WO2011041304A2 - Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase - Google Patents
Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase Download PDFInfo
- Publication number
- WO2011041304A2 WO2011041304A2 PCT/US2010/050532 US2010050532W WO2011041304A2 WO 2011041304 A2 WO2011041304 A2 WO 2011041304A2 US 2010050532 W US2010050532 W US 2010050532W WO 2011041304 A2 WO2011041304 A2 WO 2011041304A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- mmol
- methyl
- phenyl
- synthesis
- Prior art date
Links
- 0 CCC(c1n[n]2c(C)cc(*)nc2n1)(F)F Chemical compound CCC(c1n[n]2c(C)cc(*)nc2n1)(F)F 0.000 description 1
- SNTNVQYCKZKZOP-UHFFFAOYSA-N CCOc1n[n]2c(Nc3ccc(C(F)(F)F)cc3)cc(C)nc2n1 Chemical compound CCOc1n[n]2c(Nc3ccc(C(F)(F)F)cc3)cc(C)nc2n1 SNTNVQYCKZKZOP-UHFFFAOYSA-N 0.000 description 1
- TZMPAXWVWBRVRY-UHFFFAOYSA-N Cc1nc2nc(C3CC3)n[n]2c(Nc2ccc(C(F)(F)F)cc2)c1 Chemical compound Cc1nc2nc(C3CC3)n[n]2c(Nc2ccc(C(F)(F)F)cc2)c1 TZMPAXWVWBRVRY-UHFFFAOYSA-N 0.000 description 1
- KHDQRISTRZOHRU-UHFFFAOYSA-N Cc1nc2ncn[n]2c(NC2Cc3cc(F)ccc3C2)c1 Chemical compound Cc1nc2ncn[n]2c(NC2Cc3cc(F)ccc3C2)c1 KHDQRISTRZOHRU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to novel anti-malarial agents and inhibitors of dihydroorotate dehydrogenase.
- Malaria is a disease that is prevalent in many developing countries. Nearly 40% of the world's population is at risk for contracting this disease, which has been a major cause of mortality throughout history. In the United States travelers to these endemic regions are at risk for contracting the disease. The widespread emergence of drug resistance in many tropical countries has compromised many of the current chemotherapies and there is a continued need for new chemotherapeutic approaches.
- Malaria is a disease caused by a parasite transmitted by the bite of an infected female Anopheles mosquito. When an infecting sporozoite parasite enters the bloodstream it rapidly infects both liver and red blood cells and differentiates into merozoites. Asexual reproduction of the merozoite within erythrocytes results in the rupture and subsequent reinfection of other red blood cells. This cyclic process results in clinical symptoms, which include headaches, sweating, vomiting, malaise, delirium and acute fever and may be fatal if not treated. Malaria in humans is caused by 4 species of parasitic protozoa belonging to the genus Plasmodium. Of these, P. falciparum is the most deadly and the greatest threat to travelers abroad while P. malariae, P. vivax and P. ovale, though infrequently fatal in healthy adults, can cause morbidity in the endemic areas.
- Various medications are presently used for the treatment of malaria. However, many of these medications are costly and some exhibit significant toxicity and undesirable side effects in humans.
- the most common drug for treating malaria is chloroquine.
- Other drugs include quinine, melfloquine, atovaquone/proguanil, doxycycline, artesunate, hydroxychloroquine, halofantrine, pyrimethamine-sulfadoxine, and primaquine. Drug choice often depends on one of the four types of malaria parasites.
- Malaria parasites rely on de novo pyrimidine biosynthesis to provide precursors for DNA and R A synthesis, hence for proliferation.
- the parasite does not have pyrimidine nucleoside or base salvage pathways, thus the enzymes in the de novo pathway are essential to parasite survival.
- mammalian cells have salvage pathways that provide an alternative route to these essential metabolites.
- DHODH Dihydroorotate dehydrogenase
- a flavin-dependent mitochondrial enzyme that catalyzes the fourth reaction in the salvage pathway; coenzyme Q is utilized as the oxidant.
