WO2012033735A1 - Inhibitors of human immunodeficiency virus replication - Google Patents
Inhibitors of human immunodeficiency virus replication Download PDFInfo
- Publication number
- WO2012033735A1 WO2012033735A1 PCT/US2011/050503 US2011050503W WO2012033735A1 WO 2012033735 A1 WO2012033735 A1 WO 2012033735A1 US 2011050503 W US2011050503 W US 2011050503W WO 2012033735 A1 WO2012033735 A1 WO 2012033735A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- mmol
- pyrimidin
- alkyl
- phenyl
- Prior art date
Links
- 0 *C1c2c(*)[n]3nc(*)c(*)c3nc2*=C1O Chemical compound *C1c2c(*)[n]3nc(*)c(*)c3nc2*=C1O 0.000 description 1
- VQUPSZIBBVGQQH-UHFFFAOYSA-N CC(C)(C)OC(C(O)=O)c1c(-c(cc2)cc3c2OCCC3)[n]2nc(C)c(Cl)c2nc1C Chemical compound CC(C)(C)OC(C(O)=O)c1c(-c(cc2)cc3c2OCCC3)[n]2nc(C)c(Cl)c2nc1C VQUPSZIBBVGQQH-UHFFFAOYSA-N 0.000 description 1
- JQRMVOGGXZFNMM-UHFFFAOYSA-N CC(C)(C)OC(C(O)=O)c1c(-c(cc2)cc3c2OCCC3)[n]2ncc(-c3ccccc3)c2nc1C Chemical compound CC(C)(C)OC(C(O)=O)c1c(-c(cc2)cc3c2OCCC3)[n]2ncc(-c3ccccc3)c2nc1C JQRMVOGGXZFNMM-UHFFFAOYSA-N 0.000 description 1
- AVPUILSQJXTZEN-UHFFFAOYSA-N CC(C)(C)OC(C(OC)=O)c(c(C)nc([n]1nc2C)c2Cl)c1Cl Chemical compound CC(C)(C)OC(C(OC)=O)c(c(C)nc([n]1nc2C)c2Cl)c1Cl AVPUILSQJXTZEN-UHFFFAOYSA-N 0.000 description 1
- MWVYFMONEWLAGS-UHFFFAOYSA-N CC(C)(C)OC(C(OC)=O)c(c(C)nc1c2Br)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1ccccc1 Chemical compound CC(C)(C)OC(C(OC)=O)c(c(C)nc1c2Br)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1ccccc1 MWVYFMONEWLAGS-UHFFFAOYSA-N 0.000 description 1
- SBRJRVZWKUEVDB-UHFFFAOYSA-N CC(C)(C)OC(C(OC)=O)c1c(-c(cc2)cc3c2OCCC3)[n]2nc(C)c(Cl)c2nc1C Chemical compound CC(C)(C)OC(C(OC)=O)c1c(-c(cc2)cc3c2OCCC3)[n]2nc(C)c(Cl)c2nc1C SBRJRVZWKUEVDB-UHFFFAOYSA-N 0.000 description 1
- BBJKQLYLLCJOKA-UHFFFAOYSA-N CC(C)(C)OC(C(OC)=O)c1c(-c(cc2F)c(C)c3c2OCCC3)[n]2ncc(-c3cccc(-c4ccncc4)c3)c2nc1C Chemical compound CC(C)(C)OC(C(OC)=O)c1c(-c(cc2F)c(C)c3c2OCCC3)[n]2ncc(-c3cccc(-c4ccncc4)c3)c2nc1C BBJKQLYLLCJOKA-UHFFFAOYSA-N 0.000 description 1
- TZXRURYHFBZSGH-HHHXNRCGSA-N CC(C)(C)O[C@@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1ccccc1 Chemical compound CC(C)(C)O[C@@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1ccccc1 TZXRURYHFBZSGH-HHHXNRCGSA-N 0.000 description 1
- RUCKFHHSPMUJGE-MUUNZHRXSA-N CC(C)(C)O[C@@H](C(OC)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cc(Cl)ccc1 Chemical compound CC(C)(C)O[C@@H](C(OC)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cc(Cl)ccc1 RUCKFHHSPMUJGE-MUUNZHRXSA-N 0.000 description 1
- ANDWGHGCCMHLMZ-MHZLTWQESA-N CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3)ccc3F)[n]1nc2-c1cc(CCCC2)c2cc1 Chemical compound CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3)ccc3F)[n]1nc2-c1cc(CCCC2)c2cc1 ANDWGHGCCMHLMZ-MHZLTWQESA-N 0.000 description 1
- DEMMYAVNGRDONL-MHZLTWQESA-N CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cc(Cl)ccc1 Chemical compound CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cc(Cl)ccc1 DEMMYAVNGRDONL-MHZLTWQESA-N 0.000 description 1
- QEWYYDDMXMVYQQ-UMSFTDKQSA-N CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cccc(-c2cc(C)ccc2)c1 Chemical compound CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cccc(-c2cc(C)ccc2)c1 QEWYYDDMXMVYQQ-UMSFTDKQSA-N 0.000 description 1
- XELZRKZYTPPEIK-DHUJRADRSA-N CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2C)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cccc(-c2ccc3[n](C)ncc3c2)c1 Chemical compound CC(C)(C)O[C@H](C(O)=O)c(c(C)nc1c2C)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1cccc(-c2ccc3[n](C)ncc3c2)c1 XELZRKZYTPPEIK-DHUJRADRSA-N 0.000 description 1
- DKRLFOAVKFKDIP-VWLOTQADSA-N CC(C)(C)O[C@H](C(OC)=O)c(c(C)nc1c2)c(-c3ccc(C)cc3)[n]1nc2-c1cc(Cl)ccc1 Chemical compound CC(C)(C)O[C@H](C(OC)=O)c(c(C)nc1c2)c(-c3ccc(C)cc3)[n]1nc2-c1cc(Cl)ccc1 DKRLFOAVKFKDIP-VWLOTQADSA-N 0.000 description 1
- UMYDBUAIIBJNBV-KDXMTYKHSA-N CC(C)(C)O[C@H](C(OC)=O)c(c(C)nc1c2-c3cc(-c4ccccc4)ccc3)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1ccccc1 Chemical compound CC(C)(C)O[C@H](C(OC)=O)c(c(C)nc1c2-c3cc(-c4ccccc4)ccc3)c(-c(cc3F)c(C)c4c3OCCC4)[n]1nc2-c1ccccc1 UMYDBUAIIBJNBV-KDXMTYKHSA-N 0.000 description 1
- PCDWHCYAPZJSNC-UHFFFAOYSA-N CC(C)(C)[O-2]c(c(-c1c(CC(O)=O)c(C)nc2cc(-c3cc(-c4cncc(F)c4)ccc3)n[n]12)c(C)c1c2OCCC1)c2F Chemical compound CC(C)(C)[O-2]c(c(-c1c(CC(O)=O)c(C)nc2cc(-c3cc(-c4cncc(F)c4)ccc3)n[n]12)c(C)c1c2OCCC1)c2F PCDWHCYAPZJSNC-UHFFFAOYSA-N 0.000 description 1
- CMFLSSRHAOBOBS-UHFFFAOYSA-N CC(C)(C)[O-]c(c(-c1c(CC(O)=O)c(C)nc2cc(-c3cc(-c(cc4)c(C)cc4F)ccc3)n[n]12)c(C)c1c2OCCC1)c2F Chemical compound CC(C)(C)[O-]c(c(-c1c(CC(O)=O)c(C)nc2cc(-c3cc(-c(cc4)c(C)cc4F)ccc3)n[n]12)c(C)c1c2OCCC1)c2F CMFLSSRHAOBOBS-UHFFFAOYSA-N 0.000 description 1
- ANIROCOUJINZLZ-UHFFFAOYSA-N CC(C)(OC1c2c(-c(cc3F)c(C)c4c3OCCC4)[n]3ncc(-c4cccc(-c5ccncc5)c4)c3nc2C)OC1=O Chemical compound CC(C)(OC1c2c(-c(cc3F)c(C)c4c3OCCC4)[n]3ncc(-c4cccc(-c5ccncc5)c4)c3nc2C)OC1=O ANIROCOUJINZLZ-UHFFFAOYSA-N 0.000 description 1
- YDTHTLOOPIIDCK-UHFFFAOYSA-N CC(C)(OC1c2c(-c(cc3F)c(C)c4c3OCCC4)[n]3ncc(-c4cccc(Cl)c4)c3nc2C)[O](C)C1=O Chemical compound CC(C)(OC1c2c(-c(cc3F)c(C)c4c3OCCC4)[n]3ncc(-c4cccc(Cl)c4)c3nc2C)[O](C)C1=O YDTHTLOOPIIDCK-UHFFFAOYSA-N 0.000 description 1
- PBHURLIYTAJJNK-NRFANRHFSA-N CC(C)c1c([C@@H](C(OC)=O)OC(C)(C)C)c(C)nc2cc(-c3cc(Cl)ccc3)n[n]12 Chemical compound CC(C)c1c([C@@H](C(OC)=O)OC(C)(C)C)c(C)nc2cc(-c3cc(Cl)ccc3)n[n]12 PBHURLIYTAJJNK-NRFANRHFSA-N 0.000 description 1
- NLTIETZTDSJANS-UHFFFAOYSA-N CC1(C)OB(c2ccncc2)OC1(C)C Chemical compound CC1(C)OB(c2ccncc2)OC1(C)C NLTIETZTDSJANS-UHFFFAOYSA-N 0.000 description 1
- SWAOIACIQSGXLZ-UHFFFAOYSA-N CC1Nc2cc(-c3cc(-c4c(C)nccc4)ccc3)n[n]2C(c(c([O-2]C(C)(C)C)c2F)c(C)c3c2OCCC3)=C1CC(O)=O Chemical compound CC1Nc2cc(-c3cc(-c4c(C)nccc4)ccc3)n[n]2C(c(c([O-2]C(C)(C)C)c2F)c(C)c3c2OCCC3)=C1CC(O)=O SWAOIACIQSGXLZ-UHFFFAOYSA-N 0.000 description 1
- ZDRUFLCAOFTWPS-UHFFFAOYSA-N CCC1OC(C)(C)[O](C)C1=O Chemical compound CCC1OC(C)(C)[O](C)C1=O ZDRUFLCAOFTWPS-UHFFFAOYSA-N 0.000 description 1
- WLJMURVYJJHTCB-QXMHVHEDSA-N CCCCCCO/C(/CC)=C(/C=C)\F Chemical compound CCCCCCO/C(/CC)=C(/C=C)\F WLJMURVYJJHTCB-QXMHVHEDSA-N 0.000 description 1
- BTIBHSIHNCKBES-SFHVURJKSA-N CCOC([C@H](c(c(C)nc1c(C)c(-c2cccc(Cl)c2)n[n]11)c1Cl)OC(C)(C)C)=O Chemical compound CCOC([C@H](c(c(C)nc1c(C)c(-c2cccc(Cl)c2)n[n]11)c1Cl)OC(C)(C)C)=O BTIBHSIHNCKBES-SFHVURJKSA-N 0.000 description 1
- PBRWJKYEARJTLN-UHFFFAOYSA-N Cc(cc1)ccc1-c1c(C(C(O)=O)O)c(C)nc2cc(-c3ccccc3)n[n]12 Chemical compound Cc(cc1)ccc1-c1c(C(C(O)=O)O)c(C)nc2cc(-c3ccccc3)n[n]12 PBRWJKYEARJTLN-UHFFFAOYSA-N 0.000 description 1
- MYKKVDKQAUREFK-UHFFFAOYSA-N Cc(cc1)ccc1-c1c(C=O)c(C)nc2cc(-c3ccccc3)n[n]12 Chemical compound Cc(cc1)ccc1-c1c(C=O)c(C)nc2cc(-c3ccccc3)n[n]12 MYKKVDKQAUREFK-UHFFFAOYSA-N 0.000 description 1
- DOHZVIPKLYOJFW-UHFFFAOYSA-N Cc1n[n]2c(Cl)c(C(C(OC)=O)O)c(C)nc2c1Cl Chemical compound Cc1n[n]2c(Cl)c(C(C(OC)=O)O)c(C)nc2c1Cl DOHZVIPKLYOJFW-UHFFFAOYSA-N 0.000 description 1
- IIBYTDPCPGBVPV-UHFFFAOYSA-N Cc1nc2cc(-c3ccccc3)n[n]2c(Cl)c1CC(OC)=O Chemical compound Cc1nc2cc(-c3ccccc3)n[n]2c(Cl)c1CC(OC)=O IIBYTDPCPGBVPV-UHFFFAOYSA-N 0.000 description 1
- SDTFPDVWFKWNCR-UHFFFAOYSA-N Nc([nH]nc1)c1-c1cccc(-c2ccncc2)c1 Chemical compound Nc([nH]nc1)c1-c1cccc(-c2ccncc2)c1 SDTFPDVWFKWNCR-UHFFFAOYSA-N 0.000 description 1
- UPAOOWFSVZQAFL-UHFFFAOYSA-N OBc(cc1)cc2c1OCCC2 Chemical compound OBc(cc1)cc2c1OCCC2 UPAOOWFSVZQAFL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- the disclosure generally relates to compounds, compositions, and methods for the treatment of human immunodeficiency virus (HIV) infection.
- HIV human immunodeficiency virus
- the disclosure provides novel inhibitors of HIV, pharmaceutical compositions containing such compounds, and methods for using these compounds in the treatment of HIV infection.
- HIV Human immunodeficiency virus
- AIDS acquired immune deficiency syndrome
- antiviral drugs available to combat the infection. These drugs can be divided into classes based on the viral protein they target or their mode of action. In particular, saquinavir, indinavir, ritonavir, nelfinavir atazanavir darunavir, amprenavir, fosamprenavir, lopinavir and tipranavir are competitive inhibitors of the aspartyl protease expressed by HIV.
- Zidovudine, didanosine, stavudine, lamivudine, zalcitabine, emtricitibine, tenofovir and abacavir are nucleos(t)ide reverse transcriptase inhibitors that behave as substrate mimics to halt viral cDNA synthesis.
- the non-nucleoside reverse transcriptase inhibitors nevirapine, delavirdine, efavirenz and etravirine inhibit the synthesis of viral cDNA via a non-competitive (or uncompetitive) mechanism.
- Enfuvirtide and maraviroc inhibit the entry of the virus into the host cell.
- HIV integrase inhibitor MK-0518, Isentress ®
- Isentress ® An HIV integrase inhibitor, raltegravir
- the invention provides technical advantages, for example, the compounds are novel and are useful in the treatment of HIV. Additionally, the compounds provide advantages for pharmaceutical uses, for example, with regard to one or more of their mechanism of action, binding, inhibition efficacy, target selectivity, solubility, safety profiles, or bioavailability.
- the invention encompasses compounds of Formula I, including
- One aspect of the invention is a compound of Formula I
- R 1 is H, alkyl, cycloalkyl, or Ar 1
- R 2 is H, alkyl, cycloalkyl, or Ar 1
- R 3 is alkyl or Ar 2 ;
- R 4 is alkyl or haloalkyl
- R 5 is alkyl
- Ar 1 is phenyl, pyridinyl, tetralinyl, indazolyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, benzyloxy, halobenzyloxy, (alkoxy )benzyloxy, phenoxyalkyl, CONH(phenyl), CONH(benzyl), and Ar 3 ;
- Ar 2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido;
- Ar 3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 0-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl; or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a compound of formula I where:
- R 1 is H, alkyl, cycloalkyl, or Ar 1 ;
- R 2 is H, alkyl, cycloalkyl, or Ar 1 ;
- R 3 is alkyl or Ar 2 ;
- R 4 is alkyl or haloalkyl;
- R 5 is alkyl;
- Ar 1 is phenyl, pyridinyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, and phenoxyalkyl; and
- Ar 2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido;
- R 1 is Ar 1 ;
- R 2 is H;
- R 3 is Ar 2 ;
- R 4 is alkyl;
- R 5 is methyl;
- Ar 1 is phenyl, pyridinyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, and phenoxyalkyl;
- Ar 2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl,
- tetrahydroquinolinyl isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido; or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a compound of formula I where R 1 is Ar 1 ; R 2 is H; R 3 is Ar 2 ; R 4 is alkyl; R 5 is methyl; Ar 1 is phenyl substituted with 0-1 alkyl substituents; and Ar 2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido; or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a compound of formula I where R 1 is H and R 2 is Ar 1 .
- Another aspect of the invention is a compound of formula I where R 1 is Ar 1 and R 2 is H.
- Another aspect of the invention is a compound of formula I where R 3 is Ar 2 .
- Ar 2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido.
- Another aspect of the invention is a compound of formula I where R 4 is alkyl.
- Another aspect of the invention is a compound of formula I where R 5 is methyl.
- Another aspect of the invention is a compound of formula I where Ar 1 is phenyl substituted with 0-1 alkyl substituents.
- Another aspect of the invention is a compound of formula I where where R 1 is
- Ar ; R is H; R is Ar ; R is alkyl; R is methyl; Ar is phenyl or pyridinyl, and is substituted with 1 Ar 3 substituent and 0-2 substituents selected from halo, alkyl, haloalkyl, alkoxy, and haloalkoxy; and Ar 2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0
- Another aspect of the invention is a compound of formula I where R 1 is Ar 1 ; R 2 is H; and Ar 1 is phenyl or pyridinyl, and is substituted with 1 Ar 3 substituent and 0-2 substituents selected from halo, alkyl, haloalkyl, alkoxy, and haloalkoxy.
- Ar 2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido.
- Another aspect of the invention is a compound of formula I where Ar 3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 0-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl.
- Another aspect of the invention is a compound of formula I where Ar 3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 1-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl.
- Another aspect of the invention is a compound of formula I where Ar 3 is phenyl, pyridinyl, or pyrazolyl, and is substituted with 1-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl.
- Another aspect of the invention is a compound of formula I where Ar 3 is phenyl, pyridinyl, or pyrazolyl, and is substituted with 1-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, and haloalkoxy.
- variable substituent including R , R , R , R , R , Ar , Ar , and Ar
- R , R , R , R , Ar , Ar can be used independently with the scope of any other instance of a variable substituent.
- the invention includes combinations of the different aspects. Unless specified otherwise, these terms have the following meanings.
- Alkyl means a straight or branched alkyl group composed of 1 to 6 carbons.
- Alkenyl means a straight or branched alkyl group composed of 2 to 6 carbons with at least one double bond.
- Alkynyl means a straight or branched alkyl group composed of 2 to 6 carbons with at least one triple bond.
- Cycloalkyl means a monocyclic ring system composed of 3 to 7 carbons.
- Haloalkyl and haloalkoxy include all halogenated isomers from monohalo to perhalo.
- Tetralin means tetrahydronaphthalene. Terms with a hydrocarbon moiety (e.g. alkoxy) include straight and branched isomers for the hydrocarbon portion. Parenthetic and multiparenthetic terms are intended to clarify bonding relationships to those skilled in the art. For example, a term such as ((R)alkyl) means an alkyl substituent further substituted with the substituent R. "Chroman” means
- the invention includes all pharmaceutically acceptable salt forms of the compounds.
- Pharmaceutically acceptable salts are those in which the counter ions do not contribute significantly to the physiological activity or toxicity of the compounds and as such function as pharmacological equivalents. These salts can be made according to common organic techniques employing commercially available reagents. Some anionic salt forms include acetate, acistrate, besylate, bromide, chloride, citrate, fumarate, glucouronate, hydrobromide, hydrochloride, hydroiodide, iodide, lactate, maleate, mesylate, nitrate, pamoate, phosphate, succinate, sulfate, tartrate, tosylate, and xinofoate.
- Some cationic salt forms include ammonium, aluminum, benzathine, bismuth, calcium, choline, diethylamine, diethanolamine, lithium, magnesium, meglumine, 4-phenylcyclohexylamine, piperazine, potassium, sodium, tromethamine, and zinc.
- the invention includes all stereoisomeric forms of the compounds including enantiomers and diastereromers. Methods of making and separating stereoisomers are known in the art.
- the invention includes all tautomeric forms of the compounds.
- the invention includes atropisomers and rotational isomers.
- the invention is intended to include all isotopes of atoms occurring in the present compounds.
- Isotopes include those atoms having the same atomic number but different mass numbers.
- isotopes of hydrogen include deuterium and tritium.
- Isotopes of carbon include 13 C and 14 C.
- Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed. Such compounds may have a variety of potential uses, for example as standards and reagents in determining biological activity. In the case of stable isotopes, such compounds may have the potential to favorably modify biological, pharmacological, or pharmacokinetic properties.
- a recombinant NL-Rluc virus was constructed in which a section of the nef gene from NL4-3 was replaced with the Renilla Luciferase gene.
- the NL-RLuc virus was prepared by co-transfection of two plasmids, pNLRLuc and pVSVenv.
- the pNLRLuc contains the NL-Rluc DNA cloned into pUC18 at the Pvull site, while the pVSVenv contains the gene for VSV G protein linked to an LTR promoter.
- Transfections were performed at a 1 :3 ratio of pNLRLuc to pVSVenv in 293T cells using the LipofectAMINE PLUS kit from Invitrogen (Carlsbad, CA) according to the manufacturer, and the pseudotype virus generated was titered in MT-2 cells.
- the titrated virus was used to infect MT-2 cells in the presence of compound, and after 5 days of incubation, cells were processed and quantitated for virus growth by the amount of expressed luciferase. This provides a simple and easy method for quantitating the extent of virus growth and consequently, the antiviral activity of test compounds.
- Luciferase was quantitated using the Dual Luciferase kit from Promega (Madison, WI).
- Results are shown in Table 1.
- Activity equal to A refers to a compound having an EC5 0 ⁇ 100 nM
- B and C denote compounds having an EC 50 between 100 nM and luM (B) or >luM (C).
- Table 1 Activity equal to A refers to a compound having an EC5 0 ⁇ 100 nM
- B and C denote compounds having an EC 50 between 100 nM and luM (B) or >luM (C).
- Another aspect of the invention is a method for treating HIV infection in a human patient comprising administering a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, with a pharmaceutically acceptable carrier.
- Another aspect of the invention is the use of a compound of formula I in the manufacture of a medicament for the treatment of AIDS or HIV infection.
- Another aspect of the invention is a method for treating HIV infection in a human patient comprising the administration of a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, with a therapeutically effective amount of at least one other agent used for treatment of AIDS or HIV infection selected from the group consisting of nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV protease inhibitors, HIV fusion inhibitors, HIV attachment inhibitors, CCR5 inhibitors, CXCR4 inhibitors, HIV budding or maturation inhibitors, and HIV integrase inhibitors.
- Another aspect of the invention is a method wherein the agent is a nucleoside HIV reverse transcriptase inhibitor.
- nucleoside HIV reverse transcriptase inhibitor is selected from the group consisting of abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine, and zidovudine, or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a method wherein the agent is a non- nucleoside HIV reverse transcriptase inhibitor.
- Another aspect of the invention is a method wherein the non-nucleoside HIV reverse transcriptase inhibitor is selected from the group consisting of delavirdine, efavirenz, and nevirapine, or a pharmaceutically acceptable thereof.
- Another aspect of the invention is a method wherein the agent is an HIV protease inhibitor.
- HIV protease inhibitor is selected from the group consisting of amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and fosamprenavir, or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a method wherein the agent is an HIV fusion inhibitor.
- Another aspect of the invention is a method wherein the HIV fusion inhibitor is enfuvirtide or T-1249, or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a method wherein the agent is an HIV attachment inhibitor.
- Another aspect of the invention is a method wherein the agent is a CCR5 inhibitor.
- Another aspect of the invention is a method wherein the CCR5 inhibitor is selected from the group consisting of Sch-C, Sch-D, TAK-220, PRO- 140, and UK- 427,857, or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a method wherein the agent is a CXCR4 inhibitor.
- Another aspect of the invention is a method wherein the CXCR4 inhibitor is AMD-3100, or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a method wherein the agent is an HIV budding or maturation inhibitor.
- Another aspect of the invention is a method wherein the budding or maturation inhibitor is PA-457, or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a method wherein the agent is an HIV integrase inhibitor.
- Another aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, with at least one other agent used for treatment of AIDS or HIV infection selected from the group consisting of nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV protease inhibitors, HIV fusion inhibitors, HIV attachment inhibitors, CCR5 inhibitors, CXCR4 inhibitors, HIV budding or maturation inhibitors, and HIV integrase inhibitors, and a pharmaceutically acceptable carrier.
- the agent is a nucleoside HIV reverse transcriptase inhibitor.
- nucleoside HIV transcriptase inhibitor is selected from the group consisting of abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine, and zidovudine, or a pharmaceutically acceptable salt thereof.
- composition wherein the agent is a non- nucleoside HIV reverse transcriptase inhibitor.
- composition wherein the non- nucleoside HIV reverse transcriptase inhibitor is selected from the group consisting of delavirdine, efavirenz, and nevirapine, or a pharmaceutically acceptable salt thereof.
- compositions wherein the agent is an HIV protease inhibitor.
- the HIV protease inhibitor is selected from the group consisting of amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and fosamprenavir, or a pharmaceutically acceptable salt thereof.
- the agent is an HIV protease inhibitor.
- HIV fusion inhibitor HIV fusion inhibitor
- Another aspect of the invention is the composition method wherein the HIV fusion inhibitor is enfuvirtide or T-1249, or a pharmaceutically acceptable salt thereof.
- compositions wherein the agent are an HIV attachment inhibitor.
- composition wherein the agent is a CCR5 inhibitor.
- composition wherein the CCR5 inhibitor is selected from the group consisting of Sch-C, Sch-D, TAK-220, PRO- 140, and UK-427,857, or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is a method wherein the agent is a CXCR4 inhibitor.
- Another aspect of the invention is a method wherein the CXCR4 inhibitor is AMD-3100 or a pharmaceutically acceptable salt thereof.
- composition wherein the agent is an HIV budding or maturation inhibitor.
- compositions wherein the budding or maturation inhibitor is PA-457, or a pharmaceutically acceptable salt thereof.
- composition wherein the agent is an
- HIV integrase inhibitor HIV integrase inhibitor
- Combination means that the components are part of a combination antiretroviral therapy or highly active antiretroviral therapy (HAART) as understood by practitioners in the field of AIDS and HIV infection.
- HAART highly active antiretroviral therapy
- “Therapeutically effective” means the amount of agent required to provide a meaningful patient benefit as understood by practitioners in the field of AIDS and HIV infection. In general, the goals of treatment are suppression of viral load, restoration and preservation of immunologic function, improved quality of life, and reduction of HIV-related morbidity and mortality.
- "Patient” means a person infected with the HIV virus and suitable for therapy as understood by practitioners in the field of AIDS and HIV infection.
- compositions comprised of a therapeutically effective amount of a compound of Formula I or its pharmaceutically acceptable salt and a pharmaceutically acceptable carrier and may contain conventional excipients.
- a therapeutically effective amount is that which is needed to provide a meaningful patient benefit.
- Pharmaceutically acceptable carriers are those conventionally known carriers having acceptable safety profiles.
- Compositions encompass all common solid and liquid forms including capsules, tablets, losenges, and powders as well as liquid suspensions, syrups, elixers, and solutions. Compositions are made using common formulation techniques, and conventional excipients (such as binding and wetting agents) and vehicles (such as water and alcohols) are generally used for compositions. See, for example,
- Solid compositions are normally formulated in dosage units and compositions providing from about 1 to 1000 mg of the active ingredient per dose are preferred. Some examples of dosages are 1 mg, 10 mg, 100 mg, 250 mg, 500 mg, and 1000 mg. Generally, other antiretroviral agents will be present in a unit range similar to agents of that class used clinically. Typically, this is 0.25-1000 mg/unit.
- Liquid compositions are usually in dosage unit ranges. Generally, the liquid composition will be in a unit dosage range of 1-100 mg/mL. Some examples of dosages are 1 mg/mL, 10 mg/mL, 25 mg/mL, 50 mg/mL, and 100 mg/mL.
- antiretroviral agents will be present in a unit range similar to agents of that class used clinically. Typically, this is 1-100 mg/mL.
- the invention encompasses all conventional modes of administration; oral and parenteral methods are preferred.
- the dosing regimen will be similar to other antiretroviral agents used clinically. Typically, the daily dose will be 1-100 mg kg body weight daily. Generally, more compound is required orally and less parenterally. The specific dosing regime, however, will be determined by a physician using sound medical judgement.
- the invention also encompasses methods where the compound is given in combination therapy. That is, the compound can be used in conjunction with, but separately from, other agents useful in treating AIDS and HIV infection. Some of these agents include HIV attachment inhibitors, CCR5 inhibitors, CXCR4 inhibitors, HIV cell fusion inhibitors, HIV integrase inhibitors, HIV nucleoside reverse transcriptase inhibitors, HIV non-nucleoside reverse transcriptase inhibitors, HIV protease inhibitors, budding and maturation inhibitors, immunomodulators, and anti- infectives.
- the compound of Formula I will generally be given in a daily dose of 1-100 mg/kg body weight daily in conjunction with other agents.
- the other agents generally will be given in the amounts used therapeutically. The specific dosing regime, however, will be determined by a physician using sound medical judgement.
- the compounds of this invention can be made by various methods known in the art including those of the following schemes and in the specific embodiments section.
- the structure numbering and variable numbering shown in the synthetic schemes are distinct from, and should not be confused with, the structure or variable numbering in the claims or the rest of the specification.
- the variables in the schemes are meant only to illustrate how to make some of the compounds of this invention.
- (2S)- 2-tert-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo [1,5 -a] pyrimidin-6-yl) acetic acid The title compound was synthesized using a two step method starting from the racemic ester precursor for Example 39. The racemic ester was separated into two enantiomers using a chiral column and (2S)-methyl 2-tert-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5- methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate was isolated.
- Chiral separation method Chiralpak AD-H preparative column, 20 x 250mm, 5 ⁇ .
- Mobile Phase 15% MeOH in C0 2 @ 150Bar. Temp: 35°C.
- Flow rate 45.0 mL/min. for 14 min.
- UV was monitored @ 254nm.
- Hydrolysis of (2S)-methyl 2-ter/-butoxy-2-(7- (8-fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6- yl)acetate as described in Example 1 provided the title compound with 100% enantiomeric excess. Retention time: 4.38min.
- Chiral SFC method Chiralpak AD-H analytical column, 4.6 x 250mm, 5 ⁇ .
- Mobile Phase 15% MeOH in C0 2 .Temp: 35°C.Flow rate: 2.0 mL/min. for 14 min. UV monitored @ 254nm.
- Methyl 2-hydroxy-2-( 5-methyl-2-phenyl- 7-p-tolylpyrazolof 1, 5-a]pyrimidin-6- yl)acetate To a solution of 2-hydroxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5- a]pyrimidin-6-yl)acetic acid (6 mg, 0.016 mmol) in methanol (2 mL) was added thionyl chroride (0.0023 mL, 0.032 mmol). The reaction mixture was stirred at 40°C for 16 hrs. The solvent was evaporated to give the title compound. The crude product was used directly for next step. Methyl 2-hydroxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetate.
- Methyl 2-tert-butoxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetate To a solution of methyl 2 -hydroxy -2-(5-methyl-2-phenyl-7-p- tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (6.20 mg, 0.016 mmol) in tert-butyl acetate (0.3 mL) at room temperature was added perchloric acid (0.008 mL, 0.128 mmol). The reaction mixture was stirred for 2 h at room temperature. The reaction mixture was quenched with water and diluted with ethyl acetate.
- X H-NMR 400 MHz, CDCh) ⁇ ppm 1.28 (9 H, s), 2.85 (3 H, s), 3.10 (3 H, s), 3.75 (3 H, s), 5.41 (1 H, s), 7.06 (1 H, s), 7.38 - 7.56 (3 H, m), 8.02 - 8.04 (2 H, m).
- reaction mixture was heated in a microwave reactor at 70°C for 60min.
- the reaction mixture was filtered and the filtrate purified by preparative HPLC to afford (16mg, 43.3%) of the title compound as TFA salt.
- reaction mixture was heated in a microwave reactor at 130°C for 15min.
- the reaction mixture was filtered and the filtrate purified by preparative HPLC to afford the title compound as TFA salt (5mg, 49.9%).
- Examples 92 - 114 were synthesized using the procedure described for Example 46.
- (2S)-2-(2-(3-(Benzyloxy)phenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl- pyrazolo[l,5-a]pyrimidin-6-yl)-2-tert-butoxy acetic acid The title compound was separated from the racemic compound Example 93 using a chiral column and (2S)-2- (2-(3-(benzyloxy)phenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-pyrazolo[l,5- a]pyrimidin-6-yl)-2-tert-butoxyacetic acid was isolated.
- Chiral separation method Chiralpak AD-H preparative column, 20 x 250mm, 5 ⁇ .
- Mobile Phase 30% MeOH in C0 2 @ 1 lOBar. Temp: 35°C.
- Flow rate 45.0 mL/min. for 15 min. UV was monitored @ 266nm. Retention time: 4.82min.
- dicyclohexyl(2',6'-dimethoxy-[l,r-biphenyl]-2- yl)phosphine 595 mg, 1.449 mmol
- PALLADIUM(II) ACETATE 163 mg, 0.725 mmol
- o-tolylboronic acid 296 mg, 2.174 mmol
- (2S)-methyl 2-(tert-butoxy)- 2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate 400 mg, 0.725 mmol) in DMF (1.5 mL), followed by 2M K3PO4 8 ⁇ (200 ⁇ 1).
- Example 116- 120 were synthesized using the procedure described above for example 115.
- (2S)-Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6- yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate The title compound was separated from the racemic ester using a chiral column and (2S)-methyl 2-(tert- butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate was isolated with 100% enantiomeric excess.
- Chiral separation method Chiralpak AD-H preparative column, 30 x 250mm, 5 ⁇ .
- Mobile Phase 15% MeOH in C0 2 @ 150Bar. Temp: 35°C.
- Flow rate 70.0 mL/min. for 13 min.
- UV was monitored @ 266nm. provided the title compound with 100% enantiomeric excess.
- Retention time 5.02min.
- Enantiomeric Excess was determined by Chiral SFC method: Chiralpak AD-H analytical column, 4.6 x 250mm, 5 ⁇ .
- Mobile Phase 15% MeOH in C0 2 . Temp: 35°C.
- Flow rate 2.0 mL/min. for 10 min.
- UV monitored @ 266nm.
- Injection 5uL of ⁇ 2.0mg/mL solution in 50:50 MeOH:CHC13.
- the title compound was synthesized from (2S)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro- 5-methylchroman-6-yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate using the procedure described for example 115.
- the title compound was hydrolyzed from (2S)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5- methylchroman-6-yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate using the procedure described for example 115.
- Example 124 - 131 were synthesized using the procedure described above for Example 121.
- dicyclohexyl(2',6'-dimethoxy-[l,l'-biphenyl]-2- yl)phosphine (12 mg, 0.029 mmol), palladium acetate (6.59 mg, 0.029 mmol), (4- fluorophenyl)boronic acid (16.42 mg, 0.117 mmol) and methyl 2-(3-bromo-7-(8- fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2- (tert-butoxy)acetate (35 mg, 0.059 mmol), DMF (1 mL), followed by 2M K 3 P0 4 (50 ⁇ 1).
- Preparative chiral SFC condition Chiralpak AD-H preparative column, 30 x 250mm, 5 ⁇ , Mobile Phase: 15% MeOH in C0 2 @ 150Bar, Temp: 35°C , Flow rate: 70 mL/min for 10 min. UV monitored at 264 nm. Injection 0.35 mL of 35 mg/mL solution in 2: 1 chloroform: methanol using stacked injections.
- (2S)- 2-(3-Bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetic acid To a solution of (2S)-methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate.
- TFA salt (10 mg, 0.017 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 60°C for 48 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (5 mg, 50 %) of the title compound as the TFA salt.
- Examples 140-142 were prepared using the procedure similar to example 139
- Solvent B 95 % Acetonitrile: 5% Water : lOmM ammonium acetate
- Solvent B 95 % Acetonitrile: 5% Water : lOmM ammonium acetate
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Molecular Biology (AREA)
- AIDS & HIV (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The disclosure generally relates to compounds of formula (I), including compositions and methods for treating human immunodeficiency virus (HIV) infection. The disclosure provides novel inhibitors of HIV, pharmaceutical compositions containing such compounds, and methods for using these compounds in the treatment of HIV infection.
Description
INHIBITORS OF HUMAN IMMUNODEFICIENCY VIRUS REPLICATION
CROSS REFERENCE TO RELATED APPLICATIONS
This patent application claims the benefit of U.S. provisional patent application no. 61/380,759 filed September 8, 2010.
BACKGROUND OF THE INVENTION
The disclosure generally relates to compounds, compositions, and methods for the treatment of human immunodeficiency virus (HIV) infection. The disclosure provides novel inhibitors of HIV, pharmaceutical compositions containing such compounds, and methods for using these compounds in the treatment of HIV infection.
Human immunodeficiency virus (HIV) has been identified as the etiological agent responsible for acquired immune deficiency syndrome (AIDS), a fatal disease characterized by destruction of the immune system and the inability to fight off life threatening opportunistic infections. Recent statistics indicate that as many as 33 million people worldwide are infected with the virus (UNAIDS: Report on the Global HIV/AIDS Epidemic, December 1998). In addition to the large number of individuals already infected, the virus continues to spread. Estimates from 1998 point to close to 6 million new infections in that year alone. In the same year there were approximately 2.5 million deaths associated with HIV and AIDS.
There are currently a number of antiviral drugs available to combat the infection. These drugs can be divided into classes based on the viral protein they target or their mode of action. In particular, saquinavir, indinavir, ritonavir, nelfinavir atazanavir darunavir, amprenavir, fosamprenavir, lopinavir and tipranavir are competitive inhibitors of the aspartyl protease expressed by HIV. Zidovudine, didanosine, stavudine, lamivudine, zalcitabine, emtricitibine, tenofovir and abacavir are nucleos(t)ide reverse transcriptase inhibitors that behave as substrate mimics to halt viral cDNA synthesis. The non-nucleoside reverse transcriptase inhibitors
nevirapine, delavirdine, efavirenz and etravirine inhibit the synthesis of viral cDNA via a non-competitive (or uncompetitive) mechanism. Enfuvirtide and maraviroc inhibit the entry of the virus into the host cell. An HIV integrase inhibitor, raltegravir (MK-0518, Isentress®), has also been approved for use in treatment experienced patients, and it is clear that this class of inhibitors is very effective as part of a combination regimen containing HIV inhibitors of different classes.
Used alone, these drugs are effective in reducing viral replication: however, the effect is only temporary as the virus readily develops resistance to all known agents used as monotherapy. However, combination therapy has proven very effective at both reducing virus and suppressing the emergence of resistance in a number of patients. In the US, where combination therapy is widely available, the number of HIV-related deaths has dramatically declined (Palella, F. J.; Delany, K. M.; Moorman, A. C; Loveless, M. O.; Furher, J.; Satten, G. A.; Aschman, D. J.; Holmberg, S. D. N. Engl. J. Med. 1998, 338, 853-860).
Unfortunately, not all patients are responsive and a large number fail this therapy. In fact, initial studies suggest that approximately 30-50% of patients ultimately fail at least one drug in the suppressive combination. Treatment failure in most cases is caused by the emergence of viral resistance. Viral resistance in turn is caused by the replication rate of HIV- 1 during the course of infection combined with the relatively high viral mutation rate associated with the viral polymerase and the lack of adherence of HIV-infected individuals in taking their prescribed medications. Clearly, there is a need for new antiviral agents, preferably with activity against viruses already resistant to currently approved drugs. Other important factors include improved safety and a more convenient dosing regimen than many of the currently approved drugs.
Compounds which inhibit HIV replication have been disclosed. See
WO2007131350, WO2009062285, WO2009062288, WO2009062289, and
WO2009062308.
The invention provides technical advantages, for example, the compounds are novel and are useful in the treatment of HIV. Additionally, the compounds provide advantages for pharmaceutical uses, for example, with regard to one or more of their mechanism of action, binding, inhibition efficacy, target selectivity, solubility, safety profiles, or bioavailability.
DESCRIPTION OF THE INVENTION
The invention encompasses compounds of Formula I, including
pharmaceutically acceptable salts, their pharmaceutical compositions, and their inhibiting HIV integrase and treating those infected with HIV or AIDS.
One aspect of the invention is a compound of Formula I
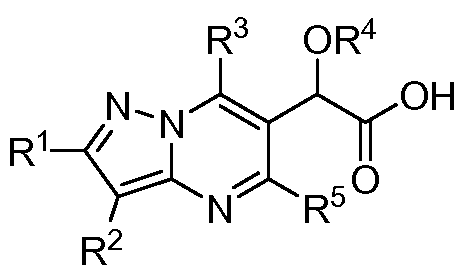
I
where:
R1 is H, alkyl, cycloalkyl, or Ar1
R2 is H, alkyl, cycloalkyl, or Ar1
R3 is alkyl or Ar2;
R4 is alkyl or haloalkyl;
R5 is alkyl;
Ar1 is phenyl, pyridinyl, tetralinyl, indazolyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, benzyloxy, halobenzyloxy, (alkoxy )benzyloxy, phenoxyalkyl, CONH(phenyl), CONH(benzyl), and Ar3;
Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido;

