WO2011106039A1 - Arylpiperazone opioid receptor antagonists - Google Patents
Arylpiperazone opioid receptor antagonists Download PDFInfo
- Publication number
- WO2011106039A1 WO2011106039A1 PCT/US2010/052311 US2010052311W WO2011106039A1 WO 2011106039 A1 WO2011106039 A1 WO 2011106039A1 US 2010052311 W US2010052311 W US 2010052311W WO 2011106039 A1 WO2011106039 A1 WO 2011106039A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- hydrogen
- opioid receptor
- receptor antagonist
- independently
- Prior art date
Links
- 229940123257 Opioid receptor antagonist Drugs 0.000 title claims abstract description 46
- 125000000217 alkyl group Chemical group 0.000 claims description 98
- 229910052739 hydrogen Inorganic materials 0.000 claims description 67
- 239000001257 hydrogen Substances 0.000 claims description 67
- 150000002431 hydrogen Chemical group 0.000 claims description 49
- 238000000034 method Methods 0.000 claims description 44
- 239000003401 opiate antagonist Substances 0.000 claims description 32
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 28
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 claims description 20
- 150000003839 salts Chemical class 0.000 claims description 19
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 18
- 102000003840 Opioid Receptors Human genes 0.000 claims description 17
- 108090000137 Opioid Receptors Proteins 0.000 claims description 17
- 125000003118 aryl group Chemical group 0.000 claims description 16
- 125000003342 alkenyl group Chemical group 0.000 claims description 14
- 229910052731 fluorine Inorganic materials 0.000 claims description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 12
- 125000001424 substituent group Chemical group 0.000 claims description 11
- 125000001188 haloalkyl group Chemical group 0.000 claims description 10
- 125000000304 alkynyl group Chemical group 0.000 claims description 8
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 8
- 229960003920 cocaine Drugs 0.000 claims description 8
- 125000004648 C2-C8 alkenyl group Chemical group 0.000 claims description 7
- 208000008589 Obesity Diseases 0.000 claims description 7
- 208000035475 disorder Diseases 0.000 claims description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 7
- 235000020824 obesity Nutrition 0.000 claims description 7
- 208000011117 substance-related disease Diseases 0.000 claims description 7
- 208000019901 Anxiety disease Diseases 0.000 claims description 6
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 6
- 230000036506 anxiety Effects 0.000 claims description 6
- 125000000623 heterocyclic group Chemical group 0.000 claims description 6
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 6
- 229910052760 oxygen Inorganic materials 0.000 claims description 6
- 229910052717 sulfur Inorganic materials 0.000 claims description 6
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 6
- 208000007848 Alcoholism Diseases 0.000 claims description 5
- 125000004649 C2-C8 alkynyl group Chemical group 0.000 claims description 5
- 230000006399 behavior Effects 0.000 claims description 5
- 208000030814 Eating disease Diseases 0.000 claims description 4
- 208000019454 Feeding and Eating disease Diseases 0.000 claims description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 4
- 235000014632 disordered eating Nutrition 0.000 claims description 4
- 201000009032 substance abuse Diseases 0.000 claims description 4
- 201000001320 Atherosclerosis Diseases 0.000 claims description 3
- 206010007559 Cardiac failure congestive Diseases 0.000 claims description 3
- 206010008111 Cerebral haemorrhage Diseases 0.000 claims description 3
- 206010010904 Convulsion Diseases 0.000 claims description 3
- 208000002249 Diabetes Complications Diseases 0.000 claims description 3
- 206010012655 Diabetic complications Diseases 0.000 claims description 3
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 3
- 208000003870 Drug Overdose Diseases 0.000 claims description 3
- 206010019280 Heart failures Diseases 0.000 claims description 3
- 206010020772 Hypertension Diseases 0.000 claims description 3
- 206010033296 Overdoses Diseases 0.000 claims description 3
- 208000006011 Stroke Diseases 0.000 claims description 3
- 230000003042 antagnostic effect Effects 0.000 claims description 3
- 231100000867 compulsive behavior Toxicity 0.000 claims description 3
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 3
- 206010012601 diabetes mellitus Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 206010013663 drug dependence Diseases 0.000 claims description 3
- 231100000725 drug overdose Toxicity 0.000 claims description 3
- 206010015037 epilepsy Diseases 0.000 claims description 3
- 208000028329 epileptic seizure Diseases 0.000 claims description 3
- 201000001421 hyperglycemia Diseases 0.000 claims description 3
- 208000020346 hyperlipoproteinemia Diseases 0.000 claims description 3
- 239000008194 pharmaceutical composition Substances 0.000 claims description 3
- 208000020016 psychiatric disease Diseases 0.000 claims description 3
- 230000001850 reproductive effect Effects 0.000 claims description 3
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 3
- 201000000980 schizophrenia Diseases 0.000 claims description 3
- 230000001568 sexual effect Effects 0.000 claims description 3
- 231100000736 substance abuse Toxicity 0.000 claims description 3
- 125000006592 (C2-C3) alkenyl group Chemical group 0.000 claims description 2
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 claims description 2
- 206010013654 Drug abuse Diseases 0.000 claims description 2
- 206010057852 Nicotine dependence Diseases 0.000 claims description 2
- 208000025569 Tobacco Use disease Diseases 0.000 claims description 2
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 2
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 2
- 208000029790 metamphetamine dependence Diseases 0.000 claims description 2
- 229930192474 thiophene Natural products 0.000 claims description 2
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 81
- 239000000243 solution Substances 0.000 description 52
- 150000001875 compounds Chemical class 0.000 description 43
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 34
- 238000005481 NMR spectroscopy Methods 0.000 description 32
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 31
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 26
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 26
- 230000002829 reductive effect Effects 0.000 description 25
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 23
- 239000002904 solvent Substances 0.000 description 22
- 239000000203 mixture Substances 0.000 description 18
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 18
- -1 N-substituted 3,4-dimethyl-4-(3-hydroxyphenyl)piperidines Chemical class 0.000 description 16
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 16
- 239000007787 solid Substances 0.000 description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 14
- XEKOWRVHYACXOJ-UHFFFAOYSA-N ethyl acetate Substances CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 14
- 239000003921 oil Substances 0.000 description 14
- 235000019198 oils Nutrition 0.000 description 14
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 102000048260 kappa Opioid Receptors Human genes 0.000 description 12
- 238000002390 rotary evaporation Methods 0.000 description 12
- 238000011282 treatment Methods 0.000 description 12
- 108020001588 κ-opioid receptors Proteins 0.000 description 12
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 239000005557 antagonist Substances 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 238000004587 chromatography analysis Methods 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- 229960001866 silicon dioxide Drugs 0.000 description 10
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 10
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Natural products C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 9
- PLDWAJLZAAHOGG-UHFFFAOYSA-N 1-bromo-3-methoxybenzene Chemical compound COC1=CC=CC(Br)=C1 PLDWAJLZAAHOGG-UHFFFAOYSA-N 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 8
- 102000051367 mu Opioid Receptors Human genes 0.000 description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 8
- 102000005962 receptors Human genes 0.000 description 8
- 108020003175 receptors Proteins 0.000 description 8
- YGCZTXZTJXYWCO-UHFFFAOYSA-N 3-phenylpropanal Chemical compound O=CCCC1=CC=CC=C1 YGCZTXZTJXYWCO-UHFFFAOYSA-N 0.000 description 7
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 239000000460 chlorine Substances 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- UZHSEJADLWPNLE-GRGSLBFTSA-N naloxone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(O)C2=C5[C@@]13CCN4CC=C UZHSEJADLWPNLE-GRGSLBFTSA-N 0.000 description 7
- 150000004885 piperazines Chemical class 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 7
- 108020001612 μ-opioid receptors Proteins 0.000 description 7
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 235000019439 ethyl acetate Nutrition 0.000 description 6
- 239000012458 free base Substances 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 6
- 238000005932 reductive alkylation reaction Methods 0.000 description 6
- 238000010898 silica gel chromatography Methods 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 0 C*C*c1cc(C*=O)ccc1 Chemical compound C*C*c1cc(C*=O)ccc1 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- 108700023159 delta Opioid Receptors Proteins 0.000 description 5
- 102000048124 delta Opioid Receptors Human genes 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 229960004127 naloxone Drugs 0.000 description 5
- 229960003086 naltrexone Drugs 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 229910015845 BBr3 Inorganic materials 0.000 description 4
- 206010012335 Dependence Diseases 0.000 description 4
- 101100244562 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) oprD gene Proteins 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 4
- 229960002715 nicotine Drugs 0.000 description 4
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- SZXBQTSZISFIAO-ZETCQYMHSA-N (2s)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoic acid Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)OC(C)(C)C SZXBQTSZISFIAO-ZETCQYMHSA-N 0.000 description 3
- MCMMCRYPQBNCPH-WMIMKTLMSA-N DPDPE Chemical compound C([C@H](N)C(=O)N[C@@H]1C(C)(C)SSC([C@@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)CNC1=O)C(O)=O)(C)C)C1=CC=C(O)C=C1 MCMMCRYPQBNCPH-WMIMKTLMSA-N 0.000 description 3
- 235000011054 acetic acid Nutrition 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 230000008485 antagonism Effects 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 230000017858 demethylation Effects 0.000 description 3
- 238000010520 demethylation reaction Methods 0.000 description 3
- 238000002825 functional assay Methods 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 3
- 230000003595 spectral effect Effects 0.000 description 3
- DVGWFQILDUEEGX-UUOKFMHZSA-N (2r,3r,4s,5r)-2-(6,8-diaminopurin-9-yl)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound NC1=NC2=C(N)N=CN=C2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O DVGWFQILDUEEGX-UUOKFMHZSA-N 0.000 description 2
- HPZJMUBDEAMBFI-WTNAPCKOSA-N (D-Ala(2)-mephe(4)-gly-ol(5))enkephalin Chemical compound C([C@H](N)C(=O)N[C@H](C)C(=O)NCC(=O)N(C)[C@@H](CC=1C=CC=CC=1)C(=O)NCCO)C1=CC=C(O)C=C1 HPZJMUBDEAMBFI-WTNAPCKOSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- ONIKNECPXCLUHT-UHFFFAOYSA-N 2-chlorobenzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1Cl ONIKNECPXCLUHT-UHFFFAOYSA-N 0.000 description 2
- HXZDAOSDNCHKFE-UHFFFAOYSA-N 3-(3,4-dimethylpiperidin-4-yl)phenol Chemical class CC1CNCCC1(C)C1=CC=CC(O)=C1 HXZDAOSDNCHKFE-UHFFFAOYSA-N 0.000 description 2
- KTVXOUHJOOZWFP-QGZVFWFLSA-N 3-[(2r)-2-methyl-4-(3-phenylpropyl)piperazin-1-yl]phenol Chemical compound C([C@H](N(CC1)C=2C=C(O)C=CC=2)C)N1CCCC1=CC=CC=C1 KTVXOUHJOOZWFP-QGZVFWFLSA-N 0.000 description 2
- UXPDMYIUDIUZIW-SECBINFHSA-N 3-[(2r)-2-methylpiperazin-1-yl]phenol Chemical compound C[C@@H]1CNCCN1C1=CC=CC(O)=C1 UXPDMYIUDIUZIW-SECBINFHSA-N 0.000 description 2
- HXZDAOSDNCHKFE-GWCFXTLKSA-N 3-[(3r,4s)-3,4-dimethylpiperidin-4-yl]phenol Chemical compound C[C@H]1CNCC[C@]1(C)C1=CC=CC(O)=C1 HXZDAOSDNCHKFE-GWCFXTLKSA-N 0.000 description 2
- WORNGLHQKRZDIL-UHFFFAOYSA-N 3-[4-(3-phenylpropyl)piperazin-1-yl]phenol Chemical compound OC1=CC=CC(N2CCN(CCCC=3C=CC=CC=3)CC2)=C1 WORNGLHQKRZDIL-UHFFFAOYSA-N 0.000 description 2
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 2
- 108700022183 Ala(2)-MePhe(4)-Gly(5)- Enkephalin Proteins 0.000 description 2
- 108700022182 D-Penicillamine (2,5)- Enkephalin Proteins 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- 108010016626 Dipeptides Proteins 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 2
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 2
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 description 2
- 208000003698 Heroin Dependence Diseases 0.000 description 2
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 description 2
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 description 2
- APSUXPSYBJVPPS-YAUKWVCOSA-N Norbinaltorphimine Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CC=2C=3C[C@]4(O)[C@]67CCN(CC8CC8)[C@@H]4CC=4C7=C(C(=CC=4)O)O[C@H]6C=3NC=25)O)CC1)O)CC1CC1 APSUXPSYBJVPPS-YAUKWVCOSA-N 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 2
- 229910000564 Raney nickel Inorganic materials 0.000 description 2
- 229910007161 Si(CH3)3 Inorganic materials 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 201000007930 alcohol dependence Diseases 0.000 description 2
- UPNUIXSCZBYVBB-JVFUWBCBSA-N alvimopan Chemical compound C([C@@H](CN1C[C@@H]([C@](CC1)(C)C=1C=C(O)C=CC=1)C)C(=O)NCC(O)=O)C1=CC=CC=C1 UPNUIXSCZBYVBB-JVFUWBCBSA-N 0.000 description 2
- 229960004516 alvimopan Drugs 0.000 description 2
- 230000008503 anti depressant like effect Effects 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 230000003542 behavioural effect Effects 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 229960002069 diamorphine Drugs 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- LJQKCYFTNDAAPC-UHFFFAOYSA-N ethanol;ethyl acetate Chemical compound CCO.CCOC(C)=O LJQKCYFTNDAAPC-UHFFFAOYSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N hexane Substances CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000002618 kappa opiate receptor antagonist Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229960005181 morphine Drugs 0.000 description 2
- 229940127240 opiate Drugs 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 150000002989 phenols Chemical class 0.000 description 2
- 150000003053 piperidines Chemical class 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- QVHJQCGUWFKTSE-RXMQYKEDSA-N (2r)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound OC(=O)[C@@H](C)NC(=O)OC(C)(C)C QVHJQCGUWFKTSE-RXMQYKEDSA-N 0.000 description 1
- 125000006593 (C2-C3) alkynyl group Chemical group 0.000 description 1
- 125000006650 (C2-C4) alkynyl group Chemical group 0.000 description 1
- 125000006564 (C4-C8) cycloalkyl group Chemical group 0.000 description 1
- ZWAQJGHGPPDZSF-UHFFFAOYSA-N 1-prop-2-enylpiperazine Chemical compound C=CCN1CCNCC1 ZWAQJGHGPPDZSF-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- VRPJIFMKZZEXLR-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid Chemical compound CC(C)(C)OC(=O)NCC(O)=O VRPJIFMKZZEXLR-UHFFFAOYSA-N 0.000 description 1
- UYVKBQMEHGCCDT-UHFFFAOYSA-N 2-aminoacetic acid;2-aminobutanoic acid Chemical compound NCC(O)=O.CCC(N)C(O)=O UYVKBQMEHGCCDT-UHFFFAOYSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- DXOHZOPKNFZZAD-UHFFFAOYSA-N 2-ethylpiperazine Chemical compound CCC1CNCCN1 DXOHZOPKNFZZAD-UHFFFAOYSA-N 0.000 description 1
- ILSNIEQODZEBKZ-UHFFFAOYSA-N 2-ethylpiperazine;dihydrobromide Chemical compound Br.Br.CCC1CNCCN1 ILSNIEQODZEBKZ-UHFFFAOYSA-N 0.000 description 1
- LSBDFXRDZJMBSC-UHFFFAOYSA-N 2-phenylacetamide Chemical compound NC(=O)CC1=CC=CC=C1 LSBDFXRDZJMBSC-UHFFFAOYSA-N 0.000 description 1
- XVTYIPADYOBHDQ-NXEZZACHSA-N 3-[(2r,5r)-2,5-dimethylpiperazin-1-yl]phenol Chemical compound C[C@@H]1CN[C@H](C)CN1C1=CC=CC(O)=C1 XVTYIPADYOBHDQ-NXEZZACHSA-N 0.000 description 1
- XVTYIPADYOBHDQ-VHSXEESVSA-N 3-[(2r,5s)-2,5-dimethylpiperazin-1-yl]phenol Chemical compound C[C@@H]1CN[C@@H](C)CN1C1=CC=CC(O)=C1 XVTYIPADYOBHDQ-VHSXEESVSA-N 0.000 description 1
- UXPDMYIUDIUZIW-VIFPVBQESA-N 3-[(2s)-2-methylpiperazin-1-yl]phenol Chemical class C[C@H]1CNCCN1C1=CC=CC(O)=C1 UXPDMYIUDIUZIW-VIFPVBQESA-N 0.000 description 1
- UPNZWPCGQYKAGS-WWPIYYJJSA-N 3-[(2s)-2-methylpiperazin-1-yl]phenol;dihydrobromide Chemical compound Br.Br.C[C@H]1CNCCN1C1=CC=CC(O)=C1 UPNZWPCGQYKAGS-WWPIYYJJSA-N 0.000 description 1
- QWYLZMWLVSMZHG-NEPJUHHUSA-N 3-[(2s)-4-[(2r)-2-aminopropyl]-2-methylpiperazin-1-yl]phenol Chemical compound C[C@H]1CN(C[C@H](N)C)CCN1C1=CC=CC(O)=C1 QWYLZMWLVSMZHG-NEPJUHHUSA-N 0.000 description 1
- GJLGVVUWOQECLH-XJKSGUPXSA-N 3-[(2s)-4-[(2s)-2-amino-3-methylbutyl]-2-methylpiperazin-1-yl]phenol Chemical compound C[C@H]1CN(C[C@@H](N)C(C)C)CCN1C1=CC=CC(O)=C1 GJLGVVUWOQECLH-XJKSGUPXSA-N 0.000 description 1
- RNCUEOOFKVDGTE-MSOLQXFVSA-N 3-[(2s,5r)-2,5-dimethyl-4-(3-phenylpropyl)piperazin-1-yl]phenol Chemical compound C([C@H](C)N(C[C@H]1C)C=2C=C(O)C=CC=2)N1CCCC1=CC=CC=C1 RNCUEOOFKVDGTE-MSOLQXFVSA-N 0.000 description 1
- XVTYIPADYOBHDQ-UWVGGRQHSA-N 3-[(2s,5s)-2,5-dimethylpiperazin-1-yl]phenol Chemical compound C[C@H]1CN[C@@H](C)CN1C1=CC=CC(O)=C1 XVTYIPADYOBHDQ-UWVGGRQHSA-N 0.000 description 1
- LVVHEFJXPXAUDD-BULFRSBZSA-N 3-[(3r,4r)-1-[(3s)-3-cyclohexyl-3-hydroxypropyl]-3,4-dimethylpiperidin-4-yl]phenol Chemical compound C1([C@@H](O)CCN2C[C@@H]([C@](CC2)(C)C=2C=C(O)C=CC=2)C)CCCCC1 LVVHEFJXPXAUDD-BULFRSBZSA-N 0.000 description 1
- ZGUVWVUDGDYLFD-UHFFFAOYSA-N 3-[2-ethyl-4-(3-phenylpropyl)piperazin-1-yl]phenol;dihydrochloride Chemical compound Cl.Cl.C1CN(C=2C=C(O)C=CC=2)C(CC)CN1CCCC1=CC=CC=C1 ZGUVWVUDGDYLFD-UHFFFAOYSA-N 0.000 description 1
- QZUYFNRMMUUWLX-UHFFFAOYSA-N 3-[3,4-dimethyl-1-(3-phenylpropyl)piperidin-4-yl]phenol Chemical compound C1CC(C=2C=C(O)C=CC=2)(C)C(C)CN1CCCC1=CC=CC=C1 QZUYFNRMMUUWLX-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000000103 Anorexia Nervosa Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 208000032841 Bulimia Diseases 0.000 description 1
- 206010006550 Bulimia nervosa Diseases 0.000 description 1
- QUMCIHKVKQYNPA-RUZDIDTESA-N C1(CCCCC1)CN1[C@@H](C=2N(C=3C=NC(=NC1=3)NC1=C(C=C(C(=O)NC3CCN(CC3)C)C=C1)OC)C(=NN=2)C)CC Chemical compound C1(CCCCC1)CN1[C@@H](C=2N(C=3C=NC(=NC1=3)NC1=C(C=C(C(=O)NC3CCN(CC3)C)C=C1)OC)C(=NN=2)C)CC QUMCIHKVKQYNPA-RUZDIDTESA-N 0.000 description 1
- RESLGEHROKIGBM-UHFFFAOYSA-N CC(C)CC(C)c1ncccn1 Chemical compound CC(C)CC(C)c1ncccn1 RESLGEHROKIGBM-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-UWTATZPHSA-N D-alanine Chemical compound C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 208000012661 Dyskinesia Diseases 0.000 description 1
- 108010092674 Enkephalins Proteins 0.000 description 1
- 229940124602 FDA-approved drug Drugs 0.000 description 1
- 208000001613 Gambling Diseases 0.000 description 1
- 208000017228 Gastrointestinal motility disease Diseases 0.000 description 1
- 208000004454 Hyperalgesia Diseases 0.000 description 1
- 208000035154 Hyperesthesia Diseases 0.000 description 1
- URLZCHNOLZSCCA-VABKMULXSA-N Leu-enkephalin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=CC=C1 URLZCHNOLZSCCA-VABKMULXSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- SCIFESDRCALIIM-VIFPVBQESA-N N-methyl-L-phenylalanine Chemical compound C[NH2+][C@H](C([O-])=O)CC1=CC=CC=C1 SCIFESDRCALIIM-VIFPVBQESA-N 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102000015336 Nerve Growth Factor Human genes 0.000 description 1
- 208000026251 Opioid-Related disease Diseases 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- 206010038678 Respiratory depression Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102000003566 TRPV1 Human genes 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 206010001584 alcohol abuse Diseases 0.000 description 1
- 208000025746 alcohol use disease Diseases 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000000954 anitussive effect Effects 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000001773 anti-convulsant effect Effects 0.000 description 1
- 230000002686 anti-diuretic effect Effects 0.000 description 1
- 230000002932 anti-schizophrenic effect Effects 0.000 description 1
- 230000002738 anti-smoking effect Effects 0.000 description 1
- 239000000043 antiallergic agent Substances 0.000 description 1
- 229940124346 antiarthritic agent Drugs 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 239000001961 anticonvulsive agent Substances 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 229940124538 antidiuretic agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 229940125684 antimigraine agent Drugs 0.000 description 1
- 239000002282 antimigraine agent Substances 0.000 description 1
- 239000000164 antipsychotic agent Substances 0.000 description 1
- 229940005529 antipsychotics Drugs 0.000 description 1
- 239000003435 antirheumatic agent Substances 0.000 description 1
- 239000003434 antitussive agent Substances 0.000 description 1
- 229940124584 antitussives Drugs 0.000 description 1
- 230000000949 anxiolytic effect Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000009699 differential effect Effects 0.000 description 1
- 235000005686 eating Nutrition 0.000 description 1
- 238000005265 energy consumption Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 229960004756 ethanol Drugs 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000021824 exploration behavior Effects 0.000 description 1
- PJMPHNIQZUBGLI-UHFFFAOYSA-N fentanyl Chemical compound C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 PJMPHNIQZUBGLI-UHFFFAOYSA-N 0.000 description 1
- 229960002428 fentanyl Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 238000012048 forced swim test Methods 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 150000004674 formic acids Chemical class 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 238000011990 functional testing Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 231100000225 lethality Toxicity 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 229960001252 methamphetamine Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000003887 narcotic antagonist Substances 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 229940053128 nerve growth factor Drugs 0.000 description 1
- 230000004112 neuroprotection Effects 0.000 description 1
- AKRYBBWYDSDZHG-UHFFFAOYSA-N nitrosobis(2-oxopropyl)amine Chemical compound CC(=O)CN(N=O)CC(C)=O AKRYBBWYDSDZHG-UHFFFAOYSA-N 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 201000005040 opiate dependence Diseases 0.000 description 1
- 239000000014 opioid analgesic Substances 0.000 description 1
- 229940005483 opioid analgesics Drugs 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 125000005004 perfluoroethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 230000000079 pharmacotherapeutic effect Effects 0.000 description 1
- 238000001050 pharmacotherapy Methods 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- YZTJYBJCZXZGCT-UHFFFAOYSA-N phenylpiperazine Chemical compound C1CNCCN1C1=CC=CC=C1 YZTJYBJCZXZGCT-UHFFFAOYSA-N 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 108010074732 preproenkephalin Proteins 0.000 description 1
- 230000006977 prepulse inhibition Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 230000005588 protonation Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 239000002469 receptor inverse agonist Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000000021 stimulant Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- QQWYQAQQADNEIC-RVDMUPIBSA-N tert-butyl [(z)-[cyano(phenyl)methylidene]amino] carbonate Chemical compound CC(C)(C)OC(=O)O\N=C(/C#N)C1=CC=CC=C1 QQWYQAQQADNEIC-RVDMUPIBSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- NZIQBDROTUFRHZ-UHFFFAOYSA-N tritert-butyl phosphite Chemical compound CC(C)(C)OP(OC(C)(C)C)OC(C)(C)C NZIQBDROTUFRHZ-UHFFFAOYSA-N 0.000 description 1
- BWHDROKFUHTORW-UHFFFAOYSA-N tritert-butylphosphane Chemical compound CC(C)(C)P(C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000012873 virucide Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/08—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms
- C07D295/096—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms with the ring nitrogen atoms and the oxygen or sulfur atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to 4-arylpiperazine compounds. These compounds function as opioid receptor antagonists, and can be used to treat a variety of disease states.
- the opioid receptors, ⁇ , ⁇ , ⁇ , and the opioid-like receptor ORL- 1 belong to the super family of G-protein coupled receptors (GPCRs) that possess seven helical trans-membrane spanning domains in their architecture. 1
- GPCRs G-protein coupled receptors
- the majority of research efforts focused upon this group of proteins has been directed toward the ⁇ receptor since it mediates the actions of both the opiate and opioid analgesics such as morphine and fentanyl, respectively.
- the entire family of proteins is actively involved in a host of biological processes.
- selective antagonists has demonstrated that pharmacotherapeutic opportunities exist via both negative and positive modulation of this receptor family.
- the opioid receptor system has been extensively studied, and thousands of compounds have been synthesized and evaluated by in vitro binding and functional assays as well as by animal models.
- An integral part of the effort to characterize the opioid receptor system has been the discovery of potent, pure antagonists.
- Naloxone (la) and naltrexone (lb), both competitive antagonists at ⁇ , ⁇ , and ⁇ opioid receptors, 9 have been extensively used as pharmacological tools to identify and characterize opioid systems (see Figure 1 for structures). Additionally, naloxone is approved to treat heroin overdose and to reverse respiratory depression caused by morphine. 9 Naltrexone is used to treat heroin and alcohol abuse.
- alvimopan (3) which is an FDA-approved drug for GI motility disorder, 1 5 LY255,582 (2d), 13 ' 16 which was developed to treat obesity, and the selective ⁇ opioid receptor antagonist
- opioid receptor antagonists such as LY255582 have been found to increase metabolic energy consumption and reduce the weight in obese rats while maintaining muscle mass. These reports suggest that opioid receptor antagonists may be useful in preventing, treating, and/or ameliorating the effect of obesity. Eli Lilly and Company has developed new classes of opioid receptor antagonists that interact with the ⁇ , ⁇ , and K receptors (termed non-selective) as potential pharmacotherapies to treat obesity and related diseases.
- Aryl-substituted piperazines (5) are a new class of opioid receptor antagonists (see the Examples section below for representative structures). Similar to the N-substituted 3,4- dimethyl-4-(3-hydroxyphenyl)piperidines, even the N-methyl substituted analog 5f is a pure opioid antagonist. Changing the N-substituent to an N-phenylpropyl group gives 5b, which has K c values of 0.88, 13.4, and 4.09 nM at the ⁇ , ⁇ , and ⁇ opioid receptors, which are similar to the c values of N-phenylpropyl 3,4-dimethyl-4-(3-hydroxyphenyl)piperidine 2c (RTI- 5989-264).
- the JDTic-like analog from this class 5j has K e values of 22, 274, and 2.7 nM at the ⁇ , ⁇ , and k opioid receptors, respectively (see Table 1 ). All compounds of this class thus far synthesized are relatively nonselective opioid receptor antagonists. Thus, their opioid receptor properties are more like those of naloxone (l a), naltrexone (lb), and the originally reported N-substituted 3,4-dimethyl-4-(3-hydroxyphenyl)piperidines. 13
- the present invention is directed to aryl-substituted piperazine opioid receptor antagonists represented by the formula (I):
- R is hydrogen, OH, OCi_ alkyl, Ci -8 alkyl, C
- Y 3 is hydrogen, Br, CI, F, CN, CF 3 , N0 2 , OR 8 , C0 2 R 9 , C , -6 alkyl, NR, 0 Rn , NHCOR, 2 , NHC0 2 R i 2, CONRi 3 R, 4 or CH 2 (CH 2 ) n Y 2 ;
- Ri , R 2 , R 3 and R are each, independently, one of the following structures:
- Ri and R 2 , R 2 and R 3 and/or R 3 and R4 are bonded together to form a cyclo alkyl group or a bridged heterocyclic ring;
- each Yi is, independently, hydrogen, OH, Br, CI, F, CN, CF 3 , N0 2 , N 3 , OR 8 , C0 2 R 9 , Cue alkyl, NR 10 Rn , NHCOR, 2 , NHC0 2 R, 2 , CONRi 3 R, 4 , or CH 2 (CH 2 ) n Y 2 , or two adjacent Y, groups form a -0-CH 2 -0- or -0-CH 2 CH 2 -0- group;
- each Y 2 is, independently, hydrogen, CF 3 , C0 2 R9, C
- each n is, independently, 0, 1 , 2 or 3;
- each o is, independently, 0, 1 , 2 or 3;
- each R 8 , R9, Rio, Rn , Ri 2 , Ri 3 and R ) 4 is, independently, hydrogen, Ci -8 alkyl, CH 2 - aryl wherein the aryl group is substituted by one or more substituents OH, Br, CI, F, CN, CF 3 , N0 2 , N 3 , C ,. 6 alkyl, or CH 2 (CH 2 ) n Y 2 ' ;
- each Y 2 ' is, independently, hydrogen, CF3, or Ci -6 alkyl
- R-6 is C 1-8 alkyl, C 2 . 8 alkenyl, C alkyl substituted C 4-8 cycloalkyl, Ci -4 alkyl substituted C 4 . 8 cycloalkenyl, or thiophene;
- X is a single bond, -C(O)- or -CH(ORi 5 )-;
- Ri5 hydrogen, Cj- 6 alkyl, -(CH 2 ) q -phenyl or -C(0)-Ri 6 ;
- Ri 6 is Ci-4 alkyl or -(CH 2 ) q -phenyl
- each q is, independently, 1, 2 or 3;
- Ri7 is hydrogen, Ci -8 alkyl, C0 2 Ci-8 alkylaryl substituted by one or more groups Yi, CH 2 -aryl substituted by one or more groups Yi, or C0 2 Ci -8 alkyl;
- Rig is hydrogen, Ci -8 alkyl, C 2 _8 alkenyl, C3.8 alkynyl, CH 2 C0 2 C
- each Y 4 is, independently, Br, CI, F, CN, CF 3 , N0 2 , N 3 , OR 22 , C0 2 R 23 , C ,. 6 alkyl, NR 24 R 25 , NHCOR 26 , NHC0 2 R 27 , CONR 28 R 29 , or CH 2 (CH 2 ) n Y 2 ,
- Y 4 groups form a -0-CH 2 -0- or -0-CH 2 CH 2 -0- group;
- p 0, 1 , 2, or 3 ;
- R 2 o is hydrogen, C i_ 8 alkyl, C 2 _ 8 alkenyl, C 2-8 alkenyl, CH 2 OR 3 o, or CH 2 -aryl substituted by one or more substituents Yj ;
- each R 2 i is, independently, hydrogen, Ci -8 alkyl, CH 2 -aryl substituted by one or more substituents Y, , NR 31 R 32 , NHCOR 33 , NHC0 2 R 34 , CONR 35 R 36 , CH 2 (CH 2 ) n Y 2 , or
- R30 is hydrogen Ci_ 8 alkyl, C 2 _ 8 alkenyl, C 2 . 8 alkenyl, CH 2 0 2 C i_8 alkyl, C0 2 Ci -8 alkyl, or CH 2 -aryl substituted by one or more substituents Yj ;
- R-22, R23, R24, R25, R26, R27, 28, R29, R31 , R32, R33» R-34, R35, R36, R37 and R 38 are, independently, hydrogen, Ci_ 8 alkyl, CH 2 -aryl substituted by one or more substituents OH, Br, CI, F, CN, CF 3 , N0 2 , N 3 , C,. 6 alkyl, or CH 2 (CH 2 ) n Y 2 ' ;
- Z is N, O or S, wherein when Z is O or S, there is no Ri 8 ;
- Xi is hydrogen, C i -8 alkyl, C 2-8 alkenyl, or C 2-8 alkynyl;
- X 2 is hydrogen, C i -8 alkyl, C 2-8 alkenyl, or C 2-8 alkynyl;
- At least one of Ri , R 2 , R 3 and R4 is other than hydrogen as defined above;
- the present invention also includes pharmaceutical compositions, which comprise the opioid receptor antagonist described above and a pharmaceutically acceptable carrier.
- the present invention also includes a method of antagonizing opioid receptors, comprising administering an effective amount of the opioid receptor antagonist discussed above to a subject in need thereof.
- the present invention also includes a method of treating drug addiction, drug abuse, depression, anxiety, schizophrenia, obesity and eating disorders, comprising administering an effective amount of the opioid receptor antagonist discussed above to a subject in need thereof.
- the present invention also includes a method of treating alcohol addiction, nicotine addiction, cocaine addition and methamphetamine addiction, comprising administering an effective amount of the opioid receptor antagonist discussed above to a subject in need thereof.
- the present invention also includes a method of treating diabetes, diabetic complications, diabetic retinopathy, sexual/reproductive disorders, epileptic seizure, hypertension, cerebral hemorrhage, congestive heart failure, sleeping disorders,
- Atherosclerosis rheumatoid arthritis
- stroke hyperlipidemia
- hypertriglycemia hypertriglycemia
- hyperglycemia hyperlipoproteinemia, substance abuse, drug overdose, compulsive behavior disorders and addictive behaviors, comprising administering an effective amount of the opioid receptor antagonist discussed above to a subject in need thereof.
- Figure 1 chemical structure of compounds 1 -6.
- R is hydrogen, OH, OC 1.3 alkyl, Ci -4 alkyl, C
- Y 3 is hydrogen, Br, CI, F, CN, CF 3 , N0 2 , OR 8 , C0 2 R 9 , C , -3 alkyl, NR, 0 Ri i ,
- NHCOR 2 , NHC0 2 Ri 2, CONR 13 R, 4 or CH 2 (CH 2 ) n Y 2 ;
- R ⁇ , R 2 , R 3 and R are each, independently, one of the following structures:
- Ri and R 2 , R 2 and R 3 and/or R 3 and R4 are bonded together to 5 to 7 membered alkyl group or a bridged heterocyclic ring.
- At least one of Ri , R 2 , R 3 and R4 is other than hydrogen.
- R is hydrogen, OH, OC 1 - 2 alkyl, Q -2 alkyl, Q. 2 haloalkyl, C 2-3 alkenyl, C 2-3 alkynyl, aryl substituted by one or more groups Yi, C3 ⁇ 4-aryl wherein the aryl group is substituted by one or more groups Yi, COCi -2 alkyl, CONH 2 , NHCHO, NH 2 , NHSO 2 C 1 - 2 alkyl, or NHC0 2 Ci. 2 alkyl.
- R is hydrogen, OH, OCH3, OCF3, COCH3, OCOCH 3 , CONH 2 , NHCHO, NH 2 , NHS0 2 CH 3 , or NHC0 2 CH 3 .
- R is hydrogen, OH, OCH3, or OCF3.
- Y 3 is hydrogen
- Ri, R 2 , R3 and R 4 are each, independently, one of the following structures:
- Ri and R 2 , R 2 and R3 and/or R3 and R4 are bonded together to 5 to 7 membered alkyl group or a bridged heterocyclic ring.
- Ri , R 2 , R3 and R4 are each, independently, hydrogen, methyl or ethyl.
- R] , R 2 , R 3 and R4 are each, independently, hydrogen or methyl.
- Ri , R 2 , R 3 and R 4 are each, independently, hydrogen or methyl, wherein at least one of Ri , R 2 , R3 and R4 is methyl.
- R 5 is hydrogen, Ci -4 alkyl or -(CH 2 ) n -phenyl.
- R $ is
- R is hydrogen, OH, OCH 3 , or OCF 3 ;
- Y 3 is hydrogen
- Ri , R.2, R 3 and R4 are each, independently, hydrogen, methyl or ethyl
- R 5 is hydrogen, Ci -4 alkyl or -(CH 2 ) n -phenyl.
- R 2 is other than hydrogen as defined above. This substitution may increase opioid efficacy by an order of magnitude.
- the chiralty at the resulting stereocenter may be (R) or (S).
- Preferred substituents are C i. 8 alkyl, preferably methyl, ethyl and propyl.
- At least one of Ri , R 2 , R 3 and R4 is other than hydrogen as defined above when
- the opioid receptor antagonists are as described in the following Examples section.
- the present invention includes any and all combination of the different structural groups defined above, including those combinations not specifically set forth above.
- alkyl group or “alkyl radical” encompass all structural isomers thereof, such as linear, branched and cyclic alkyl groups and moieties. Unless stated otherwise, all alkyl groups described herein may have 1 to 8 carbon atoms, inclusive of all specific values and subranges therebetween, such as 2, 3, 4, 5, 6, or 7 carbon atoms. Representative examples include methyl, ethyl, propyl and cyclohexyl.
- haloalkyl group or “haloalkyl radical” encompass all structural isomers thereof, such as linear, branched and cyclic groups and moieties. Unless stated otherwise, all haloalkyl groups described herein may have 1 to 8 carbon atoms, inclusive of all specific values and subranges therebetween, such as 2, 3, 4, 5, 6, or 7 carbon atoms. A Ci -2 haloalkyl group is particularly preferred. At least one hydrogen atom is replaced by a halogen atom, i.e., fluorine, chlorine, bromine or iodine. In one embodiment, all of the hydrogen atoms are replaced with halogen atoms. Fluorine is preferred. Perfluoroalkyl groups are particularly preferred. Examples of haloalkyl groups include trifluoromethyl (-CF 3 ) and perfluoroethyl (-CF 2 CF3).
- the alkenyl group or alkynyl group may have one or more double or triple bonds, respectively.
- a double or triple bond is not formed with the carbon atom bonded directly to the heteroatom.
- all alkenyl and alkynyl groups described herein may have 2 to 8 carbon atoms, inclusive of all specific values and subranges therebetween, such as 3, 4, 5, 6, or 7 carbon atoms.
- the aryl group is a hydrocarbon aryl group, such as a phenyl, naphthyl, phenanthryl, anthracenyl group, which may have one or more C alkyl group substituents.
- the compounds of the present invention may be in the form of a pharmaceutically acceptable salt via protonation of the amines with a suitable acid.
- the acid may be an inorganic acid or an organic acid.
- Suitable acids include, for example, hydrochloric, hydroiodic, hydrobromic, sulfuric, phosphoric, citric, acetic, fumaric, tartaric, and formic acids.
- the opioid receptor selectivity may be determined based on the binding affinities at the receptors indicated or their selectivity in opioid functional assays.
- the compounds of the present invention may be used to bind opioid receptors. Such binding may be accomplished by contacting the receptor with an effective amount of the inventive compound. Of course, such contacting is preferably conducted in an aqueous medium, preferably at physiologically relevant ionic strength, pH, etc. Receptor antagonism is the preferred mode of action of the compounds described herein.
- the inventive compounds may also be used to treat patients having disease states which are ameliorated by binding opioid receptors or in any treatment wherein temporary suppression of the kappa opioid receptor system is desired.
- diseases states include opiate addiction (such as heroin addiction), cocaine, nicotine, or ethanol addiction.
- the compounds of the present invention may also be used as cytostatic agents, as antimigraine agents, as immunomodulators, as immunosuppressives, as antiarthritic agents, as antiallergic agents, as virucides, to treat diarrhea, as antipsychotics, as antischizophrenics, as
- antidepressants as uropathic agents, as antitussives, as antiaddictive agents, as anti-smoking agents, to treat alcoholism, as hypotensive agents, to treat and/or prevent paralysis resulting from traumatic ischemia, general neuroprotection against ischemic trauma, as adjuncts to nerve growth factor treatment of hyperalgesia and nerve grafts, as anti-diuretics, as stimulants, as anti-convulsants, or to treat obesity.
- the present compounds can be used in the treatment of Parkinson's disease as an adjunct to L-dopa for treatment of dyskinesia associated with the L-dopa treatment.
- the compounds of the present invention are particularly useful for treating addiction, such as addiction to cocaine, alcohol, methamphetamine, nicotine, heroine, and other drugs of abuse. With respect to nicotine, the compounds of the present invention are also useful in treating nicotine withdrawal effects.
- addiction such as addiction to cocaine, alcohol, methamphetamine, nicotine, heroine, and other drugs of abuse.
- nicotine the compounds of the present invention are also useful in treating nicotine withdrawal effects.
- the compounds may be administered orally, intraveneously, or intramuscularly.
- inventive compounds may be combined with any of the well-known pharmaceutical carriers and additives that are customarily used in such pharmaceutical compositions.
- the patient is preferably a mammal, with human patients especially preferred. Effective amounts are readily determined by those of ordinary skill in the art. Studies by the present inventors show no toxicity and no lethality for the present compounds at amounts up to 300 mg/kg in mice.
- the compounds of the present invention can be administered as a single dosage per day, or as multiple dosages per day.
- the dosages can be equal doses or doses of varying amount, based upon the time between the doses (i.e. when there will be a longer time between doses, such as overnight while sleeping, the dose administered will be higher to allow the compound to be present in the bloodstream of the patient for the longer period of time at effective levels).
- the compound and compositions containing the compound are administered as a single dose or from 2-4 equal doses per day.
- compositions containing the present compounds further comprise a physiologically acceptable carrier, such as water or conventional pharmaceutical solid carriers, and if desired, one or more buffers and other excipients.
- a physiologically acceptable carrier such as water or conventional pharmaceutical solid carriers, and if desired, one or more buffers and other excipients.
- the compounds of the invention may be synthesized by, for example, the schemes shown in the following Examples. Those skilled in the art will appreciate that the synthesis of the exemplified compounds can readily be adapted for the preparation of other compounds within the scope of formula I.
- Compounds 5a-f of the present invention may be synthesized, for example, in accordance with the reaction sequence shown in Scheme 1.
- the tefY-butoxycarbonyl- protected starting piperazines 7a-e were prepared by treating the appropriate piperazine with Boc 2 0 or Boc-ON using standard conditions.
- the piperazines required for 7a-d were commercially available.
- Piperazine needed for 7e was synthesized according to reported methods. 1 ' 2
- the /er/-butoxycarbonyl-protected piperazines 7a-e were coupled to 3- bromoanisole under palladium-catalyzed conditions to give 8a-e.
- Compounds 5g,h can be synthesized by the routes shown in Scheme 2.
- Compound 10 was coupled to 3-bromoanisole under palladium-catalyzed conditions to give 11.
- Subjection of 11 to palladium on carbon in refluxing aqueous acetic acid removed the N-allyl-protecting group to give 12.
- Treatment of 12 with boron tribromide in methylene chloride at -78 °C affected demethylation of 12 to give the phenol 13.
- Reductive alkylation of 13 using 3- phenylpropionaldehyde and sodium triacetoxyborohydride in 1 ,2-dichloroethane yielded 6h.
- Measures of opioid receptor antagonism and specificity were obtained by monitoring the ability of selected test compounds to inhibit stimulation of [ S]GTPyS binding produced by the selective agonists (D-Ala 2 ,MePhe 4 ,Gly-ol 5 )enkephalin (DAMGO, mu receptor) cyclo[D-Pen 2 ,D-Pen 5 ]enkephalin (DPDPE, delta) and 5,7,8-(-)-N-methyl-N-[7-(l - pyrrolidinyl)- l -oxaspiro[4,5]dec-8-yl]benzeneacetamide (U69,593, kappa) in cloned human receptors (Table 1 ).
- Compounds 5a-j show high efficacy (low K e values) for the kappa opioid receptor in the [-"SJGTPyS in vivo functional assay, particularly 5b-e, 5g, and 5j.
- the compounds of the present invention are potent kappa opioid receptor antagonists in an in vitro functional test. Some compounds showed good selectivity for the kappa relative to the mu and delta opioid receptors.
- Reagents (a) 3-bromoanisole, Pd 2 (dba) 3 , KOtBu, P(tBu) 3 , toluene, 100 °C, 18 h; (b) KN(Si(CH 3 ) 3 ) 2 , 3-bromoanisole, 1 ,4-dioxane 100 °C, 2 h; (c) BBr 3 , CH 2 CI 2 , -78 °C, 4 h; (d) HBr (48%) reflux; (e) C 6 H 5 (CH 2 ) 2 CHO, Na(OAc) 3 BH, Et 3 N, DCE; (f) Rany Ni, H 2 CO, H 2 EtOH.
- Reagents (a) 3-bromoanisole, Pd 2 (dba) 3 , KOtBu, P(OtBu) 3 , toluene, 1 10 °C, sealed vessel; (b) Pd/C, CH 3 C0 2 H, H 2 0, reflux; (c) BBr 3 , CH 2 CI 2 , -78 °C; (d) C 6 H 5 (CH 2 ) 3 CHO,
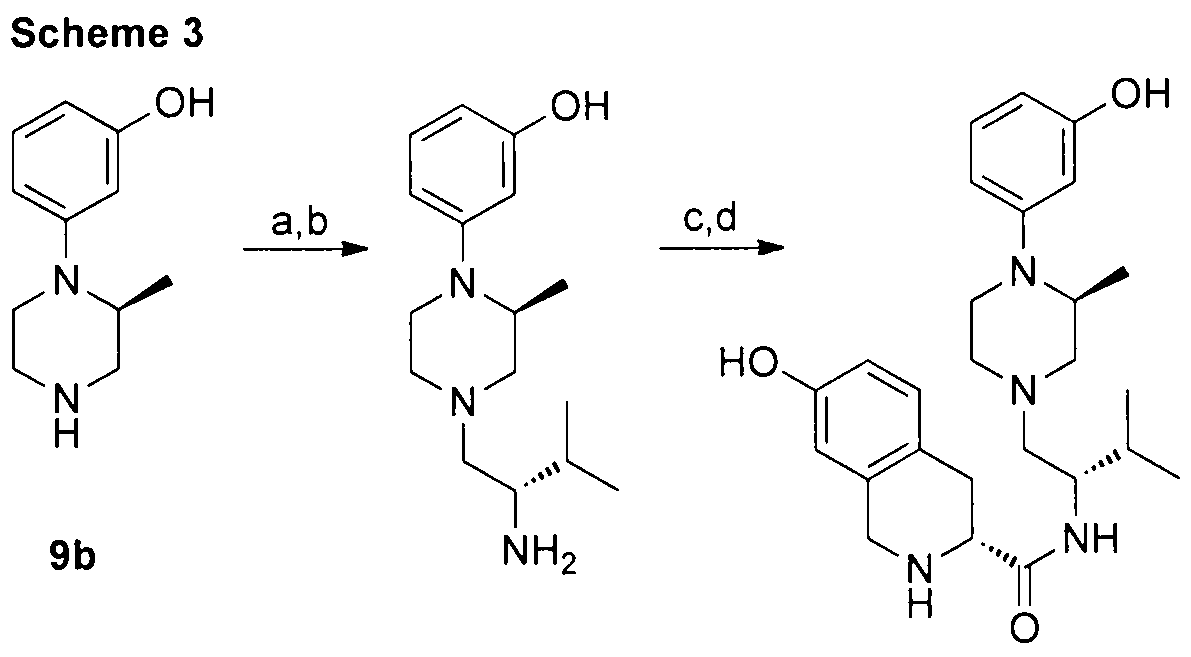
- Reagents (a) N-Boc-valine, BOP, Et 3 N, THF; (b) BH 3 , THF then cone. HCI; (c) BOP 7-HO-Boc-D-Tic, THF, Et 3 N; (d) CF 3 C0 2 H, CH 2 CI 2
- the filtered solution was reduced to a fifth of its volume by evaporation under reduced pressure.
- the remaining solution was subjected to column chromatography on silica gel eluting with hexanes-EtOAc (5 : 1 ).
- the combined fractions containing the product were subjected to rotary evaporation, and the remaining oil was dried under high vacuum.
- reaction mixture was added to a concentrated solution of NaHC0 3 (20 mL) and shaken vigorously. The layers were separated, and the organic layer was washed once with H 2 0 (5 mL) and once with brine (5 mL). The organic solution was dried (MgS0 4 ), filtered, and the solvents removed under reduced pressure to yield the product which was purified as specified.
- Trihydrochloride In a round-bottom flask, 120 mg (0.432 mmol) of 5i and 133 mg (0.454 mmol) of 7-OH-Boc-D-Tic were dissolved in dry THF (15 mL), and the solution was cooled to 0 °C. Into this solution 0.06 mL of Et 3 N were added followed by 201 mg (0.454 mmol) of BOP. The solution was warmed up to room temperature, stirred for 3 h, and then added to an ice-cold concentrated NaHC0 3 solution. The mixture was extracted three times with 5 mL of EtOAc. The pooled organic extracts were washed once with cone.
- JDTic A novel -opioid receptor antagonist. Eur. J. Pharmacol. 2004, 501, 1 1 1 - 1 19.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Addiction (AREA)
- Diabetes (AREA)
- Psychiatry (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Epidemiology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Vascular Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Ophthalmology & Optometry (AREA)
- Anesthesiology (AREA)
- Emergency Medicine (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Urology & Nephrology (AREA)
- Nutrition Science (AREA)
Abstract
Description



















Claims

















Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2787037A CA2787037C (en) | 2010-02-24 | 2010-10-12 | Arylpiperazine opioid receptor antagonists |
| ES10846785.3T ES2533990T3 (en) | 2010-02-24 | 2010-10-12 | Arylpiperazine opioid receptor antagonists |
| JP2012554983A JP6173693B2 (en) | 2010-02-24 | 2010-10-12 | Aryl piperazine opioid receptor antagonist |
| AU2010346633A AU2010346633B2 (en) | 2010-02-24 | 2010-10-12 | Arylpiperazine opioid receptor antagonists |
| EP10846785.3A EP2539706B1 (en) | 2010-02-24 | 2010-10-12 | Arylpiperazine opioid receptor antagonists |
| US13/574,179 US9273027B2 (en) | 2010-02-24 | 2010-10-12 | Arylpiperazine opioid receptor antagonists |
| US14/968,258 US9750738B2 (en) | 2010-02-24 | 2015-12-14 | Arylpiperazine opioid receptor antagonists |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US30753410P | 2010-02-24 | 2010-02-24 | |
| US61/307,534 | 2010-02-24 | ||
| US31642310P | 2010-03-23 | 2010-03-23 | |
| US61/316,423 | 2010-03-23 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/574,179 A-371-Of-International US9273027B2 (en) | 2010-02-24 | 2010-10-12 | Arylpiperazine opioid receptor antagonists |
| US14/968,258 Division US9750738B2 (en) | 2010-02-24 | 2015-12-14 | Arylpiperazine opioid receptor antagonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011106039A1 true WO2011106039A1 (en) | 2011-09-01 |
Family
ID=44507137
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/052311 WO2011106039A1 (en) | 2010-02-24 | 2010-10-12 | Arylpiperazone opioid receptor antagonists |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US9273027B2 (en) |
| EP (1) | EP2539706B1 (en) |
| JP (1) | JP6173693B2 (en) |
| AU (1) | AU2010346633B2 (en) |
| CA (1) | CA2787037C (en) |
| ES (1) | ES2533990T3 (en) |
| WO (1) | WO2011106039A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013086496A3 (en) * | 2011-12-09 | 2014-12-18 | Research Triangle Institute | 1-substituted 4-arylpiperazine as kappa opioid receptor antagonists |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3032046A1 (en) | 2016-09-16 | 2018-03-22 | Research Triangle Institute | Tetrahydroisoquinoline kappa opioid antagonists |
| WO2018129393A1 (en) * | 2017-01-06 | 2018-07-12 | The Regents Of The University Of California | Mu opioid receptor modulators |
| WO2019195634A1 (en) | 2018-04-04 | 2019-10-10 | Epiodyne, Inc. | Opioid receptor modulators and products and methods related thereto |
| TW202304869A (en) | 2021-04-05 | 2023-02-01 | 美商艾碧奧戴股份有限公司 | Opioid receptor modulators |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5658908A (en) * | 1992-02-03 | 1997-08-19 | Delta Pharmaceuticals, Inc. | Opioid diarylmethylpiperazines and piperdines |
| US20090264462A1 (en) * | 2008-04-18 | 2009-10-22 | Research Triangle Institute | Kappa opioid receptor ligands |
Family Cites Families (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB948766A (en) * | 1959-10-20 | 1964-02-05 | May & Baker Ltd | Trifluoromethylphenylpiperazine derivatives |
| FI58126C (en) * | 1973-02-03 | 1980-12-10 | Thomae Gmbh Dr K | FOERFARANDE FOER FRAMSTAELLNING AV KARDIOTONA OCH BLODTRYCKSSAENKANDE 2-PHENYL-1H-IMIDATSO (4,5-B) PYRIDINER |
| DE2456280A1 (en) * | 1974-11-28 | 1976-08-12 | Hoechst Ag | Amino-substd phenol derivs prepn - by reacting unsaturated 5-oxo-carboxylic acid esters with amines |
| FR2353291A1 (en) * | 1976-05-31 | 1977-12-30 | Parcor | NEW DERIVATIVES OF BENZOFURANNE |
| US6706880B2 (en) | 1990-08-09 | 2004-03-16 | Research Triangle Institute | Cocaine receptor binding ligands |
| US5496953A (en) | 1990-08-09 | 1996-03-05 | Research Triangle Institute | Cocaine receptor binding ligands |
| US6123917A (en) | 1990-08-09 | 2000-09-26 | Research Triangle Institute | Method for making cocaine receptor binding ligands and intermediates therefor |
| US5736123A (en) | 1990-08-09 | 1998-04-07 | Research Triangle Institute | Cocaine receptor binding ligands |
| US6531483B1 (en) | 1990-08-09 | 2003-03-11 | Research Triangle Institute | Cocaine receptor binding ligands |
| US7011813B2 (en) | 1990-08-09 | 2006-03-14 | Research Triangle Institute | Cocaine receptor binding ligands |
| US6329520B1 (en) | 1990-08-09 | 2001-12-11 | Research Triangle Institute | Cocaine receptor binding ligands |
| US7189737B2 (en) | 1991-08-09 | 2007-03-13 | Research Triangle Institute | Cocaine receptor binding ligands |
| US5380848A (en) | 1990-08-09 | 1995-01-10 | Research Triangle Institute | Cocaine receptor binding ligands |
| US5128118A (en) | 1990-08-09 | 1992-07-07 | Research Triangle Institute | Cocaine receptor binding ligands |
| US5935953A (en) | 1990-08-09 | 1999-08-10 | Research Triangle Institute | Methods for controlling invertebrate pests using cocaine receptor binding ligands |
| US5141959A (en) | 1990-09-21 | 1992-08-25 | Bristol-Myers Squibb Company | Isoprenoid phospholipase a2 inhibitors and preparations comprising same |
| US5159081A (en) * | 1991-03-29 | 1992-10-27 | Eli Lilly And Company | Intermediates of peripherally selective n-carbonyl-3,4,4-trisubstituted piperidine opioid antagonists |
| WO1993000313A2 (en) * | 1991-06-27 | 1993-01-07 | Virginia Commonwealth University | Sigma receptor ligands and the use thereof |
| US5298499A (en) | 1991-07-05 | 1994-03-29 | Research Triangle Institute | S-2-(substituted ethylamino)ethyl phosphorothioates |
| FR2722788B1 (en) * | 1994-07-20 | 1996-10-04 | Pf Medicament | NOVEL PIPERAZIDES DERIVED FROM ARYL PIPERAZINE, PROCESSES FOR THEIR PREPARATION, THEIR USE AS MEDICAMENTS AND PHARMACEUTICAL COMPOSITIONS COMPRISING THE SAME |
| US5831095A (en) | 1995-09-26 | 1998-11-03 | Research Triangle Institute | Synthesis of 5-aminocarbonyl-5H-dibenzo a,d!cyclohepten-5,10-imine |
| ES2128266B1 (en) * | 1997-07-08 | 2000-01-16 | Vita Invest Sa | THIOPHENE AND BENZOTIOFEN DERIVATIVE COMPOUNDS AND RELEVANT USE AND COMPOSITION. |
| US6358492B1 (en) | 1997-10-07 | 2002-03-19 | Research Triangle Institute | Dopamine transporter imaging ligand |
| US6900228B1 (en) | 1998-03-10 | 2005-05-31 | Research Triangle Institute | Opiate compounds, methods of making and methods of use |
| US7517884B2 (en) * | 1998-03-30 | 2009-04-14 | Kalypsys Inc. | Sulfonyl-substituted bicyclic compounds as modulators of PPAR |
| JP3390744B2 (en) * | 1998-09-22 | 2003-03-31 | 山之内製薬株式会社 | Cyanophenyl derivative |
| GB9912417D0 (en) * | 1999-05-28 | 1999-07-28 | Pfizer Ltd | Compounds useful in therapy |
| ATE241624T1 (en) * | 1999-06-30 | 2003-06-15 | Aventis Pharma Sa | BENZO(F)NAPHTHYRIDINE DERIVATIVES, THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| US6416735B1 (en) | 1999-11-08 | 2002-07-09 | Research Triangle Institute | Ligands for α-7 nicotinic acetylcholine receptors based on methyllcaconitine |
| SE9904724D0 (en) * | 1999-12-22 | 1999-12-22 | Carlsson A Research Ab | New modulators of dopamine neurotransmission I |
| US6479509B1 (en) | 2000-05-22 | 2002-11-12 | Research Triangle Institute | Method of promoting smoking cessation |
| CN1469862A (en) | 2000-10-12 | 2004-01-21 | Ss制药株式会社 | 2,2-diphenyl butanamide derivatives and medicines containing the same |
| US6538010B1 (en) | 2000-11-08 | 2003-03-25 | Research Triangle Institute | Compounds and methods for promoting smoking cessation |
| US6974824B2 (en) * | 2001-01-08 | 2005-12-13 | Research Triangle Institute | Kappa opioid receptor ligands |
| US6559159B2 (en) | 2001-02-01 | 2003-05-06 | Research Triangle Institute | Kappa opioid receptor ligands |
| DE60219521T2 (en) * | 2001-12-20 | 2007-08-16 | H. Lundbeck A/S | ARYLOXYPHENYL AND ARYLSULFANYLPHENYL DERIVATIVES |
| DE10217006A1 (en) | 2002-04-16 | 2003-11-06 | Merck Patent Gmbh | Substituted indoles |
| AU2003235256A1 (en) | 2002-04-19 | 2003-11-03 | Kyowa Hakko Kogyo Co., Ltd. | Phenylalanine derivative |
| US7842693B2 (en) * | 2002-06-12 | 2010-11-30 | Chemocentryx, Inc. | Substituted piperazines |
| US7589199B2 (en) | 2002-06-12 | 2009-09-15 | Chemocentryx, Inc. | Substituted piperazines |
| ES2393333T3 (en) * | 2002-07-12 | 2012-12-20 | Astellas Pharma Inc. | N-Phenyl- (2R, 5S) dimethylpiperazine derivative |
| KR101117061B1 (en) * | 2003-05-21 | 2012-05-16 | 아스카 세이야쿠 가부시키가이샤 | Cyclic aminophenyl sulfamate derivative |
| CA2529750A1 (en) * | 2003-06-20 | 2005-02-24 | Arena Pharmaceuticals, Inc. | N-phenyl-piperazine derivatives and methods of prophylaxis or treatment of 5ht2c receptor associated diseases |
| US20050239791A1 (en) * | 2004-04-07 | 2005-10-27 | Hutchison Alan J | Substituted 1-heteroaryl-4-substituted piperazine and piperidine analogues |
| KR101351209B1 (en) * | 2004-12-03 | 2014-02-06 | 머크 샤프 앤드 돔 코포레이션 | Substituted piperazines as CB1 antagonists |
| US7872023B2 (en) | 2005-02-17 | 2011-01-18 | Research Triangle Institute | Kappa opioid receptor ligands |
| US7399762B2 (en) | 2005-04-25 | 2008-07-15 | Research Triangle Institute | Opioid receptor agonist compounds and their use in treatment of pain |
| US7476679B2 (en) | 2005-07-26 | 2009-01-13 | Research Triangle Institute | Octahydroisoquinoline compounds as opioid receptor modulators |
| ES2458615T3 (en) * | 2006-02-03 | 2014-05-06 | Taisho Pharmaceutical Co., Ltd. | Triazole derivative |
| TW200804309A (en) * | 2006-02-28 | 2008-01-16 | Helicon Therapeutics Inc | Therapeutic piperazines |
| FR2902327B1 (en) * | 2006-06-20 | 2008-08-08 | Oreal | KERATIN FIBER COLORING COMPOSITION COMPRISING 2,3-DIAMINO-6,7-DIHYDRO-1H, 5H-PYRAZOLO-1,2-A PYRAZOL-1-ONE, PARA-PHENYLENEDIAMINE OR PARA-TOLUENEDIAMINE AND META-AMINOPHENOL SUBSTITUTES |
| WO2009001127A1 (en) * | 2007-06-26 | 2008-12-31 | Astrazeneca Ab | Cyanocyclopropylcarboxamides as cathepsin inhibitors |
| US20110015137A1 (en) * | 2007-07-05 | 2011-01-20 | Trustees Of Boston University | Reca inhibitors and their uses as microbial inhibitors or potentiators of antibiotic activity |
| AU2008345225A1 (en) * | 2007-12-21 | 2009-07-09 | University Of Rochester | Method for altering the lifespan of eukaryotic organisms |
| JO2857B1 (en) * | 2008-06-16 | 2015-03-15 | سانوفي أفينتس | Phenyl-alkylpiperazines with tnf-modulating activity |
| EP2149373A1 (en) * | 2008-08-01 | 2010-02-03 | Laboratorios Del. Dr. Esteve, S.A. | 5HT7 receptor ligands and compositions comprising the same |
| WO2010022055A2 (en) * | 2008-08-20 | 2010-02-25 | Amgen Inc. | Inhibitors of voltage-gated sodium channels |
| US20130158072A1 (en) * | 2010-01-19 | 2013-06-20 | Research Triangle Institute | Kappa opioid receptor binding ligands |
| US9512105B2 (en) * | 2011-12-09 | 2016-12-06 | Research Triangle Institute | 1-substituted 4-arylpiperazine as kappa opioid receptor antagonists |
-
2010
- 2010-10-12 US US13/574,179 patent/US9273027B2/en not_active Expired - Fee Related
- 2010-10-12 ES ES10846785.3T patent/ES2533990T3/en active Active
- 2010-10-12 CA CA2787037A patent/CA2787037C/en not_active Expired - Fee Related
- 2010-10-12 JP JP2012554983A patent/JP6173693B2/en not_active Expired - Fee Related
- 2010-10-12 EP EP10846785.3A patent/EP2539706B1/en not_active Not-in-force
- 2010-10-12 WO PCT/US2010/052311 patent/WO2011106039A1/en active Application Filing
- 2010-10-12 AU AU2010346633A patent/AU2010346633B2/en not_active Ceased
-
2015
- 2015-12-14 US US14/968,258 patent/US9750738B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5658908A (en) * | 1992-02-03 | 1997-08-19 | Delta Pharmaceuticals, Inc. | Opioid diarylmethylpiperazines and piperdines |
| US20090264462A1 (en) * | 2008-04-18 | 2009-10-22 | Research Triangle Institute | Kappa opioid receptor ligands |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2539706A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013086496A3 (en) * | 2011-12-09 | 2014-12-18 | Research Triangle Institute | 1-substituted 4-arylpiperazine as kappa opioid receptor antagonists |
| JP2015509077A (en) * | 2011-12-09 | 2015-03-26 | リサーチ トライアングル インスティテュート | 1-Substituted 4-arylpiperazines as kappa opioid receptor antagonists |
| EP2773352A4 (en) * | 2011-12-09 | 2015-12-16 | Res Triangle Inst Int | 1-substituted 4-arylpiperazine as kappa opioid receptor antagonists |
| US9512105B2 (en) | 2011-12-09 | 2016-12-06 | Research Triangle Institute | 1-substituted 4-arylpiperazine as kappa opioid receptor antagonists |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2539706A4 (en) | 2013-07-31 |
| ES2533990T3 (en) | 2015-04-16 |
| US20160095854A1 (en) | 2016-04-07 |
| US9750738B2 (en) | 2017-09-05 |
| JP2013520497A (en) | 2013-06-06 |
| US9273027B2 (en) | 2016-03-01 |
| AU2010346633B2 (en) | 2015-12-03 |
| EP2539706B1 (en) | 2015-03-04 |
| EP2539706A1 (en) | 2013-01-02 |
| US20120295919A1 (en) | 2012-11-22 |
| AU2010346633A1 (en) | 2012-07-19 |
| CA2787037A1 (en) | 2011-09-01 |
| CA2787037C (en) | 2018-04-10 |
| JP6173693B2 (en) | 2017-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5854249A (en) | Opioid diarylmethylpiperazines and piperidines | |
| US5574159A (en) | Opioid compounds and methods for making therefor | |
| AU692788B2 (en) | Piperazine compounds used in therapy | |
| US9750738B2 (en) | Arylpiperazine opioid receptor antagonists | |
| JP3205343B2 (en) | Diaryldiamine derivatives and their use as delta opioid (ant) -agonists | |
| US5681830A (en) | Opioid compounds | |
| RU2386614C2 (en) | Derivatives of n-[phenyl(pyrrolidin-2-yl)methyl]benzamide and n-[(azepan-2-yl)phenylmethyl]benzamide, method of obtaining said compounds and use thereof in therapy | |
| JP5571133B2 (en) | Kappa opioid receptor ligand | |
| KR20030059084A (en) | Carboxamide compounds and their use as antagonists of a human 11cby receptor | |
| US20130158072A1 (en) | Kappa opioid receptor binding ligands | |
| AU2009236609B2 (en) | Kappa opioid receptor ligands | |
| WO2006103342A2 (en) | Indanyl-piperazine derivatives, method for preparing same and pharmaceutical compositions containing same | |
| AU2012347416B2 (en) | 1-substituted 4-arylpiperazine as kappa opioid receptor antagonists | |
| ES2286474T3 (en) | DERIVATIVES OF 4- (FINILPIPERAZINILMETIL) BENZAMIDA AND ITS USE FOR THE TREATMENT OF PAIN OR GASTROINTESTINAL DISORDERS. | |
| JP2009526844A (en) | Piperazine derivatives | |
| PL179341B1 (en) | Diaryl methyl piperazine and diaryl methyl piperidine compounds exhibiting properties equal to those of opioides as well as method obtaining and using them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10846785 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010346633 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2787037 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2010346633 Country of ref document: AU Date of ref document: 20101012 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13574179 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010846785 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012554983 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |