. The bisulfite reads were then mapped to the converted reference separately using SOAP2Align with the parameters r = 0 (report uniquely mapped reads only), v = 2 (number of allowable mismatch), paired-end: m = 0 (minimal insert size), x = 400 (maximum insert size).
Alignment files were then combined, and one alignment per read was selected. Original C calls were placed back into the alignment information. Alignments were then converted to pileup format using SamTools. Raw SNPs and methylation frequency files were computed from pileup counts. Methylation frequencies were called using a method described
previously (Deng, J. et al. (2009) Nat. Biotechnol. 27: 353-360).
[00173] Correlation of methylation between two samples
[00174] To check whether methylation levels were similar between two samples, the Pearson's correlation was calculated on all CpG sites characterizable in both samples. First, a list of CpG sites with read depth of at least ten in both samples was generated. The
methylation frequencies at these sites were obtained from bisReadMapper output and input into the statistical package R. Finally, Pearson's correlation for the two samples was
computed using the cor() function.
[00175] Analysis of methylation
[001761 From the bisReadMapper output, the raw read counts showing methylation and lack of methylation were assembled for each line. Using these counts, a Fisher Exact Test with Benjamini-Hochberg multiple testing correction (false discovery rate = 0.01 ) was
carried out on each CpG site with minimum 10x depth coverage. This resulted in a set of DMSs between the two lines; at each of these sites, the methylation had at least 0.1 methylation level of difference. Technical replicates did not show any differential methylation, and different cell types showed a large extent of differential methylation (-33%).
Results and Discussion
[00177] These probes were used to analyze HI embryonic stem cells (HI ESCs), PGP1 fibroblasts and two technical replicates of PGP 1 fibroblast-derived induced pluripotent stem cells (PGP1 -iPSCs). For each sample, on average ~3.66 gigabases were sequenced and measured methylation for an average of 480,904 CpG sites. To assess whether these data could be used to identify potential epigenetic regulation of transcription, the Genomic Regions Enrichment of Annotations Tool ("GREAT"; McLean, C.Y. et al. (2010) Nat.
Biotechnol. 28: 495-501) was used to predict the cis-regulatory potential of regions around captured CpG sites. In total, the padlock probes captured CpG sites in regions predicted to regulate 98% of RefSeq genes (Figure 13).
[00178] The data generated with BSPPs accurately represented the methylation status of the target regions. Methylation levels for the two technical replicates of PGPl -iPSCs were consistent both within a single batch and between separate batches (Pearson's correlation coefficient R = 0.97-0.98, Figure 14A-14B). Additionally, when we compared methylation levels between technical replicates, no CpG site was different by a Fisher Exact Test with Benjamini-Hochberg multiple testing correction (false discovery rate = 0.01 , n = 439,090). In comparison, large fractions of sites were differentially methylated owing to either the process of nuclear reprogramming (27.9% DMSs between PGP l-iPSCs and PGP1 fibroblasts) or the difference in cell type (31.3% DMSs between PGP 1 fibroblasts and HI ESCs) with the same criteria (false discovery rate = 0.01 , n = 444, 1 1 1 and 359,290, respectively). Our BSPP results with I I I ESCs were consistent with the published whole- genome sequencing of bisulfite-converted DNA12 (Pearson's correlation coefficient R = 0.95, Figure 15).
[00179] This assay has very low technical variability. The assay was performed on over 150 samples in 96-well plates; the yield for each was similar (Figure 16).
Approximately 10% of CpG sites were targeted separately on each strand, allowing low-
quality datasets with poor correlation between these built-in technical replicates to be identified (Figure 14C-14E). As our BSPP assay measures absolute methylation, no normalization is necessary as long as the internal replicates are consistent. Therefore, many datasets, even those generated in different laboratories, can be directly compared without batch effects, which is important for case-control studies on large samples or for metaanalyses. Additionally, the SNP-calling feature of bisReadMapper allowed for
characterization of roughly 20,000 SNPs for each sample with an accuracy of 96% or better. This allowed unambiguous tracking of samples, which is crucial for projects involving large sample sizes.
[00180] The library-free BSPP method is flexible for different study designs. Whereas the genome-scale probe set allows global profiling on thousands of samples, a focused assay is necessary to follow up on tens to hundreds of candidate regions identified in genome-scale scanning. Such an assay needs to be customizable to different genomic targets, scalable to a very large sample size (1 ,000-100,000 samples), and inexpensive. To additionally test the flexibility, a second set of 3,918 probes (LC Sciences) was designed to evaluate the methylation state 1 kbp upstream and downstream of 120 genomic regions previously known and confirmed by BSPP to carry aberrant methylation in induced pluripotent stem cells (Lister, R. et al. (201 1 ) Nature 471 (7336): 68-73). Even with shorter capturing sequences (40 bp total for capturing arms rather than 50 bp on average, Figure 17) and a 100-fold smaller target size, an average of 56% of mappable bases were on-target, equivalent to an enrichment factor of -6,500. With the data from three cell lines (HI ESCs, PGP1 fibroblasts and PGP1 - iPSCs) regions of aberrant methylation were identified in induced pluripotent stem cells and it was found that aberrant methylation continues further upstream and downstream than observed previously.
[00181] This analysis demonstrated that a focused probe set can be used to validate specific regions of interest identified in global scanning using either our genome-wide probe set or other methods.
Example 3: Improvements to ppDesigner
[00182] Several key improvements were made to both the probe design algorithm and the ppDesigner software implementing it. These changes allow for improved generation of low-bias padlock probes for bisulfite sequencing applications and additional customizability
in probe parameters such that probes can be synthesized from additional oligonucleotide vendors and utilized in additional experimental protocols.
[00183] The previous version of the probe design algorithm utilized one neural network to generate the optimal set of padlock probes for both normal and bisulfite-converted DNA targets. However, this network was specifically designed using results from probe experiments without bisulfite conversion. In the improved algorithm, a second neural network was added specifically for bisulfite-converted DNA based solely on bisulfite probe experiments. This network contains two hidden layers with 10 and 12 nodes, respectively (Figure 18), and accepts 25 pieces of information as input rather than 7. These 25 factors (Table 8) were specifically chosen for their ability to influence padlock probe capturing efficiency on bisulfite-converted DNA.
[00184] Table 8: Factors used by neural network to predict probe capturing efficiency

[00185] Significant new factors include the counts of each DNA dinucleotide in the target region and the dinucleotides surrounding the ligation site. The use of two neural networks for the two separate capturing conditions allows for a more efficient prediction of probe efficiency, reducing capturing bias and experimental cost.
[00186] In addition, new user interface improvements were implemented to better facilitate these protocols. Several simple "default" setting profiles were added to allow the probe designer to more easily account for limitations in oligonucleotide synthesis from
various vendors (including Agilent Technologies and LC Sciences). Additional parameters were also added, allowing for easy unplementation of the new library-free padlock probe protocol, including allowing the user to set a fixed total arm size and providing the specialized library-free linker sequence.
Claims
1. A method of designing probes or primers for sequencing a target nucleic acid molecule, comprising:
selecting one or more inputs associated with efficiency of the probe or primer;
selecting a target nucleic acid sequence;
generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence;
determining the efficiency of each probe or primer sequence in the first library by using an algorithm comprising the one or more selected inputs;
ranking the probe or primer sequences in the first library by efficiency;
extracting the probe or primer sequences having the highest efficiency to generate a second library; and
adding a linker sequence to each of the probe or primer sequences in the second library.
2. The method of claim 1, further comprising synthesizing the probe or primer.
3. The method of claim 1, wherein the probe is a padlock probe.
4. The method of claim 1 , wherein the one or more inputs comprise target length, target folding energy, target GC content, extension arm A%, extension Arm G%, target A%, target T%, target G%, number of "GG" dinucleotides in ligation arm, number of "AT" dinucleotides in extension arm, number of "GG" dinucleotides in extension arm, number of "AA" dinucleotides in target, number of "AT" dinucleotides in target, number of "TA" dinucleotides in target, number of "GT" dinucleotides in target, number of "GA" dinucleotides in target, ligation arm terminal dinucleotide, extension arm terminal dinucleotide, target 5' terminal dinucleotide, ligation arm melting temperature, extension arm melting temperature, ligation arm length, extension arm length, local single-stranded folding energy of the target, and dinucleotides present at the extension site and ligation site during probe capture.
5. The method of claim 1 , wherein the target nucleic acid sequence is derived from a human.
6. The method of claim 1, wherein the target-capturing sequence includes a ligation arm and an extension arm.
7. The method of claim 1, wherein the target-capturing sequence contains one or more CpG dinucleotides.
8. The method of claim 6, wherein the target-capturing sequences in the first library contain all possible methylation state combinations of the one or more CpG dinucleotides.
9. The method of claim 5, wherein the extension arm comprises one or more priming sites for amplification of the target nucleic acid sequence.
10. The method of claim 8, wherein the one or more priming sites are universal priming sites.
11. The method of claim 1, wherein the target capturing sequence includes one or more restriction sites.
12. The method of claim 1, wherein the algorithm comprises one or more neural networks.
13. The method of claim 12, wherein the one or more neural networks comprise the one or more inputs.
14. The method of claim 13, wherein the one or more neural networks comprise seven or more inputs.
15. The method of claim 1, wherein the method further comprises, after the extracting, pooling the non-extracted probe or primer sequences and repeating the steps of generating the library of probe or primer sequences, determining the efficiency of each probe or primer sequence by using the algorithm, ranking the probe or primer sequences by efficiency, and extracting the probe or primer sequences having the highest efficiency.
16. The method of claim 1, wherein the linker sequence is common to each probe or primer sequence in the second library.
17. An apparatus comprising at least one processor and at least one memory including code which when executed by the at least one processor provides operations comprising:
selecting one or more inputs associated with efficiency of the probe or primer;
selecting a target nucleic acid sequence;
generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence;
determining the efficiency of each probe or primer sequence in the first library by using an algorithm comprising the one or more selected inputs;
ranking the probe or primer sequences in the first library by efficiency;
extracting the probe or primer sequences having the highest efficiency to generate a second library; and
adding a linker sequence to each of the probe or primer sequences in the second library.
18. The apparatus of claim 17, wherein the operations further comprise synthesizing the probe or primer.
19. The apparatus of claim 17, wherein the probe is a padlock probe.
20. The apparatus of claim 17, wherein the one or more inputs comprise target length, target folding energy, target GC content, extension arm A%, extension Arm G%, target A%, target T%, target G%, number of "GG" dinucleotides in ligation arm, number of "AT' dinucleotides in extension arm, number of "GG" dinucleotides in extension arm, number of "AA" dinucleotides in target, number of "AT" dinucleotides in target, number of "TA" dinucleotides in target, number of "GT" dinucleotides in target, number of "GA" dinucleotides in target, ligation arm terminal dinucleotide, extension arm terminal dinucleotide, target 5' terminal dinucleotide, ligation arm melting temperature, extension arm melting temperature, ligation arm length, extension arm length, local single-stranded folding energy of the target, and dinucleotides present at the extension site and ligation site during probe capture.
21. The apparatus of claim 17, wherein the algorithm comprises one or more neural networks.
22. The apparatus of claim 21, wherein the one or more neural networks comprise the one or more inputs.
23. The apparatus of claim 22, wherein the one or more neural networks comprise seven or more inputs.
24. The apparatus of claim 17, wherein the operations further comprise, after the extracting, pooling the non-extracted probe or primer sequences and repeating the steps of generating the library of probe or primer sequences, determining the efficiency of each probe or primer sequence by using the algorithm, ranking the probe or primer sequences by efficiency, and extracting the probe or primer sequences having the highest efficiency.
25. A computer-readable storage medium including code, which when executed by at least one processor provides operations comprising:
selecting one or more inputs associated with efficiency of the probe or primer;
selecting a target nucleic acid sequence;
generating a first library of probe or primer sequences that comprise a target capturing sequence that is complementary to the target nucleic acid sequence;
determining the efficiency of each probe or primer sequence in the first library by using an algorithm comprising the one or more selected inputs;
ranking the probe or primer sequences in the first library by efficiency;
extracting the probe or primer sequences having the highest efficiency to generate a second library; and
adding a linker sequence to each of the probe or primer sequences in the second library.
26. The computer-readable storage medium of claim 25, wherein the operations further comprise synthesizing the probe or primer.
27. The computer-readable storage medium of claim 25, wherein the probe is a padlock probe.
28. The computer-readable storage medium of claim 25, wherein the one or more inputs comprise target length, target folding energy, target GC content, extension arm A%, extension Arm G%, target A%, target T%, target G%, number of "GG" dinucleotides in ligation arm, number of "AT" dinucleotides in extension arm, number of "GG" dinucleotides in extension arm, number of "AA" dinucleotides in target, number of "AT" dinucleotides in target, number of "TA" dinucleotides in target, number of "GT" dinucleotides in target, number of "GA" dinucleotides in target, ligation arm terminal dinucleotide, extension arm terminal dinucleotide, target 5' terminal dinucleotide, ligation arm melting temperature, extension arm melting temperature, ligation arm length, extension arm length, local single- stranded folding energy of the target, and dinucleotides present at the extension site and ligation site during probe capture.
29. The computer-readable storage medium of claim 25, wherein the algorithm comprises one or more neural networks.
30. The computer-readable storage medium of claim 29, wherein the one or more neural networks comprise the one or more inputs.
31. The computer-readable storage medium of claim 30, wherein the one or more neural networks comprise seven or more inputs.
32. The computer-readable storage medium of claim 25, wherein the operations further comprise, after the extracting, pooling the non-extracted probe or primer sequences and repeating the steps of generating the library of probe or primer sequences, determining the efficiency of each probe or primer sequence by using the algorithm, ranking the probe or primer sequences by efficiency, and extracting the probe or primer sequences having the highest efficiency.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/113,449 US20140357497A1 (en) | 2011-04-27 | 2012-04-26 | Designing padlock probes for targeted genomic sequencing |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161479747P | 2011-04-27 | 2011-04-27 | |
| US61/479,747 | 2011-04-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012149171A1 true WO2012149171A1 (en) | 2012-11-01 |
Family
ID=47072753
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/035227 Ceased WO2012149171A1 (en) | 2011-04-27 | 2012-04-26 | Designing padlock probes for targeted genomic sequencing |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US20140357497A1 (en) |
| WO (1) | WO2012149171A1 (en) |
Cited By (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8741564B2 (en) | 2011-05-04 | 2014-06-03 | Htg Molecular Diagnostics, Inc. | Quantitative nuclease protection assay (QNPA) and sequencing (QNPS) improvements |
| WO2014160736A1 (en) * | 2013-03-29 | 2014-10-02 | University Of Washington Through Its Center For Commercialization | Systems, algorithms, and software for molecular inversion probe (mip) design |
| WO2015116837A1 (en) * | 2014-01-30 | 2015-08-06 | The Regents Of The University Of California | Methylation haplotyping for non-invasive diagnosis (monod) |
| KR20150145316A (en) * | 2014-06-18 | 2015-12-30 | 연세대학교 산학협력단 | Amplification method of probes based on programmable microarray |
| KR101608323B1 (en) * | 2014-05-28 | 2016-04-04 | 연세대학교 산학협력단 | Highly efficiently captured molecular inversion probe and method for prediction of capture efficiency of molecular inversion probe |
| WO2016115530A1 (en) * | 2015-01-18 | 2016-07-21 | The Regents Of The University Of California | Method and system for determining cancer status |
| US9403141B2 (en) | 2013-08-05 | 2016-08-02 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| EP2971159A4 (en) * | 2013-03-14 | 2017-01-25 | Good Start Genetics, Inc. | Methods for analyzing nucleic acids |
| US9677067B2 (en) | 2015-02-04 | 2017-06-13 | Twist Bioscience Corporation | Compositions and methods for synthetic gene assembly |
| US9895673B2 (en) | 2015-12-01 | 2018-02-20 | Twist Bioscience Corporation | Functionalized surfaces and preparation thereof |
| US9981239B2 (en) | 2015-04-21 | 2018-05-29 | Twist Bioscience Corporation | Devices and methods for oligonucleic acid library synthesis |
| US10053688B2 (en) | 2016-08-22 | 2018-08-21 | Twist Bioscience Corporation | De novo synthesized nucleic acid libraries |
| US10417457B2 (en) | 2016-09-21 | 2019-09-17 | Twist Bioscience Corporation | Nucleic acid based data storage |
| US10429399B2 (en) | 2014-09-24 | 2019-10-01 | Good Start Genetics, Inc. | Process control for increased robustness of genetic assays |
| US20190370164A1 (en) * | 2012-06-01 | 2019-12-05 | European Molecular Biology Laboratory | High-Capacity Storage of Digital Information in DNA |
| US10513739B2 (en) | 2017-03-02 | 2019-12-24 | Youhealth Oncotech, Limited | Methylation markers for diagnosing hepatocellular carcinoma and lung cancer |
| US10544467B2 (en) | 2016-07-06 | 2020-01-28 | Youhealth Oncotech, Limited | Solid tumor methylation markers and uses thereof |
| WO2020033238A1 (en) * | 2018-08-07 | 2020-02-13 | Singlera Genomics, Inc. | A non-invasive prenatal test with accurate fetal fraction measurement |
| US10669304B2 (en) | 2015-02-04 | 2020-06-02 | Twist Bioscience Corporation | Methods and devices for de novo oligonucleic acid assembly |
| US10696965B2 (en) | 2017-06-12 | 2020-06-30 | Twist Bioscience Corporation | Methods for seamless nucleic acid assembly |
| US10844373B2 (en) | 2015-09-18 | 2020-11-24 | Twist Bioscience Corporation | Oligonucleic acid variant libraries and synthesis thereof |
| US10851414B2 (en) | 2013-10-18 | 2020-12-01 | Good Start Genetics, Inc. | Methods for determining carrier status |
| US10894242B2 (en) | 2017-10-20 | 2021-01-19 | Twist Bioscience Corporation | Heated nanowells for polynucleotide synthesis |
| US10894959B2 (en) | 2017-03-15 | 2021-01-19 | Twist Bioscience Corporation | Variant libraries of the immunological synapse and synthesis thereof |
| US10907274B2 (en) | 2016-12-16 | 2021-02-02 | Twist Bioscience Corporation | Variant libraries of the immunological synapse and synthesis thereof |
| US10936953B2 (en) | 2018-01-04 | 2021-03-02 | Twist Bioscience Corporation | DNA-based digital information storage with sidewall electrodes |
| US11041851B2 (en) | 2010-12-23 | 2021-06-22 | Molecular Loop Biosciences, Inc. | Methods for maintaining the integrity and identification of a nucleic acid template in a multiplex sequencing reaction |
| US11149308B2 (en) | 2012-04-04 | 2021-10-19 | Invitae Corporation | Sequence assembly |
| EP3827091A4 (en) * | 2018-07-26 | 2022-04-27 | Lexent Bio, Inc. | MULTIPLE SEQUENCING USING A SINGLE-FLOW CELL |
| US11332738B2 (en) | 2019-06-21 | 2022-05-17 | Twist Bioscience Corporation | Barcode-based nucleic acid sequence assembly |
| US11352667B2 (en) | 2016-06-21 | 2022-06-07 | 10X Genomics, Inc. | Nucleic acid sequencing |
| US11377676B2 (en) | 2017-06-12 | 2022-07-05 | Twist Bioscience Corporation | Methods for seamless nucleic acid assembly |
| US11410750B2 (en) | 2018-09-27 | 2022-08-09 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US11408024B2 (en) | 2014-09-10 | 2022-08-09 | Molecular Loop Biosciences, Inc. | Methods for selectively suppressing non-target sequences |
| US11407837B2 (en) | 2017-09-11 | 2022-08-09 | Twist Bioscience Corporation | GPCR binding proteins and synthesis thereof |
| US11492727B2 (en) | 2019-02-26 | 2022-11-08 | Twist Bioscience Corporation | Variant nucleic acid libraries for GLP1 receptor |
| US11492728B2 (en) | 2019-02-26 | 2022-11-08 | Twist Bioscience Corporation | Variant nucleic acid libraries for antibody optimization |
| US11492665B2 (en) | 2018-05-18 | 2022-11-08 | Twist Bioscience Corporation | Polynucleotides, reagents, and methods for nucleic acid hybridization |
| US11512347B2 (en) | 2015-09-22 | 2022-11-29 | Twist Bioscience Corporation | Flexible substrates for nucleic acid synthesis |
| US11550939B2 (en) | 2017-02-22 | 2023-01-10 | Twist Bioscience Corporation | Nucleic acid based data storage using enzymatic bioencryption |
| US11680284B2 (en) | 2015-01-06 | 2023-06-20 | Moledular Loop Biosciences, Inc. | Screening for structural variants |
| US11840730B1 (en) | 2009-04-30 | 2023-12-12 | Molecular Loop Biosciences, Inc. | Methods and compositions for evaluating genetic markers |
| US12024750B2 (en) | 2018-04-02 | 2024-07-02 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US12091777B2 (en) | 2019-09-23 | 2024-09-17 | Twist Bioscience Corporation | Variant nucleic acid libraries for CRTH2 |
| US12110537B2 (en) | 2012-04-16 | 2024-10-08 | Molecular Loop Biosciences, Inc. | Capture reactions |
| US12173282B2 (en) | 2019-09-23 | 2024-12-24 | Twist Bioscience, Inc. | Antibodies that bind CD3 epsilon |
| US12357959B2 (en) | 2018-12-26 | 2025-07-15 | Twist Bioscience Corporation | Highly accurate de novo polynucleotide synthesis |
| US12386895B2 (en) | 2014-08-15 | 2025-08-12 | Laboratory Corporation Of America Holdings | Systems and methods for genetic analysis |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9732390B2 (en) | 2012-09-20 | 2017-08-15 | The Chinese University Of Hong Kong | Non-invasive determination of methylome of fetus or tumor from plasma |
| US10706957B2 (en) | 2012-09-20 | 2020-07-07 | The Chinese University Of Hong Kong | Non-invasive determination of methylome of tumor from plasma |
| US20150037787A1 (en) * | 2013-07-31 | 2015-02-05 | International Business Machines Corporation | Polynucleotide configuration for reliable electrical and optical sensing |
| PT3543356T (en) | 2014-07-18 | 2021-10-04 | Univ Hong Kong Chinese | Methylation pattern analysis of tissues in dna mixture |
| WO2016172442A1 (en) | 2015-04-23 | 2016-10-27 | Quest Diagnostics Investments Incorporated | Mlh1 methylation assay |
| WO2016197065A1 (en) * | 2015-06-03 | 2016-12-08 | The General Hospital Corporation | Long adapter single stranged oligonucleotide (lasso) probes to capture and clone complex libraries |
| WO2018031636A1 (en) | 2016-08-10 | 2018-02-15 | Quest Diagnostics Investments Llc | Methods of detecting mlh1 methylation |
| US11566284B2 (en) | 2016-08-10 | 2023-01-31 | Grail, Llc | Methods of preparing dual-indexed DNA libraries for bisulfite conversion sequencing |
| CA3039685A1 (en) | 2016-11-30 | 2018-06-07 | The Chinese University Of Hong Kong | Analysis of cell-free dna in urine and other samples |
| GB201621514D0 (en) | 2016-12-16 | 2017-02-01 | Q-Linea Ab | Padlock probe detection method |
| JP7047373B2 (en) * | 2017-12-25 | 2022-04-05 | トヨタ自動車株式会社 | Next-generation sequencer primer and its manufacturing method, DNA library using next-generation sequencer primer, its manufacturing method, and genomic DNA analysis method using the DNA library. |
| WO2021188395A1 (en) * | 2020-03-14 | 2021-09-23 | Colorado State University Research Foundation | Padlock probe-based rolling circle amplification paired with nuclease protection for point-of-need nucleic acid detection |
| CN111808928B (en) * | 2020-07-23 | 2024-01-05 | 生捷科技(杭州)有限公司 | SNP typing detection method |
| CN112063690A (en) * | 2020-09-18 | 2020-12-11 | 北京求臻医学检验实验室有限公司 | Construction method and application of single-molecule probe multi-target capture library |
| WO2023029044A1 (en) * | 2021-09-06 | 2023-03-09 | 百图生科(北京)智能技术有限公司 | Single-cell sequencing method and apparatus, and device, medium and program product |
| WO2023096674A1 (en) * | 2021-11-23 | 2023-06-01 | Pleno, Inc. | Encoded assays |
| CN118995902B (en) * | 2024-09-24 | 2025-08-29 | 中国医学科学院肿瘤医院 | A MIP probe and its application in capturing and building a library of free DNA in urine |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1813682A1 (en) * | 2006-01-27 | 2007-08-01 | Sysmex Corporation | Nucleic acid amplification apparatus and method |
| RU2318875C1 (en) * | 2006-04-20 | 2008-03-10 | Федеральное государственное учреждение науки "Государственный научный центр вирусологии и биотехнологии "Вектор" Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, ФГУН ГНЦ ВБ "Вектор" Роспотребнадзора | Kit of primer oligonucleotides, bioarray and method for specific identification of pathogenic for human orthopox- and herpesviruses |
-
2012
- 2012-04-26 WO PCT/US2012/035227 patent/WO2012149171A1/en not_active Ceased
- 2012-04-26 US US14/113,449 patent/US20140357497A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1813682A1 (en) * | 2006-01-27 | 2007-08-01 | Sysmex Corporation | Nucleic acid amplification apparatus and method |
