WO2010141247A1 - Hexahydrocyclopentyl[∫]indazole 5-hydroxymethyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators - Google Patents
Hexahydrocyclopentyl[∫]indazole 5-hydroxymethyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators Download PDFInfo
- Publication number
- WO2010141247A1 WO2010141247A1 PCT/US2010/035898 US2010035898W WO2010141247A1 WO 2010141247 A1 WO2010141247 A1 WO 2010141247A1 US 2010035898 W US2010035898 W US 2010035898W WO 2010141247 A1 WO2010141247 A1 WO 2010141247A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- hydroxymethyl
- indazol
- hexahydrocyclopenta
- fluorophenyl
- Prior art date
Links
- QQFYSTPPNQEHMR-UHFFFAOYSA-N CC(CC1)(C2(CC3)OCCO2)C3=CC1=O Chemical compound CC(CC1)(C2(CC3)OCCO2)C3=CC1=O QQFYSTPPNQEHMR-UHFFFAOYSA-N 0.000 description 2
- DGDAXAFEFBPWFP-STEQJIOHSA-N CC(C1)([C@@](CC(c2ccccn2)O)(CO)CC2)C2=Cc2c1cn[n]2-c(cc1)ccc1F Chemical compound CC(C1)([C@@](CC(c2ccccn2)O)(CO)CC2)C2=Cc2c1cn[n]2-c(cc1)ccc1F DGDAXAFEFBPWFP-STEQJIOHSA-N 0.000 description 1
- 0 CC(C1)([C@](C*)(CC(c2ccccn2)O)CC2)C2=Cc2c1cn[n]2-c(cc1)ccc1F Chemical compound CC(C1)([C@](C*)(CC(c2ccccn2)O)CC2)C2=Cc2c1cn[n]2-c(cc1)ccc1F 0.000 description 1
- FNYAZSZTENLTRT-UHFFFAOYSA-N CC(CC1)(C(CC2)=CC1=O)C2=O Chemical compound CC(CC1)(C(CC2)=CC1=O)C2=O FNYAZSZTENLTRT-UHFFFAOYSA-N 0.000 description 1
- SYPBNATWSLUMHH-UHFFFAOYSA-N CC(CC1C=O)(C2(CC3)OCCO2)C3=CC1=O Chemical compound CC(CC1C=O)(C2(CC3)OCCO2)C3=CC1=O SYPBNATWSLUMHH-UHFFFAOYSA-N 0.000 description 1
- UPZFLZYXYGBAPL-UHFFFAOYSA-N CCC1(C)OCCO1 Chemical compound CCC1(C)OCCO1 UPZFLZYXYGBAPL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/08—Antibacterial agents for leprosy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- Intracellular receptors are a class of structurally related proteins involved in the regulation of gene expression.
- the steroid hormone receptors are a subset of this superfamily whose natural ligands are typically comprised of endogenous steroids such as estradiol, progesterone, and Cortisol.
- Man-made ligands to these receptors play an important role in human health and, of these receptors, the glucocorticoid receptor has an essential role in regulating human physiology and immune response.
- Steroids that interact with the glucocorticoid receptor have been shown to be potent anti-inflammatory agents.
- the present invention is directed to a novel class of compounds that are selective glucocorticoid receptor modulators that have potent anti-inflammatory and immunosuppressive activity and possess advantages over steroidal glucocorticoid ligands with respect to side effects, efficacy, toxicity and/or metabolism.
- the present invention encompasses compounds of Formula I:
- compositions and methods of use are also included.
- the present invention encompasses a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, or a pharmaceutically acceptable salt of the stereoisomer thereof:
- each of X 1 and X 2 is independently hydrogen or halogen
- Y is carbon or nitrogen, provided that when Y is nitrogen, then both X 1 and X 2 are hydrogen;
- W is a bond or a methylene group
- Z is hydrogen, -ORa or -NRaRb; each of Rl, R2 and R3 is independently selected from the group consisting of:
- R4 is Cl-6alkyl or C3-6cycloalkyl; each of Ra and Rb is independently selected from the group consisting of:
- HET is selected from the group consisting of:
- each of X 1 and X 2 is independently hydrogen or halogen.
- X 2 is hydrogen.
- X 1 is halogen.
- X 1 is selected from the group consisting of fluoro, chloro, bromo, and iodo. In yet another embodiment, X 1 is fluoro.
- X 2 is hydrogen. In another embodiment, X 2 is selected from the group consisting of fluoro, chloro, bromo, and iodo. In yet another embodiment, X 2 is fluoro.
- Y is carbon or nitrogen.
- X 1 is halogen
- X 2 is hydrogen and Y is nitrogen.
- X 1 is fluoro
- X 2 is hydrogen and Y is carbon.
- W is a bond or methylene. In another embodiment, W is a bond. In yet another embodiment, W is methylene.
- Z is hydrogen, -0R a or - NR a Rb. In one subset of this embodiment, both R a and Rb are hydrogen. In another embodiment, Z is hydrogen. In another embodiment, Z is -0R a . In one subset, R a is hydrogen.
- each of Rl and R2 is independently selected from the group consisting of:
- Rl is hydrogen
- R2 is selected from the group consisting of:
- R3 is hydrogen or Cl-6alkyl. In another embodiment, R3 is hydrogen. In yet another embodiment, R3 is methyl or ethyl.
- R4 is Cl-6alkyl. In another embodiment, R4 is methyl or ethyl. In yet another embodiment, R4 is methyl.
- each of R a and Rb is independently hydrogen or Cl-6alkyl. In another embodiment, each of R a and Rb is independently hydrogen or methyl. In yet another embodiment, each of R a and Rb is hydrogen.
- aryl is phenyl
- each occurrence of HET is independently selected from the group consisting of:
- each occurrence of HET is independently a 5- or 6- membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N.
- each occurrence of HET is independently a 9- or 10- membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N.
- each occurrence of HET is independently selected from the group consisting of: pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, isooxazole, benzofuran, benzothiophene, indole, pyranopyrrole, benzopyran, quionoline, benzocyclohexyl, naphtyridine, benzimidazolyl, benzofuranyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthyridin
- each occurrence of HET is independently selected from the group consisting of: pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, and isooxazole.
- the instant invention encompasses a compound of Formula Ia, or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, or a pharmaceutically acceptable salt of the stereoisomer thereof:
- X 1 is H or halogen
- Y is carbon or nitrogen, provided that when Y is nitrogen, then X 1 is hydrogen;
- W is a bond or a methylene group
- Z is hydrogen, -OH or -NH 2 ; each of Rl and R2 is independently selected from the group consisting of:
- the compound of Formula Ia has Formula Ib:
- R 1 , R 2 , R 3 , W, X 1 , Y, and Z are as defined above under the definitions for Formula Ia.
- X 1 is fluoro and Y is carbon.
- W is a bond or a methylene group. In another embodiment, W is a bond. In another embodiment, W is a methylene group.
- Z is hydrogen or -OH. In another embodiment, Z is hydrogen. In another embodiment, Z is -OH.
- Rl is hydrogen and R2 is selected from the group consisting of:
- R3 is hydrogen or methyl. In another embodiment, R3 is hydrogen.
- each occurrence of HET is independently selected from the group consisting of pyridine, pyrimidine, pyridazine, pyrrolyl, furan, oxazole, isooxazole, benzofuran, indole, benzopyran, dihydrobenzofuranyl, dihydrobenzoxazolyl, dihydrofuranyl, dihydroindolyl, dihydroisooxazolyl, dihydropyridinyl, dihydropyrimidinyl, and dihydropyrrolyl.
- each occurrence of HET is independently selected from the group consisting of pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, and isooxazole.
- each of R a and Rb is independently hydrogen or methyl.
- the invention encompasses a pharmaceutical composition comprising a compound of Formula I, Ia, or Ib in combination with a pharmaceutically acceptable carrier.
- Another embodiment of the invention encompasses a method for treating a glucocorticoid receptor mediated disease or condition in a mammalian patient in need of such treatment comprising administering to the patient a compound of Formula I, Ia, or Ib in an amount that is effective for treating the glucocorticoid receptor mediated disease or condition.
- the glucocorticoid receptor mediated disease or condition is selected from the group consisting of: tissue rejection, leukemias, lymphomas, Cushing's syndrome, acute adrenal insufficiency, congenital adrenal hyperplasia, rheumatic fever, polyarteritis nodosa, granulomatous polyarteritis, inhibition of myeloid cell lines, immune proliferation/apoptosis, HPA axis suppression and regulation, hypercortisolemia, stroke and spinal cord injury, hypercalcemia, hypergylcemia, acute adrenal insufficiency, chronic primary adrenal insufficiency, secondary adrenal insufficiency, congenital adrenal hyperplasia, cerebral edema, thrombocytopenia, Little's syndrome, obesity, metabolic syndrome, inflammatory bowel disease, systemic lupus erythematosus, polyartitis nodosa, Wegener's granulomatosis, giant cell arteritis,
- Another embodiment of the invention encompasses the use of a compound of Formula I, Ia, or Ib for treating of a glucocorticoid receptor mediated disease or condition in a mammalian patient in need of such treatment.
- Another embodiment of the invention encompasses a method of selectively modulating the activation, repression, agonism and antagonism effects of the glucocorticoid receptor in a mammal comprising administering to the mammal a compound of Formula I, Ia, or Ib in an amount that is effective to modulate the glucocorticoid receptor.
- Another embodiment of the invention encompasses the use of a compound of Formula I, Ia, or Ib for selectively modulating the activation, repression, agonism and antagonism effects of the glucocorticoid receptor in a mammal.
- halogen or halo includes F, Cl, Br, and I.
- alkyl means linear or branched structures and combinations thereof, having the indicated number of carbon atoms.
- Cl-6alkyl includes methyl, ethyl, propyl, 2-propyl, s- and t-butyl, butyl, pentyl, hexyl, 1,1-dimethylethyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- alkoxy means alkoxy groups of a straight, branched or cyclic configuration having the indicated number of carbon atoms.
- Cl-6alkoxy for example, includes methoxy, ethoxy, propoxy, isopropoxy, and the like.
- alkenyl means linear or branched structures and combinations thereof, of the indicated number of carbon atoms, having at least one carbon-to-carbon double bond, wherein hydrogen may be replaced by an additional carbon-to-carbon double bond.
- C2-6 a lkenyl for example, includes ethenyl, propenyl, 1-methylethenyl, butenyl and the like.
- alkynyl means linear or branched structures and combinations thereof, of the indicated number of carbon atoms, having at least one carbon-to-carbon triple bond.
- C3- galkynyl for example, includes , propenyl, 1-methylethenyl, butenyl and the like.
- cycloalkyl means mono-, bi- or tri-cyclic structures, optionally combined with linear or branched structures, the indicated number of carbon atoms.
- cycloalkyl groups include cyclopropyl, cyclopentyl, cycloheptyl, adamantyl, cyclododecylmethyl, 2-ethyl-l- bicyclo[4.4.0]decyl, and the like.
- aryl is defined as a mono- or bi-cyclic aromatic ring system and includes, for example, phenyl, naphthyl, and the like.
- optionally substituted means "unsubstituted or substituted," and therefore, the generic structural formulas described herein encompass compounds containing the specified optional substituent as well as compounds that do not contain the optional substituent. Each variable is independently defined each time it occurs within the generic structural formula definitions.
- HET is defined as a 5- to 10-membered aromatic, partially aromatic or non-aromatic mono- or bicyclic ring, containing 1-4 heteroatoms selected from O, S and N, and optionally substituted with 1-2 oxo groups.
- HET is a 5- or 6-membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N, for example, pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, isooxazole and the like
- HET is a 9- or 10-membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N, for example, benzofuran, benzothiophene, indole, pyranopyrrole, benzopyran, quionoline, benzocyclohexyl, naphtyridine and the like.
- HAT also includes the following: benzimidazolyl, benzofuranyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthyridinyl, oxadiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridopyridinyl, pyridazinyl, pyridyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, thiadiazolyl, thiazolyl, thien
- each reference to a group is independent of all other references to the same group when referred to in the Specification.
- Rl and R2 are HET
- the definitions of HET are independent of each other and Rl and R2 may be different HET groups, for example furan and pyridine.
- treating encompasses not only treating a patient to relieve the patient of the signs and symptoms of the disease or condition but also prophylactically treating an asymptomatic patient to prevent the onset of the disease or condition or preventing, slowing or reversing the progression of the disease or condition.
- amount effective for treating is intended to mean that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, a system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- the term also encompasses the amount of a pharmaceutical drug that will prevent or reduce the risk of occurrence of the biological or medical event that is sought to be prevented in a tissue, a system, animal or human by a researcher, veterinarian, medical doctor or other clinician.
- Compounds described herein may contain an asymmetric center and may thus exist as enantiomers. Where the compounds according to the invention possess two or more asymmetric centers, they may additionally exist as diastereomers.
- bonds to the chiral carbon are depicted as straight lines in the formulas of the invention, it is understood that both the (R) and (S) configurations of the chiral carbon, and hence both enantiomers and mixtures thereof, are embraced within the formulas.
- the present invention includes all such possible stereoisomers as substantially pure resolved enantiomers, racemic mixtures thereof, as well as mixtures of diastereomers.
- the present invention includes all stereoisomers of Formula I and Ia and pharmaceutically acceptable salts thereof.
- Diastereoisomeric pairs of enantiomers may be separated by, for example, fractional crystallization from a suitable solvent, and the pair of enantiomers thus obtained may be separated into individual stereoisomers by conventional means, for example by the use of an optically active acid or base as a resolving agent or on a chiral HPLC column. Further, any enantiomer or diastereomer of a compound of the general Formula Ia may be obtained by stereospecif ⁇ c synthesis using optically pure starting materials or reagents of known configuration.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids.
- pharmaceutically acceptable non-toxic bases including inorganic bases and organic bases.
- Salts derived from such inorganic bases include aluminum, ammonium, calcium, copper (ic and ous), ferric, ferrous, lithium, magnesium, manganese (ic and ous), potassium, sodium, zinc and the like salts. Preferred are the ammonium, calcium, magnesium, potassium and sodium salts.
- Salts prepared from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines derived from both naturally occurring and synthetic sources.
- organic non-toxic bases from which salts can be formed include, for example, arginine, betaine, caffeine, choline, N 5 N - dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, dicyclohexylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- the compound of the present invention When the compound of the present invention is basic, its corresponding salt can be conveniently prepared from pharmaceutically acceptable non-toxic inorganic and organic acids.
- Such acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid and the like.
- Preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
- solvates of compounds of Formula I or Ia.
- the term "solvate” refers to a complex of variable stoichiometry formed by a solute (i.e., a compound of Formula I 5 Ia, or Ib) or a pharmaceutically acceptable salt thereof and a solvent that does not interfere with the biological activity of the solute.
- solvents include, but are not limited to water, ethanol, and acetic acid.
- the solvent is water
- hydrates include, but are not limited to, hemi-, mono, sesqui-, di- and trihydrates.
- the present invention includes within its scope the use prodrugs of the compounds of this invention.
- prodrugs will be functional derivatives of the compounds of this invention which are readily convertible in vivo into the required compound.
- the term "administering" shall encompass the treatment of the various conditions described with a compound of Formula I 5 Ia, or Ib or with a compound which may not be a compound of Formula I, Ia, or Ib, but which converts to a compound of Formula I, Ia, or Ib in vivo after administration to the patient.
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs," ed. H. Bundgaard, Elsevier, 1985.
- compositions of the present invention comprise a compound of Formula I, Ia, or Ib as an active ingredient or a pharmaceutically acceptable salt, thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases including inorganic bases and organic bases. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such as
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- Particularly preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
- references to the compounds of Formula I, Ia, or Ib are meant to also include the pharmaceutically acceptable salts.
- prophylactic or therapeutic dose of a compound of Formula I, Ia, or Ib will, of course, vary with the nature and the severity of the condition to be treated and with the particular compound of Formula I, Ia, or Ib and its route of administration. It will also vary according to a variety of factors including the age, weight, general health, sex, diet, time of administration, rate of excretion, drug combination and response of the individual patient. In general, the daily dose from about 0.001 mg to about 100 mg per kg body weight of a mammal, preferably 0.01 mg to about 10 mg per kg. On the other hand, it may be necessary to use dosages outside these limits in some cases.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- a formulation intended for oral administration to humans may contain from about 0.5 mg to about 5 g of active agent compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95 percent of the total composition.
- Dosage unit forms will generally contain from about 1 mg to about 2 g of an active ingredient, typically 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg, or 1000 mg.
- the compound of Formula I, Ia, or Ib may be administered orally, topically, parenterally, by inhalation spray or rectally in dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intrasternal injection or infusion techniques.
- the compound of the invention is effective in the treatment of humans.
- compositions containing the active ingredient may be in a form suitable for oral use, for example, as tablets, troches, lozenges, solutions, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups or elixirs.
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavouring agents, colouring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example, magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate may be employed.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredients is mixed with water- miscible solvents such as propylene glycol, PEGs and ethanol, or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water- miscible solvents such as propylene glycol, PEGs and ethanol
- an oil medium for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions contain the active material in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropyl methylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monoole
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl, p- hydroxybenzoate, one or more colouring agents, one or more flavouring agents, and one or more sweetening agents, such as sucrose, saccharin or aspartame.
- preservatives for example ethyl, or n-propyl, p- hydroxybenzoate, one or more colouring agents, one or more flavouring agents, and one or more sweetening agents, such as sucrose, saccharin or aspartame.
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavouring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol, mannitol,
- the pharmaceutical compositions of the invention may also be in the form of an oil- in- water emulsion.
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxy ethylene sorbitan monooleate.
- the emulsions may also contain sweetening and flavouring agents.
- Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a perservative and flavouring and colouring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butane diol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution. Cosolvents such as ethanol, propylene glycol or polyethylene glycols may also be used.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- the compounds of Formula I, Ia, or Ib may also be administered in the form of suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ambient temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- suitable non-irritating excipient which is solid at ambient temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials are cocoa butter and polyethylene glycols.
- Topical formulations may generally be comprised of a pharmaceutical carrier, cosolvent, emulsifier, penetration enhancer, preservative system, and emollient.
- the compounds of the present invention are useful to treat, prevent or ameliorate the following diseases or conditions: inflammation, tissue rejection, auto-immunity, various malianancies, such as leukemias and lymphomas, Cushing's syndrome, acute adrenal insufficiency, congenital adrenal hyperplasia, rheumatic fever, polyarteritis nodosa, granulomatous polyarteritis, inhibition of myeloid cell lines, immune proliferation/apoptosis, HPA axis suppression and regulation, hypercortisolemia, stroke and spinal cord injury, hypercalcemia, hypergylcemia, acute adrenal insufficiency, chronic primary adrenal insufficiency, secondary adrenal insufficiency, congenital adrenal hyperplasia, cerebral edema, thrombo
- the compounds of the present invention are also useful for treating, preventing or reversing the progression of disease states involving systemic inflammation such as inflammatory bowel disease, systemic lupus erythematosus, polyartitis nodosa, Wegener's granulomatosis, giant cell arteritis, rheumatoid arthritis, juvenile rheumatoid arthritis, uveitis, hay fever, allergic rhinitis, urticaria, angioneurotic edema, chronic obstructive pulmonary disease, asthma, tendonitis, bursitis, Crohn's disease, ulcerative colitis, autoimmune chronic active hepatitis, organ transplantation, hepatitis, and cirrhosis.
- systemic inflammation such as inflammatory bowel disease, systemic lupus erythematosus, polyartitis nodosa, Wegener's granulomatosis, giant cell arteritis, rheumatoi
- the compounds of the present invention are useful for treating, preventing or reversing the progression of a variety of topical diseases such as inflammatory scalp alopecia, panniculitis, psoriasis, discoid lupus erythematosus, inflamed cysts, atopic dermatitis, pyoderma gangrenosum, pemphigus vulgaris, buflous pemphigoid, systemic lupus erythematosus, dermatomyositis, herpes gestationis, eosinophilic fasciitis, relapsing polychondritis, inflammatory vasculitis, sarcoidosis, Sweet's disease, type I reactive leprosy, capillary hemangiomas, contact dermatitis, atopic dermatitis, lichen planus, exfoliative dermatitus, erythema nodosum, acne, hirsutism, toxic epidermal ne
- the compounds of the present invention are also useful in treating, preventing or reversing the progression of disease states associated with Human Immunodeficiency Virus (HIV), cell apoptosis, and cancer including, but not limited to, Kaposi's sarcoma, immune system activation and modulation, desensitization of inflammatory responses, IIL-I expression, natural killer cell development, lymphocytic leukemia, and treatment of retinitis pigmentosa.
- Cognitive and behavioral processes are also susceptible to glucocorticoid therapy where antagonists would potentially be useful in the treatment of processes such as cognitive performance, memory and learning enhancement, depression, addiction, mood disorders, chronic fatigue syndrome, schizophrenia, stroke, sleep disorders, and anxiety.
- the invention also encompasses a method for treating a glucocorticoid receptor mediated disease comprising concomitantly administering to a patient in need of such treatment a compound of Formula I, Ia, or Ib and one or additional more agents.
- a compound of Formula I, Ia, or Ib may be combined with one or more agents selected from the group consisting of: O-agonists (e.g., salmeterol), theophylline, anticholinergics (e.g., atropine and ipratropium bromide), cromolyn, nedocromil and leukotriene modifiers (e.g., montelukast).
- the compounds of Formula I, Ia, or Ib may be combined with one or the following: a salicylate, including acetylsalicylic acid, a non-steroidal antiinflammatory drug, including indomethacin, sulindac, mefenamic, meclofenamic, tolfenamic, tolmetin, ketorolac, dicofenac, ibuprofen, naproxen, fenoprofen, ketoprofen, flurbiprof ⁇ n and oxaprozin, a TNF inhibitor, including etanercept and infliximab, an IL-I receptor antagonist, a cytotoxic or immunosuppressive drug, including methotrexate, leflunomide, azathioprine and cyclosporine, a gold compound, hydroxychloroquine or sulfasalazine, penicillamine, darbufelone, and a p38 kinas
- the compound of Formula I, Ia, or Ib may also be used in combination with bisphonates such as alendronate to treat a glucocorticoid mediated disease and simultaneously inhibit osteoclast-mediated bone resorption.
- bisphonates such as alendronate
- melting points are uncorrected and M' indicates decomposition; the melting points given are those obtained for the materials prepared as described; polymorphism may result in isolation of materials with different melting points in some preparations;
- NMR data when given, NMR data is in the form of delta ( ⁇ ) values for major diagnostic protons, given in parts per million (ppm) relative to tetramethylsilane (TMS) as internal standard, determined at 500 MHz or 600 MHz using the indicated solvent, conventional abbreviations used for signal shape are: s. singlet; d. doublet; t. triplet; m. multiplet; and br. broad.
- ppm parts per million
- Tr tetramethylsilane
- reaction schemes and examples described herein illustrate the methods employed in the synthesis of the compounds of the present invention. In some cases the order of carrying out the reaction schemes may be varied to facilitate the reaction or to avoid unwanted reaction products.
- the synthesis of the novel compounds which are the subject of this invention may be accomplished by one or more of several similar routes. The following examples are provided so that the invention might be more fully understood. These examples are illustrative only and should not be construed as limiting the invention in any way.
- Step I 1-1 Ia and 1-1 Ib were separated at Step I with the TIPS (triisopropylsilyl) group still on.
- the two diastereoisomers Ex. Ia and Ex. Ib were prepared separately at Step J from 1-1 Ia and 1-1 Ib. respectively.
- Step A (7 ⁇ 'S)-7 ⁇ '-Methyl-2'.3'J'.7 ⁇ '-tetrahvdrospiro ⁇ .3-dioxolane-2J'-indenl-5 ⁇ 6 ⁇ )-one
- Step D (4 ⁇ R.5 S)-I -(4-Fluorophenyl)-4 ⁇ -methyl-lA4 ⁇ .5.6.7- hexahvdrocvclopentarflindazole-S-carbaldehvde (1-6):
- Step G (4 ⁇ t S'.5i?)-l-(4-Fluorophenvn-4a-methyl-5-(prop-2-en-l-vn-5-(r(tripropan-2- ylsilyl)oxylmethvU-l,4,4a,5,6,7-hexahydrocvclopentar/1indazole (1-9):
- Step H [(4a6',5i?)-l-(4-fluorophenyl)-4a-methyl-5- ⁇ [(tripropan-2-ylsilyl)oxylmethyU- lA4a,5A7-hexahydrocyclopenta[/1indazol-5-yl]acetaldehyde (1-10):
- Osmium tetroxide (0.12 mL, 0.394 mmol) was added to a solution of 1 ⁇ 9 (0.65 g, 1.314 mmol) and N-methylmorpholine-N-oxide (0.154 g, 1.314 mmol) in acetone:water (10:1, 3.3 mL) and the reaction was stirred at room temperature for 2 hours. The reaction was diluted with water and extracted with CH 2 Cl 2 . The combined organic extracts were dried over anhydrous MgSO 4 and the solvent removed in vacuo.
- Step I 2-r(4a6'.5i?)-l-(4-fluorophenv ⁇ -4a-methyl-5-(r(tripropan-2-ylsilv ⁇ oxy1methvU- l ⁇ a ⁇ J-hexahvdrocvclopentar/iindazol-S-yll-l-fpyridin-l-v ⁇ ethanol (1-1 Ia and 1-l lb):
- n-Butyl lithium (0.109 ml, 1.148 mmol) was added to a cooled solution of 2- bromopyridine (0.181 g, 1.148 mmol) in diethyl ether (2 mL) at -78°C and the resulting solution was stirred at that temperature for 15 min.
- a solution of 1-10 (0.285 g, 0.574 mmol) in diethyl ether (2 mL) was added and the reaction was stirred at -78°C for 1 hour and then warmed to room temperature and stirred for 1 hour.
- the reaction was quenched by the addition of a saturated solution OfNH 4 Cl and extracted with EtOAc. The combined organics were dried over anhydrous MgSO 4 and the solvent was removed in vacuo.
- Step J 2-r(4aS.5R)4-(4-Fluorophenyl)-5-(hvdroxymethyl)-4a-methyl-1.4.4a.5.6.7- hexahydrocyclopenta[/1indazol-5-yll-l-(pyridin-2-yl)ethanol (Ex. Ia and Ex. Ib):
- Ex. Ib (Isomer 2) was made from 1-1 Ib following a similar procedure as that for Ex. Ia.
- Examples 2a - 29b in Table 1 were prepared following the general synthetic schemes and procedures analogous to Examples Ia and Ib described above. In some cases (e.g. Examples 2a and 2b), the two diastereoisomers were separated at the last step after the TIPS group was removed using standard chromatographic techniques (Hexanes-Ethyl Acetate on silica gel, or Acetonitrile-water on reverse phase HPLC).
- the compounds exemplified in the present application exhibited activity in one or more of the following assays.
- TEGM (10 mM Tris-HCl, 1 mM EDTA, 10% glycerol, 1 mM beta-mecaptoethanol, 10 mM Sodium Molybdate, pH 7.2)
- Molybdate Molybdic Acid (Sigma, M 1651)
- HeLa ATCC
- RPMI 1640 Gibco 11835-055
- human insulin Sigma, 1-0259
- FBS FBS
- 20 ug/ml of Gentamicin Gibco# 15710-072
- Phenol red-free Trypsin-EDTA is diluted in the same PBS 1 :10.
- the cell layers are rinsed with IX Trypsin, extra Trypsin is poured out, and the cell layers are incubated at 37 0 C for ⁇ 2 min.
- the flask is tapped and checked for signs of cell detachment. Once the cells begin to slide off the flask, the complete media is added. The cells are counted at this point, then diluted to the appropriate concentration and split into flasks or dishes for further culturing (Usually 1 :3 to 1 :6 dilution).
- the cells When the cells are 70 to 85% confluent, they are detached as described above, and collected by centrifuging at 1000 g for 10 minutes at 4 0 C.
- the cell pellet is washed twice with TEGM (10 mM Tris-HCl, 1 mM EDTA, 10% glycerol, 1 mM beta-mercaptoethanol, 10 mM Sodium Molybdate, pH 7.2). After the final wash, the cells are resuspended in TEGM at a concentration of 107 cells/mL.
- the cell suspension is snap frozen in liquid nitrogen or ethano I/dry ice bath and transferred to -80 0 C freezer on dry ice.
- the frozen samples are left on ice-water to just thaw ( ⁇ 1 hr). Then the samples are centrifuged at 12,500 g to 20,000 g for 30 min at 4 0 C. The supernatant is used to set-up assay right away. If using 50 ⁇ L of supernatant, the test compound can be prepared in 50 ⁇ L of the TEGM buffer.
- Ix TEGM buffer is prepared, and the isotope-containing assay mixture is prepared in the following order: EtOH (2% final concentration in reaction), 3H-DEX (Amersham Biosciences) and Ix TEGM. [e.g. For 100 samples, 200 ⁇ L (100 x 2) of EtOH + 4.25 ⁇ L of 1 :10 3H-Dex stock + 2300 ⁇ L (100 x 23) Ix TEGM].
- the compound is serially diluted, e.g., if starting final cone, is 1 ⁇ M, and the compound is in 25 ⁇ L of solution, for duplicate samples, 75 ⁇ L of 4x1 ⁇ M solution is made and 3 ⁇ L of 100 ⁇ M is added to 72 ⁇ L of buffer, and 1 :5 serial dilution.
- the HAP pellet on the filter plate is incubated with 50 ⁇ L of MICRO SCINT (Packard) scintillint for 30 minutes before being counted on the TopCount micro scintillation counter (Packard).
- IC50S are calculated using DEX as a reference.
- This assay assesses the ability of test compounds to control transcription from the MMTV-LUC reporter gene in lung adenocarcinoma A549 cells or HeLa cells, a human breast cancer cell line that naturally expresses the human GR.
- the assay measures induction of a modified MMTV LTR/promoter linked to the LUC reporter gene.
- the routine transient assay consists of plating 7,000-25,000 cells/well of a white, clear-bottom 96-well plate. Alternatively, 384-well plates can be used at a cell concentration of 10,000 /well.
- the media that the cells are plated in is "exponential growth medium" which consists of phenol red-free RPMI1640 containing 10%FBS, 4mM L-glutamine, 2OmM HEPES, lOug/mL human insulin, and 20ug/mL gentamicin. Incubator conditions are 37 0 C and 5% C ⁇ 2- The transfection is done in batch mode. The cells are trypsinized and counted to the right cell number in the proper amount of fresh media.
- the transfection cocktail consists of serum- free OptiMEM, FuGene ⁇ reagent and DNA.
- the manufacturer's (Roche Biochemical) protocol for cocktail setup is as follows: The lipid to DNA ratio is approximately 2.5:1 and the incubation time is 20 min at room temperature. Sixteen to 24 hours after transfection, the cells are treated with dexamethasone to a final concentration of 1OnM as well as the compound of interest, such that final DMSO (vehicle) concentration is equal to or less than 1%.
- Each plate also contains samples that are treated with 1OnM dexamethasone alone, which is used as the 100% activity control.
- the cells are exposed to the compounds for 24 hours. After 24 hours, the cells are lysed by a Promega cell culture lysis buffer for approximately 30 min and then the lucif erase activity in the extracts is assayed in the 96-well format luminometer.
- Steady-Glo Promega
- Steady- Lite PerkinElmer
- Antagonist activity is calculated by determining the decrease in dexamethasone-induced activity in response to compound treatment relative to samples that were treated with dexamethasone alone. Results are expressed as % inhibition of 1OnM dexamethasone activity or as fold of 1OnM dexamethasone activity. This transactivation assay can be performed in an agonist and antagonist mode to identify these different activities.
- Activity of test compounds is calculated as the E ma ⁇ relative to the activity obtained with 300 nM dexamethasone.
- Activity of test compounds is calculated as the E ma ⁇ relative to the activity obtained with 300 nM DEX.
- the exemplified tissue selective glucocorticoid receptor modulators of the present invention display agonist activity in this assay of greater than 5% and less than 100%, and maximal transactivation activity less then maximal transrepression activity.
- Anti-GRAMMER an antagonist mode in which the cells are treated with medium containing an agonist such as 10 nM DEX and the ability to agents to inhibit the activation by an agonist is measured.
- This assay assesses the ability of test compounds to control transcription from the TNF ⁇ - ⁇ -lactamase reporter gene in U937 cells, a human myelomonocytic leukemia cell line that naturally expresses the human GR.
- the assay measures compound dependent-repression of the TNFa promoter linked to a reporter gene.
- U937 cells that had been stablely trans fected with the TNF- ⁇ promoter driving ⁇ -lactamase are used for this assay.
- U937 cells contain an endogenous glucocorticoid receptor (GR).
- GR glucocorticoid receptor
- Cells are maintained in RPMI 1640 Growth medium (Gibco Cat#l 1875-093) containing 25mM HEPES, 10% FBS, 2mM L-Glutamine, ImM Sodium pyruvate, 25 ⁇ g/ml Gentamicin (Gibco Cat#15710-064), 1 :1000 2-Mercaptoethanol (Gibco Cat#21985-023) and 0.8 mg/ml G418 (Gibco Cat#10131-027).
- RPMI 1640 Growth medium Gibco Cat#l 1875-093
- 25mM HEPES 10% FBS
- 2mM L-Glutamine ImM Sodium pyruvate
- the density of the cells in the flask needs to be about 1X106 - 3X106/ml at the time of harvest.
- the cells are split to 1.2-1.4x105 /ml (1 : 10) 3 days prior to the assay.
- 50,000 cells/well are plated in 96 well black-walled plates the day of assay.
- Test compounds are added 10 ⁇ L/well, and cells are incubated at 37oC for 30-45 min. For assaying compounds, first dilute 1 : 10 in DMSO to make 1 mM, then further dilute 1 : 100 in medium to make 1OX stock prior to adding to the cells.
- This assay assesses the ability of test compounds to modulate the transcription of endogenously expressed genes in a variety of cell types including but not limited to A549, HeLa or U937 cells. All cell culture reagents were purchased from Invitrogen Life Tech, Carlsbad CA. A549 cells were grown in phenol red-free DMEM/F12 medium supplemented with 10% FBS. Cells were grown at 37oC with 5% CO2. Using the RNeasy Kit (Qiagen Corp, Valencia CA.), total RNA was extracted and purified from A549 cells treated with different GC compounds for 24 hours, at a fully active dose. These cells express large amount of the GR and are very responsive to GC treatment. All samples were compared against cells treated with vehicle.
- Intact adult (6 month old) female Sprague-Dawley rats are used in the oxazolone (OX) contactdermatitis model. Rats were sensitized on the ventral abdomen with OX on Day 0. On Days 7 and 9, a randomly-selected ear was challenged (same ear each time) with OX; the other was treated with vehicle. Daily treatment begun on Day 7 and continued for 7d with test compounds at different doses and 1.3 mpk 6-methlyprednisolone or O.lmpk DEX as positive controls. The thickness of both ears are measured on Days 11 and 14. Necropsy occurred on Day 14. The rat is first weighed, then anesthetized in a CO2 chamber until near death.
- OX oxazolone
- Inter-ear thickness difference (etd) is used for the estimating the level of inflammation and effectiveness of the compounds is determined by their ability to reduce the increase the thickness of the inflamed ear.
- etd Inter-ear thickness difference
- Back of the rat skin thickness, spleen weight, serum insulin as well as the effects of gcs on the expression of molecular markers in skin inflammation, skin atrophy, muscle atrophy and glucose metabolism in liver are measured.
- Data are analyzed by anova plus fisher plsd post-hoc test to identify intergroup differences.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Virology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Dermatology (AREA)
- Psychiatry (AREA)
- Pulmonology (AREA)
- Immunology (AREA)
- Obesity (AREA)
- Molecular Biology (AREA)
- Endocrinology (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- Transplantation (AREA)
- Biotechnology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Ophthalmology & Optometry (AREA)
- Hospice & Palliative Care (AREA)
- Child & Adolescent Psychology (AREA)
Abstract
The present invention encompasses compounds of Formula (I): or pharmaceutically acceptable salts or hydrates thereof, which are useful as selective glucocorticoid receptor ligands for treating a variety of autoimmune and inflammatory diseases or conditions. Pharmaceutical compositions and methods of use are also included.
Description
TITLE OF THE INVENTION
HEXAHYDROCYCLOPENTYL[Z]INDAZOLE 5-HYDROXYMETHYL ETHANOLS AND
DERIVATIVES THEREOF AS SELECTIVE GLUCOCORTICOID RECEPTOR MODULATORS
BACKGROUND OF THE INVENTION
Intracellular receptors (IRs) are a class of structurally related proteins involved in the regulation of gene expression. The steroid hormone receptors are a subset of this superfamily whose natural ligands are typically comprised of endogenous steroids such as estradiol, progesterone, and Cortisol. Man-made ligands to these receptors play an important role in human health and, of these receptors, the glucocorticoid receptor has an essential role in regulating human physiology and immune response. Steroids that interact with the glucocorticoid receptor have been shown to be potent anti-inflammatory agents. The present invention is directed to a novel class of compounds that are selective glucocorticoid receptor modulators that have potent anti-inflammatory and immunosuppressive activity and possess advantages over steroidal glucocorticoid ligands with respect to side effects, efficacy, toxicity and/or metabolism.
SUMMARY OF THE INVENTION
The present invention encompasses compounds of Formula I:

DETAILED DESCRIPTION OF THE INVENTION
The present invention encompasses a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, or a pharmaceutically acceptable salt of the stereoisomer thereof:

Y is carbon or nitrogen, provided that when Y is nitrogen, then both X1 and X2 are hydrogen;
W is a bond or a methylene group;
Z is hydrogen, -ORa or -NRaRb; each of Rl, R2 and R3 is independently selected from the group consisting of:
(1) hydrogen,
(2) Cl-6alkyl,
(3) C3-6cycloalkyl,
(4) Cl-6alkyl-phenyl,
(5) Cl-6alkyl-HET,
(6) aryl, and
(7) HET, wherein each of items (2) to (3), items (6) to (7), the aryl portion of item (4), and the HET portion of item (5) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa,
(e) oxo,
(f) -C(O)NRaRb,
(g) -NRaRb, and (h) -SO2Ra;
R4 is Cl-6alkyl or C3-6cycloalkyl; each of Ra and Rb is independently selected from the group consisting of:
(1) hydrogen,
(2) Cl-6alkyl,
-2-
(3) aryl, and
(4) HET; and
HET is selected from the group consisting of:
(1) a 5- or 6-membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N, and
(2) a 9- or 10-membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N.
In one embodiment of compounds of Formula I, each of X1 and X2 is independently hydrogen or halogen.
In another embodiment of compounds of Formula I, X2 is hydrogen. In another embodiment, X1 is halogen. In another embodiment, X1 is selected from the group consisting of fluoro, chloro, bromo, and iodo. In yet another embodiment, X1 is fluoro.
In another embodiment of compounds of Formula I, X2 is hydrogen. In another embodiment, X2 is selected from the group consisting of fluoro, chloro, bromo, and iodo. In yet another embodiment, X2 is fluoro.
In one embodiment of compounds of Formula I, Y is carbon or nitrogen.
In another embodiment, X1 is halogen, X2 is hydrogen and Y is nitrogen. In yet another embodiment, X1 is fluoro, X2 is hydrogen and Y is carbon.
In one embodiment of compounds of Formula I, W is a bond or methylene. In another embodiment, W is a bond. In yet another embodiment, W is methylene.
In one embodiment of compounds of Formula I, Z is hydrogen, -0Ra or - NRaRb. In one subset of this embodiment, both Ra and Rb are hydrogen. In another embodiment, Z is hydrogen. In another embodiment, Z is -0Ra. In one subset, Ra is hydrogen.
In one embodiment of compounds of Formula I, each of Rl and R2 is independently selected from the group consisting of:
(1) hydrogen,
(2) Cl-6alkyl,
(3) Cl-6cycloalkyl,
(4) Cl-6alkyl-aryl,
(5) Cl_6alkyl-HET,
(6) aryl, and
(7) HET, wherein each of items (2) to (3), items (6) to (7), the aryl portion of item (4), and the HET portion of item (5) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa,
(e) oxo,
(f) -C(O)NRaRb, and
(g) -NRaRb.
In another embodiment, Rl is hydrogen, and R2 is selected from the group consisting of:
(1) Cl-6alkyl,
(2) Cl-6cycloalkyl,
(3) Cl-6alkyl-aryl,
(4) Cl-6alkyl-HET,
(5) aryl, and
(6) HET, wherein each of items (2), (5) and (6), the aryl portion of item (3), and the HET portion of item (4) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa, and
(e) oxo.
In one embodiment of compounds of Formula I, R3 is hydrogen or Cl-6alkyl. In another embodiment, R3 is hydrogen. In yet another embodiment, R3 is methyl or ethyl.
In one embodiment of compounds of Formula I, R4 is Cl-6alkyl. In another embodiment, R4 is methyl or ethyl. In yet another embodiment, R4 is methyl.
In one embodiment of compounds of Formula I, each of Ra and Rb is independently hydrogen or Cl-6alkyl. In another embodiment, each of Ra and Rb is independently hydrogen or methyl. In yet another embodiment, each of Ra and Rb is hydrogen.
In one embodiment of compounds of Formula I, aryl is phenyl.
In one embodiment of compounds of Formula I, each occurrence of HET is independently selected from the group consisting of:
(1) a 5- or 6-membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N, and
(2) a 9- or 10-membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N.
In another embodiment, each occurrence of HET is independently a 5- or 6- membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N.
In another embodiment, each occurrence of HET is independently a 9- or 10- membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N.
In yet another embodiment, each occurrence of HET is independently selected from the group consisting of: pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, isooxazole, benzofuran, benzothiophene, indole, pyranopyrrole, benzopyran, quionoline, benzocyclohexyl, naphtyridine, benzimidazolyl, benzofuranyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthyridinyl, oxadiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridopyridinyl, pyridazinyl, pyridyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, azetidinyl, 1 ,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyrrolidinyl, morpholinyl, thiomorpholinyl, dihydrobenzimidazolyl, dihydrobenzofuranyl, dihydrobenzothiophenyl, dihydrobenzoxazolyl, dihydrofuranyl, dihydroimidazolyl, dihydroindolyl, dihydroisooxazolyl, dihydroisothiazolyl, dihydrooxadiazolyl, dihydrooxazolyl, dihydropyrazinyl, dihydropyrazolyl, dihydropyridinyl, dihydropyrimidinyl, dihydropyrrolyl, dihydroquinolinyl, dihydrotetrazolyl, dihydrothiadiazolyl, dihydrothiazolyl, dihydrothienyl, dihydrotriazolyl, dihydroazetidinyl, methylenedioxybenzoyl, tetrahydrofuranyl, and tetrahydrothienyl.
In still another embodiment, each occurrence of HET is independently selected from the group consisting of: pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, and isooxazole.
In one embodiment, the instant invention encompasses a compound of Formula Ia, or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, or a pharmaceutically acceptable salt of the stereoisomer thereof:

X1 is H or halogen;
Y is carbon or nitrogen, provided that when Y is nitrogen, then X1 is hydrogen;
W is a bond or a methylene group;
Z is hydrogen, -OH or -NH2; each of Rl and R2 is independently selected from the group consisting of:
(1) hydrogen,
(2) Cl-6alkyl,
(3) C3-6cycloalkyl,
(4) Cl-6alkyl-phenyl,
(5) Cl-6alkyl-HET,
(6) phenyl, and
(7) HET, wherein each of items (2) to (3), items (6) to (7), the phenyl portion of item (4), and the HET portion of item (5) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa,
(e) oxo,
(f) -C(O)NRaRb, and
(g) -NRaRb; R3 is hydrogen or methyl; each of Ra and Rb is independently hydrogen or methyl; and each occurrence of HET is independently selected from the group consisting of:
(1) a 5- or 6-membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N, and
(2) a 9- or 10-membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N.
In one embodiment, the compound of Formula Ia has Formula Ib:

In one embodiment of compounds of Formula Ia or Ib, X1 is fluoro and Y is carbon.
In one embodiment of compounds of Formula Ia or Ib, W is a bond or a methylene group. In another embodiment, W is a bond. In another embodiment, W is a methylene group.
In one embodiment of compounds of Formula Ia or Ib, Z is hydrogen or -OH. In another embodiment, Z is hydrogen. In another embodiment, Z is -OH.
In one embodiment of compounds of Formula Ia or Ib, Rl is hydrogen and R2 is selected from the group consisting of:
(1) Cl-6alkyl,
(2) Cl-6cycloalkyl,
(3) Cl-6alkyl-phenyl,
(4) Cl-6alkyl-HET,
(5) phenyl, and
(6) HET, wherein each of items (2), (5) and (6), the phenyl portion of item (3), and the HET portion of item (4) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa, and
(e) oxo.
In one embodiment of compounds of Formula Ia or Ib, R3 is hydrogen or methyl. In another embodiment, R3 is hydrogen.
In one embodiment of compounds of Formula Ia or Ib, each occurrence of HET is independently selected from the group consisting of pyridine, pyrimidine, pyridazine, pyrrolyl, furan, oxazole, isooxazole, benzofuran, indole, benzopyran, dihydrobenzofuranyl, dihydrobenzoxazolyl, dihydrofuranyl, dihydroindolyl, dihydroisooxazolyl, dihydropyridinyl, dihydropyrimidinyl, and dihydropyrrolyl.
In another embodiment, each occurrence of HET is independently selected from the group consisting of pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, and isooxazole.
In one embodiment of compounds of Formula Ia or Ib, each of Ra and Rb is independently hydrogen or methyl.
In one embodiment, the invention encompasses a pharmaceutical composition comprising a compound of Formula I, Ia, or Ib in combination with a pharmaceutically acceptable carrier.
Another embodiment of the invention encompasses a method for treating a glucocorticoid receptor mediated disease or condition in a mammalian patient in need of such treatment comprising administering to the patient a compound of Formula I, Ia, or Ib in an amount that is effective for treating the glucocorticoid receptor mediated disease or condition.
Within this embodiment is encompassed the above method wherein the glucocorticoid receptor mediated disease or condition is selected from the group consisting of: tissue rejection, leukemias, lymphomas, Cushing's syndrome, acute adrenal insufficiency, congenital adrenal hyperplasia, rheumatic fever, polyarteritis nodosa, granulomatous polyarteritis, inhibition of myeloid cell lines, immune proliferation/apoptosis, HPA axis suppression and regulation, hypercortisolemia, stroke and spinal cord injury, hypercalcemia, hypergylcemia, acute adrenal insufficiency, chronic primary adrenal insufficiency, secondary adrenal insufficiency, congenital adrenal hyperplasia, cerebral edema, thrombocytopenia, Little's syndrome, obesity, metabolic syndrome, inflammatory bowel disease, systemic lupus erythematosus, polyartitis nodosa, Wegener's granulomatosis, giant cell arteritis, rheumatoid arthritis, juvenile rheumatoid arthritis, uveitis, hay fever, allergic rhinitis, urticaria, angioneurotic edema, chronic obstructive pulmonary disease, asthma, tendonitis, bursitis, Crohn's disease, ulcerative colitis, autoimmune chronic active hepatitis, organ transplantation, hepatitis, cirrhosis, inflammatory scalp alopecia, panniculitis, psoriasis, discoid lupus erythematosus, inflamed cysts, atopic dermatitis, pyoderma gangrenosum, pemphigus vulgaris, buflous pemphigoid, systemic lupus erythematosus, dermatomyositis, herpes gestationis, eosinophilic fasciitis, relapsing polychondritis, inflammatory vasculitis, sarcoidosis, Sweet's disease, type I reactive leprosy, capillary hemangiomas, contact dermatitis, atopic dermatitis, lichen planus, exfoliative dermatitus, erythema nodosum, acne, hirsutism, toxic
epidermal necrolysis, erythema multiform, cutaneous T-cell lymphoma, Human Immunodeficiency Virus (HIV), cell apoptosis, cancer, Kaposi's sarcoma, retinitis pigmentosa, cognitive performance, memory and learning enhancement, depression, addiction, mood disorders, chronic fatigue syndrome, schizophrenia, sleep disorders, and anxiety.
Another embodiment of the invention encompasses the use of a compound of Formula I, Ia, or Ib for treating of a glucocorticoid receptor mediated disease or condition in a mammalian patient in need of such treatment.
Another embodiment of the invention encompasses a method of selectively modulating the activation, repression, agonism and antagonism effects of the glucocorticoid receptor in a mammal comprising administering to the mammal a compound of Formula I, Ia, or Ib in an amount that is effective to modulate the glucocorticoid receptor.
Another embodiment of the invention encompasses the use of a compound of Formula I, Ia, or Ib for selectively modulating the activation, repression, agonism and antagonism effects of the glucocorticoid receptor in a mammal.
The invention is described using the following definitions unless otherwise indicated.
The term "halogen" or "halo" includes F, Cl, Br, and I.
The term "alkyl" means linear or branched structures and combinations thereof, having the indicated number of carbon atoms. Thus, for example, Cl-6alkyl includes methyl, ethyl, propyl, 2-propyl, s- and t-butyl, butyl, pentyl, hexyl, 1,1-dimethylethyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
The term "alkoxy" means alkoxy groups of a straight, branched or cyclic configuration having the indicated number of carbon atoms. Cl-6alkoxy, for example, includes methoxy, ethoxy, propoxy, isopropoxy, and the like.
The term "alkenyl" means linear or branched structures and combinations thereof, of the indicated number of carbon atoms, having at least one carbon-to-carbon double bond, wherein hydrogen may be replaced by an additional carbon-to-carbon double bond. C2-6alkenyl, for example, includes ethenyl, propenyl, 1-methylethenyl, butenyl and the like.
The term "alkynyl" means linear or branched structures and combinations thereof, of the indicated number of carbon atoms, having at least one carbon-to-carbon triple bond. C3- galkynyl, for example, includes , propenyl, 1-methylethenyl, butenyl and the like.
The term "cycloalkyl" means mono-, bi- or tri-cyclic structures, optionally combined with linear or branched structures, the indicated number of carbon atoms. Examples of cycloalkyl groups include cyclopropyl, cyclopentyl, cycloheptyl, adamantyl, cyclododecylmethyl, 2-ethyl-l- bicyclo[4.4.0]decyl, and the like.
The term "aryl" is defined as a mono- or bi-cyclic aromatic ring system and includes, for example, phenyl, naphthyl, and the like.
The term "optionally substituted" means "unsubstituted or substituted," and therefore, the generic structural formulas described herein encompass compounds containing the specified optional substituent as well as compounds that do not contain the optional substituent. Each variable is independently defined each time it occurs within the generic structural formula definitions.
The term "HET" is defined as a 5- to 10-membered aromatic, partially aromatic or non-aromatic mono- or bicyclic ring, containing 1-4 heteroatoms selected from O, S and N, and optionally substituted with 1-2 oxo groups. Preferably, "HET" is a 5- or 6-membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N, for example, pyridine, pyrimidine, pyridazine, furan, thiophene, thiazole, oxazole, isooxazole and the like, or HET is a 9- or 10-membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N, for example, benzofuran, benzothiophene, indole, pyranopyrrole, benzopyran, quionoline, benzocyclohexyl, naphtyridine and the like. "HET" also includes the following: benzimidazolyl, benzofuranyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthyridinyl, oxadiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridopyridinyl, pyridazinyl, pyridyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolyl, quinoxalinyl, thiadiazolyl, thiazolyl, thienyl, triazolyl, azetidinyl, 1 ,4-dioxanyl, hexahydroazepinyl, piperazinyl, piperidinyl, pyrrolidinyl, morpholinyl, thiomorpholinyl, dihydrobenzimidazolyl, dihydrobenzofuranyl, dihydrobenzothiophenyl, dihydrobenzoxazolyl, dihydrofuranyl, dihydroimidazolyl, dihydroindolyl, dihydroisooxazolyl, dihydroisothiazolyl, dihydrooxadiazolyl, dihydrooxazolyl, dihydropyrazinyl, dihydropyrazolyl, dihydropyridinyl, dihydropyrimidinyl, dihydropyrrolyl, dihydroquinolinyl, dihydrotetrazolyl, dihydrothiadiazolyl, dihydrothiazolyl, dihydrothienyl, dihydrotriazolyl, dihydroazetidinyl, methylenedioxybenzoyl, tetrahydrofuranyl, and tetrahydrothienyl.
For all of the above definitions, each reference to a group is independent of all other references to the same group when referred to in the Specification. For example, if both Rl and R2 are HET, the definitions of HET are independent of each other and Rl and R2 may be different HET groups, for example furan and pyridine.
The term "treating" encompasses not only treating a patient to relieve the patient of the signs and symptoms of the disease or condition but also prophylactically treating an asymptomatic patient to prevent the onset of the disease or condition or preventing, slowing or reversing the progression of the disease or condition. The term "amount effective for treating" is
intended to mean that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, a system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician. The term also encompasses the amount of a pharmaceutical drug that will prevent or reduce the risk of occurrence of the biological or medical event that is sought to be prevented in a tissue, a system, animal or human by a researcher, veterinarian, medical doctor or other clinician.
Optical Isomers - Diastereomers - Geometric Isomers - Tautomers
Compounds described herein may contain an asymmetric center and may thus exist as enantiomers. Where the compounds according to the invention possess two or more asymmetric centers, they may additionally exist as diastereomers. When bonds to the chiral carbon are depicted as straight lines in the formulas of the invention, it is understood that both the (R) and (S) configurations of the chiral carbon, and hence both enantiomers and mixtures thereof, are embraced within the formulas. The present invention includes all such possible stereoisomers as substantially pure resolved enantiomers, racemic mixtures thereof, as well as mixtures of diastereomers. The present invention includes all stereoisomers of Formula I and Ia and pharmaceutically acceptable salts thereof.
Diastereoisomeric pairs of enantiomers may be separated by, for example, fractional crystallization from a suitable solvent, and the pair of enantiomers thus obtained may be separated into individual stereoisomers by conventional means, for example by the use of an optically active acid or base as a resolving agent or on a chiral HPLC column. Further, any enantiomer or diastereomer of a compound of the general Formula Ia may be obtained by stereospecifϊc synthesis using optically pure starting materials or reagents of known configuration.
When compounds described herein contain olefϊnic double bonds, unless specified otherwise, such double bonds are meant to include both E and Z geometric isomers.
Some of the compounds described herein may exist with different points of attachment of hydrogen, referred to as tautomers. For example, compounds including carbonyl -CH2C(O)- groups (keto forms) may undergo tautomerism to form hydroxyl -CH=C(OH)- groups (enol forms). Both keto and enol forms, individually as well as mixtures thereof, are included within the scope of the present invention.
Salts
The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids. When the compound of the present invention is acidic, its corresponding salt can be conveniently prepared from pharmaceutically acceptable non-
toxic bases, including inorganic bases and organic bases. Salts derived from such inorganic bases include aluminum, ammonium, calcium, copper (ic and ous), ferric, ferrous, lithium, magnesium, manganese (ic and ous), potassium, sodium, zinc and the like salts. Preferred are the ammonium, calcium, magnesium, potassium and sodium salts. Salts prepared from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines derived from both naturally occurring and synthetic sources. Pharmaceutically acceptable organic non-toxic bases from which salts can be formed include, for example, arginine, betaine, caffeine, choline, N5N - dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, dicyclohexylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
When the compound of the present invention is basic, its corresponding salt can be conveniently prepared from pharmaceutically acceptable non-toxic inorganic and organic acids. Such acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid and the like. Preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
Solvates
The present invention includes within its scope solvates of compounds of Formula I or Ia. As used herein, the term "solvate" refers to a complex of variable stoichiometry formed by a solute (i.e., a compound of Formula I5 Ia, or Ib) or a pharmaceutically acceptable salt thereof and a solvent that does not interfere with the biological activity of the solute. Examples of solvents include, but are not limited to water, ethanol, and acetic acid. When the solvent is water, the solvate is known as hydrate; hydrates include, but are not limited to, hemi-, mono, sesqui-, di- and trihydrates.
Prodrugs
The present invention includes within its scope the use prodrugs of the compounds of this invention. In general, such prodrugs will be functional derivatives of the compounds of this invention which are readily convertible in vivo into the required compound. Thus, in the methods of treatment of the present invention, the term "administering" shall encompass the treatment of the various conditions described with a compound of Formula I5 Ia, or Ib or with a compound which
may not be a compound of Formula I, Ia, or Ib, but which converts to a compound of Formula I, Ia, or Ib in vivo after administration to the patient. Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs," ed. H. Bundgaard, Elsevier, 1985.
The pharmaceutical compositions of the present invention comprise a compound of Formula I, Ia, or Ib as an active ingredient or a pharmaceutically acceptable salt, thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases including inorganic bases and organic bases. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
When the compound of the present invention is basic, salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids. Such acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like. Particularly preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
It will be understood that in the discussion of methods of treatment which follows, references to the compounds of Formula I, Ia, or Ib are meant to also include the pharmaceutically acceptable salts.
The magnitude of prophylactic or therapeutic dose of a compound of Formula I, Ia, or Ib will, of course, vary with the nature and the severity of the condition to be treated and with the particular compound of Formula I, Ia, or Ib and its route of administration. It will also vary according to a variety of factors including the age, weight, general health, sex, diet, time of administration, rate of excretion, drug combination and response of the individual patient. In general, the daily dose from about 0.001 mg to about 100 mg per kg body weight of a mammal,
preferably 0.01 mg to about 10 mg per kg. On the other hand, it may be necessary to use dosages outside these limits in some cases.
The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. For example, a formulation intended for oral administration to humans may contain from about 0.5 mg to about 5 g of active agent compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95 percent of the total composition. Dosage unit forms will generally contain from about 1 mg to about 2 g of an active ingredient, typically 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg, or 1000 mg.
For the treatment of glucocorticoid receptor mediated diseases the compound of Formula I, Ia, or Ib may be administered orally, topically, parenterally, by inhalation spray or rectally in dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles. The term parenteral as used herein includes subcutaneous, intravenous, intramuscular, intrasternal injection or infusion techniques. In addition to the treatment of warm-blooded animals such as mice, rats, horses, cattle, sheep, dogs, cats, etc., the compound of the invention is effective in the treatment of humans.
The pharmaceutical compositions containing the active ingredient may be in a form suitable for oral use, for example, as tablets, troches, lozenges, solutions, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups or elixirs. Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavouring agents, colouring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets. These excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example, magnesium stearate, stearic acid or talc. The tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate may be employed. They may also be coated by the technique described in the U.S. Patent 4,256,108; 4,166,452; and 4,265,874 to form osmotic therapeutic tablets for control release.
Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredients is mixed with water- miscible solvents such as propylene glycol, PEGs and ethanol, or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
Aqueous suspensions contain the active material in admixture with excipients suitable for the manufacture of aqueous suspensions. Such excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropyl methylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate. The aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl, p- hydroxybenzoate, one or more colouring agents, one or more flavouring agents, and one or more sweetening agents, such as sucrose, saccharin or aspartame.
Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in mineral oil such as liquid paraffin. The oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavouring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients, for example sweetening, flavouring and colouring agents, may also be present.
The pharmaceutical compositions of the invention may also be in the form of an oil- in- water emulsion. The oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these. Suitable emulsifying agents may be naturally-occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation
products of the said partial esters with ethylene oxide, for example polyoxy ethylene sorbitan monooleate. The emulsions may also contain sweetening and flavouring agents.
Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a perservative and flavouring and colouring agents. The pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above. The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butane diol. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution. Cosolvents such as ethanol, propylene glycol or polyethylene glycols may also be used. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectables.
The compounds of Formula I, Ia, or Ib may also be administered in the form of suppositories for rectal administration of the drug. These compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ambient temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug. Such materials are cocoa butter and polyethylene glycols.
For topical use, creams, ointments, gels, solutions or suspensions, etc., containing a compound of Formula I, Ia, or Ib are employed. (For purposes of this application, topical application shall include mouth washes and gargles.) Topical formulations may generally be comprised of a pharmaceutical carrier, cosolvent, emulsifier, penetration enhancer, preservative system, and emollient.
The ability of the compounds of Formula I, Ia, or Ib to selectively modulate glucocorticoid receptors makes them useful for treating, preventing or reversing the progression of a variety of inflammatory and autoimmune diseases and conditions. Thus, the compounds of the present invention are useful to treat, prevent or ameliorate the following diseases or conditions: inflammation, tissue rejection, auto-immunity, various malianancies, such as leukemias and lymphomas, Cushing's syndrome, acute adrenal insufficiency, congenital adrenal hyperplasia, rheumatic fever, polyarteritis nodosa, granulomatous polyarteritis, inhibition of myeloid cell lines, immune proliferation/apoptosis, HPA axis suppression and regulation, hypercortisolemia, stroke and spinal cord injury, hypercalcemia, hypergylcemia, acute adrenal insufficiency, chronic primary
adrenal insufficiency, secondary adrenal insufficiency, congenital adrenal hyperplasia, cerebral edema, thrombocytopenia, Little's syndrome, obesity and metabolic syndrome.
The compounds of the present invention are also useful for treating, preventing or reversing the progression of disease states involving systemic inflammation such as inflammatory bowel disease, systemic lupus erythematosus, polyartitis nodosa, Wegener's granulomatosis, giant cell arteritis, rheumatoid arthritis, juvenile rheumatoid arthritis, uveitis, hay fever, allergic rhinitis, urticaria, angioneurotic edema, chronic obstructive pulmonary disease, asthma, tendonitis, bursitis, Crohn's disease, ulcerative colitis, autoimmune chronic active hepatitis, organ transplantation, hepatitis, and cirrhosis.
The compounds of the present invention are useful for treating, preventing or reversing the progression of a variety of topical diseases such as inflammatory scalp alopecia, panniculitis, psoriasis, discoid lupus erythematosus, inflamed cysts, atopic dermatitis, pyoderma gangrenosum, pemphigus vulgaris, buflous pemphigoid, systemic lupus erythematosus, dermatomyositis, herpes gestationis, eosinophilic fasciitis, relapsing polychondritis, inflammatory vasculitis, sarcoidosis, Sweet's disease, type I reactive leprosy, capillary hemangiomas, contact dermatitis, atopic dermatitis, lichen planus, exfoliative dermatitus, erythema nodosum, acne, hirsutism, toxic epidermal necrolysis, erythema multiform, cutaneous T-cell lymphoma.
The compounds of the present invention are also useful in treating, preventing or reversing the progression of disease states associated with Human Immunodeficiency Virus (HIV), cell apoptosis, and cancer including, but not limited to, Kaposi's sarcoma, immune system activation and modulation, desensitization of inflammatory responses, IIL-I expression, natural killer cell development, lymphocytic leukemia, and treatment of retinitis pigmentosa. Cognitive and behavioral processes are also susceptible to glucocorticoid therapy where antagonists would potentially be useful in the treatment of processes such as cognitive performance, memory and learning enhancement, depression, addiction, mood disorders, chronic fatigue syndrome, schizophrenia, stroke, sleep disorders, and anxiety.
The invention also encompasses a method for treating a glucocorticoid receptor mediated disease comprising concomitantly administering to a patient in need of such treatment a compound of Formula I, Ia, or Ib and one or additional more agents. For treating or preventing asthma or chronic obstructive pulmonary disease, the compounds of Formula I, Ia, or Ib may be combined with one or more agents selected from the group consisting of: O-agonists (e.g., salmeterol), theophylline, anticholinergics (e.g., atropine and ipratropium bromide), cromolyn, nedocromil and leukotriene modifiers (e.g., montelukast). For treating or preventing inflammation, the compounds of Formula I, Ia, or Ib may be combined with one or the following: a salicylate, including acetylsalicylic acid, a non-steroidal antiinflammatory drug, including indomethacin,
sulindac, mefenamic, meclofenamic, tolfenamic, tolmetin, ketorolac, dicofenac, ibuprofen, naproxen, fenoprofen, ketoprofen, flurbiprofϊn and oxaprozin, a TNF inhibitor, including etanercept and infliximab, an IL-I receptor antagonist, a cytotoxic or immunosuppressive drug, including methotrexate, leflunomide, azathioprine and cyclosporine, a gold compound, hydroxychloroquine or sulfasalazine, penicillamine, darbufelone, and a p38 kinase inhibitor. The compound of Formula I, Ia, or Ib may also be used in combination with bisphonates such as alendronate to treat a glucocorticoid mediated disease and simultaneously inhibit osteoclast-mediated bone resorption. Unless noted otherwise, the following abbreviations have the indicated meanings: b.p. boiling point e.q. equivalent(s)
EtOAc ethyl acetate g gram(s) mg milligram(s) min. = minute(s) mL = milliliter(s) mol. = mole(s) mmol. = millimole(s) m.p. = melting point mol. wt. = molecular weight
L liter(s) r.t. room temperature
THF tetrahydrofuran v = volume w = weight
Alkvl group abbreviations
Me methyl
Et ethyl n-Pr normal propyl i-Pr isopropyl n-Bu normal butyl i-Bu isobutyl s-Bu secondary butyl t-Bu tertiary butyl c-Pr cyclopropyl
c-Bu = cyclobutyl c-Pen = cyclopentyl c-Hex = cyclohexyl
METHODS OF SYNTHESIS
Generally, compounds of the present invention may be synthesized by using the following synthetic schemes and examples:

A was prepared as previously described:
Hajos, Z.; Parrish, D. Organic Syntheses, Coll. Vol. 7, p363; Vol. 63, p26

Unless noted otherwise, the following synthetic conditions were used for the preparation of the following examples:
(i) all operations were carried out at room or ambient temperature, that is, at a temperature in the range 18-250C,
(ii) evaporation of solvent was carried out using a rotary evaporator under reduced pressure (600-4000 pascals: 4.5-30 mm Hg) with a bath temperature of up to 6O0C,
(iii) the course of reactions was followed by thin layer chromatography (TLC) and reaction times are given for illustration only;
(iv) melting points are uncorrected and M' indicates decomposition; the melting points given are those obtained for the materials prepared as described; polymorphism may result in isolation of materials with different melting points in some preparations;
(v) the structure and purity of all final products were assured by at least one of the following techniques: TLC, mass spectrometry, nuclear magnetic resonance (NMR) spectrometry or micro-analytical data;
(vi) yields are given for illustration only;
(vii) when given, NMR data is in the form of delta (δ) values for major diagnostic protons, given in parts per million (ppm) relative to tetramethylsilane (TMS) as internal standard, determined at 500 MHz or 600 MHz using the indicated solvent, conventional abbreviations used for signal shape are: s. singlet; d. doublet; t. triplet; m. multiplet; and br. broad. In addition, "Ar" means an aromatic signal.
The reaction schemes and examples described herein illustrate the methods employed in the synthesis of the compounds of the present invention. In some cases the order of carrying out the reaction schemes may be varied to facilitate the reaction or to avoid unwanted reaction products. The synthesis of the novel compounds which are the subject of this invention may be accomplished by one or more of several similar routes. The following examples are provided so that the invention might be more fully understood. These examples are illustrative only and should not be construed as limiting the invention in any way.
EXAMPLES Ia and Ib (Diastereoisomers)
2-r(4α6'.5i?)-l-(4-Fluorophenvπ-5-(hvdroxymethvπ-4α-methyl-1.4.4α.5.6.7- hexahvdrocvclopentar/1indazol-5-yll-l-(pyridin-2-yl)ethanol (Isomer 1 and Isomer 2)
As described in more detail below, 1-1 Ia and 1-1 Ib were separated at Step I with the TIPS (triisopropylsilyl) group still on. The two diastereoisomers Ex. Ia and Ex. Ib were prepared separately at Step J from 1-1 Ia and 1-1 Ib. respectively.
Step A: (7α'S)-7α'-Methyl-2'.3'J'.7α'-tetrahvdrospiroπ.3-dioxolane-2J'-indenl-5ϊ6Η)-one
(1-2):

Ethylene glycol (12.2 niL, 219 mmol) and p-toluenesulfonic acid monohydrate (4.40 g, 25.6 mmol) were added to a solution of Hajos-Parrish Ketone (See Organic Syntheses, Coll. Vol. 7, p.363; VoI 63, p.26) CLL 60.0 g, 365 mmol) in 2-ethyl-2-methyl-l,3-dioxolan (46 mL) and the resulting solution stirred at ambient temperature for 24 hours. The reaction was poured into saturated aqueous NaHCOβ solution (1 L) and the crude product extracted with EtOAc (x3). The combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo. Purification by flash chromatography on 1.5 kg of silica, eluting with a gradient of 0-70% EtOAc in hexanes afforded 48.5 g, 64% of Ul as a clear viscous oil. MS (ESI): m/z = 209.3 (MH+).
(7α'S)-7α'-Methyl-5'-oxo-2'.3'.5'.6'.7'Jα'-hexahvdrospiroπ.3-dioxolane-2.r- indenel-6'-carbaldehyde (1-3):

12. -1:3
A 1.5 M solution of lithium diisopropylamide mono(tetrahydrofuran) in cyclohexane (465 mL, 0.698 mol) was added to a solution of Ul (48.5 g, 0.233 mol) in diethyl ether (930 mL) at -78 0C and the resulting solution stirred at this temperature for 1 hour to afford a thick suspension. Methyl formate (86.6 mL, 1.40 mol) was added dropwise over about 30 min and the resulting suspension stirred at -780C for 5 hours. The reaction was quenched at -780C with 1 M aqueous HCl solution (3 L) and the aqueous layer checked to ensure it was acidic. The crude product was extracted with EtOAc (x3) and the combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo to afford 60 g of crude K3 (74% pure) as a tan viscous oil that was used directly in the next step without purification. MS (ESI): m/z = 237.3 (MH+).
(4αS)-l-(4-Fluorophenyl)-4α-methyl-4,4α,6,7-tetrahydrocyclopenta[f|indazol- 5(lH)-one (l-5):

Sodium acetate (38.2 g, 0.465 mol) was added to a solution of crude K3 (6Og), p- fluorophenylhydrazine hydrochloride (47.3 g, 0.291 mol) and acetic acid (66.6 mL, 1.16 mol) in toluene (465 mL) and the resulting suspension heated at 1000C for 1 hour. The reaction was cooled to ambient temperature, diluted with EtOAc, and washed carefully with aqueous 5 % w/v NaHCOβ solution (2 x IL), then dried over anhydrous MgSO4 and concentrated to afford a viscous brown oil. The crude oil was dissolved in THF (1 L) and aqueous 6M HCl (155 mL) was added and the resulting solution was heated to 65°C for 3.5 hours. The resulting solution was cooled to ambient temperature, and poured slowly into aqueous 5 % w/v NaHCOβ solution (CO2 evolution!), and the crude product was extracted with EtOAc (x3). The combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo. Purification by flash chromatography on 1.5 kg of silica, eluting with a gradient of 0-50% EtOAc in hexanes afforded 48.3 g of 1^5, 74% from I^ 3_, as a viscous brown oil that solidified on standing for several days. MS (ESI): m/z = 283.3 (MH+).
Step D: (4αR.5 S)-I -(4-Fluorophenyl)-4α-methyl-lA4α.5.6.7- hexahvdrocvclopentarflindazole-S-carbaldehvde (1-6):

A 1.6 M solution of ^BuLi in hexanes (6.4 mL, 16.0 mmol) was added to a solution of diethyl isocyanomethylphosphonate (2.6 mL, 16.0 mmol) in anhydrous THF (30 mL) at -78 0C and the resulting solution stirred at this temperature for 30 min. A solution of 1^5_ (3.O g, 10.6 mmol) in anhydrous THF (10 mL) was added dropwise over about 20 min and the resulting solution was stirred at -78 0C for 1 hour, then warmed directly to ambient temperature and stirred for a further 1 hour. The reaction was quenched with saturated aqueous ammonium chloride solution, and the crude product was extracted with EtOAc (3x). The combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo. The vinyl isocyanate intermediate was
dissolved in diethyl ether (50 rnL) and 4M aqueous HCl (30 rnL) was then added and the biphasic mixture was stirred at ambient temperature for 12 hours. The reaction was quenched with IM aqueous HCl, and the crude product was extracted with EtOAc (3x). The combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo. This afforded 2.95 g, 94% of the product 1_^5 as a thick, red oil. MS (ESI): m/z = 297.2 (MH+).
(4α£5i?yi-(4-Fluorophenviy4a-methyl-5-(prop-2-en-l-ylVlA4αΛ6.7- hexahydrocyclopentar/iindazole-S-carbaldehyde d-?):

Allyl alcohol (9.36 ml, 135.0 mmol) and p-Toluenesulfonic acid monohydrate (0.26 g, 1.35 mmol) were added to a solution of aldehyde (1^6, 4.0 g, 13.50 mmol) in a sealed pressure tube and the resulting solution stirred at 165°C for 48 hours. The reaction was cooled, diluted with dichloromethane and washed with water. The combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo. Purification by flash chromatography on 120 grams of silica gel, eluting with a gradient of 0-50% EtOAc in hexanes afforded JV7 (3.05 g, 68%, mixture of diastereoisomers) as a brown oil. MS (ESI): m/z = 337.2 (MH+).
\(4aS.5R)- 1 -(4-Fluorophenyl)-4a-methyl-5-(prop-2-en- 1 -vθ- 1.4.4a.5.6.7- hexahydrocyclopenta[/1indazol-5-yllmethanol (1-8):

