WO2012003544A1 - Protein kinase inhibitors and methods of treatment - Google Patents
Protein kinase inhibitors and methods of treatment Download PDFInfo
- Publication number
- WO2012003544A1 WO2012003544A1 PCT/AU2011/000858 AU2011000858W WO2012003544A1 WO 2012003544 A1 WO2012003544 A1 WO 2012003544A1 AU 2011000858 W AU2011000858 W AU 2011000858W WO 2012003544 A1 WO2012003544 A1 WO 2012003544A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- alkyl
- compound
- aryl
- heteroaryl
- Prior art date
Links
- 0 CC(C)(C*(*)*)[n](c1ncnc(*)c11)nc1-c1ccc(C)cc1 Chemical compound CC(C)(C*(*)*)[n](c1ncnc(*)c11)nc1-c1ccc(C)cc1 0.000 description 9
- URUOLZORJBGMRA-UHFFFAOYSA-N CC(C)(C)OC(N/C(/NCC(C)(C)[n]1nc(Cc2cccc(C)c2)c2c1ncnc2N)=N/C(OC(C)(C)C)=O)=O Chemical compound CC(C)(C)OC(N/C(/NCC(C)(C)[n]1nc(Cc2cccc(C)c2)c2c1ncnc2N)=N/C(OC(C)(C)C)=O)=O URUOLZORJBGMRA-UHFFFAOYSA-N 0.000 description 1
- UGSHQEQQIXCUEO-UHFFFAOYSA-N CC(C)(C)OC(NCCc(cc1)ccc1C(NCC(C)(C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1)=O)=O Chemical compound CC(C)(C)OC(NCCc(cc1)ccc1C(NCC(C)(C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1)=O)=O UGSHQEQQIXCUEO-UHFFFAOYSA-N 0.000 description 1
- FMPJUMRHWYNLBS-UHFFFAOYSA-N CC(C)(C)OC(c1nccc(Br)c1)=O Chemical compound CC(C)(C)OC(c1nccc(Br)c1)=O FMPJUMRHWYNLBS-UHFFFAOYSA-N 0.000 description 1
- VJTWIJOULVGPTA-UHFFFAOYSA-N CC(C)(CN(C)C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 Chemical compound CC(C)(CN(C)C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 VJTWIJOULVGPTA-UHFFFAOYSA-N 0.000 description 1
- SFFNNCMHCGICCV-UHFFFAOYSA-N CC(C)(CN)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 Chemical compound CC(C)(CN)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 SFFNNCMHCGICCV-UHFFFAOYSA-N 0.000 description 1
- RYNXNYCMFWUOQZ-UHFFFAOYSA-N CC(C)(CN/C(/c1c[s]cn1)=[O]\N=C(/c1c2N)\N)[n]2nc1-c1ccc(C)cc1 Chemical compound CC(C)(CN/C(/c1c[s]cn1)=[O]\N=C(/c1c2N)\N)[n]2nc1-c1ccc(C)cc1 RYNXNYCMFWUOQZ-UHFFFAOYSA-N 0.000 description 1
- PDMIAOAHLKGAOY-UHFFFAOYSA-N CC(C)(CNC(C(F)(F)F)=O)[n](c1c2c(N)ncn1)nc2-c1ccc(C)cc1 Chemical compound CC(C)(CNC(C(F)(F)F)=O)[n](c1c2c(N)ncn1)nc2-c1ccc(C)cc1 PDMIAOAHLKGAOY-UHFFFAOYSA-N 0.000 description 1
- YJZKRIBZXFRMRJ-UHFFFAOYSA-N CC(C)(CNC(C)=O)[n](c1c2c(N)ncn1)nc2-c1ccc(C)cc1 Chemical compound CC(C)(CNC(C)=O)[n](c1c2c(N)ncn1)nc2-c1ccc(C)cc1 YJZKRIBZXFRMRJ-UHFFFAOYSA-N 0.000 description 1
- DHPKZXOKDDNCSE-UHFFFAOYSA-N CC(C)(CNC(Cc1ccccc1)=O)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 Chemical compound CC(C)(CNC(Cc1ccccc1)=O)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 DHPKZXOKDDNCSE-UHFFFAOYSA-N 0.000 description 1
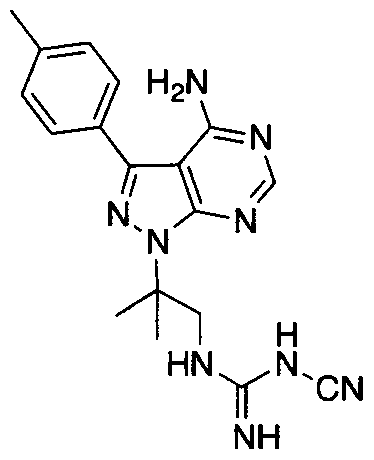
- OYBAMUHPZHMXLV-UHFFFAOYSA-N CC(C)(CNC(N)=N)[n]1nc(Cc2cccc(C)c2)c2c1ncnc2N Chemical compound CC(C)(CNC(N)=N)[n]1nc(Cc2cccc(C)c2)c2c1ncnc2N OYBAMUHPZHMXLV-UHFFFAOYSA-N 0.000 description 1
- NRKHAQWQYXPVCF-UHFFFAOYSA-N CC(C)(CNC(N)=S)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 Chemical compound CC(C)(CNC(N)=S)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 NRKHAQWQYXPVCF-UHFFFAOYSA-N 0.000 description 1
- QYKQQJRJRNFMGH-UHFFFAOYSA-N CC(C)(CNC(NC(c1ccccc1)=O)=S)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 Chemical compound CC(C)(CNC(NC(c1ccccc1)=O)=S)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 QYKQQJRJRNFMGH-UHFFFAOYSA-N 0.000 description 1
- ZEGDULMZHJLAQA-UHFFFAOYSA-O CC(C)(CNC(NC)=[NH2+])[n](c1c2c(N)ncn1)nc2-c1ccc(C)cc1 Chemical compound CC(C)(CNC(NC)=[NH2+])[n](c1c2c(N)ncn1)nc2-c1ccc(C)cc1 ZEGDULMZHJLAQA-UHFFFAOYSA-O 0.000 description 1
- ZWPZMWFJFFTDMS-UHFFFAOYSA-N CC(C)(CNC(OCc1ccccc1)=O)[n](c(N)c1C#N)nc1-c(cc1)ccc1Cl Chemical compound CC(C)(CNC(OCc1ccccc1)=O)[n](c(N)c1C#N)nc1-c(cc1)ccc1Cl ZWPZMWFJFFTDMS-UHFFFAOYSA-N 0.000 description 1
- KRJBBVLZFONYGV-UHFFFAOYSA-N CC(C)NCC(C)(C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 Chemical compound CC(C)NCC(C)(C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1 KRJBBVLZFONYGV-UHFFFAOYSA-N 0.000 description 1
- RBEYSOFJRQYEJC-UHFFFAOYSA-N CCNC(NCC(C)(C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1)=O Chemical compound CCNC(NCC(C)(C)[n](c1ncnc(N)c11)nc1-c1ccc(C)cc1)=O RBEYSOFJRQYEJC-UHFFFAOYSA-N 0.000 description 1
- RSNVAKYRWIAILY-UHFFFAOYSA-N COC(c(cc1)ccc1Cl)=C(C#N)C#N Chemical compound COC(c(cc1)ccc1Cl)=C(C#N)C#N RSNVAKYRWIAILY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates generally to chemical compounds and methods for their use and preparation.
- the invention relates to substituted pyrazolo[3,4-d]pyrimidine based compounds which can be used in treating proliferative disorders, use of these compounds in methods of therapy and the manufacture of medicaments as well as compositions containing these compounds.
- Tyrosine protein kinases are able to catalyse the transfer of the terminal phosphate of adenosine triphosphate to tyrosine residues in protein substrates.
- TPK are a subgroup of the larger protein kinase class of enzymes. The enzymes' ability to phosphorylate is an important mechanism in signal transduction for the regulation of cellular activity. Cellular proliferation in thought to rely (at least to some extent) on TKRs. Mutations can cause some TPKs to become constitutively active, and this aberrant activity has been thought to contribute to initiation or progression of proliferative disorders such as cancer.
- SFKs Src family kinases
- SFKs Src family kinases
- SFKs have provided researchers with a better understanding of the mechanism of cancer as a disease state where normally healthy cellular signalling is disrupted.
- SFKs have been observed to play a critical role in cell adhesion, invasion, proliferation, survival and angiogenesis during tumour development.
- SFKs comprise nine family members that share similar structure and function. The nine members are c-Src, Yrk, Yes, Fyn, Fgr, Lyn, Lck, Hck, and Blk. The overexpression or high activation of these SFKs has been observed in many tumours.
- SFKs can interact with tyrosine kinase receptors, such as the EGFR and the VEGF receptor.
- SFKs are thought to affect cell proliferation through the Ras/ERK/MAPK pathway and may regulate gene expression via transcription factors such as STAT molecules.
