WO2011123937A1 - Kinase inhibitors and method of treating cancer with same - Google Patents
Kinase inhibitors and method of treating cancer with same Download PDFInfo
- Publication number
- WO2011123937A1 WO2011123937A1 PCT/CA2011/000366 CA2011000366W WO2011123937A1 WO 2011123937 A1 WO2011123937 A1 WO 2011123937A1 CA 2011000366 W CA2011000366 W CA 2011000366W WO 2011123937 A1 WO2011123937 A1 WO 2011123937A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- alkoxy
- heterocycloalkyl
- halogen
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCC(C=C([C@]1NNc2c1cc(CN*)cc2)C=C*(C)C*)S(N)(=O)=O Chemical compound CCC(C=C([C@]1NNc2c1cc(CN*)cc2)C=C*(C)C*)S(N)(=O)=O 0.000 description 12
- QBEAAJOAMVJRAD-UHFFFAOYSA-N Cc1c2c(-c3cccc(S(N)(=O)=O)c3)n[n](C3OCCCC3)c2ccc1N Chemical compound Cc1c2c(-c3cccc(S(N)(=O)=O)c3)n[n](C3OCCCC3)c2ccc1N QBEAAJOAMVJRAD-UHFFFAOYSA-N 0.000 description 2
- HCOMUZCVHOCHEZ-BJMVGYQFSA-N C=C/C(/COc(cc1)cc2c1[nH]nc2)=C\C=[IH] Chemical compound C=C/C(/COc(cc1)cc2c1[nH]nc2)=C\C=[IH] HCOMUZCVHOCHEZ-BJMVGYQFSA-N 0.000 description 1
- NDMMKRLGCSNFFQ-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1)CC1c1c(CC(O)=O)cccc1)=O Chemical compound CC(C)(C)OC(N(CCC1)CC1c1c(CC(O)=O)cccc1)=O NDMMKRLGCSNFFQ-UHFFFAOYSA-N 0.000 description 1
- MNDWXTHFCWVOME-UHFFFAOYSA-N CC=[O]=C(Cc1cc(-c(c2c3)n[nH]c2ccc3NC(Cc2cc(OC)ccc2)=O)ccc1)N Chemical compound CC=[O]=C(Cc1cc(-c(c2c3)n[nH]c2ccc3NC(Cc2cc(OC)ccc2)=O)ccc1)N MNDWXTHFCWVOME-UHFFFAOYSA-N 0.000 description 1
- KEJHGIZQVCEWOP-UHFFFAOYSA-N CN(C)C(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(C)(=O)=O)c1)=O)c1c[s]cc1 Chemical compound CN(C)C(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(C)(=O)=O)c1)=O)c1c[s]cc1 KEJHGIZQVCEWOP-UHFFFAOYSA-N 0.000 description 1
- HBPYFIYSTUYCSR-UHFFFAOYSA-N CN(C)C(C(O)=O)c(cccc1)c1OC Chemical compound CN(C)C(C(O)=O)c(cccc1)c1OC HBPYFIYSTUYCSR-UHFFFAOYSA-N 0.000 description 1
- WTOKIKARUZVOHM-UHFFFAOYSA-N CN(C)CC(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(N)(=O)=O)c1)=O)c1ccccc1 Chemical compound CN(C)CC(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(N)(=O)=O)c1)=O)c1ccccc1 WTOKIKARUZVOHM-UHFFFAOYSA-N 0.000 description 1
- QKBVMGZOYPDKDZ-UHFFFAOYSA-N CN(CC1)CCN1C(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(N)(=O)=O)c1)=O)c1ccccc1 Chemical compound CN(CC1)CCN1C(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(N)(=O)=O)c1)=O)c1ccccc1 QKBVMGZOYPDKDZ-UHFFFAOYSA-N 0.000 description 1
- QERHQJUCADUIPG-UHFFFAOYSA-N CN1C(C(Nc(cc2)cc3c2[nH]nc3-c2cccc(S(N)(=O)=O)c2)=O)c2ccccc2CC1 Chemical compound CN1C(C(Nc(cc2)cc3c2[nH]nc3-c2cccc(S(N)(=O)=O)c2)=O)c2ccccc2CC1 QERHQJUCADUIPG-UHFFFAOYSA-N 0.000 description 1
- TZZYBRUEABVWGB-UHFFFAOYSA-N COc1c(C(Nc(cc23)ccc2[nH]nc3-c2cccc(S(N)(=[O-2])=O)c2)=O)[s]cc1 Chemical compound COc1c(C(Nc(cc23)ccc2[nH]nc3-c2cccc(S(N)(=[O-2])=O)c2)=O)[s]cc1 TZZYBRUEABVWGB-UHFFFAOYSA-N 0.000 description 1
- FIGFDWRNAOVLGC-UHFFFAOYSA-N CS(c1cc(-c(c2c3)n[nH]c2ccc3C(O)=O)ccc1)(=O)=O Chemical compound CS(c1cc(-c(c2c3)n[nH]c2ccc3C(O)=O)ccc1)(=O)=O FIGFDWRNAOVLGC-UHFFFAOYSA-N 0.000 description 1
- MCMARUXDPBQLMI-UHFFFAOYSA-N CS(c1cccc(-c2n[nH]c(cc3)c2cc3NC(C(c2ccc[s]2)N2CCCC2)=O)c1)(=O)=O Chemical compound CS(c1cccc(-c2n[nH]c(cc3)c2cc3NC(C(c2ccc[s]2)N2CCCC2)=O)c1)(=O)=O MCMARUXDPBQLMI-UHFFFAOYSA-N 0.000 description 1
- NXHVPIRUCUUQNP-UHFFFAOYSA-N C[O]=S(c1cc(-c2n[nH]c(cc3)c2cc3NC(c2cnccc2)=O)ccc1)(N)=O Chemical compound C[O]=S(c1cc(-c2n[nH]c(cc3)c2cc3NC(c2cnccc2)=O)ccc1)(N)=O NXHVPIRUCUUQNP-UHFFFAOYSA-N 0.000 description 1
- AIFYSIHSUXTSGT-UHFFFAOYSA-N Cc(c1c(cc2)[nH]nc1)c2[N+]([O-])=O Chemical compound Cc(c1c(cc2)[nH]nc1)c2[N+]([O-])=O AIFYSIHSUXTSGT-UHFFFAOYSA-N 0.000 description 1
- QHNIQCJTDMEPES-UHFFFAOYSA-N Cc(c1c(cc2)[nH]nc1Br)c2[N+]([O-])=O Chemical compound Cc(c1c(cc2)[nH]nc1Br)c2[N+]([O-])=O QHNIQCJTDMEPES-UHFFFAOYSA-N 0.000 description 1
- LQTBKVQSYOSNMZ-UHFFFAOYSA-N Cc1c[s]cc1C(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(N)(=O)=O)c1)=O)N1CCCC1 Chemical compound Cc1c[s]cc1C(C(Nc(cc12)ccc1[nH]nc2-c1cccc(S(N)(=O)=O)c1)=O)N1CCCC1 LQTBKVQSYOSNMZ-UHFFFAOYSA-N 0.000 description 1
- GTMBMMMOQTXXAG-UHFFFAOYSA-N Cc1cccc(CC(Nc(cc23)ccc2[nH]nc3-c2cc(S(N)(=O)=O)ccc2)=O)c1 Chemical compound Cc1cccc(CC(Nc(cc23)ccc2[nH]nc3-c2cc(S(N)(=O)=O)ccc2)=O)c1 GTMBMMMOQTXXAG-UHFFFAOYSA-N 0.000 description 1
- AGXWOMFBZJHDBF-UHFFFAOYSA-N NC(C1CCC1)c1c[s]cc1 Chemical compound NC(C1CCC1)c1c[s]cc1 AGXWOMFBZJHDBF-UHFFFAOYSA-N 0.000 description 1
- ZOEXAUMSJTTXIB-OAQYLSRUSA-N NS(c1cc(-c(c2c3)n[nH]c2ccc3C(N[C@H]2c3ccccc3CCC2)=O)ccc1)(=O)=O Chemical compound NS(c1cc(-c(c2c3)n[nH]c2ccc3C(N[C@H]2c3ccccc3CCC2)=O)ccc1)(=O)=O ZOEXAUMSJTTXIB-OAQYLSRUSA-N 0.000 description 1
- UIEKKDKARNWXFP-UHFFFAOYSA-N NS(c1cc(-c(c2c3)n[nH]c2ccc3NC(C(c2ccccc2)O)=O)ccc1)(=O)=O Chemical compound NS(c1cc(-c(c2c3)n[nH]c2ccc3NC(C(c2ccccc2)O)=O)ccc1)(=O)=O UIEKKDKARNWXFP-UHFFFAOYSA-N 0.000 description 1
- SAPNYXBZSSOJNZ-UHFFFAOYSA-N NS(c1cc(-c(c2c3)n[nH]c2ccc3OCCc2ccc[s]2)ccc1)(=O)=O Chemical compound NS(c1cc(-c(c2c3)n[nH]c2ccc3OCCc2ccc[s]2)ccc1)(=O)=O SAPNYXBZSSOJNZ-UHFFFAOYSA-N 0.000 description 1
- OYNOTANVUQGHGT-UHFFFAOYSA-N NS(c1cc(-c2n[nH]c(cc3)c2cc3NC(C(c2ccccc2)N2CCCCC2)=O)ccc1)(=O)=O Chemical compound NS(c1cc(-c2n[nH]c(cc3)c2cc3NC(C(c2ccccc2)N2CCCCC2)=O)ccc1)(=O)=O OYNOTANVUQGHGT-UHFFFAOYSA-N 0.000 description 1
- OHMNXESRQDKGMX-UHFFFAOYSA-N O=C(Cc1ccc[s]1)Nc(cc12)ccc1[nH]nc2I Chemical compound O=C(Cc1ccc[s]1)Nc(cc12)ccc1[nH]nc2I OHMNXESRQDKGMX-UHFFFAOYSA-N 0.000 description 1
- GZVYPFTYMATWJR-UHFFFAOYSA-N O=C(c1ccc2[nH]ncc2c1)NCc1ccccc1 Chemical compound O=C(c1ccc2[nH]ncc2c1)NCc1ccccc1 GZVYPFTYMATWJR-UHFFFAOYSA-N 0.000 description 1
- QNPBGCMLNTWVNJ-UHFFFAOYSA-N O=C(c1cnccc1)Nc1ccc2[nH]ncc2c1 Chemical compound O=C(c1cnccc1)Nc1ccc2[nH]ncc2c1 QNPBGCMLNTWVNJ-UHFFFAOYSA-N 0.000 description 1
- STUWJSRMOZCBIX-UHFFFAOYSA-N O=Cc(cc1)cc2c1[nH]nc2I Chemical compound O=Cc(cc1)cc2c1[nH]nc2I STUWJSRMOZCBIX-UHFFFAOYSA-N 0.000 description 1
- KRSNYIWCOPVIAV-UHFFFAOYSA-N OC(C(C1CCCC1)N1CCCC1)=O Chemical compound OC(C(C1CCCC1)N1CCCC1)=O KRSNYIWCOPVIAV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
- A61K31/522—Purines, e.g. adenine having oxo groups directly attached to the heterocyclic ring, e.g. hypoxanthine, guanine, acyclovir
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- Protein kinases have been the subject of extensive study in the search for new therapeutic agents in various diseases, for example, cancer. Protein kinases are known to mediate intracellular signal transduction by effecting a phosphoryl transfer from a nucleoside triphosphate to a protein acceptor that is involved in a signaling pathway. There are a number of kinases and pathways through which extracellular and other stimuli cause a variety of cellular responses to occur inside the cell.
- TTK protein kinase also known as tyrosine threonine kinase, dual specificity protein kinase TTK, Monopolar Spindle 1 (Mpsl) and Phosphotyrosine-Picked Threonine Kinase (PYT), is a conserved multispecific kinase that is capable of phosphorylating serine, threonine and tyrosine residues when expressed in E. coli (Mills et al., J. Biol. Chem. 22(5): 16000-16006 (1992)). TTK mRNA is not expressed in the majority of physiologically normal tissues in human (Id).
- TTK mRNA is expressed in some rapidly proliferating tissues, such as testis and thymus, as well as in some tumors (for example, TTK mRNA was not expressed in renal cell carcinoma, was expressed in 50% of breast cancer samples, was expressed in testicular tumors and ovarian cancer samples) (Id). TTK is expressed in some cancer cell lines and tumors relative to normal counterparts (Id.; see also WO 02/068444 Al).
- agents which inhibit a protein kinase have the potential to treat cancer.
- agents which inhibit a protein kinase have the potential to treat cancer.
- agents which can act as protein kinase inhibitors in particular TTK inhibitors.
- TICs tumor-initiating cells
- cancer stem cells a small subset of cells within the tumor. These cells are termed tumor-initiating cells (TICs) or cancer stem cells. It is thought that the TICs are responsible for drug resistance, cancer relapse and metastasis. Compounds that can inhibit the growth and survival of these tumor-initiating cells can be used to treat cancer, metastasis or prevent recurrence of cancer. Therefore, a need exists for new compounds that can inhibit the growth and survival of tumor- initating cells.
- indazole compounds are potent kinase inhibitors, such as TTK protein kinase, polo-like kinases 4 (PLK4), Aurora Kinases and CHK kinase (see Example B-F).
- TTK protein kinase polo-like kinases 4
- PLK4 polo-like kinases 4
- CHK kinase CHK kinase
- these indazole compounds have potent anticancer activity against breast cancer cells, colon cancer cells, lung cancer cells, melanoma cells, prostate cancer cells, ovarian cancer cells, brain cancer cells and pancreatic cancer cells in cell culture study (see Example G).
- certain indazole TTK inhibitors can inhibit the growth of colon tumor-initiating cells in a cell culture study (see Example H). Based on these discoveries, indazole compounds, pharmaceutical compositions thereof, and methods of treating cancer with the indazole compounds (including reducing the likelihood of recurrence of a
- the present teachings are directed, at least in part, to an indazole compound represented by the following structural formula:
- each R 2 is independently selected from: a)-H, -halogen, -CN, -N0 2 , -(CH 2 )o- 2 0R 10 , -(CH 2 )o- 2 NR n R 12 , -(CH 2 )o- 2 S(0)iR 10 , -(CH 2 )o- 2 NR 13 S(0)iR 10 , -(CH 2 )o- 2 NR ,3 S(0)iNR 14 R 15 , -(CH 2 ) 0 .
- alkyl alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, heterocycloalkylalkyl, heterocycloalkylalkenyl, aryl, arylalkyl, arylalkenyl, heteroaryl, heteroarylalkyl and heteroarylalkenyl, wherein each of the alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, heterocycloalkylalkyl, wherein each of the alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, heterocycloalkylalkyl,
- heterocycloalkylalkenyl, aryl, arylalkyl, arylalkenyl, heteroaryl, heteroarylalkyl and heteroarylalkenyl groups represented by R 2 is optionally substituted with 1 to 5 substituents
- R 10 is selected from -H, alkyl, cycloalkyl, cycloalkyl(Ci-C 6 )alkyl, heterocycloalkyl, heterocycloalkyl (Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl,
- R 13 is -H or an alkyl group optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy and (C]-C3)alkoxy;
- R 14 is -H or an alkyl group optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy and (Ci-C 3 )alkoxy;
- R 15 is selected from -H, alkyl, cycloalkyl, cycloalkyl(d-C 6 )alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C 6 )alkyl, aryl, aryl(d-C6)alkyl, heteroaryl and heteroaryl(Ci-C 6 )alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(Ci-C 6 )alkyl, heterocycloalkyl,
- heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci-C 6 )alkyl represented by R 15 is optionally substituted with 1 to 3 substituents independently selected from
- heterocycloalkyl heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C 3 )alkyl, and wherein each of the (Ci-C 6 )alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(C]-C3)alkyl, heteroaryl and heteroaryl(C]-C3)alkyl substituents for the groups represented by R 15 is optionally substituted with halogen, -N0 2 , -CN, (d-C 3 )alkyl, halo(C r C 3 )alkyl, (C,-C 3 )alkoxy(Ci-C 3 )alkyl, (C r C3)alkoxy or hal
- cycloalkyl, cycloalkyl(Ci-C 3 )alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C 3 )alkyl, aryl, aryl(Ci-C 3 )alkyl, heteroaryl or heteroaryl(Ci-C 3 )alkyl are optionally substituted with 1 to 3 substituents independently selected from -halogen, alkyl, -OR c and -NR a R b ;
- R a and R b are each independently -H or (C
- R° is -H, or (Ci-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NR g R h , hydroxy and (C]-C 3 )alkoxy;
- R d is -H or (Ci-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NR 8 R h , hydroxy and (Ci-C 3 )alkoxy;
- R e and R f are each independently -H or (Ci-Ce)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NR g R h , hydroxy and (Ci-C 3 )alkoxy;
- R e and R f together with the nitrogen to which they are attached, form a 3-8 membered ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NR g R h , -CN, (Ci-C 6 )alkyl, halo(Ci-C 6 )alkyl, (d-C 3 )alkoxy, halo(Ci- C 3 )alkoxy, and (Ci-C 3 )alkoxy(Ci-C 6 )alkyl;
- R g and R h are each independently selected from the group consisting of -H, (Ci-C6)alkyl, halo(Ci-C 6 )alkyl, hydroxy(Ci-C 6 )alkyl and (C 1 -C 3 )alkoxy(C 1 -C 6 )alkyl;
- i 0, 1 or 2;
- n is an integer from 1 to 4.
- n is an integer from 1 to 4.
- R 16 is alkyl, R 2 is not -CN.
- the present teachings include a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and a compound represented by Structural Formula (I) described above or a pharmaceutically acceptable salt thereof.
- Another embodiment of the present teachings provides a method of treating a subject having cancer comprising administering to the subject an effective amount of a compound of Structural Formula (I) or a pharmaceutically acceptable salt thereof.
- Another embodiment of the present teachings provides a method of inhibiting TTK activity in a subject in need of inhibition of TTK activity, comprising administering to the
- -7-4820V.1 subject an effective amount of a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof.
- Another embodiment of the present teachings includes the use of a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof in therapy.
- the therapy is for treating a subject with cancer.
- the therapy is for inhibiting TTK activity in a subject in need of inhibition of TTK activity.
- Another embodiment of the present teachings includes the use of a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating a subject with cancer.
- Another embodiment of the present teachings includes the use of a compound represented by Structural Formulas (I) or a pharamceutically acceptable salt thereof for the manufacture of a medicament for inhibiting TTK activity in a subject in need of inhibition of TTK activity.
- the present teachings provide a method of inhibiting the growth of tumor-initiating cells (or cancer stem cells) in a subject who is undergoing an anti-cancer therapy. Such method includes assessing the subject to determine whether the cancer is in remission; and, if the cancer is in remission, then administering to the subject an effective amount of a TTK inhibitor.
- the present teachings provide a method of reducing the likelihood of recurrence of a cancer in a subject who is undergoing an anti-cancer therapy. Such methods includes assessing the subject to determine whether the cancer is in remission; and, if the cancer is in remission, then administering to the subject an effective amount of a TTK inhibitor.
- the present teachings are directed to a method of inhibiting the growth of tumor-initiating cells in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhibitor.
- the present teachings are directed to a method of reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhibitor.
- the present teachings are directed to a method of treating a drug-resistant cancer in a subject comprising administering to the subject an effective amount of a TTK inhibitor. 4820V.1
- the present teachings are directed to a method of treating a subject with a cancer comprising administering to the subject an effective amount of a compound represented by Structural Formula (I) in combination with an anti-cancer therapy.
- embodiments of the present teachings include the use of a TTK inhibitor for 5 inhibiting the growth of tumor-iniating cells or reducing the likelihood of recurrence of a cancer in a subject who is undergoing an anti-cancer therapy. Further embodiments of the present teachings includes the use of a TTK inhibitor for inhibiting the growth of tumor-iniating cells or reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission. Still other embodiments of the present teachings include the use of a TTK inhibitor for treating a o subject with a drug-resistant cancer.
- the present teachings are directed to a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof.
- Structural Formula (I) or a pharmaceutically acceptable salt thereof.
- R 2 is -H.
- R 3 is cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R 6 .
- R 3 is selected from (C3-C8)cycloalkyl, phenyl, naphthyl,
- R 3 5 tetrahydropyranyl substituents on the groups represented by R 3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, - ⁇ 3 ⁇ 4, -NR a R b , (Ci-C3)alkyl, halo(Ci- C3)alkyl, (Ci-C 3 )alkoxy and (Ci-C 3 )alkoxy(Ci-C3)alkyl.
- R 3 is selected from the group consisting of phenyl, thiophenyl, pyridinyl, pyrazolyl, cyclopentyl,
- R 3 is selected from the group consisting of cyclopentyl, phenyl, thiophenyl, pyridyl, piperidinyl, tetrahydropyranyl, tetrahydrofuranyl, pyrrolidinyl, and indolyl, each of which is optionally substituted with 1 to 3 substituents independently selected from the group of substituents described above.
- R 3 is selected from5 the group consisting of phenyl, thiophenyl, pyridyl, tetrahydropyranyl and indolyl, each of which is independently selected from the group of substituents described above.
- R 3 described in the preceding paragraphs is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -OR 0 , (Ci-C3)alkyl, -(Ci-C3)alkylene-NR a R b , phenyl, pyrimidinyl, morpholinyl and benzyl,
- phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (C,-C 3 )alkyl, halo(Ci-C 3 )alkyl, (C r C 3 )alkoxy and (C,-C 3 )alkoxy(C,-C 3 )alkyl;
- R c is -H, (C]-C 3 )alkyl, pyridinyl or mo ⁇ holinyl; and
- R a and R b are each independently -H or (Ci- C 3 )alkyl.
- R 3 described in the preceding paragraphs is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -F, -CI, methyl, -CF 3 , ethyl, 2-propyl, benzyl, phenyl and -CH2NH2.
- R 4 and R 5 are each independently -H, alkyl, -OR c , -NR a R b , (C C 3 )alkylene-NR a R b , (C r C 3 )alkylene-OR c , cycloalklyl or heterocycloalkyl. In some embodiments, R 4 and R 5 are not both selected from -OR c and -NR a R b .
- R 3 and R 5 together with the carbon atom to which they are attached form a cycloalkyl or a heterocycloalkyl, wherein the cycloalkyl or heterocycloalkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Ci-C 3 )alkyl, halo(Ci-C 3 )alkyl, (C]-C 3 )alkoxy, halo(Ci- C 3 )alkoxy, (Ci-C 3 )alkoxy(Ci-C 3 )alkyl.
- R 4 and R 5 are both -H.
- R 5 is -H and R 4 is -OH, (Ci-C 3 )alkyl, -(C,-C 2 )alkylene-OH, (C,-C 3 )alkoxy, -NR a R b , -(Ci-C2)alkylene-NR a R b , pyrrolidinyl, piperidinyl, mo ⁇ holinyl or cyclopropyl, wherein R a and R b are each independently -H or (C]-C 3 )alkyl and the pyrrolidinyl, piperidinyl, morpholinyl or cyclopropyl represented by R 4 is optionally substituted with halogen, (Ci-C 3 )alkyl, halo(Ci- C 3 )alkyl, (C,-C 3 )alkoxy and (C,-C 3 )alkoxy(Ci-C 3 )alkyl.
- each R 6 is independent selected from the group consisting of halogen, -0R C , (C r C 3 )alkyl, -(C r C 3 )alkylene-NR a R b , phenyl,
- each R 6 is independently selected from the group consisting of halogen, -OR c , (Ci-C3)alkyl, -(Ci- C3)alkylene-NR a R b , phenyl, pyrimidinyl, mo holinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (Ci- C 3 )alkyl, halo(Ci-C 3 )alky., (Ci-C 3 )alkoxy and (C]-C 3 )alkoxy(Ci-C 3 )alkyl; R c is -H,
- each R 6 is independently selected from the group consisting of -F, -CI, methyl, -CF 3 , ethyl, 2-propyl, benzyl, phenyl and -CH 2 NH 2 .
- R 8 is -H or (Ci-C 3 )alkyl. Alternatively, R 8 is -H.
- each R 9 is independently selected from the group consisting of -H, halogen, -CN, -N0 2 , (Cp C 3 )alkyl, halo(Ci-C 3 )alkyl, (Ci-C 3 )alkoxy, (Ci-C 3 )alkoxy(C,-C 3 )alkyl, hydroxy(Ci-C 3 )alkyl or - (Ci-C 3 )alkylene-NR a R b .
- R is -NH 2 .
- cycloalkyl, heterocycloalkyl, aryl or heteroaryl are optionally substituted with 1 to 3 substituents independently selected from halogen, alkyl, -OR c and -NR a R b .
- each i is 0, 1 or 2. Alternatively, each i is 2.
- n is an integer from 1 to 4. Alternatively, n is 1, 2 or 3. In another alternative, n is 1 or 2. In yet another alternatively, n is i .
- n is an integer from 1 to 4. Alternatively, m is 1 or 2. In yet another alternative, m is 1. p is 0, 1 or 2. Alternatively, p is 0. In another alternative, p is 1.
- r is an integer from 1 to 4. Alternatively, r is 1 or 2.
- the present teachings provide an indazole compound represented by the following structural formula:
- the present teachings provide an indazole compound represented by the following structural formula:
- R 16 is alkyl, optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, cycloalkyl(C]-C3)alkyl, heterocycloalkyl,
- heterocycloalkyl(Ci-C 3 )alkyl, aryl, aryl(Ci-C 3 )alkyl, heteroaryl, heteroaryl(Ci-C 3 )alkyl, -CN, -N0 2 , -OR c , -NR a R b , -S(0)jR c , -NRdS(0)iRc, -S(0)iNR e R f , C( 0)OR c ,
- cycloalkyl, cycloalkyl(Ci-C 3 )alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C 3 )alkyl, aryl, aryl(C,-C 3 )alkyl, heteroaryl or heteroaryl(Ci-C 3 )alkyl are optionally substituted with 1 to 3 substituents independently selected from -halogen, alkyl, -OR c and -NR a R b .
- the present teachings provide a compound represented by a
- X is a bond or -CR 4 R 5 -, and for structural formula (II), when p is 0, X can additionally be
- Y is a bond, -NR 14 -, -CR 4 R 5 - or -NR I4 -CR 4 R 5 -;
- R 3 is a cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R 6 ;
- R 4 and R 5 are each independently selected from -H, alkyl, -OR c , -NR a R b , (Ci- C 3 )alkylene-NR a R b , -(C,-C 3 )alkylene-OR c , -(C r C 3 )alkylene-OH, cycloalklyl and
- R 4 and R 5 are not both selected from -OR c and -NR a R b ; or R 3 and R 5 together with the carbon atom to which they are attached form a cycloalkyl or a
- heterocycloalkyl wherein the cycloalkyl or heterocycloalkyl is optionally substituted with 1 to 3
- substituents independently selected from the group consisting of halogen, -CN, (Ci-C3)alkyl, halo(Ci-C 3 )alkyl, (C,-C 3 )alkoxy, halo(Ci-C 3 )alkoxy, (C r C 3 )alkoxy(Ci-C 3 )alkyl;
- p 0, 1 or 2.
- the present teachings are directed to a compound represented by a structural formula select
- compounds represented by structural formula (Ila)-(Va) are represented by a structural formula selected from:
- Y is a bond or -NR 14 ;
- R 3 is selected from (C 3 -C 8 )cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thiophenyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[cT
- the group represented by R 3 is selected from the group consisting of cyclopentyl, phenyl, thiophenyl, pyridyl, piperidinyl, benzo[c][l,2,5]oxadiazolyl, benzo[b]thiophenyl, tetrahydropyranyl, benzo[d]imidazolyl, pyrazoly, isoxazolyl, thiazolyl, quinolinyl,
- each of the (Cj-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R 3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N0 2 , -NR a R b , (d-C 3 )alkyl, halo(C r C 3 )alkyl, (C r C 3 )alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl. Values and alternative values for the remainder of the variables are as described in the first or second embodiment.
- the group represented by R 3 described in the second, third, fourth or fifth embodiment is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -OR 0 , (d-C 3 )alkyl, -(Ci-C 3 )alkylene-NR a R b , phenyl, pyrimidinyl, morpholinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (d-C 3 )alkyl, halo(Ci-C 3 )alkyl, (d-C 3 )alkoxy and (d- 4820V.1 C 3 )alkoxy(Ci-C 3 )alkyl; R c is -H, (Ci-C 3 )alkyl, pyridinyl or morpholinyl; and R a and R b are each independently -
- the group represented by R 3 described in the third, fourth or fifth embodiment is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -F, -CI, methyl, ethyl, -CF 3 , 2-propyl, benzyl, phenyl and
- compounds of the present teachings are represented by a structural formula selected from:
- W is CH or N when is a double bond, or W is -CHR 7 - or -NR 8 - when is a single bond;
- R 5 is absent when is a double bond and R 5 is -H or (Ci-C 3 )alkyl when is a single bond;
- q 1 or 2;
- R 8 is -H or a (C,-C 3 )alkyl
- each R 9 is independently selected from -H, -CN, -N0 2 , halogen, -OR c , -NR a R b , -S(0)iR c ,
- r is an integer from 1 to 4. Values and alternative values for the remainder of the variables are as described for Structural Formula (I) or in the second embodiment.
- R 7 is -H or (C,-C 6 )alkyl
- each R 9 is independently selected from the group consisting of -H, halogen, -CN, -N0 2 , (Ci-C 3 )alkyl, halo(C,-C 3 )alkyl, (d-C 3 )alkoxy, (C 1 -C 3 )alkoxy(C,-C 3 )alkyl, hydroxy(C C 3 )alkyl or -(Ci-C 3 )alkylene-NR a R b ; and
- compounds of the present teachings are represented by a structural formula selected from:
- the group represented by R 3 is selected from (C 3 -Cs)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thiophenyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[6?]imidazolyl, benzo[iflthiazolyl, benzo[b]thiophenyl, quinolinyl, tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl,
- the group represented by R 3 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -OR c , (Ci-C 3 )alkyl, - -(Ci-C 3 )alkylene-NR a R b , phenyl, pyrimidinyl, morpholinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (Q- C 3 )alkyl, haloCCrC ⁇ alkyl, (Ci-C 3 )alkoxy and (Ci-C 3 )alkoxy(Ci-C 3 )alkyl; R c is -H,
- R 3 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -F, -CI, methyl, -CF3, ethyl, 2- propyl, benzyl, phenyl and -CH2NH2.
- R 4 and R 5 are both -H.
- R 5 is -H and R 4 is -OH, (Ci-C 3 )alkyl, -(Ci-C 2 )alkylene-OH,(Ci-C 3 )alkoxy, -NR a R b , -(C,-C 2 )alkylene-NR a R b , pyrrolidinyl, piperidinyl, morpholinyl or cyclopropyl, wherein R a and R b are each independently -H or (Ci-C3)alkyl and the pyrrolidinyl, piperidinyl, morpholinyl or cyclopropyl represented by R 4 is optionally substituted with halogen, (Ci-C 3 )alkyl, halo(Ci-C 3 )alkyl, (C
- R la and R ,b are both -H and values and alternative values for the remainder of the variables are as described in the second, third, fourth, fifth, sixth, eighth, ninth, tenth, eleventh, twelfth or thirteenth embodiment.
- R la and R lb are both -H; R 2 and R 2b are both -H; and values and alternative values for the remainder of the variables are as described in the second, third, fourth, fifth, sixth, eighth, ninth, tenth, eleventh, twelfth or thirteenth embodiment.
- the present teachings provide a compound represented by a structural formula selected from:
- R 16 is (Ci- C6)alkyl, optionally substituted with -N(CH 3 ) 2 or 2,6-dimethylmorpholinyl, e.g., in some embodiments, R 16 is selected from methyl, ethyl, (2,6-dimethylmorpholinyl)propyl, and N,N- dimethylaminopropyl. In some embodiments, R 16 is methyl.
- the present teachings provide a compound represented by a structural formula s
- the present teachings provide a compound represented by a structural formula selected from:
- heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -OR c , -NR a R b , -S(0)iR c , -NR d S(0)iR c ,
- the present teachings provide a compound represented by a structural formula selected from:
- R is a cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R 6 ;
- R 4 and R 5 are each independently selected from -H, alkyl, -OR c , -NR a R b , -(C r
- R 4 and R 5 are not both selected from -OR c and -NR a R b ; or R 3 and R 5 together with the carbon atom to which they are attached form a cycloalkyl or a heterocycloalkyl, wherein the cycloalkyl or heterocycloalkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Ci-C3)alkyl, halo(Ci-C 3 )alkyl, (Cj- C 3 )alkoxy, halo(C,-C 3 )alkoxy, (Ci-C 3 )alkoxy(Ci-C 3 )alkyl;
- substituents independently selected from the group consisting of halogen, -CN, (Cj- C 6 )alkyl, halo(C,-C 6 )alkyl, (C,-C 3 )alkoxy, halo(Ci-C 3 )alkoxy and (Ci-C 3 )alkoxy(Ci-C 6 )alkyl; and
- p 0, 1 or 2;
- the group represented by R 3 is selected from (C 3 -Cs)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thienyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, i o isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[d]imidazolyl, benzo[i ]thiazolyl,
- 20 tetrahydropyranyl substituents on the groups represented by R 3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N0 2 , -NR a R b , (Ci-C )alkyl, halo(Cr C 3 )alkyl, (Ci-C 3 )alkoxy and (Ci-C 3 )alkoxy(Ci-C 3 )alkyl.
- the group represented by R 3 is selected from phenyl, pyridyl, and thienyl, each of which is optionally substituted with halogen or (C r C 3 )alkyl (e.g., chloro or methyl).
- R 4 and R 5 are each independently selected from -H, alkyl, -OR c , -NR a R b , cycloalkyl, heterocycloalkyl and -(C]-C 3 )alkylene-heterocycloalkyl, provided that R 4 and R 5 are not both selected from -OR c and -NR a R b .
- one of R 4 and R 5 is -H and the other is selected from -H, alkyl, -OR c , -NR R b , cycloalklyl, heterocycloalkyl and -(C
- R 4 and R 5 are -H and the other is selected from -H, (C]-C 3 )alkyl, -OCH 3 , -N(CH 3 ) 2 , (C 3 -C6)cycloalklyl, pyrrolidinyl, piperidinyl, - CH2-morpholinyl and -O-cyclopentyl.
- R l and R lb are each independently selected from -H and (Ci-C 3 )alkyl substituted with -N(Me)2, and values and alternative values for the remainder of the variables are as described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment.
- R la and R lb are both -H and values and alternative values for the remainder of the variables are as described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment.
- R la and R lb are both -H; R 2a and R 2b are both -H; and values and alternative values for the remainder of the variables are as described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment.
- the present teachings provide the compounds depicted and/or described by name in the Exemplification, as well as neutral forms and pharmaceutically acceptable salts thereof.
- alkyl used alone or as part of a larger moiety, such as “alkoxy”, “haloalkyl”, “cycloalkylalkyl”, “heterocycloalkylalkyl”, “aralkyl”, “heteroaralkyl” and the like, means saturated aliphatic straight-chain or branched monovalent hydrocarbon radical. Unless otherwise specified, an alkyl group typically has 1-6 carbon atoms, i.e. (Ci-C6)alkyl. As used herein, a "(Ci-C6)alkyl” group is means a radical having from 1 to 6 carbon atoms in a linear or branched arrangement.
- alkylene group is a saturated aliphatic branched or straight-chain divalent hydrocarbon radical. Unless otherwise specified, an alkylene group typically has 1-6 carbon atoms, i.e. (Ci-C6)alkylene.
- alkenyl means branched or straight-chain monovalent hydrocarbon radical containing at least one double bond. Alkenyl may be mono or polyunsaturated, and may exist in the E or Z onfiguration. Unless otherwise specified, an alkenyl group typically has 2-6 carbon
- (C2-C 6 )alkenyl means a radical having from 2-6 carbon atoms in a linear or branched arrangement.
- Alkynyl means branched or straight-chain monovalent hydrocarbon radical containing at least one triple bond. Unless otherwise specified, an alkynyl group typically has 2-6 carbon atoms, i.e. (C 2 -C 6 )alkynyl. For example, “(C 2 -C 6 )alkynyl” means a radical having from 2-6 carbon atoms in a linear or branched arrangement.
- Alkoxy means an alkyl radical attached through an oxygen linking atom, represented by -O-alkyl. For example, includes methoxy, ethoxy, propoxy, and butoxy.
- haloalkyl and “haloalkoxy” means alkyl or alkoxy, as the case may be, substituted with one or more halogen atoms.
- halogen means F, CI, Br or I.
- halogen in a haloalkyl or haloalkoxy is F.
- aryl group used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyl”, means an aromatic hydrocarbon ring system.
- aryl may be used interchangeably with the terms “aryl ring” “aromatic ring”, “aryl group” and “aromatic group”.
- An aryl group typically has six to fourteen ring atoms. Examples includes phenyl, naphthyl, anthracenyl, 1,2-dihydronaphthyl, 1,2,3,4-tetrahydronaphthyl, fluorenyl, indanyl, indenyl and the like.
- a "substituted aryl group” is substituted at any one or more substitutable ring atom, which is a ring carbon atom bonded to a hydrogen.
- Cycloalkyl means a saturated aliphatic cyclic hydrocarbon radical optionally containing one or more double bonds. It can be monocyclic, bicyclic, polycyclic (e.g., tricyclic), fused, bridged, or spiro.
- monocyclic (C3-C 8 )cycloalkyl means a radical having from 3-8 carbon atoms arranged in a monocyclic ring.
- a (C3-C8)cycloalkyl includes, but is not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctane.
- Heterocycloalkyl means a saturated or unsaturated non-aromatic 4-12 membered ring radical optionally containing one or more double bonds. It can be monocyclic, bicyclic, tricyclic, fused, bridged, or spiro.
- the heterocycloalkyl contains 1 to 4 heteroatoms, which may be the same or different, selected from N, O or S.
- the heterocycloalkyl ring optionally contains one or more double bonds and/or is optionally fused with one or more aromatic rings (e.g., phenyl ring).
- the term “heterocycloalkyl” is intended to include all the possible isomeric forms.
- heterocycloalky examples include, but are not limited to, morpholinyl, thiomorpholinyl, pyiTolidinonyl, pyrrolidinyl, piperidinyl, piperazinyl, hydantoinyl, valerolactamyl, oxiranyl, oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyrindinyl, tetrahydropyrimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl, and tetrahydroquinolinyl.
- heteroaryl "heteroaromatic”, “heteroaryl ring”, “heteroaryl group”,
- heteroaryomatic ring and “heteroaromatic group”, are used interchangeably herein.
- Heteroaryl when used alone or as part of a larger moiety as in “heteroaralkyl” or
- heteroarylalkoxy refers to aromatic ring groups having five to fourteen ring atoms selected from carbon and at least one (typically 1 to 4, more typically 1 or 2) heteroatoms (e.g., oxygen, nitrogen or sulfur).
- Heteroaryl includes monocyclic rings and polycyclic rings in which a monocyclic heteroaromatic ring is fused to one or more other aromatic or heteroaromatic rings.
- heteroaryl includes monocyclic, bicyclic or tricyclic ring systems.
- Examples of monocyclic 5-6 membered heteroaryl groups include furanyl (e.g., 2- furanyl, 3-furanyl), imidazolyl (e.g., A midazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl), isoxazolyl ( e.g., 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl), oxadiazolyl (e.g., 2-oxadiazolyl, 5- oxadiazolyl), oxazolyl (e.g., 2-oxazolyl, 4-oxazolyl, 5-oxazolyl), pyrazolyl (e.g., 3-pyrazolyl, 4- pyrazolyl), pyrrolyl (e.g., 1 -pyrrol yl, 2-pyrrolyl, 3-pyrrolyl), pyridyl (e.g., 2-pyridyl, 3-pyridyl,
- polycyclic aromatic heteroaryl groups examples include carbazolyl, benzimidazolyl, benzothienyl, benzofuranyl, indolyl, quinolinyl, benzotriazolyl, benzothiazolyl, benzoxazoiyl, benzimidazolyl, isoquinolinyl, indolyl, isoindolyl, acridinyl, or benzisoxazolyl.
- a "substituted heteroaryl group” is substituted at any one or more substitutable ring atom, which is a ring carbon or ring nitrogen atom bonded to a hydrogen.
- an "arylalkyl” moiety refers to an alkyl group substituted with an aryl group (e.g., phenylmethyl (i.e., benzyl)).
- a “heteroarylalkyl” moiety refers to an alkyl group substituted with a heteroaryl group.
- the present teachings also include various isomers and mixtures thereof.
- “Isomer” refers to compounds that have the same composition and molecular weight but differ in physical and/or chemical properties. The structural difference may be in constitution (geometric isomers) or in the ability to rotate the plane of polarized light (stereoisomers).
- Stereoisomers are compounds which differ only in their spatial arrangement.
- the present teachings encompass all such forms, including compounds in the form of essentially pure enantiomers, racemic mixtures and tautomers, which includes forms not depicted structurally.
- a disclosed compound is named or depicted by structure without indicating stereochemistry, it is understood that the name or structure encompasses all possible stereoisomers, tautomers, geometric isomers or a combination thereof.
- geometric isomeric purity of the named or depicted geometric isomer is at least 60%, 70%, 80%, 90%, 99% or 99.9% pure by weight.
- Geometric isomeric purity is determined by dividing the weight of the named or depicted geometric isomer in the mixture by the total weight of all of the geomeric isomers in the mixture.
- Racemic mixture means 50% of one enantiomer and 50% of is corresponding enantiomer.
- the present teachings encompass all enantiomerically-pure, enantiomerically- enriched, diastereomerically pure, diastereomerically enriched, and racemic mixtures, and diastereomeric mixtures of the compounds described herein.
- Enantiomeric and diastereomeric mixtures can be resolved into their component enantiomers or stereoisomers by well known methods, such as chiral-phase gas chromatography, chiral-phase high performance liquid chromatography, crystallizing the compound as a chiral salt complex, or crystallizing the compound in a chiral solvent.
- Enantiomers and diastereomers can also be obtained from diastereomerically- or enantiomerically-pure intermediates, reagents, and catalysts by well known asymmetric synthetic methods.
- a compound When a compound is designated by a name or structure that indicates a single enantiomer, unless indicated otherwise, the compound is at least 60%, 70%, 80%, 90%, 99% or 99.9% optically pure (also referred to as "enantiomerically pure").
- Optical purity is the weight in the mixture of the named or depicted enantiomer divided by the total weight in the mixture of both enantiomers.
- stereochemistry of a disclosed compound is named or depicted by structure, and the named or depicted structure encompasses more than one stereoisomer (e.g., as in a diastereomeric pair), it is to be understood that one of the encompassed stereoisomers or any mixture of the encompassed stereoisomers are included. It is to be further understood that the stereoisomeric purity of the named or depicted stereoisomers at least 60%, 70%, 80%, 90%, 99% or 99.9% by weight. The stereoisomeric purity in this case is determined by dividing the total weight in the mixture of the stereoisomers encompassed by the name or structure by the total weight in the mixture of all of the stereoisomers.
- Suitable pharmaceutically acceptable salts of the compounds disclosed herein include salts of inorganic acids (such as hydrochloric acid, hydrobromic, phosphoric, metaphosphoric, nitric, and sulfuric acids) and of organic acids (such as, acetic acid, benzenesulfonic, benzoic, citric,
- ethanesulfonic fumaric, gluconic, glycolic, isethionic, lactic, lactobionic, maleic, malic, methanesulfonic, succinic, p- toluenesulfonic, and tartaric acids).
- acidic groups such as carboxylic acids can form pharmaceutically acceptable salts with pharmaceutically acceptable base(s).
- Suitable pharmaceutically acceptable basic salts include ammonium salts, alkali metal salts (such as sodium and potassium salts) and alkaline earth metal salts (such as magnesium and calcium salts).
- Compounds with a quaternary ammonium group also contain a counteranion such as chloride, bromide, iodide, acetate, perchlorate and the like.
- a counteranion such as chloride, bromide, iodide, acetate, perchlorate and the like.
- Other examples of such salts include hydrochlorides, hydrobromides, sulfates, methanesulfonates, nitrates, maleates, acetates, citrates, fumarates, tartrates [e.g. (+)- tartrates, (-)- tartrates or mixtures thereof including racemic mixtures], succinates, benzoates and salts with amino acids such as glutamic acid.
- Compounds described herein can inhibit various kinases, including the TT , PLK (such as PLK4), Aurora A, Aurora B and CHK (such as CHK2). Thus, generally, compounds described herein are useful in the treatment of diseases or conditions associated with such kinases.
- the compounds described herein are TTK, PLK, Aurora A, Aurora B and/or CHK inhibitors, and are useful for treating diseases, such as cancer, associated with such kinase(s).
- the compounds described herein are TTK inhibitors and are useful for treating diseases associated with TTK, such as cancer.
- the compounds described herein are Aurora A and/or B inhibitors and are useful in inhibiting Aurora A and/or B activity for the treatment of various conditions such as cancers.
- the compounds described herein are PLK inhibitors and are useful in inhibiting PLK activity for the treatment of various conditions such as cancers.
- the PLK is PLK4, PLK2 and/or PLK1.
- the PLK is PLK1 and/or PLK4.
- the PLK is PLK4.
- the compounds described herein are CHK inhibitors and are useful in inhibiting CHK activity for the treatment of various conditions such as cancers.
- the compounds described herein inhibit the growth of a tumor.
- the compounds described herein inhibit the growth of a tumor that overexpresses at least one of TTK, PLK, Aurora A, Aurora B, and CHK.
- the compounds described herein inhibit the growth of a tumor that overexpresses TTK.
- Cancers that can be treated (including reduction in the likelihood of recurrence) by the methods of the present teachings include lung cancer, breast cancer, colon cancer, brain cancer, neuroblastoma, prostate cancer, melanoma, glioblastoma multiform, ovarian cancer, lymphoma, leukemia, melanoma, sarcoma, paraneoplasia, osteosarcoma, germinoma, glioma and mesothelioma.
- the cancer is selected from leukemia, acute myeloid leukemia, chronic myelogenous leukemia, breast cancer, brain cancer, colon cancer, colorectal cancer, head and neck cancer, hepatocellular carcinoma, lung adenocarcinoma, metastatic melanoma, pancreatic cancer, prostate cancer, ovanrian cancer and renal cancer.
- the cancer is lung cancer, colon cancer, brain cancer, neuroblastoma, prostate cancer, melanoma, glioblastoma mutiform or ovarian cancer.
- the cancer is lung cancer, breast cancer, colon cancer, brain cancer, neuroblastoma, prostate cancer, melanoma, glioblastoma multiform or ovarian cancer.
- the cancer is breast cancer, colon cancer and lung cancer. In yet another embodiment, the cancer is a breast cancer. In yet another embodiment, the cancer is a basal sub-type breast cancer or a luminal B sub-type breast cancer. In yet another embodiment, the cancer is a basal sub-type breast cancer that overexpresses TTK. In yet another embodiment, the basal sub-type breast cancer is ER
- the cancer is a soft tissue cancer.
- Soft tissue cancer is an art-recognized term that encompasses tumors derived from any soft tissue of the body. Such soft tissue connects, supports, or surrounds various structures and organs of the body, including, but not limited to, smooth muscle, skeletal muscle, tendons, fibrous tissues, fatty tissue, blood and lymph vessels, perivascular tissue, nerves, mesenchymal cells and synovial tissues.
- soft tissue cancers can be of fat tissue, muscle tissue, nerve tissue, joint tissue, blood vessels, lymph vessels, and fibrous tissues.
- Soft tissue cancers can be benign or malignant. Generally, malignant soft tissue cancers are referred to as sarcomas, or soft tissue sarcomas. There are many types of soft tissue
- tumors including lipoma, lipoblastoma, hibernoma, liposarcoma, leiomyoma, leiomyosarcoma, rhabdomyoma, rhabdomyosarcoma, neurofibroma, schwannoma (neurilemoma), neuroma, malignant schwannoma, neurofibrosarcoma, neurogenic sarcoma, nodular tenosynovitis, synovial sarcoma, hemangioma, glomus tumor, hemangiopericytoma, hemangioendothelioma, angiosarcoma, Kaposi sarcoma, lymphangioma, fibroma, elastofibroma, superficial fibromatosis, fibrous histiocytoma, fibrosarcoma, fibromatosis, dermatofibrosarcoma protuberans (DFSP), malignant fibrous
- the soft tissue cancer is a sarcoma selected from the group consisting of a fibrosarcoma, a gastrointestinal sarcoma, a
- a dedifferentiated liposarcoma a dedifferentiated liposarcoma, a pleomorphic liposarcoma, a malignant fibrous histiocytoma, a round cell sarcoma, and a synovial sarcoma.
- the present teachings provide methods of inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject who is undergoing an anti-cancer therapy.
- the method comprises the steps of:
- the method optionally further comprises the step of continuing the anti-cancer therapy until the cancer goes into remission and then the step b) of administering an effective amount of a TTK inhitior (e.g., a compound represented by Structural Formula (I)).
- a TTK inhibitor e.g., a compound represented by Structural Formula (I)
- the method optionally further comprises the step of continuing the anti-cancer therapy until the cancer goes into remission and then the step b) of administering an effective amount of a TTK inhitior (e.g., a compound represented by Structural Formula (I)).
- the present teachings provide methods of inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhbitior (e.g, a compound represented by Structural Formula (I)).
- a TTK inhbitior e.g, a compound represented by Structural Formula (I)
- the subject has already been treated with an anti-cancer therapy.
- the subject has already been treated with an anti-cancer therapy and the subject is in remission.
- the present teachings provide methods of treating a subject with a cancer comprising administering to the subject an effective amount of a compound represented by Structural Formula (I) in combination with an effective anti-cancer therapy.
- the cancer is a metastatic cancer.
- a "metastatic cancer” is a cancer that has spread from its primary site to other parts of the body.
- the present teachings are directed to a method of treating a subject with a drug-resistant cancer.
- a "drug-resistant cancer” is a cancer that is not responsive to one, two, three, four, five or more drugs that are typically used for the treatment of the cancer.
- the drug-resistant cancer is mediated by the growth of tumor-initiating cells.
- tumor-initiating cells refers to preventing or decreasing the rate of the proliferation and/or survival of the tumor-initiating cells.
- the term "reducing the likelihood of recurrence of a cancer” means partially or totally inhibiting, preventing or delaying the return of a cancer at or near a primary site and/or at a secondary site after a period of remission. It also means that the cancer is less likely to return with treatment described herein than in its absense.
- the term “remission” refers to a state of cancer, wherein the clinical symptoms or indicators associated with a cancer have disappeared or cannot be detected, typically after the subject has been successfully treated with an anti-cancer therapy.
- treating a subject with a cancer includes achieving, partially or substantially, one or more of the following: arresting the growth, reducing the extent of the cancer (e.g., reducing size of a tumor), inhibiting the growth rate of the cancer, ameliorating or improving a clinical symptom or indicator associated with the cancer (such as tissue or serum components) or increasing longevity of the subject; and reducing the likelihood of recurrence of the cancer.
- Suitable methods known in the art can be used for assessing a subject to determine whether the cancer is in remission.
- the size of the tumor and/or tumor markers can be monitored to determine the state of the cancer.
- Size of the tumor can be monitored with imaging devices, such as X-ray, MRI, CAT scans, ultrasound, mammography, PET and the like or via biopsy.
- TTK inhibitors for use in inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject can include any of the compounds described herein, including compounds of formulae (I)-(VIII).
- TTK inhibitors for use in inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject does not comprise administering the compound represented by Structural Formula (I) for the treatment of cancer.
- the TTK inhibitors for use in inhibiting the growth of tumor- initiating cells or reducing the likelihood of recurrence of a cancer in a subject is described in WO2009/024824, the entire teachings of which is incorporated herein by reference.
- the TTK inhibitor is presented by the followin structural formula:
- R 1 is selected from Ci ⁇ alkyl, cyclopropyl, cyclopropylmethyl and cyclobutyl; wherein said cyclopropyl may be optionally substituted by methyl; and wherein R 1 may be optionally substituted by one or more R 5 ; m is 0 or 1 ;
- R 2 is selected from C A aUcyl, C2-6alkenyl, C2-6alknyl, C3. ⁇ 3 ⁇ 4 cycloalkyl, cyclopentenyl, cyclohexenyl, oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, oxepanyl, azetidinyl, pyrrolidinyl, piperidinyl and azepanyl; wherein R 2 may be optionally substituted on carbon by one or more R 6 ; and wherein if R 2 contains a ring -NH- moiety, that nitrogen may be optionally substituted by R 7' ;
- R 3 is independently selected from fluoro, chloro, bromo, cyano, methoxy, ethoxy, trifluoromethoxy, methyl, ethyl, trifluoromethyl, ethenyl, ethynyl, cyclopropyl, methylthio, ethylthio, N-methylamino, NN-dimethylamino, amino and methylsulfonyloxy;
- n' is an integer selected from 0 to 3; wherein the values of R 3 may be the same or different;
- R 4 ' is -L-R 8' or R 9' ;
- L is selected from ethynylene, ethenylene, cyclopropyl and wherein X is a direct bond, -0-, -S-, -NH-, -OS(0) 2 -, -N(CH 3 )- or -N(CH 2 R 10' )-; and wherein L may be optionally substituted on carbon by one or more fluoro; R 5 is cyano or fluoro;
- R 6 ' is selected from Ci_ 3 alkyl, Ci_ alkoxy, N-( Ci_ 3 alkyl)amino, N,N-( Ci- 3 alkyl) 2 amino, hydroxy, amino, fluoro and cyano;
- R 7 is selected from Ci. 3 alkyl, cyclopropyl, Cj. 3 alkanoyl and Ci -3 alkylsulfonyl;
- R 8 and R 10 are each independently selected from chloro, bromo, iodo, cyano, nitro, mercapto, sulfo, hydroxy, carboxy, amino, carbamoyl, sulfamoyl, C 2 -6alkyl, C 2 _6alkenyl, C 2 -6alkynyl, C]. 6 alkoxy, Ci. 6 alkylsulfonyloxy, N-( Ci. 6 alkyl)sulfamoyloxy, N,N- (Ci.
- R 9 is selected from carboxy, carbamoyl, sulfamoyl, C 3 _6alkyl, C 3 _ 6 alkenyl, C 3 _6alkynyl, C 3 _6alkoxy, C 3 _ 6 alkylsulfinyl, C 3 ⁇ alkylsulfanyl, C 2 - 6 alkylsulfonyloxy , N- (Ci. 6 alkyl)sulfamoyloxy , N,N-(Ci.
- R 22 and R 35 are independently selected from halo, cyano, nitro, mercapto, sulfo, hydroxy, carboxy, amino, carbamoyl, sulfamoyl, C h alky 1, C2-6alkenyl, C2-6alkynyl, Ci_ 6 alkoxy, N-(C],6alkyl)sulfamoyloxy, N,N-(Ci. 6 alkyl) 2 Sulfamoyloxy, Ci. 6 alkoxycarbonyl, d-ealkanoyl, Ci.
- R" and R are independently selected from Ci ⁇ alkyl, C2-6alkenyl, C2-6alkynyl,
- Ci_ 6 alkanoyl carbamoyl, N-(Ci. 6 alkyl)carbamoyl, N,N-(
- R 20 and R 21 are each independently selected from a direct bond, -0-, -N(R 54 )-, -C(O)-,
- R 33 and R 34 ' are each independently selected from a direct bond, -0-, -N(R 63 )-, -C(O)-, -N(R 64' )C(0)-, -C(0)N(R 65' )-, -S0 2 N(R 66' )-, -N(R 6T )-C(0)-N(R 68' )-, -OS(0) 2 -, -S(0) 2 0-, -N(R 69' )S(0) 2 N(R 70' )-, -N(R 71' )S0 2 - and -S(0) a - wherein a is 0 to 2;
- R 46 and R 7 ⁇ are each independently selected from a direct bond, -0-, -N(R 72 )-, -C(O)-, - N(R 73' )C(0)-, -C(0)N(R 74' )-, -S0 2 N(R 75' )-, -N(R 76' )-C(0)-N(R 77' )-, -OS(0) 2 -, -S(0) 2 0-, - N(R 78' )S(0) 2 N(R 79' )-, -N(R 80' )SO 2 - and -S(0) a - wherein a is O to 2;
- R so and R 51' are each independently selected from a direct bond, -C(O)-, -N(R 8r )C(0)-, -N(R 82' )S0 2 -, -O-C(O)- and -S(0) a - wherein a is 1 or 2;
- R 48 ' and R 52 are each independently selected from fiuoro, chloro, cyano, nitro, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, sulfo, carbamoyl, mercapto, sulfamoyl, methyl, ethyl, ethenyl, methoxy, ethoxy, formyl, acetyl, acetoxy, N-methylamino, N-ethylamino N,N-dimethylamino, ⁇ , ⁇ -diethylamino, N-ethyl-N-methylamino, N-formylamino, N- acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, ⁇ , ⁇ -dimethylcarbamoyl, N,N- diethylcarbamoyl, N-ethyl-N-methylcarbamoyl,
- methoxycarbonyl ethoxycarbonyl, N-methylsulfamoyl, N-ethylsulfamoyl, N,N- dimethylsulfamoyl, N,N -diethylsulfamoyl and N-ethyl-N-methylsulfamoyl;
- R 49 ' and R 53 are each independently selected from Ci_6alkyl, C 3 . 6 cycloalkyl,
- R 70 ,and R 71 are each independently hydrogen or a group selected from and cyclopropyl wherein said group may be optionally substituted on carbon by one or more R 35 ;
- the TTK inhitor described in WO2009/156315 can be used for inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject.
- the entire teachings of WO2009/156315 is incorporated by reference.
- the TTK inhibitor of WO2009/156315 is represented by the following structural formula:
- Rl is an ortho-substituted-aryl group or a heterocyclyl or C3-C7 cycloalkyl group
- R2 is hydrogen atom or a straight or branched C1-C alkyl, C 2 -Ce alkenyl,
- R3 is aryl, heterocyclyl or C3-C7 cycloalkyl group
- R4 is hydrogen atom, hydroxyl or C1 -C6 alkyl group, which group may be optionally cyclized together with one of the atom of the group which R3 may represent so as to form a fused C4-C7 cyclic group;
- R5 and R6 are each independently hydrogen atom, Ci-Ce alkyl, or are optionally cyclized together with the carbon atom to which they are bonded so as to form a C3-C7
- cycloalkyl group wherein the groups ortho-substituted-aryl, aryl, heterocyclyl, C3-C7 cycloalkyl, C4-C7 cycloalkyl, Ci-C alkyl, C 2 -C 6 alkenyl and C 2 -C 6 alkynyl may be optionally (further) substituted; with the proviso that that the following compounds are excluded:
- the TTK inhibitor that can be used for inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject is a pyridine derivative described WO2010/007756, the entire teaching of which is incorported herein by reference.
- the anti-cancer therapy is selected from the group consisting of surgery, radiation therapy, immunotherapy, endocrine therapy, gene therapy and administration of an anti-cancer agent.
- the anti-cancer therapy is radiation therapy.
- the anti-cancer therapy is immunotherapy.
- the anti-cancer therapy is administration of an anti-cancer agent.
- the anti-cancer therapy is surgery.
- Radiation therapy is the use of radiation to kill, destroy or treat the cancers.
- Exemplary radiation therapy includes, but is not limited to, gamma-radiation, neutron beam radiotherapy, electron beam radiotherapy, proton therapy, brachytherapy, and radioiosotope thereapy (i.e., systemic radioactive isotopes therapy),
- An endocrine therapy is a treatment that adds, blocks or removes hormones.
- chemotherapeutic agents that can block the production or activity of estrogen have been used for treating breat cancer.
- hormonal stimulation of the immune system has been used to treat specific cancers, such as renal cell carcinoma and melanoma.
- the endocrine therapy comprises administration of natural hormones, synthetic hormones or other synthetic molecules that may block or increase the production of the body's natural hormones.
- the endocrine therapy includes removal of a gland that makes a certain hormone.
- a gene therapy is the insertion of genes into a subject's cell and biological tissues to treat diseases, such as cancer.
- exemplary gene therapy includes, but is not limited to, a germ line gene therapy and a somatic gene therapy.
- Immunotherapy also called biological response modifier therapy, biologic therapy, biotherapy, immune therapy, or biological therapy
- Immunotherapy can help the immune system recognize cancer cells, or enhance a response against cancer cells.
- Immunotherapies include active and passive immunotherapies. Active immunotherapies stimulate the body's own immune system while passive immunotherapies generally use immune system components created outside of the body.
- active immunotherapies include, but are not limited to vaccines including cancer vaccines, tumor cell vaccines (autologous or allogeneic), dendritic cell vaccines, antigen vaccines, anti-idiotype vaccines, DNA vaccines, viral vaccines, or Tumor-Infiltrating
- TIL Lymphocyte
- IL-2 Interleukin-2
- LAK Lymphokine- Activated Killer
- Examples of passive immunotherapies include but are not limited to monoclonal antibodies and targeted therapies containing toxins.
- Monoclonal antibodies include naked antibodies and conjugated monoclonal antibodies (also called tagged, labeled, or loaded antibodies). Naked monoclonal antibodies do not have a drug or radioactive material attached whereas conjugated monoclonal antibodies are joined to, for example, a chemotherapy drug (chemolabeled), a radioactive particle (radiolabeled), or a toxin (immunotoxin).
- Examples of these naked monoclonal antibody drugs include, but are not limited to Rituximab (Rituxan), an antibody against the CD20 antigen used to treat, for example, B cell non-Hodgkin lymphoma; Trastuzumab (Herceptin), an antibody against the HER2 protein used to treat, for example, advanced breast cancer; Alemtuzumab (Campath), an antibody against the CD52 antigen used to treat, for example, B cell chronic lymphocytic leukemia (B-CLL); Cetuximab (Erbitux), an antibody against the EGFR protein used, for example, in combination with irinotecan to treat, for example, advanced colorectal cancer and head and neck cancers; and Bevacizumab (Avastin) which is an antiangiogenesis therapy that works against the VEGF protein and is used, for example, in combination with chemotherapy to treat, for example, metastatic colorectal cancer.
- Rituximab an antibody against the CD20 antigen used to treat
- conjugated monoclonal antibodies include, but are not limited to Radiolabeled antibody Ibritumomab tiuxetan (Zevalin) which delivers radioactivity directly to cancerous B lymphocytes and is used to treat, for example, B cell non-Hodgkin lymphoma; radiolabeled antibody Tositumomab (Bexxar) which is used to treat, for example, certain types of non- Hodgkin lymphoma; and immunotoxin Gemtuzumab ozogamicin (Mylotarg) which contains calicheamicin and is used to treat, for example, acute myelogenous leukemia (AML).
- Zevalin Radiolabeled antibody Ibritumomab tiuxetan
- Bexxar radiolabeled antibody Tositumomab
- Mylotarg immunotoxin Gemtuzumab ozogamicin
- BL22 is a conjugated monoclonal antibody for treating, for example, hairy cell leukemia, immunotoxins for treating, for example, leukemias, lymphomas, and brain tumors, and radiolabeled antibodies such as OncoScint for example, for colorectal and ovarian cancers and ProstaScint for example, for prostate cancers.
- HERCEPTIN® Trastuzumab
- Genentech, CA which is a humanized anti-HER2 monoclonal antibody for the treatment of patients with metastatic breast cancer
- REOPRO® abciximab
- Ceentocor which is an anti-glycoprotein Ilb/IIIa receptor on the platelets for the prevention of clot formation
- ZENAPAX® (daclizumab) (Roche Pharmaceuticals, Switzerland) which is an immunosuppressive, humanized anti-CD25 monoclonal antibody for the prevention of acute renal allograft rejection
- PANOREXTM which is a murine anti-17-IA cell surface antigen IgG2a antibody (Glaxo Wellcome/Centocor)
- BEC2 which is a murine anti-idiotype (GD3 epitope) IgG antibody (ImClone System)
- IMC-C225 which is a chimeric anti-EGFR IgG antibody
- -49-4820V.1 is a primatized anti-CD4 antibody (IDEC);
- IDEC-152 is a primatized anti-CD23 antibody (IDEC/Seikagaku);
- SMART anti-CD3 is a humanized anti-CD3 IgG (Protein Design Lab);
- 5G1.1 is a humanized anti-complement factor 5 (C5) antibody (Alexion Pharm);
- D2E7 is a humanized anti-TNF- ⁇ antibody (CAT/BASF);
- CDP870 is a humanized anti-TNF-a Fab fragment (Celltech);
- IDEC- 151 is a primatized anti-CD4 IgGl antibody (IDEC)
- MDX-CD4 is a human anti-CD4 IgG antibody
- Immunotherapies that can be used in the present teachings include adjuvant
- cytokines such as granulocyte-macrophage colony- stimulating factor (GM-CSF), granulocyte-colony stimulating factor (G-CSF), macrophage inflammatory protein (MIP)-l -alpha, interleukins (including IL-1, IL-2, IL-4, IL-6, IL-7, IL-12, IL-15, IL-18, IL-21, and IL-27), tumor necrosis factors (including TNF-alpha), and interferons (including IFN-alpha, IFN-beta, and IFN-gamma); aluminum hydroxide (alum); Bacille Calmette-Guerin (BCG); Keyhole limpet hemocyanin (KLH); Incomplete Freund's adjuvant (IFA); QS-21 ; DETOX; Levamisole; and Dinitrophenyl (DNP), and combinations thereof, such as, for example, combinations of, interleukins, for example, IL-2 with other cytokines, such as
- the anti-cancer therapy described herein includes administration of an anticancer agent.
- an “anti-cancer agent” is a compound, which when administered in an effective amount to a subject with cancer, can achieve, partially or substantially, one or more of the following: arresting the growth, reducing the extent of a cancer (e.g., reducing size of a tumor), inhibiting the growth rate of a cancer, and ameliorating or improving a clinical symptom or indicator
- -50-4820V.1 associated with a cancer (such as tissue or serum components) or increasing longevity of the subject.
- the anti-cancer agent suitable for use in the methods described herein include any anticancer agents that have been approved for the treatment of cancer.
- the anti- cancer agent includes, but is not limited to, a targeted antibody, an angiogenisis inhibitor, an alkylating agent, an antimetabolite, a vinca alkaloid, a taxane, a podophyllotoxin, a topoisomerase inhibitor, a hormonal antineoplastic agent and other antineoplastic agents.
- alkylating agents useful in the methods of the present teachings include but are not limited to, nitrogen mustards (e.g. , mechloroethamine, cyclophosphamide, chlorambucil, melphalan, etc.), ethylenimine and methylmelamines (e.g., hexamethlymelamine, thiotepa), alkyl sulfonates (e.g., busulfan), nitrosoureas (e.g., carmustine, lomusitne, semustine, streptozocin, etc.), or triazenes (decarbazine, etc.).
- nitrogen mustards e.g. , mechloroethamine, cyclophosphamide, chlorambucil, melphalan, etc.
- ethylenimine and methylmelamines e.g., hexamethlymelamine, thiotepa
- antimetabolites useful in the methods of the present teachings include but are not limited to folic acid analog (e.g., methotrexate), or pyrimidine analogs (e.g., fluorouracil, floxouridine, Cytarabine), purine analogs (e.g., mercaptopurine, thioguanine, pentostatin).
- folic acid analog e.g., methotrexate
- pyrimidine analogs e.g., fluorouracil, floxouridine, Cytarabine
- purine analogs e.g., mercaptopurine, thioguanine, pentostatin
- plant alkaloids and terpenoids or derivatives thereof include, but are not limited to, vinca alkaloids (e.g., vincristine, vinblastine, vinorelbine, vindesine), podophylloto in, and taxanes (e.g., paclitaxel, docetaxel).
- topoisomerase inhibitor includes, but is not limited to, irinotecan, topotecan, amsacrine, etoposide, etoposide phosphate and teniposide.
- antineoplastic agents include, but are not limited to, actinomycin, anthracyclines (e.g., doxorubicin, daunorubicin, valrubicin, idarubicin, epirubicin), bleomycin, plicamycin and mitomycin.
- the anti-cancer agents that can be used in the present teachings include Adriamycin, Dactinomycin, Bleomycin, Vinblastine, Cisplatin, acivicin; aclarubicin; acodazole hydrochloride; acronine; adozelesin; aldesleukin; altretamine; ambomycin;
- ametantrone acetate aminoglutethimide; amsacrine; anastrozole; anthramycin; asparaginase; asperlin; azacitidine; azetepa; azotomycin; batimastat; benzodepa; bicalutamide; bisantrene hydrochloride; bisnafide dimesylate; bizelesin; bleomycin sulfate; brequinar sodium;
- bropirimine busulfan; cactinomycin; calusterone; caracemide; carbetimer; carboplatin;
- carmustine carubicin hydrochloride
- carzelesin cedefingol
- chlorambucil cirolemycin
- cladribine crisnatol mesylate; cyclophosphamide; cytarabine; dacarbazine; daunorubicin hydrochloride; decitabine; dexormaplatin; dezaguanine; dezaguanine mesylate; diaziquone; doxorubicin; doxorubicin hydrochloride; droloxifene; droloxifene citrate; dromostanolone propionate; duazomycin; edatrexate; eflornithine hydrochloride; elsamitrucin; enloplatin;
- estramustine estramustine phosphate sodium; etanidazole; etoposide; etoposide phosphate; etoprine; fadrozole hydrochloride; cambine; fenretinide; floxuridine; fludarabine phosphate; fluorouracil; flurocitabine; fosquidone; fostriecin sodium; gemcitabine; gemcitabine
- hydrochloride hydroxyurea; idarubicin hydrochloride; ifosfamide; ilmofosine; interleukin II (including recombinant interleukin II, or rIL2), interferon alfa-2a; interferon alfa-2b; interferon alfa-nl ; interferon alfa-n3; interferon beta-I a; interferon gamma-I b; iproplatin; irinotecan hydrochloride; lanreotide acetate; letrozole; leuprolide acetate; liarozole hydrochloride;
- lometrexol sodium lomustine; losoxantrone hydrochloride; masoprocol; maytansine;
- mechlorethamine hydrochloride megestrol acetate; melengestrol acetate; melphalan; menogaril; mercaptopurine; methotrexate; methotrexate sodium; metoprine; meturedepa; mitindomide; mitocarcin; mitocromin; mitogillin; mitomalcin; mitomycin; mitosper; mitotane; mitoxantrone hydrochloride; mycophenolic acid; nocodazole; nogalamycin; ormaplatin; oxisuran;
- pegaspargase peliomycin; pentamustine; peplomycin sulfate; perfosfamide; pipobroman;
- piposulfan piroxantrone hydrochloride
- plicamycin plicamycin
- plomestane porfimer sodium
- porfiromycin prednimustine; procarbazine hydrochloride; puromycin; puromycin hydrochloride; pyrazofurin; riboprine; rogletimide; safingol; safingol hydrochloride; semustine; pumprazene; sparfosate sodium; sparsomycin; spirogermanium hydrochloride; spiromustine; spiroplatin; streptonigrin; streptozocin; sulofenur; talisomycin; tecogalan sodium; tegafur; teloxantrone hydrochloride; temoporfin; teniposide; teroxirone; testolactone; thiamiprine; thioguanine;
- thiotepa tiazofurin; tirapazamine; toremifene citrate; trestolone acetate; triciribine phosphate; trimetrexate; trimetrexate glucuronate; triptorelin; tubulozole hydrochloride; uracil mustard; uredepa; vapreotide; verteporfin; vinblastine sulfate; vincristine sulfate; vindesine; vindesine sulfate; vinepidine sulfate; vinglycinate sulfate; vinleurosine sulfate; vinorelbine tartrate;
- vinrosidine sulfate vinzolidine sulfate; vorozole; zeniplatin; zinostatin; zorubicin hydrochloride.
- anti-cancer agents/drugs that can be used in the present teachings include, but are not limited to: 20-epi-l,25 dihydroxy vitamin D3; 5-ethynyl uracil; abiraterone; aclarubicin; acylfulvene; adecypenol; adozelesin; aldesleukin; ALL-TK antagonists; altretamine;
- ambamustine amidox; amifostine; aminolevulinic acid; amrubicin; amsacrine; anagrelide;
- anastrozole andrographolide; angiogenesis inhibitors; antagonist D; antagonist G; antarelix; anti-dorsalizing morphogenetic protein- 1 ; antiandrogen, prostatic carcinoma; antiestrogen;
- antineoplaston antisense oligonucleotides; aphidicolin glycinate; apoptosis gene modulators; apoptosis regulators; apurinic acid; ara-CDP-DL-PTBA; arginine deaminase; asulacrine;
- azatyrosine baccatin III derivatives; balanol; batimastat; BCR/ABL antagonists; benzochlorins; benzoylstaurosporine; beta lactam derivatives; beta-alethine; betaclamycin B; betulinic acid; bFGF inhibitor; bicalutamide; bisantrene; bisaziridinylspermine; bisnafide; bistratene A;
- bizelesin breflate; bropirimine; budotitane; buthionine sulfoximine; calcipotriol; calphostin C; camptothecin derivatives; canarypox IL-2; capecitabine; carboxamide-amino-triazole;
- carboxyamidotriazole CaRest M3; CARN 700; cartilage derived inhibitor; carzelesin; casein kinase inhibitors (ICOS); castanospermine; cecropin B; cetrorelix; chlorlns; chloroquinoxaline sulfonamide; cicaprost; cis-porphyrin; cladribine; clomifene analogues; clotrimazole;
- collismycin A collismycin B; combretastatin A4; combretastatin analogue; conagenin;
- crambescidin 816 crisnatol; cryptophycin 8; cryptophycin A derivatives; curacin A;
- cyclopentanthraquinones cycloplatam; cypemycin; cytarabine ocfosfate; cytolytic factor;
- cytostatin cytostatin; dacliximab; decitabine; dehydrodidemnin B; deslorelin; dexamethasone;
- dexifosfamide dexrazoxane; dexverapamil; diaziquone; didemnin B; didox; diethylnorspermine; dihydro-5-azacytidine; 9- dioxamycin; diphenyl spiromustine; docosanol; dolasetron;
- edrecolomab eflornithine; elemene; emitefur; epirubicin; epristeride; estramustine analogue; estrogen agonists; estrogen antagonists; etanidazole; etoposide phosphate; exemestane;
- fadrozole fadrozole; quirabine; fenretinide; filgrastim; finasteride; flavopiridol; flezelastine; fluasterone; fludarabine; fluorodaunomnicin hydrochloride; forfenimex; formestane; fostriecin; fotemustine; gadolinium texaphyrin; gallium nitrate; galocitabine; ganirelix; gelatinase inhibitors;
- hypericin ibandronic acid; idarubicin; idoxifene; idramantone; ilmofosine; ilomastat;
- imidazoacridones imiquimod; immunostimulant peptides; insulin-like growth factor- 1 receptor inhibitor; interferon agonists; interferons; interleukins; iobenguane; iododoxorubicin; ipomeanol, 4-; iroplact; irsogladine; isobengazole; isohomohalicondrin B; itasetron; jasplakinolide;
- kahalalide F lamellarin-N triacetate; lanreotide; leinamycin; lenograstim; lentinan sulfate;
- leptolstatin a leptolstatin; letrozole; leukemia inhibiting factor; leukocyte alpha interferon;
- leuprolide+estrogen+progesterone leuprorelin; levamisole; liarozole; linear polyamine analogue; lipophilic disaccharide peptide; lipophilic platinum compounds; lissoclinamide 7; lobaplatin; lombricine; lometrexol; lonidamine; losoxantrone; lovastatin; loxoribine; lurtotecan; lutetium texaphyrin; lysofylline; lytic peptides; maitansine; mannostatin A; marimastat; masoprocol; maspin; matrilysin inhibitors; matrix metalloproteinase inhibitors; menogaril; merbarone;
- meterelin methioninase; metoclopramide; MIF inhibitor; mifepristone; miltefosine; mirimostim; mismatched double stranded RNA; mitoguazone; mitolactol; mitomycin analogues; mitonafide; mitotoxin fibroblast growth factor-saporin; mitoxantrone; mofarotene; molgramostim;
- monoclonal antibody human chorionic gonadotrophin; monophosphoryl lipid A+myobacterium cell wall sk; mopidamol; multiple drug resistance gene inhibitor; multiple tumor suppressor 1- based therapy; mustard anticancer agent; mycaperoxide B; mycobacterial cell wall extract; myriaporone; N-acetyldinaline; N-substituted benzamides; nafarelin; nagrestip;
- naloxone+pentazocine napavin; naphterpin; nartograstim; nedaplatin; nemorubicin; neridronic acid; neutral endopeptidase; nilutamide; nisamycin; nitric oxide modulators; nitroxide antioxidant; nitrullyn; 06-benzylguanine; octreotide; okicenone; oligonucleotides; onapristone; ondansetron; ondansetron; oracin; oral cytokine inducer; ormaplatin; osaterone; oxaliplatin; oxaunomycin; palauamine; palmitoylrhizoxin; pamidronic acid; panaxytriol; panomifene;
- parabactin pazelliptine; pegaspargase; peldesine; pentosan polysulfate sodium; pentostatin; pentrozole; perflubron; perfosfamide; perillyl alcohol; phenazinomycin; phenylacetate;
- phosphatase inhibitors picibanil; pilocarpine hydrochloride; pirarubicin; piritrexim; placetin A; placetin B; plasminogen activator inhibitor; platinum complex; platinum compounds; platinum- triamine complex; porfimer sodium; porfiromycin; prednisone; propyl bis-acridone;
- prostaglandin J2 proteasome inhibitors; protein A-based immune modulator; protein kinase C inhibitor; protein kinase C inhibitors, microalgal; protein tyrosine phosphatase inhibitors; purine nucleoside phosphorylase inhibitors; purpurins; pyrazoloacridine; pyridoxylated hemoglobin polyoxyethylene conjugate; raf antagonists; raltitrexed; ramosetron; ras farnesyl protein transferase inhibitors; ras inhibitors; ras-GAP inhibitor; retelliptine demethylated; rhenium Re 186 etidronate; rhizoxin; ribozymes; RII retinamide; rogletimide; rohitukine; romurtide;
- roquinimex rubiginone Bl ; ruboxyl; safingol; saintopin; SarCNU; sarcophytol A; sargramostim; Sdi 1 mimetics; semustine; senescence derived inhibitor 1; sense oligonucleotides; signal transduction inhibitors; signal transduction modulators; single chain antigen-binding protein; sizofiran; sobuzoxane; sodium borocaptate; sodium phenylacetate; solverol; somatomedin binding protein; sonermin; sparfosic acid; spicamycin D; spiromustine; splenopentin;
- spongistatin 1 squalamine; stem cell inhibitor; stem-cell division inhibitors; stipiamide;
- stromelysin inhibitors sulfinosine
- superactive vasoactive intestinal peptide antagonist sulfinosine
- suradista suramin; s ainsonine; synthetic glycosaminoglycans; tallimustine; tamoxifen methiodide; tauromustine; tazarotene; tecogalan sodium; tegafur; tellurapyrylium; telomerase inhibitors; temoporfin; temozolomide; teniposide; tetrachlorodecaoxide; tetrazomine;
- thaliblastine thiocoraline; thrombopoietin; thrombopoietin mimetic; thymalfasin; thymopoietin receptor agonist; thymotrinan; thyroid stimulating hormone; tin ethyl etiopurpurin; tirapazamine; titanocene bichloride; topsentin; toremifene; totipotent stem cell factor; translation inhibitors; tretinoin; triacetyluridine; triciribine; trimetrexate; triptorelin; tropisetron; turosteride; tyrosine kinase inhibitors; tyrphostins; UBC inhibitors; ubenimex; urogenital sinus-derived growth inhibitory factor; urokinase receptor antagonists; vapreotide; variolin B; vector system, erythrocyte gene therapy; velaresol; veramine; verdins
- the anti-cancer agents that can be used in methods described herein are selected from the group consisting of paclitaxel, docetaxel, 5-fluorouracil , trastuzumab, lapatinib, bevacizumab, letrozole, goserelin, tamoxifen, cetuximab, panitumumab, gemcitabine,
- capecitabine irinotecan, oxaliplatin, carboplatin, cisplatin, doxorubicin, epirubicin,
- cyclophosphamide methotrexate, vinblastine, vincristine, melphalan and a combination thereof.
- the anti-cancer agent and the compound represented by Structural Formula (I) are administered contemporaneously.
- the anti-cancer agent and the compound can be administered in the same formulation or in different formulations.
- the compound and the additional anti-cancer agent are administered separately.
- the subject in the methods described herein has not been previously treated with a TTK inhibitor (e.g., the compound represented by Structural Formula (I)).
- a TTK inhibitor e.g., the compound represented by Structural Formula (I)
- an "effective amount” means an amount when administered to the subject which results in beneficial or desired results, including clinical results, e.g., inhibits, suppresses or reduces the cancer (e.g., as determined by clinical symptoms or the amount of cancer cells) in a subject as compared to a control.
- "treating a subject with a cancer” includes achieving, partially or substantially, one or more of the following: arresting the growth, reducing the extent of a cancer (e.g., reducing size of a tumor), inhibiting the growth rate of a cancer, and ameliorating or improving a clinical symptom or indicator associated with a cancer (such as tissue or serum components) or increasing longevity of the subject.
- an effective amount of a compound taught herein varies depending upon various factors, such as the given drug or compound, the pharmaceutical formulation, the route of administration, the type of disease or disorder, the identity of the subject or host being treated, and the like, but can nevertheless be routinely determined by one skilled in the art.
- An effective amount of a compound of the present teachings may be readily determined by one of ordinary skill by routine methods known in the art.
- an effective amount of a compound taught herein ranges from about 0.1 to about 1000 mg/kg body weight, alternatively about 1 to about 500 mg/kg body weight, and in another alternative, from about 20 to about 300 mg/kg body weight. In another embodiment, an effective amount of a compound taught herein ranges from about 0.5 to about 5000 mg/m 2 , alternatively about from 5 to about 2500 mg/m 2 , and in another alternative from about 50 to about 1000 mg/m 2 . The skilled artisan will appreciate that certain factors may influence the
- -56-4820V.1 dosage required to effectively treat a subject suffering from cancer or reduce the likelihood of recurrence of a cancer include, but are not limited to, the severity of the disease or disorder, previous treatments, the general health and/or age of the subject and other diseases present.
- a "treatment" or dosing regime of a subject with an effective amount of the compound of the present teachings may consist of a single administration, or alternatively comprise a series of applications.
- the compound of the present teachings may be administered at least once a week.
- the compound may be administered to the subject from about one time per week to once daily for a given treatment.
- the length of the treatment period depends on a variety of factors, such as the severity of the disease, the age of the patient, the concentration and the activity of the compounds of the present teachings, or a combination thereof.
- the effective dosage of the compound used for the treatment may increase or decrease over the course of a particular treatment regime. Changes in dosage may result and become apparent by standard diagnostic assays known in the art. In some instances, chronic administration may be required.
- treatment is an approach for obtaining beneficial or desired results, including clinical results.
- beneficial or desired clinical results can include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e. not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- a "subject” is a mammal, preferably a human, but can also be an animal in need of veterinary treatment, e.g., companion animals (e.g., dogs, cats, and the like), farm animals (e.g., cows, sheep, pigs, horses, and the like) and laboratory animals (e.g., rats, mice, guinea pigs, and the like).
- companion animals e.g., dogs, cats, and the like
- farm animals e.g., cows, sheep, pigs, horses, and the like
- laboratory animals e.g., rats, mice, guinea pigs, and the like.
- the compounds taught herein can be administered to a patient in a variety of forms depending on the selected route of administration, as will be understood by those skilled in the art.
- the compounds of the present teachings may be administered, for example, by oral, parenteral, buccal, sublingual, nasal, rectal, patch, pump or transdermal administration and the pharmaceutical compositions formulated accordingly.
- Parenteral administration includes intravenous, intraperitoneal, subcutaneous, intramuscular, transepithelial, nasal, intrapulmonary, intrathecal, rectal and topical modes of administration. Parenteral administration can be by continuous infusion over a selected period of time.
- the compounds taught herein can be suitably formulated into pharmaceutical compositions for administration to a subject.
- the pharmaceutical compositions of the present teachings optionally include one or more pharmaceutically acceptable carriers and/or diluents therefor, such as lactose, starch, cellulose and dextrose.
- Other excipients such as flavoring agents; sweeteners; and preservatives, such as methyl, ethyl, propyl and butyl parabens, can also be included. More complete listings of suitable excipients can be found in the Handbook of Pharmaceutical Excipients (5 th Ed., Pharmaceutical Press (2005)). A person skilled in the art would know how to prepare formulations suitable for various types of administration routes.
- a compound of the present teachings may be incorporated with excipient and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like.
- solutions of a compound of the present teachings can generally be prepared in water suitably mixed with a surfactant such as
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols, DMSO and mixtures thereof with or without alcohol, and in oils. Under ordinary
- these preparations contain a preservative to prevent the growth of microorganisms.
- sterile aqueous solutions or dispersion of, and sterile powders of, a compound described herein for the extemporaneous preparation of sterile injectable solutions or dispersions are appropriate.
- Aerosol formulations typically comprise a solution or fine suspension of the active substance in a physiologically acceptable aqueous or non-aqueous solvent and are usually presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomizing device.
- the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve which is intended for disposal after use.
- the dosage form comprises an aerosol dispenser, it will contain a propellant which can be a compressed gas such as compressed air or an organic propellant such as
- the aerosol dosage forms can also take the form of a pump-atomizer.
- the compounds of the present teachings can be formulated with with a carrier such as sugar, acacia, tragacanth, or gelatin and glycerine, as tablets, lozenges or pastilles.
- a carrier such as sugar, acacia, tragacanth, or gelatin and glycerine, as tablets, lozenges or pastilles.
- the compounds described herein can be formulated in the form of suppositories containing a conventional suppository base such as cocoa butter.
- compounds described herein can be prepared according to the following reaction Scheme 1. Halogenation of an appropriately substituted indazole wherein the indazole is substituted as defined herein provides intermediate 1 that can be reacted with a suitable cross coupling partner, ArMet, in the presence of a metal catalyst (e.g. ArBpin /PdCl 2 (dppf) / Na 2 C0 3 / EtOH / PhMe / mw / 120 °C) .
- a metal catalyst e.g. ArBpin /PdCl 2 (dppf) / Na 2 C0 3 / EtOH / PhMe / mw / 120 °C
- haloindazole 2 can be converted into a 3-(trialkylstannyl)-lH-indazole that can be subjected to Stille-type cross-coupling reaction as shown in Scheme 2 (e.g. 1. e&Sn2 / Pd(PPh 3 ) 4 / PhMe 2. Arl /Pd(PPh 3 ) 4 / Cul / THF ref. WO200102369).
- RG0 2 H / coupling reagent e.g. TBTU, EDCI, DCC
- compounds described herein, containing trisubstituted indazoles can be 5 prepared as outlined in the following Scheme 4.
- 5-Nitro-lH-indazole is halogenated with Br2, protected with a suitable indazole protecting group such as tetrahydropyranyl, and subjected to Miyaura-Suzuki cross coupling conditions (e.g. ArBpin /dioxane/H 2 0/ PdChidppf)/ Na 2 C03).
- Hydrogenation of the intermediate 1 yields lH-indazol-5-amine 2 that can be modified in a reaction with electrophilic reagents (e.g.
- PrepPak Rxn CX refers to a commercial cation-exchange resin available from Waters.
- Microwave reactions were performed with a Biotage Initiator microwave reactor.
- Optical Rotations were measured at the sodium D-line (589.44nM) using an AA-55 Polarimeter from Optical Activity Ltd with a 2.5x100mm unjacketed stainless steel tube at given sample concentrations (c, units of g/lOOmL).
- dppf 1 , 1 '- B is( dipheny Iphosphino) ferrocene
- -65-4820V.1 examples a filtration and washing (3 ⁇ 40) of the precipitate provided the desired material with the required purity.
- Aqueous sodium carbonate (0.55 mL, 2 M) was added to a mixture of 3-iodo-lH- indazole-5-carbaldehyde (153 mg, 0.56 mmol), benzenesulfonamide-3-boronic acid pinacol ester (206 mg, 0.73 mmol) and Pd(PPh 3 ) 4 (39 mg, 0.034 mmol) in toluene (1.5 mL) and ethanol (1.5 mL) under an atmosphere of argon.
- the vial was sealed and heated in the microwave at 120°C for 3h. Water (25 mL) was added and the product was extracted with ether (200 mL, then 2x75
- dichloromethane yielded a bright yellow solid (90.7mg).
- the sample was combined with an earlier batch of impure solid (31 mg, estimated 90% pure) and purified by trituration with dichloromethane (3x2 mL) to give a pale yellow solid (63.9 mg, -30%).
- Methyl 3-iodo-l H-indazole-5-carboxylate was synthesized according to the method described for N-(3-iodo-lH-indazol-6-yl)-2-(thiophen-2-yl)acetamide utilizing methyl 1H- indazole-5-carboxylate (300 mg, 1.70 mmol), K 2 C0 3 (704 mg, 5.10 mmol), I 2 (864 mg, 3.40 mmol) and DMF (8 mL). Orange solid (415 mg, 81 %).
- the title compound was synthesized according to the General Method C, utilizing 5- bromo-3-iodo-lH-indazole (150.8 mg, 0.47 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (173.8 mg, 0.61 mmol), PdCl 2 (dppf) DCM (33.8 mg, 0.041 mmol), toluene (5 mL), EtOH (5 mL), and aqueous Na 2 C0 3 (0.93 mL, 2 M, 1.86 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h.
- tert-Butyl 2-(2-isopropylphenyl)acetate was synthesized according to the method described for 2- (2,6-diethylphenyl)acetic acid utilizing 2-isopropylphenylboronic acid (200 mg, 1.2 mmol), tert- butyl-bromoacetate ( 150 pL, 1.0 mmol), P(o-tol) 3 (28 mg, 0.091 mmol), Pd 2 (dba) 3 (28 mg, 0.030 mmol), K3PO4 (1.08 g, 5.08 mmol) in THF (5 mL) to give tert-butyl 2-(2- isopropylphenyl)acetate as a yellow solid.
- reaction mixture was diluted with 20 mL of Et20 and then quenched with saturated aqueous ammonium chloride (20 mL). The reaction mixture was washed with brine, dried with MgS0 4 , and then dried under reduced pressure to give tert-butyl 2-(2,6-diethylphenyl)acetate as
- tert-Butyl 2-(2-ethyl-6-methylphenyl)acetate was synthesized according to the method described for tert-butyl 2-(2,6-diethylphenyl)acetate utilizing l-ethyl-2-iodo-3-methylbenzene (200 mg, 0.813 mmol), terf-butylacetate ( 121 ⁇ ,, 0.894 mmol), Pd(dba) 2 (23 mg, 0.041 mmol), l ,3-Bis(2,6-diisopropylphenyl)-imidazolium chloride (17 mg, 0.041 mmol), and LiHMDS (1 M in hexanes, 1.87 mL, 1.8 mmol) in toluene (6 mL) to give /erf-butyl 2-(2-ethyl-6- methylphenyl)acetate as a yellow liquid (175 mg, 92%).
- Ethyl 2-(dimethylamino)-2-(2-ethylphenyl)acetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing l-bromo-2- ethylbenzene (239 mg, 1.9 mmol), NN-dimethylglycine ethyl ester (300 L, 2.1 mmol), Pd[P(t- Bu) 3 ] 2 (49 mg, 0.096 mmol), K3PO4 (937 mg, 4.42 mmol) and toluene (4 mL) to give ethyl 2- (dimethylamino)-2-(2-ethylphenyl)acetate as a yellow oil (306 mg, 68%).
- the title compound was synthesized according to the method described for 2-(dimethylamino)-2- phenylacetic acid hydrochloride utilizing ethyl 2-(dimethylamino)-2-(2-ethylphenyl)acetate (55 mg, 0.23 mmol). A yellow solid (54 mg, 95%).
- Ethyl 2-(dimethylamino)-2-o-tolylacetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing l-bromo-2- methylbenzene (300 mg, 1.8 mmol), MN-dimethylglycine ethyl ester (274 pL, 1.9 mmol), Pd[P(t-Bu) 3 ] 2 (45 mg, 0.088 mmol), 3 P0 4 (860 mg, 4.1 mmol), and toluene (4 mL) to give ethyl 2-(dimethylamino)-2-o-tolylacetate as a yellow oil (305 mg, 78%).
- tert-Butyl 2-phenyl-2-(pyrrolidin-l-yl)acetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing bromobenzene (200 ⁇ -, 1.9 mmol), ieri-butyl 2-(pyrrolidin-l-yl)acetate (390 ⁇ , 2.1 mmol), Pd[P(t-Bu) 3 ] 2 (49 mg, 0.096 mmol), 3 P0 4 (937 mg, 4.42 mmol) and toluene (5 mL) to give ethyl 2-phenyl-2- (pyrrolidin-l-yl)acetate as a brown liquid (480 mg, 96%).
- Ethyl 2-(dimethylamino)-2-(2-(trifluoromethyl)phenyl)acetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing 1 - bromo-2-(trifluoromethyl)benzene (430 mg, 1.92 mmol), NN-dimethylglycine ethyl ester (300 2.11 mmol), Pd[P(t-Bu) 3 ] 2 (49 mg, 0.096 mmol), K 3 P0 4 (937 mg, 4.42 mmol), and toluene
- tert-Butyl 2-(2,5-dimethylphenyl)acetate was synthesized according to the method described for tert-butyl 2-(3,5-dimethoxyphenyl)acetate utilizing 2,5-dimethylphenylboronic acid (200 mg, 1.3 mL), 1 ⁇ 2rf-butyl-bromoacetate (164 ⁇ , 1.1 mmol), P(o-tol) 3 (31 mg, 0.10 mmol), Pd 2 (dba) 3 (31 mg, 0.033 mmol), K 3 P0 4 (1.18 g, 5.6 mmol) in THF (6 mL) to give tert-butyl 2-
- terf-Butyl 2-(2,6-dimethylphenyl)acetate was synthesized according to the method described for /m-butyl 2-(3,5-dimethoxyphenyl)acetate utilizing 2,6-dimethylphenylboronic acid (200 mg, 1.33 mmol), ieri-butyl-bromoacetate (164 pL, 1.1 1 mmol), P(o-tol) 3 (31 mg, 0.100 mmol), Pd 2 (dba) 3 (31 mg, 0.033 mmol), K 3 P0 4 (1.18 g, 5.56 mmol) in THF (6 mL) to give the title compound as ayellow liquid.
- Ethyl 3-morpholino-2-phenylpropanoate was synthesized according to the method described for ethyl 2-phenyl-3-(pyrrolidin-l-yl)propanoate utilizing ethyl 2-phenylacrylate (220 mg, 1.25 mmol) and morpholine (435 mL, 5 mmol) to give the title compound as a yellow oil (305 mg, 93%).
- Ethyl 2-phenyl-3-(piperidin- l-yl)propanoate was synthesized according to the method described for ethyl 2-phenyl-3-(pyrrolidin- l-yl)propanoate utilizing ethyl 2-phenylacrylate (250 mg, 1.42mol) and piperidine (0.56 mL, 5.68 mmol) to give the title compoundas a yellow oil (348 mg, 94%).
- Methyl 2-cyclopentyl-2-(pyrrolidin-l-yl)acetate (0.1 g, 0.5 mmol) was taken into MeOH (10 mL) and stirred with aq NaOH ( 2 M, 7.5 mL, 15 mmol) sealed at 90 °C overnight. Then the reaction was coolded ( 0 °C) and acidified to pH 4-5 with aq HC1 (6 M) and concentrated to dryness.
- l-Phenylprop-2-yn-l-ol (0.37mL, 3.0 mmol) was added to a suspension of NaH (60% wt, 180 mg, 4.5 mmol) in THF (10 mL) under argon atmosphere at 0°C. After 5 min, Me 2 S0 4 (0.35 mL, 3.7 mmol) was added and stirring was continued for 2.5 h. The reaction was quenched with water (20 mL) and the product was extracted with Et20 (150 mL). The organic layer was washed with brine (20 mL) and dried (Na 2 S0 4 ).
- methyl-2-(2-bromophenyl)acetate 550 mg, 2.4 mmol
- 4-pyridine boronic acid 325 mg, 2.64 mmol
- DME 8 mL
- Pd(PPh 3 ) 4 140 mg, 0.12 mmol
- the vial was charged with argon and then placed in the microwave reactor, heating at 120 °C for 20 minutes. Reaction was diluted with 10 mL of EtOAc and then washed with brine. The mixture was dried with MgS0 4 and then by reduced pressure.
- the title compound was synthesized according to the method described for 2-(pyrrolidin-l-yl)-2- o-tolylacetic acid utilizing glyoxylic acid ⁇ H 2 0 (230 mg, 2.5 mmol), cis-2,6-dimethylmorpholine (288 mg, 2.5 mmol), CH 2 C1 2 (20 mL) and 2-methoxyphenylboronic acid (238 mg, 2.5 mmol) to give the title compound; white solid (380 mg, 58%).
- the title compound was synthesized according to the method described for 2-(pyrrolidin-l-yi)-2- o-tolylacetic acid utilizing glyoxylic acid ⁇ H 2 0 (460 mg, 5 mmol), 2 M dimethylamine in THF (2.5 mL, 5 mmol), CH2CI2 (25 mL) and 2-methoxyphenylboronic acid (760 mg, 5 mmol) to give the title compound; clear oil (792 mg, 76%).
- the title compound was synthesized according to the General Method C, utilizing iodo- lH-indazole (Sinova Inc., 30 mg, 0.12 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (42 mg, 0.15 mmol), K 2 C0 3 (34 mg, 0.25 mmol), DME (1 mL), EtOH (1 mL), H 2 0 (0.5 mL) and Pd(PPh 3 ) 4 (14 mg, 0.012 mmol). The degassed solution was sealed and
- the title compound was synthesized according to the General Method B, utilizing 5- phenyl-1 H-indazole (59 mg, 0.30 mmol ), I 2 (97 mg, 0.38 mmol) and KOH (51 mg, 0.91 mmol) in DMF (2 mL). An off-white solid (81 mg, 83 %).
- the title compound was synthesized according to the General Method C, utilizing 3-iodo- 5-phenyl- l H-indazole (40 mg, 0.12 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (53 mg, 0.19 mmol), Cs 2 C0 3 (122 mg, 0.37 mmol), DMF (1.6 mL), H 2 0 (0.4 mL) and Pd(PPh 3 ) 4 (7 mg, 0.006 mmol).
- the degassed solution was sealed and heated in a microwave reactor at 120 °C for 1.5 h.
- the title compound was synthesized according to the General Method B, utilizing N-(3- iodo-lH-indazol-5-yl)acetamide (62 mg, 0.20 mmol), 3-(4,4,5,5-tetramefhyl-l,3,2-dioxaborolan- 2-yl)benzenesulfonamide (73 mg, 0.26 mmol), KF (23 mg, 0.40 mmol), DMF (1.5mL), H 2 0 (0.37 mL) and Pd(PPh 3 )4 (12 mg, 0.010 mmol).
- the degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h.
- the title compound was synthesized according to the General Method B, utilizing N N- (lH-indazol-5-yl)thiophene-2-carboxamide (0.75 g, 3.1 mmol ), (1.6 g, 6.2 mmol) and K2CO3 (1.3 g, 9.3 mmol) in DMF (11 mL). An off-white solid (0.31 g, 27 %).
- the title compound was synthesized according to the General Method C, utilizing N-(3- iodo-lH-indazol-5-yl)thiophene-2-carboxamide (63 mg, 0.17 mmol), 3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)benzenesulfonamide (58 mg, 0.20 mmol), CS2CO3 (167 mg, 0.51 mmol), DMF (1.6 mL), H 2 0 (0.4 mL) and Pd(PPh 3 ) 4 (10 mg, 0.008 mmol).
- the degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h.
- the title compound was synthesized according to the General Method C, utilizing N-(3- iodo-lH-indazol-5-yl)thiophene-2-carboxamide (74 mg, 0.20 mmol), 3-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)benzenesulfonamide (68 mg, 0.24 mmol), Cs2C03 (0.20 g, 0.60 mmol), DMF (1.6 mL), H 2 0 (0.4 mL) and Pd(PPh 3 ) 4 (12 mg, 0.010 mmol).
- the degassed solution was sealed and heated in a microwave reactor at 120 °C for 1 h.
- the title compound was synthesized according to the General Method B, utilizing N-(1H- indazol-5-yl)-2-(thiophen-2-yl)acetamide (0.51 g, 2.0 mmol ), (1.0 g, 3.9 mmol) and K2CO3 (0.83 g, 6.0 mmol) in DMF (7 mL). A gray-yellow solid (0.32 g, 42 %).
- the title compound was synthesized according to the General Method C, utilizing N-(5- iodo-lH-indazol-5-yl)-2-(thiophen-2-yl)acetamide (100 mg, 0.26 mmol), 3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)benzenesulfonamide (96 mg, 0.34 mmol), KF (45 mg, 0.78 mmol), DMF (4 mL), H 2 0 (1 mL) and Pd(PPh 3 ) 4 (15 mg, 0.013 mmol).
- the degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h.
- the title compound was synthesized according to the General Method A utilizing 1H- indazol-5-amine (0.15 g, 1.1 mmol), nicotinic acid (0.14 g, 1.1 mmol), N-ethyl-N- isopropylpropan-2-amine (0.29 mL, 1.7 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'- tetramethyluronium tetrafluoroborate (0.38 g, 1.2 mmol) in DMF (4 mL).
- the title compound was synthesized according to General Method C utilizing N-(3-iodo-lH- indazol-5-yl)nicotinamide (46 mg, 0.13 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (43 mg, 0.15 mmol), KF (15 mg, 0.25 mmol), DMF (1.25 mL), H 2 0 (0.37 mL) and Pd(PPh 3 )4 (7 mg, 0.006 mmol).
- the degassed solution was sealed and heated in a microwave reactor at 120 °C for 1.5 h.
- the title compound was synthesized according to the method for 3-(5-(3-phenylureido)- lH-indazol-3-yl)benzenesulfonamide , utilizing a DMF (2 mL) solution of 3-(5-amino-lH- indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol), N-ethyl-N- isopropylpropan-2-amine (0.17 mL, 0.99 mmol) and l ,3-dichloro-2-isocyanatobenzene (49 mg, 0.26 mmol).
- the title compound was synthesized according to the General Method A utilizing 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (0.050 g, 0.12 mmol), rac-2- methoxy-2-phenylacetic acid (0.021 g, 0.12 mmol), N-ethyl-N-isopropylpropan-2-amine (0.1 mL, 0.6 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (0.042 g, 0.13 mmol) in DMF (2 mL).
- the title compound was synthesized according to the the General Method A utilizing 3-(5- amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (0.10 g, 0.26 mmol), (R)-2- methoxy-2-phenylacetic acid (0.044 g, 0.27 mmol), N-ethyl-N-isopropylpropan-2-amine (0.1 mL, 0.6 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (0.086 g, 0.27 mmol) in DMF (2 mL).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present teachings provide a compound represented by Strutural Formula (I): or a pharmaceutically acceptable salt thereof. Also described are a pharmaceutical composition and method of use thereof.
Description
KINASE INHIBITORS AND METHOD OF TREATING CANCER WITH
SAME
RELATED APPLICATIONS
The present invention is related and claims priority to U.S. Provisional Application Serial No. 61/321 ,342, filed April 6, 2010 and U.S. Provisional Application Serial No. 61/345,849, filed May 18, 2010. The entire contents of each of these applications are hereby incorporated by this reference.
BACKGROUND OF THE INVENTION
Protein kinases have been the subject of extensive study in the search for new therapeutic agents in various diseases, for example, cancer. Protein kinases are known to mediate intracellular signal transduction by effecting a phosphoryl transfer from a nucleoside triphosphate to a protein acceptor that is involved in a signaling pathway. There are a number of kinases and pathways through which extracellular and other stimuli cause a variety of cellular responses to occur inside the cell.
Human TTK protein kinase (TTK), also known as tyrosine threonine kinase, dual specificity protein kinase TTK, Monopolar Spindle 1 (Mpsl) and Phosphotyrosine-Picked Threonine Kinase (PYT), is a conserved multispecific kinase that is capable of phosphorylating serine, threonine and tyrosine residues when expressed in E. coli (Mills et al., J. Biol. Chem. 22(5): 16000-16006 (1992)). TTK mRNA is not expressed in the majority of physiologically normal tissues in human (Id). TTK mRNA is expressed in some rapidly proliferating tissues, such as testis and thymus, as well as in some tumors (for example, TTK mRNA was not expressed in renal cell carcinoma, was expressed in 50% of breast cancer samples, was expressed in testicular tumors and ovarian cancer samples) (Id). TTK is expressed in some cancer cell lines and tumors relative to normal counterparts (Id.; see also WO 02/068444 Al).
-1-4820V.1
Therefore, agents which inhibit a protein kinase, in particular TTK, have the potential to treat cancer. There is a need for additional agents which can act as protein kinase inhibitors, in particular TTK inhibitors.
In addition, cancer recurrence, drug resistance or metastasis is one of the major challenges in cancer therapies. Cancer patients who responded favorably to the initial anticancer therapy often develop drug resistance and secondary tumors that lead to the relapse of the disease. Recent research evidences suggest that the capability of a tumor to grow and propagate is dependent on a small subset of cells within the tumor. These cells are termed tumor-initiating cells (TICs) or cancer stem cells. It is thought that the TICs are responsible for drug resistance, cancer relapse and metastasis. Compounds that can inhibit the growth and survival of these tumor-initiating cells can be used to treat cancer, metastasis or prevent recurrence of cancer. Therefore, a need exists for new compounds that can inhibit the growth and survival of tumor- initating cells.
SUMMARY OF THE INVENTION
Applicants have now discovered that certain indazole compounds are potent kinase inhibitors, such as TTK protein kinase, polo-like kinases 4 (PLK4), Aurora Kinases and CHK kinase (see Example B-F). Applicants have also now discovered that these indazole compounds have potent anticancer activity against breast cancer cells, colon cancer cells, lung cancer cells, melanoma cells, prostate cancer cells, ovarian cancer cells, brain cancer cells and pancreatic cancer cells in cell culture study (see Example G). Applicants have also discovered that certain indazole TTK inhibitors can inhibit the growth of colon tumor-initiating cells in a cell culture study (see Example H). Based on these discoveries, indazole compounds, pharmaceutical compositions thereof, and methods of treating cancer with the indazole compounds (including reducing the likelihood of recurrence of a cancer with a TTK inhibitor) are disclosed herein.
The present teachings are directed, at least in part, to an indazole compound represented by the following structural formula:
-2-4820V.1

or a pharmaceutically acceptable salt thereof, wherein:
each R1 is independently selected from -H, -halogen, -CN, -NO2, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)jNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, heterocycloalkyl and alkyl, wherein the heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -NO2, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)jNReRf, C(=0)ORc, -OC(=0)ORc,
-C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc,
-NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf,
-NRd(C=S)NReRf, -C(=S)RC and -C(=0)Rc;
each R2 is independently selected from: a)-H, -halogen, -CN, -N02, -(CH2)o-20R10, -(CH2)o-2NRnR12, -(CH2)o-2S(0)iR10, -(CH2)o-2NR13S(0)iR10, -(CH2)o-2NR,3S(0)iNR14R15, -(CH2)0.2S(O)iNR,4R15, -(CH2)o-2C(=0)OR'0, -(CH2)o-20C(=0)OR10, -(CH2)0-2C(=S)OR10,
-(CH2)o-20(C=S)R10, -(CH2)o-2C(=0)NR,4R15, -(CH2)o-2NR13C(=0)R10, -(CH2)0-2C(=S)NR14R15, -(CH2)o-2NR13C(=S)R10, -(CH2)o-2NR13(C=0)OR10, -(CH2)0-2O(C=O)NR14R15,
-(CH2)o-2NR13(C=S)OR10, -(CH2)o.20(C=S)NR14R15, -(CH2)0-2NR13(C=O)NR14R15,
-(CH2)0.2NR13(C=S)NR14R15, -(CH2)o-2C(=S)R10, and -(CH2)o-2C(=0)R10; and b) alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, heterocycloalkylalkyl, heterocycloalkylalkenyl, aryl, arylalkyl, arylalkenyl, heteroaryl, heteroarylalkyl and heteroarylalkenyl, wherein each of the alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, heterocycloalkylalkyl,
heterocycloalkylalkenyl, aryl, arylalkyl, arylalkenyl, heteroaryl, heteroarylalkyl and heteroarylalkenyl groups represented by R2 is optionally substituted with 1 to 5 substituents
-3-4820V.1
independently selected from the group consisting of -halogen, -CN, -N02, -OR0, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl substituents on the groups represented by R2 are each optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, (Cp C6)alkyl, halo(C]-C6)alkyl, (Ci-C3)alkoxy, halo(Ci-C3)alkoxy, (Ci-C3)alkoxy(Ci-C6)alkyl and -NRdC(=0)Rc;
R10 is selected from -H, alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl, heterocycloalkyl (Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl,
heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl represented by R10 is optionally substituted with 1 to 5 substituents independently selected from the group consisting of halogen, =0, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf,
-NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (Q-Cs^lkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl, and wherein each of the (Ci-C6)alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl substituents for the groups represented by R10 is optionally substituted with halogen, -OH, -N02, -CN, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (Ci-C3)alkoxy(Ci- C3)alkyl, (CrC3)alkoxy or halo(CrC3)alkoxy;
R11 and R12 are each independently selected from -H, alkyl, cycloalkyl, cycloalkyl(Ci- C6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl represented by R1 1 and R12 is optionally substituted with 1 to 5 4820V.1
substituents independently selected from the group consisting of halogen, =0, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (Ci-C6)alkyl, cycloalkyl, cycloalkyl(C]-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl, and wherein each of the (Ci- C6)alkyl, cycloalkyl, cycloalkyl(C]-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(C|-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl substituents for the groups represented by R1 1 and R12 is optionally substituted with halogen, -N02, -CN, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (C,-C3)alkoxy(C, -C3)alkyl, (Ci-C3)alkoxy or halo(Cj-C3)alkoxy; or
R1 1 and R12, together with the nitrogen to which they are attached, form a 3-8 membered ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, =0, -CN, -ORc, -NRaRb, -S(0)iRc, -S(0)iNRaRb, -NRdS(0)iRc,
-C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NRaRb, -NRdC(=0)Rc,
-C(=S)NRaRb, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NRaRb, -NRd(C=S)ORc, -0(C=S)NRaRb, -NRd(C=0)NRaRb, -NRd(C=S)NRaRb, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein each of the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl substituents on the 3-8 membered ring is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (Cp
C3)alkoxy(Ci-C3)alkyl, (CrC3)alkoxy and halo(CrC3)alkoxy;
R13 is -H or an alkyl group optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy and (C]-C3)alkoxy;
R14 is -H or an alkyl group optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy and (Ci-C3)alkoxy;
R15 is selected from -H, alkyl, cycloalkyl, cycloalkyl(d-C6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(d-C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl,
heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl represented by R15 is optionally substituted with 1 to 3 substituents independently selected from
-5-4820V.1
the group consisting of halogen, =0, -C(=0)ORc, -ORc, -SRC, -C(=0)NRaRb, -C(=0)Rc, -S(0)iRc, -N02, -CN, -NRaRb, (Ci-C6)alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl,
heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl, and wherein each of the (Ci-C6)alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(C]-C3)alkyl, heteroaryl and heteroaryl(C]-C3)alkyl substituents for the groups represented by R15 is optionally substituted with halogen, -N02, -CN, (d-C3)alkyl, halo(CrC3)alkyl, (C,-C3)alkoxy(Ci-C3)alkyl, (Cr C3)alkoxy or halo(Ci-C3)alkoxy; or
R14 and R15, together with the nitrogen to which they are attached, form a 3-8 membered ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, =0, -CN, -ORc, -NRaRb, -S(0)jRc, -S(0)iNRaRb, -NRdS(0)jRc,
-C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NRaRb, -NRdC(=0)Rc,
-C(=S)NRaRb, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NRaRb, -NRd(C=S)ORc, -0(C=S)NRaRb, -NRd(C=0)NRaRb, -NRd(C=S)NRaRb, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein each of the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl substituents on the 3-8 membered ring is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, (Ci-C3)alkyl, halo(d-C3)alkyl, (Cp
C3)alkoxy(Ci-C3)alkyl, (C,-C3)alkoxy and halo(Ci-C3)alkoxy;
R16 is selected from -NH2 and alkyl, wherein the alkyl is optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl, heteroaryl(Cr C3)alkyl, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)jNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf,
-NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf,
-NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC and -C(=0)Rc, wherein the cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl or heteroaryl(Ci-C3)alkyl are optionally substituted with 1 to 3 substituents independently selected from -halogen, alkyl, -ORc and -NRaRb;
-6-4820V.1
Ra and Rb are each independently -H or (C|-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy, -NRgRh and (C,-C3)alkoxy;
R° is -H, or (Ci-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NRgRh, hydroxy and (C]-C3)alkoxy;
Rd is -H or (Ci-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NR8Rh, hydroxy and (Ci-C3)alkoxy;
Re and Rf are each independently -H or (Ci-Ce)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NRgRh, hydroxy and (Ci-C3)alkoxy;
or Re and Rf, together with the nitrogen to which they are attached, form a 3-8 membered ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NRgRh, -CN, (Ci-C6)alkyl, halo(Ci-C6)alkyl, (d-C3)alkoxy, halo(Ci- C3)alkoxy, and (Ci-C3)alkoxy(Ci-C6)alkyl;
Rg and Rh are each independently selected from the group consisting of -H, (Ci-C6)alkyl, halo(Ci-C6)alkyl, hydroxy(Ci-C6)alkyl and (C1-C3)alkoxy(C1-C6)alkyl;
i is 0, 1 or 2;
n is an integer from 1 to 4; and
m is an integer from 1 to 4;
provided that when R16 is alkyl, R2 is not -CN.
In another embodiment, the present teachings include a pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and a compound represented by Structural Formula (I) described above or a pharmaceutically acceptable salt thereof.
Another embodiment of the present teachings provides a method of treating a subject having cancer comprising administering to the subject an effective amount of a compound of Structural Formula (I) or a pharmaceutically acceptable salt thereof.
Another embodiment of the present teachings provides a method of inhibiting TTK activity in a subject in need of inhibition of TTK activity, comprising administering to the
-7-4820V.1
subject an effective amount of a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof.
Another embodiment of the present teachings includes the use of a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof in therapy. In some embodiments, the therapy is for treating a subject with cancer. Alternatively, the therapy is for inhibiting TTK activity in a subject in need of inhibition of TTK activity.
Another embodiment of the present teachings includes the use of a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for treating a subject with cancer.
Another embodiment of the present teachings includes the use of a compound represented by Structural Formulas (I) or a pharamceutically acceptable salt thereof for the manufacture of a medicament for inhibiting TTK activity in a subject in need of inhibition of TTK activity.
In some embodiments, the present teachings provide a method of inhibiting the growth of tumor-initiating cells (or cancer stem cells) in a subject who is undergoing an anti-cancer therapy. Such method includes assessing the subject to determine whether the cancer is in remission; and, if the cancer is in remission, then administering to the subject an effective amount of a TTK inhibitor.
In other embodiments, the present teachings provide a method of reducing the likelihood of recurrence of a cancer in a subject who is undergoing an anti-cancer therapy. Such methods includes assessing the subject to determine whether the cancer is in remission; and, if the cancer is in remission, then administering to the subject an effective amount of a TTK inhibitor.
In another embodiment, the present teachings are directed to a method of inhibiting the growth of tumor-initiating cells in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhibitor. In another embodiment, the present teachings are directed to a method of reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhibitor. In yet another embodiment, the present teachings are directed to a method of treating a drug-resistant cancer in a subject comprising administering to the subject an effective amount of a TTK inhibitor. 4820V.1
In another embodiment, the present teachings are directed to a method of treating a subject with a cancer comprising administering to the subject an effective amount of a compound represented by Structural Formula (I) in combination with an anti-cancer therapy.
Other embodiments of the present teachings include the use of a TTK inhibitor for 5 inhibiting the growth of tumor-iniating cells or reducing the likelihood of recurrence of a cancer in a subject who is undergoing an anti-cancer therapy. Further embodiments of the present teachings includes the use of a TTK inhibitor for inhibiting the growth of tumor-iniating cells or reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission. Still other embodiments of the present teachings include the use of a TTK inhibitor for treating a o subject with a drug-resistant cancer.
Other embodiments of the present teachings include the use of a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof in combination with an anti-cancer therapy for treating a subject with a cancer.
DETAILED DESCRIPTION OF THE INVENTION
In one embodiment, the present teachings are directed to a compound represented by Structural Formula (I) or a pharmaceutically acceptable salt thereof. Values and alternative values for the variables in Structural Formula (I) are provided in the following paragraphs:
Each R1 is independently -H, -halogen, -CN, -ORc, -NRaRb, -C(=0)ORc,
-C(=0)NReRf, -NRdC(=0)Rc, -0(C=0)NReRf, heterocycloalkyl, or alkyl, wherein the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, -C(=0)ORc, - OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, - 0(C=0)NReRf. Alternatively, R1 is -H.
Each R2 is independently -H, halogen, -CN, -N02) -NRaRb, -ORc, -C(=0)Rc, -C(=0)NR14R15, -NR13C(=0)R10, (C,-C3)alkyl, halo(d-C3)alkyl, or (C1-C3)alkoxy(Ci-C3)alkyl, (C2-C4)alkynyl, halo(C2-C4)alkynyl, (C2-C )alkenyl, or halo(C2-C4)alkenyl. Alternatively, R2 is -H.
-9-1 1 1444820V.1
R3 is cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R6.
Alternatively, R3 is selected from (C3-C8)cycloalkyl, phenyl, naphthyl,
tetrahydronaphthyl, pyridyl, thiophenyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl,
5 isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[if]imidazolyl, benzo[i/]thiazolyl,
benzo[£>]thiophenyl, quinolinyl, tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, morpholinyl, indolinyl and indenyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, o -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc,
-NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf,
-NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci- C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and
5 tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -Ν<¾, -NRaRb, (Ci-C3)alkyl, halo(Ci- C3)alkyl, (Ci-C3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl. In another alternative, R3 is selected from the group consisting of phenyl, thiophenyl, pyridinyl, pyrazolyl, cyclopentyl,
tetrahydropyranyl, indenyl, tetrahydronaphthalenyl and indolinyl, each of which is optionally0 substituted with 1 to 3 substituents independently selected from the group of substituents
described above. In another alternative, R3 is selected from the group consisting of cyclopentyl, phenyl, thiophenyl, pyridyl, piperidinyl, tetrahydropyranyl, tetrahydrofuranyl, pyrrolidinyl, and indolyl, each of which is optionally substituted with 1 to 3 substituents independently selected from the group of substituents described above. In yet another alternative, R3 is selected from5 the group consisting of phenyl, thiophenyl, pyridyl, tetrahydropyranyl and indolyl, each of which is independently selected from the group of substituents described above.
In another alternative embodiment, R3 described in the preceding paragraphs is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -OR0, (Ci-C3)alkyl, -(Ci-C3)alkylene-NRaRb, phenyl, pyrimidinyl, morpholinyl and benzyl,
-10-1 11444820V.1
wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (C,-C3)alkyl, halo(Ci-C3)alkyl, (CrC3)alkoxy and (C,-C3)alkoxy(C,-C3)alkyl; Rc is -H, (C]-C3)alkyl, pyridinyl or moφholinyl; and Ra and Rb are each independently -H or (Ci- C3)alkyl. Alternatively, R3 described in the preceding paragraphs is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -F, -CI, methyl, -CF3, ethyl, 2-propyl, benzyl, phenyl and -CH2NH2.
R4 and R5 are each independently -H, alkyl, -ORc, -NRaRb, (C C3)alkylene-NRaRb, (Cr C3)alkylene-ORc, cycloalklyl or heterocycloalkyl. In some embodiments, R4 and R5 are not both selected from -ORc and -NRaRb. In some embodiments R3 and R5 together with the carbon atom to which they are attached form a cycloalkyl or a heterocycloalkyl, wherein the cycloalkyl or heterocycloalkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (C]-C3)alkoxy, halo(Ci- C3)alkoxy, (Ci-C3)alkoxy(Ci-C3)alkyl. Alternatively, R4 and R5 are both -H. In another alternative, R5 is -H and R4 is -OH, (Ci-C3)alkyl, -(C,-C2)alkylene-OH, (C,-C3)alkoxy, -NRaRb, -(Ci-C2)alkylene-NRaRb, pyrrolidinyl, piperidinyl, moφholinyl or cyclopropyl, wherein Ra and Rb are each independently -H or (C]-C3)alkyl and the pyrrolidinyl, piperidinyl, morpholinyl or cyclopropyl represented by R4 is optionally substituted with halogen, (Ci-C3)alkyl, halo(Ci- C3)alkyl, (C,-C3)alkoxy and (C,-C3)alkoxy(Ci-C3)alkyl.
Each R6 is independently selected from the group consisting of halogen, -CN, -NO2, =0, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)jNReRf, C(=0)0Rc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)0Rc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)R°, -C(=0)Rc, (Ci-C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci-C3)alkyl, phenyl, benzyl, mo holinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (CrC3)alkyl, halo(Ci-C3)alkyl, (C C3)alkoxy and (Ci-C3)alkoxy(C,-C3)alkyl. Alternatively, each R6 is independent selected from the group consisting of halogen, -0RC, (CrC3)alkyl, -(CrC3)alkylene-NRaRb, phenyl,
-1 1-4820V.1
pyrimidinyl, morpholinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (Ci- C3)alkoxy and (C]-C3)alkoxy(Ci-C3)alkyl; Rc is -H, (Ci-C3)alkyl, pyridinyl or morpholinyl; and Ra and Rb are each independently -H or (Ci-C3)alkyl. In another alternative, each R6 is independently selected from the group consisting of halogen, -ORc, (Ci-C3)alkyl, -(Ci- C3)alkylene-NRaRb, phenyl, pyrimidinyl, mo holinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (Ci- C3)alkyl, halo(Ci-C3)alky., (Ci-C3)alkoxy and (C]-C3)alkoxy(Ci-C3)alkyl; Rc is -H,
(Ci-C3)alkyl, pyridinyl or morpholinyl; and Ra and Rb are each independently -H or (Ci- C3)alkyl. In yet another alternative, each R6 is independently selected from the group consisting of -F, -CI, methyl, -CF3, ethyl, 2-propyl, benzyl, phenyl and -CH2NH2.
Each R7 is independently selected from -H, -CN, -N02, halogen, -ORc, -NRaRb, -S(0)tRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (CrC6)alkyl, halo(C,-C6)alkyl and (Ci-C3)alkoxy(Ci-C3)alkyl. Alternatively, each R7 is -H or (d-C6)alkyl. In another alternative, R7 is -H.
R8 is -H or (Ci-C3)alkyl. Alternatively, R8 is -H.
Each R9 is independently selected from -H, -CN, -N02, halogen, -ORc, -NRaRb, -S(0)iRc, -NR S(0)iRc, -S(0),NReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReR , -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl represented by R9 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, -N02, -OH, (Ci-C3)alkyl, halo(CrC3)alkyl, (C,-C3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl, hydroxyl(C,-C3)alkyl or -NRaRb. Alternatively, each R9 is independently selected from the group consisting of -H, halogen, -CN, -N02, (Cp C3)alkyl, halo(Ci-C3)alkyl, (Ci-C3)alkoxy, (Ci-C3)alkoxy(C,-C3)alkyl, hydroxy(Ci-C3)alkyl or - (Ci-C3)alkylene-NRaRb.
-12-4820V.1
R is -NH2. Alternatively, R is alkyl, optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -ORc, -NRaRb, -C(=0)ORc, -OC(=0)ORc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc,
-0(C=0)NReRf, -NRd(C=0)NReRf and -C(=0)R°, wherein the cycloalkyl, heterocycloalkyl, aryl or heteroaryl are optionally substituted with 1 to 3 substituents independently selected from halogen, alkyl, -ORc and -NRaRb.
Each i is 0, 1 or 2. Alternatively, each i is 2.
n is an integer from 1 to 4. Alternatively, n is 1, 2 or 3. In another alternative, n is 1 or 2. In yet another alternatively, n is i .
m is an integer from 1 to 4. Alternatively, m is 1 or 2. In yet another alternative, m is 1. p is 0, 1 or 2. Alternatively, p is 0. In another alternative, p is 1.
r is an integer from 1 to 4. Alternatively, r is 1 or 2.
In some embodiments, the present teachings provide an indazole compound represented by the following structural formula:

or pharmaceutically acceptable salts thereof.
In some embodiments, the present teachings provide an indazole compound represented by the following structural formula:

■13-44820V.1
or pharmaceutically acceptable salts thereof,
wherein R16 is alkyl, optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, cycloalkyl(C]-C3)alkyl, heterocycloalkyl,
heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl, heteroaryl(Ci-C3)alkyl, -CN, -N02, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc,
-OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf,
-NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf,
-NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC and -C(=0)Rc, wherein the cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(C,-C3)alkyl, heteroaryl or heteroaryl(Ci-C3)alkyl are optionally substituted with 1 to 3 substituents independently selected from -halogen, alkyl, -ORc and -NRaRb.
In a first embodiment, the present teachings provide a compound represented by a

-14- 4820V.1

X is a bond or -CR4R5-, and for structural formula (II), when p is 0, X can additionally be
-0-;
Y is a bond, -NR14-, -CR4R5- or -NRI4-CR4R5-;
R3 is a cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R6;
R4 and R5 are each independently selected from -H, alkyl, -ORc, -NRaRb, (Ci- C3)alkylene-NRaRb, -(C,-C3)alkylene-ORc , -(CrC3)alkylene-OH, cycloalklyl and
heterocycloalkyl, provided that R4 and R5 are not both selected from -ORc and -NRaRb; or R3 and R5 together with the carbon atom to which they are attached form a cycloalkyl or a
heterocycloalkyl, wherein the cycloalkyl or heterocycloalkyl is optionally substituted with 1 to 3
-15-4820V.1
substituents independently selected from the group consisting of halogen, -CN, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (C,-C3)alkoxy, halo(Ci-C3)alkoxy, (CrC3)alkoxy(Ci-C3)alkyl;
each R6 is independently selected from the group consisting of halogen, =0, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc,
-0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReR , -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C6)alkyl, aryl, aryl(Ci-C3)alkyl, heterocycloalkyl and heteroaryl; wherein each the (Ci-C6)alkyl, aryl, aryl(Ci-C3)alkyl, heterocycloalkyl and heteroaryl represented by R6 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Ci-C6)alkyl, halo(Ci-C6)alkyl, (Ci-C3)alkoxy, halo(Cr C3)alkoxy and (Ci-C3)alkoxy(C|-C6)alkyl; and
p is 0, 1 or 2.
Values and alternative values for the remainder of the variables are as described above for Structural Formula (I).
In a second embodiment, the present teachings are directed to a compound represented by a structural formula select

- 16-4820V.1

or a pharmaceutically acceptable salt of any one of the foregoing, wherein Y is bond or -NR -. Values and alternative values for the variables are as described for Structural Formula (I) or in the first embodiment.
In one embodiment, compounds represented by structural formula (Ila)-(Va) are represented by a structural formula selected from:
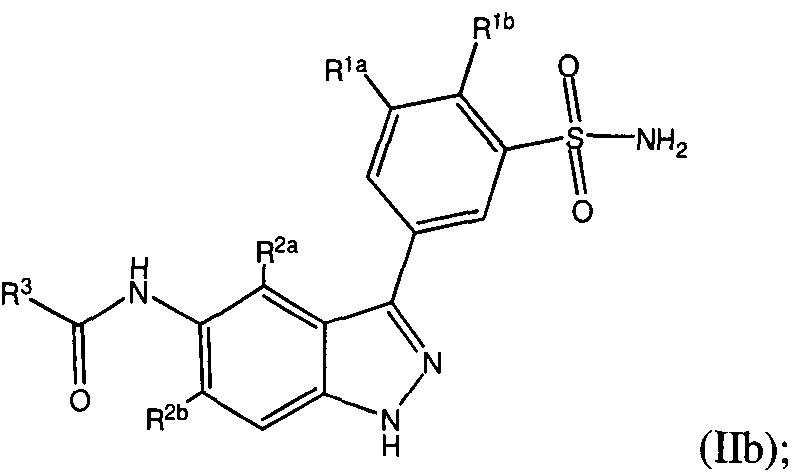
-17-4820V.1

or a pharmaceutically acceptable salt of any one of the foregoing, wherein:
Y is a bond or -NR14;
Rla and Rlb are each independently -H, -halogen, -CN, -ORc, -NRaRb, -C(=0)ORc, -C(=0)NReRf, -NRdC(=0)Rc, -0(C=0)NReRf, heterocycloalkyl, or alkyl, wherein
heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, -0(C=0)NReRf;
- 18-4820V.1
R2a and R2b each independently -H, halogen, -CN, -N02, -NRaRb, -ORc, -C(=0)Rc, (d- C3)alkyl, halo(Ci-C3)alkyl, or (C,-C3)alkoxy(Ci-C3)alkyl, (C2-C4)alkynyl, halo(C2-C4)alkynyl, (C2-C4)alkenyl, or halo(C2-C4)alkenyl; and values and alternative values for the remainder of the variables are as described above for Structural Formula (I) or for Structural Formula (Ila)-(Va).
In a third embodiment, for compounds represented by structural formulas (Ila)-(Va) and
(Ilb)-(Vb), the group represented by R3 is selected from (C3-C8)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thiophenyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[cT|imidazolyl, benzo[<i]thiazolyl,
benzo[fr]fhiophenyl, quinolinyl, tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, morpholinyl, indolinyl and indenyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)jRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc,
-NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf,
-NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (CrC3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Q- C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (Ci-C3)alkyl, halo(Cr C3)alkyl, (CrC3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl. Values and alternative values for the remainder of the variables are as described in the first or second embodiment.
In a fourth embodiment, for compounds represented by structural formulas (Ila)-(Va) and (Ilb)-(Vb), the group represented by R3 is selected from the group consisting of cyclopentyl, phenyl, thiophenyl, pyridyl, piperidinyl, benzo[c][l,2,5]oxadiazolyl, benzo[b]thiophenyl, tetrahydropyranyl, benzo[d]imidazolyl, pyrazoly, isoxazolyl, thiazolyl, quinolinyl,
tetrahydrofuranyl, pyrrolidinyl, tetrahydronaphthalenyl, indenyl, indolinyl and
tetrahydroquinolinyl, each of which is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, -N02, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)jNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, 820v.1
-C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (Ci-C3)alkyl, phenyl, benzyl, pyrimidinyl, moφholinyl, piperidinyl, pyrrolidinyl,
tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Cj-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (d-C3)alkyl, halo(CrC3)alkyl, (CrC3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl. Values and alternative values for the remainder of the variables are as described in the first or second embodiment.
In a fifth embodiment, for compounds represented by structural formulas (Ila)-(Va) and
(Ilb)-(Vb), the group represented by R3 is selected from the group consisting of phenyl, thiophenyl, pyridinyl, pyrazolyl, cyclopentyl, tetrahydropyranyl, indenyl, tetrahydronaphthalenyl and indolinyl, each of which is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (CrC3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (C]-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (C,-C3)alkyl, halo(CrC3)alkyl, (CrC3)alkoxy and (Ci-C3)alkoxy(C,-C3)alkyl. Values and alternative values for the remainder of the variables are as described in the first or second embodiment.
Alternatively, the group represented by R3 described in the second, third, fourth or fifth embodiment is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -OR0, (d-C3)alkyl, -(Ci-C3)alkylene-NRaRb, phenyl, pyrimidinyl, morpholinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (d-C3)alkyl, halo(Ci-C3)alkyl, (d-C3)alkoxy and (d- 4820V.1
C3)alkoxy(Ci-C3)alkyl; Rc is -H, (Ci-C3)alkyl, pyridinyl or morpholinyl; and Ra and Rb are each independently -H or (Ci-C3)alkyl. Alternatively, the group represented by R3 described in the third, fourth or fifth embodiment is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -F, -CI, methyl, ethyl, -CF3, 2-propyl, benzyl, phenyl and
In a sixth embodiment, compounds of the present teachings are represented by a structural formula selected from:

or a pharmaceutically acceptable salt of the foregoing, wherein
is a double bond or a single bond;
W is CH or N when is a double bond, or W is -CHR7- or -NR8- when is a single bond;
R5 is absent when is a double bond and R5 is -H or (Ci-C3)alkyl when is a single bond;
q is 1 or 2;
each R7 are independently selected from -H, -CN, -N02, halogen, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc,
-21-44820V.1
-C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (Ci-C6)alkyl, halo(Ci-C6)alkyl and (Ci-C3)alkoxy(Ci-C3)alkyl;
R8 is -H or a (C,-C3)alkyl;
each R9 is independently selected from -H, -CN, -N02, halogen, -ORc, -NRaRb, -S(0)iRc,
-NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl represented by R9 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, -NO2, -OH, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (Ci-C3)alkoxy and (C,-C3)alkoxy(CrC3)alkyl, hydroxyl(Ci-C3)alkyl or -NRaRb;
Rla and Rlb are each independently -H, -halogen, -CN, -ORc, -NRaRb, -C(=0)ORc; -C(=0)NReRf, -NRdC(=0)Rc, -0(C=0)NReRf, heterocycloalkyl, alkyl, wherein the
heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -ORc, -NRaRb, -S(0);Rc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, -0(C=0)NReRf;
R2a and R2b each independently -H, halogen, -CN, -N02, -NRaRb, -ORc, -C(=0)Rc, (C,- C3)alkyl, halo(C,-C3)alkyl, or (Ci-C3)alkoxy(Ci-C3)alkyl, (C2-C4)alkynyl, halo(C2-C4)alkynyl, (C2-C4)alkenyl, or halo(C2-C4)alkenyl; and
r is an integer from 1 to 4. Values and alternative values for the remainder of the variables are as described for Structural Formula (I) or in the second embodiment.
In one embodiment, for compounds represented by Structural Formula (lie) or (IIIc): R7 is -H or (C,-C6)alkyl;
each R9 is independently selected from the group consisting of -H, halogen, -CN, -N02, (Ci-C3)alkyl, halo(C,-C3)alkyl, (d-C3)alkoxy, (C1-C3)alkoxy(C,-C3)alkyl, hydroxy(C C3)alkyl or -(Ci-C3)alkylene-NRaRb; and
-22-4820V.1
r is 1 or 2. Values and alternative values for the remainder of the variables are as described above in the sixth embodiment.
In a seventh embodiment, compounds of the present teachings arerepresented by a

-23- 44820V.1

or a pharmaceutically acceptable salt of any one of the foregoing, wherein Y is a bond or Values and alternative values for the variables for Structural Formula (Ild)-(VId) are as described above for Structural Formula (I) or in the second embodiment.
In a eighth embodiment, compounds of the present teachings are represented by a structural formula selected from:

4820V.1

heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -ORc, -NRaRb, -S(0),Rc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, -0(C=0)NReRf; and
4820V.1
R2aand R2b are each independently -H, halogen, -CN, -N02, -ORc, -NRaRb, -C(=0)Rc, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (C,-C3)alkoxy, halo(C,-C3)alkoxy, or (Ci-C3)alkoxy(Ci-C3)alkyl, (C2-C4)alkynyl, halo(C2-C4)alkynyl, (C2-C4)alkenyl, or halo(C2-C4)alkenyl.
Values and alternative values for the remainder of the variables are as described for Structural Formula (I) or for Structural Formulas (Ild)-(VId) in the seventh embodiment.
In a ninth embodiment, for compounds represented by Structural Formulas (Ild)-(VId) and (Ile)-(VIe) described in the seventh and eight embodiments, the group represented by R3 is selected from (C3-Cs)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thiophenyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[6?]imidazolyl, benzo[iflthiazolyl, benzo[b]thiophenyl, quinolinyl, tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, morpholinyl, indolinyl and indenyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)jRc,
-NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (Ci-C3)alkoxy and (Ci-C3)alkoxy(C,-C3)alkyl. Values and alternative values for the remainder of the variables are as described in the seventh or eighth embodiment.
In a tenth embodiment, for compounds represented by Structural Formulas (Ild)-(VId) and (Ile)-(VIe) described in the seventh and eight embodiments, the group represented by R3 is selected from the group consisting of cyclopentyl, phenyl, thiophenyl, pyridyl, piperidinyl, tetrahydropyranyl, tetrahydrofuranyl, pyrrolidinyl, and indolyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)jNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc,
-26-4820V.1
-0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (Ci-C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (C|-C3)alkyl, halo(C,-C3)alkyl, (C,- C3)alkoxy and (Ci-C3)alkoxy(C)-C3)alkyl. Values and alternative values for the remainder of the variables are as described in the seventh or eighth embodiment.
In a eleventh embodiment, for compounds represented by Structural Formulas (Ild)-(VId) and (Ile)-(VIe) described in the seventh and eight embodiments, the group represented by R3 is selected from the group consisting of phenyl, thiophenyl, pyridyl, tetrahydropyranyl and indolyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc,
-OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf,
-NR C(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf,
-NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (CrC3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (Ci-C3)alkyl, halo(CrC3)alkyl, (CrC3)alkoxy and (C,-C3)alkoxy(C C3)alkyl.
In a twelfth embodiment, for compounds described in the seventh, eighth, ninth, tenth or eleventh embodiments, the group represented by R3 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -ORc, (Ci-C3)alkyl, - -(Ci-C3)alkylene-NRaRb, phenyl, pyrimidinyl, morpholinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (Q- C3)alkyl, haloCCrC^alkyl, (Ci-C3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl; Rc is -H,
(Ci-C3)alkyl, pyridinyl or morpholinyl; and Ra and Rb are each independently -H or (C
-27-4820V.1
C3)alkyl. Alternatively, the group represented by R3 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -F, -CI, methyl, -CF3, ethyl, 2- propyl, benzyl, phenyl and -CH2NH2.
In a thirteenth embodiment, for compounds described in the seventh, eighth, ninth, tenth, eleventh or twelfth embodiment, R4 and R5 are both -H. Alternatively, R5 is -H and R4 is -OH, (Ci-C3)alkyl, -(Ci-C2)alkylene-OH,(Ci-C3)alkoxy, -NRaRb, -(C,-C2)alkylene-NRaRb, pyrrolidinyl, piperidinyl, morpholinyl or cyclopropyl, wherein Ra and Rb are each independently -H or (Ci-C3)alkyl and the pyrrolidinyl, piperidinyl, morpholinyl or cyclopropyl represented by R4 is optionally substituted with halogen, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (C]-C3)alkoxy and (Cr C3)alkoxy(Ci-C3)alkyl. Values and alternative values for the remainder of the variables are as described in the seventh, eighth, ninth, tenth, eleventh or twelfth embodiment.
In another embodiment, for compounds described in the second, third, fourth, fifth, sixth, eighth, ninth, tenth, eleventh, twelfth or thirteenth embodiment, Rla and R,b are both -H and values and alternative values for the remainder of the variables are as described in the second, third, fourth, fifth, sixth, eighth, ninth, tenth, eleventh, twelfth or thirteenth embodiment.
In another embodiment, for compounds described in the second, third, fourth, fifth, sixth, eighth, ninth, tenth, eleventh, twelfth or thirteenth embodiment, Rla and Rlb are both -H; R2 and R2b are both -H; and values and alternative values for the remainder of the variables are as described in the second, third, fourth, fifth, sixth, eighth, ninth, tenth, eleventh, twelfth or thirteenth embodiment.
In a fourteenth embodiment, the present teachings provide a compound represented by a structural formula selected from:

-28-4820V.1

or a pharmaceutically acceptable salt of any one of the foregoing, wherein:
R16 is alkyl, optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NRaRb, -C(=0)NReRf, -NRdC(=0)Rc,
-NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=0)NReRf, wherein the cycloalkyl, heterocycloalkyl, aryl or heteroaryl are optionally substituted with 1 to 3 substituents independently selected from - halogen, alkyl, -OR0 and -NRaRb; and values and alternative values for the remainder of the variables are as described above for Structural Formula (I). In some embodiments, R16 is (Ci- C6)alkyl, optionally substituted with -N(CH3)2 or 2,6-dimethylmorpholinyl, e.g., in some embodiments, R16 is selected from methyl, ethyl, (2,6-dimethylmorpholinyl)propyl, and N,N- dimethylaminopropyl. In some embodiments, R16 is methyl.
In a fifteenth embodiment, the present teachings provide a compound represented by a structural formula s
ila); and

-29-4820V.1
or a pharmaceutically acceptable salt of any one of the foregoing, wherein values for the variables are as described above for Structural Formula (I) or in the fourteenth embodiment.
In a sixteenth embodiment, the present teachings provide a compound represented by a structural formula selected from:

or a pharmaceutically acceptable salt of any one of the foregoing, wherein:
R,a and Rlb are each independently -H, -halogen, -CN, -ORc, -NRaRb, -C(=0)ORc,
-C(=0)NReRf, -NRdC(=0)Rc, -0(C=0)NReRf, heterocycloalkyl, or alkyl, wherein
heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc,
-S(0)jNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc,
-NRd(C=0)ORc, -0(C=0)NReRf;
R2a and R2b each independently -H, halogen, -CN, -N02, -NRaRb, -ORc, -C(=0)Rc, (C,-C3)alkyl, halo(C1-C3)alkyl, or (Ci-C3)alkoxy(Ci-C3)alkyl, (C2-C4)alkynyl,
halo(C2-C4)alkynyl, (C2-C4)alkenyl, or halo(C2-C4)alkenyl; and values and alternative values for the remainder of the variables are as described above for Structural Formulae (I), (VII) or (VIII).
In a seventeenth embodiment, the present teachings provide a compound represented by a structural formula selected from:
-30- 4820V.1

or a pharmaceutically acceptable salt of any one of the foregoing, wherein:
R is a cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R6;
R4 and R5 are each independently selected from -H, alkyl, -ORc, -NRaRb, -(Cr
C3)alkylene-NRaRb, -(Ci-C3)alkylene-ORc , -(C,-C3)alkylene-OH, cycloalklyl,
-(Ci-C3)alkylene-cycloalkyl, heterocycloalkyl and -(Ci-C3)alkylene-heterocycloalkyl, provided that R4 and R5 are not both selected from -ORc and -NRaRb; or R3 and R5 together with the carbon atom to which they are attached form a cycloalkyl or a heterocycloalkyl, wherein the cycloalkyl or heterocycloalkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (Cj- C3)alkoxy, halo(C,-C3)alkoxy, (Ci-C3)alkoxy(Ci-C3)alkyl;
each R6 is independently selected from halogen, =0, -ORc, -NRaRb, -S(0)iRc,
-NRdS(0)jRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C6)alkyl, aryl, aryl(Cj-C3)alkyl, heterocycloalkyl and heteroaryl; wherein each the (C]-C6)alkyl, aryl, aryl(Ci-C3)alkyl, heterocycloalkyl and heteroaryl represented by R6 is optionally substituted with
-31-4820V.1
1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Cj- C6)alkyl, halo(C,-C6)alkyl, (C,-C3)alkoxy, halo(Ci-C3)alkoxy and (Ci-C3)alkoxy(Ci-C6)alkyl; and
p is 0, 1 or 2; and
5 values and alternative values for the remainder of the variables are as described above for
Structural Formulae (I), (VII), (VIII), (Vllb) or (Vlllb).
In an eighteenth embodiment, for compounds represented by structural formulas (VIIc)- (VIIIc), the group represented by R3 is selected from (C3-Cs)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thienyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, i o isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[d]imidazolyl, benzo[i ]thiazolyl,
benzo[&]fhiophenyl, quinolinyl, tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, mo holinyl, indolinyl and indenyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc,
15 -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc,
-NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf,
-NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (CrC3)alkyl, phenyl, benzyl, pyrimidinyl, moφholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci- C3)alkyl, phenyl, benzyl, ιηοφηοΐϊηγΐ, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and
20 tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (Ci-C )alkyl, halo(Cr C3)alkyl, (Ci-C3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl. For example, in some embodiments, the group represented by R3 is selected from phenyl, pyridyl, and thienyl, each of which is optionally substituted with halogen or (CrC3)alkyl (e.g., chloro or methyl).
25 In a nineteenth embodiment, for compounds represented by structural formulas (VIIc)-
(VIIIc), R4 and R5 are each independently selected from -H, alkyl, -ORc, -NRaRb, cycloalkyl, heterocycloalkyl and -(C]-C3)alkylene-heterocycloalkyl, provided that R4 and R5 are not both selected from -ORc and -NRaRb. For example, in some embodiments, one of R4 and R5 is -H and the other is selected from -H, alkyl, -ORc, -NR Rb, cycloalklyl, heterocycloalkyl and -(C
-32-E1 1 1444820V.1
C3)alkylene-heterocycloalkyl. In other embodiments, one of R4 and R5 is -H and the other is selected from -H, (C]-C3)alkyl, -OCH3, -N(CH3)2, (C3-C6)cycloalklyl, pyrrolidinyl, piperidinyl, - CH2-morpholinyl and -O-cyclopentyl.
In another embodiment, for compounds described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment, Rl and Rlb are each independently selected from -H and (Ci-C3)alkyl substituted with -N(Me)2, and values and alternative values for the remainder of the variables are as described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment. In another embodiment, for compounds described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment, Rla and Rlb are both -H and values and alternative values for the remainder of the variables are as described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment. In another embodiment, for compounds described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment, Rla and Rlb are both -H; R2a and R2b are both -H; and values and alternative values for the remainder of the variables are as described in the sixteenth, seventeenth, eighteenth or nineteenth embodiment.
In certain embodiments, the present teachings provide the compounds depicted and/or described by name in the Exemplification, as well as neutral forms and pharmaceutically acceptable salts thereof.
The term "alkyl" used alone or as part of a larger moiety, such as "alkoxy", "haloalkyl", "cycloalkylalkyl", "heterocycloalkylalkyl", "aralkyl", "heteroaralkyl" and the like, means saturated aliphatic straight-chain or branched monovalent hydrocarbon radical. Unless otherwise specified, an alkyl group typically has 1-6 carbon atoms, i.e. (Ci-C6)alkyl. As used herein, a "(Ci-C6)alkyl" group is means a radical having from 1 to 6 carbon atoms in a linear or branched arrangement.
An "alkylene group" is a saturated aliphatic branched or straight-chain divalent hydrocarbon radical. Unless otherwise specified, an alkylene group typically has 1-6 carbon atoms, i.e. (Ci-C6)alkylene.
An "alkenyl" means branched or straight-chain monovalent hydrocarbon radical containing at least one double bond. Alkenyl may be mono or polyunsaturated, and may exist in the E or Z onfiguration. Unless otherwise specified, an alkenyl group typically has 2-6 carbon
-33-4820V.1
atoms, i.e. (C2-C6)alkenyl. For example, "(C2-C6)alkenyl" means a radical having from 2-6 carbon atoms in a linear or branched arrangement.
"Alkynyl" means branched or straight-chain monovalent hydrocarbon radical containing at least one triple bond. Unless otherwise specified, an alkynyl group typically has 2-6 carbon atoms, i.e. (C2-C6)alkynyl. For example, "(C2-C6)alkynyl" means a radical having from 2-6 carbon atoms in a linear or branched arrangement.
"Alkoxy" means an alkyl radical attached through an oxygen linking atom, represented by -O-alkyl. For example,
 includes methoxy, ethoxy, propoxy, and butoxy.
includes methoxy, ethoxy, propoxy, and butoxy.
The terms "haloalkyl" and "haloalkoxy" means alkyl or alkoxy, as the case may be, substituted with one or more halogen atoms. The term "halogen" means F, CI, Br or I.
Preferably the halogen in a haloalkyl or haloalkoxy is F.
The term "aryl group" used alone or as part of a larger moiety as in "aralkyl", "aralkoxy", or "aryloxyalkyl", means an aromatic hydrocarbon ring system. The term "aryl" may be used interchangeably with the terms "aryl ring" "aromatic ring", "aryl group" and "aromatic group". An aryl group typically has six to fourteen ring atoms. Examples includes phenyl, naphthyl, anthracenyl, 1,2-dihydronaphthyl, 1,2,3,4-tetrahydronaphthyl, fluorenyl, indanyl, indenyl and the like. A "substituted aryl group" is substituted at any one or more substitutable ring atom, which is a ring carbon atom bonded to a hydrogen.
"Cycloalkyl" means a saturated aliphatic cyclic hydrocarbon radical optionally containing one or more double bonds. It can be monocyclic, bicyclic, polycyclic (e.g., tricyclic), fused, bridged, or spiro. For example, monocyclic (C3-C8)cycloalkyl means a radical having from 3-8 carbon atoms arranged in a monocyclic ring. A (C3-C8)cycloalkyl includes, but is not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctane.
"Heterocycloalkyl" means a saturated or unsaturated non-aromatic 4-12 membered ring radical optionally containing one or more double bonds. It can be monocyclic, bicyclic, tricyclic, fused, bridged, or spiro. The heterocycloalkyl contains 1 to 4 heteroatoms, which may be the same or different, selected from N, O or S. The heterocycloalkyl ring optionally contains one or more double bonds and/or is optionally fused with one or more aromatic rings (e.g., phenyl ring). The term "heterocycloalkyl" is intended to include all the possible isomeric forms.
-34-4820V.1
Examples of heterocycloalky include, but are not limited to, morpholinyl, thiomorpholinyl, pyiTolidinonyl, pyrrolidinyl, piperidinyl, piperazinyl, hydantoinyl, valerolactamyl, oxiranyl, oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyrindinyl, tetrahydropyrimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl, and tetrahydroquinolinyl.
The term "heteroaryl", "heteroaromatic", "heteroaryl ring", "heteroaryl group",
"heteroaromatic ring", and "heteroaromatic group", are used interchangeably herein.
"Heteroaryl" when used alone or as part of a larger moiety as in "heteroaralkyl" or
"heteroarylalkoxy", refers to aromatic ring groups having five to fourteen ring atoms selected from carbon and at least one (typically 1 to 4, more typically 1 or 2) heteroatoms (e.g., oxygen, nitrogen or sulfur). "Heteroaryl" includes monocyclic rings and polycyclic rings in which a monocyclic heteroaromatic ring is fused to one or more other aromatic or heteroaromatic rings. As such, "5- 14 membered heteroaryl" includes monocyclic, bicyclic or tricyclic ring systems.
Examples of monocyclic 5-6 membered heteroaryl groups include furanyl (e.g., 2- furanyl, 3-furanyl), imidazolyl (e.g., A midazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl), isoxazolyl ( e.g., 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl), oxadiazolyl (e.g., 2-oxadiazolyl, 5- oxadiazolyl), oxazolyl (e.g., 2-oxazolyl, 4-oxazolyl, 5-oxazolyl), pyrazolyl (e.g., 3-pyrazolyl, 4- pyrazolyl), pyrrolyl (e.g., 1 -pyrrol yl, 2-pyrrolyl, 3-pyrrolyl), pyridyl (e.g., 2-pyridyl, 3-pyridyl, 4-pyridyl), pyrimidinyl (e.g., 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl), pyridazinyl (e.g., 3- pyridazinyl), thiazolyl (e.g., 2-thiazolyl, 4-thiazolyl, 5-thiazolyl), triazolyl (e.g., 2-triazolyl, 5- triazolyl), tetrazolyl (e.g., tetrazolyl), thienyl (e.g., 2-thienyl, 3-thienyl), pyrimidinyl, pyridinyl and pyridazinyl. Examples of polycyclic aromatic heteroaryl groups include carbazolyl, benzimidazolyl, benzothienyl, benzofuranyl, indolyl, quinolinyl, benzotriazolyl, benzothiazolyl, benzoxazoiyl, benzimidazolyl, isoquinolinyl, indolyl, isoindolyl, acridinyl, or benzisoxazolyl. A "substituted heteroaryl group" is substituted at any one or more substitutable ring atom, which is a ring carbon or ring nitrogen atom bonded to a hydrogen.
Unless otherwise indicated, suitable substituents for a substituted alkyl, cycloalkyl, heterocycloalkyl, aryl group and heteroaryl group include the groups represented by halogen, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc,
-35-4820V.1
-0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (Ci-C6)alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl, wherein Ra, Rb, Rc, Rd, Re and Rf are described above for Structural Formula (I). Each of the (Ci-C6)alkyl, cycloalkyl, cycloalkyl(C] -C3)alkyl, heterocycloalkyl, heterocycloalkyl(C]-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl substituents is optionally substituted with halogen, -N02, -CN, -NRdC(=0)Rc, -NRgRh, (Ci-C3)alkyl, halo(C,-C3)alkyl, (C,- C3)alkoxy(Ci-C3)alkyl, (Ci-C3)alkoxy and halo(Ci-C3)alkoxy, wherein Rg and Rh are as described above for Structural Formula (I). Suitable substituents for a substituted alkyl, cycloalkyl, heterocycloalkyl can also include =0.
Regarding connectivity, an "arylalkyl" moiety, for example, refers to an alkyl group substituted with an aryl group (e.g., phenylmethyl (i.e., benzyl)). Similarly, a "heteroarylalkyl" moiety refers to an alkyl group substituted with a heteroaryl group.
The present teachings also include various isomers and mixtures thereof. "Isomer" refers to compounds that have the same composition and molecular weight but differ in physical and/or chemical properties. The structural difference may be in constitution (geometric isomers) or in the ability to rotate the plane of polarized light (stereoisomers).
Certain of the compounds described herein may exist in various stereoisomeric or tautomeric forms. Stereoisomers are compounds which differ only in their spatial arrangement. The present teachings encompass all such forms, including compounds in the form of essentially pure enantiomers, racemic mixtures and tautomers, which includes forms not depicted structurally. When a disclosed compound is named or depicted by structure without indicating stereochemistry, it is understood that the name or structure encompasses all possible stereoisomers, tautomers, geometric isomers or a combination thereof.
When a geometric isomer is depicted by name or structure, it is to be understood that the geometric isomeric purity of the named or depicted geometric isomer is at least 60%, 70%, 80%, 90%, 99% or 99.9% pure by weight. Geometric isomeric purity is determined by dividing the weight of the named or depicted geometric isomer in the mixture by the total weight of all of the geomeric isomers in the mixture.
-36-4820V.1
Racemic mixture means 50% of one enantiomer and 50% of is corresponding enantiomer. The present teachings encompass all enantiomerically-pure, enantiomerically- enriched, diastereomerically pure, diastereomerically enriched, and racemic mixtures, and diastereomeric mixtures of the compounds described herein.
Enantiomeric and diastereomeric mixtures can be resolved into their component enantiomers or stereoisomers by well known methods, such as chiral-phase gas chromatography, chiral-phase high performance liquid chromatography, crystallizing the compound as a chiral salt complex, or crystallizing the compound in a chiral solvent. Enantiomers and diastereomers can also be obtained from diastereomerically- or enantiomerically-pure intermediates, reagents, and catalysts by well known asymmetric synthetic methods.
When a compound is designated by a name or structure that indicates a single enantiomer, unless indicated otherwise, the compound is at least 60%, 70%, 80%, 90%, 99% or 99.9% optically pure (also referred to as "enantiomerically pure"). Optical purity is the weight in the mixture of the named or depicted enantiomer divided by the total weight in the mixture of both enantiomers.
When the stereochemistry of a disclosed compound is named or depicted by structure, and the named or depicted structure encompasses more than one stereoisomer (e.g., as in a diastereomeric pair), it is to be understood that one of the encompassed stereoisomers or any mixture of the encompassed stereoisomers are included. It is to be further understood that the stereoisomeric purity of the named or depicted stereoisomers at least 60%, 70%, 80%, 90%, 99% or 99.9% by weight. The stereoisomeric purity in this case is determined by dividing the total weight in the mixture of the stereoisomers encompassed by the name or structure by the total weight in the mixture of all of the stereoisomers.
Included in the present teachings are pharmaceutically acceptable salts of the compounds disclosed herein. The disclosed compounds have basic amine groups and therefore can form pharmaceutically acceptable salts with pharmaceutically acceptable acid(s). Suitable pharmaceutically acceptable acid addition salts of the compounds described herein include salts of inorganic acids (such as hydrochloric acid, hydrobromic, phosphoric, metaphosphoric, nitric, and sulfuric acids) and of organic acids (such as, acetic acid, benzenesulfonic, benzoic, citric,
-37-4820V.1
ethanesulfonic, fumaric, gluconic, glycolic, isethionic, lactic, lactobionic, maleic, malic, methanesulfonic, succinic, p- toluenesulfonic, and tartaric acids). Compounds of the present teachings with acidic groups such as carboxylic acids can form pharmaceutically acceptable salts with pharmaceutically acceptable base(s). Suitable pharmaceutically acceptable basic salts include ammonium salts, alkali metal salts (such as sodium and potassium salts) and alkaline earth metal salts (such as magnesium and calcium salts). Compounds with a quaternary ammonium group also contain a counteranion such as chloride, bromide, iodide, acetate, perchlorate and the like. Other examples of such salts include hydrochlorides, hydrobromides, sulfates, methanesulfonates, nitrates, maleates, acetates, citrates, fumarates, tartrates [e.g. (+)- tartrates, (-)- tartrates or mixtures thereof including racemic mixtures], succinates, benzoates and salts with amino acids such as glutamic acid.
Compounds described herein can inhibit various kinases, including the TT , PLK (such as PLK4), Aurora A, Aurora B and CHK (such as CHK2). Thus, generally, compounds described herein are useful in the treatment of diseases or conditions associated with such kinases.
In one embodiment, the compounds described herein are TTK, PLK, Aurora A, Aurora B and/or CHK inhibitors, and are useful for treating diseases, such as cancer, associated with such kinase(s). Alternatively, the compounds described herein are TTK inhibitors and are useful for treating diseases associated with TTK, such as cancer. In another alternative embodiment, the compounds described herein are Aurora A and/or B inhibitors and are useful in inhibiting Aurora A and/or B activity for the treatment of various conditions such as cancers. In yet another specific embodiment, the compounds described herein are PLK inhibitors and are useful in inhibiting PLK activity for the treatment of various conditions such as cancers. Typically, the PLK is PLK4, PLK2 and/or PLK1. In one example, the PLK is PLK1 and/or PLK4. In another example, the PLK is PLK4. In another alternative embodiment, the compounds described herein are CHK inhibitors and are useful in inhibiting CHK activity for the treatment of various conditions such as cancers.
Another aspect of the present teachings relates to a method of treating a subject with cancer comprising administering to the subject an effective amount of a compound described
-38-4820V.1
herein. In one embodiment, the compounds described herein inhibit the growth of a tumor. For example, the compounds described herein inhibit the growth of a tumor that overexpresses at least one of TTK, PLK, Aurora A, Aurora B, and CHK. In one embodiment, the compounds described herein inhibit the growth of a tumor that overexpresses TTK.
Cancers that can be treated (including reduction in the likelihood of recurrence) by the methods of the present teachings include lung cancer, breast cancer, colon cancer, brain cancer, neuroblastoma, prostate cancer, melanoma, glioblastoma multiform, ovarian cancer, lymphoma, leukemia, melanoma, sarcoma, paraneoplasia, osteosarcoma, germinoma, glioma and mesothelioma. In one embodiment, the cancer is selected from leukemia, acute myeloid leukemia, chronic myelogenous leukemia, breast cancer, brain cancer, colon cancer, colorectal cancer, head and neck cancer, hepatocellular carcinoma, lung adenocarcinoma, metastatic melanoma, pancreatic cancer, prostate cancer, ovanrian cancer and renal cancer. In one embodiment, the cancer is lung cancer, colon cancer, brain cancer, neuroblastoma, prostate cancer, melanoma, glioblastoma mutiform or ovarian cancer. In another embodiment, the cancer is lung cancer, breast cancer, colon cancer, brain cancer, neuroblastoma, prostate cancer, melanoma, glioblastoma multiform or ovarian cancer. In yet another embodiment, the cancer is breast cancer, colon cancer and lung cancer. In yet another embodiment, the cancer is a breast cancer. In yet another embodiment, the cancer is a basal sub-type breast cancer or a luminal B sub-type breast cancer. In yet another embodiment, the cancer is a basal sub-type breast cancer that overexpresses TTK. In yet another embodiment, the basal sub-type breast cancer is ER
(estrogen receptor), HER2 and PR (progesterone receptor) negative breast cancer. In yet another embodiment, the cancer is a soft tissue cancer. A "soft tissue cancer" is an art-recognized term that encompasses tumors derived from any soft tissue of the body. Such soft tissue connects, supports, or surrounds various structures and organs of the body, including, but not limited to, smooth muscle, skeletal muscle, tendons, fibrous tissues, fatty tissue, blood and lymph vessels, perivascular tissue, nerves, mesenchymal cells and synovial tissues. Thus, soft tissue cancers can be of fat tissue, muscle tissue, nerve tissue, joint tissue, blood vessels, lymph vessels, and fibrous tissues. Soft tissue cancers can be benign or malignant. Generally, malignant soft tissue cancers are referred to as sarcomas, or soft tissue sarcomas. There are many types of soft tissue
-39-4820V.1
tumors, including lipoma, lipoblastoma, hibernoma, liposarcoma, leiomyoma, leiomyosarcoma, rhabdomyoma, rhabdomyosarcoma, neurofibroma, schwannoma (neurilemoma), neuroma, malignant schwannoma, neurofibrosarcoma, neurogenic sarcoma, nodular tenosynovitis, synovial sarcoma, hemangioma, glomus tumor, hemangiopericytoma, hemangioendothelioma, angiosarcoma, Kaposi sarcoma, lymphangioma, fibroma, elastofibroma, superficial fibromatosis, fibrous histiocytoma, fibrosarcoma, fibromatosis, dermatofibrosarcoma protuberans (DFSP), malignant fibrous histiocytoma (MFH), myxoma, granular cell tumor, malignant
mesenchymomas, alveolar soft-part sarcoma, epithelioid sarcoma, clear cell sarcoma, and desmoplastic small cell tumor. In a particular embodiment, the soft tissue cancer is a sarcoma selected from the group consisting of a fibrosarcoma, a gastrointestinal sarcoma, a
leiomyosarcoma, a dedifferentiated liposarcoma, a pleomorphic liposarcoma, a malignant fibrous histiocytoma, a round cell sarcoma, and a synovial sarcoma.
In some embodiments, the present teachings provide methods of inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject who is undergoing an anti-cancer therapy. The method comprises the steps of:
a) assessing the subject to determine whether the cancer is in remission; and
b) if the cancer is in remission; then administering to the subject an effective amount of a TTK inhibitor (e.g., a compound represented by Structural Formula (I)). If the cancer is not in remission, the method optionally further comprises the step of continuing the anti-cancer therapy until the cancer goes into remission and then the step b) of administering an effective amount of a TTK inhitior (e.g., a compound represented by Structural Formula (I)).
In some embodiments, the present teachings provide methods of inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhbitior (e.g, a compound represented by Structural Formula (I)).
In some embodiments, e.g., where the subject is being treated to reduce the likelihood of recurrence of a cancer, the subject has already been treated with an anti-cancer therapy.
Alternatively, the subject has already been treated with an anti-cancer therapy and the subject is in remission.
-40-4820V.1
In some embodiments, the present teachings provide methods of treating a subject with a cancer comprising administering to the subject an effective amount of a compound represented by Structural Formula (I) in combination with an effective anti-cancer therapy. In one embodiment, the cancer is a metastatic cancer. A "metastatic cancer" is a cancer that has spread from its primary site to other parts of the body.
In another embodiment, the present teachings are directed to a method of treating a subject with a drug-resistant cancer. A "drug-resistant cancer" is a cancer that is not responsive to one, two, three, four, five or more drugs that are typically used for the treatment of the cancer. In one embodiment, the drug-resistant cancer is mediated by the growth of tumor-initiating cells.
The term "inhibiting the growth of tumor-initiating cells" refers to preventing or decreasing the rate of the proliferation and/or survival of the tumor-initiating cells.
As used herein, the term "reducing the likelihood of recurrence of a cancer" means partially or totally inhibiting, preventing or delaying the return of a cancer at or near a primary site and/or at a secondary site after a period of remission. It also means that the cancer is less likely to return with treatment described herein than in its absense.
As used herein, the term "remission" refers to a state of cancer, wherein the clinical symptoms or indicators associated with a cancer have disappeared or cannot be detected, typically after the subject has been successfully treated with an anti-cancer therapy.
As used herein, "treating a subject with a cancer" includes achieving, partially or substantially, one or more of the following: arresting the growth, reducing the extent of the cancer (e.g., reducing size of a tumor), inhibiting the growth rate of the cancer, ameliorating or improving a clinical symptom or indicator associated with the cancer (such as tissue or serum components) or increasing longevity of the subject; and reducing the likelihood of recurrence of the cancer.
Suitable methods known in the art can be used for assessing a subject to determine whether the cancer is in remission. For example, the size of the tumor and/or tumor markers, usually proteins associated with tumors, can be monitored to determine the state of the cancer. Size of the tumor can be monitored with imaging devices, such as X-ray, MRI, CAT scans, ultrasound, mammography, PET and the like or via biopsy.
-41-4820V.1
In some embodiments, TTK inhibitors for use in inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject can include any of the compounds described herein, including compounds of formulae (I)-(VIII). In one embodiment, TTK inhibitors for use in inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject does not comprise administering the compound represented by Structural Formula (I) for the treatment of cancer.
In some embodiments, the TTK inhibitors for use in inhibiting the growth of tumor- initiating cells or reducing the likelihood of recurrence of a cancer in a subject is described in WO2009/024824, the entire teachings of which is incorporated herein by reference. The TTK inhibitor is presented by the followin structural formula:

wherein:
R1 is selected from Ci^alkyl, cyclopropyl, cyclopropylmethyl and cyclobutyl; wherein said cyclopropyl may be optionally substituted by methyl; and wherein R1 may be optionally substituted by one or more R5 ; m is 0 or 1 ;
R2 is selected from CAaUcyl, C2-6alkenyl, C2-6alknyl, C3.<¾cycloalkyl, cyclopentenyl, cyclohexenyl, oxetanyl, tetrahydropyranyl, tetrahydrofuranyl, oxepanyl, azetidinyl, pyrrolidinyl, piperidinyl and azepanyl; wherein R2 may be optionally substituted on carbon by one or more R6 ; and wherein if R2 contains a ring -NH- moiety, that nitrogen may be optionally substituted by R7';
R3 is independently selected from fluoro, chloro, bromo, cyano, methoxy, ethoxy, trifluoromethoxy, methyl, ethyl, trifluoromethyl, ethenyl, ethynyl, cyclopropyl, methylthio, ethylthio, N-methylamino, NN-dimethylamino, amino and methylsulfonyloxy;
-42-4820V.1
n' is an integer selected from 0 to 3; wherein the values of R3 may be the same or different;
R4' is -L-R8' or R9';
L is selected from ethynylene, ethenylene, cyclopropyl and wherein X is a direct bond, -0-, -S-, -NH-, -OS(0)2-, -N(CH3)- or -N(CH2R10')-; and wherein L may be optionally substituted on carbon by one or more fluoro; R5 is cyano or fluoro;
R6' is selected from Ci_3alkyl, Ci_ alkoxy, N-( Ci_3alkyl)amino, N,N-( Ci-3alkyl)2amino, hydroxy, amino, fluoro and cyano;
R7 is selected from Ci.3alkyl, cyclopropyl, Cj.3alkanoyl and Ci-3alkylsulfonyl;
R8 and R10 are each independently selected from chloro, bromo, iodo, cyano, nitro, mercapto, sulfo, hydroxy, carboxy, amino, carbamoyl, sulfamoyl, C2-6alkyl, C2_6alkenyl, C2-6alkynyl, C].6alkoxy, Ci.6alkylsulfonyloxy, N-( Ci.6alkyl)sulfamoyloxy, N,N- (Ci.6alkyl)2sulfamoyloxy, Ci_6alkoxycarbonyl, Ci_6alkanoyl, Ci-6alkanoyloxy, N- (C].6alkyl)amino, N,N-(Ci-6alkyl)2amino, N-(Ci^alkanoyl)-N-(Ru )amino, N- (Ci.6alkoxycarbonyl)-N-(R'2 )amino, N-(Ci-6alkyl)carbamoyl, N,N-(Ci.6alkyl)2carbamoyl, N- (C,.6alkyl)sulfamoyl, N,N-(C,^alkyl)2sulfamoyl, N-[(C,.6alkyl)sulfonyl]-N-(R13')amino, (N,N- (R14)(R15')sulfamoyl)-N-(R,6')amino, 3,3-(R17')(R18')-l -(R19')ureido, carbocyclyl-R20'-, heterocyclyl-R21 - and (Ci_6alkyl)-S(0)a- wherein a is 0 to 2; wherein R8 and R10 may be optionally substituted on carbon by one or more R22 ; and wherein if said heterocyclyl has an -NH- moiety, that nitrogen may be optionally substituted by R23 ;
R9 is selected from carboxy, carbamoyl, sulfamoyl, C3_6alkyl, C3_6alkenyl, C3_6alkynyl, C3_6alkoxy, C3_6alkylsulfinyl, C3^alkylsulfanyl, C2-6alkylsulfonyloxy , N- (Ci.6alkyl)sulfamoyloxy , N,N-(Ci.6alkyl)2sulfamoyloxy , Ci^alkoxycarbonyl, C|_6alkanoyl, Ci-6alkanoyloxy, N-(C2-6alkyl)amino, N,N-(C2^alkyl)2amino, N-(Ci_6alkanoyl)-N-(R24 )amino, N-(Ci^alkoxycarbonyl)-N-(R25 )amino, N-(Ci.6alkyl)carbamoyl, N,N-(Ci.6alkyl)2carbamoyl, N- (C,_6alkyl)sulfamoyl, N,N-(Ci.6alkyl)2sulfamoyl, N-[(Ci.6alkyl)sulfonyl]-N-(R26')amino, (N,N- (R27')(R28')sulfamoyl)-N-(R 9')arnino, 3,3-(R30')(R31')-l-(R32')ureido, C4_i2carbocyclyl-R33'- and heterocyclyl-R34 -; wherein R9 may be optionally substituted on carbon by one or more R35 , and
-43-4820V.1
wherein if said heterocyclyl has an -NH- moiety, that nitrogen may be optionally substituted by
R36 ;
R22 and R35 are independently selected from halo, cyano, nitro, mercapto, sulfo, hydroxy, carboxy, amino, carbamoyl, sulfamoyl, Chalky 1, C2-6alkenyl, C2-6alkynyl, Ci_6alkoxy, N-(C],6alkyl)sulfamoyloxy, N,N-(Ci.6alkyl)2Sulfamoyloxy, Ci.6alkoxycarbonyl, d-ealkanoyl, Ci.6alkanoyloxy, N-(Ci-6alkyl)amino, N,N-(Ci-6alkyl)2 amino, N-(C]_6alkanoyl)-N-(R37 )amino, N-(Ci.6alkoxycarbonyl)-N-(R38 )amino, N-(Ci.6alkyl)carbamoyl,
N,N-(Ci-6alkyl)2carbamoyl, N-(Ci-6alkyl)sulfamoyl, N,N-(Ci.6alkyl)2Sulfamoyl, N- [(Ci_6alkyl)sulfonyl]-N-(R 9')amino, (N,N-(R40')(R41 ')sulfamoyl)-N-(R42')amino,
3,3-(R43')(R44')-l-(R45')ureido, carbocyclyl-R46'-, heterocyclyl-R47'-, and (Ci.6alkyl)-S(0)a- wherein a is 0 to 2; wherein R22 and R35 may be optionally substituted on carbon by one or more R48 ; and wherein if said heterocyclyl has an -NH- moiety, that nitrogen may be optionally substituted by R49 ;
23' 36'
R" and R are independently selected from Ci^alkyl, C2-6alkenyl, C2-6alkynyl,
C^ealkoxycarbonyl, Ci_6alkanoyl, carbamoyl, N-(Ci.6alkyl)carbamoyl, N,N-(
Ci_6alkyl)2carbamoyl, sulfamoyl, N-(Ci-6alkyl)sulfamoyl, N,N-(Ci-6alkyl)2Sulfamoyl,
carbocyclyl-R50 -, heterocyclyl-R51 -, and (C!.6alkyl)-S(0)a- wherein a is 1 or 2; wherein R23 and R36 may be independently optionally substituted on carbon by one or more R52 ; and wherein if said heterocyclyl contains an -NH- moiety that nitrogen may be optionally substituted by R53 ;
R20 and R21 are each independently selected from a direct bond, -0-, -N(R54 )-, -C(O)-,
-N(R55')C(0)-, -C(0)N(R56')-, -S02N(R57')-, -N(R58')-C(0)-N(R59')-, -OS(0)2-, -S(0)20-, -N(R60')S(O)2N(R6r)-, -N(R62')S02- and -S(0)a- wherein a is 0 to 2;
R33 and R34' are each independently selected from a direct bond, -0-, -N(R63 )-, -C(O)-, -N(R64')C(0)-, -C(0)N(R65')-, -S02N(R66')-, -N(R6T)-C(0)-N(R68')-, -OS(0)2-, -S(0)20-, -N(R69')S(0)2N(R70')-, -N(R71')S02- and -S(0)a- wherein a is 0 to 2;
R46 and R 7< are each independently selected from a direct bond, -0-, -N(R72 )-, -C(O)-, - N(R73')C(0)-, -C(0)N(R74')-, -S02N(R75')-, -N(R76')-C(0)-N(R77')-, -OS(0)2-, -S(0)20-, - N(R78')S(0)2N(R79')-, -N(R80')SO2- and -S(0)a- wherein a is O to 2;
-44-4820V.1
Rso and R51' are each independently selected from a direct bond, -C(O)-, -N(R8r)C(0)-, -N(R82')S02-, -O-C(O)- and -S(0)a- wherein a is 1 or 2;
R48' and R52 are each independently selected from fiuoro, chloro, cyano, nitro, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, sulfo, carbamoyl, mercapto, sulfamoyl, methyl, ethyl, ethenyl, methoxy, ethoxy, formyl, acetyl, acetoxy, N-methylamino, N-ethylamino N,N-dimethylamino, Ν,Ν-diethylamino, N-ethyl-N-methylamino, N-formylamino, N- acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, Ν,Ν-dimethylcarbamoyl, N,N- diethylcarbamoyl, N-ethyl-N-methylcarbamoyl, methylsulfanyl, ethylsulfanyl, methylsulfinyl, ethylsulfinyl, methylsulfonyl, methylsufonyloxy, ethylsulfonyl, ethylsulfonyloxy,
methoxycarbonyl, ethoxycarbonyl, N-methylsulfamoyl, N-ethylsulfamoyl, N,N- dimethylsulfamoyl, N,N -diethylsulfamoyl and N-ethyl-N-methylsulfamoyl;
R49' and R53 are each independently selected from Ci_6alkyl, C3.6cycloalkyl,
C].6alkanoyl, Ci^alkylsulfonyl, Ci_6alkoxycarbonyl, carbamoyl, N-(Ci.6alkyl)carbamoyl, N,N-( Ci_6alky)2carbamoyl, benzyl, benzyloxycarbonyl, benzoyl and phenylsulfonyl;
pir w 12' τ»13' r.14' „15' „16' D17' r.18' D19' τ·54' „55' D56' τ 57' τ>58' D59' τ»60' ρ61' IV , IV , Γν , IV , IV , IV ,Κ , XV , XV , XV , IV ,Κ , IV , XV , IV , IV , JV and R62 are each independently hydrogen or a group selected from and cyclopropyl wherein said group may be optionally substituted on carbon by one or more R22 ;
„24' ρ25' ρ26' „27' „28' β29' τ.30' D31' „32· ρ63' η64' τ·65' Ώ66' τ»67' ρ68' Β69'
IV , XV , XV , IV , Κ , XV , XV , XV , Κ , IV , IV , XV , XV , Κ , XV , XV ,
R70 ,and R71 are each independently hydrogen or a group selected from
 and cyclopropyl wherein said group may be optionally substituted on carbon by one or more R35 ;
and cyclopropyl wherein said group may be optionally substituted on carbon by one or more R35 ;
D37' „38' ρ39' ρ40' „41' D42' D43' D44' „45' r.72' D73' D74' „75' η76' B77' p78' p79' IV , K , IV , IV , XV , IV , IV , IV , IV , IV , IV , IV , IV , IV , IV , XV , IV and R ' are each independently hydrogen or a group selected from
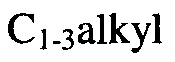 and cyclopropyl wherein said group may be optionally substituted on carbon by one or more R48';
and cyclopropyl wherein said group may be optionally substituted on carbon by one or more R48';
wherein the compound is other than:
-45-4820V.1
2- { [4-(4-acetylpiperazin-l-yl)phenyl]amino } -7-methyl-9-pentan-3-yl-7,9-dihydro-8H- purin-8- one or 7-methyl-2-{ [4-(4-methylpiperazin-l-yl)phenyl]amino}-9-pentan-3-yl-7,9-dihydro-8H- purin-8-one; or a phamaceutically acceptable salt thereof.
Alternatively, the TTK inhitor described in WO2009/156315 can be used for inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject. The entire teachings of WO2009/156315 is incorporated by reference.
The TTK inhibitor of WO2009/156315 is represented by the following structural formula:

wherein:
Rl is an ortho-substituted-aryl group or a heterocyclyl or C3-C7 cycloalkyl group;
R2 is hydrogen atom or a straight or branched C1-C alkyl, C2-Ce alkenyl,
C2-C6 alkynyl, C3-C7 cycloalkyl or heterocyclyl group;
R3 is aryl, heterocyclyl or C3-C7 cycloalkyl group;
R4 is hydrogen atom, hydroxyl or C1 -C6 alkyl group, which group may be optionally cyclized together with one of the atom of the group which R3 may represent so as to form a fused C4-C7 cyclic group;
R5 and R6: are each independently hydrogen atom, Ci-Ce alkyl, or are optionally cyclized together with the carbon atom to which they are bonded so as to form a C3-C7
cycloalkyl group; wherein the groups ortho-substituted-aryl, aryl, heterocyclyl, C3-C7 cycloalkyl, C4-C7 cycloalkyl, Ci-C alkyl, C2-C6 alkenyl and C2-C6 alkynyl may be optionally (further) substituted; with the proviso that that the following compounds are excluded:
-46-4820V.1
1 H-pyrazolo[4,3-h]quinazoline-3-carboxamide, N-cyclopropyl-4,5-dihydro- 1 - methyl-8- [( 1 -methyl-4-piperidinyl)amino] and
lH-pyrazolo[4,3-h]quinazoline-3-carboxamide, N-cyclohexyl-8- (cyclopentylamino)-4,5-dihydro-N-hydroxy-l -methyl; and the pharmaceutically acceptable salts thereof.
In another alternative, the TTK inhibitor that can be used for inhibiting the growth of tumor-initiating cells or reducing the likelihood of recurrence of a cancer in a subject is a pyridine derivative described WO2010/007756, the entire teaching of which is incorported herein by reference.
For methods described herein, e.g., coadministration methods, the anti-cancer therapy is selected from the group consisting of surgery, radiation therapy, immunotherapy, endocrine therapy, gene therapy and administration of an anti-cancer agent. Alternatively, the anti-cancer therapy is radiation therapy. In another alternative, the anti-cancer therapy is immunotherapy. In another alternative, the anti-cancer therapy is administration of an anti-cancer agent. In yet another alternative, the anti-cancer therapy is surgery.
Radiation therapy is the use of radiation to kill, destroy or treat the cancers. Exemplary radiation therapy includes, but is not limited to, gamma-radiation, neutron beam radiotherapy, electron beam radiotherapy, proton therapy, brachytherapy, and radioiosotope thereapy (i.e., systemic radioactive isotopes therapy),
An endocrine therapy is a treatment that adds, blocks or removes hormones. For example, chemotherapeutic agents that can block the production or activity of estrogen have been used for treating breat cancer. In addition, hormonal stimulation of the immune system has been used to treat specific cancers, such as renal cell carcinoma and melanoma. In one embodiment, the endocrine therapy comprises administration of natural hormones, synthetic hormones or other synthetic molecules that may block or increase the production of the body's natural hormones. In another embodiment, the endocrine therapy includes removal of a gland that makes a certain hormone.
-47-4820V.1
As use herein, a gene therapy is the insertion of genes into a subject's cell and biological tissues to treat diseases, such as cancer. Exemplary gene therapy includes, but is not limited to, a germ line gene therapy and a somatic gene therapy.
Immunotherapy (also called biological response modifier therapy, biologic therapy, biotherapy, immune therapy, or biological therapy) is treatment that uses parts of the immune system to fight disease. Immunotherapy can help the immune system recognize cancer cells, or enhance a response against cancer cells. Immunotherapies include active and passive immunotherapies. Active immunotherapies stimulate the body's own immune system while passive immunotherapies generally use immune system components created outside of the body.
Examples of active immunotherapies include, but are not limited to vaccines including cancer vaccines, tumor cell vaccines (autologous or allogeneic), dendritic cell vaccines, antigen vaccines, anti-idiotype vaccines, DNA vaccines, viral vaccines, or Tumor-Infiltrating
Lymphocyte (TIL) Vaccine with Interleukin-2 (IL-2) or Lymphokine- Activated Killer (LAK) Cell Therapy,.
Examples of passive immunotherapies include but are not limited to monoclonal antibodies and targeted therapies containing toxins. Monoclonal antibodies include naked antibodies and conjugated monoclonal antibodies (also called tagged, labeled, or loaded antibodies). Naked monoclonal antibodies do not have a drug or radioactive material attached whereas conjugated monoclonal antibodies are joined to, for example, a chemotherapy drug (chemolabeled), a radioactive particle (radiolabeled), or a toxin (immunotoxin). Examples of these naked monoclonal antibody drugs include, but are not limited to Rituximab (Rituxan), an antibody against the CD20 antigen used to treat, for example, B cell non-Hodgkin lymphoma; Trastuzumab (Herceptin), an antibody against the HER2 protein used to treat, for example, advanced breast cancer; Alemtuzumab (Campath), an antibody against the CD52 antigen used to treat, for example, B cell chronic lymphocytic leukemia (B-CLL); Cetuximab (Erbitux), an antibody against the EGFR protein used, for example, in combination with irinotecan to treat, for example, advanced colorectal cancer and head and neck cancers; and Bevacizumab (Avastin) which is an antiangiogenesis therapy that works against the VEGF protein and is used, for example, in combination with chemotherapy to treat, for example, metastatic colorectal cancer.
-48-4820V.1
Examples of the conjugated monoclonal antibodies include, but are not limited to Radiolabeled antibody Ibritumomab tiuxetan (Zevalin) which delivers radioactivity directly to cancerous B lymphocytes and is used to treat, for example, B cell non-Hodgkin lymphoma; radiolabeled antibody Tositumomab (Bexxar) which is used to treat, for example, certain types of non- Hodgkin lymphoma; and immunotoxin Gemtuzumab ozogamicin (Mylotarg) which contains calicheamicin and is used to treat, for example, acute myelogenous leukemia (AML). BL22 is a conjugated monoclonal antibody for treating, for example, hairy cell leukemia, immunotoxins for treating, for example, leukemias, lymphomas, and brain tumors, and radiolabeled antibodies such as OncoScint for example, for colorectal and ovarian cancers and ProstaScint for example, for prostate cancers.
Further examples of therapeutic antibodies that can be used include, but are not limited to, HERCEPTIN® (Trastuzumab) (Genentech, CA) which is a humanized anti-HER2 monoclonal antibody for the treatment of patients with metastatic breast cancer; REOPRO® (abciximab) (Centocor) which is an anti-glycoprotein Ilb/IIIa receptor on the platelets for the prevention of clot formation; ZENAPAX® (daclizumab) (Roche Pharmaceuticals, Switzerland) which is an immunosuppressive, humanized anti-CD25 monoclonal antibody for the prevention of acute renal allograft rejection; PANOREX™ which is a murine anti-17-IA cell surface antigen IgG2a antibody (Glaxo Wellcome/Centocor); BEC2 which is a murine anti-idiotype (GD3 epitope) IgG antibody (ImClone System); IMC-C225 which is a chimeric anti-EGFR IgG antibody (ImClone System); VITAXIN™ which is a humanized anti-aVp3 integrin antibody (Applied Molecular Evolution/Medlmmune); Campath 1H/LDP-03 which is a humanized anti CD52 IgGl antibody (Leukosite); Smart M195 which is a humanized anti-CD33 IgG antibody (Protein Design Lab/Kanebo); RITUXAN™ which is a chimeric anti-CD20 IgGl antibody (IDEC Pharm/Genentech, Roche/Zettyaku); LYMPHOCIDE™ which is a humanized anti-CD22 IgG antibody (Immunomedics); LYMPHOCIDE™ Y-90 (Immunomedics); Lymphoscan (Tc- 99m-labeled; radioimaging; Immunomedics); Nuvion (against CD3; Protein Design Labs); CM3 is a humanized anti-ICAM3 antibody (ICOS Pharm); IDEC- 1 14 is a primatied anti-CD80 antibody (IDEC Pharm/Mitsubishi); ZEVALIN™ is a radiolabeled murine anti-CD20 antibody (IDEC/Schering AG); IDEC- 131 is a humanized anti-CD40L antibody (IDEC Eisai); IDEC- 151
-49-4820V.1
is a primatized anti-CD4 antibody (IDEC); IDEC-152 is a primatized anti-CD23 antibody (IDEC/Seikagaku); SMART anti-CD3 is a humanized anti-CD3 IgG (Protein Design Lab); 5G1.1 is a humanized anti-complement factor 5 (C5) antibody (Alexion Pharm); D2E7 is a humanized anti-TNF-α antibody (CAT/BASF); CDP870 is a humanized anti-TNF-a Fab fragment (Celltech); IDEC- 151 is a primatized anti-CD4 IgGl antibody (IDEC
Pharm/SmithKline Beecham); MDX-CD4 is a human anti-CD4 IgG antibody
(Medarex/Eisai/Genmab); CD20-sreptdavidin (+biotin- yttrium 90; NeoRx); CDP571 is a humanized anti-TNF-α IgG4 antibody (Celltech); LDP-02 is a humanized anti-a4p7 antibody (LeukoSite/Genentech); OrthoClone OKT4A is a humanized anti-CD4 IgG antibody (Ortho Biotech); ANTOVA™ is a humanized anti-CD40L IgG antibody (Biogen); ANTEGREN™ is a humanized anti-VLA-4 IgG antibody (Elan); and CAT- 152 is a human anti-TGF-p2 antibody (Cambridge Ab Tech).
Immunotherapies that can be used in the present teachings include adjuvant
immunotherapies. Examples include cytokines, such as granulocyte-macrophage colony- stimulating factor (GM-CSF), granulocyte-colony stimulating factor (G-CSF), macrophage inflammatory protein (MIP)-l -alpha, interleukins (including IL-1, IL-2, IL-4, IL-6, IL-7, IL-12, IL-15, IL-18, IL-21, and IL-27), tumor necrosis factors (including TNF-alpha), and interferons (including IFN-alpha, IFN-beta, and IFN-gamma); aluminum hydroxide (alum); Bacille Calmette-Guerin (BCG); Keyhole limpet hemocyanin (KLH); Incomplete Freund's adjuvant (IFA); QS-21 ; DETOX; Levamisole; and Dinitrophenyl (DNP), and combinations thereof, such as, for example, combinations of, interleukins, for example, IL-2 with other cytokines, such as IFN-alpha.
Alternatively, the anti-cancer therapy described herein includes administration of an anticancer agent.
An "anti-cancer agent" is a compound, which when administered in an effective amount to a subject with cancer, can achieve, partially or substantially, one or more of the following: arresting the growth, reducing the extent of a cancer (e.g., reducing size of a tumor), inhibiting the growth rate of a cancer, and ameliorating or improving a clinical symptom or indicator
-50-4820V.1
associated with a cancer (such as tissue or serum components) or increasing longevity of the subject.
The anti-cancer agent suitable for use in the methods described herein include any anticancer agents that have been approved for the treatment of cancer. In one embodiment, the anti- cancer agent includes, but is not limited to, a targeted antibody, an angiogenisis inhibitor, an alkylating agent, an antimetabolite, a vinca alkaloid, a taxane, a podophyllotoxin,a topoisomerase inhibitor, a hormonal antineoplastic agent and other antineoplastic agents.
Examples of alkylating agents useful in the methods of the present teachings include but are not limited to, nitrogen mustards (e.g. , mechloroethamine, cyclophosphamide, chlorambucil, melphalan, etc.), ethylenimine and methylmelamines (e.g., hexamethlymelamine, thiotepa), alkyl sulfonates (e.g., busulfan), nitrosoureas (e.g., carmustine, lomusitne, semustine, streptozocin, etc.), or triazenes (decarbazine, etc.). Examples of antimetabolites useful in the methods of the present teachings include but are not limited to folic acid analog (e.g., methotrexate), or pyrimidine analogs (e.g., fluorouracil, floxouridine, Cytarabine), purine analogs (e.g., mercaptopurine, thioguanine, pentostatin). Examples of plant alkaloids and terpenoids or derivatives thereof include, but are not limited to, vinca alkaloids (e.g., vincristine, vinblastine, vinorelbine, vindesine), podophylloto in, and taxanes (e.g., paclitaxel, docetaxel). Examples of a topoisomerase inhibitor includes, but is not limited to, irinotecan, topotecan, amsacrine, etoposide, etoposide phosphate and teniposide. Examples of antineoplastic agents include, but are not limited to, actinomycin, anthracyclines (e.g., doxorubicin, daunorubicin, valrubicin, idarubicin, epirubicin), bleomycin, plicamycin and mitomycin.
In one embodiment, the anti-cancer agents that can be used in the present teachings include Adriamycin, Dactinomycin, Bleomycin, Vinblastine, Cisplatin, acivicin; aclarubicin; acodazole hydrochloride; acronine; adozelesin; aldesleukin; altretamine; ambomycin;
ametantrone acetate; aminoglutethimide; amsacrine; anastrozole; anthramycin; asparaginase; asperlin; azacitidine; azetepa; azotomycin; batimastat; benzodepa; bicalutamide; bisantrene hydrochloride; bisnafide dimesylate; bizelesin; bleomycin sulfate; brequinar sodium;
bropirimine; busulfan; cactinomycin; calusterone; caracemide; carbetimer; carboplatin;
carmustine; carubicin hydrochloride; carzelesin; cedefingol; chlorambucil; cirolemycin;
-51 -4820V.1
cladribine; crisnatol mesylate; cyclophosphamide; cytarabine; dacarbazine; daunorubicin hydrochloride; decitabine; dexormaplatin; dezaguanine; dezaguanine mesylate; diaziquone; doxorubicin; doxorubicin hydrochloride; droloxifene; droloxifene citrate; dromostanolone propionate; duazomycin; edatrexate; eflornithine hydrochloride; elsamitrucin; enloplatin;
enpromate; epipropidine; epirubicin hydrochloride; erbulozole; esorubicin hydrochloride;
estramustine; estramustine phosphate sodium; etanidazole; etoposide; etoposide phosphate; etoprine; fadrozole hydrochloride; fazarabine; fenretinide; floxuridine; fludarabine phosphate; fluorouracil; flurocitabine; fosquidone; fostriecin sodium; gemcitabine; gemcitabine
hydrochloride; hydroxyurea; idarubicin hydrochloride; ifosfamide; ilmofosine; interleukin II (including recombinant interleukin II, or rIL2), interferon alfa-2a; interferon alfa-2b; interferon alfa-nl ; interferon alfa-n3; interferon beta-I a; interferon gamma-I b; iproplatin; irinotecan hydrochloride; lanreotide acetate; letrozole; leuprolide acetate; liarozole hydrochloride;
lometrexol sodium; lomustine; losoxantrone hydrochloride; masoprocol; maytansine;
mechlorethamine hydrochloride; megestrol acetate; melengestrol acetate; melphalan; menogaril; mercaptopurine; methotrexate; methotrexate sodium; metoprine; meturedepa; mitindomide; mitocarcin; mitocromin; mitogillin; mitomalcin; mitomycin; mitosper; mitotane; mitoxantrone hydrochloride; mycophenolic acid; nocodazole; nogalamycin; ormaplatin; oxisuran;
pegaspargase; peliomycin; pentamustine; peplomycin sulfate; perfosfamide; pipobroman;
piposulfan; piroxantrone hydrochloride; plicamycin; plomestane; porfimer sodium;
porfiromycin; prednimustine; procarbazine hydrochloride; puromycin; puromycin hydrochloride; pyrazofurin; riboprine; rogletimide; safingol; safingol hydrochloride; semustine; simtrazene; sparfosate sodium; sparsomycin; spirogermanium hydrochloride; spiromustine; spiroplatin; streptonigrin; streptozocin; sulofenur; talisomycin; tecogalan sodium; tegafur; teloxantrone hydrochloride; temoporfin; teniposide; teroxirone; testolactone; thiamiprine; thioguanine;
thiotepa; tiazofurin; tirapazamine; toremifene citrate; trestolone acetate; triciribine phosphate; trimetrexate; trimetrexate glucuronate; triptorelin; tubulozole hydrochloride; uracil mustard; uredepa; vapreotide; verteporfin; vinblastine sulfate; vincristine sulfate; vindesine; vindesine sulfate; vinepidine sulfate; vinglycinate sulfate; vinleurosine sulfate; vinorelbine tartrate;
vinrosidine sulfate; vinzolidine sulfate; vorozole; zeniplatin; zinostatin; zorubicin hydrochloride.
-52-4820v.1
Yet other anti-cancer agents/drugs that can be used in the present teachings include, but are not limited to: 20-epi-l,25 dihydroxy vitamin D3; 5-ethynyl uracil; abiraterone; aclarubicin; acylfulvene; adecypenol; adozelesin; aldesleukin; ALL-TK antagonists; altretamine;
ambamustine; amidox; amifostine; aminolevulinic acid; amrubicin; amsacrine; anagrelide;
anastrozole; andrographolide; angiogenesis inhibitors; antagonist D; antagonist G; antarelix; anti-dorsalizing morphogenetic protein- 1 ; antiandrogen, prostatic carcinoma; antiestrogen;
antineoplaston; antisense oligonucleotides; aphidicolin glycinate; apoptosis gene modulators; apoptosis regulators; apurinic acid; ara-CDP-DL-PTBA; arginine deaminase; asulacrine;
atamestane; atrimustine; axinastatin 1 ; axinastatin 2; axinastatin 3; azasetron; azatoxin;
azatyrosine; baccatin III derivatives; balanol; batimastat; BCR/ABL antagonists; benzochlorins; benzoylstaurosporine; beta lactam derivatives; beta-alethine; betaclamycin B; betulinic acid; bFGF inhibitor; bicalutamide; bisantrene; bisaziridinylspermine; bisnafide; bistratene A;
bizelesin; breflate; bropirimine; budotitane; buthionine sulfoximine; calcipotriol; calphostin C; camptothecin derivatives; canarypox IL-2; capecitabine; carboxamide-amino-triazole;
carboxyamidotriazole; CaRest M3; CARN 700; cartilage derived inhibitor; carzelesin; casein kinase inhibitors (ICOS); castanospermine; cecropin B; cetrorelix; chlorlns; chloroquinoxaline sulfonamide; cicaprost; cis-porphyrin; cladribine; clomifene analogues; clotrimazole;
collismycin A; collismycin B; combretastatin A4; combretastatin analogue; conagenin;
crambescidin 816; crisnatol; cryptophycin 8; cryptophycin A derivatives; curacin A;
cyclopentanthraquinones; cycloplatam; cypemycin; cytarabine ocfosfate; cytolytic factor;
cytostatin; dacliximab; decitabine; dehydrodidemnin B; deslorelin; dexamethasone;
dexifosfamide; dexrazoxane; dexverapamil; diaziquone; didemnin B; didox; diethylnorspermine; dihydro-5-azacytidine; 9- dioxamycin; diphenyl spiromustine; docosanol; dolasetron;
doxifluridine; droloxifene; dronabinol; duocarmycin SA; ebselen; ecomustine; edelfosine;
edrecolomab; eflornithine; elemene; emitefur; epirubicin; epristeride; estramustine analogue; estrogen agonists; estrogen antagonists; etanidazole; etoposide phosphate; exemestane;
fadrozole; fazarabine; fenretinide; filgrastim; finasteride; flavopiridol; flezelastine; fluasterone; fludarabine; fluorodaunomnicin hydrochloride; forfenimex; formestane; fostriecin; fotemustine; gadolinium texaphyrin; gallium nitrate; galocitabine; ganirelix; gelatinase inhibitors;
-53-4820V.1
gemcitabine; glutathione inhibitors; hepsulfam; heregulin; hexamethylene bisacetamide;
hypericin; ibandronic acid; idarubicin; idoxifene; idramantone; ilmofosine; ilomastat;
imidazoacridones; imiquimod; immunostimulant peptides; insulin-like growth factor- 1 receptor inhibitor; interferon agonists; interferons; interleukins; iobenguane; iododoxorubicin; ipomeanol, 4-; iroplact; irsogladine; isobengazole; isohomohalicondrin B; itasetron; jasplakinolide;
kahalalide F; lamellarin-N triacetate; lanreotide; leinamycin; lenograstim; lentinan sulfate;
leptolstatin; letrozole; leukemia inhibiting factor; leukocyte alpha interferon;
leuprolide+estrogen+progesterone; leuprorelin; levamisole; liarozole; linear polyamine analogue; lipophilic disaccharide peptide; lipophilic platinum compounds; lissoclinamide 7; lobaplatin; lombricine; lometrexol; lonidamine; losoxantrone; lovastatin; loxoribine; lurtotecan; lutetium texaphyrin; lysofylline; lytic peptides; maitansine; mannostatin A; marimastat; masoprocol; maspin; matrilysin inhibitors; matrix metalloproteinase inhibitors; menogaril; merbarone;
meterelin; methioninase; metoclopramide; MIF inhibitor; mifepristone; miltefosine; mirimostim; mismatched double stranded RNA; mitoguazone; mitolactol; mitomycin analogues; mitonafide; mitotoxin fibroblast growth factor-saporin; mitoxantrone; mofarotene; molgramostim;
monoclonal antibody, human chorionic gonadotrophin; monophosphoryl lipid A+myobacterium cell wall sk; mopidamol; multiple drug resistance gene inhibitor; multiple tumor suppressor 1- based therapy; mustard anticancer agent; mycaperoxide B; mycobacterial cell wall extract; myriaporone; N-acetyldinaline; N-substituted benzamides; nafarelin; nagrestip;
naloxone+pentazocine; napavin; naphterpin; nartograstim; nedaplatin; nemorubicin; neridronic acid; neutral endopeptidase; nilutamide; nisamycin; nitric oxide modulators; nitroxide antioxidant; nitrullyn; 06-benzylguanine; octreotide; okicenone; oligonucleotides; onapristone; ondansetron; ondansetron; oracin; oral cytokine inducer; ormaplatin; osaterone; oxaliplatin; oxaunomycin; palauamine; palmitoylrhizoxin; pamidronic acid; panaxytriol; panomifene;
parabactin; pazelliptine; pegaspargase; peldesine; pentosan polysulfate sodium; pentostatin; pentrozole; perflubron; perfosfamide; perillyl alcohol; phenazinomycin; phenylacetate;
phosphatase inhibitors; picibanil; pilocarpine hydrochloride; pirarubicin; piritrexim; placetin A; placetin B; plasminogen activator inhibitor; platinum complex; platinum compounds; platinum- triamine complex; porfimer sodium; porfiromycin; prednisone; propyl bis-acridone;
-54-4820V.1
prostaglandin J2; proteasome inhibitors; protein A-based immune modulator; protein kinase C inhibitor; protein kinase C inhibitors, microalgal; protein tyrosine phosphatase inhibitors; purine nucleoside phosphorylase inhibitors; purpurins; pyrazoloacridine; pyridoxylated hemoglobin polyoxyethylene conjugate; raf antagonists; raltitrexed; ramosetron; ras farnesyl protein transferase inhibitors; ras inhibitors; ras-GAP inhibitor; retelliptine demethylated; rhenium Re 186 etidronate; rhizoxin; ribozymes; RII retinamide; rogletimide; rohitukine; romurtide;
roquinimex; rubiginone Bl ; ruboxyl; safingol; saintopin; SarCNU; sarcophytol A; sargramostim; Sdi 1 mimetics; semustine; senescence derived inhibitor 1; sense oligonucleotides; signal transduction inhibitors; signal transduction modulators; single chain antigen-binding protein; sizofiran; sobuzoxane; sodium borocaptate; sodium phenylacetate; solverol; somatomedin binding protein; sonermin; sparfosic acid; spicamycin D; spiromustine; splenopentin;
spongistatin 1 ; squalamine; stem cell inhibitor; stem-cell division inhibitors; stipiamide;
stromelysin inhibitors; sulfinosine; superactive vasoactive intestinal peptide antagonist;
suradista; suramin; s ainsonine; synthetic glycosaminoglycans; tallimustine; tamoxifen methiodide; tauromustine; tazarotene; tecogalan sodium; tegafur; tellurapyrylium; telomerase inhibitors; temoporfin; temozolomide; teniposide; tetrachlorodecaoxide; tetrazomine;
thaliblastine; thiocoraline; thrombopoietin; thrombopoietin mimetic; thymalfasin; thymopoietin receptor agonist; thymotrinan; thyroid stimulating hormone; tin ethyl etiopurpurin; tirapazamine; titanocene bichloride; topsentin; toremifene; totipotent stem cell factor; translation inhibitors; tretinoin; triacetyluridine; triciribine; trimetrexate; triptorelin; tropisetron; turosteride; tyrosine kinase inhibitors; tyrphostins; UBC inhibitors; ubenimex; urogenital sinus-derived growth inhibitory factor; urokinase receptor antagonists; vapreotide; variolin B; vector system, erythrocyte gene therapy; velaresol; veramine; verdins; verteporfm; vinorelbine; vinxaltine; vitaxin; vorozole; zanoterone; zeniplatin; zilascorb; and zinostatin stimalamer. Preferred additional anti-cancer drugs are 5-fluorouracil and leucovorin.
In one embodiment, the anti-cancer agents that can be used in methods described herein are selected from the group consisting of paclitaxel, docetaxel, 5-fluorouracil , trastuzumab, lapatinib, bevacizumab, letrozole, goserelin, tamoxifen, cetuximab, panitumumab, gemcitabine,
-55-4820V.1
capecitabine, irinotecan, oxaliplatin, carboplatin, cisplatin, doxorubicin, epirubicin,
cyclophosphamide, methotrexate, vinblastine, vincristine, melphalan and a combination thereof.
In one embodiment, the anti-cancer agent and the compound represented by Structural Formula (I) are administered contemporaneously. When administered contemporaneously, the anti-cancer agent and the compound can be administered in the same formulation or in different formulations. Alternatively, the compound and the additional anti-cancer agent are administered separately.
In one embodiment, the subject in the methods described herein has not been previously treated with a TTK inhibitor (e.g., the compound represented by Structural Formula (I)).
The term an "effective amount" means an amount when administered to the subject which results in beneficial or desired results, including clinical results, e.g., inhibits, suppresses or reduces the cancer (e.g., as determined by clinical symptoms or the amount of cancer cells) in a subject as compared to a control. Specifically, "treating a subject with a cancer" includes achieving, partially or substantially, one or more of the following: arresting the growth, reducing the extent of a cancer (e.g., reducing size of a tumor), inhibiting the growth rate of a cancer, and ameliorating or improving a clinical symptom or indicator associated with a cancer (such as tissue or serum components) or increasing longevity of the subject.
Generally, an effective amount of a compound taught herein varies depending upon various factors, such as the given drug or compound, the pharmaceutical formulation, the route of administration, the type of disease or disorder, the identity of the subject or host being treated, and the like, but can nevertheless be routinely determined by one skilled in the art. An effective amount of a compound of the present teachings may be readily determined by one of ordinary skill by routine methods known in the art.
In an embodiment, an effective amount of a compound taught herein ranges from about 0.1 to about 1000 mg/kg body weight, alternatively about 1 to about 500 mg/kg body weight, and in another alternative, from about 20 to about 300 mg/kg body weight. In another embodiment, an effective amount of a compound taught herein ranges from about 0.5 to about 5000 mg/m2, alternatively about from 5 to about 2500 mg/m2, and in another alternative from about 50 to about 1000 mg/m2. The skilled artisan will appreciate that certain factors may influence the
-56-4820V.1
dosage required to effectively treat a subject suffering from cancer or reduce the likelihood of recurrence of a cancer. These factors include, but are not limited to, the severity of the disease or disorder, previous treatments, the general health and/or age of the subject and other diseases present.
Moreover, for methods described herein (including treating a subject with a cancer or reducing the likelihood of recurrence of a cancer), a "treatment" or dosing regime of a subject with an effective amount of the compound of the present teachings may consist of a single administration, or alternatively comprise a series of applications. For example, the compound of the present teachings may be administered at least once a week. However, in another embodiment, the compound may be administered to the subject from about one time per week to once daily for a given treatment. The length of the treatment period depends on a variety of factors, such as the severity of the disease, the age of the patient, the concentration and the activity of the compounds of the present teachings, or a combination thereof. It will also be appreciated that the effective dosage of the compound used for the treatment may increase or decrease over the course of a particular treatment regime. Changes in dosage may result and become apparent by standard diagnostic assays known in the art. In some instances, chronic administration may be required.
As used herein, "treatment" is an approach for obtaining beneficial or desired results, including clinical results. Beneficial or desired clinical results can include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e. not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable. "Treatment" can also mean prolonging survival as compared to expected survival if not receiving treatment.
A "subject" is a mammal, preferably a human, but can also be an animal in need of veterinary treatment, e.g., companion animals (e.g., dogs, cats, and the like), farm animals (e.g., cows, sheep, pigs, horses, and the like) and laboratory animals (e.g., rats, mice, guinea pigs, and the like).
-57-4820V.1
The compounds taught herein can be administered to a patient in a variety of forms depending on the selected route of administration, as will be understood by those skilled in the art. The compounds of the present teachings may be administered, for example, by oral, parenteral, buccal, sublingual, nasal, rectal, patch, pump or transdermal administration and the pharmaceutical compositions formulated accordingly. Parenteral administration includes intravenous, intraperitoneal, subcutaneous, intramuscular, transepithelial, nasal, intrapulmonary, intrathecal, rectal and topical modes of administration. Parenteral administration can be by continuous infusion over a selected period of time.
The compounds taught herein can be suitably formulated into pharmaceutical compositions for administration to a subject. The pharmaceutical compositions of the present teachings optionally include one or more pharmaceutically acceptable carriers and/or diluents therefor, such as lactose, starch, cellulose and dextrose. Other excipients, such as flavoring agents; sweeteners; and preservatives, such as methyl, ethyl, propyl and butyl parabens, can also be included. More complete listings of suitable excipients can be found in the Handbook of Pharmaceutical Excipients (5th Ed., Pharmaceutical Press (2005)). A person skilled in the art would know how to prepare formulations suitable for various types of administration routes. Conventional procedures and ingredients for the selection and preparation of suitable formulations are described, for example, in Remington's Pharmaceutical Sciences (2003 - 20th edition) and in The United States Pharmacopeia: The National Formulary (USP 24 NF19) published in 1999. The carriers, diluents and/or excipients are "acceptable" in the sense of being compatible with the other ingredients of the pharmaceutical composition and not deleterious to the recipient thereof.
Typically, for oral therapeutic administration, a compound of the present teachings may be incorporated with excipient and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like.
Typically for parenteral administration, solutions of a compound of the present teachings can generally be prepared in water suitably mixed with a surfactant such as
hydroxypropylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols, DMSO and mixtures thereof with or without alcohol, and in oils. Under ordinary
-58-4820V.1
conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
Typically, for injectable use, sterile aqueous solutions or dispersion of, and sterile powders of, a compound described herein for the extemporaneous preparation of sterile injectable solutions or dispersions are appropriate.
For nasal administration, the compounds of the present teachings can be formulated as aerosols, drops, gels and powders. Aerosol formulations typically comprise a solution or fine suspension of the active substance in a physiologically acceptable aqueous or non-aqueous solvent and are usually presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomizing device. Alternatively, the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve which is intended for disposal after use. Where the dosage form comprises an aerosol dispenser, it will contain a propellant which can be a compressed gas such as compressed air or an organic propellant such as
fluorochlorohydrocarbon. The aerosol dosage forms can also take the form of a pump-atomizer.
For buccal or sublingual administration, the compounds of the present teachings can be formulated with with a carrier such as sugar, acacia, tragacanth, or gelatin and glycerine, as tablets, lozenges or pastilles.
For rectal administration, the compounds described herein can be formulated in the form of suppositories containing a conventional suppository base such as cocoa butter.
Compounds described herein may be prepared using the reaction routes and syntheses described below, employing the techniques available in the art using starting materials that are readily available.
In one general synthetic process, compounds described herein can be prepared according to the following reaction Scheme 1. Halogenation of an appropriately substituted indazole wherein the indazole is substituted as defined herein provides intermediate 1 that can be reacted with a suitable cross coupling partner, ArMet, in the presence of a metal catalyst (e.g. ArBpin /PdCl2(dppf) / Na2C03/ EtOH / PhMe / mw / 120 °C) .
Scheme 1
-59-4820V.1

e.g. Met = BR2"" SnR3"", ZnX, MgX
catalyst = Pd , Ni
Alternatively, haloindazole 2 can be converted into a 3-(trialkylstannyl)-lH-indazole that can be subjected to Stille-type cross-coupling reaction as shown in Scheme 2 (e.g. 1. e&Sn2 / Pd(PPh3)4 / PhMe 2. Arl /Pd(PPh3)4 / Cul / THF ref. WO200102369).
Scheme 2

ROH/triphosgene, RNHS02NHC(=0)CH2CH2C1, RS02C1, RC(=0)R7reducing agent, RC02C1 or RC02H/coupling reagent: TBTU, EDC, DCC, HATU, pyBOP) leading to preparation of substituted anilines, ureas, sulfonamides, sulfamides amides and carbamates.
-60-4820V.1
Scheme 3

RC(=0)R' or RCHO / reducing agent (e.g NaBH(OAc)3)
RNHS02NHC(=0)CH2CH2CI
RC02CI
RG02H / coupling reagent (e.g. TBTU, EDCI, DCC)
Alternatively, compounds described herein, containing trisubstituted indazoles can be 5 prepared as outlined in the following Scheme 4. 5-Nitro-lH-indazole is halogenated with Br2, protected with a suitable indazole protecting group such as tetrahydropyranyl, and subjected to Miyaura-Suzuki cross coupling conditions (e.g. ArBpin /dioxane/H20/ PdChidppf)/ Na2C03). Hydrogenation of the intermediate 1 yields lH-indazol-5-amine 2 that can be modified in a reaction with electrophilic reagents (e.g. R-NCO, R'R"NH/triphosgene, ROH/triphosgene, 1 o RNHS02NHC(=0)CH2CH2C1, RS02C1, RC(=0)R'/reducing agent, RC02C1 or RC02H/coupling reagent: TBTU, EDC, DCC, HATU, pyBOP).
-61-E1 1 1444820V.1
Scheme 4

Compounds described herein having an carboxamide group can be synthesized from 1H- indazole-5-carboxylic acid that can be derived in a two-step sequence outlined below.
Scheme 5

coupling reagent e.g.
DCC, EDC, DIC, HBTU, HATU, HCTU, pyBOP
SOCI2 or (COCI)2 4820V.1
Compounds described herein can be prepared in a manner analogous to the general procedures described above or the detailed procedures described in the examples herein.
The invention is illustrated by the following examples which are not intended to be limiting in any way.
EXEMPLIFICATION
Preparation of Starting Materials
General Methods
Commercially available starting materials, reagents, and solvents were used as received. In general, anhydrous reactions were performed under an inert atmosphere such as nitrogen or argon. PrepPak Rxn CX refers to a commercial cation-exchange resin available from Waters.
Microwave reactions were performed with a Biotage Initiator microwave reactor.
Reaction progress was generally monitored by TLC using Merck silica gel plates with visualization by UV at 254 nm, by analytical HPLC or by LCMS (Bruker Exquire 4000). Flash column chromatographic purification of intermediates or final products was performed using 230-400 mesh silica gel 60 from EMD chemicals. Final products were sometimes purified by preparative reverse-phase HPLC. Purification was performed on a Varian PrepStar model SD-1 HPLC system with a Varian Monochrom lOu C-18 reverse-phase column using a gradient of about 5-30% acetonitrile or MeOH/ 0.05% TFA water to 70-90% acetonitrile or MeOH/0.05% TFA water over a 20-40-min period at a flow rate of 30-50 mlJmin. Proton NMRs were recorded on a Bruker 400 MHz spectrometer, and mass spectra were obtained using a Bruker Esquire 4000 spectrometer. Optical Rotations were measured at the sodium D-line (589.44nM) using an AA-55 Polarimeter from Optical Activity Ltd with a 2.5x100mm unjacketed stainless steel tube at given sample concentrations (c, units of g/lOOmL).
Compound names were generated using the software built into ChemBioDraw Ultra version 11.0.
-63-4820V.1
Abbreviations:
aq aqueous
anh anhydrous
br. broad
calcd calculated
d doublet
DCM dichloromethane
DIPEA diisopropylethylamine
DME 1 ,2-dimefhoxyethane
DMF N,N-dimethylformamide
DMSO dimethyl sulfoxide
dppf 1 , 1 '- B is( dipheny Iphosphino) ferrocene
e.e. enantiomeric excess
h hours
HPLC high performance liquid chromatography
HATU 2-(lH-7-azabenzotriazol-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate
LC-MS liquid chromatography coupled to mass spectrometry min minutes
m multiplet
MS ESI mass spectra, electrospray ionization
NBS N-Bromosuccinimide
NMR nuclear magnetic resonance
O/N overnight
pin pinacol
prep preparative
RBF round bottomed flask
rt room temperature
Rt retention time
-64-44820V.1
s singlet
satd saturated
SPE solid phase extraction
t triplet
TBTU 0-(benzoijiazol- l-yl)-N,N ',N'-tetrarnethyluronium tetrafluoroborate temp. temperature
TFA trifluoroacetic acid
TLC thin layer chromatography
THF tetrahydrofuran
xs excess
Preparation of Startin Materials
General Methods
Method A (amide coupling)
A DMF solution of 3-(5-amino- 1 H-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate ( 1.0 equiv), DIPEA (3 equiv) and RC02H (1.05 equiv) at 0 °C was treated with 0-(benzotriazol-l- yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (1.05 equiv) added in one portion. The reaction was stirred allowing slowly to warm to rt. After several hours or overnight stirring the crude reaction was purified directly by prepHPLC.
Alternatively, a DMF solution of 3-(3-sulfamoylphenyl)- 1 H-indazole-5-carboxylic acid ( 1.0 equiv), DIPEA (3 equiv) and RR'NH (1.05 equiv) at 0 °C or rt was treated with 0-(benzotriazol- l-yl)-N,M N'.N'-tetramethyluronium tetrafluoroborate (1.05 equiv) added in one portion. The reaction was stirred allowing slowly to warm to rt. After several hours or overnight stirring the crude reaction was purified directly by prepHPLC.
Method B (iodination)
To a cooled (0 °C) DMF solution indazole (1.0 equiv) and K2CO3 or KOH (~3 equiv) was added I2 (2-4 equiv) in one portion. The reaction was stirred with cooling or rt for several h and then was treated with xs 10 % aq NaHS03 and subsequently diluted with H20. In the majority of
-65-4820V.1
examples a filtration and washing (¾0) of the precipitate provided the desired material with the required purity.
Method C (Suzuki-Miyaura cross coupling)
A mixture of 3-iodo-lH-indazole (1.0 equiv), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (1.2 equiv), base and palladium catalyst (0.05 equiv) in solvents was degassed with Ar and heated sealed in a Biotage microwave reactor. The crude material after filtration through Celite using MeOH to rinse the pad. In the majority of examples, purification by preparative HPLC provided the target material.

To a mixture of arylboronic acid (5 mmol) and glyoxylic acid monohydrate (460 mg, 5 mmol) in CH2CI2 (25 mL) was added dialkylamine (5 mmol). The resulting mixture was stirred overnight at rt. After evaporation of solvents, it was used as crude or purified by column chromatography.
Synthesis of 3-(5-Formyl-lH-indazol-3-yl)benzenesulfonamide

Aqueous sodium carbonate (0.55 mL, 2 M) was added to a mixture of 3-iodo-lH- indazole-5-carbaldehyde (153 mg, 0.56 mmol), benzenesulfonamide-3-boronic acid pinacol ester (206 mg, 0.73 mmol) and Pd(PPh3)4 (39 mg, 0.034 mmol) in toluene (1.5 mL) and ethanol (1.5 mL) under an atmosphere of argon. The vial was sealed and heated in the microwave at 120°C for 3h. Water (25 mL) was added and the product was extracted with ether (200 mL, then 2x75
-66-4820V.1
mL); the combined organic layer was washed with brine (25 mL) and dried over sodium sulfate. Purification by chromatography (Biotage SNAP25 HP-SIL, 10-50% ethyl acetate in
dichloromethane) yielded a bright yellow solid (90.7mg). The sample was combined with an earlier batch of impure solid (31 mg, estimated 90% pure) and purified by trituration with dichloromethane (3x2 mL) to give a pale yellow solid (63.9 mg, -30%). Ή NMR (400 MHz, DMSO-d6) δ 13.90 (br. s, 1H), 10.10 (s, 1H), 8.77 (s, 1H), 8.48 (s, 1H), 8.31 (d, / =7.6 Hz, 1H), 7.91 (m, 2H), 7.78 (m, 2H), 7.53 (s, 2H).
A. 3-Iodo-lH-indazole-5-carbaldehyde

To a solution of lH-indazole-5-carbaldehyde (315.2 mg, 2.16 mmol), K2C03 (598.8 mg, 4.33 mmol) in DMF (2.5 mL) was added dropwise a solution of I2 (938 mg, 3.7 mmol) in DMF (2.5 mL) and the reaction allowed to stir for three h. An aqueous solution consisting of Na2S204 (51 1 mg) / K2C03 (35 mg) / H20 (3.5 mL) was then added and the solution stirred for 2 h. Water (30 mL) and aqueous sodium hydrogen sulfate (1M, 10 mL) was added and the product was extracted with ethyl acetate (350 mL); this organic layer was washed with brine (3x 25 mL). The aqueous layer was then extracted with dichloromethane (3x 75 mL), this second organic layer was also washed with brine (25 mL). TLC indicated product present in both, so the residues were combined and purified by chromatography (lOg silica SPE tube, Silicycle, 5% ethyl acetate in dichloromethane) to yield a beige solid (203 mg, 35%). A precipitate that formed in the original aqueous layer was collected by vacuum filtration to give after drying a beige solid (first crop 135.6 mg, 23%; second crop 147 mg, 25%). Ή NMR (400 MHz, CDCl} plus a drop of CD3OD) δ 10.03 (s, 1H), 8.00 (s, 1H), 7.95 (d, J =8.8 Hz, 1H), 7.54 (d, J =8.8 Hz, 1H).
-67-4820V.1
Syntheisis of 3-(3-SulfamoylphenvD-lH indazole-5-carboxylic

Methyl 3-iodo-l H-indazole-5-carboxylate was synthesized according to the method described for N-(3-iodo-lH-indazol-6-yl)-2-(thiophen-2-yl)acetamide utilizing methyl 1H- indazole-5-carboxylate (300 mg, 1.70 mmol), K2C03 (704 mg, 5.10 mmol), I2 (864 mg, 3.40 mmol) and DMF (8 mL). Orange solid (415 mg, 81 %). Ή NMR (400 MHz, METHANOLS) δ ppm 8.18 (s, 1 H), 8.07 (d, 7=8.78 Hz, 1 H), 7.58 (d, 7=8.78 Hz, 1 H), 3.95 (s, 3 H); MS ESI [M + H]+ 302.8, calcd for [C9H7IN2O2 + Hf 302.96.
A sealed degassed mixture of methyl 3-iodo-l H-indazole-5-carboxylate (800 mg, 2.65 mmol), benzenesulfonamide-3-boronic acid pinacol ester (896 mg, 3.18 mmol), PddppfC¾ (108 mg, 0.130 mmol), saturated aqueous Na2C03 (2 mL) in toluene / EtOH (8 mL / 8 mL) under argon was heated under microwave irradiation at 125°C for 3 h. The reaction mixture was filtered, diluted with 20 mL of water and then acidified with 2 M HCl. The solid was filtered out and triturated with methanol to give the title compound at 90% purity (780 mg, 93%). Ή NMR (400 MHz, DMSO-d6) δ 13.76 (s, 1 H), 12.95 (bs, 1 H), 8.68 (s, 1 H), 8.44 (s, 1 H), 8.21 (d, 7=8.78 Hz, 1 H), 8.00 (d, 7=9.03 Hz, 1 H), 7.89 (d, 7=8.0 Hz, 1 H), 7.78 (t, 7=7.65 Hz, 1 H), 7.70 (d, 7=9.03 Hz, 1H), 7.52 (s, 2H); MS ESI [M + H]+ 318.0, calcd for [CMHUNSC S + H]+ 318.0.
Synthesis of 3-(3-(methylsulfonyl)phenyl)-lH-indazole-5-carboxylic acid

The title compound was synthesized according to the General Method C, utilizing 3-iodo- lH-indazole-5-carboxylic acid (1.002 g, 3.47 mmol), (3-(methylsulfonyl)phenyl)boronic acid
-68-4820V.1
(891.7 mg, 4.46 mmol), Pd(PPh3)4 (107.0 mg, 0.093 mmol), toluene (7 mL), EtOH (7 mL), and saturated aqueous Na2C03 (3.5 mL). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 6 h. After cooling to room temperature, the mixture was diluted with Et20 (300 mL) and acidified with aqueous HC1 (1M, 25 mL) and water (50 mL).
Undissolved solid was collected by filtration and rinsed with water (10 mL) and 25% EtOH / Et20 (10 mL). The separated Et20 layer was discarded since it contained only traces of product. The wet solid was transferred using a mixture of acetone, THF and DMF, and evaporated in vacuo. MeOH in Et20 (50%, 10 mL) was added and the suspension was sonicated, and filtered. The solid was rinsed with MeOH in Et20 (50%, 10 mL) to provide the title compound as an pale grey solid (946.1 mg, 86 %). Ή NMR (400 MHz, DMSO-d6) 8 ppm 13.79 (s, 1 H), 12.95 (br.s, 1H), 8.68 (s, 1 H), 8.47 (s, 1 H), 8.35 (d, J=8.0 Hz, 1 H), 8.00 (d, J=8.5 Hz, 2 H), 7.86 (t, J=8.0 Hz, 1 H), 7.71 (d, J=9.3 Hz, 1 H), 3.31 (s, 3 H); MS ESI 317.2 [M + H]+, calcd for [Ci5H12N204S + H]+ 317.0. Synthesis of 3-(5-bromo- lH-indazol-3-yl)benzenesulfonamide

The title compound was synthesized according to the General Method C, utilizing 5- bromo-3-iodo-lH-indazole (150.8 mg, 0.47 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (173.8 mg, 0.61 mmol), PdCl2(dppf) DCM (33.8 mg, 0.041 mmol), toluene (5 mL), EtOH (5 mL), and aqueous Na2C03 (0.93 mL, 2 M, 1.86 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h. The product was partitioned between water (20 mL) and EtOAc (200 mL). Solid was still present so the aqueous layer was extracted with ether (200 mL), and the remaining solid was collected by filtration. The combined organic layers were washed with brine (15 mL x 2), dried (Na2S04), filtered and evaporated in vacuo. The residue was combined with the solid and purified by column (Biotage
-69-4820V.1
Isolera 25g HP-SIL plus samplet, 10-50% EtOAc in DCM), followed by trituration with DCM (3 mL x 2) provided the title compound as an yellow solid (74.0 mg, 45 %). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.65 (br. s., 1 H), 8.39 (s, 1 H), 8.30 (s, 1 H), 8.22 (d, J=8.3 Hz, 1 H), 7.85 (d, J=7.5 Hz, 1 H), 7.72 (t, J=7.8 Hz, 1 H), 7.63 (d, J=10.0 Hz, 1 H), 7.56 (d, J=9.3 Hz, 1 H), 7.52 (br. s., 2 H) ; MS ESI 352.0 / 354.0 [M + H]+, calcd for [CuH.oBrNaC^S + H]+ 352.0/354.0.
In a repeat experiment, utilizing 5-bromo-3-iodo- lH-indazole (223 mg, 0.69 mmol), water (10 mL) was added to the cooled reaction mixture and the precipitate was collected, rinsing with water (10 mL x 3). The sticky solid was transferred using acetone and methanol and the solvent was evaporated in vacuo. The aqueous layer was extracted with EtOAc, and found not to contain product. The precipitate was purified by column as above and triturated with DCM / hexane to give a tan solid (44.2mg, 18%).
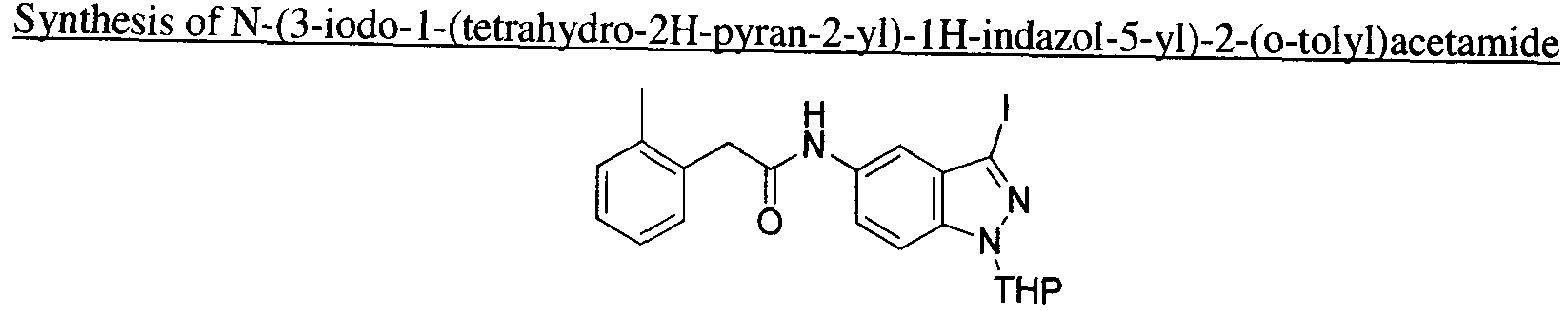
3,4-Dihydro-2H-pyran (0.20 mL, 2.2 mmol) was added to a solution of N-(3-iodo-lH-indazol-5- yl)-2-(o-tolyl)acetamide (249.6 mg, 0.64 mmol) and 4-toluenesulfonic acid monohydrate (16.9 mg, 0.089 mmol) in DCM (3 mL) and the resulting mixture was stirred at rt for 2 h, then partitioned between EtOAc (200 mL) and NaHC03(aq) (30 mL). The organic layer was washed with water (30 mL) and brine (30 mL), dried (Na2S04) and filtered. The solvent was removed in vacuo, and the residue was purified by column (Biotage Isolera 25g HP-SIL plus samplet, 20- 70% EtOAc in hexane) to provide the title compound as a yellow solid (244 mg, 80 %). Ή NMR (400 MHz, CDClj) δ ppm 7.55 (s, 1 H), 7.46 (d, J=9.0 Hz, 1 H), 7.40 (d, J=9.8 Hz, 1 H), 7.29 (br. s., 3 H), 7.19 (br. s., 1 H), 5.64 (d, J=8.8 Hz, 1 H), 3.98 (d, J=l 1.3 Hz, 1 H), 3.78 (s, 2 H), 3.66 - 3.75 (m, 1 H), 2.50 (d, J=8.5 Hz, 1 H), 2.37 (s, 3 H), 2.12 (br. s., 1 H), 2.04 (d, J=l 1.8 Hz, 1 H), 1.77 - 1.50 (m, 3 H).
-70-4820V.1
ynthesis of 2-cvclopropyl-N-(lH-indazol-5-yl)-2-phenylacetamide

The title compound was synthesized according to the method described for N-benzyl-lH- indazole-5-carboxamide utilizing cyclopropylphenylmethylamine · HCl (183 mg, 1 mmol), 1H- indazole-5-carboxylic acid (162 mg, 1 mmol), DIPEA (174 ί, 3 mmol), TBTU (321 mg, 1 mmol) and DMF (4 mL). 20 mL of water was added and the precipitate was filtered and dried to give the title compound: orange solid (154 mg, 53%). MS ESI [M + H]+ 292.3, calcd for
[C18H17N30 + H]+ 292.1 Synthesis of 2-cvclopropyl-N-(3-io - 1 H-indazol-5- yl)-2-phenylacetamide

The title compound was synthesized according to the method described for N-(3-iodo-lH- indazol-6-yl)-2-(thiophen-2-yl)acetamide utilizing 2-cyclopropyl-N-(lH-indazol-5-yl)-2- phenylacetamide (154 mg, 0.53 mmol), K2C03 (219 mg, 1.59 mmol), I2 (269 mg, 1.03 mmol) and DMF (8 mL). A yellow solid (1 19 mg, 46%). MS ESI [M + H]+ 418.13, calcd for
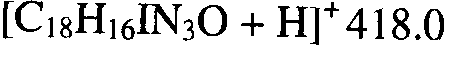
Synthesis of (S)-N-(lH-indazol-5-yl)-2-phenylpropanamide

The title compound was synthesized according to the method described for N-benzyl-lH- indazole-5-carboxamide utilizing (s)-(-)-l-phenylethylamine (150 mg, 1.23 mmol), lH-indazole- 5-carboxylic acid (200 mg, 1.23 mmol), DIPEA (478 μί, 3.70 mmol), TBTU (395 mg, 1.23
-71-4820V.1
mmol), and DMF (4 mL). 20 mL of water was added and the precipitate was filtered and dried to give the title compound: orange solid (275 mg, 84%). Ή NMR (400 MHz, METHANOL-d*) δ ppm 8.79 (d, 7=7.03 Hz, 1 H), 8.35 (s, 1 H), 8.16 (s, 1 H), 7.89 (d, 7=8.78 Hz, 1 H), 7.59 (d, 7=9.79 Hz, 1 H), 7.43 (d, 7=7.00 Hz, 2 H), 7.34 (t, 7=7.40 Hz, 2 H), 7.23 (t, 7=7.00 Hz, 1 H), 5.22 - 5.32 (m, 1 H), 1.59 (d, 7=7.03 Hz, 3 H). MS ESI [M + H]+ 266.1, calcd for [C16Hi5N30 + Hf 266.1

The title compound was synthesized according to the method described for N-(3-iodo-lH- indazol-6-yl)-2-(thiophen-2-yl)acetamide utilizing (S)-N-( 1 H-indazol-5-yl)-2- phenylpropanamide (270 mg, 1.02 mmol), K2C03 (422 mg, 3.06 mmol), I2 (518 mg, 2.04 mmol) and DMF (10 mL). Yellow solid (191 mg, 48%). Ή NMR (400 MHz, METHANOL-d*) 6 ppm 8.07 (s, 1 H), 7.90 - 8.00 (m, 1 H), 7.57 (m, 1 H), 7.42 (d, 7=7.53 Hz, 2 H), 7.33 (t, 7=7.28 Hz, 2 H), 7.23 (t, 7=6.80 Hz, 1 H), 5.23 - 5.31 (m, 1 H), 1.59 (d, 7=6.27 Hz, 3 H). MS ESI [M + H]+ 392.0, calcd for [C^H^O + H]+ 392.0
Synthesis of 3-(5-Amino-6-methyl-l-(tetrahvdro-2H-pyran-2-yl)-lH-indazol-3- vDbenzenesulfonamide

-72-4820V.1

3-bromo-6-methyl-5-nitro-lH-indazole
To a heated at reflux solution of compound 6-methyl-5-nitro-lH-indazole (3.38 g, 19 mmol) in MeOH (60 mL) was added Br2 (3.1 g, 19 mmol) slowly . The mixture was refluxed for 1 h, concentrated, and the residue was washed with aq Na2C03, extracted with CH2C12. The organic layer was washed with brine twice, dried over Na2SC"4, concentrated to give compound 3-bromo-6-methyl-5-nitro-lH-indazole as a yellow solid (3 g, 61 %). Ή NMR (300 MHz, DMSO-d6) δ 13.84 (br. s., 1 H), 8.28 (s, 1 H), 7.63 (s, 1 H), 2.62 (s, 3 H).
3-bromo-6-methyl-5-nitro-l-(tetrahydro-2H-pyran-2-yl)-lH-indazole
A solution of compound 3-bromo-6-methyl-5-nitro-lH-indazole (5.5 g, 21.6 mmol) , TsOH (0.8 g, 4.2 mmol) and 2H-3,4-dihydropyran (2.7 g, 32 mmol) in DCM (100 mL) was stirred at rt for 2 h. After the mixture was concentrated, a saturated aq NaHC03 aq was added thereto, followed by extraction with chloroform. The organic layer was dried over Na2S04, concentrated to give the tilte compound as a yellow solid (4.7 g, 64%).
-73-4820V.1
Ή NMR (300 MHz, CDCh) δ 8.37 (s, 1 H), 7.49 (s, 1 H), 5.68 (dd, 7=8.90, 2.64 Hz, 1 H), 3.90 - 4.07 (m, 1 H), 3.65 - 3.90 (m, 1 H), 2.74 (d, 7=0.82 Hz, 3 H), 2.35 - 2.60 (m, 1 H), 1.93 - 2.24 (m, 2 H), 1.62 - 1.87 (m, 3 H).
3-(5-amino-6-methyl-l-(tetrahydro-2H^yran-2-yl)-lH-indazol-3-yl)benzenesulfonamide
A solution of compound 3-bromo-6-methyl-5-nitro-l-(tetrahydro-2H-pyran-2-yl)-lH- indazole (2 g, 5.9 mmol) and benzenesulfonamide-3-boronic acid pinacol ester (2 g, 7.0 mmol) in dioxane (40 mL) and water (4 mL) was added PdCl2(dppf) (240 mg, 0.3 mmol) and Na2C03 (1.8 g, 17.6 mmol), and the mixture was stirred at 80oC under N2 overnight . The mixture was added water, and extracted with CH2CI2. The organic layer was washed with brine twice, dried over Na2S04, and concentrated. The residue was purified by flash chromatography (S1O2,
CH2Cl2/MeOH 20: 1) to give 3-(6-methyl-5-nitro-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3- yl)benzenesulfonamide as a yellow solid (0.5 g, 20%). The material was taken into MeOH (40 mL) and stirred with Pd/C (50 % H20, 100 mg, 0.05 mmol) at 30 °C under H2 (1 atm) overnight. The hot solution was filtered though a pad of celite and the filtrate was concentrated. The solvent was removed in vacuo to give compound the title compound as a yellow solid (0.4 g, 86%). Ή
NMR (300 MHz, DMSO-d6) 5 8.37 (s, 1 H), 8.08 (d, J=7.79, Hz, 1 H), 7.80 (d, J =7.88 Hz, 1 H), 7.70 (t, J =7.88 Hz, 1 H), 7.44 (s, 3 H), 7.22 (s, 1 H), 5.77 (dd, J=9.70, 2.40 Hz, 1 H), 4.82 (br.s, 2 H), 3.87-3.95 (m, 1H), 3.67 - 3.81 (m, 1 H), 2.26 (s, 3 H), 2.13-1.90 (m, 3 H), 1.52 - 1.60 (m, 2 H).
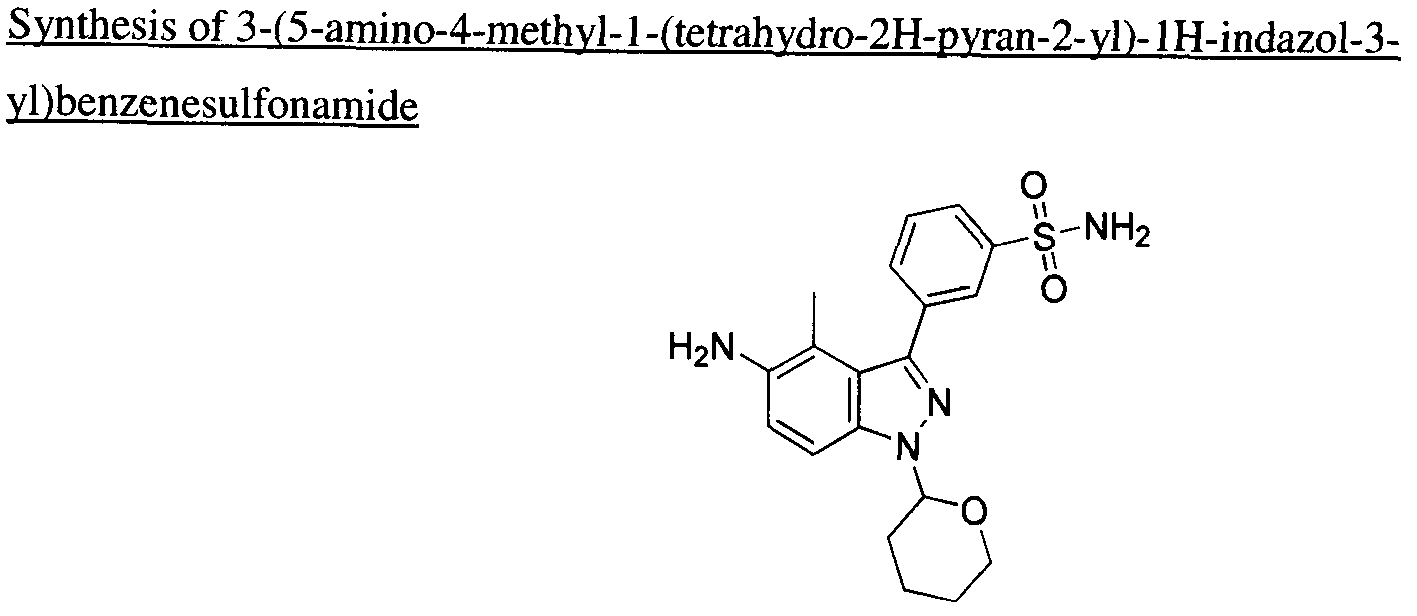
-74-4820V.1

3-bromo-4-methyl-5-nitro-lH-indazole
To a heated at reflux solution of of 4-methyl-5-nitro-lH-indazole (1.77 g, 10 mmol) in MeOH (30 mL) was added Br2 (1.6 g, 10 mmol) slowly. The reaction was refluxed for 1 h, concentrated, and the residue was washed with Na2C03 aq., extracted with (¾(-¾. The organic layer was washed with brine twice, dried over Na2S04, concentrated to give 3-bromo-4-methyl- 5-nitro-lH-indazole as a yellow solid (2.5 g, 97%). Ή NMR (300 MHz, DMSO-d6) δ 7.89 (d, 7=9.09 Hz, 1 H), 7.53 (d, 7=9.09 Hz, 1 H), 2.87 (s, 3 H).
3-bromo-4-methyl-5-nitro-l-(tetrahydro-2H-pyran-2-yl)-lH-indazole
A solution of 3-bromo-4-methyl-5-nitro-lH-indazole (8.2 g, 32 mmol), TsOH (1.2 g, 6.4mmol) and 2H-3,4-dihydropyran (4 g, 48 mmol) in DCM (100 mL) was stirred at rt for 2 h. After the mixture was concentrated, a saturated aq NaHC(¾ was added thereto, followed by extraction with chloroform. The organic layer was dried over Na2S04, concentrated to give the title compound as a yellow solid (8 g, 74%). Ή NMR (300 MHz, DMSO-d6) δ ppm 8.00 (d, 7=9.16 Hz, 1 H), 7.82 (d, 7=9.16 Hz, 1 H), 5.92 (dd, 7=9.44, 2.47 Hz, 1 H), 3.68 - 3.91 (m, 2 H), 2.90 (s, 3 H), 1.92 - 2.07 (m, 2 H), 1.51 - 1.83 (m, 4 H).
-75-4820V.1
3-(4-methyl-5-nitro-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3-yl)benzenesulfonamide
A solution of compound 3-bromo-4-methyl-5-nitro-l-(tetrahydro-2H-pyran-2-yl)-lH-indazole (2 g, 5.9 mmol) and compound benzenesulfonamide-3-boronic acid pinacol ester (2 g, 7.0 mmol) in dioxane (40 mL) and water (4 mL) was added PdCl2(dppf) (240 mg, 0.3 mmol) and
Na2C03(1.8 g, 17.6 mmol), and the mixture was stirred at 80 °C under N2 overnight . The mixture was added water, and extracted with CH2CI2. The organic layer was washed with brine twice, dried over Na2S04, and concentrated. The residue was purified by flash chromatography (Si02, CH2Cl2 MeOH 20: 1) to give the title compound as a yellow solid (1.5 g, 61%). Ή NMR (300 MHz, CDC13) £8.17 (s, 1 H), 7.99 - 8.06 (m, 2 H), 7.79 (d, 7=7.79 Hz, 1 H), 7.64 (t, 7=8.10 Hz, 1 H), 7.57 (d, 7=9.40 Hz, 1 H), 5.78 (dd, 7=9.40, 2.60 Hz, 1 H), 4.96 (br. s., 2 H), 4.02 - 4.09 (m, 1 H), 3.68 - 3.86 (m, 1 H), 2.52 (s, 3 H), 2.06 - 2.24 (m, 2 H), 1.65 - 1.87 (m, 2 H), 1.53 - 1.63 (m, 2 H).
Synthesis of 3-(5-amino-4-methyl-l-(tetrahydro-2H-pyran-2-yl)- lH-indazol-3- vDbenzenesulfonamide
Ή NMR (300 MHz, CDC13) δ 8.16 (s, 1 H), 7.94 (d, 7=7.91 Hz, 1 H), 7.81 (d, 7=7.74 Hz, 1 H), 7.58 (t, 7=7.70 Hz, 1 H), 7.34 (d, 7=8.70 Hz, 1 H), 6.92 (d, 7=8.90 Hz, 1 H), 5.67 (dd, 7=9.64, 2.55 Hz, 1 H), 4.89 (s, 2 H), 4.02 - 4.11 (m, 1 H), 3.67 - 3.80 (m, 1 H), 2.09 (s, 3H), 2.05 - 2.21 (m, 2 H), 1.57 - 1.84 (m, 4 H).

To a solution of chlorosulfonyl isocyanate (0.86 mL, 10 mmol) in CH2C12 (10 mL) at 0 °C was added 2-chloroethanol (0.67 mL, 10 mmol) dropwise. The resulting mixture was stirred for 5 min at rt, then cooled to 0 °C and added dropwise to a round bottom flask charged with 2- ethylaniline (1.24 mL, 10 mmol) and Et3N (1.54 mL, 11 mmol) in CH2C12 (40 mL) at 0 °C. After
-76-4820v.1
addition, the resulting mixture was stirred for 2 h at 0 °C then quenched with H20 (40 mL) and extracted with CH2C12 (40 mL x 2). The combined extracts were washed with H20 (30 mL x 2), dried (Na2SC>4) and concentrated to give a pinkish white solid, which was triturated with H20 (30 mL) to give the title compound as a pinkish white solid (2.76 g, 94%). 'H NMR (400 MHz, DMSO-d6) δ 1 1.52 (s, 1H), 9.75 (s, 1H), 7.30-7.15 (m, 4H), 4.37 (t, J =5.0 Hz, 2H), 3.83 (t, J =4.8 Hz, 2H), 2.69 (q, J =7.6 Hz, 2H), 1.12 (t, J =7.6 Hz, 3H).
Synthesis of 2-Chloroethyl N-(2,6-diethylphenyl)sulfamoylcarbamate

To a solution of chlorosulfonyl isocyanate (2.83 g, 20 mmol) in CH2C12 (10 mL) was added 2- chloroethanol (1.34 mL, 20 mmol) dropwise over 2 min (exothermic). The resulting mixture was stirred for 5 min at it, then cooled to 0 °C and added dropwise to a round bottom flask charged with 2,6-diethylaniline (2.98 g, 20 mmol) and Et3N (3.08 mL, 22 mmol) in CH2C12 (30 mL) at 0 °C. After addition, the resulting mixture was stirred for 2h at 0 °C then quenched with H20 and extracted with CH2C12. The combined extracts were concentrated to give a white solid, which was triturated with H20 (100 mL) to give the title compound as a white solid (6.66 g, 100%) after drying. NMR indicated the presence of trace amount of Et3N-HCl. Ή NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 9.63 (s, 1H), 7.21 (t, / =7.6 Hz, 1H), 7.1 1 (d, J = 8.0 Hz, 2H), 4.37 (t, J = 4.8 Hz, 2 H), 3.84 (t, J =5.0 Hz, 2 H), 2.67 (q, J =7.5 Hz, 2 H), 1.14 (t, / =8.0 Hz, 3 H).
Synthesis of 2-(dimethylamino)-2-phenylacetic acid hydrochloride

A sealed degassed mixture of bromobenzene (200 μί, 1.92 mmol), NN-dimethylglycine ethyl ester (300 ί, 2.1 1 mmol), Pd[P(t-Bu)3]2 (49 mg, 0.096 mmol), K3P04 (937 mg, 4.42 mmol),
-77-4820V.1
toluene (6 mL) under argon was heated at 120 °C for 20 h. The reaction mixture was diluted with 20 mL of Et20, washed with brine, and then dried under reduced pressure to give ethyl 2- (dimethylamino)-2-phenylacetate as a yellow liquid (386 mg, 95%). Ή NMR (400 MHz,
CDCh) δ 7.44 (d, /=7.28 Hz, 2 H), 7.30 - 7.38 (m, 3 H), 4.08 - 4.26 (m, 2 H), 3.85 (s, 1 H), 2.26 (s, 6 H), 1.22 (t, 7=7.15 Hz, 3 H)
A sealed mixture of ethyl 2-(dimethylamino)-2-phenylacetate (130 mg, 0.63 mmol), and 6 M HC1 (2 mL) was heated under microwave irradiation for 120 minutes at 120 °C. The reaction mixture was dried under reduced pressure to give the title compound as a yellow solid (135 mg, 100 %). Ή NMR (400 MHz, CD3OD) δ 7.49 - 7.59 (m, 5 H), 5.11 (s, 1 H), 3.07 (s, 3 H), 2.60 (s, 3 H).
Synthesis of 2-(3.5-dimethoxyphenyl)

A sealed degassed mixture of 3,5-dimefhoxyphenylboronic acid pinacol ester (50 mg, 0.177 mmol), teri-butyl-bromoacetate (22 μί, 0.148 mmol), P(o-tol)3 (4.1 mg, 0.0133 mmol),
Pd2(dba)3 (4.1 mg, 0.0044 mmol), K3PO4 (157 mg, 0.739 mmol) in THF (3 mL) under argon was stirred at room temperature for 16 h. The reaction mixture was diluted with 20 mL of Et20 and then washed with brine. The reaction mixture was dried with MgSC , dried under reduced pressure and purified by flash chromatography to give tert-butyl 2-(3,5-dimethoxyphenyl)acetate as a yellow liquid (18 mg, 48%). Ή NMR (400 MHz, CDCh) δ 6.43 (d, J = 2.0 Hz, 2 H), 6.36 (t, J = 2.3 Hz, 1 H), 3.78 (s, 6 H), 3.45 (s, 2 H), 1.44 (s, 9 H).
teri-Butyl 2-(3,5-dimethoxyphenyl)acetate (18 mg, 0.071 mmol) was dissolved in CH2CI2 (3 mL) and cooled to 0 °C. Triethylsilane (28 μί, 0.18 mmol) was added, followed by trifluoroacetic acid (60 μί, 0.92 mmol) and the mixture was stirred at 0 °C for 3 h. The reaction mixture was dried under reduced pressure to give the title compound as a yellow oil (14 mg, 100%). Ή NMR (400 MHz, CDCh) δ 6.73 (d, / = 2.3 Hz, 2 H), 6.48 (t, J = 2.01 Hz, 1 H), 3.86 (s, 6 H), 3.77 (s, 2 H).
-78-4820V.1
ynthesis of 2-(2-Isopropylphenyl)acetic acid

tert-Butyl 2-(2-isopropylphenyl)acetate was synthesized according to the method described for 2- (2,6-diethylphenyl)acetic acid utilizing 2-isopropylphenylboronic acid (200 mg, 1.2 mmol), tert- butyl-bromoacetate ( 150 pL, 1.0 mmol), P(o-tol)3 (28 mg, 0.091 mmol), Pd2(dba)3 (28 mg, 0.030 mmol), K3PO4 (1.08 g, 5.08 mmol) in THF (5 mL) to give tert-butyl 2-(2- isopropylphenyl)acetate as a yellow solid. Ή NMR (400 MHz, CDC13) δ ppm 7.1 1 - 7.32 (m, 4 H), 3.60 (s, 2 H), 3.09 - 3.18 (m, 1 H), 1.44 (s, 9 H), 1.23 (d, J = 6.8 Hz, 6 H). The title compound was synthesized according to the method described for 2-(2,6-diethylphenyl)acetic acid utilizing tert-butyl 2-(2,6-dimethylphenyl)acetate (44 mg, 0.187 mmol) to give the title compound as a yellow oil (30 mg, 90%). Ή NMR (400 MHz, CDCI3) δ 7.27 - 7.36 (m, 2 H), 7.13 - 7.22 (m, 2 H), 3.74 (s, 2 H), 3.07 - 3.16 (m, 1 H), 1.24 (d, /= 7.0 Hz, 6 H). Synthesis of 2-(2,6-Diethylphenyl)acetic acid

A sealed degassed mixture of 2-bromo- l,3-diethylbenzene (400 mg, 1.89 mmol), tert- butylacetate (280 pL, 2.08 mmol), Pd(dba)2 (54 mg, 0.09 mmol), l ,3-bis(2,6- diisopropylphenyl)imidazolium chloride (40 mg, 0.09 mmol) and 1 M LiHMDS (1 M in hexanes, 4.3 mL, 4.3 mmol) in toluene (8 mL) under argon was stirred at room temperature for 16 h. The reaction mixture was diluted with 20 mL of Et20 and then quenched with saturated aqueous ammonium chloride (20 mL). The reaction mixture was washed with brine, dried with MgS04, and then dried under reduced pressure to give tert-butyl 2-(2,6-diethylphenyl)acetate as
-79-4820V.1
a yellow liquid (322 mg, 69 %). Ή NMR (400 MHz, CDCl3) 5 7.13 (t, J = 7.5 Hz, 1 H),7.08 (d, 7 = 7.5 Hz, 2 H), 3.67 (s, 2 H), 2.68 (q, 7 = 7.5 Hz, 4 H), 1.43 (s, 9 H), 1.23 (t, 7 = 7.5 Hz, 6 H).
A solution of teri-butyl 2-(2,6-diethylphenyl)acetate (161 mg, 0. 65 mmol) in (¾(¾ (5 mL) and cooled to 0 °C. Triethylsilane (255 μί, 1.6 mmol) was added, followed by
trifluoroacetic acid (550 ί, 8.4 mmol) and the mixture was stirred at 0 °C for 3 h. The reaction mixture was dried under reduced pressure to give the title compound as a yellow solid (124 mg, 100 %). Ή NMR (400 MHz, CD3OD) δ 7.13 (t, J = 7.3 Hz, 1 H), 7.04 (d, J = 7.8 Hz, 2 H), 3.74 (s, 2 H), 2.65 (q, 7 = 7.6 Hz, 4 H), 1.19 (t, 7 = 7.5 Hz, 6 H). Synthesis of 2-(2-Ethyl-6-methylphenyl) acid

tert-Butyl 2-(2-ethyl-6-methylphenyl)acetate was synthesized according to the method described for tert-butyl 2-(2,6-diethylphenyl)acetate utilizing l-ethyl-2-iodo-3-methylbenzene (200 mg, 0.813 mmol), terf-butylacetate ( 121 μΐ,, 0.894 mmol), Pd(dba)2 (23 mg, 0.041 mmol), l ,3-Bis(2,6-diisopropylphenyl)-imidazolium chloride (17 mg, 0.041 mmol), and LiHMDS (1 M in hexanes, 1.87 mL, 1.8 mmol) in toluene (6 mL) to give /erf-butyl 2-(2-ethyl-6- methylphenyl)acetate as a yellow liquid (175 mg, 92%). Ή NMR (400 MHz, CDCl3) δ 6.99 - 7.19 (m, 5 H), 3.63 (s, 2 H), 2.76 - 2.85 (m, 2 H), 2.49 (s, 3 H), 1.44 (s, 9 H), 1.22 (t, 7=7.50 Hz, 3 H). The title compound was synthesized according to the method described for 2-(2,6- diethylphenyl)acetic acid utilizing ieri-butyl 2-(2-ethyl-6-methylphenyl)acetate (175 mg, 0.75 mmol) to give the title compound as a yellow solid (127 mg, 95%). Ή NMR (400 MHz, CDCl3) δ 6.99 - 7.20 (m, 3 H), 3.76 (s, 2 H), 2.64 - 2.85 (m, 2 H), 2.49 (s, 3 H), 1.22 (td, 7 = 7.3, 5.3 Hz, 3 H).
-80-4820V.1
ynthesis of 2-(dimethylamino)-2-(2-ethylphenyl)acetic acid hydrochloride

Ethyl 2-(dimethylamino)-2-(2-ethylphenyl)acetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing l-bromo-2- ethylbenzene (239 mg, 1.9 mmol), NN-dimethylglycine ethyl ester (300 L, 2.1 mmol), Pd[P(t- Bu)3]2 (49 mg, 0.096 mmol), K3PO4 (937 mg, 4.42 mmol) and toluene (4 mL) to give ethyl 2- (dimethylamino)-2-(2-ethylphenyl)acetate as a yellow oil (306 mg, 68%).Ή NMR (400 MHz, CDC13) δ ppm 7.60 (d, 7=7.53 Hz, 1 H), 7.15 - 7.25 (m, 3 H), 4.18 (s, 1 H), 4.06 - 4.23 (m, 2 H), 2.80 (q, 7=7.50 Hz, 2 H), 2.27 (s, 6 H), 1.26 (t, 7=7.65 Hz, 3 H), 1.20 (t, 7=7.15 Hz, 3 H). The title compound was synthesized according to the method described for 2-(dimethylamino)-2- phenylacetic acid hydrochloride utilizing ethyl 2-(dimethylamino)-2-(2-ethylphenyl)acetate (55 mg, 0.23 mmol). A yellow solid (54 mg, 95%). Ή NMR (400 MHz, CD3OD) δ 7.50 (d, 7 = 7.8 Hz, 1 H), 7.40 - 7.48 (m, 2 H), 7.36 (t, 7 = 7.3 Hz, 1 H), 5.35 (s, 1 H), 3.20 (s, 3 H), 2.92 - 3.03 (m, 1 H), 2.76 - 2.88 (m, 1 H), 2.60 (s, 3 H), 1.31 (t, 7 = 7.5 Hz, 3 H).
Synthesis of 2-(dimethylamino)-2-(2-ethylphenyl)acetic acid hydrochloride

Ethyl 2-(dimethylamino)-2-o-tolylacetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing l-bromo-2- methylbenzene (300 mg, 1.8 mmol), MN-dimethylglycine ethyl ester (274 pL, 1.9 mmol), Pd[P(t-Bu)3]2 (45 mg, 0.088 mmol), 3P04 (860 mg, 4.1 mmol), and toluene (4 mL) to give ethyl 2-(dimethylamino)-2-o-tolylacetate as a yellow oil (305 mg, 78%). ]H NMR (400 MHz, CDCI3) δ 7.55 (d, 7 = 5.0 Hz, 1 H), 7.13 - 7.23 (m, 3 H), 4.15 (s, 1 H), 4.06 - 4.25 (m, 2 H), 2.44 (s, 3 H), 2.28 (s, 6 H), 1.20 (t, 7 = 7.0 Hz, 3 H). The title compound was synthesized according
-81-4820V.1
to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing ethyl 2-(dimethylamino)-2-o-tolylacetate (250 mg, 1.13 mmol); yellow solid (258 mg, 100%) Ή
NMR (400 MHz, CD3OD) δ 7.32 - 7.48 (m, 4 H), 5.31 (s, 1 H), 3.17 (s, 3 H), 2.60 (s, 3 H), 2.53 (s, 3 H).
Synthesis of tert-Butyl 2-(pyrrolidin- l-vDacetate

Pyrrolidine (1.6 mL, 20 mmol) was dissolved in THF (10 mL) and cooled to 0 °C. tert- butyl bromoacetate (739 μί, 5 mmol) was slowly added to the mixture, which was then stirred at room temperature for 4 h. The reaction mixture was concentrated under reduced pressure and leached using EtOAc. EtOAc was removed by reduced pressure to give the title compound as a yellow liquid (3.20 g, 86 %) Ή NMR (400 MHz, CDCh) δ 3.21 (s, 2 H), 2.61 (br. s., 4 H), 1.78 (br. s., 4 H), 1.45 (s, 9 H). Synthesis of l-(carboxy(phenyl)methyl)pyrrolidinium 2.2.2-trifluoroacetate
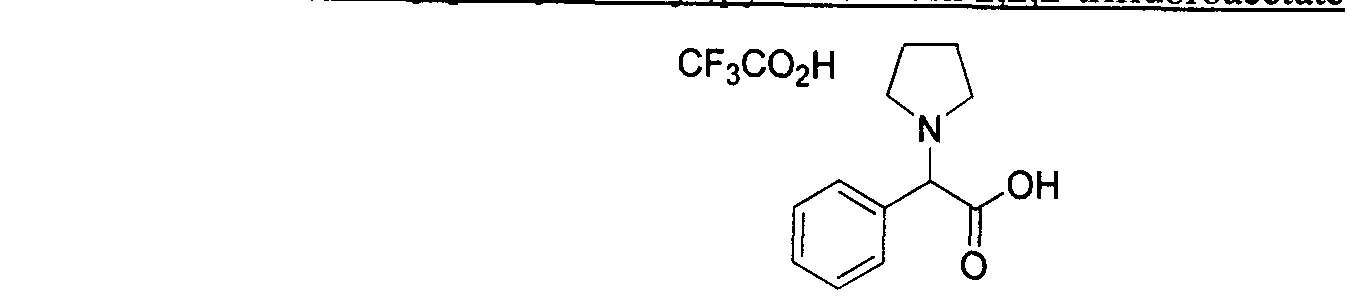
tert-Butyl 2-phenyl-2-(pyrrolidin-l-yl)acetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing bromobenzene (200 μΐ-, 1.9 mmol), ieri-butyl 2-(pyrrolidin-l-yl)acetate (390 ί, 2.1 mmol), Pd[P(t-Bu)3]2 (49 mg, 0.096 mmol), 3P04 (937 mg, 4.42 mmol) and toluene (5 mL) to give ethyl 2-phenyl-2- (pyrrolidin-l-yl)acetate as a brown liquid (480 mg, 96%). Ή NMR (400 MHz, CDCh) δ 7.48 (d, / = 7.5 Hz, 2 H), 7.30 - 7.37 (m, 3 H), 3.83 (s, 1 H), 2.56 - 2.64 (m, 2 H), 2.43 - 2.51 (m, 2 H), 1.77 - 1.85 (m, 4 H), 1.41 (s, 9 H).
The title compound was synthesized according to the method described for 2-(2,6- diethylphenyl)acetic acid utilizing tert-butyl 2-phenyl-2-(pyrrolidin-l-yl)acetate (175 mg, 0.67
-82-4820V.1
mmol) to give the title compound as a yellow solid (223 mg, 100 ). Ή NMR (400 MHz, CDC ) δ 12.68 (br. s, 1 H), 10.84 (br. s., 1 H), 6.95 - 7.81 (m, 5 H), 4.96 (s, 1 H), 3.86 (br. s., 1 H), 3.25 (br. m., 2 H), 2.85 (br. s., 1 H), 1.97 (br. s., 4 H). Synthesis of 2-(dimethylaniino)-2-(2-(trifluoromethyl)phenyl)acetic acid hydrochloride

Ethyl 2-(dimethylamino)-2-(2-(trifluoromethyl)phenyl)acetate was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing 1 - bromo-2-(trifluoromethyl)benzene (430 mg, 1.92 mmol), NN-dimethylglycine ethyl ester (300 2.11 mmol), Pd[P(t-Bu)3]2 (49 mg, 0.096 mmol), K3P04 (937 mg, 4.42 mmol), and toluene
(5 mL) to give ethyl 2-(dimethylamino)-2-(2-(trifluoromethyl)phenyl)acetate as a yellow oil (370 mg, 70 %). Ή NMR (400 MHz, CDC13) 5 ppm 8.02 (d, 7=7.78 Hz, 1 H), 7.62 - 7.75 (m, 3 H), 4.07 - 4.28 (m, 3 H), 2.24 (s, 6 H), 1.28 (t, 7=7.15 Hz, 3 H). The title compound was synthesized according to the method described for 2-(dimethylamino)-2-phenylacetic acid hydrochloride utilizing ethyl 2-(dimethylamino)-2-(2-(trifluoromethyl)phenyl)acetate (125 mg, 0.45 mmol); a brown solid (122 mg, 96%) Ή NMR (400 MHz, CD3OD) δ 7.91-7.98 (m, 2 H), 7.87 (t, 7 = 7.6 Hz, 1 H), 7.79 (t, 7 = 7.5 Hz, 1 H), 5.27 (s, 1 H), 3.25 (br. s., 3 H), 2.62 (br. s., 3 H).
Synthesis of 2-(2,5-Dimethylphenyl)acet

tert-Butyl 2-(2,5-dimethylphenyl)acetate was synthesized according to the method described for tert-butyl 2-(3,5-dimethoxyphenyl)acetate utilizing 2,5-dimethylphenylboronic acid (200 mg, 1.3 mL), ½rf-butyl-bromoacetate (164 μί, 1.1 mmol), P(o-tol)3 (31 mg, 0.10 mmol), Pd2(dba)3 (31 mg, 0.033 mmol), K3P04 (1.18 g, 5.6 mmol) in THF (6 mL) to give tert-butyl 2-
-83-4820v.1
J
(2,5-dimethylphenyDacetate as a yellow oil (272 mg,' 93%). Ή NMR (400 MHz, CDCh) δ 7.03 -7.09 (m, 2 H), 7.00 (s, 1 H), 3.51 (s, 2 H), 2.30 (s, 3 H), 2.26 (s, 3 H), 1.45 (s, 9 H). The title compound was synthesized according to the method described for 2-(2,6-diethylphenyl)acetic acid utilizing im-butyl 2-(2,5-dimethylphenyl)acetate (219 mg, 1.00 mmol). Trituration with hexanes afforded the title compound as an orange solid (50 mg, 31%). Ή NMR (400 MHz, CDCh) δ 7.09 (d, 7=7.28 Hz, 1 H), 7.03 (s, 1 H), 7.02 (d, 7=6.78 Hz, 1 H), 3.65 (s, 2 H), 2.31 (s, 6 H), 2.29 (s, 6 H).
2-(2,6-dimethylphenyl)acetic acid

terf-Butyl 2-(2,6-dimethylphenyl)acetate was synthesized according to the method described for /m-butyl 2-(3,5-dimethoxyphenyl)acetate utilizing 2,6-dimethylphenylboronic acid (200 mg, 1.33 mmol), ieri-butyl-bromoacetate (164 pL, 1.1 1 mmol), P(o-tol)3 (31 mg, 0.100 mmol), Pd2(dba)3 (31 mg, 0.033 mmol), K3P04 (1.18 g, 5.56 mmol) in THF (6 mL) to give the title compound as ayellow liquid. Ή NMR (400 MHz, CD3OD) δ 6.99 (d, 2 H), 6.90 (t, 7 = 7.0 Hz, 1 H), 3.62 (s, 2 H), 2.29 (s, 6 H), 1.42 (s, 9 H). The title compound was synthesized according to the method described for 2-(2,6-diethylphenyl)acetic acid utilizing tert-butyl 2-(2,6- dimethylphenyl)acetate (30 mg, 0.136 mol) to give the title compound as a yellow oil (20 mg, 90 %) Ή NMR (400 MHz, CDCh) δ 7.05 (d, 7 = 6.8 Hz, 2 H), 6.99 (t, 7 = 7.0 Hz, 1 H), 3.74 (s, 2 H), 2.34 (s, 6 H).
Syntheisis of 2-(Pyrrolidin-l-yl)-2-o-tolylacetic acid

Glyoxylic acid■ H20 (460 mg, 5 mmol) and pyrrolidine (413 pL, 5 mmol) was combined with CH2C12 (25 mL) and sonicated for 20 minutes. 2-Methylphenylboronic acid (680 mg, 5
-84-4820V.1
mmol) was added and the mixture was stirred at room temperature for 16 h. The solid was filtered out and dried under reduced pressure to give the title compound as a white solid (906 mg, 83%). Ή NMR (400 MHz, CD3OD) δ 7.61 (d, J =13 Hz, 1 H), 7.19-7.32 (m, 3 H), 4.82 (s, 1 H), 3.20 (br. t, / = 6.0, 1 H), 3.1 1 (br. s., 2 H), 2.53 (s, 3 H), 2.05 (br. s., 4 H), 1.96 (br. t, J = 7.5 Hz, 1 H).
Synthesis of 2-(piperidin-l-yl)-2-o-tolyl

The title compound was synthesized according to the General Method D utilizing glyoxylic acid · H20 (460 mg, 5 mmol), piperidine (493 μ!_, 5 mmol), CH2C12 (25 mL) and 2- methylphenylboronic acid (675 mg, 5 mmol) to give the title compound as a white solid (1.16 g, 100%). Ή NMR (400 MHz, CD3OD) δ 7.66 (d, J = 8.0 Hz, 1 H), 7.27 - 7.30 (m, 3 H), 4.73 (s, 1 H), 3.93 (br. s, 1 H), 2.98 (br. s., 2 H), 2.71 (br. s, 1 H), 2.52 (s, 3 H), 1.42 - 2.02 (br. m., 6 H). Synthesis of ethyl 2-phenylacrylate

A DMF (90 mL) solution of ethyl phenylacetate (1.59 mL, 10 mmol), K2C03 (1.38 g, 10 mmol), and 37% formalin (1.15 mL, 15 mmol) was stirred at 1 10°C for 3 h and then cooled to room temperature. Water was added (400 mL) and the reaction mixture was extracted with Et20 (3 x 175 mL each). The Et20 layers were pooled together, dried with MgS04 and then concentrated under reduced pressure to give the title compound as a yellow liquid (1.45 g, 82%). Ή NMR (400 MHz, CDCl3) δ 7.23 - 7.41 (m, 5 H), 6.31 (s, 1 H), 5.85 (s, 1 H), 4.25 (q, J = 7.0 Hz, 2 H), 1.29 (t, J = 7.0 Hz, 3 H).
-85-4820V.1
Synthesis of 2-phenyl-3-(pyirolidin- l-v0propanoic acid hydrochloride

Ethyl 2-phenylacrylate (1 10 mg, 0.62 mmol) was dissolved in DMF (2 mL) and then pyrrolidine (206 μί, 2.5 mmol) was added. The reaction was stirred for 60 minutes and then diluted with 10 mL of Et20 and washed with water. The ether layer was dried with MgSC and then dried under reduced pressure to give ethyl 2-phenyl-3-(pyrrolidin-l-yl)propanoate as a yellow oil (142 mg, 92%). Ή NMR (400 MHz, CDCl3) δ 7.20 - 7.41 (m, 5 H), 4.03 - 4.24 (m, 2 H), 3.81 (dd, J = 9.8, 4.77 Hz, 1 H), 3.34 (t, J = 10.9 Hz, 1 H), 2.41 - 2.67 (m, 5 H), 1.74 (s, 4 H), 1.22 (t, J = 7.2 Hz, 3 H). MS ESI [M + H]+ 248.1 , calcd for [C5H21NO2 + H]+ 248.2.
The title compound was synthesized according to the method described for 2-
(dimethylamino)-2-phenylacetic acid hydrochloride utilizing ethyl 2-phenyl-3-(pyrrolidin- l- yl)propanoate (80 mg, 0.32 mmol); a yellow solid ( 106 mg, 100%) Ή NMR (400 MHz, CD3OD) δ 7.35 - 7.44 (m, 5 H), 4.16 (t, J = 7.2 Hz, 1 H), 3.96 (dd, J = 12.9, 8.7 Hz, 1 H), 3.68 (m, 1 H), 3.48 - 3.61 (m, 2 H), 3.07 - 3.23 (m, 2 H), 2.09 - 2.17 (m, 2 H), 1.94 - 2.05 (m, 2 H).
Synthesis of 3-morpholino-2-phenylpro acid hydrochloride

Ethyl 3-morpholino-2-phenylpropanoate was synthesized according to the method described for ethyl 2-phenyl-3-(pyrrolidin-l-yl)propanoate utilizing ethyl 2-phenylacrylate (220 mg, 1.25 mmol) and morpholine (435 mL, 5 mmol) to give the title compound as a yellow oil (305 mg, 93%). Ή NMR (400 MHz, CDCl3) δ 7.28 - 7.38 (m, 5 H), 4.07-4.24 (m, 2 H), 3.83 (dd, / = 10.8, 4.8 Hz, 1 H), 3.68 (br. s., 4 H), 3.17 (t, J = 1 1. 7 Hz, 1 H), 2.51-2.64 (m, 3 H), 2.45
-86-4820V.1
(m, / = 5.0 Hz, 2 H), 1.24 (t, J = 7.2 Hz, 3 H). MS ESI [M + H]+ 264.1 , calcd for [Ci5H2iN03 + H]+ 264.2.
The title compound was synthesized according to the method described for 2- (dimethylamino)-2-phenylacetic acid utilizing ethyl 3-morpholino-2-phenylpropanoate
(200 mg, 0.76 mmol); yellow solid (205 mg, 100%). Ή NMR (400 MHz, CD3OD) δ 7.33-7.45 (m, 5 H), 4.25 (dd, 7 = 9.2, 4.1 Hz, 1 H), 3.93 - 4.09 (m, 3 H), 3.80 (t, J = 11.9 Hz, 2 H), 3.49- 3.60 (m, 2 H), 3.45 (dd, J = 12.7, 4.4 Hz, 1 H), 3.15 - 3.28 (m, 2 H).
Synthesis of 2-phenyl-3-(piperidin-l-yl)propanoic acid hydrochloride

Ethyl 2-phenyl-3-(piperidin- l-yl)propanoate was synthesized according to the method described for ethyl 2-phenyl-3-(pyrrolidin- l-yl)propanoate utilizing ethyl 2-phenylacrylate (250 mg, 1.42mol) and piperidine (0.56 mL, 5.68 mmol) to give the title compoundas a yellow oil (348 mg, 94%). Ή NMR (400 MHz, CDCh) δ 7.20 - 7.39 (m, 5 H), 4.03 - 4.24 (m, 2 H), 3.84 (dd, / = 10.2, 4.6 Hz, 1 H), 3.16 (t, J = 1 1.4 Hz, 1 H), 2.52 (m, 3 H), 2.30-2.42 (m, 1 H), 1.53 (br. s., 4 H), 1.40 (m, 2 H), 1.23 (t, / = 7.2 Hz, 3 H).
The title compound was synthesized according to the method described for 2- (dimethylamino)-2-phenylacetic acid hydrochloride utilizing ethyl 2-phenyl-3-(piperidin-l- yl)propanoate (348 mg, 1.33 mmol); yellow solid (347 mg, 97%). Ή NMR (400 MHz, CD3OD) δ ppm 7.30 - 7.45 (m, 5 H), 4.21 (s, 1 H), 3.90 (dd, J = 13.0, 9.5 Hz, 1 H), 3.56 (d, J = 1 1.5 Hz, 2 H), 3.35 (dd, J = 14.6, 3.0 Hz, 1 H), 2.93 - 3.1 1 (m, 2 H), 1.73 - 1.99 (m, 5 H), 1.43 - 1.58 (m, 1 H).
-87-4820v.1
Syntheisis of 1-Phenylcyclopropanamine

A sealed degassed mixture of 1-phenylcyclopropane carboxylic acid (500 mg, 3.09 mmol), triethylamine (429 μί,, 3.09 mmol), and toluene (2 mL) was charged with argon.
Diphenylphosphoryl azide (668 μί, 3.09 mmol) was slowly added and then the reaction mixture was heated to 80 °C and stirred for 120 minutes. ierf-Butanol (586 μί, 6.2 mmol) was added and the reaction was continued to stir at 80 °C for 6 h. The reaction mixture was diluted with Et20 (20 mL) and washed with 0.33 M aq NaOH, dried (MgS04) and concentrated under reduced pressure to give tert-butyl 1-phenylcyclopropylcarbamate as a white solid (448 mg, 62%). This material was dissolved in CH2CI2 (5 mL), cooled to 0 °C and treated with TFA (1 mL). The reaction was stirred at 0 °C for 30 minutes and then at room temperature for 60 minutes. The reaction mixture was then dried under reduced pressure to give the title compound as a white solid (475 mg, 62%). !H NMR (400 MHz, CDCl3) δ 7.95 (br. s., 2 H), 7.28 - 7.49 (m, 5 H), 1.35 (br. s., 2 H), 1.13 (br. s, 2 H).
Synthesis of 2-(2,6-Diethylphenyl)ethan

A solution of tert-butyl 2-(2,6-diethylphenyl)acetate (0.39 g, 1.6 mmol) )(described in synthesis of 2-(2,6-diethylphenyl)acetic acid) in anh THF (24 mL) was treated with LiAlH* (1.0 M in THF, 12 mL, 12 mmol) added dropwise at 0 °C under Ar. The reaction allowed to warm to rt over 1 h and then was stirred overnight. Later the reaction was quenched by slow addition of satd aq Na2C03 at 0 °C, filtered through Celite (EtOAc), diluted with EtOAc, washed (H20, satd NaHC03, brine) and dried (Na2S04). Purification by column chromatography (Si02, 0-30 % EtOAc hexanes) provided the title compound as a pale yellow oil (0.25 g, 88 %). Ή NMR (400
-88- 820v.1
MHz, CDCls) δ 7.14-7.22 (m, 1 H), 7.01 - 7.1 1 (m, 2 H), 3.78 (t, J
Hz, 2H), 2.72 (q, J = 7.4 Hz, 4 H), 1.21 - 1.27 (t, J = 7.5 Hz, 6 H).
Synthesis of 2-(2-(dimethylamino)ethoxy)-2-phenylacetic acid

A solution of KOH (2.6 g, 47.0 mmol) in 2-(dimethylamino)ethanol (1 1 mL, 110 mmol) was added over 45 min to a cooled at 0 °C, stirred solution of benzaldehyde (1.0 g, 9.4 mmol) and bromoform (2.9 g, 11.3 mmol) in 2-(dimethylamino)ethanol (1 mL, 10 mmol). The reaction was later stirred allowing to slowly warm to it. After overnight stirring the reaction was diluted o with H20 and extracted with Et20/EtOAc and then with EtOAc. The remaining aq layer was acidified to pH 5.5, diluted with 1 : 1 H20-brine and extracted with CH2C12 (2x) and EtOAc (lx). The aq layer was concentrated under reduced pressure to yield a viscous oil (13 g). A portion of the resulting material (73 %) was purified by reverse-phase preparative HPLC yielding a TFA salt of the target material as a clear gum 2.03 g (88 %) Ή NMR (400 MHz, METHANOL-di) δ5 ppm'H NMR (400 MHz, ACETONITRILE-<½) δ ppm 3.10 (s, 3 H) 3.25 (s, 3 H) 3.42-3.51 (m., 1 H) 3.92 (m, 1 H) 4.02 (br. s., 2 H) 5.30 (s, 1 H) 7.38 - 7.76 (m, 5 H); MS ESI [M + H]+ 224.0 (100), calcd for [C12H17N03 + H]+ 223.1.
Synthesis of 2-(pyridin-2-yl)-2-(pyrrolidin-l-yl)acetic acid
0

To a cold ( 0 °C) THF (75 mL) solution of ethyl 2-(pyridin-2-yl)acetate (0.20 g, 1.2 mmol) was added DBU (0.36 mL, 2.4 mmol). The reaction was stirred with cooling for 10 min and then 30 min without cooling. CBr4 (0.80 g, 2.4 mmol) in THF (6 mL) over 10 min at -78 °C. Stirring
-89-1 11444820V.1
was continued at the temperature for 10 min and then between -10 and 0 °C for 70 min. The reaction was quenched with satd aq NH4CI. The separated aq layer was extracted with EtOAc (2 x). The organic layers were combined and washed with brine, dried (Na2S04) and concentrated under reduced pressure. The material was later stirred with pyrrolidine ( 4 .2 mL, 51 mmol) at rt overnight. The reaction miture was concentrated under reduced pressure and purified by flash chromatography (Biotage S1O2) using 10-80% EtOAc -hexanes to afford ethyl 2-(pyridin-2-yl)- 2-(pyrrolidin-l-yl)acetate (0.21 g, 72 %) a pale yellow oil. MS ESI [M + H]+ 235.1(100), calcd for [Ci3Hi8N202+ H]+ 235.1.
Ethyl 2-(pyridin-2-yl)-2-(pyrrolidin-l-yl)acetate (100 mg, 0.43 mmol) was stirred with aq NaOH (2 M, 2.5 mL) at rt overnight. The reacton mixture was then cooled ( 0 °C) and acidified with aq 6 M HC1 to pH 4. Concentration under reduced pressure provided 2-(pyridin-2-yl)-2-(pyrrolidin-
1- yl)acetic acid a light beige solid that was taken into DCM with sonication, filtered to afford the
2- (pyridin-2-yl)-2-(pyrrolidin- l-yl)acetic acid hydrochloride as pale yellow gum (59.1 mg, 57 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 2.02 (brs, 4 H), 3.10 - 3.20 (m, 2 H), 3.31-3.43 (m, 2 H), (4.66 (s, 1 H), 7.35 - 7.41 (m, 1 H), 7.60 (d, =7.53 Hz, 1 H), 7.84 (t, 7=7.53 Hz, 1 H), 8.60 (d, J = 4.77 Hz, 1H)
Synthesis of 2-cvclopentyl-2-(pyrrolidin-l-yl)acetic acid

A DCM (10 mL) solution of 2-cyclopentylacetic acid (5.0 g, 39.0 mmol) under N2 was treated DMF (1 drop) and (COCl)2 ( 2 M in DCM, 19.5 mL, 39 mmmol) at rt. The reaction was stirred for 2 h and then diluted with DCM (20 mL) and treated with NBS (8.3 g, 46.8 mmol) followed by HBr (~ 5.7 M in AcOH, 3 drops) at rt. The reaction was heated at reflux overnight then concentrated under reduced pressure. 1 ,2-Dichloroethane (30 mL) was added followed by NBS (3.4 g, 19.2 mmOL) and HBr (~ 5.7 M in AcOH, 5 drops). The reaction mixture was heated at 80 °C for 2.5 h and then cooled to rt. A white precipitate was remove by filtration. The filtrate
-90-4820V.1
was collected into a flask containg cold (0°C) MeOH (65 niL) and stirred for 30 min before it was concentrated under reduced pressure. The residue was taken into three portions of Et20 decanting the liquid each time. The Et20 solutions were combined , washed (H20 2x), dried (Na2S04) and concentrated under reduced pressure. Purification by flash chromatography (100 Si02 Biotage, 0 -10 % EtOAc in hexanes) afforded methyl 2-bromo-2-cyclopentylacetate as vary pale yellow-orange oil (1.62 g, 45 %). An anh MeCN (4 mL) solution of methyl 2-bromo-2- cyclopentylacetate (0.4 g, 1.8 mmol), DIPEA (0.96 mL, 5.5 mmol) and pyrrolidine (0.14 g, 2.0 mmol) was heated sealed with microwave irradiation for 1 h at 150 °C. The reaction was concentrated under reduced pressure then portioned between Et02-DCM and H20. The organic layer was washed (H20, brine), dried (Na2S04) and concentrated under reduced pressure.
Purification by flash chromatography (50 Si02 Biotage, 0 - 100 % EtOAc in hexanes) afforded methyl 2-cyclopentyl-2-(pyrrolidin-l-yl)acetate a pale orange gum (0.1 1 g, 29 %) Ή NMR (400 MHz, ACETONITRILE-i/3) δ ppm 1.46 - 1.91 (m, 10 H), 2.05-2.33 (m, 3 H), 2.56 - 2.65 (m, 2 H), 2.65 - 2.73 (m, 2 H), 3.11 (d, 7=10.54 Hz, 1 H), 3.64 (s, 3 H).
Methyl 2-cyclopentyl-2-(pyrrolidin-l-yl)acetate (0.1 g, 0.5 mmol) was taken into MeOH (10 mL) and stirred with aq NaOH ( 2 M, 7.5 mL, 15 mmol) sealed at 90 °C overnight. Then the reaction was coolded ( 0 °C) and acidified to pH 4-5 with aq HC1 (6 M) and concentrated to dryness. The residue was taken into DCM and 2 % MeOH in DCM with sonication and filtered to afford 2-cyclopentyl-2-(pyrrolidin-l-yl)acetic acid hydrochloride as a lightly tan solid (38 mg, 33 %); MS ESI [M + H]+ 198.1 (100), calcd for [CnHi9N02+ H]+ 198.1.
Synthesis of N-f2-(dimethylamino)ethyl)-3-(4,4,5.5-tetramethyl-l,3,2-dioxaborolan-2- vDbenzenesulfonamide
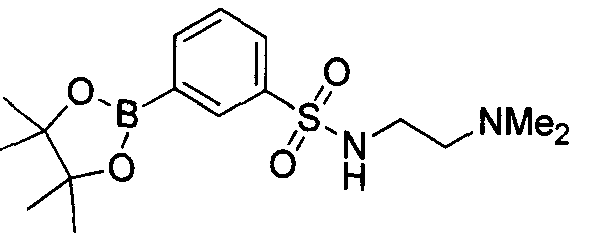
A DCM (5 mL) of 3-bromobenzene-l-sulfonyl chloride (0.5 g, 2.0 mmol) at 0 °C was treated with Et3N (0.28 mL, 2.0 mmol) followed by Nl,Nl-dimethylethane-l,2-diamine (0.17 g, 2.0 mmol). The reaction was stirred with cooling for 1.5 h then dry-loaded on silica gel and purified
-91-4820V.1
by flash chromatography (Si02 Biotage 100 g, 0-20 % MeOH in DCM) to afford 3-bromo-N-(2- (dimethylamino)ethyl)benzenesulfonamide as a clear oil (0.95 g, 90 %). MS ESI [M + H]+ 307.2 (98)/ 308.9 (100), calcd for [C,0Hi5BrN2O2S + H]+ 307.0/309.0.
3-bromo-N-(2-(dimethylamino)ethyl)benzenesulfonamide (0.20 g, 0.65 mmol), 4,4,4',4',5,5,5',5'- octamethyl-2,2'-bi( 1 ,3,2-dioxaborolane) (0.20 g, 0.78 mmol), KOAc (0.19 g, 1.9 mmol) in anh DMF (3 mL) were degassed with Ar, charged with ), PdCl2dppf*CH2Cl2 (23.8 mg, 0.032 mmol) and heated sealed in a microwave reactor at 100 °C for 3 h. The crude DMF solution was used without futher purification. ESI [M + H]+ 355.1, calcd for [C,6H27BN204S+ H]+ 355.2. Synthesis of N,N-dimethyl-3-(2-(methylsulfonyl)-4-(4,4.5.5-tetramethyl-l,3,2-dioxaborolan-2- vDphen vDpropan- 1 -amine

A stirred PhCF3 (5 mL) solution of 4-bromo-2-(methylsulfonyl)benzaldehyde (0.50 g, 1.9 mmol) EtONa (0.26 g, 3.8 mmol) was treated with triethyl phosphonoacetate (0.64 g, 0.57 mmol) at 0 °C. The reaction was warmed to rt and later heated under microwave irradiation for 10 min at 120 °C. After cooling the reaction was partitioned between EtOAc and H20. The organic layer was washed (H20, brine), dried (Na2S04) and concentrated under reduced pressure to provide (E)-ethyl 3-(4-bromo-2-(methylsulfonyl)phenyl)acrylate as white powder (0.67 g),in mixture (70 %w) with triethyl phosphonoacetate. Ή NMR (400 MHz, METHANOL-d4) δ ppm'H NMR (400 MHz, ACETONITRILE-rfj) δ ppm 1.24 (t, 7=7.03 Hz, 3 H), 3.11 (s, 3 H), 4.26 (q, 7=7.19 Hz, 2 H), 6.53 (d, 7=15.81 Hz, 1 H), 7.76 (d, 7=8.28 Hz, 1 H), 7.88 (d, 7=8.53 Hz, 1 H), 8.15 (s, 1 H), 8.40 (d, 7=15.81 Hz, 1 H). ESI [M + H]+ 333.1, 335.1, calcd for [C12H13B1O4S+ H]+
333.0/335.0.
-92-4820V.1
To a solution of (E)-ethyl 3-(4-bromo-2-(methylsulfonyl)phenyl)acrylate (0.47 g, 70 %w, 0.99 mmol) in anh THF (20 mL) was added L1BH4 (108 mg, 4.9 mmol) at 0 °C. The reaction was allowed to warm to rt overnight. Later the reaction was heated at 50 °C for 1 h before it was cooled to rt and quenched with satd aq NH4CI. The mixture was diluted with DCM, the layers were separated. The aq. layer was extracted with DCM and combined organics extracts were washed (brine), dried and concentrated under reduced pressure. Purification by flash chromatography (50 g Si02 Biotage, 0-20 % MeOH in DCM) afforded 3-(4-bromo-2- (methylsulfonyl)phenyl)propan-l-ol as a clear oil (0.23 g, 79 %). ESI [M + H]+ 293.0/295.0, calcd for [Ci0H,3BrO3S + H]+ 293.0/295.0.
This material (230 mg, 0.78 mmol) in DCM ( 5mL) was treated with Dess-Martin periodinane (0.33 g, 0.77 mmol) at rt. The reaction was stirred for 1.5 h, diluted with Et20 and treated with xs aq NaOH (1 M). After stirring for 5 min at rt, the layers were separated. The organic phase was washed (¾0, brine), dried (Na2SC<4) and concentrated under reduced pressure to yield 3-(4- bromo-2-(methylsulfonyl)phenyl)propanal a clear gum (0.19 g, 86 %). Ή NMR (400 MHz, CHLOROFORM- ) δ ppm 2.94 (t, 7=7.15 Hz, 2 H), 3.13 (s, 3 H), 3.25 (t, 7=7.53 Hz, 2 H), 7.28 (d, 7=7.53 Hz, 1 H), 7.68 (d, 7=8.03 Hz, 1 H), 8.17 (s, 1 H), 9.81 (s, 1 H).
A mixture of 3-(4-bromo-2-(methylsulfonyl)phenyl)propanal (89 mg, 0.31 mmol) in DMF (1 mL) and THF ( 5 mL) was treated with Me2NH (2 M in THF, 0.6 mL, 1.2 mmol) and
NaBH(OAc)3 (0.19 g, 0.92 mmol) at rt. Stirred for 1.5 h at rt, concentrated under reduced pressure to remove THF. Purification by preparative HPLC provided 3-(4-bromo-2-
(methylsulfonyl)phenyl)-N,N-dimethylpropan-l -amine 2,2,2-trifluoroacetate as a clear gum (64 mg, 48 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 1.96 - 2.13 (m, 2 H), 2.91 (s, 6 H), 3.06 (t, J = 8.28 Hz, 2 H), 3.21 (s , 3H), 3.17 - 3.27 (m, 2 H), 7.48 (d, 7=8.03 Hz, 1 H), 7.84 (d, 7=8.03 Hz, 1 H), 8.14 (s, 1 H). ESI [M + H]+ 320.2(98)/322.0(100), calcd for [Ci2H,8BrN02S+ Hf 322.0/320.0
A mixture of 3-(4-bromo-2-(methylsulfonyl)phenyl)-N,N-dimethylpropan-l -amine (64 mg, 0.15 mmol), ), 4,4,4',4,,5,5,5',5'-octamethyl-2,2'-bi(l,3,2-dioxaborolane) (45 mg, 0.18 mmol), KOAc (58 mg, 0.59 mmol) in anh DMF (2.5 mL) was degassed with Ar, charged with ,
PdCl2dppf*CH2Cl2 (6 mg, 0.007 mmol) and heated sealed in a microwave reactor at 100 °C for 2
-93-4820v.1
h. The crude DMF solution was used without further purification. ESI [M + H]+ 368.1 (100), calcd for [C18H30BNO4S+ H]+ 368.2.
Synthesis of (E)-2-(3-methoxy-3-phenylprop-l-en-l-yl)-4.4,5,5-tetramethyl-l,3,2-dioxaborolane
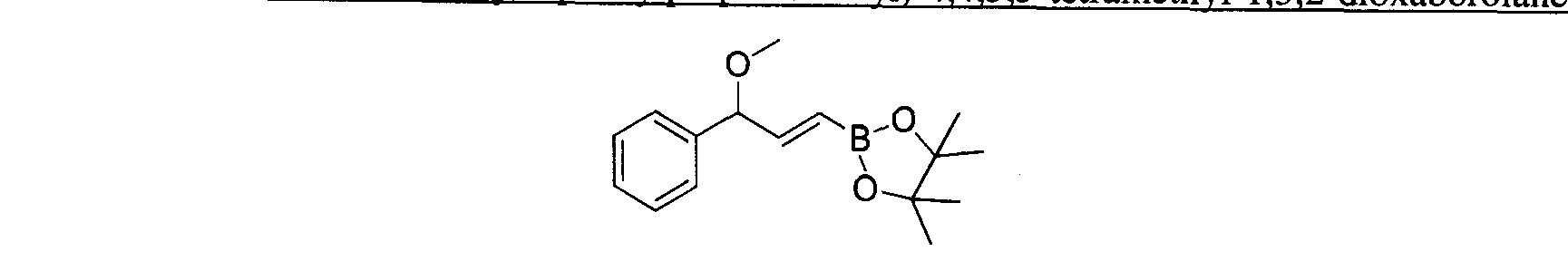
A. (l-methoxyprop-2-yn-l-yl)benzene

l-Phenylprop-2-yn-l-ol (0.37mL, 3.0 mmol) was added to a suspension of NaH (60% wt, 180 mg, 4.5 mmol) in THF (10 mL) under argon atmosphere at 0°C. After 5 min, Me2S04 (0.35 mL, 3.7 mmol) was added and stirring was continued for 2.5 h. The reaction was quenched with water (20 mL) and the product was extracted with Et20 (150 mL). The organic layer was washed with brine (20 mL) and dried (Na2S04). Evaporation of he solvent in vacuo and purification by column (Silicycle lOg SPE, silica, 0-15% DCM in hexane) gave the title compound (396.7 mg, 89 %). Ή NMR (400 MHz, CDCl3) δ ppm 7.53 (d, J=7.3 Hz, 2 H), 7.32 - 7.46 (m, 3 H), 5.11 (s, 1 H), 3.46 (s, 3 H), 2.67 (s, 1 H) .
B. (E)-2-(3-methoxy-3-phenylprop-l- -l-yl)-4,4,5,5-tetramethyl-],3,2-dioxaborolane

4,4,5,5-Tetramethyl- 1 ,3,2-dioxaborolane ( 1.0 mL, 6.9 mmol) was added to an argon- purged solution of (l-methoxyprop-2-yn-l-yl)benzene (374.5 mg, 2.56 mmol) and
HRuCl(CO)(PPh3)3 (55.1 mg, 0.057 mmol) in toluene (10 mL). The resulting mixture was heated
-94-4820v.1
at 50 °C for 21 h. The product was extracted into 50% DCM in hexane (300 mL), and the organic layer was washed sequentially with NaHC03(aq) (30 mL), water (20 mL) and brine (20 mL), dried (Na2SC>4) and evaporated in vacuo. Purification by column (Biotage Isolera, 25g HP-SIL plus samplet, 10-100% DCM in hexane) gave the title compound (337.6 mg, 48% yield). Ή NMR (400 MHz, DMSO-d6) δ ppm 7.39 - 7.29 (m, 5 H), 6.65 (dd, J=18.3, 5.8 Hz, 1 H), 5.69 (d, J=18.1 Hz, 1 H), 4.67 (d, J=6.5 Hz, 1 H), 3.34 (s, 3 H), 1.26 (s, 13 H).
Synthesis of 3-((3-bromophenyl)sulfonyl)-N,N-dimefhylpropan- 1 -amine

Dimethylamine (0.35mL, 2 M in THF, 0.70 mmol) was added to a mixture of l-bromo-3- ((3-bromopropyl)sulfonyl)benzene (50.3 mg, 0.147 mmol) and K2C03 (61.1 mg, 0.44 mmol) in CH3CN (1 mL) under argon atmosphere. In a separate vial, dimethylamine (0.35mL, 2 M in THF, 0.70 mmol) was added to a mixture of l-bromo-3-((3-bromopropyl)sulfonyl)benzene (50.3 mg, 0.147 mmol) and K2C03 (68.1 mg, 0.49 mmol) in DMF (1 mL) under argon atmosphere. The vials were sealed and heated in a reaction block at 70 °C for 16 h. TLC for both reactions was identical, so the reaction mixtures were combined. The product was partitioned between EtOAc (150 mL) and water (20 mL). The organic layer was washed with water (20 mL) and brine (20 mL), dried (Na2S04) and filtered. Evaporation of he solvent in vacuo and purification by column (Silicycle 5g SPE, silica, 0-5% 2 M NH3-MeOH in DCM) gave the title compound
(83.1 mg, 92 %). Ή NMR (400 MHz, CDC ) δ ppm 8.07 (s, 1 H), 7.85 (d, J=7.8 Hz, 1 H), 7.78 (d, J=8.3 Hz, 1 H), 7.45 (t, J=7.9 Hz, 1 H), 3.10 - 3.24 (m, 2 H), 2.32 (s, 2 H), 2.15 (s, 6 H), 1.89
(s, 2 H).
-95-4820V.1
Synthesis of Cvclobutyl(thiophen-3-yl)m

The title compound was synthesized according to the method described for tert-butyl 4- (amino(phenyl)methyl)piperidine-l-carboxylate utilizing cyclobutyl-3-thienyl ketone (500 mg, 3.01 mmol), MeOH (15 mL), NH4OAc (2.78 g, 36 mmol), and NaCNBH3 (759 mg, 12 mmol) to give the title compound: clear oil (468 mg, 93%). MS ESI [M + H]+ 168.2, calcd for [C9H13NS + H]+ 168.1.
Synthesis of cvclopropyl(thiophen-3-yl)m
The title compound was synthesized according to the method described for tert-butyl 4- (amino(phenyl)methyl)piperidine-l-carboxylate utilizing cyclopropyl-3-thienyl ketone (500 mg, 3.29 mmol), MeOH (15 mL), NH4OAc (3.04 g, 40 mmol), and NaCNBH3 (829 mg, 13 mmol) to give the title compound: clear oil (460 mg, 91%). MS ESI [M + H]+ 153.2, calcd for [C8H, ,NS + H]+ 153.1
Synthesis of 2-(2-(l-(tert-butoxycarbonyl)piperidin-3-yl)phenyl)acetic acid

The title compound was synthesized according to the method described for 2-(2-(l-(tert- butoxycarbonyl)piperidin-4-yl)phenyl)acetic acid utilizing methyl 2-(2-(piperidin-3-
-96-4820v.1
yl)phenyl)acetate (146 mg, 0.63 mmol), MeOH (2 mL), 1M NaOH (5 mL), and di-tert-butyl dicarbonate (302 mg, 1.39 mmol). Clear oil (166 mg, 83%). MS ESI [M + H]+ 320.1, calcd for [C,8H25N04 + H]+ 320.2 Synthesis of methyl 2-(2-(piperidin-3-yl

The title compound was synthesized according to the method described for methyl 2-(2- (piperidin-4-yl)phenyl)acetate utilizing methyl 2-(2-(pyridin-3-yl)phenyl)acetate (440 mg, 1.93 mmol), and Pt02 (44 mg, 0.19 mmol) and 20 mL of methanol. Purification using a PoraPak column gave the title compound: clear oil (146 mg, 32%). MS ESI [M + H]+ 234.0, calcd for [C,4H,9N02 + H]+ 234.1
Synthesis of methyl 2-(2-(pyridin-3-yl)p

The title compound was synthesized according to the method described for methyl 2-(2-(pyridin- 4-yl)phenyl)acetate utilizing methyl-2-(2-bromophenyl)acetate (550 mg, 2.4 mmol), 3-pyridine boronic acid (325 mg, 2.64 mmol), 2 M Na2C03 (4 mL), DME (8 mL), and Pd(PPh3)4 (140 mg, 0.12 mmol). Yellow oil (445 mg, 82%). MS ESI [M + H]+ 228.0, calcd for [Ci4H,3N02 + H]+ 228.1
-97-4820V.1
Synthesis of methyl 2-(2-(pyridin-4-yl)p

In a sealed degassed vial, methyl-2-(2-bromophenyl)acetate (550 mg, 2.4 mmol), 4-pyridine boronic acid (325 mg, 2.64 mmol), 2 M Na2C03 (4 mL), DME (8 mL), Pd(PPh3)4 (140 mg, 0.12 mmol) was combined. The vial was charged with argon and then placed in the microwave reactor, heating at 120 °C for 20 minutes. Reaction was diluted with 10 mL of EtOAc and then washed with brine. The mixture was dried with MgS04 and then by reduced pressure. The title compound was obtained after purification by flash chromatography (100% hexanes to 50% EtOAc/hexanes): yellow oil (330 mg, 60%). MS ESI [M + H]+ 228.0, calcd for [Ci4H,3N02 + H]+ 228.1
Synthesis of methyl 2-(2-(piperidin-4-yl

In a pressurized chamber, methyl 2-(2-(pyridin-4-yl)phenyl)acetate (227 mg, 1 mmol), and PtCh (23 mg, 0.1 mmol) was combined in 10 mL of methanol. 2 drops of concentrated HCl was added and the chamber was pressurized to 100 psi and stirred overnight at room temperature. The mixture was then filtered and the filtrate was concentrated under reduced pressure and purified using a PoraPak column to give to the title compound: yellow oil (153 mg, 66%). MS ESI [M + H]+ 234.0, calcd for [Ci4H19N02 + H]+ 234.1
-98-4820v.1

Methyl 2-(2-(piperidin-4-yl)phenyl)acetate (153 mg, 0.66 mmol) was dissolved in 2 mL of MeOH and then 1M NaOH (5 mL) was added. The reaction was stirred at 25 °C for 30 minutes and then di-tert-butyl dicarbonate (289 mg, 1.32 mmol) was added. The mixture was stirred at 25 °C for 2 days and then concentrated by reduced pressure. Mixture was diluted with d¾0 (10 mL), and extracted with EtOAc (10 mL). The organic layer was discarded and the aqueous was acidified with 1 M HC1, extracted three times with EtOAc (3 x 15 mL) and dried with MgS04. Solvent was removed under reduced pressure to give the title compound: clear oil (82 mg, 39%). MS ESI [M + H]+ 320.1, calcd for [C18H25N04 + H]+ 320.2.
Synthesis of dicvclohexylmethanamine

The title compound was synthesized according to the method described for tert-butyl 4- (amino(phenyl)methyl)piperidine-l-carboxylate utilizing dicyclohexylketone (970 mg, 5 mmol), MeOH (40 mL), NH4OAc (4.62 g, 60 mmol), and NaCNBH3 (1.26 g, 20 mmol) to give the title compound: clear oil (887 mg, 91%). MS ESI [M + H]+ 196.1 , calcd for [Ci3H25N + H]+ 196.2
-99-4820V.1
Synthesis of cvclohexyKphenvDmethana

The title compound was synthesized according to the method described for tert-butyl 4- (amino(phenyl)methyl)piperidine-l-carboxylate utilizing cyclohexyl phenyl ketone (940 mg, 5 mmol), MeOH (40mL), NH4OAc (4.62 g, 60 mmol), and NaCNBH3 ( 1.26 mg, 20 mmol) to give the title compound: clear oil (868 mg, 92%). Ή NMR (400 MHz, CHLOROFORM-d) δ ppm 7.30 - 7.37 (m, 2 H), 7.22 - 7.29 (m, 3 H), 3.61 (d, .7=7.78 Hz, 1 H), 2.77 (br. s., 2 H), 1.96 (d, 7=12.80 Hz, 1 H), 1.78 (d, 7=13.05 Hz, 1 H), 1.64 (d, 7=7.03 Hz, 2 H), 1.51 - 1.60 (m, 1 H), 1.38 (d, 7=12.30 Hz, 1 H), 1.18 - 1.31 (m, 1 H), 1.12 (dd, 7=18.32, 8.78 Hz, 3 H), 1.00 (dd, 7=24.09, 12.05 Hz, 1 H), 0.85 (q, 7=11.88 Hz, 1 H)
Synthesis of 2-(dimethylaminoV2-(thiophen-3-yl)acetic acid

The title compound was synthesized according to the method described for 2-(pyrrolidin-l-yl)-2- o-tolylacetic acid utilizing glyoxylic acid · H20 (460 mg, 5 mmol), 2 M dimethylamine in THF (2.5 mL, 5 mmol), CH2CI2 (25 mL) and thiophene-3-boronic acid (640 mg, 5 mmol) to give the title compound; yellow oil (744 mg, 84%). Ή NMR (400 MHz, METHANOL-d4) δ ppm 7.67 (br. s., 1 H), 7.55 (br. s., 1 H), 7.25 (d, 7=5.27 Hz, 1 H), 4.64 (s, 1 H), 2.75 (br. s., 6 H).
-100-4820V.1
Synthesis of (l-methylpiperidin-4-yl)(phenyl)methanone

In a microwave vial, 4-benzylpiperidine■ HCl (650 mg, 2.89 mmol) was dissolved in formic acid (3 mL) and then formalin (937 μί, 11.6 mmol) was added. The vial was sealed and placed in microwave reactor and heated at 150 °C for 5 minutes. LC-MS was checked and then solvent was removed under reduced pressure. The residue was partitioned between EtOAC (20 mL) and 0.5 M NaOH (30 mL), washed with brine and then dried using MgS04 and under reduced pressure to give the title compound: clear oil. (520 mg, 89%). Ή NMR (400 MHz, METHANOL- d4) δ ppm 7.97 - 8.06 (m, 2 H), 7.65 (t, 7=7.50 Hz, 1 H), 7.54 (t, 7=7.30 Hz, 2 H), 3.69 - 3.81 (m, 1 H), 3.61 (d, 7=1 1.54 Hz, 2 H), 3.18 (t, 7=13.55 Hz, 2 H), 2.92 (s, 3 H), 2.16 (d, 7=14.81 Hz, 2 H), 1.92 (q, 7=13.55 Hz, 2 H)
Synthesis of (l-methylpiperidin-4-yl)(phenyl)methanamine

-101-4820V.1
Synthesis of 2-(4-methylpiperazin-l-yl)- acid

The title compound was synthesized according to the method described for 2-(4- (hydroxymethyl)piperidin-l-yl)-2-phenylacetic acid, utilizing 1-methyl piperizine (1.0 g, 10 mmol), and THF (5 mL) to give the title compound: white solid (394 mg, 24%). MS ESI [M + Hf 235.0, calcd for [Ci3H18N202 + H]+ 235.1
Synthesis of 2-(4-hydroxypiperidin-l-yl)-2-phenylacetic acid

4-Hydroxypiperidine · HCl (1.04 g, 7.5 mmol) was dissolved in THF (5 mL) and cooled to 0 °C. Na2C03 (1.59 g, 15 mmol) was added, followed by the dropwise addition of (methyl-a- bromophenylacetate (1.15 g, 5 mmol), and then the reaction mixture was warmed to 25 °C. The mixture was stirred at 25 °C overnight. Completion of the reaction was monitored by LC-MS. Saturated aqueous sodium bicarbonate (20 mL) was then added, and the reaction was extracted three times with EtOAc (15 mL). The reaction was dried with MgSCu, filtered, and dried under reduced pressure. The residue was dissolved in 0.5 mL of MeOH and 1M NaOH (4 mL) was added. The mixture was stirred at 25 °C for 16 hours, at which time it was acidified using 1M HCl. Mixture was extracted with EtOAc and dried with MgS04, and then by reduced pressure to give the title compound: clear oil 1.13 g, 96%). MS ESI [M + H]+ 236.0, calcd for [C13H17NO3 + H]+ 236.1
-102-4820V.1
Synthesis of 2-(4-(hvdroxymethyl)piperidin-l-yl)-2-phenylacetic acid

4-piperidine methanol (1.15 g, 10 mmol) was dissolved in THF (5 mL) and cooled to 0 °C. Methyl-a-bromophenylacetate (572 mg, 2.5 mmol) was added dropwise and then the reaction mixture was warmed to 25 °C. The mixture was stirred at 25 °C overnight. Completion of the reaction was monitored by LC-MS. The solvent was removed under reduced pressure, redissolved in saturated aqueous sodium bicarbonate (50 mL), and extracted three times with EtOAc (20 mL). The reaction was dried with MgS0 , filtered, and dried under reduced pressure. The residue was dissolved in 0.5 mL of MeOH and 1M NaOH (4 mL) was added. The mixture was stirred at 25 °C for 16 hours, at which time it was acidified using 1M HC1. Mixture was extracted with EtOAc and dried with MgSC>4, and then by reduced pressure to give the title compound: clear oil (392 mg, 63%). MS ESI [M + H]+ 250.1, calcd for [Ci4H19N03 + H]+ 250.3
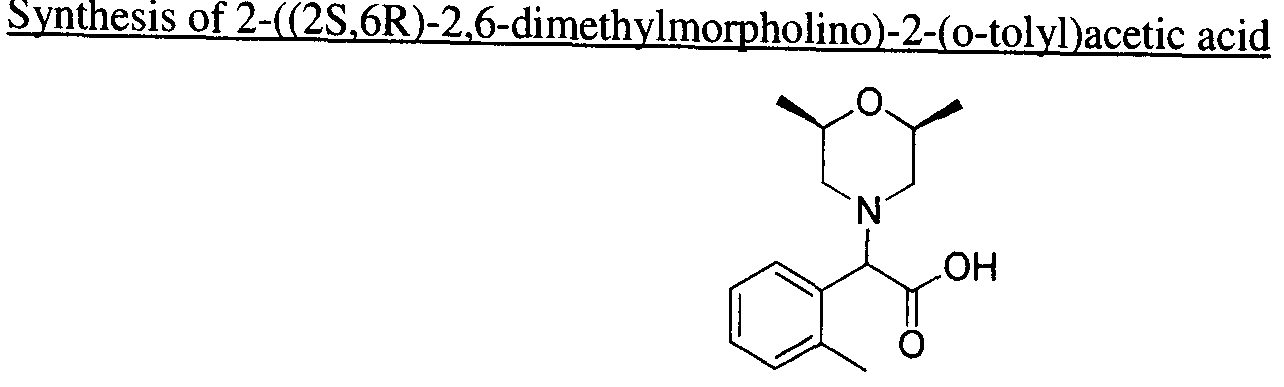
The title compound was synthesized according to the method described for 2-(pyrrolidin-l-yl)-2- o-tolylacetic acid utilizing glyoxylic acid · H20 (230 mg, 2.5 mmol), cis-2,6-dimethylmorpholine (288 mg, 2.5 mmol), CH2C12 (20 mL) and 2-methoxyphenylboronic acid (238 mg, 2.5 mmol) to give the title compound; white solid (380 mg, 58%). Ή NMR (400 MHz, METHANOLS) δ ppm 7.65 (d, 7=6.27 Hz, 1 H), 7.20 - 7.31 (m, 3 H), 4.58 - 4.65 (m, 1 H), 3.91 - 4.01 (m, 1 H), 3.77 - 3.88 (m, 1 H), 3.68 (d, 7=11.54 Hz, 1 H), 2.60 - 2.73 (m, 2 H), 2.50 (s, 3 H), 2.26 - 2.37 (m, 1 H), 1.22 (d, 7=6.02 Hz, 3 H), 1.06 (d, 7=6.27 Hz, 3 H)
-103-4820V.1
ynthesis of 2-(dimethylar no)-2-(2-methoxyphenyl)acetic acid

The title compound was synthesized according to the method described for 2-(pyrrolidin-l-yi)-2- o-tolylacetic acid utilizing glyoxylic acid · H20 (460 mg, 5 mmol), 2 M dimethylamine in THF (2.5 mL, 5 mmol), CH2CI2 (25 mL) and 2-methoxyphenylboronic acid (760 mg, 5 mmol) to give the title compound; clear oil (792 mg, 76%). Ή NMR (400 MHz, METHANOL-d4) 5 ppm 7.40 - 7.52 (m, 2 H), 7.10 (d, 7=8.03 Hz, 1 H), 7.00 - 7.06 (m, 1 H), 4.90 (s, 1 H), 3.90 (s, 3 H), 2.76 (br. s., 6 H)
Synthesis of tert-butyl 4-(amino(phenyl)mefhyl)piperidine-l-carboxylate

½r/-butyl 4-benzoylpiperidine-l-carboxylate (484 mg, 1.67 mmol) was dissolved in MeOH (15mL) and then NH4OAc (1.54 g, 20 mmol) was added. Reaction was stirred for 10 minutes at 25 °C. NaCNBH3 (420 mg, 6.68 mmol) was then added and the reaction was heated to 60 °C and stirred at that temperature for 16 hours. The solvent was removed under reduced pressure and the reaction mixture was suspended in 0.5 M NaOH (75 mL). Extracted with EtOAc (3 x 20 mL) and then dried with MgS04 and then under reduced pressure to give the title compound: clear oil (435 mg, 90%) Ή NMR (400 MHz, CHLOROFORM-d) δ ppm 7.33 (m, .7=7.30 Hz, 2 H), 7.28 (d, 7=4.77 Hz, 3 H), 4.19 (br. s, 1 H), 4.02 (br. s, 1 H), 3.62 (d, 7=7.78 Hz, 1 H), 2.62 - 2.74 (m, 1 H), 2.48 - 2.61 (m, 1 H), 1.93 (d, 7=12.55 Hz, 1 H), 1.50 - 1.69 (m, 4 H), 1.44 (s, 9 H), 1.25 - 1.33 (m, 1 H), 1.00 - 1.14 (m, 1 H)
-104-4820V.1
Synthesis of tert-butyl 4-benzoylpiperidi -carboxylate

4-Benzoylpiperidine · HC1 (400 mg, 1.78 mmol) was dissolved in acetonitrile (8 mL).
Triethylamine (1.25 mL, 8.9 mmol) and di-tert-butyl dicarbonate (465 mg, 2.13 mmol) was added and the reaction was stirred at 25 °C for 16 hours. Concentrated reaction mixture under reduced pressure and partitioned between EtOAc (40 mL) and 0.5 M HC1 (20 mL). Discarded aqueous layer and then washed with saturated aqueous sodium bicarbonate (20 mL) followed by brine (20 mL). Dried with MgSC and then under reduced pressure to give the title compound; white solid (484 mg, 94%) Ή NMR (400 MHz, CHLOROFORM-d) δ ppm 7.94 (d, 7=7.53 Hz, 2 H), 7.58 (t, 7=7.00 Hz, 1 H), 7.48 (t, 7=7.50 Hz, 2 H), 4.17 (br. s, 2 H), 3.35 - 3.47 (m, 1 H), 2.79 - 2.99 (m, 2 H), 1.78 - 1.91 (m, 2 H), 1.65 - 1.78 (m, 2 H), 1.47 (s, 9 H)
Preparation of Exemplary Compounds of the Invention
Example Al . 3-(lH-Indazol-3-yl)benzenesulfonamide

The title compound was synthesized according to the General Method C, utilizing iodo- lH-indazole (Sinova Inc., 30 mg, 0.12 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (42 mg, 0.15 mmol), K2C03 (34 mg, 0.25 mmol), DME (1 mL), EtOH (1 mL), H20 (0.5 mL) and Pd(PPh3)4 (14 mg, 0.012 mmol). The degassed solution was sealed and
-105-4820V.1
heated in a microwave reactor at 120 °C for 1 h. Purification by preparative HPLC provided the title compound as a white solid (21 mg, 63 %). Ή NMR (400 MHz, CD3OD) δ 8.51 (s, 1 H), 8.20 (d, J = 7.8 Hz, 1 H) 8.09 (d, J = 8.3 Hz, 1 H), 7.94 (d, J = 8.0 Hz, 1 H), 7.70 (t, J = 7.8 Hz, 1 H), 7.60 (d, J = 8.5 Hz, 1 H), 7.46 (t, J = 7.4 Hz, 1 H), 7.27 (t, J = 7.2 Hz, 1 H); MS ESI 274.0 [M + H]+, calcd for [C,3H, IN302S + H]+ 274.1.

A. 5-Phenyl-l H-indazole

The title compound was synthesized according to the General Method C, utilizing bromobenzene (50 mg, 0.0.32 mmol), tert-butyl 5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-
1 H-indazole- 1-carboxylate (Ryan Scientific, 1 10 mg, 0.32 mmol), Cs2C03 (291 mg, 0.89 mmol), DME (3 mL), H20 (0.75 mL) and Pd(PPh3)4 ( 17 mg, 0.014 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 1.5 h. The reaction mixture was concentrated and purified by preparative TLC (Si02, 10 % MeOH/DCM). The material was later taken into DCM (6 mL) and treated with TFA (0.5 mL). After stirring for 2.5 h at rt the reaction mixture was concentrated under reduced pressure to provide the tiltle material as a white solid (59 mg, 60 ). Ή NMR (400 MHz, CDCl3) δ 13.08 (brs, 1H), 8.52 (s, 1 H), 8.07 (s, 1 H), 8.00 (dd, 7 = 1.5 Hz, 9.0 Hz, 1 H), 7.90 (d, J = 9.0 Hz , 1 H), 7.62 - 7.67 (m, 2 H), 7.49 - 7.55 (m,
2 H), 7.44 (t, / = 7.5 Hz, 1 H),; MS ESI 194.9 [M + H]+, calcd for [CI 3H10N2 + Hf 195.0.
-106-4820V.1
B. 3-Iodo-5-phenyl-l H-indazole

The title compound was synthesized according to the General Method B, utilizing 5- phenyl-1 H-indazole (59 mg, 0.30 mmol ), I2 (97 mg, 0.38 mmol) and KOH (51 mg, 0.91 mmol) in DMF (2 mL). An off-white solid (81 mg, 83 %). Ή NMR (400 MHz, CD3OD/CDCI3) δ 7.67 (dd, J = 8.7, 1.6 Hz, 1 H), 7.59-7.63 (m, 3 H), 7.52 (dd, J = 8.7, 0.6 Hz, 1 H), 7.40-7.46 (m, 2 H), 7.29 - 7.35 (m, 1 H); MS ESI 320.9 [M + H]+, calcd for [C13H9IN2 + H]+ 321.0.
3-(5-Phenyl-lH-indazol-3-yl)benzenesulfonamide
The title compound was synthesized according to the General Method C, utilizing 3-iodo- 5-phenyl- l H-indazole (40 mg, 0.12 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (53 mg, 0.19 mmol), Cs2C03 (122 mg, 0.37 mmol), DMF (1.6 mL), H20 (0.4 mL) and Pd(PPh3)4 (7 mg, 0.006 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 1.5 h. Purification by preparative HPLC provided the title compound as a white solid (10 mg, 23 %). Ή NMR (400 MHz, CD3OD) δ 8.54 (s, 1 H), 8.23 (d, J = 7.8 Hz, 1 H), 8.20 (s, 1 H), 7.96 (d, J = 7.5 Hz, 1 H), 7.71 - 7.77 (m, 2 H), 7.65 - 7.71 (m, 3 H), 7.46 (m, 2 H), 7.34 (t, J = 7.3 Hz, 1 H); MS ESI 350.0 [M + H]+, calcd for
 + H]+ 350.1.
+ H]+ 350.1.

The title compound was synthesized according to the General Method C, utilizing 3-iodo- 5-methoxy- l H-indazole (Sinova Inc., 40 mg, 0.15 mmol), 3-(4,4,5,5-tetramethyl- 1,3,2-
- 107-4820V.1
dioxaborolan-2-yl)benzenesulfonamide (54 mg, 0.19 mmol), CS2CO3 (143 mg, 0.44 mmol), DMF (lmL), H20 (0.25 mL) and Pd(PPh3)4 (8 mg, 0.007 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 1.5 h. Purification by preparative HPLC provided the title compound as a white solid (12 mg, 27 %). Ή NMR (400 MHz, METHANOL- d ) δ ppm 8.46 (s, 1 H), 8.15 (d, 7=7.78 Hz, 1 H), 7.93 (d, 7=7.78 Hz, 1 H), 7.70 (d, 7=8.03 Hz 1 H), 7.50 (d, 7=9.03 Hz, 1 H), 7.38 (d, 7=1.76 Hz, 1 H), 7.13 (dd, 7=9.03, 2.26 Hz, 1 H), 3.89 (s, 3 H); MS ESI 304.0 [M + H]+, calcd for [C14H13N3O3S + H]+ 304.1.
Example A4. N-(3-(3-Sulfamoylphenyl)-lH-indazol-5-yl)acetamide

A DMF (2 mL) solution of Ac20 (82 mg, 0.80 mmol) N/V-dimethylpyridin-4-amine (5.5 mg, 0.045 mmol) and DIPEA (0.26 mL, 1.5 mmol) was treated lH-indazol-5-amine (0.10 g, 0.75 mmol) added in one portion at 0 °C. The reaction was stirred with cooling for 3 h then treated with H2O (10 mL) and concentrated under reduced pressure and then quenched by addition of xs H20. Trituration with DCM provided the title compound as a red solid (0.11 g, 84 %). Ή NMR (400 MHz, DMSO-d6) δ ppm 12.94 (br. s., 5 H), 9.91 (s, 1 H), 8.11 (s, 1 H), 8.00 (s, 1 H), 7.45 (d, 7=9.00 Hz, 1 H), 7.37 (d, 7=8.53 Hz, 1 H), 2.05 (s, 3 H); MS ESI 175.8 [M + H]+, calcd for [C9H9N30 + H]+ 176.1.
-108-44820V.1
B. N-( 3-Iodo- lH-indazol-5-yl )acetamide

The title compound was synthesized according to the General Method B, utilizing N- ( lH-indazol-5-yl)acetamide (105 mg, 0.60 mmol ), I2 (304 mg, 1.2 mmol) and K2C03 (330 mg, 2.4 mmol) in DMF (2 mL). A red solid (124 mg, 69 %). Ή NMR (400 MHz, DMS0-d6) δ ppm 13.41 (s, 1 H), 10.03 (s, 1 H), 7.89 (s, 1 H), 7.42 - 7.50 (m, 2 H), 2.06 (s, 3 H).
C. ( 3-( 3-Sulfamoylphenyl)-lH-indazol-5-yl)acetamide
The title compound was synthesized according to the General Method B, utilizing N-(3- iodo-lH-indazol-5-yl)acetamide (62 mg, 0.20 mmol), 3-(4,4,5,5-tetramefhyl-l,3,2-dioxaborolan- 2-yl)benzenesulfonamide (73 mg, 0.26 mmol), KF (23 mg, 0.40 mmol), DMF (1.5mL), H20 (0.37 mL) and Pd(PPh3)4 (12 mg, 0.010 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h. Purification by preparative HPLC provided the title compound as an off-white solid (29 mg, 44 %). Ή NMR (400 MHz, METHANOLS) δ ppm 10.03 (s, 1 H), 8.47 (s, 1 H), 8.35 (br. s., 1 H), 8.16 (d, 7=7.03 Hz, 1 H), 7.94 (d, 7=8.28 Hz, 1 H), 7.70 (t, 7=7.78 Hz, 1 H), 7.56 (d, 7=8.50 Hz, 1 H), 7.51 (d, 7=9.00 Hz, 1 H), 2.17 (s, 3 H); MS ESI 331.2 [M + H]+, calcd for [C15H14N4O3S + H]+ 331.2.

-109-44820V.1
5-ylcarbamate

A DMF (15 mL) solution of aminoindazole (1.0 g, 7.5 mmol) and DIPEA (2.0 mL, 1 1 mmol) was treated with B0C2O (1.7 g, 7.7 mmol) (50 % added in one portion as a solid, and 50 % as a solution in anh DMF (1 mL)) at 0 °C. The reaction was stirred with the cooling for 1.5 h and then allowed to warm slowly to rt overnight. Later it was diluted with ¾0 to ~ 100 mL. A tan precipitate was collected by filtration, washed with ¾0 and dried to afford the product (2) as a light tan solid (1.7 g, 94 %). Ή NMR (400 MHz, DMSO-d6) δ ppm 12.90 (s., 1 H), 9.27 (s, 1 H), 7.94 (s, 1 H), 7.87 (br.s, 1 H), 7.41 (d, /=8.80 Hz,l H), 7.33 (d, 7=9.2 Hz, 1 H), 1.47 (s, 9H). MS ESI 233.9 [M + H]+, calcd for [Ci2H15N302 + H]+ 234.0 . indazol-5-ylcarbamate

To a cooled (0 0 C) DMF (30 mL) solution t-butyl lH-indazol-5-ylcarbamate (1.6 g, 6.9 mmol) and 2CO3 (3.8 g, 27.6 mmol) was added I2 (1.8 g, 7.1 mmol) in one portion.
The reaction was stirred with cooling for 3 h and then treated with 10 % aq NaHS03 (50 mL) and subsequently with H2O ( 150 mL). A filtration and washing (¾0) of the ppt provided crude material which after purification by flash chromatography (S1O2, 70 g, 0 to 6 % MeOH in DCM) and recrystallization from EtOAc/hexanes yielded tert-butyl 3-iodo- lH-indazol-5-ylcarbamate (3) as an off-white solid ( 1.9 g, 78 %). Ή NMR (400 MHz, Acetone-d6) δ ppm 12.54 (br. s., 1 H), 8.51 (br. s., 1 H), 7.87 (br.s., 1 H), 7.24 - 7.66 (m, 2 H), 1.51 (s, 9 H); MS ESI [M + H]+ 359.9 (100) ]+, calcd for [C2H14IN3O2 + H]+ 360.0 .
44820V.1
C. tert-butyl 3-(3-sulfamoylphenyl)-lH-indazol-5-ylcarbamate
t-Butyl 3-iodo-lH-indazol-5-ylcarbamate (100 mg, 0.28 mmol), 3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)benzenesulfonamide (118 mg, 0.42 mmol) and KF (50 mg, 0.86 mmol) in DMF (2 mL) and H20 (0.7 mL) were degassed by evacuation and refill with Ax (3x).
Pd(PPh3)4 (16 mg, 0.014 mmol) was then added and the degassing was repeated. The reaction mixture was heated (sealed, microwave reactor) at 120 °C for 1.5 h. The crude material was concentrated under reduced pressure and then purified by flash chromatography (S1O2, 50 g, 0 to 10 % MeOH in DCM) to provide tert-butyl 3-(3-sulfamoylphenyl)-lH-indazol-5-ylcarbamate as a white solid (76 mg, 70 %). Ή NMR (400 MHz, CD3OD) δ ppm 8.48 (s, 1 H), 8.21-8.14 (m, 2 H), 7.94 (d, 7 =8.0 Hz, 1 H), 7.72 (t, 7 =7.8 Hz, 1 H), 7.52 (d, 7=9.2 Hz,l H), 7.42 (d, 7=9.2 Hz, 1 H), 1.55 (s, 9H); MS ESI [M + H]+ 389.2 (100), ]+, calcd for [C18H20N4O4S + H]+ 389.2.
Example A6. N-(3-(3-Sulfamoylphenyl)- lH-indazol-5-yl)thiophene-2-carboxamide

-(lH-lndazol-5-yl)thiophene-2-carboxamide

A DMF (5 mL) solution of lH-indazol-5-amine (0.67 g, 5.0 mmol) and Et3N (0.84 mL, 6.0 mmol) an 0°C was treated dropwise with thiophene-2-carbonyl chloride (0.53 mL, 5.0 mmol) in DMF ( 1 mL). The reaction was stirred with cooling for 60 min and then quenched by addition of xs H20. The collected by filtration solid was washed with H2O to provide the title compound as a yellow solid (0.73 g, 57 %). Ή NMR (400 MHz, METHANOL-d*) δ ppm 8.11 (s, 1 H), 8.04 (s, 1 H), 7.92 (d, 7=3.51 Hz, 1 H), 7.73 (d, 7=5.02 Hz, 1 H), 7.61 (d, 7=8.50 Hz, 1
-1 11-4820V.1
H), 7.55 (d, 7=9.00 Hz, 1 H), 7.20 (t, 7=5.00 Hz, 1 H); MS ESI 243.9 [M + H]\ calcd for
[C12H9N3OS + H]+ 244.0. -lodo-lH-indazol-5-yl)thiophene-2-carboxamide

The title compound was synthesized according to the General Method B, utilizing N N- (lH-indazol-5-yl)thiophene-2-carboxamide (0.75 g, 3.1 mmol ), (1.6 g, 6.2 mmol) and K2CO3 (1.3 g, 9.3 mmol) in DMF (11 mL). An off-white solid (0.31 g, 27 %). Ή NMR (400 MHz, METHANOL-dA) δ ppm 7.94 (d, 7=3.76 Hz, 1 H), 7.91 (s, 1 H), 7.75 (d, 7=4.77 Hz, 1 H), 7.71 (dd, 7=2.01, 9.03 Hz, 1 H), 7.53 (d, 7=9.03 Hz, 1 H), 7.21 (t, 7=4.39 Hz, 1 H); MS ESI 369.9 [M + H]+, calcd for [Ci2H8IN3OS + H]+ 370.0.
N-( 3-( 3-Sulfamoylphenyl)-lH-indazol-5-yl)thiophene-2-carboxamide
The title compound was synthesized according to the General Method C, utilizing N-(3- iodo-lH-indazol-5-yl)thiophene-2-carboxamide (63 mg, 0.17 mmol), 3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)benzenesulfonamide (58 mg, 0.20 mmol), CS2CO3 (167 mg, 0.51 mmol), DMF (1.6 mL), H20 (0.4 mL) and Pd(PPh3)4 (10 mg, 0.008 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h. Purification by column chromatography (Si02, 3-10 % MeOH/DCM) provided the title compound as a yellow solid (4.6 mg, 7 Ή NMR (400 MHz, METHANOLS) δ ppm 8.51 (s, 1 H), 8.44 (s, 1 H), 8.20 (d, 7=8.53 Hz, 1 H), 7.93 - 7.97 (m, 2 H), 7.68 - 7.76 (m, 3 H), 7.61 (d, 7=9.00 Hz, 1 H), 7.21 (t, 7=4.50 Hz, 1 H); MS ESI 399.1 [M + H]+, calcd for [C18H14N4O3S2+ H]+ 399.1.
-112-4820V.1

-( lH-Indazol-5-yl)thiophene-3-carboxamide

A DMF (5 mL) solution of lH-indazol-5-amine (0.33 g, 2.5 mmol) and Et3N (0.42 mL,
3.0 mmol) an 0 °C was treated dropwise with thiophene-2-carbonyl chloride (0.36 g, 2.5 mmol) in DMF (1 mL). The reaction was stirred with cooling for 60 min and then quenched by addition of xs H20. The solid collected by filtration was washed with H20 to provide the title compound as a brown solid (0.55 g, 90 ). Ή NMR (400 MHz, METHANOL-dA) δ ppm 8.24 (d, 7=1.76 Hz, 1 H), 8.13 (s, 1 H), 8.04 (s, 1 H), 7.66 (d, 7=5.02 Hz, 1 H), 7.62 (d, 7=9.50 Hz, 1 H), 7.52 - 7.56 (m, 2 H); MS ESI 243.9 [M + H]\ calcd for [Ci2H9N3OS + H]+ 244.0.
B. N-( 3-Iodo-lH-indazol-5-yl )thiophene-3-carboxamide

The title compound was synthesized according to the General Method B, utilizing N N-
(lH-indazol-5-yl)thiophene-3-carboxamide (0.24 g, 1.0 mmol ), (0.51 g, 2.0 mmol) and K2C03 (0.41 g, 3.0 mmol) in DMF (5 mL). A light brown solid (0.20 g, 54 %). Ή NMR (400 MHz, METHANOLS) δ ppm 8.26 (br. s., 1 H), 7.94 (s, 1 H), 7.70 (d, 7=9.03 Hz, 1 H), 7.67 (d, 7=5.02 Hz, 1 H), 7.50 - 7.57 (m, 2 H); MS ESI 369.9 [M + H]+, calcd for [CnHglNaOS + H]+ 369.9.
-1 13-4820V.1
C. N-(3-(3-S lfamoylphenyl)-lH-indazol-5-yl)thiophene-3-carboxamide
The title compound was synthesized according to the General Method C, utilizing N-(3- iodo-lH-indazol-5-yl)thiophene-2-carboxamide (74 mg, 0.20 mmol), 3-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)benzenesulfonamide (68 mg, 0.24 mmol), Cs2C03 (0.20 g, 0.60 mmol), DMF (1.6 mL), H20 (0.4 mL) and Pd(PPh3)4 (12 mg, 0.010 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 1 h. Purification by preparative HPLC provided the title compound as a light tan solid (20 mg, 25 %). Ή NMR (400 MHz, DMF-dT) δ ppm 13.55 (br. s., 1 H) 10.34 (s, 1 H), 8.66 (s, 1 H), 8.61 (s, 1 H), 8.48 (dd, 7=1.25 Hz, 2.75 Hz, 1 H), 8.26 (d, 7=7.78 Hz, 1 H), 7.98-7.91 (m, 2 H), 7.77 - 7.83 (m, 2 H), 7.74-7.69 (m, 2 H), 7.53 (s, 2 H); MS ESI 399.1 [M + H]+, calcd for [C18H14N403S2 + H]+ 399.1.

-( 1 H-Indazol-5-yl)-2-( thiophen-2-yl)acetamide

A DMF (5 mL) solution of lH-indazol-5-amine (0.67 g, 5.0 mmol) and Et3N (0.84 mL,
6.0 mmol) an 0 °C was treated dropwise with 2-(thiophen-2-yl)acetyl chloride (0.62 mL g, 5.0 mmol) in DMF ( 1 mL). The reaction was stirred with cooling for 60 min and then quenched by addition of xs H20. The collected by filtration solid was washed with H20 and then purified by column chromatography (Si02, 50-100 % EtOAc/hexanes) to provide the title compound as a light brown solid (0.73 g, 57 %). Ή NMR (400 MHz, METHANOLS) 6 ppm 8.07 (s, 1 H),
8.01 (s, 1 H), 7.50 (d, 7=9.03 Hz, 1 H), 7.47 (d, 7=8.78 Hz, 1 H), 7.30 (d, 7=5.02 Hz, 1 H), 7.02
-1 14-4820V.1
- 7.05 (m, 1 H), 6.99 (t, 7=4.10 Hz, 1 H), 3.92 (s, 2 H); MS ESI 257.9 [M + H]+, calcd for [Ci3H,iN3OS + H]+ 258.1. -(3-Iodo-lH-indazol-5-yl)-2-(thiophen-2-yl)acetamide

The title compound was synthesized according to the General Method B, utilizing N-(1H- indazol-5-yl)-2-(thiophen-2-yl)acetamide (0.51 g, 2.0 mmol ), (1.0 g, 3.9 mmol) and K2CO3 (0.83 g, 6.0 mmol) in DMF (7 mL). A gray-yellow solid (0.32 g, 42 %). Ή NMR (400 MHz, METHAN OL-di δ ppm 7.86 (s, 1 H), 7.52 (d, 7= 8.41 Hz, 1 H), 7.50 (d d, 7= 8.78 Hz, 1 H), 7.31 (d, 7= 4.99 Hz, 1 H), 7.02 - 7.05 (m, 1 H), 6.99 (t, 7=4.10 Hz, 1 H), 3.94 (s, 2 H); MS ESI 384.0 [M + H]+, calcd for [C13H10IN3OS + H]+ 384.0.
N-(3-(3-Sulfamoylphenyl)-lH-ind zol-5-yl)-2-(thiophen-2-yl)acetamide
The title compound was synthesized according to the General Method C, utilizing N-(5- iodo-lH-indazol-5-yl)-2-(thiophen-2-yl)acetamide (100 mg, 0.26 mmol), 3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)benzenesulfonamide (96 mg, 0.34 mmol), KF (45 mg, 0.78 mmol), DMF (4 mL), H20 (1 mL) and Pd(PPh3)4 (15 mg, 0.013 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h. Purification by preparative HPLC followed by preparative TLC (Si02, 10 % MeOH/DCM) and trituration (DCM, 2 % MeOH, DCM) provided the title compound as an off-white solid (35 mg, 32 Ή NMR (400 MHz, METHANOL^) δ ppm 8.47 (s, 1 H), 8.40 (s, 1 H), 8.16 (d, 7=7.53 Hz, 1 H), 7.94 (d, 7=8.03 Hz, 1 H), 7.71 (t, 7=7.80 Hz, 1 H), 7.55 (d, 7=6.78 Hz, 2 H), 7.30 (d, 7=4.52 Hz, 1 H), 7.05 (d., 7=3.26 Hz, 1 H), 6.99 (dd, 7=3.63 Hz, 4.77 Hz 1 H), 3.95 (s, 3 H); MS ESI 413.2 [M + H]+, calcd for [C9H16N4O3S2 + H]+ 413.2.
-1 15-4820V.1

-( 1 H-Indazol-5-y I Nicotinamide

The title compound was synthesized according to the General Method A utilizing 1H- indazol-5-amine (0.15 g, 1.1 mmol), nicotinic acid (0.14 g, 1.1 mmol), N-ethyl-N- isopropylpropan-2-amine (0.29 mL, 1.7 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'- tetramethyluronium tetrafluoroborate (0.38 g, 1.2 mmol) in DMF (4 mL). Purification by column chromatography (Si02, 10-30 % MeOH/DCM) followed by trituration with DCM provided the title compound as a grey-light tan solid (0.16 g, 60 %). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.04 (br. s., 1 H), 10.46 (s, 1 H), 9.12 (d, 7=1.51 Hz, 1 H), 8.76 (d, 7=5.02 Hz, 1 H), 8.31 (d, 7=7.78 Hz, 1 H), 8.25 (s, 1 H), 8.07 (s, 1 H), 7.62 (dd, 7=1.51, 8.78 Hz, 1 H), 7.51 - 7.60 (m, 2 H) ; MS ESI 238.9 [M + H]+, calcd for [Ci3H10N4O + H]+ 239.1. -( 3-lodo-lH-indazol-5-yl)nicotinamide

The title compound was synthesized according to General Method A
utilizing 1 N-(lH-indazol-5-yl)nicotinamide (0.058 g, 0.24 mmol ), I2 (0.080 g, 0.31 mmol) and K2CO3 (0.080 g, 0.58 mmol) in DMF (1.5 mL). A tan solid (0.043 g, 49 %). Ή NMR (400 MHz, METHANOLS) 8 ppm 9.13 (s, 1 H), 8.75 (d, 7=4.77 Hz, 1 H), 8.41 (d, 7=8.03 Hz, 1 H),
-1 16-44820v.1
8.00 (s, 1 H), 7.72 (d, 7=8.78 Hz, 1 H), 7.62 (dd, 7=8.28, 4.52 Hz, 1 H), 7.55 (d, 7=8.78 Hz, 1 H); MS ESI 364.9 [M + H]+, calcd for [C13H9IN4O + H]+ 365.0.
C. N-(3-(3-sulfamoylphenyl)-]H-indazol-5-yl)nicotinamide
The title compound was synthesized according to General Method C utilizing N-(3-iodo-lH- indazol-5-yl)nicotinamide (46 mg, 0.13 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzenesulfonamide (43 mg, 0.15 mmol), KF (15 mg, 0.25 mmol), DMF (1.25 mL), H20 (0.37 mL) and Pd(PPh3)4 (7 mg, 0.006 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 1.5 h. Purification by preparative HPLC provided the title compound as a white solid (23 mg, 36 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 9.24 (s, 1 H), 8.85 (d, 7=5.52 Hz, 1 H), 8.67 (d, 7=7.78 Hz, 1 H), 8.52 (br. s., 2 H), 8.20 (d, 7=7.28 Hz, 1 H), 7.95 (d, 7=7.03 Hz, 1 H), 7.84 (dd, 7=8.53, 5.77 Hz, 1 H), 7.68 - 7.77 (m, 2 H), 7.64 (d, 7=9.29 Hz, 1 H); MS ESI 394.1 [M + H]+, calcd for [Ci9HisNs03S + H]+ 394.1.
Example A10: 3-(5-Amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate

A suspension of tert-butyl 3-(3-sulfamoylphenyl)-lH-indazol-5-ylcarbamate (0.56 g, 1.4 mmol) in DCM (30 mL) was treated with TFA (10 mL) at 0 °C. The reaction mixture was stirred for 2 h at 0 °C and then at rt for 1 h. Concentration under reduced pressure and drying in vacuo afforded the title compound as a light beige solid (0.58 g, quant). 1H NMR (400 MHz,
METHANOLS) d ppm 8.47 (s, 1 H), 8.17 (d, J = 8.28 Hz, 1 H), 8.02 (s, 1 H), 7.98 (d, J=7.53 Hz, 1 H), 7.70 - 7.78 (m, 2 H), 7.42 (d, J=9.29 Hz, 1 H). MS ESI [M + H]+ 288.9, calcd for [Ci3H|2N402S + H]+ 289.1.
-1 17-4820V.1

A DMF (2 mL) solution of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate (39 mg, 0.098 mmol) and N-ethyl-N-isopropylpropan-2-amine (0.05 mL, 0.3 mmol) was treated witha DMF (0.5 mL) solution of (isocyanatomethyl)benzene (13 mg, 0.10 mmol) at 0 °C. The reaction mixture was stirred for 3 h before being stirring at rt overnight. The reaction was quenched by an addition of MeN¾ (2 M in MeOH, 0.3 mL, 0.6 mmol) at rt. The solvents were removed under reduced pressure. The residue was taken into MeOH (4 mL) and stirred with MeONa (25 wt in MeOH, 0.1 mL, 0.4 mmol) at 50-60 oC overnight. Purification by preparative HPLC provided the title compound as a white powder (8.2 mg, 20 ). Ή NMR (400 MHz, METHANOL-^) 5 ppm 8.48 (s, 1 H), 8.17 (br. s., 2 H), 7.93 (d, 7=7.78 Hz, 1 H), 7.70 (t, 7=7.91 Hz, 1 H), 7.53 (d, 7=8.78 Hz, 1 H), 7.20 - 7.42 (m, 6 H), 4.43 (s, 2 H); MS ESI 422.2 [M + H]+, calcd for
 + H]+ 422.2.
+ H]+ 422.2.

A DMF (2 mL) solution of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate (50 mg, 0.12 mmol) and N-ethyl-N-isopropylpropan-2-amine (0.1 mL, 0.6 mmol) was treated with a DMF (0.5 mL) solution of isocyanatobenzene (16 mg, 0.13 mmol) at - 20 °C. The reaction mixture was stirred with cooling for 2 h before being allowed to warm to 0 oC for 1 h and later to rt. The reaction was quenched by an addition of MeN¾ (2 M in MeOH, 0.3 mL, 0.6 mmol) at rt. Purification by preparative HPLC provided the title compound as a
-1 18-4820V.1
white powder (15 mg, 30 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.50 (s, 1 H), 8.23 (s, 1 H), 8.20 (d, 7=8.03 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.72 (t, 7=7.91 Hz, 1 H), 7.57 (d, 7=8.78 Hz, 1 H), 7.41 - 7.49 (m, 3 H), 7.31 (t, 7=7.65 Hz, 2 H), 7.04 (t, 7=7.15 Hz, 1 H); MS ESI 408.1 [M + H]+, calcd for [C20H17N5O3S+ H]+ 408.1.

A DMF (2 mL) solution of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate (50 mg, 0.12 mmol) and N-ethyl-N-isopropylpropan-2-amine (0.1 mL, 0.6 mmol) was treated with a DMF (0.5 mL) solution of l,3-diethyl-2-isocyanatobenzene (23 mg, 0.13 mmol) at 0 °C. The reaction mixture was stirred with cooling for 2 h before being allowed to warm to rt overnight. The reaction was quenched by an addition of MeN¾ (2 M in MeOH, 0.3 mL, 0.6 mmol) at rt. Purification by preparative HPLC provided the title compound as a white powder (18 mg, 31 %). Ή NMR (400 MHz, METHANOL-d4) δ ppm 8.47 (s, 1 H), 8.22 (br. s, 1 H), 8.17 (d, 7=7.28 Hz, 1 H), 7.93 (d, 7=7.80 Hz, 1 H), 7.69 (t, 7=7.78 Hz, 1 H), 7.55 (d, 7=8.03 Hz, 1 H), 7.35 - 7.50 (m, 1 H), 7.11 - 7.28 (m, 3 H), 2.72 (q, 7=7.53 Hz, 4 H), 1.24 (t, 7=7.53 Hz, 6 H); MS ESI 464.2 [M + H]+, calcd for [C24H25N503S + H]+ 464.2.

A DMF (2 mL) solution of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate (100 mg, 0.25 mmol) and N-ethyl-N-isopropylpropan-2-amine (0.17 mL, 0.99
-1 19-4820V.1
mmol) was treated with 2-isocyanato-l,3-dimethoxybenzene (52 mg, Maybridge 90 %, 0.26 mmol) at 0 oC. The reaction mixture was stirred for 3 h before being stirring at rt overnight. The reaction was quenched by an addition of MeN¾ (2 M in MeOH, 0.3 mL, 0.6 mmol) at rt. Purification by preparative HPLC provided the title compound as a white powder (28 mg, 24 %). Ή NMR (400 MHz, METHANOL-dA) δ ppm 8.47 (s, 1 H), 8.24 (s, 1 H), 8.17 (d, 7=7.78 Hz, 1 H), 7.92 (d, 7=8.03 Hz, 1 H), 7.68 (t, 7=7.78 Hz, 1 H), 7.54 (d, 7=8.53 Hz, 1 H), 7.40 (d, 7=9.03 Hz, 1 H), 7.23 (t, 7=8.41 Hz, 1 H), 6.71 (d, 7=8.28 Hz, 2 H), 3.86 (s, 6 H); MS ESI 468.2 [M + H]+, calcd for [C22H21N5O5S + H]+ 468.1. Example Al 5. 3-(5-(3-(2,6-Dichlorophenyl)ureido)- 1 H-indazol-3-yl)benzenesulfonamide

The title compound was synthesized according to the method for 3-(5-(3-phenylureido)- lH-indazol-3-yl)benzenesulfonamide , utilizing a DMF (2 mL) solution of 3-(5-amino-lH- indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol), N-ethyl-N- isopropylpropan-2-amine (0.17 mL, 0.99 mmol) and l ,3-dichloro-2-isocyanatobenzene (49 mg, 0.26 mmol). Purification by preparative HPLC provided the title compound as a white powder (15 mg, 13 %). 'H NMR (400 MHz, METHANOL-^) δ ppm 8.48 (s, 1 H), 8.23 (s, 1 H), 8.18 (d, 7=7.28 Hz, 1 H), 7.93 (d, 7=7.78 Hz, 1 H), 7.69 (t, 7=7.65 Hz, 1 H), 7.53 - 7.60 (m, 1 H), 7.43 - 7.51 (m, 3 H), 7.29 (t, 7= 8.15 Hz, 1 H) δ ppm; MS ESI 476.2 [M + H]+, calcd for
[C2oH15Cl2N503S + H]+ 476.0.
-120-4820V.1

To a solution of 1 -benzyl- lH-pyrazol-3-amine (50 mg, 0.29 mmol) in 3 mL dry DCM was added Et3N (88 mg, 0.87mmol), then the mixture was treated with bis(trichloromethyl) carbonate (37 mg, 0.125 mmol) added in one portion at -78 °C. The mixture was stirred for 1 hour at this temperature, then a solution of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide hydrochloride (94 mg, 0.29 mmol) in 3 mL DMF and TEA (58 mg, 0.57 mmol) was slowly added. The mixture was stirred with the cooling for 8 h and then allowed to warm slowly to room temperature. The solvent was evaporated to dryness in vacuum and then dissolved in DMF. The solution was filtered and then purified by preparative HPLC. Ή NMR (500 MHz, DMSO- d6) 5 ppm 13.37 ( s, 1 H), 9.21 (br. s., 1 H), 9.12 ( s, 1 H), 8.44 (s, 1 H), 8.22 (s, 1 H), 8.16 (d, J=7.00 Hz, 1 H), 7.86 (d, J=8.00 Hz, 1 H), 7.68-7.79 (m, 2 H), 7.43 - 7.62 (m, 4 H), 7.21 - 7.43 (m, 5 H), 6.28 (s, 1 H), 5.25 (s, 2 H). MS ESI 488.3 [M + H]+, calcd for [C24H2iN703S+ H]+ 488.3.
-121-44820V.1
Table 1
Ureas prepared according to the method described for 3-(5-(3-Phenylureido)-lH-indazol-3- yl)benzenesulfonamide or 3-(5-(3-(l-Benzyl-lH-pyrazol-3-yl)ureido)-lH-indazol-3- yl)benzenesulfonamide


-122- 4820V.1

-123- 0V.1
A22 508.2 [C25H25N505S+ Ή NMR (500 MHz,
H]+ 508.2 DM50-i 6) 6 ppm 13.33
(s, 1 H), 8.70 (s, 1 H),
3-(5-(3-(4-
8.53 (s, 1 H), 8.43 (s, 1
(tetrahydro-2H- H), 8.20 (s, 1 H), 8.13 (d, pyran-4- 7=7.57 Hz, 1 H), 7.84 (d, yloxy)phenyl)ureido
7=8.20 Hz, 1 H), 7.75 (t, )-lH-indazol-3- 7=7.90 Hz, 1 H), 7.57 (d, yl)benzenesulfonami
7=8.83 Hz, 1 H), 7.45 (s, 3 de
H), 7.37 (d, 7=8.80 Hz, 2 H), 6.92 (d, 7=9.14 Hz, 2 H), 4.44 - 4.51 (m, 1 H), 3.82 - 3.89 (m, 2 H), 3.44
- 3.50 (m, 2 H), 1.92 - 1.98 (m, 2 H), 1.52 - 1.61 (m, 2 H)
A23 459.2 [C23Hi8N603S+ 'H NMR (400 MHz,
H]+ 459.2 DMSO-d6) δ ppm 13.36
(s, 1 H), 9.13 (s, 1 H), 8.94 (s, 1 H), 8.73 (dd,
3-(5-(3-quinolin-6- 7=4.14, 1.63 Hz, 1 H), ylureido)-lH- 8.43 (s, 1 H), 8.23 - 8.27 indazol-3- (m, 2 H), 8.19 (d, 7=1.76 yl)benzenesulfonami Hz, 1 H), 8.14 (d, 7=7.50 de Hz, 1 H), 7.94 (d, 7=9.00
Hz, 1 H), 7.84 (d, 7=8.03 Hz, 1 H), 7.76 (d, 7=7.80 Hz, 1 H), 7.71 - 7.75 (m, 1 H), 7.59 (d, 7=8.80 Hz, 1 H), 7.44 - 7.52 (m, 4 H)
A24 494.2 [C23H23N704S+ Ή NMR (400 MHz,
H]+ 494.2 DMSO-d6) δ ppm 13.32
(s, 1 H), 8.78 (s, 1 H), 8.49 (s, 1 H), 8.41 (s, 1
3-(5-(3-(6- H), 8.18 (m, 2 H), 8.1 1 (d, morpholinopyridin- 7=7.80 Hz, 1 H), 7.83 (d, 3-yl)ureido)-lH- 7=8.00 Hz, 1 H), 7.71 - indazol-3- 7.76 (m, 2 H), 7.55 (d, yl)benzenesulfonami 7=9.00 Hz, 1 H), 7.44 - de 7.48 (m, 3 H), 6.82 (d,
7=9.03 Hz, 1 H), 3.65 - 3.73 (m, 4 H), 3.35 (m, 4
H)
-124-0V.1

-125- 20V.1

0v.1
A31 486.2 [C24H19N703S+ Ή NMR (500 MHz,
H]+ 486.2 DMS0-d6) δ ppm 13.38
(s, 1 H), 9.03 (s, 1 H), 8.89 (s, 1 H), 8.86 (d,
3-(5-(3-(4- 7=5.04 Hz, 2 H), 8.44 (s, 1
(pyrimidin-2- H), 8.34 (d, 7=8.51 Hz, 2 yl)phenyl)ureido)- H), 8.25 (s, 1 H), 8.15 (d, lH-indazol-3- 7=7.88 Hz, 1 H), 7.86 (d, yl)benzenesulfonami 7=8.20 Hz, 1 H), 7.77 (t, de 7=7.80 Hz, 1 H), 7.65 (d,
7=8.83 Hz, 2 H), 7.60 (d, 7=8.83 Hz, 1 H), 7.47 - 7.51 (m, 3 H), 7.37 (t, 7=4.70 Hz, 1 H)
A32 478.3 [C25H27N503S+ Ή NMR (500 MHz,
H]+ 478.3 DMS0-d6) δ ppm 13.31
(s, 1 H), 8.99 (br.s, 1 H),
8.42 (s, 1 H), 8.19 (s, 1 H), 8.10 (d, 7=7.57 Hz, 1
3-(5-(3-(2-ethyl-6- H), 7.77 - 7.83 (m, 2 H), isopropylphenyl)urei 7.71 (t, 7=7.90 Hz, 1 H), do)-lH-indazol-3- 7.50 - 7.57 (m, 2 H), 7.47 yl)benzenesulfonami (s, 2 H), 7.22 (t, 7=7.60 de Hz, l H), 7.17 (br. d,
7=6.90 Hz, 1 H), 7.10 (d,
7=6.90 Hz, 1 H), 3.17 -
3.25 (m, 1 H), 2.60 (q, 7=7.30 Hz, 2 H), 1.16 (m, 7=7.30 Hz, 9 H)
A33 464.1 [C24H25N503S+ Ή NMR (500 MHz,
H]+ 464.1 DMSO-d6) 8 ppm 13.30
(s., 1 H), 8.89 (br. s, 1 H), 8.42 (s., 1 H), 8.20 (br. s.,
3-(5-(3-(2-isopropyl- 1 H), 8.10 (d, 7=7.88 Hz, 6- 1 H), 7.81 (d, 7=7.25 Hz, methylphenyl)ureido 1 H), 7.69 - 7.76 (m, 2 H), )-lH-indazol-3- 7.55 (d, 7=8.80 Hz, 1 H), yl)benzenesulfonami 7.51 (d, 7=9.10 Hz, 1 H), de 7.46 (s., 2 H), 7.14 - 7.17
(m, 2 H), 7.09 (br. s., 1 H), 3.18 - 3.26 (m, 1 H), 2.23 (s, 3 H), 1.16 (d, 7=6.62 Hz, 6 H)
-127-0V.1

-128- 20V.1
W

44820V.1

A DMF (2 mL) solution of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate (39 mg, 0.098 mmol) and N-ethyl-N-isopropylpropan-2-amine (0.05 mL, 0.3 mmol) was treated with a DMF (0.5 mL) solution of thiophene-2-sulfonyl chloride (18 mg, 0.10 mmol) at 0 °C. The reaction mixture was stirred for 3 h before being stirring at rt overnight. The reaction was quenched by an addition of MeN¾ (2 M in MeOH, 0.3 mL, 0.6 mmol) at rt.
Purification by preparative HPLC provided the title compound as a white powder (19 mg, 45 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.40 (s, 1 H), 8.05 (d, 7=8.03 Hz, 1 H), 7.95 (d, 7=8.28 Hz, 1 H), 7.67 - 7.74 (m, 3 H), 7.51 (d, 7=8.78 Hz, 1 H), 7.47 (dd, 7=1.25, 3.76 Hz, 1 H), 7.26 (dd, 7=1.76, 8.78 Hz, 1 H), 7.05 (t, 7=4.52 Hz, 1 H); MS ESI 435.1 [M + H]+, calcd for [C17H14N4O4S3 + H]+ 435.1.
Table 2
Sulfonomides prepared according to the method for. N-(3-(3-sulfamoylphenyl)-lH-indazol-5- yl)thiophene-2-sulfonamide


-130- 4820V.1

-131- 20V.1

-132- 20V.1

To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (101 mg, 0.25 mmol) and 2-chloroethyl N-phenylsulfamoylcarbamate (70 mg, 0.25 mmol) in CH3CN (30 mL) was added Et3N (0.1 1 mL, 0.75 mmol). The resulting mixture was refluxed (oil temp. 90 °C) for 2 h. It was concentrated to dryness, redissolved in DMF (4 mL), filtered through microfilter and purified by preperative HPLC. The title compound was obtained as a white solid (33 mg, 30 %) after trituration with MeOH. Ή NMR (400 MHz, CD3OD) δ 8.43 (s, 1H), 7.96 (t, 7 = 8.8 Hz, 2H), 7.68 (pseudo t, 7 =7.8 Hz, 2H), 7.48 (d, 7 =8.8 Hz, 1H), 7.27-7.21 (m, 3 H), 7.13 (d, 7 =8.4 Hz, 2H), 7.04 (t, 7 =7.2 Hz, 1H); MS ESI 444.2 [M + H]+, calcd for
[C,9H17N504S2 + H]+ 444.1

-133- 4820v.1
To a mixture of 3-(5-amino- 1 H-indazol-3-yl)benzenesulfonamide trifluoroacetate (101 mg, 0.25 mmol) and 2-chloroethyl N-(2-ethylphenyl)sulfamoylcarbamate (77 mg, 0.25 mmol) in CH3CN (40 mL) was added Et3N (0.07 mL, 0.5 mmol). The resulting mixture was refluxed (oil temp. 85 °C) for 2 h. Later the reaction mixture was concentrated to dryness, redissolved in DMF (4 mL), filtered through microfilter and purified by preparative HPLC. The title compound was obtained as a white solid (30 mg, 26%) after trituration with CH2C12. Ή NMR (400 MHz, CD3OD) δ 8.44 (s, 1H), 8.01 (d, J =8.0 Hz, 1 H), 7.95 (d, J =8.4 Hz, 1H), 7.71-7.60 (m, 2H), 7.51 (d, J =8.8 Hz, 1H), 7.44 (d, J =8.0 Hz, 1H), 7.27 (d, =9.2 Hz, 1H), 7.17 (t, J =1.4 Hz, 1H), 7.1 1-7.05 (m, 2H), 2.32 (q, =7.8 Hz, 2H), 0.85 (t, J =7.4 Hz, 3H); MS ESI 472.2 [M + H]+, calcd for [C21H21N5O4S2 + H]+ 472.1
Example A51. 3-(5-(N-(2,6-Diethylphenyl)sulfamoylamino)- lH-indazol-3-yl)benzene sulfonamide

To a mixture of 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (101 mg, 0.25 mmol) and 2-chloroethyl N-(2,6-diethylphenyl)sulfamoylcarbamate (100 mg, 0.3 mmol) in CH3CN (10 mL) was added Et3N (0.1 1 mL, 0.75 mmol). The resulting mixture was refluxed (oil temp. 90 °C) for 2h. It was concentrated to dryness, redissolved in DMF (4 mL), filtered through microfilter and purified by preparative HPLC. The title compound was obtained as a light gray solid (24 mg, 19%) after trituration with CH2C12. Ή NMR (400 MHz, CD3OD) δ 8.48 (s, 1H), 8.07 (d, J =7.2 Hz, 1H), 7.94 (d, =7.6 Hz, 1H), 7.90 (s, 1 H), 7.70 (t, / =7.4 Hz, 1H), 7.55 (d, J =9.2 Hz, 1H), 7.36 (d, J =8.4 Hz, 1H), 7.13 (t, J =7.4 Hz, 1H), 7.04 (d, J =7.2 Hz, 2H), 2.68 (q, J =7.2 Hz, 4H), 1.03 (t, J =7.6 Hz, 6H). MS ESI 500.3 [M + H]+, calcd for [C23H25N504S2 + H]+ 500.1.
-134-4820V.1

The title compound was synthesized according to the General Method A utilizing 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (0.050 g, 0.12 mmol), rac-2- methoxy-2-phenylacetic acid (0.021 g, 0.12 mmol), N-ethyl-N-isopropylpropan-2-amine (0.1 mL, 0.6 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (0.042 g, 0.13 mmol) in DMF (2 mL). Purification by preparative HPLC provided the title compound as a white powder (13 mg, 24 Ή NMR (400 MHz, METHANOL-dA) δ ppm 10.00 (s, 1 H), 8.47 (s, 1 H), 8.37 (s, 1 H), 8.16 (d, 7=7.78 Hz, 1 H), 7.94 (d, 7=7.28 Hz, 1 H), 7.69 (t, 7=7.91 Hz, 1 H), 7.62 (dd, 7 = 1.51 , 9.03 Hz, 1 H), 7.50 - 7.59 (m, 3 H), 7.29 - 7.46 (m, 3 H), 4.85 (s, 1 H), 3.47 (s, 3 H); MS ESI 437.2 [M + H]+, calcd for [C22H2oN404S+ Hf 437.2.

The title compound was synthesized according to the the General Method A utilizing 3-(5- amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (0.10 g, 0.26 mmol), (R)-2- methoxy-2-phenylacetic acid (0.044 g, 0.27 mmol), N-ethyl-N-isopropylpropan-2-amine (0.1 mL, 0.6 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (0.086 g, 0.27 mmol) in DMF (2 mL). Purification by preparative HPLC provided the title compound as a white powder (67 mg, 60 %). Ή NMR (400 MHz, METHANOLS) δ ppm 9.99 (s, 1 H), 8.47 (s, 1 H), 8.38 (s, 1 H), 8.15 (d, 7=7.78 Hz, 1 H), 7.93 (d, 7=8.03 Hz, 1 H), 7.68 (t,
-135-4820v.1
7=7.78 Hz, 1 H), 7.62 (d, 7 =9.03 Hz, 1 H), 7.50 - 7.58 (m, 3 H), 7.31 - 7.46 (m, 3 H), 4.85 (s, 1 H), 3.47 (s, 3 H); MS ESI 437.3 [M + H]+, calcd for [C22H20N4O4S+ H]+ 437.3.
Example A54. 3-Methoxy-N-(3-(3-sulfamoylphenyl)- lH-indazol-5-yl)thiophene-2-carboxamide

The title compound was synthesized according to the General Method A, utilizing 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (0.050 g, 0.12 mmol), 3- methoxythiophene-2-carboxylic acid (0.020 g, 0.12 mmol), N-ethyl-N-isopropylpropan-2-amine (0.1 mL, 0.6 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (0.042 g, 0.13 mmol) in DMF (2 mL). Purification by preparative HPLC provided the title compound as a white powder (19 mg, 35 %). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.45 (br. s., 1 H), 9.40 (s, 1 H), 8.47 (s, 1 H), 8.44 (s, 1 H), 8.17 (d, 7=8.03 Hz, 1 H), 7.81 - 7.87 (m, 2 H), 7.75 (t, 7= 7.78 Hz, 1 H), 7.66 (d, 7=8.03 Hz, 1 H), 7.61 (d, 7=8.78 Hz, 1 H), 7.50 (s, 2 H), 7.22 (d, 7=5.52 Hz, 1 H), 4.09 (s, 3 H); MS ESI 429.2 [M + H]+, calcd for [C19H16N404S2+ H]+ 429.2.
Example A55. l-Benzyl-N-(3-(3-sulfamoylphenyl)- lH-indazol-5-yl)piperidine-4-carboxamide hydrochloride
Λ. 1 -benzyl-N-( lH-ind

-136-4820V.1
The title compound was synthesized according to the General Method A, utilizing lH-indazol-5- amine (50 mg, 0.38 mmol), l-benzylpiperidine-4-carboxylic acid hydrochloride (96 mg, 0.38 mmol), DIPEA (0.16 mL, 0.92 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (127 mg, 0.39 mmol) in DMF (1.3 mL). The crude material was isolated by filtration of the precipitate after an addition of xs H20. The collected solid was washed with H20 to provide the title compound as a tan solid (0.098 g, 78 %). Ή NMR (400 MHz, DMSO- d6) δ ppm 12.94 (s, 1 H), 9.83 (s, 1 H), 8.12 (s, 1H), 7.98 (s, 1H), 7.44 (d, 7=8.28 Hz, 1 H), 7.40 (d, 7= 8.53 Hz, 1 H), 7.20 - 7.34 (m, 6 H), 3.46 (s, 2 H), 2.87 (d, 7=11.29 Hz, 2 H), 2.32 (br. s., 1 H), 1.96 (t, 7=10.54 Hz, 2 H), 1.60 - 1.80 (m, 4 H) ; MS ESI 335.1 [M + H]+, calcd for

B. l-Benzyl-N-(3-iodo-lH-indazol-5-yl)piperidine-4-carboxamide

The title compound was synthesized according to the General Method B utilizing 1- benzyl-N-(lH-indazol-5-yl)piperidine-4-carboxamide (0.048 g, 0.14 mmol ), I2 (0.072 g, 0.28 mmol) and K2C03 (0.090 g, 0.65 mmol) in DMF (1 mL). A white powder (0.062 g, 90 %). MS ESI 461.0 [M + H]+, calcd for [C2oH21rN40 + H]+ 461.3.
C. 1 -benzyl-N-( 3-( 3-sulfamoylphenyl)-lH-indazol-5-yl)piperidine-4-carboxamide hydrochloride
The title compound was synthesized according to the General Method C utilizing 1- Benzyl-N-(3-iodo-lH-indazol-5-yl)piperidine-4-carboxamide (60 mg, 0.13 mmol), 3-(4,4,5,5- tetramethyl-l ,3,2-dioxaborolan-2-yl)benzenesulfonamide (44 mg, 0.16 mmol), KF (15 mg, 0.26 mmol), DMF (2.5 mL), H20 (0.5 mL) and Pd(PPh3)4 (8 mg, 0.006 mmol). The degassed solution was sealed and heated in a microwave reactor at 120 °C for 2 h. The crude material was concentrated under reduced pressure and purified by column chromatography (Si02, 0-30 % 7 M NH3 MeOH in DCM) to afford a tan solid which was taken into MeCN ( 4mL), cooled to 0 °C
-137-4820V.1
and treated with 1 M ΗΟ/Ι¾0. A cream-colored precipitate was collected by filtration and rinsed with MeCN to provide the title compound as an off-white solid (16 mg, 24 %). 'H NMR (400 MHz, METHANOL-^) δ ppm 8.46 (s, 1 H), 8.40 (s, 1 H), 8.15 (d, 7=8.28 Hz, 1 H), 7.94 (d, 7=7.28 Hz, 1 H), 7.69 (t, 7=7.78 Hz, 1 H), 7.46 - 7.59 (m, 7 H), 4.36 (s, 2 H), 3.57 - 3.64 (m, 2 H), 3.08 - 3.18 (m, 2 H), 2.76 (m, 1 H), 2.04 - 2.30 (m, 4 H); MS ESI 490.3 [M + H]+, calcd for [C26H27N503S + H]+ 490.3.
Example A56. N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-lH-indene-3-carboxamide

To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (201 mg, 0.5 mmol), lH-indene-3-carboxylic acid (80 mg, 0.5 mmol) and TBTU (161 mg, 0.5 mmol) in DMF (5 mL) at 0 °C was added 'Pr2NEt (0.26 mL, 1.5 mmol). The resulting mixture was stirred for lh at 0 °C, then filtered through microfilter and purified by preparative HPLC. The titel compound was obtained as a beige solid (68 mg, 32%) after trituration with MeOH. Ή NMR (400 MHz, DMSO-d6) δ 13.43 (s, IH), 10.32 (s, IH), 8.53 (s, IH), 8.44 (s, IH), 8.16 (d, 7 =8.0 Hz, IH), 7.96 (d, 7 =7.6 Hz, IH), 7.85 (d, 7 =7.6 Hz, I H), 7.79 (d, 7 =7.6 Hz, IH, partially overlapping with the doublet at 7.75 ppm), 7.75 (d, 7 =8.0 Hz, IH, partially overlapping with the doublet at 7.79 ppm), 7.63 (d, 7 =8.8 Hz, IH), 7.54 (d, 7 =7.6 Hz, IH), 7.48 (s, 2H, NH2), 7.44 (s, 1H), 7.31 (t, 7 =7.6 Hz, I H), 7.26 (t, 7 =7.6 Hz, I H), 3.64 (s, 2H); MS ESI 431.3 [M + H]+, calcd for [C23H18N4O3S + H]+ 431.1.
-138-4820V.1
Example A57. N-(3-(3-sulfamoylphenyl)- 1 H-indazol-5-yl)-2,3-dihvdro- 1 H-indene- 1 - carboxamide

To a solution of N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-lH-indene-3-carbox-amide (46 mg) in THF (4 niL) and MeOH (4 niL) was added 10% Pd/C (9.2 mg, 20 %wt). The resulting mixture was vacuumed and refilled with H2. This process was repeated 3 times then it was stirred for 2 h under ¾ balloon. Pd catalyst was removed by microfilter filtration and the filtrate was purified by preparative HPLC to give the title compound as a white solid (32 mg, 70%). Ή NMR (400 MHz, DMSO-d6) δ 13.40 (s, IH), 10.40 (s, IH), 8348 (s, IH), 8.40 (s, IH), 8.09 (d, J =6.8 Hz, IH), 7.82 (d, J =6.8 Hz, IH), 7.73 (t, J =7.6 Hz, IH), 7.64 (d, J =8.8 Hz, IH, partially overlapping with the doublet at 7.59 ppm), 7.59 (d, J =9.2 Hz, IH, partially overlapping with the doublet at 7.64 ppm), 7.46 (s, 2H, NH2), 7.34 (d, =6.0 Hz, IH), 7.27 (d, J =7.2 Hz, IH), 7.22-7.12 (m, 2H), 4.14 (t, / =6.8 Hz, IH), 3.12-3.02 (m, IH), 2.96-2.86 (m, IH), 2.40-2.25 (m, 2H); MS ESI 433.3 [M + H]+, calcd for [C23H20N4O3S + H]+ 433.1.
Example A58. N-(3-(3-sulfamoylphenvD- lH-indazol-5-yl)- 1 ,2.3.4-tetrahydronaphthalene- 1- carboxamide

To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (120 mg, 0.3 mmol), 1 ,2,3,4-tetrahydronaphthalene-l -carboxylic acid (53 mg, 0.3 mmol) and TBTU (96 mg, 0.3 mmol) in DMF (4 mL) at 0 °C was added 'Pr2NEt (0.16 mL, 0.9 mmol). The resulting mixture was stirred for lh at 0 °C, then filtered through microfilter and purified by preparative HPLC. The title compound was obtained as a white solid (8 mg, 6%) after trituration
-139-44820V.1
with MeOH. Ή NMR (400 MHz, DMSO-d6) δ 13.40 (s, IH), 10.36 (s, IH), 8.50 (s, IH), 8.41 (s, IH), 8.08 (d, 7 =6.8 Hz, IH), 7.82 (d, 7 =8.0 Hz, IH), 7.72 (t, 7 =8.0 Hz, IH), 7.63 (d, 7 =8.0 Hz, IH, partially overlapping with the doublet at 7.59 ppm), 7.59 (d, 7 =8.0 Hz, IH, partially overlapping with the doublet at 7.63 ppm), 7.48 (s, 2H, NH2), 7.16-7.08 (m, 4H), 3.89 (t, 7 =4.8 Hz, IH), 2.82-2.68 (m, 2H), 2.10-2.00 (m, 3H), 1.73-1.63 (m, IH); MS ESI 447.3 [M + H]+, calcd for [C24H22N4O3S + H]+ 447.1.

3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate(51 mg, 0.127 mmol), 2- phenylacetic acid (17 mg, 0.127 mmol), and DIPEA (66 μί, 0.381 mmol) was dissolved in 2 mL of DMF and cooled to 0 °C. TBTU (41 mg, 0.127 mmol) was added and the reaction was stirred at 0 °C for 90 minutes. Purification by preparative HPLC gave the title compound as a white solid (24 mg, 47%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.46 (s, 1 H), 8.39 (s, 1 H), 8.14 (d, 7=8.03 Hz, 1 H), 7.93 (d, 7=8.03 Hz, 1 H), 7.68 (t, 7=7.91 Hz, 1 H), 7.54 (m, 2 H), 7.36 (m, 4 H), 7.26 (t, 7=7.03 Hz, 1 H), 3.73 (s, 2 H) ); MS ESI [M + H]+ 407.2, calcd for
[C2iH18N403S + H]+ 407.1.

The title compound was synthesized according the General Method A, utilizing 3- phenylpropanoic acid (1 1 mg, 0.075 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide
-140-44820V.1
2,2,2-trifluoroacetate(30 mg, 0.075 mmol), DIPEA (39 μΐ,, 0.224 mmol), TBTU (24 mg, 0.075 mmol) and 2 mL of DMF. The majority of impurities were removed by an application of preparative HPLC, however some di-acetylated byproduct co-eluted with the title compound. The mixture was then dissolved in 2 mL of methanol and sodium methanolate (2 M in methanol, 0.10 mL) was added. The mixture was stirred at 40 °C overnight and then triturated with Et20 to give the title compound as a white solid (4.9 mg, 16%). *H NMR (400 MHz, METHANOL-d4) δ ppm 8.48 (s, 1 H), 8.25 (s, 1 H), 8.08 (d, 7=7.78 Hz, 1 H), 7.94 (d, 7=7.78 Hz, 1 H), 7.65 (t, 7=7.78 Hz, 1 H), 7.53 (s, 2 H), 7.26 - 7.33 (m, 4 H), 7.16 - 7.24 (m, 1 H), 3.04 (t, 7=7.78 Hz, 2 H), 2.72 (t, 7=7.28 Hz, 2 H) ); MS ESI [M + H]+ 421.2, calcd for [C22H20N4O3S + H]+ 421.1.

The title compound was synthesized according the General Method A, utilizing cyclopentylacetic acid (16 mg, 0.12 mmol) and 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate(50 mg, 0.12 mmol), DIPEA (65 ί, 0.37 mmol), TBTU (40 mg, 0.12 mmol) and 2 mL of DMF. The majority of impurities were removed by preparative HPLC, the rest by triturating with Et20, followed by CH2Cl2 to give the title compound as a white solid (18 mg, 36 %). ]H NMR (400 MHz, METHANOLS) δ ppm 8.47 (s, 1 H), 8.38 (s, 1 H), 8.15 (d, 7=7.78 Hz, 1 H), 7.93 (d, 7=7.53 Hz, 1 H), 7.68 (t, 7=7.78 Hz, 1 H), 7.52 (m, 7=6.27 Hz, 2 H), 2.41 (d, 7=8.00 Hz, 2 H), 2.29 - 2.39 (m, 1 H), 1.82 - 1.92 (m, 2 H), 1.70 (br. s., 2 H), 1.54 - 1.65 (m, 2 H), 1.21 - 1.33 (m, 2 H); MS ESI [M + H]+ 399.2, calcd for [CzoH^OsS + H]+ 399.1.
-141-4820V.1
Example A62. N-(3-(3-SulfamoylphenvD- 1 H-indazol-5-yl)benzo[c1 [ 1 ,2,51oxadiazole-5- carboxamide

A DMF (2 mL) solution of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate(35 mg, 0.087 mmol), DIPEA (46 μΐ-, 0.26 mmol) cooled to 0 °C was treated with 2,l,3-benzoxadiazole-5-carbonyl chloride (16 mg, 0.087 mmol). The reaction mixture was stirred at 0 °C for 120 minutes. Purification by preparative HPLC gave the title compound as a yellow solid (19 mg, 50%). Ή NMR (400 MHz, DMS0-d6) δ ppm 13.49 (s, 1 H), 10.80 (s, 1 H), 8.73 (s, 1 H), 8.57 (s, 1 H), 8.44 (s, 1 H), 8.21 (d, 7=9.79 Hz, 1 H), 8.15 (d, 7=6.27 Hz, 1 H), 8.05 (d, 7=9.03 Hz, 1 H), 7.86 (d, 7=7.78 Hz, 1 H), 7.73 - 7.83 (m, 2 H), 7.67 (d, 7=9.03 Hz, 1 H), 7.48 (s, 2 H);MS ESI [M + H]+ 435.1, calcd for [C20H14N6O4S + H]+ 435.1.
Example A63. N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-2-(thiophen-3-yl)acetamide

The title compound was synthesized according to the General Method A, utilizing 3- thiophene acetic acid (11 mg, 0.075 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate(30 mg, 0.075 mmol), DIPEA (39 μί, 0.225 mmol), TBTU (24 mg, 0.075 mmol) and 2 mL of DMF. The majority of impurities were removed by preparative HPLC and then the title compound was precipitated out of methanol on addition of H20 as a white solid (3.5 mg, 1 1%). Ή NMR (400 MHz, METHANOLS) 8 ppm 8.46 (s, 1 H), 8.39 (s, 1 H), 8.15 (d, 7=7.28 Hz, 1 H), 7.93 (d, 7=7.53 Hz, 1 H), 7.69 (t, 7=7.65 Hz, 1 H), 7.49 - 7.58 (m, 2 H), 7.36 - 7.41 (m, 1 H), 7.29 (br. s., 1 H), 7.14 (d, 7=5.02 Hz, 1 H), 3.76 (s, 2 H); MS ESI [M + H]+413.1, calcd for [Ci9Hi6N403S2 + H]+ 413.1.
-142-4820V.1

The title compound was synthesized according to the method described for N-(3-(3- Sulfamoylphenyl)-lH-indazol-5-yl)thiophene-2-carboxamide utilizing benzo [b]thiophene-2- carbonyl chloride (17 mg, 0.087 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate(35 mg, 0.087 mmol), DIPEA (45 pL, 0.261 mmol), and 2 mL of DMF.
Preparative HPLC removed most of the impurities, leaving two isomers. The mixture was dissolved in 2 mL of methanol and then 2 M sodium methoxide (100 pL) was added. The mixture was stirred at 40 °C overnight, followed by cooling at 4°C to precipitate out a yellow solid. The solid was washed with 0.1 M aq HCl and dried under reduced pressure to give the title compound as a beige solid (6.8 mg, 17%). Ή NMR (400 MHz, DMS0-d6) δ ppm 13.52 (s, 1 H), 10.72 (s, 1 H), 8.58 (s, 1 H), 8.48 (s, 1 H), 8.43 (s, 1 H), 8.21 (d, 7=7.53 Hz, 1 H), 8.1 1 (d, 7=7.53 Hz, 1 H), 8.07 (d, 7=8.03 Hz, 1 H), 7.90 (d, 7=7.53 Hz, 1 H), 7.78 - 7.86 (m, 2 H), 7.70 (d, 7=9.03 Hz, 1 H), 7.50 - 7.56 (m, 4 H); MS ESI [M + H]+ 449.1, calcd for [C22H16N403S2 + H]+ 448.1

The title compound was synthesized according to the General Method A, utilizing 2- pyridylacetic acid (26 mg, 0.19 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate(75 mg, 0.19 mmol), DIPEA (97 pL, 0.56 mmol), TBTU (60 mg, 0.19 mmol) and
-143-4820v.1
4 mL of DMF. The reaction mixture was purified using preparative HPLC, followed by triturating with CH2C12 to give the title compound as a white solid (33 mg, 43%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.77 (d, 7=6.27 Hz, 1 H), 8.46 (s, 1 H), 8.44 (s, 1 H), 8.40 (t, 7=8.66 Hz, 1 H), 8.14 (d, 7=8.53 Hz, 1 H), 7.93 (br. d, 7=6.80 Hz, 2 H), 7.84 (t, 7=6.30 Hz, 1 H), 7.69 (t, 7=8.03 Hz, 1 H), 7.52 - 7.61 (m, 2 H), 4.21 (s, 2 H); MS ESI [M + H]+ 408.1 , calcd for [C2oHi7N503S + H]+ 408.1
Example A66. 2-(3,5-Dimethoxyphenyl -N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)acetamide

The title compound was synthesized according to the method described the General Method A, utilizing 2-(3,5-dimethoxyphenyl)acetic acid ( 14 mg, 0.072 mmol), 3-(5-amino-lH- indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate(29 mg, 0.072 mmol), DIPEA (38 μί, 0.219 mmol), TBTU (23 mg, 0.072 mmol) and 2 mL of DMF. The reaction mixture was purified using preparative HPLC, followed by triturating with CH2C12 to give the title compound as a white solid (5.3 mg, 16%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.40 (s, 1 H), 10.27 (s, 1 H), 8.42 (s, 1 H), 8.40 (s, 1 H), 8.09 (d, 7=7.78 Hz, 1 H), 7.83 (d, 7=7.78 Hz, 1 H), 7.74 (t, 7=7.80 Hz, 1 H), 7.57 (s, 2 H), 7.47 (s, 2 H), 6.53 (s, 2 H), 6.39 (s, 1 H), 3.73 (s, 6 H), 3.59 (s, 2 H); MS ESI [M + H]+ 467.2, calcd for [C23H22N405S + H]+ 467.1.
Example A67. 2-(Pyridin-4-vn-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)acetamide
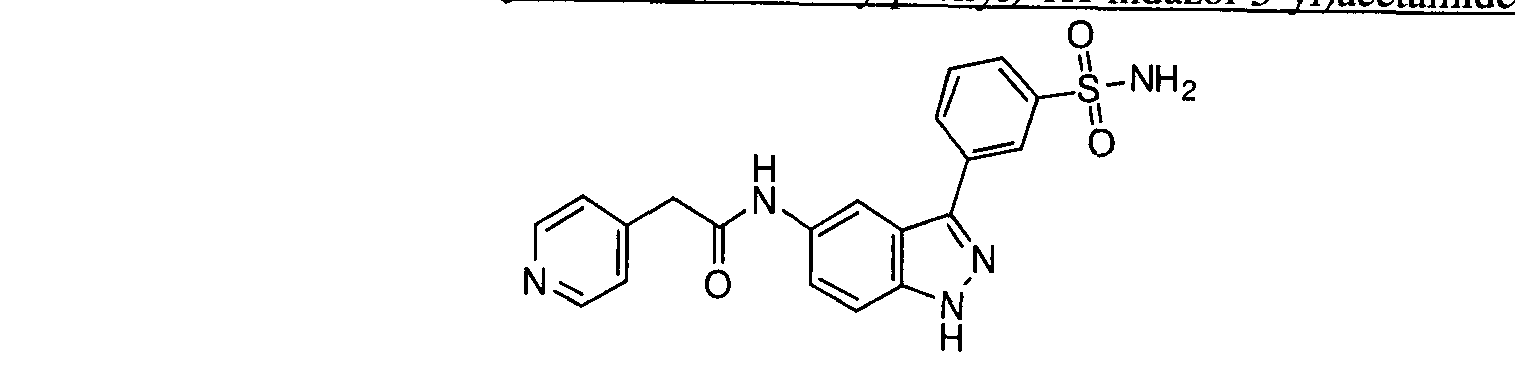
- 144-4820V.1
The title compound was synthesized according to the General Method A, utilizing 4- pyridylacetic acid · HCl (22 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (108 pL, 0.622 mmol), TBTU (40 mg, 0.124 mmol) and 2 mL of DMF. The reaction mixture was purified using preparative HPLC, followed by triturating with CH2CI2 to give the title compound as a white solid (40 mg, 79%). Ή NMR (400 MHz, DMSO-d6) 5 ppm 13.43 (br. s., 1 H), 10.47 (s, 1 H), 8.72 (d, 7=6.02 Hz, 2 H), 8.42 (s, 1 H), 8.39 (s, 1 H), 8.07 (d, 7=7.28 Hz, 1 H), 7.83 (d, 7=8.03 Hz, 1 H), 7.69 - 7.78 (m, 3 H), 7.62 - 7.54 (m, 2 H), 7.47 (s, 2 H), 3.96 (s, 2 H); MS ESI [M + H]+ 408.1, calcd for
[C2oHi7N503S + H]+ 408.1.

The title compound was synthesized according to the General Method A, utilizing α,α- dimethylphenylacetic acid (25 mg, 0.149 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate(60 mg, 0.149 mmol), DIPEA (78 pL, 0.448 mmol), TBTU (48 mg, 0.148 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC, followed by triturating with CH2C12 to give the title compound as a white solid (32 mg, 49%). *H NMR (400 MHz, DMSO-d6) δ ppm 13.37 (br. s., 1 H), 9.24 (s, 1 H), 8.41 (s, 1 H), 8.36 (s, 1 H), 8.09 (d, 7=7.28 Hz, 1 H), 7.83 (d, 7=7.53 Hz, 1 H), 7.75 (t, 7=7.78 Hz, 1 H), 7.61 (d, 7=9.54 Hz, 1 H), 7.53 (d, 7=9.03 Hz, 1 H), 7.47 (s, 2 H), 7.39 (m, 4 H), 7.24 (t, 7=7.30 Hz, 1 H), 1.59 (s, 6 H); MS ESI [M + H]+ 435.2, calcd for [C23H22N4O3S + H]+ 435.1.
-145-4820V.1
xample A69. 2-(2,6-DifluorophenylVN-(3-(3-$ulfamoylp enylVlH-indazol-5-yl')acetarnide

The title compound was synthesized according to the General Method A, utilizing 2,6- difluorophenylacetic acid (17 mg, 0.099 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate(40 mg, 0.099 mmol), DIPEA (52 pL, 0.298 mmol), TBTU (32 mg, 0.099 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC, followed by triturating with CH2CI2 to give the title compound as a white solid (19 mg, 43%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.40 (br. s., 1 H), 10.43 (s, 1 H), 8.44 (s, 1 H), 8.39 (s, 1 H), 8.07 (d, 7=7.53 Hz, 1 H), 7.79 - 7.84 (m, 1 H), 7.72 (t, 7=7.80 Hz, 1 H), 7.51 - 7.61 (m, 2 H), 7.46 (s, 2 H), 7.39 (t, 7=7.53 Hz, 1 H), 7.11 (t, 7=7.65 Hz, 2 H), 3.80 (s, 2 H); MS ESI [M + H]+ 443.2, calcd for [C2iH16F2N403S + H]+ 443.1.

The title compound was synthesized according to the method for N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)thiophene-2-carboxamide, utilizing 3-(5-amino- 1 H-indazol- 3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (30 mg, 0.075 mmol), DIPEA (39 pL, 0.22 mmol), 3-methoxyphenylacetyl chloride (12 pL, 0.075 mmol) and DMF (2 mL). Purification by preparative HPLC followed by triturating with CH2CI2 gave the title compound as a white solid (6 mg, 18%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.39 (s, 1 H), 10.29 (s, 1 H), 8.42 (s, 1 H), 8.39 (s, 1 H), 8.08 (d, 7=8.53 Hz, 1 H), 7.83 (d, 7=7.78 Hz, 1 H), 7.73 (t, 7=7.50 Hz, 1 H), 7.57
-146-4820V.1
(s, 2 H), 7.46 (s, 2 H), 7.24 (t, 7=7.80 Hz, 1 H), 6.90 - 6.95 (m, 2 H), 6.82 (d, 7=7.78 Hz, 1 H), 3.74 (s, 3 H), 3.63 (s, 2 H); MS ESI [M + H]+ 437.2, calcd for [C22H20N4O4S + H]+ 437.1.
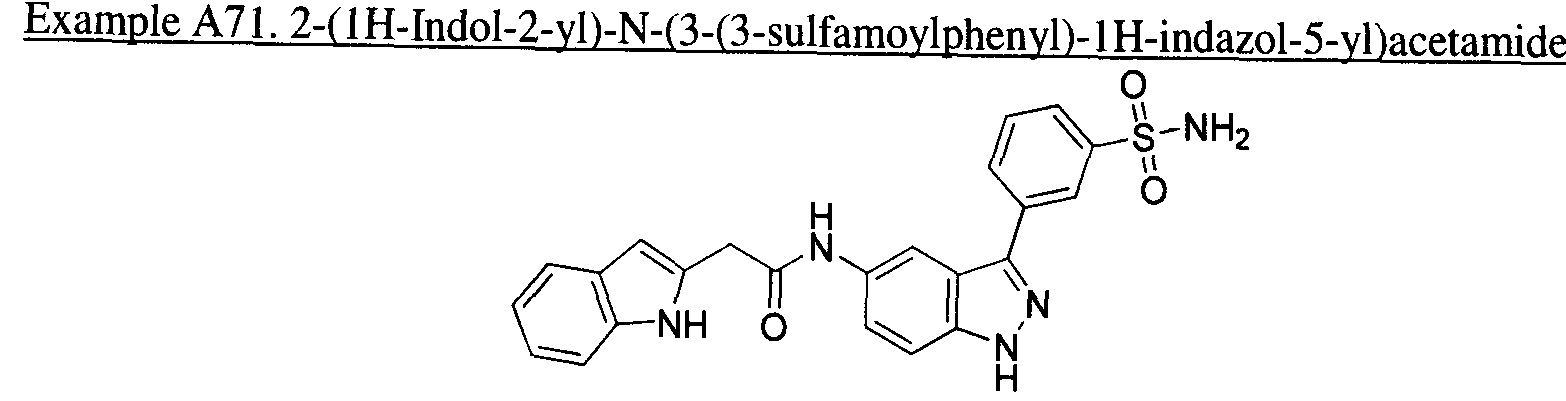
The title compound was synthesized according to the General Method A, utilizing indole-3- acetic acid (13 mg, 0.075 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate (30 mg, 0.075 mmol), DIPEA (39 μί, 0.224 mmol), TBTU (24 mg, 0.075 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2C12 to give the title compound as a white solid (16 mg, 48%) Ή NMR (400 MHz,
DMSO-d6) δ ppm 13.37 (s, 1 H), 10.92 (s, 1 H), 10.23 (s, 1 H), 8.43 (s, 1 H), 8.39 (s, 1 H), 8.07 (d, 7=7.53 Hz, 1 H), 7.81 (d, 7=7.78 Hz, 1 H), 7.71 (t, 7=8.30 Hz, 1 H), 7.62 (d, 7=8.03 Hz, 1 H), 7.56 - 7.59 (m, 1 H), 7.45 (s, 2 H), 7.34 (d, 7=7.53 Hz, 1 H), 7.27 (s, 1 H), 7.03 - 7.09 (m, 1 H), 6.94 - 7.01 (m, 1 H), 3.75 (s, 2 H); MS ESI [M + H]+ 446.2, calcd for [C^H^NJOJS + H]+ 446.1.
Example A72. 2-(2,6-diethylphenyl)-N-(3-(3-sulfamoylphenyl)- lH-indazol-5-yl)acetamide
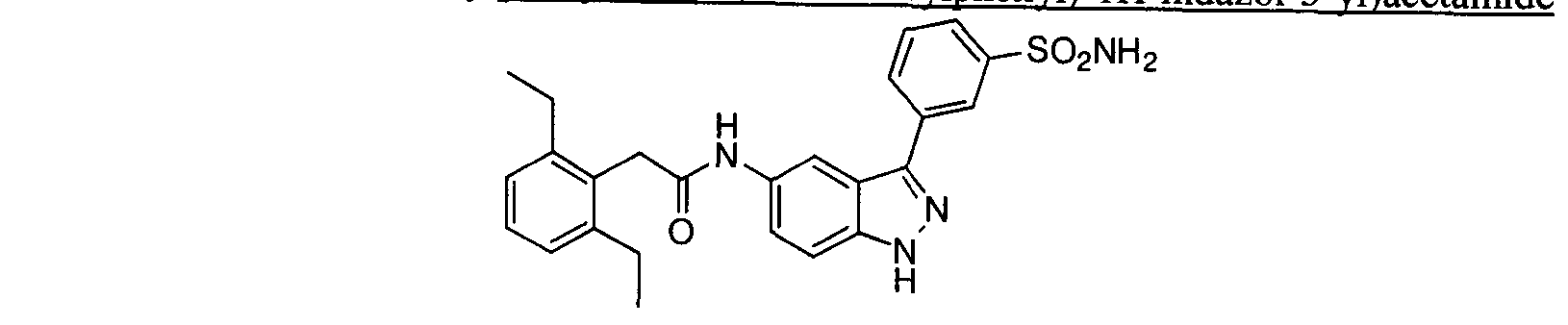
The title compound was synthesized according to the the General Method A, utilizing 2- (2,6-diethylphenyl)acetic acid (20 mg, 0.104 mmol), 3-(5-amino- 1 H-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate(42 mg, 0.104 mmol), DIPEA (55 μί, 0.313 mmol), TBTU (34 mg, 0.104 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2C12 to give the title compound as a white solid (1 1 mg,
- 147-4820V.1
23%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.45 (s, 1 H), 8.37 (s, 1 H), 8.14 (d, 7=7.53 Hz, 1 H), 7.91 (d, 7=8.28 Hz, 1 H), 7.67 (t, 7=8.03 Hz, 1 H), 7.47 - 7.59 (m, 2 H), 7.16 (t, 7=7.00 Hz, 1 H), 7.08 (d, 7=8.03 Hz, 2 H), 3.92 (s, 2 H), 2.73 (q, 7=7.53 Hz, 4 H), 1.23 (t, 7=7.53 Hz, 6 H); MS ESI [M + H]+463.3, calcd for [Cz^^OsS + H]+ 463.2.
Example A73. 2-Phenyl-N-(3-(3-sulfamoylphenyD- lH-indazol-5-yl)butanamide

The title compound was synthesized according to the General Method A, utilizing 2- phenylbutanoic acid (16 mg, 0.10 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate(40 mg, 0.10 mmol), DIPEA (52 pL, 0.30 mmol), TBTU (32 mg, 0.10 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2CI2 to give the title compound as a white solid (19 mg, 44%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.46 (s, 1 H), 8.37 (s, 1 H), 8.15 (d, 7=8.03 Hz, 1 H), 7.93 (d, 7=7.53 Hz, 1 H), 7.69 (t, 7=7.91 Hz, 1 H), 7.52 (m, 2 H), 7.44 (d, 7=7.28 Hz, 2 H), 7.33 (t, 7=7.50 Hz, 2 H), 7.24 (t, 7=7.50 Hz, 1 H), 3.58 (t, 7=7.40 Hz, 1 H), 2.12 - 2.24 (m, 1 H), 1.78 - 1.90 (m, 1 H), 0.98 (t, 7=7.28 Hz, 3 H); MS ESI [M + H]+ 435.2, calcd for [C23H22N4O3S + H]+ 435.1.

The title compound was synthesized according the General Method A, utilizing 2-(2,6- dimethylphenyl)acetic acid (20 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate(50 mg, 0.124 mmol), DIPEA (65 pL, 0.373 mmol),
-148-44820V.1
TBTU (40 mg, 0.124 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2CI2 to give the title compound as a white solid (8 mg, 15%). 'H NMR (400 MHz, DMSO-d6) δ ppm 13.39 (s, 1 H), 8.46 (s, 1 H), 8.40 (s, 1 H), 8.07 (d, 7=9.54 Hz, 1 H), 7.82 (d, 7=7.78 Hz, 1 H), 7.72 (t, 7=7.80 Hz, 1 H), 7.57 (br. s., 2 H), 7.46 (br. s., 2 H), 7.02 (br. s., 3 H), 3.78 (s, 2 H), 2.29 (s, 3 H); MS ESI [M + H]+ 435.2, calcd for
[C23H22N4O3S + H]+ 435.1.

The title compound was synthesized according to the General Method A, utilizing 2-hydroxy-2- phenylacetic acid (15 mg, 0.10 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate(40 mg, 0.10 mmol), DIPEA (52 ί, 0.30 mmol), TBTU (32 mg, 0.10 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (10 mg, 24%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.47 (s, 1 H), 8.41 (s, 1 H), 8.17 (d, 7=7.53 Hz, 1 H), 7.94 (d, 7=7.78 Hz, 1 H), 7.69 (t, 7=7.65 Hz, 1 H), 7.53 - 7.65 (m, 4 H), 7.39 (t, 7=7.50 Hz, 2 H), 7.33 (t, 7=7.50 Hz, 1 H), 5.22 (s, 1 H); MS ESI [M + H]+ 423.2, calcd for [C2iH18N404S + H]+ 423.1.
ExampleA76. N-(3-(3-Sulfamoylphenyl)-lH-indazol-5-yl)-2-o-tolylacetamide

The title compound was synthesized according to the General Method A, utilizing 2- tolylacetic acid (12 mg, 0..075 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate(30 mg, 0.075 mmol), DIPEA (39 μί, 0.225 mmol), TBTU (24 mg, 0.075 mmol)
-149-44820v.1
and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2C12 to give the title compound as a white solid (10 mg, 32%). Ή NMR (400 MHz, METHANOL-^) 8 ppm 8.46 (s, 1 H), 8.39 (s, 1 H), 8.14 (d, 7=7.53 Hz, 1 H), 7.92 (d, 7=7.53 Hz, 1 H), 7.68 (t, 7=7.78 Hz, 1 H), 7.50 - 7.52 (m, 2 H), 7.25 - 7.30 (m, 1 H), 7.13 - 7.22 (m, 3 H), 3.79 (s, 2 H), 2.38 (s, 3 H); MS ESI [M + H]+421.2, calcd for [C22H20N4O3S + H]+ 421.1.

The title compound was synthesized according to the General Method A, utilizing a- isopropylphenylacetic acid (22 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 μί, 0.372 mmol), TBTU (40 mg, 0.124 mmol) and 2 mL of DMF. The reaction mixture was purified using preparative HPLC to give the desired product and some di-acetylated byproduct. The di- acetylated byproduct was then dissolved in 2 mL of methanol and then 2 M of sodium methanolate in methanol (100 μί) was added and then stirred at 45°C for 4 h. This was purified by preparative HPLC, comebined with the pure product from the first HPLC run and triturated with CH2C12 to give the title compound as a white solid (5.8 mg, 10 %). Ή NMR (400 MHz, METHANOLS) δ ppm 8.45 (s, 1 H), 8.36 (s, 1 H), 8.15 (d, 7=7.03 Hz, 1 H), 7.93 (d, 7=8.28 Hz, 1 H), 7.67 - 7.73 (m, 1 H), 7.49 - 7.56 (m, 2 H), 7.44 - 7.48 (m, 2 H), 7.33 (t, 7=7.10 Hz, 2 H), 7.25 (t, 7=8.00 Hz, 1 H), 3.23 (d, 7=10.79 Hz, 1 H), 2.42 - 2.52 (m, 1 H), 1.13 (d, 7=6.27 Hz, 3 H), 0.75 (d, 7=6.27 Hz, 3 H); MS ESI [M + H]+ 449.2, calcd for [C24H24N4O3S + H]+ 449.2.
-150-4820V.1

The title compound was synthesized according to the General Method A, utilizing 2-(2- isopropylphenyl)acetic acid (26 mg, 0..15 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (60 mg, 0.15 mmol), DIPEA (78 μί, 0.45 mmol), TBTU (48 mg, 0.15 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2C12 to give the title compound as a white solid (33 mg, 49%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.39 (s, 1 H), 10.29 (s, 1 H), 8.41 (s, 1 H), 8.39 (s, 1 H), 8.07 (d, 7=7.53 Hz, 1 H), 7.82 (d, 7=7.53 Hz, 1 H), 7.72 (t, 7=7.20 Hz, 1 H), 7.58 (s, 2 H), 7.46 (s, 2 H), 7.18 - 7.33 (m, 3 H), 7.13 (t, 7=8.50 Hz, 1 H), 3.76 (s, 2 H), 3.19 - 3.28 (m, 1 H), 1.18 (d, 7=6.78 Hz, 6 H); MS ESI [M + H]+ 449.3, calcd for [C^H^N^S + H]+ 449.2.
Example 79. 2-(2-Ethyl-6-methylphenyl)-N-(3-(3-sulfamoylphenyl)- lH-indazol-5-yl)acetamide

The title compound was synthesized according the General Method A, utilizing 2-(2- ethyl-6-methylphenyl)acetic acid (22 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 L, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2C12, followed by dissolving in MeOH and precipitating on addition of water to give the title compound as a white solid (7 mg, 13%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.45 (s, 1 H), 8.39 (s, 1 H), 8.14 (d, 7=7.03 Hz, 1 H), 7.92 (d, 7=7.53 Hz, 1 H), 7.68 (t, 7=8.03 Hz, 1 H), 7.48 - 7.59 (m, 2 H), 7.03 - 7.13 (m, 3 H), 3.90 (s, 2
-151-4820V.1
H), 2.73 (q, 7=7.78 Hz, 2 H), 2.37 (s, 3 H), 1.22 (t, 7=8.03 Hz, 3 H); MS ESI [M + H]+ 449.2, calcd for [C24H24N4O3S + H]+ 449.2.

The title compound was synthesized according to the General Method A utilizing 2-(2,5- dimethylphenyl)acetic acid (20 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 μί, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with (¾<-¾, followed by dissolving in MeOH and precipitating on addition of water to give the title compound as a white solid (17 mg, 32%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.39 (s, 1 H), 10.26 (s, 1 H), 8.45 (s, 1 H), 8.40 (s, 1 H), 8.08 (d, 7=8.28 Hz, 1 H), 7.82 (d, 7=6.78 Hz, 1 H), 7.73 (t, 7=7.65 Hz, 1 H), 7.57 (s, 2 H), 7.46 (s, 2 H), 7.03 - 7.09 (m, 2 H), 6.96 (d, 7=7.28 Hz, 1 H), 3.67 (s, 2 H), 2.26 (s, 3 H), 2.25 (s, 3 H); MS ESI [M + H]+ 435.3, calcd for [C23H22N4O3S + H]+ 435.1.
Example A81. 3-(5-(Benzyloxy)-lH-indazol-3-yl)benzenesulfonamide
-(Benzyloxy)-l H-indazole

-152-4820V.1
A degassed DMF (30 mL) suspension of lH-indazol-5-ol (1.0 g, 7.5 mmol) and K2CO3 (2.0 g, 15 mmol) was treated with benzylbromide (0.98 mL, 8.20 mmol) at 0 oC. The reaction was stirred with cooling for 2 h and then allowed slowly to warm to rt overnight. Later the reaction mixture was diluted with H20 ( 100 mL). A precipitate was collected filtration, rinsed with H20 then suspended in Et20 and isolated by a filtration to provide the title compound as white solid (0.77 g, 46 %). Ή NMR (400 MHz, METHANOLS) δ ppm 7.93 (s, 1 H), 7.43 - 7.50 (m, 3 H), 7.36 - 7.42 (m, 2 H), 7.33 (d, 7=7.03 Hz, 1 H), 7.26 (s, 1 H), 7.15 (d, 7=8.78 Hz, 1 H), 5.12 (s, 2 H); MS ESI 225.0 [M + H]+, calc for [C,4Hi2N20+H]+ 225.1. -(Benzyloxy)-3-iodo-lH-indazole

The title compound was synthesized according to the General Method B, utilizing
5-(benzyloxy)-lH-indazole (0.77 g, 3.4 mmol), K2C03 (1.4 g, 10 mmol) and I2 (1.7 g, 6.9 mmol) in DMF ( 10 mL). An off-white solid (0.88 g, 73 %). Ή NMR (400 MHz, METHANOLS) δ ppm 7.52-7.36 (m, 5 H), 7.34 (d, 7=7.28 Hz, 1 H), 7.21 (d, 7=8.78 Hz, 1 H), 6.91 (s, 1 H), 5.15 (s, 2 H); MS ESI 351.0 [M + H]+, calcd for [C14H, ,ΓΝ20 + H]+ 351.2.
3-(5-(Benzyloxy)-JH-indazol-3-yl)benzenesulfonamide
The title compound was synthesized according to the General Method C, utilizing
5-(benzyloxy)-3-iodo-lH-indazole (0.50 g, 1.4 mmol), 3-(4,4,5,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)benzenesulfonamide (0.53 mg, 1.9 mmol), satd aq Na2C03 (3 mL), PhMe (9 mL), EtOH (9 mL) and Pd(dppf)Cl2 (52 mg, 0.071 mmol). The crude material was in sequence taken into MeOH and then H20 and filtered through Celite using MeOH and subsequently to transfer the crude material. The filtrate was concentratd to dryness affording a solid which was then suspended into H20. Filteration and rinsing with MeCN yieled the title compound to as a light tan solid ( 0.5 g, 92 %). Ή NMR (400 MHz, METHANOL-d4) δ ppm 8.46 (s, 1 H), 8.12 (d,
-153-4820V.1
7=8.53 Hz, 1 H), 7.94 (d, 7=6.78 Hz, 1 H), 7.70 (t, 7=7.53 Hz, 1 H), 7.46 - 7.55 (m, 4 H), 7.40 (m, 2 H), 7.32 (t, 7=7.30 Hz, 1 H), 7.22 (d, 7=8.53 Hz, 1 H), 5.18 (s, 2 H); MS ESI 380.2 [M + H]+, calcd for [C2oHnN303S + H]+ 380.2.

-(2-(Thiophen-2-yl)ethoxy)-lH-indazole

DIAD (0.09 mL, 0.47 mmol) was added dropwise to a solution of lH-indazol-5-ol (50 mg, 0.37 mmol), 2-(thiophen-2-yl)ethanol (58 mg, 0.47 mmol) and PPh3 (0.12 g, 0.47 mmol) and) in anh THF (2 mL) and PhMe (2 mL) at 0 oC. The reaction was stirred with cooling for 4 h and then concentrated under reduced pressure and purified by column chromatography twice (Biotage Si02, 0-12 % MeOH/DCM) and (Si02, 20-50 % EtOAc/hexanes) to provide the title compound a white solid (0.047 g, 52 %). ]H NMR (400 MHz, CDC13) δ ppm 8.12 (br. s., 1 H), 7.57 (d, 7=9.03 Hz, 1 H), 7.20 (d, 7=5.02 Hz, 1 H), 7.1 1 (s, 1 H), 6.92 - 7.01 (m, 3 H), 4.25 (t, 7=6.53 Hz, 2 H), 3.37 (t, 7=6.53 Hz, 2 H); MS ESI 244.9 [M + H]+, calcd for [C^HnNzOS + H]+ 245.1.
3-Iodo-5-(2-(thiophen-2-yl)ethoxy)-lH-indazole

The title compound was synthesized according to the General Method B, utilizing
5-(2-(thiophen-2-yl)ethoxy)-lH-indazole (0.049 g, 0.20 mmol), KOH (47 mg, 0.84 mmol) and 12 (0.078 g, 0.31 mmol) in DMF ( 10 mL). A white solid (0.059 g, 81 %). Ή NMR (400 MHz,
- 154-4820V.1
METHANOL-dA) 8 ppm 7.32 (d, 7=9.03 Hz, 1 H), 7.23 (d, 7=4.77 Hz, 1 H), 6.92 - 6.99 (m, 3 H), 6.89 (dd, 7=8.91, 1.88 Hz, 1 H), 4.26 (t, 7=6.40 Hz, 2 H), 3.35 (t, 7=6.40 Hz, 2 H); MS ESI 370.9 [M + H]+, calcd for [C13H„IN20S + H]+ 371.2.
3-(5-(2-(Thiophen-2-yl)ethoxy)-lH-indazol-3-yl)benzenesulfonamide
The title compound was synthesized according to the General Method C, utilizing
A degassed mixture of 3-iodo-5-(2-(thiophen-2-yl)ethoxy)-lH-indazole (0.030 g, 0.081 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzenesulfonamide (0.030 mg, 0.1 1 mmol) KF (14 mg, 0.24 mmol) and Pd(PPh3)4 (5 mg, 0.004),) in DMF (1.5 mL)/H20 (0.5 mL) was heated sealed in a microwave reactor at 120 oC for 2 h. The crude material filtered through Celite (MeOH), concentrated under reduced pressure and subsequently purified by column
chromatography (Biotage Si02, 0-9 % MeOH/DCM) and later by preparative TLC (Si02, 10 % MeOH/DCM). The matial was tritrated with hexanes, then dissolved in 2% MeOH/DCM and filtered to afford the title compound as a white powder (10 mg, 32 %). Ή NMR (400 MHz, METHANOLS) δ ppm 8.46 (s, 1 H), 8.14 (d, 7=7.78 Hz, 1 H), 7.93 (d, 7=8.03 Hz, 1 H), 7.70 (t, 7=7.78 Hz 1 H), 7.51 (d, 7=9.03 Hz, 1 H), 7.40 (s, 1 H), 7.22 (d, 7=5.02 Hz, 1 H), 7.17 (dd, 7=9.03, 2.01 Hz, 1 H), 6.91 - 7.00 (m, 2 H), 4.29 (t, 7=6.40 Hz, 2 H), 3.34 (t, 7=6.40 Hz, 2 H); MS ESI 400.0 [M + H]+, calcd for [C19H17N3O3S2 + H]+ 400.1.
Example A83. 3-(5-(2-Methoxy-2-phenylethoxy)- 1 H-indazol-3-yl)benzenesulfonamide

B. 3 ethoxy)-lH-indazole

The title compound was synthesized according to the General Method B, utilizing 5-(2-methoxy-2-phenylethoxy)-lH-indazole (98 mg, 0.36 mmol), K2C03 (150 mg, 1.1 mmol) and I2 (185 mg, 0.73 mmol) in DMF ( 3 mL). The reaction was quenched by an addition of xs 10 % aq NaHSC>3, partitioned between EtOAc and brine. The organic layer was washed with brine, dried (Na2S04) and concentrated under reduced pressure The precipitate was isolated by filtration, washed by H20 and purified by column chromatography (Si02, 0-8 % MeOH/DCM) to provide the title compound to as a pale orange foam (144 mg, quant). Ή NMR (400 MHz, METHANOLS) δ ppm 7.29 - 7.49 (m, 6 H), 7.13 (d, 7=9.03 Hz, 1 H), 6.77 (s, 1 H), 4.64 (dd, 7=7.15, 3.64 Hz, 1 H), 4.19 (t, 7=7.78 Hz, 1 H), 4.08 (dd, 7=10.04, 3.51 Hz, 1 H), 3.34 (s, 3 H),; MS ESI 395.0 [M + H]+, calcd for [Ci6Hi5IN202 + H]+ 395.2. C. 3-( 5-(2-methoxy-2-phenylethoxy)-lH-indazol-3-yl)benzenesulfonamide
The title compound was synthesized according to the General Method C, utilizing 3-iodo-5-(2- methoxy-2-phenylethoxy)-lH-indazole (80 mg, 0.20 mmol), 3-(4,4,5,5-tetramethyl- 1,3,2-
-156-4820V.1
dioxaborolan-2-yl)benzenesulfonamide (75 mg, 0.26 mmol), satd aq Na2C03 (0.5 mL), PhMe (1.5 mL), EtOH (1.5 mL) and Pd(dppf)Cl2 (7.4 mg, 0.010 mmol). The crude material after filtration through Celite using MeOH was purified by preparative HPLC followed by column chromatography (Biotage Si02, 5-100 % EtOAc/hexanes), trituration with hexanes, preparative TLC (Si02, 2: 1 EtOAc/hexanes) and repurification by preparative HPLC provided the title compound to as a white powder (40 mg, 47 %). Ή NMR (400 MHz, METHAN OL-d^) δ ppm 8.44 (s, 1 H), 8.12 (d, 7=7.78 Hz, 1 H), 7.92 (d, 7=8.03 Hz, 1 H), 7.68 (t, 7=7.28 Hz, 1 H), 7.26 - 7.55 (m, 7 H), 7.15 (d, 7=9.03 Hz, 1 H), 4.65 (dd, 7=7.40, 3.64 Hz, 1 H), 4.23 (dd, 7=7.78, 7.28 Hz, 1 H), 4.14 (dd, 7=10.54, 4.02 Hz, 1 H), 3.34 (s, 3 H); MS ESI 424.3 [M + H]+, calcd for [C22H2iN304S + Hf 424.3.
Example A84. 3-(5-(2,6-Diethylphenethoxy)- 1 H-indazol-3- vDbenzenesulfonamide

5 -(2 -diethylphenethoxy )- lH-indazole
The title compound was synthesized according to the method for 3-(5-(2-(thiophen-2-yl)ethoxy)- lH-indazol-3-yl)benzenesulfonamide, utilizing lH-indazol-5-ol (60 mg, 0.45 mmol), 2-(2,6- diethylphenyDethanol (100 mg, 0.56 mmol), PPh3 (0.17 g, 0.56 mmol) and DIAD (0.1 1 mL, 0.56 mmol) in anh THF (2 mL) and PhMe (2 mL). The reaction was taken into EtOAc, washed (satd aq NaHC03, brine), dried (Na2S04), concentrated under reduced pressure and purified by column chromatography (Biotage silica gel, 5-65 % EtOAc in hexanes) to provide the title compound as a white solid (0.13 g, 95 %). Ή NMR (400 MHz, METHANOLS) δ ppm 7.90 (s,
-157-4820V.1
1 H), 7.43 (d, 7=9.03 Hz, 1 H), 7.12 (br. s., 1 H), 7.08 (d, 7=7.03 Hz, 1 H), 7.01 - 7.06 (m, 3 H), 4.09 (t, 7=7.28 Hz, 2 H), 3.21 (t, 7=7.65 Hz, 2 H), 2.74 (q, 7=7.53 Hz, 4 H), 1.13 - 1.39 (m, 6 H); MS ESI 295.1 [M + H]+, calcd for [C19H22N2O + H]+ 295.4. C. 5- -Diethylphenethoxy)-3-iodo-lH-indazole

The title compound was synthesized according to the General Method B, utilizing crude 5- (2,6-diethylphenethoxy)-lH-indazole (0.14 g, 0.46 mmol ), I2 (0.31 g, 1.2 mmol) and K2CO3 (0.19 g, 1.4 mmol) in DMF (3 mL). A white powder (0.078 g, 40 ). Ή NMR (400 MHz, METHANOLS) δ ppm 7.43 (d, 7=9.03 Hz, 1 H), 7.10 - 7.17 (m, 2 H), 7.04 - 7.09 (m, 2 H), 6.79 (s, 1 H), 4.14 (t, 7=7.65 Hz, 1 H), 3.25 (t, 7=7.53 Hz, 1 H), 2.79 (q, 7 =7.53 Hz, 4 H), 1.27 (t, 7=7.53 Hz, 6 H); MS ESI 421.1 [M + H]+, calcd for [C19H21IN2O + H]+ 421.3.
D. 3-(5-(2,6-diethylphenethoxy)-lH-indazol-3-yl)benzenesulfonamide
The title compound was synthesized according to the General Method C, utilizing 5-(2,6- diethylphenethoxy)-3-iodo-lH-indazole (78 mg, 0.19 mmol), 3-(4,4,5,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)benzenesulfonamide (68 mg, 0.24 mmol), satd aq Na2C03 (0.66 mL), PhMe (2 mL), EtOH (2 mL) and Pd(dppf)Cl2 (6.8 mg, 0.0093 mmol). A white powder (42 mg, 50 %). Ή NMR (400 MHz, METHANOLS) δ ppm 8.44 (s, 1 H), 8.10 (d, 7=7.78 Hz, 1 H), 7.92 (d, 7=8.03 Hz, 1 H), 7.68 (t, 7=7.78 Hz, 1 H), 7.50 (d, 7=9.29 Hz, 1 H), 7.35 (s, 1 H), 7.14 (d, 7=9.29 Hz, 1 H), 7.10 (d, 7=7.03 Hz, 1 H), 7.00 - 7.06 (m, 2 H), 4.16 (t, 7=7.53 Hz, 2 H), 3.23 (t, 7=7.28 Hz, 2 H), 2.76 (q, 7=7.28 Hz, 4 H), 1.23 (t, 7=7.53 Hz, 7 H); MS ESI 450.3 [M + H]+, calcd for [C25H27N303S + H]+ 450.3.
-158-4820V.1

3-(5-Amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol) and 2-phenylpropanal (33 μί, 0.25 mmol) was dissolved in DCE / THF (4 mL / 4 mL) and stirred for 10 minutes. NaBH(OAc)3 (159 mg, 0.75 mmol) was then added and the reaction was stirred at room temperature for 1.5 h. The reaction mixture was dried under reduced pressure, purified by preparative HPLC, and triturated with CH2CI2 to give the title compound as a white solid ( 13 mg, 13%). Ή NMR (400 MHz, METHANOL-^) 5 ppm 8.46 (s, 1 H), 8.14 (d, 7=7.53 Hz, 1 H), 7.97 (d, 7=7.53 Hz, 1 H), 7.82 (s, 1 H), 7.72 (t, 7=7.91 Hz, 1 H), 7.69 (d, 7=9.03 Hz, 1 H), 7.29 - 7.39 (m, 5 H), 7.20 - 7.27 (m, 1 H), 3.61 - 3.76 (m, 2 H), 3.15 - 3.25 (m, 1 H), 1.38 (d, 7=7.03 Hz, 3 H); MS ESI [M + H]+ 407.1 , calcd for [C22H22N4O2S + H]+ 407.1
Example A86. 3-(5-( 1 ,2,3,4-Tetrahydronaphthalen-2-ylamino)- 1 H-indazol-3-yl)benzene sulfonamide trifluoroacetate

To a mixture of 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (101 mg, 0.25 mmol) and 3,4-dihydronaphthalen-2(lH)-one (40 mg, 0.275 mmol) in THF (8 mL) was added DCE (8 mL), followed by NaBH(OAc)3 (85 mg, 0.4 mmol), The resulting mixture was stirred for 2 h at rt. After quenching with ¾0 ( 10 mL), satd. aq NaHC03 (10 mL), it was extracted with CH2C12 (20 mL x 2), the dried over Na2S04. After removal of solvent, the residue was redissolved in DMF and purifed by preparative HPLC to give the title compound as a light yellow solid (50 mg, 38%). Ή NMR (400 MHz, CD3OD) δ 8.50 (s, 1H), 8.24 (s, 1H), 8.20 (d, 7 =7.6 Hz, 1H), 7.98 (d, 7 =7.6 Hz, 1H), 7.86 (d, 7 =8.8 Hz, 1H), 7.72 (t, 7 =8.0 Hz, 1H), 7.60 (d,
- 159-4820V.1
J =8.8 Hz, IH), 7.15-7.05 (m, 4H), 4.12-4.02 (m, 1 H), 3.14 (dd, / = 16.0 Hz, J =4.8 Hz, 1 H), 3.10-2.88 (m, 3 H), 2.40-2.30 (m, 1 H), 2.05-1.93 (m, 1 H); MS ESI 419.2 [M + H]+, calcd for [C23H22N4O2S + H]+ 419.1. Example A87. 3-(5-((2,6-Diethylbenzylamino)methyl)- 1 H-indazol-3-yl)benzenesulfonamide
2,2,2-trifluoroacetate

1 ,2-Dichloroethane (0.50 mL) and THF (0.25 mL) were added to a mixture of sodium triacetoxyborohydride (32 mg, 0.15 mmol), 3-(5-formyl- lH-indazol-3-yl)benzenesulfonamide (26.2 mg, 0.087 mmol) and (2,6-diethylphenyl)methanamine** ( 16.2 mg, 0.10 mmol) under an atmosphere of argon. The mixture was stirred at room temperature for 18h. Water (lmL) was added to quench excess reagent. Methanol was added, and the crude mixture was poured onto a Waters Porapak Rxn-CX cartridge, rinsing first with methanol (20 mL), then the product was eluted with 2 M ammonia in methanol (25 mL). Purification by preparative HPLC yielded the product as a white solid (35.7 mg, 73%). Ή NMR (400 MHz, CD3OD) δ 8.53 (s, IH), 8.35 (s,
IH) , 8.25 (d, J =12 Hz, IH), 7.99 (d, J =7.2 Hz, IH), 7.77 (d, J =8.0 Hz, IH), 7.74 (t, J =7.6 Hz, IH), 7.65 (d, J =8.4 Hz, IH), 7.30 (t, J =7.6 Hz, IH), 7.13 (d, J =7.6 Hz, 2 H), 4.58 (s, 2 H), 4.28 (s, 2H), 2.54 (q, J =7.6 Hz, 4H), 1.08 (t, J =7.6 Hz, 6H); MS ESI 449.2 [M + H]+, calcd for [C25H28N402S + H]+ 449.2.
** (2,6-diethylphenyl)methanamine was prepared as described by V. Bertini, A. De Munno, F. Lucchesini, F. Buffoni, and B. Bertocci, in US Patent number 4,888,283 (example
I I) .
-160-4820V.1
Example A.88. 3-(5-((Benzhvdrylarrdno methyl)-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate

According to example A87, using 3-(5-formyl-lH-indazol-3-yl)benzenesulfonamide (25.5 mg, 0.085 mmol), diphenylmethanamine (17.7 mg, 0.096 mmol) and sodium
triacetoxyborohydride (29 mg, 0.14 mmol), the title compound was prepared as a white solid (31.0 mg, 63%). Ή NMR (400 MHz, CD3OD plus few drops of CDC13) δ 8.48 (s, 1H), 8.16 (d, J =8.0 Hz, 1H), 8.06 (s, 1H), 7.99 (d, J =7.6 Hz, 1H), 7.67-7.73 (m, 2H), 7.41-7.54 (m, 11H), 5.53 (s, 1H), 4.36 (s, 2H); MS ESI 469.2 [M + H]+, calcd for [C27H24N4O2S + H]+ 469.2.
Example

The title compound was synthesized according to the General Method C, utilizing 5- bromo-3-iodo-lH-indazole (40 mg, 0.02 mmol), 3-(4,4,5,5-tetramethyl-l,3,2-diox borolan-2- yl)benzenesulfonamide (35 mg, 0.12 mmol), satd aq Na2CC>3 (0.5 mL), PhMe (1.5 mL), EtOH (1.5 mL) and Pd(dppf)Cl2 (5 mg, 0.006 mmol).
Degassed and sealed reaction mixture was heated in a microwave reactor at 120 oC for 1 h. The reaction vial was then cooled to rt, briefly opened and charged with (E)-3-phenylprop-l- enylboronic acid (26 mg, 0.16 mmol) and Pd(dppf)Cl2 (8 mg, 0.01 1 mmol). After repeated degassing, the reaction mixture was sealed and heated in a microwave reactor at 130 °C for 2 h. The crude material after filtration through Celite using MeOH was purified by preparative HPLC to provide the title compound to as a white powder (8 mg, 17 %). . Ή NMR (400 MHz,
-161-4820V.1
METHANOL-^) δ ppm 8.50 (s, 1 H), 8.19 (d, 7=7.03 Hz, 1 H), 7.94 (s, 2 H), 7.71 (t, 7=7.78 Hz, 1 H), 7.64 (d, 7=8.78 Hz, 1 H), 7.53 (d, 7=9.29 Hz, 1 H), 7.25 - 7.34 (m, 4 H), 7.16 - 7.23 (m, 1 H), 6.66 (d, 7=15.31 Hz, 1 H), 6.37 - 6.49 (m, 1 H), 3.59 (d, 7=7.03 Hz, 2 H); MS ESI 390.2 [M + H]+, calcd for [C22H19N3O2S + H]+ 390.2.

-((3-Iodo-lH-indazol-5-yl)methyl)benzamide

TFA (0.075 mL, 1.0 mmol) was added dropwise to a solution of lH-indazole-5- carbaldehyde (50 mg, 0.34 mmol), benzamide ( 124 mg, 1.0 mmol) and Et3SiH (0.16 mL, 1.0 mmol) in anh MeCN (6 mL) at it. The reaction was stirred at rt for 1.5 h and then heated at 50- 65 oC for 4 d. The reaction was later concentrated under reduced pressure and purified by column chromatography (Biotage Si02, 0-8 % MeOH/DCM).; MS ESI 251.9 [M + H]+, calcd for [C15H13N3O+ H]+ 252.3. The material was subjected to condition described by the general method for iodination using K2C03 ( 190 mg, 1 .4 mmol), 12 ( 1 17 mg, 2.2 mmol) and DMF (2 mL) to afford the title compound as pale yellow solid (66 mg, 51 %). Ή NMR (400 MHz, METHANOL-dn) δ ppm 7.88 (d, 7=7.53 Hz, 2 H), 7.41 - 7.59 (m, 6 H), 4.71 (s, 2 H). N-((3-( 3-sulfamoylphenyl )-1Η- indazol-5-yl jmethyl )benzamide
The title compound was synthesized according to the General Method C utilizing N-((3- iodo-l H-indazol-5-yl)methyl)benzamide (40 mg, 0.1 1 mmol), 3-(4,4,5,5-tetramethyl- 1 ,3,2- dioxaborolan-2-yl)benzenesulfonamide (39 mg, 0.14 mmol), satd aq Na2C03 (0.5 mL), PhMe
- 162-4820V.1
(1.5 mL), EtOH (1.5 mL) and Pd(dppf)Cl2 (4 mg, 0.005 mmol). Purification by preparative HPLC provided the title compound to as a white powder (20 mg, 28 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.51 (s, 1 H), 8.19 (d, 7=7.78 Hz, 1 H), 8.07 (s, 1 H), 7.94 (d, 7=8.03 Hz, 1 H), 7.86 (d, 7=7.28 Hz, 2 H), 7.69 (t, 7=7.91 Hz, 1 H), 7.42 - 7.62 (m, 5 H), 4.73 (s, 2 H); MS ESI 407.1 [M + H]+, calcd for
 + H]+ 407.1.
+ H]+ 407.1.
Example A91. 2-πιοφΙιο1ΐηο-2-ρ1ΐ6ην1-Ν-(3-(3-5ϋ^πΐον1ρ1ΐ6ην1)- lH-indazoI-5-yl)acetamide 2,2,2-trifluoroacetate

The title compound was synthesized according to the General Method A, utilizing 3-(5- amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (80 mg, 0.20 mmol), 2- morpholino-2-phenylacetic acid (48 mg, 0.22 mmol), N-ethyl-N-isopropylpropan-2-amine (0.09 mL, 0.5 mmol) and 0-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (70 mg, 0.22 mmol) in DMF (3 mL). The crude material was purified by preparative HPLC to provide 2-moφholino-2-phenyl-N-(3-(3-sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide 2,2,2- trifluoroacetate to as a white powder (27 mg, 23 %). Ή NMR (400 MHz, METHANOL-d4) δ ppm 8.49 (s, 1 H), 8.46 (s, 1 H), 8.15 (d, 7=7.28 Hz, 1 H), 7.96 (d, 7=8.03 Hz, 1 H), 7.66 - 7.77 (m, 3 H), 7.58 (m, 4 H), 7.51 (d, 7 =8.78, 1 H), 5.09 (s, 1 H), 3.95 (br. s., 4 H) *; MS ESI 492.3 [M + H]+, calcd for [C25H25N504S + H]+ 492.3.
*4 H hidden under H20 or METHANOLS peak.
-163-4820V.1
Example A92. 2-(2-6ΐ1ιν1ρ1ΐ6ην1)-2-ηιο 1ιο1ϊηο-Ν-(3-(3-5ο1ί3ηΐον1ρ1ΐ6ην1')- 1 H-indazol-5-yl) acetamide trifluoroacetate

To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (100 mg, 0.25 mmol), 2-(2-ethylphenyl)-2-morpholinoacetic acid (63 mg, 0.25 mmol) and TBTU (81 mg, 0.25 mmol) in DMF (5 mL) at 0 °C was added 'Pr2NEt (0.13 mL, 0.75 mmol). The resulting mixture was stirred for lh at 0 °C then purified by preparative HPLC to give the title compound as a white solid (61 mg, 39%). Ή NMR (400 MHz, CD3OD) δ 8.48 (s, 1H), 8.41 (s, 1H), 8.13 (d, J =7.6 Hz, 1H), 7.94 (d, J =7.6 Hz, 1H), 7.88 (d, J =8.0 Hz, 1H), 7.68 (t, J =7.6 Hz, 1H), 7.60-7.36 (m, 5H), 5.47 (s, 1H), 3.96 (s, 4H), 3.80-2.90 (m, 6H), 1.39 (t, J =7.4 Hz, 3H); MS ESI 520.3 [M + H]+, calcd for [C27H29N504S + H]+ 520.2.
Example A93. 2-(2,6-Dimethylphenyl)-2-morpholino-N-(3-(3-sulfamoylphenyl -lH-indazol-5-

To a mixture of 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (100 mg, 0.25 mmol), 2-(2,6-dimethylphenyl)-2-morpholinoacetic acid (63 mg, 0.25 mmol) and TBTU (81 mg, 0.25 mmol) in DMF (5 mL) at 0 °C was added 'Pr2NEt (0.13 mL, 0.75 mmol). After addition, the resulting mixture was stirred O/N at rt. It was purified by preparative HPLC, biotage column then second preparative HPLC to give the title compound as a white solid (6.9 mg, 4%). Ή NMR (400 MHz, CD3OD) δ 8.48 (s, 1H), 8.35 (s, 1H), 8.14 (d, J =7.6 Hz, 1H), 7.95 (d, J =8.8 Hz, 1H), 7.69 (t, J =8.0 Hz, 1H), 7.58 (d, / =9.2 Hz, 1H), 7.53 (d, / =8.4 Hz, 1H), 7.36 (t, J =7.2 Hz, 1H), 7.27 (d, J =6.4 Hz, 2H), 5.65 (s, 1H), 4.00 (s, 4H), 3.32 (s, 4H,
-164-4820V.1
overlapping with MeOH), 2.56 (s, 6H); MS ESI 520.3 [M + H]+, calcd for [C27H29N5O4S + H]+ 520.2.
Example A94. N-(3-(3-Sulfamoylphenyl)- lH-indazol-5-yl)-2-m-tolylacetamide

The title compound was synthesized according to the method the General Method A, utilizing m-tolylacetic acid (19 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 μί, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2C12 to give the title compound as a white solid (30 mg, 58%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.39 (s, 1 H), 10.28 (s, 1 H), 8.43 (s, 1 H), 8.39 (s, 1 H), 8.08 (d, 7=6.78 Hz, 1 H), 7.83 (d, 7=7.78 Hz, 1 H), 7.73 (t, 7=7.00 Hz, 1 H), 7.57 (s, 2 H), 7.46 (s, 2 H), 7.1 1 - 7.24 (m, 3 H), 7.06 (d, 7=8.03 Hz, 1 H), 3.62 (s, 2 H), 2.29 (s, 3 H). MS ESI [M + H]+ 421.2, calcd for [C22H20N4O3S + H]+ 421.1.

The title compound was synthesized according the General Method A, utilizing mesitylacetic acid (22 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide
2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 μί, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC to give the title compound as a white solid (31 mg, 56%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.38 (s, 1
-165-4820V.1
H), 10.26 (s, 1 H), 8.44 (s, 1 H), 8.40 (s, 1 H), 8.07 (d, 7=7.53 Hz, 1 H), 7.82 (d, 7=8.28 Hz, 1 H), 7.72 (t, 7=8.30 Hz, 1 H), 7.56 (m, 2 H), 7.46 (s, 2 H), 6.83 (s, 2 H), 3.72 (s, 2 H), 2.25 (s, 6 H), 2.21 (s, 3 H). MS ESI [M + H]+ 449.3, calcd for [C24H24N4O3S + H]+ 449.2.

The title compound was synthesized according to the method described for the General Method A, utilizing p-tolylacetic acid (11 mg, 0.075 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (30 mg, 0.075 mmol), DIPEA (39 μί, 0.224 mmol), TBTU (24 mg, 0.075 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with CH2CI2 to give the title compound as a white solid (4 mg, 13%). Ή NMR (400 MHz, Acetone-d6) δ ppm 12.50 (br. s., 1 H), 9.48 (br. s., 1 H), 8.54 (s, 1 H), 8.51 (s, 1 H), 8.19 (d, 7=8.03 Hz, 1 H), 7.91 (d, 7=8.03 Hz, 1 H), 7.66 - 7.76 (m, 2 H), 7.59 (d, 7=8.78 Hz, 1 H), 7.29 (d, 7=7.53 Hz, 2 H), 7.14 (d, 7=8.03 Hz, 2 H), 6.71 (br. s., 2 H), 3.68 (s, 2 H), 2.30 (s, 3 H). MS ESI [M + H]+ 421.2, calcd for [C22H20N4O3S + H]+ 421.1.

2-(2-((tert-butoxycarbonylamino)methyl)phenyl)acetic acid (41 mg, 0.25 mmol), 3-(5- amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol), DIPEA (130 xL, 0.75 mmol) was dissolved in DMF (6 mL) and cooled to 0 °C. TBTU (80 mg, 0.25 mmol) was added and the reaction was stirred for 60 minutes. Water (60 mL) was added to
-166-4820V.1
precipitate out the compound. The brown precipitate was filtered and dried under reduced pressure. CH2CI2 (6 mL) was added and the mixture was cooled to 0 °C. TFA (4 mL) was added and the reaction was stirred at 0 °C for 60 minutes. The reaction mixture was dried under reduced pressure, redissolved in MeOH, purified by preparative HPLC, and triturated with Et20 to give the title compound as an off-white solid (30 mg, 22%). Ή NMR (400 MHz, METHANOL-d*) δ ppm 8.66 (s, 1 H), 8.52 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.92 (d, 7=7.53 Hz, 1 H), 7.67 (t, 7=7.91 Hz, 1 H), 7.51 - 7.60 (m, 2 H), 7.48 (d, 7=8.28 Hz, 4 H), 4.28 (s, 2 H), 3.97 (s, 2 H). MS ESI [M + H]+ 436.2, calcd for [C22H2iN503S + H]+ 436.1.

The title compound was synthesized according to the General Method A, utilizing 2- (dimethylamino)-2-phenylacetic acid■ HC1 (32 mg, 0.15 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (60 mg, 0.15 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (48 mg, 0.15 mmol) and 5 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (38 mg, 45%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.48 (s, 1 H), 8.41 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.95 (d, 7=8.03 Hz, 1 H), 7.65 - 7.73 (m, 3 H), 7.50 - 7.60 (m, 5 H), 5.04 (s, 1 H), 3.10 (br. s, 3 H), 2.65 (br. s, 3 H) MS ESI [M + H]+ 450.2, calcd for [C23H23N5O3S + H]+ 450.1.
Example A99. (S)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)pyrrolidine-2-carboxamide 2,2,2- trifluoroacetate

-167-4820V.1
The title compound was synthesized according to the General Method A utilizing (S)-l - (tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (32 mg, 0.15 mmol), 3-(5-amino-lH-indazol- 3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (60 mg, 0.15 mmol), DIPEA (78 μί, 0.45 mmol), TBTU (48 mg, 0.15 mmol) and 4 mL of DMF. The crude product was precipitated out with water, deprotected using TFA / CH2C12 (3 mL / 1 mL) and purified by preparative HPLC. Triturating with CH2C12 gave the title compound as a white solid (9 mg, 12%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.49 (s, 1 H), 8.43 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.70 (t, 7=7.78 Hz, 1 H), 7.58 (m, 2 H), 4.45 (t, 7=7.15 Hz, 1 H), 3.45 - 3.54 (m, 1 H), 3.35 - 3.45 (m, 1 H), 2.51 - 2.62 (m, 1 H), 2.15 (m, 3 H). MS ESI [M + H]+ 386.1 , calcd for [Ci8H,9N503S + Hf 386.1.

The title compound was synthesized according to the General Method A, utilizing 2- (dimethylamino)-2-(2-ethylphenyl)acetic acid · HC1 (30 mg, 0.124 mmol), 3-(5-amino- lH- indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (108 xL, 0.621 mmol), TBTU (40 mg, 0.124 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 and CH2C12 to give the title compound as a white solid (15 mg, 20%).Ή NMR (400 MHz, METHANOL-^) δ ppm 8.47 (s, 1 H), 8.38 (s, 1 H), 8.14 (d, 7=7.28 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.77 (d, 7=8.03 Hz, 1 H), 7.70 (t, 7=8.16 Hz, 1 H), 7.56 (s, 2 H), 7.50 (d, 7=4.02 Hz, 2 H), 7.35 - 7.42 (m, 1 H), 5.36 (s, 1 H), 2.91 - 3.10 (m, 2 H), 3.07 (br. s, 3 H), 2.65 (br. s, 3 H), 1.39 (t, 7=7.40 Hz, 3 H). MS ESI [M + H]+ 478.3, calcd for [C25H27N503S + H]+ 478.2.
- 168-4820V.1
Example A1Q1. (R)-2-arnino-2-phenyl-N-(3-(3-sulfamoylphenyl)- lH-indazol-5-yl)acetamide 2.2,2-trifluoroacetate

The title compound was synthesized according to the General Method A utilizing (R)-2- (tert-butoxycarbonylamino)-2-phenylacetic acid (31 mg, 0.124 mmol), 3-(5-amino-lH-indazol- 3-yl)benzenesulfonamide 2,2,2-trifluoroacetate(50 mg, 0.124 mmol), DIPEA (65 μί, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 4 mL of DMF. The crude product was precipitated with water, deprotected using TFA / CH2C12 (3 mL / 1 mL) and purified by preparative HPLC. The undesired (N-indazole) isomer was hydrolyzed using 2 M sodium methoxide (0.5 mL) and heating at 40 °C for 16 h. A second purification by preparative HPLC gave the title compound as a white solid (25 mg, 38%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.48 (s, 1 H), 8.40 (s, 1 H), 8.14 (d, 7=7.78 Hz, 1 H), 7.95 (d, 7=8.03 Hz, 1 H), 7.69 (t, 7=7.91 Hz, 1 H), 7.64 (d, 7=6.78 Hz, 2 H), 7.45 - 7.59 (m, 5 H), 5.16 (s, 1 H). MS ESI [M + H]+ 422.2, calcd for [C21H19N5O3S + H]+ 422.1.
Example A 102. N-(3-(3-Sulfamoylphenyl)-lH-indazol-5-yl)isoindoline-l-carboxamide 2.2,2- trifluoroacetate

The title compound was synthesized according to the the General Method A utilizing 2- (tert-butoxycarbonyl)isoindoline-l-carboxylic acid (39 mg, 0.15 mmol), 3-(5-amino-lH-indazol- 3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (60 mg, 0.15 mmol), DIPEA (78 iL, 0.45 mmol), TBTU (48 mg, 0.15 mmol) and 4 mL of DMF. The crude product was precipitated out with water, deprotected using TFA / CH2C12 (3 mL / 1 mL) and purified by preparative HPLC. Triturating with CH2C12 and Et20 gave the title compound as a white solid (12 mg, 15%). Ή
- 169-4820V.1
NMR (400 MHz, METHANOL-^) δ ppm 8.47 (s, 1 H), 8.45 (s, 1 H), 8.14 (d, 7=6.52 Hz, 1 H), 7.93 (d, 7=7.03 Hz, 1 H), 7.57 - 7.73 (m, 4 H), 7.50 (m, 3 H), 5.72 (s, 1 H), 4.74 (d, 7=14.56 Hz, 2 H). MS ESI [M + H]+ 434.2, calcd for [C22Hi9N503S + H]+ 434.1.

The title compound was synthesized according to the General Method A, utilizing 2- (dimethylamino)-2-o-tolylacetic acid · HC1 (28 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (108 ί, 0.621 mmol), TBTU (40 mg, 0.124 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 and CH2C12 to give the title compound as a white solid (15 mg, 21%).Ή NMR (400 MHz, METHANOL-^) δ ppm 8.48 (br. s., 1 H), 8.37 (br. s., 1 H), 8.14 (d, 7=7.03 Hz, 1 H), 7.95 (d, 7=6.02 Hz, 1 H), 7.65 - 7.77 (m, 2 H), 7.51 - 7.62 (m, 2 H), 7.32 - 7.47 (m, 3 H), 5.28 (s, 1 H), 2.83 (br. s., 6 H), 2.65 (s, 3 H). MS ESI [M + H]+ 464.2 calcd for [C24H25N5O3S + H]+ 464.2.
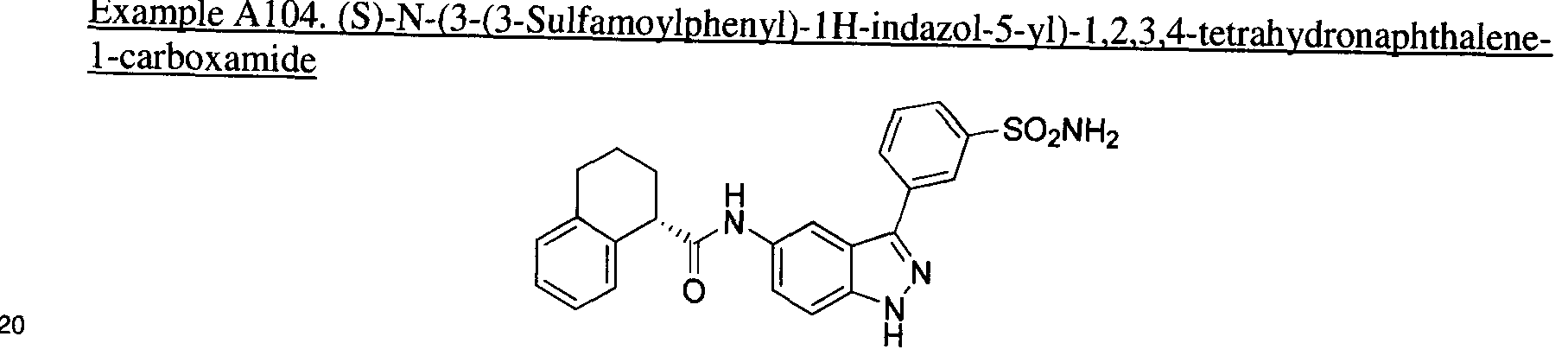
The title compound was synthesized according to the General Method A, utilizing (S)- 1 ,2,3,4-tetrahydronaphthalene-l-carboxylic acid (22 mg, 0.124 mmol), 3-(5-amino-lH-indazol- 3-yl)benzenesulfonamide 2,2,2-trifluoroacetate(50 mg, 0.124 mmol), DIPEA (65 μί, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 3 mL of DMF. The reaction mixture was purified using
-170- 4820V.1
preparative HPLC and triturated with Et20 to give the title compound as a white solid (11 mg, 20%). 'H NMR (400 MHz, DMSO-d6) δ ppm 13.39 (br. s., 1 H), 10.36 (br. s., 1 H), 8.50 (s, 1 H), 8.41 (s, 1 H), 8.08 (d, 7=7.78 Hz, 1 H), 7.82 (d, 7=8.03 Hz, 1 H), 7.72 (t, 7=7.40 Hz, 1 H), 7.56 - 7.65 (m, 2 H), 7.46 (s, 2 H), 7.08 - 7.16 (m, 4 H), 3.89 (t, 7=6.15 Hz, 1 H), 2.71 - 2.82 (m, 2 H), 1.97 - 2.10 (m, 3 H), 1.60 - 1.73 (m, 1 H) MS ESI [M + H]+ 447.3, calcd for [C24H22N4O3S + H]+ 447.1.

The title compound was synthesized according to the General Method A, utilizing 2-phenyl-2- (pyrrolidin-l-yl)acetic acid · TFA (48 mg, 0.15 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (60 mg, 0.15 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (48 mg, 0.15 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (21 mg, 24%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.48 (s, 1 H), 8.42 (s, 1 H), 8.14 (d, 7=7.53 Hz, 1 H), 7.95 (d, 7=7.53 Hz, 1 H), 7.66 - 7.73 (m, 3 H), 7.49 - 7.59 (m, 5 H), 5.06 (s, 1 H), 3.92 (br. s, 1 H), 2.86 - 3.21 (m, 2 H), 1.86 - 2.31 (m, 5 H). MS ESI [M + H]+ 476.2, calcd for
[C25H25N5O3S + H]+ 476.2.
Example A 106. l-Phenyl-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)cvclopropanecarboxamide

-171-4820V.1
The title compound was synthesized according to the General Method A, utilizing 1- phenylcyclopropanecarboxylic acid (22 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 μί, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (50 mg, 93%). Ή NMR (400 MHz, Acetone-6) δ ppm 12.50 (br. s, 2 H), 8.51 (s, 1 H), 8.35 (s, 1 H), 8.14 (d, 7=7.53 Hz, 1 H), 8.09 (br. s., 1 H), 7.89 (d, 7=7.78 Hz, 1 H), 7.70 (t, 7=7.65 Hz, 1 H), 7.54 (t, 7=8.03 Hz, 3 H), 7.42 (t, 7=7.30 Hz, 3 H), 7.35 (t, 7=7.30 Hz, 1 H), 6.71 (br. s., 1 H), 1.59 (s, 2 H), 1.13 (s, 2 H). MS ESI [M + H]+ 433.3, calcd for [C23H20N4O3S + H]+ 433.1.
Example A107. 4-Phenyl-N-(3-(3-sulfamoylphenvn-lH-indazol-5-yl)tetrahvdro-2H-pyran-4- carboxamide

The title compound was synthesized according to the General Method A, utilizing 4- phenyltetrahydro-2H-pyran-4-carboxylic acid (26 mg, 0.124 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 xL, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 and CH2CI2 to give the title compound as a pink solid (8 mg, 14%). Ή NMR (400 MHz, METHANOL-di) 5 ppm 8.45 (s, 1 H), 8.10 - 8.18 (m, 2 H), 7.94 (d, 7=8.28 Hz, 1 H), 7.70 (t, 7=7.91 Hz, 1 H), 7.52 (d, 7=8.03 Hz, 3 H), 7.36 - 7.46 (m, 3 H), 7.29 (t, 7=6.50 Hz, 1 H), 3.90 (d, 7=11.90 Hz, 2 H), 3.79 (t, 7=10.92 Hz, 2 H), 2.62 (d, 7=13.30 Hz, 2 H), 2.17 (t, 7=10.79 Hz, 2 H). MS ESI [M + H]+ 477.3, calcd for [C25H24N4O4S + H]+ 477.1.
-172-4820V.1
Example A108. (^-N-O-O-sulfamoylphenylVlH-indazol-S-vn-l.ZS^-tetrahvdronaphthalene- 1-carboxamide

The title compound was synthesized according to the General Method A, utilizing (R)- 1,2,3,4-tetrahydronaphthalene-l-carboxylic acid (22 mg, 0.124 mmol), 3-(5-amino-lH-indazol- 3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), DIPEA (65 pL, 0.373 mmol), TBTU (40 mg, 0.124 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with acetone to give the title compound as a white solid (13 mg, 24%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.40 (br. s, 1 H), 10.36 (br. s., 1 H), 8.51 (s, 1 H), 8.41 (s, 1 H), 8.09 (d, 7=8.28 Hz, 1 H), 7.83 (d, 7=8.28 Hz, 1 H), 7.73 (t, 7=8.00 Hz, 1 H), 7.56 - 7.65 (m, 2 H), 7.46 (s, 2 H), 7.07 - 7.17 (m, 4 H), 3.90 (t, 7=6.27 Hz, 1 H), 2.72 - 2.81 (m, 2 H), 1.98 - 2.09 (m, 3 H), 1.62 - 1.72 (m, 1 H). MS ESI [M + H]+ 447.3, calcd for [Q-^N^S + H]+ 447.1.

The title compound was synthesized according to the General Method A, utilizing 2- (dimethylamino)-2-(2-(trifluoromethyl)phenyl)acetic acid · HC1 (49 mg, 0.174 mmol), 3-(5- amino- lH-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (70 mg, 0.17 mmol), DIPEA (151 pL, 0.87 mmol), TBTU (56 mg, 0.17 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (22 mg, 20%).Ή NMR (400 MHz, METHANOL- dA) δ ppm 8.47 (s, 1 H), 8.41 (s, 1 H), 8.24 (d, 1 H), 8.14 (d, 1 H), 7.95 (d, 2 H), 7.89 (t, 1 H), 7.77 (m, 1 H), 7.65 - 7.73 (m, 1 H), 7.55
-173-44820V.1
- 7.63 (m, 1 H), 7.50 (d, J=6.78 Hz, 1 H), 5.29 (s, 1 H), 2.91 (br. s., 6 H). MS ESI [M 518.3, calcd for [C24H22F3N5O3S + H]+ 518.1.

The title compound was synthesized according to the General Method A, utilizing 2- (pyrrolidin- l -yl)-2-o-tolylacetic acid (54 mg, 0.249 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.249 mmol), DIPEA ( 130 μΐ,, 0.747 mmol), TBTU (80 mg, 0.249 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (36 mg, 24%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.48 (s, 1 H), 8.37 (s, 1 H), 8.14 (d, 7=7.53 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.74 (d, 7=7.78 Hz, 1 H), 7.70 (t, 7=7.65 Hz, 1 H), 7.52 - 7.59 (m, 2 H), 7.33 - 7.45 (m, 3 H), 5.41 (s, 1 H), 3.95 (br. s, 1 H), 3.41 (br. s, 1 H), 3.02 (br. s, 2 H), 2.66 (s, 3 H), 2.26 (br. s, 1 H), 2.13 (br. s, 2 H), 1.98 (br. s, 1 H). MS ESI [M + H]+ 490.3, calcd for [C26H27N5O3S + H]+ 490.2.
Example Al 1 1. 3-amino-2-phenyl-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)propanamide

The title compound was synthesized according to the General Method A, utilizing 3-(tert- butoxycarbonylamino)-2-phenylpropanoic acid (50 mg, 0.186 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (75 mg, 0.186 mmol), DIPEA (195 μί, 0.560 mmol), TBTU (60 mg, 0.186 mmol) and 4 mL of DMF. The reaction mixture was purified using
- 174- 4820V.1
preparative HPLC and dried under reduced pressure. CH2CI2 (4 mL) was added and then cooled to 0 °C. TFA (1 mL) was added and the reaction was stirred at 0 °C for 15 minutes, followed by 45 minutes at room temperature. The reaction mixture was dried by reduced pressure and then triturated with Et20 to give the title compound as a white solid (27 mg, 26%).]H NMR (400 MHz, METHANOLS) δ ppm 8.47 (s, 1 H), 8.44 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.94 (d, 7=8.03 Hz, 1 H), 7.69 (t, 7=7.50 Hz, 1 H), 7.51 - 7.56 (m, 1 H), 7.40 - 7.50 (m, 5 H), 7.37 (t, 7=6.50 Hz, 1 H), 4.08 - 4.15 (m, 1 H), 3.58 - 3.66 (m, 1 H), 3.23 - 3.27 (m, 1 H). MS ESI [M + H]+ 436.2, calcd for [C22H21N5O3S + H]+ 436.1. Example Al 12.. (S)-2-Met oxy-2-phenyl-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl acetamide

The title compound was synthesized according to the General Method A, utilizing (S)-2- methoxy-2-phenylacetic acid (83 mg, 0.50 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (200 mg, 0.50 mmol), DIPEA (260 μί, 1.50 mmol), TBTU ( 160 mg, 0.50 mmol) and 64 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (129 mg, 59%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.46 (s, 1 H), 8.37 (s, 1 H), 8.15 (d, 7=8.28 Hz, 1 H), 7.93 (d, 7=8.03 Hz, 1 H), 7.68 (t, 7=7.78 Hz, 1 H), 7.62 (d, 7=9.03 Hz, 1 H), 7.49 - 7.58 (m, 3 H), 7.40 (d, 7=7.28 Hz, 3 H), 4.84 (s, 1 H), 3.46 (s, 3 H). MS ESI [M + H]+ 437.3, calcd for ^H^N^S + H]+ 437.1.
-175-4820V.1

title compound was synthesized according to the General Method A, utilizing 3-
(dimethylamino)-2-phenylpropanoic acid (48 mg, 0.249 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate(100 mg, 0.249 mmol), DIPEA (130 μΐ., 0.747 mmol), TBTU (80 mg, 0.249 mmol) and 4 mL of DMF. The reaction mixture was purified using preparative HPLC, triturated with EtiO, and then purified by flash chromatography (5% MeOH / CH2C12 to 25% MeOH / CH2C12) to give the title compound as a white solid (14 mg, 12%).Ή NMR (400 MHz, METHANOL-^) δ ppm 8.47 (s, 1 H), 8.44 (s, 1 H), 8.16 (d, 7=7.28 Hz, 1 H), 7.94 (d, 7=8.53 Hz, 1 H), 7.70 (t, 7=7.28 Hz, 1 H), 7.42 - 7.57 (m, 4 H), 7.37 (t, 7=7.65 Hz, 2 H), 7.29 (t, 7=6.80 Hz, 1 H), 4.00 - 4.07 (m, 1 H), 3.45 - 3.54 (m, 1 H), 2.61 - 2.72 (m, 1 H), 2.47 (s, 6 H) MS ESI [M + H]+ 464.3, calcd for [C24H25N5O3S + H]+ 464.17.
Example Al 14. 2-phenyl-3-(pyrrolidin-l-yl)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5- vDpropanamide 2,2,2-trifluoroacetate

The title compound was synthesized the General Method A, utilizing 2-phenyl-3- (pyrrolidin-l-yl)propanoic acid · HC1 (50 mg, 0.196 mmol), 3-(5-amino- lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (79 mg, 0.196 mmol), DIPEA (170 μί, 0.98 mmol), TBTU (63 mg, 0.196 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (15 mg, 13 %). Ή NMR (400 MHz, METHANOL-^) 6 ppm 8.47 (s, 1 H), 8.46 (s, 1 H), 8.15 (d, 7=7.03 Hz,
-176-4820V.1
1 H), 7.95 (d, 7=7.03 Hz, 1 H), 7.70 (t, 7=7.78 Hz, 1 H), 7.47 - 7.58 (m, 4 H), 7.45 (t, 7=7.28 Hz,
2 H), 7.38 (t, 7=7.50 Hz, 1 H), 4.24 (dd, 7=9.66, 4.89 Hz, 1 H), 4.09 (dd, 7=12.55, 8.53 Hz, 1 H), 3.66 (br. s, 2 H), 3.51 (dd, 7=13.05, 5.27 Hz, 1 H), 3.22 (br. s, 2 H), 1.96 - 2.22 (br. s., 4 H) MS ESI [M + H]+ 490.3, calcd for [C26H27N5O3S + H]+ 490.2.

The title compound was synthesized according to the General Method A, utilizing 3- morpholino-2-phenylpropanoic acid · HC1 (68 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol), DIPEA (217 μί, 1.25 mmol), TBTU (80 mg, 0.25 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC and triturated with Et20 to give the title compound as a white solid (28 mg, 18 %). Ή NMR (400 MHz, METHANOL-d4) 5 ppm 8.48 (br. s., 2 H), 8.15 (d, 7=7.78 Hz, 1 H), 7.95 (d, 7=8.03 Hz, 1 H), 7.70 (t, 7=8.41 Hz, 1 H), 7.55 (d, 7=9.03 Hz, 1 H), 7.41 - 7.52 (m, 5 H), 7.38 (t, 7=6.80 Hz, 1 H), 4.32 (dd, 7=9.66, 3.14 Hz, 1 H), 4.10 (m, 1 H), 3.73 - 4.04 (m, 4 H), 3.42 (dd, 7=12.55, 3.01 Hz, 1 H), 3.42 (br. s, 4 H). MS ESI [M + H]+ 506.3, calcd for
[C26H27N504S + H]+ 506.2.
Example Al 16.(S)-N-(l-phenylethyl)-3-(3-sulfamoylphenyl)-lH-indazole-5-carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (50 mg, 0.158 mmol), (S)-l-phenylethanamine
-177-4820V.1
(19 mg, 0.158 mmol), DIPEA (41 μί, 0.237 mmol), TBTU (51 mg, 0.158 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC. A trituration with Et20 gave the title compound as a pale yellow solid (19 mg, 18 %). Ή NMR (400 MHz, METHANOL-d ) δ ppm 8.63 (s, 1 H), 8.54 (s, 1 H), 8.26 (d, 7=6.53 Hz, 1 H), 7.92 - 8.00 (m, 2 H), 7.73 (t, 7=7.40 Hz, 1 H), 7.65 (d, 7=8.28 Hz, 1 H), 7.43 (d, 7=7.03 Hz, 2 H), 7.34 (t, 7=6.53 Hz, 2 H), 7.23 (t, 7=6.50 Hz, 1 H), 5.29 (q, 7=6.78 Hz, 1 H), 1.60 (d, 7=6.02 Hz, 3 H). MS ESI [M + H]+ 421.2, calcd for [C22H20N4O3S + H]+ 421.1.

The title compound was synthesized according to the General Method A, utilizing 2- phenyl-3-(piperidin-l-yl)propanoic acid hydrochloride (67 mg, 0.25 mmol), 3-(5-amino-lH- indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol), DIPEA (217 μί, 1.25 mmol), TBTU (80 mg, 0.25 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC. A trituration with Et20 provided the title compound as a white solid (46 mg, 30 %). Ή NMR (400 MHz, METHANOLS) δ ppm 8.48 (s, 1 H), 8.45 (s, 1 H), 8.16 (d, 7=6.78 Hz, 1 H), 7.95 (d, 7=8.53 Hz, 1 H), 7.70 (t, 7=8.03 Hz, 1 H), 7.55 (d, 7=9.00 Hz, 1 H), 7.49 (d, 7=8.28 Hz, 3 H), 7.44 (t, 7=7.40 Hz, 2 H), 7.37 (t, 7=7.20 Hz, 1 H), 4.29 (dd, 7=9.54, 3.76 Hz, 1 H), 4.05 (t, 7=12.00 Hz, 1 H), 3.61 (br. s, 2 H), 3.34 - 3.38 (m, 1 H), 3.07 (br. s, 2 H), 1.72 - 2.02 (m, 5 H), 1.54 (br. s, 1 H). MS ESI [M + H]+ 504.3, calcd for ^H^NsC^S + H]+ 504.2.
-178-4820V.1
Example Al 18. (R)-N-(2-hvdroxy-l-phenylethyl)-3-(3-sulfamoylphenyl')-lH-indazole-5- carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (50 mg, 0.158 mmol), (R)-2-amino-2- phenylethanol (22 mg, 0.158 mmol), DIPEA (41 pL, 0.237 mmol), TBTU (51 mg, 0.158 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC. A trituration with Et20 gave the title compound as a white solid (22 mg, 32 %). %). Ή NMR (400 MHz,
METHANOLS) δ ppm 8.69 (s, 1 H), 8.55 (s, 1 H), 8.28 (d, 7=7.60 Hz, 1 H), 8.00 - 7.96 (m, 2 H), 7.74 (t, 7=8.00 Hz, 1 H), 7.66 (d, 7=8.80 Hz, 1 Hz), 7.44 (d, 7=7.60 Hz, 2 H), 7.36 (t, 7=6.80 Hz, 2H), 7.26 (t, 7=7.20, 1 H), 5.26 (t, 7=6.15 Hz, 1 H), 3.87 - 3.92 (m, 2 H) ). MS ESI [M + H]+ 437.2 calcd for [C22H20N4O4S + H]+ 437.1.

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (50 mg, 0.158 mmol), (R)-Nl,Nl-dimethyl-2- phenylethane- l,2-diamine (26 mg, 0.158 mmol), DIPEA (41 pL, 0.237 mmol), TBTU (51 mg, 0.158 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC followed by a trituration with Et20 to give the title compound as a white solid (9 mg, 10 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.71 (s, 1 H), 8.55 (s, 1 H), 8.25 (d, 7=7.78 Hz, 1 H), 8.00 (t, 7=9.54 Hz, 2 H), 7.73 (t, 7=7.91 Hz, 1 H), 7.68 (d, 7=8.53 Hz, 1 H), 7.54 (d, 7=7.28 Hz, 2 H), 7.46 (t, 7=7.40 Hz, 2 H), 7.38 (t, 7=6.80 Hz, 1 H), 5.78 (dd, 7=10.54, 3.26 Hz, 1 H), 3.76 (t,
- 179-4820V.1
7=12.55 Hz, 1 H), 3.59 - 3.67 (m, 1 H), 3.06 (br. s., 6 H). MS ESI [M + H]+ 464.3, calcd for [C24H25N5O3S + H]+ 464.2.
Example A120. 3-hvdroxy-2-phenyl-N-(3-(3-sulfamoylphenvn- lH-indazol-5-yl)propanamide

The title compound was synthesized according to the General Method A, utilizing DL- tropic acid (42 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide 2,2,2- trifluoroacetate (100 mg, 0.25 mmol), DIPEA (217 pL, 1.25 mmol), TBTU (80 mg, 0.25 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC followed by a trituration with Et20 to give the title compound as a white solid (28 mg, 26 %). Ή NMR (400 MHz, DMSO-d6) 8 ppm 13.38 (s, 1 H), 10.26 (s, 1 H), 8.49 (s, 1 H), 8.40 (s, 1 H), 8.09 (d, 7=7.78 Hz, 1 H), 7.84 (d, 7=7.53 Hz, 1 H), 7.76 (t, 7=7.30 Hz, 1 H), 7.52 - 7.61 (m, 2 H), 7.47 (s 2 H), 7.40 (d, 7=7.03 Hz, 2 H), 7.33 (t, 7=7.28 Hz, 2 H), 7.25 (t, 7=7.30 Hz, 1 H), 4.93 - 5.04 (br s., 1 H), 4.10 (t, 7=10.50 Hz, 1 H), 3.83 - 3.89 (m, 1 H), 3.54 - 3.61 (m, 1 H). MS ESI [M + H]+ 437.2, calcd for [C22H20N4O4S + H]+ 437.1.

The title compound was synthesized according to the General Method A, utilizing 2-
(piperidin-l-yl)-2-o-tolylacetic acid (58 mg, 0.25 mmol), 3-(5-amino- lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol), DEPEA (217 pL, 1.25 mmol), TBTU (80 mg, 0.25 mmol) and 3 mL of DMF. The reaction mixture was purified using
-180- 4820V.1
preparative HPLC followed by a trituration with Et20 to give the title compound as a white solid (83 mg, 54%). Ή NMR (400 MHz, METHANOL-d4) δ ppm 8.48 (s, 1 H), 8.39 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.95 (d, 7=8.53 Hz, 1 H), 7.80 (d, 7=7.78 Hz, 1 H), 7.70 (t, 7=8.03 Hz, 1 H), 7.59 - 7.52 (m, 2 H), 7.34 - 7.44 (m, 3 H), 5.25 (s, 1 H), 3.88 (br. s, 1 H), 3.23 (br. s, 1 H), 2.96 (br. s, 2 H), 2.65 (s, 3 H), 1.98 (br. s, 2 H), 1.82 (br. s, 3 H), 1.56 (br. s, 1 H). MS ESI [M + H]+ 504.3, calcd for [C27H29N5O3S + H]+ 504.2.
Example A 122. 3-(3-sulfamoylphenyl)-N-(2,2,2-trifluoro- 1 -phenylethvD- 1 H-indazole-5- carboxamide

3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.315 mmol) was dissolved in DMF (4 mL) and cooled to 0°C. DIPEA (110 μΐ., 0.630 mmol), and isobutyl chloroformate (82 pL, 0.630 mmol) was added. The reaction was stirred for 10 minutes and then 2,2,2-trifluoro-l-phenylethanamine (67 mg, 0.315 mmol) was added. The reaction mixture was stirred at room temperature for 1 h and then 30 mL of water was added. The reaction mixture was extracted with EtOAc and then dried under reduced pressure. The brownish residue was dissolved in MeOH (4 mL) and then NaOMe (600 μί, 25 %wt in MeOH) was added. The reaction mixture was stirred for 1 h and then neutralized with 2 M aq HCl. The reaction mixture was purified by preparative HPLCf ollowed by flash chromatorgraphy (Si02, 100% CH2Cl2 to 20% MeOH/CH2Cl2) to give the title compound as a beige solid (14 mg, 9 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.66 (s, 1 H), 8.54 (s, 1 H), 8.25 (d, 7=7.53 Hz, 1 H), 7.98 - 7.95 (m, 2 H), 7.73 (t, 7=7.78 Hz, 1 H), 7.66 (d, 7=8.78 Hz, 1 H), 7.62 (d, 7=7.28 Hz, 2 H), 7.47 - 7.41 (m, 3 H), 6.03 (q, 7=8.53 Hz, 1 H). MS ESI [M + H]+ 475.2 calcd for K^HnFa^OsS + H]+ 474.1.
-181-4820V.1
Example A123. 2-(pyrrolidin-l-ylVN-(3-(3-sulfamoylphenvn-lH-indazol-5-vn-2-(thioDhen-2- vDacetamide trifluoroacetate

To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (161 mg, 0.4 mmol), crude 2-(pyrrolidin-l-yl)-2-(thiophen-2-yl)acetic acid (88 mg, 0.4 mmol) and TBTU (129 mg, 0.4 mmol) in DMF (6 mL) at 0 °C was added 'Pr2NEt (0.35 mL, 2.0 mmol). After addition, the resulting mixture was stirred for lh at 0 °C. It was purified by preparative HPLC, biotage column (to remove HOBt) then second preparative HPLC to give the title compound as an off white solid (97 mg, 41 %). ]H NMR (400 MHz, CD3OD) δ 8.48 (s, IH), 8.40 (s, IH), 8.12 (d, J =7.6 Hz, IH), 7.94 (d, J =7.6 Hz, IH), 7.69-7.65 (m, 2H), 7.57-7.52 (m, 3H), 7.16 (t, J =4.0 Hz, IH), 5.53 (s, IH), 4.00-3.10 (m, 4H), 2.30-1.90 (m, 4H); MS ESI 482.2 [M + H]+, calcd for
 + H]+ 482.1.
+ H]+ 482.1.
Example A 124. 2-(pyrrolidin- 1 -viVN-(3-(3-sulf amoylphenvD- 1 H-indazol-5-ylV2-(thiophen-3- vDacetamide trifluoroacetate

To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide trifluoroacetate (100 mg, 0.25 mmol), crude 2-(pyrrolidin-l-yl)-2-(thiophen-3-yl)acetic acid (55 mg, 0.25 mmol) and TBTU (80 mg, 0.25 mmol) in DMF (4 mL) at 0 °C was added 'Pr2NEt (0.22 mL, 1.25 mmol). After addition, the resulting mixture was stirred for lh at 0 °C. After removal of DMF, the residue was diluted with H20 (20 mL), satd. NaHC03 (5 mL) and extracted with EtOAc (30 mL
-182-44820V.1
x 2). Combined extracts were washed with H20 (20 mL x 2) and concentrated to dryness. It was purified by preparative HPLC to give the title compound as an off white solid (58 mg, 39%). Ή NMR (400 MHz, CD3OD) δ 8.49 (s, IH), 8.42 (s, IH), 8.13 (d, J =7.6 Hz, IH), 7.94 (d, J =7.6 Hz, IH), 7.88 (s, IH), 7.68 (t, J =7.8 Hz, IH), 7.65-7.61 (m, IH), 7.55 (pseudo t, 2H), 7.38 (d, J =5.2 Hz, IH), 5.31 (s, IH), 3.88 (br.s, 1 H), 3.40-3.05 (m, 3H), 2.30-1.95 (m, 4H); MS ESI 482.2. [M + H]+, calcd for [C23H23N5O3S2 + H]+ 482.1.

1 -( tert-butoxycarbonyl)-4-phenylpiperidine-4-carboxylic acid
Solution of 4-phenylpiperidine-4-carboxylic acid hydrochloride (0.30 g, 1.2 mmol) and DIPEA (0.65 mL, 3.7 mmol) in DCM (10 mL) and DMF (4 mL) was treated with di-tert-butyl dicarbonate (0.34 g, 1.6 mmol) and stirred at rt for 3 h. Later the reaction was concentrated under reduced pressure and triturated with hexanes to afford the title compound containing 1 equiv of DIPEA as a (0.2g). The material was used in the following step without further purification. 4-phenyl-N-( 3-( 3-sulfamoylphenyl)-lH-indazol-5-yl)piperidine-4-carboxamide 2,2,2- trifluoroacetate
The title compound was synthesized according the General Method A., utilizing 3-(5- amino- l H-indazol-3-yl)benzenesulfonamide 2,2,2-trifluoroacetate (80 mg, 0.20 mmol), crude 1- (tert-butoxycarbonyl)-4-phenylpiperidine-4-carboxylic acid (153 mg, 0.22 mmol), N-ethyl-N- isopropylpropan-2-amine (0.1 mL, 0.6 mmol) and 0-(Benzotriazol- 1 -yl)-N,N,N',N'- tetramethyluronium tetrafluoroborate (70 mg, 0.22 mmol) in DMF (2 mL). The crude material
- 183-4820V.1
was purified by preparative HPLC to provide tert-butyl 4-phenyl-4-(3-(3-sulfamoylphenyl)-lH- indazol-5-ylcarbamoyl)piperidine-l-carboxylate (MS ESI 576.2 [M + H]+, calcd for
[C30H33N5O5S + H]+ 576.7) to as a pale red solid. The material was then taken in DCM (10 mL), treated with Et3SiH (0.07 mL, 0.44 mmol) and TFA (2 mL) at 0 °C allowing after 1 h to warm to rt (2h). Later the reaction mixture was concentrated and purified by preparative HPLC. A single trituration with Et20 provided the title compound as an off-white solid (30 mg, 26 %). Ή NMR (400 MHz, METHANOL-d δ ppm 8.47 (s, 1 H), 8.23 (s, 1 H), 8.15 (d, 7 =7.78 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.70 (t, 7 =7.78 Hz, 1 H), 7.51 - 7.57 (m, 3 H), 7.50 - 7.57 (m, 3 H), 7.36 (t, 7=7.16 Hz, 1 H), 3.34 - 3.51 (m, 4 H), 2.79 - 2.92 (m, 2 H), 2.25 - 2.37 (m, 2 H); MS ESI 476.3 [M + H]+, calcd for [C25H25N5O3S + H]+ 476.3.
Example A126. 3-(3-Sulfamoylphenyl)-N-((tetrahvdro-2H-pyran-4-yl)methyl)-lH-indazole-5- carboxamide

3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (30 mg, 0.095 mmol), (tetrahydro- 2H-pyran-4-yl)methanamine (11 mg, 0.095 mmol), and DIPEA (25 μί, 0.14 mmol) was dissolved in 2 mL of DMF and cooled to 0 °C. TBTU (30 mg, 0.095 mmol) was added and the reaction was stirred at 0 °C for 90 minutes. Completion of the reaction was monitored by LC- MS. Purification by preparative HPLC gave the title compound as a white solid (21 mg, 53%). Ή NMR (400 MHz, DMS0-d6) δ ppm 13.64 (s, 1 H), 8.59 - 8.64 (m, 1 H), 8.56 (s, 1 H), 8.47 (s, 1 H), 8.27 (d, 7=7.53 Hz, 1 H), 7.93 (d, 7=8.78 Hz, 1 H), 7.88 (d, 7=7.78 Hz, 1 H), 7.78 (t, 7=7.53 Hz, 1 H), 7.66 (d, 7=8.28 Hz, 1 H), 7.50 (s, 2 H), 3.85 (d, 7=10.04 Hz, 2 H), 3.18 - 3.31 (m, 4 H), 1.77 - 1.87 (m, 1 H), 1.62 (d, 7=12.30 Hz, 2 H), 1.15 - 1.28 (m, 2 H); MS ESI [M + H]+ 415.2, calcd for [C20H22N4O4S + H]+ 415.1.
-184-4820V.1
xample A127. N-(2,6-Dimethoxybenzyl -3-(3-sulfamoylphenyl)-lH-indazole-5-carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)- lH-indazole-5-carboxylic acid (30 mg, 0.095 mmol), (2,6- dimethoxyphenyl)methanamine (16 mg, 0.095 mmol), DIPEA (20 μ]_, 0.1 15 mmol), TBTU (31 mg, 0.095 mmol) and DMF (2 mL). The reaction mixture was purified by preparative HPLC and precipitated with water to give the title compound as a white solid (15 mg, 35%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.58 (s, 1 H), 8.49 (s, 1 H), 8.43 (s, 1 H), 8.28 (br. s., 1 H), 8.24 (d, 7=7.78 Hz, 1 H), 7.90 (d, 7=8.78 Hz, 1 H), 7.83 (d, 7=7.78 Hz, 1 H), 7.72 (t, 7=7.30 Hz, 1 H), 7.60 (d, 7=8.28 Hz, 1 H), 7.45 (s, 2 H), 7.25 (t, 7=8.16 Hz, 1 H), 6.66 (d, 7=8.28 Hz, 2 H), 4.47 (d, 7=4.52 Hz, 2 H), 3.77 (s, 6 H); MS ESI [M + H]+467.2, calcd for [C23H22N4O5S + H]+ 467.1.
Example A128. N-(2,6-Diethylbenzyl)-3-(3-sulfamoylphenyl)-lH-indazole-5-carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (25 mg, 0.079 mmol), (2,6-diethylphenyl) methanamine (13 mg, 0.079 mmol), DIPEA (20 μί, 0.118 mmol), TBTU (25 mg, 0.079 mmol) and DMF (3 mL). The reaction mixture was purified by preparative HPLC, redissolved in a small amount of MeOH and precipitated out of water to give the title compound as a white solid (7.5 mg, 21 %). Ή NMR (400 MHz, METHANOLS) δ ppm 8.58 (s, 1 H), 8.51 (s, 1 H), 8.22 (d, 7=7.53 Hz, 1 H), 7.94 (t, 7=9.16 Hz, 2 H), 7.69 (t, 7=7.65 Hz, 1 H), 7.62 (d, 7=8.53 Hz, 1 H),
-185-4820V.1
7.20 (t, 7=7.80 Hz, 1 H), 7.11 (d, 7=7.78 Hz, 2 H), 4.69 (s, 2 H), 2.79 (q, 7=7.53 Hz, 4 H), 1.25 (t, 7=7.53 Hz, 6 H). MS ESI [M + H]+ 463.3, calcd for [C25H26N4O3S + H]+ 463.2.

Benzylamine (150 mg, 1.40 mmol), lH-indazole-5-carboxylic acid (227 mg, 1.40 mmol), and DIPEA (293 μΐ., 1.68 mmol) was dissolved in 5 mL of DMF and cooled to 0 °C. TBTU (297 mg, 0.925 mmol) was added and the reaction was stirred at 0 °C for 120 minutes. Completion of the reaction was monitored by LC-MS and purification by flash chromatography (50%
EtOAc hexanesto 100% EtOAc) gave the title compound as a white solid (31 mg, 9%) Ή NMR (400 MHz, METHANOLS) δ ppm 8.35 (s, 1 H), 8.16 (s, 1 H), 7.91 (d, 7=8.78 Hz, 1 H), 7.60 (d, 7=9.03 Hz, 1 H), 7.38 (d, 7=7.28 Hz, 2 H), 7.33 (t, 7=7.30 Hz, 2 H), 7.25 (t, 7=7.00 Hz, 1 H), 4.61 (s, 2 H); MS ESI [M + H]+ 251.9, calcd for [Ci5H13N30 + H]+ 252.1.
B. Synthesis of N-Benzyl-3-iodo-l H-indazole-5-carboxamide

The title compound was synthesized according to the General Method B utilizing N- benzyl- lH-indazole-5-carboxamide (30 mg, 0.120 mmol), K2C03 (50 mg, 0.358 mmol), (60 mg, 0.240 mmol) and DMF (2 mL). An orange solid (32 mg, 71 %). Ή NMR (400 MHz,
-186- 4820V.1
METHANOLS) δρρηι 8.08 (s, 1 H), 7.96 (d, 7=9.03 Hz, 1 H), 7.58 (d, 7=9.03 Hz, 1 H), 7.38 (d, 7=7.30 Hz, 2 H), 7.33 (t, 7=7.30 Hz, 2 H), 7.25 (m, 7=6.80, 6.80 Hz, 1 H), 4.61 (s, 2 H); MS ESI [M + H]+ 377.9, calcd for [C] 5H12IN30 + H]+ 378.0.
C. N-benzyl-3-(3-sulfamoylphenyl)-lH-indazole-5-carboxamide
A sealed degassed mixture of N-benzyl-3-iodo-lH-indazole-5-carboxamide (30 mg, 0.080 mmol), benzenesulfonamide-3-boronic acid pinacol ester (27 mg, 0.095 mmol), PddppfCl2 (4 mg, 0.0040 mmol), saturated aqueous Na2C03 (400 ί) in toluene / EtOH (0.8 mL / 0.8 mL) under argon was heated under microwave irradiation at 125°C for 120 minutes. The reaction mixture was purified by preparative HPLC to give the title compound as a white solid (3.4 mg, 10%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.64 (s, 1 H), 8.53 (s, 1 H), 8.25 (d, 7=7.03 Hz, 1 H), 7.97 (d, 7=7.78 Hz, 2 H), 7.72 (t, 7=7.78 Hz, 1 H), 7.66 (d, 7=9.54 Hz, 1 H), 7.39 (d, 7=8.00 Hz, 2 H), 7.34 (t, 7=7.40 Hz, 2 H), 7.25 (t, 7=7.30 Hz, 1 H), 4.63 (s, 2 H); MS ESI [M + H]+ 407.1, calcd for [C21H18N4O3S + H]+ 407.1.
Example A130. N-Benzyl-N-methyl-3-(3-sulfamoylphenyl)-lH-indazole-5-carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoyIphenyl)-lH-indazole-5-carboxylic acid (60 mg, 0.189 mmol), N-methyl-1- phenylmethanamine (23 mg, 0.189 mmol), DIPEA (49 μί, 0.284 mmol), TBTU (61 mg, 0.189 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC, and triturating with Et20 to give the title compound as a white solid (43 mg, 54%). Ή NMR (400 MHz, METHANOLS) 8 ppm 8.48 (br. s, 2 H), 7.62 - 8.31 (m, 4 H), 7.58 (d, 7=8.28 Hz, 2 H), 7.17 - 7.46 (m, 5 H), 4.81 (br. s., 1 H), 4.65 (br. s., 1 H), 3.05 (br. d., 3 H). MS ESI [M + H]+ 421.3, calcd for + H]+ 421.1.
-187-4820V.1

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (40 mg, 0.126 mmol), 1- phenylcyclopropanamine- TFA (31 mg, 0.126 mmol), DIPEA (66 pL, 0.378 mmol), TBTU (41 mg, 0.126 mmol) and DMF (3 mL). The reaction mixture was purified by preparative HPLC, and triturating with Et20 to give the title compound as a white solid (14 mg, 26%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.66 (s, 1 H), 9.32 (s, 1 H), 8.66 (s, 1 H), 8.48 (s, 1 H), 8.31 (d, 7=8.53 Hz, 1 H), 7.97 (d, 7=8.03 Hz, 1 H), 7.88 (d, 7=7.28 Hz, 1 H), 7.78 (t, 7=7.78 Hz, 1 H), 7.68 (d, 7=8.28 Hz, 1 H), 7.50 (s, 2 H), 7.20 - 7.31 (m, 4 H), 7.15 (t, 7=6.80 Hz, 1 H), 1.25 - 1.35 (m, 4 H). MS ESI [M + H]+ 433.2 calcd for [C23H20N4O3S + H]+ 433.1.
Example A132. N-(cvclopropyl(phenyl)methyl)-3-(3-sulfamoylphenyl)-lH-indazole-5- carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (40 mg, 0.13 mmol),
cyclopropyl(phenyl)methanamine (23 mg, 0.13 mmol), DIPEA (66 pL, 0.38 mmol), TBTU (41 mg, 0.13 mmol) and DMF (3 mL). The reaction mixture was purified by preparative HPLC, and triturating with Et20 to give the title compound as a white solid (21 mg, 37%). Ή NMR (400 MHz, METHANOL-^) 5 ppm 8.65 (s, 1 H), 8.55 (s, 1 H), 8.27 (d, 7=8.28 Hz, 1 H), 7.98 (d, 7=8.03 Hz, 2 H), 7.73 (t, 7=7.50 Hz, 1 H), 7.66 (d, 7=9.29 Hz, 1 H), 7.49 (d, 7=7.28 Hz, 2 H), 7.34 (t, 7=7.65 Hz, 2 H), 7.24 (t, 7=8.00 Hz, 1 H), 4.48 (d, 7=8.78 Hz, 1 H), 1.36 - 1.45 (m, 1 H),
-188-4820V.1
0.64 - 0.70 (m, 2 H), 0.45 - 0.52 (m, 2 H). MS ESI [M + H]+ 447.3, calcd for [C24H22N4O3S + H]+ 447.1.
ExampleA133. N-(2,6-diethylbenzyl)-N-methyl-3-(3-sulfamoylphenylVlH-indazole-5- carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)- lH-indazole-5-carboxylic acid (35 mg, 0.11 mmol), l-(2,6-diethylphenyl)-N- methylmethanamine (19 mg, 0.11 mmol), DIPEA (29 ML, 0.17 mmol), TBTU (35 mg, 0.1 1 mmol) and DMF (3 mL). The reaction mixture was purified by preparative HPLC, redissolved in a small amount of THF and precipitated out of water to give the title compound as a white solid (14 mg, 27%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.64 (s, 1 H), 8.43 (s, 1 H), 8.21 (d, 7=7.53 Hz, 1 H), 8.14 (br. s, 1 H), 7.86 (d, 7=8.03 Hz, 1 H), 7.66 - 7.77 (m, 2 H), 7.51 (s, 2 H), 7.45 (br. s, 1 H), 7.21 (t, 7=8.00 Hz, 1 H), 7.04 - 7.13 (m, 2 H), 4.82 (br. s, 2 H), 2.70 (br. s, 4 H), 2.61 (s, 3 H), 1.1 1 (br. s, 6 H). MS ESI [M + H]+ 477.3, calcd for [C26H28N4O3S + H]+ 477.2.

A DMF (12 mL) solution of 3-(3-sulfamoylphenyl)- lH-indazole-5-carboxylic acid (34 mg, 0.107 mmol), and TBTU (35 mg, 0.1 1 mmol) was cooled to 0 °C. 1 ,2- Phenylenedimethanamine · HC1 ( 185 mg, 0.107 mmol) and DIPEA (223 μΐ., 1.29 mmol) was dissolved in DMF (3 mL) and added dropwise to the first mixture. The reaction was stirred at 0 °C for 60 minutes. The excess 1 ,2-phenylenedimethanamine had precipitated out of solution. The
- 189-44820V.1
reaction mixture was centrifuged and the supernatant was purified by preparative HPLC.
Triturating in Et20 gave the title compound as a white solid (24 mg, 40%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.64 (s, 1 H), 8.52 (s, 1 H), 8.24 (d, 7=8.53 Hz, 1 H), 7.97 (t, 7=8.80 Hz, 2 H), 7.74 (t, 7=7.78 Hz, 1 H), 7.66 (d, 7=8.28 Hz, 1 H), 7.56 (d, 7=7.28 Hz, 1 H), 7.42 - 7.49 (m, 2 H), 7.37 (t, 7=7.30 Hz, 1 H), 4.65 (s, 2 H), 4.39 (s, 2 H). MS ESI [M + H]+ 436.2 calcd for [C22H21N5O3S + H]+ 436.1.
Example A 135. (S)-N-(2,3-dihvdro- 1 H-inden- 1 - yl)-3-(3-sulfamoylphenyl)- 1 H-indazole-5- carboxamide
The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (50 mg, 0.16 mmol), (s)-(+)-l-aminoindane (21 mg, 0.16 mmol), DIPEA (55 μί, 0.32 mmol), TBTU (51 mg, 0.079 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC, and triturating with MeOH and acetone to give the title compound as a white solid (8 mg, 12%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.65 (s, 1 H), 8.91 (d, 7=8.03 Hz, 1 H), 8.64 (s, 1 H), 8.48 (s, 1 H), 8.28 (d, 7=7.78 Hz, 1 H), 8.02 (d, 7=8.28 Hz, 1 H), 7.86 (d, 7=7.78 Hz, 1 H), 7.75 (t, 7=7.80 Hz, 1 H), 7.68 (d, 7=8.78 Hz, 1 H), 7.49 (s, 2 H), 7.16 - 7.31 (m, 4 H), 5.63 (q, 7=7.78 Hz, 1 H), 2.97 - 3.06 (m, 1 H), 2.82 - 2.93 (m, 1 H), 1.96 - 2.07 (m, 1 H) *a signal corresponding to one proton is bscured by the solvent peak. MS ESI [M + H]+ 433.2 calcd for [CzsHzoN^S + H]+ 433.1.
-190-4820V.1
Example A136. (S)-3-(3-sulfamoylphenyl -N-(l,2,3,4-tetrahydronaphthalen-l-yl)-lH-indazole- 5-carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (50 mg, 0.16 mmol), (S)-l,2,3,4- tetrahydronaphthalen- 1 -amine (23 mg, 0.16 mmol), DIPEA (65 μΐ,, 0.237 mmol), TBTU (51 mg, 0.16 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC, and triturating with Et20 to give the title compound as a white solid (9 mg, 13%).Ή NMR (400 MHz, METHANOL-^) δ ppm 8.65 (s, 1 H), 8.53 (s, 1 H), 8.25 (d, 7=7.78 Hz, 1 H), 7.97 (t, 7=9.16 Hz, 2 H), 7.71 (t, 7=7.91 Hz, 1 H), 7.65 (d, 7=8.53 Hz, 1 H), 7.26 - 7.32 (m, 1 H), 7.10 - 7.19 (m, 3 H), 5.39 (t, 7=6.53 Hz, 1 H), 2.76 - 2.96 (m, 2 H), 2.11 - 2.21 (m, 1 H), 1.81 - 2.10 (m, 3 H) MS ESI [M + H]+ 447.2, calcd for [C24H22N4O3S + H]+ 447.1.
Example A137. (R)-3-(3-sulfamoylphenyl)-N-(l,2,3,4-tetrahydronaphthalen-l-yl)-lH-indazole- 5-carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (50 mg, 0.16 mmol), (R)- 1,2,3,4- tetrahydronaphthalen-1 -amine (23 mg, 0.16 mmol), DIPEA (65 μΐ,, 0.24 mmol), TBTU (51 mg, 0.16 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC, and triturating with Et20 to give the title compound as a white solid (3.5 mg, 5%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.64 (s, 1 H), 8.53 (s, 1 H), 8.24 (d, 7=7.78 Hz, 1 H), 7.97 (t, 7=9.16 Hz, 2 H), 7.71 (t, 7=7.91 Hz, 1 H), 7.65 (d, 7=8.53 Hz, 1 H), 7.26 - 7.32 (m, 1 H), 7.10 -
-191-4820V.1
7.19 (m, 3 H), 5.39 (t, 7=6.53 Hz, 1 H), 2.76 - 2.96 (m, 2 H), 2.11 - 2.21 (m, 1 H), 1.81 - 2.10 (m, 3 H). MS ESI [M + H]+ 447.2 calcd for [C24H22N4O3S + H]+ 447.1.

A. 3-bromophenyl- (3-bromopropyl) sulfide
To a solution of 3-bromothiophenol (2.5 g, 13.22 mmol) in acetonitrile ( 25 mL) was added K2C03 (2.74 g, 19.83 mmol).the mixture was stirred for 15 min at room temperature and was added 1 ,3-dibiomopropane (13.35 g, 66.1 1 mmol) at the same temperature. The reaction mixture was stirred at 70°C for 18 h and then filtered salt, washed it with little fresh solvent. The volatile were removed under reduced pressure and the resultant oily residue purified using BiotageSNAP 50 g silica column (gradient 0-5 % EtOAc in hexane) to give 3-bromophenyl- (3- bromopropyDsulfide as a colorless thick oil (3 g, 73%). Ή NMR (400 MHz, CDCl3) δ 7.48 (t, 7 = 1.6 Hz, 1H), 7.33 (dd, 7 =2.8, 0.8 Hz, 1H), 7.31 (dd, 7 = 1.6, 0.8 Hz, 1H), 7.16 (t, 7 =8 Hz, 1H), 3.53 (t, 7 =6.4 Hz, 2H), 3.09 (t, 7 =8 Hz, 2H), 2.2-2.13 (m, 2H).
B. l-bromo-3-(3-bromopropyl) sulfonyl) benzene
A solution of 3-bromophenyl- (3-bromopropyl) sulfide (1 g, 3.23 mol) in dry DCM (30 mL) was cooled in an ice bath to 0°C and treated with mCPBA (0.825 g, 3.63 mmol) at 0°C for 1 h. another portion of mCPBA (0.825 g, 3.63 mmol) at same temperature and the stirring was continued for 1 h. The reaction mixture was allowed to room temperature and stirred for 2 h.the reaction mixture was diluted with DCM and EtOAc (15 mL each) and washed twice 1M Na2CO? (40 mL).the organic phase was dried over anh Na2S04 and concentrated to dryness and purified
-192-4820V.1
by BiotageSNAP 25 g silica column (gradient 0-35 % EtOAc in hexane) to give l-bromo-3-(3- bromopropyl) sulfonyl) benzene as a colorless thick oil (0.75 g, 68% ). Ή NMR (400 MHz, CDCl3) δ 8.07 (t, J = 1.8 Hz, 1H), 7.88 (dd, J =1.6, 0.8 Hz, 1H), 7.83 (dd, J =2 , 1.2 Hz, 1H), 7.48 (t, J =8 Hz, 1H), 3.50 (t, J =6.4 Hz, 2H), 3.31-3.27 (m, 2H), 2.36-2.29 (m, 2H). MS ESI 343. [M + H]+, calcd for [C9HioBr202S + H]+ 342.8.
C. ( 2S,6R)-4-(3-( ( 3-bromophenyl)sulfonyl) propyl))-2,6-dimethylmorpholine
To a solution of l-bromo-3-(3-bromopropyl) sulfonyl) benzene (150 mg, 4.38 mmol) in acetonitrile (3 mL) was added K2CO3 (67 mg, 4.82 mmol) and cis-2,6-Dimethylmorpholine (76 mg, 6.57 mmol) at the room temperature. The reaction mixture was stirred at 80°C for 18 h. after that reaction mixture was diluted using EtOAc (3 mL), filtered the salt and washed it with little fresh solvent. The volatile were removed under reduced pressure to give (2S,6R)-4-(3-((3- bromophenyl) sulfonyl) propyl))-2,6-dimethyl morpholine as a pale yellow thick oil which was used without purification (145 mg, 87%).Ή NMR (400 MHz, CDCI3) δ 8.06 (t, J = 1.6 Hz, 1H), 7.87 (d, J =8 Hz, 1H), 7.78 (d, J =8 Hz, 1H), 7.46 (t, J =7.6 Hz, 1H), 3.62-3.56 (m, 2H), 3.56- 3.19 (m, 2H), 2.58 (d, J = 10.4 Hz, 2H), 2.35 (t, J =6.8 Hz, 2H), 1.91-1.87 (m, 2H),1.71-1.65 (m, 2H), 1.14 (d, J =6.4 Hz, 6H) MS ESI 377.9. [M + H]+, calcd for [Ci5H22BrN03S + H]+ 378. D. (2S,6R)-2,6-dimethyl-4-(3-((3-trimethylstannyl)phenyl)sulfonyl) propyl)-morpholine
To a mixture of (2S,6R)-4-(3-((3-bromophenyl)sulfonyl)propyl))-2,6-dimethyl morpholine (125 mg, 0.33 mmol) in toluene (3 mL) was added (Me)3Sn2 (152 mg, 0.46 mmol) and Pd(PPh3)4 (27 mg, 0.023 mmol).The degassed solution was sealed and heated in a pie block at 1 10°C for overnight. Then reaction mixture was diluted using EtOAc (15 mL) and 20% aq, KF (20 mL) solution and stirred for 1 h at room tempratue, filtered the solution through silica pad and washed it with EtOAc (20 mL).combined filtratet were concentrated under reduced pressure at 30°C/80 mbar. The residual oily mass was purified by using short silica silycycle column to give title compound as thick colorless oil (140 mg, 91 %).Ή NMR (400 MHz, CDCI3) δ 8.00 (s, 1H), 7.85 (d, / = 8 Hz, 1H), 7.77 (d, J =6.8 Hz, 1H), 7.45-7.47 (m, 1H), 3.59-3.55 (m,
-193-4820V.1
2H), 3.21-3.17 (m, 2H), 2.57 (d, / = 10.4 Hz, 2H), 2.35 (t, J =6.8 Hz, 2H), 1.94-1.89 (m, 2H), 1.66 (t, J =10.8 Hz, 2H), 1.13 (d, J =6.4 Hz, 6H), 0.35 (t, J =21.6 Hz, 9H); MS ESI 462. [M + H]+, calcd for [Ci8H3iN03SSn + H]+ 461.1.
E. N-(3-(3-((3-((2S, 6R-2, 6-dimethylmorpholino) propyl) sulfonyl) phenyl)- lH-indazol-5 -yl)-2- (pyrrolidin-l-yl)-2-( thiophen-3-yl) acetamide trifluoroacetate
Mixed 2S,6R)-2,6-dimethyl-4-(3-((3-trimethylstannyl)phenyl)sulfonyl) propyl)- morpholine (53 mg, 0.1 16 mmol), N-(3-iodo-lH-indazol-5-yl)-2-(pyrrolidin-l-yl)-2-(thiophene- 3-yl) acetamide (44 mg, 0.097 mmol), DMF (3 mL) and Pd(PPh3)4 (6 mg, 0.005 mmol) in clean and dry 5 mL scillilation vial at room temprature.The degassed solution was sealed and heated in a pie block at 135°C for 3.5 h. after reaction completion cooled to room temprature and diluted it with DCM(2 mL) and filtered the insoluble mass and Washed it with little DCM. combined filtratet were concentrated under reduced pressure to give brown oil. Crude product was purified using BiotageSNAP 25 g silica column (gradient 0-10% MeOH in DCM) and repurified by Reverse phase Biotage Ci8, 50 g column chromatography (gradient 90: 10-20:80 % 0.1% TFA- H20: MeOH) to give the title compound as a white solid (9 mg, 11 %).Ή NMR (400 MHz, CD3OD) δ 8.51 (s, IH), 8.45 (s, IH), 8.34 (d, J =8.0 Hz, IH), 8.02 (d, J =6.4 Hz, IH), 7.87-7.83 (m, 2H), 7.66-7.55 (m, 3H),7.38 (d, J =4.4 Ηζ,ΙΗ), 5.24 (s, IH), 3.81 (br.m, 2H), 3.48-3.42 (m, 4H), 3.18-3.11 (m, 4H), 2.68 (t, J =12.8 Hz ,2H), 2.23-1.99 (br.m, 6H),1.22 (d, J =6.4 Hz, 6H), 2H merged with solvent peak, MS ESI 622.4. [M + H]+, calcd for [C32H39N5O4S2 + H]+ 622.2.

25 The title compound was synthesized according to the General Method A utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (35 mg, 0.11 mmol), 1,2,3,4-
-194-
NIE1 11 4820V.1
tetrahydroisoquinolin-4-amine (244 mg, 1.1 1 mmol), DIPEA (325 μΐ,, 1.9 mmol), TBTU (35 mg, 0.1 1 mmol) and DMF (4 mL). The reaction mixture was purified by preparative HPLC, and triturating with Et20 to give the title compound as a white solid (5 mg, 8%).Ή NMR (400 MHz, Acetone-d6) δ ppm 12.82 (br. s, 1 H), 9.31 (br. d, 7=8.00 Hz, 1 H), 8.79 (s, 1 H), 8.60 (s, 1 H), 8.30 (d, 7=7.78 Hz, 1 H), 8.08 (d, 7=9.03 Hz, 1 H), 7.92 (d, 7=7.53 Hz, 1 H), 7.60 - 7.75 (m, 3 H), 7.47 - 7.57 (m, 1 H), 7.32 (br. s., 3 H), 6.76 (br. s, 1 H), 5.58 (br. s., 1 H), 4.52 (q, 7=16.06 Hz, 2 H), 3.72 - 3.92 (m, 2 H). MS ESI [M + H]+ 448.2, calcd for
 + H]+ 448.1.
+ H]+ 448.1.
Example A140. 2-hvdroxy-N-(4-methyl-3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-2- phenyl acetamide

To a solution of 3-(5-amino-4- methyl- l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3- yl)benzenesulfonamide (100 mg, 0.26mmol) and 2-hydroxy-2-phenylacetic acid (60 mg, 0.39mmol) in DMF (5 mL) was added HATU (145 mg, 0.52mmol) and DIPEA (68 mg, 0.52 mmol), and the mixture was stirred at 28 °C overnight. The mixture was poured into water, and extracted with CH C12. The organic layer was washed with brine twice, dried over Na2S04, concentrated. The residue was purified by preparative TLC (Si02, CH2Cl2/MeOH 20: 1) to give the title compound as a yellow solid (25 mg, 18 %). MS ESI [M + H]+ 521, calcd for
[C27H28N4O5S+ H]+ 521. The compound was stirred in 3 M HCl/dioxane overnight and concentrated. The residue was treated with Na2C03 in MeOH and the filtrate was purified by preparativeTLC (Si02, CH2Cl2/MeOH 20: 1) to give compound the title compound as a yellow solid (17 mg, 81 %). Ή NMR (300 MHz, METHANOL-^) δ ppm 8.09 (t, 7=1.61 Hz, 1 H), 7.98 (dq, 7=7.88, 0.99 Hz, 1 H), 7.79 (dt, 7=7.84, 1.36 Hz, 1 H), 7.65 (t, 7=7.80 Hz, 1 H), 7.57 (d, 7=7.04 Hz, 2 H), 7.27 - 7.44 (m, 5 H), 5.20 (s, 1 H), 2.08 (s, 3 H). MS ESI [M + H]+ 437.2, calcd for [C22H2oN404S+H]+ 437.1.
-195-4820v.1
Table 3
General Method B, 2-hydroxy-N-(4-methyl-3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-2- phenylacetamide


-196- 4820V.1

0V.1

-198- 0V.1
152 463.2 [C25H26N403S+H]+ Ή NMR (300 MHz, METHANOL-
463.2 t¾) 5 ppm 8.10 (t, 7=1.50 Hz, 1 H),
2r* 7.99 (dt, 7=7.60, 1.50 Hz, 1 H),
7.79 (dt, 7=7.90, 1.20 Hz, 1 H), 7.66 (t, 7=7.60 Hz, 1 H), 7.41 (d, 7=8.50 Hz, 1 H), 7.20 - 7.36 (m, 4 H), 7.14 (td, 7=7.30, 1.50 Hz, 1 H), 3.83 (s, 2 H), 2.13 (s, 3 H), 1.25 (d, 7=6.74 Hz, 6 H) *(CHMe2 is obscured by the slovent peak)
153 \ 424.1 Ή NMR (300 MHz, METHANOL-
[C2iH2iN503S+H]+ d ) δ ppm 8.09 (t, 7=1.76 Hz, 1H), ' 424.1 7.99 (dq, 7=7.84, 1.10 Hz, 1 H),
7.79 (dt, 7=7.60, 1.20 Hz, 1 H), 7.66 (t, 7=7.90 Hz, 1 H), 7.40 (d, 7=8.80 Hz, 1 H), 7.30 (d, 7=8.80 Hz, 1 H), 6.62 (dd, 7=2.70, 2.00 Hz, 1 H), 6.06 (dd, 7=3.66, 1.91 Hz, 1 H), 5.98 (dd, 7=3.70, 2.90 Hz, 1 H), 3.74 (s, 2 H), 3.64 (s, 3 H), 2.11 (s,
3 H)
Example A 154. (R -2-methoxy-N-(6-methoxy-3-(3-sulfamoylphenyl')- lH-indazol-5-vD-2- phenylacetamide

-199-4820V.1

Synthesis of(R)-2-methoxy-N-(6-methoxy-lH-indazol-5-yl)-2-phenylacetamide
The title compound was synthesized according to General Method A, utilizing lH-indazol-5- amine (0.040 g, 0.24 mmol), (R)-2-methoxy-2-phenylacetic acid (0.084 g, 0.50 mmol), N-ethyl- N-isopropylpropan-2-amine (0.17 mL, 0.98 mmol) and 0-(Benzotxiazol-l-yl)-N,N,N',N'- tetramethyluronium tetrafluoroborate (0.16 g, 0.50 mmol) in DMF (3 mL). Later H20 was added and the reaction concentrated to dryness under reduced pressure. The residue was taken into anh MeOH (3 mL) and heated with MeONa (25 %wt in MeOH, 0.3 mL, 1.3 mmol) at 65 oC for 1.5 h. After concentration under reduced pressure, the crude material was purifed by column chromatography (Si02, 0-10 % MeOH/DCM) to afford a tan solid (70 mg, 92 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.44 (s, 1 H), 7.90 (s, 1 H), 7.31 - 7.53 (m, 5 H), 7.08 (s, 1 H), 4.83 (s, 1 H), 4.01 (s, 3 H), 3.45 (s, 3 H) δ ppm; MS ESI 312.1 [M + H]+, calcd for
[Ci7H17N303 + H]+ 312.3.
Syntheisis of(R)-N-(3-iodo-6-methoxy-lH-indazol-5-yl)-2-methoxy-2-phenylacetamide
The title compound was synthesized according to the General Method B, utilizing crude (R)-2- methoxy-N-(6-methoxy- lH-indazol-5-yl)-2-phenylacetamide (0.070 g, 0.22 mmol ), iodine (0.1 1 g, 0.45 mmol) and K2C03 (0.09 g, 0.7 mmol) in DMF (3 mL). The reaction was quenched by an addition of xs 10 % aq NaHS03, diluted with H20. The precipitate was isolated by filtration, washed by H20 to provide the title compound to as a light tan solid (0.065 g, 68 %). Ή NMR
-200-4820V.1
(400 MHz, METHANOL-^) δ ppm 8.18 - 8.21 (s, 1 H), 7.31 - 7.53 (m, 5 H), 7.06 (s, 1 H), 4.86 (s, 1 H), 4.02 (s, 3 H), 3.46 (s, 3 H).
Synthesis of (R)-2-methoxy-N-(6-methoxy-3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-2- phenylacetamide
The title compound was synthesized according to the General Method C, utilizing (R)-N-(3- iodo-6-methoxy-lH-indazol-5-yl)-2-methoxy-2-phenylacetamide (65 mg, 0.15 mmol), 3- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzenesulfonamide (55 mg, 0.19 mmol), satd aq Na2C03 (0.5 mL), PhMe (1.5 mL), EtOH (1.5 mL) and Pd(dppf)Cl2 (5.5 mg, 0.0074 mmol). The crude material after filtration through Celite using MeOH was purified by preparative HPLC followed by preparative TLC (Si02, 5 % MeOH/DCM) and recrystallization from MeOH/DCM to provide the title compound to as a white powder (17 mg, 23 ). Ή NMR (400 MHz, CD3CN) δ ppm 11.29 (s, 1 H), 9.32 (br. s., 1 H), 8.95 (s, 1 H), 8.45 (s, 1 H), 8.15 (d, 7=8.28 Hz, 1 H), 7.86 (d, 7=7.53 Hz, 1 H), 7.69 (t, 7=8.10 Hz, 1 H), 7.33 - 7.56 (m, 5 H), 7.17 (s, 1 H), 5.74 (br. s., 2 H), 4.85 (s, 1 H), 4.06 (s, 3 H), 3.46 (s, 3 H); MS ESI 467.2 [M + H]+, calcd for
[C23H22N4O5S + H]+ 467.5.
Example A155. N-(6-methyl-3-(3-sulfamoylphenyl)- l-(tetrahvdro-2H-pyran-2-yl)-lH-indazol-
5-yl)-2-o-tolylacetamide

To a solution of 3-(5-amino-6-methyl-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-3- yl)benzenesulfonamide (50 mg, 0.13mmol) and 2-(2-methylphenyl)acetic acid (29 mg, 0.20mmol) in DMF (5 mL) was added HATU (73 mg, 0.26mmol) and DIEA(34 mg,
0.26mmol). The mixture was stirred at 28°C overnight, poured into water, and extracted with CH2CI2. The organic layer was washed with brine twice, dried over Na2S04> concentrated. The residue was purified by preparation TLC (Si02, CH2Cl2/MeOH 20: 1) to give the title compound
-201- 820v.1
N-(6-methyl-3-(3-sulfamoylphenyl)-l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-5-yl)-2-o- tolylacetamide as yellow solid (27 mg, 37%). The material was taken in 3 M HCl/dioxane and stirred overnight. After concentratinon under reduced pressure, the residue was treated with Na2C03 in MeOH, filtered and the filtrate purified by preparative TLC (Si02, CH2Cl2/MeOH 20: 1) to give the title compound as yellow solid (17 mg, 75%). Ή NMR (300MHz/ CD3OD): Ή NMR (300 MHz, METHANOL-^) δ ppm 8.45 (s, 1 H), 8.14 (d, 7=7.80 Hz, 1 H), 8.01 (s, 1 H), 7.92 (d, 7=7.80 Hz, 1 H), 7.67 (t, 7=7.80 Hz, 1 H), 7.44 (s, 1 H), 7.31 - 7.36 (m, 1 H), 7.15 - 7.23 (m, 3 H), 3.83 (s, 2 H), 2.42 (s, 3 H), 2.30 - 2.35 (s, 3 H). MS ESI 435.1 [M + H]+, calcd for [C23H22N403S+H]+ 435.1.
Table 4
Other examples synthesized according to the method for N-(6-methyl-3-(3-sulfamoylphenyl)-l- (tetrahydro-2H-pyran-2-yl)- 1 H-indazol-5-yl)-2-o-tolylacetamide


-202- 4820V.1
157 461.2 [C25H24N403S+H]+ Ή NMR (300 MHz, METHANOL- 461.2 <¾ δ ppm 8.46 (s, 1 H), 8.14 (d,
7=8.20 Hz, 1 H), 8.00 (s, 1 H), 7.92 (d, 7=7.90 Hz, 1 H), 7.67 (t, 7=7.60
(S)-N &-(6-methyl-3- Hz, 1 H), 7.44 (s, 1 H), 7.25 - 7.31 (3- (m, 1 H), 7.09 - 7.22 (m, 3 H), 3.99 sulfamoylphenyl)- (t, 7=6.70 Hz, 1 H), 2.69-2.97 (m, 2 lH-indazol-5-yl)- H), 2.37 (s, 3 H), 2.16 - 2.25 (m, 2 1,2,3,4- H), 2.02 - 2.16 (m, 1 H), 1.71 - 1.88 tetrahydronaphthale (m, 1 H) ne-l-carboxamide
158 461.1 [C25H24N403S+H]+ Ή NMR (300 MHz, DMS0-d6) δ
& 461.2 ppm 13.31 (s, 1 H), 9.64 (s, 1 H),
8.42 (s, 1 H), 8.13 (d, 7=7.84 Hz, 1 H), 8.00 (s, 1 H), 7.83 (d, 7=7.92
(S)-N-(6-methyl-3- Hz, 1 H), 7.73 (t, 7=7.60 Hz, 1 H), (3- 7.48 (s, 2 H), 7.22 - 7.31 (m, 1 H), sulfamoylphenyl)- 7.05 - 7.21 (m, 3 H), 3.96 (t, lH-indazol-5-yl)- 7=6.50 Hz, 1 H), 2.67 - 2.83 (m, 2 1,2,3,4- H), 2.37 (s, 3 H), 1.96 - 2.16 (m, 3 tetrahydronaphthale H), 1.61 - 1.77 (m, 1 H) ne-l-carboxamide
159 CF3 489.2 [C23H19F3N403S+H
]+ 489.1 Ή NMR (300 MHz, DMSO-d6) δ ppm 13.31 (s, 1 H), 9.67 (s, 1 H), 8.40 (s, 1 H), 8.10 (d, 7=7.5 Hz, 1
N-(6 &-methyl-3-(3- H), 7.97 (s, 1 H), 7.79 - 7.85 (m, 2 sulfamoylphenyl)- H), 7.68 - 7.76 (m, 3 H), 7.65 (d, lH-indazol-5-yl)-2- 7=7.3 Hz, 1 H), 7.58 (d, 7=7.5 Hz, (2- 1 H), 7.43 - 7.53 (m, 3 H), 3.98 (s,
(trifluoromethyl)ph 2 H), 2.35 (s, 3 H)
enyl)acetamide
V.1

v.1
163 OMe 451.1 [C23H22N404S+H]+ Ή NMR (300 MHz, DMSO-d6) δ
451.1 ppm 13.36 (s, 1 H), 9.68 (s, 1 H),
8.40 (t, 7= 1.6 Hz, 1 H), 8.1 1 (dd, 7=7.8, 1.2 Hz, 1 H), 7.97 (s, 1 H),
(R) σ-2-methoxy-N- 7.80 - 7.86 (m, 1 H), 7.67 - 7.75 (m,
(6-methyl-3-(3- 1 H), 7.49 - 7.56 (m, 2 H), 7.31 - sulfamoylphenyl)- 7.48 (m, 6 H), 4.91 (s, 1 H), 3.42 (s, lH-indazol-5-yl)-2- 3H), ? 2.21 (s, 3 H)
phenylacetamide
164 OH 437.1 [C22H2oN404S+H]+
437.1 Ή NMR (300 MHz, DMSO-d6) δ
ppm 13.34 (s, 1 H), 9.56 (s, 1 H), 8.40 (t, 7=1.5 Hz, 1 H), 8.07 - 8.15
2-hydroxy-N-(6- (m, 2 H), 7.83 (ddd, 7=8.2, 1.4, 1.1 methyl-3-(3- Hz, 1 H), 7.68 - 7.75 (m, 1 H), 7.52 sulfamoylphenyl)- - 7.58 (m, 2 H), 7.46 (d, 7=4.5 Hz, 3 lH-indazol-5-yl)-2- H), 7.26 - 7.42 (m, 3 H), 6.57 (d, phenylacetamide
7=4.0 Hz, 1 H), 5.16 (d, 7=2.8 Hz, 1 H), 2.26 (s, 3 H)
165 NH2 450.2 [C23H23N503S+H]+
450.1 Ή NMR (300 MHz, DMSO-d6) δ
ppm 13.37 (br. s., 1 H), 9.89 (br. s.,
Of *'" 1 H), 8.41 (s, 1 H), 8.12 (d, 7=7.9
3-amino-N-(6- Hz, 1 H), 8.02 (s, 1 H), 7.84 (d, methyl-3-(3- 7=7.9 Hz, 1 H), 7.73 (m, 7=7.9, 7.9 sulfamoylphenyl)- Hz, 1 H), 7.29 - 7.53 (m, 8 H), 4.15 lH-indazol-5-yl)-2- - 4.27 (m, 1 H), 3.45 - 3.57 (m, 1 phenylpropanamide H), 3.07 - 3.20 (m, 1 H), 2.08 (s, 3
H)
166 cr 427.1 [C2oH18N403S2+H]+
II 427.1 Ή NMR (300 MHz, METHANOL-
N-(6-methyl-3-(3- d4) δ ppm 8.45 (t, 7=1.6 Hz, 1 H), sulfamoylphenyl)- 8.14 (d, 7=7.9 Hz, 1 H), 7.98 (s, 1 lH-indazol-5-yl)-2- H), 7.92 (d, 7=7.9 Hz, 1 H), 7.67 (t, (thiophen-2- 7=7.8 Hz, 1 H), 7.44 (s, 1 H), 7.32 yl)acetamide (dd, 7=5.2, 1.2 Hz, 1 H), 7.07 (d,
7=2.5 Hz, 1 H), 7.00 (dd, 7=5.1, 3.4 Hz, 1 H), 3.98 (s, 2 H), 2.33 (s, 3 H)
v.1

Example A168. 2-(2-(Dimethylamino ethoxy)-2-phenyl-N-(3-(3-sulfamoylphenyl)-lH-indazol-

The title compound was synthesized according the General Method A, utilizing (2- (dimethylamino)ethoxy)-2-phenylacetic acid (85 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (100 mg, 0.25 mmol), Et3N (0.14 mL, 1.0 mmol), TBTU (81 mg, 0.25 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC to yield a TFA salt of the title compound as a white solid (28 mg, 18%). Ή NMR (400 MHz, METHANOL-^) δ ppm 3.48 (s, 3 H)**, 3.65-3.70 (m., 1 H) 3.95-4.03 (m, 1 H) 4.13-4.21 (m., 2 H) 5.61 (s, 1 H) 7.50 (d, 8.78 Hz, 1H) 7.53 - 7.68 (m, 4 H) 7.72 (t, 7=7.53 Hz, 1 H) 7.84 (d, 7=6.53 Hz, 2 H) 7.97 (d, 7=8.03 Hz, 1 H) 8.16 (d, 7=8.03 Hz, 1 H) 8.39 (br. s., 1 H) 8.49 (s, 1 H); MS ESI [M + H]+ 494.2, calcd for [C25H27N5O4S + H]+ 494.3.
** one signal (s, 3 H) is obscured by the solvent peak at 3.31 ppm
-206- 4820V.1
Example A169. 2-Ethoxy-2-phenyl-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)acetamide

The title compound was synthesized according the General Method A, utilizing 2-ethoxy-2- phenylacetic acid (47 mg, 0.26 mmol), 3-(5-amino- 1 H-indazol-3-yl)benzenesulfonamide 2,2,2- tnfluoroacetate ( 100 mg, 0.25 mmol), DIPEA (0.12 mL, 0.74 mmol), TBTU (81 mg, 0.25 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC to give the title compound as a white solid (43 mg, 38%). Ή NMR (400 MHz, METHANOL-^) δ ppm 1.33 (t, 7=7.03 Hz, 3 H) 3.53 - 3.74 (m, 2 H) 4.95 (s, 1 H) 7.29 - 7.45 (m, 3 H) 7.48 - 7.62 (m, 3 H) 7.63 - 7.71 (m, 1 H) 7.93 (d, 7=7.78 Hz, 1 H) 8.14 (d, 7=7.78 Hz, 1 H) 8.37 (s, 1 H) 8.46 (s, 1 H); MS ESI [M + H]+ 451.3 (100), calcd for [C23H22N4O4S + H]+ 451.1.

The title compound was synthesized according the General Method A, utilizing 2-(ethylamino)- 2-phenylacetic acid hydrochloride (59 mg, 0.27 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide hydrochloride ( (90 mg, 0.28 mmol), DIPEA (0.12 mL, 0.74 mmol), TBTU (81 mg, 0.25 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC to give the title compound as a white solid (6 mg, 4 %).
Ή NMR (400 MHz, METHANOL- 4) 8 ppm 1.37 (t, 7=6.80 Hz, 3 H), 2.95-3.08 (m, 1 H), 3.09- 3.18 (m, 1 H), 5.07 (s, 1 H), 7.50-7.60 (m, 5 H), 7.64-7.69 (m, 2 H), 7.72 (d, J = 8.00 Hz, 1H) 7.96 (d, J = 7.20 Hz, 1 H), 8.15 (d, J = 7.40 Hz, 1 H), 8.42 (s, 1H), 8.49 (s, 1H),; MS ESI [M + H]+ 450.3(100), calcd for ^Hzs S + H]+ 450.1.
-207-4820V.1
Example A171. 2-(cyclopropylamino')-2-phenyl-N-(3-(3-sulfamoylphenyl)-lH-indazol-5- vDacetamide

A mixture of c-PrNH2 (0.75 mL) and 2-bromo-2-phenylacetate (0.3 g, 1.3 mmol) in anh THF (1 mL) was stirred in a sealed vial at rt for 4 d. The reaction mixture was diluted with EtOAc , washed (brine), dried (Na2S04) and concentrated under reduced pressure to yield a colorless gum (0.3 g).
The material was stirred with NaOH (0.87 g, 22 mmol) in H20 (5 mL) and MeOH ( 10 mL) at rt overnight. The reaction mixture was cooled (0 °C), acidified with 6 M aq HC1 to pH ~ 6-7 and concentrated under reduced pressure to yield methyl 2-(cyclopropylamino)-2-phenylacetate: MS ESI [M + H]+ 206.0 (100), calcd for [C12H15NO2 + H]+ 206.1.
The solid residue was taken into MeOH and filtered. Concentration of the filtrate provided crude material that was taken into CH2C12 (24 mL) and DMF ( 6mL). DIPEA (0.74 mL, 4.2 mmol) and Boc20 (0.32 g, 1.5 mml) was added a solution in DMF (4 mL). The reaction was stirred overnight at rt later filtered rinsing the filtrate cake with 10 % MeOH/DCM. Concentration under reduced pressure provided 2-((tert-butoxycarbonyl)(cyclopropyl)amino)-2-phenylacetic acid (0.39 g, 95 %) that was used without further purification.
The title compound was synthesized according the General Method A, utilizing 2-((tert- butoxycarbonyl)(cyclopropyl)amino)-2-phenylacetic acid (112 mg, 0.31 mmol), 3-(5-amino-lH- indazol-3-yl)benzenesulfonamide hydrochloride (100 mg, 0.31 mmol), DIPEA (0.14 mL, 0.92 mmol), TBTU (99 mg, 0.31 mmol) and 3 mL of DMF. The reaction mixture was diluted with DCM and washed with (satd aq NaHC03, brine), driend (Na2S04) and concentrated under redcuced pressure. The resulting material was tatek into CH2C12 (40 mL) and treated with Et3SiH (0.1 mL) and TFA (2mL) at 0 °C. After 3 h of stirring with cooling, more TFA was added (3 mL) ant the reaction was allowed to warm to rt. After additional 1.5 the reaction concentrated under redueced pressure and purified by preparative HPLC to provide 2-
-208-4820V.1
(cyclopropylarmno)-2-phenyl-N-(3-(3-sulfamoylphenyl)- iH-indazol-5-yl)acetamide 2,2,2- trifluoroacetate as a white solid (32 mg, 18 %)
Ή NMR (400 MHz, METHANOL-^) δ ppm 0.75 - 1.08 (m, 4 H), 2.70 (br. s., 1 H), 5.16 (s, 1 H), 7.43 - 7.63 (m, 5 H), 7.64 - 7.76 (m, 3 H), 7.96 (d, 7=7.78 Hz, 1 H), 8.16 (d, 7=7.78 Hz, 1 H), 8.42 (s, 1 H), 8.49 (s, 1 H); MS ESI [M + H]+462.3 (100), calcd for [C24H23N503S + H]+ 462.1.
Example A 172. 2-(pyridin-2- yl)-2-(pyrrolidin- 1 - yl)-N-(3-(3-sulfamoylphenyl )- 1 H-indazol-5- vDacetamide

The title compound was synthesized according the General Method A, utilizing 2-(pyridin-2-yl)- 2-(pyrrolidin-l-yl)acetic acid hydrochloride (59 mg, 0.24 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide 2,2,2-trifluoroacetate (70 mg, 0.17 mmol), DIPEA (0.2 mL, 1.1 mmol), TBTU (59 mg, 0.18 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC, filtered though a PoraPak Rxn CX column (Waters, 2 g) loading and washing with MeOH and later eluting with 2 M NH3 in MeOH. The material was later repurified using flash chromatography (Si02, Biotage 50 g, 0-> 15 % MeOH in CH2C12) to give the title compound as a white solid (11.1 mg, 13 %). Ή NMR (400 MHz, METHANOL-^) 6 ppm 1.88 (brs, 4 H), 2.52-2.61 (m., 2 H), 2.67-2.75 (m., 2 H), 4.20 (s, 1 H), 7.37-7.43 (m, 1 H), 7.54-7.63 (m, 2 H), 7.71 (t, 7=7.28 Hz, 1 H), 7.75 (d, 7=7.78 Hz, 1 H), 7.88 (t, 7=7.78 Hz, 1 H), 7.94 (d, 7=7.78 Hz, 1 H), 8.16 (d, 7=7.53 Hz, 1 H), 8.41 (s, 1 H), 8.47 (s, 1 H), 8.58 (d, 7=4.52 Hz, 1 H); MS ESI [M + H]+ 477.3(100), calcd for [C Ca^N^S + H]+ 477.2.
Example A173. 2-Cvclopentyl-2-(pyrrolidin-l-yl)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5- vDacetamide

-209-4820V.1
The title compound was synthesized according the General Method A, utilizing 2-cyclopentyl-2- (pyrrolidin-l-yl)acetic acid hydrochloride (38 mg, 0.16 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide hydrochloride ( (55 mg, 0.0.17 mmol),, DIPEA (0.15 mL, 0.99 mmol), HATU (65 mg, 0.17 mmol) and 6 mL of DMF. The reaction mixture was purified using preparative HPLC to give 2-cyclopentyl-2-(pyrrolidin- 1 -yl)-N-(3-(3-sulfamoylphenyl)- 1 H- indazol-5-yl)acetamide 2,2,2-trifluoroacetate as a white solid (4.4 mg, 5 ). Ή NMR (400 MHz, METHANOL-^) δ ppm 1.50-1.90 (br.m., 6 H), 2.01 - 2.30 (br.m, 6 H), 2.50 - 2.63 (m, 1 H), 3.30-3.41 (brs, 2 H), 3.70 - 3.81 (brs, 2 H), 3.97 (d, 7=7.00 Hz, 1 H), 7.62 (m, 2 H), 7.72 (t, 7=7.78 Hz, 1 H), 7.97 (d, 7= 8.03 Hz, 1 H), 8.18 (d, 7=7.78 Hz, 1 H), 8.45 (s, 1 H), 8.50 (s, 1 H); MS ESI [M + H]+ 468.3 (100), calcd for [C24H29N5O3S + H]+ 468.2.
Example Al 74. ( S)-2-amino-3,3-dimethyl-N-(3-(3-sulfamoylphenyl)- 1 H-indazol-5- vDbutanamide

The title compound was synthesized according the General Method A, utilizing (S)-2-((tert- butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (98 mg, 0.42 mmol), 3-(5-amino-lH-indazol- 3-yl)benzenesulfonamide hydrochloride ( (200 mg, 0.62 mmol),, DIPEA (0.29 mL, 1.8 mmol), TBTU (198 mg, 0.62 mmol) and 3 mL of DMF. The reaction mixture was purified using preparative HPLC to give tert-butyl (3,3-dimethyl-l-oxo-l-((3-(3-sulfamoylphenyl)-lH-indazol- 5-yl)amino)butan-2-yl)carbamate. This material was taken into DCM (23 mL) and CDC13 ( 2mL) and treated with TFA (5 mL) at it. The reaction was stirred for 2 h, concentrated under reduced pressure and purified using preparative HPLC to give 2-amino-3,3-dimethyl-N-(3-(3- sulfamoylphenyl)-lH-indazol-5-yl)butanamide 2,2,2-trifluoroacetate as white powder (17 mg, 8 %). 'H NMR (400 MHz, METHANOL-^) δ ppm 1.20 (s, 9 H), 3.74 (s, 1 H), 7.51 - 7.65 (m, 2 H), 7.72 (t, 7=7.80 Hz, 1 H), 7.96 (d, 7=8.53 Hz, 1 H), 8.17 (d, 7=7.28 Hz, 1 H), 8.43 (s, 1 H), 8.49 (s, 1 H); MS ESI [M + H]+402.1 (100), calcd for [C]9H23N503S+ H]+ 402.1.
-210-4820V.1
Example A 175. N-(( 1 -Γηο 1ιο1ίη(χ:νο1ο1ΐ6χν1)ηΐ6ί1ιν1)-3-(3-8υ1ί ηΐον1ρ116ην1')- 1 H-indazole-5- carboxamide
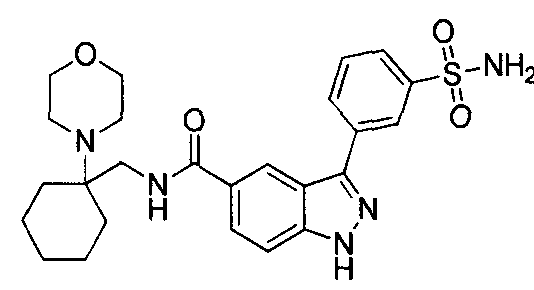
The title compound was synthesized according the General Method A, utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.31 mmol), (1- morpholinocyclohexyl)methanamine (63 mg, 0.31 mmol), DIPEA (0.1 1 mL, 0.64 mmol), TBTU (101 mg, 0.31 mmol) and 5 mL of DMF. The reaction mixture was purified using preparative HPLC followed by flash chromatography (Si02 Biotage 50 g, 3-30 % MeOH in DCM to afford the title compound as a white powder (45 mg, 29 %).
Ή NMR (400 MHz, METHANOL-^) δ ppm 1.47 (br.m, 6 H), 1.63 - 1.87 (br.m, 4 H), 2.78 (br. s., 4 H), 3.53 (s, 2 H), 3.70 (br. s., 4 H), 7.68 (d, 7=8.78 Hz, 1 H), 7.74 (t, 7=7.78 Hz, 1 H), 7.91 (dd, 7=8.78, 1.51 Hz, 1 H), 7.98 (dq, 7=7.50, 1.00 Hz, 1 H), 8.25 (dt, J = 1.6 Hz, 7.8 Hz, 1 H), 8.54 (t, 7= 1.63 1 H), 8.56 (s, 1 H); MS ESI [M + H]+ 498.3 (100), calcd for [CJSHS!NSC S + H]+ 498.2.
Example A176. lN-((l-(piperidin-l-yl)cvclohexyl)methyl)-3-(3-sulfamoylphenyl)-lH-indazole- 5-carboxamide

The title compound was synthesized according the General Method A, utilizing 3-(3- sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.31 mmol), (l-(piperidin-l- yl)cyclohexyl)methanamine 62 mg, 0.31 mmol), DIPEA (0.11 mL, 0.64 mmol), TBTU (101 mg, 0.31 mmol) and 5 mL of DMF. The reaction mixture was purified using preparative HPLC to afford N-(( l-(piperidin- l-yl)cyclohexyl)methyl)-3-(3-sulfamoylphenyl)-lH-indazole-5- carboxamide 2,2,2-trifluoroacetate as a white powder (63 mg, 33 %).
-211-4820V.1
lH NMR (400 MHz, METHANOL-^) δ ppm, 1.46 - 1.97 (m, 12 H), 2.05 (m, 4 H), 3.07 (t, 7=1 1.50 Hz, 2 H), 3.80 (d, 7=1 1.80 Hz, 2 H), 3.93 (s, 2 H), 7.70 (dd, 7=8.80, 0.80 Hz, 1 H), 7.74 (t, 7=7.78 Hz, 1 H), 7.92 - 8.04 (m, 2 H), 8.24 (dt, J = 1.26 Hz, 7.78 Hz, 1 H), 8.53 (t, 7=1.63 Hz, 1 H), 8.66 (s, 1 H); MS ESI [M + H]+ 496.3, calcd for [C26H33N5O3S + H]+ 496.2.

The title compound was synthesized according to the General Method C, utilizing 3-(5- bromo- lH-indazol-3-yl)benzenesulfonamide (67.2 mg, 0.19 mmol), (E)-2-(3-methoxy-3- phenylprop-l-en- l -yl)-4,4,5,5-tetramethyl- l,3,2-dioxaborolane (68.4 mg, 0.25 mmol),
PdCl2(dppf).DCM (21.5 mg, 0.026 mmol), toluene (2 mL), EtOH (2 mL), and aqueous Na2C03 (0.40 mL, 2 M, 0.80 mmol). The degassed solution was sealed and heated in a microwave reactor at 125 °C for 2 h. Purification by column (Silicycle lOg SPE, silica gel, 20-40% EtOAc in DCM), followed by a second column with half of this material (Biotage Isolera, lOg HP-SIL plus samplet, 30-70% EtOAc in hexane) provided the title compound as an off-white solid (20.3 mg, 51 %). ]H NMR (400 MHz, CDCh) δ ppm 1 1.0 (m, 1 H), 8.37 (s, 1 H), 7.89 (d, J=7.8 Hz, 1 H), 7.83 (d, J=8.3 Hz, 1 H), 7.65 (s, 1 H), 7.47 - 7.35 (m, 6 H), 7.33 - 7.27 (m, 2 H), 6.64 (d, J=15.8 Hz, 1 H), 6.23 (dd, J=15.8, 6.8 Hz, 1 H), 5.83 (br. s., 2 H), 4.81 (d, J=7.0 Hz, 1 H), 3.38 (s, 3 H); MS ESI 420.2 [M + H]+, calcd for [C23H21N3O3S + H]+ 420.1.

-212-4820V.1
Palladium on carbon (10%, 5.5mg, 0.005 mmol) was added to a solution of (E)-3-(5-(3- methoxy-3-phenylprop- l-en-l-yl)- lH-indazol-3-yl)benzenesulfonamide (24.2mg, 0.058 mmol) in EtOAc (2 mL) and the resulting mixture was stirred under an atmosphere of H2(g) for 1 h, then filtered through a celite depth filter using EtOAc (50 mL). NMR showed a mixture of starting olefin and product, so the sample was stirred with Palladium on carbon (10%, 8.7 mg, 0.008 mmol) in EtOAc (2 mL) for a further 1 h under an atmosphere of H2(g), and isolated as above. NMR indicated starting olefin was consumed. Purification by column (Biotage Isolera, lOg HP-SIL plus samplet, 40-70% EtOAc in hexane) provided the title compound as a sticky white film (5.1 mg, -80% pure by NMR, 14 %). Ή NMR (400 MHz, CD3OD) 8 ppm 8.49 (s, 1 H), 8.17 (d, J=7.5 Hz, 1 H), 7.94 (d, J=8.0 Hz, 1 H), 7.82 (s, 1 H), 7.67 - 7.73 (m, 1 H), 7.53 (d, J=8.5 Hz, 1 H), 7.25 - 7.38 (m, 6 H), 4.12 - 4.18 (m, 1 H), 3.21 (s, 3 H), 2.74 - 2.93 (m, 2 H), 2.09 - 2.22 (m, 1 H), 1.93 - 2.00 (m, 1 H); MS ESI 422.3 [M + H]+, calcd for ^H^NaOsS + H]+ 422.2.
Example A 179. N-(phen yl(piperidin-4-yl)methyl)-3-(3-sulf amoylphenyl)- 1 H-indazole-5- carboxamide

The title compound was synthesized using the method described for 3-(3-sulfamoylphenyl)-N- (2,2,2-trifluoro- 1 -phenylethyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)- 1 H- indazole-5-carboxylic acid (100 mg, 0.315 mmol, DMF (4 mL), DIPEA (110 pL, 0.630 mmol), isobutyl chloroformate (82 pL, 0.630 mmol), and teri-butyl 4-(amino(phenyl)methyl)piperidine- 1-carboxylate (91 mg, 0.315 mmol). The brownish residue was dissolved in MeOH (4 mL) and then 25% NaOMe (600 pL) was added. The reaction mixture was stirred for 60 minutes and then acidified with 2 M HCl.The reaction was stirred for 5 hours at which point it was purified by prep-HPLC and triturated with Et20 to give a TFA salt of the title compound: pale yellow solid
-213-4820V.1
(62 mg, 40%). Ή NMR (400 MHz, METHANOL-d4) δ ppm 9.00 (d, 7=9.29 Hz, 1 H), 8.60 (d, 7=12.05 Hz, 2 H), 8.25 (d, 7=7.03 Hz, 1 H), 7.98 (d, 7=7.53 Hz, 1 H), 7.88 (d, 7=9.03 Hz, 1 H), 7.74 (t, 7=7.91 Hz, 1 H), 7.65 (d, 7=8.03 Hz, 1 H), 7.45 (d, 7=7.03 Hz, 2 H), 7.39 (t, 7=6.65 Hz, 2 H), 7.30 (t, 7=8.00 Hz, 1 H), 4.92 - 4.95 (m, 1 H), 3.47 - 3.57 (m, 1 H), 3.33 - 3.39 (m, 1 H), 3.04 (t, 7=12.30 Hz, 1 H), 2.91 (t, 7=12.20 Hz, 1 H), 2.36 (d, 7=14.56 Hz, 1 H), 2.19 - 2.30 (m, 1 H), 1.52 - 1.70 (m, 2 H), 1.37 - 1.50 (m, 1 H) MS ESI [M + H]+ 490.3 calcd for [C26H27N5O3S + H]+ 490.2
Example A180. 2-phenyl-2-(piperidin-l-yl)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5- vDacetamide

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)-lH-indazol-5-yl)acetamide, utilizing 2-phenyl-2-(piperidin-l-yl)acetic acid (55 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated with Et20 to give a TFA salt of the title compound: white solid (96 mg, 64%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.48 (s, 1 H), 8.43 (s, 1 H), 8.15 (d, 7=7.28 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.67 - 7.74 (m, 3 H), 7.48 - 7.60 (m, 5 H), 4.97 (s, 1 H), 3.83 (br. s., 1 H), 3.17 (br. s., 2 H), 2.83 - 3.02 (m, 2 H), 1.71 - 2.06 (m, 5 H), 1.56 (br. s., 1 H) MS ESI [M + H]+ 490.2 calcd for ^H^OsS + H]+ 490.2
-214-4820V.1
Example A 181. 2-(dimethylarmno)-2-(2-methoxyphenyl)-N-(3-(3-sulfamoylphenyl)- 1 H-indazol- 5-yl)acetamide

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing 2-(dimethylamino)-2-(2- methoxyphenyl)acetic acid (52 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated with Et20 to give the title compound as a TFA salt: white solid (129 mg, 87%). !H NMR (400 MHz, METHANOLS) δ ppm 8.46 (s, 1 H), 8.37 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.93 (d, 7=7.78 Hz, 1 H), 7.68 (t, 7=7.65 Hz, 1 H), 7.52 - 7.60 (m, 3 H), 7.32 (t, 7=8.00 Hz, 1 H), 7.04 (d, 7=7.78 Hz, 1 H), 6.99 (t, 7=7.65 Hz, 1 H), 4.54 (s, 1 H), 3.89 (s, 3 H), 2.31 (s, 6 H). MS ESI [M + H]+ 480.3, calcd for [C24H25N5O4S + H]+ 480.2

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing 2-((3R,5S)-3,5-dimethylpiperidin- 1 -yl)- 2-(o-tolyl)acetic acid (66 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 ML, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated with Et20 to give the title compound: white solid (22 mg, 14%). Ή NMR (400 MHz, METHANOL-dA) 5 ppm 8.48 (s,
-215-4820V.1
1 H), 8.41 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.77 (d, 7=7.28 Hz, 1 H), 7.70 (t, 7=7.78 Hz, 1 H), 7.55 (m, 7=7.50 Hz, 2 H), 7.35 - 7.46 (m, 3 H), 5.15 (br. s, 1 H), 4.01 (br. t, 7=7.80, 7.80 Hz, 1 H), 3.89 (br. t, 7=9.00, 9.00 Hz, 1 H), 3.65 (br. s, 1 H), 2.90 - 3.00 (m, 1 H), 2.75 (d, 7=12.30 Hz, 1 H), 2.63 (s, 3 H), 2.51 - 2.61 (m, 1 H), 1.25 (d, 7=5.77 Hz, 3 H), 1.12 (d, 7=5.77 Hz, 3 H). MS ESI [M + H]+ 534.4, calcd for [C28H3iN504S + H]+ 534.2

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing 2-(4-(hydroxymethyl)piperidin- 1 -yl)-2- phenylacetic acid (62 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA ( 130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC, redissolved in MeOH (2 mL) and 6 equivalents of NaOMe was added and the reaction was stirred at 25 °C for 16 hours. The reaction was neutralized using 2 N NaOH and then repurified using prep-HPLC. Trituration with Et20 gave the title compound: white solid (21 mg, 13%). Ή NMR (400 MHz, METHANOL-d4) 6 ppm 8.48 (s, 1 H), 8.43 (s, 1 H), 8.14 (d, 7=7.78 Hz, 1 H), 7.95 (d, 7=8.28 Hz, 1 H), 7.70 (s, 3 H), 7.48 - 7.59 (m, 5 H), 5.01 (br. s., 1 H), 4.34 (br. s., 1 H), 3.92 (d, 7=1 1.54 Hz, 1 H), 3.46 (d, 7=4.52 Hz, 1 H), 3.22 (t, 7=12.67 Hz, 1 H), 2.92 - 3.05 (m, 2 H), 2.04 - 2.04 (m, 1 H), 2.07 (br. s., 1 H), 1.91 (d, 7=14.56 Hz, 1 H), 1.75 - 1.84 (m, 1 H), 1.62 - 1.75 (m, 1 H), 1.46 - 1.61 (m, 1 H). MS ESI [M + H]+ 520.4, calcd for
 + H]+ 520.2
+ H]+ 520.2
-216-4820V.1

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)- lH-indazole-5-carboxylic acid (100 mg, 0.32 mmol), (2- aminomethylphenyl)-methanol (43 mg, 0.32 mmol), DDPEA (83 pL, 0.48 mmol), TBTU (103mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-HPLC, and then triturated with Et20 and acetone to give the title compound: white solid (57 mg, 41%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.66 (s, 1 H), 9.02 - 9.08 (m, 1 H), 8.65 (s, 1 H), 8.47 (s, 1 H), 8.28 (d, 7=6.78 Hz, 1 H), 7.98 (d, 7=8.53 Hz, 1 H), 7.87 (d, 7=7.78 Hz, 1 H), 7.77 (t, 7=7.65 Hz, 1 H), 7.68 (d, 7=9.29 Hz, 1 H), 7.49 (s, 2 H), 7.38 - 7.43 (m, 1 H), 7.29 - 7.34 (m, 1 H), 7.20 - 7.27 (m, 2 H), 5.22 (t, 7=5.65 Hz, 1 H), 4.64 (d, 7=4.52 Hz, 2 H), 4.58 (d, 7=5.52 Hz, 2 H). MS ESI [M + H]+ 437.2, calcd for [C22H20N4O4S + H]+ 437.1

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)-l H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)- l H-indazole-5-carboxylic acid (100 mg, 0.32 mmol), 2- thiophenemethylamine (36 mg, 0.32 mmol), DIPEA (83 pL, 0.48 mmol), TBTU (103mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-HPLC, and then triturated
-217-4820V.1
with Et20 and acetone to give the title compound: white solid (65 mg, 49%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.67 (s, 1 H), 9.27 (t, 7=5.65 Hz, 1 H), 8.62 (s, 1 H), 8.47 (s, 1 H), 8.28 (d, 7=6.27 Hz, 1 H), 7.97 (d, 7=9.03 Hz, 1 H), 7.88 (d, 7=6.78 Hz, 1 H), 7.77 (t, 7=7.30 Hz, 1 H), 7.68 (d, 7=8.53 Hz, 1 H), 7.50 (s, 2 H), 7.38 (d, 7=4.77 Hz, 1 H), 7.02 - 7.07 (m, 1 H), 6.95 - 6.99 (m, 1 H), 4.69 (d, 7=5.77 Hz, 2 H). MS ESI [M + H]+ 413.2, calcd for [Ci9H,6N403S2 + H]+ 413.1
Example A186. N-(2-methylbenzyl)-3-(3-sulfamoylphenyl)-lH-indazole-5-carboxamide

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (200 mg, 0.63 mmol), 2- methylbenzylamine (76 mg, 0.63 mmol), DIPEA ( 164 pL, 0.95 mmol), TBTU (202 mg, 0.63 mmol) and DMF (6 mL). The reaction mixture was purified by prep-HPLC, and then triturated with Et20 and acetone to give the title compound: beige solid (157 mg, 59%). Ή NMR (400 MHz, DMSO-de) δ ppm 13.65 (s, 1 H), 9.03 (t, 7=5.40 Hz, 1 H), 8.65 (s, 1 H), 8.47 (s, 1 H), 8.28 (d, 7=6.53 Hz, 1 H), 7.99 (d, 7=9.03 Hz, 1 H), 7.87 (d, 7=8.28 Hz, 1 H), 7.77 (t, 7=8.00 Hz, 1 H), 7.68 (d, 7=9.03 Hz, 1 H), 7.49 (s, 2 H), 7.26 - 7.30 (m, 1 H), 7.13 - 7.20 (m, 3 H), 4.51 (d, 7=4.77 Hz, 2 H), 2.35 (s, 3 H). MS ESI [M + H]+ 421.2 calcd for [C22H20N4O3S + H]+ 421.1
-218-4820V.1

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- lH-indazol-5-yl)acetamide, utilizing 2-(4-Boc-piperazinyl)-2-phenylacetic acid (96 mg, 0.30 mmol), 3-(5-amino-l H-indazol-3-yl)benzenesulfonamide · TFA (120 mg, 0.30 mmol), DIPEA (156 μί, 0.90 mmol), TBTU (96 mg, 0.30 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and then redissolved in DCM (5 mL). Reaction was cooled to 0 °C and then TFA (2 mL) was added. Mixture was stirred at 0 °C for one hour, at which point the solvent was removed under reduced pressure. Redissolved the residue in MeOH and then purified by prep-HPLC to give the title compound: white solid (80 mg, 44%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.45 (s, 1 H), 8.39 (s, 1 H), 8.1 1 (d, 7=7.78 Hz, 1 H), 7.92 (d, 7=7.78 Hz, 1 H), 7.66 (t, 7=7.65 Hz, 1 H), 7.61 (d, 7=7.28 Hz, 2 H), 7.48 - 7.56 (m, 2 H), 7.38 - 7.48 (m, 3 H), 4.48 (s, 1 H), 3.36 - 3.42 (m, 4 H), 2.99 - 3.09 (m, 2 H), 2.90 - 2.99 (m, 2 H). MS ESI [M + H]+ 491.3, calcd for [C25H26N603S + H]+ 491.2
Example A 188. 2-(4-hvdroxypiperidin- 1 -yl)-2-phenyl-N-(3-(3-sulfamoylphenyl)- lH-indazol-5-

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing 2-(4-hydroxypiperidin- 1 -yl)-2- phenylacetic acid (59 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA
-219-4820V.1
(100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated with Et20 to give the title compound: white solid (24 mg, 16%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.48 (s, 1 H), 8.43 (s, 1 H), 8.15 (d, 7=8.78 Hz, 1 H), 7.96 (d, 7=7.28 Hz, 1 H), 7.70 (br. s., 3 H), 7.54 - 7.60 (m, 4 H), 7.51 (d, 7=9.29 Hz, 1 H), 5.02 (br. s., 1 H), 4.10 (br. s., 1 H), 3.88 (br. s., 1 H), 3.70 (br. s., 1 H), 3.55 (br. s., 1 H), 3.02 (br. s., 1 H), 2.82 (br. s., 1 H), 1.67 - 2.25 (m, 4 H). MS ESI [M + H]+ 506.3, calcd for [C26H27N504S + H]+ 506.2
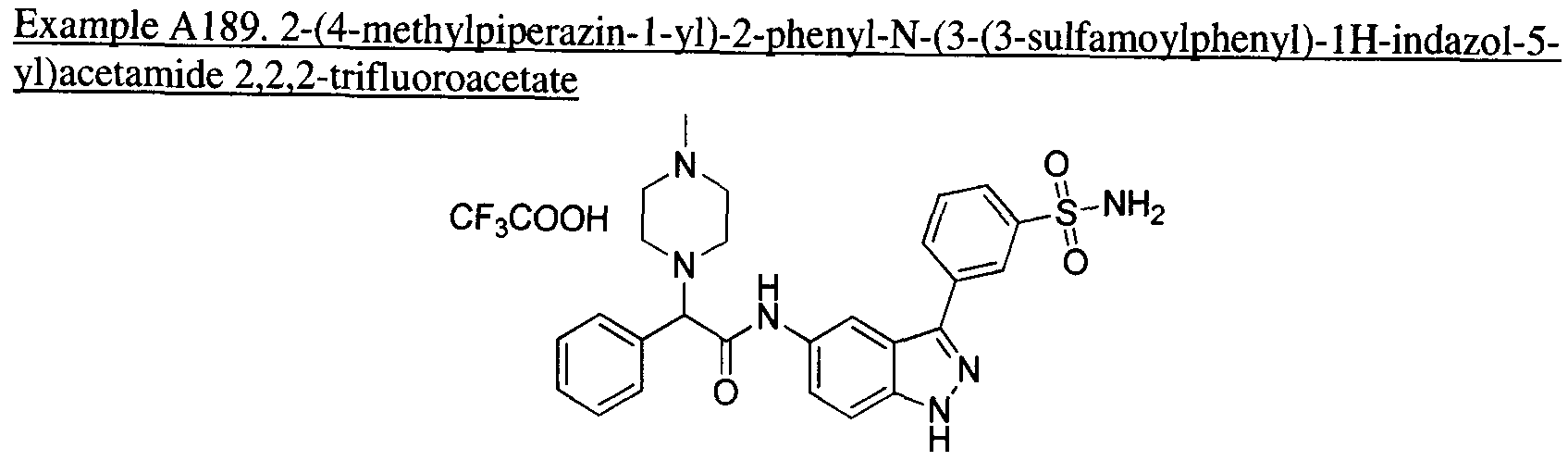
The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing 2-(4-methylpiperazin- 1 -yl)-2- phenylacetic acid (59 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated with Et20 to give the title compound: white solid (43 mg, 28%). Ή NMR (400 MHz, METHANOL-d*) δ ppm 8.47 (s, 1 H), 8.38 (s, 1 H), 8.15 (d, 7=8.78 Hz, 1 H), 7.94 (d, 7=8.28 Hz, 1 H), 7.69 (t, 7=7.53 Hz, 1 H), 7.51 - 7.60 (m, 4 H), 7.35 - 7.46 (m, 3 H), 4.24 (s, 1 H), 3.40 - 3.56 (m, 2 H), 3.14 - 3.28 (m, 3 H), 2.92 - 3.01 (m, 1 H), 2.90 (s, 3 H), 2.62 - 2.77 (m, 3 H), 2.32 - 2.46 (m, 1 H). MS ESI [M + H]+ 505.3, calcd for [CaeHasNfAS + H]+ 505.2
-220-4820V.1

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.32 mmol), 2-morpholin-4-yl- 1-phenyl-ethylamine (65 mg, 0.32 mmol), DIPEA (1 1 1 μί, 0.64 mmol), TBTU (103mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-HPLC, and then triturated with Et20 to give the title compound: white solid (74 mg, 37%). Ή NMR (400 MHz,
METHANOL-^) 5 ppm 8.68 (s, 1 H), 8.54 (s, 1 H), 8.23 (d, 7=7.53 Hz, 1 H), 7.98 (t, 7=7.91 Hz, 2 H), 7.72 (t, 7=8.03 Hz, 1 H), 7.66 (d, 7=8.78 Hz, 1 H), 7.54 (d, 7=8.78 Hz, 2 H), 7.45 (t, 7=7.53 Hz, 2 H), 7.37 (t, 7=7.00 Hz, 1 H), 5.79 (d, 7=9.03 Hz, 1 H), 3.95 (br. s., 4 H), 3.79 (t, 7=12.42 Hz, 1 H), 3.64 (d, 7=1 1.54 Hz, 1 H), 3.40 (br. s., 4 H). MS ESI [M + H]+ 506.4, calcd for
[C26H27N504S + H]+ 506.2

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)-lH-indazol-5-yl)acetamide, utilizing a-cyclopentylphenylacetic acid (51 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated with Et20 to give the title compound: white
-221-4820V.1
solid (33 mg, 28%). Ή NMR (400 MHz, METHANOL-^) 5 ppm 8.46 (s, 1 H), 8.35 (s, 1 H), 8.15 (d, 7=8.53 Hz, 1 H), 7.93 (d, 7=7.28 Hz, 1 H), 7.70 (t, 7=7.53 Hz, 1 H), 7.45 - 7.56 (m, 4 H), 7.32 (t, 7=7.78 Hz, 2 H), 7.25 (t, 7=6.80 Hz, 1 H), 3.37 (t, 7=11.30 Hz, 1 H), 2.72 (br. s, 1 H), 1.98 (br. s, 1 H), 1.33 - 1.80 (m, 6 H), 1.08 (br. s, 1 H). MS ESI [M + H]+ 475.3, calcd for
[C26H26N403S + H]+ 475.2

The title compound was synthesized using the method described for 3-(3-sulfamoylphenyl)-N- (2,2,2-trifluoro- 1 -phenylethyl)- lH-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)- 1 H- indazole-5-carboxylic acid (100 mg, 0.315 mmol, DMF (4 mL), DIPEA (1 10 μί, 0.630 mmol), isobutyl chloroformate (82 μί, 0.630 mmol), and ( 1 -methylpiperidin-4-yl)(phenyl)methanamine (60 mg, 0.315 mmol). The reaction was purified by prep-HPLC and triturated with ET20 to give the title compound: white solid (16 mg, 10%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.59 (d, 7=10.54 Hz, 2 H), 8.24 (d, 7=8.03 Hz, 1 H), 7.98 (d, 7=8.53 Hz, 1 H), 7.88 (d, 7=8.53 Hz, 1 H), 7.73 (t, 7=7.78 Hz, 1 H), 7.64 (d, 7=8.78 Hz, 1 H), 7.45 (d, 7=7.28 Hz, 2 H), 7.38 (t, 7=7.15 Hz, 2 H), 7.30 (t, 7=7.30 Hz, 1 H), 4.90 - 4.94 (m, 1 H), 3.63 (d, 7=12.55 Hz, 1 H), 3.46 (d, 7=12.55 Hz, 1 H), 3.03 (t, 7=14.05 Hz, 1 H), 2.88 - 2.97 (m, 1 H), 2.87 (s, 3 H), 2.38 (d, 7=15.31 Hz, 1 H), 2.16 - 2.30 (m, 1 H), 1.43 - 1.73 (m, 3 H). MS ESI [M + H]+ 504.5, calcd for
[C27H29N503S + H]+ 504.2
-222-4820V.1
xample A193. 2-cvclohexyl-2-phenyl-N-(3-(3-sulfamoylphenyl -lH-indazol-5-vnacetamide

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)-lH-indazol-5-yl)acetamide, utilizing cyclohexylphenylacetic acid (55 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 pL, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated with Et20 to give the title compound: white solid (71 mg, 58%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.45 (s, 1 H), 8.35 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.93 (d, 7=7.28 Hz, 1 H), 7.70 (t, 7=7.91 Hz, 1 H), 7.47 - 7.55 (m, 2 H), 7.45 (d, 7=7.03 Hz, 2 H), 7.32 (t, 7=6.90 Hz, 2 H), 7.24 (t, 7=7.30 Hz, 1 H), 3.33 - 3.37 (m, 1 H), 2.10 - 2.22 (m, 1 H), 1.97 (d, 7=1 1.80 Hz, 1 H), 1.79 (d, 7=13.55 Hz, 1 H), 1.67 (br. s., 2 H), 1.30 - 1.42 (m, 2 H), 1.10 - 1.27 (m, 3 H), 0.76 - 0.91 (m, 1 H). MS ESI [M + H]+ 489.3, calcd for [C27H28N403S + H]+ 489.2 Example A194. 2-(dimethylamino)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-2-(fhiophen-3- vDacetamide

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing 2-(dimethylamino)-2-(thiophen-3- yl)acetic acid (93 mg, 0.50 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (200 mg, 0.50 mmol), DIPEA (260 pL, 1.50 mmol), TBTU (160 mg, 0.50 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and then eluted from PoraPak column to give the title compound: pink solid (74 mg, 32%). Ή NMR (400 MHz, METHANOL-^) δ ppm
-223- 820V.1
8.48 (s, 1 H), 8.41 (s, 1 H), 8.15 (d, 7=6.27 Hz, 1 H), 7.95 (d, 7=7.78 Hz, 1 H), 7.66 - 7.77 (m, 2 H), 7.52 - 7.61 (m, 3 H), 7.34 (d, 7=4.52 Hz, 1 H), 4.78 (br. s., 1 H), 2.66 (br. s., 6 H). MS ESI [M + H]+ 456.2, calcd for
 + H]+ 456!
+ H]+ 456!

