WO2012018540A1 - 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof - Google Patents
2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof Download PDFInfo
- Publication number
- WO2012018540A1 WO2012018540A1 PCT/US2011/044807 US2011044807W WO2012018540A1 WO 2012018540 A1 WO2012018540 A1 WO 2012018540A1 US 2011044807 W US2011044807 W US 2011044807W WO 2012018540 A1 WO2012018540 A1 WO 2012018540A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- cancer
- oxo
- cyclopentyl
- Prior art date
Links
- 0 CSc1nc(N(*)C(C(C#N)=C2)=O)c2cn1 Chemical compound CSc1nc(N(*)C(C(C#N)=C2)=O)c2cn1 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the invention relates to compounds, methods for their preparation, compositions including them and methods for the treatment of cellular proliferative disorders, including, but not limited to, cancer.
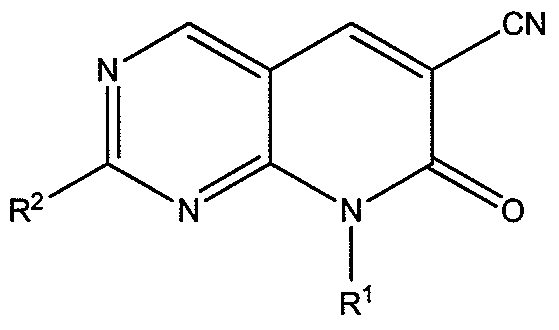
- the biologically active compounds of the invention are 2-substittued-8-alkyl-7-oxo-7,8-dihydropyrido[2,3- cT
- compounds according to Formula I, or a salt thereof, are 2-substittued-8-alkyl-7-oxo-7,8-dihydropyrido[2,3- cT
- R 1 is (C C 6 )alkyl or (C 3 -C 8 )cycloalkyl
- R 2 is 4-(4-methylpiperazin-l-yl)anilinyl, 4-morpholinoanilinyl, or
- R 3 is independently at each occurrence (C 1 -C 6 )alkoxy
- R 4 is H or (Ci-Qdalkoxy.
- R is 4-(4-methylpiperazin-l-yl)anilinyl.
- R 1 is (C 3 -C8)cycloalkyl.
- R 1 is cyclopentyl or cyclohexyl. In some embodiments, R is
- R is cyclopentyl
- each occurrence of R is methoxy.
- R 4 is hydrogen
- R 4 is methoxy
- R 2 is 4-morpholinoanilinyl.
- R 1 is cyclopentyl
- the compound of Formula I is selected from the group consisting of 8-cyclopentyl-2-((4-(4-methylpiperazin- 1 -yl)phenyl)amino)-7-oxo-7,8- dihydropyrido[2,3- J]pyrimidine-6-carbonitrile; 8-cyclohexyl-2-((4-(4-methylpiperazin- 1 ⁇ yl)phenyl)amino)-7-oxo-7,8-dihydropyrido[2,3- ]pyrimidine-6-carbonitrile; 8- cyclopentyl-2-((3 , 5 -dimethoxyphenyl)amino)-7-oxo-7, 8 -dihydropyrido [2,3 -i jpyrimidine 6-carbonitrile; 8-cyclopentyl-7-oxo-2-((3,4,5-trimethoxyphenyl)amino)-7,8- dihydro
- the compound according to Formula I is 8- cyclopentyl-2-((4-(4-methylpiperazin-l-yl)phenyl)amino)-7-oxo-7,8-dihydropyrido[2,3- J]pyrimidine-6-carbonitrile or a salt thereof.
- the present invention further provides a pharmaceutical composition comprising pharmaceutically acceptable carrier and a compound according to Formula I, or a pharmaceutically acceptable salt thereof.
- the pharmaceutical composition includes a compound of Formula I selected from the group consisting of 8-cyclopentyl-2-((4-(4-methylpiperazin- l-yl)phenyl)amino)-7-oxo-7,8-dihydropyrido[2,3- ]pyrimidine-6-carbonitrile; 8- cyclohexyl-2-((4-(4-methylpiperazin-l-yl)phenyl)amino)-7-oxo-7,8-dihydropyrido[2,3- ]pyrimidine-6-carbonitrile; 8-cyclopentyl-2-((3,5-dimethoxyphenyl)amino)-7-oxo-7,8- dihydropyrido[2,3- ]pyrimidine-6-carbonitrile; 8-cyclopentyl-7-(
- the present invention also provides a method of treating an individual for a cellular proliferative disorder, comprising administering to the individual an effective amount of at least one compound according to Formula I, or a salt thereof.
- the cellular proliferative disorder is selected from the group consisting of hemangiomatosis in newborn, secondary progressive multiple sclerosis, atherosclerosis, chronic progressive myelodegenerative disease,
- lymphocellular proliferative disorders neurofibromatosis, ganglioneuromatosis, keloid formation, Paget's disease of the bone, fibrocystic disease of the breast, uterine fibroids, Peyronie's disease, Dupuytren's disease, restenosis, benign proliferative breast disease, benign prostatic hyperplasia, X linked lymphocellular proliferative disorder, post transplantation lymphocellular proliferative disorder, macular degeneration, retinopathies, proliferative vitreoretinopathy and non cancerous lymphocellular proliferative disorders.
- the cellular proliferative disorder is cancer.
- the cancer is selected from the group consisting of ovarian cancer; cervical cancer; breast cancer; prostate cancer; testicular cancer, lung cancer, renal cancer;
- colorectal cancer skin cancer; brain cancer; leukemia, including acute myeloid leukemia, chronic myeloid leukemia, acute lymphoid leukemia, and chronic lymphoid leukemia.
- the present invention further provides a method of inducing apoptosis of cancer cells in an individual afflicted with cancer, comprising administering to the individual an effective amount of at least one compound according to Formula I, or a salt thereof.
- the cancer cells are tumor cells.
- the cancer cells are tumor cells.
- the tumor cells are selected from the group consisting of ovarian, cervical, uterine, vaginal, breast, prostate, testicular, lung, renal, colorectal, stomach, adrenal, mouth, esophageal, hepatic, gall bladder, bone, lymphatic, eye, skin, and brain tumor cells.
- the present invention further provides a process for the preparation of a compound according to Formula I.
- the process comprises treating a compound of Formula II
- an amine selected from the group consisting of 4-(4-methylpiperazin-l-yl)aniline, 4- morpholinoaniline, and
- R 1 is (C 1 -C 6 )alkyl or (C 3 -C 8 )cycloalkyl; R 3 is independently at each occurrence (C ! -C 6 )alkoxy; and R 4 is H or (d-C 6 )alkoxy.
- the process further includes obtaining a compound according to Formula I.
- a compound of Formula II is prepared by reacting a compound of Formula III
- a compound of Formula III is prepared by reacting a compound of Formula IV
- a compound of Formula IV is prepared by selectively oxidizing a compound of Formula V
- a compound of Formula V is prepared by reducing a compound of Formula VI
- a compound of Formula VI is prepared by reacting a compound of Formula VII
- the present invention further provides a method of inhibiting kinase activity in mammal in need of such treatment, said method comprising administering a
- the compounds and compositions of the invention are believed to selectively inhibit proliferation of cancer cells, and kill various tumor cell types.
- the compounds of the invention inhibit various protein kinases. Although similar compounds have been reported to inhibit kinase activity ⁇ see, for example U.S. 6,498,163), the compounds of the present invention have a surprisingly different kinase inhibition profile and inhibit a wider range of protein kinases.
- the compounds of the invention are believed to inhibit the proliferation of tumor cells, and for some compounds, induce cell death. Cell death results from the induction of apoptosis.
- the compounds are believed effective against a broad range of tumor types, including but not limited to the following: ovarian cancer, breast cancer, prostate cancer, lung cancer, renal cancer, colorectal cancer, brain cancer and leukemia.
- the compounds are also believed useful in the treatment of non-cancer cellular proliferative disorders, including but not limited to the following: hemangiomatosis in newborn, secondary progressive multiple sclerosis, chronic progressive myelodegenerative disease, neurofibromatosis, ganglioneuromatosis, keloid formation, Paget' s disease of the bone, fibrocystic disease of the breast, uterine fibroids, Peyronie's disease, Dupuytren's disease, restenosis and cirrhosis.
- non-cancer cellular proliferative disorders including but not limited to the following: hemangiomatosis in newborn, secondary progressive multiple sclerosis, chronic progressive myelodegenerative disease, neurofibromatosis, ganglioneuromatosis, keloid formation, Paget' s disease of the bone, fibrocystic disease of the breast, uterine fibroids, Peyronie's disease, Dupuytren's disease, restenosis and cirrhosis.