- SFKs like some other TPKs can also affect cell adhesion and migration.
- the SFKs are thought to act via interaction with integrins, actins, GTPase-activating proteins, scaffold proteins, such as pl30 CAS and paxillin, and kinases such as focal adhesion kinases.
- SFKs have also been shown to regulate angiogenesis via gene expression of angiogenic growth factors, such as VEGF, interleukin 8, and fibroblast growth factor. Due to this recognition and better understanding as to role being played by TPKs in general and SFKs in particular, small-molecule SFK inhibitors are being developed for the treatment of hyperproliferative disorders such as cancer. At this stage however of a number of promising SFK inhibitors (e.g., Bosutinib, AZ0530, and Desatinib as shown below) only Desatinib is approved whereas the others are presently still undergoing clinical trials and as such there is no guarantee that any further acceptable SFK inhibitor (based on the currently recognised compounds) will reach the market.
- SFK inhibitors e.g., Bosutinib, AZ0530, and Desatinib as shown below
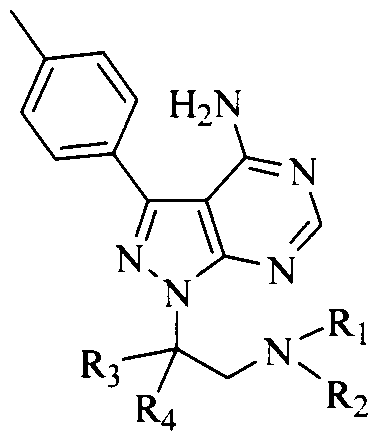
- the invention provides compounds of formula (I) or salts thereof,
- R 2 is selected from hydrogen and C [-C3 alkyl;
- R 3 and R4 are independently C i -C 3 alkyl
- L is selected from a bond, -0-, -S-, -N(R )-, optionally substituted alkylene, - N(R9)C(X')-N(R9')-, where each of R and R9' is independently hydrogen or C 1 -C4 alkyl;
- R 5 is selected from optionally substituted aryl, optionally substituted heteroaryl, optionally substituted heterocyclyl, and optionally substituted cycloalkyl;
- X and X' are independently selected from O, S and NR 7 ;
- R6 is selected from optionally substituted C i-C 6 alkyl, optionally substituted C)-C alkoxy, optionally substituted C 2 -C 6 alkenyl, optionally substituted C 2 -C 6 alkynyl, optionally substituted heteroaryl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted cycloalkenyl, optionally substituted amino, optionally substituted acylamino, optionally substituted arylacyl, optionally substituted heteroarylacyl, optionally substituted heterocyclylacyl, optionally substituted cycloalkylacyl, and trihalomethyl;
- R 7 is selected from hydrogen, cyano, optionally substituted C i-C 6 alkyl, optionally substituted aryl, S(0) 2 Rg, and optionally substituted aminoacyl;
- R 8 is selected from optionally substituted C i -C 6 alkyl and optionally substituted aryl.
- compositions comprising a compound of formula (I) or a salt thereof, together with at least one pharmaceutically acceptable adjuvant, carrier or diluent.
- the invention provides a method of treating a disease or condition characterised by cell proliferation including the step of administering an effective amount of a compound of formula (I) or a salt thereof to a patient in need thereof.
- the invention provides the use of a compound of formula (I) or a salt thereof in the manufacture of a medicament for the treatment of a disease or condition characterised by cell proliferation, including cell hyperproliferation.
- the invention also provides the use of a compound of formula (I) or a salt thereof for the treatment of a disease or condition characterised by cell proliferation, including cell hyperproliferation.
- alkyl refers to monovalent alkyl groups which may be straight chained or branched and preferably have from 1 to 10 carbon atoms, more preferably 1 to 6 carbon atoms and most preferably 1 to 4 carbon atoms. Examples of such alkyl groups include methyl, ethyl, w-propyl, wo-propyl, «-butyl, wobutyl, and the like.
- alkylene refers to divalent alkyl groups.
- alkylene groups examples include methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), and the propylene isomers (e.g., - CH 2 CH 2 CH 2 - and -CH(CH 3 )CH 2 -), and the like.
- aryl refers to an unsaturated aromatic carbocyclic group having a single ring (eg., phenyl) or multiple condensed rings (eg., naphthyl or anthryl), preferably having from 6 to 14 carbon atoms.
- aryl groups include phenyl, naphthyl and the like.
- acyl refers to groups H-C(O)-, alkyl-C(O)-, cycloalkyl-C(O)-, aryl-C(O)-, heteroaryl-C(O)- and heterocyclyl-C(O)-, where alkyl, cycloalkyl, aryl, heteroaryl and heterocyclyl are as described herein.
- alkenyl refers to a monovalent alkenyl groups which may be straight chained or branched and preferably have from 2 to 10 carbon atoms, more preferably 2 to 6 carbon atoms and most preferably 2 to 4 carbon atoms and have at least 1 and preferably from 1-2, carbon to carbon, double bonds.
- alkynyl refers to monovalent alkynyl groups which may be straight chained or branched and preferably have from 2 to 10 carbon atoms, more preferably 2 to 6 carbon atoms and most preferably 2 to 4 carbon atoms and have at least 1 , and preferably from 1-2, carbon to carbon, triple bonds.
- alkynyl groups include ethynyl (-C ⁇ CH), propargyl (-CH 2 C ⁇ CH), pent-2-ynyl (-CH 2 C ⁇ CCH 2 -CH 3 ), and the like.
- amino refers to the group -NR*R* where each R* is independently hydrogen, alkyl, cycloalkyl, aryl, heteroaryl, and heterocyclyl and where each of alkyl, cycloalkyl, aryl, heteroaryl and heterocyclyl is as described herein.
- aminoacyl refers to the group -C(0)NR*R* where each R* is independently hydrogen, alkyl, cycloalkyl and aryl, and where each of alkyl, aryl, and cycloalkyl, is as described herein.
- Acylamino refers to the group -NR*C(0)R* where each R* is independently hydrogen, alkyl, cycloalkyl, aryl, heteroaryl and heterocyclyl and where each of alkyl, cycloalkyl, aryl, heteroaryl, and heterocyclyl are as described herein, and preferably hydrogen and C 1 -C4 alkyl.
- Oxyacylamino refers to the group -NR*C(0)OR* where each R* is independently hydrogen, alkyl, cycloalkyl, aryl, heteroaryl and heterocyclyl and where each of alkyl, cycloalkyl, aryl, heteroaryl, and heterocyclyl are as described herein, and preferably hydrogen and C 1-C4 alkyl.
- cycloalkyl refers to cyclic alkyl groups having a single cyclic ring or multiple condensed rings, preferably incorporating 3 to 8 carbon atoms.
- Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclooctyl, and the like, or multiple ring structures such as adamantanyl, and the like.
- Cycloalkenyl refers to cyclic alkenyl groups having a single cyclic ring and at least one point of internal unsaturation, preferably incorporating 4 to 8 carbon atoms.
- suitable cycloalkenyl groups include, for instance, cyclobut-2-enyl, cyclopent-3-enyl, cyclohex-4-enyl, cyclooct-3-enyl and the like.
- heteroaryl refers to a monovalent aromatic carbocyclic group, preferably of from 2 to 10 carbon atoms and 1 to 4 heteroatoms selected from oxygen, nitrogen and sulfur within the ring. The most preferred heteroatoms are nitrogen, oxygen, and sulfur.
- heteroaryl groups can have a single ring (e.g., pyridyl, pyrrolyl, imididazolyl, thienyl, or furanyl) or multiple condensed rings (e.g., indolizinyl or benzothienyl).
- Heteroarylacyl refers to the group -C(0)heteroaryl where heteroaryl is given the meaning referred to above.
- Heteroarylthio refers to the group -S-heteroaryl wherein the heteroaryl group is as described above.
- Heterocyclyl refers to a monovalent saturated or unsaturated group having a single ring or multiple condensed rings, preferably from 1 to 8 carbon atoms and from 1 to 4 hetero atoms selected from nitrogen, sulfur, oxygen, selenium or phosphorous within the ring. The most preferred heteroatoms are nitrogen and oxygen. Examples of heterocyclyl groups include morpholinyl, piperidinyl and piperazinyl.
- Heterocyclylacyl refers to the group -C(0)heterocyclyl where heterocyclyl is given the meaning referred to above.
- a group may or may not be further substituted or fused (so as to form a condensed polycyclic group) with one or more groups.
- Substituents may be selected from hydroxyl, acyl, alkyl, alkoxy, alkenyl, alkenyloxy, alkynyl, alkynyloxy, amino, aminoacyl, oxyacylamino, thio, arylalkyl, arylalkoxy, aryl, aryloxy, carboxyl, cycloalkyl, cyano, halogen, nitro, sulphate, phosphate, heterocyclyl, heteroaryl, heterocyclyloxy, heteroaryloxy, trihalomethyl, and trialkylsilyl.
