WO2011053241A1 - Multiplex detection - Google Patents
Multiplex detection Download PDFInfo
- Publication number
- WO2011053241A1 WO2011053241A1 PCT/SE2010/051184 SE2010051184W WO2011053241A1 WO 2011053241 A1 WO2011053241 A1 WO 2011053241A1 SE 2010051184 W SE2010051184 W SE 2010051184W WO 2011053241 A1 WO2011053241 A1 WO 2011053241A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- probe
- target
- primer
- sequence
- nucleic acid
- Prior art date
Links
Classifications
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
- G16B25/20—Polymerase chain reaction [PCR]; Primer or probe design; Probe optimisation
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
Definitions
- the present invention relates to multiplex detection of nucleic-acids, (including such from microbiological entities), and in particular to a specific and multiplex detection method comprising mismatch-tolerant capture and amplification steps.
- the present invention also relates to probes and kits for use in such a method.
- infectious disease is a major cause of morbidity and mortality.
- Pneumonia HIV/ AIDS, diarrhea, malaria and tuberculosis are dominant causes of death and disease. They are caused by microbes (bacteria, viruses, fungi and protozoa). An exact diagnosis is necessary for successful antimicrobial treatment. Also from a national perspective, infectious diseases constitute an often preventable source of suffering and death in Sweden. However, the increasing spread of resistance to antimicrobials threatens to reverse many decades of biomedical progress. Better diagnostic techniques may reduce the development of resistance by allowing a more precise use of antimicrobials. Techniques for detection and quantification of microbes and their antimicrobial resistance genes are improving but are still limited to a single or a few pathogens. Techniques which can simultaneously detect many pathogens, and still be sensitive, rational and inexpensive are needed. Ideally, such a technique should allow detection of antibiotic resistance concomitant with the detection of the microbe.
- RNA viruses like HIV, other retroviruses, caliciviruses and influenza are highly variable and can easily escape detection.
- Another is the high diversity within families of pathogens like retroviruses, coronaviruses, enteroviruses and flaviviruses, as well as the plethora of antibiotic resistance genes. To detect many strains of such diverse families and resistance gene groups call for broadly targeted techniques.
- Another problem is the large sample volumes (10-100 mL) encountered during diagnosis of septicaemia (pneumococci, staphylococci etc), or surveillance of food-borne pathogens (E Coli 0157, Calicivirus, Sapovirus, Salmonella, Campylobacter, Listeria, Botulinus etc). Conversely, small sample volumes (50-200 ⁇ ; e.g. cerebrospinal fluid or secretions) must also be handled. Meningoencephalitis must be urgently diagnosed, which calls for
- PCR polymerase chain reaction
- any multiplex nucleic acid detection technique should not go above this limit.
- Techniques where the concentration of each oligonucleotide can be kept below 1 nM seem to be possible to multiplex up to very high levels (17). Higher concentrations may often be tolerated, but call for special optimization and control.
- Detection systems based on padlock probes or related technologies (1, 3, 16, 39) can be highly multiplex.
- Padlock probes (also called molecular inversion probes)(l, 4, 38) and Connectors (2) depend on the connection of two target- specific oligonucleotides at the ends of a linear sequence, which can be circularized after binding to a target and being ligated. The advantage of the padlock probe is that the two probe shanks are connected and cannot diffuse away from each other. Thus, when one shank encounters a target sequence, the other shank will also directly find the target sequence.
- padlock probes are in solution. Given a high enough padlock probe concentration (e.g. >10 nM) also they may have spurious interactions with each other.
- PCR primer target sequences with limited variation, separated by a more variable stretch, can be used to detect many microbes with a single or a few primer pairs. The intervening variable stretch is then used for typing, with the help of a type-specific hybridization probe.
- Such probes may be bound to a solid phase, or to microparticles (8-10, 15, 20, 21, 25).
- microparticles with hybridization probes for identification of the target nucleic acid offers rapid kinetics, which make them suitable for many clinical applications (13, 19).
- Multiplex detection is rapidly becoming the preferred way of diagnosing infections.
- detection of respiratory pathogens (6, 7, 32, 33, 35)
- papilloma virus detection and typing (2, 18, 34)
- detection and typing of fungal infections (5, 9, 11, 12, 27, 28, 31).
- PCR-based Tempi ex 14
- Megaplex 2
- MME Multiplex Microarray Enhanced PCR
- Megaplex relies on a strict localisation of both forward and reverse primers, which makes its amplification inefficient and slow. It depends on a microarray format for identification of amplified target nucleic acid(s).
- Megaplex PCR (24) depends on binding of both forward and reverse primers to a solid phase (surface). Upon binding of target to the forward primer and extension, the copied strand is covalently bound to the surface. In the next PCR run, the copied strand can only bind to the reverse primer, which then extends to the start of the forward primer. After a number of cycles, many of the target molecules have given rise to double-stranded arcs between certain forward and certain reverse primers. The surface-bound primers are locally restricted, and therefore cannot interact with other primers. The amount of illegitimate primer interaction is very low. However, the kinetics of such a solid phase PCR is slow.
- MME PCR uses gel-immobilized specific primers plus generic primers in solution, to counteract the inefficiency of the reactions of the immobilized primers.
- the method involves laborious manual steps, and the readout has to be microarray-based, with rather restricted options of detection technologies. In Templex PCR, all reactions occur in solution, and include both specific and generic primers. Probes bound to colour-coded beads are used to identify amplimers.
- the present invention relates to multiplex detection of nucleic acids, including such from microbiological entities, and in particular to a specific and multiplex detection method comprising mismatch-tolerant capture and amplification steps.
- the present invention also relates to primer/probes and kits for use in such a method, as well as a computer program for determining how the primer/probes should be constructed for an optimised result.
- the present invention relates to multiplex detection of nucleic acids, including such from microbiological entities, and in particular to a sensitive, specific and multiplex detection method which comprises mismatch-tolerant capture and amplification steps that fulfil the requirements mentioned above.
- the present invention joins several features in a novel way, . mismatch- tolerant
- primer/probes a combination of capture probe and primer, Hi. an initially locally restricted target-specific (multiplex) amplification followed by a generic amplification in solution, giving a moderate (VOCMA1, Variation-tOlerant Capture Multiplex Assay) to large
- VOCMA2 and VOCMA3/HC VOCMA potential for multiplexing and iv. a simple and specific readout.
- VOCMA2 and VOCMA3 are alternative embodiments of VOCMA1.
- nucleic acid capture, amplification and detection using mismatch-tolerant primer/probes according to the present invention can alleviate some of these problems.
- the common denominator of targeted pathogens is the presence of pathogen-specific nucleic acid.
- the present invention builds on this. Thus, one technique is used for all pathogens.
- the mismatch-tolerant probes according to the present invention are the result of a study of probes capable of detecting variable target sequences, while maintaining its specificity.
- a perfectly matching stretch of minimum length is required for hybridization, i.e. that it was more important how the mismatching nucleotides (nts) were located to each rather than the number of mismatches.
- the hybridisation is thus dependent on the length and number of stretches of matching nucleotides at the actual site of initial hybridization, "nucleation", i.e. where two DNA strands meet and are then progressively fitted together, forming a two-way "zipper" effect.
- dlnosine-probe has a lower affinity than a dlnosine-free probe, but when a regular probe fails to hybridise to a mutation-containing stretch, the probe containing dlnosine in positions overlapping said mutation(s) will still hybridise, dlnosine is a representative of the "universal" base analogues (UBAs). What is said here about dlnosine can be extended to several UBAs.
- UBAs are 3-nitropyrrole and 5- nitroindole bound to deoxyribose.
- the base Guanosine (G) may also act as a UBA.
- the hybridization prediction algorithm balances the contribution of UBAs versus the known variation of the target sequence to obtain hybridization even for highly variable targets. It is very important to balance the content of degenerated positions to the content of UBAs. It is advantageous to utilize the high likelihood of long perfectly matching segments offered by long oligonucleotides with several degenerate positions (see example 7).
- the dlnosines are thus suitably inserted at every third position.
- Such probes have been shown to hybridise well (when a complete overlap between dinosine and the mismatch exists) and rescue hybridisation where normal probes fail due to a multitude of mismatching nucleotides.
- a dlnosine-containing probe is sensitive to mmoi when the mm is positioned next to a dinosine. Further, a neighbouring mm is reducing the hybridization capacity more than a mm several nt apart from a dinosine.
- a target with as many as 30 mm can be efficiently hybridized to a dlnosine-containing probe as long as the amount of mismatches neighbouring the dinosine are few.
- a 70-mer probe with 18 dinosines in every third position could only tolerate as many as 4-5 dinosine neighbouring mismatches. This means that the location of dinosine insertions in a probe in relation to mutations in a target affects the binding affinity.
- the invention relates to a method for designing such a sequence.
- the method can be described as a method for designing the nucleotide sequence of a
- polynucleotide primer/probe capable of specific and mismatch-tolerant hybridization to a group of target polynucleotides, comprising identifying a nucleation site of 6-9 nucleotides where all or substantially all of the target polynucleotides have identical sequences and building a nucleotide sequence by
- nucleotides - positioning nucleotides A, C, T and G in positions outside the nucleation site where all or substantially all target polynucleotides have the same nucleotide;
- N is an integer between 0 and 1/3 of the length of the primer/probe, so that there are no more than N/3 mismatches between said primer/probe and said target in positions next to a position of an UBA.
- M degenerate nucleotide positions at sites of variation between target polynucleotides in a way that maximizes the likelihood of, and number of, perfectly matching stretches longer than four nucleotides, wherein M is an integer between 0 and 1/3 of the length of the primer/probe;
- a suitable length of the primer/probe used in the methods according to the invention is at least 30 nucleotides, such as between 30 and 100 nucleotides, such as 60-80 nucleotides.
- the primer/probes used in the illustrative examples are about 70 nucleotides in length.
- N and M are not both zero.
- N and M may independently be 1, 2, 3, etc. up to one third of the number of nucleotides in the primer/probe sequence.
- the invention also relates to a computer program product comprising instructions to perform the above method and a polynucleotide having a nucleotide sequence obtained or obtainable with the above method.
- the invention further relates to polynucleotides capable of specific and mismatch-tolerant hybridization to a target polynucleotide.
- a suitable length of the polynucleotides as used in the methods according to the invention is at least 30 nucleotides, such as between 30 and 100 nucleotides, such as 60-80 nucleotides.
- the primer/probes used in the illustrative examples are about 70 nucleotides in length.
- N and M are not both zero. N and M may independently be 1, 2, 3, etc. up to one third of the number of nucleotides in the primer/probe sequence.
- a polynucleotide according to this aspect may further include a primer motif for use in nucleic acid amplification reactions, such as PCR. Such polynucleotides are used in the methods (VOCMA) according to the invention.
- the mismatch-tolerant target specific part is preferably 30-100 nucleotides long, whereas the generic primer motifs are preferably of standard primer length, i.e 10-20 nucleotides preferably 14, 15 or 16 nucleotiodes.
- the present invention relates to a detection method (VOCMAl) for detecting a target nucleic acid molecule in a sample, comprising the steps
- a target specific mismatch tolerant reverse primer and a second forward primer capable of hybridizing to said generic primer sequence, a RNA reverse transcriptase or a DNA polymerase, nucleotides and optionally other amplification additives;
- VOCMAl typically comprises the following steps: After a nucleic acid extraction, performed according to conventional and suitable methods,
- a collection and washing step may or may not be performed
- help primers target-specific solution primers
- a first target-specific primer/probe (to which a first generic nucleotide stretch is covalently bound) binds a target nucleic acid, in a variation- tolerant way, i.e. it allows mismatches between the primer/probe and the target nucleic acid. A first strand copy of the bound target nucleic acid is then initiated from the first
- the second target-specific solution primer binds to the newly copied target nucleic acid in a variation-tolerant way, i.e. it allows mismatches between the primer and the target nucleic acid.
- a second strand copy of the copied target nucleic acid is then initiated from the second primer using a DNA-dependent DNA polymerase.
- the second strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer/probe.
- the second strand copy then binds a first generic primer which binds to the copy of the generic stretch.
- a third strand copy, of anti-sense orientation relative to the original target nucleic acid is then initiated from the generic primer using a DNA-dependent DNA polymerase.
- the third strand copy will in the following fourth copying round once again bind to the second target-specific primer.
- the first target-specific primer/probe or the first generic primer will be extended by a DNA-dependent DNA polymerase to produce the anti sense strand orientation relative to the original target nucleic acid, and the second target specific primer will produce the sense strand orientation relative to the original target nucleic acid.
- One or more of the primer/probes may be mismatch-tolerant as described under "Mismatch- tolerant primer/probes", or may or may not be bead-bound before binding to the target nucleic acid.
- One or more of said primer/probes may contain a dlnosine or other UBA.
- the detection may for example be performed by adding a fluorescence indicator, such as phycoerythrin bound to streptavidin, incubating the mixture, and then running it in a flow fluorescence meter.
- said method comprises the following steps:
- beads which can bind to the primer/probes through an affinity label such as the biotin/streptavidin system, can optionally be added to force primer/probes with their bound target nucleic acids to be bound to beads;
- a collection step may be performed
- the beads may or may not be separated from molecules in solution thereafter;
- the generic primers are added together with the target-specific primers in solution, allowing the preceding two steps to be performed without interruption and opening of the reaction vessel, wherein the method proceeds directly to the detection step;
- reaction solution is then separated from the beads;
- nucleic acid-binding detection probe which may or may not be mismatch-tolerant, and which probe is covalently bound to, or may via an affinity link be non-covalently bound to, a separately addressable (e.g. a colour-coded) bead, a fluorophore, or to a solid phase;
- the first target-specific primer/probe (to which a first generic nucleotide stretch is covalently bound) binds a target nucleic acid, in a variation- tolerant way, i.e. it allows mismatches between the primer/probe and the target nucleic acid.
- a first strand copy of the bound target nucleic acid is then initiated using a DNA- or RNA- dependent DNA polymerase from the first primer/probe.
- the second target-specific solution primer (to which a second generic nucleotide stretch is covalently bound) binds to the newly copied target nucleic acid in a variation-tolerant way.
- a second strand copy of the copied target nucleic acid is then initiated from the second solution primer using a DNA-dependent DNA polymerase.
- the second strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer/probe.
- An optional number of amplification rounds involving the first target-specific primer/probe and the second target- specific solution primer are then performed.
- the first target-specific primer/probe, the second target-specific solution primer, and the two generic primers may all four be present in the sample from the start.
- the amplification process is divided in two separate phases; a specific amplification phase followed by a generic amplification phase, each with an optional number of amplification rounds.
- the first specific amplification phase in which the amplification conditions are optimized for the target-specific primer/probe and the target- specific solution primer, e.g. a high annealing temperature, not allowing amplification from the first and second generic primers.
- the generic amplification the amplification conditions are shifted towards those of the first and second generic primer by e.g. decreasing the annealing temperature.
- This third strand copy will end with a copy of the second generic nucleotide stretch covalently bound to the second target-specific primer.
- the third strand copy then binds the second generic primer which binds to the copy of the second generic stretch.
- a fourth strand copy, of sense orientation relative to the original target nucleic acid, is then initiated from the second generic primer using a DNA-dependent DNA polymerase.
- the fourth strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer. In subsequent amplification steps, all copying is initiated from the first and second generic sequences and their complementary first and second generic primers.
- One or more of the primer/probes may be mismatch-tolerant as described under "Mismatch- tolerant primer/probes", or may or may not be bead-bound before binding to the target nucleic acid.
- One or more of said primer/probes may contain a dlnosine or other UBA.
- the collection step may include a washing step, where beads may be collected either through magnetism, filtration, gravity or centrifugation.
- the detection may for example be performed by adding a fluorescence indicator (in case the final amplimer is not fluorescently labelled), such as phycoerythrin bound to streptavidin, incubating the mixture, and then running it in a flow fluorescence meter.
- a fluorescence indicator in case the final amplimer is not fluorescently labelled
- VOCMA3 (Fig. 3-5), also referred to as the Hermit Crab technique (HC), said method being a method for detecting a target nucleic acid molecule in a sample, comprising bringing said sample in contact with
- nanodevice a molecular construct comprising a first and a second polynucleotide primer/probe joined, optionally through a linker, at their 5 'ends wherein said first polynucleotide primer/probe comprises a first generic primer sequence and a first target specific primer sequence, and said second polynucleotide primer/probe comprise a second generic primer sequence and a second target complement specific primer sequence; and
- RNA reverse transcriptase or a DNA polymerase nucleotides and optionally other amplification additives
- VOCMA3 comprises the following steps:
- -primer/probes specific for the target nucleic acid covalently bound to each other via their 5 'ends (a "nanodevice” as described under “Nanodevice”), are added to the extract, and are allowed to bind target nucleic acid (the primer/probe arms of the nanodevice may or may not be mismatch-tolerant as described under "Mismatch-tolerant specific primer/probes" and the nanodevice may or may not be bound to a bead or a solid phase before binding to the target nucleic acid);
- the nanodevice may be bound to beads or a solid phase via an affinity label, such as the biotin/streptavidin system, in order to force the nanodevice with its bound target nucleic acid to be bound to beads or solid phase;
- an affinity label such as the biotin/streptavidin system
- a collection step may be performed.
- a first amplification reaction, optimized for the specific primers, is subsequently conducted;
- reaction solution is then separated from the beads;
- the reaction solution containing amplimers in case the target nucleic acid was present in the nucleic acid extract, is then incubated with a nucleic acid-binding detection probe, which may or may not be mismatch-tolerant.
- This probe is covalently bound to, or may via an affinity link be non-covalently bound to, a separately addressable (e.g. a colour-coded) bead, a fluorophore, or to a solid phase;
- the target recognition and amplification is almost identical to that of VOCMA2.
- the first and second primers may both function as probes for capture and enrichment of target nucleic acid from a nucleic acid extract.
- a first target-specific primer/probe (to which a first generic nucleotide stretch is covalently bound) binds a target nucleic acid, in a variation-tolerant way, i.e. it allows mismatches between the primer/probe and the target nucleic acid.
- a first strand copy of the bound target nucleic acid is then initiated using a DNA- or RNA-dependent DNA polymerase from the first primer/probe.
- the second target specific primer binds to the newly copied target nucleic acid in a variation-tolerant way.
- a second strand copy of the copied target nucleic acid is then initiated from the second primer using a DNA-dependent DNA polymerase.
- the second strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer/probe.
- the first strand copy and its antisense second strand copy created by the elongation of the covalently coupled bound specific primer/probes may optionally be separated from the each other, by enzymatic, chemical or physical means.
- First and second, not covalently linked, target-specific prime/probes may then be added.
- An optional number of amplification rounds involving the first target-specific primer/probe and the second target-specific primer, covalently linked or not, are then performed.
- the amplification conditions are optimized for the target-specific primer/probe(s) and primer(s), and do not allow
- the generic amplification the amplification conditions are shifted towards those of the first and second generic primer by e.g. decreasing the annealing temperature. These conditions are more permissive than the previous amplification conditions allowing the generic primers, which may be shorter than the specific primer/probe arms in the nanodevice (to which generic complementary nucleotide stretch are covalently bound), to anneal and amplify.
- a second strand copy binds a first generic primer which binds to the copy of the generic stretch.
- a third strand copy, of sense orientation relative to the original target nucleic acid, is then initiated from the generic primer using a DNA-dependent DNA polymerase.
- This third strand copy will end with a copy of a second generic nucleotide stretch covalently bound to the second target-specific primer.
- the third strand copy then binds a second generic primer which binds to the copy of the second generic stretch.
- a fourth strand copy, of antisense orientation relative to the original target nucleic acid, is then initiated from the generic primer using a DNA-dependent DNA polymerase.
- the fourth strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer.
- One or more of said primer/probes may contain a dlnosine or other UBA.
- the collection step may or may not comprise a washing step.
- Beads may for example be collected through magnetism, filtration, gravity or centrifugation.
- Nanodevices bound to a solid phase may be washed directly on the solid phase.
- target-specific solution and/or generic solution primers may then optionally be added to the collected beads or solid phase bearing the nanodevice molecules.
- several steps from the collection step and up to the second amplication step may be performed without interruption and opening of the reaction vessel.
- the reactants or products bound by beads or solid phase may or may not be separated from reactants or products in solution thereafter.
- the separation may or may not be made specific for nanodevices which have undergone a complete amplification reaction with the help of a specific separation reaction such as a restriction enzyme which requires a double-stranded substrate, or a uracil-N-glycosylase enzyme which cleaves at a Uracil added to the nanodevice to facilitate specific separation of it and its amplimers from a bead or solid phase.
- the addition of generic primers may also be performed in the step of adding target-specific solution primers or in the first amplification step.
- the detection may for example be performed by adding a fluorescence indicator (in case the final amplimer is not fluorescently labelled), such as phycoerythrin bound to streptavidin, incubating the mixture, followed by an optional washing step, and then running the reaction mixture in a flow fluorescence meter, a solid phase fluorescence scanner or other fluorescence measuring device.
- a fluorescence indicator in case the final amplimer is not fluorescently labelled
- phycoerythrin bound to streptavidin incubating the mixture, followed by an optional washing step, and then running the reaction mixture in a flow fluorescence meter, a solid phase fluorescence scanner or other fluorescence measuring device.
- target-specific and variation-tolerant primer/probes, primers and probes here collectively called target-specific VOCMA binders, see below
- Variation-tolerant target-specific primer/probes and primers are Variation-tolerant target-specific primer/probes and primers:
- An oligonucleotide or other sequence-specific nucleic acid-binding molecule ("probe"), suitably designed according to a mathematical formula or a computer program which estimates the binding contribution from both local and more distant binding within in the probe in its interaction with a target molecule (Fig. 24 and 25).
- the target molecule which the probe can bind to can be an oligonucleotide, a longer nucleic acid or another sequence-specific nucleic acid-binding molecule.
- the mathematical formula or the computer program adds the binding contributions of perfectly matching (e.g.
- A means an Adenine, T a Thymine, G a Guanine and C a Cytosine base, respectively) pairs as well as contributions from UBAs or base analogues (e.g. inosine, nitroindole or a peptide nucleic acid analogue).
- base analogues e.g. inosine, nitroindole or a peptide nucleic acid analogue.
- “Local binding” here means the match of a single base or base analogue in the probe to a single base or base analogue in the target and the match between the neighbouring bases or base analogues, upstream and downstream of the single base (a triplet).
- the binding contribution from interactions occurring at bases or base analogues within the same probe or target molecule situated more than one base or base analogue away from the single base comprises both continuous stretches of perfectly matching bases longer than three bases (a “matching run"), and of cooperative binding between several matching runs.
- the mathematical formula or computer program can approximately predict the likelihood that a certain probe-target combination will bind under given conditions (probe and target concentrations, buffer and temperature).
- a primer or probe constructed according to the above-mentioned principles, to withstand target variation, is referred to as "mismatch-tolerant”.
- Said specific primer/probes suitably has a length of at least 30 nucleotides or nucleotide analogues and comprise at least one nucleation region of 4 to 9 nucleotides which is essentially free of variation and optionally one or several regions wherein inosine residues or other UBAs have been inserted at positions of expected target variation, providing mismatch (i.e. variation) tolerance.
- a preferred pattern of UBA insertion is every third nucleotide, to encompass synonymous mutations in the target nucleic acid.
- primer/probe sequence contains at least 17 matching nucleotides, contiguous or divided into a maximum of four non-contiguous stretches without mismatching neighbours, where a "universal" base analogue (UBA) in the probe counts as a matching nucleotide.
- UAA universal base analogue
- This molecule is designed to provide a high binding affinity to enhance capture of target nucleic acid from a relatively large volume of nucleic acid extract which may contain many other non-target nucleic acids. It may either be bound to a carrier (microparticle or solid phase) and/or be free in solution.
- the target-specific primer/probe may or may not possess variation (mismatch) tolerance as defined below.
- the target-specific primers may or may not possess variation (mismatch) tolerance as defined below.
- primer molecules used for the final generic amplification of all amplimers generated by the target-specific primer/probes and primers in the first round of amplification.
- Mismatch- tolerance is the ability to bind to target nucleic acid in spite of one or several mismatching nucleotides. Mismatch tolerance is achieved through i) a probe length exceeding 30 nucleotides, and/or ii) a probe sequence which contains at least 17 matching nucleotides, contiguous or divided into a maximum of four non-contiguous stretches without mismatching neighbours, where a "universal" base analogue (UBA) in the probe counts as a matching nucleotide.
- UBA can be the deoxyinosine UBA.
- UBAs An efficient pattern of such UBAs is at every third nucleotide.
- a stretch of three nucleotides which ends with a UBA is subsequently called the "synonymous mutation configuration".
- the target binding properties of the primer/probe can be approximately predicted using the "NucZip” algorithm. It combines prediction of binding at or next to a matching nucleotide (the “local” binding, embodied in triplet sequences) with binding arising from interaction of several contiguous or noncontiguous binding triplets ("distal" binding).
- the mismatch-tolerance prediction algorithm is detailed in the "Examples”.
- the primer/probes may or may not be mismatch-tolerant as defined under "Mismatch- tolerant nucleic acid-binding primer/probes".
- the connection between the primer/probes may or may not contain a cleavable bridge, also termed linker.
- the cleavage may or may not be cleavable only if the nanodevice has participated in an amplification reaction. This property may be achieved if an originally single-stranded nanodevice has become double-stranded after the first rounds of amplification.
- the nanodevice may or may not be bound to bead or solid phase.
- kits that comprises combinations of said specific primer/probes, and general primers, bound or not to beads or other solid support.
- the kit may also comprise probe-binding tag sequences, hybridization, wash and amplification solutions, or other aid substances or solutions
- Target- specific VOCMA binders are designed according to the NucZip, or a similar algorithm which estimates both local and distal contributions to binding between binder and target.
- the algorithm estimates optimal binder designs for capture, amplification and detection of target nucleic acid.
- the three binding situations require somewhat separate properties. Capture requires high affinity under moderate hybridization stringency. Target-specific amplification requires a moderate overall affinity, but a high affinity at the 3 ' end, under amplification conditions. Detection requires a high affinity under low hybridization stringency.
- the algorithm suggests suitable target-specific VOCMA binders of the three types from a variable target nucleotide sequence for which the variation is known or can be predicted.
- One embodiment of this method is a method for predicting mismatch-tolerance of a probe nucleic acid molecule, optionally comprising at least one UBA, when hybridising to a target nucleic acid molecule, comprising the steps
- nucleation score for each stretch of nucleotides in the probe, said nucleation score equalling the number of continuous base pairs perfectly matching between the probe and target sequence
- steps c)-f) optionally repeating steps c)-f) for other nucleation sites in the probe sequence.
- One embodiment of this method is the NucZip procedure.
- the NucZip procedure starts by identifying potential nucleation sites. From these, the zipping of the two hybridizing strands is modelled.
- the NucZip model scores overlapping matching tri- to pentadekamers, and the effect of inosines.
- the algorithm extends nearest neighbour theory to longer hybridizing stretches, and includes the effects of UBAs in the long oligonucleotides.
- the basis of NucZip is a scoring system for matching stretches. The program first tests for possible nucleation sites. It selects the two highest scoring ones for further evaluation. From each suggested nucleation site, zipping is performed up- and downstream. The zipping algorithm evaluates matching stretches, from tri- to pentadekamers.
- a method of producing primer/probes in which the NucZip algorithm, or computer program comprising said algorithm, is used to identify stretches in sequences of interest to the user that can be used as the basis for constructing mismatch-tolerant primer/probes and probes.
- Fig. 1 shows the general principle of the multiplex detection method according to the present invention (VOCMAl).
- VOCMAl the multiplex detection method according to the present invention
- a first primer/probe is bound to a colour-coded or otherwise separately measurable bead.
- 2. and 3. is bead-bound primer/probe, where 2. is a generic sequence and 3. is a target specific sequence.
- Fig. 2 shows the general principle of an alternative embodiment of the detection method described in Fig. 1.
- VOCMA2 the first primer/probe is bound to a magnetic bead. Magnetic bead are fishing at high bead concentration. Most targets are caught. First there is an on bead amplification, followed by amplification in solution with the general primers. Detection with Luminex beads after addition of Phycoerythrin-Streptavidin.
- 2. and 3. is a bead-bound primer/probe, where 2. is a generic sequence and 3. is a target specific sequence.
- PE is Phycoerythrin-Streptavidin
- Fig. 3 shows the double-membered first primer/probe of the capturing step of yet an alternative embodiment (VOCMA3/HC) of the detection method described in Fig. 1.
- the VOCMA3/HC has two 3 'ends.
- Figs. 4A-C show the general principle of an embodiment of the VOCMA3/HC detection method.
- Capture of target strand by major primer/probe occurs free or bead-bound to magnetic beads.
- Fig. 4B Selective liberation of newly synthesized hermit crab copies by restriction enzyme cleavage or UNG digestion.
- Fig. 5 shows the readout phase in the VOCMA3/HC method described in Figs. 4A-C general principle of an embodiment of the multiplex detection method according to the present invention.
- Fig. 6 shows results of a VOCMA1 experiment when read in a Luminex flow particle meter (see MFI in table 1 , and primer and probe in table 7).
- Fig. 7 shows results of a VOCMA 2 experiment when read in a Luminex flow particle meter(see MFI in table 2, and primer and probe in table 3).
- Fig. 8 shows results of a VOCMA2 experiment with synthetic target of Noro virus genotype 2
- Fig. 9 shows results of a 22-plex VOCMA2 experiment utilizing one synthetic target in the
- Fig. 10 shows alignment of 7333 genomes of Influenza A segment 7 in ConSort ⁇ ; most subtypes are represented. The variation was mapped, searching for a suitable region for a 70- mer probe, by using BLASTn, and ConSort ⁇ (unpubl., Blomberg J.) further used in the experiments described in Figs. 11-15, 17, 19-22. Grey bars represent conserved nucleotides (nts); Black bars represent variation of base composition; y-axes represent frequency of variation.
- Figs. 11A-C show hybridization of Influenza A probe and several Influenza A probes with increasing amount of dlnosines, to synthetic biotinylated complementary ssDNA with increasing amount of mismatching nucleotides.
- the InflA probe sequence was designed from a region in the matrix 2 gene in segment 7 of subtype H2N3 of Influenza A (sequence in table 8).
- the complementary target of InflA probe is named InflA target.
- the number of dinosines is indicated in the name of the probe; as in Ino3 probe containing three dinosines.
- Number of point mutations in target molecules is indicated in their names; like the 3pm target containing three point mutations compared to the InflA target.
- Hybridization was performed at 45 °C (Fig. 11 A and table 9A) and at 55 °C (Fig.1 IB and table 9B). Probe and target sequences are described in table 10A and 10B.
- the upper panel of Fig. 11C graphically illustrates the distribution of dinosines (grey boxes) in the probes (grey line), and the lower panel illustrates the mismatching bases (light grey boxes) in the target molecules (black line), used in Figs. HA and 11B.
- Figs. 12A-C show the length of nucleation region necessary to induce hybridization, and the the rescuing effect of dlnosine-containing probes hybridizing to targets containing
- the biotinylated targets had different percentage of mismatch, 26 %, 33 %, or 74 % mismatch, and one or two perfect matching regions of different length, 5-22 nt, compared to the InflA probe. Hybridization was performed at 45 °C (Fig. 12A) and at 55 °C (Fig.l2B). The sequence of the set of probes, with a region of different length containing dinosines in every third positioned, and the set of targets are displayed in Table 12A and 12B. The upper panel of Fig. 12C graphically illustrates the distribution of dinosines (grey boxes) in the probes (grey lines) and the lower panel illustrates the mismatching bases (light grey boxes) in the target molecules (black lines), used in Fig. 12A and 12B.
- Figs. 13A-D show analysis of specificity of hybridization comparing a Norovirus- with an Infl Avirus-probe, with or without 18 dinosines, hybridizing to targets containing different amount and distribution of both Norovirus and InflA virus-sequence.
- the sequences of the panel of probes and targets are displayed in Table 16A and B.
- the names of the targets are based on matching and mismatching nt against a Norovirus probe, the Inol 8 probe, and the InflA probe: (Norovirus matching nt in 5' and 3') mismatching nt compared to
- Norovirus probe_Inol 8 probe_InflA probe (InflA matching nt in 5' and 3').
- Fig. 13C the graphic illustration shows the three probes; Norovirus (white line), the InflA probe (grey line), and the Inol 8 probe (grey line and dark grey boxes as dlnosine nt).
- the three panels of the thirteen target molecules black lines is gradually going from the top with a Noro target sequence complementary to the Noro probe, towards the InflA target, matching the InflA probe in the bottom of the panel.
- the upper left target panel demonstrates the amount of mismatches (light grey boxes) in each target compared to the Noro-probe, and the amount of MFI the Noro-probe and resp.
- the middle left target panel demonstrates the pattern of mismatches (light grey boxes) in the target compared to the In 18 probe, and the signal of MFI received during hybridization of inol8-probe and resp. target (dark grey bars, 45°C).
- the lower left target panel shows the thirteen targets and their mismatching nt (light grey boxes) compared to the InflA probe, and the MFI received during hybridization of InflA and resp. target (light grey bars, 45°C).
- Fig. 13D demonstrates the number of nt in the position outside the pattern of 18sm dlnosines in each target that are of either Noro or InflA origin, compared with the MFI of the Inol 8 probe.
- Fig. 14 describes the predicted AG versus % MFI for all 70-mer probes and 70-mer targets from three panels presented in the table 10A and B, table 12A and B, and table 16A and B , at (45°C).
- the probes hybridizing to 70 nt long targets are visualized as black boxes.
- Visual OMP DNA Software was used to calculate the predicted AG for the interaction between probes and targets in 3M TMAC buffer conditions and hybridization temperature of 45°C.
- the % MFI is the MFI signal of a probe with an intended target divided with the
- Fig. 15 The % MFI (45°C) of all dlnosine-free combinations of probe and target, i.e. InflA probe-target and Noro probe-target, (Figs. 11 A- 13 A) versus the score for perfectly matching overlapping trimers is shown in the right panel.
- Fig. 16 demonstrates schematically the overlapping tri-, quadra-, penta- and hexamer scores.
- Figs. 17A-H demonstrate the overlapping tri (A), quadra (B), penta- (C) and hexamer scores (D) versus % MFI at 45 °C
- Fig. 17E-H demonstrates the overlapping tri (E), quadra (F), penta- (G) and hexamer scores (H) versus % MFI at 55 °C.
- Fig. 18 panels a-d. Each neighbouring trimer is given a score according
- Fig. 19 shows the TriHexa scoring system versus the % MFI for the same target and probe combinations that were demonstrated in Fig. 14 with predicted AG from Visual OMP vs % MFI.
- the TriHexa system takes all described features of scoring described in Fig. 16 and 18 into account.
- Fig. 20 shows the result of the NucZip scoring system versus the % MFI for the same target and probe combinations that were demonstrated in Fig. 14 with predicted AG from Visual OMP vs % MFI.
- the NucZip system is a further development of the TriHexa system, which adds the nucleation and zipping parts to the hybridization prediction.
- Fig. 21 Hybridization of the InflA, the Ino21 and the wobbN_21 probes to the InflA target, the 21pm target (21 point mutations) and the 21gm target, which has seven groups of three mismatches (Fig. 11C, table 18A and B). Hybridization was performed at 45°C and 55°C (table 18A and B).
- Fig.l 1C describes the distribution of dlnosines (orange/light blue/green boxes) and N wobbles (light grey boxes) in the probes (grey bar), and the mismatching bases (magenta boxes) in the target molecules (black bar).
- Fig. 22_The InflA, Inol8, 5-nitroindo_l 8, Nwobb_18 and Nwobb_24 probes were hybridized at 45 °C or 55 °C with one specific target containing 33% mismatch and one or two perfectly matching regions of different length (9-15 nt) compared with the InflA probe (table 12B).
- Fig. 12C, and table 12A shows the distribution of dlnosines, 5-nitroindole and N-wobbles in the probes used in Fig. 22.
- Fig. 23 The zipping component of NucZip
- the figure shows the downstream zipping process, with successive accumulation of score within a matching segment arising from the trimers, tetramers, etc. up to pentadecamers (each scoring equally) which fit into it. In this way, a longer matching segment gets more than a linear increase in score relative to a shorter one.
- Zipping extends from the potential nucleation site, terminating with a mismatch or the end of one of the strands, dlnosines are counted as intermediate between a match and a mismatch. If several consecutive matching segments are encountered, their scores are added.
- Fig. 24 shows a flow chart over the design process of making a VOCMA panel.
- Fig. 25 shows a flowchart over the NucZip algorithm. Brief description of the tables
- Table 1 shows results of a VOCMAl experiment when read in a Luminex flow particle meter (see also in Fig. 6).
- Table 2 shows results of a VOCMA 2 experiment when read in a Luminex flow particle meter (see also in Fig. 7).
- Table 3 shows oligonucleotides used in the VOCMAl and VOCMA2 experiments (Table 1 and 2, Figs. 6 and 7).
- Table 4 shows results of a VOCMA2 experiment with synthetic target of Norovirus genotype
- Table 5 shows oligonucleotides used in the VOCMA2 Norovirus genotype 2 experiment (Fig. 8).
- Table 6 shows results of a 22-plex VOCMA2 experiment utilizing one synthetic target in the
- Table 7 shows oligonucleotides utilized in the 22-plex VOCMA2 experiments (Fig. 9).
- Table 8 shows the variation in a rather conserved region of the segment 7, the matrix gene, between the different sub types H3N2, H5N1 and H1N1 of Influenza A virus.
- the grey nucleotides display the differences between the sequences.
- the H5N1 virus subtype differed in five nt positions and the H1N1 virus in three additional nt positions, compared with the H3N2 virus.
- the 9 pm (point mutation) sequence matches 64 different subtypes to 100% (HxNy*). Thus, if mismatches in the indicated nine positions would be tolerated by a detecting probe it will detect at least 67 Influenza A subtypes.
- Result from analysing 7333 genomes of Influenza A by using BLASTn, and ConSort ⁇ unpubl., Blomberg J.
- Table 9A shows results of hybridization at 45 °C (see also Fig. 11 A) and Table 9B at 55 °C (Fig.l lB).
- Table 10A shows the sequence of the 70-mer probes (5'-3') with increasing amount of dlnosines (I) and Table 10B shows the sequence of the 70-mer targets (5'-3') utilized in Figs. HA and 1 IB, and Fig. 21.
- Table 11A shows results of hybridization, at 45 °C (Fig. 12A) and Table 11B at 55 °C (Fig.l2B)
- Table 12 A shows the sequence of the 70-mer probes (5 '-3')
- Table 12B shows the sequence of the 70-mer targets (5 '-3') utilized in Figs. 12A and B, and in Fig. 22.
- Table 13 is a summary of the result in Fig 12A and B and Table 11 A and B, showing the nt length of the long perfectly matching regions, in either the 5' and 3' end or both ends, of the target needed to get hybridization.
- Table 14 compares the number and length of matching regions and the ability to hybridize (% MFI) for the three targets having 14 mismatches, the 14pm, 26%9F and 33%15F target; and the three targets having 16 mismatches, the 16pm, 26%5F, and 33%15F target, when they bind to the InflA probe, from the data of Fig 12A and B and Table 1 1A and B.
- Table 15A shows results of hybridization at 45 °C (Fig. 13 A) and Table 15B at 55 °C (Fig.l3B)
- Table 16A show the sequences of the three 70-mer probes (5'-3') and Table 16B show the thirteen 70-mer targets (5'-3') utilized in Fig. 13A-D.
- Table 17 demonstrates each hybridization of Inol 8 probe and the different targets and the amount of mismatches outside the position of the dlnosines of the Inol8 probe, based on results from Fig 13A-B and table 15A and B.
- Table 18A shows results of hybridization of the probes InflA, Ino21 and wobbN_21 (table 10A) and the targets InflA, 21pm, and 21 gm (table 10B) at 45 °C (Fig. 21) and Table 18B at 55 °C (Fig.21).
- Table 19A shows results of hybridization, at 45 °C (Fig. 22) and Table 19B at 55 °C
- Table 20 shows the probability of getting matching region/s of a certain length in the outer segments of the wobbN_18 probe.
- the sequences of the wobbN_18 probe are displayed with x as the nt matching the target sequence and N as the wobbling nt A, C, G or T.
- the probability of increasing the length of matching flanking 5' and 3' regions of the probe is calculated for a few examples.
- a polynucleotide primer/probe is a DNA or RNA polynucleotide that can be used either as a primer or a probe, or both.
- Mismatch-tolerant hybridization is the ability of a polynucleotide to hybridize through standard base pairing to a not fully complementary target polynucleotide.
- mismatch-tolerant and variation-tolerant shall be considered equivalent, unless specifically stated otherwise.
- UBA Universal Base Analogue
- a degenerated position in a polynucleotide is a position which has several alternative nucleotides, generally from two to four.
- the nucleotides may be chosen from A, C, G, T and UBAs.
- the terms "degenerated position” and “degeneration” shall be considered as equivalent, unless otherwise indicated.
- a perfect match between to nucleic acid molecules is when a nucleotide A in the first nucleic acid molecule pairs with a nucleotide T in the second nucleic acid molecule, or a C in the first nucleic acid molecule pairs with a nucleotide G in the second nucleic acid molecule, in a certain position of the nucleic acid molecules.
- a perfect match between two sequences means that all the nucleotide positions in that sequence are perfect matches.
- a match between two nucleic acid molecules includes perfect matches, but also the situation when a nucleotide A, T, C, G or an UBA in the first nucleic acid molecule pairs with an UBA in the second nucleic acid molecule, in a certain position of the nucleic acid molecules.
- a match between two sequences means that all the nucleotide positions in that sequence are matches, whereof some may be perfect matches.
- a mismatch between to nucleic acid molecules, in a certain position, is the situation when there is no match.
- a mismatch between two sequences means that not all the nucleotide positions in that sequence are matches.
- An analytic tag is a moiety that can be covalently or non-covalently bound to a molecule and is analytically detectable.
- One example is phycoerythrin.
- the techniques must be able to work with large extraction volumes, and both high and low target nucleic acid concentrations.
- the capture primer/probe should be mismatch-tolerant, yet have a high affinity for the target.
- the principles of achieving a mismatch-tolerant capture are described below.
- a further requirement is that the capture should be fast. It must then occur in solution, because hybridization of free probe is about 1000 fold faster than hybridization to bead-bound probe.
- a two-step capture scheme first hybridizing to free probe for 30 minutes and then "mopping up" all or most of the probes using magnetic beads (23) with shaking for 30 minutes, is more efficient than a single step with bead-bound probe.
- the beads should be small enough to easily stay in suspension, and move quickly in the solution, but be large enough to be easily collected by commercially available magnetic separators.
- Hybridization conditions may typically be at least 1 M salt and 40°C. Typical concentrations of probe are 100 million molecules/mL, typical magnetic bead concentrations are 1-10 million beads/mL. If a biotinylated probe is "mopped up" by streptavidin-coated magnetic beads, the high biotin-streptavidin dissociation constant (10 "15 M) makes the binding especially efficient. These conditions combine economy with efficiency. Magnetic beads and magnetic separation are suitable means for concentrating the probes, of which some may have found target molecules when concentrated.
- a solid phase which binds the probes (with or without attached target nucleic acid), or other kinds of beads than magnetic beads may also be used in alternative embodiments of the invention.
- MyOne streptavidin beads from Dynal/Invitrogen come at 10 10 beads/mL. They are 1 micrometer in diameter. If 1 million beads/mL are used, 1 mL of beads can be used for 10000 experiments of 1 mL. 10 million beads provide a thousandful binding excess if 100 million probe molecules/mL are used.
- Capture of target nucleic acid gives three advantages. Firstly, it increases the concentration of target nucleic acid. Secondly, it removes irrelevant nucleic acid. Thus, sample extracts with a high nucleic acid concentration can still be analyzed. Thirdly, inhibitors of ensuing enzymatic reactions will be removed.
- This can be RNA-dependent DNA-copying from + and - stranded target RNA via reverse transcriptase, or DNA-dependent DNA copying of target via DNA polymerase.
- An enzyme mixture may provide both functions.
- a further desirable specificity enhancement can be achieved by schemes to selectively remove only successfully copied nucleic acid.
- the VOCMA3/HC scheme provides this option, through the use of a restriction enzyme which only cleaves double-stranded nucleic acid. When the beads are removed, only the successful copies are left, removing all primer/probes which did not find targets. The remaining solution is thus highly enriched of successfully copied target molecules.
- Target variation is a major problem in molecular diagnostics.
- RNA viruses are especially problematic in this regard.
- a thorough bioinformatic analysis of the distribution of variation across the target sequence is necessary, and computer-based support is normally required when performing said analysis.
- the present inventors have developed a computer program, ConSort (J Blomberg, unpublished), which is optimized for this purpose and greatly facilitates this work (Fig 24).
- ConSort J Blomberg, unpublished
- the primer/probes are typically 50-70 nt long, to provide a high affinity to the target, and to provide tolerance for mismatch between primer/probe and target sequence. The greatest conservation is placed at the 3 'end.
- a primer/probe suitably has at least one stretch of nine perfectly matching nucleotides, or at least two stretches of five perfectly matching ones. Match to a UBA in the probe is here counted as a perfect match. Variable positions can be covered by UBAs in the primer/probe. However, UBA tend to decrease the affinity of the primer/probe for its target sequence. Therefore, UBAs cannot be used too generously. Above a threshold of UBA content of around 30% of the primer/probe, more UBA may make the primer/probe bind non-specifically and weakly. In order to reduce the UBA content somewhat, guanosine can be inserted at positions where guanosine is one of several naturally occurring bases.
- guanosine can base-pair weakly but non- canonically with the bases adenosine, thymine and guanosine, i.e. it has properties akin to a UBA.
- the primer/probe design algorithm must balance the content of UBA, guanosine and variable positions against each other to obtain maximal mismatch tolerance, yet high affinity to target and high specificity.
- All candidate primer/probe sequences should be analyzed for tendency to form unspecific hybrids, which may lead to primer-dimers which reduce the PCR efficiency in all three VOCMA schemes, and may cause false positivity in the VOCMAl scheme.
- concentration of solution second target-specific primer in VOCMAl should be 100 nM, and of the first target-specific primer/probes and the second target-specific primers in VOCMA2 should be around 1-10 nM, respectively.
- the first target-specfic primer/probe may also be entirely bound to bead or to solid phase in VOCMA2.
- the function of the primer/probes is to start the copying of target in a specific way.
- the degree of amplification that they give is only 1-1000 times.
- the second target-specific primer participates in the amplification up to the detection step.
- the generic primers The generic, third and fourth, primers, also referred to as “help” primers, or to as “first and second generic primers", have artificial sequences which do not have homologous or heterologous complementarity to the other primers and probes. They are entirely in solution. In VOCMA1, only one generic primer, with same sense as the first target-specific
- VOCMA2 primer/probe
- two generic primers are typically used, one forward and one reverse, in a PCR amplification.
- the VOCMA2 concept can also be used with other types of amplification, which may be isothermal.
- the function of the generic primers is to finalise the amplification, i.e. to take the brunt of amplification. Amplification factors of
- the generic primer pair could also amplify falsely.
- the very high amplification factors and their generic nature could be suspected to cause non-specificity. However, several factors counteract such non-specific amplification.
- the first specificity enhancement occurs when the third generic primer binds to its target. This can only occur if a complementary sequence was synthesized by the successive action of the first target-specific primer/probes and second target-specific primer. Likewise, the fourth generic primer can only start amplifying when the third generic prime has made a copy. Thus, each target-specific primer/probe, target-specific primer and each generic primer must act in succession to start the second, most productive, phase of amplification, provided by the generic primer(s).
- the amplification must somehow give a detection signal.
- a further specificity enhancement is caused by the placement of the label (either an affinity label, which indirectly can generate signal through binding of a signal molecule such as a fluorophore, or a signal molecule directly), which generates the readout signal, on the fourth generic primer (VOCMA2/3)
- VOCMA2/3 fourth generic primer
- a central issue is to locally restrict target-specific primer/probes. This prevents unwanted primer/probe interaction. Ideally, only the primer/probes which are specific to a target sequence should be located close to each other. This can be achieved in several ways.
- Standard PCR involves one target sequence and two primer sequences, all in solution. All possible interactions between primers can occur. If the concentration of each primer is high (>200 nM), and/or there are many different primers, the likelihood of an unwanted interaction increases.
- padlock probes depend on the connection of two target-specific oligonucleotides as the ends of a linear sequence, which can be circularized after binding to target, and ligation. When one shank encounters a target sequence, the other shank also very easily will find the target sequence. However, padlock probes are in solution. Given a high enough padlock probe concentration (e.g. >10nM), also they may have spurious interactions with each other.
- the advantages of solid phase amplification i.e. low primer/primer interactions
- those of solution amplification i.e. high efficiency.
- An initial local restriction of the target-specific primer/probes is achieved in several ways.
- the forward primer/probe may in one embodiment of said methods be bound to microparticles. This component thus cannot diffuse in solution and has a limited capacity to contribute to illegitimate interactions (leading to "primer dimers").
- the two target-specific primer/probes (forward and reverse) are covalently linked to each other via their 5' ends, thus spatially restricting the two extension reactions versus each other.
- the initial copying thus takes place in a soluble molecule. It is a "nanodevice” which seeks target molecules and tethers them to a magnetic bead which can be "fished out” from the extract.
- the two initial extension reactions can then take place free of contaminating nucleic acid. Thereafter, it is possible to increase specificity further in that only HC nanodevices which have found and appropriately extended target sequence are cleaved off from the magnetic beads or solid surface.
- the low efficiency of PCR with microparticle-bound primers, and even lower of PCR with solid-phase-bound primers may according to the present invention be counteracted in two ways.
- the target sequence may first be captured by a target-specific bead or solid phase bound primer/probe.
- the second, target-specific primer resides in solution. It is therefore free to find its specific target, which initially protrudes from the microparticle or solid phase.
- a third generic primer in solution binds to the copy of a common sequence engineered into the first target-specific microparticle-bound primer/probe.
- This third generic primer can only amplify when two strands have been copied from the original target strand. This restriction enhances specificity.
- the first specific primer/probe may be present both on a bead/solid surface and free in solution. In this case, the more efficient free primer/probe can aid the bead/solid surface bound primer/prober in the further amplification.
- VOCMA2 and VOCMA3/HC an additional fourth generic reverse primer in solution has been added.
- This additional fourth generic primer can only amplify when the longer second target-specific primer has been copied.
- VOCMA2 and VOCMA3/HC they are the only oligonucleotides which are present in high concentration.
- VOCMAl the second target-specific primers and third generic primers are the only ones present in the highest concentration.
- VOCMA and VOCMA/HC systems have been designed to combine molecular localization and mobility in an optimal way, and to increase mismatch tolerance while still preserving specificity.
- VOCMAl vs VOCMAl vs VOCMA3/HC.
- the reporting bead also carries the first target-specific primer/probe. Any mispriming involving both this primer/probe and the biotinylated second primer/probe will give a false signal. Such mispriming may arise because of the relatively high concentrations of free second target-specific primer. In practice, it turns out to be possible to multiplex up to tenfold (but it may be possible to go higher in multiplexity) with this system. The sensitivity so far settles at 10 4 - 10 5 molecules per reaction, which is enough for many purposes. VOCMA1 seems suited for applications demanding medium multiplexity, moderate sensitivity and relative simplicity.
- VOCMA2 two major differences increase the sensitivity and specificity; Firstly, the use of two generic primers allows the concentration of free first target-specific primer/probes and second target-specific primer to be decreased by a factor of 200, to 1 nM. This greatly reduces the likelihood of mispriming. Second, the readout is through hybridization to a detection sequence bound to a specific Luminex bead or any signalling molecule or bead. This will selectively detect only specific amplimers. Mispriming, if it occurs, cannot give spurious signals. However, if primer dimer formation occurs, it might reduce the efficiency of the PCR and thus reduce its sensitivity.
- VOCMA3/HC the situation is rather similar to that in VOCMA2.
- the HC construct only allows one round of specific copying. All other amplification is performed with the generic primers in solution. This may or may not be a problem which needs to be studied.
- the greatest advantages of VOCMA3/HC over VOCMA2 may be its more efficient capture of target in a large extract volume and the greater spatial isolation of the primer/probes from those of other primer/probes in a multiplex system.
- -Soluble (sense) target-specific primers e.g. 100 nM
- vs each other, vs generic primer, and vs bead-bound primer/probe e.g. 100 nM
- beads are washed pre PCR only bound targets (maybe only 1/100 of all) will be amplified, thus the sensitivity may be smaller by several logs, compared to if no washing is done.
- primers target-specific second sense primers, or target-specific second sense primers and target-specific first antisense primer/probe, plus generic primers; sense and antisense
- master mix RT-Taq mix may be used if RNA targets are included
- a paramount problem for nucleic acid-based diagnostic techniques is to overcome target variation.
- 70 nt DNA probes from the matrix- gene of H3N2 Influenza A, InflA were analyzed in the aspect of hybridization tolerance to different amount and distribution of mismatch in target DNA, and with varying number and positions of deoxyribose-Inosine (dlnosine) in the probe.
- the probes were linked to xMAP polystyrene microspheres, hybridized against biotinylated complementary single-stranded (ss) DNA, and further analyzed in the Luminex ® flow-cytometer.
- nucleation regions minimally comprise at least three regions of at least 6 nucleotides, two regions of 12-15 nt, or one region of 15-18 nt, dependent of hybridization temperature, in a 70-mer.
- probes with dlnosines in the same position as the pm of the target, like in every third position were able to rescue hybridization to targets to which a dlnosine-free probe failed to bind.
- an nt mismatch in the target neighbouring the position of dlnosine in the probe is reducing the hybridization capacity more than a mismatch several nt apart from a dlnosine.
- Specificity of high dlnosine-containing probes was tested with a 18 dlnosines InflA probe against a Norovirus target which was gradually mutated towards a InflA target sequence.
- a 50-mer probe should not contain more than 16 nt, and a 70-mer not more than 20 nt, of contiguous stretches complementary to non-targets.
- the probe length of 70 nt in this study was chosen since this length is more tolerant to a larger number of mismatches than shorter probes, although it carries a somewhat higher risk of nonspecific hybridization.
- Influenza virus is a negative ssRNA virus, whose RNA genome is highly variable due both to genetic shift, where whole segments of the genome can be exchanged, and genetic drift, caused by its error-prone RNA-dependent RNA polymerase. The latter mechanism causes the diagnostic problems which are the direct reason for of this paper.
- a 70-mer probe was designed against a conserved portion of the matrix segment 7 of Influenza A H3N2 by utilizing the programs BLASTn, ClustalX and ConSort ® (unpublished, Blomberg J. et al), in alignments with over 7000 Influenza A sequences.
- Inosine occurs naturally in tRNA, DNA repair and in RNA editing, dlnosine is one of many, more or less, generally hybridizing nucleotides. However, it is readily available and can be recognized as a G by polymerases, dlnosine can also be used to make broadly matching primers for PCR. Thus, inosine can act as a partially universal base under PCR buffer conditions (10 mM Tris, 50 mM potassium chloride, 1.5 mM magnesium chloride, 0.001% w/v gelatin, pH 8.3 at 25°C). As mentioned, 3M TMAC hybridization buffer increases Tm by increasing the binding strength of A:T base pairs, making it a platform suitable for further attempts to increase mismatch tolerance and Tm of probes towards variable targets.
- the hybridization tests were performed both against targets which had an increasing number of random and ordered pms. Of special interest were targets with regions where every third base was mutated, to resemble the common phenomenon of synonymous mutations. Much of the variation in coding viral sequences is of this kind.
- the Influenza A H3N2 70-mer probe failed to hybridize both to a target containing 21 random and evenly distributed mismatches and to a target with 18 synonymous mutations.
- introduction of dinosines in the probe, at the sites of mismatch rescues hybridization even with high numbers of mismatches. Probes with different amounts of dinosines were tested. The more dinosines the probe contains the more the MFI is reduced, showing that dlnosine destabilizes the duplex. This destabilization can to a large extent be compensated by reducing the
- mismatches greatly affected the hybridization, illustrating that hybridization predictions based on % of mismatches are imprecise.
- a minimum number of contiguous perfectly matching stretches are needed. These probably correspond to what has been termed "nucleation sites", which initiate hybridization.
- Probes with a high amount of dlnosines positioned at sites of variation needs shorter nucleation regions than dinosine free probes.
- a probe with 18 dlnosines needs only i. two regions of 9 nt (55°C), or one region of 9 perfectly matching nt (45°C), depending on hybridization temperature.
- a 70-mer probe with 18 dinosines in every third position could tolerate as many as 4-5 dinosine neighbouring mismatches.
- dinosine can be used in 3M TMAC to establish tolerance to high target variation, like in the genome of an RNA virus.
- Each VOCMA1 reaction contains a mix of all three microsphere-bound primer/probes, a generic forward solution primer, a mix of the three biotinylated target-specific reverse solution primers and one of the specific synthetic targets of 140-200 nt, all described in table 3. All three bead-bound primer/probes comprise a 5' generic sequence of 30 nt complementary to the generic forward primer, and a target-specific 3' end of 50 nt.
- Each synthetic forward target-specific primer/probe with a 5' C-12 amino modification was covalently coupled to a specific set of 5.6 ⁇ carboxylated polystyrene microspheres (Ramcon A/S, Birkerod, Denmark). 2.5 * 10 6 microspheres were pelleted at 13000 x g for 2 min and re-suspended in 25 ⁇ 0.1 M 2- Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication.
- MES 2- Morpholinoethane sulfonic acid
- microspheres were then pelleted by centrifugation at 13000 x g for 2 min and re-suspended in 1 ml of 0.1 % SDS by vortexing. Finally, the microspheres were pelleted and re-suspended to a final concentration of 50000 microspheres/ ⁇ by adding 50 ⁇ Tris-EDTA (TE, pH 8.0) buffer. Coupled microspheres were stored in darkness at 8 °C.
- Tris-EDTA TE, pH 8.0
- TMAC tetramethyl ammonium chloride
- Luminex 200 Purchased through Ramcon A/S, Birkerod, Denmark
- MFI mean fluorescence intensity
- the supernatant of the samples in VOCMA1 showed specific amplicons of correct size and the mix of beads in each sample was further analysed in the xMAP/Luminex system.
- the Luminex flow cytometer identifies each bead and measures the fluorescence of the probe bound amplicons, resulting in a MFI signal for each bead type.
- all three samples showed a significant specificity from the complementary bead-bound probe for the specific synthetic target. 100 beads of each probe specificity were analysed in every sample.
- Staphyloccocus epidermidis gseA gave a 15 fold MFI signal compared to the signal in the negative control
- the Staphyloccocus aureus MecA gave 6 fold MFI compared to negative control
- Enterococcus faecium ddl gave 10 fold compared to negative control.
- Each VOCMA2 reaction comprises a mix of all five microsphere-bound forward primer/probes, a generic forward primer, a mix of the five target- specific reverse primers, a generic biotinylated reverse primer, and one of the specific synthetic targets of 140-200 nt, all described in Fig. 7 and table 3.
- All five bead-bound probes comprise a 5' generic forward primer sequence of 30 nt, and a target-specific 3' end of 50 nt.
- the five target-specific reverse primers comprise all a 50 nt specific sequence in the 3' end and a 30 nt generic reverse primer sequence in the 5' end.
- Target genes in the example of VOCMA2 demonstrated in Fig 7 were the GseA-gene of Staphyloccocus epidermidis, the Nwc-gene of Staphyloccocus aureus the MecA-gene of Staphyloccocus aureus, the ddl-gme from Enterococcus faecium, and the i -gene from Enterococcus faecalis.
- Synthetic targets were diluted to 200 pM with 0.1 * Denhardt's solution (Sigma- Aldrich Sweden AB, Sweden) (table 3).
- PCR Gold Buffer (Applied Biosystems, Sweden), 800 ⁇ dNTP Mix, 2 mM MgCl 2 , 1 U AmpliTaq Gold ® DNA Polymerase, 30 nM generic forward primer, 300 M of biotinylated generic reverse primer, 5 nM of each of the five target-specific reverse primers and 5000 of each Luminex microsphere coupled to a target-specific primer/probe.
- Amplification was carried out on a MJ Research PTC- 100TM Peltier thermal cycler (SDS Scandinavian Diagnostic Services, Falkenberg, Sweden) as follows: 95 °C for 10 min, followed by 4 cycles of 94 °C for 30 s, 52 °C for 2 min, 72 °C for 2 min, 40 cycles of 94 °C for 30 s, 56 °C for 45 s, 72 °C for 45 s a last step with 72 °C for 7 min and the samples were kept at 4 °C until next step.
- Each synthetic target-specific forward primer/probe and detection probe oligonucleotide with a 5 ' C-12 amino modification were covalently coupled to specific sets of 5.6 ⁇ carboxylated polystyrene microspheres (Ramcon A/S, Birkerad, Denmark).
- 2.5 x lO 6 microspheres were pelleted at 13000 x g for 2 min and re-suspended in 25 ⁇ 0.1 M 2-Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication.
- MES 2-Morpholinoethane sulfonic acid
- microspheres were then pelleted by centrifugation at 13000 x g for 2 min and re-suspended in 1 ml of 0,1 % SDS by vortexing. Finally the microspheres were pelleted and re-suspended to a final concentration of 50000 microspheres/ ⁇ by adding 50 ⁇ Tris-EDTA (TE, pH 8.0) buffer. Coupled
- microspheres were stored in darkness at 8 °C.
- TMAC tetramethyl ammonium chloride
- microspheres were pelleted by centrifugation on a table centrifuge for 2 min. The supernatant were removed and the microspheres re-suspended by pipette mixing 45 ⁇ 1 ⁇ TMAC hybridization buffer comprising 3 M TMAC, 0.1 % sarkosyl, 50 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0 (Sigma- Aldrich Sweden AB, Sweden). 2 ⁇ (0.05 ⁇ g/ ⁇ l) of Streptavidin-R- Phycoerytrin (Qiagen AB, Sweden) was added to the reaction and the samples were incubated for another 15 min at 50°C.
- Luminex 200 purchased through Ramcon A/S, Birkerod, Denmark
- MFI median fluorescence intensity
- VOCMA2 is a highly specific and multiplexable detection technique. Signal to noise ratios were very low, around 1/1000 or better. The analytical sensitivity was 10 3 target copies per reaction, or better.
- Caliciviridae including Norovirus and Sapovirus
- Target-specific primer/probes for Norovirus genogroup 2 were designed targeting the POL gene of the virus.
- the VOCMA2 reaction contains microsphere-bound target-specific reverse primer/probes, generic reverse primers, biotinylated target-specific forward primers, biotinylated generic forward primers, and a synthetic target of 177 nt, all described in table 5.
- the bead-bound reverse primer comprised a 5' generic reverse primer sequence of 16 nt, and a target-specific 3' end of 71 nt.
- the biotinylated target-specific forward primer comprised a 41 nt specific sequence in the 3' end and a 16 nt generic forward sequence in the 5' end.
- the specific reverse primer includes Inosine as shown in table 5.
- Table 5 shows oligonucleotides used in VOCMA2 for detection of Norovirus genogroup 2.
- Synthetic targets (Biomers.net GMbH, Ulm, Germany) were serial-diluted from 0.,5 ⁇ 10 7 to 0.,5 l0 3 in 20 ng20ng ⁇ l yest RNA (Ambion, Austin, USA) (table 5).
- the target-specific reverse primer/probe oligonucleotide with a 5' C-12 amino modification (Biomers.net GMbH, Ulm, Germany) were covalently coupled to carboxylated Dynabeads ® MyOneTM microspheres (Invitrogen AS, Oslo, Norway). 2.0x 10 9 microspheres were pelleted by magnetism and the supernatant removed. Michrospheres were resuspend in 91 ⁇ 0.1 M 2- Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication. The pelletation and resupendation were repeated once.
- MES 2- Morpholinoethane sulfonic acid
- microspheres were then pelleted by magnetism and resuspended in 1 ml of 0, 1 % SDS by vortexing. Finally the microspheres were pelleted and resuspended to a final concentration of 10 7 microspheres/ ⁇ by adding 200 ⁇ Tris-EDTA (TE, pH 8.0) buffer.
- Tris-EDTA TE, pH 8.0
- Coupled microspheres were stored in darkness at 8°C.
- Amplification was carried out on a MJ Research PTC- 100TM Peltier thermal cycler (SDS Scandinavian Diagnostic Services, Falkenberg, Sweden) as follows: 50°C for 20 min, 95 °C for 5 min; followed by 10 cycles of 95 °C for 15 s, 56 °C for 1 min; 40 cycles of 95 °C for 15 s, 52 °C for 30 s, 60 °C for 30 s; a last step with 60 °C for 5 min before denaturation at 95 °C for 1 min. The samples were kept at 4 °C until the next step. A non-template control (NTC) was added in each run as contamination control.
- NTC non-template control
- the detection probe oligonucleotide with a 5 ' C-12 amino modification (Biomers.net GMbH, Ulm, Germany) were covalently coupled to a specific set of 5.6 ⁇ carboxylated polystyrene microspheres (Ramcon A/S, Birkerod, Denmark). 2.5 ⁇ ⁇ 6 microspheres were pelleted at 13000 x g for 2 min and resuspended in 25 ⁇ 0.1 M 2-Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication.
- MES 2-Morpholinoethane sulfonic acid
- microspheres were then pelleted by centrifugation at 13000 x g for 2 min and resuspended in 1 ml of 0.1 % SDS by vortexing. Finally the microspheres were pelleted and resuspended to a final concentration of 50000 microspheres/ ⁇ by adding 50 ⁇ Tris-EDTA (TE, pH 8.0) buffer. Coupled microspheres were stored in darkness at 8 °C.
- Samples were denatured at 95 °C for 2 min followed by hybridization at 50 °C for 20 min under shaking on a Thermostar (BMG Lab Technologies, Germany) microplate incubator. After hybridization the microspheres were pelleted by centrifugation on a table centrifuge for 2 min. The supernatant were removed and the microspheres resuspended by pipette mixing 45 ⁇ 1 x TMAC hybridization buffer comprising 3 M TMAC, 0.1 % sarkosyl, 50 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0 (Sigma-Aldrich Sweden AB, Sweden).
- FIG. 9 A demonstration of the sensitivity of a 22-plex VOCMA2 method containing 22 bacteria, fungi or antibiotic-resistance gene specific primers/probes in solution is shown in Fig. 9.
- Each VOCMA2 reaction contains a mix of 21 target-specific primer pairs, a generic forward primer and a generic biotinylated reverse primer, all described in table 7.
- the detection in Luminex is able to detect 22 different genes due to 22 different target-specific detection probes.
- the 21 target-specific forward primer/probes all comprise a 5 ' generic forward primer sequence of 16 nt, and a target-specific 3' end of 50 nt.
- the 21 target-specific reverse primers all comprise a 47-50 nt target-specific sequence in the 3' end and a 16 nt generic reverse primer sequence in the 5' end. Although all components of the 22-plex system were present in the reactions, the target gene tested in this example of VOCMA2 was the Streptococcus pneumoniae spn9802 gene. An amount of these synthetic targets of 10 to 10 molecules/reaction were added to each reaction.
- targets were serially diluted to 0,5x l0 7 -0,5 l0° molecules/ ⁇ with 0.1 x Denhardt's solution (Sigma- Aldrich Sweden AB, Sweden). As no template control a sample of 0.1 ⁇ Denhardt's solution was added.
- Amplification was carried out on a iCycler (Bio-Rad Laboratories AB, Sundbyberg, Sweden) as follows: 95 °C for 9 min; followed by 8 cycles of 94 °C for 30 s, 75 °C for 0 s, ramp with 0,1 °C/s to 58 °C for 2 min, 72 °C for 30 s; 52 cycles of 94 °C for 30 s, 53 °C for 45 s, 72 °C for 30 s; a step with 72 °C for 7 min, and a last denaturation at 95 °C for 2 min, then the samples were kept at 4 °C until next step. Coupling of microspheres
- microspheres were pelleted by centrifugation on a table centrifuge for 2 min. The supernatant were removed and the microspheres resuspended by pipette mixing 45 ⁇ 1 x TMAC hybridization buffer comprisingcomprising 3 M TMAC, 0.1 % sarkosyl, 50 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0 (Sigma-Aldrich Sweden AB, Sweden). 2 ⁇
- PCR was conducted on various combinations of coupled and not coupled Hermit Crab (HC) "arms".
- the HC arm combinations were subjected to a first PCR round using a synthetic target molecule, then amplified using generic primers. 1 ⁇ primary PCR product was used as target.
- Fig. 14 demonstrates the predicted AG (Visual OMP, DNA Software) versus % MFI (the MFI value of a modified probe relative to that of the unmodified parent probe) for different 70-mer probe and 70-mer target combinations.
- Most probe/target combinations having a predicted AG ⁇ -50 will hybridize, while most probe/target combinations with a AG > -17 do not hybridize.
- the hybridization (measured as % MFI) and the predicted AG do not correlate in the region of -17 > AG > -50, and thereby we conclude that the predicted AG is difficult to use to predict hybridization.
- a scoring system was developed in which long uninterrupted perfectly matching sequences and the neighbouring of these was taken into account.
- the Nearest-Neighbour algorithm (Santa Lucia et al.) is taking the closest neighbouring nucleotides effect on hybridization for a specific nt into account (see Fig. 18a). Thus, the Nearest-Neighbour algorithm represents local contributions to binding. It is included in the Visual OMP program. The partial failure of Visual OMP to predict hybridization mentioned in the previous paragraph made us attempt to construct a new algorithm.
- the first step in development of a new scoring system is shown in Fig. 17A and in the right panel of Fig. 15, where a score for all perfectly matching overlapping trimers in all dinosine-free combinations of probe and target, i.e.
- InflA probe-target and Noro probe-target are plotted against % MFI (45 °C).
- the Overlapping Trimer Score is schematically visualized in the upper panel of Fig. 16.
- the scoring system was further developed by using overlapping quadramers, overlapping pentamers, and overlapping hexamers, displayed in Fig. 16 and in the eight graphs of Fig. 17A-H.
- 17A-D demonstrates the overlapping tri- (A), quadra- (B), penta- (C) and hexamer scores (D) versus % MFI in 45 °C
- Fig. 17E-H demonstrates the overlapping tri- (E), quadra- (F), penta- (G) and hexamer scores (H) versus % MFI at 55 °C.
- the probes hybridizing to 70 nt long targets are visualized as black triangles, while probes hybridizing to targets of the length 12, 15, 18, and 22 nt are shown as open triangles.
- Neighbouring trimers a representation of distal but close contributions, into the scoring system. As shown in Fig. 18b, each Neighbouring trimer is given a score. Further, the Neighbouring trimers was scored according the algorithm: Pyramid factor * (n(n-l))/2); where n is the number of neighbouring trimers and the pyramid factor is decreasing the impact for each level (according to the distance between the trimers) in Fig. 18b.
- Fig. 18 demonstrates the computational work in which more distal binding contributions is taken into account.
- Each region of perfect match is weighted against a neighbouring region of perfect match and given a score, cooperativity factor 1, see Fig. 18c.
- cooperativity factor 2 see Fig. 18d.
- the TriHexa model accounts for both local (trimer) and more distal contributions to hybridization.
- Fig. 19 shows the TriHexa scoring system versus the % MFI for all target and probe combinations that were demonstrated in Fig. 14 (AG Visual OMP versus % MFI).
- the local and distal contributions to hybridization are shown symbolically in Fig. 19.
- the local binding (the binding of a matching base pair plus the binding of the next and previous base pairs) is calculated in analogy with the deltaG calculations according to nearest neighbour theory (Santa Lucia et al).
- matches to UBA are not counted as mismatches. Instead, they are given values according to the pattern of UBA distribution, and the nucleotide opposing the UBA, with "C” being the most favourable, and "G” the least favourable.
- the distal contributions are of three kinds;
- the hybridization process proceeds in two stages, nucleation and zipping.
- the nucleated 5-9 matches are further extended up- and downstream. For each turn (10 nt) of more or less perfect matches, the two strands become more and more entangled, and less likely to separate.
- the zipping process proceeds approximately as described in the TriHexa model. However, we experimented with different developments of it. It turned out that a model where the number of successive perfectly matching tri-, terra-, penta-, hexa-, hepta-, okta-, nona- and decamers were counted better explained experimental data than previous models. This is the "NucZip" model. A schematic model of NcZip is shown in Fig. 23. Its results are shown in Fig. 20.
- Single-stranded oligonucleotides of 70 nucleotides with a C12 amino link in the 5' end, with or without dlnosines, were obtained from Biomers.net (Ulm, Germany). The
- oligonucucleotides were used as probes after linkage to xMAP microspheres (Luminex Corp., Austin TX, USA).
- the probes were designed based on the programs BLASTn, ClustalX and ConSort ⁇ (unpublished, Blomberg J. et at). Briefly, query viral sequences were retrieved from the GeneBank database at NCBI, NIH. Alignments were performed by a BLASTn search. In order to identify potentially problematic sequences, the probe sequence was analyzed for sequence similarities against the human genome sequence database hgl 8. 70-mer sequences with a BLASTn score of ⁇ 37.5 (approximately corresponding to 70% mismatches or more) were accepted for further work.
- ConSort visualizes the frequency of variation and the variation of nucleotide composition in each base position, as well as the number of aligned sequences. It also provides a majority consensus sequence.
- the proposed probe sequence was further analyzed for its predicted Tm. A search for probable homodimer and hairpin interactions, using cut-offs of AG>-11.5 kcal/mol, and AG>-5 kcal/mol, respectively, was made by using Mfold at the site
- Synthetic targets complementary to the consensus sequence of InflA or Noro probe, with various numbers of mismatches, were purchased as ssDNA of 70 nucleotides with biotin attached to a 2-aminoethoxy-ethoxyethanol linker in their 5' end (Biomers.net, Ulm,
- hybridization buffer comprising 33 ⁇ 3 M TMAC buffer (3 M tetramethylammonium chloride, 0.1% Sarkosyl, 50mM Tris-HCl, pH 8.0, 4 mM EDTA, pH 8.0; Sigma) and 12 ⁇ lx TE-buffer pH 8.0 and 0.05 ⁇ (-2500 microspheres/beads) of each probe-coupled Luminex microsphere bead.
- the mixture was heated at 95°C for 2 min, to denature the DNA targets and probes, followed by hybridization at 45 °C or 55 °C for 30 min under shaking on the Thermostar (BMG LabTech; Offenburg, Germany) microplate incubator.
- RNA viruses have a high frequency of variation and almost no conserved regions in their genome.
- a broad, but yet specific, probe should detect all types or subtypes of the specific microbe it is designed for, and also be functional if the genome sequence is changed due to genetic drift.
- a 70-mer probe from the Influenza A segment 7 and a set of target molecules with increasing amounts of pm were allowed to hybridize in 3 M TMAC.
- Influenza virus is an RNA virus with a high mutation rate due to both genetic shift and genetic drift. It is divided into three types, Influenza A, B and C. The types are further divided into serotypes depending on the variation in its surface molecules haemagglutinin (HA or H) and neuraminidase (NA or N).
- Fig. 10 demonstrates the variation in an alignment of 7333 genomes of segment 7, the gene of the matrix proteins, of Influenza A; all H and N influenza A types were represented. The variation was mapped, searching for a suitable region for a 70- mer probe, by using BLASTn, and ConSort ⁇ (unpubl., Blomberg J.).
- a probe was designed to match a region of 70 nt in the gene for matrix protein 2 in segment 7 of the Influenza A H3N2 virus.
- the H5N1 virus differed in five nt and the H1N1 virus in three other nt positions, compared to the H3N2 virus (table 8). If mismatches in nine positions, including the variant nt positions in segment 7 of the H IN 1 and the H5N1 viruses, could be tolerated by a detecting probe, the probe would fully cover 67 different H and N combinations of Influenza A, i.e. nearly all variants in the chosen stretch, as demonstrated in a BLASTn search. A few subtypes would give 2 additional mismatches (table 8.).
- the Influenza A H3N2 probe from segment 7 was tested against target molecules with 3, 5, 7, 9, 1 1, 12, 13, 14, 15, 16 and 21 pms (Fig. 11 A and B) The positions of the pms were based on the variation seen comparing H3N2, H5N1 and H1N1 viruses.
- Two different targets were designed with 21 mutated nucleotides; one with 21 point mutations evenly distributed along the 70-mer, the 21rpm target, and one with seven groups of three mutations interspersed by five to seven nucleotides, the 21gm target.
- the InflA H3N2 probe was coupled to Luminex beads and subsequently allowed to hybridize to the specific biotinylated ssDNA targets in 3M TMAC in three different temperatures 45°C (Fig. 11 A), 50 °C (data not shown) and 55 °C (Fig. 1 IB).
- the hybridization was analysed in a Luminex flow cytometer where the hybridization signal is given as Median Fluorescence Intensity, MFI.
- a target containing as many as 9 pm still hybridized to the InflA probe with an MFI of 80% (45°C, Fig. 1 1 A) and 72% (55°C, Fig. 1 IB) compared to the MFI of the perfectly complementary target and probe (InflA target/InflA probe).
- MFI 80% (45°C, Fig. 1 1 A) and 72% (55°C, Fig. 1 IB)
- the MFI was reduced to 39% (45°C) and 11% (55°C) of the InflA probe/InflA target.
- the MFI was 21% (45°C) and 3% (55°C).
- the InflA probe hybridization to the 16 pm target one stretch of 6 perfectly matching nt, three regions of 5 nt, and two region of 4 nt was still detectible (12%), but at 55°C the hybridization was totally abolished (0.7%).
- the more pms that were introduced into the target the less efficient was the hybridization between target and probe.
- utilizing a lower hybridization temperature like 45°C compared to 55°C, allowed the probe to be more forgiving to mismatches.
- the MFI signal abolished in the 21rpm target increased to 52% and 25% compared to the MFI of the InflA probe/InflA target combination, at 45 °C and 55 °C, respectively.
- 1-11 evenly distributed mismatches (preserving multiple contiguous matching stretches of at least 5 nt) have little reducing effect on hybridization between probe and target, while a 70-mer probe fails to hybridize efficiently to a target containing 15-16 evenly distributed pms, although the hybridization can be rescued to a slight degree by reducing the hybridization temperature.
- seven stretches of three nonmatching nucleotides destabilize less than 21 single pms (as in 21rpm).
- the distribution of the mismatching nucleotides affects the hybridization more than the number of mismatches, indicating that the length and number of perfectly matching stretches are of greatest importance.
- the probe and target sequence will have more or less mismatch.
- Our probes were designed as 70-mers to minimize the negative effect that mismatching nt have on hybridization.
- dinosine was introduced into the probe, dinosine can bind to all four normally occurring DNA bases, but binds strongest to Cytidine
- a set of 70-mer probes were designed containing 3, 5, 7, 9 or 21 dlnosines (Fig. 11 A and B).
- the dlnosines in the probes were placed in the same positions as the pms in the Influenza targets (Fig. 11C).
- Hybridization of the 3, 5, 7 and 9 dinosine probes to the InflA target gave a small reduction of the MFI signal compared to the MFI of the normal InflA probe
- dinosine base pairs was not enhanced by TMAC as much as it enhanced A:T base pairs, and is in line with the smaller binding contribution of dlnosine-containing pairs compared to canonical base pairs.
- dinosine matches cause less destabilization than mismatches, and allows probe/target combinations with many short matching stretches, like 21 rpm, to hybridize. The dinosine thus bridges adjacent matching stretches, increasing their likelihood to nucleate.
- the dlnosine-containing probes were also analysed against the targets with different amount of pm (Fig.l 1A and B, 45°C and 55°C, respectively).
- the probes containing 3-9 dinosine gave rise to a small reduction of MFI compared to the normal InflA probe, but each dinosine probe kept its own level of MFI with targets having up to the same amount of pm as the number of matching dlnosines, plus 2 mismatches not covered by dlnosines.
- Introduction of additional mismatching nt, not covered by the dlnosine interaction reduced the level of MFI to a similar extent as seen when hybridizing the InflA probe to targets with increasing amount of pm.
- the 3-9 dlnosine probes and the InflA probe were equally efficient against the 9, 1 1 and 12 pm targets.
- the 3-9 dlnosine probes worked better than the InflA probe; 9dIno>7dIno>5dIno>3dIno> InflA.
- the 7 and 9 dlnosine probe gave rise to 1,5-1,8 times (45°C) and 4.1-7.5 times (55°C) higher MFI than the normal InflA probe during hybridization to targets with 15pm and 16 pm, although with a much lower MFI, 21-32% (45°C) and 5-14% (55°C), compared with the InflA probe/InflA target.
- the 21 dlnosine probe could rescue the hybridization to the 21rpm target, to which the InflA probe totally failed to bind (Fig. 1 1A and B).
- the signal of 3853 MFI (38% of InflA probe/InflA target) and 1381 MFI (14% of InflA probe/InflA target), at 45°C and 55°C, respectively, achieved with the 21 dlnosine probe against the 21rpm target, is in the same range as the 21 dlnosine probe binding to the other pm targets and the InflA target (calculated mean MFI is 34% of InflA probe/InflA target).
- dinosines at these positions can save the hybridization.
- the 33% mutated stretch of nucleotides (in total 18, 16, and 14 sm) was centrally placed between flanking regions of 9, 12, and 15 perfectly matching nt at 5' and the 3 end; creating the 33%9F, 33%12F, and 33%15F targets (Fig. 12A-E).
- the hybridization was analyzed at 45 °C and 55 °C (Fig. 12A and B, respectively).
- the InflA probe was shown to hybridize strongly (MFI 2259 at 45 °C and MFI 957 at 55 °C) to a target containing a centrally placed region of 14 sms and 15 perfect nt in the 5' and 3' flanks, 33%15F, at both hybridization temperatures, while 18 sm and two flanking regions of 9 perfectly matching nt, 33%9F, abolished hybridization (MFI 85 at 45 °C and MFI 37 at 55 °C) presumably due to a disturbed nucleation process even at the lower hybridization temperature, 45 °C.
- targets with approximately 74% mismatch in the centrally placed region were constructed.
- InflA probe against the 74%5F, 74%7F, 74%9F, 74%12F and 74%15F targets demonstrated that the 74%15F could hybridize to the InflA probe both at 45°C and 55°C, while the 74%12F was able to bind at 45°C (MFI 1617) but not at 55°C (MFI 73).
- Inosine-containing probe rescues hybridization to targets with synonymous mutations
- the different sm containing targets was tested against a set of probes, including the InflA probe (described above); one probe with 18 dlnosines matching the sm in 33%9F target, therefore called 18ino_33%_9F; two probes with 21 dlnosines positioned in every third base leaving either a flank of matching 9 nt in the 5 'end or in the 3' end, mo21_33%_9nt5' and Ino21_33%9nt3'; and one probe with 24 dlnosine in every third base all throughout the whole 70-mer probe, Ino24_33%.
- the 18Ino probe hybridized less well to the 33%9nt compared to the 33%9F because the two extra mutations in the 3' end of the 33%9nt target were not covered by a dlnosine in the 18Ino_33%_9F probe.
- the experiment demonstrates that when there are no suitable nucleation sequences for a dlnosine free probe, a probe with dinosines at the positions of variation will rescue the hybridization to the same target.
- ino21_33%_9nt5' could rescue binding to the 33%9F target (MFI 1017) as well as the 33%9nt target( MFI 333), even though the dinosines did not cover the two sm in the 5'end of the target.
- the ino21_33%_9nt3' that did cover the two sm in the 3' of 33%9nt, 33%12nt and 33%15nt targets gave a higher MFI than the ino21_33%_9nt5 'probe, and this resulted in a rescue of these targets with MFI of 815, 373 and 705, respectively.
- the amount of Inosines can be very high (21 dinosines in a 70 mer probe) and still give rise to MFI signals at the level of a normal probe hybridizing to a perfectly matching sequence (MFI 1179) at 45°C, as long as there are at least 9 uninterrupted matching nts without dinosines being involved, like for the ino21_33%_9nt5' probe against the 33%9F target (MFI 1017) or the ino21_33%_9nt3' probe against the 33%9nt target (MFI 815).
- MFI 1179 perfectly matching sequence
- Hybridization of a dlnosine-containing probe is sensitive to mismatches outside and with close proximity to the dlnosine position
- a probe with many dlnosines can rescue hybridization to a target with many mismatches when the dlnosine and the mismatch have the same distribution pattern and thereby the dlnosine masks the mismatch (Fig. 1 1 A and B, and Fig.12A and B).
- a test was set up to analyse how many mismatches outside the rescuing position of Inosine, mmoi (mismatch outside dlnosine), a probe can tolerate.
- the names of the targets are based on matching and mismatching nt against a Norovirus probe, the Inol8 probe, and the InflA probe:
- Norovirus probe_Inol8 probe_InflA probe (Matching nt in 5' and 3' compared to InflA probe) (Fig. 13C, Table 16A and B).
- hybridization utilizing the Inol 8 probe shows that a target with 1 mmoi ((0.10) 35_1_17 (9.8)) or even 10 mmoi ((10.10) 26 10 26 (0.8)) in the flanking sequences outside the region of the synonymous distributed 18 dlnosines had none or little inhibiting effect on hybridization (MFI 6459 and MFI 3835, resp.).
- MFI 6459 and MFI 3835, resp. Interestingly, when redistributing the 10 mmoi evenly within the 18 sm dlnosine region as in the
- the result demonstrates that the nucleation capacity of the dlnosine-containing probe Ino 18 is sensitive to mmoi when mm is positioned next to a dlnosine. Comparing the InflA probes and the Inol8 probes sensitivity to mismatch shows that the InflA probe can even hybridize to the (0.10) 35_1_17 (9.8) target, which have 17 evenly distributed mismatches in between two perfect flanks of 9 nt resp. 8 nt (MFI 2495), while 7 mmoi totally prohibits the Inol8 probe to bind to the (10.10) 19 17 33 (0.8) target (MFI 51).
- the experiment described above was performed at 45°C. In 55°C, shown in Fig. 13B, the Inol8 probe failed to hybridize already when 2 mmoi was present in the
- Norovirus sequence was constructed. Norovirus is a highly variable positive sense RNA virus, belonging to the Caliciviridae, which causes winter vomiting disease.
- the Norovirus sequence chosen (capsid gene of Norwalk-like virus, acc nr AY274264), after Blastn with the InflA-probe sequence, has a short region of 8nt that is perfectly matching the very end of the InflA probe and 10 nt dispersed spread matching nt (Fig. 13C).
- the target (10.10) 22_14_30 (0.8) and the InflA probe without dinosines could not hybridize since the target contains as many as 30 mismatches in total. Covering 16nt of these mismatches with dinosines using the Influenza based probe Inol8, gave a significant signal of 1827 MFI (27%).
- the Inol8 probe gave some but weak signal (MFI 765, 1 1%) even with a target with 31 mm, (10.10) 21 15 31 (0.8).
- the (10.10)_22_14_30_(0.8) target has less mm against the Noro probe than the InflA probe and still the InflA based Inol 8 probe gives a higher MFI than the Noro probe, MFI 1827 compared to MFI 897.
- the Fig. 13D shows the origin of the nt not covered by the dinosines when using the Inol 8 probe (MFI values from Fig. 13 A and Fig. 13C).
- the graph demonstrates how many InflA matching nt outside the dlnosine postions that are needed for hybridization and how many Norovirus matching nt that could cause a crosshybridization. 16 nt is in common of the two virus specific targets, InflA and Noro.
- Single stranded 70-mer oligonucleotides with a C12 aminolink at the 5' end, with or without dlnosines / 5 ' -nitroindole / N wobbles, were obtained from Biomers.net (Ulm, Germany).
- the synthetic 5' amine-C12 modified 70-mer probes were coupled to xMAP carboxylated colour- coded microspheres (Luminex Corp., Austin TX, USA), according to the protocol of Luminex corp (Austin TX, USA).
- the probes were hybridized to biotinylated targets as described in Example 6, and the MFI was measured in a Luminex®200TM flow cytometer (Luminex corporation, Austin Tx).
- 5-Nitroindole is a second-generation universal base nt analogue that was chosen for comparison with the first-generation dlnosine with respect to hybridization properties in 3M TMAC.
- a probe with 18 5-nitroindole residues (5-NitroInd_l 8) was designed; the nt analogues were distributed to match the pattern of the dlnosines in the Inol 8 probe (Fig. 12C).
- the probes were allowed to hybridize with the InflA target and the set of targets with synonymous mutations, 33%_xF and 33%_xnt (Fig 22).
- NucZip is performed in two steps. Nucleation is first tried. Candidates for nucleation are then used for zipping, up- and downstream of the nucleation candidate sequence. Parameter values were reached by recursive testing against measurements reported in the Ohrmalm et al., Nucleic Acids Research, 2010, 1-23.
- Fig. 23 shows a schematic model over the NucZip algorithm and
- Fig. 25 shows a schematic flowchart over the NucZip algorithm.
- the score for the 'Zip' portion of the model is obtained from the number of consecutive matching trimers, tetramers etc, up to pentadecamers, each counted with equal weight, within a contiguous matching segment.
- the same Zip scoring system was performed in two modes, counting dlnosines either as matching (mode 1) or as mismatching (mode 2). In the second mode, dlnosines will shorten the length of matching segments, decreasing the score.
- the final ZipScore was calculated as a weighted mean of ZipScore m0 d e i and ZipScore mo de2> where the weighting factor was based on the empirical data presented in the current report (Fig. 2, 3 and 4). Since dlnosine hybridizes more strongly to C than to the other nts, the algorithm adds a contribution based on the number of dI:C pairs weighted by an InoCfactor.
- PathogenMip assay a multiplex pathogen detection assay.
- PathogenMIPer a tool for the design of molecular inversion probes to detect multiple pathogens. BMC Bioinformatics 7:500.
Abstract
The present invention relates to a method for designing polynucleotide primer/probes capable of specific and mismatch tolerant hybridization useful in multiplex detection and comprising capture and amplification steps. Said method comprises identifying a nucleation site of 6-9 nucleotides, and building a nucleotide sequence by positioning nucleotides substantially complementary to said nucleation site and positioning nucleotides outside said site and introducing either universal base analogues such as deoxyinosine at sites of variation and/or positioning degenerate nucleotide positions at sites of variation thereby obtaining a continous primer/probe. The present invention also relates multiplex detection of nucleic acids and primer/probes and kits for use in such a method, as well as a computer program for determining how the primer/probes should be constructed for an optimised result.
Description
MULTIPLEX DETECTION
The present invention relates to multiplex detection of nucleic-acids, (including such from microbiological entities), and in particular to a specific and multiplex detection method comprising mismatch-tolerant capture and amplification steps. The present invention also relates to probes and kits for use in such a method.
Background
In a global perspective, infectious disease is a major cause of morbidity and mortality.
Pneumonia, HIV/ AIDS, diarrhea, malaria and tuberculosis are dominant causes of death and disease. They are caused by microbes (bacteria, viruses, fungi and protozoa). An exact diagnosis is necessary for successful antimicrobial treatment. Also from a national perspective, infectious diseases constitute an often preventable source of suffering and death in Sweden. However, the increasing spread of resistance to antimicrobials threatens to reverse many decades of biomedical progress. Better diagnostic techniques may reduce the development of resistance by allowing a more precise use of antimicrobials. Techniques for detection and quantification of microbes and their antimicrobial resistance genes are improving but are still limited to a single or a few pathogens. Techniques which can simultaneously detect many pathogens, and still be sensitive, rational and inexpensive are needed. Ideally, such a technique should allow detection of antibiotic resistance concomitant with the detection of the microbe.
However, such a technique meets several problems:
One is variable pathogen nucleic acid. RNA viruses like HIV, other retroviruses, caliciviruses and influenza are highly variable and can easily escape detection. Another is the high diversity within families of pathogens like retroviruses, coronaviruses, enteroviruses and flaviviruses, as well as the plethora of antibiotic resistance genes. To detect many strains of such diverse families and resistance gene groups call for broadly targeted techniques.
Another problem is the large sample volumes (10-100 mL) encountered during diagnosis of septicaemia (pneumococci, staphylococci etc), or surveillance of food-borne pathogens (E Coli 0157, Calicivirus, Sapovirus, Salmonella, Campylobacter, Listeria, Botulinus etc).
Conversely, small sample volumes (50-200 μί; e.g. cerebrospinal fluid or secretions) must also be handled. Meningoencephalitis must be urgently diagnosed, which calls for
simultaneous detection of herpesviruses, meningococci, pneumococci etc from a limited amount of cerebrospinal fluid sample. Likewise, respiratory pathogens like legionella, pneumoccci, influenza and coronavirus may have to be diagnosed with small volumes of secretion.
Since the end of the 1980s, the polymerase chain reaction (PCR) has revolutionized biomedicine (26, 30). Its diagnostic application made it possible to detect down to single molecules of nucleic acid from viruses, bacteria, protozoa and fungi. Real-time PCR is a variant of PCR which can not only detect nucleic acid, it can also quantify it, with a very large dynamic range. However, PCR is limited with regard to the number of different nucleic acids that can be detected simultaneously, i.e. the multiplexity. This is because above a total summative threshold concentration of 50-500 nM, oligonucleotides start to bind and react with each other, causing mispriming and so-called primer-dimers. In fact, any multiplex nucleic acid detection technique should not go above this limit. Techniques where the concentration of each oligonucleotide can be kept below 1 nM seem to be possible to multiplex up to very high levels (17). Higher concentrations may often be tolerated, but call for special optimization and control. Detection systems based on padlock probes or related technologies (1, 3, 16, 39) can be highly multiplex. Padlock probes (also called molecular inversion probes)(l, 4, 38) and Connectors (2) depend on the connection of two target- specific oligonucleotides at the ends of a linear sequence, which can be circularized after binding to a target and being ligated. The advantage of the padlock probe is that the two probe shanks are connected and cannot diffuse away from each other. Thus, when one shank encounters a target sequence, the other shank will also directly find the target sequence.
However, the short shanks of the padlock probes (15-25 nucleotides) make them vulnerable to pathogen variation, with the possibility to create false negative results due to the inability of short probe sequences to tolerate mismatches. Moreover, padlock probes are in solution. Given a high enough padlock probe concentration (e.g. >10 nM) also they may have spurious interactions with each other. PCR primer target sequences with limited variation, separated by a more variable stretch, can be used to detect many microbes with a single or a few primer pairs. The intervening variable stretch is then used for typing, with the help of a type-specific
hybridization probe. Such probes may be bound to a solid phase, or to microparticles (8-10, 15, 20, 21, 25). The use of microparticles with hybridization probes for identification of the target nucleic acid offers rapid kinetics, which make them suitable for many clinical applications (13, 19). Multiplex detection is rapidly becoming the preferred way of diagnosing infections. Especially advanced presently are the detection of respiratory pathogens (6, 7, 32, 33, 35), papilloma virus detection and typing (2, 18, 34), and detection and typing of fungal infections (5, 9, 11, 12, 27, 28, 31).
As illustrative examples of multiplexing techniques, the PCR-based Tempi ex (14), Megaplex (24) and MME PCR (Multiplex Microarray Enhanced PCR) (29) systems may be mentioned.
Megaplex relies on a strict localisation of both forward and reverse primers, which makes its amplification inefficient and slow. It depends on a microarray format for identification of amplified target nucleic acid(s).
Megaplex PCR (24) depends on binding of both forward and reverse primers to a solid phase (surface). Upon binding of target to the forward primer and extension, the copied strand is covalently bound to the surface. In the next PCR run, the copied strand can only bind to the reverse primer, which then extends to the start of the forward primer. After a number of cycles, many of the target molecules have given rise to double-stranded arcs between certain forward and certain reverse primers. The surface-bound primers are locally restricted, and therefore cannot interact with other primers. The amount of illegitimate primer interaction is very low. However, the kinetics of such a solid phase PCR is slow.
Although the initial hybridization-driven events are very specific, both padlock probe and Megaplex PCR detection schemes need a major amplification of the initial products to make them visible by various readout techniques. Typically, amplification factors of 107 to 109 are needed. Such a strong amplification is not trivial. False positive and false negative results may occur.
MME PCR uses gel-immobilized specific primers plus generic primers in solution, to counteract the inefficiency of the reactions of the immobilized primers. The method involves laborious manual steps, and the readout has to be microarray-based, with rather restricted options of detection technologies.
In Templex PCR, all reactions occur in solution, and include both specific and generic primers. Probes bound to colour-coded beads are used to identify amplimers.
Summary of the invention:
The present invention relates to multiplex detection of nucleic acids, including such from microbiological entities, and in particular to a specific and multiplex detection method comprising mismatch-tolerant capture and amplification steps. The present invention also relates to primer/probes and kits for use in such a method, as well as a computer program for determining how the primer/probes should be constructed for an optimised result.
The following properties are desired in a universal nucleic acid-based pathogen detection system:
be able to handle sample volumes of from 10 microliters to 100 milliliters (a 10 000 fold range),
- handle RNA as well as DNA
simultaneously detect all likely pathogens for a certain type of sample
- be sensitive down to a few pathogen nucleic acid molecules per reaction,
be specific and true to the intended range of detected pathogens,
be rational and rapid enough to enable a report within a few hours,
- tolerate the sometimes high target nucleic acid variation, especially of viral pathogens, allow quantification of the concentration of target nucleic acid, and thereby the concentration of the pathogen which carries the nucleic acid,
be able to use easily available equipment.
The present invention relates to multiplex detection of nucleic acids, including such from microbiological entities, and in particular to a sensitive, specific and multiplex detection method which comprises mismatch-tolerant capture and amplification steps that fulfil the requirements mentioned above.
The present invention joins several features in a novel way, . mismatch- tolerant
primer/probes, . a combination of capture probe and primer, Hi. an initially locally restricted target-specific (multiplex) amplification followed by a generic amplification in solution,
giving a moderate (VOCMA1, Variation-tOlerant Capture Multiplex Assay) to large
(VOCMA2 and VOCMA3/HC VOCMA) potential for multiplexing and iv. a simple and specific readout. VOCMA2 and VOCMA3 are alternative embodiments of VOCMA1.
Many known techniques produce false negative results due to pathogen (i.e. target sequence) variation or inhibitors of amplification. The nucleic acid capture, amplification and detection using mismatch-tolerant primer/probes according to the present invention can alleviate some of these problems.
The common denominator of targeted pathogens is the presence of pathogen-specific nucleic acid. The present invention builds on this. Thus, one technique is used for all pathogens.
The mismatch-tolerant probes according to the present invention are the result of a study of probes capable of detecting variable target sequences, while maintaining its specificity. In this study, it was noticed that a perfectly matching stretch of minimum length is required for hybridization, i.e. that it was more important how the mismatching nucleotides (nts) were located to each rather than the number of mismatches. The hybridisation is thus dependent on the length and number of stretches of matching nucleotides at the actual site of initial hybridization, "nucleation", i.e. where two DNA strands meet and are then progressively fitted together, forming a two-way "zipper" effect.
In this study it was surprisingly discovered that a probe containing dlnosines designed according to a hybridization prediction algorithm ("NucZip") based on the number and length of matching stretches, presented in this invention, could rescue hybridization to variable targets like a RNA virus. The dlnosine-probe has a lower affinity than a dlnosine-free probe, but when a regular probe fails to hybridise to a mutation-containing stretch, the probe containing dlnosine in positions overlapping said mutation(s) will still hybridise, dlnosine is a representative of the "universal" base analogues (UBAs). What is said here about dlnosine can be extended to several UBAs. Further examples of UBAs are 3-nitropyrrole and 5- nitroindole bound to deoxyribose. The base Guanosine (G) may also act as a UBA. The hybridization prediction algorithm balances the contribution of UBAs versus the known variation of the target sequence to obtain hybridization even for highly variable targets. It is very important to balance the content of degenerated positions to the content of UBAs. It is
advantageous to utilize the high likelihood of long perfectly matching segments offered by long oligonucleotides with several degenerate positions (see example 7).
It is very common that every third nucleotide varies in a variable genome (like in a virus), since that variation does not affect the virus when the code of the genome is read in order to produce the proteins which make up the virus (synonymous mutations that lead to a shift in nucleotide sequence but not in amino acid sequence). According to the present invention, the dlnosines are thus suitably inserted at every third position. Such probes have been shown to hybridise well (when a complete overlap between dinosine and the mismatch exists) and rescue hybridisation where normal probes fail due to a multitude of mismatching nucleotides.
How many mismatches outside the rescuing position of dinosine, mmoi (mismatch outside dinosine), can a probe tolerate? The result demonstrates that a dlnosine-containing probe is sensitive to mmoi when the mm is positioned next to a dinosine. Further, a neighbouring mm is reducing the hybridization capacity more than a mm several nt apart from a dinosine. A target with as many as 30 mm can be efficiently hybridized to a dlnosine-containing probe as long as the amount of mismatches neighbouring the dinosine are few. A 70-mer probe with 18 dinosines in every third position could only tolerate as many as 4-5 dinosine neighbouring mismatches. This means that the location of dinosine insertions in a probe in relation to mutations in a target affects the binding affinity.
This gives a versatile probe, which at the same time is sensitive to mismatches in the vicinity of the dinosine, thus reducing its degree of cross-hybridisation.
Thus, in one aspect, the invention relates to a method for designing such a sequence. The method can be described as a method for designing the nucleotide sequence of a
polynucleotide primer/probe capable of specific and mismatch-tolerant hybridization to a group of target polynucleotides, comprising identifying a nucleation site of 6-9 nucleotides where all or substantially all of the target polynucleotides have identical sequences and building a nucleotide sequence by
- positioning nucleotides A, C, T and G in positions within the nucleation site so as to form a sequence complementary to all or substantially all of the target
polynucleotides;
- positioning nucleotides A, C, T and G in positions outside the nucleation site where all or substantially all target polynucleotides have the same nucleotide;
- positioning N Universal Base Analogues (UBAs) at sites of sequence variations
between the target polynucleotides, wherein N is an integer between 0 and 1/3 of the length of the primer/probe, so that there are no more than N/3 mismatches between said primer/probe and said target in positions next to a position of an UBA.
- positioning M degenerate nucleotide positions at sites of variation between target polynucleotides in a way that maximizes the likelihood of, and number of, perfectly matching stretches longer than four nucleotides, wherein M is an integer between 0 and 1/3 of the length of the primer/probe;
thereby obtaining a continuous polynucleotide primer/probe sequence.
As discussed above, a suitable length of the primer/probe used in the methods according to the invention is at least 30 nucleotides, such as between 30 and 100 nucleotides, such as 60-80 nucleotides. The primer/probes used in the illustrative examples are about 70 nucleotides in length.
Preferably, N and M are not both zero. N and M may independently be 1, 2, 3, etc. up to one third of the number of nucleotides in the primer/probe sequence.
The invention also relates to a computer program product comprising instructions to perform the above method and a polynucleotide having a nucleotide sequence obtained or obtainable with the above method.
The invention further relates to polynucleotides capable of specific and mismatch-tolerant hybridization to a target polynucleotide. A suitable length of the polynucleotides as used in the methods according to the invention is at least 30 nucleotides, such as between 30 and 100 nucleotides, such as 60-80 nucleotides. The primer/probes used in the illustrative examples are about 70 nucleotides in length.
Preferably, N and M are not both zero. N and M may independently be 1, 2, 3, etc. up to one third of the number of nucleotides in the primer/probe sequence.
A polynucleotide according to this aspect may further include a primer motif for use in nucleic acid amplification reactions, such as PCR. Such polynucleotides are used in the methods (VOCMA) according to the invention. The mismatch-tolerant target specific part is preferably 30-100 nucleotides long, whereas the generic primer motifs are preferably of standard primer length, i.e 10-20 nucleotides preferably 14, 15 or 16 nucleotiodes.
According to one aspect, the present invention relates to a detection method (VOCMAl) for detecting a target nucleic acid molecule in a sample, comprising the steps
- bringing said sample in contact with a polynucleotide according to any of claims 5-6, a target specific mismatch tolerant reverse primer and a second forward primer capable of hybridizing to said generic primer sequence, a RNA reverse transcriptase or a DNA polymerase, nucleotides and optionally other amplification additives;
- optionally, enrichment of target nucleic acid molecule bound to a polynucleotide
according to any of claims 5-6 which is bound to a solid phase, such as a magnetic bead, and subsequently separated from other sample nucleic acids by displacement of the solid phase on which subsequent detection steps take place;
- optionally, increasing the specificity of detection by spatial restriction of the binding of target specific and generic primers, described in the following points, by initiating the first round of amplification on the solid phase;
- initiating a first forward replication round primed by the complex formed by the target nucleic acid molecule and the specific and mismatch-tolerant part of the primer/probe, thereby forming a first replicated nucleic acid molecule;
- initiating a first reverse replication round primed by the complex formed by the first replicated nucleic acid molecule and the target specific reverse primer, thereby forming a second replicated nucleic acid molecule;
- initiating a second forward replication round primed by the complex formed by the second replicated nucleic acid molecule and the second forward primer;
- amplifying said replicated nucleic acid molecules forming amplimers;
- detecting said amplimers.
In one embodiment, VOCMAl typically comprises the following steps:
After a nucleic acid extraction, performed according to conventional and suitable methods,
- primer/probes specific for the target nucleic acid are added to the extract, and allowed to bind target nucleic acid;
- at this stage, a collection and washing step may or may not be performed;
- generic primers ("help" primers) and target-specific solution primers are then added;
- an amplification reaction is subsequently conducted;
- the complex formed between the primers and the target nucleic acid is then washed, with or without the presence of an antisense blocking oligonucleotide complementary to the generic primer;
- the presence of nucleic acid is detected.
In one embodiment of VOCMAl (Fig. 1) , a first target-specific primer/probe (to which a first generic nucleotide stretch is covalently bound) binds a target nucleic acid, in a variation- tolerant way, i.e. it allows mismatches between the primer/probe and the target nucleic acid. A first strand copy of the bound target nucleic acid is then initiated from the first
primer/probe. In the subsequent copying round, the second target-specific solution primer binds to the newly copied target nucleic acid in a variation-tolerant way, i.e. it allows mismatches between the primer and the target nucleic acid. A second strand copy of the copied target nucleic acid is then initiated from the second primer using a DNA-dependent DNA polymerase. The second strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer/probe. The second strand copy then binds a first generic primer which binds to the copy of the generic stretch. A third strand copy, of anti-sense orientation relative to the original target nucleic acid, is then initiated from the generic primer using a DNA-dependent DNA polymerase. The third strand copy will in the following fourth copying round once again bind to the second target-specific primer. In subsequent amplification steps, the first target-specific primer/probe or the first generic primer will be extended by a DNA-dependent DNA polymerase to produce the anti sense strand orientation relative to the original target nucleic acid, and the second target specific primer will produce the sense strand orientation relative to the original target nucleic acid.
One or more of the primer/probes may be mismatch-tolerant as described under "Mismatch- tolerant primer/probes", or may or may not be bead-bound before binding to the target nucleic acid. One or more of said primer/probes may contain a dlnosine or other UBA.
The detection may for example be performed by adding a fluorescence indicator, such as phycoerythrin bound to streptavidin, incubating the mixture, and then running it in a flow fluorescence meter.
According to an embodiment of the detection method (VOCMA2) (Fig. 2), said method comprises the following steps:
After a nucleic acid extraction, performed according to conventional and suitable methods,
- primer/probes specific for the target nucleic acid are added to the extract, and allowed to bind target nucleic acid;
- in the case the primer/probes are not initially bead-bound, beads which can bind to the primer/probes through an affinity label, such as the biotin/streptavidin system, can optionally be added to force primer/probes with their bound target nucleic acids to be bound to beads;
- at this stage, a collection step may be performed;
- target-specific solution primers are then added to the collected beads;
- a first amplification reaction optimized for the target-specific primers is subsequently conducted , and the beads may or may not be separated from molecules in solution thereafter;
- subsequently, generic primers which bind to sequences in the specific primer/probes are added;
- a second amplification reaction in solution, optimized for the generic primers, is
subsequently conducted;
- optionally, the generic primers are added together with the target-specific primers in solution, allowing the preceding two steps to be performed without interruption and opening of the reaction vessel, wherein the method proceeds directly to the detection step;
- in case beads are present, the reaction solution is then separated from the beads;
- the reaction solution, containing amplimers in case the target nucleic acid was present in the nucleic acid extract, is then incubated with a nucleic acid-binding detection probe, which may or may not be mismatch-tolerant, and which probe is covalently bound to, or may via an affinity link be non-covalently bound to, a separately addressable (e.g. a colour-coded) bead, a fluorophore, or to a solid phase;
- the presence of nucleic acid is detected.
In one embodiment of VOCMA2, the first target-specific primer/probe (to which a first generic nucleotide stretch is covalently bound) binds a target nucleic acid, in a variation-
tolerant way, i.e. it allows mismatches between the primer/probe and the target nucleic acid. A first strand copy of the bound target nucleic acid is then initiated using a DNA- or RNA- dependent DNA polymerase from the first primer/probe.
In the subsequent copying round, the second target-specific solution primer (to which a second generic nucleotide stretch is covalently bound) binds to the newly copied target nucleic acid in a variation-tolerant way. A second strand copy of the copied target nucleic acid is then initiated from the second solution primer using a DNA-dependent DNA polymerase. The second strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer/probe. An optional number of amplification rounds involving the first target-specific primer/probe and the second target- specific solution primer are then performed. The first target-specific primer/probe, the second target-specific solution primer, and the two generic primers may all four be present in the sample from the start. The amplification process is divided in two separate phases; a specific amplification phase followed by a generic amplification phase, each with an optional number of amplification rounds. The first specific amplification phase in which the amplification conditions are optimized for the target-specific primer/probe and the target- specific solution primer, e.g. a high annealing temperature, not allowing amplification from the first and second generic primers. In the second amplification phase, the generic amplification, the amplification conditions are shifted towards those of the first and second generic primer by e.g. decreasing the annealing temperature. These conditions are more permissive than the previous amplification conditions allowing the generic primers, which may be shorter than the specific primer/probe (to which a first generic complementary nucleotide stretch is covalently bound) and the specific solution primer (to which a second generic complementary nucleotide stretch is covalently bound), to anneal and amplify. In this generic amplification, a second strand copy binds the first generic primer which binds to the copy of the first generic stretch. A third strand copy, of anti-sense orientation relative to the original target nucleic acid, is then initiated from the first generic primer using a DNA-dependent DNA polymerase. This third strand copy will end with a copy of the second generic nucleotide stretch covalently bound to the second target-specific primer. The third strand copy then binds the second generic primer which binds to the copy of the second generic stretch. A fourth strand copy, of sense orientation relative to the original target nucleic acid, is then initiated from the second generic primer using a DNA-dependent DNA polymerase. The fourth strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer.
In subsequent amplification steps, all copying is initiated from the first and second generic sequences and their complementary first and second generic primers.
One or more of the primer/probes may be mismatch-tolerant as described under "Mismatch- tolerant primer/probes", or may or may not be bead-bound before binding to the target nucleic acid. One or more of said primer/probes may contain a dlnosine or other UBA.
The collection step may include a washing step, where beads may be collected either through magnetism, filtration, gravity or centrifugation.
The detection may for example be performed by adding a fluorescence indicator (in case the final amplimer is not fluorescently labelled), such as phycoerythrin bound to streptavidin, incubating the mixture, and then running it in a flow fluorescence meter.
According to a further embodiment of the detection method (VOCMA3) (Fig. 3-5), also referred to as the Hermit Crab technique (HC), said method being a method for detecting a target nucleic acid molecule in a sample, comprising bringing said sample in contact with
- a molecular construct ("nanodevice") comprising a first and a second polynucleotide primer/probe joined, optionally through a linker, at their 5 'ends wherein said first polynucleotide primer/probe comprises a first generic primer sequence and a first target specific primer sequence, and said second polynucleotide primer/probe comprise a second generic primer sequence and a second target complement specific primer sequence; and
- a RNA reverse transcriptase or a DNA polymerase, nucleotides and optionally other amplification additives;
in a suitable buffer solution; said method further comprising the steps
- initiating a first replication round primed by the complex formed by the target nucleic acid molecule and the target specific primer sequence part of the first polynucleotide primer/probe, thereby forming a first replicated nucleic acid molecule;
- initiating a second replication round primed by the complex formed by the first
replicated nucleic acid molecule and the target complement specific primer sequence part of the second polynucleotide primer/probe, thereby forming a second replicated nucleic acid molecule;
- adding a third generic primer molecule with the same sequence the first generic primer sequence, and a fourth generic primer molecule with the same sequence as the second generic primer sequence;
- amplifying said replicated nucleic acid molecules forming amplimers;
- detecting said amplimers.
In one embodiment VOCMA3 comprises the following steps:
After a nucleic acid extraction, performed according to conventional and suitable methods, -primer/probes specific for the target nucleic acid, covalently bound to each other via their 5 'ends (a "nanodevice" as described under "Nanodevice"), are added to the extract, and are allowed to bind target nucleic acid (the primer/probe arms of the nanodevice may or may not be mismatch-tolerant as described under "Mismatch-tolerant specific primer/probes" and the nanodevice may or may not be bound to a bead or a solid phase before binding to the target nucleic acid);
- In case the nanodevice is not initially bound to a bead or a solid phase, the nanodevice may be bound to beads or a solid phase via an affinity label, such as the biotin/streptavidin system, in order to force the nanodevice with its bound target nucleic acid to be bound to beads or solid phase;
- At this stage, a collection step may be performed.
- A first amplification reaction, optimized for the specific primers, is subsequently conducted;
- Subsequently, generic primers which bind to sequences in the specific primer/probes are added to the reaction products;
- A second amplification reaction in solution, optimized for the generic primers, is
subsequently conducted;
- In case beads are present, the reaction solution is then separated from the beads;
- The reaction solution, containing amplimers in case the target nucleic acid was present in the nucleic acid extract, is then incubated with a nucleic acid-binding detection probe, which may or may not be mismatch-tolerant. This probe is covalently bound to, or may via an affinity link be non-covalently bound to, a separately addressable (e.g. a colour-coded) bead, a fluorophore, or to a solid phase;
- The presence of nucleic acid is detected.
In one embodiment of VOCMA3/HC, the target recognition and amplification is almost identical to that of VOCMA2. However, the first and second primers (both included in the nanodevice) may both function as probes for capture and enrichment of target nucleic acid from a nucleic acid extract. A first target-specific primer/probe (to which a first generic nucleotide stretch is covalently bound) binds a target nucleic acid, in a variation-tolerant way, i.e. it allows mismatches between the primer/probe and the target nucleic acid. A first strand copy of the bound target nucleic acid is then initiated using a DNA- or RNA-dependent DNA polymerase from the first primer/probe. In the subsequent copying round, the second target specific primer binds to the newly copied target nucleic acid in a variation-tolerant way. A second strand copy of the copied target nucleic acid is then initiated from the second primer using a DNA-dependent DNA polymerase. The second strand copy will end with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer/probe. At this stage, the first strand copy and its antisense second strand copy created by the elongation of the covalently coupled bound specific primer/probes may optionally be separated from the each other, by enzymatic, chemical or physical means. First and second, not covalently linked, target-specific prime/probes may then be added. An optional number of amplification rounds involving the first target-specific primer/probe and the second target- specific primer, covalently linked or not, are then performed. The amplification conditions are optimized for the target-specific primer/probe(s) and primer(s), and do not allow
amplification from the first and second generic primers. In the second amplification phase, the generic amplification, the amplification conditions are shifted towards those of the first and second generic primer by e.g. decreasing the annealing temperature. These conditions are more permissive than the previous amplification conditions allowing the generic primers, which may be shorter than the specific primer/probe arms in the nanodevice (to which generic complementary nucleotide stretch are covalently bound), to anneal and amplify. In this generic amplification, a second strand copy binds a first generic primer which binds to the copy of the generic stretch. A third strand copy, of sense orientation relative to the original target nucleic acid, is then initiated from the generic primer using a DNA-dependent DNA polymerase. This third strand copy will end with a copy of a second generic nucleotide stretch covalently bound to the second target-specific primer. The third strand copy then binds a second generic primer which binds to the copy of the second generic stretch. A fourth strand copy, of antisense orientation relative to the original target nucleic acid, is then initiated from the generic primer using a DNA-dependent DNA polymerase. The fourth strand copy will end
with a copy of the first generic nucleotide stretch covalently bound to the first target-specific primer.
In subsequent amplification steps, all copying is initiated from the first and second generic sequences and their complementary first and second generic primers.
One or more of said primer/probes may contain a dlnosine or other UBA.
The collection step may or may not comprise a washing step. Beads may for example be collected through magnetism, filtration, gravity or centrifugation. Nanodevices bound to a solid phase may be washed directly on the solid phase. At this stage, target-specific solution and/or generic solution primers may then optionally be added to the collected beads or solid phase bearing the nanodevice molecules. Optionally, several steps from the collection step and up to the second amplication step may be performed without interruption and opening of the reaction vessel.
Subsequent to the first amplification step, the reactants or products bound by beads or solid phase may or may not be separated from reactants or products in solution thereafter. The separation may or may not be made specific for nanodevices which have undergone a complete amplification reaction with the help of a specific separation reaction such as a restriction enzyme which requires a double-stranded substrate, or a uracil-N-glycosylase enzyme which cleaves at a Uracil added to the nanodevice to facilitate specific separation of it and its amplimers from a bead or solid phase.
The addition of generic primers may also be performed in the step of adding target-specific solution primers or in the first amplification step.
The detection may for example be performed by adding a fluorescence indicator (in case the final amplimer is not fluorescently labelled), such as phycoerythrin bound to streptavidin, incubating the mixture, followed by an optional washing step, and then running the reaction mixture in a flow fluorescence meter, a solid phase fluorescence scanner or other fluorescence measuring device.
According to another aspect, there are provided target-specific and variation-tolerant primer/probes, primers and probes (here collectively called target-specific VOCMA binders, see below) to be used in the method according to the present invention.
Variation-tolerant target- specific primer/probes and primers:
An oligonucleotide or other sequence-specific nucleic acid-binding molecule ("probe"), suitably designed according to a mathematical formula or a computer program which estimates the binding contribution from both local and more distant binding within in the probe in its interaction with a target molecule (Fig. 24 and 25). The target molecule ("target") which the probe can bind to can be an oligonucleotide, a longer nucleic acid or another sequence-specific nucleic acid-binding molecule. The mathematical formula or the computer program adds the binding contributions of perfectly matching (e.g. A*T or G*C, where A means an Adenine, T a Thymine, G a Guanine and C a Cytosine base, respectively) pairs as well as contributions from UBAs or base analogues (e.g. inosine, nitroindole or a peptide nucleic acid analogue). "Local binding" here means the match of a single base or base analogue in the probe to a single base or base analogue in the target and the match between the neighbouring bases or base analogues, upstream and downstream of the single base (a triplet). The binding contribution from interactions occurring at bases or base analogues within the same probe or target molecule situated more than one base or base analogue away from the single base (the "distal binding") comprises both continuous stretches of perfectly matching bases longer than three bases (a "matching run"), and of cooperative binding between several matching runs. The mathematical formula or computer program can approximately predict the likelihood that a certain probe-target combination will bind under given conditions (probe and target concentrations, buffer and temperature). A primer or probe constructed according to the above-mentioned principles, to withstand target variation, is referred to as "mismatch-tolerant".
Said specific primer/probes suitably has a length of at least 30 nucleotides or nucleotide analogues and comprise at least one nucleation region of 4 to 9 nucleotides which is essentially free of variation and optionally one or several regions wherein inosine residues or other UBAs have been inserted at positions of expected target variation, providing mismatch (i.e. variation) tolerance. A preferred pattern of UBA insertion is every third nucleotide, to encompass synonymous mutations in the target nucleic acid. A variation-tolerant
primer/probe sequence contains at least 17 matching nucleotides, contiguous or divided into a
maximum of four non-contiguous stretches without mismatching neighbours, where a "universal" base analogue (UBA) in the probe counts as a matching nucleotide.
Target-specific primer/probes:
This is a molecule which can function for the dual purpose of binding and amplifying target nucleic acid. This molecule is designed to provide a high binding affinity to enhance capture of target nucleic acid from a relatively large volume of nucleic acid extract which may contain many other non-target nucleic acids. It may either be bound to a carrier (microparticle or solid phase) and/or be free in solution. The target-specific primer/probe may or may not possess variation (mismatch) tolerance as defined below.
Target-specific primers:
This is a molecule which primarily functions as a primer in the amplification of target nucleic acid. It must not have the high affinity of the primer/probes mentioned above. It is used in solution. The target-specific primers may or may not possess variation (mismatch) tolerance as defined below.
Generic primers:
These are short primer molecules used for the final generic amplification of all amplimers generated by the target-specific primer/probes and primers in the first round of amplification.
Detection probe:
This is a signal-generating molecule which binds to the amplimers from the target-specific and -generic amplification rounds. It may or may not be bound to a carrier (microparticle or solid phase) or be free in solution. It may or may not possess variation (mismatch) tolerance as defined below.
Mismatch-tolerant nucleic acid-binding primer/probes:
This is a molecule which is extendable by a nucleic acid polymerase enzyme which in addition can bind specifically to a target nucleic acid in a mismatch-tolerant way. Mismatch- tolerance is the ability to bind to target nucleic acid in spite of one or several mismatching nucleotides. Mismatch tolerance is achieved through i) a probe length exceeding 30 nucleotides, and/or ii) a probe sequence which contains at least 17 matching nucleotides, contiguous or divided into a maximum of four non-contiguous stretches without mismatching
neighbours, where a "universal" base analogue (UBA) in the probe counts as a matching nucleotide. A UBA can be the deoxyinosine UBA. An efficient pattern of such UBAs is at every third nucleotide. A stretch of three nucleotides which ends with a UBA is subsequently called the "synonymous mutation configuration". The target binding properties of the primer/probe can be approximately predicted using the "NucZip" algorithm. It combines prediction of binding at or next to a matching nucleotide (the "local" binding, embodied in triplet sequences) with binding arising from interaction of several contiguous or noncontiguous binding triplets ("distal" binding). The mismatch-tolerance prediction algorithm is detailed in the "Examples".
Nanodevice:
This is a molecule built from primer/probes which are bound to each other through their 5 '- ends. The primer/probes may or may not be mismatch-tolerant as defined under "Mismatch- tolerant nucleic acid-binding primer/probes". The connection between the primer/probes may or may not contain a cleavable bridge, also termed linker. The cleavage may or may not be cleavable only if the nanodevice has participated in an amplification reaction. This property may be achieved if an originally single-stranded nanodevice has become double-stranded after the first rounds of amplification. The nanodevice may or may not be bound to bead or solid phase.
According to a further aspect, there is provided a kit that comprises combinations of said specific primer/probes, and general primers, bound or not to beads or other solid support. The kit may also comprise probe-binding tag sequences, hybridization, wash and amplification solutions, or other aid substances or solutions
According to yet another aspect, there is provided a method, optionally implemented in a computer program, which identifies what stretches in sequences of interest to the user, that can be used as the basis for producing mismatch-tolerant primer/probes and probes. Target- specific VOCMA binders are designed according to the NucZip, or a similar algorithm which estimates both local and distal contributions to binding between binder and target. The algorithm estimates optimal binder designs for capture, amplification and detection of target nucleic acid. The three binding situations require somewhat separate properties. Capture requires high affinity under moderate hybridization stringency. Target-specific amplification requires a moderate overall affinity, but a high affinity at the 3 'end, under amplification
conditions. Detection requires a high affinity under low hybridization stringency. The algorithm suggests suitable target-specific VOCMA binders of the three types from a variable target nucleotide sequence for which the variation is known or can be predicted.
One embodiment of this method is a method for predicting mismatch-tolerance of a probe nucleic acid molecule, optionally comprising at least one UBA, when hybridising to a target nucleic acid molecule, comprising the steps
a) aligning the probe and target sequences;
b) localizing possible nucleation sites by assigning a nucleation score for each stretch of nucleotides in the probe, said nucleation score equalling the number of continuous base pairs perfectly matching between the probe and target sequence;
c) assigning a first zip score to the sequence downstream from a first nucleation site, said first zip score equalling the number of consecutive matching 3-15-mers;
d) assigning a second zip score to the sequence downstream from the first nucleation site, said second zip score equalling the number of consecutive perfectly matching 3-15- mers;
e) repeating steps c) and d) for the sequence upstream from the first nucleation site;
f) assigning a total zip score to the nucleation site, said total zip score being a weighted mean of said first and second zip scores for said upstream and downstream sequences; g) assigning a combined nucleation and zip score to the nucleation site;
h) optionally repeating steps c)-f) for other nucleation sites in the probe sequence.
One embodiment of this method is the NucZip procedure.
The NucZip procedure starts by identifying potential nucleation sites. From these, the zipping of the two hybridizing strands is modelled. The NucZip model scores overlapping matching tri- to pentadekamers, and the effect of inosines. The algorithm extends nearest neighbour theory to longer hybridizing stretches, and includes the effects of UBAs in the long oligonucleotides. The basis of NucZip is a scoring system for matching stretches. The program first tests for possible nucleation sites. It selects the two highest scoring ones for further evaluation. From each suggested nucleation site, zipping is performed up- and downstream. The zipping algorithm evaluates matching stretches, from tri- to pentadekamers. It scores both stretches uninterrupted by UBAs and mismatches (Ipoints), and stretches interrupted by mismatches, where UBAs are counted as matches (Mpoints). The UBA
contribution (Mpoints-Ipoints) is counted less than a proper match. The NucZip procedure is used for prediction of optimal target-specific primerand primer/probe designs. Different sets of parameters are used for prediction of binding under amplification buffer and 3M TMAC conditions.
According to a still further aspect, there is provided a method of producing primer/probes in which the NucZip algorithm, or computer program comprising said algorithm, is used to identify stretches in sequences of interest to the user that can be used as the basis for constructing mismatch-tolerant primer/probes and probes.
Further advantages and applications of the present invention will be apparent for a skilled person when reading the detailed description.
Brief description of the drawings
Fig. 1 shows the general principle of the multiplex detection method according to the present invention (VOCMAl). In this embodiment, a first primer/probe is bound to a colour-coded or otherwise separately measurable bead.
Explanation of symbols:
1. Luminex bead
2. and 3. is bead-bound primer/probe, where 2. is a generic sequence and 3. is a target specific sequence.
4. A biotinylated target specific primer in solution
5. A generic primer in solution.
6. Target
7. x cycles of amplification
8. Addition of Phycoerythrin-Streptavidin and readout in Luminex
Fig. 2 shows the general principle of an alternative embodiment of the detection method described in Fig. 1. In this embodiment (VOCMA2), the first primer/probe is bound to a magnetic bead. Magnetic bead are fishing at high bead concentration. Most targets are caught. First there is an on bead amplification, followed by amplification in solution with the general primers. Detection with Luminex beads after addition of Phycoerythrin-Streptavidin.
Explanation of symbols:
1. Magnetic bead
2. and 3. is a bead-bound primer/probe, where 2. is a generic sequence and 3. is a target specific sequence.
4. target
5. and 6. A biotinylated primer/probe in solution where 5. is a target specific sequence, and 6. is a generic sequence with a biotin in 5'end..
7. Generic primer in solution
8. Biotinylated generic primer in solution
9. Luminex bead
10. Target specific probe
11. PE is Phycoerythrin-Streptavidin
Fig. 3 shows the double-membered first primer/probe of the capturing step of yet an alternative embodiment (VOCMA3/HC) of the detection method described in Fig. 1. The VOCMA3/HC has two 3 'ends.
Explanation of symbols:
1. Minor arm, ~30 nt , ending with dlnosine
2. Major arm, -50 nt, ending with dlnosine
3. Target for left generic primer
4. Target for right generic primer.
5. Cleavage site
6. Cleavage site
7. Biotin for binding to magnetic bead
8. Target
Figs. 4A-C show the general principle of an embodiment of the VOCMA3/HC detection method.
Fig. 4A
Capture of target strand by major primer/probe occurs free or bead-bound to magnetic beads.
9. Magnetic bead with Avidin
10. First strand synthesis by RT-Pol or Pol
11. Bead-bound
12. Second strand synthesis by Klenow polymerase
Fig. 4B
Selective liberation of newly synthesized hermit crab copies by restriction enzyme cleavage or UNG digestion.
13. Specific oligo to create a restriction site
14. Restriction site cleavage
15. Removal of beads with magnet
Fig. 4C
S YBRGreen PCR with generic primers
16. Biotinylated generic primers
17. Taq polymerase
Fig. 5 shows the readout phase in the VOCMA3/HC method described in Figs. 4A-C general principle of an embodiment of the multiplex detection method according to the present invention.
18. Luminex bead with target specific probe
19. Biotinylated amplimer
20. Free universal biotinylated primers
21. Addidion of Phycoerythrin-Streptavidin and readout in Luminex
Fig. 6 shows results of a VOCMA1 experiment when read in a Luminex flow particle meter (see MFI in table 1 , and primer and probe in table 7).
Fig. 7 shows results of a VOCMA 2 experiment when read in a Luminex flow particle meter(see MFI in table 2, and primer and probe in table 3).
Fig. 8 shows results of a VOCMA2 experiment with synthetic target of Noro virus genotype 2
7
in the titer of 10 -10 copies per sample (see MFI in table 4, and primer and probe in table 5) Fig. 9 shows results of a 22-plex VOCMA2 experiment utilizing one synthetic target in the
7 1
titer of 10 -10 copies per sample (see MFI in table 6, and primer and probe in table 7)
Fig. 10 shows alignment of 7333 genomes of Influenza A segment 7 in ConSort©; most subtypes are represented. The variation was mapped, searching for a suitable region for a 70- mer probe, by using BLASTn, and ConSort© (unpubl., Blomberg J.) further used in the experiments described in Figs. 11-15, 17, 19-22. Grey bars represent conserved nucleotides (nts); Black bars represent variation of base composition; y-axes represent frequency of variation.
Figs. 11A-C show hybridization of Influenza A probe and several Influenza A probes with increasing amount of dlnosines, to synthetic biotinylated complementary ssDNA with increasing amount of mismatching nucleotides. The InflA probe sequence was designed from a region in the matrix 2 gene in segment 7 of subtype H2N3 of Influenza A (sequence in table
8). The complementary target of InflA probe is named InflA target. The number of dinosines is indicated in the name of the probe; as in Ino3 probe containing three dinosines. Number of point mutations in target molecules is indicated in their names; like the 3pm target containing three point mutations compared to the InflA target. Hybridization was performed at 45 °C (Fig. 11 A and table 9A) and at 55 °C (Fig.1 IB and table 9B). Probe and target sequences are described in table 10A and 10B. The upper panel of Fig. 11C graphically illustrates the distribution of dinosines (grey boxes) in the probes (grey line), and the lower panel illustrates the mismatching bases (light grey boxes) in the target molecules (black line), used in Figs. HA and 11B.
Figs. 12A-C show the length of nucleation region necessary to induce hybridization, and the the rescuing effect of dlnosine-containing probes hybridizing to targets containing
synonymous mutations. The biotinylated targets had different percentage of mismatch, 26 %, 33 %, or 74 % mismatch, and one or two perfect matching regions of different length, 5-22 nt, compared to the InflA probe. Hybridization was performed at 45 °C (Fig. 12A) and at 55 °C (Fig.l2B). The sequence of the set of probes, with a region of different length containing dinosines in every third positioned, and the set of targets are displayed in Table 12A and 12B. The upper panel of Fig. 12C graphically illustrates the distribution of dinosines (grey boxes) in the probes (grey lines) and the lower panel illustrates the mismatching bases (light grey boxes) in the target molecules (black lines), used in Fig. 12A and 12B.
Figs. 13A-D show analysis of specificity of hybridization comparing a Norovirus- with an Infl Avirus-probe, with or without 18 dinosines, hybridizing to targets containing different amount and distribution of both Norovirus and InflA virus-sequence. The sequences of the panel of probes and targets are displayed in Table 16A and B. The names of the targets are based on matching and mismatching nt against a Norovirus probe, the Inol 8 probe, and the InflA probe: (Norovirus matching nt in 5' and 3') mismatching nt compared to
Norovirus probe_Inol 8 probe_InflA probe (InflA matching nt in 5' and 3'). In Fig. 13C the graphic illustration shows the three probes; Norovirus (white line), the InflA probe (grey line), and the Inol 8 probe (grey line and dark grey boxes as dlnosine nt). The three panels of the thirteen target molecules (black lines) is gradually going from the top with a Noro target sequence complementary to the Noro probe, towards the InflA target, matching the InflA probe in the bottom of the panel. The upper left target panel demonstrates the amount of mismatches (light grey boxes) in each target compared to the Noro-probe, and the amount of MFI the Noro-probe and resp. target (white bars) accomplish during hybridization at 45°C.
The middle left target panel demonstrates the pattern of mismatches (light grey boxes) in the target compared to the In 18 probe, and the signal of MFI received during hybridization of inol8-probe and resp. target (dark grey bars, 45°C). The lower left target panel shows the thirteen targets and their mismatching nt (light grey boxes) compared to the InflA probe, and the MFI received during hybridization of InflA and resp. target (light grey bars, 45°C). Fig. 13D demonstrates the number of nt in the position outside the pattern of 18sm dlnosines in each target that are of either Noro or InflA origin, compared with the MFI of the Inol 8 probe. White triangles represent the number of nt, outside the position of dlnosines, in resp target matching the Noro sequence. Grey squares represent the number of nt, outside the position of dlnosines, in resp target matching the InflA sequence. The MFI of each target hybridizing to the Inol 8 probe in Fig. 13D is from experiment displayed with the Inol 8 probe in Fig. 13 A. Fig. 14 describes the predicted AG versus % MFI for all 70-mer probes and 70-mer targets from three panels presented in the table 10A and B, table 12A and B, and table 16A and B , at (45°C). The probes hybridizing to 70 nt long targets are visualized as black boxes. Visual OMP (DNA Software) was used to calculate the predicted AG for the interaction between probes and targets in 3M TMAC buffer conditions and hybridization temperature of 45°C. The % MFI is the MFI signal of a probe with an intended target divided with the
hybridization of same probe against its perfectly matching target in the same hybridization temperature (e.g. InflA probe against InflA target). The value of % MFI for each
hybridization is from Fig. 11 A, 12A and 13A (45°C).
Fig. 15 The % MFI (45°C) of all dlnosine-free combinations of probe and target, i.e. InflA probe-target and Noro probe-target, (Figs. 11 A- 13 A) versus the score for perfectly matching overlapping trimers is shown in the right panel. The small inserted graph in the left panel, demonstrates the Overlapping Trimer Score correlation with the predicted AG (Visual OMP), left panel, with an R2=0.906.
Fig. 16 demonstrates schematically the overlapping tri-, quadra-, penta- and hexamer scores. Figs. 17A-H demonstrate the overlapping tri (A), quadra (B), penta- (C) and hexamer scores (D) versus % MFI at 45 °C, while Fig. 17E-H demonstrates the overlapping tri (E), quadra (F), penta- (G) and hexamer scores (H) versus % MFI at 55 °C.
Fig. 18 panels a-d. Each neighbouring trimer is given a score according
Pyramidfactor*(n(n-l)y2), where n is the number of neighbouring trimers and the pyramid factor is decreasing the impact for each level in Fig. 18 panel b. Each region of perfect match is weighted against the neighbouring region of perfect match and given a score, cooperativity
factor 1, Fig. 18 panel c. The score of the neighbouring regions are then weighted against each other by using the cooperativity factor 2, Fig.18 panel d.
Fig. 19 shows the TriHexa scoring system versus the % MFI for the same target and probe combinations that were demonstrated in Fig. 14 with predicted AG from Visual OMP vs % MFI. The TriHexa system takes all described features of scoring described in Fig. 16 and 18 into account.
Fig. 20 shows the result of the NucZip scoring system versus the % MFI for the same target and probe combinations that were demonstrated in Fig. 14 with predicted AG from Visual OMP vs % MFI. The NucZip system is a further development of the TriHexa system, which adds the nucleation and zipping parts to the hybridization prediction.
Fig. 21 Hybridization of the InflA, the Ino21 and the wobbN_21 probes to the InflA target, the 21pm target (21 point mutations) and the 21gm target, which has seven groups of three mismatches (Fig. 11C, table 18A and B). Hybridization was performed at 45°C and 55°C (table 18A and B). Fig.l 1C describes the distribution of dlnosines (orange/light blue/green boxes) and N wobbles (light grey boxes) in the probes (grey bar), and the mismatching bases (magenta boxes) in the target molecules (black bar).
Fig. 22_The InflA, Inol8, 5-nitroindo_l 8, Nwobb_18 and Nwobb_24 probes were hybridized at 45 °C or 55 °C with one specific target containing 33% mismatch and one or two perfectly matching regions of different length (9-15 nt) compared with the InflA probe (table 12B). Fig. 12C, and table 12A shows the distribution of dlnosines, 5-nitroindole and N-wobbles in the probes used in Fig. 22.
Fig. 23 The zipping component of NucZip When a 6-9 base pair sequence fulfilling the nucleation criteria has been detected, hybridization up- and downstream of the nucleating site is attempted (zipping). The figure shows the downstream zipping process, with successive accumulation of score within a matching segment arising from the trimers, tetramers, etc. up to pentadecamers (each scoring equally) which fit into it. In this way, a longer matching segment gets more than a linear increase in score relative to a shorter one. Zipping extends from the potential nucleation site, terminating with a mismatch or the end of one of the strands, dlnosines are counted as intermediate between a match and a mismatch. If several consecutive matching segments are encountered, their scores are added.
Fig. 24 shows a flow chart over the design process of making a VOCMA panel.
Fig. 25 shows a flowchart over the NucZip algorithm.
Brief description of the tables
Table 1 shows results of a VOCMAl experiment when read in a Luminex flow particle meter (see also in Fig. 6).
Table 2 shows results of a VOCMA 2 experiment when read in a Luminex flow particle meter (see also in Fig. 7).
Table 3 shows oligonucleotides used in the VOCMAl and VOCMA2 experiments (Table 1 and 2, Figs. 6 and 7).
Table 4 shows results of a VOCMA2 experiment with synthetic target of Norovirus genotype
7 3
2 in the titer of 10 -10 copies per sample (see also Fig 8)
Table 5 shows oligonucleotides used in the VOCMA2 Norovirus genotype 2 experiment (Fig. 8).
Table 6 shows results of a 22-plex VOCMA2 experiment utilizing one synthetic target in the
7 1
titer of 10 -10 copies per sample (see also Fig. 9)
Table 7 shows oligonucleotides utilized in the 22-plex VOCMA2 experiments (Fig. 9).
Table 8 shows the variation in a rather conserved region of the segment 7, the matrix gene, between the different sub types H3N2, H5N1 and H1N1 of Influenza A virus. The grey nucleotides display the differences between the sequences. The H5N1 virus subtype differed in five nt positions and the H1N1 virus in three additional nt positions, compared with the H3N2 virus. The 9 pm (point mutation) sequence matches 64 different subtypes to 100% (HxNy*). Thus, if mismatches in the indicated nine positions would be tolerated by a detecting probe it will detect at least 67 Influenza A subtypes. Result from analysing 7333 genomes of Influenza A by using BLASTn, and ConSort© (unpubl., Blomberg J.)
Table 9A shows results of hybridization at 45 °C (see also Fig. 11 A) and Table 9B at 55 °C (Fig.l lB).
Table 10A shows the sequence of the 70-mer probes (5'-3') with increasing amount of dlnosines (I) and Table 10B shows the sequence of the 70-mer targets (5'-3') utilized in Figs. HA and 1 IB, and Fig. 21.
Table 11A shows results of hybridization, at 45 °C (Fig. 12A) and Table 11B at 55 °C (Fig.l2B)
Table 12 A shows the sequence of the 70-mer probes (5 '-3') and Table 12B shows the sequence of the 70-mer targets (5 '-3') utilized in Figs. 12A and B, and in Fig. 22.
Table 13 is a summary of the result in Fig 12A and B and Table 11 A and B, showing the nt length of the long perfectly matching regions, in either the 5' and 3' end or both ends, of the target needed to get hybridization.
Table 14 compares the number and length of matching regions and the ability to hybridize (% MFI) for the three targets having 14 mismatches, the 14pm, 26%9F and 33%15F target; and the three targets having 16 mismatches, the 16pm, 26%5F, and 33%15F target, when they bind to the InflA probe, from the data of Fig 12A and B and Table 1 1A and B.
Table 15A shows results of hybridization at 45 °C (Fig. 13 A) and Table 15B at 55 °C (Fig.l3B)
Table 16A show the sequences of the three 70-mer probes (5'-3') and Table 16B show the thirteen 70-mer targets (5'-3') utilized in Fig. 13A-D.
Table 17 demonstrates each hybridization of Inol 8 probe and the different targets and the amount of mismatches outside the position of the dlnosines of the Inol8 probe, based on results from Fig 13A-B and table 15A and B.
Table 18A shows results of hybridization of the probes InflA, Ino21 and wobbN_21 (table 10A) and the targets InflA, 21pm, and 21 gm (table 10B) at 45 °C (Fig. 21) and Table 18B at 55 °C (Fig.21).
Table 19A shows results of hybridization, at 45 °C (Fig. 22) and Table 19B at 55 °C
(Fig.22).
Table 20 shows the probability of getting matching region/s of a certain length in the outer segments of the wobbN_18 probe.The sequences of the wobbN_18 probe are displayed with x as the nt matching the target sequence and N as the wobbling nt A, C, G or T. The probability of increasing the length of matching flanking 5' and 3' regions of the probe is calculated for a few examples.
Definitions
All terms used in the present specification shall be considered as having the meaning usually given to them by a person skilled in the art. For reasons of clarity, some terms are however further defined below.
A polynucleotide primer/probe is a DNA or RNA polynucleotide that can be used either as a primer or a probe, or both.
Mismatch-tolerant hybridization is the ability of a polynucleotide to hybridize through standard base pairing to a not fully complementary target polynucleotide. The terms mismatch-tolerant and variation-tolerant shall be considered equivalent, unless specifically stated otherwise.
An Universal Base Analogue, UBA, is a nucleotide capable of being incorporated in a polynucleotide and base pairing with all four naturally occurring nucleotides, i.e. A, C, T and G. Examples of UBAs are dlnosine, 3-nitropyrrole and 5-Nitroindole bound to deoxyrobose deoxyribose and peptide nucleotide analogue.
A degenerated position in a polynucleotide is a position which has several alternative nucleotides, generally from two to four. The nucleotides may be chosen from A, C, G, T and UBAs. The terms "degenerated position" and "degeneration" shall be considered as equivalent, unless otherwise indicated.
A perfect match between to nucleic acid molecules is when a nucleotide A in the first nucleic acid molecule pairs with a nucleotide T in the second nucleic acid molecule, or a C in the first nucleic acid molecule pairs with a nucleotide G in the second nucleic acid molecule, in a certain position of the nucleic acid molecules. As applied to a stretch or segment of nucleotides, i.e. a nucleotide sequence, a perfect match between two sequences means that all the nucleotide positions in that sequence are perfect matches.
A match between two nucleic acid molecules includes perfect matches, but also the situation when a nucleotide A, T, C, G or an UBA in the first nucleic acid molecule pairs with an UBA in the second nucleic acid molecule, in a certain position of the nucleic acid molecules. As applied to a stretch of nucleotides, i.e. a nucleotide sequence, a match between two sequences means that all the nucleotide positions in that sequence are matches, whereof some may be perfect matches.
A mismatch between to nucleic acid molecules, in a certain position, is the situation when there is no match. As applied to a stretch of nucleotides, i.e. a nucleotide sequence, a
mismatch between two sequences means that not all the nucleotide positions in that sequence are matches.
An analytic tag is a moiety that can be covalently or non-covalently bound to a molecule and is analytically detectable. One example is phycoerythrin.
Detailed description of the invention
In the development of the multiplex detection method according to the present invention, many of the requirements mentioned have been met. The key steps of said method are outlined and discussed below. These steps apply to all VOCMA methods disclosed herein unless otherwise indicated or clearly contradictory.
Capture of target nucleic acid from an extract
The techniques must be able to work with large extraction volumes, and both high and low target nucleic acid concentrations. The capture primer/probe should be mismatch-tolerant, yet have a high affinity for the target. The principles of achieving a mismatch-tolerant capture are described below. A further requirement is that the capture should be fast. It must then occur in solution, because hybridization of free probe is about 1000 fold faster than hybridization to bead-bound probe. Thus, a two-step capture scheme, first hybridizing to free probe for 30 minutes and then "mopping up" all or most of the probes using magnetic beads (23) with shaking for 30 minutes, is more efficient than a single step with bead-bound probe. The beads should be small enough to easily stay in suspension, and move quickly in the solution, but be large enough to be easily collected by commercially available magnetic separators.
In the following the MyOne® beads from Dynal/Invitrogen have been used as examples. Hybridization conditions may typically be at least 1 M salt and 40°C. Typical concentrations of probe are 100 million molecules/mL, typical magnetic bead concentrations are 1-10 million beads/mL. If a biotinylated probe is "mopped up" by streptavidin-coated magnetic beads, the high biotin-streptavidin dissociation constant (10"15 M) makes the binding especially efficient. These conditions combine economy with efficiency. Magnetic beads and magnetic separation are suitable means for concentrating the probes, of which some may have found target molecules when concentrated. However, a solid phase which binds the probes (with or without attached target nucleic acid), or other kinds of beads than magnetic beads, may also be used in alternative embodiments of the invention.
MyOne streptavidin beads from Dynal/Invitrogen come at 1010 beads/mL. They are 1 micrometer in diameter. If 1 million beads/mL are used, 1 mL of beads can be used for 10000 experiments of 1 mL. 10 million beads provide a thousandful binding excess if 100 million probe molecules/mL are used. MyOne streptavidin beads can bind 100 pmol (2 μg; 6* 1013 molecules) biotinylated 50nt ssDNA per mg bead. 1 mg of beads=109 beads. One bead can bind around 100 000 DNA molecules.
Enrichment of target nucleic acid relative to other nucleic acids
Capture of target nucleic acid gives three advantages. Firstly, it increases the concentration of target nucleic acid. Secondly, it removes irrelevant nucleic acid. Thus, sample extracts with a high nucleic acid concentration can still be analyzed. Thirdly, inhibitors of ensuing enzymatic reactions will be removed.
The first rounds of copying from target and primer/probes
This can be RNA-dependent DNA-copying from + and - stranded target RNA via reverse transcriptase, or DNA-dependent DNA copying of target via DNA polymerase. An enzyme mixture may provide both functions.
Selective release of hybridized and copied target from capture beads or solid phase
A further desirable specificity enhancement can be achieved by schemes to selectively remove only successfully copied nucleic acid. The VOCMA3/HC scheme provides this option, through the use of a restriction enzyme which only cleaves double-stranded nucleic acid. When the beads are removed, only the successful copies are left, removing all primer/probes which did not find targets. The remaining solution is thus highly enriched of successfully copied target molecules.
Target-specific mismatch-tolerant primer/probes
Target variation is a major problem in molecular diagnostics. As mentioned above, RNA viruses are especially problematic in this regard. A thorough bioinformatic analysis of the distribution of variation across the target sequence is necessary, and computer-based support is normally required when performing said analysis. The present inventors have developed a computer program, ConSort (J Blomberg, unpublished), which is optimized for this purpose and greatly facilitates this work (Fig 24).
Typically, two mismatch-tolerant primer/probes per target sequence, one forward (number one) and one reverse (number two), are used. The primer/probes are typically 50-70 nt long, to provide a high affinity to the target, and to provide tolerance for mismatch between primer/probe and target sequence. The greatest conservation is placed at the 3 'end.
The following features enhance the mismatch tolerance of a primer/probe:
Under the conditions specified in example 6 (result), a primer/probe suitably has at least one stretch of nine perfectly matching nucleotides, or at least two stretches of five perfectly matching ones. Match to a UBA in the probe is here counted as a perfect match. Variable positions can be covered by UBAs in the primer/probe. However, UBA tend to decrease the affinity of the primer/probe for its target sequence. Therefore, UBAs cannot be used too generously. Above a threshold of UBA content of around 30% of the primer/probe, more UBA may make the primer/probe bind non-specifically and weakly. In order to reduce the UBA content somewhat, guanosine can be inserted at positions where guanosine is one of several naturally occurring bases. This is because guanosine can base-pair weakly but non- canonically with the bases adenosine, thymine and guanosine, i.e. it has properties akin to a UBA. Thus, the primer/probe design algorithm must balance the content of UBA, guanosine and variable positions against each other to obtain maximal mismatch tolerance, yet high affinity to target and high specificity.
All candidate primer/probe sequences should be analyzed for tendency to form unspecific hybrids, which may lead to primer-dimers which reduce the PCR efficiency in all three VOCMA schemes, and may cause false positivity in the VOCMAl scheme. The
concentration of solution second target-specific primer in VOCMAl should be 100 nM, and of the first target-specific primer/probes and the second target-specific primers in VOCMA2 should be around 1-10 nM, respectively. The first target-specfic primer/probe may also be entirely bound to bead or to solid phase in VOCMA2.
The function of the primer/probes is to start the copying of target in a specific way. The degree of amplification that they give is only 1-1000 times. However, in VOCMAl, the second target-specific primer participates in the amplification up to the detection step.
The generic primers
The generic, third and fourth, primers, also referred to as "help" primers, or to as "first and second generic primers", have artificial sequences which do not have homologous or heterologous complementarity to the other primers and probes. They are entirely in solution. In VOCMA1, only one generic primer, with same sense as the first target-specific
primer/probe, is used. In VOCMA2, two generic primers are typically used, one forward and one reverse, in a PCR amplification. However, the VOCMA2 concept can also be used with other types of amplification, which may be isothermal. The function of the generic primers is to finalise the amplification, i.e. to take the brunt of amplification. Amplification factors of
1(T - 10° are typically achieved with these primers.
At first sight, the generic primer pair could also amplify falsely. The very high amplification factors and their generic nature could be suspected to cause non-specificity. However, several factors counteract such non-specific amplification.
The first specificity enhancement occurs when the third generic primer binds to its target. This can only occur if a complementary sequence was synthesized by the successive action of the first target-specific primer/probes and second target-specific primer. Likewise, the fourth generic primer can only start amplifying when the third generic prime has made a copy. Thus, each target-specific primer/probe, target-specific primer and each generic primer must act in succession to start the second, most productive, phase of amplification, provided by the generic primer(s).
The amplification must somehow give a detection signal. A further specificity enhancement is caused by the placement of the label (either an affinity label, which indirectly can generate signal through binding of a signal molecule such as a fluorophore, or a signal molecule directly), which generates the readout signal, on the fourth generic primer (VOCMA2/3) Thus, only newly synthesized strands complementary to the target molecule will be detected.
Solid phase PCR, solution PCR, or both?
A central issue is to locally restrict target-specific primer/probes. This prevents unwanted primer/probe interaction. Ideally, only the primer/probes which are specific to a target sequence should be located close to each other. This can be achieved in several ways.
Standard PCR involves one target sequence and two primer sequences, all in solution. All possible interactions between primers can occur. If the concentration of each primer is high
(>200 nM), and/or there are many different primers, the likelihood of an unwanted interaction increases.
As mentioned above, padlock probes depend on the connection of two target-specific oligonucleotides as the ends of a linear sequence, which can be circularized after binding to target, and ligation. When one shank encounters a target sequence, the other shank also very easily will find the target sequence. However, padlock probes are in solution. Given a high enough padlock probe concentration (e.g. >10nM), also they may have spurious interactions with each other.
The very slow kinetics of solid phase PCR, e.g. Megaplex PCR (24) is a substantial drawback. However, when the surface-bound primers are locally restricted, the illegitimate primer interactions are rare. Although the initial hybridization-driven events are very specific, both padlock probe and Megaplex PCR detection schemes need a major amplification of the initial products to make them visible by various readout techniques. Typically, amplification factors of 10' to 10' are needed. Such a strong amplification is not trivial. False and negative positivity may occur.
In the present invention, the advantages of solid phase amplification, i.e. low primer/primer interactions, are combined with those of solution amplification, i.e. high efficiency. An initial local restriction of the target-specific primer/probes is achieved in several ways. In VOCMA1 and VOCMA2, the forward primer/probe may in one embodiment of said methods be bound to microparticles. This component thus cannot diffuse in solution and has a limited capacity to contribute to illegitimate interactions (leading to "primer dimers").
In the VOCMA3/HC system, the two target-specific primer/probes (forward and reverse) are covalently linked to each other via their 5' ends, thus spatially restricting the two extension reactions versus each other. The initial copying thus takes place in a soluble molecule. It is a "nanodevice" which seeks target molecules and tethers them to a magnetic bead which can be "fished out" from the extract. The two initial extension reactions can then take place free of contaminating nucleic acid. Thereafter, it is possible to increase specificity further in that only HC nanodevices which have found and appropriately extended target sequence are cleaved off from the magnetic beads or solid surface.
The low efficiency of PCR with microparticle-bound primers, and even lower of PCR with solid-phase-bound primers (our observations, and (22, 36, 37)) may according to the present invention be counteracted in two ways. In both VOCMAl and VOCMA2, the target sequence may first be captured by a target-specific bead or solid phase bound primer/probe. Then, the second, target-specific primer resides in solution. It is therefore free to find its specific target, which initially protrudes from the microparticle or solid phase. Second, a third generic primer in solution binds to the copy of a common sequence engineered into the first target-specific microparticle-bound primer/probe. This third generic primer can only amplify when two strands have been copied from the original target strand. This restriction enhances specificity. In VOCMA2, the first specific primer/probe may be present both on a bead/solid surface and free in solution. In this case, the more efficient free primer/probe can aid the bead/solid surface bound primer/prober in the further amplification.
In VOCMA2 and VOCMA3/HC, an additional fourth generic reverse primer in solution has been added. This additional fourth generic primer can only amplify when the longer second target-specific primer has been copied.
The major part of the amplification is thus done by the generic primers. In VOCMA2 and VOCMA3/HC, they are the only oligonucleotides which are present in high concentration. In VOCMAl, the second target-specific primers and third generic primers are the only ones present in the highest concentration.
Thus, the VOCMA and VOCMA/HC systems have been designed to combine molecular localization and mobility in an optimal way, and to increase mismatch tolerance while still preserving specificity.
VOCMAl vs VOCMAl vs VOCMA3/HC.
In VOCMAl, the reporting bead also carries the first target-specific primer/probe. Any mispriming involving both this primer/probe and the biotinylated second primer/probe will give a false signal. Such mispriming may arise because of the relatively high concentrations of free second target-specific primer. In practice, it turns out to be possible to multiplex up to tenfold (but it may be possible to go higher in multiplexity) with this system. The sensitivity so far settles at 104 - 105 molecules per reaction, which is enough for many purposes.
VOCMA1 seems suited for applications demanding medium multiplexity, moderate sensitivity and relative simplicity.
In VOCMA2, two major differences increase the sensitivity and specificity; Firstly, the use of two generic primers allows the concentration of free first target-specific primer/probes and second target-specific primer to be decreased by a factor of 200, to 1 nM. This greatly reduces the likelihood of mispriming. Second, the readout is through hybridization to a detection sequence bound to a specific Luminex bead or any signalling molecule or bead. This will selectively detect only specific amplimers. Mispriming, if it occurs, cannot give spurious signals. However, if primer dimer formation occurs, it might reduce the efficiency of the PCR and thus reduce its sensitivity.
In VOCMA3/HC, the situation is rather similar to that in VOCMA2. However, the HC construct only allows one round of specific copying. All other amplification is performed with the generic primers in solution. This may or may not be a problem which needs to be studied. The greatest advantages of VOCMA3/HC over VOCMA2 may be its more efficient capture of target in a large extract volume and the greater spatial isolation of the primer/probes from those of other primer/probes in a multiplex system.
Examples of the three detections schemes according to the present invention are outlined below.
VOCMA1
Scheme:
1. (crude) NA extract.
2. Addition of beads (optional) containing target-specific primer/probe, generic primer (same sense as target-specific primer/probe) and target-specific solution primers
3. Amplification
4. Washing in presence of antisense-blocking oligo complementary to the generic primer
5. Addition of Phycoerythrin-Streptavidin
6. Run Luminex
Likelihood of false priming:
-Soluble (sense) target-specific primers (e.g. 100 nM) must be tested for complementarity vs each other, vs generic primer, and vs bead-bound primer/probe.
-Bead-bound primers cannot prime on each other.
-Soluble primer-bound primer heterodimers will occur sometimes, and may give false positives.
Sensitivity
If beads are washed pre PCR only bound targets (maybe only 1/100 of all) will be amplified, thus the sensitivity may be smaller by several logs, compared to if no washing is done.
VOCMA2
Typical scheme:
I . Nucleic acid extraction. Addition of magnetic-bead-bound, or free, antisense target-specific primer/probe, either affinity labelled or not.
3. Hybridization to targets in extract
4. In case free affinity labelled primer/probes were used, ligand-bound magnetic beads are added.
5. In case beads were used, they are collected by magnet and washed
6. Addition of primers (target-specific second sense primers, or target-specific second sense primers and target-specific first antisense primer/probe, plus generic primers; sense and antisense), and master mix (RT-Taq mix may be used if RNA targets are included)
7. Single tube-nested PCR
8. Removal of magnetic beads
9. Transfer of aliquot to detection Luminex beads
10. Hybridization
I I . Collection of supernatant, washing
12. Addition of Phycoerythrin-streptavidin
13. Hybridization
14. Run in Luminex
The VOCMA3 or Hermit Crab (HO system
Scheme:
1. Nucleic acid extraction
2. Addition of HC nanodevice
3. Hybridization to targets in extract
4. Addition of streptavidin bound to magnetic beads
4. Collection of beads by magnet
5. Washing
6. Addition of master mix.
7. Extension reactions (RT+Taq)
8. Addition of UNG or restriction enzyme
9. Removal of beads, collection of supernatant
10. Add generic primers and Taq master mix
11. Standard PCR w SYBRgreen
12. Addition of detection Luminex beads
13. Hybridization
14. Collection supernatant, wash
15. Add Phyocerythrin-streptavidin
16. Hybridization
17. Run in Luminex
Overcoming target variation
A paramount problem for nucleic acid-based diagnostic techniques is to overcome target variation. Utilizing a 3M TMAC hybridization buffer, 70 nt DNA probes from the matrix- gene of H3N2 Influenza A, InflA, were analyzed in the aspect of hybridization tolerance to different amount and distribution of mismatch in target DNA, and with varying number and positions of deoxyribose-Inosine (dlnosine) in the probe. The probes were linked to xMAP polystyrene microspheres, hybridized against biotinylated complementary single-stranded (ss) DNA, and further analyzed in the Luminex® flow-cytometer. Redistribution of evenly dispersed point mutations (pms) to stretches of contiguous mutations demonstrated the importance of mismatch distribution and the necessity of a minimum length and number of perfectly matching "nucleation" regions. Under our conditions, the nucleation regions minimally comprise at least three regions of at least 6 nucleotides, two regions of 12-15 nt, or one region of 15-18 nt, dependent of hybridization temperature, in a 70-mer. Interestingly,
probes with dlnosines in the same position as the pm of the target, like in every third position (analogous to synonymous mutations), were able to rescue hybridization to targets to which a dlnosine-free probe failed to bind. Further, an nt mismatch in the target neighbouring the position of dlnosine in the probe is reducing the hybridization capacity more than a mismatch several nt apart from a dlnosine. Specificity of high dlnosine-containing probes was tested with a 18 dlnosines InflA probe against a Norovirus target which was gradually mutated towards a InflA target sequence. In conclusion, probe designs and conditions for mismatch- tolerant hybridization to variable target sequences, which still preserve hybridization specificity, were defined.
Studies on the mismatch tolerance with and without dlnosine, mismatch distribution, and specificity of long (50 nt and higher) probes in 3M TMAC have not been reported previously. In other buffer systems, containing SSC and 50% formamide, commonly used in microarray hybridizations, it has previously been suggested that not more that 15 contiguous nt complementary to non-targets should be present in a 50-mer probe used in a microarray assay, highlighting the specificity limits of long probes. Further, He at al expanded the study by comparing the hybridization properties of 50-mer and 70-mer probes in a microarray assay. They concluded that a 50-mer probe should not contain more than 16 nt, and a 70-mer not more than 20 nt, of contiguous stretches complementary to non-targets. The probe length of 70 nt in this study was chosen since this length is more tolerant to a larger number of mismatches than shorter probes, although it carries a somewhat higher risk of nonspecific hybridization.
To study the properties of mismatch-tolerant probe design the Influenza virus was chosen. Influenza virus is a negative ssRNA virus, whose RNA genome is highly variable due both to genetic shift, where whole segments of the genome can be exchanged, and genetic drift, caused by its error-prone RNA-dependent RNA polymerase. The latter mechanism causes the diagnostic problems which are the direct reason for of this paper. A 70-mer probe was designed against a conserved portion of the matrix segment 7 of Influenza A H3N2 by utilizing the programs BLASTn, ClustalX and ConSort® (unpublished, Blomberg J. et al), in alignments with over 7000 Influenza A sequences. The majority consensus of the chosen 70- mer region was used to construct probes and biotinylated target sequences with various patterns of mismatches for the tolerance studies.
To overcome the problem with variation in the target sequence and to avoid degenerated probes, which tend to give low signal strength due to a high number of mismatches, we investigated how the introduction of dlnosine in the nucleotide positions of variation in the probe affected the hybridization capacity. All four normally occurring DNA bases can bind to dlnosine. The general trend in decreasing stability is I:C > I:A > I:T ~ I:G > 1:1 when using 1 M NaCl, 10 mM sodium cacodylate and 0,5 M EDTA (pH 7). Inosine occurs naturally in tRNA, DNA repair and in RNA editing, dlnosine is one of many, more or less, generally hybridizing nucleotides. However, it is readily available and can be recognized as a G by polymerases, dlnosine can also be used to make broadly matching primers for PCR. Thus, inosine can act as a partially universal base under PCR buffer conditions (10 mM Tris, 50 mM potassium chloride, 1.5 mM magnesium chloride, 0.001% w/v gelatin, pH 8.3 at 25°C). As mentioned, 3M TMAC hybridization buffer increases Tm by increasing the binding strength of A:T base pairs, making it a platform suitable for further attempts to increase mismatch tolerance and Tm of probes towards variable targets.
The hybridization tests were performed both against targets which had an increasing number of random and ordered pms. Of special interest were targets with regions where every third base was mutated, to resemble the common phenomenon of synonymous mutations. Much of the variation in coding viral sequences is of this kind. The Influenza A H3N2 70-mer probe failed to hybridize both to a target containing 21 random and evenly distributed mismatches and to a target with 18 synonymous mutations. Here we demonstrate that introduction of dinosines in the probe, at the sites of mismatch, rescues hybridization even with high numbers of mismatches. Probes with different amounts of dinosines were tested. The more dinosines the probe contains the more the MFI is reduced, showing that dlnosine destabilizes the duplex. This destabilization can to a large extent be compensated by reducing the
hybridization temperature. Introduction of as many as 18 dinosines in every third position was able to rescue an otherwise abolished signal to the level of the signal that the normal probe received with a target without any mismatches.
Further, the distribution of mismatches greatly affected the hybridization, illustrating that hybridization predictions based on % of mismatches are imprecise. A minimum number of contiguous perfectly matching stretches are needed. These probably correspond to what has been termed "nucleation sites", which initiate hybridization. The study shows that a dlnosine free probe of 70 nt needs either i. at least three regions of at least 6, ii. two stretches of 12-15,
/■. or one stretch of 15-18 perfectly matching nucleotides to give a measurable hybridization, when the rest of the target contains 14 or more mutations. Probes with a high amount of dlnosines positioned at sites of variation needs shorter nucleation regions than dinosine free probes. As an example, a probe with 18 dlnosines needs only i. two regions of 9 nt (55°C), or one region of 9 perfectly matching nt (45°C), depending on hybridization temperature. Mismatch studies with utilizing a probe with many dlnosines, Inol8, shows that the dinosine containing probe is sensitive to mismatches positioned next to the dinosine, but is not affected to the same extent against mm several positions away from the dinosine. A 70-mer probe with 18 dinosines in every third position could tolerate as many as 4-5 dinosine neighbouring mismatches. Thus, utilization of a dinosine containing probe will be beneficial if the target contains many variations and especially if there are no long uninterrupted stretches suitable for nucleation. Hence, dinosine can be used in 3M TMAC to establish tolerance to high target variation, like in the genome of an RNA virus.
Examples
Example 1 - VOCMA1
Two bacteria-specific genes and one gene that cause antibiotic-resistance were chosen for demonstrating the VOCMA1 method. Each VOCMA1 reaction contains a mix of all three microsphere-bound primer/probes, a generic forward solution primer, a mix of the three biotinylated target-specific reverse solution primers and one of the specific synthetic targets of 140-200 nt, all described in table 3. All three bead-bound primer/probes comprise a 5' generic sequence of 30 nt complementary to the generic forward primer, and a target-specific 3' end of 50 nt. Target genes in the example of VOCMA1 demonstrated in Fig. 6 and table 1, were the GseA-gene of Staphyloccocus epidermidis, the ec^l-gene of Staphyloccocus aureus, and the ddl-gene from Enterococcus faecium (sequences in table 3).
Materials and methods
Synthetic targets and primer/probes
Synthetic targets were diluted to 200 pM with 0.1 χ Denhardt's solution (Sigma- Aldrich Sweden AB, Stockholm, Sweden). See table 3 .
Coupling of microspheres
Each synthetic forward target-specific primer/probe with a 5' C-12 amino modification (Biomers.net GMbH, Ulm, Germany) was covalently coupled to a specific set of 5.6 μιτι carboxylated polystyrene microspheres (Ramcon A/S, Birkerod, Denmark). 2.5 * 106 microspheres were pelleted at 13000 x g for 2 min and re-suspended in 25 μΐ 0.1 M 2- Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication. 2 μΐ of 100 μΜ amino-modified forward primer/probe oligonucleotide was added to the microsphere suspension followed by addition of 2.5 μΐ of 10 mg/ml l-Ethyl-3-3-dimethylaminopropyl carbodiimide (EDC) (Nordic Biolabs, Taby, Sweden), the coupling reaction was incubated at room temperature in the dark for 30 min. Addition of 2,5 μΐ of 10 mg/ml EDC and incubation was then repeated once. After the second incubation 1 ml 0.02 % Tween-20 was added to the reaction and vortexed. The microspheres were then pelleted by centrifugation at 13000 x g for 2 min and re-suspended in 1 ml of 0.1 % SDS by vortexing. Finally, the microspheres were pelleted and re-suspended to a final concentration of 50000 microspheres/μΐ by adding 50 μΐ Tris-EDTA (TE, pH 8.0) buffer. Coupled microspheres were stored in darkness at 8 °C.
Amplification
2.5 μΐ of the synthetic target was added to a 22.5 μΐ PCR mixture for a final concentration of l xPCR Gold Buffer (Applied Biosystems, Stockholm, Sweden), 800 μΜ dNTP Mix, 2 mM MgCl2, 1U AmpliTaq Gold® DNA Polymerase, 30 nM generic forward solution primer, 50 nM of each of the three reverse target-specific primers and 5000 of each coupled microsphere. Amplification was carried out on a MJ Research PTC- 100™ Peltier thermal cycler (Sds Scandinavian Diagnostic Services, Falkenberg, Sweden) as follows: 95 °C for 10 min;
followed by 4 cycles of 94 °C for 30 s, 52 °C for 2 min, 72 °C for 2 min; 40 cycles of 94 °C for 30 s, 56 °C for 45 s, 72 °C for 45 s; a last step with 72 °C for 7 min and the samples were kept at 4 °C until next step.
Detection in Luminex 200
Samples were taken to new PCR tube strips and centrifuged for 2 min on a table centrifuge to pellet the microspheres with target-specific detection probes. The supernatant was removed and the microspheres re-suspended by pipette mixing 50 μΐ 1 χ tetramethyl ammonium chloride (TMAC) hybridization buffer comprising 3 M TMAC, 0.1 % sarkosyl, 50 mM Tris- HC1 pH 8.0 and 6 mM EDTA pH 8.0 (Sigma-Aldrich Sweden AB, Stockholm, Sweden). Samples were then centrifuged on the table centrifuge for 2min and the supernatant removed
except approximately 5 μΐ. 45 μΐ 1 * TMAC with 330 nM of blocker oligonucleotide were added to re-suspend the microspheres by pipette mixing. Samples were denatured at 95 °C for 2 min followed by hybridization at 50°C for 30 min under shaking on a Thermostar (BMG Lab Technologies, Germany) microplate incubator. 2 μΐ (0.05 μg/ l) of Streptavidin-R- Phycoerytrin (Qiagen AB, Stockholm, Sweden) was added to the reaction and incubated for another 15 min. Detection in the Luminex 200 (Purchased through Ramcon A/S, Birkerod, Denmark) system was carried out at 50°C and the mean fluorescence intensity (MFI) was based on 100 measurements from each microsphere set and calculated by the STarStation software (Applied Cytometry System, Sheffield, UK).
Results
The supernatant of the samples in VOCMA1 showed specific amplicons of correct size and the mix of beads in each sample was further analysed in the xMAP/Luminex system. The Luminex flow cytometer identifies each bead and measures the fluorescence of the probe bound amplicons, resulting in a MFI signal for each bead type. As shown in Fig. 6 and table 1 , all three samples showed a significant specificity from the complementary bead-bound probe for the specific synthetic target. 100 beads of each probe specificity were analysed in every sample. Staphyloccocus epidermidis gseA gave a 15 fold MFI signal compared to the signal in the negative control, the Staphyloccocus aureus MecA gave 6 fold MFI compared to negative control, and Enterococcus faecium ddl gave 10 fold compared to negative control.
Conclusions
Three targets were detected in the VOCMA1 experiment. Specific signals were around 10 times higher than the signal in the negative control. Sensitivity was moderate, requiring 104- 105 target copies for a clear signal.
Example 2 - VOCMA2
Four bacteria-specific genes and one gene that cause antibiotic resistance were chosen for demonstrating the VOCMA2 method. Each VOCMA2 reaction comprises a mix of all five microsphere-bound forward primer/probes, a generic forward primer, a mix of the five target- specific reverse primers, a generic biotinylated reverse primer, and one of the specific synthetic targets of 140-200 nt, all described in Fig. 7 and table 3. All five bead-bound probes comprise a 5' generic forward primer sequence of 30 nt, and a target-specific 3' end of 50 nt. The five target-specific reverse primers comprise all a 50 nt specific sequence in the 3' end
and a 30 nt generic reverse primer sequence in the 5' end. Target genes in the example of VOCMA2 demonstrated in Fig 7 were the GseA-gene of Staphyloccocus epidermidis, the Nwc-gene of Staphyloccocus aureus the MecA-gene of Staphyloccocus aureus, the ddl-gme from Enterococcus faecium, and the i -gene from Enterococcus faecalis.
Materials and methods
Synthetic targets
Synthetic targets were diluted to 200 pM with 0.1 * Denhardt's solution (Sigma- Aldrich Sweden AB, Stockholm, Sweden) (table 3).
Amplification
2.5 μΐ of the synthetic target was added to a 22.5 μΐ PCR mixture for a final concentration of 1 PCR Gold Buffer (Applied Biosystems, Stockholm, Sweden), 800 μΜ dNTP Mix, 2 mM MgCl2, 1 U AmpliTaq Gold® DNA Polymerase, 30 nM generic forward primer, 300 M of biotinylated generic reverse primer, 5 nM of each of the five target-specific reverse primers and 5000 of each Luminex microsphere coupled to a target-specific primer/probe.
Amplification was carried out on a MJ Research PTC- 100™ Peltier thermal cycler (SDS Scandinavian Diagnostic Services, Falkenberg, Sweden) as follows: 95 °C for 10 min, followed by 4 cycles of 94 °C for 30 s, 52 °C for 2 min, 72 °C for 2 min, 40 cycles of 94 °C for 30 s, 56 °C for 45 s, 72 °C for 45 s a last step with 72 °C for 7 min and the samples were kept at 4 °C until next step.
Coupling of microspheres
Each synthetic target-specific forward primer/probe and detection probe oligonucleotide with a 5 ' C-12 amino modification (Biomers.net GMbH, Ulm, Germany) were covalently coupled to specific sets of 5.6 μπι carboxylated polystyrene microspheres (Ramcon A/S, Birkerad, Denmark). 2.5 x lO6 microspheres were pelleted at 13000 x g for 2 min and re-suspended in 25 μΐ 0.1 M 2-Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication. 2 μΐ of 100 μΜ amino-modified target-specific forward primer/probe or detection probe oligonucleotide was added to the bead suspension followed by addition of 2.5 μΐ of 10 mg/ml l-Ethyl-3-3-dimethylaminopropyl carbodiimide (EDC) (Nordic biolabs, Taby, Sweden), the coupling reaction was incubated at room temperature in the dark for 30 min. Addition of 2.5 μΐ of 10 mg/ml EDC and incubation were then repeated once. After the second incubation 1 ml 0.02 % Tween-20 was added to the reaction and vortexed. The microspheres were then
pelleted by centrifugation at 13000 x g for 2 min and re-suspended in 1 ml of 0,1 % SDS by vortexing. Finally the microspheres were pelleted and re-suspended to a final concentration of 50000 microspheres/μΐ by adding 50 μΐ Tris-EDTA (TE, pH 8.0) buffer. Coupled
microspheres were stored in darkness at 8 °C.
Detection in Luminex
Samples were centrifuged for 2 min on a table centrifuge to pellet the microspheres. Five μΐ of the supernatant was added to 45 μΐ hybridization buffer containing 33 μΐ 1.5χ tetramethyl ammonium chloride (TMAC) (4.5 M TMAC, 0.15 % sarkosyl, 75 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0), 1 1.5 μΐ 1 χ TE-buffer pH 8.0, and 0.1 μΐ (-5000 microspheres) of each catch probe coupled microsphere. Samples were denatured at 95 °C for 2 min followed by hybridization at 50 °C for 30 min under shaking on a Thermostar (BMG Lab Technologies, Germany) microplate incubator. After hybridization the microspheres were pelleted by centrifugation on a table centrifuge for 2 min. The supernatant were removed and the microspheres re-suspended by pipette mixing 45 μΐ 1 χ TMAC hybridization buffer comprising 3 M TMAC, 0.1 % sarkosyl, 50 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0 (Sigma- Aldrich Sweden AB, Stockholm, Sweden). 2 μΐ (0.05 μg/μl) of Streptavidin-R- Phycoerytrin (Qiagen AB, Stockholm, Sweden) was added to the reaction and the samples were incubated for another 15 min at 50°C. Detection in the Luminex 200 (purchased through Ramcon A/S, Birkerod, Denmark) system was carried out at 50 °C. The calculation of median fluorescence intensity (MFI) was based on 100 measurements from each microsphere set and calculated by the StarStation software (Applied Cytometry System, Sheffield, UK).
Results
An aliquot of the supernatant of each VOCMA2 was added to new tubes containing a mix of five target-specific detection probes coupled to new Luminex beads. The samples were analysed in the Luminex, in which 100 beads of each detection probe was analysed in all samples. The five different detection probes were all of 50 nt length and designed to fit the target sequence in same sense as the first target-specific forward primer/probe used in the amplification step. As shown in Fig. 7 and table 2„ all five samples showed a significant MFI detected from the complementary bead-bound detection probe specific for the synthetic target added. The VOCMA2 gave even a lower background than VOCMA1. All detection probes were able to detect their correct target without any cross-hybridization.
Conclusions
VOCMA2 is a highly specific and multiplexable detection technique. Signal to noise ratios were very low, around 1/1000 or better. The analytical sensitivity was 103 target copies per reaction, or better.
Example 3 - VOCMA 2 for detection of Norovims and Sapovirus
One of the most common causing agents of gastroenteritis is the Caliciviridae (including Norovirus and Sapovirus). In this example it is shown promising results of inosine-containing primer/probes for mismatch-tolerant detection of these highly variable microbes. Target- specific primer/probes for Norovirus genogroup 2 were designed targeting the POL gene of the virus. The VOCMA2 reaction contains microsphere-bound target-specific reverse primer/probes, generic reverse primers, biotinylated target-specific forward primers, biotinylated generic forward primers, and a synthetic target of 177 nt, all described in table 5.
The bead-bound reverse primer comprised a 5' generic reverse primer sequence of 16 nt, and a target-specific 3' end of 71 nt. The biotinylated target-specific forward primer comprised a 41 nt specific sequence in the 3' end and a 16 nt generic forward sequence in the 5' end. The specific reverse primer includes Inosine as shown in table 5. Table 5 shows oligonucleotides used in VOCMA2 for detection of Norovirus genogroup 2.
Materials and methods
Synthetic target
Synthetic targets (Biomers.net GMbH, Ulm, Germany) were serial-diluted from 0.,5 < 107 to 0.,5 l03 in 20 ng20ng^l yest RNA (Ambion, Austin, USA) (table 5).
Coupling of target-specific reverse primer/probe to MyOne magnetic beads
The target-specific reverse primer/probe oligonucleotide with a 5' C-12 amino modification (Biomers.net GMbH, Ulm, Germany) were covalently coupled to carboxylated Dynabeads® MyOne™ microspheres (Invitrogen AS, Oslo, Norway). 2.0x 109 microspheres were pelleted by magnetism and the supernatant removed. Michrospheres were resuspend in 91 μΐ 0.1 M 2- Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication. The pelletation and resupendation were repeated once. 36 μΐ of 100 μΜ amino-modified target-specific reverse primer/probe oligonucleotides were added to the microsphere suspension followed by
addition of 6.66 μΐ of 20 mg/ml l-Ethyl-3-3-dimethylaminopropyl carbodiimide (EDC) (Nordic biolabs, Taby, Sweden), the coupling reaction was incubated at room temperature in the dark for 30 min. Addition of 6.66 μΐ of 20 mg/ml EDC and incubation were then repeated once. After the second incubation 1 ml 0.02 % Tween-20 was added to the reaction and vortexed. The microspheres were then pelleted by magnetism and resuspended in 1 ml of 0, 1 % SDS by vortexing. Finally the microspheres were pelleted and resuspended to a final concentration of 107 microspheres/μΐ by adding 200 μΐ Tris-EDTA (TE, pH 8.0) buffer.
Coupled microspheres were stored in darkness at 8°C.
Amplification
2 μΐ of the synthetic target was added to a 23 μΐ PCR mixture for a final concentration of 1 x iScript™ buffer (Bio-Rad, Hercules, USA), 0.5 μ1,5μ1 iScript reverse transcriptase for one- step RT-PCR, 300 nM generic reverse primer, 300 nM of biotinylated generic forward primer, 50 nM of the biotinylated specific forward primer and 5.0x 105 Dynabeads® MyOne™ microspheres (Invitrogen AS, Oslo, Norway) magnetic microspheres coupled with target- specific reverse primer/probe. Amplification was carried out on a MJ Research PTC- 100™ Peltier thermal cycler (SDS Scandinavian Diagnostic Services, Falkenberg, Sweden) as follows: 50°C for 20 min, 95 °C for 5 min; followed by 10 cycles of 95 °C for 15 s, 56 °C for 1 min; 40 cycles of 95 °C for 15 s, 52 °C for 30 s, 60 °C for 30 s; a last step with 60 °C for 5 min before denaturation at 95 °C for 1 min. The samples were kept at 4 °C until the next step. A non-template control (NTC) was added in each run as contamination control.
Coupling of detection probe to microspheres
The detection probe oligonucleotide with a 5 ' C-12 amino modification (Biomers.net GMbH, Ulm, Germany) were covalently coupled to a specific set of 5.6 μιη carboxylated polystyrene microspheres (Ramcon A/S, Birkerod, Denmark). 2.5 χ ΐθ6 microspheres were pelleted at 13000 x g for 2 min and resuspended in 25 μΐ 0.1 M 2-Morpholinoethane sulfonic acid (MES, pH 4.5) by vortexing and sonication. 2 μΐ of 100 μΜ amino-modified reverse primer or detection probe oligonucleotides were added to the bead suspension followed by addition of 2.5 μΐ of 10 mg/ml l-Ethyl-3-3-dimethylaminopropyl carbodiimide (EDC) (Nordic Biolabs, Taby, Sweden), the coupling reaction was incubated at room temperature in the dark for 30 min. Addition of 2,5 μΐ of 10 mg/ml EDC and incubation were then repeated once. After the second incubation 1 ml 0.02 % Tween-20 was added to the reaction and vortexed. The microspheres were then pelleted by centrifugation at 13000 x g for 2 min and resuspended in
1 ml of 0.1 % SDS by vortexing. Finally the microspheres were pelleted and resuspended to a final concentration of 50000 microspheres/μΐ by adding 50 μΐ Tris-EDTA (TE, pH 8.0) buffer. Coupled microspheres were stored in darkness at 8 °C.
Detection in Luminex
Samples were centrifuged for 2 min on a table centrifuge to pellet the microspheres. 5 μΐ of the supernatant was added to 45 μΐ hybridization buffer containing 33 μΐ 1.5 χ tetramethyl ammonium chloride (TMAC) (4.5 M TMAC, 0.15 % sarkosyl, 75 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0), 1 1.5 μΐ l x TE-buffer pH 8.0, and 0.1 μΐ (-5000 microspheres) of each target-specific detection probe coupled microsphere. A NTC containing 5μ1 of 1 χ TE-buffer pH 8.0 instead of sample was added as a Luminex control. Samples were denatured at 95 °C for 2 min followed by hybridization at 50 °C for 20 min under shaking on a Thermostar (BMG Lab Technologies, Germany) microplate incubator. After hybridization the microspheres were pelleted by centrifugation on a table centrifuge for 2 min. The supernatant were removed and the microspheres resuspended by pipette mixing 45 μΐ 1 x TMAC hybridization buffer comprising 3 M TMAC, 0.1 % sarkosyl, 50 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0 (Sigma-Aldrich Sweden AB, Stockholm, Sweden). 2 μΐ (0.05 μ^μΐ) of Streptavidin-R-Phycoerytrin (Qiagen AB, Stockholm, Sweden) was added to the reaction and the samples were incubated for another 10 min at 50°C. Detection in the Luminex 200 (Purchased through Ramcon A/S, Birkerad, Denmark) system was carried out at 50 °C and the mean fluorescence intensity (MFI) was based on minimum 100 measurements from the microsphere set and calculated by the STarStation software (Applied Cytometry System, Sheffield, UK).
Results
As shown in Fig. 8 and table 4„ all samples from VOCMA2, when read in the Luminex flow particle meter, showed a significant MFI detected from the complementary bead-bound target-specific detection probe for the synthetic target added. The serial dilution shows a analytical sensitivity, with significant MFI, with as low as 103 targets present.
Conclusions
These initial results hold promise for a high sensitivity and specificity of the concept. Capture of target sequence to magnetic beads was demonstrated.
Example 4 - VOCMA2
Introduction
A demonstration of the sensitivity of a 22-plex VOCMA2 method containing 22 bacteria, fungi or antibiotic-resistance gene specific primers/probes in solution is shown in Fig. 9. Each VOCMA2 reaction contains a mix of 21 target-specific primer pairs, a generic forward primer and a generic biotinylated reverse primer, all described in table 7. The detection in Luminex is able to detect 22 different genes due to 22 different target-specific detection probes. The 21 target-specific forward primer/probes all comprise a 5 ' generic forward primer sequence of 16 nt, and a target-specific 3' end of 50 nt. The 21 target-specific reverse primers all comprise a 47-50 nt target-specific sequence in the 3' end and a 16 nt generic reverse primer sequence in the 5' end. Although all components of the 22-plex system were present in the reactions, the target gene tested in this example of VOCMA2 was the Streptococcus pneumoniae spn9802 gene. An amount of these synthetic targets of 10 to 10 molecules/reaction were added to each reaction.
Materials and methods
Synthetic targets
Synthetic Streptococcus pneumoniae, shown in table 7, targets were serially diluted to 0,5x l07-0,5 l0° molecules/μΐ with 0.1 x Denhardt's solution (Sigma- Aldrich Sweden AB, Stockholm, Sweden). As no template control a sample of 0.1 χ Denhardt's solution was added.
Amplification
2 μΐ of the diluted synthetic target was added to a 23 μΐ PCR mixture, resulting in 1 x 10 - 1 10° molecules/reaction. The final concentration of l x PCR Gold Buffer (Applied
Biosystems, Stockholm, Sweden), 800 μΜ dNTP Mix, 2.5 mM MgCl2, 1 U AmpliTaq Gold® DNA Polymerase, 200 nM generic forward primer, 300 M of biotinylated generic reverse primer, 5 nM of each of the target-specific forward and reverse primers was used in each reaction. Amplification was carried out on a iCycler (Bio-Rad Laboratories AB, Sundbyberg, Sweden) as follows: 95 °C for 9 min; followed by 8 cycles of 94 °C for 30 s, 75 °C for 0 s, ramp with 0,1 °C/s to 58 °C for 2 min, 72 °C for 30 s; 52 cycles of 94 °C for 30 s, 53 °C for 45 s, 72 °C for 30 s; a step with 72 °C for 7 min, and a last denaturation at 95 °C for 2 min, then the samples were kept at 4 °C until next step.
Coupling of microspheres
22 different Luminex beads were coupled with target-specific detection probes, shown in table 7, in the same way as in VOCMA2 example 1.
Detection in Luminex
5 μΐ of the reaction was added to 45 μΐ hybridization buffer containing 33 μΐ 1.5χ tetramethyl ammonium chloride (TMAC) (4.5 M TMAC, 0.15 % sarkosyl, 75 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0), 9.8 μΐ 1 χ TE-buffer pH 8.0, and 0.1 μΐ (-5000 microspheres) of each of the 22 target-specific detection probes coupled microspheres. Samples were denatured at 95 °C for 2 min followed by hybridization at 50°C for 30 min under shaking on a Thermostar (BMG Lab Technologies, Germany) microplate incubator. After hybridization the
microspheres were pelleted by centrifugation on a table centrifuge for 2 min. The supernatant were removed and the microspheres resuspended by pipette mixing 45 μΐ 1 x TMAC hybridization buffer comprisingcomprising 3 M TMAC, 0.1 % sarkosyl, 50 mM Tris-HCl pH 8.0 and 6 mM EDTA pH 8.0 (Sigma-Aldrich Sweden AB, Stockholm, Sweden). 2 μΐ
(0.05 g μl) of Streptavidin-R-Phycoerytrin (Qiagen AB, Stockholm, Sweden) was added to the reaction and the samples were incubated for another 15 min at 50 °C. Detection in the Luminex 200 (purchased through Ramcon A/S, Birkerod, Denmark) system was carried out at 50 °C and the mean fluorescence intensity (MFI) was based on a minimum of 100
measurements from each microsphere set and calculated by the StarStation software (Applied Cytometry System, Sheffield, UK).
Results
In this demonstration a sensitivity of 10 molecules/reaction could be detected in the Luminex with a low background signal, as shown in Fig. 9 and table 6. All primer/probes and primers (specific and generic) were in solution in the capture and amplification step and only the detection probes were bound to beads..
Example 5 - VOCMA3/. HC:
Materials and methods:
PCR was conducted on various combinations of coupled and not coupled Hermit Crab (HC) "arms". The HC arm combinations were subjected to a first PCR round using a synthetic
target molecule, then amplified using generic primers. 1 \ primary PCR product was used as target. Program: 95C° 10 min , followed by 40 cycles of 95C° 30sek 52C° 30sek 72C° 30sek.
Results
In this experiment, the specific product appeared only after addition of free arms, regardless of whether the two PCR rounds were run separately or together.
Mismatch-tolerance prediction algorithms
Introduction
The examples of probe and target hybridization demonstrated in Figs. 1 1-13 A and B resulted in the conclusion that the position of mismatches in the nucleotide sequence affects the binding more than the actual number of mismatches. Thus, the length of the perfect matching regions in between mismatches is of great importance for hybridization. A target containing one longer stretch of perfect match is binding better than a target with the same amount of matching nt in short regions interspersed by mismatches. In an attempt to predict
hybridization based on Gibbs free energy, AG, the computer program Visual OMP (DNA Software) was utilized. Fig. 14 demonstrates the predicted AG (Visual OMP, DNA Software) versus % MFI (the MFI value of a modified probe relative to that of the unmodified parent probe) for different 70-mer probe and 70-mer target combinations. Most probe/target combinations having a predicted AG < -50 will hybridize, while most probe/target combinations with a AG > -17 do not hybridize. However, the hybridization (measured as % MFI) and the predicted AG do not correlate in the region of -17 > AG > -50, and thereby we conclude that the predicted AG is difficult to use to predict hybridization. To be able to predict hybridization a scoring system was developed in which long uninterrupted perfectly matching sequences and the neighbouring of these was taken into account.
Trimers, a development of the Nearest-Neighbour algorithm, represent local
contributions to binding.
The Nearest-Neighbour algorithm (Santa Lucia et al.) is taking the closest neighbouring nucleotides effect on hybridization for a specific nt into account (see Fig. 18a). Thus, the Nearest-Neighbour algorithm represents local contributions to binding. It is included in the Visual OMP program. The partial failure of Visual OMP to predict hybridization mentioned in the previous paragraph made us attempt to construct a new algorithm. The first step in
development of a new scoring system is shown in Fig. 17A and in the right panel of Fig. 15, where a score for all perfectly matching overlapping trimers in all dinosine-free combinations of probe and target, i.e. InflA probe-target and Noro probe-target, are plotted against % MFI (45 °C). As shown in the small inserted graph in the left panel, the Overlapping Trimer Score correlated very well with the predicted AG (Visual OMP), shown in the left panel, with an R2=0.906. The Overlapping Trimer Score is schematically visualized in the upper panel of Fig. 16. The scoring system was further developed by using overlapping quadramers, overlapping pentamers, and overlapping hexamers, displayed in Fig. 16 and in the eight graphs of Fig. 17A-H. The Fig. 17A-D demonstrates the overlapping tri- (A), quadra- (B), penta- (C) and hexamer scores (D) versus % MFI in 45 °C, while Fig. 17E-H demonstrates the overlapping tri- (E), quadra- (F), penta- (G) and hexamer scores (H) versus % MFI at 55 °C. The probes hybridizing to 70 nt long targets are visualized as black triangles, while probes hybridizing to targets of the length 12, 15, 18, and 22 nt are shown as open triangles.
Neighbouring trimers, a representation of distal but close contributions
To further improve the ability to predict hybridization we included Neighbouring trimers, a representation of distal but close contributions, into the scoring system. As shown in Fig. 18b, each Neighbouring trimer is given a score. Further, the Neighbouring trimers was scored according the algorithm: Pyramid factor *(n(n-l))/2); where n is the number of neighbouring trimers and the pyramid factor is decreasing the impact for each level (according to the distance between the trimers) in Fig. 18b.
Combining local and distal contributions, TriHexa model
Further, Fig. 18 demonstrates the computational work in which more distal binding contributions is taken into account. Each region of perfect match is weighted against a neighbouring region of perfect match and given a score, cooperativity factor 1, see Fig. 18c. In case there are severalse closely situated neighbouring regions their cooperative contribution to binding is then weighted using a second factor, cooperativity factor 2, Fig.18d. Thus, the TriHexa model accounts for both local (trimer) and more distal contributions to hybridization. Fig. 19 shows the TriHexa scoring system versus the % MFI for all target and probe combinations that were demonstrated in Fig. 14 (AG Visual OMP versus % MFI).
The local and distal contributions to hybridization are shown symbolically in Fig. 19. The local binding (the binding of a matching base pair plus the binding of the next and previous
base pairs) is calculated in analogy with the deltaG calculations according to nearest neighbour theory (Santa Lucia et al). In the following, matches to UBA (inosine) are not counted as mismatches. Instead, they are given values according to the pattern of UBA distribution, and the nucleotide opposing the UBA, with "C" being the most favourable, and "G" the least favourable. The distal contributions are of three kinds;
One is the contribution of uninterrupted runs of matching nucleotides, until the occurrence of a mismatch, or the end of the hybridizing molecules. This is calculated as a "pyramid" of binding interactions (Fig 18b). It contributes the so-called pyramid factor. Another is the cooperative binding of two stretches of matching nucleotides, separated by one or several mismatches (Fig 18c), which contributes the so-called cooperativity factor 1. A third kind of cooperativity is between pairs of stretches of matching nucleotides (Fig 18d), which contributes the so-called cooperativity factor 2. All four kinds of binding are included in the "trihexa" function which balances these contributions according to optimized values of the respective factors. The resulting hybridization prediction values are shown in Fig 19. It can be seen that the prediction is precise for trihexa hybridization values up to 20, and for values above 100. Values between 20 and 65 are imprecise predictors of hybridization. Values between 65 and 100 are reasonably accurate predictors of hybridization. Using the trihexa function, the degree of hybridization (i.e. mismatch tolerance) for various probe-target combinations can be predicted. The model has been set up for 70-mers in 3M TMAC at 45 °C, but it is likely to be extendable to other conditions also.
Including the Nucleation and Zipping stages in the calculations, the NucZip model
The hybridization process proceeds in two stages, nucleation and zipping.
The first engages a limited number of nucleotides (5-9) which can make contact without much torsional strain on the long nucleotide. All contacts are held within a turn (10 nt) of the helix. This is as long as two nucleotide strands can make contact without becoming entangled with each other. Nucleation allows nucleic acid strands to "sniff on each other without deeper commitment. Nucleation depends on perfectly matching stretches 5-10 nucleotides.
In Zipping, the nucleated 5-9 matches are further extended up- and downstream. For each turn (10 nt) of more or less perfect matches, the two strands become more and more entangled, and less likely to separate. The zipping process proceeds approximately as described in the TriHexa model. However, we experimented with different developments of it. It turned out that a model where the number of successive perfectly matching tri-, terra-, penta-, hexa-, hepta-, okta-, nona- and decamers were counted better explained experimental data than
previous models. This is the "NucZip" model. A schematic model of NcZip is shown in Fig. 23. Its results are shown in Fig. 20.
Conclusion
To improve the predictability of hybridization we developed a scoring system in which uninterrupted regions of perfectly matching nucleotides and neighbouring regions of perfect match are given a great impact. Together the different local and distal scoring is creating the TriHexa and the NucZip scoring system, which are used for a more accurate prediction of long oligomer hybridization.
Material and Methods
Example 6- construction and study of dlnosine probes
Single-stranded oligonucleotides of 70 nucleotides with a C12 amino link in the 5' end, with or without dlnosines, were obtained from Biomers.net (Ulm, Germany). The
oligonucucleotides were used as probes after linkage to xMAP microspheres (Luminex Corp., Austin TX, USA). The probes were designed based on the programs BLASTn, ClustalX and ConSort© (unpublished, Blomberg J. et at). Briefly, query viral sequences were retrieved from the GeneBank database at NCBI, NIH. Alignments were performed by a BLASTn search. In order to identify potentially problematic sequences, the probe sequence was analyzed for sequence similarities against the human genome sequence database hgl 8. 70-mer sequences with a BLASTn score of <37.5 (approximately corresponding to 70% mismatches or more) were accepted for further work. BLASTn and ClustalX alignments were analyzed in ConSort to define the most suitable probe sequence. ConSort visualizes the frequency of variation and the variation of nucleotide composition in each base position, as well as the number of aligned sequences. It also provides a majority consensus sequence. The proposed probe sequence was further analyzed for its predicted Tm. A search for probable homodimer and hairpin interactions, using cut-offs of AG>-11.5 kcal/mol, and AG>-5 kcal/mol, respectively, was made by using Mfold at the site
http ://eu. idtdna. com/ analyzer/ Applications/ Oli go Analyzer/ and http://mfold.bioinfo.rpi.edu/ . Synthetic targets, complementary to the consensus sequence of InflA or Noro probe, with various numbers of mismatches, were purchased as ssDNA of 70 nucleotides with biotin attached to a 2-aminoethoxy-ethoxyethanol linker in their 5' end (Biomers.net, Ulm,
Germany).
Probe coupling to beads
Specific synthetic 5' amine-C12 modified 70-mer probes for respiratory tract viruses and histon 3.3 were designed and coupled to xMAP carboxylated colour-coded microspheres (Luminex Corp., Austin TX, USA), according to the protocol of Luminex corp (Austin TX, USA). An aliquot of 2.5* 106 stock microspheres were collected by centrifugation and resuspended in 25 μΐ of 0.1M MES, pH 4.5 (2N-morpholino-ethanesulfonic acid, Sigma). 0.2 nmol probe and freshly made 2 μΐ 10 mg/ml EDC (l-ethyl-3-(3-dimethylaminopropyl) carbodiimide; "water-soluble carbodiimide"; Pierce; sold by Nordic Biolabs AB, Sweden) in H20 were added to the beads and incubated in the dark for 30 min at room temperature. Care was taken to store the EDC in a dry condition, in aliquots. After another addition of a fresh 2 μΐ (lOmg/ml) EDC in H20 and repeated incubation the beads were washed with 0.5 ml of 0.02 % Tween-20. The pellet was collected by centrifugation at 8000xg for 2 min. The collected pellet was resuspended in 0.5 ml of 0.1 % SDS and again centrifugated at 8000xg for 2 min. The pellet was resuspended in 50 μΐ of TE, pH 8.0.
Hybridization ofbiotinylated target DNA to probe linked beads
5 μΐ of 2.0 nM synthetic biotin -labelled target was mixed with hybridization buffer comprising 33 μΐ 3 M TMAC buffer (3 M tetramethylammonium chloride, 0.1% Sarkosyl, 50mM Tris-HCl, pH 8.0, 4 mM EDTA, pH 8.0; Sigma) and 12 μΐ lx TE-buffer pH 8.0 and 0.05 μΐ (-2500 microspheres/beads) of each probe-coupled Luminex microsphere bead. The mixture was heated at 95°C for 2 min, to denature the DNA targets and probes, followed by hybridization at 45 °C or 55 °C for 30 min under shaking on the Thermostar (BMG LabTech; Offenburg, Germany) microplate incubator. 2 μΐ (0.05 mg/ml) of Streptavidin-R- phycoerythrin (QIAGEN, Hilden, Germany) was added to the mixture and further incubated at 45 °C or 55 °C for 15 min before analysed for internal bead and R-phycoerythrin reporter fluorescence on the Luminex®200™ flow cytometer (Luminex corporation, Austin Tx). The quantification of the amount of biotinylated target that hybridized to probe linked bead is given by the unit MFI, Median Fluorescence Intensity. In our assay the minimum amount of beads analysed per type of bead was set to 100. All experiments were judged by using a limit of detection of MFI <20% of probe hybridizing to a perfect target as a "weak" hybridization signal, >20%-<50% as "intermediate", and >50% as "high" hybridization. As lower limit of detection (LLOD) a MFI of 100 was used.
Result
A perfectly matching stretch of minimum length is required for hybridisation.
Many viruses, especially RNA viruses, have a high frequency of variation and almost no conserved regions in their genome. A broad, but yet specific, probe should detect all types or subtypes of the specific microbe it is designed for, and also be functional if the genome sequence is changed due to genetic drift. To reveal the tolerance to pm, a 70-mer probe from the Influenza A segment 7 and a set of target molecules with increasing amounts of pm were allowed to hybridize in 3 M TMAC.
Influenza virus is an RNA virus with a high mutation rate due to both genetic shift and genetic drift. It is divided into three types, Influenza A, B and C. The types are further divided into serotypes depending on the variation in its surface molecules haemagglutinin (HA or H) and neuraminidase (NA or N). Fig. 10 demonstrates the variation in an alignment of 7333 genomes of segment 7, the gene of the matrix proteins, of Influenza A; all H and N influenza A types were represented. The variation was mapped, searching for a suitable region for a 70- mer probe, by using BLASTn, and ConSort© (unpubl., Blomberg J.). A probe was designed to match a region of 70 nt in the gene for matrix protein 2 in segment 7 of the Influenza A H3N2 virus. In the chosen region, the H5N1 virus differed in five nt and the H1N1 virus in three other nt positions, compared to the H3N2 virus (table 8). If mismatches in nine positions, including the variant nt positions in segment 7 of the H IN 1 and the H5N1 viruses, could be tolerated by a detecting probe, the probe would fully cover 67 different H and N combinations of Influenza A, i.e. nearly all variants in the chosen stretch, as demonstrated in a BLASTn search. A few subtypes would give 2 additional mismatches (table 8.).
The Influenza A H3N2 probe from segment 7 was tested against target molecules with 3, 5, 7, 9, 1 1, 12, 13, 14, 15, 16 and 21 pms (Fig. 11 A and B) The positions of the pms were based on the variation seen comparing H3N2, H5N1 and H1N1 viruses. Two different targets were designed with 21 mutated nucleotides; one with 21 point mutations evenly distributed along the 70-mer, the 21rpm target, and one with seven groups of three mutations interspersed by five to seven nucleotides, the 21gm target. The InflA H3N2 probe, called InflA probe, was coupled to Luminex beads and subsequently allowed to hybridize to the specific biotinylated ssDNA targets in 3M TMAC in three different temperatures 45°C (Fig. 11 A), 50 °C (data not shown) and 55 °C (Fig. 1 IB). The hybridization was analysed in a Luminex flow cytometer where the hybridization signal is given as Median Fluorescence Intensity, MFI. In this study,
all experiments were judged by using a limit of detection of MFI <20% of InflA probe hybridizing to a perfect InflA target as a "weak" hybridization signal, >20%-<50% as "intermediate", and >50% as "high" hybridization.
Interestingly, a target containing as many as 9 pm (one stretch of 15 perfectly matching nt, one region of 12 nt, one region of 7 nt, and four regions of 5 nt) still hybridized to the InflA probe with an MFI of 80% (45°C, Fig. 1 1 A) and 72% (55°C, Fig. 1 IB) compared to the MFI of the perfectly complementary target and probe (InflA target/InflA probe). At 14 pm (one stretch of 6 perfectly matching nt, five regions of 5 nt, and two region of 4 nt) the MFI was reduced to 39% (45°C) and 11% (55°C) of the InflA probe/InflA target. At 15 pm (one stretch of 6 perfectly matching nt, four regions of 5 nt, and two region of 4 nt) the MFI was 21% (45°C) and 3% (55°C). At 45°C the InflA probe hybridization to the 16 pm target (one stretch of 6 perfectly matching nt, three regions of 5 nt, and two region of 4 nt) was still detectible (12%), but at 55°C the hybridization was totally abolished (0.7%). As expected, the more pms that were introduced into the target the less efficient was the hybridization between target and probe. Thus, utilizing a lower hybridization temperature, like 45°C compared to 55°C, allowed the probe to be more forgiving to mismatches.
Increasing the amount of mismatches to 21 evenly distributed pm, 21rpm, caused a total loss of hybridization at both temperatures (Fig. 1 1 A and B). The longest perfectly matching sequence in this combination was 3 nt. The presence of one stretch of three and eight of two perfectly matching stretches was not enough for the hybridization, even at 45°C. Importantly, the hybridization of the InflA probe to a 21 pm containing target could be rescued when redistributing the 21 point mutations into groups, thereby creating seven stretches of 5-7 matching nt between the mutations, in the 21gm target. The MFI signal abolished in the 21rpm target increased to 52% and 25% compared to the MFI of the InflA probe/InflA target combination, at 45 °C and 55 °C, respectively. In conclusion, 1-11 evenly distributed mismatches (preserving multiple contiguous matching stretches of at least 5 nt) have little reducing effect on hybridization between probe and target, while a 70-mer probe fails to hybridize efficiently to a target containing 15-16 evenly distributed pms, although the hybridization can be rescued to a slight degree by reducing the hybridization temperature. Thus, seven stretches of three nonmatching nucleotides (as in 21 gm) destabilize less than 21 single pms (as in 21rpm). Further, the distribution of the mismatching nucleotides affects the
hybridization more than the number of mismatches, indicating that the length and number of perfectly matching stretches are of greatest importance.
Hybridization against the InflA sequence, using a probe with the nucleotide analogue dinosine at known sites of variation
Depending on how variable the genomic sequence is in a microbe, the probe and target sequence will have more or less mismatch. Our probes were designed as 70-mers to minimize the negative effect that mismatching nt have on hybridization. In an attempt to make a probe with an extended mismatch tolerance, dinosine was introduced into the probe, dinosine can bind to all four normally occurring DNA bases, but binds strongest to Cytidine
To analyse dinosine' s effect on the probe binding capacity in 3M TMAC a set of 70-mer probes were designed containing 3, 5, 7, 9 or 21 dlnosines (Fig. 11 A and B). The dlnosines in the probes were placed in the same positions as the pms in the Influenza targets (Fig. 11C). Hybridization of the 3, 5, 7 and 9 dinosine probes to the InflA target gave a small reduction of the MFI signal compared to the MFI of the normal InflA probe
(InflA~3dIno>5dIno>7dIno>9dIno), while the 21 dinosine probe bound less efficiently, 49% (45 °C) and 27% (55 °C), compared to the InflA probe (Fig. 11 A and B). The 3M TMAC in the hybridization buffer will cause an increase of Tm of the A:T base pairs, resulting in similar Tm as G:C base pair binding. A general experience from the use of dinosine is that the MFI tended to decrease when it was introduced. This is an indication that the binding strength of dinosine base pairs was not enhanced by TMAC as much as it enhanced A:T base pairs, and is in line with the smaller binding contribution of dlnosine-containing pairs compared to canonical base pairs. Obviously, dinosine matches cause less destabilization than mismatches, and allows probe/target combinations with many short matching stretches, like 21 rpm, to hybridize. The dinosine thus bridges adjacent matching stretches, increasing their likelihood to nucleate.
Introducing dlnosines in the probe at sites of variation rescues the hybridization of targets containing mismatches covered by dlnosines
Further, the dlnosine-containing probes were also analysed against the targets with different amount of pm (Fig.l 1A and B, 45°C and 55°C, respectively). The probes containing 3-9 dinosine gave rise to a small reduction of MFI compared to the normal InflA probe, but each dinosine probe kept its own level of MFI with targets having up to the same amount of pm as the number of matching dlnosines, plus 2 mismatches not covered by dlnosines. Introduction
of additional mismatching nt, not covered by the dlnosine interaction, reduced the level of MFI to a similar extent as seen when hybridizing the InflA probe to targets with increasing amount of pm. Interestingly, the 3-9 dlnosine probes and the InflA probe, were equally efficient against the 9, 1 1 and 12 pm targets. When the targets had more than 12 pm the 3-9 dlnosine probes worked better than the InflA probe; 9dIno>7dIno>5dIno>3dIno> InflA. The 7 and 9 dlnosine probe gave rise to 1,5-1,8 times (45°C) and 4.1-7.5 times (55°C) higher MFI than the normal InflA probe during hybridization to targets with 15pm and 16 pm, although with a much lower MFI, 21-32% (45°C) and 5-14% (55°C), compared with the InflA probe/InflA target.
Importantly, the 21 dlnosine probe could rescue the hybridization to the 21rpm target, to which the InflA probe totally failed to bind (Fig. 1 1A and B). The signal of 3853 MFI (38% of InflA probe/InflA target) and 1381 MFI (14% of InflA probe/InflA target), at 45°C and 55°C, respectively, achieved with the 21 dlnosine probe against the 21rpm target, is in the same range as the 21 dlnosine probe binding to the other pm targets and the InflA target (calculated mean MFI is 34% of InflA probe/InflA target). Thus, when a probe fails to hybridize due to a high number of pms in the target molecule, dinosines at these positions can save the hybridization.
The importance of relatively long uninterrupted matching sites in hybridization for the "nucleation process "
Many viruses harbor synonymous mutations (sm), often meaning that the third base in a codon can "wobble" without changing the amino acid in the protein. To test the sensitivity of the 70-mer probe against this type of sms, 70 nt long targets were designed with every third nt harboring a sm compared to the reading frame of matrix 2 protein in the sequence of H3N2 Influenza A segment 7. Since a large amount of mismatches distributed evenly along the target was shown to abolish hybridization (Fig. 1 1 A and B), the 33% mutated stretch of nucleotides (in total 18, 16, and 14 sm) was centrally placed between flanking regions of 9, 12, and 15 perfectly matching nt at 5' and the 3 end; creating the 33%9F, 33%12F, and 33%15F targets (Fig. 12A-E). Three targets with synonymous mutations starting close to their 3' end (20, 19 and 18 sm) and stretching towards 9, 12 and 15 perfectly matching nt at the 5' end; 33%9nt, 33%12nt and 33%15nt targets, were also created.
The hybridization was analyzed at 45 °C and 55 °C (Fig. 12A and B, respectively). The InflA probe was shown to hybridize strongly (MFI 2259 at 45 °C and MFI 957 at 55 °C) to a target
containing a centrally placed region of 14 sms and 15 perfect nt in the 5' and 3' flanks, 33%15F, at both hybridization temperatures, while 18 sm and two flanking regions of 9 perfectly matching nt, 33%9F, abolished hybridization (MFI 85 at 45 °C and MFI 37 at 55 °C) presumably due to a disturbed nucleation process even at the lower hybridization temperature, 45 °C. At 45 °C the hybridization of InflA to the 33%12F target (MFI 1193), with its two flanking sequences of 12 nt and 16 sm, was weaker compared to the 33%15F (MFI 2259). At the more stringent hybridization temperature, 55 °C, the InflA probe and 33%12F target hybridization is abolished (MFI 103), but interaction with the 33%15F remains (MFI 957). Interestingly, at the lower hybridization temperature (45 °C) the 33%15nt, having only one stretch of 15 uninterrupted nt, binds almost as well as the 33%12F, which has two stretches of 12 nt (compare MFI 801 with MFI 1 193). However, the 33%12nt hybridized poorly (MFI 215). At 55 °C the 33%15nt could not hybridize at all. In conclusion, if a target has a very long stretch of sm but also contains a region of at least 15 uninterrupted nt it can be detected if a less stringent hybridization temperature is used. In addition, in a target with a high amount of sm and two uninterrupted matching stretches [12 nt (45 °C) or 15 nt (55 °C)], to give an easily detectible hybridization.
By making targets with perfectly matching sequences in the 5' and 3' end and a central region of either 26% or 74% randomly distributed mutations, the importance of long nucleation sites was analyzed more thoroughly. The differences between having a long region regularly interrupted by mutations (like synonymous mutations) or having shorter sequences (4-6 nt) of perfect match in between the mutations were as well investigated. As expected, when hybridizing to InflA probe the 26% mutated targets: 26%5F, 26%7F, 26%9F, and 26%15F, with 26% random mutations in a region placed in between two flanks of 5, 7, 9 and 15 perfectly matching nt, showed that the shorter the flanking regions of perfect match, the less the MFI signal. All 26% targets tested were able to hybridize to the InflA probe at 45 °C, but the 26%5F target failed to bind at 55 °C. Interestingly, 26%5F contains, just like the 33%12F and the 16pm target, 16 mutations. All failed to bind to the InflA probe at 55 °C (Fig. 1 1 A and B, Fig. 12A and B), while at 45 °C the targets succeeded. At 45 °C, two regions of 12 uninterrupted nt in the 33%12F gave 100%) of the MFI of InflA probe/InflA target
hybridization, while the two very similar targets 26%5F and 16 pm, having several shorter matching regions of 6, 5, 4 and 3 matching nt, gave 56%, and 12%, respectively. The same effect, i.e. that a few long regions of perfect match gives better hybridization than several
shorter, were shown with the targets which had 14 pms; the 26%9F, 33%15F and 14pm target (Figs. 11 A and B, and Figs. 12 A and B).
These results confirm what was shown with the 21rpm and the 21gm target in Fig. 1 1A and B, where redistribution of the 21 pm to create longer nucleation sites rescued the abolished hybridization to the InflA probe. Conclusively, these experiments together confirm strongly that the amount of mismatches is less important than the distribution of the mismatching nt, and that the long perfectly matching sequences probably function as nucleation sites. The long nucleation sites are necessary to overcome the fact that the target contains a large amount of evenly spaced mutations.
To test if a long nucleation sequence could cause hybridization to a target with an otherwise very low percentage of matching nucleotides, targets with approximately 74% mismatch in the centrally placed region were constructed. Using the InflA probe against the 74%5F, 74%7F, 74%9F, 74%12F and 74%15F targets demonstrated that the 74%15F could hybridize to the InflA probe both at 45°C and 55°C, while the 74%12F was able to bind at 45°C (MFI 1617) but not at 55°C (MFI 73). The hybridization probably occurred exclusively in the perfect matching 5' and 3' flanks, since the 40 nt central region of 74%15F only had 9 matching nt randomly spread over it, while 74%12F had 11 matching nts in its central region of 46 nt. Thus, two regions of 12 perfectly matching nt are enough to give a high degree of hybridization at 45°C, while two regions of 15 nt are needed at the higher temperature, just as shown with the 33% sm containing targets. The hybridization of 74%5F, 74%7F, and 74%9F was totally abolished, which shows that a 70-mer with two flanks of 9 nt has to have help of the region in between, compare with 26%9F (MFI 777 and 1655, at 55 °C and 45 °C, respectively), to give a detectible MFI. Targets having 74% mismatch through the whole oligo and one matching sequence in its 5' end were used to test how short the nucleation site can be to give hybridization to a 70-mer probe. At 55 °C, 18 nt was enough to give a hybridization signal, (74%18nt, MFI 941), while 15 nt (74%15nt, MFI 839) was enough at 45 °C.
Inosine-containing probe rescues hybridization to targets with synonymous mutations The different sm containing targets was tested against a set of probes, including the InflA probe (described above); one probe with 18 dlnosines matching the sm in 33%9F target, therefore called 18ino_33%_9F; two probes with 21 dlnosines positioned in every third base
leaving either a flank of matching 9 nt in the 5 'end or in the 3' end, mo21_33%_9nt5' and Ino21_33%9nt3'; and one probe with 24 dlnosine in every third base all throughout the whole 70-mer probe, Ino24_33%.
Importantly, a probe with 18 dinosines (18Ino_33%_9F probe) in the central region, positioned at the sms within 33%9F target, was able to rescue the hybridization of all targets with more or less sms occurring at dlnosine sites (45°C; Fig. 12A). The 33%9F target was rescued from no signal at all to an MFI of 1609 and 1031 (45 °C and 55 °C, respectively). Even the 33%9nt, with its 20 sm and only one region of perfect 9 nt, could be detected (MFI 1589 at 45 C and MFI 339 at 55 °C). The 18Ino probe hybridized less well to the 33%9nt compared to the 33%9F because the two extra mutations in the 3' end of the 33%9nt target were not covered by a dlnosine in the 18Ino_33%_9F probe. Thus, the experiment demonstrates that when there are no suitable nucleation sequences for a dlnosine free probe, a probe with dinosines at the positions of variation will rescue the hybridization to the same target.
Introducing dlnosine in every third position throughout the whole probe, 24Ino_33%, caused a general decrease of MFI compared to the 18Ino_33%_9F probe. At 55 °C the 24Ino_33% probe did not hybridize to any of the targets. At 45°C the 24Ino_33% gave rise to a weak hybridization signal of 105-589 MFI. The two 21ino_33% probes worked better than the 24Ino_33% but gave lower signal than the 18ino_33% (compared with binding to InflA target). Just like shown in Fig. 1 1 A and B, the more dinosines the probe contains the less efficiently it binds to its target (Fig. 12A and B). At 45°C ino21_33%_9nt5' could rescue binding to the 33%9F target (MFI 1017) as well as the 33%9nt target( MFI 333), even though the dinosines did not cover the two sm in the 5'end of the target. The ino21_33%_9nt3' that did cover the two sm in the 3' of 33%9nt, 33%12nt and 33%15nt targets gave a higher MFI than the ino21_33%_9nt5 'probe, and this resulted in a rescue of these targets with MFI of 815, 373 and 705, respectively. Importantly, the amount of Inosines can be very high (21 dinosines in a 70 mer probe) and still give rise to MFI signals at the level of a normal probe hybridizing to a perfectly matching sequence (MFI 1179) at 45°C, as long as there are at least 9 uninterrupted matching nts without dinosines being involved, like for the ino21_33%_9nt5' probe against the 33%9F target (MFI 1017) or the ino21_33%_9nt3' probe against the 33%9nt target (MFI 815).
To summarize the results when utilizing probes rich in dlnosines, . uninterrupted matching stretches of at least 9 nt can override the fact that the rest of a target has many random mutations ii. Introduction of dlnosines in the probe in the mismatching positions can rescue hybridization when the variation in sequence is distributed over the whole target and causes many short matching stretches, dlnosine does not work as well as the original nucleotides in a matching stretch, but a probe with dlnosines in the site of variation, even placed as narrowly as synonymous mutations, will rescue hybridization where normal probes fail.
Hybridization of a dlnosine-containing probe is sensitive to mismatches outside and with close proximity to the dlnosine position
A probe with many dlnosines can rescue hybridization to a target with many mismatches when the dlnosine and the mismatch have the same distribution pattern and thereby the dlnosine masks the mismatch (Fig. 1 1 A and B, and Fig.12A and B). A test was set up to analyse how many mismatches outside the rescuing position of Inosine, mmoi (mismatch outside dlnosine), a probe can tolerate. The names of the targets are based on matching and mismatching nt against a Norovirus probe, the Inol8 probe, and the InflA probe:
(Matching nt in 5' and 3' compared to Norovirus) Mismatching nt compared to
Norovirus probe_Inol8 probe_InflA probe (Matching nt in 5' and 3' compared to InflA probe) (Fig. 13C, Table 16A and B).
As shown in Fig. 13 A, hybridization utilizing the Inol 8 probe shows that a target with 1 mmoi ((0.10) 35_1_17 (9.8)) or even 10 mmoi ((10.10) 26 10 26 (0.8)) in the flanking sequences outside the region of the synonymous distributed 18 dlnosines had none or little inhibiting effect on hybridization (MFI 6459 and MFI 3835, resp.). Interestingly, when redistributing the 10 mmoi evenly within the 18 sm dlnosine region as in the
(0.11) 26_10_26 (9.8) target the hybridization caused a total loss of hybridization (MFI 93). To investigate how sensitive the dlnosines are to neighbouring mismatches the Ino 18-probe was hybridized against targets having lOmmoi in the flanking regions and increasing amount of mmoi within the region of 18 sm dlnosine. The introduction of additional 2, 4 or 5 mmoi gave hybridization signals of 64% (MFI 4283, (10.10)_24_12_28_(0.8)), 27% (MFI 1827, (10.10)_22_14_30_(0.8)), and 11% (MFI 765, (10.10)_21_15 31 (0.8)), compared to InflA probe/InflA target (table 17). The result demonstrates that the nucleation capacity of the dlnosine-containing probe Ino 18 is sensitive to mmoi when mm is positioned next to a dlnosine.
Comparing the InflA probes and the Inol8 probes sensitivity to mismatch shows that the InflA probe can even hybridize to the (0.10) 35_1_17 (9.8) target, which have 17 evenly distributed mismatches in between two perfect flanks of 9 nt resp. 8 nt (MFI 2495), while 7 mmoi totally prohibits the Inol8 probe to bind to the (10.10) 19 17 33 (0.8) target (MFI 51). The experiment described above was performed at 45°C. In 55°C, shown in Fig. 13B, the Inol8 probe failed to hybridize already when 2 mmoi was present in the
(10.10) 24 12 28 (0.8) target. Thus, the nucleation capacity of a dlnosine-containing sequence is severely reduced when a mismatch is neighbouring the dlnosine, and the destabilising effect of the mismatch next to dlnosine are affecting the probes hybridization capacity more severely than a mismatches against a dlnosine free region. Conclusively, using the probe containing 18 dlnosines in a synonymous pattern at a non-stringent temperature (45°C), an intermediate signal of 27% and a low signal of 1 1% were achieved with targets containing 4 mmoi and 5 mmoi , respectively, that are distributed in between the positions of the dlnosines. dlnosine-containing probe is able to rescue mismatching target and still show specificity
The advantage of introducing many dlnosine in a probe designed against a target with large variation is the ability to rescue hybridization. To further test specificity and cross- hybridization of the Inol8 and the consensus InflA-probe a set of targets based on a
Norovirus sequence was constructed. Norovirus is a highly variable positive sense RNA virus, belonging to the Caliciviridae, which causes winter vomiting disease. The Norovirus sequence chosen (capsid gene of Norwalk-like virus, acc nr AY274264), after Blastn with the InflA-probe sequence, has a short region of 8nt that is perfectly matching the very end of the InflA probe and 10 nt dispersed spread matching nt (Fig. 13C). The Norovirus target
(70) 0_36_52 (0.8) was gradually changed toward the resemblance of the Influenza target, InflA (0.8) 51_0_0 (70), by altering the nt sequence in the central part of the target oligomers. A similar set of targets was also created where the nine nt in the 5 'end were changed into an InflA sequence and the central region was gradually changed from Norovirus towards Influenza sequence. Two targets have exactly the same amount of mismatches compared to the Noro- or the InflA probe but the (10.10) 26 10 26 (0.8) target differs in the distribution of mismatches compared to the (11.0) 26_10_26 (9.8) (see Fig. 13C).
The InflA probe and (0.10) 35 1 17 (9.8) target with its 17 mm compare to the InflA probe hybridized, although with a reduced capacity (MFI 2495, 37%) (45°C; Fig.13 A and C).
Increasing the amount of mm to 26 mm abolished the signal between the
(0.11) 26 10 26 (9.8) target and the InflA probe (MFI 33), while the Norovirus probe, also with 26 mm, still gave a weak signal (MFI 449, 6,7%) against the (0.1 1) 26_10_26 (9.8) target. Introduction of dlnosine in the Influenza probe, Inol 8, could not rescue the binding to (0.11) 26_10_26 (9.8) (MFI 93, 1%) since even though the dinosines mask 16 mismatches 10 mmoi still remain in between the dinosines. To the other 26 mismatching nt containing target (10.10) 26 10 26 (0.8), which has nine mismatches placed in the 5' end and one in the 3'end (10 mmoi) and 16 mm distributed in the central region, compared to the InflA probe, gave a weak signal of 301 MFI (4,5%) when utilizing the InflA probe. Further, these centrally placed 16 mm are matching the position of dlnosine in the Inol 8 probe which caused Inol 8 to hybridize with great affinity (MFI 3835, 57%). The Noro probe having two nucleation sites of 10 nt in the 5' and 3' end flanking the central region of 26 mm in the (10.10) 26_10_26 (0.8) target gave only a weak hybridization signal of MFI 849 (13%). Thus the crosshybridization of a foreign dlnosine-containing probe is dependent of the amount of mm, but even more of the distribution of mmoi here demonstrated with the different distribution of 26 mm causing either zero or 10 mmoi placed in between the position of the dinosines resulting in either good hybridization or no hybridization, respectively.
The target (10.10) 22_14_30 (0.8) and the InflA probe without dinosines could not hybridize since the target contains as many as 30 mismatches in total. Covering 16nt of these mismatches with dinosines using the Influenza based probe Inol8, gave a significant signal of 1827 MFI (27%). The Inol8 probe gave some but weak signal (MFI 765, 1 1%) even with a target with 31 mm, (10.10) 21 15 31 (0.8). The (10.10)_22_14_30_(0.8) target has less mm against the Noro probe than the InflA probe and still the InflA based Inol 8 probe gives a higher MFI than the Noro probe, MFI 1827 compared to MFI 897.
The Fig. 13D, shows the origin of the nt not covered by the dinosines when using the Inol 8 probe (MFI values from Fig. 13 A and Fig. 13C). The graph demonstrates how many InflA matching nt outside the dlnosine postions that are needed for hybridization and how many Norovirus matching nt that could cause a crosshybridization. 16 nt is in common of the two virus specific targets, InflA and Noro. At least 37-38 InflA matching nt seems to be needed to get an increase of the MFI signal utilizing the Inol 8 probe, but as shown previously (see
above) the distribution of matching and mismatching nt are of more importance than the actual number, demonstrated with the two targets (10.10) 26_10_26 (0.8) and
(0.1 1) 26_10_26 (9.8). Both of these targets contain 42 Influenza matching nt outside the dinosine position but hybridize with very different efficiency. The number of Noro virus-like nt giving raise to signal with the InflA based Inol8-probe was shown to be 30-31 nt, where of 16 of these are shared with the InflA sequence and thereby still matches the probe and only 14-15nt are mismatching.
Conclusively, here we demonstrate that a target with as many as 30 mm can be efficiently hybridized to a dlnosine-containing probe, Inol 8, as long as the amount of mismatches neighbouring the dinosine are few.
Example 7 - construction and study of 5'-nitroindole and degenerated probes
Single stranded 70-mer oligonucleotides with a C12 aminolink at the 5' end, with or without dlnosines / 5 ' -nitroindole / N wobbles, were obtained from Biomers.net (Ulm, Germany). The synthetic 5' amine-C12 modified 70-mer probes were coupled to xMAP carboxylated colour- coded microspheres (Luminex Corp., Austin TX, USA), according to the protocol of Luminex corp (Austin TX, USA). The probes were hybridized to biotinylated targets as described in Example 6, and the MFI was measured in a Luminex®200™ flow cytometer (Luminex corporation, Austin Tx).
Result
The hybridization of a long probe containing dlnosines is comparable with that of a long degenerated probe with the same number of N wobbles, under lower stringency conditions The effects of probes with dinosine or wobbles in the same positions were also investigated in 3M TMAC. The presence of a dinosine in a specific position instead of a wobble would theoretically decrease the degeneration of the probe and subsequently increase the
concentration of the particular probe variant. A probe with 21 N wobbles, wobbN_21, at the same positions as the dlnosines in the Ino21 probe, was tested (Fig 21 and Fig. 11C). The surprising result was that the probe containing N wobbles hybridized very well with the InflA target (29% of the total MFI, 2220 MFI, Fig. 21) and the 21pm target (29% of the total MFI, 2190 MFI, Fig. 21). This is in the same range as hybridization of the Ino21 probe with the InflA (26% of the total MFI, 1976 MFI, Fig. 21) and 21pm (1 1% of the total MFI, 794 MFI,
Fig. 21) targets (39% and 37% of the total MFI versus the InflA and the 21pm targets, see Fig 11 A). These results also demonstrate that the wobb_N21 probe is not affected to the same extent as the Ino21 probe by increasing the hybridization temperature from 45 °C to 55 °C (Fig. 21). The test was repeated by comparing an Inol8 with a wobbN_18 probe (Fig. 22 and Fig. 12C). At 45 °C, the Inol 8 probe hybridized at least as well as the wobbN_18 probe while, at 55 °C, the wobbN_18 probe hybridized better than the Ino 18 probe. Interestingly, a probe containing 24 wobbles still hybridized better with all 33%xnt and 33%xF targets (30- 45% of the total MFI at 45 °C; 13-22% of total MFI at 55 °C; Fig 22) compared with Ino24 (6-16% of the total MFI at 45 °C; 1% of the total MFI at 55 °C; Fig 12A and B). Obviously, the 70mer probes can accommodate multiple degenerate positions and still hybridize because the majority of probe molecules will contain several long perfectly matching stretches created by chance.
The hybridization of a long probe containing dlno sines is stronger than that of a long probe containing the same amount of 5-Nitroindole, at either high or low temperatures
5-Nitroindole is a second-generation universal base nt analogue that was chosen for comparison with the first-generation dlnosine with respect to hybridization properties in 3M TMAC. A probe with 18 5-nitroindole residues (5-NitroInd_l 8) was designed; the nt analogues were distributed to match the pattern of the dlnosines in the Inol 8 probe (Fig. 12C). The probes were allowed to hybridize with the InflA target and the set of targets with synonymous mutations, 33%_xF and 33%_xnt (Fig 22). At 45 °C, hybridization of 5- NitroInd_18 with the InflA (44% of the total MFI), 33%_xF (23-37% of the total MFI), and 33%_xnt (4-7% of the total MFI) targets resulted in hybridization signals that were much lower than those seen with the Inol 8/InflA (73% of the total MFI), Inol8/33%_xF (54-78% of the total MFI), and Inol8/33%_xnt (47-59% of the total MFI) probes (Fig. 22, 45°C). Increasing the temperature to 55°C destabilized the 5-NitroInd_l 8 probe even more, resulting in hybridization of only 1-8% of the total MFI. In conclusion, dlnosine functions much better than 5-nitroindole as a universal nt analogue, under 3 M TMAC buffer conditions
The effects of the universal base dlnosine were compared with those of N wobbles. Thus, at the lower temperature, a dlnosine-containing probe hybridized more strongly and, at the higher temperature, the N wobble probe hybridized more strongly. To understand how the highly degenerated probes, wobbN_21, wobbN_18 and wobb_N24, hybridized so well, we calculated the probability of randomly achieving an extension of the matching regions at the
5' and 3' ends of the wobbN_l 8 probe (Table 20). The probability that the closest N wobble to either the 5' or 3' end would be a perfect match is 0.5. Thus, 50% of the pool of degenerated probes have a 3nt longer perfect match (12 + 9 or 9 + 12 matching nt at the 5' and 3' flanking regions) which, according to our results with nondegenerated probe/target combinations, should lead to rather good hybridization of the wobbN_18. In fact, wobbN_18 hybridization was similar in strength to that of the InflA probe to the 33%12F or 33%15nt targets. Further, the probability of having several additional 5nt matching regions in the central region is also high, probably giving rise to many more combinations in the same pool that matched even better. By restricting the wobbles to 3 (e.g a D or a B) or 2 (e.g. a Y or a T) nt, the probability of a match becomes even greater. Thus, in a highly degenerated probe with at least one continuous region of perfectly matching nt, a large part of the pool will extend this region and contribute to nucleation, zipping and hybridization. The behaviour of the highly degenerated probes is encouraging and in accordance with the NucZip model, which predicts that the high likelihood of several matching stretches of 5 nt or longer will result in significant hybridization.
Example 8 - NucZip
NucZip is performed in two steps. Nucleation is first tried. Candidates for nucleation are then used for zipping, up- and downstream of the nucleation candidate sequence. Parameter values were reached by recursive testing against measurements reported in the Ohrmalm et al., Nucleic Acids Research, 2010, 1-23. Fig. 23 shows a schematic model over the NucZip algorithm and Fig. 25 shows a schematic flowchart over the NucZip algorithm.
The score for the 'Zip' portion of the model is obtained from the number of consecutive matching trimers, tetramers etc, up to pentadecamers, each counted with equal weight, within a contiguous matching segment. Thus, ZipScoremodex =∑k lk=kmax(∑n 3 n=i5 Sn), where kmax is the number of uninterrupted matching segments, including the chosen nucleation site, and Sn is the number of successive segments of length n (varying from 3-15), i.e. the number of full length trimers, full length tetramers etc. up to full length pentadecamers, which fit into the matching segment. The same Zip scoring system was performed in two modes, counting dlnosines either as matching (mode 1) or as mismatching (mode 2). In the second mode, dlnosines will shorten the length of matching segments, decreasing the score. The final ZipScore was calculated as a weighted mean of ZipScorem0dei and ZipScoremode2> where the weighting factor was based on the empirical data presented in the current report (Fig. 2, 3 and 4). Since dlnosine hybridizes more strongly to C than to the other nts, the algorithm adds a contribution based on the number of dI:C pairs weighted by an InoCfactor. The upstream and downstream ZipScores were obtained as: ZipScore downstream = ZipScoremodei - dlnosinefactor (ZipScoremodei - ZipScoremode2) + (dlnoCnr * dlnoCfactor) and ZipScore upstream = ZipScoremodei - dlnosinefactor (ZipScoremodei - ZipScoremode2) + (dlnoCnr * dlnoCfactor). The ZipScores from up- and downstream zipping were then added; the final NucZipScore = ZipScore downstream + ZipScore upstream.





1. Akhras, M. S., S. Thiyagarajan, A. C. Villablanca, R. W. Davis, P. Nyren, and N.
Pourmand. 2007. PathogenMip assay: a multiplex pathogen detection assay. PLoS ONE 2:e223.
2. Akhras, M. S., M. Unemo, S. Thiyagarajan, P. Nyren, R. W. Davis, A. Z. Fire, and N. Pourmand. 2007. Connector inversion probe technology: a powerful one- primer multiplex DNA amplification system for numerous scientific applications. PLoS ONE 2:e915.
3. Baner, J., P. Gyarmati, A. Yacoub, M. Hakhverdyan, J. Stenberg, O. Ericsson, M. Nilsson, U. Landegren, and S. Belak. 2007. Microarray-based molecular detection of foot-and-mouth disease, vesicular stomatitis and swine vesicular disease viruses, using padlock probes. J Virol Methods 143:200-6.
4. Blab, G. A., T. Schmidt, and M. Nilsson. 2004. Homogeneous detection of single rolling circle replication products. Anal Chem 76:495-8.
5. Bovers, M., M. R. Diaz, F. Hagen, L. Spanjaard, B. Duim, C. E. Visser, H. L.
Hoogveld, J. Scharringa, I. M. Hoepelman, J. W. Fell, and T. Boekhout. 2007. Identification of genotypically diverse Cryptococcus neoformans and Cryptococcus gattii isolates by Luminex xMAP technology. J Clin Microbiol 45: 1874-83.
6. Brown, I. H. 2006. Advances in molecular diagnostics for avian influenza. Dev Biol (Basel) 124:93-7.
7. Chiu, C. Y., A. Urisman, T. L. Greenhow, S. Rouskin, S. Yagi, D. Schnurr, C.
Wright, W. L. Drew, D. Wang, P. S. Weintrub, J. L. Derisi, and D. Ganem. 2008. Utility of DNA microarrays for detection of viruses in acute respiratory tract infections in children. J Pediatr 153:76-83.
8. Dankbar, D. M., E. D. Dawson, M. Mehlmann, C. L. Moore, J. A. Smagala, M.
W. Shaw, N. J. Cox, R. D. Kuchta, and K. L. Rowlen. 2007. Diagnostic microarray for influenza B viruses. Anal Chem 79:2084-90.
9. Das, S., T. M. Brown, K. L. Kellar, B. P. Holloway, and C. J. Morrison. 2006.
DNA probes for the rapid identification of medically important Candida species using a multianalyte profiling system. FEMS Immunol Med Microbiol 46:244-50.
10. Deregt, D., S. A. Gilbert, S. Dudas, J. Pasick, S. Baxi, K. M. Burton, and M. K.
Baxi. 2006. A multiplex DNA suspension microarray for simultaneous detection and differentiation of classical swine fever virus and other pestiviruses. J Virol Methods 136: 17-23.
11. Diaz, M. R., T. Boekhout, B. Theelen, M. Bovers, F. J. Cabanes, and J. W. Fell.
2006. Microcoding and flow cytometry as a high-throughput fungal identification system for Malassezia species. J Med Microbiol 55: 1197-209.
12. Diaz, M. R., and J. W. Fell. 2004. High-throughput detection of pathogenic yeasts of the genus trichosporon. J Clin Microbiol 42:3696-706.
13. Dunbar, S. A. 2006. Applications of Luminex xMAP technology for rapid, high- throughput multiplexed nucleic acid detection. Clin Chim Acta 363:71-82.
14. Han, J., D. C. Swan, S. J. Smith, S. H. Lum, S. E. Sefers, E. R. Unger, and Y. W.
Tang. 2006. Simultaneous amplification and identification of 25 human
papillomavirus types with Templex technology. J Clin Microbiol 44:4157-62.
15. Han, X., X. Lin, B. Liu, Y. Hou, J. Huang, S. Wu, J. Liu, L. Mei, G. Jia, and Q.
Zhu. 2008. Simultaneously subtyping of all influenza A viruses using DNA microarrays. J Virol Methods 152: 117-21.
16. Hardenbol, P., J. Baner, M. Jain, M. Nilsson, E. A. Namsaraev, G. A. Karlin- Neumann, H. Fakhrai-Rad, M. Ronaghi, T. D. Willis, U. Landegren, and R. W.
Davis. 2003. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat Biotechnol 21:673-8.
Hardenbol, P., F. Yu, J. Belmont, J. Mackenzie, C. Bruckner, T. Brundage, A. Boudreau, S. Chow, J. Eberle, A. Erbilgin, M. Falkowski, R. Fitzgerald, S.
Ghose, O. lartchouk, M. Jain, G. Karlin-Neumann, X. Lu, X. Miao, B. Moore, M. Moorhead, E. Namsaraev, S. Pasternak, E. Prakash, K. Tran, Z. Wang, H. B. Jones, R. W. Davis, T. D. Willis, and R. A. Gibbs. 2005. Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res 15:269-75.
Jiang, H. L., H. H. Zhu, L. F. Zhou, F. Chen, and Z. Chen. 2006. Genotyping of human papillomavirus in cervical lesions by LI consensus PCR and the Luminex xMAP system. J Med Microbiol 55:715-20.
Kohara, Y. 2003. Hybridization reaction kinetics of DNA probes on beads arrayed in a capillary enhanced by turbulent flow. Anal Chem 75:3079-85.
Kumar, S., L. Wang, J. Fan, A. Kraft, M. E. Bose, S. Tiwari, M. Van Dyke, R. Haigis, T. Luo, M. Ghosh, H. Tang, M. Haghnia, E. L. Mather, W. G. Weisburg, and K. J. Henrickson. 2008. Detection of 11 Common Viral and Bacterial Pathogens Causing Community-Acquired Pneumonia or Sepsis in Asymptomatic Patients Using a Multiplex Reverse Transcriptase PCR Assay with Manual (Enzyme Hybridization) or Automated Detection (Electronic Microarray). J Clin Microbiol.
Li, H., M. A. McCormac, R. W. Estes, S. E. Sefers, R. K. Dare, J. D. Chappell, D. D. Erdman, P. F. Wright, and Y. W. Tang. 2007. Simultaneous detection and high- throughput identification of a panel of RNA viruses causing respiratory tract infections. J Clin Microbiol 45:2105-9.
Lund, V., R. Schmid, D. Rickwood, and E. Homes. 1988. Assessment of methods for covalent binding of nucleic acids to magnetic beads, Dynabeads, and the characteristics of the bound nucleic acids in hybridization reactions. Nucleic Acids Res 16: 10861-80.
Lundeberg, J., and F. Larsen. 1995. Solid-phase technology: magnetic heads to improve nucleic acid detection and analysis. Biotechnol Annu Rev 1:373-401.
Meuzelaar, L. S., O. Lancaster, J. P. Pasche, G. Kopal, and A. J. Brookes. 2007. MegaPlex PCR: a strategy for multiplex amplification. Nat Methods 4:835-7.
Millard, P. J., L. E. Bickerstaff, S. E. LaPatra, and C. H. Kim. 2006. Detection of infectious haematopoietic necrosis virus and infectious salmon anaemia virus by molecular padlock amplification. J Fish Dis 29:201-13.
Mullis, K., F. Faloona, S. Scharf, R. Saiki, G. Horn, and H. Erlich. 1986. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51 Pt 1:263-73.
Page, B. T., and C. P. Kurtzman. 2005. Rapid identification of Candida species and other clinically important yeast species by flow cytometry. J Clin Microbiol 43:4507- 14.
Page, B. T., C. E. Shields, W. G. Merz, and C. P. Kurtzman. 2006. Rapid identification of ascomycetous yeasts from clinical specimens by a molecular method based on flow cytometry and comparison with identifications from phenotypic assays. J Clin Microbiol 44:3167-71.
Pemov, A., H. Modi, D. P. Chandler, and S. Bavykin. 2005. DNA analysis with multiplex microarray-enhanced PCR. Nucleic Acids Res 33:el 1.
Saiki, R. K., D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis, and H. A. Erlich. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487-91.
31. Segal, B. H., and T. J. Walsh. 2006. Current approaches to diagnosis and treatment of invasive aspergillosis. Am J Respir Crit Care Med 173:707-17.
32. Sengupta, S., K. Onodera, A. Lai, and U. Melcher. 2003. Molecular detection and identification of influenza viruses by oligonucleotide microarray hybridization. J Clin Microbiol 41:4542-50.
33. Striebel, H. M., E. Birch-Hirschfeld, R. Egerer, and Z. Foldes-Papp. 2003. Virus diagnostics on microarrays. Curr Pharm Biotechnol 4:401-15.
34. Thiyagarajan, S., M. Karhanek, M. Akhras, R. W. Davis, and N. Pourmand.
2006. PathogenMIPer: a tool for the design of molecular inversion probes to detect multiple pathogens. BMC Bioinformatics 7:500.
35. Wang, L. C, C. H. Pan, L. L. Severinghaus, L. Y. Liu, C. T. Chen, C. E. Pu, D.
Huang, J. T. Lir, S. C. Chin, M. C. Cheng, S. H. Lee, and C. H. Wang. 2008. Simultaneous detection and differentiation of Newcastle disease and avian influenza viruses using oligonucleotide microarrays. Vet Microbiol 127:217-26.
36. Wilkins Stevens, P., M. R. Henry, and D. M. Kelso. 1999. DNA hybridization on microparticles: determining capture-probe density and equilibrium dissociation constants. Nucleic Acids Res 27: 1719-27.
37. Wolf, S. F., L. Haines, J. Fisch, J. N. Kremsky, J. P. Dougherty, and K. Jacobs.
1987. Rapid hybridization kinetics of DNA attached to submicron latex particles. Nucleic Acids Res 15:291 1-26.
38. Zhang, D., J. Wu, F. Ye, T. Feng, I. Lee, and B. Yin. 2006. Amplification of
circularizable probes for the detection of target nucleic acids and proteins. Clin Chim Acta 363:61-70.
39. Zhang, D. Y., and B. Liu. 2003. Detection of target nucleic acids and proteins by amplification of circularizable probes. Expert Rev Mol Diagn 3:237-48.
Tables

















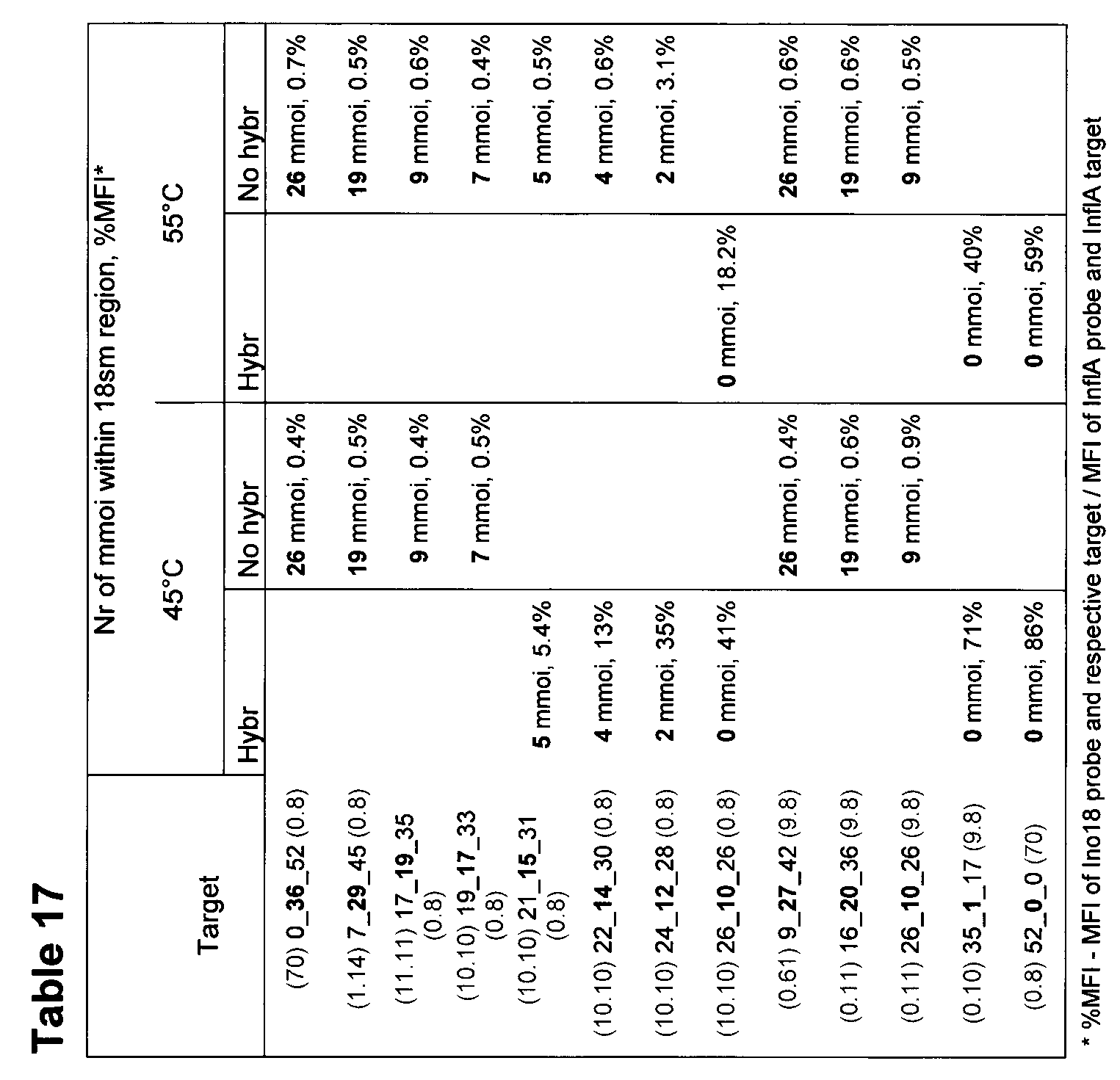



Claims
1. A method for designing the nucleotide sequence of a polynucleotide primer/probe capable of specific and mismatch-tolerant hybridization to a group of target polynucleotides, comprising identifying a nucleation site of 6-9 nucleotides where all or substantially all of the target polynucleotides have identical sequences and building a nucleotide sequence by
- positioning nucleotides A, C, T and G in positions within the nucleation site so as to form a sequence complementary to all or substantially all of the target
polynucleotides;
- positioning nucleotides A, C, T and G in positions outside the nucleation site where all or substantially all target polynucleotides have the same nucleotide;
- positioning N Universal Base Analogues (UBAs) at sites of sequence variations
between the target polynucleotides, wherein N is an integer between 0 and 1/3 of the length of the primer/probe, so that there are no more than N/3 mismatches between said primer/probe and said target in positions next to a position of an UBA.
- positioning M degenerate nucleotide positions at sites of variation between target polynucleotides in a way that maximizes the likelihood of, and number of, perfectly matching stretches longer than four nucleotides, wherein M is an integer between 0 and 1/3 of the length of the primer/probe;
thereby obtaining a continuous polynucleotide primer/probe sequence.
2. Computer program product comprising instructions to perform the method of claim 1.
3. Polynucleotide molecule having a sequence obtainable with the method of claim 1.
4. A polynucleotide primer/probe capable of specific and mismatch-tolerant hybridization to a target polynucleotide, wherein said primer/probe comprises
- at least one nucleation site of 6-9 nucleotides being complementary to a site in the target polynucleotide and
- N Universal Base Analogues (UBAs) at positions of sequence variations between the target and the primer/probe, wherein N is an integer between 0 and 1/3 of the length of the primer/probe; - M degenerate positions positioned to create a high likelihood of random creation of at least one contiguous complementary stretch of more than four nucleotides, wherein M is an integer between 0 and 1/3 of the length of the primer/probe;
5. Polynucleotide primer/probe comprising one part capable of specific and mismatch-tolerant hybridization to a target polynucleotide and one part comprising a generic forward primer sequence, said generic forward primer sequence not being present in, or complementary to, said target polynucleotide.
6. Polynucleotide according to any of claims 3-6, wherein said polynucleotide is bound to a solid phase, such as a magnetic bead.
7. Method for detecting a target nucleic acid molecule in a sample, comprising the steps
- bringing said sample in contact with a polynucleotide according to any of claims 5-6, a target specific mismatch tolerant reverse primer and a second forward primer capable of hybridizing to said generic primer sequence, a RNA reverse transcriptase or a DNA polymerase, nucleotides and optionally other amplification additives;
- optionally, enrichment of target nucleic acid molecule bound to a polynucleotide
according to any of claims 5-6 which is bound to a solid phase, such as a magnetic bead, and subsequently separated from other sample nucleic acids by displacement of the solid phase on which subsequent detection steps take place;
- optionally, increasing the specificity of detection by spatial restriction of the binding of target specific and generic primers, described in the following points, by initiating the first round of amplification on the solid phase;
- initiating a first forward replication round primed by the complex formed by the target nucleic acid molecule and the specific and mismatch-tolerant part of the primer/probe, thereby forming a first replicated nucleic acid molecule;
- initiating a first reverse replication round primed by the complex formed by the first replicated nucleic acid molecule and the target specific reverse primer, thereby forming a second replicated nucleic acid molecule;
- initiating a second forward replication round primed by the complex formed by the second replicated nucleic acid molecule and the second forward primer;
- amplifying said replicated nucleic acid molecules forming amplimers; - detecting said amplimers.
8. Method according to claim 7, wherein said target specific mismatch tolerant reverse primer further comprises a generic reverse primer sequence and said amplification forming amplimers is performed with a primer comprising a sequence complementary to said generic reverse primer sequence.
9. Method according to claim 7 or 8, wherein said forward and/or reverse primers are labelled with an analytic tag or an affinity label.
10. Method according to any of claims 7-9, wherein the detection of the amplimers is performed by contacting the amplimers with a target specific mismatch tolerant
polynucleotide probe bound to a capture agent, allowing said probe to hybridize to the amplimers, separating the probe/amplimer-complex and detecting said complex.
11. Method according to claim 10, wherein the capture-agent is a colour-coded microsphere.
12. Method according to claim 10 or 11, wherein said forward and/or reverse primers are labelled with an affinity label, such as biotin, and further comprising contacting the amplimers with an affinity label binding partner, such as streptavidin, wherein said affinity label binding partner is labelled with an analytic tag, such as phycoerythrin.
13. Method for detecting a target nucleic acid molecule in a sample, comprising bringing said sample in contact with
-a molecular construct ("nanodevice") comprising a first and a second polynucleotide primer/probe joined, optionally through a linker, at their 5 'ends wherein said first polynucleotide primer/probe comprises a first generic primer sequence and a first target specific primer sequence, and said second polynucleotide primer/probe comprise a second generic primer sequence and a second target complement specific primer sequence; and -a R A reverse transcriptase or a DNA polymerase, nucleotides and optionally other amplification additives;
in a suitable buffer solution; said method further comprising the steps
-initiating a first replication round primed by the complex formed by the target nucleic acid molecule and the target specific primer sequence part of the first polynucleotide primer/probe, thereby forming a first replicated nucleic acid molecule;
-initiating a second replication round primed by the complex formed by the first
replicated nucleic acid molecule and the target complement specific primer sequence part of the second polynucleotide primer/probe, thereby forming a second replicated nucleic acid molecule;
-adding a third generic primer molecule with the same sequence the first generic primer sequence, and a fourth generic primer molecule with the same sequence as the second generic primer sequence;
-amplifying said replicated nucleic acid molecules forming amplimers;
-detecting said amplimers.
14. Method according to claim 13, wherein said first target specific primer sequence in said first polynucleotide primer/probe and/or said second target complement specific primer sequence in said second polynucleotide primer/probe is mismatch tolerant, such as a polynucleotide primer/probe according to claim 3 or 4.
15. Method according to claim 13 or 14, further comprising cleaving, by enzymatic, chemical or physical means, the first replicated nucleic acid molecule from the second replicated nucleic acid molecule before amplifying said replicated nucleic acid molecules.
16. Method according to any of claim 13-15, wherein said molecular construct further comprises said linker comprising a first and a second site susceptible to enzymatic, chemical or physical cleavage, said sites flanking an affinity label, such as biotin.
17. Method according to claim 16, further comprising contacting the molecular construct with at least one cleaving compound and allowing said cleaving compound to cleave the molecular construct at the sites susceptible to cleavage, and removing the part of the molecular construct comprising said affinity label, e.g. with a magnetic bead having bound to it a moiety with affinity for said affinity label, wherein said cleavage is performed after the second
amplification round and before amplifying said replicated nucleic acid molecules.
18. Method according to any of claims 13-17, wherein said third and/or fourth primer molecules are labelled with an affinity label.
19. Method according to claim 18, wherein the detection of the amplimers is performed by contacting the amplimers with a target specific mismatch tolerant polynucleotide probe bound to a capture agent, allowing said probe to hybridize to the amplimers, separating the probe/amplimer-complex and detecting said complex.
20. Method according to claim 19, wherein the capture-agent is a colour-coded microsphere.
21. Method according to claim 18- 20, wherein said forward and/or reverse primers are labelled with an affinity label, such as biotin, and further comprising contacting the amplimers with an affinity label binding partner, such as streptavidin, wherein said affinity label binding partner is labelled with an analytic tag, such as phycoerythrin.
22. Molecular construct comprising a first and a second polynucleotide primer/probe joined, optionally through a linker, at their 5 'ends wherein said first polynucleotide primer/probe comprises a first generic primer sequence and a first target specific primer sequence, and said second polynucleotide primer/probe comprise a second generic primer sequence and a second target complement specific primer sequence.
23. Molecular construct according to claim 22, wherein said linker comprises a first and a second site susceptible to enzymatic, chemical or physical cleavage, said sites flanking an affinity label, such as biotin.
24. A method for predicting mismatch-tolerance of a probe nucleic acid molecule, optionally comprising at least one UBA, when hybridising to a target nucleic acid molecule, comprising the steps
a) aligning the probe and target sequences;
b) localizing possible nucleation sites by assigning a nucleation score for each
stretch of nucleotides in the probe, said nucleation score equalling the number of continuous base pairs perfectly matching between the probe and target sequence; c) assigning a first zip score to the sequence downstream from a first nucleation site, said first zip score equalling the number of consecutive matching 3-15-mers; d) assigning a second zip score to the sequence downstream from the first
nucleation site, said second zip score equalling the number of consecutive perfectly matching 3-15-mers;
e) repeating steps c) and d) for the sequence upstream from the first nucleation site; f) assigning a total zip score to the nucleation site, said total zip score being a weighted mean of said first and second zip scores for said upstream and downstream sequences;
g) assigning a combined nucleation and zip score to the nucleation site;
h) optionally repeating steps c)-f) for other nucleation sites in the probe sequence.
25. Computer program product comprising instructions to perform the method of claim 24.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US25614409P | 2009-10-29 | 2009-10-29 | |
| US61/256,144 | 2009-10-29 | ||
| US37977410P | 2010-09-03 | 2010-09-03 | |
| US61/379,774 | 2010-09-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011053241A1 true WO2011053241A1 (en) | 2011-05-05 |
Family
ID=43922351
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2010/051184 WO2011053241A1 (en) | 2009-10-29 | 2010-10-29 | Multiplex detection |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011053241A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012153153A1 (en) * | 2011-05-11 | 2012-11-15 | Diagon Kft. | Procedure for rapid determination of viruses using nucleic acid-based molecular diagnostics, and a kit for this purpose |
| WO2013049891A1 (en) | 2011-10-04 | 2013-04-11 | Genera Biosystems Limited | Compositions and methods of detecting respiratory pathogens using nucleic acid probes and subsets of beads |
| WO2013134341A1 (en) | 2012-03-07 | 2013-09-12 | Dow Agrosciences Llc | Primer designing pipeline for targeted sequencing |
| US8883423B2 (en) * | 2010-10-22 | 2014-11-11 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| US9488648B2 (en) | 2010-10-22 | 2016-11-08 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| US9562271B2 (en) | 2012-04-20 | 2017-02-07 | T2 Biosystems, Inc. | Compositions and methods for detection of Candida species |
| WO2017035347A1 (en) * | 2015-08-25 | 2017-03-02 | The Broad Institute, Inc. | Screening of nucleic acid sequence variations via random loading of optically-encoded particles |
| US11519016B2 (en) | 2016-01-21 | 2022-12-06 | T2 Biosystems, Inc. | NMR methods and systems for the rapid detection of bacteria |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003107144A2 (en) * | 2002-06-18 | 2003-12-24 | Epoch Biosciences, Inc. | Systems and methods for predicting oligonucleotide melting temperatures (tms) |
| WO2006095881A1 (en) * | 2005-03-07 | 2006-09-14 | Ebara Corporation | Substrate processing method and substrate processing apparatus |
-
2010
- 2010-10-29 WO PCT/SE2010/051184 patent/WO2011053241A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003107144A2 (en) * | 2002-06-18 | 2003-12-24 | Epoch Biosciences, Inc. | Systems and methods for predicting oligonucleotide melting temperatures (tms) |
| WO2006095881A1 (en) * | 2005-03-07 | 2006-09-14 | Ebara Corporation | Substrate processing method and substrate processing apparatus |
Non-Patent Citations (8)
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9360457B2 (en) | 2010-10-22 | 2016-06-07 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| US9714940B2 (en) | 2010-10-22 | 2017-07-25 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| US9702852B2 (en) | 2010-10-22 | 2017-07-11 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| US8883423B2 (en) * | 2010-10-22 | 2014-11-11 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| US9488648B2 (en) | 2010-10-22 | 2016-11-08 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| US9046493B2 (en) | 2010-10-22 | 2015-06-02 | T2 Biosystems, Inc. | NMR systems and methods for the rapid detection of analytes |
| WO2012153153A1 (en) * | 2011-05-11 | 2012-11-15 | Diagon Kft. | Procedure for rapid determination of viruses using nucleic acid-based molecular diagnostics, and a kit for this purpose |
| JP2015506666A (en) * | 2011-10-04 | 2015-03-05 | ジェネラ バイオシステムズ リミテッド | Compositions and methods for detecting respiratory pathogens using nucleic acid probes and bead subsets |
| EP2764125A1 (en) * | 2011-10-04 | 2014-08-13 | Genera Biosystems Limited | Compositions and methods of detecting respiratory pathogens using nucleic acid probes and subsets of beads |
| EP2764125A4 (en) * | 2011-10-04 | 2015-06-10 | Genera Biosystems Ltd | Compositions and methods of detecting respiratory pathogens using nucleic acid probes and subsets of beads |
| WO2013049891A1 (en) | 2011-10-04 | 2013-04-11 | Genera Biosystems Limited | Compositions and methods of detecting respiratory pathogens using nucleic acid probes and subsets of beads |
| WO2013134341A1 (en) | 2012-03-07 | 2013-09-12 | Dow Agrosciences Llc | Primer designing pipeline for targeted sequencing |
| US9562271B2 (en) | 2012-04-20 | 2017-02-07 | T2 Biosystems, Inc. | Compositions and methods for detection of Candida species |
| US11098378B2 (en) | 2012-04-20 | 2021-08-24 | T2 Biosystems, Inc. | Compositions and methods for detection of candida species |
| WO2017035347A1 (en) * | 2015-08-25 | 2017-03-02 | The Broad Institute, Inc. | Screening of nucleic acid sequence variations via random loading of optically-encoded particles |
| US11421270B2 (en) | 2015-08-25 | 2022-08-23 | The Broad Institute, Inc. | Screening of nucleic acid sequence variations via random loading of optically-encoded particles |
| US11519016B2 (en) | 2016-01-21 | 2022-12-06 | T2 Biosystems, Inc. | NMR methods and systems for the rapid detection of bacteria |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011053241A1 (en) | Multiplex detection | |
| US9434997B2 (en) | Methods, compounds and systems for detecting a microorganism in a sample | |
| Kostyusheva et al. | CRISPR-Cas systems for diagnosing infectious diseases | |
| Urisman et al. | E-Predict: a computational strategy for species identification based on observed DNA microarray hybridization patterns | |
| Terribilini et al. | Prediction of RNA binding sites in proteins from amino acid sequence | |
| JP6270786B2 (en) | Chimeric primer with hairpin conformation and use thereof | |
| US8232058B2 (en) | Multiplex detection of respiratory pathogens | |
| CN101292044B (en) | Method and/or apparatus of oligonucleotide design and/or nucleic acid detection | |
| Wong et al. | Optimization and clinical validation of a pathogen detection microarray | |
| Sharma et al. | Recombinase‐based isothermal amplification of nucleic acids with self‐avoiding molecular recognition systems (SAMRS) | |
| Yang et al. | Conversion strategy using an expanded genetic alphabet to assay nucleic acids | |
| CA2768478A1 (en) | Detection of short rna sequences | |
| AU2016324473B2 (en) | Virome capture sequencing platform, methods of designing and constructing and methods of using | |
| WO2012037531A1 (en) | Capture probes immobilizable via l-nucleotide tail | |
| CA2539703A1 (en) | Detection of human papilloma virus (hpv) utilizing invasive cleavage structure assays | |
| Bertilsson et al. | Sequencing-independent method to generate oligonucleotide probes targeting a variable region in bacterial 16S rRNA by PCR with detachable primers | |
| Hansen et al. | Combination random isothermal amplification and nanopore sequencing for rapid identification of the causative agent of an outbreak | |
| Roth et al. | Expression profiling using a hexamer-based universal microarray | |
| Wang et al. | Resequencing microarray probe design for typing genetically diverse viruses: human rhinoviruses and enteroviruses | |
| Öhrmalm et al. | Variation-tolerant capture and multiplex detection of nucleic acids: application to detection of microbes | |
| Yaren et al. | Standard and AEGIS nicking molecular beacons detect amplicons from the Middle East respiratory syndrome coronavirus | |
| JP2008514236A (en) | Assay for detecting and quantifying HIV-1 | |
| Chhabra et al. | Molecular detection methods of foodborne viruses | |
| WO2010016071A2 (en) | Identification of genomic signature for differentiating highly similar sequence variants of an organism | |
| Hyman et al. | Competitive SNP-LAMP probes for rapid and robust single-nucleotide polymorphism detection |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10827248 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10827248 Country of ref document: EP Kind code of ref document: A1 |