Ar3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 0-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl; or a pharmaceutically acceptable salt thereof. Another aspect of the invention is a compound of formula I where:
R1 is H, alkyl, cycloalkyl, or Ar1;
R2 is H, alkyl, cycloalkyl, or Ar1;
R3 is alkyl or Ar2; R4 is alkyl or haloalkyl; R5 is alkyl;
Ar1 is phenyl, pyridinyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, and phenoxyalkyl; and
Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido;

or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a compound of formula I where R1 is Ar1; R2 is H; R3 is Ar2; R4 is alkyl; R5 is methyl; Ar1 is phenyl, pyridinyl, or chromanyl,
and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, and phenoxyalkyl; and Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl,
tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido; or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a compound of formula I where R1 is Ar1; R2 is H; R3 is Ar2; R4 is alkyl; R5 is methyl; Ar1 is phenyl substituted with 0-1 alkyl substituents; and Ar2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido; or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a compound of formula I where R1 is H and R2 is Ar1.
Another aspect of the invention is a compound of formula I where R1 is Ar1 and R2 is H. Another aspect of the invention is a compound of formula I where R3 is Ar2.
Another aspect of the invention is a compound of formula I where Ar2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido.
Another aspect of the invention is a compound of formula I where R4 is alkyl.
Another aspect of the invention is a compound of formula I where R5 is methyl.
Another aspect of the invention is a compound of formula I where Ar1 is phenyl substituted with 0-1 alkyl substituents.
Another aspect of the invention is a compound of formula I where where R1 is
Ar ; R is H; R is Ar ; R is alkyl; R is methyl; Ar is phenyl or pyridinyl, and is substituted with 1 Ar3 substituent and 0-2 substituents selected from halo, alkyl, haloalkyl, alkoxy, and haloalkoxy; and Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido; or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a compound of formula I where R1 is Ar1; R2 is H; and Ar1 is phenyl or pyridinyl, and is substituted with 1 Ar3 substituent and 0-2 substituents selected from halo, alkyl, haloalkyl, alkoxy, and haloalkoxy.
Another aspect of the invention is a compound of formula I where Ar2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido.
Another aspect of the invention is a compound of formula I where Ar3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 0-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl.
Another aspect of the invention is a compound of formula I where Ar3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 1-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl.
Another aspect of the invention is a compound of formula I where Ar3 is phenyl, pyridinyl, or pyrazolyl, and is substituted with 1-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl.
Another aspect of the invention is a compound of formula I where Ar3 is phenyl, pyridinyl, or pyrazolyl, and is substituted with 1-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, and haloalkoxy.
For a compound of Formula I, the scope of any instance of a variable substituent, including R , R , R , R , R , Ar , Ar , and Ar , can be used independently with the scope of any other instance of a variable substituent. As such, the invention includes combinations of the different aspects. Unless specified otherwise, these terms have the following meanings.
"Alkyl" means a straight or branched alkyl group composed of 1 to 6 carbons.
"Alkenyl" means a straight or branched alkyl group composed of 2 to 6 carbons with at least one double bond. "Alkynyl" means a straight or branched alkyl group composed of 2 to 6 carbons with at least one triple bond. "Cycloalkyl" means a monocyclic ring system composed of 3 to 7 carbons. "Haloalkyl" and "haloalkoxy" include all halogenated isomers from monohalo to perhalo. "Tetralin" means tetrahydronaphthalene. Terms with a hydrocarbon moiety (e.g. alkoxy) include straight and branched isomers for the hydrocarbon portion. Parenthetic and multiparenthetic terms are intended to clarify bonding relationships to those skilled in the art. For example, a term such as ((R)alkyl) means an alkyl substituent further substituted with the substituent R.
"Chroman" means

The invention includes all pharmaceutically acceptable salt forms of the compounds. Pharmaceutically acceptable salts are those in which the counter ions do not contribute significantly to the physiological activity or toxicity of the compounds and as such function as pharmacological equivalents. These salts can be made according to common organic techniques employing commercially available reagents. Some anionic salt forms include acetate, acistrate, besylate, bromide, chloride, citrate, fumarate, glucouronate, hydrobromide, hydrochloride, hydroiodide, iodide, lactate, maleate, mesylate, nitrate, pamoate, phosphate, succinate, sulfate, tartrate, tosylate, and xinofoate. Some cationic salt forms include ammonium, aluminum, benzathine, bismuth, calcium, choline, diethylamine, diethanolamine, lithium, magnesium, meglumine, 4-phenylcyclohexylamine, piperazine, potassium, sodium, tromethamine, and zinc.
Some of the compounds of the invention exist in stereoisomeric forms. The invention includes all stereoisomeric forms of the compounds including enantiomers and diastereromers. Methods of making and separating stereoisomers are known in the art. The invention includes all tautomeric forms of the compounds. The invention includes atropisomers and rotational isomers.
The invention is intended to include all isotopes of atoms occurring in the present compounds. Isotopes include those atoms having the same atomic number but different mass numbers. By way of general example and without limitation, isotopes of hydrogen include deuterium and tritium. Isotopes of carbon include 13C and 14C. Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed. Such compounds may have a variety of potential uses, for example as standards and reagents in determining biological activity. In the case of stable isotopes, such compounds may have the
potential to favorably modify biological, pharmacological, or pharmacokinetic properties.
Biological Methods Inhibition of HIV replication. A recombinant NL-Rluc virus was constructed in which a section of the nef gene from NL4-3 was replaced with the Renilla Luciferase gene. The NL-RLuc virus was prepared by co-transfection of two plasmids, pNLRLuc and pVSVenv. The pNLRLuc contains the NL-Rluc DNA cloned into pUC18 at the Pvull site, while the pVSVenv contains the gene for VSV G protein linked to an LTR promoter. Transfections were performed at a 1 :3 ratio of pNLRLuc to pVSVenv in 293T cells using the LipofectAMINE PLUS kit from Invitrogen (Carlsbad, CA) according to the manufacturer, and the pseudotype virus generated was titered in MT-2 cells. For susceptibility analyses, the titrated virus was used to infect MT-2 cells in the presence of compound, and after 5 days of incubation, cells were processed and quantitated for virus growth by the amount of expressed luciferase. This provides a simple and easy method for quantitating the extent of virus growth and consequently, the antiviral activity of test compounds. Luciferase was quantitated using the Dual Luciferase kit from Promega (Madison, WI).
Susceptibility of viruses to compounds was determined by incubation in the presence of serial dilutions of the compound. The 50% effective concentration (EC50) was calculated by using the exponential form of the median effect equation where (Fa) = 1/[1+ (ED50/drug conc.)m] (Johnson VA, Byington RT. Infectivity Assay. In Techniques in HIV Research, ed. Aldovini A, Walker BD. 71-76. New York: Stockton Press.1990). The anti-viral activity of compounds was evaluated under three serum conditions, 10% FBS, 15mg/ml human serum albumin/10% FBS or 40% human serum/5% FBS, and the results from at least 2 experiments were used to calculate the EC50 values. Results are shown in Table 1. Activity equal to A refers to a compound having an EC50 < 100 nM, while B and C denote compounds having an EC50 between 100 nM and luM (B) or >luM (C).
Table 1.





47 A 0.08














Pharmaceutical Composition and Methods of Use
The compounds of this invention inhibit HIV replication. Accordingly, another aspect of the invention is a method for treating HIV infection in a human patient comprising administering a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, with a pharmaceutically acceptable carrier. Another aspect of the invention is the use of a compound of formula I in the manufacture of a medicament for the treatment of AIDS or HIV infection.
Another aspect of the invention is a method for treating HIV infection in a human patient comprising the administration of a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, with a therapeutically effective amount of at least one other agent used for treatment of AIDS or HIV infection selected from the group consisting of nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV protease inhibitors, HIV fusion inhibitors, HIV attachment inhibitors, CCR5
inhibitors, CXCR4 inhibitors, HIV budding or maturation inhibitors, and HIV integrase inhibitors.
Another aspect of the invention is a method wherein the agent is a nucleoside HIV reverse transcriptase inhibitor.
Another aspect of the invention is a method wherein the nucleoside HIV reverse transcriptase inhibitor is selected from the group consisting of abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine, and zidovudine, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method wherein the agent is a non- nucleoside HIV reverse transcriptase inhibitor. Another aspect of the invention is a method wherein the non-nucleoside HIV reverse transcriptase inhibitor is selected from the group consisting of delavirdine, efavirenz, and nevirapine, or a pharmaceutically acceptable thereof.
Another aspect of the invention is a method wherein the agent is an HIV protease inhibitor.
Another aspect of the invention is a method wherein the HIV protease inhibitor is selected from the group consisting of amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and fosamprenavir, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method wherein the agent is an HIV fusion inhibitor. Another aspect of the invention is a method wherein the HIV fusion inhibitor is enfuvirtide or T-1249, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method wherein the agent is an HIV attachment inhibitor.
Another aspect of the invention is a method wherein the agent is a CCR5 inhibitor.
Another aspect of the invention is a method wherein the CCR5 inhibitor is selected from the group consisting of Sch-C, Sch-D, TAK-220, PRO- 140, and UK- 427,857, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method wherein the agent is a CXCR4 inhibitor.
Another aspect of the invention is a method wherein the CXCR4 inhibitor is AMD-3100, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method wherein the agent is an HIV budding or maturation inhibitor. Another aspect of the invention is a method wherein the budding or maturation inhibitor is PA-457, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method wherein the agent is an HIV integrase inhibitor.
Another aspect of the invention is a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, with at least one other agent used for treatment of AIDS or HIV infection selected from the group consisting of nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV protease inhibitors, HIV fusion inhibitors, HIV attachment inhibitors, CCR5 inhibitors, CXCR4 inhibitors, HIV budding or maturation inhibitors, and HIV integrase inhibitors, and a pharmaceutically acceptable carrier.
Another aspect of the invention is the composition wherein the agent is a nucleoside HIV reverse transcriptase inhibitor.
Another aspect of the invention is the composition wherein the nucleoside HIV transcriptase inhibitor is selected from the group consisting of abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine, and zidovudine, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is the composition wherein the agent is a non- nucleoside HIV reverse transcriptase inhibitor.
Another aspect of the invention is the composition wherein the non- nucleoside HIV reverse transcriptase inhibitor is selected from the group consisting of delavirdine, efavirenz, and nevirapine, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is the composition wherein the agent is an HIV protease inhibitor. Another aspect of the invention is the composition wherein the HIV protease inhibitor is selected from the group consisting of amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and fosamprenavir, or a pharmaceutically acceptable salt thereof. Another aspect of the invention is the composition wherein the agent is an
HIV fusion inhibitor.
Another aspect of the invention is the composition method wherein the HIV fusion inhibitor is enfuvirtide or T-1249, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is the composition wherein the agent is an HIV attachment inhibitor.
Another aspect of the invention is the composition wherein the agent is a CCR5 inhibitor.
Another aspect of the invention is the composition wherein the CCR5 inhibitor is selected from the group consisting of Sch-C, Sch-D, TAK-220, PRO- 140, and UK-427,857, or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is a method wherein the agent is a CXCR4 inhibitor.
Another aspect of the invention is a method wherein the CXCR4 inhibitor is AMD-3100 or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is the composition wherein the agent is an HIV budding or maturation inhibitor.
Another aspect of the invention is the composition wherein the budding or maturation inhibitor is PA-457, or a pharmaceutically acceptable salt thereof. Another aspect of the invention is the composition wherein the agent is an
HIV integrase inhibitor.
"Combination," "coadministration," "concurrent" and similar terms referring to the administration of a compound of Formula I with at least one anti-HIV agent mean that the components are part of a combination antiretroviral therapy or highly active antiretroviral therapy (HAART) as understood by practitioners in the field of AIDS and HIV infection.
"Therapeutically effective" means the amount of agent required to provide a meaningful patient benefit as understood by practitioners in the field of AIDS and HIV infection. In general, the goals of treatment are suppression of viral load, restoration and preservation of immunologic function, improved quality of life, and reduction of HIV-related morbidity and mortality.
"Patient" means a person infected with the HIV virus and suitable for therapy as understood by practitioners in the field of AIDS and HIV infection.
"Treatment," "therapy," "regimen," "HIV infection," "ARC," "AIDS" and related terms are used as understood by practitioners in the field of AIDS and HIV infection.
The compounds of this invention are generally given as pharmaceutical compositions comprised of a therapeutically effective amount of a compound of Formula I or its pharmaceutically acceptable salt and a pharmaceutically acceptable carrier and may contain conventional excipients. A therapeutically effective amount is that which is needed to provide a meaningful patient benefit. Pharmaceutically acceptable carriers are those conventionally known carriers having acceptable safety profiles. Compositions encompass all common solid and liquid forms including capsules, tablets, losenges, and powders as well as liquid suspensions, syrups, elixers, and solutions. Compositions are made using common formulation techniques, and conventional excipients (such as binding and wetting agents) and vehicles (such as water and alcohols) are generally used for compositions. See, for example,
Remington 's Pharmaceutical Sciences, 17th edition, Mack Publishing Company, Easton, PA (1985).
Solid compositions are normally formulated in dosage units and compositions providing from about 1 to 1000 mg of the active ingredient per dose are preferred. Some examples of dosages are 1 mg, 10 mg, 100 mg, 250 mg, 500 mg, and 1000 mg. Generally, other antiretroviral agents will be present in a unit range similar to agents of that class used clinically. Typically, this is 0.25-1000 mg/unit.
Liquid compositions are usually in dosage unit ranges. Generally, the liquid composition will be in a unit dosage range of 1-100 mg/mL. Some examples of dosages are 1 mg/mL, 10 mg/mL, 25 mg/mL, 50 mg/mL, and 100 mg/mL.
Generally, other antiretroviral agents will be present in a unit range similar to agents of that class used clinically. Typically, this is 1-100 mg/mL.
The invention encompasses all conventional modes of administration; oral and parenteral methods are preferred. Generally, the dosing regimen will be similar to other antiretroviral agents used clinically. Typically, the daily dose will be 1-100 mg kg body weight daily. Generally, more compound is required orally and less parenterally. The specific dosing regime, however, will be determined by a physician using sound medical judgement.
The invention also encompasses methods where the compound is given in combination therapy. That is, the compound can be used in conjunction with, but separately from, other agents useful in treating AIDS and HIV infection. Some of these agents include HIV attachment inhibitors, CCR5 inhibitors, CXCR4 inhibitors, HIV cell fusion inhibitors, HIV integrase inhibitors, HIV nucleoside reverse transcriptase inhibitors, HIV non-nucleoside reverse transcriptase inhibitors, HIV protease inhibitors, budding and maturation inhibitors, immunomodulators, and anti- infectives. In these combination methods, the compound of Formula I will generally be given in a daily dose of 1-100 mg/kg body weight daily in conjunction with other agents. The other agents generally will be given in the amounts used therapeutically. The specific dosing regime, however, will be determined by a physician using sound medical judgement.
Synthetic Methods
The compounds of this invention can be made by various methods known in the art including those of the following schemes and in the specific embodiments section. The structure numbering and variable numbering shown in the synthetic schemes are distinct from, and should not be confused with, the structure or variable numbering in the claims or the rest of the specification. The variables in the schemes are meant only to illustrate how to make some of the compounds of this invention.
The disclosure is not limited to the foregoing illustrative examples and the examples should be considered in all respects as illustrative and not restrictive, reference being made to the appended claims, rather than to the foregoing examples,
and all changes which come within the meaning and range of equivalency of the claims are therefore intended to be embraced.
Abbreviations used in the schemes generally follow conventions used in the art. Chemical abbreviations used in the specification and examples are defined as follows: "NaHMDS" for sodium bis(trimethylsilyl)amide; "DMF" for N,N- dimethylformamide; "MeOH" for methanol; "NBS" for N-bromosuccinimide; "Ar" for aryl; "TFA" for trifluoroacetic acid; "LAH" for lithium aluminum hydride;
"BOC" for t-butoxycarbonate, "DMSO" for dimethylsulfoxide; "h" for hours; "rt" for room temperature or retention time (context will dictate); "min" for minutes;
"EtOAc" for ethyl acetate; "THF" for tetrahydrofuran; "EDTA" for
ethylenediaminetetraacetic acid; "Et20" for diethyl ether; "DMAP" for 4- dimethylaminopyridine; "DCE" for 1 ,2-dichloroethane; "ACN" for acetonitrile; "DME" for 1,2-dimethoxy ethane; "HOBt" for 1-hydroxybenzotriazole hydrate; "DIEA" for diisopropylethylamine, "Nf ' for CF3(CF2)3S02-; and "TMOF" for trimethylorthoformate.
Abbreviations as used herein, are defined as follows: "1 x" for once, "2 x" for twice, "3 x" for thrice, "°C" for degrees Celsius, "eq" for equivalent or equivalents, "g" for gram or grams, "mg" for milligram or milligrams, "L" for liter or liters, "mL" for milliliter or milliliters, "μΕ" for microliter or microliters, "N" for normal, "M" for molar, "mmol" for millimole or millimoles, "min" for minute or minutes, "h" for hour or hours, "rt" for room temperature, "RT" for retention time, "arm" for atmosphere, "psi" for pounds per square inch, "cone." for concentrate, "sat" or "sat'd " for saturated, "MW" for molecular weight, "mp" for melting point, "ee" for enantiomeric excess, "MS" or "Mass Spec" for mass spectrometry, "ESI" for electrospray ionization mass spectroscopy, "HR" for high resolution, "HRMS" for high resolution mass spectrometry , "LCMS" for liquid chromatography mass spectrometry, "HPLC" for high pressure liquid chromatography, "RP HPLC" for reverse phase HPLC, "TLC" or "tic" for thin layer chromatography, "NMR" for nuclear magnetic resonance spectroscopy, for proton, "δ" for delta, "s" for singlet, "d" for doublet, "t" for triplet, "q" for quartet, "m" for multiplet, "br" for
broad, "Hz" for hertz, and "α", "β", "R", "S", "E", and "Z" are stereochemical designations familiar to one skilled in the art.
Scheme 1.


LC/MS Method A
Column Supelco Ascentis Express 4.6x50mm 2.7 um CI 8 Flow Rate 2 mL/min
Solvent A 5% Acetonitrile - 95% H20 - lOmM NH4OAc Solvent B 95% Acetonitrile - 5% H20 - lOmM NH4OAc Gradient %B 0-100
Gradient Time 8 min.
Wavelength 220nm
LC/MS Method B
Column PHENOMENEX-LUNA 2.0 x 30mm 3um Flow Rate 1 mL/min
Solvent A 10% MeOH-90% H20 - 0.1% TFA Solvent B 90% MeOH-10% H20 -0.1%TFA
Gradient %B 0-100
Gradient Time 2 min.
Wavelength 220nm LC/MS Method C
Column PHENOMENEX-LUNA 2.0 x 30mm 3um Flow Rate 1 mL/min
Solvent A 10% Acetonitrile-90% H20 - 0.1% TFA
Solvent B 90% Acetonitrile-10% H20 -0.1%TFA Gradient %B 0-100
Gradient Time 2 min.
Wavelength 220nm
LC/MS Method D
Column Waters Xbridge 4.6x100mm 3.5 um CI 8
Flow Rate 1 mL/min
Solvent A H20 - 1 OmM NH4OAc
Solvent B Acetonitrile - lOmM NH4OAc
Gradient %B 30-95
Gradient Time 11 min.
Wavelength 220nm
LC/MS Method E
Column Waters Sunfire 4.6x 100mm 3.5 um C 18 Flow Rate 1 mL/min
Solvent A H20 - 0.1 % TFA
Solvent B Acetonitrile - 0.1 % TFA
Gradient %B 30-95
Gradient Time 6 min.
Wavelength 220nm
Description of Specific Embodiments
Scheme 3.



Methyl 2-(7-chloro-5-methyl-2-phenylpyrazolo[ 1, 5-a]pyrimidin-6-yl)acetate. To methyl 2-(5-methyl-7-oxo-2-phenyl-4,7-dihydropyrazolo[l,5-a]pyrimidin-6- yl)acetate (3 g, 10.09 mmol) was added POCI3 (25 mL, 268 mmol). The reaction mixture was heated at reflux for 1 h. After cooling, the reaction mixture was added drop-wise to ice-water. A brown solid precipitated. The solid was filtered and washed with water, then dissolved in ethyl acetate. The organic solution was washed with saturated aHC03 and dried over sodium sulfate. The solvent was evaporated to give the title compound (2.77 g, 84%). XH-NMR (400 MHz, DMSO-d6) δ ppm 2.58 (3 H, s), 3.71 (3 H, s), 4.04 (2 H, s), 7.29 (1 H, s), 7.43 - 7.58 (3 H, m), 8.07 (2
H, d, J=7.0 Hz).


Methyl 2-(7-chloro-5-methyl-2-phenylpyrazolo[ 1, 5-a]pyrimidin-6-yl)-2- hydroxyacetate. To a stirred solution of KHMDS (0.5 M in toluene, 9.50 mL, 4.75 mmol) in THF (24 mL) at -78°C was added a solution of methyl 2-(7-chloro-5- methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (1 g, 3.17 mmol) in THF (24 mL) dropwise over 40 min. The mixture was stirred at -78°C for 30 min. A solution of 3-phenyl-2-(phenylsulfonyl)-l,2-oxaziridine (1.241 g, 4.75 mmol) in THF (24 mL) was added over 20 min and the reaction mixture was stirred for additional 30 min at - 78 °C. The reaction mixture was quenched with saturated NH4C1 aqueous solution (4 mL). The reaction mixture was allowed to warm to room temperature and then diluted with ethyl acetate (100 mL). The organic phase was washed with water and brine and dried with sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (535mg, 50.9%). XH-NMR
(500 MHz, CDCk) δ ppm 2.62 (3 H, s), 3.83 (3 H, s), 5.29 (1 H, s), 5.76 (1 H, s), 6.94 (1 H, s), 7.38 - 7.50 (3 H, m), 8.00 - 8.02 (2 H, m).


Methyl 2-tert-butoxy-2-(7-chloro-5-methyl-2-phenylpyrazolo[ 1, 5- aJpyrimidin-6-yl) acetate. To a suspension of methyl 2-(7-chloro-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (100 mg, 0.301 mmol) in tert-butyl acetate (2 mL) at room temperature was added CH2CI2 (2 mL) followed by perchloric acid (0.027 mL, 0.452 mmol). The reaction mixture was stirred for 2 h at room temperature. The reaction mixture was quenched with water and diluted with ethyl acetate. The organic phase was washed with saturated NaHC(¾ and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel
chromatography provided the title compound (71 mg, 60.7%). XH-NMR (500 MHz, CDCh) δ ppm 1.27 (9 H, s), 2.66 (3 H, s), 3.73 (3 H, s), 5.66 (1 H, s), 6.93 (1 H, s),
7.34 - 7.52 (3 H, m), 8.01 (2 H, d, J=7.3 Hz).


Methyl 2-tert-butoxy-2-(7-(3, 4-dimethylphenyl)-5-methyl-2-phenylpyrazolo[ 1, 5- a]pyrimidin-6-yl)acetate, TFA salt. To a 0.5-2 mL microwave tube was added methyl 2-tert-butoxy-2-(7-chloro-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6- yl)acetate (20 mg, 0.052 mmol), tetrakis(triphenylphosphine)palladium(0) (8.94 mg, 7.73 μιηοΐ), 3,4-dimethylphenylboronic acid (11.60 mg, 0.077 mmol), DMF (1.5 mL), followed by 2M K3PO4 8θΓΐιίϊοη(100μι). The reaction mixture was heated in a microwave reactor at 130°C for 15min. The reaction mixture was filtered and the filtrate purified by preparative HPLC to afford (16mg, 54.3%) of the title compound as TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S10, 50 to 100% B over 22 minute gradient, 8 minute hold time, A = 10% methanol 90%
water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40ml/min. ¾-NMR (300 MHz, CDCh, 60°C) δ ppm 0.97 (9 H, s), 2.34 (3 H, s), 2.39 (3 H, s), 2.74 (3 H, s), 3.78 (3 H, s), 5.14 (1 H, s), 7.00 (1 H, s), 7.27 - 7.48 (6 H, m), 7.82 (2 H, dd, J=7.9, 1.6 Hz).

Example 1

2-tert-Butoxy-2-(7-(3,4-dimethylphenyl)-5-methyl-2-phenylpyrazolo[l,5- a] pyrimidin-6-yl) acetic acid, TFA salt. To a solution of methyl 2-tert-butoxy-2-(7- (3,4-dimethylphenyl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt (15 mg, 0.026 mmol) in dioxane (0.5 mL) was added 1.5 N LiOH aqueous
solution (0.5 niL, 0.750 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (lOmg, 67.7%) of the title compound as TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 minute gradient, 6 minute hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40ml/min. ¾-NMR (400 MHz, MeOD) δ ppm 0.97 (9 H, s), 2.40 (3 H, s), 2.45 (3 H, s), 2.68 (3 H, s), 5.18 (1 H, s), 6.93 (1 H, s), 7.32 - 7.57 (6 H, m), 7.80 - 7.94 (2 H, m).

Compounds in the Table 2 (Examples 2-38) were synthesized using the procedure described above using the appropriate boronic acids.
Table2.





2, 3-dihydropyrano[ 4, 3, 2-de] quinolin-7 -ylboronic acid.
The title compound was prepared from the known procedure as described in Reference: WO 2009/062285.

2-(8-fluoro-5-methylchroman-6-yl)-4, 4, 5, 5-tetramethyl-l, 3, 2 dioxaborolane. The title compound was prepared from the known procedure as described in reference WO 2009/062285.
Example 39

(2S)- 2-tert-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo [1,5 -a] pyrimidin-6-yl) acetic acid. The title compound was synthesized using a two step method starting from the racemic ester precursor for Example 39. The racemic ester was separated into two enantiomers using a chiral
column and (2S)-methyl 2-tert-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5- methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate was isolated. Chiral separation method: Chiralpak AD-H preparative column, 20 x 250mm, 5μιη. Mobile Phase: 15% MeOH in C02 @ 150Bar. Temp: 35°C. Flow rate: 45.0 mL/min. for 14 min. UV was monitored @ 254nm. Hydrolysis of (2S)-methyl 2-ter/-butoxy-2-(7- (8-fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6- yl)acetate as described in Example 1 provided the title compound with 100% enantiomeric excess. Retention time: 4.38min. Chiral SFC method: Chiralpak AD-H analytical column, 4.6 x 250mm, 5μιη. Mobile Phase: 15% MeOH in C02.Temp: 35°C.Flow rate: 2.0 mL/min. for 14 min. UV monitored @ 254nm.
Example 40

(2R)- 2-tert-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo [1,5 -a] pyrimidin-6-yl) acetic acid. The title compound was synthesized using a two step method starting from the racemic ester precursor for Example 39. The racemic ester was separated into two enantiomers using a chiral column and (2R)-methyl 2-ter?-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5- methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate was isolated. Chiral separation method: Chiralpak AD-H preparative column, 20 x 250mm, 5μιη to give two enantiomers. Mobile Phase: 15% MeOH in C02 @ 150Bar. Temp: 35°C. Flow rate: 45.0 mL/min. for 14 min. UV was monitored @ 254nm. Hydrolysis of (2R)- methyl 2-tert-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate as described in Example 1 provided the title compound with 100% enantiomeric excess. Retention time: 9.94min. Chiral
SFC method: Chiralpak AD-H analytical column, 4.6 x 250mm, 5μιη. Mobile Phase: 15% MeOH in C02.Temp: 35°C.Flow rate: 2.0 mL/min. for 14 min. UV monitored @ 254nm.
Scheme 4.