- Figure 1 is a graph showing the efficacy of 8-cyclopentyl-2-((4-(4- methylpiperazin-l-yl)phenyl)amino)-7-oxo-7,8-dihydropyrido[2,3- ⁇ pyrimidine-6- carbonitrile at inhibiting growth of Colo-205 tumor fragments implanted in female athymic nude mice.
- the terms “treat” and “treatment” are used interchangeably and are meant to indicate a postponement of development of a disorder and/or a reduction in the severity of symptoms that will or are expected to develop.
- the terms further include ameliorating existing symptoms, preventing additional symptoms, and ameliorating or preventing the underlying metabolic causes of symptoms.
- mammals as used herein, means both mammals and non-mammals.
- Mammals include, for example, humans; non-human primates, e.g. apes and monkeys; cattle; horses; sheep; and goats.
- Non-mammals include, for example, fish and birds.
- an effective amount when used to describe therapy to an individual suffering from a cancer or other cellular proliferative disorder, refers to the amount of a compound according to Formula I that inhibits the abnormal growth or proliferation, or alternatively induces apoptosis of cancer cells, preferably tumor cells, resulting in a therapeutically useful and selective cytotoxic effect on proliferative cells.
- cellular proliferative disorder means a disorder wherein unwanted cell proliferation of one or more subsets of cells in a multicellular organism occurs. In some such disorders, cells are made by the organism at an atypically accelerated rate.
- alkyl by itself or as part of another substituent means, unless otherwise stated, a straight, branched or cyclic chain hydrocarbon (cycloalkyl) having the number of carbon atoms designated (i.e. d-C 6 means one to six carbons) and includes straight, branched chain or cyclic groups. Examples include: methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, neopentyl, hexyl, cyclohexyl and cyclopropylmethyl.
- Preferred alkyl groups are (C 1 -C 3 )alkyl, particularly methyl, ethyl and isopropyl.
- Preferred cycloalkyl groups include (C 3 -C 8 )cyclo alkyl, with the most preferred (C 3 -C 8 )cycloalkyl groups being cyclopentyl and cyclohexyl.
- alkoxy employed alone or in combination with other terms means, unless otherwise stated, an alkyl group having the designated number of carbon atoms, as defined above, connected to the rest of the molecule via an oxygen atom, such as, for example, methoxy, ethoxy, 1-propoxy, 2-propoxy (isopropoxy) and the higher homologs and isomers. Preferred are (C 1 -C 3 )alkoxy, particularly ethoxy and methoxy.
- cyano refers to a -C ⁇ N group.
- halo or halogen by themselves or as part of another substituent mean, unless otherwise stated, a monovalent fluorine, chlorine, bromine, or iodine atom, preferably, fluorine, chlorine, or bromine, more preferably, fluorine or chlorine.
- aromatic generally refers to a carbocycle or heterocycle having one or more polyunsaturated rings having aromatic character (i.e. having (4n + 2) delocalized ⁇ (pi) electrons where n is an integer).
- R 1 is (Q-C ⁇ alkyl or (C 3 -C 8 )cycloalkyl.
- R 1 can be cyclopentyl or cyclohexyl, most preferably cyclopentyl.
- R 3 is independently at each occurrence (C C 6 )alkoxy, preferably methoxy or ethoxy, most preferably methoxy.
- R 4 is H or (CrC ⁇ alkoxy.
- R 4 is (Ci-C 6 )alkoxy, preferably it is either ethoxy or methoxy, most preferably methoxy.
- each occurrence of R 3 is methoxy and R 4 is hydrogen. In other embodiments, each occurrence of R 3 as well as R 4 are methoxy.
- exemplary compounds within the scope of the present invention include the following, and salts thereof: 8-cyclohexyl-2-((4-morpholinophenyl)amino)-7-oxo-7,8- dihydropyrido[2,3-(fJpyrimidine-6-carbonitrile, 8-cyclohexyl-2-((3,5- dimethoxyphenyl)amino)-7-oxo-7,8-dihydropyrido[2,3-(i]pyrimidine-6-carbonitrile, 8- cyclohexyl-7-oxo-2-((3,4,5-1ximethoxyphenyl)amino)-7,8-dihydropyrido[2,3- ii]pyrimidine-6-carbonitrile, 8-cyclopropyl-7-oxo-2-((3,4,5-trimethoxyphenyl)amino)- 7,8-dihydropyrido[2,3- ]pyrimidine-6-carbonit
- the compound of Formula I is an isolated compound.
- the compound of Formula I, and compositions containing the compounds, including pharmaceutical compositions are substantially free of pharmaceutically unacceptable contaminants.
- a pharmaceutically unacceptable contaminant is a substance which, if present in more than an insubstantial amount, would render the compound or composition unsuitable for use as a pharmaceutical for therapeutic administration.
- the present invention provides processes for preparing compounds according to Formula I, intermediates that are useful in the preparation of such compounds, and processes for preparing such intermediates.
- the compounds can be prepared by a variety of synthetic routes. Representative procedures are shown in Schemes 1-5. It will be readily apparent that the compounds can be synthesized by substitution of the appropriate starting materials, reactants, and reagents in the syntheses shown below, with R 1 , R 2 , R 3 , and R 4 defined as previously set forth herein. It will also be apparent that the order of the steps themselves can be changed, depending on the nature of the reactions. Precursor compounds, intermediates, and reagents are commercially available or can be prepared from commercially available starting materials. The following schemes are representative, and are in no way intended to limit the scope of the compounds in the embodiments of the present invention.
- a synthesis of compounds of formula (2) is shown in Scheme 1.
- Compounds of formula (2) can be prepared by reacting a commercially available 4-halopyrimidine carboxylate such as a compound of formula (1) with an amine, R ! -NH 2 , in the presence of a base in a polar or aprotic solvent.
- Useful bases include organic bases, for example, tertiary amines such as diisopropylethylamine (DIPEA) or triethylamine (TEA).
- Useful solvents can include tetrahydrofuran (THF), acetonitrile, ?-dioxane, or ⁇ , ⁇ ,- dimethylformamide (DMF). The reaction can be heated, to the extent necessary, at a temperature appropriate for a given solvent.
- LAH lithium aluminum hydride
- Other useful reducing agents include diisobutylaluminum hydride (DIBAL-H, 2 equivalents), borane-THF complex, and the like.
- Useful solvents include tetrahydrofuran (THF), diethyl ether, and the like.
- the intermediate alcohol can be oxidized to aldehyde (3) using an oxidizing agent such as manganese dioxide in a halogenated solvent.
- oxidizing agent capable of oxidizing an alcohol to an aldehyde, such as for example only, Dess-Martin periodinane, are well known in the art.
- Useful halogenated solvents include dichloromethane, chloroform, and the like.
- ester (2) can be converted directly to aldehyde (3) by treatment with 1 equivalent of DEBAL-H at an appropriate temperature in a solvent such as dichloromethane, THF, or toluene.
- a solvent such as dichloromethane, THF, or toluene.
- a synthesis of compounds according to formula (4) is provided in Scheme 3.
- compounds of formula (3) can be condensed with cyanoacetic acid in acetic acid, to provide a compound of formula (4).
- a catalytic amount of benzylamine can be used in the condensation reaction.
- Temperatures for the condensation reaction can range from about 100 °C to about 120 °C (reflux).
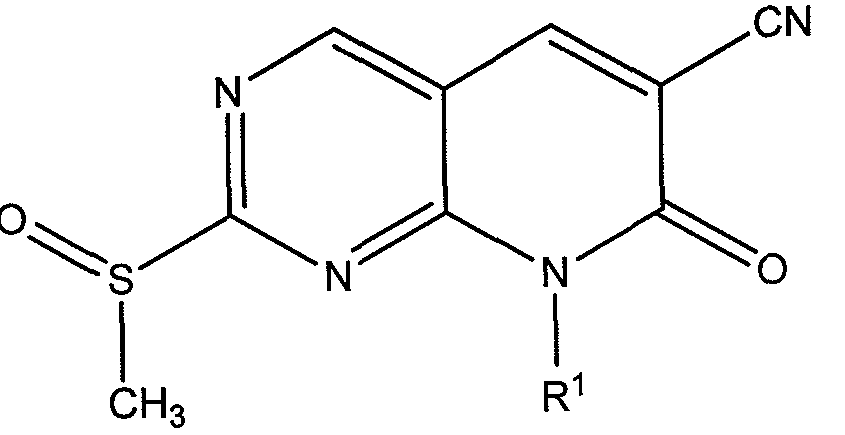
- the compound of formula (4) can be oxidized to a sulfoxide by treating (4) with an oxidizing agent.
