WO2011133444A1 - Substituted pyrimidines - Google Patents
Substituted pyrimidines Download PDFInfo
- Publication number
- WO2011133444A1 WO2011133444A1 PCT/US2011/032829 US2011032829W WO2011133444A1 WO 2011133444 A1 WO2011133444 A1 WO 2011133444A1 US 2011032829 W US2011032829 W US 2011032829W WO 2011133444 A1 WO2011133444 A1 WO 2011133444A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydroxy
- pyrazol
- pyrimidin
- alkyl
- compound
- Prior art date
Links
- 150000003230 pyrimidines Chemical class 0.000 title abstract description 5
- 208000007502 anemia Diseases 0.000 claims abstract description 25
- 108010043005 Prolyl Hydroxylases Proteins 0.000 claims abstract description 7
- 102000004079 Prolyl Hydroxylases Human genes 0.000 claims abstract description 7
- 150000001875 compounds Chemical class 0.000 claims description 169
- 125000000217 alkyl group Chemical group 0.000 claims description 128
- -1 trifluoromethoxy, trifluoroethoxy Chemical group 0.000 claims description 93
- 125000001424 substituent group Chemical group 0.000 claims description 71
- 238000000034 method Methods 0.000 claims description 44
- 125000003118 aryl group Chemical group 0.000 claims description 39
- 125000000623 heterocyclic group Chemical group 0.000 claims description 39
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 32
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 31
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 claims description 30
- 150000003839 salts Chemical class 0.000 claims description 29
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 claims description 26
- 150000002367 halogens Chemical group 0.000 claims description 25
- 229910052736 halogen Inorganic materials 0.000 claims description 24
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 20
- 239000001257 hydrogen Substances 0.000 claims description 20
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 19
- 125000003545 alkoxy group Chemical group 0.000 claims description 18
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 18
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 17
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 17
- 125000004644 alkyl sulfinyl group Chemical group 0.000 claims description 16
- 125000001072 heteroaryl group Chemical group 0.000 claims description 16
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 16
- 102000003951 Erythropoietin Human genes 0.000 claims description 15
- 108090000394 Erythropoietin Proteins 0.000 claims description 15
- 229940105423 erythropoietin Drugs 0.000 claims description 15
- 239000012453 solvate Substances 0.000 claims description 15
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 claims description 14
- 238000011282 treatment Methods 0.000 claims description 14
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- 239000003814 drug Substances 0.000 claims description 13
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 13
- 241000124008 Mammalia Species 0.000 claims description 12
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims description 11
- 125000005842 heteroatom Chemical group 0.000 claims description 11
- 125000004076 pyridyl group Chemical group 0.000 claims description 11
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- 125000000304 alkynyl group Chemical group 0.000 claims description 9
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 9
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 claims description 9
- 125000000335 thiazolyl group Chemical group 0.000 claims description 9
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 claims description 8
- 239000004202 carbamide Substances 0.000 claims description 8
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 8
- BXCQRPSSKGRMDD-UHFFFAOYSA-N pyrimidin-5-yl carbamate Chemical compound NC(=O)OC1=CN=CN=C1 BXCQRPSSKGRMDD-UHFFFAOYSA-N 0.000 claims description 8
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 8
- 125000001113 thiadiazolyl group Chemical group 0.000 claims description 8
- 125000001544 thienyl group Chemical group 0.000 claims description 8
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 claims description 8
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 7
- 125000003342 alkenyl group Chemical group 0.000 claims description 7
- 125000004429 atom Chemical group 0.000 claims description 7
- 125000002541 furyl group Chemical group 0.000 claims description 7
- 125000002883 imidazolyl group Chemical group 0.000 claims description 7
- 125000001041 indolyl group Chemical group 0.000 claims description 7
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 claims description 7
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 7
- 125000004043 oxo group Chemical group O=* 0.000 claims description 7
- 125000005010 perfluoroalkyl group Chemical group 0.000 claims description 7
- 230000002265 prevention Effects 0.000 claims description 7
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 claims description 7
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 6
- 125000004104 aryloxy group Chemical group 0.000 claims description 6
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 claims description 6
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 claims description 6
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 6
- 150000002431 hydrogen Chemical group 0.000 claims description 6
- 125000005956 isoquinolyl group Chemical group 0.000 claims description 6
- 125000002971 oxazolyl group Chemical group 0.000 claims description 6
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 6
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 6
- 125000004414 alkyl thio group Chemical group 0.000 claims description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 5
- 230000002708 enhancing effect Effects 0.000 claims description 5
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 5
- 125000001715 oxadiazolyl group Chemical group 0.000 claims description 5
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 claims description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 5
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 4
- USPCABJDZSAIQX-UHFFFAOYSA-N 3-chloro-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC(Cl)=C1 USPCABJDZSAIQX-UHFFFAOYSA-N 0.000 claims description 4
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 claims description 4
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 4
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims description 4
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 claims description 4
- 125000005436 dihydrobenzothiophenyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 claims description 4
- 125000005435 dihydrobenzoxazolyl group Chemical group O1C(NC2=C1C=CC=C2)* 0.000 claims description 4
- 125000005844 heterocyclyloxy group Chemical group 0.000 claims description 4
- UVNXNSUKKOLFBM-UHFFFAOYSA-N imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=CSC2=NC=CN21 UVNXNSUKKOLFBM-UHFFFAOYSA-N 0.000 claims description 4
- 125000005493 quinolyl group Chemical group 0.000 claims description 4
- 125000005958 tetrahydrothienyl group Chemical group 0.000 claims description 4
- 125000005940 1,4-dioxanyl group Chemical group 0.000 claims description 3
- FTUWMMSBJRCOSN-UHFFFAOYSA-N 1-benzhydryl-3-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)thiourea Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=S)NC(C=1C=CC=CC=1)C1=CC=CC=C1 FTUWMMSBJRCOSN-UHFFFAOYSA-N 0.000 claims description 3
- YEEUMCVDURSKGV-UHFFFAOYSA-N 1-benzyl-3-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)urea Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)NCC1=CC=CC=C1 YEEUMCVDURSKGV-UHFFFAOYSA-N 0.000 claims description 3
- TZBPDFHIFDICJU-UHFFFAOYSA-N 2-chloro-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC=C1Cl TZBPDFHIFDICJU-UHFFFAOYSA-N 0.000 claims description 3
- 125000002393 azetidinyl group Chemical group 0.000 claims description 3
- 125000004069 aziridinyl group Chemical group 0.000 claims description 3
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 claims description 3
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000004598 dihydrobenzofuryl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 claims description 3
- FMXYILQDSZVUHP-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)heptanamide Chemical compound N1=C(O)C(NC(=O)CCCCCC)=CN=C1N1N=CC=C1 FMXYILQDSZVUHP-UHFFFAOYSA-N 0.000 claims description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 3
- AOBHPJCBKDYSRA-UHFFFAOYSA-N 1-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-3-(1-phenylethyl)urea Chemical compound C=1C=CC=CC=1C(C)NC(=O)NC(C(=N1)O)=CN=C1N1C=CC=N1 AOBHPJCBKDYSRA-UHFFFAOYSA-N 0.000 claims description 2
- CAFPUIIZCWTDIY-UHFFFAOYSA-N 2-(1,3-benzodioxol-5-yl)-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound N=1C=C(NC(=O)CC=2C=C3OCOC3=CC=2)C(O)=NC=1N1C=CC=N1 CAFPUIIZCWTDIY-UHFFFAOYSA-N 0.000 claims description 2
- VMODKVBDJQPONZ-UHFFFAOYSA-N 2-(2-chlorophenyl)-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CC1=CC=CC=C1Cl VMODKVBDJQPONZ-UHFFFAOYSA-N 0.000 claims description 2
- KIHGBWHUDFTZLR-UHFFFAOYSA-N 2-methylsulfonyl-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound CS(=O)(=O)C1=CC=CC=C1C(=O)NC1=CN=C(N2N=CC=C2)N=C1O KIHGBWHUDFTZLR-UHFFFAOYSA-N 0.000 claims description 2
- YCZSTZJMQLQQFD-UHFFFAOYSA-N 2-naphthalen-1-yl-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound N=1C=C(NC(=O)CC=2C3=CC=CC=C3C=CC=2)C(O)=NC=1N1C=CC=N1 YCZSTZJMQLQQFD-UHFFFAOYSA-N 0.000 claims description 2
- VGOWQMKHVULTGQ-UHFFFAOYSA-N 2-naphthalen-2-yl-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound N=1C=C(NC(=O)CC=2C=C3C=CC=CC3=CC=2)C(O)=NC=1N1C=CC=N1 VGOWQMKHVULTGQ-UHFFFAOYSA-N 0.000 claims description 2
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 claims description 2
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 claims description 2
- HGYKKQHKQYZGKV-UHFFFAOYSA-N 5-(1-phenylpyrazol-4-yl)-2-pyrazol-1-yl-1h-pyrimidin-6-one Chemical compound OC1=NC(N2N=CC=C2)=NC=C1C(=C1)C=NN1C1=CC=CC=C1 HGYKKQHKQYZGKV-UHFFFAOYSA-N 0.000 claims description 2
- ADAHIAMLANIRHR-UHFFFAOYSA-N 5-phenyl-2-pyrazol-1-yl-1h-pyrimidin-6-one Chemical compound OC1=NC(N2N=CC=C2)=NC=C1C1=CC=CC=C1 ADAHIAMLANIRHR-UHFFFAOYSA-N 0.000 claims description 2
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 claims description 2
- 229920001774 Perfluoroether Polymers 0.000 claims description 2
- 125000004949 alkyl amino carbonyl amino group Chemical group 0.000 claims description 2
- 125000004471 alkyl aminosulfonyl group Chemical group 0.000 claims description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 2
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 claims description 2
- 125000005553 heteroaryloxy group Chemical group 0.000 claims description 2
- FWGCGZPLILONLV-UHFFFAOYSA-N n-(4-chlorophenyl)-2-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1CC(=O)NC1=CC=C(Cl)C=C1 FWGCGZPLILONLV-UHFFFAOYSA-N 0.000 claims description 2
- HCRYOWKKXODXON-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-(trifluoromethyl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC=C1C(F)(F)F HCRYOWKKXODXON-UHFFFAOYSA-N 0.000 claims description 2
- ZNEVDPZZUSBUMP-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-[2-(trifluoromethyl)phenyl]acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CC1=CC=CC=C1C(F)(F)F ZNEVDPZZUSBUMP-UHFFFAOYSA-N 0.000 claims description 2
- WOOICZQLUMSXLU-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-[3-(trifluoromethyl)phenyl]acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CC1=CC=CC(C(F)(F)F)=C1 WOOICZQLUMSXLU-UHFFFAOYSA-N 0.000 claims description 2
- WDKGWAHEEWZPEH-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-phenylbutanamide Chemical compound C=1C=CC=CC=1C(CC)C(=O)NC(C(=N1)O)=CN=C1N1C=CC=N1 WDKGWAHEEWZPEH-UHFFFAOYSA-N 0.000 claims description 2
- IROFDFPUKAYAKO-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-phenylpropanamide Chemical compound C=1C=CC=CC=1C(C)C(=O)NC(C(=N1)O)=CN=C1N1C=CC=N1 IROFDFPUKAYAKO-UHFFFAOYSA-N 0.000 claims description 2
- YLUJJBJPSFPOPX-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-3-(trifluoromethyl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC(C(F)(F)F)=C1 YLUJJBJPSFPOPX-UHFFFAOYSA-N 0.000 claims description 2
- OLIXQBZETBVSEU-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-4-phenoxybenzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C(C=C1)=CC=C1OC1=CC=CC=C1 OLIXQBZETBVSEU-UHFFFAOYSA-N 0.000 claims description 2
- KSXHZZIRPFLQMM-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-4-phenylbutanamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CCCC1=CC=CC=C1 KSXHZZIRPFLQMM-UHFFFAOYSA-N 0.000 claims description 2
- SJVUDDARXSLYDM-UHFFFAOYSA-N n-cyclohexyl-2-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1CC(=O)NC1CCCCC1 SJVUDDARXSLYDM-UHFFFAOYSA-N 0.000 claims description 2
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 claims description 2
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 claims description 2
- OUUUKGXDHJZWPO-UHFFFAOYSA-N tert-butyl 3-[(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)carbamoyl]piperidine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC1C(=O)NC1=CN=C(N2N=CC=C2)N=C1O OUUUKGXDHJZWPO-UHFFFAOYSA-N 0.000 claims description 2
- MNYCZTMXYXZNIJ-UHFFFAOYSA-N naphthalene-1-carboxamide Chemical compound C1=CC=C[C]2C(C(=O)N)=C=CC=C21 MNYCZTMXYXZNIJ-UHFFFAOYSA-N 0.000 claims 2
- QYHJIVXSURAZMO-UHFFFAOYSA-N tert-butyl 4-[(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)carbamoyl]piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1C(=O)NC1=CN=C(N2N=CC=C2)N=C1O QYHJIVXSURAZMO-UHFFFAOYSA-N 0.000 claims 2
- QPJIWUQZOQUODD-UHFFFAOYSA-N 1-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-3-(1-phenylethyl)thiourea Chemical compound C=1C=CC=CC=1C(C)NC(=S)NC(C(=N1)O)=CN=C1N1C=CC=N1 QPJIWUQZOQUODD-UHFFFAOYSA-N 0.000 claims 1
- ZLQLNQLKWHEBTI-UHFFFAOYSA-N 2-(3-chlorophenyl)-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CC1=CC=CC(Cl)=C1 ZLQLNQLKWHEBTI-UHFFFAOYSA-N 0.000 claims 1
- XPCYVMANYJFREI-UHFFFAOYSA-N 2-(3-cyanophenyl)-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CC1=CC=CC(C#N)=C1 XPCYVMANYJFREI-UHFFFAOYSA-N 0.000 claims 1
- YGBKHYHPOBEMRZ-UHFFFAOYSA-N 2-cyano-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC=C1C#N YGBKHYHPOBEMRZ-UHFFFAOYSA-N 0.000 claims 1
- SJVCYGWUPNQMJQ-UHFFFAOYSA-N 2-methoxy-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound COC1=CC=CC=C1C(=O)NC1=CN=C(N2N=CC=C2)N=C1O SJVCYGWUPNQMJQ-UHFFFAOYSA-N 0.000 claims 1
- AZDWRJXHQXSJGT-UHFFFAOYSA-N 2-methyl-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-phenylpropanamide Chemical compound C=1C=CC=CC=1C(C)(C)C(=O)NC(C(=N1)O)=CN=C1N1C=CC=N1 AZDWRJXHQXSJGT-UHFFFAOYSA-N 0.000 claims 1
- AJURDLXCCQYSGB-UHFFFAOYSA-N 3-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-1,1-diphenylurea Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)N(C=1C=CC=CC=1)C1=CC=CC=C1 AJURDLXCCQYSGB-UHFFFAOYSA-N 0.000 claims 1
- YZQQAAGYYGBACB-UHFFFAOYSA-N 3-cyano-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC(C#N)=C1 YZQQAAGYYGBACB-UHFFFAOYSA-N 0.000 claims 1
- YQOXIOQFTGTUKE-UHFFFAOYSA-N 3-methyl-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-phenylbutanamide Chemical compound C=1C=CC=CC=1C(C(C)C)C(=O)NC(C(=N1)O)=CN=C1N1C=CC=N1 YQOXIOQFTGTUKE-UHFFFAOYSA-N 0.000 claims 1
- QXGVZMLLRROQCG-UHFFFAOYSA-N 4-cyano-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=C(C#N)C=C1 QXGVZMLLRROQCG-UHFFFAOYSA-N 0.000 claims 1
- RDWBKPPJEHKQRD-UHFFFAOYSA-N 5-(1-benzylpyrazol-4-yl)-2-pyrazol-1-yl-1h-pyrimidin-6-one Chemical compound OC1=NC(N2N=CC=C2)=NC=C1C(=C1)C=NN1CC1=CC=CC=C1 RDWBKPPJEHKQRD-UHFFFAOYSA-N 0.000 claims 1
- 125000005045 dihydroisoquinolinyl group Chemical group C1(NC=CC2=CC=CC=C12)* 0.000 claims 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims 1
- 230000001404 mediated effect Effects 0.000 claims 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 claims 1
- KZNUMAVBIPNQJV-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-1-phenylcyclopropane-1-carboxamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1(C=2C=CC=CC=2)CC1 KZNUMAVBIPNQJV-UHFFFAOYSA-N 0.000 claims 1
- KNHWVYJZKZIPTJ-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-phenoxybenzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC=C1OC1=CC=CC=C1 KNHWVYJZKZIPTJ-UHFFFAOYSA-N 0.000 claims 1
- UNHDAPBLIZGOOB-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-phenylbenzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=CC=C1C1=CC=CC=C1 UNHDAPBLIZGOOB-UHFFFAOYSA-N 0.000 claims 1
- XNFXGTACLNNMFT-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-pyridin-4-ylacetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CC1=CC=NC=C1 XNFXGTACLNNMFT-UHFFFAOYSA-N 0.000 claims 1
- ZGBXOXIFWNQGGT-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-3-phenoxybenzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C(C=1)=CC=CC=1OC1=CC=CC=C1 ZGBXOXIFWNQGGT-UHFFFAOYSA-N 0.000 claims 1
- NMGWIMOITSAFBD-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-3-phenylpropanamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CCC1=CC=CC=C1 NMGWIMOITSAFBD-UHFFFAOYSA-N 0.000 claims 1
- VTQGITMMUDWSQX-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)cyclohexanecarboxamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1CCCCC1 VTQGITMMUDWSQX-UHFFFAOYSA-N 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 2
- 239000000203 mixture Substances 0.000 description 79
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 68
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 45
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 43
- 239000000243 solution Substances 0.000 description 42
- 238000006243 chemical reaction Methods 0.000 description 40
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 33
- 229910001868 water Inorganic materials 0.000 description 32
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 31
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 28
- 235000019439 ethyl acetate Nutrition 0.000 description 24
- 239000007787 solid Substances 0.000 description 24
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 23
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 21
- 238000004128 high performance liquid chromatography Methods 0.000 description 21
- 239000004480 active ingredient Substances 0.000 description 20
- 239000002253 acid Substances 0.000 description 18
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 17
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 15
- 230000015572 biosynthetic process Effects 0.000 description 14
- 239000011541 reaction mixture Substances 0.000 description 14
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 14
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- 229920006395 saturated elastomer Polymers 0.000 description 13
- 239000003643 water by type Substances 0.000 description 13
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- 239000000741 silica gel Substances 0.000 description 12
- 229910002027 silica gel Inorganic materials 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 238000010992 reflux Methods 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 10
- 125000004122 cyclic group Chemical group 0.000 description 10
- 150000002148 esters Chemical class 0.000 description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 150000001409 amidines Chemical class 0.000 description 9
- 238000002514 liquid chromatography mass spectrum Methods 0.000 description 9
- 125000002950 monocyclic group Chemical group 0.000 description 9
- 229910052760 oxygen Inorganic materials 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 8
- 125000002619 bicyclic group Chemical group 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- 239000003480 eluent Substances 0.000 description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 239000003208 petroleum Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- 206010021143 Hypoxia Diseases 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 150000001721 carbon Chemical group 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 238000000605 extraction Methods 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 238000002953 preparative HPLC Methods 0.000 description 7
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- LTMHNWPUDSTBKD-UHFFFAOYSA-N diethyl 2-(ethoxymethylidene)propanedioate Chemical compound CCOC=C(C(=O)OCC)C(=O)OCC LTMHNWPUDSTBKD-UHFFFAOYSA-N 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 229910052742 iron Inorganic materials 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- 102100037249 Egl nine homolog 1 Human genes 0.000 description 5
- 101710111663 Egl nine homolog 1 Proteins 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 125000003710 aryl alkyl group Chemical group 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 238000000132 electrospray ionisation Methods 0.000 description 5
- 239000004615 ingredient Substances 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 150000002500 ions Chemical class 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 229910052717 sulfur Inorganic materials 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 5
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N 1,1-dimethoxyethane Chemical compound COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 4
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 230000007812 deficiency Effects 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 210000003743 erythrocyte Anatomy 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 230000007954 hypoxia Effects 0.000 description 4
- 208000028867 ischemia Diseases 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 239000011777 magnesium Substances 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 125000003386 piperidinyl group Chemical group 0.000 description 4
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 235000019445 benzyl alcohol Nutrition 0.000 description 3
- 229960004217 benzyl alcohol Drugs 0.000 description 3
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 3
- 239000006196 drop Substances 0.000 description 3
- 230000010437 erythropoiesis Effects 0.000 description 3
- 238000005755 formation reaction Methods 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 150000002357 guanidines Chemical class 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 3
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000008297 liquid dosage form Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004193 piperazinyl group Chemical group 0.000 description 3
- 125000003367 polycyclic group Polymers 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- GMHCEDDZKAYPLB-UHFFFAOYSA-N pyridine-2-carboximidamide;hydrochloride Chemical compound [Cl-].NC(=[NH2+])C1=CC=CC=N1 GMHCEDDZKAYPLB-UHFFFAOYSA-N 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000010189 synthetic method Methods 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 238000000844 transformation Methods 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- DNCYBUMDUBHIJZ-UHFFFAOYSA-N 1h-pyrimidin-6-one Chemical compound O=C1C=CN=CN1 DNCYBUMDUBHIJZ-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- FFNVQNRYTPFDDP-UHFFFAOYSA-N 2-cyanopyridine Chemical compound N#CC1=CC=CC=N1 FFNVQNRYTPFDDP-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- VWVNPWMYGKMFNX-UHFFFAOYSA-N 5-amino-2-pyrazol-1-yl-1h-pyrimidin-6-one Chemical compound N1=C(O)C(N)=CN=C1N1N=CC=C1 VWVNPWMYGKMFNX-UHFFFAOYSA-N 0.000 description 2
- JKDACPUPAMICIA-UHFFFAOYSA-N 6-oxo-1h-pyrimidine-5-carboxylic acid Chemical class OC(=O)C1=CN=CNC1=O JKDACPUPAMICIA-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 229930194542 Keto Natural products 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 102100037247 Prolyl hydroxylase EGLN3 Human genes 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- MJBWDEQAUQTVKK-IAGOWNOFSA-N aflatoxin M1 Chemical compound C=1([C@]2(O)C=CO[C@@H]2OC=1C=C(C1=2)OC)C=2OC(=O)C2=C1CCC2=O MJBWDEQAUQTVKK-IAGOWNOFSA-N 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 125000002877 alkyl aryl group Chemical group 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 125000003725 azepanyl group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 229940073608 benzyl chloride Drugs 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000036770 blood supply Effects 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- 125000004623 carbolinyl group Chemical group 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 150000003857 carboxamides Chemical class 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 239000007891 compressed tablet Substances 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 235000008504 concentrate Nutrition 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 238000006880 cross-coupling reaction Methods 0.000 description 2
- 125000002944 cyanoaryl group Chemical group 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 150000008049 diazo compounds Chemical class 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 2
- 125000005047 dihydroimidazolyl group Chemical group N1(CNC=C1)* 0.000 description 2
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 2
- 125000005049 dihydrooxadiazolyl group Chemical group O1N(NC=C1)* 0.000 description 2
- 125000005050 dihydrooxazolyl group Chemical group O1C(NC=C1)* 0.000 description 2
- 125000005051 dihydropyrazinyl group Chemical group N1(CC=NC=C1)* 0.000 description 2
- 125000005052 dihydropyrazolyl group Chemical group N1(NCC=C1)* 0.000 description 2
- 125000005053 dihydropyrimidinyl group Chemical group N1(CN=CC=C1)* 0.000 description 2
- 125000005054 dihydropyrrolyl group Chemical group [H]C1=C([H])C([H])([H])C([H])([H])N1* 0.000 description 2
- 125000005044 dihydroquinolinyl group Chemical group N1(CC=CC2=CC=CC=C12)* 0.000 description 2
- 125000005056 dihydrothiazolyl group Chemical group S1C(NC=C1)* 0.000 description 2
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 2
- PYHXGXCGESYPCW-UHFFFAOYSA-N diphenylacetic acid Chemical compound C=1C=CC=CC=1C(C(=O)O)C1=CC=CC=C1 PYHXGXCGESYPCW-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 150000002085 enols Chemical group 0.000 description 2
- 238000010931 ester hydrolysis Methods 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 125000003106 haloaryl group Chemical group 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 2
- 125000004634 hexahydroazepinyl group Chemical group N1(CCCCCC1)* 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 229910052748 manganese Inorganic materials 0.000 description 2
- 239000011572 manganese Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 125000003566 oxetanyl group Chemical group 0.000 description 2
- 238000006213 oxygenation reaction Methods 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 239000003182 parenteral nutrition solution Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- ODLMAHJVESYWTB-UHFFFAOYSA-N propylbenzene Chemical compound CCCC1=CC=CC=C1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 2
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 238000006462 rearrangement reaction Methods 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- NDVLTYZPCACLMA-UHFFFAOYSA-N silver oxide Chemical compound [O-2].[Ag+].[Ag+] NDVLTYZPCACLMA-UHFFFAOYSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical class ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 1
- 125000006705 (C5-C7) cycloalkyl group Chemical group 0.000 description 1
- YVFQIFGPPPSDQZ-UHFFFAOYSA-N 1-cyclopentyl-3-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)urea Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)NC1CCCC1 YVFQIFGPPPSDQZ-UHFFFAOYSA-N 0.000 description 1
- QQCJPTVZIZVKEZ-UHFFFAOYSA-N 1-isothiocyanatoethylbenzene Chemical compound S=C=NC(C)C1=CC=CC=C1 QQCJPTVZIZVKEZ-UHFFFAOYSA-N 0.000 description 1
- XFNJVKMNNVCYEK-UHFFFAOYSA-N 1-naphthaleneacetamide Chemical compound C1=CC=C2C(CC(=O)N)=CC=CC2=C1 XFNJVKMNNVCYEK-UHFFFAOYSA-N 0.000 description 1
- RBZRMBCLZMEYEH-UHFFFAOYSA-N 1h-pyrazol-1-ium-1-carboximidamide;chloride Chemical compound Cl.NC(=N)N1C=CC=N1 RBZRMBCLZMEYEH-UHFFFAOYSA-N 0.000 description 1
- CNKAYTUDVAZFPB-UHFFFAOYSA-N 2-(4-chlorophenyl)-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)CC1=CC=C(Cl)C=C1 CNKAYTUDVAZFPB-UHFFFAOYSA-N 0.000 description 1
- PBVWJFRIHHXOIY-UHFFFAOYSA-N 2-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-n-propan-2-ylacetamide Chemical compound N1=C(O)C(CC(=O)NC(C)C)=CN=C1N1N=CC=C1 PBVWJFRIHHXOIY-UHFFFAOYSA-N 0.000 description 1
- WRJBOTVNQOFASA-UHFFFAOYSA-N 2-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)acetic acid Chemical compound N1=C(O)C(CC(=O)O)=CN=C1N1N=CC=C1 WRJBOTVNQOFASA-UHFFFAOYSA-N 0.000 description 1
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 description 1
- OIALIKXMLIAOSN-UHFFFAOYSA-N 2-Propylpyridine Chemical compound CCCC1=CC=CC=N1 OIALIKXMLIAOSN-UHFFFAOYSA-N 0.000 description 1
- SXAMGRAIZSSWIH-UHFFFAOYSA-N 2-[3-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,2,4-oxadiazol-5-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NOC(=N1)CC(=O)N1CC2=C(CC1)NN=N2 SXAMGRAIZSSWIH-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- ADSOSINJPNKUJK-UHFFFAOYSA-N 2-butylpyridine Chemical compound CCCCC1=CC=CC=N1 ADSOSINJPNKUJK-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- DERAACKMMNJAFU-UHFFFAOYSA-N 2-ethoxy-1,3-dioxane-4,6-dione Chemical compound CCOC1OC(=O)CC(=O)O1 DERAACKMMNJAFU-UHFFFAOYSA-N 0.000 description 1
- NRGGMCIBEHEAIL-UHFFFAOYSA-N 2-ethylpyridine Chemical compound CCC1=CC=CC=N1 NRGGMCIBEHEAIL-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 1
- GTKIGDZXPDCIKR-UHFFFAOYSA-N 2-phenylbenzamide Chemical compound NC(=O)C1=CC=CC=C1C1=CC=CC=C1 GTKIGDZXPDCIKR-UHFFFAOYSA-N 0.000 description 1
- KDFDOINBXBEOLZ-UHFFFAOYSA-N 2-phenylpropan-2-amine Chemical compound CC(C)(N)C1=CC=CC=C1 KDFDOINBXBEOLZ-UHFFFAOYSA-N 0.000 description 1
- RWBSNBWWYMFNMM-UHFFFAOYSA-N 2-pyrazol-1-yl-1h-pyrimidin-6-one Chemical compound OC1=CC=NC(N2N=CC=C2)=N1 RWBSNBWWYMFNMM-UHFFFAOYSA-N 0.000 description 1
- NBAPQKANGFQKGW-UHFFFAOYSA-N 3-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-1-phenyl-1-propan-2-ylurea Chemical compound C=1C=CC=CC=1N(C(C)C)C(=O)NC(C(=N1)O)=CN=C1N1C=CC=N1 NBAPQKANGFQKGW-UHFFFAOYSA-N 0.000 description 1
- PAKVPDACZNEDHT-UHFFFAOYSA-N 3-methyl-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-2-phenylpentanamide Chemical compound C=1C=CC=CC=1C(C(C)CC)C(=O)NC(C(=N1)O)=CN=C1N1C=CC=N1 PAKVPDACZNEDHT-UHFFFAOYSA-N 0.000 description 1
- WEQPBCSPRXFQQS-UHFFFAOYSA-N 4,5-dihydro-1,2-oxazole Chemical compound C1CC=NO1 WEQPBCSPRXFQQS-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- JSXOJGJLXAZXSP-UHFFFAOYSA-N 4-chloro-n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=C(Cl)C=C1 JSXOJGJLXAZXSP-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- QTOFUNCFTWOXKH-UHFFFAOYSA-N 4-phenylmethoxy-2-pyrazol-1-ylpyrimidine Chemical compound C(C1=CC=CC=C1)OC1=NC(=NC=C1)N1N=CC=C1 QTOFUNCFTWOXKH-UHFFFAOYSA-N 0.000 description 1
- RNDRKWAFCUAJHN-UHFFFAOYSA-N 6-oxo-2-pyridazin-3-yl-1h-pyrimidine-5-carboxylic acid Chemical compound N1=C(O)C(C(=O)O)=CN=C1C1=CC=CN=N1 RNDRKWAFCUAJHN-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 208000030760 Anaemia of chronic disease Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 108090000448 Aryl Hydrocarbon Receptors Proteins 0.000 description 1
- 102100026792 Aryl hydrocarbon receptor Human genes 0.000 description 1
- 102100030907 Aryl hydrocarbon receptor nuclear translocator Human genes 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 201000006474 Brain Ischemia Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- 0 CCN(c(nc1)nc(O)c1NC(*)**)N=CC Chemical compound CCN(c(nc1)nc(O)c1NC(*)**)N=CC 0.000 description 1
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 1
- 101000690445 Caenorhabditis elegans Aryl hydrocarbon receptor nuclear translocator homolog Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102100035882 Catalase Human genes 0.000 description 1
- 108010053835 Catalase Proteins 0.000 description 1
- 206010008120 Cerebral ischaemia Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 108010028143 Dioxygenases Proteins 0.000 description 1
- 102000016680 Dioxygenases Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 206010018910 Haemolysis Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 101000793115 Homo sapiens Aryl hydrocarbon receptor nuclear translocator Proteins 0.000 description 1
- 101000987586 Homo sapiens Eosinophil peroxidase Proteins 0.000 description 1
- 101000920686 Homo sapiens Erythropoietin Proteins 0.000 description 1
- 101000881650 Homo sapiens Prolyl hydroxylase EGLN2 Proteins 0.000 description 1
- 101000881678 Homo sapiens Prolyl hydroxylase EGLN3 Proteins 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 206010033546 Pallor Diseases 0.000 description 1
- 206010034576 Peripheral ischaemia Diseases 0.000 description 1
- 238000003482 Pinner synthesis reaction Methods 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 102100037248 Prolyl hydroxylase EGLN2 Human genes 0.000 description 1
- 101710170720 Prolyl hydroxylase EGLN3 Proteins 0.000 description 1
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 1
- 241000720974 Protium Species 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric Acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GNVMUORYQLCPJZ-UHFFFAOYSA-M Thiocarbamate Chemical compound NC([S-])=O GNVMUORYQLCPJZ-UHFFFAOYSA-M 0.000 description 1
- 208000001435 Thromboembolism Diseases 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- FIULGFJIHJJXMT-UHFFFAOYSA-N [C]1[N]C=CC=C1 Chemical class [C]1[N]C=CC=C1 FIULGFJIHJJXMT-UHFFFAOYSA-N 0.000 description 1
- LHZHYALHRDISND-UHFFFAOYSA-O [amino-(3,5-dimethylpyrazol-3-yl)methylidene]azanium;nitrate Chemical compound [O-][N+]([O-])=O.CC1=CC(C)(C(N)=[NH2+])N=N1 LHZHYALHRDISND-UHFFFAOYSA-O 0.000 description 1
- WDOSFTZMBFYTED-UHFFFAOYSA-N [isothiocyanato(phenyl)methyl]benzene Chemical compound C=1C=CC=CC=1C(N=C=S)C1=CC=CC=C1 WDOSFTZMBFYTED-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 238000010976 amide bond formation reaction Methods 0.000 description 1
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical compound [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 208000022400 anemia due to chronic disease Diseases 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 230000003064 anti-oxidating effect Effects 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 238000007080 aromatic substitution reaction Methods 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 125000002529 biphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C12)* 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 125000004452 carbocyclyl group Chemical group 0.000 description 1
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 210000003855 cell nucleus Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 230000003920 cognitive function Effects 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- RWGFKTVRMDUZSP-UHFFFAOYSA-N cumene Chemical compound CC(C)C1=CC=CC=C1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 229960000958 deferoxamine Drugs 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- BQHDXNZNSPVVKB-UHFFFAOYSA-N diethyl 2-methylidenepropanedioate Chemical class CCOC(=O)C(=C)C(=O)OCC BQHDXNZNSPVVKB-UHFFFAOYSA-N 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 125000005058 dihydrotriazolyl group Chemical group N1(NNC=C1)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229910001882 dioxygen Inorganic materials 0.000 description 1
- QILSFLSDHQAZET-UHFFFAOYSA-N diphenylmethanol Chemical compound C=1C=CC=CC=1C(O)C1=CC=CC=C1 QILSFLSDHQAZET-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- UTDHXSQPMAVKNL-UHFFFAOYSA-N ethyl 6-oxo-2-pyrazol-1-yl-1h-pyrimidine-5-carboxylate Chemical compound N1=C(O)C(C(=O)OCC)=CN=C1N1N=CC=C1 UTDHXSQPMAVKNL-UHFFFAOYSA-N 0.000 description 1
- UREBWPXBXRYXRJ-UHFFFAOYSA-N ethyl acetate;methanol Chemical compound OC.CCOC(C)=O UREBWPXBXRYXRJ-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 206010016256 fatigue Diseases 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 235000003891 ferrous sulphate Nutrition 0.000 description 1
- 239000011790 ferrous sulphate Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 230000034659 glycolysis Effects 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000003278 haem Chemical class 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 230000008588 hemolysis Effects 0.000 description 1
- 150000002390 heteroarenes Chemical group 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 230000009097 homeostatic mechanism Effects 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 102000044890 human EPO Human genes 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- SURQXAFEQWPFPV-UHFFFAOYSA-L iron(2+) sulfate heptahydrate Chemical compound O.O.O.O.O.O.O.[Fe+2].[O-]S([O-])(=O)=O SURQXAFEQWPFPV-UHFFFAOYSA-L 0.000 description 1
- 229910000359 iron(II) sulfate Inorganic materials 0.000 description 1
- 208000023589 ischemic disease Diseases 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000005906 menstruation Effects 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- HNQIVZYLYMDVSB-NJFSPNSNSA-N methanesulfonamide Chemical compound [14CH3]S(N)(=O)=O HNQIVZYLYMDVSB-NJFSPNSNSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- NTDIUKIRYXCCIU-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-1,2,3,4-tetrahydronaphthalene-1-carboxamide Chemical compound N=1C=C(NC(=O)C2C3=CC=CC=C3CCC2)C(O)=NC=1N1C=CC=N1 NTDIUKIRYXCCIU-UHFFFAOYSA-N 0.000 description 1
- ZFMLFBOXHWEFCW-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-4-(trifluoromethyl)benzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C1=CC=C(C(F)(F)F)C=C1 ZFMLFBOXHWEFCW-UHFFFAOYSA-N 0.000 description 1
- LMECQLLOAKGERW-UHFFFAOYSA-N n-(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)-4-phenylbenzamide Chemical compound OC1=NC(N2N=CC=C2)=NC=C1NC(=O)C(C=C1)=CC=C1C1=CC=CC=C1 LMECQLLOAKGERW-UHFFFAOYSA-N 0.000 description 1
- KUDPGZONDFORKU-UHFFFAOYSA-N n-chloroaniline Chemical compound ClNC1=CC=CC=C1 KUDPGZONDFORKU-UHFFFAOYSA-N 0.000 description 1
- HTKWJLCTSHNBEL-UHFFFAOYSA-N naphthalene-2-carboxamide Chemical compound C1=C(C=CC2=CC=CC=C12)C(=O)N.C1=C(C=CC2=CC=CC=C12)C(=O)N HTKWJLCTSHNBEL-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 238000006396 nitration reaction Methods 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229940054534 ophthalmic solution Drugs 0.000 description 1
- 239000002997 ophthalmic solution Substances 0.000 description 1
- 229940100654 ophthalmic suspension Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000017448 oviposition Effects 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 235000019629 palatability Nutrition 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 238000010651 palladium-catalyzed cross coupling reaction Methods 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 238000011458 pharmacological treatment Methods 0.000 description 1
- 125000004344 phenylpropyl group Chemical group 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000009758 senescence Effects 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229910001923 silver oxide Inorganic materials 0.000 description 1
- 235000010378 sodium ascorbate Nutrition 0.000 description 1
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 1
- 229960005055 sodium ascorbate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 1
- WGRULTCAYDOGQK-UHFFFAOYSA-M sodium;sodium;hydroxide Chemical compound [OH-].[Na].[Na+] WGRULTCAYDOGQK-UHFFFAOYSA-M 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- IPPRNSMNEYCPPL-UHFFFAOYSA-N tert-butyl 4-[2-oxo-2-[(6-oxo-2-pyrazol-1-yl-1h-pyrimidin-5-yl)amino]ethyl]piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1CC(=O)NC1=CN=C(N2N=CC=C2)N=C1O IPPRNSMNEYCPPL-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- UEUXEKPTXMALOB-UHFFFAOYSA-J tetrasodium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O UEUXEKPTXMALOB-UHFFFAOYSA-J 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- ZWZVWGITAAIFPS-UHFFFAOYSA-N thiophosgene Chemical compound ClC(Cl)=S ZWZVWGITAAIFPS-UHFFFAOYSA-N 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 150000008648 triflates Chemical group 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 1
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 239000000225 tumor suppressor protein Substances 0.000 description 1
- 230000034512 ubiquitination Effects 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 235000012141 vanillin Nutrition 0.000 description 1
- MWOOGOJBHIARFG-UHFFFAOYSA-N vanillin Chemical compound COC1=CC(C=O)=CC=C1O MWOOGOJBHIARFG-UHFFFAOYSA-N 0.000 description 1
- FGQOOHJZONJGDT-UHFFFAOYSA-N vanillin Natural products COC1=CC(O)=CC(C=O)=C1 FGQOOHJZONJGDT-UHFFFAOYSA-N 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Definitions
- anemia which is defined as a deficiency in the blood's oxygen-carrying capacity, and ischemia, in which restrictions in blood supply are caused by a constriction or blockage of blood vessels.
- Anemia can be caused by the loss of red blood cells (hemorrhage), excessive red blood cell destruction (hemolysis) or deficiencies in erythropoiesis (production of red blood cells from precursors found in the bone marrow).
- the symptoms of anemia can include weakness, dizziness, fatigue, pallor, impairment of cognitive function and a genera] reduction in quality of life. Chronic and/or severe anemia can lead to the exacerbation of myocardial, cerebral or peripheral ischemia and to heart failure.
- Ischemia is defined as an absolute or relative shortage of oxygen to a tissue or organ and can result from disorders such as atherosclerosis, diabetes, thromboembolisms, hypotension, etc.
- the heart, brain and kidney are especially sensitive to ischemic stress caused by low blood supply.
- the primary pharmacological treatment for anemia is administration of some variant of recombinant human erythropoietin (EPO).
- EPO human erythropoietin
- recombinant EPO is administered to enhance the supply of the hormone, correct the shortage of red blood cells and increase the blood's oxygen-carrying capacity.
- EPO replacement is not always sufficient to stimulate optimal erythropoiesis (e.g., in patients with iron processing deficiencies) and has associated risks.
- HTF Hypoxia-inducible factor
- HEF-a highly regulated a-subunit
- HIF- ⁇ constitutively expressed ⁇ -subunit
- ARNT aryl hydrocarbon receptor nuclear transporter
- HTF target genes are reported to be associated with various aspects of erythropoiesis (e.g., erythropoietin (EPO) and EPO receptor), glycolysis and angiogenesis (e.g., vascular endothelial growth factor (VEGF)).
- EPO erythropoietin
- VEGF vascular endothelial growth factor
- HTF-a is a substrate in a reaction with molecular oxygen, which is catalyzed by a family of iron(TJ)-, 2-ketoglutarate- and ascorbate-dependent dioxygenase enzymes called PHD-1 (EGLN2, or egg laying abnormal 9 homolog 2, PHD2 (EGLN1), and PHD3 (EGLN3).
- PHD-1 family of iron(TJ)-, 2-ketoglutarate- and ascorbate-dependent dioxygenase enzymes called PHD-1 (EGLN2, or egg laying abnormal 9 homolog 2, PHD2 (EGLN1), and PHD3 (EGLN3).
- Proline residues of HTF-a are hydroxylated (e.g., Pro-402 and Pro-564 of HIF- ⁇ ) and the resulting product is a target of the tumor suppressor protein von-Hippel Lindau, a component of an E3 ubiquhin ligase multiprotein complex involved in protein ubiquitination.
- HEF-a hydroxylation reaction is less efficient and HIF- a is available to dimerize with HIF- ⁇ .
- HEF dimers are translocated to the cell nucleus where they bind to a hypoxia-responsive enhancer element of HIF target genes.
- HIF HIF prolyl hydroxylases
- the present invention concerns compounds of formula I
- the present invention provides compounds of formula I and pharmaceutically acceptable salts and solvates thereof:
- A is heteroaryl optionally substituted with one or more R? substituents
- L is chosen from a bond, aryl, heteroaryl, -(C1.3 alkyl)o-iS(Ci-3 alkyl)o-l-, -(C1.3 alkylJo-lNRaCO-, (Ci-3 alkyl)0-lNRaCO2-, -(C1.3 alkyl)0-lCO2-, -(Cj.3 alkyl)0-lNRaSO2-, -(C1.3 alkylJo.
- R2 and are independently selected from hydrogen, halogen, hydroxy!, Cj-g alkyl, C3-6 heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxy I, -SR.*, -NR*2 or-C02-R*;
- R1 is selected from
- Rl said alkyl, alkenyl, alkynyl, cycloalkenyl, aryl, perfluoralkyl, perfluoroalkoxy
- heterocyclyl, and cycloalkyl are each optionally substituted with one or more R? substituents; optionally, R* and L are linked together to form a ring of 5 to 8 atoms optionally substituted with one or more substituents R?; where said ring has 0, 1 , or 2 heteroatoms independently selected from -NR -, -O- and -S(0) n -;
- halogen is selected from halogen, hydroxy, oxo, cyano, aryl, heterocyclyl, cycloalkyl, -Ci-6 alkyl, -Cl-6 alkoxy, aryloxy, heterocyclyloxy, -0(0-l)(Ci-io)perfluorc*lkyl, -C02 a , -NRbRc, -CONRbR C
- RlO is selected from hydroxy, aryl, heterocycloalkyl, heteroaryl, halogen, oxo, -Ci-6 alkyl, Cj-6 alkoxy, halogen, CO2H, cyano, O(00)o.i C 1 -6 alkyl, NO2, trifluoromethoxy, trifluoroethoxy, -O(0- l)(Ci.io)perfluoroalkyl, Cfj-lO alkylaminocarbonylamino, Q)-10 alkyloxycarbonylaminoCo-io alkyl, Crj-io alkylcarbonylaminoCo-io alkyl, Co-10 alkylaminosulfonylaminoCo-io alkyl, Cfj-io aJkylsulfonylaminoCo-lO alkyl, Co-io alkylsulfonyl, Co-10 alkylaminosulfonyl, Co.10
- n i or 2;
- Ra is chosen from hydrogen; -Cl-10 alkyl, -(Cj-6 alkyl)C3-8 cycloalkyl;
- Rb, Rc, and Rd are each independently chosen from hydrogen, -Cl-10 alkyl,C3-8 cycloalkyl, aryl, and heterocyclyl.
- Illustrative but nonlimiting examples of compounds of the invention are the following: N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2,2-diphenylacetarnide;
- naphthalen-2-yl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
- alkyl is intended to include both branched- and straight-chain saturated aliphatic hydrocarbon groups, including all isomers, having the specified number of carbon atoms. Commonly used abbreviations for alkyl groups are used throughout the specification, e.g. methyl may be represented by “Me” or CH 3) ethyl may be represented by “Et” or CH 2 CH 3 , propyl may be represented by “Pr” or CH 2 CH 2 CH ⁇ , butyl may be represented by "Bu” or CH2CH2CH2CH 3 , etc.
- Ci_6 alkyl (or “Cj-Cg alkyl”) for example, means linear or branched chain alkyl groups, including all isomers, having the specified number of carbon atoms.
- Ci-6 alkyl includes all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl.
- C1-4 alkyl means n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl.
- alkylene refers to both branched- and straight-chain saturated aliphatic hydrocarbon groups, including all isomers, having the specified number of carbons, and having two terminal end chain attachments.
- ⁇ nsubstitoted A-Qalkylene-B represents A-CH2-CH2-CH2-CH2-B.