Methyl 5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6-carboxylate. To a stirred solution of 4-methylbenzaldehyde (1.2 g, 9.99 mmol), 3 -phenyl- lH-pyrazol-5- amine (1.6 g, 9.99 mmol), and methyl 3-oxobutanoate (1.3 g, 10.99 mmol) in THF (80 mL) and heptane (20 mL) was added piperidine (30μΙ,, 0.303 mmol). The
reaction mixture was heated at reflux for 20 h. The solvent was evaporated and the crude material was dissolved in CH2CI2. DDQ (2041 mg, 8.99 mmol) was added and the mixture was stirred at room temperature for 1 h. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (2.3 g, 64.4%). ¾-NMR (500 MHz, CDCI3) δ ppm 2.47 (3 H, s), 2.65 (3 H, s), 3.64 (3 H, s), 6.93 (1 H, s), 7.31 - 7.47 (5 H, m), 7.63 (2 H, d, J=7.9 Hz), 7.86 - 7.99 (2 H, m).


(5-Methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)methanol. To a stirred solution of methyl 5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6- carboxylate (800 mg, 2.238 mmol) in CH2C12 (50 mL) was added DIBAL-H, 1M in THF (6.72 mL, 6.72 mmol) dropwise. The reaction mixture was stirred at room temperature for 1 h before being quenched with saturated NH4CI solution. The
aqueous layer was extracted with CH2CI2 and the combined organic layer was dried over anhydrous a2S04, filtered and concentrated. The residue was purified by silica gel chromatography to give the title compound (344 mg, 46.7%). XH-NMR (500 MHz, CDCh) δ ppm 1.61 (1 H, t, J=5.0 Hz), 2.49 (3 H, s), 2.78 (3 H, s), 4.59 (2 H, d, J=5.0 Hz), 6.88 (1 H, s), 7.29 - 7.42 (5 H, m), 7.56 (2 H, d, J=8.2 Hz), 7.81 - 7.96 (2 H, m).


5-Methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6-carbaldehyde. To a stirred solution of (5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)methanol (100 mg, 0.304 mmol) in CH2C12 (8 mL) was added PCC (98 mg, 0.455 mmol). The reaction mixture was stirred at room temperature for 16 h. The solvent was evaporated and the residue was purified by silica gel chromatography to give the title
compound (82 mg, 83%). XH-NMR (500 MHz, CDCk) δ ppm 2.52 (3 H, s), 2.89 (3 H, s), 6.98 (1 H, s), 7.33 - 7.50 (5 H, m), 7.60 (2 H, d, J=7.9 Hz), 7.91 (2 H, dd, J=8.1, 1.4 Hz), 9.80 (1 H, s).


2-Hydroxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetonitrile. To a solution of 5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6- carbaldehyde (30 mg, 0.092 mmol) in CH2CI2 (2 mL) at 0°C was added zinc iodide (14.63 mg, 0.046 mmol) followed by TMS-CN (0.049 mL, 0.367 mmol). The reaction mixture was stirred at room temperature for 4 h and diluted with CH2CI2 (100ml), washed with water, and dried over Na2S04. The solvent was evaporated and the residue was purified by preparative HPLC to give the title compound (15mg, 46.2%) as TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x
100mm S10, 40 to 100% B over 22 minute gradient, 7 minute hold time, A=10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40ml/min. XH-NMR (500 MHz, CDCl3) δ ppm 2.46 (3 H, s), 2.89 (3 H, s), 5.39 (1 H, s), 6.85 (1 H, s), 7.16 - 7.25 (3 H, m), 7.30 - 7.44 (4 H, m), 7.63 - 7.76 (2 H, m).


2-Hydroxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid. A solution of 2-hydroxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetonitrile (28 mg, 0.079 mmol) in cone. HCl (400 iL, 4.87 mmol) was heated at 90°C for 3 h. The solvent was evaporated and the residue was purified by preparative HPLC to afford (14mg, 47.5%) of the title compound. XH-NMR (400
MHz, MeOD) δ ppm 2.51 (3 H, s), 2.67 (3 H, s), 5.19 (1 H, s), 6.95 (1 H, s), 7.29 - 7.51 (5 H, m), 7.57 (2 H, d, J=8.3 Hz), 7.80 - 7.93 (2 H, m).


Methyl 2-hydroxy-2-( 5-methyl-2-phenyl- 7-p-tolylpyrazolof 1, 5-a]pyrimidin-6- yl)acetate. To a solution of 2-hydroxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5- a]pyrimidin-6-yl)acetic acid (6 mg, 0.016 mmol) in methanol (2 mL) was added thionyl chroride (0.0023 mL, 0.032 mmol). The reaction mixture was stirred at 40°C for 16 hrs. The solvent was evaporated to give the title compound. The crude product was used directly for next step.
Methyl 2-hydroxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetate.
MS (M+H)+ Calcd. 388
MS (M+H)+ Observ. 388
Retention Time 2.15 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-tert-butoxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetate. To a solution of methyl 2 -hydroxy -2-(5-methyl-2-phenyl-7-p- tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (6.20 mg, 0.016 mmol) in tert-butyl acetate (0.3 mL) at room temperature was added perchloric acid (0.008 mL, 0.128 mmol). The reaction mixture was stirred for 2 h at room temperature. The reaction mixture was quenched with water and diluted with ethyl acetate. The organic phase was washed with saturated aHC03 and dried over sodium sulfate. The solvent was evaporated to give the title compound. The crude product was used directly for next step.
Methyl 2-tert-butoxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetate.
MS (M+H)+ Calcd. 444
MS (M+H)+ Observ. 444
Retention Time 2.25 min
LC Condition
Solvent A 10 % Acetonitrile: 90% Water : 0.1% TFA
Solvent B 90 % Acetonitrile: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Acetonitrile: Water: TFA
Column Phenomenex Luna CI 8, 30x2, 3u
Example 41

2-tert-Butoxy-2-(5-methyl-2-phenyl-7-p-tolylpyrazolofl,5-aJpyrimidin-6-yl)acetic acid, TFA salt. To a solution of methyl 2-tert-butoxy -2-(5-methyl-2-phenyl-7-p- tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (7.10 mg, 0.016 mmol) in dioxane (0.5 mL) was added 1.5 N LiOH aqueous solution (0.5 mL, 0.750 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (4mg, 43.7% for 3 steps) of the title compound as TFA salt. Preparative HPLC condition: Waters Sunfire C18 30 x 100mm 5u, 50 to 100% B over 22 minute gradient, 6 minute hold time, A=10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40ml/min. XH-NMR (400 MHz, MeOD) δ ppm 0.97 (9 H, s), 2.53 (3 H, s), 2.69
(3 H, s), 5.19 (1 H, s), 6.94 (1 H, s), 7.28 - 7.43 (3 H, m), 7.49 (2 H, d, J=8.0 Hz), 7.67 (2 H, dd, J=7.8, 2.8 Hz), 7.82 - 7.91 (2 H, m).

Scheme 5.

Methyl 5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6-carboxylate. To a stirred solution of 4-methylbenzaldehyde (723 mg, 6.02 mmol), lH-pyrazol-5 -amine (500 mg, 6.02 mmol), and methyl 3-oxobutanoate (769 mg, 6.62 mmol) in THF (80 mL) and heptane (20 mL) was added piperidine (ΙΟμί, 0.101 mmol). The reaction mixture was heated at reflux for 20 h. The solvent was evaporated and the crude material was dissolved in (¾(¾. DDQ (1229 mg, 5.42 mmol) was added and the mixture was stirred at room temperature for 1 h. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (1120mg, 66.2%). XH-NMR (500 MHz, CDCl3) δ ppm 2.44 (3 H, s), 2.65 (3 H, s), 3.63 (3 H,
s), 6.65 (1 H, d, J=2.4 Hz), 7.34 (2 H, d, J=8.1 Hz), 7.52 (2 H, d, J=8.1 Hz), 8.10 (1 H, d, J=2.4 Hz).


(5-Methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)methanol. To a stirred solution of methyl 5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6-carboxylate (950 mg, 3.38 mmol) in CH2C12 (50 mL) was added DIBAL-H, 1M in THF (16.9 mL, 16.9 mmol) dropwise. The reaction mixture was stirred at room temperature for 1 h before being quenched with saturated NH4C1 solution. The aqueous layer was extracted with CH2CI2 and the combined organic layer was dried over anhydrous a2S04, filtered and concentrated. The residue was purified by silica gel chromatography to give the title compound (482 mg, 56.3%). ¾-NMR (500 MHz, CDCI3) δ ppm 1.64 (1 H, t,
J=4.9 Hz), 2.45 (3 H, s), 2.78 (3 H, s), 4.58 (2 H, d, J=4.9 Hz), 6.60 (1 H, d, J=2.4 Hz), 7.34 - 7.40 (2 H, m), 7.42 - 7.49 (2 H, m), 8.00 (1 H, d, J=2.4 Hz).


5-Methyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6-carbaldehyde. To a stirred solution of (5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)methanol (300mg, 1.18 mmol) in CH2CI2 (24 mL) was added PCC (383 mg, 1.78 mmol). The reaction mixture was stirred at room temperature for 16 h. The solvent was evaporated and the residue was purified by silica gel chromatography to give the title compound (212 mg, 71.2%). ¾-NMR (500 MHz, CDCI3) δ ppm 2.48 (3 H, s), 2.89 (3 H, s), 6.69 (1 H, d, J=2.4 Hz), 7.43 (2 H, d, J=7.9 Hz), 7.51 (2 H, d, J=7.9 Hz), 8.18 (1 H, d, J=2.4 Hz), 9.82 (1 H, s).
5-Methyl- 7-p-tolylpyrazolo[ 1, 5-a]pyrimidine-6-carbaldehyde.
MS (M+H)+ Calcd. 252
MS (M+H)+ Observ. 252
Retention Time 1.98 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

2-(5-Methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(trimethylsilyloxy)a^ To a solution of 5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidine-6-carbaldehyde (200 mg, 0.796 mmol) in CH2C12 (30 mL) at 0°C was added zinc iodide (127 mg, 0.398 mmol) followed by TMS-CN (0.427 mL, 3.18 mmol). The mixture was stirred at 0°C for 1.5 h and at room temp for 2 h. The reaction mixture was diluted with CH2CI2 (lOOmL), washed with water, and dried over Na2S04. The solvent was evaporated to give the title compound (234mg, 84%) without further purification.
2-( 5-Methyl- 7-p-tolylpyrazolo[ 1, 5-a]pyrimidin-6-yl)-2- (trimethylsilyloxy)acetonitrile.
MS (M+H)+ Calcd. 362
MS (M+H)+ Observ. 362
Retention Time 2.17 min
LC Condition
Solvent A 5 % methanol: 95% Water : lOmM Ammonium Acetate
Solvent B 95 % methanol: 5% Water : lOmM Ammonium Acetate
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: Ammonium Acetate
Column Phenomenex Luna 2.0 x 30mm 3um

2-hydroxy-2-(5-methyl- 7-p-tolylpyrazolof 1, 5-a]pyrimidin-6-yl)acetic acid, TFA salt. A mixture of 2-(5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)-2- (trimethylsilyloxy)acetonitrile (238 mg, 0.679 mmol) and cone. HC1 (400 iL, 4.87 mmol) was heated in a sealed tube at 110°C for 3 h. The solvent was evaporated and the residue was purified by preparative HPLC to afford (134mg, 47.5%) of the title compound as TFA salt. XH-NMR (400 MHz, MeOD) δ ppm 2.48 (3 H, s), 2.67 (3 H, s), 5.15 (1 H, s), 6.62 (1 H, d, J=2.4 Hz), 7.38 - 7.52 (4 H, m), 8.02 (1 H, d, J=2.4 Hz).
2-hydroxy-2-( 5 -methyl- 7-p-tolylpyrazolo[ 1, 5-a] pyrimidin-6-yl)acetic acid, TFA salt.
MS (M+H)+ Calcd. 298
MS (M+H)+ Observ. 298
Retention Time 1.46 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-hydroxy-2-(5-methyl-7-p-tolylpyrazolo [ 1 ,5-a] pyrimidin-6-yl)acetate, HCl salt.
To a solution of 2-hydroxy-2-(5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetic acid, TFA salt (60 mg, 0.146 mmol) in MeOH (2 mL) was added thionyl chroride (0.021 mL, 0.292 mmol). The reaction mixture was stirred at 40°C for 6 h. The solvent was evaporated to give the title compound (50mg, 99%). XH-NMR (500 MHz, CDCh) δ ppm 2.45 (3 H, s), 2.58 (3 H, s), 3.77 (3 H, s), 5.20 (1 H, s), 6.62 (1 H, d, J=2.4 Hz), 7.33 - 7.50 (4 H, m), 8.02 (1 H, d, J=2.4 Hz).
Methyl 2-hydroxy-2-(5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetate,
HCl salt.
MS (M+H)+ Calcd. 312
MS (M+H)+ Observ. 312
Retention Time 1.71 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-tert-butoxy-2-(5-methyl- 7-p-tolylpyrazolof 1, 5-aj 'pyrimidin-6-y I) acetate. To a suspension of methyl 2-hydroxy-2-(5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetate (40 mg, 0.128 mmol) in tert-butyl acetate (1 mL) at room temperature was added perchloric acid (0.015 mL, 0.257 mmol). The reaction mixture was stirred for 2 h at room temperature before being quenched with water and diluted with ethyl acetate. The organic phase was washed with saturated aHC03 and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel
chromatography provided the title compound (26 mg, 55.1%>). XH-NMR (500 MHz, CDCh) δ ppm 0.96 (9 H, m), 2.46 (3 H, s), 2.66 (3 H, s), 3.78 (3 H, s), 5.10 (1 H, s), 6.59 (1 H, d, J=2.4 Hz), 7.37 - 7.40 (2 H, m), 7.43 - 7.55 (2 H, m), 7.99 (1 H, d, J=2.4 Hz).
Methyl 2-tert-butoxy-2-(5-methyl- 7-p-tolylpyrazolo[ 1, 5-a]pyrimidin-6-yl)acetate.
MS (M+H)+ Calcd. 368
MS (M+H)+ Observ. 368
Retention Time 2.21 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Example 42

2-tert-Butoxy-2-(5-methyl-7-p-tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid, TFA salt. To a solution of methyl 2-tert-butoxy-2-(5-methyl-7-p-tolylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (19 mg, 0.052 mmol) in dioxane (0.5 mL) was added 1.5 N LiOH aqueous solution (0.5 mL, 0.750 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (22mg, 91%) of the title compound as TFA salt.
Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 30 to 100% B over 15 minute gradient, 6 minute hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 35mL/min. XH-NMR
00 MHz, MeOD) δ ppm 0.98 (9 H, s), 2.52 (3 H, s), 2.70 (3 H, s), 5.16 (1 H, s), 63 (1 H, d, J=2.4 Hz), 7.42 - 7.66 (4 H, m), 8.03 (1 H, d, J=2.4 Hz).

Scheme 6

Preparative HPLC condition: Waters Sunfire CI 8 30 x 100mm 5u, 50 to 100% B over 20 min gradient, 6 min hold time, A=10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 35mL/min. XH-NMR (400 MHz, CDCh) δ ppm 1.28 (9 H, s), 2.85 (3 H, s), 3.10 (3 H, s), 3.75 (3 H, s), 5.41 (1 H, s), 7.06 (1 H, s), 7.38 - 7.56 (3 H, m), 8.02 - 8.04 (2 H, m).


2-tert-Butoxy-2-(5, 7-dimethyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid, TFA salt. To a solution of methyl 2-tert-butoxy-2-(5, 7-dimethyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA (15 mg, 0.031 mmol) in dioxane (0.5 mL) was added 1.5 N LiOH aqueous solution (0.5 mL, 0.750 mmol). The reaction mixture was stirred at room temperature for 4 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (13mg, 89%) of the title compound as the TFA salt. Preparative HPLC condition: Waters Sunfire C18 30 x 100mm 5u, 50 to 100% B over 20 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. ¾-NMR (500 MHz, CDCl3) δ ppm 1.27 (9 H, s), 2.85 (3 H, s), 3.10 (3 H, s), 5.41 (1 H, s), 7.07 (1 H, s), 7.34 - 7.55 (3 H, m), 7.89 - 8.06 (2 H, m), 9.92 (1 H, br. s.).

TFA salt.
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Scheme 7


Methyl2-(7-hydroxy-5-methyl-3-phenyl-4, 7-dihydropyrazolof 1, 5-a]pyrimidin-6- yl) acetate. To a solution of 4-phenyl-lH-pyrazol-5-amine (0.5g, 3.14 mmol) and dimethyl 2-acetylsuccinate (1.77 g, 9.4 mmol) in xylene (100 mL) was added p- toluenesulfonic acid monohydrate (5 mg, 0.263 mmol). The reaction mixture was heated at reflux under a Dean-Stark trap for 20 hrs. The solid was filtered and
washed by hexanes to afford (0.75 g, 80%) of the title compound. XH-NMR (500 MHz, DMSO-de) δ ppm 2.38 (3 H, s), 3.39 (2 H, s), 3.59 (3 H, s), 7.33 (1 H, S), 7.47 (2 H, m), 7.57 (2 H, m)8.13 (1 H, S), 11.87 (1 H, s).

Methyl 2-(7-chloro-5-methyl-3-phenylpyrazolo[ 1, 5-a]pyrimidin-6-yl)acetate. To methyl 2-(7-hydroxy-5-methyl-3-phenyl-4,7-dihydropyrazolo[l,5-a]pyrimidin-6- yl)acetate (0.5 g, 1.68 mmol) was added POCI3 (1 mL). The reaction mixture was heated at reflux for 1 h. After cooling, the reaction mixture was added drop-wise to ice-water. A brown solid precipitated. The solid were filtered and washed with water, then dissolved in ethyl acetate. The organic solution was washed with saturated NaHCC and dried over sodium sulfate. The solvent was evaporated to give the title compound (0.48 g, 90%). XH-NMR (500 MHz, MeOD) δ ppm 2.66 (3
H, s), 3.77 (3 H, s), 4.04 (2 H, s), 7.26 (1 H, s), 7.42 (2 H, s), 8.09 (2 H, s), 8.57 (1 H, s).


Methyl 2-(7-chloro-5-methyl-3-phenylpyrazolo[ 1, 5-a]pyrimidin-6-yl)-2- hydroxy acetate.
To a stirred solution of KHMDS (0.5 M in toluene, 4.6 mL, 4.75 mmol) in THF (12 mL) at -78°C was added a solution of methyl 2-(7-chloro-5-methyl-3- phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.48 g, 1.5 mmol) in THF (12 mL) over 20 mins. The reaction mixture was stirred at -78°C for 30 min. A solution of 3-phenyl-2-(phenylsulfonyl)-l,2-oxaziridine (0.6 g, 2.3 mmol) in THF (12 mL) was added over 10 min and the resulted reaction mixture was stirred for an additional 30 min at -78 °C. The reaction mixture was quenched with saturated NH4C1 aqueous solution (2 mL). The mixture was allowed to warm up to room temperature and
diluted with EtOAc (100 mL). The organic phase was washed with water and brine and dried with sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (250mg, 51%). ^- MR (500 MHz, DMSO-d6) δ ppm 2.65 (3 H, s), 3.7 (3 H, s), 5.79 (1 H, s), 6.62 (1 H, s), 7.35 - 7.59 (3 H, m), 8.95 (1 H, s).


Methyl 2-tert-butoxy-2-(7-chloro-5-methyl-3-phenylpyrazolo[ 1, 5-a]pyrimidin-6- yl) acetate. To a suspension of methyl 2-(7-chloro-5-methyl-2-phenylpyrazolo[l,5- a]pyrimidin-6-yl)-2-hydroxyacetate (250 mg, 0.75 mmol) in tert-butyl acetate (8 mL) at room temperature was added CH2CI2 (15 mL) followed by perchloric acid (1 14 mg, 1.13 mmol). The reaction mixture was stirred for 2 h at room temperature. The
reaction mixture was diluted with ethyl acetate (15 mL). The organic phase was washed with saturated NaHCCh (2 X 10 mL), followed by water (1 X 10 mL) and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (110 mg, 38%). 1H-NMR (500 MHz, DMSO-de) δ ppm 1.23 (9 H, s), 3.33 (3 H, s), 3.69 (3 H, s), 5.72 (1 H, s), 7.29 (1 H, s), 7.47 (2 H, s), 8.14 (2 H, s), 8.85 (1 H, s).


tetrakis(triphenylphosphine)palladium(0) (8.94 mg, 7.73 μιηοΐ), 8-fluoro-5- methylchroman-6-yl boronic acid (16 mg, 0.077 mmol), dioxane (1.5 mL), followed by 2M K3PO4 solution(77uL). The reaction mixture was heated in a microwave reactor at 130° C for 30 min. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (13mg, 38%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 30 to 100% B over 17 min gradient, 5 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. ¾- NMR (500 MHz, MeOD) δ ppm 1.18 (9 H, s), 1.86 (3 H, m), 2.2 (2 H, m), 2.81 (3 H, s), 2.74 (2 H, s), 3.68 (3 H, s), 4.30 (2 H, m), 5.09 (1 H, s), 6.90 (1 H, s), 7.21 - 7.42 (4 H, m), 8.11 (1 H, s), 8.38 (1 H, s).


2-tert-Butoxy-2-(7-( 8-fluoro-5-methylchroman-6-yl)-5-methyl-3-phenylpyrazolo[ 1, 5- a] pyrimidin-6-yl) acetic acid, TFA salt. To a solution of methyl 2-tert-butoxy-2-(7-(8- fluoro-5-methylchroman-6-yl)-5-methyl-3-phenylpyrazolo[l,5-a]pyrimidin-6- yl)acetate, TFA salt (9 mg, 0.017 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (5 mg, 43%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH-NMR (500 MHz, MeOD) 8 ppm 1.19 (9 H, s), 1.57 (1 H, s), 1.86 (3 H, s), 2.14 (2 H, br. s.), 2.78 (2 H, s), 2.84 (3 H, s), 4.30 (1 H, s), 5.04 (1 H, s), 6.92 (1 H, s), 7.26 (1 H, s), 7.42 (1 H, s), 8.04 - 8.13 (1 H, m), 8.37 (1 H, s).

Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Scheme 8.

Methyl 2-tert-butoxy-2-(7-(chroman-6-yl)-5-methyl-3-phenylpyrazolo[l,5- a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5 mL microwave tube was added methyl 2-tert-butoxy-2-(7-chloro-5-methyl-3-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate
(20 mg, 0.052 mmol), tetrakis(triphenylphosphine)palladium(0) (6 mg, 0.005 μιηοΐ), 2-(chroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (20 mg, 0.077 mmol), dioxane (1.5 mL), followed by 2M K3PO4 solution(77uL). The reaction mixture was heated in a microwave reactor at 130°C for 30 min. The reaction was filtered and the filtrate was purified by preparative HPLC to afford (13mg, 52%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S 10, 30 to 100% B over 17 mingradient, 5 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. ¾-NMR (500 MHz, MeOD) δ ppm 0.99 (9 H, s), 2.2 (2 H, m), 2.74 (2 H, s), 2.81 (3 H, s), 3.8 (3 H, s), 4.26 (2 H, m), 5.24 (1 H, s), 6.97 (1 H, s), 7.22 (1 H, s), 7.32 - 7.42 (3 H, m), 8.10 (2 H, s), 8.37 (1 H, s).


2-tert-Butoxy-2-(7-(chroman-6-yl)-5-methyl-3-phenylpyrazolofl,5-aJpyrimidin-6- yl) acetic acid, TFA salt. To a solution of methyl 2-tert-butoxy-2-(7-chroman-6-yl)- 5-methyl-3-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt (13 mg, 0.027 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (12 mg, 76%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH-NMR (500 MHz, MeOD) δ ppm 0.99 (9 H, br. s.), 2.07 (2 H, br. s.), 2.74 (3 H, br. s.), 2.82 - 2.96 (2 H, m), 4.29 (2 H, br. s.), 5.21 (1 H, s), 6.98 (1 H, s), 7.24 (1 H, s), 7.41 (4 H, s), 8.11 (2 H, s), 8.38 (1 H, s).

Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Scheme 9.

3-Bromo-lH-pyrazol-5-amine was prepared as described in reference: Journal of Medicinal Chemistry, 2010, 53, 3, 1245.

Methyl 2-(2-bromo-7-hydroxy-5-methylpyrazolo / Ί ,5-aj 'pyrimidin-6-y I) acetate. To a solution of 3-bromo-lH-pyrazol-5-amine (0.2 g, 1.235 mmol) and dimethyl 2- acetylsuccinate (0.697 g, 3.70 mmol) in xylene (10 mL) was added -toluenesulfonic acid monohydrate (2 mg, 10.51 μιηοΐ). The reaction mixture was heated at reflux under a Dean-Stark trap for 8 h. The solid was filtered and washed with hexanes to afford the title compound (0.201 g, 54.2%). ¾ NMR (400 MHz, MeOD) δ ppm 2.37 (3 H, s), 3.65 (2 H, s), 3.71 (3 H, s), 6.20 (1 H, s).


Methyl 2-(2-bromo-7-chloro-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. To methyl 2-(2-bromo-5-methyl-7-oxo-4,7-dihydropyrazolo[ 1 ,5-a]pyrimidin-6- yl)acetate (180 mg, 0.600 mmol) was added POCl3 (1 mL, 10.73 mmol). The
reaction mixture was heated at reflux for 1 h. After cooling, the reaction mixture was added drop-wise to ice-water. A brown solid precipitated. The solid was filtered and washed with water to give the title compound (158 mg, 83%). XH NMR (500 MHz, DMSO-de) δ ppm 2.56 (3 H, s), 3.69 (3 H, s), 4.01 (2 H, s), 6.99 (1 H, s).


Methyl 2-(2-bromo-7-chloro-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2- hydroxy acetate. To a stirred solution of KHMDS (0.5 M in toluene, 2.83 mL, 1.413 mmol) in THF (6 mL) at -78°C was added a solution of methyl 2-(2-bromo-7-chloro- 5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (300mg, 0.942 mmol) in THF (6 mL) dropwise over 20 min. The mixture was stirred at -78°C for 30 min. A solution of 3- phenyl-2-(phenylsulfonyl)-l,2-oxaziridine (369 mg, 1.413 mmol) in THF (6 mL) was added over 15 min and the reaction mixture was stirred for additional 60 min at -78 °C. The reaction mixture was quenched with saturated NH4C1 aqueous solution (4 mL). The reaction mixture was allowed to warm to room temperature and then
diluted with ethyl acetate (100 mL). The organic phase was washed with water and brine and dried with sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (85mg, 27%). lH NMR (400 MHz, CHLOROFORM-d) δ ppm 2.63 (3 H, s), 3.84 (3 H, s), 5.74 (1 H, s), 6.71 (1 H, s).


Methyl 2-(2-bromo-7-chloro-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-tert- butoxy acetate. To a suspension of methyl 2-(2-bromo-7-chloro-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (80 mg, 0.239 mmol) in tert- butyl acetate (2 mL) at room temperature was added CH2CI2 (2 mL) followed by perchloric acid (0.022 mL, 0.359 mmol). The reaction mixture was stirred for 4 h at room temperature. The reaction mixture was quenched with water and diluted with
ethyl acetate. The organic phase was washed with saturated aHC03 and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel
chromatography provided the title compound (56 mg, 59.9%). XH NMR (500 MHz, MeOD) δ ppm 1.27 (9 H, s), 2.62 (3 H, s), 3.74 (3 H, s), 5.75 (1 H, s), 6.75 (1 H, s).


Methyl 2-tert-butoxy-2-(7-chloro-5-methyl-2-p-tolylpyrazolo[ 1, 5-a]pyrimidin-6- yl)acetate, TFA salt. To a 2-5 mL microwave tube was added methyl 2-(2-bromo-7- chloro-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-tert-butoxyacetate (28 mg, 0.072 mmol), tetrakis(triphenylphosphine)palladium(0) (12.42 mg, 10.75 μναο\), ρ- tolylboronic acid (10.72 mg, 0.079 mmol), DMF (3 mL), followed by 2M K2C03 solution (ΙΟΟμΙ). The reaction mixture was heated in a microwave reactor at 70°C for 60min. The reaction mixture was filtered and the filtrate purified by preparative
HPLC to afford (16mg, 43.3%) of the title compound as TFA salt. Preparative HPLC condition: Waters Atlantis OBD 30 x 100mm 5u, 50 to 100% B over 20 minute gradient, 6 minute hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 35ml/min. ¾ NMR (500 MHz, MeOD) δ ppm 1.29 (9 H, s), 2.40 (3 H, s), 2.62 (3 H, s), 3.75 (3 H, s), 5.79 (1 H, s), 6.98 (1 H, s), 7.30 (2 H, d, J=7.9 Hz), 7.92 (2 H, d, J=7.9 Hz).


Methyl 2-tert-butoxy-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-p- tolylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. To a 0.5-2 mL microwave was added methyl 2-tert-butoxy-2-(7-chloro-5-methyl-2-p-tolylpyrazolo[l,5- a]pyrimidin-6-yl)acetate, TFA (8 mg, 0.016 mmol),
tetrakis(triphenylphosphine)palladium(0) (2.69 mg, 2.326 μηιοΐ), 2-(8-fluoro-5- methylchroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (9.06 mg, 0.031 mmol), DMF (0.7 mL), followed by 2M K3PO4 solution (50μ1). The reaction mixture was heated in a microwave reactor at 130°C for 15min. The reaction mixture was filtered and the filtrate purified by preparative HPLC to afford the title compound as TFA salt (5mg, 49.9%). Preparative HPLC condition: Waters Atlantis OBD 30 x 100mm 5u, 50 to 100% B over 20 minute gradient, 6 minute hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 35ml/min. 'H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.19 (9 H, s), 1.86 (3 H, s), 2.16 - 2.23 (2 H, m), 2.38 (3 H, s), 2.76 - 2.81 (2 H, m), 2.86 (3 H, s), 3.68 (3 H, s), 4.33 - 4.40 (2 H, m), 5.06 (1 H, s), 6.89 (1 H, d, J=10.5 Hz), 7.01 (1 H, s), 7.21 (2 H, d, J=8.0 Hz), 7.72 (2 H, d, J=8.0 Hz).


2-tert-Butoxy-2-(7-( 8-fluoro-5-methylchroman-6-yl)-5-methyl-2-p-tolylpyrazolo[ 1, 5- a] pyrimidin-6-yl) acetic acid, TFA salt. To a solution of methyl 2-tert-butoxy-2-(7- (8-fluoro-5-methylchroman-6-yl)-5-methyl-2-p-tolylpyrazolo[l,5-a]pyrimidin-6- yl)acetate, TFA salt (5 mg, 7.74 μιηοΐ) in dioxane (0.5 mL) was added 1 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 50°C for 1 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (4mg, 82%) of the title compound as TFA salt. Preparative HPLC condition: Waters Atlantis OBD 30 x 100mm 5u, 50 to 100% B over 22 minute gradient, 6 minute hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40ml/min. ¾ NMR (500 MHz, MeOD) 8 ppm 1.17 (9 H, s), 1.87 (3 H, s), 2.10 - 2.22 (2 H, m), 2.35 (3 H, s), 2.76 (3 H, s), 2.78 - 2.86 (2 H, m), 4.24 - 4.40 (2 H, m), 5.01 (1 H, s), 6.88 (1 H, s), 6.94 (1 H, d, J=10.7 Hz), 7.21 (2 H, d, J=7.9 Hz), 7.71 (2 H, d, J=8.2 Hz).

Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Compounds shown in the Table 3 were synthesized using the method described for example 46 with appropriate boronic acids.
Table 3.

Compounds in the Table 4 (Examples 48-91) were synthesized using the procedure described for Example 1 using the appropriate boronic acids.
Table 4.






Examples 92 - 114 were synthesized using the procedure described for Example 46.
Example 92

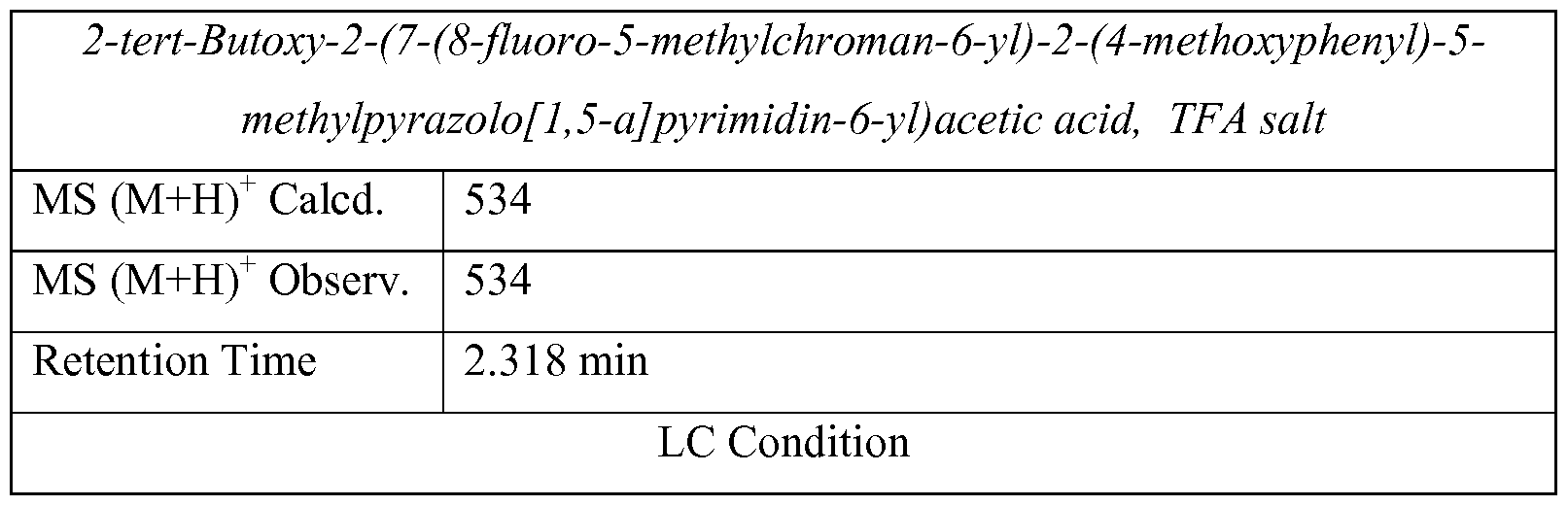
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (400MHz, CHLOROFORM-d) δ 7.77 (d, J=8.8 Hz, 2H), 7.05 - 6.76 (m, 4H), 5.11 (s, 1H), 4.34 (t, J=4.5 Hz, 2H), 3.84 (s, 3H), 2.91 - 2.66 (m, 5H), 2.17 (dd, J=6.1, 4.4 Hz, 2H), 1.91 (s, 3H), 1.22 (s, 9H).
Example 93


Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (500MHz, CHLOROFORM-d) δ 7.48 - 7.27 (m, 8H), 6.96 (dd, J=8.2, 1.8 Hz, 1H), 6.93 - 6.88 (m, 2H), 5.08 (s, 3H), 4.46 - 4.12 (m, 2H), 2.92 - 2.73 (m, 5H), 2.26 - 2.08 (m, 2H), 1.91 (s, 3H), 1.20 (s, 9H).
Example 94

(2S)-2-(2-(3-(Benzyloxy)phenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl- pyrazolo[l,5-a]pyrimidin-6-yl)-2-tert-butoxy acetic acid. The title compound was separated from the racemic compound Example 93 using a chiral column and (2S)-2- (2-(3-(benzyloxy)phenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-pyrazolo[l,5- a]pyrimidin-6-yl)-2-tert-butoxyacetic acid was isolated. Chiral separation method: Chiralpak AD-H preparative column, 20 x 250mm, 5μιη. Mobile Phase: 30% MeOH in C02 @ 1 lOBar. Temp: 35°C. Flow rate: 45.0 mL/min. for 15 min. UV was monitored @ 266nm. Retention time: 4.82min.
Example 95
MS (M+H)+ Calcd. 610
MS (M+H)+ Observ. 610
Retention Time 2.467 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (400MHz, CHLOROFORM-d) δ 7.76 (d, J=8.8 Hz, 2H), 7.47 - 7.32 (m, 5H), 7.04 - 6.95 (m, 3H), 6.91 (d, J=10.5 Hz, 1H), 5.11 (d, J=2.0 Hz, 3H), 4.44 - 4.25 m, 2H), 2.92 - 2.59 (m, 5H), 2.28 - 2.10 (m, 2H), 1.89 (s, 3H), 1.22 (s, 9H).


Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (400MHz, CHLOROFORM-d) δ 7.87 - 7.71 (m, 2H), 7.45 - 7.33 (m, 2H), 7.02 - 6.82 (m, 2H), 5.12 (s, 1H), 4.48 - 4.23 (m, 2H), 2.82 - 2.75 (m, 5H), 2.18 (dd, =6.0, 3.8 Hz, 2H), 1.93 (s, 3H), 1.23 (s, 9H).


Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (400MHz, CHLOROFORM-d) δ 7.62 - 7.49 (m, 2H), 7.41 - 7.31 (m, 3H), 7.17 - 6.95 (m, 5H), 6.90 (d, J=10.8 Hz, IH), 5.12 (s, IH), 4.50 - 4.22 (m, 2H), 2.89 - 2.66 (m, 5H), 2.17 (dd, J=6.0, 4.0 Hz, 2H), 1.90 (s, 3H), 1.28 - 1.09 (m, 9H).


Example 99


¾ NMR (400MHz, CHLOROFORM-d) δ 7.55 - 7.29 (m, 4H), 6.98 - 6.84 (m, 2H), 5.27 (s, IH), 4.45 - 4.25 (m, 2H), 3.86 (s, 3H), 2.86 - 2.62 (m, 5H), 2.31 - 2.09 (m, 2H), 1.85 (s, 3H), 1.10 (s, 9H).
Example 100


¾ NMR (500MHz, CHLOROFORM-d) δ 7.73 (d, J=7.9 Hz, 2H), 7.25 - 7.18 (m, 2H), 6.99 - 6.80 (m, 2H), 5.09 (s, 1H), 4.34 -4.31 (m, 2H), 2.75 - 2.63 (m, 7H), 1.92 (s, 3H), 1.24 - 1.21 (m, 12H).
Example 101


¾ NMR (500MHz, CHLOROFORM-d) δ 7.46 - 7.37 (m, 4H), 7.30 (t, J=8.2 Hz, 1H), 7.11 - 7.02 (m, 2H), 6.97 - 6.87 (m, 3H), 5.08 (s, 1H), 5.04 (s, 2H), 4.37 - 4.28 (m, 2H), 2.78 - 2.68 (m, 5H), 2.21 - 2.11 (m, J=4.6 Hz, 2H), 1.92 (s, 3H), 1.20 (s, 9H).
Example 102

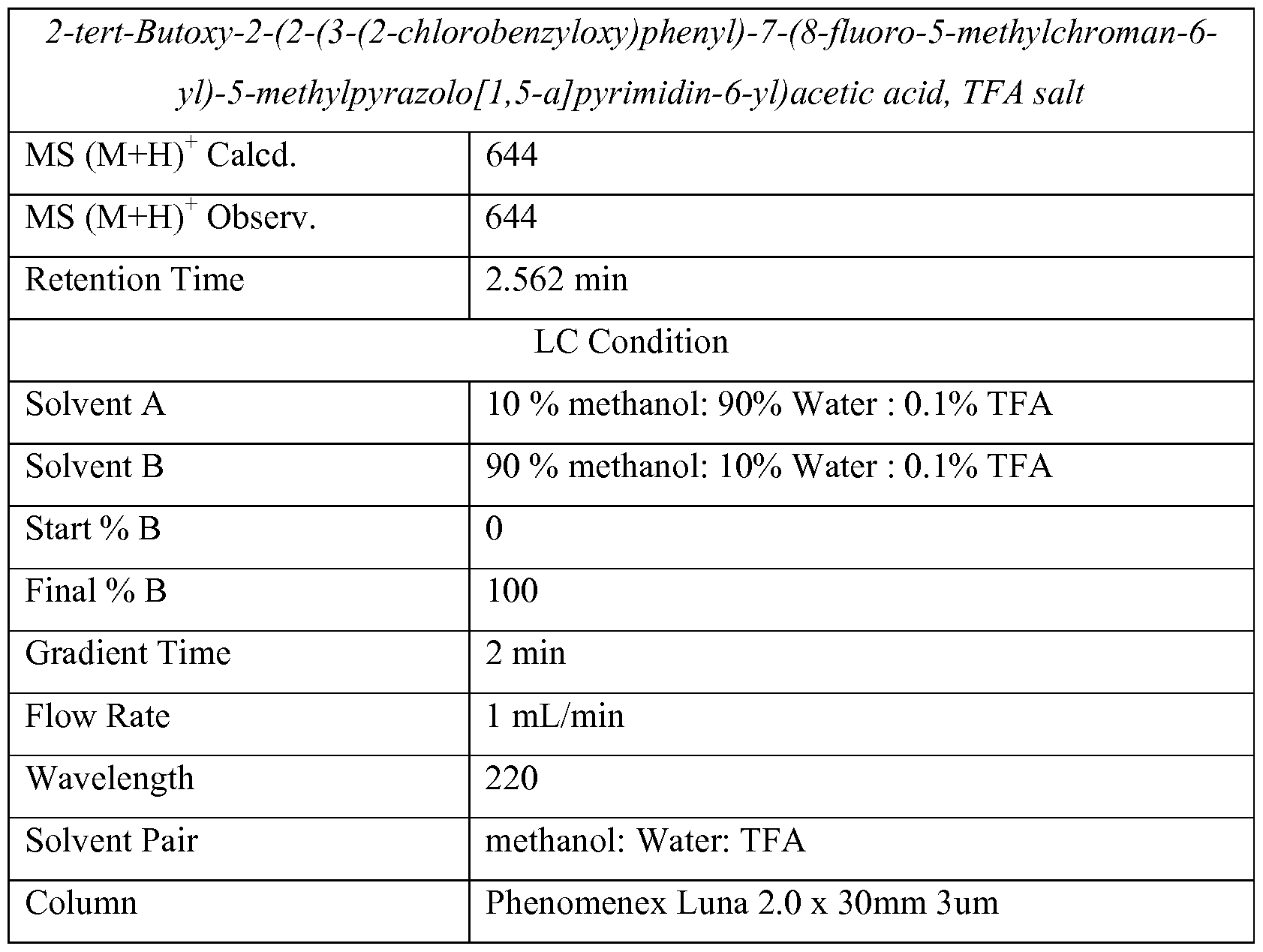
¾ NMR (500MHz, CHLOROFORM-d) δ 7.54 (dd, J=7.0, 2.1 Hz, IH), 7.48 - 7.27 (m, 6H), 7.02 - 6.79 (m, 3H), 5.20 (s, 2H), 5.09 (s, IH), 4.33 (dt, J=6.6, 3.5 Hz, 2H), 2.84 - 2.65 (m, 5H), 2.15 (d, J=5.8 Hz, 2H), 1.89 (s, 3H), 1.21 (s, 9H).
Example 103


¾ NMR (400MHz, CHLOROFORM-d) δ 7.50 - 7.29 (m, 5H), 6.99 - 6.85 (m, 5H), 5.10 (s, 1H), 5.02 (s, 2H), 4.41 - 4.27 (m, 2H), 3.83 (s, 3H), 2.87 - 2.63 (m, 5H), 2.17 (d, J=5.8 Hz, 2H), 1.95 (s, 3H), 1.23 (s, 9H).
Example 104


¾ NMR (400MHz, CHLOROFORM-d) δ 7.50 - 7.41 (m, 3H), 7.38 - 7.29 (m, 4H), 7.01 - 6.86 (m, 3H), 5.10 (s, 1H), 5.07 (s, 2H), 4.48 - 4.24 (m, 2H), 2.81 - 2.64 (m, 5H), 2.27 - 2.07 (m, 2H), 1.92 (s, 3H), 1.22 (s, 9H).
Example 105




¾ NMR (400MHz, CHLOROFORM-d) δ 7.48 - 7.29 (m, 7H), 7.05 - 6.87 (m, 3H), 5.11 (s, 1H), 5.07 (s, 2H), 4.41 - 4.29 (m, 2H), 2.86 - 2.70 (m, 5H), 2.23 - 2.10 (m, 2H), 1.91 (s, 3H), 1.23 (s, 9H).
Example 107


¾ NMR (500MHz, CHLOROFORM-d) δ 8.28 (d, J=5.5 Hz, IH), 7.56 - 7.30 (m, 7H), 7.00 (s, IH), 6.88 (d, J=10.4 Hz, IH), 5.40 (s, 2H), 5.10 (s, IH), 4.47 - 4.20 (m, 2H), 2.82 - 2.66 (m, 5H), 2.17 (dd, J=11.0, 6.4 Hz, 2H), 1.90 (s, 3H), 1.21 (s, 9H).
Example 108


¾ NMR (500MHz, CHLOROFORM-d) δ 8.65 (d, J=2.1 Hz, 1H), 8.04 (dd, J=8.5, 2.4 Hz, 1H), 7.53 - 7.28 (m, 5H), 6.98 - 6.74 (m, 3H), 5.40 (s, 2H), 5.10 (s, 1H), 4.47 - 4.10 (m, 2H), 2.89 - 2.66 (m, 5H), 2.16 (d, J=6.4 Hz, 2H), 1.92 (s, 3H), 1.22 (s, 9H).
Example 109




¾ NMR (400MHz, CHLOROFORM-d) δ 8.36 - 8.13 (m, 2H), 8.07 - 7.83 (m, 2H), 7.75 - 7.48 (m, 3H), 7.40 (t, J=7.9 Hz, 2H), 7.23 - 7.14 (m, 1H), 7.09 (s, 1H), 6.93 (d, J=10.3 Hz, 1H), 5.12 (s, 1H), 4.45 - 4.25 (m, 2H), 2.97 - 2.67 (m, 5H), 2.27 - 2.08 (m, 2H), 1.94 (s, 3H), 1.23 (s, 9H).
Example 111


¾ NMR (500MHz, CHLOROFORM-d) δ 8.15 (s, IH), 8.05 - 7.90 (m, IH), 7.78 (d, J=7.9 Hz, IH), 7.48 (t, J=7.8 Hz, IH), 7.40 - 7.28 (m, 5H), 7.01 (s, IH), 6.88 (d, J=10.4 Hz, IH), 6.69 (t, J=5.3 Hz, IH), 5.08 (s, IH), 4.77 - 4.56 (m, 2H), 4.40 - 4.23 (m, 2H), 2.87 - 2.57 (m, 5H), 2.25 - 2.03 (m, 2H), 1.88 (s, 3H), 1.20 (s, 9H).
Example 112


¾ NMR (500MHz, CHLOROFORM-d) δ 7.85 (d, J=8.9 Hz, 2H), 7.57 - 7.51 (m, 2H), 7.36 - 7.33 (m, 2H), 7.14 - 7.11 (m, 1H), 7.05 - 6.79 (m, 6H), 5.33 (s, 1H), 4.45 - 4.18 (m, 2H), 2.85 (t, J=6.0 Hz, 2H), 2.73 (s, 3H), 2.23 - 1.95 (m, 2H), 1.11 - 0.87 (m, 9H).
Example 113


¾ NMR (400MHz, CHLOROFORM-d) δ 7.73 - 7.46 (m, 4H), 7.10 - 6.94 (m, IH), 6.89 (s, IH), 6.85 - 6.77 (m, IH), 5.34 (s, IH), 4.49 - 4.03 (m, 4H), 2.94 - 2.84 (m, 4H), 2.68 (s, IH), 2.22 - 1.89 (m, 4H), 1.04 (s, 9H).

MS (M+H)+ Calcd. 486
MS (M+H)+ Observ. 486
Retention Time 2.390 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (500MHz, CHLOROFORM-d) δ 7.68 - 7.67 (m, 2H), 7.57 - 7.51 (m, 2H), 7.32 - 7.27 (m, IH), 7.19 (t, J=7.0 Hz, IH), 7.06 - 6.96 (m, 2H), 5.33 (s, IH), 4.38 - 4.26 (m, 2H), 2.86 - 2.84 (m, 2H), 2.73 (s, 3H), 2.38 (s, 3H), 2.10 - 2.05 (m., 2H), 1.02 (s, 9H).
Scheme 10

5

Methyl 2-(2-(3-chlorophenyl)-7-hydroxy-5-methylpyrazolo [ 1 ,5-a] pyrimidin-6- yl)acetate. To a solution of 3-(3-chlorophenyl)-lH-pyrazol-5-amine (23 g, 119 mmol), dimethyl 2-acetylsuccinate (22.35 g, 1 19 mmol) in o-Xylene (200 mL) was added -toluenesulfonic acid monohydrate (100 mg, 0.526 mmol). The reaction mixture was heated at reflux under a Dean-Stark trap for 2 h. The solid was filtered and washed with hexanes to afford (39 g, 99%) of the title compound. XH NMR (500MHz, METHANOL-d4) δ 8.14 - 7.95 (m, 1H), 7.93 - 7.79 (m, 1H), 7.51 - 7.27 (m, 2H), 6.62 - 6.35 (m, 1H), 3.72 (s, 3H), 3.67 (s, 2H), 2.39 (s, 3H).


Methyl 2-(7-chloro-2-(3-chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6- yl)acetate. To 2-(2-(3-chlorophenyl)-5-methyl-7-oxo-4,7-dihydropyrazolo[l,5-
a]pyrimidin-6-yl)acetate (12 g, 36.2 mmol) was added POCI3 (50 niL). The reaction mixture was heated at reflux for 2.5 h. After cooling, the reaction mixture was added drop-wise to ice-water. A brown solid precipitated. The solid was filtered and washed with water, then dissolved in ethyl acetate. The organic solution was washed with saturated NaHCC and dried over sodium sulfate. The solvent was evaporated to give the title compound (11.9 g, 94%). XH NMR (400MHz, CHLOROFORM-d) δ 8.03 (t, J=1.8 Hz, 1H), 7.94 - 7.82 (m, 1H), 7.48 - 7.34 (m, 2H), 6.94 (s, 1H), 3.93 (s, 2H), 3.78 (s, 3H), 2.63 (s, 3H).


Methyl 2-(7-chloro-2-(3-chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2- hydroxyacetate. To a stirred solution of methyl 2-(7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (7.8 g, 22.27 mmol) in THF (40 mL) at -78°C was added KHMDS (44.5 mL, 22.27 mmol) dropwise over 30 min. The mixture was stirred at -78°C for 30 min. A solution of 3-phenyl-2-(phenylsulfonyl)- 1,2-oxaziridine (8.73 g, 33.4 mmol) in THF (50 mL) was added over 30 min and the
reaction mixture was stirred for additional 2 h at -78 °C. The reaction mixture was quenched with saturated NH4C1 aqueous solution (40 mL). The reaction mixture was allowed to warm to room temperature and then diluted with ethyl acetate (200 mL). The organic phase was washed with water and brine and dried with sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (4.2g, 51.5%). 'H NMR (500MHZ, CHLOROFORM-d) δ 8.05 -
7.97 (m, 1H), 7.88 (dt, J=6.7, 1.8 Hz, 1H), 7.44 - 7.38 (m, 2H), 6.93 (s, 1H), 5.76 (s, 1H), 3.84 (s, 3H), 2.62 (s, 3H).


Methyl 2-(7-chloro-2-(3-chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2- oxoacetate. To a mixture of methyl 2-(7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (5 g, 13.65 mmol) in CH2CI2 (20 mL) was added Dess-MartinPeriodinane (6.37 g, 15.02 mmol) and the reaction mixture was stirred at room temp for 1 h. The reaction mixture was diluted with
ethyl acetate (100 mL). The organic layer was washed with saturated aHC03 solution (100 mL ) and dried ( a2S04). The solvent was evaporated and the residue was purified by a quick silica gel chromatography to afford (3.8g, 76%) of the title compound. 1H NMR (500MHz, CHLOROFORM-d) δ 8.10 - 7.98 (m, 1H), 7.89 (td, J=4.4, 1.5 Hz, 1H), 7.51 - 7.36 (m, 2H), 7.00 (s, 1H), 4.00 (s, 3H), 2.62 (s, 3H).


(S) -methyl 2-(7-chloro-2-(3-chlorophenyl)-5-methylpyrazolo[ 1, 5-a]pyrimidin-6-yl)- 2-hydroxy acetate. To a stirred solution of methyl 2-(7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (1.8 g, 4.94 mmol) in anhydrous toluene (30 mL) was added 1.1M (R)-l-methyl-3,3-diphenylhexahydropyrrolo[l,2- c][l,3,2]oxazaborole/toluene (1.797 mL, 1.977 mmol). The mixture was cooled to - 40°C (acetonitrile/dry ice bath) and a solution of 50% (by weight) catechoborane in toluene (1.695 mL, 6.92 mmol) was added over 30 min. After stirred at -45 - -35°C for 2hrs, the reaction mixture was stirred at -25°C - -15°C for additional 1 h.
Saturated a2C03 solution (20 mL) was added to quench the reaction. The mixture was stirred vigorously for 30 min and extracted with EtOAc. The organic layer was washed with saturated a2C03 solution and dried ( a2S04). The solvent was evaporated and the residue was purified by silica gel chromatography (15-50% EtOAc/hexane) to afford (1.5g, 83%) of the title compound. 1H NMR (500MHz, CHLOROFORM-d) δ 8.05 - 7.97 (m, 1H), 7.88 (dt, J=6.7, 1.8 Hz, 1H), 7.44 - 7.38 (m, 2H), 6.93 (s, 1H), 5.76 (s, 1H), 3.84 (s, 3H), 2.62 (s, 3H).


(S)-Methyl 2-(tert-butoxy)-2-(7-chloro-2-(3-chlorophenyl)-5-methylpyrazolof 1, 5- a] pyrimidin-6-yl) acetate. To a solution of (S)-methyl 2-(7-chloro-2-(3- chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (1.0 g, 2.73 mmol) in CH2CI2 (100 mL) at room temperature was added tert-butyl acetate (20 mL) followed by perchloric acid (0.282 mL, 3.28 mmol). The reaction mixture was
stirred for 16 h at room temperature. The reaction mixture was quenched with water and diluted with ethyl acetate. The organic phase was washed with saturated aHC03 and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound ( 520mg, 50%). XH NMR (400MHz, CHLOROFORM-d) δ 8.07 - 8.00 (m, 1H), 7.89 (dt, J=6.7, 1.9 Hz, 1H),
7.48 - 7.37 (m, 2H), 6.93 (s, 1H), 5.68 (s, 1H), 3.75 (s, 3H), 2.69 (s, 3H), 1.29 (s, 9H).


(2S) -Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6- yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. To a 2-5ml microwave tube was added (S)-methyl 2-(tert-butoxy)-2-(7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (420mg, 0.995mmol),
tetrakis(triphenylphosphine)palladium(0) (115 mg, 0.099 mmol), 2-(8-fluoro-5- methylchroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (320 mg, 1.094 mmol), DMF (4 mL), followed by 2M K2CO3 solution (400μ1). The reaction mixture was heated in a microwave reactor at 125°C for 45min. The reaction mixture was filtered and the filtrate was purified by silica gel chromatography to afford (204mg, 37.2%) of the title compound. Enantiomeric Excess was determined by Chiral SFC method: Chiralpak AD-H analytical column, 4.6 x 250mm, 5μιη.
Mobile Phase: 15% MeOH in C02. Temp: 35°C. Flow rate: 2.0 mL/min. for 10 min. UV monitored @ 266nm. Injection: 5uL of ~2.0mg/mL solution in 50:50
MeOH:CHC13. The enantiomeric Excess is 93.0%. ¾ NMR (500MHz,
CHLOROFORM-d) δ 7.83 - 7.78 (m, 1H), 7.69 (dt, J=6.7, 1.8 Hz, 1H), 7.35 - 7.28 (m, 2H), 6.87 (d, J=10.7 Hz, 1H), 6.84 (s, 1H), 5.00 (s, 1H), 4.48 - 4.26 (m, 2H), 3.64 (s, 3H), 2.85 - 2.67 (m, 5H), 2.30 - 2.13 (m, 2H), 1.84 (s, 3H), 1.16 (s, 9H).


(2S) -Methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-(2'- methyl-[l, 1 '-biphenyl] -3-yl)pyrazolo[l ,5-a]pyrimidin-6-yl)acetate. To a 2-5ml microwave tube was added dicyclohexyl(2',6'-dimethoxy-[l,r-biphenyl]-2- yl)phosphine (595 mg, 1.449 mmol), PALLADIUM(II) ACETATE (163 mg, 0.725 mmol), o-tolylboronic acid (296 mg, 2.174 mmol) and (2S)-methyl 2-(tert-butoxy)- 2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (400 mg, 0.725 mmol) in DMF (1.5 mL), followed by 2M K3PO4 8θΓιπιοη(200μ1). The reaction mixture was heated in a microwave reactor at 130°C for 30min. The reaction mixture was filtered and the filtrate was purified by silica gel chromatography to afford (225.6mg, 51.2%) of the title compound. 1H NMR (500MHz, CHLOROFORM-d) δ 7.86 - 7.76 (m, 2H), 7.42 (t, J=7.8 Hz, 1H), 7.29 - 7.26 (m, 5H), 6.88 - 6.86 (m, 2H), 4.99 (s, 1H), 4.39 - 4.27 (m, 2H), 3.64 (s, 3H), 2.83 - 2.70 (m, 5H), 2.26 (s, 3H), 2.22 - 2.13 (m, 2H), 1.84 (s, 3H), 1.16 (s, 9H).

Column Phenomenex Luna 2.0 x 30mm 3um
Example 1 15


Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Example 116- 120 were synthesized using the procedure described above for example 115.
Example 116

¾ NMPv (500MHz, CHLOROFORM-d) δ 8.00 - 7.95 (m, IH), 7.79 (d, J=7.6 Hz, IH), 7.51 - 7.47 (m, IH), 7.45 - 7.39 (m, IH), 7.37 - 7.31 (m, 2H), 7.08 - 6.97 (m, 2H), 6.93 (d, J=10.7 Hz, IH), 6.90 (s, IH), 5.08 (s, IH), 4.39 - 4.24 (m, 2H), 3.79 (s, 3H), 2.82 - 2.64 (m, 5H), 2.15 (d, J=3.7 Hz, 2H), 1.92 (s, 3H), 1.21 (s, 9H).

Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Example 1 17




¾ NMR (500MHz, CHLOROFORM-d) δ 7.95 (d, J=1.2 Hz, IH), 7.88 - 7.75 (m, IH), 7.57 - 7.40 (m, 3H), 7.37 - 7.29 (m, IH), 7.24 - 7.12 (m, 2H), 7.01 - 6.82 (m, 2H), 5.06 (s, IH), 4.36 - 4.24 (m, 2H), 2.80 - 2.63 (m, 5H), 2.13 (dd, J=6.1, 3.1 Hz, 2H), 1.89 (s, 3H), 1.17 (s, 9H).
Example 119


¾ NMR (400MHz, CHLOROFORM-d) δ 8.87 (d, J=1.8 Hz, 1H), 8.63 (dd, J=4.8, 1.5 Hz, 1H), 8.07 - 7.83 (m, 3H), 7.61 - 7.37 (m, 3H), 7.03 - 6.81 (m, 2H), 5.10 (s, 1H), 4.43 - 4.25 (m, 2H), 2.86 - 2.69 (m, 5H), 2.18 - 2.16 (m, 2H), 1.96 (s, 3H), 1.23 (s, 9H).
Example 120


¾ NMR (500MHZ, CHLOROFORM-d) δ 8.09 - 8.00 (m, 2H), 7.93 (s, IH), 7.81 (d, J=7.9 Hz, IH), 7.67 (dd, J=8.5, 1.5 Hz, IH), 7.59 (d, J=7.6 Hz, IH), 7.52 - 7.42 (m, 2H), 7.00 - 6.90 (m, 2H), 5.09 (s, IH), 4.37 - 4.27 (m, 2H), 4.11 (s, 3H), 2.80 - 2.67 (m, 5H), 2.21 - 2.11 (m, 2H), 1.94 (s, IH), 1.21 (s, 9H).
Example 121- 132
Scheme 1 1

Methyl 2-(tert-butoxy)-2-(7-chloro-2-(3-chlorophenyl)-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate. To a suspension of methyl 2-(7-chloro-2-(3-chlorophenyl)- 5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (3550 mg, 9.69 mmol) in tert-butyl acetate (50 mL, 9.69 mmol) at room temperature was added CH2C12 (30 mL) followed by perchloric acid (1.250 mL, 14.54 mmol). The reaction mixture was stirred for 5 h at room temperature. The reaction mixture was quenched with water and diluted with ethyl acetate. The organic phase was washed with saturated
aHC03 and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound ( 2.7mg, 66%). XH NMR (400MHz, CHLOROFORM-d) δ 8.07 - 8.00 (m, 1H), 7.89 (dt, J=6.7, 1.9 Hz, 1H), 7.48 - 7.37 (m, 2H), 6.93 (s, 1H), 5.68 (s, 1H), 3.75 (s, 3H), 2.69 (s, 3H), 1.29 (s, 9H).


Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. To a 2-5ml microwave tube was added methyl 2-(tert-butoxy)-2-(7-chloro-2-(3-chlorophenyl)-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (200mg, 0.474mmol),
tetrakis(triphenylphosphine)palladium(0) (55 mg, 0.047 mmol), 2-(8-fluoro-5- methylchroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (180 mg, 0.616
mmol), DMF (3 mL), followed by 2M K2CO3 solution (300μ1). The reaction mixture was heated in a microwave reactor at 1 15°C for 45min. The reaction mixture was filtered and the filtrate was purified by silica gel chromatography to afford (128mg, 48.9%) of the title compound. XH NMR (400MHz, CHLOROFORM-d) δ 7.82 (s, 1H), 7.76 - 7.65 (m, 1H), 7.41 - 7.29 (m, 2H), 7.01 - 6.73 (m, 2H), 5.02 (s, 1H), 4.37 (dt, J=6.5, 3.5 Hz, 2H), 3.66 (s, 3H), 2.93 - 2.69 (m, 5H), 2.21 (dd, J=5.6, 2.4 Hz, 2H), 1.86 (s, 3H), 1.18 (s, 9H).


(2S)-Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6- yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. The title compound was separated from the racemic ester using a chiral column and (2S)-methyl 2-(tert-
butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate was isolated with 100% enantiomeric excess. Chiral separation method: Chiralpak AD-H preparative column, 30 x 250mm, 5μιη. Mobile Phase: 15% MeOH in C02 @ 150Bar. Temp: 35°C. Flow rate: 70.0 mL/min. for 13 min. UV was monitored @ 266nm. provided the title compound with 100% enantiomeric excess. Retention time: 5.02min. Enantiomeric Excess was determined by Chiral SFC method: Chiralpak AD-H analytical column, 4.6 x 250mm, 5μιη. Mobile Phase: 15% MeOH in C02. Temp: 35°C. Flow rate: 2.0 mL/min. for 10 min. UV monitored @ 266nm. Injection: 5uL of ~2.0mg/mL solution in 50:50 MeOH:CHC13.

(2R)-Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6- yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. The title compound was separated from the racemic ester using a chiral column and (2R)-methyl 2-(tert- butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate was isolated with 100% enantiomeric excess. Chiral separation method: Chiralpak AD-H preparative column, 30 x 250mm, 5μιη. Mobile Phase: 15% MeOH in C02 @ 150Bar. Temp: 35°C. Flow rate: 70.0 mL/min. for 13 min. UV was monitored @ 266nm. provided the title compound with 100% enantiomeric excess. Retention time: 8.41 min. Enantiomeric Excess was determined by Chiral SFC method: Chiralpak AD-H analytical column, 4.6 x 250mm, 5μιη. Mobile Phase: 15% MeOH in C02. Temp: 35°C. Flow rate: 2.0 mL/min. for 10 min. UV monitored @ 266nm. Injection: 5uL of ~2.0mg/mL solution in 50:50 MeOH:CHC13.
Example 121

(2S)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-(3-(pyridin-4- yl)phenyl)pyrazolo[ 1,5 -a] pyrimidin-6-yl) acetic acid, TFA salt. The title compound was synthesized from (2S)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro- 5-methylchroman-6-yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate using the procedure described for example 115.

¾ NMR (500 MHz, CHLOROFORM-d) δ 8.91 (d, J=6.41 Hz, 2H), 8.13-8.05 (m, 4H), 7.75-7.60 (m, 2H), 7.00 (s, IH), 6.92 (d, J=10.38 Hz, IH), 5.09 (s, IH), 4.33 (t, J=5.04 Hz, 2H), 2.79-2.69 (m, 5H), 2.22-2.11 (m, 2H), 1.95 (s, 3H), 1.21 (s, 9H).
Example 122

(2R)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-(3-(pyridin-4- yl)phenyl)pyrazolo[ 1,5 -a] pyrimidin-6-yl) acetic acid, TFA salt. The title compound was synthesized from (2R)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro- 5-methylchroman-6-yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate using the procedure described for example 115.

¾ NMR (500MHz, CHLOROFORM-d) δ 8.95 (d, J=5.2 Hz, 2H), 8.33 - 7.94 (m, 4H), 7.83 - 7.53 (m, 2H), 7.05 (s, 1H), 6.92 (d, J=10.7 Hz, 1H), 5.10 (s, 1H), 4.33 (t, J=4.9 Hz, 2H), 2.93 - 2.58 (m, 5H), 2.31 - 2.05 (m, 2H), 1.94 (s, 3H), 1.21 (s, 9H).
Example 123

(2S)-2-(tert-Butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5- methylpyrazolo[ 1,5 -a] pyrimidin-6-yl) acetic acid, TFA salt. The title compound was hydrolyzed from (2S)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5- methylchroman-6-yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate using the procedure described for example 115.