- oxidizing agents can include, but are not limited to, meta- chloroperoxybenzoic acid (m-CPBA), hydrogen peroxide, sodium hypochlorite, sodium periodate, tert-butyl hypochlorite, and peracids such as peracetic acid. Stoichiometric use of the oxidizing agent can be employed if necessary to control the oxidation state of sulfur.
- Useful solvents include acetic acid and halogenated solvents such as chloroform or dichloromethane, and the like.
- a preferred oxidizing reagent is m-CPBA in dichloromethane.
- Synthesis of a compound according to Formula I is shown in Scheme 5.
- the compound of Formula (5) can be treated with an amine compound having the formula R 2 - H 2 to provide a compound of Formula I.
- R can be as defined previously herein.
- Exemplary solvents suitable for this reaction include benzenoid solvents such as toluene, o-xylene, m-xylene, 7-xylene, xylene mixtures, anisole, and mixtures thereof.
- Other useful solvents include /?-dioxane, 1,2-dimethoxyethane (DME), THF, and the like.
- Useful temperatures to affect reaction can range from about 65 °C to about 150 °C, dependent upon the solvent used.
- a molar excess of the amine R 2 -NH 2 can be used, including anywhere from about 1.05 to about 2.0 equivalents.
- the above-described reactions are usually conducted at a pressure of about one to about three atmospheres, preferably at ambient pressure (about one atmosphere).
- the present invention further embraces isolated compounds according to Formula I.
- isolated compound refers to a preparation of a compound of Formula I, or a mixture of compounds according to Formula I, wherein the isolated compound has been separated from the reagents used, and/or byproducts formed, during the synthesis of the compound or compounds. "Isolated” does not mean that the preparation is technically pure (homogeneous), but it is sufficiently pure to compound in a form in which it can be used therapeutically.
- an "isolated compound” refers to a preparation of a compound of Formula I or a mixture of compounds according to Formula I, which contains the named compound or mixture of compounds according to Formula I in an amount of at least 10 percent by weight of the total weight.
- the preparation contains the named compound or mixture of compounds in an amount of at least 50 percent by weight of the total weight; more preferably at least 80 percent by weight of the total weight; and most preferably at least 90 percent, at least 95 percent or at least 98 percent by weight of the total weight of the preparation.
- the compounds of the invention and intermediates may be isolated from their reaction mixtures and purified by standard techniques such as filtration, liquid-liquid extraction, solid phase extraction, distillation, recrystallization or chromatography, including flash column chromatography, or HPLC.
- the preferred method for purification of the compounds according to Formula I or salts thereof comprises crystallizing the compound or salt from a solvent to form, preferably, a crystalline form of the compounds or salts thereof. Following crystallization, the crystallization solvent is removed by a process other than evaporation, for example filtration or decanting, and the crystals are then preferably washed using pure solvent (or a mixture of pure solvents).
- Preferred solvents for crystallization include water, alcohols, particularly alcohols containing up to four carbon atoms such as methanol, ethanol, isopropanol, and butan-l-ol, butan-2-ol, and 2-methyl-2-propanol, ethers, for example diethyl ether, diisopropyl ether, t-butyl methyl ether, 1,2-dimethoxyethane, tetrahydrofuran and 1,4-dioxane, carboxylic acids, for example formic acid and acetic acid, and hydrocarbon solvents, for example pentane, hexane, toluene, and mixtures thereof, particularly aqueous mixtures such as aqueous ethanol.
- alcohols particularly alcohols containing up to four carbon atoms such as methanol, ethanol, isopropanol, and butan-l-ol, butan-2-ol, and 2-methyl-2-propanol
- ethers
- the compounds of the invention according to Formula I or salt thereof, and pharmaceutical compositions thereof are preferably in or prepared from a crystalline form, preferably prepared according to such a process.
- the synthetic methods described above reflect a convergent synthesis strategy.
- two components may be synthesized and elaborated separately prior to condensing or coupling the two components to form the target compounds.
- These convergent synthetic schemes allow for arrangement of the assembly steps of the backbone of the target compounds and derivatization of derivatizable functionalities to accommodate functional group sensitivity and/or to allow for functional groups or elements to be introduced either before or after the assembly of the backbone of the target compounds via the condensation or coupling reactions described.
- aromatic substituents in the compounds of the invention, intermediates used in the processes described above, or precursors thereto may be introduced by employing aromatic substitution reactions to introduce or replace a substituent, or by using functional group transformations to modify an existing substituent, or a combination thereof.
- aromatic substitution reactions may be effected either prior to or immediately following the processes mentioned above, and are included as part of the process aspect of the invention.
- the reagents and reaction conditions for such procedures are known in the art.
- procedures which may be employed include, but are not limited to, electrophilic functionalization of an aromatic ring, for example via nitration, halogenation, or acylation; transformation of a nitro group to an amino group, for example via reduction, such as by catalytic hydrogenation; acylation, alkylation, or sulfonylation of an amino or hydroxyl group; replacement of an amino group by another functional group via conversion to an intermediate diazonium salt followed by nucleophilic or free radical substitution of the diazonium salt; or replacement of a halogen by another group, for example via nucleophilic or organometallically-catalyzed substitution reactions.
- a protecting group is a derivative of a chemical functional group which would otherwise be incompatible with the conditions required to perform a particular reaction which, after the reaction has been carried out, can be removed to re-generate the original functional group, which is thereby considered to have been "protected".
- Any chemical functionality that is a structural component of any of the reagents used to synthesize compounds of this invention may be optionally protected with a chemical protecting group if such a protecting group is useful in the synthesis of compounds of this invention.
- sensitive functional groups may be introduced as synthetic precursors to the functional group desired in the intermediate or final product.
- An example of this is an aromatic nitro (-N0 2 ) group.
- the aromatic nitro group does not undergo any of the nucleophilic reactions of an aromatic amino group.
- the nitro group can serve as the equivalent of a protected amino group because it is readily reduced to the amino group under mild conditions that are selective for the nitro group over most other functional groups.
- a method of treating an individual suffering from a cellular proliferative disorder, particularly cancer comprising administering to said individual an effective amount of at least one compound according to Formula I, or a pharmaceutically acceptable salt thereof, either alone, or in combination with a pharmaceutically acceptable carrier.
- a method of inducing apoptosis of cancer cells, preferably tumor cells, in an individual afflicted with cancer comprising administering to said individual an effective amount of at least one compound according to Formula I, or a pharmaceutically acceptable salt thereof, either alone, or in combination with a pharmaceutically acceptable carrier.
- the invention is also directed to the use in medicine of a compound according to
- the invention is also directed to compounds of Formula I, and pharmaceutically acceptable salts thereof, for treating a proliferative disorder, or for inducing apoptosis of tumor cells.
- the invention is also directed to a medicament comprising a compound of
- Formula I for use in treating a proliferative disorder, or for inducing apoptosis of tumor cells.
- the invention is also directed to the use of a compound according to Formula I, or a pharmaceutically acceptable salt thereof in the preparation of a medicament for treatment of a cellular proliferative disorder, particularly cancer, or for inducing apoptosis of tumor cells in an individual affected with cancer.
- Particular and preferred embodiments of this aspect of the invention are those wherein the compound of Formula I used in the method of treatment, either alone or as part of a composition, is a particular or preferred embodiment of the compound of Formula I in the description of the compounds and compositions of the invention as provided herein.
- the compounds according to the invention may be administered to individuals (mammals, including animals and humans) afflicted with a cellular proliferative disorder such as cancer, malignant and benign tumors, blood vessel proliferative disorders, autoimmune disorders, and fibrotic disorders.
- a cellular proliferative disorder such as cancer, malignant and benign tumors, blood vessel proliferative disorders, autoimmune disorders, and fibrotic disorders.
- the individual treated is a human.
- the compounds are believed effective against a broad range of tumor types, including but not limited to the following: ovarian cancer; cervical cancer; breast cancer; prostate cancer; testicular cancer, lung cancer, renal cancer; colorectal cancer; skin cancer; brain cancer; leukemia, including acute myeloid leukemia, chronic myeloid leukemia, acute lymphoid leukemia, and chronic lymphoid leukemia.