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.32 mmol),
cyclohexylmethylamine (36 mg, 0.32 mmol), DIPEA ( 1 1 1 μΙ_, 0.64 mmol), TBTU (103mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-HPLC, and then triturated with Et20 to give the title compound: white solid (75 mg, 57%). Ή NMR (400 MHz, DMSO-df,) δ ppm 13.63 (br. s., 1 H), 8.56 (br. s., 2 H), 8.47 (s, 1 H), 8.27 (d, 7=8.78 Hz, 1 H), 7.92 (d, 7=9.29 Hz, 1 H), 7.88 (d, 7=7.53 Hz, 1 H), 7.78 (t, 7=7.53 Hz, 1 H), 7.65 (d, 7=9.29 Hz, 1 H), 7.49 (s, 3 H), 3.15 (br. s., 2 H), 1.71 (t, 7=14.80 Hz, 4 H), 1.50 - 1.65 (m, 2 H), 1.10 - 1.28 (m, 3 H), 0.85 - 1.02 (m, 2 H). MS ESI [M + H]+ 413.3, calcd for [C^H^OaS + H]+ 413.2
Example A196. N-(cvclohexyl(phenyl)methyl)-3-(3-sulfamoylphenyl)-lH-indazole-5- carboxamide

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-l H-indazole-5-carboxylic acid (100 mg, 0.32 mmol),
-224-4820V.1
cyclohexyl(phenyl)methanamine (60 mg, 0.32 mmol), DIPEA (1 1 1 pL, 0.64 mmol), TBTU (103mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-HPLC, and then triturated with Et20 to give the title compound: white solid (60 mg, 38%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.57 (s, 1 H), 8.53 (s, 1 H), 8.23 (d, 7=7.28 Hz, 1 H), 7.97 (d, 7=7.28 Hz, 1 H), 7.90 (d, 7=8.28 Hz, 1 H), 7.71 (t, 7=7.03 Hz, 1 H), 7.63 (d, 7=9.03 Hz, 1 H), 7.40 (d, 7=6.02 Hz, 2 H), 7.33 (t, 7=6.53 Hz, 2 H), 7.23 (t, 7=7.00 Hz, 1 H), 4.80 (d, 7=8.78 Hz, 1 H), 2.10 (d, 7=1 1.04 Hz, 1 H), 1.85 - 1.96 (m, 1 H), 1.76 - 1.85 (m, 1 H), 1.67 (d, 7=7.53 Hz, 2 H), 1.07 - 1.41 (m, 6 H), 0.93 (dd, 7=23.59, 13.05 Hz, 1 H). MS ESI [M + H]+ 489.3, calcd for [C27H28N4O3S + H]+ 489.2

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.32 mmol), (S)-(+)-l- cyclohexylethylamine (41 mg, 0.32 mmol), DIPEA (1 1 1 pL, 0.64 mmol), TBTU (103 mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-HPLC, and then triturated with Et20 to give the title compound: white solid (57 mg, 42 %). Ή NMR (400 MHz, DMSO-db) δ ppm 13.62 (s, 1 H), 8.52 (s, 1 H), 8.47 (s, 1 H), 8.25 (d, 7=7.78 Hz, 1 H), 8.21 (d, 7=8.28 Hz, 1 H), 7.92 (d, 7=8.78 Hz, 1 H), 7.87 (d, 7=7.53 Hz, 1 H), 7.77 (t, 7=7.50 Hz, 1 H), 7.65 (d, 7=8.28 Hz, 1 H), 7.49 (s, 2 H), 3.83 - 3.92 (m, 1 H), 1.64 - 1.81 (m, 4 H), 1.60 (d, 7=8.28 Hz, 1 H), 1.38 - 1.49 (m, 1 H), 1.04 - 1.25 (m, 6 H), 0.89 - 1.03 (m, 2 H). MS ESI [M + H]+ 427.4, calcd for [C22H26N4O3S + H]+ 427.2
-225-4820V.1
Example A 198. 2-(4-aminopiperidin- 1 -yl)-2-phenyl-N-(3-(3-sulfamoylphenyl)- 1 H-indazol-5- vDacetamide bis(2.2,2-trifluoroacetate )

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing (4-N-Boc-amino-piperidin- 1 -yl)-phenyl acetic acid (84 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. Saturated aqueous sodium bicarbonate (15 mL) was added. The precipitate was filtered out, dried and redissolved in DCM (6 mL). Reaction mixture was cooled to 0 °C and TFA (2 mL) was added. The mixture was stirred for 20 minutes at 0 °C and then 40 minutes at 25 °C. The solvent was removed under reduced pressure and the residue was purified using prep-HPLC and triturated with Et20 to give the title compound: pink solid (112 mg, 61 %).Ή NMR (400 MHz, METHANOL-^) δ ppm 8.47 (s, 1 H), 8.45 (s, 1 H), 8.14 (d, 7=7.53 Hz, 1 H), 7.94 (d, 7=6.78 Hz, 1 H), 7.63 - 7.72 (m, 3 H), 7.46 - 7.58 (m, 5 H), 4.81 - 4.85 (m, 1 H), 3.70 (br. s, 1 H), 3.35 - 3.44 (m, 1 H), 3.00 - 3.14 (m, 2 H), 2.88 (br. s, 1 H), 2.23 (d, 7=1 1.29 Hz, 1 H), 2.13 (d, 7=13.80 Hz, 1 H), 1.85 - 2.08 (m, 2 H). MS ESI [M + H]+ 505.2, calcd for [C26H28N603S + H]+ 505.2

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.32 mmol),
-226-4820V.1
dicyclohexylmethanamine (62 mg, 0.32 mmol), DIPEA (1 11 pL, 0.64 mmol), TBTU (103 mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-HPLC, and then triturated with Et20 to give the title compound: white solid (34 mg, 22 %). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.62 (s, 1 H), 8.53 (s, 1 H), 8.48 (s, 1 H), 8.26 (d, 7=7.78 Hz, 1 H), 7.94 (d, 7=9.03 Hz, 1 H), 7.88 (d, 7=7.78 Hz, 2 H), 7.78 (t, 7=8.00 Hz, 1 H), 7.66 (d, 7=8.53 Hz, 1 H), 7.48 (s, 2 H), 3.73 - 3.83 (m, 1 H), 1.53 - 1.76 (m, 12 H), 0.92 - 1.29 (m, 10 H). MS ESI [M + H]+ 495.4, calcd for [C27H34N4O3S + H]+ 495.2

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, 2-(2-( 1 -(tert-butoxycarbonyl)piperidin-4- yl)phenyl)acetic acid (80 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. Saturated aqueous sodium bicarbonate (15 mL) was added. The precipitate was filtered out, dried and purified by flash chromatography ( 100% DCM to 20% MeOH in DCM). Fractions were pooled and concentrated under reduced pressure to give a yellow residue and then redissolved in DCM (6 mL). Reaction was cooled to 0 °C and TFA (2 mL) was added. The mixture was stirred for 20 minutes at 0 °C and then 40 minutes at 25 °C. The solvent was removed under reduced pressure and the residue was purified using prep-HPLC and triturated with Et20 to give the title compound: white solid (15 mg, 10%). Ή NMR (400 MHz,
METHANOL-^) δ ppm 8.45 (br. s., 2 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.92 (d, 7=8.28 Hz, 1 H), 7.67 (t, 7=7.78 Hz, 1 H), 7.57 (d, 7=8.53 Hz, 1 H), 7.47 (d, 7=8.03 Hz, 1 H), 7.28 - 7.38 (m, 3 H), 7.23 (t, 7=6.50 Hz, 1 H), 3.90 (s, 2 H), 3.51 (d, 7=12.05 Hz, 3 H), 3.15 (t, 7=12.67 Hz, 2 H), 2.03
-227-4820V.1
- 2.14 (m, 2 H), 1.88 - 2.02 (m, 2 H). MS ESI [M + H]+ 490.3 calcd for [C26H27N5O3S + H]+
490.2

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, 2-(2-( 1 -(tert-butoxycarbonyl)piperidin-3- yl)phenyl)acetic acid (80 mg, 0.25 mmol), 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide · TFA (100 mg, 0.25 mmol), DIPEA (130 μί, 0.75 mmol), TBTU (80 mg, 0.25 mmol) and 4 mL of DMF. Saturated aqueous sodium bicarbonate (15 mL) was added. The precipitate was filtered out, dried and purified by flash chromatography (100% DCM to 20% MeOH in DCM). Fractions were pooled and concentrated under reduced pressure to give a white residue and then redissolved in DCM (6 mL). Reaction was cooled to 0 °C and TFA (2 mL) was added. The mixture was stirred for 20 minutes at 0 °C and then 40 minutes at 25 °C. The solvent was removed under reduced pressure and the residue was purified using prep-HPLC and triturated with Et20 to give the title compound: white solid (49 mg, 33%).Ή NMR (400 MHz,
METHANOLS) δ ppm 8.52 (s, 1 H), 8.48 (s, 1 H), 8.15 (d, 7=7.53 Hz, 1 H), 7.92 (d, 7=8.28 Hz, 1 H), 7.67 (t, 7=7.78 Hz, 1 H), 7.57 (d, 7=8.78 Hz, 1 H), 7.46 (d, 7=8.78 Hz, 1 H), 7.30 - 7.43 (m, 3 H), 7.23 - 7.29 (m, 1 H), 3.81 - 3.99 (m, 2 H), 3.58 (d, 7=1 1.80 Hz, 1 H), 3.39 - 3.52 (m, 3 H), 3.02 - 3.18 (m, 2 H), 1.97 - 2.10 (m, 2 H), 1.79 - 1.94 (m, 2 H). MS ESI [M + H]+ 490.3, calcd for [C26H27N5O3S + H]+ 490.2
-228- 4820V.1
Example A202. N-(cvclopropyl(thiophen-3-yl)methyl)-3-(3-sulfamoylphenyl)- 1 H-indazole-5- carboxamide

The title compound was synthesized according to the method described for 3-(3- sulf amoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.32 mmol),
cyclopropyl(thiophen-3-yl)methanamine (48 mg, 0.32 mmol), DIPEA (1 1 1 μΐ.., 0.64 mmol), TBTU (103 mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep- HPLC, and then triturated with Et20 to give the title compound: white solid (70 mg, 48%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.64 (s, 1 H), 8.54 (s, 1 H), 8.26 (d, 7=7.78 Hz, 1 H), 7.94 - 8.00 (m, 2 H), 7.73 (t, 7=7.78 Hz, 1 H), 7.66 (d, 7=9.03 Hz, 1 H), 7.34 - 7.39 (m, 2 H), 7.20 (m, 7=3.26 Hz, 1 H), 4.59 - 4.66 (m, 1 H), 1.38 - 1.49 (m, 1 H), 0.68 - 0.76 (m, 1 H), 0.60 - 0.68 (m, 1 H), 0.44 - 0.54 (m, 2 H). MS ESI [M + H]+ 453.2, calcd for [C22H20N4O3S2 + H]+ 453.1
Example A203. N-(cvclobutyl(thiophen-3-yl)methyl)-3-(3-sulfamoylphenyl)-lH-indazole-5- carboxamide

The title compound was synthesized according to the method described for 3-(3- sulfamoylphenyl)-N-((tetrahydro-2H-pyran-4-yl)methyl)- 1 H-indazole-5-carboxamide utilizing 3-(3-sulfamoylphenyl)-lH-indazole-5-carboxylic acid (100 mg, 0.32 mmol),
cyclobutyl(thiophen-3-yl)methanamine (53 mg, 0.32 mmol), DIPEA (1 1 1 μΐ., 0.64 mmol), TBTU (103 mg, 0.32 mmol) and DMF (4 mL). The reaction mixture was purified by prep-
-229-4820V.1
HPLC, and then triturated with Et20 to give the title compound: white solid (29 mg, 19%). Ή NMR (400 MHz, METHANOL-^) δ ppm 8.74 (d, 7=8.28 Hz, 1 Η), 8.58 (s, 1 H), 8.53 (s, 1 Η), 8.23 (d, 7=7.53 Hz, 1 H), 7.87 - 8.00 (m, 2 H), 7.71 (t, 7=7.65 Hz, 1 H), 7.63 (d, 7=8.28 Hz, 1 H), 7.31 - 7.38 (m, 1 H), 7.28 (br. s., 1 H), 7.13 (d, 7=4.77 Hz, 1 H), 5.26 (t, 7=9.03 Hz, 1 H), 2.87 - 2.99 (m, 1 H), 2.12 - 2.24 (m, 1 H), 1.80 - 2.06 (m, 5 H). MS ESI [M + H]+ 467.1, calcd for [C23H22N403S2 + H]+ 466.11
Example A204. (S)-2-(dimethylamino)-2-phenyl - V-(3-(3-sulfonylphenylVl /-indazol-5-yl) acetamide trifluoroacetate
The title compound was synthesized according to the General Method A, utilizing (S)-l- (dimethylamino)-2-phenylaceticacid hydrochloride (30 mg, 0.139 mmol), 3-(5-amino-lH- indazol-3-yl) benzenesulfonamide hydrochloride (45 mg, 0.139 mmol), N-ethyl-N-isopropyl propan-2-amine (0.12 mL, 0.695 mmol) and TBTU (45 mg, 0.139 mmol) in DMF (1.2 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0-10% MeOH in DCM) and repurified by Reverse phase Biotage Cis, 50 g column chromatography (gradient 90: 10-20:80 % 0.1% TFA- H20: MeOH) to give the title compound as a white solid (15 mg, 19 %, 86.2% ee by chiral HPLC Rt = 9.8 min, Chiralpak AS-H (25 x 0.46 cm), 3.0 mL/min, isocratic 30% isobutanol(0.1 % DEA)/C02, 100 bar). Ή NMR (400 MHz, CD3OD) δ 8.48 (s, 1H), 8.41 (s, 1H), 8.16 (d, 7 =8.0 Hz, 1H), 7.94 (d, 7 =7.6 Hz, 1H), 7.28-7.68 (m, 3H), 7.58- 7.52 (m, 5H), 5.01 (s, 1H), 3.13 (br.s, 3H), 2.70-2.64 (br.m, 3H); MS ESI 450.3. [M + H]+, calcd for [C23H23N503S + H]+ 450.1.
-230-4820V.1
Example A205. (RV2-(pyrrolidin- 1 -yl)-N-(3-(3-sulf amoylphen ylV 1 H-indazol-5-ylV2-(thiophen- 3-vD acetamide trifluoroacetate

A. (R,S)-]-phenylethyl-2-(pyrrolidin-l-yl)-2-(thiophen-3-yl) acetate
To a mixture of 2-(pyrrolidin-l-yl)-2-(thiophen-3-yl)acetic acid (2 g, 9.46 mmol), (S)-
(-)-l-phenylethanol ( 1.45 g, 1 1.86 mmol) and DMAP (116 mg, 0.946 mmol) in dry DCM (24 mL) was added solid EDCI hydrochloride (2.26 g, 11.82 mmol) at 0°C all at once. The resulting solution was stirred at room temperature for 24 hrs and then diluted it with DCM (48 mL), washed with water (24 mL) followed by satd. NaHC03 (24 mL), dried over anh Na2S04, filtered and concentrated to give yellowish oil, the crude product was purified using hexane:
ethyalacetate (0-15%) gradient on Biotage Isolera with SNAP 100 g column to give the title compound as a cream solid approximately 2: 1 diastereomeric ratio bet, (R,S) and (S,S) by NMR (1.98 g, 66 %).Ή NMR (400 MHz, CDCl3) δ 7.13-7.26 (m, 7H), 7.21 (d, J =4.8 Hz, IH), 5.95-5.90 (m, IH), 4.15 (s, IH), 2.52-2.48 (m, 4H), 1.77 (br.s, 4H), 1.45 (d, J =6.4 Hz, 3H); MS ESI 316. [M + H]+, calcd for [C,8H2iN02S + H]+ 316.1.
Recrystalization of above distereomeric pair (1.98 g) by hexane (80 mL) at 55°C.cooled to 5°C for 1 h and solid was filtered .washed with little hexane to give a white solid (0.57 g, crop-1) as (R,S) diasteromer. Purity by NMR :>98%. Filtrate and washes were combined and concentrated under reduced pressure to give 1.3 g viscous oil (approximate 2: 1 diastereomeric ratio bet, (R, S) and (S, S) by NMR). The residue was purified hexane (50 mL) as earlier to give an additional (0.245 g, crop-2) as a (R, S) diasteromer. Total yield : 0.57+0.245 = 0.815, 27 % , Purity by NMR :>98%.Ή NMR (400 MHz, CDCl3) δ 7.13-7.26 (m, 7H), 7.21 (d, J =4.8 Hz, IH), 5.95-5.90 (m, IH), 4.15 (s, IH), 2.52-2.48 (m, 4H), 1.77 (br.s, 4H), 1.45 (d, J =6.4 Hz, 3H); MS ESI 316. [M + H]+, calcd for [Ci8H2,N02S + H]+ 316.1.
Filtrate and washes were combined and concentrated under reduced pressure to give 1.06 g viscous oil. The residue was purified using hexane: ethyalacetate (0-20%) gradient on Biotage
-231-4820v.1
Isolera with SNAP 100 g column 2" eluted peak to give the title compound as a thick colorless oil as a (S,S) diasteromer (0.464 g, 15.5 %). Purity by NMR :>98%.Ή NMR (400 MHz, CDC ) δ 7.13-7.26 (m, 7H), 7.21 (d, =4.8 Hz, 1H), 5.95-5.90 (m, 1H), 4.18 (s, 1H), 2.52-2.48 (m, 4H), 1.77 (br.s, 4H), 1.45 (d, J =6.4 Hz, 3H); MS ESI 316. [M + H]+, calcd for [C18H2iN02S + H]+ 316.1.
B. (R)-2-(pyrrolidin-l-yl)-2-(thiophen-3-yl) acetic acid trifluoroacetate
To a solution of (R,S)-l-phenylethyl-2-(pyrrolidin-l-yl)-2-(thiophen-3-yl) acetate (200 mg, 0.634 mmol) in dry DCM (4 mL) was added trifluoroaceticacid (1.2 mL 15.5 mmol) and the mixture stirred at room temperature for 2 h. The volatile were subsequently removed under reduced pressure and residual oil was purified by Reverse phase Biotage Ci8, 25 g column chromatography (gradient 90:10-20:80 % 0.1 % TFA- H20: MeCN) to give the title compound (as a TFA salt) after trituration with Et20 as a white solid (0.140 mg, 68 %). Optical Rotation:
[OC]22D = -96.8° (c 0.65, MeOH).
C. (R)-2-(pyrrolidin-l-yl)-N-(3-(3-sulfamoylphenyl)-lH ndazol-5-yl)-2-(thiophen-3-yl) acetamide trifluoroacetate
The title compound was synthesized according to the General Method A, utilizing (R)- 2-(pyrrolidin-l-yl)-2-(thiophen-3-yl) acetic acid trifluoroacetate (60 mg, 0.184 mmol), 3-(5- amino- lH-indazol-3-yl)benzenesulfonamide hydrochloride (60 mg, 0.0.184 mmol), N-ethyl-N- isopropyl propan-2-amine (0.16 mL, 0. 92 mmol) and TBTU (59 mg, 0.184 mmol) in DMF (1.8 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0-10% MeOH in DCM) and repurified by Reverse phase Biotage C]8, 50 g column chromatography (gradient 90: 10-20:80 % 0.1% TFA- H20: MeOH) to give the title compound as a off white solid (40 mg, 36 %).Ή NMR (400 MHz, CD3OD) δ 8.49 (s, 1H), 8.42 (s, 1H), 8.13 (d, J =7.6 Hz, 1H), 7.94 (d, J =7.6 Hz, 1H), 7.88 (s, 1H), 7.68 (t, J =7.8 Hz, 1H), 7.65-7.61 (m, 1H), 7.55 (pseudo t, 2H), 7.38 (d, / =5.2 Hz, 1H), 5.31 (s, 1H), 3.88 (br.s, 1 H), 3.40-3.05 (m, 3H), 2.30- 1.95 (m, 4H); MS ESI 482.2. [M + H]+, calcd for
 + H]+ 482.1. Optical Rotation: [a]22 D = -108.7° (c 0.285, MeOH).
+ H]+ 482.1. Optical Rotation: [a]22 D = -108.7° (c 0.285, MeOH).
-232-4820V.1
Example A206. ( SV2-(pyrrolidin- 1 -yl)-N-(3-(3-sulfamoylphenyl)- lH-indazol-5- yl)-2-( thiophen- 3-vD acetamide trifluoroacetate

A. (S)-2-(pyrrolidin-l-yl)-2-(thiophen-3-yl) acetic acid trifluoroacetate
To a solution of (S,S)-l-phenylethyl-2-(pyrrolidin-l-yl)-2-(thiophen-3-yl) acetate (460 mg, 0.634 mmol) in dry DCM (4 mL) was added trifluoroaceticacid (2.5 mL 32.45 mmol) and the mixture stirred at room temperature for 2 h. The volatile were subsequently removed under reduced pressure and residual oil was purified by Reverse phase Biotage Ci8,25 g column chromatography (gradient 90: 10-20:80 % 0.1 % TFA- H20: MeCN) to give the title compound (as a TFA salt) after trituration with EtOAc as a white solid (0.185 mg, 39 %). Optical Rotation:
[OC]22D = 93.33° (c 0.6, MeOH).
B. (S)-2-(pyrrolidin-l-yl)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl)-2-(thiophen-3-yl) acetamide trifluoroacetate
The title compound was synthesized according to the General Method A, utilizing (S)- 2-(pyrrolidin-l-yl)-2-(thiophen-3-yl) acetic acid trifluoroacetate (50 mg, 0.153 mmol), 3-(5- amino-lH-indazol-3-yl)benzenesulfonamide hydrochloride (50 mg, 0.153 mmol), N-ethyl-N- isopropyl propan-2-amine (0.13 mL, 0. 92 mmol) and TBTU (49 mg, 0.153 mmol) in DMF (1.5 mL). The reaction mixture was purified by reverse phase column chromatography (Biotage Ci8, 50 g gradient 90: 10-20:80 % 0.1% TFA- H20: MeOH) to give the title compound as a off white solid (50 mg, 54%).Ή NMR (400 MHz, CD3OD) δ 8.49 (s, 1H), 8.42 (s, 1H), 8.13 (d, / =7.6 Hz, 1H), 7.94 (d, J =7.6 Hz, 1H), 7.88 (s, 1H), 7.68 (t, J =7.8 Hz, 1H), 7.65-7.61 (m, 1H), 7.55 (pseudo t, 2H), 7.38 (d, J =5.2 Hz, 1H), 5.31 (s, 1H), 3.88 (br.s, 1 H), 3.40-3.05 (m, 3H), 2.30- 1.95 (m, 4H); MS ESI 482.2. [M + H]+, calcd for [C23H23N503S2 + H]+ 482.1. Optical Rotation: [ct]22D = 104° (c 0.375, MeOH).
-233-4820V.1

The title compound was synthesized according to the General Method A, utilizing 2-(4- methylthiophen-3-yl)-2-(pyrrolidin-l-yl) acetic acid trifluoroacetate (62 mg, 0.184 mmol), 3-(5- amino- lH-indazol-3-yl)benzenesulfonamide hydrochloride (60 mg, 0.184 mmol), N-ethyl-N- isopropyl propan-2-amine (0.16 mL, 0.923 mmol) and TBTU (59 mg, 0.184 mmol) in DMF (2 mL). The reaction mixture was purified by preparative HPLC to give title compound as a white solid (37 mg, 33 ). Ή NMR (400 MHz, CD3OD) δ 8.49 (s, 1 H), 8.43 (s, 1H), 8.15 (d, J =7.6 Hz, 1H), 7.97 (d, J =7.6 Hz, 1H ), 7.71 (t, J =8 Hz, 1H ), 7.61-7.55 (m, 3H), 7.32 (d, J =6 Hz, 1H), 5.35 (s, 1H), 3.97-3.81 (br.m, 2H), 3.14-3.28 (br.m, 2H), 2.10-2.04 (br.s, 4H); MS ESI 516.4. [M + H]+, calcd for [C23H22CIN5O3S + H]+ 516.1. Example A208. 2-(pyridin-3-yl)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl) acetamide trifluoroacetate

The title compound was synthesized according to the General Method A, utilizing 3- Pyridineacetic acid hydrochloride (34 mg, 0.194 mmol), 3-(5-amino- lH-indazol-3- yl)benzenesulfonamide hydrochloride (60 mg, 0.184 mmol), Ν-ethyl-N-isopropyl propan-2- amine (0.16 mL, 0.923 mmol) and TBTU (62 mg, 0.194 mmol) in DMF (2 mL). The reaction mixture was purified by preparative HPLC to give title compound as a white solid (26 mg, 27 %). Ή NMR (400 MHz, CD3OD) δ 8.86 (s, 1H), 8.75 (d, J =4.4 Hz, 1 H), 8.51-8.47 (m, 2H),
-234-4820V.1
8.42 (s, IH), 8.16 (d, / =6.8 Hz, IH ), 7.98-7.93 (m, 2H), 7.69 (t, J =8.0 Hz, IH), 7.60 (m, 2H), 4.05 (s, 2H); MS ESI 408.1. [M + H]+, calcd for [C20H17N5O3S + H]+ 408.1.

The title compound was synthesized according to the General Method A, utilizing 2-(2- chlorothiophen-3-yl)-2-(pyrrolidin-l-yl)acetic acid (50 mg, 0.153 mmol), 3-(5-amino-lH- indazol-3-yl)benzenesulfonamide hydrochloride (38 mg, 0.153 mmol), N-ethyl-N- o isopropylpropan-2-amine (0.13 mL, 0.77 mmol) and HATU (58 mg, 0.1 3 mmol) in DMF (2 mL). The reaction mixture was purified by preparative HPLC to give title compound as a light pink solid (24 mg, 63 %). Ή NMR (400 MHz, CD3OD) δ 8.49 (s, IH), 8.43 (s, IH), 8.17 (d, J =8.0 Hz, IH), 7.97 (d, J =7.6 Hz, IH), 7.87 (s, IH), 7.71 (t, J =8.4 Hz, IH ), 7.61-7.55 (m, 2H), 7.30 (s, IH), 5.18 (s, IH), 3.90 (br.s, IH), 3.35 (s, 3H), 3.12-3.1 1 (m, 2H), 2.48 (m,lH), 2.27- 5 2.13 (br.m, 3H), 2.05-2.0 l(br.m, I H); MS ESI 496.3.[M+H]+, calcd for [C24H25N5O3S2 + H]+ 496.1.

The title compound was synthesized according to the General Method A, utilizing 2- methoxy)-2-(thiophen-2-yl)acetic acid (40 mg, 0.232 mmol), 3-(5-amino- lH-indazol-3- yl)benzenesulfonamide hydrochloride (75 mg, 0.232 mmol), Ν-ethyl-N-isopropyl propan-2-
-235-1 11444820V.1
amine (0.2 mL, 1.16 mmol) and TBTU (75 mg, 0.232 mmol) in DMF ( 1.2 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0-10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Ci8, 60 g gradient 90: 10-20:80 % 0.1% TFA- H20: MeOH) to give the title compound as a light pink solid (40 mg, 39%). Ή NMR (400 MHz, CD3OD) δ 8.47 (s, 1H), 8.38 (s, 1H), 8.17 (d, J =7.6 Hz, 1H), 7.94 (d, J =7.2 Hz, 1H), 7.69 (t, J =8.0 Hz, 1H), 7.62 (d, J =9.6 Hz, 1H), 7.56 (d, / =8.8 Hz, 1H), 7.45 (d, / =4.0 Hz, 1H), 7.25 (d, / =2.8 Hz, 1H), 7.05 (br.s, lH), 5.14 (s, 1H), 3.60 (s, 3H), MS ESI 443.2. [M + H]+, calcd for [C2oH,gN404S2 + H]+ 443.1.
Example A2 U . 2-(piperidin- l-yl)-N-(3-(3-sulfamoylphenyl)-lH-indazol-5-yl) - 2-(thiophen-3- yl) acetamide trifluoroacetate

The title compound was synthesized according to the General Method A, utilizing 2- (Piperidin- l-yl)-2-(thiophen-3-yl) acetic acid (40 mg, 0.177 mmol), 3-(5-amino-lH-indazol-3- yl)benzenesulfonamide hydrochloride (57 mg, 0.177 mmol), Ν-ethyl-N-isopropyl propan-2- amine (0.15 mL, 0.886 mmol) and TBTU (57 mg, 0.177 mmol) in DMF (1.2 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0- 10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Ci8, 60 g, gradient 90: 10-20:80 % 0.1 % TFA- H20: MeOH) to give the title compound as a light pink solid (20 mg, 17.5 %).Ή NMR (400 MHz, CD3OD) δ 8.48 (s, 1H), 8.44 (s, 1H), 8.16 (d, J =7.6 Hz, 1H), 7.96 (d, J =7.6 Hz, 1H), 7.87 (s, 1H), 7.72-7.65 (m, 2H), 7.59-7.52 (m, 2H), 7.38 (d, J =4.8 Hz, 1H), 5.20 (s, 1 H), 3.78 (br.s, lH), 3.26-3.07 (br.m, 2H), 2.9-2.91 (br.m, 1 H), 2.02-1.807 (br.m, 5H), 1.56-1.54 (br.s, 1 H), MS ESI 496.4. [M + H]+, calcd for [C24H25N503S2 + H]+ 496.14.
-236-4820V.1
Example A212. 2-(cvclopentyloxy) - 2-phenyl -N-(3-(3-sulfamoylphenyl)- 1 //-indazol-5-yl) acetamide
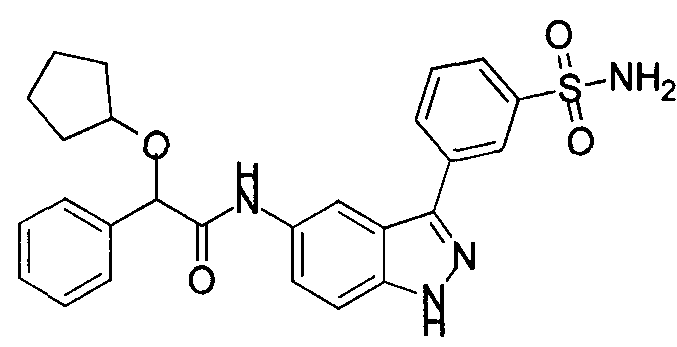
The title compound was synthesized according to the General Method A, utilizing 2-(Cyclopentyloxy)-2-phenylacetic (40 mg, 0.181 mmol), 3-(5-amino- lH-indazol-3-yl)benzene sulfonamide hydrochloride (59 mg, 0.181 mmol), N-ethyl-N-isopropyl propan-2-amine (0.15 mL, 0.908 mmol) and TBTU (58 mg, 0.181 mmol) in DMF (1.2 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0- 10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Cig, 60 g, gradient 90: 10-20:80 % 0.1 % TFA- H20: MeOH) to give the title compound as a light pink solid (24 mg, 27 %).Ή NMR (400 MHz, CDjOD) δ 8.47 (s, 1H), 8.37 (d, J =3.2 Hz, 1H), 8. 14 (d, / =7.6 Hz, 1H), 7.92 (d, J =7.6 Hz, 1H), 7.68 (t, J =8.0 Hz, 1H), 7.57-7.54 (m, 4H), 7.41-7.32 (m, 3H), 5.00 (s, 1H), 4.12 (s, 1H), 1.92- 1.78 (br.m, 6H), 1.65- 1.58 (br.m, 2H), MS ESI 490.4. [M + H]+, calcd for
[C26H26N404S + H]+ 491.1.
Example A213. (5)-2-phenyl-2-(piperidin- 1 -yl)-N-(3-(3-sulf amoylphenvD- 1 H-indazol-5- vDacetamide trifluoroacetate

To a mixture of L-(-)-phenylglycine ( 1.51 g, 10 mmol) and 1,5-dirbromopentane (2.53 g, 1 1 mmol) in EtOH (120 mL) was added NaHC03 (2.52 g, 30 mmol). The resulting mixture was refluxed overnight (oil temp. 90 °c). After filtering off insoluble materials, the filtrate was purified by Biotage column system (gradient: MeOH/DCM 0 to 20%) to give crude (S)-2- phenyl-2-(piperidin- l-yl)acetic acid as a gel-like semi solid (3.90 g).
-237-4820V.1
To a mixture of (S)-2-phenyl-2-(piperidin-l-yl)acetic acid (390 mg, considered as 1 mmol) and 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide trifluroacetate (402 mg, 1 mmol), TBTU (321 mg, 1 mmol) in DMF (6 mL) at 0 °c was added 'Pr2NEt (0.70 mL, 4 mmol). After addition, the resulting mixture was stirred for 2 h at 0 c. After filtering through HG microfilter, the residue was purified by prep-HPLC, porapak, then prep-HPLC twice to give the title compound as a white solid (20 mg, 3%). Ή NMR (400 MHz, CD3OD) δ 8.49 (s, IH), 8.44 (s, IH), 8.15 (d, J = 7.6 Hz, IH), 7.96 (d, J = 7.6 Hz, IH), 7.75-7.68 (m, 3H), 7.60-7.50 (m, 5H), 5.05 (s, IH), 3.84 (d, J = 10.0 Hz, IH), 3.21 (t, J = 10.8 Hz, IH), 3.02-2.87 (m, 2H), 2.07-1.75 (m, 5H), 1.65-1.50 (m, IH); MS ESI. [M + H]+ 490.3, calcd for [C26H27N5O3S + H]+ 490.2.

A 100 mL steel bomb was charged with isoquinoline- 1 -carboxylic acid (4.00 g), HOAc (30 mL), Pt02 (200 mg, 5 wt.%) and stirred bar. The resulting mixture was hydrogenated under 100 psi H2. Pressure went down to 0 after 2 h. Refilled with ¾ 4 times every 45 min then stirred overnight. LC-MS showed full conversion. It was diluted with MeOH (40 mL), filtered through celite and rinsed with MeOH, HOAc. Filtrate was concentrated to dryness go give a light grey solid. Trituration with MeOH gave 1,2,3,4-tetrahydroisoquinoline-l-carboxylic acid as a light grey solid (2.54 g). Mother liquor was concentrated to dryness and triturated with Μ6θΗ/¾0 to give additional 0.04 g as a light grey solid. Total 2.58 g. Ή NMR (400 MHz, DMSO-d6) δ 8.70 (s, br, IH), 7.68 (s, IH), 7.17-7.06 (m, 3H), 4.42 (s, IH), 3.30-3.10 (m, 2H, partially overlapped with MeOH residue), 3.00-3.28 (m, 2H).
To a suspension of 1,2,3,4-tetrahydroisoquinoline-l -carboxylic acid (354 mg, 2 mmol) in MeOH ( 15 mL) was added HOAc (2 mL), 6 M HC1 (0.34 mL, 2 mmol), followed by 10% Pd C (35 mg) and formalin (37% aq., 0.18 mL, 2.5 mmol). The resulting mixture was vacuumed,
-238-4820V.1
refilled with ¾; repeated 3 times and then stirred overnight under H2 balloon. After filtering through celite, the filtrate was concentrated to give 2-methyl-l,2,3,4-tetrahydroisoquinoline-l- carboxylic acid hydrochloride as a white solid (0.46 g). Ή NMR (400 MHz, DMSO-d6) δ 7.45- 7.15 (m, 4H), 5.32 (s, 1H), 4.85-4.70 (m, 2H), 3.15-3.00 (m, 2H), 2.87 (s, 3H).
To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide hydrochloride (81 mg, 0.25 mmol), 2-methyl-l,2,3,4-tetrahydroisoquinoline-l-carboxylic acid hydrochloride (1 14 mg, 0.5 mmol) and TBTU (81 mg, 0.25 mmol) in DMF (6 mL) at 0 °c was added 'Pr2NEt (0.18 mL, 1 mmol). After addition, the resulting mixture was stirred for 30 min at 0 c. After removal of 'Pr2NEt, the residue was filtered through microfilter and purified by preparative HPLC to give the title compound as a white solid (52 mg, 36%). Ή NMR (400 MHz, CD3OD) δ 8.48 (s, 2H), 8.13 (d, J = 7.6 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.69-7.60 (m, 3H), 7.48 (d, J = 7.2 Hz, 1H), 7.39-7.30 (m, 3H), 5.33 (s, 1H), 4.01 (s, br, 1H), 3.63-3.55 (m, 1H), 3.40-3.30 (m, 1H, partially overlapped with MeOH residue), 3.20-3.10 (4H; m, 1H and s, 3H at 3.13 partially overlapped); MS ESI. [M + H]+ 462.3, calcd for [C24H23N5O3S + H]+ 462.2.
Example A215. 2-methyl-N-(3-(3-sulfamoylphenyl)- 1 H-indazol-5-yl)isoindoline- 1 -carboxamide trifluoroacetate

To a solution of 2-(tert-butoxycarbonyl)isoindoline-l-carboxylic acid (526 mg, 2 mmol) in DCM (10 mL) was added TFA (2 mL). The resulting mixture was stirred for 2 h at rt.
Additional TFA (1 mL) was added and it was stirred for 2 h at rt. It was refluxed (oil temp 50 °C) to complete. Removal of solvents yielded a pale yellow liquid which was redissolved in MeOH (10 mL). Formalin (37% aq., 0.18 mL, 2.5 mmol) was added followed by 10% Pd/C (32 mg). The resulting mixture was vacuumed, refilled with H2; repeated twice. It was then hydrogenated under ¾ balloon overnight. After filtering through celite and rinsing with MeOH, the filtrate was concentrated to dryness and redissolved in MeOH (10 mL). 2 M NaOH (2 mL, 4
-239-4820V.1
mmol) was added and the resulting mixture was stirred overnight. Removal of solvents gave a white solid containing inorganic salts. Ή NMR (400 MHz, DMSO-d6) δ 7.31 (s, 1H), 7.13-7.03 (m, 3H), 4.20 (d = 12.8 Hz, 1 H), 3.91 (s, 1 H), 3.50 (d, J = 1 1.2 Hz, 1H), 2.47 (s, 3H). It was suspended in MeOH ( 10 mL) and treated with TFA (3 mL). Removal of solvents gave crude 2- methylisoindoline- 1 -carboxylic acid trifluoroacetate as a pink purple solution (2.60 g).
To a mixture of crude acid (0.65 g, assumed 0.5 mmol) obtained above and 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide hydrochloride ( 162 mg, 0.5 mmol) in DMF (6 mL) at 0 °C was added 'Pr2NEt (0.36 mL, 2 mmol). After addition, the resulting mixture was stirred for 30 min at 0 °C, then overnight at rt. LC-MS showed little formation of product. Additional 'Pr2NEt (0.36 mL, 2 mmol) was added and it was stirred for 1 h at rt. After removing volatile solvents, it was filtered through microfilter and purified directly by preparative HPLC twice to give the title compound as a pink solid (67 mg, 12%). Ή NMR (400 MHz, CD3OD) δ 8.47 (s, 2H), 8.1 1 (d, J = 7.2 Hz, 1 H), 7.92 (d, J = 7.6 Hz, 1H), 7.68-7.58 (m, 4H), 7.48 (s, 3H), 5.65 (s, 1 H), 5.10 (d, J = 14.4 Hz, 1H, partially overlapped with H20 residue), 3.23 (s, 3H); MS ESI. [M + H]+ 448.2, calcd for [C23H2iN503S + H]+ 448.1.

To a mixture of (S)-2-methylpyrrolidine (1.07 g, 12.6 mmol), glyoxylic acid hydrate (1.16 g, 12.6 mmol) in DCM (50 mL) was added thiophen-3-ylboronic acid (1.59 g, 12.6 mmol). The resulting mixture was stirred for 3 h at rt. Oil formed on the sides of flask and MeOH (5 mL) was added to make a clear solution. Reaction was stirred overnight ar rt. After removing all the solvents, the residue was purified by Biotage column system (gradient: MeOH/DCM 0 to 30%) to give (5)-2-((S)-2-methylpyrrolidin- l-yl)-2-(thiophen-3-yl)acetic acid as a beige solid ( 1.294 g, 46%). Ή NMR (400 MHz, CD3OD) δ 7.68 (dd, J = 2.8, 1.2 Hz, 1 H), 7.52 (dd, J = 5.0, 3.0 Hz,
-240-4820V.1
1H), 7.29 (dd, J = 5.0, 0.8 Hz, 1 H), 4.71 (s, 1 H), 3.71-3.60 (m, 1H), 3.25-3.16 (m, 1H), 3.06-2.88 (s, br, 1H), 2.37-2.25 (m, 1H), 2.07-1.89 (m, 2H), 1.84-1.74 (m, 1H), 1.50 (pseudo s, 3H).
To a mixture of 3-(5-arnino-lH-indazol-3-yl)benzenesulfonamide hydrochloride (163 mg, 0.5 mmol), (5)-2-((5)-2-methylpyrrolidin-l-yl)-2-(thiophen-3-yl)acetic acid (1 13 mg, 0.5 mmol) and TBTU (161 mg, 0.5 mmol) in DMF (5 mL) at 0 °C was added 'Pr2NEt (0.27 mL, 1.5 mmol). After addition, the resulting mixture was stirred for 30 min at 0 °C. The reaction mixture was purified directly by preparative HPLC twice to give the title compound as a white solid (65 mg, 21 %, 99.9% e.e., Rt = 5.4 min, by chiral HPLC using Chiralpak IC (15 x 0.46 cm), 3.0 mL/min, isocratic 35% methanol/(0.1 % DEA)/C02, 100 bar). *H NMR (400 MHz, CD3OD) δ 8.49 (s, 1H), 8.42 (s, 1H), 8.13 (d, J = 7.2 Hz, 1H), 7.95 (d, J = 7.6 Hz, 1H), 7.89 (s, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.65-7.60 (m, 1H), 7.59-7.52 (m, 2H), 7.40 (d, J = 4.0 Hz, 1H), 5.33 (s, 1H), 3.88-3.78 (m, 1H), 3.30-3.22 (m, 1H), 3.18-3.08 (m, 1H), 2.40 (sextet, J = 6.4, 1H), 2.04 (quintet, J = 6.8 Hz, 2H), 1.84 (sextet, J = 6.4 Hz, 1H), 1.53 (d, J = 6.0 Hz, 3H); MS ESI. [M + H]+ 496.3, calcd for [C^^NsC^ + H]+ 496.1.
Example A217. (R)-2-(dimethylamino)-2-phenyl-N-(3-(3-sulfamoylphenyl)-lH-indazol-5- vDacetamide trifluoroacetate

To a suspension of (D)-phenylglycine (4.53 g, 30 mmol) in MeOH (60 mL) was added 6 M HCl (5.5 mL, 33 mmol) dropwise. The resulting colorless clear solution was treated with 10% Pd/C (272 mg, 6 wt.%). It was vacuumed, refilled with H2; repeated twice. The resulting mixture was stirred 24 h under H2 balloon. After filtering through celite and rinsing with MeOH, the filtrate was concentrated to dryness to give a white solid. Recrystallization from 'PrOH/MeOH (60 mL/5 mL) in a freezer gave (R)-2-(dimethylamino)-2-phenylacetic acid hycrochloride as white pellets (2.01 g, 33%). Ή NMR (400 MHz, DMSO-d6) δ 14.10 (s, br, 1H), 1 1.00 (s, br, 1H), 7.58-7.45 (m, 5H), 5.25 (s, 1H), 2.69 (s, 6H).
-241-4820V.1
To a mixture of 3-(5-amino- lH-indazol-3-yl)benzenesulfonamide hydrochloride (163 mg, 0.5 mmol), (R)-2-(dimethylamino)-2-phenylacetic acid hydrochloride (108 mg, 0.5 mmol) and TBTU (161 mg, 0.5 mmol) in DMF (6 mL) at 0 °C was added 'Pr2NEt (0.35 mL, 2. mmol). After addition, the resulting mixture was stirred for 1 h at 0 °C. The reaction mixture was purified directly by preparative HPLC followed by trituration with Et20 to give the title compound as a light beige solid ( 173 mg, 61 %, 94.7% e.e., Rt = 8.3 min, by chiral HPLC using Chiralpak AS-H (25 x 0.46 cm), 3.0 mL/min, isocratic 30% isobutanol(0.1 % DEA)/C02, 100 bar). Ή NMR (400 MHz, CD3OD) δ 8.49 (t, J = 1.8 Hz, IH), 8.42 (s, IH), 8.16 (d, J = 7.6 Hz, IH), 7.96 (d, J = 8.0 Hz, IH), 7.74-7.67 (m, 3H), 7.61-7.56 (m, 4H), 7.54 (dd, J = 9.0, 1.8 Hz, 1 H), 5.03 (s, 1 H), 3.20-2.60 (m, br, 6H); MS ESI. [M + H]+ 450.2, calcd for [C23H23N5O3S + H]+ 450.2.

To a mixture of (R)-2-methylpyrrolidine (1.02 g, 12 mmol), glyoxylic acid hydrate (1.10 g, 12 mmol) in DCM (50 mL) was added thiophen-3-ylboronic acid (1.51 g, 12 mmol). The resulting mixture was stirred for 3 h at rt. Lots of oil formed on the sides of flask and MeOH (5 mL) was added to make a clear solution. Reaction was stirred overnight ar rt. After removing all the solvents, the residue was purified by column chromatography (Si02, gradient: MeOH/DCM 0 to 30%) to give (R)-2-((R)-2-methylpyrrolidin-l-yl)-2-(thiophen-3-yl)acetic acid as a beige solid (1.18 g, 44%). Ή NMR (400 MHz, CD3OD) δ 7.68 (dd, J = 3.2, 1.2 Hz, IH), 7.52 (dd, J = 5.0, 3.0 Hz, IH), 7.29 (dd, J = 5.0, 0.8 Hz, IH), 4.71 (s, IH), 3.71-3.60 (m, IH), 3.25-3.16 (m, IH), 3.06-2.88 (s, br, I H), 2.37-2.25 (m, IH), 2.07- 1.89 (m, 2H), 1.84- 1.74 (m, IH), 1.50 (pseudo s, 3H).
-242-4820V.1
To a mixture of 3-(5-amino-lH-indazol-3-yl)benzenesulfonamide hydrochloride (163 mg, 0.5 mmol), (R)-2-((R)-2-methylpyrrolidin-l-yl)-2-(thiophen-3-yl)acetic acid (113 mg, 0.5 mmol) and TBTU (161 mg, 0.5 mmol) in DMF (5 mL) at 0 °C was added 'Pr2NEt (0.27 mL, 1.5 mmol). After addition, the resulting mixture was stirred for 30 min at 0 °C. The reaction mixture was purified directly by preparative HPLC followed by trituration with Et20 to give the title compound as a white solid (183.5 mg, 60%, 99.9% e.e., Rt = 6.7 min, by chiral HPLC using Chiralpak IC ( 15 x 0.46 cm), 3.0 mL/min, isocratic 35% methanol/(0.1 % DEA)/C02, 100 bar). Ή NMR (400 MHz, CD3OD) δ 8.50 (s, IH), 8.43 (s, IH), 8.16 (d, J = 8.0 Hz, IH), 7.96 (d, J = 8.0 Hz, IH), 7.89 (d, J = 1.6 Hz, I H), 7.72 (t, J = 7.6 Hz, IH), 7.66 (dd, J = 4.8, 2.8 Hz, IH), 7.60 (d, J = 8.8 Hz, IH), 7.55 (dd, J = 8.8, 1.6 Hz, IH), 7.40 (d, J = 4.8 Hz, IH), 5.27-5.22 (m, I H), 3.84 (sextet, J = 6.4 Hz, I H), 3.35-3.25 (m, IH, partially overlapped with MeOH residue), 3.20-3.12 (m, IH), 2.41 (sextet, J = 7.0 Hz, IH), 2.06 (quintet, J = 7.0 Hz, 2H), 1.87 (sextet, J = 6.6 Hz, IH), 1.56 (d, J = 6.4 Hz, 3H); MS ESI. [M + H]+496.3, calcd for [C24H25N5O3S2 + H]+ 496.1.
Example A219. N-(3-(3-methylsulfonyl) phenyl)-lH-indazol-5-yl)-2-(pyridin-3-yl) acetamide trifluoroacetate

The title compound was synthesized according to the General Method A, utilizing 3- pyridineacetic acid hydrochloride (28 mg, 0.156 mmol), 3-(3-(methylsulfonyl) phenyl)- 1 H- indazole-5-amine 2,2,2-trifluoroacetate (60 mg, 0.156 mmol), N-ethyl-N-isopropylpropan-2- amine (0.13 mL, 0.77 mmol) and TBTU (50 mg, 0.156 mmol) in DMF (2 mL). The reaction mixture was purified by preparative HPLC to give title compound as a light pink solid (45 mg,
57 %). Ή NMR (400 MHz, CD3OD) δ 8.93 (s, IH), 8.80 (d, J =5.2 Hz, IH), 8.65 (d, J =8 Hz, IH), 8.47 (s, I H), 8.39 (s, IH), 8.24 (d, J =8 Hz, IH ), 8.07 (t, J =6.8 Hz, IH), 7.96 (d, /
-243-4820V.1
=7.6 Hz, IH), 7.88-7.72 (m, 2H), 7.57-7.54 (m, 3H), 4.10 (s, 2H), 3.19 (s, 3H), MS ESI 407.2. [M + H]+, calcd for [C2iHi8N403S + H]+ 407.1.

A. 2-(2-chlorothiophen-3-yl)-2-(pyrrolidin-l-yl) acetic acid
The title compound was synthesized according the General Method D using glyoxylic acid monohydrate (70.8 mg, 0.77 mmol) and pyrrolidine (64 μί, 0.77 mmol) was combined with CH2C12 (6.25 mL) and sonicated for 15 minutes. 2-Chlorothiophene-3-boronic acid (125 mg, 0.77 mmol) was added and the mixture was stirred at room temperature for 24 h. Then concentrated the reaction mass under reduced pressure to dryness. Purification by BiotageSNAP 25 g silica column (gradient 0-50% MeOH in DCM) gave the title compound as a cream solid (1 10 mg, 58%). 1 H NMR (400 MHz, CDjOD) δ 7.43 (d, J =5.6 Hz, IH), 7.15 (d, / =5.6 Hz, IH), 4.77 (s, IH), 3.55-3.51 (br.m, 2H), 3.31 (br.s, 2H), 2.10 (br.s, 4H); MS ESI 246.0 [M + H]+, calcd for [C10H12ClNO2S+ H]+ 246.0.
B. 2-(2-Chlorothiophen-3-yl)-N-(3-(3-methylsulfonyl) phenyl)-lH-indazol-5-yl)-2-(pyrrolidin-l- yl) acetamide trifluoroacetate
The title compound was synthesized according to the General Method A, utilizing 2-(2- chlorothiophen-3-yl)-2-(pyrrolidin-l-yl)acetic acid (46mg, 0.186 mmol), 3-(3- (methylsulfonyl)phenyl)-lH-indazole-5-amine 2,2,2-trifluoroacetate (75 mg, 0.186 mmol), N- ethyl-N-isopropylpropan-2-amine (0.16 mL, 0.92 mmol) and TBTU (62 mg, 0.195 mmol) in DMF (2 mL). the reaction mixture was purified by preparative HPLC to give title compound as a white solid (74 mg, 63 %). Ή NMR (400 MHz, CD3OD) δ 8.50 (d, 7 =19.2 Hz, 2H), 8.28 (d, J =7.2 Ηζ,ΙΗ), 8.00 (d, J =8 Hz, IH), 7.78 (t, J =8 Hz, IH ), 7.59-7.54 (m, 3H), 7.32 (d, J =6
-244-4820V.1
Hz, 1H), 5.38 (s, 1H), 3.21 (s, 3H), 3.40-3.32 (br.m, 4H), 2.30-2.00 (br.m, 4H); MS ESI 515.4. [M + H]+, calcd for [C24H23CIN4O3S2 + H]+ 515.1.
Example A221. N-(3-(4-(3-(dimethylamino propyl)-3-(methylsulfonyl)phenyl')-lH-indazol-5- yl)-2-(pyrrolidin- 1 - yl)-2-(thiophen-3-yl)acetamide

The title compound was synthesized according the General Method C, utilizing N-(3-iodo-lH- indazol-5-yl)-2-(pyrrolidin-l-yl)-2-(thiophen-3-yl)acetamide (60 mg, 0.13 mmol), N,N- dimethyl-3-(2-(methylsulfonyl)-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl)propan- 1 - amine (54 mg, 0.15 mmol), PdCl2dppf*CH2Cl2 (5.4 mg, 0.007 mmol), Cs2C03 (130 mg, 0.4 mmol) in DMF (2.5 mL) and H20 (0.6 mL) heated in microwave under Ar at 130 °C for 2 h. The reaction mixture was purified using preparative HPLC followed by preparative TLC (S1O2 1mm, 10 % 7 M Ν¾-ΜεΟΗ in DCM),silica gel plug (2 g) using the following solvents to elute: DCM then EtOAc followed by 2 to 10 % MeOH in DCM and finally a mixture of solvents: 0.35 M NH3/10 % MeOH/90 % DCM. The material was triturated with Et20 to give the title compound as off white powder (10.8 mg, 15 %). Ή NMR (400 MHz, METHANOL-^) δ ppm 1.86 (br. m., 4 H), 1.93 - 2.03 (m, 2 H), 2.37 (s, 6 H), 2.48-2.56 (br. m„ 2 H), 2.56 - 2.62 (m, 2 H), 2.65 - 2.74 (m, 2 H), 3.07 - 3.14 (m, 2 H), 3.25 (s, 3 H), 4.14 (s, 1 H), 7.35 (dd, 7=5.14, 1.13 Hz, 1 H), 7.43 (dd, 7=5.02, 3.01 Hz, 1 H), 7.52 (dd, 7=3.01, 1.25 Hz, 1 H), 7.55 - 7.57 (m, 2 H), 7.65 (d, 7=8.03 Hz, 1 H), 8.20 (dd, 7=8.03, 2.01 Hz, 1 H), 8.36 (s, 1 H), 8.56 (d, 7=2.01 Hz, 1 H). MS ESI [M + H]+ 566.1 (11), calcd for
 + H]+ 566.2.
+ H]+ 566.2.
-245-4820V.1

The title compound was synthesized according the General Method C, utilizing (R)-N-(3-iodo- lH-indazol-5-yl)-2-methoxy-2-phenylacetamide (200 mg, 0.42 mmol) (3- (methylsulfonyl)phenyl)boronic acid (130 mg, 0.67 mmol), PdCl2dppf*CH2Cl2 (15 mg, 0.02 mmol), satd aq Na2C03 (0.5 mL) in PhMe (1.5 mL) and EtOH ( 1.5 mL) sealed under Ar in a microwave reactor (130 °C / 2 h ). The reaction mixture was purified using preparative HPLC to afford the title compound as a white powder (141 mg, 77 %). Ή NMR (400 MHz,
METHANOL-^) δ ppm 3.21 (s, 3 H), 3.48 (s, 3 H), 4.85 (s, 1 H), 7.30 - 7.45 (m, 3 H), 7.53- 7.59 (m, 3 H), 7.64 (d, J = 9.03 Hz, 1 H), 7.79 (t, 7=7.50 Hz, 1 H), 7.99 (d, 7=7.78 Hz, 1 H), 8.29 (d, 7=7.78 Hz, 1 H), 8.38 (s, 1 H), 8.51 (s, 1 H), 10.02 (br. s., 1 H); MS ESI [M + H]+ 436.4, calcd for [C23H2,N304S + H]+ 436.1.
Example A223. 3-(3-(methylsulfonyl phenyl -N-(2-mo holino-l- henylethyl -lH-indazole-5- carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- (methylsulfonyl)phenyl)-lH-indazole-5-carboxylic acid (50.8 mg, 0.16 mmol), 2-morpholino- 1- phenylethanamine (34.0 mg, 0.165 mmol), N-ethyl-N-isopropylpropan-2-amine (0.06 mL, 0.34 mmol) and 0-(benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (52.0 mg, 0.162 mmol) in DMF (3.5 mL). Water (15mL) and NaHC03(aq) (5 mL) were added and the resulting precipitate was collected by filtration. Further precipitate that formed in the filtrate was collected by extraction into EtOAc (150 mL). The organic layer was washed sequentially with
-246-4820V.1
water (15 mL) and brine (15 mL), and dried (Na2S04). The residue after filtration and evaporation in vacuo was combined with the first precipitate using a mixture of methanol, acetone and DMF and poured into a preconditioned 20 mL PoraPak Rxn CX cartridge (Waters). The methanol (30 mL) rinse was discarded and the product was eluted with 2 M ammonia in methanol (30 mL). Purification by preparative HPLC provided the title compound as a white solid (63.7 mg, 64 %). Ή NMR (400 MHz, CD3OD) δ ppm 8.69 (br. s., 1 H), 8.59 (br. s., 1 H), 8.39 (d, J=8.0 Hz, 1 H), 8.03 (t, J=8.7 Hz, 2 H), 7.89 - 7.78 (m, 1 H), 7.69 (d, J=8.3 Hz, 1 H), 7.52 - 7.60 (m, 2 H), 7.43 - 7.52 (m, 2 H), 7.34 - 7.43 (m, 1 H), 5.83 (d, J=8.8 Hz, 1 H), 3.77 - 4.17 (m, 7 H), 3.71 (d, J=13.1 Hz, 1 H), 3.42 - 3.62 (m, 2 H), 3.24 (br. s., 3 H); MS ESI 505.4 [M + H]+, calcd for [C27H28N4O4S + H]+ 505.2.
Example A224. N-(2-methylbenzyl)-3-(3-(methylsulfonyl)phenyl)- lH-indazole-5-carboxamide

The title compound was synthesized according to the General Method A utilizing 3-(3- (methylsulfonyl)phenyl)-lH-indazole-5-carboxylic acid (50.5 mg, 0.16 mmol), 0- tolylmethanamine (0.03 mL, 0.24 mmol), N-ethyl-N-isopropylpropan-2-amine (0.06 mL, 0.34 mmol) and 0-(benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate (51.5 mg, 0.160 mmol) in DMF (3.5 mL). Water (15mL) and NaHC03(aq) (5 mL) were added and the product was extracted into EtOAc (150 mL). The organic layer was washed sequentially with water (15 mL) and brine (15 mL), and dried (Na2S04), filtered and evaporated in vacuo.
Purification by preparative HPLC provided the title compound as an off-white solid (42.4 mg, 63 %). Ή NMR (400 MHz, DMSO-d6) 8 ppm 13.69 (br. s.( 1 H), 9.1 1 - 8.99 (m, 1 H), 8.65 (s, 1 H), 8.49 (br. s., 1 H), 8.41 (d, J=8.0 Hz, 1 H), 7.98 (d, J=8.0 Hz, 2 H), 7.85 (t, J=7.8 Hz, 1 H), 7.69 (d, J=8.5 Hz, 1 H), 7. 32 - 7.23 (m, 1 H), 7.16 (br. s., 3 H), 4.51 (d, J=4.8 Hz, 2 H), 3.31 (s, 3 H), 2.34 (s, 3 H); MS ESI 420.3 [M + H]+, calcd for [C23H21N3O3S + H]+ 420.1.
-247-4820v.1
xample A225, N-(3-(3-(methylsulfonyl)phenyl)-lH-indazol-5-ylV2-(o-tolyl)acetamide

The title compound was synthesized according to the General Method C, utilizing N-(3- iodo-lH-indazol-5-yl)-2-(o-tolyl)acetamide (51.1 mg, 0.13 mmol), (3-(methylsulfonyl) phenyl)boronic acid (36.2 mg, 0.18 mmol), Pd(PPh3)4 (10.5 mg, 0.009 mmol), toluene (1.5 mL), EtOH (1.5 mL), and aqueous Na2CC»3 (0.20 mL, 2 M, 0.40 mmol). The degassed solution was sealed and heated in a microwave reactor at 125 °C for 2 h. Purification by preparative HPLC followed by column (Silicycle 10 g SPE, silica gel, 0-50 % EtOAc in DCM) and trituration with 50% DCM in hexane (2mL x 3) provided the title compound as an off-white solid (27.3 mg, 50 %). Ή NMR (400 MHz, Acetone-d6) δ ppm 12.57 (br. s., 1 H), 9.45 (br. s., 1 H), 8.57 (br. s., 2 H), 8.32 (d, J=7.8 Hz, 1 H), 7.97 (d, J=7.5 Hz, 1 H), 7.83 (t, J=7.8 Hz, 1 H), 7.70 (d, J=9.0 Hz, 1 H), 7.62 (d, J=9.3 Hz, 1 H), 7.33 (br. s., 1 H), 7.24 - 7.1 1 (m, 3 H), 3.80 (s, 2 H), 3.23 (s, 3 H), 2.39 (s, 3 H); MS ESI 420.3 [M + H]+, calcd for
 + H]+ 420.1.
+ H]+ 420.1.
Example A226. N-(3-(3-((dimethylamino)methyl)-5-( methylsulfonvDphenyl)- 1 H-indazol-5-vD- 2-(o-tolyl)acetamide
A. Methyl 3-bromo-5-iodobenz

-248-4820V.1
Acetyl chloride (2.25 mL, 31.6 mmol) was added in a dropwise manner to a suspension of 3-bromo-5-iodobenzoic acid (1.9841 g, 6.07 mmol) in MeOH (25 mL) under argon atmosphere at 0°C. After 5 min, the argon flow was replaced with a drying tube and the mixture was warmed to rt and stirred for 15h. LC-MS indicated incomplete reaction. The mixture was cooled to at 0°C and a second portion of acetyl chloride (1.1 mL, 15.5 mmol) was added and stirring was continued for 28 h. EtOAc (300 mL) was added and the organic layer was washed with water (50 mL), NaHC03(aq) (50 mL x 2), and brine (50 mL) and dried (Na2S04).
Evaporation of the solvent in vacuo gave the title compound (1.98 g, 96 %). Ή NMR (400 MHz, CDCh) 8 ppm 8.31 (s, 1 H), 8.14 (s, 1 H), 8.05 (s, 1 H), 3.94 (s, 3 H); MS ESI 340.9 / 342.9 [M + H]+, calcd for [C8H6BrI02 + H]+ 340.9 / 342.9.
B. Methyl 3-bromo-5-(me ethylsulfonyl)benzoate
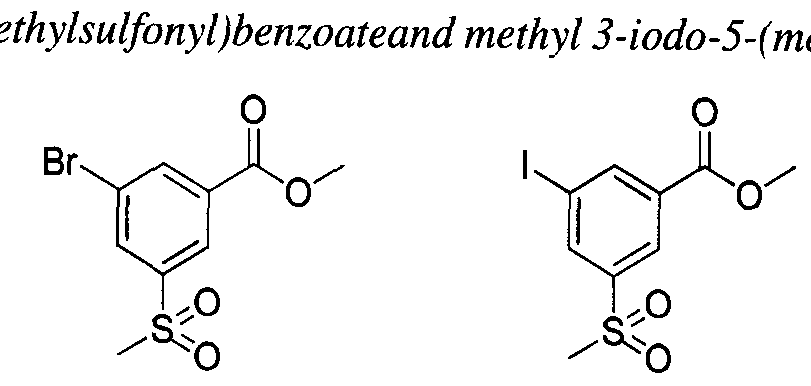
N,N'-Dimethylethylenediamine (30 μί, 0.279 mmol) was added to a mixture of methyl 3-bromo-5-iodobenzoate (754.0 mg, 2.21 mmol), sodium methanesulfinate (389.0 mg, 3.8 mmol) and copper(I) trifluoromefhanesulfonate benzene complex (68.0mg, 0.135 mmol) in DMSO (4.5 mL) under argon atmosphere. The resulting mixture was heated in a 100 °C reaction block for 2 days. EtOAc (50 mL) was added and the mixture was passed through a celite depth plug to remove solids, rinsing with EtOAc (50 mL). The organic layer was washed with water (15 mL x 2) and brine (15 mL), dried (Na2S04) and filtered. Evaporation of the solvent in vacuo and purification by column (Biotage Isolera, 25g HP-SIL plus samplet, 10-40% EtOAc in hexane) gave the title compound as a beige solid (200.2 mg, 70:30 mixture of methyl 3-bromo-5- (methylsulfonyl)benzoate and methyl 3-iodo-5-(methylsulfonyl)benzoate). Ή NMR (400 MHz, CDCh) δ ppm 8.60 (s, 0.3 H), 8.51 (s, 0.3 H), 8.48 (s, 0.7 H), 8.42 (s, 0.3 H), 8.40 (s, 0.7 H), 8.24 (s, 0.7 H), 3.96 (s, 3 H), 3.09 (m, 3 H).
-249-4820V.1
C. l-(3-Bromo-5-(methylsulfonyl)phenyl)-N,N-dimethylmethanamine and l-(3-iodo-5- ( methylsulfonyl )phenyl )-N,N-dimethylmethanamine

A mixture of methyl 3-bromo-5-(methylsulfonyl)benzoate and methyl 3-iodo-5- (methylsulfonyl)benzoate (200 mg, 70:30 mixture) and lithium borohydride (48.2 mg, 2.2 mmol) in THF (12 mL) under argon atmosphere was stirred at it for 5 h. Aqueous HC1 (2 M, l mL) was added in a dropwise manner until gas evolution ceased, then excess acid was quenched by addition of NaHC03(aq) (1.5mL). EtOAc (10 mL) was added and the organic layer was passed through a Varian ChemElut (Hydromatrix, 3 mL) cartridge, rinsing with EtOAc (5 mL x 5). Evaporation of the solvent in vacuo gave a 70:30 mixture of (3-bromo-5-
(methylsulfonyl)phenyl)methanol and (3-iodo-5-(methylsulfonyl)phenyl)methanol which was used without further purification. Ή NMR (400 MHz, CD3OD) δ ppm 8.16 (s, 0.3 H), 8.07 (s, 0.3 H), 7.99 (s, 0.7 H), 7.94 (s, 0.3 H), 7.92 (s, 0.7 H), 7.87 (s, 0.3 H), 4.71 (s, 1.4 H), 4.68 (s, 0.6 H), 3.16 (s, 2.1 H), 3.15 (s, 0.9 H).
Dess Martin Periodinane (4.0 mL, 1.2 mmol) was added to a solution of the mixture of
(3-Bromo-5-(methylsulfonyl)phenyl)methanol and (3-iodo-5-(methylsulfonyl)phenyl)methanol in DCM ( 16.0 mL) under argon atmosphere. The resulting cloudy solution was stirred at rt for 19 h. Aqueous NaOH ( 1M, 8mL) was added and the resulting solution was stirred at rt for 1 h. The product was partitioned between DCM (200 mL) and water (20 mL), and the organic layer was washed with water (20 mL) and brine (20 mL), and dried (Na2SC»4). Filtration and evaporation of the solvent in vacuo gave a 70:30 mixture of bromo-5-(methylsulfonyl)benzaldehyde and 3-iodo- 5-(methylsulfonyl)benzaldehyde (184.7 mg) which was used without further purification. Ή NMR (400 MHz, CDClj) δ ppm 10.05 (s, 0.7 H), 10.01 (s, 0.31 H), 8.51 t, J=1.5 Hz, 0.3 H), 8.48 (t, J= 1.5 Hz, 0.3 H), 8.40 (t, J= 1.6 Hz, 0.3 H), 8.37 (t, J= 1.5 Hz, 0.7 H), 8.33 (t, J=1.8 Hz, 0.7 H), 8.29 (t, J=1.5 Hz, 0.7 H), 3.14 (s, 2.1 H), 3.13 (s, 0.9 H)
Dimethylamine (1.20 mL, 2 M in THF, 2.40 mmol) was added to a solution of the mixture of 3-bromo-5-(methylsulfonyl)benzaldehyde and 3-iodo-5-
-250-4820V.1
(methylsulfonyl)benzaldehyde (184.7 mg) and NaBH(OAc)3 (282.1 mg, 1.33 mmol) in 1,2-DCE ( 10 mL) under argon atmosphere. Glacial acetic acid (4 drops) was added and the reaction mixture was stirred at it for 16 h. The product was partitioned between DCM (175 mL) and NaHC03(aq) (30 mL), and the organic layer was washed with water (30 mL) and brine (30 mL), and dried (Na2S04), filtered and evaporated in vacuo. Purification by column (Biotage Isolera, 25g HP-SIL plus samplet, 2-7% 2 M NH3-MeOH in DCM) gave a mixture of the title compounds as a pale yellow solid (181.2 mg, 70:30 mixture). Ή NMR (400 MHz, CDCl3) δ ppm 8.15 (s, 0.3 H), 7.99 (s, 0.3 H), 7.97 (s, 0.7 H), 7.86 (s, 0.3 H), 7.83 (s, 0.7 H), 7.79 (s, 0.7 H), 3.47 (s, 1.4 H), 3.45 (s, 0.6 H), 3.07 (overlapping singlets, 3 H), 2.25 (overlapping singlets, 6 H).
D. N N-(3-(3-((dimethylamino)methyl)-5-(methylsulfonyl)phenyl)-lH-in^
tolyljacetamide

Hexabutyl ditin (0.11 mL, 0.21 mmol) was added to a solution of the 70:30 mixture of 1-
(3-bromo-5-(methylsulfonyl)phenyl)-N,N-dimethylmethanamine and l-(3-iodo-5- (methylsulfonyl)phenyl)-N,N-dimethylmethanamine (45.9 mg) in toluene (3 mL) under argon bubbling. Pd(PPh3)4 (24.1 mg, 0.021 mmol) was added, and the vial was capped and heated at 110 °C for 15 h. The product was diluted with DCM (2 mL) and purified by column (Silicycle 5g SPE, silica, 0-100% Et20 in DCM) to give the title compound (39.8 mg). Ή NMR indicated mixture including Ν,Ν-dimethyl- 1 -(3-(methylsulfonyl)-5-(tributylstannyl)phenyl)methanamine; complex in aryl region, 80:20 ratio in aliphatic singlet peaks, Bu3Sn present; MS ESI 504.2 [M + H]+, calcd for [C22H4iN02SSn + H]+ 504.2. The crude product was carried forward without further purification.
-251-4820v.1
Pd(PPh3)4 (11.2 mg, 0.0097 mmol) was added to a degassed solution of N-(3-iodo-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-5-yl)-2-(o-tolyl)acetamide (37.1 mg, 0.078 mmol) and Ν,Ν-dimethyl- 1 -(3-(methylsulfonyl)-5-(tributylstannyl)phenyl)methanamine (39mg, 0.077 mmol) in DMF (2.5 mL) and the resulting mixture was heated in a microwave reactor at 120 °C for 4 h, and then heated in a 120 °C reaction block for a further 13h. The mixture was cooled to rt and the solvent was evaporated in vacuo. MeOH was added and the mixture was poured into a preconditioned 20 mL PoraPak Rxn Cx cartridge (Waters). The methanol (30 mL) rinse was discarded and the product was eluted with 2 M ammonia in methanol (30 mL). The solvent was removed in vacuo, and the residue was purified by column (Silicycle 5g SPE, silica, 50-100% EtOAc in DCM, then 2-6% MeOH in EtOAc) provided N-(3-(3-((dimethylamino)methyl)-5- (methylsulfonyl)phenyl)- l-(tetrahydro-2H-pyran-2-yl)-lH-indazol-5-yl)-2-(o-tolyl)acetamide (20.2 mg, 46 %). MS ESI 561.4 [M + H]+, calcd for [C31H36N4O4S + H]+ 561.3.
A solution of N-(3-(3-((dimethylamino)methyl)-5-(methylsulfonyl)phenyl)- l-(tetrahydro- 2H-pyran-2-yl)-lH-indazol-5-yl)-2-(o-tolyl)acetamide (20mg, 0.036 mmol) and 4- toluenesulfonic acid monohydrate (30.5 mg, 0.16 mmol) in MeOH (2.5 mL) was heated in a microwave reactor at 100 °C for 15 min. The mixture was cooled to rt and the solvent was evaporated. EtOAc (5 mL) and NaHC03(aq) (3 mL) were added and the resulting solution was poured into a Varian ChemElut cartridge (Hydromatrix, 3 mL) and rinsed with EtOAc (5mL x 5). The resulting cloudy extracts were passed through a second ChemElut cartridge
(Hydromatrix, 3 mL). The solvent was removed in vacuo, and the residue was purified by column (Silicycle 5g SPE, silica, 2-5% 2 M NH3-MeOH in DCM) and triturated with ether (1 mL x 3) to provide the title compound as a white filmy solid (8.0 mg, 47 %). Ή NMR (400 MHz, CD3OD) δ ppm 8.41 (s, 1 H), 8.37 (s, 1 H), 8.22 (s, 1 H), 7.95 (s, 1 H), 7.57 (m, 2 H), 7.28 (d, J=3.5 Hz, 1 H), 7.18 (m, 3 H), 3.79 (s, 2 H), 3.69 (s, 2 H), 3.21 (s, 3 H), 2.38 (s, 3 H), 2.32 (s, 6 H); MS ESI 477.4 [M + H]+, calcd for [C26H28N4O3S + H]+ 477.2.
-252-4820V.1
Example A227. N-(3-(4-((dimemylanuno)methyl)-3-(methylsulfonyl')phenyl -lH-indazol-5-yl)- 2-(p yrrolidin- 1 -yl)-2-(thiophen-3-yl)acetamide

A. N -dimethyl-l-(2-(methylsulfonyl)-4-(trimethylstannyl)phenyl)methanamine

Hexamethyl ditin (141 mg, 0.43 mmol) in toluene (1.5 mL) was added to l-(4-bromo-2- (methylsulfonyl)phenyl)-N,N-dimethylmethanamine (101.6 mg, 0.348 mmol) and Pd(PPh3)4 (28.5 mg, 0.025 mmol) in toluene (1.5 mL) under argon, and the vial was capped and heated at 110 °C for 6 h. The reaction mixture was cooled to rt and aqueous KF (1M, 2 mL) and EtOAc (10 mL) were added and the mixture was stirred for 90 min, then passed through a Varian ChemElut (Hydromatrix, 3 mL) cartridge, rinsing with EtOAc (5 mL x 5). The solvent was removed in vacuo, and the residue was dissolved in MeOH (5 mL) and passed through a preconditioned 20 mL PoraPak Rxn Cx cartridge (Waters). The methanol (30 mL) rinse was discarded and the product was eluted with 2 M ammonia in methanol (30 mL). Evaporation and purification by column (Silicycle lOg SPE, silica, 2.5-5% MeOH in DCM) gave the title compound (71 mg, 54%). Ή NMR (400 MHz, CDCl3) δ ppm 8.19 (m, 1 H), 7.67 (m, 1 H), 7.36 (d, J=7.3 Hz, 1 H), 3.84 (m, 2 H), 3.40 (m, 3 H), 2.25 (s, 6 H), 0.35 (m, 9 H); MS ESI 378.0 [M + H]+, calcd for [Ci3H23N02SSn + H]+ 378.2.
-253-4820V.1
β. N-(3-(4-((dimethylamino)methyl)-3-(methylsulfonyl)phenyl)-lH nd^
yl)-2-(thiophen-3-yl)acetamide

DMF (2 mL) was added under argon to a mixture of N-(3-iodo-lH-indazol-5-yl)-2-(pyrrolidin-l- yl)-2-(thiophen-3-yl)acetamide (60.5 mg, 0.134 mmol), Ν,Ν-dimethyl- 1 -(2-(methylsulfonyl)-4- (trimethylstannyl)phenyl)methanamine (70 mg, 0.15 mmol) and Pd(PPh3)4 (15.3 mg, 0.0134 mmol) and the resulting mixture was heated in a sealed vial in a 120 °C reaction block for 15 h. The mixture was cooled to rt and MeOH (5 mL) was added and the mixture was poured into a preconditioned 20 mL PoraPak Rxn Cx cartridge (Waters). The methanol (30 mL) rinse was discarded and the product was eluted with 2 M ammonia in methanol (30 mL). After the solvent was removed in vacuo, purification by column (Biotage Isolera, 25g HP-SIL plus samplet, 5- 15% MeOH in DCM) followed by reverse phase purification (Biotage Isolera, 60g RP-C18-HS plus samplet, 0.1%TFA-H2O in MeOH, gradient 10-80%) provided the title compound as a white solid (26.5 mg, 26 %). Ή NMR (400 MHz, CD3OD) δ ppm 8.78 (d, J=2.0 Hz, 1 H), 8.52 (d, J=1.3 Hz, 1 H), 8.44 (dd, J=8.0, 1.8 Hz, 1 H), 7.91 (d, J=7.8 Hz, 1 H), 7.89 (dd, J=3.0, 1.3 Hz, 1 H), 7.66 (dd, J=5.1, 2.9 Hz, 1 H), 7.63 (d, J=9.0 Hz, 1 H), 7.56 (dd, J=9.0, 2.0 Hz, 1 H), 7.39 (dd, J=5.3, 1.3 Hz, 1 H), 5.29 (s, 1 H), 4.73 (s, 2 H), 3.89 (br. s., 1 H), 3.37 (s, 3 H), 3.20 (br. s., 1 H), 3.12 (br. s, 1 H), 2.99 (s, 6 H), 2.24 (br. s., 1 H), 2.16 (m, 2 H), 2.01 (br. s., 1 H); MS ESI 538.0 [M + H]+, calcd for [C27H3 iN503S2 + H]+ 538.2.
-254-4820V.1
xample A228. 2-(dimethylamino)-N-(3-(3-(methylsulfonyl)phenyl)-lH-indazol-5-yl)-2-

The title compound was synthesized according to the method described for 2-phenyl-N-(3-(3- sulfamoylphenyl)- 1 H-indazol-5-yl)acetamide, utilizing 2-(dimethylamino)-2-(thiophen-3- yl)acetic acid (31 mg, 0.17 mmol), 3-(3-(methylsulfonyl)phenyl)-lH-indazol-5-amine■ TFA (50 mg, 0.17 mmol), DIPEA (60 μί, 0.35 mmol), TBTU (55 mg, 0.17 mmol) and 4 mL of DMF. The reaction mixture was purified using prep-HPLC and triturated in Et20 to give the title compound: a white solid (7.2 mg, 9%). Ή NMR (400 MHz, METHANOL-d*) δ ppm 8.50 (s, 1 H), 8.45 (s, 1 H), 8.29 (d, 7=7.03 Hz, 1 H), 8.00 (d, 7=7.78 Hz, 1 H), 7.88 (br. s., 1 H), 7.80 (t, 7=7.65 Hz, 1 H), 7.68 (br. s., 1 H), 7.51 - 7.62 (m, 2 H), 7.35 (d, 7=5.27 Hz, 1 H), 5.19 (s, 1 H), 3.21 (s, 3 H), 3.08 (br. s., 3 H), 2.69 (br. s., 3 H). MS ESI [M + H]+ 455.2, calcd for
[C22H22N403S2 + H]+ 455.1
Example A229. N-(cvclopropyl(phenyl)methyl)-3-(3-(methylsulfonyl phenyl)- 1 H-indazole-5- carboxamide

The title compound was synthesized according to the method described for N-benzyl-3-(3- sulfamoylphenyl)- 1 H-indazole-5-carboxamide utilizing 2-cyclopropyl-N-(3-iodo- 1 H-indazol-5- yl)-2-phenylacetamide (1 10 mg, 0.26 mmol), (3-(methylsulfonyl)phenyl)boronic acid (64 mg, 0.32 mmol), PddppfCl2 (11 mg, 0.013 mmol), saturated aqueous Na2C03 (1.25 mL), and toluene / EtOH (1.9 mL / 1.9 mL). The reaction mixture was purified by prep-HPLC and triturated with MeOH to give the title compound: white solid (14 mg, 12%). Ή NMR (400 MHz, METHANOL-
-255-4820V.1
d ) δ ppm 9.17 (d, 7=7.53 Hz, 1 H), 8.62 (s, 1 H), 8.56 (s, 1 H), 8.37 (d, 7=8.03 Hz, 1 H), 8.00 (d, 7=8.03 Hz, 1 H), 7.96 (d, 7=8.53 Hz, 1 H), 7.79 (t, 7=7.91 Hz, 1 H), 7.64 (d, 7=9.03 Hz, 1 H), 7.49 (d, 7=7.53 Hz, 2 H), 7.34 (t, 7=7.15 Hz, 2 H), 7.24 (t, 7=7.00 Hz, 1 H), 4.49 (t, 7=8.41 Hz, 1 H), 3.21 (s, 3 H), 1.35 - 1.45 (m, 1 H), 0.66 (dd, 7=17.82, 8.03 Hz, 2 H), 0.48 (ddd, 7=17.82, 10.67, 4.39 Hz, 2 H). MS ESI [M + H]+ 446.3, calcd for [C25H23N3O3S + H]+ 446.2

In a sealed degassed vial, N-(3-iodo- lH-indazol-5-yl)-2-(thiophen-2-yl)acetamide (50 mg, 0.13 mmol), 3-methylsulfonylphenylboronic acid (31 mg, 0.16 mmol), Pd(PPh3)4 (7.5 mg, 0.0065 mmol), KF (23 mg, 0.39 mmol), DMF (1.6 mL), and H20 (0.4 mL) was combined. The vial was charged with argon and the mixture was placed in the microwave reactor heating at 120 °C for 2 hours. The mixture was purified by prep-HPLC and triturated with Et20 to give the title compound: white solid (6.2 mg, 12%). Ή NMR (400 MHz, METHANOLS) δ ppm 8.50 (s, 1 H), 8.39 (s, 1 H), 8.28 (d, 7=7.78 Hz, 1 H), 7.98 (d, 7=8.53 Hz, 1 H), 7.79 (t, 7=7.91 Hz, 1 H), 7.56 (s, 2 H), 7.29 (d, 7=5.27 Hz, 1 H), 7.02 - 7.06 (m, 1 H), 6.96 - 7.01 (m, 1 H), 3.94 (s, 2 H), 3.21 (s, 3 H). MS ESI [M + H]+ 412.1, calcd for ^Hn^C^ + H]+412.1
Example A231. rSI^^S^methylsulfonvnphenvn-N-d-phenylethvD-lH-indazole-S- carboxamide

The title compound was synthesized according to the method described for N-benzyl-3-(3- sulfamoylphenyl)- 1 H-indazole-5-carboxamide utilizing (S)-N-(3-iodo- 1 H-indazol-5-yl)-2-
-256-4820V.1
phenylpropanamide (190 mg, 0.48 mmol), (3-(methylsulfonyl)phenyl)boronic acid (1 16 mg, 0.58 mmol), PddppfCl2 (20 mg, 0.024 mmol), saturated aqueous Na2CC>3 (1.25 mL), and toluene / EtOH (1.9 mL / 1.9 mL). The reaction mixture was purified by prep-HPLC and triturated with MeOH to give the title compound: white solid (54 mg, 27%). Ή NMR (400 MHz, DMSO-d6) δ ppm 13.69 (s, 1 H), 8.94 (d, 7=7.78 Hz, 1 H), 8.61 (s, 1 H), 8.51 (s, 1 H), 8.40 (d, 7=7.53 Hz, 1 H), 7.93 - 8.02 (m, 2 H), 7.87 (t, 7=7.78 Hz, 1 H), 7.68 (d, 7=8.03 Hz, 1 H), 7.43 (d, 7=8.03 Hz, 2 H), 7.34 (t, 7=7.15 Hz, 2 H), 7.23 (t, 7=7.00 Hz, 1 H), 5.18 - 5.27 (m, 1 H), 1.51 (d, 7=7.03 Hz, 3 H). MS ESI [M + H]+ 420.3, calcd for [C23H2,N303S + H]+420.1 Example A232. (S)-2-(dimethylamino)-N-(3-(3-methylsulfonvf) phenyl)- lH-indazol-5-yl)-2- phenyl acetamide trifluoroaceta

A. ethyl 2-bromo-2-phenylacetate
To a solution of alpha-Bromophenylacetic Acid (5.0 g, 0.023 mol) in EtOH (50 mL) at was added cone, H2S04 (0.5 g, 0.005 mol) drop wise in 15 min at room temperature. After addition, the resulting mixture was stirred at 75°C for 18 h. The solvent was removed under reduced pressure and dissolved residue in EtOAc (50 mL) and washed with 1M Na2C03 (50 mL).the phases were separated and dried over anh Na2SC>4, filtered and concentrated to afford the title compound as colorless oil (5.25 g, 93%). Ή NMR (400 MHz, CDCh) δ 7.56 (dd, 7 =7.7, 1.9 Hz, 2H), 7.4-7.34 (m, 3H), 5.35 (s, 1Η),4.29-4.20 (m, 2H), 1.29 (t, 7 =7.2 Hz, 3H).
B. ethyl 2-(dimethylamino)-2-phenylacetate
To a solution of ethyl 2-bromo-2-phenylacetate (5.25 g, 0.021 mol) in THF (26 mL) at room temperature was added Na2C03 (2.29 g, 0.021 mol) followed by 1M (CH3)2NH in THF (24 mL) drop wise in 15 min at room temperature. After addition, the resulting mixture was stirred at same temperature for 18 h. Filtered the insoluble salt and washed it with EtOAc (10 mL * 2).
-257-4820V.1
The combined filtrate concentrated under reduced pressure to give thick oil. The oily product was dissolved in DCM (25 mL) and washed it with water (15 mL) and dried over anh Na2S04, filtered and concentrated to afford the title compound as yellowish oil (4.25 g, 95%). Ή NMR (400 MHz, CDCh) δ 7.56 (dd, J =7.7, 1.9 Hz, 2H), 7.4-7.34 (m, 3H), 5.35 (s, 1Η),4.29-4.20 (m, 2H), 2.26(s, 6H), 1.29 (t, / =7.2 Hz, 3H); MS ESI 208. [M + H]+, calcd for [Ci2H17N02 + H]+ 208.1.
C. (S)-l-(dimethylamino)-2-phenylaceticacid hydrochloride
To a solution of ethyl 2-(dimethylamino)-2-phenylacetate (1.5 g, 0.007 mol) in EtOH (15 mL) at room temperature was added (+)-Dibenzoyl-D-tartaric acid (1.84 g, 0.005 mol). The solution was heated to 75°C for 1 h. after stirring 1 h at room temperature cooled to 5°C and agitated for 1 h. The precipitated insoluble salt was filtered, well pressed and washed it with EtOH (1.5 mL * 2) and dried under vacuum at room temperature for 24 h to give (-)-ethyl 2- (dimethylamino)-2-phenylacetate (+)-Dibenzoyl-D-tartaric acid salt. Dry wt: 1.87 g. The filtrate and washing were combined and to that 2nd portion of (+)-dibenzoyl-D-tartaric acid (0.62 g,
0.0017 mol) and water (25 mL) added at room temperature. The solution was heated to 75°C for 1 h and kept in a freezer overnight at 0°C. The solid was filtered and washed it with EtOH ( 1.5 mL * 2) and dried under vacuum at room temperature for 24 h to give a white solid as (+)-ethyl 2-(dimethylamino)-2-phenylacetate (+)-Dibenzoyl-D-tartaric acid salt 1.67 g. Optical Rotation: [oc]22D = 124° (c 0.5, MeOH).
Dissolved 1.67 g of above crude salt in a mixture of EtOH (5.85 mL) and water (2.5 mL) at 65°C.then gradually cooled to room temperature and stirred at 0°C for 1 h. The solid was filtered and suck dried to give pure white solid 1.08 g. Optical Rotation: [oc]22 D = 125.4° (c 0.63, MeOH). Ή NMR (400 MHz, CD3OD) δ 8.14 (d, J =1.2 Hz, 4H), 7.64-7.60 (m, 2H), 7.51-7.47 (m, 9H), 5.93 (s, 2H), 4.28-4.20 (m, 2H), 2.68 (s, 6H), 1.18 (t, J =6.8 Hz, 3H),1H merged with solvent peak.
1.08 g solid was stirred with Cone, HC1 (30 mL) at room temperature for 15 min and then extracted with DCM (40 mL).the phases were separated and aqu,layer refluxed for 32 h at 95°C. Removal of solvents under high vacuum at 35°C/2 mbar to gave the title compound as a
-258-4820V.1
white solid 300 mg. Optical Rotation: [a]22 D = 89.13° (c 0.46, H20). After trituration of 300 mg salt with acetonitrile (6 mL) at room temperature for 15 min to gave title compound as a white hydrochloride salt (127 mg, 8%). Optical Rotation: [a]22 D = 107.8°(c 0.51, H20). Reported value: 117°(c 0.99, H20). Ή NMR (400 MHz, DMSO-d6) δ 7.55-7.49 (m, 5H), 5.21 (s, 1H), 2.68 (s, 6H); MS ESI 180. [M + H]+, calcd for [C10H13NO2 + H]+ 180.1
D. (S)-2-(dimethylamino)-N-(3-(3-methylsulfonyl) phenyl)-lH-indazol-5-yl)-2-phenyl acetamide trifluoroacetate
The title compound was synthesized according to the General Method A, utilizing (S)-l- (dimethylamino)-2-phenylaceticacid hydrochloride (40 mg, 0.185 mmol), 3-(3-
(methylsulfonyl)phenyl)-lH-indazole-5-amine (53 mg, 0.185 mmol), N-ethyl-N-isopropyl propan-2-amine (0.16 mL, 0.925 mmol) and TBTU (59 mg, 0.185 mmol) in DMF (1.2 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0-10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Ci8, 60 g, gradient 90: 10-20:80 % 0.1% TFA- H20: MeOH) to give the title compound as a white solid (37 mg, 35 %).'H NMR (400 MHz, CD3OD) δ 8.49 (d, J = 19.2 Hz, 2H), 8.28 (d, J =7.6 Hz, 1H), 8.00 (t, J =8.0 Hz, lH),7.80-7.75 (m, 1H), 7.69-7.68 (br.s, 2H), 7.58-7.52 (m, 5H), 5.06 (s, 1H), 3.21 (s, 3H), 3.09-2.94 (br.m, 3H), 2.81-2.73 (br.m, 3H); MS ESI 449.2. [M + H]+, calcd for
[C24H24N403S -i- H]+ 449.1.

The title compound was synthesized according to the General Method A, utilizing (S)-2- (pyrrolidin-l-yl)-2-(thiophen-3-yl) acetic acid trifluoroacetate (50 mg, 0.153 mmol),
3-(3-(methylsulfonyl)phenyl)- lH-indazole-5-amine trifluoroacetate (61 mg, 0.153 mmol), N-ethyl-N-isopropyl propan-2-amine (0.13 mL, 0.77 mmol) and TBTU (49 mg, 0.153 mmol) in
-259-4820V.1
DMF ( 1.5 mL). The reaction mixture was purified by reverse phase column chromatography (Biotage d8, 60 g, gradient 90: 10-20:80 % 0.1 % TFA- H20: MeOH) to give the title compound as a white solid (39 mg, 42%, 78.5% e.e. by chiral HPLC using ( S, S) Whelk-01 ( 10 x 0.46 cm), 40% methanol/(0.1 % DEA)/C02, 100 bar,3 mL min, Rt = 4.5 min, 220 and 254 nm.
Samples dissolved in 0.5mL ethanol. Ή NMR (400 MHz, CD3OD) 8 8.48 (m, 2H), 8.29 (d, / =8 Hz, I H), 8.0 (d, J =8.3 Hz, IH), 7.88 (d, J = 1.8 Hz, IH), 7.88 (t, J =7.8 Hz, IH), 7.66-7.53 (m, 3H), 7.39 (dd, J =5, 1 Hz, IH), 5.25 (s, IH), 3.88 (br.s, IH), 3.22 (s, 3H), 3.40-3.05 (m, 2H), 2.30- 1.95 (m, 5H); MS ESI 481.3. [M + H]+, calcd for [C24H2 N4O3S2 + H]+ 481.1.

The title compound was synthesized according to the General Method A, utilizing (R)-2- (pyrrolidin- l-yl)-2-(thiophen-3-yl) acetic acid trifluoroacetate (60 mg, 0.184 mmol), 3-(3- (methylsulfonyl)phenyl)- lH-indazole-5-amine (53 mg, 0.184 mmol), N-ethyl-N-isopropyl propan-2-amine (0.16 mL, 0. 92 mmol) and TBTU (59 mg, 0.184 mmol) in DMF (1.8 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0- 10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Ci8, 60 g, gradient 90: 10-20:80 % 0.1 % TFA- H20: MeOH) to give the title compound as a off white solid (19 mg, 17 %, 74 % e.e. by chiral HPLC ( S, S) Whelk-01 ( 10 x 0.46 cm), 40% methanol/ (0.1 % DEA)/C02, 100 bar,3 mL/min, Rt = 2.7 min, 220 and 254 nm.Samples dissolved in 0.5mL ethanol.
Ή NMR (400 MHz, CD3OD) δ 8.48 (m, 2H), 8.29 (d, J =8 Hz, I H), 8.0 (d, / =8.3 Hz, IH), 7.88 (d, J = 1.8 Hz, I H), 7.88 (t, J =7.8 Hz, I H), 7.66-7.53 (m, 3H), 7.39 (dd, J =5, 1 Hz, IH), 5.25 (s, IH), 3.88 (br.s, I H), 3.22 (s, 3H), 3.40-3.05 (m, 2H), 2.30-1.95 (m, 5H); MS ESI 481.3. [M + H]+, calcd for [C24H24N4O3S2 + H]+ 481.1.
-260-4820V.1
Example A235. N-(3-(4-(4-methylpiperazin-l-yl)-3-methylsulfonyl) phenyl)-lH-indazol-5-yl)-2- (pyrrolidin-l-ylV2-(thiophen-3-yl) acetamide trifluoroacetate

A. l-(4-chloro-2-(methylsulfonyl) phenyl)-4-methyl piperazine
To a solution of l-(4-chloro-2-(methylsulfonyl) phenyl) piperazine (800 mg, 2.91 mmol) in formic acid (15 mL) was added formalin (0.92 mL, 11.33 mmol). The solution was heated to 150°C for 15 min under microwave irradiation.the solvent was removed in vacuo and the residue was dissolved in EtOAc ( 50 mL),washed with satd, NaHC03 (10 mL * 2) , brine (10 mL) and dried over anh Na2SC>4 and concentrated to dryness. Purification by BiotageSNAP 25 g silica column (gradient 0- 10% MeOH in DCM) gave the title compound as a cream solid (720 mg, 86%). Ή NMR (400 MHz, CDCh) δ 8.07 (d, J =2.4 Hz, IH), 7.58 (dd, / =8.4, 2.4 Hz, IH), 7.37 (d, J = 8.8 Hz, IH), 3.59 (s, 3H), 3.11-3.08 (br.s, 4H), 2.64-2.62 (br.s, 4H), 2.38 (s, 3H), ESI 289.1.[M + H]+, calcd for [C12H17C1N202S + H]+ 289.07 B. l-methyl-4-(2-(methylsulfonyl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl) phenyl) piperazine
A mixture of /-(4-chloro-2-(methylsulfonyl) phenyl)-4-methyl piperazine (700 mg, 2.42 mmol), pinacolato diboron (666 mg, 2.67 mmol), KOAc (713 mg, 7.27 mmol) and dioxane (21 mL) was purged with argon for 10-min. Pddba2 (69 mg,0.121 mmol) and tricyclohexyl phosphine tetrafluoroborate (89 mg, 0.242 mmol) was added, the vial sealed and heated it in oil bath to 80°C for 18 hrs. The reaction mass was diluted using EtOAc (100 mL),washed with satd, NaHC03 (20 mL * 2) , brine (20 mL) and dried over anh Na2S04 and concentrated to dryness. Purification by BiotageSNAP 50 g silica column (gradient 0-30% MeOH in DCM) gave the title compound as a light brown solid (270 mg, 29 %).Ή NMR (400 MHz, CDCh) δ 8.52 (s, IH),
-261-4820V.1
8.02 (dd, J =8 Hz, 1H), 7.38 (d, J = 8 Hz, 1H), 3.32 (s, 3H), 3.12-3.08 (br.s, 4H), 2.67-2.62 (br.s, 4H), 2.38 (s, 3H), 1.33 (s, 12H), ESI 381.1.[M + H]+, calcd for [C18H29BN2O4S + H]+ 381.2
C. N-(3-(4-(4-methylpiperazin-l-yl)-3-methylsulfonyl) phenyl)-! H-indazol-5-yl)-2-(pyrrolidin-l - yl)-2-(thiophen-3-yl) acetamide trifluoroacetate
The title compound was synthesized according to the General Method C utilizing N-(3- iodo-lH-indazol-5-yl)-2-(pyrrolidin-l-yl)-2-(thiophene-3-yl)acetarnide (75 mg, 0.165 mmol), l-methyl-4-(2-(methylsulfonyl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl) phenyl) piperazine (76 mg, 0.199 mmol), 1M Na2C03 (0.33 mL, 0.33 mmol), PhMe (2.25 mL), EtOH (1.12 mL) and Pd(PPh3)4 (10 mg, 0.008 mmol).The degassed solution was sealed and heated in a microwave reactor at 125°C for 2 h. The reaction was taken into EtOAc (25 mL), washed with H20 (10 mL * 2), brine (10 mL) and dried over anh Na2S04 and concentrated under reduced pressure. Most of the impurities were removed by purification using BiotageSNAP 25 g silica column (gradient 0-35% MeOH in DCM) and finally purified by reverse phase column chromatography (Biotage Ci8, 60 g, gradient 90: 10-20:80 % 0.1 % TFA- H20 : MeOH) to give the title compound white solid as a trifluoroacetate salt (52 mg, 49 %).Ή NMR (400 MHz, CD3OD) δ 8.63 (d, J =2.0 Hz, 1H), 8.43 (d, J =0.8 Hz, 1H), 8.32 (dd, J = 8.3, 2.3 Hz, 1H), 7.89- 7.88 (m, 1H), 7.73 (d, J =8.4 Hz, 1H), 7.66-7.64 (m, 1H), 7.60-7.53 (m, 2H), 7.39 (d, J = 1.2, 1H), 5.27 (s, 1H), 3.91-3.88 (br.m, 1H), 3.68 (d, J =12 Hz , 2H), 3.63 (d, J =1 1.6 Hz , 2H), 3.55- 3.41 (m, 5H), 3.28-3.13 (br.m, 2H), 3.01 (s, 3H), 2.18-2.13 (br.m, 3H), 2.01 (br.s, 1H), 2H merged with solvent peak, MS ESI 579.2. [M + H]+, calcd for [C29H34N6O3S2 + H]+ 579.2.
Example A236. 2-(cvclopentyloxy)-N-(3-(3-methylsulfonyl) phenyl)- lH-indazol-5-yl)-2-phenyl acetamide

-262-4820V.1
The title compound was synthesized according to the General Method A, utilizing 2- (Cyclopentyloxy)-2-phenylacetic (40 mg, 0.181 mmol), 3-(3-(methylsulfonyl)phenyl)-lH- indazole-5-amine (52 mg, 0.181 mmol), N-ethyl-N-isopropyl propan-2-amine (0.16 mL, 0. 908 mmol) and TBTU (58 mg, 0.181 mmol) in DMF (1.2 mL). The reaction mixture was purified using Biotage SNAP 25 g silica column (gradient 0- 10% MeOH in DCM) and repurified by Reverse phase column chromatography (Biotage Ci8, 60 g, gradient 90: 10-20:80 % 0.1 % TFA- H20: MeOH) to give the title compound as a light brown solid (7.5 mg, 8.4 %).!H NMR (400 MHz, CD3OD) δ 8.50 (d, J = 1.6 Hz, IH), 8.38 (s, IH), 8.30 (d, J =8.0 Hz, IH), 7.99 (d, J =7.6 Hz, IH), 7.78 (t, J =7.6 Hz, IH), 7.61 -7.54 (m, 4H), 7.41-7.32 (m, 3H), 5.00 (s, I H), 4.13 (br.m, IH), 3.21 (s, 3H), 1.92- 1.76 (m, 6H), 1.61 (br.t, J = 10.4 Hz , 2H); MS ESI 490.4. [M + H]+, calcd for [C27H27N3O4S + H]+ 490.1.

A.2-(Piperidin-J-yl)-2-(thiophen-3-yl) acetic acid
The title compound was synthesized according the General Method D using Glyoxylic acid monohydrate ( 1.8 g, 0.019 mmol) and piperidine (1.93 mL, 0.019 mmol) was combined with CH2CI2 (75 mL) and sonicated for 15 minutes. Thiophene-3-boronic acid (2.5 g, 0.019 mmol) was added and the mixture was stirred at room temperature for 24 h. after that concentrated the reaction mass under reduced pressure completely and purified by Biotage SNAP 50 g silica column (gradient 0-30% MeOH in DCM) gave the title compound as a cream solid (3.4 g, 77%). Ή NMR (400 MHz, CD3OD) δ 7.58 (s, IH), 7.33 (s, 2H), 4.70 (s, I H), 3.71-3.63 (br.m, 2H), 3.17 (br.s, 2H), 1.94- 1.93 (br.m, 2H), 1.83- 1.80 (br.m, 2H), 1.55-1.53 (br.s, 2H); MS ESI 226 [M + H]+, calcd for [Q ,H15N02S+ H]+ 226.1.
-263-4820V.1
B. N-(3-(3-methylsulfonyl) phenyl)- lH-indazol-5-yl)-2-(piperidin-l-yl)-2-(thiophen-3-yl) acetamide trifluoroacetate
The title compound was synthesized according to the General Method A, utilizing 2- (Piperidin-l-yl)-2-(thiophen-3-yl)acetic acid (40mg, 0.177 mmol), 3-(3-(methylsulfonyl)phenyl)- lH-indazole-5 -amine (51 mg, 0.177 mmol), N-ethyl-N-isopropyl propan-2-amine (0.15 mL, 0.886 mmol) and TBTU (57 mg, 0.177 mmol) in DMF (1.2 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0-10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Cig, 60 g, gradient 90: 10-20:80 % 0.1% TFA- H20: MeOH) to give the title compound as a light pink solid (20 mg, 17.5 %).Ή NMR (400 MHz, CD3OD) 8 8.51 (s, IH), 8.47 (s, IH), 8.30 (d, J =7.6 Hz, IH), 8.01 (d, J =8.4 Hz, IH), 7.87 (s, IH), 7.80 (t, J =7.6 Hz, 1Η),7.68-7.66 (m, IH), 7.60 (d, J =8.8 Hz, IH), 7.54 (d, J =8.8 Hz, IH), 7.38 (d, J =4.4 Hz, IH), 5.17 (s, IH), 3.80 (br.m,lH), 3.22 (s, 3H), 3.16-3.13 (m, 2H), 2.95 (br.m,lH), 2.00-1.82 (br.m,6H); MS ESI 495.3. [M + H]+, calcd for [C25H26N403S2 + H]+ 495.1.
Example A238. 2-methoxy-N-(3-(3-methylsulfonyl) phenyl)-lH-indazol-5-yl)-2-(thiophen-2-yl) acetamide

The title compound was synthesized according to the General Method A, utilizing 2- methoxy)-2-(thiophen-2-yl)acetic acid (40 mg, 0.232 mmol), 3-(3-(methylsulfonyl)phenyl)-lH- indazole-5-amine (67 mg, 0.232 mmol), N-ethyl-N-isopropylpropan-2-amine (0.2 mL, 0.1.16 mmol) and TBTU (74 mg, 0.232 mmol) in DMF (1.2 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0-10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Ci8, 60 g, gradient 90: 10-20:80 % 0.1 % TFA- H20 : MeOH) to give the title compound as a light brown solid (17 mg, 16%). Ή NMR (400 MHz, CD3OD) δ 8.50 (s, IH), 8.39 (s, IH), 8.29 (d, / =7.2 Hz, IH), 7.98 (d, / =7.6 Hz,
-264-4820V.1
IH), 7.78 (t, J =7.6 Hz, IH), 7.65 (d, J =9.2 Hz, IH), 7.58 (d, / =8.8 Hz, 1H),7.45 (d, J =7.8 Hz, IH), 7.25 (d, J =3.2 Hz, IH), 7.04 (t, J =4.8 Hz, IH), 5.13 (s, IH), 3.50 (s, 3H), 3.21 (s, 3H), MS ESI 442.3.[M + H]+,calcd for [C21H19N3O4S2 + H]+ 442.1.

The title compound was synthesized according to the General Method A, utilizing
2- (pyrrolidin-l-yl)-2-(thiophen-2-yl) acetic acid (40 mg, 0.189 mmol),
3- (3-(methylsulfonyl)phenyl)- lH-indazole-5-amine (54 mg, 0.189 mmol),
N-ethyl-N-isopropylpropan-2-amine (83 uL, 0.62 mmol) and TBTU (61 mg, 0.19 mmol) in DMF (1.5 mL). The reaction mixture was purified using BiotageSNAP 25 g silica column (gradient 0- 10% MeOH in DCM) and repurified by reverse phase column chromatography (Biotage Ci8, 60 g, gradient 90: 10-20:80 % 0.1% TFA- H20 : MeOH) to give the title compound as a light brown solid (1 1 mg, 9.8%).Ή NMR (400 MHz, CD3OD) 5 8.51 (d, J =22.8 Hz, 2H), 8.30 (d, J =7.6 Hz, IH), 8.02 (d, J =6.8 Hz, I H), 7.82 (t, J =3.6 Hz, IH), 7.73 (d, J =9.2 Hz, IH), 7.62- 7.49 (m, 3H), 7.20 (m, 1H),5.43 (s, IH), 3.62 (s, 3H), 3.40-3.05 (m, 4H), 2.23-2.02 (m, 4H); MS ESI 481.3. [M + H]+, calcd for [C24H24N4O3S2 + Hf 481.1.
Example A240. 2-Cvclopentyl-N-(3-(3-methylsulfonyl) phenyl)- lH-indazol-5-yl)-2- phenylacetamide
-265-4820V.1
The title compound was synthesized according to the General Method A, utilizing alpha- Phenylcyclopentylacetic acid (20 mg, 0.099 mmol), 3-(3-(methylsulfonyl)phenyl) -1H- indazole-5-amine 2,2,2-trifluoroacetate (40 mg, 0.099 mmol), N-ethyl-N-isopropylpropan-2- amine (0.05 mL, 0.29 mmol) and TBTU (32 mg, 0.099 mmol) in DMF (1.2 mL). The reaction mixture was purified by preparative HPLC to give title compound as a white solid (13 mg, 27%). Ή NMR (400 MHz, CD3OD) δ 8.54 (d, / =20.4 Hz, 1H), 8.35-815 (m, 2H), 7.99-7.76_(m, 2H), 7.57-7.47 (m, 4H), 7.34-7.31 (m, 2H), 7.26-7.20 (m, 1H), 5.38 (s, 1H), 3.41 (d, J =11.2 Hz, 1H), 3.21 (s, 3H),1.70-1.07 (br.m, 8H); MS ESI 474.4. [M + H]+, calcd for [C27H27N3O3S + H]+ 474.1.
Example A241. Ai-(3-(3-methylsulfonyl) phenyl)- lH-indazol-5-yl)-2-(4-methylthiophen-3-yl)-2- (pyrrolidin-l-yl) acetamide trifluoroacetate

A. 2-(4-methylthiophen-3-yl)-2-(pyrrolidin-l-yl) acetic acid trifluoroacetate
The title compound was synthesized according the General Method D Glyoxylic acid monohydrate (130 mg, 1.41 mmol) and pyrrolidine (167 μί, 1.41 mmol) was combined with CH2CI2 (10 mL) and 4-Methylthiophene-3-boronic acid (200 mg, 1.41 mmol) was added and the mixture was stirred at room temperature for 24 h. after that concentrated the reaction mass under reduced pressure and purified by reverse phase preparative HPLC to give trifluoroacetate salt (229 mg, 48%). Ή NMR (400 MHz, CD3OD) δ 7.67 (s, 1H), 7.23 (s, 1H), 5.15 (s, 1H), 3.25 (br.s, 4H), 2.37 (s, 3H), 2.10 (br.s, 4H);MS ESI 226.0 [M + H]+,calcd for [CuHi5N02S+ H]+ 226.0.
-266-4820V.1
B. N-(3-(3-methylsulfonyl) phenyl)-lH-indazol-5-yl)-2-(4-methylthiophen-3-yl)-2-(pyrrolidin-l- yl) acetamide trifluoroacetate
The title compound was synthesized according to the General Method A, utilizing 2-(4- methylthiophen-3-yl)-2-(pyrrolidin-l-yl) acetic acid trifluoroacetate (42 mg, 0.124 mmol), 3-(3- (methylsulfonyl)phenyl)-lH-indazole-5-amine 2,2,2-trifluoroacetate (50 mg, 0.124 mmol), N- ethyl-N-isopropylpropan-2-amine (0.1 1 mL, 0.62 mmol) and TBTU (40 mg, 0.124 mmol) in DMF (1.5 mL). The reaction mixture was purified by preparative HPLC to give title compound as a white solid (50 mg, 66%).Ή NMR (400 MHz, CD3OD) δ 8.51 (s, 1H), 8.45 (s, 1H), 8.28 (d, J =7.6 Hz, 1H), 8.00 (d, J =7.6 Hz, 1H), 7.88 (s, 1H ), 7.78 (t, J =7.6 Hz, 1H), 7.58 (t, J =10.8 Hz, 2H), 7.28 (s, 1H ), 5.23 (s, 1H), 3.38-3.36 (m, 2H), 3.21 (s, 3H), 3.18-3.11 (m, 2H), 2.48 (s, 3H), 2.26-2.00 (br.m, 4H); MS ESI 495.3. [M + H]+, calcd for [C25H26N4O3S2 + H]+ 495.1.
Example A242. N-(3-(3-(methylsulfonyl)phenvn- lH-indazol-5-yl)-2-(pyrrolidin- l-yl)-2- (thiophen-3-yl)acetamide trifluoroacetate

To a mixture of N-(3-iodo-lH-indazol-5-yl)-2-(pyrrolidin-l-yl)-2-(thiophen-3- yl)acetamide trifluoroacetate (290 mg, 0.51 mmol) and (3-(methylsulfonyl)phenyl)boronic acid (120 mg, 0.6 mmol) in PhCH3/EtOH (3 mL/9 mL) in a 20 mL microwave vial was added 1 M Na2C03 (1.5 mL, 1.5 mmol), followed by Pd(PPh3)4 (1 1.6 mg, 0.01 mmol, 2 mol%). The resulting mixture was purged with Ar, then microwaved 1 h at 125 °C. It was diluted with ¾0 (30 mL) and extracted with EtOAc (30 mL x 2). Combined extracts were concentrated and purified by prep-HPLC three times to give the title compound as a light brown solid (176 mg, 58%). Ή NMR (400 MHz, CDjOD) 5 8.44 (s, 1H), 8.38 (s, 1H), 8.17 (d, J = 7.6 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.87 (s, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.62-7.57 (m, 1H), 7.51 (s, 2H), 7.39
-267-4820V.1
(d, J = 5.2 Hz, 1H), 5.34 (s, 1H), 3.92-3.88 (s, br, 1H), 3.36-3.00 (m, 6H), 2.25-1.90 (m, 4H; including s, 3H at 3.17); MS ESI. [M + H]+ 481.3, calcd for [C24H24N4O3S2 + H]+ 481.1.

To a mixture of 4-bromo-2-(methylsulfonyl)benzaldehyde (1.315 g, 5 mmol) and dimethylamine (2 M in THF, 4 mL, 8 mmol) in DCE (20 mL) was added HOAc (0.2 mL), followed by NaBH(OAc)3 (1.378 g, 6.5 mmol). The resulting mixture was stirred for 2 h at it. After quenching with satd. NaHC03 (5 mL), H20 (10 mL), it was extracted with DCM (20 mL x 2) and dried (Na2S04). Removal of solvents gave crude l-(4-bromo-2-(methylsulfonyl)phenyl)- Ν,Ν-dimethylmethanamine as a yellow solid (1.421 g, 97%). Ή NMR (400 MHz, CDC ) δ 8.15 (s, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 3.74 (s, 2H), 3.32 (s, 3H), 2.18 (s, 6H).
To a mixture of Bis(pinacolato)diboron (279 mg, 1.1 mmol), l-(4-bromo-2- (methylsulfonyl)phenyl)-N,N-dimethylmethanamine (292 mg, 1 mmol) and KOAc (294 mg, 3 mmol) in DMSO (10 mL) was added Pd(dppf)Cl2-DCM (41 mg, 0.05 mmol, 5 mol%). The resulting mixture was microwaved 2 h at 90 °C. LC-MS showed major product being homo- coupling of bromide and minor product being the desired boronic acid. Redid this reaction on 0.5 mmol scale at 80 °C for 2 h and 0.2 mmol scale overnight at it. LC-MS showed all three were similar. All three reactions were combined, diluted with H20 (50 mL), extracted with EtOAc (30 mL x 3), washed with H20 (30 mL x 2), brine (20 mL) and dried over Na2S04. Removal of solvents gave crude (4-((dimethylamino)methyl)-3-(methylsulfonyl)phenyl)boronic acid as a brown solid with major contamination being the homo-coupled byproduct.
To a mixture of above crude boronic acid and N-(3-iodo-lH-indazol-5-yl)-2-(o- tolyl)acetamide (156.4 mg, 0.4 mmol) in PhCH3/EtOH (4 mL/8 mL) in a 20 mL microwave vial
-268-4820V.1
was added 1 M Na2C03 (1 mL, 1 mmol), followed by Pd(PPh3)4 (23 mg, 0.02 mmol). The resulting mixture was purged with Ar, then microwaved 1 h at 125 °C. After diluting with H20 (20 mL), it was extracted with EtOAc (30 mL x 2), concentrated and passed through microfilter using DMF and then purified by prep-HPLC twice to give the title compound as a light brown solid (18 mg, 8% based on iodide). Ή NMR (400 MHz, CD}OD) δ 8.74 (s, 1H), 8.55 (s, 1H), 8.39 (d, J = 8.0 Hz, 1H), 8.66 (d, J = 7.6 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 9.2 Hz, 1 H), 7.32-7.27 (m, 1H), 7.24-7.15 (m, 3H), 4.92 (s, 2H), 3.81 (s, 2H), 3.35 (s, 3H), 2.96 (s, 6H), 2.39 (s, 3H); MS ESI. [M + H]+ 477.3, calcd for [C26H28N403S + H]+ 477.2. Example A244 N-(3-(3-ethylsulfonyl) phenyl)-lH-indazol-5-yl)-2-(pyrrolidin- l-yl)-2-(thiophen- 3-yl) acetamide trifluoroacetat

The title compound was synthesized according to the General Method C utilizing N-(3- iodo- 1 H-indazol-5-yl)-2-(pyrrolidin- 1 -yl)-2-(thiophene-3-yl)acetamide (40 mg, 0.088 mmol), 3-Ethylsulfonylphenylboronic acid (24 mg, 0.1 1 mmol), 1M Na2C03 (0.27 mL, 0.26 mmol), PhMe (1.5 mL), EtOH (1.5 mL) and Pd(PPh3)4 (10 mg, 0.008 mmol). The degassed solution was sealed and heated in a microwave reactor at 125°C for 2 h. The reaction was diluted using MeOH, filtered it and concentrated under reduced pressure to dryness. Crude product was purified by preparative HPLC provided the title compound to as a white powder (23 mg, 34 %).'H NMR (400 MHz, CD3OD) δ 8.46 (s, 2H), 8.31 (d, J =7.6 Hz, 1H), 8.14 (d, J =6.8 Hz, 1H), 7.97 (d, J =7.6 Hz, 1H), 7.90 (s, 1H ), 7.80 (t, J =7.2 Hz, 1H), 7.66 (d, J =9.2 Hz, 1H), 7.54 (d, / =9.2 Hz, 1H), 7.39 (d, J =4.8 Hz, 1H), 5.24 (s, 1H),3.89 (br.s, 1H), 3.22-3.09 (m, 2H), 2.21- 1.99 (br.m, 4H), 1.30 (t, J =8.0 Hz, 3H), 3H merged with solvent peak, MS ESI 495.3. [M + H]+, calcd for [C25H26N403S2 + H]+ 495.1.
-269- 820v.1
ExampleA245. N-(3-(3-((3-(dimethylamino')propyl sulfonyl)phenyl)-lH-indazol-5-yl)-2-(o- tolvDacetamide

A. N,N-dimethyl-3-( ( 3-( trimethylstannyl)phenyl)sulfonyl)propan-l -amine

Hexamethyl ditin (0.07 mL, 0.33 mmol) in toluene (2.5 mL) was added to 3-((3- bromophenyl)sulfonyl)-N,N-dimethylpropan-l -amine (82 mg, 0.27 mmol) under argon bubbling. Pd(PPh3)4 (21.8 mg, 0.019 mmol) was added, and the vial was capped and heated at 110 °C for 2 h. The reaction mixture was cooled to rt and 3-((3-bromophenyl)sulfonyl)-N,N-dimethylpropan- 1-amine (129.4 mg, 0.27 mmol) and Pd(PPh3)4 (19.5 mg, 0.017 mmol) were added under argon bubbling, followed by DMF (1.25 mL). Aqueous KF (1M, 1 mL) and EtOAc (15 mL) were added and the mixture was stirred for 30 min, then filtered under vacuum, rinsing with EtOAc (20 mL). The solvent was removed in vacuo, and the residue was dissolved in MeOH and passed through a preconditioned 20 mL PoraPak Rxn Cx cartridge (Waters). The methanol (30 mL) rinse was discarded and the product was eluted with 2 M ammonia in methanol (30 mL).
Evaporation and purification by column (Silicycle 5g SPE, silica, 0-10% 2 M NH3-MeOH in DCM) gave the title compound (74.5 mg, 71%) along with a small amount of impure coupling product. Ή NMR (400 MHz, CDCl3) δ ppm 8.00 (m, 1 H), 7.83 (d, J=7.8 Hz, 1 H), 7.75 (d, J=7.3 Hz, 1 H), 7.50 (t, J=7.4 Hz, 1 H), 3.16 (m, 2 H), 2.32 (m, 2 H), 2.15 (s, 6 H), 1.90 (m, 2 H), 1.67 (br. s., 2 H), 0.35 (m, 9 H); MS ESI 392.0 [M + H]+, calcd for [C^sNOaSSn + H]+ 392.2.
-270-4820V.1
B. N-(3-(3-(( 3-(dimethylamino )propyl )sulfonyl)phenyl)-lH-indazol-5-yl)-2-( o-tolyl)acetamide

DMF (2.5 mL) was added under argon to a mixture of N-(3-iodo-l-(tetrahydro-2H-pyran- 2-yl)-lH-indazol-5-yl)-2-(o-tolyl)acetamide (29.0 mg, 0.074 mmol), N,N-dimethyl-3-((3- (trimethylstannyl)phenyl)sulfonyl)propan- 1 -amine (34.9mg, 0.073 mmol) and Pd(PPh3)4 (1 1.5 mg, 0.0099 mmol) and the resulting mixture was heated in a sealed vial in a 135 °C reaction block for 22 h. The mixture was cooled to rt and MeOH (5 mL) was added and the mixture was poured into a preconditioned 20 mL PoraPak Rxn Cx cartridge (Waters). The methanol (30 mL) rinse was discarded and the product was eluted with 2 M ammonia in methanol (30 mL). The solvent was removed in vacuo, and the residue was purified by column (Silicycle 5g SPE, silica, 0-5% MeOH in DCM) to provide N-(3-(3-((3-(dimethylamino)propyl)sulfonyl)phenyl)-l- (tetrahydro-2H-pyran-2-yl)-lH-indazol-5-yl)-2-(o-tolyl)acetamide (26.6 mg, 63 %). MS ESI 575.4 [M + H]+, calcd for [C32H38N4O4S + H]+ 575.3.
A solution of N-(3-(3-((3-(dimethylamino)propyl)sulfonyl)phenyl)-l-(tetrahydro-2H- pyran-2-yl)- 1 H-indazol-5-yl)-2-(o-tolyl)acetamide (26mg, 0.045 mmol) and 4-toluenesulfonic acid monohydrate (39.3 mg, 0.21 mmol) in MeOH (3 mL) was heated in a microwave reactor at 100 °C for 15 min. The mixture was cooled to rt and the solvent was evaporated. EtOAc (5 mL) and NaHC03(aq) (3 mL) were added and the resulting solution was poured into a Varian
ChemElut cartridge (Hydromatrix, 3 mL) and rinsed with EtOAc (5mL x 5). The resulting cloudy extracts were passed through a second ChemElut cartridge (Hydromatrix, 3 mL). The solvent was removed in vacuo, and the residue was purified by column (Silicycle 5g SPE, silica, 5-8% MeOH in DCM) and triturated with a mixture of EtOAc / Et20 / hexane (2 mL) to provide the title compound as an off-white solid (12.0 mg, 72 %). Ή NMR (400 MHz, CD3OD) 5 ppm 8.47 (s, 1 H), 8.41 (s, 1 H), 8.29 (d, J=7.3 Hz, 1 H), 7.94 (d, J=7.8 Hz, 1 H), 7.78 (t, J=7.8 Hz, 1 H), 7.55 (m, 2 H), 7.28 (m, 1 H), 7.16 (m, 3 H), 3.78 (s, 2 H), 3.34 (m, 2 H), 2.41 (m, 2 H), 2.38
-271-4820v.1
(s, 3 H), 2.17 (s, 6 H), 1.90 (m, 2 H); MS ESI 491.4 [M + H]+, calcd for [C27H30N4O3S + H]+ 491.2.

DMF (2.5 mL) was added under argon to a mixture of N-(3-iodo-lH-indazol-5-yl)-2- (pyrrolidin-l-yl)-2-(thiophen-3-yl)acetamide (53.6 mg, 0.119 mmol), N,N-dimethyl-3-((3- (trimethylstannyl)phenyl)sulfonyl)propan-l-amine (45.6mg, 0.117 mmol) and Pd(PPb.3)4 (16.0 mg, 0.014 mmol) and the resulting mixture was heated in a sealed vial in a 135 °C reaction block for 22 h. The mixture was cooled to rt and MeOH (5 mL) was added and the mixture was poured into a preconditioned 20 mL PoraPak Rxn Cx cartridge (Waters). The methanol (30 mL) rinse was discarded and the product was eluted with 2 M ammonia in methanol (30 mL). The solvent was removed in vacuo, and the residue was purified by column (Silicycle 5g SPE, silica, 0-15% MeOH in DCM), by on reverse phase purification (Biotage Isolera, 60g Rp-C18-HS plus samplet, 0.1%TFA-H2O in MeOH, gradient 10-80%). Conversion to the free base using another PoraPak Rxn Cx cartridge (Waters) as described above, and a second column (Silicycle 5g SPE, silica, 3-5% 2 M NH3-MeOH in DCM) provided the title compound as an off-white solid (13.1 mg, 20 %). Ή NMR (400 MHz, CD3OD) δ ppm 8.48 (s, 1 H), 8.38 (s, 1 H), 8.30 (d, J=7.3 Hz, 1 H), 7.95 (d, J=8.0 Hz, 1 H), 7.79 (t, J=7.9 Hz, 1 H), 7.57 (m, 2 H), 7.51 (br. s., 1 H), 7.41 (m, 1 H), 7.34 (d, J=4.8 Hz, 1 H), 4.13 (s, 1 H), 3.34 (m, 2 H), 2.68 (m, 2 H), 2.52 (m, 2 H), 2.40 (t, J=7.4 Hz, 2 H), 2.17 (s, 6 H), 1.90 (m, 2 H), 1.85 (m, 4 H); MS ESI 552.1 [M + H]+, calcd for [C28H33N5O3S2 + H]+ 552.2.
-272-4820V.1
Example B: TTK Inhibition Assay
Active TTK was purchased from Invitrogen as an amino terminal GST fusion of full length human TTK. Amino terminal 6 histidine, sumo tagged human TTK (residues 1-275) was expressed in E. coli, and purified to >95% homogeneity by Ni2+ agarose, gel filtration, and ion exchange chromatography.
TTK activity was measured using an indirect ELISA detection system. GST-TTK (0.15 nM) was incubated in the presence of 3 μΜ ATP (Sigma cat# A7699), 50mM Hepes pH 7.5, ImM EGTA, lOmM MgCl2, and 0.1% Pluronic in a 96 well microtitre plate pre-coated with amino terminal 6 histidine, sumo tagged TTK (amino acid residues 1-275). The reaction was allowed to proceed for 30 minutes, followed by 5 washes of the plate with Wash Buffer
(phosphate buffered saline supplemented with 0.2% Tween 20), and incubation for 30 minutes with a 1:3000 dilution of primary antibody (Cell Signaling cat# 9381). The plate was washed 5 times with Wash Buffer, incubated for 30 minutes in the presence of secondary antibody coupled to horse radish peroxidase (BioRad cat# 1721019, 1 :3000 concentration), washed an additional 5 times with Wash Buffer, and incubated in the presence of TMB substrate (Sigma cat# T0440). The colourimetric reaction was allowed to continue for 5 minutes, followed by addition of stop solution (0.5 N sulphuric acid), and quantified by detection at 450 nm with either a
monochromatic or filter based plate reader (Molecular Devices M5 or Beckman DTX880, respectively).
Compound inhibition was determined at either a fixed concentration (10 μΜ) or at a variable inhibitor concentration (typically 50 μΜ to 0.1 μΜ in a 10 point dose response titration). Compounds were pre-incubated in the presence of enzyme for 15 minutes prior to addition of ATP and the activity remaining quantified using the above described activity assay. The % Inhibition of a compound was determined using the following formula; % Inhibition = 100 x (1 - (experimental value - background value)/(high activity control - background value)). The IC50 value was determined using a non-linear 4 point logistic curve fit (XLfit4, IDBS) with the formula; (A+(B/( l+((x/C)AD)))), where A = background value, B = range, C = inflection point, D = curve fit parameter.
-273-4820V.1
Example C: PLK4 Inhibition Assay
Active PLK4 was purified from an E. coli expression system as an amino terminal GST fusion of residues 1-3 1 of human PLK4. The protein was purified from clarified cell extracts after induction at 15 °C overnight using glutathione sepharose, gel permeation chromatography, and ion exchange (Resource Q). The resulting protein was dephosphorylated with lambda phosphatase (NEB cat# P0753), and resolved from the phosphatase using gluthione sepharose. The dephosphorylated GST-PLK4 was stored in aliquots at -80°C until use.
PLK4 activity was measured using an indirect ELISA detection system.
Dephosphorylated GST-PLK4 (4 nM) was incubated in the presence of 15 μΜ ATP (Sigma cat# A7699), 50 mM HEPES-Na2+pH 7.4, 10 mM MgCl2, 0.01% Brij 35 (Sigma cat# 03-3170), in a 96 well microtitre plate pre-coated with MBP (Millipore cat# 30-01 1). The reaction was allowed to proceed for 30 minutes, followed by 5 washes of the plate with Wash Buffer (50 mM TRIS-Cl pH 7.4 and 0.2% Tween 20), and incubation for 30 minutes with a 1 :3000 dilution of primary antibody (Cell Signaling cat# 9381). The plate was washed 5 times with Wash Buffer, incubated for 30 minutes in the presence of secondary antibody coupled to horse radish peroxidase (BioRad cat# 1721019, 1 :3000 concentration), washed an additional 5 times with Wash Buffer, and incubated in the presence of TMB substrate (Sigma cat# T0440). The colourimetric reaction was allowed to continue for 5 minutes, followed by addition of stop solution (0.5 N sulphuric acid), and quantified by detection at 450 nm with either a monochromatic or filter based plate reader (Molecular Devices M5 or Beckman DTX880, respectively).
Compound inhibition was determined at either a fixed concentration (10 μΜ) or at a variable inhibitor concentration (typically 50 μΜ to 0.1 μΜ in a 10 point dose response titration). Compounds were pre-incubated in the presence of enzyme for 15 minutes prior to addition of ATP and the activity remaining quantified using the above described activity assay. The % Inhibition of a compound was determined using the following formula; % Inhibition = 100 x (1 - (experimental value - background value)/(high activity control - background value)). The IC5o value was determined using a non-linear 4 point logistic curve fit (XLfit4, IDBS) with the formula; (A+(B/(l+((x/C)AD)))), where A = background value, B = range, C = inflection point, D = curve fit parameter.
-274-4820V.1
Example D: Aurora A Inhibition Assay
Aurora A inhibition was determined using the Z-Lyte assay kit from Invitrogen. The assay was performed using the recommended manufacturer's instructions with 20 μΜ ATP and 12 nM Aurora A (Invitrogen cat # PV3612). The % inhibition values were determined according to the manufacturer's directions and IC50 values were obtained using a non-linear 4 point logistic curve fit (XLfit4, IDBS)
Example E: Aurora B Inhibition Assay
Aurora B inhibition was determined using the Z-Lyte assay kit from Invitrogen. The assay was performed using the recommended manufacturer's instructions with 128 μΜ ATP and 28 nM Aurora B (Invitrogen cat # PV3970). The % inhibition values were determined according to the manufacturer's directions and IC50 values were obtained using a non-linear 4 point logistic curve fit (XLfit4, IDBS)
In Table 5 , IC50 values for PLK4, PLK1 , TTK, Aurora A and Aurora B Kinases are indicated as "A," "B," and "C," for those less than or equal to 5 μΜ; those greater than 5 μΜ and less than or equal to 50 μΜ; and those greater than 50 μΜ, respectively. As shown in Table 5, numerous compounds described herein are effective TTK inhibitors. In addition, a number of compounds described herein also inhibit other kinases, including for instance, PLK4, Aurora A and B kinases.
Example F: Chk2 Inhibition Assay
CHK2 inhibition was determined using the Z-Lyte assay kit from Invitrogen (cat# PV3180). The assay was performed using the recommended manufacturer's instructions with 180 μΜ ATP and 7.6 nM CHK2 (Invitrogen cat # PV3367). The % inhibition values were determined according to the manufacturer's directions and IC50 values were obtained using a non-linear 4 point logistic curve fit (XLfit4, IDBS)
-275-4820V.1
Table 5: Inhibition Data of PLK4, TTK, Aurora A and Aurora B Kinases

-276-4820V.1
A37 A
A38 A
A39 A
A40 A A
A41 A
A42 A
A43 A
A44 A
A45 A
A46 A
A47 A
A48 A
A49 A A
A50 A A
A51 A A
A52 A A A A
A53 A A A A
A54 A A
A55 A A
A56 A A C
A57 A A
A58 A A A A
A59 A A A
A60 A A
A61 A A
A62 A A
A63 A A A
A64 A B
A65 A A A
A66 A A
A67 A A
A68 A A A
A69 A A
A70 A A
A71 A A
A72 A A
A73 A A
A74 A A
A75 A A
A76 A A A A A
A77 A A
A78 A A
A79 A A A B A
A80 A A
A81 A B A A A
A82 A A
A83 A A A
A84 A A
A85 A A
A86 A A B
A87 B A
A88 B A
A89 A A
A90 A A
A91 A A A A A
A92 A A B
A93 A A
A94 A A
A95 A
A96 A A
A97 A A A A
A98 A A A A
A99 A C
A100 A A A A
A101 A A A
A102 A A A
A 103 A A A A A
A104 A A A A
A105 A A A A A
A106 A A A A
A107 A A
A108 A A A
A109 A A A A A
A110 A A
Al 11 A A A A A
A112 A A
A1 13 A
A114 A A
A115 A A A
A116 A
A1 17 A A A
ΑΠ8 A
A119 A
A120 A A
A121 A A
A122
A123 A A
A124 A A A
A125 A A A A A
A126 A A A
A127 A A A
A128 A A
A129 A A
A130 A A
A131 A A
A132 A A A B
A133 B B
A134 A A A
A135 A A
A136 A A
A137 A A
A138 A A
A139 A A
A140 A
A141 A
A142 A
A143 A
A144 A
A 145 A
A146 A
A147 A
A148 A C
A 149 A
A150 A C
A151 A
A152 A
A153 A
A154 A D
A155 A
A156 A
A157 A
A158 A
A159 A
A160 A
A161 A
A162 A
A163 A A
A164 A
A165 A
A166 A A
A167 A
A168 A A
A169 A A A A
A170 A A A A
A171 A A A A
A172 A A A A
A173 A A
A174 A A A B
A175 A A B B
A176 A A B
A177 A A
A178 A A
A179 A A A
A180 A A A A
A181 A A A A
A182 A A B B
A183 A A
A184 A A
A185 A A
A186 A A A
A187 A A A A
A188 A A A A
A189 A A A A
A190 A A A B
A191 A A A B
A192 A A
A193 A A B
A194 A A A A
A195 A A B C
A196 A C C
A197 A A B C
A198 A A A A
A199 A A
A200 A A A A
A201 A A A A
A202 A A A B
A203 A A B
A204 A A
A205 A A
A206 A A A A
A207 A A A A
A208 A A
A209 A A A A
A210 A A A A
A21 1 A A A A
A212 A A B A
A213 A A A A
A214 A A A A
A215 A A A B
A216 A A A A
A217 A A
A218 A A
A219 A A
A220 A A A B
A221 A A A A A
A222 A A A B
A223 A A B C
A224 A A C
A225 A A A B
A226 A B
A227 A A
A228 A A A A
A229 A A B
A230 A A A
A231 A A C
A232 A A B A
A233 A A A A
A234 A A A B
A235 A A B
A236 A A B
A237 A A A B
A238 A A A A
A239 A A A B
A240 A A
A241 A A A B
A242 A A A A
A243 A A B
A244 A A
A245 A A
A246 A A B B
A247 A A
In addition, TTK inhibition acitivity of indazole compounds represented by the following structural formula:

were tested according to the procedures described above and IC50 values are listed in Table 5a.
Table 5a

Example G: Cancer Cell Line Data on Exemplary Compounds of the Invention
Breast cancer cells (MCF-7, MDA-MB-468, HCC 1954), colon cancer cells (SW620) and lung cancer cells (A549), together with human mammary epithelial primary cells (HMEC), were seeded (1000 to 4000 per 80 μΐ per well depending on the cell growth rate) into 96 well plates,
-282-4820V.1
24 hours before compound overlay. Compounds were prepared as lOmM stock solutions in 100% DMSO which were diluted with DMEM (Dulbecco's Modified Eagle's Medium) cell growth Medium (Invitrogen, Burlington, ON, Canada) containing 10% FBS (Fetal Bovine Serum) to concentrations ranging from 50 nM to 250 μΜ. Aliquots (20 μΐ) from each concentration were overlaid to 80 μΐ of the pre-seeded cells in the 96 well plates to make final concentrations of 10 nM to 50 μΜ. The cells were cultured for 5 days before the Sulforhodamine B assay (SRB) was performed to determine the compound's cell growth inhibition activity.
Cancer cells including Melanoma cells 518- A2, A375, Prostate cancer cells PC3, DU- 145, Ovarian cancer cells S OV-3, OVCAR-3, Brain cancer cell A172, and Pancreatic cancer cell PANC-1 were seeded at various numbers, ranging from 3500 to 5000 per 80μ1 each well according to the cell growth rate, into 96 well plates, 24 hours before compound overlay.
Compounds were prepared in lOmM stock with 100% DMSO. The lOmM compounds stock was diluted with culture medium to 250μΜ. Different concentration titrations were made from 125nM to 250μΜ. 20μ1 compound dilutions were overlaid to cells to obtain the final concentration ranging from 25nM to 50μΜ. The cells were cultured for 5 days in the presence of compounds before SRB (Sulforhodamine B) assay was conducted to determine compounds cell growth inhibition activity.
Sulforhodamine B (purchased from Sigma, Oakville, ON, Canada) is a water-soluble dye that binds to the basic amino acids of the cellular proteins. Thus, colorimetric measurement of the bound dye provides an estimate of the total protein mass that is related to the cell number. The cells are fixed in situ by gently aspirating off the culture media and adding 50 μΐ ice cold 10% Trichloroacetic Acid (TCA) per well and incubate at 4 °C for 30-60 min, The plates are washed with water five times and allowed to air dry for 5 min. Addition of 50 μΐ 0.4%(w/v) SRB solution in 1 % (v/v) acetic acid to each well and incubatation for 30 min at rt completes the staining reaction. Following staining, plates are washed four times with 1 % acetic acid to remove unbound dye and then allowed to air dry for 5 min. The stain is solubilized with 100 μΐ of 10 mM Tris pH 10.5 per well. Absorbance is read at 570 nm.
The percentage (%) of relative growth inhibition was calculated by comparing to DMSO treated only cells (100%), which were growing in the presence of 0.1% DMSO (0.5% in the case
-283-4820V.1
of 50μΜ compounds). GI50's were determined for compounds with cytotoxic activity. The GI50 was calculated using GraphPad PRISM software (GraphPad Software, Inc., San Diego, CA, USA). GI50 (growth inhibition) is the compound concentration that causes 50% inhibition of cell growth.
In Table 6 below, GI50 value ranges for several compound examples against a luminal breast cancer cell line (MCF-7), two basal breast cancer cell line (MDA-MB-468, HCC1954), a lung cancer cell line (A549), a colon cancer cell line (SW-620), two melanoma cell lines (518- A2 and A375), two prostate cell lines (PC3 and DU-145), two ovarian cancer cell lines (SKOV- 3, OVCAR-3), a brain cancer cell line (A172) and a pancreatic cancer cell line (PANC- 1) and primary breast cells (HMEC) are given. The example compounds demonstrated varying growth inhibition/cell killing activity against cancer cells of luminal breast cancer and basal breast cancer cell, lung cancer and colon cancer. In general, these compounds showed less or little activity against normal cells as exemplified by HMEC. The GI50 ranges are indicated as "A," "B," "C," and "D," for values less than or equal to 5 μΜ; those greater than 5 μΜ and less than or equal to 20 μΜ; those greater than 20 μΜ and less than or equal to 50 μΜ; and those greater than 50 μΜ, respectively.
Table 6: Cell Growth Inhibition Data

A132 B A A A A ~ A A B A C A C B
A135 A A A A A ~ — — ~ — ~ - ~ ~
A189 D A B B A ~ — — — ~ - - - ~
A195 B A B A A ~ — — — ~ - - -
A213 A A A A A ~ — ~ - ~ ~ - -
A221 A A A A A ~ — - - ~ ~ ~ -
A222 A A A A A — — ~ ~ ~
A230 A A A A A ~ ~ - ~ - ~ ~ ~
A240 A B A A A ~ — — - ~ - ~ ~ ~
A242 A A A A A D A -- A
A246 A A A A A ~ ~ ~ ~ ~ ~ ~
Example H: Colon and Ovarian Cancer Tumor-Initiating Cell Data of Exemplary Compounds
Materials and Methods: Non-tissue or tissure cultured treated T-75 flask and 96-well plates were purchased from VWR. Vitamin B-27 supplement, MEM NEAA (minimum essential medium non essential amino acids), sodium pyruvate, L-glutamine, N2 supplement, penicillin- streptomycin and fungizone/amphotericin B were obtained from Invitrogen. Lipid mixture, heparin and EGF were purchased from Sigma; bFGF from BD Biosciences. Colon tumor initiating cells (TICs) were routinely maintained using non-tissue cultured treated T-75 flasks in DMEM:F12 medium containing 0.2XB-27 supplement, 4ug/ml heparin, 1XMEM NEAA, lXsodium pyruvate, 1 mM glutamine, 10 pg/ul bFGF, 20 pg/ul EGF, IX N2 supplement, lipid mixture, penicillin-streptomycin and fungizone/amphotericin B. Ovarian TICs were were routinely maintained using tissue cultured treated T-75 flasks in DMEM:F12 medium containing 1XB-27 supplement, 4ug/ml heparin, 20 pg/ul bFGF, 20 pg/ul EGF and penicillin-streptomycin.
Assay Protocol: Compounds described herein were dissolved in DMSO and further diluted in cell culture medium for GI50 determination. Colon TICs were trypsinized and seeded into non-tissue cultured treated 96-well plates with 4,000 cells/well. After 24 h, compound was added into the cell culture at different concentrations, and the final concentration of DMSO was adjusted to 0.1 %. Cells were then cultured at 37°C for 9 days. Ovarian TICs were trypsinized and seeded into tissue cultured treated 96-well plates with 1,000 cells/well. After 24 h,
-285-4820V.1
compound was added into the cell culture at different concentrations, and the final concentration of DMSO was adjusted to 0.1%. Cells were then cultured at 37°C for 6 days. Cell viability was assessed by Alamar Blue assay: 10 ul of Alamar Blue was added into each well. After 4 hours incubation at 37°C, fluorescence was recorded at excitation 544 and emission 590. GI5o (Growth inhibition) was calculated using GraphPad Prism 4.0 software. Cell growth inhibition data for compounds described herein is tabulated below (Table 7). The GI50 ranges are indicated as "A," "B," "C," and "D," for values less than or equal to 5 μΜ; those greater than 5 μΜ and less than or equal to 20 μΜ; those greater than 20 μΜ and less than or equal to 50 μΜ; and those greater than 50 μΜ, respectively.
Table 7: Colon Tumor-Initiating Cell Growth Inhibition Data

While this invention has been particularly shown and described with references to example embodiments thereof, it will be understood by those skilled in the art that various changes in form and details may be made therein without departing from the scope of the invention encompassed by the appended claims.
-286-4820V.1
Claims
WHAT IS CLAIMED IS:
1. A compound represented by the following structural formula:

or a pharmaceutically acceptable salt thereof, wherein:
each R1 is independently -H, -halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)jNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)R°, -C(=0)Rc, heterocycloalkyl or alkyl, wherein the heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -N02, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)jNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC and -C(=0)Rc;
each R2 is independently selected from: a)-H, -halogen, -CN, -N02, -(CH2)o- 2OR10, -(CH2)o.2NRuR12, -(CH2)o-2S(0)1R,°,
 -(CH2)0.
-(CH2)0.
2NR13S(0)iNR,4R15, -(ayo-aSCC iNR' 5, -(CH2)0-2C(=O)OR'°, -(CH2)0- 2OC(=0)OR10, -(CH2)o.2C(=S)OR10, -(CH2)0-2O(C=S)R10, -(CH2)o.2C(=0)NR14R15, -(CH2)o-2NRl3C(=0)Ri0, -(CH2)o.2C(=S)NR,4R15, -(CH2)0-2NR13C(=S)R10, -(CH2)0. 2NR13(C=O)OR!0, -(CH2)o-20(C=0)NR,4R15, -(CH2)0.2NR,3(C=S)OR10, -(CH2)0- 20(C=S)NR,4R15, -(CH2)0.2NR13(C=O)NR14R15, -(CH2)0.2NR13(C=S)NR,4R15, -(CH2)0. 2C(=S)R10, and -(CH2)0.2C(=O)R10; and b) alkyl, alkenyl, alkynyl, cycloalkyl,
-287-
ME1 1 1444820V.1
heterocycloalkyl, heterocycloalkylalkyl, heterocycloalkylalkenyl, aryl, arylalkyl, arylalkenyl, heteroaryl, heteroarylalkyl and heteroarylalkenyl, wherein each of the alkyl, alkenyl, alkynyl, cycloalkyl, heterocycloalkyl, heterocycloalkylalkyl,
heterocycloalkylalkenyl, aryl, arylalkyl, arylalkenyl, heteroaryl, heteroarylalkyl and heteroarylalkenyl groups represented by R2 is optionally substituted with 1 to 5
substituents independently selected from the group consisting of -halogen, -CN, -N02, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)jNReRf, C(=0)ORc, -OC(=0)ORc,
-C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl substituents on the groups represented by R2 are each optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, (Ci-C6)alkyl, halo(Ci-C6)alkyl, (C,-C3)alkoxy, halo(C,-C3)alkoxy, (C,-C3)alkoxy(C,-C6)alkyl and -NRdC(=0)Rc
R10 is -H, alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl or heteroaryl(Ci-C6)alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(CrC6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(Ci- C6)alkyl represented by R10 is optionally substituted with 1 to 5 substituents
independently selected from the group consisting of halogen, =0, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc,
-0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C6)alkyl, cycloalkyl, cycloalkyl(C,-C3)alkyl, heterocycloalkyl, heterocycloalkyl(CrC3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Cr C3)alkyl, and wherein each of the (Ci-Ce)alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(CrC3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C )alkyl substituents for the groups represented by R10 is optionally
substituted with halogen, -OH, -N02, -CN, (C,-C3)alkyl, halo(Ci-C3)alkyl, (Cp
C3)alkoxy(Ci-C3)alkyl, (C,-C3)alkoxy and halo(Ci-C3)alkoxy;
R11 and R12 are each independently -H, alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl or heteroaryl(Ci-C6)alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(C] -C6)alkyl, heteroaryl and heteroaryl(Ci-C6)alkyl represented by R11 and R12 is optionally substituted with 1 to 5 substituents independently selected from the group consisting of halogen, =0, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C6)alkyl, cycloalkyl, cycloalkyl(CrC3)alkyl, heterocycloalkyl, heterocycloalkyl (Ci-C3)alkyl, aryl, aryl(CpC3)alkyl, heteroaryl and heteroaryl(Ci- C3)alkyl, and wherein each of the (Ci-Ce)alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl substituents for the groups represented by R11 and R12 is optionally substituted with halogen, -N02, -CN, (CrC3)alkyl, halo(CrC3)alkyl, (Q- C3)alkoxy(Ci-C3)alkyl, (C,-C3)alkoxy and halo(C,-C3)alkoxy; or
R1 1 and R12, together with the nitrogen to which they are attached, form a 3-8 membered ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, =0, -CN, -0RC, -NRaRb, -S(0)iRc, -S(0)iNRaRb, -NRdS(0),Rc, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NRaRb, -NRdC(=0)Rc, -C(=S)NRaRb, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NRaRb, -NRd(C=S)ORc, -0(C=S)NRaRb, -NRd(C=0)NRaRb, -NRd(C=S)NRaRb, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein each of the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl substituents on the 3-8 membered ring is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, (Ci-C3)alkyl, halo(C,-C3)alkyl, (C,-C3)alkoxy(Ci-C3)alkyl, (Cr C3)alkoxy and halo(Ci-C3)alkoxy;
R13 is -H or an alkyl group optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy and (Ci-C3)alkoxy;
R14 is -H or an alkyl group optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy and (Ci-C3)alkoxy;
R15 is independently -H, alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl,
heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl or heteroaryl(Ci-C6)alkyl, wherein each of the alkyl, cycloalkyl, cycloalkyl(Ci-C6)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C6)alkyl, aryl, aryl(Ci-C6)alkyl, heteroaryl and heteroaryl(C] -C6)alkyl represented by R15 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, =0,
-C(=0)ORc, -0RC, -SRC, -C(=0)NRaRb, -C(=0)Rc, -SCO^R', -N02, -CN, -NRaRb, (Q- C6)alkyl, cycloalkyl, cycloalkyl(C] -C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci- C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci-C3)alkyl, and wherein each of the (C|-C6)alkyl, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl,
heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl and heteroaryl(Ci- C3)alkyl substituents for the groups represented by R15 is optionally substituted with halogen, -N02, -CN, (Ci-C3)alkyl, halo(C1-C3)alkyl, (C,-C3)alkoxy(Ci-C3)alkyl, (Ci- C3)alkoxy and halo(C]-C3)alkoxy; or
R14 and R15, together with the nitrogen to which they are attached, form a 3-8 membered ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, =0, -CN, -ORc, -NRaRb, -S(0)iRc, -S(0)jNRaRb, -NRdS(0)iRc, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NRaRb, -NRdC(=0)Rc, -C(=S)NRaRb, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NRaRb,
-NRd(C=S)ORc, -0(C=S)NRaRb, -NRd(C=0)NRaRb, -NRd(C=S)NRaRb, -C(=S)RC, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl or heteroaryl, wherein each of the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl substituents on the 3-8 membered ring is optionally substituted with 1 to 3 substituents independently selected from
halogen, -CN, -N02, (CrC3)alkyl, halo(CrC3)alkyl, (Ci-C3)alkoxy(Ci-C3)alkyl, (Ci- C3)alkoxy and halo(Ci-C3)alkoxy;
R16 is selected from -N¾ and alkyl, wherein the alkyl is optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, cycloalkyl(Ci- C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl, heteroaryl(C,-C3)alkyl, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf,
-NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC and -C(=0)Rc, wherein the cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl or heteroaryl(Ci-C3)alkyl are optionally substituted with 1 to 3 substituents independently selected from -halogen, alkyl, -ORc and -NRaRb;
Ra and Rb are each independently -H or (Ci-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, hydroxy, -NR8Rh and (C,-C3)alkoxy;
Rc is -H, or (Ci-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NR8Rh, hydroxy and (Ci- C3)alkoxy;
Rd is -H or (Ci-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NRgRh, hydroxy and (Ci- C3)alkoxy;
Re and Rf are each independently -H or (C)-C6)alkyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NR8Rh, hydroxy and (Ci-C3)alkoxy;
or Re and Rf, together with the nitrogen to which they are attached, form a 3-8 membered ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -NRgRh, -CN, (C,-C6)alkyl, halo(Ci-C6)alkyl, (Q- C3)alkoxy, halo(C,-C3)alkoxy, and (C|-C3)alkoxy(CrC6)alkyl;
Rg and Rh are each independently selected from the group consisting of -H, (Ci- C6)alkyl, halo(Ci-C6)alkyl, hydroxy(Ci-C6)alkyl and (Ci-C3)alkoxy(Ci-C6)alkyl;
i is 0, 1 or 2;
n is an integer from 1 to 4; and
m is an integer from 1 to 4;
provided that when R16 is alkyl, R2 is not -CN.
2. The compound of Claim by a structural formula:

or pharmaceutically acceptable salts thereof.
3. The compound of Claim 1 or Claim 2 wherein the compound is represented by a
structural formula selected from:

-292-
ME1 1 1444820V.1

X is a bond or -CR R5-, and for structural formula (II), when p is 0, X can additionally be -0-;
Y is a bond, -NR14-,-CR4R5- or -NR14-CR4R5-;
R3 is a cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R6;
R4 and R5 are each independently -H, alkyl, -ORc, -NRaRb,
-(C,-C3)alkylene-NRaRb, -(C,-C3)alkylene-ORc , -(C,-C3)alkylene-OH, cycloalklyl or heterocycloalkyl, provided that R4 and R5 are not both selected from -ORc and -NRaRb; or R3 and R5 together with the carbon atom to which they are attached form a cycloalkyl or a heterocycloalkyl, wherein the cycloalkyl or heterocycloalkyl is optionally substituted
with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Ci-C3)alkyl, halo(d-C3)alkyl, (C,-C3)alkoxy, halo(Ci-C3)alkoxy, and (C,-C3)alkoxy(C1-C3)alkyl;
each R6 is independently selected from the group consisting of halogen, -CN, -N02, =0, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C6)alkyl, aryl, aryl(Ci-C3)alkyl, heterocycloalkyl and heteroaryl; wherein each the (Ci-C6)alkyl, aryl, aryl(d-C3)alkyl, heterocycloalkyl and heteroaryl represented by R6 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, (Ci-C6)alkyl, halo(CrC6)alkyl, (Ci-C3)alkoxy, halo(C,-C3)alkoxy and (C1-C3)alkoxy(C)-C6)alkyl; and
p is 0, 1 or 2.
4. The compound of Claim 3 wherein the compound is represented by a structural formula selected from:

-294-E1 1 1444820V.1

5. The compound of Claim 3 or Claim 4, wherein the group represented by R3 is selected from (C3-Cg)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thiophenyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[<i]imidazolyl, benzo[i/]thiazolyl, benzo[b]thiophenyl, quinolinyl, tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, morpholinyl, indolinyl and indenyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -NO2, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (CrC3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (C!-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is
-295-E1 1 1444820V.1
optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (Ci-C3)alkyl, halo(C,-C3)alkyl, (C,-C3)alkoxy and (C,-C3)alkoxy(Ci- C3)alkyl.
The compound of Claim 5, wherein the group represented by R3 is selected from the group consisting of phenyl, thiophenyl, pyridinyl, pyrazolyl, cyclopentyl,
tetrahydropyranyl, indenyl, tetrahydronaphthalenyl and indolinyl, each of which is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (d- C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Cj-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (C]-C3)alkyl, halo(Ci-C3)alkyl, (C,-C )alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl.
The compound of any one of Claims 4-6, the group represented by R3 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -ORc, (CrC3)alkyl, -(C,-C3)alkylene-NRaRb, phenyl, pyrimidinyl, morpholinyl and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (Ci-C3)alkyl, halo(Ci-C3)alkyl, (Ci-C3)alkoxy and (CI-C3)alkoxy(C,-C3)alkyl; Rc is -H, (C C3)alkyl, pyridinyl or morpholinyl; and Ra and Rb are each independently -H or (Ci-C3)alkyl.
The compound of Claim 3, wherein the compound is represented by a structural formula selected from:

is a double bond or a single bond;
W is CH or N when = is a double bond, or W is -CHR7- or -NR8- when is a single bond;
R5 is absent when is a double bond and R5 is -H or (Ci-C3)alkyl when is a single bond;
q is 1 or 2;
each R7 are independently selected from -H, -CN, -N02, halogen, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc,
-0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C6)alkyl, halo(Ci-C6)alkyl and (Ci-C3)alkoxy(Ci-C3)alkyl;
R8 is -H or a (C,-C3)alkyl;
each R9 is independently selected from -H, -CN, -N02, halogen, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)tNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc,
-C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc,
-0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)R°, -C(=0)Rc, alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, wherein the alkyl, cycloalkyl, heterocycloalkyl, aryl and heteroaryl represented by R9 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, -N02, -OH, (C,-C3)alkyl, halo(d-C3)alkyl, (Ci-C3)alkoxy and
(C1-C3)alkoxy(Ci-C3)alkyl, hydroxyl(C,-C3)alkyl or -NRaRb;
Rla and Rlb are each independently -H, -halogen, -CN, -ORc, -NRaRb,
-C(=0)ORc, -C(=0)NReRf, -NRdC(=0)Rc, -0(C=0)NReRf, heterocycloalkyl, or alkyl, wherein the heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -OR0, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, -0(C=0)NReRf;
R2a and R2b each independently -H, halogen, -CN, -N02, -NRaRb, -ORc, -C(=0)Rc, (C,-C3)alkyl, halo(C,-C3)alkyl, or (C1-C3)alkoxy(Ci-C3)alkyl, (C2-C4)alkynyl, halo(C2-C4)alkynyl, (C2-C4)alkenyl, or halo(C2-C4)alkenyl; and
r is an integer from 1 to 4.
The compound of Claim 8, wherein:
R7 is -H or (Ci-C6)alkyl;
each R is independently selected from the group consisting of -H, halogen, -CN, -N02, (C,-C3)alkyl, halo(CrC3)alkyl, (C,-C3)alkoxy, (Ci-C3)alkoxy(C,-C3)alkyl, hydroxy(C,-C3)alkyl or -(C,-C3)alkylene-NRaRb ; and
r is 1 or 2.
The compound of Claim 3, wherein the compound is represented by a structural formula selected from:

ME111444820V.1

11. The compound of Claim 10, wherein the compound is represented by the following structural formula:

Rla and Rl b are each independently -H, -halogen, -CN, -ORc, -NRaRb,
-C(=0)ORc, -C(=0)NReRf, -NRdC(=0)Rc, -0(C=0)NReRf, heterocycloalkyl, or alkyl, wherein the heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents
-300-
ME1 11444820V.1
independently selected from the group consisting of -halogen, -CN, -OR0, -NRaRb, -S(0)iRc, -NRdS(0)jRc, -S(0)jNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, -0(C=0)NReRf;
R2a and R2b are each independently -H, halogen, -CN, -N02, -ORc, -NRaRb, -C(=0)Rc, (CrC3)alkyl, halo(C,-C3)alkyl, (CrC3)alkoxy, halo(C,-C3)alkoxy, or (Cr C3)alkoxy(C,-C3)alkyl, (C2-C4)alkynyl, halo(C2-C4)alkynyl, (C2-C4)alkenyl, or halo(C2- C4)alkenyl.
The compound of Claim 10 or 1 1, wherein the group represented by R3 is selected from (C3-Cs)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thiophenyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, isoxazolyl, indolyl, imidazolyl, thiazolyl, benzofcGimidazolyl, benzo[i/]thiazolyl, benzo[b]thiophenyl, quinolinyl,
tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, morpholinyl, indolinyl and indenyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C3)alkyl, phenyl, benzyl, pyrimidinyl, moφholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (C,-C3)alkyl, halo(C,-C3)alkyl, (C,-C3)alkoxy and (Ci-C3)alkoxy(Cr C3)alkyl.
The compound of Claim 12, wherein the group represented by R3 is selected from the group consisting of cyclopentyl, phenyl, thiophenyl, pyridyl, piperidinyl,
tetrahydropyranyl, tetrahydrofuranyl, pyrrolidinyl, and indolyl, each of which is
optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)jRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (C|-C3)alkyl, phenyl, benzyl, mo holinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (C,-C3)alkyl, halo(Ci-C3)alkyl, (C,-C3)alkoxy and (CrC3)alkoxy(Ci- C3)alkyl.
The compound of Claim 13, wherein the group represented by R3 is selected from the group consisting of phenyl, thiophenyl, pyridyl, tetrahydropyranyl and indolyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS((¾Rc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and
tetrahydropyranyl, wherein each of the (Ci-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (CrC3)alkyl, halo(CrC3)alkyl, (C,- C3)alkoxy and (Ci-C3)alkoxy(C,-C3)alkyl.
The compound of any one of Claims 10-14, the group represented by R3 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -OR0, (C,-C3)alkyl, -(CrC3)alkylene-NRaRb, phenyl, pyrimidinyl, morpholinyl
and benzyl, wherein the phenyl, pyrimidinyl, morpholinyl and benzyl substituents are optionally substituted with halogen, (C] -C3)alkyl, halo(Ci-C3)alkyl, (Ci-C3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl; Rc is -H, (d-C3)alkyl, pyridinyl or morpholinyl; and Ra and Rb are each independently -H or (Ci-C3)alkyl.
16. The compound of any one of Claims 10-14, the group represented by R3 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of- F, -CI, methyl, -CF3, ethyl, 2-propyl, benzyl, phenyl and -CH2NH2.
17. The compound of any one of Claims 10-16, wherein R4 and R5 are both -H.
18. The compound of any one of Claims 10-16, wherein R5 is -H and R4 is -OH, (Ci- C3)alkyl, -(C,-C2)alkylene-OH,(C,-C3)alkoxy, -NRaRb, -(Ci-C2)alkylene-NRaRb, pyrrolidinyl, piperidinyl, moφholinyl or cyclopropyl, wherein Ra and Rb are each independently -H or (Ci-C3)alkyl and the pyrrolidinyl, piperidinyl, morpholinyl or cyclopropyl represented by R4 is optionally substituted with halogen, (Ci-C3)alkyl, halo(C,-C3)alkyl, (C,-C3)alkoxy and (Ci-C3)alkoxy(Ci-C3)alkyl.
19. The compound of any one of Claims 8-18, wherein Rla and Rlb are both -H.
20. The compound of any one of Claims 8-19, wherein R2a and R2b are both -H.
21. The compound of Cla ted by a structural formula:

ME1 11444820V.1
or a pharmaceutically acceptable salt thereof,
wherein R16 is alkyl, optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Ci-C3)alkyl, heteroaryl, heteroaryl(Ci-C3)alkyl, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc,
-OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC and -C(=0)Rc, wherein the cycloalkyl, cycloalkyl(Ci-C3)alkyl, heterocycloalkyl, heterocycloalkyl(Ci-C3)alkyl, aryl, aryl(Cr C3)alkyl, heteroaryl or heteroaryl(Ci-C3)alkyl are optionally substituted with 1 to 3 substituents independently selected from -halogen, alkyl, -ORc and -NRaRb.
22. The compound of Claim 1 or Claim 21 , wherein the compound is represented by a structural formula selected from:

or a pharmaceutically acceptable salt thereof, wherein:
R16 is alkyl, optionally substituted with 1 to 3 substituents independently selected from -halogen, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, -NRaRb, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=0)NReRf, wherein the
-304-
ME1 11444820V.1
cycloalkyl, heterocycloalkyl, aryl or heteroaryl are optionally substituted with 1 to 3 substituents independently selected from -halogen, alkyl, -OR0 and -NRaRb.
23. The compound of claim 22, wherein R16 is (CrC6)alkyl, optionally substituted with -N(CH3)2 or 2,6-dimefhylmorpholinyl.
24. The compound of claim 22, wherein R16 is selected from methyl, ethyl, (2,6- dimethylmorpholinyl)propyl, and N,N-dimethylaminopropyl.
25. The compound of any one of Claims 22-24, wherein the compound is represented by a structural formula selected from:

or a pharmaceutically acceptable salt thereof.
The compound of any one of Claims 22-25, wherein the compound is represented by structural formula selected from:
-305-
ME1 11444820V.1

or a pharmaceutically acceptable salt thereof, wherein:
Rla and Rlb are each independently -H, -halogen, -CN, -ORc, -NRaRb,
-C(=0)ORc, -C(=0)NReRf, -NRdC(=0)Rc, -0(C=0)NReRf, heterocycloalkyl, or alkyl, wherein heterocycloalkyl or the alkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of -halogen, -CN, -OR0, -NRaRb, - S(0)iRc, -NRdS(0)jRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -NRd(C=0)ORc, -0(C=0)NReRf; and
R2a and R2b each independently -H, halogen, -CN, -N02, -NRaRb, -ORc, - C(=0)Rc, (C,-C3)alkyl, halo(C,-C3)alkyl, or (C1-C3)alkoxy(C,-C3)alkyl, (C2-C4)alkynyl, halo(C2-C4)alkynyl, (C2-C4)alkenyl, or halo(C2-C4)alkenyl.
The compound of any one of Claims 22-26, wherein the compound is represented by a structural formula selected from:

or a pharmaceutically acceptable salt thereof, wherein:
R3 is a cycloalkyl, heterocycloalkyl, aryl or heteroaryl, each of which is optionally substituted with 1 to 3 groups represented by R6;
R4 and R5 are each independently selected from -H, alkyl, -ORc, -NRaRb, -(C,- C3)alkylene-NRaRb, -(Ci-C3)alkylene-ORc, -(Ci-C3)alkylene-OH, cycloalklyl,
-(Ci-C3)alkylene-cycloalkyl, heterocycloalkyl and -(Ci-C3)alkylene-heterocycloalkyl, provided that R4 and R5 are not both selected from -ORc and -NRaRb; or R3 and R5 together with the carbon atom to which they are attached form a cycloalkyl or a heterocycloalkyl, wherein the cycloalkyl or heterocycloalkyl is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (C,-C3)alkyl, halo(C,-C3)alkyl, (C,-C3)alkoxy, halo(C,-C3)alkoxy, (Cr
C3)alkoxy(CrC3)alkyl;
each R6is independently selected from halogen, =0, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, -C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc, -C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc,
-0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (C,-C6)alkyl, aryl, aryl(C,-C3)alkyl, heterocycloalkyl and
heteroaryl; wherein each the (Ci-C6)alkyl, aryl, aryl(Ci-C3)alkyl, heterocycloalkyl and heteroaryl represented by R6 is optionally substituted with 1 to 3 substituents independently selected from the group consisting of halogen, -CN, (Ci-Ce)alkyl, halo(C,-C6)alkyl, (CrC3)alkoxy, halo(CrC3)alkoxy and (Ci-C3)alkoxy(CrC6)alkyl; and p is 0, 1 or 2.
28. The compound of Claim 27, wherein R3 is selected from (C3-C8)cycloalkyl, phenyl, naphthyl, tetrahydronaphthyl, pyridyl, thienyl, pyrazinyl, pyrimidyl, pyridazinyl, pyrazolyl, oxazolyl, isoxazolyl, indolyl, imidazolyl, thiazolyl, benzo[if|imidazolyl, benzofi jthiazolyl, benzo[£]thiophenyl, quinolinyl, tetrahydronaphthalenyl, tetrahydroquinolinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, morpholinyl, indolinyl and indenyl, each of which is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -ORc, -NRaRb, -S(0)iRc, -NRdS(0)iRc, -S(0)iNReRf, C(=0)ORc, -OC(=0)ORc, -C(=S)ORc, -0(C=S)Rc,
-C(=0)NReRf, -NRdC(=0)Rc, -C(=S)NReRf, -NRdC(=S)Rc, -NRd(C=0)ORc, -0(C=0)NReRf, -NRd(C=S)ORc, -0(C=S)NReRf, -NRd(C=0)NReRf, -NRd(C=S)NReRf, -C(=S)RC, -C(=0)Rc, (Ci-C3)alkyl, phenyl, benzyl, pyrimidinyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl, wherein each of the (Ci-C3)alkyl, phenyl, benzyl, morpholinyl, piperidinyl, pyrrolidinyl, tetrahydrofuranyl and tetrahydropyranyl substituents on the groups represented by R3 is optionally substituted with 1 to 3 substituents independently selected from halogen, -CN, -N02, -NRaRb, (d- C3)alkyl, halo(CrC3)alkyl, (C,-C3)alkoxy and (C1-C3)alkoxy(C C3)alkyl
29. The compound of Claim 27 or Claim 28 wherein R3 is selected from phenyl, pyridyl, and thienyl, each of which is optionally substituted with halogen or (Ci-C3)alkyl.
30. The compound of any one of Claims 27-29 wherein R4 and R5 are each independently selected from -H, alkyl, -ORc, -NRaRb, cycloalkyl, heterocycloalkyl and -(Ci- C3)alkylene-heterocycloalkyl.
-308-E1 11444820V.1
31. The compound of any one of Claims 27-30 wherein one of R4 and R5 is -H and the other is selected from -H, (C,-C3)alkyl, -OCH3, -N(CH3)2, (C3-C6)cycloalklyl, pyrrolidinyl, piperidinyl, -CH2-morpholinyl and -O-cyclopentyl.
32. The compound of any one of Claims 26-31 , wherein Rla and Rlb are each independently selected from -H and (Ci-C3)alkyl substituted with -N(Me)2.
33. The compound of any one of Claims 26-32, wherein R2a and R2b are both -H.
34. A pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and a compound of any one of Claims 1-33.
35. A method of treating a subject with cancer comprising administering to the subject an effect amount of the compound of any one of Claims 1-33 or a pharmaceutically acceptable salt thereof.
36. The method of Claim 35, wherein the cancer is selected from the group consisting of lung cancer, breast cancer, colon cancer, brain cancer, neuroblastoma, prostate cancer, melanoma, glioblastoma multiform, ovarian cancer, lymphoma, leukemia, melanoma, sarcoma, paraneoplasia, osteosarcoma, germinoma, gliomas, pancreatic cancer and mesothelioma.
37. The method of Claim 36, wherein the cancer is selected from the group consisting of breast cancer, colon cancer, lung cancer, melanoma, prostate cancer, ovarian cancer, brain cancer and pancreatic cancer.
38. The method of Claim 37, wherein the cancer is breast cancer.
-309-
ME1 1144482QV.1
39. The method of Claim 38, wherein the cancer is a basal sub-type breast cancer or a luminal B sub-type breast cancer.
40. The method of Claim 39, wherein the cancer is a basal sub-type breast cancer that
overexpresses TTK.
41. The method of Claim 40, wherein the basal sub-type breast cancer is ER, HER2 and PR negative breast cancer.
42. The method of Claim 35, wherein the cancer is a soft tissue cancer.
43. The method of Claim 42, wherein the soft tissue cancer is a sarcoma selected from the group consisting of a fibrosarcoma, a gastrointestinal sarcoma, a leiomyosarcoma, a dedifferentiated liposarcoma, a pleomorphic liposarcoma, a malignant fibrous histiocytoma, a round cell sarcoma, and a synovial sarcoma.
44. A method of inhibiting the growth of tumor-initiating cells in a subject who is undergoing an anti-cancer therapy, comprising the steps of:
a) assessing the subject to determine whether the cancer is in remission; and b) if the cancer is in remission, then administering to the subject an effective amount of a TTK inhibitor.
45. A method of reducing the likelihood of recurrence of a cancer in a subject who is
undergoing an anti-cancer therapy comprising the steps of:
a) assessing the subject to determine whether the cancer is in remission; and b) if the cancer is in remission, then administering to the subject an effective amount of a TTK inhibitor.
46. The method of Claims 44 or Claim 45, wherein if the cancer is not in remission, the method further comprises the step of continuing the anti-cancer therapy until the cancer
-310-E1 11444820V.1
goes into remission and then the step b) of administering an effective amount of the
compound.
47. A method of inhibiting the growth of tumor-initiating cells in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhibitor.
48. A method of reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission comprising administering to the subject an effective amount of a TTK inhibitor.
49. The method of Claim 47 or Claim 48, wherein the subject has been treated with an anticancer therapy.
50. The method of any one of Claims 44-46 and 49, wherein the anti-cancer therapy is
selected from the group consisting of surgery, radiation therapy, immunotherapy, endocrine therapy, gene therapy and administration of an anti-cancer agent.
51. A method of treating a subject with a drug-resistant cancer comprising administering to the subject an effective amount of a TTK inhibitor.
52. A use of an effect amount of the compound of any one of Claims 1-33 or a
pharmaceutically acceptable salt thereof for treating a subject with cancer.
53. A use of an effect amount of the compound of any one of Claims 1-33 or a
pharmaceutically acceptable salt thereof for the preparation of a medicament for treating a subject with cancer.
54. A use of an effective amount of a TTK inhibitor for inhibiting the growth of tumor- initiating cells in a subject whose cancer is in remission.
-31 1-
ME1 11444820V.1
55. A use of an effective amount of a TTK inhibitor for the preparation of a medicament for inhibiting the growth of tumor-initiating cells in a subject whose cancer is in remission.
56. A use of an effective amount of a TTK inhibitor for reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission.
57. A use of an effective amount of a TTK inhibitor for the preparation of a medicament for reducing the likelihood of recurrence of a cancer in a subject whose cancer is in remission.
58. A use of an effective amount of a TTK inhibitor for treating a subject with a drug- resistant cancer.
59. A use of an effective amount of a TTK inhibitor for the preparation of a medicament for treating a subject with a drug-resistant cancer.
-312-
ME1 1 1444820V.1
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US32134210P | 2010-04-06 | 2010-04-06 | |
| US61/321,342 | 2010-04-06 | ||
| US34584910P | 2010-05-18 | 2010-05-18 | |
| US61/345,849 | 2010-05-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011123937A1 true WO2011123937A1 (en) | 2011-10-13 |
Family
ID=44761959
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2011/000366 Ceased WO2011123937A1 (en) | 2010-04-06 | 2011-04-06 | Kinase inhibitors and method of treating cancer with same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011123937A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013053051A1 (en) * | 2011-10-12 | 2013-04-18 | University Health Network | Indazole compounds as kinase inhibitors and method of treating cancer with same |
| WO2012176856A3 (en) * | 2011-06-24 | 2013-06-13 | Ishihara Sangyo Kaisha, Ltd. | 3-arylphenyl sulfide compounds and their use as pesticides |
| US8980934B2 (en) | 2012-06-22 | 2015-03-17 | University Health Network | Kinase inhibitors and method of treating cancer with same |
| WO2017121699A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Cropscience Aktiengesellschaft | Method for the preparation of substituted 2-aryl-ethanols |
| CN108368057A (en) * | 2015-08-20 | 2018-08-03 | 常州捷凯医药科技有限公司 | The heterocyclic compound condensed as the pyrazolo of ERK inhibitor |
| JP2020520890A (en) * | 2017-05-19 | 2020-07-16 | 厦▲門▼大学 | Aza-cyclic aromatic compound in which 5-membered ring and 6-membered ring are condensed, method for producing the same, pharmaceutical composition and application thereof |
| CN111662232A (en) * | 2019-03-06 | 2020-09-15 | 中国科学院上海药物研究所 | Small molecular compounds with 2H-indazole structure and their synthesis and applications |
| WO2020185044A1 (en) * | 2019-03-13 | 2020-09-17 | 주식회사 보로노이 | Heteroaryl derivatives and pharmaceutical composition comprising same as active ingredient |
| US11208696B2 (en) | 2015-04-17 | 2021-12-28 | Netherlands Translational Research Center B.V. | Prognostic biomarkers for TTK inhibitor chemotherapy |
| US11427558B1 (en) | 2019-07-11 | 2022-08-30 | ESCAPE Bio, Inc. | Indazoles and azaindazoles as LRRK2 inhibitors |
| WO2023279041A1 (en) * | 2021-06-30 | 2023-01-05 | Tyra Biosciences, Inc. | Indazole compounds |
| US11858915B2 (en) | 2021-05-11 | 2024-01-02 | Oric Pharmaceuticals, Inc. | Polo like kinase 4 inhibitors |
| CN117486730A (en) * | 2023-10-20 | 2024-02-02 | 泰州精英化成医药科技有限公司 | Preparation method of dicyclohexyl methylamine |
| CN119118956A (en) * | 2024-09-11 | 2024-12-13 | 上海电力大学 | A phenylglycine derivative and its synthesis method and application |
| EP4259118A4 (en) * | 2020-12-11 | 2025-01-01 | The Scripps Research Institute | COMPOUNDS AND THEIR USE FOR THE TREATMENT OF NEURODEGENERATIVE, DEGENERATIVE AND METABOLIC DISORDERS |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2633023A1 (en) * | 2005-12-13 | 2007-06-21 | Schering Corporation | Polycyclic indazole derivatives that are erk inhibitors |
| JP2010111624A (en) * | 2008-11-06 | 2010-05-20 | Shionogi & Co Ltd | Indazole derivative having ttk inhibitory action |
| WO2010115279A1 (en) * | 2009-04-06 | 2010-10-14 | University Health Network | Kinase inhibitors and method of treating cancer with same |
-
2011
- 2011-04-06 WO PCT/CA2011/000366 patent/WO2011123937A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2633023A1 (en) * | 2005-12-13 | 2007-06-21 | Schering Corporation | Polycyclic indazole derivatives that are erk inhibitors |
| JP2010111624A (en) * | 2008-11-06 | 2010-05-20 | Shionogi & Co Ltd | Indazole derivative having ttk inhibitory action |
| WO2010115279A1 (en) * | 2009-04-06 | 2010-10-14 | University Health Network | Kinase inhibitors and method of treating cancer with same |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012176856A3 (en) * | 2011-06-24 | 2013-06-13 | Ishihara Sangyo Kaisha, Ltd. | 3-arylphenyl sulfide compounds and their use as pesticides |
| US9580390B2 (en) | 2011-10-12 | 2017-02-28 | University Health Network | Indazole compounds as kinase inhibitors and method of treating cancer with same |
| WO2013053051A1 (en) * | 2011-10-12 | 2013-04-18 | University Health Network | Indazole compounds as kinase inhibitors and method of treating cancer with same |
| US8980934B2 (en) | 2012-06-22 | 2015-03-17 | University Health Network | Kinase inhibitors and method of treating cancer with same |
| US11208696B2 (en) | 2015-04-17 | 2021-12-28 | Netherlands Translational Research Center B.V. | Prognostic biomarkers for TTK inhibitor chemotherapy |
| US12054784B2 (en) | 2015-04-17 | 2024-08-06 | Netherlands Translational Research Center B.V. | Prognostic biomarkers for TTK inhibitor chemotherapy |
| CN108368057A (en) * | 2015-08-20 | 2018-08-03 | 常州捷凯医药科技有限公司 | The heterocyclic compound condensed as the pyrazolo of ERK inhibitor |
| US20180237450A1 (en) * | 2015-08-20 | 2018-08-23 | Changzhou Jiekai Pharmatech Co., Ltd. | Pyrazolo fused heterocyclic compounds as erk inhibitors |
| US10696687B2 (en) * | 2015-08-20 | 2020-06-30 | Changzhou Jiekai Pharmatech Co., Ltd. | Pyrazolo fused heterocyclic compounds as ERK inhibitors |
| CN108368057B (en) * | 2015-08-20 | 2022-03-15 | 捷思英达医药技术(上海)有限公司 | Pyrazolo-fused heterocyclic compounds as ERK inhibitors |
| WO2017121699A1 (en) | 2016-01-15 | 2017-07-20 | Bayer Cropscience Aktiengesellschaft | Method for the preparation of substituted 2-aryl-ethanols |
| US10519085B2 (en) | 2016-01-15 | 2019-12-31 | Bayer Cropscience Aktiengesellschaft | Process for preparing substituted 2-arylethanols |
| JP7145873B2 (en) | 2017-05-19 | 2022-10-03 | 厦▲門▼大学 | Azacyclic aromatic compound with condensed 5-membered ring and 6-membered ring, method for producing the same, pharmaceutical composition and application thereof |
| JP2020520890A (en) * | 2017-05-19 | 2020-07-16 | 厦▲門▼大学 | Aza-cyclic aromatic compound in which 5-membered ring and 6-membered ring are condensed, method for producing the same, pharmaceutical composition and application thereof |
| CN111662232B (en) * | 2019-03-06 | 2022-08-02 | 中国科学院上海药物研究所 | Small molecule compound with 2H-indazole structure and synthesis and application thereof |
| CN111662232A (en) * | 2019-03-06 | 2020-09-15 | 中国科学院上海药物研究所 | Small molecular compounds with 2H-indazole structure and their synthesis and applications |
| JP2022523351A (en) * | 2019-03-13 | 2022-04-22 | ボロノイ・カンパニー・リミテッド | Heteroaryl derivative and pharmaceutical composition containing it as an active ingredient |
| CN113574057A (en) * | 2019-03-13 | 2021-10-29 | 株式会社沃若诺伊 | Heteroaryl derivatives and pharmaceutical compositions containing them as active ingredients |
| WO2020185044A1 (en) * | 2019-03-13 | 2020-09-17 | 주식회사 보로노이 | Heteroaryl derivatives and pharmaceutical composition comprising same as active ingredient |
| US11427558B1 (en) | 2019-07-11 | 2022-08-30 | ESCAPE Bio, Inc. | Indazoles and azaindazoles as LRRK2 inhibitors |
| EP4259118A4 (en) * | 2020-12-11 | 2025-01-01 | The Scripps Research Institute | COMPOUNDS AND THEIR USE FOR THE TREATMENT OF NEURODEGENERATIVE, DEGENERATIVE AND METABOLIC DISORDERS |
| US11858915B2 (en) | 2021-05-11 | 2024-01-02 | Oric Pharmaceuticals, Inc. | Polo like kinase 4 inhibitors |
| US12312339B2 (en) | 2021-05-11 | 2025-05-27 | Oric Pharmaceuticals, Inc. | Polo like kinase 4 inhibitors |
| WO2023279041A1 (en) * | 2021-06-30 | 2023-01-05 | Tyra Biosciences, Inc. | Indazole compounds |
| CN117486730A (en) * | 2023-10-20 | 2024-02-02 | 泰州精英化成医药科技有限公司 | Preparation method of dicyclohexyl methylamine |
| CN117486730B (en) * | 2023-10-20 | 2024-10-22 | 泰州精英化成医药科技有限公司 | Dicyclohexyl group Process for the preparation of methylamines |
| CN119118956A (en) * | 2024-09-11 | 2024-12-13 | 上海电力大学 | A phenylglycine derivative and its synthesis method and application |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011123937A1 (en) | Kinase inhibitors and method of treating cancer with same | |
| CA2889919C (en) | Pyrazolopyrimidine compounds | |
| EP2766352B1 (en) | Indazole compounds as kinase inhibitors and method of treating cancer with same | |
| AU2017260367B2 (en) | Modulators of the integrated stress pathway | |
| CA3023261A1 (en) | Modulators of the integrated stress pathway | |
| TW202311262A (en) | Inhibitors of the menin-mll interaction | |
| CA2956360A1 (en) | Piperazine derivatives as liver x receptor modulators | |
| US8980934B2 (en) | Kinase inhibitors and method of treating cancer with same | |
| HK1225027A1 (en) | Pyrazolopyrimidine compounds | |
| HK1225027B (en) | Pyrazolopyrimidine compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11764979 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11764979 Country of ref document: EP Kind code of ref document: A1 |