¾ NMR (500MHz, CHLOROFORM-d) δ 7.81 (s, 1H), 7.74 - 7.61 (m, 1H), 7.41 - 7.28 (m, 2H), 7.03 - 6.81 (m, 2H), 5.09 (s, 1H), 4.56 - 4.21 (m, 2H), 2.88 - 2.61 (m, 5H), 2.30 - 2.07 (m, 2H), 1.91 (s, 3H), 1.21 (s, 9H).
Example 124 - 131 were synthesized using the procedure described above for Example 121.
Example 124


¾ NMR (500MHz, CHLOROFORM-d) δ 7.99 - 7.88 (m, 1H), 7.80 (d, J=7.6 Hz, 1H), 7.64 - 7.39 (m, 4H), 7.19 - 7.08 (m, 2H), 7.00 (s, 1H), 6.93 (d, J=10.7 Hz, 1H), 5.10 (s, 1H), 4.32 (dt, J=6.8, 3.2 Hz, 2H), 2.79 - 2.66 (m, 5H), 2.15 (dd, J=6.3, 4.1 Hz, 2H), 1.92 (s, 3H), 1.21 (s, 9H).
Example 125


¾ NMR (500MHz, CHLOROFORM-d) δ 8.05 - 7.98 (m, IH), 7.81 (d, J=7.6 Hz, IH), 7.65 - 7.58 (m, 2H), 7.55 (d, J=7.6 Hz, IH), 7.46 (t, J=7.5 Hz, 3H), 7.40 - 7.33 (m, IH), 6.98 - 6.90 (m, 2H), 5.09 (s, IH), 4.38 - 4.27 (m, 2H), 2.82 - 2.66 (m, 5H), 2.15 (d, J=6.1 Hz, 2H), 1.93 (s, 3H), 1.21 (s, 9H).
Example 126

MS (M+H)+ Calcd. 594
MS (M+H)+ Observ. 594
Retention Time 2.570 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (500MHz, CHLOROFORM-d) δ 7.98 (t, J=1.5 Hz, 1H), 7.78 (d, J=7.9 Hz, 1H), 7.60 - 7.38 (m, 4H), 7.29 - 7.23 (m, 2H), 7.03 - 6.97 (m, 1H), 6.96 - 6.88 (m, 1H), 5.09 (s, 1H), 4.45 - 4.20 (m, 2H), 2.79 - 2.67 (m, 5H), 2.40 (s, 3H), 2.14 (d, J=5.8 Hz, 2H), 1.90 (s, 3H), 1.21 (s, 9H).
Example 127


Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (400MHz, CHLOROFORM-d) δ 7.97 (t, J=1.5 Hz, 1H), 7.77 (d, J=7.5 Hz, 1H), 7.63 - 7.39 (m, 4H), 7.11 - 6.81 (m, 4H), 5.11 (s, 1H), 4.43 - 4.30 (m,2H), 3.88 (s, 3H), 2.80 (s, 3H), 2.72 (t, J=6.4 Hz, 2H), 2.15 (dd, J=6.3, 4.3 Hz, 2H), 1.89 (s,


Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
¾ NMR (500MHz, CHLOROFORM-d) δ 7.97 (t, J=1.5 Hz, IH), 7.86 - 7.79 (m, IH), 7.55 (dt, J=7.9, 1.4 Hz, IH), 7.51 - 7.35 (m, 3H), 7.32 - 7.27 (m, IH), 7.10 - 7.03 (m, IH), 7.01 (s, IH), 6.93 (d, J=10.7 Hz, IH), 5.10 (s, IH), 4.32 (m, 2H), 2.82 - 2.66 (m, 5H), 2.14 (dd, J=6.4, 3.7 Hz, 2H), 1.90 (s, 3H), 1.21 (s, 9H).
Example 129


Example 130


¾ NMR (500MHz, CHLOROFORM-d) δ 7.98 (s, IH), 7.79 (d, J=7.6 Hz, IH), 7.55 (d, J=7.6 Hz, IH), 7.48 - 7.31 (m, 4H), 7.19 (d, J=7.3 Hz, IH), 7.00 (s, IH), 6.93 (d, J=10.7 Hz, IH), 5.10 (s, IH), 4.37 - 4.27 (m, 2H), 2.78 - 2.68 (m, 5H), 2.43 (s, 3H), 2.14 (d, J=3.4 Hz, 2H), 1.90 (s, 3H), 1.22 (s, 9H).
Example 131


Scheme 12

Methyl 2( 3-(3-chlorophenyl)-(7-hydroxy-5-methylpyrazolo[ 1, 5-a]pyrimidin-6- yl)acetate. To a solution of 4-(3-chlorophenyl)-lH-pyrazol-5-amine (lg, 5.2 mmol) and dimethyl 2-acetylsuccinate (2.92 g, 15.5 mmol) in xylene (100 mL) was added p- toluenesulfonic acid monohydrate (10 mg, 0.052 mmol). The reaction mixture was heated at reflux under a Dean-Stark trap for 2 hrs. The solid was filtered and washed by hexanes to afford (1.3 g, 76%) of the title compound. ^- MR (500 MHz, DMSO-d6) δ ppm 2.39 (s, 3H), 3.59 (s, 2H), 3.63 (s, 3H), 7.37 (s, IH), 7.48 (s, IH), 7.54 (s, IH), 7.56 (s, IH), 7.64 (d, IH), 8.19 (s, IH), 11.94 (s, IH).
Methyl2(3-(3-chlorophenyl)-(7-hydroxy-5-methylpyrazolo[l,5-a]pyrimidin-6- yl)acetate
Methyl2(3-(3-chlorophenyl)-(7-hydroxy-5-methylpyrazolo[l,5-a]pyrimidin-6- yl)acetate
MS (M+H)+ Calcd. 332
MS (M+H)+ Observ. 332
Retention Time 1.81 min
LC Condition
Solvent A 10 % Methanol: 90% Water : 0.1% TFA
Solvent B 90 % Methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-(7-chloro-3-(3-chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6- yl)acetate. To methyl 2-(3-(3-chlorophenyl)-(7-hydroxy-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (1.3 g, 3.92 mmol) was added POCI3 (4 mL). The reaction mixture was heated at reflux for 1 h. After cooling, the reaction mixture was added drop-wise to ice-water. A brown solid precipitated. The solid were filtered and washed with water, then dissolved in ethyl acetate. The organic solution was washed with saturated aHC03 and dried over sodium sulfate. The solvent was evaporated to give the title compound (1.3 g, 90%). Used as is in the next step.

MS (M+H)+ Observ. 351
Retention Time 2.1 min
LC Condition
Solvent A 10 % Methanol: 90% Water : 0.1% TFA
Solvent B 90 % Methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-(7-chloro-3-(3-chlorophenyl)-5-methyl-3-phenylpyrazolo[ 1, 5-aJpyrimidin- 6-yl)-2-hydroxyacetate.
To a stirred solution of KHMDS (0.5 M in toluene, 7.4 mL) in THF (20 mL) at -78°C was added a solution of methyl 2-(7-chloro-3-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (1.3 g, 3.7 mmol) in THF (20 mL) over 20 mins. The reaction mixture was stirred at -78°C for 30 min. A solution of 3- phenyl-2-(phenylsulfonyl)-l,2-oxaziridine (1.16 g, 4.45 mmol) in THF (20 mL) was added over 10 min and the resulted reaction mixture was stirred for an additional 30 min at -78 °C. The reaction mixture was quenched with saturated NH4C1 aqueous solution (2 mL). The mixture was allowed to warm up to room temperature and diluted with EtOAc (100 mL). The organic phase was washed with water and brine and dried with sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (0.4 mg, 30%). Used as is in the next
step.


Methyl 2-tert-butoxy-2-(7-chloro-3-(3chlorophenyl)-5-methylpyrazolo[ 1, 5- aJpyrimidin-6-yl) acetate. To a suspension of methyl 2-(7-chloro-3-(3-chlorophenyl)- 5-methyl-3-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (400 mg, 1.09 mmol) in tert-butyl acetate (5 mL) at room temperature was added CH2CI2 (15 mL) followed by perchloric acid (165 mg, 1.6 mmol). The reaction mixture was stirred for 2 h at room temperature. The reaction mixture was diluted with ethyl acetate (15 mL). The organic phase was washed with saturated aHC03 (2 X 10 mL), followed by water (1 X 10 mL) and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (300 mg,
65%). Used as is in the next step.


Methyl 2-tert-butoxy-2-(3-(3-chorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5 mL microwave tube was added methyl 2-tert-butoxy-2-(7-chloro-3-(3chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (25 mg, 0.059 mmol),
tetrakis(triphenylphosphine)palladium(0) (10 mg, 8.88 μιηοΐ), 8-fluoro-5- methylchroman-6-yl boronic acid (17 mg, 0.059 mmol), dioxane (1.5 mL), followed
by 2M K3PO4 solution(77uL). The reaction mixture was heated in a microwave reactor at 130°C for 30 min. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (13mg, 38%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 30 to 100% B over 17 min gradient, 5 min hold time, A = 10% methanol 90% water
0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. Used as is in the next step.

Example 132



Methyl 2-tert-butoxy-2-(3-(3-chlorophenyl)-7-(chroman-6-yl)-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5 mL microwave tube was added methyl 2-tert-butoxy-2-(7-chloro-3-(3chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6- yl)acetate (25 mg, 0.059 mmol), tetrakis(triphenylphosphine)palladium(0) (6 mg, 0.005 μιηοΐ), 2-(chroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (15 mg, 0.059 mmol), dioxane (1.5 mL), followed by 2M K3P04 solution(77uL). The reaction mixture was heated in a microwave reactor at 130°C for 30 min. The reaction was filtered and the filtrate was purified by preparative HPLC to afford (12mg, 39%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 30 to 100% B over 17 mingradient, 5 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. Compound used as is in the next step.
Methyl 2-tert-butoxy-2-(3-(3-chlorophenyl)-7-(chroman-6-yl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt.
MS (M+H)+ Calcd. 520
MS (M+H)+ Observ. 520
Retention Time 2.56 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Example 133


Scheme 14

Methyl 2-tert-butoxy-2-(7-(2-chloro-4-methylphenyl)-3-(3chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5 mL microwave tube was added methyl 2-tert-butoxy-2-(7-chloro-3-(3chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (25 mg, 0.059 mmol),
tetrakis(triphenylphosphine)palladium(0) (10 mg, 0.008 μιηοΐ), 2-(2-chloro-4- methylphenyl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (15 mg, 0.059 mmol), dioxane (1.5 mL), followed by 2M K3PO4 solution(77uL). The reaction mixture was heated in a microwave reactor at 130°C for 30 min. The reaction was filtered and the filtrate was purified by preparative HPLC to afford (12mg, 39%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S 10, 30 to 100% B over 17 mingradient, 5 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow
rate: 40mL/min. Compound used as is in the next step.

Example 134

2-tert-Butoxy-2-(7-(2-chloro-4-methylphenyl)-3-(3chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid. To a solution of methyl 2-tert- butoxy-2-(7-(2-chloro-4-methylphenyl)-3-(3chlorophenyl)-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate, TFA salt (10 mg, 0.027 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified
by preparative HPLC to afford (3 mg, 30%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH-NMR (400 MHz, DMSO-d6) δ ppm 1.02 (9 H, br. s.), 2.54 (3 H, br. s.), 2.82 - 2.96 (3 H, m),
4.77 (1 H, br. s.), 7.28 (1 H, s), 7.41 (3 H, s), 8.1 1 (1 H, s), 8.16 (1 H, s), 8.17 (1 H, s), 8.26 (1 H, s).

Scheme 15

Methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-3-(3-(pyridin- 4-yl)phenyl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate. To a 2-5ml microwave tube was added methyl 2-tert-butoxy-2-(3-(3-chorophenyl)-7-(8-fluoro-5-methylchroman-6- yl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. (45mg, 0.082mmol), dicyclohexyl(2',6'-dimethoxy-[l,l'-biphenyl]-2-yl)phosphine (16.73mg, 0.045 mmol), palladium (II) acetate (9.15 mg, 0.045 mmol), and 4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)pyridine (37.1 mg, 0.181 mmol) in DMF (2 mL), followed by 2M K3PO4 (50 uL). The reaction mixture was heated by microwave at 130°C for
30mins. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (lOmg, 20%) of the title compound as the TFA salt. Preparative
HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. Compound is used as is in the next step.


2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-3-(3-(pyridin-4- yl)phenyl)pyrazolo[ 1,5 -a] pyrimidin-6-yl) acetic acid. To a solution of methyl 2-(tert- butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-3-(3-(pyridin-4- yl)phenyl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate.TFA salt (8 mg, 0.013mmol) in
dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (4 mg, 50%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH-NMR (400 MHz, DMSO-d6) δ ppm 1.1 (9 H, br. s.), 2.51 (4 H, br. s.), 2.74 (3 H, br. s.), 2.82 - 3.1 (5 H, m), 4.29 (1 H, br. s.), 6.98 (1 H, s), 7.61 (2 H, m), 7.81 (2 H, m), 8.31 (1 H, m), 8.5 (1 H, s), 8.7 (3 H, m).


( tert-butoxy) acetate
MS (M+H)+ Calcd. 466
MS (M+H)+ Observ. 466
Retention Time 2.457 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate. To a 2-5ml microwave tube was added methyl 2-(3-bromo-7-chloro-5-methyl-2-phenylpyrazolo[l,5- a]pyrimidin-6-yl)-2-(tert-butoxy)acetate (290 mg, 0.621 mmol),
tetrakis(triphenylphosphine)palladium(0) (71.8 mg, 0.062 mmol), 2-(8-fluoro-5- methylchroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (200 mg, 0.683 mmol), DMF (2 mL) followed by 2M K2CO3 solution (200μ1). The reaction mixture was heated in a microwave reactor at 1 10°C for 40min. The reaction mixture was filtered and the filtrate was purified by silica gel chromatography to afford (35mg, 9.4%) of the title compound. XH NMR (500MHz, CHLOROFORM-d) δ
7.90 (dd, J=8.1, 1.4 Hz, 2H), 7.53 - 7.32 (m, 3H), 6.86 (d, J=10.7 Hz, 1H), 5.02 (s, 1H), 4.48 - 4.18 (m, 2H), 3.64 (s, 3H), 2.82 (s, 3H), 2.75 (t, J=6.4 Hz, 2H), 2.20 - 2.12 (m, 2H), 1.84 (s, 3H), 1.16 (s, 9H).


Methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-3-(4-fluorophenyl)-5- methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5ml microwave tube was added dicyclohexyl(2',6'-dimethoxy-[l,l'-biphenyl]-2- yl)phosphine (12 mg, 0.029 mmol), palladium acetate (6.59 mg, 0.029 mmol), (4- fluorophenyl)boronic acid (16.42 mg, 0.117 mmol) and methyl 2-(3-bromo-7-(8- fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-
(tert-butoxy)acetate (35 mg, 0.059 mmol), DMF (1 mL), followed by 2M K3P04 (50μ1). The reaction mixture was heated in a microwave reactor at 120°C for 20min. The reaction mixture was filtered and the filtrate was purified by PrepHPLC to afford (24mg, 56.4%) of the title compound as TFA salt. XH NMR (400MHz,
CHLOROFORM-d) δ 7.58 - 7.44 (m, 4H), 7.35 - 7.29 (m, 3H), 7.12 - 7.02 (m, 2H), 6.93 (d, J=10.8 Hz, 1H), 5.04 (s, 1H), 4.38 - 4.30 (m, 2H), 3.66 (s, 3H), 2.83 - 2.72 (m, 5H), 2.25 - 2.12 (m, 2H), 1.92 (s, 3H), 1.19 (s, 9H).

Example 136

Preparative HPLC condition: Waters Sunfire OBD CI 8 30 x 100mm 5u, 15 to 60% B over 18 minute gradient, 2 minute hold time, A = 5% acetonitrile 95% water lOmM Ammonium Acetate, B = 95% acetonitrile 5% waterlOmM Ammonium Acetate. Flow rate: 40ml/min. XH NMR (400MHz, DMSO-d6) δ 7.97 (s, 1H), 7.48 (dd, J=8.5, 5.5 Hz, 2H), 7.37 (s, 5H), 7.24 (t, J=8.8 Hz, 2H), 7.13 (d, J=11.3 Hz, 1H), 4.83 (s, 1H), 4.27 (t, J=4.9 Hz, 2H), 2.70 (s, 3H), 2.16 - 1.96 (m, J=4.5 Hz, 2H), 1.86 (s, 3H), 1.09 (s, 9H).


Methyl 2-(3, 7-bis( 8-fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[ 1, 5- a]pyrimidin-6-yl)-2-(tert-butoxy)acetate, TFA salt. To a 2-5ml microwave tube was added dicyclohexyl(2',6'-dimethoxy-[l,r-biphenyl]-2-yl)phosphine (12 mg, 0.029 mmol), palladium acetate (6.59 mg, 0.029 mmol), 8-fluoro-5-methylchroman-6-yl boronic acid (17 mg, 0.059 mmol), and methyl 2-(3-bromo-7-(8-fluoro-5- methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert- butoxy)acetate (35 mg, 0.059 mmol), DMF (1 mL), followed by 2M Κ3Ρ04 (50μ1). The reaction mixture was heated in a microwave reactor at 120°C for 20min. The reaction mixture was filtered and the filtrate was purified by PrepHPLC to afford (24mg, 56.4%) of the title compound as TFA salt. Used as is in the next step.


2-(3 , 7-Bis(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo / Ί ,5- a]pyrimidin-6-yl)-2-(tert-butoxy)acetic acid. To a solution of methyl 2-(3,7-bis(8- fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2- (tert-butoxy)acetate, TFA salt., (10 mg, 0.030 mmol) in Dioxane (0.8 mL) was added 1 N Li OH aqueous solution (0.8 mL, 0.8 mmol). The reaction mixture was stirred at 50°C for 4 h. The reaction mixture was filtered and purified by preparative HPLC to afford (5 mg, 94%) of the title compound. Preparative HPLC condition: Waters Sunfire OBD C18 30 x 100mm 5u, 15 to 60% B over 18 minute gradient, 2 minute hold time, A = 5% acetonitrile 95% waterlOmM Ammonium Acetate, B = 95% acetonitrile 5% waterlOmM Ammonium Acetate. Flow rate: 40ml/min. XH NMR (400MHz, DMSO-d6) δ 7.97 (s, IH), 7.34 (dd, J=8.5, 5.5 Hz, IH), 7.30 (s, 3H), 7.29 (t, J=8.8 Hz, IH), 6.95 (d, J=l 1.3 Hz, IH), 4.83 (s, IH), 4.27 (t, J=4.9 Hz, 4H), 2.70 (s, 6H), 2.5 - 1.96 (m, J=4.5 Hz, 8H), 1.86 (s, 3H), 1.09 (s, 9H).

Ammonium Acetate
Start % B 0
Final % B 100
Gradient Time 4 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair acetonitrile : Water : Ammonium Acetate
Column Waters BEH CI 8, 2.0 x 50 mm, 1.7-μιη particles
Scheme 17


Methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate. To a solution of methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-3,5-dimethyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (350 mg, 0.676 mmol) in acetonitrile (20 mL) was added N-bromosuccinimide (119 mg, 0.676 mmol). The reaction mixture was stirred at r.t. for 1 h. The solvent was evaported and the residue was purified by silica gel chromatography to afford (210 mg, 52%) of the title compound. ¾ NMR (500MHz, CHLOROFORM-d) δ 1.16 (s, 9H), 1.56 (s, 2H), 1.84 (s, 3H), 2.15 (br. s., 2H), 2.77 (d, 5H), 3.64 (s, 3H), 4.32 (s, 2H), 5.02 (s, IH), 6.85 (s, IH), 7.90 (s, 2H).


(2S)-Methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate. Chiral separation of methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[ 1 ,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate using preparative chiral SFC resulted in the title compound 100 mg, RT 5.43 min (50% yield). Preparative chiral SFC condition: Chiralpak AD-H preparative column, 30 x 250mm, 5μιη, Mobile Phase: 15% MeOH in C02 @ 150Bar, Temp: 35°C , Flow rate: 70 mL/min for 10 min. UV monitored at 264 nm. Injection 0.35 mL of 35 mg/mL solution in 2: 1 chloroform: methanol using stacked injections.
¾ NMR (500MHz, CHLOROFORM-d) δ 1.16 (s, 9H), 1.56 (s, 2H), 1.84 (s, 3H), 2.15 (br. s., 2H), 2.77 (d, 5H), 3.64 (s, 3H), 4.32 (s, 2H), 5.02 (s, 1H), 6.85 (s, 1H), 7.90 (s, 2H).
Example 138
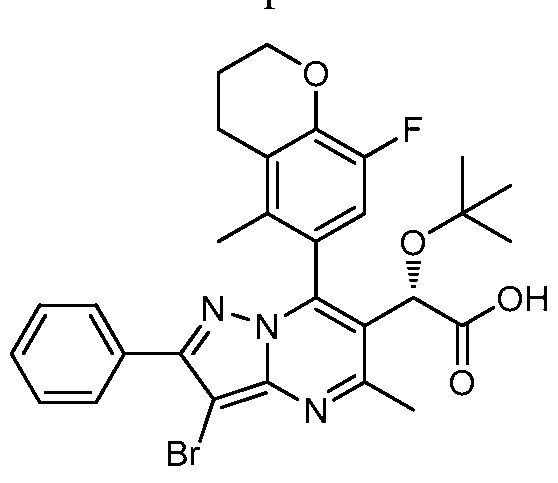
(2S)- 2-(3-Bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetic acid. To a solution of (2S)-methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate. TFA salt, (10 mg, 0.017 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 60°C for 48 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (5 mg, 50 %)
of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. 1H NMR (400MHz, DMSO-d6) δ 1.16 (s, 9H), 1.56 (s, 2H), 1.84 (s, 3H), 2.15 (br. s., 2H), 2.77 (d, 5H), 4.32 (s, 2H), 5.02 (s, IH), 6.85 (s, IH), 7.90 (s, 2H).


(2S)-Methyl 2-(3-(fl, l '-biphenyl]-3-yl)-7-(8-fluoro-5-methylchroman-6-yl)- 5- methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate, TFA salt. To
a 2-5ml microwave tube was added tetrakis(triphenylphosphine)palladium(0) (5.8 mg, 0.029 mmol), (l,l '-biphenyl)-3-ylboronic acid (7 mg, 0.117 mmol) and (2S)- methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate (20 mg, 0.034 mmol), DMF (1 mL), followed by 2M Κ3Ρ04 (50μ1). The reaction mixture was heated in a microwave reactor at 130°C for 20min. The reaction mixture was filtered and the filtrate was purified by PrepHPLC to afford (20mg, 77%) of the title compound as TFA salt. 'H NMR (400MHZ, CHLOROFORM-d) δ 7.7 (s, 1H), 7.5 - 7.25 (m, 13H), 6.93 (s, 1H), 5.04 (s, 1H), 4.31 - 4.30 (m, 2H), 3.66 (s, 3H), 2.74 - 2.79 (m, 5H), 2.25 - 2.12 (m, 2H), 1.92 (s, 3H), 1.17 (s, 9H).


(2S)-2-(3-(fl,l '-Biphenyl]-3-yl)-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- phenyipyrazoiof 1, 5-a]pyrimidin-6-yl)-2-( tert-butoxy) acetic acid.
To a solution of (2S)-methyl 2-(3-([l,l'-biphenyl]-3-yl)-7-(8-fluoro-5- methylchroman-6-yl)- 5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert- butoxy)acetate,TFA salt. (20 mg, 0.030 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 60°C for 48 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (5 mg, 25 %) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min.
¾ NMR (400MHz, DMSO-d6) δ 7.7 (s, 1H), 7.5 - 7.25 (m, 13H), 6.93 (s, 1H), 5.04 (s, 1H), 4.31 - 4.30 (m, 2H), 2.74 - 2.79 (m, 5H), 2.25 - 2.12 (m, 2H), 1.92 (s, 3H), 1.17 (s, 9H).

Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Acetonitrile: Water: ammonium acetate
Column Waters BEH CI 8, 2.0 x 50 mm
Examples 140-142 were prepared using the procedure similar to example 139

(2S) -Methyl 2-(tert-butoxy)-2-(3-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6- yl)- 5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5ml microwave tube was added tetrakis(triphenylphosphine)palladium(0) (5.8 mg, 0.005mmol), (3-chlorphenyl)-boronic acid (5.2 mg, 0.034 mmol) and (2S)-methyl 2- (3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5- a]pyrimidin-6-yl)-2-(tert-butoxy)acetate (20 mg, 0.034 mmol), DMF (1 mL), followed by 2M Κ3Ρ04 (50μ1). The reaction mixture was heated in a microwave reactor at 130°C for 30min. The reaction mixture was filtered and the filtrate was purified by PrepHPLC to afford (lOmg, 48%) of the title compound as TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. Product used as is in the next reaction.
(2S)-Methyl 2-(tert-butoxy)-2-(3-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6- yl)- 5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt.
MS (M+H)+ Calcd. 628
MS (M+H)+ Observ. 628
Retention Time 2.59 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Example 140

(2S)-2-(tert-Butoxy)-2-(3-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)- 5- methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid
To a solution of (2S)-methyl 2-(3-([l,l'-biphenyl]-3-yl)-7-(8-fluoro-5- methylchroman-6-yl)- 5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert- butoxy)acetate, TFA salt (10 mg, 0.016 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 60°C for 48 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (3 mg, 30 %) of the title compound as the TFA salt.
Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH-NMR (400 MHz, DMSO-d6) δ ppm 1.084 (9 H, br. s.), 1.866 (2 H, br. s.), 2.51 (2 H, br. s.), 2.71 (3H, br. s.) 2.82 - 2.96 (2 H, m), 4.29 (2 H, br. s.), 4.6 (1 H, s), 7.06 (1 H, s), 7.33- 7.39 (8 H, m), 7.54 (1H, s).


tetrakis(triphenylphosphine)palladium(0) (5.8 mg, 0.005mmol), (3-((4- fluorobenzyl)oxy)phenyl)boronic acid (8.3 mg, 0.034 mmol) and (2S)-methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-phenylpyrazolo[l,5- a]pyrimidin-6-yl)-2-(tert-butoxy)acetate (20 mg, 0.034 mmol), DMF (1 mL), followed by 2M Κ3Ρ04 (50μ1). The reaction mixture was heated in a microwave reactor at 130°C for 30min. The reaction mixture was filtered and the filtrate was purified by PrepHPLC to afford (lOmg, 48%) of the title compound as TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. Product used as is in the next reaction.


(2S)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-3-(3-((4- fluorobenzyl)oxy)phenyl)- 5-methyl-2-phenylpyrazolo[ 1, 5-a]pyrimidin-6-yl)acetic acid To a solution of (2S)-methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman- 6-yl)-3-(3-((4-fluorobenzyl)oxy)phenyl)- 5-methyl-2-phenylpyrazolo[ 1 ,5- a]pyrimidin-6-yl)acetate TFA salt (10 mg, 0.014 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 60°C for 48 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (5 mg, 50 %) of the title compound as the
TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH- NMR (400 MHz, DMSO-d6) δ ppm 1.087 (9 H, br. s.), 1.88 (2 H, br. s.), 2.51 (2 H, br. s.), 2.71 (3H, br. s.) 2.82 - 2.96 (2 H, m), 4.29 (2 H, br. s.), 4.6 (1 H, s), 5.02 (2H, s), 6.9 (1 H, s), 7.14-7.47 (12 H, m), 7.97 (1H, s).

Solvent B 95 % Acetonitrile: 5% Water : lOmM ammonium acetate
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Acetonitrile: Water: ammonium acetate
Column Waters BEH CI 8, 2.0 x 50 mm

(2S) -Methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)- 5-methyl-3-(l- methyl-lH-indazol-5-yl)-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5ml microwave tube was added tetrakis(triphenylphosphine)palladium(0) (5.8 mg, 0.005mmol), (1 -methyl- lH-indazol-5-yl)boronic acid
(5.2 mg, 0.034 mmol) and (2S)-methyl 2-(3-bromo-7-(8-fluoro-5-methylchroman-6- yl)-5-methyl-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)-2-(tert-butoxy)acetate (20 mg, 0.034 mmol), DMF (1 mL), followed by 2M Κ3Ρ04 (50μ1). The reaction mixture was heated in a microwave reactor at 130°C for 30min. The reaction mixture was filtered and the filtrate was purified by PrepHPLC to afford (lOmg, 48%) of the title compound as TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10%
methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min.

Example 142

(2S)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)- 5-methyl-3-(l-methyl- lH-indazol-5-yl)-2-phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid. To a solution of (2S)-methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)- 5-methyl-3-(l- methyl- lH-indazol-5-yl)-2-phenylpyrazolo[ 1 ,5-a]pyrimidin-6-yl)acetate, TFA salt (10 mg, 0.014 mmol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution
(0.5 mL, 0.5 mmol). The reaction mixture was stirred at 80°C for 3 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (3 mg, 30 %) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S 10, 50 to 100% B over 22 min gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH-NMR (400 MHz, DMSO-d6) δ ppm 1.09 (9 H, br. s.), 1.88 (2 H, br. s.), 2.51 (2 H, br. s.), 2.71 (3H, br. s.) 2.82 - 2.96 (2 H, m), 4.29 (2 H, br. s.), 4.6 (1 H, s), 5.02 (2H, s), 6.9 (1 H, s), 7.14-7.47 (8 H, m), 7.97 (1H, s).


Methyl 2-(3chloro-7-hydroxy-2, 5-di-methylpyrazolof 1, 5-a]pyrimidin-6-yl)acetate. To a solution of 4-chloro-3-methyl-lH-pyrazol-5-amine (0.263g, 2 mmol) and dimethyl 2-acetylsuccinate (1.3 g, 6 mmol) in xylene (50 mL) was added -toluenesulfonic acid monohydrate (4 mg, 0.02 mmol). The reaction mixture was heated at reflux under a Dean-Stark trap for 2 hrs. The solid was filtered and washed by hexanes to afford (0.31 g, 58%) of the title compound. ¾-NMR (500 MHz, DMSO-d6) δ ppm 2.28 (s, 3H), 2.33 (s, 3H), 3.54 (s, 2H), 3.61 (s, 3H).
Methyl 2-(3chloro-7-hydroxy-2,5-di-methylpyrazolo[l,5-a]pyrimidin-6- yl)acetate.
MS (M+H)+ Calcd. 270
MS (M+H)+ Observ. 270
Retention Time 1.4 min
LC Condition
Solvent A 10 % Methanol: 90% Water : 0.1% TFA
Solvent B 90 % Methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
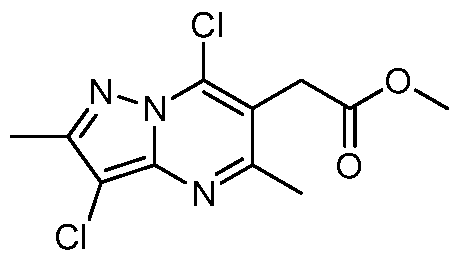
Methyl 2-(3, 7-dichloro-2,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. Toa solution of methyl 2-(3chloro-7-hydroxy-2,5-di-methylpyrazolo[l,5-a]pyrimidin-6- yl)acetate (0.160 g, 0.6 mmol) was added POCI3 (1 mL). The reaction mixture was heated at reflux for 1 h. After cooling, the reaction mixture was added drop-wise to ice-water. A brown solid precipitated. The solid were filtered and washed with water, then dissolved in ethyl acetate. The organic solution was washed with saturated aHC03 and dried over sodium sulfate. The solvent was evaporated to give the title compound (0.1 g, 59%). NMR (500 MHz, DMSO-d6) δ ppm 2.44 (s, 3H), 2.57 (s, 3H), 3.68 (s, 3H), 4.00 (s, 2H). Used as is in the next step.

Retention Time 1.82 min
LC Condition
Solvent A 10 % Methanol: 90% Water : 0.1% TFA
Solvent B 90 % Methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-(3, 7-dichloro-2, 5-dimethylpyrazolof 1, 5-a]pyrimidin-6-yl)-2-hydroxyacetate To a stirred solution of KHMDS (0.5 M in toluene, 1.04 mL) in THF (5 mL) at -78°C was added a solution of methyl 2-(3,7-dichloro-2,5-dimethylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (0.15 g, 0.5 mmol) in THF (5 mL) over 20 mins. The reaction mixture was stirred at -78°C for 30 min. A solution of 3-phenyl-2- (phenylsulfonyl)-l,2-oxaziridine (0.16 g, 0.63 mmol) in THF (5 mL) was added over 10 min and the resulted reaction mixture was stirred for an additional 30 min at -78 °C. The reaction mixture was quenched with saturated NH4C1 aqueous solution (2 mL). The mixture was allowed to warm up to room temperature and diluted with EtOAc (100 mL). The organic phase was washed with water and brine and dried with sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (80 mg, 30%). Used as is in the next step.
Methyl 2-(3, 7-dichloro-2, 5-dimethylpyrazolo[ 1, 5-a]pyrimidin-6-yl)-2- hydroxy acetate
MS (M+H)+ Calcd. 304
MS (M+H)+ Observ. 304
Retention Time 1.73 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-tert-butoxy-2-(3, 7-dichloro-2, 5-dimethylpyrazolof 1, 5-a]pyrimidin-6- yl) acetate. To a suspension of methyl 2-(3,7-dichloro-2,5-dimethylpyrazolo[l,5- a]pyrimidin-6-yl)-2-hydroxyacetate (80 mg, 0.26 mmol) in tert-butyl acetate (1.5 mL) at room temperature was added CH2CI2 (1.5 mL) followed by perchloric acid (40 mg, 2.1 mmol). The reaction mixture was stirred for 2 h at room temperature. The reaction mixture was diluted with ethyl acetate (15 mL). The organic phase was washed with saturated aHC03 (2 X 10 mL), followed by water (1 X 10 mL) and dried over sodium sulfate. The solvent was evaporated. Purification by silica gel chromatography provided the title compound (85 mg, 90%). Used as is in the next step.
Methyl 2-tert-butoxy-2-(3, 7-dichloro-2, 5-dimethylpyrazolo[ 1, 5-a]pyrimidin-6- yl)acetate
MS (M+H)+ Calcd. 360
MS (M+H)+ Observ. 360
Retention Time 2.2 min
LC Condition
Solvent A 10 % methanol: 90% Water : 0.1% TFA
Solvent B 90 % methanol: 10% Water : 0.1% TFA
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um

Methyl 2-tert-butoxy-2-(3-chloro-7-(8-fluoro-5-methylchroman-6-yl)-2,5- dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5 mL microwave tube was added methyl 2-tert-butoxy-2-(3,7-dichloro-2,5-dimethylpyrazolo[l,5- a]pyrimidin-6-yl)acetate. (25 mg, 0.069 mmol),
tetrakis(triphenylphosphine)palladium(0) (10 mg, 8.88 μιηοΐ), 8-fluoro-5- methylchroman-6-yl boronic acid (20 mg, 0.069 mmol), dioxane (1.5 mL), followed by 2M K3PO4 solution(77uL). The reaction mixture was heated in a microwave reactor at 130°C for 30 min. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (7 mg, 21%) of the title compound as the TFA salt. Preparative HPLC condition: Phenomenex Luna CI 8 30 x 100mm S 10, 30 to 100% B over 17 min gradient, 5 min hold time, A = 10% methanol 90% water
0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min.

Example 143

2-(tert-Butoxy)-2-( 3-chloro- 7-( 8-fluoro-5-methylchroman-6-yl)-2, 5- dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid. To a solution of methyl 2-tert- butoxy-2-(3-chloro-7-(8-fluoro-5-methylchroman-6-yl)-2,5-dimethylpyrazolo[l,5- a]pyrimidin-6-yl)acetate, (5 mg, 10 umol) in dioxane (0.5 mL) was added 1.0 N LiOH aqueous solution (0.5 mL, 0.5 mmol). The reaction mixture was stirred at 50°C for 2 h. The reaction mixture was filtered and the filtrate was purified by preparative HPLC to afford (3 mg, 60%) of the title compound. Preparative HPLC condition: Phenomenex Luna C18 30 x 100mm S10, 50 to 100% B over 22 min
gradient, 6 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. XH-NMR (400 MHz, DMSO- d6) δ ppm 1.03 (9 H, br. s.), 1.8 (2H, s), 2.28 (2 H, br. s.), 2.52 (3H, s), 2.74 (3 H, br. s.), 2.96 (3 H, s), 4.29 (2 H, br. s.), 4.7 (1 H, s), 6.98 (1 H, s), 7.96 (1 H, s).