- cancers that may be treated by the compounds, compositions and methods of the invention include, but are not limited to, the following:
- cardiac cancers including, for example sarcoma, e.g., angiosarcoma, fibrosarcoma, rhabdomyosarcoma, and liposarcoma; myxoma; rhabdomyoma; fibroma; lipoma and teratoma;
- sarcoma e.g., angiosarcoma, fibrosarcoma, rhabdomyosarcoma, and liposarcoma
- myxoma rhabdomyoma
- fibroma fibroma
- sarcoma e.g., angiosarcoma, fibrosarcoma, rhabdomyosarcoma, and liposarcoma
- myxoma rhabdomyoma
- fibroma fibroma
- lipoma and teratoma teratoma
- lung cancers including, for example, bronchogenic carcinoma, e.g., squamous cell, undifferentiated small cell, undifferentiated large cell, and adenocarcinoma; alveolar and bronchiolar carcinoma; bronchial adenoma; sarcoma; lymphoma; chondromatous hamartoma; and mesothelioma;
- bronchogenic carcinoma e.g., squamous cell, undifferentiated small cell, undifferentiated large cell, and adenocarcinoma
- alveolar and bronchiolar carcinoma bronchial adenoma
- sarcoma sarcoma
- lymphoma chondromatous hamartoma
- mesothelioma mesothelioma
- cancers of the esophagus e.g., squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, and lymphoma
- cancers of the stomach e.g., carcinoma, lymphoma, and leiomyosarcoma
- cancers of the pancreas e.g., ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, and vipoma
- cancers of the small bowel e.g., adenocarcinoma, lymphoma, carcinoid tumors, Kaposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, and fibroma
- cancers of the large bowel e.g., adenocarcinoma, tubular adenoma, villous ade
- liver cancers including, for example, hepatoma, e.g., hepatocellular carcinoma; cholangiocarcinoma; hepatoblastoma; angiosarcoma; hepatocellular adenoma; and hemangioma;
- hepatoma e.g., hepatocellular carcinoma
- cholangiocarcinoma e.g., hepatocellular carcinoma
- hepatoblastoma hepatoblastoma
- angiosarcoma hepatocellular adenoma
- hemangioma hemangioma
- bone cancers including, for example, osteogenic sarcoma (osteosarcoma), fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma), multiple myeloma, malignant giant cell tumor chordoma, osteochrondroma (osteocartilaginous exostoses), benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma and giant cell tumors;
- osteogenic sarcoma osteosarcoma
- fibrosarcoma malignant fibrous histiocytoma
- chondrosarcoma chondrosarcoma
- Ewing's sarcoma malignant lymphoma (reticulum cell sarcoma)
- multiple myeloma malignant giant cell tumor chordoma
- nervous system cancers including, for example, cancers of the skull, e.g., osteoma, hemangioma, granuloma, xanthoma, and osteitis deformans; cancers of the meninges, e.g., meningioma, meningiosarcoma, and gliomatosis; cancers of the brain, e.g., astrocytoma, medulloblastoma, glioma, ependymoma, germinoma
- cancers of the skull e.g., osteoma, hemangioma, granuloma, xanthoma, and osteitis deformans
- cancers of the meninges e.g., meningioma, meningiosarcoma, and gliomatosis
- cancers of the brain e.g., astrocytoma, medulloblastoma,
- spinaloma glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, and congenital tumors; and cancers of the spinal cord, e.g., neurofibroma, meningioma, glioma, and sarcoma;
- gynecological cancers including, for example, cancers of the uterus, e.g., endometrial carcinoma; cancers of the cervix, e.g., cervical carcinoma, and pre-tumor cervical dysplasia; cancers of the ovaries, e.g., ovarian carcinoma, including serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma, cone-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, and malignant teratoma; cancers of the vulva, e.g., squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, and melanoma; cancers of the vagina, e.g., clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma, and embryonal rhabdomyosarcoma; and cancers of
- hematologic cancers including, for example, cancers of the blood, e.g., acute myeloid leukemia, chronic myeloid leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, and myelodysplastic syndrome, Hodgkin's lymphoma, non-Hodgkin's lymphoma (malignant lymphoma) and Waldenstrom's macroglobulinemia;
- skin cancers including, for example, malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and
- adrenal gland cancers including, for example, neuroblastoma.
- Cancers may be solid tumors that may or may not be metastatic. Cancers may also occur, as in leukemia, as a diffuse tissue. Thus, the term "tumor cell”, as provided herein, includes a cell afflicted by any one of the above identified disorders.
- the compounds are also believed useful in the treatment of non-cancer cellular proliferative disorders, that is, cellular proliferative disorders which are characterized by benign indications. Such disorders may also be known as "cytoproliferative” or “hyperproliferative” in that cells are made by the body at an atypically elevated rate.
- Non-cancer cellular proliferative disorders believed treatable by compounds according to the invention include, for example: hemangiomatosis in newborn, secondary progressive multiple sclerosis, atherosclerosis, chronic progressive myelodegenerative disease, neurofibromatosis, ganglioneuromatosis, keloid formation, Paget' s disease of the bone, fibrocystic disease of the breast, uterine fibroids, Peyronie's disease, Dupuytren's disease, restenosis, benign proliferative breast disease, benign prostatic hyperplasia, X-linked lymphocellular proliferative disorder (Duncan disease), post-transplantation lymphocellular proliferative disorder (PTLD), macular degeneration, and retinopathies, such as diabetic retinopathies and proliferative vitreoretinopathy (PVR)
- hemangiomatosis in newborn secondary progressive multiple sclerosis, atherosclerosis, chronic progressive myelodegenerative disease, neurofibromatosis, gang
- non-cancer cellular proliferative disorders believed treatable by compounds according to the invention include the presence of pre-cancerous lymphoproliferative cells associated with an elevated risk of progression to a cancerous disorder.
- Many non-cancerous lymphocellular proliferative disorders are associated with latent viral infections such as Epstein-Barr virus (EBV) and Hepatitis C. These disorders often begin as a benign pathology and progress into lymphoid neoplasia as a function of time.
- EBV Epstein-Barr virus
- Hepatitis C Hepatitis C
- the compounds of the present invention may take the form of salts.
- salts embraces addition salts of free acids or free bases which are compounds of the invention.
- pharmaceutically-acceptable salt refers to salts which possess toxicity profiles within a range that affords utility in pharmaceutical applications. Pharmaceutically unacceptable salts may nonetheless possess properties such as high crystallinity, which have utility in the practice of the present invention, such as for example utility in process of synthesis, purification or formulation of compounds of the invention.
- Suitable pharmaceutically-acceptable acid addition salts may be prepared from an inorganic acid or from an organic acid.
- inorganic acids include hydrochloric, hydrobromic, hydriodic, nitric, carbonic, sulfuric, and phosphoric acids.
- Appropriate organic acids may be selected from aliphatic, cycloaliphatic, aromatic, araliphatic, heterocyclic, carboxylic and sulfonic classes of organic acids, examples of which include formic, acetic, propionic, succinic, glycolic, gluconic, lactic, malic, tartaric, citric, ascorbic, glucuronic, maleic, fumaric, pyruvic, aspartic, glutamic, benzoic, anthranilic, 4-hydroxybenzoic, phenylacetic, mandelic, embonic (pamoic), methanesulfonic, ethanesulfonic, benzenesulfonic, pantothenic, trifiuoroacetic, trifluoromethanesulfonic, 2-hydroxyethanesulfonic, p-toluenesulfonic, sulfanilic, cyclohexylaminosulfonic, stearic, alginic
- All of these salts may be prepared by conventional means from the corresponding compound according to Formula I and the appropriate acid.
- the salts are in crystalline form, and preferably prepared by crystallization of the salt from a suitable solvent.
- suitable salt forms for example, as described in Handbook of Pharmaceutical Salts: Properties, Selection, and Use By P. H. Stahl and C. G. Wermuth (Wiley-VCH 2002).
- the compounds of the invention may be administered in the form of a pharmaceutical composition, in combination with a pharmaceutically acceptable carrier.
- the active ingredient in such formulations may comprise from 0.1 to 99.99 weight percent.
- “Pharmaceutically acceptable carrier” means any carrier, diluent or excipient which is compatible with the other ingredients of the formulation and not deleterious to the recipient.
- the active agent is preferably administered with a pharmaceutically acceptable carrier selected on the basis of the selected route of administration and standard pharmaceutical practice.
- the active agent may be formulated into dosage forms according to standard practices in the field of pharmaceutical preparations. See Alphonso Gennaro, ed., Remington 's Pharmaceutical Sciences, 18 th Edition (1990), Mack Publishing Co., Easton, PA. Suitable dosage forms may comprise, for example, tablets, capsules, solutions, parenteral solutions, troches, suppositories, or suspensions.
- the active agent may be mixed with a suitable carrier or diluent such as water, an oil (particularly a vegetable oil), ethanol, saline solution, aqueous dextrose (glucose) and related sugar solutions, glycerol, or a glycol such as propylene glycol or polyethylene glycol.
- Solutions for parenteral administration preferably contain a water soluble salt of the active agent.
- Stabilizing agents, antioxidant agents and preservatives may also be added. Suitable antioxidant agents include sulfite, ascorbic acid, citric acid and its salts, and sodium EDTA. Suitable preservatives include benzalkonium chloride, methyl- or propyl-paraben, and chlorbutanol.
- the composition for parenteral administration may take the form of an aqueous or non-aqueous solution, dispersion, suspension or emulsion.
- the active agent may be combined with one or more solid inactive ingredients for the preparation of tablets, capsules, pills, powders, granules or other suitable oral dosage forms.
- the active agent may be combined with at least one excipient such as fillers, binders, humectants, disintegrating agents, solution retarders, absorption accelerators, wetting agents absorbents or lubricating agents.
- the active agent may be combined with carboxymethylcellulose calcium, magnesium stearate, mannitol and starch, and then formed into tablets by conventional tableting methods.
- the specific dose of a compound according to the invention to obtain therapeutic benefit for treatment of a cellular proliferative disorder will, of course, be determined by the particular circumstances of the individual patient including the size, weight, age and sex of the patient, the nature and stage of the cellular proliferative disorder, the aggressiveness of the cellular proliferative disorder, and the route of administration of the compound.
- a daily dosage from about 0.05 to about 50 mg/kg/day may be utilized, more preferably from about 0.1 to about 10 mg kg day. Higher or lower doses are also contemplated as it may be necessary to use dosages outside these ranges in some cases.
- the daily dosage may be divided, such as being divided equally into two to four times per day daily dosing.
- the compositions are preferably formulated in a unit dosage form, each dosage containing from about 1 to about 500mg, more typically, about 10 to about lOOmg of active agent per unit dosage.
- unit dosage form refers to physically discrete units suitable as a unitary dosage for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient.
- compositions of the present invention may also be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydropropylmethyl cellulose in varying proportions to provide the desired release profile, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes and/or microspheres.
- a controlled-release preparation is a pharmaceutical composition capable of releasing the active ingredient at the required rate to maintain constant pharmacological activity for a desirable period of time.
- dosage forms provide a supply of a drug to the body during a predetermined period of time and thus maintain drag levels in the therapeutic range for longer periods of time than conventional non-controlled formulations.
- U.S. Patent No. 5,674,533 discloses controlled-release pharmaceutical compositions in liquid dosage forms for the administration of moguisteine, a potent peripheral antitussive.
- U.S. Patent No. 5,059,595 describes the controlled-release of active agents by the use of a gastro-resistant tablet for the therapy of organic mental disturbances.
- U.S. Patent No. 5,591,767 describes a liquid reservoir transdermal patch for the controlled administration of ketorolac, a non-steroidal anti-inflammatory agent with potent analgesic properties.
- U.S. Patent No. 5,120,548 discloses a controlled-release drug delivery device comprised of swellable polymers.
- U.S. Patent No. 5,639,476 discloses a stable solid controlled-release formulation having a coating derived from an aqueous dispersion of a hydrophobic acrylic polymer. Biodegradable microp articles are known for use in controlled-release formulations.
- U.S. Patent No. 5,354,566 discloses a controlled-release powder that contains the active ingredient.
- U.S. Patent No. 5,733,566 describes the use of polymeric microparticles that release antiparasitic compositions.
- the controlled-release of the active ingredient may be stimulated by various inducers, for example pH, temperature, enzymes, water, or other physiological conditions or compounds.
- various mechanisms of drug release exist.
- the controlled-release component may swell and form porous openings large enough to release the active ingredient after administration to a patient.
- the term "controlled-release component" in the context of the present invention is defined herein as a compound or compounds, such as polymers, polymer matrices, gels, permeable membranes, liposomes and/or microspheres, that facilitate the controlled-release of the active ingredient in the pharmaceutical composition.
- the controlled-release component is biodegradable, induced by exposure to the aqueous environment, pH, temperature, or enzymes in the body.
- sol-gels may be used, wherein the active ingredient is incorporated into a sol-gel matrix that is a solid at room temperature.
- This matrix is implanted into a patient, preferably a mammal, having a body temperature high enough to induce gel formation of the sol-gel matrix, thereby releasing the active ingredient into the patient.
- the components used to formulate the pharmaceutical compositions are of high purity and are substantially free of potentially harmful contaminants (e.g., at least National Food grade, generally at least analytical grade, and more typically at least pharmaceutical grade).
- the composition is preferably manufactured or formulated under Good Manufacturing Practice standards as defined in the applicable regulations of the U.S. Food and Drug Administration.
- suitable formulations may be sterile and/or substantially isotonic and/or in full compliance with all Good Manufacturing Practice regulations of the U.S. Food and Drug Administration. VII. Routes of Administration of Compounds and Compositions of the Invention
- the compounds may be administered by any route, including but not limited to oral, rectal, sublingual, buccal, ocular, pulmonary, and parenteral administration, or as an oral or nasal spray (e.g. inhalation of nebulized vapors, droplets, or solid particles).
- Parenteral administration includes, for example, intravenous, intramuscular, intraarterial, intraperitoneal, intranasal, intravaginal, intravesical (e.g., to the bladder), intradermal, transdermal, topical or subcutaneous administration.
- parenteral administration includes, for example, intravenous, intramuscular, intraarterial, intraperitoneal, intranasal, intravaginal, intravesical (e.g., to the bladder), intradermal, transdermal, topical or subcutaneous administration.
- the instillation of a drug in the body of the patient in a controlled formulation with systemic or local release of the drag to occur at a later time.
- the drug may be localized in
- One or more compounds useful in the practice of the present inventions may be administered simultaneously, by the same or different routes, or at different times during treatment.
- the compounds may be administered before, along with, or after other medications, including other antiproliferative compounds.
- the treatment may be carried out for as long a period as necessary, either in a single, uninterrupted session, or in discrete sessions.
- the treating physician will know how to increase, decrease, or interrupt treatment based on patient response.
- treatment is carried out for from about four to about sixteen weeks.
- the treatment schedule may be repeated as required. VIII. Examples
- Lithium aluminum hydride (lOg, 35.5 mmol) was suspended in THF under a nitrogen atmosphere and cooled with dry ice.
- the reaction was subsequently brought to room temperature and stirred for 5 h. After stirring, the reaction was quenched by the addition of water (5 ml), 15% NaOH (10 ml) and then water (15ml) again.
- a white solid that precipitated was filtered away and the filtrate evaporated in vacuo to provide product (8) as a yellow solid (7.2 g).
- Product (8) was used without further purification or characterization.
- Example 7 Cytotoxicity Assay For 8-cyclopentyl-2-((4-(4-methylpiperaziii-l- yl)phenyl)amino)-7-oxo-7.i8-dihvdropyrido[2,3- ⁇ inpyrimidine-6-carboiiitrile
- kinase buffer 25 mM HEPES pH 7.5, 10 mM MgCl 2 , 0.5 mM EGTA, 0.5 mM Na 3 V0 4 , 5 mM ⁇ Glycerophosphate, 2.5 mM DTT, 0.01% Triton X-100
- kinase reactions were then initiated by the addition of (3.28 ⁇ ) CHKtide substrate peptide (Upstate 12-414), 1 ⁇ ATP and 10 ⁇ ⁇ 32 P-ATP. The reactions were incubated at 30°C for 10 minutes.
- Example 9 In vivo efficacy of 8-cvclopentyl-2-((4-(4-methylpiperazin-l- yl)phenyl)ammo)-7-oxo-7,8-dihvdropyridof2J- ⁇ /1pyrimidiiie-6-carboiiitrile against human colon cancer.