- L is -0-, -S-, -N(R.9)-, optionally substituted alkylene (preferably C 1 -C3 alkylene), or -N(R9)C(X')-N(R 9 )-, where each of R9 and is independently hydrogen or C ⁇ - C 4 alkyl.
- L is a bond.
- R 5 is selected from optionally substituted aryl, optionally substituted heteroaryl, optionally substituted heterocyclyl, and optionally substituted cycloalkyl.
- L is a bond and R 5 is an optionally substituted aryl or optionally substituted heteroaryl.
- L is a bond and R 5 is an optionally substituted phenyl.
- L is a bond and R 5 is a phenyl group substituted one to three times with substitutent groups independently selected from halo, hydroxyl, acyl, Ci-C 8 alkyl, C 2 -C6 alkenyl, C 2 -C 6 alkenyloxy, amino, oxyacylamino, C i-C 8 alkoxy, aryl, aryloxy, carboxyl, cycloalkyl, cycloalkyloxy, cyano, sulphate, phosphate, heterocyclyl, heterocyclyloxy, heteroaryl, heteroaryloxy, trihalomethyl, and trialkylsilyl.
- substitutent groups independently selected from halo, hydroxyl, acyl, Ci-C 8 alkyl, C 2 -C6 alkenyl, C 2 -C 6 alkenyloxy, amino, oxyacylamino, C i-C 8 alkoxy, aryl, aryloxy, carboxyl, cycloal
- L is a bond and R 5 is a phenyl group substituted one or two times with substituent groups independently selected from halo, hydroxyl, acyl, C C 8 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkenyloxy, amino, oxyacylamino, Ci-C 8 alkoxy, aryl, aryloxy, carboxyl, cycloalkyl, cycloalkyloxy, cyano, sulphate, phosphate, heterocyclyl, heterocyclyloxy, heteroaryl, heteroaryloxy, trihalomethyl, and trialkylsilyl, and preferably C 1 -C4 alkyl, hydroxy, oxyacylamino, heteroaryl, aryloxy, and halo.
- substituent groups independently selected from halo, hydroxyl, acyl, C C 8 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkenyloxy, amino, oxyacy
- L is a bond and R 5 is a phenyl group substituted with hydroxyl.
- L is a bond and R 5 is a phenyl group substituted with Ci-C 4 alkoxy and - NHC(0)OC ,-C 4 alkyl.
- L is a bond and R 5 is a phenyl group substituted with halo and hydroxy. In an embodiment L is a bond and R 5 is a heteroaryl group.
- L is a bond and R 5 is a phenyl group substituted with aryloxy.
- L is -CH 2 - and R 5 is a phenyl group substituted with halo, hydroxy, and C i- C 3 alkyl.
- L is -CH 2 - and R 5 is a phenyl group substituted with hydroxy.
- -LR5 is .
- L is -CH 2 - and R 5 is a phenyl group substituted with C 1-C3 alkyl.
- -LR5 is In an embodiment, L is -CH 2 - and R 5 is a phenyl group substituted with chloro and C 1 -C4 alkyl. In an embodiment -LR 5 is
- -LR 5 is phenyl
- L is a bond and R 5 is a phenyl group substituted with a substituent group selected from halo, hydroxyl, acyl, C i-C 8 alkyl, C 2 -C6 alkenyl, C 2 -C 6 alkenyloxy, amino, d- C 8 alkoxy, aryl, aryloxy, carboxyl, cycloalkyl, cycloalkyloxy, cyano, sulphate, phosphate, heterocyclyl, trihalomethyl, and trialkylsilyl.
- a substituent group selected from halo, hydroxyl, acyl, C i-C 8 alkyl, C 2 -C6 alkenyl, C 2 -C 6 alkenyloxy, amino, d- C 8 alkoxy, aryl, aryloxy, carboxyl, cycloalkyl, cycloalkyloxy, cyano, sulphate, phosphate, heterocyclyl, trihal
- the invention provides compounds of formula (la) or salts thereof,
- R.2 is selected from hydrogen and C 1 -C3 alkyl;
- R 3 and R4 are independently C 1 -C3 alkyl
- Rio is selected from hydrogen, halo, C 1 -C4 alkyl, C2-C4 alkynyl, C 2 -C 4 alkenyl, arylalkyl, OR 1 (where R 1 is H, C ,-C 3 alkyl or aryl), COOR 2 (where R 2 is H, C 1-C3 alkyl or aryl), nitro, cyano, amino, trihalomethyl, thio, and thio C 1 -C3 alkyl;
- X is selected from O, S and NR 7 ;
- R3 ⁇ 4 is selected from optionally substituted C) -C 6 alkyl, optionally substituted C]-C 6 alkoxy, optionally substituted C 2 -C 6 alkenyl, optionally substituted C 2 -C 6 alkynyl, optionally substituted heteroaryl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted cycloalkenyl, optionally substituted amino, optionally substituted acylamino, optionally substituted arylacyl, optionally substituted heteroarylacyl, optionally substituted heterocyclylacyl, optionally substituted cycloalkylacyl, and trihalomethyl;
- R 7 is selected from hydrogen, cyano, optionally substituted C i-C 6 alkyl, optionally substituted aryl, S(0) 2 R 8 , and optionally substituted aminoacyl;
- R 8 is selected from optionally substituted C i -C 6 alkyl and optionally substituted aryl.
- Rio is C 1 -C4 alkyl or halo.
- R3 and R4 are independently C 1-C3 alkyl, and Rio is C1-C4 alkyl or halo.
- R 2 is selected from hydrogen and C1-C3 alkyl
- R 3 and R4 are independently C1 -C3 alkyl
- Rio is C1 -C4 alkyl or halo
- X is selected from O, S and NR 7 ;
- R6 is selected from optionally substituted C) -C 6 alkyl, optionally substituted C
- R 7 is selected from hydrogen, cyano, acyl, optionally substituted C i -C 6 alkyl, optionally substituted aryl, and optionally substituted aminoacyl;
- R 8 is selected from optionally substituted Ci-C 6 alkyl, and optionally substituted aryl.
- Rio is methyl or chloro. In a further embodiment and with reference to formula (la) or (lb), Rio is methyl.
- R 2 is selected from hydrogen and C 1-C3 alkyl
- R 3 and R4 are independently C 1-C3 alkyl
- X is selected from O, S and NR 7 ;
- Rf is selected from optionally substituted C i -C 6 alkyl, optionally substituted C i-C 6 alkoxy, optionally substituted C 2 -C 6 alkenyl, optionally substituted C 2 -C 6 alkynyl, optionally substituted heteroaryl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted cycloalkenyl, optionally substituted amino, optionally substituted acylamino, optionally substituted arylacyl, optionally substituted heteroarylacyl, optionally substituted heterocyclylacyl, optionally substituted cycloalkylacyl, and trihalomethyl; R 7 is selected from hydrogen, cyano, optionally substituted C i-C 6 alkyl, optionally substituted aryl, S(0) 2 R8, and optionally substituted aminoacyl; and R 8 is selected from optionally substituted C i -C 6 alkyl and optionally substituted aryl
- R 3 and R4 are independently selected from C 1-C2 alkyl.
- R 3 and j are methyl.
- R 2 is selected from hydrogen and C pC 3 alkyl;
- X is selected from O, S and NR 7 ;
- R is selected from optionally substituted C i-C 6 alkyl, optionally substituted Ci -C 6 alkoxy, optionally substituted C 2 -C 6 alkenyl, optionally substituted C 2 -C 6 alkynyl, optionally substituted heteroaryl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted cycloalkenyl, optionally substituted amino, optionally substituted acylamino, optionally substituted arylacyl, optionally substituted heteroarylacyl, optionally substituted heterocyclylacyl, optionally substituted cycloalkylacyl, and trihalomethyl;
- R 7 is selected from hydrogen, cyano, optionally substituted Ci-C 6 alkyl, optionally substituted aryl, S(0) 2 R 8 , and optionally substituted aminoacyl;
- R 8 is selected from optionally substituted Ci-C 6 alkyl and optionally substituted aryl.
- R 2 is hydrogen or methyl.
