WO2011020861A1 - Heterocyclic oxime compounds - Google Patents
Heterocyclic oxime compounds Download PDFInfo
- Publication number
- WO2011020861A1 WO2011020861A1 PCT/EP2010/062057 EP2010062057W WO2011020861A1 WO 2011020861 A1 WO2011020861 A1 WO 2011020861A1 EP 2010062057 W EP2010062057 W EP 2010062057W WO 2011020861 A1 WO2011020861 A1 WO 2011020861A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- alkyl
- compound
- methyl
- optionally substituted
- Prior art date
Links
- 0 CC(c1cnc2nn[n](Cc(cc(cccn3)c3c3)c3F)c2n1)=N*[C@]1CNCC1 Chemical compound CC(c1cnc2nn[n](Cc(cc(cccn3)c3c3)c3F)c2n1)=N*[C@]1CNCC1 0.000 description 2
- KJZCMRGSXUDMRS-RPPGKUMJSA-N C/C(/c(cc1)n[n]2c1ncc2Cc(c(F)cc1c2cccn1)c2F)=N\OCCO Chemical compound C/C(/c(cc1)n[n]2c1ncc2Cc(c(F)cc1c2cccn1)c2F)=N\OCCO KJZCMRGSXUDMRS-RPPGKUMJSA-N 0.000 description 1
- ADCSWIVUDXJHKA-WYMPLXKRSA-N C/C(/c(cc1)n[n]2c1ncc2Cc(cc(cccn1)c1c1)c1F)=N\OC Chemical compound C/C(/c(cc1)n[n]2c1ncc2Cc(cc(cccn1)c1c1)c1F)=N\OC ADCSWIVUDXJHKA-WYMPLXKRSA-N 0.000 description 1
- LUCHSWBIHFKBSN-AFUMVMLFSA-N C/C(/c(cc1)n[n]2c1ncc2Cc(cc1)cc2c1nccc2)=N\OCC(CO)O Chemical compound C/C(/c(cc1)n[n]2c1ncc2Cc(cc1)cc2c1nccc2)=N\OCC(CO)O LUCHSWBIHFKBSN-AFUMVMLFSA-N 0.000 description 1
- VTRDOGKAJOPEHB-LSFURLLWSA-N C/C(/c1cnc2nn[n](Cc(cc(cccn3)c3c3)c3F)c2n1)=N\O Chemical compound C/C(/c1cnc2nn[n](Cc(cc(cccn3)c3c3)c3F)c2n1)=N\O VTRDOGKAJOPEHB-LSFURLLWSA-N 0.000 description 1
- IYDJLLFRPDINJR-RWPZCVJISA-N C/C(/c1cnc2nn[n](Cc(cc3)cc4c3nccc4)c2n1)=N\OCC1CCNCC1 Chemical compound C/C(/c1cnc2nn[n](Cc(cc3)cc4c3nccc4)c2n1)=N\OCC1CCNCC1 IYDJLLFRPDINJR-RWPZCVJISA-N 0.000 description 1
- FKDCFTCWGQQZSP-WYMPLXKRSA-N C/C(/c1cnc2nn[n](Cc(cc3)cc4c3nccc4)c2n1)=N\OCCN Chemical compound C/C(/c1cnc2nn[n](Cc(cc3)cc4c3nccc4)c2n1)=N\OCCN FKDCFTCWGQQZSP-WYMPLXKRSA-N 0.000 description 1
- SSXORFSXSFSXSD-NLOAGFHQSA-N C/C(/c1cnc2nn[n](Cc(cc3)cc4c3nccc4)c2n1)=N\O[C@H]1CNCC1 Chemical compound C/C(/c1cnc2nn[n](Cc(cc3)cc4c3nccc4)c2n1)=N\O[C@H]1CNCC1 SSXORFSXSFSXSD-NLOAGFHQSA-N 0.000 description 1
- FYVCBFXRWYNZNX-DHRITJCHSA-N CC(c1cnc(cc2)[n]1nc2/C(/C)=N/OCCO)c(c(F)c1)cc2c1[n](C)nc2 Chemical compound CC(c1cnc(cc2)[n]1nc2/C(/C)=N/OCCO)c(c(F)c1)cc2c1[n](C)nc2 FYVCBFXRWYNZNX-DHRITJCHSA-N 0.000 description 1
- YKYVROGUOHEXED-UHFFFAOYSA-N CC(c1cnc(cc2)[n]1nc2C(C)=O)c(cc(cccn1)c1c1)c1F Chemical compound CC(c1cnc(cc2)[n]1nc2C(C)=O)c(cc(cccn1)c1c1)c1F YKYVROGUOHEXED-UHFFFAOYSA-N 0.000 description 1
- UCVNKHOURHKXRE-UHFFFAOYSA-N CC(c1cnc(cc2)[n]1nc2C(C)=O)c(cc(cn[n]1C)c1c1)c1F Chemical compound CC(c1cnc(cc2)[n]1nc2C(C)=O)c(cc(cn[n]1C)c1c1)c1F UCVNKHOURHKXRE-UHFFFAOYSA-N 0.000 description 1
- LPBQYCQCBLELPY-UHFFFAOYSA-N CC(c1cnc(cc2)[n]1nc2Cl)(c(c(F)c1)cc2c1nccc2)N Chemical compound CC(c1cnc(cc2)[n]1nc2Cl)(c(c(F)c1)cc2c1nccc2)N LPBQYCQCBLELPY-UHFFFAOYSA-N 0.000 description 1
- SBEBYFOHKKDHGZ-UHFFFAOYSA-N CC(c1cnc(cc2)[n]1nc2Cl)(c(cc(cn[n]1C)c1c1)c1F)O Chemical compound CC(c1cnc(cc2)[n]1nc2Cl)(c(cc(cn[n]1C)c1c1)c1F)O SBEBYFOHKKDHGZ-UHFFFAOYSA-N 0.000 description 1
- ODNBVEIAQAZNNM-UHFFFAOYSA-N CC(c1cnc(cc2)[n]1nc2Cl)=O Chemical compound CC(c1cnc(cc2)[n]1nc2Cl)=O ODNBVEIAQAZNNM-UHFFFAOYSA-N 0.000 description 1
- FEHQAVXMYVVATN-UHFFFAOYSA-N CC(c1cnc(cc2)[n]1nc2Cl)c(c(F)c1)cc2c1nccc2 Chemical compound CC(c1cnc(cc2)[n]1nc2Cl)c(c(F)c1)cc2c1nccc2 FEHQAVXMYVVATN-UHFFFAOYSA-N 0.000 description 1
- SNQNODAYAFHXOU-UHFFFAOYSA-N CC(c1cnc(cc2)[n]1nc2Cl)c(cc(cn[n]1C)c1c1)c1F Chemical compound CC(c1cnc(cc2)[n]1nc2Cl)c(cc(cn[n]1C)c1c1)c1F SNQNODAYAFHXOU-UHFFFAOYSA-N 0.000 description 1
- JPFPUIVEKFDIOV-UHFFFAOYSA-N CCOC(c(cc1)n[n]2c1ncc2C(C)c(cc(cn[n]1C)c1c1)c1F)=C Chemical compound CCOC(c(cc1)n[n]2c1ncc2C(C)c(cc(cn[n]1C)c1c1)c1F)=C JPFPUIVEKFDIOV-UHFFFAOYSA-N 0.000 description 1
- GDAYFDQSHBRCBT-KGGZJZRKSA-N C[C@@H](c1cnc(cc2)[n]1nc2/C(/C)=N/OCCO)c(cc(cccn1)c1c1)c1F Chemical compound C[C@@H](c1cnc(cc2)[n]1nc2/C(/C)=N/OCCO)c(cc(cccn1)c1c1)c1F GDAYFDQSHBRCBT-KGGZJZRKSA-N 0.000 description 1
- GDAYFDQSHBRCBT-VLJKMFQDSA-N C[C@H](c1cnc(cc2)[n]1nc2/C(/C)=N/OCCO)c(cc(cccn1)c1c1)c1F Chemical compound C[C@H](c1cnc(cc2)[n]1nc2/C(/C)=N/OCCO)c(cc(cccn1)c1c1)c1F GDAYFDQSHBRCBT-VLJKMFQDSA-N 0.000 description 1
- LILHEKOPCWVHAA-UHFFFAOYSA-N C[n](c1c2)ncc1cc(Br)c2F Chemical compound C[n](c1c2)ncc1cc(Br)c2F LILHEKOPCWVHAA-UHFFFAOYSA-N 0.000 description 1
- AKVDMQHGNSYFAG-UHFFFAOYSA-N C[n]1ncc2cc(C=O)ccc12 Chemical compound C[n]1ncc2cc(C=O)ccc12 AKVDMQHGNSYFAG-UHFFFAOYSA-N 0.000 description 1
- GEOOVYBHZDGENN-UHFFFAOYSA-N NC(CON(C(c1ccccc11)=O)C1=O)=O Chemical compound NC(CON(C(c1ccccc11)=O)C1=O)=O GEOOVYBHZDGENN-UHFFFAOYSA-N 0.000 description 1
- JNZSBFJKDDTGFN-UHFFFAOYSA-N O=C(c1cnc(cc2)[n]1nc2Cl)c(c(F)c1)cc2c1nccc2 Chemical compound O=C(c1cnc(cc2)[n]1nc2Cl)c(c(F)c1)cc2c1nccc2 JNZSBFJKDDTGFN-UHFFFAOYSA-N 0.000 description 1
- NJQHABZKCJRYDM-UHFFFAOYSA-N OC(c1cnc(cc2)[n]1nc2Cl)c1cc2cccnc2cc1F Chemical compound OC(c1cnc(cc2)[n]1nc2Cl)c1cc2cccnc2cc1F NJQHABZKCJRYDM-UHFFFAOYSA-N 0.000 description 1
- CFMZSMGAMPBRBE-UHFFFAOYSA-N ON(C(c1ccccc11)=O)C1=O Chemical compound ON(C(c1ccccc11)=O)C1=O CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/5025—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/06—Peri-condensed systems
Definitions
- the invention relates to bicyclic compounds of formula (I) and salts thereof, the uses of such compounds to treat the human or animal body, in particular with regard to a proliferative disease, pharmaceutical compositions comprising such compounds, combinations comprising a compound of formula (I), and processes for the preparation of such
- the Hepatocyte Growth Factor Receptor herein referred to as c-Met, is a receptor tyrosine kinase that has been shown to be over-expressed and/or genetically altered in a variety of malignancies, specifically, gene amplification and a number of c-Met mutations are found in various solid tumors, see e.g. WO2007/126799. Further, the receptor tyrosine kinase c-Met is involved in the processes of migration, invasion and morphogenesis that accompany embryogenesis and tissue regeneration. C-met is also involved in the process of metastasis. Several lines of evidence have indicated that c-Met plays a role in tumor pathogenesis.
- Gain of function germ line mutations in c-Met is associated with development of hereditary papillary renal cell carcinoma (PRCC). Amplification or mutations in c-Met have also been reported in sporadic forms of PRCC, in head and neck squamous cell carcinoma, in gastric carcinoma, in pancreatic carcinoma and in lung cancer. Such alterations have been shown in selected instances to confer dependence of the tumor on c-Met and/or resistence to other targeted therapies. Elevated levels of c-Met, together with its unique ligand HGF/SF, are observed at high frequency in multiple clinically relevant tumors. A correlation between increased expression and disease progression, metastases and patient mortality has been reported in several cancers, including bladder, breast, squamous cell carcinoma and gastric carcinoma as well as leiomyosarcoma and glioblastoma.
- PRCC hereditary papillary renal cell carcinoma
- WO 2008/008539 discloses certain fused heterocyclic derivatives which are useful in the treatment of HGF mediated diseases.
- WO 2007/013673 discloses fused heterocyclic derivatives as Lck inhibitors which are useful as immunosuppressive agents.
- EP0490587 discloses certain pyrazolopyrimidines which are useful as angiotensin Il antagonists.
- the disclosures of the publications cited in this specification are herein incorporated by reference. It is an aim of the present invention to provide further compounds that modulate, and in particular inhibit, c-Met.
- the compounds of the formula (I) described herein are inhibitors of c-Met and have a number of therapeutic applications.
- the compounds of formula (I) are suitable for use in the treatment of diseases dependent on c-Met activity, especially solid tumors or metastasis derived therefrom.
- compounds of the invention also have utility as antiinflammatory agents, for example for the treatment of an inflammatory condition which is due to an infection.
- the compounds of the invention are metabolically stable, are non-toxic and demonstrate few side-effects.
- preferred compounds of the invention exist in a physical form that is stable, non-hygroscopic and easily formulated.
- One aspect of the invention is directed to compounds of formula (I) having an activity that is superior to the activity of compounds of the prior art, or other similar compounds.
- Another aspect of the invention is directed to compounds of formula (I) having have good kinase selectivity.
- Preferred compounds of the invention posses favourable pharmacokinetic properties, such as good in-vivo exposure and/or solubility.
- the present invention relates to a compound of the formula (I)
- Y is C or N
- X is CH or N
- B is CH or N
- A is a ring; such that when X is CH and B is N, ring A is ring Ai or ring Aii;
- R 1 is a group selected from i, ii and iii: V - A - wherein R 5 is heteroaryl 1 ,
- R 6 is hydrogen, deuterium, OH, methyl or halo
- R 7 is hydrogen, deuterium, halo, or (d-C 3 )alkyl, wherein said (d-C 3 )alkyl is optionally substituted by one or more substituents independently selected from OH and halo; or R 6 and R 7 , together with the carbon to which they are attached, form cyclopropyl
- n is O, 1 or 2;
- R 2 is hydrogen, NH 2 , or (d-C 4 )alkyl, wherein said (d-C 4 )alkyl is optionally substituted by one or more substituents independently selected from OH, NH 3 and halo; and R 3 is
- phenyl wherein said phenyl is optionally substituted by one or more substituents independently selected from halo, hydroxy, (d-C 3 )alkyl and (d-C 3 )alkoxy, wherein said (C r C 3 )alkyl and (C r C 3 )alkoxy are each optionally substituted by one or more halo substituents,
- a “compound of the invention”, or “compounds of the invention”, or “a compound of the present invention” means a compound or compounds of formula (I) as described herein.
- the terms “including”, “containing” and “comprising” are used herein in their open, non-limiting sense.
- Treatment includes prophylactic (preventive) and therapeutic treatment as well as the delay of progression of a disease, disorder or condition. Any non-cyclic carbon containing group or moiety with more than 1 carbon atom is straight- chain or branched.
- Alkyl refers to a straight-chain or branched-chain alkyl group. For example, (d-C 4 )alkyl includes methyl, ethyl, n- or iso-propyl, and n-, iso-, sec- or tert-butyl.
- heteroaryl 1 is a 9- or 10- membered, unsaturated or partially unsaturated bicyclic group comprising 1 , 2, 3 or 4 ring heteroatoms independently selected from N, O and S, wherein the total number of ring S atoms does not exceed 1 , and the total number of ring O atoms does not exceed 1 , wherein heteroaryl 1 is optionally substituted by one or more substituents, preferably 1 , 2 or 3 substituents, independently selected from:
- heteroaryl 1 is optionally substituted by one or more substituents, preferably 1 , 2 or 3 substituents, independently selected from:
- heteroaryl 1 include, but are not limited to, quinolinyl, isoquinolinyl, cinnolinyl, quinazolinyl, quinoxalinyl, phthalazinyl, 1 ,5-naphthyridinyl, 1 ,6-naphthyridinyl, 1 ,7- naphthyridinyl, 1 ,8-naphthyridinyl, 2,6-naphthyridinyl, 2,7-naphthyridinyl, pyrido[3,2- d]pyrimidinyl, pyrido[4,3-d]pyrimidinyl, pyrido[3,4-d]pyrimidinyl, pyrido[2,3-d]pyrimidinyl, pyrido[2,3-d]pyrimidinyl, pyrido[2,3-b]pyrazinyl, pyrido[
- benzimidazolyl benzoxazolyl, indazoyl, benzotriazolyl, pyrrolo[2,3-b]pyridinyl, pyrrolo[2,3- c]pyridinyl, pyrrolo[3,2-c]pyridinyl, pyrrolo[3,2-b]pyridinyl, imidazo[4,5-b]pyridinyl, imidazo[4,5- c]pyridinyl, pyrazolo[4,3-d]pyridinyl, pyrazolo[4,3-c]pyridinyl, pyrazolo[3,4-c]pyridinyl, pyrazolo[3,4-b]pyridinyl, isoindolyl, indazolyl, purinyl, indolizinyl, imidazo[1 ,2-a]pyridinyl, imidazo[1 ,5-a]pyridinyl, pyrazolo[1
- heteroaryl 3 means a 9- or 10- membered, unsaturated or partially unsaturated bicyclic group comprising 1 or 2 ring N heteroatoms. Heteroaryl 3 is optionally substituted by one or more substituents independently selected from:
- heteroaryl 3 is optionally substituted by one or more substituents independently selected from:
- heteroaryl 3 include, but are not limited to, quinolinyl, isoquinolinyl, cinnolinyl, azaquinazolinyl, quinoxalinyl, phthalazinyl, 1 ,5-naphthyridinyl, 1 ,6-naphthyridinyl, 1 ,7-naphthyridinyl, 1 ,8-naphthyridinyl, 2,6-naphthyridinyl, 2,7-naphthyridinyl, indolyl, benzimidazolyl, indazolyl, pyrrolo[2,3-b]pyridinyl, pyrrolo[2,3-c]pyridinyl, pyrrolo[3,2- c]pyridinyl, pyrrolo[3,2-b]pyridinyl, isoindolyl, indazolyl, indolininyl,
- heterocyclyl 1 means a 4, 5 or 6 membered saturated or partially unsaturated monocyclic group comprising 1 or 2 ring heteroatoms independently selected from N, O and S.
- Heterocyclyl 1 is optionally substituted by CONH 2 or (d-C 3 )alkyl.
- Specific examples of heterocyclyl 1 include, but are not limited to, oxetanyl, thiatanyl, azetidinyl, tetrahydrofuranyl, tetrahydrothiophenyl, 1 ,2,3,4-tetrahydropyridinyl, 1 ,2,5,6-tetrahydropyridinyl, pyrrolidinyl, piperidinyl and piperazinyl.
- heterocyclyl 2 means a 5 or 6 membered saturated, unsaturated or partially unsaturated monocyclic group comprising 1 , 2 or 3 ring heteroatoms independently selected from N, O and S, wherein the total number of ring S atoms does not exceed 1 , and the total number of ring O atoms does not exceed 1 .
- Heterocyclyl 2 is optionally substituted by (d-C 3 )alkyl.
- heterocyclyl 2 include, but are not limited to, tetrahydrofuranyl, tetrahydrothiophenyl, 1 ,2,3,4-tetrahydropyridinyl, 1 ,2,5,6-tetrahydropyridinyl, pyrrolidinyl, piperidinyl, piperazinyl, pyrrolyl, furanyl, thiophenyl, pyrazolyl, imidazolyl, isoxazolyl, oxazolyl, isothiazolyl, thiazolyl, 1 ,2,3-triazolyl, 1 ,3,4-triazolyl, 1 -oxa-2,3-diazolyl, 1 -oxa-2,4-diazolyl, 1 - oxa-2,5-diazolyl, 1 -oxa-3,4-diazolyl, 1 -thia-2,3-diazolyl, 1 -thia
- halo means fluoro, chloro, bromo or iodo. In a particular embodiment of the invention, halo is fluoro or chloro. Preferably, halo is fluoro.
- the compound of formula (I) is selected from any one of the following structures (Ia) to (Ie):
- Y is C or N
- X is CH or N
- Such compounds have the structure (Ia), (Ib), (Ic) or (Ie) as disclosed herein.
- B is N;
- Y is C or N;
- X is CH or N;
- Such compounds have the structure (Ia), (Ib) or (Ic) as disclosed herein.
- R 1 is i or ii:
- R 1 is
- R 1 is a group selected from i, ii and iii: and R 5 is heteroaryl 3 . In another embodiment of the invention, R 1 is
- R 5 is heteroaryl 3 .
- heteroaryl 1 and heteroaryl 3 are each optionally substituted by one or more substituents independently selected from halo, 4-methylpiperazin- 1 -yl, ethanone O-(2-hydroxyethyl)oxime and (CrC 2 )alkyl.
- R 5 is indazolyl or quinolinyl, each optionally substituted by one or more substituents independently selected from halo, (d-C 3 )alkyl, 4- methylpiperazin-1 -yl and ethanone O-(2-hydroxyethyl)oxime.
- R 5 is indazolyl or quinolinyl optionally substituted by one, two or three substituents independently selected from halo and (CrC 3 )alkyl.
- R 5 is indazolyl optionally substituted by one, two or three substituents independently selected from methyl and fluoro, or R 5 is quinolinyl optionally substituted by one or two fluoro substituents.
- R 5 is indazol-5-yl substituted at the 1 position by a methyl substituent and optionally further substituted by one or two fluoro substituents, or R 5 is quinolin-6-yl optionally substituted by one or two fluoro substituents.
- said fluoro substituents are present at the 5 and/or 7 positions of the quinolinyl group.
- R 5 is quinolin-6-yl substituted by
- R 5 is quinolin-6-yl substituted by 3-(4-Methylpiperazin-1 -yl) or quinolin-6-yl substituted by 3-ethanone O-(2-hydroxy-ethyl)oxime.
- R 5 is 7-fluoro-quinolin-6-yl, quinolin-6-yl, 5,7- difluoroquinolin-6-yl, 1 -methyl-1 H-indazol-5-yl, 6-fluoro-1 -methyl-1 H-indazol-5-yl, 4,6-difluoro- 1 -methyl-1 H-indazol-5-yl, 5-Fluoroquinolin-6-yl, 3-(4-Methylpiperazin-1 -yl)quinolin-6-yl or 6- quinolin-3-yl-ethanone O-(2-hydroxy-ethyl)oxime.
- R 6 is hydrogen, deuterium, OH or halo, particularly hydrogen, deuterium or halo, and in a preferred embodiment, R 6 is hydrogen.
- R 7 is hydrogen, deuterium, halo, or methyl, wherein said methyl is optionally substituted by one or more substituents independently selected from OH and halo.
- R 7 is hydrogen, deuterium, halo, or methyl.
- R 7 is hydrogen or methyl, more preferably hydrogen.
- the compound of formula (I) contains an asymmetric carbon atom at R 1 .
- R 1 is i, and R 6 and R 7 are not both hydrogen
- the compound of formula (I) contains an asymmetric carbon atom at R 1 .
- R 1 is i, and R 6 and R 7 are not both hydrogen
- the compound of formula (I) contains an asymmetric carbon atom at R 1 .
- n is O.
- R 2 is hydrogen or methyl.
- R 2 is methyl.
- R 3 is selected from:
- phenyl wherein said phenyl is optionally substituted by one or more substituents independently selected from halo, hydroxy, (d-C 3 )alkyl and (d-C 3 )alkoxy, wherein said (CrC 3 )alkyl and (d-C 3 )alkoxy are each optionally substituted by one or more halo substituents,
- R 3 is selected from:
- phenyl wherein said phenyl is optionally substituted by one or more substituents independently selected from halo, hydroxy, (d-C 3 )alkyl and (d-C 3 )alkoxy, wherein said (Ci-C 3 )alkyl and (d-C 3 )alkoxy are each optionally substituted by one or more halo substituents,
- R 3 is ethyl, methyl, 2-hydroxyethyl, 1 ,3- dihydroxypropan-2-yl-, 2,3-dihydroxypropyl-, 2-hydroxy-1 ,1 -dimethyl-ethyl-, 2-hydroxy-2- methyl-propyl-, pyrrolidin-3-yl-, -3-pyrrolidine-1 -carboxamide, hydrogen, benzyl, A- methoxybenzyl-, piperidin-4-yl-, piperidin-4-ylmethyl-, -CH 2 CO 2 CH 3 , -CH 2 CONH 2 2- aminoethyl-, 1 -hydroxypropan-2-yl-, -CH 2 CH 2 NHCO 2 CH 3 , -CH 2 CH 2 NHCONH 2 ; 3- trifluoromethylphenyl-, phenyl, 2-fluoroethyl-, 3-hydroxypropyl, 2-methoxyethyl, A- hydroxybutyl
- R 3 is hydroxyethyl, particularly 2- hydroxyethyl, or hydrogen.
- Y is C or N
- X is CH or N
- B is CH or N
- A is a ring; such that when X is CH and B is N, ring A is ring Ai or ring Aii;
- R 1 is i:
- R 5 is 7-fluoro-quinolin-6-yl, quinolin-6-yl, 5,7-difluoroquinolin-6-yl, 1 -methyl-1 H-indazol-5-yl, 6- fluoro-1 -methyl-1 H-indazol-5-yl, 4,6-difluoro-1 -methyl-1 H-indazol-5-yl, 5-Fluoroquinolin-6-yl, (4-Methylpiperazin-1 -yl)quinolin-6-yl or 6-quinolin-3-yl-ethanone O-(2-hydroxy-ethyl)oxime
- R 6 is hydrogen, deuterium, OH, methyl or halo
- R 7 is hydrogen, deuterium, halo, or (d-C 3 )alkyl, wherein said (d-C 3 )alkyl is optionally substituted by one or more substituents independently selected from OH and halo; or R 6 and R 7 , together with the carbon to which they are attached form cyclopropyl;
- R 2 is hydrogen, NH 2 , or (CrC 4 )alkyl
- R 3 is ethyl, methyl, 2-hydroxyethyl, 1 ,3-dihydroxypropan-2-yl-, 2,3-dihydroxypropyl-, 2- hydroxy-1 ,1 -dimethyl-ethyl-, 2-hydroxy-2-methyl-propyl-, pyrrolidin-3-yl-, -3-pyrrolidine-1 - carboxamide, hydrogen, benzyl, 4-methoxybenzyl-, piperidin-4-yl-, piperidin-4-ylmethyl-, - CH 2 CO 2 CH 3 , -CH 2 CONH 2 2-aminoethyl-, 1 -hydroxypropan-2-yl-, -CH 2 CH 2 NHCO 2 CH 3 , - CH 2 CH 2 NHCONH 2 ; 3-trifluoromethylphenyl-, phenyl, 2-fluoroethyl-, 3-hydroxypropy
- the invention provides a compound of formula (I) having a structure selected from (Ia), (Ib) or (Ic) above, wherein
- R 1 is i:
- R 5 is quinolin-6-yl optionally substituted by one or two fluoro substituents, the fluoro substituents being preferably in the 5 and/or 7 positions;
- R 6 is hydrogen
- R 7 is hydrogen or methyl
- R 2 is methyl
- R 3 is hydroxyethyl, more particularly 2-hydroxyethyl, or R 3 is hydrogen;
- the invention provides one or more compounds selected from the Example compounds disclosed herein, or a pharmaceutically acceptable salt thereof.
- Figure 1 herein shows the X-ray diffractogram of Example 53S amorphous form.
- Figure 2 shows the X-ray diffractogram of Example 53S crystalline form I.
- Figure 3 shows the X-ray diffractogram of Example 53S crystalline form II.
- isomers refers to different compounds that have the same molecular formula but differ in arrangement and configuration of the atoms.
- an optical isomer or "a stereoisomer” refers to any of the various stereo isomeric configurations which may exist for a given compound of the present invention and includes geometric isomers. It is understood that a substituent may be attached at a chiral center of a carbon atom.
- the invention includes enantiomers, diastereomers or racemates of the compound.
- Enantiomers are a pair of stereoisomers that are non- superimposable mirror images of each other.
- a 1 :1 mixture of a pair of enantiomers is a "racemic” mixture.
- the term is used to designate a racemic mixture where appropriate.
- “Diastereoisomers” are stereoisomers that have at least two asymmetric atoms, but which are not mirror-images of each other. The absolute stereochemistry is specified according to the Cahn- Ingold- Prelog R-S system. When a compound is a pure enantiomer the stereochemistry at each chiral carbon may be specified by either R or S.
- Resolved compounds whose absolute configuration is unknown can be designated (+) or (-) depending on the direction (dextro- or levorotatory) which they rotate plane polarized light at the wavelength of the sodium D line.
- Certain of the compounds described herein contain one or more asymmetric centers or axes and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-.
- the present invention is meant to include all such possible isomers, including racemic mixtures, optically pure forms and intermediate mixtures.
- Optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques.
- the substituent may be E or Z configuration. If the compound contains a disubstituted cycloalkyl, the cycloalkyl substituent may have a cis- or trans-configuration. All tautomeric forms are also intended to be included.
- Any asymmetric atom (e.g., carbon or the like) of the compound(s) of the present invention can be present in racemic or enantiomerically enriched, for example the (R)-, (S)- or (R,S)- configuration, such as for the asymmetric carbon atom which may be present within the R 1 group (i) defined herein.
- each asymmetric atom has at least 50 % enantiomeric excess, at least 60 % enantiomeric excess, at least 70 % enantiomeric excess, at least 80 % enantiomeric excess, at least 90 % enantiomeric excess, at least 95 % enantiomeric excess, or at least 99 % enantiomeric excess in the (R)- or (S)- configuration.
- the (S) enantiomer is in excess, in amounts as described above.
- Substituents at atoms with unsaturated bonds may, if possible, be present in cis- (Z)- or trans- (E)- form.
- the hydrazones of the present invention have the trans-(£)- form.
- a compound of the present invention can be in the form of one of the possible isomers, rotamers, atropisomers, tautomers or mixtures thereof, for example, as substantially pure geometric (cis or trans) isomers, diastereomers, optical isomers (antipodes), racemates or mixtures thereof.
- Any resulting mixtures of isomers can be separated on the basis of the physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
- Any resulting racemates of final products or intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound.
- a basic moiety may thus be employed to resolve the compounds of the present invention into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl tartaric acid, diacetyl tartaric acid, di-0,0'-p-toluoyl tartaric acid, mandelic acid, malic acid or camphor-10-sulfonic acid. Racemic products can also be resolved by chiral
- isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 F 31 P, 32 P, 35 S, 36 CI, 125 I respectively.
- isotopically labeled compounds of the present invention for example those into which radioactive isotopes such as 3 H, 13 C, and 14 C are incorporated.
- isotopically labelled compounds are useful in metabolic studies (preferably with 14 C), reaction kinetic studies (with, for example 2 H or 3 H), detection or imaging techniques [such as positron emission tomography (PET) or single-photon emission computed tomography
- PET positron emission tomography
- SPECT single photoelectron emission tomography
- an 18 F or labeled compound may be particularly preferred for PET or SPECT studies.
- substitution with heavier isotopes such as deuterium (i.e., 2 H) may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements.
- isotopically labeled compounds of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the schemes or in the examples and preparations described below by substituting a. readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- isotopic enrichment factor means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
- any atom not specifically designated as a particular isotope is meant to represent any stable isotope of that atom.
- a position is designated specifically as “H” or “hydrogen”, the position is understood to have hydrogen at its natural abundance isotopic composition.
- any atom specifically designated as a deuterium (D) is meant to represent deuterium, for example in the ranges given above.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagent ⁇ in place of the non-labeled reagent previously employed.
- the term "pharmaceutically acceptable salts” refers to salts that retain the biological effectiveness and properties of the compounds of this invention and, which typically are not biologically or otherwise undesirable.
- the salt can be present alone or in mixture with free compound of the formula (I).
- the compounds of the present invention are capable of forming acid salts by virtue of the presence of amino groups or groups similar thereto.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfornate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, , hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/di
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- the pharmaceutically acceptable salts of the present invention can be synthesized from a parent compound, a basic or acidic moiety, by conventional chemical methods. Generally, such salts can be prepared by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid. Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two. Generally, use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable.
- any reference to the free compounds hereinbefore and hereinafter is to be understood as referring also to the corresponding salts, as appropriate and expedient.
- the compounds of the invention may exist in both unsolvated and solvated forms.
- the term 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- the term 'hydrate' is employed when said solvent is water.
- Pharmaceutically acceptable solvates include hydrates and other solvates wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 O, d 6 -acetone, d 6 -DMSO. Any formula given herein is intended to represent hydrates, solvates, and polymorphs of such compounds, and mixtures thereof.
- Compounds of the invention i.e. compounds of formula (I) that contain groups capable of acting as donors and/or acceptors for hydrogen bonds may be capable of forming co-crystals with suitable co-crystal formers.
- These co-crystals may be prepared from compounds of formula (I) by known co-crystal forming procedures. Such procedures include grinding, heating, co-subliming, co-melting, or contacting in solution compounds of formula (I) with the co-crystal former under crystallization conditions and isolating co-crystals thereby formed.
- Suitable co-crystal formers include those described in WO 2004/078163.
- the invention further provides co-crystals comprising a compound of formula (I).
- the compounds of the invention therefore include compounds of formula I, polymorphs, and isomers thereof (including optical, geometric and tautomeric isomers) and isotopically- labelled compounds of formula I, as defined herein.
- the invention relates to a compound of the formula (I), in free base form or in acid addition salt form, wherein the substituents are as defined herein.
- the compounds of the present invention may be administered as prodrugs.
- certain derivatives of compounds of formula (I) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) having the desired activity, for example, by hydrolytic cleavage.
- prodrugs Such derivatives are referred to as 'prodrugs'.
- 'prodrugs' Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) and 'Bioreversible Carriers in Drug Design', Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association).
- Prodrugs can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro- moieties' as described, for example, in "Design of Prodrugs" by H Bundgaard (Elsevier, 1985).
- prodrugs include:
- Certain compounds of formula (I) may also themselves act as prodrugs of other compounds of formula (I).
- C-Met tyrosine kinase mediated diseases are especially such disorders that respond in a beneficial way (e.g. amelioration of one or more symptoms, delay of the onset of a disease, up to temporary or complete cure from a disease) to the inhibition of a protein tyrosine kinase, especially inhibition of a c-Met kinase.
- These disorders include proliferative diseases such as tumor diseases, in particular solid tumors and metastasis derived thereof, e.g.
- PRCC hereditary papillary renal cell carcinoma
- sporadic forms of PRCC head and neck cancer
- squamous cell carcinoma gastric carcinoma
- pancreatic carcinoma lung cancer, bladder cancer, breast cancer
- leiomyosarcoma glioblastoma
- melanoma alveolar soft part sarcoma.
- inflammatory conditions such as inflammatory conditions due to an infection.
- Combination refers to either a fixed combination in one dosage unit form, or a kit of parts for the combined administration where a compound of the formula (I) and a combination partner (e.g. an other drug as explained below, also referred to as “therapeutic agent” or “co- agent”) may be administered independently at the same time or separately within time intervals, especially where these time intervals allow that the combination partners show a cooperative, e.g. synergistic effect.
- a combination partner e.g. an other drug as explained below, also referred to as “therapeutic agent” or “co- agent”
- administration or the like as utilized herein are meant to encompass administration of the selected combination partner to a single subject in need thereof (e.g. a patient), and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
- pharmaceutical combination as used herein means a product that results from the mixing or combining of more than one active ingredient and includes both fixed and non-fixed combinations of the active ingredients.
- fixed combination means that the active ingredients, e.g. a compound of formula (I) and a combination partner, are both administered to a patient simultaneously in the form of a single entity or dosage.
- non-fixed combination means that the active ingredients, e.g.
- a compound of formula (I) and a combination partner are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the two compounds in the body of the patient.
- cocktail therapy e.g. the administration of three or more active ingredients.
- the invention relates especially to a compound of the formula (I) as provided in the
- the compounds of formula (I) have valuable pharmacological properties, as described hereinbefore and hereinafter.
- a method for treating a c-Met related disorder or condition is preferably a proliferative disease such as a cancer or an inflammatory condition.
- Compounds of formula (I) are further useful for treating diseases associated with a c-Met-related condition.
- Compounds of formula (I) are useful in the treatment of cancer wherein the cancer is selected from the group consisting of brain cancer, stomach cancer, genital cancer, urinary cancer, prostate cancer, bladder cancer (superficial and muscle invasive), breast cancer, cervical cancer, colon cancer, colorectal cancer, glioma (including glioblastoma, anaplastic astrocytoma, oligoastrocytoma, oligodendroglioma), esophageal cancer, gastric cancer, gastrointestinal cancer, liver cancer, hepatocellular carcinoma (HCC) including childhood HCC, head and neck cancer (including head and neck squamous-cell carcinoma,
- HCC hepatocellular carcinoma
- nasopharyngeal carcinoma Hurthle cell carcinoma, epithelial cancer, skin cancer, melanoma (including malignant melanoma), mesothelioma, lymphoma, myeloma (including multiple myeloma), leukemias, lung cancer (including non-small cell lung cancer (including all histological subtypes: adenocarcinoma, squamous cell carcinoma, bronchoalveolar carcinoma, large-cell carcinoma, and adenosquamous mixed type), small-cell lung cancer), ovarian cancer, pancreatic cancer, prostate cancer, kidney cancer (including but not limited to papillary renal cell carcinoma), intestine cancer, renal cell cancer (including hereditary and sporadic papillary renal cell cancer, Type I and Type II, and clear cell renal cell cancer); sarcomas, in particular osteosarcomas, clear cell sarcomas, and soft tissue sarcomas (including alveolar and embryonal r
- Compounds of formula (I) are useful in the treatment of cancer wherein the cancer is stomach, colon, liver, genital, urinary, melanoma, or prostate.
- the cancer is liver or esophageal.
- Compounds of formula (I) are useful in the treatment of colon cancer, including metastases, e.g. in the liver, and of non-small-cell lung carcinoma.
- Compounds of formula (I) may also be used in the treatment of hereditary papillary renal carcinoma (Schmidt, L. et al. Nat. Genet. 16, 68-73, 1997) and other proliferative diseases in which c-MET is overexpressed or constitutively activated by mutations (Jeffers and Vande Woude. Oncogene 18, 5120-5125, 1999; and reference cited therein) or chromosomal rearrange-ments (e.g. TPR-MET; Cooper et al. Nature 31 1 , 29-33, 1984; Park, et al. Cell 45, 895-904, 1986).
- Compounds of formula (I) are further useful in the treatment of additional cancers and conditions as provided herein or known in the art.
- the inflammatory condition is due to an infection.
- the method of treatment would be to block pathogen infection.
- the infection is a bacterial infection, e.g., a Listeria infection. See, e.g., Shen et al. Cell 103: 501 -10, (2000) whereby a bacterial surface protein activates c-Met kinase through binding to the extracellular domain of the receptor, thereby mimicking the effect of the cognate ligand HGF/SF.
- any of the above methods involve further administering a chemotherapeutic agent.
- the chemotherapeutic agent is an anti-cancer agent. Specific combinations are provided throughout the application.
- any of the above methods involve further administering a pathway specific inhibitor.
- the pathway specific inhibitor may be a chemotherapeutic agent or may be a biologic agent, e.g., such as antibodies.
- Pathway specific inhibitors include, but are not limited to, inhibitors of EGFR, Her-2, Her-3, VEGFR, Ron, IGF-IR, PI-3K, mTOR,
- the invention relates to a method of treating a c-Met related disorder or condition which involves administering to a subject in need thereof an effective amount of a compound of formula (I).
- the invention relates to a compound of formula (I) or a
- the invention relates to the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of one or more C-Met tyrosine kinase mediated diseases.
- the invention relates to a method for the treatment of a disease or disorder which responds to an inhibition of C-Met tyrosine kinase, which comprises administering a compound of formula (I) or a pharmaceutically acceptable salt thereof, especially in a quantity effective against said disease, to a warm-blooded animal requiring such treatment.
- the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) as active ingredient in association with at least one pharmaceutical carrier or diluent.
- the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising: (a) an effective amount of compound of formula (I) and pharmaceutically acceptable salts, pharmaceutically acceptable prodrugs, and/or pharmaceutically active metabolites thereof; and (b) one or more pharmaceutically acceptable excipients and / or diluents.
- a pharmaceutical preparation comprising a compound of formula (I) as defined herein, or a pharmaceutically acceptable salt of such a compound, or a hydrate or solvate thereof, and at least one pharmaceutically acceptable carrier and / or diluents and optionally one or more further therapeutic agents.
- the term "pharmaceutically acceptable carrier” includes any and all solvents, dispersion media, coatings, surfactants, antioxidants, preservatives ⁇ e.g., antibacterial agents, antifungal agents), isotonic agents, absorption delaying agents, salts, preservatives, drugs, drug stabilizers, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, and the like and combinations thereof, as would be known to those skilled in the art (see, for example, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289- 1329). Except insofar as any conventional carrier is incompatible with the active ingredient, its use in the therapeutic or pharmaceutical compositions is contemplated.
- a therapeutically effective amount of a compound of the present invention refers to an amount of the compound of the present invention that will elicit the biological or medical response of a subject, for example, reduction or inhibition of an enzyme or a protein activity, or ameliorate symptoms, alleviate conditions, slow or delay disease progression, or prevent a disease, etc.
- the term "a therapeutically effective amount” refers to the amount of the compound of the present invention that, when administered to a subject, is effective to (1 ) at least partially alleviating, inhibiting, preventing and/or
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a cell, or a tissue, or a non-cellular biological material, or a medium, is effective to at least partially reducing or inhibiting the activity of cMet; or at least partially reducing or inhibiting the expression of cMet.
- the term "subject" refers to an animal. Typically the animal is a mammal. A subject also refers to for example, primates ⁇ e.g., humans), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice, fish, birds and the like. In certain embodiments, the subject is a primate. In yet other embodiments, the subject is a human.
- the term “inhibit”, “inhibition” or “inhibiting” refers to the reduction or suppression of a given condition, symptom, or disorder, or disease, or a significant decrease in the baseline activity of a biological activity or process.
- the term “treat”, “treating” or “treatment” of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat”, “treating” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- “treat”, “treating” or “treatment” refers to modulating the disease or disorder, either physically, ⁇ e.g., stabilization of a discernible symptom), physiologically, ⁇ e.g., stabilization of a physical parameter), or both.
- “treat”, “treating” or “treatment” refers to preventing or delaying the onset or development or progression of the disease or disorder.
- a subject is "in need of” a treatment if such subject would benefit biologically, medically or in quality of life from such treatment.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the present invention and a pharmaceutically acceptable carrier.
- the pharmaceutical composition can be formulated for particular routes of administration such as oral administration, parenteral administration, and rectal administration, etc.
- the pharmaceutical compositions of the present invention can be made up in a solid form
- compositions can be subjected to conventional pharmaceutical operations such as sterilization and/or can contain conventional inert diluents, lubricating agents, or buffering agents, as well as adjuvants, such as preservatives, stabilizers, wetting agents, emulsifers and buffers, etc.
- the pharmaceutical compositions are tablets or gelatin capsules comprising the active ingredient together with a) diluents, e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b) lubricants, e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethyleneglycol; for tablets also c) binders, e.g., magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired d) disintegrants, e.g., starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and/or e) absorbents, colorants, flavors and sweeteners.
- diluents e.g., lactose, dextrose, sucrose
- Tablets may be either film coated or enteric coated according to methods known in the art.
- compositions for oral administration include an effective amount of a compound of the invention in the form of tablets, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
- Compositions intended for oral use are prepared according to any method known in the art for the manufacture of pharmaceutical compositions and such compositions can contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets may contain the active ingredient in admixture with nontoxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients are, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example, starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets are uncoated or coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate can be employed.
- Formulations for oral use can be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example, peanut oil, liquid paraffin or olive oil.
- compositions are aqueous isotonic solutions or suspensions, and suppositories are advantageously prepared from fatty emulsions or suspensions.
- Said compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances.
- Said compositions are prepared according to conventional mixing, granulating or coating methods, respectively, and contain about 0.1 -75%, or contain about 1 -50%, of the active ingredient.
- compositions for transdermal application include an effective amount of a compound of the invention with a suitable carrier.
- Carriers suitable for transdermal delivery include absorbable pharmacologically acceptable solvents to assist passage through the skin of the host.
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound of the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- compositions for topical application include aqueous solutions, suspensions, ointments, creams, gels or sprayable formulations, e.g., for delivery by aerosol or the like.
- topical delivery systems will in particular be appropriate for dermal application, e.g., for the treatment of skin cancer, e.g., for prophylactic use in sun creams, lotions, sprays and the like. They are thus particularly suited for use in topical, including cosmetic, formulations well-known in the art.
- Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
- a topical application may also pertain to an inhalation or to an intranasal application. They may be conveniently delivered in the form of a dry powder (either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids) from a dry powder inhaler or an aerosol spray presentation from a pressurised container, pump, spray, atomizer or nebuliser, with or without the use of a suitable propellant.
- a dry powder either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids
- the invention relates also to a pharmaceutical composition
- a pharmaceutical composition comprising an effective amount, especially an amount effective in the treatment of one of the above-mentioned diseases, disorders or conditions, of a compound of formula (I) or a pharmaceutically acceptable salt thereof, together with one or more pharmaceutically acceptable carriers that are suitable for topical, enteral, for example oral or rectal, or parenteral administration and that may be inorganic or organic, solid or liquid.
- the dosage of the active ingredient to be applied to a warm-blooded animal depends upon a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; the renal and he- patic function of the patient; and the particular compound employed.
- a physician, clinician or veterinarian of ordinary skill can readily determine and prescribe the effective amount of the drug required to prevent, counter or arrest the progress of the condition.
- Optimal precision in achieving concentration of drug within the range that yields efficacy without toxicity requires a regimen based on the kinetics of the drug's availability to target sites. This involves a con- sideration of the distribution, equilibrium, and elimination of a drug.
- the dose of a compound of the formula (I) or a pharmaceutically acceptable salt thereof to be administered to warmblooded animals, for example humans of approximately 70 kg body weight, is preferably from approximately 3 mg to approximately 5 g, more preferably from approximately 10 mg to approximately 1 .5 g per person per day, divided preferably into 1 to 3 single doses which may, for example, be of the same size. Usually, children receive half of the adult dose.
- the pharmaceutical composition or combination of the present invention can be in unit dosage of about 1 -1000 mg of active ingredient(s) for a subject of about 50-70 kg, or about 1 -500 mg or about 1 -250 mg or about 1 -150 mg or about 0.5-100 mg, or about 1 -50 mg of active ingredients.
- the therapeutically effective dosage of a compound, the pharmaceutical composition, or the combinations thereof is dependent on the species of the subject, the body weight, age and individual condition, the disorder or disease or the severity thereof being treated. A physician, clinician or veterinarian of ordinary skill can readily determine the effective amount of each of the active ingredients necessary to prevent, treat or inhibit the progress of the disorder or disease.
- the above-cited dosage properties are demonstrable in vitro and in vivo tests using advantageously mammals, e.g., mice, rats, dogs, monkeys or isolated organs, tissues and preparations thereof.
- the compounds of the present invention can be applied in vitro in the form of solutions, e.g., aqueous solutions, and in vivo either enterally, parenterally, advantageously intravenously, e.g., as a suspension or in aqueous solution.
- the dosage in vitro may range between about 10 3 molar and 10 9 molar concentrations.
- a therapeutically effective amount in vivo may range depending on the route of administration, between about 0.1 -500 mg/kg, or between about 1 -100 mg/kg.
- a combination of a compound of formula (I) with one or more other therapeutically active agents in another embodiment, there is provided a combination of a compound of formula (I) with one or more other therapeutically active agents.
- a compound of formula (I) can be administered alone or in combination with one or more other therapeutic agents, possible combination therapy taking the form of fixed combinations or the administration of a compound of the invention and one or more other therapeutic agents being staggered or given independently of one another, or the combined administration of fixed combinations and one or more other therapeutic agents.
- a compound of formula (I) can besides or in addition be administered especially for tumor therapy in combination with chemotherapy, radiotherapy, immunotherapy, surgical intervention, or a combination of these.
- Long-term therapy is equally possible as is adjuvant therapy in the context of other treatment strategies, as described above.
- Other possible treatments are therapy to maintain the patient's status after tumor regression, or even chemopreventive therapy, for example in patients at risk.
- a compound of the formula (I) may be used in combination with other antiproliferative compounds.
- antiproliferative compounds include, but are not limited to aromatase inhibitors; antiestrogens; topoisomerase I inhibitors; topoisomerase Il inhibitors; microtubule active compounds; alkylating compounds; histone deacetylase inhibitors; compounds which induce cell differentiation processes; cyclooxygenase inhibitors; MMP inhibittors; mTOR inhibitors; antineoplastic antimetabolites; platin compounds; compounds targeting/decreasing a protein or lipid kinase activity; anti-angiogenic compounds; compounds which target, decrease or inhibit the activity of a protein or lipid phosphatase; gonadorelin agonists; anti- androgens; methionine aminopeptidase inhibitors; bisphosphonates; biological response modifiers; antiproliferative antibodies; heparanase inhibitors; inhibitors of Ras oncogenic isoforms; telomerase inhibitors; proteasome inhibitors; compounds used in the treatment of hematologic malignancies;com
- tumor treatment approaches including surgery, ionizing radiation, photodynamic therapy, implants, e.g. with corticosteroids, hormones, or they may be used as radiosensitizers.
- aromatase inhibitor as used herein relates to a compound which inhibits the estrogen production, i.e. the conversion of the substrates androstenedione and testosterone to estrone and estradiol, respectively.
- the term includes, but is not limited to steroids, especially atamestane, exemestane and formestane and, in particular, non-steroids, especially aminoglutethimide, roglethimide, pyridoglutethimide, trilostane, testolactone, ketokonazole, vorozole, fadrozole, anastrozole and letrozole.
- Exemestane can be administered, e.g., in the form as it is marketed, e.g.
- Form- estane can be administered, e.g., in the form as it is marketed, e.g. under the trademark LENTARON.
- Fadrozole can be administered, e.g., in the form as it is marketed, e.g. under the trademark AFEMA.
- Anastrozole can be administered, e.g., in the form as it is marketed, e.g. under the trademark ARIMIDEX.
- Letrozole can be administered, e.g., in the form as it is marketed, e.g. under the trademark FEMARA or FEMAR.
- Aminoglutethimide can be administered, e.g., in the form as it is marketed, e.g.
- a combination of the invention comprising a chemotherapeutic agent which is an aromatase inhibitor is particularly useful for the treatment of hormone receptor positive tumors, e.g. breast tumors.
- antiestrogen as used herein relates to a compound which antagonizes the effect of estrogens at the estrogen receptor level.
- the term includes, but is not limited to tamoxifen, fulvestrant, raloxifene and raloxifene hydrochloride. Tamoxifen can be administered, e.g., in the form as it is marketed, e.g. under the trademark NOLVADEX.
- Raloxifene hydrochloride can be administered, e.g., in the form as it is marketed, e.g. under the trademark EVISTA.
- Fulvestrant can be formulated as disclosed in US 4,659,516 or it can be administered, e.g., in the form as it is marketed, e.g. under the trademark FASLODEX.
- a combination of the invention comprising a chemotherapeutic agent which is an antiestrogen is particularly useful for the treatment of estrogen receptor positive tumors, e.g. breast tumors.
- anti-androgen as used herein relates to any substance which is capable of inhibiting the biological effects of androgenic hormones and includes, but is not limited to, bicalutamide (CASODEX), which can be formulated, e.g. as disclosed in US 4,636,505.
- bicalutamide CASODEX
- gonadorelin agonist as used herein includes, but is not limited to abarelix, go- serelin and goserelin acetate. Goserelin is disclosed in US 4,100,274 and can be administered, e.g., in the form as it is marketed, e.g. under the trademark ZOLADEX.
- Abarelix can be formulated, e.g. as disclosed in US 5,843,901.
- topoisomerase I inhibitor includes, but is not limited to topotecan, gimatecan, irinotecan, camptothecian and its analogues, 9-nitrocamptothecin and the macromolecular camptothecin conjugate PNU-166148 (compound A1 in WO99/ 17804).
- Irinotecan can be administered, e.g. in the form as it is marketed, e.g. under the trademark CAMPTOSAR.
- Topotecan can be administered, e.g., in the form as it is marketed, e.g. under the trademark HYCAMTIN.
- topoisomerase Il inhibitor includes, but is not limited to the an- thracyclines such as doxorubicin (including liposomal formulation, e.g. CAELYX), dauno- rubicin, epirubicin, idarubicin and nemorubicin, the anthraquinones mitoxantrone and lo- soxantrone, and the podophillotoxines etoposide and teniposide.
- Etoposide can be ad- ministered, e.g. in the form as it is marketed, e.g. under the trademark ETOPOPHOS.
- Teniposide can be administered, e.g. in the form as it is marketed, e.g. under the trademark VM 26-BRISTOL.
- Doxorubicin can be administered, e.g. in the form as it is marketed, e.g. under the trademark ADRIBLASTIN or ADRIAMYCIN.
- Epirubicin can be administered, e.g. in the form as it is marketed, e.g. under the trademark FARMORUBICIN.
- Idarubicin can be administered, e.g. in the form as it is marketed, e.g. under the trademark ZAVEDOS.
- Mitoxantrone can be administered, e.g. in the form as it is marketed, e.g. under the trademark NOVANTRON.
- microtubule active compound relates to microtubule stabilizing, microtubule destabilizing compounds and microtublin polymerization inhibitors including, but not limited to taxanes, e.g. paclitaxel and docetaxel, vinca alkaloids, e.g., vinblastine, especially vinblastine sulfate, vincristine especially vincristine sulfate, and vinorelbine, discodermolides, cochicine and epothilones and derivatives thereof, e.g. epothilone B or D or derivatives thereof.
- taxanes e.g. paclitaxel and docetaxel
- vinca alkaloids e.g., vinblastine, especially vinblastine sulfate, vincristine especially vincristine sulfate, and vinorelbine
- discodermolides cochicine and epothilones and derivatives thereof, e.g. epothilone B or D or derivative
- Paclitaxel may be administered e.g. in the form as it is marketed, e.g. TAXOL.
- Docetaxel can be administered, e.g., in the form as it is marketed, e.g. under the trademark TAXOTERE.
- Vinblastine sulfate can be administered, e.g., in the form as it is marketed, e.g. under the trademark VINBLASTIN R. P..
- Vincristine sulfate can be administered, e.g., in the form as it is marketed, e.g. under the trademark FARMISTIN.
- Discodermolide can be obtained, e.g., as disclosed in US 5,010,099.
- Epothilone derivatives which are disclosed in WO 98/10121 , US 6,194,181 , WO 98/25929, WO 98/08849, WO 99/43653, WO 98/22461 and WO 00/31247. Especially preferred are Epothilone A and/or B.
- alkylating compound includes, but is not limited to, cyclophospha- mide, ifosfamide, melphalan or nitrosourea (BCNU or Gliadel).
- Cyclophosphamide can be administered, e.g., in the form as it is marketed, e.g. under the trademark CYCLOSTIN.
- Ifosfamide can be administered, e.g., in the form as it is marketed, e.g. under the trademark HOLOXAN.
- histone deacetylase inhibitors or "HDAC inhibitors” relates to compounds which inhibit the histone deacetylase and which possess antiproliferative activity.
- SAHA Suberoylanilide hydroxamic acid
- HDAC histone deacetylase
- SAHA suberoylanilide hydroxamic acid
- HDAC inhibitors include MS275, SAHA, FK228 (formerly FR901228), Trichostatin A and compounds disclosed in US 6,552,065, in particular, ⁇ /-hydroxy-3-[4-[[[2-(2-methyl-1 H-indol-3-yl)-ethyl]-amino]me- thyl]phenyl]-2£-2-propenamide, or a pharmaceutically acceptable salt thereof and ⁇ /-hydroxy- 3-[4-[(2-hydroxyethyl) ⁇ 2-(1 H-indol-3-yl)ethyl]-amino]methyl]phenyl]-2E-2-propenamide, or a pharmaceutically acceptable salt thereof, especially the lactate salt.
- antimetabolite includes, but is not limited to, 5-Fluorouracil or 5-FU, capecitabine, gemcitabine, DNA demethylating compounds, such as 5-azacytidine and decitabine, methotrexate and edatrexate, and folic acid antagonists such as pemetrexed.
- Capecitabine can be administered, e.g., in the form as it is marketed, e.g. under the trademark XELODA.
- Gemcitabine can be administered, e.g., in the form as it is marketed, e.g. under the trademark GEMZAR.
- platinum compound as used herein includes, but is not limited to, carboplatin, cis- platin, cisplatinum and oxaliplatin.
- Carboplatin can be administered, e.g., in the form as it is marketed, e.g. under the trademark CARBOPLAT.
- Oxaliplatin can be administered, e.g., in the form as it is marketed, e.g. under the trademark ELOXATIN.
- compounds targeting/decreasing a protein or lipid kinase activity includes, but is not limited to, c-Met tyrosine kinase and/or serine and/or threonine kinase inhibitors or lipid kinase inhibitors, e.g.,
- PDGFR platelet-derived growth factor-receptors
- compounds which target, decrease or inhibit the activity of PDGFR especially compounds which inhibit the PDGF receptor, e.g. a N-phenyl-2- pyrimidine-amine derivative, e.g. imatinib, SU101 , SU6668 and GFB-1 1 1 ;
- FGFR fibroblast growth factor- receptors
- IGF-IR insulin-like growth factor receptor I
- IR especially compounds which inhibit the kinase activity of IGF-I receptor, such as those compounds disclosed in WO 02/092599, or antibodies that target the extracellular domain of IGF-I receptor or its growth factors;
- Kit/SCFR receptor tyrosine kinase e.g. imatinib
- gene-fusion products e.g. BCR-AbI kinase
- mutants such as compounds which target decrease or inhibit the activity of c-Abl family members and their gene fusion products, e.g. a N-phenyl-2-pyrimidine-amine derivative, e.g. imatinib or nilotinib (AMN107)
- PD180970 AG957; NSC 680410; PD173955 from ParkeDavis; or dasatinib (BMS-354825) j) compounds targeting, decreasing or inhibiting the activity of members of the protein kinase C (PKC) and Raf family of serine/threonine kinases, members of the MEK, SRC, JAK, FAK, PDK1 , PKB/Akt, and Ras/MAPK family members, and/or members of the cyclin- dependent kinase family (CDK) and are especially those staurosporine derivatives disclosed in US 5,093,330, e.g. midostaurin; examples of further compounds include e.g.
- UCN-01 safingol, BAY 43-9006, Bryostatin 1 , Perifosine; llmofosine; RO 318220 and RO 320432; GO 6976; lsis 3521 ; LY333531/LY379196; isochinoline compounds such as those disclosed in WO 00/09495; FTIs; PD184352 or QAN697 (a P13K inhibitor) or AT7519 (CDK inhibitor); k) compounds targeting, decreasing or inhibiting the activity of protein-tyrosine kinase inhibitors, such as compounds which target, decrease or inhibit the activity of protein-tyrosine kinase inhibitors include imatinib mesylate (GLEEVEC) or tyrphostin.
- GLEEVEC imatinib mesylate
- tyrphostin tyrphostin.
- a tyrphostin is preferably a low molecular weight (Mr ⁇ 1500) compound, or a pharmaceutically acceptable salt thereof, especially a compound selected from the benzylidenemalonitrile class or the S- arylbenzenemalonirile or bisubstrate quinoline class of compounds, more especially any compound selected from the group consisting of Tyrphostin A23/RG-50810; AG 99;
- adaphostin (4- ⁇ [(2,5-dihydroxyphenyl)methyl]amino ⁇ -benzoic acid adamantyl ester; NSC 680410, adaphostin);
- compounds targeting, decreasing or inhibiting the activity of the epidermal growth factor family of receptor tyrosine kinases (EGFR, ErbB2, ErbB3, ErbB4 as homo- or heterodimers) and their mutants, such as compounds which target, decrease or inhibit the activity of the epidermal growth factor receptor family are especially compounds, proteins or antibodies which inhibit members of the EGF receptor tyrosine kinase family, e.g. EGF receptor, ErbB2, ErbB3 and ErbB4 or bind to EGF or EGF related ligands, and are in particular those compounds, proteins or monoclonal antibodies generically and specifically disclosed in WO 97/02266, e.g. the compound of ex.
- trastuzumab HerceptinTM
- cetuximab ErbituxTM
- Iressa Tarceva
- OSI- 774 Tarceva
- OSI- 774 OSI- 774
- EKB-569 E1 .1 , E2.4, E2.5, E6.2, E6.4, E2.1 1 , E6.3 or E7.6.3, and 7H-pyrrolo-[2,3-d]pyrimidine derivatives which are disclosed in WO 03/013541 ; and m) compounds targeting, decreasing or inhibiting the activity of the c-Met receptor, such as compounds which target, decrease or inhibit the activity of c-Met, especially compounds which inhibit the kinase activity of c-Met receptor, or antibodies that target the extracellular domain of c-Met or bind to HGF;
- n) compounds targeting, decreasing or inhibiting the activity of the Ron receptor tyrosine kinase.
- anti-angiogenic compounds include compounds having another mechanism for their activity, e.g. unrelated to protein or lipid kinase inhibition e.g. thalidomide (THALOMID) and TNP-470.
- TAALOMID thalidomide
- TNP-470 TNP-470.
- Compounds which target, decrease or inhibit the activity of a protein or lipid phosphatase includes, but is not limited to inhibitors of phosphatase 1 , phosphatase 2A, or CDC25, e.g. okadaic acid or a derivative thereof.
- Compounds which induce cell differentiation processes includes, but is not limited to e.g. retinoic acid, ⁇ - ⁇ - or ⁇ -tocopherol or ⁇ - ⁇ - or ⁇ -tocotrienol.
- cyclooxygenase inhibitor as used herein includes, but is not limited to, e.g. Cox-2 inhibitors, 5-alkyl substituted 2-arylaminophenylacetic acid and derivatives, such as celecoxib (CELEBREX), rofecoxib (VIOXX), etoricoxib, valdecoxib or a 5-alkyl-2- arylaminophenylacetic acid, e.g. 5-methyl-2-(2'-chloro-6'-fluoroanilino)phenyl acetic acid, lumiracoxib.
- bisphosphonates as used herein includes, but is not limited to, etridonic, clodronic, tiludronic, pamidronic, alendronic, ibandronic, risedronic and zoledronic acid.
- Etridonic acid can be administered, e.g., in the form as it is marketed, e.g. under the trademark DIDRONEL.
- Clodronic acid can be administered, e.g., in the form as it is marketed, e.g. under the trademark BONEFOS.
- Teiludronic acid can be administered, e.g., in the form as it is marketed, e.g. under the trademark SKELID.
- Pamidronic acid can be administered, e.g. in the form as it is marketed, e.g. under the trademark AREDIATM.
- “Alendronic acid” can be administered, e.g., in the form as it is marketed, e.g. under the trademark FOSAMAX.
- “Ibandronic acid” can be administered, e.g., in the form as it is marketed, e.g. under the trademark BONDRANAT.
- “Risedronic acid” can be administered, e.g., in the form as it is marketed, e.g. under the trademark ACTONEL.
- “Zoledronic acid” can be administered, e.g. in the form as it is marketed, e.g. under the trademark ZOMETA.
- the term “mTOR inhibitors” relates to compounds which inhibit the mammalian target of rapamycin (mTOR) and which possess antiproliferative activity such as sirolimus
- heparanase inhibitor refers to compounds which target, decrease or inhibit heparin sulfate degradation.
- the term includes, but is not limited to, PI-88.
- biological response modifier refers to a lymphokine or interferons, e.g. interferon ⁇ .
- inhibitor of Ras oncogenic isoforms e.g. H-Ras, K-Ras, or N-Ras
- H-Ras, K-Ras, or N-Ras refers to compounds which target, decrease or inhibit the oncogenic activity of Ras e.g. a "farnesyl transferase inhibitor” e.g. L-744832, DK8G557 or R1 15777 (Zarnestra).
- telomerase inhibitor refers to compounds which target, decrease or inhibit the activity of telomerase.
- Compounds which target, decrease or inhibit the activity of telomerase are especially compounds which inhibit the telomerase receptor, e.g.
- methionine aminopeptidase inhibitor refers to compounds which target, decrease or inhibit the activity of methionine aminopeptidase.
- Compounds which target, decrease or inhibit the activity of methionine aminopeptidase are e.g. bengamide or a derivative thereof.
- proteasome inhibitor refers to compounds which target, decrease or inhibit the activity of the proteasome.
- Compounds which target, decrease or inhibit the activity of the proteasome include e.g. Bortezomid (VelcadeTM)and MLN 341.
- matrix metalloproteinase inhibitor or (“MMP” inhibitor) as used herein includes, but is not limited to, collagen peptidomimetic and nonpeptidomimetic inhibitors, tetracycline derivatives, e.g.
- FMS-like tyrosine kinase inhibitors e.g. compounds targeting, decreasing or inhibiting the activity of FMS-like tyrosine kinase receptors (Flt-3R); interferon, 1 -b-D-arabinofuransylcytosine (ara-c) and bisulfan; and ALK inhibitors e.g. compounds which target, decrease or inhibit anaplastic lymphoma kinase.
- FMS-like tyrosine kinase receptors are especially compounds, proteins or antibodies which inhibit members of the Flt-3R receptor kinase family, e.g. PKC412, midostaurin, a staurosporine derivative, SU1 1248 and MLN518.
- HSP90 inhibitors includes, but is not limited to, compounds targeting, decreasing or inhibiting the intrinsic ATPase activity of HSP90; degrading, targeting, decreasing or inhibiting the HSP90 client proteins via the ubiquitin proteosome pathway.
- Compounds targeting, decreasing or inhibiting the intrinsic ATPase activity of HSP90 are especially compounds, proteins or antibodies which inhibit the ATPase activity of HSP90 e.g., 17-allylamino,17-demethoxygeldanamycin (17AAG, 17-DMAG), a geldanamycin derivative; other geldanamycin related compounds; radicicol and HDAC inhibitors;IPI-504, CNF1010, CNF2024, CNF1010 from Conforma Therapeutics; temozolomide (TEMODAL®), AUY922 from Novartis.
- antiproliferative antibodies includes, but is not limited to, trastuzumab (HerceptinTM), Trastuzumab-DM1 ,erbitux, bevacizumab (AvastinTM), rituximab (Rituxan ® ), PRO64553 (anti-CD40) and 2C4 Antibody.
- antibodies is meant e.g. intact monoclonal antibodies, polyclonal antibodies, multispecific antibodies formed from at least 2 intact antibodies, and antibodies fragments so long as they exhibit the desired biological activity.
- antigenemic compounds includes, for example, Ara-C, a pyrimidine analog, which is the 2 ' -alpha-hydroxy ribose (arabinoside) derivative of deoxycytidine. Also included is the purine analog of hypoxanthine, 6-mercaptopurine (6-MP) and fludarabine phosphate.
- Ara-C Ara-C
- pyrimidine analog which is the 2 ' -alpha-hydroxy ribose (arabinoside) derivative of deoxycytidine.
- purine analog of hypoxanthine 6-mercaptopurine (6-MP)
- 6-MP 6-mercaptopurine
- fludarabine phosphate fludarabine phosphate.
- compounds of formula (I) can be used in combination with standard leukemia therapies, especially in combination with therapies used for the treatment of AML.
- compounds of formula (I) can be administered in combination with, e.g., farnesyl transferase inhibitors and/or other drugs useful for the treatment of AML, such as Daunorubicin, Adriamycin, Ara-C, VP-16, Teniposide,
- Somatostatin receptor antagonists refers to compounds which target, treat or inhibit the somatostatin receptor such as octreotide, and SOM230.
- Tumor cell damaging approaches refer to approaches such as ionizing radiation.
- ionizing radiation means ionizing radiation that occurs as either electromagnetic rays (such as X-rays and gamma rays) or particles (such as alpha and beta particles). Ionizing radiation is provided in, but not limited to, radiation therapy and is known in the art. See Hellman, Principles of Radiation Therapy, Cancer, in Principles and Practice of Oncology, Devita et al., Eds., 4 th Edition, Vol. 1 , pp. 248-275 (1993).
- EDG binders refers a class of immunosuppressants that modulates lymphocyte recirculation, such as FTY720.
- kinesin spindle protein inhibitors is known in the field and includes SB715992 or SB743921 from GlaxoSmithKline, pentamidine/chlorpromazine from CombinatoRx;
- MEK inhibitors is known in the field and includes ARRY142886 from Array PioPharma, AZD6244 from AstraZeneca, PD181461 from Pfizer, leucovorin.
- ribonucleotide reductase inhibitors includes, but is not limited to to pyrimidine or purine nucleoside analogs including, but not limited to, fludarabine and/or cytosine arabinoside (ara-C), 6-thioguanine, 5-fluorouracil, cladribine, 6-mercaptopurine (especially in combination with ara-C against ALL) and/or pentostatin.
- Ribonucleotide reductase inhibitors are especially hydroxyurea or 2-hydroxy-1 H-isoindole-1 ,3-dione derivatives, such as PL-1 , PL-2, PL-3, PL-4, PL-5, PL-6, PL-7 or PL-8 mentioned in Nandy et al., Acta Oncologica, Vol. 33, No. 8, pp. 953-961 (1994).
- S-adenosylmethionine decarboxylase inhibitors includes, but is not limited to the compounds disclosed in US 5,461 ,076.
- VEGF / VEGFR disclosed in WO 98/35958, e.g. 1 -(4-chloroanilino)-4-(4-pyridylmethyl)phthalazine or a pharmaceutically acceptable salt thereof, e.g. the succinate, or in WO 00/09495, WO 00/27820, WO 00/59509, WO 98/1 1223, WO 00/27819 and EP 0 769 947; those as described by Prewett et al, Cancer Res, Vol. 59, pp. 5209-5218 (1999); Yuan et al., Proc Natl Acad Sci U S A, Vol.
- Angiozyme RPI 4610
- Bevacizumab AvastinTM
- Photodynamic therapy refers to therapy which uses certain chemicals known as photosensitizing compounds to treat or prevent cancers. Examples of
- photodynamic therapy includes treatment with compounds, such as e.g. VISUDYNE and porfimer sodium.
- Angiostatic steroids refers to compounds which block or inhibit
- angiogenesis such as, e.g., anecortave, triamcinolone, hydrocortisone, 1 1 - ⁇ -epihydrocotisol, cortexolone, 17 ⁇ -hydroxyprogesterone, corticosterone, desoxycorticosterone, testosterone, estrone and dexamethasone.
- Corticosteroids as used herein includes, but is not limited to compounds, such as e.g.
- fluocinolone, dexamethasone in particular in the form of implants.
- “Other chemotherapeutic compounds” include, but are not limited to, plant alkaloids, hormonal compounds and antagonists; biological response modifiers, preferably lymphokines or interferons; antisense oligonucleotides or oligonucleotide derivatives; shRNA or siRNA; or miscellaneous compounds or compounds with other or unknown mechanism of action.
- a compound of formula (I) may also be used in combination with one or more further drug substances selected from the group of anti-inflammatory drug substances; antihistamine drug substances; bronchodilatatory drug substances, NSAID; antagonists of chemokine receptors.
- the compounds of the invention are also useful as co-therapeutic compounds for use in combination with such further drug substances, particularly in the treatment of inflammatory diseases such as those mentioned hereinbefore, for example as potentiators of therapeutic activity of such drugs or as a means of reducing required dosaging or potential side effects of such drugs.
- a compound of the invention may be mixed with such other drug substance in a fixed pharmaceutical composition or it may be administered separately (i.e. before, simultaneously with or after the other drug substance).
- the invention includes a combination of a compound of formula (I) with one or more further drug substance selected from the group of anti-inflammatory drug substances; antihistamine drug substances;
- bronchodilatatory drug substances NSAID antagonists of chemokine receptors
- said compound of the formula(l) and said drug substance being in the same or different pharmaceutical composition.
- Suitable anti-inflammatory drugs include steroids, in particular glucocorticosteroids such as budesonide, beclamethasone dipropionate, fluticasone propionate, ciclesonide or
- Suitable bronchodilatory drugs include anticholinergic or antimuscarinic compounds, in particular ipratropium bromide, oxitropium bromide, tiotropium salts and CHF 4226 (Chiesi), and glycopyrrolate, but also those described in WO 01/041 18, WO 02/51841 , WO 02/53564, WO 03/00840, WO 03/87094, WO 04/05285, WO 02/00652, WO 03/53966, EP 424021 , US 5171744, US 3714357, WO 03/33495 and WO 04/018422.
- Suitable chemokine receptors include, e.g. CCR-1 , CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 and CCR10, CXCR1 , CXCR2, CXCR3, CXCR4, CXCR5, particularly CCR-5 antagonists such as Schering-Plough antagonists SC-351 125, SCH-55700 and SCH- D, Takeda antagonists such as N-[[4-[[[[6,7-dihydro-2-(4-methylphenyl)-5H-benzo- cyclohepten-8-yl]carbonyl]amino]phenyl]-methyl]tetrahydro-N,N-dimethyl-2H-pyran-4-amin- ium chloride (TAK-770), and CCR-5 antagonists described in US 6166037 (particularly claims 18 and 19), WO 00/66558 (particularly claim 8), WO 00/66559 (particularly claim 9),
- Suitable antihistamine drug substances include cetirizine hydrochloride, acetaminophen, clemastine fumarate, promethazine, loratidine, desloratidine, diphenhydramine and fexofena- dine hydrochloride, activastine, astemizole, azelastine, ebastine, epinastine, mizolastine and tefenadine as well as those disclosed in WO 03/099807, WO 04/026841 and JP
- Therapeutic agents for possible combination are especially one or more antiproliferative, cytostatic or cytotoxic compounds, for example one or several agents selected from the group which includes, but is not limited to, an inhibitor of polyamine biosynthesis, an inhibitor of a protein kinase, especially of a serine/threonine protein kinase, such as protein kinase C, or of a tyrosine protein kinase, such as the EGF receptor tyrosine kinase, e.g. Iressa®, the VEGF receptor tyrosine kinase, e.g.
- PTK787 or Avastin® an antibody against the ligand VEGF, or the PDGF receptor tyrosine kinase, e.g. STI571 (Glivec®), PI3K (such as BEZ235 from Novartis) and mToR inhibitors, such as rapamycin, RAD001 , a cytokine, a negative growth regulator, such as TGF- ⁇ or IFN- ⁇ , an aromatase inhibitor, e.g.
- letrozole (Femara®) or anastrozole, an inhibitor of the interaction of an SH2 domain with a phosphorylated protein, antiestrogens, topoisomerase I inhibitors, such as irinotecan, topoisomerase Il inhibitors, microtubule active agents, e.g. paclitaxel or an epothilone, alkylating agents, antiproliferative antimetabolites, such as gemcitabine or capecitabine, platin compounds, such as carboplatin or cis-platin, bisphosphonates, e.g. AREDIA® or ZOMET A®, and monoclonal antibodies, e.g. against HER2, such as trastuzumab.
- a phosphorylated protein e.g. paclitaxel or an epothilone
- alkylating agents e.g. paclitaxel or an epothilone
- antiproliferative antimetabolites such as gemcitabine or cap
- the structure of the active agents identified by code nos., generic or trade names may be taken from the actual edition of the standard compendium "The Merck Index” or from databases, e.g. Patents International (e.g. IMS World Publications). The corresponding content thereof is hereby incorporated by reference.
- the above-mentioned compounds, which can be used in combination with a compound of the formula (I), can be prepared and administered as described in the art, such as in the documents cited above.
- the combination therapies of the invention the compound of the invention and the other therapeutic agent may be manufactured and/or formulated by the same or different manufacturers.
- the compound of the invention and the other therapeutic may be brought together into a combination therapy: (i) prior to release of the combination product to physicians (e.g.
- kits comprising the compound of the invention and the other therapeutic agent
- the physician themselves or under the guidance of the physician
- shortly before administration or in the patient themselves, e.g. during sequential administration of the compound of the invention and the other therapeutic agent.
- the invention provides the use of a compound of formula (I) for treating a disease or condition mediated by cMet, wherein the medicament is prepared for
- the invention also provides the use of another therapeutic agent for treating a disease or condition mediated cMet,, wherein the
- the invention relates in a further embodiment to a combination, particularly a pharmaceutical composition) comprising a therapeutically effective amount of a compound of formula (I) in free form or in pharmaceutically acceptable salt form and a second
- the additional therapeutic agent is preferably selected from the group consisting of an anti-cancer agent; an anti-inflammatory agent.
- the invention further relates to a method for the treatment of a disease or disorder which responds to a C-Met tyrosine kinase, especially a proliferative disorder or disease, in particular a cancer, said method comprises administration of an effective amount of a combination of pharmaceutical agents which comprise: (a) a compound of formula (I); and (b) one or more pharmaceutically active agents, to a subject in need thereof, especially human.
- a combination of pharmaceutical agents which comprise: (a) a compound of formula (I); and (b) one or more pharmaceutically active agents, to a subject in need thereof, especially human.
- the invention further relates to the use of a combination of pharmaceutical agents which comprise: (a) a compound of formula (I); and (b) one or more pharmaceutically active agents for the treatment of a disease or disorder which responds to a C-Met tyrosine kinase, especially a proliferative disorder or disease, in particular a cancer.
- a combination of pharmaceutical agents which comprise: (a) a compound of formula (I); and (b) one or more pharmaceutically active agents for the treatment of a disease or disorder which responds to a C-Met tyrosine kinase, especially a proliferative disorder or disease, in particular a cancer.
- the invention further relates to the use of a combination of pharmaceutical agents which comprise: (a) a compound of formula (I); and (b) one or more pharmaceutically active agents for themanufacture of a medicament for the treatment of a disease or disorder which responds to a C-Met tyrosine kinase, especially a proliferative disorder or disease, in particular a cancer.
- the invention further relates to pharmaceutical compositions comprising (a) a compound of formula (I) and (b) a pharmaceutically active agent; and (c) a pharmaceutically acceptable carrier; wherein at least one pharmaceutically active agent is an anti-cancer therapeutic.
- the present invention further relates to a commercial package or product comprising:
- the invention also provides the use of a compound of formula (I) for treating a disease or condition mediated by cMet, wherein the patient has previously (e.g. within 24 hours) been treated with another therapeutic agent.
- the invention also provides the use of another therapeutic agent for treating a disease or condition mediated by cMet, wherein the patient has previously (e.g. within 24 hours) been treated with a compound of formula (I).
- delay of progression means administration of the combination to patients being in a pre-stage or in an early phase, of the first manifestation or a relapse of the disease to be treated, in which patients, e.g., a pre-form of the corresponding disease is diagnosed or which patients are in a condition, e.g., during a medical treatment or a condition resulting from an accident, under which it is likely that a corresponding disease will develop.
- joint therapeutically active or “joint therapeutic effect” means that the compounds may be given separately (in a chronically staggered manner, especially a sequence-specific manner) in such time intervals that they preferably, in the warm-blooded animal, especially human, to be treated, still show a (preferably synergistic) interaction (joint therapeutic effect).
- a joint therapeutic effect can, inter alia, be determined by following the blood levels, showing that both compounds are present in the blood of the human to be treated at least during certain time intervals.
- the term "Pharmaceutically effective” preferably relates to an amount that is therapeutically or in a broader sense also prophylactically effective against the progression of a disease or disorder as disclosed herein.
- a commercial package or "a product”, as used herein defines especially a "kit of parts” in the sense that the components (a) and (b) as defined above can be dosed independently or by use of different fixed combinations with distinguished amounts of the components (a) and (b), i.e., simultaneously or at different time points.
- these terms comprise a commercial package comprising (especially combining) as active ingredients components (a) and (b), together with instructions for simultaneous, sequential (chronically staggered, in time-specific sequence, preferentially) or (less preferably) separate use thereof in the delay of progression or treatment of a proliferative disease.
- the parts of the kit of parts can then, e.g., be administered simultaneously or chronologically staggered, that is at different time points and with equal or different time intervals for any part of the kit of parts.
- the time intervals are chosen such that the effect on the treated disease in the combined use of the parts is larger than the effect which would be obtained by use of only any one of the combination partners (a) and (b) (as can be determined according to standard methods.
- the ratio of the total amounts of the combination partner (a) to the combination partner (b) to be administered in the combined preparation can be varied, e.g., in order to cope with the needs of a patient sub-population to be treated or the needs of the single patient which different needs can be due to the particular disease, age, sex, body weight, etc. of the patients.
- there is at least one beneficial effect e.g., a mutual enhancing of the effect of the combination partners (a) and (b), in particular a more than additive effect, which hence could be achieved with lower doses of each of the combined drugs, respectively, than tolerable in the case of treatment with the individual drugs only without combination, producing additional advantageous effects, e.g., less side effects or a combined therapeutic effect in a non-effective dosage of one or both of the combination partners (components) (a) and (b), and very preferably a strong synergism of the
- any combination of simultaneous, sequential and separate use is also possible, meaning that the components (a) and (b) may be administered at one time point simultaneously, followed by administration of only one component with lower host toxicity either chronically, e.g., more than 3-4 weeks of daily dosing, at a later time point and subsequently the other component or the combination of both components at a still later time point (in subsequent drug combination treatment courses for an optimal effect) or the like.
- a method of manufacturing a compound of formula (I) and intermediates thereof A compound of the formula (I) may be prepared by processes that, though not applied hitherto for the new compounds of the present invention where they thus form new processes, are known per se.
- the schemes provide a general overview of synthetic strategies to obtain a compound of formula (I). All methods described can be performed in any suitable order unless otherwise indicated herein or otherwise clearly contradicted by context. The use of any and all examples, or exemplary language ⁇ e.g. "such as”) provided herein is intended merely to better illuminate the invention and does not pose a limitation on the scope of the invention otherwise claimed.
- the invention relates in a further aspect to a manufacturing process (a method for manufacturing) a compound of formula (I) comprising at least one reaction step as disclosed herein, and intermediates thereof.
- Z 1 is selected from Cl, Br and I
- Scheme 2 provides details for a synthetic strategy to obtain preferred compounds of formula (IA, IB, IC) through (NA, NB and NC)
- Zi and Z 2 are independently selected from Cl, Br and I
- Scheme 3 provides details for a synthetic strategy to obtain preferred compounds of formula (ID) through (ND).
- MD Z 1 is selected from Cl, Br and I
- Scheme 4 provides details for a synthetic strategy to obtain preferred compounds of formula (IE) through (NE).
- Zi is selected from Cl, Br and I Scheme 5 provides details for a synthetic strategy to obtain preferred compounds of formula (IF) through (NF).
- Z 1 and Z 2 are independently selected from Cl, Br and I
- Scheme 6 provides details for a synthetic strategy to obtain preferred compounds of formula (IG) and (IH) through (NG) and (NH) respectively.
- Z 1 is selected from Cl, Br and I Scheme 7 provides alternative details for a synthetic strategy to obtain preferred compounds of formula (IH) through (NH).
- Scheme 8 provides details for a synthetic strategy to obtain preferred compounds of formula (IK) through (NK).
- Z 1 is selected from Cl, Br and I
- Scheme 9 provides details for a synthetic strategy to obtain preferred compounds of formula (IL).
- the flow is 0.5 mL/min of methanol and water (with 0.5% acetic acid)
- the flow is 1.2 mL/min of methanol and water (with 0.5% acetic acid)
- the flow is 0.5 mL/min of methanol and water (with 0.5% acetic acid)
- the flow is 0.5 mL/min of methanol and water (with 0.5% acetic acid)
- the flow is 0.5 mL/min of methanol and water (with 0.5 % acetic acid)
- the flow is 1 ml_/min of Hexane/Ethanol/Diethyleamine 80/20/0.1 , v/v/v
- the flow is 1 ml_/min of Hexane/Ethanol/Diethyleamine 70/30/0.1 , v/v/v
- the flow is 1 ml_/min of Hexane/lsopropanol/Diethylamine 70/30/0.1 , v/v/v Column: CHIRALPAK OD-H
- the flow is 45 g/min of Methanol/CO 2 70/30
- the flow is 1.2 mL/min of methanol and water (with 0.5% acetic acid) 0 - 3.0 min: 60% to 90% of methanol
- the flow is 45 g/min of Methanol/CO 2 75/25
- 6-chloroimidazo[1 ,2-b]pyridazine (1 .5 g, 9.8 mmol) in AcOH (50 ml_) was added NaOAc (1 .4 g, 17.1 mmol) and paraformaldehyde (1.5 g). The mixture was heated at reflux overnight and concentrated under reduced pressure. The residue was basified to pH 12. Then the mixture was filtered and the solid was washed with EtOH to afford (6-chloro- imidazo[1 ,2-b]pyridazin-3-yl)-methanol (1.3 g) in 72% yield. 6-Chloro-imidazo[1 ,2-b]pyridazine-3-carbaldehyde (intermediate N)
- the title compound was prepared according to the procedure described in WO2006/10094. To a mixture of /-butyl hydroxycarbamate (1 1.27 g, 85 mmol) and DBU (12.8 ml_, 85 mmol) was added 2-bromoethanol (15 ml_, 212 mmol) slowly, and the reaction mixture was stirred 30 at 40 0 C for 2.5 days. After cooling, the reaction mixture was diluted with DCM, and washed successively with cold 1 N HCI aqueous solution, water, and brine. The organic layer was dried over anhydrous Na 2 SO 4 , purified by column chromatography (38% EtOAc in hexane) to afford 12.6 g (70%) of the title compound as a colorless syrup.
- Example 7A To a mixture of 2-(2,3-dihydroxypropoxy)isoindoline-1 ,3-dione and 2-(1 ,3-dihydroxypropan-2- yloxy)isoindoline-1 ,3-dione (1 :1 mixture) (intermediate B) in MeOH was added hydrazine monohydrate. The reaction mixture was allowed to stir at reflux for two h and then the solvent was removed under reduced pressure. The residue was washed with EtOAc and filtered. The filtrate was collected, evaporated and redissolved in MeOH.
- Example 11 (example 11 -R, example 11 -S)
- Example 12 (example 12-R, example 12-S)
- the title compound (390.0 mg, 91 %) was synthesized from 1 -(6-chloro-imidazo[1 ,2- b]pyridazin-3-yl)-1 -(1 -methyl-1 H-indazol-5-yl)-ethanol (430.0 mg, 1 .31 mmol), I 2 (832.0 mg, 3.28 mmol) and H 3 PO 2 (0.72 mL, 6.56 mmol) using the same procedure as described in the synthesis of compound 13.2.
- the title compound (12.0 mg, 89%) was synthesized from 1 -[3-(7-fluoro-quinolin-6-ylmethyl)- 3H-[1 ,2,3]triazolo[4,5-b]pyrazin-5-yl]-ethanone (15.4) (10.0 mg, 0.03 mmol) and 2-aminooxy- 2-methyl-propan-1 -ol hydrochloride (8.8 mg, 0.06 mmol) using the same procedure as described in the synthesis of example 15.
- the title compound (12.0 mg, 89%) was synthesized from 1 -[3-(7-fluoro-quinolin-6-ylmethyl)- 3H-[1 ,2,3]triazolo[4,5-b]pyrazin-5-yl]-ethanone (15.4) (10.0 mg, 0.03 mmol) and 1 -aminooxy- 2-methyl-propan-2-ol (6.5 mg, 0.06 mmol) using the same procedure as described in the synthesis of example 15.
- the title compound (7.0 mg, 50%) was synthesized from 1-[3-(7-fluoro-quinolin-6-ylmethyl)- 3H-[1,2,3]triazolo[4,5-b]pyrazin-5-yl]-ethanone (15.4) (10.0 mg, 0.03 mmol) and [R)-O- pyrrolidin-3-yl-hydroxylamine hydrochloride (10.8 mg, 0.06 mmol) using the same procedure as described in the synthesis of example 15.
- the title compound was prepared from 1 -(1 -(quinolin-6-ylmethyl)-1 H-[1 ,2,3]triazolo[4,5- b]pyrazin-6-yl)ethanone (22.4) and 1 -(aminooxy)-2-methylpropan-2-ol in 85% yield using the same procedure as described in the synthesis of example 22.
- Example 39 (example 39-R, example 39-S)
- the title compound (12 mg, 28.6%) was synthesized from 2-(1 ,3-dihydroxypropan-2- yloxy)isoindoline-1 ,3-dione (intermediate B) (152 mg, 0.643 mmol, isomer mixture), hydrazine monohydrate (14 mg, 0.27 mmol) and 1 -(3-(quinolin-6-ylmethyl)-[1 ,2,4]triazolo[4,3- b]pyridazin-6-yl)ethanone (41.2) (30 mg, 0.1 1 mmol) using the same procedure as described in the synthesis of example 44.
- the title compound (6 mg, 14.3%) was synthesized from 2-(2,3-dihydroxypropoxy)iso- indoline-1 ,3-dione (intermediate B) (152 mg, 0.643 mmol, isomer mixture), hydrazine monohydrate (14 mg, 0.27 mmol) and 1 -(3-(quinolin-6-ylmethyl)-[1 ,2,4]triazolo[4,3- b]pyridazin-6-yl)ethanone (41.2) (30 mg, 0.1 1 mmol) using the same procedure as described in the synthesis of example 44. After purification by HPLC (acidic with 0.05%TFA, then neutralized to basic), the title compound was obtained as a white solid.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description




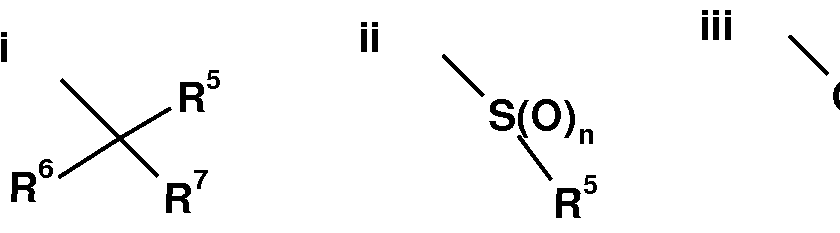




















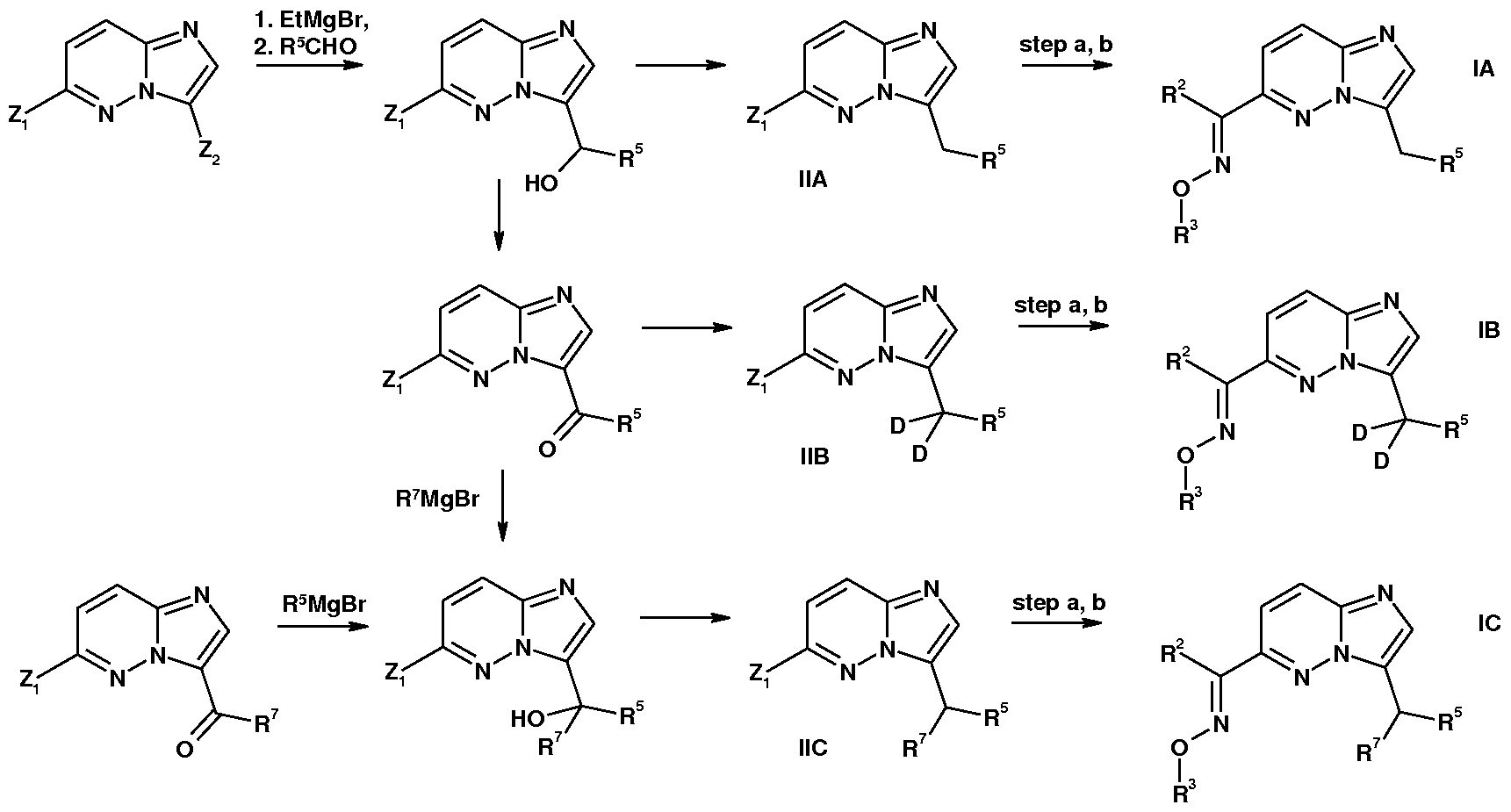











































































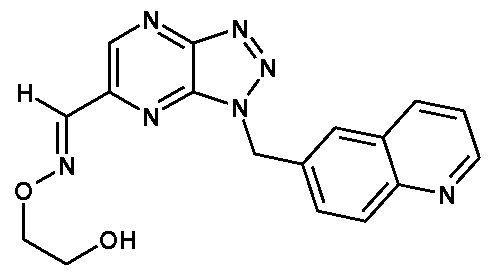


























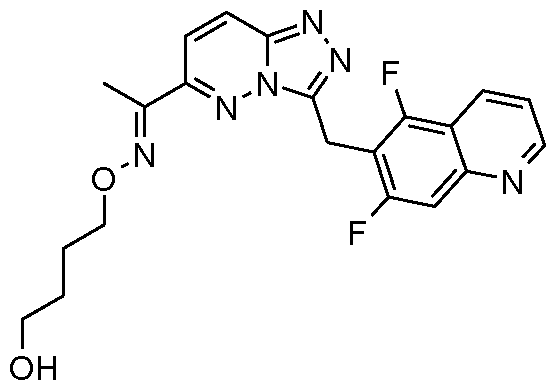
















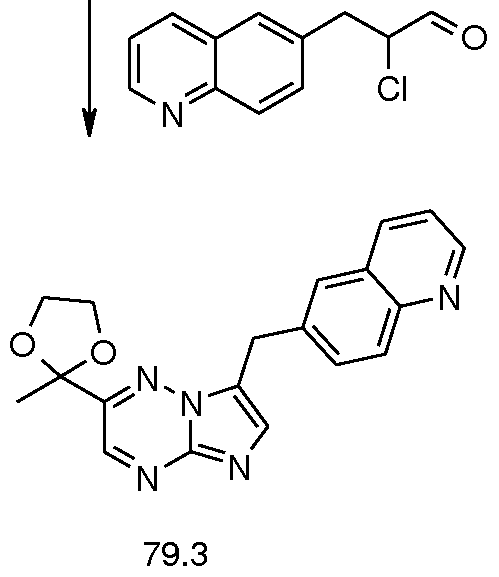




Claims






Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10762878A EP2467383A1 (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds |
| MX2012002179A MX2012002179A (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds. |
| JP2012525167A JP5775871B2 (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds |
| CA2771432A CA2771432A1 (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds |
| BR112012008061A BR112012008061A2 (en) | 2009-08-20 | 2010-08-18 | heterocyclic oxime compounds |
| AU2010284972A AU2010284972A1 (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds |
| CN201080047132.XA CN102574853B (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds |
| KR1020127007032A KR20120089463A (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds |
| EA201200318A EA201200318A1 (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oximes |
| IN1453DEN2012 IN2012DN01453A (en) | 2009-08-20 | 2010-08-18 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US23544009P | 2009-08-20 | 2009-08-20 | |
| US61/235,440 | 2009-08-20 | ||
| CNPCT/CN2010/071978 | 2010-04-21 | ||
| CN2010071978 | 2010-04-21 | ||
| CN2010074089 | 2010-06-18 | ||
| CNPCT/CN2010/074089 | 2010-06-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011020861A1 true WO2011020861A1 (en) | 2011-02-24 |
Family
ID=43243152
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2010/062057 WO2011020861A1 (en) | 2009-08-20 | 2010-08-18 | Heterocyclic oxime compounds |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US8410264B2 (en) |
| EP (1) | EP2467383A1 (en) |
| JP (1) | JP5775871B2 (en) |
| KR (1) | KR20120089463A (en) |
| AR (1) | AR079486A1 (en) |
| AU (1) | AU2010284972A1 (en) |
| BR (1) | BR112012008061A2 (en) |
| CA (1) | CA2771432A1 (en) |
| EA (1) | EA201200318A1 (en) |
| IN (1) | IN2012DN01453A (en) |
| MX (1) | MX2012002179A (en) |
| TW (1) | TW201113284A (en) |
| UY (1) | UY32848A (en) |
| WO (1) | WO2011020861A1 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011145035A1 (en) * | 2010-05-17 | 2011-11-24 | Indian Incozen Therapeutics Pvt. Ltd. | Novel 3,5-disubstitued-3h-imidazo[4,5-b]pyridine and 3,5- disubstitued -3h-[1,2,3]triazolo[4,5-b] pyridine compounds as modulators of protein kinases |
| WO2012107500A1 (en) * | 2011-02-10 | 2012-08-16 | Novartis Ag | [1, 2, 4] triazolo [4, 3 -b] pyridazine compounds as inhibitors of the c-met tyrosine kinase |
| WO2013038362A1 (en) | 2011-09-15 | 2013-03-21 | Novartis Ag | 6 - substituted 3 - (quinolin- 6 - ylthio) - [1,2,4] triazolo [4, 3 -a] pyradines as tyrosine kinase |
| WO2013098229A2 (en) | 2011-12-27 | 2013-07-04 | Bayer Intellectual Property Gmbh | Heteroarylpiperidine and piperazine derivatives as fungicides |
| CN103459396A (en) * | 2011-02-10 | 2013-12-18 | 诺瓦提斯公司 | [1, 2, 4] triazolo [4, 3 -b] pyridazine compounds as inhibitors of c-met tyrosine kinase |
| EP2719699A1 (en) * | 2009-12-31 | 2014-04-16 | Hutchison Medipharma Limited | Certain triazolopyrazines, compositions thereof and methods of use therefor |
| CN103958509A (en) * | 2011-09-15 | 2014-07-30 | 诺华股份有限公司 | 6 - substituted 3 - (quinolin- 6 - ylthio) - [1,2,4] triazolo [4, 3 -a] pyradines as tyrosine kinase |
| US9206166B2 (en) | 2012-11-06 | 2015-12-08 | SHANGHAI iNSTITUTE OF MATERIA MEDICA ACADEMY OF SCIENCES | Certain protein kinase inhibitors |
| US9221804B2 (en) | 2013-10-15 | 2015-12-29 | Janssen Pharmaceutica Nv | Secondary alcohol quinolinyl modulators of RORγt |
| US9284308B2 (en) | 2013-10-15 | 2016-03-15 | Janssen Pharmaceutica Nv | Methylene linked quinolinyl modulators of RORγt |
| US9290476B2 (en) | 2012-10-16 | 2016-03-22 | Janssen Pharmaceutica Nv | Methylene linked quinolinyl modulators of RORγt |
| US9303015B2 (en) | 2012-10-16 | 2016-04-05 | Janssen Pharmaceutica Nv | Heteroaryl linked quinolinyl modulators of RORγt |
| US9309222B2 (en) | 2012-10-16 | 2016-04-12 | Janssen Pharmaceutica Nv | Phenyl linked quinolinyl modulators of RORγt |
| US9328095B2 (en) | 2013-10-15 | 2016-05-03 | Janssen Pharmaceutica Nv | Heteroaryl linked quinolinyl modulators of RORgammat |
| US9346782B2 (en) | 2013-10-15 | 2016-05-24 | Janssen Pharmaceutica Nv | Alkyl linked quinolinyl modulators of RORγt |
| US9403816B2 (en) | 2013-10-15 | 2016-08-02 | Janssen Pharmaceutica Nv | Phenyl linked quinolinyl modulators of RORγt |
| US9624225B2 (en) | 2013-10-15 | 2017-04-18 | Janssen Pharmaceutica Nv | Quinolinyl modulators of RORγt |
| EP3241830A1 (en) | 2016-05-04 | 2017-11-08 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pesticides |
| US10555941B2 (en) | 2013-10-15 | 2020-02-11 | Janssen Pharmaceutica Nv | Alkyl linked quinolinyl modulators of RORγt |
| WO2020260857A1 (en) | 2019-06-13 | 2020-12-30 | AdoRx Therapeutics Limited | Hydroxamate compounds as antagonists of the adenosine a2a receptor |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2010283806A1 (en) | 2009-08-12 | 2012-03-01 | Novartis Ag | Heterocyclic hydrazone compounds and their uses to treat cancer and inflammation |
| GB201321736D0 (en) * | 2013-12-09 | 2014-01-22 | Ucb Pharma Sa | Therapeutic agents |
| IL266955B2 (en) * | 2016-11-29 | 2023-09-01 | Ptc Therapeutics Mp Inc | Polymorphs of sepiapterin and salts thereof |
| JP2020532535A (en) | 2017-09-01 | 2020-11-12 | ピーティーシー セラピューティクス エムピー,インコーポレイテッド | Pharmaceutical composition containing sepiapterin and its use |
Citations (145)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3714357A (en) | 1968-07-15 | 1973-01-30 | Rech D Applic Scient Sogeras S | Pharmaceutical compositions comprising quinuclidinol derivatives |
| US4100274A (en) | 1976-05-11 | 1978-07-11 | Imperial Chemical Industries Limited | Polypeptide |
| US4636505A (en) | 1982-07-23 | 1987-01-13 | Imperial Chemical Industries Plc | Amide derivatives |
| US4659516A (en) | 1983-10-12 | 1987-04-21 | Imperial Chemical Industries Plc | Steroid derivatives |
| EP0409595A2 (en) | 1989-07-19 | 1991-01-23 | Glaxo Group Limited | Process for the preparation of a carbocyclic nucleoside analogue |
| US5010099A (en) | 1989-08-11 | 1991-04-23 | Harbor Branch Oceanographic Institution, Inc. | Discodermolide compounds, compositions containing same and method of preparation and use |
| EP0424021A1 (en) | 1989-10-19 | 1991-04-24 | Pfizer Limited | Antimuscarinic bronchodilators |
| US5093330A (en) | 1987-06-15 | 1992-03-03 | Ciba-Geigy Corporation | Staurosporine derivatives substituted at methylamino nitrogen |
| EP0490587A1 (en) | 1990-12-07 | 1992-06-17 | Merck & Co. Inc. | Substituted pyrazolopyrimidines and imidazopyridazines as angiotensin II antagonists |
| WO1992019594A1 (en) | 1991-05-02 | 1992-11-12 | Smithkline Beecham Corporation | Pyrrolidinones |
| EP0520722A1 (en) | 1991-06-28 | 1992-12-30 | Zeneca Limited | Therapeutic preparations containing quinazoline derivatives |
| EP0564409A1 (en) | 1992-04-03 | 1993-10-06 | Ciba-Geigy Ag | Pyrimidin derivatives and process for their preparation |
| WO1993019750A1 (en) | 1992-04-02 | 1993-10-14 | Smithkline Beecham Corporation | Compounds useful for treating allergic or inflammatory diseases |
| WO1993019749A1 (en) | 1992-04-02 | 1993-10-14 | Smithkline Beecham Corporation | Compounds useful for treating allergic and inflammatory diseases |
| WO1993019751A1 (en) | 1992-04-02 | 1993-10-14 | Smithkline Beecham Corporation | Compounds useful for treating inflammatory diseases and inhibiting production of tumor necrosis factor |
| EP0566226A1 (en) | 1992-01-20 | 1993-10-20 | Zeneca Limited | Quinazoline derivatives |
| WO1994010202A1 (en) | 1992-10-28 | 1994-05-11 | Genentech, Inc. | Vascular endothelial cell growth factor antagonists |
| WO1994017090A1 (en) | 1993-01-20 | 1994-08-04 | Glaxo Group Limited | 2,6-diaminopurine derivatives |
| WO1995003283A1 (en) | 1993-07-19 | 1995-02-02 | Zeneca Limited | Quinazoline derivatives and their use as anti-cancer agents |
| US5451700A (en) | 1991-06-11 | 1995-09-19 | Ciba-Geigy Corporation | Amidino compounds, their manufacture and methods of treatment |
| US5461076A (en) | 1990-05-07 | 1995-10-24 | Stanek; Jaroslav | Hydrazones |
| WO1996002553A2 (en) | 1994-07-14 | 1996-02-01 | Glaxo Group Limited | AMINO PURINE-β-D-RIBOFURANURONAMIDE DERIVATIVES |
| WO1996002543A1 (en) | 1994-07-14 | 1996-02-01 | Glaxo Group Limited | 2,3-dihydroxy cyclopentane derivatives of purines |
| WO1996030347A1 (en) | 1995-03-30 | 1996-10-03 | Pfizer Inc. | Quinazoline derivatives |
| WO1996033980A1 (en) | 1995-04-27 | 1996-10-31 | Zeneca Limited | Quinazoline derivatives |
| WO1997002266A1 (en) | 1995-07-06 | 1997-01-23 | Novartis Ag | Pyrrolopyrimidines and processes for the preparation thereof |
| EP0769947A1 (en) | 1995-06-07 | 1997-05-02 | Sugen, Inc. | Indolinone compounds for the treatment of disease |
| EP0787722A1 (en) | 1996-02-05 | 1997-08-06 | American Cyanamid Company | Substituted quinazoline derivatives |
| WO1997030034A1 (en) | 1996-02-14 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as antitumor agents |
| WO1997038983A1 (en) | 1996-04-12 | 1997-10-23 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| WO1997049688A1 (en) | 1996-06-24 | 1997-12-31 | Pfizer Inc. | Phenylamino-substituted tricyclic derivatives for treatment of hyperproliferative diseases |
| WO1998008849A1 (en) | 1996-08-30 | 1998-03-05 | Novartis Aktiengesellschaft | Method for producing epothilones, and intermediate products obtained during the production process |
| WO1998010121A1 (en) | 1996-09-06 | 1998-03-12 | Obducat Ab | Method for anisotropic etching of structures in conducting materials |
| WO1998010767A2 (en) | 1996-09-13 | 1998-03-19 | Sugen, Inc. | Use of quinazoline derivatives for the manufacture of a medicament in the treatment of hyperproliferative skin disorders |
| WO1998011223A1 (en) | 1996-09-11 | 1998-03-19 | Schering Aktiengesellschaft | Monoclonal antibodies against the extracellular domain of human vegf-receptor protein (kdr) |
| EP0837063A1 (en) | 1996-10-17 | 1998-04-22 | Pfizer Inc. | 4-Aminoquinazoline derivatives |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| WO1998018796A1 (en) | 1996-10-28 | 1998-05-07 | Novartis Ag | Naphthyridine derivatives |
| WO1998022461A1 (en) | 1996-11-18 | 1998-05-28 | GESELLSCHAFT FüR BIOTECHNOLOGISCHE FORSCHUNG MBH (GBF) | Epothilone c, d, e and f, production process, and their use as cytostatic as well as phytosanitary agents |
| WO1998025929A1 (en) | 1996-12-13 | 1998-06-18 | Novartis Ag | Epothilone analogs |
| WO1998028319A1 (en) | 1996-12-24 | 1998-07-02 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1998035958A1 (en) | 1997-02-13 | 1998-08-20 | Novartis Ag | Phthalazines with angiogenesis inhibiting activity |
| US5843901A (en) | 1995-06-07 | 1998-12-01 | Advanced Research & Technology Institute | LHRH antagonist peptides |
| WO1999003854A1 (en) | 1997-07-18 | 1999-01-28 | Novartis Ag | Crystal modification of a n-phenyl-2-pyrimidineamine derivative, processes for its manufacture and its use |
| WO1999016766A1 (en) | 1997-10-01 | 1999-04-08 | Kyowa Hakko Kogyo Co., Ltd. | Benzodioxole derivatives |
| WO1999017804A1 (en) | 1997-10-03 | 1999-04-15 | Pharmacia & Upjohn S.P.A. | Polymeric derivatives of camptothecins |
| WO1999024451A2 (en) | 1997-11-08 | 1999-05-20 | Glaxo Group Limited | Adenosine a1 receptor agonists |
| WO1999024449A2 (en) | 1997-11-08 | 1999-05-20 | Glaxo Group Limited | Adenosine a1 receptor agonists |
| WO1999024450A2 (en) | 1997-11-08 | 1999-05-20 | Glaxo Group Limited | Adensine a1 receptor agonists |
| WO1999038877A2 (en) | 1998-01-31 | 1999-08-05 | Glaxo Group Limited | 2-(PURIN-9-yl)-TETRAHYDROFURAN-3,4-DIOL DERIVATIVES |
| WO1999041267A1 (en) | 1998-02-14 | 1999-08-19 | Glaxo Group Limited | 2-(purin-9-yl) -tetrahydrofuran-3, 4-diol derivatives |
| WO1999043653A1 (en) | 1998-02-25 | 1999-09-02 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues therof |
| WO1999067263A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1999067266A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1999067264A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1999067265A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO2000000531A1 (en) | 1998-06-30 | 2000-01-06 | The Dow Chemical Company | Polymer polyols and a process for the production thereof |
| WO2000009495A1 (en) | 1998-08-11 | 2000-02-24 | Novartis Ag | Isoquinoline derivatives with angiogenesis inhibiting activity |
| WO2000023457A1 (en) | 1998-10-16 | 2000-04-27 | Pfizer Limited | Adenine derivatives |
| WO2000027819A2 (en) | 1998-11-10 | 2000-05-18 | Schering Aktiengesellschaft | Antrhranilic acid amides and the use thereof as medicaments |
| WO2000027820A1 (en) | 1998-11-10 | 2000-05-18 | Novartis Ag | N-aryl(thio)anthranilic acid amide derivatives, their preparation and their use as vegf receptor tyrosine kinase inhibitors |
| WO2000031247A2 (en) | 1998-11-20 | 2000-06-02 | Kosan Biosciences, Inc. | Recombinant methods and materials for producing epothilone and epothilone derivatives |
| WO2000037502A2 (en) | 1998-12-22 | 2000-06-29 | Genentech, Inc. | Vascular endothelial cell growth factor antagonists and uses thereof |
| WO2000059509A1 (en) | 1999-03-30 | 2000-10-12 | Novartis Ag | Phthalazine derivatives for treating inflammatory diseases |
| WO2000066559A1 (en) | 1999-05-04 | 2000-11-09 | Schering Corporation | Piperidine derivatives useful as ccr5 antagonists |
| WO2000066558A1 (en) | 1999-05-04 | 2000-11-09 | Schering Corporation | Piperazine derivatives useful as ccr5 antagonists |
| EP1052264A2 (en) | 1999-05-11 | 2000-11-15 | Pfizer Products Inc. | Process for the synthesis of nucleoside analogs |
| WO2000075114A1 (en) | 1999-06-04 | 2000-12-14 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2000077018A2 (en) | 1999-06-15 | 2000-12-21 | Pfizer Limited | Purine derivatives |
| US6166037A (en) | 1997-08-28 | 2000-12-26 | Merck & Co., Inc. | Pyrrolidine and piperidine modulators of chemokine receptor activity |
| WO2000078774A2 (en) | 1999-06-18 | 2000-12-28 | University Of Virginia Patent Foundation | Induction of pharmacological stress with adenosine receptor agonists |
| WO2001004118A2 (en) | 1999-07-14 | 2001-01-18 | Almirall Prodesfarma S.A. | Quinuclidine derivatives and their use as muscarinic m3 receptor ligands |
| US6194181B1 (en) | 1998-02-19 | 2001-02-27 | Novartis Ag | Fermentative preparation process for and crystal forms of cytostatics |
| WO2001013953A2 (en) | 1999-08-21 | 2001-03-01 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Synergistic combination of pde inhibitors and beta 2 adrenoceptor agonist |
| WO2001023399A1 (en) | 1999-09-30 | 2001-04-05 | Pfizer Products Inc. | Compounds for the treatment of ischemia |
| WO2001027131A1 (en) | 1999-10-14 | 2001-04-19 | Pfizer Limited | Purine derivatives |
| WO2001027130A1 (en) | 1999-10-14 | 2001-04-19 | Pfizer Limited | Purine derivatives |
| WO2001060835A1 (en) | 2000-02-18 | 2001-08-23 | Pfizer Limited | Purine derivatives |
| WO2001094368A1 (en) | 2000-06-06 | 2001-12-13 | Pfizer Limited | 2-aminocarbonyl-9h-purine derivatives |
| WO2002000652A1 (en) | 2000-06-27 | 2002-01-03 | Laboratorios S.A.L.V.A.T., S.A. | Carbamates derived from arylalkylamines |
| WO2002000679A2 (en) | 2000-06-28 | 2002-01-03 | Novartis Ag | 9.alpha.-chloro-6.alpha.-fluoro-17.alpha.-hydroxy-16-methyl-17-beta-methoxycarbonyl-androst-1,4-dienes esterified in position 17.alpha. by a cyclic acyl group |
| WO2002010143A1 (en) | 2000-07-28 | 2002-02-07 | Schering Aktiengesellschaft | Non-steroidal inflammation inhibitors |
| WO2002012266A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 17.beta.-carbothioate 17.alpha.-arylcarbonyloxyloxy androstane derivative as anti-inflammatory agents |
| WO2002022630A1 (en) | 2000-09-15 | 2002-03-21 | Pfizer Limited | Purine derivatives |
| WO2002022577A2 (en) | 2000-09-01 | 2002-03-21 | Novartis Ag | Hydroxamate derivatives useful as deacetylase inhibitors |
| WO2002042298A1 (en) | 2000-11-21 | 2002-05-30 | Novartis Ag | Aminothiazoles and their use as adenosine receptor antagonists |
| WO2002051841A1 (en) | 2000-12-22 | 2002-07-04 | Almirall Prodesfarma Ag | Quinuclidine carbamate derivatives and their use as m3 antagonists |
| WO2002053564A2 (en) | 2000-12-28 | 2002-07-11 | Almirall Prodesfarma Ag | Quinuclidine derivatives and their use as m3 antagonists |
| EP1241176A1 (en) | 2001-03-16 | 2002-09-18 | Pfizer Products Inc. | Purine derivatives for the treatment of ischemia |
| WO2002088167A1 (en) | 2001-04-30 | 2002-11-07 | Glaxo Group Limited | Anti-inflammatory 17.beta.-carbothioate ester derivatives of androstane with a cyclic ester group in position 17.alpha |
| WO2002092599A1 (en) | 2001-05-14 | 2002-11-21 | Novartis Ag | 4-amino-5-phenyl-7-cyclobutyl-pyrrolo (2,3-d) pyrimidine derivatives |
| WO2002096462A1 (en) | 2001-05-25 | 2002-12-05 | Pfizer Inc. | An adenosine a2a receptor agonist and an anticholinergic agent in combination for treating obstructive airways diseases |
| WO2002100676A1 (en) | 2001-06-11 | 2002-12-19 | Ts Tech Co., Ltd. | Operating lever, and seat having the same |
| WO2002100879A1 (en) | 2001-06-12 | 2002-12-19 | Glaxo Group Limited | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| WO2003000840A2 (en) | 2001-06-21 | 2003-01-03 | Diversa Corporation | Nitrilases |
| WO2003013541A1 (en) | 2001-08-07 | 2003-02-20 | Novartis Ag | 4-amino-6-phenyl-pyrrolo[2,3-d]pyrimidine derivatives |
| WO2003033495A1 (en) | 2001-10-17 | 2003-04-24 | Ucb, S.A. | Quinuclidine derivatives, processes for preparing them and their uses as m2 and/or m3 muscarinic receptor inhibitors |
| WO2003035668A2 (en) | 2001-10-20 | 2003-05-01 | Glaxo Group Limited | Novel anti-inflammatory androstane derivatives -17-carboxy-lactone substituted steroids with an aryl-carboxylic ester in position 17.alpha |
| WO2003039544A1 (en) | 2001-11-05 | 2003-05-15 | Novartis Ag | Naphtyridine derivatives, their preparation and their use as phosphodiesterase isoenzyme 4 (pde4) inhibitors |
| WO2003048181A1 (en) | 2001-12-01 | 2003-06-12 | Glaxo Group Limited | 17.alpha. -cyclic esters of 16-methylpregnan-3,20-dione as anti-inflammatory agents |
| WO2003053966A2 (en) | 2001-12-20 | 2003-07-03 | Laboratorios S.A.L.V.A.T., S.A. | 1-alkyl-1-azoniabicyclo [2.2.2] octane carbamate derivatives and their use as muscarinic receptor antagonists |
| WO2003062259A2 (en) | 2002-01-21 | 2003-07-31 | Glaxo Group Limited | Non-aromatic 17.alpha.-esters of androstane-17.beta.-carboxylate esters as anti-inflammatory agents |
| WO2003072592A1 (en) | 2002-01-15 | 2003-09-04 | Glaxo Group Limited | 17.alpha-cycloalkyl/cycloylkenyl esters of alkyl-or haloalkyl-androst-4-en-3-on-11.beta.,17.alpha.-diol 17.beta.-carboxylates as anti-inflammatory agents |
| WO2003082280A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003082787A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003087094A2 (en) | 2002-04-16 | 2003-10-23 | Almirall Prodesfarma Ag | Pyrrolidinium derivatives as antagonists of m3 muscarinic receptors |
| WO2003086408A1 (en) | 2002-04-10 | 2003-10-23 | University Of Virginia Patent Foundation | Use of a2a adenosine receptor agonists for the treatment of inflammatory diseases |
| WO2003099807A1 (en) | 2002-05-29 | 2003-12-04 | Almirall Prodesfarma S.A. | New indolylpiperidine derivatives as potent antihistaminic and antiallergic agents |
| WO2003104204A1 (en) | 2002-06-05 | 2003-12-18 | Merck Patent Gmbh | Pyridazine derivatives |
| WO2003104195A1 (en) | 2002-06-06 | 2003-12-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | 4-(aryl or heteroaryl) -2-butylamine derivatives and their use as glucocorticoid ligans |
| WO2003104205A1 (en) | 2002-06-10 | 2003-12-18 | Merck Patent Gmbh | Aryloximes |
| WO2003106444A1 (en) | 2002-06-14 | 2003-12-24 | Recordati S.A. | N,n-disubstituted diazocycloalkanes useful for the treatment of cns disorders due to serotonergic dysfunction |
| WO2004000839A1 (en) | 2002-06-19 | 2003-12-31 | Merck Patent Gmbh | Thiazole derivatives as phosphodiesterase iv inhibitors |
| WO2004000814A1 (en) | 2002-06-25 | 2003-12-31 | Merck Frosst Canada & Co. | 8-(biaryl) quinoline pde4 inhibitors |
| WO2004005258A1 (en) | 2002-07-02 | 2004-01-15 | Merck Frosst Canada & Co. | Di-aryl-substituted-ethane pyridone pde4 inhibitors |
| WO2004005229A1 (en) | 2002-07-08 | 2004-01-15 | Pfizer Products Inc. | Modulators of the glucocorticoid receptor |
| WO2004005285A1 (en) | 2002-07-02 | 2004-01-15 | Almirall Prodesfarma Ag | New quinuclidine amide derivatives |
| WO2004016601A1 (en) | 2002-08-09 | 2004-02-26 | Novartis Ag | Benzothiazole derivatives having beta-2-adrenoreceptor agonist activity |
| WO2004018449A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Piperidine-derivatives as pde4 inhibitors |
| WO2004018457A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Pyrrolidinedione substituted piperidine-phthalazones as pde4 inhibitors |
| WO2004018422A1 (en) | 2002-08-23 | 2004-03-04 | Ranbaxy Laboratories Limited | Fluoro and sulphonylamino containing 3,6-disubstituted azabicyclo (3.1.0) hexane derivatives as muscarinic receptor antagonists |
| WO2004018465A2 (en) | 2002-08-17 | 2004-03-04 | Altana Pharma Ag | Benzonaphthyridines with pde 3/4 inhibiting activity |
| WO2004018451A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Pyridazinone-derivatives as pde4 inhibitors |
| WO2004018425A1 (en) | 2002-08-21 | 2004-03-04 | Astrazeneca Ab | N-4-piperidinyl compounds as ccr5 modulators |
| WO2004018450A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Piperidine-n-oxide-derivatives |
| WO2004019944A1 (en) | 2002-08-29 | 2004-03-11 | Altana Pharma Ag | 2-hydroxy-6-phenylphenanthridines as pde-4 inhibitors |
| WO2004026841A1 (en) | 2002-09-18 | 2004-04-01 | Sumitomo Pharmaceuticals Co., Ltd. | Novel 6-substituted uracil derivative and therapeutic agent for allergic disease |
| WO2004026873A1 (en) | 2002-09-18 | 2004-04-01 | Ono Pharmaceutical Co., Ltd. | Triazaspiro[5.5]undecane derivatives and drugs comprising the same as the active ingredient |
| JP2004107299A (en) | 2002-09-20 | 2004-04-08 | Japan Energy Corp | New 1-substituted urasil derivative and therapeutic agent for allergic disease |
| WO2004033412A1 (en) | 2002-10-04 | 2004-04-22 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel beta mimetics with extended duration of action, method for production and use thereof as medicaments |
| WO2004037805A1 (en) | 2002-10-23 | 2004-05-06 | Glenmark Pharmaceuticals Ltd. | Novel tricyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| WO2004039766A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenylethanolamine derivatives for the treatment of respiratory diseases |
| WO2004039762A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenethanolamine derivatives for the treatment of respiratory diseases |
| WO2004045607A1 (en) | 2002-11-15 | 2004-06-03 | Elbion Ag | Novel hydroxyindoles, use as inhibitors of phosphodiesterase 4 and method for production thereof |
| WO2004046083A1 (en) | 2002-11-15 | 2004-06-03 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel dihydroxy-methylphenyl derivatives, method for the production and use thereof as medicaments |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004101994A1 (en) | 2003-05-19 | 2004-11-25 | Federalnoe Gosudarstvennoe Unitarnoe Predpriiatie 'rossiiskii Federalnyi Iadernyi Centr - Vserossiiskii Nauchno-Issledovatelskii Institut Tehnicheskoi Fisiki Imeni Akademika E.I.Zababahina' | Ultrahigh pressure plunger pump |
| WO2004101846A1 (en) | 2003-05-16 | 2004-11-25 | Sandvik Intellectual Property Ab | Cutting tool for metalworking as well as method in the production of cutting tools |
| WO2004104561A1 (en) | 2003-05-21 | 2004-12-02 | Bayer Technology Services Gmbh | Method and device for measuring the content of ferromagnetic particles in liquid suspensions, aerosol dispensers or solid mixtures |
| WO2006110094A1 (en) | 2005-04-14 | 2006-10-19 | Gyros Patent Ab | Upward microconduits |
| WO2007013673A1 (en) | 2005-07-29 | 2007-02-01 | Astellas Pharma Inc. | Fused heterocycles as lck inhibitors |
| WO2007126799A2 (en) | 2006-03-30 | 2007-11-08 | Novartis Ag | Compositions and methods of use for antibodies of c-met |
| WO2008008539A2 (en) * | 2006-07-14 | 2008-01-17 | Amgen Inc. | Fused heterocyclic derivatives useful as inhibitors of the hepatocyte growth factor receptor |
| WO2008100853A1 (en) | 2007-02-12 | 2008-08-21 | Wm. Wrigley Jr. Company | Confectionery products comprising polyols |
| WO2008144767A1 (en) | 2007-05-21 | 2008-11-27 | Sgx Pharmaceuticals, Inc. | Heterocyclic kinase modulators |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU6031898A (en) | 1997-01-22 | 1998-08-07 | Eli Lilly And Company | Anti-viral compounds |
| US6346534B1 (en) | 1998-09-23 | 2002-02-12 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| ES2244613T3 (en) | 2000-04-27 | 2005-12-16 | Astellas Pharma Inc. | IMIDAZOPIRIDINE DERIVATIVES |
| CA2530589A1 (en) | 2003-07-02 | 2005-01-20 | Sugen Inc. | Arylmethyl triazolo and imidazopyrazines as c-met inhibitors |
| US7122548B2 (en) | 2003-07-02 | 2006-10-17 | Sugen, Inc. | Triazolotriazine compounds and uses thereof |
| CA2537916A1 (en) | 2003-09-03 | 2005-03-31 | Neurogen Corporation | 5-aryl-pyrazolo[4,3-d]pyrimidines, pyridines, and pyrazines and related compounds |
| WO2006101456A1 (en) | 2005-03-21 | 2006-09-28 | S*Bio Pte Ltd | Bicyclic heterocycles hydroxamate compounds useful as histone deacetylase (hdac) inhibitors |
| US7572807B2 (en) | 2005-06-09 | 2009-08-11 | Bristol-Myers Squibb Company | Heteroaryl 11-beta-hydroxysteroid dehydrogenase type I inhibitors |
| EP1904501A2 (en) | 2005-07-11 | 2008-04-02 | Smithkline Beecham Corporation | Chemical compounds |
| WO2007064797A2 (en) | 2005-11-30 | 2007-06-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of c-met and uses thereof |
| US8030305B2 (en) | 2005-12-21 | 2011-10-04 | Janssen Pharmaceutica N.V. | Triazolopyridazines as kinase modulators |
| NL2000613C2 (en) | 2006-05-11 | 2007-11-20 | Pfizer Prod Inc | Triazole pyrazine derivatives. |
| WO2007138472A2 (en) | 2006-05-30 | 2007-12-06 | Pfizer Products Inc. | Triazolopyridazine derivatives |
| WO2008003753A1 (en) | 2006-07-07 | 2008-01-10 | Biovitrum Ab (Publ) | Pyrazolo [1,5-a] pyrimidine analogs for use as inhibitors of stearoyl-coa desaturase (scd) activity |
| JP5378222B2 (en) | 2006-10-23 | 2013-12-25 | エスジーエックス ファーマシューティカルズ、インコーポレイテッド | Bicyclic triazoles as protein kinase modulators |
| ES2393132T3 (en) | 2006-10-23 | 2012-12-18 | Sgx Pharmaceuticals, Inc. | Triazolopyridazine protein kinase modules |
| HUE026659T2 (en) | 2006-11-22 | 2016-07-28 | Incyte Holdings Corp | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| WO2008116898A1 (en) | 2007-03-28 | 2008-10-02 | Biovitrum Ab (Publ) | Pyrazolo [1,5-a]pyrimidines as inhibitors of stearoyl-coa desaturase |
| JP5488778B2 (en) | 2007-07-02 | 2014-05-14 | 日産化学工業株式会社 | Controlled release particulates and solid agrochemical formulations containing the granules |
| MX2010009414A (en) | 2008-02-28 | 2010-09-24 | Novartis Ag | Imidazo [1,2-b] pyridazine derivatives for the treatment of c-met tyrosine kinase mediated disease. |
| US8389526B2 (en) | 2009-08-07 | 2013-03-05 | Novartis Ag | 3-heteroarylmethyl-imidazo[1,2-b]pyridazin-6-yl derivatives |
| AU2010283806A1 (en) | 2009-08-12 | 2012-03-01 | Novartis Ag | Heterocyclic hydrazone compounds and their uses to treat cancer and inflammation |
-
2010
- 2010-08-18 BR BR112012008061A patent/BR112012008061A2/en not_active Application Discontinuation
- 2010-08-18 AU AU2010284972A patent/AU2010284972A1/en not_active Abandoned
- 2010-08-18 KR KR1020127007032A patent/KR20120089463A/en not_active Application Discontinuation
- 2010-08-18 JP JP2012525167A patent/JP5775871B2/en not_active Expired - Fee Related
- 2010-08-18 IN IN1453DEN2012 patent/IN2012DN01453A/en unknown
- 2010-08-18 EP EP10762878A patent/EP2467383A1/en not_active Withdrawn
- 2010-08-18 EA EA201200318A patent/EA201200318A1/en unknown
- 2010-08-18 MX MX2012002179A patent/MX2012002179A/en not_active Application Discontinuation
- 2010-08-18 CA CA2771432A patent/CA2771432A1/en not_active Abandoned
- 2010-08-18 WO PCT/EP2010/062057 patent/WO2011020861A1/en active Application Filing
- 2010-08-19 TW TW099127805A patent/TW201113284A/en unknown
- 2010-08-19 AR ARP100103038A patent/AR079486A1/en unknown
- 2010-08-19 US US12/859,653 patent/US8410264B2/en not_active Expired - Fee Related
- 2010-08-19 UY UY0001032848A patent/UY32848A/en unknown
-
2012
- 2012-08-06 US US13/567,386 patent/US8507676B2/en not_active Expired - Fee Related
Patent Citations (147)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3714357A (en) | 1968-07-15 | 1973-01-30 | Rech D Applic Scient Sogeras S | Pharmaceutical compositions comprising quinuclidinol derivatives |
| US4100274A (en) | 1976-05-11 | 1978-07-11 | Imperial Chemical Industries Limited | Polypeptide |
| US4636505A (en) | 1982-07-23 | 1987-01-13 | Imperial Chemical Industries Plc | Amide derivatives |
| US4659516A (en) | 1983-10-12 | 1987-04-21 | Imperial Chemical Industries Plc | Steroid derivatives |
| US5093330A (en) | 1987-06-15 | 1992-03-03 | Ciba-Geigy Corporation | Staurosporine derivatives substituted at methylamino nitrogen |
| EP0409595A2 (en) | 1989-07-19 | 1991-01-23 | Glaxo Group Limited | Process for the preparation of a carbocyclic nucleoside analogue |
| US5010099A (en) | 1989-08-11 | 1991-04-23 | Harbor Branch Oceanographic Institution, Inc. | Discodermolide compounds, compositions containing same and method of preparation and use |
| US5171744A (en) | 1989-10-19 | 1992-12-15 | Pfizer Inc. | Antimuscarinic bronchodilators |
| EP0424021A1 (en) | 1989-10-19 | 1991-04-24 | Pfizer Limited | Antimuscarinic bronchodilators |
| US5461076A (en) | 1990-05-07 | 1995-10-24 | Stanek; Jaroslav | Hydrazones |
| EP0490587A1 (en) | 1990-12-07 | 1992-06-17 | Merck & Co. Inc. | Substituted pyrazolopyrimidines and imidazopyridazines as angiotensin II antagonists |
| WO1992019594A1 (en) | 1991-05-02 | 1992-11-12 | Smithkline Beecham Corporation | Pyrrolidinones |
| US5451700A (en) | 1991-06-11 | 1995-09-19 | Ciba-Geigy Corporation | Amidino compounds, their manufacture and methods of treatment |
| EP0520722A1 (en) | 1991-06-28 | 1992-12-30 | Zeneca Limited | Therapeutic preparations containing quinazoline derivatives |
| EP0566226A1 (en) | 1992-01-20 | 1993-10-20 | Zeneca Limited | Quinazoline derivatives |
| WO1993019750A1 (en) | 1992-04-02 | 1993-10-14 | Smithkline Beecham Corporation | Compounds useful for treating allergic or inflammatory diseases |
| WO1993019749A1 (en) | 1992-04-02 | 1993-10-14 | Smithkline Beecham Corporation | Compounds useful for treating allergic and inflammatory diseases |
| WO1993019751A1 (en) | 1992-04-02 | 1993-10-14 | Smithkline Beecham Corporation | Compounds useful for treating inflammatory diseases and inhibiting production of tumor necrosis factor |
| EP0564409A1 (en) | 1992-04-03 | 1993-10-06 | Ciba-Geigy Ag | Pyrimidin derivatives and process for their preparation |
| WO1994010202A1 (en) | 1992-10-28 | 1994-05-11 | Genentech, Inc. | Vascular endothelial cell growth factor antagonists |
| WO1994017090A1 (en) | 1993-01-20 | 1994-08-04 | Glaxo Group Limited | 2,6-diaminopurine derivatives |
| WO1995003283A1 (en) | 1993-07-19 | 1995-02-02 | Zeneca Limited | Quinazoline derivatives and their use as anti-cancer agents |
| WO1996002553A2 (en) | 1994-07-14 | 1996-02-01 | Glaxo Group Limited | AMINO PURINE-β-D-RIBOFURANURONAMIDE DERIVATIVES |
| WO1996002543A1 (en) | 1994-07-14 | 1996-02-01 | Glaxo Group Limited | 2,3-dihydroxy cyclopentane derivatives of purines |
| WO1996030347A1 (en) | 1995-03-30 | 1996-10-03 | Pfizer Inc. | Quinazoline derivatives |
| WO1996033980A1 (en) | 1995-04-27 | 1996-10-31 | Zeneca Limited | Quinazoline derivatives |
| EP0769947A1 (en) | 1995-06-07 | 1997-05-02 | Sugen, Inc. | Indolinone compounds for the treatment of disease |
| US5843901A (en) | 1995-06-07 | 1998-12-01 | Advanced Research & Technology Institute | LHRH antagonist peptides |
| WO1997002266A1 (en) | 1995-07-06 | 1997-01-23 | Novartis Ag | Pyrrolopyrimidines and processes for the preparation thereof |
| EP0787722A1 (en) | 1996-02-05 | 1997-08-06 | American Cyanamid Company | Substituted quinazoline derivatives |
| WO1997030034A1 (en) | 1996-02-14 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as antitumor agents |
| WO1997038983A1 (en) | 1996-04-12 | 1997-10-23 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| WO1997049688A1 (en) | 1996-06-24 | 1997-12-31 | Pfizer Inc. | Phenylamino-substituted tricyclic derivatives for treatment of hyperproliferative diseases |
| WO1998008849A1 (en) | 1996-08-30 | 1998-03-05 | Novartis Aktiengesellschaft | Method for producing epothilones, and intermediate products obtained during the production process |
| WO1998010121A1 (en) | 1996-09-06 | 1998-03-12 | Obducat Ab | Method for anisotropic etching of structures in conducting materials |
| WO1998011223A1 (en) | 1996-09-11 | 1998-03-19 | Schering Aktiengesellschaft | Monoclonal antibodies against the extracellular domain of human vegf-receptor protein (kdr) |
| WO1998010767A2 (en) | 1996-09-13 | 1998-03-19 | Sugen, Inc. | Use of quinazoline derivatives for the manufacture of a medicament in the treatment of hyperproliferative skin disorders |
| EP0837063A1 (en) | 1996-10-17 | 1998-04-22 | Pfizer Inc. | 4-Aminoquinazoline derivatives |
| WO1998018796A1 (en) | 1996-10-28 | 1998-05-07 | Novartis Ag | Naphthyridine derivatives |
| WO1998022461A1 (en) | 1996-11-18 | 1998-05-28 | GESELLSCHAFT FüR BIOTECHNOLOGISCHE FORSCHUNG MBH (GBF) | Epothilone c, d, e and f, production process, and their use as cytostatic as well as phytosanitary agents |
| WO1998025929A1 (en) | 1996-12-13 | 1998-06-18 | Novartis Ag | Epothilone analogs |
| WO1998028319A1 (en) | 1996-12-24 | 1998-07-02 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1998035958A1 (en) | 1997-02-13 | 1998-08-20 | Novartis Ag | Phthalazines with angiogenesis inhibiting activity |
| WO1999003854A1 (en) | 1997-07-18 | 1999-01-28 | Novartis Ag | Crystal modification of a n-phenyl-2-pyrimidineamine derivative, processes for its manufacture and its use |
| US6166037A (en) | 1997-08-28 | 2000-12-26 | Merck & Co., Inc. | Pyrrolidine and piperidine modulators of chemokine receptor activity |
| WO1999016766A1 (en) | 1997-10-01 | 1999-04-08 | Kyowa Hakko Kogyo Co., Ltd. | Benzodioxole derivatives |
| WO1999017804A1 (en) | 1997-10-03 | 1999-04-15 | Pharmacia & Upjohn S.P.A. | Polymeric derivatives of camptothecins |
| WO1999024451A2 (en) | 1997-11-08 | 1999-05-20 | Glaxo Group Limited | Adenosine a1 receptor agonists |
| WO1999024449A2 (en) | 1997-11-08 | 1999-05-20 | Glaxo Group Limited | Adenosine a1 receptor agonists |
| WO1999024450A2 (en) | 1997-11-08 | 1999-05-20 | Glaxo Group Limited | Adensine a1 receptor agonists |
| WO1999038877A2 (en) | 1998-01-31 | 1999-08-05 | Glaxo Group Limited | 2-(PURIN-9-yl)-TETRAHYDROFURAN-3,4-DIOL DERIVATIVES |
| WO1999041267A1 (en) | 1998-02-14 | 1999-08-19 | Glaxo Group Limited | 2-(purin-9-yl) -tetrahydrofuran-3, 4-diol derivatives |
| US6194181B1 (en) | 1998-02-19 | 2001-02-27 | Novartis Ag | Fermentative preparation process for and crystal forms of cytostatics |
| WO1999043653A1 (en) | 1998-02-25 | 1999-09-02 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues therof |
| WO1999067263A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1999067264A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1999067265A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO1999067266A1 (en) | 1998-06-23 | 1999-12-29 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| WO2000000531A1 (en) | 1998-06-30 | 2000-01-06 | The Dow Chemical Company | Polymer polyols and a process for the production thereof |
| WO2000009495A1 (en) | 1998-08-11 | 2000-02-24 | Novartis Ag | Isoquinoline derivatives with angiogenesis inhibiting activity |
| WO2000023457A1 (en) | 1998-10-16 | 2000-04-27 | Pfizer Limited | Adenine derivatives |
| WO2000027819A2 (en) | 1998-11-10 | 2000-05-18 | Schering Aktiengesellschaft | Antrhranilic acid amides and the use thereof as medicaments |
| WO2000027820A1 (en) | 1998-11-10 | 2000-05-18 | Novartis Ag | N-aryl(thio)anthranilic acid amide derivatives, their preparation and their use as vegf receptor tyrosine kinase inhibitors |
| WO2000031247A2 (en) | 1998-11-20 | 2000-06-02 | Kosan Biosciences, Inc. | Recombinant methods and materials for producing epothilone and epothilone derivatives |
| WO2000037502A2 (en) | 1998-12-22 | 2000-06-29 | Genentech, Inc. | Vascular endothelial cell growth factor antagonists and uses thereof |
| WO2000059509A1 (en) | 1999-03-30 | 2000-10-12 | Novartis Ag | Phthalazine derivatives for treating inflammatory diseases |
| WO2000066558A1 (en) | 1999-05-04 | 2000-11-09 | Schering Corporation | Piperazine derivatives useful as ccr5 antagonists |
| WO2000066559A1 (en) | 1999-05-04 | 2000-11-09 | Schering Corporation | Piperidine derivatives useful as ccr5 antagonists |
| EP1052264A2 (en) | 1999-05-11 | 2000-11-15 | Pfizer Products Inc. | Process for the synthesis of nucleoside analogs |
| WO2000075114A1 (en) | 1999-06-04 | 2000-12-14 | Novartis Ag | Beta2-adrenoceptor agonists |
| WO2000077018A2 (en) | 1999-06-15 | 2000-12-21 | Pfizer Limited | Purine derivatives |
| WO2000078774A2 (en) | 1999-06-18 | 2000-12-28 | University Of Virginia Patent Foundation | Induction of pharmacological stress with adenosine receptor agonists |
| WO2001004118A2 (en) | 1999-07-14 | 2001-01-18 | Almirall Prodesfarma S.A. | Quinuclidine derivatives and their use as muscarinic m3 receptor ligands |
| WO2001013953A2 (en) | 1999-08-21 | 2001-03-01 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Synergistic combination of pde inhibitors and beta 2 adrenoceptor agonist |
| WO2001023399A1 (en) | 1999-09-30 | 2001-04-05 | Pfizer Products Inc. | Compounds for the treatment of ischemia |
| WO2001027131A1 (en) | 1999-10-14 | 2001-04-19 | Pfizer Limited | Purine derivatives |
| WO2001027130A1 (en) | 1999-10-14 | 2001-04-19 | Pfizer Limited | Purine derivatives |
| WO2001060835A1 (en) | 2000-02-18 | 2001-08-23 | Pfizer Limited | Purine derivatives |
| WO2001094368A1 (en) | 2000-06-06 | 2001-12-13 | Pfizer Limited | 2-aminocarbonyl-9h-purine derivatives |
| WO2002000652A1 (en) | 2000-06-27 | 2002-01-03 | Laboratorios S.A.L.V.A.T., S.A. | Carbamates derived from arylalkylamines |
| WO2002000679A2 (en) | 2000-06-28 | 2002-01-03 | Novartis Ag | 9.alpha.-chloro-6.alpha.-fluoro-17.alpha.-hydroxy-16-methyl-17-beta-methoxycarbonyl-androst-1,4-dienes esterified in position 17.alpha. by a cyclic acyl group |
| WO2002010143A1 (en) | 2000-07-28 | 2002-02-07 | Schering Aktiengesellschaft | Non-steroidal inflammation inhibitors |
| WO2002012266A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 17.beta.-carbothioate 17.alpha.-arylcarbonyloxyloxy androstane derivative as anti-inflammatory agents |
| WO2002022577A2 (en) | 2000-09-01 | 2002-03-21 | Novartis Ag | Hydroxamate derivatives useful as deacetylase inhibitors |
| US6552065B2 (en) | 2000-09-01 | 2003-04-22 | Novartis Ag | Deacetylase inhibitors |
| WO2002022630A1 (en) | 2000-09-15 | 2002-03-21 | Pfizer Limited | Purine derivatives |
| WO2002042298A1 (en) | 2000-11-21 | 2002-05-30 | Novartis Ag | Aminothiazoles and their use as adenosine receptor antagonists |
| WO2002051841A1 (en) | 2000-12-22 | 2002-07-04 | Almirall Prodesfarma Ag | Quinuclidine carbamate derivatives and their use as m3 antagonists |
| WO2002053564A2 (en) | 2000-12-28 | 2002-07-11 | Almirall Prodesfarma Ag | Quinuclidine derivatives and their use as m3 antagonists |
| EP1241176A1 (en) | 2001-03-16 | 2002-09-18 | Pfizer Products Inc. | Purine derivatives for the treatment of ischemia |
| WO2002088167A1 (en) | 2001-04-30 | 2002-11-07 | Glaxo Group Limited | Anti-inflammatory 17.beta.-carbothioate ester derivatives of androstane with a cyclic ester group in position 17.alpha |
| WO2002092599A1 (en) | 2001-05-14 | 2002-11-21 | Novartis Ag | 4-amino-5-phenyl-7-cyclobutyl-pyrrolo (2,3-d) pyrimidine derivatives |
| WO2002096462A1 (en) | 2001-05-25 | 2002-12-05 | Pfizer Inc. | An adenosine a2a receptor agonist and an anticholinergic agent in combination for treating obstructive airways diseases |
| WO2002100676A1 (en) | 2001-06-11 | 2002-12-19 | Ts Tech Co., Ltd. | Operating lever, and seat having the same |
| WO2002100879A1 (en) | 2001-06-12 | 2002-12-19 | Glaxo Group Limited | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| WO2003000840A2 (en) | 2001-06-21 | 2003-01-03 | Diversa Corporation | Nitrilases |
| WO2003013541A1 (en) | 2001-08-07 | 2003-02-20 | Novartis Ag | 4-amino-6-phenyl-pyrrolo[2,3-d]pyrimidine derivatives |
| WO2003033495A1 (en) | 2001-10-17 | 2003-04-24 | Ucb, S.A. | Quinuclidine derivatives, processes for preparing them and their uses as m2 and/or m3 muscarinic receptor inhibitors |
| WO2003035668A2 (en) | 2001-10-20 | 2003-05-01 | Glaxo Group Limited | Novel anti-inflammatory androstane derivatives -17-carboxy-lactone substituted steroids with an aryl-carboxylic ester in position 17.alpha |
| WO2003039544A1 (en) | 2001-11-05 | 2003-05-15 | Novartis Ag | Naphtyridine derivatives, their preparation and their use as phosphodiesterase isoenzyme 4 (pde4) inhibitors |
| WO2003048181A1 (en) | 2001-12-01 | 2003-06-12 | Glaxo Group Limited | 17.alpha. -cyclic esters of 16-methylpregnan-3,20-dione as anti-inflammatory agents |
| WO2003053966A2 (en) | 2001-12-20 | 2003-07-03 | Laboratorios S.A.L.V.A.T., S.A. | 1-alkyl-1-azoniabicyclo [2.2.2] octane carbamate derivatives and their use as muscarinic receptor antagonists |
| WO2003072592A1 (en) | 2002-01-15 | 2003-09-04 | Glaxo Group Limited | 17.alpha-cycloalkyl/cycloylkenyl esters of alkyl-or haloalkyl-androst-4-en-3-on-11.beta.,17.alpha.-diol 17.beta.-carboxylates as anti-inflammatory agents |
| WO2003062259A2 (en) | 2002-01-21 | 2003-07-31 | Glaxo Group Limited | Non-aromatic 17.alpha.-esters of androstane-17.beta.-carboxylate esters as anti-inflammatory agents |
| WO2003082280A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003082787A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2003086408A1 (en) | 2002-04-10 | 2003-10-23 | University Of Virginia Patent Foundation | Use of a2a adenosine receptor agonists for the treatment of inflammatory diseases |
| WO2003087094A2 (en) | 2002-04-16 | 2003-10-23 | Almirall Prodesfarma Ag | Pyrrolidinium derivatives as antagonists of m3 muscarinic receptors |
| WO2003099807A1 (en) | 2002-05-29 | 2003-12-04 | Almirall Prodesfarma S.A. | New indolylpiperidine derivatives as potent antihistaminic and antiallergic agents |
| WO2003104204A1 (en) | 2002-06-05 | 2003-12-18 | Merck Patent Gmbh | Pyridazine derivatives |
| WO2003104195A1 (en) | 2002-06-06 | 2003-12-18 | Boehringer Ingelheim Pharmaceuticals, Inc. | 4-(aryl or heteroaryl) -2-butylamine derivatives and their use as glucocorticoid ligans |
| WO2003104205A1 (en) | 2002-06-10 | 2003-12-18 | Merck Patent Gmbh | Aryloximes |
| WO2003106444A1 (en) | 2002-06-14 | 2003-12-24 | Recordati S.A. | N,n-disubstituted diazocycloalkanes useful for the treatment of cns disorders due to serotonergic dysfunction |
| WO2004000839A1 (en) | 2002-06-19 | 2003-12-31 | Merck Patent Gmbh | Thiazole derivatives as phosphodiesterase iv inhibitors |
| WO2004000814A1 (en) | 2002-06-25 | 2003-12-31 | Merck Frosst Canada & Co. | 8-(biaryl) quinoline pde4 inhibitors |
| WO2004005285A1 (en) | 2002-07-02 | 2004-01-15 | Almirall Prodesfarma Ag | New quinuclidine amide derivatives |
| WO2004005258A1 (en) | 2002-07-02 | 2004-01-15 | Merck Frosst Canada & Co. | Di-aryl-substituted-ethane pyridone pde4 inhibitors |
| WO2004005229A1 (en) | 2002-07-08 | 2004-01-15 | Pfizer Products Inc. | Modulators of the glucocorticoid receptor |
| WO2004016601A1 (en) | 2002-08-09 | 2004-02-26 | Novartis Ag | Benzothiazole derivatives having beta-2-adrenoreceptor agonist activity |
| WO2004018449A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Piperidine-derivatives as pde4 inhibitors |
| WO2004018457A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Pyrrolidinedione substituted piperidine-phthalazones as pde4 inhibitors |
| WO2004018451A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Pyridazinone-derivatives as pde4 inhibitors |
| WO2004018450A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Piperidine-n-oxide-derivatives |
| WO2004018465A2 (en) | 2002-08-17 | 2004-03-04 | Altana Pharma Ag | Benzonaphthyridines with pde 3/4 inhibiting activity |
| WO2004018425A1 (en) | 2002-08-21 | 2004-03-04 | Astrazeneca Ab | N-4-piperidinyl compounds as ccr5 modulators |
| WO2004018422A1 (en) | 2002-08-23 | 2004-03-04 | Ranbaxy Laboratories Limited | Fluoro and sulphonylamino containing 3,6-disubstituted azabicyclo (3.1.0) hexane derivatives as muscarinic receptor antagonists |
| WO2004019944A1 (en) | 2002-08-29 | 2004-03-11 | Altana Pharma Ag | 2-hydroxy-6-phenylphenanthridines as pde-4 inhibitors |
| WO2004026841A1 (en) | 2002-09-18 | 2004-04-01 | Sumitomo Pharmaceuticals Co., Ltd. | Novel 6-substituted uracil derivative and therapeutic agent for allergic disease |
| WO2004026873A1 (en) | 2002-09-18 | 2004-04-01 | Ono Pharmaceutical Co., Ltd. | Triazaspiro[5.5]undecane derivatives and drugs comprising the same as the active ingredient |
| JP2004107299A (en) | 2002-09-20 | 2004-04-08 | Japan Energy Corp | New 1-substituted urasil derivative and therapeutic agent for allergic disease |
| WO2004033412A1 (en) | 2002-10-04 | 2004-04-22 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel beta mimetics with extended duration of action, method for production and use thereof as medicaments |
| WO2004037805A1 (en) | 2002-10-23 | 2004-05-06 | Glenmark Pharmaceuticals Ltd. | Novel tricyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| WO2004039766A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenylethanolamine derivatives for the treatment of respiratory diseases |
| WO2004039762A1 (en) | 2002-11-01 | 2004-05-13 | Glaxo Group Limited | Phenethanolamine derivatives for the treatment of respiratory diseases |
| WO2004045607A1 (en) | 2002-11-15 | 2004-06-03 | Elbion Ag | Novel hydroxyindoles, use as inhibitors of phosphodiesterase 4 and method for production thereof |
| WO2004046083A1 (en) | 2002-11-15 | 2004-06-03 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel dihydroxy-methylphenyl derivatives, method for the production and use thereof as medicaments |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004101846A1 (en) | 2003-05-16 | 2004-11-25 | Sandvik Intellectual Property Ab | Cutting tool for metalworking as well as method in the production of cutting tools |
| WO2004101994A1 (en) | 2003-05-19 | 2004-11-25 | Federalnoe Gosudarstvennoe Unitarnoe Predpriiatie 'rossiiskii Federalnyi Iadernyi Centr - Vserossiiskii Nauchno-Issledovatelskii Institut Tehnicheskoi Fisiki Imeni Akademika E.I.Zababahina' | Ultrahigh pressure plunger pump |
| WO2004104561A1 (en) | 2003-05-21 | 2004-12-02 | Bayer Technology Services Gmbh | Method and device for measuring the content of ferromagnetic particles in liquid suspensions, aerosol dispensers or solid mixtures |
| WO2006110094A1 (en) | 2005-04-14 | 2006-10-19 | Gyros Patent Ab | Upward microconduits |
| WO2007013673A1 (en) | 2005-07-29 | 2007-02-01 | Astellas Pharma Inc. | Fused heterocycles as lck inhibitors |
| WO2007126799A2 (en) | 2006-03-30 | 2007-11-08 | Novartis Ag | Compositions and methods of use for antibodies of c-met |
| WO2008008539A2 (en) * | 2006-07-14 | 2008-01-17 | Amgen Inc. | Fused heterocyclic derivatives useful as inhibitors of the hepatocyte growth factor receptor |
| WO2008100853A1 (en) | 2007-02-12 | 2008-08-21 | Wm. Wrigley Jr. Company | Confectionery products comprising polyols |
| WO2008144767A1 (en) | 2007-05-21 | 2008-11-27 | Sgx Pharmaceuticals, Inc. | Heterocyclic kinase modulators |
Non-Patent Citations (17)
| Title |
|---|
| "Remington's Pharmaceutical Sciences, 18th Ed.", 1990, MACK PRINTING COMPANY, pages: 1289 - 1329 |
| "Remington's Pharmaceutical Sciences, 20th ed.,", 1985, MACK PUBLISHING COMPANY |
| COOPER ET AL., NATURE, vol. 311, 1984, pages 29 - 33 |
| HELLMAN ET AL.: "Principles and Practice of Oncology., 4th Edition", vol. 1, 1993, article "Principles of Radiation Therapy, Cancer", pages: 248 - 275 |
| JEFFERS; VANDE WOUDE, ONCOGENE, vol. 18, 1999, pages 5120 - 5125 |
| MORDENTI ET AL., TOXICOL PATHOL, vol. 27, no. 1, 1999, pages 14 - 21 |
| NANDY ET AL., ACTA ONCOLOGICA, vol. 33, no. 8, 1994, pages 953 - 961 |
| O'REILLY ET AL., CELL, vol. 79, 1994, pages 315 - 328 |
| O'REILLY ET AL., CELL, vol. 88, 1997, pages 277 - 285 |
| ORGANIC LETT., vol. 31, 2001, pages 139 |
| PARK. ET AL., CELL, vol. 45, 1986, pages 895 - 904 |
| PREWETT ET AL., CANCER RES, vol. 59, 1999, pages 5209 - 5218 |
| SCHMIDT, L. ET AL., NAT. GENET., vol. 16, 1997, pages 68 - 73 |
| SHEN ET AL., CELL, vol. 103, 2000, pages 501 - 10 |
| STAHL AND WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| YUAN ET AL., PROC NATL ACAD SCI U S A, vol. 93, 1996, pages 14765 - 14770 |
| ZHU ET AL., CANCER RES, vol. 58, 1998, pages 3209 - 3214 |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2719699A1 (en) * | 2009-12-31 | 2014-04-16 | Hutchison Medipharma Limited | Certain triazolopyrazines, compositions thereof and methods of use therefor |
| US9956218B2 (en) | 2009-12-31 | 2018-05-01 | Hutchison Medipharma Limited | Certain triazolopyridines and triazolopyrazines, compositions thereof and methods of use therefor |
| US11896592B2 (en) | 2009-12-31 | 2024-02-13 | Hutchison Medipharma Limited | Certain triazolopyridines and triazolopyrazines, compositions thereof and methods of use therefor |
| US8987269B2 (en) | 2009-12-31 | 2015-03-24 | Hutchison Medipharma Limited | Certain triazolopyridines and triazolopyrazines, compositions thereof and methods of use therefor |
| US10946014B2 (en) | 2009-12-31 | 2021-03-16 | Hutchison Medipharma Limited | Certain triazolopyridines and triazolopyrazines, compositions thereof and methods of use therefor |
| US10512645B2 (en) | 2009-12-31 | 2019-12-24 | Hutchinson Medipharma Limited | Certain triazolopyridines and triazolopyrazines, compositions thereof and methods of use therefor |
| US10590129B2 (en) | 2010-05-17 | 2020-03-17 | Rhizen Pharmaceuticals Sa | 3,5-disubstituted-3H-imidazo[4,5-B]pyridine and 3,5-disubstituted-3H-[1,2,3]triazolo[4,5-B] pyridine compounds as modulators of protein kinases |
| EA025281B1 (en) * | 2010-05-17 | 2016-12-30 | Инкозен Терапьютикс Пвт. Лтд. | 3,5-DISUBSTITUED-3H-IMIDAZO[4,5-b]PYRIDINE AND 3,5-DISUBSTITUED-3H-[1,2,3]TRIAZOLO[4,5-b]PYRIDINE COMPOUNDS AS MODULATORS OF PROTEIN KINASES |
| US8481739B2 (en) | 2010-05-17 | 2013-07-09 | Incozen Therapeutics Pvt. Ltd. | 3,5-Disubstituted-3H-imidazo[4,5-b]pyridine and 3,5-disubstituted-3H[1,2,3]triazolo [4,5-b] Pyridine Compounds as Modulators of protein kinases |
| US8912331B2 (en) | 2010-05-17 | 2014-12-16 | Rhizen Pharmaceuticals Sa | 3,5-disubstituted-3H-imidazo[4,5-B]pyridine and 3,5-disubstituted-3H-[1,2,3]triazolo[4,5-B] pyridine compounds as modulators of protein kinases |
| CN103068824A (en) * | 2010-05-17 | 2013-04-24 | 印蔻真治疗公司 | Novel 3,5-disubstitued-3h-imidazo[4,5-b]pyridine and 3,5- disubstitued -3h-[1,2,3]triazolo[4,5-b] pyridine compounds as modulators of protein kinases |
| WO2011145035A1 (en) * | 2010-05-17 | 2011-11-24 | Indian Incozen Therapeutics Pvt. Ltd. | Novel 3,5-disubstitued-3h-imidazo[4,5-b]pyridine and 3,5- disubstitued -3h-[1,2,3]triazolo[4,5-b] pyridine compounds as modulators of protein kinases |
| US10087182B2 (en) | 2010-05-17 | 2018-10-02 | Incozen Therapeutics Pvt. Ltd. | 3,5-disubstituted-3H-imidazo[4,5-B]pyridine and 3,5-disubstituted-3H-[1,2,3]triazolo[4,5-B] pyridine compounds as modulators of protein kinases |
| CN103068824B (en) * | 2010-05-17 | 2017-09-08 | 印蔻真治疗公司 | It is used as new 3,5 2 substitution 3H imidazos [4,5 B] pyridine and 3,5 2 substitution 3H [1,2,3] triazol [4,5 B] pyridine compounds of protein kinase modulators |
| EA025281B9 (en) * | 2010-05-17 | 2017-08-31 | Инкозен Терапьютикс Пвт. Лтд. | 3,5-DISUBSTITUED-3H-IMIDAZO[4,5-b]PYRIDINE AND 3,5-DISUBSTITUED-3H-[1,2,3]TRIAZOLO[4,5-b]PYRIDINE COMPOUNDS AS MODULATORS OF PROTEIN KINASES |
| CN103459396A (en) * | 2011-02-10 | 2013-12-18 | 诺瓦提斯公司 | [1, 2, 4] triazolo [4, 3 -b] pyridazine compounds as inhibitors of c-met tyrosine kinase |
| CN103459396B (en) * | 2011-02-10 | 2015-08-19 | 诺瓦提斯公司 | As [1,2,4] triazolo [4,3-b] pyridazine compound of c-Met tyrosine kinase inhibitor |
| WO2012107500A1 (en) * | 2011-02-10 | 2012-08-16 | Novartis Ag | [1, 2, 4] triazolo [4, 3 -b] pyridazine compounds as inhibitors of the c-met tyrosine kinase |
| US9062045B2 (en) | 2011-09-15 | 2015-06-23 | Novartis Ag | Triazolopyridine compounds |
| JP2014526499A (en) * | 2011-09-15 | 2014-10-06 | ノバルティス アーゲー | 6-Substituted 3- (quinolin-6-ylthio)-[1,2,4] triazolo [4,3-A] pyrazine as tyrosine kinase |
| US9474762B2 (en) | 2011-09-15 | 2016-10-25 | Novartis Ag | Triazolopyridine compounds |
| EA026655B1 (en) * | 2011-09-15 | 2017-05-31 | Новартис Аг | 6-SUBSTITUTED 3-(QUINOLIN-6-YLTHIO)[1,2,4]TRIAZOLO[4,3-a]PYRIDINES AS c-Met TYROSINE KINASE INHIBITORS |
| WO2013038362A1 (en) | 2011-09-15 | 2013-03-21 | Novartis Ag | 6 - substituted 3 - (quinolin- 6 - ylthio) - [1,2,4] triazolo [4, 3 -a] pyradines as tyrosine kinase |
| CN103958509A (en) * | 2011-09-15 | 2014-07-30 | 诺华股份有限公司 | 6 - substituted 3 - (quinolin- 6 - ylthio) - [1,2,4] triazolo [4, 3 -a] pyradines as tyrosine kinase |
| WO2013098229A2 (en) | 2011-12-27 | 2013-07-04 | Bayer Intellectual Property Gmbh | Heteroarylpiperidine and piperazine derivatives as fungicides |
| EP2921494A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | Heteroarylpiperidine and heteroarylpiperazine derivatives |
| EP2921485A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | Isoxazole derivatives |
| EP2921492A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | Heteroarylpiperidine and heteroarylpiperazine derivatives |
| EP2921484A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | Oxazole derivatives |
| EP2921493A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | Heteroarylpiperidine and heteroarylpiperazine derivatives |
| EP2921481A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | 4-piperidine carboxylic acid derivatives |
| EP2921491A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | Intermediates for the production of heteroarylpiperidine and heteroarylpiperazine derivatives as fungicides |
| EP2921495A1 (en) | 2011-12-27 | 2015-09-23 | Bayer Intellectual Property GmbH | Heteroarylpiperidine and heteroarylpiperazine derivatives as fungicides |
| US9303015B2 (en) | 2012-10-16 | 2016-04-05 | Janssen Pharmaceutica Nv | Heteroaryl linked quinolinyl modulators of RORγt |
| US9290476B2 (en) | 2012-10-16 | 2016-03-22 | Janssen Pharmaceutica Nv | Methylene linked quinolinyl modulators of RORγt |
| US9309222B2 (en) | 2012-10-16 | 2016-04-12 | Janssen Pharmaceutica Nv | Phenyl linked quinolinyl modulators of RORγt |
| US9567342B2 (en) | 2012-11-06 | 2017-02-14 | Shanghai Fochon Pharmaceutical Co., Ltd. | Certain protein kinase inhibitors |
| US9206166B2 (en) | 2012-11-06 | 2015-12-08 | SHANGHAI iNSTITUTE OF MATERIA MEDICA ACADEMY OF SCIENCES | Certain protein kinase inhibitors |
| US10369146B2 (en) | 2013-10-15 | 2019-08-06 | Janssen Pharmaceutica Nv | Phenyl linked quinolinyl modulators of RORγt |
| US9328095B2 (en) | 2013-10-15 | 2016-05-03 | Janssen Pharmaceutica Nv | Heteroaryl linked quinolinyl modulators of RORgammat |
| US10201546B2 (en) | 2013-10-15 | 2019-02-12 | Janssen Pharmaceutica Nv | Quinolinyl modulators of RORγt |
| US9403816B2 (en) | 2013-10-15 | 2016-08-02 | Janssen Pharmaceutica Nv | Phenyl linked quinolinyl modulators of RORγt |
| US9624225B2 (en) | 2013-10-15 | 2017-04-18 | Janssen Pharmaceutica Nv | Quinolinyl modulators of RORγt |
| US10555941B2 (en) | 2013-10-15 | 2020-02-11 | Janssen Pharmaceutica Nv | Alkyl linked quinolinyl modulators of RORγt |
| US9284308B2 (en) | 2013-10-15 | 2016-03-15 | Janssen Pharmaceutica Nv | Methylene linked quinolinyl modulators of RORγt |
| US9221804B2 (en) | 2013-10-15 | 2015-12-29 | Janssen Pharmaceutica Nv | Secondary alcohol quinolinyl modulators of RORγt |
| US9346782B2 (en) | 2013-10-15 | 2016-05-24 | Janssen Pharmaceutica Nv | Alkyl linked quinolinyl modulators of RORγt |
| EP3241830A1 (en) | 2016-05-04 | 2017-11-08 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pesticides |
| WO2020260857A1 (en) | 2019-06-13 | 2020-12-30 | AdoRx Therapeutics Limited | Hydroxamate compounds as antagonists of the adenosine a2a receptor |
Also Published As
| Publication number | Publication date |
|---|---|
| US8410264B2 (en) | 2013-04-02 |
| BR112012008061A2 (en) | 2016-03-01 |
| TW201113284A (en) | 2011-04-16 |
| IN2012DN01453A (en) | 2015-06-05 |
| JP5775871B2 (en) | 2015-09-09 |
| US20110065708A1 (en) | 2011-03-17 |
| KR20120089463A (en) | 2012-08-10 |
| AU2010284972A1 (en) | 2012-03-08 |
| US20120302570A1 (en) | 2012-11-29 |
| AR079486A1 (en) | 2012-02-01 |
| EP2467383A1 (en) | 2012-06-27 |
| EA201200318A1 (en) | 2012-09-28 |
| CA2771432A1 (en) | 2011-02-24 |
| UY32848A (en) | 2011-03-31 |
| JP2013502396A (en) | 2013-01-24 |
| MX2012002179A (en) | 2012-03-16 |
| US8507676B2 (en) | 2013-08-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8507676B2 (en) | Heterocyclic oxime compounds | |
| US8497368B2 (en) | Heterocyclic hydrazone compounds | |
| TWI383982B (en) | Imidazoquinolines as lipid kinase inhibitors | |
| US9474762B2 (en) | Triazolopyridine compounds | |
| US20100305113A1 (en) | Substituted Imidazopyridazines as Lipid Kinase Inhibitors | |
| JP2010504933A (en) | Pyrazolopyrimidines as PI3K lipid kinase inhibitors | |
| EP2462143B1 (en) | 3-heteroarylmethyl-imidazo[1,2-b]pyridazin-6-yl derivatives as c-met tyrosine kinase modulators | |
| US20130324526A1 (en) | [1,2,4] triazolo [4,3-b] pyridazine compounds as inhibitors of the c-met tyrosine kinase | |
| CN102548995B (en) | Heterocyclic hydrazone compounds and their uses to treat cancer and inflammation | |
| CN102574853B (en) | Heterocyclic oxime compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080047132.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10762878 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010284972 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1453/DELNP/2012 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2771432 Country of ref document: CA Ref document number: 2012525167 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/002179 Country of ref document: MX |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010762878 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010762878 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2010284972 Country of ref document: AU Date of ref document: 20100818 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201200318 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20127007032 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012008061 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012008061 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120217 |