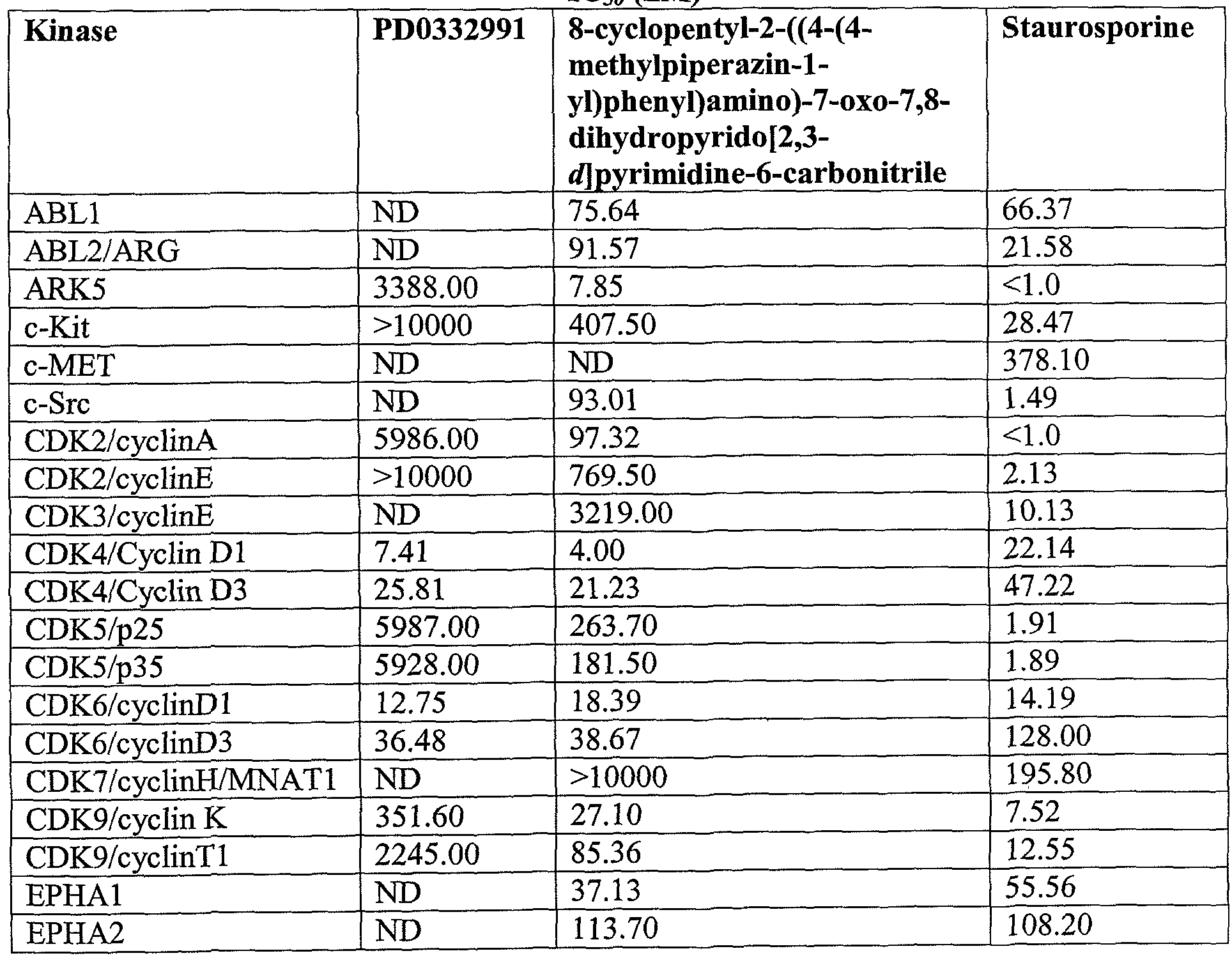
- Example 10 Comparison of Kinase Inhibition of 8-cyclopentyl-2-((4-(4- methylpiperazin-l-yl)phenyl)amino)-7-oxo-7,8-dihydropyrido[2,3- ⁇ /1pyrimidine-6- carbonitrile to Kinase Inhibition of PD0332991
- PD0332991 has the structure:
- PD0332991 is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity.
- CDK pyridopyrimidine-derived cyclin-dependent kinase
- PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin Dl kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation, which prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the Gl phase). This results in suppression of DNA replication and decreased tumor cell proliferation.
- the compounds in Table 4 were tested for the ability to inhibit the kinase activity of the listed protein kinases.
- Compounds were tested in 5 dose IC5 0 mode with 10-fold serial dilution starting at 10 ⁇ .
- Staurosporine a known protein kinase inhibitor, was tested in 5 dose IC 5 0 mode with 3-fold serial dilution starting at 20 ⁇ . Reactions were carried out in 10 ⁇ ATP.
- 8-cyclopentyl-2-((4-(4-methylpiperazin-l- yl)phenyl)amino)-7-oxo-7,8-dihydropyrido[2,3- ⁇ pyrimidine-6-carbonitrile has a significantly different protein kinase inhibition profile as compared to PD0332991.
- the compound of the invention is a multi-specific protein kinase inhibitor, being inhibitory against a broader range of protein kinases than PD0332991. All references cited herein are incorporated by reference in their entirety.
- the present invention may be embodied in other specific forms without departing from the spirit or essential attributes thereof and, accordingly, reference should be made to the appended claims, rather than to the foregoing specification, as indicating the scope of the invention.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Epidemiology (AREA)
- Ophthalmology & Optometry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Vascular Medicine (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description



























Claims








Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2807498A CA2807498C (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carbonitriles and uses thereof |
| AU2011286282A AU2011286282B2 (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof |
| EA201390199A EA022527B1 (en) | 2010-08-05 | 2011-07-21 | 2-SUBSTITUTED-8-ALKYL-7-OXO-7,8-DIHYDROPYRIDO[2,3-d]PYRIMIDINE-6-CARBONITRILES AND USES THEREOF |
| NZ606281A NZ606281A (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carbonitriles and uses thereof |
| KR1020137004979A KR101434841B1 (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof |
| MX2013001427A MX2013001427A (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof. |
| JP2013523185A JP5512894B2 (en) | 2010-08-05 | 2011-07-21 | 2-Substituted-8-alkyl-7-oxo-7,8-dihydropyrido [2,3d] pyrimidine-6-carbonitrile and uses thereof |
| DK11815012.7T DK2600719T3 (en) | 2010-08-05 | 2011-07-21 | 2-Substituted 8-alkyl-7-oxo-7,8-dihydropyrido [2,3-d] pyrimidine-6-carbon nitriles and uses thereof |
| ES11815012.7T ES2525866T3 (en) | 2010-08-05 | 2011-07-21 | 8-alkyl-7-oxo-7,8-dihydropyrid [2,3-d] pyrimidine-6-carbonitriles substituted in 2 and uses thereof |
| BR112013002375-9A BR112013002375B1 (en) | 2010-08-05 | 2011-07-21 | COMPOUND, SAME PREPARATION PROCESS, PHARMACEUTICAL COMPOSITION, AND, USES OF A COMPOUND |
| CN201180038325.3A CN103200822B (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof |
| PL11815012T PL2600719T3 (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carbonitriles and uses thereof |
| EP11815012.7A EP2600719B1 (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carbonitriles and uses thereof |
| US13/813,023 US8987267B2 (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-OXO-7,8-dihydropyrido[2,3-D]pyrimidine-6-carbonitriles and uses thereof in treating proliferative disorders |
| IL224555A IL224555A (en) | 2010-08-05 | 2013-02-03 | 2-(primary amino)-8-(alkyl/cycloalkyl)-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles, process for their preparation and pharmaceutical compositions comprising them |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37094610P | 2010-08-05 | 2010-08-05 | |
| US61/370,946 | 2010-08-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012018540A1 true WO2012018540A1 (en) | 2012-02-09 |
Family
ID=45559737
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/044807 WO2012018540A1 (en) | 2010-08-05 | 2011-07-21 | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US8987267B2 (en) |
| EP (1) | EP2600719B1 (en) |
| JP (1) | JP5512894B2 (en) |
| KR (1) | KR101434841B1 (en) |
| CN (1) | CN103200822B (en) |
| AU (1) | AU2011286282B2 (en) |
| BR (1) | BR112013002375B1 (en) |
| CA (1) | CA2807498C (en) |
| DK (1) | DK2600719T3 (en) |
| EA (1) | EA022527B1 (en) |
| ES (1) | ES2525866T3 (en) |
| IL (1) | IL224555A (en) |
| MX (1) | MX2013001427A (en) |
| NZ (1) | NZ606281A (en) |
| PL (1) | PL2600719T3 (en) |
| PT (1) | PT2600719E (en) |
| WO (1) | WO2012018540A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130131058A1 (en) * | 2010-08-05 | 2013-05-23 | E. Premkumar Reddy | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof |
| WO2014031571A1 (en) * | 2012-08-21 | 2014-02-27 | Icahn School Of Medicine At Mount Sinai | Treatment of viral infections |
| CN104447743A (en) * | 2014-11-26 | 2015-03-25 | 苏州明锐医药科技有限公司 | Preparation method for palbociclib |
| CN104447739A (en) * | 2014-11-07 | 2015-03-25 | 郑州泰基鸿诺药物科技有限公司 | Deuterated palbociclib derivative, and preparation method and application thereof |
| CN104478874A (en) * | 2014-12-08 | 2015-04-01 | 新发药业有限公司 | Preparation method of palbociclib |
| CN104610254A (en) * | 2015-01-26 | 2015-05-13 | 新发药业有限公司 | Low-cost preparation method for palbociclib |
| US9815847B2 (en) | 2013-03-14 | 2017-11-14 | Icahn School Of Medicine At Mount Sinai | Pyrimidine compounds as kinase inhibitors |
| WO2018108167A1 (en) | 2016-12-16 | 2018-06-21 | 基石药业 | Cdk4/6 inhibitor |
| US10005778B2 (en) | 2014-11-26 | 2018-06-26 | Suzhou Miracpharma Technology Co., Ltd. | Method for preparing Palbociclib |
| WO2020101638A1 (en) * | 2018-11-12 | 2020-05-22 | Onconova Therapeutics, Inc. | 8-cyclopentyl-7-oxo-2-(4-piperazin-1-yl-phenylamino)-7, 8-dihydro-pyrido [2,3-d]pyrimidine-6-carbonitrile and uses thereof in treating proliferative disorders |
| WO2020140053A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| WO2020140055A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| US11174252B2 (en) | 2018-02-15 | 2021-11-16 | Nuvation Bio Inc. | Heterocyclic compounds as kinase inhibitors |
| US11697648B2 (en) | 2019-11-26 | 2023-07-11 | Theravance Biopharma R&D Ip, Llc | Fused pyrimidine pyridinone compounds as JAK inhibitors |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3125920B1 (en) | 2014-04-04 | 2020-12-23 | Del Mar Pharmaceuticals | Dianhydrogalactitol, diacetyldianhydrogalactitol or dibromodulcitol to treat non-small-cell carcinoma of the lung and ovarian cancer |
| EP3172214B1 (en) | 2014-07-26 | 2020-05-13 | Sunshine Lake Pharma Co., Ltd. | 2-amino-pyrido[2,3-d]pyrimidin-7(8h)-one derivatives as cdk inhibitors and uses thereof |
| CN105111205B (en) * | 2015-09-12 | 2017-01-04 | 山东罗欣药业集团股份有限公司 | A kind of preparation method of Pa Boxini |
| KR102286701B1 (en) * | 2018-09-28 | 2021-08-06 | 재단법인 대구경북첨단의료산업진흥재단 | Pyrido[2,3-d]pyrimidine derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating protein kinase related disease as an active ingredient |
| TWI818930B (en) * | 2018-11-12 | 2023-10-21 | 美商昂可諾法治療股份有限公司 | 8-cyclopentyl-7-oxo-2-(4-piperazin-1-yl-phenyl amino)-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile and uses thereof in treating proliferative disorders |
| JP2022539259A (en) * | 2019-07-02 | 2022-09-07 | ニューベイション・バイオ・インコーポレイテッド | Heterocyclic compounds as kinase inhibitors |
| WO2021242770A1 (en) * | 2020-05-28 | 2021-12-02 | University Of Washington | Drug-like molecules and methods for the therapeutic targeting of viral rna structures |
| CA3237696A1 (en) | 2021-11-08 | 2023-05-11 | Progentos Therapeutics, Inc. | Platelet-derived growth factor receptor (pdgfr) alpha inhibitors and uses thereof |
| WO2023091724A1 (en) * | 2021-11-18 | 2023-05-25 | Onconova Therapeutics, Inc. | Methods and compositions for treating cancer |
| WO2023092104A1 (en) * | 2021-11-18 | 2023-05-25 | Onconova Therapeutics, Inc. | Methods and compositions for treating cancer |
| WO2023230288A1 (en) * | 2022-05-25 | 2023-11-30 | Onconova Therapeutics, Inc. | Methods and compositions for treating cancer |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20010027196A1 (en) * | 2000-02-25 | 2001-10-04 | Borroni Edilio Maurizio | Adenosine receptor ligands and their use in the treatment of disease |
| US20080004285A1 (en) * | 2004-12-30 | 2008-01-03 | De Jonghe Steven C A | Pyrido(3,2-d)pyrimidines and pharmaceutical compositions useful for medical treatment |
| US20090062274A1 (en) * | 2005-10-07 | 2009-03-05 | Exelixis, Inc | Pyridopyrimidinone inhibitors of pi3kalpha |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5620981A (en) | 1995-05-03 | 1997-04-15 | Warner-Lambert Company | Pyrido [2,3-D]pyrimidines for inhibiting protein tyrosine kinase mediated cellular proliferation |
| US6498163B1 (en) * | 1997-02-05 | 2002-12-24 | Warner-Lambert Company | Pyrido[2,3-D]pyrimidines and 4-aminopyrimidines as inhibitors of cellular proliferation |
| EP1080092B1 (en) * | 1998-05-26 | 2008-07-23 | Warner-Lambert Company LLC | Bicyclic pyrimidines and bicyclic 3,4-dihydropyrimidines as inhibitors of cellular proliferation |
| JP2003523358A (en) | 2000-01-27 | 2003-08-05 | ワーナー−ランバート・カンパニー、リミテッド、ライアビリティ、カンパニー | Pyridopyrimidinone derivatives for the treatment of neurodegenerative diseases |
| IL151480A0 (en) | 2000-03-06 | 2003-04-10 | Warner Lambert Co | 5-alkylpyrido[2,3-d] pyrimidines tyrosine kinase inhibitors |
| CN101001857B (en) | 2002-01-22 | 2011-06-22 | 沃尼尔·朗伯有限责任公司 | 2-(pyridin-2-ylamino)-pyrido [2,3-d]pyrimidin-7-ones |
| JP2007523151A (en) | 2004-02-18 | 2007-08-16 | ワーナー−ランバート カンパニー リミテッド ライアビリティー カンパニー | 2- (Pyridin-3-ylamino) -pyrido [2,3-d] pyrimidin-7-one |
| WO2005105097A2 (en) | 2004-04-28 | 2005-11-10 | Gpc Biotech Ag | Pyridopyrimidines for treating inflammatory and other diseases |
| US20060142312A1 (en) | 2004-12-23 | 2006-06-29 | Pfizer Inc | C6-aryl and heteroaryl substituted pyrido[2,3-D] pyrimidin-7-ones |
| JP2008526704A (en) * | 2004-12-30 | 2008-07-24 | 4・アー・ゼット・アー・アイ・ピー・ナムローゼ・フエンノートシャップ | Pyrido (3,2-d) pyrimidine and pharmaceutical composition useful for medical treatment |
| US8232278B2 (en) * | 2005-06-24 | 2012-07-31 | Gilead Sciences, Inc. | Pyrido(3,2-D)pyrimidines and pharmaceutical compositions useful for treating hepatitis C |
| WO2007044813A1 (en) * | 2005-10-07 | 2007-04-19 | Exelixis, Inc. | PYRIDOPYRIMIDINONE INHIBITORS OF PI3Kα |
| US20090191193A1 (en) | 2006-08-02 | 2009-07-30 | Temple University-Of The Commonwealth System Of Higher Education | Aryl Vinyl Sulfides, Sulfones, Sulfoxides and Sulfonamides, Derivatives Thereof and Therapeutic Uses Thereof |
| JP2010506902A (en) | 2006-10-16 | 2010-03-04 | ジーピーシー・バイオテック・インコーポレーテッド | Pyrido [2,3-D] pyrimidines and their use as kinase inhibitors |
| WO2008150260A1 (en) | 2007-06-06 | 2008-12-11 | Gpc Biotech, Inc. | 8-oxy-2-aminopyrido (2, 3-d) pyrimidin-7-one derivatives as kinase inhibitors and anticancer agents |
| CA2784749C (en) | 2009-12-18 | 2017-12-12 | E. Premkumar Reddy | Substituted pyrido[2,3-d]pyrimidin-7(8h)-ones and therapeutic uses thereof |
| EA022527B1 (en) * | 2010-08-05 | 2016-01-29 | Темпл Юниверсити - Оф Дзе Коммонвелт Систем Оф Хайер Эдьюкейшн | 2-SUBSTITUTED-8-ALKYL-7-OXO-7,8-DIHYDROPYRIDO[2,3-d]PYRIMIDINE-6-CARBONITRILES AND USES THEREOF |
-
2011
- 2011-07-21 EA EA201390199A patent/EA022527B1/en not_active IP Right Cessation
- 2011-07-21 MX MX2013001427A patent/MX2013001427A/en active IP Right Grant
- 2011-07-21 EP EP11815012.7A patent/EP2600719B1/en active Active
- 2011-07-21 ES ES11815012.7T patent/ES2525866T3/en active Active
- 2011-07-21 WO PCT/US2011/044807 patent/WO2012018540A1/en active Search and Examination
- 2011-07-21 CA CA2807498A patent/CA2807498C/en active Active
- 2011-07-21 DK DK11815012.7T patent/DK2600719T3/en active
- 2011-07-21 PT PT118150127T patent/PT2600719E/en unknown
- 2011-07-21 JP JP2013523185A patent/JP5512894B2/en active Active
- 2011-07-21 AU AU2011286282A patent/AU2011286282B2/en active Active
- 2011-07-21 CN CN201180038325.3A patent/CN103200822B/en active Active
- 2011-07-21 US US13/813,023 patent/US8987267B2/en active Active
- 2011-07-21 PL PL11815012T patent/PL2600719T3/en unknown
- 2011-07-21 BR BR112013002375-9A patent/BR112013002375B1/en active IP Right Grant
- 2011-07-21 KR KR1020137004979A patent/KR101434841B1/en active IP Right Grant
- 2011-07-21 NZ NZ606281A patent/NZ606281A/en unknown
-
2013
- 2013-02-03 IL IL224555A patent/IL224555A/en active IP Right Grant