- the enzyme has a number of properties that make it a particularly strong candidate as a new drug target in the parasite.
- Inhibitors of human DHODH have proven efficacy for the treatment of rheumatoid arthritis demonstrating that the target pathway can be effectively blocked in vivo.
- the X-ray structures of DHODH reveal that the inhibitor binding pocket of the enzyme is highly variable between species, providing a structural basis for the design of species-specific inhibitors.
- this invention provides novel potent anti-malarial agents and methodology of treating malaria using novel potent anti-malarial agents.
- the invention also provides potent anti-malarial agents that are selective inhibitors of P. falciparum dihydroorotate dehydrogenase and active against chloroquine-sensitive and resistant malarial strains.
- the present invention relates to novel compounds for inhibiting the activity of Plasmodium falciparum dihydroorotate dehydrogenase.
- the compounds display selective inhibition of Plasmodium falciparum dihydroorotate dehydrogenase over human
- the present invention also relates to methods for preventing or treating diseases associated with the action of Plasmodium falciparum dihydroorotate dehydrogenase, such as malaria.
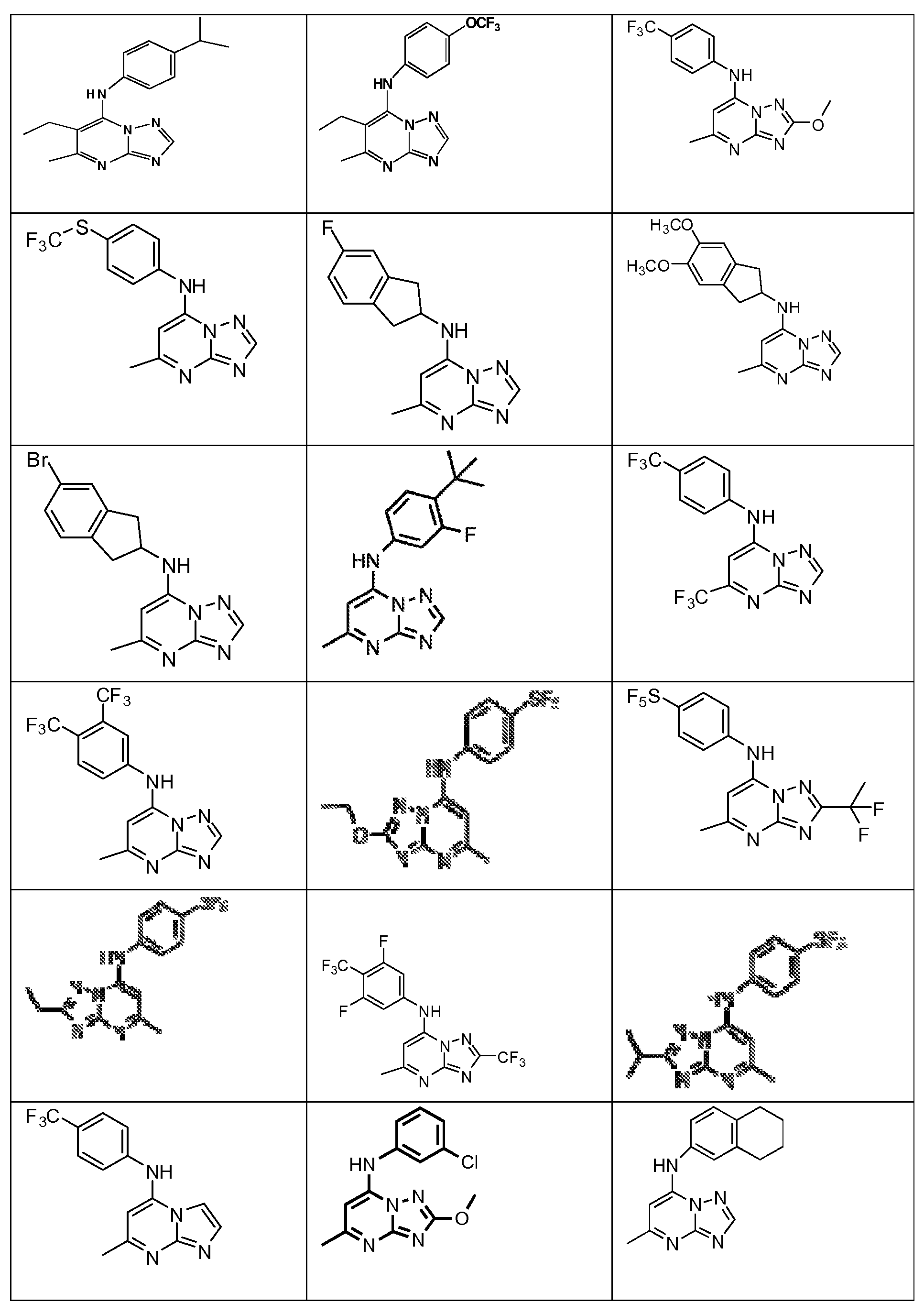
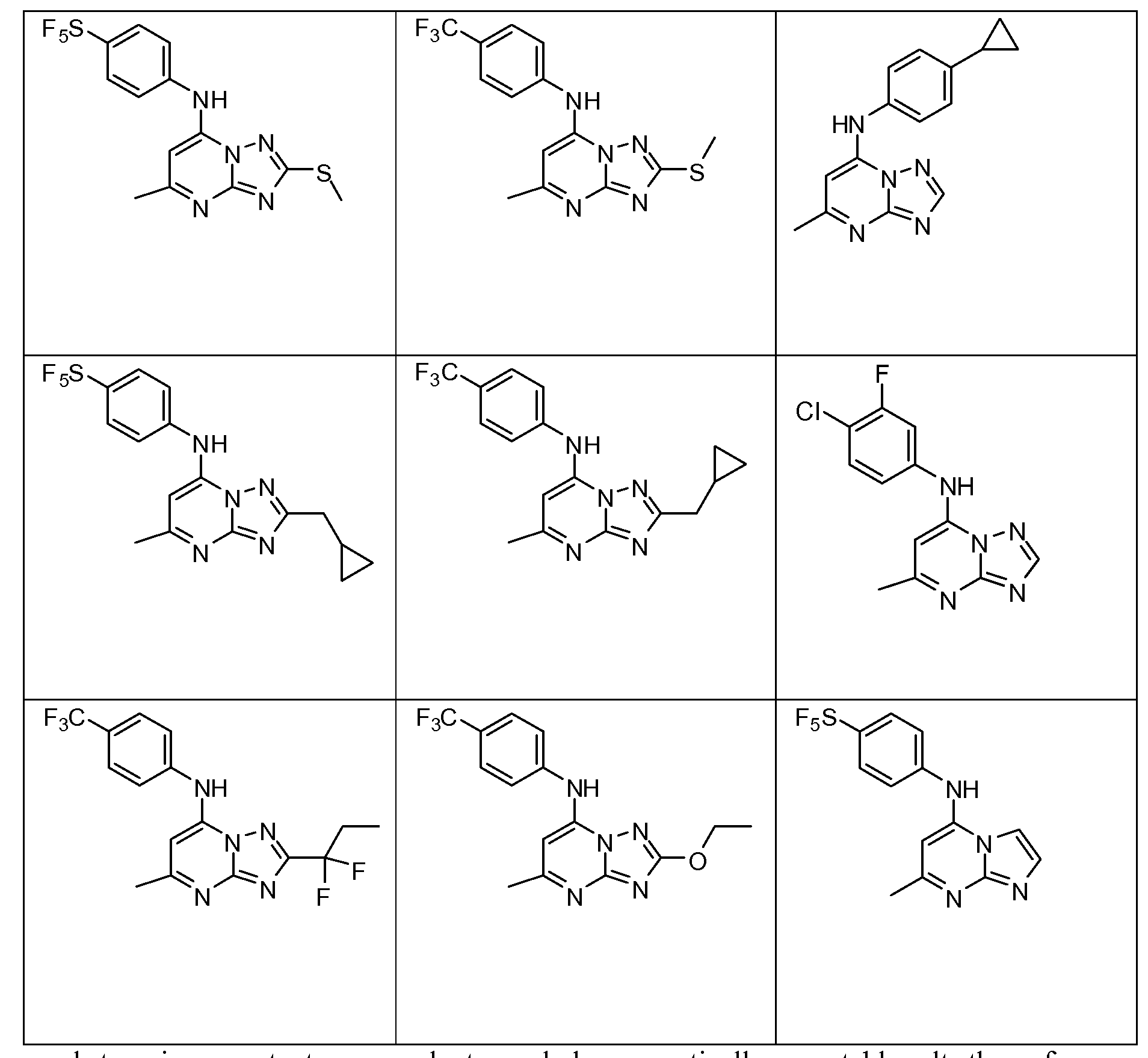
- One embodiment of the invention provides a compound that is selected from the followin table:
- the invention also encompasses pharmaceutically acceptable salts, solvates, stereoisomers, tautomers, and prodrugs of such compounds.
- composition that comprises a compound that is selected from the following table:
- R is selected from the group consisting of phenyl, aryl, 6- to 14- membered heterocycloalkyl, 6- to 14- membered arylcycloalkylene, 6- to 14- membered cycloalkylarylene, 3- to 8- membered cycloalkyl and 6- to 14-membered heteroaryl.
- R 34 is a phenyl, an aryl, a heterocycloalkyl, an arylcycloalkylene, a cycloalkylarylene, or a heteroaryl group
- R 34 is optionally substituted with one or more members selected from the group consisting of F, I, Br, CI, CN, N0 2 , -NR a R b , (Ci- Cg)haloalkyl, (Ci-Cg)haloalkoxy, (Ci-Cg)alkoxy, oxaaryl, (Ci-Cg)alkyl, (Ci-Cg)-alkylarylene, (C 2 -Cg)alkenyl, (C 2 -Cg)alkynyl, (C 3 -Cg)aryl(Ci-Cg)alkylene, and aryl.
- Substituent R 35 is selected from the group consisting of hydrogen, halogen, (Ci- Cg)alkyl, (C 2 -Cg)alkenyl, (C 2 -Cg)alkynyl, (Ci-Cg)alkoxy, and (Ci-Cg)haloalkyl, phenyl, (C 3 -Cg)cycloalkyl, (C 3 -Cg)heteroaryl, (C 3 -Cg)heterocycloalkyl, (C 3 -Cg)aryl(Ci-Cg)alkylene, and (C 3 -Cg)cycloalkyl(Ci-Cg)alkylene.
- the cycloalkyl ring can be either 5- 6- or 7-membered, depending on the integer value of "n". In one embodiment n is an integer from 0 to 2.
- Substituents R a and R b are independently selected from the group H, (Ci-Cg)alkyl, aryl, heteroaryl, heterocycloalkyl, (Ci-Cg)haloalkoxy, (Ci-Cg)haloalkyl, (C 3 -Cg)cycloalkyl and (Ci-C6)hydroxyalkyl.
- the -C0 2 H group can be replaced with bioisosteric replacements such as:
- the invention provides a pharmaceutical composition of a compound as defined herein, or a pharmaceutically acceptable salt, solvate, stereoisomer, tautomer, or prodrug thereof in a pharmaceutically acceptable carrier.
- Another embodiment of the invention is a method of inhibiting dihydroororate dehydrogenase in a parasite.
- the method comprises contacting the parasite with a compound as defined herein or a pharmaceutically acceptable salt, solvate, stereoisomer, tautomer, or prodrug thereof.
- alkyl refers to a straight or branched chain, saturated hydrocarbon having the indicated number of carbon atoms.
- (Ci-Cg)alkyl is meant to include but is not limited to methyl, ethyl, propyl, isopropyl, butyl, sec -butyl, tert- vXy ⁇ , pentyl, isopentyl, neopentyl, hexyl, isohexyl, and neohexyl, etc.
- An alkyl group can be unsubstituted or optionally substituted with one or more substituents as described herein below.
- alkoxy refers to an -O-alkyl group having the indicated number of carbon atoms.
- a (Ci-Ce)alkoxy group includes -O-methyl, -O-ethyl, -O-propyl, -O-isopropyl, -O-butyl, -O-sec -butyl, -O-tert-butyl, -O-pentyl, -O-isopentyl, -O-neopentyl, - O-hexyl, -O-isohexyl, and -O-neohexyl.
- aryl refers to a 6- to 18-membered bicyclic, tricyclic, or polycyclic aromatic hydrocarbon ring system.
- Examples of an aryl group include phenyl, naphthyl, pyrenyl, and anthracyl.
- An aryl group can be unsubstituted or optionally substituted with one or more substituents as described herein below.
- halogen and "halo" refers to -F, -CI, -Br or -I.
- heteroatom is meant to include oxygen (O), nitrogen (N), and sulfur (S).
- cycloalkyl refer to monocyclic, bicyclic, tricyclic, or polycyclic, 3- to 14-membered ring systems, which are either saturated, unsaturated or aromatic moieties.
- a heterocycle may be attached to the cycloalkyl via any heteroatom or carbon atom.
- Cycloalkyl include aryls and hetroaryls.
- Representative examples of cycloalkyl include, but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclopentenyl, cyclohexenyl, phenyl, 2,3-dihydro-lH-indenyl, naphthyl, 1,2,3,4-tetrahydronaphthl, anthracyl, benzofuranyl, and benzothiophenyl.
- a cycloalkyl group can be unsubstituted or optionally substituted with one or more substituents as described herein below.
- 'nitrile or cyano can be used interchangeably and refer to a -CN group which is bound to a carbon atom of an (Ci-C 8 )alkyl group, a heteroaryl ring, aryl ring, cycloalkyl ring, and a heterocycloalkyl ring.
- haloalkoxy refers to an -0-(Ci-C 8 )alkyl group wherein one or more hydrogen atoms in the Ci-C 8 alkyl group is replaced with a halogen atom, which can be the same or different.
- haloalkyl groups include, but are not limited to,
- alkylarylene refers to Ci-C 8 alkyl group in which at least one hydrogen atom of the Ci-C 8 alkyl chain is replaced by an aryl atom, which may be optionally substituted with one or more substituents as described herein below.
- alkylarylene groups include, but are not limited to, methylphenylene, ethylnaphthylene, propylphenylene, and butylphenylene groups.
- nitro refers to a -N0 2 group which is bound to a carbon atom of an (Ci- C 8 )alkyl group, a heteroaryl ring, aryl ring, cycloalkyl ring, and a heterocycloalkyl ring.
- arylalkylene refers to an aryl group having the indicated number of carbon atoms, in which one or more carbon atoms of the aryl group are bound to a carbon of a (Ci-C 8 )alkyl group which can be linear or branched.
- arylalkylene groups include, but are not limited to, phenylmethylene or benzyl, phenylethylene,
- arylalkoxylene refers to an aryl group having the indicated number of carbon atoms, in which one or more carbon atoms of the aryl group are bound to a carbon of a -0-(Ci-C 8 )alkyl group.
- arylcycloalkylene refers to an aryl group having the indicated number of carbon atoms, in which one or more carbon atoms of the aryl group are bound to a carbon of a (C3-Ci4)cycloalkyl group.
- two of the substituents on adjacent atoms of the aryl ring may optionally be replaced with a substituent of the formula -A-(CH 2 ) r -B-, where each of A and B are independently -CH 2 -, or a single bond, and r is an integer of from 1 to 5.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- arylcycloalkylene groups include, but are not limited to,
- cycloalkylalkylene refers to an cycloalkyl group having the indicated number of carbon atoms, in which one or more carbon atoms of the cycloalkyl group are bound to a carbon of a (Ci-C 8 )alkyl group.
- cycloalkylarylene groups include, but are not limited to, cycloproylethylene, cyclopropylmethylene and cyclobutylpropylene groups.
- cycloalkylarylene refers to an cycloalkyl group having the indicated number of carbon atoms, in which one or more carbon atoms of the cycloalkyl group are bound to a carbon of a (C3-Ci 4 )aryl group.
- Examples of cycloalkylarylene groups include, but are not limited to, cycloproylphenylene,
- haloalkyl refers to a Ci-C 8 alkyl group wherein one or more hydrogen atoms in the Ci-C 8 alkyl group is replaced with a halogen atom, which can be the same or different.
- haloalkyl groups include, but are not limited to, -CH 2 C1, -CH 2 F, - CH 2 CH 2 C1, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl,
- heteroaryl denotes a monocyclic or polycyclic aromatic heterocyclic ring system ring of 5 to 14 members, having at least one heteroatom selected from nitrogen, oxygen and sulfur, and containing at least 1 carbon atom, including bicyclic, and tricyclic ring systems.
- heteroaryls include, but are not limited to, benzofuranyl, benzothiophenyl, quinolinyl, indolyl, benzoxazolyl, benzimidazolyl, benzothiazolyl, indole, oxazole, imidazole, thiazole, pyrimidinyl, cinnolinyl, phthalazinyl, quinazolinyl, pyrimidyl, chromenonyl, quinoxalinyl.
- a heteroaryl group can be unsubstituted or optionally substituted with one or more substituents as described herein below.
- heteroarylalkylene denotes a monocyclic or polycyclic aromatic heterocyclic ring system ring of 5 to 14 members, having at least one heteroatom selected from nitrogen, oxygen and sulfur, and containing at least 1 carbon atom, including bicyclic, and tricyclic ring systems.
- the heteroaryl group may be attached via any heteroatom or carbon atom to a Ci-C 8 alkyl group.
- Examples of heteroarylalkylene include, but are not limited to, methylenethiazole, ethylenethiazole, methyleneimidazole and methylenethiophene groups.
- R', R" and R'" each independently refer to hydrogen, unsubstituted (Ci-Cg)alkyl, unsubstituted hetero(Ci-Cg)alkyl, unsubstituted aryl and aryl substituted with one to three substituents selected from -halo, unsubstituted alkyl, unsubstituted alkoxy, unsubstituted thioalkoxy and unsubstituted aryl(Ci-C4)alkyl.
- R' and R" When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6- or 7-membered ring.
- -NR'R is meant to include 1 -pyrrolidinyl and 4-morpholinyl.
- An alkyl, aryl or heteroaryl group will have from zero to three substituents.
- an alkyl or heteroalkyl radical is unsubstituted or monosubstituted.
- An alkyl or heteroalkyl radical can be unsubstituted.
- compounds of the present invention can have asymmetric centers they can occur, except when specifically noted, as mixtures of enantiomers, diastereoisomers or as individual diastereomers and enantiomers, with all isomeric forms being contemplated by the present invention.
- the present invention also encompasses racemic mixtures.
- Compounds of the present invention embrace all conformational isomers, including, for example, cis- and trans- conformations.
- the separation of diasteroisomers or enantiomers can be achieved by known analytical methods, such as chiral chromatography, crystallization, or through the employment of optically active resolving agents.
- stereoisomer means one stereoisomer of a compound that is substantially free of other stereoisomers of that compound.
- a stereomerically pure compound having one chiral center will be substantially free of the opposite enantiomer of the compound.
- a stereomerically pure compound having two chiral centers will be substantially free of other diastereomers of the compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, for example greater than about 90% by weight of one stereoisomer of the compound and less than about 10%) by weight of the other stereoisomers of the compound, or greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, or greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound.