- the terra "alkoxy” represents a linear or branched alkyl group of indicated number of carbon atoms attached through an oxygen bridge.
- alkyl (either as a stand alone radical or as part of a radical such as alkoxy, alkylthio and aralkyl) groups are unsubstituted or substituted with 1 to 3 substituents on each carbon atom, with halo, C1-C20 CF 3» N3 ⁇ 4 N(Ci-C6 alkyl)2, ⁇ 3 ⁇ 4, oxo, CN, N3, -OH, -0(Ci-C6 alkyl), C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, (Cfj-Cg alkyl) S(O)0-2-, (Co-C 6 alkyl)S(O)0- 2(Co-C 6 alkyl)-, (C0-C6 alkyl)C(0)NH-, H 2 N-C(NH)
- heterocyclylalkyl halo-aryl, halo-aralkyl, halo-heterocycle, halo-heterocyclylalkyl, cyano-aryl, cyano- aralkyl, cyano-heterocycle and cyano-heterocyclylalkyl.
- u CQ as employed in expressions such as "Co_6 alkyl” means a direct covalent bond; or when the term appears at the terminus of a substituent, Co-6 alkyl means hydrogen or Cl-6alkyl.
- an integer defining the presence of a certain number of atoms in a group is equal to zero, it means that the atoms adjacent thereto are connected directly by a bond.
- s is an integer equal to zero, 1 or 2
- C3-8 cycloalkyl (or “C3-Cg cycloalkyl”) means a cyclic ring of an alkane having three to eight total carbon atoms (i.e., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, or cyclooctyl).
- C3-7 cycloalkyl “C3-6 cycloalkyl”, “C5-7 cycloalkyl” and the like have analogous meanings.
- halogen (or “halo" refers to fluorine, chlorine, bromine and iodine
- fluoro F
- chloro CI
- bromo Br
- iodo I
- aryl refers to aromatic mono- and poly-carbocyclic ring systems, wherein the individual carbocyclic rings in the polyring systems are fused or attached to each other via a single bond.
- Suitable aryl groups include phenyl, naphthyl, and biphenylenyl.
- carbocycle (and variations thereof such as “carbocyclic” or “carbocyclyl”) as used herein, unless otherwise indicated, refers to (i) a C3 to Cg monocyclic, saturated or unsaturated ring or (ii) a C7 to C12 bicyclic saturated or unsaturated ring system. Each ring in (ii) is either independent of, or fused to, the other ring, and each ring is saturated or unsaturated.
- the carbocycle may be attached to the rest of the molecule at any carbon atom which results in a stable compound.
- fused bicyclic carbocycles are a subset of the carbocycles; i.e., the term "fused bicyclic carbocycle” generally refers to a C7 to C] 0 bicyclic ring system in which each ring is saturated or unsaturated and two adjacent carbon atoms are shared by each of the rings in the ring system.
- a fused bicyclic carbocycle in which one ring is saturated and the other is saturated is a saturated bicyclic ring system.
- a fused bicyclic carbocycle in which one ring is benzene and the other is saturated is an unsaturated bicyclic ring system.
- a fused bicyclic carbocycle in which one ring is benzene and the other is unsaturated is an unsaturated ring system.
- Saturated carbocyclic rings are also referred to as cycloalkyl rings, e.g., cyclopropyl, cyclobutyl, etc.
- carbocycle is unsubstituted or substituted with C]_6 alkyl, Ci-6 alkenyl, C]-6 alkynyl, aryl, halogen, N3 ⁇ 4 or OH.
- a subset of the fused bicyclic unsaturated carbocycles are those bicyclic carbocycles in which one ring is a benzene ring and the other ring is saturated or unsaturated, with attachment via any carbon atom that results in a stable compound.
- Representative examples of this subset include the following:
- heterocycle broadly refers to (i) a stable 4- to 8-membered, saturated or unsaturated monocyclic ring, or (ii) a stable 7- to 12-membered bicyclic ring system, wherein each ring in (ii) is independent of, or fused to, the other ring or rings and each ring is saturated or unsaturated, and the monocyclic ring or bicyclic ring system contains one or more heteroatoms (e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms) selected from N, O and S and a balance of carbon atoms (the monocyclic ring typically contains at least one carbon atom and the ring systems typically contain at least two carbon atoms); and wherein any one or more of the nitrogen and sulfur h t t i ti ll idi d d e or more of the nitrogen heteroatoms is optionally qua
- the heterocyclic ring may be attached at any heteroatom or carbon atom, provided that attachment results in the creation of a stable structure.
- the heterocyclic ring has substituents, it is understood that the substituents may be attached to any atom in the ring, whether a heteroatom or a carbon atom, provided that a stable chemical structure results.
- heterocyclylic moieties include, but are not limited to, the following: azepanyl, azabenzimidazole, benzoimidazolyl, benzofuryl, benzofurazanyl, benzopyrazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, chromanyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuryl, isochromanyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazoryl, naphthpyridinyl, oxadiazolyl, oxazoryl, oxazolinyl, isooxazolinyl, oxetanyl,
- saturated heterocyclics form a subset of the heterocycles; i.e., the term “saturated heterocyclic” generally refers to a heterocycle as defined above in which the entire ring system (whether mono- or poly-cyclic) is saturated.
- saturated heterocyclic ring refers to a 4- to 8-membered saturated monocyclic ring or a stable 7- to 12-membered bicyclic ring system which consists of carbon atoms and one or more heteroatoms selected from N, O and S.
- Representative examples include piperidinyl, piperazinyl, azepanyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl,
- Heteroaromatics form another subset of the heterocycles; i.e., the term “heteroaromatic” (alternatively “heteroary ) generally refers to a heterocycle as defined above in which the entire ring system (whether mono- or poly-cyclic) is an aromatic ring system.
- the term “heteroaromatic ring” refers a 5- or 6-membered monocyclic aromatic ring or a 7- to 12-membered bicyclic which consists of carbon atoms and one or more heteroatoms selected from N, O and S.
- substituted heteroaryl rings containing at least one nitrogen atom e.g., pyridine
- substitutions can be those resulting in N-oxide formation.
- heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl (or thiophcnyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl.
- bicyclic heterocycles include benzotriazolyl, indolyl, isoindolyl, indazolyl, indolinyl, isoindolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, chromanyl, isochromanyl, tetrahydroquinolinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl,
- 2,3-dihydroDenzofuryl, 2 r 3-dihydrobenzo-l,4-dioxinyI i.e., imia3 ⁇ 4zo(2,l-b)(l,3)thiazole, (i.e., and benzo-l,3-dioxolyl (i.e., In certain contexts herein, is alternatively
- phenyl having as a substituent methylenedioxy attached to two adjacent carbon atoms.
- arylalkyl and “alkylaryl” include an alkyl portion where alkyl is as defined above and include an aryl portion where aryl is as defined above.
- arylalkyl include, but are not limited to, benzyl, phenylethyl, phenylpropyl, naphthylmethyl, and naphthyletbyl.
- alkylaryl include, but are not limited to, toluene, ethylbenzene, propylbenzene, methylpyridine, ethylpyridine, propylpyridine and butylpyridine.
- substituted C3- Cjo cycloalky As used herein, the terms “substituted C3- Cjo cycloalky , "substituted aryl (including phenyl)” and “substituted heterocycle” are intended to include the cyclic group containing from 1 to 3 substituents in addition to the point of attachment to the rest of the compound.
- the substituents are selected from the group which includes, but are not limited to, halo, C] -C20 alkyl, CF3, NH2, (Ci-C6 alkyl)2, ⁇ 2» oxo, CN, N3, -OH, -0(Ci-C6 alkyl), C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C alkynyl, (Q)-C 6 alkyl) S(0)o-2- > aryl-S(O)0-2- > (Co-C 6 alk l)S(0)o-2(Co-C 6 alkyl)-, (Co-C 6 alkyl)C(0)NH-, H2N-C(NH , -0(C -C6 alkyl)CF3, (Co-Q, alkyl)C(O)-, (C0-C6 alkyl)OC(0)-, (C0-C alkyl)2NC(0)- (C()-C6alkyl)0
- an “unsaturated” ring is a partially or fully unsaturated ring.
- an “unsaturated monocyclic C5 carbocycle” refers to cyclohexene, cyclohexadiene, and benzene.
- heterocycle described as containing from “1 to 4 heteroatoms” means the heterocycle can contain 1, 2, 3 or 4 heteroatoms.
- substituted e.g., as in "aryl which is optionally substituted with one or more substituents "
- substituents include mono- and poly-substitution by a named substituent to the extent such single S and multiple substitution (including multiple substitution at the same site) is chemically allowed.
- oxy means an oxygen (O) atom.
- thio means a sulfur (S) atom.
- carbonyl means "CO.”
- any variable e.g., R 2 , R.3, etc.
- its definition in each occurrence is independent of its definition at every other occurrence. 10
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- Ci-5 alkylcarbonylamino Ci_ ⁇ ; alkyl substituent is equivalent to j 5
- the bond be attached to any of the substitutable ring atoms. If the ring system is polycyclic, it is intended mat the bond be attached to any of the suitable carbon atoms on the proximal ring only.
- A includes, but is not limited to, the following: azabenzimidazole, benzoiraidazolyl, benzofuryl, benzofurazanyl, benzopyrazolyl, benzothiazolyl, benzothienyl, benzotriazoryl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, chromanyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuryl, isochromanyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxazoline, isoxazoline,
- 35 oxetanyl, pyranyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, quinoxalinyl, tetrahydropyranyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, azetidinyl, aziridinyl, 1,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyrrolidinyl, morpholinyl, thiomo hol ⁇ n l,
- tbe heterocyclyl moiety in A includes azabenzimidazole, benzoimidazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiopbenyl, benzoxazolyl, furanyl, imidazolyl, indolyl, indazolyl, isobenzofuryl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazoly], oxazolyl, pyrazinyJ, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, quinazolinyl, quinolyl, quinoxalinyl, quinoxalinyl, tetrahydroisoquinolinyl, tetra
- A is selected from: benzoimidazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, furanyl, imidazolyl, indolyl, indazolyl, isobenzofuryl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, and thienyl, wherein A is optionally substituted with one or more R9 substituents.
- A is selected from: pyrazinyl, pyrazolyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, and thienyl, wherein A is optionally substituted with one or more R9 substituents.
- A is selected from pyrazinyl, pyrazolyl, pyridazinyl, pyridinyl, and pyrimidyl, wherein A is optionally substituted with one or more R9 substituents.
- A is selected from pyrazolyl, pyridinyl, pyridazinyl, and pyrimidyl, wherein A is optionally substituted with one or more R? substituents.
- A is chosen from pyrazolyl and pyridazinyl, wherein A is optionally substituted with one or more 9 substituents.
- Rl is selected from halogen, C ⁇ . ⁇ Q alkyl, -C2_i o alkenyl, -C2-10 alkynyl, cycloalkyl Co-ifj alkyl(oxy)o_i, heterocyclyl Co-lO alkyl(oxy)0-l» arylCfj-lO alkyl(oxy)o-i, Ci-io alkoxy (ca carboxyl aryl, carboxylcycloalkyl, carboxylheterocyclyl, Ci-ioalkyloxy Co-ioaltyl * hydroxy Co-ioalkyl, hydroxycarbonylCo-loalkoxy, Ci-io alkylthio, Cj-io alkylsulfinyl, aryl Crj-io alkylsulfinyl, heterocyclyl Co_io alkylsulfinyl, cycloalkyl Co-10 alkylsulfin
- Rl is selected from halogen, Ci-io alkyl, -C2-io alkenyl, - ⁇ - 10 alkynyl, cycloalkyl Co-io alkyl(oxy)o-l, heterocyclyl Co-ifj alkyl(oxy)fj-l, arylCrj-lO alkyl(°xy)0-l, Ci-io alkoxy (carbonyl)0-lCo-io alkyl, carboxyl Co_io alkyl, carboxyl aryl, carboxylcycloalkyl, carboxylheterocyclyl, C i . i oalkyloxy Co.
- i oalkyl hydroxy Cfj- 1 Oalkyl, hydroxycarbonylCo- 1 oalkoxy, Ci-io alkylthio, Ci_]0 alkylsulfinyl, aryl Co-io alkylsulfinyl, heterocyclyl Cfj-io alkylsulfinyl, cycloalkyl Co-io alkylsulfinyl, Ci-io alkylsulfonyl, aryl Co-io alkylsulfonyl, heterocyclylCo-io alkylsulfonyl, cycloalkyl Co_io alkylsulfonyl, nitro, perfluoroC i-6alkyl, and perfluoroC i_6alkoxy;
- Rl is optionally substituted with one or more R9 substituents.
- Rl is selected from halogen, phenylmethyl, pyridinyL, phenyl,
- Rl is selected from: cycloalkyl Co-10
- R 2 is selected from hydrogen, halogen, hydroxy!, - Ci- ⁇ 5 alkyl, C3-6 heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxy!, -SR ⁇ - R ⁇ or-CC ⁇ -R*.
- R3 is selected from hydrogen, halogen, hydroxy!, -Cj-6 alkyl, C3-6 heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxyl, -SR ⁇ -NR*2 or -C02-R'-
- L is selected from: is chosen from -(C1.3 alkyl)o- i RaCO-, -(C1.3 alkyl)0-l RaCO2-, -(Ci-3 alkyl)0-lCO2-, - ⁇ C ⁇ . 3 alkyl)0-l RaSO2-, -(C1.3 alkyl)0- iNRaCSNRb-, and -(C1.3 alkyl)o-lNRaCONRb.
- L is selected from -(CI -3 alkyl)0-lNRaCO-,
- L is—(CI -3 aIkyl) -lNRaCO-.
- R9 is selected from halogen, hydroxy, oxo, cyano, aryl, heterocyclyl, cycloalkyl, -Cl-6 alkyl, -Cl-6 alkoxy, aryloxy, heterocyclyloxy, -0(0-1 XC 1-10)perfluoroalkyl, -C02Ra, -OC02Ra, -OCO RbRc, , -SCO-6 alkyl and -S(0)nRjd, wherein R9 is optionally substituted by one or more substituents RIO.
- R9 is selected from halogen, hydroxy, cyano, aryl, heterocyclyl, cycloalkyl, -Cl-6 alkyl, -Cl-6 alkoxy, aryloxy, heterocyclyloxy, -O(0-lXCl-10)perfluoroalkyl, -C02Ra, and -OC02Ra wherein R9 is optionally substituted by one or more substituents RIO.
- R 10 is selected from hydroxy, aryl, heterocycloalkyl, heteroaryl, halogen, oxo, -C ⁇ -6 alkyl, Cj-6 alkoxy,C02H, cyano, 0(00)o.iCi-6 alkyl, trifluoromethoxy, trifluoroethoxy, -0(0-l)(Ci_io)perfluoroalkyl, Crj-io alkylaminocarbonyl, and H2.
- Another embodiment of the invention includes the following compounds: N- ⁇ 4-hydroxy-2-( 1 H-pyr3 ⁇ 4zol-l-yl)pyrimidm-5-yl)-2,2-diphenylacetamide;
- Structural representations of compounds having substhuents terminating with a methyl group may display the terminal methyl group either using the characters "CH3", e.g. "-CH3" or using a straight line representing the presence of the methyl group, e.g.
- Ri is a defined variable
- Rj is a defined variable
- the value of Ri may differ in each instance in which it occurs, and the value of Rj may differ in each instance in which it occurs.
- Ri and Ri are independently selected from the group consisting of methyl, ethyl, propyl and butyl, then (CRiR!2 can be
- Compounds described herein may contain an asymmetric center and may thus exist as enantiomers. Where the compounds according to the invention possess two or more asymmetric centers, they may additionally exist as diastereomers.
- the present invention includes all such possible stereoisomers as substantially pure resolved enantiomers, racemic mixtures thereof, as well as mixtures of diastereomers.
- the above Formula I is shown without a definitive stereochemistry at certain positions, The present invention includes all stereoisomers of Formula I and pharmaceutically acceptable salts and solvates thereof. Unless specifically mentioned otherwise, reference to one isomer applies to any of the possible isomers. Whenever the isomeric composition is unspecified, all possible isomers are included.
- Diastereoisomeric pairs of enantiomers may be separated by, for example, fractional crystallization from a suitable solvent, and the pair of enantiomers thus obtained may be separated into individual stereoisomers by conventional means, for example by the use of an optically active acid or base as a resolving agent or on a chiral HPLC column. Further, any enantiomer or diastereomer of a compound of the general Formula I may be obtained by stereospeciMc synthesis using optically pure starting materials or reagents of known configuration .
- Pharmaceutically acceptable salts include both the metallic (inorganic) salts and organic salts; a list of which is given in Remington's Pharmaceutical Sciences, 17th Edition, pg. 1418 (1985). It is well known to one skilled in the art that an appropriate salt form is chosen based on physical and chemical stability, flowabilhy, hydro-scopicity and solubility.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids. When the compound of the present invention is acidic, its corresponding salt can be conveniently prepared from inorganic bases or organic bases.
- Salts derived from such inorganic bases include aluminum, ammonium, calcium, copper (ic and ous), ferric, ferrous, lithium, magnesium, manganese (ic and ous), potassium, sodium, zinc and the like salts. Preferred are the ammonium, calcium, magnesium, potassium and sodium salts.
- Salts prepared from organic bases include salts of primary, secondary, and tertiary amines derived from both naturally occurring and synthetic sources.
- organic non-toxic bases from which salts can be formed include, for example, arginine, betaine, caffeine, choline, N,N -dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimeraylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, dicyclohexylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- the compound of the present invention When the compound of the present invention is basic, its corresponding salt can be conveniently prepared from inorganic or organic acids.
- Such acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid and the like.
- solvates of compounds of Formula I.
- the term "solvate” refers to a complex of variable stoichiometry formed by a solute (i.e., a compound of Formula I) or a pharmaceutically acceptable salt thereof and a solvent that does not interfere with the biological activity of the solute.
- solvents include, but are not limited to water, ethanol, and acetic acid.
- the solvent is water, the solvate is known as hydrate; hydrate includes, but is not limited to, hemi-, mono, sesqui-, di- and trihydrates.
- the present invention includes within its scope the use of prodrugs of the compounds of this invention.
- prodrugs will be functional derivatives of the compounds of mis invention which are readily convertible in vivo into the required compound.
- the term "administering" shall encompass the treatment of the various conditions described with a compound of formula I or with a compound which may not be a compound of formula L, but which converts to a compound of formula I in vivo after administration to the patient
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs," ed. H. Bundgaard, Elsevier, 1985.
- the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature.
- the present invention is meant to include all suitable isotopic variations of the compounds of generic Formula I.
- different isotopic forms of hydrogen (H) include protium (lH) and deuterium (2H).
- Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
- Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- Compounds of the present invention are inhibitors of hypoxia-inducible factor (RTF) prolyl hydroxylases, and as such are useful in the treatment and prevention of diseases and conditions in which HIF modulation is desirable, such as anemia and ischemia.
- Compounds of the invention can be used in a selective and controlled manner to induce hypoxia-inducible factor stabilization and to rapidly and reversibly stimulate erythropoietin production and secretion.
- another aspect of the present invention provides a method of treating or preventing a disease or condition in a mammal, the treatment or prevention of which xylase inhibition, which comprises administering an amount of a compound of Formula I that is effective for inhibiting HGDF prolyl hydroxylase.
- This aspect of the present invention further includes the use of a compound of Formula I in the manufacture of a medicament for the treatment or prevention of a disease or condition modulated by HIF prolyl hydroxylase.
- In one embodiment is a method of enhancing endogenous production of erythropoietin in a mammal which comprises administering to said mammal an amount of a compound of Formula I that is effective for enhancing endogenous production of erythropoietin.
- Another embodiment is a method of treating anemia in a mammal which comprises administering to said mammal a therapeutically effective amount of a compound of Formula I.
- Anemia includes, but is not limited to, chronic kidney disease anemia, chemotherapy-induced anemia (e.g., anemia resulting from antiviral drug regimens for infectious diseases, such as HTV and hepatitis C virus), anemia of chronic disease, anemia associated with cancer conditions, anemia resulting from radiation treatment for cancer, anemias of chronic immune disorders such as rheumatoid arthritis, inflammatory bowel disease, and lupus, and anemias due to menstruation or of senescence or in other individuals with iron processing deficiencies such as those who are iron-replete but unable to utilize iron properly.
- chemotherapy-induced anemia e.g., anemia resulting from antiviral drug regimens for infectious diseases, such as HTV and hepatitis C virus
- anemia of chronic disease e.g., anemia resulting from antiviral drug regimens
- Another embodiment is a method of treating ischemic diseases in a mammal, which comprises administering to said mammal a therapeutically effective amount of a compound of Formula I.
- Compounds of Formula I may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds of Formula I are useful. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I.
- a pharmaceutical composition containing such other drugs in addition to the compound of Formula I is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of Formula I.
- the compounds of this invention can be administered for the treatment or prevention of afflictions, diseases and illnesses according to the invention by any means that effects contact of the active ingredient compound with the site of action in the body of a warm-blooded animal.
- administration can be oral, topical, including transdermal, ocular, buccal, intranasal, inhalation, intravagjnal, rectal, intracisternal and parenteral.
- parenteral refers to modes of administration which include subcutaneous, intravenous, intramuscular, intraarticular injection or infusion, intrastemal and intraperitoneal.
- a warm-blooded animal is a member of the animal kingdom possessed of a homeo static mechanism and includes mammals and birds.
- the compounds can be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be tered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- the dosage administered will be dependent on the age, health and weight of the recipient, the extent of disease, kind of concurrent treatment, if any, frequency of treatment and the nature of the effect desired.
- a daily dosage of active ingredient compound will be from about 0.1-2000 milligrams per day. Ordinarily, from 10 to 500 milligrams per day in one or more applications is effective to obtain desired results.
- These dosages are the effective amounts for the treatment and prevention of afflictions, diseases and illnesses described above, e.g., anemia.
- compositions which comprises a compound of Formula I and a pharmaceutically acceptable carrier.
- composition as in pharmaceutical composition, is intended to encompass a product comprising the active
- compositions of the present invention encompass any composition made by admixing a compound of Formula I, additional active ingredients), and pharmaceutically acceptable excipients.
- compositions of the present invention comprise a compound represented by Formula I (or a pharmaceutically acceptable salt or solvate thereof) as an active ingredient, a pharmaceutically acceptable carrier and optionally other therapeutic ingredients or adjuvants.
- the compositions include compositions suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration, although the most suitable route in any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered.
- the pharmaceutical compositions may be
- the active ingredient can be administered orally in solid dosage forms, such as capsules, tablets, troches, dragees, granules and powders, or in liquid dosage forms, such as elixirs, syrups, , emulsions, dispersions, and suspensions.
- the active ingredient can also be administered parenteral ly, in sterile liquid dosage forms, such as dispersions, suspensions or solutions.
- dosages forms that can also be used to administer the active ingredient as an ointment, cream, drops, transdermal patch or powder for topical administration, as an ophthalmic solution or suspension formation, i.e., eye drops, for ocular administration, as an aerosol spray or powder composition for inhalation or intranasal administration, or as a cream, ointment, spray or suppository for rectal or vaginal administration.
- Gelatin capsules contain the active ingredient and powdered carriers, such as lactose, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets. Bo sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric coated for selective disintegration in the gastrointestinal tract.
- powdered carriers such as lactose, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets. Bo sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric coated for selective disintegration in the gastrointestinal tract.
- Liquid dosage forms for oral administration can contain coloring and flavoring to increase patient acceptance.
- water a suitable oil, saline, aqueous dextrose (glucose), and related sugar solutions and glycols such as propylene glycol or polyethylene gycols are suitable carriers for parenteral solutions.
- Solutions for parenteral administration preferably contain a water soluble salt of the active ingredient, suitable stabilizing agents, and if necessary, buffer substances.
- Antioxidizing agents such as sodium bisulfite, sodium sulfite, or ascorbic acid, either alone or combined, are suitable stabilizing agents.
- citric acid and its salts and sodium EDTA are also used.
- parenteral solutions can contain preservatives, such as benzalkonium chloride, methyl- or propylparaben, and chlorobutanol.
- Suitable pharmaceutical carriers are described in Remington's Pharmaceutical Sciences, A. Osol, a standard reference text in Ihis field.
- the compounds of the present invention may be conveniently delivered in the form of an aerosol spray presentation from pressurized packs or nebulisers.
- the compounds may also be delivered as powders which may be formulated and the powder composition may be inhaled with the aid of an insufflation powder inhaler device.
- the preferred delivery system for inhalation is a metered dose inhalation (MDI) aerosol, which may be formulated as a suspension or solution of a compound of Formula I in suitable propellents, such as fluorocarbons or hydrocarbons.
- MDI metered dose inhalation
- an ophthalmic preparation may be formulated with an appropriate weight percent solution or suspension of the compounds of Formula I in an appropriate ophthalmic vehicle, such that the compound is maintained in contact with the ocular surface for a sufficient time period to allow the compound to penetrate the corneal and internal regions of the eye.
- Useful pharmaceutical dosage-forms for administration of the compounds of this invention include, but are not limited to, hard and soft gelatin capsules, tablets, parenteral injectables, and oral suspensions.
- a large number of unit capsules are prepared by filling standard two-piece hard gelatin capsules each with 100 milligrams of powdered active ingredient, 150 milligrams of lactose, 50 milligrams of cellulose, and 6 milligrams magnesium stearate.
- a mixture of active ingredient in a digestible oil such as soybean oil, cottonseed oil or olive oil is prepared and injected by means of a positive displacement pump into gelatin to form soft gelatin capsules containing 100 milligrams of the active ingredient The capsules are washed and dried.
- a digestible oil such as soybean oil, cottonseed oil or olive oil
- a large number of tablets are prepared by conventional procedures so that the dosage unit is 100 milligrams of active ingredient, 0.2 milligrams of colloidal silicon dioxide, 5 milligrams of magnesium stearate, 275 milligrams of microcrystalline cellulose, 1 1 milligrams of starch and 98.8 milligrams of lactose. Appropriate coatings may be applied to increase palatability or delay absorption.
- a parenteral composition suitable for administration by injection is prepared by stirring 1.5% by weight of active ingredient in 10% by volume propylene glycol. The solution is made to volume with water for injection and sterilized.
- An aqueous suspension is prepared for oral administration so that each 5 milliliters contain 100 milligrams of finely divided active ingredient, 100 milligrams of sodium carboxymethyl cellulose, 5 milligrams of sodium benzoate, 1.0 grams of sorbitol solution, U.S.P., and 0.02S milliliters of vanillin.
- the same dosage forms can generally be used when the compounds of this invention are administered stepwise or in conjunction w h another therapeutic agent.
- the dosage form and administration route should be selected depending on the compatibility of the combined drugs.
- coadministration is understood to include the administration of the two agents concomitantly or sequentially, or alternatively as a fixed dose combination of the two active components.
- Compounds of the invention can be administered as the sole active ingredient or in combination with a second active ingredient, including other active ingredients known to be useful for improving the level of erythropoietin in a patient.
- the compounds of mis invention may be prepared by employing reactions as shown in the following schemes, in addition to other manipulations that are known in the literature or exemplified in the experimental procedures.
- Substituent numbering as shown in the schemes does not necessarily correlate to that used in the claims and often, for clarity, a single substituent is shown attached to the compound in place of multiple substituents which are allowed under the definitions of Formula I defined previously.
- This method involves the initial synthesis of substituted 4-hydroxypyrimidine-5- carboxylates of general formula 1S-4 and 1S-6.
- the synthesis of 4-hydroxypyrimidine-5-carboxylates exemplified in scheme 1 is based upon reported methods (Dostert, P.; Imbert, T.; Ancher, J.F.; Langlois, M.; Bucher, B.; Mocquet, G. Eur. J. Med Chem. 1982, 77, 437-44. Juby, P.F.; Hudyma, T.W.; Brown, M.; Essery, J.M.; Partyka, R.A. J. Med Chem. 1979, 22, 263-9).
- an amidine or a suitable salt thereof of general formula lS-1 is reacted with an optionally substituted diethyl methylenemalonate of general formula 1S-2.
- This reaction is usually conducted using a suitable base such as sodium or potassium ethoxide in ethanol, although other reaction conditions may also be applied.
- the alkoxide base and the alcohol solvent are chosen to correspond to the esters present in reagent 1S-2 to prevent the formation of mixtures of esters in the product of general formula 1S-3.
- the reaction is conducted at elevated temperature, typically at the reflux temperature of the solvent until reaction is complete (generally within 1-4 hours). It is also convenient to conduct this reaction under microwave heating in sealed reaction vessels. In this instance, the reaction is generally conducted at temperatures between 80 and 120 °C and the reactions are typically completed in 5-30 minutes.
- compounds of general formula 1S-3 are useful intermediates to prepare compounds of formula I of the present invention.
- compounds of general formula 1S-3 may be hydrolyzed using a suitable base (e.g. sodium or potassium hydroxide) to give acids of formula 1S-4; alternatively, they are converted to compounds of formula 1S-5, in which the hydroxy! group of the pyrimidine core is protected with a desired protecting group (e.g. PG is benzyl, para-methoxybenzyl, trityl, or tert-butyl- dimethyl silyl).
- a suitable base e.g. sodium or potassium hydroxide
- a desired protecting group e.g. PG is benzyl, para-methoxybenzyl, trityl, or tert-butyl- dimethyl silyl.
- amidines of general formula lS-1 When amidines of general formula lS-1 are not commercially available, they may be prepared by a variety of method k i h li A idi only prepared from nitriles using the Pinner reaction and variations thereof (see Amidines and N-substituted amidines. Dunn, Peter J. in Comprehensive Organic Functional Group Transformations 1995, 5, 741-82, 1161-308 Editors): atrizky, Alan R.; Meth-Cohn, Otto; Rees, Charles Wayne. Publisher: Elsevier, Oxford, UK). Amidines may also be prepared from esters using the method reported by Gielen et al. (Gielen, H.; Alonso-Alija, C; Hendrix, M.; Nie serveer, U.; Schauss, D. Tetrahedron Lett. 2002, 43, 419-21).
- the substituent A is selected to be a five-membered heterocyclic ring, it is possible that this heterocyclic group be bonded to the carbon atom at the 2-posttion of the pyrimidine ring through either a carbon-carbon or a carbon-nitrogen bond.
- the precursor for the substituent A is an amidine of general formula lS-1 and the method using lS-1 for the synthesis of the title compound of general formula I is as described in the preceding reaction schemes.
- the precursor for the substituent A is a guanidine of general formula 2S-7.
- Ester hydrolysis as described above affords compounds of formula 2S-10, which is useful to prepare compounds of general formula I wherein the group A is a five-membered heterocyclic group attached to the pyrimidine 2- position with a carbon-nitrogen bond.
- guanidine derivative (2S-7) bearing the desired substituents is not commercially available, h may be synthesized using reported methods for guanidine synthesis (e.g. the guanidinylation of amines). Numerous methods for the guanidinylation of amines are reported (see Katritzky, A.R.; Rogovoy, B.V. ARKIVOC 2005, 4, 49-87; http://www.arkat- usa.org ark/journal 2005 I04 Zefirov/ 1256/1256.pdf).
- Scheme 3 One general method is shown in Scheme 3, which entails the reaction of compounds of formula 3S-11 with 3,5-dimethyl-l-pyrazolylformamidinium nitrate to afford a guanidine of general formula 3S-12 using the method described by Fletcher et al. (Fletcher, D.L; Ganellin, C.R.; Piergentili, A.; Dunn, P.M.; Jenkinson, D.H. Bioorg. Med Chem. 2007, 15, 5457- 79).
- the title compounds of general formula I prepared as described above may be further modified using known methods and that the starting materials selected for use in the reaction schemes above may contain functional groups to enable said further transformation.
- aromatic rings in the title compounds of general formula I may be subjected to a variety of aromatic substitution reactions such as nitration, halogenation and the like.
- Aromatic substituent groups in the title compounds of general formula I bearing leaving groups such as halogens, triflates or the like can be employed in a variety of metal-catalyzed cross coupling reactions to incorporate new substitution patterns.
- palladium-catalyzed cross coupling reactions such as those described by Suzuki, Stille, Buchwald and others, may be used to introduce a variety of new substituent groups.
- Substituent groups that may be introduced using such cross-coupling methods include, but are not limited to, alkyl, alkenyl, alkynyl and aryl groups as well as acyl groups (e.g. carboxylic acids, esters, amides, or ketones), hydroxyl and amino or substituted amino groups.
- acyl groups e.g. carboxylic acids, esters, amides, or ketones
- the esters 4S-14 can be prepared by reaction of esters 4S-13 with a substituted carboxamide dimethyl acetal.
- reaction Schemes 1 and 2 may be further generalized when it is desired to prepare compounds of general formula I where neither of the R.3 or R4 substiruents are hydroxyl groups.
- Reaction Scheme 5 illustrates the process beginning with a beta-ketoester of general formula 5S-16 bearing the R.3 substituent.
- the ester of general formula 5S-16 is condensed with a carboxamide dimethyl acetal of general formula 2S-8 to afford the vinylogous amide of general formula 5S-17.
- the intermediate 5S-17 is then reacted with an amidine derivative of general formula lS-1 using the method of Schenone et al. (Schenone, P.; Sansebastiano, L.; Mosti, L. J. Heterocyclic Chem. 1990, 27, 295) to afford the alkyl pyrimidine-5-carboxylate of general formula 5S-18.
- Hydrolysis of compounds of formula 5S-18 under suitable conditions e.g. KOH or NaOH in EtOH-H 2 0, heating when necessary
- the compounds of general formula 5S-19 are then converted to the title compounds of general formula I using the methods described previously.
- compounds of formula I are prepared via pyrimidine ring formation reactions (e.g. Schemes 1, 2, 4, 5) with the desired substituents (e.g. A, R 2 , R 3 and L) at various positions.
- the desired substituents on the pyrimidine core can be introduced after the pyrimidine ring is formed, which can be achieved using synthetic methods reported in the literature.
- the hydroxy! group present at the pyrimidine 4-position in compounds of general formulae 1S-3, 1S-4, or 4S-15 may be converted to a halogen substituent upon reaction with a suitable halogenating reagent (e.g. POCb, BBr 3 , etc.).
- carboxylic acids such as compounds of formula 5S-19 can be converted to their corresponding acyl azide such as compounds of formula 6S-22, and under suitable thermal rearrangement reaction conditions acyl azides such as 6S-22 can be converted to their corresponding isocyanate, and subsequent reactions of the resulting isocyanate with various nucleophiles such as alcohols, amines and thiols can produce compounds of formula 6S-23 wherein L is a carbamate (-NHCOO-), urea (-NHCONH-), or thiocarbamate (-NHCOS-).
- HPLC High performance liquid chromatography
- Agilent 1100 series HPLC Waters C18 XTerra 3.5 urn 3.0 x50 mm column with gradient 10:90-100 v/v CH 3 C H 2 0 + v 0.05 % TFA over 3.75 min then hold at 100 C3 ⁇ 4CN + v 0.05 % TFA for 1.75 min; flow rate 1.0 mL/min, UV wavelength 254 nm (all HPLC MS data was generated with this method unless indicated otherwise).
- Analytical HPLC/MS - Basic Method Mass analysis was performed on a Waters Micromass* ZQTM with electrospray ionization in positive ion detection mode.
- HPLC High performance liquid chromatography
- Flash chromatography was performed using a Biotage Horizon or SP1 Flash Chromatography apparatus (Dyax Corp.) on silica gel (32-63 uM particle size, KP-Sil 60 A packing material type) in pre-packed cartridges or using an ISCO CombiFlashTM Sq 16x or CombiFlash* CompanionTM apparatus on silica gel (32-63 ⁇ , 60 A) in pre-packed cartridges.
- Microwave reactions were carried out on a Biotage InitiatorTM 2.0 or CEM DiscoverTM system.
- Preparative HPLC/MS - Standard Method Mass analysis was performed on a Waters Micromass* ZQTM with electrospray ionization in positive ion detection mode.
- HPLC High performance liquid chromatography
- Step A 2-Tosv ⁇ 23 iihvdn)Pvridazine-3-carbonitrile
- Step C Pvridazine-3- ⁇ rhn 3 ⁇ 4 imiH fl niide hydrochloride ⁇ U-d)
- Step E 4-fbeiizyloxy 2 ⁇ Dyridazin-3-vnpYriinidine-S-carboiyllc acid ( 1-f)
- Step B Benzvl 4 ⁇ iizvloxvV2 1H-Dvrazo ⁇ l-vnDvrimidine-S-carboxvlatc (2-d)
- Step D 4-(b nzvloiv>-2-ilH-pvrazQl-l-vnDvrimidin-5-amine (2-f)
- 2,2-diphenylacetic acid 110 mg, 0.52 mmol was dissolved in DMF (10 mL) and EDC (100 mg, 0.52 mmol) and HOBt (70 mg, 0.52 mmol) were added. The mixture was stirred at rt for 5 mins before Et3N (130 mg, 1.3 mmol) and compound 2-f (115 mg, 0.43 mmol) were added. The resulting mixture was stirred at the ambient temperature for 24 h. The resulting mixture was diluted with water followed by extraction with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated under vacuum.
- Step A Ethvl 4-hvdrorv-2 1H-Dvrazol-l-vnDvrimidine-5-carboxvlate (3-a3 ⁇ 4
- Step C 4-fbeiizvloxvV2 ⁇ 1H-DVi ⁇ l-l-vnDvrimidine-S-carboxvlic acid (3-ci)
- Step B Preparation of amides (Reversed amide 3-f)
- reaction mixture was concentrated under vacuum and the residue was taken into THF (10ml) and treated with 2N lithium hydroxide (10ml) The mixture was stirred at room temperature for 30 min to 12 hours. For some of the compounds (ca. 20%) a solid precipitated out, thus the solid was collected via filtration to provide the product For compounds that did not precipitate out, the THF layer was separated and concentrated to afford the crude product, which was further purified by recrystallization using a mixed solvent system of petroleum ether/ethyl acetate/methanol (solvent ratio was optimized for each compound to produce the best crystallization conditions).
- Table 1 discloses compounds 3-1 through 3-55 that were made in accordance to the general procedure outlined in Example 3 and by utilizing the appropriate carboxylic acid. Table 1
- Table 3 discloses compounds 5-1 through 5-11 that were made in accordance to the general procedure outlined in Example 5 and by utilizing the appropriate urea or carbamate.
- Step B 4-hvdroiY-2 Dvridiji-2-vnDvrimidine-5-carhoxvlic acid (10-c)
- Table 5 discloses compounds 12-2 and 12-3 that were made in accordance to the general procedure outlined in Example 13 and by utilizing the appropriate amine.
- Examples 1 through 13 of the present invention have been found to inhibit the interaction between PHD2 and a HIF peptide and exhibit IC50 values ranging between 0.1 nanomolar to 10 micromolar.
- assays that may be useful to detect favorable activity are disclosed in the following publications: Oehme, F., et al., Anal. Biochem. 330:74- 80 (2004); Hirsilft, M, et al., J. Bio. Chem. 278 (33): 30772-30780 (2005); Hyunju, C, et al., Biochem. Bjophys, Res, Comm. 330 (2005) 275-280; and Hewitson, K. S., et al., Methods in Enzvmologv. (Oxygen Biology and Hypoxia); Elsevier Publisher (2007), pg. 25-42 (ISSN: 0076-6879).
- the biological activity of the present compounds may be evaluated using assays described herein below:
- test compound in DMSO and 20 ⁇ of assay buffer (50 mM Tris pH 7.4/0.01% Tween-20/0.1 mg ml bovine serum albumin 10 ⁇ ferrous sulfate/1 mM sodium ascorbate/20 ⁇ catalase) containing 0.15 g ml FLAG-tagged full length PHD2 expressed in and purified from baculovirus-infected Sf9 cells.
- assay buffer 50 mM Tris pH 7.4/0.01% Tween-20/0.1 mg ml bovine serum albumin 10 ⁇ ferrous sulfate/1 mM sodium ascorbate/20 ⁇ catalase
- Inhibition of the catalytic activity of HlP-PHDl and HIF-PHD3 can be determined similarly.
- Table 6 depicts the inhibition of HIF PHD2 activity expressed as IC50 (nM), for the exemplified compounds, 1-1, 1-2, 2-1, 3-1 to 3-6, 4-1, and 5-1 of the present invention.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to substituted pyrimidines useful as HIF prolyl hydroxylase inhibitors to treat anemia and like conditions.
Description
TITLE OF THE INVENTION
SUBSTITUTED PYRJMIDINES
CROSS-REFERENCE TO RELATED APPLICATIONS
This application claims the benefit of International Application No.
PCT/CN2010/071974, filed April 21, 2010, the disclosure of which is incorporated by reference herein in its entirety.
BACKGROUND OF THE INVENTION
The insufficient delivery of oxygen to cells and tissues is associated with anemia, which is defined as a deficiency in the blood's oxygen-carrying capacity, and ischemia, in which restrictions in blood supply are caused by a constriction or blockage of blood vessels. Anemia can be caused by the loss of red blood cells (hemorrhage), excessive red blood cell destruction (hemolysis) or deficiencies in erythropoiesis (production of red blood cells from precursors found in the bone marrow). The symptoms of anemia can include weakness, dizziness, fatigue, pallor, impairment of cognitive function and a genera] reduction in quality of life. Chronic and/or severe anemia can lead to the exacerbation of myocardial, cerebral or peripheral ischemia and to heart failure. Ischemia is defined as an absolute or relative shortage of oxygen to a tissue or organ and can result from disorders such as atherosclerosis, diabetes, thromboembolisms, hypotension, etc. The heart, brain and kidney are especially sensitive to ischemic stress caused by low blood supply.
The primary pharmacological treatment for anemia is administration of some variant of recombinant human erythropoietin (EPO). For anemias associated with kidney disease, chemotherapy- induced anemia, anemia from HTV-therapy or anemia due to blood loss, recombinant EPO is administered to enhance the supply of the hormone, correct the shortage of red blood cells and increase the blood's oxygen-carrying capacity. EPO replacement is not always sufficient to stimulate optimal erythropoiesis (e.g., in patients with iron processing deficiencies) and has associated risks.
Hypoxia-inducible factor (HTF) has been identified as a primary regulator of the cellular response to low oxygen. HTJF is a heterodimeric gene transcription factor consisting of a highly regulated a-subunit (HEF-a) and a constitutively expressed β-subunit (HIF-β, also known as ARNT, or aryl hydrocarbon receptor nuclear transporter). HTF target genes are reported to be associated with various aspects of erythropoiesis (e.g., erythropoietin (EPO) and EPO receptor), glycolysis and angiogenesis (e.g., vascular endothelial growth factor (VEGF)). Genes for proteins involved in iron absorption, transport and utilization as well as heme synthesis are also targets of H1F.
Under normal oxygenation, HTF-a is a substrate in a reaction with molecular oxygen, which is catalyzed by a family of iron(TJ)-, 2-ketoglutarate- and ascorbate-dependent dioxygenase enzymes called PHD-1 (EGLN2, or egg laying abnormal 9 homolog 2, PHD2 (EGLN1), and PHD3 (EGLN3). Proline residues of HTF-a are hydroxylated (e.g., Pro-402 and Pro-564 of HIF-Ια) and the resulting product is a target of the tumor suppressor protein von-Hippel Lindau, a component of an E3
ubiquhin ligase multiprotein complex involved in protein ubiquitination. Under low oxygenation, the HEF-a hydroxylation reaction is less efficient and HIF- a is available to dimerize with HIF-β. HEF dimers are translocated to the cell nucleus where they bind to a hypoxia-responsive enhancer element of HIF target genes.
Cellular levels of HIF are known to increase under conditions of hypoxia and after exposure to hypoxia mimetic agents. The latter includes, but is not limited to, specific metal ions (e.g., cobalt, nickel, manganese), iron chelators (e.g., desferrioxamine) and analogs of 2-ketoglurate (e.g., N- oxaryl glycine). The compounds of the present invention inhibit the HIF prolyl hydroxylases (PHD-1 , PHD-2, PHD-3) and can also serve to modulate HIF levels. These compounds therefore have utility for the treatment and/or prevention of disorders or conditions where HIF modulation is desirable, such as anemia and ischemia. As an alternative to recombinant erythropoietin therapy, the compounds of the present invention provide a simpler and broader method for the management of anemia.
SUMMARY OF THE INVENTION
The present invention concerns compounds of formula I

