WO2011038293A1 - Cyclic peptide inhibitors of hepatitis c virus replication - Google Patents
Cyclic peptide inhibitors of hepatitis c virus replication Download PDFInfo
- Publication number
- WO2011038293A1 WO2011038293A1 PCT/US2010/050298 US2010050298W WO2011038293A1 WO 2011038293 A1 WO2011038293 A1 WO 2011038293A1 US 2010050298 W US2010050298 W US 2010050298W WO 2011038293 A1 WO2011038293 A1 WO 2011038293A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- alkyl
- group
- cycloalkyl
- heteroaryl
- Prior art date
Links
- 0 CC(C)=NN(C(CC1C(NC(C2)(C*2CCCCCCCC2N*)C(*)=O)=O)CN1C2=O)C=C(C)* Chemical compound CC(C)=NN(C(CC1C(NC(C2)(C*2CCCCCCCC2N*)C(*)=O)=O)CN1C2=O)C=C(C)* 0.000 description 12
- AZALJBCEJYFKTP-UUMQRWBGSA-N CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)C(N(CCC1)[C@@H]1C(N)=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)C(N(CCC1)[C@@H]1C(N)=O)=O)=O AZALJBCEJYFKTP-UUMQRWBGSA-N 0.000 description 2
- XJKGGTFRFIREAB-UHFFFAOYSA-N [O-2]C(N(C1)Cc2c1cccc2F)=O Chemical compound [O-2]C(N(C1)Cc2c1cccc2F)=O XJKGGTFRFIREAB-UHFFFAOYSA-N 0.000 description 2
- JLKJMBAGVSNLRT-UHFFFAOYSA-N CC(C)(C)C1=Nc(cccc2)c2NC1=O Chemical compound CC(C)(C)C1=Nc(cccc2)c2NC1=O JLKJMBAGVSNLRT-UHFFFAOYSA-N 0.000 description 1
- FLBFNDLKHJYHCD-WIQOXFDPSA-N CC(C)(C)OC(N(C)[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(C1(C)CC1)(=O)=O)=O)C(N(CCC1)[C@@H]1C(N)=O)=O)=O Chemical compound CC(C)(C)OC(N(C)[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(C1(C)CC1)(=O)=O)=O)C(N(CCC1)[C@@H]1C(N)=O)=O)=O FLBFNDLKHJYHCD-WIQOXFDPSA-N 0.000 description 1
- QTNNCICZGXJIAJ-QMMMGPOBSA-N CC(C)(C)OC(NCC(N(CCC1)[C@@H]1C(N)=O)=O)=O Chemical compound CC(C)(C)OC(NCC(N(CCC1)[C@@H]1C(N)=O)=O)=O QTNNCICZGXJIAJ-QMMMGPOBSA-N 0.000 description 1
- ISHQLINSLGTMLL-QGQFBJCESA-N CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)C(N(CC(C1)Oc2nc(cccc3)c3[n]2C(/C=C\C)=C)[C@@H]1C(N)=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)C(N(CC(C1)Oc2nc(cccc3)c3[n]2C(/C=C\C)=C)[C@@H]1C(N)=O)=O)=O ISHQLINSLGTMLL-QGQFBJCESA-N 0.000 description 1
- FYWVJGVONGGTKA-WIRVNAGMSA-N CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(OC1CC1)(=O)=O)=O)C(N(CCC1)[C@@H]1C(N)=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(OC1CC1)(=O)=O)=O)C(N(CCC1)[C@@H]1C(N)=O)=O)=O FYWVJGVONGGTKA-WIRVNAGMSA-N 0.000 description 1
- NADBADKLQJBOLR-PUGRFQAISA-N CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@]1(C(NS(N(C)C)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1O)=O)C2=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CCCCC/C=C\[C@H](C1)[C@]1(C(NS(N(C)C)(=O)=O)=O)NC([C@H](C1)N2C[C@@H]1O)=O)C2=O)=O NADBADKLQJBOLR-PUGRFQAISA-N 0.000 description 1
- CWFPSKVBKAPBPV-UHFFFAOYSA-N CC(C)C1=Nc2ccccc2NC1=O Chemical compound CC(C)C1=Nc2ccccc2NC1=O CWFPSKVBKAPBPV-UHFFFAOYSA-N 0.000 description 1
- JATXMLLIFGZVIX-AKRCKQFNSA-N CC(C)[n]1c(OC(C[C@H]2C(N)=O)CN2C(CNC(OC(C)(C)C)=O)=O)nc2c1cccc2NS(Cc1ccccc1)(=O)=O Chemical compound CC(C)[n]1c(OC(C[C@H]2C(N)=O)CN2C(CNC(OC(C)(C)C)=O)=O)nc2c1cccc2NS(Cc1ccccc1)(=O)=O JATXMLLIFGZVIX-AKRCKQFNSA-N 0.000 description 1
- LEGMNHGUYHAFBJ-UHFFFAOYSA-M CC(C)[n]1c([O-])nc2c1cccc2N Chemical compound CC(C)[n]1c([O-])nc2c1cccc2N LEGMNHGUYHAFBJ-UHFFFAOYSA-M 0.000 description 1
- CTOKCNOIYVKBFN-UHFFFAOYSA-N CC(C)c(cc1)cc2c1[n](C(C)C)c(Cl)n2 Chemical compound CC(C)c(cc1)cc2c1[n](C(C)C)c(Cl)n2 CTOKCNOIYVKBFN-UHFFFAOYSA-N 0.000 description 1
- ALXPJGJXDOLQCE-UHFFFAOYSA-N CC(C)c1c(C)[s]c(-c(c(F)ccc2[n]3C(C)C)c2nc3Cl)n1 Chemical compound CC(C)c1c(C)[s]c(-c(c(F)ccc2[n]3C(C)C)c2nc3Cl)n1 ALXPJGJXDOLQCE-UHFFFAOYSA-N 0.000 description 1
- WHJJWCCTQKMBBY-JRDGRLKISA-N CC(C)c1c(C)[s]c(-c(c2c(cc3)[n](C(C)C)c(O[C@H](C[C@H]4C(N[C@](C5)([C@@H]5/C=C\CCCCC[C@@H]5NC(OC(C)(C)C)=O)C(NS(N(C)C)(=O)=O)=O)=O)CN4C5=O)n2)c3F)n1 Chemical compound CC(C)c1c(C)[s]c(-c(c2c(cc3)[n](C(C)C)c(O[C@H](C[C@H]4C(N[C@](C5)([C@@H]5/C=C\CCCCC[C@@H]5NC(OC(C)(C)C)=O)C(NS(N(C)C)(=O)=O)=O)=O)CN4C5=O)n2)c3F)n1 WHJJWCCTQKMBBY-JRDGRLKISA-N 0.000 description 1
- HFZXHVSEFPSFPE-UHFFFAOYSA-N CC(C)c1c[s]c(-c2cc(OC)c(ccc(OC)c3C)c3n2)n1 Chemical compound CC(C)c1c[s]c(-c2cc(OC)c(ccc(OC)c3C)c3n2)n1 HFZXHVSEFPSFPE-UHFFFAOYSA-N 0.000 description 1
- CLNMJNWAUQBABM-AWJINBMRSA-N CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N)=O)CN2C([C@H](CCCCC/C=C\[C@H](C2)[C@H]2C(NS(C2(C)CC2)(=O)=O)=O)N)=O)n1 Chemical compound CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N)=O)CN2C([C@H](CCCCC/C=C\[C@H](C2)[C@H]2C(NS(C2(C)CC2)(=O)=O)=O)N)=O)n1 CLNMJNWAUQBABM-AWJINBMRSA-N 0.000 description 1
- HURXWSKNKIKNFV-AWJINBMRSA-N CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N)=O)CN2C([C@H](CCCCC/C=C\[C@H](C2)[C@H]2C(NS(C2(C)CC2)(=O)=O)=O)NC(N)=S)=O)n1 Chemical compound CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N)=O)CN2C([C@H](CCCCC/C=C\[C@H](C2)[C@H]2C(NS(C2(C)CC2)(=O)=O)=O)NC(N)=S)=O)n1 HURXWSKNKIKNFV-AWJINBMRSA-N 0.000 description 1
- JQBQBTOZHJUPID-QCSGBROFSA-N CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N[C@](C3)([C@@H]3/C(/C)=C\CCCCC[C@@H]3Nc4ncc[s]4)C(NS(C4(C)CC4)(=O)=O)=O)=O)CN2C3=O)n1 Chemical compound CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N[C@](C3)([C@@H]3/C(/C)=C\CCCCC[C@@H]3Nc4ncc[s]4)C(NS(C4(C)CC4)(=O)=O)=O)=O)CN2C3=O)n1 JQBQBTOZHJUPID-QCSGBROFSA-N 0.000 description 1
- ZMXUJWPPJVIDKC-QJQXIEHNSA-N CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N[C@](C3)([C@@H]3/C=C\CCCCC[C@@H]3NC(OC4C[C@H](C5)[C@H]5C4)=O)C(NS(C4(C)CC4)(=O)=O)=O)=O)CN2C3=O)n1 Chemical compound CC(C)c1c[s]c(-c2cccc([n]3C(C)C)c2nc3O[C@H](C[C@H]2C(N[C@](C3)([C@@H]3/C=C\CCCCC[C@@H]3NC(OC4C[C@H](C5)[C@H]5C4)=O)C(NS(C4(C)CC4)(=O)=O)=O)=O)CN2C3=O)n1 ZMXUJWPPJVIDKC-QJQXIEHNSA-N 0.000 description 1
- RMCBYNGFQXXISV-UHFFFAOYSA-N CC(C)c1c[s]c(NC(c(cccc2[n]3C(C)C)c2nc3Cl)=O)n1 Chemical compound CC(C)c1c[s]c(NC(c(cccc2[n]3C(C)C)c2nc3Cl)=O)n1 RMCBYNGFQXXISV-UHFFFAOYSA-N 0.000 description 1
- VXDCASZZOCAPPK-SNYNKORZSA-N CC(C1)(C2)[C@@H]1CC2OC(ON(C(CC1)O)C1=O)=O Chemical compound CC(C1)(C2)[C@@H]1CC2OC(ON(C(CC1)O)C1=O)=O VXDCASZZOCAPPK-SNYNKORZSA-N 0.000 description 1
- ULSRZZYREOKGFZ-WNBKYALVSA-N CC(CCCCCC[C@@H](C(N(CCC1)[C@@H]1C(N)=O)=O)NC(OC(C)(C)C)=O)[C@H](C1)[C@H]1C(O)=O Chemical compound CC(CCCCCC[C@@H](C(N(CCC1)[C@@H]1C(N)=O)=O)NC(OC(C)(C)C)=O)[C@H](C1)[C@H]1C(O)=O ULSRZZYREOKGFZ-WNBKYALVSA-N 0.000 description 1
- APQKBHZPQFDFGX-YFKZHCIASA-N CC1(CC1)S(NC([C@@](C1)([C@@H]1/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1O)=O)Nc1ccccc1)NC2=O)=O)(=O)=O Chemical compound CC1(CC1)S(NC([C@@](C1)([C@@H]1/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1O)=O)Nc1ccccc1)NC2=O)=O)(=O)=O APQKBHZPQFDFGX-YFKZHCIASA-N 0.000 description 1
- BMIMNRPAEPIYDN-UHFFFAOYSA-N CC1=Nc2ccccc2NC1=O Chemical compound CC1=Nc2ccccc2NC1=O BMIMNRPAEPIYDN-UHFFFAOYSA-N 0.000 description 1
- MIIFHRBUBUHJMC-UHFFFAOYSA-N CCOC(C1=Nc2ccccc2NC1=O)=O Chemical compound CCOC(C1=Nc2ccccc2NC1=O)=O MIIFHRBUBUHJMC-UHFFFAOYSA-N 0.000 description 1
- XPPPIOIAMJFNMI-UHFFFAOYSA-N CCc1cccc([n]2C(C)C)c1nc2Cl Chemical compound CCc1cccc([n]2C(C)C)c1nc2Cl XPPPIOIAMJFNMI-UHFFFAOYSA-N 0.000 description 1
- CPPBTWLTZUTPII-NQHIMBQPSA-N CCc1cccc([n]2C(C)C)c1nc2[O-]C(C[C@H]1C(N)=O)CN1C([C@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)N)=O Chemical compound CCc1cccc([n]2C(C)C)c1nc2[O-]C(C[C@H]1C(N)=O)CN1C([C@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)N)=O CPPBTWLTZUTPII-NQHIMBQPSA-N 0.000 description 1
- XNFWQZHNVQLRPL-QMTFLEAVSA-N CCc1cccc([n]2C(C)C)c1nc2[O-]C(C[C@H]1C(N)=O)CN1C([C@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)NC(OC(C)(C)C)=O)=O Chemical compound CCc1cccc([n]2C(C)C)c1nc2[O-]C(C[C@H]1C(N)=O)CN1C([C@H](CCCCC/C=C\[C@H](C1)[C@H]1C(NS(N(C)C)(=O)=O)=O)NC(OC(C)(C)C)=O)=O XNFWQZHNVQLRPL-QMTFLEAVSA-N 0.000 description 1
- FUSFWUFSEJXMRQ-UHFFFAOYSA-N COC(CBr)OC Chemical compound COC(CBr)OC FUSFWUFSEJXMRQ-UHFFFAOYSA-N 0.000 description 1
- USQLAJAZCXMOPJ-UHFFFAOYSA-N COc1c(-c2ccc[s]2)nc(cccc2)c2n1 Chemical compound COc1c(-c2ccc[s]2)nc(cccc2)c2n1 USQLAJAZCXMOPJ-UHFFFAOYSA-N 0.000 description 1
- BAXABXIZQPAJEF-UHFFFAOYSA-N COc1cnc(cccc2)c2n1 Chemical compound COc1cnc(cccc2)c2n1 BAXABXIZQPAJEF-UHFFFAOYSA-N 0.000 description 1
- JZMUUDCDJJOJSW-UHFFFAOYSA-N Clc1nc2ccccc2[n]1-c1ccccc1 Chemical compound Clc1nc2ccccc2[n]1-c1ccccc1 JZMUUDCDJJOJSW-UHFFFAOYSA-N 0.000 description 1
- XPCCMGJNLNAIKA-UHFFFAOYSA-N Clc1ncc2nc[n](Cc3ccccc3)c2n1 Chemical compound Clc1ncc2nc[n](Cc3ccccc3)c2n1 XPCCMGJNLNAIKA-UHFFFAOYSA-N 0.000 description 1
- XXKQACFOJGQZTB-UHFFFAOYSA-N NS(OC1CC1)(=O)=O Chemical compound NS(OC1CC1)(=O)=O XXKQACFOJGQZTB-UHFFFAOYSA-N 0.000 description 1
- XUBHLQIHDDSOME-UHFFFAOYSA-N O=C1Nc(cccc2)c2N=C1c(cc1)ccc1F Chemical compound O=C1Nc(cccc2)c2N=C1c(cc1)ccc1F XUBHLQIHDDSOME-UHFFFAOYSA-N 0.000 description 1
- ZBBQSGVRBQKLLE-UHFFFAOYSA-N O=C1Nc(cccc2)c2N=C1c1ccccc1 Chemical compound O=C1Nc(cccc2)c2N=C1c1ccccc1 ZBBQSGVRBQKLLE-UHFFFAOYSA-N 0.000 description 1
- NOGLKXWLUDJZDQ-UHFFFAOYSA-N O=C1Nc2ccccc2N=C1C(F)(F)F Chemical compound O=C1Nc2ccccc2N=C1C(F)(F)F NOGLKXWLUDJZDQ-UHFFFAOYSA-N 0.000 description 1
- OAHKWDDSKCRNFE-UHFFFAOYSA-N O=S(Cc1ccccc1)(Cl)=O Chemical compound O=S(Cc1ccccc1)(Cl)=O OAHKWDDSKCRNFE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- the present invention relates to compounds, processes for their synthesis, compositions and methods for the treatment of hepatitis C virus (HCV) infection.
- HCV hepatitis C virus
- HCV infection is the most common chronic blood borne infection in the United States. Although the numbers of new infections have declined, the burden of chronic infection is substantial, with Centers for Disease Control estimates of 3.9 million (1.8%) infected persons in the United States.
- Chronic liver disease is the tenth leading cause of death among adults in the United States, and accounts for approximately 25,000 deaths annually, or approximately 1% of all deaths. Studies indicate that 40% of chronic liver disease is HCV-related, resulting in an estimated 8,000- 10,000 deaths each year. HCV-associated end-stage liver disease is the most frequent indication for liver transplantation among adults.
- Antiviral therapy of chronic hepatitis C has evolved rapidly over the last decade, with significant improvements seen in the efficacy of treatment. Nevertheless, even with combination therapy using pegylated IFN-0C plus ribavirin, 40% to 50% of patients fail therapy, i.e., are nonresponders (NR) or relapsers. These patients currently have no effective therapeutic alternative. In particular, patients who have advanced fibrosis or cirrhosis on liver biopsy are at significant risk of developing complications of advanced liver disease, including ascites, jaundice, variceal bleeding, encephalopathy, and progressive liver failure, as well as a markedly increased risk of hepatocellular carcinoma.
- HCV is an enveloped positive strand RNA virus in the Flaviviridae family.
- the single strand HCV RNA genome is approximately 9500 nucleotides in length and has a single open reading frame (ORF) encoding a single large polyprotein of about 3000 amino acids. In infected cells, this polyprotein is cleaved at multiple sites by cellular and viral proteases to produce the structural and non-structural (NS) proteins of the virus.
- ORF open reading frame
- NS structural and non-structural
- the generation of mature nonstructural proteins (NS2, NS3, NS4, NS4A, NS4B, NS5A, and NS5B) is effected by two viral proteases.
- the first viral protease cleaves at the NS2-NS3 junction of the polyprotein.
- the second viral protease is serine protease contained within the N-terminal region of NS3 (herein referred to as "NS3 protease").
- NS3 protease mediates all of the subsequent cleavage events at sites downstream relative to the position of NS3 in the polyprotein (i.e., sites located between the C-terminus of NS3 and the C-terminus of the polyprotein).
- NS3 protease exhibits activity both in cis, at the NS3-NS4 cleavage site, and in trans, for the remaining NS4A- NS4B, NS4B-NS5A, and NS5A-NS5B sites.
- the NS4A protein is believed to serve multiple functions, acting as a cofactor for the NS3 protease and possibly assisting in the membrane localization of NS3 and other viral replicase components.
- the formation of the complex between NS3 and NS4A is necessary for NS3-mediated processing events and enhances proteolytic efficiency at all sites recognized by NS3.
- the NS3 protease also exhibits nucleoside triphosphatase and RNA helicase activities.
- NS5B is an RNA-dependent RNA polymerase involved in the replication of HCV RNA.
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl.
- R lc and R ld are each separately selected from the group consisting of - H, Ci-4 alkoxy, C 1-6 alkyl, C3-7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2 is selected from the group consisting of
- X, Y, and 2 are each independently selected from -CH- or -N-, wherein X and Y are not both -CH-, and X, Y 1 , and Y 2 are not all -CH-; Z is O (oxygen) or S (sulfur); V and W are each independently selected from -CR 2k - or -N-, wherein V and W are not both -CR 2k -; n is 1, 2 or 3; and R 2j and R 2k are each independently selected from the group consisting of H, halo, optionally substituted aryl, optionally substituted heteroaryl; or R 2j and R 2k together form an aryl ring optionally substituted by 1-3 R 2g .
- R 2a , R 2e and R 2g are each independently selected from the group consisting of halo, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , Ci-6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C3-7 cycloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl and optionally substituted heteroaryl.
- Each R 2c is independently selected from the group consisting of halo, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , Ci_6 alkyl, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl, wherein said Ci_6 alkyl, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, Ci_6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl each optionally substituted with one or more R 12.
- Each R 12 is independently selected from the group consisting of Ci_6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, heteroaryl, arylalkyl, aryl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , -C(0)NR'R” and -NR'R", wherein said Ci_ 6 alkyl, C 3-7 cycloalkyl, Ci_ 6 alkoxy, heteroaryl, arylalkyl, cycloalkylalkyl, and aryl are each optionally substituted with one or more R 12a .
- Each R 12a is independently selected from the group consisting of -F, -CI, - CF 3 , -OCF 3 , Ci_6 alkyl, Ci_ 6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- Each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), halo, -C(0)NR'R", optionally substituted C 1-6 alkyl, optionally substituted C 2 _ 6 alkenyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- R 2b , R 2d , and R 2 are each independently selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl;
- R 2h is selected from the group consisting of propyl, butyl and phenyl;
- R 1 is Ci_ 6 alkyl optionally substituted with up to 5 fluoro.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond. [0020] Provided that if R 2 is or , then R 1 is not phenyl.
- R 2 is ⁇ N ⁇ ⁇ s t en
- R 1 is not -C(0)0-t-butyl, phenyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro, chloro and -CF 3 .
- R 1 is not -C(0)0-t-butyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro and -CF 3 .
- R i is not -C(0)0-t-butyl or phenyl.
- R 1 is not -C(0)0-t- butyl, benzoxazyl, t-butylthiazyl, phenyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro, chloro, methyl, -CF 3 and - OCF 3 .
- Some embodiments provide a compound having the structure of Formula IIa-1: (IIa-1) or a pharmaceutically acceptable salt or prodrug thereof wherein R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alk
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- R 7 is selected from the group consisting of -NH 2 , -NH 2 HC1, -COOH, -C(0)NR la R lb , -NHC(0)NR la R lb and heteroaryl containing 1-3 heteroatoms independently selected from N or O;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl;
- R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C3_7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- Some embodiments provide a compound having the structure of Formula ⁇ or IV:
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl
- R lc and R ld are each separately selected from the group consisting of - H, Ci-4 alkoxy, Ci_6 alkyl, C 3 -7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- X, Y, Y , and Y are each independently selected from -CH- or -N-, wherein X and Y are not both -CH-, and X, Y 1 , and Y 2 are not all -CH-;
- R 2b is selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 -7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl.
- Each R c is independently selected from the group consisting of halo, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , C 2 _ 6 alkyl, C 2 _ 6 alkenyl, C3-7 cycloalkyl, C 1-6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl, said C 2 _ 6 alkyl, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl each optionally substituted with one or more R 1 ⁇ 2.
- Each R 1"2 is independently selected from the group consisting of Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, heteroaryl, arylalkyl, aryl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , - C(0)NR'R" and -NR'R", wherein said C 2 _ 6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, heteroaryl, arylalkyl, cycloalkylalkyl, and aryl are each optionally substituted with one or more R 12a .
- Each R 12a is independently selected from the group consisting of -F, -CI, - CF 3 , -OCF 3 , Ci- 6 alkyl, C 1-6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- Each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), halo, -C(0)NR'R", optionally substituted Ci_ 6 alkyl, optionally substituted C 2 _ 6 alkenyl, optionally substituted Ci- 6 alkoxy, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- R 1 is Ci_ 6 alkyl optionally substituted with up to 5 fluoro.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond.
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl.
- R lc and R ld are each separately selected from the group consisting of - H, Ci_4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2a is selected from the group consisting of -H, -C(0)OR lc , Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, optionally substituted aryl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 )tC3_ 7 cycloalkyl, C 2 - 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C3_ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 )tC3_ 7 cycloalkyl, C 2 - 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond.
- Some embodiments provide a compound having the structure of Formulas VI- 1 or I-2:
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 - 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from N and O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- X is -N- or -CH-;
- R 2d is selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 o r l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected from the
- Some embodiments provide a compound having the structure of Formula Vila or llb:
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from N and O; R lc and R ld are each separately selected from the group consisting of -H, C 1-4 alkoxy, C 1-6 alkyl, C 3 -7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2e is selected from the group consisting of -H, -Br, -CI, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", Ci_6 alkyl optionally substituted with up to 5 fluoro, C 2 _6 alkenyl, C 3 -7 cycloalkyl, optionally substituted C 1-6 alkoxy, optionally substituted aryl and optionally substituted heteroaryl; wherein R' and R" are each independently selected from the group consisting of -H, optionally substituted C 1-6 alkyl, optionally substituted C 2 - 6 alkenyl, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Some embodiments provide a compound having the structure of Formula Villa:
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 - 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from N and O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2 is selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 o r l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Some embodiments provide a compound having the structure of Formula IX:
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from N and O; R lc and R ld are each separately selected from the group consisting of -H, C 1-4 alkoxy, C 1-6 alkyl, C 3 -7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2g is selected from the group consisting of -H, -Br, -CI, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", Ci_6 alkyl optionally substituted with up to 5 fluoro, C 2 _6 alkenyl, C 3 -7 cycloalkyl, optionally substituted C 1-6 alkoxy, optionally substituted aryl and optionally substituted heteroaryl; wherein R' and R" are each independently selected from the group consisting of -H, optionally substituted Ci_ 6 alkyl, optionally substituted C 2 - 6 alkenyl, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Some embodiments provide a compound having the structure of Formula X:
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from N and O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl , arylalkyl and heteroaryl.
- R 2h is selected from the group consisting of n-propyl, cyclopropyl, n- butyl, t-butyl, 1 -sec-butyl and phenyl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Some embodiments provide a pharmaceutical composition comprising a pharmaceutically acceptable excipient and a compound of any one of Formulas I, la, II, ⁇ , IV, V, VI- 1, VI-2, Vn, Vni, IX, X, XI, and ⁇ , or any compounds disclosed herein.
- Some embodiments provide a method of inhibiting NS3/NS4 protease activity comprising contacting a NS3/NS4 protease with a compound of any one of Formulas I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, and XII, any compounds disclosed herein, or a pharmaceutical composition disclosed herein.
- Some embodiments provide a method of treating liver fibrosis in an individual, the method comprising administering to the individual an effective amount of a compound of any one of Formulas I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, and ⁇ , any compounds disclosed herein, or a pharmaceutical composition disclosed herein.
- Some embodiments provide a method of increasing liver function in an individual having a hepatitis C virus infection, the method comprising administering to the individual an effective amount of a compound of any one of Formulas I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, and ⁇ , any compounds disclosed herein, or a pharmaceutical composition disclosed herein.
- hepatic fibrosis used interchangeably herein with “liver fibrosis,” refers to the growth of scar tissue in the liver that can occur in the context of a chronic hepatitis infection.
- the terms "individual,” “host,” “subject,” and “patient” are used interchangeably herein, and refer to a mammal, including, but not limited to, primates, including simians and humans.
- liver function refers to a normal function of the liver, including, but not limited to, a synthetic function, including, but not limited to, synthesis of proteins such as serum proteins (e.g., albumin, clotting factors, alkaline phosphatase, aminotransferases (e.g., alanine transaminase, aspartate transaminase), 5'- nucleosidase, ⁇ -glutaminyltranspeptidase, etc.), synthesis of bilirubin, synthesis of cholesterol, and synthesis of bile acids; a liver metabolic function, including, but not limited to, carbohydrate metabolism, amino acid and ammonia metabolism, hormone metabolism, and lipid metabolism; detoxification of exogenous drugs; a hemodynamic function, including splanchnic and portal hemodynamics; and the like.
- serum proteins e.g., albumin, clotting factors, alkaline phosphatase, aminotransferases (e.g., alanine transa
- sustained viral response refers to the response of an individual to a treatment regimen for HCV infection, in terms of serum HCV titer.
- a sustained viral response refers to no detectable HCV RNA (e.g., less than about 500, less than about 200, or less than about 100 genome copies per milliliter serum) found in the patient's serum for a period of at least about one month, at least about two months, at least about three months, at least about four months, at least about five months, or at least about six months following cessation of treatment.
- Treatment failure patients generally refers to HCV- infected patients who failed to respond to previous therapy for HCV (referred to as “non- responders") or who initially responded to previous therapy, but in whom the therapeutic response was not maintained (referred to as “relapsers").
- the previous therapy generally can include treatment with IFN-a monotherapy or IFN-a combination therapy, where the combination therapy may include administration of IFN-a and an antiviral agent such as ribavirin.
- treatment refers to obtaining a desired pharmacologic and/or physiologic effect.
- the effect may be prophylactic in terms of completely or partially preventing a disease or symptom thereof and/or may be therapeutic in terms of a partial or complete cure for a disease and/or adverse affect attributable to the disease.
- Treatment covers any treatment of a disease in a mammal, particularly in a human, and includes: (a) preventing the disease from occurring in a subject which may be predisposed to the disease but has not yet been diagnosed as having it; (b) inhibiting the disease, i.e., arresting its development; and (c) relieving the disease, i.e., causing regression of the disease.
- a Type I interferon receptor agonist refers to any naturally occurring or non-naturally occurring ligand of human Type I interferon receptor, which binds to and causes signal transduction via the receptor.
- Type I interferon receptor agonists include interferons, including naturally-occurring interferons, modified interferons, synthetic interferons, pegylated interferons, fusion proteins comprising an interferon and a heterologous protein, shuffled interferons; antibody specific for an interferon receptor; non-peptide chemical agonists; and the like.
- Type II interferon receptor agonist refers to any naturally occurring or non-naturally occurring ligand of human Type ⁇ interferon receptor that binds to and causes signal transduction via the receptor.
- Type ⁇ interferon receptor agonists include native human interferon- ⁇ , recombinant IFN- ⁇ species, glycosylated IFN- ⁇ species, pegylated IFN- ⁇ species, modified or variant IFN- ⁇ species, IFN- ⁇ fusion proteins, antibody agonists specific for the receptor, non-peptide agonists, and the like.
- a Type ⁇ interferon receptor agonist refers to any naturally occurring or non-naturally occurring ligand of humanIL-28 receptor a ("IL-28R”), the amino acid sequence of which is described by Sheppard, et al., infra., that binds to and causes signal transduction via the receptor.
- IL-28R humanIL-28 receptor a
- interferon receptor agonist refers to any Type I interferon receptor agonist, Type ⁇ interferon receptor agonist, or Type ⁇ interferon receptor agonist.
- dosing event refers to administration of an antiviral agent to a patient in need thereof, which event may encompass one or more releases of an antiviral agent from a drug dispensing device.
- dosing event includes, but is not limited to, installation of a continuous delivery device (e.g., a pump or other controlled release injectible system); and a single subcutaneous injection followed by installation of a continuous delivery system.
- Continuous delivery as used herein (e.g., in the context of “continuous delivery of a substance to a tissue”) is meant to refer to movement of drug to a delivery site, e.g., into a tissue in a fashion that provides for delivery of a desired amount of substance into the tissue over a selected period of time, where about the same quantity of drug is received by the patient each minute during the selected period of time.
- substantially continuous as used in, for example, the context of “substantially continuous infusion” or “substantially continuous delivery” is meant to refer to delivery of drug in a manner that is substantially uninterrupted for a pre-selected period of drug delivery, where the quantity of drug received by the patient during any 8 hour interval in the pre-selected period never falls to zero.
- substantially continuous drug delivery can also encompass delivery of drug at a substantially constant, pre-selected rate or range of rates (e.g., amount of drug per unit time, or volume of drug formulation for a unit time) that is substantially uninterrupted for a pre-selected period of drug delivery.
- substantially steady state as used in the context of a biological parameter that may vary as a function of time, it is meant that the biological parameter exhibits a substantially constant value over a time course, such that the area under the curve defined by the value of the biological parameter as a function of time for any 8 hour period during the time course (AUC8hr) is no more than about 20% above or about 20% below, and preferably no more than about 15% above or about 15% below, and more preferably no more than about 10% above or about 10% below, the average area under the curve of the biological parameter over an 8 hour period during the time course (AUC8hr average).
- the serum concentration of the drug is maintained at a substantially steady state during a time course when the area under the curve of serum concentration of the drug over time for any 8 hour period during the time course (AUC8hr) is no more than about 20% above or about 20% below the average area under the curve of serum concentration of the drug over an 8 hour period in the time course (AUC8hr average), i.e., the AUC8hr is no more than 20% above or 20% below the AUC8hr average for the serum concentration of the drug over the time course.
- AUC8hr area under the curve of serum concentration of the drug over time for any 8 hour period during the time course
- AUC8hr average the average area under the curve of serum concentration of the drug over an 8 hour period in the time course
- hydrogen bond refers to an attractive force between an electronegative atom (such as oxygen, nitrogen, sulfur or halogen) and a hydrogen atom which is linked covalently to another electronegative atom (such as oxygen, nitrogen, sulfur or halogen). See, e.g., Stryer et. al. "Biochemistry", Fith Edition 2002, Freeman & Co. N.Y. Typically, the hydrogen bond is between a hydrogen atom and two unshared electrons of another atom.
- a hydrogen bond may be present when the distance between the electronegative atom to which the hydrogen is covalently bonded, and the other electronegative atom to which the hydrogen is attracted, is 2.2 angstroms to about 3.8 angstroms, and the angle formed by the three atoms (electronegative atom covalently bound to hydrogen, hydrogen, and electronegative atom not-covalently bound) deviates from 180 degrees by about 60 degrees or less.
- the distance between the two electronegative atoms may be referred to herein as the "hydrogen bond length,” and the angle formed by the three atoms (electronegative atom covalently bound to hydrogen, hydrogen, and electronegative atom not-covalently bound) may be referred to herein as the "hydrogen bond angle", as shown in Figure X:
- hydrogen bond lengths may range from about 2.4 angstroms to about 3.6 angstroms, or about 2.5 angstroms to about 3.4 angstroms.
- stronger hydrogen bonds are formed when the hydrogen bond angle is closer to being linear; thus, in some instances, hydrogen bond angles may deviate from 180 degrees by about 25 degrees or less, or by about 10 degrees or less.
- non-polar interaction refers to the proximity of a non-polar atom, molecule or moiety to another atom, molecule or moiety, or the proximity of an atom, molecule or moiety with low polarity to another atom, molecule or moiety, sufficient for van der Waals interaction between the atoms/molecules. See, e.g., Stryer et. al. "Biochemistry", Fifth Edition 2002, Freeman & Co. N.Y. Typically, the distance between heavy (non-hydrogen) atoms of non-polar interacting moieties is sufficiently close to exclude polar solvent molecules, such as water molecules.
- Non-polar interactions may range from about 2.5 angstroms to about 4.8 angstroms, from about 2.5 angstroms to about 4.3 angstroms, or from about 2.5 angstroms to about 3.8 angstroms.
- a non-polar moiety or moiety with low polarity refers to moieties with low dipolar moments (typically dipolar moments less than the dipolar moment of O-H bonds of H 2 0 and N-H bonds of NH 3 ), and/or moieties that are not typically present in hydrogen bonding or electrostatic interactions. Examples of moieties with low polarity are alkyl, alkenyl, and unsubstituted aryl moieties.
- the term "non-polar interactions” refers to "hydrophobic interactions" and/or "van der Waals Interactions.”
- an NS3 protease SI' pocket moiety refers to a moiety of the NS3 protease that interacts with the amino acid positioned one residue C-terminal to the cleavage site of the substrate polypeptide cleaved by NS3 protease as described in paragraph [0066] of WO 2007/015824 incorporated herein in its entirety.
- exemplary moieties include, but are not limited to, atoms of the peptide backbone or side chains of amino acids Lysl36, Glyl37, Serl39, His57, Gly58, Gln41, Ser42, and Phe43, see Yao. et. al., Structure 1999, 7, 1353, incorporated herein in its entirety.
- an NS3 protease S2 pocket moiety refers to a moiety of the NS3 protease that interacts with the amino acid positioned two residues N-terminal to the cleavage site of the substrate polypeptide cleaved by NS3 protease as described in paragraph [0067] of WO 2007/015824, incorporated herein in its entirety.
- exemplary moieties include, but are not limited to, atoms of the peptide backbone or side chains of amino acids Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81, see Yao. et. al., Structure 1999, 7, 1353.
- alkyl refers to a radical of a fully saturated hydrocarbon, including, but not limited to, methyl, ethyl, n-propyl, isopropyl (or i-propyl),
- alkyl as used herein includes radicals of fully saturated hydrocarbons defined by the following general formula's: the general formula for linear or branched fully saturated hydrocarbons not containing a cyclic structure is C n H 2n+ 2; the general formula for a fully saturated hydrocarbon containing one ring is C n H 2n ; the general formula for a fully saturated hydrocarbon containing two rings is C n H 2 ( n -i); the general formula for a saturated hydrocarbon containing three rings is C n H 2 (n- 2 )- When a more specific term for alkyl (such as propyl, butyl, etc.) is used without specifying linear or branched, the term is to be interpreted to include linear and branched alkyl.
- alkyl such as propyl, butyl, etc.
- halo refers to fluoro, chloro, bromo, or iodo.
- alkoxy refers to straight or branched chain alkyl radical covalently bonded to the parent molecule through an — O— linkage.
- alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, butoxy, n-butoxy, sec-butoxy, t-butoxy and the like.
- a more specific term for alkoxy such as propoxy, butaoxy, etc.
- the term is to be interpreted to include linear and branched alkoxy.
- alkenyl used herein refers to a monovalent straight or branched chain radical of from two to twenty carbon atoms containing a carbon double bond including, but not limited to, 1-propenyl, 2-propenyl, 2-methyl-l-propenyl, 1- butenyl, 2-butenyl, and the like.
- alkynyl used herein refers to a monovalent straight or branched chain radical of from two to twenty carbon atoms containing a carbon triple bond including, but not limited to, 1-propynyl, 1-butynyl, 2-butynyl, and the like.
- polycyclic moiety refers a bicyclic moiety or tricyclic moiety optionally containing one or more heteroatoms wherein at least one of the rings is an aryl or heteroaryl ring and at least one of the rings is not an aryl or heteroaryl ring.
- the bicyclic moiety contains two rings wherein the rings are fused. The bicyclic moiety can be appended at any position of the two rings. For example, bicyclic moiety
- aryl used herein refers to homocyclic aromatic radical whether one ring or multiple fused rings. Examples of aryl groups include, but are not limited to, phenyl, naphthyl, phenanthrenyl, naphthacenyl, and the like.
- cycloalkyl used herein refers to saturated aliphatic ring system radical having three to twenty carbon atoms including, but not limited to, cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like.
- cycloalkenyl refers to aliphatic ring system radical having three to twenty carbon atoms having at least one carbon-carbon double bond in the ring.
- Examples of cycloalkenyl groups include, but are not limited to, cyclopropenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, bicyclo[3.1.0]hexyl, and the like.
- heterocyclic or “heterocyclyl” or “heterocycloalkyl” used herein refers to cyclic non-aromatic ring system radical having at least one ring in which one or more ring atoms are not carbon, namely heteroatom. In fused ring systems, the one or more heteroatoms may be present in only one of the rings.
- heterocyclic groups include, but are not limited to, morpholinyl, tetrahydrofuranyl, dioxolanyl, pyrolidinyl, pyranyl, piperidyl, piperazyl, oxetanyl and the like.
- heteroaryl refers to an aromatic group comprising one or more heteroatoms, whether one ring or multiple fused rings. When two or more heteroatoms are present, they may be the same or different. In fused ring systems, the one or more heteroatoms may be present in only one of the rings. Examples of heteroaryl groups include, but are not limited to, benzothiazyl, benzoxazyl, quinazolinyl, quinolinyl, isoquinolinyl, quinoxalinyl, pyridinyl, pyrrolyl, oxazolyl, indolyl, thiazyl and the like.
- heteroatom refers to S (sulfur), N (nitrogen), and O (oxygen).
- arylalkyl refers to one or more aryl groups appended to an alkyl radical.
- arylalkyl groups include, but are not limited to, benzyl, phenethyl, phenpropyl, phenbutyl, and the like.
- cycloalkylalkyl refers to one or more cycloalkyl groups appended to an alkyl radical.
- examples of cycloalkylalkyl include, but are not limited to, cyclohexylmethyl, cyclohexylethyl, cyclopentylmethyl, cyclopentylethyl, and the like.
- heteroarylalkyl refers to one or more heteroaryl groups appended to an alkyl radical.
- heteroarylalkyl include, but are not limited to, pyridylmethyl, furanylmethyl, thiopheneylethyl, and the like.
- aryloxy used herein refers to an aryl radical covalently bonded to the parent molecule through an— O— linkage.
- alkylthio refers to straight or branched chain alkyl radical covalently bonded to the parent molecule through an — S— linkage.
- alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, butoxy, n-butoxy, sec-butoxy, t-butoxy and the like.
- arylthio refers to an aryl radical covalently bonded to the parent molecule through an— S— linkage.
- alkylamino refers to nitrogen radical with one or more alkyl groups attached thereto.
- monoalkylamino refers to nitrogen radical with one alkyl group attached thereto and dialkylamino refers to nitrogen radical with two alkyl groups attached thereto.
- cyanoamino used herein refers to nitrogen radical with nitrile group attached thereto.
- hydroxyalkyl refers to one or more hydroxy groups appended to an alkyl radical.
- aminoalkyl refers to one or more amino groups appended to an alkyl radical.
- arylalkyl refers to one or more aryl groups appended to an alkyl radical.
- sulfamyl used herein refers to -SO 2 NH 2 .
- thiocarboxy used herein refers to CSOH.
- a radical indicates species with one or more, unpaired electron such that the species containing the radical can be covalently bonded to one or more other species.
- a radical is not necessarily a free radical. Rather, a radical indicates a specific portion of a larger molecule.
- the term "radical” can be used interchangeably with the terms "group” and "moiety.”
- a substituted group is derived from the unsubstituted parent structure in which there has been an exchange of one or more hydrogen atoms for another atom or group.
- the substituent group(s) is (are) one or more group(s) individually and independently selected from CrC 6 alkyl, Ci-C 6 alkenyl, Ci-C 6 alkynyl, C 3 -C7 cycloalkyl (optionally substituted with halo, alkyl, alkoxy, carboxyl, CN, -S0 2 -alkyl, -CF 3 , and -OCF 3 ), C 3 -C 6 heterocycloalkyl (e.g., tetrahydrofuryl) (optionally substituted with halo, alkyl, alkoxy, carboxyl, CN, -S0 2 - alkyl, -CF 3 , and -OCF 3 ), aryl (optionally substituted with
- Asymmetric carbon atoms may be present in the compounds described. All such isomers, including diastereomers and enantiomers, as well as the mixtures thereof are intended to be included in the scope of the recited compound. In certain cases, compounds can exist in tautomeric forms. All tautomeric forms are intended to be included in the scope. Likewise, when compounds contain an alkenyl or alkenylene group, there exists the possibility of cis- and trans- isomeric forms of the compounds. Both cis- and trans- isomers, as well as the mixtures of cis- and trans- isomers, are contemplated. Thus, reference herein to a compound includes all of the aforementioned isomeric forms unless the context clearly dictates otherwise.
- Isotopes may be present in the compounds described. Each chemical element as represented in a compound structure may include any isotope of said element.
- a hydrogen atom may be explicitely disclosed or understood to be present in the compound.
- the hydrogen atom can be any isotope of hydrogen, including but not limited to hydrogen-1 (protium) and hydrogen-2 (deuterium).
- reference herein to a compound encompasses all potential isotopic forms unless the context clearly dictates otherwise.
- a substituent is depicted as a di-radical (i.e., has two points of attachment to the rest of the molecule), it is to be understood that the substituent can be attached in any directional configuration unless otherwise indicated.
- a substituent depicted as -AE- or Y 3 ⁇ 4 A ⁇ C tA includes the substituent being oriented such that the A is attached at the leftmost attachment point of the molecule as well as attached at the rightmost attachment point of the molecule.
- radical naming conventions can include either a mono-radical or a di-radical, depending on the context. For example, where a substituent requires two points of attachment to the rest of the molecule, it is understood that the substituent is a di-radical.
- a substituent identified as alkyl, that requires two points of attachment includes di-radicals such as -CH 2 -, -CH 2 CH 2 -, - CH 2 CH(CH 3 )CH 2 -, and the like; a substituent depicted as alkoxy that requires two points of attachment, includes di-radicals such as -OCH 2 - -OCH 2 CH 2 - -OCH 2 CH(CH 3 )CH 2 - and the like: and a substituent depict - that requires two points of
- attachment includes di-radicals such as
- a polymorph is a composition having the same chemical formula, but a different structure.
- a solvate is a composition formed by solvation (the combination of solvent molecules with molecules or ions of the solute).
- a hydrate is a compound formed by an incorporation of water.
- a conformer is a structure that is a conformational isomer. Conformational isomerism is the phenomenon of molecules with the same structural formula but different conformations (conformers) of atoms about a rotating bond. Salts of compounds can be prepared by methods known to those skilled in the art.
- salts of compounds can be prepared by reacting the appropriate base or acid with a stoichiometric equivalent of the compound.
- a prodrug is a compound that undergoes biotransformation (chemical conversion) before exhibiting its pharmacological effects.
- a prodrug can thus be viewed as a drug containing specialized protective groups used in a transient manner to alter or to eliminate undesirable properties in the parent molecule.
- reference herein to a compound includes all of the aforementioned forms unless the context clearly dictates otherwise.
- the present embodiments provide compounds of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VT2, Vn, VIE, IX, X, XI, and ⁇ , as well as pharmaceutical compositions and formulations comprising any compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VT2, Vn, Vni, IX, X, XI, and ⁇ .
- a subject compound is useful for treating HCV infection and other disorders, as discussed below.
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc .
- said heteroaryl contains 1-3 heteroatoms independently selected from S, N, or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O.
- R lc and R ld are each separately selected from the group consisting of -H, Ci_4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2 is selected from the group consisting of :
- X, Y, Y 1 , and ⁇ 2 are each independently selected from -CH- or -N-, wherein X and Y are not both -CH-, and X, Y 1 , and Y 2 are not all -CH-; Z is O or S; V and W are each independently selected from -CR 2k - or -N-, wherein V and W are not both -CR 2k -; n is 1, 2 or 3; and R 2j and R 2k are each independently selected from the group consisting of H, halo, optionally substituted aryl, optionally substituted heteroaryl; or R 2j and R 2k together form an aryl ring optionally substituted by 1-3 R 2g .
- R 2a , R 2e and R 2g are each independently selected from the group consisting of halo, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , Ci-6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C3-7 cycloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl and optionally substituted heteroaryl.
- Each R 2c is independently selected from the group consisting of halo, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl, wherein said Ci_6 alkyl, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, Ci_6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl each optionally substituted with one or more R 12.
- Each R 12 is independently selected from the group consisting of Ci_6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, heteroaryl, arylalkyl, aryl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , -C(0)NR'R” and -NR'R", wherein said Ci_ 6 alkyl, C 3-7 cycloalkyl, Ci_ 6 alkoxy, heteroaryl, arylalkyl, cycloalkylalkyl, and aryl are each optionally substituted with one or more R 12a .
- Each R 12a is independently selected from the group consisting of -F, -CI, - CF 3 , -OCF 3 , Ci_6 alkyl, Ci_ 6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- Each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), halo, -C(0)NR'R", optionally substituted C 1-6 alkyl, optionally substituted C 2 _ 6 alkenyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- R 2b , R 2d , and R 2 are each independently selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl;
- R 2h is selected from the group consisting of propyl, butyl and phenyl;
- R 1 is Ci_ 6 alkyl optionally substituted with up to 5 fluoro.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring bonded to the parent structure through a nitrogen, and where the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group
- R 1 is not -C(0)0-t-butyl, phenyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro, chloro and -CF 3 .
- R 1 is not -C(0)0-t-butyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro and -CF 3 .
- R 1 is not -C(0)0-t-butyl or phenyl.
- R 1 is not -C(0)0-t- butyl, benzoxazyl, t-butylthiazyl, phenyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro, chloro, methyl, -CF 3 and - OCF 3. .
- compounds of Formula I have the structure of Formula la:
- R 1 , R2 , and R 3 are the same as defined above.
- Some embodiments provide compounds of Formula I or Formula la, in which R 1 is selected from the group consisting of -C(0)0-R le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substitutents each independently selected from the group consisting of Ci_ 6 alkyl, fluoro, amino, -CF 3 , - OCF 3 , -C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OH, and oxazolyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more lc substituents independently selected from Ci_ 6 alkyl, C 2 - 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR
- heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; and R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- Some embodiments provide compounds of Formula I or Formula la, in which R 1 is aryl optionally substituted with one or more substitutents each independently selected from the group consisting of -C(0)NR la R lb and -NHC(0)NR la R lb , wherein R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with Ci_ 6 alkyl, hydroxy-Ci_ 6 alkyl, amino-Ci_ 6 alkyl, aryl-C 1-6 alkyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently sseelleecctteedd ffrroomm NN oorr OO.. IInn ssoommee eemmbbooddiimmfents
- R 4 is selected from the group consisting of -H, Ci_ 6 alkyl optionally substituted with one or more amine, aryl or hydroxy, aryl optionally substituted with C 1-4 alkyl, -CF 3 , or -OCF 3 , and -C(0)R 4a , where R 4a is selected from the group consisting of Ci_ 4 alkoxy, C 3 _7 cycloalkyl and aryl; and R 5 and R 6 are each independently -H or C 1-6 alkyl optionally substituted with phenyl.
- Some embodiments provide compounds of Formula I or Formula la, in which R is selected from the group consisting of
- each R c is independently selected from the group consisting of -CF 3 , -Br, -CI, - C(0)OH, -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , Ci_6 alkyl, C 2 _ 6 alkenyl, Ci_ 6 alkoxy, polycyclic moiety, phenyl, and heteroaryl, said C 1-6 alkyl, C 2 _ 6 alkenyl, C 1-6 alkoxy, polycyclic moiety, aryl, and heteroaryl each optionally substituted with one or more R 12 ; and in some embodiments, said heteroaryl may be selected from the group consisting of furanyl, thiazolyl, oxazolyl, thiophenyl, pyrazolyl, and benzo thiazolyl.
- Each R 12 is independently selected from the group consisting of C 1-6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , -C(0)NR'R", morpholinyl, pyrrolidinyl, piperidiny, C 3 _ 7 cycloalkyl-alkyl, wherein said Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, morpholinyl, pyrrolidinyl, piperidiny, are each optionally substituted with one or more R 12a .
- Each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), -F, -Clford - C(0)NR'R", Ci-6 alkyl, C 2 _ 6 alkenyl, C 1-6 alkoxy, phenyl, phenylalkyl, and heteroaryl; and each R 12a is independently selected from the group consisting of -F, -CI, Ci_ 6 alkyl, Ci_ 6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- R 2d is selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 3 _ 7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl; and R 1 is ethyl or i-propyl.
- Some embodiments provide compounds of Formula I or Formula la, in
- each R c is independently selected from the group consisting of -CF 3 , -Br, -CI, -C(0)OH, -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , Ci_ 6 alkyl, C 2 _ 6 alkenyl, Ci_ 6 alkoxy, polycyclic moiety, phenyl, and heteroaryl, said C 1-6 alkyl, C 2 _ 6 alkenyl, C 1-6 alkoxy, polycyclic moiety, aryl, and heteroaryl each optionally substituted with one or more R 12 ; and in some embodiments, said heteroaryl may be selected from the group consisting of furanyl, thiazolyl, oxazolyl, thiophenyl, pyrazolyl, and benzothiazolyl.
- each R 12 is independently selected from the group consisting of Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , -C(0)NR'R” and morpholinyl, pyrrolidinyl, piperidiny, C 3 _ 7 cycloalkyl-alkyl, wherein said Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, morpholinyl, pyrrolidinyl, piperidiny, are each optionally substituted with one or more R 12a .
- each R 12a is independently selected from the group consisting of -F, -CI, Ci_ 6 alkyl, Ci_ 6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), -F, - CI, -C(0)NR'R", Ci-6 alkyl, C 2 -6 alkenyl, Ci_6 alkoxy, phenyl, phenylalkyl and heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- each R 2c is independently aryl optionally substituted with halo, cyano, Ci_6 alkyl optionally substituted with up to 5 fluoro, or Ci_6 alkoxy optionally substituted with up to 5 fluoro, C(0)NR'R", wherein R' and R" are independently optionally substituted Ci_6 alkyl.
- each R 2c is independently heteroaryl or polycyclic moiety, each optionally substituted with aryl, arylalkyl, Ci_6 alkyl optionally substituted with up to 5 fluoro, heteroaryl, heterocyclyl, C 3 _ 7 cycloalkyl, or . C 3 _ 7 cycloalkyl-alkyl; wherein said aryl, heteroaryl, and heterocyclyl may be further substituted with C 1-6 alkyl, C 1-6 alkoxy, halo, or phenyl.
- R 1 is selected from the group consisting of - C(0)OR le , or optionally substituted heteroaryl and optionally substituted aryl
- R 3a is - NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci-6 alkyl and -(CH 2 ) q C 3 _ 7 cycloalkyl, each optionally substituted with C 1-6 alkyl.
- Some embodiments provide compounds of Formula I or Formula la, in which R 3 is -NHS(0) 2 R 3a or -NHS(0) 2 OR 3a , wherein R 3a is C 3-77 cycloalkyl optionally substituted with Ci_ 6 alkyl.
- Some embodiments provide compounds of Formula I or Formula la, in which R 1 is aryl substituted with one or more substitutents each independently selected from the group consisting of halo, amino, C 1-6 alkoxy optionally substituted with up to 5 fluoro, -COOH, -C(0)NR la R lb , -NHC(0)NR la R lb and heteroaryl containing 1-3
- R is and R 3 is
- R 3a is selected from the group consisting of Ci_ 6 alkyl and -(CH 2 ) q C 3 _ 7 cycloalkyl, each optionally substituted with Ci- 6 alkyl.
- R 1 is aryl substituted with one or more substitutents each independently selected from the group consisting of -C(0)NR la R lb and -NHC(0)NR la R lb ; R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , hydroxy-Ci- 6 alkyl, amino-Ci_ 6 alkyl, aryl-Ci_ 6 alkyl, aryl optionally substituted with Ci_ 6 alkyl or Ci_ 6 alkyl substituted with up to 5 fluoro, and heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; and R l
- R 1 is phenyl substituted with one or more substitutents each independently selected from the group consisting of -C(0)NR la R lb , - NHC(0)NR la R lb and heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O; and R 3 is -NHS(0) 2 R 3a or - NHS(0) 2 NR 3b R 3c , where R 3a is C 3 _ 7 cycloalkyl optionally substituted with methyl, and R 3b and R 3c are methyl.
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; R lc and R ld are each separately selected from the group consisting of -H, C 1-4 alkoxy, C 1-6 alkyl, C3-7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ;
- R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3-7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 )tC 3-7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-C 1-6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro; and R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_6 al
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond. Provided that if R is
- R 1 is not -C(0)0-t-butyl, benzoxazyl, t-butylthiazyl, phenyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro, chloro, methyl, -CF 3 and -OCF 3 .
- R 1 is selected from the group consisting of - C(0)0-t-butyl and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- the compound of Formula ⁇ is selected from the group consisting of Compounds 901, 101-129, 601-602, 1001-1002, and 1733 as shown in the Examples below.
- Some embodiments provide compounds of Formula IIa-1 :
- R is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 )tC3_ 7 cycloalkyl, C 2 - 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C3_ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 )tC3_ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring bonded to the parent structure through a nitrogen, and where the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group consisting of
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- R 7 is selected from the group consisting of -NH 2 , -NH 2 HC1, -COOH, -C(0)NR la R lb , -NHC(0)NR la R lb and heteroaryl containing 1-3 heteroatoms independently selected from N or O;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl,
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or - NHS(0) 2 NR 3b R 3c , where R 3a is C 3 _ 7 cycloalkyl optionally substituted with methyl, and R 3b and R 3c are methyl; and R 7 is selected from the group consisting of -NH 2 , -NH 2 HC1, -COOH, -C(0)NR la R lb , -NHC(0)NR la R lb and heteroaryl.
- said heteroaryl may contain 1-3 heteroatoms independently selected from N or O, wherein R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from Ci_ 6 alkyl, -C(0)OR lc , -C(0)R , hydroxy-Ci_ 6 alkyl, amino-Ci_ 6 alkyl, aryl-Ci_ 6 alkyl, phenyl optionally substituted with Ci_ 6 alkyl or -CF 3 , and heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O.
- Some embodiments provide a compound having the structure of Formula ⁇ :
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl.
- said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; and R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 1 is Ci_ 6 alkyl optionally substituted with up to 5 fluoro.
- Each R 2c is independently selected from the group consisting of halo, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , C 2 _ 6 alkyl, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, C 1-6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl, said C 2 - 6 alkyl, C 2 _ 6 alkenyl, C 3 -7 cycloalkyl, C 1-6 alkoxy, arylalkyl, polycyclic moiety, aryl, and heteroaryl, each optionally substituted with one or more R 1 ⁇ 2.
- Each IT 12" is independently selected from the group consisting of C 1-6 alkyl, C 3 -7 cycloalkyl, C 1-6 alkoxy, heteroaryl, arylalkyl, aryl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , - C(0)NR'R", and -NR'R", wherein said C 2-6 alkyl, C 3-7 cycloalkyl, Ci_ 6 alkoxy, heteroaryl, arylalkyl, cycloalkylalkyl, and aryl are each optionally substituted with one or more R 12a .
- Each R 12a is independently selected from the group consisting of -F, -CI, - CF 3 , -OCF 3 , Ci_ 6 alkyl, Ci_ 6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- Each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), halo, -C(0)NR'R", optionally substituted C 1-6 alkyl, optionally substituted C 2 _ 6 alkenyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro; wherein R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- n is 1, 2 or 3. Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond. Provided that if R is , then R is not -C(0)0-t-butyl, phenyl or phenyl substituted with one or more substitu consisting of fluoro, chloro and -CF 3 ; and
- R 2c is -F or methyl, then R 1 is not -C(0)0-t- butyl or phenyl.
- the compound of Formula ⁇ is selected from the group consisting of Compounds 201-204, 210-293, 1201-1222, 1401-1436, 1701- 1732, and 1734-1778 as shown in the Examples below.
- each R 2c is independently selected from the group consisting of -CF 3 , -Br (bromo), -CI (chloro),, -C(0)OH, -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , C 2 _ 6 alkyl, C 2 _ 6 alkenyl, Ci_ 6 alkoxy, polycyclic moiety, phenyl, and heteroaryl, said C 2 _ 6 alkyl, C 2 _ 6 alkenyl, C 1-6 alkoxy, polycyclic moiety, aryl, and heteroaryl, each optionally substituted with one or more R 12 ; and in some embodiments, said heteroaryl may be selected from the group consisting of furanyl, thiazolyl, oxazolyl, thiophenyl, pyrazolyl, and benzothiazolyl.
- Each R 12 is independently selected from the group consisting of C 1-6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , -C(0)NR'R", -NR'R", C 3-7 cycloalkyl-alkyl, wherein said Ci_6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, and -NR'R" are each optionally substituted with one or more R 12a .
- each R 12a is independently selected from the group consisting of -F, -CI, Ci_6 alkyl, Ci_6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), -F, - CI, -C(0)NR'R", Ci_6 alkyl, C 2 _ 6 alkenyl, Ci_ 6 alkoxy, phenyl, phenylalkyl and heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- said heterocyclyl may be morpholinyl, pyrrolidinyl, or piperidinyl.
- each R 2c is independently aryl optionally substituted with halo, cyano, Ci_6 alkyl optionally substituted with up to 5 fluoro, or Ci_6 alkoxy optionally substituted with up to 5 fluoro, C(0)NR'R", wherein R' and R" are independently optionally substituted Ci_ 6 alkyl.
- each R 2c is independently heteroaryl or polycyclic moiety, each optionally substituted with aryl, arylalkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, heteroaryl, heterocyclyl, C 3 _ 7 cycloalkyl, or . C 3 _ 7 cycloalkyl-alkyl; wherein said aryl, heteroaryl, and heterocyclyl may be further substituted with Ci_ 6 alkyl, Ci_ 6 alkoxy, halo, or phenyl.
- R 1 is selected from the group consisting of - C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 - 6 alkynyl, -C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O; and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C 3 _ 7 cycloalkyl optionally substituted with Ci_ 6 alkyl, and R 3b and R 3c are independently selected from -H
- R 1 is selected from the group consisting of - C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, and C 2 _ 6 alkynyl; and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C 3 _ 7 cycloalkyl optionally substituted with C 1-6 alkyl, and R 3b and R 3c are independently selected from -H or Ci_ 6 alkyl.
- Some embodiments provide a compound having the structure of Formula Ilia or nib:
- each R 2c is independently selected from the group consisting of -CF 3 , -Br (bromo), -CI (chloro),, -C(0)OH, -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , C 2 _ 6 alkyl, C 2 _ 6 alkenyl, Ci_ 6 alkoxy, polycyclic moiety, phenyl, and heteroaryl, said C 2 _ 6 alkyl, C 2 _ 6 alkenyl, Ci_6 alkoxy, polycyclic moiety, aryl, and heteroaryl, each optionally substituted with one or more R 12 ; and in some embodiments, said heteroaryl may be selected from the group consisting of furanyl, thiazolyl, oxazolyl, thiophenyl, pyrazolyl, and benzothiazolyl.
- Each R 12 is independently selected from the group consisting of Ci_6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , -C(0)NR'R", -NR'R", C 3-7 cycloalkyl-alkyl, wherein said Ci_6 alkyl, C 3 _ 7 cycloalkyl, Ci_ 6 alkoxy, pyridinyl, phenylalkyl, phenyl, and -NR'R" are each optionally substituted with one or more R 12a .
- each R 12a is independently selected from the group consisting of -F, -CI, Ci_6 alkyl, Ci_6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), -F, - CI, -C(0)NR'R", Ci_6 alkyl, C 2 _ 6 alkenyl, Ci_ 6 alkoxy, phenyl, phenylalkyl and heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- said heterocyclyl may be morpholinyl, pyrrolidinyl, or piperidinyl.
- each R 2c is independently aryl optionally substituted with one or more substituents selected from the group consisting of halo, cyano, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, or Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C(0)NR'R", wherein R' and R" are independently optionally substituted Ci_ 6 alkyl.
- each R 2c is independently heteroaryl or polycyclic moiety, each optionally substituted with -CF 3 , aryl, arylalkyl, Ci_6 alkyl optionally substituted with up to 5 fluoro, heteroaryl, heterocyclyl, C 3 _ 7 cycloalkyl, or C 3 _ 7 cycloalkyl-alkyl; wherein said aryl, heteroaryl, and heterocyclyl may be further substituted with Ci_ 6 alkyl, Ci_ 6 alkoxy, halo, or phenyl.
- the compound may have the structure of formula (IIIa-1):
- R 2c in Formula Ilia or nib is selected from the group consisting of -CF 3 , -Br (bromo), -CI (chloro),, -C(0)OH, -C(0)NR'R", -NR'R", -NHC(0)NR'R", -NHC(0)OR lc , -NHS(0) 2 R lc , C 2-6 alkyl, C 2-6 alkenyl, Ci_ 6 alkoxy, polycyclic moiety, phenyl, and heteroaryl, said C 2 _ 6 alkyl, C 2 _ 6 alkenyl, Ci_ 6 alkoxy, polycyclic moiety, aryl, and heteroaryl, each optionally substituted with one or more R 12 ; and in some embodiments, said heteroaryl may be selected from the group consisting of furanyl, thiazolyl, oxazolyl, thiophenyl, pyrazolyl, and benzothiazolyl.
- Each R 12 is independently selected from the group consisting of Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, Ci_6 alkoxy, pyridinyl, phenylalkyl, phenyl, -F (fluoro), -CI (Chloro), -CN, -CF 3 , -OCF 3 , -C(0)NR'R", -NR'R", C 3 _ 7 cycloalkyl-alkyl, wherein said Ci-6 alkyl, C 3 _ 7 cycloalkyl, Ci_6 alkoxy, pyridinyl, phenylalkyl, phenyl, and -NR'R" are each optionally substituted with one or more R 12a .
- each R 12a is independently selected from the group consisting of -F, -CI, Ci_ 6 alkyl, Ci_ 6 alkoxy, C 3 _ 7 cycloalkyl, and aryl.
- each NR'R" is separately selected wherein R' and R" are each independently selected from the group consisting of -H (hydrogen), -F, - CI, -C(0)NR'R", Ci-6 alkyl, C 2 _ 6 alkenyl, Ci_6 alkoxy, phenyl, phenylalkyl and heteroaryl; or R' and R" are taken together with the nitrogen to which they are attached to form heterocyclyl.
- said heterocyclyl may be morpholinyl, pyrrolidinyl, or piperidinyl.
- R lc is selected from the group consisting of Ci_6 alkyl, aryl and arylalkyl.
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O;
- R lc and R ld are each separately selected from the group consisting of -H, C 1-4 alkoxy, C 1-6 alkyl, C 3 -7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- X and Y are each independently selected from -CH- or -N-, wherein X and Y are not both -CH-.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _7cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _7cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_6alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro; wherein R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond.
- Some embodiments provide a compound having the structure selected from the group consisting of Compounds 209 and 501-504.
- Some embodiments provide a compound having the structure of Formula IVa or IVb:
- R 1 and R 3 are as defined above.
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl containing 1-3 heteroatoms independently selected from N or O; and R 3 is -OH, -NHS(0) 2 R 3a or - NHS(0) 2 NR 3b R 3c , where R 3a is C 3 - 7 cycloalkyl optionally substituted with methyl, and R 3b and R 3
- Some embodiments provide a compound having the structure of Formula ⁇ or IV
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from S, N or O, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl containing 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl containing 1-3 heteroatoms independently selected from N, O and S.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , - C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from N and O.
- R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, linear and branched Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl , arylalkyl and heteroaryl containing 1-3 heteroatoms independently selected from N, O and S.
- X and Y are each independently selected from -CH- or -N-, wherein X and Y are not both -CH-;(c) R 2b is selected from the group consisting of linear and branched C 1-6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from S, N or O.
- Each R 2c is independently selected from the group consisting of -Br, - CI, -CF 3 , C 2 _6 alkyl, C 2-6 alkenyl, -C(0)NR'R", -NR'R", -NHC(0)NR'R", - NHC(0)OR lc , -NHS(0) 2 R lc , -C(0)OH, aryl and heteroaryl containing 1-3 heteroatoms independently selected from S, N or O, wherein the heteroaryl is optionally substituted with one or more substituents selected from the group consisting of -CF 3 , linear and branched Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, arylalkyl and aryl, and the aryl is optionally substituted with one or more substituents selected from the group consisting of -F, -CN, -CF 3 , -OCF 3 , Ci_ 6 alkyl, Ci_ 6 alkoxy, and C(0)NR'
- R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or io aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring containing 1-3 heteroatoms independently selected from S, N or O, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- n is 1, 2 or 3; and any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond.
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 - 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O;
- R lc and R ld are each separately selected from the group consisting of -H, C 1-4 alkoxy, C 1-6 alkyl, C3-7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2a is selected from the group consisting of -H, -C(0)OR lc , Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _7 cycloalkyl, optionally substituted aryl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro; wherein R 3b and R 3c are each separately a hydrogen atom, or separately selected from the
- each t is independent each q is independently 0, 1 or 2.
- R 1 is not phenyl.
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond.
- R 1 is selected from the group consisting of - C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, and C 2 _ 6 alkynyl; and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C3_ 7 cycloalkyl optionally substituted with C 1-6 alkyl, and R 3b and R 3c are independently selected from -H or Ci_ 6 alkyl.
- Some embodiments provide a compound of Formula V selected from the group consisting of Compounds 301-312.
- Some embodiments provide a compound having the structure of Formula VI- 1 or I-2:
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci-6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 _ 6 alkynyl, - C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O;
- R lc and R ld are each separately selected from the group consisting of -H, C 1-4 alkoxy, C 1-6 alkyl, C3-7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2d is selected from the group consisting of C 1-6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3-7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3-7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-C 1-6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro; wherein R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C
- Each t is independently 0, 1 or 2; and each q is independently 0, 1 or 2.
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond.
- the compound may have the structure of one of the followin formulas:
- R 1 may be selected from the group consisting of -C(0)0-t-butyl, and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C 3-7 cycloalkyl optionally substituted with methyl, and R 3b and R 3c are methyl.
- R 2d is selected from the group consisting of Ci_6 alkyl optionally substituted with up to 5 fluoro and optionally substituted aryl. In some embodiments, R 2d is methyl, ethyl, i-propyl or phenyl.
- Some embodiments provide a compound of Formula VI selected from the group consisting of Compounds 294-299 and 701-702.
- R 1 is selected from the group consisting of - C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, and C 2 _ 6 alkynyl; and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C3_ 7 cycloalkyl optionally substituted with C 1-6 alkyl, and R 3b and R 3c are independently selected from -H or Ci_ 6 alkyl.
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, - C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2e is selected from the group consisting of -H, halo, -C(0)OR lc , - C(0)NR'R", -NR'R", -NHC(0)NR'R", Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl and optionally substituted heteroaryl; wherein R' and R" are each independently selected from the group consisting of -H, optionally substituted Ci_ 6 alkyl, optionally substituted C 2 - 6 alkenyl, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro; wherein R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting
- the compound may have the structure of one of the following formulas
- Some embodiments provide a compound of Formula VII selected from the group consisting of Compounds 1251-1253.
- R 1 is selected from the group consisting of -C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_6 alkyl optionally substituted with up to 5 fluoro, Ci_6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, and C 2 _ 6 alkynyl; and R is -OH, - NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C 3 - 7 cycloalkyl optionally substituted with Ci_6 alkyl, and R 3b and R 3c are independently selected from -H or Ci_ 6 alkyl.
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_6 alkyl optionally substituted with up to 5 fluoro, Ci_6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; R lc and R ld are each separately selected from the group consisting of -H, C 1-4 alkoxy, C 1-6 alkyl, C 3 -7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- X and Y are each independently selected from -CH- or -N-, wherein X and Y are not both -CH-;
- R is selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 -7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl;
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3-7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3-7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-C 1-6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring bonded to the parent structure through a nitrogen, and where the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group consisting of
- Some embodiments provide a compound having the structure of Formula Villa:
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O;
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2 is selected from the group consisting of Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 )tC3_ 7 cycloalkyl, C 2 - 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C3_ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 )tC3_ 7 cycloalkyl, C 2 - 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, Ci_ 6 alkyl substituted with up to 5 fluoro, and Ci_ 6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring bonded to the parent structure through a nitrogen, and where the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group consist
- Some embodiments provide a compound of Formula VIII selected from Compound 505 or 506.
- R 1 is selected from the group consisting of - C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 - 6 alkenyl, and C 2 - 6 alkynyl; and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C3_ 7 cycloalkyl optionally substituted with Ci_ 6 alkyl, and R 3b and R 3c are independently selected from -H or C 1-6 alkyl.
- V and W are each independently selected from -CR 2k - or -N-, wherein V and W are not both -CR 2k -;
- R 2j and R 2k are each independently selected from the group consisting of H, halo, optionally substituted aryl, optionally substituted heteroaryl; or R 2j and R 2k together form an aryl ring optionally substituted by 1-3 R 2g .
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O; and R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2g is selected from the group consisting of -H, halo, -C(0)OR lc , - C(0)NR'R", -NR'R", -NHC(0)NR'R", Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _6 alkenyl, C 3 _ 7 cycloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl and optionally substituted heteroaryl; R' and R" are each independently selected from the group consisting of -H, optionally substituted C 1-6 alkyl, optionally substituted C 2 - 6 alkenyl, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring bonded to the parent structure through a nitrogen, and where the heterocylic ring is optionally substituted with one or more substituents each independently selected from
- Some embodiments provide a compound of Formula IX selected from the follo ing formulae:
- Some embodiments provide a compound of Formula IX selected from the group consisting of Compounds 801-805 and 1501-1506.
- R 1 is selected from the group consisting of - C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, C 1-6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 - 6 alkenyl, and C 2-6 alkynyl; and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C3_ 7 cycloalkyl optionally substituted with Ci_ 6 alkyl, and R 3b and R 3c are independently selected from -H or C 1-6 alkyl.
- Some embodiments provide a compound having the structure of Formula X:
- R 1 is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O;.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 2h is selected from the group consisting of n-propyl, cyclopropyl, n- butyl, t-butyl, 1 -sec-butyl and phenyl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocyclic ring, bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Some embodiments provide a compound of Formula X selected from the group consisting of Compounds 200 and 205-208.
- R 1 is selected from the group consisting of - C(0)0-t-butyl and phenyl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, and C 2 _ 6 alkynyl; and R 3 is -OH, -NHS(0) 2 R 3a or -NHS(0) 2 NR 3b R 3c , where R 3a is C3_ 7 cycloalkyl optionally substituted with C 1-6 alkyl, and R 3b and R 3c are independently selected from -H or Ci_ 6 alkyl.
- Z is a group configured to hydrogen bond to an NS3 protease His57 imidazole moiety, and to hydrogen bond with the hydrogen and nitrogen of the backbone amide group of the NS3 amino acid at position 137;
- Pi' is a group configured to form a non-polar interaction with at least one NS3 protease SI' pocket moiety selected from the group consisting of Lysl36, Glyl37, Serl39, His57, Gly58, Gln41, Ser42, and Phe43;
- (g) L is a linker group consisting of from 1 to 5 atoms selected from the group consisting of carbon, oxygen, nitrogen, hydrogen, and sulfur;
- P 2 is selected from the group consisting of unsubstituted aryl, substituted aryl, unsubstituted heteroaryl, substituted heteroaryl, unsubstituted heterocyclic and substituted heterocyclic; P 2 being configured to form a non-polar interaction with at least one NS3 protease S2 pocket moiety selected from the group consisting of Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81, and P 2 being configured so that no atom of P2 makes a nonpolar interaction with an epsilon, zeta, or eta sidechain atom of the amino acid at position 155;
- Pv 5 is selected from the group consisting of H, C(0)NR 6 R 7 and C(0)OR 8 ;
- R 6 and R 7 are each independently H, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, C 4 _io alkylcycloalkyl or phenyl, said phenyl optionally substituted by up to three halo, cyano, nitro, hydroxy, C 3 _ 7 cycloalkyl, C 4 _io alkylcycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro; or R 6 and R 7 are taken together with the nitrogen to which they are attached to form indolinyl, pyrrolidinyl, piperidinyl, piperazinyl, or morpholinyl;
- R is Ci_6 alkyl, C 3 _ 7 cycloalkyl, C 4 _io alkylcycloalkyl, which are all optionally substituted from one to three times with halo, cyano, nitro, hydroxy, C 1-6 alkoxy, or phenyl; or R is C 6 or 10 aryl which is optionally substituted by up to three halo, cyano, nitro, hydroxy, C 3 _ 7 cycloalkyl, C 4-1 o alkylcycloalkyl, C 2 -6 alkenyl, C 1-6 alkoxy, hydroxy- Ci_6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro; or R is C 1-6 alkyl optionally substituted with up to 5 fluoro groups; or R is a tetrahydrofuran ring linked through the C 3 or C 4 position of the tetrahydro groups;
- Y is is a C 5 _ 7 saturated or unsaturated chain optionally containing one or two heteroatoms selected from O, S, or NR 9 R 10 ;
- R 9 and R 10 are each independently H, C 1-6 alkyl, C 3 _ 7 cycloalkyl, C 4-1 o cycloalkyl-alkyl, or substituted or unsubstituted phenyl; or R 9 and R 10 are taken together with the nitrogen to which they are attached to form indolinyl, pyrrolidinyl, piperidinyl, piperazinyl, or morpholinyl.
- the present embodiments also provide compounds having the formula (XI) or a pharmaceutically acceptable salt, prodrug, or ester thereof wherein:
- Z is a group configured to hydrogen bond to an NS3 protease His57 imidazole moiety, and to hydrogen bond with the hydrogen and nitrogen of the backbone amide group of the NS3 amino acid at position 137;
- Pi' is a group configured to form a non-polar interaction with at least one NS3 protease SI' pocket moiety selected from the group consisting of Lysl36, Glyl37, Serl39, His57, Gly58, Gln41, Ser42, and Phe43;
- (g) L is a linker group consisting of from 1 to 5 atoms selected from the group consisting of carbon, oxygen, nitrogen, hydrogen, and sulfur;
- P 2 is selected from the group consisting of unsubstituted aryl, substituted aryl, unsubstituted heteroaryl, substituted heteroaryl, unsubstituted heterocyclic and substituted heterocyclic; P 2 being configured to form a non-polar interaction with at least one NS3 protease S2 pocket moiety selected from the group consisting of Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81, and P 2 being configured so that no atom of P2 makes a nonpolar or polar interaction with an epsilon, zeta, or eta sidechain atom of the amino acid at position 155;
- R 5 is selected from the group consisting of H, C(0)NR 6 R 7 and C(0)OR 8 ;
- R 6 and R 7 are each independently H, Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, C 4 _io alkylcycloalkyl or phenyl, said phenyl optionally substituted by up to three halo, cyano, nitro, hydroxy, C 3 _ 7 cycloalkyl, C 4 _io alkylcycloalkyl, C 2 -6 alkenyl, hydroxy-Ci_6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 1-6 alkoxy optionally substituted with up to 5 fluoro; or R 6 and R 7 are taken together with the nitrogen to which they are attached to form indolinyl, pyrrolidinyl, piperidinyl, piperazinyl, or morpholinyl;
- R is Ci_6 alkyl, C 3 _ 7 cycloalkyl, C 4 _io alkylcycloalkyl, which are all optionally substituted from one to three times with halo, cyano, nitro, hydroxy, C 1-6 alkoxy, or phenyl; or R is C 6 or 10 aryl which is optionally substituted by up to three halo, cyano, nitro, hydroxy, C 3 _ 7 cycloalkyl, C 4-1 o alkylcycloalkyl, C 2 _ 6 alkenyl, C 1-6 alkoxy, hydroxy- Ci_6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro; or R is C 1-6 alkyl optionally substituted with up to 5 fluoro groups; or R is a tetrahydrofuran ring linked through the C 3 or C 4 position of the tetrahydr
- Y is is a C 5 _ 7 saturated or unsaturated chain optionally containing one or two heteroatoms selected from O, S, or NR 9 R 10 ;
- R 9 and R 10 are each independently H, C 1-6 alkyl, C 3 _ 7 cycloalkyl, C 4-1 o cycloalkyl-alkyl, or substituted or unsubstituted phenyl; or R 9 and R 10 are taken together with the nitrogen to which they are attached to form indolinyl, pyrrolidinyl, piperidinyl, piperazinyl, or morpholinyl.
- the compound having the general Formula XI may contain one or more moieties that form a hydrogen bond with a peptide backbone atom or side chain moiety located in the substrate binding pocket of NS3 protease. In another example, the compound having the general Formula XI may contain one or more moieties that form non-polar interactions with peptide backbone or side chain atom or atoms located in the substrate binding pocket of NS3 protease.
- Z may be configured to form a hydrogen bond with a peptide backbone atom or side chain moiety located in the substrate binding pocket of NS3 protease, including, but not limited to, NS3 protease His57 imidazole moiety and hydrogen and nitrogen atoms of the amino acid at position 137 of NS3 protease. In some instances, Z may be configured to form a hydrogen bond with both the NS3 protease His57 imidazole moiety and hydrogen and nitrogen atoms of the amino acid at position 137 of NS3 protease.
- the ⁇ group of the compound having the general Formula XI may be configured to form a non-polar interaction with peptide backbone or side chain atom or atoms located in the substrate binding pocket of NS3 protease, including, but not limited to amino acid residues that form the NS3 protease SI' pocket.
- the ⁇ group may form a non-polar interaction with at least one amino acid selected from Lysl36, Glyl37, Serl39, His57, Gly58, Gln41, Ser42, and Phe43.
- the P 2 group of the compound having the general Formula XI may be configured to form a non-polar interaction with peptide backbone or side chain atom or atoms located in the substrate binding pocket of NS3 protease, including, but not limited to amino acid residues that form the NS3 protease S2 pocket.
- the P 2 group may form a non-polar interaction with at least one amino acid selected from Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81.
- the P 2 group also may be configured to form a polar interaction with peptide backbone or side chain atom or atoms located in the substrate binding pocket of NS3 protease, including, but not limited to amino acid residues that form the NS3 protease S2 pocket.
- the P 2 group may form a polar interaction with at least one amino acid selected from Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81.
- the P 2 group also may be configured to form a hydrogen bond with peptide backbone or side chain atom or atoms located in the substrate binding pocket of NS3 protease, including, but not limited to amino acid residues that form the NS3 protease S2 pocket.
- the P 2 group may form a hydrogen bond with at least one amino acid selected from Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81.
- P 2 may form two or more of a non-polar interaction, polar interactin, and a hydrogen bond with peptide backbone or side chain moieties or atoms located in the substrate binding pocket of NS3 protease, such amino acids selected from Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81.
- a hydrogen bond with peptide backbone or side chain moieties or atoms located in the substrate binding pocket of NS3 protease, such amino acids selected from Tyr56, Gly58, Ala59, Gly60, Gln41, His57, Val78, Asp79, Gln80 and Asp81.
- Such hydrogen bond, polar interaction and non- polar interaction may occur with the same amino acid residue or with different amino acid residues in the NS3 protease S2 pocket.
- P 2 may be selected from the group consisting of unsubstituted aryl, substituted aryl, unsubstituted heteroaryl, substituted heteroaryl, unsubstituted heterocyclic and substituted heterocyclic.
- the P 2 group of the compound having the general Formula XI may be configured so that no atom of P 2 makes a nonpolar or polar interaction with an epsilon, zeta, or eta sidechain atom of the amino acid at position 155.
- the P 2 group may be configured so that no atom of P 2 makes a nonpolar or polar interaction with an epsilon, zeta, or eta sidechain atom Argl55.
- the P 2 group may be configured so that no atom of P 2 makes a nonpolar or polar interaction with an epsilon, zeta, or eta sidechain atom of a non-arginine amino acid at 155.
- non- arginine amino acids at 155 include Lysl55 and Gin 155.
- L may be a linker group that links P 2 to the heterocyclic backbone of the compound of Formula XL
- Linker L may contain any of a variety of atoms and moieties suitable for positioning P 2 in the NS3 protease substrate binding pocket.
- L may contain 1 to 5 atoms selected from the group consisting of carbon, oxygen, nitrogen, hydrogen, and sulfur.
- L may contain 2 to 5 atoms selected from the group consisting of carbon, oxygen, nitrogen, hydrogen, and sulfur.
- Specific exemplary groups for L include, but are not limited to, ester, amide, carbamate, thioester, and thioamide.
- the compound of Formula XI also may contain an R 5 group, where the R 5 group may contain a carboxyl moiety.
- exemplary carboxyl moieties of R 5 include C(0)NR 6 R 7 and C(0)OR 8 where R 6 and R 7 are each independently H, Ci_ 6 alkyl, C 3 -7 cycloalkyl, C 4 _io alkylcycloalkyl or phenyl, said phenyl optionally substituted by up to three halo, cyano, nitro, hydroxy, C 3 -7 cycloalkyl, C 4 _io alkylcycloalkyl, C2-6 alkenyl, hydroxy-Ci_6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro; or R 6 and R 7 are taken together with the nitrogen to which they are attached to form indolinyl, pyrrolidinyl, piperidinyl, piperaz
- R 8 is 8
- Ci-6 alkyl optionally substituted with up to 5 fluoro groups; or R is a tetrahydrofuran ring linked through the C 3 or C 4 position of the tetrahydrofuran ring; or R is a tetrapyranyl ring linked through the C 4 position of the tetrapyranyl ring.
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 -6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- R le is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl;
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted Ci_ 6 alkyl, C 2 - 6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , -C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl, wherein in some embodiments, said heteroaryl may contain 1-3 heteroatoms indepedently selected from N or O; and R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, Ci_ 6 alkyl, C 3 _7 cycloalkyl, aryl, arylalkyl and heteroaryl.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of C 1-6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, and C 1-6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or 10 aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c together with N form a three- to six- membered heterocyclic ring , bonded to the parent structure through a nitrogen, and the heterocylic ring is optionally substituted with one or more substituents each independently selected from the group consisting of halo,
- R 1 may be selected from -C(0)0-t-butyl or aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, - C(0)NR la R lb , -NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl; in some embodiments, said heteroaryl may contain 1-3 heteroatoms independently selected from N or O.
- compounds of Formula I have the structure of Formula Xlla:
- Some embodiments provide a compound having the structure of Formula I or ⁇ :
- R is selected from the group consisting of -C(0)OR le , optionally substituted heteroaryl, and aryl optionally substituted with one or more substituents each independently selected from the group consisting of halo, amino, Ci_ 6 alkyl optionally substituted with up to 5 fluoro, Ci_ 6 alkoxy optionally substituted with up to 5 fluoro, C 2 -6 alkenyl, C 2 _ 6 alkynyl, -C(0)NR la R lb , - NHC(0)NR la R lb , -C(0)OR lc , and heteroaryl containing 1-3 heteroatoms independently selected from N or O.
- R e is selected from the group consisting of t-butyl, cycloalkyl, and heterocyclyl containing 1-3 heteroatoms independently selected from N, O and S.
- R la and R lb are taken together with the nitrogen to which they are attached to form piperazinyl or morpholinyl, each optionally substituted with one or more substituents independently selected from optionally substituted C 1-6 alkyl, C 2 -6 alkenyl, C 2 _ 6 alkynyl, -C(0)OR lc , - C(0)R ld , optionally substituted aryl, and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from N and O.
- R lc and R ld are each separately selected from the group consisting of -H, Ci_ 4 alkoxy, linear and branched Ci_ 6 alkyl, C 3 _ 7 cycloalkyl, aryl, arylalkyl and heteroaryl containing 1-3 heteroatoms independently selected from N, O and S.
- R 2 ⁇ and R 2k are each independently selected from the group consisting of H, halo, optionally substituted aryl, optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from S, N or O; or R 2 ⁇ and R 2k together form an aryl ring optionally substituted by 1-3 R 2 .
- R 2a , each R 2c , R 2e and R 2 are each independently selected from the group consisting of halo, -C(0)OR lc , -C(0)NR'R", -NR'R", -NHC(0)NR'R", - NHC(0)OR lc , -NHS(0) 2 R lc , linear and branched Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 _ 6 alkenyl, C 3 _ 7 cycloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted aryl and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from S, N or O.
- R 2b , R 2d and R 2f are each independently selected from the group consisting of linear and branched Ci_ 6 alkyl optionally substituted with up to 5 fluoro, C 2 - 6 alkenyl, C 3 -7 cycloalkyl, arylalkyl, optionally substituted aryl and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from S, N or O.
- R 2h is selected from the group consisting of propyl, butyl and phenyl;
- R' is Ci_ 6 alkyl optionally substituted with up to 5 fluoro;
- R' and R" are each independently selected from the group consisting of -H, optionally substituted linear and branched Ci_ 6 alkyl, optionally substituted C 2 _ 6 alkenyl, optionally substituted aryl, optionally substituted arylalkyl and optionally substituted heteroaryl containing 1-3 heteroatoms independently selected from S, N or O.
- R 3 is -OH, -NHS(0) 2 R 3a , -NHS(0) 2 OR 3a or -NHS(0) 2 NR 3b R 3c ; where R 3a is selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, -(CH 2 ) q C 6 or l oaryl, and a heteroaryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -COOH, -(CH 2 ) t C 3 _ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, C 1-6 alkyl optionally substituted with up to 5 fluoro, and Ci_ 6 alkoxy optionally substituted with up to 5 fluoro.
- R 3b and R 3c are each separately a hydrogen atom, or separately selected from the group consisting of Ci_ 6 alkyl, -(CH 2 ) q C 3 _ 7 cycloalkyl, and C 6 or io aryl, each optionally substituted with one or more substituents each independently selected from the group consisting of halo, cyano, nitro, hydroxy, -(CH 2 ) t C3_ 7 cycloalkyl, C 2 _ 6 alkenyl, hydroxy-Ci_ 6 alkyl, phenyl, C 1-6 alkyl substituted with up to 5 fluoro, and C 1-6 alkoxy substituted with up to 5 fluoro; or R 3b and R 3c are taken together with the nitrogen to which they are attached to form a three- to six- membered heterocylic ring containing 1-3 heteroatoms independently selected from S, N or O, and the heterocylic ring is optionally substituted with one or more substituents each independently selected
- Any bond represented by a dashed and solid line represents a bond selected from the group consisting of a single bond and a double bond. [0326] Provided that if R 2 is or , then R 1 is not phenyl.
- R 1 is not -C(0)0-t-butyl, phenyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro, chloro and -CF 3 .
- R 1 is not -C(0)0-t-butyl or phenyl.
- R 1 is not -C(0)0-t-butyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro and -CF 3 .
- R 1 is not -C(0)0-t- butyl, benzoxazyl, t-butylthiazyl, phenyl or phenyl substituted with one or more substituents selected from the group consisting of fluoro, chloro, methyl, -CF 3 and - OCF 3 .
- Some embodiments provide a compound selected from the group consisting of:
- C 1-6 alkyl may include linear and branched Ci_ 6 alkyl
- Ci_ 6 alkoxy may include linear and branched Ci-6 alkoxy
- compositions including pharmaceutical compositions, comprising compounds of the general Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIE, IX, X, XI, and XII, or any compounds disclosed herein.
- a subject pharmaceutical composition comprises a subject compound; and a pharmaceutically acceptable excipient.
- a pharmaceutically acceptable excipient A wide variety of pharmaceutically acceptable excipients is known in the art and need not be discussed in detail herein. Pharmaceutically acceptable excipients have been amply described in a variety of publications, including, for example, A. Gennaro (2000) "Remington: The Science and Practice of Pharmacy," 20th edition, Lippincott, Williams, & Wilkins; Pharmaceutical Dosage Forms and Drug Delivery Systems (1999) H.C. Ansel et al., eds., 7 th ed., Lippincott, Williams, & Wilkins; and Handbook of Pharmaceutical Excipients (2000) A.H. Kibbe et al., eds., 3 ed. Amer. Pharmaceutical Assoc.
- compositions such as vehicles, adjuvants, carriers or diluents
- pharmaceutically acceptable auxiliary substances such as pH adjusting and buffering agents, tonicity adjusting agents, stabilizers, wetting agents and the like, are readily available to the public.
- the present embodiments provide for a method of inhibiting NS3/NS4 protease activity comprising contacting a NS3/NS4 protease with a compound disclosed herein.
- the present embodiments provide for a method of treating hepatitis by modulating NS3/NS4 protease comprising contacting a NS3/NS4 protease with a compound disclosed herein.
- Example compounds of Formulae I, la, ⁇ , ⁇ , W, V, VI- 1, VI-2, VII, Vni, IX, X, XI, and XII include Compound Numbers 101-129, 200-299, 301-312, 401, 501-506, 601-602, 701-702, 801-805, 901, 1001-1003, 1102-1103, 1201-1224, 1251- 1253, 1401-1436, and 1701-1780 as set forth herein.
- Compounds 401, 1004, 1005, 1005S, 1101, 1101S are also disclosed.
- Preferred embodiments provide a method of treating a hepatitis C virus infection in an individual, the method comprising administering to the individual an effective amount of a composition comprising a preferred compound.
- Preferred embodiments provide a method of treating liver fibrosis in an individual, the method comprising administering to the individual an effective amount of a composition comprising a preferred compound.
- Preferred embodiments provide a method of increasing liver function in an individual having a hepatitis C virus infection, the method comprising administering to the individual an effective amount of a composition comprising a preferred compound.
- a subject compound inhibits the enzymatic activity of a hepatitis virus C (HCV) NS3 protease. Whether a subject compound inhibits HCV NS3 protease can be readily determined using any known method. Typical methods involve a determination of whether an HCV polyprotein or other polypeptide comprising an NS3 recognition site is cleaved by NS3 in the presence of the agent.
- HCV hepatitis virus C
- a subject compound inhibits NS3 enzymatic activity by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, or at least about 90%, or more, compared to the enzymatic activity of NS3 in the absence of the compound.
- a subject compound inhibits enzymatic activity of an HCV NS3 protease with an IC 50 of less than about 50 ⁇ , e.g., a subject compound inhibits an HCV NS3 protease with an IC 50 of less than about 40 ⁇ , less than about 25 ⁇ , less than about 10 ⁇ , less than about 1 ⁇ , less than about 100 nM, less than about 80 nM, less than about 60 nM, less than about 50 nM, less than about 25 nM, less than about 10 nM, less than about 5 nM, less than about 1 nM, or less than about 0.5 nM, or less.
- a subject compound inhibits the enzymatic activity of a hepatitis virus C (HCV) NS3 helicase. Whether a subject compound inhibits HCV NS3 helicase can be readily determined using any known method. In many embodiments, a subject compound inhibits NS3 enzymatic activity by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, or at least about 90%, or more, compared to the enzymatic activity of NS3 in the absence of the compound.
- HCV hepatitis virus C
- a subject compound inhibits HCV viral replication.
- a subject compound inhibits HCV viral replication by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, or at least about 90%, or more, compared to HCV viral replication in the absence of the compound.
- Whether a subject compound inhibits HCV viral replication can be determined using methods known in the art, including an in vitro viral replication assay.
- Whether a subject method is effective in treating an HCV infection can be determined by a reduction in viral load, a reduction in time to seroconversion (virus undetectable in patient serum), an increase in the rate of sustained viral response to therapy, a reduction of morbidity or mortality in clinical outcomes, or other indicator of disease response.
- an effective amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to reduce viral load or achieve a sustained viral response to therapy.
- Whether a subject method is effective in treating an HCV infection can be determined by measuring viral load, or by measuring a parameter associated with HCV infection, including, but not limited to, liver fibrosis, elevations in serum transaminase levels, and necroinflammatory activity in the liver. Indicators of liver fibrosis are discussed in detail below.
- the method involves administering an effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein, optionally in combination with an effective amount of one or more additional antiviral agents.
- an effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to reduce viral titers to undetectable levels, e.g., to about 1000 to about 5000, to about 500 to about 1000, or to about 100 to about 500 genome copies/mL serum.
- an effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, Vn, VIE, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to reduce viral load to lower than 100 genome copies/mL serum.
- an effective amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to achieve a 1.5-log, a 2-log, a 2.5-log, a 3-log, a 3.5-log, a 4-log, a 4.5-log, or a 5 -log reduction in viral titer in the serum of the individual.
- an effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to achieve a sustained viral response, e.g., non-detectable or substantially non- detectable HCV RNA (e.g., less than about 500, less than about 400, less than about 200, or less than about 100 genome copies per milliliter serum) is found in the patient's serum for a period of at least about one month, at least about two months, at least about three months, at least about four months, at least about five months, or at least about six months following cessation of therapy.
- a sustained viral response e.g., non-detectable or substantially non- detectable HCV RNA (e.g., less than about 500, less than about 400, less than about 200, or less than about 100 genome copies per milliliter serum) is
- liver fibrosis As noted above, whether a subject method is effective in treating an HCV infection can be determined by measuring a parameter associated with HCV infection, such as liver fibrosis. Methods of determining the extent of liver fibrosis are discussed in detail below. In some embodiments, the level of a serum marker of liver fibrosis indicates the degree of liver fibrosis.
- ALT levels of serum alanine aminotransferase are measured, using standard assays. In general, an ALT level of less than about 45 international units is considered normal.
- an effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount effective to reduce ALT levels to less than about 45 IU/mL serum.
- a therapeutically effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to reduce a serum level of a marker of liver fibrosis by at least about 10%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, or at least about 80%, or more, compared to the level of the marker in an untreated individual, or to a placebo-treated individual.
- Methods of measuring serum markers include immunological-based methods, e.g., enzyme-linked immunosorbent assays (ELISA), radioimmunoassays, and the like, using antibody specific for a
- an effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein and an additional antiviral agent is a synergistic amount.
- the additional antiviral agent may itself be a combination of antiviral agents, e.g., a combination of pegylated interferon- alf a and ribavirin.
- a "synergistic combination” or a “synergistic amount" of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein and an additional antiviral agent is a combined dosage that is more effective in the therapeutic or prophylactic treatment of an HCV infection than the incremental improvement in treatment outcome that could be predicted or expected from a merely additive combination of (i) the therapeutic or prophylactic benefit of the compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein when administered at that same dosage as a monotherapy and (ii) the therapeutic or prophylactic benefit of the additional antiviral agent when administered at the same dosage as a monotherapy.
- a selected amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein and a selected amount of an additional antiviral agent are effective when used in combination therapy for a disease, but the selected amount of the compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein and/or the selected amount of the additional antiviral agent is ineffective when used in monotherapy for the disease.
- the embodiments encompass (1) regimens in which a selected amount of the additional antiviral agent enhances the therapeutic benefit of a selected amount of the compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, Vin, IX, X, XI, or, XII, or any compounds disclosed herein when used in combination therapy for a disease, where the selected amount of the additional antiviral agent provides no therapeutic benefit when used in monotherapy for the disease (2) regimens in which a selected amount of the compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein enhances the therapeutic benefit of a selected amount of the additional antiviral agent when used in combination therapy for a disease, where the selected amount of the compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, Vn, Vni, IX, X, XI, or,
- a "synergistically effective amount" of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein and an additional antiviral agent, and its grammatical equivalents, shall be understood to include any regimen encompassed by any of (l)-(3) above.
- the embodiments provides methods for treating liver fibrosis (including forms of liver fibrosis resulting from, or associated with, HCV infection), generally involving administering a therapeutic amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, Vn, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents.
- Effective amounts of compounds of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein, with and without one or more additional antiviral agents, as well as dosing regimens, are as discussed below.
- liver fibrosis reduction is determined by any of a number of well-established techniques for measuring liver fibrosis and liver function. Liver fibrosis reduction is determined by analyzing a liver biopsy sample.
- An analysis of a liver biopsy comprises assessments of two major components: necroinflammation assessed by "grade” as a measure of the severity and ongoing disease activity, and the lesions of fibrosis and parenchymal or vascular remodeling as assessed by "stage” as being reflective of long-term disease progression. See, e.g., Brunt (2000) Hepatol. 31:241-246; and METAVIR (1994) Hepatology 20:15-20. Based on analysis of the liver biopsy, a score is assigned. A number of standardized scoring systems exist which provide a quantitative assessment of the degree and severity of fibrosis. These include the METAVIR, Knodell, Scheuer, Ludwig, and Ishak scoring systems.
- the METAVIR scoring system is based on an analysis of various features of a liver biopsy, including fibrosis (portal fibrosis, centrilobular fibrosis, and cirrhosis); necrosis (piecemeal and lobular necrosis, acidophilic retraction, and ballooning degeneration); inflammation (portal tract inflammation, portal lymphoid aggregates, and distribution of portal inflammation); bile duct changes; and the Knodell index (scores of periportal necrosis, lobular necrosis, portal inflammation, fibrosis, and overall disease activity).
- each stage in the METAVIR system is as follows: score: 0, no fibrosis; score: 1, stellate enlargement of portal tract but without septa formation; score: 2, enlargement of portal tract with rare septa formation; score: 3, numerous septa without cirrhosis; and score: 4, cirrhosis.
- Knodell's scoring system also called the Hepatitis Activity Index, classifies specimens based on scores in four categories of histologic features: I. Periportal and/or bridging necrosis; II. Intralobular degeneration and focal necrosis; ⁇ . Portal inflammation; and IV. Fibrosis.
- scores are as follows: score: 0, no fibrosis; score: 1, mild fibrosis (fibrous portal expansion); score: 2, moderate fibrosis; score: 3, severe fibrosis (bridging fibrosis); and score: 4, cirrhosis. The higher the score, the more severe the liver tissue damage.
- the Ishak scoring system is described in Ishak (1995) J. Hepatol. 22:696-699. Stage 0, No fibrosis; Stage 1, Fibrous expansion of some portal areas, with or without short fibrous septa; stage 2, Fibrous expansion of most portal areas, with or without short fibrous septa; stage 3, Fibrous expansion of most portal areas with occasional portal to portal (P-P) bridging; stage 4, Fibrous expansion of portal areas with marked bridging (P-P) as well as portal-central (P-C); stage 5, Marked bridging (P-P and/or P-C) with occasional nodules (incomplete cirrhosis); stage 6, Cirrhosis, probable or definite.
- the benefit of anti-fibrotic therapy can also be measured and assessed by using the Child-Pugh scoring system which comprises a multicomponent point system based upon abnormalities in serum bilirubin level, serum albumin level, prothrombin time, the presence and severity of ascites, and the presence and severity of encephalopathy. Based upon the presence and severity of abnormality of these parameters, patients may be placed in one of three categories of increasing severity of clinical disease: A, B, or C.
- a therapeutically effective amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that effects a change of one unit or more in the fibrosis stage based on pre- and post-therapy liver biopsies.
- a therapeutically effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents reduces liver fibrosis by at least one unit in the METAVIR, the Knodell, the Scheuer, the Ludwig, or the Ishak scoring system.
- indices of liver function can also be used to evaluate the efficacy of treatment with a compound of Formulae I, la, II, ⁇ , ⁇ , V, VI- 1, VI-2, Vn, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein.
- Morphometric computerized semi- automated assessment of the quantitative degree of liver fibrosis based upon specific staining of collagen and/or serum markers of liver fibrosis can also be measured as an indication of the efficacy of a subject treatment method.
- Secondary indices of liver function include, but are not limited to, serum transaminase levels, prothrombin time, bilirubin, platelet count, portal pressure, albumin level, and assessment of the Child-Pugh score.
- An effective amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to increase an index of liver function by at least about 10%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, or at least about 80%, or more, compared to the index of liver function in an untreated individual, or to a placebo-treated individual.
- Those skilled in the art can readily measure such indices of liver function, using standard assay methods, many of which are commercially available, and are used routinely in clinical settings.
- Serum markers of liver fibrosis can also be measured as an indication of the efficacy of a subject treatment method.
- Serum markers of liver fibrosis include, but are not limited to, hyaluronate, N-terminal procollagen ⁇ peptide, 7S domain of type IV collagen, C-terminal procollagen I peptide, and laminin.
- Additional biochemical markers of liver fibrosis include ⁇ -2-macroglobulin, haptoglobin, gamma globulin, apolipoprotein A, and gamma glutamyl transpeptidase.
- a therapeutically effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective to reduce a serum level of a marker of liver fibrosis by at least about 10%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, or at least about 80%, or more, compared to the level of the marker in an untreated individual, or to a placebo-treated individual.
- ELISA enzyme-linked immunosorbent assays
- radioimmunoassays radioimmunoassays
- Quantitative tests of functional liver reserve can also be used to assess the efficacy of treatment with an interferon receptor agonist and pirfenidone (or a pirfenidone analog). These include: indocyanine green clearance (ICG), galactose elimination capacity (GEC), aminopyrine breath test (ABT), antipyrine clearance, monoethylglycine-xylidide (MEG-X) clearance, and caffeine clearance.
- a "complication associated with cirrhosis of the liver” refers to a disorder that is a sequellae of decompensated liver disease, i.e., or occurs subsequently to and as a result of development of liver fibrosis, and includes, but it not limited to, development of ascites, variceal bleeding, portal hypertension, jaundice, progressive liver insufficiency, encephalopathy, hepatocellular carcinoma, liver failure requiring liver transplantation, and liver-related mortality.
- a therapeutically effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount that is effective in reducing the incidence (e.g., the likelihood that an individual will develop) of a disorder associated with cirrhosis of the liver by at least about 10%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, or at least about 80%, or more, compared to an untreated individual, or to a placebo-treated individual.
- the embodiments provide methods for increasing liver function, generally involving administering a therapeutically effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents.
- Liver functions include, but are not limited to, synthesis of proteins such as serum proteins (e.g., albumin, clotting factors, alkaline phosphatase, aminotransferases (e.g., alanine transaminase, aspartate transaminase), 5 '-nucleosidase, ⁇ -glutaminyltranspeptidase, etc.), synthesis of bilirubin, synthesis of cholesterol, and synthesis of bile acids; a liver metabolic function, including, but not limited to, carbohydrate metabolism, amino acid and ammonia metabolism, hormone metabolism, and lipid metabolism; detoxification of exogenous drugs; a hemodynamic function, including splanchnic and portal hemodynamics; and the like.
- proteins such as serum proteins (e.g., albumin, clotting factors, alkaline phosphatase, aminotransferases (e.g., alanine transaminase, aspartate transaminase), 5
- liver function is increased is readily ascertainable by those skilled in the art, using well-established tests of liver function.
- markers of liver function such as albumin, alkaline phosphatase, alanine transaminase, aspartate transaminase, bilirubin, and the like, can be assessed by measuring the level of these markers in the serum, using standard immunological and enzymatic assays.
- Splanchnic circulation and portal hemodynamics can be measured by portal wedge pressure and/or resistance using standard methods.
- Metabolic functions can be measured by measuring the level of ammonia in the serum.
- Whether serum proteins normally secreted by the liver are in the normal range can be determined by measuring the levels of such proteins, using standard immunological and enzymatic assays. Those skilled in the art know the normal ranges for such serum proteins. The following are non-limiting examples.
- the normal level of alanine transaminase is about 45 IU per milliliter of serum.
- the normal range of aspartate transaminase is from about 5 to about 40 units per liter of serum.
- Bilirubin is measured using standard assays. Normal bilirubin levels are usually less than about 1.2 mg/dL.
- Serum albumin levels are measured using standard assays. Normal levels of serum albumin are in the range of from about 35 to about 55 g/L.
- Prolongation of prothrombin time is measured using standard assays. Normal prothrombin time is less than about 4 seconds longer than control.
- a therapeutically effective amount of a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents, is one that is effective to increase liver function by at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, or more.
- a therapeutically effective amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, Vn, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agents is an amount effective to reduce an elevated level of a serum marker of liver function by at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, or more, or to reduce the level of the serum marker of liver function to within a normal range.
- a therapeutically effective amount of a compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents is also an amount effective to increase a reduced level of a serum marker of liver function by at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, or more, or to increase the level of the serum marker of liver function to within a normal range.
- the active agent(s) e.g., compound of Formulae I, la, ⁇ , ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, XII, or any compounds disclosed herein, and optionally one or more additional antiviral agents
- the agent may be administered to the host using any convenient means capable of resulting in the desired therapeutic effect.
- the agent can be incorporated into a variety of formulations for therapeutic administration.
- the agents of the embodiments can be formulated into pharmaceutical compositions by combination with appropriate, pharmaceutically acceptable carriers or diluents, and may be formulated into preparations in solid, semi-solid, liquid or gaseous forms, such as tablets, capsules, powders, granules, ointments, solutions, suppositories, injections, inhalants and aerosols.
- compositions are provided in formulation with a pharmaceutically acceptable excipient(s).
- a pharmaceutically acceptable excipient A wide variety of pharmaceutically acceptable excipients is known in the art and need not be discussed in detail herein.
- Pharmaceutically acceptable excipients have been amply described in a variety of publications, including, for example, A. Gennaro (2000) "Remington: The Science and Practice of Pharmacy," 20 th edition, Lippincott, Williams, & Wilkins; Pharmaceutical Dosage Forms and Drug Delivery Systems (1999) H.C.
- compositions such as vehicles, adjuvants, carriers or diluents
- pharmaceutically acceptable auxiliary substances such as pH adjusting and buffering agents, tonicity adjusting agents, stabilizers, wetting agents and the like, are readily available to the public.
- an agent is formulated in an aqueous buffer.
- Suitable aqueous buffers include, but are not limited to, acetate, succinate, citrate, and phosphate buffers varying in strengths from about 5 mM to about 100 mM.
- the aqueous buffer includes reagents that provide for an isotonic solution. Such reagents include, but are not limited to, sodium chloride; and sugars e.g., mannitol, dextrose, sucrose, and the like.
- the aqueous buffer further includes a non-ionic surfactant such as polysorbate 20 or 80.
- the formulations may further include a preservative.
- Suitable preservatives include, but are not limited to, a benzyl alcohol, phenol, chlorobutanol, benzalkonium chloride, and the like. In many cases, the formulation is stored at about 4°C. Formulations may also be lyophilized, in which case they generally include cryoprotectants such as sucrose, trehalose, lactose, maltose, mannitol, and the like. Lyophilized formulations can be stored over extended periods of time, even at ambient temperatures.
- administration of the agents can be achieved in various ways, including oral, buccal, rectal, parenteral, intraperitoneal, intradermal, subcutaneous, intramuscular, transdermal, intratracheal, etc., administration.
- administration is by bolus injection, e.g., subcutaneous bolus injection, intramuscular bolus injection, and the like.
- compositions of the embodiments can be administered orally, parenterally or via an implanted reservoir. Oral administration or administration by injection is preferred.
- Subcutaneous administration of a pharmaceutical composition of the embodiments is accomplished using standard methods and devices, e.g., needle and syringe, a subcutaneous injection port delivery system, and the like. See, e.g., U.S. Patent Nos. 3,547,119; 4,755,173; 4,531,937; 4,311,137; and 6,017,328.
- a combination of a subcutaneous injection port and a device for administration of a pharmaceutical composition of the embodiments to a patient through the port is referred to herein as "a subcutaneous injection port delivery system.”
- subcutaneous administration is achieved by bolus delivery by needle and syringe.
- the agents may be administered in the form of their pharmaceutically acceptable salts, or they may also be used alone or in appropriate association, as well as in combination, with other pharmaceutically active compounds.
- the following methods and excipients are merely exemplary and are in no way limiting.
- the agents can be used alone or in combination with appropriate additives to make tablets, powders, granules or capsules, for example, with conventional additives, such as lactose, mannitol, corn starch or potato starch; with binders, such as crystalline cellulose, cellulose derivatives, acacia, corn starch or gelatins; with disintegrators, such as corn starch, potato starch or sodium carboxymethylcellulose; with lubricants, such as talc or magnesium stearate; and if desired, with diluents, buffering agents, moistening agents, preservatives and flavoring agents.
- conventional additives such as lactose, mannitol, corn starch or potato starch
- binders such as crystalline cellulose, cellulose derivatives, acacia, corn starch or gelatins
- disintegrators such as corn starch, potato starch or sodium carboxymethylcellulose
- lubricants such as talc or magnesium stearate
- the agents can be formulated into preparations for injection by dissolving, suspending or emulsifying them in an aqueous or nonaqueous solvent, such as vegetable or other similar oils, synthetic aliphatic acid glycerides, esters of higher aliphatic acids or propylene glycol; and if desired, with conventional additives such as solubilizers, isotonic agents, suspending agents, emulsifying agents, stabilizers and preservatives.
- an aqueous or nonaqueous solvent such as vegetable or other similar oils, synthetic aliphatic acid glycerides, esters of higher aliphatic acids or propylene glycol
- solubilizers isotonic agents
- suspending agents emulsifying agents
- stabilizers and preservatives such as solubilizers, isotonic agents, suspending agents, emulsifying agents, stabilizers and preservatives.
- the agents can be made into suppositories by mixing with a variety of bases such as emulsifying bases or water-soluble bases.
- bases such as emulsifying bases or water-soluble bases.
- the compounds of the embodiments can be administered rectally via a suppository.
- the suppository can include vehicles such as cocoa butter, carbowaxes and polyethylene glycols, which melt at body temperature, yet are solidified at room temperature.
- Unit dosage forms for oral or rectal administration such as syrups, elixirs, and suspensions may be provided wherein each dosage unit, for example, teaspoonful, tablespoonful, tablet or suppository, contains a predetermined amount of the composition containing one or more inhibitors.
- unit dosage forms for injection or intravenous administration may comprise the inhibitor(s) in a composition as a solution in sterile water, normal saline or another pharmaceutically acceptable carrier.
- unit dosage form refers to physically discrete units suitable as unitary dosages for human and animal subjects, each unit containing a predetermined quantity of compounds of the embodiments calculated in an amount sufficient to produce the desired effect in association with a pharmaceutically acceptable diluent, carrier or vehicle.
- the specifications for the novel unit dosage forms of the embodiments depend on the particular compound employed and the effect to be achieved, and the pharmacodynamics associated with each compound in the host.
- compositions such as vehicles, adjuvants, carriers or diluents
- pharmaceutically acceptable auxiliary substances such as pH adjusting and buffering agents, tonicity adjusting agents, stabilizers, wetting agents and the like, are readily available to the public.
- a subject method will in some embodiments be carried out by administering an NS3 inhibitor that is a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agent(s).
- an NS3 inhibitor that is a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, Vni, IX, X, XI, or, ⁇ , or any compounds disclosed herein, and optionally one or more additional antiviral agent(s).
- the method further includes administration of one or more interferon receptor agonist(s).
- Interferon receptor agonists are described herein.
- the method further includes administration of pirfenidone or a pirfenidone analog. Pirfenidone and pirfenidone analogs are described herein.
- Additional antiviral agents that are suitable for use in combination therapy include, but are not limited to, nucleotide and nucleoside analogs.
- Non-limiting examples include azidothymidine (AZT) (zidovudine), and analogs and derivatives thereof; 2',3'-dideoxyinosine (DDI) (didanosine), and analogs and derivatives thereof; 2',3'-dideoxycytidine (DDC) (dideoxycytidine), and analogs and derivatives thereof; 2'3,'-didehydro-2' ,3'-dideoxythymidine (D4T) (stavudine), and analogs and derivatives thereof; combivir; abacavir; adefovir dipoxil; cidofovir; ribavirin; ribavirin analogs; and the like.
- the method further includes administration of ribavirin.
- Ribavirin, l- -D-ribofuranosyl-lH-l,2,4-triazole-3-carboxamide available from ICN Pharmaceuticals, Inc., Costa Mesa, Calif., is described in the Merck Index, compound No. 8199, Eleventh Edition. Its manufacture and formulation is described in U.S. Pat. No. 4,211,771. Some embodiments also involve use of derivatives of ribavirin (see, e.g. , U.S. Pat. No. 6,277,830).
- the ribavirin may be administered orally in capsule or tablet form, or in the same or different administration form and in the same or different route as the NS-3 inhibitor compound.
- other types of administration of both medicaments as they become available are contemplated, such as by nasal spray, transdermally, intravenously, by suppository, by sustained release dosage form, etc. Any form of administration will work so long as the proper dosages are delivered without destroying the active ingredient.
- the method further includes administration of ritonavir.
- Ritonavir 10-hydroxy-2-methyl-5-( 1 -methylethyl)- 1 - [2-( 1 -methylethyl)-4- thiazolyl]-3,6-dioxo-8,l l-bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic acid, 5- thiazolylmethyl ester ⁇ - ⁇ , ⁇ , ⁇ , ⁇ ⁇ *)], available from Abbott Laboratories, is an inhibitor of the protease of the human immunodeficiency virus and also of the cytochrome P450 3A and P450 2D6 liver enzymes frequently involved in hepatic metabolism of therapeutic molecules in man.
- ritonavir at doses below the normal therapeutic dosage may be combined with other protease inhibitors to achieve therapeutic levels of the second protease inhibitor while reducing the number of dosage units required, the dosing frequency, or both.
- Coadministration of low-dose ritonavir may also be used to compensate for drug interactions that tend to decrease levels of a protease inhibitor metabolized by CYP3A. Its structure, synthesis, manufacture and formulation are described in U.S. Pat. No. 5,541,206 U.S. Pat. No. 5,635,523 U.S. Pat. No. 5,648,497 U.S. Pat. No. 5,846,987 and U.S. Pat. No. 6,232,333.
- the ritonavir may be administered orally in capsule or tablet or oral solution form, or in the same or different administration form and in the same or different route as the NS-3 inhibitor compound.
- an additional antiviral agent is administered during the entire course of NS3 inhibitor compound treatment.
- an additional antiviral agent is administered for a period of time that is overlapping with that of the NS3 inhibitor compound treatment, e.g., the additional antiviral agent treatment can begin before the NS3 inhibitor compound treatment begins and end before the NS3 inhibitor compound treatment ends; the additional antiviral agent treatment can begin after the NS3 inhibitor compound treatment begins and end after the NS3 inhibitor compound treatment ends; the additional antiviral agent treatment can begin after the NS3 inhibitor compound treatment begins and end before the NS3 inhibitor compound treatment ends; or the additional antiviral agent treatment can begin before the NS3 inhibitor compound treatment begins and end after the NS3 inhibitor compound treatment ends.
- the NS3 inhibitor compounds described herein may be used in acute or chronic therapy for HCV disease.
- the NS3 inhibitor compound is administered for a period of about 1 day to about 7 days, or about 1 week to about 2 weeks, or about 2 weeks to about 3 weeks, or about 3 weeks to about 4 weeks, or about 1 month to about 2 months, or about 3 months to about 4 months, or about 4 months to about 6 months, or about 6 months to about 8 months, or about 8 months to about 12 months, or at least one year, and may be administered over longer periods of time.
- the NS3 inhibitor compound can be administered 5 times per day, 4 times per day, tid, bid, qd, qod, biw, tiw, qw, qow, three times per month, or once monthly. In other embodiments, the NS3 inhibitor compound is administered as a continuous infusion.
- an NS3 inhibitor compound of the embodiments is administered orally.
- an NS3 inhibitor compound as described herein may be administered to the patient at a dosage from about 0.01 mg to about 100 mg/kg patient bodyweight per day, in 1 to 5 divided doses per day.
- the NS3 inhibitor compound is administered at a dosage of about 0.5 mg to about 75 mg/kg patient bodyweight per day, in 1 to 5 divided doses per day.
- the amount of active ingredient that may be combined with carrier materials to produce a dosage form can vary depending on the host to be treated and the particular mode of administration.
- a typical pharmaceutical preparation can contain from about 5% to about 95% active ingredient (w/w). In other embodiments, the pharmaceutical preparation can contain from about 20% to about 80% active ingredient.
- dose levels can vary as a function of the specific NS3 inhibitor compound, the severity of the symptoms and the susceptibility of the subject to side effects.
- Preferred dosages for a given NS3 inhibitor compound are readily determinable by those of skill in the art by a variety of means.
- a preferred means is to measure the physiological potency of a given interferon receptor agonist.
- multiple doses of NS3 inhibitor compound are administered.
- an NS3 inhibitor compound is administered once per month, twice per month, three times per month, every other week (qow), once per week (qw), twice per week (biw), three times per week (tiw), four times per week, five times per week, six times per week, every other day (qod), daily (qd), twice a day (qid), or three times a day (tid), over a period of time ranging from about one day to about one week, from about two weeks to about four weeks, from about one month to about two months, from about two months to about four months, from about four months to about six months, from about six months to about eight months, from about eight months to about 1 year, from about 1 year to about 2 years, or from about 2 years to about 4 years, or more.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of ribavirin.
- Ribavirin can be administered in dosages of about 400 mg, about 800 mg, about 1000 mg, or about 1200 mg per day.
- One embodiment provides any of the above-described methods modified to include co-administering to the patient a therapeutically effective amount of ribavirin for the duration of the desired course of NS3 inhibitor compound treatment.
- Another embodiment provides any of the above-described methods modified to include co-administering to the patient about 800 mg to about 1200 mg ribavirin orally per day for the duration of the desired course of NS3 inhibitor compound treatment.
- any of the above-described methods may be modified to include co-administering to the patient (a) 1000 mg ribavirin orally per day if the patient has a body weight less than 75 kg or (b) 1200 mg ribavirin orally per day if the patient has a body weight greater than or equal to 75 kg, where the daily dosage of ribavirin is optionally divided into to 2 doses for the duration of the desired course of NS3 inhibitor compound treatment.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of levovirin.
- Levovirin is generally administered in an amount ranging from about 30 mg to about 60 mg, from about 60 mg to about 125 mg, from about 125 mg to about 200 mg, from about 200 mg to about 300 gm, from about 300 mg to about 400 mg, from about 400 mg to about 1200 mg, from about 600 mg to about 1000 mg, or from about 700 to about 900 mg per day, or about 10 mg/kg body weight per day.
- levovirin is administered orally in dosages of about 400, about 800, about 1000, or about 1200 mg per day for the desired course of NS3 inhibitor compound treatment.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of viramidine.
- Viramidine is generally administered in an amount ranging from about 30 mg to about 60 mg, from about 60 mg to about 125 mg, from about 125 mg to about 200 mg, from about 200 mg to about 300 mg, from about 300 mg to about 400 mg, from about 400 mg to about 1200 mg, from about 600 mg to about 1000 mg, or from about 700 to about 900 mg per day, or about 10 mg/kg body weight per day.
- viramidine is administered orally in dosages of about 800 mg, or about 1600 mg per day for the desired course of NS3 inhibitor compound treatment.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of ritonavir.
- Ritonavir is generally administered in an amount ranging from about 50 mg to about 100 mg, from about 100 mg to about 200 mg, from about 200 mg to about 300 mg, from about 300 mg to about 400 mg, from about 400 mg to about 500 mg, or from about 500 mg to about 600 mg, twice per day.
- ritonavir is administered orally in dosages of about 300 mg, or about 400 mg, or about 600 mg twice per day for the desired course of NS3 inhibitor compound treatment.
- Suitable oc-glucosidase inhibitors include any of the above-described imino-sugars, including long-alkyl chain derivatives of imino sugars as disclosed in U.S. Patent Publication No. 2004/0110795; inhibitors of endoplasmic reticulum-associated oc- glucosidases; inhibitors of membrane bound oc-glucosidase; miglitol (Glyset®), and active derivatives, and analogs thereof; and acarbose (Precose®), and active derivatives, and analogs thereof.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of an oc-glucosidase inhibitor administered for a period of about 1 day to about 7 days, or about 1 week to about 2 weeks, or about 2 weeks to about 3 weeks, or about 3 weeks to about 4 weeks, or about 1 month to about 2 months, or about 3 months to about 4 months, or about 4 months to about 6 months, or about 6 months to about 8 months, or about 8 months to about 12 months, or at least one year, and may be administered over longer periods of time.
- An oc-glucosidase inhibitor can be administered 5 times per day, 4 times per day, tid (three times daily), bid, qd, qod, biw, tiw, qw, qow, three times per month, or once monthly. In other embodiments, an oc-glucosidase inhibitor is administered as a continuous infusion.
- an oc-glucosidase inhibitor is administered orally.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of oc-glucosidase inhibitor administered to the patient at a dosage of from about 10 mg per day to about 600 mg per day in divided doses, e.g., from about 10 mg per day to about 30 mg per day, from about 30 mg per day to about 60 mg per day, from about 60 mg per day to about 75 mg per day, from about 75 mg per day to about 90 mg per day, from about 90 mg per day to about 120 mg per day, from about 120 mg per day to about 150 mg per day, from about 150 mg per day to about 180 mg per day, from about 180 mg per day to about 210 mg per day, from about 210 mg per day to about 240 mg per day, from about 240 mg per day to about 270 mg per day, from about
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of oc-glucosidase inhibitor administered in a dosage of about 10 mg three times daily.
- an a-glucosidase inhibitor is administered in a dosage of about 15 mg three times daily.
- an ⁇ -glucosidase inhibitor is administered in a dosage of about 20 mg three times daily.
- an a- glucosidase inhibitor is administered in a dosage of about 25 mg three times daily.
- an ⁇ -glucosidase inhibitor is administered in a dosage of about 30 mg three times daily.
- an ⁇ -glucosidase inhibitor is administered in a dosage of about 40 mg three times daily. In some embodiments, an a-glucosidase inhibitor is administered in a dosage of about 50 mg three times daily. In some embodiments, an ⁇ -glucosidase inhibitor is administered in a dosage of about 100 mg three times daily. In some embodiments, an ⁇ -glucosidase inhibitor is administered in a dosage of about 75 mg per day to about 150 mg per day in two or three divided doses, where the individual weighs 60 kg or less. In some embodiments, an a-glucosidase inhibitor is administered in a dosage of about 75 mg per day to about 300 mg per day in two or three divided doses, where the individual weighs 60 kg or more.
- the amount of active ingredient (e.g., ⁇ -glucosidase inhibitor) that may be combined with carrier materials to produce a dosage form can vary depending on the host to be treated and the particular mode of administration.
- a typical pharmaceutical preparation can contain from about 5% to about 95% active ingredient (w/w). In other embodiments, the pharmaceutical preparation can contain from about 20% to about 80% active ingredient.
- dose levels can vary as a function of the specific ⁇ -glucosidase inhibitor, the severity of the symptoms and the susceptibility of the subject to side effects.
- Preferred dosages for a given a-glucosidase inhibitor are readily determinable by those of skill in the art by a variety of means. A typical means is to measure the physiological potency of a given active agent.
- multiple doses of an ⁇ -glucosidase inhibitor are administered.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of ⁇ -glucosidase inhibitor administered once per month, twice per month, three times per month, every other week (qow), once per week (qw), twice per week (biw), three times per week (tiw), four times per week, five times per week, six times per week, every other day (qod), daily (qd), twice a day (qid), or three times a day (tid), over a period of time ranging from about one day to about one week, from about two weeks to about four weeks, from about one month to about two months, from about two months to about four months, from about four months to about six months, from about six months to about eight months, from about eight months to about 1 year, from about 1 year to about 2 years, or from about 2 years to about 4 years, or more.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of thymosin-oc.
- Thymosin-oc ZadaxinTM
- Thymosin-oc can be administered tid, bid, qd, qod, biw, tiw, qw, qow, three times per month, once monthly, substantially continuously, or continuously for the desired course of NS3 inhibitor compound treatment.
- thymosin-oc is administered twice per week for the desired course of NS3 inhibitor compound treatment.
- Effective dosages of thymosin-oc range from about 0.5 mg to about 5 mg, e.g., from about 0.5 mg to about 1.0 mg, from about 1.0 mg to about 1.5 mg, from about 1.5 mg to about 2.0 mg, from about 2.0 mg to about 2.5 mg, from about 2.5 mg to about 3.0 mg, from about 3.0 mg to about 3.5 mg, from about 3.5 mg to about 4.0 mg, from about 4.0 mg to about 4.5 mg, or from about 4.5 mg to about 5.0 mg.
- thymosin-oc is administered in dosages containing an amount of 1.0 mg or 1.6 mg.
- Thymosin-oc can be administered over a period of time ranging from about one day to about one week, from about two weeks to about four weeks, from about one month to about two months, from about two months to about four months, from about four months to about six months, from about six months to about eight months, from about eight months to about 1 year, from about 1 year to about 2 years, or from about 2 years to about 4 years, or more.
- thymosin-oc is administered for the desired course of NS3 inhibitor compound treatment.
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of an interferon receptor agonist.
- a compound of Formulae I, la, II, ⁇ , IV, V, VI- 1, VI-2, VII, VIII, IX, X, XI, or, ⁇ , or any compounds disclosed herein and a Type I or ⁇ interferon receptor agonist are co-administered in the treatment methods described herein.
- Type I interferon receptor agonists suitable for use herein include any interferon-oc (IFN-Oc).
- the interferon-oc is a PEGylated interferon-oc.
- the interferon-oc is a consensus interferon, such as INFERGEN® interferon alfacon-1. In still other embodiments, the interferon-oc is a monoPEG (30 kD, linear)-ylated consensus interferon.
- Effective dosages of an IFN-OC range from about 3 ⁇ g to about 27 ⁇ g, from about 3 MU to about 10 MU, from about 90 ⁇ g to about 180 ⁇ g, or from about 18 ⁇ g to about 90 ⁇ g.
- Effective dosages of Infergen® consensus IFN-OC include about 3 ⁇ g, about 6 ⁇ g, about 9 ⁇ g, about 12 ⁇ g, about 15 ⁇ g, about 18 ⁇ g, about 21 ⁇ g, about 24 ⁇ g, about 27 ⁇ g, or about 30 ⁇ g, of drug per dose.
- Effective dosages of IFN-0c2a and IFN- 0c2b range from 3 million Units (MU) to 10 MU per dose.
- Effective dosages of PEGASYS®PEGylated IFN-0c2a contain an amount of about 90 ⁇ g to 270 ⁇ g, or about 180 ⁇ g, of drug per dose.
- Effective dosages of PEG-INTRON®PEGylated IFN-0c2b contain an amount of about 0.5 ⁇ g to 3.0 ⁇ g of drug per kg of body weight per dose.
- Effective dosages of PEGylated consensus interferon (PEG-CIFN) contain an amount of about 18 ⁇ g to about 90 ⁇ g, or from about 27 ⁇ g to about 60 ⁇ g, or about 45 ⁇ g, of CIFN amino acid weight per dose of PEG-CIFN.
- Effective dosages of monoPEG (30 kD, linear) -ylated CIFN contain an amount of about 45 ⁇ g to about 270 ⁇ g, or about 60 ⁇ g to about 180 ⁇ g, or about 90 ⁇ g to about 120 ⁇ g, of drug per dose.
- IFN-OC can be administered daily, every other day, once a week, three times a week, every other week, three times per month, once monthly, substantially continuously or continuously.
- the Type I or Type ⁇ interferon receptor agonist and/or the Type II interferon receptor agonist is administered for a period of about 1 day to about 7 days, or about 1 week to about 2 weeks, or about 2 weeks to about 3 weeks, or about 3 weeks to about 4 weeks, or about 1 month to about 2 months, or about 3 months to about 4 months, or about 4 months to about 6 months, or about 6 months to about 8 months, or about 8 months to about 12 months, or at least one year, and may be administered over longer periods of time.
- Dosage regimens can include tid, bid, qd, qod, biw, tiw, qw, qow, three times per month, or monthly administrations.
- Some embodiments provide any of the above-described methods in which the desired dosage of IFN-OC is administered subcutaneously to the patient by bolus delivery qd, qod, tiw, biw, qw, qow, three times per month, or monthly, or is administered subcutaneously to the patient per day by substantially continuous or continuous delivery, for the desired treatment duration.
- any of the above-described methods may be practiced in which the desired dosage of PEGylated IFN-a (PEG-IFN-a) is administered subcutaneously to the patient by bolus delivery qw, qow, three times per month, or monthly for the desired treatment duration.
- an NS3 inhibitor compound and a Type ⁇ interferon receptor agonist are co-administered in the treatment methods of the embodiments.
- Type II interferon receptor agonists suitable for use herein include any interferon- ⁇ (IFN- ⁇ ).
- Effective dosages of IFN- ⁇ can range from about 0.5 g/m 2 to about
- IFN- ⁇ can be administered daily, every other day, three times a week, or substantially continuously or continuously.
- IFN- ⁇ is administered to an individual in a unit dosage form of from about 25 ⁇ g to about 500 ⁇ g, from about 50 ⁇ g to about 400 ⁇ g, or from about 100 ⁇ g to about 300 ⁇ g. In particular embodiments of interest, the dose is about 200 ⁇ g IFN- ⁇ . In many embodiments of interest, IFN-ylb is administered.
- the amount of IFN- ⁇ per body weight (assuming a range of body weights of from about 45 kg to about 135 kg) is in the range of from about 4.4 ⁇ g IFN- ⁇ per kg body weight to about 1.48 ⁇ g IFN- ⁇ per kg body weight.
- an IFN- ⁇ dosage ranges from about 150 ⁇ g/m 2 to about 20 ⁇ g/m 2.
- an IFN- ⁇ dosage ranges from about 20 ⁇ g/m 2 to about 30 ⁇ g/m 2 , from about 30 ⁇ g/m 2 to about 40 ⁇ g/m 2 , from about 40 ⁇ g/m 2 to about 50 ⁇ g/m 2 , from about 50 ⁇ g/m 2 to about 60 ⁇ g/m 2 , from about 60 ⁇ g/m 2 to about 70 ⁇ g/m 2 , from about 70 ⁇ g/m 2 to about 80 ⁇ g/m 2 , from about 80 ⁇ g/m 2 to about 90 ⁇ g/m 2 , from about 90 ⁇ g/m 2 to about 100 ⁇ g/m 2 , from about 100 ⁇ g/m 2 to about 110 ⁇ g/m 2 , from about 110
- a Type I or a Type ⁇ interferon receptor agonist is administered in a first dosing regimen, followed by a second dosing regimen.
- the first dosing regimen of Type I or a Type ⁇ interferon receptor agonist generally involves administration of a higher dosage of the Type I or Type ⁇ interferon receptor agonist.
- the first dosing regimen comprises administering CIFN at about 9 ⁇ g, about 15 ⁇ g, about 18 ⁇ g, or about 27 ⁇ g.
- the first dosing regimen can encompass a single dosing event, or at least two or more dosing events.
- the first dosing regimen of the Type I or Type ⁇ interferon receptor agonist can be administered daily, every other day, three times a week, every other week, three times per month, once monthly, substantially continuously or continuously.
- the first dosing regimen of the Type I or Type ⁇ interferon receptor agonist is administered for a first period of time, which time period can be at least about 4 weeks, at least about 8 weeks, or at least about 12 weeks.
- the second dosing regimen of the Type I or Type ⁇ interferon receptor agonist generally involves administration of a lower amount of the Type I or Type ⁇ interferon receptor agonist.
- the second dosing regimen comprises administering CIFN at a dose of at least about 3 ⁇ g, at least about 9 ⁇ g, at least about 15 ⁇ g, or at least about 18 ⁇ g.
- the second dosing regimen can encompass a single dosing event, or at least two or more dosing events.
- the second dosing regimen of the Type I or Type ⁇ interferon receptor agonist can be administered daily, every other day, three times a week, every other week, three times per month, once monthly, substantially continuously or continuously.
- a "priming" dose of a Type II interferon receptor agonist e.g., IFN- ⁇
- IFN- ⁇ is administered for a period of time from about 1 day to about 14 days, from about 2 days to about 10 days, or from about 3 days to about 7 days, before the beginning of treatment with the Type I or Type ⁇ interferon receptor agonist. This period of time is referred to as the "priming" phase.
- the Type ⁇ interferon receptor agonist treatment is continued throughout the entire period of treatment with the Type I or Type ⁇ interferon receptor agonist.
- the Type II interferon receptor agonist treatment is discontinued before the end of treatment with the Type I or Type ⁇ interferon receptor agonist.
- the total time of treatment with Type ⁇ interferon receptor agonist (including the "priming" phase) is from about 2 days to about 30 days, from about 4 days to about 25 days, from about 8 days to about 20 days, from about 10 days to about 18 days, or from about 12 days to about 16 days.
- the Type ⁇ interferon receptor agonist treatment is discontinued once Type I or a Type ⁇ interferon receptor agonist treatment begins.
- the Type I or Type ⁇ interferon receptor agonist is administered in single dosing regimen.
- the dose of CIFN is generally in a range of from about 3 ⁇ g to about 15 ⁇ g, or from about 9 ⁇ g to about 15 ⁇ g.
- the dose of Type I or a Type ⁇ interferon receptor agonist is generally administered daily, every other day, three times a week, every other week, three times per month, once monthly, or substantially continuously.
- the dose of the Type I or Type ⁇ interferon receptor agonist is administered for a period of time, which period can be, for example, from at least about 24 weeks to at least about 48 weeks, or longer.
- a "priming" dose of a Type ⁇ interferon receptor agonist (e.g., IFN- ⁇ ) is included.
- IFN- ⁇ is administered for a period of time from about 1 day to about 14 days, from about 2 days to about 10 days, or from about 3 days to about 7 days, before the beginning of treatment with the Type I or Type ⁇ interferon receptor agonist. This period of time is referred to as the "priming" phase.
- the Type II interferon receptor agonist treatment is continued throughout the entire period of treatment with the Type I or Type ⁇ interferon receptor agonist.
- the Type II interferon receptor agonist treatment is discontinued before the end of treatment with the Type I or Type ⁇ interferon receptor agonist.
- the total time of treatment with the Type II interferon receptor agonist (including the "priming" phase) is from about 2 days to about 30 days, from about 4 days to about 25 days, from about 8 days to about 20 days, from about 10 days to about 18 days, or from about 12 days to about 16 days.
- Type ⁇ interferon receptor agonist treatment is discontinued once Type I or a Type ⁇ interferon receptor agonist treatment begins.
- an NS3 inhibitor compound, a Type I or ⁇ interferon receptor agonist, and a Type ⁇ interferon receptor agonist are co-administered for the desired duration of treatment in the methods described herein.
- an NS3 inhibitor compound, an interferon- oc, and an interferon- ⁇ are coadministered for the desired duration of treatment in the methods described herein.
- the invention provides methods using an amount of a Type I or Type ⁇ interferon receptor agonist, a Type II interferon receptor agonist, and an NS3 inhibitor compound, effective for the treatment of HCV infection in a patient. Some embodiments provide methods using an effective amount of an IFN-a, IFN- ⁇ , and an NS3 inhibitor compound in the treatment of HCV infection in a patient. One embodiment provides a method using an effective amount of a consensus IFN-a, IFN- ⁇ and an NS3 inhibitor compound in the treatment of HCV infection in a patient.
- an effective amount of a consensus interferon (CIFN) and IFN- ⁇ suitable for use in the methods of the embodiments is provided by a dosage ratio of 1 ⁇ g CIFN: 10 ⁇ g IFN- ⁇ , where both CIFN and IFN- ⁇ are unPEGylated and unglycosylated species.
- the invention provides any of the above-described methods modified to use an effective amount of INFERGEN®consensus IFN-a and IFN- ⁇ in the treatment of HCV infection in a patient comprising administering to the patient a dosage of INFERGEN® containing an amount of about 1 ⁇ g to about 30 ⁇ g, of drug per dose of INFERGEN®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 10 ⁇ g to about 300 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of INFERGEN®consensus IFN-a and IFN- ⁇ in the treatment of virus infection in a patient comprising administering to the patient a dosage of INFERGEN® containing an amount of about 1 ⁇ g to about 9 ⁇ g, of drug per dose of INFERGEN®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 10 ⁇ g to about 100 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of INFERGEN®consensus IFN-OC and IFN- ⁇ in the treatment of virus infection in a patient comprising administering to the patient a dosage of INFERGEN® containing an amount of about 1 ⁇ g of drug per dose of INFERGEN®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 10 ⁇ g to about 50 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of INFERGEN®consensus IFN-OC and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of INFERGEN® containing an amount of about 9 ⁇ g of drug per dose of INFERGEN®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 90 ⁇ g to about 100 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of INFERGEN®consensus IFN-OC and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of INFERGEN® containing an amount of about 30 ⁇ g of drug per dose of INFERGEN®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 200 ⁇ g to about 300 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGylated consensus IFN-a and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGylated consensus IFN-a (PEG-CIFN) containing an amount of about 4 ⁇ g to about 60 ⁇ g of CIFN amino acid weight per dose of PEG-CIFN, subcutaneously qw, qow, three times per month, or monthly, in combination with a total weekly dosage of IFN- ⁇ containing an amount of about 30 ⁇ g to about 1,000 ⁇ g of drug per week in divided doses administered subcutaneously qd, qod, tiw, biw, or administered substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- PEG-CIFN PEGylated consensus IFN-a
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGylated consensus IFN-a and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGylated consensus IFN-a (PEG-CIFN) containing an amount of about 18 ⁇ g to about 24 ⁇ g of CIFN amino acid weight per dose of PEG-CIFN, subcutaneously qw, qow, three times per month, or monthly, in combination with a total weekly dosage of IFN- ⁇ containing an amount of about 100 ⁇ g to about 300 ⁇ g of drug per week in divided doses administered subcutaneously qd, qod, tiw, biw, or substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- PEG-CIFN PEGylated consensus IFN-a
- an effective amount of IFN-a 2a or 2b or 2c and IFN- ⁇ suitable for use in the methods of the embodiments is provided by a dosage ratio of 1 million Units (MU) IFN-a 2a or 2b or 2c : 30 ⁇ g IFN- ⁇ , where both IFN-a 2a or 2b or 2c and IFN- ⁇ are unPEGylated and unglycosylated species.
- MU 1 million Units
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN-a 2a or 2b or 2c and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of IFN-a 2a, 2b or 2c containing an amount of about 1 MU to about 20 MU of drug per dose of IFN-a 2a, 2b or 2c subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 30 ⁇ g to about 600 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN-a 2a or 2b or 2c and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of IFN-a 2a, 2b or 2c containing an amount of about 3 MU of drug per dose of IFN-a 2a, 2b or 2c subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 100 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN-a 2a or 2b or 2c and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of IFN-a 2a, 2b or 2c containing an amount of about 10 MU of drug per dose of IFN-a 2a, 2b or 2c subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, in combination with a dosage of IFN- ⁇ containing an amount of about 300 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGASYS®PEGylated IFN-a2a and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGASYS® containing an amount of about 90 ⁇ g to about 360 ⁇ g, of drug per dose of PEGASYS®, subcutaneously qw, qow, three times per month, or monthly, in combination with a total weekly dosage of IFN- ⁇ containing an amount of about 30 ⁇ g to about 1,000 ⁇ g, of drug per week administered in divided doses subcutaneously qd, qod, tiw, or biw, or administered substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGASYS®PEGylated IFN-a2a and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGASYS® containing an amount of about 180 ⁇ g of drug per dose of PEGASYS®, subcutaneously qw, qow, three times per month, or monthly, in combination with a total weekly dosage of IFN- ⁇ containing an amount of about 100 ⁇ g to about 300 ⁇ g, of drug per week administered in divided doses subcutaneously qd, qod, tiw, or biw, or administered substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEG-INTRON®PEGylated IFN-Cc2b and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEG-INTRON® containing an amount of about 0.75 ⁇ g to about 3.0 ⁇ g of drug per kilogram of body weight per dose of PEG-INTRON®, subcutaneously qw, qow, three times per month, or monthly, in combination with a total weekly dosage of IFN- ⁇ containing an amount of about 30 ⁇ g to about 1,000 ⁇ g of drug per week administered in divided doses subcutaneously qd, qod, tiw, or biw, or administered substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEG-INTRON®PEGylated IFN-Cc2b and IFN- ⁇ in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEG-INTRON® containing an amount of about 1.5 ⁇ g of drug per kilogram of body weight per dose of PEG-INTRON®, subcutaneously qw, qow, three times per month, or monthly, in combination with a total weekly dosage of IFN- ⁇ containing an amount of about 100 ⁇ g to about 300 ⁇ g of drug per week administered in divided doses subcutaneously qd, qod, tiw, or biw, or administered substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-OC administered subcutaneously qd or tiw, and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; and 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; and 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; 25 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; 200 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; and 25 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 9 ⁇ g INFERGEN® consensus IFN-a administered subcutaneously qd or tiw; and 200 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 100 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-a administered subcutaneously every 10 days or qw, and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 100 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 100 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 100 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; and 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 100 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; and 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 150 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw, and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 150 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 150 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 150 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; and 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 150 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; and 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 200 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw, and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 200 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 200 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw; and ribavirin administered orally qd, where the duration of therapy is 48 weeks.
- ribavirin is administered in an amount of 1000 mg for individuals weighing less than 75 kg, and 1200 mg for individuals weighing 75 kg or more.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 200 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; and 50 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- One embodiment provides any of the above-described methods modified to comprise administering to an individual having an HCV infection an effective amount of an NS3 inhibitor; and a regimen of 200 ⁇ g monoPEG(30 kD, linear) -ylated consensus IFN-OC administered subcutaneously every 10 days or qw; and 100 ⁇ g Actimmune® human IFN-ylb administered subcutaneously tiw, where the duration of therapy is 48 weeks.
- any of the above-described methods involving administering an NS3 inhibitor, a Type I interferon receptor agonist (e.g., an IFN-Oc), and a Type II interferon receptor agonist (e.g., an IFN- ⁇ ), can be augmented by administration of an effective amount of a TNF-oc antagonist (e.g., a TNF-oc antagonist other than pirfenidone or a pirfenidone analog).
- a TNF-oc antagonists e.g., a TNF-oc antagonist other than pirfenidone or a pirfenidone analog.
- Exemplary, non-limiting TNF-oc antagonists that are suitable for use in such combination therapies include ENBREL®, REMICADE®, and HUMIRATM.
- One embodiment provides a method using an effective amount of ENBREL®; an effective amount of IFN-OC; an effective amount of IFN- ⁇ ; and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage ENBREL® containing an amount of from about 0.1 ⁇ g to about 23 mg per dose, from about 0.1 ⁇ g to about 1 ⁇ g, from about 1 ⁇ g to about 10 ⁇ g, from about 10 ⁇ g to about 100 ⁇ g, from about 100 ⁇ g to about 1 mg, from about 1 mg to about 5 mg, from about 5 mg to about 10 mg, from about 10 mg to about 15 mg, from about 15 mg to about 20 mg, or from about 20 mg to about 23 mg of ENBREL®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or once every other month, or per day substantially continuously or continuously, for the desired duration of treatment.
- One embodiment provides a method using an effective amount of REMICADE®, an effective amount of IFN-OC; an effective amount of IFN- ⁇ ; and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage of REMICADE® containing an amount of from about 0.1 mg/kg to about 4.5 mg/kg, from about 0.1 mg/kg to about 0.5 mg/kg, from about 0.5 mg/kg to about 1.0 mg/kg, from about 1.0 mg/kg to about 1.5 mg/kg, from about 1.5 mg/kg to about 2.0 mg/kg, from about 2.0 mg/kg to about 2.5 mg/kg, from about 2.5 mg/kg to about 3.0 mg/kg, from about 3.0 mg/kg to about 3.5 mg/kg, from about 3.5 mg/kg to about 4.0 mg/kg, or from about 4.0 mg/kg to about 4.5 mg/kg per dose of REMICADE®, intravenously qd, qod, tiw, biw, q
- One embodiment provides a method using an effective amount of HUMIRATM, an effective amount of IFN-OC; an effective amount of IFN- ⁇ ; and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage of HUMIRATM containing an amount of from about 0.1 ⁇ g to about 35 mg, from about 0.1 ⁇ g to about 1 ⁇ g, from about 1 ⁇ g to about 10 ⁇ g, from about 10 ⁇ g to about 100 ⁇ g, from about 100 ⁇ g to about 1 mg, from about 1 mg to about 5 mg, from about 5 mg to about 10 mg, from about 10 mg to about 15 mg, from about 15 mg to about 20 mg, from about 20 mg to about 25 mg, from about 25 mg to about 30 mg, or from about 30 mg to about 35 mg per dose of a HUMIRATM, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or once
- the methods provide for combination therapy comprising administering an NS3 inhibitor compound as described above, and an effective amount of pirfenidone or a pirfenidone analog.
- an NS3 inhibitor compound, one or more interferon receptor agonist(s), and pirfenidone or pirfenidone analog are co-administered in the treatment methods of the embodiments.
- an NS3 inhibitor compound, a Type I interferon receptor agonist, and pirfenidone (or a pirfenidone analog) are co-administered.
- an NS3 inhibitor compound, a Type I interferon receptor agonist, a Type II interferon receptor agonist, and pirfenidone (or a pirfenidone analog) are co-administered.
- Type I interferon receptor agonists suitable for use herein include any IFN-a, such as interferon alfa-2a, interferon alfa-2b, interferon alfacon-1, and PEGylated IFN-a' s, such as peginterferon alfa-2a, peginterferon alfa-2b, and PEGylated consensus interferons, such as monoPEG (30 kD, linear)-ylated consensus interferon.
- Type II interferon receptor agonists suitable for use herein include any interferon- ⁇ .
- Pirfenidone or a pirfenidone analog can be administered once per month, twice per month, three times per month, once per week, twice per week, three times per week, four times per week, five times per week, six times per week, daily, or in divided daily doses ranging from once daily to 5 times daily over a period of time ranging from about one day to about one week, from about two weeks to about four weeks, from about one month to about two months, from about two months to about four months, from about four months to about six months, from about six months to about eight months, from about eight months to about 1 year, from about 1 year to about 2 years, or from about 2 years to about 4 years, or more.
- Effective dosages of pirfenidone or a specific pirfenidone analog include a weight-based dosage in the range from about 5 mg/kg/day to about 125 mg/kg/day, or a fixed dosage of about 400 mg to about 3600 mg per day, or about 800 mg to about 2400 mg per day, or about 1000 mg to about 1800 mg per day, or about 1200 mg to about 1600 mg per day, administered orally in one to five divided doses per day.
- Other doses and formulations of pirfenidone and specific pirfenidone analogs suitable for use in the treatment of fibrotic diseases are described in U.S. Pat. Nos., 5,310,562; 5,518,729; 5,716,632; and 6,090,822.
- One embodiment provides any of the above-described methods modified to include co-administering to the patient a therapeutically effective amount of pirfenidone or a pirfenidone analog for the duration of the desired course of NS3 inhibitor compound treatment.
- the methods provide for combination therapy comprising administering an effective amount of an NS3 inhibitor compound as described above, and an effective amount of TNF-oc antagonist, in combination therapy for treatment of an HCV infection.
- Effective dosages of a TNF-oc antagonist range from 0.1 ⁇ g to 40 mg per dose, e.g., from about 0.1 ⁇ g to about 0.5 ⁇ g per dose, from about 0.5 ⁇ g to about 1.0 ⁇ g per dose, from about 1.0 ⁇ g per dose to about 5.0 ⁇ g per dose, from about 5.0 ⁇ g to about 10 ⁇ g per dose, from about 10 ⁇ g to about 20 ⁇ g per dose, from about 20 ⁇ g per dose to about 30 ⁇ g per dose, from about 30 ⁇ g per dose to about 40 ⁇ g per dose, from about 40 ⁇ g per dose to about 50 ⁇ g per dose, from about 50 ⁇ g per dose to about 60 ⁇ g per dose, from about 60 ⁇ g per dose to about 70 ⁇ g per dose, from about 70 ⁇ g to about 80 ⁇ g per dose, from about 80 ⁇ g per dose to about 100 ⁇ g per dose, from about 100 ⁇ g to about 150 ⁇ g per dose, from about 150 ⁇ g per
- effective dosages of a TNF-oc antagonist are expressed as mg/kg body weight.
- effective dosages of a TNF-oc antagonist are from about 0.1 mg/kg body weight to about 10 mg/kg body weight, e.g., from about 0.1 mg/kg body weight to about 0.5 mg/kg body weight, from about 0.5 mg/kg body weight to about 1.0 mg/kg body weight, from about 1.0 mg/kg body weight to about 2.5 mg/kg body weight, from about 2.5 mg/kg body weight to about 5.0 mg/kg body weight, from about 5.0 mg/kg body weight to about 7.5 mg/kg body weight, or from about 7.5 mg/kg body weight to about 10 mg/kg body weight.
- a TNF-oc antagonist is administered for a period of about 1 day to about 7 days, or about 1 week to about 2 weeks, or about 2 weeks to about 3 weeks, or about 3 weeks to about 4 weeks, or about 1 month to about 2 months, or about 3 months to about 4 months, or about 4 months to about 6 months, or about 6 months to about 8 months, or about 8 months to about 12 months, or at least one year, and may be administered over longer periods of time.
- the TNF-oc antagonist can be administered tid, bid, qd, qod, biw, tiw, qw, qow, three times per month, once monthly, substantially continuously, or continuously.
- a TNF-oc antagonist is administered once per month, twice per month, three times per month, every other week (qow), once per week (qw), twice per week (biw), three times per week (tiw), four times per week, five times per week, six times per week, every other day (qod), daily (qd), twice a day (bid), or three times a day (tid), substantially continuously, or continuously, over a period of time ranging from about one day to about one week, from about two weeks to about four weeks, from about one month to about two months, from about two months to about four months, from about four months to about six months, from about six months to about eight months, from about eight months to about 1 year, from about 1 year to about 2 years, or from about 2 years to about 4 years, or more.
- a TNF-oc antagonist and an NS3 inhibitor are generally administered in separate formulations.
- a TNF-oc antagonist and an NS3 inhibitor may be administered substantially simultaneously, or within about 30 minutes, about 1 hour, about 2 hours, about 4 hours, about 8 hours, about 16 hours, about 24 hours, about 36 hours, about 72 hours, about 4 days, about 7 days, or about 2 weeks of one another.
- One embodiment provides a method using an effective amount of a TNF-oc antagonist and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage of a TNF-oc antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF- OC antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- One embodiment provides a method using an effective amount of ENBREL® and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage ENBREL® containing an amount of from about 0.1 ⁇ g to about 23 mg per dose, from about 0.1 ⁇ g to about 1 ⁇ g, from about 1 ⁇ g to about 10 ⁇ g, from about 10 ⁇ g to about 100 ⁇ g, from about 100 ⁇ g to about 1 mg, from about 1 mg to about 5 mg, from about 5 mg to about 10 mg, from about 10 mg to about 15 mg, from about 15 mg to about 20 mg, or from about 20 mg to about 23 mg of ENBREL®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or once every other month, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- One embodiment provides a method using an effective amount of REMICADE® and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage of REMICADE® containing an amount of from about 0.1 mg/kg to about 4.5 mg/kg, from about 0.1 mg/kg to about 0.5 mg/kg, from about 0.5 mg/kg to about 1.0 mg/kg, from about 1.0 mg/kg to about 1.5 mg/kg, from about 1.5 mg/kg to about 2.0 mg/kg, from about 2.0 mg/kg to about 2.5 mg/kg, from about 2.5 mg/kg to about 3.0 mg/kg, from about 3.0 mg/kg to about 3.5 mg/kg, from about 3.5 mg/kg to about 4.0 mg/kg, or from about 4.0 mg/kg to about 4.5 mg/kg per dose of REMICADE®, intravenously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or once every other month
- One embodiment provides a method using an effective amount of HUMIRATM and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage of HUMIRATM containing an amount of from about 0.1 ⁇ g to about 35 mg, from about 0.1 ⁇ g to about 1 ⁇ g, from about 1 ⁇ g to about 10 ⁇ g, from about 10 ⁇ g to about 100 ⁇ g, from about 100 ⁇ g to about 1 mg, from about 1 mg to about 5 mg, from about 5 mg to about 10 mg, from about 10 mg to about 15 mg, from about 15 mg to about 20 mg, from about 20 mg to about 25 mg, from about 25 mg to about 30 mg, or from about 30 mg to about 35 mg per dose of a HUMIRATM, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or once every other month, or per day substantially continuously or continuously, for the desired duration of treatment with
- the methods provide for combination therapy comprising administering an effective amount of an NS3 inhibitor compound as described above, and an effective amount of thymosin-oc, in combination therapy for treatment of an HCV infection.
- Effective dosages of thymosin-oc range from about 0.5 mg to about 5 mg, e.g., from about 0.5 mg to about 1.0 mg, from about 1.0 mg to about 1.5 mg, from about 1.5 mg to about 2.0 mg, from about 2.0 mg to about 2.5 mg, from about 2.5 mg to about 3.0 mg, from about 3.0 mg to about 3.5 mg, from about 3.5 mg to about 4.0 mg, from about 4.0 mg to about 4.5 mg, or from about 4.5 mg to about 5.0 mg.
- thymosin-oc is administered in dosages containing an amount of 1.0 mg or 1.6 mg.
- One embodiment provides a method using an effective amount of ZADAXINTM thymosin-oc and an effective amount of an NS3 inhibitor in the treatment of an HCV infection in a patient, comprising administering to the patient a dosage of ZADAXINTM containing an amount of from about 1.0 mg to about 1.6 mg per dose, subcutaneously twice per week for the desired duration of treatment with the NS3 inhibitor compound.
- Some embodiments provide a method of treating an HCV infection in an individual having an HCV infection, the method comprising administering an effective amount of an NS3 inhibitor, and effective amount of a TNF-a antagonist, and an effective amount of one or more interferons.
- One embodiment provides any of the above-described methods modified to use an effective amount of IFN- ⁇ and an effective amount of a TNF-a antagonist in the treatment of HCV infection in a patient comprising administering to the patient a dosage of IFN- ⁇ containing an amount of about 10 ⁇ g to about 300 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- One embodiment provides any of the above-described methods modified to use an effective amount of IFN- ⁇ and an effective amount of a TNF-a antagonist in the treatment of HCV infection in a patient comprising administering to the patient a dosage of IFN- ⁇ containing an amount of about 10 ⁇ g to about 100 ⁇ g of drug per dose of IFN- ⁇ , subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN- ⁇ and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a total weekly dosage of IFN- ⁇ containing an amount of about 30 ⁇ g to about 1,000 ⁇ g of drug per week in divided doses administered subcutaneously qd, qod, tiw, biw, or administered substantially continuously or continuously, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN- ⁇ and an effective amount of a TNF-oc antagonist in the treatment of a virus infection in a patient comprising administering to the patient a total weekly dosage of IFN- ⁇ containing an amount of about 100 ⁇ g to about 300 ⁇ g of drug per week in divided doses administered subcutaneously qd, qod, tiw, biw, or administered substantially continuously or continuously, in combination with a dosage of a TNF-oc antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-oc antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- One embodiment provides any of the above-described methods modified to use an effective amount of INFERGEN® consensus IFN-OC and a TNF-oc antagonist in the treatment of HCV infection in a patient comprising administering to the patient a dosage of INFERGEN® containing an amount of about 1 ⁇ g to about 30 ⁇ g, of drug per dose of INFERGEN®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of a TNF-oc antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-oc antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- One embodiment provides any of the above-described methods modified to use an effective amount of INFERGEN® consensus IFN-OC and a TNF-oc antagonist in the treatment of HCV infection in a patient comprising administering to the patient a dosage of INFERGEN® containing an amount of about 1 ⁇ g to about 9 ⁇ g, of drug per dose of INFERGEN®, subcutaneously qd, qod, tiw, biw, qw, qow, three times per month, once monthly, or per day substantially continuously or continuously, in combination with a dosage of a TNF-oc antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-oc antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGylated consensus IFN-a and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGylated consensus IFN-a (PEG-CIFN) containing an amount of about 4 ⁇ g to about 60 ⁇ g of CIFN amino acid weight per dose of PEG-CIFN, subcutaneously qw, qow, three times per month, or monthly, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- PEG-CIFN PEGylated consensus IFN-a
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGylated consensus IFN-a and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGylated consensus IFN-a (PEG-CIFN) containing an amount of about 18 ⁇ g to about 24 ⁇ g of CIFN amino acid weight per dose of PEG-CIFN, subcutaneously qw, qow, three times per month, or monthly, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- PEG-CIFN PEGylated consensus IFN-a
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN-a 2a or 2b or 2c and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of IFN-a 2a, 2b or 2c containing an amount of about 1 MU to about 20 MU of drug per dose of IFN-a 2a, 2b or 2c subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN-a 2a or 2b or 2c and an effective amount of a TNF-oc antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of IFN-OC 2a, 2b or 2c containing an amount of about 3 MU of drug per dose of IFN-OC 2a, 2b or 2c subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF- ⁇ antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of IFN-a 2a or 2b or 2c and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of IFN-a 2a, 2b or 2c containing an amount of about 10 MU of drug per dose of IFN-a 2a, 2b or 2c subcutaneously qd, qod, tiw, biw, or per day substantially continuously or continuously, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF- ⁇ antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGASYS®PEGylated IFN-a2a and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGASYS® containing an amount of about 90 ⁇ g to about 360 ⁇ g, of drug per dose of PEGASYS®, subcutaneously qw, qow, three times per month, or monthly, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEGASYS®PEGylated IFN-a2a and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEGASYS® containing an amount of about 180 ⁇ g, of drug per dose of PEGASYS®, subcutaneously qw, qow, three times per month, or monthly, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEG-INTRON®PEGylated IFN-Cc2b and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEG-INTRON® containing an amount of about 0.75 ⁇ g to about 3.0 ⁇ g of drug per kilogram of body weight per dose of PEG-INTRON®, subcutaneously qw, qow, three times per month, or monthly, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- Another embodiment provides any of the above-described methods modified to use an effective amount of PEG-INTRON®PEGylated IFN-a2b and an effective amount of a TNF-a antagonist in the treatment of a virus infection in a patient comprising administering to the patient a dosage of PEG-INTRON® containing an amount of about 1.5 ⁇ g of drug per kilogram of body weight per dose of PEG-INTRON®, subcutaneously qw, qow, three times per month, or monthly, in combination with a dosage of a TNF-a antagonist containing an amount of from about 0.1 ⁇ g to about 40 mg per dose of a TNF-a antagonist, subcutaneously qd, qod, tiw, or biw, or per day substantially continuously or continuously, for the desired duration of treatment with an NS3 inhibitor compound.
- HCV NS3 helicase Other agents such as inhibitors of HCV NS3 helicase are also attractive drugs for combinational therapy, and are contemplated for use in combination therapies described herein.
- Ribozymes such as HeptazymeTM and phosphorothioate oligonucleotides which are complementary to HCV protein sequences and which inhibit the expression of viral core proteins are also suitable for use in combination therapies described herein.
- the additional antiviral agent(s) is administered during the entire course of treatment with the NS3 inhibitor compound described herein, and the beginning and end of the treatment periods coincide. In other embodiments, the additional antiviral agent(s) is administered for a period of time that is overlapping with that of the NS3 inhibitor compound treatment, e.g., treatment with the additional antiviral agent(s) begins before the NS3 inhibitor compound treatment begins and ends before the NS3 inhibitor compound treatment ends; treatment with the additional antiviral agent(s) begins after the NS3 inhibitor compound treatment begins and ends after the NS3 inhibitor compound treatment ends; treatment with the additional antiviral agent(s) begins after the NS3 inhibitor compound treatment begins and ends before the NS3 inhibitor compound treatment ends; or treatment with the additional antiviral agent(s) begins before the NS3 inhibitor compound treatment begins and ends after the NS3 inhibitor compound treatment ends.
- the NS3 inhibitor compound can be administered together with (i.e., simultaneously in separate formulations; simultaneously in the same formulation; administered in separate formulations and within about 48 hours, within about 36 hours, within about 24 hours, within about 16 hours, within about 12 hours, within about 8 hours, within about 4 hours, within about 2 hours, within about 1 hour, within about 30 minutes, or within about 15 minutes or less) one or more additional antiviral agents.
- any of the above-described methods featuring an IFN-a regimen can be modified to replace the subject IFN-a regimen with a regimen of monoPEG (30 kD, linear)-ylated consensus IFN-a comprising administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-a containing an amount of 100 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a regimen can be modified to replace the subject IFN-a regimen with a regimen of monoPEG (30 kD, linear)-ylated consensus IFN-a comprising administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-a containing an amount of 150 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a regimen can be modified to replace the subject IFN-a regimen with a regimen of monoPEG (30 kD, linear)-ylated consensus IFN-a comprising administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-a containing an amount of 200 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-OC regimen can be modified to replace the subject IFN-OC regimen with a regimen of INFERGEN® interferon alfacon-1 comprising administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily or three times per week for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-OC regimen can be modified to replace the subject IFN-OC regimen with a regimen of INFERGEN® interferon alfacon-1 comprising administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily or three times per week for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN- ⁇ regimen can be modified to replace the subject IFN- ⁇ regimen with a regimen of IFN- ⁇ comprising administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN- ⁇ regimen can be modified to replace the subject IFN- ⁇ regimen with a regimen of IFN- ⁇ comprising administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN- ⁇ regimen can be modified to replace the subject IFN- ⁇ regimen with a regimen of IFN- ⁇ comprising administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-OC and IFN- ⁇ combination regimen can be modified to replace the subject IFN-OC and IFN- ⁇ combination regimen with an IFN-OC and IFN- ⁇ combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 100 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring a TNF antagonist regimen can be modified to replace the subject TNF antagonist regimen with a TNF antagonist regimen comprising administering a dosage of a TNF antagonist selected from the group of: (a) etanercept in an amount of 25 mg of drug per dose subcutaneously twice per week, (b) infliximab in an amount of 3 mg of drug per kilogram of body weight per dose intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter, or (c) adalimumab in an amount of 40 mg of drug per dose subcutaneously once weekly or once every 2 weeks; for the desired treatment duration with an NS3 inhibitor compound.
- a TNF antagonist selected from the group of: (a) etanercept in an amount of 25 mg of drug per dose subcutaneously twice per week, (b) infliximab in an amount of 3 mg of drug per kilogram of body weight per dose intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter, or (c) adalimumab in
- any of the above-described methods featuring an IFN-OC and IFN- ⁇ combination regimen can be modified to replace the subject IFN-0C and IFN- ⁇ combination regimen with an IFN-OC and IFN- ⁇ combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 100 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-OC and IFN- ⁇ combination regimen can be modified to replace the subject IFN-OC and IFN- ⁇ combination regimen with an IFN-OC and IFN- ⁇ combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 150 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-OC and IFN- ⁇ combination regimen can be modified to replace the subject IFN-OC and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-a containing an amount of 150 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-a containing an amount of 200 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-a containing an amount of 200 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-OC and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily; and (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily; and (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily; and (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily; and (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily; and (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a and IFN- ⁇ combination regimen can be modified to replace the subject IFN-a and IFN- ⁇ combination regimen with an IFN-a and IFN- ⁇ combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily; and (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-a, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-a, IFN- ⁇ and TNF antagonist combination regimen with an IFN- a, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-a containing an amount of 100 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii)
- any of the above-described methods featuring an IFN-a, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-a, IFN- ⁇ and TNF antagonist combination regimen with an IFN- ⁇ , IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 100 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii)
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 150 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii)
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 150 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii)
- any of the above-described methods featuring an IFN-0C, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-0C, IFN- ⁇ and TNF antagonist combination regimen with an IFN- oc, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-0C containing an amount of 200 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (
- any of the above-described methods featuring an IFN-0C, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-0C, IFN- ⁇ and TNF antagonist combination regimen with an IFN- oc, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-0C containing an amount of 200 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (
- any of the above-described methods featuring an IFN-0C, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- oc, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously three times per week; (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg sub
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously three times per week; (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously three times per week; (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily; (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily; (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily; (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly
- any of the above-described methods featuring an IFN-0C, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-0C, IFN- ⁇ and TNF antagonist combination regimen with an IFN- oc, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously three times per week; (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg
- any of the above-described methods featuring an IFN-0C, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-0C, IFN- ⁇ and TNF antagonist combination regimen with an IFN- oc, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously three times per week; (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg
- any of the above-described methods featuring an IFN-0C, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- oc, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously three times per week; (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg sub
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily; (b) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily; (b) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly
- any of the above-described methods featuring an IFN-OC, IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN-OC, IFN- ⁇ and TNF antagonist combination regimen with an IFN- OC, IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily; (b) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (c) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly
- any of the above-described methods featuring an IFN-OC and TNF antagonist combination regimen can be modified to replace the subject IFN-OC and TNF antagonist combination regimen with an IFN-OC and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 100 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-OC and TNF antagonist combination regimen can be modified to replace the subject IFN-OC and TNF antagonist combination regimen with an IFN-OC and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-OC containing an amount of 150 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-0C and TNF antagonist combination regimen can be modified to replace the subject IFN-0C and TNF antagonist combination regimen with an IFN-0C and TNF antagonist combination regimen comprising: (a) administering a dosage of monoPEG (30 kD, linear) -ylated consensus IFN-0C containing an amount of 200 ⁇ g of drug per dose, subcutaneously once weekly, once every 8 days, or once every 10 days; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN-0C and TNF antagonist combination regimen can be modified to replace the subject IFN-0C and TNF antagonist combination regimen with an IFN-0C and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, subcutaneously once daily or three times per week; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- a dosage of INFERGEN® interferon alfacon-1 containing an amount of 9 ⁇ g of drug per dose, sub
- any of the above-described methods featuring an IFN-0C and TNF antagonist combination regimen can be modified to replace the subject IFN-0C and TNF antagonist combination regimen with an IFN-0C and TNF antagonist combination regimen comprising: (a) administering a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, subcutaneously once daily or three times per week; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- a dosage of INFERGEN® interferon alfacon-1 containing an amount of 15 ⁇ g of drug per dose, sub
- any of the above-described methods featuring an IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN- ⁇ and TNF antagonist combination regimen with an IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of IFN- ⁇ containing an amount of 25 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN- ⁇ and TNF antagonist combination regimen with an IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of IFN- ⁇ containing an amount of 50 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an IFN- ⁇ and TNF antagonist combination regimen can be modified to replace the subject IFN- ⁇ and TNF antagonist combination regimen with an IFN- ⁇ and TNF antagonist combination regimen comprising: (a) administering a dosage of IFN- ⁇ containing an amount of 100 ⁇ g of drug per dose, subcutaneously three times per week; and (b) administering a dosage of a TNF antagonist selected from (i) etanercept in an amount of 25 mg subcutaneously twice per week, (ii) infliximab in an amount of 3 mg of drug per kilogram of body weight intravenously at weeks 0, 2 and 6, and every 8 weeks thereafter or (iii) adalimumab in an amount of 40 mg subcutaneously once weekly or once every other week; for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods that includes a regimen of monoPEG (30 kD, linear)-ylated consensus IFN-0C can be modified to replace the regimen of monoPEG (30 kD, linear)-ylated consensus IFN-0C with a regimen of peginterferon alfa-2a comprising administering a dosage of peginterferon alfa- 2a containing an amount of 180 ⁇ g of drug per dose, subcutaneously once weekly for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods that includes a regimen of monoPEG (30 kD, linear)-ylated consensus IFN-0C can be modified to replace the regimen of monoPEG (30 kD, linear)-ylated consensus IFN-OC with a regimen of peginterferon alfa-2b comprising administering a dosage of peginterferon alfa- 2b containing an amount of 1.0 ⁇ g to 1.5 g of drug per kilogram of body weight per dose, subcutaneously once or twice weekly for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods can be modified to include administering a dosage of ribavirin containing an amount of 400 mg, 800 mg, 1000 mg or 1200 mg of drug orally per day, optionally in two or more divided doses per day, for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods can be modified to include administering a dosage of ribavirin containing (i) an amount of 1000 mg of drug orally per day for patients having a body weight of less than 75 kg or (ii) an amount of 1200 mg of drug orally per day for patients having a body weight of greater than or equal to 75 kg, optionally in two or more divided doses per day, for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods can be modified to replace the subject NS3 inhibitor regimen with an NS3 inhibitor regimen comprising administering a dosage of 0.01 mg to 0.1 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with the NS3 inhibitor compound.
- any of the above-described methods can be modified to replace the subject NS3 inhibitor regimen with an NS3 inhibitor regimen comprising administering a dosage of 0.1 mg to 1 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with the NS3 inhibitor compound.
- any of the above-described methods can be modified to replace the subject NS3 inhibitor regimen with an NS3 inhibitor regimen comprising administering a dosage of 1 mg to 10 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with the NS3 inhibitor compound.
- any of the above-described methods can be modified to replace the subject NS3 inhibitor regimen with an NS3 inhibitor regimen comprising administering a dosage of 10 mg to 100 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with the NS3 inhibitor compound.
- any of the above-described methods featuring an NS5B inhibitor regimen can be modified to replace the subject NS5B inhibitor regimen with an NS5B inhibitor regimen comprising administering a dosage of 0.01 mg to 0.1 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an NS5B inhibitor regimen can be modified to replace the subject NS5B inhibitor regimen with an NS5B inhibitor regimen comprising administering a dosage of 0.1 mg to 1 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an NS5B inhibitor regimen can be modified to replace the subject NS5B inhibitor regimen with an NS5B inhibitor regimen comprising administering a dosage of 1 mg to 10 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with an NS3 inhibitor compound.
- any of the above-described methods featuring an NS5B inhibitor regimen can be modified to replace the subject NS5B inhibitor regimen with an NS5B inhibitor regimen comprising administering a dosage of 10 mg to 100 mg of drug per kilogram of body weight orally daily, optionally in two or more divided doses per day, for the desired treatment duration with an NS3 inhibitor compound.
- the present embodiments provide for a method of treating a hepatitis C virus infection comprising administering to a human dosages of peginterferon alfa-2a and ribavirin under a standard of care protocol (SOC) in combination with ITMN-191 or a pharmaceutically acceptable salt thereof.
- SOC standard of care protocol
- ITMN-191 or a pharmaceutically acceptable salt thereof The chemical structure of ⁇ -191 is shown below.
- the peginterferon alfa-2a and ribavirin in combination with ⁇ -191 or a pharmaceutically acceptable salt thereof are administered in combination and provide HCV RNA levels below about 43 IU/mL, below about 25 IU/mL, or below about 9.3 IU/mL after 14 days of treatment.
- the dosage of peginterferon alfa-2a can be about 180 ⁇ g of peginterferon alfa-2a per dose, administered subcutaneously once weekly for the desired treatment duration. In some embodiments, the dosage of peginterferon alfa-2a can be an amount in the range of about 1.0 ⁇ g to about 1.5 g of drug per kilogram of body weight per dose, subcutaneously once or twice weekly for the desired treatment duration with the ITMN-191 and the ribavarin.
- the dosage of ribavirin can be about 400 mg, about 800 mg, about 1000 mg or about 1200 mg of drug orally per day, optionally in two or more divided doses per day, for the desired treatment duration with the peginterferon alfa-2a and ⁇ -191.
- the dosage of ribavirin can be an amount of about 1000 mg of drug orally per day for patients having a body weight of less than 75 kg or an amount of about 1200 mg of drug orally per day for patients having a body weight of greater than or equal to 75 kg, optionally in two or more divided doses per day, for the desired treatment duration with the peginterferon alfa-2a and ITMN-191.
- the amounts of peginterferon alfa-2a and ribavirin administered in the SOC protocol can be lowered due to combination with ⁇ -191.
- the amounts of peginterferon alfa-2a and ribavirin can be reduced below the SOC by about 10% to about 75% during the combination treatment.
- the specific regimen of drug therapy used in treatment of the HCV patient is selected according to certain disease parameters exhibited by the patient, such as the initial viral load, genotype of the HCV infection in the patient, liver histology and/or stage of liver fibrosis in the patient.
- some embodiments provide any of the above-described methods for the treatment of HCV infection in which the subject method is modified to treat a treatment failure patient for a duration of 48 weeks.
- HCVL high viral load
- One embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having advanced or severe stage liver fibrosis as measured by a Knodell score of 3 or 4 and then (2) administering to the patient the drug therapy of the subject method for a time period of about 24 weeks to about 60 weeks, or about 30 weeks to about one year, or about 36 weeks to about 50 weeks, or about 40 weeks to about 48 weeks, or at least about 24 weeks, or at least about 30 weeks, or at least about 36 weeks, or at least about 40 weeks, or at least about 48 weeks, or at least about 60 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having advanced or severe stage liver fibrosis as measured by a Knodell score of 3 or 4 and then (2) administering to the patient the drug therapy of the subject method for a time period of about 40 weeks to about 50 weeks, or about 48 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 infection and an initial viral load of greater than 2 million viral genome copies per mL of patient serum and then (2) administering to the patient the drug therapy of the subject method for a time period of about 24 weeks to about 60 weeks, or about 30 weeks to about one year, or about 36 weeks to about 50 weeks, or about 40 weeks to about 48 weeks, or at least about 24 weeks, or at least about 30 weeks, or at least about 36 weeks, or at least about 40 weeks, or at least about 48 weeks, or at least about 60 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 infection and an initial viral load of greater than 2 million viral genome copies per mL of patient serum and then (2) administering to the patient the drug therapy of the subject method for a time period of about 40 weeks to about 50 weeks, or about 48 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 infection and an initial viral load of greater than 2 million viral genome copies per mL of patient serum and no or early stage liver fibrosis as measured by a Knodell score of 0, 1, or 2 and then (2) administering to the patient the drug therapy of the subject method for a time period of about 24 weeks to about 60 weeks, or about 30 weeks to about one year, or about 36 weeks to about 50 weeks, or about 40 weeks to about 48 weeks, or at least about 24 weeks, or at least about 30 weeks, or at least about 36 weeks, or at least about 40 weeks, or at least about 48 weeks, or at least about 60 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 infection and an initial viral load of greater than 2 million viral genome copies per mL of patient serum and no or early stage liver fibrosis as measured by a Knodell score of 0, 1, or 2 and then (2) administering to the patient the drug therapy of the subject method for a time period of about 40 weeks to about 50 weeks, or about 48 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 infection and an initial viral load of less than or equal to 2 million viral genome copies per mL of patient serum and then (2) administering to the patient the drug therapy of the subject method for a time period of about 20 weeks to about 50 weeks, or about 24 weeks to about 48 weeks, or about 30 weeks to about 40 weeks, or up to about 20 weeks, or up to about 24 weeks, or up to about 30 weeks, or up to about 36 weeks, or up to about 48 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 infection and an initial viral load of less than or equal to 2 million viral genome copies per mL of patient serum and then (2) administering to the patient the drug therapy of the subject method for a time period of about 20 weeks to about 24 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 infection and an initial viral load of less than or equal to 2 million viral genome copies per mL of patient serum and then (2) administering to the patient the drug therapy of the subject method for a time period of about 24 weeks to about 48 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 2 or 3 infection and then (2) administering to the patient the drug therapy of the subject method for a time period of about 24 weeks to about 60 weeks, or about 30 weeks to about one year, or about 36 weeks to about 50 weeks, or about 40 weeks to about 48 weeks, or at least about 24 weeks, or at least about 30 weeks, or at least about 36 weeks, or at least about 40 weeks, or at least about 48 weeks, or at least about 60 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 2 or 3 infection and then (2) administering to the patient the drug therapy of the subject method for a time period of about 20 weeks to about 50 weeks, or about 24 weeks to about 48 weeks, or about 30 weeks to about 40 weeks, or up to about 20 weeks, or up to about 24 weeks, or up to about 30 weeks, or up to about 36 weeks, or up to about 48 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 2 or 3 infection and then (2) administering to the patient the drug therapy of the subject method for a time period of about 20 weeks to about 24 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 2 or 3 infection and then (2) administering to the patient the drug therapy of the subject method for a time period of at least about 24 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV genotype 1 or 4 infection and then (2) administering to the patient the drug therapy of the subject method for a time period of about 24 weeks to about 60 weeks, or about 30 weeks to about one year, or about 36 weeks to about 50 weeks, or about 40 weeks to about 48 weeks, or at least about 24 weeks, or at least about 30 weeks, or at least about 36 weeks, or at least about 40 weeks, or at least about 48 weeks, or at least about 60 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV infection characterized by any of HCV genotypes 5, 6, 7, 8 and 9 and then (2) administering to the patient the drug therapy of the subject method for a time period of about 20 weeks to about 50 weeks.
- Another embodiment provides any of the above-described methods for the treatment of an HCV infection, where the subject method is modified to include the steps of (1) identifying a patient having an HCV infection characterized by any of HCV genotypes 5, 6, 7, 8 and 9 and then (2) administering to the patient the drug therapy of the subject method for a time period of at least about 24 weeks and up to about 48 weeks.
- Any of the above treatment regimens can be administered to individuals who have been diagnosed with an HCV infection. Any of the above treatment regimens can be administered to individuals who have failed previous treatment for HCV infection ("treatment failure patients," including non-responders and relapsers).
- Individuals who have been clinically diagnosed as infected with HCV are of particular interest in many embodiments.
- Individuals who are infected with HCV are identified as having HCV RNA in their blood, and/or having anti-HCV antibody in their serum.
- Such individuals include anti-HCV ELISA-positive individuals, and individuals with a positive recombinant immunoblot assay (RIBA).
- RIBA positive recombinant immunoblot assay
- Individuals who are clinically diagnosed as infected with HCV include naive individuals (e.g., individuals not previously treated for HCV, particularly those who have not previously received IFN-OC-based and/or ribavirin-based therapy) and individuals who have failed prior treatment for HCV ("treatment failure" patients).
- naive individuals e.g., individuals not previously treated for HCV, particularly those who have not previously received IFN-OC-based and/or ribavirin-based therapy
- treatment failure individuals who have failed prior treatment for HCV
- Treatment failure patients include non-responders (i.e., individuals in whom the HCV titer was not significantly or sufficiently reduced by a previous treatment for HCV, e.g., a previous IFN-a monotherapy, a previous IFN-a and ribavirin combination therapy, or a previous pegylated IFN-a and ribavirin combination therapy); and relapsers (i.e., individuals who were previously treated for HCV, e.g., who received a previous IFN-a monotherapy, a previous IFN-a and ribavirin combination therapy, or a previous pegylated IFN-a and ribavirin combination therapy, whose HCV titer decreased, and subsequently increased).
- non-responders i.e., individuals in whom the HCV titer was not significantly or sufficiently reduced by a previous treatment for HCV, e.g., a previous IFN-a monotherapy, a previous IFN-a and ribavirin combination therapy,
- individuals have an HCV titer of at least about 10 5 , at least about 5 x 10 5 , or at least about 10 6 , or at least about 2 x 10 6 , genome copies of HCV per milliliter of serum.
- the patient may be infected with any HCV genotype (genotype 1, including la and lb, 2, 3, 4, 6, etc. and subtypes (e.g., 2a, 2b, 3a, etc.)), particularly a difficult to treat genotype such as HCV genotype 1 and particular HCV subtypes and quasispecies.
- HCV-positive individuals are HCV-positive individuals (as described above) who exhibit severe fibrosis or early cirrhosis (non-decompensated, Child' s-Pugh class A or less), or more advanced cirrhosis (decompensated, Child' s-Pugh class B or C) due to chronic HCV infection and who are viremic despite prior anti-viral treatment with IFN-0C- based therapies or who cannot tolerate IFN-OC-based therapies, or who have a contraindication to such therapies.
- HCV-positive individuals with stage 3 or 4 liver fibrosis according to the METAVIR scoring system are suitable for treatment with the methods described herein.
- individuals suitable for treatment with the methods of the embodiments are patients with decompensated cirrhosis with clinical manifestations, including patients with far- advanced liver cirrhosis, including those awaiting liver transplantation.
- individuals suitable for treatment with the methods described herein include patients with milder degrees of fibrosis including those with early fibrosis (stages 1 and 2 in the METAVIR, Ludwig, and Scheuer scoring systems; or stages 1, 2, or 3 in the Ishak scoring system.).
- HCV protease inhibitors in the following sections can be prepared according to the procedures and schemes shown in each section.
- the numberings in each of the following Preparation of NS3 Inhibitor sections including the General Method or General Procedure designations, are meant for that specific section only, and should not be construed or confused with the same numberings, if any, in other sections.
- 3-Alkoxy- 2-alkyl-anilines such as 3-methoxy-2-methyl-aniline
- Lewis acids for example boron trichloride and aluminum trichloride
- the 2-alkyl-3-alkoxy-6-acetyl-anilines can be coupled to an an optionally substituted thiazole-2-carboxylic acid chloride, such as 4- isopropylthiazole-2-carbonyl chloride to provide an optionally substiuted l-acetyl-2- [(thiazol-2-yl)-carbonylamino]-3-alkyl-4-alkoxy-benzene, such as l-acetyl-2-[(4- isopropyl-thiazol-2-yl)-carbonylamino] -3-methyl-4-methoxy-benzene.
- thiazole-2-carboxylic acid chloride such as 4- isopropylthiazole-2-carbonyl chloride to provide an optionally substiuted l-acetyl-2- [(thiazol-2-yl)-carbonylamino]-3-alkyl-4-alkoxy-benzene, such as l-acetyl-2
- the optionally substiuted l-acetyl-2-[(thiazol-2-yl)-carbonylamino]-3-alkyl-4-alkoxy-benzene such as 1- acetyl-2-[(4-isopropyl-thiazol-2-yl)-carbonylamino]-3-methyl-4-methoxy-benzene, can be cyclized under basic conditions, for example sodium tert-butoxide in tert-butanol, to provide an optionally substituted 2-(thiazol-2-yl)-4-hydroxy-7-alkoxy-8-alkyl-quinoline, such as 2-(4-isopropylthiazol-2-yl)-4-hydroxy-7-methoxy-8-methyl-quinoline.
- an optionally substituted 2-(thiazol-2-yl)-4-hydroxy-7-alkoxy-8-alkyl-quinoline such as 2-(4-isopropylthiazol-2-yl)-4-hydroxy-7-methoxy-8-methyl-quinoline
- a chlorinating agent for example phosphorous oxychloride, oxalyl chloride, thionyl chloride and the like
- 2-(thiazol-2-yl)-4-chloro-7- alkoxy-8-alkyl-quinoline 2-phenyl-4-chloro-7-alkoxy-quinolines, such as 2-(4- isopropylthiazol-2-yl)-4-chloro-7-methoxy-8-methyl-quinoline.
- reaction mixture was stirred for 30 minutes at 0 °C, then acetonitrile (4.06 mL, 77.71 mmol., 2.6 eq.) was added dropwise keeping the reaction mixture in the range 0-10 °C. Stirring was continued for a further 30 minutes keeping the temperature bellow 10 °C.
- the reaction mixture was transferred to a dropping funnel, using dichloromethane (20 mL) to rinse the initial reaction flask. This solution was added dropwise to a stirred suspension of aluminium trichloride (4.18 g, 31.38 mmol., 1.05 eq.) in dichloromethane (10 mL) at 0 °C. The resulting reaction mixture was then heated under reflux for 15 hours.
- reaction mixture was cooled to 0 °C and ice cold 2M hydrochloric acid (120 mL) was slowly added giving a light yellow suspension. The suspension was then stirred at 80 °C for around 90 minutes until a clear yellow solution was obtained. The reaction mixture was left to cool to ambient temperature and extracted with dichloromethane (3 x 100 mL). The organic extracts were combined, dried over sodium sulphate, filtered and the solvent removed under vacuum. The obtained solid was washed with diethyl ether (2 x 5 mL) and collected by filtration to give 2.31 g (43%) of the title compound as a beige solid.
- a tube (40 mL) was charged with compound 30 (850 mg, 1.5 mmol), Cul (57 mg, 0.3 mmol), L-proline (69 mg, 0.6 mmol) and K 2 CO 3 (1.24 g, 9 mmol), evacuated and backfilled with argon.
- DMSO (10 mL) and l-tert-butyl-3-iodobenzene 31 (1.95 g, 7.5 mmol) were added successively.
- the tube was sealed and heated at 70°C for 48 hours.
- LCMS monitored the reaction, after material was consumed, the reaction mixture was cooled to r.t. and diluted with ethyl acetate (200 mL), filtered.
- the isoindoline carbamate 16 can be synthesized according to WO 2008/137779.
- Compound 16 can be treated with acid, for example TFA in DCM, to remove the Boc protecting group thereby providing compound 17.
- Compound 17 can be treated with optionally substituted aryl boronic acids under Cu 2+ -catalyzed conditions thereby providing isoindoline carbamates having general structure 18.
- the isoindoline carbamate having general structure 18 can be treated under basic conditions, for example aqueous sodium hydroxide in methanol, to hydrolyse the isoindoline carbamate thereby providing alcohols having general structure Formula 2 A.
- Compound 77 was prepared using methods described in PCT Publication No. WO 2007/015824, which is incorporated herein by reference in its entirety. To a solution of compound 77 (leq) in 4mL of DMSO was added t-BuOK (5 eq.). The resulting mixture was stirred at room temperature for 1.5h before the addition of compound 56 (1.5 eq.), and it was stirred overnight. The reaction was quenched with water (10 mL), extracted with ethyl acetate, washed with brine, dried over Na 2 S0 4 , concentrated to get a residue, which was purified by prep-HPLC to give target compound. 71.3 mg, 28.0 %. MS (ESI) m / z (M+H) + 728.1.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Engineering & Computer Science (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Gastroenterology & Hepatology (AREA)
- Biotechnology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description












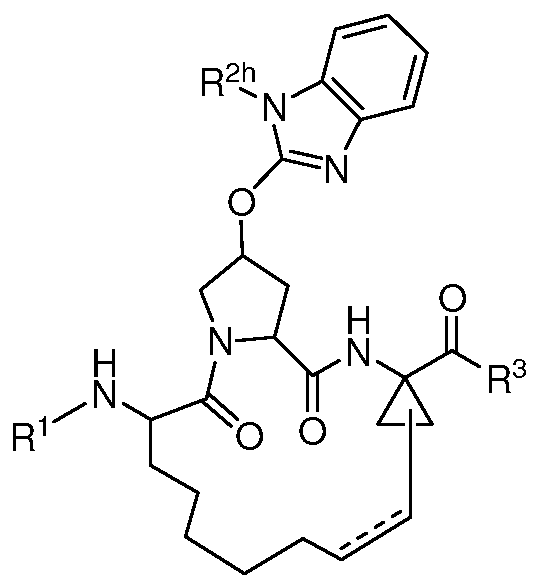





















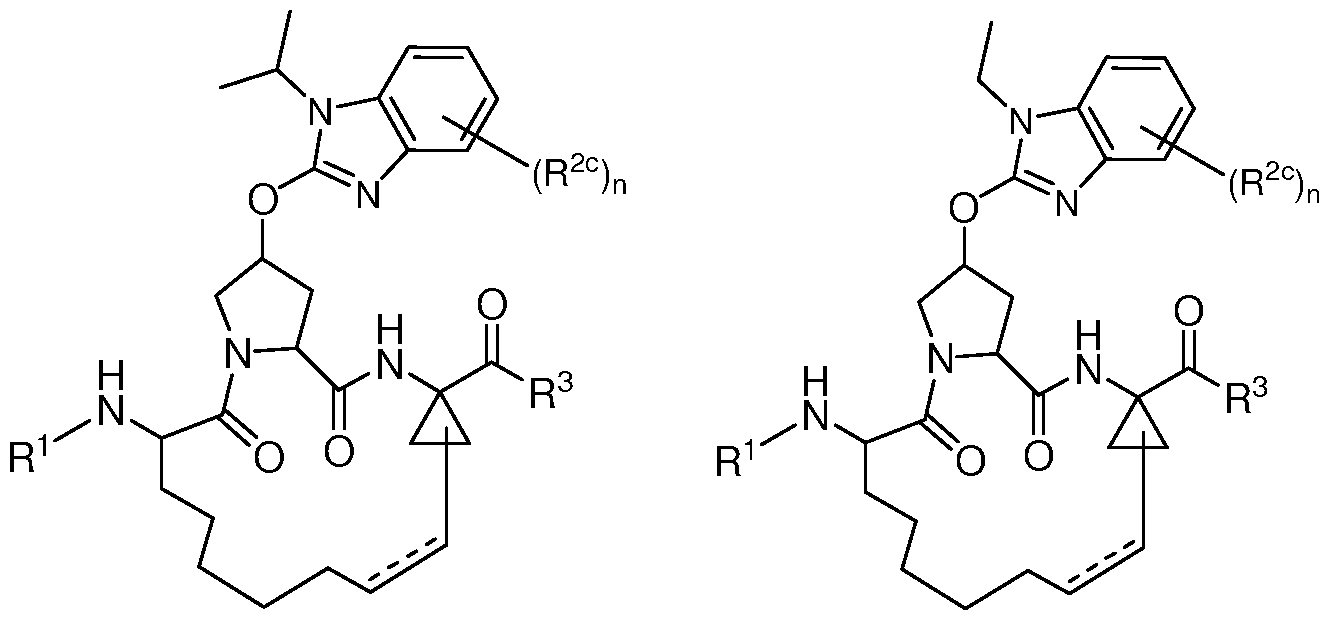









































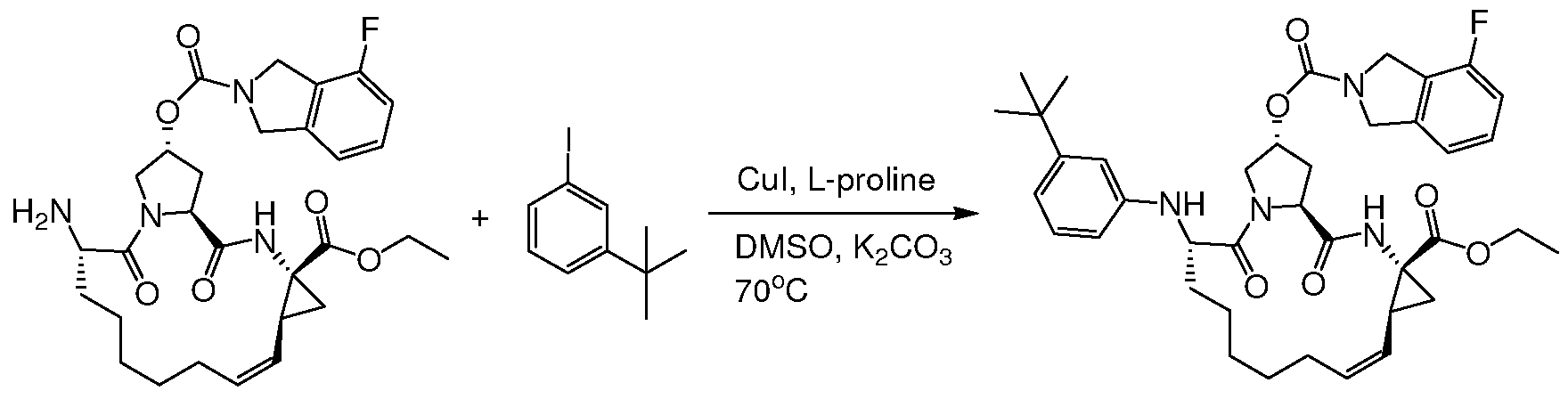



























































































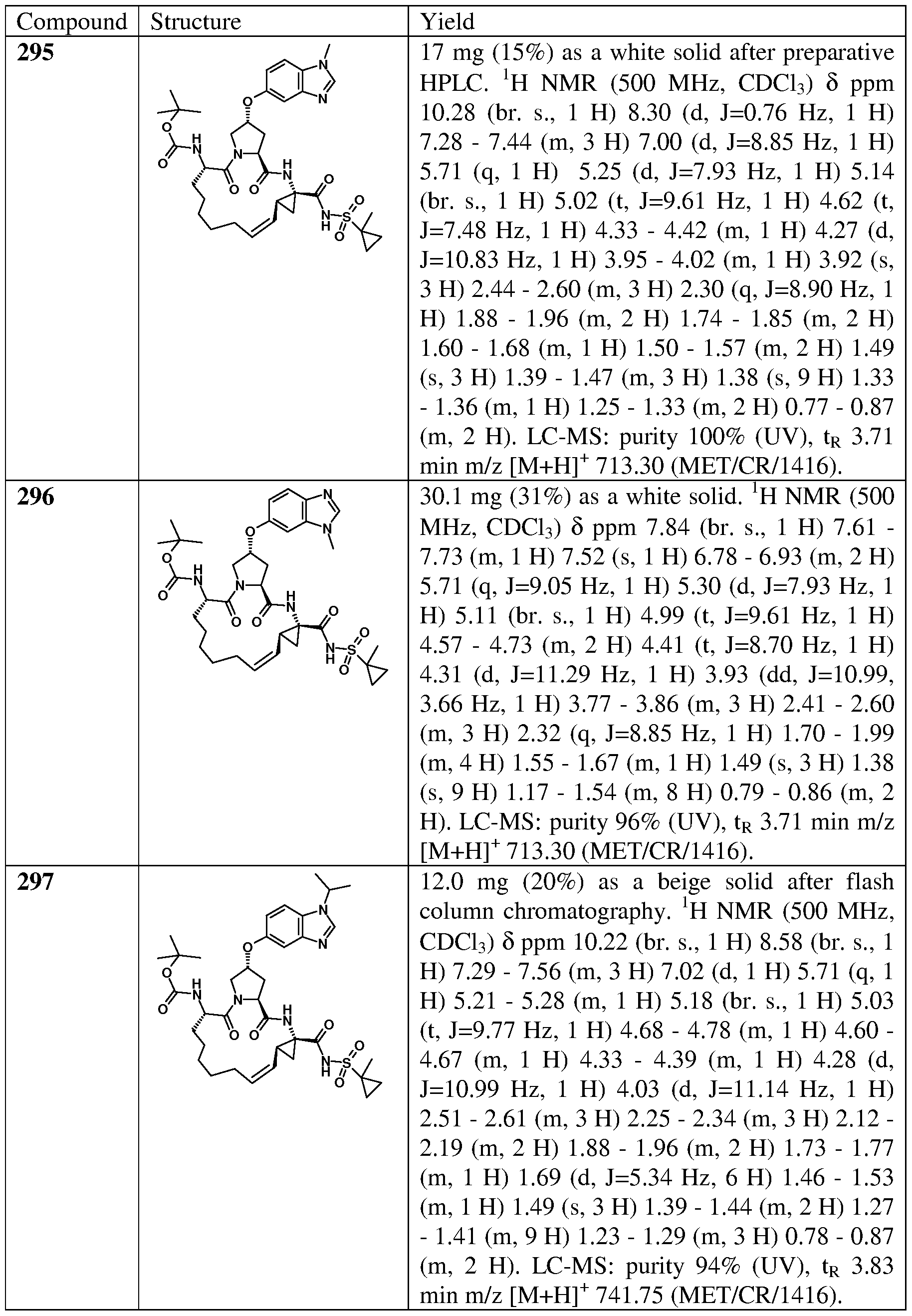






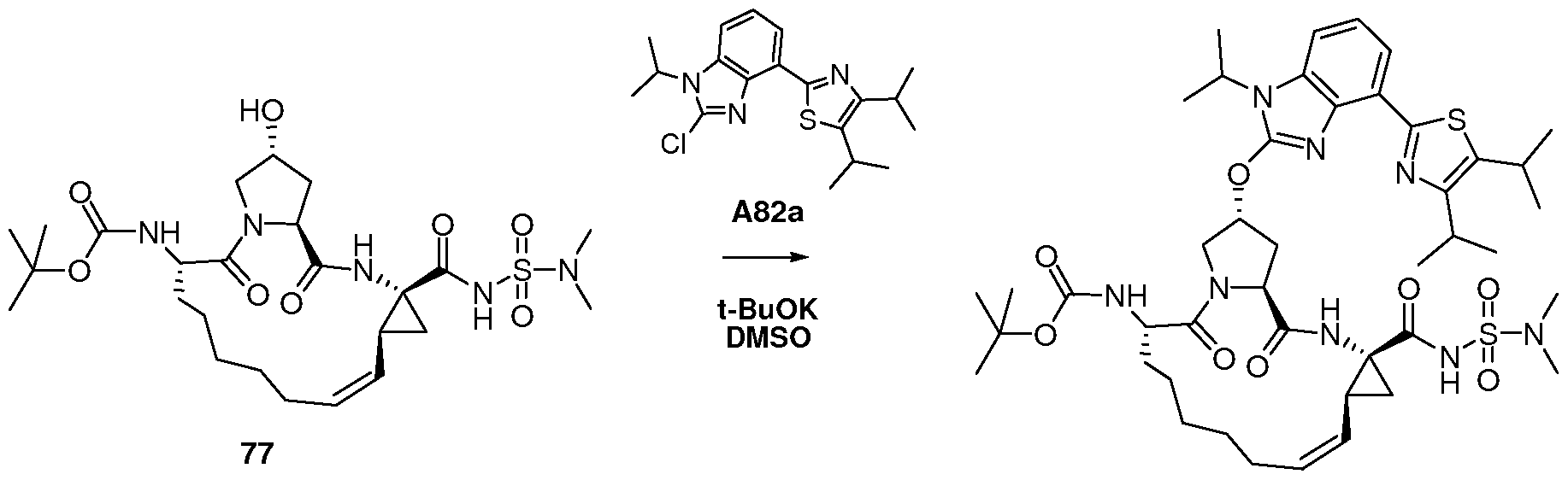


















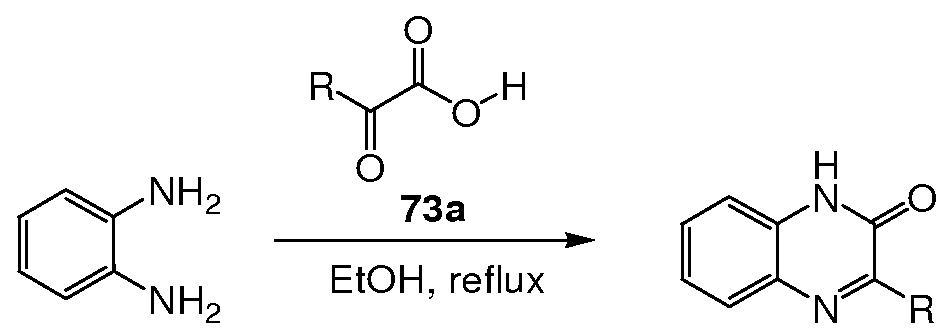





























































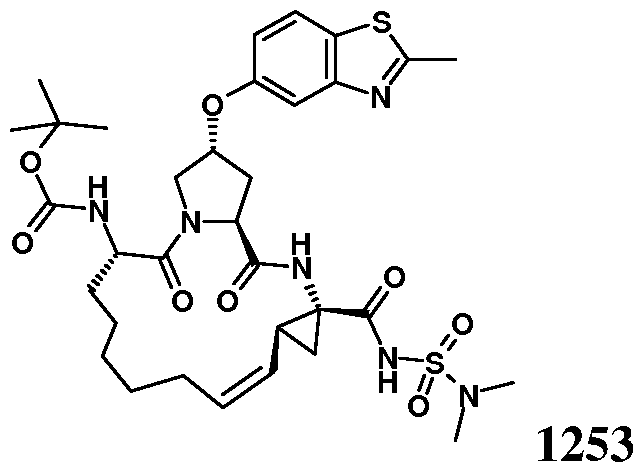




























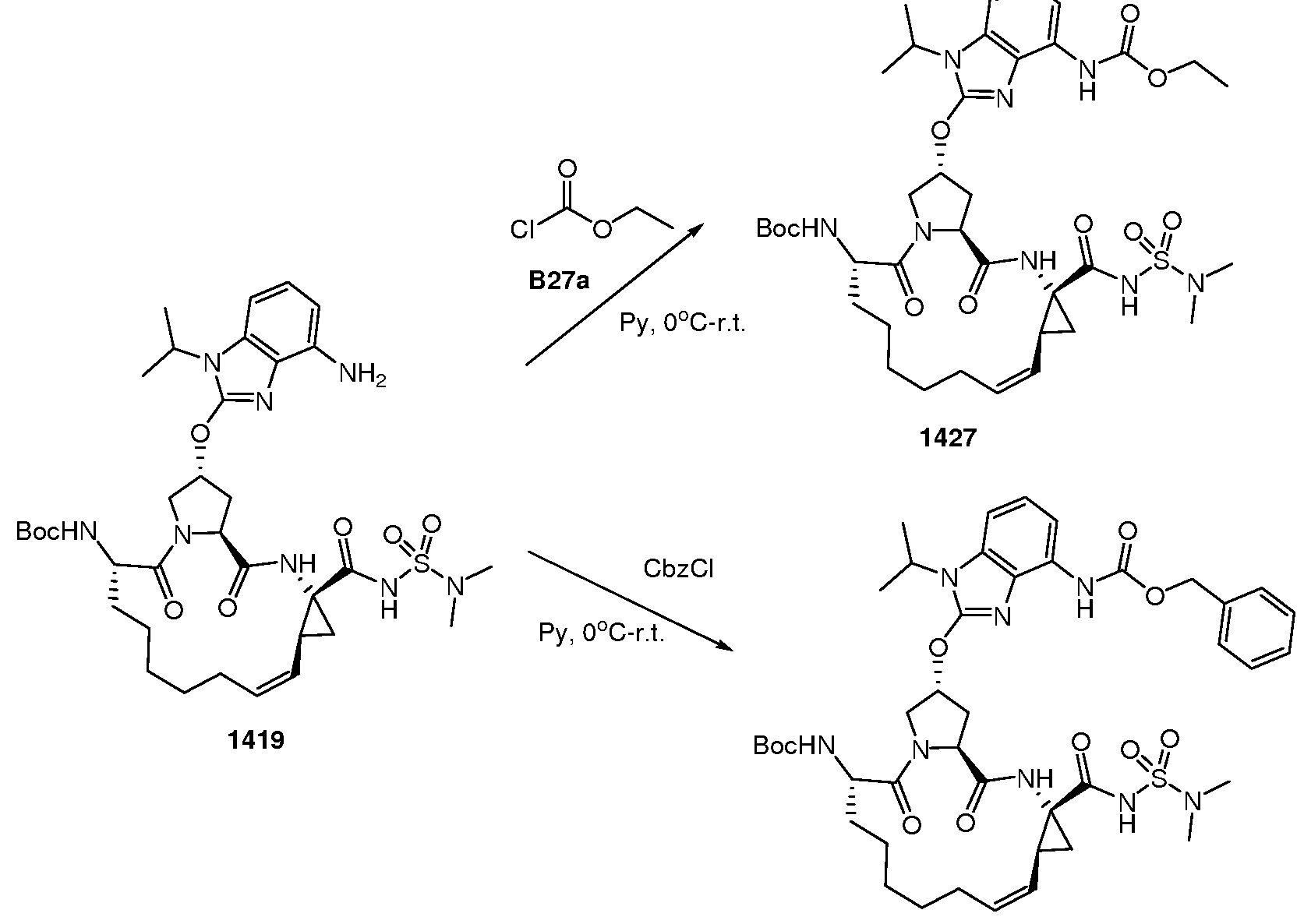













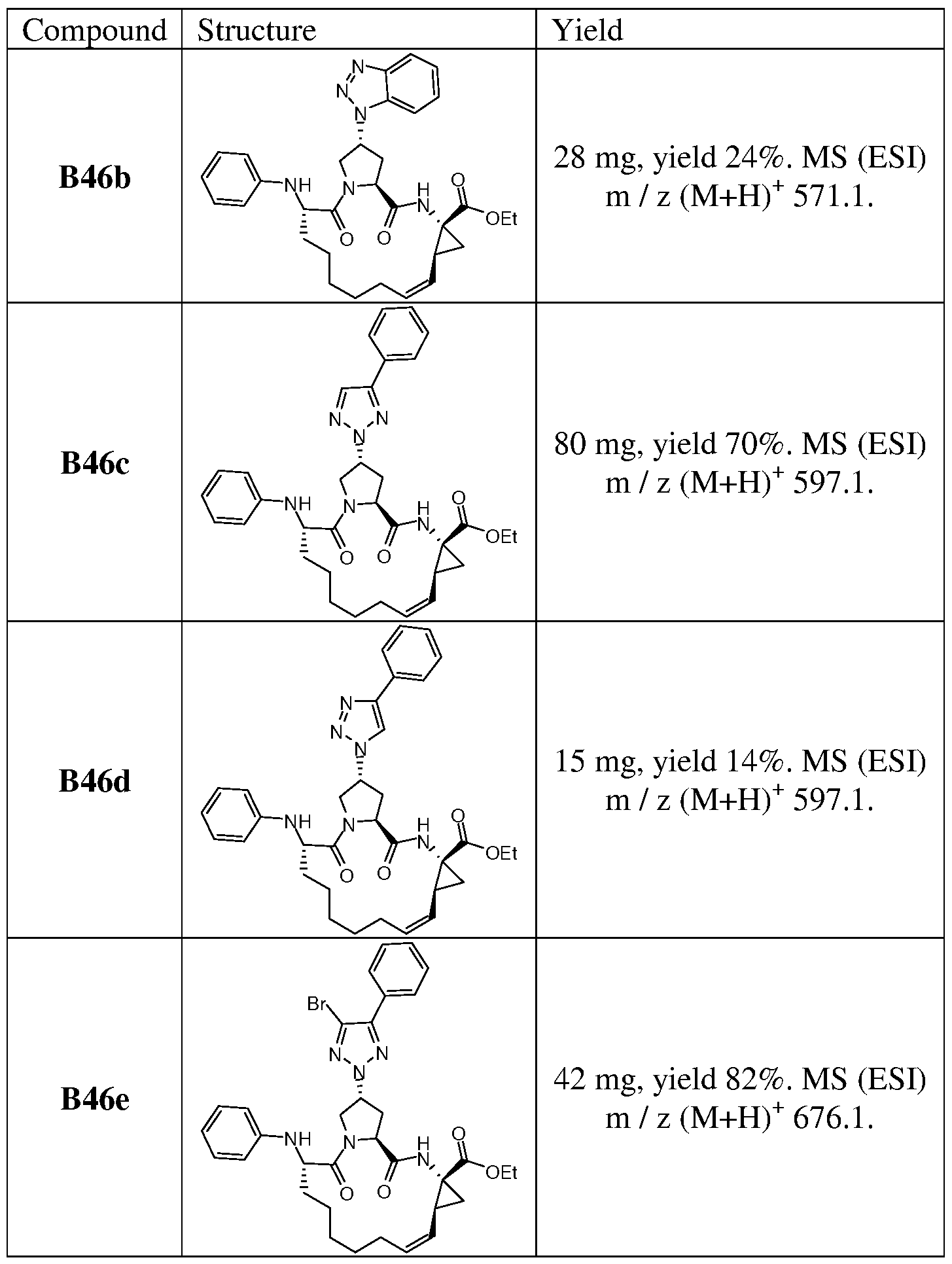





















































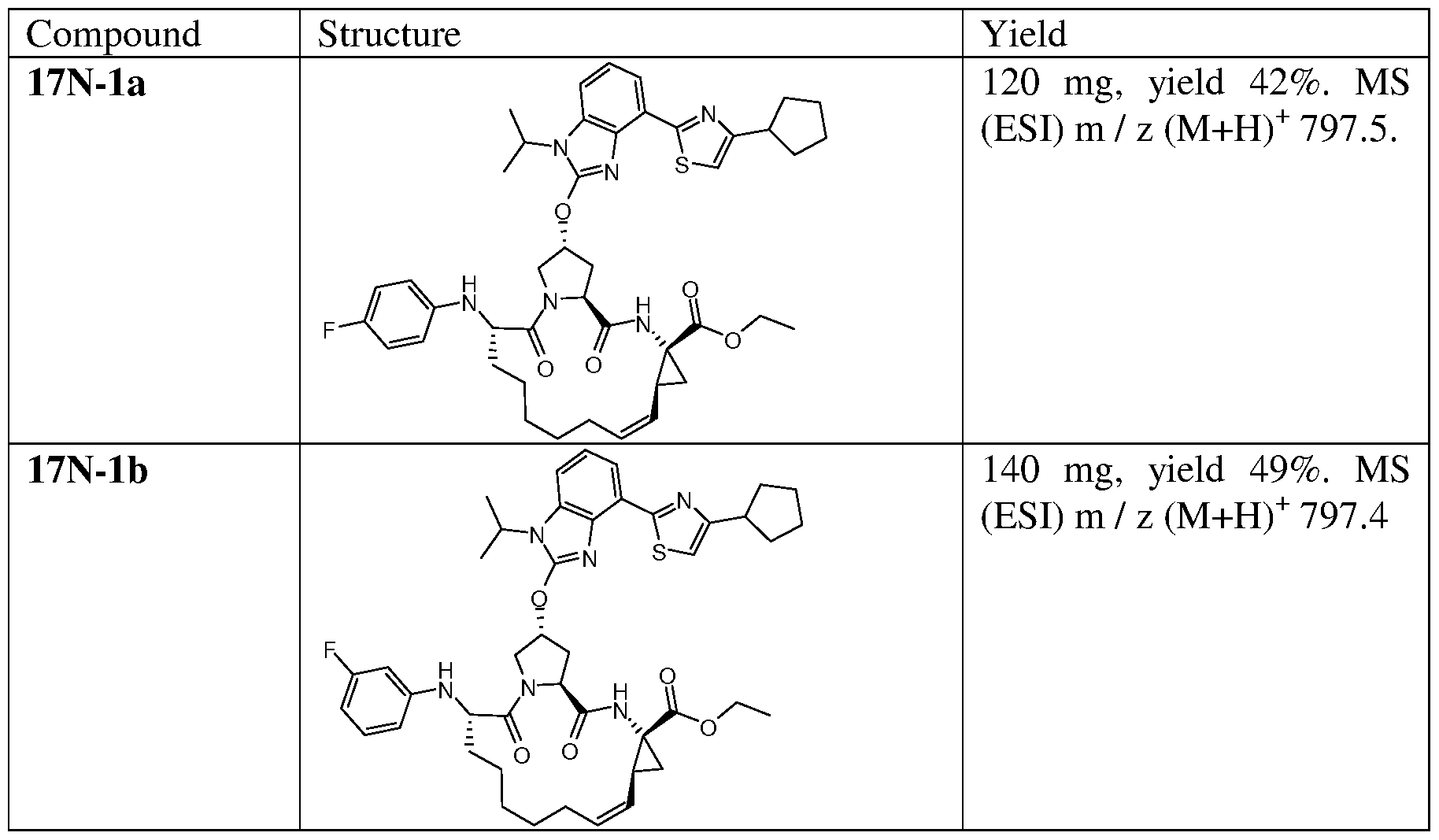












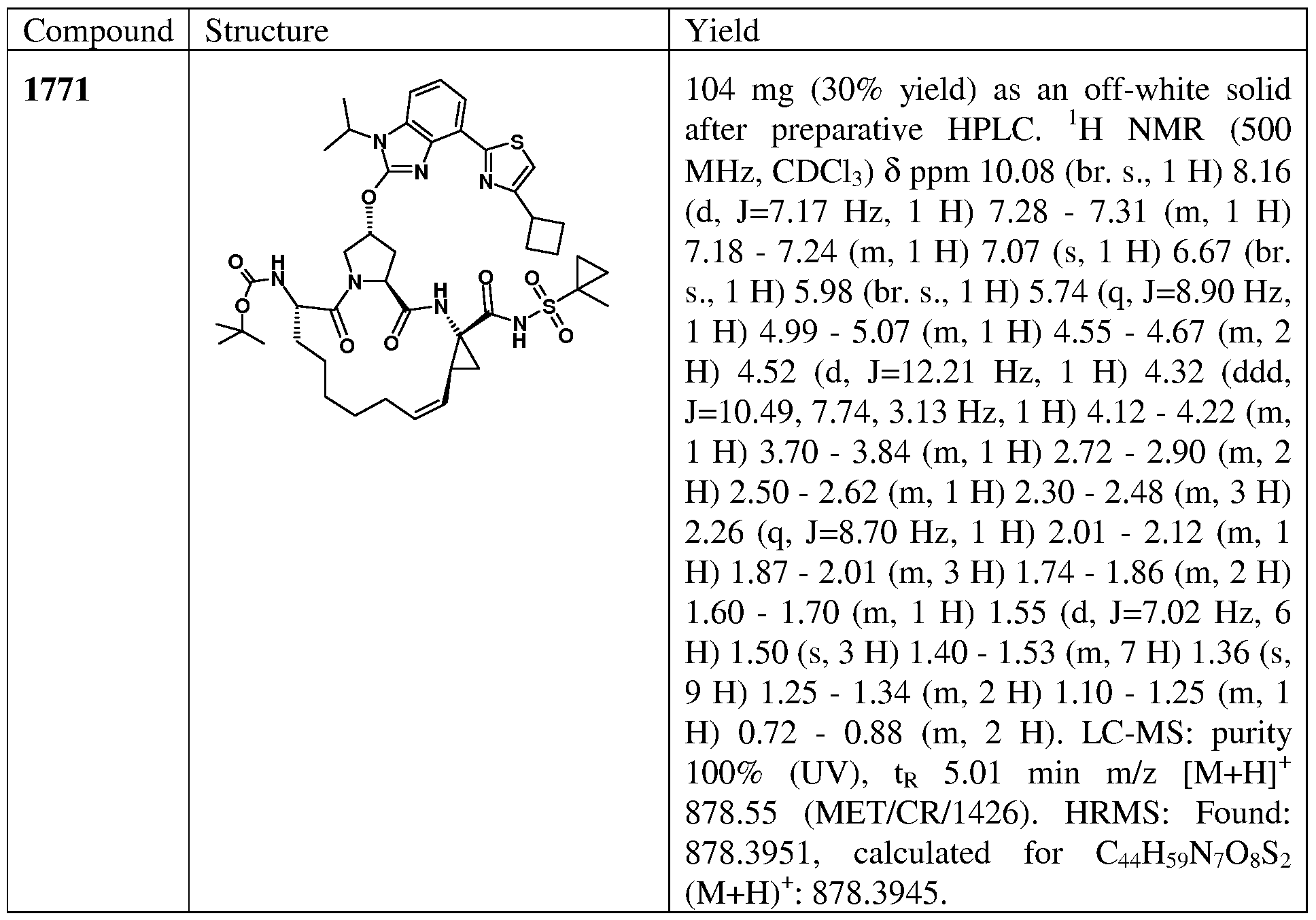










Claims



























Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2693DEN2012 IN2012DN02693A (en) | 2009-09-28 | 2010-09-24 | |
| EA201290128A EA201290128A1 (en) | 2009-09-28 | 2010-09-24 | NEW MACROCYCLIC INHIBITORS OF HEPATITIS C VIRUS REPLICATION |
| KR1020127010337A KR20130026410A (en) | 2009-09-28 | 2010-09-24 | Cyclic peptide inhibitors of hepatitis c virus replication |
| EP10819571.0A EP2483290A4 (en) | 2009-09-28 | 2010-09-24 | Cyclic peptide inhibitors of hepatitis c virus replication |
| AU2010298028A AU2010298028A1 (en) | 2009-09-28 | 2010-09-24 | Cyclic peptide inhibitors of hepatitis C virus replication |
| JP2012531086A JP2013505952A (en) | 2009-09-28 | 2010-09-24 | New macrocyclic inhibitor of hepatitis C virus replication |
| MX2012003500A MX2012003500A (en) | 2009-09-28 | 2010-09-24 | Cyclic peptide inhibitors of hepatitis c virus replication. |
| CA2775697A CA2775697A1 (en) | 2009-09-28 | 2010-09-24 | Cyclic peptide inhibitors of hepatitis c virus replication |
| CN201080053601.9A CN102741270B (en) | 2009-09-28 | 2010-09-24 | Cyclic peptide inhibitors of hepatitis C virus replication |
| IL218766A IL218766A0 (en) | 2009-09-28 | 2012-03-21 | Cyclic peptide inhibitors of hepatitis c virus replication |
| TNP2012000135A TN2012000135A1 (en) | 2009-09-28 | 2012-03-27 | Cyclic peptide inhibitors of hepatitis c virus replication |
| MA34813A MA33720B1 (en) | 2009-09-28 | 2012-04-26 | Cyclic digestive inhibitors of hepatitis C virus replication c |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US24646509P | 2009-09-28 | 2009-09-28 | |
| US61/246,465 | 2009-09-28 | ||
| US32425110P | 2010-04-14 | 2010-04-14 | |
| US61/324,251 | 2010-04-14 | ||
| US34573710P | 2010-05-18 | 2010-05-18 | |
| US61/345,737 | 2010-05-18 | ||
| US34623810P | 2010-05-19 | 2010-05-19 | |
| US61/346,238 | 2010-05-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011038293A1 true WO2011038293A1 (en) | 2011-03-31 |
Family
ID=43796242
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/050298 WO2011038293A1 (en) | 2009-09-28 | 2010-09-24 | Cyclic peptide inhibitors of hepatitis c virus replication |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US20110081315A1 (en) |
| EP (1) | EP2483290A4 (en) |
| JP (1) | JP2013505952A (en) |
| KR (1) | KR20130026410A (en) |
| CN (2) | CN105001302A (en) |
| AR (1) | AR078462A1 (en) |
| AU (1) | AU2010298028A1 (en) |
| CA (1) | CA2775697A1 (en) |
| CO (1) | CO6531497A2 (en) |
| EA (1) | EA201290128A1 (en) |
| EC (1) | ECSP12011845A (en) |
| IL (1) | IL218766A0 (en) |
| IN (1) | IN2012DN02693A (en) |
| MA (1) | MA33720B1 (en) |
| MX (1) | MX2012003500A (en) |
| TN (1) | TN2012000135A1 (en) |
| TW (1) | TW201124137A (en) |
| WO (1) | WO2011038293A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012054874A1 (en) * | 2010-10-22 | 2012-04-26 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| EP2576564A2 (en) * | 2010-06-07 | 2013-04-10 | Abbvie Inc. | Macrocyclic hepatitis c serine protease inhibitors |
| EP2617722A1 (en) * | 2010-09-10 | 2013-07-24 | Shionogi & Co., Ltd. | Hetero ring-fused imidazole derivative having ampk activating effect |
| WO2013186089A2 (en) | 2012-06-14 | 2013-12-19 | Basf Se | Pesticidal methods using substituted 3-pyridyl thiazole compounds and derivatives for combating animal pests |
| US8691757B2 (en) | 2011-06-15 | 2014-04-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CN103906739A (en) * | 2011-09-22 | 2014-07-02 | 杨森制药公司 | Processes and intermediates for preparing a macrocyclic protease inhibitor of HCV |
| WO2014140365A1 (en) * | 2013-03-15 | 2014-09-18 | Syngenta Participations Ag | Microbicidally active imidazopyridine derivatives |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9296782B2 (en) | 2012-07-03 | 2016-03-29 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9617310B2 (en) | 2013-03-15 | 2017-04-11 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CN110305018A (en) * | 2019-06-06 | 2019-10-08 | 浙江普洛家园药业有限公司 | A kind of preparation method of the bromo- 2- fluoronitrobenzene of 3- |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2305697A3 (en) | 2005-07-25 | 2011-07-27 | Intermune, Inc. | Macrocyclic inhibitors of Hepatitis C virus replication |
| JP2009511595A (en) * | 2005-10-11 | 2009-03-19 | インターミューン・インコーポレーテッド | Compounds and methods for inhibiting replication of hepatitis C virus |
| MX2010011306A (en) * | 2008-04-15 | 2010-11-09 | Intermune Inc | Novel macrocyclic inhibitors of hepatitis c virus replication. |
| EP2358736A1 (en) * | 2008-10-15 | 2011-08-24 | Intermune, Inc. | Therapeutic antiviral peptides |
| TW201116540A (en) * | 2009-10-01 | 2011-05-16 | Intermune Inc | Therapeutic antiviral peptides |
| WO2011107530A2 (en) * | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| CN105884779B (en) * | 2015-02-13 | 2018-06-12 | 广东东阳光药业有限公司 | Application as the compound of hepatitis c inhibitor and its in drug |
| CN108314648A (en) * | 2018-04-12 | 2018-07-24 | 苏州康润医药有限公司 | The synthetic method of the bromo- 7- fluorine isoquinolin of 4- |
| CN112358447B (en) * | 2020-11-16 | 2022-04-12 | 苏州康润医药有限公司 | Synthesis method of 7-fluoroisoquinoline-1-carboxylic acid |
| CN114105800B (en) * | 2021-11-25 | 2023-09-01 | 杭州国瑞生物科技有限公司 | Preparation method of 2, 3-diaminomethyl benzoate |
Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3547119A (en) | 1967-12-08 | 1970-12-15 | Baxter Laboratories Inc | Catheter assembly |
| US4211771A (en) | 1971-06-01 | 1980-07-08 | Robins Ronald K | Treatment of human viral diseases with 1-B-D-ribofuranosyl-1,2,4-triazole-3-carboxamide |
| US4311137A (en) | 1980-04-30 | 1982-01-19 | Sherwood Medical Industries Inc. | Infusion device |
| US4531937A (en) | 1983-01-24 | 1985-07-30 | Pacesetter Systems, Inc. | Introducer catheter apparatus and method of use |
| US4755173A (en) | 1986-02-25 | 1988-07-05 | Pacesetter Infusion, Ltd. | Soft cannula subcutaneous injection set |
| US5310562A (en) | 1989-11-22 | 1994-05-10 | Margolin Solomon B | Composition and method for reparation and prevention of fibrotic lesions |
| US5518729A (en) | 1989-11-22 | 1996-05-21 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| US5541206A (en) | 1989-05-23 | 1996-07-30 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| US5648497A (en) | 1989-05-23 | 1997-07-15 | Abbott Laboraotries | Retroviral protease inhibiting compounds |
| US5716632A (en) | 1989-11-22 | 1998-02-10 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| US5846987A (en) | 1992-12-29 | 1998-12-08 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| US6017328A (en) | 1993-01-21 | 2000-01-25 | Magnolia Medical, Llc | Device for subcutaneous medication delivery |
| US6090822A (en) | 1995-03-03 | 2000-07-18 | Margolin; Solomon B. | Treatment of cytokine growth factor caused disorders |
| US6232333B1 (en) | 1996-11-21 | 2001-05-15 | Abbott Laboratories | Pharmaceutical composition |
| US6277830B1 (en) | 1998-10-16 | 2001-08-21 | Schering Corporation | 5′-amino acid esters of ribavirin and the use of same to treat hepatitis C with interferon |
| US20040110795A1 (en) | 1999-08-10 | 2004-06-10 | United Therapeutics Corp. | Use of iminosugar derivatives to inhibit ion channel activity |
| WO2004093798A2 (en) * | 2003-04-18 | 2004-11-04 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis c serine protease inhibitors |
| WO2007014926A1 (en) * | 2005-07-29 | 2007-02-08 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis c virus |
| WO2007015824A2 (en) | 2005-07-25 | 2007-02-08 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| WO2008137779A2 (en) | 2007-05-03 | 2008-11-13 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US20090005387A1 (en) * | 2007-06-26 | 2009-01-01 | Deqiang Niu | Quinoxalinyl macrocyclic hepatitis c virus serine protease inhibitors |
| WO2010030359A2 (en) * | 2008-09-11 | 2010-03-18 | Enanta Pharjmaceuticals, Inc. | Macrocyclic hepatitis c serine protease inhibitors |
Family Cites Families (98)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3798209A (en) * | 1971-06-01 | 1974-03-19 | Icn Pharmaceuticals | 1,2,4-triazole nucleosides |
| CS263951B1 (en) * | 1985-04-25 | 1989-05-12 | Antonin Holy | 9-(phosponylmethoxyalkyl)adenines and method of their preparation |
| GB8918806D0 (en) * | 1989-08-17 | 1989-09-27 | Shell Int Research | Chiral compounds,their preparation and use |
| US5624949A (en) * | 1993-12-07 | 1997-04-29 | Eli Lilly And Company | Protein kinase C inhibitors |
| US6693072B2 (en) * | 1994-06-02 | 2004-02-17 | Aventis Pharmaceuticals Inc. | Elastase inhibitors |
| US5756466A (en) * | 1994-06-17 | 1998-05-26 | Vertex Pharmaceuticals, Inc. | Inhibitors of interleukin-1β converting enzyme |
| US6323180B1 (en) * | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| US6608027B1 (en) * | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
| CA2405521C (en) * | 2000-04-05 | 2010-06-29 | Schering Corporation | Macrocyclic ns3-serine protease inhibitors of hepatitis c virus comprising n-cyclic p2 moieties |
| US6914122B2 (en) * | 2000-04-19 | 2005-07-05 | Schering Corporation | Macrocyclic NS-3 serine protease inhibitors of hepatitis C virus comprising alkyl and aryl alanine P2 moieties |
| MY143322A (en) * | 2000-07-21 | 2011-04-15 | Schering Corp | Peptides as ns3-serine protease inhibitors of hepatitis c virus |
| US7244721B2 (en) * | 2000-07-21 | 2007-07-17 | Schering Corporation | Peptides as NS3-serine protease inhibitors of hepatitis C virus |
| CN100391967C (en) * | 2000-11-20 | 2008-06-04 | 布里斯托尔-迈尔斯斯奎布公司 | Hepatitis C tripeptide inhibitors |
| WO2002089738A2 (en) * | 2001-05-08 | 2002-11-14 | Yale University | Proteomimetic compounds and methods |
| US6867185B2 (en) * | 2001-12-20 | 2005-03-15 | Bristol-Myers Squibb Company | Inhibitors of hepatitis C virus |
| CN100381440C (en) * | 2002-04-11 | 2008-04-16 | 沃泰克斯药物股份有限公司 | Inhibitors of serine proteases, particularly HCV NS3-NS4A protease |
| DE60336550D1 (en) * | 2002-05-20 | 2011-05-12 | Bristol Myers Squibb Co | INHIBITORS OF HEPATITIS C VIRUS |
| DE60324552D1 (en) * | 2002-05-20 | 2008-12-18 | Bristol Myers Squibb Co | Substituierte cycloalkyl p1' hepatitis c virus inhibitoren |
| US6869964B2 (en) * | 2002-05-20 | 2005-03-22 | Bristol-Myers Squibb Company | Heterocyclicsulfonamide hepatitis C virus inhibitors |
| MY140680A (en) * | 2002-05-20 | 2010-01-15 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| PL199412B1 (en) * | 2002-10-15 | 2008-09-30 | Boehringer Ingelheim Int | Ruthenium new complexes as (pre) catalytic agents of permutation reaction, new derivatives of 2-alkoxy-5-nitrostyrene as intermediate compounds and method of their receiving |
| EP1615947A2 (en) * | 2003-04-10 | 2006-01-18 | Boehringer Ingelheim International GmbH | Process for preparing macrocyclic compounds |
| TW200510391A (en) * | 2003-04-11 | 2005-03-16 | Vertex Pharma | Inhibitors of serine proteases, particularly HCV NS3-NS4A protease |
| EP1629000B1 (en) * | 2003-04-16 | 2009-02-18 | Bristol-Myers Squibb Company | Macrocyclic isoquinoline peptide inhibitors of hepatitis c virus |
| US7125845B2 (en) * | 2003-07-03 | 2006-10-24 | Enanta Pharmaceuticals, Inc. | Aza-peptide macrocyclic hepatitis C serine protease inhibitors |
| AU2004282148A1 (en) * | 2003-10-10 | 2005-04-28 | Vertex Pharmaceuticals Incoporated | Inhibitors of serine proteases, particularly HCV NS3-NS4A protease |
| US7491794B2 (en) * | 2003-10-14 | 2009-02-17 | Intermune, Inc. | Macrocyclic compounds as inhibitors of viral replication |
| PT1680137E (en) * | 2003-10-14 | 2013-02-21 | Hoffmann La Roche | Macrocyclic carboxylic acid and acylsulfonamide compound as inhibitor of hcv replication |
| US7939538B2 (en) * | 2004-06-28 | 2011-05-10 | Amgen Inc. | Compounds, compositions and methods for prevention and treatment of inflammatory and immunoregulatory disorders and diseases |
| DE102004033312A1 (en) * | 2004-07-08 | 2006-01-26 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Continuous metathesis process with ruthenium catalysts |
| ES2366478T3 (en) * | 2004-07-20 | 2011-10-20 | Boehringer Ingelheim International Gmbh | PEPTIDE ANALOGS INHIBITORS OF HEPATITIS C. |
| CN101068828A (en) * | 2004-08-27 | 2007-11-07 | 先灵公司 | Acylsulfonamide compounds as inhibitors of hepatitis c virus NS3 serine protease |
| EP1794179A1 (en) * | 2004-09-17 | 2007-06-13 | Boehringer Ingelheim International Gmbh | Ring-closing metathesis process in supercritical fluid |
| EP1879607B1 (en) * | 2005-05-02 | 2014-11-12 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| US7608592B2 (en) * | 2005-06-30 | 2009-10-27 | Virobay, Inc. | HCV inhibitors |
| US7601686B2 (en) * | 2005-07-11 | 2009-10-13 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| TWI389908B (en) * | 2005-07-14 | 2013-03-21 | Gilead Sciences Inc | Antiviral compounds |
| AR057456A1 (en) * | 2005-07-20 | 2007-12-05 | Merck & Co Inc | HCV PROTEASA NS3 INHIBITORS |
| PL1912997T3 (en) * | 2005-07-29 | 2012-02-29 | Tibotec Pharm Ltd | Macrocyclic inhibitors of hepatitis c virus |
| EP2402331A1 (en) * | 2005-08-02 | 2012-01-04 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases |
| JP2009511595A (en) * | 2005-10-11 | 2009-03-19 | インターミューン・インコーポレーテッド | Compounds and methods for inhibiting replication of hepatitis C virus |
| US7772183B2 (en) * | 2005-10-12 | 2010-08-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CA2629080A1 (en) * | 2005-11-16 | 2007-05-24 | F. Hoffmann-La Roche Ag | Novel pyrrolidine derivatives as inhibitors of coagulation factor xa |
| US7728148B2 (en) * | 2006-06-06 | 2010-06-01 | Enanta Pharmaceuticals, Inc. | Acyclic oximyl hepatitis C protease inhibitors |
| KR20090024834A (en) * | 2006-07-05 | 2009-03-09 | 인터뮨, 인크. | Novel inhibitors of hepatitis c virus replication |
| WO2008008776A2 (en) * | 2006-07-11 | 2008-01-17 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US7906619B2 (en) * | 2006-07-13 | 2011-03-15 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US20090035267A1 (en) * | 2007-07-31 | 2009-02-05 | Moore Joel D | Acyclic, pyridazinone-derived hepatitis c serine protease inhibitors |
| CN101674844A (en) * | 2006-08-04 | 2010-03-17 | 英安塔制药有限公司 | tetrazolyl macrocyclic hepatitis c serine protease inhibitors |
| US7635683B2 (en) * | 2006-08-04 | 2009-12-22 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl tripeptide hepatitis C virus inhibitors |
| US7718612B2 (en) * | 2007-08-02 | 2010-05-18 | Enanta Pharmaceuticals, Inc. | Pyridazinonyl macrocyclic hepatitis C serine protease inhibitors |
| US20080038225A1 (en) * | 2006-08-11 | 2008-02-14 | Ying Sun | Triazolyl acyclic hepatitis c serine protease inhibitors |
| US20090035268A1 (en) * | 2007-08-01 | 2009-02-05 | Ying Sun | Tetrazolyl acyclic hepatitis c serine protease inhibitors |
| US7582605B2 (en) * | 2006-08-11 | 2009-09-01 | Enanta Pharmaceuticals, Inc. | Phosphorus-containing hepatitis C serine protease inhibitors |
| US7687459B2 (en) * | 2006-08-11 | 2010-03-30 | Enanta Pharmaceuticals, Inc. | Arylalkoxyl hepatitis C virus protease inhibitors |
| US7662779B2 (en) * | 2006-08-11 | 2010-02-16 | Enanta Pharmaceuticals, Inc. | Triazolyl macrocyclic hepatitis C serine protease inhibitors |
| US7605126B2 (en) * | 2006-08-11 | 2009-10-20 | Enanta Pharmaceuticals, Inc. | Acylaminoheteroaryl hepatitis C virus protease inhibitors |
| US20090098085A1 (en) * | 2006-08-11 | 2009-04-16 | Ying Sun | Tetrazolyl acyclic hepatitis c serine protease inhibitors |
| US8343477B2 (en) * | 2006-11-01 | 2013-01-01 | Bristol-Myers Squibb Company | Inhibitors of hepatitis C virus |
| US20080107625A1 (en) * | 2006-11-01 | 2008-05-08 | Bristol-Myers Squibb Company | Inhibitors of Hepatitis C Virus |
| US7772180B2 (en) * | 2006-11-09 | 2010-08-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| AU2007321182B2 (en) * | 2006-11-17 | 2013-11-14 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis C virus |
| EP2125870A4 (en) * | 2007-02-16 | 2011-04-06 | Boehringer Ingelheim Int | Inhibitors of hepatitis c ns3 protease |
| CN101679240A (en) * | 2007-03-23 | 2010-03-24 | 先灵公司 | p1-nonepimerizable ketoamide inhibitors of hcv ns3 protease |
| US20090123423A1 (en) * | 2007-04-26 | 2009-05-14 | Yonghua Gai | Hydroxyamic analogs as hepatitis c virus serine protease inhibitor |
| PE20090213A1 (en) * | 2007-05-04 | 2009-02-28 | Bristol Myers Squibb Co | AGONISTS OF THE RECEPTOR COUPLED TO PROTEIN G GPR119 [6,5] -BICYCLIC |
| EA200971041A1 (en) * | 2007-05-10 | 2010-08-30 | Интермьюн, Инк. | NEW PEPTIDE HEPATITIS C VIRUS REPLICATION INHIBITORS |
| BRPI0813733A2 (en) * | 2007-06-29 | 2019-11-05 | Gilead Sciences Inc | compound with hcv inhibitory activity, its pharmaceutical composition and its use. |
| WO2009014730A1 (en) * | 2007-07-26 | 2009-01-29 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US20090053175A1 (en) * | 2007-08-24 | 2009-02-26 | Yat Sun Or | Substitute pyrrolidine derivatives |
| US20090060874A1 (en) * | 2007-09-05 | 2009-03-05 | Yao-Ling Qiu | Bicyclic pyrrolidine derivatives |
| WO2009042668A2 (en) * | 2007-09-24 | 2009-04-02 | Achillion Pharmaceuticals, Inc. | Urea-containing peptides as inhibitors of viral replication |
| US20090082366A1 (en) * | 2007-09-26 | 2009-03-26 | Protia, Llc | Deuterium-enriched telaprevir |
| EP2205584A1 (en) * | 2007-10-10 | 2010-07-14 | Novartis Ag | Spiropyrrolidines and their use against hcv and hiv infection |
| US20090111757A1 (en) * | 2007-10-25 | 2009-04-30 | Taigen Biotechnology Co., Ltd. | Hcv protease inhibitors |
| US8383583B2 (en) * | 2007-10-26 | 2013-02-26 | Enanta Pharmaceuticals, Inc. | Macrocyclic, pyridazinone-containing hepatitis C serine protease inhibitors |
| WO2009067225A2 (en) * | 2007-11-20 | 2009-05-28 | Concert Pharmaceuticals, Inc. | Boceprevir derivatives for the treatment of hcv infections |
| MX2010006659A (en) * | 2007-12-21 | 2010-07-05 | Hoffmann La Roche | Process for the preparation of a macrocycle. |
| MX2010011306A (en) * | 2008-04-15 | 2010-11-09 | Intermune Inc | Novel macrocyclic inhibitors of hepatitis c virus replication. |
| CA2729168A1 (en) * | 2008-07-02 | 2010-02-04 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of viral infections |
| WO2010015545A1 (en) * | 2008-08-07 | 2010-02-11 | F. Hoffmann-La Roche Ag | Process for the preparation of a macrocycle |
| US7906655B2 (en) * | 2008-08-07 | 2011-03-15 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| PE20110343A1 (en) * | 2008-09-17 | 2011-06-25 | Boehringer Ingelheim Int | COMBINATION OF HCV PROTEASE NS3 INHIBITOR WITH INTERFERON AND RIBAVIRIN |
| US8603737B2 (en) * | 2008-09-19 | 2013-12-10 | Celgene Avilomics Research, Inc. | Methods for identifying HCV protease inhibitors |
| US8044087B2 (en) * | 2008-09-29 | 2011-10-25 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20100080770A1 (en) * | 2008-09-29 | 2010-04-01 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| US8563505B2 (en) * | 2008-09-29 | 2013-10-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EP2358736A1 (en) * | 2008-10-15 | 2011-08-24 | Intermune, Inc. | Therapeutic antiviral peptides |
| US8445430B2 (en) * | 2008-11-20 | 2013-05-21 | Achillion Pharmaceuticals, Inc. | Cyclic carboxamide compounds and analogues thereof as of hepatitis C virus |
| US20110064694A1 (en) * | 2009-09-09 | 2011-03-17 | Yale University | Anti-hepatitis c activity of meso-tetrakis-porphyrin analogues |
| US8815928B2 (en) * | 2009-09-11 | 2014-08-26 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| EP2475256A4 (en) * | 2009-09-11 | 2013-06-05 | Enanta Pharm Inc | Hepatitis c virus inhibitors |
| US8927709B2 (en) * | 2009-09-11 | 2015-01-06 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8759332B2 (en) * | 2009-09-11 | 2014-06-24 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8703938B2 (en) * | 2009-09-11 | 2014-04-22 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8822700B2 (en) * | 2009-09-11 | 2014-09-02 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| AU2009352688B2 (en) * | 2009-09-15 | 2014-04-17 | Taigen Biotechnology Co., Ltd. | HCV protease inhibitors |
| TW201116540A (en) * | 2009-10-01 | 2011-05-16 | Intermune Inc | Therapeutic antiviral peptides |
-
2010
- 2010-09-24 JP JP2012531086A patent/JP2013505952A/en not_active Withdrawn
- 2010-09-24 US US12/890,475 patent/US20110081315A1/en not_active Abandoned
- 2010-09-24 CA CA2775697A patent/CA2775697A1/en not_active Abandoned
- 2010-09-24 MX MX2012003500A patent/MX2012003500A/en not_active Application Discontinuation
- 2010-09-24 WO PCT/US2010/050298 patent/WO2011038293A1/en active Application Filing
- 2010-09-24 KR KR1020127010337A patent/KR20130026410A/en not_active Application Discontinuation
- 2010-09-24 CN CN201510341151.8A patent/CN105001302A/en active Pending
- 2010-09-24 EP EP10819571.0A patent/EP2483290A4/en not_active Withdrawn
- 2010-09-24 EA EA201290128A patent/EA201290128A1/en unknown
- 2010-09-24 AU AU2010298028A patent/AU2010298028A1/en not_active Abandoned
- 2010-09-24 CN CN201080053601.9A patent/CN102741270B/en not_active Expired - Fee Related
- 2010-09-24 IN IN2693DEN2012 patent/IN2012DN02693A/en unknown
- 2010-09-28 AR ARP100103512A patent/AR078462A1/en unknown
- 2010-09-28 TW TW099132976A patent/TW201124137A/en unknown
-
2012
- 2012-03-21 IL IL218766A patent/IL218766A0/en unknown
- 2012-03-27 TN TNP2012000135A patent/TN2012000135A1/en unknown
- 2012-04-26 MA MA34813A patent/MA33720B1/en unknown
- 2012-04-27 EC ECSP12011845 patent/ECSP12011845A/en unknown
- 2012-04-27 CO CO12069712A patent/CO6531497A2/en not_active Application Discontinuation
Patent Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3547119A (en) | 1967-12-08 | 1970-12-15 | Baxter Laboratories Inc | Catheter assembly |
| US4211771A (en) | 1971-06-01 | 1980-07-08 | Robins Ronald K | Treatment of human viral diseases with 1-B-D-ribofuranosyl-1,2,4-triazole-3-carboxamide |
| US4311137A (en) | 1980-04-30 | 1982-01-19 | Sherwood Medical Industries Inc. | Infusion device |
| US4531937A (en) | 1983-01-24 | 1985-07-30 | Pacesetter Systems, Inc. | Introducer catheter apparatus and method of use |
| US4755173A (en) | 1986-02-25 | 1988-07-05 | Pacesetter Infusion, Ltd. | Soft cannula subcutaneous injection set |
| US5635523A (en) | 1989-05-23 | 1997-06-03 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| US5648497A (en) | 1989-05-23 | 1997-07-15 | Abbott Laboraotries | Retroviral protease inhibiting compounds |
| US5541206A (en) | 1989-05-23 | 1996-07-30 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| US5716632A (en) | 1989-11-22 | 1998-02-10 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| US5518729A (en) | 1989-11-22 | 1996-05-21 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| US5310562A (en) | 1989-11-22 | 1994-05-10 | Margolin Solomon B | Composition and method for reparation and prevention of fibrotic lesions |
| US5846987A (en) | 1992-12-29 | 1998-12-08 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| US6017328A (en) | 1993-01-21 | 2000-01-25 | Magnolia Medical, Llc | Device for subcutaneous medication delivery |
| US6090822A (en) | 1995-03-03 | 2000-07-18 | Margolin; Solomon B. | Treatment of cytokine growth factor caused disorders |
| US6232333B1 (en) | 1996-11-21 | 2001-05-15 | Abbott Laboratories | Pharmaceutical composition |
| US6277830B1 (en) | 1998-10-16 | 2001-08-21 | Schering Corporation | 5′-amino acid esters of ribavirin and the use of same to treat hepatitis C with interferon |
| US20040110795A1 (en) | 1999-08-10 | 2004-06-10 | United Therapeutics Corp. | Use of iminosugar derivatives to inhibit ion channel activity |
| WO2004093798A2 (en) * | 2003-04-18 | 2004-11-04 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis c serine protease inhibitors |
| WO2007015824A2 (en) | 2005-07-25 | 2007-02-08 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| WO2007014926A1 (en) * | 2005-07-29 | 2007-02-08 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis c virus |
| WO2008137779A2 (en) | 2007-05-03 | 2008-11-13 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US20090005387A1 (en) * | 2007-06-26 | 2009-01-01 | Deqiang Niu | Quinoxalinyl macrocyclic hepatitis c virus serine protease inhibitors |
| WO2010030359A2 (en) * | 2008-09-11 | 2010-03-18 | Enanta Pharjmaceuticals, Inc. | Macrocyclic hepatitis c serine protease inhibitors |
Non-Patent Citations (16)
| Title |
|---|
| "Handbook of Pharmaceutical Excipients", 2000, AMER. PHARMACEUTICAL ASSOC. |
| "Merck Index" |
| "Pharmaceutical Dosage Forms and Drug Delivery Systems", 1999, LIPPINCOTT, WILLIAMS, & WILKINS |
| A. GENNARO ET AL.: "Pharmaceutical Dosage Forms and Drug Delivery Systems", 1999, LIPPINCOTT, WILLIAMS, & WILKINS, article "Remington: The Science and Practice of Pharmacy" |
| A. GENNARO: "Remington: The Science and Practice of Pharmacy", 2000, LIPPINCOTT, WILLIAMS, & WILKINS |
| BRUNT, HEPATOL., vol. 31, 2000, pages 241 - 246 |
| GREENE; WUTS: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY AND SONS |
| ISHAK, J. HEPATOL., vol. 22, 1995, pages 696 - 699 |
| KNODELL, HEPATOL., vol. 1, 1981, pages 431 |
| METAVIR, HEPATOLOGY, vol. 20, 1994, pages 15 - 20 |
| SCHEUER, J. HEPATOL., vol. 13, 1991, pages 372 |
| See also references of EP2483290A4 |
| STRYER: "Biochemistry", 2002, FREEMAN & CO. |
| THIBEAULT, D. ET AL.: "Use of the fused NS4A Peptide -NS3 protease domain to study the importance of the helicase domain for protease inhibitor binding to hepatitis C virus N3-NS4A", BIOCHEMISTRY, vol. 48, 2009, pages 744 - 753, XP008159450 * |
| YAO, STRUCTURE, vol. 7, 1999, pages 1353 |
| YAO., STRUCTURE, vol. 7, 1999, pages 1353 |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2576564A4 (en) * | 2010-06-07 | 2014-01-22 | Abbvie Inc | Macrocyclic hepatitis c serine protease inhibitors |
| EP2576564A2 (en) * | 2010-06-07 | 2013-04-10 | Abbvie Inc. | Macrocyclic hepatitis c serine protease inhibitors |
| JP2013528217A (en) * | 2010-06-07 | 2013-07-08 | アッヴィ・インコーポレイテッド | Macrocyclic hepatitis C serine protease inhibitor |
| US9133186B2 (en) | 2010-09-10 | 2015-09-15 | Shionogi & Co., Ltd. | Hetero ring-fused imidazole derivative having AMPK activating effect |
| AU2011299894B2 (en) * | 2010-09-10 | 2015-08-06 | Shionogi & Co., Ltd. | Hetero ring-fused imidazole derivative having AMPK activating effect |
| EP2617722A4 (en) * | 2010-09-10 | 2013-11-13 | Shionogi & Co | Hetero ring-fused imidazole derivative having ampk activating effect |
| EP2617722A1 (en) * | 2010-09-10 | 2013-07-24 | Shionogi & Co., Ltd. | Hetero ring-fused imidazole derivative having ampk activating effect |
| WO2012054874A1 (en) * | 2010-10-22 | 2012-04-26 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US9527885B2 (en) | 2011-05-05 | 2016-12-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8691757B2 (en) | 2011-06-15 | 2014-04-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CN103906739A (en) * | 2011-09-22 | 2014-07-02 | 杨森制药公司 | Processes and intermediates for preparing a macrocyclic protease inhibitor of HCV |
| WO2013186089A2 (en) | 2012-06-14 | 2013-12-19 | Basf Se | Pesticidal methods using substituted 3-pyridyl thiazole compounds and derivatives for combating animal pests |
| US10603318B2 (en) | 2012-07-03 | 2020-03-31 | Gilead Pharmasset Llc | Inhibitors of hepatitis C virus |
| US9296782B2 (en) | 2012-07-03 | 2016-03-29 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US10335409B2 (en) | 2012-07-03 | 2019-07-02 | Gilead Pharmasset Llc | Inhibitors of hepatitis C virus |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9617310B2 (en) | 2013-03-15 | 2017-04-11 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9512120B2 (en) | 2013-03-15 | 2016-12-06 | Syngenta Participations Ag | Microbicidally active imidazopyridine derivatives |
| WO2014140365A1 (en) * | 2013-03-15 | 2014-09-18 | Syngenta Participations Ag | Microbicidally active imidazopyridine derivatives |
| CN110305018A (en) * | 2019-06-06 | 2019-10-08 | 浙江普洛家园药业有限公司 | A kind of preparation method of the bromo- 2- fluoronitrobenzene of 3- |
| CN110305018B (en) * | 2019-06-06 | 2022-07-15 | 浙江普洛家园药业有限公司 | Preparation method of 3-bromo-2-fluoronitrobenzene |
Also Published As
| Publication number | Publication date |
|---|---|
| ECSP12011845A (en) | 2012-06-29 |
| AU2010298028A2 (en) | 2012-10-04 |
| TW201124137A (en) | 2011-07-16 |
| CA2775697A1 (en) | 2011-03-31 |
| IN2012DN02693A (en) | 2015-09-04 |
| MA33720B1 (en) | 2012-11-01 |
| JP2013505952A (en) | 2013-02-21 |
| IL218766A0 (en) | 2012-06-28 |
| EP2483290A1 (en) | 2012-08-08 |
| AR078462A1 (en) | 2011-11-09 |
| MX2012003500A (en) | 2012-08-01 |
| CO6531497A2 (en) | 2012-09-28 |
| CN102741270B (en) | 2015-07-22 |
| KR20130026410A (en) | 2013-03-13 |
| EP2483290A4 (en) | 2013-05-01 |
| AU2010298028A1 (en) | 2012-04-19 |
| CN105001302A (en) | 2015-10-28 |
| EA201290128A1 (en) | 2013-01-30 |
| CN102741270A (en) | 2012-10-17 |
| US20110081315A1 (en) | 2011-04-07 |
| TN2012000135A1 (en) | 2013-09-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011038293A1 (en) | Cyclic peptide inhibitors of hepatitis c virus replication | |
| US8048862B2 (en) | Macrocyclic inhibitors of hepatitis C virus replication | |
| EP1999129B1 (en) | Compounds and methods for inhibiting hepatitis c viral replication | |
| EP2177523A1 (en) | Novel macrocyclic inhibitors of hepatitis c virus replication | |
| KR20100027134A (en) | Novel peptide inhibitors of hepatitis c virus replication | |
| WO2011041551A1 (en) | Therapeutic antiviral peptides | |
| EP2044032A2 (en) | Novel inhibitors of hepatitis c virus replication | |
| WO2011146401A1 (en) | Novel inhibitors of hepatitis c virus replication | |
| WO2010045266A1 (en) | Therapeutic antiviral peptides | |
| WO2011038283A1 (en) | Novel macrocyclic inhibitors of hepatitis c virus replication | |
| WO2012054874A1 (en) | Novel macrocyclic inhibitors of hepatitis c virus replication | |
| WO2012037259A1 (en) | Novel inhibitors of hepatitis c virus replication | |
| WO2012047764A1 (en) | Therapeutic antiviral peptides | |
| WO2011057215A1 (en) | Novel inhibitors of hepatitis c virus replication |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080053601.9 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10819571 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 218766 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12012500602 Country of ref document: PH Ref document number: MX/A/2012/003500 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010298028 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2775697 Country of ref document: CA Ref document number: 2012531086 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2693/DELNP/2012 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010819571 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201290128 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2010298028 Country of ref document: AU Date of ref document: 20100924 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20127010337 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201205153 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12069712 Country of ref document: CO |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012007396 Country of ref document: BR |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: 112012007396 Country of ref document: BR |
|
| ENPW | Started to enter national phase and was withdrawn or failed for other reasons |
Ref document number: 112012007396 Country of ref document: BR |