Sodium borohydride (0.686 g, 18.1 mmol) was added to a cooled solution of 1-7 (5.60 g, 15.1 mmol) in methanol and CH2Cl2 (2:1) and the resulting solution stirred at 00C. The reaction was quenched by the addition of a saturated solution of ammonium chloride and extracted with EtOAc. The combined organic extracts were dried over anhydrous MgSO4 and the solvent
removed in vacuo. Purification by flash chromatography on 120 grams of silica gel, eluting with a gradient of 0-80% EtOAc in hexanes afforded the lower spot as a single diastereoisomer of I1S (2.3 g, 45%) as a yellow foam. MS (ESI): m/z = 339.2 (MH+).
Step G: (4αtS'.5i?)-l-(4-Fluorophenvn-4a-methyl-5-(prop-2-en-l-vn-5-(r(tripropan-2- ylsilyl)oxylmethvU-l,4,4a,5,6,7-hexahydrocvclopentar/1indazole (1-9):

2,6-Lutidene (1.731 ml, 14.86 mmol) and Triisopropylsilyl trifluoromethanesulfonate (4.03 ml, 14.86 mmol) were added to a cooled solution of 1^8 (5.03 g, 14.86 mmol) in anhydrous CH2Cl2 and the resulting solution stirred at 00C for 15 mins. The reaction was warmed to room temperature and stirred for 1 hour. The reaction was quenched by the addition of water and the aqueous was extracted with CH2Cl2. The combined organics were washed with IN HCl and brine and then the combined extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo to afford 8.06 g (110%) of crude U9. MS (ESI): m/z = 495.3 (MH+).
Step H: [(4a6',5i?)-l-(4-fluorophenyl)-4a-methyl-5-{[(tripropan-2-ylsilyl)oxylmethyU- lA4a,5A7-hexahydrocyclopenta[/1indazol-5-yl]acetaldehyde (1-10):

Osmium tetroxide (0.12 mL, 0.394 mmol) was added to a solution of 1^9 (0.65 g, 1.314 mmol) and N-methylmorpholine-N-oxide (0.154 g, 1.314 mmol) in acetone:water (10:1, 3.3 mL) and the reaction was stirred at room temperature for 2 hours. The reaction was diluted with water and extracted with CH2Cl2. The combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo. Sodium periodate (0.281 g, 1.314 mmol) was added to a solution of the crude residue (0.7 g, 1.314 mmol) in methanol (3 mL) and the resulting solution was stirred at room temperature for 2 hours. The reaction was diluted with water and extracted with
dichloromethane. The combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in vacuo. Purification by flash chromatography on 40 grams of silica gel, eluting with a gradient of 0-50% EtOAc in hexanes afforded KlO (0.56 g, 86%) as a yellow foam. MS (ESI): m/z = 497.3 (MH+).
Step I: 2-r(4a6'.5i?)-l-(4-fluorophenvπ-4a-methyl-5-(r(tripropan-2-ylsilvπoxy1methvU- lΛΛa^^J-hexahvdrocvclopentar/iindazol-S-yll-l-fpyridin-l-vπethanol (1-1 Ia and 1-l lb):

Step J: 2-r(4aS.5R)4-(4-Fluorophenyl)-5-(hvdroxymethyl)-4a-methyl-1.4.4a.5.6.7- hexahydrocyclopenta[/1indazol-5-yll-l-(pyridin-2-yl)ethanol (Ex. Ia and Ex. Ib):

Tetrabutylammonium fluoride (0.26 ml, 0.26 mmol) was added to a solution of 1-1 Ia (0.15 g, 0.26 mmol) in tetrahydrofuran (3 mL) and the resulting solution was stirred at room temperature for 1 hour. The reaction was quenched by the addition of water and extracted with
CH2Cl2. The combined organics were dried over anhydrous MgSO4 and the solvent was removed in vacuo. Purification by reverse phase chromatography afforded Ex. Ia (Isomer 1) (0.087 g, 80 %) as a white solid. HRMS (APCI): m/z = 420.1781 (MH+).
Similarly, Ex. Ib (Isomer 2) was made from 1-1 Ib following a similar procedure as that for Ex. Ia.
The following Examples 2a - 29b in Table 1 were prepared following the general synthetic schemes and procedures analogous to Examples Ia and Ib described above. In some cases (e.g. Examples 2a and 2b), the two diastereoisomers were separated at the last step after the TIPS group was removed using standard chromatographic techniques (Hexanes-Ethyl Acetate on silica gel, or Acetonitrile-water on reverse phase HPLC).
Table 1






