WO2011048112A1 - Glycoside derivatives and uses thereof - Google Patents
Glycoside derivatives and uses thereof Download PDFInfo
- Publication number
- WO2011048112A1 WO2011048112A1 PCT/EP2010/065747 EP2010065747W WO2011048112A1 WO 2011048112 A1 WO2011048112 A1 WO 2011048112A1 EP 2010065747 W EP2010065747 W EP 2010065747W WO 2011048112 A1 WO2011048112 A1 WO 2011048112A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- tetrahydro
- phenyl
- pyran
- ylmethyl
- Prior art date
Links
- 229930182470 glycoside Natural products 0.000 title description 4
- 150000002338 glycosides Chemical class 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 256
- 238000000034 method Methods 0.000 claims abstract description 67
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 54
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims abstract description 52
- 201000010099 disease Diseases 0.000 claims abstract description 45
- 239000011734 sodium Substances 0.000 claims abstract description 27
- 238000011282 treatment Methods 0.000 claims abstract description 27
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims abstract description 26
- 206010012601 diabetes mellitus Diseases 0.000 claims abstract description 25
- 229910052708 sodium Inorganic materials 0.000 claims abstract description 25
- 102000003673 Symporters Human genes 0.000 claims abstract description 24
- 108090000088 Symporters Proteins 0.000 claims abstract description 24
- 230000001404 mediated effect Effects 0.000 claims abstract description 20
- 125000005843 halogen group Chemical group 0.000 claims description 159
- 125000001424 substituent group Chemical group 0.000 claims description 127
- -1 Ci-ealkyl Chemical group 0.000 claims description 125
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 125
- 150000003839 salts Chemical class 0.000 claims description 106
- 229910052739 hydrogen Inorganic materials 0.000 claims description 94
- 239000001257 hydrogen Substances 0.000 claims description 93
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 claims description 85
- 125000000623 heterocyclic group Chemical group 0.000 claims description 85
- 229960002429 proline Drugs 0.000 claims description 77
- 125000000217 alkyl group Chemical group 0.000 claims description 75
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical group C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 68
- 239000013078 crystal Substances 0.000 claims description 65
- 229930182821 L-proline Natural products 0.000 claims description 56
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 55
- 150000002431 hydrogen Chemical group 0.000 claims description 53
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 50
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 49
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 46
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 44
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 44
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 40
- 229910052757 nitrogen Inorganic materials 0.000 claims description 38
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 37
- 239000008103 glucose Substances 0.000 claims description 37
- 125000001072 heteroaryl group Chemical group 0.000 claims description 37
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 37
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 36
- 125000003118 aryl group Chemical group 0.000 claims description 33
- 229910052799 carbon Inorganic materials 0.000 claims description 33
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 32
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 31
- 125000004429 atom Chemical group 0.000 claims description 31
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 31
- 239000003446 ligand Substances 0.000 claims description 30
- 239000000843 powder Substances 0.000 claims description 30
- XFJAMQQAAMJFGB-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3OCCOC3=CC=2)C(CC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O XFJAMQQAAMJFGB-ZQGJOIPISA-N 0.000 claims description 28
- 125000004043 oxo group Chemical group O=* 0.000 claims description 27
- 239000003112 inhibitor Substances 0.000 claims description 25
- 125000003003 spiro group Chemical group 0.000 claims description 25
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 claims description 22
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 claims description 22
- 239000003795 chemical substances by application Substances 0.000 claims description 20
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 19
- 150000001721 carbon Chemical group 0.000 claims description 19
- 125000002950 monocyclic group Chemical group 0.000 claims description 19
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims description 18
- 150000003527 tetrahydropyrans Chemical group 0.000 claims description 18
- 239000003814 drug Substances 0.000 claims description 17
- 102000004877 Insulin Human genes 0.000 claims description 16
- 108090001061 Insulin Proteins 0.000 claims description 16
- 206010022489 Insulin Resistance Diseases 0.000 claims description 16
- 125000005842 heteroatom Chemical group 0.000 claims description 16
- 229940125396 insulin Drugs 0.000 claims description 16
- 229910052760 oxygen Inorganic materials 0.000 claims description 16
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 13
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 12
- 208000008589 Obesity Diseases 0.000 claims description 12
- 238000000113 differential scanning calorimetry Methods 0.000 claims description 12
- 235000020824 obesity Nutrition 0.000 claims description 12
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 12
- YNCHESVKDKRUDJ-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-cyclopropyl-3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C(C=C1CC=2C=C3OCCOC3=CC=2)=CC=C1C1CC1 YNCHESVKDKRUDJ-SJSRKZJXSA-N 0.000 claims description 11
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 claims description 11
- 229940123208 Biguanide Drugs 0.000 claims description 11
- 229920001268 Cholestyramine Polymers 0.000 claims description 11
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 claims description 11
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 claims description 11
- 101000684208 Homo sapiens Prolyl endopeptidase FAP Proteins 0.000 claims description 11
- 229940122199 Insulin secretagogue Drugs 0.000 claims description 11
- 239000003888 alpha glucosidase inhibitor Substances 0.000 claims description 11
- 229940125753 fibrate Drugs 0.000 claims description 11
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 11
- 229960003512 nicotinic acid Drugs 0.000 claims description 11
- 235000001968 nicotinic acid Nutrition 0.000 claims description 11
- 239000011664 nicotinic acid Substances 0.000 claims description 11
- ABADUMLIAZCWJD-UHFFFAOYSA-N 1,3-dioxole Chemical compound C1OC=CO1 ABADUMLIAZCWJD-UHFFFAOYSA-N 0.000 claims description 10
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 claims description 10
- XNCOSPRUTUOJCJ-UHFFFAOYSA-N Biguanide Chemical compound NC(N)=NC(N)=N XNCOSPRUTUOJCJ-UHFFFAOYSA-N 0.000 claims description 10
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims description 10
- 229940122355 Insulin sensitizer Drugs 0.000 claims description 10
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 claims description 10
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 claims description 10
- 102000018692 Sulfonylurea Receptors Human genes 0.000 claims description 10
- 108010091821 Sulfonylurea Receptors Proteins 0.000 claims description 10
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 10
- 229960001138 acetylsalicylic acid Drugs 0.000 claims description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 10
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims description 10
- 239000004026 insulin derivative Substances 0.000 claims description 10
- 230000002473 insulinotropic effect Effects 0.000 claims description 10
- 239000004059 squalene synthase inhibitor Substances 0.000 claims description 10
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 9
- 208000035475 disorder Diseases 0.000 claims description 9
- BOIWCSUQYQXIIP-DFLSAPQXSA-N (2s,3r,4r,5s,6r)-2-[3-[(4-benzyl-2,3-dihydro-1,4-benzoxazin-6-yl)methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3N(CC=4C=CC=CC=4)CCOC3=CC=2)=C1 BOIWCSUQYQXIIP-DFLSAPQXSA-N 0.000 claims description 8
- 208000001145 Metabolic Syndrome Diseases 0.000 claims description 8
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 8
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 claims description 8
- 125000002619 bicyclic group Chemical group 0.000 claims description 8
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 8
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 8
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 8
- 238000002560 therapeutic procedure Methods 0.000 claims description 8
- 206010020772 Hypertension Diseases 0.000 claims description 7
- 125000003342 alkenyl group Chemical group 0.000 claims description 7
- RHXPDNGQJSXOMW-OIIXUNCGSA-N (2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6r)-6-(2-cyclohexylethoxy)-4,5-dihydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)O[C@@H](OCCC2CCCCC2)[C@H](O)[C@H]1O RHXPDNGQJSXOMW-OIIXUNCGSA-N 0.000 claims description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical group C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 6
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 6
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 6
- 208000032928 Dyslipidaemia Diseases 0.000 claims description 6
- 125000004171 alkoxy aryl group Chemical group 0.000 claims description 6
- 125000003545 alkoxy group Chemical group 0.000 claims description 6
- 125000002947 alkylene group Chemical group 0.000 claims description 6
- 125000000304 alkynyl group Chemical group 0.000 claims description 6
- 230000003247 decreasing effect Effects 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 208000011580 syndromic disease Diseases 0.000 claims description 6
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 6
- 208000002249 Diabetes Complications Diseases 0.000 claims description 5
- 201000001431 Hyperuricemia Diseases 0.000 claims description 5
- 208000017170 Lipid metabolism disease Diseases 0.000 claims description 5
- 241000124008 Mammalia Species 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 5
- VDGBFZJBLBGSAP-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[4-cyclopropyl-3-(3,4-dihydro-2h-chromen-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C(C=C1CC=2C=C3CCCOC3=CC=2)=CC=C1C1CC1 VDGBFZJBLBGSAP-RXFVIIJJSA-N 0.000 claims description 4
- PGCVETRMLZSUHR-OOIHMXSWSA-N 4-benzyl-6-[[2-cyclopropyl-5-[(2s,3s,4r,5r,6r)-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-yl]phenyl]methyl]-2,3-dihydro-1,4-benzoxazine Chemical compound C([C@@H]1[C@H]([C@H](OCC=2C=CC=CC=2)[C@@H](OCC=2C=CC=CC=2)[C@@H](O1)C=1C=C(CC=2C=C3N(CC=4C=CC=CC=4)CCOC3=CC=2)C(C2CC2)=CC=1)OCC=1C=CC=CC=1)OCC1=CC=CC=C1 PGCVETRMLZSUHR-OOIHMXSWSA-N 0.000 claims description 4
- 206010012655 Diabetic complications Diseases 0.000 claims description 4
- 102000016267 Leptin Human genes 0.000 claims description 4
- 108010092277 Leptin Proteins 0.000 claims description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 4
- ZECVUQDNEANOBO-LUTHVSDMSA-N [(2r,3r,4r,5s)-3,4,5-triacetyloxy-6-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]oxan-2-yl]methyl acetate Chemical compound C1=C(CC=2C=C3OCCOC3=CC=2)C(CC)=CC=C1C1O[C@H](COC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O ZECVUQDNEANOBO-LUTHVSDMSA-N 0.000 claims description 4
- WGTFHBOPFPZZFH-BXZFMWHFSA-N [(2r,3r,4r,5s)-3,4,5-triacetyloxy-6-[4-cyclopropyl-3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)OC1C(C=C1CC=2C=C3OCCOC3=CC=2)=CC=C1C1CC1 WGTFHBOPFPZZFH-BXZFMWHFSA-N 0.000 claims description 4
- DQXUKCYHHRKAIR-ZCCUTQAASA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-bromo-3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Br)C(CC=2C=C3OCCOC3=CC=2)=C1 DQXUKCYHHRKAIR-ZCCUTQAASA-N 0.000 claims description 4
- 230000037396 body weight Effects 0.000 claims description 4
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 claims description 4
- 229940039781 leptin Drugs 0.000 claims description 4
- 125000003386 piperidinyl group Chemical group 0.000 claims description 4
- 125000004187 tetrahydropyran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 4
- 201000005665 thrombophilia Diseases 0.000 claims description 4
- 230000004580 weight loss Effects 0.000 claims description 4
- WRMXDAVTEAMGLW-RQKPWJHBSA-N (2s,3r,4r,5s,6r)-2-[3-[(4-benzylspiro[3h-1,4-benzoxazine-2,1'-cyclopropane]-6-yl)methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3N(CC=4C=CC=CC=4)CC4(CC4)OC3=CC=2)=C1 WRMXDAVTEAMGLW-RQKPWJHBSA-N 0.000 claims description 3
- GPAOTEWJXDLZRN-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(1,2,3,4-tetrahydroquinolin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CCCNC3=CC=2)=C1 GPAOTEWJXDLZRN-BDHVOXNPSA-N 0.000 claims description 3
- JYOHAXKLLUXGRH-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-cyclopropyl-3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C(C=C1CC=2C=C3NCCOC3=CC=2)=CC=C1C1CC1 JYOHAXKLLUXGRH-SJSRKZJXSA-N 0.000 claims description 3
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 3
- BAAZPPLWASSJBL-BDHVOXNPSA-N 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-2,3-dihydrochromen-4-one Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3C(=O)CCOC3=CC=2)=C1 BAAZPPLWASSJBL-BDHVOXNPSA-N 0.000 claims description 3
- MZSXGYZCLGHHIZ-AOALBDIGSA-N 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-3,4-dihydro-2h-1,4-benzoxazine-2-carbonitrile Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCC(OC3=CC=2)C#N)=C1 MZSXGYZCLGHHIZ-AOALBDIGSA-N 0.000 claims description 3
- FSRJARNFARVFPC-BDHVOXNPSA-N 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]chromen-4-one Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3C(=O)C=COC3=CC=2)=C1 FSRJARNFARVFPC-BDHVOXNPSA-N 0.000 claims description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 claims description 3
- 239000007983 Tris buffer Substances 0.000 claims description 3
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 claims description 3
- 125000005055 alkyl alkoxy group Chemical group 0.000 claims description 3
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Chemical group C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 claims description 3
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 3
- 125000001624 naphthyl group Chemical group 0.000 claims description 3
- RUVCJLHNBYZJHZ-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-methoxyphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3OCCOC3=CC=2)C(OC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O RUVCJLHNBYZJHZ-BDHVOXNPSA-N 0.000 claims description 2
- KSOOOXHCWVAMAA-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-4-propan-2-ylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCOC3=CC=2)C(C(C)C)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O KSOOOXHCWVAMAA-SJSRKZJXSA-N 0.000 claims description 2
- QNNLYRNKHQVOPL-VJLURNNQSA-N (2s,3r,4r,5s,6r)-2-[3-[(1-benzyl-3,4-dihydro-2h-quinolin-7-yl)methyl]-4-propan-2-ylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound CC(C)C1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(C=C12)=CC=C1CCCN2CC1=CC=CC=C1 QNNLYRNKHQVOPL-VJLURNNQSA-N 0.000 claims description 2
- XTTIEOZDXAMWBO-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(1,2,3,4-tetrahydroquinolin-7-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCCCC3=CC=2)=C1 XTTIEOZDXAMWBO-BDHVOXNPSA-N 0.000 claims description 2
- YCKYXZOCAFMWQE-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(spiro[3,4-dihydrochromene-2,1'-cyclobutane]-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CCC4(CCC4)OC3=CC=2)=C1 YCKYXZOCAFMWQE-SJSRKZJXSA-N 0.000 claims description 2
- UVWJFVUMYXJKLK-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-cyclopropyl-2,3-dihydro-1,4-benzoxazin-7-yl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3OCCN(C3=CC=2)C2CC2)=C1 UVWJFVUMYXJKLK-SJSRKZJXSA-N 0.000 claims description 2
- OWJOIZYZAKGOPN-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-methyl-2,3-dihydro-1,4-benzoxazin-7-yl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C2N(C)CCOC2=CC=1CC(C(=CC=1)Cl)=CC=1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O OWJOIZYZAKGOPN-BDHVOXNPSA-N 0.000 claims description 2
- RRHQFFQLFNUECC-RQKPWJHBSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-[(4-methoxyphenyl)methyl]-2,3-dihydro-1,4-benzoxazin-6-yl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=CC(OC)=CC=C1CN1C2=CC(CC=3C(=CC=C(C=3)[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)Cl)=CC=C2OCC1 RRHQFFQLFNUECC-RQKPWJHBSA-N 0.000 claims description 2
- BXPOXWBDDUIEHX-VOFPXKNKSA-N 7-[[2-methoxy-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-4h-1,4-benzoxazin-3-one Chemical compound C1=C(CC=2C=C3OCC(=O)NC3=CC=2)C(OC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O BXPOXWBDDUIEHX-VOFPXKNKSA-N 0.000 claims description 2
- UJNRUYFASNGLPC-GGINGMSCSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[(2-cyano-3,4-dihydro-2h-1,4-benzoxazin-6-yl)methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCC(OC3=CC=2)C#N)=C1 UJNRUYFASNGLPC-GGINGMSCSA-N 0.000 claims description 2
- POCFBDFTJMJWLG-UHFFFAOYSA-N dihydrosinapic acid methyl ester Natural products COC(=O)CCC1=CC(OC)=C(O)C(OC)=C1 POCFBDFTJMJWLG-UHFFFAOYSA-N 0.000 claims description 2
- 102100025101 GATA-type zinc finger protein 1 Human genes 0.000 claims 5
- IUYYXBYVTKKXSS-ZQGJOIPISA-N (2r,3s,4r,5r,6s)-2-(hydroxymethyl)-6-[4-methoxy-3-(1,2,3,4-tetrahydroquinolin-7-ylmethyl)phenyl]oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCCC3=CC=2)C(OC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O IUYYXBYVTKKXSS-ZQGJOIPISA-N 0.000 claims 1
- VNOFFBXXFWYKLQ-RXFVIIJJSA-N (2r,3s,4r,5r,6s)-2-(hydroxymethyl)-6-[4-propan-2-yl-3-(1,2,3,4-tetrahydroquinolin-6-ylmethyl)phenyl]oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3CCCNC3=CC=2)C(C(C)C)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VNOFFBXXFWYKLQ-RXFVIIJJSA-N 0.000 claims 1
- IEUXUJIJOYVFAY-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-4-ethylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCOC3=CC=2)C(CC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O IEUXUJIJOYVFAY-ZQGJOIPISA-N 0.000 claims 1
- MSRYKMIVHLHUSC-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-4-methoxyphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCOC3=CC=2)C(OC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O MSRYKMIVHLHUSC-BDHVOXNPSA-N 0.000 claims 1
- WKVLZIFCTJQOME-RQKPWJHBSA-N (2s,3r,4r,5s,6r)-2-[3-[(2-benzyl-3,4-dihydro-1h-isoquinolin-7-yl)methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CN(CC=4C=CC=CC=4)CCC3=CC=2)=C1 WKVLZIFCTJQOME-RQKPWJHBSA-N 0.000 claims 1
- YMBKJXJZFLKDDI-ADAARDCZSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(2,3-dihydro-1-benzofuran-5-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CCOC3=CC=2)=C1 YMBKJXJZFLKDDI-ADAARDCZSA-N 0.000 claims 1
- RHYPECPJIIJPFS-ADAARDCZSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(2,3-dihydro-1h-indol-5-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CCNC3=CC=2)=C1 RHYPECPJIIJPFS-ADAARDCZSA-N 0.000 claims 1
- BRPWQKCNFXULRV-ADAARDCZSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(3,4-dihydro-2h-1,4-benzoxazin-7-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3OCCNC3=CC=2)=C1 BRPWQKCNFXULRV-ADAARDCZSA-N 0.000 claims 1
- GHNZUEJRBDGPBK-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(3,4-dihydro-2h-chromen-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CCCOC3=CC=2)=C1 GHNZUEJRBDGPBK-BDHVOXNPSA-N 0.000 claims 1
- YZJQYLKXKKISBW-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[4-(cyclopropylmethyl)-2,3-dihydro-1,4-benzoxazin-6-yl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3N(CC4CC4)CCOC3=CC=2)=C1 YZJQYLKXKKISBW-RXFVIIJJSA-N 0.000 claims 1
- OBEMIDSFGQEIKV-OKNYXOPQSA-N 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-3,4-dihydro-2h-1,4-benzoxazine-2-carboxylic acid Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCC(OC3=CC=2)C(O)=O)=C1 OBEMIDSFGQEIKV-OKNYXOPQSA-N 0.000 claims 1
- FBHUAAQASTXZAS-RXFVIIJJSA-N 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]spiro[3h-chromene-2,4'-piperidine]-4-one Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3C(=O)CC4(CCNCC4)OC3=CC=2)=C1 FBHUAAQASTXZAS-RXFVIIJJSA-N 0.000 claims 1
- RXBYZHBPWFNHJO-RQXATKFSSA-N 7-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-4h-1,4-benzoxazin-3-one Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3OCC(=O)NC3=CC=2)=C1 RXBYZHBPWFNHJO-RQXATKFSSA-N 0.000 claims 1
- 229940123934 Reductase inhibitor Drugs 0.000 claims 1
- PWDJCVFKMIQLDE-ZQGJOIPISA-N ethyl 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-2,3-dihydro-1,4-benzoxazine-4-carboxylate Chemical compound C1=C2N(C(=O)OCC)CCOC2=CC=C1CC(C(=CC=1)Cl)=CC=1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O PWDJCVFKMIQLDE-ZQGJOIPISA-N 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 119
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 204
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 161
- 239000000243 solution Substances 0.000 description 156
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 87
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 85
- 239000011541 reaction mixture Substances 0.000 description 84
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 80
- 235000019439 ethyl acetate Nutrition 0.000 description 69
- 229940093499 ethyl acetate Drugs 0.000 description 65
- 239000012267 brine Substances 0.000 description 63
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 63
- 238000006243 chemical reaction Methods 0.000 description 62
- 229910052938 sodium sulfate Inorganic materials 0.000 description 62
- 235000011152 sodium sulphate Nutrition 0.000 description 62
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 58
- 239000012044 organic layer Substances 0.000 description 52
- 239000000543 intermediate Substances 0.000 description 51
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 46
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 45
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 43
- 238000003756 stirring Methods 0.000 description 43
- 238000005160 1H NMR spectroscopy Methods 0.000 description 42
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 41
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 41
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 40
- 230000002829 reductive effect Effects 0.000 description 37
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 36
- 238000010898 silica gel chromatography Methods 0.000 description 35
- 239000002904 solvent Substances 0.000 description 33
- 239000012043 crude product Substances 0.000 description 31
- 229920006395 saturated elastomer Polymers 0.000 description 29
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 28
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 28
- 239000002253 acid Substances 0.000 description 26
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 25
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 22
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 21
- 239000003039 volatile agent Substances 0.000 description 21
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 20
- 125000004432 carbon atom Chemical group C* 0.000 description 18
- 238000004440 column chromatography Methods 0.000 description 18
- 238000009472 formulation Methods 0.000 description 18
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 17
- 235000017557 sodium bicarbonate Nutrition 0.000 description 17
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 15
- 210000004027 cell Anatomy 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 13
- 239000003153 chemical reaction reagent Substances 0.000 description 13
- 238000002953 preparative HPLC Methods 0.000 description 13
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- 108091006277 SLC5A1 Proteins 0.000 description 12
- 102000058090 Sodium-Glucose Transporter 1 Human genes 0.000 description 12
- 239000002585 base Substances 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 11
- 108091006269 SLC5A2 Proteins 0.000 description 11
- 102000058081 Sodium-Glucose Transporter 2 Human genes 0.000 description 11
- 229960000583 acetic acid Drugs 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 229910052736 halogen Inorganic materials 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 11
- OXMCCMNQOLPNQU-RXFVIIJJSA-N (2r,3s,4r,5r,6s)-2-(hydroxymethyl)-6-[4-propan-2-yl-3-(1,2,3,4-tetrahydroquinolin-7-ylmethyl)phenyl]oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCCC3=CC=2)C(C(C)C)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O OXMCCMNQOLPNQU-RXFVIIJJSA-N 0.000 description 10
- FTNIERGYEVLCRV-ADAARDCZSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCCOC3=CC=2)=C1 FTNIERGYEVLCRV-ADAARDCZSA-N 0.000 description 10
- 229910015900 BF3 Inorganic materials 0.000 description 10
- 238000001704 evaporation Methods 0.000 description 10
- 230000008020 evaporation Effects 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- 238000001228 spectrum Methods 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- 0 CCCC*CC1=*CC*1 Chemical compound CCCC*CC1=*CC*1 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 9
- 150000002367 halogens Chemical class 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- RAFKCLFWELPONH-UHFFFAOYSA-N acetonitrile;dichloromethane Chemical compound CC#N.ClCCl RAFKCLFWELPONH-UHFFFAOYSA-N 0.000 description 8
- 238000004587 chromatography analysis Methods 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 229910052744 lithium Inorganic materials 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- 229940098779 methanesulfonic acid Drugs 0.000 description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 description 8
- 229910052717 sulfur Inorganic materials 0.000 description 8
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 7
- 235000011054 acetic acid Nutrition 0.000 description 7
- 239000012298 atmosphere Substances 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 7
- 239000011777 magnesium Substances 0.000 description 7
- 238000002360 preparation method Methods 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- 239000012279 sodium borohydride Substances 0.000 description 7
- 229910000033 sodium borohydride Inorganic materials 0.000 description 7
- 102100040918 Pro-glucagon Human genes 0.000 description 6
- 150000007513 acids Chemical class 0.000 description 6
- 235000019270 ammonium chloride Nutrition 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- 238000012512 characterization method Methods 0.000 description 6
- 125000001183 hydrocarbyl group Chemical group 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 239000012453 solvate Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 6
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 5
- 239000000556 agonist Substances 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 150000002430 hydrocarbons Chemical class 0.000 description 5
- 201000001421 hyperglycemia Diseases 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 229910052749 magnesium Inorganic materials 0.000 description 5
- GNWXVOQHLPBSSR-UHFFFAOYSA-N oxolane;toluene Chemical compound C1CCOC1.CC1=CC=CC=C1 GNWXVOQHLPBSSR-UHFFFAOYSA-N 0.000 description 5
- 230000002265 prevention Effects 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 150000003254 radicals Chemical group 0.000 description 5
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- KQNDZYRBEHYEQA-RLXMVLCYSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[[1-[(4-methoxyphenyl)methyl]-3,4-dihydro-2h-quinolin-6-yl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=CC(OC)=CC=C1CN1C2=CC=C(CC=3C(=CC=C(C=3)[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)Cl)C=C2CCC1 KQNDZYRBEHYEQA-RLXMVLCYSA-N 0.000 description 4
- DTALCVXXATYTQJ-UHFFFAOYSA-N 2-propan-2-ylbenzaldehyde Chemical compound CC(C)C1=CC=CC=C1C=O DTALCVXXATYTQJ-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- HQFJGVBQTPLJJG-UHFFFAOYSA-N 4-benzyl-6-[(5-bromo-2-chlorophenyl)methyl]-2,3-dihydro-1,4-benzoxazine Chemical compound ClC1=CC=C(Br)C=C1CC1=CC=C(OCCN2CC=3C=CC=CC=3)C2=C1 HQFJGVBQTPLJJG-UHFFFAOYSA-N 0.000 description 4
- HHVHJYWNGBGSJF-UHFFFAOYSA-N 5-bromo-2-propan-2-ylbenzaldehyde Chemical compound CC(C)C1=CC=C(Br)C=C1C=O HHVHJYWNGBGSJF-UHFFFAOYSA-N 0.000 description 4
- ZTYHULJFRDFCNI-UHFFFAOYSA-N 6-[(5-bromo-2-chlorophenyl)methyl]-3,4-dihydro-2h-1,4-benzoxazine Chemical compound ClC1=CC=C(Br)C=C1CC1=CC=C(OCCN2)C2=C1 ZTYHULJFRDFCNI-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 239000004215 Carbon black (E152) Substances 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- 229940123518 Sodium/glucose cotransporter 2 inhibitor Drugs 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 210000004204 blood vessel Anatomy 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 238000003745 diagnosis Methods 0.000 description 4
- 235000005911 diet Nutrition 0.000 description 4
- 230000037213 diet Effects 0.000 description 4
- 108010037444 diisopropylglutathione ester Proteins 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 230000003914 insulin secretion Effects 0.000 description 4
- 210000003734 kidney Anatomy 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 4
- 239000007921 spray Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 230000032258 transport Effects 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 3
- LEWYCBRUDUVOLG-UHFFFAOYSA-N 1,1,2,2-tetrafluoro-2-(1,1,2,2,3,3,4,4-octafluoro-4-iodobutoxy)ethanesulfonyl fluoride Chemical compound FC(F)(I)C(F)(F)C(F)(F)C(F)(F)OC(F)(F)C(F)(F)S(F)(=O)=O LEWYCBRUDUVOLG-UHFFFAOYSA-N 0.000 description 3
- MPCAJMNYNOGXPB-UHFFFAOYSA-N 1,5-anhydrohexitol Chemical compound OCC1OCC(O)C(O)C1O MPCAJMNYNOGXPB-UHFFFAOYSA-N 0.000 description 3
- ZKUCVUOHZFWFNV-UHFFFAOYSA-N 1-(2,3-dihydro-1,4-benzoxazin-4-yl)-2,2,2-trifluoroethanone Chemical compound C1=CC=C2N(C(=O)C(F)(F)F)CCOC2=C1 ZKUCVUOHZFWFNV-UHFFFAOYSA-N 0.000 description 3
- VKZPZYVLZRZKED-UHFFFAOYSA-N 1-[6-(5-bromo-2-chlorobenzoyl)-2,3-dihydro-1,4-benzoxazin-4-yl]-2,2,2-trifluoroethanone Chemical compound C1=C2N(C(=O)C(F)(F)F)CCOC2=CC=C1C(=O)C1=CC(Br)=CC=C1Cl VKZPZYVLZRZKED-UHFFFAOYSA-N 0.000 description 3
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 3