Methyl 2-(tert-butoxy)2-(3-chloro- 7-(chroman-6-yl)-2, 5-dimethylpyrazolof 1, 5- a]pyrimidin-6-yl)acetate, TFA salt. To a 2-5 mL microwave tube was added added methyl 2-tert-butoxy-2-(3,7-dichloro-2,5-dimethylpyrazolo[l,5-a]pyrimidin-6- yl)acetate. (25 mg, 0.069 mmol), tetrakis(triphenylphosphine)palladium(0) (6 mg, 0.005 μιηοΐ), 2-(chroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (18 mg, 0.069 mmol), dioxane (1.5 mL), followed by 2M K3P04 solution(77uL). The reaction mixture was heated in a microwave reactor at 130°C for 30 min. The reaction was filtered and the filtrate was purified by preparative HPLC to afford (7 mg, 22%) of the title compound as the TFA salt. Preparative HPLC condition:
Phenomenex Luna C18 30 x 100mm S 10, 30 to 100% B over 17 mingradient, 5 min hold time, A = 10% methanol 90% water 0.1% TFA, B = 90% methanol 10% water 0.1% TFA. Flow rate: 40mL/min. Compound used as is in the next step.

Solvent Pair methanol: Water: TFA
Column Phenomenex Luna 2.0 x 30mm 3um
Example 144
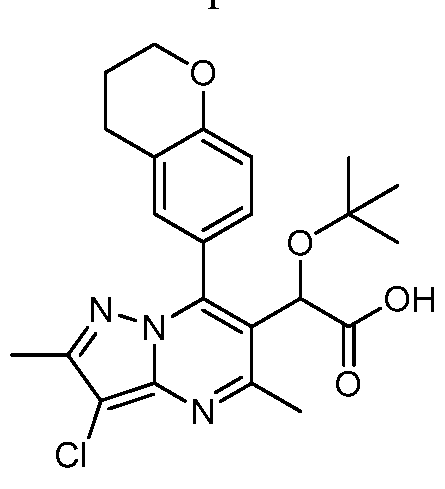

Solvent A 5 % Acetonitrile: 95% Water : lOmM ammonium acetate
Solvent B 95 % Acetonitrile: 5% Water : lOmM ammonium acetate
Start % B 0
Final % B 100
Gradient Time 2 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair Acetonitrile: Water: ammonium acetate
Column Waters BEH CI 8, 2.0 x 50 mm
Examples 145-149 were prepared using the following intermediates
 2-(3-Chlorophenyl)-5-methylpyrazolofl,5-aJpyrimidin-7-ol. A suspension of 3-(3- chlorophenyl)-lH-pyrazol-5 -amine (10 g, 51.6 mmol) and ethyl 3-oxobutanoate (26.3 ml, 207 mmol) in o-xylene (200 mL) was heated at reflux (oil bath temp: 155-160 °C) for 20 h. Note: The reaction turned clear solution between 60-70 °C and solids started crashing out at 130 °C. Then, cooled, diluted with hexanes (100 mL), filtered, washed with hexanes (100 mL) and dried to afford 2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-7-ol (1 1.8 g, 45.4 mmol, 88 % yield) as off-white solid. 'H NMR (400MHZ, DMSO-d6) δ: 12.46 (s, 1H), 8.05 (s, 1H), 8.00 - 7.95 (m, 1H), 7.56 - 7.45 (m, 2H), 6.70 (s, 1H), 5.64 (s, 1H), 2.33 (s, 3H). LCMS (M+H) calcd for Ci3HnClNnO: 260.06; found: 260.1.
2-(3-Chlorophenyl)-5-methylpyrazolofl,5-aJpyrimidin-7-ol. A suspension of 3-(3- chlorophenyl)-lH-pyrazol-5 -amine (10 g, 51.6 mmol) and ethyl 3-oxobutanoate (26.3 ml, 207 mmol) in o-xylene (200 mL) was heated at reflux (oil bath temp: 155-160 °C) for 20 h. Note: The reaction turned clear solution between 60-70 °C and solids started crashing out at 130 °C. Then, cooled, diluted with hexanes (100 mL), filtered, washed with hexanes (100 mL) and dried to afford 2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-7-ol (1 1.8 g, 45.4 mmol, 88 % yield) as off-white solid. 'H NMR (400MHZ, DMSO-d6) δ: 12.46 (s, 1H), 8.05 (s, 1H), 8.00 - 7.95 (m, 1H), 7.56 - 7.45 (m, 2H), 6.70 (s, 1H), 5.64 (s, 1H), 2.33 (s, 3H). LCMS (M+H) calcd for Ci3HnClNnO: 260.06; found: 260.1.

monobrominated and dibrominated products. So, this solid resuspended in
MeOH/CH2Cl2 (1 : 1, 100 mL) and a solution of briomine in CH2C12 (1.4 mL) was added over 5 min. After 1 h, the reaction mixture was concentrated and the resulting tan solid was used in the next step without purification.
To the above crude product was added N,N-diethylaniline (16.23 ml, 102 mmol) and POCI3 (47.5 ml, 510 mmol) and the mixture was stirred at 120 °C for 16 h. Then, cooled, concentrated and the dark residue taken up in EtOAc (250 mL) and stirred with ice-water for 30 min. Aqueous layer separated and organic layer washed with water (2 X 50 mL). The combine aqueous layers extracted with EtOAc (100 mL) and the combine organic layers washed with brine (50 mL), dried (Na2S04/C), filtered and concentrated to give dark paste. Purification by flash column chromatography on silica gel column using 5-10% EtOAc/Hex/5% CH2C12 (2.5% increment/lit) provided 6-bromo-7-chloro-2-(3-chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidine (2.003 g, 5.61 mmol, 16.50 % yield) as pale yellow solid. XH NMR (500MHz, CDCI3) δ: 8.00 (s, 1H), 7.89 - 7.83 (m, 1H), 7.43 - 7.36 (m, 2H), 6.94 (s, 1H), 2.78 (s, 3H). LCMS (M+H) calcd for Ci3H9BrCl2N3: 355.94; found: 358.0.

Methyl 2-(2-(3-chlorophenyl)-7-isopropyl-5-methylpyrazolo [ 1 ,5-a] pyrimidin-6-yl)-2- oxoacetate. A 100 mL RB-flask was charged with 6-bromo-7-chloro-2-(3- chlorophenyl)-5-methylpyrazolo[l,5-a]pyrimidine (0.918 g, 2.57 mmol) and
copper(I) bromide (0.092 g, 0.643 mmol) was added anhydrous THF (30 mL). To the resulting mixture was added 1M iPrMgCl-LiCl/THF (3.34 ml, 3.34 mmol) over 5 min. After 1 h, methyl 2-chloro-2-oxoacetate (0.501 ml, 5.45 mmol) was added at once to the dark reaction mixture and stirred for additional 1 h. Then, the resulting homogeneous orange brown reaction mixture was quenched with sat aHC03 (1 mL), diluted with Et20 (75 mL), washed with water (2 X 25 mL), brine (25 mL), dried ( a2S04/C), filtered and concentrated to give yellow residue. This residue was purified on silica gel column using 5-30% EtOAc/Hex (5% increment per 500 mL) to give methyl 2-(2-(3-chlorophenyl)-7-isopropyl-5-methylpyrazolo[l,5-a]pyrimidin-6- yl)-2-oxoacetate (0.1402 g, 0.377 mmol, 14.67 % yield) as yellow solid; 'H NMR (500MHz, CDC13) δ: 8.01 - 7.98 (m, 1H), 7.87 (dt, J= 7.0, 1.7 Hz, 1H), 7.43 - 7.38 (m, 2H), 6.87 (s, 1H), 3.98 (s, 3H), 3.43 - 3.34 (m, 1H), 2.47 (s, 3H), 1.62 (d, J= 7.0 Hz, 6H). LCMS (M+H) calcd for Ci7H16 302: 372.11; found: 372.2.

(S) -Methyl 2-(2-(3-chlorophenyl)-7-isopropyl-5-methylpyrazolo[l,5-a]pyrimidin-6- yl)-2-hydroxy acetate. To a stirred yellow solution of methyl 2-(2-(3-chlorophenyl)- 7-isopropyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (0.103 g, 0.277 mmol) in anhydrous toluene (5 mL) was added 1.1M (R)-l-methyl-3,3- diphenylhexahydropyrrolo[l,2-c][l,3,2]oxazaborole/toluene (0.101 ml, 0.111 mmol). The mixture was cooled to -35 °C and a solution of 1M catechoborane/THF (0.388 ml, 0.388 mmol) was added over 10 min. After 30 min, the reaction mixture was slowly warmed to -15 C and diluted with EtOAc (5 mL) and sat. a2C03 (2 mL). The mixture was stirred vigorously for 30 min, and the organic phase washed with sat a2C03 (2 X 5 mL), dried (Na2S04), filtered, concentrated and the residue was purified by prep-HPLC to give (S)-methyl 2-(2-(3-chlorophenyl)-7-isopropyl-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (0.0693 g, 0.185 mmol, 66.9 % yield) as white solid. XH NMR (500MHz, CDC13) δ: 7.99 (t, J= 1.5 Hz, 1H), 7.86 (dt, J= 7.4, 1.5 Hz, 1H), 7.41 - 7.33 (m, 2H), 6.80 (s, 1H), 5.63 (s, 1H), 3.81 (s, 3H),
3.40 (br. s., 1H), 2.62 (s, 3H), 1.68 (d, J= 7.0 Hz, 3H), 1.59 (d, J=7.0 Hz, 3H).
LCMS (M+H) calcd for C19H21CIN3O3: 374.13; found: 374.2.

(S) -Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-isopropyl-5-methylpyrazolo [ 1 ,5- aJpyrimidin-6-yl) acetate. To a stirred solution of (S)-methyl 2-(2-(3-chlorophenyl)- 7-isopropyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (0.069 g, 0.185 mmol) and tert-butyl acetate (1.247 ml, 9.23 mmol) in CH2CI2 (5 mL) was added 70% perchloric acid (0.048 ml, 0.554 mmol) at rt. After 3 h, the reaction mixture was dilued with Et20 (35 mL), washed with sat a2C03 (2 X 5 mL), brine (5 mL), dried (Na2S04), filtered and concentrated to give colorless paste which was purified by prep-HPLC to afford (S)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7- isopropyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.0496 g, 0.244 mmol, 62.5 % yield) as white solid. ¾ NMR (500MHz, CDC13) δ: 7.98 (t, J= 1.7 Hz, 1H), 7.87 (dt, J= 7.6, 1.4 Hz, 1H), 7.41 - 7.32 (m, 2H), 6.77 (s, 1H), 5.42 (s, 1H), 4.07 (dt, J= 13.6, 6.9 Hz, 1H), 3.71 (s, 3H), 2.73 (s, 3H), 1.68 (d, J= 7.0 Hz, 3H), 1.56 (d, J = 7.0 Hz, 3H), 1.26 (s, 9H). LCMS (M+H) calcd for CzsH^Cl^C : 430.19; found: 430.3.
Example 145

C22H27C1 303: 416.17; found: 416.3.

Methyl 2-(2-(3-chlorophenyl)-7-isobutyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2- oxoacetate. To a stirred solution of 6-bromo-7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidine (0.179 g, 0.5 mmol), copper(I) bromide (0.036 g, 0.250 mmol) and 0.5M LiCl/THF (8 ml, 4.00 mmol) was added 2M z'-BuMgCl/THF (2.000 ml, 4.00 mmol) over 5 min at rt. After 2 h, LCMS indicated presence of mostly the unreacted starting material. More iBuMgCl (1 ml) was added and continued stirring at rt for additional 4 h and methyl 2-chloro-2-oxoacetate (0.736 ml, 8.00 mmol) added at once to the reaction mixture, and stirred for additional 1 h. Then, the reaction was diluted with Et20 (50 mL), quenched with a2C03 (1 mL), washed with 10 mL each water and brine, dried (Na2S04), filtered and concentrated to give orange paste which was purified by prep-HP LC to afford methyl 2-(2-(3- chlorophenyl)-7-isobutyl-5-methylpyrazolo[ 1 ,5-a]pyrimidin-6-yl)-2-oxoacetate (0.0772 g, 0.170 mmol, 34.0 % yield) as yellow oil which turned to yellow solid overtime and contaminated with about 15% of 2-(3-chlorophenyl)-7-isobutyl-5- methylpyrazolo[l,5-a]pyrimidine. 'H NMR (500MHZ, CDC13) δ: 7.98 (d, J= 1.8 Hz, 1H), 7.87 - 7.83 (m, 1H), 7.41 - 7.31 (m, 2H), 6.87 (s, 1H), 3.97 (s, 3H), 3.13 (d, J=
7.3 Hz, 2H), 2.51 (s, 3H), 2.44 (dt, J= 13.7, 6.8 Hz, 1H), 0.97 (d, J
LCMS (M+H) calcd for C20H21CIN3O3: 386.13; found: 386.3.

(S) -Methyl 2-(2-(3-chlorophenyl)-7-isobutyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)- 2-hydroxy acetate. To a stirred solution of methyl 2-(2-(3-chlorophenyl)-7-isobutyl- 5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (0.077 g, 0.170 mmol) in toluene (3 mL) was added 1M (R)-l-methyl-3,3-diphenylhexahydropyrrolo[l,2- c][l,3,2]oxazaborole/toluene (0.034 ml, 0.034 mmol) at rt and cooled to -35 °C. To this was added dropwise 1M catecholborane/THF (0.237 ml, 0.237 mmol) over 10 min. After stirring 30 min, the reaction mixture was slowly warm to -15 °C over 30 min and diluted with EtOAc (15 mL). Then, the mixture was vigorously stirred with sat a2C03 (2 mL) for 45 min. The aqueous layer separated and organic layer washed with sat sat a2C03 (2 X 5 mL), dried (Na2S04), filtered, concentrated and the resulting yellow residue purified by prep-HPLC to afford (S)-methyl 2-(2-(3- chlorophenyl)-7-isobutyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (0.0592 g, 0.153 mmol, 90 % yield) as white solid. ¾ NMR (500MHz, CDC13) δ: 7.98 (t, J= 1.7 Hz, 1H), 7.85 (dt, J= 7.3, 1.5 Hz, 1H), 7.40 - 7.32 (m, 2H), 6.80 (s,
1H), 5.52 (s, 1H), 3.81 (s, 3H), 3.66 (br. s., 1H), 3.34 - 3.23 (m, 2H), 2.51 (s, 3H), 2.43 (dt, J= 13.6, 6.9 Hz, 1H), 1.05 (d, J= 6.7 Hz, 6H). LCMS (M+H) calcd for C2oH23Cl 303: 388.14; found: 388.3.

(S)-Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-isobutyl-5-methylpyrazolo[l,5- aJpyrimidin-6-yl) acetate. To a stirred solution of (S)-methyl 2-(2-(3-chlorophenyl)-
7-isobutyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (0.059 g, 0.152 mmol) and tert-butyl acetate (1 ml, 7.40 mmol) in CH2CI2 (3 mL) was added 70% perchloric aci (0.039 ml, 0.456 mmol) at rt. After 2 h, the reaction was diluted with Et20 (50 mL), washed with sat. Na2C03 (2 x 5 mL), dried (Na2S04), filtered and concentrated to give yellow solid which was purified by prep-HPLC to afford (S)- methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-isobutyl-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (0.0357 g, 0.080 mmol, 52.9 % yield) as brown paste. XH NMR (500MHz, CDC13) δ: 7.97 (t, J= 1.7 Hz, 1H), 7.86 (dt, J= 7.6, 1.3 Hz, 1H), 7.40 - 7.31 (m, 2H), 6.79 (s, 1H), 5.36 (s, 1H), 3.70 (s, 3H), 3.53 (dd, J= 13.4, 7.6 Hz, 1H), 3.14 (dd, J= 13.4, 7.0 Hz, 1H), 2.69 (s, 3H), 2.65 - 2.54 (m, 1H), 1.25 (s, 9H), 1.03 (d, J= 6.7 Hz, 3H), 0.98 (d, J= 6.7 Hz, 3H). LCMS (M+H) calcd for C24H3iCl 303: 444.21 ; found: 444.3.
Example 146

(S)-2-(tert-Butoxy)-2-(2-(3-chlorophenyl)-7-isobutyl-5-methylpyrazolo [ 1 ,5- aJpyrimidin-6-yi) acetic acid. A solution of (S)-methyl 2-(tert-butoxy)-2-(2-(3- chlorophenyl)-7-isobutyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.035 g, 0.079 mmol) and 1M NaOH (0.788 ml, 0.788 mmol) in MeOH (5 mL) was heated at reflux for 4 h. Then, cooled, nuetralized with 1M HCl (0.8 mL), concentrated and the residue was taken up in Et20 (25 mL), washed with water (5 mL), brine (5 mL), dried (MgS04), filtered and concentrated to give (S)-2-(tert-butoxy)-2-(2-(3-chlorophenyl)- 7-isobutyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid (0.0303 g, 0.069 mmol, 88 % yield) as white solid. XH NMR (500MHz, CDC13) δ: 7.99 - 7.96 (m, 1H), 7.85 (dt, J= 7.3, 1.4 Hz, 1H), 7.41 - 7.33 (m, 2H), 6.82 (s, 1H), 5.41 (s, 1H), 3.75 - 3.56 (m, 1H), 3.05 - 2.92 (m, 1H), 2.65 (s, 3H), 2.55 (br. s., 1H), 1.29 (s, 9H), 1.09 (d, J= 6.7 Hz, 3H), 1.02 (d, J= 6.4 Hz, 3H). LCMS (M+H) calcd for

430.19; found: 430.3.

Methyl 2-(2-(3-chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin- 6-yl)-2-oxoacetate. To a stirred mixture of 6-bromo-7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidine (0.179 g, 0.5 mmol) and copper(I) bromide (0.072 g, 0.500 mmol) in THF (3 mL) was added dropwise a pre-mixed solution of 2M 4- fluorophenylmagnesium bromide/ether (3.00 ml, 6.00 mmol) and 0.5M LiCl/THF (6.00 ml, 3.00 mmol) over 15 min. After 2 h, added at once methyl 2-chloro-2- oxoacetate (0.368 ml, 4.00 mmol) to the dark solution. After 1 h, the reaction was diluted with Et20 (50 mL), quenched with a2C03 (1 mL), washed with 10 mL each water and brine, dried ( a2S04), filtered and concentrated to give orange paste which was purified by prep-HPLC to afford methyl 2-(2-(3-chlorophenyl)-7-(4- fluorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (0.0644 g, 0.152 mmol, 30.4 % yield) as yellow solid contaminated with some impurity. lH NMR (500MHz, CDC13) δ: 7.88 (s, 1H), 7.81 - 7.74 (m, 1H), 7.73 - 7.67 (m, 2H), 7.35 (d, J = 5.2 Hz, 2H), 7.31 - 7.24 (m, 2H), 6.97 (s, 1H), 3.44 (s, 3H), 2.67 (s, 3H). LCMS (M+H) calcd for C22H16C1F 303: 424.09; found: 424.3. LCMS (M+H) calcd for C22H16C1FN303: 424.09; found: 424.3.

(S) -Methyl 2-(2-(3-chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo [ 1 ,5- a]pyrimidin-6-yl)-2-hydroxyacetate. To a stirred solution of methyl 2-(2-(3- chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-
oxoacetate (0.083 g, 0.196 mmol) in toluene (5 mL) was added 1M (R)-l-methyl-3,3- diphenylhexahydropyrrolo[l,2-c][l,3,2]oxazaborole/toluene (0.039 ml, 0.039 mmol) and cooled to -35 °C. To this was added dropwise 1M catecholborane/THF (0.294 ml, 0.294 mmol) over 10 min and stirred for additional 20 min. Then, the reaction was slowly warmed to -15 C over 30 min, diluted with EtOAc (10 ml) and quenched with sat. a2C03 (5 mL). The resulting mixture was vigarously stirred for 30 min, aq. larer removed and organic layer washed with sat. a2C03 (2 X 5 mL), dried ( a2S04), filtered, concentrated to yellow residue which was purified by prep-HPLC to afford (S)-methyl 2-(2-(3-chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)-2-hydroxyacetate (0.08 g, 0.184 mmol, 94 % yield) as solid. XH NMR (500MHz, CDC13) δ: 7.82 (s, 1H), 7.75 - 7.68 (m, 1H), 7.68 - 7.58 (m, 2H), 7.34 - 7.24 (m, 4H), 6.88 (s, 1H), 5.16 (s, 1H), 3.78 (s, 3H), 3.69 - 3.34 (br.s., 1H), 2.58 (s, 3H). LCMS (M+H) calcd for C22H18C1F 303: 426.1; found: 426.2.

(S) -Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(4-fluorophenyl)-5- methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. To a stirred solution of (S)-methyl 2- (2-(3-chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2- hydroxyacetate (0.06 g, 0.141 mmol) and tert-butyl acetate (0.952 ml, 7.04 mmol) in CH2C12 (3 mL) was added 70% perchloric acid (0.036 ml, 0.423 mmol) at rt. After 2 h, the reaction was diluted with Et20 (50 mL), washed with sat. a2C03 (2 x 5 mL), dried ( a2S04), filtered and concentrated to give yellow solid which was purified by prep-HPLC to afford (S)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(4- fluorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.0478 g, 0.099 mmol, 70.4 % yield) as whie solid. ¾ NMR (500MHz, CDC13) δ: 7.83 (s, 1H), 7.77 - 7.69 (m, 2H), 7.67 - 7.62 (m, 1H), 7.35 - 7.26 (m, 4H), 6.87 (s, 1H), 5.07 (s, 1H), 3.81 (s,
3H), 2.66 (s, 3H), 0.96 (s, 9H). LCMS (M+H) calcd for C26H26C1F 303: 482.16; found: 482.3.
Example 147

(S)-2-(tert-Butoxy)-2-(2-(3-chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo [ 1 ,5- aJpyrimidin-6-yl) acetic acid. A solution of (S)-methyl 2-(tert-butoxy)-2-(2-(3- chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetate
(0.0475 g, 0.099 mmol) and 1M NaOH (0.986 ml, 0.986 mmol) in MeOH was heated at reflux for 3 h. Then, cooled, nuetralized with 1M HC1 (1 mL), concontrated and the residue was taken up in Et20 (25 mL), washed with water (5 mL), brine (5 mL), dried (MgS04), filtered and concentrated to give (S)-2-(tert-butoxy)-2-(2-(3- chlorophenyl)-7-(4-fluorophenyl)-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid (0.0407 g, 0.086 mmol, 87 % yield) as whie solid. ¾ NMR (500MHz, 13) δ: 7.87 - 7.83 (m, 2H), 7.82 - 7.78 (m, 1H), 7.75 - 7.71 (m, 1H), 7.36 - 7.28 (m, 4H), 6.90 (s, 1H), 5.19 (s, 1H), 2.67 (s, 3H), 1.02 (s, 9H). LCMS (M+H) calcd for
C25H24C1F 303: 468.15; found: 468.2.

Methyl 2-(2-(3-chlorophenyl)-5-methyl-7-(p-tolyl)pyrazolofl,5-aJpyrimidin-6-yl)-2- oxoacetate. To a stirred mixture of 6-bromo-7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidine (0.179 g, 0.5 mmol) and copper(I) bromide (0.036
g, 0.250 mmol) in THF (3 mL) was added dropwise over 10 min a pre-mixed solution of 1M p-tolylmagnesiumbromide/THF (3.00 ml, 3.00 mmol) and 0.5M LiCl/THF (6.00 ml, 3.00 mmol) at rt. After 2.5 h, methyl 2-chloro-2-oxoacetate (0.552 ml, 6 mmol) was added at once and stirred overnight at rt. Then, the reaction was diluted with Et20 (50 mL), quenched with a2C03 (1 mL), washed with 10 mL each water and brine, dried (Na2S04), filtered and concentrated to give orange paste which was purified by prep-HPLC to afford methyl 2-(2-(3-chlorophenyl)-5-methyl-7-(p- tolyl)pyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (0.103 g, 0.123 mmol, 24.53 % yield) as yellow solid which is contaminated with impurity. XH NMR (500MHz, CDC13) δ: 7.95 (d, J= 0.9 Hz, 1H), 7.81 (td, J= 4.4, 1.5 Hz, 1H), 7.62 (d, J= 8.2 Hz, 2H), 7.40 (d, J= 7.6 Hz, 2H), 7.38 - 7.36 (m, 2H), 6.99 (s, 1H), 3.39 (s, 3H), 2.71 (s, 3H), 2.71 (s, 3H). LCMS (M+H) calcd for CMHWCIF SOS: 420.1 1; found: 420.2.

(S)-Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-5-methyl-7-(p-tolyl)pyrazolo[l,5- aJpyrimidin-6-yl) acetate. To a stirred solution of methyl 2-(2-(3-chlorophenyl)-5- methyl-7-(p-tolyl)pyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (0.103 g, 0.123 mmol) and 1M (R)-l-methyl-3,3-diphenylhexahydropyrrolo[l,2- c][l,3,2]oxazaborole/toluene (0.025 ml, 0.025 mmol) in toluene (5 mL) at -35 C was added dropwise 1M catecholoborane/THF (0.245 ml, 0.245 mmol) over 5 min. After 30 min, the reaction mixture was warmed to -15 °C, diluted with EtOAc (10 ml) and quenched with sat. a2C03 (5 mL). The resulting mixture was vigarously stirred for 30 min, aqueous layer removed and organic layer washed with sat. a2C03 (2 X 5 mL), dried (Na2S04), filtered, concentrated to yellow residue which was purified by prep-HPLC to afford (S)-methyl 2-(2-(3-chlorophenyl)-5-methyl-7-(p- tolyl)pyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate as white solid.
To a stirred solution above solid (18 mg) in CH2C12 (3 mL) and tert-BuOAc (1 mL) was added 70% perchloric acid (0.025 mL) at rt and sealed for 1.5 h. Then, the
reaction was diluted with Et20 (25 mL), washed with sat. a2C03 (2 x 5 mL), dried ( a2S04), filtered and concentrated to give yellow solid which was purified by prep- HPLC to afford (S)-methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-5-methyl-7-(p- tolyl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.01 g, 0.021 mmol, 17.06 % yield) as pale yellow solid. XH NMR (500MHz, CDC13) δ 7.86 - 7.83 (m, 1H), 7.73 (dt, J = 6.6, 1.7 Hz, 1H), 7.62 (d, J= 7.9 Hz, 1H), 7.50 (d, J= 7.6 Hz, 1H), 7.40 (dd, J= 12.5, 7.9 Hz, 2H), 7.32 - 7.27 (m, 2H), 6.86 (s, 1H), 5.14 (s, 1H), 3.80 (s, 3H), 2.65 (s, 3H), 2.51 (s, 3H), 0.95 (s, 9H). LCMS (M+H) calcd for C^H^Cl sOs: 478.19; found: 480.3.
Example 148

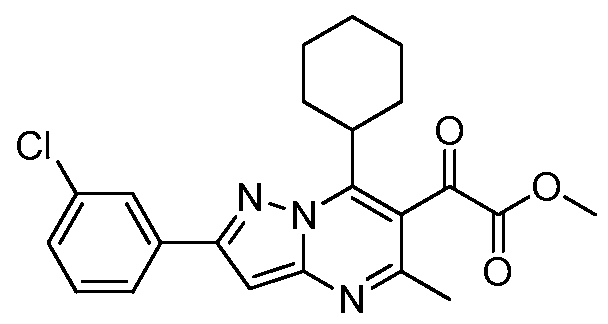
Methyl 2-(2-(3-chlorophenyl)-7-cyclohexyl-5-methylpyrazolofl,5-aJpyrimidin-6-yl)- 2-oxoacetate. To a stirred mixture of 6-bromo-7-chloro-2-(3-chlorophenyl)-5- methylpyrazolo[l,5-a]pyrimidine (0.179 g, 0.5 mmol) and copper(I) bromide (0.036 g, 0.250 mmol) in THF (5 mL) was added dropwise a pre- mixed solution of 0.5M LiCl/THF (6.00 ml, 3.00 mmol) and 2M cyclohexylmagnesium chloride/ether (1.500 ml, 3.00 mmol) over 10 min at rt. After 1.5 h, methyl 2-chloro-2-oxoacetate (0.368 ml, 4.00 mmol) was added at once to the dark reaction mixture and stirred for 1 h. Then, the reaction was diluted with Et20 (50 mL), quenched with a2C03 (1 mL), washed with 10 mL each water and brine, dried (Na2S04), filtered and concentrated to give orange paste which was purified by prep-HP LC to afford methyl 2-(2-(3- chlorophenyl)-7-cyclohexyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (0.0943 g, 0.206 mmol, 41.2 % yield) as yellow solid and contaminated with -10% of 2-(3-chlorophenyl)-7-cyclohexyl-5-methylpyrazolo[l,5-a]pyrimidine. 1H NMR (500MHz, CDC13) δ: 7.98 (s, 1H), 7.89 - 7.85 (m, 1H), 7.43 - 7.38 (m, 2H), 6.86 (s, 1H), 3.98 (s, 3H), 3.11 - 2.95 (m, 1H), 2.46 (s, 3H), 1.95 - 1.88 (m, 2H), 1.82 - 1.7 (m, 2H), 1.46 - 1.23 (m, 6H). LCMS (M+H) calcd for C22H23C1 303: 412.14; found: 412.3.

(S)-Methyl 2-(2-( 3-chlorophenyl)-7-cyclohexyl-5-methylpyrazolo[ 1, 5-aJpyrimidin- yl)-2-hydroxy acetate. To a stirred solution ofmethyl 2-(2-(3-chlorophenyl)-7- cyclohexyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (0.094 g, 0.205 mmol) in toluene (3 mL) was added 1M (R)- 1-methy 1-3,3 -
diphenylhexahydropyrrolo[l,2-c][l,3,2]oxazaborole/toluene (0.041 ml, 0.041 mmol) at rt and cooled to -35 °C. To this was added dropwise 1M catecholborane/THF (0.288 ml, 0.288 mmol) over 10 min. After stirring 30 min, the reaction mixture was slowly warm to -15 °C over 30 min and diluted with EtOAc (15 mL). Then, the mixture was vigorously stirred with sat a2C03 (2 mL) for 45 min. The aq layer separated and org layer washed with sat sat a2C03 (2 X 5 mL), dried (Na2S04), filtered, concentrated and the resulting yellow residue purified by prep-HPLC to afford (S)-methyl 2-(2-(3-chlorophenyl)-7-cyclohexyl-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)-2-hydroxyacetate (0.0705 g, 0.170 mmol, 83 % yield) as yellow solid. LCMS (M+H) calcd for C22H25C1 303: 414.16; found: 414.3.

(S)-Methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-cyclohexyl-5-methylpyrazolo[l,5- a] pyrimidin-6-yl) acetate. To a stirred solution of (S)-methyl 2-(2-(3-chlorophenyl)- 7-cyclohexyl-5-methylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (0.067 g, 0.162 mmol) and tert-butyl acetate (1 ml, 7.40 mmol) in CH2CI2 (3 mL) was added 70% perchloric acid (0.042 ml, 0.486 mmol) at rt. After 2 h, the reaction was diluted with Et20 (50 mL), washed with sat. Na2C03 (2 x 5 mL), dried (Na2S04), filtered and concentrated to give yellow solid which was purified by prep-HPLC to afford (S)- methyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-cyclohexyl-5-methylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (0.0481 g, 0.102 mmol, 63.2 % yield) as whie solid. XH NMR (500MHz, CDC13) δ: 7.96 (t, J= 1.5 Hz, 1H), 7.87 (dt, J= 7.6, 1.3 Hz, 1H), 7.41 - 7.31 (m, 2H), 6.75 (s, 1H), 5.40 (s, 1H), 3.87 - 3.77 (m, 1H), 3.70 (s, 3H), 2.93 - 2.79 (m, 2H), 2.73 (s, 3H), 1.96 - 1.85 (m, 2H), 1.77 - 1.68 (m, 2H), 1.54 - 1.38 (m, 4H), 1.25 (s, 9H). LCMS (M+H) calcd for C26H33CI 3O3: 470.22; found: 472.4.
Example 149

4H), 1.28 (br. s., 9H). LCMS (M+H) calcd for C25H3iCl 303: 456.21 ; found: 456.3.
Examples 150-157 were prepared using the synthetic route similar to Scheme 10.

3-(5, 6, 7, 8-Tetrahydronaphthalen-2-yl)-lH-pyrazol-5-amine. Acetonitrile (21.48 mL, 41 1 mmol) was added to a stirred suspension of 60% NaH (7.05 g, 176 mmol) in dioxane (200 mL) and the resulting mixture was stirred at rt for 20 min. Solution of ethyl 5,6,7,8-tetrahydronaphthalene-2-carboxylate (12 g, 58.7 mmol) in dioxane (50 mL) was then added and the mixture was heated at reflux for 4 h. After cooling to rt, water followed by IN HC1 (100 mL) was added and the mixture was extracted twice
with dichloromethane, dried ( a2S04), filtered and concentrated to afford 3-oxo-3- (5,6,7,8-tetrahydronaphthalen-2-yl)propanenitrile as dark solid. A mixture of this syrup and hydrazine hydrate (2.77 mL, 88 mmol) in ethanol (200 mL) was refluxed for 16 h. The reaction mixture was then cooled to rt and concentrated in vacuo. The resulting crude was diluted with dichloromethane and washed with water, dried ( a2S04), filtered, concentrated and purified by silica gel chromatography (5-10% CH2C12/ MeOH) to afford desired 3-(5,6,7,8-tetrahydronaphthalen-2-yl)-lH-pyrazol- 5-amine (6.1 g, 28.6 mmol, 48.7 % yield) as yellow solid. 'H NMR (400MHZ, CDCI3) δ: 7.26 (d, J = 3.5 Hz, 2H), 7.12 (d, J = 7.8 Hz, 1H), 5.89 (s, 1H), 4.14 (br. s., 3H), 2.88 - 2.75 (m, 4H), 1.83 (dt, J = 6.1, 3.4 Hz, 4H). LCMS (M+H) = 214.2.
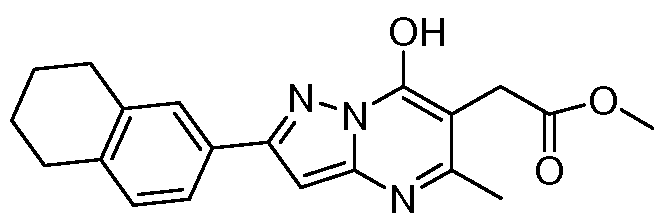
Methyl 2-(7-hydroxy-5-methyl-2-( 5, 6, 7, 8-tetrahydronaphthalen-2-yl)pyrazolo[ 1, 5- a] pyrimidin-6-yl) acetate. A suspension of 3-(5,6,7,8-tetrahydronaphthalen-2-yl)-lH- pyrazol-5 -amine (6 g, 28.1 mmol), 1-ethyl 4-methyl 2-acetylsuccinate (24.52 mL, 141 mmol) and Ts-OH»H20 (0.096 g, 0.506 mmol) in o-xylene (200 mL) was heated at 150 °C (oil bath temp) for 16 h. (Note: Mixture became homogeneous and in about 15 min slowly yellow solid started crashing out of the reaction.). Then, the reaction mixture was cooled, diluted with hexanes (300 mL), filtered, washed with hexanes and dried to afford methyl 2-(7-hydroxy-5-methyl-2-(5, 6,7,8- tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate (8.4 g, 23.90 mmol, 85 % yield) as light yellow solid. XH NMR (400MHz, DMSO-d6) δ: 12.35 (s, 1H),
7.78 - 7.62 (m, 2H), 7.15 (d, J = 8.5 Hz, 1H), 6.52 (s, 1H), 3.64 (s, 3H), 3.58 (s, 2H), 2.82-2.77 (m, 4H), 2.33 (s, 3H), 1.78 (t, J = 3.0 Hz, 4H). LCMS (M+H) = 352.3.