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20010027196A1 (en) * | 2000-02-25 | 2001-10-04 | Borroni Edilio Maurizio | Adenosine receptor ligands and their use in the treatment of disease |
| US20080004285A1 (en) * | 2004-12-30 | 2008-01-03 | De Jonghe Steven C A | Pyrido(3,2-d)pyrimidines and pharmaceutical compositions useful for medical treatment |
| US20090062274A1 (en) * | 2005-10-07 | 2009-03-05 | Exelixis, Inc | Pyridopyrimidinone inhibitors of pi3kalpha |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2600719A4 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130131058A1 (en) * | 2010-08-05 | 2013-05-23 | E. Premkumar Reddy | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine-6-carbonitriles and uses thereof |
| US8987267B2 (en) * | 2010-08-05 | 2015-03-24 | Temple University—Of the Commonwealth System of Higher Education | 2-substituted-8-alkyl-7-OXO-7,8-dihydropyrido[2,3-D]pyrimidine-6-carbonitriles and uses thereof in treating proliferative disorders |
| WO2014031571A1 (en) * | 2012-08-21 | 2014-02-27 | Icahn School Of Medicine At Mount Sinai | Treatment of viral infections |
| US9815847B2 (en) | 2013-03-14 | 2017-11-14 | Icahn School Of Medicine At Mount Sinai | Pyrimidine compounds as kinase inhibitors |
| CN104447739A (en) * | 2014-11-07 | 2015-03-25 | 郑州泰基鸿诺药物科技有限公司 | Deuterated palbociclib derivative, and preparation method and application thereof |
| US10005778B2 (en) | 2014-11-26 | 2018-06-26 | Suzhou Miracpharma Technology Co., Ltd. | Method for preparing Palbociclib |
| CN104447743B (en) * | 2014-11-26 | 2016-03-02 | 苏州明锐医药科技有限公司 | The preparation method of Pa Boxini |
| CN104447743A (en) * | 2014-11-26 | 2015-03-25 | 苏州明锐医药科技有限公司 | Preparation method for palbociclib |
| US9850244B2 (en) | 2014-11-26 | 2017-12-26 | Suzhou Miracpharma Technology Co., Ltd. | Method for preparing Palbociclib |
| CN104478874A (en) * | 2014-12-08 | 2015-04-01 | 新发药业有限公司 | Preparation method of palbociclib |
| CN104610254A (en) * | 2015-01-26 | 2015-05-13 | 新发药业有限公司 | Low-cost preparation method for palbociclib |
| WO2018108167A1 (en) | 2016-12-16 | 2018-06-21 | 基石药业 | Cdk4/6 inhibitor |
| US10676474B2 (en) | 2016-12-16 | 2020-06-09 | Cstone Pharmaceuticals | 1,6-naphthyridine derivatives as CDK4/6 inhibitor |
| US11174252B2 (en) | 2018-02-15 | 2021-11-16 | Nuvation Bio Inc. | Heterocyclic compounds as kinase inhibitors |
| WO2020101638A1 (en) * | 2018-11-12 | 2020-05-22 | Onconova Therapeutics, Inc. | 8-cyclopentyl-7-oxo-2-(4-piperazin-1-yl-phenylamino)-7, 8-dihydro-pyrido [2,3-d]pyrimidine-6-carbonitrile and uses thereof in treating proliferative disorders |
| WO2020140053A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| WO2020140055A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| US11697648B2 (en) | 2019-11-26 | 2023-07-11 | Theravance Biopharma R&D Ip, Llc | Fused pyrimidine pyridinone compounds as JAK inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2011286282B2 (en) | 2014-08-21 |
| EP2600719B1 (en) | 2014-10-08 |
| EA201390199A1 (en) | 2013-07-30 |
| JP2013538196A (en) | 2013-10-10 |
| PT2600719E (en) | 2014-12-22 |
| EP2600719A4 (en) | 2014-01-01 |
| US20130131058A1 (en) | 2013-05-23 |
| IL224555A (en) | 2016-03-31 |
| KR20130098314A (en) | 2013-09-04 |
| CN103200822A (en) | 2013-07-10 |
| BR112013002375A2 (en) | 2016-05-24 |
| CA2807498C (en) | 2017-02-07 |
| AU2011286282A1 (en) | 2013-02-14 |
| US8987267B2 (en) | 2015-03-24 |
| MX2013001427A (en) | 2013-06-13 |
| PL2600719T3 (en) | 2015-03-31 |
| CA2807498A1 (en) | 2012-02-09 |
| CN103200822B (en) | 2014-12-24 |
| BR112013002375B1 (en) | 2020-05-12 |
| NZ606281A (en) | 2014-09-26 |
| EP2600719A1 (en) | 2013-06-12 |
| EA022527B1 (en) | 2016-01-29 |
| ES2525866T3 (en) | 2014-12-30 |
| JP5512894B2 (en) | 2014-06-04 |
| DK2600719T3 (en) | 2014-12-15 |
| KR101434841B1 (en) | 2014-08-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2807498C (en) | 2-substituted-8-alkyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carbonitriles and uses thereof | |
| AU2015335783B2 (en) | Tricyclic atropisomer compounds | |
| KR20190104632A (en) | Tricyclic gyrase inhibitors | |
| WO2016192131A1 (en) | Kinase inhibitor and application thereof | |
| ES2260300T3 (en) | TETRACICLIC COMPOUNDS REPLACED WITH AMINO USEFUL AS ANTI-INFLAMMATORY AGENTS AND PHARMACEUTICAL COMPOSITIONS THAT INCLUDE THE SAME. | |
| JP3734180B2 (en) | New pyrazole derivatives | |
| WO2023284537A1 (en) | Kras g12d inhibitors and uses thereof | |
| US8889696B2 (en) | Substituted pyrido[2,3-d]pyrimidin-7(8H)-ones and therapeutic uses thereof | |
| TW201300390A (en) | Quinoxaline compounds | |
| EP3328862A1 (en) | Novel fused pyrimidinone and triazinone derivatives, their process of preparation and their therapeutic uses as antifungal and/or antiparasitic agents. | |
| WO2017181974A1 (en) | Five-membered heterocyclic compound, preparation method therefor, pharmaceutical composition and use | |
| EP3880206A1 (en) | 8-cyclopentyl-7-oxo-2-(4-piperazin-1-yl-phenylamino)-7, 8-dihydro-pyrido [2,3-d]pyrimidine-6-carbonitrile and uses thereof in treating proliferative disorders | |
| JP2010504943A (en) | Pyridoxazepine progesterone receptor modulator | |
| AU2010330863B2 (en) | Substituted pyrido[2,3-d]pyrimidin-7(8H)-ones and therapeutic uses thereof | |
| WO2013142010A1 (en) | 3-AMINOTHIENO[3,2-c]QUINOLINE DERIVATIVES, METHODS OF PREPARATION AND USES | |
| WO2017181973A1 (en) | 5-member heterocycle and manufacturing method, pharmaceutical composition, and application thereof | |
| CN114380841A (en) | Tricyclic or dihydroquinazoline compounds as AKT inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11815012 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13813023 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2013523185 Country of ref document: JP Kind code of ref document: A Ref document number: 2807498 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2013/001427 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011815012 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2011286282 Country of ref document: AU Date of ref document: 20110721 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20137004979 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201390199 Country of ref document: EA |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013002375 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013002375 Country of ref document: BR Kind code of ref document: A2 Effective date: 20130130 |