- R 2 is hydrogen
- X is selected from O, S and NR 7 ;
- R6 is selected from optionally substituted Ci-C 6 alkyl, optionally substituted Ci-C 6 alkoxy, optionally substituted C 2 -C 6 alkenyl, optionally substituted C 2 -C 6 alkynyl, optionally substituted heteroaryl, optionally substituted heterocyclyl, optionally substituted aryl, optionally substituted cycloalkyl, optionally substituted cycloalkenyl, optionally substituted amino, optionally substituted acylamino, optionally substituted arylacyl, optionally substituted heteroarylacyl, optionally substituted heterocyclylacyl, optionally substituted cycloalkylacyl, and trihalomethyl;
- R 7 is selected from hydrogen, cyano, optionally substituted Ci-C 6 alkyl, optionally substituted aryl, S(0) 2 R 8 , and optionally substituted aminoacyl;
- R 8 is selected from optionally substituted Ci-C 6 alkyl and optionally substituted aryl. Still a further embodiment and with reference to formula (I),(Ia), (lb), (Ic), (Id) or (le) Ri is lected from the following preferred groups:
- Ci-C 6 alkyl optionally substituted Ci-C 6 alkyl, and more preferably Ci-C alkyl;
- heteroaryl preferably represents: (i) a 5-membered heteroaryl group selected from pyrrole, 2H-pyrrole, furan, pyrazole, thiophene, isothiazole, thiazole, 1 ,2,3-triazole, 1 ,2,4-triazole, 1 ,2,3-oxadiazole, 1 ,2,5-oxadiazole, 1 ,3,4-thiadiazole, tetrazole, imidazole, oxazole, and isoxazole; or
- heterocyclyl preferably represents:
- a 6-membered heterocyclyl group selected from 2H-pyran, 4H-pyran, 3,4-dihydro- 2H-pyran, piperidine, 1 ,4-oxazine, 1 ,4-dioxine, piperazine, morpholine, 1 -4- dioxane, 1 ,4-thazine, thiomo holine, 1 ,4-oxathane, 1 ,4-dithane, 1 ,3,5-trioxane, 6H- l ,2,5-thiadiazine, 2H- 1 ,5,2-dithiazine, and 1 ,3,5-trithiane; and wherein preferably aryl is selected from phenyl, napthyl and anthracenyl; and where the heteroaryl, heterocyclyl or aryl group may be substituted from 1 to 4 times by the group consisting of hydro xyl, acyl, C 1 -C4
- -C(0)-optionally substituted C i-C alkyl includes: -C(0)-(CH 2 ) n -substituent,
- n is an integer from 1 to 6
- Ri is C(0)R 6 where R6 is defined above.
- the optionally substituted heteroaryl group is selected from optionally substituted pyridyl or optionally substituted thiazolyl.
- Preferred substituents, when present, include -(CH 2 ) n -heterocyclyl, optionally substituted C 1 -C4 alkoxy and optionally substituted phenyl, where n is an integer from 0-4.
- the compounds of the present invention may be prepared by the following general reaction sequence depicted in the Schemes below:
- Step A Addition of the 2,2-disubstituted nitroethene (1) to protected carbazate (2) may be facilitated by mixing the starting materials (in preferably equimolar amounts) in an aqueous solvent system.
- the solvent system is 1 : 1 water/acetonitrile.
- the reaction is conducted at room temperature. The reaction progress can be monitored by layer chromatography (TLC) [e.g., CH 2 Cl2/MeOH 90: 10].
- Step B The reduction of (3) to primary amine (4) may be facilitated by any suitable reducing agent known in the art such as FeCl 3 , and sodium dithionite.
- the reduction may also be facilitated by hydrogenation using pallasium (Pd) or Raney nickel as catalysts. More preferably the reduction process is catalysed by 10% Pd/C in the presence of ammonium formate in a polar protic solvent such as methanol.
- This reaction is preferably conducted at room temperature and the reaction progress may be monitored by TLC (e.g., CH 2 Cl 2 /MeOH 95 :5).
- TLC e.g., CH 2 Cl 2 /MeOH 95 :5
- the crude reaction product may be separated from the catalyst by filtration and used in the next step without any further purification.
- Step C Involves t rotection of the primary amine group.
- Suitable nitrogen protecting groups for are known to those skilled in the art of organic synthesis and include acyl groups (e.g., acetyl, trifluoroacetyl, and benzoyl), acyloxy groups (e.g., benzylester), aryl (e.g., phenyl), alkylaryl (e.g., benzyl), etc.
- Other nitrogen protecting groups may be found in Protective Groups in Organic Synthesis, T.W. Greene and P. W John Wiley & Son, 3 rd edition. In a preferred embodiment the protecting group is t-butyloxy carbonyl (BOC).
- the protecting group is the N-benzyloxy carbonyl group (Cbz). Protection may be facilitated by reacting the amine (4) in acetonitrile with triethylamine and N- (Benzyloxycarbonyloxy)succinimide.
- Step D Involves the deprotection of the ® group of (5) to prepare the salt (6).
- Step E In a preferred embodiment, the reaction product (6) from step D is not purified but instead directly reacted (in situ) with 2-(methoxy-optionally substituted aryl- methylene)-malonitrile (7) to afford the substituted pyrazole (8).
- This cyclisation step may be facilitated with the use of a suitable non-nucleophilic base (such as Hiinigs base, or TEA).
- the reaction is preferably conducted at elevated temperatures (e.g., between 50°-70°C).
- Step F The subsequent ring forming step to prepare the pyrazole-[3,4,d]-pyrimidine (9)
- Step G Involves a deprotection step which may be facilitated by a suitable deprotection agent known in the art.
- suitable deprotection agent known in the art.
- Such agents include those discussed in Protective Groups in Organic Synthesis, T.W. Greene and P. Wutz, John Wiley & Son, 3 rd edition.
- deprotection agents include inorganic and organic acids and accordingly ⁇ ⁇ represents an anion which has been exchanged during the deprotection step using the inorganic or organic acid. Accordingly, depending on the deprotection agents employed in the process, both ⁇ ⁇ and ⁇ ⁇ may be the same or different. Preferably, ⁇ ⁇ and ⁇ ⁇ are different.
- the deprotection agent may be a HBr solution. In this instance, ⁇ ⁇ would be represented as ⁇ ⁇ (bromide).
- the invention provides a process for preparing a compound of formula (8):
- the invention also provides a process for preparing a compound of formula (9): said process comprising the step of: a) reacting a compound of formula (8)
- reaction is conducted at elevated temperatures, preferably between 130°-160°C.
- reaction is conducted at elevated temperatures, using methoxyethanol as solvent.
- the invention also provides a process for preparing a compound of formula (10) said process comprising the step of:
- the deprotection step involves treating a compound of formula (10) with an acid, and preferably an inorganic acid selected from HBr or HC1, more preferably an aqueous solution of HBr.
- the compounds of formula (I) with variable and R 2 groups may be prepared, for instance, using conventional nucleophilic chemistry by initially treating the salt (9) with a suitable non-nucleophilic base (e.g., Hunigs base or TEA) and then reacting with a desired electrophilic group (e.g, a substituted anhydride).
- a suitable non-nucleophilic base e.g., Hunigs base or TEA
- a desired electrophilic group e.g, a substituted anhydride
- the invention also provides a process for preparing a compound of formula (I):
- Step A' Uses hydrazine or salt thereof maybe in presence of base in EtOH at reflux
- Step B' Starting material heated at, for instance, 180°C in formamide or reacted with formamidine acetate in ethoxyethanol at 120°C. Other classical methods of pyrrazolopyrimidine synthesis can be used.
- Step C Starting material and substituted nitroalkene stirred in, for instance, DMF at 95°C for 48 hrs.
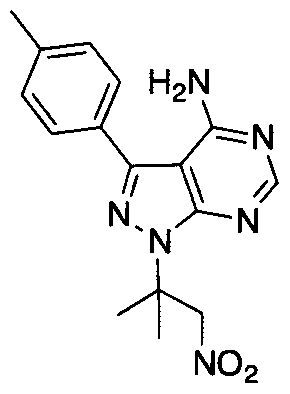
- Step D' Aliphatic nitro reduction methods apply here. Used for this work: Zn, 6M HCl, EtOH or Pd(OH) 2 in MeOH under hydrogen.
- Step A Usual iodination methods. Methods used: N-iodosuccinimide in DMF at 80°C.
- Step B Metal mediated coupling reactions such as Suzuki or Negishi coupling reactions can be applied here.
- Metal mediated coupling reactions such as Suzuki or Negishi coupling reactions can be applied here.
- Step C" and D" As with step C and D' above.
- Another variation is to add, remove or modify the substituents of the product to form new derivatives which fall within the scope of the compounds of the present invention. This could be achieved again by using standard techniques for functional group inter-conversion, well known in the industry such as those described in Comprehensive Organic Transformations: a guide to functional group preparations by Larock R C, New York, VCH Publishers, Inc. 1989.
- Examples of possible functional group inter-conversions are: -C(0)NRR' from -C0 2 CH 3 by heating with or without catalytic metal cyanide, e.g. NaCN, and HNRR' in CH 3 OH; -OC(0)R from -OH with e.g., C1C(0)R' in pyridine; -NR-C(S)NR'R" from -NHR with an alkylisothiocyanate or thiocyanic acid; -NRC(0)OR from -NHR with alkyl chloroformate; - NRC(0)NR'R" from -NHR by treatment with an isocyanate, e.g.
- the preferred compounds of the invention are inhibitors of tyrosine kinase and in particular Src kinase and therefore can be useful in methods of therapy.
- the compounds of the present invention have also been observed to target serine/threonin protein kinases, and in particular RIP 2 and ME 5. As such these compounds may be used for treating tumours.