Methyl 2-(7-chloro-5-methyl-2-(5, 6, 7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5- a]pyrimidin-6-yl)-2-hydroxyacetate. To a stirred solution of 0.9M KHMDS/THF (9.76 mL, 8.79 mmol) in THF (25 mL) at -78 °C was added dropwise a THF (25 mL) solution of methyl 2-(7-chloro-5-methyl-2-(5,6,7,8-tetrahydronaphthalen-2- yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate (2.5 g, 6.76 mmol) over 5 min. After 30 min, a THF (20 mL) solution of 3-phenyl-2-(phenylsulfonyl)-l,2-oxaziridine (2.296 g, 8.79 mmol) was added to the resulting red reaction mixture and stirred for additional 30 min at -78 °C. Then, the resulting orange reaction mixture was quenched with sat. NH4C1 (50 mL), diluted with EtOAc (200 mL), washed with water (100 mL), brine (100 mL), dried (Na2S04), filtered and concentrated to give yellow solid. This was purified by flash column chromatograpgy on silica gel column (5-40 % EtOAc/hexane) to afford the 2.2 g desired methyl 2-(7-chloro-5- methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)-2- hydroxyacetate as off-white solid. Impurities were present by NMR and LCMS.
Used in the next step without further purification. XH NMR (500MHz, CDC13) δ: 7.77 - 7.71 (m, 2H), 7.19 (d, J = 7.6 Hz, 1H), 6.93 (s, 1H), 5.78 (d, J = 2.7 Hz, 1H), 3.86 (s, 3H), 3.56 (d, J = 2.7 Hz, 1H), 2.89-1.81 (m, 4H), 2.64 (s, 3H), 1.86 (dt, J = 6.5, 3.3 Hz, 4H). LCMS (M+H) = 386.3.

Methyl 2-(7-chloro-5-methyl-2-(5,6, 7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5- a]pyrimidin-6-yl)-2-oxoacetate. To a mixture of methyl 2-(7-chloro-5-methyl-2- (5,6,7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (2.5 g, 6.48 mmol) in CH2CI2 (70 mL) was added Dess-Martin periodinane (3.02 g, 7.13 mmol) and the resulting mixture was stirred at rt for 1 hr. Then diluted with ethyl acetate (500 mL), washed with sat. NaHC03 ( 100 mL), dried (Na2S04), filtered and concentrated. The residue was purified by silica gel chromatography (5-30 % EtOAc/hexane) to afford desired methyl 2-(7-chloro-5-methyl-2-(5, 6,7,8- tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (1.1 g, 2.87 mmol, 44.2 % yield) as off-white solid. 44 % yield based on 2 steps. XH NMR (500MHz, CDCI3) δ: 7.77 - 7.71 (m, 2H), 7.20 (d, J = 7.6 Hz, 1H), 7.00 (s, 1H), 4.02 (s, 3H), 2.89-2.83 (m, 4H), 2.64 (s, 3H), 1.86 (dt, J = 6.5, 3.3 Hz, 4H). LCMS (M+H) = 384.3.

(S)-Methyl 2-(7-chloro-5-methyl-2-( 5, 6, 7, 8-tetrahydronaphthalen-2-yl)pyrazolo[ 1, 5- a]pyrimidin-6-yl)-2-hydroxyacetate. To a stirred yellow solution of methyl 2-(7- chloro-5-methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6- yl)-2-oxoacetate (1 g, 2.61 mmol) in anhydrous toluene (25 mL) was added 1.1M (R)- 1 -methyl-3 ,3 -diphenylhexahydropyrrolo[ 1 ,2-c] [ 1 ,3,2]oxazaborole/toluene (0.947 mL, 1.042 mmol). The mixture was cooled to -35 °C and a solution of 1M
catechoborane/THF (3.65 mL, 3.65 mmol) was added over 10 min. After 30 min, the reaction mixture was slowly warmed to -15 °C and stirred for additional 30 min. Then, diluted with EtOAc (30 mL) and sat. a2C03 (10 mL), and the mixture was stirred vigorously for 30 min. The organic phase washed with sat a2C03 (2 X 5 mL), dried (Na2S04), filtered, concentrated and the residue was purified by silica gel chromatography (5-70% EtOAc/hexane) to afford desired (S)-methyl 2-(7-chloro-5- methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)-2- hydroxyacetate (888 mg, 2.301 mmol, 88 % yield) as off-white solid. EE = 95.4%. ¾ NMR (500MHz, CDC13) δ: 7.77 - 7.71 (m, 2H), 7.19 (d, J = 7.6 Hz, 1H), 6.93 (s, 1H), 5.78 (d, J = 2.7 Hz, 1H), 3.86 (s, 3H), 3.56 (d, J = 2.7 Hz, 1H), 2.89-1.81 (m, 4H), 2.64 (s, 3H), 1.86 (dt, J = 6.5, 3.3 Hz, 4H). LCMS (M+H) = 386.3.

(5,6,7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate (735 mg, 1.663 mmol, 72.3 % yield) as white solid. 150 mg of starting material was also recovered. XH NMR (500MHz, CDC13) δ: 7.76 - 7.70 (m, 2H), 7.18 (d, J = 7.9 Hz, 1H), 6.91 (s, 1H), 5.68 (s, 1H), 3.76 (s, 3H), 2.89-2.84 (m, 4H), 2.68 (s, 3H), 1.89 - 1.83 (m, 4H), 1.30 (s, 9H). LCMS (M+H) = 444.3.

(2S) -Methyl 2-(tert-butoxy)-2-(7-( 8-fluoro-5-methylchroman-6-yl)-5-methyl-2- (5,6, 7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate. A mixture of (S)-methyl 2-(tert-butoxy)-2-(7-chloro-5-methyl-2-(5,6,7,8- tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate (50 mg, 0.113 mmol), 2-(8-fluoro-5-methylchroman-6-yl)-4,4,5,5-tetramethyl-l,3,2-dioxaborolane (36.4 mg, 0.124 mmol) and 2M a2C03 (0.113 mL, 0.226 mmol) in DMF (1.5 mL) was added tetrakis(triphenylphosphine)pallafium(0) (13.07 mg, 0.011 mmol) and the mixture was subjected to microwave heating at 120 °C for lh. The mixture was then filtered and purified by prep HPLC to afford atrope isomer 1 (major, first eluting): (2S)-methyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2- (5,6,7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate (27 mg, 0.047 mmol, 41.7 % yield) as white solid. ¾ NMR (500MHz, CDC13) δ: 7.56 (d, J = 7.9 Hz, 1H), 7.52 (s, 1H), 7.09 (d, J = 7.9 Hz, 1H), 6.92 (d, J = 10.7 Hz, 1H), 6.84 (s, 1H), 5.03 (s, 1H), 4.41 - 4.32 (m, 2H), 3.66 (s, 3H), 2.88 - 2.78 (m, 6H), 2.77 (s, 3H), 2.26 - 2.17 (m, 2H), 1.86 (s, 3H), 1.85-1.79 (m, 4H), 1.19 (s, 9H).
LCMS(M+H) = 572.4. 5 mg of atrope isomer-2 (minor, second eluting) was also isolated. XH NMR (500MHz, CDC13) δ: 7.59 (s, 1H), 7.54 (s, 1H), 7.20 (d, J = 11.0 Hz, 1H), 7.11 (s, 1H), 6.85 (s, 1H), 5.08 (s, 1H), 4.42 - 4.32 (m, 2H), 3.81 (s, 3H), 2.86 - 2.78 (m, 6H), 2.68 (s, 3H), 2.23 - 2.16 (m, 2H), 1.84 (s, 3H), 1.83-1.78 (m, 4H), 1.01 (s, 9H). LCMS (M+H) = 572.4.
The following intermediates were prepared according to the above procedure using appropriate boronate reagents.

(S) -Methyl 2-(tert-butoxy)-2-(7-(chroman-6-yl)-5-methyl-2-(5, 6, 7,8- tetrahydronaphthalen-2-yl)pyrazolo[ 1, 5-a]pyrimidin-6-yl)acetate XH NMR
(500MHz, CDC13) δ: 7.65 - 7.57 (m, 2H), 7.54 - 7.46 (m, 1H), 7.41 - 7.31 (m, 1H), 7.10 (t, J = 7.0 Hz, 1H), 7.00 (dd, J = 13.1, 8.2 Hz, 1H), 6.85 (s, 1H), 5.22 (d, J = 5.2 Hz, 1H), 4.33 (t, J = 5.0 Hz, 2H), 3.83 (d, J = 5.8 Hz, 3H), 2.90 - 2.74 (m, 6H), 2.66 (d, J = 4.6 Hz, 3H), 2.15 - 2.06 (m, 2H), 1.88 - 1.75 (m, 4H), 1.02 - 0.94 (m, 9H). LCMS (M+H) = 540.30.

(S)-Methyl 2-(tert-butoxy)-2-(7-(4-fluorophenyl)-5-methyl-2-(5, 6, 7,8- tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate: ¾ NMR
(500MHz, CDCI3) δ: 7.82 - 7.75 (m, 1H), 7.71 - 7.67 (m, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.56 (s, 1H), 7.36 - 7.30 (m, 2H), 7.10 (d, J = 7.9 Hz, 1H), 6.87 (s, 1H), 5.09 (s, 1H), 3.83 (s, 3H), 2.81 (d, J = 14.0 Hz, 4H), 2.68 (s, 3H), 1.86 - 1.78 (m, 4H), 1.01 - 0.96 (m, 9H). LCMS (M+H) = 502.25.



(S)-Methyl 2-(tert-butoxy)-2-(7-(4-chlorophenyl)-5-methyl-2-(5, 6, 7,8- tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate: XH NMR
(500MHz, CDC13) δ: 7.72 (d, J = 7.6 Hz, 1H), 7.64 - 7.54 (m, 5H), 7.10 (d, J = 7.9 Hz, 1H), 6.87 (s, 1H), 5.07 (s, 1H), 3.83 (s, 3H), 2.86 - 2.77 (m, 4H), 2.68 (s, 3H), 1.86 - 1.78 (m, 4H), 0.99 (s, 9H). LCMS (M+H) = 518.4.
Example 150

The following examples were prepared according to the procedure for Example 150 using appropriate esters.
Example 151

Example 152

(S)-2-( tert-Butoxy)-2-(7-( 4-fluorophenyl)-5-methyl-2-( 5, 6, 7, 8-tetrahydronaphthalen- 2-yl)pyrazolo[ 1,5 -a] pyrimidin-6-yl) acetic acid: 'H NMR (400MHZ, DMSO-d6) δ: 7.91 - 7.83 (m, 1H), 7.78 - 7.69 (m, 1H), 7.60 - 7.50 (m, 4H), 7. 12 (s, 1H), 7.09 (s, 1H), 4.91 (s, 1H), 2.91 (s, 3H), 2.78 - 2.71 (m, 4H), 1.81 - 1.68 (m, 4H), 0.91 (s, 9H). LCMS (M+H) = 488. 16.
Example 153

(S)-2-(tert-Butoxy)-2-(5-methyl-2-(5, 6, 7,8-tetrahydronaphthalen-2-yl)-7-(p- tolyl)pyrazolo[l,5-a]pyrimidin-6-yl)acetic acid: XH NMR (400MHz, DMSO-d6) δ: 7.67 (d, J = 7.8 Hz, 1H), 7.63 - 7.54 (m, 3H), 7.49 (d, J = 8.3 Hz, 2H), 7.10 (d, J =
7.8 Hz, IH), 7.06 (s, IH), 4.95 (s, IH), 2.91 (s, 3H), 2.80-2.71 (m, 4H), 2.48 (s, 3H), 1.82-1.72 (m, 4H), 0.88 (s, 9H). LCMS (M+H) = 484.29.
Example 154

(2S)-2-(tert-Butoxy)-2-(7-(2,3-dihydropyrano[4,3,2-de]quinolin-7-yl)-5-methyl-2- (5,6, 7,8-tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetic acid: XH NMR (500MHz, DMSO-d6) δ: 8.61 (d, J = 4.3 Hz, IH), 7.99 - 7.94 (m, IH), 7.35 - 7.31 (m, 2H), 7.30 - 7.26 (m, IH), 7.24 (d, J = 7.9 Hz, IH), 7.00 - 6.93 (m, 2H), 4.82 (s, IH), 4.57 (t, J = 5.8 Hz, 2H), 3.39 - 3.32 (m, 4H), 2.69 - 2.67 (m, 2H), 2.66 (s, 3H), 1.70 (t, J = 2.9 Hz, 4H), 0.68 (s, 9H). LCMS (M+H) = 563.4.
Example 155

(S)-2-(tert-Butoxy)-2-(7-(4-chlorophenyl)-5-methyl-2-(5, 6, 7,8-tetrahydronaphthalen- 2-yl)pyrazolo[ 1,5 -a] pyrimidin-6-yl) acetic acid: XH NMR (400MHz, DMSO-d6) δ: 7.87 - 7.73 (m, 4H), 7.60 - 7.54 (m, 2H), 7.10 (d, J = 7.8 Hz, IH), 7.07 (s, IH), 4.83 (s, IH), 2.82 - 2.71 (m, 4H), 2.62 (s, 3H), 1.72-1.79 (m., 4H), 0.90 (s, 9H). LCMS (M+H) = 504.4.
Example 156

Example 157

(2S)-2-(tert-Butoxy)-2-(7-(4-chloro-2-fluorophenyl)-5-methyl-2-(5, 6, 7,8- tetrahydronaphthalen-2-yl)pyrazolo[l,5-a]pyrimidin-6-yl)acetic acid: XH NMR (400MHz, DMSO-d6) δ: 7.85 (dd, J = 9.7, 1.9 Hz, 1H), 7.65 (dd, J = 8.3, 1.8 Hz, 1H), 7.61 - 7.52 (m, 3H), 7.15 - 7.06 (m, 2H), 4.78 (s, 1H), 2.82 - 2.70 (m, 4H), 2.64 (s, 3H), 1.72-1.78 (m, 4H), 0.94 (s, 9H). LCMS (M+H) = 522.14.
Examples 158-164 were prepared using the synthetic route similar to scheme 10.

Ethyl 2-(2-( 3-chlorophenyl)- 7-hydroxy-3, 5-dimethylpyrazolof 1, 5-a]pyrimidin-6- yl)acetate: A mixture of 3-(3-chlorophenyl)-4-methyl-lH-pyrazol-5-amine (10 g, 48.2 mmol), diethyl 2-acetylsuccinate (28.9 ml, 144 mmol) and TsOH.H20 (100 mg) in o-xylene (200 mL) was heated at 120 C for 2 h. After this, the resulting reaction slurry was cooled, filtered, washed with hexanes and dried to afford ethyl 2-(2-(3- chlorophenyl)-7-hydroxy-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (13.48 g, 37.5 mmol, 78 % yield) as light brown solid which was used in the next step without purification. ¾ NMR (500MHz, DMSO-d6) δ: 1 1.98 (s, 1H), 7.79 (t, J = 1.1 Hz, 1H), 7.74 (dt, J= 7.4, 1.5 Hz, 1H), 7.58 - 7.49 (m, 2H), 4.09 (q, J= 7.0 Hz, 2H), 3.56 (s, 2H), 2.37 (s, 3H), 2.32 (s, 3H), 1.20 (t, J= 7.2 Hz, 3H). LCMS (M+H) calcd for Ci8H19Cl 303: 360.1 ; found: 360.3.

Ethyl 2-(7-chloro-2-( 3-chlorophenyl) -3, 5-dimethylpyrazolof 1, 5-a]pyrimidin-6- yl) acetate. A suspension of ethyl 2-(2-(3-chlorophenyl)-7-hydroxy-3,5- dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (1 1.5 g, 20.78 mmol), N,N- dimethylaniline (5.04 g, 41.6 mmol) and POCl3 (40 ml, 429 mmol) was heated (120 °C oil bath) for 3 hrs. The reaction was then concentrated in-vacuo and the dark residue taken up in EtOAc (75 mL) and stirred with ice-water (75 mL) for 30 min. The organic layer was washed with water (2 x 50 mL). The combined aqueous layers were extracted with EtOAc (50 mL) and the combined organic layers washed with brine (50 mL), dried (Na2S04), filtered and concentrated to give a brown solid. The crude product was purified by silica gel flash column chromatography, eluting with 10% - 30% EtOAc in hexane. Product fractions were pooled and concentrated under reduced pressure, affording the purified product, ethyl 2-(7-chloro-2-(3-
chlorophenyl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (8.3 g, 20.45 mmol, 98 % yield) as an off-white powdery solid. XH NMR (500MHz, CDC13) δ: 7.87 (t, J = 1.7 Hz, 1H), 7.74 (dt, J = 7.3, 1.5 Hz, 1H), 7.44 - 7.36 (m, 2H), 4.21 (q, J = 7.1 Hz, 2H), 3.89 (s, 2H), 2.62 (s, 3H), 2.52 (s, 3H), 1.28 (t, J = 7.2 Hz, 3H). LC/MS (M+H) = 378.2.

Ethyl 2-(7-chloro-2-( 3-chlorophenyl)-3, 5-dimethylpyrazolof 1, 5-a] pyrimidin-6-yl)-2- hydroxy acetate. To a cold (-78°C dry ice/acetone) stirred solution of 0.91 M
KHMDS in THF (32 ml, 29.1 mmol) in additional THF (100 ml) was added ethyl 2- (7-chloro-2-(3-chlorophenyl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (8.3 g, 21.94 mmol) in THF (100 ml), by dropwise addition over 30 min. The mixture was stirred for 20 min, and then a solution of 3-phenyl-2-(phenylsulfonyl)-l,2- oxaziridine (7.45 g, 28.5 mmol) in THF (50 ml) was added. The reaction was stirred for 2.5 hrs at -78 °C. The orange reaction mixture was quenched with sat. NH4C1 (100 mL), diluted with EtOAc (300 mL), washed with water (200 mL), brine (100 mL), dried (Na2S04), filtered and concentrated to give a yellow solid. The crude residue was loaded onto a flash silica gel column and eluted with 10% - 30% EtOAc in hexanes. Product fractions were pooled and concentrated under reduced pressure, affording ethyl 2-(7-chloro-2-(3-chlorophenyl)-3,5-dimethylpyrazolo[l,5- a]pyrimidin-6-yl)-2-hydroxyacetate (5.4 g, 11.40 mmol, 51.9 % yield) as a yellow solid. 'H NMR (500MHZ, CDCI3) δ: 7.89 - 7.84 (m, 1H), 7.74 (dt, J = 7.2, 1.6 Hz,
1H), 7.44 - 7.36 (m, 2H), 5.73 (s, 1H), 4.32 - 4.26 (m, 2H), 3.65 (br. s., 1H), 2.63 (s, 3H), 2.51 (s, 3H), 1.24 (t, J = 7.02 Hz, 3H). LCMS (M+H) = 394.06.


(S)-Ethyl 2-(7-chloro-2-(3-chlorophenyl)-3, 5-dimethylpyrazolof 1, 5-a]pyrimidin-6- yl)-2-hydroxy acetate. To a stirred suspension of ethyl 2-(7-chloro-2-(3- chlorophenyl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)-2-oxoacetate (3.45 g, 8.80 mmol) in anhydrous toluene (200 ml) was added 1.1 M (R)- 1-methy 1-3,3 - diphenylhexahydropyrrolo[l,2-c][l,3,2]oxazaborole in toluene (3.20 ml, 3.52 mmol). The mixture was cooled (-40°C, dry ice/acetonitrile) and a solution of 1.0 M catecholborane in THF (17.59 ml, 17.59 mmol) was added over 1 min. The mixture was maintained at -40°C for 2 hrs, and then warmed to room temperature with stirring for 16 hrs. The reaction was diluted with EtOAc (600 mL) and sat. a2CC>3 (200 mL). The mixture was stirred vigorously for 30 min, the layers were separated, and the organic layer washed with sat a2C03 (5 x 100 mL), dried ( a2S04), filtered, and concentrated under reduced pressure. The crude product was purified by flash silica gel column chromatography, eluting with 10% - - 30% EtOAc in hexanes,
affording the product, (S)-ethyl 2-(7-chloro-2-(3-chlorophenyl)-3,5- dimethylpyrazolo[l,5-a]pyrimidin-6-yl)-2-hydroxyacetate (3.08 g, 7.81 mmol, 84 % yield) as a white solid. XH NMR (500MHz, CDC13) δ: 7.85 - 7.79 (m, 1H), 7.69 (dt, J = 7.1, 1.6 Hz, 1H), 7.40 - 7.32 (m, 2H), 5.71 (s, 1H), 4.26 (q, J = 7.1 Hz, 2H), 4.02 (br. s., 1H), 2.60 (s, 3H), 2.47 (s, 3H), 1.21 (t, J = 7.2 Hz, 3H). LCMS (M+H) = 394.2. Chiral column analysis indicated 98.1% chiral purity (ee: 96.2%).

(7-chloro-2-(3-chlorophenyl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (2.05 g, 4.42 mmol, 56.5 % yield) as a viscous oil, which began to crystallize upon standing. XH NMR (500MHz, CDC13) δ: 7.87 (t, J = 1.5 Hz, 1H), 7.74 (dt, J = 7.3, 1.5 Hz, 1H), 7.46 - 7.36 (m, 2H), 5.63 (s, 1H), 4.19 (q, J = 7.0 Hz, 2H), 2.68 (s, 3H), 2.51 (s, 3H), 1.26 (s, 9H), 1.21 (t, J = 7.0 Hz, 3H).

(2 S) -Ethyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)- 3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. A solution of (S)-ethyl 2-(tert- butoxy)-2-(7-chloro-2-(3-chlorophenyl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6- yl)acetate (0.207 g, 0.460 mmol), 2-(8-fluoro-5-methylchroman-6-yl)-4,4,5,5- tetramethyl-l,3,2-dioxaborolane (0.151 g, 0.517 mmol) and
tetrakis(triphenylphosphine)palladium(0) (0.081 g, 0.070 mmol) in anhydrous DMF (4 mL) was treated with 2.0 M K2CO3 (0.55 mL, 1.100 mmol), degassed by nitrogen stream, then sealed and heated (110°C, microwave) for 60 min. The reaction slurry was filtered though a 0.45 micron syringe tip filter, and the filtrate was purified by biotage silica gel column, eluting with 0% - 20% ethyl acetate in hexanes. Individual atropisomers eluted separately, and the early eluting isomer was concentrated under reduced pressure, affording the single isomer, (2S)-ethyl 2-(tert-butoxy)-2-(2-(3- chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-3,5-dimethylpyrazolo[l,5- a]pyrimidin-6-yl)acetate (0.095 g, 0.164 mmol, 35.7 % yield) as a white glassy solid. ¾ NMR (500MHz, CDC13) δ: 7.66 (t, J = 1.5 Hz, 1H), 7.57 (dt, J = 7.2, 1.6 Hz, 1H), 7.36 - 7.27 (m, 2H), 6.87 (d, J = 10.7 Hz, 1H), 4.97 (s, 1H), 4.32 (dd, J = 5.8, 4.3 Hz, 2H), 4.11 (q, J = 7.3 Hz, 2H), 2.77 (s, 3H), 2.77 - 2.72 (m, 2H), 2.52 - 2.48 (m, 3H), 2.20 - 2.12 (m, 2H), 1.86 (s, 3H), 1.18 (t, J = 7.3 Hz, 3H), 1.16 (s, 9H). LCMS (M+H) = 580.4.


(2S)-Ethyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-2-(3-(4- methoxypyridin-3-yl)phenyl)-3, 5-dimethylpyrazolo[ 1, 5-a]pyrimidin-6-yl)acetate. To a 2-5 ml microwave tube was added dicyclohexyl(2',6'-dimethoxy-[l,l'-biphenyl]-2- yl)phosphine (0.061 g, 0.147 mmol), palladium(II) acetate (0.022 g, 0.098 mmol), and (2S)-ethyl 2-(tert-butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman- 6-yl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.057 g, 0.098 mmol) in DMF (1 mL), followed by an aqueous solution of 2.0 M K3P04 (0.098 mL, 0.197 mmol). The reaction was degassed using nitrogen stream and and then heated
(130°C, microwave) for 60 min. The reaction was filtered through a 0.45 micron syringe tip filter, and the residue was purified by preparative HPLC. Product fractions were pooled and concentrated under reduced pressure for afford the product, (2S)-ethyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-2-(3-(4- methoxypyridin-3-yl)phenyl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.022 g, 0.033 mmol, 33.8 % yield), as an oily film. ¾ NMR (500MHz, CDC13) δ: 8.48 (d, J= 5.8 Hz, 1H), 8.46 - 8.44 (m, 1H), 7.81 (s, 1H), 7.68 (dt, J= 6.6, 1.9 Hz, 1H), 7.50 - 7.45 (m, 2H), 6.90 (d, J= 5.8 Hz, 1H), 6.88 (d, J= 11.0 Hz, 1H), 4.97 (s,
1H), 4.32 - 4.28 (m, 2H), 4.11 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 2.77 (s, 3H), 2.74 (t, J= 6.6 Hz, 2H), 2.53 (s, 3H), 2.17 - 2.10 (m, 2H), 1.87 (s, 3H), 1.18 (t, J= 7.2 Hz, 3H), 1.16 (s, 9H). LCMS (M+H) = 653.6. The following interemediates were prepared according to the above procedure.

(2S)-Ethyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-3,5-dimethyl-2- phenylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. XH NMR (500MHz, CDCI3) δ: 7.71 - 7.66 (m, 2H), 7.42 - 7.36 (m, 2H), 7.36 - 7.30 (m, 1H), 6.88 (d, J= 10.7 Hz, 1H), 4.97 (s, 1H), 4.34 - 4.28 (m, 2H), 4.10 (q, J= 7.2 Hz, 2H), 2.77 (s, 3H), 2.76 - 2.72 (m, 2H), 2.51 (s, 3H), 2.19 - 2.11 (m, 2H), 1.87 (s, 3H), 1.17 (t, J= 7.0 Hz, 3H), 1.16 (s, 9H). LCMS (M+H) = 546.5.

(2S)-Ethyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-2-(2'-fluoro-[l, 1 '- biphenyl]-3-yl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. LCMS (M+H) = 640.5.

(2 S) -Ethyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-2-(3 '-fluoro-f 1, 1 '- biphenyl]-3-yl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate. LCMS (M+H) = 640.6.

(2 S) -Ethyl 2-(tert-butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-3,5-dimethyl-2-(3- (l-methyl-lH-indazol-5-yl)phenyl)pyrazolo[l,5-a]pyrimidin-6-yl)acetate. LCMS (M+H) = 676.6
Example 158

(2S)-2-(tert-Butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-3,5- dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid. A solution of (2S)-ethyl 2-(tert- butoxy)-2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-3,5-
dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetate (0.025 g, 0.043 mmol) in MeOH (2 niL) was treated with 1.0 M NaOH (0.215 mL, 0.215 mmol) and the reaction was stirred with heating (75°C oil bath) for 16 hrs. The reaction was concentrated under reduced pressure to give a white paste. This was partitioned between 0.1 N HC1 (5 mL) and (¾(¾ (5 mL). The layers were separated and the organic layer was dried (MgS04), filtered, and concentrated under reduced pressure, affording an off-white solid. The product was purified by preparative-HPLC. Product fractions were pooled and concentrated to 1/2 volume and the resulting solid was extracted with CH2CI2 (2 x 10 mL). The combined organic extracts were dried (Na2S04), filtered, and concentrated under reduced pressure, affording the product, (2S)-2-(tert-butoxy)- 2-(2-(3-chlorophenyl)-7-(8-fluoro-5-methylchroman-6-yl)-3,5-dimethylpyrazolo[l,5- a]pyrimidin-6-yl)acetic acid (0.014 g, 0.024 mmol, 55.9 % yield), as an off-white solid. 'H NMR (500MHZ, CDCI3) d: 7.69 - 7.64 (m, 1H), 7.57 (dt, J=7.0, 1.7 Hz, 1H), 7.36 - 7.28 (m, 2H), 6.89 (d, J=10.7 Hz, 1H), 5.06 (s, 1H), 4.30 (t, J=5.2 Hz, 2H), 2.77 - 2.69 (m, 5H), 2.53 - 2.48 (m, 3H), 2.14 (m, 2H), 1.91 (s, 3H), 1.21 - 1.17 (m, 9H). LCMS (M+H) = 552.4.
The following examples 159- 164 were prepared according to the procedure for Example 158 using appropriate esters.
Example 159

Example 160

(2S)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-2-(3-(4-methoxypyridin- 3-yl)phenyl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid: XH NMR (500MHz, CDC13) δ: 8.56 (d, J=6.1 Hz, IH), 8.32 (s, IH), 7.75 - 7.64 (m, 2H), 7.52 7.43 (m, 2H), 7.01 (d, J=6.1 Hz, IH), 6.91 (d, J=10.7 Hz, IH), 5.00 (s, IH), 4.30 - 4.20 (m, 2H), 3.92 (s, 3H), 2.77 (s, 3H), 2.67 (t, J=6.4 Hz, 2H), 2.48 (s, 3H), 2.07 (bi s, 2H), 1.90 (s, 3H), 1.19 (s, 9H). LCMS (M+H) = 625.5.
Example 161
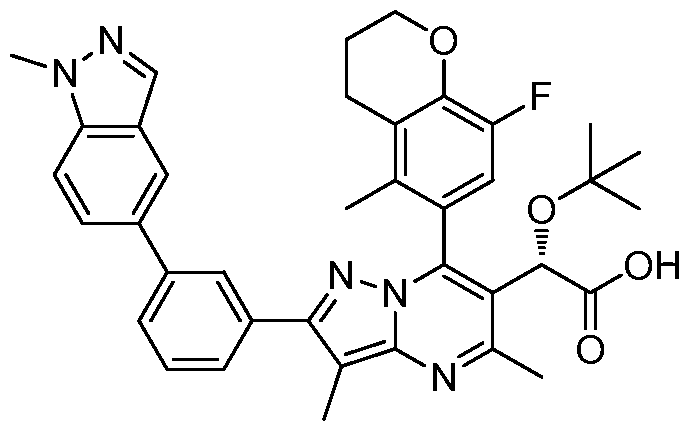
Example 162

(2S)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-2-(2'-fluoro-[l,r- biphenyl]-3-yl)-3,5-dimethylpyrazolo[l,5-a]pyrimidin-6-yl)acetic acid: XH NMR (500MHz, CDC13) δ: 7.76 (d, J=1.2 Hz, IH), 7.60 (d, J=7.6 Hz, IH), 7.59 - 7.54 (m, IH), 7.52 - 7.46 (m, IH), 7.45 - 7.40 (m, IH), 7.36 - 7.29 (m, IH), 7.24 - 7.18 (m, IH), 7.18 - 7.12 (m, IH), 6.90 (d, J=10.7 Hz, IH), 5.09 (s, IH), 4.32 - 4.22 (m, 2H), 2.80 (s, 3H), 2.68 (t, J=6.6 Hz, 2H), 2.51 (s, 3H), 2.14 - 2.06 (m, 3H), 1.90 (s, 2H), 1.25 (s, IH), 1.21 (s, 9H). LCMS ( M+H) = 612.5.
Example 163


Examples 165-175 were synthesized using the procedure described above for example 1 15.
Example 165


Acetate
Start % B 0
Final % B 100
Gradient Time 4 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair acetonitrile : Water : Ammonium Acetate
Column Waters BEH C18, 2.0 x 50 mm, 1.7-μιη particles
¾ NMR (500MHz, DMSO-d6) δ 8.06 (s, IH), 7.78 (d, J=7.9 Hz, IH), 7.69 - 7.61 (m, 2H), 7.56 (ddd, J=8.0, 5.1, 2.4 Hz, IH), 7.50 (t, J=7.6 Hz, IH), 7.31 - 7.21 (m, 2H), 7.1 1 (d, J=1 1.0 Hz, IH), 4.84 (s, IH), 4.36 - 4.23 (m, 2H), 2.79 - 2.72 (m, 2H), 2.71 (s, 3H), 2.33 (s, 3H), 2.10 - 2.06 (m, 2H), 1.83 (s, 3H), 1.10 (s, 9H).
Example 166


Gradient Time 4 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair acetonitrile : Water : Ammonium Acetate
Column Waters BEH CI 8, 2.0 x 50 mm, 1.7-μιη particles
¾ NMR (500MHz, DMSO-d6) δ 7.82 (d, J=7.6 Hz, IH), 7.76 (s, IH), 7.50 (t, J=7.8 Hz, IH), 7.39 - 7.24 (m, 2H), 7.23 - 7.16 (m, 2H), 7.14 - 7.04 (m, 2H), 4.82 (s, IH), 4.33 - 4.20 (m, 2H), 2.77 - 2.66 (m, 5H), 2.24 (s, 3H), 2.12 - 2.00 (m, 2H), 1.82 (s, 3H), 1.09 (s, 9H).
Example 167

(2S)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-5-methyl-2-(3-(2- methylpyridin-3-yl)phenyl)pyrazolo[ 1, 5 -aJpyrimidin-6-yi) acetic acid
MS (M+H)+ Calcd. 595
MS (M+H)+ Observ. 595
Retention Time 2.57 min
LC Condition
Solvent A 5 % acetonitrile : 95% Water : lOmM
Ammonium Acetate
Solvent B 95 % acetonitrile : 5% Water : lOmM
Ammonium Acetate
Start % B 0
Final % B 100
Gradient Time 4 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair acetonitrile : Water : Ammonium Acetate
Column Waters BEH CI 8, 2.0 x 50 mm, 1.7-μιη particles
¾ NMR (500MHz, DMSO-d6) δ 8.50 (dd, J=4.7, 1.7 Hz, IH), 7.90 - 7.81 (m, 2H), 7.67 (dd, J=7.6, 1.5 Hz, IH), 7.53 (t, J=7.6 Hz, IH), 7.41 (d, J=7.6 Hz, IH), 7.33 (dd, J=7.6, 4.9 Hz, IH), 7.22 (s, IH), 7.10 (d, J=l 1.3 Hz, IH), 4.83 (s, IH), 4.37 - 4.20 (m, 2H), 2.78 - 2.71 (m, 2H), 2.69 (s, 3H), 2.44 (s, 3H), 2.13 - 2.02 (m, 2H), 1.82 (s, 3H), 1.09 (s, 9H).
Example 168

(2S)-2-(tert-Butoxy)-2-(7-(8-fluoro-5-methylchroman-6-yl)-2-(3-(2-methoxypyridin- 4-yl)phenyl)-5-methylpyrazolo[ 1, 5 -a]pyrimidin-6-yl) acetic acid
MS (M+H)+ Calcd. 61 1
MS (M+H)+ Observ. 61 1
Retention Time 2.80 min
^C Condition
Solvent A 5 % acetonitrile : 95% Water : lOmM
Ammonium Acetate
Solvent B 95 % acetonitrile : 5% Water : lOmM
Ammonium Acetate
Start % B 0
Final % B 100
Gradient Time 4 min
Flow Rate 1 mL/min
Wavelength 220
Solvent Pair acetonitrile : Water : Ammonium Acetate
Column Waters BEH C18, 2.0 x 50 mm, 1.7-μιη
particles
¾ NMR (500MHz, DMSO-d6) δ 8.27 (d, J=5.5 Hz, IH), 8.19 (s, IH), 7.89 (d, J=7.9 Hz, IH), 7.79 (d, J=7.9 Hz, IH), 7.56 (t, J=7.6 Hz, IH), 7.38 (dd, J=5.5, 1.5 Hz, IH), 7.33 (s, IH), 7.19 (s, IH), 7.1 1 (d, J=1 1.3 Hz, IH), 4.84 (s, IH), 4.36 - 4.22 (m, 2H), 3.93 (s, 3H), 2.79 - 2.73 (m, 2H), 2.71 (s, 3H), 2.08 (t, J=5.6 Hz, 2H), 1.83 (s, 3H), 1.10 (s, 9H).
Example 169


¾ NMR (500MHz, DMSO-d6) δ 8.55 (d, J=2.4 Hz, IH), 8.13 - 8.05 (m, 2H), 7.79 (d, J=7.9 Hz, IH), 7.66 (d, J=7.9 Hz, IH), 7.51 (t, J=7.6 Hz, IH), 7.26 (s, IH), 7.09 (d, J=11.3 Hz, IH), 6.95 (d, J=8.5 Hz, IH), 4.78 (s, IH), 4.34 - 4.21 (m, 2H), 3.92 (s,
2.75 - 2.73 (m, 2H), 2.71 (s, 3H), 2.08 (t, J=5.6 Hz, 2H), 1.83 (s, 3H), 1.09 (s
Example 170


¾ NMR (500MHz, DMSO-d6) δ 8.54 (d, J=5.2 Hz, IH), 8.20 (s, IH), 7.89 (d, J=7.9 Hz, IH), 7.79 (d, J=7.9 Hz, IH), 7.66 (s, IH), 7.61 - 7.52 (m, 2H), 7.33 (s, IH), 7.1 1 (d, J=11.3 Hz, IH), 4.86 (s, IH), 4.35 - 4.23 (m, 2H), 2.79 - 2.73 (m, 2H), 2.71 (s, 3H), 2.56 (s, 3H), 2.08 (t, J=5.5 Hz, 2H), 1.83 (s, 3H), 1.10 (s, 9H).
Example 171


¾ NMR (500MHz, DMSO-d6) δ 8.89 (s, IH), 8.63 (d, J=2.7 Hz, IH), 8.28 - 8.11 (m, 2H), 7.88 (d, J=7.6 Hz, IH), 7.81 (d, J=7.6 Hz, IH), 7.57 (t, J=7.8 Hz, IH), 7.33 (s, IH), 7.10 (d, J=11.3 Hz, IH), 4.81 (s, IH), 4.35 - 4.19 (m, 2H), 2.79 - 2.67 (m, 5H), 2.08 (t, J=5.8 Hz, 2H), 1.83 (s, 3H), 1.09 (s, 9H).
Example 172


¾ NMR (500MHz, DMSO-d6) δ 9.32 (d, J=2.4 Hz, IH), 8.74 (d, J=2.1 Hz, IH),
8.33 (s, IH), 8.10 (d, J=8.5 Hz, 2H), 7.90 (t, J=7.5 Hz, 2H), 7.84 - 7.78 (m, IH), 7.73 - 7.65 (m, IH), 7.62 (t, J=7.8 Hz, IH), 7.35 (s, IH), 7.12 (d, J=11.3 Hz, IH), 4.84 (s, IH), 4.37 - 4.20 (m, 2H), 2.78 - 2.68 (m, 5H), 2.14 - 2.02 (m, 2H), 1.84 (s, 3H), 1.10 (s, 9H).
Example 173


¾ NMR (500MHz, DMSO-d6) δ 7.88 (dd, J=11.9, 7.9 Hz, 2H), 7.79 (s, IH), 7.78 - 7.72 (m, IH), 7.69 - 7.62 (m, IH), 7.56 - 7.44 (m, 2H), 7.33 (d, J=7.6 Hz, IH), 7.17 (s, IH), 7.09 (d, J=11.3 Hz, IH), 4.81 (s, IH), 4.33 - 4.20 (m, 2H), 2.74 - 2.66 (m, 5H), 2.06 (br. s., 2H), 1.82 (s, 3H), 1.08 (s, 9H).
Example 174


¾ NMR (500MHz, DMSO-d6) δ 8.02 - 7.92 (m, 3H), 7.78 - 7.70 (m, IH), 7.63 7.56 (m, 3H), 7.54 - 7.45 (m, 3H), 7.20 (s, IH), 7.10 (d, J=l 1.3 Hz, IH), 4.85 (s, 4.34 - 4.21 (m, 2H), 2.79 - 2.67 (m, 5H), 2.42 (s, 3H), 2.09 (d, J=5.2 Hz, 2H), 1.83 (s, 3H), 1.10 (s, 9H).
Example 175


¾ NMR (500MHz, DMSO-d6) δ 8.52 (d, J=2.4 Hz, IH), 8.04 (s, IH), 7.92 (dd, J=8.9, 2.7 Hz, IH), 7.73 (d, J=7.6 Hz, IH), 7.63 (d, J=7.9 Hz, IH), 7.48 (t, J=7.6 Hz, IH), 7.27 (s, IH), 7.11 (d, J=1 1.0 Hz, IH), 6.96 (d, J=9.2 Hz, IH), 4.84 (s, IH), 4.35 - 4.22 (m, 2H), 3.58 (br. s., 4H), 2.78 - 2.66 (m, 5H), 2.48 (br. s., 4H), 2.28 (s, 3H), 2.08 (t, J=5.5 Hz, 2H), 1.82 (s, 3H), 1.10 (s, 9H).
It will be evident to one skilled in the art that the present disclosure is not limited to the foregoing illustrative examples, and that it can be embodied in other specific forms without departing from the essential attributes thereof. It is therefore
desired that the examples be considered in all respects as illustrative and not restrictive, reference being made to the appended claims, rather than to the foregoing examples, and all changes which come within the meaning and range of equivalency of the claims are therefore intended to be embraced therein.
Claims
1. A compound of Formula I

I
where:
R1 is H, alkyl, cycloalkyl, or Ar1;
R2 is H, alkyl, cycloalkyl, or Ar1; R3 is alkyl or Ar2;
R4 is alkyl or haloalkyl;
R5 is alkyl;
Ar1 is phenyl, pyridinyl, tetralinyl, indazolyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, benzyloxy, halobenzyloxy, (alkoxy )benzyloxy, phenoxyalkyl, CONH(phenyl), CONH(benzyl), and Ar3;
Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido;

Ar3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 0-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl; or a pharmaceutically acceptable salt thereof. 2. A compound of claim 1 where: R1 is H, alkyl, cycloalkyl, or Ar1; R2 is H, alkyl, cycloalkyl, or Ar1; R3 is alkyl or Ar2;
R4 is alkyl or haloalkyl; R5 is alkyl; Ar1 is phenyl, pyridinyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, and phenoxyalkyl; Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl,
dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido;

1 1 2 3 2 4 5
3. A compound of claim 1 where R is Ar ; R is H; R is Ar ; R is alkyl; R is methyl; Ar1 is phenyl, pyridinyl, or chromanyl, and is substituted with 0-3 substituents selected from halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, benzyl, phenoxy, and phenoxyalkyl; and Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido; or a pharmaceutically acceptable salt thereof.
4. A compound of claim 1 where R is Ar ; R is H; R is Ar ; R is alkyl; R is methyl; Ar1 is phenyl or pyridinyl, and is substituted with 1 Ar3 substituent and 0-2 substituents selected from halo, alkyl, haloalkyl, alkoxy, and haloalkoxy; and Ar2 is phenyl, pyridinyl, indanyl, naphthyl, tetrahydronaphthalenyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, isochromanyl, benzodioxanyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl,
dihydrobenzoxazinyl, indolyl, dihydroindolyl, benzthiazolyl, or benzothiazolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, cycloalkyl, halocycloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, thioalkyl, and acetamido; or a pharmaceutically acceptable salt thereof.
5. A compound of claim 1 where R1 is Ar1; R2 is H; and Ar1 is phenyl or pyridinyl, and is substituted with 1 Ar3 substituent and 0-2 substituents selected from halo, alkyl, haloalkyl, alkoxy, and haloalkoxy.
6. A compound of claim 5 where Ar2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido.
7. A compound of claim 1 where R4 is alkyl.
8. A compound of claim 1 where R5 is methyl.
9. A compound of claim 1 where Ar2 is phenyl, pyridinyl, indanyl, naphthyl, benzofuranyl, dihydrobenzofuranyl, benzodioxyl, chromanyl, benzodioxanyl, or indolyl, and is substituted with 0-3 substituents selected from halo, cyano, alkyl, haloalkyl, hydroxy, alkoxy, haloalkoxy, phenoxy, benzyloxy, and acetamido.
10. A compound of claim 1 where Ar3 is phenyl, pyridinyl, pyrazolyl, quinolinyl, chromanyl, or indazolyl, and is substituted with 0-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, haloalkoxy, phenyl, and methylpiperazinyl.
11. A compound of claim 1 where Ar3 is phenyl, pyridinyl, or pyrazolyl, and is substituted with 1-3 substituents selected from the group consisting of halo, alkyl, haloalkyl, alkoxy, and haloalkoxy.
12. A composition useful for treating HIV infection comprising a therapeutic amount of a compound of claim 1 and a pharmaceutically acceptable carrier.
13. The composition of claim 12 further comprising a therapeutically effective amount at least one other agent used for treatment of AIDS or HIV infection selected from the group consisting of nucleoside HIV reverse transcriptase inhibitors, non- nucleoside HIV reverse transcriptase inhibitors, HIV protease inhibitors, HIV fusion inhibitors, HIV attachment inhibitors, CCR5 inhibitors, CXCR4 inhibitors, HIV budding or maturation inhibitors, and HIV integrase inhibitors, and a
pharmaceutically acceptable carrier.
14. A method for treating HIV infection comprising administering a
therapeutically effective amount of a compound of claim 1, or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
15. The method of claim 14 further comprising administering a therapeutically effective amount of at least one other agent used for treatment of AIDS or HIV infection selected from the group consisting of nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV protease inhibitors, HIV fusion inhibitors, HIV attachment inhibitors, CCR5 inhibitors, CXCR4 inhibitors, HIV budding or maturation inhibitors, and HIV integrase inhibitors.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11758033.2A EP2614064B8 (en) | 2010-09-08 | 2011-09-06 | Inhibitors of human immunodeficiency virus replication |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US38075910P | 2010-09-08 | 2010-09-08 | |
| US61/380,759 | 2010-09-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012033735A1 true WO2012033735A1 (en) | 2012-03-15 |
Family
ID=44653572
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/050503 WO2012033735A1 (en) | 2010-09-08 | 2011-09-06 | Inhibitors of human immunodeficiency virus replication |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US8633200B2 (en) |
| EP (1) | EP2614064B8 (en) |
| WO (1) | WO2012033735A1 (en) |
Cited By (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013025584A1 (en) * | 2011-08-18 | 2013-02-21 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2013062028A1 (en) | 2011-10-25 | 2013-05-02 | 塩野義製薬株式会社 | Hiv replication inhibitor |
| WO2013134113A1 (en) * | 2012-03-05 | 2013-09-12 | Bristol-Myers Squibb Company | Fused pyrimidines as inhibitors of immunodeficiency virus replication |
| WO2013134142A1 (en) * | 2012-03-06 | 2013-09-12 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| US20140051692A1 (en) * | 2012-08-16 | 2014-02-20 | Bristol-Myers Squibb Company | Inhibitors of Human Immunodeficiency Virus Replication |
| WO2014119636A1 (en) | 2013-01-31 | 2014-08-07 | 塩野義製薬株式会社 | Hiv replication inhibitor |
| WO2014159959A1 (en) | 2013-03-14 | 2014-10-02 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014159076A1 (en) | 2013-03-14 | 2014-10-02 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014164409A1 (en) | 2013-03-13 | 2014-10-09 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014164428A1 (en) | 2013-03-13 | 2014-10-09 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014164467A1 (en) | 2013-03-13 | 2014-10-09 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| EP2821082A1 (en) | 2013-07-05 | 2015-01-07 | Laboratoire Biodim | Method of producing an inactivated lentivirus, especially HIV, vaccine, kit and method of use |
| US8987250B2 (en) | 2012-04-20 | 2015-03-24 | Gilead Sciences, Inc. | Therapeutic compounds |
| US9006229B2 (en) | 2011-04-21 | 2015-04-14 | Gilead Sciences, Inc. | Benzothiazole compounds and their pharmaceutical use |
| JP2015522061A (en) * | 2012-07-12 | 2015-08-03 | ヴィーブ ヘルスケア ユーケー リミテッド | Compounds and methods for treating HIV |
| US9102614B2 (en) | 2010-07-02 | 2015-08-11 | Gilead Sciences, Inc. | Naphth-2-ylacetic acid derivatives to treat AIDS |
| WO2015123230A1 (en) | 2014-02-12 | 2015-08-20 | Bristol-Myers Squibb Company | Benzothiazole macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015123182A1 (en) * | 2014-02-12 | 2015-08-20 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126743A1 (en) | 2014-02-18 | 2015-08-27 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126765A1 (en) | 2014-02-19 | 2015-08-27 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2015126737A1 (en) * | 2014-02-19 | 2015-08-27 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126376A1 (en) * | 2014-02-19 | 2015-08-27 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126726A1 (en) | 2014-02-20 | 2015-08-27 | Bristol-Myers Squibb Company | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2015126751A1 (en) | 2014-02-18 | 2015-08-27 | Bristol-Myers Squibb Company | Imidazopyridine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126758A1 (en) | 2014-02-18 | 2015-08-27 | Bristol-Myers Squibb Company | Imidazopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015179448A1 (en) * | 2014-05-21 | 2015-11-26 | Gilead Sciences, Inc. | Therapeutic compounds |
| US9284323B2 (en) | 2012-01-04 | 2016-03-15 | Gilead Sciences, Inc. | Naphthalene acetic acid derivatives against HIV infection |
| US9296758B2 (en) | 2010-07-02 | 2016-03-29 | Gilead Sciences, Inc. | 2-quinolinyl-acetic acid derivatives as HIV antiviral compounds |
| US9376392B2 (en) | 2012-01-04 | 2016-06-28 | Gilead Sciences, Inc. | 2-(tert-butoxy)-2-(7-methylquinolin-6-yl) acetic acid derivatives for treating AIDS |
| WO2017006261A1 (en) | 2015-07-06 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017006280A1 (en) | 2015-07-09 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017006281A1 (en) | 2015-07-09 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017006260A1 (en) | 2015-07-08 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025917A1 (en) | 2015-08-11 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | 5-(n-benzyl tetrahydroisoquinolin-6-yl) pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025864A1 (en) | 2015-08-07 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025916A1 (en) | 2015-08-12 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | 5-(n-[6,5]-fused bicyclic aryl tetrahydroisoquinolin-6-yl) pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025913A1 (en) | 2015-08-10 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | Imidazopyridine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2017025915A1 (en) | 2015-08-12 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025914A1 (en) | 2015-08-12 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | 5-(n-fused tricyclic aryl tetrahydroisoquinolin-6-yl) pyridin-3- yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017029631A1 (en) | 2015-08-20 | 2017-02-23 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| JP2017526748A (en) * | 2014-08-27 | 2017-09-14 | ヴィーブ ヘルスケア ユーケー(ナンバー5)リミテッド | Imidazo [1,2-A] pyridine derivatives for use as inhibitors of human immunodeficiency virus replication |
| WO2017195111A1 (en) | 2016-05-11 | 2017-11-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017195113A1 (en) | 2016-05-11 | 2017-11-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017195112A1 (en) | 2016-05-11 | 2017-11-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| EP3144311A4 (en) * | 2014-05-16 | 2018-01-03 | Shionogi & Co., Ltd. | Tricyclic heterocyclic derivative having hiv replication-inhibiting effect |
| WO2018127801A1 (en) | 2017-01-03 | 2018-07-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2018127800A1 (en) | 2017-01-03 | 2018-07-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| US10112960B2 (en) | 2013-09-05 | 2018-10-30 | Dow Agrosciences Llc | Methods for producing borylated arenes |
| US10166533B2 (en) | 2014-06-16 | 2019-01-01 | Dow Agrosciences Llc | Methods for producing borylated arenes |
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10494380B2 (en) | 2015-05-29 | 2019-12-03 | Shionogi & Co., Ltd. | Nitrogen-containing tricyclic derivatives having HIV replication inhibitory activity |
| WO2019244066A2 (en) | 2018-06-19 | 2019-12-26 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2020003093A1 (en) | 2018-06-25 | 2020-01-02 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2719685A1 (en) | 2012-10-11 | 2014-04-16 | Laboratoire Biodim | Inhibitors of viral replication, their process of preparation and their therapeutical uses |
| ES2728932T3 (en) * | 2014-01-09 | 2019-10-29 | Intra Cellular Therapies Inc | Organic compounds |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1375486A1 (en) * | 2001-03-01 | 2004-01-02 | Shionogi & Co., Ltd. | Nitrogen-containing heteroaryl compounds having hiv integrase inhibitory activity |
| WO2004052315A2 (en) * | 2002-12-11 | 2004-06-24 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| WO2007131350A1 (en) | 2006-05-16 | 2007-11-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062288A1 (en) | 2007-11-15 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062285A1 (en) | 2007-11-16 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062308A1 (en) | 2007-11-16 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062289A1 (en) | 2007-11-15 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| EP2085398A1 (en) * | 2008-02-01 | 2009-08-05 | Merz Pharma GmbH & Co. KGaA | Pyrazolopyrimidines, a process for their preparation and their use as medicine |
| WO2010032195A1 (en) * | 2008-09-16 | 2010-03-25 | Csir | Imidazopyridines and imidazopyrimidines as hiv-i reverse transcriptase inhibitors |
| WO2011076765A1 (en) * | 2009-12-23 | 2011-06-30 | Katholieke Universiteit Leuven | Novel antiviral compounds |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5331404B2 (en) | 2008-08-01 | 2013-10-30 | 国立大学法人 東京医科歯科大学 | Method for detecting chromosomal deletions in congenital anomalies |
| US8338441B2 (en) | 2009-05-15 | 2012-12-25 | Gilead Sciences, Inc. | Inhibitors of human immunodeficiency virus replication |
| GB0913636D0 (en) | 2009-08-05 | 2009-09-16 | Univ Leuven Kath | Novel viral replication inhibitors |
-
2011
- 2011-09-02 US US13/224,802 patent/US8633200B2/en not_active Expired - Fee Related
- 2011-09-06 EP EP11758033.2A patent/EP2614064B8/en not_active Not-in-force
- 2011-09-06 WO PCT/US2011/050503 patent/WO2012033735A1/en active Application Filing
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1375486A1 (en) * | 2001-03-01 | 2004-01-02 | Shionogi & Co., Ltd. | Nitrogen-containing heteroaryl compounds having hiv integrase inhibitory activity |
| WO2004052315A2 (en) * | 2002-12-11 | 2004-06-24 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| US20060025426A1 (en) * | 2002-12-11 | 2006-02-02 | Fraley Mark E | Tyrosine kinase inhibitors |
| WO2007131350A1 (en) | 2006-05-16 | 2007-11-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062288A1 (en) | 2007-11-15 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062289A1 (en) | 2007-11-15 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062285A1 (en) | 2007-11-16 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| WO2009062308A1 (en) | 2007-11-16 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Inhibitors of human immunodeficiency virus replication |
| EP2085398A1 (en) * | 2008-02-01 | 2009-08-05 | Merz Pharma GmbH & Co. KGaA | Pyrazolopyrimidines, a process for their preparation and their use as medicine |
| WO2010032195A1 (en) * | 2008-09-16 | 2010-03-25 | Csir | Imidazopyridines and imidazopyrimidines as hiv-i reverse transcriptase inhibitors |
| WO2011076765A1 (en) * | 2009-12-23 | 2011-06-30 | Katholieke Universiteit Leuven | Novel antiviral compounds |
Non-Patent Citations (4)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| JOHNSON VA, BYINGTON RT.: "Techniques in HIV Research", 1990, STOCKTON PRESS, article "Infectivity Assay", pages: 71 - 76 |
| PALELLA, F. J., DELANY, K. M., MOORMAN, A. C., LOVELESS, M. 0., FURHER, J., SATTEN, G. A., ASCHMAN, D. J., HOLMBERG, S. D., N. ENGL. J. MED., vol. 338, 1998, pages 853 - 860 |
| UNAIDS: REPORT ON THE GLOBAL HIV/AIDS EPIDEMIC, December 1998 (1998-12-01) |
Cited By (108)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9296758B2 (en) | 2010-07-02 | 2016-03-29 | Gilead Sciences, Inc. | 2-quinolinyl-acetic acid derivatives as HIV antiviral compounds |
| US9102614B2 (en) | 2010-07-02 | 2015-08-11 | Gilead Sciences, Inc. | Naphth-2-ylacetic acid derivatives to treat AIDS |
| US9006229B2 (en) | 2011-04-21 | 2015-04-14 | Gilead Sciences, Inc. | Benzothiazole compounds and their pharmaceutical use |
| WO2013025584A1 (en) * | 2011-08-18 | 2013-02-21 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2013062028A1 (en) | 2011-10-25 | 2013-05-02 | 塩野義製薬株式会社 | Hiv replication inhibitor |
| US9199959B2 (en) | 2011-10-25 | 2015-12-01 | Shionogi & Co., Ltd. | HIV replication inhibitor |
| US9284323B2 (en) | 2012-01-04 | 2016-03-15 | Gilead Sciences, Inc. | Naphthalene acetic acid derivatives against HIV infection |
| US9376392B2 (en) | 2012-01-04 | 2016-06-28 | Gilead Sciences, Inc. | 2-(tert-butoxy)-2-(7-methylquinolin-6-yl) acetic acid derivatives for treating AIDS |
| WO2013134113A1 (en) * | 2012-03-05 | 2013-09-12 | Bristol-Myers Squibb Company | Fused pyrimidines as inhibitors of immunodeficiency virus replication |
| US9034882B2 (en) | 2012-03-05 | 2015-05-19 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| US9006235B2 (en) | 2012-03-06 | 2015-04-14 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2013134142A1 (en) * | 2012-03-06 | 2013-09-12 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| US8987250B2 (en) | 2012-04-20 | 2015-03-24 | Gilead Sciences, Inc. | Therapeutic compounds |
| US9096586B2 (en) | 2012-04-20 | 2015-08-04 | Gilead Sciences, Inc. | Therapeutic compounds |
| JP2015522061A (en) * | 2012-07-12 | 2015-08-03 | ヴィーブ ヘルスケア ユーケー リミテッド | Compounds and methods for treating HIV |
| US8906929B2 (en) | 2012-08-16 | 2014-12-09 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014028384A1 (en) | 2012-08-16 | 2014-02-20 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| JP2015524841A (en) * | 2012-08-16 | 2015-08-27 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Human immunodeficiency virus replication inhibitor |
| CN104583214A (en) * | 2012-08-16 | 2015-04-29 | 百时美施贵宝公司 | Inhibitors of human immunodeficiency virus replication |
| AU2013302873B2 (en) * | 2012-08-16 | 2017-02-02 | VIIV Healthcare UK (No.5) Limited | Inhibitors of human immunodeficiency virus replication |
| US20140051692A1 (en) * | 2012-08-16 | 2014-02-20 | Bristol-Myers Squibb Company | Inhibitors of Human Immunodeficiency Virus Replication |
| WO2014119636A1 (en) | 2013-01-31 | 2014-08-07 | 塩野義製薬株式会社 | Hiv replication inhibitor |
| CN105189510A (en) * | 2013-03-13 | 2015-12-23 | 百时美施贵宝公司 | Inhibitors of human immunodeficiency virus replication |
| WO2014164409A1 (en) | 2013-03-13 | 2014-10-09 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014164467A1 (en) | 2013-03-13 | 2014-10-09 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| US9663536B2 (en) | 2013-03-13 | 2017-05-30 | VIIV Healthcare UK (No.5) Limited | Inhibitors of human immunodeficiency virus replication |
| CN105189511B (en) * | 2013-03-13 | 2017-05-24 | 百时美施贵宝公司 | Inhibitors of human immunodeficiency virus replication |
| CN105189510B (en) * | 2013-03-13 | 2017-05-17 | 百时美施贵宝公司 | Inhibitors of human immunodeficiency virus replication |
| JP2016512511A (en) * | 2013-03-13 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| CN105189511A (en) * | 2013-03-13 | 2015-12-23 | 百时美施贵宝公司 | Inhibitors of human immunodeficiency virus replication |
| JP2016516692A (en) * | 2013-03-13 | 2016-06-09 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014164428A1 (en) | 2013-03-13 | 2014-10-09 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| CN105008369A (en) * | 2013-03-14 | 2015-10-28 | 百时美施贵宝公司 | Inhibitors of human immunodeficiency virus replication |
| US9540393B2 (en) | 2013-03-14 | 2017-01-10 | VIIV Healthcare UK (No.5) Limited | Inhibitors of human immunodeficiency virus replication |
| JP2016512501A (en) * | 2013-03-14 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| JP2016512558A (en) * | 2013-03-14 | 2016-04-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014159076A1 (en) | 2013-03-14 | 2014-10-02 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2014159959A1 (en) | 2013-03-14 | 2014-10-02 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| CN105008369B (en) * | 2013-03-14 | 2017-07-11 | 百时美施贵宝公司 | human immunodeficiency virus replication inhibitor |
| EP2821082A1 (en) | 2013-07-05 | 2015-01-07 | Laboratoire Biodim | Method of producing an inactivated lentivirus, especially HIV, vaccine, kit and method of use |
| US10112960B2 (en) | 2013-09-05 | 2018-10-30 | Dow Agrosciences Llc | Methods for producing borylated arenes |
| WO2015123230A1 (en) | 2014-02-12 | 2015-08-20 | Bristol-Myers Squibb Company | Benzothiazole macrocycles as inhibitors of human immunodeficiency virus replication |
| US9932356B2 (en) | 2014-02-12 | 2018-04-03 | Viiv Healthcare Uk (No. 5) Limited | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015123182A1 (en) * | 2014-02-12 | 2015-08-20 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126751A1 (en) | 2014-02-18 | 2015-08-27 | Bristol-Myers Squibb Company | Imidazopyridine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126743A1 (en) | 2014-02-18 | 2015-08-27 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126758A1 (en) | 2014-02-18 | 2015-08-27 | Bristol-Myers Squibb Company | Imidazopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| US9834566B2 (en) | 2014-02-18 | 2017-12-05 | VIIV Healthcare UK (No.5) Limited | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| US10125148B2 (en) | 2014-02-19 | 2018-11-13 | Viiv Healthcare Uk (No. 5) Limited | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| US9273067B2 (en) | 2014-02-19 | 2016-03-01 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126765A1 (en) | 2014-02-19 | 2015-08-27 | Bristol-Myers Squibb Company | Inhibitors of human immunodeficiency virus replication |
| WO2015126737A1 (en) * | 2014-02-19 | 2015-08-27 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126376A1 (en) * | 2014-02-19 | 2015-08-27 | Bristol-Myers Squibb Company | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2015126726A1 (en) | 2014-02-20 | 2015-08-27 | Bristol-Myers Squibb Company | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| EP3144311A4 (en) * | 2014-05-16 | 2018-01-03 | Shionogi & Co., Ltd. | Tricyclic heterocyclic derivative having hiv replication-inhibiting effect |
| US9975906B2 (en) | 2014-05-16 | 2018-05-22 | Shionogi & Co., Ltd. | Tricyclic heterocycle derivatives having HIV replication inhibitory effect |
| WO2015179448A1 (en) * | 2014-05-21 | 2015-11-26 | Gilead Sciences, Inc. | Therapeutic compounds |
| US10166533B2 (en) | 2014-06-16 | 2019-01-01 | Dow Agrosciences Llc | Methods for producing borylated arenes |
| JP2017526748A (en) * | 2014-08-27 | 2017-09-14 | ヴィーブ ヘルスケア ユーケー(ナンバー5)リミテッド | Imidazo [1,2-A] pyridine derivatives for use as inhibitors of human immunodeficiency virus replication |
| US10494380B2 (en) | 2015-05-29 | 2019-12-03 | Shionogi & Co., Ltd. | Nitrogen-containing tricyclic derivatives having HIV replication inhibitory activity |
| US10870661B2 (en) | 2015-05-29 | 2020-12-22 | Shionogi & Co., Ltd. | Nitrogen-containing tricyclic derivatives having HIV replication inhibitory activity |
| WO2017006261A1 (en) | 2015-07-06 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017006260A1 (en) | 2015-07-08 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017006280A1 (en) | 2015-07-09 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017006281A1 (en) | 2015-07-09 | 2017-01-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025864A1 (en) | 2015-08-07 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025913A1 (en) | 2015-08-10 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | Imidazopyridine macrocycles as inhibitors of human immunodeficiency virus replication |
| WO2017025917A1 (en) | 2015-08-11 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | 5-(n-benzyl tetrahydroisoquinolin-6-yl) pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025916A1 (en) | 2015-08-12 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | 5-(n-[6,5]-fused bicyclic aryl tetrahydroisoquinolin-6-yl) pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025915A1 (en) | 2015-08-12 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017025914A1 (en) | 2015-08-12 | 2017-02-16 | VIIV Healthcare UK (No.5) Limited | 5-(n-fused tricyclic aryl tetrahydroisoquinolin-6-yl) pyridin-3- yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017029631A1 (en) | 2015-08-20 | 2017-02-23 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11866435B2 (en) | 2015-12-22 | 2024-01-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2017195113A1 (en) | 2016-05-11 | 2017-11-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017195112A1 (en) | 2016-05-11 | 2017-11-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2017195111A1 (en) | 2016-05-11 | 2017-11-16 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11339149B2 (en) | 2016-12-22 | 2022-05-24 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| US10800768B2 (en) | 2016-12-22 | 2020-10-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11566026B2 (en) | 2016-12-22 | 2023-01-31 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11787793B2 (en) | 2016-12-22 | 2023-10-17 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2018127801A1 (en) | 2017-01-03 | 2018-07-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2018127800A1 (en) | 2017-01-03 | 2018-07-12 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| US11124511B2 (en) | 2018-03-30 | 2021-09-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10906920B2 (en) | 2018-05-11 | 2021-02-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11414433B2 (en) | 2018-05-11 | 2022-08-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| WO2019244066A2 (en) | 2018-06-19 | 2019-12-26 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| WO2020003093A1 (en) | 2018-06-25 | 2020-01-02 | VIIV Healthcare UK (No.5) Limited | Pyridin-3-yl acetic acid derivatives as inhibitors of human immunodeficiency virus replication |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| US12084443B2 (en) | 2020-11-06 | 2024-09-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
Also Published As
| Publication number | Publication date |
|---|---|
| US20120225888A1 (en) | 2012-09-06 |
| EP2614064B8 (en) | 2015-01-14 |
| EP2614064B1 (en) | 2014-07-16 |
| EP2614064A1 (en) | 2013-07-17 |
| US8633200B2 (en) | 2014-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2614064B1 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP2822950B1 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP2822949B1 (en) | Fused pyrimidines as inhibitors of immunodeficiency virus replication | |
| EP2814490B1 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP2888266B1 (en) | Inhibitors of human immunodeficiency virus replication | |
| US9580431B2 (en) | Inhibitors of human immunodeficiency virus replication | |
| WO2013025584A1 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP3186254B1 (en) | Imidazo[1,2-a]pyridine derivatives for use as inhibitors of human immunodeficiency virus replication | |
| EP3107913B1 (en) | Inhibitors of human immunodeficiency virus replication | |
| WO2015126751A1 (en) | Imidazopyridine macrocycles as inhibitors of human immunodeficiency virus replication | |
| EP3105236B1 (en) | Benzothiazole macrocycles as inhibitors of human immunodeficiency virus replication | |
| EP2970297A1 (en) | Inhibitors of human immunodeficiency virus replication | |
| WO2015126743A1 (en) | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication | |
| US9932353B2 (en) | Imidazopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication | |
| WO2015126376A1 (en) | Pyrazolopyrimidine macrocycles as inhibitors of human immunodeficiency virus replication |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11758033 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011758033 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |