WO2011112499A1 - Compositions, methods, and devices for the treatment of dysmenorrhea - Google Patents
Compositions, methods, and devices for the treatment of dysmenorrhea Download PDFInfo
- Publication number
- WO2011112499A1 WO2011112499A1 PCT/US2011/027370 US2011027370W WO2011112499A1 WO 2011112499 A1 WO2011112499 A1 WO 2011112499A1 US 2011027370 W US2011027370 W US 2011027370W WO 2011112499 A1 WO2011112499 A1 WO 2011112499A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- subject
- compound
- uterine
- group
- administration
- Prior art date
Links
- 0 C*COc1cc(CC(CC2)NCC(c(cc3CCO)ccc3O)O)c2cc1 Chemical compound C*COc1cc(CC(CC2)NCC(c(cc3CCO)ccc3O)O)c2cc1 0.000 description 2
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/075—Ethers or acetals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/01—Hydrocarbons
- A61K31/015—Hydrocarbons carbocyclic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/196—Carboxylic acids, e.g. valproic acid having an amino group the amino group being directly attached to a ring, e.g. anthranilic acid, mefenamic acid, diclofenac, chlorambucil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P23/00—Anaesthetics
- A61P23/02—Local anaesthetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/70—Web, sheet or filament bases ; Films; Fibres of the matrix type containing drug
- A61K9/7023—Transdermal patches and similar drug-containing composite devices, e.g. cataplasms
Definitions
- Uterine contractility disorders are significant health problems. Dysmenorrhea, painful uterine contractions or cramping during the menstrual period, affects gonadal women. The etiology of uterine contractility disorders are largely unknown and effective therapy to inhibit uterine contractility and prevent the symptoms associated with these diseases are unknown.
- Dysmenorrhea which may be primary or secondary, is the occurrence of painful uterine cramps during menstruation. In secondary dysmenorrhea, there is a visible pelvic lesion to account for the pain, whereas a biochemical imbalance is responsible for primary dysmenorrhea.
- Primary dysmenorrhea affects 50 percent of post-pubescent women, and absenteeism among severe dysmenorrheics has been estimated to cost about several million lost working hours or billions of dollars annually.
- a method for a prevention and/or treatment of dysmenorrhea or amelioration of a symptom thereof in a subject comprising administering to said sub ect an effective amount of a compound of formula I:
- n is an integer selected from 1 or 2;
- X is a Ci-C 6 alkylene group
- Y is -N(R) 2 wherein each R is independently selected from hydrogen or C r C6 alkyl, or two R along with the nitrogen bound thereto join together to form a 3 to 7 membered heterocyclic ring optionally containing an oxygen atom; and
- a method for a prevention and/or treatment of dysmenorrhea or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a compound of formula V:
- a method for a prevention and/or treatment of uterine contraction or cramping or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a compound of formula I:
- n is an integer selected from 1 or 2;
- X is a C]-C 6 alkylene group
- Y is -N(R) 2 wherein each R is independently selected from hydrogen or C]-C 6 alkyl, or two R along with the nitrogen bound thereto join together to form a 3 to 7 membered heterocyclic ring optionally containing an oxygen atom; and
- a method for uterine contraction or cramping or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a compound of formula V:
- Figure 1 illustrates the effects of MN-221, ritodrine hydrochloride, and isoproterenol bitartrate on spontaneous contractions of uterine muscle isolated from pregnant rats.
- the data represent the mean ⁇ standard error of 10 samples.
- Figure 2 illustrates the effect of CGP 20712A on inhibitory effect of MN-221 on uterine contraction. Each point represents the mean ⁇ standard error of 10 samples.
- Figure 3 illustrates the antagonistic effect of ICI 1 18,551 on inhibitory effect of MN- 221 on uterine contraction.
- Figure 3 A Concentration response curve: Each point represents the mean ⁇ standard error of 10 samples.
- Figure 3B Schild regression: Each point represents the results of 8 to 10 samples (a total of 28 samples).
- Figure 4 illustrates the effect of SR 59230A on inhibitory effect of MN-221 on uterine contraction. Each point represents the mean ⁇ standard error of 10 to 12 samples.
- Figure 5 illustrates the effect of MN-221 , ritodrine hydrochloride, and isoproterenol bitartrate on PGF 2a rinduced contraction of uterine muscle isolated from pregnant rats. Uterine contraction was induced with the addition of 5 g/mL of PG F 2a . Each data point indicates a mean ⁇ standard error of 10 samples.
- Figure 6 illustrates the Effect of MN-221 , ritodrine hydrochloride, and isoproterenol bitartrate on oxytocin-induced contraction of uterine muscle isolated from pregnant rats. Uterine contraction was induced with the addition of 1 mU/mL of oxytocin. Each data point indicates a mean ⁇ standard error of 10 samples.
- Figure 7 illustrates the effects of MN-221 on uterine activity, heart rate, and blood pressure of anesthetized pregnant rats.
- Figure 8 illustrates the effects of MN-221 and other 32-adrenoceptor agonists on uterine activity of anesthetized pregnant rats (Figure 8A), increases in heart rate of dam (Figure 8B), and mean blood pressure of dam (Figure 8C).
- Figure 9 illustrates representative recordings of the effect of MN-221 on the oxytocin-induced uterine contraction in the sheep.
- Figure 10 illustrates comparison of changes over time in intrauterine pressure between the MN-221 group and the control group.
- MN-221 group is represented as closed circle; control group is represented as open circle; asterisk, P ⁇ 0.05 compared with the pre- infusion value.
- Figure 11 illustrates comparison of changes over time in maternal and fetal heart rate and blood pressure between the MN-221 group and the control group during the experiment.
- Figure HA maternal heart rate
- Figure 11B fetal heart rate
- Figure 11C maternal systolic blood pressure
- Figure 11D maternal diastolic blood pressure
- Figure HE maternal mean blood pressure
- Figure 11F fetal mean blood pressure.
- MN-221 group is represented as closed circle & triangle
- control group is represented as open circle & triangle
- asterisk P ⁇ 0.05 compared with the pre-infusion value.
- Figure 12 illustrates comparison of changes over time in maternal respiratory parameters between the MN-221 group and the control group.
- Figure 12 A pH
- Figure 12B Pco 2
- Figure 12C Po 2
- Figure 12D base excess.
- MN-221 group is represented as closed circle
- control group is represented as open circle.
- Figure 13 illustrates comparison of changes over time in fetal respiratory parameters between the MN-221 group and the control group.
- Figure 13A pH
- Figure 13B Pco 2
- Figure 13C P 02
- Figure 13D base excess.
- MN-221 group is represented as closed triangle and control group is represented as open triangle.
- Figure 14 illustrates comparison of changes over time in maternal metabolic parameters between the MN-221 group and the control group.
- Figure 14 A blood Na +
- Figure 14B blood K +
- Figure 14C blood CI "
- Figure 14D blood Ca 2+
- Figure 14E plasma glucose
- Figure 14F blood lactate
- Figure 14G plasma insulin
- Figure 14H plasma NEFA levels.
- MN-221 group is represented as closed circle and the control group is represented as open circle.
- Figure 15 illustrates comparison of changes over time in fetal metabolic parameters between the MN-221 group and the control group.
- Figure 15A blood Na + ;
- Figure 15B blood K + ;
- Figure 15C blood CI " ;
- Figure 15D blood Ca 2+ ;
- Figure 15E plasma glucose;
- Figure 15F blood lactate;
- Figure 15G plasma insulin.
- MN-221 group is represented as closed triangle and the control group is represented as open triangle. Asterisk, P ⁇ 0.05 compared with the pre-infusion value.
- Figure 16 illustrates the effects of MN-221 and other /32-adrenoceptor agonists on spontaneous contractions of uterine muscles isolated from pregnant rabbits.
- Figure 17 illustrates the effects of MN-221 and other (32-adrenoceptor agonists on oxytocin-induced contractions of uterine muscles isolated from pregnant rabbits.
- an “administration” or “administering,” refers to the delivery of a medication, such as the composition used according to the invention to an appropriate location of the subject or in vitro, where a desired effect is achieved.
- a medication such as the composition used according to the invention
- Non-limiting examples include topical, oral, parenteral, direct application to target area or proximal areas on the skin, or applied transdermally such as a patch.
- Various physical and/or mechanical technologies are available to permit the sustained or immediate release of the composition after administration.
- CpCe alkyl refers to saturated monovalent hydrocarbyl groups having from 1 to 6 carbon atoms, more particularly from 1 to 5 carbon atoms, and even more particularly 1 to 3 carbon atoms. This term is exemplified by groups such as methyl, ethyl, n-propyl, iso- propyl, n-butyl, i-butyl, n-pentyl, and the like.
- a "Ci-C 6 alkylene” refers to divalent saturated aliphatic hydrocarbyl groups having from 1 to 6 carbon atoms and, in some embodiments, from 1 to 3 carbon atoms.
- the alkylene groups include branched and straight chain hydrocarbyl groups. Examples include methylene (-CH 2 -), ethylene, propylene, 2-methypropylene, pentylene, and the like.
- a "compound” herein refers to a compound used according to the invention, a pharmaceutically acceptable salt thereof, a metabolite thereof, a prodrug thereof, a pharmaceutically acceptable salt of the metabolite thereof, or a pharmaceutically acceptable salt of the prodrug thereof.
- the compounds include stereoisomeric forms and the tautomeric forms of the compounds.
- compositions and methods include the recited elements, but not excluding others.
- Consisting essentially of when used to define compositions and methods shall mean excluding other elements of any essential significance to the combination for the stated purpose. Thus, a composition consisting essentially of the elements as defined herein would not exclude trace contaminants from the isolation and purification method and pharmaceutically acceptable carriers, such as phosphate buffered saline, preservatives and the like.
- Consisting of shall mean excluding more than trace elements of other ingredients and substantial method steps for administering the compositions of this invention or process steps to produce a composition or achieve an intended result. Embodiments defined by each of these transition terms are within the scope of this invention.
- an "effective amount” or a “therapeutically effective amount” is an amount sufficient to effect beneficial or desired results, e.g., alleviation, amelioration, palliation or elimination of one or more manifestations of dysmenorrhea in the subject.
- the full therapeutic effect may occur in one dose; may not necessarily occur by administration of one dose (or dosage); and may occur only after administration of a series of doses.
- a therapeutically effective amount may be administered in one or more administrations, applications or dosages.
- a “heterocycle” or “heterocyclic” refers to a saturated or unsaturated (but not aromatic) group having a single ring or multiple condensed rings, from 3 to 6 carbon atoms, and from 1 to 4 hetero atoms selected from the group consisting of nitrogen or oxygen within the ring wherein, in fused ring systems, one or more of the rings can be aryl or heteroaryl provided that the point of attachment is at the heterocycle.
- the nitrogen ring atoms can optionally be oxidized to provide for the N-oxide derivatives.
- heterocycles include, but are not limited to, azetidine, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, etc.
- a "metabolite” refers to any substance that is produced as an intermediate or a product after the metabolism of the compound used according to the invention.
- metabolites include, but are not limited to, acid metabolized from the amide moiety, amine metabolized from the substituted amide moiety, alcohol metabolized from alkoxy moiety, and the like.
- a carboxylic acid, representative of a metabolite, is described in US Patent No. 6,136,852, the disclosure of which is incorporated herein by reference in its entirety.
- a "subject” or “patient” is a female mammal, including a human.
- Non-human animals subject to diagnosis or treatment include, for example, murine, such as rats, mice, canine, such as dogs, leporids, such as rabbits, livestock, sport animals, and pets.
- a "pharmaceutically acceptable carrier” encompasses any of the standard pharmaceutical carriers, such as a phosphate buffered saline solution, water, and emulsions, such as an oil/water or water/oil emulsion, and various types of wetting agents.
- the compositions also can include stabilizers and preservatives.
- stabilizers and adjuvants see Martin, Remington's Pharm. Sci., 15th Ed. (Mack Publ. Co., Easton (1975)).
- the term includes carriers that facilitate controlled release of the active agent as well as immediate release.
- a "pharmaceutically acceptable salt” refers to pharmaceutically acceptable salts of a compound, which salts are derived from a variety of organic, and inorganic counter ions well known in the art.
- salts include, by way of example only, sodium, potassium, calcium, magnesium, ammonium, tetraalkyl ammonium, and the like.
- salts of organic or inorganic acids include, such as hydrochloric, sulfuric, phosphoric, acetic, citric, oxalic, malonic, salicyclic, malic, gluconic, fumaric, succinic, ascorbic, maleic, methanesulfonic acid, etc.
- a "prodrug”, as used herein, refers to any covalently bonded carrier which releases the active parent drug in vivo when such prodrug is administered to a subject.
- Prodrugs of a compound are prepared by modifying functional groups present in the compounds in such a way that the bonds are cleaved, either in routine manipulation or in vivo, to the parent compounds.
- Prodrugs include, but are not limited to, compounds wherein hydroxyl or amine groups are bonded to any group that, when administered to a subject, cleave to form a free hydroxyl or amino, group, respectively.
- prodrugs include, but are not limited to, acetate, formate, benzoate and phosphate ester derivatives of hydroxyl functional groups, especially the hydroxyl group on the phenyl ring of formula I, and acetyl and benzoyl derivatives of amine functional groups in the compounds of the invention and the like.
- a “treating,” “treatment” and the like refer to obtaining a desired pharmacologic and/or physiologic effect.
- the effect can be prophylactic in terms of completely or partially preventing a disease or disorder or sign or symptom thereof, and/or can be therapeutic in terms of a partial or complete cure for a disorder and/or adverse effect attributable to the disorder.
- Examples of “treatment” include but are not limited to: preventing a disease from occurring in a subject that may be predisposed or at risk of a disease, but has not yet been diagnosed as having it; inhibiting a disease, i.e., arresting its development; and/or relieving or ameliorating the symptoms of disease or reducing the likelihood of recurrence of the disease, such as dysmenorrhea.
- “treatment” can include systemic amelioration of the symptoms associated with the pathology and/or a delay in onset of symptoms.
- Dysmenorrhea is a condition that refers to the pain or discomfort associated with menstruation in female subjects. Dysmenorrhea may be classified as primary or secondary. Primary dysmenorrhea is a severe and frequent menstrual cramping caused by severe and abnormal uterine contractions. Secondary dysmenorrhea is a painful menstrual period caused by another medical condition present in the body (e.g., pelvic inflammatory disease, endometriosis).
- Endometriosis is a condition in which tissue that appears and acts like endometrial tissue becomes implanted outside the uterus, typically on other reproductive organs inside the pelvis or in the abdominal cavity, resulting in internal bleeding, infection, and pelvic pain.
- Other possible causes of secondary dysmenorrhea include, but are not limited to, pelvic inflammatory disease (PID), pelvic congestion syndrome, pelvic infection, cervical stenosis, uterine fibroids, adenomyosis, abnormal pregnancy (i.e., miscarriage, ectopic), and infection, tumors, or polyps in the pelvic cavity.
- dysmenorrhea resemble symptoms of other conditions or medical problems, such as, but are not limited to, cramping in the lower abdomen; pain in the lower abdomen; low back pain; pain radiating down the legs; nausea; vomiting; diarrhea; fatigue; weakness; fainting; and headaches.
- the dosage and the regimen for the treatment for dysmenorrhea using the compositions and methods of the invention can depend on age, overall health, and medical history; extent of the condition; cause of the condition (primary or secondary); and tolerance for specific medications, procedures, or therapies.
- a method for a prevention and/or treatment of dysmenorrhea or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a compound of formula I:
- n is an integer selected from 1 or 2;
- X is a C]-C 6 alkylene group
- Y is -N(R) 2 wherein each R is independently selected from hydrogen or Cj-C 6 alkyl, or two R along with the nitrogen bound thereto join together to form a 3 to 7 membered heterocyclic ring optionally containing an oxygen atom; and
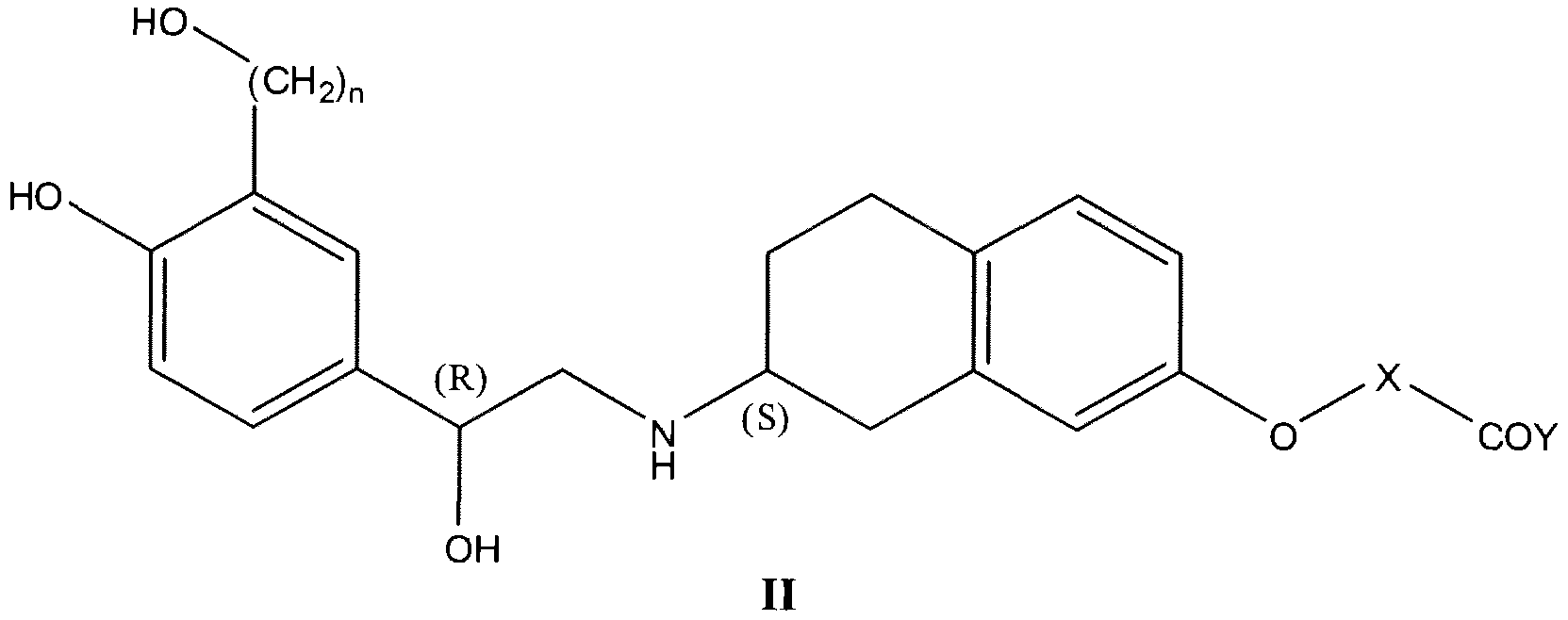
- a method for a prevention and/or treatment of dysmenorrhea or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a compound of formula II:
- n is an integer selected from 1 or 2;
- X is a Cj-C 6 alkylene group
- Y is — N(R) 2 wherein each R is independently selected from hydrogen or Cj-C 6 alkyl, or two R along with the nitrogen bound thereto join together to form a 3 to 7 membered heterocyclic ring optionally containing an oxygen atom;
- a method for a prevention and/or treatment of dysmenorrhea or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a compound of formula III:
- a method for a prevention and/or treatment of dysmenorrhea or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a metabolite of formula IV:
- n is an integer selected from 1 or 2;
- X is a C
- a method for a prevention and/or treatment of dysmenorrhea or amelioration of a symptom thereof in a subject comprising administering to said subject an effective amount of a compound of formula V:
- the present invention provides a method for treating a human female suffering from dysmenorrhea.
- the methods provided herein further comprise administering to the subject a drug selected from the group consisting of non-steroidal anti-inflammatory drugs, anti-prostaglandins, COX-2 inhibitors, local anesthetics, calcium channel blockers, potassium channel blockers, leukotriene blocking agents, smooth muscle inhibitors, vasodilators, and drugs capable of inhibiting dyskinetic muscle contraction.
- a drug selected from the group consisting of non-steroidal anti-inflammatory drugs, anti-prostaglandins, COX-2 inhibitors, local anesthetics, calcium channel blockers, potassium channel blockers, leukotriene blocking agents, smooth muscle inhibitors, vasodilators, and drugs capable of inhibiting dyskinetic muscle contraction.
- Non-limiting examples of non-steroidal anti-inflammatory drugs suitable for use in the method of the invention include, but are not limited to, aspirin, ibuprofen, indomethacin, phenylbutazone, bromfenac, fenamate, sulindac, nabumetone, ketorolac, and naproxen.
- Examples of local anesthetics include, but are not limited to, lidocaine, mepivacaine, etidocaine, bupivacaine, 2-chloroprocaine hydrochloride, procaine, and tetracaine hydrochloride.
- Examples of calcium channel blockers include, but are not limited to, diltiazem, israpidine, nimodipine, felodipine, verapamil, nifedipine, nicardipine, and bepridil.
- Examples of potassium channel blockers include, but are not limited to, dofetilide, E-4031 , almokalant, sematilide, ambasilide, azimilide, tedisamil, RP58866, sotalol, piroxicam, and ibutilide.
- Vasodilators which are believed to relieve muscle spasm in the uterine muscle, include, but are not limited to, nitroglycerin, isosorbide dinitrate and isosorbide mononitrate.
- COX-2 inhibitors include, but are not limited to, celecoxib, meloxicam and flosulide.
- a synergistic effect may be achieved by using a combination of the compound used according to the invention (e.g., those encompassed by formulas I, II, III, IV, V, and metabolites, isomers, and prodrugs of each thereof) with the drugs recited above.
- the compound used according to the invention and optionally the above recited drug is in combination with a biocompatible excipient provided herein.
- the compound used according to the invention is present in an amount sufficient to attain a therapeutically effective amount of the compound in the uterine muscle of the subject upon administration.
- the drug is absorbable through the vaginal mucosa and thereby transmitted via venous and lymphatic channels to the uterus.
- a subject need not wait until the onset of menses and the occurrence of pain to begin treatment.
- the present invention comprises administration of the compound or the composition as soon as the subject realizes that she is nearing the onset of menses, for example within a day or two.
- the method disclosed herein prevent the process of dyskinetic contractions from occurring, including treating them once the contractions have already begun.
- the compositions provided herein can treat dysmenorrhea and its dyskinetic contractions, without interfering with the normal contractions and bleeding during menstruation.
- Dysmenorrhea involves dyskinetic contractions, which are erratic and abnormal with an increase in the amplitude and frequency of contraction.
- Dysmenorrhea includes, without limitation, antegrade contractions (fundus to cervix), retrograde contractions (cervix to fundus), and non- functional fibrillations.
- the composition of the present invention treats dysmenorrhea by selective action on the dyskinetic contractions without preventing the normal, regularized contractions necessary for menstruation. As menstrual blood does not clot, normal, regularized contractions are helpful to stop the bleeding. If there are no contractions, then the patient may not stop bleeding and may hemorrhage.
- the compound of the present invention interferes with the dyskinetic contractions causing dysmenorrhea, without stopping contractions entirely.
- compositions and/or devices of the invention or the compositions and/or devices used according to the invention are applied several hours before or just after onset of menstruation in order to treat or prevent dysmenorrhea.
- the treatment would continue for a few hours up to 6 days, as needed, to alleviate and prevent painful menstruation and symptoms such as nausea, fatigue, diarrhea, lower backache, and headache.
- the administration of the compound according to the invention to the subject results in reduced, negligible, or no adverse side effects.
- the side effects of common ⁇ -adrenergic agonists include, but are not limited to, cardiovascular such as palpitations, peripheral tremors, high heart rate, and low blood pressure; pulmonary edema and hypoglycemia; aggravation of preexisting diabetes and keto acidosis; tremors; nervousness; increased heart rate; palpitations; dizziness; headaches; drowsiness; vomiting; nausea; sweating; muscle cramps; and ECG changes.
- the use of the compounds according to the invention reduces or eliminates one or more of the above-noted side effects.
- the administration of the compounds reduce the incidence of one or more adverse side effects in the subject.
- the number of incidences of the one or more of adverse side effects in the subject is reduced with the administration of the compound according to the invention as compared to the number of such incidences, which would have been observed in the subject with the administration of terbutaline, ritodrine, or meluadrine.
- the ⁇ - adrenergic agonist is terbutaline, ritodrine hydrochloride, or HSR-81.
- the administration of a compound according to the invention reduces the incidence of one or more adverse side effects in the subject as compared to terbutaline.
- the number of incidences of increased heart rate, decrease in mean blood pressure, or both in the subject after the administration of the compound according to the invention is reduced compared to the number of such incidences, which would have been observed in the subject with the administration of terbutaline.
- the reduction of one or more of the adverse side effects by the compound used according to the invention is more than 10% reduction; or alternatively more than 20% reduction; or alternatively more than 30% reduction; or alternatively more than 40% reduction; or alternatively more than 50% reduction; or alternatively more than 60% reduction; or alternatively more than 70% reduction; or alternatively more than 80% reduction; or alternatively more than 90% reduction; or alternatively more than 99% reduction; or alternatively complete reduction of the adverse side effect.
- the above recited reduction in the one or more of the adverse side effects is as compared to the adverse side effects of other ⁇ -adrenergic agonists.
- the above recited reduction in the one or more of the adverse side effects is as compared to the adverse side effects of terbutaline.
- the ⁇ -adrenergic agonists suffer from a short half life or low bioavailability.
- the compounds used according to the invention have a longer half life or higher bioavailability as compared to other ⁇ -adrenergic agonists, such as but not limited to, terbutaline.
- the compound is of formula I:
- n is an integer selected from 1 or 2;
- X is a C C alkylene group
- Y is -N(R) 2 wherein each R is independently selected from hydrogen or Q-Q alkyl, or two R along with the nitrogen bound thereto join together to form a 3 to 7 membered heterocyclic ring optionally containing an oxygen atom; and
- the compound is of formula II:
- n is an integer selected from 1 or 2;
- X is a Ci-C 6 alkylene group
- Y is -N(R) 2 wherein each R is independently selected from hydrogen or Ci-C 6 alkyl, or two R along with the nitrogen bound thereto join together to form a 3 to 7 membered heterocyclic ring optionally containing an oxygen atom;
- the compound is of formula III:
- the metabolite is of formula IV:
- n is an integer selected from 1 or 2;
- X is a Ci-C 6 alkylene group
- the compound is of formula V:
- X is a C 1 -C3 alkylene group. In some embodiments of the above recited aspects, X is a -CH 2 - group.
- Y is -N(R) 2 wherein each R is hydrogen.
- Y is -N(R) 2 wherein each R is C ⁇ - C 6 alkyl. In some embodiments of the above recited aspects, Y is -N(R) 2 wherein each R is Ci-C 2 alkyl. In some embodiments of the above recited aspects, Y is -N(R) 2 wherein each R is methyl. In some embodiments of the above recited aspects, Y is -NHR wherein R is Ci-C 2 alkyl.
- Y is -N(R) 2 wherein two R along with the nitrogen bound thereto join together to form a 3 to 7 membered heterocyclic ring optionally containing an oxygen atom.
- * represents a carbon atom in R configuration. In some embodiments of the above recited aspects, * represents a carbon atom in S configuration. In some embodiments of the above recited aspects, * represents a carbon atom which is a mixture of R and S configuration.
- n is 1. In some embodiments of the above recited aspects, n is 2.
- the compounds of the invention can exist in unsolvated as well as solvated forms, including hydrated forms.
- the solvated forms, including hydrated forms and the like are equivalent to the unsolvated forms for purposes of the invention.
- the compound is in a form of a prodrug wherein the prodrug is selected from the group consisting of compounds wherein hydroxyl or amine groups are bonded to a group that, when administered to a subject, cleaves to form a free hydroxyl or amine group, respectively.
- the prodrug is selected from the group consisting of acetate, formate, benzoate and phosphate ester derivatives of hydroxyl functional group, and acetyl and benzoyl derivatives of amine functional group.
- the compound or the prodrug thereof is in a form of a pharmaceutically acceptable salt thereof wherein the pharmaceutically acceptable salt thereof is an acid addition salt wherein the acid is selected from the group consisting of hydrochloric, sulfuric, phosphoric, acetic, citric, oxalic, malonic, salicyclic, malic, gluconic, fumaric, succinic, ascorbic, maleic, and methanesulfonic acid.
- the pharmaceutically acceptable salt thereof is sulfuric acid.
- the compound is a metabolite thereof where metabolites are as described herein.
- the compound is a pharmaceutically acceptable salt of the metabolite of the compound, where pharmaceutically acceptable salts are as described herein.
- the compounds used according to the invention can be synthesized using routine synthetic chemistry known to one skilled in the art. For example, the syntheses of the compounds used according to the invention and their experimental data are described in US Patent No. 6,133,266 and US Patent No. 6,136,852, which are incorporated herein by reference in their entirety.
- composition comprising a compound used according to the invention and a pharmaceutically acceptable carrier, wherein the composition is suitable for use according to the invention.
- the compounds used according to the invention can be administered admixed with conventional excipients, such as, pharmaceutically acceptable liquid, semi-liquid or solid organic or inorganic carriers, which do not deleteriously react with the active compound in admixture therewith.
- Suitable pharmaceutically acceptable carriers include but are not limited to water, salt solutions, alcohols, vegetable oils, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin, perfume oil, fatty acid monoglycerides and diglycerides, pentaerythritol fatty acid esters, hydroxy methylcellulose, polyvinyl pyrrolidone, etc.
- the pharmaceutical preparations can be sterilized and if desired mixed with auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, coloring, flavoring, and/or aromatic substances and the like which do not deleteriously react with the active compounds.
- auxiliary agents e.g., lubricants, preservatives, stabilizers, wetting agents, emulsifiers, salts for influencing osmotic pressure, buffers, coloring, flavoring, and/or aromatic substances and the like which do not deleteriously react with the active compounds.
- Various delivery systems are known and can be used to administer the compounds or compositions according to the invention, including, for example, encapsulation in liposomes, microbubbles, emulsions, microparticles, microcapsules and the like.
- the required dosage can be administered as a single unit or in a sustained release form.
- the composition is administered as a formulation suitable for parenteral routes of administration, such as intravenous, intramuscular, percutaneous, and subcutaneous administration.
- parenteral routes of administration such as intravenous, intramuscular, percutaneous, and subcutaneous administration.
- suitable are solutions, preferably oily or aqueous solutions, as well as suspensions, emulsions, or implants, including suppositories.
- the intravenous formulation comprises approximately 0.20 mg to about 20 mg; or alternatively about 0.20 mg to about 10 mg; or alternatively about 0.20 mg to about 5 mg; or alternatively about 0.20 mg to about 3 mg; or alternatively about 0.20 mg to about 2 mg; or alternatively about 0.20 mg to about 1 mg; of the compound used according to the invention in an aqueous delivery system.
- the aqueous delivery system may comprise about 0.02% to about 0.5% (w/v) of an acetate, phosphate, or citrate buffer.
- the formulation has a pH of about 3.0 to about 7.0.
- the concentration of the compound in the intravenous formulation falls in the range of about 0.15 ⁇ ⁇ /mL to about 0.25 ⁇ /mL.
- the subject is administered an amount of the compound useful according to the invention in the range of about 3 igT ⁇ ig patient (or about 200 ⁇ g per patient) to about 60 patient (or about 4 mg per patient).
- the dosage may be administered intravenously as a single bolus injection to the subject, or as single bolus injection followed by a constant infusion for up to 24, 36, 48, or 72 hours, or as a constant infusion for up to 24, 36, 48, or 72 hours.
- the dosage may be administered subcutaneously or intravenously at intervals not less than 4 hours and for up to 24, 36, 48, or 72 hours.
- the subject is administered intravenously for 15 minutes at about 40 ⁇ g/min and then about 45 minutes at about 13 ⁇ .
- the subjects are those who have been admitted to an emergency room.
- the intravenous formulation is reconstituted from a freeze- dried drug product comprising the compound used according to the invention.
- the freeze-dried drug product further comprises carbohydrate and/or polyhydric alcohols.
- the carbohydrate may be mannose, ribose, trehalose, maltose, inositol, lactose, or the like.
- the polyhydric alcohols may be sorbitol, mannitol, or the like.
- the compound is administered by infusion.
- the infusion is performed at a rate of about 3 ⁇ g ⁇ gm or ⁇ g)/minute to about 60 ⁇ g/min; about 6 ⁇ g/minute to about 30 ⁇ ⁇ ; about 12 ⁇ to about 15 ⁇ ⁇ ; about 7 ⁇ to about 18 /minute; about 9 ⁇ ; about 13 ⁇ ' ⁇ : and about 16 ⁇ / ⁇ .
- the compound is formulated as a liquid formulation for administration in accordance with the various aspects and embodiments of the present invention.
- the liquid formulation comprises the compound in an amount of about 3 ⁇ g/mL to about 60 ⁇ g/mL, about 6 ⁇ g/mL to about 30 ⁇ g/mL, and about 12 ⁇ g/mL to about 30 ⁇ g/mL, and about 15 ⁇ g/mL to about 20 g/mL.
- the liquid formulation further comprises dextrose.
- the liquid formulation is an aqueous formulation.
- the liquid formulation is suitable for intravenous injection or infusion.
- the compound is used as a 2 mg, unit dose, lyophilized drug product.
- Other unit dose forms in the range of about 0.2 mg to about 20 mg are also contemplated.
- the lyophilized drug product further comprises lactose.
- compositions of the invention are delivered topically. Topical administration can involve the use of transdermal administration such as transdermal patches or iontophoresis devices.
- Dosage forms for topical administration of the compounds and compositions can include creams, sprays, lotions, gels, ointments, and the like.
- the compositions of the invention can be mixed to form white, smooth, homogeneous, opaque cream or lotion with, for example, benzyl alcohol 1 % or 2% (wt/wt) as a preservative, emulsifying wax, glycerin, isopropyl palmitate, lactic acid, purified water and sorbitol solution.
- compositions can contain polyethylene glycol 400. They can be mixed to form ointments with, for example, benzyl alcohol 2% (wt/wt) as preservative, white petrolatum, emulsifying wax, and tenox II (butylated hydroxyanisole, propyl gallate, citric acid, propylene glycol).
- benzyl alcohol 2% wt/wt
- tenox II butylated hydroxyanisole, propyl gallate, citric acid, propylene glycol.
- compositions can also be applied topically using a transdermal system, such as one of an acrylic-based polymer adhesive with a resinous cross-linking agent impregnated with the composition and laminated to an impermeable backing.
- a transdermal system such as one of an acrylic-based polymer adhesive with a resinous cross-linking agent impregnated with the composition and laminated to an impermeable backing.
- the compositions of the present invention are administered in the form of a transdermal patch, such as in the form of a sustained-release transdermal patch.
- the compositions of the present invention are administered in a form of a five day transdermal patch.
- the transdermal patches of the present invention can include any conventional form such as, for example, adhesive matrix, polymeric matrix, reservoir patch, matrix or monolithic-type laminated structure, and are generally comprised of one or more backing layers, adhesives, penetration enhancers, an optional rate controlling membrane and a release liner which is removed to expose the adhesives prior to application.
- Polymeric matrix patches also comprise a polymeric-matrix forming material.
- the transdermal patches comprise a therapeutically effective amount of the composition of the invention and optionally an antioxidant.
- antioxidants include, but are not limited to, hydralazine compounds, glutathione, vitamin C, vitamin E, cysteine, N-acetyl-cysteine, ⁇ -carotene, ubiquinone, ubiquinol-10, tocopherols, coenzyme Q, and the like.
- Suitable antioxidant enzymes include, but are not limited to, superoxide dismutase, catalase, glutathione peroxidase, and the like.
- Suitable antioxidants are described more fully in the literature, such as in Goodman and Gilman, The Pharmacological Basis of Therapeutics (9th Edition), McGraw-Hill, 1995; and the Merck Index on CD-ROM, Twelfth Edition, Version 12: 1 , 1996).
- the composition, the transdermal patch, or the delivery device can be a controlled release composition.
- a suitable biocompatible excipient for applying the compound include a lipophilic carrier or a hydrophilic carrier.
- Non-limiting examples of a lipophilic carrier include semi-synthetic glycerides of saturated fatty acids.
- Non-limiting examples of a hydrophilic carrier include polyethylene glycol having an average molecular weight of 6000, polyethylene glycol having an average molecular weight of 1500, polyethylene glycol having an average molecular weight of 400 or mixtures thereof.
- the biocompatible excipient can also include a muco-adhesive agent such as alginate, pectin, or cellulose derivative.
- the biocompatible excipient can also include a penetration enhancer such as bile salts, organic solvents, ethoxydiglycol, or interested fied stone oil.
- the excipient comprises between about 60 to 90% by weight lipophilic carrier, between about 5 to 25% mucoadhesive agent, and between about 5 to 20% penetration enhancer.
- the excipient comprises between about 60 to 90% by weight hydrophilic carrier, between about 5 to 25% muco-adhesive agent, and between about 5 to 20% penetration enhancer.
- the patch or the drug delivery device comprises a standard fragrance free lotion formulation.
- the biocompatible excipient can include glycerin, mineral oil, polycarbophil, carbomer 934P, hydrogenated palm oil, glyceride, sodium hydroxide, sorbic acid, and purified water.
- the transdermal patch contains, about 5-5000 mg; or alternatively about 5-4000 mg; or alternatively about 5-3000 mg; or alternatively about 5- 2000 mg; or alternatively about 5-1000 mg; or alternatively about 5-500 mg; or alternatively about 5-100 mg; or alternatively about 5-50 mg, of the compound used according to the invention.
- the transdermal patch administers a sustained release of the compound used according to the invention over a period of 6 days; or 5 days; or 4 days; or 3 days; or 2 days; or 1 day.
- unit dosage forms e.g., tablets, dragees or capsules having talc and/or a carbohydrate carrier or binder or the like, the carrier preferably being lactose and/or corn starch and/or potato starch; particulate solids, e.g., granules; and liquids and semi-liquids, e.g., syrups and elixirs or the like, wherein the active compound is protected with differentially degradable coatings, e.g., by microencapsulation, multiple coatings, etc.
- suitable for oral administration are, inter alia, tablets, dragees, capsules, pills, granules, suspensions and solutions.
- Each unit dose, e.g., each tablespoon of liquid or each tablet, or dragee contains, for example, 5-5000 mg of each active agent or the compound used according to the invention.
- the pharmaceutically acceptable carrier is a bioadhesive carrier.
- the bioadhesive carrier is a cross-linked water-insoluble but water- swellable polycarboxylic acid polymer.
- the cross-linked polycarboxylic acid polymer formulation is generally described in U.S. Pat. No. 4,615,697 (hereinafter "the '697 patent"), which is incorporated herein by reference. In general, at least about eighty percent of the monomers of the polymer in such a formulation may contain at least one carboxyl functionality.
- the cross-linking agent may be present at such an amount as to provide enough bioadhesion to allow the system to remain attached to the target epithelial surfaces for a sufficient time to allow the desired dosing to take place.
- This preferred level of bioadhesion can be attained when the cross-linking agent is present at about 0.1 to 6.0 weight percent of the polymer, with about 1.0 to 2.0 weight percent being most preferred, as long as the appropriate level of bioadhesion results.
- Bioadhesion can also be measured by commercially available surface tensiometers utilized to measure adhesive strength.
- the polymer formulation can be adjusted to control the release rate of the compounds used according to the invention, by varying the amount of cross-linking agent in the polymer.
- Suitable cross-linking agents include divinyl glycol, divinylbenzene, N,N-diallylacrylamide, 3,4-dihydroxy-l ,5-hexadiene, 2,5-dimethyl-l ,5-hexadiene and similar agents.
- a preferred polymer for use in such a formulation is Polycarbophil, U.S.P., which is commercially available from B.F. Goodrich Speciality Polymers of Cleveland, Ohio under the trade name NOVEON®-Ml .
- polycarbophil is a polyacrylic acid, cross-linked with divinyl glycol.
- Polycarbophil is a main ingredient in the vaginal moisturizer Replens®. It has also been used as a base for compositions with other active substances such as progesterone (Crinone®) (see U.S. Pat. No. 5,543,150) and Nonoxynol-9 (Advantage-S) (see U.S. Pat. No. 5,667,492).
- Other useful bioadhesive polymers that may be used in such a drug delivery system formulation are mentioned in the '697 patent. For example, these include polyacrylic acid polymers cross- linked with, for example, 3,4-dihydroxy-l,5-hexadiene, and polymefhacrylic acid polymers cross-linked with, for example, divinyl benzene.
- bioadhesive polymers may not be used in their salt form, because this would decrease their bioadhesive capability.
- bioadhesive polymers may be prepared by conventional free radical polymerization techniques utilizing initiators such as benzoyl peroxide, azobisisobutyronitrile, and the like. Exemplary preparations of useful bioadhesives are provided in the '697 patent.
- the bioadhesive formulation may be in the form of a gel, cream, tablet, pill, capsule, suppository, film, or any other pharmaceutically acceptable form that adheres to the mucosa and does not wash away easily.
- Different formulations are further described in the '697 Patent, which is incorporated herein by reference.
- the additives taught in the '697 patent may be mixed in with the cross- linked polymer in the formulation for maximum or desired efficacy of the delivery system or for the comfort of the patient.
- Such additives include, for example, lubricants, plasticizing agents, preservatives, gel formers, tablet formers, pill formers, suppository formers, film formers, cream formers, disintegrating agents, coatings, binders, vehicles, coloring agents, taste and/or odor controlling agents, humectants, viscosity controlling agents, pH-adjusting agents, and similar agents.
- the compounds used according to the invention or the other optional drug can be administered as an admixture or as a separate unit dosage form, either simultaneously therewith or at different times during the day from each other.
- the compound and the optional drug are preferably administered at least once daily (unless administered in a dosage form which delivers the active agents continuously) and more preferable several times daily, e.g., in 2 to 6 divided doses.
- the typical dose is about 0.5 to 1000 mg of each active agent.
- the method of the invention comprises intravaginal insertion of a device comprising a compound used according to the invention for treatment of dysmenorrhea in a pharmaceutically acceptable, non-toxic carrier.
- the composition is combined with a suitable delivery device or system which permits the transvaginal delivery of the drug to the uterus through the vaginal mucosa.
- Examples of the drug delivery system include, but are not limited to, a tampon device, vaginal ring, pessary, tablet, vaginal suppository, vaginal sponge, bioadhesive tablet, bioadhesive microparticle, cream, lotion, foam, ointment, solution and gel.
- a tampon device vaginal ring, pessary, tablet, vaginal suppository, vaginal sponge, bioadhesive tablet, bioadhesive microparticle, cream, lotion, foam, ointment, solution and gel.
- it can be a coating on a suppository wall or a sponge or other absorbent material impregnated with a liquid drug containing solution, lotion, or suspension of bioadhesive particles.
- Any form of drug delivery system which will effectively deliver the treatment agent to the uterus or the vaginal epithelium is intended to be included within the scope of this invention.
- the device is an absorbent vaginal tampon device having a proximal and a distal end. Located at the distal end is a means for delivery of the compound to the epithelium of the vagina.
- the device also includes a means for preferentially conveying fluid discharged from the uterus near the proximal end to the tampon and thereby preventing contact of the fluid with the compound.
- the device also has a means for retrieval of the device, such as a string or tape as used in tampons, vaginal rings and diaphragms.
- the drug delivery device can be a vaginal suppository.
- the compound and an optional drug are in the form of a microsphere for enhancing uptake of the compound and the drug.
- the microparticles have a diameter of 10-100 pm and can be prepared from starch, gelatin, albumin, collagen, or dextran.
- the compound can also be incorporated into creams, lotions, foams, paste, ointments, and gels which can be applied to the vagina using an applicator.
- Processes for preparing pharmaceuticals in cream, lotion, foam, paste, ointment and gel formats can be found throughout the literature.
- An example of a suitable system is a standard fragrance free lotion formulation containing glycerol, ceramides, mineral oil, petrolatum, parabens, fragrance and water.
- Suitable nontoxic pharmaceutically acceptable systems for use in the compositions of the present invention will be apparent to those skilled in the art of pharmaceutical formulations and examples are described in REMINGTON'S Pharmaceutical Sciences, 19th Edition, A. R. Gennaro, ed., 1995.
- suitable carriers will depend on the exact nature of the particular dosage form desired, e.g., whether the active ingredient(s) is/are to be formulated into a cream, lotion, foam, ointment, paste, solution, or gel, as well as on the compound.
- the excipient can be an inert or inactive substance used in the production of pharmaceutical products or other tablets, including without limitation any substance used as a binder, disintegrant, coating, compression/encapsulation aid, cream or lotion, lubricant, parenteral, sweetener or flavoring, suspending/gelling agent, or wet granulation agent.
- Binders include, e.g., carbopol, povidone, xanthan gum, etc.; coatings include, e.g., cellulose acetate phthalate, ethylcellulose, gellan gum, maltodextrin, etc.; compression/encapsulation aids include, e.g., calcium carbonate, dextrose, fructose dc, honey dc, lactose (anhydrate or monohydrate; optionally in combination with aspartame, cellulose, or microcrystalline cellulose), starch dc, sucrose, etc. ; disintegrants include, e.g., croscarmellose sodium, gellan gum, sodium starch glycolate, etc.; creams and lotions include, e.g.
- lubricants include, e.g., magnesium stearate, stearic acid, sodium stearyl fumarate, etc.
- materials for chewable tablets include, e.g., dextrose, fructose dc, lactose (monohydrate, optionally in combination with aspartame or cellulose), etc.
- parenterals include, e.g., mannitol, povidone, etc.
- plasticizers include, e.g., dibutyl sebacate, polyvinylacetate phthalate, etc.
- suspending/gelling agents include, e.g., carrageenan, sodium starch glycolate, xanthan gum, etc.
- sweeteners include, e.g., aspartame, dextrose, fructose dc, sorbitol, sucrose dc, etc
- wet granulation agents include, e.g., calcium carbonate, maltodextrin, microcrystalline cellulose, etc.
- the compound is administered in an amount of about 2000 ⁇ g (or 2 mg), about 1200 ⁇ , about 1000 ⁇ , about 800 ⁇ , about 600 ⁇ , about 450 ⁇ g, about 400 ⁇ g, about 250 ⁇ g, and about 200 ⁇ g (or 0.2 mg). In other embodiments, the compound is administered in an amount of about 200 ⁇ g to about 2000 ⁇ g.
- the compound is administered for a period of time up to about 6 days, up to about 5 days, up to about 4 days, up to about 3 days, up to about 2 days, up to about 1 day, up to about 8 hours (h), up to about 2 h, up to about 1 h, up to about 45 min; up to about 30 min, and up to about 15 min.
- the compound may be administered at various rates of administration, for various periods of time.
- MN-221 can be synthesized according to methods reported in literature. See, e.g., US Patent No. 6,133,266, which is incorporated herein by reference in its entirety.
- the test equipment used in the studies below are: Tension transducer, 45196A, Force displacement transducer 45196 A, FD Pick-up SB- IT (force displacement transducer), FD Pick-up TB-61 1T (force displacement transducer), Amplification unit for conversion 1829 (Strain pressure amplifier), Amplifier case 7747, Amplifier case 7903, Strain pressure amplifier, AP-601 G; Amplifier case, RMP-6004; Pen-writing recorder: RECTI-HORIZ 8K10 (Rectigraph), Pen-writing recorder: RECTI-HORIZ 8K20 (Rectigraph), Thermostatic chamber: Thermominder DX-10, Electronic balance PG3001-S, Pan electronic balance MC210S, Electronic balance 1412MP8, Refrigerated counter for drugs: MPR-1010R, medical freezer, MDF-U332, and Water purification system: Autostil WG-75.
- test substance was MN-221 ; the control substance was ritodrine hydrochloride (( ⁇ )-ery ⁇ ra-l-(/7-hydroxyphenyl)-2-[2-(/7-hydroxyphenyl)ethyl amino] -1-propanol
- a quarantine period of at least 1 week was set. Body weight was measured and general condition observed at the start and end of the quarantine period. Each animal was identified by writing an animal number at the root of the tail with Magic Ink during the quarantine period.
- the animals were housed in cages as a group of 5 or less. They were allowed to take feed (Rodent diet CE-2 solid food; Clea Japan, Inc.) and drink water (ultraviolet-irradiated tap water of Hotaka-cho) ad libitum. The temperature and humidity of the animal room was kept constant (23°C ⁇ 3°C and 50 ⁇ 10%, respectively). An illumination cycle with a room light being on for 12 hours (from 8:00 am to 8:00 pm) was used.
- Each of the substances was weighed and dissolved in distilled water to have a concentration of 1 x 10 "2 mol/L. Each solution was diluted as required in series (1 to 10) to 1 x 10 "8 mol/L for MN-221, 1 x 10 "7 mol/L for ritodrine hydrochloride, and to 1 x 10 "9 mol/L for isoproterenol bitartrate.
- Locke-Ringer solution The following substances were weighed and dissolved in distilled water to make 10 L: 90.0 g of NaCl, 4.2 g of C1, 2.85 g of CaCl 2 , 4.25 g of MgCl 2 ⁇ 6H 2 0, 5.0 g of glucose, and 5.0 g of NaHC0 3 .
- Each myometrial strip was suspended in an organ bath containing 10 mL of a Locke- Ringer solution at 37°C (aerated with 95% 0 2 + 5% C0 2 gas) with a load of about 1.0 g. After the amplitude and frequency of the muscular spontaneous contractions became stable, distilled water was added. Five minutes later, the test, control, or positive control substance was added cumulatively with intervals of 5 minutes. The myometrial contractile force was delivered to a strain pressure amplifier through a force displacement transducer and recorded on a Rectigraph.
- MN-221 inhibited the spontaneous contractions of the uterine muscle isolated from pregnant rats in a concentration-dependent manner ( Figure 1).
- the EC 5 0 and pD 2 values of the substance were 1.01 ⁇ 0.27 nmol/L and 9.16 ⁇ 0.14, respectively (Table 2).
- Ritodrine hydrochloride and isoproterenol bitartrate also inhibited the spontaneous contractions in a concentration-dependent manner ( Figure 1).
- the EC 50 (pD 2 ) value of the ritodrine hydrochloride and isoproterenol bitartrate was 39.81 ⁇ 13.28 nmol/L (7.59 ⁇ 0.13) and 0.42 ⁇ 0.10 nmol/L (9.52 ⁇ 0.13), respectively (Table 2).
- the inhibitory effect on the spontaneous contractions of the uterine muscle isolated from pregnant rats was observed in the following order: isoproterenol bitartrate >MN-221 > ritodrine hydrochloride.
- the data represent the mean + standard error.
- This study demonstrates the action mechanism of MN-221 by examining the interaction between the inhibitory effect of MN-221 on the spontaneous contraction of uterine muscle isolated from pregnant rats and the effect of various 3-adrenoceptor antagonists including CGP 20712A 1 (selective ⁇ -adrenoceptor antagonist), ICI 1 18,551 2 (selective 02- adrenoceptor antagonist), and SR 59230A 3 (selective ⁇ 3 -adrenoceptor antagonist).
- CGP 20712A 1 selective ⁇ -adrenoceptor antagonist
- ICI 1 18,551 2 selective 02- adrenoceptor antagonist
- SR 59230A 3 selective ⁇ 3 -adrenoceptor antagonist
- test substance was MN-221.
- Other chemicals used in the study were obtained from Nacalai Tesque, Inc.; SIGMA; Otsuka Pharmaceutical Factory, Inc.; and Yoneyama Yakuhin ogyo Co., Ltd.
- a quarantine period of at least 1 week was set. Body weight of each animal was measured and general condition observed at the start and end of the quarantine period. Each animal was identified by writing an animal number at the root of the tail with Magic Ink during the quarantine period.
- the animals were housed in cages as a group of 5 or less. They were allowed to take feed (Rodent diet CE-2 solid food; Clea Japan, Inc.) and drink water (ultraviolet-irradiated tap water of Hotaka-cho) ad libitum. The temperature and humidity of the animal room was kept constant (23°C ⁇ 3°C and 50 ⁇ 10%, respectively). An illumination cycle with a room light being on for 12 hours (from 8:00 am to 8:00 pm) was used.
- MN-221 was weighed and dissolved in distilled water to prepare a solution at 1 ⁇ 10 ⁇ 2 mol/L, which was diluted with distilled water in series (at a ratio of 1 to 10) to prepare solutions up to 1 ⁇ 10 "8 mol/L as required. Then, MN-221 was cumulatively added in the following concentration ranges.
- Locke-Ringer solution The following substances were weighed and dissolved in distilled water to make 10 L: 90.0 g of NaCl, 4.2 g of KC1, 2.85 g of CaCl 2 , 4.25 g of MgCl 2 -6H20, 5.0 g of glucose, and 5.0 g of NaHC0 3 .
- CGP 20712A solution CGP 20712A was weighed and dissolved in distilled water to prepare a solution at 1 x 10 "3 mol/L. This solution was used as a stock solution and divided into several portions for cryopreservation. An appropriate portion was diluted with distilled water in series (at a ratio of 1 to 10) to 1 ⁇ 10 "7 mol/L on the experimental day.
- ICI 1 18,551 solution ICI 118,551 was weighed and dissolved in distilled water to prepare a solution at 1 x 10 "3 mol/L. This solution was used as a stock solution and divided into several portions for cryopreservation. An appropriate portion was diluted with distilled water in series (at a ratio of 1 to 10) to 1 10 "6 mol/L on the experimental day.
- SR 59230A solution SR 59230A was weighed and dissolved in DMSO to prepare a solution at 1 x 10 "3 mol/L. This solution was used as a stock solution and divided into several portions for cryopreservation. An appropriate portion was diluted with distilled water in series (at a ratio of 1 to 10) to 1 ⁇ 10 "6 mol/L on the experimental day.
- each antagonist or distilled water was added to the bath to pretreat them for about 15 minutes.
- a solution of MN-221 in a concentration range of 3 ⁇ 10 "1 1 to 1 x 10 "6 mol/L as described above) was cumulatively added at intervals of 5 minutes.
- the contractile force of each sample was outputted through a force displacement transducer to a strain pressure amplifier and recorded with a rectigraph.
- a response rate to each concentration of MN-221 was calculated as a ratio of the sum of uterine contraction for 5 minutes after it was added to that for 5 minutes before it was added: the sum of uterine contraction before it was added was considered as 100%. Any variable point (peak) with amplitude (tension) of 0.2g or lower was excluded from analysis. Samples meeting any of the following criteria were rejected or removed from analysis.
- This EC 5 0 value was used to calculate the concentration ratio (CR) of EC 50 when an antagonist was added (for each sample) to that when no antagonist was added (mean value of the whole control group) and the logarithmic value of CR-1 (log [CR-1 ]).
- the added concentration of the antagonist was plotted on the X axis (logarithmic value) and log (CR-1) on the Y axis (Schild plot) to calculate the value of the X-axis intercept (pA 2 ) and slope of the line using linear approximation (Schild regression 5 ).
- the slope was statistically compared with a slope of 1 (paired t-test; a probability level of less than 5% was considered significant).
- no Schild regression was performed for the CGP and SR treatment groups because of no evident shift of the concentration response curve to the right.
- the mean and standard error were calculated for each data point obtained and indicated to two decimal places (or in 3 significant digit).
- CGP 20712A a selective ⁇ -adrenoceptor antagonist, had no evident antagonistic effect on the effect of MN-221 at a concentration of up to 1 x 10 "8 mol/L ( Figure 2).
- SR 59230A a selective /3 ⁇ 4- adrenoceptor antagonist, had no evident antagonistic effect on the effect of MN-221 at a concentration of up to 3 x 10 " mol/L ( Figure 4).
- ICI 1 18,551 a selective /3 ⁇ 4- adrenoceptor antagonist, had a concentration-dependent, antagonistic effect on the inhibitory effect of MN- 221 on uterine contraction (Figure 3A).
- the results of the Schild regression showed that the antagonistic effect of ICI 118,551 had a pA 2 value of 9.30 ⁇ 0.1 1 and a slope of 0.87 ⁇ 0.23, which was not significantly different from a slope of 1 (Figure 3B).
- Table 3 Effect of various ⁇ -adrenoceptor antagonists on inhibitory effect of MN-221 on spontaneous contraction of uterine muscle isolated from pregnant rat
- Each data indicates a mean ⁇ standard error.
- the inhibitory effect of MN-221 on the spontaneous contraction of the uterine muscle isolated from pregnant rats may be a response via ⁇ -adrenoceptors.
- test substance was MN-221 ; the control substance was ritodrine hydrochloride (( ⁇ )-erytAro-l-(p-hydroxyphenyl)-2-[2-(p-hydroxyphenyl)ethylamino]-l -propanol
- a quarantine period of at least 3 days was set. Body weight was measured and general condition observed at the start and end of the quarantine period. Each animal was identified by writing an animal number at the root of the tail with Magic Ink during the quarantine period.
- the animals were housed in cages as a group of 5 or less. They were allowed to take feed (Rodent diet CE-2 solid food; Clea Japan, Inc.) and drink water (ultraviolet-irradiated tap water of Hotaka-cho) ad libitum. The temperature and humidity of the animal room was kept constant (23 °C ⁇ 3°C and 50 ⁇ 10%, respectively). An illumination cycle with a room light being on for 12 hours (from 8:00 am to 8:00 pm) was used.
- Each of the substances was weighed and dissolved in distilled water to have a concentration of 1 ⁇ 10 "2 mol/L. Each solution was diluted as required in series (1 to 10) to 1 x 10 "8 mol/L for MN-221 , 1 ⁇ 10 "7 mol/L for ritodrine hydrochloride, and to 1 ⁇ 10 "9 mol/L for isoproterenol bitartrate.
- Modified Locke-Ringer solution The following substances were weighed and dissolved in distilled water to make 10 L: 88.0 g of NaCl, 4.0 g of KCl, 0.4 g of CaCl 2 , 0.38 g of MgCl 2 -6H 2 0, 0.2 g of KH 2 P0 4 , 2.02 g of Na 2 HP0 4 12H 2 0, 5.0 g of glucose, and 4.0 g of NaHC0 3 .
- PG F? record solution Prostarmon®-F Injection 1000 (containing 2000 g of PG F 2a in a 2 mL-ampoule) was diluted with distilled water to prepare a 500 ig/mL solution, as required.
- Oxytocin solution Five units of Atonin®-0 (containing 5 units of oxytocin in a 1 mL-ampoule) was diluted with distilled water to prepare a 100 mU/mL solution, as required.
- Forskolin solution An appropriate amount of forskolin was weighed and dissolved in DMSO to prepare a 1 x 10 "2 mol/L solution, which was stored at room temperature in the shade before use.
- test, control, or positive control substance solution was added cumulatively at intervals of 5 minutes.
- 1 x 10 ⁇ 5 mol/L of forskolin was added to obtain maximal relaxation.
- the contractile force of the uterine sample was delivered via a force displacement transducer to a strain pressure amplifier and recorded on a Rectigraph.
- a mean value and its standard error were calculated for the contraction by each concentration of the test, control, and positive control substance solutions as well as their pEC 50 and EC 50 values: the mean and standard error of the pEC 50 value were expressed to 2 decimal places, and those of the EC 50 value as 3 effective digits.
- variance was examined with Bartlett's test. When the variance was equal, parametric Tukey multiple comparison test was performed. When the variance was not equal, non-parametric Tukey multiple comparison test was performed. In either case, a probability level of less than 5% for both sides was considered to indicate a significant difference.
- MN-221 inhibited the PG F 2cr induced contraction of the uterine muscle isolated from pregnant rats in a concentration-dependent manner ( Figure 5), with EC 5 o and pEC 50 values of 66.4 ⁇ 19.3 nmol/L and 7.29 ⁇ 0.10, respectively (Table 3).
- Ritodrine hydrochloride and isoproterenol tartrate also inhibited the PG F 2a -induced contraction of the uterine muscle isolated from pregnant rats in a concentration-dependent manner ( Figure 5), with an EC 50 (pECso) value of 3430 ⁇ 720 nmol/L (5.58 ⁇ 0.1 1) and 5.10 ⁇ 0.633 nmol/L (8.32 ⁇ 0.05), respectively (Table 3).
- the potency of inhibitory effect of MN-221 on the PG F 2cr induced uterine contraction was significantly different from that of both ritodrine hydrochloride and isoproterenol bitartrate.
- the data represent the mean ⁇ standard error of 10 samples.
- MN-221 inhibited the oxytocin-induced contraction of the uterine muscle isolated from pregnant rats in a concentration-dependent manner ( Figure 6), with EC 50 and pEC 50 values of 2.25 ⁇ 0.440 nmol/L and 8.73 ⁇ 0.09, respectively (Table 4).
- Ritodrine hydrochloride and isoproterenol bitartrate also inhibited the oxytocin-induced contraction of the uterine muscle isolated from pregnant rats in a concentration-dependent manner, with an EC 50 (pEC 50 ) value of 133 ⁇ 16.8 nmol/L (6.92 ⁇ 0.07) and 0.556 ⁇ 0.0412 nmol/L (9.27 ⁇ 0.03), respectively (Table 4).
- the data represent the mean ⁇ standard error of 10 samples.
- MN-221 was dosed at 0.1 , 0.3, 1.0, 3.0, and 10.0 ⁇ g/kg/min.
- Ritodrine hydrochloride was dosed at 3.0, 10.0, 30.0, 100.0, and 300.0 ⁇ g/kg/min.
- Meluadrine tartrate (HSR-81 ) was dosed at 0.3, 1.0, 3.0, 10.0, and 30.0 /ig/kg/min.
- Terbutaline sulfate was dosed at 0.3, 1.0, 3.0, 10.0, and 30.0 ⁇ g/kg/min.
- Rats were anesthetized with urethane, and experiments were conducted based on balloon method.
- Uterine activity and mean blood pressure of dam were led to a pressure amplifier via a pressure transducer.
- heart rate pulse waves were led to tachometer. Recti-graphs were used for recording.
- the test substance, control substance, or positive control substance was administered intravenously and cumulatively every 15 minutes, while doses were increased gradually.
- MN-221 and other 32-adrenoceptor stimulants inhibited uterine motility dose-dependently ( Figures 7 and 8A, Table 1).
- MN-221 results in negligible or no adverse side effects in the subject as compared to other 3-adrenoceptor stimulants such as terbutaline, ritodrine or HSR-81.
- MN-221 at dose that sufficiently inhibits uterine activity has weak actions on heart rate and mean blood pressure of dam, showing that the agent is superior in organ selectivity to other ⁇ - adrenoceptor stimulants.
- MN-221 suppressed oxytocin-induced uterine contractions more than 90% at doses over 0.03 ⁇ g/kg/min ( Figures 9 & 10). Significant differences between the two groups were found for the following parameters: maternal heart rate, diastolic blood pressure, mean blood pressure, base excess, blood K+, blood lactate, plasma glucose, plasma insulin, plasma non- esterified fatty acid levels, and fetal plasma glucose and plasma insulin levels ( Figures 11, 12, 13, 14 & 15). MN-221 significantly inhibited oxytocin-induced uterine contractions at doses over 0.03 ⁇ g kg/min and showed reduced cardiovascular and metabolic side effects.
- test substances MN-221 ; ritodrine hydrochloride; meluadrine tartrate (HSR-81); isoproterenol tartrate (Sigma); and terbutaline sulfate (Sigma), were weighed and dissolved in distilled water, respectively. Further dilution was performed using distilled water considering administration dose concentrations.
- MN-221 was dosed at 10 "10 , 3x10 "10 , 10 ⁇ 9 , 3xl 0 "9 , 10 ⁇ 8 , 3x10 ⁇ 8 , 10 "7 , 3xl 0 “7 , 10 "6 mol/L.
- Isoproterenol tartrate was dosed at 10 "10 , 3xl0 “10 , 10 "9 , 3xl0 “9 , 10 ⁇ 8 , 3xlO ⁇ 8 , 10 ⁇ 7 , 3x10 " 10 "6 mol/L.
- MN-221 and all other drugs used in the test demonstrated inhibitory effect against oxytocin-induced contractions of isolated uterine muscles. Inhibitory effect of MN-221 against spontaneous contractions of uterine muscles was clearly stronger than that of HSR- 81, ritodrine and terbutaline ( Figure 16 & Table 1). It is contemplated that MN-221 inhibited spontaneous contractions of isolated uterine muscles of pregnant rabbits through ⁇ 2- adrenoceptor.
- Table 1 pD 2 values for the inhibitory effects of MN-221 and other P2-adrenocepter agonists on spontaneous contractions of uterine muscles isolated from pregnant rabbits
- test substances MN-221 ; ritodrine hydrochloride; meluadrine tartrate (HSR-81 ); isoproterenol tartrate (Sigma); and terbutaline sulfate (Sigma), were weighed and dissolved in distilled water, respectively. Further dilution was performed using distilled water considering administration dose concentrations.
- MN-221 was dosed at 1(T 10 , 3xl0 ⁇ 10 , 10 ⁇ 9 , 3 ⁇ 10 ⁇ 9 , 10 "8 , 3xl0 “8 , 1(T 7 , 3xl0 "7 , 10 "6 mol/L.
- Isoproterenol tartrate was dosed at 10 "10 , 3xl0 “10 , 10 “9 , 3xl0 ⁇ 9 , 10 "8 , 3xl0 “8 , 10 ⁇ 7 , 3x10 " 10 "6 mol/L.
- MN-221 and all other drugs used in the test demonstrated inhibitory effect against oxytocin-induced contractions of isolated uterine muscles. Inhibitory effect of MN-221 against oxytocin-induced contractions of uterine muscles was clearly stronger than that of HSR-81, ritodrine and terbutaline ( Figure 17 & Table 1). It is contemplated that MN-221 inhibited oxytocin-induced contractions of isolated uterine muscles of pregnant rabbits through ?2-adrenoceptor. Table 1: pD 2 values for the inhibitory effects of MN-221 and other P2-adrenocepter agonists on oxytocin-induced contractions of uterine muscles isolated from pregnant rabbits
- Myometrial activity and uterine blood flow is recorded in a group of women, aged 16 to 39 years. All have regular cycles of 25 to 32 days. They suffer regularly from menstrual pain. All are so disabled by the condition that they have to abstain from work for one to three days a month, even if they use non-narcotic analgesics. At least during the first menstrual day, all have a continuous lower abdominal pain, which varies in intensity; most of them also complain of symptoms such as nausea and vomiting. The symptoms during the recordings conform to those experienced during previous menstruations.
- Myometrial activity is recorded as changes in intrauterine pressure by a microtransducer catheter.
- the transducer is connected to an amplifier and a potentiometer recorder.
- Uterine blood flow is recorded by a technique based on measuring thermodilution from a heated thermistor to blood flow in the surrounding tissue.
- the thermistor is placed in contact with the endometrium of the fundus and, consequently, the recordings mainly reflect the blood flow at that site.
- the thermistor and the pressure transducer are inserted through the cervical canal into the uterus.
- the receptors are kept in position by the rigidity of the transducer catheter and by use of sterile paste around the catheters in the vagina.
- MN-221 is given as a single bolus intravenous injection of 0.30 mg, 0.60 mg, or 0.90 mg. Before and after the administration of MN-221 , pulse rate and arterial blood pressure (measured by auscultation) are registered at 5-minute intervals.
- the maximum intensity of the intrauterine pressure in the different women varies between 200 and 350 mm Hg.
- the duration of the contractions namely the time when the intrauterine pressure is higher than the basal tone, generally varies between 1.5 and 3 minutes, and contractions occurr with a frequency of about 20 to 40 per hour.
- MN-221 decreases the myometrial activity where the uterine contractions are either inhibited by the drug or appear with a lower frequency and amplitude. Furthermore, there are well defined periods of relaxation between the contractions. The local uterine blood flow generally increases after the drug is given.
- MN-221 causes substantially no increase in heart rate, blood pressure, palpitations, tremors, and/or flushes.
- MN-221 gives a positive response with relief in women as compared to placebo. The difference is statistically significant.
- the MN-221 transdermal patch provides relief to menstruating women throughout their menstrual cycle. In all subjects, MN-221 causes substantially no increase in heart rate, blood pressure, palpitations, tremors, and/or flushes.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Anesthesiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Gynecology & Obstetrics (AREA)
- Pregnancy & Childbirth (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description


























Claims





Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2011800127257A CN102791127A (en) | 2010-03-08 | 2011-03-07 | Compositions, methods, and devices for the treatment of dysmenorrhea |
| JP2012557149A JP2013522206A (en) | 2010-03-08 | 2011-03-07 | Composition for the treatment of dysmenorrhea, treatment method thereof and treatment device thereof |
| KR1020127025995A KR20130049178A (en) | 2010-03-08 | 2011-03-07 | Compositions, methods, and devices for the treatment of dysmenorrhea |
| EP11753866.0A EP2544534A4 (en) | 2010-03-08 | 2011-03-07 | Compositions, methods, and devices for the treatment of dysmenorrhea |
| BR112012022619A BR112012022619A2 (en) | 2010-03-08 | 2011-03-07 | compositions, methods, and means for treating dysmenorrhea |
| CA2791984A CA2791984A1 (en) | 2010-03-08 | 2011-03-07 | Compositions, methods, and devices for the treatment of dysmenorrhea |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US31167610P | 2010-03-08 | 2010-03-08 | |
| US61/311,676 | 2010-03-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011112499A1 true WO2011112499A1 (en) | 2011-09-15 |
Family
ID=44563793
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/027370 WO2011112499A1 (en) | 2010-03-08 | 2011-03-07 | Compositions, methods, and devices for the treatment of dysmenorrhea |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20120035265A1 (en) |
| EP (1) | EP2544534A4 (en) |
| JP (1) | JP2013522206A (en) |
| KR (1) | KR20130049178A (en) |
| CN (1) | CN102791127A (en) |
| BR (1) | BR112012022619A2 (en) |
| CA (1) | CA2791984A1 (en) |
| TW (1) | TW201136581A (en) |
| WO (1) | WO2011112499A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3687520A4 (en) * | 2017-09-28 | 2021-06-23 | Nevakar, Inc | Fixed dose combination formulations for treating pain |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387710A (en) * | 1992-02-03 | 1995-02-07 | Fujisawa Pharmaceutical Co., Ltd. | Ethanolamine derivatives having sympathomimetic and anti-pollakiuria activities |
| US6133266A (en) * | 1996-02-19 | 2000-10-17 | Kissei Pharmaceutical Co., Ltd. | 3,4-disubstituted phenylethanolaminotetralincarboxamide derivatives |
| US7030160B2 (en) * | 2000-06-06 | 2006-04-18 | Sanofi-Aventis | Propanolaminotetralines, preparation thereof and compositions containing same |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3708624B2 (en) * | 1996-03-27 | 2005-10-19 | キッセイ薬品工業株式会社 | 3,4-disubstituted phenylethanolaminotetralin carboxylic acid derivative |
| US6197327B1 (en) * | 1997-06-11 | 2001-03-06 | Umd, Inc. | Device and method for treatment of dysmenorrhea |
-
2011
- 2011-03-07 EP EP11753866.0A patent/EP2544534A4/en not_active Withdrawn
- 2011-03-07 CA CA2791984A patent/CA2791984A1/en not_active Abandoned
- 2011-03-07 CN CN2011800127257A patent/CN102791127A/en active Pending
- 2011-03-07 KR KR1020127025995A patent/KR20130049178A/en not_active Application Discontinuation
- 2011-03-07 WO PCT/US2011/027370 patent/WO2011112499A1/en active Application Filing
- 2011-03-07 US US13/042,214 patent/US20120035265A1/en not_active Abandoned
- 2011-03-07 JP JP2012557149A patent/JP2013522206A/en active Pending
- 2011-03-07 BR BR112012022619A patent/BR112012022619A2/en not_active IP Right Cessation
- 2011-03-08 TW TW100107725A patent/TW201136581A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387710A (en) * | 1992-02-03 | 1995-02-07 | Fujisawa Pharmaceutical Co., Ltd. | Ethanolamine derivatives having sympathomimetic and anti-pollakiuria activities |
| US6133266A (en) * | 1996-02-19 | 2000-10-17 | Kissei Pharmaceutical Co., Ltd. | 3,4-disubstituted phenylethanolaminotetralincarboxamide derivatives |
| US7030160B2 (en) * | 2000-06-06 | 2006-04-18 | Sanofi-Aventis | Propanolaminotetralines, preparation thereof and compositions containing same |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2544534A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2791984A1 (en) | 2011-09-15 |
| TW201136581A (en) | 2011-11-01 |
| JP2013522206A (en) | 2013-06-13 |
| KR20130049178A (en) | 2013-05-13 |
| EP2544534A4 (en) | 2015-05-13 |
| CN102791127A (en) | 2012-11-21 |
| BR112012022619A2 (en) | 2015-10-20 |
| EP2544534A1 (en) | 2013-01-16 |
| US20120035265A1 (en) | 2012-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20030104976A1 (en) | Analgesic methods using endothelin receptor ligands | |
| US10632086B2 (en) | Methods and compositions for treating Raynaud's disease | |
| US20020187169A1 (en) | Kavalactone compositions | |
| Van Laar et al. | Pharmacokinetics and clinical efficacy of rectal apomorphine in patients with Parkinson's disease: a study of five different suppositories | |
| PT996447E (en) | Combination therapy for modulating the human sexual response | |
| Janssen et al. | Use of topical ketanserin in the treatment of skin ulcers: a double-blind study | |
| EP2219447B1 (en) | S-alkylisothiouronium derivatives for treating uterine hypercontractility disorders | |
| US20120035265A1 (en) | Compositions, methods, and devices for the treatment of dysmenorrhea | |
| US20090291140A1 (en) | Treatment of dysmenorrhea via transdermal administration of nonsteroidal anti-inflammatory drugs | |
| US20110319490A1 (en) | Methods and compositions for the treatment of irritable bowel syndrome | |
| US12011423B2 (en) | S-alkylisothiouronium derivatives for treating uterine hypercontractility disorders | |
| JP4176358B2 (en) | Pharmaceutical composition for hemorrhoids | |
| US9889126B2 (en) | Use of naratriptan in the treatment of rosacea | |
| WO2003097021A2 (en) | Use of transdermal topical systems for the prevention and treatment of primary dysmenorrhea | |
| MXPA99010400A (en) | Combination therapy for modulating the human sexual response |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180012725.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11753866 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2791984 Country of ref document: CA |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011753866 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012557149 Country of ref document: JP Ref document number: 2011753866 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20127025995 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012022619 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012022619 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120906 |