which inhibit HIF prolyl hydroxylase, their use for enhancing endogenous production of erythropoietin, and for treating conditions associated with reduced endogenous production of erythropoietin such as anemia and like conditions, as well as pharmaceutical compositions comprising such a compound and a pharmaceutical carrier.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides compounds of formula I and pharmaceutically acceptable salts and solvates thereof:
wherein

A is heteroaryl optionally substituted with one or more R? substituents
L is chosen from a bond, aryl, heteroaryl, -(C1.3 alkyl)o-iS(Ci-3 alkyl)o-l-, -(C1.3 alkylJo-lNRaCO-, (Ci-3 alkyl)0-lNRaCO2-, -(C1.3 alkyl)0-lCO2-, -(Cj.3 alkyl)0-lNRaSO2-, -(C1.3 alkylJo. iNRaO-, -(C1.3 alky o-lNRaCsNRb., - Ci-3 alkyl)0-lNRaCONRb-, and -(C1.3 alkyl)o- iNRaCOS-;
R2 and are independently selected from hydrogen, halogen, hydroxy!, Cj-g alkyl, C3-6 heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxy I, -SR.*, -NR*2 or-C02-R*;
R1 is selected from
halogen,
Ci-io alkyl,
-C2_io alkenyl,
-C2-IO alkynyl,
cycloalkyl Co-io alkyl(oxy)0-l,
heterocyclyl Co_io alkyl(oxy)o-],
arylCo-io alkyl(oxy)0-l,
Cj-io alkoxy (cxarbonyl)o.iCo.io alkyl,
carboxyl Qj-io alkyl,
carboxyl aryl,
carboxylcycloalkyl,
carboxylheterocyclyl,
Ci-ioalkyloxy Co-ioalkyl,
hydroxy Co-ioalkyl,
hydrox carbonylCo- 1 oalkoxy,
Ci-io alkylthio,
Ci-io alkylsulfinyl,
aryl Cfj-10 alkylsulfinyl,
heterocyclyl Cfj-io alkylsulfinyl,
cycloalkyl Cfj-io alkylsulfinyl,
Ci-io alkylsulfonyl,
aryl Co-10 alkylsulfonyl,
heterocyclylCo-10 alkylsulfonyl,
cycloalkyl Co-10 alkylsulfonyl,
nitro,
perfluoroCmsalkyl, and
perfluoroC 1 ^alkoxy ;
wherein in Rl said alkyl, alkenyl, alkynyl, cycloalkenyl, aryl, perfluoralkyl, perfluoroalkoxy,
heterocyclyl, and cycloalkyl are each optionally substituted with one or more R? substituents;
optionally, R* and L are linked together to form a ring of 5 to 8 atoms optionally substituted with one or more substituents R?; where said ring has 0, 1 , or 2 heteroatoms independently selected from -NR -, -O- and -S(0)n-;
is selected from halogen, hydroxy, oxo, cyano, aryl, heterocyclyl, cycloalkyl, -Ci-6 alkyl, -Cl-6 alkoxy, aryloxy, heterocyclyloxy, -0(0-l)(Ci-io)perfluorc*lkyl, -C02 a, -NRbRc, -CONRbRC
-OC02Ra, -OCONRbRc, - dC02Ra, - dCO bRc -SQ)-6 alkyl and -S(0)nRd, wherein said aryl, heteroaryl, heterocycloaJkyl, alkoxy, aryloxy, heteroaryloxy, heterocycloalkyloxy are optionally substituted by one or more substituents RlO;
RlO is selected from hydroxy, aryl, heterocycloalkyl, heteroaryl, halogen, oxo, -Ci-6 alkyl, Cj-6 alkoxy, halogen, CO2H, cyano, O(00)o.i C 1 -6 alkyl, NO2, trifluoromethoxy, trifluoroethoxy, -O(0- l)(Ci.io)perfluoroalkyl, Cfj-lO alkylaminocarbonylamino, Q)-10 alkyloxycarbonylaminoCo-io alkyl, Crj-io alkylcarbonylaminoCo-io alkyl, Co-10 alkylaminosulfonylaminoCo-io alkyl, Cfj-io aJkylsulfonylaminoCo-lO alkyl, Co-io alkylsulfonyl, Co-10 alkylaminosulfonyl, Co.10
alkylaminocarbonyl, -(C=0)N(Co-6 alkyl^ -S(C()-6 alkyl), and H2;
n is i or 2;
Ra is chosen from hydrogen; -Cl-10 alkyl, -(Cj-6 alkyl)C3-8 cycloalkyl; and
-(Ci-6 alkyl)phenyl; and
Rb, Rc, and Rd are each independently chosen from hydrogen, -Cl-10 alkyl,C3-8 cycloalkyl, aryl, and heterocyclyl.
Illustrative but nonlimiting examples of compounds of the invention are the following: N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2,2-diphenylacetarnide;
2- chloro-N-(4-hydiOxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
3- chloro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
4-chloro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
3- (trifluoromethyl)-N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidjn-5-yl)benz
4- (trifluoromethyl)-N-{4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
N-(4-hydroxy-2-(l H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(naphthalen-3-yl)acetamide;
2-cyano-N-(4-hydroxy-2-< 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
3-cyano-N-(4-hydroxy-2-(lH-pyrazol-l-yl)ryrimidin-5-yl)benzami
4-cyano-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -y l)pyrimidin-5 -y1)benzamide;
2-(trifluoromethyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -y l)pyrimidin-5 -yl 2-methoxybenzamide;
N-<4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-methoxybenzamide;
N-(4-hydroxy-2-(lH-pyra2ol-l-yl)pyrimidin-5-yl)-4-methoxybenzamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(methylsulfonyl)benzamide;
N-(4-hydroxy-2-( I H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-(methylsulfonyl)benzamide;
N 4-hydroxy-2-{ 1 H-pyra∞l- 1 -y l)pyri
N-[4-hydroxy-2-( 1 H-pyrazoJ- 1 -yl)pyrimidm-5-yl]biphenyl-2-carboxam ide;
N-[4-hydroxy-2-( 1 H-pyra∞l- 1 -yl )pyri^
N-[4-hydroxy-2-(lH-pyi¾-∞l-l-yl)pyrimidin-5-yl]biphenyl^^^
N 4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl 2-phenoxybenzamW
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyiimidin-5-yl)-3-phenoxybenzamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimW^
2-(2-chlorophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yI)pyrimidin-5-yl)acetamide;
2-(3-chlorophenyl)-N-(4-hydroxy-2-(lH-pyra∞^
2-(4-chlorophenyl)-N-(4-hydiOxy-2-(lH-pyrazol-l-yl)pyrimidi
2-(3-cyanophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -y1)pyrimidin-5-yl)acetamide;
2-(2-(trifluoromethyl)phenyl N-(4-hydrox^^
2-(3-{trifluoromethyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(4-(trifluoronieithyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yI)pyrimidin-5-yl)acetam.de;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(2-rnethoxyphenyl)acetamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-2-(3-me1 oxyph
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl 2-(4-me1hoxyphenyl)acetarnide;
N-(4-hydroxy-2-(lH-pyrazol-l-yI)rjyrimidin-5-yl 2-phenylpropan
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl 2-phenylbutai-am
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl>-3-me1hyl-2-phenylbutanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-methyl-2-phenylpentanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl 2-methyl-2-phenylpropanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)- 1 -phenylcycloproDanecarboxam.de;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(naphthalen- 1 -yl)acetamide;
2-(benzo[d] [ 1 ]dioxol-5-yl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetaniide;
N-(4-hydroxy-2-(lH-pyn^l-l-yl)pyrimid»n-5-yl)-2-(pyridin-4-yl)a∞
2-(biphenyl-4-yl)-N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)a(»tami
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)heptanamide;
N-(4-hydroxy-2-(l H-pyrazol- l-yl)pyrimid^
N-(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-4-phenylbutanamide;
N-(4-hydroxy-2-(l H-pyrazol- 1 -yl)pyrimidin-5-yl)cyclobexanecarboxamide;
tert-buty 14-(4-hydroxy-2-( 1 Η-pyrazol·
2-cyclohexy l-N-(4-hydroxy-2-( 1 H-pyrazo 101.01- 1 -yl)pyrimidin-5-yl)acetamide;
tert-butyl 3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-ylcarbamoyl)piperidine- 1-carboxylate;
tert-butyl4-((4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-ylcarbamoyl)methyl)piperidine- 1 -carboxylate; tert-butyl 3-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-ylc^^
1 ^2^,4-tetrahydro-N-(4-hydroxy-2-( 1 H-pyrazol-1 -yI)pyrimidin-5-yl)naphthalenfr-l-<¾irt)Oxamide; 2,3-dihydro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)- 1 H-indene- 1 -car oxamide;
1 ^,3,4-tetrahydro-N-(4-hydroxy-2-( ] H-pyrazol- 1 -yl)pyrimidin-5-yl)naphthalene-2-carboxamide; 2 -dihydro-N-(4-hydroxy-2-( I H-pyrazol- 1 -yl)pyrimidin-5-yl)- 1 H-indene-2-carboxamide;
4-chlon N-[4-hydroxy-2-(lH-pyra∞
1 -(4-chlorophenyl)-N-[4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidm-5-yl]methanesulfonamide;
3-chloro-N-[4-hydroxy-2- H-pyrazol-l-yl)pyr^
N-[4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl]- 1 -[4-(trifluoromethyl)phenyl]methanesulfonaniide; l-(4-hydroxy-2-{l H-pyrazol- l-yl)pyrimM
1 -benzyl-3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea;
1 -(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidirj-5-yl)-3-( 1 -phenylethyl)urea;
1 -cycIohexyl-3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea;
1 -cyclopentyl-3-{4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea;
ert-buty ] -3 -(4-hy drox -2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea;
3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyriinidin-5-yl)- 1 , 1 -diphenylurea;
3- (4-hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl)- 1 -isopropyl- 1 -phenylurea;
phenyl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
cyclohexyl 4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl carbamate;
cyclopentyl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
tert-butyl 4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl carbamate;
butyl 4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-y] carbamate;
4- methoxyphenyl 4-hydroxy-2-(lH-r¾ azol-l-yl)pyrimidjn-5-yl carbamate;
3,3-dimethylburyl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
phenethyl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
biphenyl-4-yl 4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl carbamate;
naphthalen-2-yl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
1 -benzhydryl-3-(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)thiourea;
1 -(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-( 1 -pbenylethyl)th iourea;
1 -(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-(2-pbenylpropan-2-yl)thiourea;
benzhydryl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidine-5-carboxylate;
N^hydroxy^-ipyridin^-y^pyrimidm-S-yl^^-dipbenylacetamide;
5- ( 1 -phenyl- 1 H-pyrazol-4-yl)-2-( 1 H-pyrazol- 1 -yl)pyrimidin-4-ol;
5-( 1 -benzyl- 1 H-pyrazoI-4-yl)-2-( 1 H-pyrazol- 1 -yl)pyrimidin-4-ol;
5-phenyl-2-( 1 H-pyrazol-1 -yl)pyrimidin-4-ol;
N-(4-chlorophenyl)-2-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)acetamide;
N-cyclohexyl-2-(4-hydroxy-2-{lH-pyrazol-l-yl)pyrimidin-5-yl)acetamide;
2-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-N-isopropylacetamide;
4-hydroxy-2^yridazin-3-yI N-tosylpyrimidine-5-cai^
and pharmaceutically acceptable salts and solvates thereof.
As used herein except where noted, "alkyl" is intended to include both branched- and straight-chain saturated aliphatic hydrocarbon groups, including all isomers, having the specified number of carbon atoms. Commonly used abbreviations for alkyl groups are used throughout the specification, e.g. methyl may be represented by "Me" or CH3) ethyl may be represented by "Et" or CH2CH3, propyl may be represented by "Pr" or CH2CH2CH}, butyl may be represented by "Bu" or CH2CH2CH2CH3 , etc. "Ci_6 alkyl" (or "Cj-Cg alkyl") for example, means linear or branched chain alkyl groups, including all isomers, having the specified number of carbon atoms. Ci-6 alkyl includes all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. "C1-4 alkyl" means n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. The term "alkylene" refers to both branched- and straight-chain saturated aliphatic hydrocarbon groups, including all isomers, having the specified number of carbons, and having two terminal end chain attachments. For illustration, the term ^nsubstitoted A-Qalkylene-B" represents A-CH2-CH2-CH2-CH2-B. The terra "alkoxy" represents a linear or branched alkyl group of indicated number of carbon atoms attached through an oxygen bridge.
Unless otherwise specifically noted as only "unsubstituted" or only "substituted", or when substituents are enumerated, alkyl (either as a stand alone radical or as part of a radical such as alkoxy, alkylthio and aralkyl) groups are unsubstituted or substituted with 1 to 3 substituents on each carbon atom, with halo, C1-C20 CF3» N¾ N(Ci-C6 alkyl)2, Ν<¾, oxo, CN, N3, -OH, -0(Ci-C6 alkyl), C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, (Cfj-Cg alkyl) S(O)0-2-, (Co-C6 alkyl)S(O)0- 2(Co-C6 alkyl)-, (C0-C6 alkyl)C(0)NH-, H2N-C(NH , -0(Ci-C6 alkyl)CF3, (C0-C6 alkyl)C(0)-, (Co- C6 alkyl)OC(0)-, (Q)-C6 alkyl)0(Ci-C6 alkyl)-, (C0-C6 alkyl)C(0)i.2(Co-C6 alkyl)-, (Co-C* alkyl)OC(0)NH-, -NH(C]-C6 alkyl)NHC(0)NH(Ci-C6 alkyl), NHC(0)OCi-C6 alkyl, -NH(Ci-C6 alkyl) HS02(Ci-C6 alkyl), - C0-C6 alkyl)NHS02(Ci-C6 alkyl), aryl, aralkyl, heterocycle,
heterocyclylalkyl, halo-aryl, halo-aralkyl, halo-heterocycle, halo-heterocyclylalkyl, cyano-aryl, cyano- aralkyl, cyano-heterocycle and cyano-heterocyclylalkyl.
The term uCQ" as employed in expressions such as "Co_6 alkyl" means a direct covalent bond; or when the term appears at the terminus of a substituent, Co-6 alkyl means hydrogen or Cl-6alkyl. Similarly, when an integer defining the presence of a certain number of atoms in a group is equal to zero, it means that the atoms adjacent thereto are connected directly by a bond. For example, in the structure , wherein s is an integer equal to zero, 1 or 2, the structure is

The term "C3-8 cycloalkyl" (or "C3-Cg cycloalkyl") means a cyclic ring of an alkane having three to eight total carbon atoms (i.e., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, or cyclooctyl). The terms "C3-7 cycloalkyl", "C3-6 cycloalkyl", "C5-7 cycloalkyl" and the like have analogous meanings.
The term "halogen" (or "halo") refers to fluorine, chlorine, bromine and iodine
(alternatively referred to as fluoro (F), chloro (CI), bromo (Br), and iodo (I)).
The term "aryl" refers to aromatic mono- and poly-carbocyclic ring systems, wherein the individual carbocyclic rings in the polyring systems are fused or attached to each other via a single bond. Suitable aryl groups include phenyl, naphthyl, and biphenylenyl.
The term "carbocycle" (and variations thereof such as "carbocyclic" or "carbocyclyl") as used herein, unless otherwise indicated, refers to (i) a C3 to Cg monocyclic, saturated or unsaturated ring or (ii) a C7 to C12 bicyclic saturated or unsaturated ring system. Each ring in (ii) is either independent of, or fused to, the other ring, and each ring is saturated or unsaturated. The carbocycle may be attached to the rest of the molecule at any carbon atom which results in a stable compound. The fused bicyclic carbocycles are a subset of the carbocycles; i.e., the term "fused bicyclic carbocycle" generally refers to a C7 to C] 0 bicyclic ring system in which each ring is saturated or unsaturated and two adjacent carbon atoms are shared by each of the rings in the ring system. A fused bicyclic carbocycle in which one ring is saturated and the other is saturated is a saturated bicyclic ring system. A fused bicyclic carbocycle in which one ring is benzene and the other is saturated is an unsaturated bicyclic ring system. A fused bicyclic carbocycle in which one ring is benzene and the other is unsaturated is an unsaturated ring system. Saturated carbocyclic rings are also referred to as cycloalkyl rings, e.g., cyclopropyl, cyclobutyl, etc. Unless otherwise noted, carbocycle is unsubstituted or substituted with C]_6 alkyl, Ci-6 alkenyl, C]-6 alkynyl, aryl, halogen, N¾ or OH. A subset of the fused bicyclic unsaturated carbocycles are those bicyclic carbocycles in which one ring is a benzene ring and the other ring is saturated or unsaturated, with attachment via any carbon atom that results in a stable compound. Representative examples of this subset include the following:

The term "heterocycle" (and variations thereof such as "heterocyclic" or "heterocycly ) broadly refers to (i) a stable 4- to 8-membered, saturated or unsaturated monocyclic ring, or (ii) a stable 7- to 12-membered bicyclic ring system, wherein each ring in (ii) is independent of, or fused to, the other ring or rings and each ring is saturated or unsaturated, and the monocyclic ring or bicyclic ring system contains one or more heteroatoms (e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms) selected from N, O and S and a balance of carbon atoms (the monocyclic ring typically contains at least one carbon atom and the ring systems typically contain at least two carbon atoms); and wherein any one or more of the nitrogen and sulfur h t t i ti ll idi d d e or more of the nitrogen
heteroatoms is optionally quaternized. Unless otherwise specified, the heterocyclic ring may be attached at any heteroatom or carbon atom, provided that attachment results in the creation of a stable structure. Unless otherwise specified, when the heterocyclic ring has substituents, it is understood that the substituents may be attached to any atom in the ring, whether a heteroatom or a carbon atom, provided that a stable chemical structure results.
Non limiting examples of heterocyclylic moieties include, but are not limited to, the following: azepanyl, azabenzimidazole, benzoimidazolyl, benzofuryl, benzofurazanyl, benzopyrazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, chromanyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuryl, isochromanyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazoryl, naphthpyridinyl, oxadiazolyl, oxazoryl, oxazolinyl, isooxazolinyl, oxetanyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, quinoxalinyl, tetrahydropyranyl, tetrahydroisoquinolinyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, azetidinyl, aziridinyl, 1,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyrrolidinyl, mo holinyl, thiomo holinyl, dihydrobenzoimidazolyl, dihydrobenzofuryl, dihydrobenzothiophenyl, dihydrobenzoxazolyl, dihydrofuryl, dihydroimidazolyl, dihydroindolyl, dihydroisooxazolyl, dihydroisothiazolyl, dihydrooxadiazolyl, dihydrooxazolyl, dihydropyrazinyl, dihydropyrazolyl, dibydropyridinyl, tetrabydroquinolinyl, tetrahydroisoquinolinyl, dihydropyrimidinyl, dihydropyrrolyl, dihydroquinolinyl, dihydrotetrazolyl, dihydrothiadiazolyl, dihydrothiazolyl, dihydrothienyl, dihydrotriazolyl, dihydroazetidinyl, methylenedioxybenzoyl, tetrahydrofuryl, tetrahydrothienyl, tetrahidroquinolinyl, 2,3-dihydrobenzofuryl, 2,3-dihydrobenzo-l,4-dioxinyl, imidazo(2,l-bXl,3)thiazole, and benzo-l,3-dioxoryl.
Saturated heterocyclics form a subset of the heterocycles; i.e., the term "saturated heterocyclic" generally refers to a heterocycle as defined above in which the entire ring system (whether mono- or poly-cyclic) is saturated. The term "saturated heterocyclic ring" refers to a 4- to 8-membered saturated monocyclic ring or a stable 7- to 12-membered bicyclic ring system which consists of carbon atoms and one or more heteroatoms selected from N, O and S. Representative examples include piperidinyl, piperazinyl, azepanyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl,
isoxazolidinyl, morpholinyl, thiomorpholiny], thiazolidinyl, isothiazolidinyl, and tetrahydrofuryl (or tetrahydrofuranyl).
Heteroaromatics form another subset of the heterocycles; i.e., the term "heteroaromatic" (alternatively "heteroary ) generally refers to a heterocycle as defined above in which the entire ring system (whether mono- or poly-cyclic) is an aromatic ring system. The term "heteroaromatic ring" refers a 5- or 6-membered monocyclic aromatic ring or a 7- to 12-membered bicyclic which consists of carbon atoms and one or more heteroatoms selected from N, O and S. In the case of substituted heteroaryl rings containing at least one nitrogen atom (e.g., pyridine), such substitutions can be those resulting in N-oxide formation. Representative examples of heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl,
pyrimidinyl, pyridazinyl, thienyl (or thiophcnyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl.
Representative examples of bicyclic heterocycles include benzotriazolyl, indolyl, isoindolyl, indazolyl, indolinyl, isoindolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, chromanyl, isochromanyl, tetrahydroquinolinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl,
2,3-dihydroDenzofuryl, 2r3-dihydrobenzo-l,4-dioxinyI (i.e., imia¾zo(2,l-b)(l,3)thiazole, (i.e.,
 and benzo-l,3-dioxolyl (i.e., In certain contexts herein, is alternatively
and benzo-l,3-dioxolyl (i.e., In certain contexts herein, is alternatively



referred to as phenyl having as a substituent methylenedioxy attached to two adjacent carbon atoms.
The terms "arylalkyl" and "alkylaryl" include an alkyl portion where alkyl is as defined above and include an aryl portion where aryl is as defined above. Examples of arylalkyl include, but are not limited to, benzyl, phenylethyl, phenylpropyl, naphthylmethyl, and naphthyletbyl. Examples of alkylaryl include, but are not limited to, toluene, ethylbenzene, propylbenzene, methylpyridine, ethylpyridine, propylpyridine and butylpyridine.
Unless otherwise specifically noted as only "unsubstituted" or only "substituted", or when substituents are specifically enumerated, cycloalkyl, aryl (including phenyl) and heterocycle
(including heteroaryl) groups are unsubstituted or substituted. As used herein, the terms "substituted C3- Cjo cycloalky , "substituted aryl (including phenyl)" and "substituted heterocycle" are intended to include the cyclic group containing from 1 to 3 substituents in addition to the point of attachment to the rest of the compound. Preferably, the substituents are selected from the group which includes, but are not limited to, halo, C] -C20 alkyl, CF3, NH2, (Ci-C6 alkyl)2, ΝΟ2» oxo, CN, N3, -OH, -0(Ci-C6 alkyl), C3-C10 cycloalkyl, C2-C6 alkenyl, C2-C alkynyl, (Q)-C6 alkyl) S(0)o-2-> aryl-S(O)0-2-> (Co-C6 alk l)S(0)o-2(Co-C6 alkyl)-, (Co-C6 alkyl)C(0)NH-, H2N-C(NH , -0(C -C6 alkyl)CF3, (Co-Q, alkyl)C(O)-, (C0-C6 alkyl)OC(0)-, (C0-C alkyl)2NC(0)- (C()-C6alkyl)0(Ci-C6 alkyl , (Q>-C6 alkyl)C(0)i.2(Co-C6 alkyl)-, (Co-C¾ alkyl)OC(0)NH-, aryl, aralkyl, heteroaryl, heterocyclylalkyl, halo- aryl, halo-aralkyl, halo-heterocycle, halo-heterocyclylalkyl, cyano-aryl, cyano-aralkyl, cyano-heterocycle and cyano-heterocyclylalkyl.
Unless expressly stated to the contrary, an "unsaturated" ring is a partially or fully unsaturated ring. For example, an "unsaturated monocyclic C5 carbocycle" refers to cyclohexene, cyclohexadiene, and benzene.
Unless expressly stated to the contrary, all ranges cited herein are inclusive. For example, a heterocycle described as containing from "1 to 4 heteroatoms" means the heterocycle can contain 1, 2, 3 or 4 heteroatoms.
When any variable occurs more than one time in any constituent or in any formula depicting and describing compounds of the invention, its definition on each occurrence is independent of
its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
The term "substituted" (e.g., as in "aryl which is optionally substituted with one or more substituents ...") includes mono- and poly-substitution by a named substituent to the extent such single S and multiple substitution (including multiple substitution at the same site) is chemically allowed.
The term "oxy" means an oxygen (O) atom. The term "thio" means a sulfur (S) atom. The term "oxo" means "=0". The term "carbonyl" means "CO."
When any variable (e.g., R2, R.3, etc.) occurs more than one time in any substituent or in formulas Ι-ΠΪ, its definition in each occurrence is independent of its definition at every other occurrence. 10 Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
Under standard nomenclature used throughout this disclosure, the terminal portion of the designated side chain is described first, followed by the adjacent functionality toward the point of attachment For example, a Ci-5 alkylcarbonylamino Ci_<; alkyl substituent is equivalent to j 5

In choosing compounds of the present invention, one of ordinary skill in the art will recognize that the various substituents, i.e. Ill, R^, R , etc., are to be chosen in conformity with well- known principles of chemical structure connectivity.
Lines drawn into the ring systems from substituents indicate mat the indicated bond can
20 be attached to any of the substitutable ring atoms. If the ring system is polycyclic, it is intended mat the bond be attached to any of the suitable carbon atoms on the proximal ring only.
It is understood that substituents and substitution patterns on the compounds of the instant invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those
25 methods set form below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups can be on the same carbon or on different carbons, so long as a stable structure results. The phrase "optionally substituted with one or more substituents" should be taken to be equivalent to the phrase "optionally substituted with at least one substituent" and in such cases one embodiment will have from zero to three substituents.
30 In one embodiment, A, includes, but is not limited to, the following: azabenzimidazole, benzoiraidazolyl, benzofuryl, benzofurazanyl, benzopyrazolyl, benzothiazolyl, benzothienyl, benzotriazoryl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, chromanyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuryl, isochromanyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxazoline, isoxazoline,
35 oxetanyl, pyranyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl,
pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, quinoxalinyl, tetrahydropyranyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, azetidinyl, aziridinyl, 1,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyrrolidinyl, morpholinyl, thiomo holίn l,
dihydrobenzoimidazolyl, dihydrobenzofuryl, dihydrobenzothiophenyl, dihydrobenzoxazolyl, dihydrofuryl, dihydroimidazolyl, dihydroindolyl, dihydroisooxazolyl, dihydroisothiazolyl,
dihydrooxadiazolyl, dihydrooxazolyl, dihydropyrazinyl, dihydropyrazolyl, dihydropyridinyl,
tetrahydroquinolinyl, tetrahydroisoquinolinyl, dihydropyrimidinyl, dihydropyrrolyl, dihydroquinolinyl, dihydrotetrazolyl, dihydrothiadiazolyl, dihydrothiazolyl, dihydrothienyl, dihydrotriazoly],
dihydroazetidinyl, methylenedioxybenzoyl, tetrahydrofuryl, tetrahydrothienyl, tetrahidroquinolinyl, 2,3-dihydrobenzofuryl, 2,3^ihydrobenzo-l,4-dioxinyl, imidazo(2,l-bXl,3)<hiazole, and benzo-1,3- dioxolyl, wherein A is optionally substituted with one or more R9 substituents.
In a variant of this embodiment, tbe heterocyclyl moiety in A includes azabenzimidazole, benzoimidazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiopbenyl, benzoxazolyl, furanyl, imidazolyl, indolyl, indazolyl, isobenzofuryl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazoly], oxazolyl, pyrazinyJ, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, quinazolinyl, quinolyl, quinoxalinyl, quinoxalinyl, tetrahydroisoquinolinyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, dihydrobenzoimidazolyl, phenothiazinyl, dihydrobenzofiiryl, dihydrobenzothiophenyl, dihydrobenzoxazolyl, methylenedioxybenzoyl,
tetrahydrofuryl, tetrahydrothienyl, 2,3-dihydrobenzofuryl, 2,3-dihydrobenzo-l,4-dioxinyl, imidazo(2,l- bXl,3)thiazole, and benzo-l,3-dioxolyl, wherein A is optionally substituted with one or more R9 substituents.
In another embodiment, A is selected from: benzoimidazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, furanyl, imidazolyl, indolyl, indazolyl, isobenzofuryl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, and thienyl, wherein A is optionally substituted with one or more R9 substituents.
In another embodiment, A is selected from: pyrazinyl, pyrazolyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, and thienyl, wherein A is optionally substituted with one or more R9 substituents.
In another embodiment, A is selected from pyrazinyl, pyrazolyl, pyridazinyl, pyridinyl, and pyrimidyl, wherein A is optionally substituted with one or more R9 substituents. In a variant of this embodiment, A is selected from pyrazolyl, pyridinyl, pyridazinyl, and pyrimidyl, wherein A is optionally substituted with one or more R? substituents. In yet another variant of this invention, A is chosen from pyrazolyl and pyridazinyl, wherein A is optionally substituted with one or more 9 substituents.
In one embodiment of the invention, Rl is selected from halogen, C\.\Q alkyl, -C2_i o alkenyl, -C2-10 alkynyl, cycloalkyl Co-ifj alkyl(oxy)o_i, heterocyclyl Co-lO alkyl(oxy)0-l» arylCfj-lO alkyl(oxy)o-i, Ci-io alkoxy (ca carboxyl aryl,
carboxylcycloalkyl, carboxylheterocyclyl, Ci-ioalkyloxy Co-ioaltyl* hydroxy Co-ioalkyl, hydroxycarbonylCo-loalkoxy, Ci-io alkylthio, Cj-io alkylsulfinyl, aryl Crj-io alkylsulfinyl, heterocyclyl Co_io alkylsulfinyl, cycloalkyl Co-10 alkylsulfinyl, Ci-io alkylsulfonyl, aryl Co-io alkylsulfonyl, heterocyclylCo-io alkylsulfonyl, cycloalkyl Co-io alkylsulfonyl, n ro, perfluoroC] .
6*lkyl> 811(1 perfluoroC i-galkoxy; wherein in Rl is optionally substituted with one or more R9 substituents.
In another embodiment, Rl is selected from halogen, Ci-io alkyl, -C2-io alkenyl, - ∑- 10 alkynyl, cycloalkyl Co-io alkyl(oxy)o-l, heterocyclyl Co-ifj alkyl(oxy)fj-l, arylCrj-lO alkyl(°xy)0-l, Ci-io alkoxy (carbonyl)0-lCo-io alkyl, carboxyl Co_io alkyl, carboxyl aryl, carboxylcycloalkyl, carboxylheterocyclyl, C i . i oalkyloxy Co. i oalkyl, hydroxy Cfj- 1 Oalkyl, hydroxycarbonylCo- 1 oalkoxy, Ci-io alkylthio, Ci_]0 alkylsulfinyl, aryl Co-io alkylsulfinyl, heterocyclyl Cfj-io alkylsulfinyl, cycloalkyl Co-io alkylsulfinyl, Ci-io alkylsulfonyl, aryl Co-io alkylsulfonyl, heterocyclylCo-io alkylsulfonyl, cycloalkyl Co_io alkylsulfonyl, nitro, perfluoroC i-6alkyl, and perfluoroC i_6alkoxy;
wherein in Rl is optionally substituted with one or more R9 substituents.
In another embodiment, Rl is selected from halogen, phenylmethyl, pyridinyL, phenyl,
Ci-io alkyl, naphtholinyl Ci-6 alkyl, cyclopropyl, phenyl Cl-6 alkyl, cyclohexyl, piperidinyl, pyrrolidinyl, 1^,3,4-tetrahydronaphthalinenyl, 2,3-dihydro-lH-indinyl, benzyl, phenylethyl, cyclopentyl, tertbutyl, wherein in Rl is optionally substituted with one or more R9 substituents.
In another embodiment of the invention, Rl is selected from: cycloalkyl Co-10
alkyl(oxy)o-i, heterocyclyl Co-iO alkyl(oxy)0-l, arylCo-io alkyl(oxy)0-l, perfluorCi-6 alkyl, perfluoro Ci^alkoxy, and Ci-io alkoxy (carbonyl)o_iCo_io al » wherein Rl is optionally substituted with one or more R9 substituents.
In one embodiment of the invention, R2 is selected from hydrogen, halogen, hydroxy!, - Ci-<5 alkyl, C3-6 heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxy!, -SR\ - R^ or-CC^-R*.
In another embodiment, R3 is selected from hydrogen, halogen, hydroxy!, -Cj-6 alkyl, C3-6 heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxyl, -SR\ -NR*2 or -C02-R'- In one emobidiment of the invention L is selected from: is chosen from -(C1.3 alkyl)o- i RaCO-, -(C1.3 alkyl)0-l RaCO2-, -(Ci-3 alkyl)0-lCO2-, -<C\.3 alkyl)0-l RaSO2-, -(C1.3 alkyl)0- iNRaCSNRb-, and -(C1.3 alkyl)o-lNRaCONRb..
In another embodiment, L is selected from -(CI -3 alkyl)0-lNRaCO-,
-{Cl-3 alkyl)0-l RaCO2-, and -(Cl-3 alkyl)0-lCO2-. In a variant of this embodiement, L is—(CI -3 aIkyl) -lNRaCO-.
In one embodiment, R9 is selected from halogen, hydroxy, oxo, cyano, aryl, heterocyclyl, cycloalkyl, -Cl-6 alkyl, -Cl-6 alkoxy, aryloxy, heterocyclyloxy, -0(0-1 XC 1-10)perfluoroalkyl, -C02Ra,
-OC02Ra, -OCO RbRc, , -SCO-6 alkyl and -S(0)nRjd, wherein R9 is optionally substituted by one or more substituents RIO.
In another embodiment, R9 is selected from halogen, hydroxy, cyano, aryl, heterocyclyl, cycloalkyl, -Cl-6 alkyl, -Cl-6 alkoxy, aryloxy, heterocyclyloxy, -O(0-lXCl-10)perfluoroalkyl, -C02Ra, and -OC02Ra wherein R9 is optionally substituted by one or more substituents RIO.
In one embodiment, RlO is selected from hydroxy, aryl, heterocycloalkyl, heteroaryl, halogen, oxo, -Cl-6 alkyl, C -6 alkoxy,C02H, cyano, 0(C=0)o.iCi-6 alkyl, trifluoromethoxy, trifluoroethoxy, -0(0-l)(Ci.io)perfluoroalkyl, Cfj-io alkylaminocarbonyl, -(C=0)N(Co-6 a]kyl)2, -S(Co- 6 alkyl), and N¾.
In another embodiment, R10 is selected from hydroxy, aryl, heterocycloalkyl, heteroaryl, halogen, oxo, -C\-6 alkyl, Cj-6 alkoxy,C02H, cyano, 0(00)o.iCi-6 alkyl, trifluoromethoxy, trifluoroethoxy, -0(0-l)(Ci_io)perfluoroalkyl, Crj-io alkylaminocarbonyl, and H2.
Another embodiment of the invention includes the following compounds: N-{4-hydroxy-2-( 1 H-pyr¾zol-l-yl)pyrimidm-5-yl)-2,2-diphenylacetamide;
2-chloro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
3-chloro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
3-(trifluoromethyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(naphthalen-3-yl)acetamide;
2-(trifluoromethyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yI)benzamide;
N-[4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl]biphenyl-4-carboxamide;
N-(4-hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl 3-phenoxybenzamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-4-phenoxybenzamide;
2-(2-chlorophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(3-chlorophenyI)-N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)acetamide
2-(4-chlorophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(3-cyanopheny l)-N-(4-hydroxy-2-( 1 H-pyra^
2-(2-(trifluoromethyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(3-{trifluoromemyl)phenyl)-N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-^
2-(4-(trifluoromethyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetarnide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-2-(2-memoxyphenyl)acetami
N-(4-hyd^xy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-2-(3-memoxyphenyl)acetamid^
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-2-(4-methoxyphenyl)acetomide^
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-phenylpropanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-phenylbutanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-methyl-2-pheny lpentanamide;
N-(4-hydroxy-2-(l H-pyrazol-l-yl)pyrimidin-5-yl)-2-methyl-2-phenylpropanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yI)pyrimidin 5 yl) 2 (naphthalen 1 y l)acetamide;
2-(benzo[d] [ 1 ,3 ]dioxol-5-yl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(biphenyl-4-yl N-(4-hydroxy-2-(lH-p^
N-(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)heptanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-4-pheDylbutanamide;
tert-buty 14-(4-hydroxy-2-(l H-pyrazol-1 -yl)pyrimidin-5-ylcarbamoyl)piperidine- 1 -carboxylate;
2-cyclohexyl-N-(4-hydroxy-2-( 1 H-pyrazo 101.01-1 -yl)pyrimidin-5-yI)acetamide;
1 ,2,3,4-tetrahydro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)naphthalene- 1 -carboxamide;
2,3-dihydro-N-(4-hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl)-l H-indene- 1 -carboxamide;
2,3-dihydro-N-(4-hydroxy-2-(IH-pyrazol-l-yl)pyrimidin-5-yl)-lH-inden
1 -benzyl-3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea;
1 -(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-( 1 -phenylethyl)urea;
S-i^hydroxy^-ilH-pyi-azol-l-yl^yrimidin-S-ylJ-l.I-diphenylurea;
1 -benzhydryl-3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)thiourea;
1 -{4-hydroxy-2-(lH-pyra2»l-l-yl pyrimidin-5-yl)-3-(l -phenylethyl)thiourea;
and pharmaceutically acceptable salts and solvates thereof.
Structural representations of compounds having substhuents terminating with a methyl group may display the terminal methyl group either using the characters "CH3", e.g. "-CH3" or using a straight line representing the presence of the methyl group, e.g.
For variable definitions containing terms having repeated terms, e.g., (CRiRJ)r, where r is the integer 2, Ri is a defined variable, and Rj is a defined variable, the value of Ri may differ in each instance in which it occurs, and the value of Rj may differ in each instance in which it occurs. For example, if Ri and Ri are independently selected from the group consisting of methyl, ethyl, propyl and butyl, then (CRiR!)2 can be

Optical Isomers - Diastereomers - Geometric Isomers - Tautomers
Compounds described herein may contain an asymmetric center and may thus exist as enantiomers. Where the compounds according to the invention possess two or more asymmetric centers, they may additionally exist as diastereomers. The present invention includes all such possible stereoisomers as substantially pure resolved enantiomers, racemic mixtures thereof, as well as mixtures of diastereomers. The above Formula I is shown without a definitive stereochemistry at certain positions, The present invention includes all stereoisomers of Formula I and pharmaceutically acceptable salts and solvates thereof. Unless specifically mentioned otherwise, reference to one isomer applies to any of the possible isomers. Whenever the isomeric composition is unspecified, all possible isomers are included. Diastereoisomeric pairs of enantiomers may be separated by, for example, fractional crystallization from
a suitable solvent, and the pair of enantiomers thus obtained may be separated into individual stereoisomers by conventional means, for example by the use of an optically active acid or base as a resolving agent or on a chiral HPLC column. Further, any enantiomer or diastereomer of a compound of the general Formula I may be obtained by stereospeciMc synthesis using optically pure starting materials or reagents of known configuration .
When compounds described herein contain olefinic double bonds, unless specified otherwise, such double bonds are meant to include both E and Z geometric isomers.
Some of the compounds described herein may exist with different points of attachment of hydrogen, referred to as tautomers. For example, compounds including carbonyl -CH2C(0)- groups (keto forms) may undergo tautomerism to form hydroxyl -CH=C(OH groups (enol forms). Both keto and enol forms, individually as well as mixtures thereof, are included within the scope of the present invention.
Salts
Pharmaceutically acceptable salts include both the metallic (inorganic) salts and organic salts; a list of which is given in Remington's Pharmaceutical Sciences, 17th Edition, pg. 1418 (1985). It is well known to one skilled in the art that an appropriate salt form is chosen based on physical and chemical stability, flowabilhy, hydro-scopicity and solubility. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids. When the compound of the present invention is acidic, its corresponding salt can be conveniently prepared from inorganic bases or organic bases. Salts derived from such inorganic bases include aluminum, ammonium, calcium, copper (ic and ous), ferric, ferrous, lithium, magnesium, manganese (ic and ous), potassium, sodium, zinc and the like salts. Preferred are the ammonium, calcium, magnesium, potassium and sodium salts. Salts prepared from organic bases include salts of primary, secondary, and tertiary amines derived from both naturally occurring and synthetic sources. Pharmaceutically acceptable organic non-toxic bases from which salts can be formed include, for example, arginine, betaine, caffeine, choline, N,N -dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimeraylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, dicyclohexylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
When the compound of the present invention is basic, its corresponding salt can be conveniently prepared from inorganic or organic acids. Such acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid and the like.
Preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
Solvates
The present invention includes within its scope solvates of compounds of Formula I. As used herein, the term "solvate" refers to a complex of variable stoichiometry formed by a solute (i.e., a compound of Formula I) or a pharmaceutically acceptable salt thereof and a solvent that does not interfere with the biological activity of the solute. Examples of solvents include, but are not limited to water, ethanol, and acetic acid. When the solvent is water, the solvate is known as hydrate; hydrate includes, but is not limited to, hemi-, mono, sesqui-, di- and trihydrates.
Prodrugs
The present invention includes within its scope the use of prodrugs of the compounds of this invention. In general, such prodrugs will be functional derivatives of the compounds of mis invention which are readily convertible in vivo into the required compound. Thus, in the methods of treatment of the present invention, the term "administering" shall encompass the treatment of the various conditions described with a compound of formula I or with a compound which may not be a compound of formula L, but which converts to a compound of formula I in vivo after administration to the patient Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs," ed. H. Bundgaard, Elsevier, 1985.
In the compounds of generic Formula I, the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of generic Formula I. For example, different isotopic forms of hydrogen (H) include protium (lH) and deuterium (2H). Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
Utilities
Compounds of the present invention are inhibitors of hypoxia-inducible factor (RTF) prolyl hydroxylases, and as such are useful in the treatment and prevention of diseases and conditions in which HIF modulation is desirable, such as anemia and ischemia. Compounds of the invention can be used in a selective and controlled manner to induce hypoxia-inducible factor stabilization and to rapidly and reversibly stimulate erythropoietin production and secretion. Accordingly, another aspect of the present invention provides a method of treating or preventing a disease or condition in a mammal, the treatment or prevention of which xylase inhibition, which
comprises administering an amount of a compound of Formula I that is effective for inhibiting HGDF prolyl hydroxylase. This aspect of the present invention further includes the use of a compound of Formula I in the manufacture of a medicament for the treatment or prevention of a disease or condition modulated by HIF prolyl hydroxylase.
In one embodiment is a method of enhancing endogenous production of erythropoietin in a mammal which comprises administering to said mammal an amount of a compound of Formula I that is effective for enhancing endogenous production of erythropoietin.
Another embodiment is a method of treating anemia in a mammal which comprises administering to said mammal a therapeutically effective amount of a compound of Formula I. "Anemia" includes, but is not limited to, chronic kidney disease anemia, chemotherapy-induced anemia (e.g., anemia resulting from antiviral drug regimens for infectious diseases, such as HTV and hepatitis C virus), anemia of chronic disease, anemia associated with cancer conditions, anemia resulting from radiation treatment for cancer, anemias of chronic immune disorders such as rheumatoid arthritis, inflammatory bowel disease, and lupus, and anemias due to menstruation or of senescence or in other individuals with iron processing deficiencies such as those who are iron-replete but unable to utilize iron properly.
Another embodiment is a method of treating ischemic diseases in a mammal, which comprises administering to said mammal a therapeutically effective amount of a compound of Formula I. Combination Therapy
Compounds of Formula I may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds of Formula I are useful. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I. When a compound of Formula I is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound of Formula I is preferred. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of Formula I.
Route of Administration/Dosage
The compounds of this invention can be administered for the treatment or prevention of afflictions, diseases and illnesses according to the invention by any means that effects contact of the active ingredient compound with the site of action in the body of a warm-blooded animal. For example, administration can be oral, topical, including transdermal, ocular, buccal, intranasal, inhalation, intravagjnal, rectal, intracisternal and parenteral. The term "parenteral" as used herein refers to modes of administration which include subcutaneous, intravenous, intramuscular, intraarticular injection or infusion, intrastemal and intraperitoneal. For the purpose of this disclosure, a warm-blooded animal is a member of the animal kingdom possessed of a homeo static mechanism and includes mammals and birds.
The compounds can be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be tered with a
pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
The dosage administered will be dependent on the age, health and weight of the recipient, the extent of disease, kind of concurrent treatment, if any, frequency of treatment and the nature of the effect desired. Usually, a daily dosage of active ingredient compound will be from about 0.1-2000 milligrams per day. Ordinarily, from 10 to 500 milligrams per day in one or more applications is effective to obtain desired results. These dosages are the effective amounts for the treatment and prevention of afflictions, diseases and illnesses described above, e.g., anemia.
Pharmaceutical Composition
Another aspect of the present invention provides pharmaceutical compositions which comprises a compound of Formula I and a pharmaceutically acceptable carrier. The term "composition", as in pharmaceutical composition, is intended to encompass a product comprising the active
ingredients), and the inert ingredients) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of Formula I, additional active ingredients), and pharmaceutically acceptable excipients.
The pharmaceutical compositions of the present invention comprise a compound represented by Formula I (or a pharmaceutically acceptable salt or solvate thereof) as an active ingredient, a pharmaceutically acceptable carrier and optionally other therapeutic ingredients or adjuvants. The compositions include compositions suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration, although the most suitable route in any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered. The pharmaceutical compositions may be
conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy.
The active ingredient can be administered orally in solid dosage forms, such as capsules, tablets, troches, dragees, granules and powders, or in liquid dosage forms, such as elixirs, syrups, , emulsions, dispersions, and suspensions. The active ingredient can also be administered parenteral ly, in sterile liquid dosage forms, such as dispersions, suspensions or solutions. Other dosages forms that can also be used to administer the active ingredient as an ointment, cream, drops, transdermal patch or powder for topical administration, as an ophthalmic solution or suspension formation, i.e., eye drops, for ocular administration, as an aerosol spray or powder composition for inhalation or intranasal administration, or as a cream, ointment, spray or suppository for rectal or vaginal administration.
Gelatin capsules contain the active ingredient and powdered carriers, such as lactose, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets. Bo sustained release products
to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric coated for selective disintegration in the gastrointestinal tract.
Liquid dosage forms for oral administration can contain coloring and flavoring to increase patient acceptance.
In general, water, a suitable oil, saline, aqueous dextrose (glucose), and related sugar solutions and glycols such as propylene glycol or polyethylene gycols are suitable carriers for parenteral solutions. Solutions for parenteral administration preferably contain a water soluble salt of the active ingredient, suitable stabilizing agents, and if necessary, buffer substances. Antioxidizing agents such as sodium bisulfite, sodium sulfite, or ascorbic acid, either alone or combined, are suitable stabilizing agents. Also used are citric acid and its salts and sodium EDTA. In addition, parenteral solutions can contain preservatives, such as benzalkonium chloride, methyl- or propylparaben, and chlorobutanol.
Suitable pharmaceutical carriers are described in Remington's Pharmaceutical Sciences, A. Osol, a standard reference text in Ihis field.
For administration by inhalation, the compounds of the present invention may be conveniently delivered in the form of an aerosol spray presentation from pressurized packs or nebulisers. The compounds may also be delivered as powders which may be formulated and the powder composition may be inhaled with the aid of an insufflation powder inhaler device. The preferred delivery system for inhalation is a metered dose inhalation (MDI) aerosol, which may be formulated as a suspension or solution of a compound of Formula I in suitable propellents, such as fluorocarbons or hydrocarbons.
For ocular administration, an ophthalmic preparation may be formulated with an appropriate weight percent solution or suspension of the compounds of Formula I in an appropriate ophthalmic vehicle, such that the compound is maintained in contact with the ocular surface for a sufficient time period to allow the compound to penetrate the corneal and internal regions of the eye.
Useful pharmaceutical dosage-forms for administration of the compounds of this invention include, but are not limited to, hard and soft gelatin capsules, tablets, parenteral injectables, and oral suspensions.
A large number of unit capsules are prepared by filling standard two-piece hard gelatin capsules each with 100 milligrams of powdered active ingredient, 150 milligrams of lactose, 50 milligrams of cellulose, and 6 milligrams magnesium stearate.
A mixture of active ingredient in a digestible oil such as soybean oil, cottonseed oil or olive oil is prepared and injected by means of a positive displacement pump into gelatin to form soft gelatin capsules containing 100 milligrams of the active ingredient The capsules are washed and dried.
A large number of tablets are prepared by conventional procedures so that the dosage unit is 100 milligrams of active ingredient, 0.2 milligrams of colloidal silicon dioxide, 5 milligrams of magnesium stearate, 275 milligrams of microcrystalline cellulose, 1 1 milligrams of starch and 98.8 milligrams of lactose. Appropriate coatings may be applied to increase palatability or delay absorption.
A parenteral composition suitable for administration by injection is prepared by stirring 1.5% by weight of active ingredient in 10% by volume propylene glycol. The solution is made to volume with water for injection and sterilized.
An aqueous suspension is prepared for oral administration so that each 5 milliliters contain 100 milligrams of finely divided active ingredient, 100 milligrams of sodium carboxymethyl cellulose, 5 milligrams of sodium benzoate, 1.0 grams of sorbitol solution, U.S.P., and 0.02S milliliters of vanillin.
The same dosage forms can generally be used when the compounds of this invention are administered stepwise or in conjunction w h another therapeutic agent. When drugs are administered in physical combination, the dosage form and administration route should be selected depending on the compatibility of the combined drugs. Thus the term coadministration is understood to include the administration of the two agents concomitantly or sequentially, or alternatively as a fixed dose combination of the two active components.
Compounds of the invention can be administered as the sole active ingredient or in combination with a second active ingredient, including other active ingredients known to be useful for improving the level of erythropoietin in a patient.
Abbreviations Used in the Description of the Preparation of the Compounds of the Present Invention:
AcOH Acetic acid
AgjO Silver oxide
Aq Aqueous
Bn Benzyl
BnBr benzylbroraide
BnCl benzylchloride
BnOH benzylalcohol
Brine Saturated aqueous sodium chloride solution
CDI Carbony] diimidazole
DBU 1 ,8-diazabicyclo[5.4.0]undec-7-ene
DEAD diethylazodicarboxylate
DCM Dichloromethane
DIPEA N,N-diisopropylethylaime
DMAP 4-.V,#-dimemylaminopyridine
DMF N,tf-dimethylformamide
DMSO Dimethyl sulfoxide

EtOAc or EA Ethyl acetate
Et (et) Ethyl
EtOH Ethanol
Et20 or ether Diethyl ether
Etj triethylamine
G Grains
h or hr Hour
HATU CK7-Azaben-»1riazol-l-yl -V,-V,^N'-tetrame ^
hexafluorophosphate
HC1 Hydrochloric acid
HOBt 1-hydroxybenzatriazole
HPLC High-performance liquid chromatography
j'-PrOH or IPA Isopropyl alcohol
K2CO3 Potassium carbonate
KOH Potassium hydroxide
LiOH Lithium hydroxide
Mg Milligrams
mL Milliliters
Mmol Millimole
MeOH Methanol
Min Minutes
ms or MS Mass spectrum
Mg Microgram(s)
uL Microliters
NaOEt Sodium ethoxide
NaOMe Sodium methoxide
Na2S04 Sodium sulfate
NHAc Acetamido
NHCbz Benz loxycarboxamido
NaOH Sodium hydroxide
NaN3 Sodium azide
N¾OH ammonium hydroxide
Pd/C Palladium on carbon
Pd(OH>2 Palladium hydroxide
Ph Phenyl group
Triphenyphosphine
Retention time
Rt Room temperature
TEA Triethylamine
TFA Trifluoroacetic acid
THF Tetrahydrofuran
TMSCHNj (trimethylsilyl)diazomethane
v v Volume/Volume
V Volume
Synthesis
The compounds of mis invention may be prepared by employing reactions as shown in the following schemes, in addition to other manipulations that are known in the literature or exemplified in the experimental procedures. The illustrative schemes below, therefore, are not limited by the compounds listed or by any particular substituents employed for illustrative purposes. Substituent numbering as shown in the schemes does not necessarily correlate to that used in the claims and often, for clarity, a single substituent is shown attached to the compound in place of multiple substituents which are allowed under the definitions of Formula I defined previously.
The following schemes and descriptions illustrate methods which may be employed for the synthesis of the novel compounds described in this invention. All substituents are as defined above unless indicated otherwise. Several strategies based upon synthetic transformations known in the literature of organic synthesis may be employed for the preparation of the title compounds of general formula I. The choice of the method employed is influenced by the selection of the desired substituent groups (R1 through R3, L and A) in the title of formula I.

Pyrimidine intermediates useful for the preparation of compounds of formula I of the present invention are either purchased or prepared using suitable procedures reported in the literature (sometimes with minor modifications). One generally useful method for the synthesis of pyrimidines suitable for the preparation of the title compounds of general formula I wherein the substituent R3 is a hydroxy] group is illustrated in Scheme 1.
Scheme ¾

This method involves the initial synthesis of substituted 4-hydroxypyrimidine-5- carboxylates of general formula 1S-4 and 1S-6. The synthesis of 4-hydroxypyrimidine-5-carboxylates exemplified in scheme 1 is based upon reported methods (Dostert, P.; Imbert, T.; Ancher, J.F.; Langlois, M.; Bucher, B.; Mocquet, G. Eur. J. Med Chem. 1982, 77, 437-44. Juby, P.F.; Hudyma, T.W.; Brown, M.; Essery, J.M.; Partyka, R.A. J. Med Chem. 1979, 22, 263-9).
In this method, an amidine or a suitable salt thereof of general formula lS-1 is reacted with an optionally substituted diethyl methylenemalonate of general formula 1S-2. This reaction is usually conducted using a suitable base such as sodium or potassium ethoxide in ethanol, although other reaction conditions may also be applied. The alkoxide base and the alcohol solvent are chosen to correspond to the esters present in reagent 1S-2 to prevent the formation of mixtures of esters in the product of general formula 1S-3. When required, the reaction is conducted at elevated temperature, typically at the reflux temperature of the solvent until reaction is complete (generally within 1-4 hours). It is also convenient to conduct this reaction under microwave heating in sealed reaction vessels. In this instance, the reaction is generally conducted at temperatures between 80 and 120 °C and the reactions are typically completed in 5-30 minutes.
Compounds of general formula 1S-3 are useful intermediates to prepare compounds of formula I of the present invention. For example, compounds of general formula 1S-3 may be hydrolyzed using a suitable base (e.g. sodium or potassium hydroxide) to give acids of formula 1S-4; alternatively, they are converted to compounds of formula 1S-5, in which the hydroxy! group of the pyrimidine core is protected with a desired protecting group (e.g. PG is benzyl, para-methoxybenzyl, trityl, or tert-butyl- dimethyl silyl). Hydrolysis of compounds of formula 1S-5 gives acids of general formula 1S-6, which is readily achieved under suitable ester hydrolysis reaction conditions (Wuts, P.G.M.; Greene, T.W., Protecting Groups in Organic Synmesis, John Wiley and Sons, 4* Edition, 2007).
When amidines of general formula lS-1 are not commercially available, they may be prepared by a variety of method k i h li A idi only prepared from nitriles
using the Pinner reaction and variations thereof (see Amidines and N-substituted amidines. Dunn, Peter J. in Comprehensive Organic Functional Group Transformations 1995, 5, 741-82, 1161-308 Editors): atrizky, Alan R.; Meth-Cohn, Otto; Rees, Charles Wayne. Publisher: Elsevier, Oxford, UK). Amidines may also be prepared from esters using the method reported by Gielen et al. (Gielen, H.; Alonso-Alija, C; Hendrix, M.; Niewohner, U.; Schauss, D. Tetrahedron Lett. 2002, 43, 419-21).
In instances where the substituent A is selected to be a five-membered heterocyclic ring, it is possible that this heterocyclic group be bonded to the carbon atom at the 2-posttion of the pyrimidine ring through either a carbon-carbon or a carbon-nitrogen bond. In the case of attachment through a carbon-carbon bond, the precursor for the substituent A is an amidine of general formula lS-1 and the method using lS-1 for the synthesis of the title compound of general formula I is as described in the preceding reaction schemes.
When a substituent A is attached through a carbon-nitrogen bond, the precursor for the substituent A is a guanidine of general formula 2S-7. In this example, the synthesis begins with the condensation of the guanidine derivative of general formula 2S-7 with compounds of general formula 2S- 8 (or a diethyl ethoxymethylenemalonate of general formula 1S-2 when it is desired that R3 = OH) to afford the substituted pyrimidine-5-carboxylate derivative of general formula 2S-9. Ester hydrolysis as described above affords compounds of formula 2S-10, which is useful to prepare compounds of general formula I wherein the group A is a five-membered heterocyclic group attached to the pyrimidine 2- position with a carbon-nitrogen bond.

In cases when the guanidine derivative (2S-7) bearing the desired substituents is not commercially available, h may be synthesized using reported methods for guanidine synthesis (e.g. the guanidinylation of amines). Numerous methods for the guanidinylation of amines are reported (see Katritzky, A.R.; Rogovoy, B.V. ARKIVOC 2005, 4, 49-87; http://www.arkat- usa.org ark/journal 2005 I04 Zefirov/ 1256/1256.pdf). One general method is shown in Scheme 3, which
entails the reaction of compounds of formula 3S-11 with 3,5-dimethyl-l-pyrazolylformamidinium nitrate to afford a guanidine of general formula 3S-12 using the method described by Fletcher et al. (Fletcher, D.L; Ganellin, C.R.; Piergentili, A.; Dunn, P.M.; Jenkinson, D.H. Bioorg. Med Chem. 2007, 15, 5457- 79).

It is recognized that the title compounds of general formula I prepared as described above may be further modified using known methods and that the starting materials selected for use in the reaction schemes above may contain functional groups to enable said further transformation. For instance, aromatic rings in the title compounds of general formula I may be subjected to a variety of aromatic substitution reactions such as nitration, halogenation and the like. Aromatic substituent groups in the title compounds of general formula I bearing leaving groups such as halogens, triflates or the like, can be employed in a variety of metal-catalyzed cross coupling reactions to incorporate new substitution patterns. For example, palladium-catalyzed cross coupling reactions such as those described by Suzuki, Stille, Buchwald and others, may be used to introduce a variety of new substituent groups. Substituent groups that may be introduced using such cross-coupling methods include, but are not limited to, alkyl, alkenyl, alkynyl and aryl groups as well as acyl groups (e.g. carboxylic acids, esters, amides, or ketones), hydroxyl and amino or substituted amino groups.
Other pyrimidines with various substituents at the 5-position (wherein R" is not a carboxylate) are also prepared via cyclization reactions described in Schemes 1 & 2, which are exemplified in Scheme 4.

For example, compounds of formula 4S-15 are prepared when amidines lS-1 are cyclized with esters 4S-14 wherein X = O-alkyl under reaction conditions exemplified as in Scheme 1. Alternatively, compounds of formula 4S-14 wherein X = N(alkyl)2 are also cyclized with amidines lS-1 to give compounds of formula 4S-15 (Chen, W.; Feng, J.; Tu, H. Huaxue Tongbao, 2006, 69, 623-6). The esters 4S-14 can be prepared by reaction of esters 4S-13 with a substituted carboxamide dimethyl acetal.
The methods presented in reaction Schemes 1 and 2 may be further generalized when it is desired to prepare compounds of general formula I where neither of the R.3 or R4 substiruents are hydroxyl groups.
Scheme 5 R*

In one aspect, compounds of formula I are prepared via pyrimidine ring formation reactions (e.g. Schemes 1, 2, 4, 5) with the desired substituents (e.g. A, R2, R3 and L) at various positions. In another aspect, the desired substituents on the pyrimidine core can be introduced after the pyrimidine ring is formed, which can be achieved using synthetic methods reported in the literature. For example, the hydroxy! group present at the pyrimidine 4-position in compounds of general formulae 1S-3, 1S-4, or 4S-15 may be converted to a halogen substituent upon reaction with a suitable halogenating reagent (e.g. POCb, BBr3, etc.).
Compounds of general formula I wherein L is an amino or an amino derivative can be prepared from suitable pyrimidine derivatives such as 1S-4, 1S-6, 2S-10 and 5S-19 using synthetic methods reported in the literature. For example, carboxylic acids of formula 5S-19 are converted to their corresponding amines of formula 6S-20 using suitable methods such as Curtis rearrangement reactions, Scheme 6. The amino group in compounds of formula 6S-20 is further derivatized using common synthetic methods such as amide bond formation reactions (e.g. CDI couplings, EDCI couplings, reactions with acyl chlorides, etc.), sulfonamide bond formation reactions (e.g. reactions with sulfonyl chlorides in the presence of a suitable base), reductive amination reactions with a suitable carbonyl compounds (e.g. aldehydes and ketones). For example, compounds of formula I wherein L is -NHCO- and/or -NHSOr are prepared from compounds of formula 6S-20 as depicted in Scheme 6. In another aspect, carboxylic acids such as compounds of formula 5S-19 can be converted to their corresponding acyl azide such as compounds of formula 6S-22, and under suitable thermal rearrangement reaction conditions acyl azides such as 6S-22 can be converted to their corresponding isocyanate, and subsequent reactions of the resulting isocyanate with various nucleophiles such as alcohols, amines and thiols can produce compounds of formula 6S-23 wherein L is a carbamate (-NHCOO-), urea (-NHCONH-), or thiocarbamate (-NHCOS-). (March's advanced organic chemistry, Wiley-interscience, 2007;
Comprehensive Organic Transformations: a guide to functional group preparetions by Richard Larock, Wiley-VCH, 2000)

General Methods
Reactions sensitive to moisture or air were performed under nitrogen using anhydrous solvents and reagents. The progress of reactions was determined by either analytical thin layer chromatography (TLC) performed with E. Merck precoated TLC plates, silica gel 60F-254, layer thickness 0.2S mm or liquid chromatography-mass spectrum (LC-MS). Analytical HPLC/MS - Standard Method: Mass analysis was performed on a Waters Micromass* ZQ™ with electrospray ionization in positive ion detection mode. High performance liquid chromatography (HPLC) was conducted on an Agilent 1100 series HPLC on Waters C18 XTerra 3.5 urn 3.0 x50 mm column with gradient 10:90-100 v/v CH3C H20 + v 0.05 % TFA over 3.75 min then hold at 100 C¾CN + v 0.05 % TFA for 1.75 min; flow rate 1.0 mL/min, UV wavelength 254 nm (all HPLC MS data was generated with this method unless indicated otherwise). Analytical HPLC/MS - Basic Method: Mass analysis was performed on a Waters Micromass* ZQ™ with electrospray ionization in positive ion detection mode. High performance liquid chromatography (HPLC) was conducted on an Agilent 1100 series HPLC on Waters C 18 XBridge 3.5 urn 3.0 x 50 mm column with gradient 10:90-98:2 v/v CH3CN H2O + v 0.025 % H4OH over 3.25 min then hold at 98:2 CH3C + v 0.025 % NH4OH for 2.25 min; flow rate 1.0 mUmin, UV wavelength 254 nm. Concentration of solutions was carried out on a rotary evaporator under reduced pressure. Flash chromatography was performed using a Biotage Horizon or SP1 Flash Chromatography apparatus (Dyax Corp.) on silica gel (32-63 uM particle size, KP-Sil 60 A packing material type) in pre-packed cartridges or using an ISCO CombiFlash™ Sq 16x or CombiFlash* Companion™ apparatus on silica gel (32-63 μΜ, 60 A) in pre-packed cartridges. Microwave reactions were carried out on a Biotage Initiator™ 2.0 or CEM Discover™ system. Preparative HPLC/MS - Standard Method: Mass analysis was performed on a Waters Micromass* ZQ™ with electrospray ionization in positive ion detection mode. High performance liquid chromatography (HPLC) was conducted on a Waters Prep. HPLC System on Waters C18 Sunfire 5 um 30 x 100 mm column with gradient 10:90-100 v/v CH3CN/H20 + v 0.1 % TFA over 12 min; flow rate 50 mL min, UV wavelength 210-400 nm.
Preparative HPLC/MS - Non-Polar Method
Mass analysis was performed on a Waters Micromass* ZQ™ with electrospray ionization in positive ion detection mode. High performance liquid chromatography (HPLC) was conducted on a Waters Prep. HPLC System on Waters C18 Sunfire 5 um 30 x 100 mm column with gradient 40:60-100 v/v CH3CN H20 + v 0.1 % TFA over 10 min then hold at 100 C¾CN + v 0.1 % TFA for 4 min; flow rate 50 mL min, UV wavelength 210-400 nm.
Preparative HPLC/MS - Basic Method
Mass analysis was performed on a Waters Micromass* ZQ™ with electrospray ionization in positive ion detection mode. High performance liquid chromatography (HPLC) was conducted on a Waters Prep. HPLC System on Waters C18 XBridge 5 um 50 x 150 mm column with gradient 10:90-35:65 v/v CH3CN/¾0 (pH = 10 with NH4OH) over 10 min; flow rate 120 mL min, UV wavelength 210-400 nm.

Step A: 2-Tosv^23 iihvdn)Pvridazine-3-carbonitrile
A solution of pyridazine (5 mL, 69.12 mmol) (Sigma-Aldrich), aluminum chloride (28 mg, 0.21 mmol) and TMSCN (16.75 mmol) in DCM (60 mL) was stirred under a nitrogen atomosphere at 0 •C for 20 min. A solution of TsCl (22.75g, 119.7 mmol) in DCM (40 mL) was added dropwise. The reaction was warmed to room temperature, stirred for an additional 60h, and concentrated in vacuo. The residue ws washed with EtOH (150 mL) and the resulting solids were filtered to afford the title
compound 1-b (15 g, 83%). Ή NMR (300 MHz, CDC13) 57.93 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 1.8 Hz, 2H), 6.23-6.21 (m, 2H), 5.79 (d,J- 6.3 Hz, 1H), 2.44 (s, 3H). LC-MS: (M + H)+= 262.
Step B: PvridazlBe-3-carbonttrile (1-c)
To a solution of compound 1-b (9.0 g, 34.4 mmol) in THF (100 mL) was added DBU
(6.5ml, 68.9mmol). The reaction was stirred at room temperature under a nitrogen atmosphere for 1 h. Saturated aqueous NH CI (100 mL) was poured into water (150 mL). The aqueous medium was extracted with EtOAc, dried, filtered and concentrated. The residue was washed with hexane (100 mL) to afford the title compound 1-c (2.7 g, 75%). Ή NMR (300 MHz, CDC13) δ 9.40 (dd, J - 1.5, 5.1 Hz), 7.88 (dd, J~ 1.8, 8.4 Hz), 7.70 (dd, J - 5.1, 8.4 Hz). LC-MS: (M + H)+=106.
Step C: Pvridazine-3-^rhn¾imiHflniide hydrochloride <U-d)
To a solution of compound 1-c (2.7 g, 25.7 mmol) in MeOH (25 mL) was added sodium methoxide (139 mg, 0.257 mmol). The reaction was stirred at room temperature overnight when ammonium chloride (1.65 g, 30.8 mmol) was added. The reaction was refluxed for 3 h, cooled to room temperature and concentrated to afford the crude product of 1-d (3.8 g, 93%).Ή NMR (300 MHz, DMSOt¾) δ 9.38 (dd, J = 1.5, 5.1 Hz, 1H), 8.30 (dd, J = 1.5, 8.7 Hz , 1H), 7.92 (dd, J = 5.1, 8.4 Hz, 1H). LC-MS: (M + H)+ 123. Sfrp Pt Methvl 4-hvdrory-2 DYridaan-3-vnDvrimidine-5-carb xvlate
A mixture of crude compound 1-d (20.0 g, 34.4 mmol), diethyl ethoxymethylenemalonate (35.0 g 162 mmol) and sodium methoxide (10.0 g, 185 mmol) in MeOH (1000 mL) was refluxed overnight. The reaction mixture was cooled and concentrated to remove the solvent and the residue was added to water (500 mL), extracted with EtOAc (500 mL). The organic phase was adjusted to pH= 2 by addition of aq. HC1. The solids were filtered to afford the product 11-e (15.0 g, 39%). LC-MS: (M + H) + =233.
Step E: 4-fbeiizyloxy 2^Dyridazin-3-vnpYriinidine-S-carboiyllc acid ( 1-f)
A mixture of compound 11-e ( 1.5g, 6.316 mmol) and PPh3 (3.39 g, 12.98 mmol) in THF ( 50 mL) was drop wise added with DEAD ( 2.26 g, 12.98 mmol) and stirred at room temperature for 30 min. Then BnOH (0.84 g, 7.75 mmol) was added and the mixture was stirred for additional 12 h at room temperature. The reaction mixture was concentrated. The residue was purified by chromatography column with eluent PE: EA = 2:1 to afford crude 11-f.
A solution of the crude compound (2.1 g, crude) in MeOH (30 mL) was added with aqueous LiOH (0.55 g, 12.9 mmol, 1M) and stirred at room temperature for overnight. The reaction mixture was concentrated, dissolved in water (30 mL), and extracted with EtOAc (50 mL). The aqueous
layer was adjusted to pH= 2 by addition of aq. HC1. The solids were filtered to afford the product 1-f (400 mg, 21%) LC-MS: (M+H)+ - 309.
Step F; 4-flieiizyloiv 2- PYridaziiH3-yl)pyrimidine-5-carborylic acid ( 1-g)
A mixture of compound 1-f (200 mg, 0.649 mmol), DPPA ( 196 mg, 0.71 mmol), Et3N ( 197 mg, 1.95 mmol) and BnOH (105 mg, 0.97 mmol) in THF/ toluene (20 mL, 1:1) was stirred at 65 Ό overnight The reaction mixture was concentrated and purified by chromatography column with eluent PE: EA = 2:1 to afford the product 1-g (160 mg, 60%). LC-MS: (M+H)+ = 414.
Steo G: 4^benzvloxvV2-fDvridazin-3-vriDvrimidine-S-carboxvUc add (1-V
A solution of compound 1-g (160 mg, 0.387 mmol) in MeOH (5 mL) was added with Pd/ C (10%, 0.15 g) and stirred under hydrogen for 5 h. The mixture was filtered and the solution was concentrated to afford the crude product 11-h (450 mg). LC-MS: (M+H)+ - 190. Example 2

Step A; 4-hvdroxy-2-f lH-pYrazoI-l-yl)pyrimidine-S-carboxylic acid (2-c)
To lH-pyrarole-l-carboximidamide hydrochloride (Sigma-Aldrich), 2-a, (44.22 g, 299 mmol) in EtOH (500 mL) was added sodium methoxide ( 102 mL, 448 mmo), 25 wt% in MeOH) and diethyl ethoxymethylenemalonate (Sigma-Aldrich), 2-b, (61.0 mL, 299 mmol, 99%). The reaction was heated for about 40 min at 75 °C and then cooled slightly (71 °C) before adding potassium hydroxide
(33.5 g, 597) in water (] 25 mL). The reaction was heated to 75 °C for lh. During this time an additional portion of EtOH (100 mL) was added to improve mixing. The reaction was cooled to 40 °C before adding aqueous HC1 (81.3 mL, 991 mmol, 37%) in portions. The reaction aged for 1 h 40 min and then Et20 (180 mL) was added. The solids were filtered and rinsed with EtOH, Et20, and then hexane. The solid was men suspended in aqueous HC1 (300 mL, 0.67 M), filtered and washed with aqueous HC1 (300 mL, 1 ), 2:1 Et20:EtOH (350 mL), 1:1 Et EtOH (200 mL), Et20 (150 mL) and then hexane (150 mL) to afford the compound, 2-c. HPLC/MS: 207.2 (M+l); Rt - 0.61 min.
Step B; Benzvl 4^iizvloxvV2 1H-Dvrazo^l-vnDvrimidine-S-carboxvlatc (2-d)
To a solution of 4-hydroxy-2^1H-pyrazol-l-yl)pyrimidine-5-<iariwxylic acid, 2-c, (lg, 4.85mmol) in DMF (40ml) was added K2CO3 (2.34g, 16.98mmol) and benzyl bromide ( 1.83g,
10.67mmol). The mixture was heated at 70°C for 16 hours. The solution was then cooled to room temperature and diluted with water followed by extraction with ethyl acetate. The combined organic layer was washed with water, dried over sodium sulfate and concentrated under vacuum to provide the crude product, 2-d, as a solid (1.2g). Step C; 4^η.^οχν 2 1Η-ρνΓ¾^1-1-νηρντΰηίάίηΒ-5-θ8Γ5ο νΗ€ acid (2-e)
To a solution of benzyl 4-(benzyloxy 2-(lH-pyraz l-l-yl)ryrimidine-5-carb^ 2-d, (2g crude) in EtOH(20mJ)/THF (20ml) water (20ml) was added KOH (0.87g, 15.5mmol). The mixture was stirred at room temperature for 12 hours. The mixture was then concentrated under vacuum and the residue was diluted with water and basified to pH=2 with HC1 followed by extraction with ethyl acetate. The organic layer was brine washed, dried over sodium sulfate and concentrated under vacuum. The residue was purified on a silica gel column with the eluent of petroleum ether/ethyl acetate=7/l to afford the compound 2-e (200mg, 13%).
Step D: 4-(b nzvloiv>-2-ilH-pvrazQl-l-vnDvrimidin-5-amine (2-f)
4-(benzyloxy 2-(lH-pyra2ol-l-yl)pyrimidine-5-carboxylic acid, 2-e, (200mg, 0.68mmol) was dissolved in THF (20ml) and treated with Et3N (204mg, 2.02mmol) and DPPA (204mg, 0.74mmol). The mixture was heated at 66°C for 12 hours. Then water was added and the resulting mixture was continued to stir at reflux for 2h. After that the mixture was concentrated under vacuum. The residue was diluted with aq. 2C03 followed by extraction with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under vacuum. The residue was purified on silica gel column with the eluent of dichloromethane/methanol=50/l(Rf=0.7) to provide compound 2-f (72mg, 40%).

Steo E; N^4-hvdro3cv-2^1H-DYrazo^l-vl Dvrimidin-5.vlV-2^iDhenvIacetamide r2-n
2,2-diphenylacetic acid (110 mg, 0.52 mmol) was dissolved in DMF (10 mL) and EDC (100 mg, 0.52 mmol) and HOBt (70 mg, 0.52 mmol) were added. The mixture was stirred at rt for 5 mins before Et3N (130 mg, 1.3 mmol) and compound 2-f (115 mg, 0.43 mmol) were added. The resulting mixture was stirred at the ambient temperature for 24 h. The resulting mixture was diluted with water followed by extraction with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated under vacuum. The residue was purified on silica gel column with the eluent of ethyl acetate/petroleum ether=l/3 (Rf= 0.7) to afford an intermediate (80 mg, 40%). The intermediate, prepared as above, was hydrogenated with hydrogen balloon in methanol
(15 mL) and Pd/C (20 mg) at room temperature for 16 hours. The mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by prep-HPLC to provide the product, 2-1, as a white solid (10 mg). Ή NMR (300 MHz, CDjOD): δ 8.78(s, lH), 8.48(d, J 2A Hz, 1H), 7.83(s, 1H), 7.24(m, 10H), 6.58(s, IH), 5.37(s, 1H). LC-MS: (M+H)+ =372.

1 H-pyrazole- 1 -carboximidamide hydrochloride, compound 2-a from Example 2, (22.11 g, O.lSmol) in EtOH (250ml) was added sodium methoxide (12.3g, 0.23mol) and diethyl
ethoxymethylenemalonate, compound 2-b from Example 2, (30ml, 0.1 Smol). The reaction was heated for about 40 min at 75 °C and then cooled to room temperature. The mixture was filtered and the solid was washed with ethanol and ether and dried under vacuum to give compound 3-a (21 g, 60%). !H NMR (DMSO-d6, 300ΜΗζ,):δ 8.50(m, 2H), 7.70(s, 1H), 6.47(m, 1H), 4.13(m, 2H), 1.24(m, 3H).
SteP B; Ethvl 4-H enzvlo¾vV2-riH-Dvrazol-l-vnpvrimidine-S-carboivlate (3-b) To a solution of ethyl 4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidine-5-carboxylate, 3-a,
(0.62g, 2.7mmol) in DMF (7ml) were added benzyl chloride (0.5g, 3.99mmol) and Et3N (0.8g,
7.97mmol). The resulting mixture was then heated at 60°C for 12 hours. Then the mixture was cooled to room temperature and diluted with water followed by extraction with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated under vacuum. The residue was purified on silica gel column with the eluent of ethyl acetate petroleum ether=l/l (Rf=0.7) to afford the compound, 3-b, (0.36g, 42%).
Step C: 4-fbeiizvloxvV2^1H-DVi^l-l-vnDvrimidine-S-carboxvlic acid (3-ci
Ethyl 4-(benzyloxy)-2-(lH-pyraz»l-l-yl)pyi½^ 3-b, (150g, 0.46mol) in THF (1.5L) was treated with LiOH monohydrate (58g, 1.39mol) in water (60ml) at room temperature for 3 hours. The mixture was concentrated under vacuum and diluted with water (200ml) followed by extraction with ethyl acetate. The aqueous layer was acidified to pH=2 with 10% HC1 and solid crushed out from the solution which was then filtered and washed with water to afford the compound, 3-c. (40g, 29%).
Ste D; Benzvl 4-H>enCTloxvV2-f lH-pvrazol-l-vnpYrimidin-5-vlcarbamate (3-d¾ To a solution of 4-(benzyloxy)-2-(lH-pyrazol-l-yl)pyrimidin acid, 3-c,
(2g, 6.76 mmol) in dry THF (40ml) and toluene (40ml) were added Et3N (2.04g, 20.3mmol), DPPA (2.04g, 7.43 mmol) and benzyl alcohol (l.lg, 10.1 mmol). The resulting mixture was heated at 60°C for 12 hours. The mixture was concentrated under vacuum and the residue was purified on silica gel column with the eluent of ethyl acetate petroleum ether=l/5 (Rf=0.6) to provide the compound, 3-d (1.96g, 72.6%). Ή NMR (DMSO-d*, 300ΜΗζ,):δ 9.39(br, 1H), 8.73(s, IH), 8.62(s, 1H), 7.83(s, 1 H), 7.56(m, 2H), 7.33(m, 8H), 6.57(m, IH), 5.58(s, 2H), 5.16(s, 2H).
Steo E: S-amino-2 1H-Dvrazol-l-vnDvrimidin-4-ol (3-ei
Benzyl 4^benzyloxy 2 lH-pyrazol-l-yl)pyrimidin-5-ylcarbam^ 3-d, (1.96g, 4.9mmol) was hydrogen ated with hydrogen ballon in THF(20ml) and Pd/C (200mg) at room temperature for 12 hours. The mixture was filtered through a celite pad and the filtrate was concentrated under vacuum to provide the compound, 3-e (0.8g, 93%). Ή NMR (DMSO-de, 300ΜΗζ,):δ 8.39(s, 1H), 7.80(s, 1H), 7.20(s, 1H), 6.56(m, 1H), 5.00(br, 2H).
Example 3

Step A: Preparation of acid chloride
An appropriate carboxylic acid (commercially available, purchased and used as received, 1 mmol) containing the desired R group of compounds 3-f was heated at reflux in excess thionylchloride for 2 hours. The cooled reaction mixture was concentrated under vacuum and the residue was co- evaporated with anhydrous dichloromethane to give the desired acid chloride.
Step B: Preparation of amides (Reversed amide 3-f)
5-amino-2 lH-pyrazol-l-yl)pyrimidin-4-ol, 3-e, (1 mmol) and Et3N (2mmols) were dissolved in dichloromethane(5ml) and cooled to 0 °C. The appropriate concentrated acid chloride (1 mmol) made in Step A was further diluted in dichloromethane(5ml) to form a mixture. This acid chloride mixture was then added dropwise to the solution containing 3-e and Et3N to form a reaction mixture. The reaction mixture was allowed to stir at room temperature for additional 3 hours. The reaction mixture was concentrated under vacuum and the residue was taken into THF (10ml) and treated with 2N lithium hydroxide (10ml) The mixture was stirred at room temperature for 30 min to 12 hours.
For some of the compounds (ca. 20%) a solid precipitated out, thus the solid was collected via filtration to provide the product For compounds that did not precipitate out, the THF layer was separated and concentrated to afford the crude product, which was further purified by recrystallization using a mixed solvent system of petroleum ether/ethyl acetate/methanol (solvent ratio was optimized for each compound to produce the best crystallization conditions).
Table 1 discloses compounds 3-1 through 3-55 that were made in accordance to the general procedure outlined in Example 3 and by utilizing the appropriate carboxylic acid. Table 1




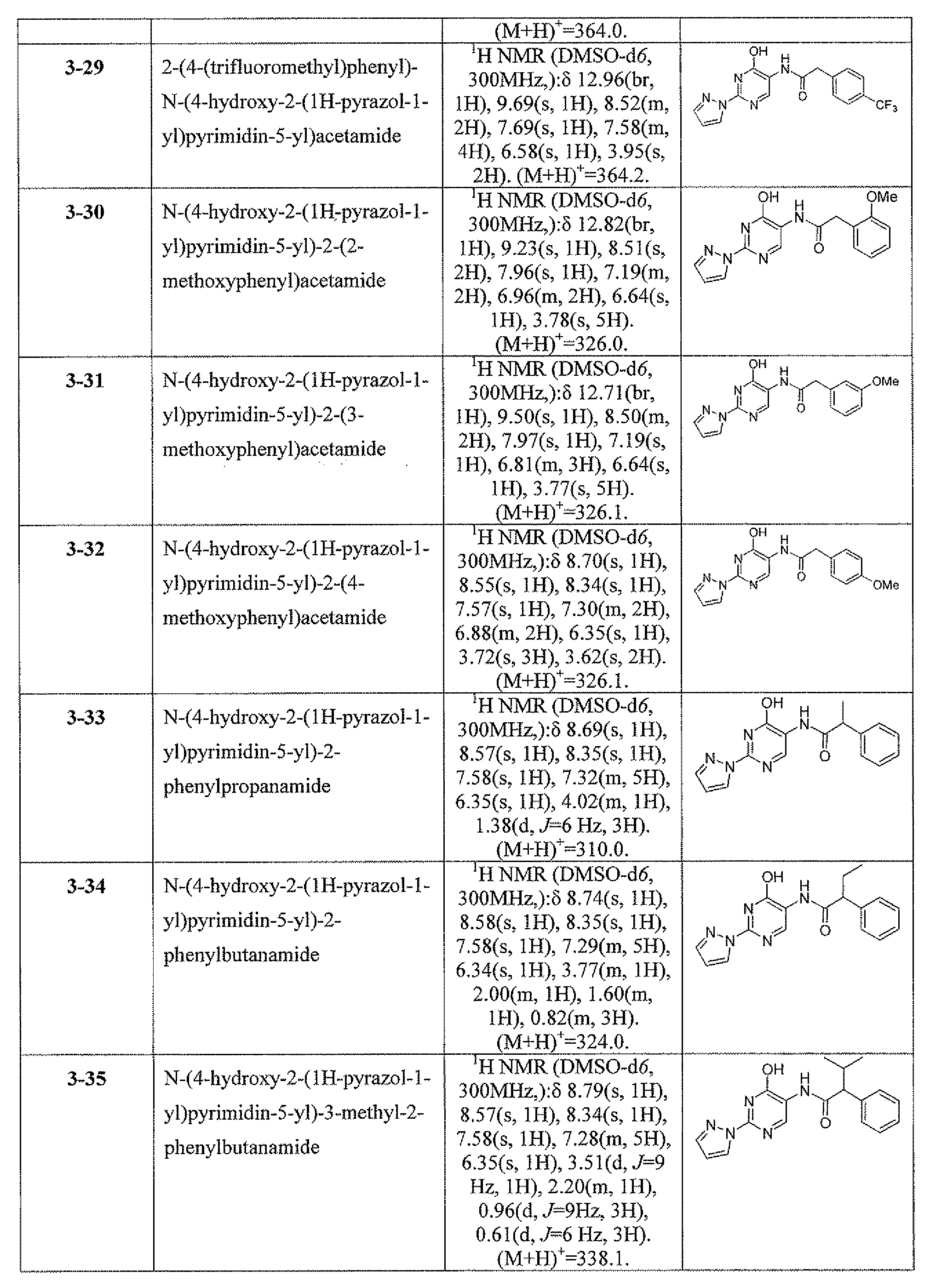



General procedure for Compound 4-a
5-amino-2-( 1 H-pyrazol- 1 -yl) pyrimidin-4-ol (1.44mmol) (3-e) was dissolved pyridine(5ml) and cooled to 0°C. Sulfonyl chloride (1.58minol) was added dropwise. The resulting
mixture was allowed to stir at room temperature for additional 3 hours. The mixture was concentrated under vacuum and the residue was diluted with formic acid (10ml) and water (10ml). A solid precipitated out of solution. The solid was then filtered out of solution. The crude solid was recrystallized (methanol, ethyl acetate or tetrafuran) to provide the product.
Compounds 4-1 through 4-4 were prepared in a manner similar to that described in the general procedure of Example 4-a by substituting the appropriate R group. Table 2 shows compounds 4-1 through 4-4.
Table 2



Azido^4-n egzYloxvV2-riH-Dvrazol-l-vnDvrimidin-S-TDmethanone f5-a
4-(beiizyloxy}-2-(lH-pyrazol-l-yl)pyrimidm acid (3-c) was synthesized in a manner outlined in Steps A through Step C of the synthesis of Intermediate 3-e.
To a solution of 4-(benzyloxy)-2-(lH-pyrazol-l-yl)pyrimW^ acid, 3-c, (500 mg, 1.69 mmol) in dry dichloromethane (10 ml) was added dropwise oxalyl chloride (0.29 ml, 3.38 mmol) at 0 °C and a catalytic amount of DMF (3-5 drops). The mixture was stirred at room temperature for 3 hours. Then the mixture was concentrated under vacuum and the residue was dissolved in acetone (10 ml). Et3N (205mg, 2.03 mmol) was added at 0 °C and the mixture was stirred for 5 mins before sodium azide (988 mg, 15.2 mmol) in water (1 ml) was added. The resulting mixture was stirred at room temperature for lh. Subsequently, the mixture was diluted with water and filtered. The solid was washed with water (5-10 ml) and then taken into approximately 20-30 mL of dichloromethane. The organic layer was dried over sodium sulfate and concentrated under vacuum to provide compound 5-a as a solid(130 mg) which was used in Step B without any further purification. (M+H)+=322.
The yyp f hesfo of fte Carb rnqteftTrea ff-b)

Sft Pf?)} Pe-benzvlationfCarbamate/Urea 1) The desired urea or carbamate obtained in Step B( 1 ) was dissolved in THF( 10ml) and treated with Pd(OH)2(10% amount). The mixture underwent hydrogenation at room temperature and 1 atm (a balloon filled with hydrogen) of hydrogen for 5 hours. The mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by prep-HPLC to provide the desired product.
Table 3 discloses compounds 5-1 through 5-11 that were made in accordance to the general procedure outlined in Example 5 and by utilizing the appropriate urea or carbamate.
Table 3
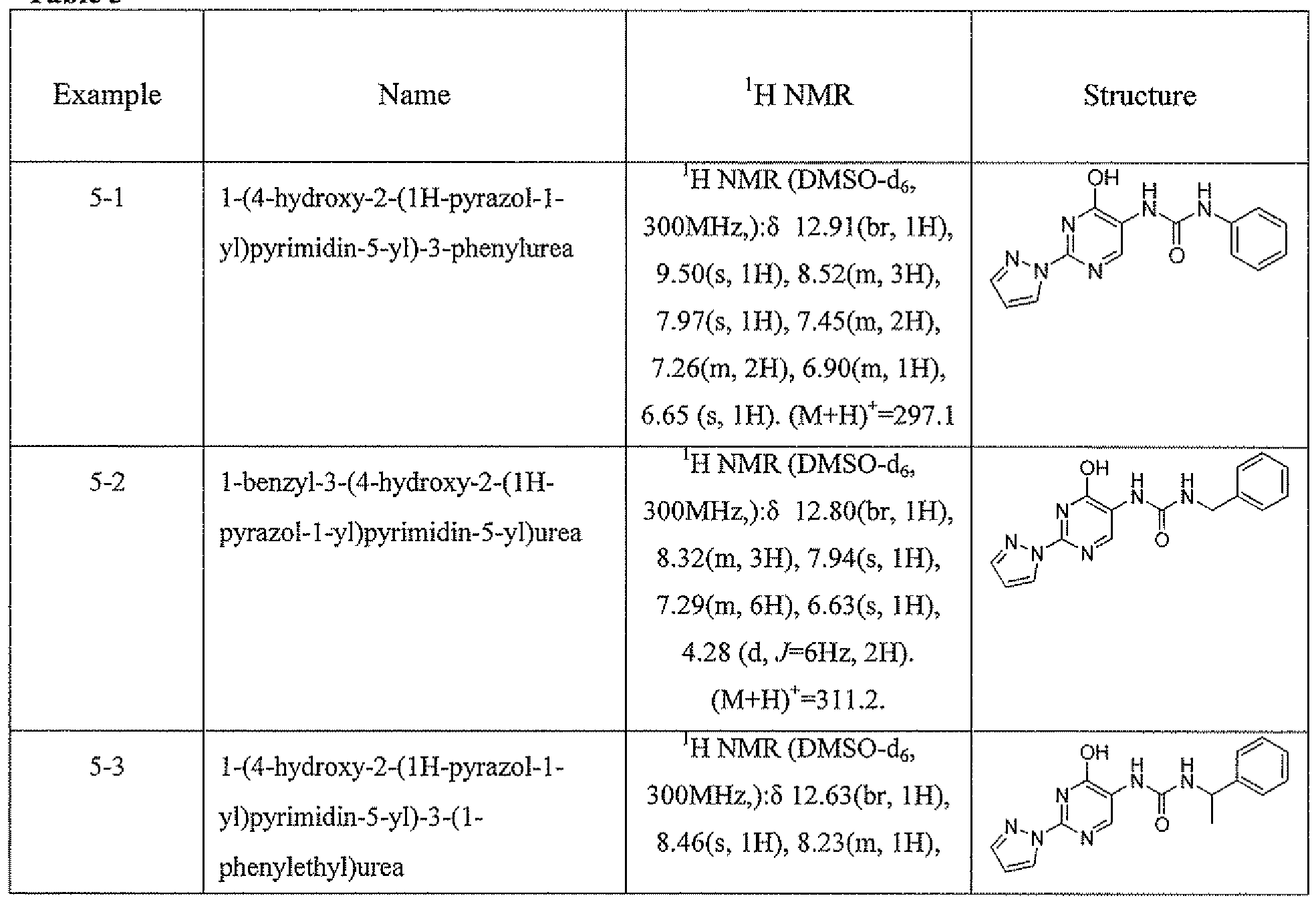




l-benzhvdrvl-3-f4-hvdroiv-2-ilH-Dvra2ol-l-vnDvrimidiD-5-vnthionrea(6-l)
5-amino-2-( 1 H-pyrazol- 1 -yl)pyTimidin-4-ol, 3-e(Sigma-Aldricb), (44mg, 0.25mmol) and isothiocyanatodiphenylmethane (56mg, 0.25mmol) were dissolved in dry THF and the mixture was stirred at reflux for 60 hours. Then the mixture was filtered. The solid was washed with MeOH and purified by pre-HPLC to give the title compound, 6-1. Ή NMR (DMSO-d«, 300MHz,): δ 12.72(br, 1H), 9.2 (br, 3H), 8.50(d, lH), 7.96(s, lH), 7.23(m, lOH), 6.65(m, 2H). (M+H)+=403.1.
Example 7-1
l-(4-hvdroxv-2^1H-Dvrazo l-vnDv^^
5-amino-2-(lH-pyrazol-l-yl)pyrimidin-4-ol, 3-e, (lOOmg, 0.56mmol) and alpha- methylbenzyl isothiocyanate (Sigma-Aldrich) (92mg, 0.56mmol) were dissolved in dry THF and the mixture was stirred at reflux for 60 hours. Then the mixture was filtered. The solid was washed with MeOH and purified by pre-HPLC to give the title compound, 7-1. Ή NMR (DMSO-d«, 300MHz,): δ 9.21(br, 3H), 8.50(d, 1H), 7.95(s, 1H), 7.22<m, SH), 6.65(s, 2H), 5.41(m, 1H), 1.41(d, 3H).
(M+H)+=341.1.

l-f4-hvdroiy-2-aH-Pvi^l-l-vnDvrim^
2-phenylpropan-2-amine (500mg, 3.7mmol) and Et3 (747mg, 7.4mmol) was dissolved in dry ethyl acetate (10ml), thiophosgene (425mg, 3.7mmol) was added dropwise to the mixture at 0°C.
The reaction mixture was warmed to room temperature and stirred for 2 hours. The mixture was washed with water and brine, dried and concentrated. The residue was dissolved in THF (10ml) and 5-amino-2-
(1 H-pyrazol- l-yl)pyrimidin-4-ol, 3-e, (655mg, 3.7mmol) was added. The resulting mixture was reflux ed
for 60 hours. Then it was filtered and the solid was washed with MeOH and purified by pre-HPLC to give the title compound, 8-1. Ή NMR (DMSO-de, 300MHz,): δ I2.62(br, lH), 9.23(br, IH), 8.49(m, lH), 7.93(s, IH), 7.13(m, 5H), 6.64(m, 1H), 1.70(s, 6H). (M+H)+=355.1.

benzhvdryl 4-hvdroxv-2-ClH-Dvrazol-l-Ynpvrimidine-5-€arboxvlate (9-1)
To a solution of 4-(benzyloxy)-2-(lH-pyrazol-l-yl)pyrimidme-5-car^ acid, 2-e, (Ig, 3.38mmol) in dry DCM (20ml) was added dropwise oxalyl chloride (0.44ml, 5.07mmol) at 0°C and catalytic DMF. The reaction mixture was stirred for 3 hours at room temperature. Then it was concentrated under vacuum and the residue was dissolve in DCM (10ml). To the mixture was added dropwise a solution of diphenylmethanol (685mg, 3.72mmol) and E.3N(1.02g, lO.lmmol) in DCM (20ml) at 0°C. The resulting mixture was stirred at room temperature for 3 hours. The resulting mixture was then concentrated and the residue was purified on silica gel (petroleum ether/ethyl acetate=3/l) to give an ester (0.9g, 58%).
The ester (200mg, 0.43mmol) was dissolved in dry THF (10ml) and Pd(OHyC (30mg) was added. The reaction mixture was stirred under ¾ balloon at room temperature for S hours. The mixture was filtered and the filtrate was concentrate under vacuum. The residue was purified by pre- HPLC to give the title compound, 9-1. Ή NMR (DMSO-de, 300MHz,): δ 8.76(br, lH), 8.60(d, IH), 7.99(s, IH), 7.27(m, 1 IH), 6.98(s, IH), 6.68(m, IH). (M+Na)4=395.1.
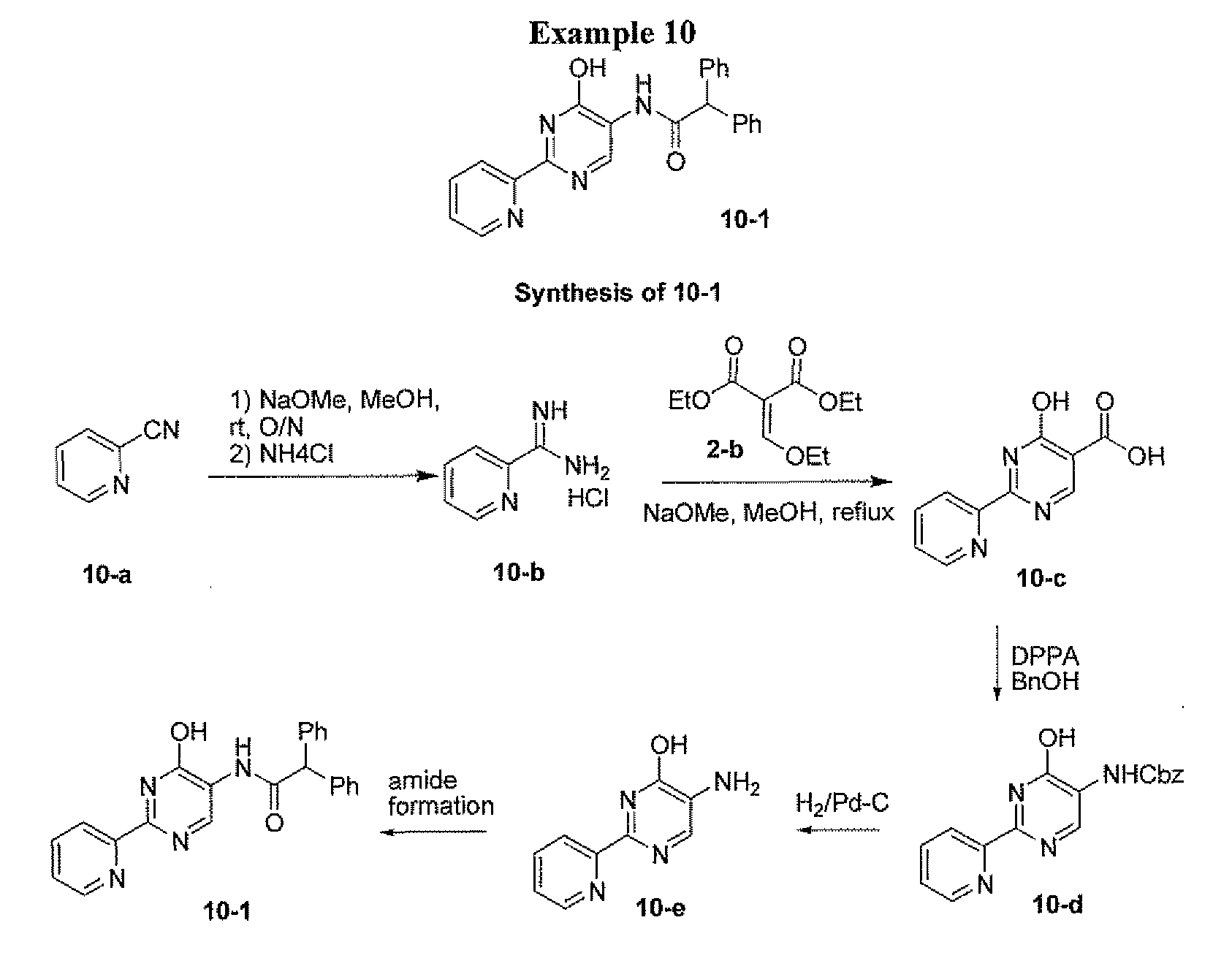
SSSBLAI Pfco)in¾mjd-ne hydrochloride flo-b)
To a solution of 2-cyanopyridine (10-a) (Sigma-Aldrich) (lOOg, 0.95mol) in methanol (1.5L) under nitrogen was added sodium methoxide (2.5g, 44mmoJ). The reaction mixture was stirred at room temperature for 24 hours. Ammonium chloride (53.5g, lmol) was added to the reaction mixture. The mixture was stirred at room temperature for 4h and the solvent was removed in vacuo. The residue was washed with ipropanol/ethyl acetate=l/10 and dried in vacuo to provide compound 10-b (lOOg, 66%).
Step B: 4-hvdroiY-2 Dvridiji-2-vnDvrimidine-5-carhoxvlic acid (10-c)
To picolinamidine hydrochloride (10-b) (112 g, 0.71mol) in MeOH (1.1L) was added sodium methoxide (39.4g, 0.7 lmol) and diethyl ethoxymethylenemalonate (2-b) (142g, 0.71 mol). The reaction was heated at reflux overnight and then cooled to room temperature. The resulting mixture was filtered and to the filtrate was added potassium hydroxide (71.4 g, 1.37 mol) in water (265 mL). The reaction was heated at reflux for 2h. The reaction mixture was cooled to room temperature before adding cone. HC1 (37 %, 180 mL, 2.35 mol) in portions. The reaction aged for 2 h. The solids were filtered and rinsed with EtOH, Et20 and then hexane to provide compound 10-c (HOg, 65%).
Steo C: Benzvl 4-hvdroxY-2-fDvridin-2-vnDvrimidin-5-vlcarfaamate fl0-d¾
To a solution of 4-hydroxy-2-(pyridin-2-yl)pyrimidine-5-cart)oxylic acid (10-c) (10g, 46mmol) in toluene (150ml) was added DPPA(13.94g, 50.6mmol), Et3N (9.31, 92.1mmol) and benzyl alcohol (14.93g, 0.14mol). The resulting mixture was heated at reflux overnight Then the mixture was concentrated under vacuum and the residue was purified on silica gel chromatography with the eluent of petroleum ether/ethyl acetate=l/5, v/v, (Rf«=0.3) to afford the crude compound 10-d (3.2g). The sample delivered was re-purified on prep-HPLC. Ή R (DMSO-d6, 300ΜΗζ,):δ 8.73(m, 1H), 8.40(m, 2H), 8.00(m, 1H), 7.37(m, 6H), 5.19(s, 1H). (M+H)+=323.1.
5-atnino-2-('pvridin-2-vnDvriinldin-4-ol (10-e)
Benzyl 4-hydroxy-2-(pyridin-2-yI)pyrimidin-5-ylcarbamate (10-d) (Ig, 3.1mmol) in methanol (20ml) was hydrogenated under 1 atm of hydrogen in the presence of Pd C (500mg) overnight. The mixture was filtered and the filtrate was concentrated to provide the crude product, 10-e (240mg).
Steo E; N-f4-hvdroxv-2-fDvridin-2-vnDvr^ To a solution of 5-ammo-2-(pyridin-2-yl)pyrimidin-4-ol (10-e) (120mg, 0.64mmol) in MeCN (5ml) were added HATU (254mg, 0.67mmol), Et3N (130mg, 1.28mmol) and 2^-dipbenylacetic acid (135mg, 0.64mmol). The resulting mixture was stirred at room temperature for lhour. Then the mixture was diluted with water and followed by extraction with dichloromethane. The organic layer was washed with brine, dried over sodium sulfate and concentrated under vacuum. The residue was purified by prep- HPLC to provide compound 10-1. Ή NMR (DMSO-d*, 300ΜΗζ,):δ 12.50(br, 1H), 9.85(br, 1H), 8.73(m, 2H), 8.10(m, 1H), 8.00(m, 1H), 7.60(m, 1H), 7.35(m, 10H), 5.75 (s, 1H). (M+H)+-383.2.
Example 11
Synthetic route


Example 12

Step A Ethvl 2^4^benzvlo¾vV-2-flH-pvimol-l-vnpvrimidin-S-vnacetate (12-b)
To a solution of 4-(benzyloxy)-2-(lH-pyrazol-l-yl)pyrim acid, (3- c),(500mg, 1.69mmol) in dry DCM (10ml) was added dropwise oxalyl chloride (0.29ml, 3.38 mmol) at 0°C. Subsequently, one drop of DMF was added. The reaction mixture was stirred for 3 hours at room temperature. The mixture was concentrated under vacuum and the residue was dissolved in DCM (Sml). The resulting solution was added dropwise to a solution of TMSCHN2(193mg, 8.45mmol) and
Et3N(512mg, 5.07mmol) in DCM(20ml) at 0°C. The mixture was stirred at room temperature overnight. The mixture was then concentrated under vacuum to give the diazo compound as a brown solid (crude product).
To a solution of the diazo compound (397mg, 1.24mmol) in EtOH (5ml) was added Ag20 (287mg, 1.24mmol). The mixture was then stirred at room temperature for 2 hours. The mixture was heated to SO °C and stirred for 1 hour. The mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by silica gel (PE EA=5: 1 ) to give compound 12-b as a yellow oil (90mg, 21.5%). Ή MR (CDCl3-<¼, 300 MHz,): δ 8.51 (s, 1 H), 8.34 (s, 1 H), 7.81 (s, 1 H), 7.32-7.44 (m, 5H), 6.46 (s, 2H), 5.54 (s, 2H), 4.07-4.14 (q, 2H), 3.58 (s, 2H), 1.15-1.20 (t, 3H). (M+H)+=339.2.
Ste B 2^4 beiizyloivV-2^1H-pyrazo---l-vnpyrimidin-S-yl)acetlc acid (12-c
To a solution of ethyl 2-(4-(benzyloxy)-2-(lH-pynizol-l-yl)pyrimidin-5-yl)acetate, 12-b, (90mg, 0.27mmol) in THF (6ml) was added LiOH monohydrate (19mg, 0.8mmol) in H20(3ml). The mixture was stirred at room temperature for 3 hours. The mixture was concentrated under vacuum and the remaining aqueous solution was extracted with ethyl acetate (5ml). The separated aqueous phase was adjusted to pH=2 with Cone. HC1. The solid crashed out and was filtered to give compound 12-c(40mg, 48.5%). ¾ NMR (DMSO-tfc, 300 MHz,): δ 12.56 (s, 1H), 8.66 (s, 1H), 8.47 (s, 1H), 7.84 (S, 1H),7.32- 7.52 (M, 5H), 6.59 (s, 1H), 5.57 (S, 2H), 3.61 (s, 2H). (M+H)+=311.1.
SiSEjQ 2^4-hvdrory-2-ilH-Dvrazo^l-vnDvrinildin-S-vnacetic acid (12-d
To a solution of 2-(4-(benzyloxy)-2-(lH-pyra-a)l-l-yl)pyruOidm-5-yl)acetic acid, 12-c, (1.239g, 4mmol) in THF (40ml) was added PdiOHVC (0.14g). The resulting mixture was hydrogenated at room temperature at 1 atm overnight The mixture was filtered and the filtrate was concentrate under vacuum to give compound 12-d (800mg, 91%). Ή NMR (DMSO-tfi, 300 MHz,): δ 12.59 (s, 2H), 8.53 (s, 1H), 7.89 (s, 2H), 6.61 (s, 1H), 3.37 (s, 2H). (M+H)+=221.1.
?tep P N-^4-ch oroDhenvl -2-f4-hvdroiv-2-flH-Dvrazo^l-Yr Dvrimidin-5-vn
ace^nn^e AH)
To a solution of 2-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetic acid, 12-d,
(150mg, 0.68mmol) and HOBt (101.2mg, 0.75mmol) in DMF(5ml) was added EDC.HC1 (144mg, 0.75mmol) and DIEPA(264mg, 2.05mmol). The solution was stirred at room temperature for 5 mins before the addition of the chlorophenyl amine (0.75mmol) (Sigma-Aldrich). The mixture was stirred at room temperature for 24 hours. The mixture was then diluted with water and extracted with ethyl acetate. The organic layer was concentrated under vacuum and the residue was purified by silica gel column or Prep-HPLC to give the final compound 12-1. Ή NMR (DMSO-c , 300 MHz,): δ 10.23 (s, 1H), 8.56 (d, 1H), 7.92 (s, 1H), 7.63 (d, 2H), 7.37 (d, 2H), 6.64 (s, 1H), 3.53 (s, 2H). (M+H)+=330.1.
In addition to compound 12-1, Table 5 discloses compounds 12-2 and 12-3 that were made in accordance to the general procedure outlined in Example 13 and by utilizing the appropriate amine.
Table 5

Example 13

4-hvdroxY-2^Dvridaztn-3-vlVN-tosvlDvrimidine-5-carboiamide Q3-1)
4-hydroxy-2-(pyridazin-3-yl) pyrimidine-5-carboxylic acid, 13-b, (350mg, 1.6 mmol, which was prepared f om compound 1-e via sodium hydroxide hydrolysis in a similar manner as the conversion of 3-b to 3-c) in thionyl chloride (10 ml) was heated at reflux for 4h. The mixture was concentrated under vacuum and the residue was co-evaporated with dichloromethane twice. The remaining oil was dissolved in dichloromethane (10ml). /7-toluenesulfonamide (329mg, 1.93 mmol), triethylamine (486mg, 4.82mmol) and DMAP (390mg, 3.2mmol) were added to the solution at room temperature. The mixture was subsequently stirred at room temperature overnight. The mixture was then concentrated under vacuum and the residue was diluted with water (10ml). The solid crashed out and then filtered out of the solution. The solids were then recrystallization in methanol ethyl acetate to afford compound 13-1 (300mg, 50%). ]H NMR (DMSO^j, 300 MHz,): δ 12.92 (br, 1H), 9.51 (s, 1H), 8.53 (s, 2H), 8.00 (m, 3H), 7.46 (s, 2H), 2.41 (s, 3H). (M+H)+=372.1.
Biological Away?
The exemplified compounds, Examples 1 through 13 of the present invention, have been found to inhibit the interaction between PHD2 and a HIF peptide and exhibit IC50 values ranging between 0.1 nanomolar to 10 micromolar. Non-limiting examples of assays that may be useful to detect favorable activity are disclosed in the following publications: Oehme, F., et al., Anal. Biochem. 330:74- 80 (2004); Hirsilft, M, et al., J. Bio. Chem. 278 (33): 30772-30780 (2005); Hyunju, C, et al., Biochem. Bjophys, Res, Comm. 330 (2005) 275-280; and Hewitson, K. S., et al., Methods in Enzvmologv. (Oxygen Biology and Hypoxia); Elsevier Publisher (2007), pg. 25-42 (ISSN: 0076-6879).
The biological activity of the present compounds may be evaluated using assays described herein below:
To each well of a 96-well plate was added 1 uL of test compound in DMSO and 20 μΐ of assay buffer (50 mM Tris pH 7.4/0.01% Tween-20/0.1 mg ml bovine serum albumin 10 μΜ ferrous sulfate/1 mM sodium ascorbate/20 μ^πιΐ catalase) containing 0.15 g ml FLAG-tagged full length PHD2 expressed in and purified from baculovirus-infected Sf9 cells. After a 30 min preincubation at room temperature, the enzymatic reactions were initiated by the addition of 4 μΐ, of substrates (final concentrations of 0.2 μΜ 2-oxoglutarate and 0.5 μΜ HIF- la peptide biotinyl- DLDLEMLAPYIPMDDDFQL (SEQ ID NO: 1)). After 2 hr at room temperature, the reactions were terminated and signals were developed by the addition of a 25 μί, quench detection mix to a final concentration of 1 mM ortho-phenanthroline, 0.1 mM EDTA, 0.5 nM anti-{His)6 LANCE reagent (Perkin-Elmer Life Sciences), 100 nM AF647-labeled streptavidin (Invitrogen), and 2 μg/ml (His)6-VHL complex (S. Tan (2001) Protein Expr. Purif. 21, 224-234). The ratio of time resolved fluorescence signals at 665 and 620 nm was determined, and percent inhibition was calculated relative to an uninhibited control sample run in parallel.
Inhibition of the catalytic activity of HlP-PHDl and HIF-PHD3 can be determined similarly.
Table 6 depicts the inhibition of HIF PHD2 activity expressed as IC50 (nM), for the exemplified compounds, 1-1, 1-2, 2-1, 3-1 to 3-6, 4-1, and 5-1 of the present invention.
TABLE 6
PHD2 Inhibition Activity
+ = 0.5 < IC5o < 20 (nM)
++ = 20 < IC5o < 100 (nM)
+++ = 100 <IC50 < ΙΟΟΟΟ(ηΜ)





Claims
1. A compound of formula I and pharmaceutically acceptable salts and solvates thereof

wherein
A is heteroaryl, optionally substituted with one or more R9 substituents;
L is chosen from a bond, aryl, heteroaryl, -(C1-3 alkyl)o-]S(Ci_3 alkyl)0-i-, -(C1-3 alkyl)o- iNRaCO-, -(Ci-3 alkyl)0-l RaCO2-, -(C1.3 alkyl)o-iC02-, -(C1.3 alkyl)o- lNRaS02-, -(C1-3 alkylto-l RaO-, <Cl-3 alkyl)o-iNRaCSNRb-, -{C1.3 alkyl)0- iNRaCONRb-, and -(C1.3 alky o-iNRaCOS-;
R2 and R3 are independently selected from hydrogen, halogen, hydroxyl, C\- alkyl, C3-6
heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxyl, -SR\ -NR*2 or -C02-R*;
R1 is selected from
halogen,
Ci-10 alkyl,
-C2-10 alkenyl,
-C2-IO alkynyl,
cycloalkyl Co_io alkyl(oxy)o_i,
heterocyclyl Co-]0 alkyl(oxy)o-l,
arylCo-io alkyl(oxy)o.i,
C]-io alkoxy (carbonyl)0-iCo-10 alkyl,
carboxyl Co-10 alkyl.
carboxyl aryl,
carboxylcycloalkyl,
carboxylheterocyclyl,
Ci-ioalkyloxy Co-ioalkyl,
hydroxy Cn-loal .
hydroxycarbonylCo- 1 oalkoxy,
Ci-io alkylthio,
Ci-io alkylsulfinyl,
aryl Cn_io alkylsulfinyl,
heterocyclyl Co-10 alkylsulfinyl,
cycloalkyl Co-io alkylsulfinyl,
Q-io alkylsulfonyl,
aryl Co-10 alkylsulfonyl,
heterocyclylCo-io alkylsulfonyl,
cycloalkyl Cn-io alkylsulfonyl,
nitro,
perfiuoroCi-6alkyl, and
perfluoroC i -^alkoxy;
wherein in Rl said alkyl, alkenyl, alkynyl, cycloalkenyl, aryl, perfluoralkyl, perfluoroalkoxy,
heterocyclyl, and cycloalkyl are each optionally substituted with one or more R9
substituents;
optionally, R* and L are linked together to form a ring of 5 to 8 atoms optionally substituted with one or more substituents R9; where said ring has 0, 1 , or 2 heteroatoms independently selected from -NRb-, -O- and -S(0)n-;
R9 is selected from halogen, hydroxy, oxo, cyano, aryl, heterocyclyl, cycloalkyl, -C\-6 alkyl, -C\-6 alkoxy, aryloxy, heterocyclyloxy, -0(0-l)(Ci.io)perfluoroalkyl, -C02Ra, -NRbRc -CONRbRc -OC02Ra, -OCONRbRC -NRdC02Ra, -NRdcO RbRc, -SQ)-6 alkyl and -S(0)nRd, wherein said aryl, heteroaryl, heterocycloalkyl, alkoxy, aryloxy, heteroaryloxy, heterocycloalkyloxy are optionally substituted by one or more substituents RlO;
RlO is selected from hydroxy, aryl, heterocycloalkyl, heteroaryl, halogen, oxo, -Cl-6 alkyl, C\s alkoxy, halogen, CO2H, cyano, 0(C=0)o.iCl-6 alkyl, NO2, trifluoromethoxy, trifluoroethoxy, -O(0- l)(Cl-l0)perfluoroalkyl, Co-io alkylaminocarbonylamino, Q)_io alkyloxycarbonylaminoCo-io alkyl, Co-io alkylcarbonylaminoCo-10 alkyl, Co-io alkylaminosulfonylaminoCo-10 alkyl, Co-10 alkylsulfonylaminoCo-io alkyl, Co-10 alkylsulfonyl, Q)_io alkylaminosulfonyl, Co_io
alkylaminocarbonyl, -(C=0)N(Co-6 alkyl)2, -S(Q)-6 alkyl), and NH2;
n is 1 or 2;
Ra is chosen from hydrogen; -Ci-10 alkyl, -(Cl-6 alkyl)C3-8 cycloalkyl; and
-(Ci-6 alkyl)phenyl; and
Rb, Rc, and Rd are each independently chosen from hydrogen, -Ci-10 alkyl,C3-g cycloalkyl, aryl, and heterocyclyl.
2. A compound of Claim 1 wherein Rl is selected from: cycloalkyl Co-10 alk l(oxy)o- J, heterocyclyl Co-io alkyl(oxy)o-l, arylCo-iO alkyl(oxy)o-l, perfluorCi-6 alkyl, perfluoro Ci^alkoxy, and Ci-10 alkoxy (carbonyl)O-lCo-lO alkyl, wherein Rl is optionally substituted with one or more R^ substituents.
3. A compound of Claim 1 wherein R2 is selected from hydrogen, halogen, hydroxy], -Cj-6 alkyl, C3-6 heterocycloalkyl and C3- cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxyl, - SR'. -NR^ or-CC^-R*.
4. A compound of Claim 1 wherein R.3 is selected from hydrogen, halogen, hydroxyl, -Ci- alkyl, C3-6 heterocycloalkyl and C3-6 cycloalkyl wherein said alkyl, heterocycloalkyl and cycloalkyl are each optionally substituted with one or more substituents selected from: hydroxy], - SRVNR^ or-CC^-R*.
5. A compound of Claim 1, wherein L is selected from -(Ci-3 alkyl)o_iNRaCO-, _ (Ci-3 allcy])o_iNRaC02-. -(C1.3 alkyl)0-lCO2-, -(C1-3 alkyl)0-lNRaSO2-, -<Ci_3 alkyl)0- i RacSNRb-, and -(Ci-3 alkyl)o-lNRaCONRb-.
6. A compound of Claim 1, wherein L is selected from -{Ci_3 alkyl)o-iNRaCO-,
-{Ci-3 alkyl)o-iNRaC02-, and -(Ci.3 alkyl)0-lCO2-.
7. A compound of Claim 1, wherein L is -(Ci-3 alky o-lNRaCO-.
8. A compound of Claim 1, wherein A is selected from azabenzimidazole, benzoimidazolyl, benzofuryl, benzofurazanyl, benzopyrazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, cinnolinyl, furanyl, imidazolyl, indolyl, indazolyl, isobenzofuryl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, dihydroisoquinolinyl,
tetrahydroisoquinolinyl, naphthpyridinyl, dihydronaphthyridinyl, tetrahydronaphthyridinyl, oxadiazoh l, oxazolyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl, pyrimidyl, quinazolinyl, quinolyl, quinoxalinyl, quinoxalinyl, tetrahydropyranyl, tetrahydroisoquinolinyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, thienyl, triazoly], azetidinyl, aziridinyl, 1,4-dioxanyl, dihydrobenzoimidazolyl, phenothiazinyl, dihydrobenzofuryl, dihydrobenzothiophenyl,
dihydrobenzoxazolyl, methylenedioxybenzoyl, tetrahydrofuryl, tetrahydrothienyl, 2 ,3 -dihydrobenzofuryl, 2,3-dihydrobenzo-l,4-dioxiny], imidazo(2,l-bXl,3)thiazole, and benzo-1 -dioxolyl, wherein A is optionally substituted with one or more R9 substituents.
9. A compound of Claim 8, wherein A is selected from benzoimidazolyl, benzothiazolyl, benzothienyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, furanyl, imidazolyl, indolyl, indazolyl, isobenzofuryl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridopyridinyl, pyridazinyl, pyridinyl,
pyrimidyl, tetrazolyl, tetrazolopyridyl, thiadiazolyl, thiazolyl, and thienyl, wherein A is optionally substituted with one or more R9 substituents.
10. A compound of Claim 9, wherein A is selected from pyrazinyl,
pyrazolyl, pyridazinyl, pyridinyl, and pyrimidyl, wherein A is optionally substituted with one or more substituents..
11. A compound of Claim 9, wherein A is selected from pyrazolyl and pyridazinyl, wherein A is optionally substituted with one or more substituents.
12. A compound selected from:
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2,2-diphenylacetaraide;
2- chloro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
3- chloro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
4-chloro-N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidm-5-yl)benzamide;
3- (trifluoromemyI)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
4- (trifluoromemyl)-N-(4^iydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(naphthalen-3-yl)acetamide;
2-cyano-N-(4-hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl)benzamide;
3-cyano-N-(4-hydroxy-2-(lH-pyrazol-l -yl)pyrimidin-5-yl)benzamide;
4-cyano-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
2-(trifluoromethyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-2-methoxybenzamide;
N-(4-hydroxy-2-(lH-pyn^l-l-yl)pyrimidm-5-yl)-3-methoxybenzamide;
N-(4-hydroxy-2-(] H-pyrazol- l-yl)pyrimidin-5-yl)-4-methoxybenzamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-2-(methylsulfonyl)benzamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-3-(methylsulfonyl)ben-Eami
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-4-(methylsulfonyl)be
N-[4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5-yl]biphenyl-2-carboxamide;
N-[4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl]biphenyl-3-<»rbox^ide;
N-[4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-S-yl]biphenyl-4-carb^
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-2-phenoxybenzamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-phenoxybenzamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-4-phenoxybenzamide;
2-(2-chlorophenyl)-N-(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(3-chlorophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(4-chlorophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetam ide;
2-(3-cyanophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(2-(trifluoromethyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(3-(trifluoromethyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetaniide;
2-(4-(trifluoromethyl)plienyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-^^
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrim
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -ylJpyrimidiii-S-yl^^methoxyphenyl^cetainide;
N-(4-hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl)-2-phenylpropanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-phenylbutanamide;
N-(4-hydroxy-2-( 1 H-pyrazol-l-yl)pyrimidin-5-yl)-3-methyl-2-phenylbutanamide;
N-(4-hydroxy-2-( lH-pyrazol-1 -yl)pyrimidin-5-yl)-3-metbyl-2-phenylpentanarnide;
N-(4-hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl)-2-methyl-2-phenylpropanamide;
N-(4-hydroxy-2-( lH-pyrazol- 1 -yl)pyrimidin-5-yl)-l-phenylcyclopropanecarboxamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -ylJpyrimidin-S-yl^^naphthalen- 1 -yl)acetamide;
2-(benzo[d][l ,3]dioxol-5-yl)-N-(4-hydroxy-2-(lH-pynrol-l^^
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(pyridin-4-yl)acetamide;
2-(biphenyl-4-yl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetairjide;
N-{4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)heptanamide;
N-{4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-phenylpropanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-4-phenylbutanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)cyclohexanecarboxamide;
tert-butyl4-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-ylcarbamoyl)piperidine- 1 -carboxylate;
2- cyclohexyl-N-(4-hydiK>xy-2-( lH-py razo 101.01- 1 -yl)pyrimidin-5-yl)acetamide;
tert-butyl 3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-ylcarbamoyl)piperidine- 1 -carboxylate;
tert-buryl4-((4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5-ylcarbamoyl)me1hyl)piperidine- 1 -carboxylate; tert-butyl 3-(4-hydroxy-2-( lH-pyrazol-l-yl)pyrimidin-5-ylcarbamoyl)py^
1 ,2,3,4-tetrahydix>-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)naphthalene- 1 -carboxamide;
2^-dihydro-N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)-lH-indene-l
1 ,2,3 ,4-tetrahydro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -y l)pyrimidin-5-yl)naphthalenc-2-carboxamide; 2,3-dihydro-N-(4-hydroxy-2-(l H-pyrazol-1 -yl)py^
4-chloro-N-[4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl]benzenesulfonam
l-(4-chlorophenyl)-N-[4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl]meth
3- chlorc)-N-[4-hydi xy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl]benzenesulfonam
N-[4-bydroxy-2-( 1 H-pyrazol-1 -yI)pyrimidin-5-yl]- 1 -[4-{trifluoromethyl)phenyl]methanesulfonamide; 1 -(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrim idin-5-yl)-3-phenylurea;
1 -benzy l-3-(4-bydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea
1 -(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-( 1 -phenylethyl)urea;
1 -cyclohexyl-3 -(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea
1 -cyclopentyl-3-(4-hydroxy-2-( lH-pyrazol-1 -yl)pyrimidin-5-yl)urea;
ert-butyl-3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yI)urea
3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl 1 , 1 -diphenylurea
3- (4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)- 1 -isopropyl- 1 -phenylurea;
phenyl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yI carbamate;
cyclohexyl 4-hydroxy-2-{]H-pyra-K)l-l-yl)pyrimidin-5-yl carbamate;
cyclopentyl 4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl carbamate;
tert-butyl 4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl carbamate;
butyl 4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl carbamate;
4- methoxyphenyl 4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl carbamate;
3,3-dimethylbutyl 4-hydroxy-2-{l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
phenethyl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidin-5-yl carbamate;
biphenyl-4-yl 4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl carbamate;
naphthalen-2-yl 4-hydroxy-2-(l H-pyrazol- l-yl)pyrimidiri-5-yl carbamate;
1 -benzhydryl-3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)thiourea;
1 -(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-( 1 -phenylethyl)thiourea
1- (4-hydroxy-2-( 1 H-pyrazol- 1 -yl)r>yrimidm-5-yl)-3-(2-phenylpropan-2-yl)thiourea; benzhydryl 4-hydroxy-2-(lH-py-,azol-l-yl)pyrimidme-5-carboxylate
N-(4-hydroxy-2-(pyridin-2-yl)pyrimidin-5-yl)-2^-diphenylacetamide;
5- ( 1 -phenyl- 1 H-pyrazol-4-yl)-2-( 1 H-pyrazol- 1 -yl)pyrimidin-4-ol;
5-( 1 -benzyl- 1 H-pyrazol-4-yl)-2-( 1 H-pyrazol- 1 -yl)pyrimidin-4-ol;
5-phenyl-2-( 1 H-pyrazol- 1 -yl)pyrimidin-4-ol;
N-(4-chlorophenyl)-2-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide; N-cyclohexyl-2-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2- (4-hydroxy-2-(lH-pyra--ol-l-yl)pyrimidin-5-yl)-N-isopiOpylacetamide;
4-hydroxy-2-(pyridazm-3-yl)-N-tosylpyrimidme-5-carboxamide;
and pharmaceutically acceptable salts and solvates thereof.
13. A compound selected from:
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2^-diphenylacetamide;
2- chloroN-(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)benzamide;
3-chloro-N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)benzamide;
3- (trifluoromethyl)-N-(4-hydroxy-2-( 1 H-pyrazol- l-yl)pyrimidin-5-yl)benzamide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrim
2-(trifluoromethyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidm-5-yl)benzamide;
N-[4-hydroxy-2-(lH-pyrazol-l-yl)pvrimidin^
N-(4-hydiOxy-2-(lH-pyrazol-l-yl)pyrimidin^^
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-4-phenoxyben2amide;
2-(2-chIorophenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(3-chloropbenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyii-nidin-5-yl)acetamide;
2-(4-chlorophenyl N^4-hydroxy-2-(lH-p^
2-(3-cyanophenyl)-N-(4-hydroxy-2-(lH-pyrazol-^^
2-(2-{trifluoromethyl)plienyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-(3-(trifluoromethyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetamide;
2-{4-{trifluoromethyl)phenyl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetaniide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(2-methoxyphenyl)a 5etamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(3-methoxyphenyl)a«tarnide;
N-(4-hydroxy-2-(l H-pyrazol-l-yl)pyrimidin-5-yl 2-(4-meth
N-(4-hydroxy-2-(l H-pyrazol-1 -yl)pyrimidin-5-yl)-2-phenylpropanamide;
N-(4~hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl)-2-phenylbutanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-3-rnethyl-2-phenylpentanamide;
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yljpyrimidm
N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)-2-(naphthalen- 1 -yl)acetamide;
2-(benzo[d][ 1 ,3]dioxol-5-yl)-N-(4-hydroxy-2-( lH-pyrazol-1 -yl)pyrimidin-5-yl)acetamide;
2-(biphenyl-4-yl)-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)acetarnide;
N-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimidin-5-yl)heptanarnide;
N-(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrirnldiri-5-yl)-4-phenylbutanarnide;
tert-butyl4-(4-hydroxy-2-( lH-pyrazol-1 -yl)pyrimidin-5-ylcarbamoyl)piperidine-l -carboxylate;
2- cycIohexyl-N-(4-hydroxy-2-( 1 H-pyrazol 01.01- 1 -yl)pyrimidin-5-yl)acetamide;
1,2,3 ,4-tetrahy dro-N-(4-hydroxy-2-{ 1 H-pyrazol- 1 -yl)pyrimidin-5 -y l)naphthalene- 1 -carboxamide; 2,3-dihydro-N-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidiii-5-yl)- lH-indene- 1 -carboxamide;
2,3-dihydrc-N-<4-hydroxy-2-( lH-pyrazol- 1 -yl)pyrimidin-5-yl)- 1 H-indene-2-carboxamide;
1 -benzyl-3-(4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl)urea;
l-(4-hydroxy-2-(lH-pyrazol-l-yl)pyrimid^
3- (4-hydroxy-2-( 1 H-pyrazol- 1 -yl)pyrimidin-5-yl 1 , 1 -diphenylurea
l-beiizhydr l-S-^hydroxy^-OH-pyniro ^
l-(4-hydroxy-2-( 1 H-pyrazol-1 -yl)pyrimidin-5-yl)-3-(l -phenylethyl)thiourea;
and pharmaceutically acceptable salts and solvates thereof.
14. A pharmaceutical composition comprising a compound of Claim 1 and pharmaceutically acceptable carri
IS. A method of enhancing endogenous production of erythropoietin in a mammal which comprises administering to the mammal an amount of a compound of Claim 1, or a
pharmaceutically acceptable salt or solvate thereof, that is effective for enhancing endogenous production of erythropoietin.
1 . A method for the prevention or treatment of anemia in a mammal which comprises administering to the mammal an effective amount of a compound of Claim 1, or a pharmaceutically acceptable salt or solvate thereof.
17. Use of a compound of Claim 1 , or a pharmaceutically acceptable salt or solvate thereof, in the manufacture of medicaments for the treatment of conditions mediated by HIF prolyl hydroxylase.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11772489.8A EP2560655B1 (en) | 2010-04-21 | 2011-04-18 | Substituted pyrimidines |
| US13/635,275 US9006433B2 (en) | 2010-04-21 | 2011-04-18 | Substituted pyrimidines |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2010/071974 WO2011130908A1 (en) | 2010-04-21 | 2010-04-21 | Substituted pyrimidines |
| CNPCT/CN2010/071974 | 2010-04-21 | ||
| US33443210P | 2010-05-13 | 2010-05-13 | |
| US61/334,432 | 2010-05-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011133444A1 true WO2011133444A1 (en) | 2011-10-27 |
Family
ID=44834465
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/032829 WO2011133444A1 (en) | 2010-04-21 | 2011-04-18 | Substituted pyrimidines |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9006433B2 (en) |
| EP (1) | EP2560655B1 (en) |
| WO (1) | WO2011133444A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014184234A1 (en) * | 2013-05-14 | 2014-11-20 | Active Biotech Ab | N-(heteroaryl)-sulfonamide derivatives useful as s100-inhibitors |
| WO2016054804A1 (en) * | 2014-10-10 | 2016-04-14 | Merck Sharp & Dohme Corp. | Substituted pyrimidines as inhibitors of hif prolyl hydroxylase |
| WO2019105275A1 (en) | 2017-11-29 | 2019-06-06 | 沈阳中化农药化工研发有限公司 | Substituted pyrimidine compound and preparation method therefor and use thereof |
| EP3702355A1 (en) | 2015-05-18 | 2020-09-02 | Shenyang Sinochem Agrochemicals R&D Co., Ltd. | Substituted pyrazole compounds containing pyrimidine, the preparation and application thereof |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3237413A4 (en) | 2014-12-23 | 2018-09-12 | FGH Biotech | Compositions of fatostatin based heterocyclic compounds and uses thereof |
| CN104961686B (en) * | 2015-05-18 | 2017-12-05 | 华中师范大学 | The synthetic method and its application in five kinds of common causative fungi growth activities suppress of 1,6 dihydrogen dazins and pyridazine compound |
| US20170020769A1 (en) * | 2015-07-20 | 2017-01-26 | Prs Medical Technologies, Inc. | Cushion support insert |
| KR102498741B1 (en) | 2016-04-29 | 2023-02-10 | 에프지에이치 바이오테크 인코포레이티드 | Di-substituted pyrazole compounds for the treatment of disease |
| CN110072861B (en) | 2016-09-07 | 2022-11-11 | Fgh生物科技公司 | Disubstituted pyrazoles for the treatment of diseases |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090239876A1 (en) | 2008-03-18 | 2009-09-24 | Clements Matthew J | Substituted 4-Hydroxypyrimidine-5-Carboxamides |
| US20100093803A1 (en) | 2006-10-26 | 2010-04-15 | Bayer Scherring Pharma Aktiengellschaft | Substituted dipyridyl-dihydropyrazolones and use thereof |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL114673A (en) | 1995-07-19 | 2000-12-06 | Hadasit Med Res Service | Pharmaceutical compositions containing protein for oral administration |
| DE60327640D1 (en) | 2002-10-08 | 2009-06-25 | Scripps Research Inst | |
| CA2766056A1 (en) * | 2009-06-30 | 2011-01-06 | Merck Sharp & Dohme Corp. | Substituted 4-hydroxypyrimidine-5-carboxamides |
| US20110021531A1 (en) | 2009-07-27 | 2011-01-27 | Chobanian Harry | Oxazole derivatives useful as inhibitors of faah |
| MX2012008801A (en) | 2010-01-28 | 2012-08-17 | Merck Sharp & Dohme | Pharmaceutical compositions for the treatment of pain and other indicatons. |
| AU2011238487B2 (en) | 2010-04-08 | 2015-10-01 | Merck Sharp & Dohme Corp. | Oxazole derivatives useful as modulators of FAAH |
| WO2011130908A1 (en) * | 2010-04-21 | 2011-10-27 | Merck Sharp & Dohme Corp. | Substituted pyrimidines |
-
2011
- 2011-04-18 US US13/635,275 patent/US9006433B2/en active Active
- 2011-04-18 EP EP11772489.8A patent/EP2560655B1/en active Active
- 2011-04-18 WO PCT/US2011/032829 patent/WO2011133444A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100093803A1 (en) | 2006-10-26 | 2010-04-15 | Bayer Scherring Pharma Aktiengellschaft | Substituted dipyridyl-dihydropyrazolones and use thereof |
| US20090239876A1 (en) | 2008-03-18 | 2009-09-24 | Clements Matthew J | Substituted 4-Hydroxypyrimidine-5-Carboxamides |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2560655A4 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014184234A1 (en) * | 2013-05-14 | 2014-11-20 | Active Biotech Ab | N-(heteroaryl)-sulfonamide derivatives useful as s100-inhibitors |
| CN105377837A (en) * | 2013-05-14 | 2016-03-02 | 活跃生物技术有限公司 | N-(heteroaryl)-sulfonamide derivatives useful as S100-inhibitors |
| CN105377837B (en) * | 2013-05-14 | 2017-08-04 | 活跃生物技术有限公司 | N (heteroaryl) sulfamide derivative as S100 inhibitor |
| EA028512B1 (en) * | 2013-05-14 | 2017-11-30 | Эктив Байотек Аб | N-(heteroaryl)sulfonamide derivatives useful as s100-inhibitors |
| US9873687B2 (en) | 2013-05-14 | 2018-01-23 | Active Biotech Ab | N-(heteroaryl)-sulfonamide derivatives useful as S100-inhibitors |
| US10125125B2 (en) | 2013-05-14 | 2018-11-13 | Active Biotech Ab | N-(heteroaryl)-sulfonamide derivatives useful as S100-inhibitors |
| WO2016054804A1 (en) * | 2014-10-10 | 2016-04-14 | Merck Sharp & Dohme Corp. | Substituted pyrimidines as inhibitors of hif prolyl hydroxylase |
| EP3702355A1 (en) | 2015-05-18 | 2020-09-02 | Shenyang Sinochem Agrochemicals R&D Co., Ltd. | Substituted pyrazole compounds containing pyrimidine, the preparation and application thereof |
| WO2019105275A1 (en) | 2017-11-29 | 2019-06-06 | 沈阳中化农药化工研发有限公司 | Substituted pyrimidine compound and preparation method therefor and use thereof |
| US11457628B2 (en) | 2017-11-29 | 2022-10-04 | Shenyang Sinochem Agrochemicals R&D Co., Ltd. | Substituted pyrimidine compound and preparation method and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US9006433B2 (en) | 2015-04-14 |
| EP2560655B1 (en) | 2016-08-24 |
| EP2560655A4 (en) | 2013-09-04 |
| US20130018053A1 (en) | 2013-01-17 |
| EP2560655A1 (en) | 2013-02-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2718528C (en) | Substituted 4-hydroxypyrimidine-5-carboxamides | |
| EP2560655A1 (en) | Substituted pyrimidines | |
| EP2257170B1 (en) | Tetrahydrofuropyridones | |
| EP2257171B1 (en) | Tetrahydro-1h-pyrrolo-fused pyridones | |
| EP3197455B1 (en) | Inhibitors of hif prolyl hydroxylase | |
| EP2257169B1 (en) | Tetrahydrothieno pyridines | |
| EP2758058B1 (en) | Substituted pyrimidines | |
| JP2012532127A (en) | Substituted 4-hydroxypyrimidine-5-carboxamide | |
| WO2013040789A1 (en) | Substituted pyrimidines | |
| WO2011130908A1 (en) | Substituted pyrimidines | |
| US10208060B2 (en) | Inhibitors of HIF prolyl hydroxylase | |
| WO2016057753A1 (en) | Substituted pyrimidines as inhibitors of hif prolyl hydroxylase | |
| EP3204384A1 (en) | Substituted pyridine inhibitors of hif prolyl hydroxylase | |
| WO2016054804A1 (en) | Substituted pyrimidines as inhibitors of hif prolyl hydroxylase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11772489 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13635275 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011772489 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011772489 Country of ref document: EP |