| RU2318875C1 (en) * | 2006-04-20 | 2008-03-10 | Федеральное государственное учреждение науки "Государственный научный центр вирусологии и биотехнологии "Вектор" Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, ФГУН ГНЦ ВБ "Вектор" Роспотребнадзора | Kit of primer oligonucleotides, bioarray and method for specific identification of pathogenic for human orthopox- and herpesviruses |
Cited By (98)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11840730B1 (en) | 2009-04-30 | 2023-12-12 | Molecular Loop Biosciences, Inc. | Methods and compositions for evaluating genetic markers |
| US11041851B2 (en) | 2010-12-23 | 2021-06-22 | Molecular Loop Biosciences, Inc. | Methods for maintaining the integrity and identification of a nucleic acid template in a multiplex sequencing reaction |
| US11768200B2 (en) | 2010-12-23 | 2023-09-26 | Molecular Loop Biosciences, Inc. | Methods for maintaining the integrity and identification of a nucleic acid template in a multiplex sequencing reaction |
| US11041852B2 (en) | 2010-12-23 | 2021-06-22 | Molecular Loop Biosciences, Inc. | Methods for maintaining the integrity and identification of a nucleic acid template in a multiplex sequencing reaction |
| US8741564B2 (en) | 2011-05-04 | 2014-06-03 | Htg Molecular Diagnostics, Inc. | Quantitative nuclease protection assay (QNPA) and sequencing (QNPS) improvements |
| US11149308B2 (en) | 2012-04-04 | 2021-10-19 | Invitae Corporation | Sequence assembly |
| US11667965B2 (en) | 2012-04-04 | 2023-06-06 | Invitae Corporation | Sequence assembly |
| US11155863B2 (en) | 2012-04-04 | 2021-10-26 | Invitae Corporation | Sequence assembly |
| US12110537B2 (en) | 2012-04-16 | 2024-10-08 | Molecular Loop Biosciences, Inc. | Capture reactions |
| US20190370164A1 (en) * | 2012-06-01 | 2019-12-05 | European Molecular Biology Laboratory | High-Capacity Storage of Digital Information in DNA |
| EP2971159A4 (en) * | 2013-03-14 | 2017-01-25 | Good Start Genetics, Inc. | Methods for analyzing nucleic acids |
| US10202637B2 (en) | 2013-03-14 | 2019-02-12 | Molecular Loop Biosolutions, Llc | Methods for analyzing nucleic acid |
| US9677124B2 (en) | 2013-03-14 | 2017-06-13 | Good Start Genetics, Inc. | Methods for analyzing nucleic acids |
| EP2979168A4 (en) * | 2013-03-29 | 2016-11-23 | Univ Washington Ct Commerciali | MOLECULAR INVERSION (MIP) DESIGN SYSTEMS, ALGORITHMS AND SOFTWARE DESIGN SOFTWARE |
| WO2014160736A1 (en) * | 2013-03-29 | 2014-10-02 | University Of Washington Through Its Center For Commercialization | Systems, algorithms, and software for molecular inversion probe (mip) design |
| US9409139B2 (en) | 2013-08-05 | 2016-08-09 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US10272410B2 (en) | 2013-08-05 | 2019-04-30 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US9839894B2 (en) | 2013-08-05 | 2017-12-12 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US10773232B2 (en) | 2013-08-05 | 2020-09-15 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US10639609B2 (en) | 2013-08-05 | 2020-05-05 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US9833761B2 (en) | 2013-08-05 | 2017-12-05 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US11559778B2 (en) | 2013-08-05 | 2023-01-24 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US9889423B2 (en) | 2013-08-05 | 2018-02-13 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US9555388B2 (en) | 2013-08-05 | 2017-01-31 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US10384188B2 (en) | 2013-08-05 | 2019-08-20 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US11185837B2 (en) | 2013-08-05 | 2021-11-30 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US10632445B2 (en) | 2013-08-05 | 2020-04-28 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US11452980B2 (en) | 2013-08-05 | 2022-09-27 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US10583415B2 (en) | 2013-08-05 | 2020-03-10 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US10618024B2 (en) | 2013-08-05 | 2020-04-14 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US9403141B2 (en) | 2013-08-05 | 2016-08-02 | Twist Bioscience Corporation | De novo synthesized gene libraries |
| US12077822B2 (en) | 2013-10-18 | 2024-09-03 | Molecular Loop Biosciences, Inc. | Methods for determining carrier status |
| US10851414B2 (en) | 2013-10-18 | 2020-12-01 | Good Start Genetics, Inc. | Methods for determining carrier status |
| WO2015116837A1 (en) * | 2014-01-30 | 2015-08-06 | The Regents Of The University Of California | Methylation haplotyping for non-invasive diagnosis (monod) |
| KR101608323B1 (en) * | 2014-05-28 | 2016-04-04 | 연세대학교 산학협력단 | Highly efficiently captured molecular inversion probe and method for prediction of capture efficiency of molecular inversion probe |
| KR101660496B1 (en) * | 2014-06-18 | 2016-09-28 | 연세대학교 산학협력단 | Amplification method of probes based on programmable microarray |
| KR20150145316A (en) * | 2014-06-18 | 2015-12-30 | 연세대학교 산학협력단 | Amplification method of probes based on programmable microarray |
| US12386895B2 (en) | 2014-08-15 | 2025-08-12 | Laboratory Corporation Of America Holdings | Systems and methods for genetic analysis |
| US11408024B2 (en) | 2014-09-10 | 2022-08-09 | Molecular Loop Biosciences, Inc. | Methods for selectively suppressing non-target sequences |
| US10429399B2 (en) | 2014-09-24 | 2019-10-01 | Good Start Genetics, Inc. | Process control for increased robustness of genetic assays |
| US11680284B2 (en) | 2015-01-06 | 2023-06-20 | Moledular Loop Biosciences, Inc. | Screening for structural variants |
| US9984201B2 (en) | 2015-01-18 | 2018-05-29 | Youhealth Biotech, Limited | Method and system for determining cancer status |
| EA036566B1 (en) * | 2015-01-18 | 2020-11-24 | Зе Реджентс Оф Зе Юниверсити Оф Калифорния | Method and system for determining cancer status |
| WO2016115530A1 (en) * | 2015-01-18 | 2016-07-21 | The Regents Of The University Of California | Method and system for determining cancer status |
| US9677067B2 (en) | 2015-02-04 | 2017-06-13 | Twist Bioscience Corporation | Compositions and methods for synthetic gene assembly |
| US11697668B2 (en) | 2015-02-04 | 2023-07-11 | Twist Bioscience Corporation | Methods and devices for de novo oligonucleic acid assembly |
| US10669304B2 (en) | 2015-02-04 | 2020-06-02 | Twist Bioscience Corporation | Methods and devices for de novo oligonucleic acid assembly |
| US11691118B2 (en) | 2015-04-21 | 2023-07-04 | Twist Bioscience Corporation | Devices and methods for oligonucleic acid library synthesis |
| US9981239B2 (en) | 2015-04-21 | 2018-05-29 | Twist Bioscience Corporation | Devices and methods for oligonucleic acid library synthesis |
| US10744477B2 (en) | 2015-04-21 | 2020-08-18 | Twist Bioscience Corporation | Devices and methods for oligonucleic acid library synthesis |
| US11807956B2 (en) | 2015-09-18 | 2023-11-07 | Twist Bioscience Corporation | Oligonucleic acid variant libraries and synthesis thereof |
| US10844373B2 (en) | 2015-09-18 | 2020-11-24 | Twist Bioscience Corporation | Oligonucleic acid variant libraries and synthesis thereof |
| US11512347B2 (en) | 2015-09-22 | 2022-11-29 | Twist Bioscience Corporation | Flexible substrates for nucleic acid synthesis |
| US10384189B2 (en) | 2015-12-01 | 2019-08-20 | Twist Bioscience Corporation | Functionalized surfaces and preparation thereof |
| US9895673B2 (en) | 2015-12-01 | 2018-02-20 | Twist Bioscience Corporation | Functionalized surfaces and preparation thereof |
| US10987648B2 (en) | 2015-12-01 | 2021-04-27 | Twist Bioscience Corporation | Functionalized surfaces and preparation thereof |
| US11352667B2 (en) | 2016-06-21 | 2022-06-07 | 10X Genomics, Inc. | Nucleic acid sequencing |
| US10544467B2 (en) | 2016-07-06 | 2020-01-28 | Youhealth Oncotech, Limited | Solid tumor methylation markers and uses thereof |
| US10975372B2 (en) | 2016-08-22 | 2021-04-13 | Twist Bioscience Corporation | De novo synthesized nucleic acid libraries |
| US10053688B2 (en) | 2016-08-22 | 2018-08-21 | Twist Bioscience Corporation | De novo synthesized nucleic acid libraries |
| US10754994B2 (en) | 2016-09-21 | 2020-08-25 | Twist Bioscience Corporation | Nucleic acid based data storage |
| US10417457B2 (en) | 2016-09-21 | 2019-09-17 | Twist Bioscience Corporation | Nucleic acid based data storage |
| US11263354B2 (en) | 2016-09-21 | 2022-03-01 | Twist Bioscience Corporation | Nucleic acid based data storage |
| US12056264B2 (en) | 2016-09-21 | 2024-08-06 | Twist Bioscience Corporation | Nucleic acid based data storage |
| US11562103B2 (en) | 2016-09-21 | 2023-01-24 | Twist Bioscience Corporation | Nucleic acid based data storage |
| US10907274B2 (en) | 2016-12-16 | 2021-02-02 | Twist Bioscience Corporation | Variant libraries of the immunological synapse and synthesis thereof |
| US11550939B2 (en) | 2017-02-22 | 2023-01-10 | Twist Bioscience Corporation | Nucleic acid based data storage using enzymatic bioencryption |
| US10513739B2 (en) | 2017-03-02 | 2019-12-24 | Youhealth Oncotech, Limited | Methylation markers for diagnosing hepatocellular carcinoma and lung cancer |
| US10894959B2 (en) | 2017-03-15 | 2021-01-19 | Twist Bioscience Corporation | Variant libraries of the immunological synapse and synthesis thereof |
| US11377676B2 (en) | 2017-06-12 | 2022-07-05 | Twist Bioscience Corporation | Methods for seamless nucleic acid assembly |
| US11332740B2 (en) | 2017-06-12 | 2022-05-17 | Twist Bioscience Corporation | Methods for seamless nucleic acid assembly |
| US12270028B2 (en) | 2017-06-12 | 2025-04-08 | Twist Bioscience Corporation | Methods for seamless nucleic acid assembly |
| US10696965B2 (en) | 2017-06-12 | 2020-06-30 | Twist Bioscience Corporation | Methods for seamless nucleic acid assembly |
| US11407837B2 (en) | 2017-09-11 | 2022-08-09 | Twist Bioscience Corporation | GPCR binding proteins and synthesis thereof |
| US11745159B2 (en) | 2017-10-20 | 2023-09-05 | Twist Bioscience Corporation | Heated nanowells for polynucleotide synthesis |
| US10894242B2 (en) | 2017-10-20 | 2021-01-19 | Twist Bioscience Corporation | Heated nanowells for polynucleotide synthesis |
| US10936953B2 (en) | 2018-01-04 | 2021-03-02 | Twist Bioscience Corporation | DNA-based digital information storage with sidewall electrodes |
| US12086722B2 (en) | 2018-01-04 | 2024-09-10 | Twist Bioscience Corporation | DNA-based digital information storage with sidewall electrodes |
| US12024750B2 (en) | 2018-04-02 | 2024-07-02 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US12435375B2 (en) | 2018-04-02 | 2025-10-07 | Grail, Inc. | Methylation markers and targeted methylation probe panel |
| US12522868B2 (en) | 2018-05-18 | 2026-01-13 | Twist Bioscience Corporation | Polynucleotides, reagents, and methods for nucleic acid hybridization |
| US11732294B2 (en) | 2018-05-18 | 2023-08-22 | Twist Bioscience Corporation | Polynucleotides, reagents, and methods for nucleic acid hybridization |
| US11492665B2 (en) | 2018-05-18 | 2022-11-08 | Twist Bioscience Corporation | Polynucleotides, reagents, and methods for nucleic acid hybridization |
| EP3827091A4 (en) * | 2018-07-26 | 2022-04-27 | Lexent Bio, Inc. | MULTIPLE SEQUENCING USING A SINGLE-FLOW CELL |
| US12435366B2 (en) | 2018-07-26 | 2025-10-07 | Lexent Bio, Inc. | Multiple sequencing using a single flow cell |
| WO2020033238A1 (en) * | 2018-08-07 | 2020-02-13 | Singlera Genomics, Inc. | A non-invasive prenatal test with accurate fetal fraction measurement |
| US11685958B2 (en) | 2018-09-27 | 2023-06-27 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US11725251B2 (en) | 2018-09-27 | 2023-08-15 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US12410482B2 (en) | 2018-09-27 | 2025-09-09 | Grail, Inc. | Methylation markers and targeted methylation probe panel |
| US11410750B2 (en) | 2018-09-27 | 2022-08-09 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US11795513B2 (en) | 2018-09-27 | 2023-10-24 | Grail, Llc | Methylation markers and targeted methylation probe panel |
| US12357959B2 (en) | 2018-12-26 | 2025-07-15 | Twist Bioscience Corporation | Highly accurate de novo polynucleotide synthesis |
| US11492728B2 (en) | 2019-02-26 | 2022-11-08 | Twist Bioscience Corporation | Variant nucleic acid libraries for antibody optimization |
| US12331427B2 (en) | 2019-02-26 | 2025-06-17 | Twist Bioscience Corporation | Antibodies that bind GLP1R |
| US11492727B2 (en) | 2019-02-26 | 2022-11-08 | Twist Bioscience Corporation | Variant nucleic acid libraries for GLP1 receptor |
| US11332738B2 (en) | 2019-06-21 | 2022-05-17 | Twist Bioscience Corporation | Barcode-based nucleic acid sequence assembly |
| US12173282B2 (en) | 2019-09-23 | 2024-12-24 | Twist Bioscience, Inc. | Antibodies that bind CD3 epsilon |
| US12091777B2 (en) | 2019-09-23 | 2024-09-17 | Twist Bioscience Corporation | Variant nucleic acid libraries for CRTH2 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140357497A1 (en) | 2014-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20140357497A1 (en) | Designing padlock probes for targeted genomic sequencing | |
| TWI832482B (en) | Determination of base modifications of nucleic acids | |
| Yong et al. | Profiling genome-wide DNA methylation | |
| Pomraning et al. | Genome-wide high throughput analysis of DNA methylation in eukaryotes | |
| Zilberman et al. | Genome-wide analysis of DNA methylation patterns | |
| US20220042090A1 (en) | PROGRAMMABLE RNA-TEMPLATED SEQUENCING BY LIGATION (rSBL) | |
| Ludwig et al. | Mapping chromatin modifications at the single cell level | |
| Buermans et al. | Next generation sequencing technology: advances and applications | |
| CN103917654B (en) | Methods and systems for sequencing long nucleic acids | |
| US11473140B2 (en) | Highly selective omega primer amplification of nucleic acid sequences | |
| Shin et al. | Decoding neural transcriptomes and epigenomes via high-throughput sequencing | |
| CN103233072B (en) | High-flux mythelation detection technology for DNA (deoxyribonucleic acid) of complete genome | |
| JP2023508795A (en) | Methods and Kits for Enrichment and Detection of DNA and RNA Modifications, and Functional Motifs | |
| Good | Reduced representation methods for subgenomic enrichment and next-generation sequencing | |
| WO2021097252A1 (en) | Methylation assays and uses thereof | |
| Reinders et al. | Bisulfite methylation profiling of large genomes | |
| Myllykangas et al. | Targeted deep resequencing of the human cancer genome using next-generation technologies | |
| Barbaro | Overview of NGS platforms and technological advancements for forensic applications | |
| Zıplar et al. | Genomic and Transcriptomic Sequencing and Analysis Approaches | |
| US20220157469A1 (en) | Methods of predicting age, and identifying and treating conditions associated with aging using spectral clustering and discrete cosine transform | |
| TURAN | 2. INTERNATIONAL PARIS CONGRESS ON AGRICULTURE & ANIMAL HUSBANDRY | |
| WO2026022472A1 (en) | Profiling method | |
| HK40044336A (en) | Determination of base modifications of nucleic acids | |
| CA2375433A1 (en) | Transcription-based gene mapping | |
| Mikkelsen | Mammalian comparative genomics and epigenomics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12777068 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12777068 Country of ref document: EP Kind code of ref document: A1 |