WO2010151595A1 - Pyrrolo-benzo-1,4-diazines useful as sodium channel blockers - Google Patents
Pyrrolo-benzo-1,4-diazines useful as sodium channel blockers Download PDFInfo
- Publication number
- WO2010151595A1 WO2010151595A1 PCT/US2010/039669 US2010039669W WO2010151595A1 WO 2010151595 A1 WO2010151595 A1 WO 2010151595A1 US 2010039669 W US2010039669 W US 2010039669W WO 2010151595 A1 WO2010151595 A1 WO 2010151595A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- quinoxaline
- pyrrolo
- piperidine
- spiro
- methoxy
- Prior art date
Links
- 0 *[N+](CC1)(CCC1=O)[O-] Chemical compound *[N+](CC1)(CCC1=O)[O-] 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/20—Spiro-condensed systems
Definitions
- the present invention relates to compounds useful as inhibitors of ion channels.
- the invention also provides pharmaceutically acceptable compositions comprising the compounds of the invention and methods of using the compositions in the treatment of various disorders.
- Voltage-gated ion channels allow electrically excitable cells to generate and propagate action potentials and therefore are crucial for nerve and muscle function.
- Sodium channels play a special role by mediating rapid depolarization, which constitutes the rising phase of the action potential and in turn activates voltage-gated calcium and potassium channels.
- Voltage-gated sodium channels represent a multigene family. Nine sodium channel subtypes have been cloned and functionally expressed to date. [Clare, J. J. s et at, Drug Discovery Today, 2000, 5: 506-520]. They are differentially expressed throughout muscle and nerve tissues and show distinct biophysical properties.
- All voltage-gated sodium channels are characterized by a high degree of selectivity for sodium over other ions and by their voltage-dependent gating. [Catterall, W. A., Current Opinion in Neurobiology, 1991, 1: 5-13].
- sodium channels are closed. Following membrane depolarization, sodium channels open rapidly and then inactivate. Sodium channels only conduct currents in the open state and, once inactivated, have to return to the resting state, favored by membrane hyperpolarization, before they can reopen.
- Different sodium channel subtypes vary in the voltage range over which they activate and inactivate as well as in their activation and inactivation kinetics.
- Sodium channels are the target of a diverse array of pharmacological agents, including neurotoxins, antiarrhythmics, anticonvulsants and local anesthetics. [Clare, J. J., et al., supra]. Several regions in the sodium channel secondary structure are involved in interactions with these blockers and most are highly conserved. Indeed, most sodium channel blockers known to date interact with similar potency with all channel subtypes. Nevertheless, it has been possible to produce sodium channel blockers with therapeutic selectivity and a sufficient therapeutic window for the treatment of epilepsy (e.g. lamotrigine, phenytoin and carbamazepine) and certain cardiac arrhythmias (e.g. lignocaine, tocainide and mexiletine).
- epilepsy e.g. lamotrigine, phenytoin and carbamazepine
- cardiac arrhythmias e.g. lignocaine, tocainide and mexiletine.
- neuropathic pain persisting long after the initial injury resolves.
- Examples of neuropathic pain include, but are not limited to, postherpetic neuralgia, trigeminal neuralgia, diabetic neuropathy, chronic lower back pain, phantom limb pain, pain resulting from cancer and chemotherapy, chronic pelvic pain, complex regional pain syndrome and related neuralgias. It has been shown in human patients as well as in animal models of neuropathic pain, that damage to primary afferent sensory neurons can lead to neuroma formation and spontaneous activity, as well as evoked activity in response to normally innocuous stimuli.
- lidocaine administered in the form of a dermal patch, is currently the only FDA approved treatment for PHN. [Devers, A. and Galer, B.S., Clinical J. Pain, 2000. 16(3): 205- 208].
- sodium channel blockers In addition to neuropathic pain, sodium channel blockers have clinical uses in the treatment of epilepsy and cardiac arrhythmias. Recent evidence from animal models suggests that sodium channel blockers may also be useful for neuroprotection under ischaemic conditions caused by stroke or neural trauma and in patients with multiple sclerosis (MS). [Clare, J. J., et al. and Anger, T., et al., supra].
- the present invention is directed to pyrrolo-benzo-l,4 ⁇ diazine compounds which are sodium channel blockers useful for the treatment of chronic and neuropathic pain.
- the compounds of the present invention are also useful for the treatment of other conditions, including disorders of the CNS such as epilepsy, manic depression and bipolar disorder.
- This invention also provides pharmaceutical compositions comprising a compound of the present invention, either alone, or in combination with one or more therapeutically active compounds, and a pharmaceutically acceptable carrier.
- This invention further comprises methods for the treatment of acute pain, chronic pain, visceral pain, inflammatory pain, neuropathic pain and disorders of the CNS including, but not limited to, epilepsy, manic depression, depression, anxiety and bipolar disorder comprising administering the compounds and pharmaceutical compositions of the present invention.
- the present invention is directed to sodium channel blockers and derivatives of structural formula I
- W is pyrazolyl, phenyl or pyridinyl, each optionally substituted with.0 to 3 groups of R a ;
- R x and Ry are independently selected from the group consisting of
- Z is selected from the group consisting of (1) a bond, (2) -Ci-C6 alkylene,
- Rl is selected from the group consisting of
- R3 and R.4 are independently selected from the group consisting of (1) hydrogen, (2)-Ci-C6alkyl, (3) -(CH2)nC6 ⁇ Cio aryl, which may be optionally substituted with 1 to 4 groups of R a ,
- R a is selected from the group consisting of
- R x and RY form a 5 to 8 membered carbocylic ring substituted with Z — Rl .
- R x and R v form a 5 to 8 membered carbocylic ring of which one of the ring atoms is ntrogen.
- a sub-embodiment of this invention is realized when Z — Rl is attached to the ntrogen atom.
- Another embodiment of the present invention is realized when Z is C(O). Another embodiment of the present invention is realized when Z is SO2-
- Rl is (CH2)nC6 ⁇ ClO sryh where said aryl is selected from the group consisting of phenyl and napthyl.
- J is 1, which makes a six member nitrogen containing ring.
- Rl is optionally substituted heteroaryl, where said heteroaryl is selected from the group consisting of benzylfuranyl, qumolinyl, indolyl, benzodioxolyl, benzothiazolyl, benzoxazmyl, benoxazolyl, and benzoxazine.
- heteroaryl is selected from the group consisting of benzylfuranyl, qumolinyl, indolyl, benzodioxolyl, benzothiazolyl, benzoxazmyl, benoxazolyl, and benzoxazine.
- Rl is selected from the group consisting of
- R a is selected from the group consisting of
- Specific embodiments of the present invention include a compound of the Examples or made according to the methods therein and pharmaceutically acceptable salts thereof and individual enantiomers and diastereomers thereof.
- any variable e.g. aryl, heterocycle, Ra etc.
- its definition on each occurrence is independent at every other occurrence.
- combinations of substituents or variables are permissible only if such combinations result in stable compounds.
- R a When R a is -O- and attached to a carbon it is referred to as a carbonyl group and when it is attached to a nitrogen (e.g., nitrogen atom on a pyridyl group) or sulfur atom it is referred to a N-oxide and sulfoxide group, respectively.
- a nitrogen e.g., nitrogen atom on a pyridyl group
- sulfur atom it is referred to a N-oxide and sulfoxide group, respectively.
- alkyl encompasses groups having the prefix “alk” such as, for example, alkylenyl, alkoxy, alkanoyl, alkenyl, and alkynyl and means carbon chains which may be linear or branched or combinations thereof.
- alk such as, for example, alkylenyl, alkoxy, alkanoyl, alkenyl, and alkynyl and means carbon chains which may be linear or branched or combinations thereof.
- Cl-C6 includes alkyls containing 6, 5, 4, 3, 2, or 1 carbon atoms. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tejrt-butyl, pentyl, hexyl, and heptyl.
- Alkylenyl refers to an alkyl have substitutions at both ends.
- Alkoxy refers to an alkyl group connected to the oxy connecting atom and also includes alkyl ether groups, where the term 'alkyl' is defined above, and 'ether' means two alkyl groups with an oxygen atom between them.
- alkoxy groups include methoxy, ethoxy, n- propoxy, i-propoxy, n-butoxy, s-butoxy, t-butoxy > methoxymethane (also referred to as 'dimethyl ether'), and methoxyethane (also referred to as 'ethyl methyl ether').
- alkenyl refers to a hydrocarbon radical straight, branched or cyclic containing from 2 to 10 carbon atoms and at least one carbon to carbon double bond.
- alkenyl groups include ethenyl, propenyl, butenyl and cyclohexenyl.
- fluoroalkyl refers to an alkyl substituent as described herein containing at least one fluorine substituent.
- cycloalkyl refers to a saturated hydrocarbon containing one ring having a specified number of carbon atoms. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- C3-C10 cycloalkyl refers to cycloalkyl having 3, 4, 5, 6, 7,8, 9, or 10 carbon atoms.
- aryl is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 members in each ring, wherein at least one ring is aromatic. Examples of such aryl elements include phenyl, napthyl, tetrahydronapthyl, indanyl, or biphenyl.
- heterocycle refers to a stable 5- to 7- membered monocyclic or stable 8- to 11-membered bicyclic heterocyclic ring which is either saturated or unsaturated, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O, and S, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure.
- heterocycle or heterocyclic includes heteroaryl moieties.
- heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydrobenzothienyl, dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, 1,3-dioxolanyl, furyl, imidazolidinyl, imidazolinyl, imidazolyl, indolinyl, indolyl, isochromanyl, isomdolinyl, isoquinolinyl, isothiazolidinyl, isothiazolyl, isothiazolidinyl, morpholinyl, naphthyrid
- heteroaryl represents a stable 5- to 7- membered monocyclic- or stable 9- to 10-membered fused bicyclic heterocyclic ring system which contains an aromatic ring, any ring of which may be saturated, such as piperidinyl, partially saturated, or unsaturated, such as pyridinyl, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O and S, and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quaternized, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure.
- heteroaryl groups include, but are not limited to, benzimidazole, benzisothiazole, benzisoxazole, benzofuran, benzothiazole, benzothiophene, benzotriazole, benzoxazole, carboline, cinnoline, furan, furazan, imidazole, indazole, indole, indolizine, isoquinoline, isothiazole, isoxazole, naphthyridine, oxadiazole, oxazole, phthalazine, pteridine, purine, pyran, pyrazine, pyrazole, pyridazine, pyridine, pyriniidine, pyrrole, qumazoline, quinoline, quinoxaline, tetrazole, thiadiazole, thiazole
- heterocycloalkyls examples include azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, tetrahydrofuranyl, imidazolinyl, pyrolidin-2 ⁇ one, piperidin-2-one, and thiomorpholinyl.
- heteroatom means O, S or N, selected on an independent basis.
- a group which is designated as being substituted with substituents may be substituted with multiple numbers of such substituents, which may be the same or different, for example, with one to four groups of R a .
- a group which is designated as being independently substituted with substituents may be independently substituted with multiple numbers of such substituents.
- the compounds of the invention may have one or more asymmetric centers.
- Compounds with asymmetric centers give rise to enantiomers (optical isomers), diastereomers (configurational isomers) or both, and it is intended that all of the possible enantiomers and diastereomers in mixtures and as pure or partially purified compounds are included within the scope of this invention.
- the present invention is meant to encompass all such isomeric forms of the compounds of formulae (I) and (II).
- Formulae (I) and (II) are shown above without a definite stereochemistry.
- the present invention includes all stereoisomers of formulae (I) and (II), and pharmaceutically acceptable salts thereof.
- the independent syntheses of the enantiomerically or diastereomerically enriched compounds, or their chromatographic separations, may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration and by appropriate modification of the methodology disclosed herein as known in the art.
- Their absolute stereochemistry may be determined by the x-ray crystallography of crystalline products or crystalline intermediates that are derivatized, if necessary, with a reagent containing an asymmetric center of known absolute configuration.
- racemic mixtures of the compounds may be separated so that the individual enantiomers or diastereomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diastereomeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods using chiral stationary phases, which methods are well known in the art.
- the protecting groups may be removed at a convenient sequent stage using methods known from the art, hi the compounds of formulas (I )- (IV), the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature.
- the present invention is meant to include all suitable isotopic variations of the compounds of generic formulas (I) - (IV).
- different isotopic forms of hydrogen (H) include protium (lH) and deuterium ( ⁇ H). Protium is the predominant hydrogen isotope found in nature.
- Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
- Isotopically-enriched compounds within generic formulas (I) - (IV) can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- the term "substantially pure” means that the isolated material is at least 90% pure, and preferably 95% pure, and even more preferably 99% pure as assayed by analytical techniques known in the art.
- substituted means that one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- stable compound' or “stable structure” is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- purified refers to the physical state of said compound after being isolated from a synthetic process (e.g. from a reaction mixture), or natural source or combination thereof.
- purified refers to the physical state of said compound after being obtained from a purification process or processes described herein or well known to the skilled artisan (e.g., chromatography, recrystallization and the like), in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan.
- the compounds of the present invention may be administered in the form of a pharmaceutically acceptable salt.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- the compounds of the invention may be mono, di or tris salts, depending on the number of acid functionalities present in the free base form of the compound.
- Free bases and salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylene-diamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such as
- the compound of the present invention When the compound of the present invention is basic, its corresponding salt may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- Such acids include acetic, trifluoroacetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, para-toluenesulfonic acid, and the like. It will be understood that, as used herein, references to the compounds of the present invention are meant to also include the pharmaceutically acceptable salts.
- composition as used herein is intended to encompass a product comprising specified ingredients in predetermined amounts or proportions, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- This term in relation to pharmaceutical compositions is intended to encompass a product comprising one or more active ingredients, and an optional carrier comprising inert ingredients, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- the term "pharmaceutical composition” of the present invention encompasses any composition made by combining a compound of the present invention and a pharmaceutically acceptable agent, including another active agent, carrier or diluent.
- a pharmaceutically acceptable agent including another active agent, carrier or diluent.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- compositions of the present invention comprise a compound of the invention (or pharmaceutically acceptable salts thereof) as an active ingredient, a pharmaceutically acceptable carrier, and optionally one or more additional therapeutic agents or adjuvants.
- additional therapeutic agents can include, for example, i) opiate agonists or antagonists, ii) calcium channel antagonists, iii) 5HT receptor agonists or antagonists, iv) sodium channel antagonists, v) NMDA receptor agonists or antagonists, vi) COX-2 selective inhibitors, v ⁇ ) NKl antagonists, viii) non-steroidal anti-inflammatory drugs ("NSAID”), ix) selective serotonin reuptake inhibitors ("SSRI”) and/or selective serotonin and norepinephrine reuptake inhibitors (“SSNRI”), x) tricyclic antidepressant drugs, xi) norepinephrine modulators, xii) lithium, xi ⁇ ) valproate, and xiv) neurontin
- the pharmaceutical compositions may be conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy.
- pharmaceutical composition is also intended to encompass both the bulk composition and individual dosage units comprised of more than one (e.g., two) pharmaceutically active agents such as, for example, a compound of the present invention and an additional agent selected from the lists of the additional agents described herein, along with any pharmaceutically inactive excipients.
- the bulk composition and each individual dosage unit can contain fixed amounts of the afore-said "more than one pharmaceutically active agents".
- the bulk composition is material that has not yet been formed into individual dosage units.
- An illustrative dosage unit is an oral dosage unit such as tablets, pills and the like.
- the herein-described method of treating a patient by administering a pharmaceutical composition of the present invention is also intended to encompass the administration of the afore-said bulk composition and individual dosage units,
- a pharmaceutically acceptable derivative includes, but is not limited to, pharmaceutically acceptable salts, esters, salts of such esters, or any other adduct or derivative which upon administration to a patient in need is capable of providing, directly or indirectly, a compound as otherwise described herein, or a metabolite or residue thereof.
- pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active compound which is a compound of formulae (I) to (IV) 5 is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- the pharmaceutical compositions of the present invention can be presented as discrete units suitable for oral administration such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient.
- the compositions can be presented as a powder, as granules, as a solution, as a suspension in an aqueous liquid, as a non-aqueous liquid, as an oil- in- water emulsion or as a water-in-oil liquid emulsion.
- the compounds of the invention, or pharmaceutically acceptable salts thereof may also be administered by controlled release means and/or delivery devices.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets may contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a tablet containing the composition of this invention may be prepared by compression or molding, optionally with one or more accessory ingredients or adjuvants.
- Compressed tablets may be prepared by compressing, in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent.
- Each tablet preferably contains from about 0.1 mg to about 500 mg of the active ingredient and each cachet or capsule preferably containing from about 0.1 mg to about 500 mg of the active ingredient.
- compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example peanut oil, liquid paraffin, or olive oil.
- Other pharmaceutical compositions include aqueous suspensions, which contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example aracbis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. Oily suspensions may also contain various excipients.
- the pharmaceutical compositions of the invention may also be in the form of
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleaginous suspension, or in the form of sterile powders for the extemporaneous preparation of such sterile injectable solutions or dispersions.
- the final injectable form must be sterile and must be effectively fluid for easy syringability.
- the pharmaceutical compositions must be stable under the conditions of manufacture and storage; thus, preferably should be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- compositions of the present invention can be in a form suitable for topical use such as, for example, an aerosol, cream, ointment, lotion, dusting powder, or the like. Further, the compositions can be in a form suitable for use in transdermal devices. These formulations may be prepared via conventional processing methods. As an example, a cream or ointment is prepared by mixing hydrophilic material and water, together with about 5 wt% to about 10 wt% of the compound, to produce a cream or ointment having a desired consistency. Pharmaceutical compositions of this invention can also be in a form suitable for rectal administration wherein the carrier is a solid. It is preferable that the mixture forms unit dose suppositories.
- Suitable carriers include cocoa butter and other materials commonly used in the art.
- the compounds and pharmaceutically acceptable compositions of the present invention can be employed in combination therapies, that is, the compounds and pharmaceutically acceptable compositions can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutics or medical procedures.
- the particular combination of therapies (therapeutics or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutics and/or procedures and the desired therapeutic effect to be achieved.
- the therapies employed may achieve a desired effect for the same disorder (for example, an inventive compound may be administered concurrently with another agent used to treat the same disorder), or they may achieve different effects (e.g., control of any adverse effects).
- additional therapeutic agents which are normally administered to treat or prevent a particular disease, or condition, are known as "appropriate for the disease, or condition, being treated".
- additional therapeutic agents include, but are not limited to: nonopioid analgesics (indoles such as Etodolac, Indomethacin, Sulindac, Tolmetin; naphthylalkanon.es such as Nabumetone; oxicams such as Piroxicam; para-aminophenol derivatives, such as
- Acetaminophen propionic acids such as Fenoprofen, Flurbiprofen, Ibuprofen, Ketoprofen, Naproxen, Naproxen sodium, Oxaprozin; salicylates such as Aspirin, Choline magnesium trisalicylate, Diflunisal; fenamates such as meclofenamic acid, Mefenamic acid; and pyrazoles such as Phenylbutazone); or opioid (narcotic) agonists (such as Codeine, Fentanyl, Hydromorphone, Levorphanol, Meperidine, Methadone, Morphine, Oxycodone, Oxymorphone, Propoxyphene, Buprenorphine, Butorphanol, Dezocine, Nalbuphine, and Pentazocine).
- opioid (narcotic) agonists such as Codeine, Fentanyl, Hydromorphone, Levorphanol, Meperidine, Methadone, Morphine, Oxycodone, Ox
- nondrug analgesic approaches may be utilized in conjunction with administration of one or more compounds of the invention.
- anesthesiologic intraspinal infusion, neural blocade
- neurosurgical neurolysis of CNS pathways
- neurostimulatory transcutaneous electrical nerve stimulation, dorsal column stimulation
- physiatric physical therapy, orthotic devices, diathermy
- psychologic psychologic
- the amount of additional therapeutic agent present in the compositions of this invention will be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent.
- the amount of additional therapeutic agent in the presently disclosed compositions will range from about 50% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent.
- the present invention additionally provides any one of the aforementioned methods further comprising administering to said patient an amount effective to treat said disease or disorder of at least one additional therapeutic agent, wherein the additional therapeutic agent(s) is/are selected from the group consisting of therapeutic agents known to be useful to treat said disease or disorder.
- the additional therapeutic agent(s) is/are selected from the group consisting of opiate agonists, opiate antagonists, calcium channel antagonists, 5HT receptor agonists, 5HT receptor antagonists, sodium channel antagonists, NMDA receptor agonists, NMDA receptor antagonists, COX-2 selective inhibitors, NKl antagonists, non-steroidal anti-inflammatory drugs, selective serotonin reuptake inhibitors, selective serotonin and norepinephrine reuptake inhibitors, tricyclic antidepressants, norephinephrine modulators, lithium, valproate, neurontin and pregabalin.
- administering a should be understood to mean providing a compound of the invention to the individual in need of treatment in a form that can be introduced into that individual's body in a therapeutically useful form and therapeutically useful amount, including, but not limited to: oral dosage forms, such as tablets, capsules, syrups, suspensions, and the like; injectable dosage forms, such as IV, IM, or IP, and the like; transdermal dosage forms, including creams, jellies, powders, or patches; buccal dosage forms; inhalation powders, sprays, suspensions, and the like; and rectal suppositories.
- oral dosage forms such as tablets, capsules, syrups, suspensions, and the like
- injectable dosage forms such as IV, IM, or IP, and the like
- transdermal dosage forms including creams, jellies, powders, or patches
- buccal dosage forms inhalation powders, sprays, suspensions, and the like
- rectal suppositories rectal suppositories.
- an effective amount or “therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- treatment means any administration of a compound of the present invention and includes (1) inhibiting the disease in an animal that is experiencing or displaying the pathology or symptomatology of the diseased (i.e., arresting further development of the pathology and/or symptomatology), or (2) ameliorating the disease in an animal that is experiencing or displaying the pathology or symptomatology of the diseased
- compositions containing compounds of the present invention may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
- unit dosage form is taken to mean a single dose wherein all active and inactive ingredients are combined in a suitable system, such that the patient or person administering the drug to the patient can open a single container or package with the entire dose contained therein, and does not have to mix any components together from two or more containers or packages.
- Typical examples of unit dosage forms are tablets or capsules for oral administration, single dose vials for injection, or suppositories for rectal administration. This list of unit dosage forms is not intended to be limiting in any way, but merely to represent typical examples of unit dosage forms.
- compositions containing compounds of the present invention may conveniently be presented as a kit, whereby two or more components, which may be active or inactive ingredients, carriers, diluents, and the like, are provided with instructions for preparation of the actual dosage form by the patient or person administering the drug to the patient.
- kits may be provided with all necessary materials and ingredients contained therein, or they may contain instructions for using or making materials or components that must be obtained independently by the patient or person administering the drug to the patient.
- the compounds of the present invention are administered at a daily dosage of from about 0.1 mg to about 100 mg per kg of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 mg to about 2000 mg, preferably from about 0.1 mg to about 20 mg per kg of body weight. In the case of a 70 kg adult human, the total daily dose will generally be from about 7 mg to about 1 ,400 mg. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- a formulation intended for the oral administration to humans may conveniently contain from about 0.005 mg to about 2.5 g of active agent, compounded with an appropriate and convenient amount of carrier material.
- Unit dosage forms will generally contain between from about 0.005 mg to about 1000 mg of the active ingredient,, typically 0.005, 0.01 mg, 0.05 mg, 0.25 mg, 1 mg, 5 mg, 25 rag, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg or 1000 mg, administered once, twice or three times a day.
- compositions of this invention can be administered to humans and other animals orally , rectally, parenterally, intracistemally, intra vaginally, intraperitoneally, topically (as by powders, ointments, or drops), bucally, as an oral or nasal spray, or the like, depending on the severity of the infection being treated.
- the compounds of the invention may be administered orally or parenterally at dosage levels of about 0.01 mg/kg to about 50 mg/kg and preferably from about 1 mg/kg to about 25 mg/kg, of subject body weight per day, one or more times a day, to obtain the desired therapeutic effect.
- an aspect of this invention is the treatment of mammals of maladies that are amenable to amelioration through blocking, inhibiting, or any other form of modulating a voltage-gated sodium channel for therapeutic effect, including, but not limited to, acute pain, chronic pain, neuropathic pain, or inflammatory pain, arthritis, migraine, cluster headaches, trigeminal neuralgia, herpetic neuralgia, general neuralgias, epilepsy or epilepsy conditions, neurodegenerative disorders, psychiatric disorders, such as, anxiety and depression, myotonia, arrhythmia, movement disorders, neuroendocrine disorders, ataxia, multiple sclerosis, irritable bowel syndrome, incontinence, visceral pain, osteoarthritis pain, postherpetic neuralgia, diabetic neuropathy, radicular pain, sciatica, back pain, head or neck pain, severe or intractable pain, nociceptive pain, breakthrough pain, postsurgical
- this invention provides a method of treating a disease or disorder, including those disorders and conditions listed above, in a patient in need of such treating, wherein the method comprises administering to said patient an amount effective to treat said disease or disorder of a compound of this invention, or a pharmaceutically acceptable salt, solvate or prodrug thereof, or a pharmaceutically acceptable salt or solvate of said prodrug.
- the present invention is further directed to a method for that manufacture of a medicament for treating a disease or disorder, including those disorders and conditions listed above, in humans and animals comprising a compound of the present invention either alone or in combination with one or more additional therapeutic agents, carriers, or diluents.
- the present invention is also directed to compounds of the invention for use in the treatment of a disease or disorder associated with the dysfunction of a voltage-gated sodium channel, including those disorders and conditions listed above, in humans and animals comprising a compound of the present invention either alone or in combination with one or more additional therapeutic agents, carriers, or diluents.
- the compounds of the invention are useful as blockers of voltage-gated sodium channels.
- the compounds and pharmaceutical compositions of the invention are blockers of one or more of NaV 1.1, NaV 1.2, NaV 1.3, NaV 1.4, NaV 1.5, NaV 1.6, NaVl .7, NaV 1.8, or NaVl .9, and thus, without wishing to be bound by any particular theory, the compounds and pharmaceutical compositions are particularly useful for treating or lessening the severity of a disease, condition, or disorder where activation or hyperactivity of one or more of NaVl.1, NaVl.2, NaVl .3, NaV 1.4, NaVLS 5 NaVl .6, NaV 1.7, NaV 1.8, or NaV 1.9 is implicated in the disease, condition, or disorder.
- this invention provides a method for treating or lessening the severity of a disease, condition, or disorder where activation or hyperactivity of one or more of NaV 1.1, NaV 1.2, NaVU, NaVl .4, NaVl.5, NaVI .6, NaVl.7, NaV1.8 , or is implicated in the disease state.
- NaV 1.3, NaVl .4, NaV 1.5, NaV 1.6, NaV 1.7, NaV 1.8, or NaV 1.9 may be assayed according to methods described generally in the Examples herein, or according to methods available to one of ordinary skill in the ail.
- compounds of the invention are useful as blockers of HaV 1.7.
- the compounds of the invention may be prepared according to the following reaction schemes, in which variables are as defined before or are derived, using readily available starting materials, from reagents and conventional synthetic procedures. It is also possible to use variants which are themselves known to those of ordinary skill in organic synthesis art, but are not mentioned in greater detail.
- NMR spectra were acquired on the following instrument: 300 MHZ NMR (Bruker) using CD 3 OD, CDCl 3 or DMSOd 6 as the solvent.
- LC-MS data were obtained using a PESciex API 150EX quadropole mass spectrometer using eleclrospray ionization or a Waters Acquity UPLC system with a Waters SQ Detector BEH Cl 8 1.7 urn 2.1 x 50 mm.
- Purification via reverse phase chromatography (Varian) was accomplished using a C 18 reverse phase column with a gradient of (0.1 % TFA) 5:95 to 90:10 acetonitrile: water, at a flow rate of 25 mL/min. Samples were collected using UV detection.
- 2-(lH-pyrrol-l-yl)aniline and 5-melhoxy-2-(lH-pyrrol-l-yl)aniline were commercially available from Aldrich and ABCR, respectively, and were used as received.
- Suzuki Reaction Procedure A 5 mL microwave vial was charged with chloride 4-150 (0.200 g, 0.456 mmol), phenylboronic acid (0.067 g, 0.548 mmol), Pd 2 (dba) 3 (0.042 g 5 0.0456 mmol), X-Phos (0.044 g, 0.0912 mmol) and potassium acetate (0.090 g, 0.912 mmol) and sealed. Separately, 10 mL of 1,4-dioxane was dried over molecular sieves and was sparged with argon gas for 30 minutes with stirring.
- Suzuki Reaction Procedure A 5 mL microwave vial was charged with bromide 4-171 (0.05 g, 0.104 mmol), phenylboronic acid (0.019 g, 0.156 mmol), Pd(dppf)Cl 2 (0.042 g, 0.0456 mmol) and sodium carbonate (0.024 g, 0.228 mmol) and sealed. Separately, 10 mL of dimethoxyethane and 5 mL of water was sparged with argon gas for 30 minutes with stirring. This dried solvent (3 mL) was added to the reaction mixture under argon, and then the sealed tube was heated to 100°C in an oil bath for 24 hours. The reaction was filtered through Celite to remove the catalyst and concentrated under reduced pressure.
- Suzuki reaction procedure To a sealed Biotage microwave vial was added DMF (3 niL) and 2 drops Of H 2 O and the system was sparged with argon for 10 minutes. A second vial was charged with 4-64 (0.08 g, 0.177 mmol), 5-(4,4,5 ⁇ 5-tetramethyl-l,3,2-dioxaboroIan-2- yl)mcotinonitrile (0.049 g, 0.212 mmol), Cs 2 CO 3 (0.1 ISg 5 0.354 mmol), and Pd(PPh 3 ) 4 (0.03Og, 0.027 mmol).
- a Biotage microwave vial with magnetic stir bar was charged with Pd(OAc) 2 (0.016 g, 5 mol%), RuPhos (0.017 g, 10 mol%), 4-64 (0.056 g, 0.123 mmol), 2 (0.027 g, 0J36mmol), and Na 2 CO 3 (0.026 g, 0.247 mmol).
- the tube was sealed with a cap lined with a Teflon septum, evacuated, and purged with N 2 (x3).
- Ethanol 1.5 mL was added via syringe, and the reaction was heated to 85 °C for 12 hours. The reaction was allowed to cool to it and filtered through a thin pad of silica gel and filter agent.
- Trifluoroacetic anhydride (0.050 mL, 0.36 mmol) was added to a solution of 4-5 (50 mg, 0.12 mmol) in benzene (4 mL) at room temperature. The reaction was stirred at room temperature for 7 hours, heated to 80 0 C for 30 minutes, and then stirred at room temperature overnight. The reaction was partitioned between methylene chloride and saturated aqueous sodium bicarbonate. The organic layer was dried with sodium sulfate and purified by silica gel chromatography to afford 7-3 (23 mg, 38%) as a bright yellow solid. MS (ESI): 514 (M + H) +
- Compound 6 can be prepared according to the procedures for the preparation of Example 1, by hydrogenation of 4-171 with 2 H 2 in the presence of Pd/C at 1 atm.
- the pore-forming subunits of human voltage gated sodium channels were stably expressed in either HEK 293 cells of CHO-Kl cells.
- Cells were maintained in standard growth media containing 10% heat-inactivated fetal bovine serum and a selection antibiotic. The cells were grown at 37°C in a humidified tissue culture incubator with the carbon dioxide concentration regulated at 5%.
- a FLIPR assay using a membrane potential dye (Molecular Devices Corporation, blue no wash voltage-sensitive dye) was used to screen for sodium channel activity of compounds.
- Cells were plated onto black- walled 384 well plates with poly-lysine-coated glass bottoms 24 hours prior to evaluation on the FLIPR.
- the growth medium was removed and replaced with a physiological saline solution containing voltage-sensitive dye and compounds of interest. After dye loading for 60 minutes, cell depolarization was evoked on the FLIPR by the addition of veratridine. Maximum veratridine-induced increase in fluorescence intensity from baseline was used to plot concentration-effect curves. Non-linear regression analysis was used to generate IC 50 values.
- Standard ruptured whole cell patch clamp techniques were used to measure sodium channel blocking activity. All studies were conducted at room temperature using a flowing extracellular solution containing (niM concentrations): 129 NaCl 5 20 letraethylammonium chloride, 3.25 KCl, 2 CaCl 2 , 2 MgCl 2 , 10 glucose, 10 HEPES-NaOH (pH 7.35).
- the glass whole cell patch electrodes for these studies had tip resistances of approximately 1.5 M ⁇ when filled with the following intracellular solution (mM concentrations): 120 CsF, 10 NaCl 5 10 tetraethylammonium chloride, 11 EGTA, 1 CaCl 2 , 1 MgCl 2 , 10 HEPES-CsOH (pH 7.3).
- FLIPR activity for the compounds of the invention fell into one of six ranges: InM to 200 nM (A); 200 nM to 1000 nM (B); 1000 nM to 5000 nM (C); 5000 to 10000 11M (D); 10000 to 15000 (E); 15000 to 30000 (F); and 30000 or greater (G).
- Compounds of present invention made according to the schemes and examples above showed activity in the foregoing assays.
- the activity of representative compounds of the invention is shown in Table 11. Table 1 1
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Pyrrolo-benzo-1,4-diazine compounds represented by Formula I or II, or pharmaceutically acceptable salts thereof, are blockers of voltage-gated sodium channels. Pharmaceutical compositions comprise an effective amount of the instant compounds, either alone, or in combination with one or more other therapeutically active compounds, and a pharmaceutically acceptable carrier. Methods of treating conditions associated with, or caused by, voltage-gated sodium channel activity, including, for example, acute pain, chronic pain, visceral pain, inflammatory pain, neuropathic pain, epilepsy or epilepsy conditions, irritable bowel syndrome, depression, anxiety, bipolar disorder, neurodegenerative disorders, psychiatric disorders, myotonia, arrhythmia, movement disorders, neuroendocrine disorders, ataxia, multiple sclerosis, irritable bowel syndrome, incontinence, neuropathy and tinnitus, comprise administering an effective amount of the present compounds, either alone, or in combination with one or more other therapeutically active compounds.
Description
TITLE OF THE INVENTION
PYRROLO-BENZO- 1,4-DIAZINES USEFUL AS SODIUM CHANNEL BLOCKERS
FIELD OF THE INVENTION The present invention relates to compounds useful as inhibitors of ion channels. The invention also provides pharmaceutically acceptable compositions comprising the compounds of the invention and methods of using the compositions in the treatment of various disorders.
BACKGROUND OF THE INVENTION Voltage-gated ion channels allow electrically excitable cells to generate and propagate action potentials and therefore are crucial for nerve and muscle function. Sodium channels play a special role by mediating rapid depolarization, which constitutes the rising phase of the action potential and in turn activates voltage-gated calcium and potassium channels. Voltage-gated sodium channels represent a multigene family. Nine sodium channel subtypes have been cloned and functionally expressed to date. [Clare, J. J.s et at, Drug Discovery Today, 2000, 5: 506-520]. They are differentially expressed throughout muscle and nerve tissues and show distinct biophysical properties. All voltage-gated sodium channels are characterized by a high degree of selectivity for sodium over other ions and by their voltage-dependent gating. [Catterall, W. A., Current Opinion in Neurobiology, 1991, 1: 5-13]. At negative or hyperpolarized membrane potentials, sodium channels are closed. Following membrane depolarization, sodium channels open rapidly and then inactivate. Sodium channels only conduct currents in the open state and, once inactivated, have to return to the resting state, favored by membrane hyperpolarization, before they can reopen. Different sodium channel subtypes vary in the voltage range over which they activate and inactivate as well as in their activation and inactivation kinetics. Sodium channels are the target of a diverse array of pharmacological agents, including neurotoxins, antiarrhythmics, anticonvulsants and local anesthetics. [Clare, J. J., et al., supra]. Several regions in the sodium channel secondary structure are involved in interactions with these blockers and most are highly conserved. Indeed, most sodium channel blockers known to date interact with similar potency with all channel subtypes. Nevertheless, it has been possible to produce sodium channel blockers with therapeutic selectivity and a sufficient therapeutic window for the treatment of epilepsy (e.g. lamotrigine, phenytoin and carbamazepine) and certain cardiac arrhythmias (e.g. lignocaine, tocainide and mexiletine).
It is well known that the voltage-gated Na+ channels in nerves play a critical role in neuropathic pain. Injuries of the peripheral nervous system often result in. neuropathic pain persisting long after the initial injury resolves. Examples of neuropathic pain include, but are not limited to, postherpetic neuralgia, trigeminal neuralgia, diabetic neuropathy, chronic lower back pain, phantom limb pain, pain resulting from cancer and chemotherapy, chronic pelvic pain, complex regional pain syndrome and related neuralgias. It has been shown in human patients as well as in animal models of neuropathic pain, that damage to primary afferent sensory neurons can lead to neuroma formation and spontaneous activity, as well as evoked activity in response to normally innocuous stimuli. [Carter, G.T. and Galer, B. S., Physical Medicine and Rehabilitation Clinics of North America, 2001, 12(2): 447-459]. The ectopic activity of normally silent sensory neurons is thought to contribute to the generation and maintenance of neuropathic pain. Neuropathic pain is generally assumed to be associated with an increase in sodium channel activity in the injured nerve. [Baker, M.D. and Wood, J.N., TRENDS in Pharmacological Sciences. 2001, 22(1); 27-31]. Indeed, in rat models of peripheral nerve injury, ectopic activity in the injured nerve corresponds to the behavioral signs of pain. In these models, intravenous application of the sodium channel blocker and local anesthetic lidocaine can suppress the ectopic activity and reverse the tactile allodynia at concentrations that do not affect general behavior and motor function. [Mao, J. and Chen, L.L., Pain, 2000, 87: 7-17]. These effective concentrations were similar to concentrations shown to be clinically efficacious in humans. [Tanelian, D.L. and
Brose, W.G., Anesthesiology, 1991, 74(5): 949-951]. In a placebo-controlled study, continuous infusion of lidocaine caused reduced pain scores in patients with peripheral nerve injury, and in a separate study, intravenous lidocaine reduced pain intensity associated with postherpetic neuralgia (PHN). [Mao, J. and Chen, L.L., supra; Anger, T., et a!., J. Med. Chem.. 2001, 44(2): 115-137]. Lidoderm®, lidocaine applied in the form of a dermal patch, is currently the only FDA approved treatment for PHN. [Devers, A. and Galer, B.S., Clinical J. Pain, 2000. 16(3): 205- 208].
In addition to neuropathic pain, sodium channel blockers have clinical uses in the treatment of epilepsy and cardiac arrhythmias. Recent evidence from animal models suggests that sodium channel blockers may also be useful for neuroprotection under ischaemic conditions caused by stroke or neural trauma and in patients with multiple sclerosis (MS). [Clare, J. J., et al. and Anger, T., et al., supra].
International Patent Publication WO 00/57877; International Patent Publication WO 01/68612; International Patent Publication WO 99/32462; International Patent Publication WO 03/095427; US Patent 3,939,159; Artico, et al., J. Heterocyclic Chem, 1992, 29:241-245.
However, there remains a need for novel compounds and compositions that therapeutically block neuronal sodium channels with less side effects and higher potency than currently known compounds.
SUMMARY OF THE INVENTION
The present invention is directed to pyrrolo-benzo-l,4~diazine compounds which are sodium channel blockers useful for the treatment of chronic and neuropathic pain. The compounds of the present invention are also useful for the treatment of other conditions, including disorders of the CNS such as epilepsy, manic depression and bipolar disorder. This invention also provides pharmaceutical compositions comprising a compound of the present invention, either alone, or in combination with one or more therapeutically active compounds, and a pharmaceutically acceptable carrier.
This invention further comprises methods for the treatment of acute pain, chronic pain, visceral pain, inflammatory pain, neuropathic pain and disorders of the CNS including, but not limited to, epilepsy, manic depression, depression, anxiety and bipolar disorder comprising administering the compounds and pharmaceutical compositions of the present invention.
DETAILED DESCRIPTION OF THE INVENTION
The present invention is directed to sodium channel blockers and derivatives of structural formula I

or pharmaceutically acceptable salts and individual enantiomers and diastereomers thereof, wherein:
W is pyrazolyl, phenyl or pyridinyl, each optionally substituted with.0 to 3 groups of Ra; Rx and Ry are independently selected from the group consisting of
(1) hydrogen,
(2) -C(O)R1, (3) -(CH2)nC5-Cl0 heterocyclyl, and
(4) -Ci-Cio alkyl, wherein said, heterocyclyl and Ci-Cio alkyl may be optionally substituted with one to four groups of Ra, orRx and RY together with the carbon atom to which they are attached can form a 5 to 8 mernbered carbocyclic ring of which one of the ring atoms is optionally nitrogen, said ring substituted with Z — Rl and optionally substituted with 1 to 3 groups of R , '
Z is selected from the group consisting of (1) a bond, (2) -Ci-C6 alkylene,
(3) -C(O)-, (4) -C(O)O-, and
(5) -SO2-, wherein when Z is a bond or alkylene, Rl cannot be hydrogen; Rl is selected from the group consisting of
(1) hydrogen, (2) -{CHR2)nC6-Cio aryl,
(3) "(CH2)UCs-CiO hCtCrOCyCIyI,
(4) -(CH2)nC3-C6 cycloaklyl, and
 (6) -Ci-C6 alkyl, and (7) -CH(NHBoc)(CH2)nC6-Ci o aryl wherein said heteroaryl is not thienyl and may be optionally substituted with 1 to 4 groups of Ra, and said aryl and cycloaklyl are substituted with 1 to 4 groups of Ra; R2 is selected from the group consisting of
(6) -Ci-C6 alkyl, and (7) -CH(NHBoc)(CH2)nC6-Ci o aryl wherein said heteroaryl is not thienyl and may be optionally substituted with 1 to 4 groups of Ra, and said aryl and cycloaklyl are substituted with 1 to 4 groups of Ra; R2 is selected from the group consisting of
(1) hydrogen, (2) -Ci-C6 alkyl, and
(3) -(CH2)nC6-Cio aryl> which may be optionally substituted with 1 to 4 groups of Ra;
R3 and R.4 are independently selected from the group consisting of (1) hydrogen, (2)-Ci-C6alkyl, (3) -(CH2)nC6~Cio aryl, which may be optionally substituted with 1 to 4 groups of Ra,
(4) -F, and
(5) -(CH2)nCF3
Ra is selected from the group consisting of
(1) hydrogen, (2)hydroxyl,
(3) halogen, (4) -Ci-Cio alkyl,
(5) -C2-C6 alkenyl, (6)-(CH2)nC6-Cioaryl, (7) ~(CH2)nC5-Cl0 heterocyclyl,
(8) -O- (9)-OR3, (10) -O(CH2)nOR4 (ll)-(CH2)nN(R3)2, (12) -NR3C(O)R4,
(13) -C(O)5
(14)-C(O)R3,
(15) -(CH2)nCN,
(16)-Ci-C3haloalkyl, (17)-NO2,
(18)-NR3C(O)OR4,
(19)-C(O)OR4,

(21)-O(CH2)nN(R3),and (22) ™(CH2)nNHC(O)OR3,
wherein said aryl and heteroaryl may be optionally substituted with 1 to 4 groups of Rb, which is selected from the group consisting of
(1) -Ci-C6 alkyl, (2) halogen,
(3) -CN,
(4) -NH2, and .
(5) -OR3, j is 1 to 3; n is O to 4; p is 0 to 3; q is 1 to 3; and wherein the compounds of said formula I do not include the compounds of Table 1. Table 1





One embodiment of the present invention is realized when Rx and RY form a 5 to 8 membered carbocylic ring substituted with Z — Rl .
Another embodiment of the present invention is realized when Rx and Rv form a 5 to 8 membered carbocylic ring of which one of the ring atoms is ntrogen. A sub-embodiment of this invention is realized when Z — Rl is attached to the ntrogen atom.
Another embodiment of the present invention is realized when Z is C(O). Another embodiment of the present invention is realized when Z is SO2-
Another embodiment of the present invention is realized when Rl is (CH2)nC6~ClO sryh where said aryl is selected from the group consisting of phenyl and napthyl.
Another embodiment of the present invention is realized when J is 1, which makes a six member nitrogen containing ring.
Another embodiment of the present invention is realized when Rl is optionally substituted heteroaryl, where said heteroaryl is selected from the group consisting of benzylfuranyl, qumolinyl, indolyl, benzodioxolyl, benzothiazolyl, benzoxazmyl, benoxazolyl, and benzoxazine.
Another embodiment of the present invention is realized by compounds of structural formula II:

II and pharmaceutically aceptable salts and individual enantiomers and diasteromers thereof, wherein, Rl is selected from the group consisting of
(1) -Ce-Cio aryl, and
(2) ~{CH2)nC6-ClO heteroaiyl, wherein said heteroaryl is not thienyl and may be optionally substituted with 1 to 4 groups of Ra, and said aryl is substituted with 1 to 4 groups of Ra. In another embodiment of formula I or II, Ra is selected from the group consisting of
(1) hydrogen,
(2) halogen,
(3) -Ci-Cβ alkyl,
(4) -(CH2)nC6-C 10 heteroaryl, (5) -OR3,
(6) -N(R3)2,
(7) -CN, and
(8) -CF3.
Specific embodiments of the compounds of the invention and methods of making them as described in the schemes and examples that follow include the compounds in Table 2. FLIPR activity for the compounds of the invention fell into one of six ranges: InM to 200 nM (A); 200 nM to 1000 nM (B); 1000 nM to 5000 nM (C); 5000 to 10000 nM (D); 10000 to 15000 (E); 15000 to 30000 (F); and 30000 or greater (G). Table 2






Specific embodiments of the present invention include a compound of the Examples or made according to the methods therein and pharmaceutically acceptable salts thereof and individual enantiomers and diastereomers thereof. When any variable (e.g. aryl, heterocycle, Ra etc.) occurs more than one time in any constituent, its definition on each occurrence is independent at every other occurrence. Also, combinations of substituents or variables are permissible only if such combinations result in stable compounds.
When Ra is -O- and attached to a carbon it is referred to as a carbonyl group and when it is attached to a nitrogen (e.g., nitrogen atom on a pyridyl group) or sulfur atom it is referred to a N-oxide and sulfoxide group, respectively.
As used herein, "alkyl" encompasses groups having the prefix "alk" such as, for example, alkylenyl, alkoxy, alkanoyl, alkenyl, and alkynyl and means carbon chains which may be linear or branched or combinations thereof. The term "Cl-C6" includes alkyls containing 6, 5, 4, 3, 2, or 1 carbon atoms. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tejrt-butyl, pentyl, hexyl, and heptyl. "Alkylenyl" refers to an alkyl have substitutions at both ends. "Alkoxy" refers to an alkyl group connected to the oxy connecting atom and also includes alkyl ether groups, where the term 'alkyl' is defined above, and 'ether' means two alkyl groups with an oxygen atom between them. Examples of alkoxy groups include methoxy, ethoxy, n- propoxy, i-propoxy, n-butoxy, s-butoxy, t-butoxy> methoxymethane (also referred to as 'dimethyl ether'), and methoxyethane (also referred to as 'ethyl methyl ether'). "Alkenyl" refers to a
hydrocarbon radical straight, branched or cyclic containing from 2 to 10 carbon atoms and at least one carbon to carbon double bond. Exemplary alkenyl groups include ethenyl, propenyl, butenyl and cyclohexenyl.
As used herein, "fluoroalkyl" refers to an alkyl substituent as described herein containing at least one fluorine substituent.
The term "cycloalkyl" refers to a saturated hydrocarbon containing one ring having a specified number of carbon atoms. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. The term C3-C10 cycloalkyl refers to cycloalkyl having 3, 4, 5, 6, 7,8, 9, or 10 carbon atoms. As used herein, "aryl" is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 members in each ring, wherein at least one ring is aromatic. Examples of such aryl elements include phenyl, napthyl, tetrahydronapthyl, indanyl, or biphenyl.
The term "heterocycle," "heterocyclyl," or "heterocyclic," refers to a stable 5- to 7- membered monocyclic or stable 8- to 11-membered bicyclic heterocyclic ring which is either saturated or unsaturated, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O, and S, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring. The heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure. The term heterocycle or heterocyclic includes heteroaryl moieties. Examples of such heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydrobenzothienyl, dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, 1,3-dioxolanyl, furyl, imidazolidinyl, imidazolinyl, imidazolyl, indolinyl, indolyl, isochromanyl, isomdolinyl, isoquinolinyl, isothiazolidinyl, isothiazolyl, isothiazolidinyl, morpholinyl, naphthyridinyl, oxadiazolyl, 2-oxoazepinyl, oxazolyl, 2-oxopiperazinyl, 2-oxopiperdinyl, 2-oxopyrrolidinyl, piperidyl, piperazinyl, pyridyl, pyrazinyl, pyrazolidinyl, pyrazolyl, pyridazinyl, pyrimidinyl, pyrrolidinyl, pyrrolyl, quinazolinyl, quinolinyl, quinoxalinyl, tetrahydrofuryl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, thiamoφholinyl, thiamorpholinyl sulfoxide, thiazolyl, thiazolinyl, thienofuryl, thienothienyl, triazolyl and thienyl.
The term "heteroaryl", as used herein except where noted, represents a stable 5- to 7- membered monocyclic- or stable 9- to 10-membered fused bicyclic heterocyclic ring system which contains an aromatic ring, any ring of which may be saturated, such as piperidinyl,
partially saturated, or unsaturated, such as pyridinyl, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O and S, and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quaternized, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring. The heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure. Examples of such heteroaryl groups include, but are not limited to, benzimidazole, benzisothiazole, benzisoxazole, benzofuran, benzothiazole, benzothiophene, benzotriazole, benzoxazole, carboline, cinnoline, furan, furazan, imidazole, indazole, indole, indolizine, isoquinoline, isothiazole, isoxazole, naphthyridine, oxadiazole, oxazole, phthalazine, pteridine, purine, pyran, pyrazine, pyrazole, pyridazine, pyridine, pyriniidine, pyrrole, qumazoline, quinoline, quinoxaline, tetrazole, thiadiazole, thiazole, thiophene, triazine, triazole, and N~oxides thereof.
Examples of heterocycloalkyls include azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, tetrahydrofuranyl, imidazolinyl, pyrolidin-2~one, piperidin-2-one, and thiomorpholinyl.
The term "heteroatom" means O, S or N, selected on an independent basis.
A group which is designated as being substituted with substituents, may be substituted with multiple numbers of such substituents, which may be the same or different, for example, with one to four groups of Ra. A group which is designated as being independently substituted with substituents may be independently substituted with multiple numbers of such substituents.
The compounds of the invention may have one or more asymmetric centers. Compounds with asymmetric centers give rise to enantiomers (optical isomers), diastereomers (configurational isomers) or both, and it is intended that all of the possible enantiomers and diastereomers in mixtures and as pure or partially purified compounds are included within the scope of this invention. The present invention is meant to encompass all such isomeric forms of the compounds of formulae (I) and (II).
Formulae (I) and (II) are shown above without a definite stereochemistry. The present invention includes all stereoisomers of formulae (I) and (II), and pharmaceutically acceptable salts thereof. The independent syntheses of the enantiomerically or diastereomerically enriched compounds, or their chromatographic separations, may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration and by appropriate modification of the methodology disclosed herein as known in the art. Their absolute
stereochemistry may be determined by the x-ray crystallography of crystalline products or crystalline intermediates that are derivatized, if necessary, with a reagent containing an asymmetric center of known absolute configuration.
If desired, racemic mixtures of the compounds may be separated so that the individual enantiomers or diastereomers are isolated. The separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography. The coupling reaction is often the formation of salts using an enantiomerically pure acid or base. The diastereomeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue. The racemic mixture of the compounds can also be separated directly by chromatographic methods using chiral stationary phases, which methods are well known in the art.
During any of the above synthetic sequences it may be necessary or desirable to protect sensitive or reactive groups on any of the molecules concerned. This may be achieved by means of conventional protecting groups, such as those described in Protective Groups in Organic Chemistry, ed. J .F. W. McOmie, Plenum Press, 1973, and T. W. Greene & P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1999. The protecting groups may be removed at a convenient sequent stage using methods known from the art, hi the compounds of formulas (I )- (IV), the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of generic formulas (I) - (IV). For example, different isotopic forms of hydrogen (H) include protium (lH) and deuterium (^H). Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples. Isotopically-enriched compounds within generic formulas (I) - (IV) can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
The term "substantially pure" means that the isolated material is at least 90% pure, and preferably 95% pure, and even more preferably 99% pure as assayed by analytical techniques known in the art.
The term "substituted" means that one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
By "stable compound' or "stable structure" is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
The term "optionally substituted" means optional substitution with the specified groups, radicals or moieties.
The term "purified", "in purified form" or "in isolated and purified form" for a compound refers to the physical state of said compound after being isolated from a synthetic process (e.g. from a reaction mixture), or natural source or combination thereof. Thus, the term "purified", "in purified form" or "in isolated and purified form" for a compound refers to the physical state of said compound after being obtained from a purification process or processes described herein or well known to the skilled artisan (e.g., chromatography, recrystallization and the like), in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan.
The compounds of the present invention may be administered in the form of a pharmaceutically acceptable salt. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. The compounds of the invention may be mono, di or tris salts, depending on the number of acid functionalities present in the free base form of the compound. Free bases and salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates. Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine,
betaine, caffeine, choline, N,N'-dibenzylethylene-diamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
When the compound of the present invention is basic, its corresponding salt may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids. Such acids include acetic, trifluoroacetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, para-toluenesulfonic acid, and the like. It will be understood that, as used herein, references to the compounds of the present invention are meant to also include the pharmaceutically acceptable salts.
The term "composition" as used herein is intended to encompass a product comprising specified ingredients in predetermined amounts or proportions, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts. This term in relation to pharmaceutical compositions is intended to encompass a product comprising one or more active ingredients, and an optional carrier comprising inert ingredients, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. In general, pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation. In the pharmaceutical composition the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
Accordingly, the term "pharmaceutical composition" of the present invention encompasses any composition made by combining a compound of the present invention and a pharmaceutically acceptable agent, including another active agent, carrier or diluent. By "pharmaceutically acceptable" it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
The pharmaceutical compositions of the present invention comprise a compound of the invention (or pharmaceutically acceptable salts thereof) as an active ingredient, a
pharmaceutically acceptable carrier, and optionally one or more additional therapeutic agents or adjuvants. Such additional therapeutic agents can include, for example, i) opiate agonists or antagonists, ii) calcium channel antagonists, iii) 5HT receptor agonists or antagonists, iv) sodium channel antagonists, v) NMDA receptor agonists or antagonists, vi) COX-2 selective inhibitors, vϋ) NKl antagonists, viii) non-steroidal anti-inflammatory drugs ("NSAID"), ix) selective serotonin reuptake inhibitors ("SSRI") and/or selective serotonin and norepinephrine reuptake inhibitors ("SSNRI"), x) tricyclic antidepressant drugs, xi) norepinephrine modulators, xii) lithium, xiϋ) valproate, and xiv) neurontin (gabapentin). The instant compositions include compositions suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration, although the most suitable route in any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered. The pharmaceutical compositions may be conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy. The term "pharmaceutical composition" is also intended to encompass both the bulk composition and individual dosage units comprised of more than one (e.g., two) pharmaceutically active agents such as, for example, a compound of the present invention and an additional agent selected from the lists of the additional agents described herein, along with any pharmaceutically inactive excipients. The bulk composition and each individual dosage unit can contain fixed amounts of the afore-said "more than one pharmaceutically active agents". The bulk composition is material that has not yet been formed into individual dosage units. An illustrative dosage unit is an oral dosage unit such as tablets, pills and the like. Similarly, the herein-described method of treating a patient by administering a pharmaceutical composition of the present invention is also intended to encompass the administration of the afore-said bulk composition and individual dosage units,
It will also be appreciated that certain of the compounds of present invention can exist in free form for treatment, or where appropriate, as a pharmaceutically acceptable derivative thereof. According to the present invention, a pharmaceutically acceptable derivative includes, but is not limited to, pharmaceutically acceptable salts, esters, salts of such esters, or any other adduct or derivative which upon administration to a patient in need is capable of providing, directly or indirectly, a compound as otherwise described herein, or a metabolite or residue thereof.
In general, pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation. In the pharmaceutical composition the active compound, which is a compound of formulae (I) to (IV)5 is included in an amount sufficient to produce the desired effect upon the process or condition of diseases. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
The carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous). Thus, the pharmaceutical compositions of the present invention can be presented as discrete units suitable for oral administration such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient. Further, the compositions can be presented as a powder, as granules, as a solution, as a suspension in an aqueous liquid, as a non-aqueous liquid, as an oil- in- water emulsion or as a water-in-oil liquid emulsion. In addition to the common dosage forms set out above, the compounds of the invention, or pharmaceutically acceptable salts thereof, may also be administered by controlled release means and/or delivery devices.
Pharmaceutical compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets may contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets. These excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc. The tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
A tablet containing the composition of this invention may be prepared by compression or molding, optionally with one or more accessory ingredients or adjuvants. Compressed tablets may be prepared by compressing, in a suitable machine, the active ingredient in a free-flowing
form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent. Each tablet preferably contains from about 0.1 mg to about 500 mg of the active ingredient and each cachet or capsule preferably containing from about 0.1 mg to about 500 mg of the active ingredient. Compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil. Other pharmaceutical compositions include aqueous suspensions, which contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions. In addition, oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example aracbis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. Oily suspensions may also contain various excipients. The pharmaceutical compositions of the invention may also be in the form of oil-in- water emulsions, which may also contain excipients such as sweetening and flavoring agents.
The pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleaginous suspension, or in the form of sterile powders for the extemporaneous preparation of such sterile injectable solutions or dispersions. In all cases, the final injectable form must be sterile and must be effectively fluid for easy syringability. The pharmaceutical compositions must be stable under the conditions of manufacture and storage; thus, preferably should be preserved against the contaminating action of microorganisms such as bacteria and fungi.
Pharmaceutical compositions of the present invention can be in a form suitable for topical use such as, for example, an aerosol, cream, ointment, lotion, dusting powder, or the like. Further, the compositions can be in a form suitable for use in transdermal devices. These formulations may be prepared via conventional processing methods. As an example, a cream or ointment is prepared by mixing hydrophilic material and water, together with about 5 wt% to about 10 wt% of the compound, to produce a cream or ointment having a desired consistency. Pharmaceutical compositions of this invention can also be in a form suitable for rectal administration wherein the carrier is a solid. It is preferable that the mixture forms unit dose suppositories. Suitable carriers include cocoa butter and other materials commonly used in the art.
It will also be appreciated that the compounds and pharmaceutically acceptable compositions of the present invention can be employed in combination therapies, that is, the compounds and pharmaceutically acceptable compositions can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutics or medical procedures. The particular combination of therapies (therapeutics or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutics and/or procedures and the desired therapeutic effect to be achieved. It will also be appreciated that the therapies employed may achieve a desired effect for the same disorder (for example, an inventive compound may be administered concurrently with another agent used to treat the same disorder), or they may achieve different effects (e.g., control of any adverse effects). As used herein, additional therapeutic agents which are normally administered to treat or prevent a particular disease, or condition, are known as "appropriate for the disease, or condition, being treated". For example, exemplary additional therapeutic agents include, but are not limited to: nonopioid analgesics (indoles such as Etodolac, Indomethacin, Sulindac, Tolmetin; naphthylalkanon.es such as Nabumetone; oxicams such as Piroxicam; para-aminophenol derivatives, such as
Acetaminophen; propionic acids such as Fenoprofen, Flurbiprofen, Ibuprofen, Ketoprofen, Naproxen, Naproxen sodium, Oxaprozin; salicylates such as Aspirin, Choline magnesium trisalicylate, Diflunisal; fenamates such as meclofenamic acid, Mefenamic acid; and pyrazoles such as Phenylbutazone); or opioid (narcotic) agonists (such as Codeine, Fentanyl, Hydromorphone, Levorphanol, Meperidine, Methadone, Morphine, Oxycodone, Oxymorphone, Propoxyphene, Buprenorphine, Butorphanol, Dezocine, Nalbuphine, and Pentazocine). Additionally, nondrug analgesic approaches may be utilized in conjunction with administration of one or more compounds of the invention. For example, anesthesiologic (intraspinal infusion, neural blocade), neurosurgical (neurolysis of CNS pathways), neurostimulatory (transcutaneous electrical nerve stimulation, dorsal column stimulation), physiatric (physical therapy, orthotic devices, diathermy), or psychologic (cognitive methods-hypnosis, biofeedback, or behavioral methods) approaches may also be utilized. Additional appropriate therapeutic agents or approaches are described generally in The Merck Manual, Seventeenth Edition, Ed. Mark H. Beers and Robert Berkow, Merck Research Laboratories, 1999, and the Food and Drug Administration website, www.fda.gov, the entire contents of which are hereby incorporated by reference.
The amount of additional therapeutic agent present in the compositions of this invention will be no more than the amount that would normally be administered in a composition
comprising that therapeutic agent as the only active agent. Preferably, the amount of additional therapeutic agent in the presently disclosed compositions will range from about 50% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent. Accordingly, the present invention additionally provides any one of the aforementioned methods further comprising administering to said patient an amount effective to treat said disease or disorder of at least one additional therapeutic agent, wherein the additional therapeutic agent(s) is/are selected from the group consisting of therapeutic agents known to be useful to treat said disease or disorder. In one embodiment of this combination therapy method, the additional therapeutic agent(s) is/are selected from the group consisting of opiate agonists, opiate antagonists, calcium channel antagonists, 5HT receptor agonists, 5HT receptor antagonists, sodium channel antagonists, NMDA receptor agonists, NMDA receptor antagonists, COX-2 selective inhibitors, NKl antagonists, non-steroidal anti-inflammatory drugs, selective serotonin reuptake inhibitors, selective serotonin and norepinephrine reuptake inhibitors, tricyclic antidepressants, norephinephrine modulators, lithium, valproate, neurontin and pregabalin.
The terms "administration of or "administering a" compound should be understood to mean providing a compound of the invention to the individual in need of treatment in a form that can be introduced into that individual's body in a therapeutically useful form and therapeutically useful amount, including, but not limited to: oral dosage forms, such as tablets, capsules, syrups, suspensions, and the like; injectable dosage forms, such as IV, IM, or IP, and the like; transdermal dosage forms, including creams, jellies, powders, or patches; buccal dosage forms; inhalation powders, sprays, suspensions, and the like; and rectal suppositories.
The terms "effective amount" or "therapeutically effective amount" means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
As used herein, the term "treatment" or "treating" means any administration of a compound of the present invention and includes (1) inhibiting the disease in an animal that is experiencing or displaying the pathology or symptomatology of the diseased (i.e., arresting further development of the pathology and/or symptomatology), or (2) ameliorating the disease in an animal that is experiencing or displaying the pathology or symptomatology of the diseased
(i.e., reversing the pathology and/or symptomatology).
The compositions containing compounds of the present invention may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. The term "unit dosage form" is taken to mean a single dose wherein all active and inactive ingredients are combined in a suitable system, such that the patient or person administering the drug to the patient can open a single container or package with the entire dose contained therein, and does not have to mix any components together from two or more containers or packages. Typical examples of unit dosage forms are tablets or capsules for oral administration, single dose vials for injection, or suppositories for rectal administration. This list of unit dosage forms is not intended to be limiting in any way, but merely to represent typical examples of unit dosage forms.
The compositions containing compounds of the present invention may conveniently be presented as a kit, whereby two or more components, which may be active or inactive ingredients, carriers, diluents, and the like, are provided with instructions for preparation of the actual dosage form by the patient or person administering the drug to the patient. Such kits may be provided with all necessary materials and ingredients contained therein, or they may contain instructions for using or making materials or components that must be obtained independently by the patient or person administering the drug to the patient.
When treating or ameliorating a disorder or disease for which compounds of the present invention are indicated, generally satisfactory results are obtained when the compounds of the present invention are administered at a daily dosage of from about 0.1 mg to about 100 mg per kg of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form. The total daily dosage is from about 1.0 mg to about 2000 mg, preferably from about 0.1 mg to about 20 mg per kg of body weight. In the case of a 70 kg adult human, the total daily dose will generally be from about 7 mg to about 1 ,400 mg. This dosage regimen may be adjusted to provide the optimal therapeutic response. The compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. For example, a formulation intended for the oral administration to humans may conveniently contain from about 0.005 mg to about 2.5 g of active agent, compounded with an appropriate and convenient amount of carrier material. Unit dosage forms will generally contain between from about 0.005 mg to about 1000 mg of the active ingredient,, typically 0.005,
0.01 mg, 0.05 mg, 0.25 mg, 1 mg, 5 mg, 25 rag, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg or 1000 mg, administered once, twice or three times a day.
It will be understood, however, that the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
The pharmaceutically acceptable compositions of this invention can be administered to humans and other animals orally , rectally, parenterally, intracistemally, intra vaginally, intraperitoneally, topically (as by powders, ointments, or drops), bucally, as an oral or nasal spray, or the like, depending on the severity of the infection being treated. In certain embodiments, the compounds of the invention may be administered orally or parenterally at dosage levels of about 0.01 mg/kg to about 50 mg/kg and preferably from about 1 mg/kg to about 25 mg/kg, of subject body weight per day, one or more times a day, to obtain the desired therapeutic effect.
The inventive compounds and pharmaceutical composition of the invention have been found to modulate voltage-gated sodium channels. Accordingly, an aspect of this invention is the treatment of mammals of maladies that are amenable to amelioration through blocking, inhibiting, or any other form of modulating a voltage-gated sodium channel for therapeutic effect, including, but not limited to, acute pain, chronic pain, neuropathic pain, or inflammatory pain, arthritis, migraine, cluster headaches, trigeminal neuralgia, herpetic neuralgia, general neuralgias, epilepsy or epilepsy conditions, neurodegenerative disorders, psychiatric disorders, such as, anxiety and depression, myotonia, arrhythmia, movement disorders, neuroendocrine disorders, ataxia, multiple sclerosis, irritable bowel syndrome, incontinence, visceral pain, osteoarthritis pain, postherpetic neuralgia, diabetic neuropathy, radicular pain, sciatica, back pain, head or neck pain, severe or intractable pain, nociceptive pain, breakthrough pain, postsurgical pain, tinnitus, or cancer pain by administering an effective amount of a compound of this invention, The term "mammal" includes humans, as well as other animals, such as, for example, dogs, cats, horses, pigs and cattle. Accordingly, it is understood that the treatment of mammals other than humans refers to the treatment of clinical conditions in non-human mammals that correlate to the above recited conditions.
In another aspect, this invention provides a method of treating a disease or disorder, including those disorders and conditions listed above, in a patient in need of such treating, wherein the method comprises administering to said patient an amount effective to treat said disease or disorder of a compound of this invention, or a pharmaceutically acceptable salt, solvate or prodrug thereof, or a pharmaceutically acceptable salt or solvate of said prodrug.
The present invention is further directed to a method for that manufacture of a medicament for treating a disease or disorder, including those disorders and conditions listed above, in humans and animals comprising a compound of the present invention either alone or in combination with one or more additional therapeutic agents, carriers, or diluents. The present invention is also directed to compounds of the invention for use in the treatment of a disease or disorder associated with the dysfunction of a voltage-gated sodium channel, including those disorders and conditions listed above, in humans and animals comprising a compound of the present invention either alone or in combination with one or more additional therapeutic agents, carriers, or diluents. As described generally above, the compounds of the invention are useful as blockers of voltage-gated sodium channels. In one embodiment, the compounds and pharmaceutical compositions of the invention are blockers of one or more of NaV 1.1, NaV 1.2, NaV 1.3, NaV 1.4, NaV 1.5, NaV 1.6, NaVl .7, NaV 1.8, or NaVl .9, and thus, without wishing to be bound by any particular theory, the compounds and pharmaceutical compositions are particularly useful for treating or lessening the severity of a disease, condition, or disorder where activation or hyperactivity of one or more of NaVl.1, NaVl.2, NaVl .3, NaV 1.4, NaVLS5 NaVl .6, NaV 1.7, NaV 1.8, or NaV 1.9 is implicated in the disease, condition, or disorder. When activation or hyperactivity of NaVl.1, NaVl .2, NaVU, NaVl .4, NaV1.5, NaV1.6, NaVl .7, NaVl .8, or NaVl .9 is implicated in a particular disease, condition, or disorder, the disease, condition, or disorder may also be referred to as a "NaV 1.1, NaV 1.2, NaVl .3, NaV1.4, NaVLS, NaVl.6, NaVl .7, NaV 1.8 or NaVl .9-mediated disease, condition or disorder." Accordingly, in another aspect, this invention provides a method for treating or lessening the severity of a disease, condition, or disorder where activation or hyperactivity of one or more of NaV 1.1, NaV 1.2, NaVU, NaVl .4, NaVl.5, NaVI .6, NaVl.7, NaV1.8 , or is implicated in the disease state. The activity of a compound utilized in this invention as a blocker of NaV 1.1, NaV 1.2,
NaV 1.3, NaVl .4, NaV 1.5, NaV 1.6, NaV 1.7, NaV 1.8, or NaV 1.9 may be assayed according to methods described generally in the Examples herein, or according to methods available to one of
ordinary skill in the ail. In certain exemplary embodiments, compounds of the invention are useful as blockers of HaV 1.7.
While the invention has been described and illustrated with reference to certain particular embodiments thereof, those skilled in the art will appreciate that various adaptations, changes, modifications, substitutions, deletions, or additions of procedures and protocols may be made without departing from the spirit and scope of the invention.
The compounds of the invention may be prepared according to the following reaction schemes, in which variables are as defined before or are derived, using readily available starting materials, from reagents and conventional synthetic procedures. It is also possible to use variants which are themselves known to those of ordinary skill in organic synthesis art, but are not mentioned in greater detail.
Several methods for preparing the compounds of this invention are illustrated in the schemes and examples herein. The following examples are provided so that the invention might be more fully understood.
EXAMPLES
The invention disclosed herein is exemplified by the following preparations and examples which should not be construed to limit the scope of the disclosure. Alternative mechanistic pathways and analogous structures will be apparent to those skilled in the art. The following abbreviations are used in the procedures and schemes: ACN: acetonitrile;
AcOH: acetic acid; Aq: aqueous; Boc: lert-butoxycarbonyl; BoC2O: boc anhydride; ° C: degrees Celsius; Calcd: calculated; Cbz: carbobenzloxy; CbzCl: benzyl chloroformate; CDI: carbonyldiimidazole; DCE: 1,2-dichloroethane; DCM: dichloromethane; DIPEA: diisopropyletrrylamine; DMAP: 4-dimelhyIaminopyridine; DME: dimethoxyethane; DMF: N5N- dimethylformamide; DMSO: methylsulfoxide; EDCI: l-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride; EI: electron ionization; Eq: equivalents; EtOAc: ethyl acetate; EtOH: ethanol; g: grams; h: hours; 1H: proton; HCl: hydrogen chloride; Hex: hexanes; HOAT: 1- hydroxy-7-aza-benzotriazole; HOBT: 1-hydroxybenzotriazole; HPLC: high pressure liquid chromatography; LAH: lithium aluminum hydride; LCMS: liquid chromatography mass spectroscopy; M: molar; mM: milϊimolar; mmol: millimoles; Me: methyl; MeCN: acetonitrile; MeOH: methanol; min: minutes; mg: milligrams; MHz: megahertz; mL: milliliter; MPLC: medium pressure liquid chromatography; MS: mass spectroscopy; N: normal; NaHCθ3: sodium
bicarbonate; Na2SO4: sodium sulfate; NMR: nuclear magnetic resonance; Obsd: observed; ON: overnight; Ph: phenyl; Py: pyridine; rt: room temperature; Satd: saturated; sgc: silica gel 60 chromatography; soln: solution; TEA: triethylamine; TFA: trifluoroacetic acid; THF: tetrahydrofuran; TLC: thin layer chromatography; t^: retention time; UV254 run- ultraviolet light 254 nm.
NMR spectra were acquired on the following instrument: 300 MHZ NMR (Bruker) using CD3OD, CDCl3 or DMSOd6 as the solvent. LC-MS data were obtained using a PESciex API 150EX quadropole mass spectrometer using eleclrospray ionization or a Waters Acquity UPLC system with a Waters SQ Detector BEH Cl 8 1.7 urn 2.1 x 50 mm. Purification via reverse phase chromatography (Varian) was accomplished using a C 18 reverse phase column with a gradient of (0.1 % TFA) 5:95 to 90:10 acetonitrile: water, at a flow rate of 25 mL/min. Samples were collected using UV detection.
Normal phase silica gel chromatography was either accomplished by hand-packed silica columns or on an ISCO CorabiFlash Rf system using RediSep Rf silica gel columns. A general preparatory scheme for preparing the compounds of the present invention is shown below in Scheme 1.
Scheme 1


2-(lH-pyrrol-l-yl)aniline and 5-melhoxy-2-(lH-pyrrol-l-yl)aniline were commercially available from Aldrich and ABCR, respectively, and were used as received.
The intermediates used herein were prepared according to the procedures in: Guillon, J. et al; J. Med, Chem. 47, 2004, 1997-2009. A general scheme for the preparation of the intermediates herein is given in Scheme 2 for the preparation of Ib.
Scheme 2

S-methoxy-N-methyl-2-flH-pynOl-l-yl)aniline 1-lb
A round bottom 0ask was charged with 1-1 (0.20 g, 1.08 mmol) in pyridine (3.0 mL) was cooled in an ice bath before adding drop-wise via syringe ethyl chloroformate (0.1 mL, 1.08 mmol). The reaction mixture was stirred under N2 for 4 hours then poured over crushed ice.
Acidification with 2N HCl (5 mL) gave a yellow precipitate which was filtered, rinsed with water and allowed to dry to give the carbamate, 1-la (0.28 g, 87%). MS (ESI): 261 (M + H)+
To a stirred suspension of lithium aluminum hydride (1.2 mL, 2.35 mmol) in THF at 0 0C under N2 was added 1-2 (0.24 g, 0.94 mmol) in THF. The mixture was stirred at 0 0C for 1 hour, then at room temperature for 12 hours. The solution was cooled to 0 0C and then quenched by drop- wise addition of water and a 10% solution of sodium hydroxide. After filtration, the residue was washed with ethyl acetate (2 x 20 mL). The organic solution was dried with magnesium sulfate and filtered. Evaporation of the solvent afforded the desired product 1-lb (0.17 g, 89%) in satisfying purity. MS (ESI): 203 (M H- H)+
EXAMPLE 1

1
(3.5-Dimethoxyphenyl)(7'-methoxy-5'H-spiro|"piperidine-4,4'-ρyrrolo[ 1 ,2-a.]quinoxaline]-l - ypmethanone hydrochloride 1
A. Benzyl 7'-methoxy-5'H-spirofpiperidine-4.4'-pyrroIo[ 1.2-a]quinoxaline]- 1 -carboxylate 1-3

A 50-mL, 1 -necked round-bottomed flask was charged with aniline 1-1 (1.006 g, 5.345 mmol), piperidone 1-2 (1.124 g, 4.818 mmol), and maleic acid (0.064 g, 0.55 mmol). The flask was fitted with a water condenser and the apparatus was evacuated and flushed with nitrogen (3*). Ethanol (25 mL) was then added via syringe and the mixture was heated to reflux (80 0C) and stirred for 18 hours. The crude mixture was concentrated under reduced pressure and purified by flash chromatography (silica gel, 1 :20 EtOAc/hexanes to 10: 1 EtOAc/hexanes). The pure fractions were concentrated to yield 1-3 (1.949 g, 4.83 mmol, 90%) as a light yellow foam. MS (ESI): 404 (M H- H)+
The compounds of Table 3 were prepared according to the methods above.
Table 3




B. I 7'-methoxv-5Η-spirorpiperidine-4.4'-pvτrolo|' 1.2-aiquinρxalmei 1-4

1-3 1-4
A 50-mL, 1-necked round-bottomed flask was charged with spirocycle 1-3 (0.312 g, 0.773 mmol) and 5 % palladium on carbon (50 % by wt, 57.45 % H2O, 0.163 g). Ethanol (30 mL) was added and nitrogen bubbled through the solution with stirring. Hydrogen was bubbled through the reaction mixture for 10 minutes and the reaction was stirred over night under hydrogen atmosphere. The reaction was filtered through Celite and concentrated under reduced pressure to yield amine 1-4 (0.200 g, 0.743 mmol, 96%). MS (ESI): 270 (M + H)+
AU other -Cbz intermediates were cleaved in a similar fashion. B.2 Alternate procedure for 71-methoxy-5'H-spiro[piperidine-4,4'-pyrrolo[l:,2:a]ιquinoxaline3 1-4

A round bottomed flask was charged with (5.00 g, 26.6 mmol), 4-piperidinone hydrochloride monohydrate (4.07 g, 26.6 mmol), maleic acid (100 mg, 0.85 mmol), and elhanol (100 mL). The reaction mixture was heated to 80 0C for 16 hours, cooled to room temperature, and concentrated in vacuo. The crude mixture was triturated with methylene chloride and the solid was isolated by filtration and dried under vacuum at 40 0C to afford 1-4 (6.73 g, 94%) as an off-white solid. MS (ESI): 270 (M + H)+
C. 7'-methoxy-5Η-8-azaspiro[bicyclo[3.2.1]octane-3,4'-pyrrolo[l,2-a]qumoxaHne] l-4a

TFA (1.0 mL, 13.0 mmol) was added to a solution of tert-butyl 7'-methoxy-5'H-8- azasρiro[bicyclo[3.2.1]octane-3s4l-pyrrolo[l,2-a]quinoxaline]-8-carboxylate 3-22 (69 mg, 0.17 mmol) in methylene chloride (3 mL) at 0 0C. The solution was stirred for 1 hour at 0 0C then 1 hour at room temperature. The reaction was quenched with saturated aqueous sodium bicarbonate and extracted with methylene chloride. The combined extracts were dried with Na2SO4, filtered, and concentrated in vacuo to afford 1-4-1 (51 mg, 100%) as a light green residue. MS (ESI): 296 (M + H)+
. (3 ,5 -dimethoxyphenyl)(7'-methoxy- 5 Η-spiro f ρiperidine-4,4'-pyrrolo [ 1 ,2-a] quinoxaline] - 1 -

1-4
A 50-mL, 1 -necked round-bottomed flask was charged with benzoic acid 1-5 (0.136 g, 0.743 mmol), EDGE (0.213 g, 1.11 mmol), HOAt (0.159 g, 1.11 mmol), and DMAP (0.095 g,
0.74 mmol). The apparatus was evacuated and flushed with nitrogen (3X) and CH2Cl2 (15 mL) was added. Amine 1-4 (0.200 g, 0.743 mmol) in CH2Cl2 (10 mL) was added and the resultant solution was stirred over night at room temperature. The reaction was diluted with CH2CI2 and washed with sat. NaHCO3 (1 X 100 mL) and brine (1 X 100 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, ISCO CombiFlash Rf system, CH2Cl2Io 10:1 CH∑Cymethanol) and lyopholized from aq. HC1/CH3CN to give amide 1 (0.272 g, 0.579 mmol, 78%) as a light brown solid. MS (ESI): 434 (M + H)+
E. (4:methoxy-3 -ρropylphenyl)( 7!-methoxy-5 'H-spiro [piperidme-4,4'-pyrrolo [ 12- alquinoxaline]-l-yl)methanone hydrochloride 4-62

A solution of (3-allyl-4~methoxyphenyl)(7'-methoxy-5'H-spiro[piperidine-4,4'- pyiτolo[l ,2-a] quinoxaline] -l-yl)methanone (90 mg, 0.20 mmol) in ethanol (10 mL) was purged with nitrogen for 5 minutes. Palladium on carbon (45 mg, 10% Pd/C, 50% water) was added and the system was again flushed with nitrogen. Hydrogen was bubbled through the solution and the reaction was stirred under a hydrogen atmosphere for 5 hours. The reaction was flushed with nitrogen, filtered through Celite, and concentrated in vacuo. The residue was purified on the Isco CombiFlash followed by semi-prep. HPLC5 concentrated, and lyopholized to afford 4-62 (21 mg, 23%) as a brown solid. MS (ESI): 446 (M + H)+
F. Preparation of final compounds and analogues: Schemes A. B, C, and D


Ia Ib
1. Amide Coupling Scheme A: A 50-mL, 1 -necked round-bottomed flask was charged with benzoic acid 1-5 (0.136 g, 0.743 mmol), EDCI (0.213 g, 1.11 mmol), HOAt (0.159 g, 1.11 mmol), and DMAP (0.095 g, 0.74 mmol). The apparatus was evacuated and flushed with nitrogen (3X) and CH2Cl2 (15 mL) was added. Amine 1-4 (0.200 g, 0.743 mmol) in CH2Cl2 (10 mL) was added followed by diisopropylethylamine (0.38 mL, 2.2 mmol) and the resultant solution was stirred over night at room temperature. The reaction was diluted with CH2Cl2 and washed with sat. NaHCO3 (1 X 100 mL) and brine (1 X 100 mL). The organic layer was dried over Na2Sθ4 and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, ISCO CombiFlash Rf system, CH2Cl2 to 10:1 CH2Cl2/methanol) and
lyopholized from aq. HC1/CH3CN to give amide 1 (0.272 g, 0.579 mmol, 78%) as a light brown solid. MS (ESI): 434 (M + H)+
2. Amide coupling Scheme B: To a solution of amine 1-1 (1.0 eq) and carboxylic acid
(1.5 eq) in methylene chloride (0.2M) at 0 0C was added diisopropylethylamine (3.0 eq) followed by T3P (propylphosphonic acid anhydride, 50% solution in ethyl acetate, 1.2 eq). The reaction was allowed to warm to room temperature overnight and purified by silica gel chromatography to afford the desired amide product.
3. Spirocylization Scheme C: Utilizing the general procedure above (Scheme 2) for intermediate 1-3, the pyrole-aniline was reacted with a functionalized piperidinone to afford the desired product.
4. Alkylation of aniline nitrogen Scheme D: To a solution of the starting aniline Ia in DMF (0.2 M) was added potassium carbonate (2 eq) and either iodomethane (1.2 eq) or benzyl bromide (1.2 eq). The reaction was heated to 100-120 0C either thermally or in a microwave reactor until the reaction was complete as judged by LC-MS. Additional alkylating agent was added if necessary. Upon completion of the reaction, the mixture was diluted with ethyl acetate, washed with brine, and purified by silica gel chromatography and/or semi-preparative HPLC.
The compounds of Table 4 were prepared according to the methods above. Table 4







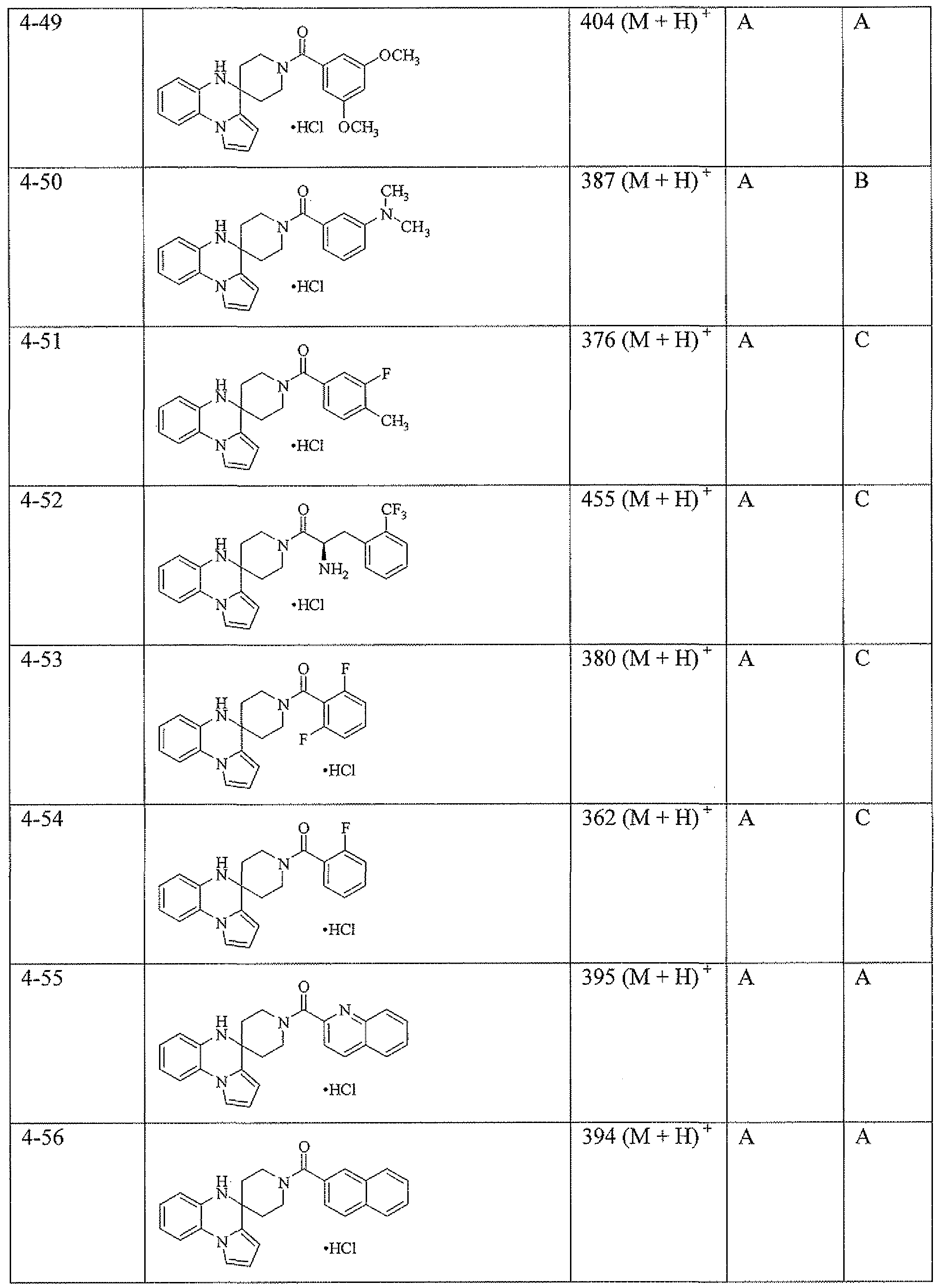
























G. (4-methoxy-3-propylphenyl')(7'-niethoxy-5Η-spiro[piperidine-4,4'-pyrroιlo[l,2- a]guinρxaline]-l-yl)methanone hydrochloride 4-214

A solution of (3-allyl-4-methoxyphenyl)(7'-methoxy-5Η~spiro[piperidine-4,4'- pyrrolo[l,2-a]quinoxaline]-l-yl)methanone 4-19 (90 mg, 0.20 mmol) in ethanol (10 mL) was purged with nitrogen for 5 minutes. Palladium on carbon (45 mg, 10% Pd/C, 50% water) was added and the system was again flushed with nitrogen. Hydrogen was bubbled through the solution and the reaction was stirred under a hydrogen atmosphere for 5 hours. The reaction was
flushed with nitrogen, filtered through Celite, and concentrated in vacuo. The residue was purified on the Isco CombiFlash followed by semi-prep. HPLC, concentrated, and lyopholized to afford 4-214 (21 mg, 23%) as a brown solid. MS (ESI): 446 (M + H)+
H. (4-hydroxyphenyl)(7'-methoxy-5'H-spiro[piperidine-4,4'-pyrrolo^L2-a]quinoxaline]-l: vDmethanone 4-215

4-41 4-215
The benzyl ether of 4-41 was cleaved using the standard hydrogenation procedure detailed above for the preparation of 1-4 to afford 4-215. MS (ESI): 390 (M + H)+
I. (3,5-dirnethoxyphenyiχ7!-phenyl-5Η-spiro[piperidine-4,4'-pyrrolo[l,2-a]qιιinoxaline]-l- vDmethanone hydrochloride 4-216

Suzuki Reaction Procedure: A 5 mL microwave vial was charged with chloride 4-150 (0.200 g, 0.456 mmol), phenylboronic acid (0.067 g, 0.548 mmol), Pd2(dba)3 (0.042 g5 0.0456 mmol), X-Phos (0.044 g, 0.0912 mmol) and potassium acetate (0.090 g, 0.912 mmol) and sealed. Separately, 10 mL of 1,4-dioxane was dried over molecular sieves and was sparged with argon gas for 30 minutes with stirring. This dried solvent (4 mL) was added to the reaction mixture under argon, and then the sealed tube was heated to 1000C in an oil bath for 72 hours. The reaction was filtered through Celite to remove the catalyst and concentrated under reduced pressure. The residue was purified by semi prep HPLC and then lyopholized from aq. HCI/CH3CN to give compound 4-216 (0.068 g, 0.131 mmol, 28%) as an off-white solid. MS (ESI): 480 (M + H)+
The compounds of Table 5 were prepared according to the methods above.
Table 5

J- (3,5-d.imethoxyphenyl)(8Lphenyl-5'H-spiro[piperidine-4.,4'-pyrrolo[l,2-a]quinoxaline]-l- ypmethanone hydrochloride 5-5

Suzuki Reaction Procedure: A 5 mL microwave vial was charged with bromide 4-171 (0.05 g, 0.104 mmol), phenylboronic acid (0.019 g, 0.156 mmol), Pd(dppf)Cl2 (0.042 g, 0.0456 mmol) and sodium carbonate (0.024 g, 0.228 mmol) and sealed. Separately, 10 mL of dimethoxyethane and 5 mL of water was sparged with argon gas for 30 minutes with stirring. This dried solvent (3 mL) was added to the reaction mixture under argon, and then the sealed tube was heated to 100°C in an oil bath for 24 hours. The reaction was filtered through Celite to remove the catalyst and concentrated under reduced pressure. The residue was purified by semi
prep HPLC and then lyopholized from aq. HC1/CH3CN to give compound 5-5 (0.017 g, 0.0325 mmol, 32%) as an off-white solid. MS (ESI): 480 (M + H)+
K. l-r33-dimethoxybenzoyl)-5Η-spiro[piperidine-4Λ'-pyrrolo[l,2-a]quinoxaline]-8'- carbonitrile 5-6

A 5 mL microwave vial was charged with bromide 4-171 (0.200 g, 0.415 mmol), Pd2(dba)3 (0.042 g, 0.0456 mmol), dppf (0.161 mg, 0.291 mmol), Zn metal (0.0065 g, 0.0996 mmol) and Zn(CN)2 (0.058 mmol, 0.498 mmol) and sealed. Separately, 10 mL of dimethylacetamide (DMA) was dried over molecular sieves and was sparged with argon gas for 30 minutes with stirring. This dried solvent (4 mL) was added to the reaction mixture under argon, and then the sealed tube was heated to 1000C in an oil bath for 72 hours. The reaction was filtered through Celite to remove the catalyst and concentrated under reduced pressure. The residue was purified by semi prep HPLC and then lyopholized from aq. HCI/CH3CN to give compound 5-6 (0.062 g, 0.133 mmol, 32%) as an off-white solid. MS (ESI): 480 (M + H)+
L. (4-( 1 H-imidazol- 1 -yPnaphthalen- 1 -vQ(7'-methoxy~5'H~spiro [piperidine-4,4!-ρyrroIo [1,2- a]quinoxaline]-l-yl)methanone hydrochloride 5-7

4-65 5-7 A suspension of 4-65 (50 mg, 0.11 mmol), imidazole (9 mg, 0.13 mmol), and potassium carbonate (31 mg, 0.22 mmol) in DMSO (0.2 mL) was heated at 100-120 0C overnight. The reaction mixture was diluted with ethyl acetate, washed with brine, and the organic layer dried with sodium sulfate. The solution was concentrated in vacuo, purified by silica gel chromatography, and lyophilized from acetonitrile/aqueous hydrochloric acid to afford 5-7 (26 mg, 47%) as a white solid. MS (ESI): 490 (M + H)+
M. 4~(7'-methoχy-5Η-spiro[piperidme-4,4'-pyrrolo[ 1 ,2-a]quinoxaline]-l -ylcarbonyl)- 1 - naphthonitrile hydrochloride 5-8

Utilizing the procedure above for the preparation of 5-6, 4-65 (50 mg, 0.10 mmol) was reacted to afford 5-8 (21 mg, 45%). MS (ESI): 449 (M + H)+
N. (7'-methoxy-5'H-spiro[piperidine-4,4'-pyrrolo[L2-a]quinoxaline]-l-yl)(4-(3-methyl-lH- pγrazol-5-yl)phenyl)methanone hydrochloride 5-9

5-9
To a suspension of NaH (100 mg, 60% dispersion, 2.5 mmol) and THF (13 mL) under N2 was added EtOAc (0.25 mL, 2.4 mmol) and the mixture was stirred for 30 minutes. To a separate RB flask was added 4-83 (0.5 g5 1.2 mmol), dibenzo-18-crown-6 (0.07 g, 0.19 mmol), 2 drops of EtOH, and THF (13 mL). Once all the solids were dissolved, the above sodium enolate solution was added drop- wise via syringe. The mixture was then heated at 80 °C for 3 hours, cooled to room temperature, and quenched with aqueous HCl (10 mL, IM). The mixture was extracted with EtOAc (2 x 20 mL) and the organic layer was dried over Na2Sθ4 and concentrated to give 4-83a (0.53 g, 96%) in satisfying purity.
To a solution of 4-83a (0.2 g, 0.44 mmol) in MeOH was added hydrazine monohydrate (0.02 mL, 0.48 mmol). The mixture was stirred for 2.5 hours at rt, then concentrated, purified by silica gel CombiFlash chromatography, followed by semi-prep HPLC and lyophilized from acetonitrile/water to afford 5-9 (83 mg, 82%) as an off-white solid. MS (ESI): 454 (M + H)+
. 5-(4-f 7'-methoχy-5Η-spiro[piperidine-4,4'-pyπ;Qlo[ 1 ,2-a]quinoxalme]- 1 -

Suzuki reaction procedure: To a sealed Biotage microwave vial was added DMF (3 niL) and 2 drops Of H2O and the system was sparged with argon for 10 minutes. A second vial was charged with 4-64 (0.08 g, 0.177 mmol), 5-(4,4,5ϊ5-tetramethyl-l,3,2-dioxaboroIan-2- yl)mcotinonitrile (0.049 g, 0.212 mmol), Cs2CO3 (0.1 ISg5 0.354 mmol), and Pd(PPh3)4 (0.03Og, 0.027 mmol). The tube was sealed with a cap lined with a Teflon septum and the DMF/H2O mixture above (3 mL) was added via a syringe. The reaction was sparged with argon for another 5 minutes, then heated to 85 0C for 12 hr. The reaction was cooled to rt and filtered through a thin pad of silica gel and filter agent. The solvent was concentrated and the crude product was purified by silica gel CombiFlash chromatography (eluting with 0 - 10% MeOH in dichioromethane). The material was then purified by semi prep HPLC and lyophilized from aq. HCI/CH3CN to give compound 5-10 ( 0.053 g, 0.104 mmol, 59%) as an off white solid. MS (ESI): 476 (M+H)+
P- 5-f4-(71-methoxy-5Η-spiro[piperidine-4,4'-pyrrolρ[1.2-a]quinoxaline]-l- ylcarbonyDphenvDnicotinic acid 5-11

5-11
Compound 5-11 was isolated as a side reaction from the preparation of 5-10.
R- (4-(5-fluoropyridin-3-yl)phertvl)(7'-methoxy-5Η-spiro[piperidine-4,4'-pyrrolρ[l,2- alquinoxalinei-l-vDmethanone hydrochloride 5-12

To a sealed Biotage microwave vial was added DME (3 mL) and 2 drops of H2O and the system was sparged with argon for 10 minutes. A second vial was charged with 4-64 (0.08 g, 0.177 mrnol), 5-iluoropyridin-3-ylboromc acid (0.030 g, 0.212 mmol), Na2CO3 (0.037g, 0.354 mmol) and Pd(PPh3)4 (0.02Og, 0.017 mmol). The tube was sealed with a Teflon septum and the DME/H2O mixture above (3 mL) was added. The reaction mixture was sparged with argon for another 5 minutes then heated to 85 °C for 12 hours. The reaction was cooled to room temperature and filtered through a thin pad of silica gel and filter agent. The solvent was concentrated and the crude product was purified by silica gel CombiFlash chromatography (eluting with 0 - 10% MeOH in dichloromethane). The material was then purified by semi prep HPLC and lyophilteed from aq. HC1/CH3CN to give compound 5-12 (0.043 g, 0.085 mmol, 49%) as an off white solid. MS (ESI): 469 (M+H)+
S. (7'-methoxv-5'H-spiro[piperidine-4,4'-pyrrolori ,2-a]quirtoxaline]- 1 -yD(4-(thiophen-2- vDphenyDmethanone hydrochloride 6-1

A Biotage microwave vial with magnetic stir bar was charged with Pd(OAc)2 (0.016 g, 5 mol%), RuPhos (0.017 g, 10 mol%), 4-64 (0.056 g, 0.123 mmol), 2 (0.027 g, 0J36mmol), and Na2CO3 (0.026 g, 0.247 mmol). The tube was sealed with a cap lined with a Teflon septum, evacuated, and purged with N2 (x3). Ethanol (1.5 mL) was added via syringe, and the reaction was heated to 85 °C for 12 hours. The reaction was allowed to cool to it and filtered through a thin pad of silica gel and filter agent. The solvent was concentrated, and the crude product was
purified by silica gel CombiFlash chromatography (eluting with 0 to 90% ethyl acetate in hexanes). The material was then purified by semi-prep HPLC and lyophilized from aq. HCI/CH3CN to give compound 6-1 ( 0.017 g, 0.041mmol, 36%) as an off white solid. MS (ESI): 456 (M+H)÷
The compound of Table 6 was prepared according to the methods above. Table 6

T. ( 1 '-( (dimethylammo)methyl>7'-methoxy-5 'H"Spiro[piperidine-4,4'-pyrrolo [1,2- a] quinoxaline] - 1 -yl)(4-ethoxyphenyl)methanone 7- 1

A solution of dimethylamine (0.042 niL, 40% aqueous solution, 0.36 mmol), formaldehyde (0.022 mL, 37% aqueous solution, 0.26 mmol), and acetic acid (0.2 mL) was prepared. This solution was added into a solution of 4-5 (50 mg, 0.12 mmol) in ethanol. The reaction was stirred at room temperature for 3 days and then saturated aqueous sodium bicarbonate was added and the mixture was extracted with methylene chloride. The combined organic layers were dried with sodium sulfate and purified by silica gel chromatography to afford 7-1 (24 mg, 42%) as an off-white solid. MS (ESI): 430 ((M~HN(CH3)2) + H)+
The compound of Table 7 was prepared according to the methods above.
Table 7

U. l-(l-(4-ethoxybenzoyl)-7'-methoxy-5'H-spiro[piperidine-4,4'-pyrrolo[L2-a]quinoxaline]-r- yl)-2.2.2-trifluoroethanone 7-3

Trifluoroacetic anhydride (0.050 mL, 0.36 mmol) was added to a solution of 4-5 (50 mg, 0.12 mmol) in benzene (4 mL) at room temperature. The reaction was stirred at room temperature for 7 hours, heated to 80 0C for 30 minutes, and then stirred at room temperature overnight. The reaction was partitioned between methylene chloride and saturated aqueous sodium bicarbonate. The organic layer was dried with sodium sulfate and purified by silica gel chromatography to afford 7-3 (23 mg, 38%) as a bright yellow solid. MS (ESI): 514 (M + H)+
EXAMPLE 2

l-(4-fluorophenyl)-7'-methoxy-5'H-spiro[piperidine-4,4'-pyrrolo[l,2-a]quinoxaline] hydrochloride 2

A solution of 5-methoxy-2-(lH-pyrrol-l-yl)aniline 1-1 (50 mg, 0.27 mmol),, l-(4- fluorophenyl)piperidin-4-one (51 mg, 0.27 mmol), and maleic acid (10 mg, 0.08 mmol) in ethanol (1.5 mL) was stirred at 80 0C overnight (16 hours). The reaction was cooled to room temperature, concentrated in vacuo, and purified by flash column chromatography. The purified product was lyophilized from acetonitrile and aqueous HCl to afford the desired product 2 (98 mg, 91%) as an off-white solid. MS (ESI): 364 (M + H)+
The compounds of Table 8 were prepared according to the methods above. Table 8



l-(3,5-bis(Mfluoromethyl)benzpy])-7'i-methoxy-5'H-spiro[piperidine-4,4'-pyrrolo[L2- a]quinoxalin]-2-one hydrochloride 3

2K
A 25-mL, 1 -necked round-bottomed flask was charged with amide 8-13 (0.063 g, 0.22 mmol) and sodium hydride (60% in mineral oil, 0.019 g, 0.26 mmol). The apparatus was evacuated and flushed with nitrogen (3X). THF/DMF (1:1, 8 mL) was added via syringe and followed by 3,5-bis(trifluoromethyl)-benzyl bromide (0.1 mL, 0.3 mmol) and the resultant solution was stirred for 3 hr at room temperature. The reaction was diluted with brine (10 mL) and extracted with EtOAc (1 X 5 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by semi prep HPLC and lyopholized from aq. HC1/CH3CN to give amide 3 (0.019 g, 0.035 mmol, 16%) as a light brown solid. MS (ESI): 510 (M + H)+ The compounds of Table 9 were prepared according to the methods above.
Table 9


EXAMPLE 4
 -(5Η-sρiro|"ρiρeridine-4,4'-pvn.'olorL2-alquinoxalinel-l-vl)benzordlthiazole hvdiOchloride 4
-(5Η-sρiro|"ρiρeridine-4,4'-pvn.'olorL2-alquinoxalinel-l-vl)benzordlthiazole hvdiOchloride 4
 l-4b 4
A 25-mL, 1-necked round-bottomed flask was charged with amine l-4b (0.081 g5 0.34 mmol) and potassium carbonate (0.130 g, 0.941 mmol). 1,4-Dioxane (10 niL) and 2- chlorobenzothiazole (0.5 mL, 0.3 mmol) were added to the flask. The mixture was stirred for 72 hours at 40 0C. The mixture was diluted with CH2CI2 and washed with H2O (2 X 20 mL). The aqueous layer was extracted with CH2Cl2 and the combined organics were dried over Na2SO4. The organic layer was concentrated and the residue was purified by semi prep HPLC and lyopholized from aq. HC1/CH3CN to give 4 (0.048 g, 0.12 mmol, 35%) as a white solid. MS (ESI): 373 (M + H)+
l-4b 4
A 25-mL, 1-necked round-bottomed flask was charged with amine l-4b (0.081 g5 0.34 mmol) and potassium carbonate (0.130 g, 0.941 mmol). 1,4-Dioxane (10 niL) and 2- chlorobenzothiazole (0.5 mL, 0.3 mmol) were added to the flask. The mixture was stirred for 72 hours at 40 0C. The mixture was diluted with CH2CI2 and washed with H2O (2 X 20 mL). The aqueous layer was extracted with CH2Cl2 and the combined organics were dried over Na2SO4. The organic layer was concentrated and the residue was purified by semi prep HPLC and lyopholized from aq. HC1/CH3CN to give 4 (0.048 g, 0.12 mmol, 35%) as a white solid. MS (ESI): 373 (M + H)+
EXAMPLE 5 l-(3,5-dimethoxybenzyl)-7'-methoxy-5'H-spiro[piperidine-4,4'-pyrrolo[1.2-a]quinoxaline] 5

A solution of 1-1 (50 mg, 0.19 mmol), l-(bromomethyl)-3,5-dirnethoxybenzene (43 mg5 0.19 mmol), and diisopropylethylamine (0.066 mL, 0.37 mmol) in DMF (1 mL) was stirred at room temperature overnight (16 hours). The reaction was diluted with ethyl acetate, washed with brine, and the organic layer was dried with sodium sulfate. The solution was concentrated in vacuo and purified by silica gel chromatography to afford 5 (49 mg, 63%) as a light brown solid. MS (ESI): 420 (M + H)+ The compounds of Table 10 were prepared according to the methods above.
Table 10



Compound 6 can be prepared according to the procedures for the preparation of Example 1, by hydrogenation of 4-171 with 2H2 in the presence of Pd/C at 1 atm.
EXAMPLE 7
Assay Methods
A. Cell Culture
The pore-forming subunits of human voltage gated sodium channels were stably expressed in either HEK 293 cells of CHO-Kl cells. Cells were maintained in standard growth media containing 10% heat-inactivated fetal bovine serum and a selection antibiotic. The cells
were grown at 37°C in a humidified tissue culture incubator with the carbon dioxide concentration regulated at 5%.
B. FLIPR evaluation of sodium channel activity A FLIPR assay using a membrane potential dye (Molecular Devices Corporation, blue no wash voltage- sensitive dye) was used to screen for sodium channel activity of compounds. Cells were plated onto black- walled 384 well plates with poly-lysine-coated glass bottoms 24 hours prior to evaluation on the FLIPR. On the day of experiment, the growth medium was removed and replaced with a physiological saline solution containing voltage-sensitive dye and compounds of interest. After dye loading for 60 minutes, cell depolarization was evoked on the FLIPR by the addition of veratridine. Maximum veratridine-induced increase in fluorescence intensity from baseline was used to plot concentration-effect curves. Non-linear regression analysis was used to generate IC 50 values.
C. Voltage clamp measurement of sodium channel activity
Standard ruptured whole cell patch clamp techniques were used to measure sodium channel blocking activity. All studies were conducted at room temperature using a flowing extracellular solution containing (niM concentrations): 129 NaCl5 20 letraethylammonium chloride, 3.25 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, 10 HEPES-NaOH (pH 7.35). The glass whole cell patch electrodes for these studies had tip resistances of approximately 1.5 MΩ when filled with the following intracellular solution (mM concentrations): 120 CsF, 10 NaCl5 10 tetraethylammonium chloride, 11 EGTA, 1 CaCl2, 1 MgCl2, 10 HEPES-CsOH (pH 7.3). Currents were evoked with 50 millisecond voltage steps from a holding potential of -120 mV to a potential corresponding to the peak of the transient inward current- voltage relationship of the subtype of voltage-gated sodium current being studied. The stimulation frequency was 10 Hz unless noted otherwise. The fraction of baseline current remaining after exposure to compound was used to construct concentration-effect curves which were fit by non-linear regression analysis to generate ICs 0 values.
FLIPR activity for the compounds of the invention fell into one of six ranges: InM to 200 nM (A); 200 nM to 1000 nM (B); 1000 nM to 5000 nM (C); 5000 to 10000 11M (D); 10000 to 15000 (E); 15000 to 30000 (F); and 30000 or greater (G).
Compounds of present invention made according to the schemes and examples above showed activity in the foregoing assays. The activity of representative compounds of the invention is shown in Table 11. Table 1 1




The present invention is not to be limited by the specific embodiments disclosed in the examples that are intended as illustrations of a few aspects of the invention and any embodiments that are functionally equivalent are within the scope of this invention. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art and are intended to fall within the scope of the appended claims.
Claims
1. A compound of structural formula I:

I
or pharmaceutically acceptable salts and individual enantiomers and diastereomers thereof, wherein: W is pyrazolyl, phenyl or pyridinyl, each optionally substituted with 0 to 3 groups of Ra; Rx and R^ are independently selected from the group consisting of
(5) hydrogen,
(6) -C(O)R1,
(7) -(CH2)nC5-Cio heterocyclyl, and (8) -Ci-CiO alkyl, wherein said, heterocyclyl and Ci-Cio alkyl may be optionally substituted with one to four groups of Ra, orRx and RY together with the carbon atom to which they are attached can form a 5 to 8 membered carbocyclic ring of which one of the ring atoms is optionally nitrogen, said ring substituted with Z — Rl and optionally substituted with 1 to 3 groups of R ; Z is selected from the group consisting of (1) a bond, (2) -Ci-C6 alkylene,
(3) -C(O)-,
(4) -C(O)O-, and (5) -SO2-, wherein when Z is a bond or alkylene, Rl cannot be hydrogen;
Rl is selected from the group consisting of (8) hydrogen, (9) -(CHR2)nC6-Cio aryl,
(10) -(CH2)nC5-Cio heterocyclyl,
(11) -(CH2)nC3-C6 cycloaklyl, and 
(13) -Ci-C6 alkyl5 and
(14) -CH(NHBoc)(CH2)nC6-Cio aryl wherein said heteroaryl is not thienyl and may be optionally substituted with 1 to 4 groups of Ra, and said aryl and cycloaklyl are substituted with 1 to 4 groups of Ra; R2 is selected from the group consisting of (1) hydrogen, (2) -Ci-C6 alkyl, and
(3) ™(CH2)nC6-ClO aryl, which may be optionally substituted with 1 to 4 groups of Ra;
R3 and R4 are independently selected from the group consisting of (1) hydrogen,
(2) -Ci-C6 alkyl,
(3) -(CH2)nC6-ClO aryl, which may be optionally substituted with 1 to 4 groups of Ra, (4) -F, and
(5MCH2)nCF3 Ra is selected from the group consisting of
(1) hydrogen,
(2) hydroxy.,
(3) halogen,
(4) -Ci-Cio alkyl, (5) -C2-C6 aϊkenyl,
(6) -(CH2)n C6-Ci0 aryl,
(7) ~(CH2)nC5-Cio heterocyclyl,
(8) -O-, (9) -OR3, (10) -O(CH2)nOR4 (llMCH2)nN(R3)2, (12)-NR3C(O)R4} (13) -C(O),
(14)~C(O)R3, (15) -(CH2)J1CN5
(16)~Ci-C3haloalkyl,
(H)-NO2, (18)-NR3C(O)OR4S (19)-C(O)OR4, 
(21)-O(CH2)nN(R3),and (22) -(CH2)nNHC(O)OR3, wherein said aryl and heteroaryl may be optionally substituted with 1 to 4 groups of Rb, which is selected from the group consisting of (l)-Ci-C6aJkyl,
(2) halogen, (3) -CN5 (4) -NH2, and (5)-OR3> j is 1 to 3; n is 0 to 4; p is 0 to 3; qis 1 to 3; and wherein the compounds of said formula I do not include the compounds of Table 1.
2. A compound of claim 1 wherein Rx and RY form a 5 to 8 membered carbocylic ring substituted with Z — Rl.
3. A compound of claim 2 wherein one of the ring atoms is nitrogen.
4. A compound of claim 3 wherein Z — Rl is attached to the nitrogen atom.
5. A compound of claim 1 wherein Z is C(O).
6. A compound of claim 1 wherein Rl is (CH2)nC6-Cio aryl> where said aryl is selected from the group consisting of phenyl and napthyl.
7. A compound of claim 1 wherein Rl is optionally substituted heteroaryl, where said heteroaryl is selected from the group consisting of benzylfuranyl, quinolinyl, indolyl, benzodioxolyl, benzothiazolyl, benzoxazinyl, benoxazolyl, and benzoxazine.
8. A compound of claim 1 represented by structural formula II:

II and pharmaceutically aceptable salts and individual enantiomers and diasteromers thereof, wherein,
Rl is selected from the group consisting of (3) -C6-Cio aryl5 and (4) -(CH2)nC6-Cl0 heteroaryl, wherein said heteroaryl is not thienyl and may be optionally substituted with 1 to 4 groups of Ra, and said aryl is substituted with 1 to 4 groups of Ra.
9. A compound of claim 8 wherein Ra is selected from the group consisting of
(1) hydrogen,
(2) halogen, (3) -Ci-C6 alkyl, (4) -(CH2)nC6-C i o heteroaryl,
(5) -OR3,
(6) -N(R3)2,
(7) -CN, and
(8) -CF3.
10. A compound of claim 1 represented by Examples 1 through 6 and Tables 2 through 11 or pharmaceutically acceptable sals and individual enantiomers and diasteromers thereof.
11. A compound of claim 1 selected from the group consisting of: l-(4-bromo-3-methoxybenzoyl)-7t-methoxysρiro[ρiperidine-454'(5'h)-pyrrolo[l,2-a] quinoxaline];
1 , 1 -dimethylethyl [4- [(7' -methoxyspiro [piperidine-4,4'(5'h)-pyrrolo [ 1 ,2-a] quinoxalin]- 1 -y 1) carbonyljcyclohexyl] carbamate; 7!-methoxy-l-[(2-methyl-6-benzothiazolyl) carbonyl] spiro [piperidine -4,4'(5'h)-pyrrolo[l,2-a] quinoxaline];
1 ~[(2-chloro-6-benzothiazolyl) carbonyl]~7'-methoxy spiro [piperidine-4,4'(5 'h)-pyrrolo [ 1 ,2~a] quinoxaline];
7'-methoxy- 1 -[(1 -methyl-lh-indol-6-yl)carbonyl]spiro[piperidine-4,4'(5'h)-pyrrolo[l 52- a] quinoxaline];
1 - [(3 ,4-dihydro-3 -oxo-2h- 1 ,4-benzoxazin-7-yl) carbonyl] -7'-methoxy spiro [piperidine-4,4'(5 'h)- pyrrolofl,2-a] quinoxaline];
7'-methoxy- 1 - [( 1 -methyl- 1 h-indol-5-yl)carbonyl] spiro [piperidine-4J4'(5 'h)-pyrrolo [ 1 ,2-a] quinoxaline]; l-[(2-amino-6-benzothiazolyl) carbonyl] -7'-methoxy spiro[piperidine-4,4'(5'h)-pyrrolo[l,2-a] quinoxaline];
7'-methoxy-l-[(3-methyl-lh-indol-5-yl)carbonyl] spiro[piperidine-4,4'(5!h)-pyrrolo[l,2-a] quinoxaline];
1 -(6-quinolinylcarbonyl)-7'-(trifluoromethyl) spiro [piperidine-4,4'(5'h)-pyrrolo[l ,2-a] quinoxaline]; l-[(l,3-dimethyHh-indol-5-yl)carbonyl]-7'-methoxy spiro [piperidine-4,4'(5'h)-pyrrolo(l,2-a] quinoxaline];
1 - [(354-dihydro-4-methyl-2h- 1 ,4-benzoxazin-7-yl)carbonyl] -7'-methoxyspiro [piperidine-
4,4'(51Ji)-PyIToIo [1,2-a] quinoxaline]; 7!-chloro-l -[3-(dimethylamino)-4-methoxybenzoyl]spiro [piperidine~4,4t(5'h)-pyrrolo[l,2-a] quinoxaline];
7*-chloro-l -(6-quinoHnylcarbonyl) spiro[piperidine-454'(5'h)-pyrrolo[l ,2-a] quinoxaline] l-tt^-chlorofl^'-biphenylJ-S-y^carbonylJ^'-methoxyspiro^iperidine^^XS'^-pyrroloEl^-a] quinoxaline]; 1 -[4-amino-3-(trifluoromethoxy) ben2oyl]-7!-methoxyspiro[piperidine-4,4'(5'h)-pyrrolo[l ,2-a] quinoxaline];
1 -(4-amino-3 -methoxybenzoyl)-7'-methoxyspiro[piperidine-4f4'(5'li)-pyrrolo [ 1 ,2-a] quinoxaline] ; 3-amino-4-methoxy-n-[2-methoxy-5-[[7'-(lrifluorometliyl)spiro [piperidine-4,4'(5'li)-pyrrolo[l,2- a] quinoxalin] - 1 -yl] carbonyl] phenyl] benzamide;
1 -(3-amino-4-methoxybenzoyl)-7'-(trifluoromethyl)spiro [ρiperidine-4541(5'h)-ρyrrolo[ 1 ,2-a] quinoxaline]; 1 -[3 -(dimethylamino)-4-methoxybenzoyl] -7'-(trifluoromethyl)spiro [piperidine-4,4'(5'h)- pyrrolo [ 1 ,2-a] quinoxaline] ;
1 -(3-bromobenzoyl)-7'-methoxyspiro[piperidine-4,4'(5'h)-pyrrolo [ 1 ,2-a] quinoxaline] ;
7'-methoxy- 1 -[ [8 -methoxy-2-(trifluoromethyl)- 5 -quinolinyl] carbonyl] spiro[piperidine-4,4'(5'h)- pyrrolo[l,2-a] quinoxaline]; l-[(2,3-dihydro-2-oxo-5-benzoxazolyl)sulfonyl]-7'-methoxyspiro [piperidine-4,4'(5'h)- pyrrolo [ 1 ,2-a] quinoxaline] ;
1 -[(3,4-dihydro-2h-l ,4-benzoxazin-6-yl) carbonyl] -7'-methoxy spiro[piperidine-4s4'(5'h)- pyrrolo [ 1 ,2-a] quinoxaline] ; n-[2-methoxy-5-[(7'-methoxyspiro[piperidine-4,41(5'h)-pyrrolo[ 1 ,2-a] quinoxalin]- 1 -yl) sulfonyljphenyl] acetamide;
7'-methoxy- 1 -[(5-methoxy-2-pyridinyl)carbonyl] spiro[piperidine-4,4<(5'h)-pyrrolo[ 1 ,2-a] quinoxaline];
1 -[(5-cyano-2-pyridinyl) carbonyl ]-7!-methoxy spiro[piperidine-4,4'(5>h)-pyrrolo[l 52-a] quinoxaline]; 7!-methoxy-l-(3-quinolinylsulfonyl) spiro[piperidine-4,4'(5'h)-pyrrolo[l,2-a] quinoxaline];
7'-methoxy-l-[(4-methoxy phenyl)sulfonyl]spiro [piperidine-4,4'(5'h)-pyrrolo[l,2-a] quinoxaline]; l-[(4~cyanophenyl) sulfonyl]-7'-methoxy spiro[pϊperidine-4?4'(5'li)~pyrrolo[l52-a] quinoxaline];
1 -(3-amino-4-methoxy benzoyl)-7'-methoxysρiro [piperidine-4)4'(5'h)-pyrrolo[l ,2-a] quinoxaline]; spiro[piρeridine-4s4'(5'h)-pyrrolo[l ,2-a] quinoxaline], 1 -(3,5-dimethylbenzoyl)-7'-methoxy; spiro [piperidine-4 ,4'(5 'h)-pyrrolo [ 1 , 2-a] quinoxaline] , 1 -(3 ,5 -dimethoxybenzoyl)-7'-methoxy ; spiro[piperidine-4,4t(5'h)-pyrrolo[ 1 ,2-a] quinoxaline], 1 -(4-cyanobenzoyl)-7'-methoxy; spiro[piperidme-4,4'(5'h)-pyrrolo[l ,2-a] quinoxaline], 8'-chloro- 1 -(4-cyanobenzoyl); l-(4-ethoxy-3-methoxybenzoyl)-7'-methoxyspiro[piperidine-4,4t(5'h)-pyrrolo [1,2- a]quinoxaline];
1 -(5-benzofuranylcarbonyl)-7'-methoxyspiro[piperidine-4,4'(5'h)-pyrrolo [1 ,2-a]quinoxaline] ; 1 -[3-(dimethylamino)-4-methoxybenzoyl]-7r-methoxyspiro[piperidine-4,4'(5'h)-pyrrolo [1,2- a]quinoxaline];
7-methoxy- 1 -(6-quinolinylcarbonyl) spiro [piperidme-4,4'(5'h)-pyrrolo [ 1 ,2-a] quinoxaline] ;
T'-methoxy-l-CS-quinolinylcarbony^ spirotpiperidine^^XS'hJ-pyiToloJl^-a] quinoxaline]; 7'-methoxy- 1 -(4-quinolinylcarbonyI) sρirojpIperidIne-4 ,4'(5'h)-pyrrolo[ 1 ,2-a] quinoxaline] ; δ'-chloro- 1 - [4-methoxy-3-(trifluoromethyl) benzoyl] spiro [piρeridine-4,4'(5 'h)-pyrrolo [1,2- a] quinoxaline];
1 -(4-cyanobenzoyl)-7'-( 1 -methylethyl) spiro[piperidine-4;4'(5'h)-pyrrolo [ 1 ,2-a] quinoxaline] ;
7'-methoxy- 1 -[4-(2,2,2-trifluoroethoxy) benzoyI]spiro[piperidine-4J4'(5'h)-ρyrrolo [1,2- a] quinoxaline];
1 ~(4~cyanobenzoyl)-7'-(trifluorometlωxy) spiro[piperidine-4,4'(5'h)-pyrrolo[l ,2-a] quinoxaline];
1 -(5-benzofuranylcarbonyl)-8'-chlorospiro [piperidme-4,4'(5'h)-pyrrolo[ 1 ,2-a] quinoxaline];
1 -(3 ,5 -dimethoxybenzoy l)-7'-( 1 h-pyrrol- 1 -yl) spiro [piperidine-4 ,4'(5'h)-pyrrolo [ 1 ,2-a] quinoxaline]; 1 -[(2,2-difluoro-l 53-benzodioxol-5-yl) carbonyl]-7'~methoxyspiro[piperidine-4J4'(5'h)- pyrrolo[ 1 ,2-a]quinoxaline] ;
1 -(I h-indol-5-ylcarbonyl)-7'-methoxyspiro [piperidine-4,4'(5'h)-pyrrolo[l ,2-a] quinoxaline] ;
7'-methoxy- 1 - [4-methoxy-3 -(trifluoromethyl)benzoyl] spiro [piperldine~4 ,4'(5'h)-pyrrolo [ 1 ,2- a] quinoxaline]; 8'-chloro-l-(lli-iiidol-5-ylcarbonyl) sρirotpipeϊridine-4J4'(5lh)-pyrrolo[l,2-a] quinoxaline];
1 -( 1 h-indazol-5 -ylcarbonyl)-7'-methoxyspiro [piperidine-454'(5 'h)-pyrrolo [ 1 ,2-a] quinoxaline] ;
7!-methoxy-l-[4-(ls2,3-thiadiazol-4-yl) benzoyl]spiro[pIperidine-4,4'(5lh)-pyrrolo [1,2- a] quinoxaline]; and
7'-methoxy-l-[4-(lh-pyrazol-l-yl)benzoyl] spiro[piperidine-4)4'(5'h)-pyrrolo[l,2-a] quinoxaline].
12. A pharmaceutical composition comprising a pharmaceutically acceptable carrier and a compound of claim 1 or a pharmaceutically acceptable salt thereof.
13. Use of a compound of claim 1 , or a pharmaceutically acceptable salt thereof, for the treatment of chronic and neuropathic pain.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22085609P | 2009-06-26 | 2009-06-26 | |
| US61/220,856 | 2009-06-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010151595A1 true WO2010151595A1 (en) | 2010-12-29 |
Family
ID=43386873
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/039672 WO2010151597A1 (en) | 2009-06-26 | 2010-06-23 | Methods for using pyrrolo-benzo-1,4-diazines as sodium channel blockers |
| PCT/US2010/039669 WO2010151595A1 (en) | 2009-06-26 | 2010-06-23 | Pyrrolo-benzo-1,4-diazines useful as sodium channel blockers |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/039672 WO2010151597A1 (en) | 2009-06-26 | 2010-06-23 | Methods for using pyrrolo-benzo-1,4-diazines as sodium channel blockers |
Country Status (1)
| Country | Link |
|---|---|
| WO (2) | WO2010151597A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011140425A1 (en) * | 2010-05-06 | 2011-11-10 | Vertex Pharmaceuticals Incorporated | Heterocyclic chromene-spirocyclic piperidine amides as modulators of ion channels |
| WO2012106499A1 (en) * | 2011-02-02 | 2012-08-09 | Vertex Pharmaceuticals Incorporated | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| WO2012112743A1 (en) * | 2011-02-18 | 2012-08-23 | Vertex Pharmaceuticals Incorporated | Chroman - spirocyclic piperidine amides as modulators of ion channels |
| WO2012125613A1 (en) * | 2011-03-14 | 2012-09-20 | Vertex Pharmaceuticals Incorporated | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| WO2013067248A1 (en) * | 2011-11-04 | 2013-05-10 | Vertex Pharmaceuticals Incorporated | Benzoxazines as modulators of ion channels |
| WO2014022639A1 (en) * | 2012-08-01 | 2014-02-06 | Vertex Pharmaceuticals Incorporated | Solid forms of (4-isopropoxy-3-methoxyphenyl)(2'-methyl-6'-(trifluoromethyl)-3',4'-dihydro-2'h-spiro[piperidine-4,1'-pyrrolo[1,2-a]pyrazine]-1-yl)methanone |
| WO2015006280A1 (en) * | 2013-07-10 | 2015-01-15 | Vertex Pharmaceuticals Incorporated | Fused piperidine amides as modulators of ion channels |
| WO2015151001A1 (en) | 2014-03-29 | 2015-10-08 | Lupin Limited | Sulfonamide compounds as voltage gated sodium channel modulators |
| WO2017037682A1 (en) | 2015-09-04 | 2017-03-09 | Lupin Limited | Sulfonamide compounds as voltage-gated sodium channel modulators |
| WO2018163077A1 (en) | 2017-03-08 | 2018-09-13 | Lupin Limited | Indanyl compounds as voltage gated sodium channel modulators |
| JP2019031531A (en) * | 2013-08-26 | 2019-02-28 | パーデュー、ファーマ、リミテッド、パートナーシップ | Azaspiro[4.5] decane derivatives and use thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013109521A1 (en) * | 2012-01-16 | 2013-07-25 | Vertex Pharmaceuticals Incorporated | Pyran-spirocyclic piperidine amides as modulators of ion channels |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040248890A1 (en) * | 2003-03-03 | 2004-12-09 | Gonzalez Jesus E. | Quinazolines useful as modulators of ion channels |
| US20040266802A1 (en) * | 1999-06-07 | 2004-12-30 | Alain Calvet | Tricyclic analgesics |
-
2010
- 2010-06-23 WO PCT/US2010/039672 patent/WO2010151597A1/en active Application Filing
- 2010-06-23 WO PCT/US2010/039669 patent/WO2010151595A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040266802A1 (en) * | 1999-06-07 | 2004-12-30 | Alain Calvet | Tricyclic analgesics |
| US20040248890A1 (en) * | 2003-03-03 | 2004-12-09 | Gonzalez Jesus E. | Quinazolines useful as modulators of ion channels |
Non-Patent Citations (1)
| Title |
|---|
| MARBAN ET AL: "Structure and function of voltage-gated sodium channels", JOURNAL OF PHYSIOLOGY, vol. 508, 1998, pages 647 - 657, XP000938782, DOI: doi:10.1111/j.1469-7793.1998.647bp.x * |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8598164B2 (en) | 2010-05-06 | 2013-12-03 | Vertex Pharmaceuticals Incorporated | Heterocyclic chromene-spirocyclic piperidine amides as modulators of ion channels |
| WO2011140425A1 (en) * | 2010-05-06 | 2011-11-10 | Vertex Pharmaceuticals Incorporated | Heterocyclic chromene-spirocyclic piperidine amides as modulators of ion channels |
| KR101923367B1 (en) | 2011-02-02 | 2018-12-04 | 버텍스 파마슈티칼스 인코포레이티드 | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| AU2012212196B2 (en) * | 2011-02-02 | 2016-10-13 | Vertex Pharmaceuticals Incorporated | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| JP2016041764A (en) * | 2011-02-02 | 2016-03-31 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| CN103403004A (en) * | 2011-02-02 | 2013-11-20 | 沃泰克斯药物股份有限公司 | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| CN103403004B (en) * | 2011-02-02 | 2016-06-01 | 沃泰克斯药物股份有限公司 | As the Pyrrolopyrazine-spirocyclic piperidine acid amides of ion channel modulators |
| EP3056495A1 (en) * | 2011-02-02 | 2016-08-17 | Vertex Pharmaceuticals Inc. | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| WO2012106499A1 (en) * | 2011-02-02 | 2012-08-09 | Vertex Pharmaceuticals Incorporated | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| RU2634900C2 (en) * | 2011-02-02 | 2017-11-08 | Вертекс Фармасьютикалз Инкорпорейтед | Spirocyclic pyrrolopyrazine(piperidine)amides as ionic channels modulators |
| JP2014504642A (en) * | 2011-02-02 | 2014-02-24 | バーテックス ファーマシューティカルズ インコーポレイテッド | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| US9511067B2 (en) | 2011-02-02 | 2016-12-06 | Vertex Pharmaceuticals Incorporated | Substituted spiro[piperidine-4,1'-pyrrolo[1,2-a]pyrazine]s as modulators of ion channels |
| CN106008504A (en) * | 2011-02-02 | 2016-10-12 | 沃泰克斯药物股份有限公司 | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| US8916565B2 (en) | 2011-02-02 | 2014-12-23 | Vertex Pharmaceuticals Incorporated | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| US10385070B2 (en) | 2011-02-18 | 2019-08-20 | Vertex Pharmaceuticals Incorporated | Chroman-spirocyclic piperidine amides as modulators of ion channels |
| CN103443105A (en) * | 2011-02-18 | 2013-12-11 | 沃泰克斯药物股份有限公司 | Chroman-spirocyclic piperidine amides as modulators of ion channels |
| WO2012112743A1 (en) * | 2011-02-18 | 2012-08-23 | Vertex Pharmaceuticals Incorporated | Chroman - spirocyclic piperidine amides as modulators of ion channels |
| CN103517910A (en) * | 2011-03-14 | 2014-01-15 | 沃泰克斯药物股份有限公司 | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| US9181273B2 (en) | 2011-03-14 | 2015-11-10 | Vertex Pharmaceuticals Incorporated | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| WO2012125613A1 (en) * | 2011-03-14 | 2012-09-20 | Vertex Pharmaceuticals Incorporated | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| US8828996B2 (en) | 2011-03-14 | 2014-09-09 | Vertex Pharmaceuticals Incorporated | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| JP2014508169A (en) * | 2011-03-14 | 2014-04-03 | バーテックス ファーマシューティカルズ インコーポレイテッド | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| US9365588B2 (en) | 2011-11-04 | 2016-06-14 | Vertex Pharmaceuticals Incorporated | Benzoxazines as modulators of ion channels |
| WO2013067248A1 (en) * | 2011-11-04 | 2013-05-10 | Vertex Pharmaceuticals Incorporated | Benzoxazines as modulators of ion channels |
| WO2014022639A1 (en) * | 2012-08-01 | 2014-02-06 | Vertex Pharmaceuticals Incorporated | Solid forms of (4-isopropoxy-3-methoxyphenyl)(2'-methyl-6'-(trifluoromethyl)-3',4'-dihydro-2'h-spiro[piperidine-4,1'-pyrrolo[1,2-a]pyrazine]-1-yl)methanone |
| CN105473554A (en) * | 2013-07-10 | 2016-04-06 | 沃泰克斯药物股份有限公司 | Fused piperidine amides as modulators of ion channels |
| JP2016526571A (en) * | 2013-07-10 | 2016-09-05 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Condensed piperidine amides as modulators of ion channels |
| WO2015006280A1 (en) * | 2013-07-10 | 2015-01-15 | Vertex Pharmaceuticals Incorporated | Fused piperidine amides as modulators of ion channels |
| CN105473554B (en) * | 2013-07-10 | 2019-08-13 | 沃泰克斯药物股份有限公司 | Condensed piperidine amide as ion channel modulators |
| US10233191B2 (en) | 2013-07-10 | 2019-03-19 | Vertex Pharmaceuticals Incorporated | Fused piperidine amides as modulators of ion channels |
| JP2019031531A (en) * | 2013-08-26 | 2019-02-28 | パーデュー、ファーマ、リミテッド、パートナーシップ | Azaspiro[4.5] decane derivatives and use thereof |
| WO2015151001A1 (en) | 2014-03-29 | 2015-10-08 | Lupin Limited | Sulfonamide compounds as voltage gated sodium channel modulators |
| US10239869B2 (en) | 2015-09-04 | 2019-03-26 | Lupin Limited | Sulfonamide compounds as voltage-gated sodium channel modulators |
| WO2017037682A1 (en) | 2015-09-04 | 2017-03-09 | Lupin Limited | Sulfonamide compounds as voltage-gated sodium channel modulators |
| WO2018163077A1 (en) | 2017-03-08 | 2018-09-13 | Lupin Limited | Indanyl compounds as voltage gated sodium channel modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010151597A1 (en) | 2010-12-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010151595A1 (en) | Pyrrolo-benzo-1,4-diazines useful as sodium channel blockers | |
| AU2019210624A1 (en) | Cyclopropylamines as lsd1 inhibitors | |
| AU2017382360A1 (en) | Compounds, compositions and methods of use | |
| KR101738866B1 (en) | Cyclic N,N'-diarylthioureas and N,N'-diarylureas as androgen receptor antagonists, anti-cancer agent, method for producing and using same | |
| WO2012047703A2 (en) | Cyclopropyl-spiro-piperidines useful as sodium channel blockers | |
| CN115697327A (en) | 5-oxo-pyrrolidine-3-carboxamides as NAV1.8 inhibitors | |
| EP1957494A2 (en) | Lactam compounds and methods of using the same | |
| CN104024251A (en) | Benzenesulfonamide compounds and their use as therapeutic agents | |
| KR20130029368A (en) | Pyrazolopyridine, pyrazolopyrazine, pyrazolopyrimidine, pyrazolothiophene and pyrazolothiazole compounds as mglur4 allosteric potentiators, compounds, and methods of treating neurological dysfunction | |
| NZ582524A (en) | Spirocycles as inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 comprising compounds with carboxamide and diazaspiro[4.5]dec-7-yl moities | |
| TW202214587A (en) | Cyclopropyl dihydroquinoline sulfonamide compounds | |
| CA2994203A1 (en) | Azaspiro compounds as muscarinic agonists | |
| CN102803268A (en) | Histamine H3 inverse agonists and antagonists and methods of use thereof | |
| JP2024501641A (en) | Substituted Macrocycles and Related Treatment Methods | |
| CN113924092A (en) | Quinoline and quinazoline compounds and methods of use thereof | |
| AU2013202973A1 (en) | Heterocyclic compounds and methods for their use | |
| WO2016062789A1 (en) | New thienopyrimidine derivatives as nik inhibitors | |
| KR20240005878A (en) | Cycloalkyl 3-oxopiperazine carboxamide and cycloheteroalkyl 3-oxopiperazine carboxamide as NAV1.8 inhibitors | |
| EP3828174A1 (en) | Pyridazinone derivative | |
| JP2009511631A (en) | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators | |
| JP5465716B2 (en) | 5- [5- [2- [3- (3,5-Bis (trifluoromethyl) phenyl) -2-methylpropanoylmethylamino] -4- (4-fluoro-2-methylphenyl)] as an NK1 receptor antagonist -2-pyridinyl-2-alkyl-prolinamide | |
| CN112313220B (en) | PD-L1 antagonist compounds | |
| EP3442958A1 (en) | Novel n-[(pyridyloxy)propanyl]benzamides | |
| CN115066423B (en) | PD-L1 antagonist compounds | |
| EP3442963A1 (en) | Novel n-[(pyrazinyloxy)propanyl]benzamides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10792611 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10792611 Country of ref document: EP Kind code of ref document: A1 |