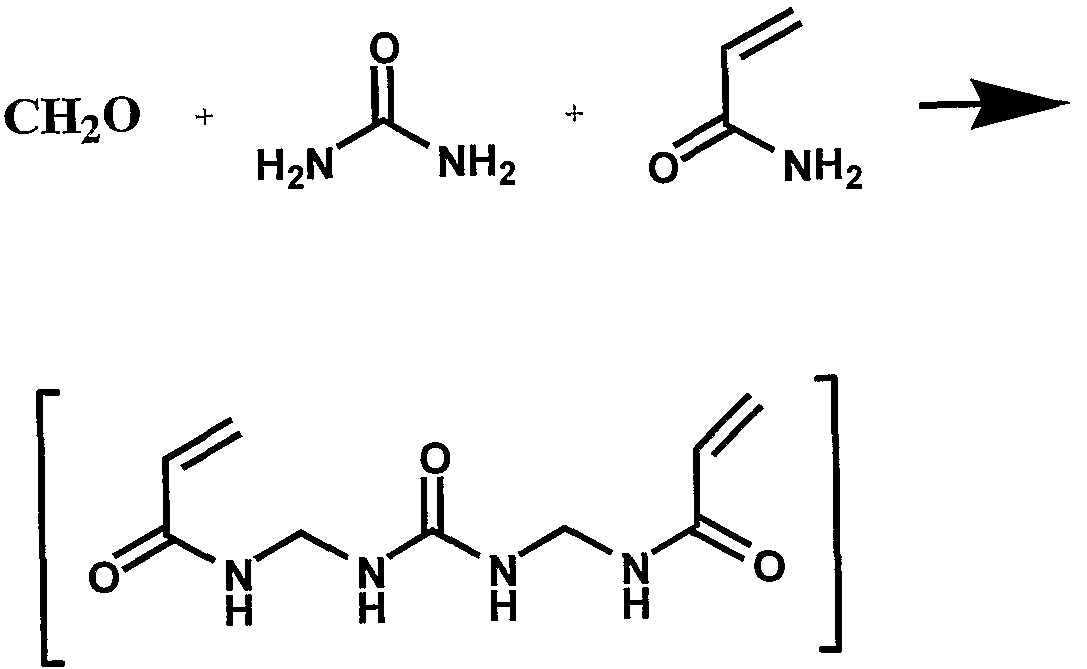
SYNTHESIS OF DEGRADABLE POLYMERS DOWNHOLE FOR
OILFIELD APPLICATIONS
Background of the Invention
This invention relates to well completion, stimulation and remediation. More specifically, it relates to a method of polymerization of monomers downhole to create responsive polymer structures to provide a temporary or reversible treatment in a wellbore or in a fracture. The method includes irreversible polymerization of monomers under downhole conditions. The resulting polymer slowly degrades after it has performed its function, such as fluid diversion, wellbore isolation, proppant aggregation, formation consolidation, and fluid loss prevention.
Degradable polymers have found many uses in the oilfield. As non-limiting examples: They may be used as diverting or isolating agents, for example in the form of ball-sealers, polymer fibers, flakes, and fine powders, and as a coating on proppant particles. They have been delivered downhole in the discontinuous (internal) phase of emulsions. They have also been used as acid precursors, and fluid loss additives in drill-in fluids. They have been used to replace some of the proppant used so that, after they degrade, the proppant placement is heterogeneous. Many materials have been used for these purposes, such as hydrolysable polyesters and oil soluble polyolefms. The degradation may be, for example, controlled hydrolysis, or dissolution in water or oil. There are several problems in common for most of the applications here. First of all, pumping of solids increases the risk of bridging and plugging in the wellbore, fracture, coiled tubing or surface equipment. Second, there are always limitations on the concentrations of solids and on the velocity that can be attained during slurry pumping. Third, placement of solids is difficult to control. Fourth, fluid-solid separation, for example by settling, during a job is possible. Fifth, if the polymer is soluble, the viscosity of the fluid may make pumping difficult and expensive. Formation of the polymer downhole alleviates these problems.
Polymerization of monomers downhole has been used, but to form polymers that are intended to remain permanently in place to perform their functions (or that
may degrade gradually but are not required to degrade to perform their function). For example, most water control treatments use polymers, such as polyacrylamides, that are polymerized in place. Most curable resin proppants have a coating containing monomers or oligomers of the final polymer. Monomers or oligomers of epoxy, melamine, urethane, phenolic and other resins have been polymerized downhole, for example to create thermoset nanocomposite particulates. All of the methods mentioned above for downhole polymerization assume permanent polymer placement, and application of the methods is limited to situations in which irreversible polymer placement is acceptable. However, for a number of applications such as fluid diversion, well isolation, multistage fracturing, fluid loss and formation control, there are more sophisticated requirements. Namely, it is preferable to pump monomers downhole to avoid incorrect placement, pumping power loss, and plugging issues, but at the same time it would be preferable to have a degradable polymer, which would disappear (degrade) after the time needed for a given purpose.
There is a need for a method of synthesizing degradable polymers downhole and for controlling their placement and the timing of their formation and degradation.
Summary of the Invention
One embodiment of the invention is a method of downhole treatment that includes pumping a fluid containing a polymerizable monomer downhole, initiating downhole polymerization, and allowing decomposition of the resulting polymer under downhole conditions. The monomer contains at least two polymerizable chemical groups and one or more degradable chemical groups and, optionally, one or more water-soluble groups linking the polymerizable and degradable groups. Examples of the water soluble linking groups include oligoethyleneglycol, glycerol, phosphate, and mixtures of these. The monomer may also include one or more precursors to water- soluble groups that link the polymerizable and degradable groups. Examples of the polymerizable groups include acrylic, methacrylic, aziridine, oxirane, isocyanate, vinyl, and mixtures of these groups. The monomer may be a Mannich reaction product.
In another embodiment, the fluid containing a first such monomer also includes a second, different, monomer that contains at least two polymerizable chemical groups and one or more degradable chemical groups. The fluid may contain acrylic acid, methacrylic acid, or both. The two different monomers may be a nucleophilic monomer and an electrophilic monomer
In a preferred embodiment, the monomers for polymerization are diacrylic ethers or anhydrides or dimethylacrylic ethers or anhydrides.
In other embodiments, the downhole polymerization of monomer is emulsion polymerization; the fluid further contains a polymerization initiator; the fluid further contains a chain transfer agent; the polymerization is nitroxide mediated free radical polymerization; the fluid further contains a free radical trap; the fluid further contains a surfactant; and/or the fluid further contains a fiber.
In yet other embodiments, the degradation of the polymer is caused by an increase in temperature; a change in fluid pH; and/or a polymer degradation agent pumped downhole before or after the fluid containing monomer. The fluid may also contain a polymer degradation agent.
In a further embodiment the fluid is an emulsion.
Detailed Description of the Invention
Although the following discussion emphasizes fracturing, the downhole polymerization method of the invention may be used in other stimulation methods, such as gravel packing, and combined fracturing and gravel packing in a single operation, and may be used in other oilfield practices such as, but not limited to, water control, well completion, scale control, acidizing, and drilling fluid loss control. The invention will be described in terms of treatment of vertical wells, but is equally applicable to wells of any orientation. The invention will be described for hydrocarbon production wells, but it is to be understood that the invention may be used for wells for production of other fluids, such as water or carbon dioxide, or, for example, for injection or storage wells. It should also be understood that throughout this specification, when a concentration or amount range is described as being useful, or suitable, or the like, it is intended that any and every concentration or amount
within the range, including the end points, is to be considered as having been stated. Furthermore, each numerical value should be read once as modified by the term "about" (unless already expressly so modified) and then read again as not to be so modified unless otherwise stated in context. For example, "a range of from 1 to 10" is to be read as indicating each and every possible number along the continuum between about 1 and about 10. In other words, when a certain range is expressed, even if only a few specific data points are explicitly identified or referred to within the range, or even when no data points are referred to within the range, it is to be understood that the inventors appreciate and understand that any and all data points within the range are to be considered to have been specified, and that the inventors have possession of the entire range and all points within the range.
The techniques of emulsion polymerization (for example batch processing), including the choices of monomers, surfactants, initiators, and chain transfer agents are well known in the art. Surfactants having low critical micelle concentrations are favored; the polymerization rate shows a dramatic increase when the surfactant level is above the critical micelle concentration, and minimization of the amount of surfactant used is preferred for economic reasons. Mixtures of surfactants are often used, including mixtures of anionic and nonionic surfactants. Examples of surfactants commonly used in emulsion polymerization include fatty acids, fatty acid salts, sodium lauryl sulfate, quaternary ammonium salts, and alpha olefin sulfonates.
The monomers may be pumped downhole in an emulsion, in a one-phase solution, in a dispersion, or in any other type of mixture.
Monomers suitable for use in the invention have two types of chemical groups: first, a polymerizable group or groups and second, a degradable group or groups which allow the polymer to degrade under downhole conditions via hydrolysis or another mechanism. This approach is different from the approach which uses reversible cross-linking of a polymer via a change in pH, or in ion concentration, such as the way polysaccharides are crosslinked with borate. In the present invention, the two chemical processes (polymerization from monomers and degradation of the polymer) take place almost independently and are controlled differently.
If the monomer is delivered to a downhole location in emulsified form, the monomer emulsion may be stabilized with surfactants (ionic or non-ionic) and stabilizers. Monomer may also be diluted with organic solvent and/or mixed with other materials such as proppants. Degradable inorganic or organic solid particulates that form a composite material with the polymer under bottomhole conditions may also be included.
Polymerization may be triggered, for example, by a radical initiator and/or by an increase in temperature. The rate of the downhole polymerization process and the length of the polymer produced may be controlled with additives that are responsible for the polymer chain length and branching.
Polymer obtained from monomers pumped downhole may be used, by non- limiting example, for fluid diversion plugs, isolation plugs, formation consolidation, flowback control, and fluid loss control. Synthesized polymer may form gels, films, solids or any other kind of structure in treated wellbores, fractures, and/or formations. After the desired effect is achieved, the deposited polymer degrades via hydrolysis or other mechanism and the polymer degradation products dissolve, leaving the wellbore, fracture and/or formation clean with no damage that might have decreased fluid flow.
Monomers
Examples of suitable monomers include polymerizable acrylic acid esters or anhydrides, which may subsequently be hydrolyzed to produce water-soluble acrylic acid derivatives; the monomers usually include an optional water-soluble linking group. A generalized formula is as follows:
The two polymerizable groups and the two decomposable (degradable) groups may each either be the same as one another or different. There may be more than two polymerizable groups in a single monomer. There may be only a single degradable group between the polymerizable groups. The water-soluble linker or its precursor may not be needed if the monomer is sufficiently water-soluble without it. The monomer used may be a mixture of monomers of this type. The polymerizable groups in the monomers may be, by non-limiting example, acrylic, methacrylic, aziridine, oxirane, isocyanate, vinyl, and mixtures of these. The water soluble linking groups may include, by non-limiting example, mono-, di-, tri-, tetraethyleneglycol (oligoethyleneglycol), glycerol, phosphate, and other units. The following are commercially available examples of suitable monomers; the first four, designated Ml- M4, were used in the examples described later. These are used as non-limiting examples of suitable monomers in the Experimental section below.

Another example of monomers suitable for downhole formation of degradable polymers is the product of certain Mannich reactions. For example, the reaction product of a basic component (for example urea, alkylamine, or melamine), formaldehyde (or a precursor, for example paraformaldehyde or hexamine), and an acrylamide is used as the monomer or as a co-monomer. For example, mixing, under basic conditions, of urea, formaldehyde and acrylamide results in a mixture of products, some of which are polymerizable-degradable monomers, such as that shown below, for further polymerization. The acrylic moieties are responsible for subsequent polymerization; the structure formed from the urea and N-CH
2-N fragments, is responsible for hydrolysis.
Yet other examples of polymerizable - degradable downhole materials are those based on nucleophile - electrophile polymerization reactions. In such a reaction, an electrophilic monomer having two or more electrophilic groups reacts with a nucleophilic monomer having two or more nucleophilic groups, resulting in a cross-linked polymer. When one or both of those monomers has one or more cleavable units (for example ester, orthoester, etc.), the polymer slowly dissolves (for example, as a result of hydrolysis).
Electrophilic groups may, by non-limiting example, be aziridine, carbodiimide, oxirane, epoxide, and molecules containing combinations of these groups; examples include ethyleneglycol bis-(2,3-epoxybutyrate); trimethylolpropane tri-[P-(N-aziridinyl)-propionate], and 2,2-bishydroxymethyl butanoltris[3-(l- aziridine) propionate].
Nucleophilic groups may, by non-limiting example, be amines, hydroxyls, thiols, urethanes, and amides. Examples include tetraethylenepentamine; ethylene diamine; cadaverine; sucrose; glycerin; pentaerythritol; pentaerythritolethoxylate; pentaerythritol propoxylate; ethylene glycol; diethylene glycol; triethylene glycol; tetraethylene glycol; polyethylene glycol; 1,2,3-propanetriol; polyglycerol; propylene glycol; 1,2-propanediol; 1,3 -propanediol; trimethylol propane; diethanolamine; triethaiiolamine; sorbitol; and molecules containing combinations of such reactions include
Also, combined mechanisms of downhole polymerizations are suitable. For example, glycidyl acrylate contains both acrylic and epoxide units; 2-isocyanatoethyl methacrylate contains an isocyanate and an acrylic group. Such molecules undergo
9 000751 both radical and electrophile-nucleophile polymerization. The ester function is responsible for the subsequent hydrolysis.
Monomers are pumped downhole in liquid, encapsulated, adsorbed or emulsified form and may be diluted with water or with organic solvents.
Monomer emulsions may be stabilized by surfactants (cationic, anionic, non- ionic, amphoteric, and zwitter ionic). Monomers may be emulsified in pure form or first dissolved in an organic solvent.
Monomers may be pre-mixed with solid particulates (including fibers) to give a composite plugging material after downhole polymerization.
Initiators
After the monomers are pumped downhole, monomer polymerization is induced, for example by temperature increase, or by chemical initiators. Examples of initiators for radical polymerization include persulfates, azo compounds (such as 2,2'- azobis(2-methylpropionitrile and 4,4-azobis(4-cyanovaleric acid)), hydroxylamine derivatives, such as (2,2,5-trimethyl-3-(l-phenylethoxy)-4-phenyl-3-azahexane, peroxide derivatives such as benzoyl peroxide and peracids; these are all well known chemical initiators for radical polymerization.
The initiator may be placed downhole before the monomer is pumped; it may be added to the fluid as a solute, as solid particles, adsorbed or absorbed by solid particulates, encapsulated, or impregnated, or in a monomer emulsion. The initiator may be pumped together with the monomer, or may be pumped after monomer placement. Initiators may be activated by temperature, shear, or chemical reaction, for example reaction on a surface. For Mannich reactions, formaldehyde or its precursor (hexamine, polyformaldehyde, etc) can be the initiator.
Polymerization
The initiator causes radical or another type (for example, ionic) of monomer polymerization downhole, resulting in a homogeneous or heterogeneous polymer mass, which provides bridging, sealing, cross-linking, plugging, particulate
9 000751 aggregation, or other effects where it is placed. As a result of these effects, the method is used for example for downhole fluid diversion, fluid loss or circulation loss control, proppant aggregation, wellbore isolation, and flowback control.
Various additives may be added to the monomer to provide polymerization control, for example chain length control. Chain length may be critical for the rate of subsequent polymer decomposition, as well as for precipitation with calcium or other ions coming from the formation or from other fluids, and for fines formation.
In the case of acrylates, chain length may be controlled by increasing or decreasing the initiator concentration. The more polymer chains are initiated simultaneously, the more chains are synthesized, and fewer monomers are involved in each single chain. Polymer chain transfer agents (such as ferric chloride, ethyl acetate and others) may be used as additives for chain growth control in radical polymerization reactions via transfer of radicals from growing polymer chains to monomers, thus initializing new chain formation and stopping another chain growth.
The effect of chain transfer agents on polymerization is based on competition between chain propagation (reaction of the chain with a monomer) (kp - rate constant of the propagation reaction) and reaction of the chain with the chain transfer agent (ktr k
- rate constant of the chain transfer). The higher the ratio C =— the more favored is the chain transfer reaction. After reaction with the polymer, the radical chain transfer agent can initiate a new chain. That is why chain transfer agents do not stop the polymerization reaction. Among others, iron chloride (III) and ethyl acetate exhibit large chain transfer constants.
Radical traps (RT) scavenge polymer radicals and reduce the number of growing polymer chains. Compounds such as 2,2,6,6-tetramethylpiperidine-l-oxyl
(TEMPO) also can act as mediators of radical polymerization (called nitroxide mediated polymerization (NMP)). TEMPO reacts reversibly with carbon centered polymer radicals, forming alkoxyamines. The latter can undergo radical C-0 bond homolysis, releasing active polymer chains and the nitroxyl radical TEMPO. The polymer obtained in this case has an alkoxyamine end-group. Thus it can act as a macromolecular polymerization initiator for the preparation of suitable co-polymers
of the invention. Such polymers are called "living" polymers. Among nitroxyl radicals available for NMP of acrylates are TEMPO, DEPN (N-tert-butyl-1- diethylphosphono-2,2-dimethylpropyl), and TIPNO (N-tert-butyl-N-(2-methyl-l- phenylpropyl)-N-oxyl) are commonly used. In some cases when the rate constant of homolysis of an alkoxyamine is too low at a given temperature, nitroxyl radical will stop the polymerization of monomer. Depending on the rate of alkoxyamine homolysis and the recombination rate constant between nitroxyl and C-centered radical for a particular monomer and nitroxide, one can predict the behavior of polymerizations.
Radical traps or their precursors (tetramethylpipiridine 1-oxyl (TEMPO), TEMPO derivatives, other stable radicals; phenyl-tert-butyl nitrone (PBN), PBN analogues, other nitrones; alkoxyamines; and other chemicals) scavenge polymer radicals and reduce the number of growing polymer chains, but they can also stop monomer conversion. TEMPO also can act as a mediator of radical polymerization (so called nitroxide mediated polymerization). It reacts reversibly with carbon- centered polymer radicals, forming alkoxyamines. The latter can undergo radical C-0 bond homolysis, releasing active polymer chains and nitroxyl radicals. The polymer obtained in this case has an alkoxyamine end-group; thus it can react as a macromolecular polymerization initiator for preparation of additional polymers. Such polymers are called "living polymers". Phenyl-tert-butyl nitrone (PBN) can also form nitroxyl radicals after reaction with C-centered radicals. This macromolecular nitroxyl radical can act as a polymerization mediator and control the polymerization of monomers such as acrylates.
Preferred monomers, that give high polymerization yields under typical downhole conditions, include but are not limited to the monomers of the type named
Ml and M2 above (derivatives of acrylate esters), and monomers of the types named
M3 and M4 above (such as methacrylic anhydride). Monomers such as M3 and M4 and their variants may be used as co-monomers with Ml and M2 monomers. In this embodiment, the time of complete polymerization (typically from about 10 minutes to several hours) is controlled by polymerization initiators or other additives and the downhole temperature. A nonlimiting example of an initiator is ammonium persulfate in concentrations of from about 0.5 to about 20 percent by weight of the monomer.
Another example of a polymerization initiator is phenyl-tert-butyl nitrone (PBN) in concentrations from about 0.5 to about 20 % percent by weight of the monomer. Other examples of radical initiators for polymerization are well known in the art. In another embodiment of the invention, the rate of polymerization and the later stability of the polymer chain are controlled with a chain transfer agent such as ferric chloride or ethyl acetate.
In a preferred embodiment, monomers of the type of Ml and M2 (derivatives of acrylate esters) are pumped in the form of an emulsion, produced by stirring with or without nonionic or ionic surfactants. The stability of the monomer-surfactant emulsion is checked easily in the laboratory before field operations. Monomers that are miscible with or soluble in water may be pumped as water solutions.
It has been found that emulsion polymerization in many cases gives a number of advantages, as follows:
1. Controlled polymer chain length;
2. Greater homogeneity of the resulting polymer;
3. Higher degradation rate of the resulting polymer;
4. Better wellsite delivery (no plugging risk); and
5. Lower viscosity (lower pump power required).
Degradation
The polymer formed should stay substantially intact downhole while it is required for bridging, sealing, cross-linking, plugging, particulate aggregation, or the like. After that, decomposition occurs via thermolysis or another chemical transformation, such as hydrolysis, with the formation of oligomeric or small molecules. All decomposition products are water- or oil-soluble. Thus the effect of the polymer is stopped or reversed and after the polymer degradation, no damage to the production is left. In one method, for example, hydrolysis of gelled polymer occurs at high pH. The high pH is provided by addition of alkaline-releasing material into the working fluid, by addition of fine slowly-dissolving solid alkaline into the slurry, or due to reaction of the polymer with a slightly alkaline (e.g., carbonate) formation. The amount of alkaline additive and the resulting pH under downhole conditions depends upon the time selected for removal of the emplaced polymer.
It has been found experimentally that diacrylic monomers polymerized in water form polymers by free radical polymerization that can easily be hydrolyzed in water. Polymers formed from dimethacrylic monomers are not as readily hydrolyzed; not to be limited by theory, but it is believed that these polymers are more highly crosslinked. Four strategies may be used to produce polymers that are more easily hydrolyzed under typical downhole conditions: (i) perform polymerization in the presence of surfactants; (ii) increase the amount of initiator; (iii) add a co-monomer; and (iv) employ chain transfer agents and radical traps. These may be used to produce polymers that can be degraded in a wellbore, fracture, or formation in a suitable time at the temperature and pH of the fluid.
For polymerization in the presence of surfactants the stability of monomer emulsions was first studied, and it was found that emulsions of acrylic monomers are stable during the time of polymerization, but emulsions of methacrylic polymers are not. It was then found that polymers obtained in the presence of surfactants undergo hydrolysis more easily. The products of hydrolysis produce relatively little precipitate with Ca2+. Precipitation of degradation products by cations in the fluid will usually, but not always, be detrimental to the use of the degradable polymers.
Again not to be limited by theory, but it is believed that polymerization with a higher concentration of initiator leads to polymers in which more monomer units have both polymerizable groups of the monomer incorporated into the chain backbone. This plus the lower polymer molecular weight makes hydrolysis easier.
It was found that copolymers of monomers Ml and M4 can be hydrolyzed under mild conditions, although the products of hydrolysis form a precipitate with Ca2+. Thus copolymerization is another way to control degradation rate.
Polymerization of Ml in the presence of chain transfer agents (iron chloride (III) and ethyl acetate) produced polymers that underwent hydrolysis relatively quickly; the products of hydrolysis formed only a light precipitate with Ca .
Polymerization of one acrylic monomer (Ml) in the presence of TEMPO failed at low temperatures and was slow at higher temperatures.
A particularly preferred polymer, based on the polymerization time, hydrolysis rate, and low precipitation by Ca2+, was a polymer of Ml obtained in the presence of tert-butyl-phenylnitrone (PBN).
Polymerization of Ml in the presence of ammonium persulfate at 50 °C with PLA fiber resulted in soft well-consolidated fiber floes. The approach of downhole delivery of monomer of the invention and fibers or other solid particles and subsequent polymerization and consequent flocculation may be used for diversion and/or isolation, for example in curing lost circulation.
The present invention can be further understood from the following examples.
General Polymerization Procedure:
A calculated amount of monomer (any one of M1-M4, or a 9:1 mixture of any two, or a 10:1 mixture of any one with acrylamide) was added to a round-bottom two- necked flask to which de-ionized water was then added. The concentration of monomer was 0.5 mol/1 and the total volume of the reaction mixture was 5 ml. Surfactants, chain transfer agents, and radical traps were added to the mixture when needed. The mixture was stirred on a magnetic stirrer (or an Ika-Werke T 10 emulsifier) for 10 minutes and then 5 or 50 mM of a polymerization initiator (for example ammonium persulfate) was added. The reaction mixture was made oxygen- free by degassing by 10 minutes of argon bubbling and then heated in an oil bath to 60 C°. The typical heating duration was from 20 min to 7 hours, depending upon the monomer: fast polymerization typically occurred for Ml and M2 (for example about 20 min for Ml, 30 min for M2), but longer polymerization was required for monomers of type M3 and M4 (for example about 7 hours). After heating, the mixture was cooled to room temperature; a portion was dried for polymer mass determination and a portion was left un-dried for hydrolysis tests. According to gravimetric analyses, at these times and temperatures, polymerizations of polymers made only from Ml or M2 were typically about 100%, from M3 about 30% and from M4 less than about 10%. These results indicate that by appropriate choices of concentration, initiator, inhibitor, temperature, co-monomers, and other factors, the timing and location of polymerization can be controlled.
General Hydrolysis Procedure:
A 0.002 g sample of the dry polymer was placed in a chromatographic vial and 1 ml of alkaline solution (for example solution of KOH in de-ionized water or buffer solution) was added. The sample was heated in an oil bath for the desired temperature and time. It was assumed that hydrolysis was finished after the solution became transparent. It was found that polymers made from only Ml, M2 or M3 were relatively difficult to hydrolyze, commonly requiring a pH of above about 12.5, but polymers made from only M4 hydrolyzed readily. These results indicate that by proper choices of monomers, pH, co-monomer and other factors, the degradation of the polymers can be controlled. For example, inclusion of some M4 in a polymer made of any of the other monomers accelerates the degradation.
After hydrolysis, neutralized reaction mixtures were mixed with Ca2+ solution (to provide final concentrations of 1% Ca2+) to see whether Ca2+ complexes precipitated. It was found that hydrolysis of polymers obtained with chain transfer agents or PBN (nitrone) result in soluble products that give no precipitation with Ca2+ ions. The polymer hydrolysis products do not cause formation damage.
Example 1:
It is well known that methacrylates cannot undergo thermal self-initiation of polymerization. Thus, for the polymerization of these monomers, radical polymerization initiators were employed. The number of polymeric chains in the polymerization is in direct proportion to the initial concentration of initiator. As polymerization proceeds up to a monomer conversion of 100%, then for a given polymer concentration, the larger the initial initiator concentration, the larger the number of growing polymer radicals that will be formed and be able to grow; thus there is a smaller number of monomeric units in each polymer chain. Thus, by varying the initiator/polymer concentration ratio, one can control the polymer molecular weight, and therefore the ease of polymer hydrolysis.
Polymerization of Ml in the presence of 10% of ammonium persulfate as the initiator was performed by the standard procedure described above, yielding white flakes of dry polymer. Hydrolysis of the polymer was performed with 0.1 M KOH solution (pH about 13.1) at 95 °C. The time for complete hydrolysis was estimated to
be about 18 hours. A "wet" sample, obtained from the reaction mixture by simple vacuum filtration through a glass filter without drying, was completely hydrolyzed under similar conditions in about 10 hours.
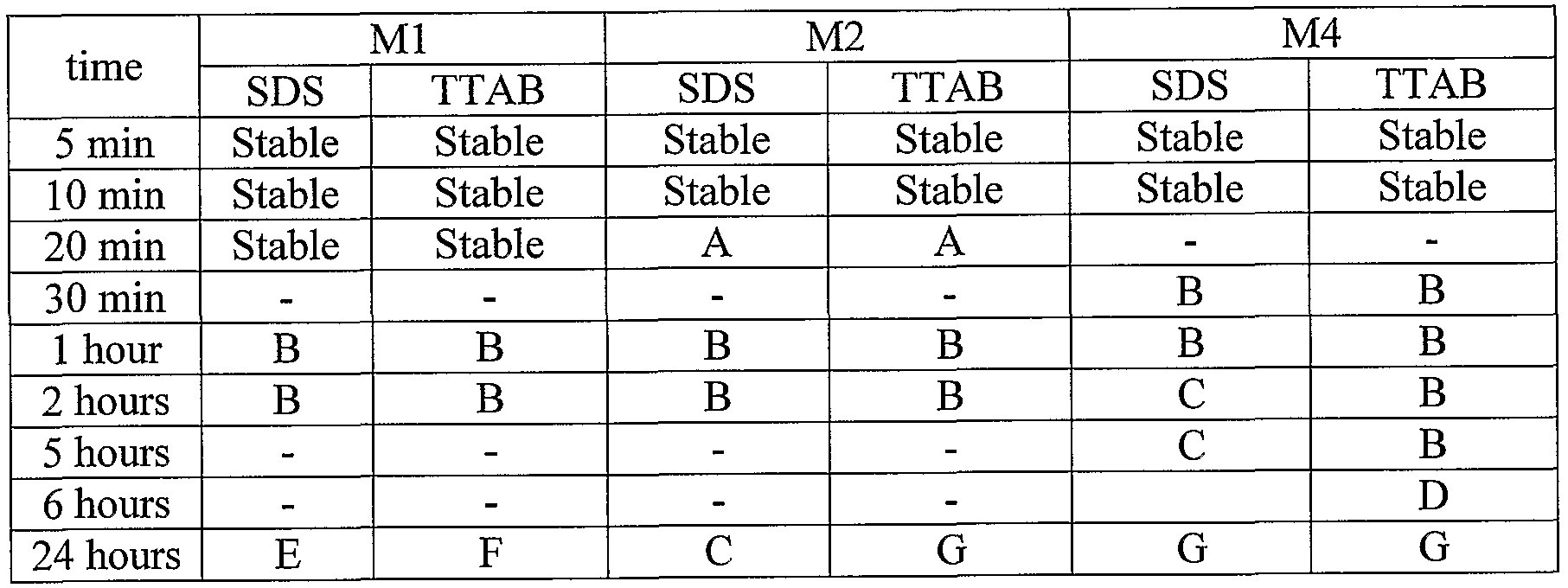
Example 2:
Samples containing 0.5 M monomer in 5 ml of water were prepared with double the critical micelle concentration of surfactant (0.015 M for SDS and 0.007 M for TTAB) and treated with an Ika-Werke T 10 basic emulsifier for 5 minutes. The stability of the emulsions vs. time was checked; the results are sown in Table 1 :
Table 1
A: White precipitate formed as emulsion begins to separate and disappears after mixing; emulsion was white.
B: White precipitate which disappeared after mixing; solution was white.
C: White precipitate which disappeared after mixing; solution was transparent.
D: Emulsion nearly completely separated. E: Emulsion started to separate.
F: Emulsion stared to separate; less stable than E (with SDS).
G: White precipitate which disappeared after mixing; solution was transparent;
emulsion was totally separated.
(Note that all these emulsions re-formed upon shaking or mixing.)
Example 3:
Emulsion polymerization experiments were carried out as follows: Samples containing 0.5 M monomer in 5 ml of de-ionized water were prepared with double the
critical micelle concentration of surfactant (0.015 M (0.0217 g/5 ml) for SDS and 0.007 M 0.0118 g/5 ml) for TTAB). The mixture was stirred with an emulsifier for 5 min. Ammonium persulfate initiator (2.5 10"3 mol/1 (0.0029 g/5 ml)) was added. The mixture was degassed by argon bubbling for 10 min and then heated in an oil bath to 60 °C for about 10 min to about 7 hours, depending on the monomer. The polymers obtained were dried at 100 °C. The results are shown I n Table 2.
* Mixture polymerized during bubbling of argon in less than 5 min.
Table 2
Example 4:
The hydrolysis of polymers prepared with surfactants was carried out by the standard procedure. The results of the hydrolysis experiments are presented in Table 3. It should be mentioned that M2 TTAB polymer did not undergo hydrolysis in 0.1 M KOH solution in 4 days at 95 °C.
temperature was increased for the sample
solution became cloudy- white without solid particles.
Table 3
Emulsions made with the surfactants and Ml or M2 were stable during the polymerization time; not shown is that emulsions made with the surfactants and M4
were not stable. The rates of polymerization wesre high for Ml and M2 and lower for M4 compared to the polymerizations in the absence of surfactants. Hydrolysis of the polymer samples made with surfactants proceeded faster than for the samples obtained without surfactants. This indicates that the polymer particles were smaller. This demonstrates how surfactants can be used to control the placement And degradation of polymers. Not to be limited by theory, but degradation kinetics and degradation product molecular weight examination suggest that the surfactant affected the amount of polymer in which only one monomer polymerizable group was included in the polymer backbone as opposed to the amount of polymer in which both monomer polymerizable groups were included in the polymer backbone (the extent of crosslinking).
Example 5:
Water-soluble products formed after hydrolysis of polymers made with Ml or M2, were checked for precipitation with Ca2+. 1% by weight of Ca2+ (in the form of CaCl2) was added to neutralized or non-neutralized solutions of hydrolyzed polymers with the results shown in Table 4.
Table 4
Example 6:
Co-polymerization of Ml and M4 was performed using the standard polymerization procedure and a Ml to M4 ratio of 1 to 9. The conversion was about
100 per cent and the dry polymer obtained was a white powder that did not hydrolyze in 0.1 M KOH solution at 95 °C in 4 days.
Co-polymerization of Ml with acrylamide (AA) was done with the standard polymerization procedure using an Ml to AA ratio of 1 to 9. The wet polymer obtained was a transparent gel; when dried this material was white particles. Part of the wet polymer was precipitated in a 9/1 acetone/methanol solution, resulting in white particles. Polymer samples were hydrolyzed in alkaline solutions, with the results shown in Table 5. In the laboratory, sometimes dry polymer samples were obtained, for example in determining yields or solubilities. Wet samples, as would be found in downhole use, degraded more rapidly than dry.
* transparent viscous solution is formed
** transparent non viscous solution is formed
pH0 is the initial pH; pHt is the pH at time t; m0 is the initial mass
"precipitated" above means precipitated from water with an acetone/methanol 9:1 mixture
Table 5
Co-polymerization of M4 with AA was performed using the standard polymerization procedure and an M4 to AA ratio of 1 to 9. The wet polymer obtained was a suspension of white particles in water; the dry polymer was transparent particles. Polymer samples were hydrolyzed in alkaline solutions, and the results are shown in Table 6. The polymers degraded at low temperatures in several minutes at relatively low pH, typical for gels, which means that degradation can be controlled by mixing these monomers and acrylamide, and that the degradation time downhole is not months or years and that the polymer disappears downhole after treatment.
In the presence of co-monomer, polymerization of M4 proceeded faster and with higher conversion. Preparation of co-polymers provided polymers having low crosslinking. That favored the hydrolysis of the polymeric samples under mild conditions.
Example 7:
Water-soluble polymers formed after hydrolysis of copolymers were checked for precipitation with Ca2+. 1% weight percent of Ca2+ (in the form of CaCl2) was added to non-neutralized solutions of hydrolyzed polymers and the results are shown
94- in Table 7. Note that the same amount of Ca dissolved in a carbonate buffer formed a white non- viscous solution without formation of flakes.
These copolymers of AA-M1 and AA-M4 hydrolyzed easily, and produced water-soluble polymers that precipitate with Ca . The sample of Ml (made with 10
3AMEH K)LU,HH JIHCT (ΠΡΑΒΚΠΟ 26)
percent ammonium persulfate) hydrolyzed in an alkaline solution formed a non- viscous solution with Ca2+; this was mainly due to the high concentration of alkaline.
Example 8:
Polymerization of Ml was carried out in the presence of either iron (III) chloride or ethyl acetate as transfer agents and TEMPO or PBN as radical traps. A calculated amount of polymer and sodium dodecyl sulfate (SDS) surfactant was put into a round bottomed flask and de-ionized water was added to give a volume of 5 ml. The mixture was mixed by an emulsifier for 5 min. A chain transfer agent (CTA) or radical trap (RT) was added and the mixture was stirred for 1 more min. The initiator was added afterwards. The mixture was bubbled with Ar for 10 min and heated in an oil bath at 80 °C (for samples with RT). For polymerization with CTA, no heating was applied; polymerization proceeded rapidly at room temperature. If heated, the mixture was then cooled to room temperature, and the polymer was dried at 100 °C to constant mass.
A typical mixture for polymerization with CTA was prepared as following:
[Ml] - 0.5 M
[APS] = 2.5 10"3 M (polymerization initiator)
[SDS] - 0.015 M
[CTA] = 5xl0-3 M ethyl acetate or 5xl0-2 M FeCl3
Water up to 5 ml
A mixture for polymerization of Ml with TEMPO was prepared as following:
[M1] = 0.5 M
[APS] = 2.5x10-3 M (polymerization initiator)
[SDS] -0 .015 M
[TEMPO] = 5xl0"3M
A mixture for polymerization with PBN was prepared according to:
[Ml] = 0.5 M
[APS] = 1.25xl0-3 M (polymerization initiator)
[SDS] = 0.015 M
[PBN] = 2.63x10_3 M
Polymerization of Ml in the presence of CTA proceeded under more mild conditions in comparison to polymerization without CTA. Conversions were high in both cases. Polymerization of acrylic monomer Ml in the presence of TEMPO stopped at low conversion due to the low homolysis rate of dormant species (intermediate alkoxyamines, formed when TEMPO quenched a polymer chain). Polymerization in the presence of PBN produced did not reduce the conversion; PBN is particularly suitable for polymerization of monomers like Ml.
Example 9:
"Wet" and "dry" samples of polymers obtained in the presence of chain transfer agents and radical traps were hydrolyzed in alkaline solutions at 95° C. The mass of "wet" samples was calculated from the monomer/water ratio in the polymerization mixture (0.011 g/ ml of alkaline solution). The results are summarized in Tables 8 (for CTA) and 9 (for RT).
Table 13
It can be seen that addition of CTA to these emulsion polymerizations produced polymers that underwent hydrolysis faster that the ones prepared in the presence of only surfactants. From GPC of hydrolyzed samples it can be seen that low molecular weight polymer molecules (103 g/mol) were formed. This may be due to the reaction of polymer radicals with CTA. GPC also shows material at high molecular weight (>106) which is believed to have been from polymerization after all the CTA had reacted. An increase in the amount of chain transfer agent (FeCl3) resulted in formation of polymer with a smaller amount of high molecular weight material.
Polymer obtained in the presence of PBN underwent hydrolysis under the most mild conditions and much faster than all other polymer samples obtained. The amount of water-soluble material obtained after hydrolysis depended strongly on the time of heating. In the case of hydrolysis in 0.02 M KOH, the polymer was heated for a longer time, so that not only could ether function react with alkaline but also alkoxyamine functions could cleave. This resulted in polymers with lower molecular weights than in the case of heating for a short time.
For both the CTA and PBN samples, the differences in the time of hydrolysis of "wet" and "dry" samples showed that for dry samples some additional time was required for the alkaline ions to reach the reaction centers in the polymer molecules.
Example 10:
The precipitation of hydrolysis products with Ca2+ was studied by adding 1% by weight of Ca2+ in the form of CaCl2 to the transparent solutions obtained after hydrolysis of Ml made with FeCl3 or Ml made with ethyl acetate.
For Ml made with FeCl3, a cloudy non- viscous solution was formed immediately after adding Ca2+. For Ml made with 1% EtOAc, a cloudy viscous solution containing several flakes was formed. For Ml made with 10% FeCl3, less cloudy solution was formed than for Ml made with 1% FeCl3. For Ml made with
PBN in 0.02 M KOH a cloudy non-viscous solution looking much the same as the initial 1 percent Ca2+ in 0.1 M KOH. The cloudiness of the solution of Ml made with
FeC13 and of the Ml made with EtOAc was roughly the same as for solutions of Ml containing 10 percent sodium dodecyl sulfate or Ml containing TTAB. Of all the polymer samples obtained in the presence of a CTA or RT, Ml made with PBN and hydrolyzed in 0.02 M KOH gave the least cloudy solution with Ca2+.
Example 11:
A bottle with 100 ppt of PL A fiber mixed with 0.5 g of ammonium persulfate and 5 g of diacrylic monomer Ml in 100 ml of DI water, and another bottle with 100 ppt of PL A fibers mixed with 5 g of diacrylic monomer Ml in 100 ml of DI water were placed in an oven at 50 °C. Two bottles with similar contents were left at ambient temperature. After 30 minutes, traces of polymerization were found in the ambient temperature bottle that included ammonium persulfate, and well-developed polymerization was seen in the bottle that included ammonium persulfate at 50 °C. After 2 hours, all the fibers inside the bottle with persulfate kept at 50 °C were flocculated, giving white, soft, but well-consolidated floes. When the fiber was mixed with the monomer alone, there was no organic/water phase separation. The monomer covered all the fibers with an even layer, showing that this is a suitable method for downhole delivery of the slurry.