- tumor is used broadly to define any malignant cancerous growth, and may include leukemias, melanomas, colon, lung, ovarian, skin, breast, prostate, CNS, and renal cancers, as well as other cancers.
- the compounds of the invention having Src kinase inhibitory activity may also be used in the treatment of tumours, and in particular colon cancer.
- the invention also provides for the use of a compound of formula (I), (la), (lb), (Ic), (Id), (Ie) or (If) in the manufacture of a medicament for treating tumours, and in particular colon cancer.
- tumours in particular colon cancer
- a method of treatment of tumours comprising the administration of an effective amount of a compound of formula (I), (la), (lb), (Ic), (Id), (Ie) or (If) to a subject in need thereof.
- the tumour is breast cancer.
- the compounds of the invention can be used in treating diseases or conditions characterised by cell proliferation (including cell hyperproliferation) which is initiated and/or progressed by aberrant TP activity and more particualrly serine/threonine protein kinase and/or Src kinase activity.
- the present compounds can also be used in treating psoriasis, immunoregulation (graft rejection), atherosclerosis, rheumatoid arthrities, acute and chronic inflammatroy conditions, Crohn's disease and the like.
- Known related pyrazolopyrimidines PPl and PP2 (as disclosed in US 5,593,997)
- PPl PP2 have been reported as potent inhibitors of SFKs, althought they do not discriminate between members of this kinase family. They also inhibit other tyrosine kinases such as EGF-R. While such activity would appear to be therapeutically useful, PP1 and PP2 are poorly soluble and accordingly these compounds are not viewed as being "drug like". It has been surprisingly found that the compounds of the present invention have improved solubility and drug-like profiles (especially under acidic conditions) while at the same time retain potent inhibitory acitivity.
- Compounds of the invention which possess bioactivity, such as Src kinase inhibitory activity, can be formulated as a composition, particularly a pharmaceutical composition, together with a pharmaceutically acceptable additive.
- a treatment effective amount is intended to include at least partially attaining the desired effect, or delaying the onset of, or inhibiting the progression of, or halting or reversing altogether the onset or progression of the particular disease of condition being treated (e.g., colon cancer).
- the term "effective amount" relates to an amount of compound which, when administered according to a desired dosing regimen, provides the desired therapeutic activity. Dosing may occur at intervals of minutes, hours, days, weeks, months or years or continuously over any one of these periods. Suitable dosages may lie within the range of about 0.1 ng per kg of body weight to 1 g per kg of body weight per dosage. A typical dosage is in the range of 1 ⁇ g to 1 g per kg of body weight per dosage, such as is in the range of 1 mg to 1 g per kg of body weight per dosage. In one embodiment, the dosage may be in the range of 1 mg to 500 mg per kg of body weight per dosage. In another embodiment, the dosage may be in the range of 1 mg to 250 mg per kg of body weight per dosage. In yet another embodiment, the dosage may be in the range of 1 mg to 100 mg per kg of body weight per dosage, such as up to 50 mg per body weight per dosage.
- Suitable dosage amounts and dosing regimens can be determined by the attending physician and may depend on the particular condition being treated, the severity of the condition as well as the general age, health and weight of the subject.
- the active ingredient may be administered in a single dose or a series of doses. While it is possible for the active ingredient to be administered alone, it is preferable to present it as a composition, preferably as a pharmaceutical composition.
- the formulation of such compositions is well known to those skilled in the art.
- the composition may contain any suitable carriers, diluents or excipients. These include all conventional solvents, dispersion media, fillers, solid carriers, coatings, antifungal and antibacterial agents, dermal penetration agents, surfactants, isotonic and absorption agents and the like. It will be understood that the compositions of the invention may also include other supplementary physiologically active agents.
- compositions include those suitable for oral, rectal, nasal, topical (including buccal and sublingual), vaginal or parental (including subcutaneous, intramuscular, intravenous and intradermal) administration.
- the compositions may conveniently be presented in unit dosage form and may be prepared by any methods well blown in the art of pharmacy. Such methods include the step of bringing into association the active ingredient with the carrier which constitutes one or more accessory ingredients.
- the compositions are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both, and then if necessary shaping the product.
- compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, sachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be presented as a bolus, electuary or paste.
- a tablet may be made by compression or moulding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder (e.g inert diluent, preservative disintegrant (e.g. sodium starch glycolate, cross-linked polyvinyl pyrrolidone, cross-linked sodium carboxymethyl cellulose) surface-active or dispersing agent.
- a binder e.g inert diluent, preservative disintegrant (e.g. sodium starch glycolate, cross-linked polyvinyl pyrrolidone, cross-linked sodium carboxymethyl cellulose) surface-active or dispersing agent.
- Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in varying proportions to provide the desired release profile. Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach.
- compositions suitable for topical administration in the mouth include lozenges comprising the active ingredient in a flavoured base, usually sucrose and acacia or tragacanth gum; pastilles comprising the active ingredient in an inert basis such as gelatine and glycerin, or sucrose and acacia gum; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- compositions suitable for topical administration to the skin may comprise the compounds dissolved or suspended in any suitable carrier or base and may be in the form of lotions, gel, creams, pastes, ointments and the like.
- suitable carriers include mineral oil, propylene glycol, polyoxyethylene, polyoxypropylene, emulsifying wax, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- Transdermal patches may also be used to administer the compounds of the invention.
- compositions for rectal administration may be presented as a suppository with a suitable base comprising, for example, cocoa butter, glycerin, gelatine or polyethylene glycol.
- compositions suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- compositions suitable for parenteral administration include aqueous and non-aqueous isotonic sterile injection solutions which may contain anti-oxidants, buffers, bactericides and solutes which render the composition isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the compositions may be presented in unit-dose or multi-dose sealed containers, for example, ampoules and vials, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Preferred unit dosage compositions are those containing a daily dose or unit, daily sub- dose, as herein above described, or an appropriate fraction thereof, of the active ingredient.
- compositions of this invention may include other agents conventional in the art having regard to the type of composition in question, for example, those suitable for oral administration may include such further agents as binders, sweeteners, thickeners, flavouring agents disintegrating agents, coating agents, preservatives, lubricants and/or time delay agents.
- suitable sweeteners include sucrose, lactose, glucose, aspartame or saccharine.
- Suitable disintegrating agents include cornstarch, methylcellulose, polyvinylpyrrolidone, xanthan guni, bentonite, alginic acid or agar.
- Suitable flavouring agents include peppermint oil, oil of wintergreen, cherry, orange or raspberry flavouring.
- Suitable coating agents include polymers or copolymers of acrylic acid and/or niethacrylic acid and/or their esters, waxes, fatty alcohols, zein, shellac or gluten.
- Suitable preservatives include sodium benzoate, vitamin E, alpha-tocopherol, ascorbic acid, methyl paraben, propyl paraben or sodium bisulphite.
- Suitable lubricants include magnesium stearate, stearic acid, sodium oleate, sodium chloride or talc.
- Suitable time delay agents include glyceryl monostearate or glyceryl distearate.
- novel bioactive compounds of the invention can be administered to a subject as a pharmaceutically acceptable salt thereof. It will be appreciated however that non- pharmaceutically acceptable salts also fall within the scope of the present invention since these may be useful as intermediates in the preparation of pharmaceutically acceptable salts.
- Suitable pharmaceutically acceptable salts include, but are not limited to salts of pharmaceutically acceptable inorganic acids such as hydrochloric, sulphuric, phosphoric, nitric, carbonic, boric, sulfamic, and hydrobromic acids, or salts of pharmaceutically acceptable organic acids such as acetic, propionic, butyric, tartaric, maleic, hydroxymaleic, fumaric, maleic, citric, lactic, mucic, gluconic, benzoic, succinic, oxalic, phenylacetic, methanesulphonic, toluenesulphonic, benezenesulphonic, salicyclic sulphanilic, aspartic, glutamic, edetic, stearic, palmitic, oleic, lauric, pantothenic, tannic, ascorbic and valeric acids.
- pharmaceutically acceptable inorganic acids such as hydrochloric, sulphuric, phosphoric, nitric
- Base salts include, but are not limited to, those formed with pharmaceutically acceptable cations, such as sodium, potassium, lithium, calcium, magnesium, ammonium and alkylammonium.
- the present invention includes within its scope cationic salts eg sodium or potassium salts, or alkyl esters (eg methyl, ethyl) of the phosphate group.
- Basic nitrogen-containing groups may be quartemised with such agents as lower alkyl halide, such as methyl, ethyl, propyl, and butyl chlorides, bromides and iodides; dialkyl sulfates like dimethyl and diethyl sulfate; and others.
- prodrug is used in its broadest sense and encompasses those derivatives that are converted in vivo to the compounds of the invention. Such derivatives would readily occur to those skilled in the art, and include, for example, compounds where a free hydroxy group is converted into an ester, such as an acetate or phosphate ester, or where a free amino group is converted into an amide (eg a-aminoacid amide).
- esterifying, eg. acylating, the compounds of the invention are well known in the art and may include treatment of the compound with an appropriate carboxylic acid, anhydride or chloride in the presence of a suitable catalyst or base.
- the compounds of the invention may be in crystalline form either as the free compounds or as solvates (e.g. hydrates) and it is intended that both forms are within the scope of the present invention.
- Methods of solvation are generally known within the art.
- compounds of the invention may possess asymmetric centres and are therefore capable of existing in more than one stereoisomeric form.
- the invention thus also relates to compounds in substantially pure isomeric form at one or more asymmetric centres eg., greater than about 90% ee, such as about 95% or 97% ee or greater than 99% ee, as well as mixtures, including racemic mixtures, thereof.
- Such isomers may be prepared by asymmetric synthesis, for example using chiral intermediates, or mixtures may be resolved by conventional methods, eg., chromatography, or use of a resolving agent.
- the compounds of the present invention may be capable of undergoing tautomerism. Accordingly, all possible tautomers of a compound of the present invention fall within the scope and spirit of the invention.
- the mode of proliferative diseases is multi-factorial.
- drugs with different mechanisms may be combined (ie combination therapies).
- the compounds of the invention may be particularly useful in combination therapy, eg. combining the treatment with other chemotherapeutic or radiation treatments.
- cytotoxic compounds including 5-FU, oxaliplatin, paclitaxel, gemcitabine, docetaxel, cisplatin, and doxorubicin may also be administered.
- the combination therapy may also include the addition of other Src kinase inhibitors such as AZD- 0530 (Saracatinib), Dasatinib (BMS-354825 or Sprycel) and Bosutinib (S I0606) or inhibitors of E K5, ME 5, RIP 5, and FA (PT 2) such as AZD6244, U0126, SB202190, and PF-562271.
- the combination partners in such therapies may be administered together, one after the other, separately in one combined unit dosage or in separate unit dosage forms.
- Example 1 Preparation of N'-(l ,l -Dimethyl-2-nitro-ethyl)-hydrazinecarboxylic acid tert- butyl ester tert-Butyl carbazate (2.61 g, 19.8 mmol) was added to mixture of 2,2-dimethyl- nitroethylene (2 g, 19.8 mmol) in 20 mL of 1 : 1 water/acetonitrile. After one hour, TLC (CH 2 Cl 2 /MeOH 90: 10) indicated complete reaction. The reaction was diluted with water. The aqueous phase was washed three times with EtOAc. The combined organic phases were rinsed with water and brine, dried over Na 2 S0 4 and concentrated.
- Compound 12 was prepared according to the procedure used to prepare compound from example 1 1 using picolinic acid (22 mg, 0.18 mmol). The off-white solids obtained were triturated with Et 2 0, collected by filtration and rinsed with more Et 2 0. A white powder was obtained (15 mg, 22%).
- Example 42 was prepared according to the procedure described for example 1 1 using 4- bromopicolinic acid (17.9 mg, 88.7 ⁇ ⁇ ). A dark cream solid was obtained ( 18.6 mg, 43%).
- ⁇ NMR (ppm, CDCI3): ⁇ 9.02 (s, 1 H), 8.41 -8.30 (m, 3H), 7.68-7.36 (m, 5H), 5.30 (s, 2H), 4.16 (d, J 6.6 Hz, 1 H), 2.45 (s, 3H), 1.75 (s, 6H).
- LCMS (+esi): 482.0 (M+H + ), RT 7.05 min.
- 69B compound 69A (100 mg, 0.58 mmol) and pipendine (171 /vL, 1.7 mmol) were heated at 1 10°C overnight. 776 L of an 8% aqueous hydrochloric acid solution (1.7 mmol) were then added and the brown solution was stirred at 1 10°C for 2 hours. The solution was then cooled at 0°C and concentrated in vacuo. The residue was dissolved in boiling water and heated at 1 10°C for 5 minutes then kept at -20°C for 1 hour and at 4°C for 72 hours. Filtration afforded compound 69B as brown crystals (18 mg, 9 % yield).
- Triethylamine (696 mg, 6.9 mmol) and compound 74A (300 mg, 1.4 mmol) were successively added. The reaction was then stirred at 60°C for 3 hours. After cooling down, the reaction was concentrated. Water and ethyl acetate were then added and the aqueous phase was extracted three times with ethyl acetate. The combine organic layers were dried over sodium sulphate and concentrated. The oily residue obtained was purified by flash chromatography on Si0 2 using 100% DCM then MeOH/DCM 1 :99 to afford compound 74C as a semi solid (387 mg, 67%).
- 82B in an oven dried Schlenk tube were added palladium acetate (13 mg, 0.02 mmol), butyl di-l -adamantyl phosphine (14 mg, 0.04 mmol), potassium (piperidin-l -yl)- methyltrifluoroborate (0.08 g, 0.4 mmol), compound 82A (0.1 g, 0.4 mmol) and cesium carbonate (0.38 g, 1.2 mmol). The tube was filled with nitrogen and evacuated three times. 2 mL of anhydrous toluene were added and the mixture stirred for 5 minutes, followed by the addition of 200 / L of water. The mixture was heated up to 95°C and left stirring for 24 hours.
- Example 84 84A: Compound 84A was prepared according to the procedure described for the preparation of compound 10A using caproic acid (200 mg, 1.32 mmol). A white solid was obtained (296 mg, 97%).
- 86B compound 86A (20 mg, 0.06 mmol) was dissolved in 2 mL of ethanol. Next, 589 / L of 6M of hydrogen chloride was added followed by zinc dust (60 mg, 0.92 mmol). The reaction was stirred at room temperature for 2 hours. The excess zinc was removed by filtration and the ethanol was concentrated in vacuo. Saturated sodium hydrogen carbonate was added until ph 9 and dichloromethane was added to the aqueous layer and was stirred for 30 minutes. The aqueous layer was further extracted with dichloromethane and the crude compound 86B was obtained. LCMS (+esi): 297 (M+H + ).
- 87D Compound 87C (200 mg, 0.77 mmol), ,P0 (488 mg, 2.30 mmol), 4-phenoxybenzene boronic acid (491 mg, 2.32 mmol) and Tetrakis-(triphenylphosphine)palladium (124 mg, 0.1 1 mmol) were dissolved in 2.5 mL of dioxane in a microwave vial. The vial was sealed and the reaction mixture was heated to 180°C for 10 min under microwave irradiation. The reaction mixture was partitioned between water and ethylacetate and the organic layer was separated, dried over anhydrous Na 2 S0 4 , filtered and concentrated.
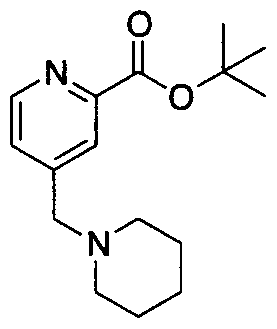
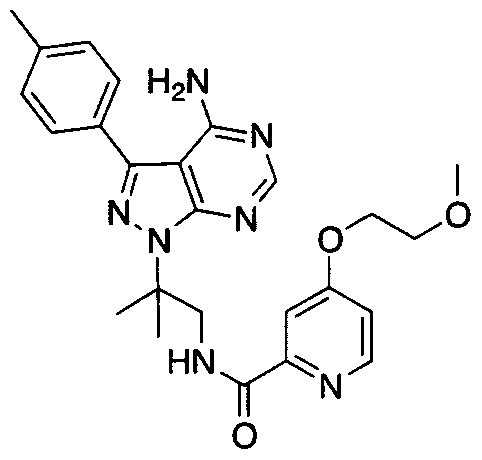
- 89A Sodium (54 mg, 2 mmol) was placed into an oven dried Schlenk tube. 5 mL of anhydrous tetrahydrofuranwere then added followed by 2-methoxy-ethanol (0.18 mL, 2 mmol). A gas evolution occurred. The reaction was stirred at room temperature for 30 minutes. It was then heated to 50°C until all the sodium had disappeared. In another flask 4- chloropicolinic tert-butyl ester (0.5 g, 2 mmol) was dissolved in 5 mL of anhydrous tetrahydrofuran. The solution of the alkoxide was then added to the reaction mixture and the reaction was stirred at reflux for 5 hours. Another solution of alkoxide was prepared and was added to the reaction mixture.
- 89B Compound 89 A (50 g, 0.18 mmol) was dissolved in 1 mL of methanol and 0.5 mL of 2M sodium hydroxide was added. The reaction was stirred at room temperature overnight. The methanol was concentrated down and the aqueous layer was acidified to pH 2 with 2 mL of 1M hydrogen chloride. The aqueous layer was placed on a freeze drier overnight. Compound 89B was used in the next step without further purification.
- 89C Compound 89C was obtained following the procedure described for the preparation of compound 82D using compound I E (67 mg, 0.17 mmol) and compound 89B (37 mg 0.19 mmol). Purification via flash chromatography on Si0 2 using methanol/dichloromethane (2:98) afforded compound 89C as a white solid (54 mg, 67%).
- a biotin labeled peptide was used as substrate (amino acid sequence: Biotin-Glu-Gly-Pro-Trp-
- Src recombinant enzyme was purchased as N-terminally His 6 tagged full-length human protein. The 15 ⁇ L ⁇ assay reactions were run in Greiner brand white 384-well low volume plates.
- All reactions contained 10 mM HEPES pH 7.4, 25 mM NaCl, 10 mM MgCl 2 , 0.01 % (v/v) Tween-20, 50 ⁇ Na 3 V0 4 , 0.01% (w/v) albumin from chicken egg white, 1 1 1 nM peptide substrate, 80 ⁇ ATP, and 0.3 ng/reaction Src enzyme, with the enzyme being omitted from negative control reactions.
- Compounds were added in a volume of 100 nL from dilution series made up in DMSO, positive and negative control reactions receiving the same volume DMSO without compounds. The plates were sealed with adhesive seals and incubated for 90 minutes at 30 degree Celsius. The reactions were stopped with the detection reagents added at the same time.
- Product formation was quantified as photochemiluminescence between PerkinElmer AlphaScreenTM beads, using Streptavidin-coated donor and anti-phosphotyrosine (P-Tyr-100) acceptor beads.
- P-Tyr-100 Streptavidin-coated donor and anti-phosphotyrosine
- 5 ⁇ containing 10 mM HEPES pH 7.4, 25 mM NaCl, lOO mM EDTA, 0.01 % (v/v) Tween-20, and 6.25 ⁇ g/mL of each bead type were added. Plates were incubated for 5 hours before being read on a PerkinElmer EnVisionTM plate reader in HTS AlphascreenTM mode.
- SRC percent inhibition
- LIM 1899 colon carcinoma derived epithelial cells were grown in RPMl + Adds 1 + 10%FCS.
- Assay medium RPMl + Adds + 5% FCS Cells are trypsinized, washed once in assay medium and brought to the required concentration as described below:
- Cells were then diluted to 10 3 cells/mL (need 10 mL minimum per plate).
- Inhibitors were dissolved in DMSO for 10 mM stocks. MW and amount of compound given was used to calculate the volume required for each.
- Rows C,D Inhibitor 2
- Rows E,F Inhibitor 3
- Row H medium was carefully removed from the cell wells with a 200 ⁇ , tip, and replaced with 200 / L serum free medium.
- the compounds in this invention display cellular activities against a range of tumour or transformed cell lines in particular colon cancer cell lines such as LIM 1215, LIM2537 and LIM 1899.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Transplantation (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description








































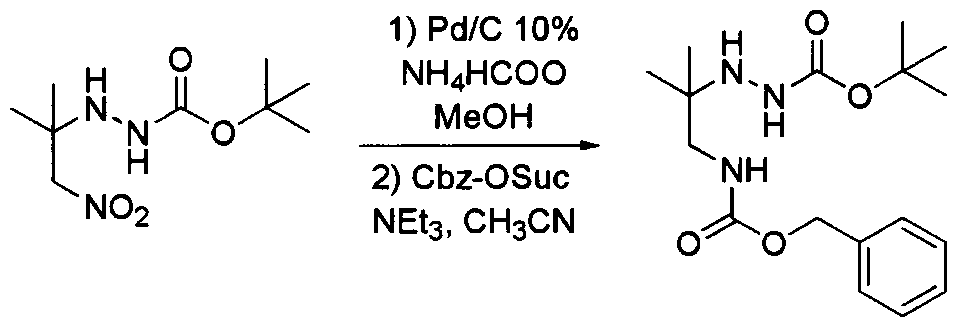








































































































Claims












Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2804648A CA2804648C (en) | 2010-07-09 | 2011-07-08 | Protein kinase inhibitors and methods of treatment |
| AU2011276955A AU2011276955B2 (en) | 2010-07-09 | 2011-07-08 | Protein kinase inhibitors and methods of treatment |
| US13/809,369 US8962830B2 (en) | 2010-07-09 | 2011-07-08 | Protein kinase inhibitors and methods of treatment |
| JP2013516912A JP5810157B2 (en) | 2010-07-09 | 2011-07-08 | Protein kinase inhibitors and methods of treatment |
| EP11803020.4A EP2590982B1 (en) | 2010-07-09 | 2011-07-08 | Protein kinase inhibitors and methods of treatment |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36273910P | 2010-07-09 | 2010-07-09 | |
| US61/362,739 | 2010-07-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012003544A1 true WO2012003544A1 (en) | 2012-01-12 |
Family
ID=45440707
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2011/000858 WO2012003544A1 (en) | 2010-07-09 | 2011-07-08 | Protein kinase inhibitors and methods of treatment |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8962830B2 (en) |
| EP (1) | EP2590982B1 (en) |
| JP (1) | JP5810157B2 (en) |
| AU (1) | AU2011276955B2 (en) |
| CA (1) | CA2804648C (en) |
| WO (1) | WO2012003544A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102977012A (en) * | 2012-12-20 | 2013-03-20 | 江苏弘和药物研发有限公司 | Synthesis method of methyl 4-bromopyridyl-2-formate |
| CN103626774A (en) * | 2013-11-20 | 2014-03-12 | 苏州明锐医药科技有限公司 | Preparation method of Ibrutinib |
| CN103965201A (en) * | 2014-04-30 | 2014-08-06 | 淮海工学院 | Method for synthesizing intermediate 4-amino-3-(4-phenoxy-phenyl)-1H-pyrazolo[3,4-d]pyrimidine of Ibrutinib |
| WO2014139970A1 (en) * | 2013-03-15 | 2014-09-18 | Janssen Pharmaceutica Nv | Processes and intermediates for preparing a medicament |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10196397B2 (en) | 2014-11-19 | 2019-02-05 | Sun Pharmaceutical Industries Limited | Process for the preparation of ibrutinib |
| US10214508B2 (en) | 2014-06-13 | 2019-02-26 | Takeda Pharmaceutical Company Limited | Nitrogen-containing heterocyclic compound |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12065494B2 (en) | 2021-04-12 | 2024-08-20 | Incyte Corporation | Combination therapy comprising an FGFR inhibitor and a Nectin-4 targeting agent |
| US12122767B2 (en) | 2020-09-30 | 2024-10-22 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5593997A (en) | 1995-05-23 | 1997-01-14 | Pfizer Inc. | 4-aminopyrazolo(3-,4-D)pyrimidine and 4-aminopyrazolo-(3,4-D)pyridine tyrosine kinase inhibitors |
| WO2002076986A1 (en) * | 2001-03-22 | 2002-10-03 | Abbott Gmbh & Co. Kg | Pyrazolopyrimidines as therapeutic agents |
| WO2002080926A1 (en) * | 2001-03-22 | 2002-10-17 | Abbott Gmbh & Co. Kg | Pyrazolopyrimidines as therapeutic agents |
| US20070293516A1 (en) | 2006-04-04 | 2007-12-20 | Regents Of The University Of California | Kinase antagonists |
| WO2008039218A2 (en) | 2006-09-22 | 2008-04-03 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2010009342A2 (en) | 2008-07-16 | 2010-01-21 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| WO2011046964A2 (en) * | 2009-10-12 | 2011-04-21 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2687402B1 (en) * | 1992-02-14 | 1995-06-30 | Lipha | NOVEL AZAINDOLES, METHODS OF PREPARATION AND MEDICAMENTS CONTAINING THEM. |
| PT1784396E (en) * | 2004-08-26 | 2011-01-27 | Pfizer | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
-
2011
- 2011-07-08 EP EP11803020.4A patent/EP2590982B1/en not_active Not-in-force
- 2011-07-08 WO PCT/AU2011/000858 patent/WO2012003544A1/en active Application Filing
- 2011-07-08 AU AU2011276955A patent/AU2011276955B2/en not_active Ceased
- 2011-07-08 US US13/809,369 patent/US8962830B2/en not_active Expired - Fee Related
- 2011-07-08 JP JP2013516912A patent/JP5810157B2/en not_active Expired - Fee Related
- 2011-07-08 CA CA2804648A patent/CA2804648C/en not_active Expired - Fee Related
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5593997A (en) | 1995-05-23 | 1997-01-14 | Pfizer Inc. | 4-aminopyrazolo(3-,4-D)pyrimidine and 4-aminopyrazolo-(3,4-D)pyridine tyrosine kinase inhibitors |
| WO2002076986A1 (en) * | 2001-03-22 | 2002-10-03 | Abbott Gmbh & Co. Kg | Pyrazolopyrimidines as therapeutic agents |
| WO2002080926A1 (en) * | 2001-03-22 | 2002-10-17 | Abbott Gmbh & Co. Kg | Pyrazolopyrimidines as therapeutic agents |
| US20070293516A1 (en) | 2006-04-04 | 2007-12-20 | Regents Of The University Of California | Kinase antagonists |
| WO2008039218A2 (en) | 2006-09-22 | 2008-04-03 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2010009342A2 (en) | 2008-07-16 | 2010-01-21 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| WO2011046964A2 (en) * | 2009-10-12 | 2011-04-21 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
Non-Patent Citations (6)
| Title |
|---|
| APSEL ET AL.: "Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases", NATURE CHEMICAL BIOLOGY, vol. 4, no. 11, 2008, pages 691 - 699, XP007909493 * |
| DATABASE CA Database accession no. 2008:1263189 * |
| FEICHTINGER ET AL., J. ORG. CHEM., vol. 63, 1998, pages 8432 |
| JOURNAL OF IMMUNOLOGICAL METHODS, vol. 65, 1983, pages 55 - 63 |
| LAROCK R C: "Comprehensive Organic Transformations: a guide to functional group preparations", 1989, VCH PUBLISHERS, INC. |
| T.W. GREENE; P. WUTZ: "Protective Groups in Organic Synthesis", JOHN WILEY & SON |
Cited By (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10813930B2 (en) | 2010-12-22 | 2020-10-27 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US10213427B2 (en) | 2010-12-22 | 2019-02-26 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11840534B2 (en) | 2012-06-13 | 2023-12-12 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11053246B2 (en) | 2012-06-13 | 2021-07-06 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US10131667B2 (en) | 2012-06-13 | 2018-11-20 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9745311B2 (en) | 2012-08-10 | 2017-08-29 | Incyte Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| CN102977012B (en) * | 2012-12-20 | 2015-06-03 | 江苏弘和药物研发有限公司 | Synthesis method of methyl 4-bromopyridyl-2-formate |
| CN102977012A (en) * | 2012-12-20 | 2013-03-20 | 江苏弘和药物研发有限公司 | Synthesis method of methyl 4-bromopyridyl-2-formate |
| KR20150132172A (en) * | 2013-03-15 | 2015-11-25 | 얀센 파마슈티카 엔.브이. | Processes and intermediates for preparing a medicament |
| CN105026400A (en) * | 2013-03-15 | 2015-11-04 | 詹森药业有限公司 | Processes and intermediates for preparing a medicament |
| WO2014139970A1 (en) * | 2013-03-15 | 2014-09-18 | Janssen Pharmaceutica Nv | Processes and intermediates for preparing a medicament |
| AU2018204086B2 (en) * | 2013-03-15 | 2020-03-12 | Janssen Pharmaceutica Nv | Processes and intermediates for preparing a medicament |
| KR102377688B1 (en) | 2013-03-15 | 2022-03-22 | 얀센 파마슈티카 엔.브이. | Processes and intermediates for preparing a medicament |
| KR102311329B1 (en) * | 2013-03-15 | 2021-10-14 | 얀센 파마슈티카 엔.브이. | Processes and intermediates for preparing a medicament |
| KR20210123429A (en) * | 2013-03-15 | 2021-10-13 | 얀센 파마슈티카 엔.브이. | Processes and intermediates for preparing a medicament |
| US11530214B2 (en) | 2013-04-19 | 2022-12-20 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10040790B2 (en) | 2013-04-19 | 2018-08-07 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10947230B2 (en) | 2013-04-19 | 2021-03-16 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10450313B2 (en) | 2013-04-19 | 2019-10-22 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| CN103626774B (en) * | 2013-11-20 | 2015-11-04 | 苏州明锐医药科技有限公司 | Yi Lu is for the preparation method of Buddhist nun |
| CN103626774A (en) * | 2013-11-20 | 2014-03-12 | 苏州明锐医药科技有限公司 | Preparation method of Ibrutinib |
| CN103965201A (en) * | 2014-04-30 | 2014-08-06 | 淮海工学院 | Method for synthesizing intermediate 4-amino-3-(4-phenoxy-phenyl)-1H-pyrazolo[3,4-d]pyrimidine of Ibrutinib |
| US10214508B2 (en) | 2014-06-13 | 2019-02-26 | Takeda Pharmaceutical Company Limited | Nitrogen-containing heterocyclic compound |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10196397B2 (en) | 2014-11-19 | 2019-02-05 | Sun Pharmaceutical Industries Limited | Process for the preparation of ibrutinib |
| US10632126B2 (en) | 2015-02-20 | 2020-04-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11667635B2 (en) | 2015-02-20 | 2023-06-06 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10251892B2 (en) | 2015-02-20 | 2019-04-09 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11014923B2 (en) | 2015-02-20 | 2021-05-25 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10214528B2 (en) | 2015-02-20 | 2019-02-26 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10016438B2 (en) | 2015-02-20 | 2018-07-10 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11173162B2 (en) | 2015-02-20 | 2021-11-16 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10738048B2 (en) | 2015-02-20 | 2020-08-11 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9801889B2 (en) | 2015-02-20 | 2017-10-31 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US11472801B2 (en) | 2017-05-26 | 2022-10-18 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US12024517B2 (en) | 2018-05-04 | 2024-07-02 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12083124B2 (en) | 2019-10-14 | 2024-09-10 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12122767B2 (en) | 2020-09-30 | 2024-10-22 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12065494B2 (en) | 2021-04-12 | 2024-08-20 | Incyte Corporation | Combination therapy comprising an FGFR inhibitor and a Nectin-4 targeting agent |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2011276955B2 (en) | 2014-11-06 |
| US20130184274A1 (en) | 2013-07-18 |
| EP2590982A1 (en) | 2013-05-15 |
| CA2804648A1 (en) | 2012-01-12 |
| JP5810157B2 (en) | 2015-11-11 |
| US8962830B2 (en) | 2015-02-24 |
| EP2590982A4 (en) | 2014-04-02 |
| EP2590982B1 (en) | 2017-08-23 |
| JP2013529649A (en) | 2013-07-22 |
| CA2804648C (en) | 2019-01-22 |
| AU2011276955A1 (en) | 2013-01-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8962830B2 (en) | Protein kinase inhibitors and methods of treatment | |
| AU2019218186B2 (en) | Urea-substituted aromatic ring-linked dioxinoquinoline compounds, preparation method and uses thereof | |
| AU2019218187B2 (en) | Dioxinoquinoline compounds, preparation method and uses thereof | |
| ES2660215T3 (en) | Antitumor effect enhancer comprising an imidazooxazine compound | |
| ES2968421T3 (en) | Atropisomerism for enhanced kinase inhibitor selectivity | |
| JPWO2005080392A1 (en) | Pyrazoloquinolone derivatives and uses thereof | |
| AU2019372121A1 (en) | Heterocyclic compounds as BET inhibitors | |
| JP2019520367A (en) | Novel Heterocyclic Derivative Compound and Use Thereof | |
| CN114437116A (en) | Heterocyclic compound and preparation method, pharmaceutical composition and application thereof | |
| WO2017191599A1 (en) | Substituted 2, 4-diamino-quinoline derivatives for use in the treatment of proliferative diseases | |
| CN117425648A (en) | Compounds as PARP7 inhibitors | |
| CN112839930B (en) | 3, 9-diazaspiro [5,5] undecanes as FLT3 and AXL inhibitors | |
| CN110167943B (en) | Pyrrolotriazine derivatives as kinase inhibitors | |
| EP3750894B1 (en) | Urea-substituted aromatic ring-linked dioxazoline compound, preparation method therefor, and uses thereof | |
| US20230416221A1 (en) | Dihydroisoquinolinone and isoindolinone derivatives and uses thereof | |
| JP7110335B2 (en) | Pyridoquinazoline derivatives useful as protein kinase inhibitors | |
| US20130085143A1 (en) | Aminoacid derivatives, their process of preparation and their therapeutical uses as inhibitors of oncogenic signals by the met family | |
| EP2578588A1 (en) | Novel 1,4-diazepam pde-5 inhibitor derivatives | |
| KR102516260B1 (en) | Compounds as a TRAP1 selective inhibitor, and composition for preventing or treating cancer comprising the same | |
| CN112209933B (en) | BTK inhibitors containing 4-azacycloheptane | |
| CN117343081A (en) | MAT2A inhibitor, pharmaceutical composition and application thereof | |
| EA043715B1 (en) | COMPOUNDS, COMPOSITIONS AND METHODS FOR MODULATING CDK9 ACTIVITY |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11803020 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013516912 Country of ref document: JP Kind code of ref document: A Ref document number: 2804648 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2011276955 Country of ref document: AU Date of ref document: 20110708 Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011803020 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011803020 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13809369 Country of ref document: US |