- KTJYTQACFZFXKR-FAZZITNRSA-N 6-[[2-cyclopropyl-5-[(3s,4r,5r,6r)-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-yl]phenyl]methyl]-3,4-dihydro-2h-chromene Chemical compound C([C@@H]1[C@H]([C@H](OCC=2C=CC=CC=2)[C@@H](OCC=2C=CC=CC=2)C(O1)C=1C=C(CC=2C=C3CCCOC3=CC=2)C(C2CC2)=CC=1)OCC=1C=CC=CC=1)OCC1=CC=CC=C1 KTJYTQACFZFXKR-FAZZITNRSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 201000004569 Blindness Diseases 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 125000000174 L-prolyl group Chemical class [H]N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C(*)=O 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical group [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- 206010054805 Macroangiopathy Diseases 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- DQXUKCYHHRKAIR-GRIBUZNISA-N [(2r,3r,4r,5s)-3,4,5-triacetyloxy-6-[4-bromo-3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)OC1C1=CC=C(Br)C(CC=2C=C3OCCOC3=CC=2)=C1 DQXUKCYHHRKAIR-GRIBUZNISA-N 0.000 description 3
- NTCKRWNHOCBQBX-ZCCUTQAASA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[(2,2-dimethyl-3-oxo-4h-1,4-benzoxazin-6-yl)methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NC(=O)C(C)(C)OC3=CC=2)=C1 NTCKRWNHOCBQBX-ZCCUTQAASA-N 0.000 description 3
- DKOQYKRDCDCNOR-ZCCUTQAASA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)=CC=C1Cl DKOQYKRDCDCNOR-ZCCUTQAASA-N 0.000 description 3
- GZCGUPFRVQAUEE-SLPGGIOYSA-N aldehydo-D-glucose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O GZCGUPFRVQAUEE-SLPGGIOYSA-N 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000009792 diffusion process Methods 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 239000002934 diuretic Substances 0.000 description 3
- 210000002919 epithelial cell Anatomy 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000012362 glacial acetic acid Substances 0.000 description 3
- 229940093915 gynecological organic acid Drugs 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 210000000936 intestine Anatomy 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 208000030159 metabolic disease Diseases 0.000 description 3
- 229920000609 methyl cellulose Polymers 0.000 description 3
- 239000001923 methylcellulose Substances 0.000 description 3
- 235000010981 methylcellulose Nutrition 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N n-propyl alcohol Natural products CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- MTYYIWGKIVFIMX-UHFFFAOYSA-N oxazine-2-carboxylic acid Chemical compound OC(=O)N1OC=CC=C1 MTYYIWGKIVFIMX-UHFFFAOYSA-N 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 210000000512 proximal kidney tubule Anatomy 0.000 description 3
- 150000003214 pyranose derivatives Chemical group 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 238000004611 spectroscopical analysis Methods 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 3
- 208000035408 type 1 diabetes mellitus 1 Diseases 0.000 description 3
- WQZGKKKJIJFFOK-SVZMEOIVSA-N (+)-Galactose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-SVZMEOIVSA-N 0.000 description 2
- RLIALKZRPQJASE-UHFFFAOYSA-N (1-benzyl-3,4-dihydro-2h-quinolin-7-yl)-(5-bromo-2-propan-2-ylphenyl)methanol Chemical compound CC(C)C1=CC=C(Br)C=C1C(O)C1=CC=C(CCCN2CC=3C=CC=CC=3)C2=C1 RLIALKZRPQJASE-UHFFFAOYSA-N 0.000 description 2
- PMFLZOKAHBEXLK-UHFFFAOYSA-N (2-bromo-5-iodophenyl)-(2,3-dihydro-1,4-benzodioxin-6-yl)methanone Chemical compound BrC1=CC=C(I)C=C1C(=O)C1=CC=C(OCCO2)C2=C1 PMFLZOKAHBEXLK-UHFFFAOYSA-N 0.000 description 2
- FQFOXFRGQNERTG-UHFFFAOYSA-N (2-bromo-5-iodophenyl)-(3,4-dihydro-2h-chromen-6-yl)methanone Chemical compound BrC1=CC=C(I)C=C1C(=O)C1=CC=C(OCCC2)C2=C1 FQFOXFRGQNERTG-UHFFFAOYSA-N 0.000 description 2
- YKXCWZVUWWQSAV-BTVCFUMJSA-N (2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O YKXCWZVUWWQSAV-BTVCFUMJSA-N 0.000 description 2
- PRLYHCILDSBOOG-VOFPXKNKSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzothiazin-6-ylmethyl)-4-methoxyphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCSC3=CC=2)C(OC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O PRLYHCILDSBOOG-VOFPXKNKSA-N 0.000 description 2
- CGRAMNVCMHDJCN-SEFGFODJSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-4-phenoxyphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C(C=C1CC=2C=C3NCCOC3=CC=2)=CC=C1OC1=CC=CC=C1 CGRAMNVCMHDJCN-SEFGFODJSA-N 0.000 description 2
- IKDUSEGUYUWRGL-SAEUYMBFSA-N (2s,3r,4r,5s,6r)-2-[3-[(4-benzyl-2,3-dihydro-1,4-benzoxazin-6-yl)methyl]-4-propan-2-ylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound CC(C)C1=CC=C([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)C=C1CC(C=C12)=CC=C1OCCN2CC1=CC=CC=C1 IKDUSEGUYUWRGL-SAEUYMBFSA-N 0.000 description 2
- ZRIDXJYDPPFGBL-RQXATKFSSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(3,4-dihydro-2h-1,4-benzothiazin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCCSC3=CC=2)=C1 ZRIDXJYDPPFGBL-RQXATKFSSA-N 0.000 description 2
- JKVDIQFAMLCYRL-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethyl-2,3-dihydro-1,4-benzoxazin-6-yl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C2N(CC)CCOC2=CC=C1CC(C(=CC=1)Cl)=CC=1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O JKVDIQFAMLCYRL-ZQGJOIPISA-N 0.000 description 2
- HIDSOKLGQJEHSF-AALIFHQRSA-N (2s,3r,4s,5s,6r)-2-[3-[(1-benzyl-3,4-dihydro-2h-quinolin-7-yl)methyl]-4-propan-2-ylphenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(C(C)C)C(CC=2C=C3N(CC=4C=CC=CC=4)CCCC3=CC=2)=CC=1[C@]1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O HIDSOKLGQJEHSF-AALIFHQRSA-N 0.000 description 2
- MZWXHHXDVQCREW-RQKPWJHBSA-N (2s,3r,4s,5s,6r)-2-[3-[(4-benzyl-2,3-dihydro-1,4-benzoxazin-6-yl)methyl]-4-chlorophenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=C3N(CC=4C=CC=CC=4)CCOC3=CC=2)=CC=1[C@]1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O MZWXHHXDVQCREW-RQKPWJHBSA-N 0.000 description 2
- OFXACHZYSMQSLL-ZQGJOIPISA-N (2s,3r,4s,5s,6r)-2-[4-chloro-3-[(4-methyl-2,3-dihydro-1,4-benzoxazin-7-yl)methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=C3OCCN(C)C3=CC=2)=CC=1[C@]1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O OFXACHZYSMQSLL-ZQGJOIPISA-N 0.000 description 2
- OEGRSOIZRVWFLK-SAEUYMBFSA-N (2s,3r,4s,5s,6r)-2-[4-chloro-3-[[1-[(4-methoxyphenyl)methyl]-3,4-dihydro-2h-quinolin-6-yl]methyl]phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C1=CC(OC)=CC=C1CN1C2=CC=C(CC=3C(=CC=C(C=3)[C@@]3(OC)[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)Cl)C=C2CCC1 OEGRSOIZRVWFLK-SAEUYMBFSA-N 0.000 description 2
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 2
- JUHGKROOWPWQOA-AWGDKMGJSA-N (3r,4r,5s,6r)-2-[4-bromo-3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)OC1C1=CC=C(Br)C(CC=2C=C3OCCOC3=CC=2)=C1 JUHGKROOWPWQOA-AWGDKMGJSA-N 0.000 description 2
- YHQWSHZCSJWFRB-KSHFZJHMSA-N (3r,4s,5s,6r)-2-[3-[(4-benzylspiro[3h-1,4-benzoxazine-2,1'-cyclopropane]-6-yl)methyl]-4-chlorophenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Cl)C(CC=2C=C3N(CC=4C=CC=CC=4)CC4(CC4)OC3=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O YHQWSHZCSJWFRB-KSHFZJHMSA-N 0.000 description 2
- SVLKBIWNNLFLBX-VEIQOZLZSA-N (3r,4s,5s,6r)-2-[4-bromo-3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)phenyl]-6-(hydroxymethyl)-2-methoxyoxane-3,4,5-triol Chemical compound C=1C=C(Br)C(CC=2C=C3OCCOC3=CC=2)=CC=1C1(OC)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O SVLKBIWNNLFLBX-VEIQOZLZSA-N 0.000 description 2
- WAPNSZCXPAOMGF-UHFFFAOYSA-N (4-benzyl-2,3-dihydro-1,4-benzoxazin-6-yl)-(5-bromo-2-propan-2-ylphenyl)methanol Chemical compound CC(C)C1=CC=C(Br)C=C1C(O)C1=CC=C(OCCN2CC=3C=CC=CC=3)C2=C1 WAPNSZCXPAOMGF-UHFFFAOYSA-N 0.000 description 2
- TZMQAULWXKMJAB-UHFFFAOYSA-N (4-benzylspiro[3h-1,4-benzoxazine-2,1'-cyclopropane]-6-yl)-(5-bromo-2-chlorophenyl)methanol Chemical compound C=1C(Br)=CC=C(Cl)C=1C(O)C(C=C1N(CC=2C=CC=CC=2)C2)=CC=C1OC12CC1 TZMQAULWXKMJAB-UHFFFAOYSA-N 0.000 description 2
- JETBEZFAOUBXRL-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-[1-[(4-methoxyphenyl)methyl]-3,4-dihydro-2h-quinolin-6-yl]methanol Chemical compound C1=CC(OC)=CC=C1CN1C2=CC=C(C(O)C=3C(=CC=C(Br)C=3)Cl)C=C2CCC1 JETBEZFAOUBXRL-UHFFFAOYSA-N 0.000 description 2
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 description 2
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 2
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical group C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 2
- LOZWAPSEEHRYPG-UHFFFAOYSA-N 1,4-dithiane Chemical group C1CSCCS1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 2
- NAHNYBZEIMXXHS-UHFFFAOYSA-N 1-[6-[(2-bromo-5-iodophenyl)methyl]-2,3-dihydro-1,4-benzoxazin-4-yl]-2,2,2-trifluoroethanone Chemical compound C1=C2N(C(=O)C(F)(F)F)CCOC2=CC=C1CC1=CC(I)=CC=C1Br NAHNYBZEIMXXHS-UHFFFAOYSA-N 0.000 description 2
- GTTWAVACSITBKO-UHFFFAOYSA-N 1-[6-[(5-bromo-2-chlorophenyl)methyl]-2,3-dihydro-1,4-benzoxazin-4-yl]-2,2,2-trifluoroethanone Chemical compound C1=C2N(C(=O)C(F)(F)F)CCOC2=CC=C1CC1=CC(Br)=CC=C1Cl GTTWAVACSITBKO-UHFFFAOYSA-N 0.000 description 2
- VTDUAVWYIBLQPS-UHFFFAOYSA-N 1-benzyl-7-[(5-bromo-2-propan-2-ylphenyl)methyl]-3,4-dihydro-2h-quinoline Chemical compound CC(C)C1=CC=C(Br)C=C1CC1=CC=C(CCCN2CC=3C=CC=CC=3)C2=C1 VTDUAVWYIBLQPS-UHFFFAOYSA-N 0.000 description 2
- HYFJSTKBJCJCFS-UHFFFAOYSA-N 1-benzyl-7-bromo-3,4-dihydro-2h-quinoline Chemical compound C12=CC(Br)=CC=C2CCCN1CC1=CC=CC=C1 HYFJSTKBJCJCFS-UHFFFAOYSA-N 0.000 description 2
- LECYCYNAEJDSIL-UHFFFAOYSA-N 1-bromo-2-propan-2-ylbenzene Chemical compound CC(C)C1=CC=CC=C1Br LECYCYNAEJDSIL-UHFFFAOYSA-N 0.000 description 2
- QPKKBDSNZFSSOD-UHFFFAOYSA-N 2-bromo-5-iodobenzoic acid Chemical compound OC(=O)C1=CC(I)=CC=C1Br QPKKBDSNZFSSOD-UHFFFAOYSA-N 0.000 description 2
- BQDRMTDACLJNPW-UHFFFAOYSA-N 2-bromo-5-iodobenzoyl chloride Chemical compound ClC(=O)C1=CC(I)=CC=C1Br BQDRMTDACLJNPW-UHFFFAOYSA-N 0.000 description 2
- CMLFRMDBDNHMRA-UHFFFAOYSA-N 2h-1,2-benzoxazine Chemical compound C1=CC=C2C=CNOC2=C1 CMLFRMDBDNHMRA-UHFFFAOYSA-N 0.000 description 2
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 2
- YRLORWPBJZEGBX-UHFFFAOYSA-N 3,4-dihydro-2h-1,4-benzoxazine Chemical compound C1=CC=C2NCCOC2=C1 YRLORWPBJZEGBX-UHFFFAOYSA-N 0.000 description 2
- HVBSAKJJOYLTQU-UHFFFAOYSA-N 4-aminobenzenesulfonic acid Chemical compound NC1=CC=C(S(O)(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-N 0.000 description 2
- IZZZPEBDTBFZHL-UHFFFAOYSA-N 4-benzyl-6-[(5-bromo-2-chlorophenyl)methyl]spiro[3h-1,4-benzoxazine-2,1'-cyclopropane] Chemical compound ClC1=CC=C(Br)C=C1CC1=CC=C(OC2(CC2)CN2CC=3C=CC=CC=3)C2=C1 IZZZPEBDTBFZHL-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- YJRQBOOMJGYUPI-UHFFFAOYSA-N 4-methyl-2,3-dihydro-1,4-benzoxazine Chemical compound C1=CC=C2N(C)CCOC2=C1 YJRQBOOMJGYUPI-UHFFFAOYSA-N 0.000 description 2
- QRCGFTXRXYMJOS-UHFFFAOYSA-N 4h-1,4-benzoxazin-3-one Chemical compound C1=CC=C2NC(=O)COC2=C1 QRCGFTXRXYMJOS-UHFFFAOYSA-N 0.000 description 2
- DPKKRQAEYWOISP-UHFFFAOYSA-N 5-bromo-2-chlorobenzaldehyde Chemical compound ClC1=CC=C(Br)C=C1C=O DPKKRQAEYWOISP-UHFFFAOYSA-N 0.000 description 2
- PNHUJQOGRWCBCR-UHFFFAOYSA-N 6-[(2-bromo-5-iodophenyl)methyl]-2,3-dihydro-1,4-benzodioxine Chemical compound BrC1=CC=C(I)C=C1CC1=CC=C(OCCO2)C2=C1 PNHUJQOGRWCBCR-UHFFFAOYSA-N 0.000 description 2
- FYMSUNTZCRDPRG-UHFFFAOYSA-N 6-[(2-bromo-5-iodophenyl)methyl]-3,4-dihydro-2h-1,4-benzoxazine Chemical compound BrC1=CC=C(I)C=C1CC1=CC=C(OCCN2)C2=C1 FYMSUNTZCRDPRG-UHFFFAOYSA-N 0.000 description 2
- ZTHBXZJZQVICER-UHFFFAOYSA-N 6-[(2-bromo-5-iodophenyl)methyl]-3,4-dihydro-2h-chromene Chemical compound BrC1=CC=C(I)C=C1CC1=CC=C(OCCC2)C2=C1 ZTHBXZJZQVICER-UHFFFAOYSA-N 0.000 description 2
- CAEJUUMEDRWHNR-UHFFFAOYSA-N 6-[(5-bromo-2-chlorophenyl)methyl]-1-[(4-methoxyphenyl)methyl]-3,4-dihydro-2h-quinoline Chemical compound C1=CC(OC)=CC=C1CN1C2=CC=C(CC=3C(=CC=C(Br)C=3)Cl)C=C2CCC1 CAEJUUMEDRWHNR-UHFFFAOYSA-N 0.000 description 2
- XOVSKRABFHBXBI-PALZRNSMSA-N 6-[[2-bromo-5-[(3r,4s,5r,6r)-2-methoxy-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-yl]phenyl]methyl]-3,4-dihydro-2h-chromene Chemical compound C([C@H]1OC([C@@H]([C@@H](OCC=2C=CC=CC=2)[C@@H]1OCC=1C=CC=CC=1)OCC=1C=CC=CC=1)(OC)C=1C=C(CC=2C=C3CCCOC3=CC=2)C(Br)=CC=1)OCC1=CC=CC=C1 XOVSKRABFHBXBI-PALZRNSMSA-N 0.000 description 2
- VBIUOJLDVPONTO-PFLUPBOVSA-N 6-[[2-bromo-5-[(3s,4r,5r,6r)-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-yl]phenyl]methyl]-3,4-dihydro-2h-chromene Chemical compound C([C@H]1OC([C@@H]([C@@H](OCC=2C=CC=CC=2)[C@@H]1OCC=1C=CC=CC=1)OCC=1C=CC=CC=1)C1=CC=C(C(=C1)CC=1C=C2CCCOC2=CC=1)Br)OCC1=CC=CC=C1 VBIUOJLDVPONTO-PFLUPBOVSA-N 0.000 description 2
- WEHMHBSITKCQBY-UHFFFAOYSA-N 6-bromo-1,2,3,4-tetrahydroquinoline Chemical compound N1CCCC2=CC(Br)=CC=C21 WEHMHBSITKCQBY-UHFFFAOYSA-N 0.000 description 2
- HLBIATYWCPRIPY-UHFFFAOYSA-N 6-bromo-1-[(4-methoxyphenyl)methyl]-3,4-dihydro-2h-quinoline Chemical compound C1=CC(OC)=CC=C1CN1C2=CC=C(Br)C=C2CCC1 HLBIATYWCPRIPY-UHFFFAOYSA-N 0.000 description 2
- MEKWQPBFYRGZDS-UHFFFAOYSA-N 6-bromospiro[3,4-dihydro-1,4-benzoxazine-2,1'-cyclopropane] Chemical compound C1NC2=CC(Br)=CC=C2OC21CC2 MEKWQPBFYRGZDS-UHFFFAOYSA-N 0.000 description 2
- WBJKUJQWVVFJAS-UHFFFAOYSA-N 6-bromospiro[4h-1,4-benzoxazine-2,1'-cyclopropane]-3-one Chemical compound O=C1NC2=CC(Br)=CC=C2OC21CC2 WBJKUJQWVVFJAS-UHFFFAOYSA-N 0.000 description 2
- GUFQZRWJOBEKRJ-UHFFFAOYSA-N 7-[(5-bromo-2-chlorophenyl)methyl]-4-methyl-2,3-dihydro-1,4-benzoxazine Chemical compound C=1C=C2N(C)CCOC2=CC=1CC1=CC(Br)=CC=C1Cl GUFQZRWJOBEKRJ-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- 101100086716 Caenorhabditis elegans ran-3 gene Proteins 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 206010007558 Cardiac failure chronic Diseases 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 2
- 208000008960 Diabetic foot Diseases 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- 108010011459 Exenatide Proteins 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- 208000002705 Glucose Intolerance Diseases 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 239000002841 Lewis acid Substances 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 208000017442 Retinal disease Diseases 0.000 description 2
- 206010057430 Retinal injury Diseases 0.000 description 2
- 206010038923 Retinopathy Diseases 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 238000006069 Suzuki reaction reaction Methods 0.000 description 2
- 108010084455 Zeocin Proteins 0.000 description 2
- MDAKIGWYNQSRRE-MMKSVXGCSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[3-[(3-amino-4-hydroxyphenyl)methyl]-4-chlorophenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C(N)C(O)=CC=2)=C1 MDAKIGWYNQSRRE-MMKSVXGCSA-N 0.000 description 2
- MFKHHBDFHOZIDG-MMKSVXGCSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[(4-hydroxy-3-nitrophenyl)methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C(C(O)=CC=2)[N+]([O-])=O)=C1 MFKHHBDFHOZIDG-MMKSVXGCSA-N 0.000 description 2
- WKIINJAQJXPHQM-ZCCUTQAASA-N [(2r,3r,4r,5s,6s)-6-[3-[(3-acetyl-4-hydroxyphenyl)methyl]-4-chlorophenyl]-3,4,5-triacetyloxyoxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C(C(O)=CC=2)C(C)=O)=C1 WKIINJAQJXPHQM-ZCCUTQAASA-N 0.000 description 2
- 230000009102 absorption Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- UCVQKYCKXLVLHU-UHFFFAOYSA-N acetonitrile;1,1-dichloroethane Chemical compound CC#N.CC(Cl)Cl UCVQKYCKXLVLHU-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 230000006978 adaptation Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 150000001447 alkali salts Chemical class 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 2
- RWZYAGGXGHYGMB-UHFFFAOYSA-N anthranilic acid Chemical compound NC1=CC=CC=C1C(O)=O RWZYAGGXGHYGMB-UHFFFAOYSA-N 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 239000002220 antihypertensive agent Substances 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- 150000001491 aromatic compounds Chemical class 0.000 description 2
- 150000005840 aryl radicals Chemical class 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000003613 bile acid Substances 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- 244000309466 calf Species 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 208000020832 chronic kidney disease Diseases 0.000 description 2
- 208000022831 chronic renal failure syndrome Diseases 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 2
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 229920006237 degradable polymer Polymers 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 208000033679 diabetic kidney disease Diseases 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 229940030606 diuretics Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- PYOSCVMKQJXRPT-UHFFFAOYSA-N ethyl 1-(4-bromo-2-nitrophenoxy)cyclopropane-1-carboxylate Chemical compound C=1C=C(Br)C=C([N+]([O-])=O)C=1OC1(C(=O)OCC)CC1 PYOSCVMKQJXRPT-UHFFFAOYSA-N 0.000 description 2
- IOLQWGVDEFWYNP-UHFFFAOYSA-N ethyl 2-bromo-2-methylpropanoate Chemical compound CCOC(=O)C(C)(C)Br IOLQWGVDEFWYNP-UHFFFAOYSA-N 0.000 description 2
- 229960001519 exenatide Drugs 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 230000037406 food intake Effects 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 239000007902 hard capsule Substances 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 150000002390 heteroarenes Chemical class 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- QWTDNUCVQCZILF-UHFFFAOYSA-N isopentane Chemical compound CCC(C)C QWTDNUCVQCZILF-UHFFFAOYSA-N 0.000 description 2
- 238000010902 jet-milling Methods 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 150000007517 lewis acids Chemical class 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- DIXJYYPYYYQVJQ-AVGHQTNXSA-N methyl 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-3,4-dihydro-2h-1,4-benzoxazine-2-carboxylate Chemical compound C=1C=C2OC(C(=O)OC)CNC2=CC=1CC(C(=CC=1)Cl)=CC=1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O DIXJYYPYYYQVJQ-AVGHQTNXSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 150000004682 monohydrates Chemical class 0.000 description 2
- 230000007823 neuropathy Effects 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 206010033675 panniculitis Diseases 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 229920000747 poly(lactic acid) Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 238000012877 positron emission topography Methods 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 2
- 201000009104 prediabetes syndrome Diseases 0.000 description 2
- 239000003380 propellant Substances 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000030558 renal glucose absorption Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- 239000007901 soft capsule Substances 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 210000004304 subcutaneous tissue Anatomy 0.000 description 2
- 125000003107 substituted aryl group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 2
- 238000001757 thermogravimetry curve Methods 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- DBGIVFWFUFKIQN-VIFPVBQESA-N (+)-Fenfluramine Chemical compound CCN[C@@H](C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-VIFPVBQESA-N 0.000 description 1
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical group C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- NAUIIWZAYNSNEP-RCMDHJDMSA-N (1r,2r,3s,4s,6r)-4-[3-(9a,10-dihydro-5ah-phenothiazin-3-ylmethyl)-4-chlorophenyl]-6-(hydroxymethyl)cyclohexane-1,2,3-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)C[C@H]1C1=CC=C(Cl)C(CC=2C=C3SC4C=CC=CC4NC3=CC=2)=C1 NAUIIWZAYNSNEP-RCMDHJDMSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- OBETXYAYXDNJHR-SSDOTTSWSA-M (2r)-2-ethylhexanoate Chemical compound CCCC[C@@H](CC)C([O-])=O OBETXYAYXDNJHR-SSDOTTSWSA-M 0.000 description 1
- GQSFEZGDYCBNSR-ZQGJOIPISA-N (2r,3s,4r,5r,6s)-2-(hydroxymethyl)-6-[4-methyl-3-(1,2,3,4-tetrahydroquinolin-7-ylmethyl)phenyl]oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCCC3=CC=2)C(C)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O GQSFEZGDYCBNSR-ZQGJOIPISA-N 0.000 description 1
- PJRSUKFWFKUDTH-JWDJOUOUSA-N (2s)-6-amino-2-[[2-[[(2s)-2-[[(2s,3s)-2-[[(2s)-2-[[2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[(2-aminoacetyl)amino]-4-methylsulfanylbutanoyl]amino]propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]propanoyl]amino]acetyl]amino]propanoyl Chemical compound CSCC[C@H](NC(=O)CN)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(N)=O PJRSUKFWFKUDTH-JWDJOUOUSA-N 0.000 description 1
- KNXWEXPHINKVRJ-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethynylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(C#C)C(CC=2C=C3OCCOC3=CC=2)=C1 KNXWEXPHINKVRJ-ZQGJOIPISA-N 0.000 description 1
- NNOSRDNUKZCUKQ-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-propan-2-ylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3OCCOC3=CC=2)C(C(C)C)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O NNOSRDNUKZCUKQ-SJSRKZJXSA-N 0.000 description 1
- DZHNOSVQMQVFNJ-ZXGKGEBGSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-4-(1,3-oxazol-4-yloxy)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C(C=C1CC=2C=C3NCCOC3=CC=2)=CC=C1OC1=COC=N1 DZHNOSVQMQVFNJ-ZXGKGEBGSA-N 0.000 description 1
- MTBYIRHJPGAVHZ-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-4-(dimethylamino)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCOC3=CC=2)C(N(C)C)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O MTBYIRHJPGAVHZ-ZQGJOIPISA-N 0.000 description 1
- DVKUSDXNSFOFKS-ADAARDCZSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-4-fluorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(F)C(CC=2C=C3NCCOC3=CC=2)=C1 DVKUSDXNSFOFKS-ADAARDCZSA-N 0.000 description 1
- UVKPLODOGCDGSG-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-chromen-6-ylmethyl)-4-(trifluoromethoxy)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(OC(F)(F)F)C(CC=2C=C3CCCOC3=CC=2)=C1 UVKPLODOGCDGSG-BDHVOXNPSA-N 0.000 description 1
- VMEMPUDQLWCKPE-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-chromen-6-ylmethyl)-4-methoxyphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3CCCOC3=CC=2)C(OC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VMEMPUDQLWCKPE-ZQGJOIPISA-N 0.000 description 1
- VAVISQBUAMWTQQ-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-chromen-6-ylmethyl)-4-methylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3CCCOC3=CC=2)C(C)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VAVISQBUAMWTQQ-ZQGJOIPISA-N 0.000 description 1
- PQOFAQUTXJXRSC-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[3-(3,4-dihydro-2h-chromen-6-ylmethyl)-4-pyrazol-1-ylphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(N2N=CC=C2)C(CC=2C=C3CCCOC3=CC=2)=C1 PQOFAQUTXJXRSC-RXFVIIJJSA-N 0.000 description 1
- GKWVZUZEJLUICL-VOFPXKNKSA-N (2s,3r,4r,5s,6r)-2-[3-[(1,1-dioxo-3,4-dihydro-2h-1$l^{6},4-benzothiazin-6-yl)methyl]-4-methoxyphenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3C(S(CCN3)(=O)=O)=CC=2)C(OC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O GKWVZUZEJLUICL-VOFPXKNKSA-N 0.000 description 1
- VLTIRLGXYRMPPJ-ADAARDCZSA-N (2s,3r,4r,5s,6r)-2-[4-amino-3-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCOC3=CC=2)C(N)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VLTIRLGXYRMPPJ-ADAARDCZSA-N 0.000 description 1
- BLCYOHUXTXFESY-PQYHXYLCSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(spiro[3,4-dihydro-1,4-benzoxazine-2,1'-cyclopropane]-6-ylmethyl)phenyl]-6-(1-hydroxyethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](C(O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCC4(CC4)OC3=CC=2)=C1 BLCYOHUXTXFESY-PQYHXYLCSA-N 0.000 description 1
- JOFXGKBUQVCDAZ-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-(spiro[3,4-dihydrochromene-2,1'-cyclopentane]-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CCC4(CCCC4)OC3=CC=2)=C1 JOFXGKBUQVCDAZ-RXFVIIJJSA-N 0.000 description 1
- UEQUAJICHIHROY-ZQGJOIPISA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-ethyl-2,3-dihydro-1,4-benzoxazin-7-yl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C=1C=C2N(CC)CCOC2=CC=1CC(C(=CC=1)Cl)=CC=1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O UEQUAJICHIHROY-ZQGJOIPISA-N 0.000 description 1
- XRNTYSCFJIAWDQ-VUNWFEDTSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-hydroxy-3,4-dihydro-2h-chromen-6-yl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3C(O)CCOC3=CC=2)=C1 XRNTYSCFJIAWDQ-VUNWFEDTSA-N 0.000 description 1
- VDMIGDKNMBPOOL-BDHVOXNPSA-N (2s,3r,4r,5s,6r)-2-[4-chloro-3-[(4-methyl-2,3-dihydro-1,4-benzoxazin-6-yl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C2N(C)CCOC2=CC=C1CC(C(=CC=1)Cl)=CC=1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VDMIGDKNMBPOOL-BDHVOXNPSA-N 0.000 description 1
- XPNFSMNTMRRDRH-RTJMFUJLSA-N (2s,3r,4r,5s,6r)-2-[4-cyclobutyl-3-(3,4-dihydro-2h-chromen-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C(C=C1CC=2C=C3CCCOC3=CC=2)=CC=C1C1CCC1 XPNFSMNTMRRDRH-RTJMFUJLSA-N 0.000 description 1
- UUUIKCYQTDINPS-RXFVIIJJSA-N (2s,3r,4r,5s,6r)-2-[4-cyclopropyl-3-(1,2,3,4-tetrahydroquinolin-7-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C(C=C1CC=2C=C3NCCCC3=CC=2)=CC=C1C1CC1 UUUIKCYQTDINPS-RXFVIIJJSA-N 0.000 description 1
- ZTAPLANSVGXCRG-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-ethyl-3-(1,2,3,4-tetrahydroquinolin-6-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3CCCNC3=CC=2)C(CC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O ZTAPLANSVGXCRG-SJSRKZJXSA-N 0.000 description 1
- DKWSDJDXYGDNPB-SJSRKZJXSA-N (2s,3r,4r,5s,6r)-2-[4-ethyl-3-(1,2,3,4-tetrahydroquinolin-7-ylmethyl)phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound C1=C(CC=2C=C3NCCCC3=CC=2)C(CC)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O DKWSDJDXYGDNPB-SJSRKZJXSA-N 0.000 description 1
- MTJPVPPGSWUZLJ-SJSRKZJXSA-N (2s,3r,4r,5s,6s)-2-[4-chloro-3-(spiro[3,4-dihydro-1,4-benzoxazine-2,1'-cyclopentane]-6-ylmethyl)phenyl]-6-(fluoromethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CF)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NCC4(CCCC4)OC3=CC=2)=C1 MTJPVPPGSWUZLJ-SJSRKZJXSA-N 0.000 description 1
- JEJWEBUJAREOPO-UTPRGODJSA-N (2s,3r,4s,5s,6r)-2-[4-chloro-3-(10h-phenoxazin-2-ylmethyl)phenyl]-6-methyloxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3NC4=CC=CC=C4OC3=CC=2)=C1 JEJWEBUJAREOPO-UTPRGODJSA-N 0.000 description 1
- HCBKACGDTLXDSZ-WAECPPJVSA-N (2s,3r,4s,5s,6r)-2-[4-chloro-3-(spiro[3,4-dihydrochromene-2,3'-pyrrolidine]-6-ylmethyl)phenyl]-6-methyloxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](C)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3CCC4(CNCC4)OC3=CC=2)=C1 HCBKACGDTLXDSZ-WAECPPJVSA-N 0.000 description 1
- GQPYTJVDPQTBQC-KLQYNRQASA-N (3r)-3-amino-1-[3-(trifluoromethyl)-6,8-dihydro-5h-[1,2,4]triazolo[4,3-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one;phosphoric acid;hydrate Chemical compound O.OP(O)(O)=O.C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F GQPYTJVDPQTBQC-KLQYNRQASA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- KPBHJRFRKPMOST-UNXRTQJZSA-N (3r,4s,5r,6r)-2-[3-[(4-benzyl-2,3-dihydro-1,4-benzoxazin-6-yl)methyl]-4-bromophenyl]-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-ol Chemical compound C([C@H]1OC([C@@H]([C@@H](OCC=2C=CC=CC=2)[C@@H]1OCC=1C=CC=CC=1)OCC=1C=CC=CC=1)(O)C=1C=C(CC=2C=C3N(CC=4C=CC=CC=4)CCOC3=CC=2)C(Br)=CC=1)OCC1=CC=CC=C1 KPBHJRFRKPMOST-UNXRTQJZSA-N 0.000 description 1
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 description 1
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- YFMFNYKEUDLDTL-UHFFFAOYSA-N 1,1,1,2,3,3,3-heptafluoropropane Chemical compound FC(F)(F)C(F)C(F)(F)F YFMFNYKEUDLDTL-UHFFFAOYSA-N 0.000 description 1
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- ILWJAOPQHOZXAN-UHFFFAOYSA-N 1,3-dithianyl Chemical group [CH]1SCCCS1 ILWJAOPQHOZXAN-UHFFFAOYSA-N 0.000 description 1
- IMLSAISZLJGWPP-UHFFFAOYSA-N 1,3-dithiolane Chemical compound C1CSCS1 IMLSAISZLJGWPP-UHFFFAOYSA-N 0.000 description 1
- NMHRMAGGRPUBCR-UHFFFAOYSA-N 1-(2,2-dimethyl-3h-1,4-benzoxazin-4-yl)-2,2,2-trifluoroethanone Chemical compound C1=CC=C2OC(C)(C)CN(C(=O)C(F)(F)F)C2=C1 NMHRMAGGRPUBCR-UHFFFAOYSA-N 0.000 description 1
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 1
- BABPQVRKCWHCMZ-UHFFFAOYSA-N 1-(hydroxymethyl)cyclohexane-1,2,3-triol Chemical compound OCC1(O)CCCC(O)C1O BABPQVRKCWHCMZ-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical group CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- SJJCQDRGABAVBB-UHFFFAOYSA-N 1-hydroxy-2-naphthoic acid Chemical compound C1=CC=CC2=C(O)C(C(=O)O)=CC=C21 SJJCQDRGABAVBB-UHFFFAOYSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- VFTFKUDGYRBSAL-UHFFFAOYSA-N 15-crown-5 Chemical compound C1COCCOCCOCCOCCO1 VFTFKUDGYRBSAL-UHFFFAOYSA-N 0.000 description 1
- PDQRQJVPEFGVRK-UHFFFAOYSA-N 2,1,3-benzothiadiazole Chemical compound C1=CC=CC2=NSN=C21 PDQRQJVPEFGVRK-UHFFFAOYSA-N 0.000 description 1
- DCYGAPKNVCQNOE-UHFFFAOYSA-N 2,2,2-triphenylacetic acid Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C(=O)O)C1=CC=CC=C1 DCYGAPKNVCQNOE-UHFFFAOYSA-N 0.000 description 1
- LJRUKUQEIHJTEL-UHFFFAOYSA-N 2,2-dimethyl-4h-1,4-benzoxazin-3-one Chemical compound C1=CC=C2NC(=O)C(C)(C)OC2=C1 LJRUKUQEIHJTEL-UHFFFAOYSA-N 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003660 2,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- CYDRLYFLACMBNA-UHFFFAOYSA-N 2-(5-bromo-2-methoxyphenyl)-1,3-dioxolane Chemical compound COC1=CC=C(Br)C=C1C1OCCO1 CYDRLYFLACMBNA-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- FHEYFIGWYQJVDR-ACJLOTCBSA-N 2-[[3-[(2r)-2-[[(2r)-2-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]-1h-indol-7-yl]oxy]acetic acid Chemical compound C1([C@@H](O)CN[C@@H](CC=2C3=CC=CC(OCC(O)=O)=C3NC=2)C)=CC=CC(Cl)=C1 FHEYFIGWYQJVDR-ACJLOTCBSA-N 0.000 description 1
- RBCPJQQJBAQSOU-UHFFFAOYSA-N 2-bromo-5-chlorobenzoic acid Chemical compound OC(=O)C1=CC(Cl)=CC=C1Br RBCPJQQJBAQSOU-UHFFFAOYSA-N 0.000 description 1
- BNQXSFFVGRPZIP-UHFFFAOYSA-N 2-bromo-5-chlorobenzoyl chloride Chemical compound ClC(=O)C1=CC(Cl)=CC=C1Br BNQXSFFVGRPZIP-UHFFFAOYSA-N 0.000 description 1
- OYUNTGBISCIYPW-UHFFFAOYSA-N 2-chloroprop-2-enenitrile Chemical compound ClC(=C)C#N OYUNTGBISCIYPW-UHFFFAOYSA-N 0.000 description 1
- WBJWXIQDBDZMAW-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carbonyl chloride Chemical compound C1=CC=CC2=C(C(Cl)=O)C(O)=CC=C21 WBJWXIQDBDZMAW-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- NKPYIZCCIFRRRC-USWKJHDZSA-N 3-(1-benzofuran-5-yl)-1-[2-hydroxy-4-methyl-6-[(2r,3r,4s,5s,6s)-3,4,5,6-tetrahydroxyoxan-2-yl]oxyphenyl]propan-1-one Chemical compound C=1C(C)=CC(O)=C(C(=O)CCC=2C=C3C=COC3=CC=2)C=1O[C@@H]1O[C@H](O)[C@@H](O)[C@H](O)[C@H]1O NKPYIZCCIFRRRC-USWKJHDZSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- QXEIZJNFPVIUQM-RQXATKFSSA-N 3-chloro-2-(3,4-dihydro-2h-1,4-benzoxazin-6-ylmethyl)-6-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]benzamide Chemical compound NC(=O)C1=C(CC=2C=C3NCCOC3=CC=2)C(Cl)=CC=C1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O QXEIZJNFPVIUQM-RQXATKFSSA-N 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000003148 4 aminobutyric acid receptor blocking agent Substances 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- HLRYHWGIUUHNIJ-UHFFFAOYSA-N 4-benzyl-6-[(2-bromo-5-iodophenyl)methyl]-2,3-dihydro-1,4-benzoxazine Chemical compound BrC1=CC=C(I)C=C1CC1=CC=C(OCCN2CC=3C=CC=CC=3)C2=C1 HLRYHWGIUUHNIJ-UHFFFAOYSA-N 0.000 description 1
- NJLXMWAIIQZZPA-UHFFFAOYSA-N 4-benzyl-6-[(5-bromo-2-propan-2-ylphenyl)methyl]-2,3-dihydro-1,4-benzoxazine Chemical compound CC(C)C1=CC=C(Br)C=C1CC1=CC=C(OCCN2CC=3C=CC=CC=3)C2=C1 NJLXMWAIIQZZPA-UHFFFAOYSA-N 0.000 description 1
- PZOFEYIKPKTRHE-UQPIPOSESA-N 4-benzyl-6-[[2-bromo-5-[(2s,3s,4r,5r,6r)-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-yl]phenyl]methyl]-2,3-dihydro-1,4-benzoxazine Chemical compound BrC1=CC=C([C@H]2[C@@H]([C@@H](OCC=3C=CC=CC=3)[C@H](OCC=3C=CC=CC=3)[C@@H](COCC=3C=CC=CC=3)O2)OCC=2C=CC=CC=2)C=C1CC(C=C12)=CC=C1OCCN2CC1=CC=CC=C1 PZOFEYIKPKTRHE-UQPIPOSESA-N 0.000 description 1
- TYGQZHKNVGOGOS-UHFFFAOYSA-N 4-benzyl-6-bromospiro[3h-1,4-benzoxazine-2,1'-cyclopropane] Chemical compound C1N(CC=2C=CC=CC=2)C2=CC(Br)=CC=C2OC21CC2 TYGQZHKNVGOGOS-UHFFFAOYSA-N 0.000 description 1
- VQSACDGRYKNYRN-UHFFFAOYSA-N 4-bromo-3-fluoro-2-nitrophenol Chemical compound OC1=CC=C(Br)C(F)=C1[N+]([O-])=O VQSACDGRYKNYRN-UHFFFAOYSA-N 0.000 description 1
- XRHGYUZYPHTUJZ-UHFFFAOYSA-N 4-chlorobenzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1 XRHGYUZYPHTUJZ-UHFFFAOYSA-N 0.000 description 1
- YJLVQDIJFGCGRW-UHFFFAOYSA-N 4-ethyl-2,3-dihydro-1,4-benzoxazine Chemical compound C1=CC=C2N(CC)CCOC2=C1 YJLVQDIJFGCGRW-UHFFFAOYSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- DBJMEBUKQVZWMD-UHFFFAOYSA-N 4-methyl-1,4-benzoxazin-3-one Chemical compound C1=CC=C2N(C)C(=O)COC2=C1 DBJMEBUKQVZWMD-UHFFFAOYSA-N 0.000 description 1
- YQDGQEKUTLYWJU-UHFFFAOYSA-N 5,6,7,8-tetrahydroquinoline Chemical compound C1=CC=C2CCCCC2=N1 YQDGQEKUTLYWJU-UHFFFAOYSA-N 0.000 description 1
- 102000006902 5-HT2C Serotonin Receptor Human genes 0.000 description 1
- 108010072553 5-HT2C Serotonin Receptor Proteins 0.000 description 1
- NFFXEUUOMTXWCX-UHFFFAOYSA-N 5-[(2,4-dioxo-1,3-thiazolidin-5-yl)methyl]-2-methoxy-n-[[4-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound C1=C(C(=O)NCC=2C=CC(=CC=2)C(F)(F)F)C(OC)=CC=C1CC1SC(=O)NC1=O NFFXEUUOMTXWCX-UHFFFAOYSA-N 0.000 description 1
- IJIBRSFAXRFPPN-UHFFFAOYSA-N 5-bromo-2-methoxybenzaldehyde Chemical compound COC1=CC=C(Br)C=C1C=O IJIBRSFAXRFPPN-UHFFFAOYSA-N 0.000 description 1
- IMVDZFOSXJSXFP-UHFFFAOYSA-N 5-chloro-2-propyl-3-[[4-[2-(2h-tetrazol-5-yl)phenyl]phenyl]methyl]imidazole-4-carbaldehyde Chemical compound CCCC1=NC(Cl)=C(C=O)N1CC1=CC=C(C=2C(=CC=CC=2)C2=NNN=N2)C=C1 IMVDZFOSXJSXFP-UHFFFAOYSA-N 0.000 description 1
- HXFLZWAZSSPLCO-UHFFFAOYSA-N 6,6-dimethylbicyclo[3.1.1]heptyl Chemical group C1[C-]2C([CH2+])([CH2-])[C+]1CCC2 HXFLZWAZSSPLCO-UHFFFAOYSA-N 0.000 description 1
- LMAYDNVIKBCSLP-UHFFFAOYSA-N 6,7-dihydro-5h-pyrrolo[3,2-d]pyrimidine Chemical compound C1=NC=C2NCCC2=N1 LMAYDNVIKBCSLP-UHFFFAOYSA-N 0.000 description 1
- CVLWKZSKUHAZSX-UHFFFAOYSA-N 6-(2-bromo-5-iodobenzoyl)-4-(2,2,2-trifluoroacetyl)-1,4-benzoxazin-3-one Chemical compound C1=C2N(C(=O)C(F)(F)F)C(=O)COC2=CC=C1C(=O)C1=CC(I)=CC=C1Br CVLWKZSKUHAZSX-UHFFFAOYSA-N 0.000 description 1
- FNGWBNFJBVVODV-ADAARDCZSA-N 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-2,2-dimethyl-4h-1,4-benzoxazin-3-one Chemical compound C1=C2NC(=O)C(C)(C)OC2=CC=C1CC(C(=CC=1)Cl)=CC=1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O FNGWBNFJBVVODV-ADAARDCZSA-N 0.000 description 1
- PAHPXCAQZOBMAO-ADAARDCZSA-N 6-[[2-chloro-5-[(2s,3r,4r,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]phenyl]methyl]-2,3-dihydro-1,4-benzoxazine-4-carboxylic acid Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=CC=C(Cl)C(CC=2C=C3N(C(O)=O)CCOC3=CC=2)=C1 PAHPXCAQZOBMAO-ADAARDCZSA-N 0.000 description 1
- DRVWZEWZXCZNAR-UHFFFAOYSA-N 7-bromo-1,2,3,4-tetrahydroquinoline Chemical compound C1CCNC2=CC(Br)=CC=C21 DRVWZEWZXCZNAR-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 208000010444 Acidosis Diseases 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 101100460776 Arabidopsis thaliana NPY2 gene Proteins 0.000 description 1
- 101100460788 Arabidopsis thaliana NPY5 gene Proteins 0.000 description 1
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 206010061666 Autonomic neuropathy Diseases 0.000 description 1
- PTQXTEKSNBVPQJ-UHFFFAOYSA-N Avasimibe Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1CC(=O)NS(=O)(=O)OC1=C(C(C)C)C=CC=C1C(C)C PTQXTEKSNBVPQJ-UHFFFAOYSA-N 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- IOWJJTQJQOATPR-UHFFFAOYSA-N C(C1)C1N1c(cccc2)c2OCC1 Chemical compound C(C1)C1N1c(cccc2)c2OCC1 IOWJJTQJQOATPR-UHFFFAOYSA-N 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 239000002081 C09CA05 - Tasosartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- ZCORIUOUDMZLSG-UHFFFAOYSA-N C1NC2=CC=CCC2OC1 Chemical compound C1NC2=CC=CCC2OC1 ZCORIUOUDMZLSG-UHFFFAOYSA-N 0.000 description 1
- LPAGFVYQRIESJQ-UHFFFAOYSA-N C1c2ccccc2NC1 Chemical compound C1c2ccccc2NC1 LPAGFVYQRIESJQ-UHFFFAOYSA-N 0.000 description 1
- PVFMWANILUSZPM-COBSHVIPSA-N C=CCC(C[C@H](CI)Cl)Br Chemical compound C=CCC(C[C@H](CI)Cl)Br PVFMWANILUSZPM-COBSHVIPSA-N 0.000 description 1
- LJHOFBQOJOQMLD-XFVOGMNUSA-N CC1(C)OC(CCC(CC(C2)C(Cl)=CCC2C([C@@H](C2O)O)OC(CO)[C@H]2O)C2)=C2NC1=O Chemical compound CC1(C)OC(CCC(CC(C2)C(Cl)=CCC2C([C@@H](C2O)O)OC(CO)[C@H]2O)C2)=C2NC1=O LJHOFBQOJOQMLD-XFVOGMNUSA-N 0.000 description 1
- ZSKTYCQHNCXLOF-FJGSZFSXSA-N CC[C@H](CC(C1)OCc2ccccc2)OC1(c(cc1)cc(Cc2ccc3OCCN(Cc4ccccc4)c3c2)c1Br)O Chemical compound CC[C@H](CC(C1)OCc2ccccc2)OC1(c(cc1)cc(Cc2ccc3OCCN(Cc4ccccc4)c3c2)c1Br)O ZSKTYCQHNCXLOF-FJGSZFSXSA-N 0.000 description 1
- YVBSECQAHGIWNF-UHFFFAOYSA-N CN1c2ccccc2CCC1 Chemical compound CN1c2ccccc2CCC1 YVBSECQAHGIWNF-UHFFFAOYSA-N 0.000 description 1
- FUFQOLKATLNVLE-UHFFFAOYSA-N COC1=CC=C(CN(CCCc2c3)c2ccc3Br)CC1 Chemical compound COC1=CC=C(CN(CCCc2c3)c2ccc3Br)CC1 FUFQOLKATLNVLE-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- GHOSNRCGJFBJIB-UHFFFAOYSA-N Candesartan cilexetil Chemical compound C=12N(CC=3C=CC(=CC=3)C=3C(=CC=CC=3)C3=NNN=N3)C(OCC)=NC2=CC=CC=1C(=O)OC(C)OC(=O)OC1CCCCC1 GHOSNRCGJFBJIB-UHFFFAOYSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- GHOKWGTUZJEAQD-UHFFFAOYSA-N Chick antidermatitis factor Natural products OCC(C)(C)C(O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-UHFFFAOYSA-N 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-YMDCURPLSA-N D-galactopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-YMDCURPLSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- JVHXJTBJCFBINQ-ADAARDCZSA-N Dapagliflozin Chemical compound C1=CC(OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)=CC=C1Cl JVHXJTBJCFBINQ-ADAARDCZSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 229940098788 GABA receptor antagonist Drugs 0.000 description 1
- 108091007911 GSKs Proteins 0.000 description 1
- 201000000628 Gas Gangrene Diseases 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- 229940122904 Glucagon receptor antagonist Drugs 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 102000003638 Glucose-6-Phosphatase Human genes 0.000 description 1
- 108010086800 Glucose-6-Phosphatase Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- 102000007390 Glycogen Phosphorylase Human genes 0.000 description 1
- 108010046163 Glycogen Phosphorylase Proteins 0.000 description 1
- 102000004103 Glycogen Synthase Kinases Human genes 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 102100030488 HEAT repeat-containing protein 6 Human genes 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000990566 Homo sapiens HEAT repeat-containing protein 6 Proteins 0.000 description 1
- 101000801684 Homo sapiens Phospholipid-transporting ATPase ABCA1 Proteins 0.000 description 1
- 101000716688 Homo sapiens Sodium/glucose cotransporter 1 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 1
- 208000013016 Hypoglycemia Diseases 0.000 description 1
- 208000007976 Ketosis Diseases 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 208000001344 Macular Edema Diseases 0.000 description 1
- 206010025415 Macular oedema Diseases 0.000 description 1
- 206010025421 Macule Diseases 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- 206010027417 Metabolic acidosis Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 206010027910 Mononeuritis Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 101150111774 NPY5R gene Proteins 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- 102000003729 Neprilysin Human genes 0.000 description 1
- 108090000028 Neprilysin Proteins 0.000 description 1
- 208000028389 Nerve injury Diseases 0.000 description 1
- 239000006057 Non-nutritive feed additive Substances 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 239000005480 Olmesartan Substances 0.000 description 1
- 208000018262 Peripheral vascular disease Diseases 0.000 description 1
- 102100038831 Peroxisome proliferator-activated receptor alpha Human genes 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000331 Polyhydroxybutyrate Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 229940123924 Protein kinase C inhibitor Drugs 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- ILRCGYURZSFMEG-RKQHYHRCSA-N Salidroside Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OCCC1=CC=C(O)C=C1 ILRCGYURZSFMEG-RKQHYHRCSA-N 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 239000005864 Sulphur Substances 0.000 description 1
- 208000002847 Surgical Wound Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 102000008316 Type 4 Melanocortin Receptor Human genes 0.000 description 1
- 108010021436 Type 4 Melanocortin Receptor Proteins 0.000 description 1
- 101710204865 Tyrosine-protein phosphatase 1 Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 1
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 1
- 229940116731 Uricosuric agent Drugs 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- IPPDHWALLUPDNH-MMKSVXGCSA-N [(2r,3r,4r,5s,6s)-3,4,5-triacetyloxy-6-[4-chloro-3-[(4-hydroxyphenyl)methyl]phenyl]oxan-2-yl]methyl acetate Chemical compound CC(=O)O[C@@H]1[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1C1=CC=C(Cl)C(CC=2C=CC(O)=CC=2)=C1 IPPDHWALLUPDNH-MMKSVXGCSA-N 0.000 description 1
- FDKIDFYIEWFERB-UHFFFAOYSA-N [2-butyl-5-chloro-3-[[4-[2-(2h-tetrazol-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol;potassium Chemical compound [K].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 FDKIDFYIEWFERB-UHFFFAOYSA-N 0.000 description 1
- WDUKZMSVLKASQH-XEEFFQQJSA-N [5-[(3r,4s,5r,6r)-2-hydroxy-3,4,5-tris(phenylmethoxy)-6-(phenylmethoxymethyl)oxan-2-yl]-2-methoxyphenyl]-spiro[3,4-dihydrochromene-2,1'-cyclobutane]-6-ylmethanone Chemical compound COC1=CC=C(C2(O)[C@@H]([C@@H](OCC=3C=CC=CC=3)[C@H](OCC=3C=CC=CC=3)[C@@H](COCC=3C=CC=CC=3)O2)OCC=2C=CC=CC=2)C=C1C(=O)C(C=C1CC2)=CC=C1OC12CCC1 WDUKZMSVLKASQH-XEEFFQQJSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Chemical group CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical group 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 208000012998 acute renal failure Diseases 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 239000000670 adrenergic alpha-2 receptor antagonist Substances 0.000 description 1
- 229940127252 advanced glycation end product inhibitor Drugs 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 239000003288 aldose reductase inhibitor Substances 0.000 description 1
- 229940090865 aldose reductase inhibitors used in diabetes Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001341 alkaline earth metal compounds Chemical class 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 102000030484 alpha-2 Adrenergic Receptor Human genes 0.000 description 1
- 108020004101 alpha-2 Adrenergic Receptor Proteins 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- PYHXGXCGESYPCW-UHFFFAOYSA-N alpha-phenylbenzeneacetic acid Natural products C=1C=CC=CC=1C(C(=O)O)C1=CC=CC=C1 PYHXGXCGESYPCW-UHFFFAOYSA-N 0.000 description 1
- 159000000013 aluminium salts Chemical class 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 239000010405 anode material Substances 0.000 description 1
- 230000003178 anti-diabetic effect Effects 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 239000003524 antilipemic agent Substances 0.000 description 1
- 239000003705 antithrombocytic agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 229950010046 avasimibe Drugs 0.000 description 1
- 108010014210 axokine Proteins 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- BNBQRQQYDMDJAH-UHFFFAOYSA-N benzodioxan Chemical compound C1=CC=C2OCCOC2=C1 BNBQRQQYDMDJAH-UHFFFAOYSA-N 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 1
- 229960000516 bezafibrate Drugs 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000000227 bioadhesive Substances 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 229940043430 calcium compound Drugs 0.000 description 1
- 150000001674 calcium compounds Chemical class 0.000 description 1
- 235000019577 caloric intake Nutrition 0.000 description 1
- 229960004349 candesartan cilexetil Drugs 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 235000015111 chews Nutrition 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 230000001906 cholesterol absorption Effects 0.000 description 1
- 239000003354 cholesterol ester transfer protein inhibitor Substances 0.000 description 1
- VZWXIQHBIQLMPN-UHFFFAOYSA-N chromane Chemical compound C1=CC=C2CCCOC2=C1 VZWXIQHBIQLMPN-UHFFFAOYSA-N 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 238000009109 curative therapy Methods 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960003834 dapagliflozin Drugs 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960004597 dexfenfluramine Drugs 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- AFABGHUZZDYHJO-UHFFFAOYSA-N dimethyl butane Natural products CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 1
- 229940113088 dimethylacetamide Drugs 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 229940112141 dry powder inhaler Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 229960000573 eprosartan mesylate Drugs 0.000 description 1
- DJSLTDBPKHORNY-XMMWENQYSA-N eprosartan methanesulfonate Chemical compound CS(O)(=O)=O.C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 DJSLTDBPKHORNY-XMMWENQYSA-N 0.000 description 1
- 230000003628 erosive effect Effects 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002169 ethanolamines Chemical class 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- DPMGQZGXWRHYPE-UHFFFAOYSA-N ethyl 1-hydroxycyclopropane-1-carboxylate Chemical compound CCOC(=O)C1(O)CC1 DPMGQZGXWRHYPE-UHFFFAOYSA-N 0.000 description 1
- PPEKFACTSZVDSK-WUWJBHAQSA-N ethyl 2-[4-[[2-chloro-5-[(2s,3s,4r,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]phenyl]methyl]-2-nitrophenoxy]-2-methylpropanoate Chemical compound C1=C([N+]([O-])=O)C(OC(C)(C)C(=O)OCC)=CC=C1CC1=CC([C@H]2[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O2)OC(C)=O)=CC=C1Cl PPEKFACTSZVDSK-WUWJBHAQSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- PAVZHTXVORCEHP-UHFFFAOYSA-N ethylboronic acid Chemical compound CCB(O)O PAVZHTXVORCEHP-UHFFFAOYSA-N 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
- 229960000815 ezetimibe Drugs 0.000 description 1
- 229960002297 fenofibrate Drugs 0.000 description 1
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- 230000009229 glucose formation Effects 0.000 description 1
- 208000018914 glucose metabolism disease Diseases 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- 230000002641 glycemic effect Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 238000000265 homogenisation Methods 0.000 description 1
- 102000052194 human SLC5A1 Human genes 0.000 description 1
- 229960002003 hydrochlorothiazide Drugs 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 230000035879 hyperinsulinaemia Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007915 intraurethral administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 230000004140 ketosis Effects 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 210000001039 kidney glomerulus Anatomy 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- ZEUXAIYYDDCIRX-UHFFFAOYSA-N losartan carboxylic acid Chemical compound CCCCC1=NC(Cl)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C2=NNN=N2)C=C1 ZEUXAIYYDDCIRX-UHFFFAOYSA-N 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000010230 macular retinal edema Diseases 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 238000004452 microanalysis Methods 0.000 description 1
- 206010062198 microangiopathy Diseases 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000003595 mist Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 208000013734 mononeuritis simplex Diseases 0.000 description 1
- 201000005518 mononeuropathy Diseases 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000003232 mucoadhesive effect Effects 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 206010028320 muscle necrosis Diseases 0.000 description 1
- 230000003387 muscular Effects 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- HHQJWDKIRXRTLS-UHFFFAOYSA-N n'-bromobutanediamide Chemical compound NC(=O)CCC(=O)NBr HHQJWDKIRXRTLS-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 230000008764 nerve damage Effects 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- VTRAEEWXHOVJFV-UHFFFAOYSA-N olmesartan Chemical compound CCCC1=NC(C(C)(C)O)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 VTRAEEWXHOVJFV-UHFFFAOYSA-N 0.000 description 1
- 229960005117 olmesartan Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- OOFGXDQWDNJDIS-UHFFFAOYSA-N oxathiolane Chemical compound C1COSC1 OOFGXDQWDNJDIS-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229940055726 pantothenic acid Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 108010021753 peptide-Gly-Leu-amide Proteins 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 229930029653 phosphoenolpyruvate Natural products 0.000 description 1
- DTBNBXWJWCWCIK-UHFFFAOYSA-N phosphoenolpyruvic acid Chemical compound OC(=O)C(=C)OP(O)(O)=O DTBNBXWJWCWCIK-UHFFFAOYSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 239000005015 poly(hydroxybutyrate) Substances 0.000 description 1
- 239000004632 polycaprolactone Substances 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 230000000291 postprandial effect Effects 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 239000003881 protein kinase C inhibitor Substances 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 1
- 229960004034 sitagliptin Drugs 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 229960004274 stearic acid Drugs 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229950000244 sulfanilic acid Drugs 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000001117 sulphuric acid Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960000651 tasosartan Drugs 0.000 description 1
- ADXGNEYLLLSOAR-UHFFFAOYSA-N tasosartan Chemical compound C12=NC(C)=NC(C)=C2CCC(=O)N1CC(C=C1)=CC=C1C1=CC=CC=C1C=1N=NNN=1 ADXGNEYLLLSOAR-UHFFFAOYSA-N 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 150000004685 tetrahydrates Chemical class 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229940086542 triethylamine Drugs 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 210000004926 tubular epithelial cell Anatomy 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 229940116269 uric acid Drugs 0.000 description 1
- 230000003424 uricosuric effect Effects 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- ACWBQPMHZXGDFX-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=NN1 ACWBQPMHZXGDFX-QFIPXVFZSA-N 0.000 description 1
- SYOKIDBDQMKNDQ-XWTIBIIYSA-N vildagliptin Chemical compound C1C(O)(C2)CC(C3)CC1CC32NCC(=O)N1CCC[C@H]1C#N SYOKIDBDQMKNDQ-XWTIBIIYSA-N 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
- 229960001729 voglibose Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 230000003820 β-cell dysfunction Effects 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/04—Carbocyclic radicals
Definitions
- Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition.
- Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
- Type 1 diabetes also known as Insulin Dependent Diabetes Mellitus (IDDM)
- IDDM Insulin Dependent Diabetes Mellitus
- Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) - is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin).
- NIDDM Non-Insulin Dependent Diabetes Mellitus
- Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from ⁇ -cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
- microvascular disease due to damage of small blood vessels
- macro vascular disease due to damage of the arteries.
- microvascular disease include diabetic retinopathy, neuropathy and nephropathy
- macrovascular disease examples include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
- Diabetic retinopathy characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US.
- Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy.
- Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide.
- a high glycemic diet i.e., a diet that consists of meals that give high postprandial blood sugar
- a high glycemic diet i.e., a diet that consists of meals that give high postpran
- Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level.
- therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
- Glucose is unable to diffuse across the cell membrane and requires transport proteins.
- the transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase.
- SGLT sodium-D-glucose co-transporter
- Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
- SGLT belongs to the sodium/glucose co-transporter family SLCA5.
- Two different SGLT isoforms, SGLT1 and SGLT2 have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different.
- SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
- glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments.
- Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule.
- SGLT1 a high-affinity/low-capacity system
- SGLT2 It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
- SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule.
- a drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient's needs for both blood glucose control, while preserving insulin secretion.
- SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
- SGLT-2 inhibitors Various glucopyranosyl-substituted aromatic and heteroaromatic compounds have also been reported as SGLT-2 inhibitors in patent publications such as: WO 01/27128, WO 04/080990, US 06/0025349, WO 05/085265, WO 05/085237, WO 06/054629 and WO 06/01 1502.
- SGLT1 is predominantly found in the intestine and plays a major role in the absorption of D-glucose and D-galactose. Therefore, SGLT1 inhibitors have the potential to act both in the kidney as well as the intestine to reduce calorie intake and hyperglycemia.
- WO2004/018491 discloses pyrazole derivatives which are SGLT1 inhibitors.
- Glucopyranosyl-substituted aromatic or heteroaromatic compounds where, in general, the sugar moiety has been modified at C4, C5, or C6 positions of pyranose have been published (US 06/0009400, US 06/0019948, US 06/0035841 , US 06/0074031, US 08/0027014 and WO 08/016132).
- FIG. 1 is a differential scanning calorimetry thermogram of a 1 :1 L-proline co-crystal of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6- hydroxymethyl-tetrahydro-pyran-3,4,5-triol prepared by method 1.
- Figure 2 is a powder X-ray diffraction pattern for a 1 :1 L-proline co-crystal of
- Figure 3 is a differential scanning calorimetry thermogram of a 2:1 L-proline co-crystal of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6- hydroxymethyl-tetrahydro-pyran-3,4,5-triol prepared by method 3.
- Figure 4 is a powder X-ray diffraction pattern for a 2:1 L-proline co-crystal of
- This invention relates to compounds useful for treating diseases and conditions mediated by the sodium D-glucose co-transporter (SGLT), e.g. diabetes.
- SGLT sodium D-glucose co-transporter
- the invention also provides methods of treating such diseases and conditions, and compounds and compositions etc. for their treatment.
- the invention provides novel glycoside derivatives, their polymorphs, stereoisomers, pro-drugs, solvates, pharmaceutically acceptable salts and formulations thereof.
- the invention also relates to processes for the preparation of the compounds of the invention.
- the compounds of the invention possess sodium-D-glucose co-transporter (SGLT) inhibition effects, which are beneficial for the prophylaxis, management, treatment, control of progression, or adjunct treatment of diseases and/or medical conditions where the inhibition of SGLT would be beneficial, such as diabetes (including Type-I and Type-I I), obesity, dyslipidemia, insulin resistance, and other metabolic syndrome, and/or diabetes-related complications including retinopathy, nephropathy, neuropathy, ischemic heart disease, arteriosclerosis, ⁇ -cell dysfunction, and as therapeutic and/or prophylactic agents for obesity.
- SGLT sodium-D-glucose co-transporter
- Ring A is an C 6-10 aryl which is optionally substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, cyano, nitro, C -6 alkyl, C 2- ealkenyl, C 2-6 alkynyl, C -6 alkoxy, haloC ⁇ alkoxy, C 3-7 cycloalkyl, C 3- haloC 1-6 alkyl, C 6-10 aryl, -C(0)OR 3 , -C(0)R 3 , - C(0)NR 4 R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 , C ⁇ alkoxy, C 3 .
- alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heterocyclyl and heteroaryl groups may be optionally substituted with one or more substituents selected from the group consisting of halo, hydroxy, cyano, nitro, C ⁇ alkyl, -S(0) p R 3 , -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , - CH 2 NR 4 R 5 and d-ealkoxy;
- Ring A' is a 5-, 6- or 7-membered heterocycle, provided that Ring A' is not 1 ,3- dioxole;
- Y a is a bond or a (C 1 -C 6 )alkylene which is optionally substituted with one or more substituents independently selected from halo, hydroxy, C ⁇ alkyl, C lJ4 alkoxy, haloC ⁇ . 4 alkyl;
- V is hydrogen, halo or -OR 1b ; n is 0, 1 , 2, or 3; q is 0, 1 , 2, or 3; R 1 , R 1a , R 1b and R 1c are independently selected from hydrogen, C 1-6 alkyl, C 6 . 10 aryl-d- alkyl, -C(O)C 6- i 0 aryl and -C(0)C 1-e alkyl;
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, C 1-e alkyl, C 3-7 cycloalkyl, C 3-7 cycloalkylC 1-4 alkyl, C e-10 aryl, C 6- 10 arylC 1-4 alkyl, -C(0)OR 3 , -C(0)R 3 , -C(0)NR R 5 ,-NR R 5 , -CH 2 NR 4 R 5 , C 1-6 alkoxy, C3.7 cycloalkoxy, -S(0) p R 3 , -S(0) 2 NR 4 R 5 , -OS(0) 2 R 3 , -CH z C(0)OR 3 , -CH 2 C(0)NR 4 R 5 , - NR 3 C(0)NR 4 R 5 , -NR 3 C(0)OR 3 , C 6- ioaryloxy, C 2 .ioheterocyclyl, C 2-10 heterocyclylC 1-4 alkyl
- R 2a for each occurrence, is independently selected from the group consisting of oxo, halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 3 . 7 cycloalkyl, d-rcycloalkyld ⁇ alkyl, C 6 . 10 aryl, C 6- ioarylC alkyl, -C(0)OR 3 , -C(0)R 3 , -C(0)NR R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 , d.
- R 2a 6alkoxy, C 3 . 7 cycloalkoxy, -S(0) p R 3 , -S(0) 2 NR R s , -OS(0) 2 R 3 , -CH 2 C(0)OR 3 , - CH 2 C(0)NR 4 R 5 , -NR 3 C(0)NR 4 R 5 , -NR 3 C(0)OR 3 , C 6-10 aryloxy, C 2-10 heterocyclyl, C 2- l oheterocyclyld ⁇ alkyl, Ci.ioheteroarylCi -4 alkyl, d-ioheteroaryl, Ci.ioheteroaryloxy and d-ioheterocycloxy; wherein when any portion of R 2a is an alkyl, cycloalkyl, aryl, heterocyclyl or heteroaryl, for each occurrence, it may be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, cyano, nitro, d
- R 3 for each occurrence, is independently selected from hydrogen, C 1-e alkyl, C 3-7 cycloalkyl, C ⁇ cycloalkylC ⁇ alkyl, C 6- ioaryl, C 1-10 heteroaryl, and C 2 . 10 heterocyclyl; p is 0, 1 or 2; X is [C(R 6 )(R 7 )],;
- Y is H, halo, C alkyl, OR c or NR 4 R 5 ; t is 1, 2, or 3;
- R 6 and R 7 are independently selected from hydrogen, C 1-6 alkyl, OR 1e , and NR 4 R 5 ; or when t is 1 , R 6 and R 7 together may form an oxo group, or when R 6 and R 7 are on the same carbon they can be taken together to form a C 3-7 cycloalkyl or a 3- to 7-membered heterocycle;
- R e for each occurrence, is independently selected from hydrogen, alkyl, C 6 . 1 0 aryl-Ci -4 alkyl, -C(O)C 6 -i 0 aryl and -C(0)Ci ⁇ alkyl;
- R 4 and R 5 are independently selected from hydrogen, alkyl, C 3 . 7 cycloalkyl, C 3-7 cycloalkylC 1-4 alkyl, C B- ioarylCMalkyl, C 6 -i 0 aryl, Ci -10 heteroaryl, CMoheteroarylC alkyl, C 2- i 0 heterocyclyl, and C 2- i 0 heterocyclylCi-4alkyl; or
- R 4 and R 5 taken together along with the nitrogen to which they are bound may form a monocyclic or a bicyclic heteroaryl or heterocyclyl which may be optionally substituted with one or more halo or d ⁇ alkyl; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (l-a):
- Ring A is an C 6 -i 0 aryl which is optionally substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, cyano, C 1-6 alkyl, C ⁇ alkenyl, C 2 . 6 alkynyl, C ⁇ alkoxy, haloC ⁇ alkoxy, a 5-membered heteroaryl and a 6-membered heteroaryl; Ring A' is a 5- or 6-membered heterocycle, provided that Ring A' is not 1,3- dioxole;
- Y a is a bond or a ((-VCeJalkylene which is optionally substituted with one or more substituents independently selected from halo, C 1- alkyl, haloC ⁇ alkyl;
- V is hydrogen, halo or -OR 1b ; n is 0, 1 , 2, or 3; q is 0, 1 , 2, or 3;
- R 1 , R 1a , R 1 and R 1c are independently selected from hydrogen, C 1-6 alkyl, C 6 . 10 aryl-C M alkyl, -C(O)C e . 10 aryl and -C(0)C 1-6 alkyl;
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, Cj. 6 alkyl, C 3 . 7 cycloalkyl, C 3-7 cycloalkylC 1- alkyl, C 6 .ioaryl, C 6 .
- R 2 may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C ⁇ alkyl, and 4 alkoxy;
- R 2a for each occurrence, is independently selected from the group consisting of oxo, halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 3 . 7 cycloalkyl, C 3 . 7 cycloalkylCi. 4 alkyl, C 6- 10 aryl, Ce- 10 arylC 1-4 alkyl, -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR R 5 , -CH 2 NR R 5 , C,. ealkoxy, C 3 .
- R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, d. 4 alkyl, and C 1- alkoxy; or two R on adjacent atoms taken together with the atoms to which they are attached may form a fused C 3 .
- R 7 cycloalkyl which may be optionally substituted with one or more substituent independently selected from halo, hydroxy, C -4 alkyl, and C 1 4 alkoxy; and R 3 , for each occurrence, is independently selected from hydrogen, C -6 alkyl, C 3-7 cycloalkyl, C 6 .ioaryl, C 1-10 heteroaryl, and C 2 -i 0 heterocyciyl; p is 0, 1 or 2;
- X is [C(R 6 )(R 7 )] t ;
- Y is H, halo, alkyl, OR 1c or NR R 5 ; t is 1, 2, or 3;
- R 6 and R 7 are independently selected from hydrogen, C 1-6 alkyl, OR 1e , and NR R 5 ; or when t is 1 , R 6 and R 7 together may form an oxo group, or when R 6 and R 7 are on the same carbon they can be taken together to form a C 3-7 cycloalkyl or a 3- to 7-membered heterocycle;
- R 1e for each occurrence, is independently selected from hydrogen, C 1-B alkyl, C 6- 10 aryl-C 1-4 alkyl, -C(O)C6 -10 aryl and -C(0)C 1-6 alkyl;
- R 4 and R 5 are independently selected from hydrogen, C 1-6 alkyl, C 3 . 7 cycloalkyl, C 3-7 cycloalkylC ⁇ alkyl, (V ⁇ arylC ⁇ alkyl, C 6-10 aryl, C ⁇ oheteroaryl, C 2 -i 0 heterocyclyl, and or
- R 4 and R 5 taken together along with the nitrogen to which they are bound may form a monocyclic or a bicyclic heteroaryl or heterocyclyl which may be optionally substituted with one or more halo or C 1- alkyl; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (l-i):
- Ring A is a C 6 .ioaryl which is optionally substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, C h alky!, Ci.3alkoxy, haloCi. 3 alkoxy and 5-membered heteroaryl;
- Ring A' is a 5- or 6-membered heterocycle containing at least one O or N heteroatom, provided that Ring A' is not 1,3-dioxole;
- Y a is a bond or a C r3 alkylene which is optionally substituted with one or more substituents independently selected from halo,
- V is hydrogen, halo or -OR 1b ; n is 0, 1 or 2; q is 0, 1 , 2, or 3;
- R 1 , R 1a , R 1 and R 1c are independently selected from hydrogen, 0,. 6 alkyl, C 6 . i 0 aryl-C 1-4 alkyl, -C(O)C 6 -i 0 aryl and -C(0)Ci- 6 alkyl;
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, d. 6 alkyl, C 3-7 cycloalkyl, C 6- ioaryl, C 6- ioarylC 1-4 alkyl, - C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR R 5 , -CH 2 NR R 5 , C 1-6 alkoxy, C 3 . 7 cycloalkoxy, - CH 2 C(0)OR 3 , -CH 2 C(0)NR R 5 , -NR 3 C(0)NR R 5 , -NR 3 C(0)OR 3 , C 6 . 10 aryloxy, C 2 .
- R 2 may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C 1-4 alkyl, and C ⁇ alkoxy;
- R for each occurrence, is independently selected from the group consisting of oxo, hydroxy, C 1-6 alkyl, Ca ⁇ cycloalkylC ⁇ alkyl, C6 -10 arylC alkyl, -C(0)OR 3 , -C(0)R 3 , - C(0)NR 4 R 5 and CH 2 C(0)OR 3 ;
- R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C 1-4 alkyl, and C -4 alkoxy; or two R 2a on adjacent atoms taken together with the atoms to which they are attached may form a fused
- X is [C(R 6 )(R 7 )] t ;
- R 4 and R 5 are independently selected from hydrogen, C 1-6 alkyl, C3.7 cycloalkyl, C 3 . 7 cycloalkylC 1-4 alkyl, C 6 -ioarylCi. 4 alkyl, C 6 . 10 aryl, C ⁇ oheteroaryl, CL ! oheteroarylC alkyl, C 2 . 10 heterocyclyl, and C 2 .
- the compound of Formula (I) is of Formula (l-ia):
- Ring A is an C 6- i 0 aryl which is optionally substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 2 - 6 alkenyl, C 2-6 alkynyl, CVealkoxy, haloC ⁇ alkoxy, C 3-7 cycloalkyl, C 3- ⁇ cloalkylC ⁇ alkyl, haloC ⁇ alkyl, C 6 -ioaryl, C 6- ioarylC 1-4 alkyl, -C(0)OR 3 , -C(0)R 3 , - C(0)NR R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 , d-ealkoxy, C 3 - 7 cycloalkoxy, -S(0) p R 3 , -S(0) 2 NR 4 R 5 , - OS(0) 2 R 3 , -CH 2 C(0)OR
- alkyl, alkenyl, alkynyl, cycloalkyi, aryl, heterocyclyl and heteroaryl groups may be optionally substituted with one or more substituents selected from the group consisting of halo, hydroxy, cyano, nitro, C 1-6 alkyl, -S(0) p R 3 , -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , - CH 2 NR 4 R 5 and C ⁇ alkoxy;
- Ring A' is a 5- or 6-membered heterocycle containing at least one O or N heteroatom, provided that Ring A is not 1,3-dioxole;
- Y a is a bond or a d ⁇ alkylene which is optionally substituted with one or more substituents independently selected from halo, C 1- alkyl, haloC 1-4 alkyl; V is hydrogen, halo or -OR 1b ; n is 0, 1 or 2; q is 0, 1 , 2, or 3; p is 0, 1 or 2; R 1 , R 1a , R 1b and R 1c are independently selected from hydrogen, alkyl, C 6-
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, C h alky!, C ⁇ cycloalkyl, C 6-10 aryl, C 6-10 arylC 1-4 a!kyl, - C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR R 5 , -CH 2 NR 4 R 5 , C ⁇ alkoxy, C 3 . 7 cycloalkoxy, - CH 2 C(0)OR 3 , -CH 2 C(0)NR 4 R 5 , -NR 3 C(0)NR 4 R 5 , -NR 3 C(0)OR 3 , C ⁇ oaryloxy, C 2 .
- R 2 may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C 1-4 alky!, and C ⁇ alkoxy;
- R 2a for each occurrence, is independently selected from the group consisting of oxo, halo, hydroxy, cyano, nitro, C ⁇ alkyl, C 3 . 7 cycloalkyl, C 3 . 7 cycloalkylC -4 alkyl, C 6-10 aryl, C 6 .
- R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C 1-4 alkyl, and C ⁇ . 4 alkoxy; or two R 2a on adjacent atoms taken together with the atoms to which they are attached may form a fused C 3-7 cycloalkyl, C 6 aryl, 3- to 7-membered heterocyclyl, or 5- membered heteroaryl, wherein the fused cycloalkyi, aryl, heterocyclyl, and heteroary!
- cycloalkyl which may be optionally substituted with one or more substituent independently selected from halo, hydroxy, C -4 a!kyl, and Ci.
- R 3 for each occurrence, is independently selected from hydrogen, C 1-6 alkyl, C 3-7 cycloalkyl, C ⁇ cycloalkylC ⁇ alkyl, C 6 -i 0 aryl, Ci.i 0 heteroaryl, and C 2-10 heterocyclyl;
- X is [C(R 6 )(R 7 )] t ;
- Y is H, halo, C 1-4 alkyl, OR 1c or NR 4 R 5 ; t is 1, 2, or 3;
- R 6 and R 7 are independently selected from hydrogen, C 1-6 alkyl, OR e , and NR R 5 ; or when t is 1 , R 6 and R 7 together may form an oxo group, or when R 6 and R 7 are on the same carbon they can be taken together to form a C 3 . 7 cycloalkyl or a 3- to 7-membered heterocycle;
- R 1e for each occurrence, is independently selected from hydrogen, C -6 alkyl, C 6- i 0 aryl-C 1-4 alkyl, -C(O)C6. 10 aryl and -C(0)d. 6 alkyl;
- R 4 and R 5 are independently selected from hydrogen, Ci ⁇ alkyl, C 3 . cycloalkyl, C 3 . 7 cycloalkylCi. 4 alkyl, C 6 . 10 aryl, Ci. 10 heteroaryl, Ci.ioheteroarylC -4 alkyl, C 2 .ioheterocyclyl, and or
- R 4 and R 5 taken together along with the nitrogen to which they are bound may form a monocyclic or a bicyclic heteroaryl or heterocyclyl which may be optionally substituted with one or more halo or C 1-4 alkyl; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (l-ii):
- Ring A is a C 6-10 aryl which is optionally substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, d. 3 alkyl, C ⁇ alkoxy, haloCi. 3 alkoxy and 5-membered heteroaryl; Ring A' is a 5- or 6-membered heterocycle containing at least one O or N heteroatom, provided that Ring A' is not 1 ,3-dioxole;
- Y a is a bond or a Cnalkylene which is optionally substituted with one or more substituents independently selected from halo, C -4 alkyl, haloC 1 . alkyl;
- R 1 and R 1a are each hydrogen
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, Ci. 6 alkyl, C 3-7 cycloalkyl, C 6-10 aryl, C 6 . 10 arylC 1-4 alkyl, - C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 , Ci. 6 alkoxy, C 3 .
- R 2 may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C 1-4 alkyl, and C ⁇ alkoxy;
- R a for each occurrence, is independently selected from the group consisting of oxo, hydroxy, C 1-6 alkyl, C 3 .
- R a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C 1-4 alkyl, and or two R 2a on adjacent atoms taken together with the atoms to which they are attached may form a fused C 3 .
- R 4 alkyl, and C ⁇ alkoxy; or two R on the same carbon atom taken together may form a spiro 3- to 7- membered heterocyclyl or a spiro C 3-7 cycloalkyl which may be optionally substituted with one or more substituent independently selected from halo, hydroxy, C 1-4 alkyl, and C 1-4 alkoxy; and R 3 , for each occurrence, is independently selected from hydrogen, C -6 alkyl, C 3-7 cycloalkyl, C ⁇ cycloalkylC ⁇ alkyl, C 6 -i 0 aryl, C 1-10 heteroaryl, and C 2 -ioheterocyclyl;
- X is [C(R 6 )(R 7 )] t ;
- Y is H or OH; t is 1 ; R 6 and R 7 , for each occurrence, are independently selected from hydrogen and
- R 4 and R 5 are independently selected from hydrogen, C 1-6 alkyl, C 3 . 7 cycloalkyl, C 3-7 cycloalkylC 1-4 alkyl, C e . 10 aryl, C 1-10 heteroaryl, Ci.i 0 heteroarylCi_4alkyl, C 2 .io heterocyclyl, and C 2- ioheterocyclylC _ 4 alkyl; or
- R 4 and R 5 taken together along with the nitrogen to which they are bound may form a monocyclic or a bicyclic heteroaryl or heterocyclyl which may be optionally substituted with one or more halo or C 1-4 alkyl; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (I
- Ring A is an C 6 .i 0 aryl which is optionally substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 aikoxy, haloC 1-6 alkoxy, C 3 . 7 cycloalkyl, C 3 .
- Ring A' is a 5- or 6-membered heterocycle containing at least one O or N heteroatom, provided that Ring A' is not 1 ,3-dioxole;
- Y a is a bond or a ( salkylene which is optionally substituted with one or more substituents independently selected from halo, C 1-4 alkyl, haloC 1-4 a!kyl;
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 3-7 cycloalkyl, C 6 .ioaryl, - C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R s , -CH 2 NR 4 R 5 , C ⁇ alkoxy, C 3 .
- R 2 may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C ⁇ alkyl, and C ⁇ alkoxy;
- R 2a for each occurrence, is independently selected from the group consisting of oxo, halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 3 . 7 cycloalkyl, C ⁇ cycloalkylC ⁇ alkyl, C 6 . 10 aryl, C 6-10 arylC 1-4 alkyl, -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , -CH 2 NR R 5 , C,.
- R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, G,. 4 alkyl, and C ⁇ alkoxy; or two R 2a on adjacent atoms taken together with the atoms to which they are attached may form a fused C 3 .
- 7 cycloalkyl which may be optionally substituted with one or more substituent independently selected from halo, hydroxy, C ⁇ alkyl, and C 1- alkoxy; and
- R 3 for each occurrence, is independently selected from hydrogen, Ci-e alkyl, C 3- cycloalkyl, C 3 . 7 cycloalkylC 1-4 alkyl, C6 -10 aryl, C 1-10 heteroaryl, and C 2- i 0 heterocyclyl; p is 0, 1 or 2;
- X is [C(R 6 )(R 7 )] t ;
- Y is H or OH; t is 1 ; R 6 and R 7 , for each occurrence, are independently selected from hydrogen and
- R 4 and R 5 are independently selected from hydrogen, C 1-6 alkyl, C 3 . 7 cycloalkyl, C 3-7 cycloalkylC -4 alkyl, C ⁇ oarylCnalkyl, C 6 . 10 aryl, C -10 heteroaryl, C 2 .i 0 heterocyclyl, and C 2 -ioheterocyclylC 1 . alkyl; or
- R 4 and R 5 taken together along with the nitrogen to which they are bound may form a monocyclic or a bicyc!ic heteroaryl or heterocyciyi which may be optionally substituted with one or more halo or C 1- alkyl; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (l-iii):
- Ring A is a phenyl ring which is optionally substituted with one or more substituents independently selected from the group consisting of chloro, fluoro, hydroxy, methyl, methoxy, ethoxy, trifluoromethoxy and N-pyrazolyl; the structure represented by the following formula:
- Y a is CH 2 ; V is OH; n is 0 or 1 ; q is 0 or 1 ;
- R 1 and R 1a are each hydrogen; R 2 is halo; wherein R 2 may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, 4alkyl, and C ⁇ alkoxy;
- R 2a for each occurrence, is independently selected from the group consisting of hydroxy, C 1-6 alkyl, C 3-7 cycloalkylC 1-4 alkyl; wherein R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C -4 alkyl, and C 1-4 alkoxy; or two R 2a on the same carbon atom taken together may form a spiro C 3-7 cycloalkyl which may be optionally substituted with one or more substituent independently selected from halo, hydroxy, Ci ⁇ alkyl, and d ⁇ alkoxy;
- X is CH 2 ;
- Y is OH; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (l-iiia):
- Ring A is a phenyl ring which is optionally substituted with one or more substituents independently selected from the group consisting of chloro, fiuoro, hydroxy, cyano, methyl, ethyl, isopropyl, ethynyl, methoxy, ethoxy, trifluoromethoxy, amino, dimethylamino, methylsulfanyl, methylsulfonyl, carbamoyl, cyclopropyl, cyciobutyl, phenyl, toulyl, phenoxy, oxazolyloxy, and N-pyrazolyl; the structure represented by the following formula:
- R a for each occurrence, is independently selected from halo, hydroxy, cyano, nitro, C 1-6 alkyl, - S(0) p R 3 , -C ⁇ 0)OR 3 , -C(0)R 3 , -C ⁇ 0)NR R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 and C ⁇ alkoxy; and m is 0 or an integer from 1 -4; Y a is CH 2 ;
- R 1 and R 1a are each hydrogen; R 2 is halo; wherein R 2 may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, d 4 alkyl, and d ⁇ alkoxy;
- R 2a for each occurrence, is independently selected from the group consisting of oxo, halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 3-7 cycloalkyl, C 3 . 7 cycloalkylC 1- alkyl, C 6 . 10 aryl, C 6 . 10 arylC 1-4 alkyl, -C ⁇ 0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 , d- 6 alkoxy, C 3 .
- R a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, d- 4 alkyl, and d ⁇ alkoxy; or two R a on the same carbon atom taken together may form a spiro d. 7 cycloalkyl which may be optionally substituted with one or more substituent independently selected from halo, hydroxy, C 1-4 alkyl, and d ⁇ alkoxy; p is 0, 1 or 2;
- X is CH 2 ;
- Y is OH; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (l-iv):
- Ring A is a phenyl ring which is optionally substituted with one substituent independently selected from the group consisting of chloro, fluoro, methyl and methoxy; wherein Y a is situated meta to the tetrahydropyran ring and the one substituent is situated para to the tetrahydropyran ring; the structure represented by the following formula:
- R 1 and R 1a are each hydrogen; R 2 is halo; R 2a , for each occurrence, is independently selected from the group consisting of unsubstituted hydroxy and unsubstituted C 1-2 alkyl; and
- X is CH 2 ;
- Y is OH; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (l-iva):
- Ring A is a phenyl ring which is optionally substituted with one substituent independently selected from the group consisting of chloro, fluoro, hydroxy, cyano, methyl, ethyl, isopropyl, ethynyl, methoxy, ethoxy, trifluoromethoxy, amino,
- R a for each occurrance, is independently selected from halo, hydroxy, cyano, nitro, C 1-6 alkyl, - S(0) p R 3 , -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , -CH 2 NR R 5 and C ⁇ alkoxy; and m is 0 or an integer from 1-4; p is 0, 1 , or 2; Y a is CH 2 ;
- R 1 and R 1a are each hydrogen
- R 2 is halo
- R for each occurrence, is independently selected from the group consisting of oxo, halo, hydroxy, cyano, nitro, Ce- 10 aryl, Ce. 10 arylC M alkyl, -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR R 6 , -CH 2 NR 4 R 5 , d_ 6 a!koxy, C 3 .
- R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, cyano, nitro, C 1-6 alkyl, -S(0) p R 3 , -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 and d- 6 alkoxy;
- X is CH 2 ;
- Y is OH; or a pharmaceutically acceptable salt thereof.
- the compound of Formula (I) is of Formula (V):
- ring A is phenyl which is substituted with one substituent selected from halo, Ci. alkyl, and C 3 .7cycloalkyl; wherein Y a is situated meta to the tetrahydropyran ring and the one substituent is situated para to the tetrahydropyran ring; the structure represented by the following formula:
- V is -OR 1b ;
- X is CH 2 ;
- Y is OR 1c ;
- R 1 , R 1a , R 1b and R 1c are hydrogen; or a pharmaceutically acceptable salt thereof.
- V is OR 1b . In a further embodiment, V is OH. In one embodiment, R 1 and R 1a are independently selected from hydrogen, C -3 alkyl, C 6 . 10 aryl-C 1-4 alkyl, -C ⁇ O)C 6- i 0 aryl and -C(0)C -8 alkyl. In a further embodiment, R is H. In a further embodiment, R a is H. In a further embodiment, R 1 and R 1a are both H.
- t is 1 or 2. In a further embodiment, t is 1.
- X is CH 2 .
- Y is H or OR 1c .
- Y is H or OH.
- Y is OH.
- Y is a halo.
- Y is fluoro.
- the tetrahydropyran ring is a pyranose ring of the structure:
- the pyranose ring has the following stereochemistry:
- R 1b is selected from hydrogen, C 1 3 alkyl, C 6- i 0 aryl-C alkyl, - C(O)C 6 . 10 aryl and -C(0)C 1-8 alkyl. In a further embodiment, R 1b is H.
- R 1 is selected from hydrogen, Ci -3 alkyl, C 6- 0 aryl-C alkyl, - C(O)C 6- i 0 aryl and -CiOJC L aalkyl. In a further embodiment, R 1c is H.
- R 6 and R 7 are independently selected from hydrogen, C 1-3 alkyl, OR 1e , and NR R 5 ; or when t is 1 , R 6 and R 7 together may form an oxo group; or when R 6 and R 7 are on the same carbon they can be taken together to form a C 3-7 cycloalkyl or a 3- to 7-membered heterocycle.
- R and R 7 for each occurrence, are independently selected from hydrogen and Ci. 3 alkyl.
- R 1e for each occurrence, is independently selected from hydrogen, Ci-3 alkyl, -C(O)C6-i 0 aryl and -C(0)Ci- 6 alkyl. In a further embodiment, R 1e , for each occurrence, is independently selected from hydrogen and C 1 .3 alkyl.
- Ring A is substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, Ci -3 alkyl, C 1-3 alkoxy, haloC ⁇ alkoxy and 5-membered heteroaryl.
- Ring A is substituted with one or more substituents independently selected from the group consisting of halo, hydroxy, C 1-3 alkyl, C 3-7 cycloalkyl, Ci. 3 alkoxy, haloC 1-3 alkoxy and 5-membered heteroaryl.
- Ring A is substituted with one or more substituents independently selected from the group consisting of chloro, fluoro, hydroxy, methyl, methoxy, ethoxy, trifluoromethoxy and N-pyrazolyl.
- Ring A is substituted with one or more substituents independently selected from the group consisting of chloro, fluoro, hydroxy, methyl, ethyl, isopropyl, cyclopropyi, methoxy, ethoxy, trifluoromethoxy and N-pyrazolyl.
- Ring A is substituted with one or more substituents independently selected from the group consisting of chloro, fluoro, methyl and methoxy.
- Ring A is substituted with one or more chloro substituents. In a further embodiment, Ring A is substituted with one or more substituents independently selected from the group consisting of chloro, ethyl, isopropyl, and cyclopropyi. in a further embodiment, Ring A is substituted with one chloro. In a further embodiment, Ring A is substituted with one ethyl. In a further embodiment, Ring A is substituted with one isopropyl. In a further embodiment, Ring A is substituted with one cyclopropyi.
- Ring A is naphthyl which is optionally substituted. in a one embodiment, Ring A is phenyl which is optionally substituted.
- Y a is situated meta to the tetrahydropyran ring.
- Ring A has one substituent. In one aspect of this embodiment, Ring A has one substituent which is selected from the group consisting of halo, hydroxy, C 1-3 alkyl, C 3-7 cycloalkyl, Ci. 3 alkoxy, haloCi -3 alkoxy and 5-membered heteroaryl. In another aspect of this embodiment, Ring A has one substituent which is selected from the group consisting of chloro, fluoro, hydroxy, methyl, ethyl, isopropyl, cyclopropyl, methoxy, ethoxy, trifluoromethoxy and N-pyrazolyl. In another aspect of this
- Ring A is substituted with one chloro.
- Ring A is substituted with one ethyl, in another aspect of this
- Ring A is substituted with one isopropyl. in another aspect of this embodiment, Ring A is substituted with one cyclopropyl.
- Ring A is unsubstituted.
- Ring A is phenyl with one substituent
- Y a is situated meta to the tetrahydropyran ring and the one substituent is situated para to the tetrahydropyran ring.
- the substituent on Ring A is selected from the group consisting of halo, hydroxy, C 1-3 alkyl, C 3 . 7 cycloalkyl, Ci -3 alkoxy, haloCi. 3 alkoxy and 5-membered heteroaryl.
- the substituent on Ring A is selected from the group consisting of chloro, fluoro, hydroxy, methyl, ethyl, isopropyl, cyclopropyl, methoxy, ethoxy, trifluoromethoxy and N-pyrazolyl.
- the substituent on Ring A is chloro.
- the substituent on Ring A is ethyl.
- the substituent on Ring A is isopropyl. in another aspect of this embodiment, the substituent on Ring A is cyclopropyl.
- Linker Y a is a bond or a C ⁇ alkylene. In one embodiment, Y a is unsubstiuted. In one embodiment, Y a is CH 2 . The R 2 substituent(s)
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, C 1-6 alkyl, C ⁇ cycloalkyl, C 6- ioaryl, Ce-ioaryK 4 alkyl, -C(0)OR 3 , -C(0)R 3 , -C(0)NR 4 R 5 ,-NR 4 R 5 , -CH 2 NR 4 R 5 , C 1-6 alkoxy, C 3-7 cycloalkoxy, -CH 2 C(0)OR 3 , -CH 2 C(0)NR 4 R 5 , -NR 3 C(0)NR R 5 , -NR 3 C(0)OR 3 , C 6- 10 aryloxy, C 2 .i 0 heterocyclyl, Ci -10 heteroaryl, C ⁇ oheteroaryloxy and C M oheterocycloxy.
- R 2 for each occurrence, is independently selected from the group consisting of halo, hydroxy, cyano, nitro, C 1-3 alkyl, C 3-7 cycloalkyl, C 6 .ioaryl, C 6 .
- n is 0, 1 or 2. In a further embodiment, n is 0 or 1. In a further embodiment, n is 0. In one embodiment, R 2 is halo and n is 1. In a further embodiment, R 2 is fluoro and n is 1.
- Ring A' contains at least one O or N heteroatom. In a further embodiment, Ring A' contains one or two heteroatoms, wherein the heteroatoms are independently O or N.
- Ring A' contains at least one O, S or N heteroatom. In a further embodiment, Ring A' contains one or two heteroatoms, wherein the heteroatoms are independently O, S, or N.
- Ring A' is selected from the group consisting of a morpholine ring, a pipendine ring, a pyrrolidine ring, a tetrahydropyran ring, a tetrahydrofuran ring and a 1,4-dioxane ring.
- Ring A' is selected from the group consisting of a morpholine ring, a piperidine ring, and a 1 ,4-dioxane ring.
- R for each occurrence, is independently selected from the group consisting of oxo, hydroxy, C -e alkyl, zcycloalkyld ⁇ alkyl, C 6 . 10 ary1Ci-4alkyl, -C(0)OR 3 , -C(0)R 3 , -C(0)NR R 5 and CH 2 C(0)OR 3 ; wherein R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, or two R 2a on adjacent atoms taken together with the atoms to which they are attached may form a fused C 3-7 cycloalkyl, C 6 aryl, 3- to 7-membered heterocyclyl, or 5- membered heteroaryl, wherein the fused cycloalkyl, aryl, heterocyclyl, and heteroaryl may be optionally substituted with one or more substituent independently selected from halo, hydroxy, d ⁇ alkyl, and d
- R 2a for each occurrence, is independently selected from the group consisting of oxo, halo, hydroxy, cyano, nitro, C 1-6 alkyl, C 3 - 7 cycloalkyl, C3.
- R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, cyano, nitro, d.
- R 2a for each occurrence, is independently selected from the group consisting of hydroxy, d. 6 alkyl, C ⁇ cycloalkylC ⁇ alkyl; wherein R 2a may, for each occurrence, be optionally substituted with one or more substituents which are independently selected from halo, hydroxy, C -4 alkyl, and C 1-4 alkoxy; or two R 2a on the same carbon atom taken together may form a spiro C 3- 7cycloalkyl which may be optionally substituted with one or more substituent independently selected from halo, hydroxy, C ⁇ alkyl, and C 1-4 alkoxy.
- R 23 for each occurrence, is independently selected from the group consisting of unsubstituted hydroxy and unsubstituted C 1-2 alkyl.
- q is 0, 1 or 2. In a further embodiment, q is 0 or 1. In a further embodiment, q is 0.
- R 3 for each occurrence, is independently selected from hydrogen, C -3 alkyl, C 3-7 cycloalkyl, C 3 . 7 cycloalkylCi. 3 alkyl, C 6 . 10 aryl, C heteroaryl, and C 2- sheterocyclyl. In a further embodiment, R 3 , for each occurrence, is independently selected from hydrogen and C 1-3 alkyl.
- R 4 and R 5 are independently selected from hydrogen, C 6 -i 0 aryl, Ci - 0 heteroaryl, C 2-10 heterocyclyl, and C 2 -i 0 heterocyclylC 1-4 alkyl; or R 4 and R 5 taken together along with the nitrogen to which they are bound may form a monocyclic or a bicyclic heteroaryl (with 5 to 14 members and having 1 to 8 heteratoms selected from N, O and S) or heterocyclyl (which is a 4 to 7 membered monocyclic ring or a 7 to 12 membered bicyclic ring or a 10 to 15 membered tricyclic ring having at least one heteratom selected from N, O and S) which may be optionally substituted with one or more halo or C 1-4 alkyl substituent.
- R 4 and R 5 are independently selected from hydrogen, d -3 alkyl, C 3 . 7 cycloalkyl, C 3 . 7 cycloalkylC 1-3 alkyl, C e . 10 arylC alkyl, C 6 . 1 0 aryl, C 1-7 heteroaryl, C 1-7 heteroarylC 1-3 alkyl, C 2 . 8 heterocyclyl, and C 2 . 8 heterocyclylC 1 .
- R 4 and R 5 taken together along with the nitrogen to which they are bound may form a monocyclic or a bicycltc heteroaryl or heterocyclyl which may be optionally substituted with one or more halo or C 1-3 alkyl.
- R 4 and R 5 for each occurrence, are independently selected from hydrogen and C 1-3 alkyl.
- the moiety is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl moiety
- the compound is compound 40, 39, 30, 16, 14, 1 , 7, 15, 13, 27, 20, 8, 10, 21 or 1 , or a pharmaceutically acceptable salt thereof.
- the compound is example 8, or a pharmaceutically acceptable salt thereof. More preferably, the compound is example 60, or a pharmaceutically acceptable salt thereof.
- the compound is example 62, or a pharmaceutically acceptable salt thereof. More preferably, the compound is example 71 , or a pharmaceutically acceptable salt thereof.
- the terms “compound of the invention” and “compound of Formula (I)” etc. include pharmaceutically acceptable derivatives thereof and polymorphs, isomers and isotopically labelled variants thereof.
- the term “compounds of the invention” and “compound of Formula (I)” etc. include compounds of formulae (I), (l-a), (l-i), (l-ia), (l-ii), (l-iia), (l-iii), (l-iiia), (l-iv), (l-iva), and (V), and the embodiments thereof disclosed herein.
- Pharmaceutically acceptable derivatives include pharmaceutically acceptable derivatives thereof and polymorphs, isomers and isotopically labelled variants thereof.
- the term “compounds of the invention” and “compound of Formula (I)” etc. include compounds of formulae (I), (l-a), (l-i), (l-ia), (l-ii), (l-iia), (l-iiia), (l-iv
- pharmaceutically acceptable derivative includes any pharmaceutically acceptable salt, solvate, or hydrate of a compound of Formula (I).
- pharmaceutically acceptable salt includes a salt prepared from pharmaceutically acceptable non-toxic acids or bases including inorganic or organic acids and bases.
- compositions of Formula (I) which contain basic, e.g. amino, groups are capable of forming pharmaceutically acceptable salts with acids.
- pharmaceutically acceptable acid addition salts of the compounds of Formula (I) include, but are not limited to, those of inorganic acids such as hydrohalic acids (e.g. hydrochloric, hydrobromic and hydroiodic acid), sulfuric acid, nitric acid, and phosphoric acids.
- pharmaceutically acceptable acid addition salts of the compounds of Formula (I) include, but are not limited to, those of organic acids such as aliphatic, aromatic, carboxylic and sulfonic classes of organic acids, examples of which include: aliphatic monocarboxylic acids such as formic acid, acetic acid, propionic acid or butyric acid; aliphatic hydroxy acids such as lactic acid, citric acid, tartaric acid or malic acid; dicarboxylic acids such as maleic acid or succinic acid; aromatic carboxylic acids such as benzoic acid, p-chlorobenzoic acid, phenylacetic acid, diphenylacetic acid or triphenylacetic acid; aromatic hydroxyl acids such as o-hydroxybenzoic acid, p- hydroxy benzoic acid, 1-hydroxynaphthalene-2-carboxylic acid or 3-hydroxynaphthalene-2-carboxyiic acid; and sulfonic acids such as methanesulfonic acid, ethan
- Other pharmaceutically acceptable acid addition salts of the compounds of Formula (I) include, but are not limited to, those of glycolic acid, glucuronic acid, furoic acid, glutamic acid, anthranilic acid, salicylic acid, mandelic acid, embonic (pamoic) acid, pantothenic acid, stearic acid, sulfanilic acid, algenic acid, and galacturonic acid.
- compositions of Formula (I) which contain acidic, e.g. carboxyl, groups are capable of forming pharmaceutically acceptable salts with bases.
- pharmaceutically acceptable basic salts of the compounds of Formula (I) include, but are not limited to, metal salts such as alkali metal or alkaline earth metal salts (e.g. sodium, potassium, magnesium or calcium salts) and zinc or aluminium salts.
- pharmaceutically acceptable basic salts of the compounds of Formula (I) include, but are not limited to, salts formed with ammonia or pharmaceutically acceptable organic amines or heterocyclic bases such as ethanolamines (e.g. diethanolamine), benzylamines, N-methyl-glucamine, amino acids (e.g. lysine) or pyridine.
- Hemisalts of acids and bases may also be formed, e.g. hemisulphate salts.
- the compounds of the invention may exist in both unsolvated and solvated forms.
- solvate includes molecular complexes comprising a compound of the invention and one or more pharmaceutically acceptable solvent molecules such as water or Ci. 6 alcohols, e.g. ethanol.
- hydrate means a "solvate” where the solvent is water.
- the compounds of the present invention may exist as a hydrate, including a monohydrate, dihydrate, hemihydrate, sesquihydrate, trihydrate, tetrahydrate and the like, as well as the corresponding solvated forms.
- the compound of the invention may be true solvates, while in other cases, the compound of the invention may merely retain adventitious water or be a mixture of water plus some adventitious solvent.
- the compounds of the invention may exist in solid states from amorphous through to crystalline forms. All such solid forms are included within the invention.
- the compounds of the invention may exist as co-crystals. All such co-crystalline forms are included within the invention. In one embodiment, the compounds of the invention exist as co-crystals with L-proline.
- L-proline co-crystal of a compound of the invention such as an L-proline co-crystal of (2S,3R,4R,5S,6R)-2-[4-Chloro-3-(3,4- dihydro-2H-benzo[1 ,4
- these terms encompass: (i) a non-ionic association between L-proline and a compound of the invention (i.e. where no proton transfer has occurred between L-proline and a compound of the invention); or (ii) an ionic interaction where proton transfer between L- proline and a compound of the invention has occurred to form an L-proline salt of the compound of the invention, or (iii) mixtures of (i) and (ii) above.
- the L-proline co-crystal comprises is a non- ionic association between a compound of the invention and L-proline (i.e. where no proton transfer has occurred between L-proline and the compound of the invnetion).
- the L-proline co-crystal is an L-proline salt of the compound of the invention.
- the invention provides a crystalline form of L-proline co-crystal of
- the crystalline form is non- ionic.
- the crystalline form has differential scanning calorimetry endotherms at about 64 °C, about 104 °C and/or about 157 °C.
- the crystalline form has a molar ratio of L-proline to (2S,3R,4R,5S,6R)-2-[4-Chloro-3-(3,4- dihydro-2H-benzo[1 ,4]oxazin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran- 3,4,5-triol of 1 :1.
- the crystalline form has powder X-ray diffraction peak(s) at about 19.3, about 23.2, about 17.0, and/or about 5.7 degrees 2 ⁇ .
- the crystalline form has powder X-ray diffractions peaks substantially the same as those listed in Table 1A.
- the invention provides a crystalline form of L-proline co-crystal of (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1,4]dioxin-6-ylmethyl)-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol.
- the crystalline form is non- ionic.
- the crystalline form has differential scanning calorimetry endotherms at about 151 °C.
- the crystalline form has a molar ratio of L-proline to (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol of 1 : 1.
- the crystalline form has powder X-ray diffraction peak(s) at about 16.7, about 19.9, about 17.6, and/or about 21.9 degrees 2 ⁇ .
- the crystalline form has powder X-ray diffractions peaks substantially the same as those listed in Table 2A.
- the invention provides a crystalline form of L-proline co-crystal of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6- hydroxymethyl-tetrahydro-pyran-3,4,5-triol.
- the crystalline form is non- ionic.
- the crystalline form has differential scanning calorimetry endotherms at about 136 °C.
- the crystalline form has a molar ratio of L-proline to (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol of 1:1.
- the crystalline form has powder X-ray diffraction peak(s) at about 17.3, about 20.4, about 18.0, about 18.9, and/or about 23.8 degrees 2 ⁇ .
- the crystalline form has powder X-ray diffractions peaks substantially the same as those listed in Table 3A.
- the crystalline form has a powder X-ray diffraction spectrum
- the crystalline form has a molar ratio of L-proline to (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro- benzo[1,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5- triol of 2:1.
- the crystalline form has differential scanning calorimetry endotherms at about 176 °C.
- the invention provides a crystalline form of L-proline co-crystal of (2R,3S,4R,5R,6S)-2-Hydroxymethyl-6-[4-isopropyl-3-(1 ,2,3,4-tetrahydro-quinolin-7- ylmethyl)-phenyl]-tetrahydro-pyran-3,4,5-triol.
- the crystalline form is non- ionic.
- the crystalline form has differential scanning calortmetry endotherms at about 156 °C and/or about 158 °C.
- the crystalline form has a molar ratio of L-proline to (2R,3S,4R,5R,6S)-2-Hydroxymethyl-6-[4- isopropyl-3-(1 ,2,3,4-tetrahydro-quinolin-7-ylmethyl)-phenyl]-tetrahydro-pyran-3,4,5-triol of 1 :1.
- the crystalline form has powder X-ray diffractions peaks substantially the same as those listed in Table 4.
- the invention provides a crystalline form of L-pro!ine co-crystal of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6- hydroxymethyl-tetrahydro-pyran-3,4,5-triol.
- the crystalline form has a molar ratio of L-proline to (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol of 2:1.
- the crystalline form is non-ionic.
- the crystalline form has a differential scanning calorimetry endotherm at about 176 °C.
- the crystalline form has powder X-ray diffraction peak(s) at about 6.1 , 9.1 , 12.8, 15.2, 16.5, 17,8, 18.9, 20.9, and/or 28.4.
- the crystalline form has powder X-ray diffraction pattern which is substantially the same as the powder X-ray diffraction pattern shown in Fig. 4.
- the phrase "a molar ratio of about 1 :1" is used to indicate that the crystalline form has between 0.9-1.1 moles of a compound of the invention to 1 mole of L-proline.
- the phrase "a molar ratio of about 1 :2" is used to indicate that the crystalline form has between 0.9-1.1 moles of a compound of the invention to 2 moles of L-proline.
- the present invention relates to a crystalline form of an L- proline co-crystal of a compound of the invention such as, (2S,3R,4R,5S,6R)-2-[4- Chloro-3-(3,4-dihydro-2H-benzo[1 ,4]oxazin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol, (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-y!methyl)-phenyl]-6-hydroxymethy!-tetrahydro-pyran-3,4,5-triol, (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl
- the term "at about” is used to indicate that the precise position of peaks (i.e. the recited 2-theta angle values) should not be construed as being absolute values because, as will be appreciated by those skilled in the art, the precise position of the peaks may vary slightly between one machine and another, from one sample to another, or as a result of slight variations in measurement conditions utilized.
- a powder X-ray diffraction pattern may be obtained which has one or more measurement errors depending on measurement conditions (such as equipment, sample preparation or machine used).
- intensities in an X-ray powder diffraction pattern may fluctuate depending on measurement conditions and sample preparation.
- persons skilled in the art of powder X-ray diffraction will realize that the relative intensity of peaks can be affected by, for example, grains above 30 microns in size and non-unitary aspect ratios, which may affect analysis of samples.
- the skilled person will also realize that the position of reflections can be affected by the precise height at which the sample sits in the diffractometer and the zero calibration of the diffractometer.
- the surface planarity of the sample may also have a small effect.
- the diffraction pattern data presented herein is not to be construed as absolute (for further information see Jenkins, R & Snyder, R. L. ' Introduction to X- Ray Powder Diffractometry * John Wiley & Sons, 1996). Therefore, it shall be
- the crystalline form of an L-proline co-crystal of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol of the present invention is not limited to the crystals that provide powder X-ray diffraction spectra having identical peaks as shown in Fig. 2 or 4, respectively, and any crystals providing X-ray powder diffraction spectra substantially the same as that shown in Fig. 2 or 4, fall within the scope of the present invention.
- Compounds of the invention may exist in one or more geometrical, optical, enantiomeric, diastereomeric and tautomeric forms, including but not limited to c/s- and frans-forms, E- and Z-forms, R-, S- and mesoforms, keto-, and enol-forms. All such isomeric forms are included within the invention.
- the isomeric forms may be in isomerically pure or enriched form, as well as in mixtures of isomers (e.g. racemic or diastereomeric mixtures).
- the invention provides:
- isomers can be separated from their mixtures by the application or adaptation of known methods (e.g. chromatographic techniques and recrystallisation techniques). Where appropriate isomers can be prepared by the application or adaptation of known methods (e.g. asymmetric synthesis).
- Optically active (+) and (-), (f?)- and (S)-, or (D)- and (L)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques, such as HPLC using a chiral column.
- the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers. Likewise, all tautomeric forms are also intended to be included.
- stereoisomer refers to a compound made up of the same atoms bonded by the same bonds but having different three-dimensional structures, which are not interchangeable.
- the present invention contemplates various stereoisomers and mixtures thereof and includes “enantiomers”, which refers to two stereoisomers whose molecules are non-superimposeable mirror images of one another.
- the invention includes pharmaceutically acceptable isotopically-labelled compounds of Formula (I) wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 36 CI, fluorine, such as 18 F, iodine, such as 123 l and 125 l, nitrogen, such as 13 N and 15 N, oxygen, such as 15 0, 17 0 and 18 0, phosphorus, such as 32 P, and sulphur, such as 35 S.
- Certain isotopically-labelled compounds of Formula (I) for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- the radioactive isotopes 3 H and 14 C are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
- Isotopically-labelled compounds of Formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein using an appropriate isotopically-labelled reagent in place of the non-labelled reagent previously employed.
- treatment includes curative and prophylactic treatment.
- a “patient” means an animal, preferably a mammal, preferably a human, in need of treatment.
- the amount of the compound of the invention administered should be a therapeutically effective amount where the compound or derivative is used for the treatment of a disease or condition and a prophylactically effective amount where the compound or derivative is used for the prevention of a disease or condition.
- terapéuticaally effective amount refers to the amount of compound needed to treat or ameliorate a targeted disease or condition.
- prophylactically effective amount used herein refers to the amount of compound needed to prevent a targeted disease or condition.
- the exact dosage will generally be dependent on the patient's status at the time of administration. Factors that may be taken into consideration when determining dosage include the severity of the disease state in the patient, the general health of the patient, the age, weight, gender, diet, time, frequency and route of administration, drug combinations, reaction sensitivities and the patient's tolerance or response to therapy. The precise amount can be determined by routine experimentation, but may ultimately lie with the judgement of the clinician.
- an effective dose will be from 0.01 mg/kg/day (mass of drug compared to mass of patient) to 1000 mg/kg/day, e.g. 1 mg/kg/day to 100 mg/kg/day or 1 mg/kg/day to 10 mg/kg/day.
- Compositions may be administered individually to a patient or may be administered in combination with other agents, drugs or hormones.
- the terms “disease” and “condition” may be used interchangeably or may be different in that the particular malady or condition may not have a known causative agent (so that etiology has not yet been worked out) and it is therefore not yet recognized as a disease but only as an undesirable condition or syndrome, wherein a more or less specific set of symptoms have been identified by clinicians.
- the term “disorder” is synonymous with “condition”.
- inhibitors of Formula (I) have been found to be inhibitors of SGLT.
- inhibition of SGLT means inhibition exclusively of SGLT2, inhibition exclusively of SGLT1 or inhibition of both SGLT1 and SGLT2.
- the invention provides a compound of Formula (I) for use in therapy.
- the invention further provides a pharmaceutical composition comprising a compound of Formula (I) in combination with a pharmaceutically acceptable excipient.
- the invention further provides a method for the treatment of a disease or condition mediated by the sodium D-glucose co-transporter, comprising the step of administering a therapeutically effective amount of a compound of Formula (I) to a patient.
- the invention also provides the use of a compound of Formula (I) in the manufacture of a medicament for the treatment of a disease or condition mediated by the sodium D- glucose co-transporter.
- the invention also provides a compound of Formula (I) for use in treating a disease or condition mediated by the sodium D-glucose co-transporter.
- the SGLT inhibitory activity of the compounds of the invention may be demonstrated by the SGLT2 and SGLT1 assays disclosed hereinbelow.
- Preferred compounds of the invention have an ⁇ ⁇ in the SGLT2 assay of ⁇ 100 n , in one embodiment ⁇ 30 nM, in one embodiment ⁇ 20 nM, in one embodiment ⁇ 10 nM, in another embodiment ⁇ 5 nM, and in another embodiment ⁇ 1 nM, and in another embodiment ⁇ 0.5 nM.
- preferred compounds of the invention have an IC 50 in the SGLT1 assay of ⁇ 10,000 nM, in one embodiment ⁇ 1500 nM, in one embodiment ⁇ 1000 nM, in one embodiment ⁇ 700 nM, in another embodiment ⁇ 500 nM and in another embodiment ⁇ 200 nM.
- the present invention also provides a method of treating diabetes comprising administering a compound of Formula (I) to a subject in need thereof.
- the invention provides a method of treating a disease or condition mediated by the sodium D-glucose co-transporter in a mammal, comprising administering to the mammal in need thereof a therapeutically effective amount of a compound according to any one of claims 1 to 36.
- the compounds of the present invention are useful as both prophylactic and therapeutic treatments for diseases or conditions related to the inhibition of SGLT-2 and SGLT-1.
- the invention is useful for the treatment of a disease or disorder mediated by the sodium D-glucose co-transporter.
- Diseases and conditions mediated by the sodium D- glucose co-transporter include: metabolic disorders, retinopathy, nephropathy, diabetic foot, ulcers, macroangiopathies, metabolic acidosis or ketosis, reactive hypoglycaemia, hyperinsulinaemia, glucose metabolic disorder, insulin resistance, metabolic syndrome (such as dyslipidemia, obesity, insulin resistance, hypertension, microalbumtnemia, hyperuricaemia, and hypercoagulability), dyslipidaemias of different origins, atherosclerosis and related diseases, high blood pressure, chronic heart failure, edema, hyperuricaemia, Syndrome X, diabetes, insulin resistance, decreased glucose tolerance (also known as impaired glucose tolerance, IGT), non-insulin-dependent diabetes mellitus, Type II diabetes, Type I diabetes, diabetic complications, body weight disorders, weight loss, body mass index and leptin related diseases.
- metabolic disorders retinopathy, nephropathy
- the diseases and conditions include metabolic syndrome (such as dyslipidemia, obesity, insulin resistance, hypertension, microalbuminemia, hyperuricaemia, and hypercoagulability), Syndrome X, diabetes, insulin resistance, decreased glucose tolerance (also known as impaired glucose tolerance, IGT), non- 5 insulin-dependent diabetes mellitus, Type II diabetes, Type I diabetes, diabetic complications, body weight disorders, weight loss, body mass index and leptin related diseases.
- metabolic syndrome such as dyslipidemia, obesity, insulin resistance, hypertension, microalbuminemia, hyperuricaemia, and hypercoagulability
- Syndrome X diabetes
- diabetes resistance also known as impaired glucose tolerance, IGT
- non- 5 insulin-dependent diabetes mellitus also known as impaired glucose tolerance, IGT
- Type II diabetes also known as impaired glucose tolerance, IGT
- diabetes or disorder is decreased glucose tolerance, Type II diabetes or obesity.
- Compounds of formula (I) may be also suitable for preventing beta-cell degeneration 10 such as apoptosis or necrosis of pancreatic beta cells, for improving or restoring the functionality of pancreatic cells, increasing the number and size of pancreatic beta cells, for use as diuretics or antihypertensives and for the prevention and treatment of acute renal failure.
- beta-cell degeneration 10 such as apoptosis or necrosis of pancreatic beta cells
- the invention relates to a method for treating a disorder selected I S from type I and type II diabetes mellitus, complications of diabetes, comprising administration of an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- a patient is suffering from "obesity" if the patient exhibits at least one of:
- BMI body mass index
- a patient is suffering from "Type II diabetes” if they meet the World 25 Health Organisation criteria for Diabetes diagnosis (Definition and diagnosis of diabetes mellitus and intermediate hyperglycaemia, WHO, 2006), i.e. the patient exhibits at least one of:
- a venous plasma glucose ⁇ 11.1 mmol/l (200mg/dl) 2 hours after ingestion of 75g 30 oral glucose load As used herein a patient is suffering from "IGT” if they meet the World Health Organisation criteria for IGT diagnosis (Definition and diagnosis of diabetes mellitus and intermediate hyperglycaemia, WHO, 2006), i.e. the patient exhibits both of:
- the compounds of the invention may be administered as a medicament by enteral or parenteral routes, including intravenous, intramuscular, subcutaneous, transdermal, airway (aerosol), oral, intranasal, rectal, vaginal and topical (including buccal and sublingual) administration.
- enteral or parenteral routes including intravenous, intramuscular, subcutaneous, transdermal, airway (aerosol), oral, intranasal, rectal, vaginal and topical (including buccal and sublingual) administration.
- the compounds of Formula (I) should be assessed for their biopharmaceutical properties, such as solubility and solution stability (across pH), permeability, etc., in order to select the most appropriate dosage form and route of administration for treatment of the proposed indication.
- the compounds of the invention may be administered as crystalline or amorphous products.
- the compounds of the invention may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof)- Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients.
- excipient includes any ingredient other than the compound(s) of the invention which may impart either a functional (e.g drug release rate controlling) and/or a nonfunctional (e.g. processing aid or diluent) characteristic to the formulations.
- a functional e.g drug release rate controlling
- a nonfunctional e.g. processing aid or diluent
- the choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound according to Formula (I) and a pharmaceutically acceptable excipient.
- Typical pharmaceutically acceptable excipients include:
- diluents e.g. lactose, dextrose, sucrose, mannito!, sorbitol, cellulose and/or glycine
- lubricants e.g. silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethyleneglycol
- binders e.g. magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone;
- ⁇ disintegrants e.g. starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and/or
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula (I) and a pharmaceutically acceptable excipient.
- the compounds of the invention may be administered orally.
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, and/or buccal, lingual, or sublingual administration by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid plugs, solid microparticulates, semi-solid and liquid (including multiple phases or dispersed systems) such as tablets; soft or hard capsules containing multi- or nano-particulates, liquids (e.g. aqueous solutions), emulsions or powders; lozenges (including liquid-filled); chews; gels; fast dispersing dosage forms; films; ovules; sprays; and buccal/mucoadhesive patches.
- solid plugs solid microparticulates, semi-solid and liquid (including multiple phases or dispersed systems) such as tablets; soft or hard capsules containing multi- or nano-particulates, liquids (e.g. aqueous solutions), emulsions or powders; lozenges (including liquid-filled); chews; gels; fast dispersing dosage forms; films; ovules; sprays; and buccal/mucoadhesive patches.
- Formulations suitable for oral administration may also be designed to deliver the compounds of Formula (I) in an immediate release manner or in a rate-sustaining manner, wherein the release profile can be delayed, pulsed, controlled, sustained, or delayed and sustained or modified in such a manner which optimises the therapeutic efficacy of the said compounds.
- Means to deliver compounds in a rate-sustaining manner are known in the art and include slow release polymers that can be formulated with the said compounds to control their release. Examples of rate-sustaining polymers include degradable and non-degradable polymers that can be used to release the said compounds by diffusion or a combination of diffusion and polymer erosion.
- rate-sustaining polymers examples include hydroxypropyl methylcellulose, hydroxypropyl cellulose, methyl cellulose, ethyl cellulose, sodium carboxymethy! cellulose, polyvinyl alcohol, polyvinyl pyrrolidone, xanthum gum, polymethacry!ates, polyethylene oxide and polyethylene glycol.
- Liquid (including multiple phases and dispersed systems) formulations include emulsions, suspensions, solutions, syrups and elixirs. Such formulations may be presented as fillers in soft or hard capsules (made, for example, from gelatin or hydroxypropylmethylcellulose) and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- the compounds of the invention may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Liang and Chen, Expert Opinion in Therapeutic Patents 2001, 11(6): 981-986.
- the compounds of the invention can be administered parenterally.
- the compounds of the invention may be administered directly into the blood stream, into subcutaneous tissue, into muscle, or into an internal organ.
- Suitable means for administration include intravenous, intraarterial, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, intrasynovial and subcutaneous.
- Suitable devices for administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous or oily solutions. Where the solution is aqueous, excipients such as sugars (including but restricted to glucose, mannitol, sorbitol, etc.) salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile nonaqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water (WFI).
- excipients such as sugars (including but restricted to glucose, mannitol, sorbitol, etc.) salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile nonaqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water (WFI).
- WFI sterile, pyrogen-free
- Parenteral formulations may include implants derived from degradable polymers such as polyesters (i.e. polylactic acid, polylactide, polylactide-co-glycolide, polycapro- lactone, poly hydroxybuty rate), polyorthoesters and polyanhydrides. These formulations may be administered via surgical incision into the subcutaneous tissue, muscular tissue or directly into specific organs.
- degradable polymers such as polyesters (i.e. polylactic acid, polylactide, polylactide-co-glycolide, polycapro- lactone, poly hydroxybuty rate), polyorthoesters and polyanhydrides.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of compounds of Formula (I) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of co-solvents and/or solubility-enhancing agents such as surfactants, micelle structures and cyclodextrins.
- the compounds of the invention can be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler, as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1 ,1,1 ,2-tetrafluoroethane or 1,1,1 ,2,3,3,3- heptafluoropropane, or as nasal drops.
- the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
- the pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- the drug product Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- comminuting method such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- Capsules made, for example, from gelatin or hydroxypropylmethylcellulose
- blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannito!, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate, preferably the latter.
- Other suitable excipients include dextran, glucose, maltose, sorbitol, xylito!, fructose, sucrose and trehalose.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound of the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- a compound of formula (I) of the present invention may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, for use in therapy.
- a compound of the formula (I), or a pharmaceutically acceptable salt thereof, as defined above may be administered simultaneously, sequentially or separately in combination with one or more agents for the treatment of disorders previously listed.
- Therapeutic agents which are suitable for such a combination include, for example, antidiabetic agents such as metformin, sulphonylureas (e.g. glibenclamide, tolbutamide, glimepiride), nateglinide, repaglinide, thiazolidinediones (e.g. rosiglitazone, pioglitazone), PPAR-gamma-agonists (e.g. Gl 262570) and antagonists, PPAR- gamma/alpha modulators (e.g. KRP 297), alpha- glucosidase inhibitors (e.g. acarbose, voglibose), DPPIV inhibitors (e.g.
- antidiabetic agents such as metformin, sulphonylureas (e.g. glibenclamide, tolbutamide, glimepiride), nateglinide, repaglinide, thia
- LAF237, MK-431 alpha2-antagonists, insulin and insulin analogues, GLP-1 and GLP-1 analogues (e.g. exendin-4) or amy!in.
- the list also includes inhibitors of protein tyrosinephosphatase 1 , substances that affect deregulated glucose production in the liver, such as e.g.
- avasimibe or cholesterol absorption inhibitors such as, for example, ezetimibe
- bile acid-binding substances such as, for example, cholestyramine, inhibitors of ileac bile acid transport, HDL-raistng compounds such as CETP inhibitors or ABC1 regulators or active substances for treating obesity, such as sibutramine or tetrahydrolipostatin, dexfenfluramine, axokine, antagonists of the cannabinotdi receptor, MCH-1 receptor antagonists, MC4 receptor agonists, NPY5 or NPY2 antagonists or ⁇ 3- agonists such as SB-418790 or AD-9677 and agonists of the 5HT2c receptor.
- drugs for influencing high blood pressure, chronic heart failure or atherosclerosis such as e.g. A-ll antagonists or ACE inhibitors, ECE inhibitors, diuretics, ⁇ - blockers, Ca-antagonists, centrally acting antihypertensives, antagonists of the alpha-2- adrenergic receptor, inhibitors of neutral endopeptidase, thrombocyte aggregation inhibitors and others or combinations thereof are suitable.
- drugs for influencing high blood pressure, chronic heart failure or atherosclerosis such as e.g. A-ll antagonists or ACE inhibitors, ECE inhibitors, diuretics, ⁇ - blockers, Ca-antagonists, centrally acting antihypertensives, antagonists of the alpha-2- adrenergic receptor, inhibitors of neutral endopeptidase, thrombocyte aggregation inhibitors and others or combinations thereof are suitable.
- angiotensin II receptor antagonists examples include candesartan cilexetil, potassium losartan, eprosartan mesylate, valsartan, telmisartan, irbesartan, EXP-3174, L-158809, EXP- 3312, olmesartan, medoxomil, tasosartan, KT-3-671 , GA-01 13, RU-64276, EMD- 90423, BR-9701 , etc.
- Angiotensin II receptor antagonists are preferably used for the treatment or prevention of high blood pressure and complications of diabetes, often combined with a diuretic such as hydrochlorothiazide.
- a combination with uric acid synthesis inhibitors or uricosurics is suitable for the treatment or prevention of gout.
- a combination with GABA-receptor antagonists, Na-channel blockers, topiramat, protein- kinase C inhibitors, advanced glycation end product inhibitors or aldose reductase inhibitors may be used for the treatment or prevention of complications of diabetes.
- Such combinations may offer significant advantages, including synergistic activity, in therapy.
- the present invention thus provides:
- an agent selected from the group consisting of insulin, insulin derivative or mimetic; insulin secretagogue; insulinotropic sulfonylurea receptor ligand; PPAR ligand; insulin sensitizer; biguanide; alpha-glucosidase inhibitors; GLP-1 , GLP-1 analog or mimetic; DPPIV inhibitor; HMG-CoA reductase inhibitor; squalene synthase inhibitor; FXR or LXR ligand; cholestyramine; fibrates; nicotinic acid, and aspirin in the manufacture of a medicament for the treatment of a disease or condition in a subject mediated by the sodium D-giucose co-transporter, wherein the agent is administered in combination with a compound according to Formula (I);
- a compound according to Formula (I) in the manufacture of a medicament for the treatment of a disease or condition in a subject mediated by the sodium D-glucose co-transporter, wherein the compound is administered in combination with an agent selected from the group consisting of insulin, insulin derivative or mimetic; insulin secretagogue; insulinotropic sulfonylurea receptor ligand; PPAR ligand; insulin sensitizer; biguanide; alpha-glucosidase inhibitors; GLP-1 , GLP-1 analog or mimetic; DPPIV inhibitor; HMG-CoA reductase inhibitor; squalene synthase inhibitor; FXR or LXR ligand; cholestyramine; fibrates; nicotinic acid, and aspirin, and
- a compound according to any one of claims 1 to 36 in combination with an agent selected from the group consisting of insulin, insulin derivative or mimetic; insulin secretagogue; insulinotropic sulfonylurea receptor ligand; PPAR ligand; insulin sensitizer; biguanide; alpha-glucosidase inhibitors; GLP-1, GLP-1 analog or mimetic; DPPIV inhibitor; HMG-CoA reductase inhibitor; squalene synthase inhibitor; FXR or LXR ligand; cholestyramine; fibrates; nicotinic acid, and aspirin in the manufacture of a medicament for treating a disease or condition in a subject mediated by the sodium D- glucose co-transporter,
- an agent selected from the group consisting of insulin, insulin derivative or mimetic; insulin secretagogue; insulinotropic sulfonylurea receptor ligand; PPAR ligand; insulin sensitizer; biguanide; alpha-glu
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula (I) in combination with a therapeutically effective amount of insulin, insulin derivative or mimetic; insulin secretagogue; insulinotropic sulfonylurea receptor ligand; PPAR ligand; insulin sensitizer; biguanide; alpha-glucosidase inhibitors; GLP-1 , GLP-1 analog or mimetic; DPPIV inhibitor; HMG-CoA reductase inhibitor; squalene synthase inhibitor; FXR or LXR ligand; cholestyramine; fibrates; nicotinic acid; or aspirin.
- the invention provides a product comprising a compound of Formula (I) and an agent selected from the group consisting of insulin, insulin derivative or mimetic; insulin secretagogue; insulinotropic sulfonylurea receptor ligand; PPAR ligand; insulin sensitizer; biguanide; alpha-glucosidase inhibitors; GLP-1, GLP-1 analog or mimetic; DPPIV inhibitor; HMG-CoA reductase inhibitor; squalene synthase inhibitor; FXR or LXR ligand; cholestyramine; fibrates; nicotinic acid, and aspirin for simultaneous, separate or sequential use in therapy.
- an agent selected from the group consisting of insulin, insulin derivative or mimetic; insulin secretagogue; insulinotropic sulfonylurea receptor ligand; PPAR ligand; insulin sensitizer; biguanide; alpha-glucosidase inhibitors; GLP-1, GLP-1 analog or mim
- alkyl refers to a fully saturated branched or unbranched hydrocarbon moiety.
- the alkyl comprises 1 to 20 carbon atoms, more preferably 1 to 16 carbon atoms, 1 to 10 carbon atoms, 1 to 6 carbon atoms, or 1 to 4 carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, /so-propyl, n-butyl, sec-butyl, / " so-butyl, fert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl, 2,2- dimethylpentyl, 2,3-dimethylpentyl, n-heptyl, n- octyl, n-nonyl, or n-decyl.
- Alkylene refers to a straight or branched divalent hydrocarbon chain consisting solely of carbon and hydrogen atoms, having from one to twelve carbon atoms, preferably one to 6 carbon atoms, and linking the rest of the molecule to a radical group.
- alkylene groups include methylene, ethylene, propylene, n-butylene, and the like.
- the alkylene is attached to the rest of the molecule through a single bond and to the radical group through a single bond.
- the points of attachment of the alkylene to the rest of the molecule and to the radical group can be through one carbon or any two carbons within the chain.
- Halogen or “halo” may be fluoro, chloro, bromo or iodo.
- alkenyl refers to a monovalent hydrocarbon having at least one carbon- carbon double bond.
- C 2 -C 6 alkenyl refers to a monovalent hydrocarbon having two to six carbon atoms and comprising at least one carbon-carbon double bond.
- alkynyl refers to a monovalent hydrocarbon having at least one carbon- carbon triple bond.
- C 2 -C 6 -alkyny ' refers to a monovalent hydrocarbon having two to six carbon atoms and comprising at least one carbon-carbon triple bond.
- alkoxy refers to alkyl-O-, wherein alkyl is defined herein above.
- Representative examples of alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, 2-propoxy, butoxy, ie i-butoxy, pentyloxy, hexyloxy, cyclopropyloxy-, cyclohexyloxy- and the like.
- alkoxy groups have about 1-6, more preferably about 1-4 carbons.
- AlkyI, alkenyl, atkynyl, and alkoxy groups, containing the requisite number of carbon atoms can be unbranched or branched. The requisite number of carbon may be represented as C ⁇ , C , etc.
- aryl refers to monocyclic or bicyclic aromatic hydrocarbon groups having 6- 10 carbon atoms in the ring portion. Non-limiting examples include phenyl and naphthyl.
- aryl also refers to a group in which a aryl ring is fused to one or more cycloa!kyl or heterocyc!y! rings, where the radical or point of attachment is on the aryl ring.
- Nonlimiting examples include 2,3-dihydro-1H-indene, 1 ,2,3,4-tetrahydronaphthyl and 3,4-dihydro-2H-benzo[b][1 ,4]oxazinyl.
- arylalkyl refers to an aryl group which is linked to another moiety via an alkyl group which may be branched or unbranched.
- alkyl group which may be branched or unbranched.
- arylalkyl groups include benzyl, 2-phenyl-ethyl, 2-(naphth-2-yl)-butan-1-yl, and the like.
- aryloxy refers to an aryl group which is linked to another moiety through an oxygen atom, such as phenoxy.
- heterocyclyl refers to an optionally substituted, saturated or unsaturated non-aromatic ring or ring system, e.g., which is a 4-, 5-, 6-, or 7-membered monocyclic, 7-, 8-, 9-, 10-, 11-, or 12-membered bicyclic or 10-, 11-, 12-, 13-, 14- or 15- membered tricyclic ring system and contains at least one heteroatom selected from O, S and N, where the N and S can also optionally be oxidized to various oxidation states.
- the heterocyclic group can be attached at a heteroatom or a carbon atom.
- the heterocyclyl can include fused or bridged rings as well as spirocyclic rings.
- heterocycles include dihydrofuranyl, [1 ,3]dioxolane, 1 , 4-dioxane, 1 ,4-dithiane, piperazinyl, 1 ,3-dioxolane, imidazolidinyl, imidazo!inyl, pyrrolidine, dihydropyran, oxathiolane, dithiolane, l,3-dioxane, 1 ,3-dithianyl, oxathianyl, thiomorpholinyl, oxiranyl, aziridinyl, oxetanyl, azetidinyl, tetrahydrofuranyl, pyrrolidinyl, tetrahydropyranyl, piperidinyl, morpholinyl, piperazinyl, azepinyl, oxa
- cycloalkyl refers to saturated or partially unsaturated (but not aromatic) monocyclic, bicyclic or tricyclic hydrocarbon groups of 3-12 carbon atoms, preferably 3-9, or 3-7 carbon atoms, Exemplary monocyclic hydrocarbon groups include, but are not limited to, cyc!opropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl or cyclohexenyl.
- Exemplary bicyclic hydrocarbon groups include bornyl, decahydronaphthyl, bicyclo[2.1.1]hexyl, bicyclo[2.2.1]heptyl, bicyclo[2.2.1]heptenyl, 6,6- dimethylbicyclo[3.1.1]heptyl, 2,6,6-trimethylbicyclo[3.1.1]heptyl, or bicyclo[2.2.2]octyl.
- Exemplary tricyclic hydrocarbon groups include adamantyl.
- heterocycloxy refers to a heterocyclyl which is linked to another moiety through an oxygen atom, e.g. piperazin-2-yloxy.
- heteroaryl refers to a 5-14 membered monocyclic- or bicyclic- or polycyclic-aromatic ring system having 1 to 8 heteroatoms selected from N, O or S.
- the heteroaryl is a 5-10 or 5-7 membered ring system.
- monocyclic heteroaryl groups include pyridyl, thienyl, furanyl, pyrrolyl, pyrazolyl, imidazoyi, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazoiyi, oxadiazoiyi, thiadiazolyl and tetrazolyl.
- bicyclic heteroaryl groups include indoly!, benzofuranyl, quinolyl, isoquinolyl indazolyl, indolinyl, isoindolyl, indolizinyl, benzamidazolyl, and quinolinyl.
- heteroaryl also refers to a group in which a heteroaromatic ring is fused to one or more cycloalkyl, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring.
- Nonlimiting examples include 5,6,7, 8-tetrahydroquinoline and 6,7-dihydro-5H-pyrrolo[3,2-d]pyrimidine.
- a heteroaryl group may be mono-, bi-, tri-, or polycyclic, preferably mono-, bi-, or tricyclic, more preferably mono- or bicyclic.
- heteroaryialkyl refers to an heteroaryl group which is linked to another moiety via an alkyl group which may be branched or unbranched.
- heteroaryialkyl groups include 2-(pyridin-3-yl)-ethyl, 3-(quinolin-7-yl)-butan-1 -yl, and the like.
- heteroaryloxy refers to a heteroaryl group which is linked to another moiety through an oxygen atom, such as pyrtdin-3-lyoxy.
- Heteroaryl and “heterocyclyl” is also intended to include oxidized S or N, such as sulfinyl, sulfonyl and N-oxide of tertiary ring nitrogen.
- N-(alkyl)amino refers to an amino group in which one hydrogen is replaced by an alkyl group.
- N-(C . 6 alkyl)amino refers to an amino group in which one of the hydrogens has been replaced with an alkyi group having from 1 to 6 carbon atoms.
- I ⁇ N-di-talky amino refers to an amino group in which both hydrogens have been replaced by an alkyl group which may be the same or different.
- N.N-di-iC ⁇ alkylJamino refers to an amino group in which both of the hydrogens have been replaced with an alkyl group which may be the same or different having from 1 to 6 carbon atoms.
- a “carbamoyl” group as used herein refers to -C(0)NH 2 .
- the term "N-(alkyl)- carbamoyl” refers to a carbamoyl group in which one hydrogen is replaced by an alkyl group.
- N-(C 1-6 alkyl)-carbamoyl refers to a carbamoyl group in which one of the hydrogens has been replaced with an alkyl group having from 1 to 6 carbon atoms.
- N,N-di- ⁇ alkyl)-carbamoyl refers to a carbamoyl group in which both hydrogens have been replaced by an alkyl group which may be the same or different.
- N,N-di- ⁇ C 1-6 alkyl)-carbamoyl refers to a carbamoyl group in which both of the hydrogens have been replaced with an alkyl group which may be the same or different having from 1 to 6 carbon atoms.
- alkanoyl refers to a group having the formula -C(0)-R, wherein R is an alkyl group.
- C1-6alkanoyl refers to an alkanoyl group which has from one to six carbon atoms, such as acetyl, isopropyl-carbonyl, and the like.
- composition comprising
- X may consist exclusively of X or may include something additional, e.g. X + Y.
- Optional or “optionally” means that the subsequently described event of circumstances may or may not occur, and that the description includes instances where said event or circumstance occurs and instances in which it does not.
- optionally substituted aryl means that the aryl radical may or may not be substituted and that the description includes both substituted aryl radicals and aryl radicals having no substitution.
- Salts of compounds of the present invention having at least one salt-forming group may be prepared in a manner known per se.
- salts of compounds of the present invention having acid groups may be formed, for example, by treating the compounds with metal compounds, such as alkali metal salts of suitable organic carboxylic acids, e.g. the sodium salt of 2-ethylhexanoic acid, with organic alkali metal or alkaline earth metal compounds, such as the corresponding hydroxides, carbonates or hydrogen carbonates, such as sodium or potassium hydroxide, carbonate or hydrogen carbonate, with corresponding calcium compounds or with ammonia or a suitable organic amine, stoichiometric amounts or only a small excess of the salt-forming agent preferably being used.
- metal compounds such as alkali metal salts of suitable organic carboxylic acids, e.g. the sodium salt of 2-ethylhexanoic acid
- organic alkali metal or alkaline earth metal compounds such as the corresponding hydroxides, carbonates or hydrogen carbonates, such as sodium
- Acid addition salts of compounds of the present invention are obtained in customary manner, e.g. by treating the compounds with an acid or a suitable anion exchange reagent.
- Internal salts of compounds of the present invention containing acid and basic salt-forming groups, e.g. a free carboxy group and a free amino group, may be formed, e.g. by the neutralisation of salts, such as acid addition salts, to the isoelectric point, e.g. with weak bases, or by treatment with ion exchangers.
- Salts can be converted in customary manner into the free compounds; metal and ammonium salts can be converted, for example, by treatment with suitable acids, and acid addition salts, for example, by treatment with a suitable basic agent.
- Mixtures of isomers obtainable according to the invention can be separated in a manner known per se into the individual isomers; diastereoisomers can be separated, for example, by partitioning between polyphasic solvent mixtures, recrystallisation and/or chromatographic separation, for example over silica gel or by e.g.
- medium pressure liquid chromatography over a reversed phase column and racemates can be separated, for example, by the formation of salts with optically pure salt-forming reagents and separation of the mixture of diastereoisomers so obtainable, for example by means of fractional crystallisation, or by chromatography over optically active column materials.
- Intermediates and final products can be worked up and/or purified according to standard methods, e.g. using chromatographic methods, distribution methods, (re-) crystallization, and the like.
- mixtures of isomers that are formed can be separated into the individual isomers, for example diastereoisomers or enantiomers, or into any desired mixtures of isomers, for example racemates or mixtures of diastereoisomers, for example analogously to the methods described under "Additional process steps”.
- solvents from which those solvents that are suitable for any particular reaction may be selected include those mentioned specifically or, for example, water, esters, such as lower alkyl-lower alkanoates, for example ethyl acetate, ethers, such as aliphatic ethers, for example diethyl ether, or cyclic ethers, for example tetrahydrofuran or dioxane, liquid aromatic hydrocarbons, such as benzene or toluene, alcohols, such as methanol, ethanol or 1- or 2-propanol, nitriles, such as acetonitrile, halogenated hydrocarbons, such as methylene chloride or chloroform, acid amides, such as dimethylformamide or dimethyl acetamide, bases, such as heterocyclic nitrogen bases, for example pyridine or A/-methylpyrroiidin-2-one, carboxylic acid anhydrides, such as lower alkanoic acid anhydrides, for example acetic anhydr
- the compounds, including their salts, may also be obtained in the form of hydrates, or their crystals may, for example, include the solvent used for crystallization. Different crystalline forms may be present.
- the invention relates also to those forms of the process in which a compound obtainable as an intermediate at any stage of the process is used as starting material and the remaining process steps are carried out, or in which a starting material is formed under the reaction conditions or is used in the form of a derivative, for example in a protected form or in the form of a salt, or a compound obtainable by the process according to the invention is produced under the process conditions and processed further in situ.
- the compounds of Formula (I), (l-i), (l-ii), (l-iii), (l-iv), (IA), (Mb), (lie), (lid), (Me), (llf), (ilg), (ilh) and (Hi), can be prepared according to the Schemes provided infra.
- the invention provides, in another aspect, a process for preparing a compound of Formula (I).
- the schemes, outlined below, show general routes for synthesizing compounds of Formula (I).
- any reactive group present such as hydroxyl, amino, carbonyl or imino groups may be protected during the reaction by conventional protecting groups such as trimethylsilyl, tert-butyldimethylsilyl, benzyl, acetal, ketal etc., which are cleaved again after the reaction.
- Compounds of Formula (I) may be prepared from other compounds of Formula (I) by methods well known to one skilled in the art.
- Carboxylic acid of formula (llg) or corresponding acid halide, wherein Lg is a leaving group such as halogen and ring A is as defined herein above, may be reacted with compounds of formula (llh), wherein all symbols are defined herein above, to provide intermediate (II) wherein the symbols are defined herein above.
- the reaction may be carried out in the presence of a Lewis acid followed by reducing the intermediate ketone using reagent such as triethylsilane BF 3 -etherate or under hydrogenation conditions.
- Any mixtures of final products or intermediates obtained can be separated on the basis of the physico-chemical differences of the constituents, in a known manner, into the pure final products or intermediates, for example by chromatography, distillation, fractional crystallisation, or by the formation of a salt if appropriate or possible under the circumstances.
- Step I To a stirred solution of 4H-Benzo[1,4]oxazin-3-one (2.5 g, 16.77 mmol) in DMF (10 mL) was added potassium tert-butoxide (2.81 g, 25.16 mmol) at 0 °C. After stirring for 5 min, methyl iodide (3.54 g, 25.16 mmol) was added and the reaction mixture was stirred for another 3h. The reaction was quenched by addition of water and extracted with ethyl acetate (30 x 2 mL). The organic layer was washed with water (20 mL), and evaporated to get a crude product 2.2 g.
- Step II To a stirred solution of 4-methyl-4H-benzo[1,4]oxazin-3-one (2.18 g, 13.37 mmol) in THF (5 mL) was added borane-tetrahydrofuran complex (4.02 g, 46.8 mmol) at room temperature. After stirring the solution for 2h, the reaction mixture was refluxed for 4h. After complete conversion, reaction mixture was quenched by adding MeOH (10 mL) and evaporated the solvents.
- Step I To a stirred suspension UAIH 4 (7.6 g, 201 mmol) in THF at 0 °C was added 4H- benzo[1 ,4]oxazin-3-one (15 g, 100 mmol) in 30 mL of THF and stirred for 4h at room temperature. After cooling, excess of LiAIH 4 was quenched by the addition of EtOAc followed by aq. NH 4 CI solution. The residue was filtered through a celite bed and filtrate was concentrated. The residue was diluted with water and extracted with ethylacetate (200 ml_X 2), combined organic layer was washed with water (100 mL) and brine (100 mL). Evaporation of the solvent resulted in 3,4-dihydro-2H-benzo[1 > 4]oxazine (12 g) which was used as such for the next step.
- Step I To a stirred solution of 3,4-dihydro-2H-benzo[1 ,4]oxazine (5 g, 37.0 mmol) in DMF (20 mL) was added potassium tert-butoxide (6.22 g, 55.55 mmol) at 0 °C. After stirring for 5 min, bromo-cyclopropane (4.44 mL, 55.55 mmol) was added and the reaction mixture was stirred for another 4h at room temperature. The reaction was quenched by addition of water and extracted with ethyl acetate (50 x 2 mL).
- Step I To a stirred solution of 2-aminophenol (10 g, 9.2 mmol) in DCM (92 mL) at 0 °C was added 2-bromoisobutyryl bromide (11.4 mL, 9.17 mmol) followed by triethyl amine (12.7 mL, 9.2 mmol) and stirred the reaction mixture at 0 °C for 4 h. Reaction mixture was diluted with DCM 100 mL and then washed with water 100 mL dried over sodium sulfate, concentrated on rotavap to give 2-Bromo-N-(2-hydroxy-phenyl)-2-meth yl-propionamide (21.8 g) brown solid which was used for next reaction without purification
- Step II To a stirred solution of 2-Bromo-N-(2-hydroxy-phenyl)-2-methyl-propionamide (21.8 g, 84.4 mmol) in DMF (85 mL) at 25 °C was added potassium carbonate (23.32 g, 168.99 mmol) and stirred the reaction mixture at 80 °C for 4 h.
- Step III To a stirred solution of LAH (3.01 g, 79.10 mmol) in THF (80 mL) at 0 °C was added 2,2-dimethyl-4H-benzo[1 ,4]oxazin-3-one (7.00 g, 39.5 mmol) in portions and stirred the reaction mixture at 25 °C for 1 h and then 50 °C for 3 h.
- Reaction mixture was diluted with DCM (100 mL) and then washed with water (100 mL x 2), brine (100 mL), dried over anhydrous sodium sulfate, concentrated on to give 1-(2,2-dimethyl-2,3-dihydro-benzo[1 ,4]oxazin-4-yl)-2,2,2- trifluoro-ethanone (9.46 g) as brown solid which was purified by column chromatography to furnish 8.7 g pure product.
- Step I To a stirred solution of 4-methyl-3,4-dihydro-2H-benzo[1,4]oxazine (2.00 g, 13.4 mmol) in dichloromethane (30 mL) was added 5-bromo-2-chlorobenzoyl chloride (4.07 g, 16.1 mmol) in dichloromethane (20 mL) at 0 °C followed by addition of AICI 3 (2.14 g, 16.1 mmol). After 2h, the reaction mixture was brought to room temperature and stirred overnight. The reaction was quenched by pouring over crushed ice and extracted with dichloromethane (30 x 2 mL). The organic layer was washed with aq. NaHC0 3 (20 mL), H 2 0 (20mL) and to obtain a crude product 3.0 g.
- Step II To the crude product (0.9 g, 2.45 mmol) in 1 :2 mixture of 1 ,2- dichloroethane/MeCN (12 mL) was added Et 3 SiH (0.83 mL, 5.16 mmol) and BF3.0E.2
- Step III To a stirred solution of 7-(5-bromo-2-chloro-benzyl)-4-methyl-3,4-dihydro-2H- benzo[1 ,4]oxazine (0.37 g, 1.06 mmol) in THFtoluene (5 mL of 1 :2 mixture) was added 1.6 solution of nBuLi in hexanes (0.68 mL, 1.06 mmol) at 78 °C.
- reaction mixture was stirred for 1 h and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (0.49 g, 1.06 mmol) in toluene (5 mL) at -78 °C.
- 0.6 N methanesulfonic acid in methanol 5 mL was added and stirred the reaction mixture for 12 h at room temperature. Reaction was quenched by the addition of aq.
- Step IV To a stirred solution of (2S,3R,4S,5S,6R)-2-[4-chloro-3-(4-methyl-3,4-dihydro- 2H-benzo[1 ,4]oxazin-7-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (0.17 g, 0.37 mmol) in acetonitrile-dichloromethane mixture (1 :1 mixture, 6 mL) was added boron trifluoride diethyletharate complex (0.09 mL, 0.75 mmol), and triethy!silane (0.24 mL, 1.50 mmol) at " 10 °C.
- Step I To a stirred solution of 1-(2,3-dihydro-benzo[1,4]oxazin-4-yl)-2,2,2-trifluoro- ethanone (6.5 g, 28.1 mmol) in dichloromethane (45 mL) was added 5-bromo-2- chlorobenzoyl chloride (8.54 g, 33.7 mmol) in dichloromethane (35 mL) and AICI 3 (5.61 g, 42.2 mmol) at 0 °C. After 2h, the reaction mixture was brought to room temperature and stirred overnight. The reaction was quenched by pouring over crushed ice and extracted with dichloromethane (2 x 50 mL). The organic layer was washed with aq.
- Step II To a stirred solution of 1-[6-(5-bromo-2-chloro-benzoyl)-2,3-dihydro- benzo[1,4]oxazin-4-yl]-2,2,2-trifluoro-ethanone (1.0 g, 2.23 mmol) in 1:2 of 1 ,2- dichloroethane/MeCN (12 mL) was added Et 3 SiH (0.755 mL, 4.68 mmol) and BF 3 .OEt 2 (0.34 mL, 2.90 mmol) simultaneously at 20 °C. The reaction mixture was heated at 50 °C for 4h and quenched by the addition of aq. NaHC0 3 (10 mL).
- Step III To a stirred solution of 1-[6-(5-bromo-2-chloro-benzyl)-2,3-dihydro- benzo[1 ,4]oxazin-4-yl]-2,2,2-trifluoro-ethanqne (8.6 g, 19.8 mmol) in ethanol (40 mL) was added NaBH 4 portion wise and the reaction mixture was stirred overnight. The excess of NaBH 4 was quenched by adding aq. HCI. Ethanol was evaporated and the residue was partitioned between dichloromethane and water.
- Step IV To a stirred solution of 6-(5-bromo-2-chloro-benzyl)-3,4-dihydro-2H- benzo[1 ,4]oxazine (8.0 g, 23.66 mmol) in DMF (35 mL) was added potassium carbonate (6.53 g, 36.0 mmol), benzyl bromide (4.33 mL, 35.50 mmol) and heated to 50 °C for 8 h. Reaction mixture was cooled to room temperature, quenched by the addition of water (50 mL), extracted with ethyl acetate (3 X 20 mL).
- Step V To a stirred solution of 4-benzyl-6-(5-bromo-2-chloro-benzyl)-3,4-dihydro-2H- benzo[1 ,4]oxazine (7.0 g, 16.3 mmol) in THF-toluene (40 mL of 1 :2 mixture) was added 1.6 M solution of n-BuLi in hexanes (10.46 mL, 16.35 mmol) at -78 °C.
- reaction mixture was stirred for 1 h and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (7.62 g, 16.35 mmol) in toluene (25 mL) at -78 °C.
- 0.6 N methanesuifonic acid in methanol (50 mL) was added and stirred for 12 h at room temperature. Reaction was quenched by the addition of aq.
- Step VI To a stirred solution of (2S,3R,4S,5S,6R)-2-[3-(4-benzyl-3,4-dihydro-2H- benzo[1 ,4]oxazin-6-ylmethyl)-4-chloro-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro- pyran-3,4,5-triol (5.0 g, 9.24 mmol) in acetonitrile-dichloromethane mixture (1 :1 mixture, 40 mL) was added boron trifluoride diethyletharate complex (2.34 mL, 18.48 mmol), and triethylsilane (5.95 mL, 36.9 mmol) at -10 °C.
- Step I To a stirred suspension LiAIH 4 (7.6 g, 201 mmol) in THF (70 mL) at 0 °C was added 4H-benzo[1 ,4]oxazin-3-one (15 g, 100 mmol) in 30 mL of THF and the mixture was stirred for 4h at room temperature. After cooling, excess of LiAIH was quenched by the addition of ethyl acetate (30 mL) followed by aq. NH 4 CI solution. The mixture was filtered through a celite bed and the filtrate was concentrated under reduced pressure and extracted with ethyl acetate (3 x 30 mL). The combined organic layers were washed with brine (30 mL) and dried over Na 2 S0 4 . Evaporation of the solvent resulted in benzoxazine (12 g) which was used as such for the next step.
- Step II To an ice-cold solution of benzoxazine (4.5 g, 33.3 mmol) in dichloromethane (25 mL) was added trifluoroacetic anhydride (6.95 mL, 49.9 mmol) and the reaction mixture was stirred for 2h then quenched by the addition of aq. NaHC0 3 solution. The mixture was partitioned between dichloromethane and water. The organic layer was separated, and the aqueous layer was extracted with dichloromethane.
- Step IV To a stirred solution of 1-[6-(5-bromo-2-chloro-benzoyl)-2,3-dihydro- benzo[1 ,4]oxazin-4-yl]-2,2,2-trifluoro-ethanone (36 g, 80.35 mmol) in 1 ,2- dichloroethane:MeCN 1 :2 (180 mL) was added BF 3 .OEt 2 (13.2 mL, 104 mmol) and Et 3 SiH (26.9 mL, 168.7 mmol) at 0 °C. The reaction mixture was stirred overnight at room temperature then quenched by the addition of aq. NaHC0 3 ( ⁇ 200 mL).
- Step V To a stirred solution of 1-[6-(5-bromo-2-chloro-benzyl)-2,3-dihydro- benzo[1 ,4]oxazin-4-yl]-2,2,2-trifluoro-ethanone (8.6 g, 19.8 mmol) in ethanol (40 mL) was added NaBH 4 ⁇ 1.5 g, 39.0 mmol) portion wise and the reaction mixture was stirred overnight. Excess of NaBH 4 was quenched by adding aq. NH 4 CI, and ethanol was evaporated.
- Step VI To a stirred solution of 6-(5-bromo-2-chloro-benzyl)-3,4-dihydro-2H- benzo[1,4]oxazine (8.0 g, 23.66 mmol) in DMF (35 mL) was added potassium carbonate (6.53 g, 47.3 mmol), and benzyl bromide (4.33 mL, 35.50 mmol). The reaction mixture was heated to 60 °C for 8 h, then cooled to room temperature and quenched by the addition of ice-cold water (50 mL).
- Step VII To a stirred solution of 4-benzyl-6-(5-bromo-2-chloro-benzyl)-3,4-dihydro-2H- benzo[1 ,4]oxazine (7.0 g, 16.3 mmol) in THF-toluene 1 :2 (40 mL) was added 1.6 M solution of n-BuLi in hexanes (10.46 mL, 16.35 mmol) at -78 °C.
- reaction mixture was stirred for 1h and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (7.62 g, 16.35 mmol) in toluene (25 mL) at -78 °C.
- 0.6 N methanesulfonic acid in methanol (70 mL) was added and the reaction mixturer was stirred for 12h at room temperature then quenched by the addition of aq. saturated sodium bicarbonate solution ⁇ 25 mL).
- the resulting mixture was extracted with ethyl acetate (3 X 100 mL) and the combined organic layers were dried over sodium sulfate, concentrated and purified by silica gel column
- Step VIII To a stirred solution of (2S,3R,4S,5S,6R)-2-[3-(4-benzyl-3,4-dihydro-2H- benzo[1 ,4]oxazin-6-ylmethyl)-4-chloro-phenyl]-6-hydroxymethyi-2-rnethoxy-tetrahydro- pyran-3,4,5-trioi (5.0 g, 9.24 mmoi) in acetonitrile-dichloromethane mixture 1 :1 (40 mL) was added boron trifluoride diethyietharate complex (2.34 mL, 18.48 mmol), and triethyisilane (5.95 mL, 36.9 mmol) at -5 °C. After stirring for 4h at the same
- Step IX To a solution of (2S,3R,4R,5S,6R)-2-[3-(4-benzyl-3,4-dihydro-2H- benzo[ 1 , 4]oxazin-6-ylmethyl)-4-chloro-phenyl]-6-hyd roxymethyl-tetrahydro-pyran-3,4 , 5- triol ( 2.4 g, 4.68 mmoi) in methanol (15 mL) was added 10% palladium on charcoal (240 mg) and 0.05 mL cone. HCI. The reaction mixture was stirred under hydrogen atmosphere for 2h then filtered through celite bed (which was washed with methanol).
- Step I To a cooled solution of 6-bromo-1-(4-methoxy-benzyl)-1 ,2,3,4-tetrahydro- quinoline (2,5 g, 7.5 mmol) in THF (30 mL) was added 1.6 M n-butyl lithium in hexanes (4.7 mL, 7.5 mmol) at -78 °C, stirred for 30 min. This was transferred to a stirred solution of 5-bromo-2-chlorobenzaldehyde (1.73 g, 7.9 mmol) in THF (30 mL) at -78 °C.
- reaction was quenched by the addition of saturated aqueous solution of ammonium chloride and extracted with ethyl acetate (2X50 mL). The ethyl acetate layer was washed with water, brine, dried over sodium sulphate, and concentrated. The resulting residue was purified by silica gel column chromatography to give (5-bromo-2-chloro-phenyl)-[1-(4-methoxy-benzyl)-1 ,2,3,4-tetrahydro-quinolin-6- yl]-methanol (2.31 g).
- Step III To a stirred solution of 6-(5-bromo-2-chloro-benzyl)-1-(4-methoxy-benzyl)- 1 ,2,3,4-tetrahydro-quinoline (700 mg, 1.5 mmol) in THF-toluene (15 mL of 1:2 mixture) was added 1.6 M solution of n-BuLi in hexanes (1.0 mL, 1.5 mmol) at -78 °C.
- reaction mixture was stirred for 30 min., and then transferred to a stirred solution of 2,3,4,6-tetrakis-0-(trimethylsilyl)-D-glucopyranone (715 mg, 1.5 mmol) in toluene (10 mL) at -78 °C. After stirring for 40 min., 0.6 N methanesulfonic acid in methanol (30 mL) was added and stirred for 20 h at room temperature. Reaction was quenched by the addition of aq.
- Step IV To a stirred solution of (2S,3R,4S,5S,6R)-2- ⁇ 4-chloro-3-[1-(4-methoxy-benzyl)- 1 ,2,3,4-tetrahydro-quinolin-6-ylmethyl]-phenyl ⁇ -6-hydroxymethyl-2-methoxy-tetrahydro- pyran-3,4,5-triol (325 mg, 0.6 mmol) in acetonitrile-dichloromethane mixture (1 :1 mixture, 14 mL) was added triethylsilane (0.4 mL, 2.2 mmol) and boron trifluoride diethyletharate complex (0.15 mL, 1.1 mmol) at -20 °C.
- reaction was quenched with aq. saturated NaHC0 3 solution (8 mL). The volati!es were evaporated under reduced pressure; the resulting mixture was extracted with ethyl acetate (3 X 20 mL).
- AICI 3 (9.03 g, 67.68 mmol) was added over 30 min at a rate to ensure that the temperature did not exceed 4 °C. After 1h, the reaction mixture was taken to room temperature and stirred at 50 °C overnight. The reaction was quenched by pouring over ice and the resulting suspension was diluted with water (100 mL) and extracted with dichloromethane (100 x 2 mL). The organic layer was washed with water (50 mL), brine (50mL) and dried over anhydrous sodium sulfate. Solvent was removed under reduced pressure to get a crude product (12 g).
- Step II To acetic acid (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[3-(3-acetyl-4-hydroxy- benzyl)-4-chloro-phenyl]-tetrahydro-pyran-2-ylmethylester (12.00 g, 20.32 mmol) was added ⁇ , ⁇ -dimethyl formamide dimethyl acetal (3.0 mL, 22.35 mmol). The reaction mixture was stirred at 90 °C overnight. The reaction was quenched by the addition of water (30 mL) and extracted with ethyl acetate (150 mL x 3), solvent was removed under reduced pressure to get a crude product (8.12 g). The crude product was used for next reaction without any purification. MS (ES) m/z 477.9 (M+1)
- Step III To (E)-1- ⁇ 5-[2-Chloro-5-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-hydroxymethyl- tetrahydro-pyran-2-yl)-benzyl]-2-hydroxy-phenyl ⁇ -3-dimethylamino-propenone (8.00 g, 16.77 mmol) in chloroform (80 mL) was added cone. HCI (3 mL). The reaction mixture was refluxed overnight. The reaction was quenched by the addition of water (50 mL) and extracted with ethyl acetate (150 mL x 3), solvent was removed under reduced pressure to get a crude product (4.0 g).
- Step IV To a stirred solution of 6-[2-chloro-5-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6- hydroxymethyl-tetrahydro-pyran-2-yl)-benzyl]-chromen-4-one (0.10 g, 0.2315 mmol) in ethyl acetate (2.5 mL) was added 10% Palladium on C (20 mg, 20 % w/w) followed by methanol (2.5 mL). After strrring for for 18h under hydrogen atmosphere, the reaction mixture was filtered through celite and concentrated to furnish the crude product, which was further purified by Preparative HPLC to yield the title compound (41 mg)
- Step I To the acetic acid (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[3-(3-acetyl-4-hydroxy- benzyl)-4-chloro-phenyl]-tetrahydro-pyran-2-ylmethylester (0.1 g, 0.17 mmol) was added cyclopentanone (0.015 mL, 0.17 mmol) followed by pyrrolidine (0.056 mL, 0.34 mmol). The reaction mixture was subjected to microwave irradiation for 4 min.
- Step II To a stirred solution of the crude product obtained in step 1 in THF:MeOH (3:2, 5 mL) was added lithium hydroxide (0.02 g, 0.52 mmol) in water (1 mL). The reaction mixture was stirred for 3h at room temperature, diluted with water (3 mL), extracted with ethyl acetate (5 mL x 3), solvent was removed under reduced pressure to get a crude product , which was purified by column chromatography to yield the title compound (20 mg).
- Step I To a stirred solution of 5-bromo-2-methoxybenzaldehyde (5.0 g, 23.25 mmol) in toluene (50 mL) was added ethylene glycol (2.6 mL, 46.5 mmol) and p-toluenesulfonic acid monohydrate (0.45 g, 2.32 mmol) and the reaction mixture was azeotroped for 2h, quenched with sat. NaHC0 3 (50 mL).
- Step II To a stirred solution of compound prepared in step I (3.50 g, 13.51 mmol) in THF (20 mL) was added n-butyl lithium (8.5 mL, 13.51 mmol) at -78 °C and stirred for 1 h, Ttetra-OBn-glucaranolactone (7.25 g, 13.51 mmol) in toluene (20 mL) was cooled to -78 °C and Ithium salt prepared above was added to this at -78 °C and stirred for 1h, quenched with sat. NH 4 CI soln.(10 mL) and extracted with ethyl acetate (2X70 mL).
- Step III To a stirred solution of compound prepared in step II (4.80 g, 6.68 mmol) in THF (20 mL) was added 6N HCI (10 mL) and stirred for 16h. This reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate (100 mL) washed with sat.
- Step IV To a stirred solution of 6-bromospiro[chromane-2,f-cyclobutane] (0.563 g, 2.23 mmol) in THF (3 mL) at -78 °C was added n-butyl lithium (1.45 mL, 2.23 mmol) and stirred for 1h.
- Compound obtained in step III (0.3 g, 0.45 mmol) in toluene (3 mL) was cooled to -78 °C and Ithium salt prepared above was added to this at -78 °C.
- This reaction mixture was stirred for 1h, quenched with sat. NH 4 CI (10 mL) and extracted with ethyl acetate (2X20 mL). The ethyl acetate layer was washed with water, brine, dried over sodium sulphate, concentrated and purified by silica gel column
- Example 40 6- ⁇ 2-Methoxy-5-((2S,3S,4R,5R,6R)-3,4,5-tris(benzyloxy)- 6(benzyloxymethyl)tetrahydro-2H-pyran-2-yl)benzyl)spiro[chroman-2,1'- cyclobutane
- Step V To a stirred solution of compound obtained in step IV (0.250 g, 0.29 mmol) in DCE (2 mL) and acetonitrile (2 mL) at -30 °C was added triethylsilane (0.28 g, 1.03 mmol) followed by borntrifluoride.diethyletherate (0.13 g, 1.76 mmol) and stirred at -30 °C for 5h and then at 25 °C for 16h. Reaction was quenched with sat.
- Step VI To a stirred solution of (0.21 g, 0.25 mmol) in ethyl acetate (5 mL) was added Palladium on C 10 % w/w (50 mg) followed by a drop of conc.HCI was added. The reaction was stirred for 18h under hydrogen atmosphere. Reaction mixture was filtered through celite and concentrated to furnish the crude titled compound which was purified by preparative HPLC (22 mg).
- Step I To a stirred solution of acetic acid (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4- chloro-3-(4-ethoxy-benzyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (4.0 g, 6.93 mmol) prepared using the procedures described in J. Med. Chem. 2008, 51 (5), 1145-49, in dichloromethane (40 mL) was added 1 molar solution of BBr 3 (34.6 mL, 34.6 mmol) at - 78 °C under nitrogen atmosphere. Reaction was stirred at -78 °C for 1.5h and -30 °C for 1h.
- Step II To a stirred solution of compound prepared in step I (2 g, 3.64 mmol) in dichloroethane (20 mL) was added TBAB (117 mg, 0.364 mmol), 6% aqueous nitric acid (20 mmol) at 0-5 °C and stirred at room temperature for 4h. Organic layer was separated, washed with water and brine and concentrated to furnish crude acetic acid (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-chloro-3-(4-hydroxy-3-nitro-benzyl)-phenyl]- tetrahydro-pyran-2-ylmethyl ester which was further purified by column chromatography (1 -5 g)
- Step III To a stirred solution of compound prepared in step II (0.70 g, 1.178 mmol) in anhydrous acetonitrile (10 mL) was added anhydrous Cs 2 C0 3 (1.5 g, 4.71 mmol) and 2- bromo-2-methyl-propionic acid ethyl ester (0.6 mL, 5.89 mmol). Reaction was heated to reflux under nitrogen atmosphere for 15h. Additional amount of 2-bromo-2-methyl- propionic acid ethyl ester (0.6 mL, 5.89 mmol) was added at room temperature and heating continued for 15h.
- Reaction mixture was filtered, residue was washed with anhydrous acetonitrile and concentrated to obtaine crude product, which contains varying amounts of products resulting from partial hydrolyses of acetates.
- the crude product was reacetylated by using acetic anhydride, pyridine and DMAP in dichloromethane. Reaction was quenched with aq.
- Step IV To a stirred solution of compound prepared in step III (515 mg, 0.73 mmol) in glacial acetic acid (8 mL) was added iron powder (400 mg, 7.1 mmol) and stirred at 60 °C overnight. Reaction mixture was cooled to room temperature, diluted with EtOAc (15 mL) and filtered through DCite.
- Example 46 6-[2-Chloro-5-( ⁇ 2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-hydroxymethyl- tetrahydro-pyran-2-yl)-benzyl]-2,2-dimethyl-4H-benzo[1,4Joxazin-3-one
- Step t To a stirred solution of ethyl-1 -hydroxycyclopropane carboxylate (2.93 g, 20.5 mmol) in THF (50 mL) was added sodium hydride (60% in mineral oil, 981 mg, 24.5 mmol) under argon atmosphere. After 10 min, 15-crown-5 (0.2 mL) followed by 4- bromo-2-nitro-fluorphenol (4.5 g, 20.5 mmol) were added. The reaction mixture was stirred at room temperature overnight then quenched by the addition of methanol (1.5 mL) and diluted with ethyl acetate.
- Step IV To a stirred solution of 6-bromospiro[3,4-dihydro-1 ,4-benzoxazine-2,1'- cyclopropane] (3.11 g, 12.9 mmol) in DMF (20 mL) was added potassium carbonate (3.6 g, 26.0 mmol) and benzyl bromide (1.61 mL, 13.6 mmol). The reaction mixture was heated at 60 °C for 6h then cooled to room temperature and quenched by the addition of water.
- the reaction mixture was extracted with ethylacetate (2X50 mL) and the combined organic layers were washed with water (20 mL) and brine (20 mL), then dried over sodium sulfate, filtered and concentrated.
- the crude product was purified by silica gel column chromatography to furnish 4-benzy!-6-bromo-spiro[3H-1 ,4-benzoxazine-2,1'- cyclopropane] ⁇ 1.13 g).
- Step V To a stirred solution of 4-benzyl-6-bromo-spiro[3H-1 ,4-benzoxazine-2,1'- cyclopropane] (1.12 g, 3.4 mmol) in THF (10 mL) was added 1.6 M solution of n-BuLi in hexanes (2.12 mL, 3.4 mmol) at -78 °C. The reaction mixture was stirred for 30 min, and then transferred to a stirred solution of 5-bromo-2-chlorobenzaldehyde (745 mg, 3.4 mmol) in THF (10 mL) at -78 °C.
- Trifluoroacetic acid was evaporated under reduced pressure, and the resulting residue was taken in saturated aq. sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was washed with water, brine, dried over sodium sulfate, and concentrated. The resulting residue was purified by silica gel column chromatography to furnish 4-benzyl-6-[(5-bromo-2- chloro-phenyl)methyl]spiro[3H-1 ,4-benzoxazine-2,1'-cyclopropane] (715 mg).
- Step VII To a stirred solution of 4-benzyl-6-[ ⁇ 5-bromo-2-chloro-phenyl)methyl]spiro[3H- 1 ,4-benzoxazine-2,1 -cyclopropane] (710 mg, 1.6 mmol) in THF-toluene (20 mL of 1 :2 mixture) was added 1.6 M solution of n-BuLi in hexanes (1.6 mL, 1.6 mmol) at -78 °C.
- Step VIII To a stirred solution of (3R,4S,5S,6R)-2-[3-[(4-benzylspiro[3H-1 ,4- benzoxazine-2,1'-cyclopropane]-6-yl)methyl]-4-chloro-phenyl]-6-(hydroxymethyl)-2- methoxy-tetrahydropyran-3,4,5-triol (340 mg, 0.6 mmol) in acetonitrile-dichloromethane (6 mL, 1 :1 mixture) was added triethylsilane (0.4 mL, 2.4 mmol) and boron trifluoride diethyletharate complex (0.15 mL, 1.2 mmol) at -5 °C.
- Step IX To a solution of (2S,3R,4R,5S,6R)-2-[3-[(4-benzylspiro[3H-1 ,4-benzoxazine- 2,1 '-cyclopropane]-6-yl)methyl]-4-chloro-phenyl]-6-(hydroxymethyl)tetrahydropyran- 3,4,5-triol (300 mg) in methanol ⁇ 3 mL), was added ethyl acetate (0.5 mL), 10% palladium on charcoal (40 mg), and 0.3 mL cone. HCI.
- Step I To a stirred solution of acetic acid (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4- chloro-3-(4-ethoxy-benzyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (3.0 g, 5.2 mmol, prepared using the procedures described in J. Med. Chem. 2008, 51 (5), 1145-49) in dichloromethane (30 mL) was added boron tribromide solution (1.0 M in DCM, 26.0 mL, 26.0 mmol) at -78 °C.
- Step II To a stirred solution of (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-chloro-3-(4- hydroxy-benzyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g) in ethylenedichloride (55 mL) was added 6% HN0 3 (22.2 mL) and tetra-butyl ammonium bromide (324 mg). The reaction mixture was heated at 50 °C for 15 min. then cooled and diluted with dichloromethane.
- Step IV To a stirred solution of acetic acid (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[3-(3- amino-4-hydroxy-benzyl)-4-chloro-phenyl]-tetrahydro-pyran-2-ylmethyl ester (1.8 g, 3.2 mmol) in acetonitrile (15 ml_) was added 2-chloroacrylonitrile (0.35 ml_, 4.5 mmol) and potassium carbonate (882 mg, 6.4 mmol). After the reaction was refluxed overnight, the mixture was filtered through a celite bed.
- Example 51 6-[2-Chloro-5-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-hydroxymethyl- tetrahydro-pyran-2-yl)-benzyl]-3,4-dihydro-2H-benzo[1,4]oxazine-2-carbonitrile
- Step V To a stirred solution of acetic acid (2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4- chloro-3-(2-cyano-3,4-dihydro-2H-benzo[1 ,4]oxazin-6-ylmethyl)-phenyl]-tetrahydro- pyran-2-ylmethyl ester (430 mg) in THF : Methanol : water (2:1 :1 mixture, 4 ml.) was added lithium hydroxide (20 mg). After stirring at room temperature overnight, the reaction mixture was concentrated. The resulting residue was taken up in 50% methanol in ethyl acetate then filtered through celite bed.
- Example 52 6-[2-chloro-5-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-hydroxymethyl- tetrahydro-pyran-2-yl)-benzyl]-3,4-dihydro-2H-benzo[1,4]oxazine-2-carboxyltc acid methyl ester Step I.
- Step II To a stirred solution of 6-[2-chloro-5-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6- hydroxymethyl-tetrahydro-pyran-2-yl)-benzyl]-3,4-dihydro-2H-benzo[1 ,4]oxazine-2- carboxylic acid methyl ester (190 mg) in THF: Methanol: water (1 :1 :1 mixture, 1.5 mL) was added lithium hydroxide (17 mg). After stirring at room temperature overnight, the reaction mixture was concentrated, and the resulting residue was taken up in 50% methanol in ethyl acetate.
- Step I To a stirred solution of 2-bromo-5-iodobenzoic acid (1.0 g, 3.06 mmol) in DCM (5 mL) was added DMF (0.2 mL) and oxalyl chloride (0.44 mL, 4.59 mmol) at 0 °C. After complete addition, the reaction mixture was stirred at room temperature for 3h. The volatiles were evaporated under reduced pressure, and the crude product was dissolved in DCM (4 mL) and added to chroman (488 mg, 3.67 mmol) which had been cooled to 0°C. To this mixture was added aluminum chloride (488 mg, 3.67 mmol) in portions.
- Step // To a stirred solution of (2-bromo-5-iodo-phenyl)-chroman-6-yl-methanone (2.0 g, 4.51 mmol) in acetonitrile: dichloromethane (2:1 mixture, 9 mL) was added triethylsilane (2.52 mL, 15.78 mmol) and boron trifluoride diethyl etherate complex ( 1.11 mL, 9.02 mmol ) at 0 °C. After stirring overnight at room temperature, reaction was quenched by the addition of saturated aqueous sodium bicarbonate solution. Volatiles were evaporated under reduced pressure.
- Step III To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-chroman (1.0 g, 2.33 mmol) in dry THF (6 mL) was added n-BuLi (1.6 M in hexane, 1.45 mL, 2.33 mmol) at -78°C. After stirring for 45 min, the reaction mixture was transferred to a cooled solution of 2,3,4,6-tetrakis-0-(benzyl)-D-glucopyranone (1.66 g, 2.56 mmol) in THF (6 mL) at - 78°C.
- Example 54 6-[2-bromo-5-((3S,4R,5R,6R)-3,4,5-tris-benzyloxy-6-benzyloxymethyl- tetrahydro-pyran-2-yl)-benzyl]-chroman
- Step IV To a stirred solution of 6-[2-bromo-5-((3R,4S,5R,6R)-3,4,5-tris-benzyloxy-6- benzyloxymethyl-2-methoxy-tetrahydro-pyran-2-yl)-benzyl]-chroman (900 mg, 1.05 mmol) in acetonitrile : dichloromethane (1 :1 mixture, 6 mL) was added triethylsilane (0.34 mL, 2.1 mmol) and boron trifluoride diethyletharate complex (0.19 mL, 1.58 mmol), at 0 °C.
- Example 55 6-[2-cyclopropyl-5-((3S,4R,5R,6R)-3,4,5-tris-benzyloxy-6- benzyloxymethyl-tetrahydro-pyran-2-yl)-benzyl]-chroman
- Step V To a stirred solution of 6-[2-bromo-5-((3S,4R,5R,6R)-3,4,5-tris-benzyloxy-6- benzyloxymethyl-tetrahydro-pyran-2-yl)-benzyl]-chroman (300 mg, 0.363 mmol) in toluene (1.6 mL) was added tricyclohexylphosphine (10 mg), potassium phosphate (346 mg, 1.63 mmol), water (81 ⁇ ), cyclopropylboronic acid (93 mg, 1.09 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (4 mg) was added.
- Step VI To a stirred solution of 6-[2-cyclopropyl-5-((3S,4R,5R,6R)-3,4,5-tris-benzyloxy- 6-benzyloxymethyl-tetrahydro-pyran-2-yl)-benzyl]-chroman (650 mg) in THF (5 mL) was added 10 % palladium charcoal activated (dry) (100 mg), methanol (5 mL), and cone. HCI (0.2 mL). The reaction mixture was stirred under hydrogen atmosphere (bladder pressure) overnight then filtered through a celite bed.
- Step I To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
- Step II To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI 3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2).
- Step III To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure.
- Step IV To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C.
- reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C.
- 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq.
- Step V To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C.
- Example 58 [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2 ! 3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
- Step V To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature.
- Example 59 Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-cyclopropyl-3-(2,3- dihydro-benzo[1,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester
- Step I To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (100 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and cyclopropylboronic acid (4.06 g, 47.2 mmol).
- Step II To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4- cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2- ylmethyl ester (10.5 g, 17.61 mmol) in methanol:THF:water 3:2:1 (120 mL) was added lithium hydroxide (813 mg, 19.37 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure.
- Example 61 Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
- Step I To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol).
- Step II To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure.
- acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol
- Step I To a stirred solution of 1-(2,3-dihydro-benzo[1 ,4]oxazin-4-yl)-2,2,2-trifluoro- ethanone (9.2 g, 39.77 mmol) in dichloromethane (70 mL) was added 5-iodo-2- bromobenzoyl chloride (13.7 g, 39.77 mmol) in dichloromethane (30 mL) at 0 °C followed by addition of AICI 3 (13.3 g, 99.41 mmol). After 3h, the reaction mixture was brought to room temperature and stirred overnight. The reaction was quenched by pouring it over crushed ice and the resultanting mixture was extracted with
- Step II To a stirried solution of 6-(2-bromo-5-iodo-benzoyl)-4-(2,2,2-trifluoro-acetyl)-4H- benzo[1 ,4]oxazin-3-one (16.0 g, 29.252 mmol) in 1 ,2-dichloroethane/MeCN (1 :2 mixture, 60 mL) was added triethylsilane (9.9 mL, 62.43 mmol) and borontrifluoride diethyletherate complex (4.9 mL, 38.51 mmol) simultaneously at -10 °C. After stirring overnight at room temperature, the reaction was heated at 50 °C for 3h.
- Step IV To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-3,4-dihydro-2H- benzo[1 ,4]oxazine (9.4 g, 21.86 mmol) in DMF (50 mL) was added potassium
- Step V To a stirred solution of 4-benzyl-6-(2-bromo-5-iodo-benzyl)-3,4-dihydro-2H- benzo[1 ,4]oxazine (2.0 g, 3.85 mmol) in THF (20 mL) was added n-Butyl lithium (2.4 mL, 3.85 mmol) at -78 °C and the mixture was stirred for 1h. This was transferred to a solution of 2,3,4,6-tetrakis-0-(benzyl)-D-glucopyranone (2.07 g, 3.85 mmol) in THF (18 mL) at -78 °C. After stirring for 1h, the reaction was quenched with Sat.
- Step VII To a stirred solution of 4-benzyl-6-[2-bromo-5-((2S,3S,4R,5R,6R)-3,4,5-tris- benzyloxy-6-benzyloxymethyl-tetrahydro-pyran-2-yl)-benzyl]-3,4-dihydro-2H- benzo[1 ,4]oxazine (0.35 g, 0.38 mmol) in toluene : water (10:1 mixture, 10 mL) was added cyclopropylboronic acid (49.2 mg, 0.5731 mmol) tricyclohexylphosphine (26.7 mg, 0.0955 mmol), and potassium phosphate (0.28 g, 1.34 mmol).
- reaction mixture was degassed for 45 min then palladium (II) acetate (8.5 mg, 0.03821 mmol) was added. After heating overnight at 100 °C, the reaction mixture was cooled to room temperature and water (20 mL) was added.
- Step VIII To a solution of 4-benzyl-6-[2-cyclopropyl-5-((2S,3S,4R,5R,6R)-3,4,5-tris- benzyloxy-6-benzyloxymethyl-tetrahydro-pyran-2-yl)-benzyl]-3,4-dihydro-2H- benzo[1 ,4]oxazine ( 0.42 g, 0.4783 mmol) in THF (4.7 mL) was added 10% palladium on charcoal (80 mg), 0.1 mL cone. HCI followed by methanol (4.7 mL) and the mixture was stirred under hydrogen atmosphere for 18h.
- Step I To a solution of 2-bromoisopropyl benzene (2.0 g, 10.0 mmole) in dry THF (20 ml_), nBuLi (1.6 M in hexane) , (6.9 ml_,11.05 mmole) was added at -78 °C and the mixture was stirred at same temperature for one hour. DMF (0.9 g, 12.0 mmole) was added and the mixture was stirred at -78 °C for half an hour then allowed to stir at 0 °C for 15 min. The reaction mixture was diluted with saturated aqueous ammonium chloride (10 mL) and extracted with EtOAc (3X30 ml_).
- Step II A solution of 2-isopropyl benzaldehyde ⁇ 1.5 g, 10.13 mmole) in DCM ⁇ 10 mL) was added to a solution of AICI 3 (2.6g, 20.26 mmole) in DCM (10mL) at 0 °C followed by addition of a dilute solution of Br 2 ⁇ 0.67 mL, 13.1 mmole in 20 mL DCM) to the reaction mixture. The solution was stirred at 0 C C for 6 hours then stirred overnight at room temperature.
- the reaction mixture was basified using saturated aqueous sodium bicarbonate and extracted with DCM (30X2 mL). The organic layers were combined and the crude product was obtained by evaporation of the solvent. The crude product was purified by column chromatography using 1% EtOAc in Hexane to yield 5-bromo-2- isopropylbenzaldehyde ⁇ 800 mg).
- Step III To a solution of 4-Benzyl-6-bromo-3 I 4-dihydro-2H-benzo[1 ,4]oxazine ⁇ 1.5 g, 4.93 mmol) in THF (20 mL) was added 1.6 M n-butyl lithium in hexanes ⁇ 3.0 mL, 74.93 mmol) at -78 °C. The reaction was stirred for 30 min. then transferred to a stirred solution of 5-bromo-2-isopropylbenzaldehyde (1.12 g, 4.93 mmol) in THF (15 mL) at -78 °C.
- Step IV To an ice cold solution of (4-Benzyl-3,4-dihydro-2H-benzo[1 ,4]oxazin-6-yl)-(5- bromo-2-isopropyl-phenyl)-methanol (1.3 g, 2.87 mmol) in dichlorom ethane (25 mL) was added Et 3 SiH (4.8 mL, 5.70 mmol) followed by BF 3 .OEt 2 (0.74 mL, 5.7 mmol). The reaction mixture was stirred at room temperature overnight then quenched by the addition of aq. NaHC0 3 .
- reaction mixture was extracted with ethyl acetate (3 x 30 mL), and the combined organic layers were washed with brine (30 mL) and dried over sodium sulfate.
- Crude product obtained after evaporation of the solvent was purified by silica gel column chromatography to furnish 4-Benzyl-6-(5-bromo-2-isopropyl-benzyl)- 3,4-dihydro-2H-benzo[1 ,4]oxazine (1.0 g).
- Step V To a stirred solution of 4-Benzyl-6-(5-bromo-2-isopropyl-benzyl)-3,4-dihydro-2H- benzo[1 ,4]oxazine (1.0g, 2.29 mmol) in THF-toluene (15 mL of 1:2 mixture) was added 1.6 M solution of n-BuLi in hexanes (1.40 mL, 1.40 mmol) at -78 °C. The reaction mixture was stirred for 30 min.
- Step VII To a solution (2S,3R,4R,5S,6R)-2-[3-(4-benzyl-3,4-dihydro-2H- benzo[1 ,4]oxazin-6-ylmethyl)-4-isopropyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran- 3,4,5-triol (520 mg, 1.0 mmol) in methanol ( 5 mL) was added 10% palladium on charcoal (150 mg), 0.05 mL cone. HCI and the mixture was stirred under hydrogen balloon pressure for 18 h. The reaction mixture was filtered through a celite bed, and the celite was washed with methanol.
- Step I To a solution of 2-bromoisopropyl benzene (2.0 g, 10.0 mmole) in dry THF (20 mL), nBuLi (1.6 M in hexanes) (6.9 mL, 11.05 mmole) was added at -78 °C and the mixture was stirred at same temperature for one hour. DMF (0.9 g, 12.0 mmole) was added and the mixture was stirred at -78 °C for an additional half an hour, then allowed to stir at 0 °C for 15 min. The reaction mixture was diluted with saturated aqueous ammonium chloride (10 mL) and extracted with ethyl acetate (3X30 mL).
- Step II To a solution of trifluoroacetic acid (50ml) and 2-isopropylbenzaldehyde (2.0 g, 13.5 mmol) was added cone, sulphuric acid (98%) (10ml) at room temperature, followed by N-bromosuccinamide (NBS, 3.6 g 20.2 mmol) in portions. After 2 hrs, the mixture was poured into ice water and extracted with dichloromethane (3x30 mL).
- Step I To a stirred solution of 7-bromo-1 , 2,3,4-tetrahydroquinoline (7.0 g, 33.0 mmol) in DMF (50 mL) was added potassium carbonate (13.6 g, 99.0 mmol), and benzyl bromide (4.33 mL, 36.3 mmol), and the mixture was heated to 60 °C for 12 h, then cooled to room temperature and quenched by the addition of ice-cold water (150 mL).
- Step II To a solution of 1-benzyl-7-bromo-1,2,3,4-tetrahydro-quinoline (2.50 g, 8.27 mmol) in THF (20 mL) was added 1.6 M n-butyl lithium in hexanes (5.14 mL, 8.27 mmol) at -78 °C. The mixture was stirred for 45 min. then transferred to a stirred solution of 5-bromo-2-isopropylbenzaldehyde (1.87 g, 8.27 mmol) in THF (15 mL) at -78 °C.
- Step III To a solution of (1-benzyl-1 ,2,3,4-tetrahydro-quinolin-7-yl)-(5-bromo-2- isopropyl-phenyl)-methanol (2.61 g, 5.79 mmol) in TFA (7.0 mL), Et 3 SiH (4.63 mL, 28.95 mmol) was added followed by triflic acid (1.0 mL, 11.5 mmol) at room
- Step IV To a stirred solution of 1-benzyl-7-(5-bromo-2-isopropyl-benzyl)-1 , 2,3,4- tetrahydro-quinoline (1.50g, 3.45 mmol) in THF-toluene 1 :2 (15 mL) was added 1.6 M solution of n-BuLi in hexanes (2.16 mL, 3.45 mmol) at -78 °C.
- reaction mixture was stirred for 45 min., and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (1.60g, 3.45 mmol) in toluene (10 mL) at -78 °C.
- 0.6 N methanesulfonic acid in methanol 15 mL was added and the mixture was stirred for 20 h at room temperature.
- the reaction was quenched by addition of aq. saturated NaHCO 3 (10 mL) and the resulting mixture was extracted with ethyl acetate (3 X 20 mL).
- Example 70 ⁇ 2S,3R,4R,5S,6R)-2-[3-(1-Benzyl-1,2,3,4-tetrahydro-quinolin-7- ylmethyl)-4-isopropyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
- Step V To a stirred solution of (2S,3R,4S,5S,6R)-2-[3-(1-benzyl-1 ,2,3,4-tetrahydro- quinolin-7-ylmethyl)-4-isopropyl-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (1.20 g, 2.19 mmol) in acetonitrile-dichloroethane 1 :1 (20 mL) was added triethylsilane (1.39 mL, 8.76 mmol) at room temperature, then the reaction mixture cooled to -50 to -60 °C and boron trifluoride diethyletharate complex (0.55 mL, 4.38 mmol) was added dropwise, and the reaction mixture was stirred at same temperature and allowed to stir at below -30 °C for 2 hours and at -20 °C for 1 hour and then below 0 °C for 1 hour.
- Step-VI To a solution (2S,3R,4R,5S,6R)-2-[3-(1-benzyl-1 ,2,3,4-tetrahydro-quinolin-7- ylmethyl)-4-isopropyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (1.0 g, 1.0 mmol) in methanol (20 mL) was added 10% dry palladium on charcoal (200 mg) and cone. HCI (0.2 mL), and the mixture was stirred under hydrogen balloon pressure for 18 h. The reaction mixture was filtered through a celite bed which was washed with methanol and the filtrate was concentrated.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyrrole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description



















































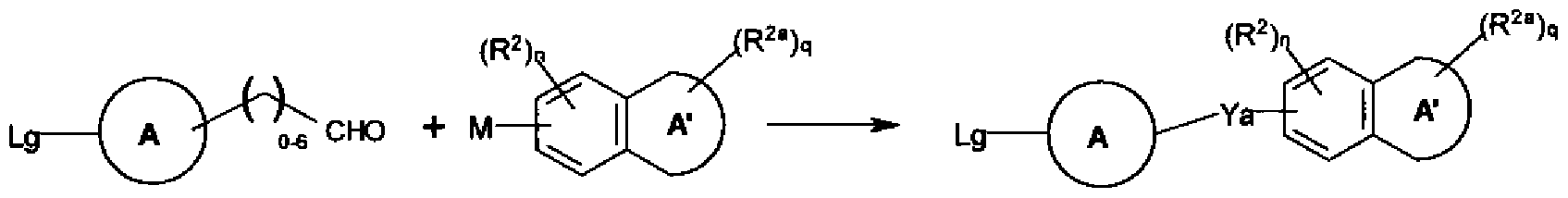





























































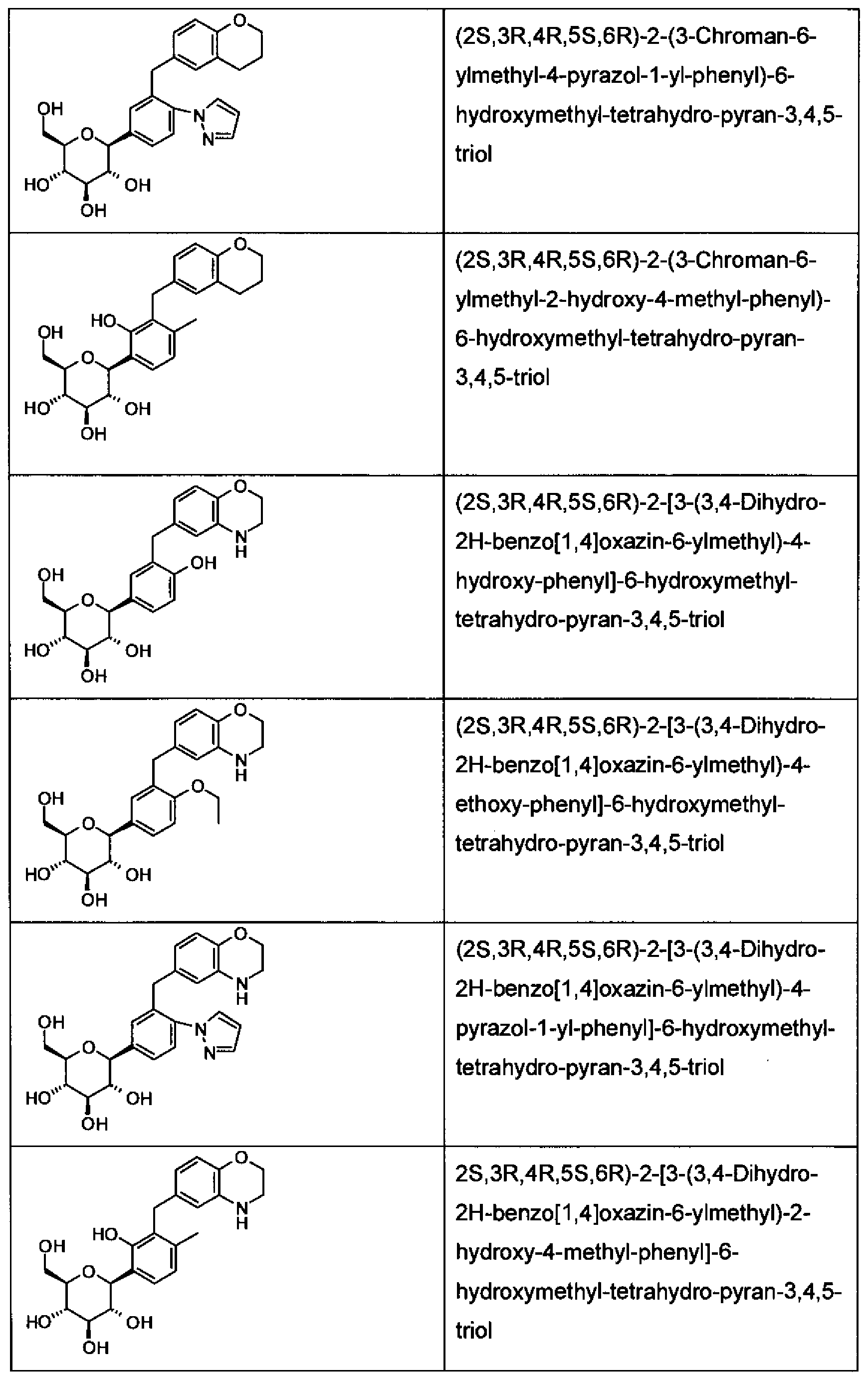
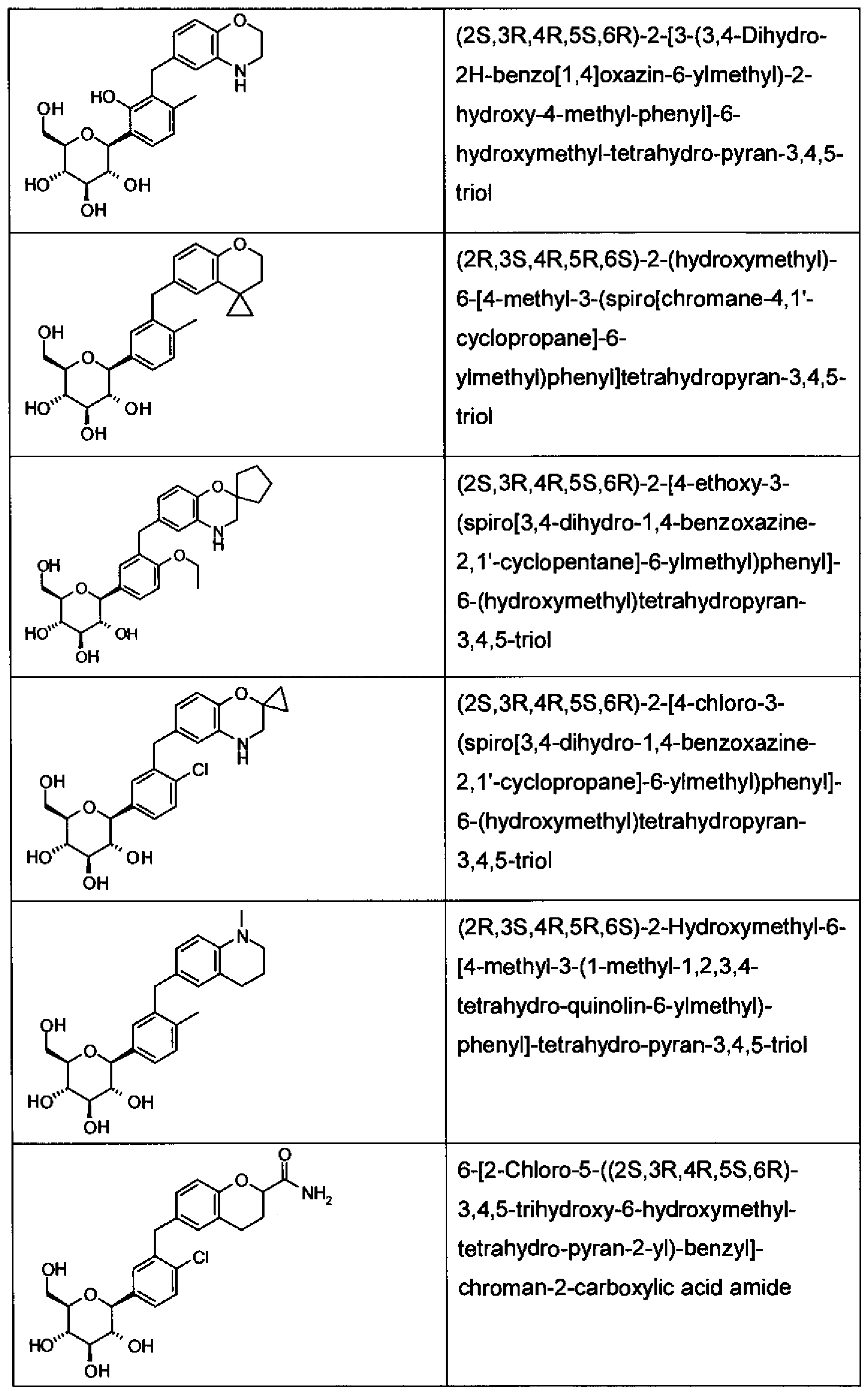

















Claims


















Priority Applications (25)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020127012873A KR101465308B1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| UAA201204674A UA109415C2 (en) | 2009-10-20 | 2010-10-19 | GLYCOSIDE DERIVATIVES AND THEIR APPLICATIONS |
| NZ598717A NZ598717A (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| AU2010309833A AU2010309833B2 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| BR112012009053-4A BR112012009053B1 (en) | 2009-10-20 | 2010-10-19 | GLYCOSIDE DERIVATIVES, THEIR USES, AND PHARMACEUTICAL COMPOSITIONS |
| CU2012000062A CU24079B1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives |
| CA2777812A CA2777812C (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| RS20160112A RS54563B1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| KR1020147008569A KR20140054415A (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| SI201031162A SI2491029T1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| SG2012016820A SG179562A1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| CN201080044483.5A CN102656165B (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| EP10768927.5A EP2491029B1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| ES10768927.5T ES2564191T3 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| DK10768927.5T DK2491029T3 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| PL10768927T PL2491029T3 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| JP2012534672A JP5384744B2 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| EA201200600A EA023781B1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
| MX2012004700A MX2012004700A (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof. |
| ZA2012/01680A ZA201201680B (en) | 2009-10-20 | 2012-03-07 | Glycoside derivatives and uses thereof |
| TNP2012000109A TN2012000109A1 (en) | 2009-10-20 | 2012-03-09 | Glycoside derivatives and uses thereof |
| IL219309A IL219309A (en) | 2009-10-20 | 2012-04-19 | Glycoside derivatives, pharmaceutical compositions comprising them and use thereof for the preparation of medicaments for treating diabetes |
| MA34854A MA33737B1 (en) | 2009-10-20 | 2012-05-11 | Antraquinone derivatives and their use |
| HK12108866.0A HK1168099A1 (en) | 2009-10-20 | 2012-09-11 | Glycoside derivatives and uses thereof |
| HRP20160436TT HRP20160436T1 (en) | 2009-10-20 | 2016-04-21 | Glycoside derivatives and uses thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2173DE2009 | 2009-10-20 | ||
| IN2173/DEL/2009 | 2009-10-20 | ||
| IN2689DE2009 | 2009-12-23 | ||
| IN2689/DEL/2009 | 2009-12-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011048112A1 true WO2011048112A1 (en) | 2011-04-28 |
Family
ID=43216622
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2010/065747 WO2011048112A1 (en) | 2009-10-20 | 2010-10-19 | Glycoside derivatives and uses thereof |
Country Status (39)
| Country | Link |
|---|---|
| US (7) | US8163704B2 (en) |
| EP (1) | EP2491029B1 (en) |
| JP (2) | JP5384744B2 (en) |
| KR (2) | KR101465308B1 (en) |
| CN (1) | CN102656165B (en) |
| AR (1) | AR078685A1 (en) |
| AU (1) | AU2010309833B2 (en) |
| BR (1) | BR112012009053B1 (en) |
| CA (1) | CA2777812C (en) |
| CL (1) | CL2012000991A1 (en) |
| CO (1) | CO6531486A2 (en) |
| CR (1) | CR20120176A (en) |
| CU (1) | CU24079B1 (en) |
| CY (1) | CY1117409T1 (en) |
| DK (1) | DK2491029T3 (en) |
| EA (1) | EA023781B1 (en) |
| EC (1) | ECSP12011909A (en) |
| ES (1) | ES2564191T3 (en) |
| GT (1) | GT201200117A (en) |
| HK (1) | HK1168099A1 (en) |
| HN (1) | HN2012000798A (en) |
| HR (1) | HRP20160436T1 (en) |
| HU (1) | HUE027531T2 (en) |
| IL (1) | IL219309A (en) |
| JO (1) | JO3152B1 (en) |
| MA (1) | MA33737B1 (en) |
| MX (1) | MX2012004700A (en) |
| MY (1) | MY158326A (en) |
| NZ (1) | NZ598717A (en) |
| PE (1) | PE20121281A1 (en) |
| PL (1) | PL2491029T3 (en) |
| RS (1) | RS54563B1 (en) |
| SG (1) | SG179562A1 (en) |
| SI (1) | SI2491029T1 (en) |
| TN (1) | TN2012000109A1 (en) |
| TW (2) | TWI594756B (en) |
| UY (1) | UY32958A (en) |
| WO (1) | WO2011048112A1 (en) |
| ZA (1) | ZA201201680B (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012140597A1 (en) * | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and their uses for the treatment of diabetes |
| WO2012140596A1 (en) * | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and uses thereof |
| EP2530079A1 (en) * | 2010-01-26 | 2012-12-05 | Tianjin Institute Of Pharmaceutical Research | Phenyl c-glucoside derivatives, preparation methods and uses thereof |
| CN102827122A (en) * | 2011-06-17 | 2012-12-19 | 山东亨利医药科技有限责任公司 | Glucoside derivate |
| CN102863417A (en) * | 2011-07-09 | 2013-01-09 | 山东轩竹医药科技有限公司 | C-indican derivative |
| WO2012165914A3 (en) * | 2011-06-01 | 2013-03-28 | Green Cross Corporation | Novel diphenylmethane derivatives as sglt2 inhibitors |
| JP2014513693A (en) * | 2011-05-09 | 2014-06-05 | ヤンセン ファーマシューティカ エヌ.ベー. | (2S, 3R, 4R, 5S, 6R) -2- (3-((5- (4-fluorophenyl) thiophen-2-yl) methyl) -4-methylphenyl) -6- (hydroxymethyl) tetrahydro- Co-crystal of 2H-pyran-3,4,5-triol with L-proline and citric acid |
| WO2014203217A1 (en) | 2013-06-21 | 2014-12-24 | Lupin Limited | Substituted heterocyclic compounds as crac modulators |
| CN104250272A (en) * | 2013-06-27 | 2014-12-31 | 上海方楠生物科技有限公司 | Method for preparing Invokana medicine intermediate by using micro-reactor |
| WO2015032272A1 (en) * | 2013-09-09 | 2015-03-12 | 江苏豪森药业股份有限公司 | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| CN105294624A (en) * | 2015-11-16 | 2016-02-03 | 山东罗欣药业集团股份有限公司 | Preparation method for dapagliflozin |
| US9296775B2 (en) | 2012-05-10 | 2016-03-29 | Eli Lilly And Company | Pyrazole compounds |
| JP2016185963A (en) * | 2011-07-15 | 2016-10-27 | ノバルティス アーゲー | Aza bicyclic diaryl ether salt, method for manufacturing the same and method for manufacturing precursor thereof |
| US9573970B2 (en) | 2013-11-08 | 2017-02-21 | Eli Lilly And Company | 4-{4-[(1E)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-1-en-1-yl]-2-methylbenzyl}-5-(propan-2-yl)-1H-pyrazol-3-yl beta-D glucopyranoside acetate |
| US9790231B2 (en) | 2013-06-24 | 2017-10-17 | Lupin Limited | Chromane and chromene derivatives and their use as CRAC modulators |
| WO2018089449A1 (en) * | 2016-11-10 | 2018-05-17 | Janssen Pharmaceutica Nv | Benzocyclobutane derivatives useful as dual sglt1/sglt2 modulators |
| WO2018158744A1 (en) * | 2017-03-03 | 2018-09-07 | Novartis Ag | Sglt1/2 inhibitor lik066 for treating obesity |
| WO2018235020A1 (en) * | 2017-06-21 | 2018-12-27 | Novartis Ag | Licofligozin for the treatment of non-alcoholic steatohepatitis |
| WO2019129588A1 (en) * | 2017-12-27 | 2019-07-04 | Bp Oil International Limited | Methods for preparing fuel additives |
| US10544135B2 (en) | 2011-04-13 | 2020-01-28 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of SGLT2 |
| WO2020021447A1 (en) | 2018-07-25 | 2020-01-30 | Novartis Ag | Nlrp3 inflammasome inhibitors |
| WO2020039394A1 (en) | 2018-08-24 | 2020-02-27 | Novartis Ag | New drug combinations |
| US10617668B2 (en) | 2010-05-11 | 2020-04-14 | Janssen Pharmaceutica Nv | Pharmaceutical formulations |
| US10640496B2 (en) | 2016-06-17 | 2020-05-05 | Daewoong Pharmaceutical Co., Ltd. | Method for producing diphenylmethane derivative |
| US10696662B2 (en) | 2017-08-21 | 2020-06-30 | Janssen Pharmaceutica Nv | 5-fluoro-C-(aryl or heterocyclyl)-glycoside derivatives useful as dual SGLT1 / SGLT2 modulators |
| WO2020234715A1 (en) | 2019-05-17 | 2020-11-26 | Novartis Ag | Nlrp3 inflammasome inhibitors |
| WO2021014350A1 (en) | 2019-07-23 | 2021-01-28 | Novartis Ag | Combination treatment of liver diseases using fxr agonists |
| US11359151B2 (en) | 2017-12-27 | 2022-06-14 | Bp Oil International Limited | Methods for preparing fuel additives |
| RU2775228C2 (en) * | 2017-06-21 | 2022-06-28 | Новартис Аг | Lycofligosin for treatment of non-alcoholic steatohepatitis |
| US11384302B2 (en) | 2017-12-27 | 2022-07-12 | Bp Oil International Limited | Methods for preparing fuel additives |
| US11384057B2 (en) | 2017-12-27 | 2022-07-12 | Bp Oil International Limited | Methods for preparing fuel additives |
| US11421168B2 (en) | 2017-12-27 | 2022-08-23 | Bp Oil International Limited | Methods for preparing fuel additives |
| US11576894B2 (en) | 2009-07-08 | 2023-02-14 | Janssen Pharmaceutica Nv | Combination therapy for the treatment of diabetes |
| WO2024028782A1 (en) | 2022-08-03 | 2024-02-08 | Novartis Ag | Nlrp3 inflammasome inhibitors |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2011009824A (en) | 2009-03-23 | 2012-01-25 | Glenmark Pharmaceuticals Sa | Fused pyrimidine-dione derivatives as trpa1 modulators. |
| US8163704B2 (en) * | 2009-10-20 | 2012-04-24 | Novartis Ag | Glycoside derivatives and uses thereof |
| AR087701A1 (en) * | 2011-08-31 | 2014-04-09 | Japan Tobacco Inc | PIRAZOL DERIVATIVES WITH SGLT1 INHIBITING ACTIVITY |
| CN105007914A (en) * | 2013-03-11 | 2015-10-28 | 詹森药业有限公司 | Dual SGLT1/SGLT2 inhibitors |
| CN104017031A (en) * | 2014-06-21 | 2014-09-03 | 李友香 | Hypoglycemic drug and composition |
| CN104031098A (en) * | 2014-06-21 | 2014-09-10 | 李友香 | Hypoglycemic medicine |
| CN108203432B (en) * | 2016-12-20 | 2021-03-02 | 宜昌东阳光长江药业股份有限公司 | Glucopyranosyl derivative and application thereof in medicine |
| WO2018189671A1 (en) * | 2017-04-12 | 2018-10-18 | Novartis Ag | Use of lik066 in heart failure patients |
| CN108863736A (en) * | 2018-07-27 | 2018-11-23 | 福州大学 | A kind of preparation method of the aromatic carboxylic acids of carbonyl functionalization |
| WO2020151623A1 (en) * | 2019-01-24 | 2020-07-30 | 北京盈科瑞创新药物研究有限公司 | Compound, preparation method therefor, and medical uses of intermediate thereof |
| WO2020151620A1 (en) * | 2019-01-24 | 2020-07-30 | 北京盈科瑞创新药物研究有限公司 | Compound, and preparation method therefor and application thereof as drug intermediate |
| CN111099975A (en) * | 2019-12-23 | 2020-05-05 | 河北合佳医药科技集团股份有限公司 | Preparation method of 5-bromo-2-chloro-4' -ethoxy benzophenone |
| WO2022105845A1 (en) * | 2020-11-19 | 2022-05-27 | 北京盈科瑞创新药物研究有限公司 | Glucoside derivative, and preparation method therefor and application thereof |
Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001016147A1 (en) | 1999-08-31 | 2001-03-08 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 (en) | 1999-10-12 | 2001-04-19 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 (en) | 2000-03-17 | 2001-09-20 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 (en) | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-aryl glucoside sglt2 inhibitors and method |
| JP2001288178A (en) * | 2000-02-02 | 2001-10-16 | Kotobuki Seiyaku Kk | C-glycoside, method for producing the same and medicine containing the same |
| WO2003020737A1 (en) | 2001-09-05 | 2003-03-13 | Bristol-Myers Squibb Company | O-pyrazole glucoside sglt2 inhibitors and method of use |
| US20040018998A1 (en) | 2000-09-29 | 2004-01-29 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| WO2004018491A1 (en) | 2002-08-23 | 2004-03-04 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004080990A1 (en) | 2003-03-14 | 2004-09-23 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| WO2004099230A1 (en) | 2003-05-12 | 2004-11-18 | Fujisawa Pharmaceutical Co., Ltd. | Monosaccharide compounds |
| WO2005011592A2 (en) | 2003-08-01 | 2005-02-10 | Janssen Pharmaceutica N.V. | Substituted indazole-o-glucosides |
| WO2005021566A2 (en) | 2003-08-26 | 2005-03-10 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085265A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085237A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| US20060009400A1 (en) | 2004-07-06 | 2006-01-12 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948A1 (en) | 2004-07-17 | 2006-01-26 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349A1 (en) | 2004-07-27 | 2006-02-02 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| WO2006011502A1 (en) | 2004-07-27 | 2006-02-02 | Chugai Seiyaku Kabushiki Kaisha | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| US20060035841A1 (en) | 2004-08-11 | 2006-02-16 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031A1 (en) | 2004-10-01 | 2006-04-06 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2006054629A1 (en) | 2004-11-18 | 2006-05-26 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| US20060293252A1 (en) | 2002-07-11 | 2006-12-28 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014A1 (en) | 2006-07-28 | 2008-01-31 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| WO2008016132A1 (en) | 2006-08-04 | 2008-02-07 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5410031A (en) * | 1992-02-24 | 1995-04-25 | The Regents Of The University Of California Office Of Technology Transfer | Nucleoside cotransporter protein cDNA |
| US5410054A (en) | 1993-07-20 | 1995-04-25 | Merck Frosst Canada, Inc. | Heteroaryl quinolines as inhibitors of leukotriene biosynthesis |
| WO1998031697A1 (en) | 1997-01-15 | 1998-07-23 | Sankyo Company, Limited | Aryl c-glycoside compounds and sulfated esters thereof |
| US6515117B2 (en) * | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| JP4272319B2 (en) * | 1999-11-30 | 2009-06-03 | アイシン精機株式会社 | Headrest device |
| US6627611B2 (en) * | 2000-02-02 | 2003-09-30 | Kotobuki Pharmaceutical Co Ltd | C-glycosides and preparation of thereof as antidiabetic agents |
| US6936590B2 (en) * | 2001-03-13 | 2005-08-30 | Bristol Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| WO2002080936A1 (en) * | 2001-04-04 | 2002-10-17 | Ortho Mcneil Pharmaceutical, Inc. | Combination therapy comprising glucose reabsorption inhibitors and ppar modulators |
| CA2443325C (en) | 2001-04-04 | 2011-06-14 | Ortho-Mcneil Pharmaceutical, Inc. | Combination therapy comprising glucose reabsorption inhibitors and retinoid-x receptor modulators |
| CA2507665A1 (en) | 2002-12-04 | 2004-06-17 | Kissei Pharmaceutical Co., Ltd. | Preventive or remedy for diseases caused by hyperglycemia |
| DE10258008B4 (en) | 2002-12-12 | 2006-02-02 | Sanofi-Aventis Deutschland Gmbh | Heterocyclic fluoroglycoside derivatives, medicaments containing these compounds and methods of making these medicaments |
| DE10258007B4 (en) | 2002-12-12 | 2006-02-09 | Sanofi-Aventis Deutschland Gmbh | Aromatic fluoroglycoside derivatives, medicaments containing these compounds and methods for the preparation of these medicaments |
| EP1444890B1 (en) | 2003-02-05 | 2009-01-07 | Hermann Koepsell | RS1 deficient transgenic animal and uses thereof |
| PT1651658E (en) * | 2003-08-01 | 2013-03-07 | Mitsubishi Tanabe Pharma Corp | Novel compounds having inhibitory activity against sodium-dependant transporter |
| DE102004028241B4 (en) | 2004-06-11 | 2007-09-13 | Sanofi-Aventis Deutschland Gmbh | New fluoroglycoside derivatives of pyrazoles, medicines containing these compounds and manufacture of these medicines |
| CA2577632C (en) | 2004-09-22 | 2014-04-01 | Soraya Shirazi-Beechey | Intestinal epithelial glucose sensor |
| UA91546C2 (en) * | 2005-05-03 | 2010-08-10 | Бьорінгер Інгельхайм Інтернаціональ Гмбх | Crystalline form of 1-chloro-4-(я-d-glucopyranos-1-yl)-2-[4-((s)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, a method for its preparation and the use thereof for preparing medicaments |
| TWI370818B (en) * | 2006-04-05 | 2012-08-21 | Astellas Pharma Inc | Cocrystal of c-glycoside derivative and l-proline |
| WO2007140191A2 (en) * | 2006-05-23 | 2007-12-06 | Theracos, Inc. | Glucose transport inhibitors and methods of use |
| WO2008115574A1 (en) | 2007-03-21 | 2008-09-25 | Reliant Pharmaceuticals, Inc. | Cb1 antagonist and a dyslipidemic agent and/or metabolic regulator, and methods of making and using same |
| TW200904454A (en) | 2007-03-22 | 2009-02-01 | Bristol Myers Squibb Co | Methods for treating obesity employing an SGLT2 inhibitor and compositions thereof |
| WO2008131224A2 (en) | 2007-04-18 | 2008-10-30 | Tethys Bioscience, Inc. | Diabetes-related biomarkers and methods of use thereof |
| EP2197433A4 (en) | 2007-08-20 | 2010-09-08 | Sinai School Medicine | Regulating glp-1 and sglt-1 in gastrointestinal cells |
| PL2187742T3 (en) | 2007-08-23 | 2018-06-29 | Theracos Sub, Llc | (2s,3r,4r,5s,6r)-2-(4-chloro-3-benzylphenyl)-6-(hydroxymethyl)tetrahydro-2h-pyran-3,4,5-triol derivatives for use in the treatment of diabetes |
| CL2008003653A1 (en) | 2008-01-17 | 2010-03-05 | Mitsubishi Tanabe Pharma Corp | Use of a glucopyranosyl-derived sglt inhibitor and a selected dppiv inhibitor to treat diabetes; and pharmaceutical composition. |
| CN101969944B (en) * | 2008-01-31 | 2013-04-10 | 安斯泰来制药有限公司 | Pharmaceutical composition for treatment of fatty liver diseases |
| BRPI0913129A2 (en) | 2008-05-22 | 2016-01-05 | Bristol Myers Squibb Co | method for treating hyperuricemia employing an sglt2 inhibitor and composition containing the same |
| WO2009143010A1 (en) | 2008-05-22 | 2009-11-26 | Bristol-Myers Squibb Company | Method for treating hyponatremia employing an sglt2 inhibitor and composition containing same |
| WO2009143021A1 (en) | 2008-05-22 | 2009-11-26 | Bristol-Myers Squibb Company | Method for treating and preventing kidney stones employing an sglt2 inhibitor and composition containing same |
| CN102159206A (en) | 2008-09-19 | 2011-08-17 | 诺瓦提斯公司 | Glucoside derivatives and uses thereof as sglt inhibitors |
| BRPI0919411A2 (en) | 2008-09-19 | 2015-12-15 | Novartis Ag | glycoside derivative and uses thereof |
| WO2010045656A2 (en) | 2008-10-17 | 2010-04-22 | Nectid, Inc. | Novel sglt2 inhibitor dosage forms |
| US9056850B2 (en) | 2008-10-17 | 2015-06-16 | Janssen Pharmaceutica N.V. | Process for the preparation of compounds useful as inhibitors of SGLT |
| WO2010048358A2 (en) | 2008-10-22 | 2010-04-29 | Auspex Pharmaceutical, Inc. | Ethoxyphenylmethyl inhibitors of sglt2 |
| WO2010074219A1 (en) | 2008-12-26 | 2010-07-01 | アステラス製薬株式会社 | Benzothiophen compound |
| US20120035105A1 (en) | 2009-01-09 | 2012-02-09 | Sdg, Inc. | Insulin Therapies for the Treatment of Diabetes, Diabetes Related Ailments, and/or Diseases or Conditions Other Than Diabetes or Diabetes Related Ailments |
| CN104906582A (en) | 2009-02-13 | 2015-09-16 | 勃林格殷格翰国际有限公司 | Pharmaceutical composition comprising a SGLT2 inhibitor, a DPP-IV inhibitor and optionally a further antidiabetic agent and uses thereof |
| MA33043B1 (en) | 2009-02-13 | 2012-02-01 | Boehringer Ingelheim Int | Sglt-2 inhibitor for type 1 diabetes, type 2 diabetes, glucose imbalance or hyperglycemia |
| WO2010128152A1 (en) * | 2009-05-07 | 2010-11-11 | Novartis Ag | Fused heterocyclic c-glycosides for the treatment of diabetes |
| US8163704B2 (en) | 2009-10-20 | 2012-04-24 | Novartis Ag | Glycoside derivatives and uses thereof |
-
2010
- 2010-10-18 US US12/906,682 patent/US8163704B2/en active Active
- 2010-10-19 WO PCT/EP2010/065747 patent/WO2011048112A1/en active Application Filing
- 2010-10-19 MY MYPI2012001056A patent/MY158326A/en unknown
- 2010-10-19 CN CN201080044483.5A patent/CN102656165B/en not_active Expired - Fee Related
- 2010-10-19 TW TW105117771A patent/TWI594756B/en not_active IP Right Cessation
- 2010-10-19 KR KR1020127012873A patent/KR101465308B1/en active IP Right Grant
- 2010-10-19 AU AU2010309833A patent/AU2010309833B2/en not_active Ceased
- 2010-10-19 EP EP10768927.5A patent/EP2491029B1/en active Active
- 2010-10-19 CU CU2012000062A patent/CU24079B1/en active IP Right Grant
- 2010-10-19 EA EA201200600A patent/EA023781B1/en not_active IP Right Cessation
- 2010-10-19 DK DK10768927.5T patent/DK2491029T3/en active
- 2010-10-19 PL PL10768927T patent/PL2491029T3/en unknown
- 2010-10-19 HU HUE10768927A patent/HUE027531T2/en unknown
- 2010-10-19 NZ NZ598717A patent/NZ598717A/en not_active IP Right Cessation
- 2010-10-19 RS RS20160112A patent/RS54563B1/en unknown
- 2010-10-19 TW TW099135639A patent/TWI558723B/en not_active IP Right Cessation
- 2010-10-19 KR KR1020147008569A patent/KR20140054415A/en not_active Application Discontinuation
- 2010-10-19 SI SI201031162A patent/SI2491029T1/en unknown
- 2010-10-19 CA CA2777812A patent/CA2777812C/en active Active
- 2010-10-19 JP JP2012534672A patent/JP5384744B2/en not_active Expired - Fee Related
- 2010-10-19 SG SG2012016820A patent/SG179562A1/en unknown
- 2010-10-19 MX MX2012004700A patent/MX2012004700A/en active IP Right Grant
- 2010-10-19 ES ES10768927.5T patent/ES2564191T3/en active Active
- 2010-10-19 BR BR112012009053-4A patent/BR112012009053B1/en not_active IP Right Cessation
- 2010-10-19 PE PE2012000511A patent/PE20121281A1/en active IP Right Grant
- 2010-10-20 UY UY0001032958A patent/UY32958A/en active IP Right Grant
- 2010-10-20 JO JOP/2010/0359A patent/JO3152B1/en active
- 2010-10-20 AR ARP100103826A patent/AR078685A1/en active IP Right Grant
-
2012
- 2012-03-07 ZA ZA2012/01680A patent/ZA201201680B/en unknown
- 2012-03-09 TN TNP2012000109A patent/TN2012000109A1/en unknown
- 2012-03-21 US US13/425,888 patent/US8466114B2/en active Active
- 2012-04-12 CR CR20120176A patent/CR20120176A/en unknown
- 2012-04-19 CL CL2012000991A patent/CL2012000991A1/en unknown
- 2012-04-19 HN HN2012000798A patent/HN2012000798A/en unknown
- 2012-04-19 IL IL219309A patent/IL219309A/en active IP Right Grant
- 2012-04-20 GT GT201200117A patent/GT201200117A/en unknown
- 2012-04-26 CO CO12068967A patent/CO6531486A2/en active IP Right Grant
- 2012-05-11 MA MA34854A patent/MA33737B1/en unknown
- 2012-05-17 EC ECSP12011909 patent/ECSP12011909A/en unknown
- 2012-09-11 HK HK12108866.0A patent/HK1168099A1/en not_active IP Right Cessation
- 2012-09-13 US US13/614,534 patent/US8828951B2/en active Active
-
2013
- 2013-10-02 JP JP2013207478A patent/JP2014040451A/en active Pending
-
2014
- 2014-05-30 US US14/291,268 patent/US20140274921A1/en not_active Abandoned
-
2015
- 2015-01-23 US US14/604,173 patent/US20150141354A1/en not_active Abandoned
-
2016
- 2016-04-19 CY CY20161100330T patent/CY1117409T1/en unknown
- 2016-04-20 US US15/134,016 patent/US9895389B2/en not_active Ceased
- 2016-04-21 HR HRP20160436TT patent/HRP20160436T1/en unknown
-
2020
- 2020-02-18 US US16/793,734 patent/USRE49080E1/en active Active
Patent Citations (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001016147A1 (en) | 1999-08-31 | 2001-03-08 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 (en) | 1999-10-12 | 2001-04-19 | Bristol-Myers Squibb Company | C-aryl glucoside sglt2 inhibitors |
| JP2001288178A (en) * | 2000-02-02 | 2001-10-16 | Kotobuki Seiyaku Kk | C-glycoside, method for producing the same and medicine containing the same |
| WO2001068660A1 (en) | 2000-03-17 | 2001-09-20 | Kissei Pharmaceutical Co., Ltd. | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 (en) | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-aryl glucoside sglt2 inhibitors and method |
| US20040018998A1 (en) | 2000-09-29 | 2004-01-29 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| WO2003020737A1 (en) | 2001-09-05 | 2003-03-13 | Bristol-Myers Squibb Company | O-pyrazole glucoside sglt2 inhibitors and method of use |
| US20060293252A1 (en) | 2002-07-11 | 2006-12-28 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| WO2004018491A1 (en) | 2002-08-23 | 2004-03-04 | Kissei Pharmaceutical Co., Ltd. | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| EP1609785A1 (en) * | 2003-03-14 | 2005-12-28 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| WO2004080990A1 (en) | 2003-03-14 | 2004-09-23 | Astellas Pharma Inc. | C-glycoside derivatives and salts thereof |
| WO2004099230A1 (en) | 2003-05-12 | 2004-11-18 | Fujisawa Pharmaceutical Co., Ltd. | Monosaccharide compounds |
| WO2005011592A2 (en) | 2003-08-01 | 2005-02-10 | Janssen Pharmaceutica N.V. | Substituted indazole-o-glucosides |
| WO2005021566A2 (en) | 2003-08-26 | 2005-03-10 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085265A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085237A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| US20060009400A1 (en) | 2004-07-06 | 2006-01-12 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948A1 (en) | 2004-07-17 | 2006-01-26 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349A1 (en) | 2004-07-27 | 2006-02-02 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| WO2006011502A1 (en) | 2004-07-27 | 2006-02-02 | Chugai Seiyaku Kabushiki Kaisha | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| EP1803721A1 (en) * | 2004-07-27 | 2007-07-04 | Chugai Seiyaku Kabushiki Kaisha | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| US20060035841A1 (en) | 2004-08-11 | 2006-02-16 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031A1 (en) | 2004-10-01 | 2006-04-06 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2006054629A1 (en) | 2004-11-18 | 2006-05-26 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| US20080027014A1 (en) | 2006-07-28 | 2008-01-31 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| WO2008016132A1 (en) | 2006-08-04 | 2008-02-07 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
Non-Patent Citations (15)
| Title |
|---|
| CURR: OPINON IN INVESTIGATIONAL DRUGS, vol. 8, no. 4, 2007, pages 285 - 292 |
| DIABETES CARE, vol. 13, 1990, pages 610 |
| DIABETES, vol. 48, 1999, pages 1794 - 1800 |
| DIABETES, vol. 57, 2008, pages 1723 - 1729 |
| GENNARO: "Remington: The Science and Practice of Pharmacy", 2000 |
| H. LIEBERMAN; L. LACHMAN: "Pharmaceutical Dosage Forms: Tablets", vol. 1, 1980, MARCEL DEKKER |
| HOUBEN-WEYL: "Methods of Organic Synthesis", vol. 21, 1952, THIEME |
| J. F. W. MCOMIE: "Protective Groups in Organic Chemistry", 1973, PLENUM PRESS |
| J. MED. CHEM., vol. 51, no. 5, 2008, pages 1145 - 49 |
| J. PARENTER. ENTERAL NUTR., vol. 28, 2004, pages 364 - 371 |
| JENKINS, R; SNYDER, R. L.: "Introduction to X-Ray Powder Diffractometry", 1996, JOHN WILEY & SONS |
| KIDNEY INTEMATIONA, vol. 72, 2007, pages S27 - S35 |
| LIANG; CHEN, EXPERT OPINION IN THERAPEUTIC PATENTS, vol. 11, no. 6, 2001, pages 981 - 986 |
| NATURE, vol. 414, 2001, pages 821 - 827 |
| STAHL; WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection and Use", 2002, WILEY |
Cited By (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11576894B2 (en) | 2009-07-08 | 2023-02-14 | Janssen Pharmaceutica Nv | Combination therapy for the treatment of diabetes |
| EP2530079A4 (en) * | 2010-01-26 | 2013-06-19 | Tianjin Inst Pharm Research | Phenyl c-glucoside derivatives, preparation methods and uses thereof |
| EP2530079A1 (en) * | 2010-01-26 | 2012-12-05 | Tianjin Institute Of Pharmaceutical Research | Phenyl c-glucoside derivatives, preparation methods and uses thereof |
| US10617668B2 (en) | 2010-05-11 | 2020-04-14 | Janssen Pharmaceutica Nv | Pharmaceutical formulations |
| US10544135B2 (en) | 2011-04-13 | 2020-01-28 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of SGLT2 |
| WO2012140597A1 (en) * | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and their uses for the treatment of diabetes |
| WO2012140596A1 (en) * | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and uses thereof |
| US8614195B2 (en) | 2011-04-14 | 2013-12-24 | Novartis Ag | Glycoside derivatives and uses thereof |
| US8722633B2 (en) | 2011-04-14 | 2014-05-13 | Novartis Ag | Glycoside derivatives and uses thereof |
| JP2014513693A (en) * | 2011-05-09 | 2014-06-05 | ヤンセン ファーマシューティカ エヌ.ベー. | (2S, 3R, 4R, 5S, 6R) -2- (3-((5- (4-fluorophenyl) thiophen-2-yl) methyl) -4-methylphenyl) -6- (hydroxymethyl) tetrahydro- Co-crystal of 2H-pyran-3,4,5-triol with L-proline and citric acid |
| US9034921B2 (en) | 2011-06-01 | 2015-05-19 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
| EP2714032A4 (en) * | 2011-06-01 | 2015-05-06 | Green Cross Corp | Novel diphenylmethane derivatives as sglt2 inhibitors |
| US9371303B2 (en) | 2011-06-01 | 2016-06-21 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
| WO2012165914A3 (en) * | 2011-06-01 | 2013-03-28 | Green Cross Corporation | Novel diphenylmethane derivatives as sglt2 inhibitors |
| JP2014515396A (en) * | 2011-06-01 | 2014-06-30 | グリーン クロス コーポレーション | Novel diphenylmethane derivatives as SGLT2 inhibitors |
| CN102827122B (en) * | 2011-06-17 | 2015-01-14 | 山东轩竹医药科技有限公司 | Glucoside derivate |
| CN102827122A (en) * | 2011-06-17 | 2012-12-19 | 山东亨利医药科技有限责任公司 | Glucoside derivate |
| CN102863417B (en) * | 2011-07-09 | 2015-01-14 | 山东轩竹医药科技有限公司 | C-indican derivative |
| CN102863417A (en) * | 2011-07-09 | 2013-01-09 | 山东轩竹医药科技有限公司 | C-indican derivative |
| US10421755B2 (en) | 2011-07-15 | 2019-09-24 | Novartis Ag | Salts of aza-bicyclic di-aryl ethers and methods to make them or their precursors |
| US9802931B2 (en) | 2011-07-15 | 2017-10-31 | Novartis Ag | Salts of aza-bicyclic di-aryl ethers and methods to make them or their precursors |
| JP2016185963A (en) * | 2011-07-15 | 2016-10-27 | ノバルティス アーゲー | Aza bicyclic diaryl ether salt, method for manufacturing the same and method for manufacturing precursor thereof |
| US9296775B2 (en) | 2012-05-10 | 2016-03-29 | Eli Lilly And Company | Pyrazole compounds |
| US9725463B2 (en) | 2013-06-21 | 2017-08-08 | Lupin Limited | Substituted heterocyclic compounds as CRAC modulators |
| WO2014203217A1 (en) | 2013-06-21 | 2014-12-24 | Lupin Limited | Substituted heterocyclic compounds as crac modulators |
| US9790231B2 (en) | 2013-06-24 | 2017-10-17 | Lupin Limited | Chromane and chromene derivatives and their use as CRAC modulators |
| CN104250272B (en) * | 2013-06-27 | 2018-10-09 | 上海方楠生物科技有限公司 | A method of it is prepared using microreactor and arranges net class pharmaceutical intermediate |
| CN104250272A (en) * | 2013-06-27 | 2014-12-31 | 上海方楠生物科技有限公司 | Method for preparing Invokana medicine intermediate by using micro-reactor |
| JP2016529298A (en) * | 2013-09-09 | 2016-09-23 | ジエンス ハンセン ファーマセウティカル グループ カンパニー リミテッド | C-aryl glucoside derivative, production method thereof and pharmaceutical application thereof |
| CN105518014B (en) * | 2013-09-09 | 2018-03-23 | 上海研健新药研发有限公司 | C aryl glucosides derivative, its preparation method and its application in medicine |
| US10011627B2 (en) | 2013-09-09 | 2018-07-03 | Youngene Therapeutics Co., Ltd | C-aryl glucoside derivative, preparation methods thereof, and medical applications thereof |
| WO2015032272A1 (en) * | 2013-09-09 | 2015-03-12 | 江苏豪森药业股份有限公司 | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| EP3056507A4 (en) * | 2013-09-09 | 2017-04-19 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| CN105518014A (en) * | 2013-09-09 | 2016-04-20 | 江苏豪森药业集团有限公司 | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9573970B2 (en) | 2013-11-08 | 2017-02-21 | Eli Lilly And Company | 4-{4-[(1E)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-1-en-1-yl]-2-methylbenzyl}-5-(propan-2-yl)-1H-pyrazol-3-yl beta-D glucopyranoside acetate |
| CN105294624A (en) * | 2015-11-16 | 2016-02-03 | 山东罗欣药业集团股份有限公司 | Preparation method for dapagliflozin |
| US10889574B2 (en) | 2016-06-17 | 2021-01-12 | Daewoong Pharmaceutical Co., Ltd. | Method for producing diphenylmethane derivative |
| US10640496B2 (en) | 2016-06-17 | 2020-05-05 | Daewoong Pharmaceutical Co., Ltd. | Method for producing diphenylmethane derivative |
| WO2018089449A1 (en) * | 2016-11-10 | 2018-05-17 | Janssen Pharmaceutica Nv | Benzocyclobutane derivatives useful as dual sglt1/sglt2 modulators |
| US10815210B2 (en) | 2016-11-10 | 2020-10-27 | Janssen Pharmaceutica Nv | Benzocyclobutane derivatives useful as dual SGLT1 / SGLT2 modulators |
| US10952989B2 (en) | 2017-03-03 | 2021-03-23 | Novartis Ag | SGLT1/2 inhibitor LIK066 for treating obesity |
| WO2018158744A1 (en) * | 2017-03-03 | 2018-09-07 | Novartis Ag | Sglt1/2 inhibitor lik066 for treating obesity |
| US11179364B2 (en) | 2017-06-21 | 2021-11-23 | Novartis Ag | Licofligozin for the treatment of non-alcoholic steatohepatitis |
| AU2018287810B2 (en) * | 2017-06-21 | 2021-03-25 | Novartis Ag | Licofligozin for the treatment of non-alcoholic steatohepatitis |
| WO2018235020A1 (en) * | 2017-06-21 | 2018-12-27 | Novartis Ag | Licofligozin for the treatment of non-alcoholic steatohepatitis |
| RU2775228C2 (en) * | 2017-06-21 | 2022-06-28 | Новартис Аг | Lycofligosin for treatment of non-alcoholic steatohepatitis |
| US10696662B2 (en) | 2017-08-21 | 2020-06-30 | Janssen Pharmaceutica Nv | 5-fluoro-C-(aryl or heterocyclyl)-glycoside derivatives useful as dual SGLT1 / SGLT2 modulators |
| US11014917B2 (en) | 2017-08-21 | 2021-05-25 | Janssen Pharmaceutica Nv | 5-fluoro-c-(aryl or heterocyclyl)-glycoside derivatives useful as dual SGLT1 / SGLT2 modulators |
| US11384057B2 (en) | 2017-12-27 | 2022-07-12 | Bp Oil International Limited | Methods for preparing fuel additives |
| WO2019129588A1 (en) * | 2017-12-27 | 2019-07-04 | Bp Oil International Limited | Methods for preparing fuel additives |
| US11230680B2 (en) | 2017-12-27 | 2022-01-25 | Bp Oil International Limited | Methods for preparing fuel additives |
| US11359151B2 (en) | 2017-12-27 | 2022-06-14 | Bp Oil International Limited | Methods for preparing fuel additives |
| US11384302B2 (en) | 2017-12-27 | 2022-07-12 | Bp Oil International Limited | Methods for preparing fuel additives |
| US11421168B2 (en) | 2017-12-27 | 2022-08-23 | Bp Oil International Limited | Methods for preparing fuel additives |
| WO2020021447A1 (en) | 2018-07-25 | 2020-01-30 | Novartis Ag | Nlrp3 inflammasome inhibitors |
| WO2020039394A1 (en) | 2018-08-24 | 2020-02-27 | Novartis Ag | New drug combinations |
| WO2020234715A1 (en) | 2019-05-17 | 2020-11-26 | Novartis Ag | Nlrp3 inflammasome inhibitors |
| WO2021014350A1 (en) | 2019-07-23 | 2021-01-28 | Novartis Ag | Combination treatment of liver diseases using fxr agonists |
| WO2024028782A1 (en) | 2022-08-03 | 2024-02-08 | Novartis Ag | Nlrp3 inflammasome inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| USRE49080E1 (en) | Glycoside derivatives and uses thereof | |
| US8394772B2 (en) | Glycoside derivative and uses thereof | |
| EP2697217B1 (en) | Glycoside derivatives and their uses for the treatment of diabetes | |
| US8722633B2 (en) | Glycoside derivatives and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080044483.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10768927 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010309833 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12012500518 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2010309833 Country of ref document: AU Date of ref document: 20101019 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2012-000176 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2777812 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 000511-2012 Country of ref document: PE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012000991 Country of ref document: CL Ref document number: 2012534672 Country of ref document: JP Ref document number: 219309 Country of ref document: IL |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201001783 Country of ref document: TH Ref document number: MX/A/2012/004700 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12068967 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010768927 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201204674 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4342/DELNP/2012 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20127012873 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201200600 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012009053 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2016/0112 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: 112012009053 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120417 |