- the term "pharmaceutically acceptable salt” is a pharmaceutically acceptable, organic or inorganic acid or base salt of a compound of the invention.
- Representative pharmaceutically acceptable salts include, e.g., alkali metal salts, alkali earth salts, ammonium salts, water-soluble and water-insoluble salts, such as the acetate, amsonate (4,4-diaminostilbene-2, 2 -disulfonate), benzenesulfonate, benzonate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, calcium, calcium edetate, camsylate, carbonate, chloride, citrate, clavulariate, dihydrochloride, edetate, edisylate, estolate, esylate, fiunarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexafluorophosphate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, iso
- a pharmaceutically acceptable salt can have more than one charged atom in its structure. In this instance the pharmaceutically acceptable salt can have multiple counterions. Thus, a pharmaceutically acceptable salt can have one or more charged atoms and/or one or more counterions.
- prodrug denotes a derivative of a compound that can hydrolyze, oxidize, or otherwise react under biological conditions, in vitro or in vivo, to provide an active compound, particularly a compound of the invention.
- prodrugs include, but are not limited to, derivatives and metabolites of a compound of the invention that include biohydrolyzable groups such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues (e.g., monophosphate, diphosphate or triphosphate).
- prodrugs of compounds with carboxyl functional groups are the lower alkyl esters of the carboxylic acid.
- the carboxylate esters are conveniently formed by esterifying any of the carboxylic acid moieties present on the molecule.
- Prodrugs can typically be prepared using well-known methods, such as those described by BURGER' S MEDICINAL CHEMISTRY AND DRUG DISCOVERY 6 th ed. (Wiley, 2001) and DESIGN AND APPLICATION OF PRODRUGS (Harwood Academic Publishers Gmbh, 1985).
- treat refers to the amelioration or eradication of a disease or symptoms associated with a disease. In certain embodiments, such terms refer to minimizing the spread or worsening of the disease resulting from the administration of one or more prophylactic or therapeutic agents to a patient with such a disease.
- prevent refers to the prevention of the onset, recurrence, or spread of the disease in a patient resulting from the administration of a prophylactic or therapeutic agent.
- a therapeutically effective amount with respect to a compound of the invention means that amount of therapeutic agent alone, or in combination with other therapies, that provides a therapeutic benefit in the treatment or prevention of a disease.
- the term can encompass an amount that improves overall therapy, reduces or avoids symptoms or causes of disease, or enhances the therapeutic efficacy of or synergies with another therapeutic agent.
- modulate refers to the ability of a compound to increase or decrease the function, or activity of, for example, DHODH.
- Module in its various forms, is intended to encompass inhibition, antagonism, partial antagonism, activation, agonism and/or partial agonism of the activity associated with DHODH.
- DHODH inhibitors are compounds that, e.g., bind to, partially or totally block stimulation, decrease, prevent, delay activation, inactivate, desensitize, or down regulate signal transduction.
- DHODH activators are compounds that, e.g., bind to, stimulate, increase, open, activate, facilitate, enhance activation, sensitize or up regulate signal transduction. The ability of a compound to modulate DHODH can be demonstrated in an enzymatic assay or a cell-based assay.
- a "patient” includes an animal (e.g., cow, horse, sheep, pig, chicken, turkey, quail, cat, dog, mouse, rat, rabbit or guinea pig), in one embodiment a mammal such as a non- primate and a primate (e.g., monkey and human), and in another embodiment a human.
- a patient is a human.
- the patient is a human infant, child, adolescent or adult.
- the present invention provides small molecule therapeutics that are potent inhibitors of the pyrimidine salvage enzyme DHOD.
- Compounds in accordance with this invention are shown in Table 1 below.
- the compounds of the inventions are triazolopyrimidine compounds that conform to Formula IX.
- R in Formula IX is selected from the group consisting of phenyl, aryl, 6- to 14- membered heterocycloalkyl, 6- to 14- membered arylcycloalkylene, 6- to 14- membered cycloalkylarylene, 3- to 8- membered cycloalkyl and 6- to 14-membered heteroaryl. Furthermore, each of these ring systems can be optionally substituted.
- R 34 is a phenyl, an aryl, a heterocycloalkyl, an arylcycloalkylene, a cycloalkylarylene, or a heteroaryl group
- R 34 is optionally substituted with one or more members selected from the group consisting of F, I, Br, CI, CN, N0 2 , -NR a R b , (Ci- Cg)haloalkyl, (Ci-Cg)haloalkoxy, (Ci-Cg)alkoxy, oxaaryl, (Ci-Cg)alkyl, (Ci-Cg)-alkylarylene, (C 2 -Cg)alkenyl, (C 2 -Cg)alkynyl, (C 3 -Cg)aryl(Ci-Cg)alkylene, and aryl.
- each of R a and R b is independently selected from the group H, (Ci- Cg)alkyl, aryl, heteroaryl, heterocycloalkyl, (Ci-Cg)haloalkoxy, (Ci-Cg)haloalkyl,
- R 34 is a phenyl and cycloalkylarylene ring substituted with halo- or alkyl.
- R 34 is a phenyl having (Ci-Cg)alkyl groups, such as methyl, ethyl propyl, isopropyl or is a phenyl group substituted with one or more halogens which may be the same or different.
- Substituent R 35 is selected from the group consisting of hydrogen, halogen, (Ci- Cg)alkyl, (C 2 -Cg)alkenyl, (C 2 -Cg)alkynyl, (Ci-Cg)alkoxy, and (Ci-Cg)haloalkyl, phenyl, (C 3 -Cg)cycloalkyl, (C 3 -Cg)heteroaryl, (C 3 -Cg)heterocycloalkyl, (C 3 -Cg)aryl(Ci-Cg)alkylene, and (C 3 -C g)cyclo alkyl(C 1 -C g)alkylene .
- the invention provides a pharmaceutical composition comprising a compound selected from Table 4 or a salt, solvate, stereoisomer, tautomer or prodrug thereof, and a pharmaceutically acceptable carrier.
- compositions of the compounds or their pharmaceutically acceptable salts further comprise a pharmaceutically acceptable carrier material.
- a pharmaceutically acceptable carrier material can be utilized.
- the carrier material can be an organic or inorganic inert carrier material, for example, one that is suitable for oral administration.
- Suitable carriers include water, gelatin, gum arabic, lactose, starch, magnesium stearate, talc, vegetable oils, polyalkylene-glycols, petroleum jelly and the like.
- the pharmaceutical preparations may also contain other pharmaceutically active agents. Additional additives such as flavoring agents, preservatives, stabilizers, emulsifying agents, buffers and the like may be added in accordance with accepted practices of pharmaceutical compounding.
- the pharmaceutical preparations can be made up in any conventional form including a solid form for oral administration such as tablets, capsules, pills, powders, granules, and the like.
- the pharmaceutical preparations may be sterilized and/or may contain adjuvants such as preservatives, stabilizers, wetting agents, emulsifiers, salts for varying the osmotic pressure and/or buffers.
- the compounds of the invention can also be administered to a patient in accordance with the invention by topical (including transdermal, buccal or sublingual), or parenteral (including intraperitoneal, subcutaneous, intravenous, intradermal or intramuscular injection) routes.
- topical including transdermal, buccal or sublingual
- parenteral including intraperitoneal, subcutaneous, intravenous, intradermal or intramuscular injection
- the compounds are administered orally.
- An oral dosage form comprises tablets, capsules of hard or soft gelatin methylcellulose or of another suitable material easily dissolved in the digestive tract.
- the oral dosages contemplated in accordance with the present invention will vary in accordance with the needs of the individual patient as determined by the prescribing physician. For instance, a daily dosage of from about 1 mg to about 50 mg per kg of body weight, such as from about 5 mg to about 25 mg per kg of body weight of the patient may be utilized.
- the therapeutically active substance enumerated herein in any desired amount for enteral administration within an oral unit dosage form.
- Particularly suitable for enteral or oral administration are tablets, dragees or capsules having talc and/or carbohydrate carrier binder or the like, the carrier could be lactose and/or corn starch and/or potato starch.
- a syrup, elixir or the like can be used where a sweetened vehicle is employed.
- Sustained release compositions can also be formulated including those where the active component is protected with differentially degradable coatings, e.g., by microencapsulation, multiple coatings, etc.
- solutions typically oily or aqueous solutions as well as suspensions, emulsions, or implants, including suppositories.
- Therapeutic compounds will be formulated in sterile form in multiple or single dose formats such as being dispersed in a fluid carrier such as sterile physiological saline or 5% saline dextrose solutions commonly used with injectables.
- the compound(s) of the invention can be suitably admixed in a pharmacologically inert topical carrier such as a gel, an ointment, a lotion or a cream.
- topical carriers include water, glycerol, alcohol, propylene glycol, fatty alcohols, triglycerides, fatty acid esters, or mineral oils.
- Other possible topical carriers are liquid petrolatum, isopropylpalmitate, polyethylene glycol, ethanol 95%, polyoxyethylene monolauriate 5% in water, sodium lauryl sulfate 5% in water, and the like.
- materials such as anti-oxidants, humectants, viscosity stabilizers and the like also may be added if desired.
- the invention provides a method for treating or preventing a condition or disorder associated with inhibition of Plasmodium dihydroorotate dehydrogenase in a subject by administering to the subject a a therapeutically effective amount of a compound of the invention.
- inventive compounds and pharmaceutically acceptable compositions of the same are candidate therapeutics for treating parasite infections in humans, such as a P. falciparum infection that causes the disease malaria.
- the invention provides a method of inhibiting dihydroororate dehydrogenase in a parasite, comprising contacting said parasite with a compound of the invention.
- the parasite is a member of the Plasmodium genus.
- the parasite is Plasmodium falciparum.
- Example 2 Synthesis of 2-(l,l-difiuoroethyl)-5-methyl[l,2,4]triazolo[l,5- a]pyrimidin-7-ol (Intermediate 2).
- Example 3 Synthesis of 7-chloro-2-( 1,1 -difiuoroethyl)-5 - methyl[l,2,4]triazolo[l,5-a] pyrimidine (Intermediate 3)
- This intermediate was synthesized by suspending Intermediate 2 in phosphorus oxychloride (ALDRICH, 21.98 ml, 236 mmol) was heating the mixture under reflux for 2 h, the starting material dissolved meanwhile. An aliquot of the reaction was partitioned between NaC0 3 10% aq.(l mL) and DCM (1 mL) and the organic phase was checked by TLC (Hexane/AcOEt 6/4) showing the reaction was complete. The reaction mixture was slowly added to a mixture of water and ice. The solution was neutralized with 10% aq. Na 2 C0 3 (800 mL approx.) and product was extracted with DCM (250 mL).
- ADRICH phosphorus oxychloride
- Example 4 Synthesis of 2-ethyl-5-methyl[l,2,4]triazolo[l,5-a]pyrimidine-7-ol (Intermediate 4).
- Intermediate 1 0.4 g, 2.85 mmol
- propionyl chloride ADRICH, 0.372 mL, 4.28 mmol
- 1 ,4-dioxane 8 mL
- N,N- dimethylformamide 2 mL
- Example 5 Synthesis of 7-chloro-2-ethyl-5-methyl[l,2,4]triazolo[l,5-a]pyrimidine (Intermediate 5).
- the titled compound (Intermediate 5), was obtained by refluxing a suspension of Intermediate 4 (0.3 g, 1.684 mmol) in phosphorus oxychloride (ALDRICH, 1.88 mL, 20.2 mmol) for 2 h and then, adding the reaction mixture slowly to a mixture of water and ice.
- the solution was neutralized with solid Na 2 C0 3 and extracted with DCM (25 mL).
- the aqueous layer was further extracted with DCM (2 x 5 mL) and the combined organic layers were washed with water (20 mL), then with brine (20 mL), and then dried over MgS0 4 .
- Solvent was removed under reduced pressure and the crude product was purified (silica gel column, eluting with Hexane/EtOAc mixtures from 100:0 to 40:60%) to obtain Intermediate 5 as a white solid.
- Example 6 Synthesis of 2-(l,l-difluoropropyl)-5-methyl[l,2,4]triazolo[l,5- a]pyrimidin-7-ol. (Intermediate 6). [0086] To a solution of NaEtO prepared from sodium (FLUKA, 0.152 g, 6.62 mmol) and ethanol (25 mL), Intermediate 1 (0.773 g, 5.52 mmol) was added, and the mixture was heated at 80 °C for 30 min.
- Example 7 Synthesis of 7-chloro-2-( 1,1 -difluoropropyl)-5 - methyl[l,2,4]triazolo[l,5-a] pyrimidine (Intermediate 7).
- Example 8 Synthesis 7-chloro-5-methyl-2-(methylthio) [l,2,4]triazolo[l,5- a]pyrimidine (Intermediate 8).
- TCI 7-hydroxy-5-methyl-2-methylthio-S-triazolo[l,5- ajpyrimidine
- ADRICH phosphorus oxychloride
- Example 9 Synthesis of 5-methyl-2-(methylthio)-N-[4-(pentafluoro- 6 - sulfanyl)phenyl] [l,2,4]triazolo[l,5-a]pyrimidin-7-amine (Intermediate 9).
- Example 10 Synthesis of 5 -methyl-2-(methylsulfonyl)-N-[4-(pentafluoro- 6 - sulfanyl)phenyl] [l,2,4]triazolo[l,5-a]pyrimidin-7-amine (Intermediate 10).
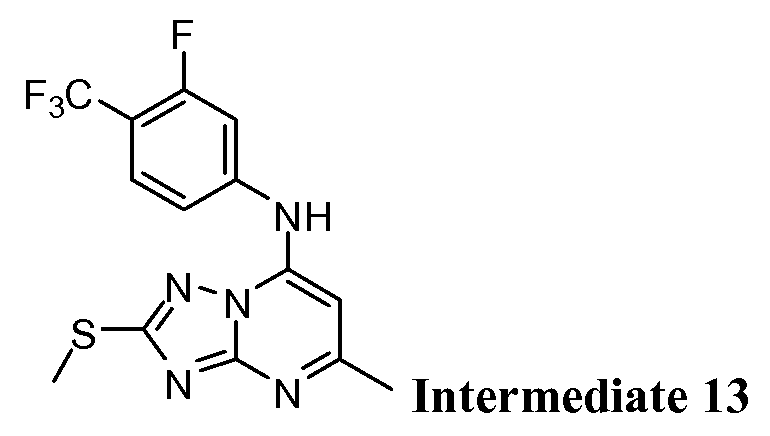
- Example 11 Synthesis of N- [3,5 -difluoro-4-(trifluoromethyl)phenyl] -5 -methyl-2- (methylthio) [l,2,4]triazolo[l,5-a]pyrimidin-7-amine (Intermediate 11).
- Example 12 Synthesis of N-[3,5-difluoro-4-(trif uoromethyl)phenyl]-5-methyl-2- (methylsulfonyl) [l,2,4]triazolo[l,5-a]pyrimidin-7-amine (Intermediate 12).
- Example 14 Synthesis of N-[3-fluoro-4-(trifluoromethyl)phenyl]-5-methyl-2- (methylsulfonyl) [l,2,4]triazolo[l,5-a]pyrimidin-7-amine (Intermediate 14).
- Example 15 Synthesis of 5 -methyl-2-(methylthio)-N- [4- (trifluoromethyl)phenyl][ 1,2,4] triazolo[l,5-a]pyrimidin-7-amine (Intermediate 15).
- Example 16 Synthesis of 5 -methyl-2-(methylsulfonyl)-N- [4- (trifluoromethyl)phenyl] [1,2,4] triazolo[l,5-a]pyrimidin-7-amine (Intermediate 16).
- Example 18 Synthesis of 7-chloro-5-methyl-2-(l-methylethyl)[l,2,4]triazolo[l,5- a] pyrimidine.
- Intermediate 18 A suspension of Intermediate 17 (0.225 g, 1.171 mmol) in phosphorous oxychloride (ALDRICH, 0.327 ml, 3.51 mmol) was heated under reflux for 1 h. The reaction mixture was added dropwise into iced water, neutralized with solid Na 2 C03 and product was extracted with DCM. The combined organic layers were washed with brine and dried over anhydrous Na 2 S0 4 . A brown oil was obtained upon solvent removal in vacuo which was purified by flash chromatography (Si, eluting with Hexane:EtOAc mixtures from 100:0 to 40:60%) to yield the title compound as a white solid.
- Example 20 Synthesis of 7-chloro-2-cyclopropyl-5-methyl[l,2,4]triazolo[l,5- a]pyrimidine (Intermediate 20) [0128] A suspension of Intermediate 19 (0.15 g, 0.789 mmol) in phosphorous oxychloride (ALDRICH, 1 ml, 10.73 mmol) was heated under reflux for 1 h. The reaction mixture was added dropwise into iced water, neutralized with solid Na 2 C03 and product was extracted with DCM. The combined organic layers were washed with brine and dried over anhydrous Na 2 S0 4 . The crude mixture was purified by flash chromatography (Si, eluting with
- Scheme I shows a representative method for synthesizing compounds that belong to the triazolopyrimidine class, particularly, the synthesis of 5-Methyl-[l,2,4]triazolo[l,5- a]pyrimidin-7-yl)-(4-trifluoromethyl-phenyl)-amine.
- Step (A) To a well-stirred solution of 5-(trifluoromethyl)-4H-l,2,4-triazol-3-amine (1 g, 6.58 mmol) in 15-20 mL glacial acetic acid, was added ethyl acetoacetate (2.57 g or 2.5 mL, 19.77 mmol) and the reaction mixture was refluxed for 6-6.5 h. Reaction progress was monitored by TLC. After completion, the reaction mixture was brought to room temperature and the excess glacial acetic acid was evaporated in vacuo. The light pinkish white solid, thus obtained, was filtered off, washed with dichloromethane and dried to yield 0.750 g (52.3 %) of the titled compound.
- Step (B) Synthesis of 7-chloro-5-methyl-2-(trifiuoromethyl)-[ 1 ,2,4]triazolo[ 1 ,5- ajpyrimidine:
- Step (C) Synthesis of N-(4-chlorophenyl)-5-methyl-2-(trifiuoromethyl)- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine:
- Example 22 Synthesis of 5 -Methyl-2-(trifluoromethyl)-N-(4- (pentafluorosulfur)phenyl)-[ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine:
- Example 23 Synthesis of N-(2,3-Dihydro-lH-inden-2-yl)-5-methyl- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 24 Synthesis of 5-Methyl-N-(l,2,3,4-tetrahydronaphthalen-2-yl)- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 25 Synthesis of 5-Methyl-N-(4-(2,2,2-trifuoroethyl)phenyl)- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 26 Synthesis of N-(4-Chloro-3-fluorophenyl)-5-methyl- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 27 Synthesis of 5-Methyl-N-(4-(trifuoromethylthio)phenyl)- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 28 Synthesis of N-(5-Fluoro-2,3-dihydro-lH-inden-2-yl)-5-methyl- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 29 Synthesis of N-(5,6-Dimethoxy-2,3-dihydro-lH-inden-2-yl)-5- methyl-[ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 30 Synthesis of N-(5-Bromo-2,3-dihydro-lH-inden-2-yl)-5-methyl- [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 31 Synthesis of 2-ethyl-5-methyl-N-[4-(pentafhioro- 6 - sulfanyl)phenyl] [ 1 ,2,4]triazolo [ 1 ,5-a]pyrimidin-7-amine.
- Example 32 Synthesis of 2-(ethyloxy)-5-methyl-N-[4-(pentafluoro- 6 - sulfanyl)phenyl] [ 1 ,2,4] triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 34 Synthesis of 2-(ethyloxy)-N-[3-fluoro-4-(trifluoromethyl)phenyl]-5- methyl[ 1 ,2,4] triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 36 Synthesis of 5-methyl-2-(l-methylethyl)-N-[4- (trifluoromethyl)phenyl] [1 ,2,4] triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 38 Synthesis of 2-cyclopropyl-5-methyl-N-[4-(trifluoromethyl)phenyl] [ 1 ,2,4]triazolo [ 1 ,5-a]pyrimidin-7-amine
- Example 39 Example 15: Synthesis of 2-cyclopropyl-5-methyl-N-[4-(pentafluoro- 6-sulfanyl)phenyl] [ 1 ,2,4] triazolo[ 1 ,5-a]pyrimidin-7-amine.
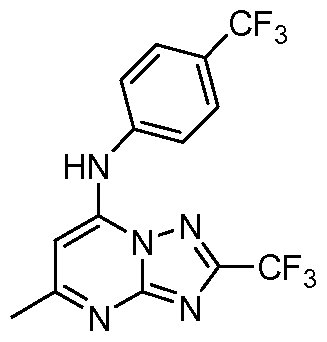
- Example 40 Synthesis of 2-(trifluoromethyl)-N-(4-(trifluoromethyl)phi methyl-[ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine
- Example 41 Synthesis of 2-(trifluoromethyl)-N-(4-(sulfurpentafluoro)phenyl)-5- methyl-[ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine
- Example 42 Synthesis of N-(3,5-difluoro-4-(trifluoromethyl)phenyl)-2- (trifluoromethyl)-5-methyl-[ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine
- Example 43 Synthesis of 7- Amino substituted 2-(difluoroethyl)-5 -methyl - [l,2,4]triazolo[l,5-a]pyrimidine compounds were prepared as illustrated in Scheme 3
- Example 44 Synthesis of 2-(l,l-difluoroethyl)-5-methyl-N-[4-(pentafluoro- 6 - sulfanyl)phenyl] [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 45 Synthesis of 2-( 1,1 -difluoroethyl)-5-methyl-N- [4- (trifluoromethyl)phenyl] [1 ,2,4] triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 46 Synthesis of 2-( 1,1 -difiuoroethyl)-N- [3 -fluoro-4- (trifluoromethyl)phenyl] -5 -methyl [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 48 Synthesis of 2-(l,l-difluoropropyl)-5-methyl-N-[4- (trifluoromethyl)phenyl] [1 ,2,4] triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Example 49 Synthesis of 2-(l,l-difluoropropyl)-5-methyl-N-[4-(pentafiuoro- 6 - sulfanyl)phenyl] [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
- Scheme 4 illustrates a protocol for synthesizing imidazo[l,2-a]pyrimidin-5- compounds of the invention.
- Example 50 Synthesis of 7-methyl-N-(4-(trifluoromethyl)phenyl)imidazo[l,2- a]pyrimidin-5-amine.
- Step (C) Synthesis of 7-methyl-N-(4-(trifiuoromethyl)phenyl)imidazo[l,2- a]pyrimidin-5 -amine :
- Example 52 7-Methyl-N-(4-(pentafluorosulfur)phenyl)imidazo[l,2-a]pyrimidin-5- amine:
- Example 54 Synthesis of N-(2,3-dihydro-lH-inden-5-yl)-7-methylimidazo[l,2- a]pyrimidin-5 -amine : Mp 255-257° C.
- 1H NMR 300 MHz, dmso-d 6 ): ⁇ 9.32 (brs, NH), 7.97 (s, 1H), 7.53 (s, 1H), 7.24 (m, 3H), 6.00 (s, 1H), 2.90 (m, 4H), 2.31 (s, 3H), 2.01 (m, 2H). MS m/z 265.0.
- DHOD plays an important role in malaria. Described here are assays useful for testing the inhibition of DHOD by compounds of the present invention.
- the assay was carried out using a solution containing 100 mM HEPES, pH 8.0, 150 mM NaCl, 10% glycerol, 0.1% Triton X-100, 20 micro molar COQ D (coenzyme Q D ), 200 micro molar L- dihydroorotate, and 120 micro molar DCIP. Reactions are initiated by addition of enzyme to a final concentration in the range of about 5 nM to about 50 nM while maintaining the temperature of a circulating water bath at 25° C.
- Assay solutions were prepared as discussed above, except that DCIP is not present and the solution is depleted of oxygen by the inclusion of an oxidase/reductase system, such as, 0.1 mg/ml of glucose oxidase, 0.02 mg/ml catalase and 50 mM glucose.
- the data obtained was fitted to equation 1, to determine the IC50 values of the representative compounds.
- Table 5 shows the IC50 and EC50 values for an illustrative set of compounds against Plasmodium falciparum.
- the compounds of the invention are potent inhibitors of the Plasmodium falciparum DHOD enzymes, with IC 50 values in the low to sub-micromolar range.
- H-hypoxanthine uptake is measured in drug-treated, P. falciparum- fectGd erythrocytes grown in culture, pursuant to the methodology of Desjardins, et al. (1979) Antimicrobial Agents and Chemotherapy 16, 710- 718, and Zhang and Rathod (2002) Science 296, 545-547.
- Inhibition of parasite growth is determined microscopically by staining a thin smear of blood obtained from the test animal using a Wright-Giemsa stain. Greater suppression of parasite growth is observed if drug is administered twice a day rather than a single dose.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description















































































Claims













Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL10821105T PL2483274T3 (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| CA2775354A CA2775354C (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| BR112012007179A BR112012007179B8 (en) | 2009-09-29 | 2010-09-28 | dihydro-orotate dehydrogenase inhibitor compound, and pharmaceutical formulation |
| MEP-2015-8A ME02020B (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| DK10821105.3T DK2483274T3 (en) | 2009-09-29 | 2010-09-28 | Antimalarials, which are inhibitors of dihydroorotate |
| CN201080043385.XA CN102741255B (en) | 2009-09-29 | 2010-09-28 | As the anti-malarial agents of two hydrogen vitamin B13 dehydrogenase inhibitor |
| JP2012532236A JP5818266B2 (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotic acid dehydrogenase |
| ES10821105.3T ES2527835T3 (en) | 2009-09-29 | 2010-09-28 | Anti malaria agents that are dihydroorotate dehydrogenase inhibitors |
| SI201030853T SI2483274T1 (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| US13/499,031 US9238653B2 (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| EP10821105.3A EP2483274B1 (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| HK13101950.1A HK1174639A1 (en) | 2009-09-29 | 2013-02-15 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| HRP20150066AT HRP20150066T1 (en) | 2009-09-29 | 2015-01-19 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
| SM201500071T SMT201500071B (en) | 2009-09-29 | 2015-03-18 | Antimalarial agents that are inhibitors of dihydro-orotate dehydrogenase |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US24686309P | 2009-09-29 | 2009-09-29 | |
| US61/246,863 | 2009-09-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011041304A2 true WO2011041304A2 (en) | 2011-04-07 |
| WO2011041304A3 WO2011041304A3 (en) | 2011-08-04 |
Family
ID=43826848
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/050532 WO2011041304A2 (en) | 2009-09-29 | 2010-09-28 | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US9238653B2 (en) |
| EP (1) | EP2483274B1 (en) |
| JP (1) | JP5818266B2 (en) |
| CN (1) | CN102741255B (en) |
| BR (1) | BR112012007179B8 (en) |
| CA (1) | CA2775354C (en) |
| DK (1) | DK2483274T3 (en) |
| ES (1) | ES2527835T3 (en) |
| HK (1) | HK1174639A1 (en) |
| HR (1) | HRP20150066T1 (en) |
| ME (1) | ME02020B (en) |
| PL (1) | PL2483274T3 (en) |
| PT (1) | PT2483274E (en) |
| SI (1) | SI2483274T1 (en) |
| SM (1) | SMT201500071B (en) |
| WO (1) | WO2011041304A2 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3072894A1 (en) | 2015-03-26 | 2016-09-28 | Board Of Regents Of the University Of Texas System | New substituted triazolopyrimidines as anti-malarial agents |
| CN110573154A (en) * | 2017-02-06 | 2019-12-13 | 卡斯西部储备大学 | Compositions and methods for modulating short-chain dehydrogenase activity |
| EP3525786A4 (en) * | 2016-10-12 | 2020-03-18 | Merck Sharp & Dohme Corp. | Kdm5 inhibitors |
| WO2021123266A1 (en) | 2019-12-20 | 2021-06-24 | The Board Of Regents Of The University Of Texas System | Anti-malarial agents |
| US11426420B2 (en) | 2017-04-07 | 2022-08-30 | Case Western Reserve University | Inhibitors of short-chain dehydrogenase activity for treating coronary disorders |
| US11690847B2 (en) | 2016-11-30 | 2023-07-04 | Case Western Reserve University | Combinations of 15-PGDH inhibitors with corticosteroids and/or TNF inhibitors and uses thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUE040254T2 (en) * | 2013-12-19 | 2019-02-28 | Novartis Ag | [1,2,4]triazolo[1,5-a]pyrimidine derivatives as protozoan proteasome inhibitors for the treatment of parasitic diseases such as leishmaniasis |
| EP3254679A1 (en) | 2016-06-09 | 2017-12-13 | Christian-Albrechts-Universität zu Kiel | Antiprotozoal and antibacterial agents |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007014211A1 (en) | 2005-07-25 | 2007-02-01 | Metal Container Corporation | Can lid closure and method of joining a can lid closure to a can body |
| WO2009082691A1 (en) | 2007-12-21 | 2009-07-02 | Board Of Regents, University Of Texas System | Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5478825A (en) | 1991-02-22 | 1995-12-26 | Egis Gyogyszergyar | 5-(substituted amino)-1,2,4-triazolo (1,5-A) pyrimidine derivatives |
| HU208693B (en) | 1991-02-22 | 1993-12-28 | Egyt Gyogyszervegyeszeti Gyar | Process for producing 1,2,4-triazolo (1,5-a) pyrimidinis derivatives and their carbicycli-tetrahydro-thiofurane-tetrahydrothiopyrane-, or tetrahydropyridine- condensated derivatives or medical preparatives containing them |
| CZ294535B6 (en) * | 2001-08-02 | 2005-01-12 | Ústav Experimentální Botaniky Avčr | Heterocyclic compounds based on N6-substituted adenine, processes of their preparation, their use in the preparation of medicaments, cosmetic compositions and growth regulators, as well as pharmaceutical preparations, cosmetic compositions and growth regulators in which these compounds are comprised |
| WO2007149211A1 (en) * | 2006-06-22 | 2007-12-27 | Board Of Regents, University Of Texas System | Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity |
| CN101622001A (en) * | 2007-01-26 | 2010-01-06 | Irm责任有限公司 | The purine compound and the compositions that are used for the treatment of plasmodium related diseases as inhibitors of kinases |
-
2010
- 2010-09-28 PL PL10821105T patent/PL2483274T3/en unknown
- 2010-09-28 ME MEP-2015-8A patent/ME02020B/en unknown
- 2010-09-28 JP JP2012532236A patent/JP5818266B2/en not_active Expired - Fee Related
- 2010-09-28 WO PCT/US2010/050532 patent/WO2011041304A2/en active Application Filing
- 2010-09-28 PT PT108211053T patent/PT2483274E/en unknown
- 2010-09-28 CA CA2775354A patent/CA2775354C/en not_active Expired - Fee Related
- 2010-09-28 ES ES10821105.3T patent/ES2527835T3/en active Active
- 2010-09-28 EP EP10821105.3A patent/EP2483274B1/en active Active
- 2010-09-28 CN CN201080043385.XA patent/CN102741255B/en not_active Expired - Fee Related
- 2010-09-28 DK DK10821105.3T patent/DK2483274T3/en active
- 2010-09-28 BR BR112012007179A patent/BR112012007179B8/en not_active IP Right Cessation
- 2010-09-28 SI SI201030853T patent/SI2483274T1/en unknown
- 2010-09-28 US US13/499,031 patent/US9238653B2/en not_active Expired - Fee Related
-
2013
- 2013-02-15 HK HK13101950.1A patent/HK1174639A1/en not_active IP Right Cessation
-
2015
- 2015-01-19 HR HRP20150066AT patent/HRP20150066T1/en unknown
- 2015-03-18 SM SM201500071T patent/SMT201500071B/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007014211A1 (en) | 2005-07-25 | 2007-02-01 | Metal Container Corporation | Can lid closure and method of joining a can lid closure to a can body |
| WO2009082691A1 (en) | 2007-12-21 | 2009-07-02 | Board Of Regents, University Of Texas System | Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity |
Non-Patent Citations (3)
| Title |
|---|
| GUJJAR ET AL., JOURNAL OF MEDICINAL CHEMISTRY, 2009 |
| PHILLIPS ET AL., JOURNAL OF MEDICINAL CHEMISTRY, 2008 |
| See also references of EP2483274A4 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3072894A1 (en) | 2015-03-26 | 2016-09-28 | Board Of Regents Of the University Of Texas System | New substituted triazolopyrimidines as anti-malarial agents |
| WO2016151521A1 (en) | 2015-03-26 | 2016-09-29 | Board Of Regents Of The University Of Texas System | New substituted triazolopyrimidines as anti-malarial agents |
| EP3525786A4 (en) * | 2016-10-12 | 2020-03-18 | Merck Sharp & Dohme Corp. | Kdm5 inhibitors |
| US11690847B2 (en) | 2016-11-30 | 2023-07-04 | Case Western Reserve University | Combinations of 15-PGDH inhibitors with corticosteroids and/or TNF inhibitors and uses thereof |
| CN110573154A (en) * | 2017-02-06 | 2019-12-13 | 卡斯西部储备大学 | Compositions and methods for modulating short-chain dehydrogenase activity |
| EP3576737A4 (en) * | 2017-02-06 | 2021-04-21 | Case Western Reserve University | Compositions and methods of modulating short-chain dehydrogenase activity |
| US11718589B2 (en) | 2017-02-06 | 2023-08-08 | Case Western Reserve University | Compositions and methods of modulating short-chain dehydrogenase |
| US11426420B2 (en) | 2017-04-07 | 2022-08-30 | Case Western Reserve University | Inhibitors of short-chain dehydrogenase activity for treating coronary disorders |
| WO2021123266A1 (en) | 2019-12-20 | 2021-06-24 | The Board Of Regents Of The University Of Texas System | Anti-malarial agents |
| US11903936B2 (en) | 2019-12-20 | 2024-02-20 | The Board Of Regents Of The University Of Texas System | Anti-malarial agents |
| EP3842100A1 (en) | 2019-12-24 | 2021-06-30 | The Board Of Regents Of The University Of Texas System | Anti-malarial agents |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2011041304A3 (en) | 2011-08-04 |
| US20120302586A1 (en) | 2012-11-29 |
| CA2775354C (en) | 2019-01-15 |
| BR112012007179B8 (en) | 2021-05-25 |
| ES2527835T3 (en) | 2015-01-30 |
| PT2483274E (en) | 2015-02-04 |
| EP2483274A2 (en) | 2012-08-08 |
| JP5818266B2 (en) | 2015-11-18 |
| EP2483274A4 (en) | 2013-03-06 |
| PL2483274T3 (en) | 2015-04-30 |
| HK1174639A1 (en) | 2013-06-14 |
| BR112012007179B1 (en) | 2020-12-01 |
| CN102741255A (en) | 2012-10-17 |
| BR112012007179A2 (en) | 2017-07-25 |
| DK2483274T3 (en) | 2015-01-19 |
| US9238653B2 (en) | 2016-01-19 |
| EP2483274B1 (en) | 2014-10-29 |
| BR112012007179A8 (en) | 2018-01-23 |
| SMT201500071B (en) | 2015-05-05 |
| CA2775354A1 (en) | 2011-04-07 |
| ME02020B (en) | 2015-05-20 |
| SI2483274T1 (en) | 2015-02-27 |
| CN102741255B (en) | 2015-10-07 |
| HRP20150066T1 (en) | 2015-02-27 |
| JP2013506004A (en) | 2013-02-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2483274B1 (en) | Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase | |
| WO2009082691A1 (en) | Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity | |
| EP2010505B1 (en) | Heterocyclic compounds and uses thereof in the treatment of sexual disorders | |
| EP0738270B1 (en) | Dihydro pyrazolopyrroles | |
| US9840515B2 (en) | Protein kinase D inhibitors | |
| US20080096903A1 (en) | Sulfamoyl-containing derivatives and uses thereof | |
| WO2016022460A1 (en) | Potent dual brd4-kinase inhibitors as cancer therapeutics | |
| US9695193B2 (en) | Inhibitors of Plasmodium falciparum equilibrative nucleoside transporter type I as anti-parasitic compounds | |
| JP2010522226A (en) | Dual inhibitory compounds, pharmaceutical compositions and methods of use of the enzymes PDE7 and / or PDE4 | |
| JPH06228108A (en) | Phthalozinone derivative | |
| US6423720B1 (en) | Pyrimidine compounds and methods for making and using the same | |
| WO2007149211A1 (en) | Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity | |
| JP2009509963A (en) | New compounds | |
| WO2002059125A1 (en) | Cyclin dependent kinase inhibiting purine derivatives | |
| FR2711992A1 (en) | New heterocyclic derivatives, process of preparation and pharmaceutical composition containing them | |
| Ramalingam et al. | Synthesis and antimicrobial activity of azasteroid-type compounds and related systems. Effect of hydrophilic and lipophilic groups on activity | |
| Dal Piaz et al. | Isoxazolo [3, 4-d] pyridazinones and analogues as Leishmania mexicana PDE inhibitors | |
| WO2014016789A1 (en) | Pyrazolo[3,4-d]pyrimidin-4(5h)-one derivatives as pde9 inhibitors | |
| AU665223B2 (en) | 4-amino-3-acyl quinoline derivatives | |
| CN114728963B (en) | Novel triazolopyridine derivatives and pharmaceutical compositions comprising the same | |
| US8815846B2 (en) | Family of antichagasics derived from imidazo[4,5-C][1,2,6]thiadiazine 2,2-dioxide | |
| US20130345420A9 (en) | Selective inhibitors of excitatory amino acid transporter subtype 1 (eaat1/glast) | |
| US20240092737A1 (en) | Substituted salicylamide compounds and use thereof | |
| WO2017090058A1 (en) | Fused pyrimidines as isoform selective phosphoinositide-3-kinase-alpha inhibitors and process for preparation thereof | |
| CA2385244A1 (en) | Novel nicotinonitrile compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080043385.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10821105 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2775354 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012532236 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010821105 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13499031 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012007179 Country of ref document: BR |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: 112012007179 Country of ref document: BR |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: ERR Ref document number: 112012007179 Country of ref document: BR Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 112012007179 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120329 |