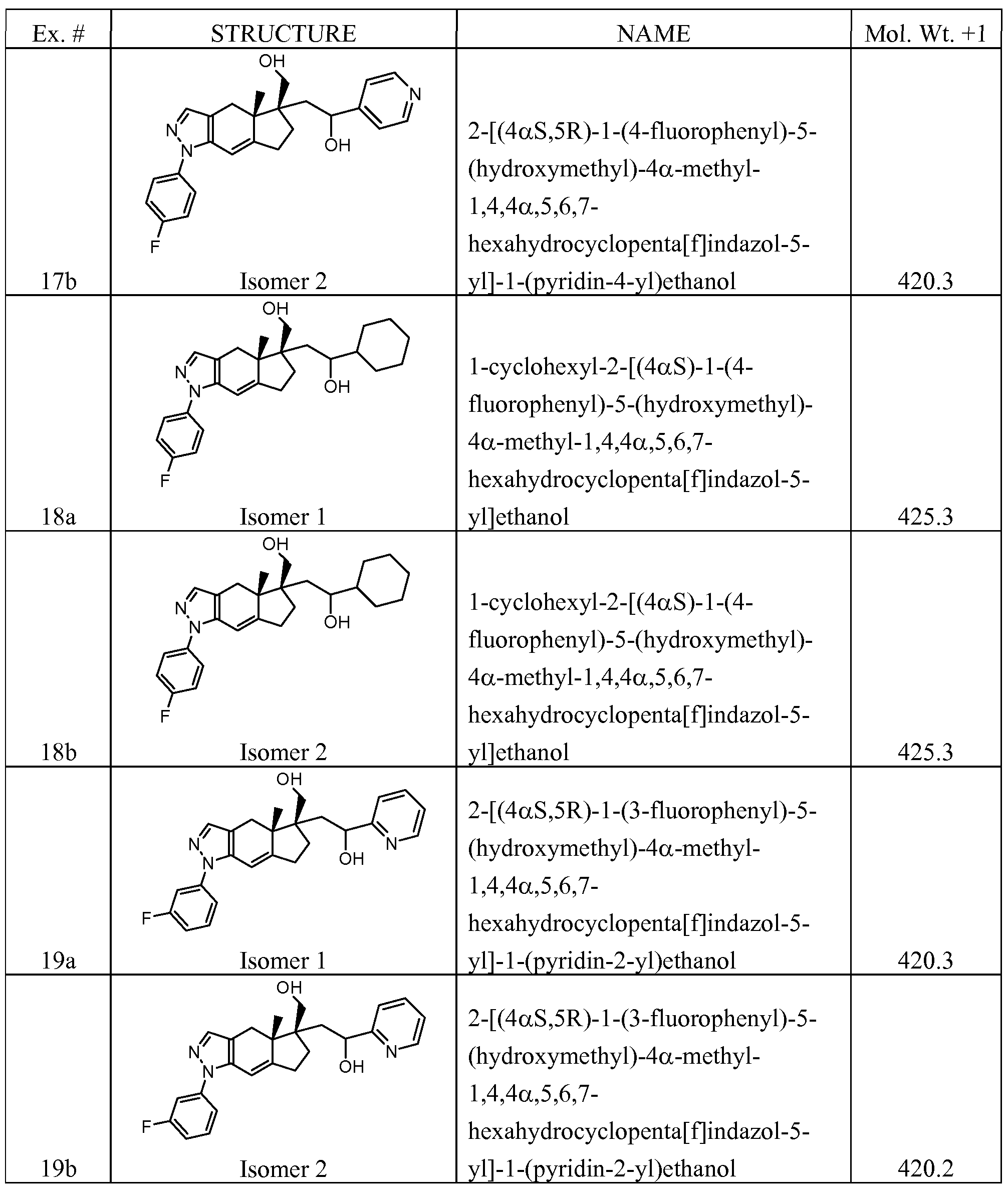





BIOLOGICAL ASSAYS
The compounds exemplified in the present application exhibited activity in one or more of the following assays.
GR Ligand Binding Assay Materials:
Binding Buffer. TEGM (10 mM Tris-HCl, 1 mM EDTA, 10% glycerol, 1 mM beta-mecaptoethanol, 10 mM Sodium Molybdate, pH 7.2)
50% HAP Slurry: Calbiochem Hydroxylapatite, Fast Flow, in 10 mM Tris, pH 8.0 and 1 mM EDTA.
Wash Buffer: 40 mM Tris, pH7.5, 100 mM KCl, 1 mM EDTA and 1 mM EGTA.
95% EtOH
Dexmethasone-methyl-3H, (DEX*); (Amersham cat# TRK645)
Dexamethasone(DEX) (Sigma, cat# D 1756):
Hydroxylapatite Fast Flow; Calbiochem Cat#391947
Molybdate = Molybdic Acid (Sigma, M 1651)
HeLa cell culture media:
RPMI 1640 (Gibco 11835-055) w/23.8 mM NaHCO3, 2 mM L-glutamine in 500 mL of complete media Final cone. lO mL (lM Hepes) 2O mM
5 mL (200 mM L-glu) 4 mM
0.5 mL (10 mg/mL human insulin) 10 μg/mL in 0.01 N HCl Calbiochem#407694-S)
50 mL FBS (Sigma F2442) 10%
1 mL (10 mg/mL Gentamicin 20 μg /mL
Gibco#15710-072) Cell Passaging
Cells (Hall R. E., et al, European Journal of Cancer. 30A: 484-490 (1994)) HeLa (ATCC) cultured in RPMI 1640 (Gibco 11835-055) containing 20 mM Hepes, 4 mM L-glu, 10 ug/ml of human insulin (Sigma, 1-0259), 10% FBS and 20 ug/ml of Gentamicin (Gibco# 15710-072) are rinsed twice in PBS. Phenol red-free Trypsin-EDTA is diluted in the same PBS 1 :10. The cell layers are rinsed with IX Trypsin, extra Trypsin is poured out, and the cell layers are incubated at 370C for ~ 2 min. The flask is tapped and checked for signs of cell detachment. Once the cells begin to slide off the flask, the complete media is added. The cells are counted at this point, then diluted to the appropriate concentration and split into flasks or dishes for further culturing (Usually 1 :3 to 1 :6 dilution).
Preparation of HeLa Cell Lysate
When the cells are 70 to 85% confluent, they are detached as described above, and collected by centrifuging at 1000 g for 10 minutes at 40C. The cell pellet is washed twice with TEGM (10 mM Tris-HCl, 1 mM EDTA, 10% glycerol, 1 mM beta-mercaptoethanol, 10 mM Sodium Molybdate, pH 7.2). After the final wash, the cells are resuspended in TEGM at a concentration of 107 cells/mL. The cell suspension is snap frozen in liquid nitrogen or ethano I/dry ice bath and transferred to -800C freezer on dry ice. Before setting up the binding assay, the frozen
samples are left on ice-water to just thaw (~1 hr). Then the samples are centrifuged at 12,500 g to 20,000 g for 30 min at 40C. The supernatant is used to set-up assay right away. If using 50 μL of supernatant, the test compound can be prepared in 50 μL of the TEGM buffer.
Procedure for Multiple Compound Screening
Ix TEGM buffer is prepared, and the isotope-containing assay mixture is prepared in the following order: EtOH (2% final concentration in reaction), 3H-DEX (Amersham Biosciences) and Ix TEGM. [e.g. For 100 samples, 200 μL (100 x 2) of EtOH + 4.25 μL of 1 :10 3H-Dex stock + 2300 μL (100 x 23) Ix TEGM]. The compound is serially diluted, e.g., if starting final cone, is 1 μM, and the compound is in 25 μL of solution, for duplicate samples, 75 μL of 4x1 μM solution is made and 3 μL of 100 μM is added to 72 μL of buffer, and 1 :5 serial dilution.
25 μL of 3H-DEX (6 nM) trace and 25 μL compound solution are first mixed together, followed by addition of 50 μL receptor solution. The reaction is gently mixed, spun briefly at about 200 rpm and incubated at 40C overnight. 100 μL of 50% HAP slurry is prepared and added to the incubated reaction which is then vortexed and incubated on ice for 5 to 10 minutes. The reaction mixture is vortexed twice more to resuspend HAP while incubating reaction. The samples in 96-well format are then washed in wash buffer using The FilterMate™ Universal Harvester plate washer (Packard). The washing process transfers HAP pellet containing ligand- bound expressed receptor to Unifilter-96 GF/B filter plate (Packard). The HAP pellet on the filter plate is incubated with 50 μL of MICRO SCINT (Packard) scintillint for 30 minutes before being counted on the TopCount micro scintillation counter (Packard). IC50S are calculated using DEX as a reference.
Trans- Activation Modulation of Glucocorticoid Receptor Assay
This assay assesses the ability of test compounds to control transcription from the MMTV-LUC reporter gene in lung adenocarcinoma A549 cells or HeLa cells, a human breast cancer cell line that naturally expresses the human GR. The assay measures induction of a modified MMTV LTR/promoter linked to the LUC reporter gene.
The routine transient assay consists of plating 7,000-25,000 cells/well of a white, clear-bottom 96-well plate. Alternatively, 384-well plates can be used at a cell concentration of 10,000 /well. The media that the cells are plated in is "exponential growth medium" which consists of phenol red-free RPMI1640 containing 10%FBS, 4mM L-glutamine, 2OmM HEPES, lOug/mL human insulin, and 20ug/mL gentamicin. Incubator conditions are 370C and 5% Cθ2- The transfection is done in batch mode. The cells are trypsinized and counted to the right cell number in the proper amount of fresh media. It is then gently mixed with the FuGene6/DNA mix and plated
onto the 96 or 384-well plate, all the wells receive 100 uL or 4OuL, respectively, of medium + lipid/DNA complex then incubated 370C overnight. The transfection cocktail consists of serum- free OptiMEM, FuGeneβ reagent and DNA. The manufacturer's (Roche Biochemical) protocol for cocktail setup is as follows: The lipid to DNA ratio is approximately 2.5:1 and the incubation time is 20 min at room temperature. Sixteen to 24 hours after transfection, the cells are treated with dexamethasone to a final concentration of 1OnM as well as the compound of interest, such that final DMSO (vehicle) concentration is equal to or less than 1%. Each plate also contains samples that are treated with 1OnM dexamethasone alone, which is used as the 100% activity control. The cells are exposed to the compounds for 24 hours. After 24 hours, the cells are lysed by a Promega cell culture lysis buffer for approximately 30 min and then the lucif erase activity in the extracts is assayed in the 96-well format luminometer. In 384-well format, Steady-Glo (Promega) or Steady- Lite (PerkinElmer) can be used by adding an equal volume of reagent to the media present in each well. Activity induced by 1OnM dexamethasone alone is set at 100% activity. Antagonist activity is calculated by determining the decrease in dexamethasone-induced activity in response to compound treatment relative to samples that were treated with dexamethasone alone. Results are expressed as % inhibition of 1OnM dexamethasone activity or as fold of 1OnM dexamethasone activity. This transactivation assay can be performed in an agonist and antagonist mode to identify these different activities.
Activity of test compounds is calculated as the Emaχ relative to the activity obtained with 300 nM dexamethasone. Activity of test compounds is calculated as the Emaχ relative to the activity obtained with 300 nM DEX. The exemplified tissue selective glucocorticoid receptor modulators of the present invention display agonist activity in this assay of greater than 5% and less than 100%, and maximal transactivation activity less then maximal transrepression activity.
The action of compounds is also tested in an antagonist mode (Anti-GRAMMER) in which the cells are treated with medium containing an agonist such as 10 nM DEX and the ability to agents to inhibit the activation by an agonist is measured.
Transrepression Assay
This assay assesses the ability of test compounds to control transcription from the TNFα-β-lactamase reporter gene in U937 cells, a human myelomonocytic leukemia cell line that naturally expresses the human GR. The assay measures compound dependent-repression of the TNFa promoter linked to a reporter gene.
The human U937cells that had been stablely trans fected with the TNF-α promoter driving β-lactamase are used for this assay. U937 cells contain an endogenous glucocorticoid receptor (GR). Cells are maintained in RPMI 1640 Growth medium (Gibco Cat#l 1875-093)
containing 25mM HEPES, 10% FBS, 2mM L-Glutamine, ImM Sodium pyruvate, 25μg/ml Gentamicin (Gibco Cat#15710-064), 1 :1000 2-Mercaptoethanol (Gibco Cat#21985-023) and 0.8 mg/ml G418 (Gibco Cat#10131-027). The density of the cells in the flask needs to be about 1X106 - 3X106/ml at the time of harvest. Usually, the cells are split to 1.2-1.4x105 /ml (1 : 10) 3 days prior to the assay. 50,000 cells/well are plated in 96 well black-walled plates the day of assay. Test compounds are added 10 μL/well, and cells are incubated at 37oC for 30-45 min. For assaying compounds, first dilute 1 : 10 in DMSO to make 1 mM, then further dilute 1 : 100 in medium to make 1OX stock prior to adding to the cells. Add 50ng/ml PMA (Sigma, cat# P8139) 10 μL/well to a final concentration 5ng/ml, and 1 μg/ml LPS (Sigma, cat# L4130) 10 μL/well to a final concentration 100ng/ml. Incubate cells at 370C overnight for ~18hr. PMA is stored frozen as 100 μg/ml stock in DMSO. Dilute 1 : 10 in DMSO for a working stock of 10 μg/ml and store at -20C. For assaying, dilute the 10 μg/ml working stock 1 :200 in medium to make a 1OX solution (50 ng/ml). Store frozen LPS at 1 mg/ml in PBS, dilute 1 :1000 in medium to make 1OX (lμg/ml) for the assay. Add 6X loading buffer (CCF2-AM) 20 μL/well, and incubate at room temperature for 70-90 min. Read plates on CytoFluor II Plate Reader according to manufacture suggested protocols. The activity repressed by 10OnM dexamethasone alone is set as 100% activity.
Microarray Analysis
This assay assesses the ability of test compounds to modulate the transcription of endogenously expressed genes in a variety of cell types including but not limited to A549, HeLa or U937 cells. All cell culture reagents were purchased from Invitrogen Life Tech, Carlsbad CA. A549 cells were grown in phenol red-free DMEM/F12 medium supplemented with 10% FBS. Cells were grown at 37oC with 5% CO2. Using the RNeasy Kit (Qiagen Corp, Valencia CA.), total RNA was extracted and purified from A549 cells treated with different GC compounds for 24 hours, at a fully active dose. These cells express large amount of the GR and are very responsive to GC treatment. All samples were compared against cells treated with vehicle. Expression levels of 23000 genes were measured using oligonucleotide microarrays purchased from Agilent Technologies, Inc. Each comparison was done on a pair of microarrays with reversed fluorophores. Raw image intensity data were processed according to the method described in Patent 6,351,712. The method was used to remove dye bias and to derive a Rosetta probability (p) and fold change value for each gene and each sample pair. Furthermore, for each gene an ANOVA model was constructed across all treatments to derive error estimates. P values for evaluating expression differences were computed using a Bayesian adjusted t-test that was developed by Lόnnstedt and Speed (2002) and extended by Smyth (2003). A gene was declared differentially expressed in any particular comparison if it satisfied two criteria:
1. The Rosetta p value had to be less than 0.1 and the Rosetta fold change value had to be greater than 1.4 in at least one of the treatments.
2. The ANOVA p value had to be less than 0.01 and the fold change greater than 2 in the comparison under consideration.
In Vivo Inflammation Assay
Intact adult (6 month old) female Sprague-Dawley rats are used in the oxazolone (OX) contactdermatitis model. Rats were sensitized on the ventral abdomen with OX on Day 0. On Days 7 and 9, a randomly-selected ear was challenged (same ear each time) with OX; the other was treated with vehicle. Daily treatment begun on Day 7 and continued for 7d with test compounds at different doses and 1.3 mpk 6-methlyprednisolone or O.lmpk DEX as positive controls. The thickness of both ears are measured on Days 11 and 14. Necropsy occurred on Day 14. The rat is first weighed, then anesthetized in a CO2 chamber until near death. Approximately 5ml whole blood is obtained by cardiac puncture. The rat is then examined for certain signs of death and completeness. Tissues are dissected in a highly stylized fashion. The the following endpoints were evaluated: a) inhibiting ear inflammation induced by oxazalone, b) raising serum insulin, c) reducing serum ACTH, d) reducing spleen weight, e) reducing skin thickness, f) reducing body weight, g) increasing expression of bone-related genes with potential relationship to negative glucocorticoid effects on bone; e) changes in molecular markers that correlate with skin inflammation, skin thinning, muscle atrophy and glucose metabolism in liver. All blood samples were collected between 1330-1530 hours, ~4-5 hrs after the last compound treatment.
Primary data for this assay are left and right ear thickness. Inter-ear thickness difference (etd) is used for the estimating the level of inflammation and effectiveness of the compounds is determined by their ability to reduce the increase the thickness of the inflamed ear. Back of the rat skin thickness, spleen weight, serum insulin as well as the effects of gcs on the expression of molecular markers in skin inflammation, skin atrophy, muscle atrophy and glucose metabolism in liver are measured. Data are analyzed by anova plus fisher plsd post-hoc test to identify intergroup differences.
Bioassay Results
The compounds described herein were tested in the Binding, GRAMMER and GITAR assays and demonstrated the activity profiles as shown in Table 2. Compounds shown in Table 2 have potencies in the GRAMMER and GITAR assays (as measured by inflection points, IP) of less than 100O nM.
Table 2
Transactivation Transrepression
A549 Cells U937 Cells
GRAMMER GITAR
Example GR
# BIND Ki (nM) i InP ( /n lMux) E (m%a) x IP (nM) Emax (%)
1a 5.2 5.6 83.9 5.6 93.0
1 b 4.0 26.4 56.0 24.0 91.7
2a 2.2 485.4 46.6 137.0 85.0
2b 2.1 316.1 45.5 235.5 94.4
3a 0.9 38.8 127.2 37.6 93.3
3b 0.9 16.7 130.3 6.0 99.6
4 0.9 39.7 80.3 28.2 104.0
5a 1.4 78.8 65.5 86.1 84.7
5b 0.9 23.6 58.4 29.0 91.8
6a 1.1 30.5 62.4 35.7 104.0
Transactivation Transrepression
A549 Cells U937 Cells
GRAMMER GITAR
Example GR
# BIND Ki (nM) i InP ( /n lMux) E (m%a) x IP (nM) Emax (%)
6b 1.3 124.3 84.7 68.8 98.3
7a 0.6 58.8 60.2 87.6 95.3
7b 0.9 39.3 55.8 44.7 98.4
8 1.2 32.2 56.0 21.4 89.9
9 2.1 3.2 80.3 0.6 101.6
10a 3.1 23.7 56.6 16.5 89.2
10b 1.8 12.4 67.0 4.0 93.9
11 19.2 332.3 41.8 240.6 84.6
12a 2.6 126.6 47.4 93.1 78.8
12b 1.5 33.3 54.2 34.9 93.0
Transactivation Transrepression
A549 Cells U937 Cells
GRAMMER GITAR
Example GR
# BIND Ki (nM) i InP ( /n lMux) E (m%a) x IP (nM) Emax (%)
13a 2.5 8.8 73.1 3.9 91.0
13b 1.9 7.2 57.5 18.0 96.4
14a 6.4 478.8 25.0 1133.0 82.2
14b 14.1 206.1 32.6 248.7 82.3
15a 10.0 68.1 52.7 56.3 87.7
15b 8.3 149.2 39.3 98.5 80.6
16 3.7 18.4 38.3 21.4 85.4
17a 6.4 101.3 26.4 75.3 86.1
17b 7.5 77.5 35.6 59.7 83.3
18a 1.7 22.3 55.3 7.9 93.1
Transactivation Transrepression
A549 Cells U937 Cells
GRAMMER GITAR
Example GR
# BIND Ki (nM) i InP ( /n lMux) E (m%a) x IP (nM) Emax (%)
18b 2.3 59.6 56.2 25.7 97.7
19a 3.7 41.9 43.7 32.7 91.2
19b 9.0 94.8 24.2 73.8 86.7
20a 22.6 187.0 23.1 132.7 79.9
20b 15.1 807.9 1 1.3 572.9 72.0
21a 7.6 151.8 32.4 100.5 85.4
21 b 3.7 40.2 64.8 18.7 91.3
22a 2.3 28.8 63.2 18.7 90.9
22b 5.1 64.2 74.3 14.1 90.6
23a 7.7 49.7 39.4 22.8 82.6
Transactivation Transrepression
A549 Cells U937 Cells
GRAMMER GITAR
Example GR
# BIND Ki (nM) i InP ( /n lMux) E (m%a) x IP (nM) Emax (%)
23b
3.3 63.2 47.7 49.6 83.7
24a
3.1 469.8 7.6 447.4 63.0
24b
7.5 1000.0 4.5 376.3 57.9
25a
26.3 291.1 7.9 293.5 61.3
25b
21.9 401.4 7.6 185.5 69.8
26a
8.6 143.9 24.8 80.8 82.1
26b
35.5 653.2 4.3 1509.0 51.8
27a
13.9 357.1 6.8 597.8 64.2
27b
13.4 633.6 15.4 334.3 71.9
28a
8.3 107.3 60.4 54.3 81.2
Transactivation Transrepression
A549 Cells U937 Cells
GRAMMER GITAR
Example GR
# BIND Ki (nM) i InP ( /n lMux) E (m%a) x IP (nM) Emax (%)
28b
5.4 107.3 42.3 90.9 80.2
29a
11.8 168.0 30.7 176.4 66.6
29b
8.2 121.1 13.7 102.9 60.9
While the invention has been described and illustrated with reference to certain particular embodiments thereof, those skilled in the art will appreciate that various changes, modifications and substitutions can be made therein without departing from the spirit and scope of the invention. For example, effective dosages other than the particular dosages as set forth herein above may be applicable as a consequence of variations in the responsiveness of the mammal being treated for any of the indications for the active agents used in the instant invention as indicated above. Likewise, the specific pharmacological responses observed may vary according to and depending upon the particular active compound selected or whether there are present pharmaceutical carriers, as well as the type of formulation employed, and such expected variations or differences in the results are contemplated in accordance with the objects and practices of the present invention. It is intended, therefore, that the invention be defined by the scope of the claims which follow and that such claims be interpreted as broadly as is reasonable.
Claims
1. A compound of Formula I, or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, or a pharmaceutically acceptable salt of the stereoisomer thereof:

Y is carbon or nitrogen, provided that when Y is nitrogen, then both X1 and X2 are hydrogen;
W is a bond or a methylene group;
Z is hydrogen, -ORa or -NRaRb; each of Rl, R2 and R3 is independently selected from the group consisting of:
(1) hydrogen,
(2) Cl-6alkyl,
(3) C3-6cycloalkyl,
(4) Cl-6alkyl-aryl,
(5) Cl-6alkyl-HET,
(6) aryl, and
(7) HET, wherein each of items (2) to (3), items (6) to (7), the aryl portion of item (4), and the HET portion of item (5) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa,
(e) oxo,
(f) -C(O)NRaRb,
(g) -NRaRb, and (h) -SO2Ra; R4 is Cl-6alkyl or C3-6cycloalkyl; each of Ra and Rb is independently selected from the group consisting of:
(1) hydrogen,
(2) Cl-6alkyl,
(3) aryl, and
(4) HET; and each occurrence of HET is independently selected from the group consisting of:
(1) a 5- or 6-membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N, and
(2) a 9- or 10-membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N.
2. The compound of Claim 1, wherein Z is hydrogen, -OH or -NH2.
3. The compound of Claim 1 , wherein Rl is hydrogen, and
R2 is selected from the group consisting of:
(1) Cl-6alkyl,
(2) Cl-6cycloalkyl,
(3) Cl-6alkyl-aryl,
(4) Cl-6alkyl-HET,
(5) aryl, and
(6) HET, wherein each of items (2), (5) and (6), the aryl portion of item (3), and the HET portion of item (4) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa, and
(e) oxo.
4. The compound of Claim 1, wherein R4 is methyl or ethyl.
5. The compound of Claim 1 , wherein each of Ra and Rb is independently hydrogen or methyl.
6. The compound of Claim 1, wherein HET is independently selected from the group consisting of: pyridine, pyrimidine, pyridazine, pyrrolyl, furan, oxazole, isooxazole, benzofuran, indole, benzopyran, dihydrobenzofuranyl, dihydrobenzoxazolyl, dihydrofuranyl, dihydroindolyl, dihydroisooxazolyl, dihydropyridinyl, dihydropyrimidinyl, and dihydropyrrolyl.
7. A compound of Formula Ia, or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, or a pharmaceutically acceptable salt of the stereoisomer thereof:

X1 is H or halogen;
Y is carbon or nitrogen, provided that when Y is nitrogen, then X1 is hydrogen; W is a bond or a methylene group; Z is hydrogen or -OH; each of Rl and R2 is independently selected from the group consisting of:
(1) hydrogen,
(2) Cl-6alkyl,
(3) C3-6cycloalkyl,
(4) Cl-6alkyl-phenyl,
(5) Cl-6alkyl-HET,
(6) phenyl, and
(7) HET, wherein each of items (2) to (3), items (6) to (7), the phenyl portion of item (4), and the HET portion of item (5) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl, (c) Cl-6alkyl-halogen,
(d) -ORa,
(e) oxo,
(f) -C(O)NRaRb, and
(g) -NRaRb; R3 is hydrogen or methyl; each of Ra and Rb is independently hydrogen or methyl; and each occurrence of HET is independently selected from the group consisting of:
(1) a 5- or 6-membered aromatic or non-aromatic monocyclic ring containing 1-3 heteroatoms selected from O, S and N, and
(2) a 9- or 10-membered aromatic or partially aromatic bicyclic ring containing 1-3 heteroatoms selected from O, S, and N.
8. The compound of Claim 7, wherein X1 is fluoro and Y is carbon.
9. The compound of Claim 8, wherein Z is -OH.
10. The compound of Claim 8, wherein Rl is hydrogen, and
R2 is selected from the group consisting of:
(1) Cl-6alkyl,
(2) Cl-6cycloalkyl,
(3) Cl-6alkyl-phenyl,
(4) Cl_6alkyl-HET,
(5) phenyl, and
(6) HET, wherein each of items (2), (5) and (6), the phenyl portion of item (3), and the HET portion of item (4) above is optionally substituted with one to three substituents independently selected from the group consisting of:
(a) halogen,
(b) Cl-6alkyl,
(c) Cl-6alkyl-halogen,
(d) -ORa, and
(e) oxo.
11. The compound of Claim 8, wherein each occurrence of HET is independently selected from the group consisting of: pyridine, pyrimidine, pyridazine, pyrrolyl, furan, oxazole, isooxazole, benzofuran, indole, benzopyran, dihydrobenzofuranyl, dihydrobenzoxazolyl, dihydrofuranyl, dihydroindolyl, dihydroisooxazolyl, dihydropyridinyl, dihydropyrimidinyl, and dihydropyrrolyl.
12. The compound of Claim 11 , wherein each occurrence of HET is independently selected from the group consisting of: pyridine, pyrimidine, pyridazine, furan, oxazole, benzofuran, indole, and benzopyran.
13. The compound of Claim 8, wherein each of Ra and Rb is independently hydrogen or methyl.
14. The compound of Claim 8, wherein the compound has Formula Ib:

15. A compound selected from the group consisting of: 2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]- 1 -(pyridin-2-yl)ethanol, 2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]- 1 -(4-methylphenyl)ethanol, l-(2-chlorophenyl)-2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]ethanol, l-(2-fluorophenyl)-2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]ethanol,
2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]- 1 -(3-methylphenyl)ethanol,
1 -(3-chloro-5-fluorophenyl)-2-[(4αS,5R)- 1 -(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl- l,4,4α,5,6,7-hexahydrocyclopenta[f]indazol-5-yl]ethanol, 2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5 -yl] - 1 -[3 -(trifluoromethyl)phenyl] ethanol, l-(3-chlorophenyl)-2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopentaffJindazol-S-yl] ethanol, l-(3,5-difluorophenyl)-2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopentaffJindazol-S-yl] ethanol,
2-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5 -yl] - 1 -(3 -fluoropyridin-2-yl)ethanol,
3-{[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7,7a,8- octahydrocyclopenta[f]indazol-5-yl]methyl} -2-benzofuran- 1 (3H)-one,
3-{[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]methyl} -2-benzofuran- 1 (3H)-one,
2-[(4αS,5R)-l-(4-fiuorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]- 1 -phenylethanol, l-[(4αS,5R)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]-3-methylbutan-2-ol,
2-[(4αS,5R)-l-(4-fiuorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]- 1 -(pyridin-3-yl)ethanol,
2-[(4α5',5i?)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[/]indazol-5-yl]-l-(5-methylpyridin-2-yl)ethanol,
2-[(4αS,5R)-l-(4-fiuorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]- 1 -(pyridin-4-yl)ethanol, l-cyclohexyl-2-[(4αS)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl] ethanol,
2-[(4αS,5R)-l-(3-fiuorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl]- 1 -(pyridin-2-yl)ethanol, l-cyclopropyl-2-[(4αS)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5-yl] ethanol,
2-[(4αS,5R)-l-(4-fiuorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5 -yl] - 1 -(3 -methoxypyridin-2-yl)ethanol,
2-[(4αS,5R)-l-(4-fiuorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[f]indazol-5 -yl] - 1 -(5 -fluoropyridin-2-yl)ethanol,
2-[(4α5',5i?)-5-(hydroxymethyl)-4α-methyl-l-phenyl-l,4,4α,5,6,7-hexahydrocyclopenta[/]indazol-
5 -yl] - 1 -(pyridin-2-yl)ethanol, 2-[(4α5',5i?)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[/]indazol-5-yl]-l-(5-methoxypyridin-2-yl)ethanol, l-[(4α5',5i?)-l-(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[/]indazol-5-yl]-2-(pyridin-3-yl)propan-2-ol,
2-[(4α5',5i?)-l-(3-fluorophenyl)-5-(hydroxymethyl)-4a-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[/]indazol-5-yl]-l-(3-methoxypyridin-2-yl)ethanol,
2-[(4α5',5i?)-l-(3-fluorophenyl)-5-(hydroxymethyl)-4a-methyl-l,4,4α,5,6,7- hexahydrocyclopenta[/]indazol-5-yl]- 1 -(pyridin-3-yl)ethanol,
1,1,1 -trifluoro-3-[(4α5',5i?)- 1 -(4-fluorophenyl)-5-(hydroxymethyl)-4α-methyl- 1 ,4,4α,5,6,7- hexahydrocyclopenta[/]indazol-5-yl]propan-2-ol, and
2-[(4α5',5i?)-5-(hydroxymethyl)-4α-methyl-l-phenyl-l,4,4α,5,6,7-hexahydrocyclopenta[/]indazol-
5 -yl] - 1 -(pyridin-3 -yl)ethanol; or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, or a pharmaceutically acceptable salt of the stereoisomer thereof.
16. A pharmaceutical composition comprising a compound of Claim 1 in combination with a pharmaceutically acceptable carrier.
17. A method for treating a glucocorticoid receptor mediated disease or condition in a mammalian patient in need of such treatment comprising administering to the patient a compound of Claim 1 in an amount that is effective for treating the glucocorticoid receptor mediated disease or condition.
18. The method of Claim 17 wherein the glucocorticoid receptor mediated disease or condition is selected from the group consisting of: tissue rejection, leukemias, lymphomas, Cushing's syndrome, acute adrenal insufficiency, congenital adrenal hyperplasia, rheumatic fever, polyarteritis nodosa, granulomatous polyarteritis, inhibition of myeloid cell lines, immune proliferation/apoptosis, HPA axis suppression and regulation, hypercortisolemia, stroke and spinal cord injury, hypercalcemia, hypergylcemia, acute adrenal insufficiency, chronic primary adrenal insufficiency, secondary adrenal insufficiency, congenital adrenal hyperplasia, cerebral edema, thrombocytopenia, Little's syndrome, obesity, metabolic syndrome, inflammatory bowel disease, systemic lupus erythematosus, polyartitis nodosa, Wegener's granulomatosis, giant cell arteritis, rheumatoid arthritis, juvenile rheumatoid arthritis, uveitis, hay fever, allergic rhinitis, urticaria, angioneurotic edema, chronic obstructive pulmonary disease, asthma, tendonitis, bursitis, Crohn's disease, ulcerative colitis, autoimmune chronic active hepatitis, organ transplantation, hepatitis, cirrhosis, inflammatory scalp alopecia, panniculitis, psoriasis, discoid lupus erythematosus, inflamed cysts, atopic dermatitis, pyoderma gangrenosum, pemphigus vulgaris, buflous pemphigoid, systemic lupus erythematosus, dermatomyositis, herpes gestationis, eosinophilic fasciitis, relapsing polychondritis, inflammatory vasculitis, sarcoidosis, Sweet's disease, type I reactive leprosy, capillary hemangiomas, contact dermatitis, atopic dermatitis, lichen planus, exfoliative dermatitus, erythema nodosum, acne, hirsutism, toxic epidermal necrolysis, erythema multiform, cutaneous T-cell lymphoma, Human Immunodeficiency Virus (HIV), cell apoptosis, cancer, Kaposi's sarcoma, retinitis pigmentosa, cognitive performance, memory and learning enhancement, depression, addiction, mood disorders, chronic fatigue syndrome, schizophrenia, sleep disorders, and anxiety.
19. A method of selectively modulating the activation, repression, agonism or antagonism effect of the glucocorticoid receptor in a mammal comprising administering to the mammal a compound of Claim 1 in an amount that is effective to modulate the glucocorticoid receptor.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10783799A EP2459190A4 (en) | 2009-06-02 | 2010-05-24 | Hexahydrocyclopentyl[¦]indazole 5-hydroxymethyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators |
| US13/375,551 US20120095055A1 (en) | 2009-06-02 | 2010-05-24 | HEXAHYDROCYCLOPENTYL[f]INDAZOLE 5-HYDROXYMETHYL ETHANOLS AND DERIVATIVES THEREOF AS SELECTIVE GLUCOCORTICOID RECEPTOR MODULATORS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18332509P | 2009-06-02 | 2009-06-02 | |
| US61/183,325 | 2009-06-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010141247A1 true WO2010141247A1 (en) | 2010-12-09 |
Family
ID=43298030
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/035898 WO2010141247A1 (en) | 2009-06-02 | 2010-05-24 | Hexahydrocyclopentyl[∫]indazole 5-hydroxymethyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20120095055A1 (en) |
| EP (1) | EP2459190A4 (en) |
| WO (1) | WO2010141247A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2493302A1 (en) * | 2009-10-30 | 2012-09-05 | Merck Sharp & Dohme Corp. | Hexahydrocyclopentyl[f]indazole pyridyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators |
| WO2020152193A1 (en) | 2019-01-22 | 2020-07-30 | Akribes Biomedical Gmbh | Selective glucocorticoid receptor modifiers for treating impaired skin wound healing |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003015692A2 (en) * | 2001-07-17 | 2003-02-27 | Smithkline Beecham Corporation | Crystallized glucocorticoid receptor ligand binding domain polypeptide and screening methods employing same |
| WO2008060391A2 (en) | 2006-10-23 | 2008-05-22 | Merck & Co., Inc. | 2- [l-phenyl-5-hydr0xy-4alpha-methyl-hexahydr0cycl0penta[f] indaz0l-5-yl] ethyl phenyl derivatives as glucocorticoid receptor ligands |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011053574A1 (en) * | 2009-10-30 | 2011-05-05 | Merck Sharp & Dohme Corp. | 2- [1-PHENYL-5-HYDROXY-4a-SUBSTITUTED-HEXAHYDROCYCLOPENTA [F] INDAZOL-5-YL] ETHYL PHENYL DERIVATIVES AS GLUCOCORTICOID RECEPTOR LIGANDS |
-
2010
- 2010-05-24 WO PCT/US2010/035898 patent/WO2010141247A1/en active Application Filing
- 2010-05-24 US US13/375,551 patent/US20120095055A1/en not_active Abandoned
- 2010-05-24 EP EP10783799A patent/EP2459190A4/en not_active Withdrawn
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003015692A2 (en) * | 2001-07-17 | 2003-02-27 | Smithkline Beecham Corporation | Crystallized glucocorticoid receptor ligand binding domain polypeptide and screening methods employing same |
| WO2008060391A2 (en) | 2006-10-23 | 2008-05-22 | Merck & Co., Inc. | 2- [l-phenyl-5-hydr0xy-4alpha-methyl-hexahydr0cycl0penta[f] indaz0l-5-yl] ethyl phenyl derivatives as glucocorticoid receptor ligands |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP2459190A4 * |
| THOMPSON ET AL., BIOORG. MED. CHEM. LETT., vol. 17, 2007, pages 3354 - 3361 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2493302A1 (en) * | 2009-10-30 | 2012-09-05 | Merck Sharp & Dohme Corp. | Hexahydrocyclopentyl[f]indazole pyridyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators |
| EP2493302A4 (en) * | 2009-10-30 | 2013-04-24 | Merck Sharp & Dohme | Hexahydrocyclopentyl[f]indazole pyridyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators |
| WO2020152193A1 (en) | 2019-01-22 | 2020-07-30 | Akribes Biomedical Gmbh | Selective glucocorticoid receptor modifiers for treating impaired skin wound healing |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2459190A1 (en) | 2012-06-06 |
| US20120095055A1 (en) | 2012-04-19 |
| EP2459190A4 (en) | 2012-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2097385B1 (en) | 2-[1-phenyl-5-hydroxy or methoxy-4alpha-methyl-hexahydroclopenta[f]indazol-5-yl]ethyl phenyl derivatives as glucocorticoid receptor ligands | |
| AU2004207482B2 (en) | 17-carbamoyloxy cortisol derivatives as selective glucocorticoid receptor modulators | |
| WO2004026248A2 (en) | Octahydro-2-h-naphtho[1,2-f] indole-4-carboxamide derivatives as selective glucocorticoid receptor modulators | |
| AU2009217493B2 (en) | Hexahydrocyclopentyl[f]indazole carboxamides and derivatives thereof as selective glucocorticoid receptor modulators | |
| US20120214847A1 (en) | 2-[1-PHENYL-5-HYDROXY-4a-SUBSTITUTED-HEXAHYDROCYCLOPENTA[F]INDAZOL-5-YL]ETHYL PHENYL DERIVATIVES AS GLUCOCORTICOID RECEPTOR LIGANDS | |
| WO2004075840A9 (en) | Selective non-steroidal glucocorticoid receptor modulators | |
| EP2265273B1 (en) | Hexahydrocyclopentyl[f] indazole sulfonamides and derivatives thereof as selective glucocorticoid receptor modulators | |
| US20120172397A1 (en) | HEXAHYDROCYCLOPENTA[f]INDAZOLE 5-YL ETHANOLS AND DERIVATIVES THEREOF AS SELECTIVE GLUCOCORTICOID RECEPTOR MODULATORS | |
| US20120095055A1 (en) | HEXAHYDROCYCLOPENTYL[f]INDAZOLE 5-HYDROXYMETHYL ETHANOLS AND DERIVATIVES THEREOF AS SELECTIVE GLUCOCORTICOID RECEPTOR MODULATORS | |
| WO2010138421A1 (en) | HEXAHYDROCYCLOPENTYL[f]INDAZOLE REVERSED AMIDES AND DERIVATIVES AS SELECTIVE GLUCOCORTICOID RECEPTOR MODULATORS | |
| US7745657B2 (en) | D-homoandrosta-17-yl carbamate derivatives as selective glucocorticoid receptor ligands | |
| US20120214846A1 (en) | Hexahydrocyclopentyl[f]indazole pyridyl ethanols and derivatives thereof as selective glucocorticoid receptor modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10783799 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010783799 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13375551 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |