WO2010118207A1 - Pyrazolo [1, 5-a] pyrimidine derivatives as mtor inhibitors - Google Patents
Pyrazolo [1, 5-a] pyrimidine derivatives as mtor inhibitors Download PDFInfo
- Publication number
- WO2010118207A1 WO2010118207A1 PCT/US2010/030350 US2010030350W WO2010118207A1 WO 2010118207 A1 WO2010118207 A1 WO 2010118207A1 US 2010030350 W US2010030350 W US 2010030350W WO 2010118207 A1 WO2010118207 A1 WO 2010118207A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- substituted
- heteroaryl
- group
- methyl
- Prior art date
Links
- LDIJKUBTLZTFRG-UHFFFAOYSA-N pyrazolo[1,5-a]pyrimidine Chemical class N1=CC=CN2N=CC=C21 LDIJKUBTLZTFRG-UHFFFAOYSA-N 0.000 title abstract description 4
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 title description 6
- 229940124302 mTOR inhibitor Drugs 0.000 title description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 303
- 238000000034 method Methods 0.000 claims abstract description 112
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 60
- 201000010099 disease Diseases 0.000 claims abstract description 58
- 238000011282 treatment Methods 0.000 claims abstract description 43
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 claims abstract description 42
- 230000002401 inhibitory effect Effects 0.000 claims abstract description 19
- 230000005764 inhibitory process Effects 0.000 claims abstract description 15
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 claims abstract description 14
- -1 Aphidicolon Chemical compound 0.000 claims description 242
- 125000000217 alkyl group Chemical group 0.000 claims description 209
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 191
- 125000001072 heteroaryl group Chemical group 0.000 claims description 172
- 125000003118 aryl group Chemical group 0.000 claims description 143
- 150000003839 salts Chemical class 0.000 claims description 91
- 239000012453 solvate Substances 0.000 claims description 85
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 79
- 150000002148 esters Chemical class 0.000 claims description 78
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 78
- 229940002612 prodrug Drugs 0.000 claims description 78
- 239000000651 prodrug Substances 0.000 claims description 78
- 125000003545 alkoxy group Chemical group 0.000 claims description 52
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 50
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 46
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 45
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 37
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 30
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims description 30
- 229910052739 hydrogen Inorganic materials 0.000 claims description 30
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 28
- 125000004104 aryloxy group Chemical group 0.000 claims description 23
- 239000002246 antineoplastic agent Substances 0.000 claims description 22
- 239000003112 inhibitor Substances 0.000 claims description 22
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 21
- 206010028980 Neoplasm Diseases 0.000 claims description 19
- 230000002062 proliferating effect Effects 0.000 claims description 18
- 125000000304 alkynyl group Chemical group 0.000 claims description 17
- 201000011510 cancer Diseases 0.000 claims description 17
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 claims description 14
- 206010025323 Lymphomas Diseases 0.000 claims description 13
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 13
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 12
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 12
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 12
- 210000001685 thyroid gland Anatomy 0.000 claims description 12
- 229960004679 doxorubicin Drugs 0.000 claims description 11
- 230000001225 therapeutic effect Effects 0.000 claims description 11
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 claims description 10
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 10
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 claims description 10
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 claims description 10
- 208000027771 Obstructive airways disease Diseases 0.000 claims description 10
- 229930012538 Paclitaxel Natural products 0.000 claims description 10
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 10
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 claims description 10
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 claims description 10
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 claims description 10
- 229960000473 altretamine Drugs 0.000 claims description 10
- 210000004027 cell Anatomy 0.000 claims description 10
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 claims description 10
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 claims description 10
- 229960001592 paclitaxel Drugs 0.000 claims description 10
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 10
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 claims description 10
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 claims description 9
- 229960003881 letrozole Drugs 0.000 claims description 9
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 claims description 9
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 9
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 claims description 9
- 229960002340 pentostatin Drugs 0.000 claims description 9
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 claims description 8
- XMYKNCNAZKMVQN-NYYWCZLTSA-N [(e)-(3-aminopyridin-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=NC=CC=C1N XMYKNCNAZKMVQN-NYYWCZLTSA-N 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 8
- 125000005312 heteroarylalkynyl group Chemical group 0.000 claims description 8
- 208000032839 leukemia Diseases 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 229960005526 triapine Drugs 0.000 claims description 8
- 206010003571 Astrocytoma Diseases 0.000 claims description 7
- 208000027866 inflammatory disease Diseases 0.000 claims description 7
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 6
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 6
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 6
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 claims description 6
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 6
- 239000012824 ERK inhibitor Substances 0.000 claims description 6
- 206010014733 Endometrial cancer Diseases 0.000 claims description 6
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 6
- 201000008808 Fibrosarcoma Diseases 0.000 claims description 6
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 claims description 6
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 claims description 6
- 208000032612 Glial tumor Diseases 0.000 claims description 6
- 206010018338 Glioma Diseases 0.000 claims description 6
- 208000017604 Hodgkin disease Diseases 0.000 claims description 6
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 6
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 6
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 6
- 239000002136 L01XE07 - Lapatinib Substances 0.000 claims description 6
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 6
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 6
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 6
- 206010029260 Neuroblastoma Diseases 0.000 claims description 6
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 6
- 201000010208 Seminoma Diseases 0.000 claims description 6
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 6
- 206010042971 T-cell lymphoma Diseases 0.000 claims description 6
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 claims description 6
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 6
- 230000001154 acute effect Effects 0.000 claims description 6
- 208000026935 allergic disease Diseases 0.000 claims description 6
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 claims description 6
- 210000000481 breast Anatomy 0.000 claims description 6
- 210000003679 cervix uteri Anatomy 0.000 claims description 6
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 claims description 6
- 229960004316 cisplatin Drugs 0.000 claims description 6
- 210000001072 colon Anatomy 0.000 claims description 6
- 239000000824 cytostatic agent Substances 0.000 claims description 6
- 208000024558 digestive system cancer Diseases 0.000 claims description 6
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 claims description 6
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 claims description 6
- 210000003238 esophagus Anatomy 0.000 claims description 6
- 230000003325 follicular Effects 0.000 claims description 6
- 229960002258 fulvestrant Drugs 0.000 claims description 6
- 210000000232 gallbladder Anatomy 0.000 claims description 6
- 201000010231 gastrointestinal system cancer Diseases 0.000 claims description 6
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 claims description 6
- 229960001101 ifosfamide Drugs 0.000 claims description 6
- 210000003734 kidney Anatomy 0.000 claims description 6
- 210000004185 liver Anatomy 0.000 claims description 6
- 210000004072 lung Anatomy 0.000 claims description 6
- 201000001441 melanoma Diseases 0.000 claims description 6
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 claims description 6
- 210000003739 neck Anatomy 0.000 claims description 6
- 208000007538 neurilemmoma Diseases 0.000 claims description 6
- 201000008968 osteosarcoma Diseases 0.000 claims description 6
- 210000001672 ovary Anatomy 0.000 claims description 6
- 210000000496 pancreas Anatomy 0.000 claims description 6
- 210000002307 prostate Anatomy 0.000 claims description 6
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 6
- 229960004641 rituximab Drugs 0.000 claims description 6
- 210000003491 skin Anatomy 0.000 claims description 6
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 6
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 6
- 210000002784 stomach Anatomy 0.000 claims description 6
- 208000001608 teratocarcinoma Diseases 0.000 claims description 6
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 claims description 5
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 claims description 5
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 claims description 5
- GCKMFJBGXUYNAG-UHFFFAOYSA-N 17alpha-methyltestosterone Natural products C1CC2=CC(=O)CCC2(C)C2C1C1CCC(C)(O)C1(C)CC2 GCKMFJBGXUYNAG-UHFFFAOYSA-N 0.000 claims description 5
- DBPWSSGDRRHUNT-CEGNMAFCSA-N 17α-hydroxyprogesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 DBPWSSGDRRHUNT-CEGNMAFCSA-N 0.000 claims description 5
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 claims description 5
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 claims description 5
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 claims description 5
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 5
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 claims description 5
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 claims description 5
- 108010024976 Asparaginase Proteins 0.000 claims description 5
- OLCWFLWEHWLBTO-HSZRJFAPSA-N BMS-214662 Chemical compound C=1C=CSC=1S(=O)(=O)N([C@@H](C1)CC=2C=CC=CC=2)CC2=CC(C#N)=CC=C2N1CC1=CN=CN1 OLCWFLWEHWLBTO-HSZRJFAPSA-N 0.000 claims description 5
- 102100028845 Biogenesis of lysosome-related organelles complex 1 subunit 2 Human genes 0.000 claims description 5
- 108010006654 Bleomycin Proteins 0.000 claims description 5
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 claims description 5
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 claims description 5
- 102100025832 Centromere-associated protein E Human genes 0.000 claims description 5
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 claims description 5
- 108010092160 Dactinomycin Proteins 0.000 claims description 5
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 claims description 5
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 claims description 5
- 101000935458 Homo sapiens Biogenesis of lysosome-related organelles complex 1 subunit 2 Proteins 0.000 claims description 5
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 claims description 5
- 239000005551 L01XE03 - Erlotinib Substances 0.000 claims description 5
- 239000002147 L01XE04 - Sunitinib Substances 0.000 claims description 5
- 239000002067 L01XE06 - Dasatinib Substances 0.000 claims description 5
- 108010000817 Leuprolide Proteins 0.000 claims description 5
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 claims description 5
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 claims description 5
- GCKMFJBGXUYNAG-HLXURNFRSA-N Methyltestosterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)CC2 GCKMFJBGXUYNAG-HLXURNFRSA-N 0.000 claims description 5
- QJZRFPJCWMNVAV-HHHXNRCGSA-N N-(3-aminopropyl)-N-[(1R)-1-[7-chloro-4-oxo-3-(phenylmethyl)-2-quinazolinyl]-2-methylpropyl]-4-methylbenzamide Chemical compound NCCCN([C@H](C(C)C)C=1N(C(=O)C2=CC=C(Cl)C=C2N=1)CC=1C=CC=CC=1)C(=O)C1=CC=C(C)C=C1 QJZRFPJCWMNVAV-HHHXNRCGSA-N 0.000 claims description 5
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 claims description 5
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 claims description 5
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 claims description 5
- 229940009456 adriamycin Drugs 0.000 claims description 5
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 claims description 5
- 229960001220 amsacrine Drugs 0.000 claims description 5
- 229960002932 anastrozole Drugs 0.000 claims description 5
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 claims description 5
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 claims description 5
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 claims description 5
- 229960001561 bleomycin Drugs 0.000 claims description 5
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 claims description 5
- 229960001467 bortezomib Drugs 0.000 claims description 5
- 229940112129 campath Drugs 0.000 claims description 5
- 229960004117 capecitabine Drugs 0.000 claims description 5
- 229960004562 carboplatin Drugs 0.000 claims description 5
- 229960005243 carmustine Drugs 0.000 claims description 5
- 108010031379 centromere protein E Proteins 0.000 claims description 5
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 claims description 5
- 229960004630 chlorambucil Drugs 0.000 claims description 5
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 claims description 5
- 229960002559 chlorotrianisene Drugs 0.000 claims description 5
- 229960004397 cyclophosphamide Drugs 0.000 claims description 5
- 229960000684 cytarabine Drugs 0.000 claims description 5
- 229960003901 dacarbazine Drugs 0.000 claims description 5
- 229960000640 dactinomycin Drugs 0.000 claims description 5
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 5
- 229960000975 daunorubicin Drugs 0.000 claims description 5
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 claims description 5
- 229960003668 docetaxel Drugs 0.000 claims description 5
- 229960001904 epirubicin Drugs 0.000 claims description 5
- 229930013356 epothilone Natural products 0.000 claims description 5
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 claims description 5
- 229960001842 estramustine Drugs 0.000 claims description 5
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 claims description 5
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 claims description 5
- 229960005420 etoposide Drugs 0.000 claims description 5
- 229960000255 exemestane Drugs 0.000 claims description 5
- 229960005304 fludarabine phosphate Drugs 0.000 claims description 5
- 229960001751 fluoxymesterone Drugs 0.000 claims description 5
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 claims description 5
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 claims description 5
- 229960002074 flutamide Drugs 0.000 claims description 5
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 claims description 5
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 claims description 5
- 229960005277 gemcitabine Drugs 0.000 claims description 5
- 210000003128 head Anatomy 0.000 claims description 5
- 229960002899 hydroxyprogesterone Drugs 0.000 claims description 5
- 229950007344 ispinesib Drugs 0.000 claims description 5
- 229960004891 lapatinib Drugs 0.000 claims description 5
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 claims description 5
- 229960004338 leuprorelin Drugs 0.000 claims description 5
- 229960001614 levamisole Drugs 0.000 claims description 5
- 229960002247 lomustine Drugs 0.000 claims description 5
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 claims description 5
- 229960002985 medroxyprogesterone acetate Drugs 0.000 claims description 5
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 claims description 5
- 229960004296 megestrol acetate Drugs 0.000 claims description 5
- 229960001924 melphalan Drugs 0.000 claims description 5
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 claims description 5
- 229960001428 mercaptopurine Drugs 0.000 claims description 5
- 229960000485 methotrexate Drugs 0.000 claims description 5
- 229960001566 methyltestosterone Drugs 0.000 claims description 5
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 claims description 5
- 229960004857 mitomycin Drugs 0.000 claims description 5
- 229960000350 mitotane Drugs 0.000 claims description 5
- 229960001156 mitoxantrone Drugs 0.000 claims description 5
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 5
- PGXYIBJJCLWJST-MUUNZHRXSA-N n-(3-aminopropyl)-n-[(1r)-1-(3-benzyl-7-chloro-4-oxochromen-2-yl)-2-methylpropyl]-4-methylbenzamide Chemical compound NCCCN([C@H](C(C)C)C1=C(C(=O)C2=CC=C(Cl)C=C2O1)CC=1C=CC=CC=1)C(=O)C1=CC=C(C)C=C1 PGXYIBJJCLWJST-MUUNZHRXSA-N 0.000 claims description 5
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 claims description 5
- 229960001756 oxaliplatin Drugs 0.000 claims description 5
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 claims description 5
- 229960000952 pipobroman Drugs 0.000 claims description 5
- 229960003171 plicamycin Drugs 0.000 claims description 5
- 229960005205 prednisolone Drugs 0.000 claims description 5
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 claims description 5
- 229960004618 prednisone Drugs 0.000 claims description 5
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 claims description 5
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 claims description 5
- 229960000624 procarbazine Drugs 0.000 claims description 5
- 229960001302 ridaforolimus Drugs 0.000 claims description 5
- 229960001052 streptozocin Drugs 0.000 claims description 5
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 claims description 5
- 229960001603 tamoxifen Drugs 0.000 claims description 5
- 229940063683 taxotere Drugs 0.000 claims description 5
- 229960001278 teniposide Drugs 0.000 claims description 5
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 claims description 5
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 claims description 5
- 229960005353 testolactone Drugs 0.000 claims description 5
- 229960003604 testosterone Drugs 0.000 claims description 5
- 229960001196 thiotepa Drugs 0.000 claims description 5
- 229960003087 tioguanine Drugs 0.000 claims description 5
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 claims description 5
- 229960000303 topotecan Drugs 0.000 claims description 5
- 229960005026 toremifene Drugs 0.000 claims description 5
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 claims description 5
- 229960005267 tositumomab Drugs 0.000 claims description 5
- 229960000575 trastuzumab Drugs 0.000 claims description 5
- 229960005294 triamcinolone Drugs 0.000 claims description 5
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 claims description 5
- 229960001055 uracil mustard Drugs 0.000 claims description 5
- 229960003048 vinblastine Drugs 0.000 claims description 5
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 claims description 5
- 229960004528 vincristine Drugs 0.000 claims description 5
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 claims description 5
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 claims description 5
- 229960004355 vindesine Drugs 0.000 claims description 5
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 claims description 5
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 claims description 4
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 claims description 4
- WHMXDBPHBVLYRC-OFVILXPXSA-N 3-chloro-n-[(2s)-1-[[2-(dimethylamino)acetyl]amino]-3-[4-[8-[(1s)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl]phenyl]propan-2-yl]-4-propan-2-yloxybenzamide Chemical compound C1=C(Cl)C(OC(C)C)=CC=C1C(=O)N[C@H](CNC(=O)CN(C)C)CC1=CC=C(C=2N=C3C([C@H](C)O)=CC=CN3C=2)C=C1 WHMXDBPHBVLYRC-OFVILXPXSA-N 0.000 claims description 4
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 claims description 4
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 claims description 4
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 claims description 4
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 claims description 4
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 claims description 4
- 108010069236 Goserelin Proteins 0.000 claims description 4
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 claims description 4
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 claims description 4
- 239000005411 L01XE02 - Gefitinib Substances 0.000 claims description 4
- 239000005536 L01XE08 - Nilotinib Substances 0.000 claims description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 4
- QJMCKEPOKRERLN-UHFFFAOYSA-N N-3,4-tridhydroxybenzamide Chemical compound ONC(=O)C1=CC=C(O)C(O)=C1 QJMCKEPOKRERLN-UHFFFAOYSA-N 0.000 claims description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 claims description 4
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 claims description 4
- 206010052779 Transplant rejections Diseases 0.000 claims description 4
- MSLJYSGFUMYUDX-UHFFFAOYSA-N Trimidox Chemical compound ON=C(N)C1=CC(O)=C(O)C(O)=C1 MSLJYSGFUMYUDX-UHFFFAOYSA-N 0.000 claims description 4
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 claims description 4
- 229960003437 aminoglutethimide Drugs 0.000 claims description 4
- 229960000397 bevacizumab Drugs 0.000 claims description 4
- 229960002092 busulfan Drugs 0.000 claims description 4
- 229960005395 cetuximab Drugs 0.000 claims description 4
- 229960002436 cladribine Drugs 0.000 claims description 4
- 229960002448 dasatinib Drugs 0.000 claims description 4
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 claims description 4
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 claims description 4
- 229960001433 erlotinib Drugs 0.000 claims description 4
- 229960000390 fludarabine Drugs 0.000 claims description 4
- 229960002584 gefitinib Drugs 0.000 claims description 4
- 229960000578 gemtuzumab Drugs 0.000 claims description 4
- 229960002913 goserelin Drugs 0.000 claims description 4
- 229960000908 idarubicin Drugs 0.000 claims description 4
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 claims description 4
- 229960004768 irinotecan Drugs 0.000 claims description 4
- DHMTURDWPRKSOA-RUZDIDTESA-N lonafarnib Chemical compound C1CN(C(=O)N)CCC1CC(=O)N1CCC([C@@H]2C3=C(Br)C=C(Cl)C=C3CCC3=CC(Br)=CN=C32)CC1 DHMTURDWPRKSOA-RUZDIDTESA-N 0.000 claims description 4
- 208000020816 lung neoplasm Diseases 0.000 claims description 4
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 claims description 4
- 229960004961 mechlorethamine Drugs 0.000 claims description 4
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 claims description 4
- 229960001346 nilotinib Drugs 0.000 claims description 4
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 claims description 4
- 229960001796 sunitinib Drugs 0.000 claims description 4
- 229960004964 temozolomide Drugs 0.000 claims description 4
- GFFXZLZWLOBBLO-ASKVSEFXSA-N tezacitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(=C/F)/[C@H](O)[C@@H](CO)O1 GFFXZLZWLOBBLO-ASKVSEFXSA-N 0.000 claims description 4
- 229950006410 tezacitabine Drugs 0.000 claims description 4
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 claims description 4
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 claims description 4
- 229950001353 tretamine Drugs 0.000 claims description 4
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 claims description 4
- 229960002066 vinorelbine Drugs 0.000 claims description 4
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 claims description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 3
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 claims description 3
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 claims description 3
- 229960000928 clofarabine Drugs 0.000 claims description 3
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 claims description 3
- 229960000452 diethylstilbestrol Drugs 0.000 claims description 3
- 229940017825 dromostanolone Drugs 0.000 claims description 3
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 claims description 3
- 229960000961 floxuridine Drugs 0.000 claims description 3
- 229960002949 fluorouracil Drugs 0.000 claims description 3
- 229950001750 lonafarnib Drugs 0.000 claims description 3
- 201000005202 lung cancer Diseases 0.000 claims description 3
- 229960004584 methylprednisolone Drugs 0.000 claims description 3
- 208000011580 syndromic disease Diseases 0.000 claims description 3
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 claims description 2
- 229960002568 ethinylestradiol Drugs 0.000 claims description 2
- 210000004765 promyelocyte Anatomy 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 8
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 claims 3
- 229950009158 tipifarnib Drugs 0.000 claims 3
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 claims 1
- 150000003883 epothilone derivatives Chemical class 0.000 claims 1
- 229960005309 estradiol Drugs 0.000 claims 1
- 229930182833 estradiol Natural products 0.000 claims 1
- 230000002265 prevention Effects 0.000 abstract description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 156
- 239000000243 solution Substances 0.000 description 106
- 239000000203 mixture Substances 0.000 description 92
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 91
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 89
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 88
- 125000005843 halogen group Chemical group 0.000 description 88
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 82
- 239000011541 reaction mixture Substances 0.000 description 68
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 64
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 63
- 235000019439 ethyl acetate Nutrition 0.000 description 63
- 229910001868 water Inorganic materials 0.000 description 63
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 62
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 60
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 58
- 238000006243 chemical reaction Methods 0.000 description 56
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 51
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 48
- 230000015572 biosynthetic process Effects 0.000 description 46
- 239000002904 solvent Substances 0.000 description 46
- 238000003786 synthesis reaction Methods 0.000 description 46
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 45
- 239000000047 product Substances 0.000 description 45
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 42
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 42
- 239000002253 acid Substances 0.000 description 41
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 40
- 125000001424 substituent group Chemical group 0.000 description 37
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 35
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 34
- 239000003921 oil Substances 0.000 description 33
- WPYMKLBDIGXBTP-UHFFFAOYSA-M benzoate Chemical compound [O-]C(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-M 0.000 description 32
- 125000004432 carbon atom Chemical group C* 0.000 description 31
- 239000000741 silica gel Substances 0.000 description 31
- 229910002027 silica gel Inorganic materials 0.000 description 31
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 30
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 30
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 29
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 29
- 102100023085 Serine/threonine-protein kinase mTOR Human genes 0.000 description 29
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 28
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 28
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 28
- 238000000746 purification Methods 0.000 description 27
- 239000007787 solid Substances 0.000 description 27
- 229910052938 sodium sulfate Inorganic materials 0.000 description 24
- 238000003756 stirring Methods 0.000 description 24
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 23
- 125000003342 alkenyl group Chemical group 0.000 description 23
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 22
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 22
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 21
- 229910052757 nitrogen Inorganic materials 0.000 description 21
- 238000004128 high performance liquid chromatography Methods 0.000 description 20
- 125000003107 substituted aryl group Chemical group 0.000 description 20
- 239000007832 Na2SO4 Substances 0.000 description 19
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 19
- 230000002829 reductive effect Effects 0.000 description 18
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 17
- 229910052786 argon Inorganic materials 0.000 description 17
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 17
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 16
- 125000004122 cyclic group Chemical group 0.000 description 16
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 16
- 229960000583 acetic acid Drugs 0.000 description 15
- 125000002877 alkyl aryl group Chemical group 0.000 description 15
- 125000006413 ring segment Chemical group 0.000 description 15
- 125000000547 substituted alkyl group Chemical group 0.000 description 15
- 238000004458 analytical method Methods 0.000 description 14
- 239000000725 suspension Substances 0.000 description 14
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- 239000012267 brine Substances 0.000 description 13
- 229910052760 oxygen Inorganic materials 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 229910052717 sulfur Inorganic materials 0.000 description 13
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 150000004702 methyl esters Chemical class 0.000 description 12
- 239000001301 oxygen Substances 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- 125000004076 pyridyl group Chemical group 0.000 description 12
- 229910000104 sodium hydride Inorganic materials 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 11
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 11
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 11
- 239000001257 hydrogen Substances 0.000 description 11
- 230000014759 maintenance of location Effects 0.000 description 11
- 238000010898 silica gel chromatography Methods 0.000 description 11
- 238000004809 thin layer chromatography Methods 0.000 description 11
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 11
- 238000003818 flash chromatography Methods 0.000 description 10
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Substances IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 9
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 9
- 125000004429 atom Chemical group 0.000 description 9
- 229910052799 carbon Inorganic materials 0.000 description 9
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 9
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 9
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 9
- 125000001544 thienyl group Chemical group 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- 125000005213 alkyl heteroaryl group Chemical group 0.000 description 8
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 8
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 8
- 125000004434 sulfur atom Chemical group 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 7
- 125000001246 bromo group Chemical group Br* 0.000 description 7
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 7
- 125000001309 chloro group Chemical group Cl* 0.000 description 7
- 235000011152 sodium sulphate Nutrition 0.000 description 7
- 239000005711 Benzoic acid Substances 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 6
- 102000001253 Protein Kinase Human genes 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- 235000010233 benzoic acid Nutrition 0.000 description 6
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 6
- 125000000392 cycloalkenyl group Chemical group 0.000 description 6
- 125000000623 heterocyclic group Chemical group 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000002480 mineral oil Substances 0.000 description 6
- 235000010446 mineral oil Nutrition 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 108060006633 protein kinase Proteins 0.000 description 6
- 125000003226 pyrazolyl group Chemical group 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 6
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 5
- 208000023275 Autoimmune disease Diseases 0.000 description 5
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 description 5
- 102100036239 Cyclin-dependent kinase 2 Human genes 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 5
- 206010003246 arthritis Diseases 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 125000001153 fluoro group Chemical group F* 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 150000002367 halogens Chemical class 0.000 description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 5
- 208000015122 neurodegenerative disease Diseases 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- WPFZGADUIUVTCF-UHFFFAOYSA-N pyrazolo[1,5-a]pyrimidin-7-amine Chemical compound NC1=CC=NC2=CC=NN12 WPFZGADUIUVTCF-UHFFFAOYSA-N 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 239000011593 sulfur Substances 0.000 description 5
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 4
- 206010061218 Inflammation Diseases 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 102000009308 Mechanistic Target of Rapamycin Complex 2 Human genes 0.000 description 4
- 108010034057 Mechanistic Target of Rapamycin Complex 2 Proteins 0.000 description 4
- 208000031888 Mycoses Diseases 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 125000002252 acyl group Chemical group 0.000 description 4
- 125000004414 alkyl thio group Chemical group 0.000 description 4
- 125000000266 alpha-aminoacyl group Chemical group 0.000 description 4
- 230000001093 anti-cancer Effects 0.000 description 4
- WPYMKLBDIGXBTP-VQEHIDDOSA-N benzoic acid Chemical compound OC(=O)C1=CC=C[13CH]=C1 WPYMKLBDIGXBTP-VQEHIDDOSA-N 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 230000004663 cell proliferation Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 4
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 125000001624 naphthyl group Chemical group 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 235000011056 potassium acetate Nutrition 0.000 description 4
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 4
- 239000011698 potassium fluoride Substances 0.000 description 4
- 238000002953 preparative HPLC Methods 0.000 description 4
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 4
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 4
- 238000004007 reversed phase HPLC Methods 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 4
- 229960002930 sirolimus Drugs 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- GMYAQHAKWKXYHG-UHFFFAOYSA-N tributylstannylformonitrile Chemical compound CCCC[Sn](CCCC)(CCCC)C#N GMYAQHAKWKXYHG-UHFFFAOYSA-N 0.000 description 4
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- JVVRJMXHNUAPHW-UHFFFAOYSA-N 1h-pyrazol-5-amine Chemical compound NC=1C=CNN=1 JVVRJMXHNUAPHW-UHFFFAOYSA-N 0.000 description 3
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 3
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo(3.3.1)nonane Chemical compound C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 3
- 201000004384 Alopecia Diseases 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 101150050673 CHK1 gene Proteins 0.000 description 3
- 208000024172 Cardiovascular disease Diseases 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- NZNMSOFKMUBTKW-UHFFFAOYSA-N Cyclohexanecarboxylic acid Natural products OC(=O)C1CCCCC1 NZNMSOFKMUBTKW-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- 108060006678 I-kappa-B kinase Proteins 0.000 description 3
- 102000001284 I-kappa-B kinase Human genes 0.000 description 3
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 3
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 3
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 3
- 150000001204 N-oxides Chemical class 0.000 description 3
- 208000012902 Nervous system disease Diseases 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- 201000004681 Psoriasis Diseases 0.000 description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 3
- 241000534944 Thia Species 0.000 description 3
- QQIRAVWVGBTHMJ-UHFFFAOYSA-N [dimethyl-(trimethylsilylamino)silyl]methane;lithium Chemical compound [Li].C[Si](C)(C)N[Si](C)(C)C QQIRAVWVGBTHMJ-UHFFFAOYSA-N 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 3
- 125000005038 alkynylalkyl group Chemical group 0.000 description 3
- 231100000360 alopecia Toxicity 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 238000011394 anticancer treatment Methods 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 125000004659 aryl alkyl thio group Chemical group 0.000 description 3
- 125000005110 aryl thio group Chemical group 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 229910002092 carbon dioxide Inorganic materials 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- OGEBRHQLRGFBNV-RZDIXWSQSA-N chembl2036808 Chemical class C12=NC(NCCCC)=NC=C2C(C=2C=CC(F)=CC=2)=NN1C[C@H]1CC[C@H](N)CC1 OGEBRHQLRGFBNV-RZDIXWSQSA-N 0.000 description 3
- 229910052681 coesite Inorganic materials 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 229910052906 cristobalite Inorganic materials 0.000 description 3
- NZNMSOFKMUBTKW-UHFFFAOYSA-M cyclohexanecarboxylate Chemical compound [O-]C(=O)C1CCCCC1 NZNMSOFKMUBTKW-UHFFFAOYSA-M 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- 239000002254 cytotoxic agent Substances 0.000 description 3
- 231100000599 cytotoxic agent Toxicity 0.000 description 3
- 230000001747 exhibiting effect Effects 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 230000001537 neural effect Effects 0.000 description 3
- 230000000926 neurological effect Effects 0.000 description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 229910000160 potassium phosphate Inorganic materials 0.000 description 3
- 235000011009 potassium phosphates Nutrition 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- YGDICLRMNDWZAK-UHFFFAOYSA-N quinolin-3-ylboronic acid Chemical compound C1=CC=CC2=CC(B(O)O)=CN=C21 YGDICLRMNDWZAK-UHFFFAOYSA-N 0.000 description 3
- 238000001959 radiotherapy Methods 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 3
- 125000003003 spiro group Chemical group 0.000 description 3
- 229910052682 stishovite Inorganic materials 0.000 description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 3
- 229910052905 tridymite Inorganic materials 0.000 description 3
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- JEJWJXKIGYZTMJ-UHFFFAOYSA-N (5-bromothieno[2,3-b]pyridin-2-yl)-trimethylsilane Chemical compound BrC1=CN=C2SC([Si](C)(C)C)=CC2=C1 JEJWJXKIGYZTMJ-UHFFFAOYSA-N 0.000 description 2
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 2
- YFTHTJAPODJVSL-UHFFFAOYSA-N 2-(1-benzothiophen-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(SC=C2)C2=C1 YFTHTJAPODJVSL-UHFFFAOYSA-N 0.000 description 2
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 2
- RGHQKFQZGLKBCF-UHFFFAOYSA-N 2-bromoethyl acetate Chemical compound CC(=O)OCCBr RGHQKFQZGLKBCF-UHFFFAOYSA-N 0.000 description 2
- XLBHFDXPYKKVFI-UHFFFAOYSA-N 2-phenyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(C=2C=CC=CC=2)N=C1 XLBHFDXPYKKVFI-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- UBUWVSMWVYLZAV-UHFFFAOYSA-N 5-(4-methylsulfanylphenoxy)-n,n-bis(2-trimethylsilylethoxymethyl)pyrazolo[1,5-a]pyrimidin-7-amine Chemical compound C1=CC(SC)=CC=C1OC1=NC2=CC=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1 UBUWVSMWVYLZAV-UHFFFAOYSA-N 0.000 description 2
- KWXLZDLROUJSOQ-UHFFFAOYSA-N 5-[(4-methylsulfanylphenyl)methyl]-n,n-bis(2-trimethylsilylethoxymethyl)pyrazolo[1,5-a]pyrimidin-7-amine Chemical compound C1=CC(SC)=CC=C1CC1=NC2=CC=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1 KWXLZDLROUJSOQ-UHFFFAOYSA-N 0.000 description 2
- YMOORGYWRBQPDZ-UHFFFAOYSA-N 5-bromothieno[2,3-b]pyridine Chemical compound BrC1=CN=C2SC=CC2=C1 YMOORGYWRBQPDZ-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 2
- 208000032467 Aplastic anaemia Diseases 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 101710118321 Casein kinase I isoform alpha Proteins 0.000 description 2
- 102100034356 Casein kinase I isoform alpha-like Human genes 0.000 description 2
- 102100032857 Cyclin-dependent kinase 1 Human genes 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- 102100028554 Dual specificity tyrosine-phosphorylation-regulated kinase 1A Human genes 0.000 description 2
- 102100022466 Eukaryotic translation initiation factor 4E-binding protein 1 Human genes 0.000 description 2
- 108050000946 Eukaryotic translation initiation factor 4E-binding protein 1 Proteins 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerol Natural products OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 101000838016 Homo sapiens Dual specificity tyrosine-phosphorylation-regulated kinase 1A Proteins 0.000 description 2
- 101001052493 Homo sapiens Mitogen-activated protein kinase 1 Proteins 0.000 description 2
- 101000628949 Homo sapiens Mitogen-activated protein kinase 10 Proteins 0.000 description 2
- 101000950695 Homo sapiens Mitogen-activated protein kinase 8 Proteins 0.000 description 2
- 101000950669 Homo sapiens Mitogen-activated protein kinase 9 Proteins 0.000 description 2
- 101000987310 Homo sapiens Serine/threonine-protein kinase PAK 2 Proteins 0.000 description 2
- 101000983111 Homo sapiens Serine/threonine-protein kinase PAK 6 Proteins 0.000 description 2
- 101001046426 Homo sapiens cGMP-dependent protein kinase 1 Proteins 0.000 description 2
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 description 2
- 102100026931 Mitogen-activated protein kinase 10 Human genes 0.000 description 2
- 102100037808 Mitogen-activated protein kinase 8 Human genes 0.000 description 2
- 102100037809 Mitogen-activated protein kinase 9 Human genes 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 108090000315 Protein Kinase C Proteins 0.000 description 2
- 102000003923 Protein Kinase C Human genes 0.000 description 2
- 102100024908 Ribosomal protein S6 kinase beta-1 Human genes 0.000 description 2
- 101710108924 Ribosomal protein S6 kinase beta-1 Proteins 0.000 description 2
- 102100029680 Serine/threonine-protein kinase Kist Human genes 0.000 description 2
- 102100031206 Serine/threonine-protein kinase N1 Human genes 0.000 description 2
- 102100027939 Serine/threonine-protein kinase PAK 2 Human genes 0.000 description 2
- 102100026840 Serine/threonine-protein kinase PAK 6 Human genes 0.000 description 2
- 241000710960 Sindbis virus Species 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 125000003158 alcohol group Chemical group 0.000 description 2
- 150000001447 alkali salts Chemical class 0.000 description 2
- 125000005206 alkoxycarbonyloxymethyl group Chemical group 0.000 description 2
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- 125000003435 aroyl group Chemical group 0.000 description 2
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 2
- 125000005015 aryl alkynyl group Chemical group 0.000 description 2
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 2
- 229940120638 avastin Drugs 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- HGXJOXHYPGNVNK-UHFFFAOYSA-N butane;ethenoxyethane;tin Chemical compound CCCC[Sn](CCCC)(CCCC)C(=C)OCC HGXJOXHYPGNVNK-UHFFFAOYSA-N 0.000 description 2
- 102100022422 cGMP-dependent protein kinase 1 Human genes 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 230000002113 chemopreventative effect Effects 0.000 description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 2
- VZFUCHSFHOYXIS-UHFFFAOYSA-N cycloheptane carboxylic acid Natural products OC(=O)C1CCCCCC1 VZFUCHSFHOYXIS-UHFFFAOYSA-N 0.000 description 2
- ZQWPRMPSCMSAJU-UHFFFAOYSA-N cyclohexanecarboxylic acid methyl ester Natural products COC(=O)C1CCCCC1 ZQWPRMPSCMSAJU-UHFFFAOYSA-N 0.000 description 2
- 238000002716 delivery method Methods 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- UXXWGBLTOSRBAY-UHFFFAOYSA-N ethyl 1,4-dioxaspiro[4.5]decane-8-carboxylate Chemical compound C1CC(C(=O)OCC)CCC21OCCO2 UXXWGBLTOSRBAY-UHFFFAOYSA-N 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 125000004475 heteroaralkyl group Chemical group 0.000 description 2
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical class I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- RCCPEORTSYDPMB-UHFFFAOYSA-N hydroxy benzenecarboximidothioate Chemical compound OSC(=N)C1=CC=CC=C1 RCCPEORTSYDPMB-UHFFFAOYSA-N 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 230000002297 mitogenic effect Effects 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 2
- 150000002923 oximes Chemical group 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- SIOXPEMLGUPBBT-UHFFFAOYSA-N picolinic acid Chemical compound OC(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-N 0.000 description 2
- HXEACLLIILLPRG-UHFFFAOYSA-N pipecolic acid Chemical compound OC(=O)C1CCCCN1 HXEACLLIILLPRG-UHFFFAOYSA-N 0.000 description 2
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- 235000003270 potassium fluoride Nutrition 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical group O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 2
- 125000004548 quinolin-3-yl group Chemical group N1=CC(=CC2=CC=CC=C12)* 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 125000005017 substituted alkenyl group Chemical group 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 238000006257 total synthesis reaction Methods 0.000 description 2
- GGUBFICZYGKNTD-UHFFFAOYSA-N triethyl phosphonoacetate Chemical compound CCOC(=O)CP(=O)(OCC)OCC GGUBFICZYGKNTD-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical class CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- PAORVUMOXXAMPL-SECBINFHSA-N (2s)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoyl chloride Chemical compound CO[C@](C(Cl)=O)(C(F)(F)F)C1=CC=CC=C1 PAORVUMOXXAMPL-SECBINFHSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000005859 (C1-C6)alkanoyloxymethyl group Chemical group 0.000 description 1
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 1
- XJLSEXAGTJCILF-RXMQYKEDSA-N (R)-nipecotic acid zwitterion Chemical compound OC(=O)[C@@H]1CCCNC1 XJLSEXAGTJCILF-RXMQYKEDSA-N 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000004530 1,2,4-triazinyl group Chemical group N1=NC(=NC=C1)* 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- YMEMAALYLSKCLM-UHFFFAOYSA-N 1,4-dioxaspiro[4.5]decane-8-carboxylic acid Chemical compound C1CC(C(=O)O)CCC21OCCO2 YMEMAALYLSKCLM-UHFFFAOYSA-N 0.000 description 1
- ZZFHYYHMRANMAT-UHFFFAOYSA-N 1,6-bis[(2-methylpropan-2-yl)oxycarbonyl]piperidine-3-carboxylic acid Chemical compound CC(C)(C)OC(=O)C1CCC(C(O)=O)CN1C(=O)OC(C)(C)C ZZFHYYHMRANMAT-UHFFFAOYSA-N 0.000 description 1
- 125000005846 1-(alkanoyloxy)ethyl group Chemical group 0.000 description 1
- 125000005848 1-(alkoxycarbonyloxy)ethyl group Chemical group 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical class CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- 125000005847 1-methyl-1-(alkanoyloxy)-ethyl group Chemical group 0.000 description 1
- 125000005849 1-methyl-1-(alkoxycarbonyloxy)ethyl group Chemical group 0.000 description 1
- 125000001088 1-naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- HAAWUJYUZIVHEE-UHFFFAOYSA-N 1-o,2-o-ditert-butyl 5-o-methyl piperidine-1,2,5-tricarboxylate Chemical compound COC(=O)C1CCC(C(=O)OC(C)(C)C)N(C(=O)OC(C)(C)C)C1 HAAWUJYUZIVHEE-UHFFFAOYSA-N 0.000 description 1
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 1
- KEZNMOUMHOZFRA-UHFFFAOYSA-N 1h-pyrazol-4-ylboronic acid Chemical compound OB(O)C=1C=NNC=1 KEZNMOUMHOZFRA-UHFFFAOYSA-N 0.000 description 1
- IAAQUOVTPAMQCR-UHFFFAOYSA-N 1h-pyrido[3,2-d]pyrimidin-2-one Chemical compound C1=CC=C2NC(=O)N=CC2=N1 IAAQUOVTPAMQCR-UHFFFAOYSA-N 0.000 description 1
- GELKGHVAFRCJNA-UHFFFAOYSA-N 2,2-Dimethyloxirane Chemical compound CC1(C)CO1 GELKGHVAFRCJNA-UHFFFAOYSA-N 0.000 description 1
- WEHMOUKAAGQMFZ-UHFFFAOYSA-N 2-[4-(7-amino-6-bromo-3-quinolin-3-ylpyrazolo[1,5-a]pyrimidin-5-yl)oxyphenyl]acetic acid Chemical compound N=1C2=C(C=3C=C4C=CC=CC4=NC=3)C=NN2C(N)=C(Br)C=1OC1=CC=C(CC(O)=O)C=C1 WEHMOUKAAGQMFZ-UHFFFAOYSA-N 0.000 description 1
- HOBBCPXBGQXEKF-UHFFFAOYSA-N 2-[4-[7-[bis(2-trimethylsilylethoxymethyl)amino]-3-iodopyrazolo[1,5-a]pyrimidin-5-yl]cyclohexyl]acetonitrile Chemical group N=1C2=C(I)C=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=CC=1C1CCC(CC#N)CC1 HOBBCPXBGQXEKF-UHFFFAOYSA-N 0.000 description 1
- GXOZDATXTYWUAC-UHFFFAOYSA-N 2-[4-[7-[bis(2-trimethylsilylethoxymethyl)amino]-3-quinolin-3-ylpyrazolo[1,5-a]pyrimidin-5-yl]cyclohexyl]acetonitrile Chemical compound N=1C2=C(C=3C=C4C=CC=CC4=NC=3)C=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=CC=1C1CCC(CC#N)CC1 GXOZDATXTYWUAC-UHFFFAOYSA-N 0.000 description 1
- WGDFSJPIEFVOJJ-UHFFFAOYSA-N 2-[4-[7-amino-6-bromo-3-(6-bromonaphthalen-2-yl)pyrazolo[1,5-a]pyrimidin-5-yl]cyclohexyl]-n-methylsulfonylacetamide Chemical compound C1CC(CC(=O)NS(=O)(=O)C)CCC1C1=NC2=C(C=3C=C4C=CC(Br)=CC4=CC=3)C=NN2C(N)=C1Br WGDFSJPIEFVOJJ-UHFFFAOYSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- SURMYNZXHKLDFO-UHFFFAOYSA-N 2-bromopropanedial Chemical compound O=CC(Br)C=O SURMYNZXHKLDFO-UHFFFAOYSA-N 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- IEFBTHROULVUAH-UHFFFAOYSA-N 2-chloro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical group O1C(C)(C)C(C)(C)OB1C1=CC=C(Cl)N=C1 IEFBTHROULVUAH-UHFFFAOYSA-N 0.000 description 1
- KWMBADTWRIGGGG-UHFFFAOYSA-N 2-diethoxyphosphorylacetonitrile Chemical group CCOP(=O)(CC#N)OCC KWMBADTWRIGGGG-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- JJKWHOSQTYYFAE-UHFFFAOYSA-N 2-methoxyacetyl chloride Chemical compound COCC(Cl)=O JJKWHOSQTYYFAE-UHFFFAOYSA-N 0.000 description 1
- XROIPUGZLLIXAO-UHFFFAOYSA-N 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)furo[2,3-b]pyridine Chemical compound C=1N=C2OC(C)=CC2=CC=1B1OC(C)(C)C(C)(C)O1 XROIPUGZLLIXAO-UHFFFAOYSA-N 0.000 description 1
- YAGYHIJKSZSNTG-UHFFFAOYSA-N 2-o-tert-butyl 5-o-methyl piperidine-2,5-dicarboxylate Chemical compound COC(=O)C1CCC(C(=O)OC(C)(C)C)NC1 YAGYHIJKSZSNTG-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- KIWODJBCHRADND-UHFFFAOYSA-N 3-anilino-4-[1-[3-(1-imidazolyl)propyl]-3-indolyl]pyrrole-2,5-dione Chemical compound O=C1NC(=O)C(C=2C3=CC=CC=C3N(CCCN3C=NC=C3)C=2)=C1NC1=CC=CC=C1 KIWODJBCHRADND-UHFFFAOYSA-N 0.000 description 1
- GAUYSLCHJGXKCX-UHFFFAOYSA-N 3-iodo-5-(4-methylsulfanylphenoxy)-n,n-bis(2-trimethylsilylethoxymethyl)pyrazolo[1,5-a]pyrimidin-7-amine Chemical compound C1=CC(SC)=CC=C1OC1=NC2=C(I)C=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1 GAUYSLCHJGXKCX-UHFFFAOYSA-N 0.000 description 1
- 101710082567 3-methylorcinaldehyde synthase Proteins 0.000 description 1
- 125000004364 3-pyrrolinyl group Chemical group [H]C1=C([H])C([H])([H])N(*)C1([H])[H] 0.000 description 1
- TVOJIBGZFYMWDT-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CNN=C1 TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 1
- IMWLLBAFSFXIBI-UHFFFAOYSA-N 4-(7-aminopyrazolo[1,5-a]pyrimidin-5-yl)cyclohexan-1-one Chemical compound N=1C2=CC=NN2C(N)=CC=1C1CCC(=O)CC1 IMWLLBAFSFXIBI-UHFFFAOYSA-N 0.000 description 1
- QASBCTGZKABPKX-UHFFFAOYSA-N 4-(methylsulfanyl)phenol Chemical compound CSC1=CC=C(O)C=C1 QASBCTGZKABPKX-UHFFFAOYSA-N 0.000 description 1
- WOJGEDMHAUFCQX-UHFFFAOYSA-N 4-[6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl]benzoic acid Chemical compound C=12N=C(C=3C=CC(=CC=3)C(O)=O)C(C(=O)C)=C(N)N2N=CC=1C(C=N1)=CC=C1C1=CC=CC=C1 WOJGEDMHAUFCQX-UHFFFAOYSA-N 0.000 description 1
- CUVLPLSVQOTZII-UHFFFAOYSA-N 4-[7-[bis(2-trimethylsilylethoxymethyl)amino]pyrazolo[1,5-a]pyrimidin-5-yl]cyclohexan-1-one Chemical compound N=1C2=CC=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=CC=1C1CCC(=O)CC1 CUVLPLSVQOTZII-UHFFFAOYSA-N 0.000 description 1
- DOWWQQGCYDTMOC-UHFFFAOYSA-N 4-[7-amino-6-cyano-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl]benzoic acid Chemical compound C1=NN2C(N)=C(C#N)C(C=3C=CC(=CC=3)C(O)=O)=NC2=C1C(C=N1)=CC=C1C1=CC=CC=C1 DOWWQQGCYDTMOC-UHFFFAOYSA-N 0.000 description 1
- ZQJNPHCQABYENK-UHFFFAOYSA-N 4-methoxycarbonylcyclohexane-1-carboxylic acid Chemical compound COC(=O)C1CCC(C(O)=O)CC1 ZQJNPHCQABYENK-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 102100036009 5'-AMP-activated protein kinase catalytic subunit alpha-2 Human genes 0.000 description 1
- PWNZOGLNPAOHSL-UHFFFAOYSA-N 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)furo[2,3-b]pyridine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN=C(OC=C2)C2=C1 PWNZOGLNPAOHSL-UHFFFAOYSA-N 0.000 description 1
- ZFALGGJIXOGZJQ-UHFFFAOYSA-N 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thieno[2,3-b]pyridine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN=C(SC=C2)C2=C1 ZFALGGJIXOGZJQ-UHFFFAOYSA-N 0.000 description 1
- NDMZZQRNZFWMEZ-UHFFFAOYSA-N 5-bromo-1h-pyridin-2-one Chemical compound OC1=CC=C(Br)C=N1 NDMZZQRNZFWMEZ-UHFFFAOYSA-N 0.000 description 1
- IXGZQYCKHWDIJC-UHFFFAOYSA-N 5-bromo-2-methylfuro[2,3-b]pyridine Chemical compound BrC1=CN=C2OC(C)=CC2=C1 IXGZQYCKHWDIJC-UHFFFAOYSA-N 0.000 description 1
- WAUPPDAXWKLNNU-UHFFFAOYSA-N 5-bromo-3-iodo-1h-pyridin-2-one Chemical compound BrC1=CNC(=O)C(I)=C1 WAUPPDAXWKLNNU-UHFFFAOYSA-N 0.000 description 1
- UILPFSXNAGPJRZ-UHFFFAOYSA-N 5-chloro-3-quinolin-3-yl-n,n-bis(2-trimethylsilylethoxymethyl)pyrazolo[1,5-a]pyrimidin-7-amine Chemical compound C1=CC=CC2=CC(C3=C4N=C(Cl)C=C(N4N=C3)N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=CN=C21 UILPFSXNAGPJRZ-UHFFFAOYSA-N 0.000 description 1
- HIOBMQBWVIPQQC-UHFFFAOYSA-N 5-chloro-n,n-bis(2-trimethylsilylethoxymethyl)pyrazolo[1,5-a]pyrimidin-7-amine Chemical compound C[Si](C)(C)CCOCN(COCC[Si](C)(C)C)C1=CC(Cl)=NC2=CC=NN12 HIOBMQBWVIPQQC-UHFFFAOYSA-N 0.000 description 1
- DGWMSNIIORGYBE-UHFFFAOYSA-N 6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thieno[3,2-b]pyridine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN=C(C=CS2)C2=C1 DGWMSNIIORGYBE-UHFFFAOYSA-N 0.000 description 1
- OAQYSYGLIZFGFU-UHFFFAOYSA-N 6-bromo-5-[(4-methylsulfonylphenyl)methyl]-3-quinolin-3-ylpyrazolo[1,5-a]pyrimidin-7-amine Chemical compound C1=CC(S(=O)(=O)C)=CC=C1CC1=NC2=C(C=3C=C4C=CC=CC4=NC=3)C=NN2C(N)=C1Br OAQYSYGLIZFGFU-UHFFFAOYSA-N 0.000 description 1
- 102100021546 60S ribosomal protein L10 Human genes 0.000 description 1
- CMUMJUNDDARAIX-UHFFFAOYSA-N 7-chloro-5-(1,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[1,5-a]pyrimidine Chemical compound N=1C2=CC=NN2C(Cl)=CC=1C(CC1)CCC21OCCO2 CMUMJUNDDARAIX-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- XUOBFFPGZFFAIT-UHFFFAOYSA-N 8-azaspiro[4.5]dec-2-ene Chemical compound C1C=CCC11CCNCC1 XUOBFFPGZFFAIT-UHFFFAOYSA-N 0.000 description 1
- AMKGKYQBASDDJB-UHFFFAOYSA-N 9$l^{2}-borabicyclo[3.3.1]nonane Chemical compound C1CCC2CCCC1[B]2 AMKGKYQBASDDJB-UHFFFAOYSA-N 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 102100036409 Activated CDC42 kinase 1 Human genes 0.000 description 1
- 239000000275 Adrenocorticotropic Hormone Substances 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 108010004586 Ataxia Telangiectasia Mutated Proteins Proteins 0.000 description 1
- 102100032306 Aurora kinase B Human genes 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 206010065687 Bone loss Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 229940126074 CDK kinase inhibitor Drugs 0.000 description 1
- 108091011896 CSF1 Proteins 0.000 description 1
- 101100015811 Caenorhabditis elegans grk-2 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 206010058019 Cancer Pain Diseases 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 108010002947 Connectin Proteins 0.000 description 1
- 102000004726 Connectin Human genes 0.000 description 1
- 101800000414 Corticotropin Proteins 0.000 description 1
- 101150069913 Csk gene Proteins 0.000 description 1
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 1
- 108010025454 Cyclin-Dependent Kinase 5 Proteins 0.000 description 1
- 108010025468 Cyclin-Dependent Kinase 6 Proteins 0.000 description 1
- 101710106279 Cyclin-dependent kinase 1 Proteins 0.000 description 1
- 102100033245 Cyclin-dependent kinase 16 Human genes 0.000 description 1
- 102100033145 Cyclin-dependent kinase 19 Human genes 0.000 description 1
- 102100034741 Cyclin-dependent kinase 20 Human genes 0.000 description 1
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 description 1
- 102100026804 Cyclin-dependent kinase 6 Human genes 0.000 description 1
- 102100026810 Cyclin-dependent kinase 7 Human genes 0.000 description 1
- 102100024457 Cyclin-dependent kinase 9 Human genes 0.000 description 1
- 102100034770 Cyclin-dependent kinase inhibitor 3 Human genes 0.000 description 1
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 1
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 1
- 102100026805 Cyclin-dependent-like kinase 5 Human genes 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 101100503636 Danio rerio fyna gene Proteins 0.000 description 1
- 101100457345 Danio rerio mapk14a gene Proteins 0.000 description 1
- 101100457347 Danio rerio mapk14b gene Proteins 0.000 description 1
- 101100481404 Danio rerio tie1 gene Proteins 0.000 description 1
- 101100481408 Danio rerio tie2 gene Proteins 0.000 description 1
- 101100314281 Danio rerio trappc11 gene Proteins 0.000 description 1
- 108010031042 Death-Associated Protein Kinases Proteins 0.000 description 1
- 102100038587 Death-associated protein kinase 1 Human genes 0.000 description 1
- 102100038605 Death-associated protein kinase 2 Human genes 0.000 description 1
- 102100038606 Death-associated protein kinase 3 Human genes 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 101001044938 Dictyostelium discoideum Diacylglycerol kinase A Proteins 0.000 description 1
- 101000876610 Dictyostelium discoideum Extracellular signal-regulated kinase 2 Proteins 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 101100181139 Drosophila melanogaster Pkcdelta gene Proteins 0.000 description 1
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 1
- 206010013774 Dry eye Diseases 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 102100023266 Dual specificity mitogen-activated protein kinase kinase 2 Human genes 0.000 description 1
- 102100023275 Dual specificity mitogen-activated protein kinase kinase 3 Human genes 0.000 description 1
- 102100023274 Dual specificity mitogen-activated protein kinase kinase 4 Human genes 0.000 description 1
- 102100023401 Dual specificity mitogen-activated protein kinase kinase 6 Human genes 0.000 description 1
- 102100023332 Dual specificity mitogen-activated protein kinase kinase 7 Human genes 0.000 description 1
- 102100040862 Dual specificity protein kinase CLK1 Human genes 0.000 description 1
- 102100036492 Dual specificity testis-specific protein kinase 1 Human genes 0.000 description 1
- 102100036498 Dual specificity testis-specific protein kinase 2 Human genes 0.000 description 1
- 102100023115 Dual specificity tyrosine-phosphorylation-regulated kinase 2 Human genes 0.000 description 1
- 102100023114 Dual specificity tyrosine-phosphorylation-regulated kinase 3 Human genes 0.000 description 1
- 206010014824 Endotoxic shock Diseases 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 108010055191 EphA3 Receptor Proteins 0.000 description 1
- 108010055179 EphA4 Receptor Proteins 0.000 description 1
- 108010055155 EphA8 Receptor Proteins 0.000 description 1
- 108010055334 EphB2 Receptor Proteins 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- 102100030324 Ephrin type-A receptor 3 Human genes 0.000 description 1
- 102100021616 Ephrin type-A receptor 4 Human genes 0.000 description 1
- 102100021601 Ephrin type-A receptor 8 Human genes 0.000 description 1
- 102100030779 Ephrin type-B receptor 1 Human genes 0.000 description 1
- 102100031968 Ephrin type-B receptor 2 Human genes 0.000 description 1
- 102100031982 Ephrin type-B receptor 3 Human genes 0.000 description 1
- 101100306202 Escherichia coli (strain K12) rpoB gene Proteins 0.000 description 1
- 101100480905 Escherichia phage P1 tec gene Proteins 0.000 description 1
- 102100034169 Eukaryotic translation initiation factor 2-alpha kinase 1 Human genes 0.000 description 1
- 101150036586 FES gene Proteins 0.000 description 1
- 101150018272 FYN gene Proteins 0.000 description 1
- 101150093530 Fer gene Proteins 0.000 description 1
- 101150040897 Fgr gene Proteins 0.000 description 1
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 1
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 1
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 1
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 1
- 102100027844 Fibroblast growth factor receptor 4 Human genes 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical class OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- 101150023186 GRK1 gene Proteins 0.000 description 1
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 1
- 102100031132 Glucose-6-phosphate isomerase Human genes 0.000 description 1
- 108010070600 Glucose-6-phosphate isomerase Proteins 0.000 description 1
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 description 1
- 102000019058 Glycogen Synthase Kinase 3 beta Human genes 0.000 description 1
- 208000002927 Hamartoma Diseases 0.000 description 1
- 206010019668 Hepatic fibrosis Diseases 0.000 description 1
- 102100035108 High affinity nerve growth factor receptor Human genes 0.000 description 1
- 102100032827 Homeodomain-interacting protein kinase 2 Human genes 0.000 description 1
- 102100032826 Homeodomain-interacting protein kinase 3 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000783681 Homo sapiens 5'-AMP-activated protein kinase catalytic subunit alpha-2 Proteins 0.000 description 1
- 101001108634 Homo sapiens 60S ribosomal protein L10 Proteins 0.000 description 1
- 101001117935 Homo sapiens 60S ribosomal protein L15 Proteins 0.000 description 1
- 101000779641 Homo sapiens ALK tyrosine kinase receptor Proteins 0.000 description 1
- 101000928956 Homo sapiens Activated CDC42 kinase 1 Proteins 0.000 description 1
- 101000798306 Homo sapiens Aurora kinase B Proteins 0.000 description 1
- 101000868333 Homo sapiens Cyclin-dependent kinase 1 Proteins 0.000 description 1
- 101000944357 Homo sapiens Cyclin-dependent kinase 16 Proteins 0.000 description 1
- 101000944345 Homo sapiens Cyclin-dependent kinase 19 Proteins 0.000 description 1
- 101000945708 Homo sapiens Cyclin-dependent kinase 20 Proteins 0.000 description 1
- 101000911952 Homo sapiens Cyclin-dependent kinase 7 Proteins 0.000 description 1
- 101000980930 Homo sapiens Cyclin-dependent kinase 9 Proteins 0.000 description 1
- 101000945639 Homo sapiens Cyclin-dependent kinase inhibitor 3 Proteins 0.000 description 1
- 101000909198 Homo sapiens DNA polymerase delta catalytic subunit Proteins 0.000 description 1
- 101000956145 Homo sapiens Death-associated protein kinase 1 Proteins 0.000 description 1
- 101000956149 Homo sapiens Death-associated protein kinase 3 Proteins 0.000 description 1
- 101001115394 Homo sapiens Dual specificity mitogen-activated protein kinase kinase 3 Proteins 0.000 description 1
- 101001115395 Homo sapiens Dual specificity mitogen-activated protein kinase kinase 4 Proteins 0.000 description 1
- 101000624426 Homo sapiens Dual specificity mitogen-activated protein kinase kinase 6 Proteins 0.000 description 1
- 101000624594 Homo sapiens Dual specificity mitogen-activated protein kinase kinase 7 Proteins 0.000 description 1
- 101000749294 Homo sapiens Dual specificity protein kinase CLK1 Proteins 0.000 description 1
- 101000714159 Homo sapiens Dual specificity testis-specific protein kinase 1 Proteins 0.000 description 1
- 101000714156 Homo sapiens Dual specificity testis-specific protein kinase 2 Proteins 0.000 description 1
- 101001049990 Homo sapiens Dual specificity tyrosine-phosphorylation-regulated kinase 2 Proteins 0.000 description 1
- 101001049991 Homo sapiens Dual specificity tyrosine-phosphorylation-regulated kinase 3 Proteins 0.000 description 1
- 101000967216 Homo sapiens Eosinophil cationic protein Proteins 0.000 description 1
- 101001064150 Homo sapiens Ephrin type-B receptor 1 Proteins 0.000 description 1
- 101001064458 Homo sapiens Ephrin type-B receptor 3 Proteins 0.000 description 1
- 101000926530 Homo sapiens Eukaryotic translation initiation factor 2-alpha kinase 1 Proteins 0.000 description 1
- 101000917134 Homo sapiens Fibroblast growth factor receptor 4 Proteins 0.000 description 1
- 101000878536 Homo sapiens Focal adhesion kinase 1 Proteins 0.000 description 1
- 101001066435 Homo sapiens Hepatocyte growth factor-like protein Proteins 0.000 description 1
- 101000596894 Homo sapiens High affinity nerve growth factor receptor Proteins 0.000 description 1
- 101001066401 Homo sapiens Homeodomain-interacting protein kinase 2 Proteins 0.000 description 1
- 101001066389 Homo sapiens Homeodomain-interacting protein kinase 3 Proteins 0.000 description 1
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 1
- 101000599951 Homo sapiens Insulin-like growth factor I Proteins 0.000 description 1
- 101001002695 Homo sapiens Integrin-linked protein kinase Proteins 0.000 description 1
- 101000926535 Homo sapiens Interferon-induced, double-stranded RNA-activated protein kinase Proteins 0.000 description 1
- 101000977771 Homo sapiens Interleukin-1 receptor-associated kinase 4 Proteins 0.000 description 1
- 101001137642 Homo sapiens Kinase suppressor of Ras 1 Proteins 0.000 description 1
- 101001005128 Homo sapiens LIM domain kinase 1 Proteins 0.000 description 1
- 101001042360 Homo sapiens LIM domain kinase 2 Proteins 0.000 description 1
- 101000763322 Homo sapiens M1-specific T cell receptor beta chain Proteins 0.000 description 1
- 101000628967 Homo sapiens Mitogen-activated protein kinase 11 Proteins 0.000 description 1
- 101000628954 Homo sapiens Mitogen-activated protein kinase 12 Proteins 0.000 description 1
- 101000628968 Homo sapiens Mitogen-activated protein kinase 13 Proteins 0.000 description 1
- 101001052490 Homo sapiens Mitogen-activated protein kinase 3 Proteins 0.000 description 1
- 101001052477 Homo sapiens Mitogen-activated protein kinase 4 Proteins 0.000 description 1
- 101000950710 Homo sapiens Mitogen-activated protein kinase 6 Proteins 0.000 description 1
- 101000950687 Homo sapiens Mitogen-activated protein kinase 7 Proteins 0.000 description 1
- 101001005602 Homo sapiens Mitogen-activated protein kinase kinase kinase 11 Proteins 0.000 description 1
- 101001005550 Homo sapiens Mitogen-activated protein kinase kinase kinase 14 Proteins 0.000 description 1
- 101001018196 Homo sapiens Mitogen-activated protein kinase kinase kinase 5 Proteins 0.000 description 1
- 101001055092 Homo sapiens Mitogen-activated protein kinase kinase kinase 7 Proteins 0.000 description 1
- 101001055091 Homo sapiens Mitogen-activated protein kinase kinase kinase 8 Proteins 0.000 description 1
- 101001059991 Homo sapiens Mitogen-activated protein kinase kinase kinase kinase 1 Proteins 0.000 description 1
- 101001059990 Homo sapiens Mitogen-activated protein kinase kinase kinase kinase 2 Proteins 0.000 description 1
- 101001059984 Homo sapiens Mitogen-activated protein kinase kinase kinase kinase 4 Proteins 0.000 description 1
- 101000613563 Homo sapiens PAS domain-containing serine/threonine-protein kinase Proteins 0.000 description 1
- 101100243116 Homo sapiens PDK1 gene Proteins 0.000 description 1
- 101100463123 Homo sapiens PDK3 gene Proteins 0.000 description 1
- 101100463125 Homo sapiens PDK4 gene Proteins 0.000 description 1
- 101000605639 Homo sapiens Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform Proteins 0.000 description 1
- 101000595741 Homo sapiens Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform Proteins 0.000 description 1
- 101000595746 Homo sapiens Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Proteins 0.000 description 1
- 101000595751 Homo sapiens Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform Proteins 0.000 description 1
- 101000945272 Homo sapiens Phosphorylase b kinase regulatory subunit alpha, liver isoform Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101000979748 Homo sapiens Protein NDRG1 Proteins 0.000 description 1
- 101000878540 Homo sapiens Protein-tyrosine kinase 2-beta Proteins 0.000 description 1
- 101000712530 Homo sapiens RAF proto-oncogene serine/threonine-protein kinase Proteins 0.000 description 1
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 101000927796 Homo sapiens Rho guanine nucleotide exchange factor 7 Proteins 0.000 description 1
- 101000669917 Homo sapiens Rho-associated protein kinase 1 Proteins 0.000 description 1
- 101000669921 Homo sapiens Rho-associated protein kinase 2 Proteins 0.000 description 1
- 101001051714 Homo sapiens Ribosomal protein S6 kinase beta-2 Proteins 0.000 description 1
- 101000648174 Homo sapiens Serine/threonine-protein kinase 10 Proteins 0.000 description 1
- 101000628647 Homo sapiens Serine/threonine-protein kinase 24 Proteins 0.000 description 1
- 101000701401 Homo sapiens Serine/threonine-protein kinase 38 Proteins 0.000 description 1
- 101000697608 Homo sapiens Serine/threonine-protein kinase 38-like Proteins 0.000 description 1
- 101000880431 Homo sapiens Serine/threonine-protein kinase 4 Proteins 0.000 description 1
- 101000695043 Homo sapiens Serine/threonine-protein kinase BRSK1 Proteins 0.000 description 1
- 101000777293 Homo sapiens Serine/threonine-protein kinase Chk1 Proteins 0.000 description 1
- 101000777277 Homo sapiens Serine/threonine-protein kinase Chk2 Proteins 0.000 description 1
- 101000885321 Homo sapiens Serine/threonine-protein kinase DCLK1 Proteins 0.000 description 1
- 101000913761 Homo sapiens Serine/threonine-protein kinase ICK Proteins 0.000 description 1
- 101000939549 Homo sapiens Serine/threonine-protein kinase Kist Proteins 0.000 description 1
- 101001129076 Homo sapiens Serine/threonine-protein kinase N1 Proteins 0.000 description 1
- 101001123846 Homo sapiens Serine/threonine-protein kinase Nek1 Proteins 0.000 description 1
- 101000601441 Homo sapiens Serine/threonine-protein kinase Nek2 Proteins 0.000 description 1
- 101000588540 Homo sapiens Serine/threonine-protein kinase Nek6 Proteins 0.000 description 1
- 101000588553 Homo sapiens Serine/threonine-protein kinase Nek9 Proteins 0.000 description 1
- 101000987315 Homo sapiens Serine/threonine-protein kinase PAK 3 Proteins 0.000 description 1
- 101000987295 Homo sapiens Serine/threonine-protein kinase PAK 5 Proteins 0.000 description 1
- 101000729945 Homo sapiens Serine/threonine-protein kinase PLK2 Proteins 0.000 description 1
- 101000691614 Homo sapiens Serine/threonine-protein kinase PLK3 Proteins 0.000 description 1
- 101000577652 Homo sapiens Serine/threonine-protein kinase PRP4 homolog Proteins 0.000 description 1
- 101000665442 Homo sapiens Serine/threonine-protein kinase TBK1 Proteins 0.000 description 1
- 101000649929 Homo sapiens Serine/threonine-protein kinase VRK1 Proteins 0.000 description 1
- 101000763321 Homo sapiens T cell receptor beta chain MC.7.G5 Proteins 0.000 description 1
- 101000823316 Homo sapiens Tyrosine-protein kinase ABL1 Proteins 0.000 description 1
- 101000823271 Homo sapiens Tyrosine-protein kinase ABL2 Proteins 0.000 description 1
- 101001009087 Homo sapiens Tyrosine-protein kinase HCK Proteins 0.000 description 1
- 101001050476 Homo sapiens Tyrosine-protein kinase ITK/TSK Proteins 0.000 description 1
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 1
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- 101000606067 Homo sapiens Tyrosine-protein kinase TXK Proteins 0.000 description 1
- 101000818543 Homo sapiens Tyrosine-protein kinase ZAP-70 Proteins 0.000 description 1
- 101000577737 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp4 Proteins 0.000 description 1
- 101000851018 Homo sapiens Vascular endothelial growth factor receptor 1 Proteins 0.000 description 1
- 101000739853 Homo sapiens [3-methyl-2-oxobutanoate dehydrogenase [lipoamide]] kinase, mitochondrial Proteins 0.000 description 1
- 101001117143 Homo sapiens [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 2, mitochondrial Proteins 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 101150026109 INSR gene Proteins 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 1
- 102100037852 Insulin-like growth factor I Human genes 0.000 description 1
- 102100020944 Integrin-linked protein kinase Human genes 0.000 description 1
- 102100034170 Interferon-induced, double-stranded RNA-activated protein kinase Human genes 0.000 description 1
- 102100023533 Interleukin-1 receptor-associated kinase 4 Human genes 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 102100021001 Kinase suppressor of Ras 1 Human genes 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- 150000000994 L-ascorbates Chemical class 0.000 description 1
- 125000003580 L-valyl group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(C([H])([H])[H])(C([H])([H])[H])[H] 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 102100026023 LIM domain kinase 1 Human genes 0.000 description 1
- 102100021756 LIM domain kinase 2 Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 101150028321 Lck gene Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 101150058160 Lyn gene Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102100026964 M1-specific T cell receptor beta chain Human genes 0.000 description 1
- 108010068342 MAP Kinase Kinase 1 Proteins 0.000 description 1
- 108010068353 MAP Kinase Kinase 2 Proteins 0.000 description 1
- 108010075654 MAP Kinase Kinase Kinase 1 Proteins 0.000 description 1
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 1
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 1
- 102100034069 MAP kinase-activated protein kinase 2 Human genes 0.000 description 1
- 102100026299 MAP kinase-interacting serine/threonine-protein kinase 1 Human genes 0.000 description 1
- 101710139011 MAP kinase-interacting serine/threonine-protein kinase 1 Proteins 0.000 description 1
- 102100033610 MAP kinase-interacting serine/threonine-protein kinase 2 Human genes 0.000 description 1
- 101710138999 MAP kinase-interacting serine/threonine-protein kinase 2 Proteins 0.000 description 1
- 108010041955 MAP-kinase-activated kinase 2 Proteins 0.000 description 1
- 108700012928 MAPK14 Proteins 0.000 description 1
- 102100028123 Macrophage colony-stimulating factor 1 Human genes 0.000 description 1
- 101150003941 Mapk14 gene Proteins 0.000 description 1
- 102000008135 Mechanistic Target of Rapamycin Complex 1 Human genes 0.000 description 1
- 108010035196 Mechanistic Target of Rapamycin Complex 1 Proteins 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102100026929 Mitogen-activated protein kinase 11 Human genes 0.000 description 1
- 102100026932 Mitogen-activated protein kinase 12 Human genes 0.000 description 1
- 102100026930 Mitogen-activated protein kinase 13 Human genes 0.000 description 1
- 102000054819 Mitogen-activated protein kinase 14 Human genes 0.000 description 1
- 102100024192 Mitogen-activated protein kinase 3 Human genes 0.000 description 1
- 102100024189 Mitogen-activated protein kinase 4 Human genes 0.000 description 1
- 102100037801 Mitogen-activated protein kinase 6 Human genes 0.000 description 1
- 102100037805 Mitogen-activated protein kinase 7 Human genes 0.000 description 1
- 102100033115 Mitogen-activated protein kinase kinase kinase 1 Human genes 0.000 description 1
- 102100025207 Mitogen-activated protein kinase kinase kinase 11 Human genes 0.000 description 1
- 102100025211 Mitogen-activated protein kinase kinase kinase 14 Human genes 0.000 description 1
- 102100033127 Mitogen-activated protein kinase kinase kinase 5 Human genes 0.000 description 1
- 102100026888 Mitogen-activated protein kinase kinase kinase 7 Human genes 0.000 description 1
- 102100026907 Mitogen-activated protein kinase kinase kinase 8 Human genes 0.000 description 1
- 102100028199 Mitogen-activated protein kinase kinase kinase kinase 1 Human genes 0.000 description 1
- 102100028192 Mitogen-activated protein kinase kinase kinase kinase 2 Human genes 0.000 description 1
- 102100028194 Mitogen-activated protein kinase kinase kinase kinase 4 Human genes 0.000 description 1
- 102100027983 Molybdenum cofactor sulfurase Human genes 0.000 description 1
- 101710132461 Molybdenum cofactor sulfurase Proteins 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 101100306001 Mus musculus Mst1r gene Proteins 0.000 description 1
- 101100405128 Mus musculus Nr4a3 gene Proteins 0.000 description 1
- 101100480907 Mus musculus Tec gene Proteins 0.000 description 1
- 101100481410 Mus musculus Tek gene Proteins 0.000 description 1
- 101100481406 Mus musculus Tie1 gene Proteins 0.000 description 1
- 102100030783 Myosin light chain kinase 3 Human genes 0.000 description 1
- 101710198035 Myosin light chain kinase, smooth muscle Proteins 0.000 description 1
- 125000005850 N-(alkoxycarbonyl)aminomethyl group Chemical group 0.000 description 1
- KFBDEKPSTRSAHD-UHFFFAOYSA-N N=1C2=CC=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=CC=1C1CCC(C(O)=O)N(C(O)=O)C1 Chemical compound N=1C2=CC=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=CC=1C1CCC(C(O)=O)N(C(O)=O)C1 KFBDEKPSTRSAHD-UHFFFAOYSA-N 0.000 description 1
- QVLRUUMOXFMGKK-UHFFFAOYSA-N N=1C2=CC=NN2C(O)=CC=1C(CC1)CCC21OCCO2 Chemical compound N=1C2=CC=NN2C(O)=CC=1C(CC1)CCC21OCCO2 QVLRUUMOXFMGKK-UHFFFAOYSA-N 0.000 description 1
- 102000004722 NADPH Oxidases Human genes 0.000 description 1
- 108010002998 NADPH Oxidases Proteins 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- 208000009905 Neurofibromatoses Diseases 0.000 description 1
- 102100026379 Neurofibromin Human genes 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 125000005861 N—(C1-C6)alkoxycarbonylaminomethyl group Chemical group 0.000 description 1
- 101100381429 Oryza sativa subsp. japonica BADH2 gene Proteins 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 101700056750 PAK1 Proteins 0.000 description 1
- 102100040902 PAS domain-containing serine/threonine-protein kinase Human genes 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 102100038332 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform Human genes 0.000 description 1
- 102100036061 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform Human genes 0.000 description 1
- 102100036056 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Human genes 0.000 description 1
- 102100036052 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform Human genes 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 102100033548 Phosphorylase b kinase regulatory subunit alpha, liver isoform Human genes 0.000 description 1
- 101000796832 Physarum polycephalum Actin-fragmin kinase Proteins 0.000 description 1
- 108010051742 Platelet-Derived Growth Factor beta Receptor Proteins 0.000 description 1
- 102000018967 Platelet-Derived Growth Factor beta Receptor Human genes 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 108010078137 Protein Kinase C-epsilon Proteins 0.000 description 1
- 102000014458 Protein Kinase C-epsilon Human genes 0.000 description 1
- 108010015499 Protein Kinase C-theta Proteins 0.000 description 1
- 102000001892 Protein Kinase C-theta Human genes 0.000 description 1
- 102100024924 Protein kinase C alpha type Human genes 0.000 description 1
- 101710109947 Protein kinase C alpha type Proteins 0.000 description 1
- 101800000618 Protein kinase C delta type catalytic subunit Proteins 0.000 description 1
- 102100021004 Protein sidekick-1 Human genes 0.000 description 1
- 102100037787 Protein-tyrosine kinase 2-beta Human genes 0.000 description 1
- 102100032315 RAC-beta serine/threonine-protein kinase Human genes 0.000 description 1
- 101710156940 RAC-beta serine/threonine-protein kinase Proteins 0.000 description 1
- 102100033479 RAF proto-oncogene serine/threonine-protein kinase Human genes 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 101150077555 Ret gene Proteins 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 208000007014 Retinitis pigmentosa Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 102100039313 Rho-associated protein kinase 1 Human genes 0.000 description 1
- 102100039314 Rho-associated protein kinase 2 Human genes 0.000 description 1
- 101710119197 Ribosomal protein S6 kinase alpha-1 Proteins 0.000 description 1
- 102100033536 Ribosomal protein S6 kinase alpha-1 Human genes 0.000 description 1
- 101710119185 Ribosomal protein S6 kinase alpha-2 Proteins 0.000 description 1
- 102100033534 Ribosomal protein S6 kinase alpha-2 Human genes 0.000 description 1
- 101710119204 Ribosomal protein S6 kinase alpha-3 Proteins 0.000 description 1
- 102100033643 Ribosomal protein S6 kinase alpha-3 Human genes 0.000 description 1
- 102100024917 Ribosomal protein S6 kinase beta-2 Human genes 0.000 description 1
- 101001117144 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) [Pyruvate dehydrogenase (acetyl-transferring)] kinase 1, mitochondrial Proteins 0.000 description 1
- 101000734335 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) [Pyruvate dehydrogenase (acetyl-transferring)] kinase 2, mitochondrial Proteins 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 102100028900 Serine/threonine-protein kinase 10 Human genes 0.000 description 1
- 102100026764 Serine/threonine-protein kinase 24 Human genes 0.000 description 1
- 102100030514 Serine/threonine-protein kinase 38 Human genes 0.000 description 1
- 102100027898 Serine/threonine-protein kinase 38-like Human genes 0.000 description 1
- 102100037629 Serine/threonine-protein kinase 4 Human genes 0.000 description 1
- 102100028623 Serine/threonine-protein kinase BRSK1 Human genes 0.000 description 1
- 102100031081 Serine/threonine-protein kinase Chk1 Human genes 0.000 description 1
- 102100031075 Serine/threonine-protein kinase Chk2 Human genes 0.000 description 1
- 102100039758 Serine/threonine-protein kinase DCLK1 Human genes 0.000 description 1
- 102100026621 Serine/threonine-protein kinase ICK Human genes 0.000 description 1
- 101710106482 Serine/threonine-protein kinase Kist Proteins 0.000 description 1
- 102100028751 Serine/threonine-protein kinase Nek1 Human genes 0.000 description 1
- 102100037703 Serine/threonine-protein kinase Nek2 Human genes 0.000 description 1
- 102100031401 Serine/threonine-protein kinase Nek6 Human genes 0.000 description 1
- 102100031398 Serine/threonine-protein kinase Nek9 Human genes 0.000 description 1
- 102100027911 Serine/threonine-protein kinase PAK 3 Human genes 0.000 description 1
- 102100031463 Serine/threonine-protein kinase PLK1 Human genes 0.000 description 1
- 102100031462 Serine/threonine-protein kinase PLK2 Human genes 0.000 description 1
- 102100026209 Serine/threonine-protein kinase PLK3 Human genes 0.000 description 1
- 102100028868 Serine/threonine-protein kinase PRP4 homolog Human genes 0.000 description 1
- 102100026715 Serine/threonine-protein kinase STK11 Human genes 0.000 description 1
- 101710181599 Serine/threonine-protein kinase STK11 Proteins 0.000 description 1
- 102100038192 Serine/threonine-protein kinase TBK1 Human genes 0.000 description 1
- 102100028235 Serine/threonine-protein kinase VRK1 Human genes 0.000 description 1
- 241000580858 Simian-Human immunodeficiency virus Species 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 101150110875 Syk gene Proteins 0.000 description 1
- 102100030168 Testis-specific serine/threonine-protein kinase 3 Human genes 0.000 description 1
- 206010043391 Thalassaemia beta Diseases 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 102100022596 Tyrosine-protein kinase ABL1 Human genes 0.000 description 1
- 102100022651 Tyrosine-protein kinase ABL2 Human genes 0.000 description 1
- 102100027389 Tyrosine-protein kinase HCK Human genes 0.000 description 1
- 102100023345 Tyrosine-protein kinase ITK/TSK Human genes 0.000 description 1
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 1
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 1
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 1
- 102100021125 Tyrosine-protein kinase ZAP-70 Human genes 0.000 description 1
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 1
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 1
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 101150060655 WNK1 gene Proteins 0.000 description 1
- 101150040313 Wee1 gene Proteins 0.000 description 1
- 101001001642 Xenopus laevis Serine/threonine-protein kinase pim-3 Proteins 0.000 description 1
- 102100037607 [3-methyl-2-oxobutanoate dehydrogenase [lipoamide]] kinase, mitochondrial Human genes 0.000 description 1
- 102100024148 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial Human genes 0.000 description 1
- 102100024150 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 2, mitochondrial Human genes 0.000 description 1
- 102100034824 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 3, mitochondrial Human genes 0.000 description 1
- 102100034825 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 4, mitochondrial Human genes 0.000 description 1
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical group C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 1
- 229950010817 alvocidib Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 150000003927 aminopyridines Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 238000011319 anticancer therapy Methods 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 230000005735 apoptotic response Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 125000005140 aralkylsulfonyl group Chemical group 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 125000005018 aryl alkenyl group Chemical group 0.000 description 1
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 1
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 239000005441 aurora Substances 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000005513 benzoazaindolyl group Chemical group 0.000 description 1
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 150000005347 biaryls Chemical class 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000004305 biphenyl Chemical group 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 1
- 150000004648 butanoic acid derivatives Chemical class 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000009134 cell regulation Effects 0.000 description 1
- 108091092328 cellular RNA Proteins 0.000 description 1
- 208000025434 cerebellar degeneration Diseases 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- FCYRSDMGOLYDHL-UHFFFAOYSA-N chloromethoxyethane Chemical compound CCOCCl FCYRSDMGOLYDHL-UHFFFAOYSA-N 0.000 description 1
- 125000006378 chloropyridyl group Chemical group 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 1
- 229960000258 corticotropin Drugs 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000004802 cyanophenyl group Chemical group 0.000 description 1
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- FNIATMYXUPOJRW-UHFFFAOYSA-N cyclohexylidene Chemical group [C]1CCCCC1 FNIATMYXUPOJRW-UHFFFAOYSA-N 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000002548 cytokinetic effect Effects 0.000 description 1
- 230000003436 cytoskeletal effect Effects 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 125000003493 decenyl group Chemical group [H]C([*])=C([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- 125000005265 dialkylamine group Chemical group 0.000 description 1
- LMEDOLJKVASKTP-UHFFFAOYSA-N dibutyl sulfate Chemical class CCCCOS(=O)(=O)OCCCC LMEDOLJKVASKTP-UHFFFAOYSA-N 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000005047 dihydroimidazolyl group Chemical group N1(CNC=C1)* 0.000 description 1
- 125000005049 dihydrooxadiazolyl group Chemical group O1N(NC=C1)* 0.000 description 1
- 125000005056 dihydrothiazolyl group Chemical group S1C(NC=C1)* 0.000 description 1
- 125000005072 dihydrothiopyranyl group Chemical group S1C(CCC=C1)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- MEQJPQWRCJBURZ-UHFFFAOYSA-N ditert-butyl 5-(3-ethoxy-3-oxopropanoyl)piperidine-1,2-dicarboxylate Chemical compound CCOC(=O)CC(=O)C1CCC(C(=O)OC(C)(C)C)N(C(=O)OC(C)(C)C)C1 MEQJPQWRCJBURZ-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229940115080 doxil Drugs 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- KXNNKMDSGZNNGL-UHFFFAOYSA-N ethyl 2-[4-[7-[bis(2-trimethylsilylethoxymethyl)amino]-6-bromo-3-quinolin-3-ylpyrazolo[1,5-a]pyrimidin-5-yl]cyclohexyl]acetate Chemical compound C1CC(CC(=O)OCC)CCC1C1=NC2=C(C=3C=C4C=CC=CC4=NC=3)C=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1Br KXNNKMDSGZNNGL-UHFFFAOYSA-N 0.000 description 1
- FVPISMANESAJQZ-UHFFFAOYSA-N ethyl 2-diethoxyphosphoryl-2-fluoroacetate Chemical compound CCOC(=O)C(F)P(=O)(OCC)OCC FVPISMANESAJQZ-UHFFFAOYSA-N 0.000 description 1
- KGGFUWDAYSUHOA-UHFFFAOYSA-N ethyl 3-(1,4-dioxaspiro[4.5]decan-8-yl)-3-oxopropanoate Chemical compound C1CC(C(=O)CC(=O)OCC)CCC21OCCO2 KGGFUWDAYSUHOA-UHFFFAOYSA-N 0.000 description 1
- ZXYAWONOWHSQRU-UHFFFAOYSA-N ethyl 4-oxocyclohexanecarboxylate Chemical compound CCOC(=O)C1CCC(=O)CC1 ZXYAWONOWHSQRU-UHFFFAOYSA-N 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000000219 ethylidene group Chemical group [H]C(=[*])C([H])([H])[H] 0.000 description 1
- 125000004705 ethylthio group Chemical group C(C)S* 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- RFHAOTPXVQNOHP-UHFFFAOYSA-N fluconazole Chemical compound C1=NC=NN1CC(C=1C(=CC(F)=CC=1)F)(O)CN1C=NC=N1 RFHAOTPXVQNOHP-UHFFFAOYSA-N 0.000 description 1
- 229960004884 fluconazole Drugs 0.000 description 1
- 238000002376 fluorescence recovery after photobleaching Methods 0.000 description 1
- 125000006379 fluoropyridyl group Chemical group 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- RCFDIXKVOHJQPP-UHFFFAOYSA-N furo[2,3-b]pyridine Chemical compound C1=CN=C2OC=CC2=C1 RCFDIXKVOHJQPP-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000005251 gamma ray Effects 0.000 description 1
- 125000005643 gamma-butyrolacton-4-yl group Chemical group 0.000 description 1
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 108010049611 glycogen synthase kinase 3 alpha Proteins 0.000 description 1
- 125000003147 glycosyl group Chemical group 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 150000002373 hemiacetals Chemical group 0.000 description 1
- 230000007866 hepatic necrosis Effects 0.000 description 1
- 206010019692 hepatic necrosis Diseases 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 125000004447 heteroarylalkenyl group Chemical group 0.000 description 1
- 125000005143 heteroarylsulfonyl group Chemical group 0.000 description 1
- 125000005368 heteroarylthio group Chemical group 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000005945 imidazopyridyl group Chemical group 0.000 description 1
- 230000006058 immune tolerance Effects 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical group II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- 208000037906 ischaemic injury Diseases 0.000 description 1
- LVPMIMZXDYBCDF-UHFFFAOYSA-N isocinchomeronic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)N=C1 LVPMIMZXDYBCDF-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 150000003903 lactic acid esters Chemical class 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 201000011614 malignant glioma Diseases 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- APXOMRFLJBRHNX-UHFFFAOYSA-N methyl 2-[4-(bromomethyl)phenyl]acetate Chemical compound COC(=O)CC1=CC=C(CBr)C=C1 APXOMRFLJBRHNX-UHFFFAOYSA-N 0.000 description 1
- PMCGLEDOEQIMQB-UHFFFAOYSA-N methyl 2-[4-[[7-[bis(2-trimethylsilylethoxymethyl)amino]-3-iodopyrazolo[1,5-a]pyrimidin-5-yl]methyl]phenyl]acetate Chemical compound C1=CC(CC(=O)OC)=CC=C1CC1=NC2=C(I)C=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1 PMCGLEDOEQIMQB-UHFFFAOYSA-N 0.000 description 1
- SVMRGBGTHRCMEG-UHFFFAOYSA-N methyl 2-[4-[[7-[bis(2-trimethylsilylethoxymethyl)amino]pyrazolo[1,5-a]pyrimidin-5-yl]methyl]phenyl]acetate Chemical compound C1=CC(CC(=O)OC)=CC=C1CC1=NC2=CC=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1 SVMRGBGTHRCMEG-UHFFFAOYSA-N 0.000 description 1
- IUTGNWFEYGGONQ-UHFFFAOYSA-N methyl 4-(7-aminopyrazolo[1,5-a]pyrimidin-5-yl)cyclohexane-1-carboxylate Chemical compound C1CC(C(=O)OC)CCC1C1=NC2=CC=NN2C(N)=C1 IUTGNWFEYGGONQ-UHFFFAOYSA-N 0.000 description 1
- PCVWIBLDHOMPFE-UHFFFAOYSA-N methyl 4-(7-chloropyrazolo[1,5-a]pyrimidin-5-yl)cyclohexane-1-carboxylate Chemical compound C1CC(C(=O)OC)CCC1C1=NC2=CC=NN2C(Cl)=C1 PCVWIBLDHOMPFE-UHFFFAOYSA-N 0.000 description 1
- YOBSOVZMYHAPGB-UHFFFAOYSA-N methyl 4-[7-[bis(2-trimethylsilylethoxymethyl)amino]-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl]oxybenzoate Chemical compound C1=CC(C(=O)OC)=CC=C1OC1=NC2=C(C=3C=NC(=CC=3)C=3C=CC=CC=3)C=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1 YOBSOVZMYHAPGB-UHFFFAOYSA-N 0.000 description 1
- JJLHHOKQRMHQKD-UHFFFAOYSA-N methyl 4-[7-[bis(2-trimethylsilylethoxymethyl)amino]-3-iodopyrazolo[1,5-a]pyrimidin-5-yl]oxybenzoate Chemical compound C1=CC(C(=O)OC)=CC=C1OC1=NC2=C(I)C=NN2C(N(COCC[Si](C)(C)C)COCC[Si](C)(C)C)=C1 JJLHHOKQRMHQKD-UHFFFAOYSA-N 0.000 description 1
- ASKDFGVMJZMYEM-UHFFFAOYSA-N methyl hex-5-enoate Chemical compound COC(=O)CCCC=C ASKDFGVMJZMYEM-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000002346 musculoskeletal system Anatomy 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 1
- JTSLALYXYSRPGW-UHFFFAOYSA-N n-[5-(4-cyanophenyl)-1h-pyrrolo[2,3-b]pyridin-3-yl]pyridine-3-carboxamide Chemical compound C=1C=CN=CC=1C(=O)NC(C1=C2)=CNC1=NC=C2C1=CC=C(C#N)C=C1 JTSLALYXYSRPGW-UHFFFAOYSA-N 0.000 description 1
- 125000004370 n-butenyl group Chemical group [H]\C([H])=C(/[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000006606 n-butoxy group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000005029 naphthylthio group Chemical group C1(=CC=CC2=CC=CC=C12)S* 0.000 description 1
- 201000004931 neurofibromatosis Diseases 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- ODUCDPQEXGNKDN-UHFFFAOYSA-N nitroxyl Chemical compound O=N ODUCDPQEXGNKDN-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 125000004365 octenyl group Chemical group C(=CCCCCCC)* 0.000 description 1
- GTVPOLSIJWJJNY-UHFFFAOYSA-N olomoucine Chemical compound N1=C(NCCO)N=C2N(C)C=NC2=C1NCC1=CC=CC=C1 GTVPOLSIJWJJNY-UHFFFAOYSA-N 0.000 description 1
- 239000003605 opacifier Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 239000012285 osmium tetroxide Substances 0.000 description 1
- 150000003901 oxalic acid esters Chemical class 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004095 oxindolyl group Chemical group N1(C(CC2=CC=CC=C12)=O)* 0.000 description 1
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 1
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 125000006678 phenoxycarbonyl group Chemical group 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 125000001476 phosphono group Chemical group [H]OP(*)(=O)O[H] 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Chemical group 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 108010056274 polo-like kinase 1 Proteins 0.000 description 1
- 208000030151 polycystic kidney disease 3 with or without polycystic liver disease Diseases 0.000 description 1
- 208000015768 polyposis Diseases 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical class [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- ALDITMKAAPLVJK-UHFFFAOYSA-N prop-1-ene;hydrate Chemical group O.CC=C ALDITMKAAPLVJK-UHFFFAOYSA-N 0.000 description 1
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- DNIAPMSPPWPWGF-UHFFFAOYSA-N propylene glycol Substances CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 108010027883 protein kinase C eta Proteins 0.000 description 1
- 108010008359 protein kinase C lambda Proteins 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- QLULGIRFKAWHOJ-UHFFFAOYSA-N pyridin-4-ylboronic acid Chemical compound OB(O)C1=CC=NC=C1 QLULGIRFKAWHOJ-UHFFFAOYSA-N 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000004943 pyrimidin-6-yl group Chemical group N1=CN=CC=C1* 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000006085 pyrrolopyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000022983 regulation of cell cycle Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000003488 releasing hormone Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 150000003902 salicylic acid esters Chemical class 0.000 description 1
- 230000036573 scar formation Effects 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 238000002603 single-photon emission computed tomography Methods 0.000 description 1
- 201000009890 sinusitis Diseases 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 208000002320 spinal muscular atrophy Diseases 0.000 description 1
- 229940068117 sprycel Drugs 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 229910000080 stannane Inorganic materials 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003900 succinic acid esters Chemical class 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 229940034785 sutent Drugs 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- CTEDVGRUGMPBHE-UHFFFAOYSA-N tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(CO)CC1 CTEDVGRUGMPBHE-UHFFFAOYSA-N 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- DBDCNCCRPKTRSD-UHFFFAOYSA-N thieno[3,2-b]pyridine Chemical compound C1=CC=C2SC=CC2=N1 DBDCNCCRPKTRSD-UHFFFAOYSA-N 0.000 description 1
- 125000004588 thienopyridyl group Chemical group S1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 150000003567 thiocyanates Chemical class 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M toluenesulfonate group Chemical group C=1(C(=CC=CC1)S(=O)(=O)[O-])C LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- INQOMBQAUSQDDS-FIBGUPNXSA-N trideuterio(iodo)methane Chemical compound [2H]C([2H])([2H])I INQOMBQAUSQDDS-FIBGUPNXSA-N 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical class OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- BWHDROKFUHTORW-UHFFFAOYSA-N tritert-butylphosphane Chemical compound CC(C)(C)P(C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-N 0.000 description 1
- 108010090229 tropomyosin kinase Proteins 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 229940094060 tykerb Drugs 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 238000007631 vascular surgery Methods 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 125000005853 β-dimethylaminoethyl group Chemical group 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- This invention is directed to pyrazolo pyrimidine derivatives as inhibitors of mammalian Target Of Rapamycin (mTOR) kinase, which are also known as FRAP, RAFT, RAPT or SEP and as a result have a role in controlling cell cycle progression and mitogenic signals involved in cell growth and proliferation and as a result are useful in the treatment of cancer.
- mTOR mammalian Target Of Rapamycin
- rapamycin The mammalian target of rapamycin (mTOR) is a central regulator of cell growth and proliferation and plays a gate keeper role in the control of cell cycle progression and mediates mitogenic signals from P13K/AKT through to the downstream targets S6K1 and 4E-BP1 and to Ser 473 on AKT.
- mTORCI raptor-mTOR complex
- mTORC2 rapamycin-sensitive complex
- mTORC2 a rapamycin-insensitive complex that signals to AKT.
- rapamycin partially inhibits mTOR function through mTORCI . It has been found that mTORC2 is involved in the regulation of cell survival and actin cytoskeletal organization in a rapamycin-independent manner, and inhibition of mTOR through inhibition of mTORCI and mTORC2 is probably important for antitumor activity and better efficacy.
- WO 2007/087395 describes unsaturated mTOR inhibitors useful in treatment of cancer.
- WO-2008/012326 describes 2,4-substituted quinozolines as lipid kinase inhibitors useful in treatment of P13K -related diseases, such as proliferative diseases, inflammatory diseases, obstructive airways disorder and transplant related diseases.
- WO 2006/090169 describes 2,4-diamineo-pyrido-pyrmidine derivatives and their use as mTOR inhibitors.
- WO 2007/066099 describes pyrimidine derivatives useful as mTOR kinase inhibitors for anticancer and various other therapeutic treatments involving mTOR kinase.
- WO 2007044813 describes pyridopyrimidinone inhibitors of PI3Ka protein kinase for anticancer or other PI3Ka protein kinase related diseases.
- US 2002/0041880 describes pyrazolo[1 ,5 a]pyrimidin-7-yl derivatives to inhibit kinase insert domain-containing receptor to block angiogenesis.
- WO 1998/003510 describes pyrazolo[1 ,5 a]pyrimidin-7-yl derivatives to treat corticotrophin releasing factor dependent diseases.
- WO 2003/091256 describes pyrazolo[1 ,5-a]pyrimidine derivatives to treat NAD(P)H oxidase dependent diseases.
- WO 2007/044449, WO 2005/077954, WO 2004/022560 and WO 2004/022561 describe pyrazolopyrimidine derivatives as cyclin-dependent kinase dependent diseases.
- WO 2004/106341 describes pyrazolopyrimidine derivatives as fungicides There is need in the art for small molecule inhibitors of mTOR kinase that block signaling through mTORCI and mT0RC2 as a potential anticancer treatment or a treatment for other cell proliferative disorders.
- the present invention discloses novel compounds having mTOR inhibitory activity, methods of preparing such compounds, pharmaceutical compositions comprising one or more of such compounds, methods of treatment or prevention of one or more diseases associated with mTOR by administering one or more of such compounds or pharmaceutical compositions, the compound being represented by the general Formula I:
- R 1 is independently selected from the group consisting of heterocycloalkyl, heterocycloalkylalkyl, spiroheterocycloalkyl, heterocyclenyl, -NR 3 R 4 , cycloalkyl, heteroaryl, aryl, alkyl, alkynyl, heterocyclenylalkyl, cycloalkylalkyl, heteroarylalkyl, heteroarylalkynyl, spiroheterocycloalkylalkyl, -N-heteroaryl, -alkyl-NH-heterocyclyl and arylalkyl, wherein each of said heterocycloalkyl, heterocycloalkylalkyl, spiroheterocycloalkyl, heterocyclenyl, cycloalkyl, heteroaryl, aryl, alkyl, alkynyl, heterocyclenylalkyl, cycloalkylalkyl, heteroarylalkyl
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl;
- R 3 is cycloalkyl or heteroaryl, wherein each of said cycloalkyl or heteroaryl can be unsubstituted or substituted with one or more moieties independently selected from the group consisting of X; and R 4 is H.
- the present invention includes a method of inhibiting mammalian Target Of Rapamycin in a patient, wherein the method comprises administering a therapeutically effective amount of at least one compound of the structural Formula I.
- the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, wherein the method comprises administering a therapeutically effective amount of at least one compound represented by the structural Formula I.
- the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, wherein the method comprises administering a therapeutically effective amount of at least one compound represented by the structural Formula I, to a patient in need thereof.
- the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, and disorders commonly occurring in connection with transplantation, diseases that respond to inhibition of mammalian Target Of Rapamycin, wherein the method comprises administering a therapeutically effective amount of compound represented by the structural Formula I, to a patient in need of such treatment.
- the present invention includes a method of treatment of a disease, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I.
- the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, and disorders commonly occurring in connection with transplantation, diseases that respond to inhibition of mammalian Target Of Rapamycin, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I, wherein the disease is a proliferative disease, autoimmune disease, viral disease, fungal disease, neurological/neurodegenerative disorder, arthritis, inflammation, neuronal, alopecia or cardiovascular disease.
- a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, and disorders commonly occurring in connection with transplantation, diseases that respond to inhibition of mammalian Target Of Rapamycin
- the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I, wherein the disease is a proliferative disease, autoimmune disease, viral disease, fungal disease, neurological/neurodegenerative disorder, arthritis, inflammation, neuro
- the present invention includes a method of treatment of a disease, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I, wherein the disease is a proliferative disease.
- the present invention includes a method of treatment of a proliferative disease, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I, wherein the proliferative disease is selected from the group consisting of: cancer of the bladder, breast, colon, kidney, liver, lung, small cell lung cancer, non- small cell lung cancer, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma, Burkett's lymphoma; acute and chronic myelogenous leukemia, myelodysplastic syndrome, promyelocytic leukemia, mye
- the present invention includes a method of treatment of a proliferative disease comprising administering a therapeutically effective amount of the compound represented by the structural Formula I, further comprising treatment with radiation therapy.
- the present invention includes a method of inhibiting a mammalian Target Of Rapamycin, wherein the method comprises administering a therapeutically effective amount of a compound represented by the structural Formula I.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R 1 is heterocycloalkyl, wherein said heterocycloalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl- C(O) 2 H, -alkyl(CO)N(CH 3 )-O-CH 3 , -alkyl(CO)-heteroaryl, -C(O) 2 -alkyl, -alkyl-C(O)-NH 2 , -NH 2 , heteroaryl, -alkyl-CN, -C(O) 2
- R 1 is heterocyclenyl, wherein said heterocyclenyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl- C(O) 2 H, -alkyl(CO)N(CH 3 )-O-CH 3 , -alkyl(CO)-heteroaryl, -C(O) 2 -alkyl, -alkyl-C(O)-NH 2 , -NH 2 , heteroaryl, -alkyl-CN, -C(O) 2
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CHa) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R 1 is heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl - C(O) 2 H, -alkyl(CO)N(CH 3 )-O-CH 3 , -alkyl(CO)-heteroaryl, -C(O) 2 -alkyl
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R 1 is cycloalkyl, wherein said cycloalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl-
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of halo, hydroxyl, amino,
- R 1 is cycloalkenyl, wherein said cycloalkenyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl- C(O) 2 H, -alkyl(CO)N(CH 3 )-O-CH 3 , -alkyl(CO)-heteroaryl, -C(O) 2 -alkyl, -alkyl-C(O)-NH 2 , -NH 2 , heteroaryl, -alkyl-CN, -C(O) 2
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heteroeycloalkyl, and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R 1 is aryl, wherein said aryl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heteroeycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl- C(O) 2 H, -alkyl(CO)N(CH 3 )-O-CH 3 , -alkyl (CO)-heteroaryl, -C(O) 2 -alkyl
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R 1 is alkyl, wherein said alkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl-
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of halo, hydroxyl, amino,
- R 1 is alkynyl, wherein said alkynyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(0) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl- C(O) 2 H, -alkyl(CO)N(CH 3 )-O-CH 3 , -alkyl(CO)-heteroaryl, -C(O) 2 -alkyl, -alkyl-C(O)-NH 2 , -NH 2 , heteroaryl, -alkyl-CN, -C(O) 2 -
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R 1 is spiroheterocycloalkyl, wherein said spiroheterocycloalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
- X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O) 2 alkyl, -C(O) 2 H, hydroxyalkyl, -S(O) 2 alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O) 2 -alkyl, -C(O)-heteroaryl, -alkyl- C(O) 2 H, -alkyl(CO)N(CH 3 )-O-CH 3 , -alkyl(CO)-heteroaryl, -C(O) 2 -alkyl, -alkyl-C(O)-NH 2 , -NH 2 , heteroaryl, -alkyl-CN, -C(O) 2
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R 1 is independently selected from the group consisting of:
- R 2 is quinolinyl, 1 -methyl-pyrazolyl, naphtyl, methoxy-naphtyl, cyano-phenyl, methoxy-phenyl, phenoxy-phenyl, biphenyl, chloro-pyridyl, methoxy-pyridyl, bromo- methyl-pyrazolyl, hydroxyl-methoxy-phenyl, di-methoxy-phenyl, 1 H-indazolyl, fluoro- hydroxy-phenyl, chloro-quinolyl, morpholino-pyridyl-, 4-(isobutyronitrilo)-phenyl, bromo-quinolyl, 4-trifluoromethyl-phenyl-, benzyl -pyrazolyl, fluoro-pyridyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1 -methyl-pyrazolyl, and thienyl;
- R 1 is independently selected from the group consisting of:
- R 2 is indazolyl, wherein said indazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R 1 is independently selected from the group consisting of:
- R 2 is phenyl, wherein said phenyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, alkoxy, -CN, hydroxyl, aryl and heteroaryl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R 1 is independently selected from the group consisting of:
- R 2 is naphthyl, wherein said naphthyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R 1 is independently selected from the group consisting of:
- R 2 is quinolinyl, wherein said quinolinyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R 1 is independently selected from the group consisting of:
- R 2 is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R 1 is independently selected from the group consisting of:
- R 2 is pyridinyl, wherein said pyridinyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, heterocyclyl, heteroaryl, aryl and alkoxy.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is halo
- R 1 is independently selected from the group consisting of:
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, heteroaryl and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is thienyl; R 1 is independently selected from the group consisting of:
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ⁇ CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, heteroaryl and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with alkyl;
- R 1 is independently selected from the group consisting of:
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is pyridinyl; R 1 is independently selected from the group consisting of:
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyljieterocycloalkyl, heteroaryl and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is methyl; R 1 is independently selected from the group consisting of:
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CHa) 2 CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
- R is phenyl, wherein said phenyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl;
- R 1 is independently selected from the group consisting of:
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is acetyl; R 1 is independently selected from the group consisting of:
- R 2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyljieterocycloalkyl, heteroaryl and arylalkyl.
- the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
- R is -CN; R 1 is independently selected from the group consisting of: and
- R 2 is h ⁇ teroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH 3 ) 2 CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
- Non-limiting examples of the compounds of the present invention include.
- the present invention relates to a compound represented by the structural Formula I or a pharmaceutically acceptable salt, ester, solvate or prodrug thereof, in purified form.
- the present invention relates to a compound represented by the structural Formula I or a pharmaceutically acceptable salt, ester, solvate or prodrug thereof, in isolated form.
- the present invention includes a composition comprising a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, in combination with at least one pharmaceutically acceptable carrier.
- the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, said method comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above, or a pharmaceutically acceptable salt, solvate, ester or prodrug of the compound, to a patient in need thereof.
- the present invention includes a composition comprising a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof, and an anti-cancer agent.
- the present invention includes a composition comprising a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof, and an anti-neoplastic, anti-tumor, anti-angiogenic, or chemotherapeutic agent.
- the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, and disorders commonly occurring in connection with transplantation, diseases that respond to inhibition of nnTOR, comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
- the present invention includes a method of treatment of a proliferative disease, autoimmune disease, viral disease, fungal disease, neurological/neurodegenerative disorder, arthritis, inflammation, neuronal, alopecia or cardiovascular disease comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
- the present invention includes a method of treatment of a proliferative disease.
- the present invention includes a method of treatment of a proliferative disease, wherein the proliferative disease is selected from the group consisting of: cancer of the bladder, breast, colon, kidney, liver, lung, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin; small cell lung cancer, non-small cell lung cancer, squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma, Burkett's lymphoma; acute and chronic myelogenous leukemia, myelodysplastic syndrome, promyelocyte leukemia; fibrosarcoma, rhabdomyosarcoma; astrocytom
- the present invention includes a method of treatment of hamartoma syndromes, transplant rejection, bowel disorders, inflammatory bowel disease, multiple sclerosis, immunosuppression, immune tolerance, autoimmune diseases, inflammation, bone loss, rheumatoid arthritis, restinosis, cardiac allograft vasculopathy, psoriasis, ocular conditions such as dry eye, hepatic fibrosis, hepatic necrosis, beta-thalassaemia, comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
- the present invention includes a method of treatment of cancer of the bladder, breast, colon, kidney, liver, lung, small cell lung cancer, non- small cell lung cancer, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma, Burkett's lymphoma; acute and chronic myelogenous leukemia, myelodysplastic syndrome, promyelocytic leukemia; fibrosarcoma, rhabdomyosarcoma; astrocytoma, neuroblastoma, glioma and schwannomas; melanoma
- the present invention includes a method of treating a disease by inhibiting a mTOR, comprising administering to a patient in need of such treatment an amount of a first compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, said second compound being an anti-cancer agent; wherein said anti-cancer agent is selected from the group consisting of Adriamycin, Altretamine, Amidox, Aminoglutethimide, Amsacrine, Anastrazole, Antibodies to EGFR, 3-AP, Aphidicolon, Ara-C, Arsenic trioxide, L-Asparaginase, Bevacizumab, Bleomycin, BMS 214662, Bortezomib, Busulfan, Campath, Camptostar, Capecitabine, Carboplatin, Carmustine, Centrosome associated protein E (“CENP-E”) inhibitors, Cetuximab, Cladrib
- the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, said method comprising administering a therapeutically effective amount of at least one compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug of the compound, to a patient in need thereof.
- the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, diseases related to transplant rejection and diseases that respond to inhibition of mTOR, comprising administering a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
- a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, diseases related to transplant rejection and diseases that respond to inhibition of mTOR
- the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, diseases related to transplant rejection and diseases that respond to inhibition of mTOR, comprising administering a therapeutically effective amount of at least one compound of Formula I or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- the present invention includes a compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof exhibiting mTOR inhibition which is at least five-fold the inhibition of CDK2 or CHK-1 by said compound.
- the present invention includes a compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof exhibiting mTOR inhibition which is at least ten-fold the inhibition of CDK2 or CHK-1 by said compound.
- the present invention includes a compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof exhibiting mTOR inhibition which is at least fifty-fold the inhibition of CDK2 or CHK-1 by said compound.
- the compounds of this invention can be used to inhibit the following kinases:
- Alkyl means an aliphatic hydrocarbon group which may be straight or branched and comprising about 1 to about 20 carbon atoms in the chain. Preferred alkyl groups contain about 1 to about 12 carbon atoms in the chain. More preferred alkyl groups contain about 1 to about 6 carbon atoms in the chain. Branched means that one or more lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkyl chain.
- Lower alkyl means a group having about 1 to about 6 carbon atoms in the chain which may be straight or branched.
- alkyl groups include methyl, ethyl, n-propyl, isopropyl and t-butyl.
- alkenyl means an aliphatic hydrocarbon group containing at least one carbon- carbon double bond and which may be straight or branched and comprising about 2 to about 15 carbon atoms in the chain. Preferred alkenyl groups have about 2 to about 12 carbon atoms in the chain; and more preferably about 2 to about 6 carbon atoms in the chain. Branched means that one or more, lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkenyl chain.
- “Lower alkenyl” means about 2 to about 6 carbon atoms in the chain which may be straight or branched.
- “Alkenyl” may be unsubstituted or optionally substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of halo, alkyl. aryl, cycloalkyl, cyano, alkoxy and -S(alkyl).
- suitable alkenyl groups include ethenyl, propenyl, n-butenyl, 3-methylbut- 2-enyl, n-pentenyl, octenyl and decenyl.
- “Alkylene” means a difunctional group obtained by removal of a hydrogen atom from an alkyl group that is defined above. Non-limiting examples of alkylene include methylene, ethylene and propylene.
- Alkynyl means an aliphatic hydrocarbon group containing at least one carbon- carbon triple bond and which may be straight or branched and comprising about 2 to about 15 carbon atoms in the chain.
- Preferred alkynyl groups have about 2 to about 12 carbon atoms in the chain; and more preferably about 2 to about 4 carbon atoms in the chain.
- Branched means that one or more, lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkynyl chain.
- Lower alkynyl means about 2 to about 6 carbon atoms in the chain which may be straight or branched.
- alkynyl groups include ethynyl, propynyl, 2-butynyl and 3- methylbutynyl.
- Alkynyl may be unsubstituted or optionally substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of alkyl, aryl and cycloalkyl.
- Aryl means an aromatic monocyclic or multicyclic ring system comprising about 6 to about 14 carbon atoms, preferably about 6 to about 10 carbon atoms. The aryl group can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein.
- Suitable aryl groups include phenyl and naphthyl.
- “Bridged cyclic ring” is a hydrocarbon ring such as cycloalkyl, cyclenyl, or aryl or heteroatom containing ring such as, heterocyclyl, heterocyclenyl, or heteroaryl as described herein, that contains a bridge, which is a valence bond or an atom or an unbranched chain of atoms connecting two different parts of the ring.
- bridgeheads The two tertiary carbon atoms connected through the bridge are termed "bridgeheads".
- Heteroaryl means an aromatic monocyclic or multicyclic ring system comprising about 5 to about 14 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the ring atoms is an element other than carbon, for example nitrogen, oxygen or sulfur, alone or in combination. Preferred heteroaryls contain about 5 to about 6 ring atoms.
- the "heteroaryl” can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein.
- the prefix aza, oxa or thia before the heteroaryl root name means that at least a nitrogen, oxygen or sulfur atom respectively, is present as a ring atom.
- heteroaryl may also include a heteroaryl as defined above fused to an aryl as defined above.
- suitable heteroaryls include pyridyl, pyrazinyl, furanyl, thienyl, pyrimidinyl, pyridone (including N-substituted pyridones), isoxazolyl, isothiazolyl, oxazolyl, thiazolyl, pyrazolyl, furazanyl, pyrrolyl, pyrazolyl, triazolyl, 1 ,2,4- thiadiazolyl, pyrazinyl, pyridazinyl, quinoxalinyl, phthalazinyl, oxindolyl, imidazo[1 ,2- a]pyridinyl, imidazo[2,1 -b]thiazolyl
- Aralkyl or “arylalkyl” means an aryl-alkyl- group in which the aryl and alkyl are as previously described. Preferred aralkyls comprise a lower alkyl group. Non-limiting examples of suitable aralkyl groups include benzyl, 2-phenethyl and naphthalenylmethyl. The bond to the parent moiety is through the alkyl.
- Alkylaryl means an alkyl-aryl- group in which the alkyl and aryl are as previously described. Preferred alkylaryls comprise a lower alkyl group. Non-limiting example of a suitable alkylaryl group is tolyl. The bond to the parent moiety is through the aryl.
- Cycloalkyl means a non-aromatic mono- or multicyclic ring system comprising about 3 to about 10 carbon atoms, preferably about 5 to about 10 carbon atoms. Preferred cycloalkyl rings contain about 5 to about 7 ring atoms.
- the cycloalkyl can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined above.
- suitable monocyclic cycloalkyls include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- suitable multicyclic cycloalkyls include 1 -decalinyl, norbomyl, adamantyl and the like.
- Cycloalkylalkyl means a cycloalkyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable cycloalkylalkyls include cyclohexylmethyl, adamantylmethyl and the like.
- Cycloalkenyl means a non-aromatic mono or multicyclic ring system comprising about 3 to about 10 carbon atoms, preferably about 5 to about 10 carbon atoms which contain at least one carbon-carbon double bond. Preferred cycloaikenyl rings contain about 5 to about 7 ring atoms.
- the cycloalkenyl can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined above.
- suitable monocyclic cycioalkenyls include cyclopentenyi, cyclohexenyl, cyclohepta-1 ,3-dienyl, and the like.
- Non-iimiting example of a suitable multicyclic cycloalkenyl is norbornylenyl.
- Cycloalkenylalkyl means a cycloalkenyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable cycloalkenyialkyls include cyclopentenylmethyl, cyclohexenylmethyl and the like.
- Halogen means fluorine, chlorine, bromine, or iodine. Preferred are fluorine, chlorine and bromine.
- Ring system substituent means a substituent attached to an aromatic or non- aromatic ring system which, for example, replaces an available hydrogen on the ring system.
- Ring system substituents may be the same or different, each being independently selected from the group consisting of alkyl, alkenyl, alkynyl, aryl, heteroaryi, araikyl, alkylaryl, heteroaralkyl, heteroarylalkenyl, heteroarylalkynyl, alkyl heteroaryl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, acyl, aroyl, halo, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyi, aralkoxycarbonyl, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaral
- Ring system substituent may also mean a single moiety which simultaneously replaces two available hydrogen on two adjacent carbon atoms (one H on each carbon) on a ring system.
- Examples of such moiety are methylene dioxy, ethyl enedioxy, -C(CH 3 J 2 - and the like which form moieties such as, for example:
- Heteroarylalky means a heteroary! moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable heteroaryis include 2-pyridinylmethyl, quinolinylmethy! and the like.
- Heterocyciyl means a non-aromatic saturated monocyclic or multicyclic ring system comprising about 3 to about 10 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is an element other than carbon, for example nitrogen, oxygen or sulfur, alone or in combination. There are no adjacent oxygen and/or sulfur atoms present in the ring system.
- Preferred heterocyclyls contain about 5 to about 6 ring atoms.
- the prefix aza, oxa or thia before the heterocyciyl root name means that at least a nitrogen, oxygen or sulfur atom respectively is present as a ring atom.
- Any -NH in a heterocyciyl ring may exist protected such as, for example, as an -N(Boc), -N(CBz), -N(Tos) group and the like; such protections are also considered part of this invention.
- the heterocyciyl can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein.
- the nitrogen or sulfur atom of the heterocyciyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S 1 S- dioxide.
- suitable monocyclic heterocyciyl rings include piperidyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyi, thiazolidinyl, 1 ,4- dioxanyl, tetrahydrofuranyl, tetrahydrothiophenyl, lactam, lactone, and the like.
- Heterocyciyl may also mean a single moiety (e.g., carbonyl) which simultaneously replaces two available hydrogen on the same carbon atom on a ring system.
- a single moiety e.g., carbonyl
- pyrrolidone e.g., pyrrolidone
- Heterocyclylalkyl means a heterocyclyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable heterocyclylaikyls include piperidinylmethyl, piperazinylmethyl and the like.
- Heterocyclenyl means a non-aromatic monocyclic or multicyclic ring system comprising about 3 to about 10 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is an element other than carbon, for example nitrogen, oxygen or sulfur atom, alone or in combination, and which contains at least one carbon-carbon double bond or carbon-nitrogen double bond. There are no adjacent oxygen and/or sulfur atoms present in the ring system.
- Preferred heterocycienyl rings contain about 5 to about 6 ring atoms.
- the prefix aza, oxa or thia before the heterocyclenyl root name means that at least a nitrogen, oxygen or sulfur atom respectively is present as a ring atom.
- the heterocycienyl can be optionally substituted by one or more ring system substituents, wherein "ring system substituent" is as defined above.
- the nitrogen or sulfur atom of the heterocyclenyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide.
- heterocyclenyl groups include 1 ,2,3,4- tetrahydropyridinyl, 1 ,2-dihydropyridinyl, 1 ,4-dihydropyridinyl, 1 ,2,3,6- tetrahydropyridinyl, 1 ,4,5,6-tetrahydropyrimidinyl, 2-pyrrolinyl, 3-pyrrolinyl, 2- imidazolinyl, 2-pyrazoiinyl, dihydroimidazolyl, dihydrooxazolyi, dihydrooxadiazolyl, dihydrothiazolyl, 3,4-dihydro ⁇ 2H-pyranyS, dihydrofuranyl, fluorodihydrofuranyi, 7- oxabicyclo[2.2.1]heptenyl, dihydrothiophenyi, dihydrothiopyranyl, and the like.
- Heterocyclenyl may also mean a single moiety
- Heterocyclenylalkyl means a heterocyclenyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core. It should be noted that in hetero-atom containing ring systems of this invention, there are no hydroxyl groups on carbon atoms adjacent to a N, O or S, as well as there are no N or S groups on carbon adjacent to another heteroatom. Thus, for example, in the ring:
- Alkynylalkyl means an alkynyi-alkyl- group in which the alkynyl and alkyl are as previously described. Preferred alkynylalkyls contain a lower alkynyi and a lower alkyl group. The bond to the parent moiety is through the alkyf. Non-limiting examples of suitable alkynylalkyl groups include propargyimethyl. "Heteroaralkyi” means a heteroaryl-alkyl- group in which the heteroaryl and alkyl are as previously described. Preferred heteroaralkyls contain a lower alky! group. Non-limiting examples of suitable araikyl groups include pyridyl methyl, and quinolin-3- ylmethyl. The bond to the parent moiety is through the alkyi.
- Spiro ring systems have two or more rings linked by one common atom.
- Preferred spiro ring systems include spiroheteroaryl, spiroheterocyclenyl, spiroheterocyclyl, spirocycloaikyl, spirocyclenyl, and spiroaryl.
- the spiro ring systems can be optionally substituted by one or more ring system substituents, wherein "ring system substituent" is as defined above.
- systems include spiro[4.5jdecane, 8-azaspiro[4.5]dec-2-ene, and ' spiro[4.4]nona-2 t 7-diene.
- ⁇ ydroxyalkyl means a HO-aikyl- group in which alkyl is as previously defined.
- Preferred hydroxyalkyls contain lower alkyl.
- suitable hydroxyalkyl groups include hydroxymethyl and 2-hydroxyethyl.
- acyl means an H-C(O)-, alkyl-C(O)- or cycloalkyl-C(O)-, group in which the various groups are as previously described.
- the bond to the parent moiety is through the carbonyl.
- Preferred acyls contain a lower alkyl.
- suitable acyl groups include formyl, acetyl and propanoyl.
- Aroyl means an aryl-C(O)- group in which the aryl group is as previously described.
- the bond to the parent moiety is through the carbonyi.
- suitable groups include benzoyl and 1- naphthoyl.
- Alkoxy means an alkyl-O- group in which the alky! group is as previously described.
- suitable alkoxy groups include methoxy, ethoxy, n-propoxy, isopropoxy and n-butoxy.
- the bond to the parent moiety is through the ether oxygen.
- Aryloxy means an aryl-O- group in which the aryl group is as previously described. Non- ⁇ miting examples of suitable aryloxy groups include phenoxy and naphthoxy. The bond to the parent moiety is through the ether oxygen.
- Aralkyloxy means an aralkyl-O- group in which the aralkyi group is as previously described. Non-limiting examples of suitable aralkyloxy groups include benzyloxy and 1- or 2-naphthalenemethoxy. The bond to the parent moiety is through the ether oxygen.
- Alkylthio means an alkyl-S- group in which the aikyl group is as previously described.
- suitable alkylthio groups include methylthio and ethylthio.
- the bond to the parent moiety is through the sulfur.
- Arylthio means an aryl-S- group in which the aryl group is as previously described.
- suitable arylthio groups include phenyithio and naphthylthio.
- the bond to the parent moiety is through the sulfur.
- Aralkylthio means an aralkyl-S- group in which the aralkyl group is as previously described.
- Non-limiting example of a suitable aralkylthio group is benzylthio. The bond to the parent moiety is through the sulfur.
- Alkoxycarbonyl means an alkyi-O-CO- group.
- suitable alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl.
- the bond to the parent moiety is through the carbonyl.
- Aryloxycarbonyl means an aryl-O-C(O)- group.
- suitable aryloxycarbonyi groups include phenoxycarbonyl and naphthoxycarbonyi.
- the bond to the parent moiety is through the carbonyl.
- Aralkoxycarbonyl means an aralkyl-O-C(O)- group.
- Non-limiting example of a suitable aralkoxycarbonyl group is benzyloxycarbonyl. The bond to the parent moiety is through the carbonyl.
- Alkylsulfonyi means an alkyi-S(O 2 )- group. Preferred groups are those in which the alkyl group is lower alkyl. The bond to the parent moiety is through the sulfonyl.
- Arylsulfonyl means an aryl-S(O 2 )- group. The bond to the parent moiety is through the sulfonyl.
- substituted means that one or more hydrogen on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- stable compound' or “stable structure” is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- purified refers to the physical state of said compound after being isolated from a synthetic process (e.g. from a reaction mixture), or natural source or combination thereof.
- purified refers to the physical state of said compound after being obtained from a purification process or processes described herein or wefl known to the skilled artisan (e.g., chromatography, recrystallization and the like) , in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan. It should aiso be noted that any carbon as well as heteroatom with unsatisfied valences in the text, schemes, examples and Tables herein is assumed to have the sufficient number of hydrogen atom(s) to satisfy the valences.
- protecting groups When a functional group in a compound is termed "protected", this means that the group is in modified form to preclude undesired side reactions at the protected site when the compound is subjected to a reaction. Suitable protecting groups will be recognized by those with ordinary skill in the art as well as by reference to standard textbooks such as, for example, T. W. Greene et a/, Protective Groups in organic Synthesis (1991), Wiley, New York.
- variable e.g., aryl, heterocycle, R 2 , etc.
- its definition on each occurrence is independent of its definition at every other occurrence.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- Prodrugs and solvates of the compounds of the invention are also contemplated herein.
- a discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press.
- the term "prodrug” means a compound (e.g., a drug precursor) that is transformed in vivo Xo yieid a compound of the present invention or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (e.g., by metabolic or chemical processes), such as, for example, through hydrolysis in blood.
- a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as, for example, (C 1 -C ⁇ jalkyl, (C 2 - Ci 2 )alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1- methyl-1 -(alkanoyloxy)-ethyl having from 5 to 10 carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1-(alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1-methyl-1-(alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N-(alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1 -(N-(alk)alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1 -(N-
- a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as, for example, (C 1 -C 6 )alkanoyloxymethyl, 1 -((Ci- C 6 )alkanoyloxy)ethyl, 1 -methyl-1 -((C 1 -C 6 )alkanoyloxy)ethyl, (C 1 - C 6 )alkoxycarbonyloxymethyl, N-(C 1 -C 6 )alkoxycarbonylaminomethyl, succinoyl, (Cr C 6 )alkanoy!, ⁇ -amino(C 1 -C 4 )alkanyl, aryiacyl and ⁇ -aminoacyl, or ⁇ -aminoacyl- ⁇ - aminoacyl, where each ⁇ -aminoacyl group is independently selected from the naturally occurring L-amino acids, P(O)
- a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as, for example, R-carbonyi, RO-carbonyl, NRR'-carbonyl where R and R' are each independently (Ci ⁇ Cio)alkyt, (C 3 -C 7 ) cycloalkyl, benzyl, or R-carbonyl is a natural ⁇ -aminoacyl or natural ⁇ -aminoacyl, -C(OH)C(O)OY 1 wherein Y 1 is H, (C 1 -Ce)alkyl or benzyl, -C(OY 2 JY 3 wherein Y 2 is (C 1 -C 4 ) alky] and Y 3 is (C 1 -C 6 )alkyl, carboxy (C 1 -C 6 )alky!, amino(C 1 -C 4 )alkyl or mono-N —
- R-carbonyi RO-carbonyl
- One or more compounds of the invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable sêts such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms.
- “Solvate” means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. "Solvate” encompasses both solution-phase and isolatable solvates. Non-limiting examples of suitable solvates include ethanoiates, methanolates, and the like.
- “Hydrate” is a solvate wherein the solvent molecule is H 2 O.
- One or more compounds of the invention may optionally be converted to a solvate.
- Preparation of solvates is generally known.
- M. Caira et al, J. Pharmaceutical Sci., 93(3), 601-611 (2004) describes the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water.
- Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS PharmSciTech., 5£1), article 12 (2004); and A. L. Bingham et al, Chem. Comm ⁇ n., 603-604 (2001).
- a typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than ambient temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods.
- Analytical techniques such as, for example I. R. spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate).
- Effective amount or “therapeutically effective amount” is meant to describe an amount of compound or a composition of the present invention effective in inhibiting the above-noted diseases and thus producing the desired therapeutic, ameliorative, inhibitory or preventative effect.
- the compounds of the present invention can form salts which are also within the scope of this invention.
- Reference to a compound of the present invention herein is understood to include reference to salts thereof, unless otherwise indicated.
- the term “saltfs)" denotes acidic salts formed with inorganic and/or organic acids, as well as basic salts formed with inorganic and/or organic bases.
- salts when a compound of the present invention contains both a basic moiety, such as, but not limited to a pyridine or imidazole, and an acidic moiety, such as, but not limited to a carboxylic acid, zwitterions ("inner salts") may be formed and are included within the term "salt(s)" as used herein.
- Pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) salts are preferred, although other salts are also useful.
- Salts of the compounds of the The present invention may be formed, for example, by reacting a compound of the present invention with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates,) and the like.
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as dicyclohexylamines, t-butyl amines, and salts with amino acids such as arginine, lysine and the like.
- Basic nitrogen-containing groups may be quartemized with agents such as lower alkyl hatides (e.g. methyl, ethyl, and butyl chlorides, bromides and iodides), dialky! sulfates (e.g.
- dimethyl, diethyl, and dibutyl sulfates dimethyl, diethyl, and dibutyl sulfates
- long chain halides e.g. decyl, iauryl, and stearyl chlorides, bromides and iodides
- aralkyl haiides e.g. benzyl and phenethyl bromides
- esters of the present compounds include the following groups: (1 ) carboxylic acid esters obtained by esterification of the hydroxy groups, in which the non-carbonyl moiety of the carboxyiic acid portion of the ester grouping is selected from straight or branched chain alkyl (for example, acetyl, n- propyl, t-butyl, or n-butyl), alkoxyalkyl (for example, methoxymethyl), araikyl (for example, benzyl), aryloxyalkyl (for example, phenoxymethyl), aryl (for example, phenyl optionally substituted with, for example, halogen, d ⁇ alkyl, or Ci.
- alkyl for example, acetyl, n- propyl, t-butyl, or n-butyl
- alkoxyalkyl for example, methoxymethyl
- araikyl for example, benzyl
- aryloxyalkyl for example,
- sulfonate esters such as alkyl- or aralkylsulfonyl (for example, methanesulfonyl); (3) amino acid esters (for example, L-valyl or L-isoieucyl); (4) phosphonate esters and (5) mono-, di- or triphosphate esters.
- the phosphate esters may be further esterified by, for example, a C 1-2 o alcohol or reactive derivative thereof, or by a 2,3-di (C 6 - 24 )acyl glycerol.
- the compounds of the present invention may contain asymmetric or chiral centers, and, therefore, exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of the present invention as well as mixtures thereof, including racemic mixtures, form part of the present invention.
- the present invention embraces all geometric and positional isomers. For example, if a compound of the present invention incorporates a double bond or a fused ring, both the cis- and trans-forms, as well as mixtures, are embraced within the scope of the invention.
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods well known to those skilled in the art, such as, for example, by chromatography and/or fractional crystallization.
- Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride), separating the diastereomers and converting (e.g., hydroiyzing) the individual diastereomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride
- converting e.g., hydroiyzing
- some of the compounds of the present invention may be atropisomers (e.g., substituted biaryls) and are considered as part of this invention.
- the compounds of the present invention may exist in different tautomeric forms, and all such forms are embraced within the scope of the invention. Also, for example, all keto-enol and imine-enamine forms of the compounds are included in the invention.
- All stereoisomers for example, geometric isomers, optical isomers and the like
- of the present compounds ⁇ including those of the salts, solvates, esters and prodrugs of the compounds as well as the salts, solvates and esters of the prodrugs), such as those which may exist due to asymmetric carbons on various substituents, including enantiomeric forms (which may exist even in the absence of asymmetric carbons), rotameric forms, atropisomers, and diastereomeric forms, are contemplated within the scope of this invention, as are positional isomers (such as, for example, 4-pyridyl and 3-pyridyl).
- a compound of the present invention incorporates a double bond or a fused ring, both the cis- and trans-forms, as well as mixtures, are embraced within the scope of the invention. Also, for example, all keto-enol and imine-enamine forms of the compounds are included in the invention.
- Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers, or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers.
- the chiral centers of the present invention can have the S or R configuration as defined by the IUPAC 1974 Recommendations.
- salt is intended to equally apply to the salt, solvate, ester and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, racemates or prodrugs of the inventive compounds.
- the present invention also embraces isotopically-labelled compounds of the present invention which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine and chlorine and iodine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 17 O, 31 P, 32 P, 35 S 1 18 F, 36 CI and 123 I, respectively.
- Certain isotopically-labelled compounds of Formula (!) are useful in compound and/or substrate tissue distribution assays. Tritiated ⁇ i.e., 3 H) and carbon-14 (i.e., 14 C) isotopes are particularly preferred for their ease of preparation and detectabiiity. Certain isotopically-labelled compounds of Formula (I) can be useful for medical imaging purposes.
- those labeled with positron-emitting isotopes like 11 C or 18 F can be useful for application in Positron Emission Tomography (PET) and those labeled with gamma ray emitting isotopes like 123 I can be useful for application in Single photon emission computed tomography (SPECT).
- PET Positron Emission Tomography
- SPECT Single photon emission computed tomography
- substitution with heavier isotopes such as deuterium (i.e., 2 H) may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances.
- isotopically labeled compounds of Formula (I) in particular those containing isotopes with longer half lives (T1/2 >1 day), can generally be prepared by following procedures analogous to those disclosed in the Schemes and/or in the Examples herein below, by substituting an appropriate isotopically labeled reagent for a non-isotopically labeled reagent.
- polymorphic forms of the compounds of the present invention, and of the salts, solvates, esters and prodrugs of the compounds of the present invention, are intended to be included in the present invention.
- the compounds according to the invention have pharmacological properties; in particular, the compounds of the present invention can be inhibitors, regulators or modulators of mTOR protein kinases.
- the compounds of the present invention can be inhibitors of protein kinases such as, for example, the inhibitors of the mTOR.
- Preferred compounds can exhibit IC 50 values of less than about 5 ⁇ m, preferably about 0.001 to about 1.0 ⁇ m, and more preferably about 0.001 to about 0.1 ⁇ m.
- the assay methods are described in the Examples set forth below.
- the compounds of the present invention can be useful in the therapy of proliferative diseases such as cancer, autoimmune diseases, viral diseases, fungal diseases, neurological/neurodegenerative disorders, arthritis, inflammation, antiproliferative (e.g., ocular retinopathy), neuronal, alopecia and cardiovascular disease.
- proliferative diseases such as cancer, autoimmune diseases, viral diseases, fungal diseases, neurological/neurodegenerative disorders, arthritis, inflammation, antiproliferative (e.g., ocular retinopathy), neuronal, alopecia and cardiovascular disease.
- proliferative diseases such as cancer, autoimmune diseases, viral diseases, fungal diseases, neurological/neurodegenerative disorders, arthritis, inflammation, antiproliferative (e.g., ocular retinopathy), neuronal, alopecia and cardiovascular disease.
- the compounds of the present invention can be useful in the treatment of a variety of cancers, including (but not limited to) the following: tumor of the bladder, breast (including BRCA-mutated breast cancer, colorectal, colon, kidney, liver, lung, small cell lung cancer, non-smail cell lung cancer, head and neck, esophagus, bladder, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, including squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma and Burkett's lymphoma; chronic lymphocytic leukemia ("CLL”), acute and chronic myelogenous leukemia, myelodysplastic syndrome and prom
- CLL chronic lympho
- inhibitors could act as reversible cytostatic agents which may be useful in the treatment of any disease process which features abnormal cellular proliferation, e.g., benign prostate hyperplasia, familial adenomatosis polyposis, neuro-fibromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, glomerulonephritis, restenosis following angioplasty or vascular surgery, hypertrophic scar formation, inflammatory bowel disease, transplantation rejection, endotoxic shock, and fungal infections.
- Compounds of the present invention may induce or inhibit apoptosis.
- apoptotic response is aberrant in a variety of human diseases.
- Compounds of the present invention, as modulators of apoptosis will be useful in the treatment of cancer (including but not limited to those types mentioned hereinabove), virai infections (including but not limited to herpevirus, poxvirus, Epstein- Barr virus, Sindbis virus and adenovirus), prevention of AIDS development in HIV-infected individuals, autoimmune diseases (including but not limited to systemic lupus, erythematosus, autoimmune mediated glomerulonephritis, rheumatoid arthritis, psoriasis, inflammatory bowel disease, and autoimmune diabetes mellitus), neurodegenerative disorders (including but not limited to Alzheimer's disease, AIDS-reiated dementia, Parkinson's disease, amyotrophic lateral sclerosis, retinitis pigmentosa, spinal muscular atrophy and cerebellar degeneration), myelodysplasia syndromes, aplastic anemia, ische
- Compounds of the present invention can modulate the level of cellular RNA and DNA synthesis. These agents would therefore be useful in the treatment of viral infections (including but not limited to HIV, human papilloma virus, herpesvirus, poxvirus, Epstein-Barr virus, Sindbis virus and adenovirus). Compounds of the present invention may also be useful in the chemoprevention of cancer. Chemoprevention is defined as inhibiting the development of invasive cancer by either blocking the initiating mutagenic event or by blocking the progression of pre-malignant cells that have already suffered an insult or inhibiting tumor relapse.
- Compounds of the present invention may also be useful in inhibiting tumor angiogenesis and metastasis.
- Another aspect of this invention is a method of treating a mammal ⁇ e.g., human) having a disease or condition associated with mTOR kinases by administering a therapeutically effective amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound to the mammal.
- a preferred dosage is about 0.001 to 1000 mg/kg of body weight/day of the compound of the present invention.
- An especially preferred dosage is about 0.01 to 25 mg/kg of body weight/day of a compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound.
- the compounds of this invention may also be useful in combination (administered together or sequentially) with one or more of anti-cancer treatments such as radiation therapy, and/or one or more anti-cancer agents different from the compound of the present invention.
- the compounds of the present invention can be present in the same dosage unit as the anti-cancer agent or in separate dosage units.
- Another aspect of the present invention is a method of treating one or more diseases associated with a mTOR protein kinase, comprising administering to a mammal in need of such treatment: an amount of a first compound, which is a compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent different from the compound of the present invention, wherein the amounts of the first compound and the second compound result in a therapeutic effect.
- suitable anti-cancer agent is selected from the group consisting of a Cytostatic agent, Cisplatin, Deforolimus (described in PCT publication No.
- Triethylenethiophosphoramine Busulfan, Carmustine, Lomustine, Streptozocin, dacarbazine, Floxuridine, Cytarabine, 6-Mercaptopurine, 6-Thioguanine, Fludarabine phosphate, Oxaliplatin, Leucovirin, ELOXATINTM, Vinblastine, Vincristine, Vindesine, Bleomycin, Dactinomycin, Daunorubicin, Doxorubicin, Epirubicin, Idarubicin, Mithramycin, Deoxycoformycin, Mitomycin-C, L-Asparaginase, Teniposide 17oc- Ethinylestradiol, Diethylstilbestroi, Testosterone, Prednisone, Fluoxymesterone, Dromostanolone propionate, Testolactone, Megestrolacetate, Methylprednisoione, Methyltestosterone, Prednisolone, Triamcino
- such combination products employ the compounds of this invention within the dosage range described herein and the other pharmaceutically active agent or treatment within its dosage range.
- the CDC2 inhibitor olomucine has been found to act synergistically with known cytotoxic agents in inducing apoptosis (J. CeII ScL, (1995) 108, 2897.
- Compounds of the present invention may also be administered sequentially with known anticancer or cytotoxic agents when a combination formulation is inappropriate.
- the invention is not limited in the sequence of administration; compounds of the present invention may be administered either prior to or after administration of the known anticancer or cytotoxic agent.
- this invention includes combinations comprising an amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof, and an amount of one or more anticancer treatments and anti-cancer agents listed above wherein the amounts of the compounds/ treatments result in desired therapeutic effect.
- Another aspect of the present invention is a method of inhibiting one or more mTOR protein kinases in a patient in need thereof, comprising administering to the patient a therapeutically effective amount of at least one compound of the present invention or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more mTOR protein kinases in a patient in need thereof, comprising administering a therapeutically effective amount of at least one compound of the present invention or a pharmaceuticaily acceptable salt, solvate, ester or prodrug thereof.
- Yet another aspect of the present invention is a method of treating one or more diseases associated with mTOR protein kinases, comprising administering to a mammai in need of such treatment an amount of a first compound, which is a compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent, wherein the amounts of the first compound and the second compound result in a therapeutic effect.
- a first compound which is a compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof
- an amount of at least one second compound the second compound being an anti-cancer agent
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more mTOR protein kinases in a patient in need thereof, comprising administering a therapeutically effective amount of a pharmaceutical composition comprising in combination at least one pharmaceutically acceptable carrier and at least one compound according to The present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- the mTOR protein kinases to be inhibited can exist in two complexes, mTORC1 and mTORC2.
- ERK inhibitors include but are not limited to the following compounds: a) Formula H
- Each Q 1 represents a ring independentiy selected from the group consisting of: cycloalkyl, substituted cycloalkyi, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted ary!, heteroaryl, and substituted heteroaryl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: halo and the R 10 moieties; provided that when Q 1 is ary!, heteroaryl, substituted aryl or substituted heteroaryl then the carbon atoms at the ring junction are not substituted;
- Q 2 represents a ring selected from the group consisting of: cycloalkyl, substituted cycloaikyl, heterocycloaikyl, and substituted heterocycloalkyl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R 10 moieties;
- Z 1 represents -(C(R 24 ) 2 ) W - wherein each R 24 is independently selected from the group consisting of: H, alkyl and F, and wherein w is 1 , 2 or 3;
- Z 2 is selected from the group consisting of: -N(R 44 )-, -O- and -C(R 46 J 2 -; m is 1 to 6; n is 1 to 6; p is 0 to 6; t is O 1 1 , or 2;
- R 1 is selected from the group consisting of: (I) -CN,
- R 2 is selected from the group consisting of: (D H 1 )
- each alkyi is independently selected
- each alkyl is independently selected
- each R 48 is independently selected from the group consisting of: H and alkyl
- each R 3 , R 4 , R 5 , R 6 and R 7 is independently selected from the group consisting of: (I) H 1
- R 11 is selected from the group consisting of: F, -OH, -CN, -OR 10 , -NHNR 1 R 10 , -SR 10 and heteroaryl;
- R 12 is selected from the group consisting of: alkyl, aryl, heteroaryl, cycloalkyi, eye! oa Iky I alkyl, heterocycloalkyl and heterocycloalkylaikyi;
- R 14 is selected from the group consisting of: alkyt, aryl, heteroaryl, cycloalkyi, cycloaikylalkyl-, heterocycloalkyl, alkylheterocycloalkyl, heterocycioalkylalkyl-, aikylheteroaryi- and aikyiaryl-;
- R 15 is selected from the group consisting of: H, -OH, alkyl, aryl, heteroaryl, cycloalkyl, cycloaikylalkyl-, heterocycloalkyl and heterocycloalkylaikyi-, alkylheteroaryl- and alkylary!-;
- R 20 represents alkyl
- R 23 is selected from the group consisting of: H, alkyl, aryl, cycloaikyl, and cycloalkylalkyi-; each R 26 is independently selected from the group consisting of: H and alkyl;
- R 28 is alkyi; each R 30 is independently selected from the group consisting of: H, alkyl, and F; each R 32 is independently selected from the group consisting of: H and alkyl, and wherein each R 32 is generally H; each R 35 is independently selected from the group consisting of: H and Ci to C 6 aikyl;
- R 36 is selected from the group consisting of: H, alkyl, and -O-alkyl; each R 38 is independently selected from the group consisting of: H, alkyl, aryi, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalky!
- R 38 substituted alkyl is substituted with 1 to 3 substituents independently selected from the group consisting of: -NH 2 , -NO 2 , -CN, -OR 26 , halo, -C(O)-NH-R 28 , -C(O)OR 28 , and -C(O)R 28 , and said R 3 ⁇ substituted aryl, substituted aryialkyl, substituted heteroaryl, substituted hetero
- R 42 is selected from the group consisting of: alkyl, aryl, heteroaryl, and cycloalkyl
- R 44 is selected from the group consisting of: H, alkyl, cycloalkyl, and cycloaikylalkyl
- Each R 46 is independently selected from the group consisting of: H, alkyi, cycloalkyl, and cycloalkylalkyl; and provided that:
- Q 1 is a substituted ring wherein at least one substitutent is halo; and/or (2) R 2 is substituted alkyl wherein said substituent is -OCH 3 , or R 2 is selected from the group consisting of: -CH 2 OH and -CH 2 OCH 3 ; and/or
- R 3 , R 4 , R 5 , R 6 , and R 7 is:
- R 5A is alkyl
- R 10 is a substituted aryl, and at least one substitutent is selected from the group consisting of: (a) -S(O) t R 38 wherein R 38 is isopropyl,
- R 20 is isopropyl
- Each Q 1 represents a ring independently selected from the group consisting of: cycioalkyl, substituted cycioaikyl, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted aryl, heteroaryl, and substituted heteroaryl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: haio and the R 10 moieties; provided that when Q 1 is ary!, heteroaryl, substituted aryl or substituted heteroaryl then the carbon atoms at the ring junction are not substituted;
- Q 2 represents a ring selected from the group consisting of: cycloalkyl, substituted cycloalkyi, heterocycloalkyl, and substituted heterocycloalkyl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R 10 moieties;
- Z 1 represents - ⁇ C(R 24 ) 2 ) W - wherein each R 24 is independently selected from the group consisting of: H, alkyl and F, and wherein w is 1 , 2 or 3;
- Z 2 is selected from the group consisting of: -N(R 44 )-, -O- and -C(R 46 ) 2 -;
- m is 1 to 6;
- n is 1 to 6;
- p is 0 to 6;
- t is 0, 1 , or 2;
- R 1 is selected from the group consisting of:
- each R 10 is independently selected, (18) wherein each R 10 is independently selected, (19)
- each R 10 is independently selected, (22) -C(O)-NR 32 -C(R 18 ) 3 ,
- R 2 is selected from the group consisting of: (I) H 1
- substituted alkyl wherein said substituted alkyl is substituted with 1 to 3 substitutents selected from the group consisting of: (a) -OH, (b) -O-alkyl, (c) -O-alkyl substituted with 1 to 3 F atoms, and (d) -N(R 40 ) 2 wherein each R 40 is independently selected from the group consisting of: (i) H, (ii) C 1 -C 3 alkyl, (iii) -CF 3 , and (e) halo, (6) alkynyl,
- each alkyl is independently selected
- each R 48 is independently selected from the group consisting of: H and aikyl
- each R 3 , R 4 , R 5 , R 6 and R 7 is independently selected from the group consisting of:
- R SA is selected from the group consisting of: halo, -OH, alkyl, -O-alkyl;
- R 8 is selected from the group consisting of: H, -OH, -N(R 10 ) 2) -NR 10 C(O)R 12 ; each R 9 is independently selected from the group consisting of :halogen, -CN,
- each R 10 is independently selected from the group consisting of: H, alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalkylalkyl, heterocycioalkyl, heterocycloalkylalkyl, alkylheteroaryl-, alkylaryl-, substituted alkyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloaikyl, substituted cycloalkylalkyl, substituted heterocycioalkyl, substituted heterocycloaikylalkyi, substituted alkylheteroaryl-, substituted alkyiaryl-, heterocycloaikenyl
- R 10 substituted alkyl is substituted with 1 to 3 substituents independently selected from the group consisting of: -NH 2 , -NHR 20 , -NO 2 , -CN, -OR 26 , halo, -C(O)-NH-R 26 , -C(O)OR 26 , and -C(O)R 26 , and said R 10 substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroaryialkyl, substituted cycloalkyl, substituted cycloalkylalkyl, substituted heterocycloalkyl, substituted heterocycloaikylalkyi, substituted alkylheteroaryl- and substituted alkyiaryl- are substituted with 1 to 3 substituents independently selected from the group consisting of: (1 ) -NH 2 , (2) -NO 2 , (3) -CN, (4) -OH, (5)
- R 11 is selected from the group consisting of: F, -OH, -CN, -OR 10 , -NHNR 1 R 10 , -SR 10 and heteroaryl;
- R 12 is selected from the group consisting of: alkyl, aryl, heteroaryl, cycioalkyl, cycloalkylalkyl, heterocycloalkyl and heterocycloaikylalkyi;
- R 14 is selected from the group consisting of: aikyl, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl-, heterocycloalkyl, alkylheterocycloalkyl, heterocycloaikylalkyi-, afkylheteroaryt- and alkyiaryl-;
- R 15 is selected from the group consisting of: H, -OH, alkyi, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl-, heterocycloaikyl and heterocycioalkylalkyl-, alkylheteroaryi- and aikylaryl-;
- R 20 represents alkyl
- R 23 is selected from the group consisting of: H, alkyl, aryl, cycloalkyl, and cycloalkylalkyl-
- each R 26 is independently selected from the group consisting of: H and alkyi;
- R 28 is alkyl; each R 30 is independently selected from the group consisting of: H 1 alkyl, and F; each R 32 is independently selected from the group consisting of: H and alkyl; each R 35 is independently selected from the group consisting of: H and Ci to C 6 alkyl; each R 38 is independently selected from the group consisting of: H, alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyi, heterocycloalkylalky], alkylheteroaryl-, alkylaryl-, substituted alkyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloalkyl, substituted cycloalkylalkyl, substituted heterocycloaikyl, substituted heterocycioalkylalkyl, substituted alkylheteroaryl-
- R 42 is selected from the group consisting of: alkyi, aryl (e.g., phenyl), heteroaryl, and cycloalkyi;
- R 44 is selected from the group consisting of: H, alkyl, cycloalkyi, and cycloalkylaikyl;
- Each R 46 is independently selected from the group consisting of: H, alkyl, cycloalkyi, and cycloalkylaikyl;
- Y 1 , Y 2 , and Y 3 are each independently selected from the group consisting of:
- Each Q 1 represents a ring independently selected from the group consisting of: cycloalkyl, substituted cycloalkyl, heterocycioalkyl, substituted heterocycloalkyi, aryl, substituted aryl, heteroaryl, and substituted heteroaryl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R 10 moieties; provided that when Q 1 is aryl, heteroaryl, substituted aryl or substituted heteroaryl then the carbon atoms at the ring junction are not substituted;
- Q 2 represents a ring selected from the group consisting of: cycloalkyi, substituted cycloalkyi, heterocycJoalkyi, and substituted heterocycioalkyl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R 10 moieties;
- Z 1 represents -(C(R 24 ) 2 )w- wherein each R 24 is independently selected from the group consisting of: H, alkyl and F, and wherein w is 1 , 2 or 3, and generally w is 1 or 2, and usually w is 1 , and wherein in one example each R 24 is H, and in another example w is 1 , and in another example each R 24 is H and w is 1 , preferably w is 1 and each R 24 is H;
- Z 2 is selected from the group consisting of: -N(R 44 )-, -O- and -C(R 46 J 2 -; m is 1 to 6; n is 1 to 6; p is 0 to 6; t is 0, 1 , or 2;
- R 1 is selected from the group consisting of: (I) -CN,
- each R 10 is independently selected, and preferably each R 10 is independently selected from the group consisting of: (a) H, (b) alkyl, (c) heteroaryl, (d) aryl, and (e) cycloalkyl, wherein for example, each R 10 is selected from the group consisting of: H, methyl, butyl, i-propyl, pyridyl, phenyl and cyclopropyl, wherein, for example, said -C(O)N(R 10 ) 2 moiety is selected from the group consisting of: -C(O)NH 2 , -C(O)NH(CH 3 ), -C(O)NH(CH)(CH 3 ) 2J -C(O)NH(C 4 H 9 ), -C(O)NH(C 6 H 5 ), -C(O)NH(C 3 H 5 ), and -C(O)NH(C 5 H 4 N);
- R 2 is selected from the group consisting of: (1) H,
- substituted alkyl wherein said substituted alkyl is substituted with 1 to 3 substitutents selected from the group consisting of: (a) -OH, (b) -O-alkyl (e.g., -0-(Cr C 3 alkyl), (c) -O-alkyi (e.g., -O-(C r C 3 alkyl)) substituted with 1 to 3 F atoms, and (d) - N(R 40 )2 wherein each R 40 is independently selected from the group consisting of: (i) H, (ii) CrC 3 alkyl and (iii) -CF 3 ,
- each R 3 , R 4 , R 5 , R 6 and R 7 is independently selected from the group consisting of:
- R 10 is selected from the group consisting of: alkyl,
- arylheteroaryl- (16) substituted arylheteroaryl-, (17) heteroarylaryi-, such as, for example, pyrimidinylphenyl-, pyrazinylphenyl-, pyridinylphenyl-, furanylphenyl-, thienylphenyl-, and thiazolylphenyl-,
- substituted heteroarylaryi- such as, for example, substituted pyrimidinylphenyl-, substituted pyrazinylphenyl-, substituted pyridinyiphenyl-, substituted furanylphenyl-, substituted thienylphenyl-, substituted thiazolylphenyl-, and substituted pyrimidinylphenyl,
- R 8 is selected from the group consisting of: H, -OH, aikyl, aryl, -N(R 10 J 2 and -NR 1 °C(O)R 12 ; each R 9 is independently selected from the group consisting of:halogen, -CN, -NO 2 , -OR 10 , -SR 10 , -N(R 10 J 2 , and R 10 ; each R 10 is independently selected from the group consisting of; H, aikyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalkyl aikyl, heterocycloalkyl, heterocycloalkylalkyl, alkylheteroaryl-, alkylaryl-, substituted aikyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloalkyS, substituted cycloalkylal
- R 1 Ms selected from the group consisting of: F, -OH, -CN, -OR 10 , -NHNR 1 R 10 ,
- R 12 is selected from the group consisting of: alkyi, aryl, heteroaryi, cycloalkyl, cyctoalkylalkyl, heterocycloalkyl and heterocycloalkylalkyl;
- R 14 is selected from the group consisting of: aikyl, aryl, heteroaryl, cycloalkyi, cycloalkylalkyl-, heterocycloalkyl, alkylheterocycloalkyl, heterocycloalkylalkyl-, alkylheteroaryl- and alkyiaryl-;
- R 15 is selected from the group consisting of: H, -OH, alkyl, aryl, heteroaryl, cycioalkyl, cycloalkylalkyl-, heterocycloalkyl and heterocycloalkylalkyl-, alkylheteroaryl- and alkyiaryl-;
- R 20 represents alkyl;
- R 23 is selected from the group consisting of: H, alkyl, aryl, cycloalkyl, and cycloalkyialkyl-; each R 26 is independently selected from the group consisting of: H and alkyl; R 28 is alkyl; each R 30 is independently selected from the group consisting of: H, afkyl, and
- R 42 is selected from the group consisting of: alkyl, aryi, heteroaryl, and cycloalkyi;
- R 44 is selected from the group consisting of: H, alkyl, cycloaikyl, and cycloalkyialkyl; and Each R 46 is independently selected from the group consisting of: H, alkyl, cycloalkyi, and cycloalkylalkyi;
- Non-limiting examples of ERK inhibitors are selected from the group consisting of:
- compositions which comprise at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and at least one pharmaceutically acceptable carrier.
- inert, pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, dispersible granules, capsules, cachets and suppositories. The powders and tablets may be comprised of from about 5 to about 95 percent active ingredient. Suitable solid carriers are known in the art, e.g., magnesium carbonate, magnesium stearate, talc, sugar or lactose.
- Tablets, powders, cachets and capsules can be used as solid dosage forms suitable for oral administration.
- Examples of pharmaceutically acceptable carriers and methods of manufacture for various compositions may be found in A. Gennaro (ed.), Remington's Pharmaceutical Sciences, 18 th Edition, (1990), Mack Publishing Co., Easton, Pennsylvania.
- Liquid form preparations include solutions, suspensions and emulsions. As an example may be mentioned water or water-propylene glycol solutions for parenteral injection or addition of sweeteners and opacifiers for oral solutions, suspensions and emulsions. Liquid form preparations may also include solutions for intranasal administration.
- Aerosol preparations suitable for inhalation may include solutions and solids in powder form, which may be in combination with a pharmaceutically acceptable carrier, such as an inert compressed gas, e.g. nitrogen. Also included are solid form preparations that are intended to be converted, shortly before use, to liquid form preparations for either oral or parenteral administration. Such liquid forms include solutions, suspensions and emulsions.
- the compounds of the invention may also be deliverable transdermally.
- the transdermal compositions can take the form of creams, lotions, aerosols and/or emulsions and can be included in a transdermal patch of the matrix or reservoir type as are conventional in the art for this purpose.
- the compounds of this invention may also be delivered subcutaneously.
- the compound is administered orally or intravenously.
- delivery methods that are combinations of the above- noted delivery methods, Such methods are typically decided by those skilled in the art.
- the pharmaceutical preparation is in a unit dosage form.
- the preparation is subdivided into suitably sized unit doses containing appropriate quantities of the active component, e.g., an effective amount to achieve the desired purpose.
- the quantity of active compound in a unit dose of preparation may be varied or adjusted from about 1 mg to about 100 mg, preferably from about 1 mg to about 50 mg, more preferably from about 1 mg to about 25 mg, according to the particular application.
- the actual dosage employed may be varied depending upon the requirements of the patient and the severity of the condition being treated. Determination of the proper dosage regimen for a particular situation is within the skill of the art. For convenience, the total daily dosage may be divided and administered in portions during the day as required.
- a typical recommended daily dosage regimen for oral administration can range from about 1 mg/day to about 500 mg/day, preferably 1 mg/day to 200 mg/day, in two to four divided doses.
- kits comprising a therapeutically effective amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and a pharmaceutically acceptable carrier, vehicle or diluent.
- kits comprising an amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and an amount of at least one anticancer therapy and/or anti-cancer agent listed above, wherein the amounts of the two or more ingredients result in desired therapeutic effect.
- Biosystems API-100 mass spectrometer and Shimadzu SCL-10A LC column Altech platinum C18, 3 micron, 33mm x 7mm ID; gradient flow: 0 min - 10% CH 3 CN, 5 min -
- NBS N-Bromosuccinimide it: room temperature
- LiHMDS lithium hexamethyldisilazane
- HMDS hexamethyldis ⁇ azane
- Pd/C palladium on carbon
- PDCI 2 (dppf): [1 ,1 '-bis(diphenylphosphino)ferrocene] dichloropalladium(il) ⁇ mol: micromole TFA: trifluoroacetic acid
- Pd(PPh 3 ) 4 tetrakis(triphenylphosphine) palladium
- N-lodosuccinimide (1.26 g, 5.61 mmol, dissolved in 25 ml acetonitrile) was added into a solution of di-tert-buty! 5-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5- a]pyrimidin-5-yl)piperidine-1 ,2-dicarboxylate (3.8 g, 5.61 mmol) in acetonitrile (26 mL). The reaction mixture was stirred at room temperature for 16 hours.
- N-lodosuccinimide (81 1 mg, 3.6 mmol) was added into a solution of 4 ⁇ 7-[bis-(2- trimethylsilanyl-ethoxymethyl)-amino]-pyrazolo[1 ,5-a]pyrimid!n-5-yl ⁇ - cyclohexanecarboxylic acid methyl ester (1.75 g, 3.27 mmol) in acetonitrile (25 mL). The resulting reaction mixture was stirred at room temperature for 16 hours.
- N-bromosuccinimide (148 mg, 0.832 mmol) was added to a solution of 4- ⁇ 7-[bis-(2- trimethy!silanyl-ethoxymethyl)-amino]-3-quinolin-3-yl-pyrazolo[1 ,5-a]pyrimidin-5-yl ⁇ - cyciohexanecarboxylic acid methyl ester (500 mg, 0.756 mmol) in acetonitrile (10 ml_). The reaction mixture was stirred at room temperature for 1 hour.
- Potassium phosphate (3 M -in H 2 O, 0.2 mmoi) was added followed by the addition of 5-chloro-3-(quinolin-3-yl)-N,N- bis((2-(trimethylsily!ethoxy)methyl) pyrazo!o[1 ,5-a]pyrimidin-7-amine (50 mg, 0.09 mmol) in DMF (0.5 ml) and Pd(dppf)CI 2 -CH 2 CI 2 (5 mg, 0.005 mmol). The reaction mixture was heated at 90 °C for 16 h under argon. The solution was cooled to room temperature and concentrated under reduced pressure.
- Part C To a EtOH (8.0 mL) solution of compound from Part B (1.12 g, 4.1 mmol) was added CuI (39 mg, 0.2 mmol) and followed with Et 3 N (3.5 mL) the resulting mixture was stirred at 70 °C for 3 hours. The reaction mixture was allowed to cool to 25 °C, concentrated to dryness and then dissolved in toluene (3OmL). The solution was washed with 0.1 N HCI (20 mL), saturated NaHCO 3 (20 mL), and brine (30 mL), dried over Na 2 SO 4 .
- Part D A mixture of the product from Part C (1 equiv), bispinacoiatodiboron (1.2 equiv), PdCI 2 dppf (0.1 equiv), and potassium acetate (3 equiv) in dioxane was heated at 85 °C until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction was allowed to cool to room temperature, filtered through a pad of celite, concentrated to dryness, treated with diethyl ether, filtered through a pad of celite, then concentrated to afford the desired product, 5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)furo[2,3-b]pyridine.
- Part A A mixture of 5-bromo-3-iodopyridin-2-ol (1 equiv), copper iodide (3 equiv), palladium catalyst (0.05 equiv), TBAF (1 equiv), triethylamine (3.3 equiv) and alkyne (1 equiv) in toluene was stirred at room temperature until the reaction was deemed complete by TLC analysis. The reaction mixture was diluted with dichloromethane, washed with water, saturated sodium bicarbonate, and brine, dried over sodium bicarbonate, filtered through celite, concentrated, and purified on silica gel to give the product,5-bromo-2-methylfuro[2,3-b]pyridine. Part B:
- reaction was allowed to cool to room temperature, filtered through a pad of celite, concentrated to dryness, treated with diethyl ether, filtered through a pad of celite, and then concentrated to afford the desired product, 2-methyl-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)furo[2,3- b]pyridine.
- Part B The 5-bromo-2-(trimethylsilyl)thieno[2,3-b]pyridine from Part A (8.99 g, 31.4 mmol) was dissolved in ethano! (70 mL), followed by addition of K 2 CO 3 (10.85g, 78.5 mmol). Reaction was stirred at 65 °C for 1 hour. The reaction mixture was allowed to cool to 25 °C, concentrated to dryness, and dissolved in EtOAc (150 mL). The solution was washed with H 2 O (80 mL), and brine (15OmL), dried over Na 3 SO 4, filtered, and concentrated to yield white solid, 5-bromothieno[2,3-b]pyridine, 6.57 g (98%).
- reaction was allowed to cool to room temperature, filtered through a pad of celite, concentrated to dryness, treated with diethyl ether, filtered through a pad of celite, and then concentrated to afford the desired product, 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)thieno[2,3-b]pyridine.
- the resulting material was stirred with a mixture of vinyl stannane (2 equiv), palladium catalyst (0.1 equiv), and potassium fluoride (3 equiv) in dioxane at 85 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis.
- the mixture was allowed to cool to room temperature, diluted with 10% aqueous potassium fluoride, and extracted three times with ethyl acetate. The combined organics were dried over sodium sulfate, filtered, concentrated, and purified on silica gel.
- the resulting material was stirred with a mixture of HCI in methanol and water at 60 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis.
- Part D methyl 2-(4-(7-(bis((2-(trimethyisilyl)ethoxy)methyl)amino)-6-bromo-3-(quinolin-3- yl)pyrazolori .5-alpyrimidin-5-yloxy)phenyl)acetate:
- Part E methyl 2-(4-(7-amino-6-bromo-3-(Quinolin-3-yl)pyrazolo[1 ,5-alpyrimidin-5- yloxy)phenvOacetate: The crude methyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyi)amino)-6-bromo-3-(quinolin- 3-yl)pyrazolo[1,5-a]pyrimidin-5-yloxy)pheny[)acetate from above step D was treated with 50% TFAZH 2 O (3 mL) and stilted at room temperature for 1 h.
- Part F 4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazoio[l,5-a]pyrimidin-5-yl)benzoic acid:
- Part C 4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolori .5-aipyrimidin-5-yl)benzoic acid: Compound methyl 4-(6-acetyf-7-amino-3-(6-phenylpyridin-3-yI)pyrazolo[ l,5-a]pyrimtdin-5- yi)benzoate was treated with 2 equivalents of LiOH and stirred at room temperature for 4hrs.
- Methyl 2-(4-(bromomethyl)phenyl)acetate (1.03 g, 4.24 mmol) was mixed with Pd(PPh3)4 (500 mg, 0.4 mmol, 4,4,4 1 ,4',5,5,5 1 ,5'-octamethyl-2,2 1 -bi(l,3,2-dioxaborolane) (1.27 g, 5 mmol), K2CO3 (1.75 g, 12.7 mmol) and dioxane (20 mL) under inert Argon atmoshphere. The resulting mixture was heated at 80 °C and stirred over night.
- Pd(PPh3)4 500 mg, 0.4 mmol, 4,4,4 1 ,4',5,5,5 1 ,5'-octamethyl-2,2 1 -bi(l,3,2-dioxaborolane)
- K2CO3 (1.75 g, 12.7 mmol
- dioxane 20 mL
- Part B 5-(4-(methylthio)benzyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5- aipyrimidin-7-amine:
- Part F ⁇ -bromo-5-(4-(methylsulfonyl)benzyl)-3-(quinoiin-3-yl)-N,N-bis((2-
- Ttile compound 6-bromo-5-(4-(methylsulfonyl)benzyl)-3-(quinolin-3-yl)pyrazolo[1,5- a]pyrimidin-7-amine was prepared with the same deprotection condition described in example I Part F.
- HPLC-MS t R 1.49 min (UV 254 nm ); mass calculated for formula C 23 Hj 8 BrN 5 O 2 S 507.0, observed LCMS m/z 5O8.O(M+H).
- step E The crude from above step E was treated with 50% TFA/H2O (3 mL) and stirred at room temperature for 1 h. Solvent was removed to yield thick oil, methyl 4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-6-(l-ethoxyvinyl)-3-(6-phenylpyridin-3-yl)pyrazolo[1,5- a]pyrimidin-5-yioxy)benzoate.
- HPLC-MS IR 1.84 min (UV 254 nm ); mass calculated for formula C 27 H 21 N 5 O 4 479.2, observed LCMS m/z 480.2 (M+H).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides methods for inhibiting mTOR using pyrazolo[1,5-a]pyrimidine compounds and methods of treatment, prevention, inhibition, or amelioration of one or more diseases associated with mTOR using such compounds.
Description
PYRAZOLO [1 , 5-A] PYRIMIDINE DERIVATIVES AS MTOR INHIBITORS
FIELD OF THE INVENTION
This invention is directed to pyrazolo pyrimidine derivatives as inhibitors of mammalian Target Of Rapamycin (mTOR) kinase, which are also known as FRAP, RAFT, RAPT or SEP and as a result have a role in controlling cell cycle progression and mitogenic signals involved in cell growth and proliferation and as a result are useful in the treatment of cancer.
BACKGROUND OF THE INVENTION
The mammalian target of rapamycin (mTOR) is a central regulator of cell growth and proliferation and plays a gate keeper role in the control of cell cycle progression and mediates mitogenic signals from P13K/AKT through to the downstream targets S6K1 and 4E-BP1 and to Ser 473 on AKT. Recently it has been shown that mTOR exists in two complexes, raptor-mTOR complex (mTORCI ), a rapamycin-sensitive complex, signaling to S6K1 and 4E-BP1 and rictor-mTOR complex (mTORC2), a rapamycin-insensitive complex that signals to AKT. Although the precise mechanism by which rapamycin inhibits mTOR function is not well understood, rapamycin partially inhibits mTOR function through mTORCI . It has been found that mTORC2 is involved in the regulation of cell survival and actin cytoskeletal organization in a rapamycin-independent manner, and inhibition of mTOR through inhibition of mTORCI and mTORC2 is probably important for antitumor activity and better efficacy.
US 2007/0112005 describes the fused bicyclic mTOR inhibitors useful in treatment of cancer.
WO 2007/087395 describes unsaturated mTOR inhibitors useful in treatment of cancer.
WO-2008/012326 describes 2,4-substituted quinozolines as lipid kinase inhibitors useful in treatment of P13K -related diseases, such as proliferative diseases, inflammatory diseases, obstructive airways disorder and transplant related diseases.
WO 2006/090169 describes 2,4-diamineo-pyrido-pyrmidine derivatives and their use as mTOR inhibitors.
WO 2007/066099 describes pyrimidine derivatives useful as mTOR kinase inhibitors for anticancer and various other therapeutic treatments involving mTOR kinase.
WO 2007044813 describes pyridopyrimidinone inhibitors of PI3Ka protein kinase for anticancer or other PI3Ka protein kinase related diseases.
US 2005/0222171 , WO 2005/070431 , WO 2007/0570431 and WO 2007/009773 describe pyrazolo[1 ,5 a]pyrimidin-7-yl amine derivatives to treat protein kinase dependent diseases.
US 2002/0041880 describes pyrazolo[1 ,5 a]pyrimidin-7-yl derivatives to inhibit kinase insert domain-containing receptor to block angiogenesis.
WO 1998/003510 describes pyrazolo[1 ,5 a]pyrimidin-7-yl derivatives to treat corticotrophin releasing factor dependent diseases.
WO 2003/091256 describes pyrazolo[1 ,5-a]pyrimidine derivatives to treat NAD(P)H oxidase dependent diseases.
WO 2007/044449, WO 2005/077954, WO 2004/022560 and WO 2004/022561 describe pyrazolopyrimidine derivatives as cyclin-dependent kinase dependent diseases.
WO 2004/106341 describes pyrazolopyrimidine derivatives as fungicides
There is need in the art for small molecule inhibitors of mTOR kinase that block signaling through mTORCI and mT0RC2 as a potential anticancer treatment or a treatment for other cell proliferative disorders.
SUMMARY OF THE INVENTION
In its many embodiments, the present invention discloses novel compounds having mTOR inhibitory activity, methods of preparing such compounds, pharmaceutical compositions comprising one or more of such compounds, methods of treatment or prevention of one or more diseases associated with mTOR by administering one or more of such compounds or pharmaceutical compositions, the compound being represented by the general Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is independently selected from the group consisting of heterocycloalkyl, heterocycloalkylalkyl, spiroheterocycloalkyl, heterocyclenyl, -NR3R4, cycloalkyl, heteroaryl, aryl, alkyl, alkynyl, heterocyclenylalkyl, cycloalkylalkyl, heteroarylalkyl, heteroarylalkynyl, spiroheterocycloalkylalkyl, -N-heteroaryl, -alkyl-NH-heterocyclyl and arylalkyl, wherein each of said heterocycloalkyl, heterocycloalkylalkyl, spiroheterocycloalkyl, heterocyclenyl, cycloalkyl, heteroaryl, aryl, alkyl, alkynyl, heterocyclenylalkyl, cycloalkylalkyl, heteroarylalkyl, heteroarylalkynyl, -N-heteroaryl and arylalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, -alkyl- C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, -alkyl(CO)NS(O)2- cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SO2-cycloalkyl, -alkyl(CO)NS(O)2-alkyl, - alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2-cycloalkyl, -CO-CO2H, - C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl;
R3 is cycloalkyl or heteroaryl, wherein each of said cycloalkyl or heteroaryl can be unsubstituted or substituted with one or more moieties independently selected from the group consisting of X; and R4 is H.
In another embodiment, the present invention includes a method of inhibiting mammalian Target Of Rapamycin in a patient, wherein the method comprises administering a therapeutically effective amount of at least one compound of the structural Formula I. In another embodiment, the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, wherein the method comprises administering a therapeutically effective amount of at least one compound represented by the structural Formula I.
In another embodiment, the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, wherein the method comprises administering a therapeutically effective
amount of at least one compound represented by the structural Formula I, to a patient in need thereof.
In another embodiment, the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, and disorders commonly occurring in connection with transplantation, diseases that respond to inhibition of mammalian Target Of Rapamycin, wherein the method comprises administering a therapeutically effective amount of compound represented by the structural Formula I, to a patient in need of such treatment. In another embodiment, the present invention includes a method of treatment of a disease, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I.
In another embodiment, the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, and disorders commonly occurring in connection with transplantation, diseases that respond to inhibition of mammalian Target Of Rapamycin, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I, wherein the disease is a proliferative disease, autoimmune disease, viral disease, fungal disease, neurological/neurodegenerative disorder, arthritis, inflammation, neuronal, alopecia or cardiovascular disease.
In another embodiment, the present invention includes a method of treatment of a disease, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I, wherein the disease is a proliferative disease.
In another embodiment, the present invention includes a method of treatment of a proliferative disease, wherein the method comprises administering a therapeutically effective amount of the compound represented by the structural Formula I, wherein the proliferative disease is selected from the group consisting of: cancer of the bladder, breast, colon, kidney, liver, lung, small cell lung cancer, non- small cell lung cancer, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, squamous cell carcinoma; leukemia,
acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma, Burkett's lymphoma; acute and chronic myelogenous leukemia, myelodysplastic syndrome, promyelocytic leukemia; fibrosarcoma, rhabdomyosarcoma; astrocytoma, neuroblastoma, glioma and schwannomas; melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderma pigmentosum, keratoctanthoma, thyroid follicular cancer, endometrial cancer, gastrointestinal tract cancer and Kaposi's sarcoma.
In another embodiment, the present invention includes a method of treatment of a proliferative disease comprising administering a therapeutically effective amount of the compound represented by the structural Formula I, further comprising treatment with radiation therapy.
In another embodiment, the present invention includes a method of inhibiting a mammalian Target Of Rapamycin, wherein the method comprises administering a therapeutically effective amount of a compound represented by the structural Formula I.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
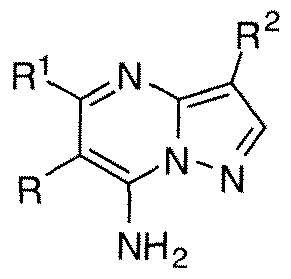
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is heterocycloalkyl, wherein said heterocycloalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, -
trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-aikyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl; R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl. In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo; R1 is heterocyclenyl, wherein said heterocyclenyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2,
-NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SOa-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CHa)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or moieties independently selected from the group X; X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl - C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2) -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, -
alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is cycloalkyl, wherein said cycloalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl-
C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2-
cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein: R is independently selected from the group consisting of halo, hydroxyl, amino,
-CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is cycloalkenyl, wherein said cycloalkenyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3) -N-alkyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said
heteroeycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heteroeycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is aryl, wherein said aryl can be unsubstituted or substituted with one or moieties independently selected from the group X; X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heteroeycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl (CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-eycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heteroeycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is alkyl, wherein said alkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl-
C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently
selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein: R is independently selected from the group consisting of halo, hydroxyl, amino,
-CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is alkynyl, wherein said alkynyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(0)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl,
-C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(C1-C6)alkyl, alkoxy, -C(=O)alkyl, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl and halo;
R1 is spiroheterocycloalkyl, wherein said spiroheterocycloalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2, -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-alkyl, -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl,
-C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R1 is independently selected from the group consisting of:





R2 is quinolinyl, 1 -methyl-pyrazolyl, naphtyl, methoxy-naphtyl, cyano-phenyl, methoxy-phenyl, phenoxy-phenyl, biphenyl, chloro-pyridyl, methoxy-pyridyl, bromo- methyl-pyrazolyl, hydroxyl-methoxy-phenyl, di-methoxy-phenyl, 1 H-indazolyl, fluoro- hydroxy-phenyl, chloro-quinolyl, morpholino-pyridyl-, 4-(isobutyronitrilo)-phenyl, bromo-quinolyl, 4-trifluoromethyl-phenyl-, benzyl -pyrazolyl, fluoro-pyridyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1 -methyl-pyrazolyl, and thienyl; R1 is independently selected from the group consisting of:





R2 is indazolyl, wherein said indazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R1 is independently selected from the group consisting of:





R2 is phenyl, wherein said phenyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, alkoxy, -CN, hydroxyl, aryl and heteroaryl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
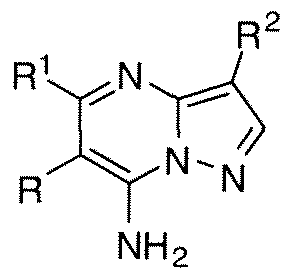
R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R1 is independently selected from the group consisting of:



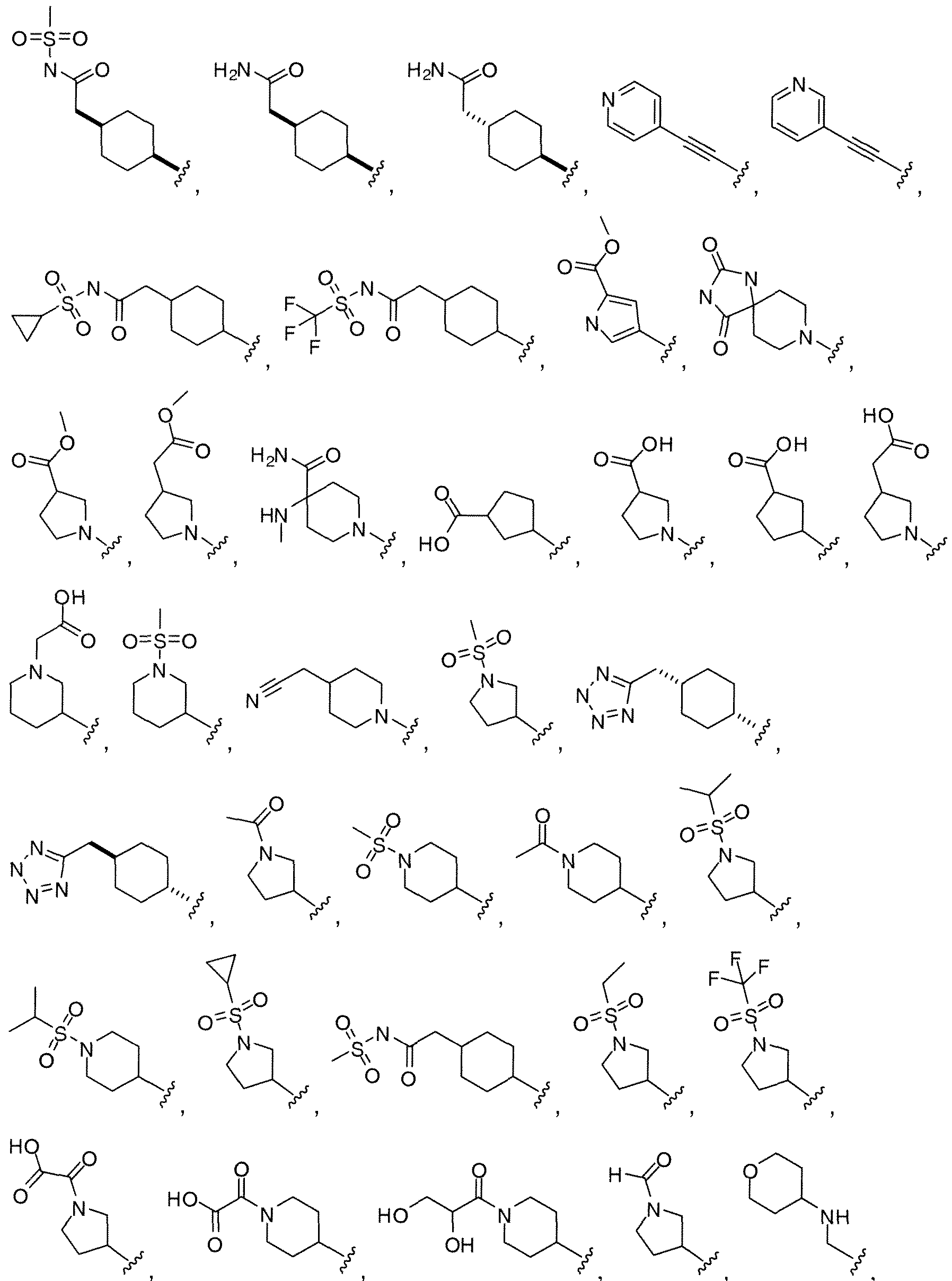

R2 is naphthyl, wherein said naphthyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R1 is independently selected from the group consisting of:




R2 is quinolinyl, wherein said quinolinyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R1 is independently selected from the group consisting of:


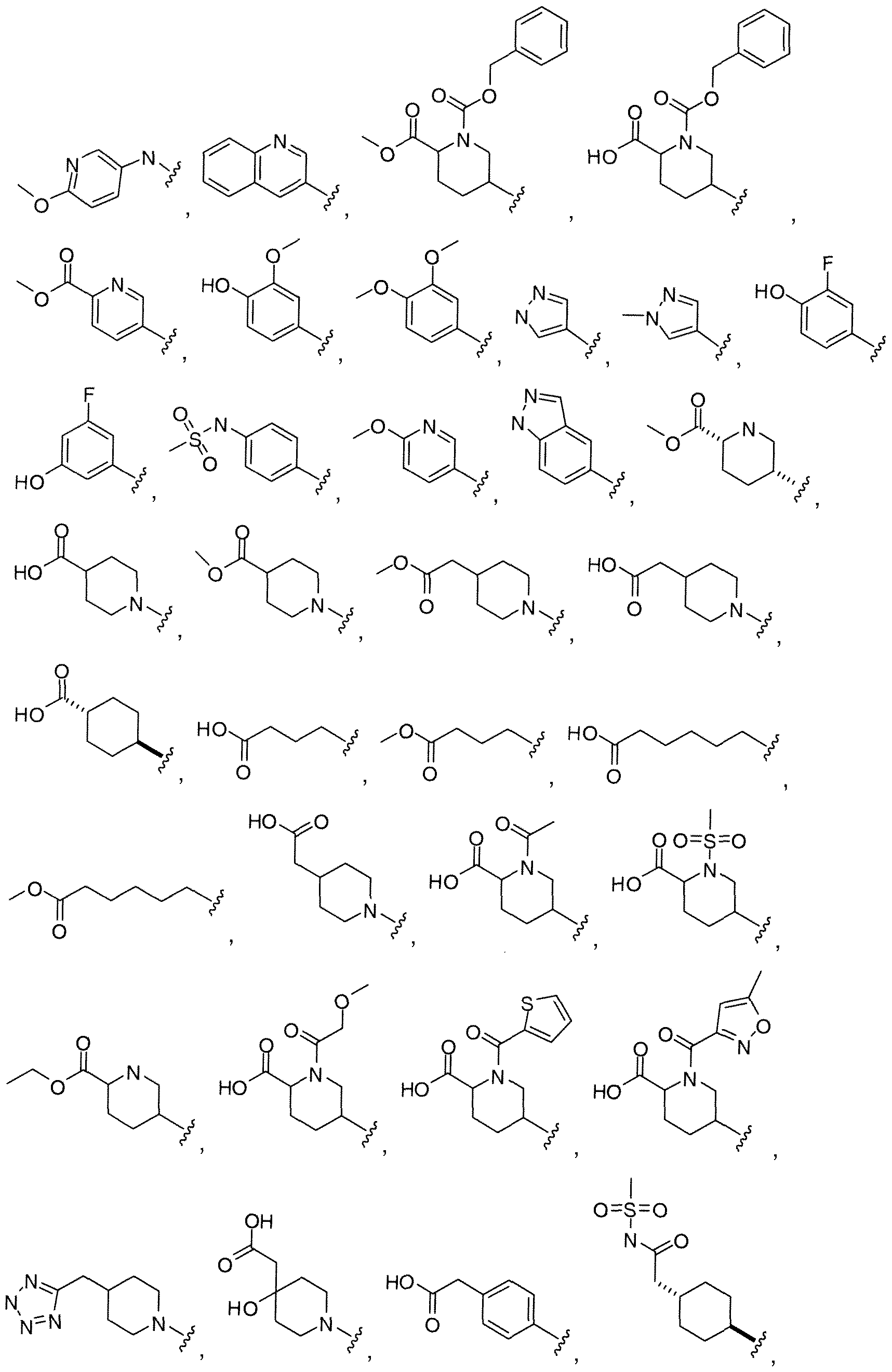


R2 is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is independently selected from the group consisting of bromo, chloro, -CN, H, methyl, acetyl, pyridyl, phenyl, 1-methyl-pyrazolyl, and thienyl; R1 is independently selected from the group consisting of:





R2 is pyridinyl, wherein said pyridinyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, heterocyclyl, heteroaryl, aryl and alkoxy.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

R is halo;
R1 is independently selected from the group consisting of:
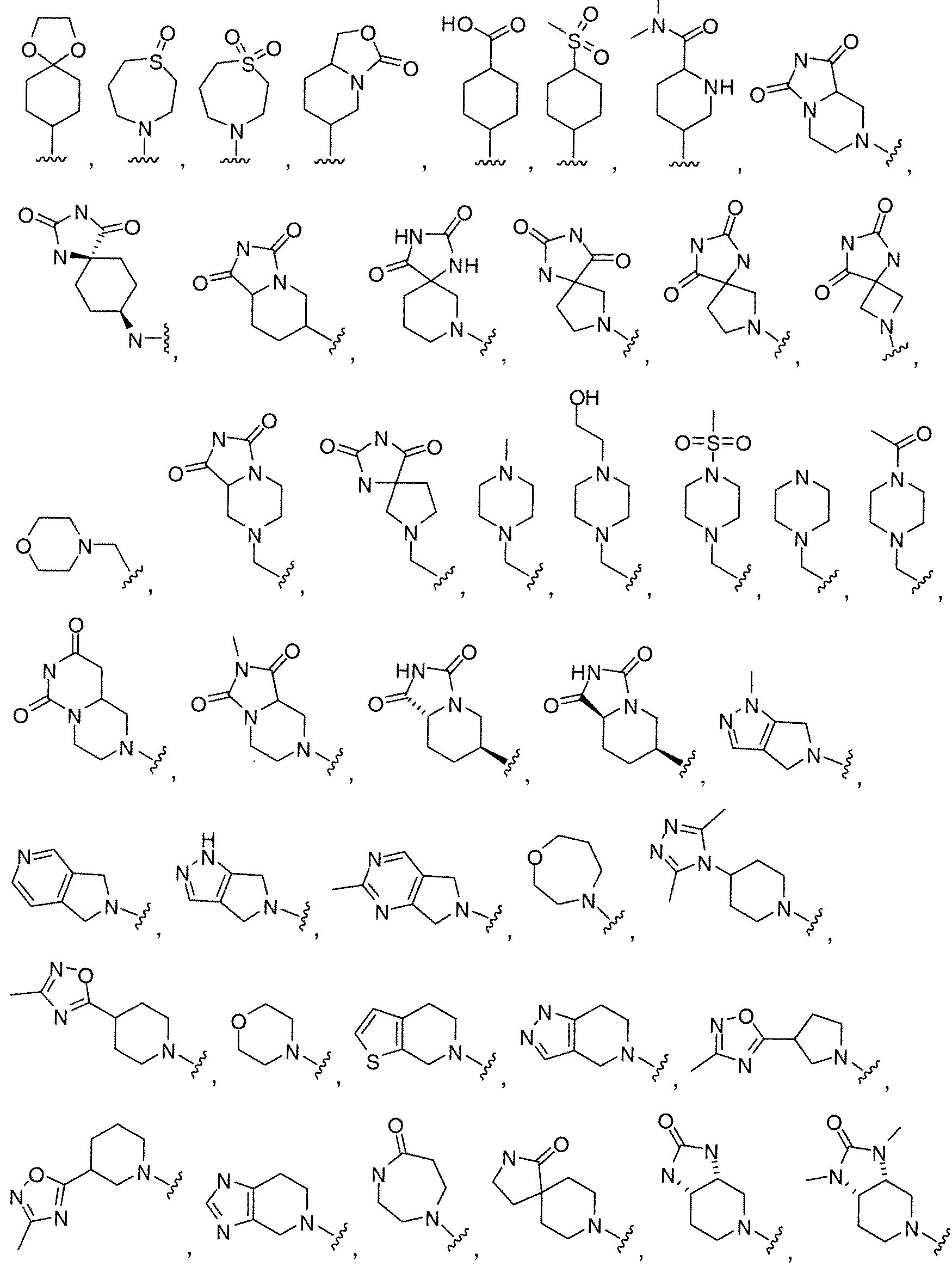




R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, heteroaryl and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is thienyl; R1 is independently selected from the group consisting of:





R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3^CN, trifluromethyl, difluromethyl, monofluromethyl, heterocycloalkyl, heteroaryl and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
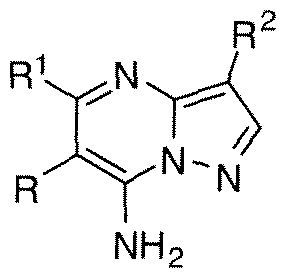
Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with alkyl;
R1 is independently selected from the group consisting of:





R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is pyridinyl; R1 is independently selected from the group consisting of:

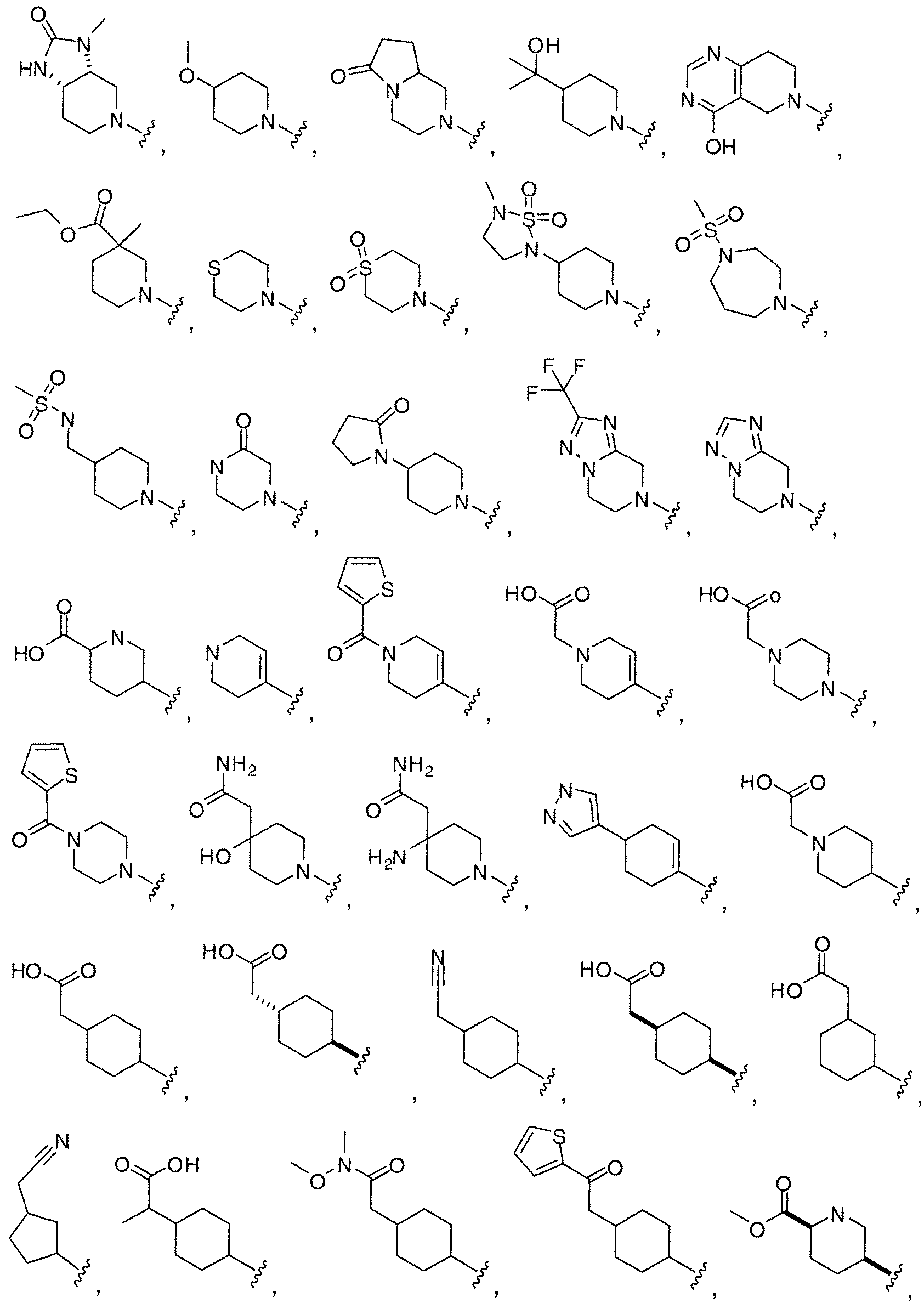



R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyljieterocycloalkyl, heteroaryl and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is methyl; R1 is independently selected from the group consisting of:

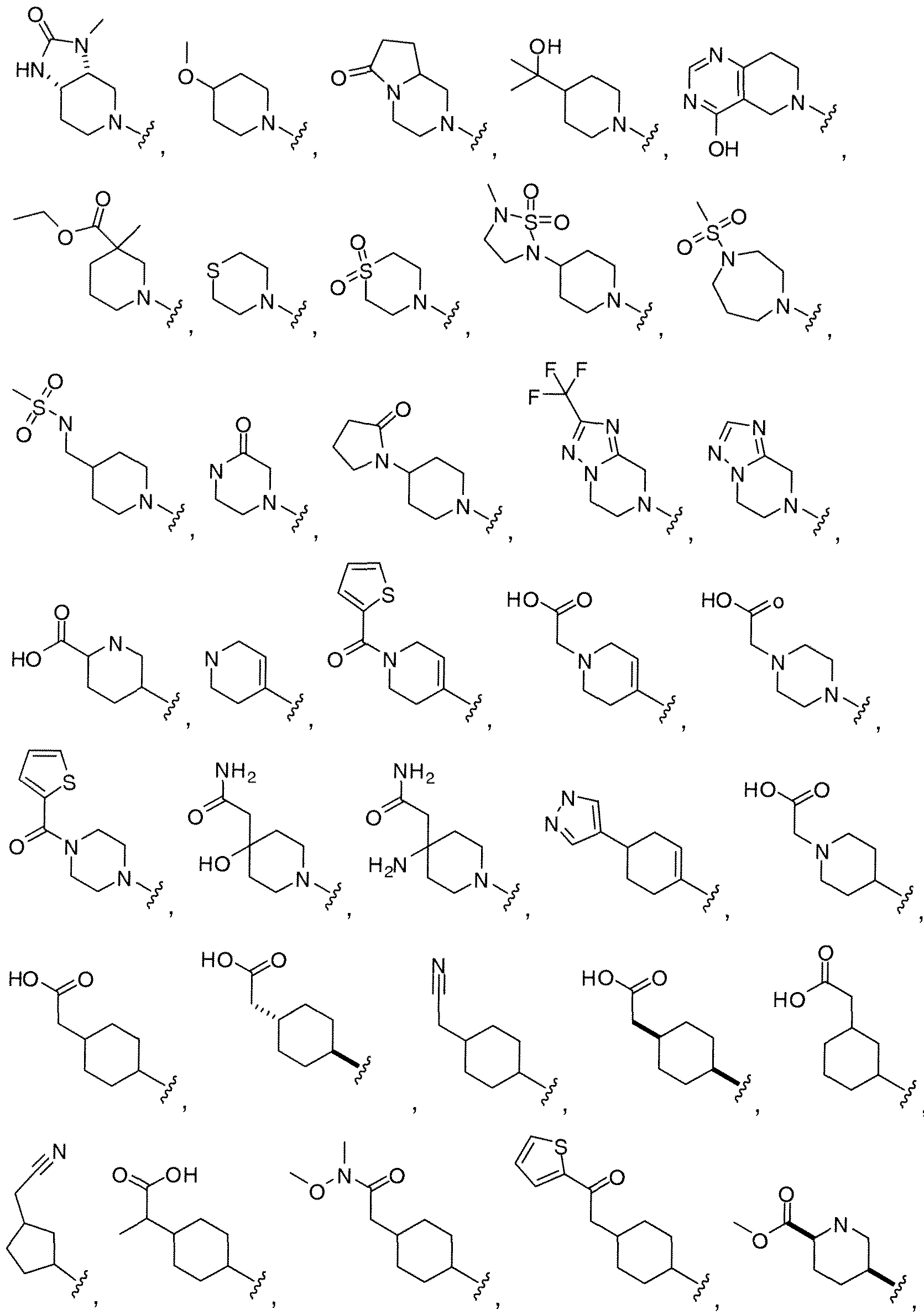



R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CHa)2CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
In another embodiment, the present invention relating to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is phenyl, wherein said phenyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo or alkyl;
R1 is independently selected from the group consisting of:





R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:

Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is acetyl; R1 is independently selected from the group consisting of:





R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyljieterocycloalkyl, heteroaryl and arylalkyl.
In another embodiment, the present invention relates to the foregoing method of using a compound represented by the structural Formula I:
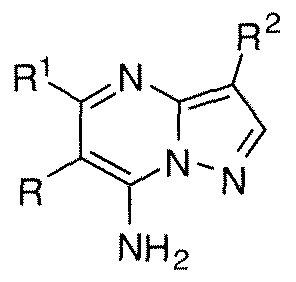
Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein:
R is -CN; R1 is independently selected from the group consisting of:




 and
and
R2 is hβteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, heteroaryl and arylalkyl.
Non-limiting examples of the compounds of the present invention include.
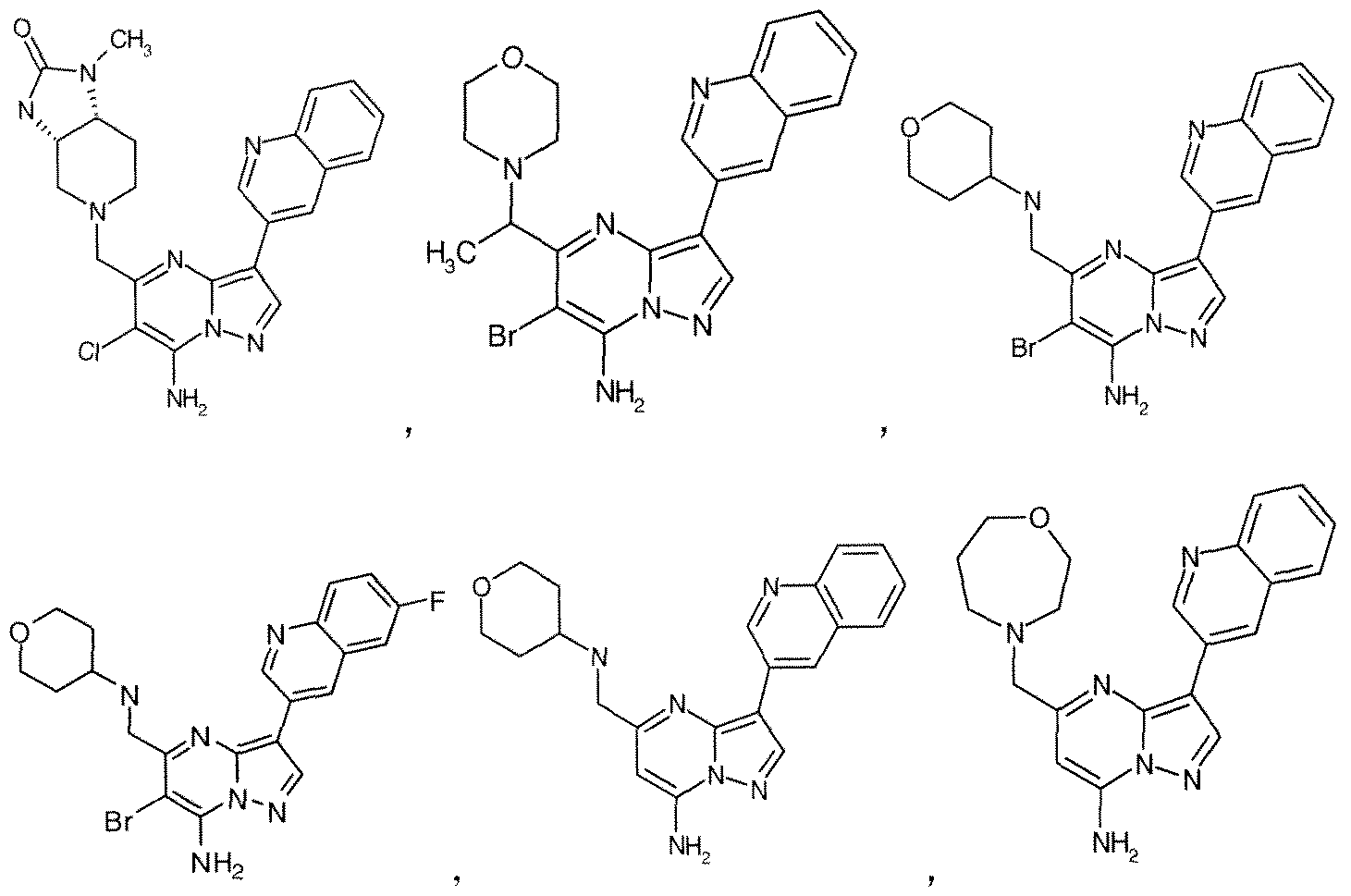


























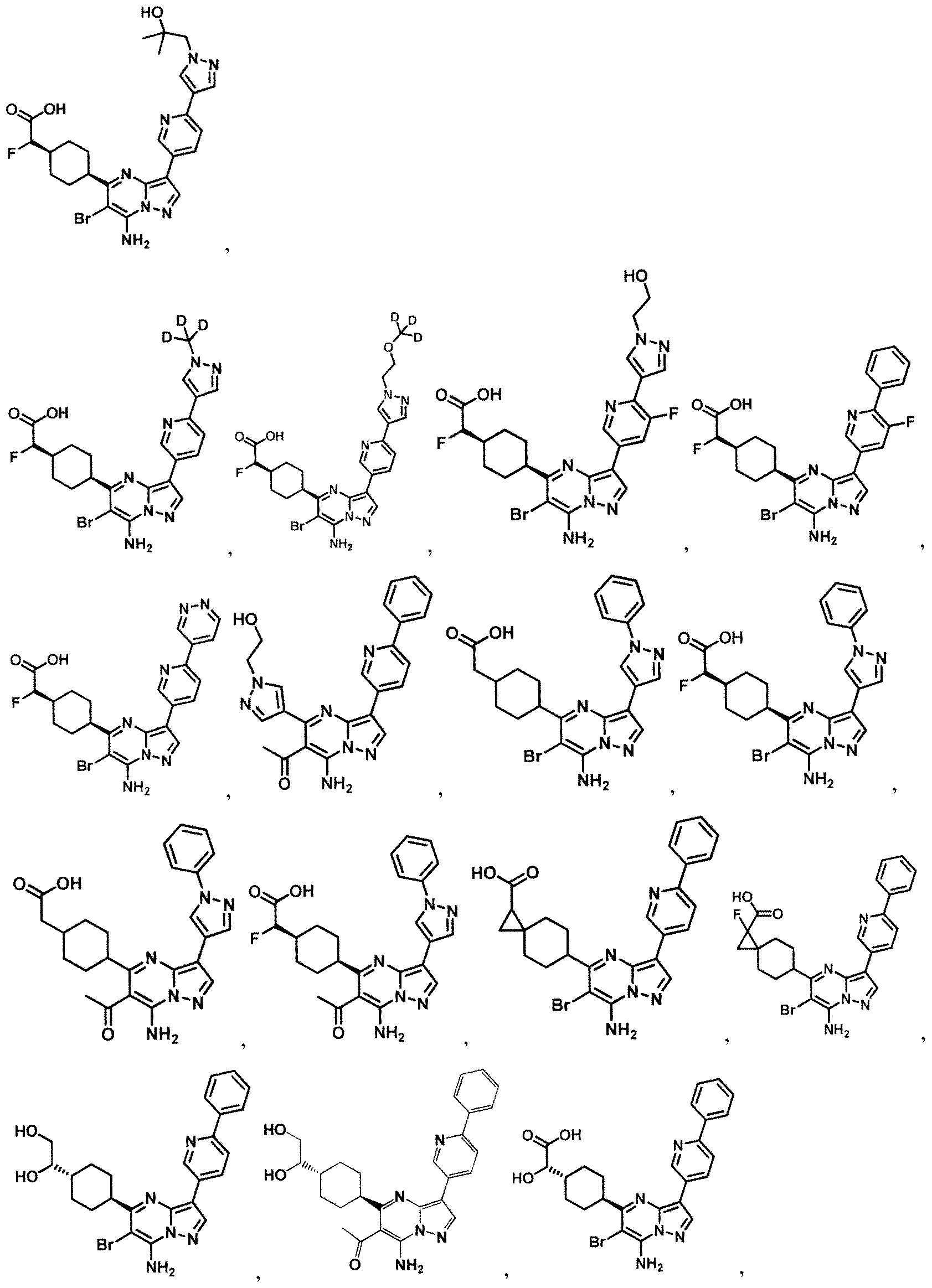


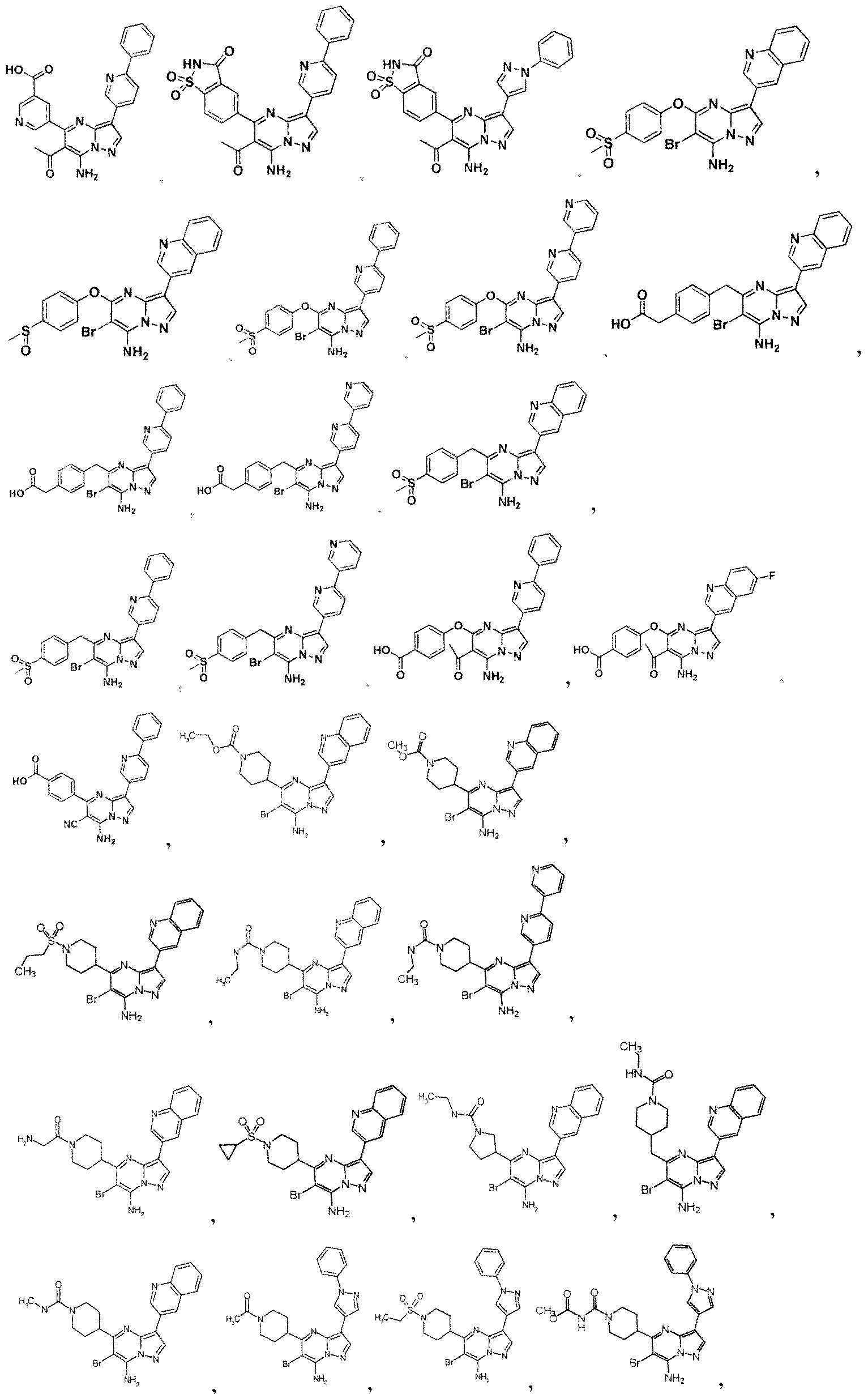






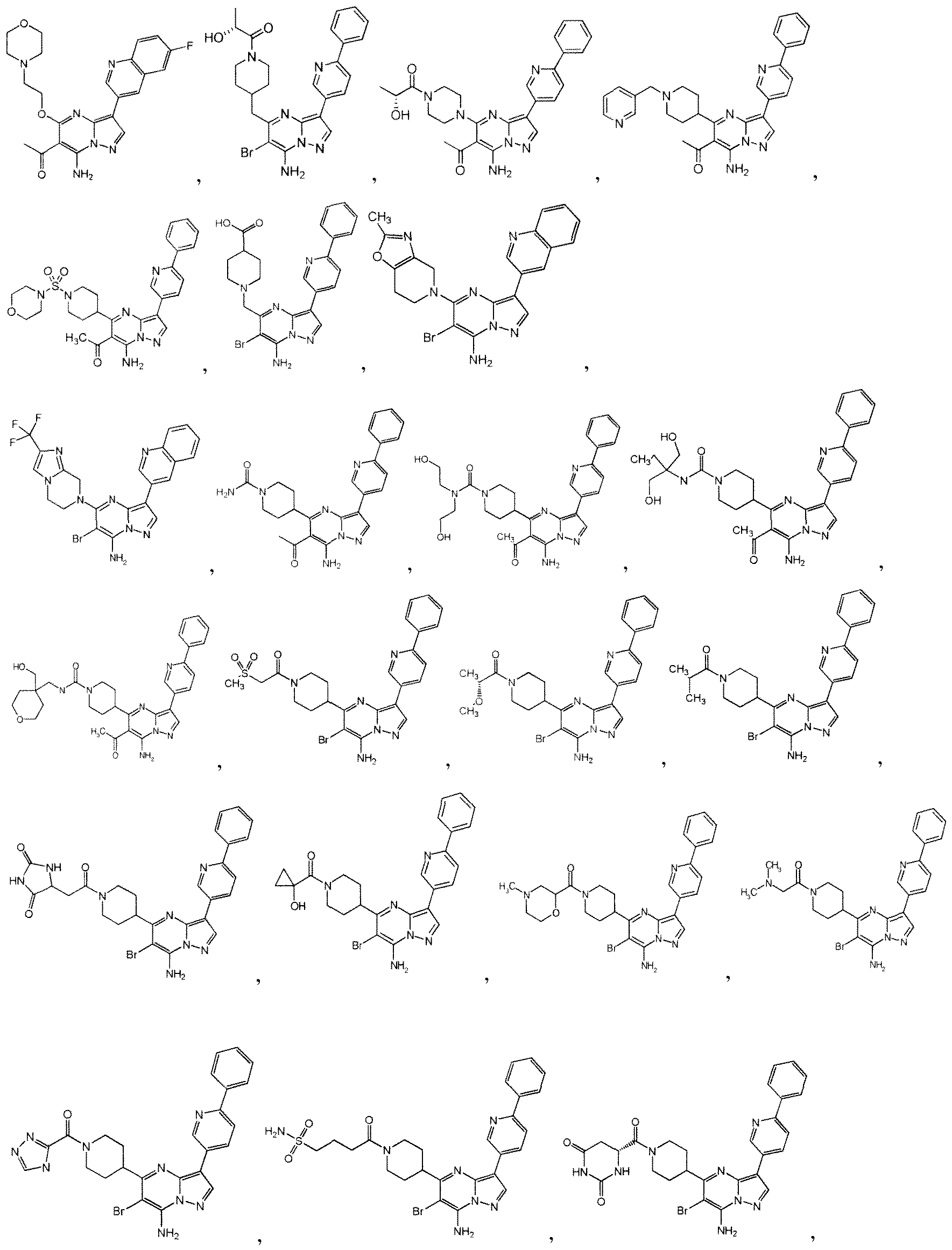
















or pharmaceutically acceptable salt, ester, solvate or prodrug thereof.
In another embodiment, the present invention relates to a compound represented by the structural Formula I or a pharmaceutically acceptable salt, ester, solvate or prodrug thereof, in purified form.
In another embodiment, the present invention relates to a compound represented by the structural Formula I or a pharmaceutically acceptable salt, ester, solvate or prodrug thereof, in isolated form.
In another embodiment, the present invention includes a composition comprising a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, in combination with at least one pharmaceutically acceptable carrier.
In another embodiment, the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, said method comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above, or a
pharmaceutically acceptable salt, solvate, ester or prodrug of the compound, to a patient in need thereof.
In another embodiment, the present invention includes a composition comprising a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof, and an anti-cancer agent.
In another embodiment, the present invention includes a composition comprising a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof, and an anti-neoplastic, anti-tumor, anti-angiogenic, or chemotherapeutic agent.
In another embodiment, the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, and disorders commonly occurring in connection with transplantation, diseases that respond to inhibition of nnTOR, comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
In another embodiment, the present invention includes a method of treatment of a proliferative disease, autoimmune disease, viral disease, fungal disease, neurological/neurodegenerative disorder, arthritis, inflammation, neuronal, alopecia or cardiovascular disease comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
In another embodiment, the present invention includes a method of treatment of a proliferative disease.
In another embodiment, the present invention includes a method of treatment of a proliferative disease, wherein the proliferative disease is selected from the group consisting of: cancer of the bladder, breast, colon, kidney, liver, lung, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin; small cell lung cancer, non-small cell lung cancer, squamous cell carcinoma;
leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma, Burkett's lymphoma; acute and chronic myelogenous leukemia, myelodysplastic syndrome, promyelocyte leukemia; fibrosarcoma, rhabdomyosarcoma; astrocytoma, neuroblastoma, glioma and schwannomas; melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderma pigmentosum, keratoctanthoma, thyroid follicular cancer, endometrial cancer, gastrointestinal tract cancer and Kaposi's sarcoma.
In another embodiment, the present invention includes a method of treatment of hamartoma syndromes, transplant rejection, bowel disorders, inflammatory bowel disease, multiple sclerosis, immunosuppression, immune tolerance, autoimmune diseases, inflammation, bone loss, rheumatoid arthritis, restinosis, cardiac allograft vasculopathy, psoriasis, ocular conditions such as dry eye, hepatic fibrosis, hepatic necrosis, beta-thalassaemia, comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
In another embodiment, the present invention includes a method of treatment of cancer of the bladder, breast, colon, kidney, liver, lung, small cell lung cancer, non- small cell lung cancer, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma, Burkett's lymphoma; acute and chronic myelogenous leukemia, myelodysplastic syndrome, promyelocytic leukemia; fibrosarcoma, rhabdomyosarcoma; astrocytoma, neuroblastoma, glioma and schwannomas; melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderma pigmentosum, keratoctanthoma, thyroid follicular cancer, endometrial cancer, gastrointestinal tract cancer and Kaposi's sarcoma comprising administering a therapeutically effective amount of at least one compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or
prodrug thereof, further comprising treatment with radiation therapy to a patient in need of such treatment.
In another embodiment, the present invention includes a method of treating a disease by inhibiting a mTOR, comprising administering to a patient in need of such treatment an amount of a first compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, said second compound being an anti-cancer agent; wherein said anti-cancer agent is selected from the group consisting of Adriamycin, Altretamine, Amidox, Aminoglutethimide, Amsacrine, Anastrazole, Antibodies to EGFR, 3-AP, Aphidicolon, Ara-C, Arsenic trioxide, L-Asparaginase, Bevacizumab, Bleomycin, BMS 214662, Bortezomib, Busulfan, Campath, Camptostar, Capecitabine, Carboplatin, Carmustine, Centrosome associated protein E ("CENP-E") inhibitors, Cetuximab, Cladribine, Chlorambucil, Chlormethine, Chlorotrianisene, Cisplatin, Clofarabine, cyclophosphamide, Cytarabine, a Cytostatic agent, Cytoxan, Dacarbazine, Dactinomycin, Daunorubicin, Dasatinib, Deforolimus (described in PCT publication No. 2003/064383), Deoxycoformycin, Didox, Diethylstilbestrol, Docetaxel, Doxorubicin, Dromostanolone, Droloxafine, Epirubicin, Epothilones, ERK inhibitors, Erlotinib, Etoposide, 17α-Ethinylestradiol, Estramustine, Exemestane, Floxuridine, Fludarabine, Fludarabine phosphate, 5-Fluorouracil, Fluoxymesterone, Flutamide, Fulvestrant, Gefitinib, Gemcitabine, Gemtuzumab ozogamcicin, Goserelin, GSK- 923295, Hexamethylmelamine, Hydroxyprogesterone, Hydroxyurea, lbritumomab Tiuxetan, Idarubicin, Ifosfamide, lmatinib mesylate, Intron, Irinotecan, ispinesib, KSP inhibitors, L778,123, Lapatinib, Leucovirin, Leuprolide, Lerozole, Letrazole, Levamisole, Liposomal Doxorubicin, Liposomal, Lomustine, Lonafarnib,
Medroxyprogesteroneacetate, Megestrolacetate, Melphalan, 6-Mercaptopurine, Methoxtrexate, Methylprednisolone, Methyltestosterone, Mithramycin, Mitomycin-C, Mitotane, Mitoxantrone, Navelbene, Nilotinib, Oxaliplatin, Paclitaxel, Panitubimab, Pentostatin, Pipobroman, Porfimer, Prednisolone, Prednisone propionate, Procarbazine, Reloxafine, Rituximab, Satriplatin, SB-743921 , SmH , Sorafinib, Streptozocin, Sunitinib, Tamoxifen, Taxotere, Taxol, Temozolomide, Teniposide, Testolactone, Testosterone, Tezacitabine, 6-Thioguanine, Thiotepa, Tipifamib,
Topotecan, Toremifene, Tositumomab, Trastuzumab, Triamcinolone, Triapine, Triethylenemelamine, Triethylenethiophosphoramine, Trimidox, Uracil mustard, Vinblastine, Vincristine, Vindesine, and Vinorelbine; wherein the amounts of the first compound and said second compound result in a therapeutic effect.
In another embodiment, the present invention includes a method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, said method comprising administering a therapeutically effective amount of at least one compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug of the compound, to a patient in need thereof.
In another embodiment, the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, diseases related to transplant rejection and diseases that respond to inhibition of mTOR, comprising administering a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
In another embodiment, the present invention includes a method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, diseases related to transplant rejection and diseases that respond to inhibition of mTOR, comprising administering a therapeutically effective amount of at least one compound of Formula I or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
In another embodiment, the present invention includes a compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof exhibiting mTOR inhibition which is at least five-fold the inhibition of CDK2 or CHK-1 by said compound.
In another embodiment, the present invention includes a compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof exhibiting mTOR inhibition which is at least ten-fold the inhibition of CDK2 or CHK-1 by said compound.
In another embodiment, the present invention includes a compound from the group of compounds listed above or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof exhibiting mTOR inhibition which is at least fifty-fold the inhibition of CDK2 or CHK-1 by said compound. The compounds of this invention can be used to inhibit the following kinases:
ABL1 , ABL2, AFK, ALK, AMPK, ATM, ATR, Aurora A, Aurora B, AxI, BCKDK, BLK, BMPR1 B, BMX, Brk, BRSK1 , BTK, CaM-Klalpha, CaM-Kllalpha, CaM-KIV, CaM- KKalpha, CaM-KKbeta, CCDPK, CCRK, CDK1 , CDK11 , CDK2, CDK4, CDK5, CDK6, CDK7, CDK9, Chaki , CHK1 , CHK2, CK1 alpha, CK1 delta, CDkI epsilon, CDK2 beta, CLK1 , CSF1 R, Csk, DAPK1 , DAPK2, DAPK3, DCAMKL1 , DNA-PK, DYRK1 A,
DYRK1 B, DYRK2, DYRK3, eEF2K, Eg3, EGFR, EIF2AK2, EphA2, EphA3, EphA4, EphA8, EphB1, EphB2, EphB3, EphB5, ErbB2, FAK, Fer, Fes, FGFR1 , FGFR3, FGFR4, Fgr, FLT1 , FLT3, FLT4, Fyn, GRK-1 , GRK-2, GRk-3, GRK-4, GRK-5, GRK-6, GSK-3alpha, GSK-3beta, HCK, HIPK2, HIPK3, HRI, ICK, IGF1 R, IKK-alpha, IKK-beta, IKK-epsilon, ILK, InsR, IPL1 , IRAKI , IRAK4, ITK, JAK1 , JAK2, JAK3, JNK1 , JNK2, JNK3, KDR, KIS, Kit, KSR1 , Lck, LIMK1 , LIMK2, LKB1 , LOK, Lyn, MAP2K1 , MAP2K2, MAP2K3, MAP2K4, MAP2K6, MAP2K7, MAP3K1 , MAP3K11 , MAP3K14, MAP3K5, MAP3K7, MAP3K8, MAP4K1 , MAP4K2, MAP4K4, MAPK1 , MAPK10, MAPK11 , MAPK12, MAPK13, MAPK14, MAPK3, MAPK4, MAPK6, MAPK7, MAPK8, MAPK9, MAPKAPK2, Mer, Met, MHCK, MLCK, Mnk1 , Mnk2, MOS, MRCKa, MST1 , MST3, NDR1 , NDR2, NEK1 , NEK2, NEK6, NEK9, NLK, NuaK1 , p37, p38, p70S6K, p70S6Kb, PAK1 , PAK2, PAK2, PAK3, PAK5, PAK6, PASK, P-CIP2, PCTAIRE1 , PDGFR alpha, PDGFR beta, PDHK1 , PDHK2, PDHK3, PDHK4, PDK-1 , PDK-2, PHK, PIK3CA, PIK3CB, PIK3CD, PIK3CG, Pim-1 , PKA alpha, PKB beta, PKC alpha, PKC beta, PKC delta, PKC epsilon, PKC eta, PKC gamma, PKC iota, PKC theta, PKC zeta, PKD1 , PKD2, PKD3, PKG1/cGK-l, PKG1/cGK-ll, PKN1 , PLK1 , PLK2, PLK3, PRP4, PYK2, RAF1 , Ret, ROCK1 , ROCK2, Ron, RPL10, RSK-1 , RSK-2, RSK-3, RSK-5, SDK1 , SIK, Sky, Src, STLK3, Syk, TBK1 , Tec, TESK1 , TESK2, TGFbRI , TGFbR2, Tie1 , Tie2, Titin kinase, TNK2, TRKA, TRB, tropomyosin kinase, TSSK3, TXK, Tyk2, VRK1 , Wee1 , Wnk1 , Yes, and ZAP70.
As used above, and throughout this disclosure, the following terms, unless otherwise indicated, shall be understood to have the following meanings, including any possible substitutions of the stated groups or moieties: "Patient" includes both human and animals.
"Mammal" means humans and other mammalian animals. "Alkyl" means an aliphatic hydrocarbon group which may be straight or branched and comprising about 1 to about 20 carbon atoms in the chain. Preferred alkyl groups contain about 1 to about 12 carbon atoms in the chain. More preferred alkyl groups contain about 1 to about 6 carbon atoms in the chain. Branched means that one or more lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkyl chain. "Lower alkyl" means a group having about 1 to about 6 carbon atoms in the chain which may be straight or branched. "Alkyl" may be unsubstituted or optionally substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of halo, alkyl, aryl, cycloalkyl, cyano, hydroxy, alkoxy, alkylthio, amino, oxime (e.g., =N-OH), - NH(alkyl), -NH(cycloalkyl), -N(alkyl)2, -O-C(O)-alkyl, -O-C(O)-aryl, -O-C(O)-cycloalkyl, carboxy and -C(O)O-alkyl. Non-limiting examples of suitable alkyl groups include methyl, ethyl, n-propyl, isopropyl and t-butyl. "Alkenyl" means an aliphatic hydrocarbon group containing at least one carbon- carbon double bond and which may be straight or branched and comprising about 2 to about 15 carbon atoms in the chain. Preferred alkenyl groups have about 2 to about 12 carbon atoms in the chain; and more preferably about 2 to about 6 carbon atoms in the chain. Branched means that one or more, lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkenyl chain. "Lower alkenyl" means about 2 to about 6 carbon atoms in the chain which may be straight or branched. "Alkenyl" may be unsubstituted or optionally substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of halo, alkyl. aryl, cycloalkyl, cyano, alkoxy and -S(alkyl). Non-limiting examples of suitable alkenyl groups include ethenyl, propenyl, n-butenyl, 3-methylbut- 2-enyl, n-pentenyl, octenyl and decenyl.
"Alkylene" means a difunctional group obtained by removal of a hydrogen atom from an alkyl group that is defined above. Non-limiting examples of alkylene include methylene, ethylene and propylene.
"Alkynyl" means an aliphatic hydrocarbon group containing at least one carbon- carbon triple bond and which may be straight or branched and comprising about 2 to about 15 carbon atoms in the chain. Preferred alkynyl groups have about 2 to about 12 carbon atoms in the chain; and more preferably about 2 to about 4 carbon atoms in the chain. Branched means that one or more, lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkynyl chain. "Lower alkynyl" means about 2 to about 6 carbon atoms in the chain which may be straight or branched. Non-limiting examples of suitable alkynyl groups include ethynyl, propynyl, 2-butynyl and 3- methylbutynyl. "Alkynyl" may be unsubstituted or optionally substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of alkyl, aryl and cycloalkyl. "Aryl" means an aromatic monocyclic or multicyclic ring system comprising about 6 to about 14 carbon atoms, preferably about 6 to about 10 carbon atoms. The aryl group can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein. Non-limiting examples of suitable aryl groups include phenyl and naphthyl. "Bridged cyclic ring" is a hydrocarbon ring such as cycloalkyl, cyclenyl, or aryl or heteroatom containing ring such as, heterocyclyl, heterocyclenyl, or heteroaryl as described herein, that contains a bridge, which is a valence bond or an atom or an unbranched chain of atoms connecting two different parts of the ring. The two tertiary carbon atoms connected through the bridge are termed "bridgeheads". "Heteroaryl" means an aromatic monocyclic or multicyclic ring system comprising about 5 to about 14 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the ring atoms is an element other than carbon, for example nitrogen, oxygen or sulfur, alone or in combination. Preferred heteroaryls contain about 5 to about 6 ring atoms. The "heteroaryl" can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein. The prefix aza, oxa or thia before the heteroaryl root name means that at least a nitrogen, oxygen or sulfur atom respectively, is present as a ring atom. A
nitrogen atom of a heteroaryl can be optionally oxidized to the corresponding N-oxide. "Heteroaryl" may also include a heteroaryl as defined above fused to an aryl as defined above. Non-limiting examples of suitable heteroaryls include pyridyl, pyrazinyl, furanyl, thienyl, pyrimidinyl, pyridone (including N-substituted pyridones), isoxazolyl, isothiazolyl, oxazolyl, thiazolyl, pyrazolyl, furazanyl, pyrrolyl, pyrazolyl, triazolyl, 1 ,2,4- thiadiazolyl, pyrazinyl, pyridazinyl, quinoxalinyl, phthalazinyl, oxindolyl, imidazo[1 ,2- a]pyridinyl, imidazo[2,1 -b]thiazolyl, benzofurazanyl, indolyl, azaindolyl, benzimidazolyl, benzothienyl, quinolinyl, imidazolyl, thienopyridyl, quinazolinyl, thienopyrimidyl, pyrrolopyridyl, imidazopyridyl, isoquinolinyl, benzoazaindolyl, 1 ,2,4-triazinyl, benzothiazolyl and the like. The term "heteroaryl" also refers to partially saturated heteroaryl moieties such as, for example, tetrahydroisoquinolyl, tetrahydroquinolyl and the like.
"Aralkyl" or "arylalkyl" means an aryl-alkyl- group in which the aryl and alkyl are as previously described. Preferred aralkyls comprise a lower alkyl group. Non-limiting examples of suitable aralkyl groups include benzyl, 2-phenethyl and naphthalenylmethyl. The bond to the parent moiety is through the alkyl.
"Alkylaryl" means an alkyl-aryl- group in which the alkyl and aryl are as previously described. Preferred alkylaryls comprise a lower alkyl group. Non-limiting example of a suitable alkylaryl group is tolyl. The bond to the parent moiety is through the aryl.
"Cycloalkyl" means a non-aromatic mono- or multicyclic ring system comprising about 3 to about 10 carbon atoms, preferably about 5 to about 10 carbon atoms. Preferred cycloalkyl rings contain about 5 to about 7 ring atoms. The cycloalkyl can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined above. Non-limiting examples of suitable monocyclic cycloalkyls include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl and the like. Non-limiting examples of suitable multicyclic cycloalkyls include 1 -decalinyl, norbomyl, adamantyl and the like.
"Cycloalkylalkyl" means a cycloalkyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core. Non-limiting examples of suitable cycloalkylalkyls include cyclohexylmethyl, adamantylmethyl and the like.
"Cycloalkenyl" means a non-aromatic mono or multicyclic ring system comprising about 3 to about 10 carbon atoms, preferably about 5 to about 10 carbon atoms which contain at least one carbon-carbon double bond. Preferred cycloaikenyl rings contain about 5 to about 7 ring atoms. The cycloalkenyl can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined above. Non-limiting examples of suitable monocyclic cycioalkenyls include cyclopentenyi, cyclohexenyl, cyclohepta-1 ,3-dienyl, and the like. Non-iimiting example of a suitable multicyclic cycloalkenyl is norbornylenyl.
"Cycloalkenylalkyl" means a cycloalkenyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core. Non-limiting examples of suitable cycloalkenyialkyls include cyclopentenylmethyl, cyclohexenylmethyl and the like.
"Halogen" means fluorine, chlorine, bromine, or iodine. Preferred are fluorine, chlorine and bromine.
"Ring system substituent" means a substituent attached to an aromatic or non- aromatic ring system which, for example, replaces an available hydrogen on the ring system. Ring system substituents may be the same or different, each being independently selected from the group consisting of alkyl, alkenyl, alkynyl, aryl, heteroaryi, araikyl, alkylaryl, heteroaralkyl, heteroarylalkenyl, heteroarylalkynyl, alkyl heteroaryl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, acyl, aroyl, halo, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyi, aralkoxycarbonyl, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio, cycloalkyl, heterocyclyl, amide, -CHO, -O-C(O)-alkyl, -O-C(O)-aryl, - O-C(O)-cycloalkyl, -C(=N-CN)-NH2l -C(=NH)-NH2, -C(=NH)-NH(alkyl), oxime (e.g., =N-OH), YiY2N-, Y1Y2N-^kVl-, Y1Y2NC(O)-, Y1Y2NSO2- and -SO2NY1Y2, wherein Y1 and Y2 can be the same or different and are independently selected from the group consisting of hydrogen, alkyl, aryl, cycloalkyi, and araikyl. "Ring system substituent" may also mean a single moiety which simultaneously replaces two available hydrogen on two adjacent carbon atoms (one H on each carbon) on a ring system. Examples of such moiety are methylene dioxy, ethyl enedioxy, -C(CH3J2- and the like which form moieties such as, for example:

"Heteroarylalky!" means a heteroary! moiety as defined above linked via an alkyl moiety (defined above) to a parent core. Non-limiting examples of suitable heteroaryis include 2-pyridinylmethyl, quinolinylmethy! and the like. "Heterocyciyl" means a non-aromatic saturated monocyclic or multicyclic ring system comprising about 3 to about 10 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is an element other than carbon, for example nitrogen, oxygen or sulfur, alone or in combination. There are no adjacent oxygen and/or sulfur atoms present in the ring system. Preferred heterocyclyls contain about 5 to about 6 ring atoms. The prefix aza, oxa or thia before the heterocyciyl root name means that at least a nitrogen, oxygen or sulfur atom respectively is present as a ring atom. Any -NH in a heterocyciyl ring may exist protected such as, for example, as an -N(Boc), -N(CBz), -N(Tos) group and the like; such protections are also considered part of this invention. The heterocyciyl can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein. The nitrogen or sulfur atom of the heterocyciyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S1S- dioxide. Non-limiting examples of suitable monocyclic heterocyciyl rings include piperidyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyi, thiazolidinyl, 1 ,4- dioxanyl, tetrahydrofuranyl, tetrahydrothiophenyl, lactam, lactone, and the like.
"Heterocyciyl" may also mean a single moiety (e.g., carbonyl) which simultaneously replaces two available hydrogen on the same carbon atom on a ring system. Example of such moiety is pyrrolidone:
"Heterocyclenyl" means a non-aromatic monocyclic or multicyclic ring system comprising about 3 to about 10 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is an element other than carbon, for example nitrogen, oxygen or sulfur atom, alone or in combination, and which contains at least one carbon-carbon double bond or carbon-nitrogen double bond. There are no adjacent oxygen and/or sulfur atoms present in the ring system. Preferred heterocycienyl rings contain about 5 to about 6 ring atoms. The prefix aza, oxa or thia before the heterocyclenyl root name means that at least a nitrogen, oxygen or sulfur atom respectively is present as a ring atom. The heterocycienyl can be optionally substituted by one or more ring system substituents, wherein "ring system substituent" is as defined above. The nitrogen or sulfur atom of the heterocyclenyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide. Non- limiting examples of suitable heterocyclenyl groups include 1 ,2,3,4- tetrahydropyridinyl, 1 ,2-dihydropyridinyl, 1 ,4-dihydropyridinyl, 1 ,2,3,6- tetrahydropyridinyl, 1 ,4,5,6-tetrahydropyrimidinyl, 2-pyrrolinyl, 3-pyrrolinyl, 2- imidazolinyl, 2-pyrazoiinyl, dihydroimidazolyl, dihydrooxazolyi, dihydrooxadiazolyl, dihydrothiazolyl, 3,4-dihydro~2H-pyranyS, dihydrofuranyl, fluorodihydrofuranyi, 7- oxabicyclo[2.2.1]heptenyl, dihydrothiophenyi, dihydrothiopyranyl, and the like. "Heterocyclenyl" may also mean a single moiety (e.g., carbonyl) which simultaneously replaces two available hydrogen on the same carbon atom on a ring system. Example of such moiety is pyrrolidinone:

"Heterocyclenylalkyl" means a heterocyclenyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
It should be noted that in hetero-atom containing ring systems of this invention, there are no hydroxyl groups on carbon atoms adjacent to a N, O or S, as well as there are no N or S groups on carbon adjacent to another heteroatom. Thus, for example, in the ring:

It should also be noted that tautomeric forms such as, for example, the moieties:
"Alkynylalkyl" means an alkynyi-alkyl- group in which the alkynyl and alkyl are as previously described. Preferred alkynylalkyls contain a lower alkynyi and a lower alkyl group. The bond to the parent moiety is through the alkyf. Non-limiting examples of suitable alkynylalkyl groups include propargyimethyl. "Heteroaralkyi" means a heteroaryl-alkyl- group in which the heteroaryl and alkyl are as previously described. Preferred heteroaralkyls contain a lower alky! group. Non-limiting examples of suitable araikyl groups include pyridyl methyl, and quinolin-3- ylmethyl. The bond to the parent moiety is through the alkyi.
"Spiro ring systems" have two or more rings linked by one common atom. Preferred spiro ring systems include spiroheteroaryl, spiroheterocyclenyl, spiroheterocyclyl, spirocycloaikyl, spirocyclenyl, and spiroaryl. The spiro ring systems can be optionally substituted by one or more ring system substituents, wherein "ring system substituent" is as defined above. Non-limiting examples of suitable spiro ring
systems include
 spiro[4.5jdecane,
spiro[4.5jdecane,
 8-azaspiro[4.5]dec-2-ene, and '
8-azaspiro[4.5]dec-2-ene, and '
 spiro[4.4]nona-2t7-diene.
spiro[4.4]nona-2t7-diene.
Ηydroxyalkyl" means a HO-aikyl- group in which alkyl is as previously defined. Preferred hydroxyalkyls contain lower alkyl. Non-limiting examples of suitable hydroxyalkyl groups include hydroxymethyl and 2-hydroxyethyl.
"Acyl" means an H-C(O)-, alkyl-C(O)- or cycloalkyl-C(O)-, group in which the various groups are as previously described. The bond to the parent moiety is through the carbonyl. Preferred acyls contain a lower alkyl. Non-limiting examples of suitable acyl groups include formyl, acetyl and propanoyl. "Aroyl" means an aryl-C(O)- group in which the aryl group is as previously described. The bond to the parent moiety is through the carbonyi. Non-limiting examples of suitable groups include benzoyl and 1- naphthoyl.
"Alkoxy" means an alkyl-O- group in which the alky! group is as previously described. Non-limiting examples of suitable alkoxy groups include methoxy, ethoxy, n-propoxy, isopropoxy and n-butoxy. The bond to the parent moiety is through the ether oxygen.
"Aryloxy" means an aryl-O- group in which the aryl group is as previously described. Non-ϋmiting examples of suitable aryloxy groups include phenoxy and naphthoxy. The bond to the parent moiety is through the ether oxygen. "Aralkyloxy" means an aralkyl-O- group in which the aralkyi group is as previously described. Non-limiting examples of suitable aralkyloxy groups include benzyloxy and 1- or 2-naphthalenemethoxy. The bond to the parent moiety is through the ether oxygen.
"Alkylthio" means an alkyl-S- group in which the aikyl group is as previously described. Non-limiting examples of suitable alkylthio groups include methylthio and ethylthio. The bond to the parent moiety is through the sulfur.
"Arylthio" means an aryl-S- group in which the aryl group is as previously described. Non-limiting examples of suitable arylthio groups include phenyithio and naphthylthio. The bond to the parent moiety is through the sulfur.
"Aralkylthio" means an aralkyl-S- group in which the aralkyl group is as previously described. Non-limiting example of a suitable aralkylthio group is benzylthio. The bond to the parent moiety is through the sulfur.
"Alkoxycarbonyl" means an alkyi-O-CO- group. Non-limiting examples of suitable alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl. The bond to the parent moiety is through the carbonyl.
"Aryloxycarbonyl" means an aryl-O-C(O)- group. Non-limiting examples of suitable aryloxycarbonyi groups include phenoxycarbonyl and naphthoxycarbonyi. The bond to the parent moiety is through the carbonyl. "Aralkoxycarbonyl" means an aralkyl-O-C(O)- group. Non-limiting example of a suitable aralkoxycarbonyl group is benzyloxycarbonyl. The bond to the parent moiety is through the carbonyl.
"Alkylsulfonyi" means an alkyi-S(O2)- group. Preferred groups are those in which the alkyl group is lower alkyl. The bond to the parent moiety is through the sulfonyl.
"Arylsulfonyl" means an aryl-S(O2)- group. The bond to the parent moiety is through the sulfonyl.
The term "substituted" means that one or more hydrogen on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. By "stable compound' or "stable structure" is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
The term "optionally substituted" means optional substitution with the specified groups, radicals or moieties.
The term "purified", "in purified form" or "in isolated and purified form" for a compound refers to the physical state of said compound after being isolated from a synthetic process (e.g. from a reaction mixture), or natural source or combination thereof. Thus, the term "purified", "in purified form" or "in isolated and purified form" for a compound refers to the physical state of said compound after being obtained from a
purification process or processes described herein or wefl known to the skilled artisan (e.g., chromatography, recrystallization and the like) , in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan. It should aiso be noted that any carbon as well as heteroatom with unsatisfied valences in the text, schemes, examples and Tables herein is assumed to have the sufficient number of hydrogen atom(s) to satisfy the valences.
When a functional group in a compound is termed "protected", this means that the group is in modified form to preclude undesired side reactions at the protected site when the compound is subjected to a reaction. Suitable protecting groups will be recognized by those with ordinary skill in the art as well as by reference to standard textbooks such as, for example, T. W. Greene et a/, Protective Groups in organic Synthesis (1991), Wiley, New York.
When any variable (e.g., aryl, heterocycle, R2, etc.) occurs more than one time in any constituent or in The present invention, its definition on each occurrence is independent of its definition at every other occurrence.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
Prodrugs and solvates of the compounds of the invention are also contemplated herein. A discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. The term "prodrug" means a compound (e.g., a drug precursor) that is transformed in vivo Xo yieid a compound of the present invention or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (e.g., by metabolic or chemical processes), such as, for example, through hydrolysis in blood. A discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series, and in
Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutica! Association and Pergamon Press, 1987.
For example, if a compound of the present invention or a pharmaceutically acceptable salt, hydrate or solvate of the compound contains a carboxyiic acid functional group, a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as, for example, (C1-Cβjalkyl, (C2- Ci2)alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1- methyl-1 -(alkanoyloxy)-ethyl having from 5 to 10 carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1-(alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1-methyl-1-(alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N-(alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1 -(N-(alkoxycarbonyl)amino)ethy! having from 4 to 10 carbon atoms, 3-phthalidyl, 4- crotonolactonyl, gamma-butyrolacton-4-yl, di-N, N-(Ci -C2)alkylamino(C2-C3)alkyl (such as β-dimethylaminoethyl), carbamoyl-fC1-C^alkyl, N,N-di (C1-C2)alkyicarbamoyi-(C1- C2)alkyl and piperidino-, pyrrolidino- or morpholino{C2-C3)alkyl, and the like.
Similarly, if a compound of the present invention contains an alcohol functional group, a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as, for example, (C1-C6)alkanoyloxymethyl, 1 -((Ci- C6)alkanoyloxy)ethyl, 1 -methyl-1 -((C1-C6)alkanoyloxy)ethyl, (C1- C6)alkoxycarbonyloxymethyl, N-(C1-C6)alkoxycarbonylaminomethyl, succinoyl, (Cr C6)alkanoy!, α-amino(C1-C4)alkanyl, aryiacyl and α-aminoacyl, or α-aminoacyl-α- aminoacyl, where each α-aminoacyl group is independently selected from the naturally occurring L-amino acids, P(O)(OH)2, -P(O)(O(Ci -C6)alkyl)2 or glycosyl (the radical resulting from the removal of a hydroxyl group of the hemiacetal form of a carbohydrate), and the like.
If a compound of the present invention incorporates an amine functional group, a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as, for example, R-carbonyi, RO-carbonyl, NRR'-carbonyl where R and R' are each independently (Ci~Cio)alkyt, (C3-C7) cycloalkyl, benzyl, or R-carbonyl is a natural α-aminoacyl or natural α-aminoacyl, -C(OH)C(O)OY1 wherein Y1 is H, (C1-Ce)alkyl or benzyl, -C(OY2JY3 wherein Y2 is (C1-C4) alky] and Y3 is (C1-C6)alkyl, carboxy (C1-C6)alky!, amino(C1-C4)alkyl or mono-N — or di~N,N-(C1-C6)alky!aminoalkyl,
-C(Y4) Y5 wherein Y4 is H or methyl and Y5 is mono-N— or di-N,N-(C1-C6)alkylamino morpholino, piperidin-1-yl or pyrroiidin-1 -yl, and the like.
One or more compounds of the invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable soivents such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms. "Solvate" means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. "Solvate" encompasses both solution-phase and isolatable solvates. Non-limiting examples of suitable solvates include ethanoiates, methanolates, and the like. "Hydrate" is a solvate wherein the solvent molecule is H2O.
One or more compounds of the invention may optionally be converted to a solvate. Preparation of solvates is generally known. Thus, for example, M. Caira et al, J. Pharmaceutical Sci., 93(3), 601-611 (2004) describes the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water. Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS PharmSciTech., 5£1), article 12 (2004); and A. L. Bingham et al, Chem. Commυn., 603-604 (2001). A typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than ambient temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods. Analytical techniques such as, for example I. R. spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate).
"Effective amount" or "therapeutically effective amount" is meant to describe an amount of compound or a composition of the present invention effective in inhibiting the above-noted diseases and thus producing the desired therapeutic, ameliorative, inhibitory or preventative effect. The compounds of the present invention can form salts which are also within the scope of this invention. Reference to a compound of the present invention herein is understood to include reference to salts thereof, unless otherwise indicated. The
term "saltfs)", as employed herein, denotes acidic salts formed with inorganic and/or organic acids, as well as basic salts formed with inorganic and/or organic bases. In addition, when a compound of the present invention contains both a basic moiety, such as, but not limited to a pyridine or imidazole, and an acidic moiety, such as, but not limited to a carboxylic acid, zwitterions ("inner salts") may be formed and are included within the term "salt(s)" as used herein. Pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) salts are preferred, although other salts are also useful. Salts of the compounds of the The present invention may be formed, for example, by reacting a compound of the present invention with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates,) and the like. Additionally, acids which are generally considered suitable for the formation of pharmaceutically useful salts from basic pharmaceutical compounds are discussed, for example, by P. Stahl et al, Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiiey-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201 -217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; and in The Orange Book (Food & Drug Administration, Washington, D. C. on their website). These disclosures are incorporated herein by reference thereto.
Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as dicyclohexylamines, t-butyl amines, and salts with amino acids such as arginine, lysine and the like. Basic nitrogen-containing groups may be quartemized with agents such as lower alkyl hatides (e.g. methyl, ethyl, and butyl chlorides, bromides and iodides), dialky! sulfates (e.g. dimethyl, diethyl, and dibutyl sulfates), long chain
halides (e.g. decyl, iauryl, and stearyl chlorides, bromides and iodides), aralkyl haiides (e.g. benzyl and phenethyl bromides), and others.
All such acid salts and base salts are intended to be pharmaceutically acceptable salts within the scope of the invention and all acid and base salts are considered equivalent to the free forms of the corresponding compounds for purposes of the invention.
Pharmaceutically acceptable esters of the present compounds include the following groups: (1 ) carboxylic acid esters obtained by esterification of the hydroxy groups, in which the non-carbonyl moiety of the carboxyiic acid portion of the ester grouping is selected from straight or branched chain alkyl (for example, acetyl, n- propyl, t-butyl, or n-butyl), alkoxyalkyl (for example, methoxymethyl), araikyl (for example, benzyl), aryloxyalkyl (for example, phenoxymethyl), aryl (for example, phenyl optionally substituted with, for example, halogen, d^alkyl, or Ci.4aikoxy or amino); (2) sulfonate esters, such as alkyl- or aralkylsulfonyl (for example, methanesulfonyl); (3) amino acid esters (for example, L-valyl or L-isoieucyl); (4) phosphonate esters and (5) mono-, di- or triphosphate esters. The phosphate esters may be further esterified by, for example, a C1-2o alcohol or reactive derivative thereof, or by a 2,3-di (C6-24)acyl glycerol.
Compounds of the present invention, and salts, solvates, esters and prodrugs thereof, may exist in their tautomeric form (for example, as an amide or imino ether). All such tautomeric forms are contemplated herein as part of the present invention.
The compounds of the present invention may contain asymmetric or chiral centers, and, therefore, exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of the present invention as weil as mixtures thereof, including racemic mixtures, form part of the present invention. In addition, the present invention embraces all geometric and positional isomers. For example, if a compound of the present invention incorporates a double bond or a fused ring, both the cis- and trans-forms, as well as mixtures, are embraced within the scope of the invention. Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods weil known to those skilled in the art, such as, for example, by chromatography and/or fractional
crystallization. Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride), separating the diastereomers and converting (e.g., hydroiyzing) the individual diastereomers to the corresponding pure enantiomers. Also, some of the compounds of the present invention may be atropisomers (e.g., substituted biaryls) and are considered as part of this invention. Enantiomers can also be separated by use of chiral HPLC column.
It is also possible that the compounds of the present invention may exist in different tautomeric forms, and all such forms are embraced within the scope of the invention. Also, for example, all keto-enol and imine-enamine forms of the compounds are included in the invention.
All stereoisomers (for example, geometric isomers, optical isomers and the like) of the present compounds {including those of the salts, solvates, esters and prodrugs of the compounds as well as the salts, solvates and esters of the prodrugs), such as those which may exist due to asymmetric carbons on various substituents, including enantiomeric forms (which may exist even in the absence of asymmetric carbons), rotameric forms, atropisomers, and diastereomeric forms, are contemplated within the scope of this invention, as are positional isomers (such as, for example, 4-pyridyl and 3-pyridyl). (For example, if a compound of the present invention incorporates a double bond or a fused ring, both the cis- and trans-forms, as well as mixtures, are embraced within the scope of the invention. Also, for example, all keto-enol and imine-enamine forms of the compounds are included in the invention.) Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers, or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers. The chiral centers of the present invention can have the S or R configuration as defined by the IUPAC 1974 Recommendations. The use of the terms "salt", "solvate", "ester", "prodrug" and the like, is intended to equally apply to the salt, solvate, ester and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, racemates or prodrugs of the inventive compounds.
The present invention also embraces isotopically-labelled compounds of the present invention which are identical to those recited herein, but for the fact that one
or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine and chlorine and iodine, such as 2H, 3H, 11C, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S1 18F, 36CI and 123I, respectively.
Certain isotopically-labelled compounds of Formula (!) (e.g., those labeled with 3H and 14C) are useful in compound and/or substrate tissue distribution assays. Tritiated {i.e., 3H) and carbon-14 (i.e., 14C) isotopes are particularly preferred for their ease of preparation and detectabiiity. Certain isotopically-labelled compounds of Formula (I) can be useful for medical imaging purposes. E.g., those labeled with positron-emitting isotopes like 11C or 18F can be useful for application in Positron Emission Tomography (PET) and those labeled with gamma ray emitting isotopes like 123I can be useful for application in Single photon emission computed tomography (SPECT). Further, substitution with heavier isotopes such as deuterium (i.e., 2H) may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances. Further, substitution with heavier isotopes such as deuterium (i.e., 2H) may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances. Additionally, isotopic substitution at a site where epimerization occurs may slow or reduce the eptmerization process and thereby retain the more active or efficacious form of the compound for a longer period of time, isotopically labeled compounds of Formula (I), in particular those containing isotopes with longer half lives (T1/2 >1 day), can generally be prepared by following procedures analogous to those disclosed in the Schemes and/or in the Examples herein below, by substituting an appropriate isotopically labeled reagent for a non-isotopically labeled reagent.
Polymorphic forms of the compounds of the present invention, and of the salts, solvates, esters and prodrugs of the compounds of the present invention, are intended to be included in the present invention.
The compounds according to the invention have pharmacological properties; in particular, the compounds of the present invention can be inhibitors, regulators or modulators of mTOR protein kinases.
The compounds of the present invention can be inhibitors of protein kinases such as, for example, the inhibitors of the mTOR. Preferred compounds can exhibit IC50 values of less than about 5μm, preferably about 0.001 to about 1.0 μm, and more preferably about 0.001 to about 0.1 μm. The assay methods are described in the Examples set forth below.
The compounds of the present invention can be useful in the therapy of proliferative diseases such as cancer, autoimmune diseases, viral diseases, fungal diseases, neurological/neurodegenerative disorders, arthritis, inflammation, antiproliferative (e.g., ocular retinopathy), neuronal, alopecia and cardiovascular disease. Many of these diseases and disorders are iisted in U.S. 6,413,974, incorporated by reference herein. More specif icaliy, the compounds of the present invention can be useful in the treatment of a variety of cancers, including (but not limited to) the following: tumor of the bladder, breast (including BRCA-mutated breast cancer, colorectal, colon, kidney, liver, lung, small cell lung cancer, non-smail cell lung cancer, head and neck, esophagus, bladder, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, including squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma and Burkett's lymphoma; chronic lymphocytic leukemia ("CLL"), acute and chronic myelogenous leukemia, myelodysplastic syndrome and promyelocytic leukemia; fibrosarcoma, rhabdomyosarcoma; head and neck, mantle cell lymphoma, myeloma; astrocytoma, neuroblastoma, glioma, glioblastoma, malignant glial tumors, astrocytoma, hepatocellular carcinoma, gastrointestinal stromal tumors ("GIST") and schwannomas;
melanoma, multiple myeloma, seminoma, teratocarcinoma, osteosarcoma, xenoderma pigmentosum, keratoctanthoma, thyroid follicular cancer, endometrial cancer, gastrointestinal tract cancer and Kaposi's sarcoma.
Due to the key roie of kinases in the reguiation of cellular proliferation in general, inhibitors could act as reversible cytostatic agents which may be useful in the treatment of any disease process which features abnormal cellular proliferation, e.g., benign prostate hyperplasia, familial adenomatosis polyposis, neuro-fibromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, glomerulonephritis, restenosis following angioplasty or vascular surgery, hypertrophic scar formation, inflammatory bowel disease, transplantation rejection, endotoxic shock, and fungal infections. Compounds of the present invention may induce or inhibit apoptosis. The apoptotic response is aberrant in a variety of human diseases. Compounds of the present invention, as modulators of apoptosis, will be useful in the treatment of cancer (including but not limited to those types mentioned hereinabove), virai infections (including but not limited to herpevirus, poxvirus, Epstein- Barr virus, Sindbis virus and adenovirus), prevention of AIDS development in HIV-infected individuals, autoimmune diseases (including but not limited to systemic lupus, erythematosus, autoimmune mediated glomerulonephritis, rheumatoid arthritis, psoriasis, inflammatory bowel disease, and autoimmune diabetes mellitus), neurodegenerative disorders (including but not limited to Alzheimer's disease, AIDS-reiated dementia, Parkinson's disease, amyotrophic lateral sclerosis, retinitis pigmentosa, spinal muscular atrophy and cerebellar degeneration), myelodysplasia syndromes, aplastic anemia, ischemic injury associated with myocardial infarctions, stroke and reperfusion injury, arrhythmia, atherosclerosis, toxin-induced or alcohol related liver diseases, hematological diseases (including but not limited to chronic anemia and aplastic anemia), degenerative diseases of the musculoskeletal system (including but not limited to osteoporosis and arthritis) aspirin-sensitive rhinosinusitis, cystic fibrosis, multiple sclerosis, kidney diseases and cancer pain.
Compounds of the present invention, as inhibitors of kinases, can modulate the level of cellular RNA and DNA synthesis. These agents would therefore be useful in the treatment of viral infections (including but not limited to HIV, human papilloma virus, herpesvirus, poxvirus, Epstein-Barr virus, Sindbis virus and adenovirus).
Compounds of the present invention may also be useful in the chemoprevention of cancer. Chemoprevention is defined as inhibiting the development of invasive cancer by either blocking the initiating mutagenic event or by blocking the progression of pre-malignant cells that have already suffered an insult or inhibiting tumor relapse.
Compounds of the present invention may also be useful in inhibiting tumor angiogenesis and metastasis.
Another aspect of this invention is a method of treating a mammal {e.g., human) having a disease or condition associated with mTOR kinases by administering a therapeutically effective amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound to the mammal.
A preferred dosage is about 0.001 to 1000 mg/kg of body weight/day of the compound of the present invention. An especially preferred dosage is about 0.01 to 25 mg/kg of body weight/day of a compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound. The compounds of this invention may also be useful in combination (administered together or sequentially) with one or more of anti-cancer treatments such as radiation therapy, and/or one or more anti-cancer agents different from the compound of the present invention. The compounds of the present invention can be present in the same dosage unit as the anti-cancer agent or in separate dosage units.
Another aspect of the present invention is a method of treating one or more diseases associated with a mTOR protein kinase, comprising administering to a mammal in need of such treatment: an amount of a first compound, which is a compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent different from the compound of the present invention, wherein the amounts of the first compound and the second compound result in a therapeutic effect. Non-limiting examples of suitable anti-cancer agent is selected from the group consisting of a Cytostatic agent, Cisplatin, Deforolimus (described in PCT publication No. 2003/064383), Doxorubicin, liposomal doxorubicin {e.g., Caelyx®, Myocet®,
Doxil®), Taxotere, Taxol, Etoposide, frinotecan, Camptostar, Topotecan, Paclitaxel, Docetaxel, Epothilones, Tamoxifen, 5-Fluorouracil, Methoxtrexate, Temozolomide, cyclophosphamide, SCH 66336, R115777®, L778.123®, BMS 214662®, iressa®, Tarceva®, Antibodies to EGFR, antibodies to IGFR (including, for example, those published in US 2005/0136063 published June 23, 2005), ESK inhibitors, KSP inhibitors (such as, for example, those published in WO 2006/098962 and WO 2006/098961 ; ispinesib, SB-743921 from Cytokinetics), Centrosome associated protein E ("CENP-E") inhibitors (e.g., GSK-923295), Gleevec®, Intron, Ara-C, Adriamycin, Cytoxan, Gemcitabine, Uracil mustard, Chlormethine, ifosfamide, Melphalan, Chlorambucil, Pipobroman, Triethylenemelamine,
Triethylenethiophosphoramine, Busulfan, Carmustine, Lomustine, Streptozocin, Dacarbazine, Floxuridine, Cytarabine, 6-Mercaptopurine, 6-Thioguanine, Fludarabine phosphate, Oxaliplatin, Leucovirin, ELOXATIN™, Vinblastine, Vincristine, Vindesine, Bleomycin, Dactinomycin, Daunorubicin, Doxorubicin, Epirubicin, Idarubicin, Mithramycin, Deoxycoformycin, Mitomycin-C, L-Asparaginase, Teniposide 17oc- Ethinylestradiol, Diethylstilbestroi, Testosterone, Prednisone, Fluoxymesterone, Dromostanolone propionate, Testolactone, Megestrolacetate, Methylprednisoione, Methyltestosterone, Prednisolone, Triamcinolone, Chlorotrianisene, Hydroxyprogesterone, Aminoglutethimide, Estramustine, Medroxyprogesteroneacetate, Leuprolide, Flutamide, Toremifene, Goserelin, Cisplatin, Carboplatin, Hydroxyurea, Amsacrine, Procarbazine, Mitotane, Mitoxantrone, Levamisole, IMavelbene, Anastrazole, Letrazole, Capecitabine, Reioxafine, Droloxafine, Hexamethylmelamine, Avastin, herceptin, Bexxar, bortezomib ("Velcade"), Zevalin, Trisenox, Xeloda, Vinorelbine, Porfimer, Erbitux, Liposomal, Thiotepa, Altretamine, Melphalan, Trastuzumab, Lerozole, Fulvestrant, Exemestane, Fulvestrant, Ifosfomide, Rituximab, C225®, Satriplatin, mylotarg, Avastin, Rituxan, Panitubimab, Sutent, Sorafinib, Sprycel (dastinib), Nilotinib, Tykerb (Lapatinib) and Campath.
If formulated as a fixed dose, such combination products employ the compounds of this invention within the dosage range described herein and the other pharmaceutically active agent or treatment within its dosage range. For example, the CDC2 inhibitor olomucine has been found to act synergistically with known cytotoxic
agents in inducing apoptosis (J. CeII ScL, (1995) 108, 2897. Compounds of the present invention may also be administered sequentially with known anticancer or cytotoxic agents when a combination formulation is inappropriate. The invention is not limited in the sequence of administration; compounds of the present invention may be administered either prior to or after administration of the known anticancer or cytotoxic agent. For example, the cytotoxic activity of the cyclin-dependent kinase inhibitor flavopiridol is affected by the sequence of administration with anticancer agents. Cancer Research, (1997) 57, 3375. Such techniques are within the skills of persons skiiled in the art as well as attending physicians. Accordingly, in an aspect, this invention includes combinations comprising an amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof, and an amount of one or more anticancer treatments and anti-cancer agents listed above wherein the amounts of the compounds/ treatments result in desired therapeutic effect. Another aspect of the present invention is a method of inhibiting one or more mTOR protein kinases in a patient in need thereof, comprising administering to the patient a therapeutically effective amount of at least one compound of the present invention or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more mTOR protein kinases in a patient in need thereof, comprising administering a therapeutically effective amount of at least one compound of the present invention or a pharmaceuticaily acceptable salt, solvate, ester or prodrug thereof.
Yet another aspect of the present invention is a method of treating one or more diseases associated with mTOR protein kinases, comprising administering to a mammai in need of such treatment an amount of a first compound, which is a compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent, wherein the amounts of the first compound and the second compound result in a therapeutic effect.
Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more mTOR protein kinases in a
patient in need thereof, comprising administering a therapeutically effective amount of a pharmaceutical composition comprising in combination at least one pharmaceutically acceptable carrier and at least one compound according to The present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
In the above methods, the mTOR protein kinases to be inhibited can exist in two complexes, mTORC1 and mTORC2.
ERK inhibitors (i.e., ERK1 inhibitors and/or ERK2 inhibitors) include but are not limited to the following compounds: a) Formula H

or the pharmaceutically acceptable salts, esters and solvates thereof, wherein:
Y1 , Y2, and Y3 are each independently selected from the group consisting of: -CH=, -N= and -CR9=; z is 1 to 3; Q is a substituent selected from the group consisting of:
(2.8)
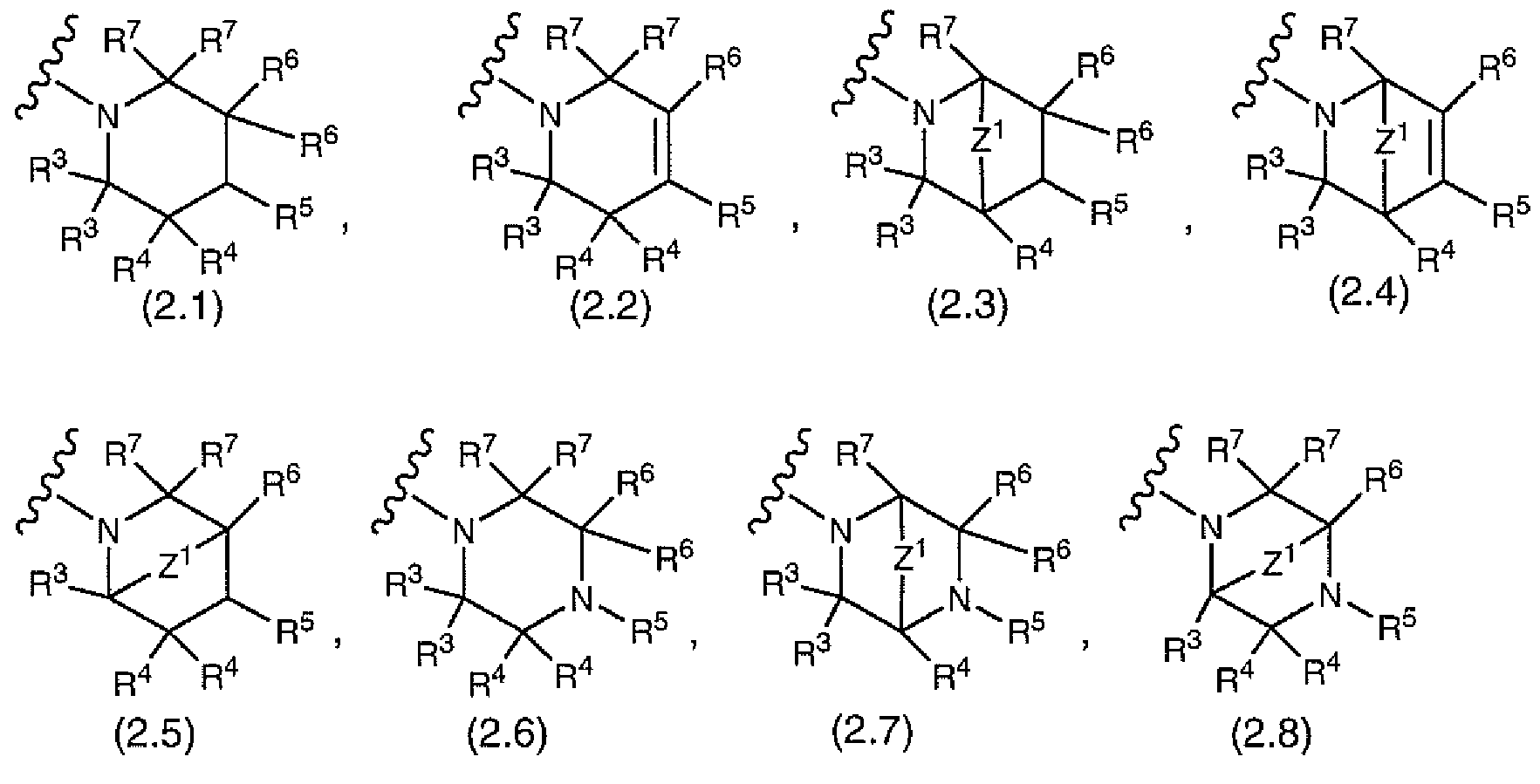 (2.13) ) (2.16)
(2.13) ) (2.16)

Each Q1 represents a ring independentiy selected from the group consisting of: cycloalkyl, substituted cycloalkyi, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted ary!, heteroaryl, and substituted heteroaryl, wherein said substituted rings
are substituted with 1 to 3 substituents independently selected from the group consisting of: halo and the R10 moieties; provided that when Q1 is ary!, heteroaryl, substituted aryl or substituted heteroaryl then the carbon atoms at the ring junction are not substituted; Q2 represents a ring selected from the group consisting of: cycloalkyl, substituted cycloaikyl, heterocycloaikyl, and substituted heterocycloalkyl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R10 moieties;
Z1 represents -(C(R24)2)W- wherein each R24 is independently selected from the group consisting of: H, alkyl and F, and wherein w is 1 , 2 or 3;
Z2 is selected from the group consisting of: -N(R44)-, -O- and -C(R46J2-; m is 1 to 6; n is 1 to 6; p is 0 to 6; t is O1 1 , or 2;
R1 is selected from the group consisting of: (I) -CN,
(2) -NO2,
(3) -OR10, (4) -SR10,
(5) -N(R10J2,
(6) R10,
(7) -C(O)R10,
(8) -(C(R30)2)n-NR32-C(O)-R10, (9) -(C(R30)2)n-NR32-S(O)rR10,
(10) -(C(R30)2)n-NR32-C(O)-N(R32)-R10,
(11 )

(12) -CF3,
(13) -C(O)OR10,
(14) -(C(R30)2)nR13 (β.g., -(CH2)nR13),
(15) alkenyl, (16) -NR32-C(O)-R14, (17)
 wherein each R10 is independently selected, (18)
wherein each R10 is independently selected, (18)
 wherein each R10 is independently selected, (19)
wherein each R10 is independently selected, (19)

(20) -C(O)-N R32-(C(R30)2)p-OR10,
(21 ) -C(O)N(R10)2 wherein each R10 is independently selected, (22) -C(O)-N R32-C(R18)3,
(23) -C(O)-NR32-(C(R30)2)n-C(O)-N(R10)2,
(24) heterocycloalkenyl,
(25)

R2 is selected from the group consisting of: (D H1
(2) -CN,
(3) halo, (4) alkyl,
(5) substituted alkyl wherein said substituted aikyl is substituted with 1 to 3 substitutents selected from the group consisting of: (a) -OH, (b) -O-alkyl (e.g., -0-(Cr C3a!ky!), (c) -O-alkyl substituted with 1 to 3 F atoms, and (d) -N(R40)2 wherein each R40 is independently selected from the group consisting of: (i) H, (ii) C1-C3 alkyl, (iii) - CF3, and (e) halo,
(6) alkynyl,
(7) alkenyl, <8) -(CHΞ)mR1\ (9) -N(R26)2> (1 O) -OR23,
(11 ) -N(R26JC(O)R42, (12) cycloaikyl, (13) cycloaikylalkyl,
 (15) -O-(substituted alkyl) wherein said substituted alkyi is substituted with
(15) -O-(substituted alkyl) wherein said substituted alkyi is substituted with
1 to 3 F atoms,
(16) -S(0)ralkyl, <17) -C(O)-alkyl,
(18)
N— O— H £ Il ξ — C— alkyl >
(19)
N— O— alkyl I — C— alkyl
wherein each alkyi is independently selected,
(20)
O
H I! N—N— C— aikyl
< — C— alkyi > which each alkyl is independently selected,
(21 )

(22) -N(R48)-C(O)-R48 wherein each R48 is independently selected from the group consisting of: H and alkyl, and
(23) -C(O)-alkyl, such as, for example, -C(O)-(C1-C6 alky!), such as, for example, -C(O)CH3; each R3, R4, R5, R6 and R7 is independently selected from the group consisting of: (I) H1
(2) alkenyl,
(3) substituted alkenyl,
(4) alkyl,
(5) substituted aikyl, (6) cycloalkyl,
(7) substituted cycioalkyl,
(8) cycloaikylalkyl-,
(9) substituted cycloaikylalkyl-,
(10) heterocycloalkyl, (11 ) substituted heterocycloalkyl,
(12) heterocycloalkylalkyl-,
(13) substituted heterocycloalkylalkyl-,
(14) -C(O)R10,
(15) arylheteroaryl-, (16) substituted arylheteroaryl-,
(17) heteroarylaryl-,
(18) substituted heteroarylaryl-, (19) aryl,
(20) substituted aryl,
(21) heteroaryl,
(22) substituted heteroaryi,
(23) heteroarylheteroaryi-,
(24) substituted heteroarylheteroaryi-, (25) arylaminoheteroaryl-,
(26) substituted arylaminoheteroaryl-,
(27) aryialkynyl-,
(28) substituted arylalkynyi-,
(29) heteroarylalkynyl-, (30) substituted heteroarylaikynyl-, and
(31) benzoheteroaryl, wherein said R3 T R4, R5, R6 and R7 substituted groups (7), (9), (11), (13), (16), (18), (20), (22), (24), (26), (28) and (30) are substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, -NHR20, -N(R20J2 wherein each R20 is independently selected, alkyl, alkenyl, halo, -C(O)-NH-R26, -C(O)OR28, -C(O)R28, and -OR20, and wherein said R3, R4, R5, R6 and R7 substituted groups (3) and (5) are substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, halo (e.g., F, Cl and Br, and in another example F), -C(O)-NH-R28 (e.g., -C(O)-NH-CH3), -C(O)OR28 (e.g., -C(O)OC2H5), and -C(O)R28 (e.g., -C(O)CH3); R5A is selected from the group consisting of: haio, -OH, alkyt, and -O-alkyl; R8 is selected from the group consisting of: H, -OH, -N(R10J2, -NR1°C(O)R12, and alkyl; each R9 is independently selected from the group consisting of:halogen, -CN, -NO2, -OR10, -SR10, -N(R10J2, and R10; each R10 is independently seiected from the group consisting of: H, alkyi, aryl, arylalkyl, heteroaryi, heteroaryl alkyl, cycioalkyl, cycloalkylalkyl, heterocycloalkyl, heterocycloalkylalkyi, alkyl heteroaryl-, alkylaryl-, substituted alkyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycioalkyl, substituted cycloalkylalkyl, substituted heterocycloalkyl, substituted heterocycloalkylalkyi, substituted alkyiheteroaryh substituted alkylaryl-, heterocycioalkenyl, and substituted heterocycloalkenyl, and wherein:
said R10 substituted alkyl is substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, -NHR20, -NO2, -CN, -OR26, halo, -C(O)-NH-R26, -C(O)OR26, and -C(O)R26, and said R10 substituted aryl, substituted arylalkyi, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloalkyi, substituted cycloaikylalkyl, substituted heterocycloalkyl, substituted heterocycloalkylaikyi, substituted aikylheteroaryl- and substituted alkyiaryl- are substituted with 1 to 3 substituents independently selected from the group consisting of: (1) -NH2, (2) -NO2, (3) -CN, (4) -OH, (5) -OR20, (6) -OCF3, (7) alkyl substituted with 1 to 3 independently selected halo atoms, (8) -C(O)R33, (9) alkyl, (10) alkenyl, (11 ) halo, (12) -C(O)-NH-R26, (13) -C(O)OR38, (14) -C(O)-NR32-(C(R30)2)n-N(R38)2, (15) -S(O)1R38, (16) -C(O)-NR32-R38, (17) -NR32-C(O)-R38,

(19) -NHR20, (20) cycloalkyi, (21 ) -O-alkyl-O-R20, (22) hydroxyalkyl, (23) -N(R20J2 wherein each R20 is independently selected, (24) -alkyl-OR20, (25) -0-alkyi-OH, (26) -NH(hydroxyalkyl), and (27) oxazolidinone;
R11 is selected from the group consisting of: F, -OH, -CN, -OR10, -NHNR1R10, -SR10 and heteroaryl;
R12 is selected from the group consisting of: alkyl, aryl, heteroaryl, cycloalkyi, eye! oa Iky I alkyl, heterocycloalkyl and heterocycloalkylaikyi;
R14 is selected from the group consisting of: alkyt, aryl, heteroaryl, cycloalkyi, cycloaikylalkyl-, heterocycloalkyl, alkylheterocycloalkyl, heterocycioalkylalkyl-, aikylheteroaryi- and aikyiaryl-;
R15 is selected from the group consisting of: H, -OH, alkyl, aryl, heteroaryl, cycloalkyl, cycloaikylalkyl-, heterocycloalkyl and heterocycloalkylaikyi-, alkylheteroaryl- and alkylary!-;
R20 represents alkyl;
R23 is selected from the group consisting of: H, alkyl, aryl, cycloaikyl, and cycloalkylalkyi-; each R26 is independently selected from the group consisting of: H and alkyl;
R28 is alkyi;
each R30 is independently selected from the group consisting of: H, alkyl, and F; each R32 is independently selected from the group consisting of: H and alkyl, and wherein each R32 is generally H; each R35 is independently selected from the group consisting of: H and Ci to C6 aikyl;
R36 is selected from the group consisting of: H, alkyl, and -O-alkyl; each R38 is independently selected from the group consisting of: H, alkyl, aryi, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalky! alkyl, heterocycloalkyl, heterocycloalkylalkyl, alkylheteroaryl-, alkylaryl-, substituted alkyl, substituted aryl, substituted arylaikyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloalkyl, substituted cycloalkylalkyl, substituted heterocycloalkyi, substituted heterocycloalkylalkyi, substituted alkylheteroaryl- and substituted alkylaryl-, and wherein: said R38 substituted alkyl is substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, -NO2, -CN, -OR26, halo, -C(O)-NH-R28, -C(O)OR28, and -C(O)R28, and said R3δ substituted aryl, substituted aryialkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloaikyl, substituted cycloalkylalkyl, substituted heterocycloalkyl, substituted heterocycloalkylalkyl, substituted alkylheteroaryl- and substituted alkylaryl- are substituted with 1 to 3 substituents independently selected from the group consisting of: (1) -NH2, (2) -NO2, (3) -CN, (4) -OH, (5) -OR20, (6) -OCF3, (7) -CF3, (8) -C(O)R26, (9) aikyl, (10) alkenyl, (11) halo, (12) -C(O)-NH-R26, (13) -C(O)OR26, (14) -C(O)-NR32-{C(R30)2)π-N(R26)2, (15) -S(O)1R26, (16) -C(O)N(R32J(R26), (17) -NR32C(O)R26,

(19) -NHR20;
R42 is selected from the group consisting of: alkyl, aryl, heteroaryl, and cycloalkyl; R44 is selected from the group consisting of: H, alkyl, cycloalkyl, and cycloaikylalkyl;
Each R46 is independently selected from the group consisting of: H, alkyi, cycloalkyl, and cycloalkylalkyl; and provided that:
(1) Q1 is a substituted ring wherein at least one substitutent is halo; and/or (2) R2 is substituted alkyl wherein said substituent is -OCH3, or R2 is selected from the group consisting of: -CH2OH and -CH2OCH3; and/or
(3) at least one of R3, R4, R5, R6, and R7 is:
(i) selected from the group consisting of: oxadiazolylphenyl-, pyridazinylphenyl-, pyrimidinylpyrazinyl-, substituted oxadiazolylphenyl-, substituted pyridazinylphenyl-, substituted pyrimidinylpyrazinyi-, and benzoheteroaryl; or (ii) selected from the group consisting of:
(a) substituted cycloalkyl,
(b) substituted cycloalkyialkyl-,
(c) substituted heterocycloaikyl, <d) substituted heterocycloalkylalkyl-,
(e) substituted arylheteroaryl-,
(f) substituted heteroarylaryl-,
(g) substituted aryl,
(h) substituted heteroaryl, (i) substituted heteroarylheteroaryl-,
G) substituted arylaminoheteroaryl-, (k) substituted arylalkynyl-, and (!) substituted heteroarylalkynyl-, and wherein at least one substitutent on at least one at least one of said (ii)(a) to (U)(I) R3, R4, R5, R6, or R7 group is selected from the group consisting of: -NHR20, -N(R20J2 (wherein each Rao is independently selected), and -OR20; and/or
(4) R5A is alkyl; and/or
(5) R10 is a substituted aryl, and at least one substitutent is selected from the group consisting of: (a) -S(O)tR38 wherein R38 is isopropyl,
(b) -O-alkyl-O-R20,
(c) hydroxyalkyl,

(e) -aikyl-OR20,
(f) -O-alkyl-OH,
(g) -NH(hydroxyalkyl), and (h) oxazolidinone; and/or
(6) R20 is isopropyl;
Formula Il is described in PCT publication No. 2007/070398, herein incorporated by reference; b) Formula III

II! or the pharmaceutically acceptable salts, esters or solvates thereof, wherein: z is 1 to 3; Q is a substituent selected from the group consisting of:

(2.20) (2.22)

Each Q1 represents a ring independently selected from the group consisting of: cycioalkyl, substituted cycioaikyl, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted aryl, heteroaryl, and substituted heteroaryl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group
consisting of: haio and the R10 moieties; provided that when Q1 is ary!, heteroaryl, substituted aryl or substituted heteroaryl then the carbon atoms at the ring junction are not substituted;
Q2 represents a ring selected from the group consisting of: cycloalkyl, substituted cycloalkyi, heterocycloalkyl, and substituted heterocycloalkyl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R10 moieties;
Z1 represents -{C(R24)2)W- wherein each R24 is independently selected from the group consisting of: H, alkyl and F, and wherein w is 1 , 2 or 3; Z2 is selected from the group consisting of: -N(R44)-, -O- and -C(R46)2-; m is 1 to 6; n is 1 to 6; p is 0 to 6; t is 0, 1 , or 2; R1 is selected from the group consisting of:
O) -CN,
(2) -NO2,
(3) -OR10,
(4) -SR10, (5) -N(R1V
(6) R10,
(7) -C(O)R10,
(8) -(C(R30)2)n-NR32-C(O)-R10, wherein in one example n is 1 , each R30 is H, R32 is H, and R10 is selected from the group consisting of: cycloalkyl and alkyl, (9) -(C(R30)2)n-NR32-S(O)rR10,
(10) -(C(R30)2)n-NR32-C(O)-N(R32}-R10,
(11)

(12) -CF3,
(13) -C(O)OR10,
(14) -(C(R30)2)nR13,
(15) alkenyl (e.g., -CH=CHCH3),
(16) -NR32-C(O)-R14, (17)



(20) -C(O)-NR32-(C(R30)2)p-OR10,
(21 ) -C(O)N(R10)2 wherein each R10 is independently selected, (22) -C(O)-NR32-C(R18)3,
(23) -C(O)-NR32-(C(R30)2)n-C(O)-N(R10)2l (24) heterocycloalkenyl, such as, for example:
 wherein r is 1 to 3, (25)
wherein r is 1 to 3, (25)
(26) arylalkenyl-, and
(27) halo;
R2 is selected from the group consisting of: (I) H1
(2) -CN,
(3) halo, (4) alkyl,
(5) substituted alkyl wherein said substituted alkyl is substituted with 1 to 3 substitutents selected from the group consisting of: (a) -OH, (b) -O-alkyl, (c) -O-alkyl substituted with 1 to 3 F atoms, and (d) -N(R40)2 wherein each R40 is independently selected from the group consisting of: (i) H, (ii) C1-C3 alkyl, (iii) -CF3, and (e) halo, (6) alkynyl,
(7) alkenyl,
(8) -(CH2UR1 \
(9) -N(R26)2) (1O) -OR23, (11 ) -N(R26)C(O)R42,
(12) cycloalkyl, (13) cycloalkylalkyl,
(14)

(15) -O-(substituted alkyl) wherein said substituted alkyl is substituted with 1 to 3 F atoms,
(16) -S(0)t-alkyl, (17) -C(O)-alkyl,
(18)
 (19) S ?
(19) S ?
 wherein each alkyl is independently selected, (20)
wherein each alkyl is independently selected, (20)
 wherein each alkyl is independently selected,
wherein each alkyl is independently selected,
(21)

(22) -N(R4^-C(O)-R48 wherein each R48 is independently selected from the group consisting of: H and aikyl, and
(23) -C(O)-alkyl; each R3, R4, R5, R6 and R7 is independently selected from the group consisting of:
O) H,
(2) aikenyl,
(3) substituted aikenyl,
(4) alkyl, (5) substituted alkyl,
(6) cycloalkyl,
(7) substituted cycloalkyl,
(8) cycloalkylalkyl-,
(9) substituted cycloalkylalkyl-, (10) heterocycloaikyl,
(1 1) substituted heterocycioalkyl,
(12) heterocydoalkyialkyl-,
(13) substituted heterocydoalkyialkyl-,
(14) -C(O)R10, (15) arylheteroaryl-,
(16) substituted arylheteroaryl-,
(17) heteroarylaryl-,
(18) substituted heteroaryiaryl-, (19) aryl,
(20) substituted aryl,
(21) heteroaryl, (22) substituted heteroaryl,
(23) heteroarylheteroaryl-,
(24) substituted heteroarylheteroaryi-,
(25) arylaminoheteroaryl-,
(26) substituted aryiaminoheteroaryl-, (27) arylalkynyl-,
(28) substituted arylalkynyl-,
(29) heteroarylalkynyl-,
(30) substituted heteroarylalkynyl-,
(31 ) benzoheteroaryl; wherein said R3, R4, R5, R6 and R7 substituted groups (7), (9), (11), (13), (16),
(18), (20), (22), (24), (26), (28) and (30) are substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, -NHR20, -N(R20)2 wherein each R20 is independently selected, alkyl, alkenyl, halo, -C(O)-NH-R28, -C(O)OR28, -C(O)R28, and -OR20, wherein said R3, R4, R5, R6 and R7 substituted groups (3) and (5) are substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, halo, -C(O)-NH-R28, -C(O)OR26, and -C(O)R28;
RSA is selected from the group consisting of: halo, -OH, alkyl, -O-alkyl;
R8 is selected from the group consisting of: H, -OH, -N(R10)2) -NR10C(O)R12; each R9 is independently selected from the group consisting of :halogen, -CN,
-NO2, -OR10, -SR10, -N(R10J2, and R10; each R10 is independently selected from the group consisting of: H, alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalkylalkyl, heterocycioalkyl, heterocycloalkylalkyl, alkylheteroaryl-, alkylaryl-, substituted alkyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloaikyl, substituted cycloalkylalkyl, substituted heterocycioalkyl, substituted
heterocycloaikylalkyi, substituted alkylheteroaryl-, substituted alkyiaryl-, heterocycloaikenyl


(19) -NHR20, (20) cycloalkyl, (21) -O-alkyi-O-R20, (22) hydroxyalkyl, (23) -N(R20J2 wherein each R20 is independently selected, (24) -alky!-OR20, (25) -O-aikyl-OH, (26) -NH (hydroxyalkyl), and (27) oxazolidinone;
R11 is selected from the group consisting of: F, -OH, -CN, -OR10, -NHNR1R10, -SR10 and heteroaryl;
R12 is selected from the group consisting of: alkyl, aryl, heteroaryl, cycioalkyl, cycloalkylalkyl, heterocycloalkyl and heterocycloaikylalkyi; R14 is selected from the group consisting of: aikyl, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl-, heterocycloalkyl, alkylheterocycloalkyl, heterocycloaikylalkyi-, afkylheteroaryt- and alkyiaryl-;
R15 is selected from the group consisting of: H, -OH, alkyi, aryl, heteroaryl, cycloalkyl, cycloalkylalkyl-, heterocycloaikyl and heterocycioalkylalkyl-, alkylheteroaryi- and aikylaryl-;
R20 represents alkyl; R23 is selected from the group consisting of: H, alkyl, aryl, cycloalkyl, and cycloalkylalkyl-; each R26 is independently selected from the group consisting of: H and alkyi;
R28 is alkyl; each R30 is independently selected from the group consisting of: H1 alkyl, and F; each R32 is independently selected from the group consisting of: H and alkyl; each R35 is independently selected from the group consisting of: H and Ci to C6 alkyl; each R38 is independently selected from the group consisting of: H, alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalkylalkyl, heterocycloalkyi, heterocycloalkylalky], alkylheteroaryl-, alkylaryl-, substituted alkyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloalkyl, substituted cycloalkylalkyl, substituted heterocycloaikyl, substituted heterocycioalkylalkyl, substituted alkylheteroaryl- and substituted alkylaryl-, and wherein: said R38 substituted alkyl is substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, -NO2, -CN, -OR26, halo, -C(O)-NH-R28, -C(O)OR28, and said R38 substituted aryl, substituted aryialkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloaikyl, substituted cycloalkyl alkyi, substituted heterocycloalkyi, substituted heterocycioalkylalkyl, substituted alkylheteroaryl- and substituted alkylaryl- are substituted with 1 to 3 substituents independently selected from the group consisting of: (1) -NH2, (2) -NO2, (3) -CN, (4) -OH, (5) -OR20, (6) -OCF3, (7) -CF3, (8) -C(O)R26, (9) alkyl, (10) alkenyi, (1 1 ) halo, (12) -C(O)-NH-R26, (13) -C(O)OR26, (14) -C(O)-NR32-(C(R30)2)n-N(R26)Ξ) (15) - S(O)1R26, (16) -C(O)N (R32KR26), (17) -NR32C(O)R26,
 and (19) -NHR20;
and (19) -NHR20;
R42 is selected from the group consisting of: alkyi, aryl (e.g., phenyl), heteroaryl, and cycloalkyi; R44 is selected from the group consisting of: H, alkyl, cycloalkyi, and cycloalkylaikyl; and
Each R46 is independently selected from the group consisting of: H, alkyl, cycloalkyi, and cycloalkylaikyl;
Formuia III is described in PCT publication No. 2008/156739, herein incorporated by reference; or c) Formula IV:

IV or the pharmaceutically acceptable salts thereof, wherein: Y1 , Y2, and Y3 are each independently selected from the group consisting of:
-CH=, -N= and -CR9=; z is 1 to 3; Q is a substituent selected from the group consisting of:

(2.9) (2. 0) (2.1 1 ) (2.12)
(2.13) (2.14) (2.15) (2.16)

Each Q1 represents a ring independently selected from the group consisting of: cycloalkyl, substituted cycloalkyl, heterocycioalkyl, substituted heterocycloalkyi, aryl, substituted aryl, heteroaryl, and substituted heteroaryl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R10 moieties; provided that when Q1 is aryl, heteroaryl, substituted aryl or substituted heteroaryl then the carbon atoms at the ring junction are not substituted;
Q2 represents a ring selected from the group consisting of: cycloalkyi, substituted cycloalkyi, heterocycJoalkyi, and substituted heterocycioalkyl, wherein said substituted rings are substituted with 1 to 3 substituents independently selected from the group consisting of: the R10 moieties;
Z1 represents -(C(R24)2)w- wherein each R24 is independently selected from the group consisting of: H, alkyl and F, and wherein w is 1 , 2 or 3, and generally w is 1 or 2, and usually w is 1 , and wherein in one example each R24 is H, and in another example w is 1 , and in another example each R24 is H and w is 1 , preferably w is 1 and each R24 is H;
Z2 is selected from the group consisting of: -N(R44)-, -O- and -C(R46J2-; m is 1 to 6; n is 1 to 6; p is 0 to 6; t is 0, 1 , or 2;
R1 is selected from the group consisting of: (I) -CN,
(2) -NO2,
(3) -OR10, (4) -SR10,
(5) -N(R10J2,
(6) R10,
(7) halo,
(8) -CF3; (9) alkenyl;
(10) -C(O)N(R10J2 wherein each R10 is independently selected, and preferably each R10 is independently selected from the group consisting of: (a) H, (b) alkyl, (c) heteroaryl, (d) aryl, and (e) cycloalkyl, wherein for example, each R10 is selected from the group consisting of: H, methyl, butyl, i-propyl, pyridyl, phenyl and cyclopropyl, wherein, for example, said -C(O)N(R10)2 moiety is selected from the group consisting of: -C(O)NH2, -C(O)NH(CH3), -C(O)NH(CH)(CH3)2J -C(O)NH(C4H9), -C(O)NH(C6H5), -C(O)NH(C3H5), and -C(O)NH(C5H4N);
(1 1) arylalkenyl-;
R2 is selected from the group consisting of: (1) H,
(2) -CN,
(3) halo,
(4) alkyi,
(5) substituted alkyl wherein said substituted alkyl is substituted with 1 to 3 substitutents selected from the group consisting of: (a) -OH, (b) -O-alkyl (e.g., -0-(Cr C3alkyl), (c) -O-alkyi (e.g., -O-(CrC3alkyl)) substituted with 1 to 3 F atoms, and (d) - N(R40)2 wherein each R40 is independently selected from the group consisting of: (i) H, (ii) CrC3 alkyl and (iii) -CF3,
(6) alkynyl,
(7) alkenyl, <8) -(CH2)mR1\ (9) -N(R26)2,
(1 O) -OR23, (1 1 ) -N(R26)C(O)R42,
(12) cycloalkyl,
(13) cycloalkylalkyl, and (14)
 each R3, R4, R5, R6 and R7 is independently selected from the group consisting of:
each R3, R4, R5, R6 and R7 is independently selected from the group consisting of:
O) H, (2) alkenyl,
(3) substituted alkenyl,
(4) alkyl,
(5) substituted aikyl,
(6) cycloalkyl, (7) substituted cycloalkyl ,
(8) cycloalkylalkyl-,
(9) substituted cycloalkylalkyl-,
(10) heterocycloalkyl,
(11 ) substituted heterocycloalkyl, (12) heterocycloalkylalkyl-,
(13) substituted heterocycloalkylalkyl-,
(14) -C(O)R10 wherein in one example R10 is selected from the group consisting of: alkyl,
(15) arylheteroary1-,
(16) substituted arylheteroaryl-, (17) heteroarylaryi-, such as, for example, pyrimidinylphenyl-, pyrazinylphenyl-, pyridinylphenyl-, furanylphenyl-, thienylphenyl-, and thiazolylphenyl-,
(18) substituted heteroarylaryi-, such as, for example, substituted pyrimidinylphenyl-, substituted pyrazinylphenyl-, substituted pyridinyiphenyl-, substituted furanylphenyl-, substituted thienylphenyl-, substituted thiazolylphenyl-, and substituted pyrimidinylphenyl,
(19) aryl,
(20) substituted aryl,
(21) heteroaryl,
(22) substituted heteroaryl, (23) heteroaryiheteroaryl-,
(24) substituted heteroaryiheteroaryl-,
(25) arylaminoheteroaryl-,
(26) substituted aryiaminoheteroaryl-,
(27) arylalkynyl-, (28) substituted arylalkynyl-,
(29) heteroaryialkynyl-,
(30) substituted heteroaryialkynyl-, (3I) -C(O)NHR28,
(32) cycloaikylheteroarylaryl-, (33) substituted arylaryh
(34) arylalkenylaryh
(35) arylary!-,
(36) substituted arylaikyl-,
(37) arylaikyl-, (38) -SO2aryl,
(39) benzoheteroaryl-C(O)-(substituted heterocycloalkyl)-,
(40) substituted heterocycloalkyl,
(41) heterocycloalkyl-C(O)-alky!-, and
(42) benzo[1 ,3]dioxolyl, wherein said R3, R4, R5, R6 and R7 substituted groups (7), (9), (11 ), (13), (16), (18), (20), (22), (24), (26), (28), (30), (33), (36), (39) and (40) are substituted with 1 to 3 substituents independently selected from the group consisting of: -CH2OH, CN, -OH1 -NH2, aikyl, alkenyl, halo, -C(O)-NH-R28, -C(O)NH2, -C(O)OR28, -C(O)R28, -C(alky!)=NOH, -C(alkyi)=NO(alkyl), alkoxy, hydroxy! substituted alkyi, dialkylamine wherein each aikyl group is independently selected, -CF3, -S02alkyl, and -NHC(O)H, wherein said R3, R4, R5, R6 and R7 substituted groups (3) and (5) are substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, halo, -C(O)-NH-R28, -C(O)OR28, and -C(O)R28;
R8 is selected from the group consisting of: H, -OH, aikyl, aryl, -N(R10J2 and -NR1°C(O)R12; each R9 is independently selected from the group consisting of:halogen, -CN, -NO2, -OR10, -SR10, -N(R10J2, and R10; each R10 is independently selected from the group consisting of; H, aikyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloalkyl, cycloalkyl aikyl, heterocycloalkyl, heterocycloalkylalkyl, alkylheteroaryl-, alkylaryl-, substituted aikyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloalkyS, substituted cycloalkylalkyl, substituted heterocycloalkyl, substituted heterocycioalkylalkyl, substituted alkylheteroaryl- and substituted alkylaryl-, and wherein: said R10 substituted aikyl is substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, -NHR20, -NO2, -CN, -OR26, halo, -C(O)-NH-R26, -C(O)OR26, and -C(O)R26, and said R10 substituted aryl, substituted aryialkyl, substituted heteroaryl, substituted heteroarylalkyi, substituted cycloalkyl, substituted cycloalkylalkyl, substituted heterocycloalkyl, substituted heterocycloalkylalkyl, substituted alkylheteroaryl- and substituted alkylaryl- are substituted with 1 to 3 substituents independently selected from the group consisting of: (1 ) -NH2, (2) -NO2, (3) -CN,
(4) -OH, (5) -OR20, (6) -OCF3, (7) -CF3, (8) -C(O)R38, (9) aikyl, (10) alkenyl, (1 1 ) halo,
(12) -C(O)-NH-R26, (13) -C(O)OR38, (14) -C(O)-NR32-(C(R30)2)π-N(R38)2, (15) -S(O)tR38, (16) -C(O)-NR32-R38, (17) -NR32-C(O)-R38,

(19) -NHR20; R1Ms selected from the group consisting of: F, -OH, -CN, -OR10, -NHNR1R10,
-SR10 and heteroaryl;
R12 is selected from the group consisting of: alkyi, aryl, heteroaryi, cycloalkyl, cyctoalkylalkyl, heterocycloalkyl and heterocycloalkylalkyl;
R14 is selected from the group consisting of: aikyl, aryl, heteroaryl, cycloalkyi, cycloalkylalkyl-, heterocycloalkyl, alkylheterocycloalkyl, heterocycloalkylalkyl-, alkylheteroaryl- and alkyiaryl-;
R15 is selected from the group consisting of: H, -OH, alkyl, aryl, heteroaryl, cycioalkyl, cycloalkylalkyl-, heterocycloalkyl and heterocycloalkylalkyl-, alkylheteroaryl- and alkyiaryl-; R20 represents alkyl;
R23 is selected from the group consisting of: H, alkyl, aryl, cycloalkyl, and cycloalkyialkyl-; each R26 is independently selected from the group consisting of: H and alkyl; R28 is alkyl; each R30 is independently selected from the group consisting of: H, afkyl, and
F; each R32 is independently selected from the group consisting of: H and alky!; each R35 is independently selected from the group consisting of: H and C1 to C6 alkyl; R36 is selected from the group consisting of: H and alkyl; each R38 is independently selected from the group consisting of: H, alkyl, aryl, arylalkyl, heteroaryl, heteroaryl alkyl, cycloalkyS, cycloalkylalkyl, heterocycloalkyl, heterocycloalkylalkyl, alkylheteroaryl-, alkyiaryl-, substituted alkyl, substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyl, substituted cycloaikyl, substituted cycloalkylalkyl, substituted heterocycloalkyl, substituted
heterocycioalkylalkyl, substituted alkylheteroaryl- and substituted alkylaryl-, and wherein: said R38 substituted alkyl is substituted with 1 to 3 substituents independently selected from the group consisting of: -NH2, -NO2, -CN, -OR26, halo, -C(O)-NH-R28 , -C(O)OR28, and -C(O)R28, and said R38 substituted aryl, substituted arylalkyl, substituted heteroaryl, substituted heteroarylalkyi, substituted cycloalkyi, substituted cycloalkylalkyl, substituted heterocycloalkyl, substituted heterocycloalkylaikyl, substituted alkylheteroaryl- and substituted alkylaryl- are substituted with 1 to 3 substituents independently selected from the group consisting of: (1) -NH2, (2) -NO2, (3) -CN, (4) -OH, (5) -OR20, (6) -OCF3, (7) -CF3, (8) -C(O)R26, (9) alkyl, (10) alkenyl, (11) halo, (12) -C(O)-NH-R26, (13) -C(O)OR26, (14) -C(O)-NR32-(C(R30)2)n-N(R26)2, (15) -S(O)1R26, (16) -C(O)N(R32J(R26), (17) -NR32C(O)R26,
 (19) -NHR20;
(19) -NHR20;
R42 is selected from the group consisting of: alkyl, aryi, heteroaryl, and cycloalkyi;
R44 is selected from the group consisting of: H, alkyl, cycloaikyl, and cycloalkyialkyl; and Each R46 is independently selected from the group consisting of: H, alkyl, cycloalkyi, and cycloalkylalkyi;
Formula IV is described in U.S. publication No. 2007/0232610, herein incorporated by reference.
Non-limiting examples of ERK inhibitors are selected from the group consisting of:































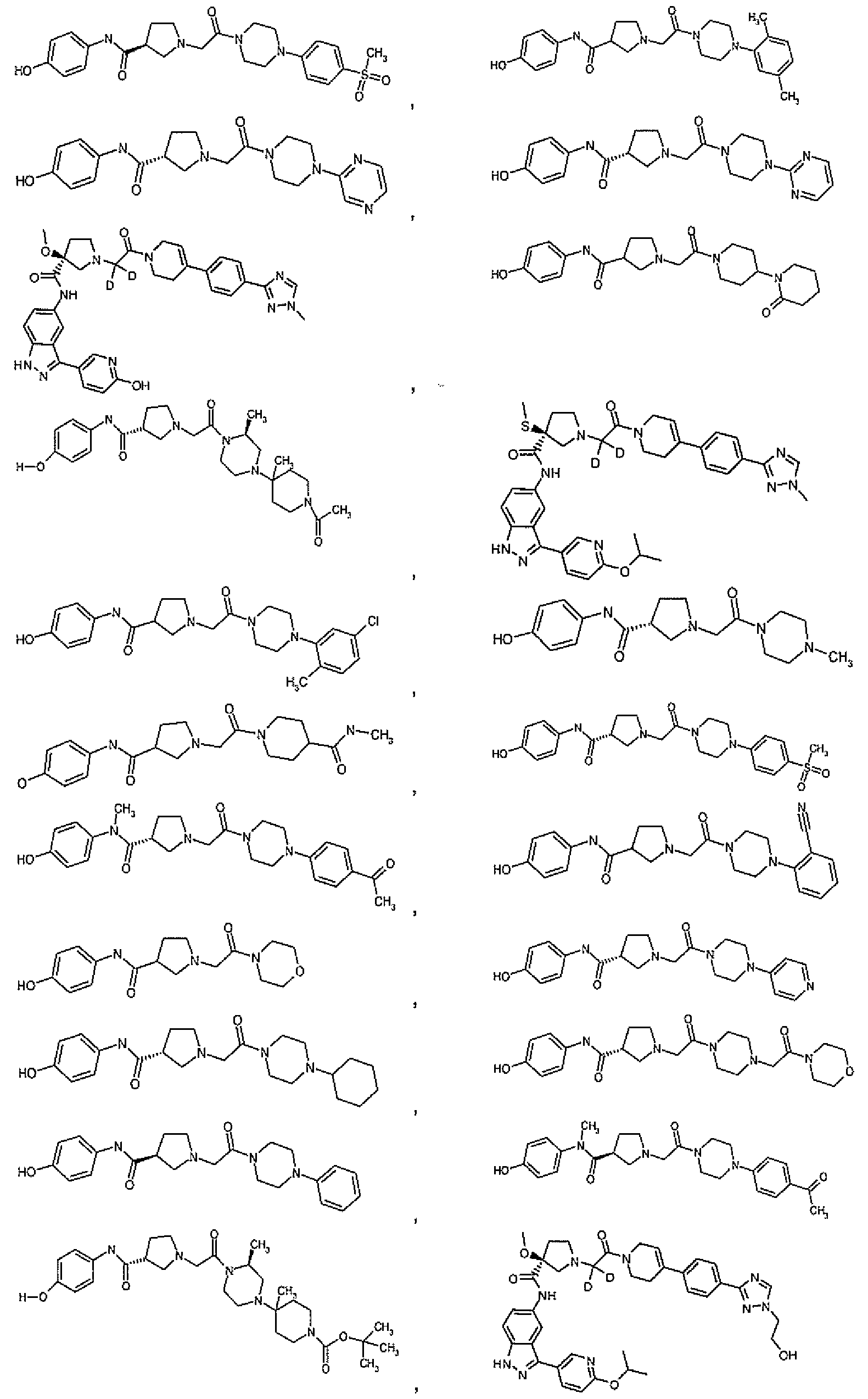


These compounds are described in PCT publication No. WO 2007/070398, U.S. application No. US 61/030407, PCT publication No. WO 2008/156739 and U.S. publication No. 2007/0232610, herein incorporated by reference. The pharmacoiogical properties of the compounds of this invention may be confirmed by a number of pharmacological assays. The exemplified pharmacological assays which are described herein below have been carried out with compounds according to the invention and their salts, solvates, esters or prodrugs.
This invention is also directed to pharmaceutical compositions which comprise at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and at least one pharmaceutically acceptable carrier.
For preparing pharmaceutical compositions from the compounds described by this invention, inert, pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, dispersible granules, capsules, cachets and suppositories. The powders and tablets may be comprised of from about 5 to about 95 percent active ingredient. Suitable solid carriers are known in the art, e.g., magnesium carbonate, magnesium stearate, talc, sugar or lactose. Tablets, powders, cachets and capsules can be used as solid dosage forms suitable for oral administration. Examples of pharmaceutically acceptable carriers and methods of manufacture for various compositions may be found in A. Gennaro (ed.), Remington's Pharmaceutical Sciences, 18th Edition, (1990), Mack Publishing Co., Easton, Pennsylvania.
Liquid form preparations include solutions, suspensions and emulsions. As an example may be mentioned water or water-propylene glycol solutions for parenteral injection or addition of sweeteners and opacifiers for oral solutions, suspensions and emulsions. Liquid form preparations may also include solutions for intranasal administration.
Aerosol preparations suitable for inhalation may include solutions and solids in powder form, which may be in combination with a pharmaceutically acceptable carrier, such as an inert compressed gas, e.g. nitrogen. Also included are solid form preparations that are intended to be converted, shortly before use, to liquid form preparations for either oral or parenteral administration. Such liquid forms include solutions, suspensions and emulsions.
The compounds of the invention may also be deliverable transdermally. The transdermal compositions can take the form of creams, lotions, aerosols and/or emulsions and can be included in a transdermal patch of the matrix or reservoir type as are conventional in the art for this purpose.
The compounds of this invention may also be delivered subcutaneously. Preferably the compound is administered orally or intravenously. Also contemplated are delivery methods that are combinations of the above- noted delivery methods, Such methods are typically decided by those skilled in the art.
Preferably, the pharmaceutical preparation is in a unit dosage form. In such form, the preparation is subdivided into suitably sized unit doses containing
appropriate quantities of the active component, e.g., an effective amount to achieve the desired purpose.
The quantity of active compound in a unit dose of preparation may be varied or adjusted from about 1 mg to about 100 mg, preferably from about 1 mg to about 50 mg, more preferably from about 1 mg to about 25 mg, according to the particular application.
The actual dosage employed may be varied depending upon the requirements of the patient and the severity of the condition being treated. Determination of the proper dosage regimen for a particular situation is within the skill of the art. For convenience, the total daily dosage may be divided and administered in portions during the day as required.
The amount and frequency of administration of the compounds of the invention and/or the pharmaceutically acceptable salts thereof will be regulated according to the judgment of the attending clinician considering such factors as age, condition and size of the patient as well as severity of the symptoms being treated. A typical recommended daily dosage regimen for oral administration can range from about 1 mg/day to about 500 mg/day, preferably 1 mg/day to 200 mg/day, in two to four divided doses.
Another aspect of this invention is a kit comprising a therapeutically effective amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and a pharmaceutically acceptable carrier, vehicle or diluent.
Yet another aspect of this invention is a kit comprising an amount of at least one compound of the present invention, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and an amount of at least one anticancer therapy and/or anti-cancer agent listed above, wherein the amounts of the two or more ingredients result in desired therapeutic effect.
The invention disclosed herein is exemplified by the following preparations and examples which should not be construed to limit the scope of the disclosure. Alternative mechanistic pathways and analogous structures will be apparent to those skilled in the art.
Where LC/MS data are presented, analyses was performed using an Applied
Biosystems API-100 mass spectrometer and Shimadzu SCL-10A LC column: Altech platinum C18, 3 micron, 33mm x 7mm ID; gradient flow: 0 min - 10% CH3CN, 5 min -
95% CH3CN, 7 min - 95% CH3CN, 7.5 min - 10% CH3CN1 9 min - stop. The retention time and observed parent ion are given.
The following solvents, reagents and reaction conditions may be referred to by their abbreviations:
Aq: aqueous g: grams psi: pounds per square inch pH: percent Hydrogen
°C: degrees Celsius h: hours
THF: Tetrahydrofuran HMDS:
Et2O: diethyloxide
SEM: 2-(trimethylsilyi)ethoxymethyl
LC-MS: Liquid chromatography mass spectrometry
DCM: dichloromethane N: Normal mS: milliliter
NBS: N-Bromosuccinimide it: room temperature
MeOH: methanol DIEA: diisopropylethylamine
EtOAc: ethyl acetate
EtOH: ethanol
DMF: dimethylformamide
WT%: weight percent m/z: mass per charge
LiOH: lithium hydroxide
DMSO: dimethylsulfoxide
HPLC: high performance liquid chromatography
Ret: retention
RP: reverse phase
CH3CN: acetonitriie MeCN: acetonitriie pTSA: para-to!uene sulfonic acid
RT: retention time
NaOH: sodium hydroxide
CDI: N,N'-carbonyldiimidazole mg: milligram
PMA: phosphomolybdic acid
CO2: carbon dioxide
LiHMDS: lithium hexamethyldisilazane
HMDS: hexamethyldisϋazane Pd/C: palladium on carbon
H2: hydrogen gas
NIS: N-iodosuccinimide
PDCI2(dppf): [1 ,1 '-bis(diphenylphosphino)ferrocene] dichloropalladium(il) μmol: micromole TFA: trifluoroacetic acid
NMP: N-methyl-2-pyrrolidone min: minute
NaIO4: sodium periodate
DME: dimethylethane OsO4: osmium tetroxide
Na2S2Og: sodium thiosulfate
AcOH: acetic acid
NaBH3CN: sodium cyanoborohydride
H2O: water BBN: 9-borabicyclo[3.3.1]nonane
CH2CI2: dichloromethane
BOC: tertiary-butyloxycarbonyi
POCI3: phosphorous oxychloride
NaHCO3: sodium bicarbonate
NH4CI: ammonium chloride
Na2SO4: sodium sulphate HC!: hydrogen chloride
M: Molar mmol: miilimolar
NH3: ammonia
DIEA; diisopropylethylamine Bu3SnCN: tributyltin cyanide
Pd[P(t-Bu)3]2: bis(tributyl)Phosphine) palladium
Pd(PPh3)4: tetrakis(triphenylphosphine) palladium
K2CO3: potassium carbonate
EDC: 1 -ethyl-3-(3-dimethylaminopropyl)-carbodiimide UV: ultraviolet
K3PO4: potassium phosphate
LDA: lithium diisopropylamide
Tf: trifluoromethanesulfonyl
NaH: sodium hydride

Scheme 1
Pvridine-2.5-dicarboxvlic acid 2-tert-butv) ester 5-methvi ester

5-{Methoxycarbony!)pyridine-2-carboxylic acid (7.72 g, 42.65 mmol) was suspended in tert-butanol {70 mL) and pyridine (25 ml_) and cooled in an ice-water bath. 4- Toluenesulfonyl chloride (19.4 g, 102 mmol) was added in one portion and the mixture was stirred 30 minutes in the ice-water bath. The reaction mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was then slowly poured into a stirring mixture of saturated aqueous sodium bicarbonate (300 mL) and ethyl ether (150 mL). The resulting two-phase mixture was then extracted with ethyl ether (3x150 mL). The extracts were combined, washed with brine, dried with anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude product (5.8 g, 51% yield) was used in the next step without further purification.
Piperidine-2,5-dicarboxylic acid 2-tert-butyl ester 5-methyl ester

Pyridine-2,5-dicarboxylic acid 2-tert-buty! ester 5-methyl ester (5.8 g, 24.45 mmol) was dissolved in glacial acetic acid (30 mL) and hydrogenated at 50-60 psi for 3 days with 10% palladium on carbon catalyst (0.6 g). The reaction mixture was filtered through a pad of Ceiite which was then washed with methanol. The filtrates were combined and concentrated under reduced pressure. The residue was dissolved in water (100 mL) and solid sodium carbonate (15 g) was added to bring the pH to 8. The solution was extracted with dichloromethane (2x100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the title compound (5.2 g, 87%).
Piperidine-1 ,2,5-tricarboxylic acid 1 ,2-di-tert-butyl ester 5-methyl ester

Piperidine-2,5-dicarboxyiic acid 2-tert-butyl ester 5-methyl ester (9.0 g, 37.0 mmol) and triethylamine (15.5 ml, 111 mmol) were combined in dichloromethane (70 ml_) and cooled to 0 °C. Di-tert-butyldicarbonate (12.2 g, 55.5 mmol) was added and reaction mixture was warmed to room temperature and stirred for 16 h. The reaction mixture was diluted with water (100 ml_) and extracted with dichloromethane (2x100 ml_). The combined extracts were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by Biotage chromatography column (hexanes/EtOAc, 9:1 to 8:2). A colorless oil was obtained (12.6 g, 99%).
1.6-bisftert-butoxycarbonyl)piperidine-3-carboxylic acid
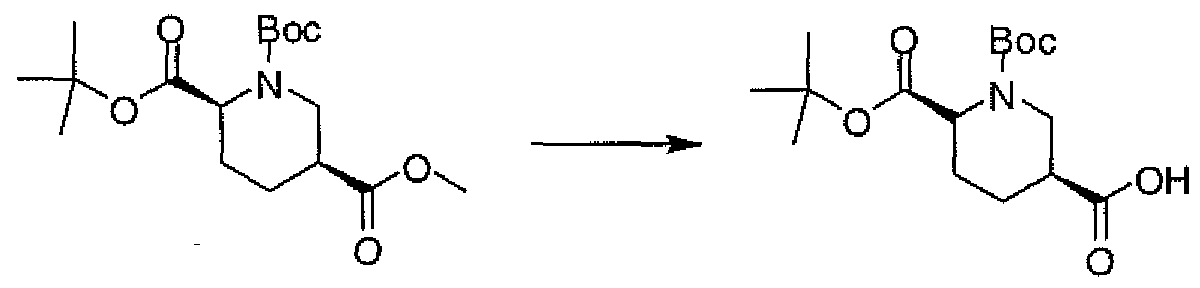
Di-tert-butyl 5-(3-ethoxy-3-oxopropanov!)piperidine-1,2-dicarboxvlate
 i jδ-bisttert-butoxycarbonylJpiperidine-3-carboxylic acid (41.4 mmol, 13.6 g) and N, N'- carbonyldiimidazole (CDI) (51.75 mmol, 8.4 g) in anhydrous THF (140 mL) were stirred 16 h at room temperature under argon. In a separate, sealed and argon- flushed flask, lithium HMDS (1.0 M in THF, 86.9 mmol) is added to 80 mL anhydrous THF stirring at -78° C. To this solution is added dropwise anhydrous ethyl acetate (89 mmol, 8.69 mL). This solution is allowed to stir at -78° C for 1 hour prior to the dropwise addition of the original CDi/acid solution that had been stirred overnight. The reaction mixture is allowed to stir and warm to room temperature overnight. The reaction is then quenched with saturated NH4C!(aq) (250 mL) and extracted with Et2O (100x2). The combined organics are then washed with water, saturated brine, and dried over Na2SO4. The solvent is removed in vacuo and the residue (12.6 g) was used in the next step without further purification.
i jδ-bisttert-butoxycarbonylJpiperidine-3-carboxylic acid (41.4 mmol, 13.6 g) and N, N'- carbonyldiimidazole (CDI) (51.75 mmol, 8.4 g) in anhydrous THF (140 mL) were stirred 16 h at room temperature under argon. In a separate, sealed and argon- flushed flask, lithium HMDS (1.0 M in THF, 86.9 mmol) is added to 80 mL anhydrous THF stirring at -78° C. To this solution is added dropwise anhydrous ethyl acetate (89 mmol, 8.69 mL). This solution is allowed to stir at -78° C for 1 hour prior to the dropwise addition of the original CDi/acid solution that had been stirred overnight. The reaction mixture is allowed to stir and warm to room temperature overnight. The reaction is then quenched with saturated NH4C!(aq) (250 mL) and extracted with Et2O (100x2). The combined organics are then washed with water, saturated brine, and dried over Na2SO4. The solvent is removed in vacuo and the residue (12.6 g) was used in the next step without further purification.

Scheme 2
Di-tert-butyl 5-(7-hydroxypyrazoio[1 ,5-alpyrimidin-5-vθpiperidine-1 ,2-dicarboxylate

Di-tert-butyl 5-(3-ethoxy-3-oxopropanoyl)piperidine-1,2-dicarboxylate (18.79 mmol, 7.5
 g) and 3-amino-1 H-pyrazole (15.0 mmol, 1.25g) were mixed and heated neat at 100° C for 16 hours. The resulting residue is dissolved in dichloromethane (100 ml.) and concentrated in vacuo to remove water formed during cyclization. LC-MS: 419 [M+H], This solid is taken forward without further purification.
g) and 3-amino-1 H-pyrazole (15.0 mmol, 1.25g) were mixed and heated neat at 100° C for 16 hours. The resulting residue is dissolved in dichloromethane (100 ml.) and concentrated in vacuo to remove water formed during cyclization. LC-MS: 419 [M+H], This solid is taken forward without further purification.
Di-tert-butyl 5-(7-chloropyrazolori ,5-a1pyrimidin-5-yl)piperidine-1 ,2-dicarboxyiate
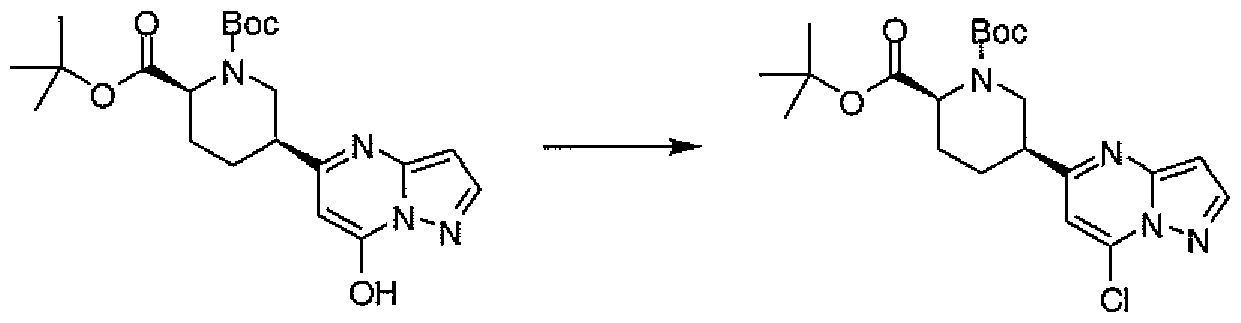
Di-tert-butyl 5-(7-aminopyrazoloH ,5-a1pyrimidin-5-yl)piperidine-1 ,2-dicarboxylate

Di-tert-butyl 5-(7-ch!oropyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1 ,2-dicarboxylate was dissolved in 30 mL ~7N ammonia in methanol in a sealed vessel. The reaction mixture
was heated at 80° C for 16 hours. After 16 hours, the reaction mixture is cooled to room temperature and concentrated in vacuo to yield a brown solid. LC-MS: 418 [M+H]. The title compound was used in the next step without further purification.
Di-tert-butyl 5-(7-(bisff2-(trimethylsiiyl)ethoxy)methyl)amino)pyrazoloπ .5-aipyrimidin- 5-yl)piperidine-1 ,2-dicarboxvlate

Di-tert-butyl 5-(7~aminopyrazolo[1 ,5-a]pyrimidin-5-yi)piperidine-1 ,2-dicarboxylate (5.5 g, 13.18 mmol) was dissolved in 1 ,2-dichloroethane {60 ml_). To this solution was added N, N'-diisopropylethylamine (92.28 mmol, 16.1 ml_). The resulting solution was stirred at room temperature while 2-(tritnethylsilyi)ethoxymethyl chloride (SEM-CI, 8.14 mL, 46.14 mmol) was added dropwise. After the addition is completed, the reaction mixture was stirred at 90° C for 2 h. The solvent is removed in vacuo and the residue is purified on silica gel column (0% to 60% ethyl acetate in hexanes gradient) to yieid the title compound (3.8 g, 34% yield over three steps) as pale yellow oil. LC-MS: 678 [M+H].
Di-tert-butyl 5-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolori ,5- aipyrimidin-5-yl)piperidine-1 ,2-dicarboxylate

N-lodosuccinimide (1.26 g, 5.61 mmol, dissolved in 25 ml acetonitrile) was added into a solution of di-tert-buty! 5-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5- a]pyrimidin-5-yl)piperidine-1 ,2-dicarboxylate (3.8 g, 5.61 mmol) in acetonitrile (26 mL).
The reaction mixture was stirred at room temperature for 16 hours. The reaction mixture was concentrated in vacuo and purified via silica gel chromatography (0% to 30% ethyl acetate in hexanes gradient) to yield the title compound (3.16 g, 70% yield) at clear oil. LC-MS: 804 [M+H].
Di-tert-butyl 5-(7-(bisf(2-(trimethylsilyl)ethoxy)methyl)amino)-3-(Quinolin-3- yl)pyrazoloM ,5-a]pyrimidin~5-yl)piperidine-1 ,2-dicarboxylate

3-Quinoline boronic acid (1.25 mmol, 216 mg), K3PO4 (1.87 mmol, 400 mg), and PdCI2(dppf) • CH2CI2 (0.062 mmol, 51 mg) was added to a solution of di-tert-butyl 5-(7- (bis((2-(trimethytsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yi)piperidine-1 ,2-dtcarboxylate (0.62 mmol, 500 mg) in dioxane (5 ml_). To this suspension was added distilled H2O (0.5 ml_). The reaction mixture was stirred at 100° C under an argon atmosphere for 18 hours. The reaction mixture was concentrated in vacuo and then purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield the title compound (450 mg, 90% yield) as yellow oil. LC-MS: 805 [M+H].
5^-amino-3-(quinolin-3-yl)pyrazolofi .5-aipyrimidin-5-yl)piperidine-2-carboxyiic acid

Di-tert-buty! 5-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)pipehdine-1 ,2-dicarboxylate (45 mg, 0.056 mmol) was
dissolved in ethanol (1 ml) and treated with 3N hydrochloride solution (1.4 ml) at 65 °C for 4 h. The reaction solution was concentrated and purified by prep-LC to afford the title compound (9.6 mg): LC/MS RT = 2.01 min. Mass calculated for, M+H 389.17, observed 389.17.
By essentially the same procedure given in Scheme 2, the compounds listed in Table 1 can be prepared.
Table 1




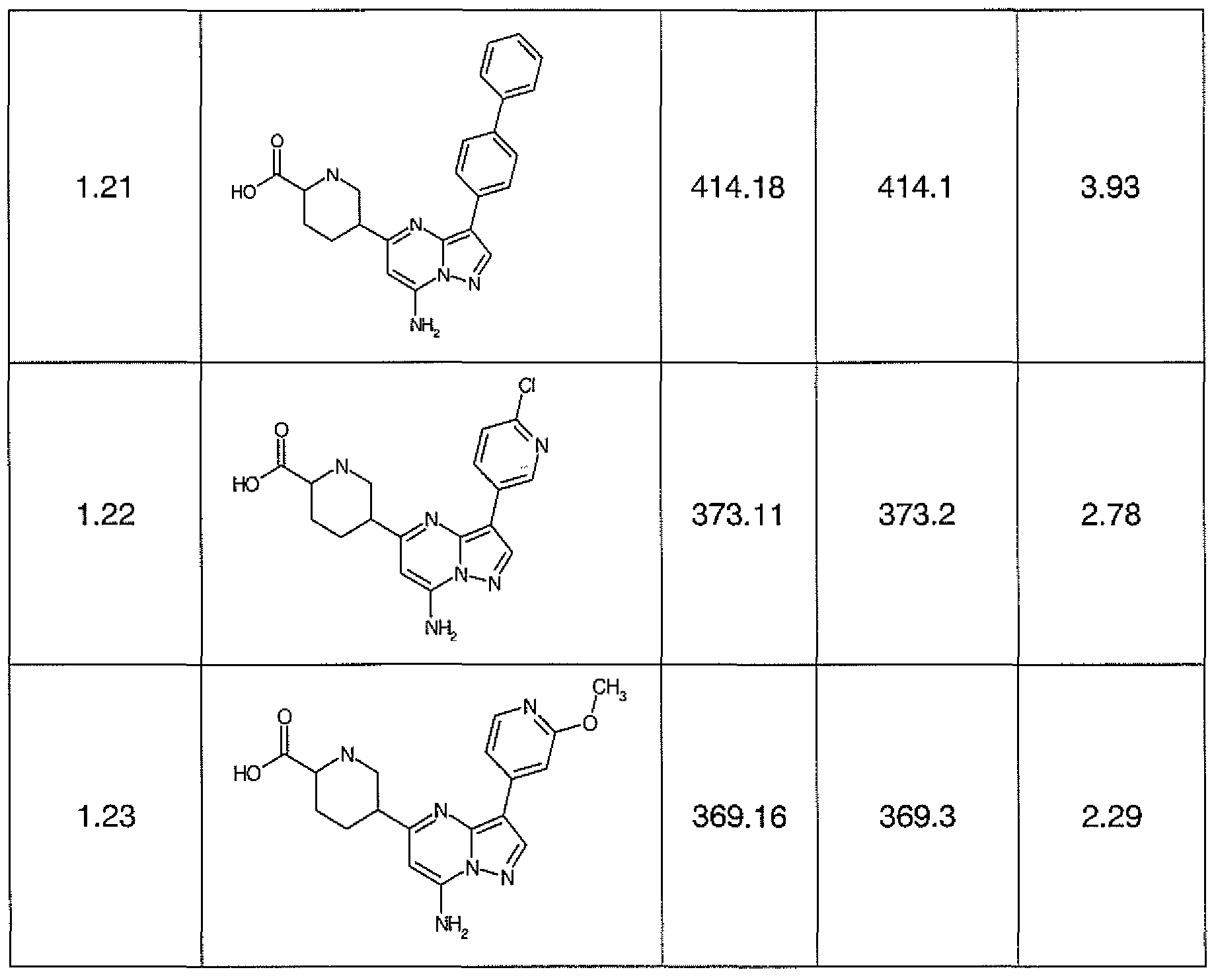

Scheme 3 Methyl 5-(7-amino-3-(quinoiin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-2-carboxylate

Di-tert-butyl 5-(7-(bis({2-(trimethylsilyi)ethoxy)m8thyl)amino)-3-(quino!in-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1,2-dicarboxylate (370 mg, 0.46 mmol) in methanol (12.6 mL) was treated with 4N hydrogen chloride in dioxane (5.4 ml). The reaction mixture was heated at 80 °C for 16 h, cooled to room temperature and concentrated in vacuo. The residue was purified by prep-LC to afford the title compound: LC/MS RT = 2.31 min. Mass calculated for, M+H 403.18, observed 403.18.
Methyl 5-(7-amino-6-bromo-3-(αuinoiin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)pipeπdine-2- carboxylate
N-bromosuccinimide (44 mg, 0.249 mmol) was added into a solution of methyl 5-(7- amino-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-2-carboxy!ate (100 mg, 0.249 mmol) in acetonitrite (1.5 mL) and methanol (0.5 ml). The resulting solution is stirred at room temperature for 1 hour. The reaction was concentrated in vacuo and purified by prep-LC to afford the title compound (22.32 mg): LC/MS RT = 2.56 min. Mass calculated for, M+H 481.09, observed 481.09.

Methyl 5-(7-amino-6-bromo-3-(quinoiin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-2- carboxylate (1 1 mg, 0.023 mmol) in 2:1 THFiH2O (1.5 ml_) was treated with 2N NaOH(aq) (0.25 ml_). The resulting solution was stirred at room temperature for 18 hours. This solution is reduced in vacuo and purified by prep-LC to afford the title compound (8.9 mg): LC/MS RT = 2.16 min. Mass calculated for, M+H 467.08, observed 467.08.
By essentially the same procedure given in Scheme 3, the compounds listed in Table 2 can be prepared.
Table 2




Scheme 4 5-(7"Amino-6-bromo-3-quinoiin-3-yl-pyrazoiof1 ,5-a1pyrjmidin-5-yl)-1 -(2-methoxy- acetyl)-piperidine-2-carboxylic acid
A mixture of 5-(7-amino-6-bromo-3-quinolin-3-yl-pyrazolo[1 ,5-a]pyrimidin-5-yi)- piperidine-2-carboxylic acid methyl ester (20 mg, 0.042 mmol), methoxy-acetyl chloride (13 mg, 0.071 mmol) and triethylamine (0.024 ml, 0.167 mmol) in THF:DMF (0.5 ml:0.5 ml) was stirred at room temperature for 16 h. The crude mixture was treated with 2.0 N sodium hydroxide solution (0.14 ml) at room temperature for 16 h. The reaction mixture was acidified with 1.0 N aqueous hydrogen chloride solution (0.5 ml), concentrated and purified by prep-LC to afford the title compound (7.94 mg): LC/MS RT = 3.06 min. Mass calculated for, M+H 539.10, observed 539.10. By essentially the same procedure given in Scheme 4, the compounds listed in Table 3 can be prepared.
Table 3





Scheme 5
4-(2-Ethoxycarbonyl-acetylVcvclohexanecarboxyiic acid methyl ester

Cyclohexane-1 ,4-dicarboxylic acid monomethyl ester (26.9 mmol, 5.0 g) and N, N'- carbonyldiimidazole (33.6 mmol, 5.44 g) in anhydrous THF (80 ml_) were stirred 16 h at room temperature under argon. In a separate, sealed and argon-flushed flask, lithium HMDS (1.0 M in THF1 56.5 ml) is added to 40 mL anhydrous THF stirring at - 78° C. To this solution is added dropwise anhydrous ethyl acetate (57.8 mmol, 5.65 mL). This solution is allowed to stir at -78° C for 1 hour prior to dropwise addition of original CDI/acid solution that had been stirring overnight. The reaction mixture was allowed to stir and warm to room temperature overnight. The reaction is then quenched with saturated NH4CI(aq) (200 mL) and extracted with Et2O (100x2). The combined organics are then washed with water, saturated brine, and dried over Na2SO4. The solvent was removed in vacuo and the crude residue (8.5 g) was used in the next step without further purification.
4-(7-Hvdroxy-pyrazoloh ,5-aipyrimidin-5-yl)-cvclohexanecarboxylic acid methyl ester

4-{2-Ethoxycarbony[-acetyl)-cyc!ohexanecarboxylic acid methyl ester (8.5 g) and 3- amino-1 H-pyrazole (26.5 mmol, 2.2 g) were mixed and heated neat at 100° C for 16 hours. The resulting residue is dissolved in dichloromethane (100 mL) and concentrated in vacuo to remove water formed during cyciization. This solid is taken forward without further purification. LC-MS: 403 [M+HJ.
4-(7-Chloro-pyrazolo[1 ,5-aipvrimidin-5-vl)-cyclohexanecarboxvlic acid methyl ester

4-(7-Amino-pyrazolori ,5-a1pyrimidin-5-yl)-cvciohexanecarboxylic acid methyl ester

4-(7-Chloro-pyrazolo[1 ,5-a]pyrimidin-5-yl)-cyclohexanecarboxylic acid methyl ester (2.5 g, 8.51 mmol) was dissolved in 15 mL ~7N ammonia in methanol in a sealed
vessel. The reaction mixture was heated at 80° C for 16 hours. After 16 hours, the reaction mixture is cooled to room temperature and concentrated in vacuo to yield a brown solid (2.58 g). The title compound was used in the next step without further purification. LC-MS: 275 [M+H].
4-(7-|-Bis-(2-trimethylsilanyl-ethoxymethyl)-amino1-pyrazolo[1 ,5-aiPyrimidin-5-yll- cvclohexanecarboxylic acid methyl ester

4-(7-Amino-pyrazolo[1 ,5-a]pyrimidin-5-yl)-cyclohexanecarboxylic acid methyl ester (2.58 g, 9.4 mmoi) was dissolved in 1 ,2-dichloroethane (20 mL). To this solution was added N, N'-diisopropylethylamine (65.8 mmol, 11.5 mL). The resulting solution was stirred at room temperature while 2-(trimethylsilyl)ethoxymethyl chloride (SEM-CI, 5.8 mL, 33.0 mmol) was added dropwise. After the addition was completed, the reaction mixture was stirred at 90° C for 2 h. The solvent is removed in vacuo and the residue is purified on silica gel column (0% to 60% ethyl acetate in hexanes gradient) to yield the title compound (2.05 g, 41% yield over two steps) as pale yellow oil. LC-MS: 535 [M+H].
4-f7-rBis-(2-trimethylsilanyl-ethoxymethyl)-aminol-3-iodo-pyrazoloπ .5-aipyrimidin-5- yl)-cvclohexanecarboxylic acid methyl ester

N-lodosuccinimide (81 1 mg, 3.6 mmol) was added into a solution of 4~{7-[bis-(2- trimethylsilanyl-ethoxymethyl)-amino]-pyrazolo[1 ,5-a]pyrimid!n-5-yl}- cyclohexanecarboxylic acid methyl ester (1.75 g, 3.27 mmol) in acetonitrile (25 mL). The resulting reaction mixture was stirred at room temperature for 16 hours. The
reaction mixture was concentrated in vacuo and purified via silica gel chromatography (0% to 30% ethyl acetate in hexanes gradient) to yield the title compound (1.79 g, 82.8% yield) at clear oil. LC-MS: 661 [M+H].
4-{7-rBis-(2-tπmethylsilanyl-ethoxymethyl)-amino1-3-quinolin-3-yl-Pyrazolo[1 ,5- aipyrimidin-5-yll-cvclohexanecarboχylic acid methyl ester

3-Quinoline boronic acid (2.42 mmol, 420 mg), K3PO4 (3.63 mmol, 771 mg), and PdCI2(dppf) ■ CH2CI2 (0.121 mmol, 100 mg) was added to a solution of 4-{7-[Bis-(2- trimethylsilanyi-ethoxymethyl)-amino]-3-iodo-pyrazolo[1 ,5-a]pyrimidin-5-yl}- cyclohexanecarboxylic acid methyl ester (1.21 mmoi, 800 mg) in dioxane (10 mL). To this suspension was added distilled H2O (1.0 mL). The resulting reaction mixture was stirred at 100° C under an argon atmosphere for 18 hours. The reaction mixture was concentrated in vacuo and then purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield the title compound (630 mg, 79% yield) as yellow oil. LC-MS: 662 [M+H].
4-(7-[Bis-(2-trimethylsilanyl-ethoxymethyi)-amino1-6-bromo-3-quinolin-3-yl- pyrazolof^5-alpyrimidin-5-yll-cvciohexanecarboxylic acid methyl ester

N-bromosuccinimide (148 mg, 0.832 mmol) was added to a solution of 4-{7-[bis-(2- trimethy!silanyl-ethoxymethyl)-amino]-3-quinolin-3-yl-pyrazolo[1 ,5-a]pyrimidin-5-yl}-
cyciohexanecarboxylic acid methyl ester (500 mg, 0.756 mmol) in acetonitrile (10 ml_). The reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo and the resulting oil is then purified via silica gel chromatography (0% to 30% ethyl acetate in hexanes gradient) to yield the title compound (530 mg, 95% yield) as yellow oil. LC-MS: 740 [M+H].
4-(7-Amino-6-bromo-3-quinolin-3-yl-pyrazoloπ .5-alpyrimidin-5-yl)- cyclohexanecarboxylic acid

Table 4




Following additional compounds in table~4a were synthesized following the general experimental procedures described in scheme-5
TABLE-4A

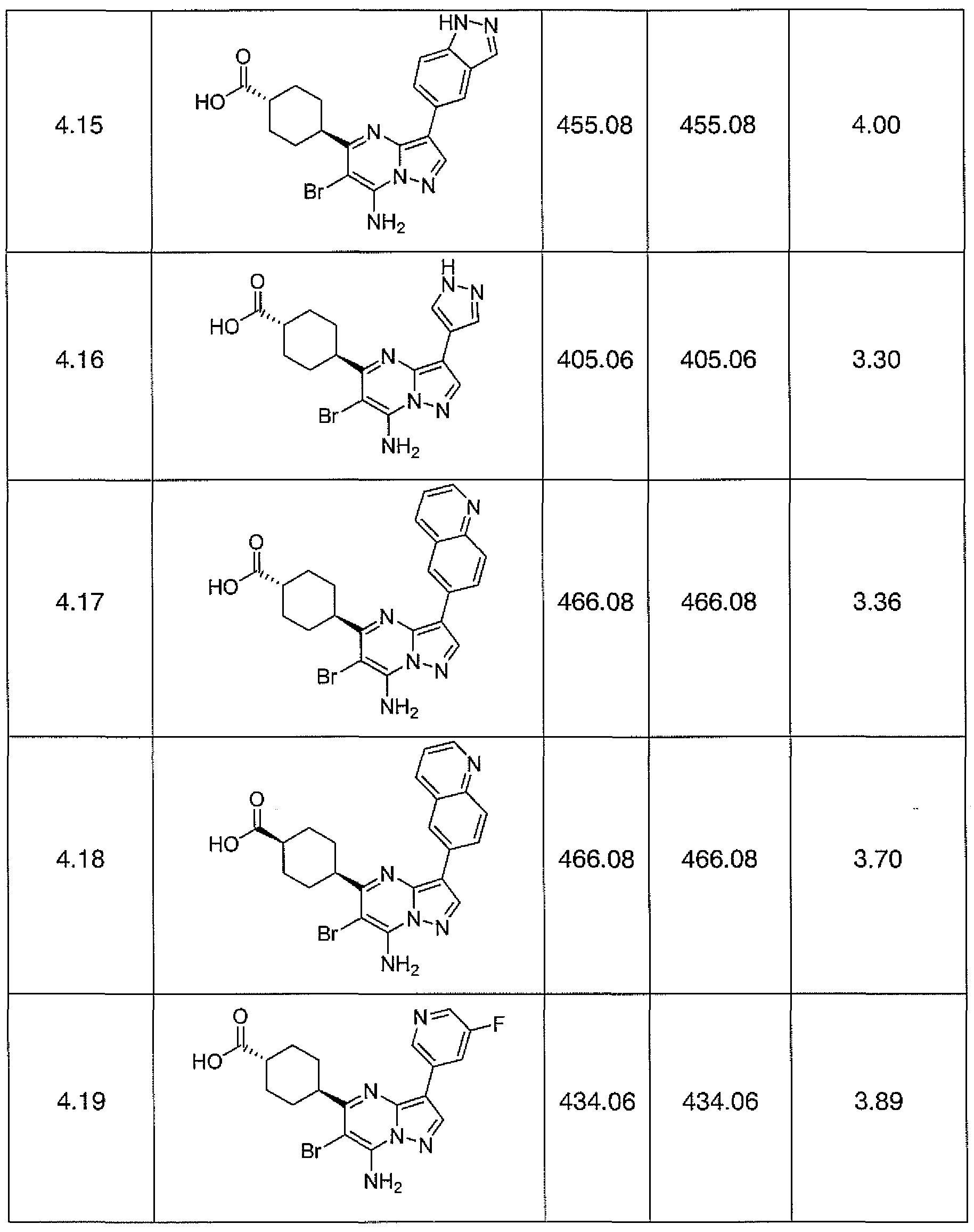


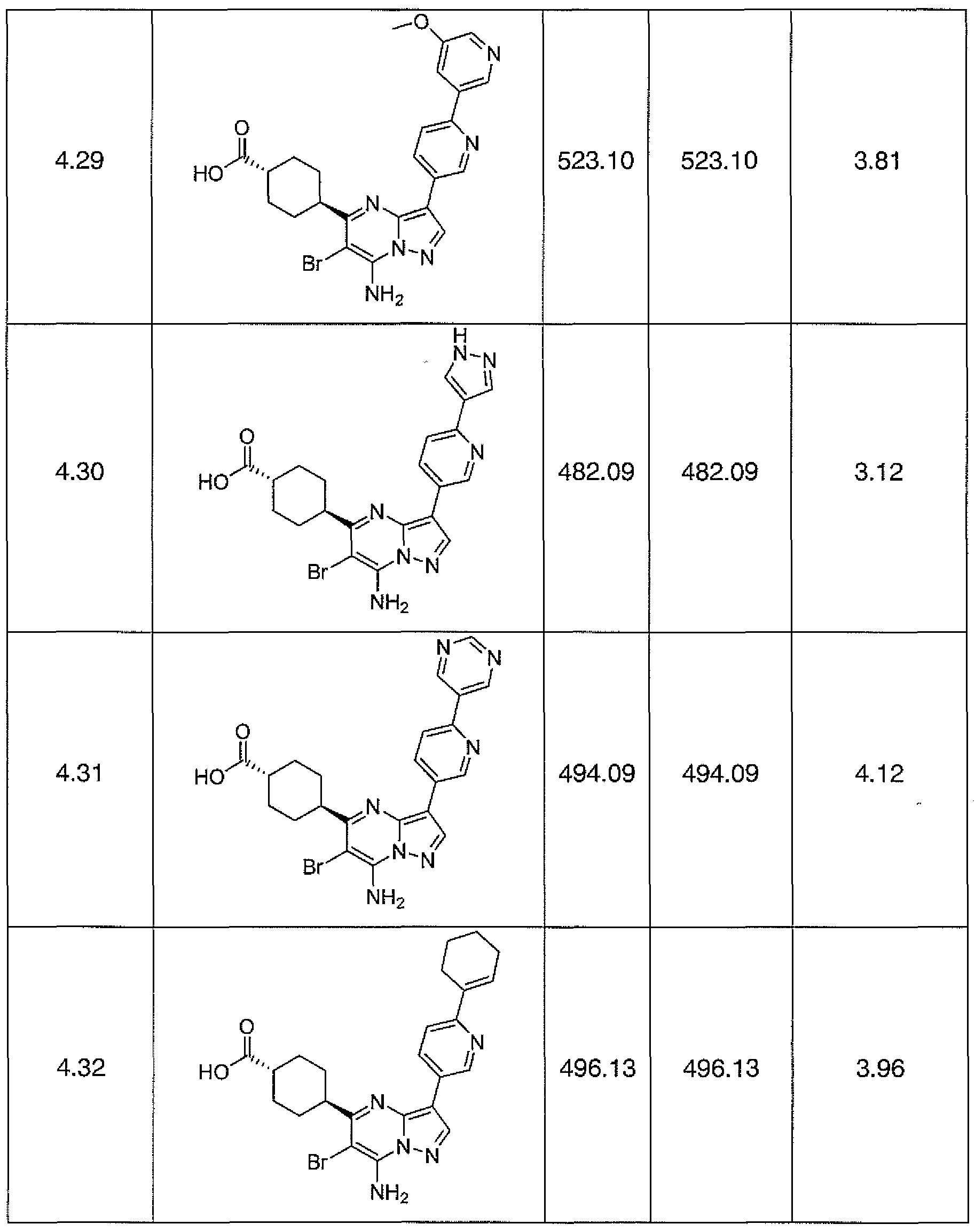













SCHEME- 5a

6-(7-amino-3-(quinolin-3-yl)pyrazok>[1 ,5-a]pyrimidin-5-yl)hexanoic acid (4.87)
To a solution of methyl hex-5-enoate (12 mg, 0.1 mmol) in THF (0.5 ml) under argon at 0 °C was added 9-BBN {0.2 mmol, 0.4 mi of 0.5 M solution in THF). The mixture was warmed to room temperature and stirred for 16 h. Potassium phosphate (3 M -in H2O, 0.2 mmoi) was added followed by the addition of 5-chloro-3-(quinolin-3-yl)-N,N- bis((2-(trimethylsily!)ethoxy)methyl) pyrazo!o[1 ,5-a]pyrimidin-7-amine (50 mg, 0.09 mmol) in DMF (0.5 ml) and Pd(dppf)CI2-CH2CI2 (5 mg, 0.005 mmol). The reaction mixture was heated at 90 °C for 16 h under argon. The solution was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in 2:1 THFiMeOH (3 ml_) and was treated with 2N NaOH(aq) (0.25 ml_). The resulting solution was stirred at room temperature for 4 hours followed by the addition of 2N hydrochloride solution (1.0 ml). The solution was heated at 65 °C for 2 h, cooled to room temperature, concentrated and purified by prep-LC to afford the title compound (8.2 mg): LC/MS RT = 2.74 min. Mass calculated for, M+H 376.17, observed 376.17.
Analogues to the compound 4.87, following compounds in the table-4C can be synthesized as described above in the scheme-5a
Table-4C



Following nucleophilic displacement reaction of 5-chloro-3-(quinolin-3-yl)-N,N-bis{(2- (trimethylsilyl)ethoxy)methyl) pyrazolo[1 ,5-a]pyrimidin-7-amine with appropriate amine or alchol ( experimental procedure described in Scheme-5a, compounds in the table- 4C can be synthesized.
Synthesis of 2-methyl-6-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-vnthienor3,2- bfoyridine:

Part A:
To a stirring solution of thiophenecarboxyfic acid (1 equiv) in DMF and f-BuOH at 0 °C was added the DPPA (1 equiv) and TEA (1 equiv). The reaction was then stirred at room temperature for 1 hour, then warmed to reflux and stirred for 15 hours. The reaction was allowed to cool to room temperature, and then evaporated to dryness.
The resulting residue was dissolved in EtOAc then washed with sat. aq. NH4CI, sat. aq. NaHCO3, and brine, dried over sodium sulfate, filtered, and purified on silica gel to afford the product.
Part B:
A solution of HCI in dioxane was added to a stirring solution of the product from Part A in methanol at 0 °C. The reaction was then stirred at reflux for 1 hour, then allowed to cool to room temperature and concentrated to afford the product.
Part C: Bromomalonaldehyde (1 equiv) was added to a solution of the product from Part B (1 equiv) in glacial acetic acid at room temperature. The mixture was stirred at reflux for 1 hour, then allowed to cool to room temperature, then evaporated to dryness. The residue was dissolved in methanol, filtered through a pad of celite, concentrated, and purified on silica gel to afford the product.
Part D:
A mixture of the product from Part C (1 equiv), bispinacolatodiboron (1.2 equiv), PdCbdppf (0.1 equiv), and potassium acetate (3 equiv) in dioxane was heated at 85 °C until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction was allowed to cool to room temperature, filtered through a pad of celite,
concentrated to dryness, treated with diethyl ether, filtered through a pad of celite, then concentrated to afford the desired product, 2-methyl-6-{4,4,5l5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)thieno[3,2-b]pyridine.
Synthesis of 6-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-yl)thienof3,2-biPyridine:
Essentially utilizing the procedure described above scheme- the title compound 6- (4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)thieno[3,2-b]pyridine can be synthesized
P N

SCHEME-7
Synthesis of 5-(4,4.5.5-tetramethyl-1 ,3,2-dioxaboroian-2-vhfurof2.3-bipyridine:
(modified procedure from the patent # GB2289276A)
H — TMS

Part A:
To a solution of 5-bromo-2-(1H)-pyridone (18.8 gf 10.8 mmol) in dry toluene {400 ml_) was added N-iodosuccinimide (24.3 g, 10.8 mmol) under nitrogen. The reaction mixture was stirred at 90 °C for 20 minutes then cooled to 25 °C. The precipitate was filtered off and washed with methanol, dried under vacuum to give the pink-orange color solid 28.0 g (86%). 1H NMR (300 MHz, CDCi3 ) δ 8.1 (s, 1 H), 7.6 (s, 1 H). Mass caicuiated for formula C5H3BrINO 298.84, observe MS ES+: 300.0/302.0.
Part B:
The iodide (6.0 g, 20.0 mmol), Pd(OAc)2 (45 mgt 0.2 mmol), PPh3 (105 mgt 0.4 mmol), CuI (76 mg, 0.4 mmol) were added to a round bottom flask under nitrogen. Dry THF (30 ml_) was added and the solution was degassed 2 minutes with nitrogen. TMS- acetylene (2.9g, 30 mmol) was added followed with n-BuNH2 (2.9 g, 40 mmol). The homogeneous green solution was heated to 38 °C for 4 hours. The reaction mixture was cool to 25 °C, concentrated to dryness and then dissolved in EtOAc (100 ml_). The solution was washed with saturated sodium potassium tartrate (50 ml_), followed with 0.1 N HCI (50 mL), saturated NaHCO3 (50 mL), and brine (5OmL), dried over NasSO4. Purification by column chromatography (SiO2, 50% ethyl acetate/hexanes) afforded compound as a green solid 4.40 g (81 %). 1H NMR (300 MHz, CDCI3) δ 7.85 (s, 1 H), 7.5 (s, 1 H), 0.2 (s, 9H).
Part C: To a EtOH (8.0 mL) solution of compound from Part B (1.12 g, 4.1 mmol) was added CuI (39 mg, 0.2 mmol) and followed with Et3N (3.5 mL) the resulting mixture was stirred at 70 °C for 3 hours. The reaction mixture was allowed to cool to 25 °C, concentrated to dryness and then dissolved in toluene (3OmL). The solution was washed with 0.1 N HCI (20 mL), saturated NaHCO3 (20 mL), and brine (30 mL), dried over Na2SO4. Purification by column chromatography (SiO2, 20% ethyl acetate/hexanes) afforded compound as a white solid 0.26 g (32%). H NMR (300 MHz, CDCl3) δ 8.4 (s, 1 H), 8.1 (s, 1H), 7.7 (s, 1H), 6.8 (s, 1 H).
Part D: A mixture of the product from Part C (1 equiv), bispinacoiatodiboron (1.2 equiv), PdCI2dppf (0.1 equiv), and potassium acetate (3 equiv) in dioxane was heated at 85 °C until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction was allowed to cool to room temperature, filtered through a pad of celite, concentrated to dryness, treated with diethyl ether, filtered through a pad of celite, then concentrated to afford the desired product, 5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)furo[2,3-b]pyridine.
2-methyl-5-(4.4.5.5-tetramethyi-1 ,3,2-dioxaborolan-2-yl)furo[2,3-blPyridine:
I- Λ ,
YV

Part B
SCHEME-8
Part A: A mixture of 5-bromo-3-iodopyridin-2-ol (1 equiv), copper iodide (3 equiv), palladium catalyst (0.05 equiv), TBAF (1 equiv), triethylamine (3.3 equiv) and alkyne (1 equiv) in toluene was stirred at room temperature until the reaction was deemed complete by TLC analysis. The reaction mixture was diluted with dichloromethane,
washed with water, saturated sodium bicarbonate, and brine, dried over sodium bicarbonate, filtered through celite, concentrated, and purified on silica gel to give the product,5-bromo-2-methylfuro[2,3-b]pyridine. Part B:
A mixture of the product, 5-bromo-2-methy[furo[2,3-b]pyridine, from Part A (1 equiv), bispinacolatodiboron (1.2 equiv), PdCi2dppf (0.1 equiv), and potassium acetate (3 equiv) in dioxane was heated at 85 °C until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction was allowed to cool to room temperature, filtered through a pad of celite, concentrated to dryness, treated with diethyl ether, filtered through a pad of celite, and then concentrated to afford the desired product, 2-methyl-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)furo[2,3- b]pyridine.
Synthesis of 5-(4,4.5,5-tetramethyl-1.3i2-dioxaborolan-2-vnthienoF2,3-biDyridine:

SCHEME-9
Part A:
To a solution of 5-bromo-3-((trimethylsiyl)ethynyl)ipyhdin-2-oI (10.63 g, 39.5 mmol) in toluene (195 mL) was added Lawesson's reagent (8.00 g, 19.8 mmol) under nitrogen. The reaction mixture was stirred at 120 °C for 1 hour, and then allowed to cool to 25 °C. Purification by column chromatography (SiO2, 20% ethyl acetate/hexanes) afforded compound as a white solid, 5-bromo-2-(trimethylsilyl)thieno[2,3-b]pyridine, 8.99 g (79%). 1H NMR (300 MHz, CDCI3) δ 8.57 (d, J= 2.2 Hz, 1 H), 8.16 (d, J = 2.2 Hz 1 H), 7.30 (s, 1H), 0.39 (s, 9H).
Part B: The 5-bromo-2-(trimethylsilyl)thieno[2,3-b]pyridine from Part A (8.99 g, 31.4 mmol) was dissolved in ethano! (70 mL), followed by addition of K2CO3 (10.85g, 78.5 mmol). Reaction was stirred at 65 °C for 1 hour. The reaction mixture was allowed to cool to 25 °C, concentrated to dryness, and dissolved in EtOAc (150 mL). The solution was washed with H2O (80 mL), and brine (15OmL), dried over Na3SO4, filtered, and concentrated to yield white solid, 5-bromothieno[2,3-b]pyridine, 6.57 g (98%). 1H NMR (300 MHz, CDCI3) δ 8.60 (d, J = 2.2 Hz, 1 H), 8.16 (d, J = 2.2 Hz 1 H), 7.57 (d, J = 6.0 Hz 1 H), 7.21 (d, J = 6.0 Hz, 1 H). Part C: A mixture of the product, 5-bromothieno[2,3-b]pyridineI from Part B (1 equiv), bispinacolatodiboron (1.2 equiv), PdCI2dppf (0.1 equiv), and potassium acetate (3 equiv) in dioxane was heated at 85 °C until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction was allowed to cool to room temperature, filtered through a pad of celite, concentrated to dryness, treated with
diethyl ether, filtered through a pad of celite, and then concentrated to afford the desired product, 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)thieno[2,3-b]pyridine.
General synthesis of (1 r,4r)-4-(7-amino-3- Aryl,6-substituted pyrazoioM ,5-alPyrimidin- 5-vOcvclohexanecarboxvlic acid:

A mixture of compound {1 r,4r)-tert-butyl 4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yljcyclohexanecarboxylate (1 equiv), boronate (1.75 equiv), palladium catalyst (0.1 equiv), and potassium carbonate (3 equiv) in water and dioxane was stirred at 40 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The mixture was allowed to cool to room temperature, diluted with water and extracted three times with dichloromethane. The combined organics were dried over sodium sulfate, filtered, concentrated, and purified on silica gel to afford compound (1r,4r)-tert- butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyI)amino)-3-aryl-pyrazolo[1 ,5-a]pyrimidin- 5-yi)cyclohexanecarboxylate.
When R = Br:
Compound , (1 r,4r)-tert-butyl 4-(7-(bis((2-{trimethylsilyl)ethoxy)methyl)amino)-3-aryl- pyrazoioJI.5-ajpyrimidin-5-yl)cyclohexanecarboxylate, was stirred in a mixture of dichloromethane, water, and TFA until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The reaction was concentrated and purified on prep-HPLC. The resulting material was stirred with NBS (1.05 equiv) in acetonitrile and DMF at room temperature until the reaction was deemed complete by HPLC1 LC-MS, or TLC analysis. The mixture was concentrated, purified on prep-HPLC, and freeze-dried to afford compound, (1 r,4r)-4-(7-amino-6-bromo-3-aryl - pyrazolo[1 ,5-a]pyrimidin-5- y!)cyclohexanecarboxy!ic acid.
When R = acetyl: Compound, (1 r,4r)-tert-buty! 4-(7-{bis{(2- {trimethylsilyi)ethoxy)methyl)amino)-3-aryl-pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexanecarboxylate , (1 equiv) was stirred with NBS (1.05 equiv) in DMF at room temperature until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The reaction was diluted with ethyl acetate, washed three times with brine, dried over sodium sulfate, filtered, concentrated, and purified on silica gel. The resulting material was stirred with a mixture of vinyl stannane (2 equiv), palladium catalyst (0.1 equiv), and potassium fluoride (3 equiv) in dioxane at 85 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The mixture was
allowed to cool to room temperature, diluted with 10% aqueous potassium fluoride, and extracted three times with ethyl acetate. The combined organics were dried over sodium sulfate, filtered, concentrated, and purified on silica gel. The resulting material was stirred with a mixture of HCI in methanol and water at 60 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The mixture was then allowed to cool to room temperature, concentrated, purified by prep-HPLC, and freeze-dried to afford compound (1 r,4r)-4-(6-acetyl-7-amino-3~aryl - pyrazolo[1 ,5-a]pyrimidin-5- yi)cyclohexanecarboxylic acid.
Essentially using the general procedure described above the compounds in the table 4-D can be synthesized.
TABLE-4D


6-Bromo-5-((1-(cvclopropylsulfonyl)pipehdin-4-yl)nnethoxy)-3-(Quinolin-3- yl)pyrazoiof1 ,5-aiPyrimidin-7-amine:

SCHEME- 11
tert-Butv! 4-(f7-(bis(f2-(trinπethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(quinoiin-3- yl)pyrazoio[1 ,5-aiPyrimidin-5-yloxy)methyl)piperidine-1-carboxylate: To a solution of tert-butyl 4-{hydroxymethyl)piperidine-1 -carboxylate (31 1 mg, 1 .44 mmol) and sodium hydride (60% in mineral oil, 58 mg, 1.44 mmol) in THF (2 mf) under argon at room temperature was added 5-chloro-3-(quinolin-3-y!)-N,N-bis((2 (trimethylsilyl)ethoxy)methyl) pyrazolo[1 ,5-a3pyrimidin-7-amine {400 mg, 0.72 mmoi). The mixture was heated at 100°C for 30 min in microwave. Ethyl acetate (50 ml) was added and the mixture was washed with brine and water, dried over Na2SO4, and concentrated in vacuo. The crude product was dissolved in acetonitrile, followed by the addition of N-bromosuccinimide (193 mg, 1.08 mmol). The reaction mixture was
stirred at room temperature for 2 h, concentrated in vacuo and purified via silica gel chromatography (10% to 50% ethyl acetate in hexanes gradient) to yield the title compound (0.56 g, 95% yield). LC-MS: 813 [M+H].
6-Bromo-5-(piperidin-4-ylmethoxy)-3-(quinolin-3-yl)pyrazolof1 ,5-a1pyrimidin-7-amine: tert-Butyl 4-({7-(bis((2-(trimethylsi[yl)ethoxy)methyl)amino)-6-bromo-3-(quinolin-3- y[)pyrazolo[1 ,5-a]pyrimidin-5-yloxy)methyl)piperidine-1-carboxylate {0.56 g, 68.9 mmol) was dissolved in methanol (6 ml) and treated with 4N hydrochloride solution in dioxane (4 ml) at 65 °C for 30 min. The reaction solution was concentrated and the crude product (0.48 g) was used in the next step without further purification. LC-MS: 453 [M+H].
6-Bromo-5-((1 -(cvclopropylsulfonyl)piperidin-4-yl)methoxy)-3-(quinolin-3- yi)pyrazolo[1 ,5-alpyrimidin-7-amine:
Cyclopropanesuifonyl chloride {24 mg, 0.167 mmoi) was added into a solution of 6- bromo-5-(piperidin-4~ylmethoxy)-3-(quinolin-3-y!)pyrazoio[1 ,5-a]pyrimidin-7-amine (50 mg, 0.11 1 mmol) and triethylamine (56 mg, 0.553 mmol) in DMF at room temperature. The resulting reaction mixture was stirred at room temperature for 16 h, concentrated and purified by prep-LC to afford the title compound (21.5 mg): LC/MS RT = 4.17 min. Mass calculated for, M+H 557.09, observed 557.09.
Compounds in the table-4-E can be synthesized as described following general procedure dscribed above scheme( 11 )
TABLE-4E






By essentially using the similar experimental conditions used in the scheme 11 , compounds in the table 4F can be synthesized.
TABLE-4F


SCHEME-12
PartA:
Methyl 2-(4-(7-(bis((2-(trimethyisilyl)ethoxy)methyhamino)pyrazoio[1 ,5-aipyrimiclin-5- yloxy)phenyl)acetate:
The compound methyl 2- (4-hydroxy phenyl) acetate (330 mg, 0.2 mmol) was dissolved in dry DMF (3 mL) and NaH (80 mg, 60% in oil, 0.2 mmol) was added and the mixture was stirred at room temperature for 5 min. Then, 5-chloro-N,N-bis((2-
(trimethylsilyl)ethoxy)methyI)pyrazolo[1,5-a]pyrimidin-7-amine (430 mg, 0.1 mmol) was added and the resulting mixture was heated up to 1300C with microwave and stirred for 30 min. After cooling to room temperature, NH4Cl (aq) was added to quench the reaction and extracted with EtOAc (50 mL x 3). The combined organics were washed with water and brine and dried over Na2SO4. After concentration, the residue was purified with column (silica gel, 20% EtOAc/Hexane) gave the product, methyl 2-(4-(7-(bis((2- (trimethylsiIyi)ethoxy)methyl)amino)pyrazolo[ L,5-a]pyrimidin-5-yIoxy)phenyl)acetate, (490
mg) as clear oil. HPLC-MS tR = 2.77 min (UV254 nm); mass calculated for formula 027H42N4O5Si2 558.3, observed LCMS m/z 559.3 (M+H).
Part B: Methyl 2-(4-(7-(bis((2-(trimethylsiiyl)ethoxy)methyl)amino)-3-iodopyrazoiori ,5- aipyrimidin-5-yloxy)phenvOacetate:
Methyl 2-(4-(7-{bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[l,5-a]pyrimidin-5- yloxy)phenyl)acetate (450 mg, 0.81 mmol) was dissolved in ACN (15 raL) and NIS (200 mg, 0.88 mmol) was added. The resulting mixture was stirred at room temperature for 2 h and then the solvent was removed under reduced pressure. The residue was purified with column (silica gel, 10% EtOAc/Hexane) gave the product, methyl 2-(4-(7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1,5-a]pyrimidin-5-yloxy)phenyl)acetate (543 mg) as yellowish oil. HPLC-MS tR = 2.96 min (UV254 nm); mass calculated for formula C27H4IlN4O5Si2684.2, observed LCMS m/z 685.2 (M+H).
Part C:
Methyl 2-(4-(7-(bisf(2-(trimethylsiiyl)ethoxy)methyπamino)-3-(quinolin-3- yl)pyrazolori ,5-a]pyrSmidin-5-yloxy)phenyl)acetate:
Under Ar, compound, methyl 2-(4-(7-(bis((2-(rrimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[1,5-a]pyrimidin-5-yloxy)phenyI)acetate (180 mg, 0.26 mmol) was mixed with Pd(dppf)Cl2 (21 mg, 0.26 mmol, K3PO4 (106 mg, 0.5 mmol), 3-quinoIine bornic acid (55 mg, 0.31 mmol) and dioxane (10 mL with 1 ml water). The resulting mixture was heated at 90 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (60 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product, methyl 2-(4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3-yl)pyrazolo[l,5-a]pyrimidin-5- yloxy)phenyl)acetate (150 mg). HPLC-MS tR = 2.60 min (UV254 nm); mass calculated for formula C36H47N5O5Si2685.3, observed LCMS m/z 686.3 (M+H).
Part D: methyl 2-(4-(7-(bis((2-(trimethyisilyl)ethoxy)methyl)amino)-6-bromo-3-(quinolin-3- yl)pyrazolori .5-alpyrimidin-5-yloxy)phenyl)acetate:
Compound, methyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinoIin-3- yl)pyrazolo[1,5-a]pyrimidin-5-yloxy)phenyl)acetate (150 mg, 0.22 mmol) was dissolved in ACN (10 mL). NBS (40 mg, 0.22 mmol) was added and the mixture was stirred at room temperature for 1 h. After concentration, the product, methyl 2-(4-(7-(bis((2-
(trimethylsilyl)ethoxyJmethyl)aminoJ-ό-bromo-3-tquinolin-3-yl)pyrazoloCU5-alpyrimidin-5- yloxy)phenyl)acetate was used in the next step without further purification. HPLC-MS tR - 2.70 min (UV254 nm); mass calculated for formula C36H46BrN5O5Si2 763.2, observed LCMS m/z 764.2 (M+H).
Part E: methyl 2-(4-(7-amino-6-bromo-3-(Quinolin-3-yl)pyrazolo[1 ,5-alpyrimidin-5- yloxy)phenvOacetate:
The crude methyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyi)amino)-6-bromo-3-(quinolin- 3-yl)pyrazolo[1,5-a]pyrimidin-5-yloxy)pheny[)acetate from above step D was treated with 50% TFAZH2O (3 mL) and stilted at room temperature for 1 h. Solvent was removed to yield thick oil, methyl 2-(4-(7-amino-ό-brorno-3-(quinolin-3-yl)pyrazolo[1,5-a]pyrimidin-5- yloxy)phenyl)acetate, which was used in the next step directly without further purification. HPLC-MS tit = 1.60 min (UV254 nm); Mass calculated for formula C24H18BrN5O3. 503.1, observed LCMS m/z 504.1 (M+ Na).
Part F:
2-(4-(7-amino-6-bromo-3-(quinolin-3-yl) pyrazoloH ,5-a]pyrimidin-5-yloxy)phenyl)acetic acid:
Crude compound , methyl 2-(4-(7-amino-6-brorno-3-(quinolin-3-yl)pyrazolo[1,5-a]pyrimidin- 5 -yloxy) phenyl) acetate from step E was dissolved in THF (5 mL) and LiOH ( IN, 1 mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed under vacuo, and the resulting residue was taken over with water and the pH value was adjusted to 5-6. The solid was collected with filtration and purified with HPLC gave the product, 2-(4-(7- amino-6-bromo-3-(quinolin-3-yl) pyrazolo[1,5-a]pyrimidin-5-yloxy)phenyl)acetic acid. HPLC-MS tR = 1.53 min (UV254 nm); Mass calculated for formula C23Hj6BrN5O3, 489.0, observed LCMS m/z 490.0 (M+H).
By essentially the same procedure given in Preparative Example given above, compounds 4.130-4.144 given in Column 2 of Table 4G can be prepared.
TABLE 4G



Synthesis of 4-(7-amino-6-bromo-3-(6-phenylpyndin-3-yl)pyrazolori ,5-a1pyrimidin-5- yl)benzoic acid:
SCHEME- 13

Part A: Syntheisis of methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methy!)amino)pyrazolori ,5- aipyrimidin-5-yl)benzoate:
Under Ar, compound, 5-chloro-N,N-bis((2-(trimemylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrirnidin-7-amine, (430 mg, 1.0 mmol) was mixed with Pd(dppf)Cl2 (82 mg, 0.1 mmol, K3PO4 (636 mg, 3.0 mmol), methyl 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzoate (200 mg, 1.1 mmol) and dioxane (20 mL with 1 mi water). The resulting mixture was heated at 90 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (60 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product, methyl 4-(7-(bis((2- (trimethy!siiyl)ethoxy)methyl)amino)pyrazolo[1,5-a]pyrimidin-5-yi)benzoate (459 mg). HPLC-MS tR = 2.89 min (UV254 πm); mass calculated for formula C26H40N4O4Si2 528.3, observed LCMS m/z 529.2 (M+H).
Part B:
Synthesis of methyl 4-(7-(bis((2-(trimethylsilvt)ethoxy)methyl)amino)-3- iodopyrazolo[1 ,5-a]pyrimidin-5-yl)benzoate:
Compound, methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyI)amino)-3-iodopyrazolo[ 1 ,5- a]pyrimidin-5-yl)benzoate was prepared with the same iodonation condition described in scheme- 12 Part B. HPLC-MS tR = 3.09 min (UV254 ,,m); mass calculated for formula C26H39IN4O4Si2 654.2, observed LCMS m/z 655.2 (M+H).
Part C:
Synthesis of methyl 4-(3-(biphenyl-4-yl)-7-(bis((2- (trimethylsilvi)ethoxy)methyl)amino)pyrazolo[1 ,5-alpyrimidin-5-yl)benzoate: Title Compound, methyl 4-(3-(biphenyl-4-yiy7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)pyrazolof 1,5-a1pyrimidin-5-yl)benzoate was prepared with the same coupling condition described in the schem-12 Part C. HPLC-MS IR = 2.98 min (UV254 nm); mass calculated for formula C37H47N5O4S12681.3, observed LCMS m/z 682.2 (M+H).
Part D:
Synthesis of methyl 4-(7-(bisf(2-(trimethylsilyl)ethoxy)methy0amino)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazolo[1 ,5-alpyrimidin-5-yl)benzoate:
Title Compound was prepared with the same bromonation condition described in Scheme- 12, Part D . HPLC-MS tR = 3.11 min (UV254 nm); mass calculated for formula C37H46BrN5O4Si2 759.2, observed LCMS m/z 760.2 (M+H).
Part E:
Synthesis of methyl 4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5- aipyrimidin-5-yl)benzoate:
Title compound, methyl 4-(7-amino-6-brorno-3-(6-phenylpyridin-3-yl)pyrazolo[l,5- a]pyrimidin-5-yl)benzoate was prepared with the same deprotection condition described in Scheme- 12 Part E. HPLC-MS tR = 1.88 min (UV254 nm); mass calculated for formula C23H18BrN5O2499.1 , observed LCMS m/z 500.1 (M+H).
Part F: 4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazoio[l,5-a]pyrimidin-5-yl)benzoic acid:
Compound, 4-(7-amino-6-bromo-3-(6-pheny!pyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5- yl)benzoic acid was prepared with the same hydrolysis condition described in Scheme- 12 Part E. HPLC-MS tR = 1.44 min (UV254 nm); mass calculated for formula C24Hi6BrN5O2 485.0, observed LCMS m/z 486.1 (M+H).
By essentially the same procedure given in Preparative scheme-33 above, compound 4.145- 4.166 given in Column 2 of Table 4 H can be prepared from compound 2.
TABLE - 4H





Synthesis of 4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-y0pyrazoloH ,5-a1pyrimidin-5- yl)benzoic acid:

SCHEME-14
Part A methyl 4-(7-(bisff2-(trimethyisilyl)ethoxy)methyl)amino)-6-(1-ethoxyylnyl)-3-(6- phenylpyridin-3-yl)pyrazolori ,5-aipyrimidin-5-yl)benzoate:
Compound, methyl 4-(7-(bis((2-(trimemylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazolo[1,5-a]pyrirnidin-5-yl)benzoate (40 mg, 0.053 mmol) under Argon, was mixed with Pd(PPh3)4 (12 mg, 0.01 mmol, (l-ethoxyvinyl)tributylstannane (85 uL, 0.25 mmol) and dioxane (3 mL). The resulting mixture was heated at 100 °C and stirred over night. After cooled to room temperature, the mixture was filtered through 10% KF on silica gel and washed with EtOAc. After concentration, the crude product, methyl 4-(7-(bis((2- (trimethylsily!)ethoxy)methyl)amino)-6-(l-ethoxyvinyl)-3-(6-phenyIpyridin-3-yl)pyrazolo[1,5- a]pyrimidin-5-yl)benzoate was used in the next step directly without further purification. HPLC-MS tR = 3.18 min (UV254 ,™); mass calculated for formula C4[H53N5O5Si2 751.4, observed LCMS m/z 752.3 (M+H).
Part B
Methyl 4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-alpyrimidin-5- yl)benzoate:
Compound, methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-(l-ethoxyvinyl)-3-(6- phenylpyridin-3-yl)pyrazoIo[1,5-a]pyrimidin-5-yl)benzoate was treated with 4N HCi in doioxane for 2hrs to result in the product, methyl 4-(6-acetyl-7-amino-3-(6-phenylpyridin-3- yl)pyrazolo[l,5-a]pyrimidin-5-y[)benzoate purified by prep.HPLC. HPLC-MS tR = 1.85 min (UV254 nm); mass calculated for formula C27H2IN5O3463.2, observed LCMS m/z 464.0 (M+H).
Part C: 4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolori .5-aipyrimidin-5-yl)benzoic acid:
Compound methyl 4-(6-acetyf-7-amino-3-(6-phenylpyridin-3-yI)pyrazolo[ l,5-a]pyrimtdin-5- yi)benzoate was treated with 2 equivalents of LiOH and stirred at room temperature for 4hrs. Neutralization with dil.HCl and extraction in to organic layer, gave the product, 4-(6-acetyl-7- amino-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl)benzoic acid in quantitavive yield. The compound dissolved in to water-acetonitrile and lyophilized to give a white powder. HPLC-MS tR = 1.49 min (UV254 nm); mass calculated for formula C26Hi9N5O3 449.1, observed LCMS m/z 450.2 (M+H).
By essentially the same procedure given in above preparative example , compounds 4.167-4.171 given in Column 2 of Table 41 can be prepared.
TABLE-41


Synthesis of 6-bronno-5-(4-(methylsu[fonyl)phenoxy)-3-(quinolin-3-yl)pyrazolori ,5- aiDyrimidin-7-amine:

SCHEME- 15
Part A:
5-(4-(methylthSo)phenoxy)-N,N-bis((2-(trimethylsilyπethoxy)methyl)pyrazolori ,5- aipyrimidin-7-amine: The 4-(methylthio)phenol (280 mg, 2.0 mmol) was dissolved in dry DMF (3 mL) and NaH (88 mg, 60% in oil, 2.2 mmol) was added and the mixture was stirred at room temperature for 5 min. Then, 5-chloro-N,N-bis((2-(trimemyIsilyl)ethoxy)methyl)pyrazolo[l ,5-a]pyrimidin-7- amine (430 mg, 0.1 mmol) was added and the resulting mixture was heated up to 130 0C with Microwave and stirred for 30 min. After cooling to room temperature, NH4C1 (aq) was added to quench the reaction and extracted with EtOAc (50 mL x 3). The combined organics were washed with water and brine and dried over Na2SO4. After concentration, the residue was purified with column (silica gel, 20% EtOAc/Hexane) gave the product 5-(4- (methylthio)phenoxy)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7- amine (490 mg) HPLC-MS .R = 2,98 min (UV254 nm); mass calculated for formula C25H40N4O3SSi2 532.2, observed LCMS m/z 533.2 (M+H).
Part B:
3-iodo-5-(4-(methyithio)pheπoxy)-N.N-bisff2-
(trimethyisilyl)ethoxy)methyi)pyrazolof1.5-aipyrimidin-7-amine:
5-(4-(methylthio)phenoxy)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5- a]pyrimidin-7-amine (490 mg, 0.81 m) was dissolved in ACN (15 mL) and NIS (200 mg, 0.88 mmol) was added. The resulting mixture was stirred at room temperature for 2 h and then the solvent was removed under reduced pressure. The residue was purified with column (silica gel, 10% EtOAc/Hexane) gave the product, 3-iodo-5-(4-(methylthio)phenoxy)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (541 mg) as yellowish oil.. HPLC-MS tR = 3.18 mln (UV254 nJ; mass calculated for formula C25H39IN4O3SSi2658.1, observed LCMS m/z 659.2 (M+H). Part C:
5-(4-(methylthio)phenoxy)-3-(αuinolin-3-yl)-N.N-bisff2- (trimethylsilvi)ethoxy)methyl)pyrazoiori ,5-aipynmidin-7-amine:
3-iodo-5-(4-(methylthio)phenoxy)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrimidin-7-amine (180 mg, 0.26 mmol) under Argon,was mixed with Pd(dppf)Cl2 (21 mg, 0.26 mmol, K3PO4 (106 mg, 0.5 mmol), 3-quinoline bornic acid (55 mg, 0.31 mmol) and dioxane (10 mL with 1 ml water). The resulting mixture was heated at 90 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (60 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product 5-(4-(methylthio)phenoxy)-3-(quirιolin-3-y I)-N1N- bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (150 mg). HPLC-MS tR = 2.85 min (UV254 nm); mass calculated for formula C34H45N5O3SSi2659.3, observed LCMS m/z 660.1 (M+H). Part D:
5-(4-(methylsuifonyl)phenoxy)-3-(quinoiin-3-yl)-N.N-bis(f2- (trimethy[silyl)ethoxy)methyl)pyrazolori .5-alpyrimidin-7-amine:
Compound, 5-(4-(methylthio)phenoxy)-3-(quinolin-3-yl)-N,N-bis((2- (trimethyIsilyl)ethoxy)methyi)pyrazolo[l,5-a]pyrimidm-7-amine (87 mg, OJ 32 mmol) was dissolved in DCM (5 mL) and m-CPBA (66 mg, -70%, 0.27 mmol) was added. The result mixture was stirred at room temperature for 1 h. NaHCO3 (aq.) was added and the water was extracted with DCM. The combined organics was dried over Na2SO4 and concentrated. The crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product, 5-(4- (methylsulfonyl)phenoxy)-3-(quinolin-3-yl)-N,N-bis((2-
(trimethylsilyI)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine (89 mg). HPLC-MS IR = 2.37 min (UV254 nm); mass calculated for formula C34H4SNsOsSSi2 691.3, observed LCMS m/z 692.2 (M+H). Part E:
6-Bromo-5-(4-(methylsulfonyl)phenoxy)-3-(quinoiin-3-yl)-N,N-bis((2- (tπmethylsiiyl)ethoxy)methyl)pyrazolo[1 ,5-alpyrimidin-7-amine:
Compound, 5-(4-(methylsulfonyl)phenoxy)-3-(quinolin-3-yl)-N,N-bis((2- (trimethyisilyl)ethoxy)methyl)pyrazolo[ 1 ,5-a]pyrimidin-7-amine (150 mg, 0.22 mmol) was dissolved in ACN (10 mL). NBS (40 mg, 0.22 mmol) was added and the mixture was stirred at room temperature for 1 h. After concentration, the residue, 6-bromo-5-(4-
(methylsulfonyl)phenoxy)-3-(quinolin-3-yi)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazoIo[1,5-a]pyrimidin-7-amine was used in the next step without further purification. HPLC-MS IR = 2.66 min (UV254 nm); mass calculated for formula
C34H44BrN5O5SSi2769.2, observed LCMS m/z 770.0 (M+H).
Part F:
6-bromo-5-(4-(methylsu[fonyl)phenoxy)-3-(quinolin-3-yl)pyrazolof1 ,5-alpyrimidin-7- amine:
The crude, 6-bromo-5-(4-(methylsulfonyl)phenoxy)-3-(quinolin-3-yl)-N,N-bis((2- (tritnethylsilyl)ethoxy)methyl)pyrazoIo[1,5-a]pyrimidin-7-amine from above step E was treated with 50% TFA/H2O (3 mL) and stirred at room temperature for 1 h. Solvent was removed to yield thick oil which was purified by prep HPLC to give the final product. 6- bromo-5-(4-(methylsuifonyl)phenoxy)-3-(quinoHn-3-yl)pyrazolo[l,5-a]pyrimidin-7-amine. HPLC-MS tR = 1.37 min (UV254 nm); mass calculated for formula C22Hi6BrN5O3S2509,0, observed LCMS m/z 510.0 (M+H).
By essentially the same procedure given in Preparative above Example, compounds 4.172- 4.174 given in Column 2 of Table 4 J can be prepared.
TABLE 4-J


Synthesis of 2-(4-((7-amino-6-bromo-3-(quinolin-3-yltoyrazolori ,5-alpyrimidin-5- yl)methyDphenvOacetic acid :

SCHEME- 16
Part A:
Methyl 2-(4-((4,4,5,5-tetramethyl-1.3.2-dioxaborolan-2-yl)methyπphenyl)acetate:
Methyl 2-(4-(bromomethyl)phenyl)acetate (1.03 g, 4.24 mmol) was mixed with Pd(PPh3)4 (500 mg, 0.4 mmol, 4,4,41,4',5,5,51,5'-octamethyl-2,21-bi(l,3,2-dioxaborolane) (1.27 g, 5 mmol), K2CO3 (1.75 g, 12.7 mmol) and dioxane (20 mL) under inert Argon atmoshphere. The resulting mixture was heated at 80 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (100 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 10~30% EA/hexane) gave the product, methyl 2-(4-((4,4,5,5-tetramethyl- [,3,2-dioxaborolan-2-yl)methyl)phenyl)acetate (721 mg) as semi-
solid. HPLC-MS tR = 2.17 min (UV254 πm); mass calculated for formula Ci6H23BO4290.2, observed LCMS m/z 291.3 (M+H).
Part B:
Methyl 2-(4-((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a1pyπmidin-5- yl)methyDphenvOacetate:
Under Ar, 5-chloro-N,N-bis((2-(trimethylsiIyl)ethoxy)methyl)pyrazolo[1,5-a]pyrirnidin-7- amine (429 mg, 1.0 mmol) was mixed with Pd(dppf)C12 (82 mg, 0.1 mmol, boronate, methyl 2-(4-((4,4,5,5-tetramethyl-I,3,2-dioxaborolan-2-yl)methyl)phenyl)acetate (4go mg, 1.65 mmol, not very pure), K3PO4 (424 mg, 2.0 mmol) and dioxane (10 mL). The resulting mixture was heated at 100 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (100 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 10-30% EA/hexane) gave the product, methyl 2-(4-((7- (bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1,5-a]pyrimidin-5- y!)methyl)phenyl)acetate (188 mg) and recovered the starting material 5-chloro-N,N-bis((2- (trimethylsilyI)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (289 mg). HPLC-MS IR = 2.50 min (UV254 nm); mass calculated for formula C28H44N4O4Si2 556.3, observed LCMS m/z 557.3 (M+H).
Part C:
Methyl 2-(4-((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5- a1pyrimidin-5-yl)methyl)phenyl)acetate: methyl 2-(4-((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[ 1 ,5-a]pyrimidin-5- yl)methyl)phenyl)acetate (450 mg, 0.81 mmol) was dissolved in ACN (15 mL) and NIS (200 mg, 0.88 mmol) was added. The resulting mixture was stirred at room temperature for 2 h and then the solvent was removed under reduced pressure. The residue was purified with column (silica gel, 10% EtOAc/Hexane) gave the product methyl 2-(4-((7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1,5-a]pyrimidin-5- yi)methyl)phenyi)acetate (541 mg) as yellowish oil. HPLC-MS .R = 2.97 min (UV2g4 nm); mass calculated for formula C28H43IN4O4Si2682.2, observed LCMS m/z 683.2 (M+H). Part D:
Methyl 2-(4-(f7-(bis(f2-(trimethylsilyl)βthoxy)methyl)amino)-3-(quinolin-3- yl)pyrazoloH ,5-a1pyrimidin-5-yl)methyl)phenyl)acetate:
Under Ar, methyl 2-(4-((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1,5- a]pyrimidin-5-yl)methyl)phenyl)acetate (180 mg, 0.26 mmol) was mixed with Pd(dppf)Cl2 (21 mg, 0.26 mmol, K3PO4 (106 mg, 0.5 mmol), 3-quinoline bornic acid (55 mg, 0.31 mmol) and dioxane (10 mL with 1 ml water). The resulting mixture was heated at 90 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (60 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product methyl 2-(4-((7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3-yl)pyrazolo[l ,5-a]pyrimidin-5- yl)methyl)phenyl)acetate (150 mg).HPLC-MS tR = 2.50 min (UV254 nm); mass calculated for formula C37H49N5O4Si2683.3, observed LCMS m/z 684.4 (M+H).
Part E:
Methyl 2-(4-((7-(bis((2-(thmethylsilyl)ethoxy)methyl)amino)-6-bromQ-3-(quinolin-3- vQpyrazoloH .5-aipyrimidin-5-yl)methyl)phenyl)acetate:
Methyl 2-(4-{(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quLnolin-3-yl)pyrazolo[l,5- a]pyrimidin-5-yI)methyl)phenyl)acetate (150 mg, 0.22 mmol) was dissolved in ACN (10 mL).
NBS (40 mg, 0.22 mmol) was added and the mixture was stirred at room temperature for 1 h, After concentration, the product that resulted, methyl 2-(4-((7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(quinolin-3-yl)pyrazoio[1,5-a]pyrimidin-5- yl)methyl)phenyl)acetate was used in the next step without further purification
HPLC-MS tR = 2.66 min (UV254 nm); mass calculated for formula C37H48BrN5O4Si2 761.2, observed LCMS m/z 762.2 (M+H).
Part F:
Methyl 2-(4-(f7-amino-6-bromo-3-(αuinolin-3-yl)pyrazoloπ .5-a1pyrimidin-5- yl)methyDphenypacetate: The crude methyl 2-(4-((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(quinolin- 3-yI)pyrazolo[1,5-a]pyrimidin-5-yl)methyl)phenyl)acetate from above step E was treated with 50% TFA/H2O (3 mL) and stirred at room temperature for 1 h. Solvent was removed to yield thick oil, Methyl 2-(4-((7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[l,5-a]pyrimidin-5- yl)methyl)phenyl)acetate which was used in the next step directly without further purification. HPLC-MS tR = 1.72 m in (UV254 nm); mass calculated for formula C25H20BrN5O2 501.1, observed LCMS m/z 502.0(M+H).
Part G:
2-(4-((7-amino-6-bromo-3-(quinolin-3-yl)pyrazoio[1 T5-alpyrimidin-5- yl)methyl)phenyl)acetic acid :
Crude Methyl 2-(4-((7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1,5-a]pyrimidin-5- yl)methy[)phenyl)acetate from step F was dissolved in THF (5 mL) and LiOH (IN, i mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed under vacuo, and the resulting residue was taken over with water and the pH value was adjusted to 5-6. The solid was collected with filtration and purified with HPLC gave the product 2-(4-((7- amino-ό-bromo-3-tquinolin-3-ylJpyrazolofU5-alpyrimidin-5-ylJmethyl)phenylJacetic acid. HPLC-MS CR = 1.53 min (UV254 nm); mass calculated for formula C24Hi8BrN5O2487.1, observed LCMS m/z 487.9 (M+H).
By essentially same procedure given, in preparative above example, compounds 4.175 & 4. 176 given in Column 2 of Table 4Kcan be prepared.
TABLE-4 K

Synthesis of 6-bromo-5-(4-(methylsulfonyl)benzvn"3-(quinoiin-3-yl)pyrazolof1 ,5- alpyrimidin-7-amine:

SCHEME-17
Essentially the steps from A through G described in the example, synthesis of 2-(4-((7-amino- 6-bromo-3-(quinolin-3-yl)pyrazolo[J,5-a]pyrimidin-5~yi)methyl)phenyI)acetic acid can be used for this synthesis of 6-bromo-5-(4-(methylsulfonyI)benzyl)-3-(quinolin-3-yl)pyrazolo[1,5- a] py rim id in-7 -am ine
Part A:
4,4,5, 5-tetramethyi-2-(4-(methylthio)benzyl)-1 ,3,2-dioxaboroiane: 4,4,5, 5-tetramethyl-2-(4-(methyIthio)benzyl)- [,3,2-dioxaborolane prepared with the same condition described in above example Part A. HPLC-MS _R = 2.23 min (UV254 nm); mass calculated for formula Ci4H2IBO2S 264.1, observed LCMS m/z 265.2 (M+H).
Part B: 5-(4-(methylthio)benzyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5- aipyrimidin-7-amine:
Compound, 5-(4-(methylthio)benzyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[ 1 ,5- a]pyrimidin-7-amine was prepared with the same coupling condition described in above example Part B. HPLC-MS ta = 2.74 min (UV254 nm); mass calculated for formula C26H42N4O2SSi2530.3, observed LCMS m/z 531.2 (M+H).
Part C:
5-(4-(methylsulfonyl)benzyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5- aipyrimidin-7-amine:
Compound, 5-(4-(methylthio)benzyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[ 1,5- a]pyrimidin-7-amine (87 mg, 0.132 mmol) was dissolved in DCM (5 niL) and m-CPBA (66 mg, -70%, 0.27 mmoi) was added. The result mixture was stirred at room temperature for 1 h. NaHCO3 (aq.) was added and the water was extracted with DCM. The combined organics was dried over Na2SO4 and concentrated. The crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product 5-(4-(methy[sulfonyl)benzyl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (89 mg). HPLC-MS tR = 2.52 min (UV2S4 nm); mass calculated for formula C26H42N4O4SSi2 562.2, observed LCMS m/z 563.3 (M+H).
Part D:
3-iodo-5-(4-(methylsulfonvhbenzyl)-N.N-bisff2-
(trimethyisilyl)ethoxy)methyl)pyrazolof1 ,5-aipyrimidin-7-anπine:
Compound, 3-iodo-5-(4-(methylsulfonyl) benzyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl) pyrazoio [1,5-a]pyrimidin-7-amine was prepared with the same iodonation condition described in above example part C. HPLC-MS tR = 2.81 min (UV254 nm); mass calculated for formula C26H4IlN4O4SSi2 688.1, observed LCMS m/z 689.2 (M+H).
Part E:
5-(4-(methylsulfonyl)benzvn-3-(αuinoiin-3-yl)-N,N-bisf(2- ftrimethvlsilvπethoxv)methvl)pvrazolof1.5-alpvrimidin-7-amine:
Compound, 5-(4-(methylsulfonyl)benzyl)-3-(quinolin-3-yϊ)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoio[1,5-a]pyrimidin-7-arnine was prepared with the same coupling condition described in the above example Part D. HPLC-MS tR = 2.35 mm (UV2S4 πm); mass calculated for formula C35H47N5O4SSi2689.3, observed LCMS m/z 690.3 (M+H).
Part F: β-bromo-5-(4-(methylsulfonyl)benzyl)-3-(quinoiin-3-yl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazoiori ,5-alpyrimidin-7-amine:
Compound, 6-bromo-5-(4-(methylsulfonyl)benzyl)-3-(quinoIin-3-yI)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[l ,5-a]pyrimidin-7-amine was prepared with the same bromonation condition described in the above example 1 Part E. HPLC-MS IR = 2.52 min (UV254 nm); mass calculated for formula C35H46BrN5O4SSi2 767.2, observed LCMS m/z 768.2 (M+H).
Part G:
6-bromo-5-(4-(methylsulfonyl)benzyl)-3-(quinolin-3-yl)pyrazolof1 ,5-alpyrimidin-7- amine:
Ttile compound, 6-bromo-5-(4-(methylsulfonyl)benzyl)-3-(quinolin-3-yl)pyrazolo[1,5- a]pyrimidin-7-amine was prepared with the same deprotection condition described in example I Part F. HPLC-MS tR = 1.49 min (UV254 nm); mass calculated for formula C23Hj8BrN5O2S 507.0, observed LCMS m/z 5O8.O(M+H).
By essentially the same procedure given in preparative above example, compound 4.177-4.178 given in Column 2 of Table 4L can be prepared.
TABLE 4L


Synthesis of 4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5- yloxy)benzoic acid:

SCHEME-18 Part A:
Methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolof1 ,5-a1pyrimidin-5- yloxy)benzoate:
The methyl 4-hydroxybenzoate (330 mg, 0.2 mmol) was dissolved in dry DMF (3 niL) and NaH (8 mg, 60% in oil, 0.2 mmol) was added and the mixture was stirred at room temperature for 5 min. Then, 5-chloro-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin- 7-amine (430 mg, 0.1 mmol) was added and the resulting mixture was heated up to 130 oC with Microwave and stirred for 30 min. After cooling Co room temperature, NH4CI (aq) was added to quench the reaction and extracted with EtOAc (50 mL x 3). The combined organics
were washed with water and brine and dried over Na2SCW. After concentration, the residue was purified with column (silica gel, 20% EtOAc/Hexane) gave the product, methyl 4-(7-(bis((2- (trimemylsilyl)ethoxy)methyl)amino)pyrazolo[1,5-a]pyrimidin-5-yloxy)benzoate (490 mg) as clear oil. HPLC-MS IR = 2.84 min (UV254 πm); mass calculated for formula C26H4ON4O5Si2 544.3, observed LCMS m/z 545.3 (M+H).
Part B:
Methyl 4-(7-{bisff2-(trimethylsiiyl)ethoxy)methynamino)-3-iodopyrazolof1 ,5- a1pyrimidin-5-yloxy)benzoate:
Methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[ 1 ,5-a]pyrimidin-5- yioxy)benzoate (450 mg, 0.81 mmol) was dissolved in ACN ( 15 mL) and NIS (200 mg, 0.88 mmol) was added. The resulting mixture was stirred at room temperature for 2 h and then the solvent was removed under reduced pressure. The residue was purified with column (silica gel, 10% EtOAc/Hexane) gave the product, methyl 4-(7-(bis((2-
(trimethylsilyl)ethoxy)methyl)ammo)-3-iodopyrazolo[1,5-a]pyrimidin-5-yloxy)benzoate (541 mg) as yellowish oil. HPLC-MS CR = 3.10 min (UV254 nm); mass calculated for formula C26H39IN4O5Si2 670.2, observed LCMS m/z 671.2 (M+H). Part C:
Methyl 4-(7-(bis(f2-(trimethylsiiyl)ethoxy)methyπamSnoV3-(6-phenylpyridin-3- v])pyrazoiof1 ,5-a1pyrimidin-5-yloxy)benzoate:
Under Ar, methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1,5- a]pyrimidin-5-yloxy)benzoate (180 mg, 0.26 mmol) was mixed with Pd(dppf)Cl2 (21 mg, 0.26 mmol, K3PO4 (106 mg, 0.5 mmol), 3-quinoline bornic acid (55 mg, 0.31 mmol) and dtoxane (10 mL with 1 ml water). The resulting mixture was heated at 90 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (60 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- (6-phenyIpyridin-3-yl)pyrazoIo[1,5-a]pyrimidin-5-yIoxy)benzoate. HPLC-MS tR = 2.77 min (UV254 nm); mass calculated for formula C37H47N5O5Si2697.3, observed LCMS m/z 698.2 (M+H). Part D:
Methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenylpyridin-3- yl)pyrazolori ,5-a1pyrimidin~5-yloxy)benzoate:
Compound, methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3- yl)pyrazolo[1,5-a]pyrimidin-5-yloxy)benzoate (150 mg, 0.22 mmol) was dissolved in ACN (10 mL). NBS (40 mg, 0.22 mmol) was added and the mixture was stirred at room temperature for
1 h. After concentration, the residue, methyl 4-(7-(bis((2-
(trimethylsiIyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[l,5- alpyrimidin-5-yloxy)benzoate was used in the next step without further purification. HPLC-MS tR = 3.06 min (UV254 πm); mass calculated for formula C37H46BrN5O5Si2775.2, observed LCMS m/z 776.0 (M+H).
Part E:
Methyl 4-(7-(bisf(2-(trimethylsiiyl)ethoxy)methyl)amino)-6-(1 -ethoxyylnyl)-3-(6- phenylpyridin-3-yl)pyrazolof1 ,5-aipyrimidin-5-yloxy)benzoate:
Under Ar, methyl 4-(7-(bis((2-(trimethylsiIyl)ethoxy)methyl)amino)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yIoxy)benzoate (180 mg, 0.26 mmoi) was mixed with Pd(dppf)Cl2 (21 mg, 0.26 mmol, K3PO4 (106 mg, 0.5 mmol), 3-phenyl pyridyl bornic acid (75 mg, 0.31 mmol) and dioxane (10 mL with 1 ml water). The resulting mixture was heated at 90 °C and stirred over night. After cooled to room temperature, the mixture was diluted with EtOAc (60 mL) and filtered through celite. After concentration, the crude was purified with column (silica gel, 0-30% EtOAc/Hexane) gave the product 5 (150 mg). which was used in the next step directly without further purification. HPLC-MS tβ = 2.96 min (UV254 πm); mass calculated for formula C4IH53N5O6Si2 767.4, observed LCMS m/z 768.4 (M+H).
Part F:
Methyl 4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5- yloxy)benzoate :
The crude from above step E was treated with 50% TFA/H2O (3 mL) and stirred at room temperature for 1 h. Solvent was removed to yield thick oil, methyl 4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-6-(l-ethoxyvinyl)-3-(6-phenylpyridin-3-yl)pyrazolo[1,5- a]pyrimidin-5-yioxy)benzoate. HPLC-MS IR = 1.84 min (UV254 nm); mass calculated for formula C27H21N5O4 479.2, observed LCMS m/z 480.2 (M+H).
Part G:
4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazoiof1 ,5-a]pyrimidin-5-yloxy)benzoic acid;
Crude from step F was dissolved in THF (5 mL) and LiOH (IN, 1 mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed under vacuo, and the resulting residue was taken over with water and the pH value was adjusted to 5-6. The solid was collected with filtration and purified with HPLC gave the product 4-(ό-acetyl-7- amino-3-(6-phenylpyridin-3-yl) pyrazolo[1,5-a]pyrimidin-5-yloxy)benzoic acid.HPLC-MS IR = 1.49 min (UV254 πm); mass calculated for formula C26H19N5O4465.1, observed LCMS m/z 466.2 (M+H).
By essentially the same procedure given in preparative above example(scheme-18), compound 4.179 given in Column 2 of Table 4M can be prepared.
TABLE 4M


Synthesis of 4-(7-amino-6-cvano-3-(6-phenylpyridin-3-yl)pyrazolori ,5-aipyrimidin-5- yl)benzoic acid:
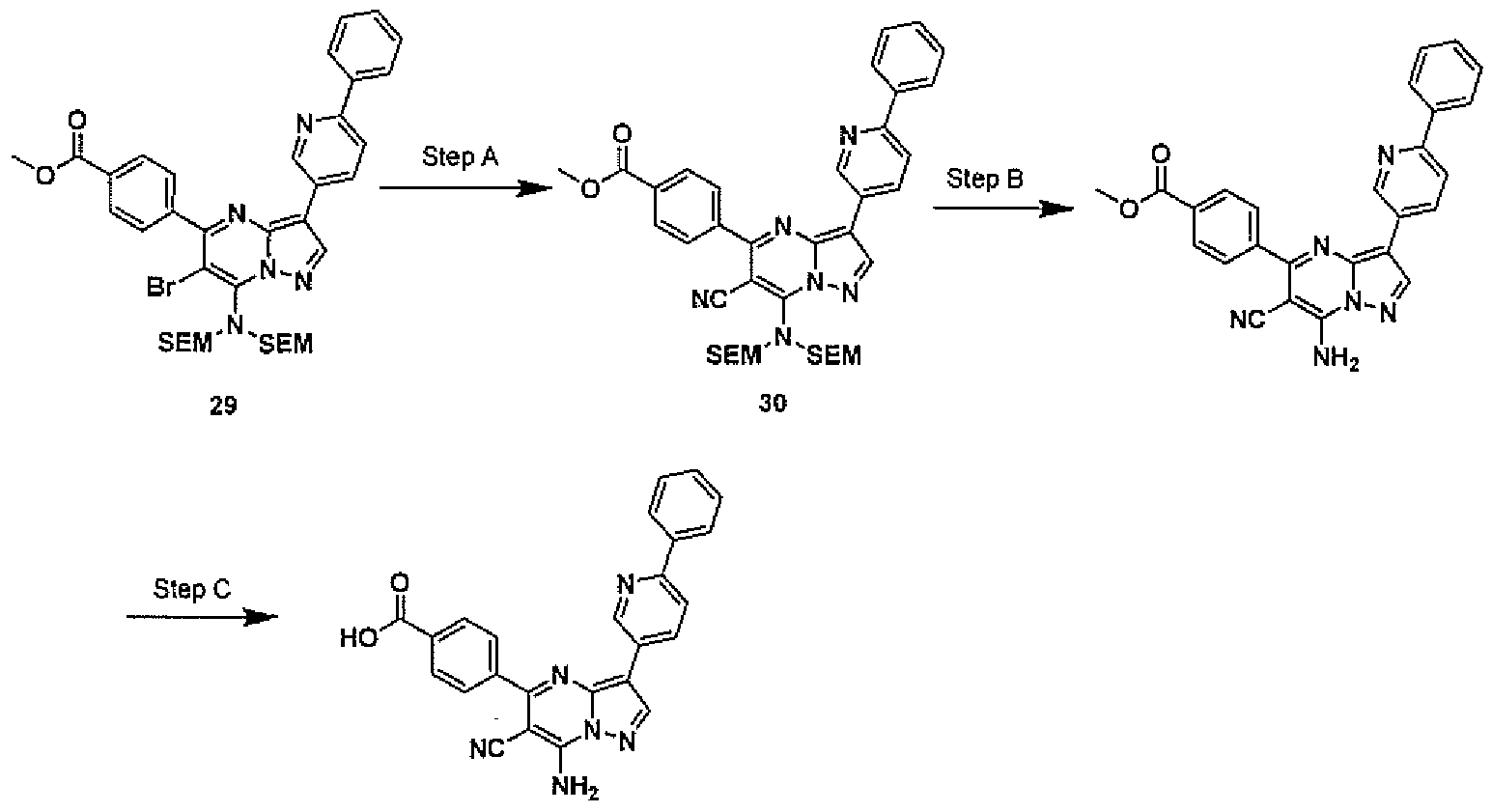
SCHEME-19
Part A
Methyl 4-(7-(bis((2-(trimethylsilyi)ethoxy)methyl)amino)-6-cyano-3-(6-phenylpyridin-3- yl)pyrazolo[1,5-a]pyrimidin-5-yl)benzoate:
Under Ar, compound, methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazolo[3 ,5-a]pyrimidin-5-yi)benzoate (80 mg, 0.1 mmol) was mixed with Pd(PPh3)4 (12 mg, 0.01 mmol, Pd(t-Bu3P)3 (5 mg, 0.01 mmol), cyanotributylstannane (85 uL, 0.3 mmol) and dioxane (3 mL) in sealed tube. The resulting mixture was heated at 160 °C and stirred 2 hrs. After cooled to room temperature, the mixture was filtered through 10% KF on silica gel and washed with EtOAc. After concentration, the crude was used in the next step directly without further purification. HPLC-MS tR = 3.18 min (UV254 nm); mass calculated for formula C38H46N6O4Si2 706.3, observed LCMS rπ/z 707.2 (M+H).
Part B
Methyl Φfy-amino-β-cvano-3-(6-phenylpyriclin-3-yl)pyrazolori .5-aipyπmidin-5- yQbenzoate: The crude from above step A was treated with 50% TFA/H2O (3 niL) and stirred at room temperature for 1 h. Solvent was removed to yield thick oil which was used in the next step directly without further purification. HPLC-MS t.R = 2.15 min (UV254 nm); mass calculated for formula C26Hi8N6O2446.1, observed LCMS m/z 447.1 (M+H). Part C:
4-(7-amino-6-cvano-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yi)benzoic acid:
Crude compound from step B was dissolved in THF (5 mL) and LiOH (IN, 1 mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed under vacuo, and the resulting residue was taken over with water and the pH value was adjusted to 5~6. The solid was collected with filtration and purified with HPLC gave the product 4-(7-amino-6- cyano-3-(6-phenylpyridin-3-yl) pyrazolo[1,5-a]pyrimidin-5-yl)benzoic acid. HPLC-MS tR - 1.40 min (UV254 nm); mass calculated for formula C25Hi6NeO2432.1, observed LCMS m/z 433.1 (MH-H).

Synthesis of Ethyl 1 ,4-dioxaspiror4.51decane-8-carboxylate:

Ethyl 4-oxocyclohexane carboxylate (122.56 mmol, 20.86 g) and para-toluenesulfonic acid monohydrate (12.56 mmol, 2.33 g) are charged to a 1000 rriL roundbottom flask. To this flask was added benzene (300 mL) followed by ethylene glycol (0.37 mol, 20.5 mL). The resulting bi-layer reaction mixture was refluxed overnight in a Dean-Stark trap. Upon cooling, the reaction mixture was diluted in 300 mL DCM and washed with 300 mL saturated NaHCO3(aq)- The acqueous layer was washed with DCM (3X) and the combined organics are dried over Na2SO4. The residue was disolved in DCM and purified by silica gel chromatography on 0% to 50% ethyl acetate in hexanes gradient. The product is tracked via PMA stain. Chromatography yields 17.05 grams (79.58 mmol, 65% yield) ethyl 1 ,4-dioxaspiro[4.5]decane-8-carboxylate.
Synthesis of 1 ,4-DioxasDiro[4.51decane-8-carboxylic acid:

Synthesis of Ethyl 3-oxo-3-(1 ,4-dioxaspirof4.51decan-8-yl)propanoate:

1 ,4-Dioxaspiro[4.5]decane-8-carboxylic acid (80.34 mmol, 14.96 g) is charged to a 500 mL roundbottom flask. To this flask is added anhydrous THF (200 ml_), followed by N, N'-carbonyldiimidazole (96.41 mmol, 15.63 g). After vigorous release of CO2 gas, the solution was flushed with argon, sealed, and allowed to stir overnight at room temperature under argon.
After 18 hours, in a separate, sealed and argon-flushed 1000 mL roundbottom flask, LiHMDS (1.0 M in THF1 168.7 mmol) is added to 200 mL anhydrous THF stirring at -78° C. To this solution is added dropwise anhydrous ethyl acetate (173 mmoi, 16.9 mL). This solution is allowed to stir at -78° C for 1 hour prior to dropwise addition of original CDi/acid solution that has been stirred overnight. The reaction mixture was allowed to stir and warm to room temperature overnight.
The reaction was then quenched with saturated NH4CI(aq) (500 mL) and extracted with Et2O (x2). The combined organics are then washed with H2O, saturated brine, and dried over Na2SO4. The solvent is removed in vacuo and the residue is taken up in DCM. The reaction mixture was purified with silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield ethyl 3-oxo-3- (1 ,4-dioxaspiro[4.5]decan-8-y!)propanoate (1 1.26 g, 55% yield) as pale yellow oil.
Synthesis of 5-( 1 ,4-dioxaspiror4.51decan-8-yl)pyrazolo[1 ,5-aipyrimidin-7-ol:

To a 20 ml_ scintillation vial containing ethyl 3-oxo-3-(1,4-dioxaspiro[4.5]decan-8- yl)propanoate {1 1.7 mmol, 3.00 g) is added 3-amino-1 H-pyrazole (11.7 mmol, 973 mg). The two oils were mixed and heated neat at 100° C for 3 hours. The resulting off-white solid was dissolved up in EtOH (100 ml_) and reduced in vacuo to remove water formed during cyclization. This solid was used in the next step without further purification.
Synthesis of 7-chloro-5-(1 ,4-dioxaspiro[4.51decan-8-yl)pyrazolo[1 ,5-aipyrimidine:

5-(1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[1 ,5-a]pyrimidin-7-ol (11.7 mmol) is charged to 250 ml_ roundbottom flask. To this fiask is added 4 mL N, N-dimethylaniline, followed by 40 mL POCl3. This suspension was sonicated to break up the starting material and stirred at room temperature for 18 hours. After 18 hours, a solution formed. The solution was reduced in vacuo and cooled to 0° C in ice bath. The reaction is then quenched with sat. NaHCO3(aq) and extracted with DCM (x3). The combined organics are dried with Na2SO4 and the solvent was removed in vacuo. The resulting oil was purified via silica gel column on 20% to 100% ethyl acetate in hexanes gradient to yield 7-chloro-5-(1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazoio[1 ,5- a]pyrimidine (2.65 grams, 77% across 2 steps) as white solid.
Synthesis of 5-( 1 ,4-dioxaspirof4.51decan-8-yi)pyrazolof 1 ,5-alpyrimidin-7-amine:

7-chloro-5-{1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[1 ,5-a]pyrimidine (5.58 mmol, 1.64 g) is charged to 10-20 ml microwave vessel. To this vessel is added 10 mL ~7N NH3 in methanol. The vessel was sealed and heated at 100° C for 18 hours. After 18 hours, the reaction mixture was cooled to room temperature and diluted with 100 mL DCM. The resulting solution is washed with saturated NaHCO3(aq) and extracted with DCM twice more. The combined organics were dried over Na2SO4 and the solvent removed in vacuo to yield 5-{1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazoto[1 ,5-a]pyrimidin-7- amine (1.52 g, 99% yield) as pale orange solid.
Synthesis of 4-(7-aminopyrazolod ^-alpyrimidin-5-vPcvclohexanone:

In a 40 mL scintillation vial is combined 5 (1 ,4 dioxaspiro[4.5]decan-8-yl)pyrazoio[1,5- a]pyrimidin-7-amine {5.54 mmol, 1.52 g), EtOH (10 mL), H2O (4 mL) and 4N
HCkdioxane (4 mL). The vial was capped, sealed and the reaction was heated to 80° C overnight. After 18 hours, the reaction mixture was cooled to room temperature and diluted with DCM (100 mL). The solution was washed with saturated NaHCO3^ and extracted with DCM twice more. The combined organics are dried over Na2SO4 and the solvent removed in vacuo to yield 4-(7-aminopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexanone (1.18 g, 93% yield) as pale orange solid.
Synthesis of 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolori ,5-aipyrimidin- 5-yl)cyclohexanone:

4-(7-aminopyrazolo[1 ,5-a]pyrimidin-5-y!)cyclohexanone (5.12 mmol, 1.18 g) was dissolved in DCM (15 mL). To this solution was added N, NJ-diisopropyiethylamine (17.94 mmol, 3.13 mL). The resulting solution was stirred at room temperature while 2-(trimethylsilyl)ethoxymethyl chloride (SEM-CI, 17.94 mL, 3.17 mL) in DCM (5 mL) was added dropwise. After the addition was completed, the reaction mixture was stirred at 50° C for 10 minutes. The solvent was removed in vacuo, and the residue was purified on silica gel column (0% to 60% ethyl acetate in hexanes gradient) to yield 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexanone (1.87 g, 74% yield) as pale yellow oil.
Synthesis of Ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5- a1pyrimidin-5-yl)cvclohexylidene)acetate:

A 20 mL scintillation vial containing 5 mL THF was charged sodium hydride (60% w/w in mineral oil, 448 μmol, 18 mg). This suspension was broken up via sonication. Triethyl phosphonoacetate (448 μmol, 89.3 μL) in THF (2 mL) was added dropwise. The resulting solution was stirred at room temperature for 10 minutes. To this solution was slowly added a solution of 4-(7-(bis((2-
(trimethy!silyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyr!midin-5-yl)cyclohexanone (408 μmol, 200 mg) in THF (2 mL). The reaction mixture was stirred for 18 hours at room temperature. After 18 hours, the reaction mixture was diluted with DCM (25 mL) and
washed with H2O. The aqueous phase was washed with DCM (x2), and the combined organics was dried over NaaSOtt. The solvent was removed in vacuo to yield ethyl 2-(4-(7-(b!s((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin- 5-yl)cyclohexyJidene)acetate (222 mg, 97% yield) as pale yellow oil.
Synthesis of 2-(4-(7-(Bis((2-(trimethy]silvOethoxy)methyhamino)pyrazolori .5- alpyrimidin-5-yl)cvclohexylidenetecetonitriie:

2-(4-(7-{Bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexy!idene)acetonitrile was synthesized in a manner similar to the synthesis of ethyl 2-{4-(7-(bis((2-{trimethy!silyi)ethoxy}nnethyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyiidene)acetate, but with diethyl cyanomethylphosphonate substituted for triethyl phosphonoacetate.
Synthesis of ethyl 2-(4-(7-(bis((2-(trimethylsiiyl)ethoxy)methyl)amino)pyrazolo[1 ,5- aipyrimidin-5-yi)cvclohexyl)acetate:

Synthesis of 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy^methyl)amino)pyrazolori ,5- alpyrimidin-5-vOcvclohexyl)acetonitriie:
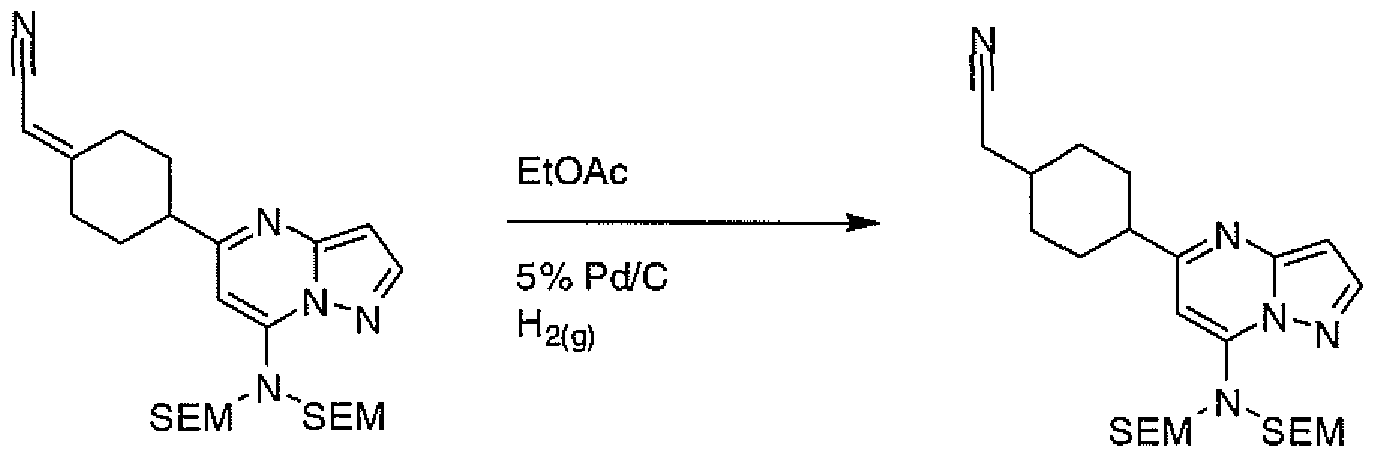
2-(4-(7-(Bis((2-{trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetonitriie was synthesized in a manner similar to the synthesis of ethyl 2-(4-(7-{bis((2-(trimethylsiIyl)ethoxy)methyl)amino)pyrazolo[1 ]5-a]pyrimidin-5- yl)cyclohexyl)acetate, but with 2-(4-{7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyc!ohexyiidene)acetonitrile substituted for ethyl 2-(4~(7-(bis((2- (trimethylsityl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexylidene)acetate.
Synthesis of ethyl 2-(4-(7-(bis((2-(trimethylsilvhethoxyϊmethyπamino)-3- iodopyrazoloπ ^-aipyrimidin-5-yl)cvclohexyl)acetate:
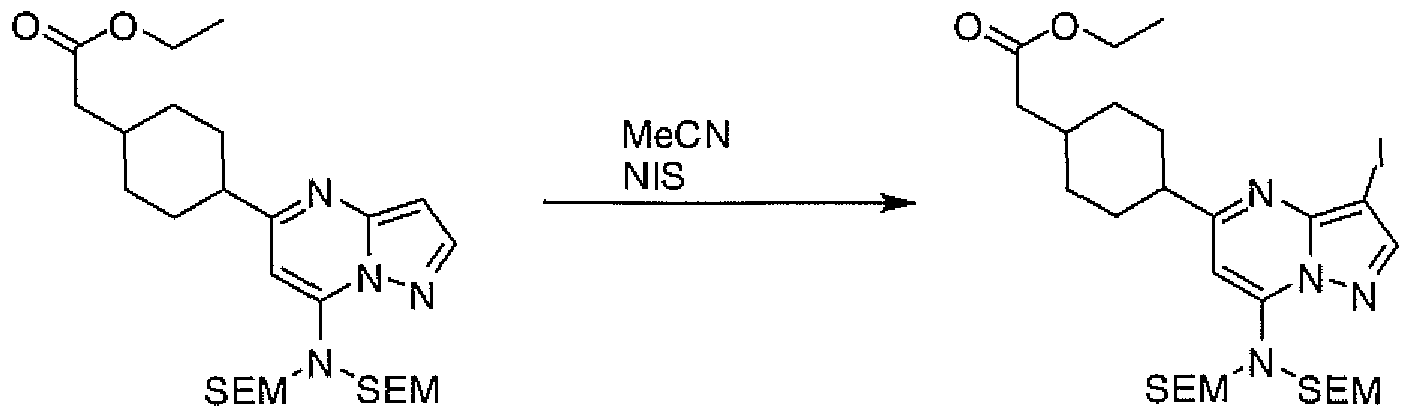
Synthesis of 2-(4-(7-(bis((2-(tπmethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolof 1 ,5- alpyrimidin-3-yl)cvclohexyl)acetonitrile:

2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 !5-a]pyrimidin-5- yl)cyclohexyi)acetonitrile was synthesized in a manner similar to the synthesis of ethyl
2-(4-(7-(bis((2-(trimethyIsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate, but with 2-(4-(7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetonitrile substituted for ethyl 2-(4-(7-(bis((2- (trimethylsilyl)ethoxyJmethylJaminoJpyrazoloJi .5-alpyrimidin-5-yl)cyclohexyl)acetate.
Synthesis of ethyl 2-(4-(7-(bis((2-(trimethylsilvnethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazoloπ .5-aipyrimidin-5-yl)cvclohexyl)acetate:

To a 40 ml_ scintillation via! was charged 3-quinoline boronic acid (0.73 mmol, 127 mg), K3PO4 (1.46 mmol, 310 mg) and PdC!2(dppf) • CH2CI2 (.049 mmol, 40 mg). To this mixture was added a solution of ethyl 2-(4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1,5-a]pyrimidin-5- yl)cyciohexyl)acetate (335 mg, 0.49 mmol) in dioxane (9 m!_). To this suspension was added distilled H2O (1 m!_). The resulting solution was stirred at 100° C for 18 hours. The reaction was concentrated in vacuo and then purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield ethyl 2-{4-(7- (bis((2-(trimethylsiiyl)ethoxy)methyl)amino)-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate (198 mg, 287 μmol, 59% yield) as yellow oil.
Synthesis of 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyπamino)-3-(αuinolin-3- yl)pyrazoloH ^-alpyrimidin-5-vOcvclohexyl)acetonitrile:

2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyI)amrno)-3-(quinolin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)acetonitrile was synthesized in a manner similar to the synthesis of ethyl 2-(4-(7-(bis((2-(thmethylsilyi)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-y!)cyclohexyl)acetate, but with 2-(4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetonitrile substituted for ethyl 2-(4-(7-(bis((2- (trimethylsily[)ethoxy)methy!)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate.
Synthesis of ethyl 2-(4-(7-(bis((2-(trimethyisilvBethoxy)methyπamino)-6-bromo-3- (quinolin-3-yl)pyrazolori ,5-aipyrirnidin-5-yltovclohexyl)acetate:

To a 25 mL roundbottom flask was charged ethyl 2-(4-(7-{bis((2-(trimethylsi!yl)ethoxy)- methyl)amιno)-3-(quinolin-3-yl}pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)acetate (198 mg, 287 μmoi) and acetonitrile (10 mL). To this solution was added N- bromosuccinimide (56 mg, 316 μmol). The resulting reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo and the resulting oil was then purified via silica gel chromatography (0% to 30% ethyl acetate in hexanes gradient) to yield ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)- 6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)acetate (205 mg, 266 μmol, 93% yield) as yellow oil.
Synthesis of 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyπamino)-6-bromo-3-(Quinolin- 3-vOpyrazoloH .5-aipyrimidin-5-yl)cvclohexyl)acetonitrile:

Synthesis of 2-(4-(7-amino-6-bromo-3-(αuinolin-3-yl)pyrazolo[1 ,5-a1pyrimidin-5- yl)cvclohexyl)acetic acid:

To a 20 nrsL scintillation vial was charged ethyl 2-(4-{7-(bis((2-(trimethy!silyl)ethoxy)- methyl)amino)-6-bromo-3-(quinolin-3-yl)pyrazoio[1 ,5-a]pyrirrιidin-5- yl)cyclohexyl)acetate (100 mg, 130 μmol) and 2:1 THF:H2O (10 ml_). To this solution was added 1 N LiOH(aq) (200 μi_). The resulting reaction mixture was stirred at room temperature for 18 hours. The reaction mixture was concentrated in vacuo and the resulting residue was dissolved in TFA and stirred at room temperature for 1 hour. This solution was concentrated in vacuo and the residue was dissolved in 3:1 DMSO:MeCN. The crude product was purified via reverse-phase preparatory HPLC to yield 2-(4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyciohexyl)acetic acid (m+H = 480.1 , retention time = 3.95 min (isomer 1) and 4.03 min (isomer 2))
Synthesis of fraπs-2-(4-(7-amino-6-bromo-3-(αuinolin-3-v[)pyrazo]oi"1 ,5-alpyrimidin-5- yl)cvclohexyPacetic acid and c/s-2-(4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5- alpyrimidin-5-yl)cvclohexyl)acetic acid:

2-(4-(7-amino-6-bromo-3-(quinolin-3-yi)pyrazoio[1 ,5-a3pyrimidin-5-yl)cyclohexyl)acetic acid is taken up in 9:1 MeOH:TFA. The mixture is etuted through chiral column to yield two single isomers. (m+H = 480.1 , 10 min. RP HPLC Ret(isoi): 4.03 min., 10 min. RP HPLC Ret(iso2): 3.95 min.)
Synthesis of 2-(4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5-alpyrimidin-5- vQcyclohexyl)acetonitrile:

2-(4-(7-amino-6-bromo-3-{quinolin-3-yi)pyrazolo[1 ,5-a3pyrimidin-5- yl)cyciohexyl)acetonitrile (44 μmol, 32 mg) is disolved in 1 mL TFA in a 20 mL scintillation viai. The reaction mixture is allowed to stir 1 hour at room temperature. The reaction mixture is concentrated in vacuo and purified via reverse-phase preparatory HPLC to yield 2-(4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyciohexyi)acetonitrile as a two-isomer mixture. (m+H = 461.2, retention time = 4.13 min (isomer 1 ) and 4.18 min (isomer 2))
5-Trøns^-(fi H-tetrazol-5-yl)methyπcvclohexyl)-β-bromo-3-(quinolin-3-yl)pyrazoloπ .5- alpyrimidin-7-amine and 5-c/s-4-((1 H-tetrazoi-5-yl)methy0cvclohexyl)-6-bromo-3- (quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine:
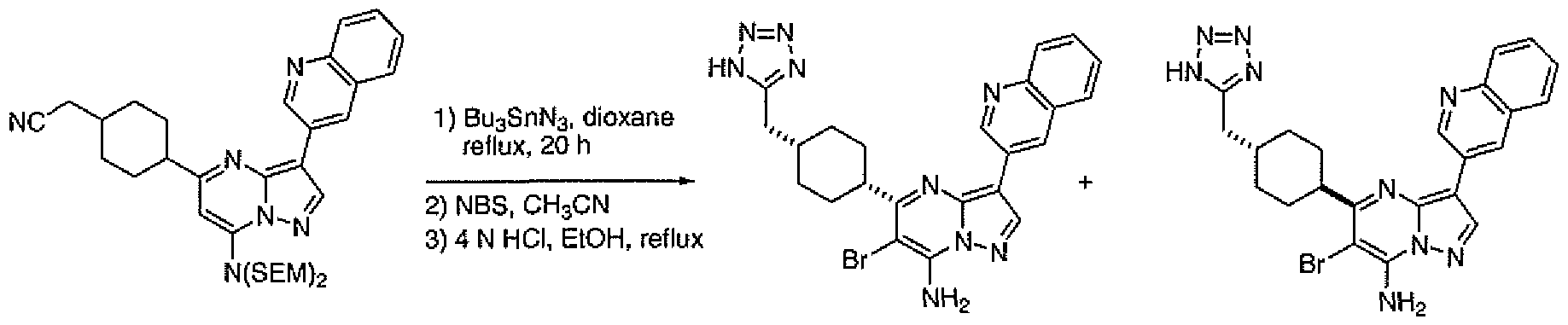
To a solution of 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)acetonitrile (28 mg, 0.044 mmol) in dioxane {0.50 mL) was added azotributyltin azide (22 mg, 0.066 mmoi). The mixture was heated at 105 °C for 20 h. The tin reagent was removed by a short column and the fraction containing the desired product was collected and concentrated in vacuo. The residue was dissolved in CH3CN and NBS (2.0 mg) was added. The mixture was stirred at rt for 1 h and concentrated in vacuo. The residue was purified by prep-LC to give the fraπ-isomer and the c/s-isomer of the title compounds. LC/MS RT = 3.53 min (trans), 3.73 min (cis). Mass calculated for, M+H 504.12, observed 504.12.
By essentially the same procedures given in the complete synthesis of 2-(4-(7-amino- 6-bromo-S^quinolin-3-yl)pyrazoloII .S-alpyrimidin-5-yl)cyclohexyl)acetonitrile, but with cis/trans isomers separated on silica gel after hydrogen reduction. By essentially the same procedure given in Scheme 6, the compounds listed in Table 5 can be prepared.
Table 5




fΦfy-Amino-6-pyridin-Φyl-S-quinolin-S-yl-pyrazolori .5-alpyrimidin-5-yl)-cvclohexyll- acetic acid:

SCHEME-21 :
4-Pyridine boronic acid (0.156 mmoi, 20 mg), K2CO3 (0.234 mmol, 33 mg), and Pd(PPh3)4 (0.008 mmol, 10 mg) was added to a solution of (4-{7-[bis-(2- trimethylsilanyl-ethoxymethyl)-aminol-e-bromo-S-quinolin-S-yl-pyrazoloCI .5- a]pyrimidin-5-yl}-cyciohexyl)-acetic acid ethyl ester (0.078 mmol, 60 mg) in dioxane (1 mL). To this suspension was added distilled H2O (0.2 ml_). The resulting reaction mixture was stirred at 100° C under argon for 18 hours. The reaction mixture was concentrated in vacuo. The crude mixture was dissolved in 2:1 MeOH:H2O (1.5 mL) and was treated with 2N NaOH(aq) {0.5 mL). The resulting reaction mixture was stirred at room temperature for 18 hours. Aqueous hydrochloride solution (1.0 N, 2 ml) was added to the reaction mixture and the resulting solution was stirred at 65 °C for 2 h. The solution was concentrated in vacuo and purified by prep-LC to afford the title compound (3.6 mg) as a mixture of cis and trans isomers: LC/MS RT = 2.84 min. Mass calculated for, M+H 479.21 , observed 479.21.
r4-(7-Amino-6-cvano-3-quinolin-3-yl-pyrazoion ,5-a1pyrimidin-5-yl)-cvclohexyl]-acetic acid
A degassed mixture of (4-{7-[bis-(2-trimethylsilanyl-ethoxymethyl)-amino3-6-bromo-3- quinolin-3-yl-pyrazolo[1 ,5-a]pyrimidin-5-yt}-cyclohexyl)-acetic acid ethyl ester {0.078 mmol, 60 mg), Bu3SnCN (50 mg, 0.156 mmoL), Pd[P{t-Bu)3]2 (8.3 mg, 0.016 mmoL) in Dioxane (1 mL) was heated at 100°C for 18 h. The reaction mixture was concentrated in vacuo. The crude mixture was dissolved in 2:1 MeOH:H2O (1.5 mL) and was treated with 2N NaOH(aq) (0.5 mL). The resulting reaction mixture was stirred at room temperature for 18 hours. Aqueous hydrochloride solution {1.0 N, 2 ml) was added to the reaction mixture and the resulting solution was stirred at 65 °C for 2 h. The solution was concentrated and purified by prep-LC to afford the title compound (8.8 mg) as a mixture of cis and trans isomers: LC/MS RT = 3.88 min. Mass calculated for, M+H 427.18, observed 427.18.
By essentially the same procedure given in Scheme 7, the compounds listed in Table 6 can be prepared.
Table 6


2-(4-(7-amino-6-bromo-3-(αuinolin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)cvclohexyl)-N- (methylsulfonvOacetamide

SCHEME-22
A mixture of 2-(4-(7-{bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(quinolin-3- yl)pyrazoio[1 ,5-a]pyrimidin-5-yi)cyclohexyl)acetic acid (60 mg, 0.081 mmo!), methanesuifonamide (12 mg, 0.122 mmol), 4-dimethy!aminopyridine (20 mg, 0.163 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (32 mg, 0.163 mmol) in dichloromethane (2.0 ml) was stirred at room temperature for 16 h. The reaction mixture was concentrated in vacuo. The crude mixture was dissolved in methanol (2 ml) and treated with 1 N hydrochloride solution (2 ml) at 65 °C for 2 h. The reaction solution was concentrated and purified by prep-LC to afford the title compound (two isomers, cis and trans). Isomer 1 : LC/MS RT = 3.63 min. Mass calculated for, M+H 557.09, observed 557.09. Isomer 2: LC/MS RT = 3.88 min. Mass calculated for, M+H 557.09, observed 557.09.
By essentially the same procedure given in Scheme 22, the compounds listed in Table 7 can be prepared.
TABLE- 7

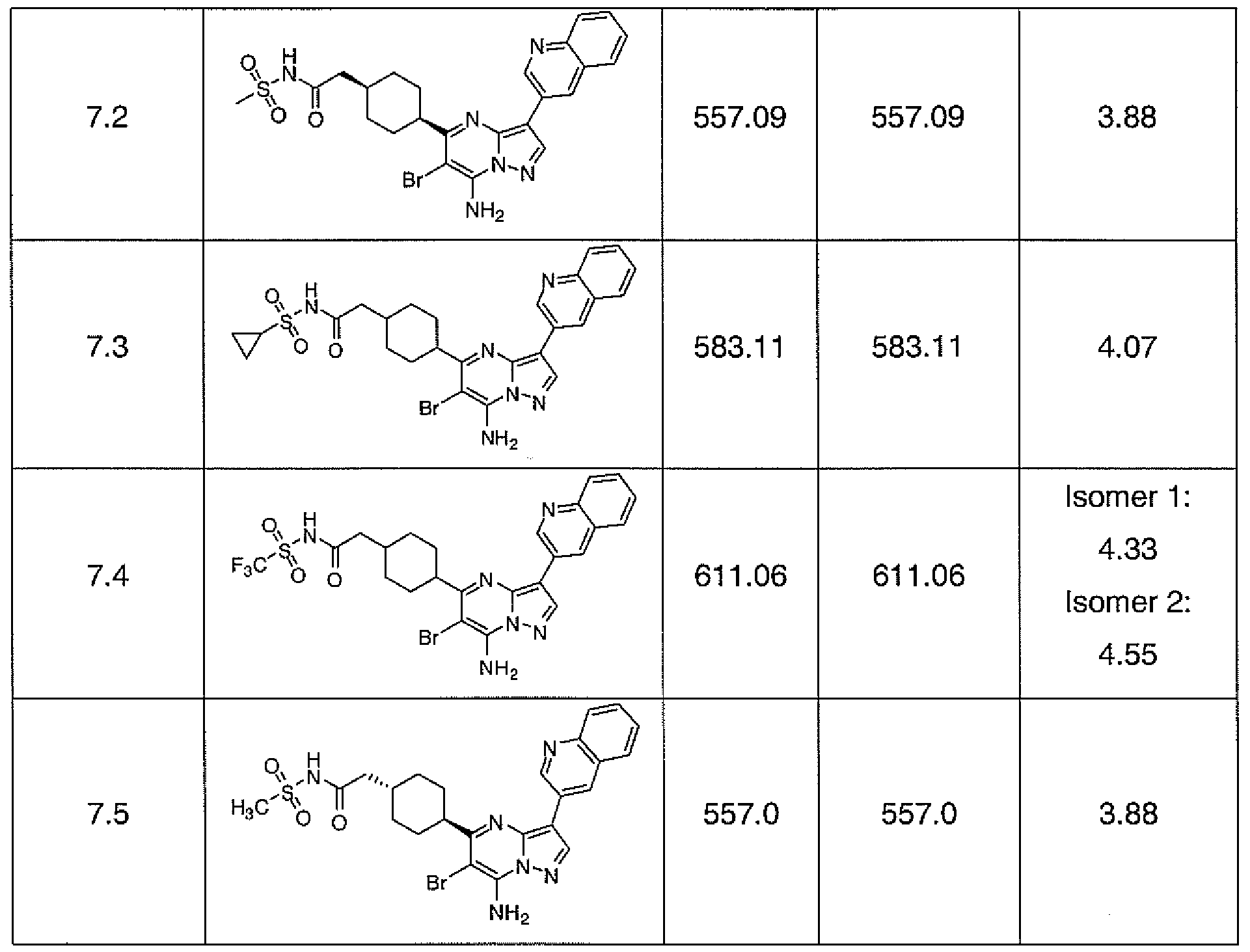
Ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)m8thyl)amino)-3-(naphthalen-2- vnpyrazoion ,5-aipyrimidin-5-yl)cvclohexyl)acetate. LC/MS RT = 3.40 min {5 min method). Mass calculated for, M+H 689.38, observed 689.38.

2-(4-(7-Amino-6-bromo-3-(6-bromonaphthalen-2-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cvclohexyl)acetic acid . LC/MS RT = 6.01 min (10 min method). Mass calculated for, M+H 557.01 , observed 557.01.

2-(4-(7-amino-6-bromo-3-(6-bromonaphthalen-2-yl)pyrazolo[1 ,5-a]pyrimidin -5- yl)cyclohexyl)-N-(methylsulfonyl)acetamide. LC/MS RT = 5.86 min (10 min method). Mass calculated for, M+H 634.01 , observed 634.01 .

fS)-2-(f1s,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-alpyhmidin-5- yl)cvclohexyl)-2-fluoroacetic acid
Part-A
Ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)m8t{iyl)amino)pyrazolo[1 ,5-a1pyrimidin-5- yl)cvclohexylidene)-2-fluoroacetate

To a 250 mL roundbottom flask containing 80 ml. THF was charged sodium hydride (60% w/w in mineral oil, 12.86 mmol, 515 mg). This suspension was broken up in the
sonicaid and then cooled to 0° C in icebath. Triethyl 2-fluoro-2-phosphonoacetate (12.86 mmol, 3.12 g) was taken up in THF (10 ml_) and added dropwise to stirring NaH suspension. The resulting solution was allowed to stir at 0° C for 10 minutes. 4- (7-(bis{(2-(trimethylsilyl)ethoxy)methyI)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexanone (6.43 mmol, 3.16 g) was taken up in THF (10 mL) and added dropwise to the stirring solution. The reaction was allowed to warm to room temperature and stir 18 hours. At 18 hours, the reaction was diluted with DCM (400 mL) and washed with H2O. The organic layer was collected and dried over NaaSO4. This solution was reduced in vacuo and purified via flash chromatography to yield the title compound (3.43 g, 5.92 mmol).
PART-B
Ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyπamino)pyrazoloπ ,5-alpyhmidin-5- yl)cvclohexylidene)-2-fluoroacetate

To a 50 mL roundbottom flask was charged ethanol (20 mL), ethyl 2-(4-(7-(bis((2- (trimethylsilyl)ethoxyJmethyl)aminoJpyrazoloCI ^-aJpyrimidin-5-yl)cyclohexylidene)-2- fiuoroacetate (1.99 mmol, 1.15 g), and 10% palladium on carbon (200 mg). The flask was sealed and degassed under vacuum. Hydrogen gas was then added via balloon and the reaction was allowed to stir at room temperature for 18 hours. At 18 hours the reaction was filtered through celite and the solvent reduced in vacuo. The title compound was then purified via flash chromatography on high-performance silica (829 mg). PART-C
(S)-ethyi 2-((1s.4R)-4-(7-(bis(f2-(trimethylsilyl)ethoxy)methynaminoV3- iodopyrazolofi ^-aipyrimidin-5-yl)cvclohexyl)-2-fluoroacetate

To a 50 ml_ roundbottom flask was charged acetonitrϋe (20 mL), ethyl 2-(4-(7-(bis({2- (trimethylsϋyl)ethoxyJmethyl)aminoJpyrazoioti .5-alpyrimidin-5-yllcyclohexylidene)-2- fluoroacetate (1.43 mmol, 828 mg), and N-lodosuccinimtde (1.57 mmol, 353 mg). The resulting solution was allowed to stir at room temperature for 18 hours. At 18 hours, the reaction was reduced in vacuo and purified via flash chromatography to yield the title compound (995 mg, 1.41 mmol). PART-D
(S)-ethyl 2-(f1s,4R)-4-(7-(bis(f2-(trimethylsilyl^ethoxy)methynaminoV3-(6- phenylpyridin-G-vOpyrazoloπ.5-aipyrimidin-5-vhcvclohexyπ-2-fluoroacetate

A pressure vial was charged with (S)-ethyl 2-((1s,4R)-4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)-2- fiuoroacetate (4.26 mmol, 3.01 g), 6-phenylpyridine-3-boronic acid pinacol ester (5.54 mmol, 1.56 g), potassium phosphate (12.78 mmol, 2.71 g), PdCI2(dppf)-CH2CI2 (0.43 mmol, 348 mg), and a solution of 9:1 1 ,4-dioxane:H20 (45 mL). The flask was flushed with argon and sealed. The reaction was stirred at 80° C for 18 hours. At 18 hours, the reaction was diluted with DCM (200 mL) and washed with H2O. The organic layer was collected and dried over Na2SO4. The resulting residue was purified via flash chromatography to yield the title compound (3.79 mmol, 2.78 g)
PART-E fS)-ethyl 2-(f1s.4RV4-(7-(bisff2-(trimethylsilyl)ethoxy)methyl)amino)-e-bromo-3-(6- phenylpyridin-3-yl)pyrazolori .5-aipyrimidin-5-yl)cvclohexyl)-Z-fluoroacetate
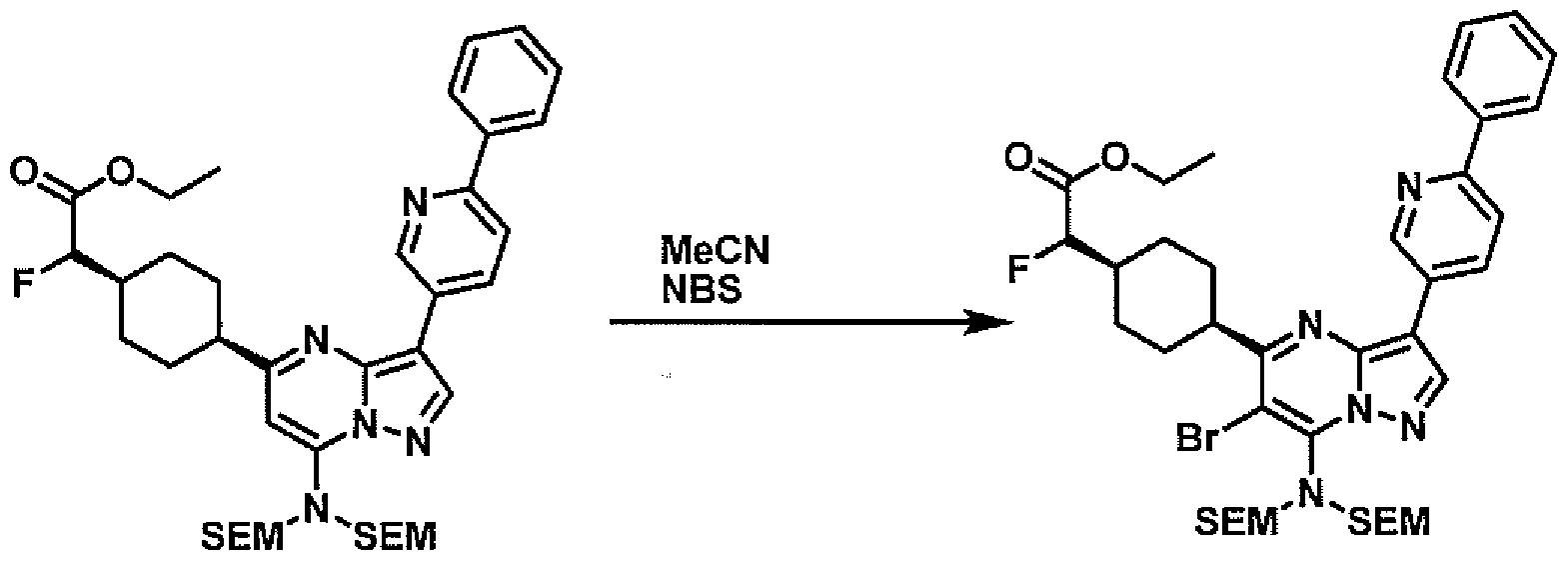
To a 100 mL roundbottom flask was charged (S)-ethyl 2-{(1s,4R)-4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yI)pyrazo[o[1 ,5--a]pyrimidin-5- yl)cyclohexyl)-2-fluoroacetate (3.51 mmol, 2.58 g), acetonitrile {40 mL) and N- bromosuccinimide (3.87 mmol, 688 mg). The resulting solution was stirred at room temperature for 18 hours. At 18 hours, the solvent was removed in vacuo and the residue was purified via flash chromatography to yield the title compound (3.21 mmol, 2.61 g) PART-F f SVethyl 2-ff1 s.4RV4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazoloπ .5- alpyrimidin-5-yl)cvciohexyl)-2-fluoroacetate

(S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6-phenylpyhdin-3-yl)pyrazoloπ .5-a1pyrimidin-5- yl)cvclohexyl)-2-f[uoroacetic acid

To a 20 mL scintillation vial was charged (S)-ethy! 2-((1 s,4R)-4-(7-amino-6-bromo-3- (6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyciohexyl)-2-fluoroacetate as a formic acid salt (0.25 mmol, 150 mg), a solution of 2:1 THF:H2O (9 mL), and 1 N NaOH(aq) (0.50 mmol, 0.50 mL). This solution was stirred at room temperature for 2 hours. At 2 hours, the solvent was removed in vacuo and the residue was taken up in 2 mL 1 :1 MeCN:H2θ. This solution was frozen in liquid nitrogen and then pumped dry on lyophilizer to yield (S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-y[)cyclohexyl)-2-fiuoroacetic acid as a sodium salt (m+H = 524.08, retention time = 4.16 min).
The following compounds ( Table-7A) were synthesized using essentially the same procedures used for the total synthesis of (S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6- phenylpyridin-3-yi)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)-2-fluoroacetic acid:
TABLE-7A
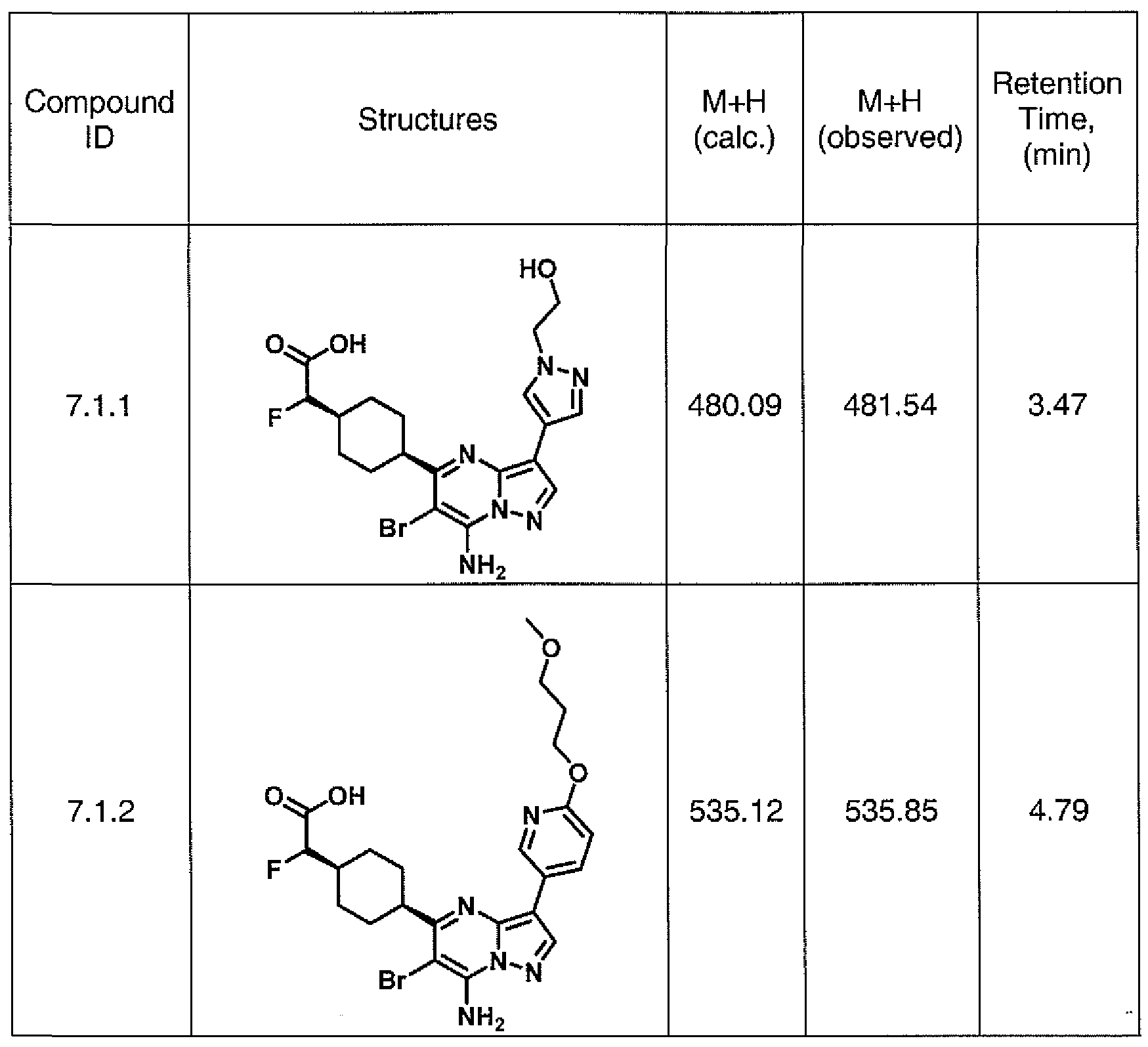
Ethyl 7-(bis((2-(trimethylsilyl)ethoxy)methynamino)-5-((1 R,4s)-4-((S)-2-ethoxy-1 - fluoro-2-oxoethyl)cyclohexyl)-3-(6-phenylpyridin-3-yl)pγrazolo[1 ,5-a1pyrimidine-6- carboxylate

(SV2-((1s,4R)-4-(6-acetyl-7-amino-3-(6-phenylpyridSn-3-yl)pyrazolo[1 ,5-aipyrimidin-5- vhcvclohexyl)-2-fluoroacetic acid

To a 50 mL roundbottom flask is charged ethyl 7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-5-((1 R,4s)-4-((S)-2-ethoxy-1 -fluoro-2~ oxoethyl)cyclohexyl)-3-tβ-phenyipyridin-3-yl)pyrazoloti ^-alpyrimidine-6-carboxylate (0.29 mmol, 236 mg), a solution of 2:1 THF:H2O (9 mL), and 1 N LiOH(aq) (0.59 mmol, 0.59 mL). The resulting solution was stirred at room temperature for 2 hours. The reaction was then acidified to pH ~ 4 with 1 N HCI(aq) and the solvent removed in vacuo. This residue was dissolved in 10 mL 1 ,4-dioxane. To this solution was added H2O (3 mL) followed by 4N HChdioxane (5 mL). This solution was stirred at room temperature for 18 hours. After 18 hours, the solvent was removed in vacuo and the residue purified via reverse-phase HPLC to yield (S)-2-((1s,4R)-4-(6-acetyl-7-amino-3- (6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cycIohexyl)-2-fluoroacetic acid (m+H = 488.62, retention time = 3.71 min).
fS^-(fisΛRM-(e-acetyl-y-amino-3-(e-(i -(a-hydroxyethyD-I H-pyrazol-Φyl^pyridin-3- yl)pyrazolof1 ,5-alpyrimidin-5-yl)cvclohexyl)-2-fluoroacetic acid
HChdioxaπe

(S)-2-((1s,4R)-4-(6-acetyl-7-amino-3-(6-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)pyridin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)-2-fluoroacetic acid was synthesized in the manner previously described. (m+H = 521.22, retention time = 3.01 min).
(S)-2-((1 s,4R)-4-(7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cvclohexyl)-2-fluoroacetic acid

To a 50 ml. roundbottom flask was charged (S)-ethyl 2-({1s,4R)-4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-(6-pheny[pyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl^-fiuoroacetate (0.27 mmol, 200 mg), 1 ,4-dioxane (5 nriL), H2O (1 mL), and 4N HCI in 1 ,4-dioxane. The resulting solution was stirred at room temperature for 5 days. At day 5, the solvent was removed in vacuo and the residue taken on without further purification.
(S)-2-((1 s,4R)-4-(7-amino-6-chloro-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-alpyrimidin-5- yl)cvclohexyl)-2-fluoroacetic acid

To a 50 mL roundbottom flask was charged (S)-2-((1s,4R)-4-(7-amino-3-(6- phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)-2-f!uoroacetic acid hydrochloride (0.27 mmol), DMF (8 mL), and N-chlorosuccinimide (0.30 mmol, 40 mg). The resulting solution was stirred at room temperature for 18 hours. At 18 hours, the solvent was removed in vacuo. The resulting residue was purified via reverse-phase HPLC to yield (S)-2-((1 s,4R)-4-(7-amino-6-chloro-3-(6-phenylpyridin-3-
yi)pyrazolo[1 ,5-a]pyrimidin-5-yi)cyclohexyI)-2-fluoroacetic acid (m+H = 480.02, retention time = 4.02 min).
fS)-ethyi 2-((1s,4RV4-(7-(bisff2-(trimethyisilyl)ethoxy)methyl)amino)-3-(6- 5 chloropyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)cvclohexyl)-2-fluoroacetate

(S)-ethy! 2-((1 s,4R)-4-(7-(bis({2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-
I O chloropyridin-3-yi)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyciohexyl)-2-fluoroacetate is synthesized in a manner similar to the sythesis of (S)-ethyl 2-((1s,4R)-4-(7-(bis((2- (trimethyIsilyl)ethoxy)methyl)amino)-3-(6-phenyipyridin-3-yl)pyrazo!o[1 ,5-a]pyrimidin-5- yl)cyclohexyl)-2-fluoroacetate, but with 6-chloropyridine-3-boronic acid pinacol ester substituted for 6-phenylpyridine-3-boronic acid pinacol ester.
15 fS)-ethyl 2-(f1 s.4R)-4-(7-(bisff2-(trimethylsilyl)ethoxy)methynamino)-3-(6-(pyrimidin-2- yl)pyridin-3-yl)pyrazolof1 ,5-aiPyrimidin-5-yl)cvdohexyl)-2-fluoroacetate

20
To a 2-5 mL microwave vessel was charged (S)-ethyl 2-((1s,4R)-4-(7-(bis{(2- (trimethylsi[yl)ethoxy)methyl)amino)-3-(6-chloropyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cycIohexyl)-2-fluoroacetate (0.22 mmol, 150 mg), acetonitrile (3 mL), 2-
(tributylstannyi)pyrimidine (0.65 mmoi, 240 mg), and tetrakis(triphenylphosphine)palladium{0) (0.02 mmol, 25 mg). The vessel was flushed with argon, sealed and heated to 150° C for 40 minutes in microwave synthesizer. Upon completion, the solvent was removed in vacuo and the residue purified by flash chromatography to yield the title compound.
(S)-2-(f1s.4R)-4-('7-amino-6-bromo-3-(6-(pyrimidin-2-yl)pyridin-3-yl)pyrazoloπ .5- a1pyrimidin-5-yl)cvclohexyl)-2-fluoroacetic acid

(S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6-(pyrimidin-2-y[)pyridin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)-2-fluoroacetic acid is synthesized in a manner similar to the sythesis of (S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3- yl)pyrazolo[1.5-aJpyrimidin-5-yl)cyclohexyl)-2-fluoroacetic acid (m+H = 524.80, retention time = 3.70 min).
The following compounds ( Table-7B) were synthesized using essentially the same procedures used for the total synthesis of (S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6- (pyrimidin-2-yl)pyridin-3-yi)pyrazolo[1,5-a]pyrimidin-5-yl)cyciohexyl)-2-fluoroacetic acid:
TABLE-7B





To a 20 ml_ pressure vial was charged pyrazole-4-boronic acid pinacoi ester (5.15 mmo!, 1.00 g) and DMF (10 ml_). NaH (60% w/w in mineral oil, 5.67 mmol, 227 mg) was then added portion-wise. The resulting solution was stirred at room temperature for 10 minutes. 2-bromoethyl acetate (5.67 mmol, 623 μl_) was then added dropwise. The vial was flushed with argon, sealed, and the reaction was stirred at 60° C for 18 hours. At 18 hours, the DMF was removed in vacuo, and the residue taken up in DCM. This suspension was filtered through celite and then purified via flash chromatography to yield the title compound.
2-(Φ-f4,4,5.5-tetramethyi-1.3.2-dioxaborolan-2-vh-1 H-pyrazoi-1-yl)ethanol

To a 50 mL roundbottom flask was charged 2-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yI)-1 H-pyrazol-1-y!)ethyl acetate (2.11 mmol, 591 mg), a solution of 2:1 THFiH2O (15 mL), and 1 N LiOH(aq) (4.82 mmol, 4.82 mL). The resulting solution was stirred at room temperature for 48 hours. At 48 hours, the reaction was quenched with 1 N HCI(aq) (4.82 mmol, 4.82 mL) and the solvent removed in vacuo. The product was taken forward without further purification.
1 -(2-trideuteromethoxyethyl)-4-(4.4.5.5-tetramethyl-1 ,3,2-dioxaborolan-2-vh-1 H- pyrazoie

To a 20 mL pressure via! was 2-(4-(4,4,5,5-tetramethy!-1 ,3,2-dioxaborolan-2-y!)-1 H- pyrazol-1-yl)ethanol (2.1 1 mmol) and DMF (10 mL). NaH (60% w/w in mineral oil, 4.22 mmol, 169 mg) was added portion-wise and the resulting soiution was allowed to stir at room temperature for 10 minutes. At 10 minutes, iodomethane-d3 (4.22 mmoi, 263 μL) was added dropwise. The vial was sealed and the reaction was stirred at 80° C for 48 hours. At 48 hours, the solvent was removed in vacuo. The residue was
taken up in DCM and filtered through celite. The title compound was then purified via fiash chromatography.
2-methy)-1 -(4-(4.4.5.5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-ρyrazoi-1 -vnpropan-2- ol

To a 20 mL pressure vial was charged pyrazole-4-boronic acid pinacol ester (5.15 mmol, 1.00 g), DMF (10 mL), and Cs2CO3 (5.67 mmoi, 1.85 g). To this suspension was added isobutylene oxide (15.45 mmol, 1.38 mL). The vial was flushed with argon and sealed. The reaction was stirred 18 hours at 80° C. After 18 hours, the reaction was filtered through celite and the solvent removed in vacuo. This residue was pumped dry under high vacuum and then purified via flash chromatography to yield the title compound.
1 -trideuteromethyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-ylV1 H-pyrazole

1 -trideuteromethyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazo!e was synthesized in the manner previously described.
3-(6-phenylpyridin-3-ylV5-(1 H-Pyrazol-4-yl)-N.N-bis((2- (trimethyisilyl)ethoxy)methyl)pyrazolo[1,5-a1pyrimidin-7-amine

3-(6-phenylpyridin-3-yl)-5-(1 H-pyrazot-4-yl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin-7-amine was synthesized in a manner similar to the sythesis of (S)-ethyl 2-((1s,4R)-4-{7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenyipyridin-3-yl)pyrazo!o[1 ,5-a]pyrimidin-5- yl)cyciohexyl)-2-fluoroacetate.
2-(4-(7-(bis{(2-(trimethylsilyl)ethoxy)methyl)anninoV3-(6-phenylpyridin-3- vOpyrazolofi ,5-aipyrimidin-5-vP-1 H-pyrazol-1 -yl)ethyl acetate

To a 2-5 mL pressure vial was charged 3-(6-phenylpyridin-3-yt)-5-(1H-pyrazol-4-yl)- N,N-bis((2-(trimethylsily!)ethoxy)methyl)pyrazolo[1 f5-a]pyrimidin-7-amine (0.38 mmol, 230 mg), DMF (4 mL), and sodium hydride (40% w/w in mineral oil, 0.56 mmol, 23
mg). This suspension was sonicaided and allowed to stir at room temperature for 15 minutes. At 15 minutes, 2-bromoethyl acetate (0.56 mmol, 62 μl_). The vial was then sealed and the reaction was stirred at 80° C for 18 hours. After 18 hours, the solvent was removed in vacuo, and the resulting residue was purified by flash chromatography to yield the title compound.
1 -(7-amino-5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)~3-(6-phenylpyridin-3- vQpyrazoio[1 ,5-alpyrimidin-6-yl)ethanone


1 -(7-amino-5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-y!)-3-(6-phenylpyridin-3- yl)pyrazolo[1 ,5-a]pyrimidin-6-yl)ethanone was synthesized in a manner similar to the sythesis of {S)-2-((1s,4R)-4-(6-acetyl-7-amino-3-(6-phenylpyπdin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)-2-fluoroacetic acid (m+H = 440.64, retention time = 2.95 min).
Ethyl 2-(4-(7-(bisf(2-(trimethylsilyl)ethoxy)methyi')aminoV3-(1 -phenyl- 1H-pyrazoi-4- vQpyrazolon .5-alpyrimidin-5-vOcvclohexynacetate

Ethyl 2-(4-(7-{bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(1 -phenyl-1 H-pyrazol-4- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)acetate was synthesized in a manner similar to the sythesis of (S)-ethyl 2-((1s,4R)-4-(7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-3-{6-phenylpyridin-3-yi)pyrazoto[1 ,5-a]pyrimidin-5- yl)cyclohexyl)-2-fluoroacetate.
2-(4-(7-amino-6-bromo-3-(1 -phenyl-1 H-pyrazol-4-yl)pyrazoloH ,5-a1pyrimidin-5- yl)cvclohexyl)acetic acid

2-(4-(7-amino-6-bromo-3-(1 -phenyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5-a]pyrimidin-5- yi)cyciohexyl)acetic acid was synthesized in the manner previously described. (m+H = 495.57, retention time = 4.78 min (isomer-!) and 4.84 min (isomer 2)).
0350
365
(SV2-((1 s,4RV4-(7-amtno-6-bromo-3-(1 -phenyl-1 H-pyrazol-4-vhpyrazoloH ,5- aipyrimidin-5-yl)cvclohexyl)-2-fiuoroacetic acid
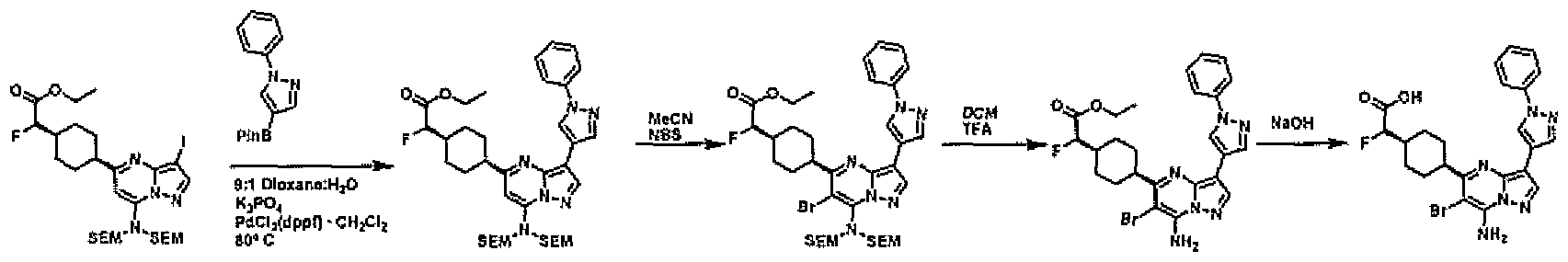
(S)-2-((1s,4R)-4-(7-aminθ"6-bromo-3-(1-phenyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5- a]pyrimidin-5-yi)cyclohexyl)-2-fluoroacetic acid was synthesized in the manner similar to the synthesis of 2-{4-(7-amino-6-bromo-3-(1-phenyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)acetic acid. (m+H = 513.54, retention time = 4.81 min).
2-(4-(6-acetyl-7-amino-3-(1 -phenyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5-alpyrimidin-5- yl)cvciohexyl)acetic acid

2-(4-{6-acetyl-7-amino-3-(1 -phenyl-1 H-pyrazol-4-yl)pyrazo!o[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetic acid was synthesized in the manner previously described. {m+H = 459.66, retention time = 4.58 min).
(SV2-((1 s,4R)-4-(6-acetyl-7-amsno-3-(1 -phenyl-1 H-pyrazoi-4-vQpyrazolo[1.5- alpyrimidin-5-vOcvclohexyπ-2-fluoroacetic acid

(S)-2-((1 s,4R)-4~(6-acetyl-7-amtno-3-(1 -phenyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)-2-fluoroacetic acid was synthesized in the manner similar to the synthesis of 2-(4-(6-acetyl-7-amino-3-(1 -phenyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)acetic acid. {m+H = 477.64, retention time = 4.53 min).
Ethyl 6-(7-(bisf(2-(trimethylsilv[)ethoxy^methyl)amino)pyrazolo[1 ,5-alpyrimidin-5- yl)spirof2.5'loctane-1-carboxylate

To 50 mL roundbottom flask containing trimethylsuifoxonium iodide (3.57 mmol, 785 mg) in DMSO (8 mL) was added potassium tert-butoxide (3.57 mmol, 401 mg). This suspension was allowed to stir at room temperature for 3 hours. After 3 hours, ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexylidene)acetate (1.78 mmol, 1.00 g) was added in 2 mL DMSO. The resulting solution was stirred at room temperature for 18 hours. After 18 hours, the reaction was diluted with saturated NaC!(aq) (30 mL). The pH was adjusted to ~7 with saturated NH4CI(aq) and extracted with DCM (50 mL) 4 times. The combined organics are dried over Na2S0_j and reduced in vacuo. The resulting residue was purified via flash chromatography to yield the title compound.
Ethyl 6-( 7-(bis(f2-(trimethyisilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-aiPyrimidin-5-y1H ■ f!uorospiro[2.51octane-1 -carboxylate

Ethyl 6-(7-(bis((2-(trinrtethylsily!)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5-y!)-1 ■ fluorospiro[2.5]octane-1 -carboxylate was synthesized in the manner similar to the synthesis of ethyl 6-(7-(bis((2-(trimethytsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5- a]pyrimidin-5-yl)spiro[2.5]octane-1 -carboxylate.
6-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-alpynmidin-5- yl)spiro[2.5]octane-1 -carboxylic acid

6-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazoio[1 ,5-alpyrimidin-5-yl)-1- f Iuorospirof2.51octane-1 -carboxylic acid

6-(7-amino-6-bromo-3-(6-pheny!pyridin-3-yi)pyrazolo[1 ,5-a]pyrimidin-5-yI)-1 - fluorospiro[2.5]octane-1 -carboxylic acid was synthesized in the manner previously described. (m+H = 435.86, retention time = 4.40 min).
ethyl 2-acetoxy-2-(4-(7-(bis((2-(trimethylsilyltethoxy)methy0amino)pyrazoloH ,5- aipyrimidin-5-yl)cvclohexylidene)acetate

Ethyl 2-acetoxy-2-(4-(7-(bisf(2-(trimethylsiiyl)ethoxy)methynamino)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)cvdohexylidene)acetate

2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenylpyridin-3- vOpyrazoioπ .5-a1pyrimidin-5-yl)cvclohexyl)-2-oxoacetic acid

To a 50 mL roundbottom flask was charged ethyl 2-acetoxy-2-(4-(7-(bis((2-
{trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5- alpyrimidin-5-yl)cyclohexylideneJacetate (0.47 mmol, 400 mg), a solution of 2:1 THF:H2O (15 mL), and 1 N LiOH(aq) (1.18 mmol, 1.18 mL). The resulting solution was stirred at room temperature for 18 hours. After 18 hours, the reaction was quenched with 1 N HCI(aq) (1.18 mmol, 1.18 mL) and the solvent removed in vacuo. The resulting residue was taken on without further purification.
(RV2-(f1 r.4R)-4-(7-(bisα2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6- phenylpyπdin-3-yl)pyrazolori ,5-alpyrimidin-5-yl)cvclohexylV2-hydroxyacetic acid

To a 50 mL roundbottom fiask was charged 2-{4-(7-(bis((2- (tπmethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6--phenylpyridin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yi)cyclohexyl)-2-oxoacetic acid (0.47 mmol) and MeOH (10 mL). To this solution was charged NaBH4 (0.71 mmol, 27 mg). This solution was allowed to stir at room temperature for 18 hours. After 18 hours, the reaction was quenched with saturated NH4CI(aq) {10 mL) and extracted with DCM twice. The combined organics were dried over Na≥SCλi, and the solvent removed in vacuo. The compound was taken on without further purification.
(R)-1-(f1 r.4R)-4-(7-(bisff2-(trimethylsiivnethoxy)methyl)amino)-3-(6-phenylpyridin-3- yl)pyrazolori ,5-aipyrimidin-5-yl)cyclohexyl)ethane-1 ,2-diol

To a 50 mL roundbottom flask was charged (R)-2-((1 r,4R)-4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenyIpyridin-3"yl)pyrazolo[1 ,5- a]pyrimidin-5-yi)cyclohexyl)-2-hydroxyacetic acid (0.47 mmol) and THF (20 mL). This solution was cooled to 0° C in an ice bath. Lithium aluminum hydride (1.0 M in
hexanes, 2.06 mmol, 2.06 mL) was added dropwise. The reaction is then allowed to slowly warm to room temperature and then stirred for 30 hours. After 30 hours, the reaction was quenched with EtOAc (20 mL) and washed with H2O. The organic layer was dried over Na2SO4, and the solvent removed in vacuo. The product was taken on without further purification.
(FO-1 -((1 r,4R)-4-{7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5- yl)cyclohexyl)ethane-1 ,2-diol
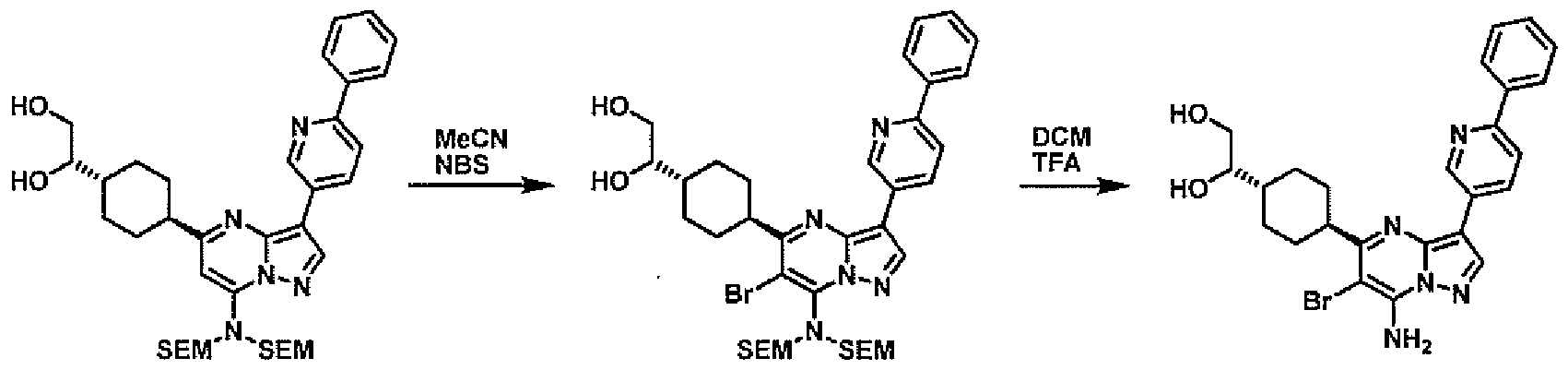
(R)-1-((1 r,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)ethane-1 ,2-diol was synthesized in the manner previously described. (m+H = 507.84, retention time = 3.56 min).
1 -(7-amino-5-((1 R,4r)-4-(f RV 1 ^-dihydroxyethyncvclohexyl)-3-(e-phenylpyridin-3- vθpyrazolori ,5-a1pyrimidin-6-yl)ethanone

1 -(7-amino-5-((1 R,4r)-4-((R)-1 ^-dihydroxyethyl)cyclohexyl)-3-^-phenyipyridin-3- yl)pyrazoio[1 ,5-a]pyrimidin-6-yl)ethanone was synthesized in the manner previously described. (m+H = 471.98, retention time = 3.45 min).
(R)-2-(f1 rτ4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5- yl)cyclohexyl)-2-hydroxyacetic acid

(R)-2-((1 r,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexy!)-2-hydroxyacetic acid was synthesized in the manner previously described. (m+H = 522.06, retention time = 3.51 min).
2-((1 rT4r)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo|-1.5-a1pyrimidin-5- yl)cvclohexyl)-2-oxoacetic acid

2-((1 r,4r)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)-2-oxoacetic acid was synthesized in the manner previously described. (m+H = 520.05, retention time = 3.79 min).
(SVethyl 2-acetoxy-2-((1 s,4R)-4-(7-(bisff2-(trimethylsilvnethoxy)methyπamino)-3-(6- phenylpyridin-3-yl)pyrazolofi .5-aipyrimidin-5-yl)cvclohexyQacetate

To a 50 ml_ roundbottom flask was charged ethyl 2-acetoxy-2-(4-(7-(bis((2- (trimethylstlyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyiidene)acetate (0.47 mmol, 400 mg), ethanol (15 ml_), di- isopropylethyl amine (0.50 mmol, 88 μL), and Raney 2800 nickel (slurry in H2O, 100 mg). The flask was sealed and degassed under vacuum for 15 minutes. After 15 minutes, the hydrogen gas was added in balloon and the reaction stirred at room temperature for 18 hours. After 18 hours, the reaction was filtered through celite,
10 being careful to not dry out Raney nickel. The filtrate was reduced in vacuo and the resulting residue was purified via flash chromatography to yield the title compound.
(S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolof1 ,5-alpyrimidin-5- vOcycloheχγQ-2-hydroxyacetic acid
\5

(S)-2-((1s,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)~2-hydroxyacetic acid was synthesized in the manner previously 0 described. (m+H = 522.10, retention time = 2.68 min).
(R)-methyl 2-(f1 r,4R)-4-(7-(b!S((2-(trimethylsilyl)ethoxy)methyπamino)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazolori .5-alPyrimidin-5-vncvciohexyl)-2-methoxyacetate
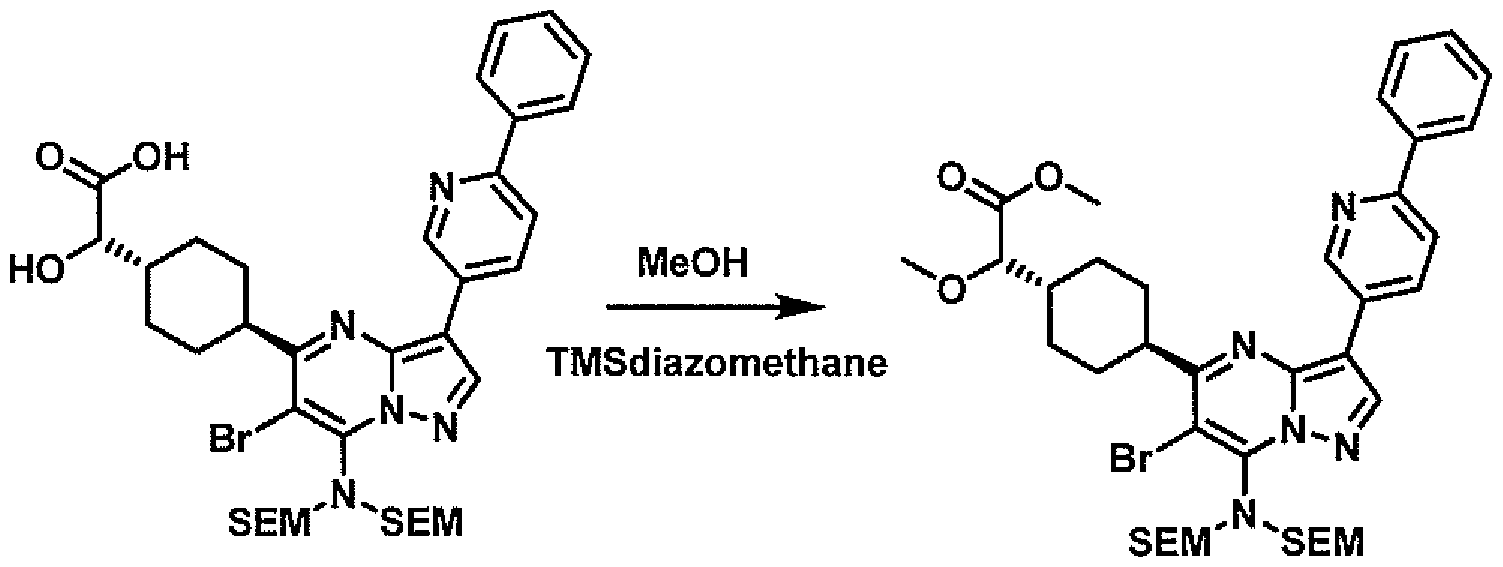
To a 50 ml_ roundbottom flask was charged (R)-2-((1 r,4R)-4-(7-(bis((2- (trimethylsi!yl)ethoxy)methyl)anrιino)-6-bromo-3-(6-phenylpyridin-3-yl)pyrazoio[1 ,5- a]pyrimidin-5-yI)cyclohexyl)-2-hydroxyacetic acid (0.40 mmol), MeOH (15 m!_), and TMS diazomethane (2.0 M in hexanes, 40 mmol, 20 mL). This solution was allowed to stir at room temperature for 72 hours. After 72 hours, the solvent was removed in vacuo and the residue was purified via flash chromatography to yield the title compound.
(R)-2-((1 rτ4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolori ,5-a1pyrimidin-5- yl)cvclohexyl)-2-methoxyacetic acid
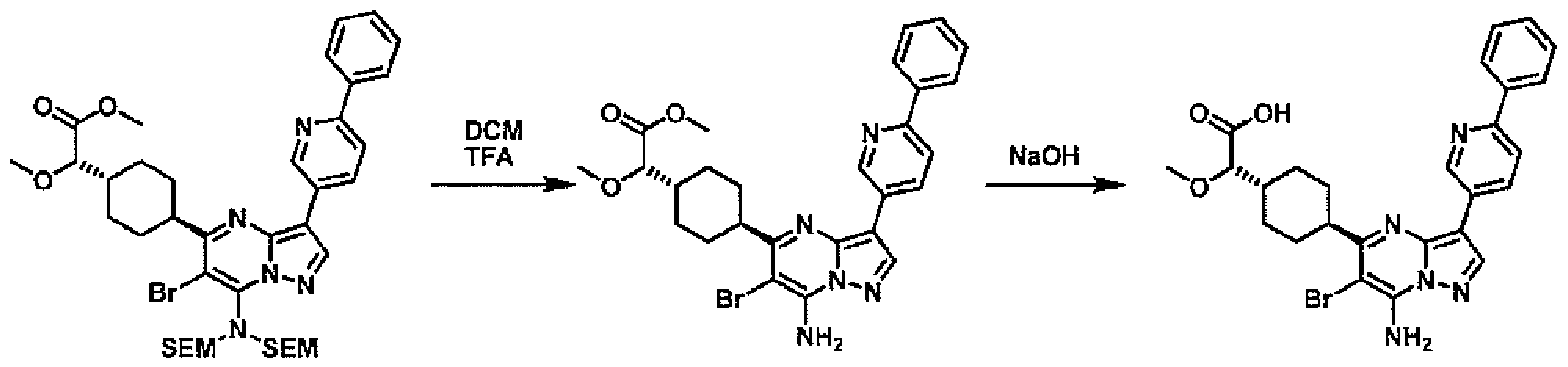
(R)-2-((1 r,4R)-4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)-2-methoxyacetic acid was synthesized in the manner previously described. (m+H = 536.56, retention time = 3.84 min).
2-(4-(7-amino-3-(6-phenylpyridin-3-yl)pyrazolof1 ,5-aipyrimidin-5- vOcyclohexyl)acetohydrazide

To a 2-5 mL pressure vial was charged ethyl 2-(4-(7-(bis{(2- (trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)acetate (0.12 rnrnol, 95 mg), DMF (1 mL). and hydrazine monohydrate (1 mL). The pressure vial was sealed and heated to 80° C for 18 hours. After 18 hours, the solvents were removed in vacuo and the product taken on without further purification.
5-((4-(7-amino-3-(6-phenylpyridin-3-yl)pyrazoloπ .5-aipyrimidin-5- yl)cvciohexyl)methyiyi .3,4-oxadiazol-2(3H)-one

To a 20 mL scintillation vial was charged 2-{4-(7-amino-3-(6-phenylρyridin-3- yl)pyrazoloti .5-atøyrimidin-5-yl)cyclohexyl)acetohydrazide (0.12 mmol), THF (5 mL), and 1 ,1'-carbonyidiimidazo!e (0.12 mmol, 25 mg). The resulting solution was stirred at room temperature for 2 hours. After 2 hours, the solvent was removed in vacuo and
the residue taken up in 3:1 DMSO: MeCN. The solids are removed via centrifugation, and the title compound was purified via reverse-phase HPLC.
5-((4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolof1.5-aipyrimidin-5- yl)cvclohexyl)methyl)-1.3.4-oxadiazol-2(3HVone

To a 20 mL scintillation vial was charged 5-((4-(7-amino-3-(6-phenylpyridin-3- yl)pyrazoio[1 ,5-a]pyhmidin-5-yt)cyclohexyl)methyl)-1 ,3,4-oxadiazol-2(3H)-one as a trifluoroacetate salt (0.035 mmol, 20 mg), MeCN (2 mL) and N-bromosuccinimide (0.035 mmol, 6.3 mg). The resulting solution was stirred at room temperature for 1 hour. After 1 hour, the solvent was removed in vacuo and the resulting residue was purified via reverse-phase HPLC to yield 5-((4-(7-amino-6-bromo-3-(6-phenylpyridin-3- yl)pyrazolo[1.5-alpyrimidin-5-yl)cyclohexyl)methylJ-i ,3,4-oxadiazol-2(3H)-one (m+H = 546.57, retention time = 4.07 min).
4-ftert-butoxycarboπylamino)cvclorιex-1-enyl trifluoromethanesulfonate

(Step 2)
To a 100 mL roundbottom flask was charged 4-N-Boc-aminocyclohexanone (4.69 mmol,
1.0Og) and THF (15 mL). The flask was flushed with argon and sealed with a rubber septum.
The solution was then cooled to -78° C in a dry ice/isopropanol bath. LiHMDS (1.0 M in hexanes, 9.61 mmol, 9.61 mL) was then added dropwise. After addition, the resulting solution was stirred at -78° C for 45 minutes. At this point, 2-[NIN-Bis(trifiuoromethanesulfonyl)amino]- 5-chloropyridine (5.16 mmol, 2.03 g) was taken up in THF (5 mL) and added dropwise to stirring solution at -78° C. The resulting solution was allowed to gently warm to room temperature over 1 hour. At 1 hour, the reaction was quenched with H2O and extracted with DCM (x2). The combined organics were dried over Na2SO4 and the solvent them removed in vacuo. The residue was then purified via flash chromatography to yield the title compound.
4-(tert-butoxycarbonylamino)cvdohex-1-enyl trifluoromethanesulfonate

To a 10-20 mL pressure vial was charged 4-(tert-butoxycarbonylamino)cyclohex-1- enyi trifluoromethanesulfonate (1.15 mmol, 397 mg), 1 ,4-dioxane (8 mL), bis(pinacolato)diboron (1.72 mmol, 438 mg), potassium acetate (3.45 mmol, 339 mg), and PdCI2(dppf) ■ CH2Cl2 (1 15 μmol, 94 mg). The via! was flushed with argon, sealed, and heated to 80° C for 5 hours. After 5 hours, the solvent removed in vacuo and the residue was taken up in DCM. This suspension was filtered through celite and then purified via flash chromatography to yield the title compound.
tert-butyl 4-(7-(bisff2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolof1 ,5-alpyrimidin-5- vOcvclohex-3-enylcarbamate

tert-butyl 4-(7-(bis({2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohex-^-enylcarbamate was synthesized in the manner previously described.
tert-fautyl f 1 s,4s)-4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolori ,5- alpyrimidin-5-vOcyclohexylcarbamate and tert-butyl (1 r,4r)-4-(7-(bis((2- ftrimethylsilyl)ethoxy^methyπamino^pyrazolofi .5-aiPyrimidin-5-yl)cvclohexylcarbamate

To a 50 mL roundbottom flask was charged tert-butyl 4-{7-{bis{(2- (trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohex-3- enylcarbamate (0.67 mmoi, 396 irng), ethanol (15 mL), and 10% palladium on carbon (100 mg). The flask was flushed with argon, sealed, and degassed under vacuum. Hydrogen gas was added in a balloon and the reaction was allowed to stir at room temperature under a hydrogen atmosphere for 18 hours. After 18 hours, the reaction was filtered though ceiite. The solvent was removed in vacuo and the resulting residue was purified to yield the title compounds (isomer 1 = cis, isomer 2 = trans).
5-(dsΛsM-aminocyclohexyl)-e-bromo-3-(6-phenylpyridin-3-yl)pyrazoloπ .5- aipyrimidin-7-amine
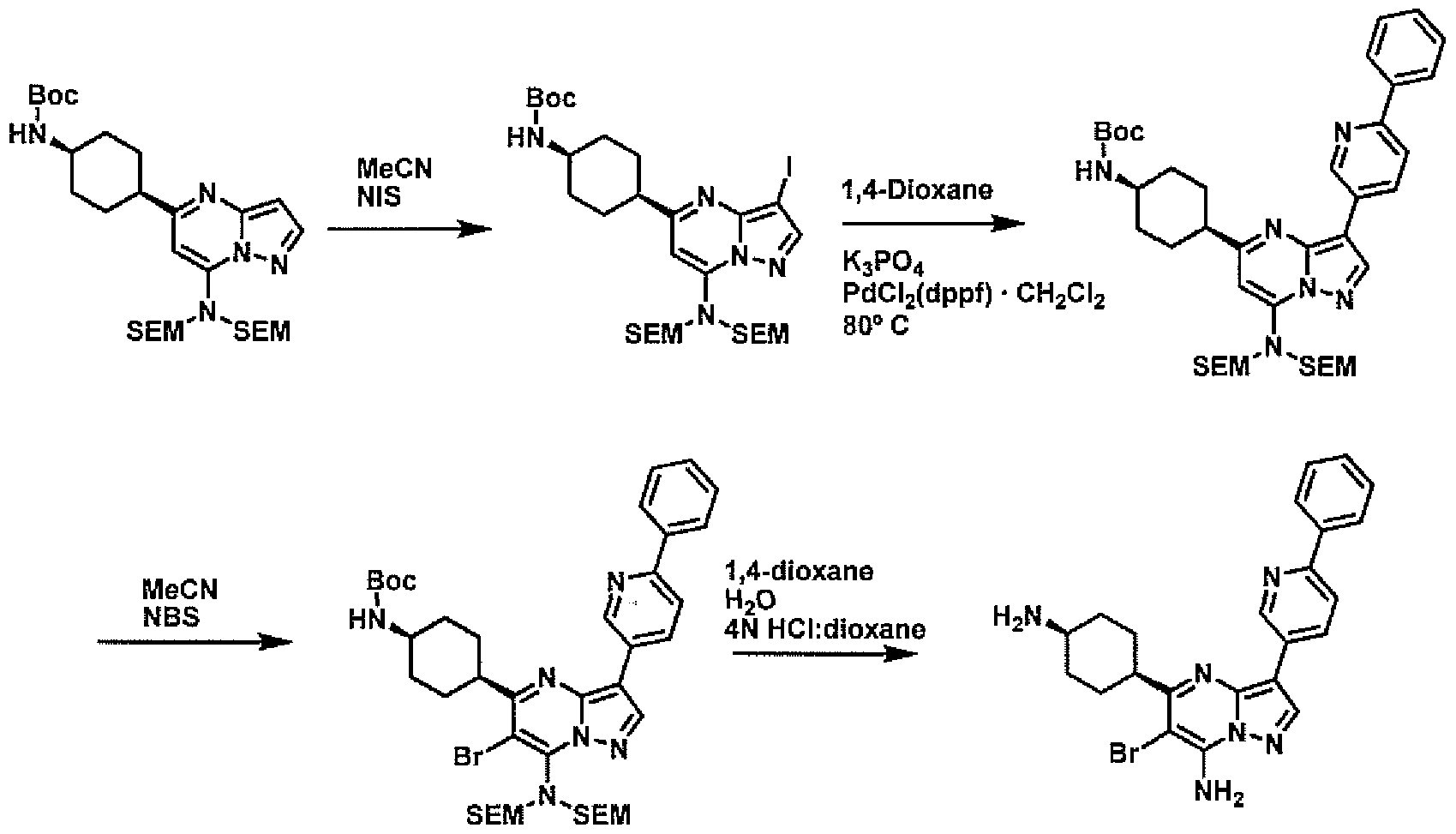
5-tCis^sJ-Φaminocyclohexyl)-6-bromo-3-tβ-phenylpyridin-3-yl)pyrazoloti .5- a]pyrimidin-7-amine was synthesized in the manner previously described.
(R)-N-(f1s,4S)-4-(7-amino-6-brQmo-3-(6-phenylpyridin-3-yl)pyrazolof1 ,5-alpyrimidin-5- vOcvclohexyl)-2-hydroxypropanamide

To a 20 mL scintillation vial was charged 5-((1 s,4s)-4-aminocyclohexyl)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazo!o[1 ,5-a]pyrimidin-7-amine as a hydrochloride salt (37 μmol), DMF (2 mL), D-!actic acid (0.08 mmol, 7.2 mg), EDC (0.08 mmol, 15.4 mg), HOBt (0.08 mmol, 10.8 mg), and DIEA (0.15 mmol, 26 μL). The resulting solution was stirred at room temperature for 4 hours. After 4 hours, the solvent was removed in
vacuo and the residue purified via reverse-phase HPLC to yield (R)-N-((1 s,4S)-4-(7- amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazoIo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)-2- hydroxypropanamide (m+H = 534.74, retention time = 3.82 min).
The following compound ( Table-7C) was synthesized using essentially the same procedures used for the total synthesis of (R)-N-((1 s,4S)-4-(7-amino-6-bromo-3-(6- phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)-2-hydroxypropanamide:
TABLE-7C

(R)-N-((1s,4S)-4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)-2-hydroxypropanamide

{R)-N-((1s,4S)-4-(6-acetyl-7-amino-3-(6-pheny!pyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- ylJcyclohexyl)-2-hydroxypropanamide was synthesized in the manner previously described (m+H = 499.11 , retention time = 3.68 min).
The following compounds( Tabie-7D) were synthesized using essentially the same procedures used for the total synthesis of (R)-N-((1 s,4S)-4-(6-acetyl-7-amino-3-(6- phenylpyridin-3-yl)pyrazoloti .5-alpyrimidin-5-yl)cyclohexyl^-hydroxypropanamide:
TABLE-7D


Synthesis of 1 -(7-amino-5-((1 r,4r)-4-( methylsulfonylmethyl)cvclohexyπ-3-(1 -phenyl- 1 H-pyrazol-4-yl)pyrazoloM ,5-atoyrimidin-6-yl)ethanone:

SCHEME- 23 Part A:
(lr.4r)-tert-butyl 4-(7-(bisf(2-(trimethylsilyl)ethoxy)methyl)amirLθ)-3-("l-phenyl-1H-pyrazol-4- yl)pyrazolori.5-alpyrimidin^-yl)cyclohexanecarboxyiate:
Compound, (lr,4r)-tert- butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(l-phenyl- 1H-pyrazol-4-y[)pyrazolo[1,5-a]pyrimidin-5-yl)cyclohexanecarboxylate was prepared from compound (lr,4r)-tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyI)amino)-3- iodopyrazolo[1,5-a]pyrimidin-5-yl)cyclohexanecarboxylate and 1 -phenyl-4-(4,4,5,5- tetramethyl- l,3,2-dioxaborolan-2-yl)-lH-pyrazoIe using the coupling conditions described in Part G of Example 8 (68%). ΗPLC-MS TR = 3.13 min (UV 254 nm, 5 min method); mass calculated for formula C38H58N6O4Si2718.4, observed LCMS m/z 719.3 (M+H).
Part B:
((1 r,4r)-4-(7-(bisf(2-(trimethylsilyl)ethoxy)methyl)amino)-3-(1 -phenyl-1 H-pyrazoi-4- yl)pyrazoiof1 ,5-alpyrimidin-5-yl)cvclohexyl)methanoi:
To a solution of compound (lr,4r)-tert-butyl 4-(7-(bis((2-
(trimethylsiIyl)ethoxy)methyl)amino)-3-(l-phenyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidin- 5-yl)cyclohexanecarboxylate (1.68 mmol, 1.21 g) in THF (20 mL) at 0 °C was added lithium aluminum hydride solution (1.68 mL, 2.0 M in THF). The resulting reaction mixture was slowly warmed to rt and stirred for 1 h. The reaction quenched with 3 N NaOH and filtered. The filtrate and washes were combined and concentrated. The crude product was purified by a SiO2 column (0-40% EtOAc/Hexanes, Rf = 0.7 in 50% EtOAc) to afford compound, ((lr,4r)-4- (7-(bis((2-(trimethylsily0ethoxy)methyl)amino)-3-(l-phenyl-1H-pyrazol-4-yl)pyrazolo[1,5- a]pyrimidin-5-yl)cyclohexyl)methanol, as a pale yellow solid (923 mg, 85%). HPLC-MS TR = 3.08 min (UV 254 nm, 5 min method); mass calculated for formula C34H52N6O3Si2648.4, observed LCMS m/z 649.3 (M+H).
Part C: f(1 r,4r)-4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(1-phenyl-1H-pyrazol-4- yl)pyrazolofi .5-alPyrimidin-5-yl)cvciohexyl)methyl methanesulfonate:
To a solution of compound, ((lr,4r)-4-(7-(bis((2-(trimethyIsilyl)ethoxy)methyl)amino)-3-(l- phenyI-1H-pyrazoi-4-yi)pyrazolo[1,5-a]pyrimidin-5-yl)cycIohexyl)methanol (1.42 mmol, 920
mg) in DCM (10 mL) was added TEA (4.26 mmol, 0.594 mL), followed by MsCl (2.13 mmol, 0.165 mL) at 0 °C and stirred for 1 h. The reaction mixture was washed with H2O and brine, dried over Na2SO4, and concentrated. The crude product was purified by a SiO2 column (0- 30% EtOAc/Hexanes, Rf = 0.8 in 50% EtOAc) to afford compound, ((lr,4r)-4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-(i-phenyl-1H-pyrazol-4-yl)pyrazolo[l,5-a]pyrimidin- 5-yl)cyclohexyl)methyl methanesulfonate, as a pale yellow oil (949 rag, 92%).
Part D: 5-(fi rΛ^^-(methylthiomethyDcvclohexyπ-3-(i -phenyl-1H-pyrazol^-vh-N.N-bisffa- (trimethylsilyl)ethoxy)iinethyl)pyrazolo[1,5-a1pyrimidin-7-amine:
A mixture compound(( 1 r,4r) -4-(7-(b is((2-(trimethy lsilyi)ethoxy)methy!)amino)-3-(l -phenyl - 1H-pyrazol-4-yl)pyrazolo[l,5-a]pyrimidin-5-yi)cyciohexyl)methyl methanesulfonate (1.31 mmol, 949 mg) and NaSMe (2.61 mmol, 183 mg) in DMF (5 mL) was heated at 80 °C for 3 d. The reaction mixture was diluted with EtOAc, washed with H2O and brine, dried over Na2SO4, and concentrated. The crude product was purified by a SiO2 column (0-20% EtOAc/Hexanes, Rf = 0.5 in 20% EtOAc) to afford compound, 5-((lr,4r)-4-(methylthiomethyl)cyclohexyl)-3-(l- phenyl-1H-pyrazol-4-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin- 7-amine, as a pale yellow oil (804 mg, 90%). HPLC-MS TR = 3.59 min (UV 254 nm, 5 min method); mass calculated for formuia C35H54N6O2SSi2 678.4, observed LCMS m/z 679.3 (M+H).
Part E:
5-((1r,4r)-4-(mθthyisulfonylmethyl)cvclohexyl)-3-(1-phenyl-1 H-pyrazol-4-yl)-N,N-bisf(2- (trimethylsiiyl)ethoxy)methyl)pyrazolon ,5-a1pyrimiciin-7-amine:
To a solution of compound, 5-((l r,4r)-4-(methylthiomethyl)cyclohexyl)-3-(l-phenyl-1H- pyrazol-4-yI)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (1.18 mmol, 804 mg) in DCM (10 mL) was added mCPBA (2.83 mmol, 699 mg) and the resulting mixture was stirred at rt for overnight. The reaction was quenched with Na2S2O3 (aq.) and diluted with DCM (10 mL). The separated organic layer was washed with NaHCO3 (2*) and brine, dried over Na2SO4, and concentrated. The crude product was purified by a SiO2
column (0-40% EtO Ac/He xanes, Rf = 0.45 in 50% EtOAc) to afford compound, 5-((lr,4r)-4- (methylsulfonylmethyl)cycϊohexy])-3-(l -phenyl- 1H-pyrazoI-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoIo[1,5-a]pyrimidin-7-amine, as a pale yellow oil (446 mg, 53%). HPLC-MS TR = 2.99 min (UV 254 nm, 5 min method); mass calculated for formula C35H54N6O4SSi2710.3, observed LCMS m/z 71 1.2 (M+H).
Part F:
1 -(7-amino-5-((1 r,4r)-4-(methylsulfonylmethyl)cyciohexyl)-3-(1 -phenyl-1 H-pyrazol-4- yl)pyrazolori ,5-aipyrimidin-6-vQethanone:
By essentially the same procedures given in Preparative earliar Example compound, l-(7- amino-5-((lr,4r)-4-(methylsulfonylmethyl)cyclohexyl)-3-(l-phenyl-iH-pyrazol-4- yl)pyrazolo[1,5-a]pyrimidm-6-yI)ethanone was prepared from compound, 5-((lr,4r)-4- (methylsulfonylmethyl)cyclohexyl)-3-(l-phenyI-1H-pyrazol-4-yl)-N,N-bis((2- (trimethylsilyi)ethoxy)methy!)pyrazolo[1,5-a]pyrimidin-7-arnine. HPLC-MS TR = 4.64 min (UV 254 nm, 10 min method); mass calculated for formula C2SH28N6O3S, 492.2, observed LCMS m/z 493.1 (M+H).

SCHEME- 24
Synthesis of ethyl 1 ,4-dioxaspiro[4.51decane-7-carboxyiate

Ethyl 1 ,4-dioxaspiro[4.5]decane-7-carboxylate was synthesized in a manner similar to the synthesis of ethyl 1 ,4-dioxasptro[4.5]decane-8-carboxyiate, but with ethyl 3- oxocyclohexanecarboxylate substituted for ethyl 4-oxocyclohexanecarboxylate.
Synthesis of ethyl 1 ,4-dioxaspiro[4.41nonane-7-carboxylate
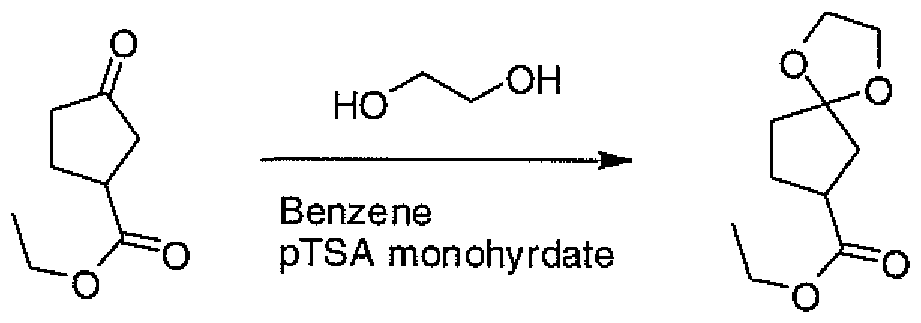
Ethyl 1 ,4-dioxaspiro[4.4]nonane-7-carboxylate was synthesized in a manner similar to the synthesis of ethyl 1 ,4-dioxaspiro[4.5]decane-8-carboxylate, but with ethyl 3- oxocyclopentanecarboxylate substituted for ethyl 4-oxocyclohexanecarboxytate.
Synthesis of 1 ,4-dioxaspirof4.51decane"7-carboxylic acid

1 ,4-Dioxaspiro[4.5]decane-7-carboxylic acid was synthesized in a manner similar to the synthesis of 1 ,4-dioxaspiro[4.5]decane-8-carboxyiic acid, but with ethyl 1 ,4- dioxaspiro[4.5]decane-7-carboxylate substituted for ethyl 1 ,4-dioxaspiro[4.5]decane-8- carboxylate.
Synthesis of 1.4-dioxaspiro[4.41nonane-7-carboxyiic acid

1 ,4-dioxaspiro[4.4]nonane-7-carboxylic acid was synthesized in a manner simiiar to the synthesis of 1 ,4-dioxaspiro[4.5]decane-8-carboxy!ic acid, but with ethyl 1 ,4- dioxaspiro[4.4]nonane-7-carboxylate substituted for ethyl 1 ,4-dioxaspiro[4.5]decane-8- carboxylate.
Synthesis of ethyl 3-oxo-3-(1.4-dioxaspiror4.51decan-7-yl)propanoate

Ethyl 3-oxo-3-(1 ,4-dioxaspiro[4.5]decan-7-yl)propanoate was synthesized in a manner similar to the synthesis of ethyl 3-oxo-3-(1 ,4-dioxaspiro[4.5]decan-8-yl)propanoate, but with 1 ,4-dioxaspiro[4.5]decane-7-carboxylic acid substituted for 1 ,4- dioxaspiro[4.5]decane-8-carboxylic acid.
Synthesis of ethyl 3-oxo-3-(1.4-dioxaspirof4.41nonan-7-yl)propanoate

Synthesis of 5-( 1.4-dioxaspirof4.5idecan-7-yl)pyrazolori ,5-aipyrimidin-7-ol

5-(1 ,4-dioxaspiro[4.5]decan-7-yl)pyrazolo[1 ,5-a]pyrimidin-7-o! was synthesized in a manner similar to the synthesis of 5-(1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[ 1 ,5- a]pyrimidin-7-ol, but with ethyl 3-oxo-3-{1 ,4-dioxaspiro[4.5]decan-7-yl)propanoate substituted for ethyl 3-oxo-3-(1 ,4-dioxaspiro[4.5]decan-8-yl)propanoate.
Synthesis of 5-(1 AdioxaspiroKΛInonan^-vQpyrazolori ^-alpyrimidin^-ol

5-(1 ,4-dioxaspiro[4.4]nonan-7-yl)pyrazolo[1 ,5-a]pyrimidin-7-o! was synthesized in a manner similar to the synthesis of 5-(1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[1,5- a]pyrimidin-7-ol, but with ethyl 3-oxo-3-(1 ,4-dioxaspiro[4.4]nonan-7-yl)propanoate substituted for ethyl 3-oxo-3-(1 ,4-dioxaspiro[4.5]decan-8-y!)propanoate.
Synthesis of 7-chloro-5-(1 ,4-dioxaspiror4.51decan-7-y])pyrazolof1.5-aipyrimidine

7-Chloro-5-(1 ,4-dioxaspiro[4.5]decan-7-yl)pyrazo!o[1 ,5-a]pyrimidine is synthesized in a manner similar to the synthesis of 7-chloro-5-(1,4-dioxaspiro[4.5]decan-8- yl)pyrazolo[1 ,5-a]pyrimidine, but with 5-(1 ,4-dioxaspiro[4.5]decan-7-yl)pyrazolo[1 ,5- a]pyrimidin-7-ol substituted for 5-(1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[1 ,5- a]pyrimidin-7-ol.
Synthesis of 7-chloro-5-( 1.4-dioxaspiroF4,41nonan-7-yl)pyrazoiof 1 ,5-aipyrimidine

Synthesis of 5-(1 ,4-dioxaspirof4.5ldecan-7-yl)pyrazoiof1 ,5-a1pyrimidin-7-amine

Synthesis of 5-(1.4-dioxaspiro[4.41nonan-7-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine

5-(1 ,4-Dioxaspiro[4.4]nonan-7-yl)pyrazo!o[1 ,5-a]pyrimidin-7-amine was synthesized in a manner similar to the synthesis of 5-(1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazo!o[1 ,5- a]pyrimidin-7-amine, but with 7-chloro-5-(1 ,4-dioxaspiro[4.4]nonan-7~yl)pyrazolo[1 ,5- a]pyrimidine substituted for 7-chloro-5-(1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[1 ,5- a]pyrimidine.
Synthesis of 3-(7-aminopyrazoiori ,5-aipyrimidin-5-yl)cyciohexanone

3-(7-Aminopyrazolo[1 ,5-a]pyrimidin-5-yl)cycIohexanone was synthesized in a manner similar to the synthesis of 4-(7-aminopyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexanone, but 5-{1 ,4-dioxaspiro[4.5]decan-7-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine substituted for 5- (1 ,4-dioxaspiro[4.5]decan-8-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine.
Synthesis of 3-(7-aminopyrazolo[1 ^-alpyrimidin-5-yl)cvclopentanone

3-(7-Aminopyrazolo[1 ,5-a]pyrimidin-5-yl)cyclopentanone was synthesized in a manner similar to the synthesis of 4-(7-aminopyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexanone, but 5-(1 ,4-dioxaspiro[4.4]nonan-7-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine substituted for 5- (1 Adioxaspiro[4.5]decan-8-yl)pyrazo]o[1 ,5-a]pyrimidin-7-amine.
Synthesis of 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-aipyrimidin- 5-yl)cvciohexanone

Synthesis of 3-{7-{ bis((2-(trimethylsilyl)ethoxy)methyi)amino)pyrazolof 1 ,5-aipyrimidin- 5-yl)cvclopentanone

3-(7-(Bis({2-{trimethylsilyl)ethoxy)methyt)amino)pyrazolo[1 ,5-a]pynmidin-5- yl)cyclopentanone was synthesized in a manner similar to the synthesis of 4-(7- (bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1,5-a]pyrimidin-5- yl)cyclohexanone, but 3-(7-aminopyrazolo[1 ,5-a]pyrimidin-5-yl)cyclopentanone substituted for 4-(7-aminopyrazolo[1 ,5-a]pyrimidin-5-yi)cyclohexanone.
Synthesis of ethyl 2-(3-(7-(bis(f2-(trimethylsilyl)ethoxy)methyl)amino)pyrazoio[1 T5- aipyrimidin-5-yl)cvclohexy!idene)acetate

Ethyl 2-(3-(7-(bis{(2-(trimethylsi!y!)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexylidene)acetate was synthesized in a manner similar to the synthesis of ethyl 2-(4-(7-{bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexylidene)acetate, but with 3-(7-(bis((2- (trimethylsilyl)ethoxyJmethyl)aminoJpyrazoloCi .5-alpyrimidin-5-ylJcyclohexanone
substituted for 4-(7-(bis{(2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5- a]pyrimidin-5-yi)cyclohexanone.
Synthesis of 2-(3-(7-(bis((2-(trimethylsilyl)ethoxy)methyπamino)pyrazolo[1 ,5- aipyrimidin-5-yl)cvclopentylidene)acetonitrile

2-(3-(7-(bis((2-{trimethylsiiy!)ethoxy)πnethyl)amino)pyrazolo[1 )5-a]pyrimidin-5- yl)cyclopentylidene)acetonitriie was synthesized in a manner similar to the synthesis of 2-(4-(7-(Bis({2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1,5-a]pyrimidin-5- yi)cyclohexylidene)acetonitrile, but with 3-(7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyhmidin-5-yl)cyclopentanone substituted for 4-{7-(bis((2-(trimethylsilyl)ethoxy)methyS)amino)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexanone.
Synthesis of ethyl 2-(3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazoloM ,5- aipyri m idi n-5-yl )cycioh exyl) acetate

Synthesis of 2-(3-(7-(bisf(2-(trimethylsiiyl)ethoxy)methyl)amino)pyrazolo[1 ,5- alpyrimidin-5-yl)cvclopentyl)acetonitrile

2-(3-(7-(Bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 I5-a]pyrimidin-5- yl)cyciopentyl)acetonitrile was synthesized in a manner similar to the synthesis of ethyl 2-{4-(7-(bis((2-{trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 I5-a]pyrimidin-5- yl)cyclohexyl)acetate, but 2-(3-(7-(bis((2- {trimethylsiiyi)ethoxy)methyl)amino)pyrazolo[1 ,5-a3pyrimidin-5- yl)cyclopentylidene)acetonitrile substituted for ethyl 2-(4-(7-(bis((2- (trimethy!silyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexylidene)acetate.
Synthesis of ethyl 2-(3-(7-(bisff2-(thmethylsilvhethoxy)methyπaminoV3- iodopyrazoloπ .5-alpyrimidin-5-yl)cvciohexyl)acetate

Synthesis of 2-(3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazoio[1 ,5- alpyrimidin-5-yl)cyclopentvPacetonitrile

2-{3-(7-(Bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclopentyl)acetonitrile was synthesized in a manner similar to the synthesis of ethyl 2-(4-(7-(bis((2-(trimeihylsilyi)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyi)acetate, but with ethyl 2-(3-(7-(bis({2- (tnmethylsilyl)ethoxyJmethylJaminoJpyrazolofi .5-aJpyrimidin-5-yl)cyclohexylJacetate substituted for ethyl 2-(4-(7-{bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexyl)acetate.
Synthesis of ethyl 2-(3-(7-(bis((2-(trimethy[silyl)ethoxy)methy[)amino)-3-(quinoiin-3- yl)pyrazolori .5-aipyrimidin-5-vOcvclohexyl)acetate

Ethyl 2-(3-(7-(bis((2-(trimethylsilyI)ethoxy)methyl)amino)-3-(quinolin-3-yl)pyrazolo[1 l5- atøyrimidin-5-yl)cyclohexyl)acetate was synthesized in a manner similar to the synthesis of ethyl 2-(4-{7-{bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quino!in-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-y!)cyclohexyi)acetate, but with ethyl 2-(3-(7-(bis({2- (trimethylsilyI)ethoxy)methy!)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate substituted for ethyl 2-(4-(7-(bis((2- (trimethylsilyi)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate.
Synthesis of 2-(3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolori ,5-aipγrimidin-5-yl)cvclopentyl)acetonitrile

2-(3-(7-(Bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclopentyl)acetonitrile was synthesized in a manner similar to the synthesis of ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolo[1.5-aføyrimidin-5-ylJcyclohexyl)acetate, but with 2-(3-(7-(bis((2- (trimethyJsilyl)ethoxy)methyl)amino)-3-iodopyrazo[o[1,5-a]pyrimidιn-5- yl)cyclopentyl)acetonitrile substituted for ethyl 2-(4-(7-(bis((2-
(trimethylsiiyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate.
Synthesis of ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(1-methyl-1 H- pyrazol^-yl)pyrazoloH .5-alpyrimidin-5-yl)cvclohexyl)acetate

Ethyl 2-(4-{7-(bis((2-(trimethylsilyI)ethoxy)methyl)amino)-3-(1-methyl-1 H-pyrazol-4- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)acetate was synthesized in a manner similar to the synthesis of ethyl 2-(4-(7-(bis{(2-(trimethylsilyl)ethoxy)methyl)amino)-3- (quinolin-3-yl)pyrazo!o[1 ,5-a3pyrimidin-5-y!)cyclohexyi)acetate, but with N-methyl pyrazo!e-4-boronic acid pinacol ester substituted for quinoline-3-boronic acid.
Synthesis of 2-(3-(7-amino-6-bromo-3-(αuinolin-3-yl)pyrazolo[1 ,5-alpyrimidin-5- vOcyclohexyltecetic acid

2-(3-(7-Amino-6-bromo-3-(quinolin-3-y!)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)acetic acid was synthesized in a manner similar to the synthesis of 2-(4-(7-amino-6-bromo-3- (quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexyl)acetic acid, but with ethyl 2-(3- {7-{bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinotin-3-yl)pyrazolo[1 t5- a]pyrimidin-5-yl)cyclohexyl)acetate substituted for ethyl 2-(4-(7-(bis((2- {trimethylsiiy!)ethoxy)methyl}amino)-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate. The reaction was concentrated in vacuo and purified via reverse-phase preparatory HPLC to yield 2-(4-(7-amino-6-bromo-3-(quinolin~3-
yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexy[)acetonitrile as a four-isomer mixture. (m+H = 480.1 , retention time = 3.51 min)
Synthesis of 2-(3-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolori ,5-a1pyrimidin-5- yl)cvclopentyl )aceton it ri Ie
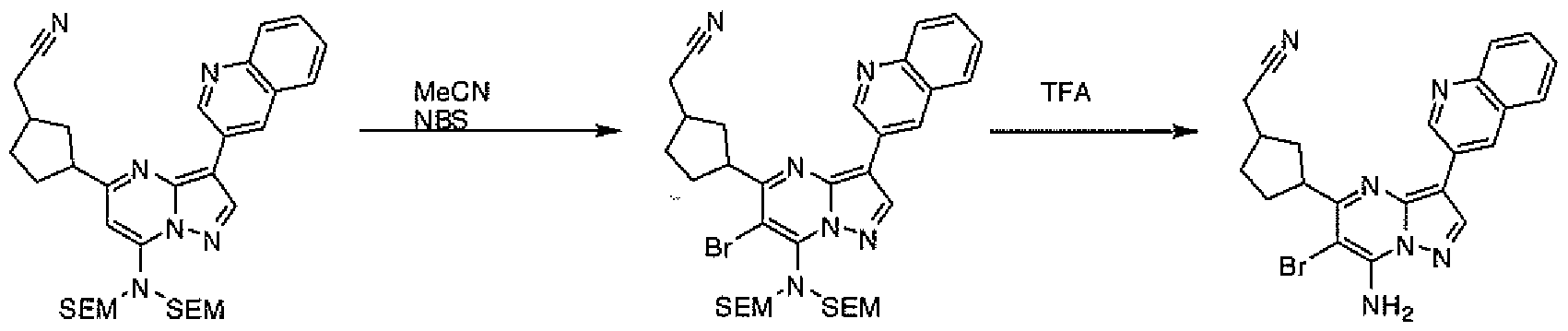
2-(3-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- y!)cyclopentyl)acetonitrile was synthesized in a manner similar to the synthesis of 2- (4-(7-amino-6-bromo-3-(quinolin-3-yI)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidin-1 -yl)acetic acid, but with 2-{3-(7-(bis((2-(trimethylsiIyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolo[1.5-alpyrimidin-5-ylJcyclopentyl)acetonitrile substituted for tert-butyl 2-(4- (7-amino-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidin-1 -yljacetate. The reaction was concentrated in vacuo and purified via reverse-phase preparatory HPLC to yield 2-(3-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclopentyl)acetonitrile as a yellow solid. (m+H = 447.22, retention time = 3.99 min).

SCHEME- 6
Synthesis of tert-butyl 4-(7-hydroxypyrazolori ,5-alpyrimidin-5-yl)piperidine-1- carboxylate

Synthesis of tert-butyl 4-(7-chloropyrazolori .5-alpyrimidin-5-vnpiperidine-1- carboxviate
 tert-Butyl 4-(7-chloropyrazo!o[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxylate was synthesized in a manner similar to the synthesis of 7-chloro-5-(1 ,4- dioxaspiro[4.5]decan-8-yI)pyrazolo[1.5-ajpyrimidine, but with te/t-butyl 4-(7- hydroxypyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxylate substituted for 5-(1 ,4- dioxaspiro[4.5]decan-8-yl)pyrazolo[1 ,5-a]pyrimidin-7-oi.
tert-Butyl 4-(7-chloropyrazo!o[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxylate was synthesized in a manner similar to the synthesis of 7-chloro-5-(1 ,4- dioxaspiro[4.5]decan-8-yI)pyrazolo[1.5-ajpyrimidine, but with te/t-butyl 4-(7- hydroxypyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxylate substituted for 5-(1 ,4- dioxaspiro[4.5]decan-8-yl)pyrazolo[1 ,5-a]pyrimidin-7-oi.
Synthesis of fert-butyl 4-(7-aminopyrazoloM ,5-aipyrimidin-5-γl)piperidine-1- carboxylate

Synthesis of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazoloF1 ,5- aipyrimidin-5-yl)pipeπdine-1 -carboxylate

Synthesis of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxyiate

Synthesis of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolo[1 ,5-aipyrimidin-5-yl)piperidine-1-carboxvlate

tert-Butyl 4-(7-(bis({2-(thmethylsilyl)ethoxy)methyl)amino)-3-(quinoiin-3- y!)pyrazo!o[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxyiate was synthesized in a manner
similar to the synthesis of ethyl 2-(4-(7-(bis({2-(trimethylsilyl)ethoxy)methyl)amino)-3- (quinolin-3-yi)pyrazolo[1.5-alpyrimidin-5-yl)cyclohexylJacetate, but with førf-butyl 4-(7- (bis((2-(trimethylsilyi)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5- yl)piperidine-1-carboxylate substituted for ethyl 2-{4-(7-(bis((2- {trimethylsilyl)ethoxy)methyl)amino)-3~iodopyrazolo[1 ,5-ajpyrimidin-5- yl)cyclohexyl)acetate.
Synthesis of te/f-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)aminoV3-(1-methyl- 1 H-pyrazol-4-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)piperidine-1 -carboxyiate

Synthesis of 5-(piperidin-4-vO-3-(cιuino[in-3-yl)pyrazolori ,5-aipyrimidin-7-amine

Synthesis of tert-butyl 2-(4-(7-amino-3-(quinolin-3-yl)pyrazolori ,5-a1pyrimidin-5- yl)piperidin-1 -yl)acetate

Synthesis of 2~(4-(7-amino-6-bromo-3-(quinolin~3-yl)pyrazolof 1 ^-alpyrimidin-5- yl)piperidin-1-yl)acetic acid

To a 20 mL scintillation vial was charged 2-(4-(7-amino-6-bromo-3-(quinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperic!in-1-yl)acetic acid (41 mg, 90 μmol) plus acetonitrile (2 mL). To this solution was added N-bromosuccinimide {90 μmol, 16 mg). This solutionwas allowed to stir at room temperature for 1 hour. The reaction was concentrated in vacuo and dissolved in TFA (2 mL). The reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo and purified via reverse-phase preparatory HPLC to yield 2-(4-(7-amino-6-bromo-3- (quino!in-3-yl)pyrazolo[1T5-a]pyrimidin-5-yi)piperidin-1-yl)acetic acid as yellow solid. (m+H = 481.22, retention time = 2.32 min)
Synthesis of 3-(1 -methyl-1 H-pyrazol-4-yl)-5-(piperidin-4-vQpyrazoloπ ,5-a1pyrimidin-7- amine

3-(1 -Methyl-1 H-pyrazol-4-yl)-5-(piperidin-4-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine was synthesized in a manner similar to the synthesis of 5-(piperidin-4-yl)-3-(quinolin-3- yl)pyrazoio[1 ,5-a]pyrimidin-7-amine, but with tert-butyl 4-(7-(bis((2- (trimethylsilyi)ethoxy)methyl)amino)-3-(1 -methyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5- a]pyrimidin-5-yi)piperidine-1-carboxylate substituted for tert-butyl 4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-(quino!in-3-yl)pyrazolo[1 ,5-a3pyrimidin-5- yl)piperidine-1 -carboxylate.
Synthesis of tert-butyl 2-(4-(7-amino-3-(1 -methyl-1 H-pyrazol-4-yl)pyrazolori ,5- aipyrimidin-5-yl)piperidin-1-yl)acetate
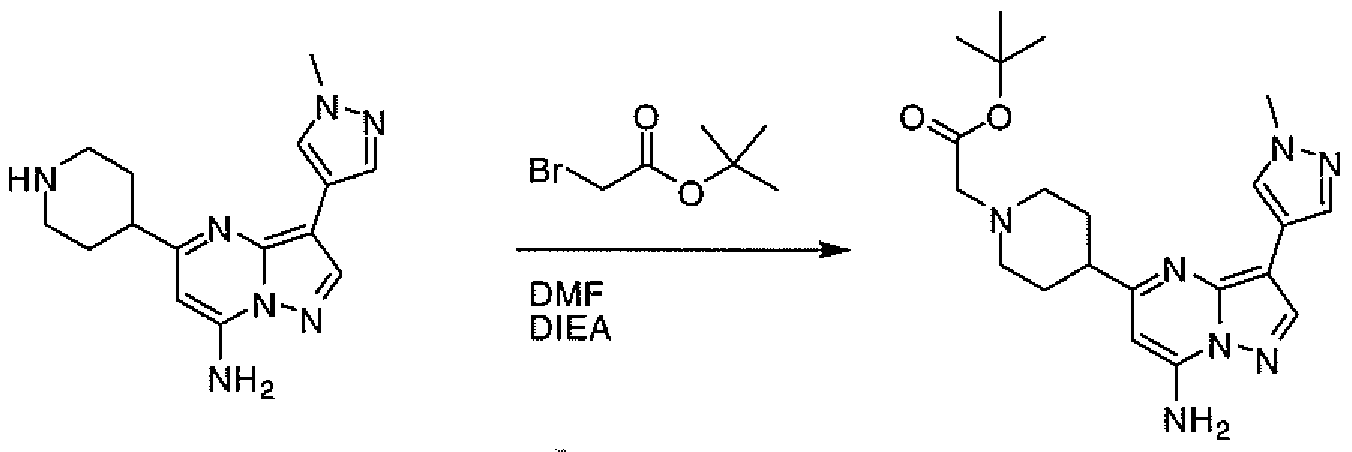
Synthesis of 2-(4-(7-amino-6-bromo-3-( 1-methyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5- alpyrimidin-5-vOpiperidin-i -ypacetic acid

2-(4-{7-amino-6-bromo-3-(1 -methyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)piperidin-1-yl)acetic acid was synthesized in a manner similar to the synthesis of 2- (4-(7-amino-6-bromo-3-(quinolin-3-yJ)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidin-1-yl)acetic acid, but with tert-butyl 2-(4-(7-amino-3-(1-methyI-1 H-pyrazol-4-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)piperidin-1-yl)acetate substituted for tert-butyl 2-{4-(7-amino-3- (quinolin-3-yi)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidin-1-yl)acetate. The reaction mixture wass reduced in vacuo and purified via reverse-phase preparatory HPLC to yieid 2-(4-(7-amino-6-bromo-3-(1-methyl-1 H-pyrazol-4-yl)pyrazolo[1 ,5-a]pyrimidin-5-
yl)piperidin-1-yl)acetic acid as off-yellow solid. (m+H = 434.19, retention time = 2.25 min)
tert-butyl 3-(7-hydroxypyrazolof 1 ,5-a1pyrimidin-5-yl)pyrrolidine-1 -carboxylate

A mixture of tert-butyl 3-(3-ethoxy-3-oxopropanoyl)pyrrolidine-1 -carboxylate (7.00 g, 25 mmol) and 1 H-pyrazol-3-amine (2.08 g, 25 mmol) in toluene 30 mL) was heated at 1 10 °C under argon for 18 h and concentrated in vacuo. The residue was triturated with EtOAc and the solid was collected by filtration and dried in high vacuum to give 6.84 g (90%) of the title compound as a white solid. LC/MS RT = 1.40 min (5 min method). Mass calculated for, M+H 305.15, observed 305.15.
tert-butyl 3-(7-chloropyrazolo|"1 ,5-a1pyrimidin-5-yl)pyrrolidine-1 -carboxylate

To a mixture of tert-butyl 3-(7-hydroxypyrazoio [1 , 5-a] pyrimidin-5-yl) pyrrolidine-1 - carboxylate (3.70 g, 12.0 mmol) and dimethylaniline (4.0 mL, 30 mml) was added phosphoryl trichloride (40 mL). The mixture was heated at 50 °C for 5 h and concentrated in vacuo. The residue was diluted with methylene chloride (100 mL) and quenched with saturated NaHCOe (100 mL). The mixture was separated and the aqueous layer was extracted with chloride (100 mL x Z). The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by Biotage (CH2Ci2/Et0Ac, 20:1 to 4:1). LC/MS RT = 2.09 min (5 min method). Mass calculated for, M+H 322.12, observed 337.14.
tert-butyl 3-( 7-aminopyrazolo[1 ,5-a1pyrimidin-5-yl)pyrroiidine-1 -carboxyiate

A mixture of tert-butyl 3-(7-chloropyrazolo [1 , 5-a] pyrimidin-5-yl) pyrroiidine-1- carboxyiate (3.40 g, 10.5 mmol) in a solution of ammonia in MeOH (7N1 20 mL) was heated in sealed vessel at 80 °C for 5 h. The reaction mixture was cooled down and concentrated in vacuo. The residue was dried in high vacuum and used for the next step without further purification. LC/MS RT = 1.30 min (5 min method). Mass calculated for, M+H 303.17, observed 318.19.
tert-butyl 3-(7-(bisf(2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-aipyrimidin-5- vQpyrrolidine-1 -carboxviate
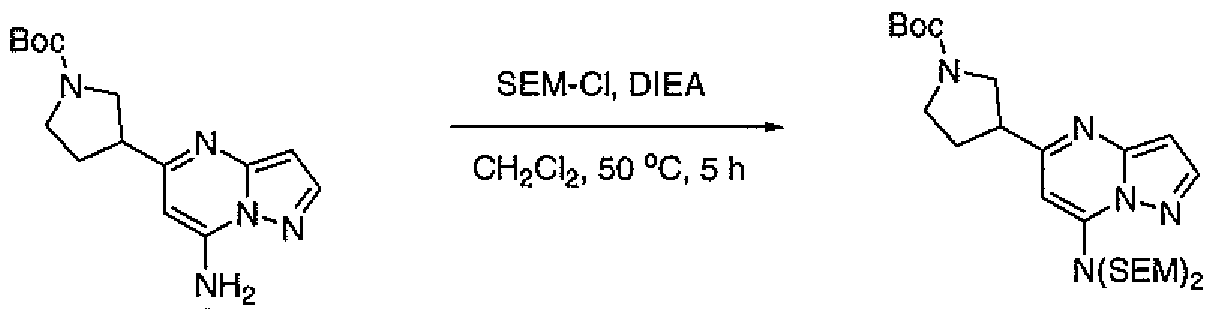
To a mixture of tert-butyl 3-(7-aminopyrazolo [1 , 5-a]pyrimidin-5-yi)pyrrolidine-1 - carboxyiate (10.5 mmol) in CH2CI2 was added DIEA (10.8 mL, 63 mmol) and SEM-CI (5.6 mL, 32 mmol). The mixture was heated at 50 °C for 5 h under an argon atmosphere. The mixture was cooled, and the reaction was quenched with saturated NaHCO3 (100 mL) and extracted with CH2CI2 (50 mL x 3). The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by Biotage (CH2CI2/EtOAc, 20:1 to 5:1 ) to give the title compound as a light brown oil. LC/MS RT - 2.90 min (5 min method). Mass calculated for, M+H 563.33, observed 578.35.
tert-butyl 3-(7-(bisf(2-(trimethylsiiyl)ethoxy)methyl)amino)-3-iodopyrazolori .5- aiPvrimidin-5-vhpvrroiidine-1 -carboxviate

To a solution of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methy!)amino)pyrazoio[1 ,5- a]pyrimidin-5-yl)pyrroliciine-1-carboxylate 3.95 g, 7.0 mmol) in CH3CN {30 mL) was added NIS {1.65 mg, 7.0 mmol). The mixture was stirred at rt for 2 h and diluted with EtOAC (50 mL) and washed with saturated sodium thiosulfate, dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by Biotage (CH2CI2/Et0Ac, 100:1 to 10:1 ) to the title compound as a light brown oil. LC/MS RT = 2.49 min. Mass calculated for, M+H 689.23, observed 389.17.
tert-butyl 3-(7-(bisf(2-(trimethylsilyl)ethoxy)methyhamino)-3-(quinolin-3- yhpyrazoioπ .5-alpvrimidin-5-vhpyrrolidine-1 -carboxylate

A mixture of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazolo[1 ,5-a]pyrirnidin-5-yl)pyrrolidine-1 -carboxylate (703 moi), quinolin-3- ylboronic acid (350 mg, 2.00 mmol), Pd(dppf)CI2-CH2CI2 (82 mg, 0.10 mmol) and potassium phosphate (636 mg, 3.0 mmol) in a mixture of dioxane/H2O (9:1 , 20 mL) was heated at 100 0C for 5 h under argon. The reaction mixture was cooled down and diluted with EtOAC, washed with Brine, dried over Na2SO4, filtered and concentrated. The residue was purified by Biotage (CH2CI2ZEtOAc, 10:1 to 3:1 ) to give the title compound as brown solid. LC/MS RT = 2.49 min. Mass calculated for, M+H 690.37, observed 389.17.
tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(αuinoiin-3- yl)pyrazoloπ ,5-aipyrimidin-5-yl)piperidine-1 -carboxyiate

To a solution of tert-butyl 3-{7-(bis((2-(trimethylsiIyl)ethoxy)methyl)amino)-3-(quinolin- 3-y!)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxyiate (350 mg, 0.50 mmol) in CH3CN (5.0 mL) was added NBS (90 mg, 0.50 mmol). The mixture was stirred at rt for 3 h and concentrated in vacuo. The residue was purified by Biotage (CH2CI2/Et0Ac, 100:1 to 10:1) to give the title compound as light yellow solid. LC/MS RT = 2.49 min. Mass calculated for, M+H 768.29, observed 389.17.
6-bromo-5-(piperidin-3-yl)-3-(quinoiin-3-yl)pyrazolo[1 ,5-aiPyrimidin-7-amine

To a solution of tert-butyl 3-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromσ-3- (quinolin-3-yl)pyrazoio[1 ,5-a3pyrimidin-5-yl)piperidine-1-carboxylate (196 mg, 0.40 mmol) in EtOH (5.0 mL) was added 4 N HCI (5.0 mL). The mixture was heated at 60 °C for 4 h under argon, cooled down and concentrated in vacuo. The residue was dried in high vacuum to give the title compound as yellow solid (HCI salt). LC/MS RT = 2.49 min. Mass calculated for, M+H 408.07, observed 389.17.
6-bromo-5-(1 -(methylsulfonyl)piperidin-3-yl)-3-(quinolin-3-yl)pyrazolo[1 ,5-aipyrimidin- 7-amine

By essentially the same procedure given in Schemes 25, the compounds listed in Table 8 can be prepared.
Table 8



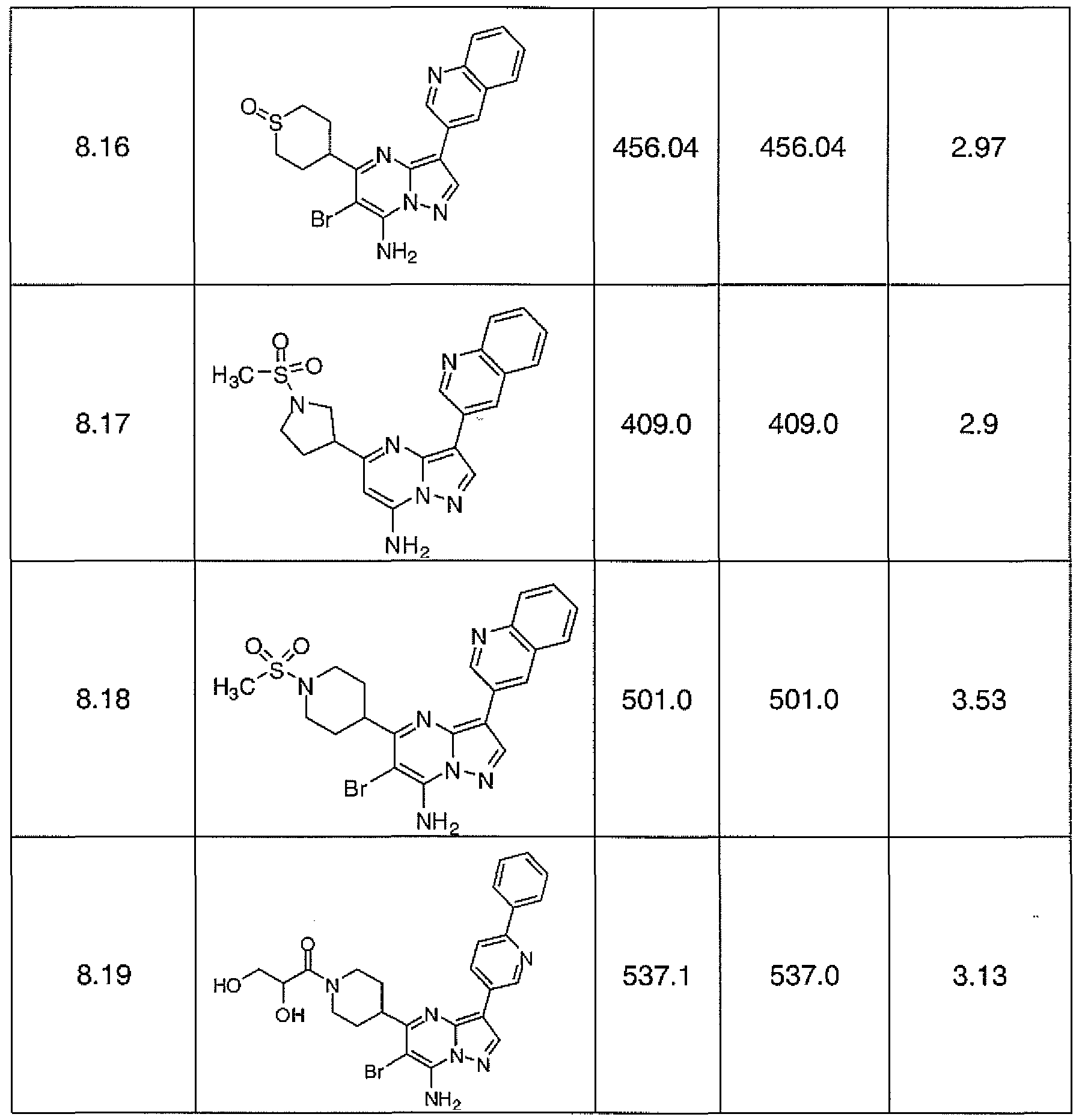
Alternative to the described procedure in the Scheme-14, these compounds can aiso be made by the scheme 25 given below. 7-Amino-3-(6-Dhenylpyridin-3-yl)pyrazoloπ .5-aipyrimidin-5-yl)piperidine-1 -carboxyiate:

SCHEME-26
Step-1
Synthesis of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amSno)pyrazoioπ ,5- alpyrimidin-5-yl)-5,6-dihydropyridine-1 (2H)-carboxylate

DME, 2M Na2CO3
100 °C
To a pressure tube were charged 5-chloro-N,N-bis((2-
(trimethylsϋylJethoxyJmethyl)pyrazoloII ^-alpyrimidin-y-amine {5.36 g, 12.5 mmols), tert-butyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaboroian-2-yl)-5,6-dihydropyridine-1 (2H)- carboxylate (4.26 g, 13.8 mmols), PdC!2(dppf).CH2Cl2 ([1 ,1 '- bis(diphenylphosphino)ferrocene]dichloropal!adium(il) complex with dichloromethane (1 :1 ) ) (510 mg, 0.62 mmol), 2M Na2CO3 (30 mL) and DME (60 ml_). The tube was degassed with Ar briefly, capped and heated at 100 0C with stirring overnight. After cooling, the reaction mixture was diluted with EtOAc and water, organic layer was isolated, washed with brine and dried (MgSO4). After solvent was removed under reduced pressure, the residue was purified on silica. Elution with EtOAc in hexanes
(0-25%) gave tert-butyl 4-(7-(bis((2-(trimethylsilyi)ethoxy)methyl)amino)pyrazolo[1 ,5- a]pyrimidin-5-yl)-5,6-dihydropyridine-1(2H)-carboxylate (5.76 g, 80%).
Step 2
Synthesis of fe/t-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolof 1 ,5- aipyrimidin-5-yl)piperidine-1-carboxyiate

A mixture of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5- a]pyrimidin-5-yl)-5,6-dihydropyridine-1 (2H)-carboxylate (5.05 g,8.78 mmoi) and 10% Pd/C (100 mg) in EtOAc was stirred at 45 °C under hydrogen (balloon pressure) for three hours. After filtration, washing with EtOAc (3x) and concentration, te/t-butyl A- (7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine- 1 -carboxyiate (5.1 g, 99.5%) was obtained as oil.
Step 3 Synthesis of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyhamino)-3- iodopyrazolori ,5-aipyrimidin-5-yl)piperidine-1-carboxviate

Step 4
Synthesis of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyQamino)-3-(6- phenv[pyridin-3-yl)pyrazoloM,5-aiPyrimidin-5-yl)piperidine-1-carboxylate
DME/water

To a pressure tube were charged te/ϊ-butyl 4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1 - carboxylate (4.2 g, 5.97 mmol), 2-phenyl-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pyridine (2 g, 7.1 mmol), PdCI2(dppf).CH2CI2 {240 mg, 0.33 mmol), DME (16 ml_) and 2M Na2Cθ3 (8 mL). The mixture was briefly degassed with Argon and the tube was capped and heated at 100 °C for 15 hours. On cooling, H2O (20 mL) and EtOAc (40 mL) and aqueous layer was extracted with EtOAc (3x) and combined organic layers were washed with brine once and dried (MgSO4). After concentration in vacuo the residue was purified on silica gel. Elution with EtOAc/Hexanes (0-40%) gave the title compound (3.24 g, 74%).
Step 5 Preparation of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyDamino)-6-bromo-3- (6-phenylpyridin-3-yl)pyrazolori,5-a1pyrimidin-5-yl)piperidine-1 -carboxvlate

To a solution of tert-buty! 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazo!o[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxylate (3.24 g, 4.44 mmo!) in DMF (20 mL) was added NBS (750 mg, 4.21 mmol). After stirring at rt for one hour, NBS (75 mg 0.42 mmol) was added and the resulting mixture was stirred for another 40 minutes to complete the reaction. The reaction mixture was diluted with EtOAc (200 mL), washed with water (2x50 mL), brine (20 mL) and dried (MgSO4).
After concentration in vacuo the residue was purified on silica gel. Elution with EtOAc/Hexanes (0-40%) gave the title compound (3 g, 83%).
Step-6 General method:

TABLE-8A














Synthesis of (RH-(4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolori ,5- a1pyrimidin-5-yl)piperidin-1 -yl)-2-hydroxypropan-1 -one

Step 6
Preparation of tert-butyl 4-(7-(bisf(2-(trimethylsilyl)ethoxy)methyπamino)-6-(1 - ethoxyylnyl)-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)piperidine-1- carboxylate

To a pressure flask were charged compound fert-buty! 4-(7-(bis((2- (trimethylsilyl}ethoxy)methy!)amino)-6-bromo-3-{6-phenylpyridin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)piperidine-1 -carboxylate ( intermediate from Scheme-10A) (13.66 g, 16.86 mmol), tributyl (i-ethoxyvinyl)tin (11.4 mL, 33.74 mmols) and dioxane (150 ml). The flask was degassed with Ar briefly, capped and heated at 100 C with stirring overnight. After cooling, the reaction mixture was concentrated. The residue was taken into ether (200 ml), washed with 0.5 M KF (50 ml), brine and dried (MgSO4). After solvent was removed under reduced pressure, the residue was purified on silica. Elution with EtOAc in hexanes (0-30%) gave compound tert-butyl 4-
(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-{1 -ethoxyvinyl)-3-(6-phenylpyridin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1-carboxylate (1 1.55 g, 85%) as yellow form.
Step 7
Preparation of 1 -(7-amino-3-(6-phenylpyridin-3-yl)-5-(piperidin-4-yl)pyrazoiori ,5- aipyhmidin-6-yl)ethanoπe

To a solution of tert-butyl 4-(7-(bis({2-(trimethylsilyl)ethoxy)methyl)amino)-6-(1- ethoxyvinyl)-3-(6-phenylpyridin-3-y!)pyrazoio[1 ,5-a]pyrimidin-5-yi)piperidine-1- carboxyiate (1 1.5 g, 14.4 mmol) in dioxane (60 mL) was added 4M HCI in water (14.4 mL) at 0 °C. After stirring for 30 min at 0 °C, the reaction mixture was allowed to warm to rt in 1.5 hours. 4M HCl in dioxane (10 mL) was added. The reaction mixture was stirred at rt overnight. Solvent was removed in vacuo to get the desired product as an HCI salt (6.97 g).
Step 8
Preparation of (R)-1-(4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolof1 ,5- alpyhmidin-5-γ[)piperidin-1 -yl)-2-hydroxypropan-1 -one

LC/MS RT = 3.22 min. Mass calculated for M+H 485.2, observed 485.4.
Synthesis of (R)-1-(4-(3-(6-( 1H-1 , 2,4~triazol-1-vnpyridin-3-ylV-6-acetyl-7- aminopyrazoioh ,5-alpyrimidin-5-yl)piperidin-1 -yl)-2-hydroxypropan-1 -one

Step 1
Preparation of 5-bromo-2-(1 H-1 ,2,4-triazol-1 -yl)pyridine:

A mixture of 1 ,2,4-triazole {1.17 g, 16.9 mmol), 2,5-dibromopyridine (2 g, 8.4 mmol) and K2CO3 (1.2 g, 8.7 mmol) in NMP (20 mL) was heated at 100 °C with stirring for 5 hours. After cooling, the reaction mixture was poured into water (100 mL) and solid was collected by filtration and further purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-50%) provided the title compound (250 mg, 13%).
Step 2
Preparation of 5-(4.4.5.5-tetramethyl-1 ,3,2-dioxaborolan-2-ylV-2-(1 H-1 ,2,4-triazol-1- vppyridine:

A mixture of 5-bromo-2-(1 H-1 ,2,4-triazol-1 -yl)pyridine (248 mg, 1.1 mmol), bis(pinacolato)diboron {337 mg, 1.4 mmol), KOAc (324 mg, 3.3 mmol), PdCI2(dppf)2.CH2CI2 (45 mg, 0.06 mmol) in dioxane (4 mL) was flushed with Argon and stirred at 80 °C for 16h. On cooling, the solvent was rotoevaporated, and the crude was redissolved in dichloromethane (20 mL), washed with water (3 x 10 ml), brine (1 x 101 ml), and dried over MgSO4. Solvent was removed in vacuo and the residue was triturated with hexanes to provide the title compound (287 mg, 96%).
Step 3
Preparation of fert-butyl 4-(3-(6-(1 H-1.2.4-triazol-1-vnpyridin-3-yl)-7-(bisff2-
(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]Dyrimidin-5-yl)piperidine-1 - carboxyiate:
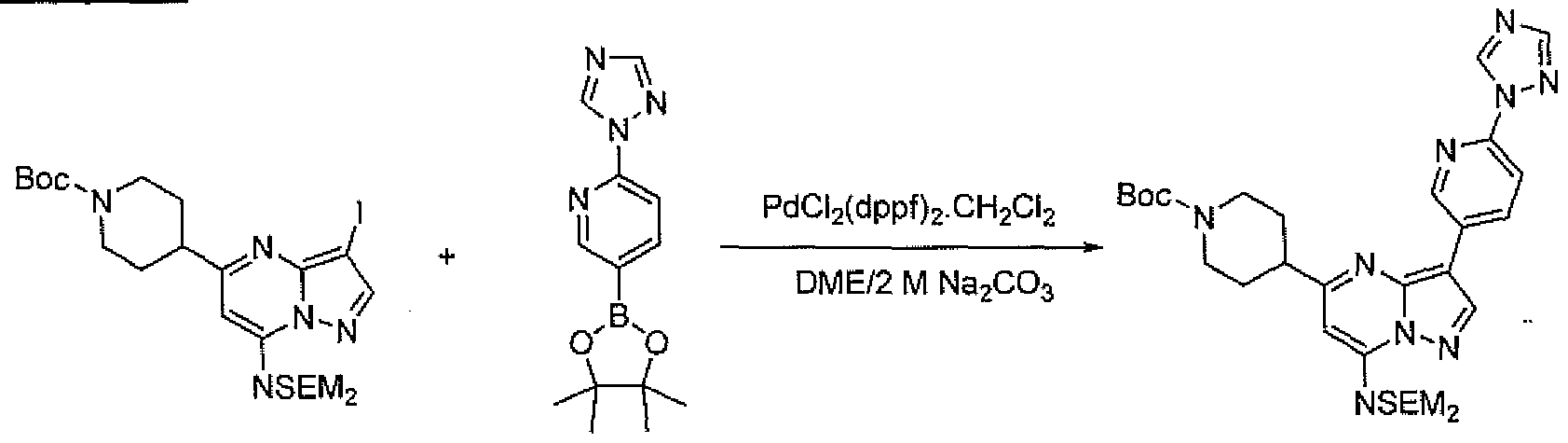
To a pressure tube were charged tert-butyl 4-(7-(bis((2- (trimethy!silyl)ethoxy)methyl)amino)-3-iodopyrazoio[1 ,5-a]pyrimidin-5-yl)piperidine-1 - carboxylate (563 mg, 0.8 mmoi), 5-{4,4,5,5-tetramethyl-1 ,3,2-dioxaboroian-2-yl)-2- (1 H-1 ,2,4-triazol-1-yl)pyridine (287 mg, 1.1 mmol), PdCI2JdPPf)2-CH2CI2 (29 mg, 0.04 mmol), DME (6 mL) and 2M Na2CO3 (2 mL). The resulting mixture was briefly degassed with Argon; the tube was capped, and heated with stirring under 80 ° C overnight. After cooling, solvent was removed. The residue was diluted with EtOAc (20 ml), and organic layer was isolated, washed with brine and dried (MgSO4). After concentration under reduced pressure, the residue was purified on silica gel eluting with EtOAc/Hexanes (0-50%) to provide the title compound (370 mg, 64%). Step 4
Synthesis of (RH -(4-(3-(6-(1 H-1.2.4-triazol-1-vθDyridin-3-yl)-6-acetyl-7- aminopyrazolof 1 ,5-a1pyrimidin-5-vPpipeπdin-1 -vO-2-hydroxypropan-1 -one:
The rest of the synthesis is similar to the synthesis of (R)-1 -(4-(6-acetyl-7-amino-3-{6- phenylpyridin-3-yi)pyrazolo[1 ,5-a]pyrimidin-5-yi)piperidin-1 -yl)-2-hydroxypropan-1 -one.
Synthesis of 1 -( 7-aminθ"5-(1 -methylpiperidin-4-vO-3-(6-phenylpyridin-3- yl)pyrazolori ,5-aipyrimidin-6-y[)ethanone

To a solution of 1-(7-amino-3-(6-phenylpyridin-3-yt)-5-(piperidin-4-yl)pyrazolo[1 ,5- a]pyrimidin-6-yl)ethanone (25 mg, 0.06 mmoi), DiEA (52 μl_, 0.3 mmol) in DMF (1 ml_) was added Mei (15 μL, 0.24 mmol). The reaction mixture was stirred for one hour and directly purified by HPLC to provide the title compound. LC/MS RT = 2.76 min. Mass calculated for M+H 427.2, observed 427.3.
General methods described above, compounds in the table 8-B can be synthesized.
TABLE- 8B









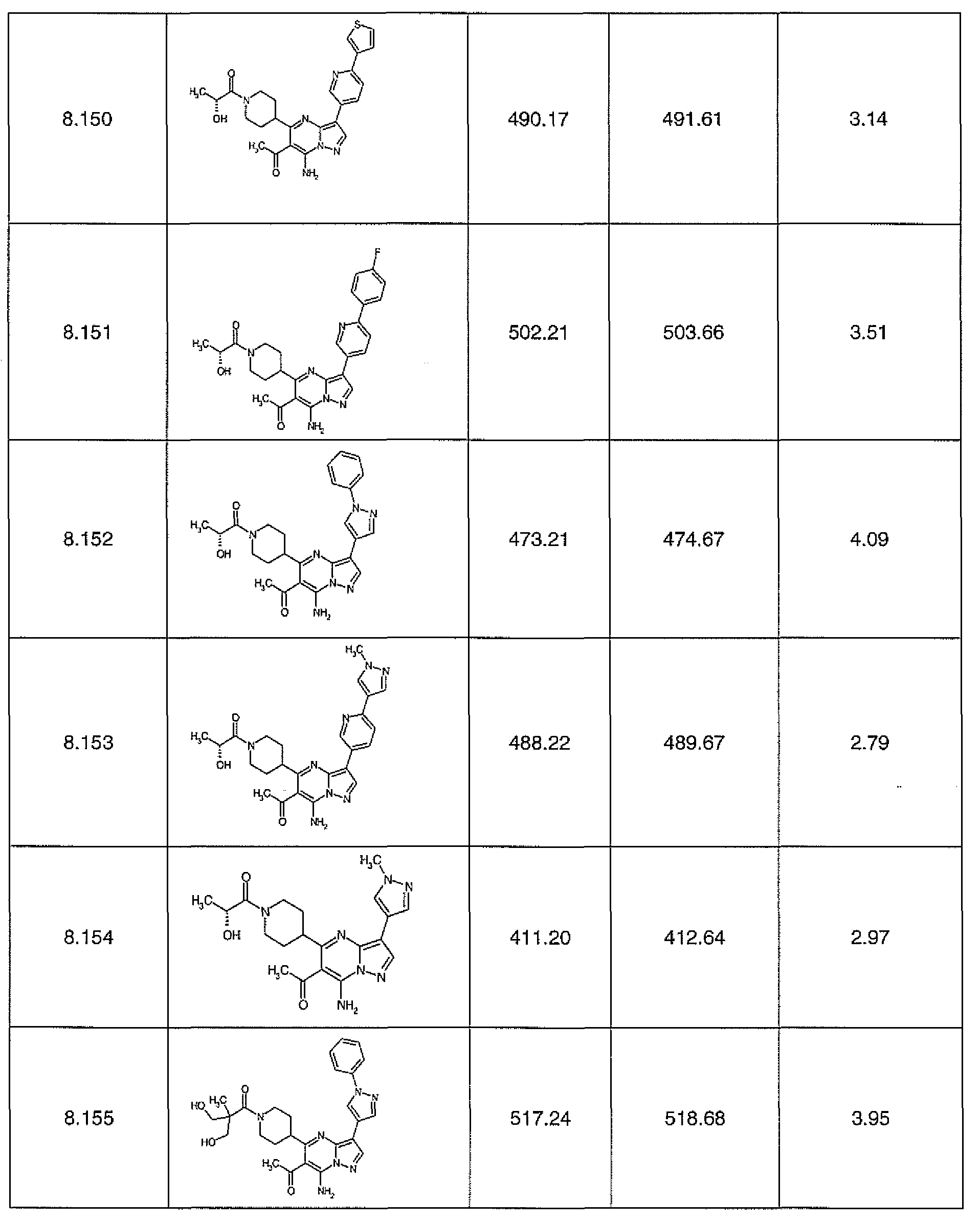





















Synthesis of tert-butyl 4-(3-(6-(1 H-Dyrazol-1-yl)pyridin-3-yl)-7-(bisff2-(trimethylsilyl) ethoxy)methyl)amino)pyrazolof1 ,5-aiPyrimidin-5-yl)piDeridine-1-carboxylate:

To tert-butyl 4-(7-(bis((2-(iximethylsilyl)ethoxy)methyl)amino)-3-(6-chloropyridin-3- yl)pyrazolo[l,5-a]pyrimidin-5-yi)piperidine-1-carboxylate (2.25g, 3.3mmol) in Dioxane (24ml) and H2O (6mi) was added the 2-fluorophenyIboronic acid (0.92g, 6.6mmol), PdCl2(dppf)2.CH2Cl2 (0.32g, 0.39mmol) and K3PO4-H2O ( L7g, 8.2mmol). The reaction was heated at 100 °C for 16 hours, at which time LC/MS analysis confirmed full consumption of starting material. On cooling, H2O (100ml) and EtOAc (100ml) were added and organics were extracted with EtOAc (4 x 50ml), dried (Na2SO4) and concentrated in vacuo to give a crude product. Gradient column chromatography on silica eluting with 0% to 100% EtOAc/hexanes gave tert-butyl 4-(7-(bis((2-(trimethylsilyl) ethoxy)methyl)amino)-3-(6-(2- fluoropheny!)pyridin-3-yi)pyrazoio[i,5-a]pyrimidin-5-yl)piperidine-l-carboxylate (2.3g, 95%).
By essentially the same procedure as to other related compound, different derivatives of above compound is prepared (Table-SB ).
Synthesis of tert-butyl 4-(3-(6-(1 H-pyrazol-1-yl)pyridin-3-yl)-7-(bisff2-(trimethylsilyl) ethoxyϊmethyltøminotøyrazoloH .5-aipyrimidin-5-vQpiperidine-i -carboxylate:

By essentially the same procedure as above, tert-butyl 4-(3-(6-(1H-pyrazol-1-yl)pyridin-3-yl)- 7-(bis((2-(trimethylsiiyl) ethoxy)methyl)amino) pyrazolo[ 1 ,5-a]pyrimidin-5-yl)piperidine-1- carboxylate is prepared.
Synthesis of (RH -(4-(7-aminQ-6-(2-hydroxyacetyl)-3-(6-phenylpyridin-3- yl)pyrazolo[1 ,5-alpyrimidin-5-yl)piperidin-1 -yl)-2-hydroxypropan-1 -one:

Step-1
Synthesis of 1 -(7-amino-3-(6-phenylpyridin-3-yl)-5-(piperidin-4-yl)pyrazoloπ .5- a1pyrimidin-6-yl)-2-hydroxyethanone:
A solution of oxone (1.9g, 3.1mmol), trifluoroacetic anhydride (1.1ml, 7.9mmoi) and H2O (5.7ml) was stirred at 40°C for 7 h, and then cooled to room temperature. To this a solution of tert-butyl 4-(6-acetyl-7-(bis((2-(trimethylsiIyi)ethoxy)methyl)amino)-3-(6-phenylpyridin-3- yl)pyrazolo[t,5-a]pyrimidin-5-yl)piperidine-l-carboxylate (0.87g, 1.13mmol) and PhI (0.03ml, 0.23mmol) in acetonitrile (17ml) was added. The resulting solution was stirred at 90°C for 15 h. On cooling, the reaction mixture was filtered and solids were washed with DCM. It was then concentrated in vacuo to give a crude product which was submitted to the analytical group for purification to afford the desired l-(7-amino-3-(6-phenyIpyridin-3-yl)-5-(piperidin-4- yl)pyrazolo[ 1,5-a]pyrimidin-6-yl)-2-hydroxyethanone (94.2mg, 17%).
Step-2:
Synthesis of (RH -(4-(7-amino-6-(2-hydroxyacetyl)-3-(6-phenylpyridin-3- yl)pyrazoioM ,5-aipyrimidin-5-yl)piperidin-1 -yl)-2-hydroxypropan-1 -one:
A mixture of (R)-2-hydroxypropanoic acid (13.8mg, O.lβmraoi), EDCI (46mg, 0.24mmol), and 1-hydroxybenzotriazole (16.22mg, 0.12mmol) in DMF (2ml) was stirred at room temperature for lOmin. To this l-(7-amino-3-(6-phenylpyridin-3-yl)-5-(piperidin-4- y!)pyrazolo[1,5-a]pyrimidin-6-yl)-2-hydroxyethanone (58.77mg, 0.12mmol) was added followed by N,N-diisopropylethylamine (0. ImI, 0.6mmol). It was stirred further for 20min at room temperature at which time LC/MS analysis confirmed full consumption of starting material. This crude compound was submitted to the analytical group for purification to afford the desired (R)-1-(4-(7-amino-6-(2-hydroxyacetyl)-3-(6-phenylpyridin-3-yl)pyrazolo[ 1 ,5- a]pyrimidin-5-yl)piperidin-l-yl)-2-hydroxypropan-l -one. LCMS: 3.27 mins, m/z = 501.0 (MH+).
Synthesis of (R)-1-(7-amino-5-(1-(5-(1 -hydroxyethyl)-1 ,3.4-oxadiazol-2-yl)piperidin-4- yl)-3-(6-phenylpyridin-3-yl)pyrazoio[1 ,5-alpyrimidin-6-yl)ethaπone:

Step 1
Preparation of benzyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3- iodopyrazoioπ ^-alpyrimidin-5-yPpiperidine-i -carboxylate:
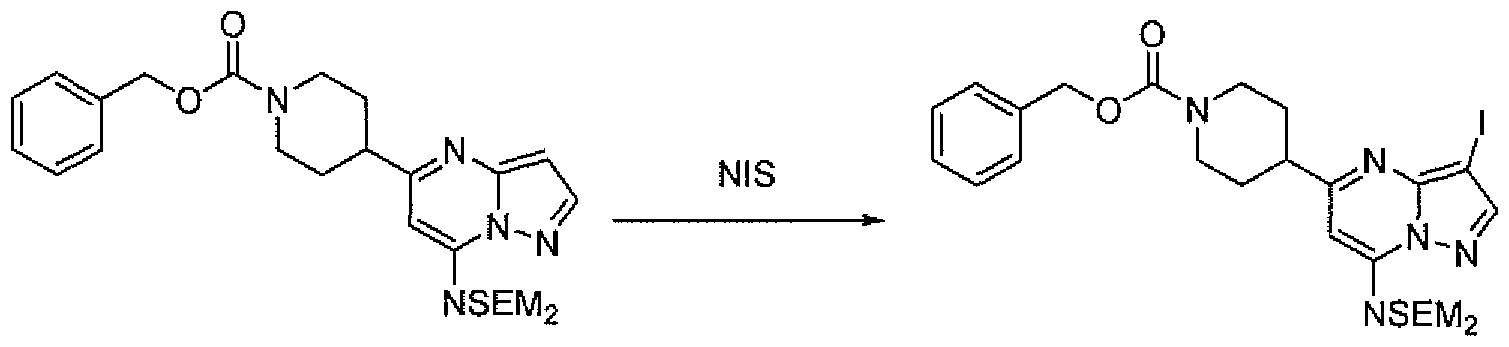
To a solution of benzyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1,5- a]pyrimidin-5-yl)piperidine-1-carboxylate (2.27g, 3.7 mmol) in acetonitriie (15 mL) was added NIS (833 mg, 1 eq}. After stirring 1.5 hour, solvent was removed and the residue was purified on silica gel. Elution with EtOAc/Hexanes (0-50%) gave the title compound (2.38 g, 86%) as gum.
Step 2
Preparation of benzyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazo[ori ,5-alpyrimidin-5-yl)piperidine-1 -carboxylate:

To a pressure tube were charged benzyl 4-(7-(bis((2-
(trimethylsilyl)ethoxyJmethyl)aminoJ-3-iodopyrazoloCI^-alpyrimidin-5-yl)piperidine-1- carboxylate (1 g, 2.85 mmol), 2-phenyl-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pyridine (1.2g, 4.1 mmol), Pd(PPh^)4 (164 mg, 0.14 mmol), DME (20 mL) and 2 M IMa2CO3 (10 mL). The mixture was briefly degassed with Argon and the tube was capped and heated at 80 °C for 15 hours. On cooling, H2O (40 mL) and EtOAc (100
mL) and aqueous layer was extracted with EtOAc (3x) and combined organic layers were washed with brine once and dried (MgSO4). After concentration in vacuo the residue was purified on silica gel. Eiution with EtOAc/Hexanes (0-30%) gave the title compound (1.33 g, 61%).
Step 3
Preparation of 3-(6-phenylpyridin-3-yl)-5-(piperidin-4-yl)-NiN-bis(f2- ftrimethylsiiyl)ethoxy)methyl)pyrazolo[1 ,5-aipyrimidin-7-amine:

A mixture of benzyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolo[1 ,5-a3pyrimidin-5-yi)piperidine-1-carboxylate (1.33 g, 1.77 mmol), 10% Pd/C (170 mg) in EtOAc (10 mL) was stirred under hydrogen (balloon pressure) for two days, and then at 45 °C for 15 hours. After filtration and washing with EtOAc 4 times the title product (1 g, 93%) was obtained.
Step 4
Preparation of (R)-2-(benzyloxy)propanehydrazide;

A mixture of (R)-methyl 2-(benzyloxy)propanoate (1g, 5.15 mmol), hydrazine monohydrate (250 ul) in MeOH ( 14 mL) was refluxed 3 hours. Solvent was removed under reduced pressure and the residue was purified on silica gel. Eiution with EtOAc yielded title compound (640 mg, 64%).
Step S
Preparation of (RV5-M -(benzyloxy)ethyh-i ,3.4-oxadiazol-2(3/-fl-one:

To a solution of (R)-2-(benzyloxy)propanehydrazide (194 mg, 1 mmol) in anhydrous THF ( 2 mL) was added CDI (178 mg, 1.1 mmol) and the resulting mixture was allowed to stir one hour. . Solvent was removed under reduced pressure and the residue was purified on silica gel. Elution with EtOAc yielded title product.
Step 6
Preparation of (R)-5-(1-(5-(1-(benzyloxy)ethyl)-1 ,3.4-oxadiazol-2-yl)piperidin-4-yl)-3-
(6-phenylpyridin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5- alpyrimidin-7-amine:

To a stirred solution of (R)-5-(1-(benzyloxy)ethyl)-1 ,3,4-oxadiazol-2(3H)-one {44 mg, 0.2 mmol), 3-(6-phenyipyridin-3-yl)-5-(piperidin-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-a3pyrimidin-7-amine (150 mg, 0.24 mmol) and DIEA {70 ul, 0.4 mmol) in DMF (1.4 mL) was added BOP (97 mg,0.22 mmol) and the resulting solution was allowed to stir overnight. Water was added and was extracted with EtOAc (3x) and combined organic layers were washed with water (3x), brine once and dried (MgSO4). After concentration in vacuo the residue was purified on silica gel. Elution with EtOAc/Hexanes (0-10%) gave the title compound (142 mg, 85 %).
Step 7
Preparation of I RH -(5-(4-( 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)pipehdin-1-yl)-1 ,3,4-oxadiazol-2-
yl)ethanol:

A mixture of (R)-5-(1-(5-(1 -(benzyloxy)ethyl)-1 ,3,4-oxadiazoi-2-yl)piperidin-4-yl)-3-{6- phenylpyπdin-3-yl)-Λ/,Λ/-bis{(2-(trimethylstlyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrirτiidin-7- amine (105 mg), 20% Pd(OH)2/C (150 mg) in MeOH {4 ml_) was stirred under hydrogen (60 psi) overnight. After filtration, washing with EtOAc 4 times and concentration in vacuo the residue was purified on siiica gel. Elution with EtOAc/Hexanes (0-100%) gave the title compound (32 mg,43%).
Step 8
Preparation of (RH -(5-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)aminoV6-bromo-3-
(6-phenylpyridin-3-yl)pyrazolof 1 ,5-a1pyrimidin-5-vQpiperidin-1 -yl)-1 ,3,4-oxadiazol-2- yl)ethanol:

To a solution of (R)-1 -(5-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidin-1 -yl)-1 ,3,4-oxadiazol-2- yl)ethanol (32 mg, 0.04 mmo!) in acetonitriie (1 mi_) was added NBS (7 mg, 0.04 mmol) and the resulting solution was stirred for 10 minutes and concentrated, the residue was purified on silica gel. Elution with EtOAc provided a relatively pure product (9 mg).

Step 9
Preparation of (R)-1-(7-amino-5-(1-(5-(1-hydroxyethyl)-1 ,3,4-oxadiazol-2-yl)piperidin-
4-yl)-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-6-yl)ethanone:

To a pressure tube were charged (R)-1-(5-(4-(7-(bis({2- (trimethylstlyl)ethoxy)methyl)amino)--6-brorno-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5- a]pyhmidin-5-yl)piperidin-1-yI)-1 ,3,4-oxadiazol-2-yl)ethanol (9 mg,~0.01 mmol), Pd(PPh3)4 (10 mg, 0.087 mmoi), tributyl{-ethoxy-vinyl)tin (8 ui, 0.02 mmol) and dioxane (2 mL). The resulting mixture was degassed with Argon briefly, capped with a Teflon cap and stirred at 100 °C overnight. On cooling, the solvent was rotoevaporated, and the crude was redissolved in EtOAc (10 ml), washed with 0.5 M KF solution (1 x 2 ml), water (1 x 3 ml), brine (1 x 3 ml), and dried over MgSO4. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-100%) gave the intermediate eno! ether which was re-dissolve in dioxane (2 mL) and cooled to 0 °C and treated aqueous 4N HCI (0.2 mL). After the reaction mixture was warmed up to room temperature and stirred for 4 hours, solvents were removed and the residue was directly purified by HPLC to provide the title compound. Synthesis of (R)-7-amino-5-(1 -(2,3-dihydroxypropanoyl)piperidin-4-yl)-3-(6- phenylpyridin-3-yl)pyrazoio, ,5-alpyrimidine-6-carbonitrile:

Step 1
Preparation of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-cvano-3-(6- phenv[pyridin-3-yl)pyrazoiori ,5-alpyrimidin-5-yl)Diperidine-1 -carboxylate:

To a Schlenk tube were charged compound tert-butyl 4-(7-{bis((2- (trimethylsilyl)ethoxy)methy!)amino)-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)piperidine-1 -carboxylate (81 mg, 0.1 mmol), Bu3SnCN (47 mg, 1.5 eq.), Pd(PPh3J4 (23 mg, 0.2 eq.), Bis(tri-f-butylphosphine)palladium (0) (10 mg, 0.2 eq.). The tube was evacuated and charged with Ar for three cycles. Dioxane (3 ml) was added; the tube was capped and heated at 160 C with stirring for one hour. After cooling, the mixture was diluted with EtOAc and washed with brine once. Organic layer separated, dried over MgSO4 and concentrated. The residue was purified on silica gel. Elution with EtOAc/hexane (0-25%) gave the desired title product (63 mg, 84%).
Step 2
Preparation of 7-amino-3-(6-phenylpyridin-3-yl)-5-(piperidin-4-yl)pyrazolof 1 ,5- aipyrimidine-6-carbonitrile:

Tert-butyl 4-{7-(bis((2-(trtmethylsilyl)ethoxy)methyl)amino)-6-cyano-3-(6-pheny!pyridin- 3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)piperidine-1 -carboxylate (190 mg) was treated with TFA/water(95:5, 3 ml) with stirring for 10 minutes. The reaction mixture was concentrated and lyophilized to yield the title compound as TFA salt.
Step 3
Preparation of (R)-7-amino-5-(1 -(2,3-dihydroxypropanoyl)piperidin-4-vO-3-(6- phenylpyridin-3-yl)pyrazoloF1 ,5-alpyrimidine-6-carbonitrile:
DMF

To a solution of 7-amino-3-(6-phenyJpyridin-3-yl)-5-(piperidin-4-yl)pyrazolo[1 ,5- a]pyrimidine-6-carbonitrile TFA salt (0.1 mmol), DIEA (87μL, 0.5 mmol) in DMF (2 ml_) was added a solution of (R)-2,3-dihydroxypropanoic acid (11 mg, 0.1 mmol), HOBt (14 mg, 0.1 mmoi), EDCl (29 mg, 0.15 mmol) in DMF (1 mL). The reaction was stirred at rt for 2.5 hour and the mixture was directly purified by HPLC to furnish the title compound. LC/MS RT = 2.86 min. Mass calculated for M+H 484.2, observed 484.3.
C

Scheme 27
Synthesis of pyrazoloH ,5-aipyrimidine-5,7-diol:
Diethyl malonate NaOEt, EtOH
 To 1 H-pyrazol-3-amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethy! malonate (25.0 mL, 164.7 mmol), 21wt% NaOEt in EtOH (1 10 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting reaction mixture was then heated at 80 °C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H2O (500 mL) was added. Vigorous stirring aided the dissolution of solids, at which time cone. HCI was added until pH~2 was attained (precipitate formed). The precipitate was collected and dried by vacuum fiitration giving pyrazolo[1 ,5-a]pyrimidine-5,7-diol as a tan solid (17.13 g, 113.4 mmol, 77%).
To 1 H-pyrazol-3-amine (12.3 g, 148.0 mmol) in EtOH (50 mL) was added diethy! malonate (25.0 mL, 164.7 mmol), 21wt% NaOEt in EtOH (1 10 mL, 294.6 mmol) and additional EtOH (50 mL). The resulting reaction mixture was then heated at 80 °C under an atmosphere of argon for 16 hours, at which time the reaction was allowed to cool to room temperature. The reaction mixture was then concentrated in vacuo until almost dry, before H2O (500 mL) was added. Vigorous stirring aided the dissolution of solids, at which time cone. HCI was added until pH~2 was attained (precipitate formed). The precipitate was collected and dried by vacuum fiitration giving pyrazolo[1 ,5-a]pyrimidine-5,7-diol as a tan solid (17.13 g, 113.4 mmol, 77%).
Synthesis of 5.7-dichloropyrazolo[1 ,5-aipyrimidine

To pyrazolo[1.5-alpyrimidine-5J-diol (9.6 g, 63.5 mmol) in a 500 mL flask was added POCI3 (125 mL, 1341 .1 mmol). The flask was then cooled to 0 °C and N, N-
dimethylaniline (22 mL, 173.6 mmol) was carefully added. On warming to room temperature, the reaction mixture was then heated at 60 °C under an atmosphere of argon for 16 hours. On cooling, the reaction mixture was concentrated in vacuo to give a brown viscous liquid. This brown viscous liquid was carefully poured onto ice and allowed to warm to room temperature overnight. To the brown solution was carefully added saturated NaHCO3 solution until no further effervescence was observed and pH ~ 8 was attained. Organics were then extracted with CH2CI2 (4 x 50 mL), dried (Na2SO4) and concentrated in vacυo to give a brown liquid (29.8 g). Gradient column chromatography on silica eluting with 50% CH2C!2/hexanes (to elute aniline) followed by 75% CH2CI2/hexanes (to elute product) gave 5,7-dichloropyrazoio[1 ,5-a]pyrimidine as a white solid (7.7 g, 40.8 mmol, 64%).
Synthesis of 5-chloropyrazoloH ,5-a]pyrimidin-7-amine

To 5,7-dichloropyrazolo[1 ,5-a]pyrimidine (7.6 g, 40.4 mmoi) in a sealed vessel was added NH4OH (100 mL). The vessel was then sealed and heated at 85 °C for 2.5 .. hours, at which time the consistency of the white solid had changed (from foamy white solid to free-flowing white solid). The vessel was removed from the heat source and allowed to cool to room temperature overnight. On cooling, the contents of the vessel were collected and dried by vacuum filtration giving 5-chloropyrazolo[1 ,5-a]pyrimidin- 7-amine as a yellow-tinged white solid (6.8 g, 40.3 mmol, 100%).
Synthesis of 5-chloro-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolof 1 ,5-aipyrimidin- 7-amine

Synthesis of 5-chloro-3-iodo-N,N-bis((2-(trimethylsi[yπethoxy)methyl)pyrazolori .5- aipyrimidin-7-amine

To crude 5-chloro-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazoIo[1 ,5-a]pyrimidin-7- amine (7.9 g) in CH3CN (100 mL) was added N-iodosuccinimide (4.3 g, 19.2 mmol) and the resulting mixture was stirred at room temperature for 30 mins, at which time LC/MS confirmed full conversion of starting material to product. Saturated sodium thiosulfate solution (-20 mL) was added and stirring continued for 5 minutes before the mixture was transferred to a separatory funnel using CH2Cb (30 mL) and H2O (30 mL). Brine (50 mL) was added and organics were extracted with CH2CI2 (4 x 40 mL), dried (Na2SO4) and concentrated in vacuo to give crude 5-chloro-3-iodo-N,N-bis({2- (trimethylsilyl)ethoxy)methyl)pyrazoio[1,5-a]pyrimidin-7-amine as a Sight brown liquid (10.4 g). LCMS: 2.95 mins, m/z = 555.1 (MH+).
Synthesis of 5-chloro-3-(quinolin-3-vO-N,N-bisf(2- ftrimethylsilyl)ethoxy)methyl)pyrazolon .5-alpyrimidin-7-amine

To crude 5-chloro-3-iodo-N,N-bis((2-(tnmethytsilyI)ethoxy)methyl)pyrazolo[1 ,5- a]pyrimi,din-7-amine (7.1 g) in DME (120 mL) and H2O (15 ml_) was added the quinoline boronic acid (2.4 g, 14.1 mmol), PdCI2(dppf)2 (1.0 g, 1.2 mmol) and K3PO4-H2O (5.4 g, 25.6 mmol). The reaction mixture was heated at 60 °C for 2 hours, at which time LC/MS analysis confirmed full consumption of starting material. On cooling, H2O (40 mL) and EtOAc (100 mL) were added and organics were extracted with EtOAc (4 x 50 mL), dried (Na2SO4) and concentrated in vacuo to give a brown oil. Gradient column chromatography on silica eluting with 10% to 60% EtOAc/hexanes gave 5-chloro-3-(quinolin-3-yl)-N,N-bis((2-(thmethylsiiyl)ethoxy)methyl)pyrazoio[1 J5- a]pyrimidin-7-amine as a light yellow solid (4.1 g, 7.4 mmol, 65% over three steps). LCMS: 2.62 mins, m/z = 556.2 (MH+).

SCHEME-28
Synthesis of 5-morpholino-3-(αuinolin-3-yl)pyrazoloM .5-aipyrimidin-7-amine

10
By essentially the same procedure given in the synthesis of 5-morpho!ino-3-(quinolin- 3-yl) pyrazolo[1 ,5-a]pyrimidin-7-amine, Schemes 27 & 28, the compounds listed in Table 9 can be prepared.
\5 Table 9



Synthesis of 1-(7-amino-3-(quinolin-3-yl)pyrazolo[1 ,5-aipyrimidin-5-yl)-1,4-ciiazepan-5- one

A mixture of 5-chIoro-3-(quinolin-3-yl)-N]N-bis((2-(trimethylsilyl)ethoxy) methyl)- pyrazolo-[1 ,5-a]pyrimidin-7-amine (50 mg, 0.09 mmoL), 1 ,4-diazepan-5-one (36 mg, 0.32 mmoL), EtN(SPr)2 (110 uL, 0.63 mmoL) in NMP (0.8 ml_) was heated at 185°C under microwave condition for 45 min. Purification by prep-LC afforded titled compound (32.3 mg, 85%) as its formic acid salt. LCMS tR = 2.49 Min. Mass calculated for, M+ 373.1 , observed LC/MS m/z 374.1 (M+H).
Synthesis of 1-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo|"1 ,5-a1pyrimidin-5-yl)-1 A- diazepan-5-one

A solution of NBS (14.6 mg, 0.082 mmoL) in CH3CN (0.5 mL) was added to a mixture of 1 -(7-amino-3-(quinolin-3-yi)pyrazolo[1 ,5-a]pyrimidin-5-yi)-1 ,4-diazepan-5-one formic acid salt (32.3 mg, 0.077 mmoL) in CH3CN (3 mL). After stirring at room temperature for 10 min, the solvent was evaporated and the residue was purified by prep-LC to afford titled compound (22.6 mg, 59%). LCMS tR =2.78 Min. Mass calculated for, M+ 451.0, observed LC/MS m/z 452.0 (M+H).
Methyi 1 -(7-amino-3-(αuinolin-3-yl)pyrazoloH ,5-aipyrimidin-5-yl)pyrrolidine-3- carboxyiate

A mixture of 5-chloro-3-(quinolin-3-yl)-N,N-bis((2-(trimethylsiiyl)ethoxy) methyl)- pyrazolo-[1 ,5-a]pyrimidin-7-amine (55 mg, 0.10 mmoL), methyl pyrrolidine-3- carboxylate (52 mg, 0.30 mmoL) and EtN(IPr)2 (86 uL, 0.50 mmoL) in DMF (1.0 ml_) was heated at 18O°C under microwave condition for 45 min. The mixture was purified by prep-LC to afford the title compound. LC/MS RT = 3.30 min. Mass calculated for, M+H 389.17, observed 389.17.
1 -(7-Amino-3-(quinolin-3-yl)pyrazo!of 1 ,5-alpyrimidin-5-yl)pyrrolidine-3-carboxylic acid

To a solution of methyl 1-(7-amino-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5- yl)pyrrolidine-3-carboxylate (19 mg, 0.050 mmol) in a 2:1 mixture of THF and H2O (3 ml_) was added 1 N LiOH solution (0.25 ml_). The reaction mixture was stirred at rt overnight and concentrated in vacuo. The residue was purified by prep-LC to afford the title compound. LC/MS RT = 2.80 min. Mass calculated for, M+H 375.16, observed 375.15.
.1-(7-Amino-6-bromo-3-(Quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)pyrroiidine-3- carboxylic acid

To a solution of 1 -(7-amino-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)pyrroiidine-3- carboxylic acid (17 mg, 0.033 mmol) in a 2:1 mixture of CH3CN/MeOH (1.0 mL) was added NBS (6.2 mg, 0.033 mmol). The mixture was stirred at rt for 1 h then concentrated in vacuo. The residue was purified by prep-LC to afford the title compound. LC/MS RT = 3.27 min. Mass calculated for, M+H 453.07, observed 453.07
Synthesis of tetrahydro-imidazoH ,5-a]pyrazine-1 ,3-dione

Stepi :
Potassium cyanate (10 mmol, 811 mg) was added to 1 -benzyi-3-methyl piperazine-
1 ,3-dicarboxy!ate ( 2 mmol, 556 mg) dissolved in methanol-water, and the reaction mixture heated at 75 °C for 2hrs until LCMS showed the completion of the reaction.
The solvent was evaporated in vacuo and the crude product was purified by silica get column chromatography to yield the product, benzyl 1 ,3- dioxohexahydroimidazo[1 ,5- a]pyrazine-7(1 H)-carboxylate, ( 290mg, 50%). LCMS tR =2.50 min. Mass calculated for, M+ 289.11, observed LC/MS m/z 288.20 (M-H negative ion mode).
Step2;
To benzyl 1 , 3- dioxohexahydroimidazo[1 ,5-a]pyrazine-7(1 H)-carboxylate ( 1.0 mmol,
290 mg) in methanol was added 50 mg of Pd/C(10%). The solution was kept under hydrogen atmospheric by a balloon for 14 hrs at which time LCMS showed the completion of the reaction. The solution was filtered through celite and the solvent was evaporated in vacuo Xo dryness to yield, tetrahydroimidazo[1 ,5-a]pyrazine-1 ,3(2H,5H)-
dione. LCMS tR =1.00 min. Mass calculated for, M+ 155.07, observed LC/MS m/z 156 (M+H).
By essentially the same procedure given in Scheme 28, the compounds listed in TablelO can be prepared. Table lO








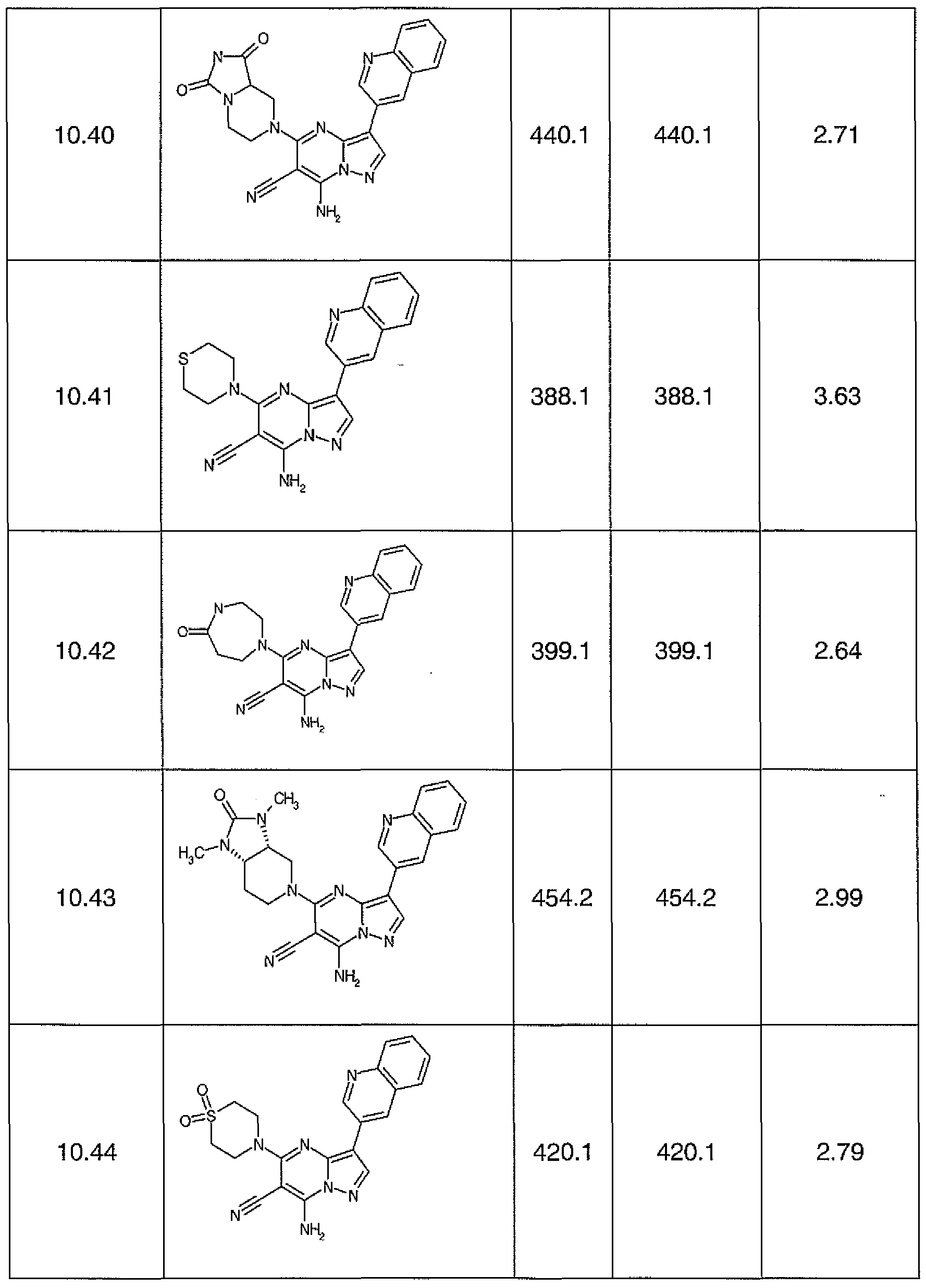




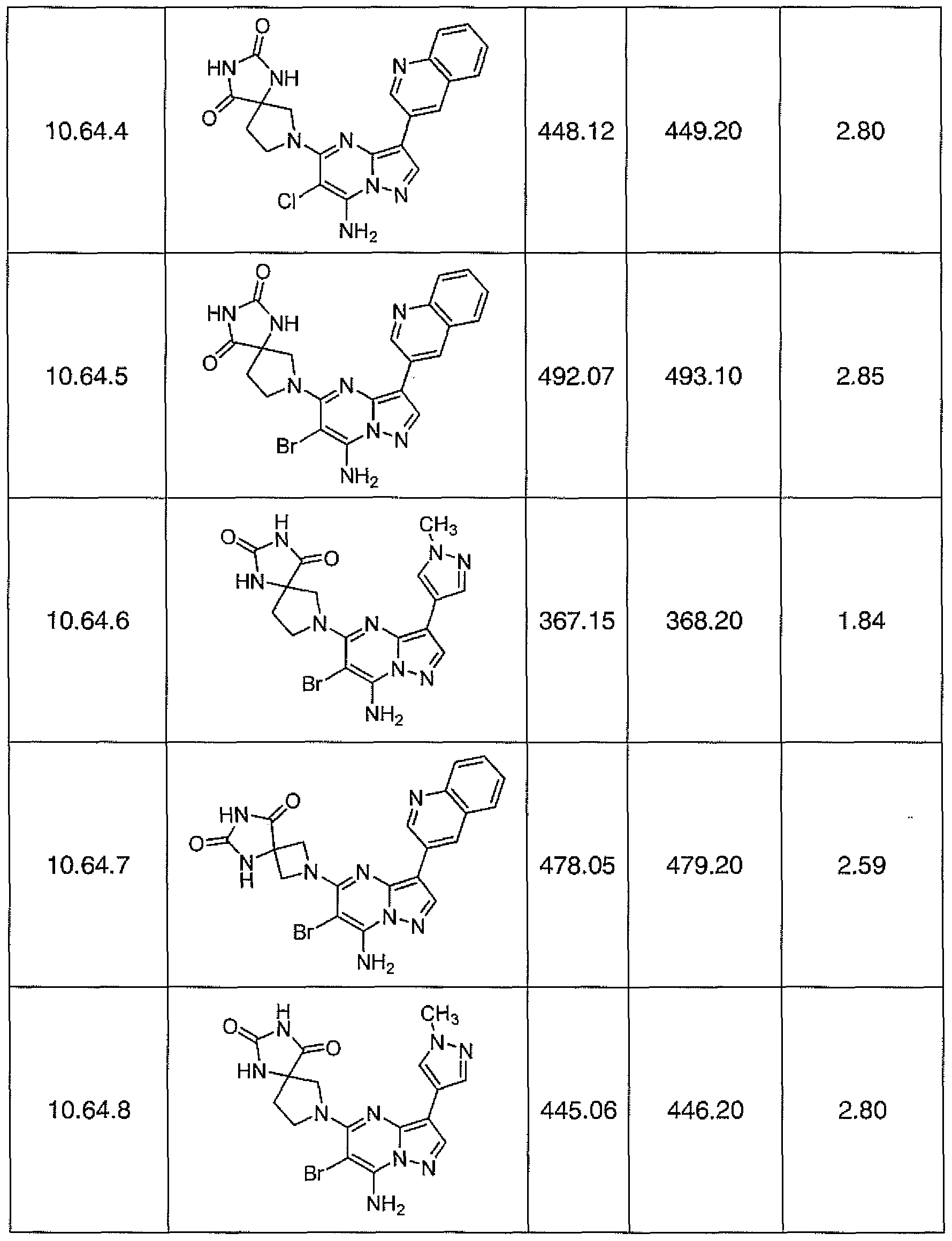
6-bromo-5-morphoiino-3-(quinolin-3-yl)-N,N-bisf(2-(trimethyisilyl) ethoxy)methyl)pyrazolo[1 ,5-aiPyrimidin-7-amine

A solution of NBS (20.6 mg, 0.116 mmoL) in CH3CN (1 mL) was added to a mixture of 5-morphoiino-3-(quinolin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyra--olo[1 ,5- a]pyrimidin-7-amine (70.0 mg, 0.1 16 mmoL) in CH3CN (6 mL). After stirring at room temperature for 10 min, the reaction mixture was concentrated and purified by column chromatography to afford the desired compound. LCMS tR =2.91 Min (5 min run, UV 254nm). Mass calculated for, M+ 684.2, observed LC/MS m/z 685.2 (M+H).
Synthesis of 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-5-morphoiino-3-fquinolin-3- yl)pyrazolori .5-aipyrimidine-6-carbonitrile

A degassed mixture of 6-bromo-5-morpholino-3-(quinolin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin-7-amine (35 mg, 0.051 mmoL), Bu3SnCN (32.3 mg, 0.10 mmoL}, Pd[P(t-Bu)3]2 (5.2 mg, 0.010 mmoL) in Dioxane (3 mL) was heated at 100°C overnight. The mixture was cooled to room temperature and the solvent was evaporated in vacuo. Purification by column chromatography afforded the titled compound. LCMS tR = 2.76 Min (5 min run, UV 254nm)- Mass calculated for, M+ 631.3, observed LC/MS m/z 632.2 (M+H).
Synthesis of 7-amino-5-morpholino-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidine-6- carbonitrile

A mixture of 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-5-morpholino-3-(quinolin-3- yi)pyrazolo[1 ,5-a]pyrimtdine-6-carbonitπle (5 mg), EtOH (1 mL) and 3N HCi (1 mL) was heated at 6O°C until LCMS indicated the completed reaction. The mixture was cooled to room temperature and the solvent was evaporated in vacuo. Purification by prep-LC afforded the desired compound. LCMS tR = 3.12 Min (UV 254nm)- Mass calculated for, M+ 371.1 , observed LC/MS m/z 372.2 (M+H).
Synthesis of 5-morpholino-3-(quinolin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-aipyrimidin-7-amine
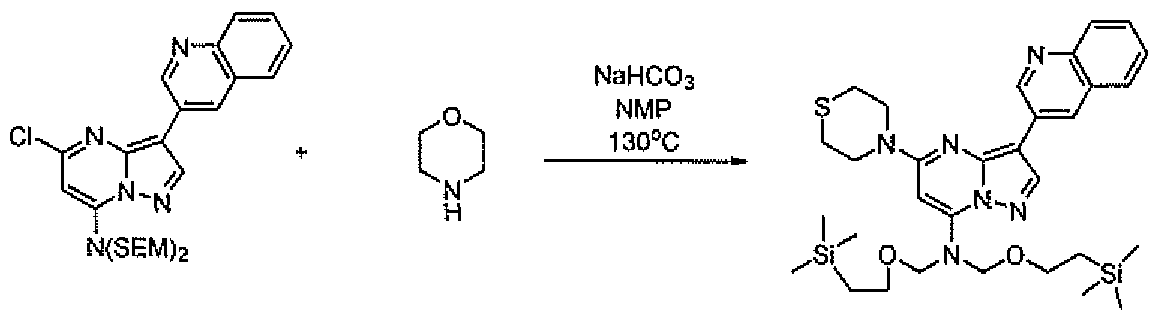
A mixture of 5-chloro-3-(quinolin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoio[l,5-a]pyrimidin-7-amine (77.9 mg, 0.14 mmoL), morpholine (36.7 mg, 0.42 mmoL), NaHCO3 (53 mg, 0.63 mmoL) in NMP (3 mL) was heated at 13O°C overnight. The mixture was cooled to room temperature and diluted with H2O and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation of solvent afforded the crude displacement compound. Purification by column chromatography afforded 5-morpholino-3-(quinolin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methy[)pyrazolo[l,5-a]pyrimidin-7-amine (70 mg, 82%). LCMS tR = 2.72 Min (5 min run, UV 2sW- Mass calculated for, M+ 606.3, observed LC/MS m/z 607.2 (M+H).
Synthesis of 5-thiomorpholinodioxo-3-(quinolin-3-yl)-N, N-bis((2- (trimethylsilyl)ethoxy)methyJ)pyrazolof1 ,5-a1pyrimidin-7-amine

A mixture of 5-chloro-3-(quinolin-3-yl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine (503.0 mg, 0.91 mmoL), thiomorpholine 1, 1-dioxide (367.1 mg, 2.72 mmoL), NaHCO3 (533.0 mg, 6.34 mmoL) in NMP (8 mL) was heated at 13O°C overnight. The mixture was cooled to room temperature and diluted with H2O and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation of solvent afforded the crude displacement compound. Purification by column chromatography afforded compound 5j; thiomorpholinodioxo-3-(quinolin-3-yl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin-7-amine LCMS tR = 2.52 Min (5 min run, UV 254nm)< Mass calculated for, M+ 654.2, observed m/z 655.2 (M+H).
Synthesis of 5-morpholino-3-(quinolin-3-yl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazo[oπ,5-aipyrimidin — 6-bromo-7-amine

A solution of NBS (20.6 mg, 0.116 mmoL) in CH3CN (1 mL) was added to a mixture of 5- morpholino-3-(quinolin-3-yl)-N,N-bis((2-(trimethy lsilyl)ethoxy)methyl)pyrazolo[ 1 ,5- a]pyrimidin-7-amine (70.0 mg, 0.116 mmoL) in CH3CN (6 mL). After stirring at room temperature for 10 min, the reaction mixture was concentrated and purified by column chromatography to afford 6-bromo-5-morphoIino-3-(quinolin-3-yl)-N,N-bis((2- (trimethyisiIyl)ethoxy)methyl)pyrazoIo[1,5-a]pyrimidin-7-amine. LCMS tR =2.91 Min (5 min run, UV 254nm). Mass calculated for, M+ 684.2, observed LC/MS m/z 685.2 (M+H).
Synthesis of 5-thiomorpholinodioxo-3-(αuinolin-3-yl)-N,N-bis((2- (trimethylsilv[)ethoxy)methyl)pyrazoio|"1 ,5-aipyrimidin-6-bromo-7-amine

By essentially the same procedure given in Preparative Example 3- 1, 6-bromo-3-(quinolin-3- yl)-5-thiomorpholino-r, r-dioxide-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[I ,5- a]pyrimidin-7-amine can be prepared. LCMS IR = 2.90 Min (S min run, UV 2S4nm)- Mass calculated for, M+ 732.2, observed LC/MS m/z 733.2 (M+H).
Synthesis of 6-bromo-3-(6-fluoroquino[in-3-yl)-5-thiomorpholin-dioxo-pyrazoiori ,5- aipyrimidin-7-amine:

SCHEME-29
Part A:
Synthesis of 3-iodo-5-thiomorpholin-4-dioxo-N,N-bisf(2-
(trimethylsilyl)ethoxy)methyQpyrazolori ,5-a]pyrimidin-7-amine:
A mixture of compound 5-chloro-3-iodo-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (10.8 mmol, 6.00 g, preparation reported previously), thiomorpholine 1,1-dioxide (32.4 mmol, 4.38 g), and NaHCO3 (48.6 mmol, 4.08 g) in NMP (50 mL) was heated at 130 °C for 28 h. LCMS showed nearly complete consumption of A. The reaction mixture was diluted with EtOAc, washed with H2O and brine, dried over Na2SO4, and concentrated. The crude product was purified by a SiO2 column (0-30% EtOAc/Hexanes) to afford compound 3-iodo-5-thiomorpholin-4-dioxo- N,N-bis((2-(trimethyisiiyI)ethoxy)methy[)pyrazoIo[1,5-a]pyrimidin-7-amine as a pale yellow solid (6.15 g, 87%). HPLC-MS TR = 2.70, min (UV 254 nm, 5 min method); mass calculated for formula C22H40IN5O4SSi2653.1 , observed LCMS m/z 654.1 (M+H).
Part B:
Synthesis of 3-(6-fluoroαuinolin-3-yl)-5-thiomorpholin-4'-dioxo-N,N-bts((2-
(trimethylsilyl)ethoxy)methyi)pyrazolori ,5-a1pyrimidin-7-amine: A mixture of compound 3-iodo-5-thiomorpholin-4-dioxo-N,N-bis((2-
(trimethylsi!yl)ethoxy)methyl)pyrazoio[1,5-a]pyrimidin-7-amine (0.551 mmol, 360 mg), boronate 7 (0.827 mmol, prepared in situ, see below), PdCl2CdPPf)CH2Cl2 (0.055 mmol, 45.0 mg), and K2CO3 (1.65 mmoi, 229 mg) in DMEyH2O (5/1 mL) was degassed and then heated at 90 °C for 24 h. The reaction mixture was diluted with EtOAc and filtered. The filtrate was concentrated and purified by a SiO2 column (0-50% EtOAc/Hexanes) to afford compound 3- (6-fluoroquinolin-3-yl)-5-thiomorphoIin-4'-dioxo-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine as a yellow solid (260 mg, 70%). HPLC-MS TR = 2.39 min (UV 254 nm, 5 min method); mass calculated for formula C3 IH45FN6O4SSi2672.3, observed LCMS m/z 673.2 (M+H).
Synthesis of 6-fluoro-3-(4,4,5,5-tetramethyl-1 ,3.2-dioxaborolan-2-yl)Quinoliπe:

A mixture of the corresponding 3-bromo-6-fluoro quinoline (0.827 mmol, 187 mg, 1.0 eq), bis(pinacolato)diboron ( 12 eq) ,PdCl2(dppf)CH2Cl2 (0.1 eq), and KOAc (3.0 eq) in dioxane (5
mL) was degassed and then heated at 80 °C for 16 h. All the volatiles were removed under reduced pressure and the crude product 6-fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)quinoline was used without further purification.
Part C:
3-(6-fluoroαuinolin-3-yl)-5-thiomorpholin-4'-dioxo- pyrazolo[1.5-a1pyrimidin-7-amine :
To a solution of compound 3-(6-fluoroquino[in-3-yl)-5-thiomorphoIin-4'-dioxo-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (0.169 mmol, 156 mg) in EtOH (4 mL) was added 4 N HCl in dioxane ( 1 mL) and the resulting mixture was heated at 65 °C for 1 h. All the volatiles were removed under reduced pressure. The residue, which has a very poor solubility in DMSO, was diluted with a small amount of DMSO and basicified with NaHCO3. The resulting slurry was stirred vigorously overnight and filtered. The precipitates were washed with H2O and MeOH to afford compound 3-(6-fluoroquinolin-3-yl)-5- thiomorphoiin-4'-dioxo- pyrazolo[l ,5-a]pyrimidin-7-amine as a pale yellow solid (60.1 mg, 86%). HPLC-MS TR = 1.17 min (UV 254 nm, 5 min method); mass calculated for formula CI9HI7FN6O2S 412.1, observed LCMS m/z 413.0 (M+H).
Part D: 3-(6-fluoroquinolin-3-yl)-5-thiornorpholin-4'-dioxo-6-brorno-pyrazolori .5-aipyrimidin-7- amine :
To a slurry of compound 3-(6-fluoroquinolin-3-yl)-5-thiomorphoUn-4'-dioxo- pyrazolo[l,5- a]pyrimidin-7-amine (0.0659 mmol, 27.2 mg) in CH3CN was added NBS (1.0 eq, in a stock solution in CH3CN) and the resulting mixture was stirred at rt for ! h. The reaction solution was concentrated and purified by prep-HPLC to afford compound 3-(6-fluoroquinolin-3-yl)-5- thiomorpholin-4'-dioxo-6-bromo-pyrazolo[1,5-a]pyrimidin-7-amine as a pale yellow solid (6.1 mg, 19%). HPLC-MS TR = 1.56 min (UV 254 nm, 5 min method); mass calculated for formula C19Hi6BrFN6O2S 490.0, observed LCMS m/z 491.0 (M+H).
By essentially the same procedures given in Preparative Example (scheme-29)Part B, Part C and Part D, compounds 10.65-10.85 given in Table 1OA can be prepared.
TABLE ■ IQA




Synthesis of 3-(2,3'-bipyridin-5-yl)-5-thiomorphoiino-4-dioxo-N, N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-alpyrimidin-7-amine:

SCHEME-30
Part A:
3-(6-chloropyridin-3-yl)-5-thionnorpholino4-dioxo-N,N-bis((2- (trimethylsilyi)ethoxy)methyl)pyrazolo[1,5-aipyrimidin-7-amine:
Title compound was prepared from compound 3-iodo-5-εhiomorpholino4'-dioxo-N,N-bis((2- (trimethy I si Iy i )ethox y ) methy l)py razolo [ 1 ,5 -a] py rimidin-7-amine and 6-chloro -3 - pyridineboronic acid pinacol ester using the coupling conditions described in Part B of
Example 1. EPLC-MS TR = 2.89 min (UV 254 ran, 5 min method); mass calculated for formula C27H43ClN6O4SSi2 638.2, observed LCMS m/z 639.1 (M+H).
Part B: 3-(2.3'-bipyridin-5-yl)-5-thiomorphoiino-4-dioxo-N.N-bisf(2- (trimethylsilyl)ethoxy)methyi)pyrazolori ,5-a1pyrinnidin"7-amine: A mixture of 3-(6-chloropyridin-3-yl)-5-thiomorpholino4-dioxo-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine (0.258 mmol, 165 mg), pyridine-3-ylboronic acid (0.516 mmol, 63.4 mg), PdCI2(dppf)CH2Cl2 (0.026 mmol, 21.2 mg), and K2CO3 (0.774 mmol, 107 mg) in OMEfH2O (2/0.4 mL) was degassed and then heated at 150 °C under microwave radiation for 1 h. The reaction mixture was diluted with EtOAc and filtered. The filtrate was concentrated and purified by a SiO2 column (20-100% EtOAc/Hexanes) to afford compound 3-(2,3'-bipyridin-5-yl)-5-thiomorpholino-4-dioxo-N,N- bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine as a pale yellow solid (161 mg, 91%). HPLC-MS TR = 2.15 min (UV 254 nm, 5 min method); mass calculated for formula C32H47N7O4SSi2681.3, observed LCMS m/z 682.3 (M+H).
By essentially the same procedures given in Preparative above Example(scheme-30) Compounds given in Table 1OB can be prepared.
Table 1OB


Synthesis of 3-(3,3'-bipyriclin-5-yl)-5-thiomorpholin-4'-dioxo-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-alpyrimidin-7-amine;

SCHEME-31
Part A:
3-(5-bromopyridin-3-yl)-5-thiomorpholin-4'-dSoxo-N,N-bis((2- ftrimethylsilyl)ethoxy)methyl)pyrazolof1 ,5-aipyrimidSn-7-amine:
Compound 3-(5-bromopyridin-3-yI)-5-thiomorpholin-4'-dioxo-N,N-bis((2- (trimethylsϋyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine was prepared from compound 3-iodo-5-thiomorphoUno4'-dioxo-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrimidin-7-amine and 5-bromo-3-pyridineboronic acid pinacol ester using the coupling conditions described in Part B of Example 1. HPLC-MS TR = 2.62 min (UV 254 nm, 5 min method); mass calculated for formula C2TH43BrN6O4SSi2 682.2, observed LCMS m/z 683.0 (M+H).
Part B: 3-(3,3'-bipyridin-5-yl)-5-thiomorDholin-4'-dioxo-N.N-bis((2-
(trimethylsilvOethoxy)nnethyl)pyrazoloπ,5-alpyrimidin-7-amine:
Compound, 3-(3,3'-bipyridin-5-yl)-5-thiomorpholin-4'-dioxo-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine was prepared from 3-(5- bromopyridin-3-yi)-5-thiomorpholin-4'-dioxo-N,N-bis((2- trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine using the coupling conditions described in Part B of Preparative Example 3. HPLC-MS TR = 2.50 min (UV 254 nm, 5 min method); mass calculated for formula C27H43BrN6O4SSi2 681.3, observed LCMS m/z 682.2 (M+H). By essentially the same procedures given in Preparative Example above( Scheme-31 ), compounds given in Table- 1OC can be prepared
Table IQC


SCHEME-32
A degassed mixture of 6-bromo-5-morpholino-3-(quinolin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)niethyl)pyrazolo[l,5-a]pyrimidin-7-amine (141 mg, 0.20mmoL), tributyl(l-ethoxyvinyl)tin (112 mg, 0.31 mmoL), Pd(PPh3)4 (24 mg, 0.02 mmoL) in Dioxane (4 mL) was heated at 100°C for 2 days. The mixture was cooled to room temperature and filtered through short plug of 10% KF/Siθ2. The crude Stille coupling product obtained after concentrating the filtrate was then treated with 3mL of 1: 1 mixture of TFA and H2O at room temperature for 30 min, Concentration and purification by prep-LC afforded l-(7-amino-5- morpholino-3-(quinolin-3-yl)pyrazolo[1,5-a]pyrimidin-6-yl)ethanone. LCMS tR = 3.32 Min. Mass calculated for, M+ 388.1, observed LC/MS mJz 389.1 (M+H).
By essentially the same procedure given in Preparative above(Scheme-32), compounds in the table- 10-D can be synthesized.
TABLE-10D



Synthesis of 1-(7-amino-3-(1-phenyl-1 H-pyrazoi-4-yl)-5-thiomorpholin-4'-dioxo- pyrazoion ,5-aipyrimidin-6--yl)etharione:

SCHEME- 33
Part A: 10 3-(1 H-pyrazol-4-ylV5-thiomorpholin-4'-dioxo-N.N-bisf(2- ftrimethvlsilvπethoxvlmethvπpvrazoloFl5-aipvrimidin-y-amine:
A mixture of compound, 3-(5-bromopyridin-3-yl)-5-thiomorpholin-4'-dioxo-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine (1.90 mmol, 1.24 g) 1-Boc- ] 5 pyrazole-4-boronic acid pinacol ester (2.85 mmol, 838 mg), Pd(PPh3)4 (0.19 mmol, 220 mg), and K2CO3 (5.70 mmol, 788 mg) in dioxane/H2O (20/5 mL) was degassed and heated at 80 °C overnight using an oil bath. Then the reaction mixture was heated at 150 °C under microwave
radiation for 1 h. After cooling to rt, the reaction mixture was diluted with EtOAc, washed with H2O and brine, dried over Na2SO4, and concentrated. The residue was purified by a SiO2 column (20-100% EtOAc/Hexanes, Rf = 0.5 in 100% EtOAc/Hexanes) to afford compound, 3- (1H-pyrazol-4-yl)-5-thiomorpholin-4'-dioxo-N,N-bis((2- (trimethylsilyl)ethoxy)methy!)pyrazolo[1,5-a]pyrirnidin-7-amine as a brown solid (358 rag, 32%). HPLC-MS TR = 2.39 min (UV 254 nm, 5 min method); mass calculated for formula C25H43N7O4SSi2593.3, observed LCMS m/z 594.2 (M+H).
Part B: 3-(1 -pheπyl-1 H-pyrazol-4-yl)-5-thiomorprιoiin-4'-dioxo-N,N-bis(f2- (trimethyisilvnethoxy)methyl)pyrazolori ,5-a1pyrimiciin-7-aiinine: A mixture compound, 3-(1H-pyrazol-4-yl)-5-thiomorpholin-4'-dioxo-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[l ,5-a]pyrimidin-7-amine (0.603 mmol, 358 mg), iodobenzene (0.904 mmol, 101 μL), CuI (0.0603 mmol, 11.5 mg), N,N'-dimethyl- ethylenediamine (0.241 mmol, 25.9 μL) and K3PO4 (1.27 mmol, 269 mg) in toluene (6 mL) was stirred at 110 °C under Ar2 for 24 h. After cooling to rt, the crude mixture was diluted with EtOAc and filtered. The filtrate was concentrated and purified by a SiO2 column (0-50% EtOAc/Hexanes, Rf = 0.7 in 50% EtOAc/Hexanes) to afford the compound, 3-(l-phenyl-1H- pyrazol-4-yl)-5-thiomorpho!in-4'-dioxo-Ν,Ν-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine, as a pale yellow solid (365 mg, 90%). HPLC-MS TR = 2.77 min (UV 254 nm, 5 min method); mass calculated for formula C31H47N7O4SSi2 669.3, observed LCMS m/z 670.3 (M+H).
Part C: 6-bromo-3-(1 -phenyl-1 H-pyrazol-4-yl)-5-thiomorpholin-4'-dioxo-N.N-bis((2- (trirπethylsilyl)ethoxy)methyhpyrazolof1,5-aipyrimidin-7-amine:
To a solution of compound 3-(l-phenyl-1H-pyrazol-4-yl)-5-thiomorpholin-4'-dioxo-N,N- bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (0.545 mmol, 365 mg) in CH3CN/DCM (3/3 mL) was added NBS (0.545 mmol, 97 mg) and stirred at rt for 1 h. All the volatiles were removed under reduced pressure and the residue was purified by a SiO2 column (0-50% EtOAc/Hexanes, Rf = 0.8 in 50% EtOAc) to afford compound, 6-Bromo-3-(l-
phenyl- 1H-pyrazoI-4-yl)-5-thiomorpholin-4'-dioxo- -N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine, as a yellow forming solid (224 mg, 55%). HPLC-MS TR = 3.01 min (UV 254 nm, 5 min method); mass calculated for formula C3IH46BrN7O4SSi2 747.2, observed LCMS m/z 748.0 (M+H).
Part D:
6-(1-ethoxyylnylV3-(1-phenyl-1 H-pyrazoi-4-yl)-5-thiomorpholin-4'"dioxo-N,N-bis(f2-
(trimethylsiiyl)ethoxy)methyl)pyrazoiof1,5-alPyrimidin-7-amine:
A mixture of compound 6-Bromo-3-(l-phenyl-1H-pyrazoI-4-yI)-5-thiomorpholin-4'-dioxo- - N,N-bis((2-(trimethyIsilyl)ethoxy)methyl)pyrazolo[ 1 ,5-a]pyrimidin-7-amine (0.299 mmol, 224 mg), tributyl(l-ethoxyviny[)tin (0.897 mmol, 0.303 mL), Pd(PPh3)4 (0.030 mmol, 34.6 mg) in dioxane (5 mL) was stirred at 100 °C under Ar2 for 16 h. After cooling to rt, the reaction mixture was passed through a short SiCVKF (9:1) plug to removed majority of the Sn species, and then purified by a SiO2 column (0-40% EtOAc/Hexanes, Rf = 0.7 in 50% EtOAc) to afford compound, 6-( L-ethoxyvinyl)-3-(l-phenyl-1H-pyrazol-4-yl)-5-thiomorpholin-4'-dioxo-N,N- bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine as a pale yellow oil (185 mg, 84%). HPLC-MS TR = 3.06 min (UV 254 nm, 5 min method); mass calculated for formula C35H53N7O5SSi2739.3, observed LCMS m/z 622.2 (M-117).
Part E:
1-(7-amino-3-(1-phenyl-1 H-pyrazo!-4-yl)-5-thiomorpholin-4'-dioxo-pyrazolo[1 ,5- alpyrimidin-6-yπethanone:
Compound , 6-(l-ethoxyvinyl)-3-(l-phenyl-1H-pyrazol-4-yl)-5-thiomorpholin-4'-dioxo-N,N- bis((2-(trimethylsi!yl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine, (0.250 mmol, 185 mg) was treated with a mixture of TFAZH2O (2/2 mL) at rt for 30 min. All the volatiles were removed under reduced pressure. The residue was dissolved in 3.0 mL of DMSO and purified by a reverse phase HPLC (H2O/CH3CN, 0.1 % TFA). The product fraction was concentrated to dryness and converted to HCl salt by adding 1 N HCl (aq) solution to its slurry in MeOH, and then concentrated to dryness. Compound, l-(7-amino-3-(l-phenyl-IH-pyrazoi-4-yl)-5- thiomorpholin-4'-dioxo-pyrazolo [1,5-a]pyrimidin-6-yl)ethanone (HCl salt) was obtained as a
pale yeliow solid (16.4 mg, 38%). HPLC-MS TR = 1.86 min (UV 254 nm, 5 min method); mass calculated for formula C2iH2!N7O3S 451.1, observed LCMS m/z 452.0 (M+H).
By essentially the same procedures given in Preparative Example above(Scheme-33), compounds given in Table- 1OE can be prepared.
TABLE-IOE




A degassed mixture of 6-bromo-3-(quinolin-3-yl)-5-thiomorpholino- f, l'-dioxide--N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (16 mg, 0,022 mmoL), tributy!(prop-1-ynyl)stannane (22 mg, 0.067 mmoL), Pd(PPh3)4 (2.6 mg, 0.002mmoL) in Dioxane (3 mL) was heated at 100°C overnight. The mixture was cooled to room temperature and filtered through short plug of 10% KFZSiO2. The crude Stille coupling product obtained after concentrating the filtrate was then treated with 1 ; 1 mixture of TFA and H2O at room temperature for 30 min. Concentration and purification by prep-LC afforded i-(7-amino-3- (quinolin-3-yl)-5-thiomorpholino-r, r-dioxide-pyrazolo[1,5-a]pyrimidin-6-yl)propan-1-one. LCMS tR = 3.20 Min ( 10 min run, UV 254nm). Mass calculated for, M+ 450.1, observed LC/MS m/z 451.1 (M+H).
7-(7-amino-6-propionyl-3-(quinolin-3-yl)pyrazolof1.5-aipyrimidin-5- yl)tetrahydroimidazofi ,5-aiPyrazine-1.S^H.SI-D-dione:

By essentially the same procedure given in Preparative Example above, 7-(7-amino-6- propionyl-3-(quinolin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl)tetrahydroimidazo[l,5-a]pyrazine-
1 ,3(2H,5H)-dione can be prepared from 7-(7-(bis((2-(trimethyIsilyl)ethoxy)methyl)amino)-6- bromo-3-(quinoIin-3-yl)pyrazoIo[1,5-a]pyrimidin-5-yl)tetrahydroimidazo[1,5-a]pyrazine- 1,3(2H,5H)-dione. LCMS tR = 3.14 Min (10 min run, UV 254nm). Mass calculated for, M+ 470.18, observed LC/MS m/z 471.28 (M+H).
Synthesis of 1-(7-amino-3-(αuinolin-3-yl)-5-thiomorpholino-1 '. 1 '-dioxide-pyrazolo[1 ,5- aipyrimidin-6-vO-2-phenylethanone:

By essentially the same procedure given in Preparative Example above, l-(7-amino-3-
(quinolin-3-yl)-5-thiomorpholino-l', r-dioxide-pyrazolo[l,5-a]pyrimidin-6-yl)-2- phenylethanone can be prepared using tributy[(phenyiethynyl)stannane instead of tributyl(prop-1-ynyl)stannane. LCMS IR = 3.74 Min (10 min run, UV 2S4nm)- Mass calculated for, M+ 512.16, observed LC/MS m/z 513.18 (M+H).
Synthesis of 1 -(7-amino-5-(3-methylthiomorpholin-4'dioxo)-3-(1 -phenyl- 1 H-pyrazol-4- yl)pyrazolol"l .5-alpyrimidin-6-vhethanone:

SCHEME-34
Part A:
Synthesis of 3-methyithiomorpholine:
To a solution of KOH (331 mmol, 21.9 g, 85%) in 250 mL of MeOH was added cystamine hydrochloride (158 mmol, 17.9 g), in small portions, followed by a solution of chloroacetone (158 mmol, 12.6 mL) in 100 mL of MeOH at 0-5 °C. After being stirred for 90 min at 0 °C, the reaction was acidified with 350 mL of ! .25 M HCI in MeOH at 0-5 °C and stirred for 1 h. NaBH4 was then added in small portions at below 5 °C and the mixture was stirred for 30 min. The reaction was quenched with 1 N HCl (aq) and filtered. The filtrate was concentrated and then dissolved in H2O and extracted with DCM (*8). The organic layers were discarded and the aqueous phase was carefully neutralized with NaOH first, and then Na2CO3. The aqueous phase was extracted with DCM, dried over Na2SO4 and concentrated to afford compound, 3- methylthiomorpholine (7.69 g, 42%), which slowly turned into a sticky solid and was used without further purification. HPLC-MS TR = 0.19 min (UV 254 nm, 5 min method); mass calculated for formula C5Hi iNS 117.1 , observed LCMS m/z 1 18.2 (M+H).
Part B:
Synthesis of tert-butyl 3-methylthiomorpholine-4-carboxylate:
To a solution of compound, 3-methylthiomorpholine (65.6 mmol, 7.69 g) and TEA (131 mmol, 18.3 mL) in THF (200 mL) was added (BoC)2O (98.4 mmol, 21.5 g), followed by DMAP (13.1 mmol, 1.60 g). The resulting reaction mixture was stirred at rt overnight. THF was removed and the residue was dissolved in DCM and directly purified by a SiO2 column (0-15% EtOAc/Hexanes, Rf = 0.55 in 20% EtOAc, KMnO4 staining) to afford compound tert-butyl 3- methylthiomorpholine-4-carboxylate as a white sticky solid (7.31 g, 51%).
Part C:
Synthesis of tert-butyl 3-methylthiomorpholine-4'-dioxo-4-carboxylate: To a solution of compound, tert-butyl 3-methylthiomorpholine-4-carboxylate (33.6 mmol, 7.33 g) in DCM (170 mL) at 0 °C was added mCPBA (101 mmol, 24.0 g) in small portions. The reaction mixture was warmed to rt and stirred for 1 h. DCM was removed and the residue was then partitioned between EtOAc and NaHCO3 (aq). The aqueous layer was extracted with EtOAc (*3). The combined organic layers were washed with brine, dried over Na2SO^ and concentrated. The crude product was purified by a SiO2 column (0-50% EtOAc/Hexanes, Rf = 0.6 in 50% EtOAc) to afford compound, tert-butyl 3-methyIthiomorpholine-4'-dioxo-4- carboxylate as a white solid (6.51 g, 78%).
Part D: Synthesis of 3-methylthiomorpholine-4'-dioxide:
Compound tert-butyl 3-methylthiomorpholine-4'-dioxo-4-carboxylate (20.1 mmol, 5.00 g) was treated with TFA/DCM (25/25 mL) at rt for 1 h. The reaction mixture was evaporated to afford compound, 3-methylthiomorpholine-4' -dioxide as a white solid (TFA salt MW:263.23, 5.18 g, 98%), which was used without further purification. HPLC-MS TR = 0.18 min (UV 254 nm, 5 min method); mass calculated for formula C5H1 1NO2S 149.1, observed LCMS m/z 150.1 (M+H).
Part E:
Synthesis of 5-(3-methylthiomorpholin-4'-dioxo)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoio[1 ,5-a1pyrimidin-7-amine:
A mixture of compound, 3-methylthiomorpholine-4' -dioxide (1.00 mmol, 263 mg), 5-chloro- N,N-bis[(2-trimethylsilylethoxy)methyl]-pyrazolo[1,5-a]pyrimidin-7-amine (0.667 mmol, 286
mg, preparation was reported previously), Pd(OAc)2 and CS2CO3 in dioxane (3 mL) was heated at 1 10 °C for 24 h. The reaction mixture was diluted with EtOAc and washed with H2O and brine, dried over Na2SO4, and concentrated. The crude product was purified by a SiO2 column (0-40% EtOAc/Hexanes, Rf = 0.5 in 50% EtOAc) to afford compound, 5-(3- methylthiomorpholin-4'-dioxo)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazoio[l,5- a]pyrimidin-7-amine, as a colorless oil (145 mg, 40%). HPLC-MS TR = 2.75 min (UV 254 nm, 5 min method); mass calculated for formula C23H43NjO4SSi2 541.3, observed LCMS m/z 542.2 (M+H).
Part F:
Synthesis of 3-iodo-5-(3-methylthiomorpholin-4'-dioxo)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazoio[1 ,5-a|pyrimidin-7-amine:
To a solution of compound, 5-(3-methylthiomorpholin-4'-dioxo)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (0.268 mmol, 145 mg) in CH3CN (5 mL) was added NIS (0.294 mmol, 66.2 mg). The resulting solution was stirred at rt for 30 min. The reaction mixture was evaporated and directly purified by a SiO2 column (0- 15% EtOAc/Hexanes, Rf = 0.55 in 20% EtOAc) to afford compound, 3-iodo-5-(3- methylthiomorpholin-4'-dioxo)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazoIo[1,5- a]pyrimidin-7-amine, as a pale yellow oil (161 mg, 90.0%). HPLC-MS TR = 7.29 min (UV 254 nm, 10 min method); mass calculated for formula C23H42INgO4SSi2 667.2, observed LCMS m/z 668.0 (M+H).
Part G:
Synthesis of 5-(3-methylthiomorphoiin-4'dioxo)-3-(1 -phenyl-1 H-pyrazol-4-yl)-NTN- bisf(2-(trimethylsilyl)ethoxy)methyl)pyrazolori .5-a]pyrimidin-7-amine:
A mixture of compound, 3-iodo-5-(3-memylthiomorpholin-4'-dioxo)-N,N-bis((2- (trimethylsilyl)ethoxy)methyf)pyrazolo[ l,5-a]pyrimidin-7-amine (0.428 mmol, 286 mg), 1- phenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)»1H-pyrazole (0.557 mmol, 150 mg) , PdCl2(dρpf)CH2Cl2, and K3PO4 in dioxane/H2O (4/0.4 mL) was degassed and then heated at 100 °C for 16 h. After cooling to r.t., the reaction mixture was diluted with EtOAc and filtered. The filtrate was concentrated and purified by a SiO2 column (0-40% EtOAc/Hexanes, Rf = 0.6
in 50% EtOAc/Hexanes) to afford compound, 5-(3-methyIthiomorpholin-4'-dioxo>3-(l- phenyl- 1H-pyrazol-4-yl)-N,N-bis((2-(trimethylsilyi)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin- 7-amine, as a pale yellow oil (142 mg, 48%). HPLC-MS TR = 2.92 min (UV 254 nm, 5 min method); mass calculated for formula C32H49N7O4SSi2 683.3, observed LCMS m/z 684.2 (M+H).
Part H:
1-(7-amino-5-(3-methylthiomorpholin-4'dioxo)-3-(1-phenyl-1 H-pyrazol-4- yl)pyrazolo[1 ,5-aipyrimidin-6-yl)ethanone:
compound, 5-(3-methylthiomorpholin-4'-dioxo)-3-(l-phenyl-1H-pyrazol-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoIo[1,5-a]pyrimidin-7-amine, bromination with NBS, foliwed by on stiile reaction with Vinyl triflate and treatment with 4N HCl in dioxane as described in the above example (scheme-???) resulted in a compound, 5-(3- methylthiomorpholin-4'-dioxo)-3-(l-phenyl-1H-pyrazoI-4-yl)pyrazolo[1,5-a]pyrimidin-7- amine as HCl salt. Evaporation of dioxane and lyophilization of the product resulted in 5-(3- methylthiomorpholin-4'-dioxo)-3-( l-phenyl-1H-pyrazol-4-yl) pyrazolo [1, 5-a] pyrimidin-7- amine as white powder. HPLC-MS TR = 4.93 min (UV 254 nm, 10 min method); mass calculated for formula C32H49N7O4S 465.16, observed LCMS m/z 466.10 (M+H).
Synthesis of 6-bromo-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyraπ-4-yl-1 ', V- dioxide)- methyQpyrazolori ,5-aipyrimidin-7-amine

SCHEME-35
Part A:
3-oxo-3-(tetrahydro-2H-thiopyran-4-yl)propanenitriie:
A solution of CH3CN (48,7 mrnol, 2.54 mL) in 10 mL of THF was added dropwise to a solution of "BuLi (48.7 mniol) in 15 mL of THF at -78 °C. After stirring for 1 h at -78 °C, a solution of methyl tetrahydro-2H-thiopyran-4-carboxylate (24.3 mmol, 3.90 g) in 10 mL of TΗF was added dropwise and the resulting reaction mixture was stirred at -78 °C for 1 h, then slowly warmed to -40 °C in 2 h. The reaction was quench with 1 N HCl (pΗ<l). THF was removed and the residue was extracted with EtOAC, washed with brine, dried over Na2SO4, and concentrated. The crude product was purified by a SiO2 column (0-50% EtOAc/Hexanes, Rf = 0.45 in 50% EtOAc) to afford compound 3-oxo-3-(tetrahydro-2H-thiopyran-4- yl)propanenitrile (3.60 g, 87.5%) as a light brownish oil. HPLC-MS TR = 1.15 min (UV 254 nm, 5 min method); mass calculated for formula CeHnNOS 169.1 observed LCMS m/z 170.1 (M+H).
Part B: 5-(tetrahydro-2H-thiopyran-4-yl)-N.N-bis((2-(trimethylsilvhethoxy)methynpyrazolori .5- aipyrimidin-7-aminβ:
A mixture of 3-aminopyrazole (19.3 mmol, 1.61 g) and compound 3-oxo-3-(tetrahydro-2H- thiopyran-4-yl) propanenitrile (21.3 mmol, 3.60 g) in acetic acid (40 mL) was heated at 100 °C in a sealed tube overnight. After cooling to rt, all the volatiles were removed under reduced pressure to afford a light brownish oil. This brownish oil was dissolve in DCM (60 mL), and then SEMCI (67.6 mmoi, 11.9 mL) and DIPEA (135 mmol, 23.5 mL) were added. The resulting reaction mixture was stirred at 45 °C for 1 h. After cooling to rt, all the volatiles were removed under reduced pressure. The residue was diluted with EtOAc (300 mL), filtered and washed with EtOAc. The filtrate was concentrated and the crude product was purified by a SiO2 column (0-30%) to afford compound, 5-(tetrahydro-2H-thiopyran-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine as a light brownish oil (9.23 g, 97%). HPLC-MS TR = 3.09 min (UV 254 nm, 5 min method); mass calculated for formula C23H42N4O2SSi2494.3, observed LCMS m/z 495.1 (M+H).
Part C: 5-(tetrahydro-2H-thiopyran-4-yl-1 '. 1 '-dioxide)-)-N,N-bis(f2- ftrimethylsiiyl)ethoxy)methyl)pyrazolof1 ,5-alpyrimidin-7-amine: To a solution of compound, 5-(tetrahydro-2H-thiopyran-4-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methy0pyrazolo[1,5-a]pyrimidin-7-amine (18.7 mmol, 9.23 g) in DCM (100 mL) was added mCPBA and stirred at rt for 1 h. The reaction was quenched with Na2S2O3 (aq.) and diluted with DCM (100 mL), The separated organic layer was washed with NaHCO3 (2*) and brine, dried over Na2SO4, and concentrated to afford crude compound, 5- (tetrahydro-2H-thiopyran-4-yl-l ', 1 '-dioxide)-)-N,N-bis((2-
(trimethylsilyi)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-arnine as a purple oil, which was used without further purification. HPLC-MS TR = 2.72 min (UV 254 nm, 5 min method); mass calculated for formula C23H42N4O4SSi2 526.2, observed LCMS m/z 527.2 (M+H).
Part D:
3-iodo-5-(tetrahydro-2H-thioDyran-4-yl-1.1-dioxideVN.N-bis(f2- (trimethylsilyl)ethoxy)methyl)pyrazolof1 ,5-aipyrimidin-7-amine: To a solution of all crude compound, 5-(tetrahydro-2H-thiopyran-4-yl- 1 ', 1 '-dioxide)-)-N,N- bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine from Part C (18.7 mmol) in CH3CN (100 mL) was added NIS (20.6 mmol, 4.63 g). The resulting solution was
stirred at it for 30 min. TLC (50% EtOAc/Hexanes) indicated complete conversion to a new spot (Rf= 0.5, less polar than SM Rf = 0.3). The reaction was quenched with Na2S2O3. CH3CN was removed under reduced pressure. The residue was dissolved in DCM, washed with H2O and brine, dried over Na2SO4, and concentrated. The crude product was purified by a SiO2 column (0-50% EtO Ac/Hex anes) to afford compound, 3-iodo-5-(tetrahydro-2H-thiopyran-4- yl- 1 , l-dioxide)-N,N-bis((2-(trimethyIsilyI)ethoxy)methyl)pyrazoIo[ 1 ,5-a]pyrimidin-7-amine: as a pale yellow oil (6.82 g, 56%).
Part E: 3-(5-phenylpyridin-2-yl)-5-(tetrahydro-2H-thiopyran-4-yl-1 ,1 -dioxide)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoiof1 ,5-alpyrimidin-7-amine:
To a solution of 2-bromo-5-phenylpyridine (0.400 mmol, 93.6 mg) and triisopropyl borate (0.440 mmol, 0.102 mL) in a mixed solvent of toluene/THF ( 1.6/0.4 mL) was added dropwise "BuLi (0.440 mmol) at -78 °C, then kept at -78 °C for 30 min (sticky mixture, stopped stirring after 5 min). The reaction mixture was allowed to warm to rt and stirred for 4 h. All the volatiles were removed under reduced pressure from rt to 90 °C. The thus formed crude lithium borate was used without further purification. A mixture of the crude lithium borate (0.400 mmol), compound, 3-iodo-5-(tetrahydro-2H-fhiopyran-4-yl-I,l-dioxide)-N,N-bis((2- (trimethytsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine (0.267 mmol, 174 mg),
PdCl2(dppf) CH2Cl2 (0.027 mmol, 22.0 mg), and KF (0.800 mmol, 46.5 mg) in dioxane (2 mL) was degassed and then heated at 90 °C for 16 h. The reaction mixture was diluted with EtOAc and filtered. The filtrate was concentrated and purified by a SiO2 column (0-40% EtOAc/Hexanes, Rf = 0.5 in 50% EtO Ac/Hex anes) to afford compound, 3-(5-phenylpyridin-2- yl)-5-(tetrahydro-2H-thiopyran-4-yl- 1 , 1 -dioxide)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazo[o[l ,5-a]pyrimidin-7-amine as a pale yellow solid (32 mg, 18%). HPLC-MS TR = 2.62 min (UV 254 nm, 5 min method); mass calculated for formula C34H49N5O4SSi2 679.3, observed LCMS m/z 680.2 (M+H).
Part- F:
6-bromo-3-(6-phenylpyridin-3-vh-5-(tetrahydro-2H-thiopyran-4-yl-1 '. 1 '-dioxideVN.N- bisf(2-(trimethylsilyl)ethoxy)methyl)pyrazolori ,5-a1pyrimidin-7-amine:
NBS (176 mg, 0.99 mmoL) was added to a solution of 3-(6-phenyipyridin-3-yl)-5-(tetrahydro- 2H-thiopyran-4-yl-l', r-dioxide)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5- a]pyrimidin-7-amine (679 mg, 0.94 mmoL) in CH3CN (10 mL). The mixture was stirred at room temperature for Ih. Concentration and purification by column chromatography afforded 6-bromo-3-(6-pheny lpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl- 1 ', 1 '-dioxide)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidirι-7-amine. LCMS .R = 2.87 Min (5 min run, UV 254nm). Mass calculated for, MH- 757, observed LC/MS m/z 758.2 (M+H). Part G:
6-bromo-3-(6-phenylpyridin-3-yl)-5-(tΘtrahydro-2H-thiopyran-4-yl-1 ', 1 -dioxide)- methyl)pyrazolof1 ,5-a]pyrimidin-7-amine:
6-bromo-3-(6-phenyIpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-l', l'-dioxide)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine was treated with 4NHC1 in dioxane at room temperature for 1 hr and reaction evaporated to dry ness and lyophilized under vacuo to give product in powder form, 6-bromo-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H- thiopyran-4-yl-T, 1 '-dioxide)- methyl)pyrazolo[i ,5-a]pyrimidin-7-amine as HCl salt. LCMS IR = 1.27 Min (5 min run, UV 254nm). Mass calculated for, M+ 497.10, observed LC/MS m/z 498.20 (M+H).
By essentially the same procedure given in Preparative above(Scheme-35), compounds 10.1 16 to 10.133 given in the table (10-F) can be prepared.




Synthesis of 3-(6-phenylpyridin-3-vO-5-(tetrahydro-2H-thiopyran-4--yl-1 M '-dioxide)- N,N-bis((2-(trimethylsilyl)ethoxy)methyl)-6-ylnylpyrazoio[1 ,5-alpyrimidin-7-amine

SCHEME-36
Part A
3-(6-phenylpyridin-3-vn-5-(tetrahydro-2H-thiopyran-4-yl-1M'-dioxide)-N,N-bis((2- (trimethyisilyl)ethoxy)methyl)-6-ylnylpyrazolori ,5-a1pyrimidin-7-amine:
A degassed mixture of 6-bromo-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl- r,r-dioxide)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (203 mg, 0.27mmol), Pd(PPh3)4 (31 mg, 0.027 mmoL), tributyl(vinyl)stannane (255 mg, 0.80 mmoL) in CH3CN (6 mL) was heated at 150°C under microwave condition for 60 min. The reaction mixture was cooled to room temperature, filtered through 9: 1 Siθ2:KF plug and concentrated in vacuo. Purification by column chromatography afforded 3-(6-phenylpyridin-3- yl)-5-(tetrahydro-2H-thiopyran-4-yl- 1 ' , r-dioxide)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)- 6-vinylpyrazolo[1,5-a]pyrimidin-7-amine: LCMS tR = 2.82 Min (5 min run, UV 254nm). Mass calculated for, M+ 705.3, observed LC/MS m/z 706.3 (M+H).
Part B
7-(bisf(2-(trimethyisiiyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-ylV5-(tetrahydro- 2H-thiopyran-4-yl-1 M '-dioxide)pyrazolori ,5-aipyrirnidine-6-carbaidehyde:
To 3-(6-phenylpyridin-3-y l)-5-(tetrahydro-2H-thiopyran-4-yl- 1 ' , 1' -dioxide)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)-6-vinylpyrazolo[1,5-a]pyrimidin-7-amine (189 mg, 0.26 mmol) in 1 , 4-doixane (3 mL) was added 2.5 wt% OsO4 in 1 , 4-dioxane ( 168 uL, 0.013 mmol), 2,6- kitidine (265 uL, 2.68 mmol) and H2O (1 mL) and the resulting mixture was stirred at room temperature for 20 minutes. NaIO4 (287 mg, 1.34 mmol) was then added and stirring at room temperature continued for 4 days. Saturated Na2S2O3 solution (5 mL) was added and the mixture stirred for 10 minutes. Organic s were then extracted with CH2Cl2 (2 x), dried (Na2SO4) and concentrated in vacuo to 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-r,r-dioxide)pyrazolo[l,5-a]pyrimidine- 6-carbaldehyde, which was used without further purification: LCMS tR = 2.87 Min (5 min run, UV 254nm). Mass calculated for, M+ 707.2, observed LC/MS m/z 708.3 (M+H).
Part C
7-amino-3-(6-pheπylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-1 ', 1 '- dioxide)pyrazoiol"1.5-aipyrimidine-6-carbaldehyde:
The crude 7-(bis((2-(ti-imethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yi)-5- (tetrahy dro-2H-thiopyran-4-y 1 - 1 ' , 1 ' -dio xide)py razo io [ 1 ,5- a]pyr i midine-6-c arbaldehy de (150 mg, 0.21 ramoL) was treated with 50% TFA in H2O (4 mL) until the disappearance of starting material in LCMS. Concentration and purification by prep-LC afforded 7-amino-3-(6- phenyIpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-l', l'-dioxide)pyrazolo[l ,5- a]pyrimidine-6-carbaldehyde, LCMS tR = 3. !0 Min (10 min run, UV 2s4nm)- Mass calculated for, M+ 447.1 , observed LC/MS m/z 448.1 (M+H).
By essentially the same procedure given in Preparative Example(Scheme-36) compounds 10.134- 10.137 ( Table-10-G) can be prepared.
TABLE- 10 G


Synthesis of i -fy-amino-3-(β-phenylpyridin-3-yl)-5-(tetrahydro^H-thiopyran-4-yl-1 ', 1 '- dioxide)pyrazolo[1 ^-alpyrimidin-6-yl)propan-i -one:

By essentially the same procedure given in Preparative Example above(Scheme-36), l-(7- amino-3-(6-phenylpyridm-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl- 1 ', l'-dioxide)pyrazolo[1,5- a]pyrimidin-6-yl)propan-1-one can be prepared from 6-bromo-3-(6-phenylpyridin-3-yl)-5- (tetrahydro-2H-thiopyran-4-yl-r, 1 '-dioxide)-N,N-bis((2 trimethylsilyl) ethoxy) methyl)pyrazolo[1,5-a]pyrimidin-7-amine. LCMS tR = 3.13 Min ( 10 min run, UV 254nm). Mass calculated for, M+ 449.15, observed LC/MS m/z 450.40 (M+H).
Synthesis of 1 -(7-amino-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-1 ', 1 '- dioxide)pyrazolo[1.5-aipyrimidin-6- yl)ethanone:

A degassed mixture of 6-bromo-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl- r,r-dioxide)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7-amine (203 mg, 0.27mmol), Pd(PPh3)4 (31 mg, 0.027 mmoL), tributyl(vinyl)stannane (255 mg, 0.80 mmoL) in CH3CN (6 mL) was heated at 15O°C under microwave condition for 60 min. The reaction mixture was cooled to room temperature, filtered through 9:1 SiO2:KF plug and concentrated in vacuo. The crude β-Cl-ethoxyvinyO-3-Cό-phenylpyridin-3-yl)-5-Ctetrahydro- 2H-thiopyran-4-yl-r,r-dioxide)-N,N-bis((2-(trimethylsUyl)ethoxy)methyI)pyrazolo[1,5- a]pyrimidϊn-7-aminewas treated with 50% TFA in H2O (2 mL) for Ih. Concentration and purification afforded l-(7-amino-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yi- Γ,1' -dioxide)pyrazolo[1,5-a]pyrimidin-6-yl)ethanone: LCMS IR = 3.18 Min (10 min run, UV 254nm). Mass calculated for, M+ 461.1, observed LC/MS m/z 462.1 (M+H).
6-(methylsulfonvh-3-(Quinolin-3-yi)-5-thiomorpholino-1 M '-dioxide-pyrazoioH ,5- aipyrimidin-7-amine:

A mixture of 6-bromo-3-(quinolin-3-yl)-5-thiomorpholino-r,r-dioxide-N,N-bis((2- (trimethyIsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (0.06J mmol, 45 mg), CuI (0.49 mmol, 94 mg) and NaSO2Me (0.49 mmol, 50 mg) in NMP (2mL) was heated at 12O°C for Ih and then 13O°C for Ih under microwave condition. Purification by prep-LC afforded 6- (methylsulfonyl)-3-(quinolin-3-yl)-5-thiomorpholino--r, l'-dioxide-pyrazolo [1,5-
a]pyrimidin-7-amine: LCMS tR = 2.94 Min (10 min run, UV 254nm)- Mass calculated for, M+ 472.0, observed LC/MS m/z 473.0 (M+H).

A degassed mixture of 6-bromo-5-thiomorpholinodioxo-3-(quinoIin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyra2olo[1,5-a]pyrimidin-7-amine (35 mg, 0.051mmoL), Bu3SnCN (32.3 mg, 0.10 mmoL), Pd[P(t-Bu)3]2 (5.2 mg, 0.010 mmoL) in Dioxane (3 mJL) was heated at 100°C overnight. The mixture was cooled to room temperature and the solvent was evaporated. Purification by column chromatography afforded 7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-5-thiomorpholinodioxo-3-(quinoiin-3-yl)pyrazolo[l,5- a]pyrimidine-6-carbonitrile. LCMS tβ = 2.76 Min (5 min run, UV 254nm). Mass calculated for, M+ 678.28, observed LC/MS m/z 679.20 (M+H).
A mixture of 7-(bis((2-(trimethylsilyL)ethoxy)methyl)araino)-5-thiomorpholinodioxo-3- (quinoiin-3-yl)pyrazolo[i,5-a]pyrimidine-6-carbonitrile (5 mg), EtOH (ImL) and 3N HCl (ImL) was heated at 6O°C until LCMS indicated complete reaction. The mixture was cooled to room temperature and the solvent was evaporated. Purification by prep-LC afforded 7-amino- 5-thiomorpholinodioxo-3-(quinolin-3-yl) pyrazolo [1, 5-a] pyrimidine-6-carbonitrile. LCMS tR = 3.12 Min (10 min run, UV 254nm)- Mass calculated for, M+ 418.12, observed LC/MS m/z 419.2 (M+H).
Following the examples described above, the compounds (Scheme-36)) given in the Table- 10- H can be synthesized
TABLE-IO-H
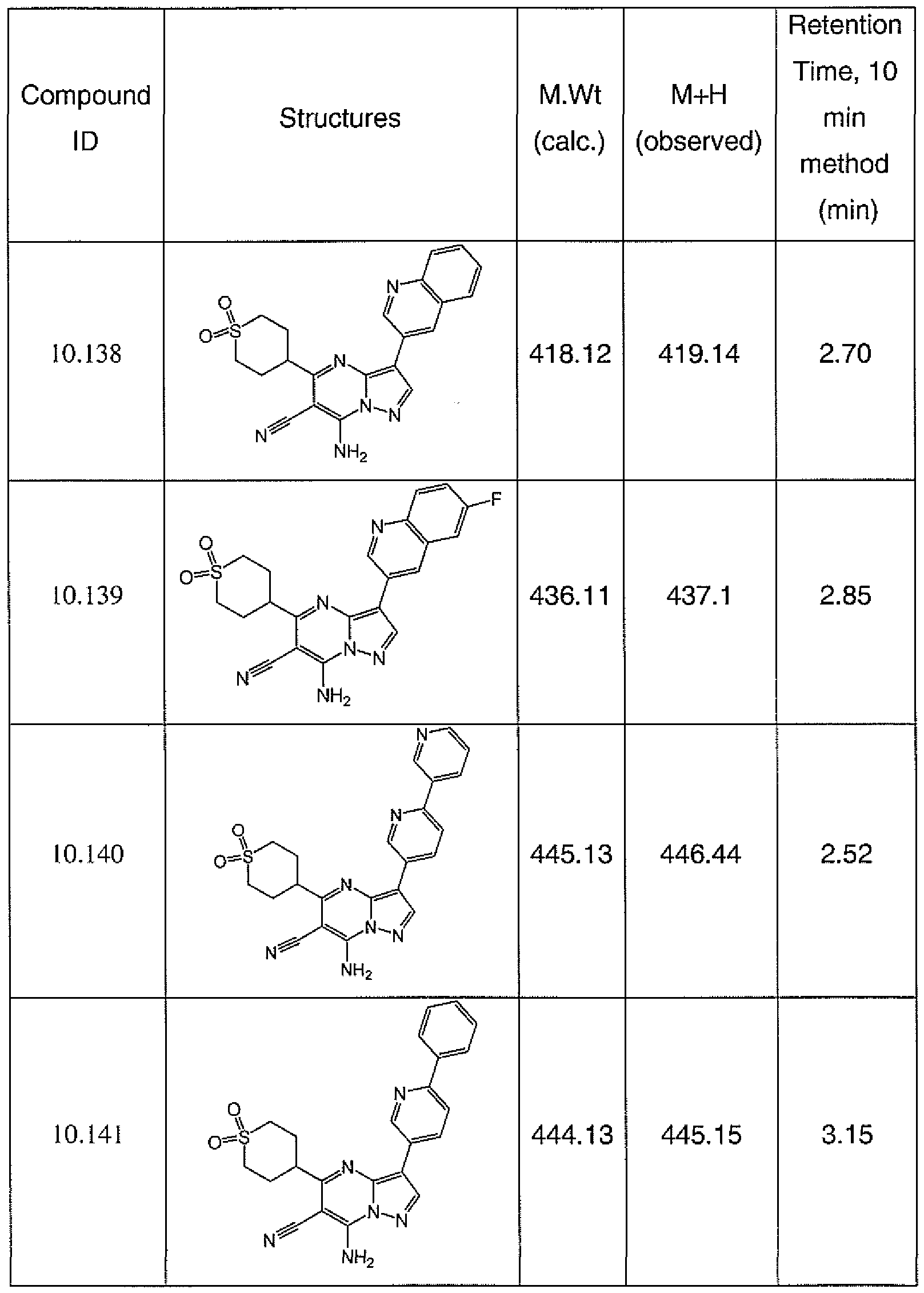
Synthesis of 7-amino-3-(Quinolin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-1 M'- dioxide)pyrazoiof1 ,5-aipyrimidine-6-carboxamide:

The 7-amino-3-(quinoUn-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl- 1', 1 '-dioxide)pyrazolo[ 1 ,5- a]pyrimidine-6-carbonitriiewas stirred in cone. H2SO4(ImL) at 6O°C until LCMS indicated complete conversion. The reaction was quenched with ice and the pH was adjusted to ~ 8 by 6 N NaOH. The mixture was extratce by 15% j-PrOH/CH2Cl2 (x3). The combined organic layers were dried with Na2SO^ Evaporation of solvent afforded the crude displacement compound. Purification by prep-LC afforded 7-amino-3-(quinolin-3-yl)-5-(tetrahydro-2H- thiopyran-4-yl- l',r-dioxide)pyrazolo[i,5-a]pyrimidine-6-carboxamide. LCMS tR = 2.27 Min (10 min run, UV 254nm). Mass calculated for, M+ 436.13, observed LC/MS m/z 437.23 (M+H).
Synthesis of 7-amino-3-(quinolin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl)pyrazolo[1 ,5- a]pyrimidine-6-carboxamide:

By essentially the same procedure given in Preparative above, 7-amino-3-(quinolin-3-yl)-5- (tetrahydro-2H-thiopyran-4-yl) pyrazolo [1,5-a]pyrimidine-6-carboxamide can be prepared. LCMS tR = 3.21 Min (10 min run, UV 2s4!im). Mass calculated for, M+ 405.13, observed LC/MS m/z 406.15 (M+H).
Synthesis of 1-(7-amino-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-1M '- dioxide)pyrazolo[1 ,5-a]pyrimidin-6-yl)ethanone oxime:

Synthesis of 1-amino-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyraπ-4-yl--1', 1'- dioxide)pyrazoio[1 ,5-a]pyrimidine-6-carbaldehyde oxirne:
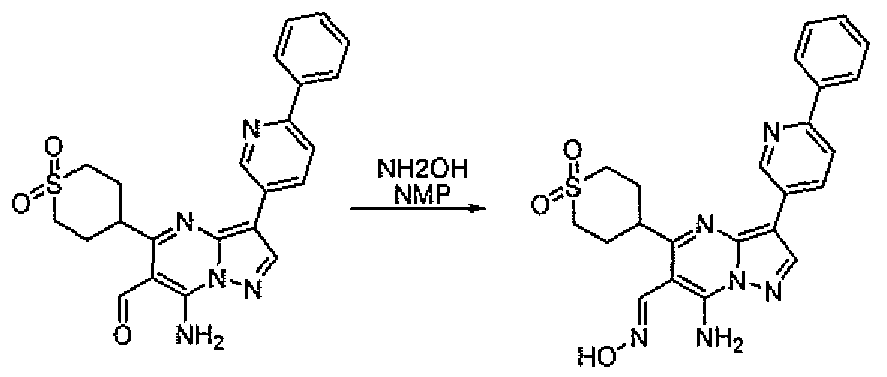
By essentially the same procedure given in Preparative Example given above l-amino-3-(6- phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl--r, l'-dioxide) pyrazolo [1,5- a]pyrimidine-6-carbaldehyde oximecan be prepared. LCMS tR = 3.15 Min (10 min run, UV 254nm)- Mass calculated for, M+ 462.14, observed LC/MS m/z 463.57 (M+H).
Synthesis of 1-(7-amino-3-(αuinoiin-3-vO-5~thiomorpholinopyrazoio-1', 1'-dioxide-f1.5- aipyrimidin-6-vhethanol:

SCHEME-37
NaBH4 (10 mg) was added to a solution of l-(7-amino-3-(quinolm-3-yl)-5-thiomorpholino-r, l'-dioxide-pyrazoio [1, 5-a]pyrimidin-6-yl)ethanone (10 mg, 0.023 mmoL) in MeOH (1 mL). After stirring at room temperature for Ih, the mixture was concentrated to dry. Purification by prep-TLC afforded l-(7-amino-3-(quinolin-3-yI)-5-thiomorpholinopyrazolo-r, l '-dioxide-
[1,5-a]pyrimidin-6-yl)ethanol: LCMS tR = 2.44 Min (5 min run, UV 254nm)- Mass calculated for, M+ 438.1, observed LC/MS m/z 439.1 (M+H).
By essentially the same procedure given in Preparative Example described above(Scheme-37), following compounds 10.142-10.148 in table-10-I can be prepared.
TABLE-IQ-I


Synthesis of 3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl--1 M '-dioxide)-6- (thiophen-2-yl)pyrazoiof1 ,5-aipyrimidin-7-amine:

By essentially the same procedure given in Preparative Example described above(Scheme-36), 3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl- 1 ' , l'-dioxide)-6-(thiophen-2- yl)pyrazolo[i,5-a]pyrimidin-7-amine be prepared. LCMS tR = 4.18 Min (10 min run, UV 254πm). Mass calculated for, M+ 501.12, observed LC/MS m/z 502.0 (M+H).
By essentially the same procedure given in Preparative Example above(36), compounds in table 10-J ( 10.149 -10.151) can be prepared.
TABLE-10-.T


Synthesis of β-cvciopropyl-3-(β-phenylpyridin-3-vh-5-ftetrahydro^H-thiopyran-Φyl-- 1 M '-dioxide)pyrazolo[1 ,5-aipyrimidin-7~amine:

SCHEME-38
A degassed mixture of 6-bronio-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl- l',i '-dioxide)-N,N-bis((2-(tπmethylsilyl)ethoxy)methyl)pyrazoio[1,5-a]pyrimidin-7-amine (53 mg, 0.07mmol), Pd(OAc)2 (3.1 mg, 0.014 mmoL), P(C6H11)3 (7.9 mg, 0.028 mmoL), 2- cyclopropyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (23.5 mg, 0.14 mmoL), K3PO4 (53 mg, 0.25 mmoL) in Toluene (0.4 mL) and H2O (0.02 mL)was heated at 10O°C overnight. The reaction mixture was cooled to room temperature, filtered through celite and concentrated in vacuo. The crude coupling product was treated with 50% TFA in H2O (4 mL) for Ih. The reaction mixture was concentrated in vacuo. Concentration and purification afforded 6- cyclopropyl-3-(6-phenylpyridin-3-yI)-5-(tetrahydro-2H-thiopyran-4-yl-- IM'- dioxide)pyrazolo[1,5-a]pyrimidin-7-amine: LCMS tβ = 3.49 Min (10 min run, UV 254nm). Mass calculated for, M+ 459.17, observed LC/MS m/z 460.22 (M+H).
By essentially the same procedure given in Preparative Example above(Scheme-38), compounds 10.152 - 10.154 can be prepared are given in Table-10-K
TABLE-IO-K


Scheme-39
Part A:
(7--(bisf(2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)-5-(tetrahydro- 2H-thiopyran~4-yl-1',1 '-dioxide) pyrazolo[1 ,5-alpyrimidin-6-yl)methyl methanesulfonate:
At O°C, Et3N (41.7 uL, 0.29 mmol) and MsCl (17.4 uL, 0.22 mmol) were added to a solution of (7-(bis((2-(trimethylsilyl)ethoxy)methyl)aniino)-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H- thiopyran-4-yl-r,r-dioxide)pyrazoIo[l,5-a]pyrimidin-6-yl)methanol (53 tng, 0.75 mmol)in THF (3mL). After stirring at room temperature for Ih, the compound, (7-(bis((2- (trimethylsilyl)ethoxy)raethyl)amino)-3-(6-phenylpyridin-3-yl)-5-(teti-ahydro-2H-thiopyran-4- y I- 1',Γ -dioxide)pyrazolo[l,5-a]pyrimidin-6-yl)methyl methanesulfonate was evenly transferred to three vials.
Part B:
6-(aminomethyπ-3-(6-phenylpyriciin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-1 M '-dioxide
)pyrazoio[1 ,5-alpyrimidin-7-amine:
7N NH3 in MeOH (ImL) added was added to vial I. 6-(aminomethyl)-3-(6-phenyipyridin-3- yl)-5-(tetrahydro-2H-thiopyran-4-yl-l', l'-dioxide) pyrazolo[l,5-a]pyrimidin-7-amine was obtained after concentration and deprotection by treating with 50% TFA in H2O and then prep- LC purification to give the product, 6-(aminomethyI)-3-(6-phenylpyridin-3-yl)-5-(tetrahydro- 2H-thiopyran-4-yl-l', l'-dioxide) pyrazolo[1,5-a]pyrimidin-7-amine. LCMS tR = 1.97 Min ( 10 min run, UV 254nm). Mass calculated for, M+ 448.16, observed LCMS m/z 449.2 (M+H).
Part C:
6-((methylamino)methyl)-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-1 M '- dioxide) pyrazoloH ,5-a1pyrimidin-7-amine:
2 N MeNH2 in THF (ImL) added was added to vial 2. 6-((methylamino)methyI)-3-(6- phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-r, r-dioxide)pyrazolo[l,5-a]pyrimidin- 7-amine was obtained after concentration and deprotection by treating with 50% TFA in H2O and then prep-LC purification resulted in pure 6-((methylamino)metb.yl)-3-(6-phenyIpyridin-3- yl)-5-(tetrahydro-2H-thiopyran-4-yl-l\t '-dioxide) pyrazolo[1,5-a]pyrimidin-7-amine as TFA salt. LCMS tR = 2.12 Min (10 min run, UV 254nm). Mass calculated for, M+ 462.18, observed LC/MS m/z 463.3 (M+H).
Part D: 6-(methoxynnethyl)-3-(6-phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4- yl)pyrazolori ,5-aipyrimidin-7-amine:
0.5 M NaOMe in MeOH (1 mL) added was added to vial 3. 6-(methoxymethyl)-3-(6- phenylpyridin-3-yl)-5-(tetrahydro-2H-thiopyran-4-yl-l ', 1 '-dioxide)pyrazoio[l ,5-a]pyrimidin- 7-aminewas obtained after concentration and deprotection by treating with 50% TFA in H2O and then prep-LC purification provided pure compound, 6-(methoxymethyl)-3-(6- phenylpyridin-3-y[)-5-(tetrahydro-2H-thiopyran-4-yl- i ', l'-dioxide)pyrazolo[l,5-a]pyrimidin-
7-amine. LCMS tR = 2.66 Min (10 min run, UV 254nm). Mass calculated for, M+ 463.1, observed LC/MS m/z 464.1 (M+H).
By essentially the same procedure given in Scheme-39, following compounds 10.155 to 10.161can be prepared. (Table-10-L)
TABLE-IO-L


Synthesis of tert-butyl 2-(7-(bis((2-(trimethyisilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yi)pyrazolo[1 ,5-a]pyrimidin-5-yl)-5-methoxy-1 H-indole-1 -carboxylate:

Htert-butoxycarbonyl)-5-methoxy-1H-indol-Z-ylboronic acid (0.24 mmol, 70 mg), K3PO4 (0.36 mmoi, 77 mg), and PdCl2(dppf>CH2Cl2 (0.012 mmol, 9.8 mg) were added to a solution of 5-chloro-3-(6-phenyIpyridin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1,5- a]pyrimidin-7-amine (0.12 mmol, 72 mg) in dioxane (4 mL) and H2O (0.5 mL). The resulting solution was stirred at 90° C under argon overnight. The mixture was diluted with H2O and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation and purification by column chromatography afforded tert- butyl 2-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)pyrazolo[1,5- a]pyrimidJn-5-yl)-5-methoxy- 1H-indoie-1-carboxylate, LCMS tR = 3.48 Min (5 min run, UV 254nm). Mass calculated for, M+ 792.3, observed LC/MS m/z 793.2 (M+H).
6-bromo-5-(5-methoxy-1 H-indol-2-yl)-3-(6-pheπylpyridin-3-yl)pyrazolof1 ,5-a1pyrimidin- 7-amine:

By essentially the same procedure given in Preparative Examples above, 6-bromo-5-(5- methoxy- 1 H-indol-2-yl)-3-(6-pheny lpyridin-3-yl)pyrazolo[ 1 ,5-a]pyrimidin-7-amine can be prepared. LCMS IR = 4.59 Min (10 min run, UV 254nm)- Mass calculated for, M+ 510.08, observed LC/MS m/z 5 U.50 (M+H).
Synthesis of 5-chloro-3-(6-phenylpyπdin-3-yl)-N,N-bis((2- (trimethyisilyl)ethoxy)methyl)pyrazolo[1 ,5-aipyrimidin-7-amine:

2-phenyl-5-(4,4,5,5-tetraraethyl-1,3,2-dioxaboroian-2-yl)pyridine (2.38 mmol, 675 mg), K3PO4 (5.96 mmol, 1264 mg), and PdCl2(dppfyCH2Cl2 (0.20 mmol, 162 mg) were added to a solution of 5-chloro-3-iodo-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[I,5-a]pyrimidin-7- amine (1.98 mmol, UOl mg) in dioxane (18 mL) and H2O (3 mL). The resulting solution was stirred at 70° C under argon overnight. The mixture was diluted with H2O and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation and purification by column chromatography afforded 5-chloro-3-(6- phenyipyridin-3-yl)-N,N-bis((2 trimethylsilyl)ethoxy)methyl)pyraεoio[l ,5-a]pyrimidin-7- amine: LCMS IR = 3.36 Min (5 min run, UV 254nm)- Mass calculated for, M+ 581.2, observed LC/MS m/z 582.2 (M+H).
Synthesis of tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazoiori .5-alPyrimidin-5-yloxy)piperidine-1-carboxyiate:
NaH (120 mg, 3 mmoL) was added to a solution of tert-butyl 4-hydroxypiperidine-1- carboxylate (522 mg, 2.60 mmoL) in THF (8 mL). After stirring at room temperature for 5 min, 5-chloro-3-(6-phenyipyridin-3-yl)-N,N-bis((2-
(trimethylsiIyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (755 mg, 1.30 mmoL) in THF (4 mL) was added dropwise. The mixture was heated under microwave condition at 100°C for 30 min, diluted with H2O and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation and purification by column chromatography afforded tert-butyl 4-(7-(bis((2-(trimetrtylsilyl)ethoxy)methyt)amino)- 3-(6-phenylpyridin-3-yl)pyrazolo[I,5-a]pyrimidin-5-yloxy)piperidine-1-carboxylate: LCMS tR
= 3.53 Min (5 min run, UV 254«m). Mass calculated for, M+ 746.4, observed LC/MS m/z 747.2 (M+H).
Synthesis of tert-butyl 4-(7-(bisf(2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6- phenylpyridin-3-yl)pyrazoloπ .5-aipyrimidin-5-yloxy)pipericiine-1-carboxylate:

NBS (246 mg, 1.38 mmoL) was added to a solution of tert-butyl 4-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5- yloxy)piperidine-1-carboxylate (1022 mg, 1.38 mmoL) in CH3CN (15 mL). After stirring at room temperature for 30 min, the mixture was concentrated in vacuo. Purification by column chromatography afforded tert-butyl 4-(7-(bis((2-(trimethy!siiyi)ethoxy)methyl)amino)-6- bromo-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yloxy)piperidine-1-carboxyIate: LCMS tR = 3.59 Min (5 min run, UV 254nm). Mass calculated for, M+ 824.3, observed LC/MS m/z 825.1 (M+H).
The tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-phenylpyridin- 3-yl)pyrazolo(1,5-a]pyrimidin-5-yloxy)piperidine-1-carboxylate (143 mg, OJ 7 mmoL) was treated with 50% TFA in H2O (6 mL) and stirred overnight. The reaction mixture was concentrated in vacuo.
(R)-1 -(4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolof1 ,5-a1pyrimidin-5- y1oxy)piperidin-1 -yl)-2-hydroxypropan-1 -one:
A mixture of the crude 6-bromo-3-(6-phenylpyridin-3-yl)-5-(piperidin-4-yloxy)pyrazolo[1,5- a]pyrimidin-7-amine, D-(-)-iactic acid (18.5 mg, 0.20 mmoL), EDC (78.7 mg, 0.41 mmoL), HOBt (27.7 mg, 0.20 mmoL) and DIEA ( 238 uL, 1.37 mmoL) in DMF ( 4 mL) was stirred at room temperature for 2 hr. Purification with prep-LC provided (R)-1-(4-(7-amino-6-bromo-3-
(6-phenylpyridin-3-yl)pyrazolo[l,5-a]pyrimidin-5-yloxy)piperidin-1-yl)-2-hydroxypropan-l- one, LCMS tR = 4.39 Min (10 min run, UV 254nm). Mass calculated for, M+ 513.0, observed LC/MS m/z 514.43 (M+H).
Synthesis of 1 -(7-amino-3-(6-phenylpyridin-3-yl)-5-(piperidin-4-yloxy)pyrazolori .5- alpyrimidin-6-yl)ethaπoπe:

A degassed mixture of tert-butyl 4-(7-(bis((2-(trimethytsilyl)ethoxy)methyl)amino)-6-bromo» 3-Cό-phenylpyridin-3-yl)pyrazolotl^-aJpyrimidin-5-yloxyJpiperidine-1-carboxylate (0.57 mmol, 486 mg), Pd(PPh3)4 (66 mg, 0.056 mmoL), tributyl(l-ethoxyvinyl)stannane (410mg, 1.13 mmoL) in Dioxane (6 mL) was heated at 100°C overnight. The reaction mixture was cooled to room temperature, filtered through 9: 1 Siθ2:KF plug and concentrated in vacuo.
The crude tert-butyl 4-(7-(bts((2-(trimethyIsilyl)ethoxy)methyl)amino)-6-( l-ethoxyvinyl)-3-(6- phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yloxy)piperidine-1-carboxylate was treated with 50% TFA in H2O (6 mL) and stirred overnight. The reaction mixture was concentrated in vacuo. Purification with prep-LC provided l-(7-amino-3-(6-phenylpyridin-3-yl)-5-(piperidin- 4-yloxy) pyrazolo [1,5-a]pyrimidin-6-yl)ethanone: LCMS IR = 1.1 IMin (5 min run, UV 254nm). Mass calculated for, M+ 428.1 , observed LC/MS m/z 429.1 (M+H).
(R)-1-(4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolori ,5-a1pyrimidin-5- y[oxy)piperidin-1 -vO-2-hydroxypropan-1 -one:
A mixture of t-(7-amino-3-(6-phenyJpyridin-3-yl)-5-(piperidm-4-yloxy)pyrazolo[1,5- a]pyrimidin-6-yl)ethanone (21 1 mg, 0.49 mmoL), D-(-)-lactic acid (53.3 mg, 0.59 mmoL), EDC (189.0 mg, 0.99 mmoL), HOBt (132.0 mg, 0.99 mmoL) and DEA ( 514 uL, 2.96 mmoL) in DMF (5 mL) was stirred at room temperature for 2 hr. Purification with prep-LC provided
(R)-I -(4-(6-acetyl-7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[ 1,5-a]pyrimidin-5- yloxy)piperidin-1-yl)-2-hydroxypropan-1-one: LCMS tR = 3.53 Min (10 min run, UV 254nm). Mass calculated for, M+ 500.21, observed LC/MS m/z 501.62 (M+H).

R = H or F
SCHEME-40
By essentially the same procedure given in Preparative Examples above and scheme-40, compounds 10.162 and 10.163 can be prepared ( Table- 10-M).
TABLE-IO-M


SCHEME-41
By essentially the same procedure given in Preparative Example(Scheme-4l), 10.164 and 10.165 can be prepared.(Table- 10-N)
TABLE-IO-N

Synthesis of tert-butyl 4-(2-methoxy-2-oxoethyl)piperidine-1-carboxylate:

At O°C, TMSCHN2 (2.0 M in ether, 15 mL) was added dropwise to a mixture of 2-(l-(tert- butoxycarbonyl)piperidin-4-yl)acetic acid (6272 mg, 25.78 mmoL) in Toluene (60 mL) and MeOH (60 mL). After stirring at room temperature for Ih, the mixture was concentrated. The crude tert-butyl 4-(2-methoxy-2-oxoethyl)piperidine-1-carboxylate was used as it is without further purification.
tert-butyl 4-(f7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolof1 ,5-aipyrimidin-5- yl)methyl)piperidine-1-carboxvlate:

tert-butyl 4-(3-cyano-2-oxopropyl)piperidine-1-carboxylate
At -78°C, CH3CN in THF (1505 uL, 28.82 mmoL) was added dropwise to BuLi (2.5 M in hexane, 11.52 mL) in THF (40 mL). After stirring at -78°C for Ih, tert-butyl 4-(2-methoxy-2- oxoethyl)piperidine-1-carboxylate (3708 mg, 14.41 mmoL) in THF (10 mL) was added dropwise in 5 min. The mixture was stirred at -78°C for Ih and -45°C for Ih. At O°C, IN HCl was added carefully to adjust the pH to about 7. The mixture was then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation afforded tert-butyl 4-(3-cyano-2-oxopropyl) piperidine-1 -carboxylate.
tert-butyl 4-((7-aminopyrazolon .5-aipyrimidin-5-yl)methyhpiperidine-1-carboxylate:
The crude tert-butyl 4-(3-cyano-2-oxopropyl)piperidine-1-carboxylate was then heated at 100°C overnight with 1 H-pyrazol-5-amine (1197 mg, 14.41 mmoL) in HOAc (25 mL). Concentration provided crude tert-butyl 4-((7-aminopyrazolo[l,5-a]pyrimidin-5- yl)methyl)piperidine- 1 -carboxylate.
tert-butyl 4-((7-(bis((2-(trimethyisilyl)ethoxy)methyπamino)pyrazo[ori ,5-alpyrimidin-5- yl)methyl)piperidine-1 -carboxylate:
The amino analog was treated with SEMCl (72.05 mmoL, 12.71 mL) and DEEA (144.4 mmoL, 25.05 mL) in DCE (100 mL) at 5O°C for 1 h. The mixture was diluted with H2O and then extracted with CH2Cl2 (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation and purification by column chromatography afforded tert-butyl 4- ((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolo[1,5-a]pyrimidin-5- yl)methyl)piperidine-1-carboxylate: LCMS tβ = 3.05 Min (5 min run, UV 254nm)- Mass calculated for, M+ 591.3, observed LC/MS m/z 592.3 (M+H).
Synthesis of tert-butyl 4-((7-(bis(f2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- phenylpyridin-3-yl)pyrazolori ,5-aipyrimidin-5-yl)methyhpiperidine-1-carboxvlate:

tert-butyl 4-((7-(bis((2-(trimethyisilyl)ethoxy)methyl)amino)-3-iodopyrazoio[1,5- a1pyrimidin-5-yl)methyl)piperidine-1-carboxylate:
NIS (288 mg, 1.28 mmoL) was added to a solution of tert-butyl 4-((7-(bis((2- (trimethyIsilyl)ethoxy)methyl)amino)pyrazolo[1,5-a]pyrimidin-5-yI)methyl)piperidine-l - carboxylate (741.6 mg, 1.25 mmoL) in CH3CN (10 mL). The mixture was stirred at room
temperature for Ih. Purification by column chromatography afforded tert-butyl 4-((7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-iodopyrazolo[1,5-a]pyrimidin-5-yl)methyl)piperidine- 1-carboxylate: LCMS tR = 3.02 Min (5 min run, UV 254nm). Mass calculated for, M+ 717.2, observed LC/MS m/z 718.3 (M+H).
tert-butyl 4-((7-(bisff2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3- yl)pyrazoiof^5-aipyrimidin-5-yl)methyl)piperidine-i -carboxylate:
2-phenyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (1.32 mmol, 370 mg), K3PO4 (3.04 mmol, 644 mg), and PdCl2(dppf) • CH2Cl2 (0.10 mmol, 83 mg) were added to a solution of tert-butyl 4-((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-todopyrazolo[1,5- a]pyrimidin-5-yl)methyl)piperidine- 1-carboxylate (1.02 mmol, 726 mg) in dioxane (6 mL) and H2O (ImL). The resulting solution was stirred at 100° C under argon for 18 hours. The mixture was diluted with H2O and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation and purification by column chromatography afforded tert-butyl 4-((7-(bis((2-
(trimethylsslyl)ethoxy)memyl)ammo)-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5- yl)methyl)piperidine- 1-carboxylate: LCMS IR = 3.47 Min (5 min run, UV 254™)- Mass calculated for, M+ 744.2, observed LC/MS m/z 745.2 (M+H).
(R)- 1 -(4-((7-amino-6-bromo-3-( 6-phenylpyridin-3-yl) pyrazoioM ,5-alpyrimidin-5- yl)methyl)piperidin-1 -vO-2-hydroxypropan-1 -one:

By essentially the same procedure given in Preparative Example above, (R)-1-(4-((7-amino-6- bromo-3-(6-phenylpyridin-3-yl) pyrazolo[l,5-a]pyrimidin-5-yl)methyl)piperidin-1-yl)-2- hydroxypropan-ϊ-one can be prepared. LCMS tβ = 3.58 Min (10 min run, UV 254nm)- Mass calculated for, M-s- 534.13, observed LC/MS m/z 535.55 (M+H).
(R)-1-(4-((6-acetyl-7-amJno-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-aipyrimidin-5- yl)methyl)piperidin-1 -yl)-2-hydroxypropan-1 -one:

By essentially the same procedure given in Preparative Example above, (R)-1-(4-((6-acetyl-7- amino-3-(6-pheny[pyriciin-3-yl)pyrazolo[l ,5-a]pyrimidin-5-yl)methyl)piperidin-1-yl)-2- hydroxypropan-1-one was synthesized in a manner similar: LCMS IR = 3.42 Min (10 min run, UV 254nm). Mass calculated for, M+ 498.23, observed LC/MS m/z 499.64 (M+H).
tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3- yl)pyrazolo[1 ,5-aipyrimidin-5-yl)piperazine-1 -carboxviate:

A mixture of 5-chloro-3-(6-phenylpyridin-3-yI)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoio[i,5-a]pyrimidin-7-amine (200 mg, 0.34 mmoL), tert- butyl piperazine-1-carboxylate (224 mg, 1.20 mmoL) and NaHCO3 (116 mg, 1.38 mmoL) in NMP (5 mL) was heated at 13O°C overnight. The mixture was diluted with H2O and then extracted with ethyl acetate (x2). The combined organic layers were washed with brine and dried with Na2SO4. Evaporation and purification by column chromatography afforded tert- butyl 4-(7-(bis((2-(trimethylsiIyl)ethoxy)methyl)amino)-3-(6-phenylpyridin-3-yi)pyrazoio[1,5- a]pyrimidin-5-yl)piperazine-1-carboxylate: LCMS tR = 3.04 Min (5 min run, UV 2S4nm). Mass calculated for, M+ 731.4, observed LC/MS m/z 732.4 (M+H).
(R)-1-(4-(6-acety[-7-amino-3-(6-phenylpyridin-3-yl)pyrazolo[1 ,5-alpyrimidin-5- yl)piperazin-1 -yl)-2-hydroxypropan-1 -one:

By essentially the same procedure given in Preparative Example above scheme, (R)- 1 -(4-(6- acetyl-V-amino-3-tό-phenylpyridin-3-ylJpyrazolotl.5-alpyrimidin-5-yl)piperazin- l-yl)-Z- hydroxypropan-1-one can be prepared in 96% yield, LCMS IR = 2.79 Min (10 min run, UV 254nm). Mass calculated for, M+ 485.2, observed LC/MS m/z 486.3 (M+H).
i -(y-amino-3-(e-phenvIpyridin-3-yl)-5-d -(pyridin-3-ylmethyDpiperidin^- yl)pyrazolon ,5-aipyrimidin-6-yl)ethanone:
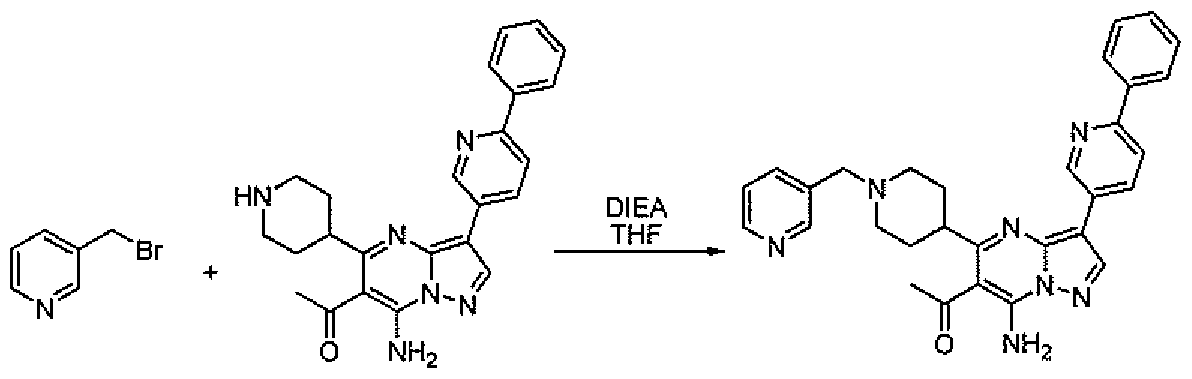
A mixture of l-(7-amino-3-(6-phenyipyridin-3-yl)-5-(piperidin-4-yl)pyrazoIo[l ,5-a]pyrimidin- 6-yI)ethanone (0.049 mmol, 20 mg), 3-(bromomethyl)pyridine (0.049 mmol, 12 mg) and DDEA (0.29 mmol, 37 mg) in THF (2mL) was stirred at room temperature overnight. Concentration and purification by prep-LC afforded l-(7-aniino-3-{6-phenylpyridin-3-yl)-5-(l-(pyridin-3- ylmethyl)piρeπdin-4-yl)pyrazoIo[1,5-a]pyrimidin-6-yl)ethanone: LCMS tn = 2.71 Min (10 min run, UV 254nm)- Mass calculated for, M+ 503.24, observed LC/MS m/z 503.99 (M+H).
1-(7-amino-5-(1 -(morpholinosulfonyl)piperidin-4-yl)-3-(6-phenylpyridin-3- yl)pyrazolo[1 ,5a1pyrimidin-6-yl)ethanone :

DIEA (121 mg, 0.94 mmoL) was added to at O°C a mixture of l-(7-amino-3-(6-phenylpyridin- 3-yl)-5-(piperidin-4-yl) pyrazolo [1,5-a]pyrimidin-6-yl)ethanone (64.3 mg, 0.15 mmoL) and morpholine-4-sulfonyl chloride (28.9 mg, 0.15 mmol) in DMF (3 mL). After stirring at O°C for 0.5h and then room temperature for 2 h, the mixture was purified by prep-LC to afford l-(7- amino»5-(l-(morpholinosulfonyl)piperidin-4-yI)-3-(6-phenylpyridin-3-yl)pyrazolo[1,5-
10 a]pyrimidin-6-yl)ethanone, LCMS tR = 3.83 Min (5 min run, UV 2S4nm)- Mass calculated for, M+ 561.66, observed LC/MS m/z 567.66 (M+H).
Synthesis of tert-butyl 4-(7-amino-6-bromo-3-(Quinoiin-3-yl)pyrazolori ,5-a1pyrimidin-5- vQpiperazine-1 -carboxylate
\ 5
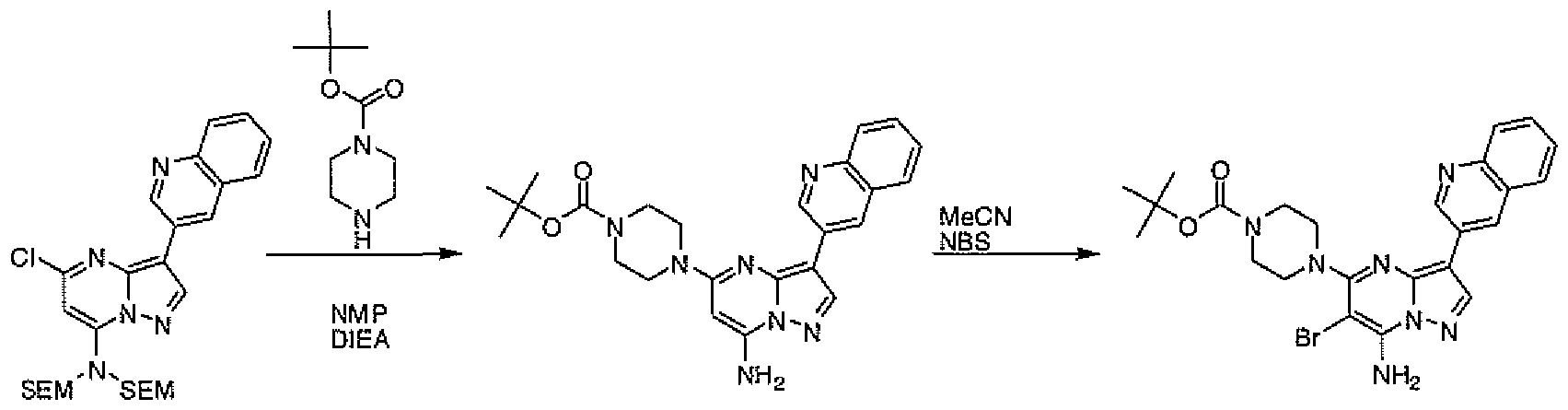
To a 20 mL scintillation via! was charged 5-chloro-3-(quinolin-3-yl)-N,N-bis((2- {trimethylsilyl)-ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidtn-7-amine (0.09 mmoi, 50 mg), NMP (1 mL), N,N-diisopropylethylamine (0.36 mmol, 63 μL), and 4-N-Boc-piperazine 0 (0.27 mmol, 50 mg). The resulting solution was heated to 200° C in microwave synthesizer for 30 minutes. Upon completion, the NMP was removed in vacuo as an azeotrope using chlorobenzene. This residue was dissolved in acetonitrile (1 mL) to which N-bromosuccinimide (0.10 mmol, 17.7 mg) was added. This reaction mixture
was allowed to stir at room temperature for 2 hours. The reaction mixture was concentrated in vacuo and tert-butyl 4-(7-amino-6-bromo-3-(quinoiin-3-yl)pyrazoio[1 ,5- a]pyrimidin-5-yl)piperazine-1-carboxylate used without further purification.
Synthesis of 6-ch[oro-5-(piperazin-1-yl)-3-(Quinolin-3-vθpyrazoiori ,5-aipyrimidin-7- amine
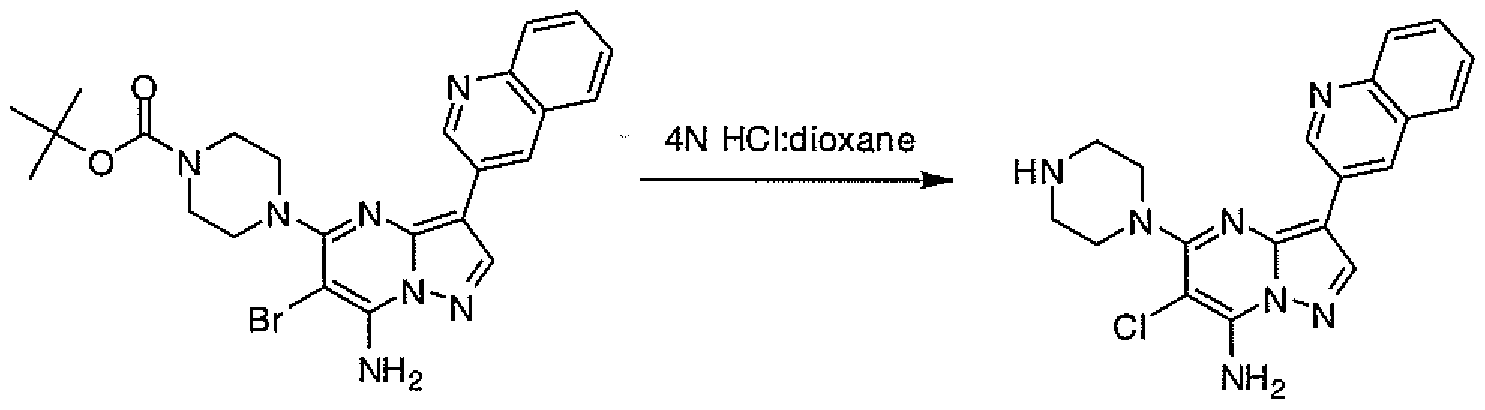
Synthesis of (4-(7-amino-6-chloro-3-(quinolin-3-yl)pyrazolof1 ,5-alpyrimidin-5- yl)piperazin-1-yl)(thiophen-2-yl)methanone


Scheme-42
[4-(7-Amino-3-auinonn-3-yl-pyrazolori ,5-a1pyrimidin-5-yl)-phenyll-acetic acid

[4-(4,4,5,5-Tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-phenyl]-acetic acid methyl ester (0.576 mmoi, 160 mg), K3PO4 (0.864 mmol, 183 mg), and PdCI2(dppf) • CH2CI2 (0.029 mmol, 24 mg) was added to a solution of (5-Chloro-3-quinolin-3-yl-pyrazolo[1 ,5- a]pyrimidin-7-yl)-bis-(2-trimethylsilanyl-ethoxymethy[)-amine (0.288 mmol, 160 mg) in dioxane (2 ml_). To this suspension was added distilled H2O (0.2 ml_). The resulting solution was stirred at 100° C under an argon atmosphere for 18 hours. The reaction mixture was concentrated in vacuo. The crude mixture was dissolved in 2:1 MeOH:H2O (1.5 ml_) and was treated with 2N NaOH(aq) (1.0 ml_). The resulting solution was stirred at room temperature for 18 hours. Aqueous hydrochloride solution (1.0 N, 4 ml) was added to the reaction mixture and the resulting solution was stirred at 65 °C for 2 h. The reaction mixture was concentrated and purified by prep-
LC to afford the title compound (70 mg, 62% yield): LC/MS RT = 3.21 min. Mass calculated for, M+H 396.14, observed 396.14.

By essentially the same procedure given in Scheme 42, the compounds listed in Table 11 can be prepared.
Table 11



Synthesis of tert-butyl 4-(7-(bis(f2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolof1 ,5-aipyrimidin-5-yl)-5,6-dihydropyridine-1 (2H)-carboxylate

To a 40 mL scintillation vial was charged 5-chloro-3-(quinoiin-3-yl)-N,N-bis((2- (trimethylsiiyl)-ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin-7-amine (0.86 mmol, 480 mg), tert-butyl 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-5,6-dihydropyrid!ne-1 (2H)- carboxylate (1.30 mmol, 400 mg), K3PO4 (2.60 mmol, 550 mg), and PdCI2(UpPf)-CH2CI2 (0.086 mmol, 70 mg). To this reacton mixture was added a 9:1 solution of 1 ,4-dioxane:H2O (10 mL). The vial was flushed with argon, sealed with Teflon tape, and stirred at 100° C 18 hours. After 18 hours, the reaction was concentrated in vacuo and the residue purified via silica gel chromatography to yield tert-butyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3- yl)pyrazolo[1 ,5-a]pyhmidin-5-yl)-5,6-dihydropyridine-1 (2H)-carboxylate (586 mg, 833 mmol, 97% yield) as yellow solid.
.Synthesis of 3-(αuinolin-3-yl)-5-(1 ,2.3.6-tetrahydropyridin-4-yl)pyrazoioM .5- aipyrimidin-7-amine
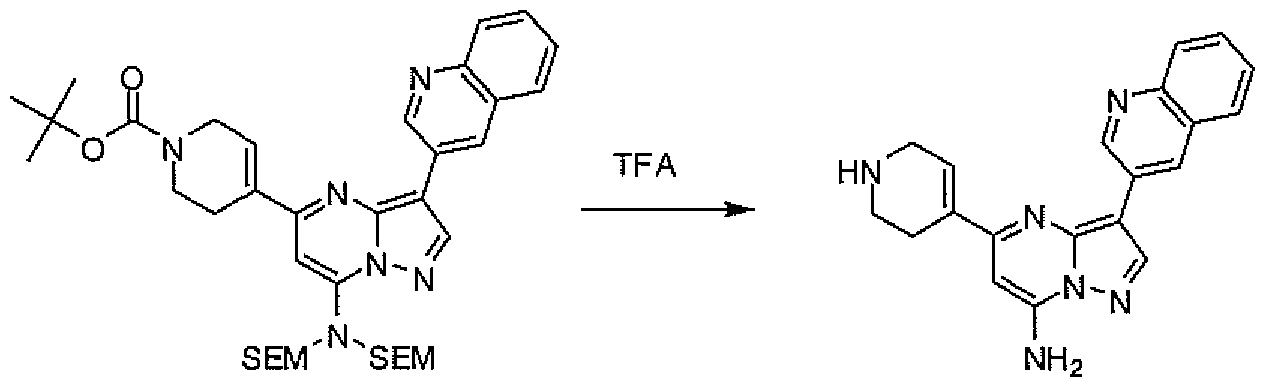

tø/t-Butyl 4-(7-(bis((2-{trimethy!silyl)ethoxy)methyI)amino)-3-(quinolin-3- yl)pyrazoio[1 ,5-a]pyrimidin-5-yl)-5,6-dihydropyridine-1 (2H)-carboxyiate (0.071 mmol, 50 mg) was charged to a 20 ml_ scintillation vial containing 1 mi_ acetonitrile. To this solution was added N-bromosuccinimide (0.078 mmoi, 14 mg). The reaction mixture was allowed to stir at room temperature for 1 hour. After 1 hour, the reaction was concentrated in vacuo and the crude intermediate was dissolved in 1 mL TFA. The reaction mixture was stirred for 1 hour at room temperature and concentrated in vacuo to yield 6-bromo-3-(quinolin-3-y!)-5-(1 ,2,3,6-tetrahydropyridin-4-yl)pyrazolo[1 ,5- a]pyrimidin-7-amine as yellow solid.
Synthesis of 6-chloro-3-(quinolin-3-yl)-5-(1 ,2,3,6-tetrahydropyridin-4-yl)pyrazolof 1 ,5- aipyrimidin-7-amine

6-Chloro-3-(quinolin-3-yl)-5-(1 ,2,3,6-tetrahydropyridin-4-yl)pyrazoio[1 ,5-a3pyrimidin-7- amine was synthesized in a manner similar to the synthesis of 6-bromo-3-(quinolin-3- yl)-5-(1 ,2,3,6-tetrahydropyridin-4-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine, but with N- chlorosuccinimide substituted for N-bromosuccinimide.
Synthesis of 2-(4-(7-amino-6-bromo-3-(αuinolin-3-yl)pyrazolof1 ,5-alpyrimidin-5-yl)-5,6- dihydropyridin-1 (2H)-yl)acetic acid

6-Bromo-3-{quinolin-3-yl)-5-{1 ,2,3,6-tetrahydropyrid!n-4-yl)pyrazo[o[1 ,5-a]pyrimidin-7- amine (0.035 mmol) was charged to a 20 mL scintillation vial. Then DMF (1 ml), N1N- diisopropylethylamine {0.14 mmo!, 25 μl_), and tert-butyl 2-bromoacetate (0.035 mmol, 5.2 μl_) were added, respectively. The resulting reaction mixture was stirred at 50Q C 18 hours. After 18 hours, the reaction was concentrated in vacuo and treated with 1 mL TFA. This reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated in vacuo and purified via reverse-phase preparatory HPLC to yield 2-(4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)-5,6-dihydropyridin-1 (2H)-yl)acetic acid as yellow solid. (m+H = 479.17, retention time = 2.09 min)
Synthesis of (4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazoiori ,5-a1pyrimidin-5-yl)-5.6- dihydropyridin-1 (2H)-yl)(thiophen-2-yl)methanone and (4-(7-amino-3-(quinolin-3- yl)pyrazolo[1 ,5-alpyrimidin-5-yl)-5,6-dihydropyridin-1(2H)-yl)(thiophen-2-yl)methanone

SCHEME-43
6-Bromo-3~(quino!in-3-yl)-5-(1 ,2,3,6-tetrahydropyridin-4-yl)pyrazolo[1 ,5-a]pyrimidin-7- amine (0.035 mmol) was charged to a 20 mL scintillation vial. Then DMF (1 ml), N1N-
diisopropylethylamine (0.14 mmol, 25 μL), thiophene-2-carboxylic acid (0.039 mmol, 5 mg), and EDC (0.039 mmol, 7.4 mg) were added, respectively. The resulting reaction mixture was stirred at room temperature for 18 hours. Upon completion, the reaction mixture was concentrated in vacuo and purified via reverse-phase preparatory HPLC to yield (4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yI)-5,6- dihydropyridin-1(2H)-yl)(thiophen-2-yl)methanone (m+H = 531.06, retention time = 3.74 min). Also recovered was the dehalogenated product, (4-(7-amino-3-(quinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl}-5,6-dihydropyridin-1 (2H)-yl)(thiophen-2-y])methanone (m+H = 453.24, retention time = 3.51 min).
Synthesis of (4-(7-amino-6-chloro-3-(quino[in-3-yl)pyrazolof1.5-alpyrimidin-5-yl)-5,6- dihydropyridin-1(2H)-yl)(thiophen-2-yl)methanone

(4-(7-amino-6-chloro-3-(quinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)-5I6-dihydropyridin- 1 (2H)-yl)(thiophen-2-yl)methanone was synthesized in a manner similar to the synthesis of (4-(7-amino-6-bromo-3-(quinolin-3-yl)pyrazolo[1 ,5~a]pyrimidin-5-yl)-5,6- dihydropyridin-1 (2H)-yl)(thiophen-2-yi)methanone, but with 6-chloro-3-(quinolin-3-yl)- 5-(1 ,2,3,6-tetrahydropyridin-4-yI)pyrazolo[1 ,5<|pyrimidin-7-amine substituted for 6- bromo-3-(quinolin-3-yl)-5-(1 ,2,3,6-tetrahydropyridin-4-yl)pyrazolo[1 ,5-a]pyrimidin-7- amine. The reaction mixture was purified via reverse-phase preparatory HPLC to yield (4-(7-amino-6-chloro-3-(quino!in-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yi)-5,6- dihydropyridin-1(2H)-yl)(thiophen-2-yl)methanone as yellow solid. (m+H = 487.17, retention time = 3.73 min)
Synthesis of 4-bromo-1 -((2-(trimethylsilyl)ethoxy)nπethyπ-1 H-pyrazole
SEM-Cl

To a 100 mL roundbottom flask was charged 4-bromopyrazole (13.6 mmol, 2.00 g), K2CO3 (20.4 mmol, 2.82 g), and DMF (20 mL). To this suspension was added 2- (trimethylsilyl)ethoxymethyl chloride (15.0 mmol, 2.64 mL). This suspension was allowed to stir at room temperature 18 hours. The reaction mixture was then concentrated in vacuo, dissolved in 20 mL DCM, and filtered through celite. The crude mixture was purified via silica gel chromatography (0% to 80% ethyl acetate in hexanes gradient) to yield 4-bromo-1 -({2-(trimethylsilyI) ethoxy) methyl)-1 H-pyrazole (2.53 g, 9.12 mmol, 67% yield) as clear oil.
Synthesis of 4-(1 ,4-dioxaspiro[4.51dec-7-en-8-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)- 1 H-pyrazole

To a 10-20 mL microwave vessel was charged 4-bromo-1 -({2- (trimethylsilyl)ethoxy)methyl)-1 H-pyrazole (0.60 mmol, 166 mg), 4,4,5,5-tetramethyl-2- (1 ,4-dioxaspiro[4.5]dec-7-en-8-yl)-1 ,3,2-dioxaborolane (0.84 mmol, 224 mg), K3PO4 (1.80 mmol, 382 mg), PdCI2{dppf) ■ CH2CI2 (0.06 mmoi, 50 mg) and 9:1 Dioxane:H2O (15 mL). The suspension was sonicated to break up salts, flushed with argon, sealed, and heated to 160° C for 45 minutes in microwave synthesizer. The reaction was concentrated in vacuo, and the residue purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes) to yield 4-(1 ,4-dioxaspiro[4.5]dec-7-en-8-yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1 H-pyrazole (115 mg, 0.34 mmol, 57% yield) as clear oil.
Synthesis of 4-(1 ,4-dioxaspiro|"4.5ldecan-8-y1H -((2-(trimethyisilyl)ethoxy)methyl)-1 H- pyrazoie
EtOAc

4-{1 ,4-Dioxasptro[4.5]dec-7-en-8-yl)-1 -((2-(trimethylsiiyl)ethoxy)methyl)-1 H-pyrazole (505 μmol, 170 mg) was dissolved in 10 ml_ EtOAc in a 50 ml_ roundbottom flask. The flask was flushed with argon and 5% palladium on carbon (50 mg) was added. The flask was sealed, degassed under vacuum, and hydrogen gas was added with a balloon. The reaction mixture was stirred at room temperature under hydrogen atmosphere for 18 hours. The reaction was then filtered through ceiite to yield 4-(1 ,A- dioxaspiro[4.5]decan-8-yl)-1 -((2-(trimethylsilyl)ethoxy)methyl)-1 H-pyrazole (165 mg, 487 μmol, 97% yield) as a clear oil.
Synthesis of 4-(1-((2-(trimethylsilyl)ethoxy)methy1H H-pyrazol-4-yl)cvclohexanone
EtOH

4-(1 ,4-dioxaspiro[4.53decan-8-yi)-1-((2-(trimethylsilyI)ethoxy)methyl)-1 H-pyrazoie (487 μmol, 165 mg) was dissolved in EtOH (10 m L) in a 20 ml_ scintillation vial. To this solution was added H2O (3 ml_) followed by 4N HCI in 1 ,4-dioxane. The resulting reaction mixture was stirred at room temperature for 60 hours. At this point, the reaction was quenched with saturated
 and extracted with DCM (x3). The combined organics were dried over Na2SO4 to yield 4-(1-((2- (trimethylsilyl)ethoxy)methyl)-1 H-pyrazol-4-yl)cyclohexanone (141 mg, 480 μmol, 98% yield) as white solid.
and extracted with DCM (x3). The combined organics were dried over Na2SO4 to yield 4-(1-((2- (trimethylsilyl)ethoxy)methyl)-1 H-pyrazol-4-yl)cyclohexanone (141 mg, 480 μmol, 98% yield) as white solid.
Synthesis of 4-(1 -((2-(trimethylsiiyl)ethoxy)methyl)-1 H-pyrazol-4-yl)cvclohex-i -enyl trifluoromethanesulfonate

4-(1 -((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazol-4-yl)cyclohexanone (480 μmol, 141 mg) was added to a 50 mi_ roundbottom flask containing anhydrous THF (5 ml_). The flask was flushed with argon, sealed, and cooled to -78° C in dry ice/isopropanol bath. To this solution was added dropwtse LDA (1.5 M is cyclohexane, 0.72 mmol, 480 μL). This solution was stirred at -78° C for 30 minutes. To this solution was added 2-[N1N- bis(trifluoromethylsulfonyl)amino]-5-chloropyridine (0.72 mmol, 282 mg). The resulting reaction mixture was allowed to gradually warm to room temperature as it stirs for 18 hours. After 18 hours, the reaction mixture was concentrated in vacuo and purified via silica gel chromatography to yield 4-(1-((2-(tπmethylsilyl)ethoxy)methyl)-1H-pyrazol-4- yl)cyclohex-1-enyl trifluoromethanesulfonate (115 mg, 270 μmol, 56% yield) as clear oil.
Synthesis of 4-(4-(4,4,5,5-tetramethyl-1.3.2-dioxaborolan-2-vncvclohex-3-enyl)-1 -((2- (trimethylsiiyl)ethoxy)methyl)-1 H-pyrazole

TFA

To a 2 ml microwave vessel was charged 5-chloro-3-(quinolin-3-yl)-N,N-bis({2- (trimethylsilyl)ethoxy)methy])pyrazolo[1 ,5-a]pyrimidin-7-amine (0.043 mmol, 23.5 mg), 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)cyclohex-3-enyl)-1 -((2- (trimethylsilyi)ethoxy)methyl)-1 H-pyrazole (0.047 mmol), K3PO4 (130 mmol, 27 mg), and PdCi≥(dppf) • CH2CI2 (0.0043 mmoi, 3.5 mg). To this mixuture was added 9:1 dioxane: H2O (1 mL). The vessel was flushed with argon and heated to 160° C for 30 minutes in microwave synthesizer. Upon completion, the reaction was concentrated in vacuo and the residue was dissolved in 1 mL TFA. This reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo and purified via reverse-phase preparatory HPLC to yield 5-(4-(1 H-pyrazo!-4-yl) cyclohex~1-enyl)-3~(quinolin-3-yl) pyrazolo [1 , 5-a] pyrimidin-7-amine as yellow solid. (m+H = 408.30, retention time = 3.16 min)
Synthesis of 5-(4-(1 H-pyrazoi-4-vOcvclohex-i -enyl)-6-bromo-3-(αuinolin-3- yl)pyrazolof1.5-aipyrimidin-7-amine

5-(4-(1 H-pyrazoi-4-yl)cyclohex-1 -enyl)-6-bromo-3-(quinolin-3-yl)pyrazo!o[1 ,5- ajpyrimidin-7-amine was synthesized in a manner similar to the synthesis of 6-bromo- 3-(quinoiin-3~yl)-5-(1 ,2,3,6-tetrahydropyridin-4-yl)pyrazolo[1 t5-a]pyrimidin-7-amine, but
with 6-bromo-3-{quinolin-3-yl)-N,N-bis((2-(trimethylsilyi)ethoxy)methyl)-5-(4-(1-((2- (trimethylsilyl)ethoxy)methyl)-1 H-pyrazo!-4-yl)cyclohex-1 -enyl)pyrazolo[1 ,5- a]pyrimidin-7-anπine substituted for tert-Butyl 4-{7-{bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-3-(quinolin-3-yl)pyrazoio[1 ,5--a]pyπmicIin-5-yl)-5,6- dihydropyridine-1 (2H)-carboxylate. The reaction mixture was purified via reverse- phase preparatory HPLC to yield 5-(4-(1 H-pyrazoi-4-yi) cyclohex-1-enyl)-3-(quinolin-3- yi) pyrazolo [1 ,5-a]pyrimidin-7-amine as yellow solid. (M+H = 486.12, retention time = 3.43 min.
By essentially the same procedure given in Scheme 43, the compounds listed in Table 12 can be prepared.
Table 12



Scheme 44
Synthesis of 3-(quinolin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)-5- vJnylpyrazolof1 ,5-alpyrinnidin-7-amine

To 5-chloro-3-tquinolin-3-yl)-N.N-bisftZ-^rimethylsilyl)ethoxyJmethyl)pyrazoloCi .5- a]pyrimidin-7-amine (0.9 g, 1.64 mmol) in a 20 ml_ vials was added 1 ,4-dioxane (10 ml_), tributyl(vinyl)tin (0.5 mL, 1.71 mmol) and Pd(PPh3J4 (0.1 g, 0.08 mmol). The vial was flushed with argon, sealed and heated at 100 °C for 16 hours, at which time LC/MS analysis confirmed full conversion of the starting material to the product. The reaction mixture was filtered through a mixture of silica (8 g) and KF (2 g), and EtOAc was used to wash the filter pad. Concentration of the filtrate gave a brown oil (1.4 g). Further purification via gradient column chromatography on silica eltuting with 5% to 40% EtOAc/hexanes gave 3-(quinoiin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)-5- viny]pyrazolo[1 ,5-a]pyrimidin-7-amine as a yellow oil (0.7 g, 1.3 mmol, 78%). LCMS: 2.55 mins, m/z = 548.2 (MH+).
Synthesis of 7-(bis((2-(tnmethylsilyl)ethoxy)methyl)amino)-3-(quinoiin-3- yl)pyrazoloH ,5-aipyrimidine-5-carbaidehyde

To 3-{quinolin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)-5-vinylpyrazolo[1 ,5- a]pyrimidin-7-amine (0.7 g, 1.3 mmol) in 1 ,4-doixane (10 ml_) was added 2.5 wt% OsO4 in 1 ,4-dioxane (1.0 g, 0.02 mmoi), 2,6-lutidine (0.6 ml_, 5.2 mmol) and H2O (2 mL), and the r esulting mixture was stirred at room temperature for 20 minutes. NaIO4 (0.8 g, 3.81 mmol) was then added and the reaction mixture was stirred at room temperature for 16 hours. LC/MS analysis at that time showed the diol intermediate still present. Therefore, more NaIO4 (0.3 g, 1 ,3 mmol) was added, and the reaction mixture was stirred for 12 hours for fuil conversion to the product. Saturated Na2S2Oa solution (10 mL) was added and the mixture was stirred for 10 minutes. Organics were then extracted with CH2CI2 (4 x 40 mL), dried (Na2SO4) and concentrated in vacuo to give 7-(bis((2-(trimethy!silyl)ethoxy)methyl)amino)-3-(quinolin-3-yi)pyrazolo[1 ,5- a]pyrimidine-5-carbaldehyde as a yellow solid (0.6 g, 1.2 mmol, 92%). LCMS: 2.67 mins, m/z = 550.0 (MH+).
Synthesis of 5-(morpholinomethyl)-3-(Quinolin-3-vS)-N, N-bis((2-(trimethylsilvh- ethoxy)methyl)pyrazolo[1 ,5-aipyrimidin-7-amine

(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin-7-amine as a brown oil (0.2 g). LCMS: 2.01 mins, m/z = 621.3 (MH+).
Synthesis of 5-(morphoJinomethyD-3-(αuinolin-3-vS)pyrazolori ,5-alpyrimidin-7-amine

To crude 5-(morpholinomethyl)-3-(quinoiin-3-yl)-N,N-bis((2-(trimethylsilyl)- ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin~7-amine (0.2 g) in a 20 mL vial was added EtOH (4 mL) and then 4M HCI/dioxane (1.5 mL, 6.0 mmol). The vial was capped and heated at 85 °C for 16 hours. On cooling, saturated NaHCO3 solution (~5 mL) was added, and the reaction mixture was stirred for 30 minutes. The reaction mixture was then transferred to a separatory funnel using CH2Cl2 (20 mL) and H2O (20 mL) and saturated NaHCO3 solution (20 mL) and brine (20 mL) were added. Organics were extracted with CH2CI2 (4 x 30 mL), dried (Na2SO4) and concentrated in vacuo to give crude 5-(morpholinomethyl)-3-(quinolin-3-yl) pyrazolo [1, 5-a] pyrimidin-7-amine as a brown solid (0.04 g). LCMS: 0.78 mins, m/z = 361.1 (MH+).
Synthesis of 6-bromo-5-(morphoiinomethyl)-3-(quinolin-3-yl)pyrazolo[1 ,5-aipyrimidin- 7-amine

To crude 5-(morpholinomethyl)-3-(quinolin-3-yl) pyrazolo[1 ,5-a]pyrimidin-7-amine (44 mg) in CH3CN {8 mL) was added N-bromosuccinimide (22 mg, 0.12 mmol), followed by additional CH3CN (4 mL). The reaction was then stirred at room temperature for 30 minutes, at which time LC/MS analysis confirmed full conversion of the starting material to the product. The reaction mixture was concentrated in vacuo and purified by preparative HPLC to give 6-bromo-5-(morpholinomethyl)-3-(quinolin-3- yi)pyrazolo[1 ,5-a]pyrimidin-7-amine as a yellow solid (8.7 mg, 0.02 mmol, 7% over three steps). LCMS: 0.82 mins, m/z = 439.0 and 441.0 (MH+).
Related reductive amination analogs were prepared as described in Preparative Example included in the following table. In most cases, reduction of the aldehyde to the primary alcohol was a competing reaction to the desired reductive amination. Generally, reactions giving >80% reductive amination product progressed without purification whereas those with >20 % primary alcohol were purified by silica gel column chromatography before continuing to the de-SEM reaction.
By essentially the same procedure given in Scheme 44, the compounds listed in Table 13 can be prepared.
Table 13















Synthesis of 6-bromo-5-ff(2R.6SV2.6-dimethyltetrahydro-2H-Dyran-4- ylamino)methyl)3-(6-fluoroQuinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-7-amine:
Part A


To 2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4-amine (crude) in CH3CN (3.2ml) was added
N,N-diisopropylethylamine (0.9ml) followed by di-tørf-butyl dicarbonate (1.9g) and the resulting mixture was stirred at room temperature for Ih, at which time LC/MS confirmed full conversion of starting material to product. Solvent was removed in vacuo and the crude was redissoived in DCM (20ml), washed with water (1 x 5ml), brine (1 x 5ml), dried over MgSO4. Solvent was removed in vacuo to give tert-butyl (2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4- ylcarbamate as a crude product which was used for the next step without any purification.
Part C:

To tert-butyl (2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4-ylcarbamate (crude, 0.6g) was added 4M HCl in 1,4-dixoane (15ml) at r.t. It was stirred further at room temperature for 2 h, at which time LC/MS analysis confirmed full consumption of starting material. Solvent was removed in vacuo to get the desired (2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4-amine as an HCl salt.
Part D:

To crude (2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4-amine (O.lδg) in DCE (4.2ml) and Ethanol (4.2ml) was added 7-(bis((2-(trimethylsilyI)ethoxy)methyl)amino)-6-bromo-3-(6- fluoroquinolin-3-yl)pyrazolo[l,5-a]pyrimidine-5-carbaldehyde (0.19g, 0.3mmol) followed by AcOH (0.03ml, O.όmrøol). After stirring for 30 minutes at room temperature, NaBH3CN
(37.8mg, O.όmmol) was added and stirring was continued further for 30 minutes more. LC/MS showed no starting material remaining. After the solvent was rotoevaporated, the crude was dissolved with DCM (20ml), washed with sat. NaHCO3 (1 x 5ml), water (1 x 5ml), brine (1 x 5ml), dried over MgSO4. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-60%) gave desired 6- bromo-5-(((2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4-ylamino)methyl)-3-(6-fluoroquinolin- 3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methy[)pyrazolo[ 1 ,5-a]pyrimidin-7-amine (68 mg, 30%).
Part E
6-bromo-5-(((2R,6S)-2.6-dimethyltetrahyclro-2H-Dyran-4-ylamino)methyπ-3-(6- fluoroquinolin-3-yl)pyrazoloπ,5-a1pyrimidin-7-amine:

To a 6-bromo-5-(((2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4-ylamino)methyl)-3-(6- fluoroquinolin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)niethyl)pyrazolo[1,5-a]pyrimidin-7- amine (69.3mg, 0.09mmol) in TFA (2ml) was added few drops of water and stirring continued for 2h at room temperature. LC/MS showed no starting material remaining. TFA along with water was rotoevaporated. This crude compound was submitted to the analytical group for purification to afford the desired 6-brorno-5-(((2R,6S)-2,6-dimethyltetrahydro-2H-pyran-4- ylamino)methyl)-3-(6-fluoroquinolin-3-yl)pyrazolo[1,5-a]pyrimidin-7-amine. LCMS: 3.157 mins, m/z = 499.0 (MH+).
Synthesis of 6-bromo-3-(6-fluoroquinolin-3-yl)-5-((3-methoxytetrahydro-2H-pyran-4- ylamino)methyθpyrazolori ,5-aipyrimidin-7-amine:
M
 SCHEME- 45
SCHEME- 45
Above compound was prepared by essentially the same procedure (1) as to compound 7 is prepared. However exact configuration has not been determined. Isomer 1: LCMS: 1.942 mins, m/z = 501.1 (MH+) Isomer 2: LCMS: 2.706 mins, m/z = 501.0 (MH+)
Synthesis of 6-bromo-3-(6-fluoroquinolin-3-ylV5-((1 -
(methylsulfonylmethyl)cvclopropylamino)methyπpyrazolof1.5-aipyrinnidin-7-amiπe:

SCHEME-46
/nthesis of ethyl 1 -(tert-butoxycarbonylamino)cvclopropanecarboxylate:
To ethyl 1-aminocyclopropane-1-carboxylic acid ethyl ester hydrochloride (7.9g, 48mmol) in DCM (400ml) was added N,N-diisopropylethylamine (21.7ml, 124.7mmol) followed by di- tert-butyl dicarbonate (1 l.5g, 52.8mmoi) at O°C, and the resulting mixture was stirred at room temperature for 16h, at which time LC/MS confirmed full conversion of starting material to product. Reaction mixture was transferred in to the seperatory funnel and washed with water (1
x 75ml), brine (1 x 75ml), dried over MgSO4. Solvent was removed in vacuo to get desired ethyl l-(tert-butoxycarbonylamino)cyclopropanecarboxylate as a crude product which was used for the next step without any purification.
Synthesis of tert-butyl Hhydroxymethyπcvclopropylcarbarnate:
While under argon atmosphere, LiBH4 (0.73g, 33.6mmol) was suspended in Et2O (18ml). To this mixture was added dropwise a solution of ethyl 1 -(tert- butoxycarbonylamino)cyclopropane carboxylate (5g, 21.8mmol) in Et2O (18ml). The reaction mixture was stirred at room temperature for 2 h and then quenched by slow addition of MeOH (18ml), After being stirred overnight, the reaction mixture was poured into an equal volume of sat. NH4Cl (54ml). The Et2O layer was separated, and the aqueous layer was extracted four times with Et2O. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (50-100%) gave desired tert-butyl 1 -(hydroxymethyl)cyclopropyl carbamate as a white solid (3.14g, 77%).
Synthesis of (1 -(tert-butoxycarbonylamino^cvclopropyl)methylmethanesulfonate:
To tert-butyl l-OαydroxymethyOcyclopropylcarbamate (1.Ig, 5.83mmo!) in DCM (47ml) was added N,N-diisopropylethylamine (1.3ml, 7.67mmol) followed by methanesulfonylchloride (0.5ml, 6.4mmol) at 0°C, and the resulting mixture was stirred at room temperature for 16h, at which time LC/MS confirmed full conversion of starting material to product. Reaction mixture was transferred in to the seperatory funnel and washed with sat. NaHCOj (1 x 10ml), water (1 x 10ml), brine (1 x 10ml), dried over MgSO4. Solvent was removed in vacuo to get desired (1- (tert-butoxycarbonylamino)cyclopropyl)methylmethanesulfonate as a crude product ( 1.38g) which was used for the next step without any purification.
Synthesis of tert-butyl i-(methylthiomethyDcvciopropylcarbamate:
A mixture of (l-(tert-butoxycarbonylamino)cyc[opropyl)methy!methanesulfonate (1.38g,
5.2mmol), and NaSCH3 (0.48g, 6.8mmol) in DMF (21ml) was stireed at room temperature for 2 h, at which time LC/MS confirmed full conversion of starting material to product (No
Starting material is present). Reaction mixture was diluted with EtOAc (250ml), and. washed with water (3 x 60ml), brine (I x 60ml) and dried over MgSO4. Solvent was removed to give the tert-butyl l-(methylthiomethyl)cyclopropylcarbamate as a crude product which was used for the next step without any purification.
Synthesis of tert-butyl i -(methyisulfonylmethyDcvciopropylcarbamate:
A mixture of tert-butyl l-tmethylthiomethyDcyclopropylcarbamate (crude), Oxone (4.8g, 7.8mmol) and NaHCO3 (2.6g, 3 L3mmol) in MeOH (28ml) and Water (7ml) was stirred at room temperature for 1.5 h, at which time LC/MS analysis confirmed full consumption of starting material. Solvent was removed in vacuo and the crude was redissolved in EtOAc (100ml), washed with water (1 x 20ml), brine (I x 20ml), dried over MgSO4. Solvent was removed in vacuo to give tert-butyl l-(methylsulfonylmethyl) cyclopropy [carbamate as a crude product (white solid, l.lg) which was used for the next step without any purification.
Synthesis of i-(methylsulfonylmethyl)cyclopropanamine:
To tert-butyl l-(methylsulfonylmethyl)cyclopropyicarbamate_(crυde, 1.05g) in DCM (7ml) was added 4M HCl in 1,4-dixoane (16ml) at r.t. It was stirred further at room temperature for 1 h, at which time LC/MS analysis confirmed full consumption of starting material. Solvent was removed in vacuo and the solid was washed with ether to get the desired 1 - (methytsulfonylmethyl)cyclopropanarnine as an HCl salt.
Synthesis of 6-bromo-3-(6-fluoroquinolin-3-yl)-5-(f 1 -(methyisulfonylmethyhcvclo propylamino)methyl)pyrazolori,5-aipyrimidin-7-amine:
6-bromo-3-(6-fluoroquinolin-3-yI)-5-((l-(methylsulfonylmethyl)cyclo propylamino)methyl)pyrazolo[1,5-a]pyrimidin-7-amine was prepared by essentially the same procedure (1) as to compound 7 is prepared. LCMS: 2.52 mins, m/z = 519.0 (MH+).
Synthesis of (1 -((7-(bis(f2-(trinnethylsilyl)ethoxy)methyπamino)-6-bromo-3-(6- fiuoroQuinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)methylamino)cyclopropyl)methanoi:

To l-aminocyclopropyl)methanol (0.22g, 2.5mmol) in DCE (7ml) and Ethanol (7ml) was added -(bis{(2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-fluoroquinolin-3- yl)pyrazoIo[1,5-a]pyrimidine-5-carbaldehyde (0.32g, O.Smmol) followed by AcOH (0.064ml, Immol). After stirring for 30 minutes at room temperature, NaBH3CN (63mg, Immol) was added and stirring was continued further for 30 minutes more. After the solvent was rotoevaporated, the crude was dissolved with DCM (40ml), washed with sat. NaHCO3 (I x 10ml), water (1 x 10ml), brine (1 x 10ml), dried over MgSO^ Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-100%) gave slightly impure desired (1 -((7 -(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-fluoroquinolin-3-yOpyrazolo[1,5- a]pyrimidin-5-yl)methylamino)cyclopropyl)methanol (320mg).
Synthesis of tert-butyl (7-(bis((2-(trimethylsilyl)ethoχy) methyl)amino)-6-bromo-3-(6- fluoroquinolin-3-yl)pyrazolori 3-a1pyrimidin-5-vOmethyl(1 - (hydroxymethyDcvciopropyl)carbamate:

To ( 1 -((7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-6-bromo-3-(6-fluoroquinolin-3- yl)pyrazolo[1,5-a]pyrimidin-5-yl)methylamino)cyclopropyl)methanol (0.17g, 0.24mmol) in dichloroethane (2ml) was added N,N-diisoproρylethyIamine (0.09ml, 0.49mmoi) followed by di-tert-butyl dicarbonate (O.lόg, 0.73mmol) and the resulting mixture was stirred at room
temperature for 16 h, at which time LC/MS confirmed full conversion of starting material to product. The mixture was diluted with DCM (20ml), washed with water (1 x 5ml), brine (1 x 5ml), dried over MgSO4. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-60%) gave desired tert- butyl (7-(bis((2-(trimethylsilyl)ethoxy) methyl)amino)-6-bromo-3-(6-fluoroquinolin-3- yl)pyrazolo[1,5-a]pyrimidin-5-yl)metliyl(l-(hydroxymethyI)cyclopropyl)carbamate (O.l9g, 95%).


Synthesis of 4-(methoxymethy0tetrahydro-2H-pyran-4-amine:
Synthesis of benzyl 4-(methoxymethyl)tetrahydro-2H-pyran-4-ylcarbamate:

To benzyl 4-(hydroxymethyl)tetrahydro-2H-pyran-4-ylcarbamate (0.59g, 2.23mraol) in DCM (24ml) was added protonsponge (0.53g, 2.5mmol) followed by trimethyloxoniumtetrafluoroborate (0.36g, 2.5mmol) at 0°C. After 20min at 0°C, reaction mixture was warmed up to room temperature and stirred further for 16 h, at which time LC/MS confirmed full conversion of starting material to product. Reaction mixture was quenched with sat. NH4Cl and with DCM (75ml). Organics were separated and washed with brine (20ml) and dried over MgSO4. Solvent was removed in vacuo and the residue was was purified by column chromatography on silica gel. Eiution with EtOAc/Hexanes (66%-i00%) gave slightly impure desired benzyl 4-(methoxymethyl)tetrahydro-2H-pyran-4-ylcarbamate (0.3 Ig, 50%).
Synthesis of 4-(methoxymethyDtetrahydro-2H-pyran-4-amine:

A mixture of benzyl 4-(methoxymethyl)tetrahydro-2H-pyran-4-ylcarbamate (0.86g, 3.1mmol), 10% Pd/C (0.2I g) in EtOAc (25ml) was stirred at room temperature under hydrogen (balloon pressure) for 16 hours. It was then filtered and concentrated to get the desired 4- (m ethoxy methy l)tetrahy dro-2H-py ran-4-am ine.

By essentially the same procedure as to compounds above is prepared. LCMS: 2.173 mins, m/z = 515.2 (MH+).
Synthesis of 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazolof 1 ,5-a]pyrimidine-5- carbonitrile:

5-chloro-N,N-bis((2-(trimethylsiIyl)ethoxy)methyl)pyrazolo[i,5-a]pyrimidin-7-arnine (1.5 g, 3.5 mmol), tributyltin cyanide (1.7 g, 5.3 mmol), tetrakis (triphenylphosphine) palladium (0.8 g, 0.7 mmol), bis(tri-^-butylphosphine) palladium (0.4 g, 0.7 mmol) were charged in a pressure tube. The tube was evacuated and charged with argon for three cycles. Dioxane (20 ml) was added, and the tube was capped and heated at 150°C with stirring for one hour. After cooling, the mixture was diluted with EtOAc (100 ml) and washed with brine (1 x 20 ml), dried over MgSO4. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with EtOAc/Hexanes (0-35%) gave desired product, 7- (bis((2-(trimethylsilyl) ethoxy)methyl)amino) pyrazolo[ l,5-a]pyrimidine-5-carbonitrile (1.3 g, 90%).
Synthesis of 5-π-aminoethyl)-N,N-bis((2~(trimethyisilyl)ethoxy)methyl) pyrazoloM ,5- a]pyrimidin-7-amine:

To a solution 7-(bis((2-(tiimethylsilyl)ethoxy)methyi)amino)pyrazolo[l ,5-a]pyrimidine-5- carbonitrile (0.6 g, i .4 mmol), copper bromide (42 mg, 0.3 mmol), and TBSCl (0.2 g, 1.4 mmol) in THF (14 m!) at O°C was added methyl magnesium bromide (3M in ether, 0.47 ml, 1.4 mmol) dropwise. Reaction mixture was warmed up to room temperature and stirred further for 6 h. LC/MS showed no starting material (M.W = 419) remaining. A borane.THF solution (IM in THF, 2.8 ml, 2.8 mmol) was then added to the reaction mixture at -78°C, and the resulting solution was stirred for 4 h and allowed to warm at room temperature overnight. Distilled water (0.5 ml) was cautiously added to the solution cooled at -20°C, followed by sat. NH4Cl (0.5 ml). After the THF was rotoevaporated, the aqueous phase was extracted with DCM (60 ml), washed with water (1 x 5 ml), brine (1 x 5 ml), dried over MgSO4. Solvent was removed in vacuo and the crude was used for the next step without any further purification.
Alternatively, the 5-(l-aminoethyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl) pyrazolo[l,5- a]pyrimidin-7-amine can also be synthesized by the following procedure.
Synthesis of 5-(1 -aminoethyl)-N,N-bis(f2-(trimethylsilyl)ethoxy)methyl) pyrazolof 1 ,5- alpyrimidin-7-amine can also be synthesized by the following procedure:
MeMgBr

Part A Synthesis of 1 -(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazoloM .5-aipyrimidin-
5-vOethanone:
An oven-dried 250 rriL 3-neck round-bottom flask fitted with a pressure-equalizing addition funnel, temperature probe and septum was charged with 7-(bis{[2- (trimemyisilyI)ethoxy]methyl}amino)pyrazolo[l,5-a]pyrimidine-5-carbonitrile (7.10 g, 16.9 mmol) in dry THF (59 niL), copper© bromide (120 mg, 0.84 mmol) and tert- butyldimethylsilyl chloride (2.80 g, 18.6 mmoi), and the stirred mixture was cooled in an ice- brine bath. 3M MeMgBr in ether (9.59 mL, 28.8 mmol) was added dropwise over 10 min via the addition funnel, and the resulting heterogeneous mixture was stirred at -5-0 °C for 1.5 h and quenched carefully by the dropwise addition of water until gas evolution ceased. The mixture was diluted with additional water (100 mL) and extracted with diethyl ether (2 x 150 mL), and the combined organic phase was washed with brine (100 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification by chromatography on silica gel (120 g), eluting with 0-20% EtOAc/hexanes, afforded the title compound, l-(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)pyrazoIo[1,5-a]pyrimidin-5-yl)ethanone (4.34 g, 59% yield) as a light yellow oil: MS (ESI+) for C20H36N4O3Si2 m/z 437 (M+H)+; R/= 0.70 (25% EtOAc/hexanes).
Part B Synthesis of 1 -(7-(bis((2-(trimethylsilyl)ethoxy)methy0amino)pyrazolof 1 ,5-aipyrimidin- 5-yl)ethanone oxime:
A solution of l-[7-(bis{[2-(trimethylsilyl)ethoxy]methyl }amino)pyrazolo[ 1 ,5-a]pyrimidin-5- yi]ethanone (4.34 g, 9.94 mmol) in 1:1 EtOH/pyridine (50 mL) was treated with hydroxyiamine hydrochloride (3.45 g, 49.7 mmol). The resulting mixture was stirred at room temperature for 1.5 h and then concentrated under reduced pressure to remove most solvent. The residue was partitioned between EtOAc (100 mL) and water (60 mL), the layers were separated, and the organic phase was washed with water (60 mL) and brine (20 mL), dried over Na2SO4 and concentrated under reduced pressure to give the title compound, l-(7-(bis((2- (trimethylsilyl)ethoxy)methyI)amino)pyrazolo[l,5-alpyrimidin-5-yl)ethanone oxime (4.90 g, 85% purity, 93% yield) as an off-white semisolid: MS (ESI+) for C20H37N5O3Si2 m/z 452 (M+H)+.
Part C Synthesis of 5-(1-aminoethyi)-N.N-bisf(2-(trimethylsilyS)ethoxy)methyl)pyrazolori ,5- aipyrimidin-7-amine:
: A solution of l-[7-(bis{[2-(trimethylsily0ethoxy]methyl}amino)pyrazolo[l,5-a]pyrimidin-5- yl]ethanone oxime (4,50 g, 8.77 mmol) in 4: 1 EtOH/ AcOH (90 mL) under nitrogen in a Parr bottle was treated with 10% Pd on carbon (933 mg, 0.877 mmol), and the mixture was shaken on the Parr apparatus under 45 psi hydrogen for 23 h. The catalyst was removed by filtration through CeI ite, and the filtrate was concentrated under reduced pressure, azeotroping with toluene at 40-45 °C to remove most AcOH. The oily residue was taken up in EtOAc (-100 mL), washed with saturated aqueous NaHCO3 (2 x 50 mL) and brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure to give the title compound, 5-(l-
aminoethyl)-N,N-bis((2-(tximethylsilyl)ethoxy)methyl)pyrazolo[l ,5-a]pyrimidin-7-amine (4.02 g, 91% purity, 95% yield) as an oil: MS (ESI+) for C20H39N5O2Si2 m/z 438 (M+H)+.
Synthesis of 5-(1 -aminoethyl)-3-(6-fiuoroquinolin-3-yl)-N,N-bis((2- (trimethylsiM)ethoxy)methyθpyrazolo[1 ,5~a1pyrimidin-7-amine:

Part A
Synthesis of 1 -(7-(bis(f2-(trimethylsiiyl)ethoxy)methyl)annino)-3-(6-fluoroauinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)ethanol:
To a solution of 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3- yl)pyrazolo[1,5-a]pyrimidine-5-carbaidehyde (11.0 g, 19.4 mmol) in anhydrous THF (200 mL, 2 mol) under nitrogen at 0 °C was added 3M methylmagnesium bromide in ether (9.69 mL, 29.0 mmol) dropwise. After 1 hour the reaction was complete by HPLC. Water (10 mL) was added over a 15 minute period to quench the reaction. Removed the flask from the ice bath and concentrated in vacuo. Dissolved the remaining residue in ethyl acetate, washed three times with water and then twice with brine. The organics were dried over sodium sulfate, filtered and concentrated in vacuo. Purified the remaining crude material by flash chromatography (5-40% EtOAc/hex) to afford l-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3- yl)pyrazolo[l ,5-a]pyrimidin-5-yl)ethanol (6.8 g; 60% yield) as a yellow solid. HPLC: 7.54 min (Method C). LCMS: ESI+ m/z of 584 (M+H)
Part B
Synthesis of 1 -(7-( bis((2-(trimethylsilyl)ethoxy)methyi)amino)-3-(6-f luoroquinoiin-3- yl)pyrazolofi ,5-a1pyrimidin-5-yl)ethyl methanesulfonate:
To a solution of l-(7-(bis((2-(trimethylsilyl)ethoxy)methy!)amino)-3-(6-fIuoroquinolin-3- yl)pyrazolo[1,5-a]pyrimidin-5-yl)ethanol (6.1 g, 10.4 mmol) in methylene chloride (100 mL) and triethytamine (7.1 mL, 50.9 mmol) under nitrogen was added methanesulfonyl chloride (2.4 mL, 31.0 mmol) dropwise. After 15 minutes the reaction was complete by HPLC. The
contents of the flask were transferred to a separatory funnel, washed twice with water and twice with brine. The organics were dried over sodium sulfate, filtered and concentrated in vacuo to afford l-(7-(bis((2-(trimethyIsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3- yl)pyrazoIo[1,5-a]pyrimidin-5-yl)ethyl methanesulfonate (6.8 g; 98% yield) as a dark yellow oil that was used as is for the following step. HPLC: 6.18 min (Method D). LCMS: ESI+ m/z of 662 (M+H)
Part C
Synthesis of 5-( 1 -azidoethyl)-3-(6-fluoroquinolin-3-yl)-N,N-bis((2- (trimethylsiiyl)ethoxy)methy0pyrazolof1 ,5-a1pyrimidin-7-amine:
To a solution of l-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinoiin-3- y!)pyrazolo[1,5-a]pyrimidin-5-yl)ethyl methanesulfonate (6.8 g, 10.3 mmol) in DMSO (100 mL) and N,N-diisopropyl-ethylamine (8.95 mL, 51.4 mmol) was added sodium azide (2.0 g, 30.8 mmol). After 4 hours the reaction was complete by HPLC. The contents of the flask were transferred to a separatory funnel, washed twice with water and twice with brine. The organics were dried over sodium sulfate, filtered and concentrated in vacuo to afford 5-(l -azidoethyl)-3- (6-fluoroquinolin-3-yl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7- amine (6.2 g; 99% yield) as a dark yellow oil. HPLC: 6.67 min (Method D). LCMS: No ion detected.
Part D
Synthesis of 5-(1 -aminoethyπ-3-(6-fluoroαuinolin-3-yl)-N,N-bisf (2- ftrimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-alpyrimidin-7-amine:
To a solution of 5-(l-azidoethyl)-3-(6-fluoroquinolin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (6.2 g, 10.3 mmol) and THF (90 mL) was added IM trimethylphosphine in THF (30.8 mL, 30.8 mmol) dropwise. After 30 minutes the reaction was complete by HPLC. Water (30 mL) was therefore added and the reaction stirred an additional hour. The contents of the flask were transferred to a separatory funnel, diluted with 300 mL of EtOAc and washed three times with water and three times with brine. The organics were dried over sodium sulfate, filtered and concentrated in vacuo. Purified the remaining crude material by flash chromatography (0-2% 0.7N NH3 in MeOH/CHC13) to afford 5-(l -aminoethyl)-3-(6-fluoroquinolin-3-yl)-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (4.9 g, 82% yield) as a yellow solid. HPLC: 6.62 min (Method C). LCMS:ESI+ m/z of 583 (M+H)
Synthesis of 5-( 1 -(tetrahydro-2H-Dyran-4-ylarπino)ethyπ-N.N-bis(f2-(trimethy!silyl) ethoxy)methyl)pyrazoloπ .5-aipyrimidin-7-amine:
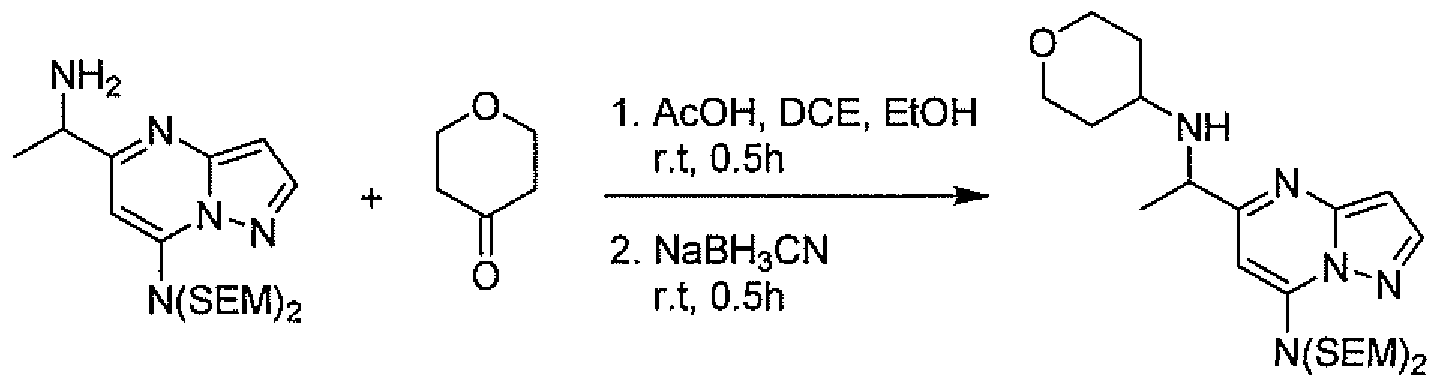
To crude 5-(l-aminoethyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5- a]pyrimidin-7-amine (0.6 g) in DCE (20 ml) and Ethanol (20 ml) was added tetrahydro-4H- pyran-4-one (0.6 ml, 7 mmol) followed by AcOH (0.08 ml, 1.4 mmol). After stirring for 30 minutes at room temperature, NaBH3CN (0.18 g, 2.8mmoi) was added and stirring was continued further for 30 minutes more. LC/MS showed no starting material (M.W = 437) remaining. After the solvent was rotoevaporated, the crude was dissolved with DCM (60ml), washed with sat. NaHCO3 (1 x 5ml), water (1 x 5ml), brine (1 x 5ml), dried over MgSO4. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Eiution with MeOH/EtOAC (0-20%) gave desired product, 5-(l -(tetrahydro-2H- pyran-4-ylamino)ethyl)-N,N-bis((2-(trimethylsilyl) ethoxy)methyl) pyrazolo[l ,5-a]pyrimidin- 7-amine (0.2 g, 27%).
Synthesis of 3-iodo-5-(1-(tetrahydro-2H-pyran-4-ylamino)ethyl)-N,N-bis((2- (trimethylsiivhethoxy)methyl)pyrazolof1 ,5-a]pyrimidin-7-amine:

To a solution of 5-(l-(tetrahydro-2H-pyran-4-ylamino)ethyl)-N,N-bis((2-(trimethylsiIy[) ethoxy) methyl) pyrazolo[1,5-a]pyrimidin-7-amine (0.17g, 0.32mmol) in acetonitrile (15 ml) at 0°C was added NIS (78mg, 0.35 mmol) and stirring continued for 10 minutes. LC/MS showed no starting material (M.W = 521) remaining. Saturated sodium thiosulfate solution (~0.5ml) was added and stirring continued for 5 minutes. After the acetonitrile was rotoevaporated, the
aqueous phase was extracted with DCM (25ml), washed with water (I x 5ml), brine ( I x 5ml), dried over MgSO4. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with MeOH/EtOAC (0-20%) gave desired product, 3- iodo-5-(l-(tetrahydro-2H-pyran-4-ylamino)ethyl)-N,N-bis((2- (trimethylsilyl)ethoxy)methy[)pyrazolo[ 1 ,5-a]pyrimidin-7-amine (0.14g, 69%).
Synthesis of 3-(6-fluoroQuinolin-3-yl)-5-(1-(tetrahydro-2H-pyran-4-ylamino)ethyh-N .N- bis(f2-(trimethylsilyl)ethoxy)methyl)pyrazolori ,5-aipyrimidin-7-amine:

To 3-iodo-5-( 1 -(tetrahydro-2H-pyran-4-ylamino)ethyl)-N,N-bis((2-(trimethylsilyl) ethoxy)methyl)pyrazolo[1,5-a]pyrimidin-7-amine (0.14g, 0.21mmol) was added 6- fluoroquinoline boronate (0.1 Ig, 0.42mmol), K2CO3 (87mg, 0.63mmol), PdC!2(dppf).CH2Cl2 (18mg, 0.02mmoi), dioxane (4ml) and H2O (ImI). The resulting mixture was heated at 100 °C for 2 hours, at which time LC/MS analysis confirmed full consumption of starting material. On cooling, the solvent was rotoevaporated, and the crude was redissoived in DCM (40ml), washed with water (1 x 5ml), brine (i x 5ml), dried over MgSO4. Solvent was removed in vacuo and the residue was purified by column chromatography on silica gel. Elution with MeOH/EtOAC (0-20%) gave desired product, 3-(6-fluoroquinolin-3-yl)-5-( 1 -(tetrahydro-2H- pyran-4-ylamino)ethyl)-N,N-bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[l,5-a]pyrimidin-7- amine (84mg, 60%).
Synthesis of 3-(6-f luoroquinolin-3-vh-5-(1 -(tetrahydro-2H-pyran-4- ylamino)ethyl)pyrazolof1 ,5-alpyrimidin-7-amine:

To a solution of 3-(6-fiuoroquinolin-3-yl)-5-(l-(tetrahydro-2H-pyran-4-ylamino)ethyl)-N,N- bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[ l,5-a]pyrimidin-7-amine (84 mg, 0.13mmol) in TFA (2 ml) was added few drops of water and stirring continued for 2h at room temperature. LC/MS showed no starting material (M. W = 666) remaining. TFA along with water was rotoevaporated, and the crude was dried under the high vacuum for 4 h, which was used without further purification for the next step.
Synthesis of 6-bromo-3-(6-fluoroQuinolin-3-yl)-5-(1 -(tetrahydro-2H-pyran-4- ylamino)ethyl)pyrazolo[1 ,5-aipyrimidin-7-amine:

To a solution of 3-(6-fluoroquinolin-3-y!)-5-(l-(tetrahydro-2H-pyran-4- ylamino)ethyl)pyrazolo[1,5-a]pyrimidin-7-amine (Crude, ~0.13mmol) in DMF (2ml) at room temperature was added NBS (25mg, 0.14mmol) and stirring continued for 10 minutes. LC/MS showed no starting material (M.W = 406) remaining. This crude compound was submitted to the analytical group for purification to afford the desired 6-bromo-3-(6-fluoroquinolin-3-yl)-5- (l-(tetrahydro-2H-pyran-4-yIamino)ethyi)pyrazolo[1,5-a]pyrimidin-7-amine.
Synthesis of H4-aminopiperidin-1-yl)-2-methoxyethanone:

Methyl chloroformate (289 μl_, 3.74 mmol) was added to a solution of 4-(N-Boc- amino)-piperidine (500 mg, 2.50 mmol) in methylene chloride (25 mL) at room temperature. N,N-diisopropylethy!amine (1.30 mL, 7.49 mmol) was added and the solution was stirred overnight. The reaction mixture was diluted with sat. ammonium chloride (30 mL) and extracted with methylene chloride (3 x 30 mL). The combined extracts were dried over sodium sulfate and concentrated. The crude material was purified by column chromatography on silica gel (0% to 100% ethyl acetate in methylene chloride) to give 566 mg of a white soiid. This solid was taken up in methylene chloride (2 mL) and trifluoroacetic acid (2 mL) was added at room temperature. After 20 min, the solution was concentrated, and then diluted with methylene chloride (2 mL). The solution was basified with sat. sodium carbonate (2 mL) and the mixture was extrated with methylene chloride (5 x 5 mL) and ethyl acetate (5 x 5 mL). The combined extracts were concentrated and the resulting soiid was freeze dried from acetonitrile and water to give 290 mg of the title compound as a white solid, 1 -{4-aminopiperidin-1-yl)-2-methoxyethanone, (73% over two steps). NMR (300 MHz, D2O) δ 4.79 (s, 2H), 4.12 (d, J= 13.5 Hz, 2H), 3.72 (s, 3H), 3.28-3.14 (m, 1 H), 2.95 (t, J = 12.6 Hz1 2H), 1.98 (d, J = 12.0 Hz1 2H)1 1.54-1.36 (m, 2H).
Synthesis of 1-(4-aminopiperidin-1-yl)-3-methoxypropan-1-one:

3-Methoxypropanoic acid (225 μL, 2.40 mmol) was added to a solution of 4-(N-Boc- amino)-piperidine (400 mg, 2.00 mmol) in methylene chloride (25 mL) at room temperature. N,N-dsisopropylethylamine (1.30 mL, 7.49 mmol) and propylphosphonic anhydride (1.78 mL, 50 wt. % in ethyl acetate, 3.00 mmol) was added, and the solution was stirred for 4.5 h. The reaction mixture was concentrated and partitioned between ethyl acetate (5 mL) and water (5 mL). The organic portion was washed with 0.2M HCl (2 mL), sat. Sodium bicarbonate (5 mL) and brine (5 mL). The solution was then dried over sodium sulfate and concentrated to a white solid. This solid was taken up in methylene chloride (6 mL), and trifluoroacetic acid (3 mL) was added at room temperature. After 1 h the solution was concentrated. The resulting solid was dissolved in methylene chloride (2 mL) and 1 M sodium hydroxide (1 mL). This mixture was extracted with methylene chloride (4 x 2 mL) and ethyl acetate (4 x 2 mL). The
combined organic extracts were dried over sodium sulfate and concentrated to give 135 mg of the title compound, 1 -(4-aminopiperidin-1-yl)-3-methoxypropan-1-one (36% over two steps). 1H NMR (300 MHz, CD3OD) δ 4.51-4.41 (m, 1H), 4.03-3.93 (m, 1 H), 3.64 (d, J = 6.3 Hz, 2H), 3.32 (s, 3H), 3.17-3.05 (m, 1 H), 2.96-2.83 (m, 1 H), 2.75-2.55 (m, 3H), 1.99-1.80 (m, 2H), 1.40-1.14 (m, 2H).
Synthesis of (R)-1-(4-aminopiperidin-1-yl)-2,3-dihydroxy-3-methyibutan-1 -one:

3,3-Dimethylacryioyl chloride (670 μl_, 5.99 mmol) was added to a solution of 4-(N- Boc-amino)-piperidine (1.00 g, 4.99 mmol) in methylene chloride (50 mi_) at room temperature. N,N-diisopropylethylamine (1.74 ml_, 9.99 mmol) was added and the solution was stirred overnight. The reaction mixture was diluted with sat. ammonium chloride (50 rriL) and extracted with methylene chloride (3 x 50 rriL). The combined extracts were dried over sodium sulfate and concentrated. The crude material was purified by column chromatography on silica gel (0% to 100% ethyl acetate in methylene chloride) to give 1.39 g of an off white solid. This solid was dissolved in t- butanol (18 mL) and water (18 ml_), and the solution was cooled to 0 °C. To this mixture were added AD-mix-α (7.50 g), methanesulfonamide (449 mg, 4.72 mmol), sodium bicarbonate (1.13 g, 13.5 mmol}, and potassium osmate dihydrate (15 mg? 0.0450 mmol). The solution was allowed to warm to room temperature overnight. A 1.9M solution of sodium sulfite (40 mL) was added and the mixture was stirred for 1 h. The resulting aqueous solution was extracted with ethyl acetate (4 x 10OmL) and dried over sodium sulfate. The solution was concentrated and the crude material was purified by column chromatography on silica gel (0% to 20% methanol in methylene chloride, and 0% to 10% methanol in methylene chloride) to give 670 mg of the diol. A portion of this diol (650 mg, 2.05 mmol) was dissolved in dioxane (10 mL) and HCl in dioxane (5.1 mL, 4M, 20.5 mmol) was added at room temperature. After stirring overnight, the reaction mixture was diluted with ether (20 mL) and filtered to give a white solid. This solid was taken up in methylene chloride (50 mL) and MP-carbonate resin (830 mg, 2.11 mmol) was added. After 1.5 h the solvent was decanted and the resin was washed with methylene chloride (20 mL). Methylene chloride (50 mL) was added to the resin along with additional fresh resin (1.0 g, 2.54 mmol). After stirring for 30 min, the solvent was decanted andthe remaining resin was washed with methylene chloride (20 mL). The combined organics were concentrated to 121 mg of the free amine, (R)-1 -(4-aminopiperidin-1-yl)-2,3-dihydroxy-3-methylbutan-1 -one (13% over three steps). 1H NMR (300 MHz, CD3OD) δ 4.54-4.40 (m, 1 H), 4.32-4.26 (m, 1 H)1 4.26-4.13 (m, 1 H), 3.16-3.03 (m, 1 H), 2.95-2.81 (m, 1 H), 2.80-2.69 (m, 1 H), 1.94-1.79 (m, 2H), 1.47-1.28 (m, 2H), 1.25 (d, J = 5.4 Hz, 3H), 1.17 (d, J = 5.1 Hz, 3H).
Synthesis of (R)-1 -(4-aminopiperidin-1-yl)-2,3-dihydroxypropan-1 -one:

N.N-diisopropylethylamine (2.54 mL, 14.6 mmol) was added to a solution of 4-(N-Boc- amino)-piperidine (1.75 g, 8.75 mmol) in tetrahydrofuran {36 mL) at O °C. Propylphosphonic anhydride (5.92 mL, 50 wt. % in ethyl acetate, 9.48 mmol) and acrylic acid (500 μL, 7.29 mmol) were added, and the solution was allowed to warm to room temperature overnight. The reaction mixture was partitioned between ethyl acetate (20 mL) and water (20 mL). The organic portion was washed with 0.2M HCI (5 mL), sat. sodium bicarbonate (20 mL), and brine (20 mL). The solution was then dried over sodium sulfate and concentrated to a white solid. A portion of this solid (800 mg, 3.15 mmol) was dissolved in f-butanol (13 mL) and water (13 mL) and cooled to 0 °C. To this mixture were added AD-mix-α (5.24 g), methanesulfonamide (314 mg, 3.30 mmol), sodium bicarbonate (793 mg, 9.44 mmol), and potassium osmate dihydrate (52 mg, 0.157 mmol). After 4.5 h, a 0.8M solution of sodium sulfite (100 mL) was added and the mixture was stirred for 1 h at room temperature. The resulting aqueous solution was extracted with ethyl acetate (4 x 10OmL) and dried over sodium sulfate. The solution was concentrated and the crude material was purified by column chromatography on silica gel (0% to 20% methanol in methylene chloride) to give a clear syrup. A portion of this syrup (790 mg, 2.74 mmol) was dissolved in dioxane (14 mL) and HCI in dioxane (6.8 mL, 4M, 27.4 mmol) was added at room temperature. After 18 h, the solution was concentrated. The resulting white solid was taken up in methylene chloride (50 mL) and MP-carbonate resin (3.2 g, 8.1 mmoi) was added. After stirring for 30 min, the solvent was decanted. The resin was washed with ethyl acetate (10 mL) and methylene chloride (10 mL), and the combined organics were discarded. The remaining resin was diluted with methanol (50 mL) and stirred for 30 min. The methanol was decanted and the resin was washed with methanol (10 mL). The combined methanol portions were concentrated to give clear oil, (R)-1 -(4- aminopiperidin-1-yl)-2,3-dihydroxypropan-1-one, 382 mg (60% over three steps). 1H NMR (300 MHz, CD3OD) δ 4.53-4.40 (m, 2H), 4.14-4.04 (m, 1 H), 3.77-3.56 (m, 2H), 3.20-3.05 (m, 1 H), 3.04-3.92 (m, 1 H), 2.84-2.69 (m, 1 H), 2.00-1.82 (m, 2H), 1.48-1.20 (m, 2H).
Synthesis of (4-aminopiperidin-1 -yl)(3-methyloxetan-3-yl)methanone:

A solution of 3-hydroxymethyI-3-methyloxetane (1.90 mL, 19.1 mmol) in methylene chloride (6 mL) was added to a solution of nickel (II) chloride hexahydrate (113 mg, 0.476 mmol) in water (2 mL). This mixture was cooled to 0 °C and bleach (127 mL) was added over a 30 min period via addition funnel. After 2 h the ice bath was removed and the reaction mixture was stirred at room temperature for an additional 2 h. 2M HCI (120 mL) was added and the solution was extracted with ether (4 x 200 mL). The combined extracts were dried over sodium sulfate and concentrated to a clear oil. A portion of this oil (1.00 g, 8.61 mmol) was dissolved in tetrahydrofuran (43 mL) and 4-(N-Boc-amino)-piperidine (2.07 g, 10.3 mmol) was added at room temperature. Propylphosphonic anhydride (6.99 mL, 50 wt. % in ethyl acetate, 11.2 mmol) and N,N-diisopropylethylamine (3.00 mL, 17.2 mmol) were added, and the reaction mixture was stirred for 24 h. The solution was partitioned between ethyl acetate (35 mL) and water (35 mL). The organic portion was washed with 0.2M HCI (10 mL), sat. sodium bicarbonate (35 mL), and brine (35 mL). The solution was then dried over sodium sulfate and concentrated. The crude material was purified by column chromatography on silica gel (0% to 100% methylene chloride in ethyl acetate). The resulting amide (213 mg, 0.714 mg) was diluted with methylene chloride (7 mL) and trifluoroacetic acid (1.8 mL) was added at room temperature. After 2 h the solution was concentrated and a 2M sodium hydroxide solution was added. The aqueous solution was extracted with ethyl acetate (3 x 10 mL). The combined extracts were dried over sodium sulfate and concetrated to give the title compound as a clear oil, (4-aminopiperidin-1-yl)(3-methyloxetan-3-yl)methanone, 68 mg (4% over three- steps). 1H NMR (300 MHz, CD3OD) δ 4.95-4.88 (m, 2H), 4.48-4.38 (m, 1 H), 4.38-4.31 (m, 2H), 3.19-2.96 (m, 2H), 2.94-2.81 (m, 1 H), 2.81-2.66 (m, 1 H), 1.93-1.81 (m, 2H), 1.67-1.60 Jm1 3H)1 1.36-1.15 (m, 2H).
Synthesis of (2R,3S)-1 -(4-aminopiperidin-1-yl)-2,3-dihydroxybutan-1~one:
1. crotyl chloride

3. HCI1 dioxane, rt
Crotyl chloride (674 μl_, 5.99 mmol) was added to a solution of 4-(N-Boc-amino)- piperidine (1.00 g, 4.99 mmol) in methylene chloride (50 ml_) at 0 °C. N, N- diisopropylethylamine (1.74 mL, 9.99 mmol) was added and the solution was stirred for 21 h. The reaction mixture was diluted with sat. ammonium chloride (50 mL) and extracted with methylene chloride (3 x 50 mL). The combined extracts were dried over sodium sulfate and concentrated. The residue was dissolved in methylene chloride (10 mL) and was triturated with hexanes (100 mL). The solvent was filtered off to give an off white solid. This solid was dissolved in f-butanoi (16 mL) and water (16 mL) and cooled to 0 °C. To this mixture were added AD-mix-α (6.83 g), methanesuifonamide (409 mg, 4.30 mmol), sodium bicarbonate (1.03 g, 12.3 mmol), and potassium osmate dihydrate (68 mg, 0.205 mmol). The solution was allowed to warm to room temperature overnight. A 0.5M solution of sodium sulfite (100 mL) was added and the mixture was stirred for 1 h. The resulting aqueous solution was extracted with ethyl acetate (4 x 10OmL) and dried over sodium sulfate. The solution was concentrated and the crude material was purified by column chromatography on silica gel (0% to 20% methanol in methylene chloride, and 0% to 20% methanol in methylene chloride) to give the diol as a white solid. A portion of the diol (450 mg, 1.49 mmol) was dissolved in dioxane (7 mL) and HCI in dioxane (3.7 mL, 4M, 27.4 mmol) was added at room temperature. After 1.5 h, the solution was concentrated. The resulting white solid was taken up in methylene chloride (30 mL) and MP-carbonate resin (2.0 g, 5.1 mmol) was added. After stirring for 45 min, the solvent was decanted, and the resin was washed with methylene chloride. The remaining resin was washed with methanol (2 x 30 mL) and the combined methanol portions were concentrated to give a clear oil, (2R,3S)-1 -(4-aminopiperidin-1-yl)-2,3-dihydroxybutan-1 -one, 160 mg (33% over three steps). 1H NMR (300 MHz, CD3OD) 54.51-4.35 (m, 1 H), 4.32-4.25 (m, 1H), 4.14-4.01 (m, 1H), 3.96-3.84 (m, 1 H), 3.19-3.02 (m, 1 H), 2.98-2.84 (m, 1 H), 2.84-2.67 (m, 1 H), 1.97-1.80 (m, 2H), 1.44-1.20 (m, 2H), 1.18 (s, 3H).
Synthesis of 1-(4-amino-4-methylpiperidin-1 -yl)ethanone:

Synthesis of 1 -(4-aminopiperidin-1 -yl)-3-hydroxy-3-methylbutan-1 -one:

3-hydroxy-3-methyl butyric acid (1.20 equiv), DIPEA (2.00 equiv), and T3P (1.50 equiv) were added to a stirring solution of compound 1 (1.00 equiv) in THF (0.2M) at room temperature under nitrogen. TLC analysis after 2 hours showed almost complete conversion, so reaction was concentrated. Residue was dissolved in ethyl acetate then washed with water, dilute HCI, saturated NaHCO3, and brine. The phases were separated and the organics dried over sodium sulfate, filtered, and concentrated. The resulting residue was purified by flash column chromatography (SiO2, 12 g; 0% to 10% methanol in methylene chloride). The resulting material was dissolved in methylene chloride (0.97M) and TFA (0.97M) was added under nitrogen and the mixture was stirred at room temperature for 1 hour at which time HPLC analysis indicated the reaction was complete. Reaction was concentrated and the resulting residue was dissolved in 1 N NaOH and stirred at room temperature for 30 minutes. The aqueous mixture was extracted with methylene chloride. The phases were separated and the organics dried over sodium sulfate, filtered, and concentrated affording the title compound, 1 -(4-aminopiperidin-1 -yl)-3-hydroxy-3-methylbutan-1 - one. 1H NMR (300 MHz1 CDCI3) δ 4.40 (d, 1H, JHz=13.9), 3.73 (d, 1 H, JHz=12.1 ), 2.99-2.95 (m, 1H), 2.90 (m, 1 H), 2.70-2.63 (m, 1 H), 2.33 (s, 2H), 1.76 (m, 2H), 1.12 (s, 8H). General synthesis of 1-(4-aminopiperidin~1-yl)-acyl derivatives:

DlPEA (3.00 equiv) and propionyl chloride {compound A), isopropyl sulfonyl chloride (compound B), dimethyl carbamyl chloride (compound C), or isobutyryl chloride (compound D) (1.50 equiv) were added to a stirring solution of tert-butyl piperidin-4- ylcarbamate (1.00 equiv) in methylene chloride (0.1 M) at 0 °C. Reaction warmed to room temperature. TLC analysis after 16 hours showed no remaining starting materia! and reaction was diluted in saturated NH4CI and extracted with methylene chloride. The phases were separated and the organics dried over sodium sulfate, filtered, and concentrated. The resulting residue was purified by flash column chromatography (SiO2, 40 g; 0% to 100% ethyl acetate in hexanes). The resulting material was dissolved in methylene chloride (1.0M) and TFA (1.0M) was added under nitrogen and the mixture was stirred at room temperature for 30 minutes at which time TLC analysis indicated the reaction was complete. Reaction was concentrated and the resulting residue was dissolved in 1 N NaOH and stirred at room temperature for 30 minutes. The aqueous mixture was extracted with methylene chloride. The phases were separated and the organics dried over sodium sulfate, filtered, and concentrated . affording the title compound.
Compound A: 1H NMR (300 MHz, CDCl3) δ 4.53 (d, 1H, JHz=13.2), 3.84 (d, 1H,
JHz=13.2), 3.11-3.02 (m, 1 H), 2.97-2.87 (m, 1H), 2.76-2.67 (m, 1 H), 2.40-2.33 (m, 2H), 1.89-1.83 (m, 2H)5 1.37-1.14 (m, 5H);
Compound B: 1H NMR (300 MHz, CDCI3) δ 3.82-3.77 (m, 2H), 3.26-3.12 (m, 1 H)1 3.02-2.93 (m, 2H), 2.90-2.81 (m, 1 H), 1.91-1.86 (m, 2H)1 1.47-1.33 (m, 8H); Compound C: 1H NMR (300 MHz, CDCi3) δ 3.54 (d, 2H1 JHz=14.1 ), 2.77-2.65 (m, 7H), 1.75-1.70 (m, 2H), 1.30-1.61 (m, 4H);
Compound D: 1H NMR (300 MHz, CDCI3) δ 4.50-4.55 (d, J= 13.5 Hz, 1H), 3.89-3.92 (d, J = 12.3, 1 H), 3.03-3.11 (t, J= 12.2 Hz, 1H), 2.76-2.95 (m, 2H), 2.64-2.73 (t, J = 12.9 Hz, 1 H) 1.82-1.90 (m, 2H), 1 .32 (s, 2H), 1.19-1.32 (m, 2H), 1.11 -1.13 (d, J= 6.3 Hz1 6H).
General Synthesis of 3-aryl-6-bromo-5-(1 -(disubstitutedamino)ethyQpyrazolori .5- a]pyrimidin-7-amine:
Scheme-XXXXXX

Synthesis of 3-aryl-5-(1-(disubstitutedamino)ethyπ-N,N-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazolori ,5-aipyrimidin-7-amine:
Sodium cyanoborohydride (3.5 equiv) was added to a stirring mixture of ketone, 1 -(3- argio-7-(bis{(2-(trimethylsiiyl)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)ethanone (1 equiv) and amine (2 equiv} in ethanol and acetic acid at room temperature. When the reaction was deemed complete by HPLC, LC-MS, or TLC analysis, the mixture was quenched with saturated aqueous sodium bicarbonate, then extracted with dichloromethane (2x). The combined organics were dried over sodium sulfate, filtered, concentrated to dryness, and purified on silica gel to afford compound 3-aryl-5-(1-(disubstitutedamino)ethyl)-N,N-bis((2- (trimethylsiIyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin-7-amine.
Synthesis of 3-Aryl -6-bromo-5-(1 -(disubstitutedamino)ethyl)pyrazolori ,5-a]pyrirnidin- 7-amine: Compound, 3-aryl-5-(1-(disubstitutedamino)ethyl)-N,N-bis((2-
(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidin-7-amine, was stirred in a mixture of TFA, dichloromethane, and water, then concentrated to dryness when the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. This material was then treated with NBS (1.05 equiv) in acetonithle. When the reaction was deemed complete by HPLC, LC-MS, or TLC analysis, the mixture was concentrated to dryness, purified by prep-HPLC, and freeze-dried to afford compound 3-Aryl -6- bromo-5-(1 -(disubstitutedamino)ethyl)pyrazolo[1 ,5-a]pyrimtdin-7-amine.
General synthesis of 3-aryl-6-acetyl-5-π-(disubstitutedamino)ethyθpyrazoiof1.5- aipyrimidin-7-amine:

A mixture of compound 3-ary!-5-(1-(disubstitutedamino)ethyl)-N,N-bis({2- {trimethylsiIyl)ethoxy)methyl)pyrazoio[1,5-a]pyrimidin-7-amine. (1 equiv) and NBS (1.05 equiv) in acetonitrile was stirred at room temperature until the reaction was deemed complete by HPLC1 LC-MS, or TLC analysis. The reaction mixture was concentrated, re-dissolved in dichloromethane, washed with saturated aqueous sodium thiosulfate, dried over sodium sulfate, filtered, concentrated, and purified on silica ge!. The resulting material was stirred with a mixture of vinyl stannane (2 equiv), palladium catalyst (0.1 equiv), and potassium fluoride (3 equiv) in dioxane at 85 °C until the reaction was deemed complete by HPLC1 LC-MS, or TLC analysis. The mixture was allowed to cool to room temperature, diluted with 10% aqueous potassium fluoride, and extracted three times with ethyl acetate. The combined organics were dried over sodium sulfate, filtered, concentrated, and purified on silica gel. The resulting material was stirred with a mixture of HCl in methanol and water at 60 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The mixture was then allowed to cool to room temperature, concentrated, purified by prep-HPLC, and freeze-dried to afford compound 3-aryl-6-acetyl-5-(1 - (disubstitutedamino)ethyl)pyrazolo[1 ,5-a]pyrimidin-7-amine.
4-((7-amino-6-bromo-3-(6-fluoroαuinolin-3-yl)pyrazolo[1 ,5-aipyrimidin-5- ylamino)methyl)-N-alkylpiperidine-1 -carboxamide:
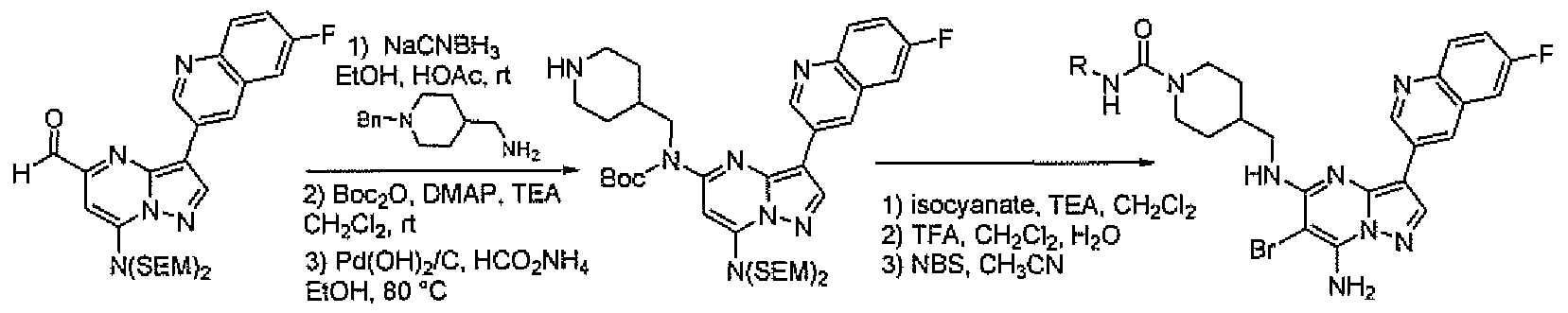
SCHEME-47
Sodium cyanoborohydride (3.5 equiv) was added to a stirring mixture of aldehyde, 7- (bis((2-{trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3-yl)pyrazoio[1 ,5- a]pyrimidine-5-carbaidehyde (1 equiv) and 1-benzylpiperidin-4-yl)methanamine (2 equiv) in ethanol and acetic acid at room temperature. When the reaction was deemed complete by HPLC, LC-MS, or TLC analysis, the mixture was quenched with saturated aqueous sodium bicarbonate, then extracted with dichloromethane (2x). The combined organics were dried over sodium sulfate, filtered, concentrated to dryness, and purified on silica gel to afford the desired product, N5-((1 -
benzylpiperidin-4-yl)methyl)-3-(6-fluoroquinolin-3-y!)-N7,N7-bis({2- (trimethylsilyl)ethoxy)methy!)pyrazolo[1 ,5-a]pyrimidine-5,7-diamine.
The N5-{(1-benzylpiperidin-4-yl)methyl)-3-{6-fluoroquinolin-3-yl)-N7,N7-bis((2- (trimethylsilyl)ethoxy)methyl)pyrazoIo[1 ,5-a3pyrimidine-5,7-diamine (1 equiv), BoC2O (2 equiv), DMAP (0.2 equiv), and triethylamine (3 equiv) in dichloromethane was stirred at room temperature until the reaction was deemed complete by TLC, HPLC, or LC- MS analysis. The mixture was diluted with additional dichloromethane, washed with saturated aqueous ammonium chloride, dried over sodium sulfate, filtered, concentrated, and purified on silica gel to give the product, tert-butyl (1- benzylpiperidin-4-yl)methyl(7-(bis({2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- fluoroquinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yi)carbamate.
This material, tert-butyl (1 -benzyipiperidin-4-yl)methyl(7-(bis((2- (trimethylsilyl)ethoxy)methyl)amino)-3-{6-fIuoroquinolin-3-yI)pyrazolo[1 ,5-a]pyrimidin- 5-yl)carbamate, was then stirred with Pd(OH)2/C (0.2 equiv) and ammonium formate (20 equiv) in ethanol at 80 °C until the reaction was deemed complete by HPLC, LC- MS, or TLC analysis. The mixture was allowed to cool to room temperature, filtered through a pad of celite, concentrated and purified on silica gel to afford compound tert- butyl 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl(piperidin-4-ylmethyl)carbamate.
6-bromo-3-(6-fluoroquinolin-3-yl)-N5-(piperidin-4-ylmethyl)pyrazolori,5-aipyrimidine- 5,7-diamine :
Compound tert-butyl 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin- 3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yS(piperidin-4~ylmethyl)carbamate was stirred in a mixture of TFA, dichloromethane, and water, then concentrated to dryness when the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. This material -was then treated with NBS (1.05 equiv) in acetonitrile. When the reaction was deemed complete by HPLC, LC-MS, or TLC analysis, the mixture was concentrated to dryness, purified by prep-HPLC, and freeze-dried to afford compound 6-bromo-3-(6- fluoroquinolin-3-yl)-N5-(piperidin-4-ylmethyl)pyrazolo[1 ,5"a]pyrimidine-5,7-diamine.
Compound, tert-butyl 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin- 3-yl)pyrazoio[1 ,5-a]pyrimidin-5-y!(piperidin-4-ylmethyl)carbamate (1 equiv) was stirred in a mixture of isocyanate (1.1 equiv), and triethylamine (1.5 equiv) in dichloromethane at 0 °C until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction mixture was poured into saturated aqueous ammonium chloride, and then extracted twice with dichloromethane. The combined organics were dried over sodium sulfate, filtered, concentrated, and purified on silica gel to give the desired product. This material was stirred in a mixture of TFA, dichloromethane, and water, and then concentrated to dryness when the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. This material was then treated with NBS (1.05 equiv) in acetonitrile. When the reaction was deemed complete by HPLC, LC- MS, or TLC analysis, the mixture was concentrated to dryness, purified by prep- HPLC, and freeze-dried to afford compound.
Synthesis of 4-((7-amino-6-cvano-3-(6-fiuoroquinoiin-3-yl)pyrazolof1 ,5-alpyrimidin-5- ylamino)methyl)-N-methylpiperidine-1-carboxamide:

SCHEME-48 Compound, tert-butyl 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinoiin- 3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yI(piperidin~4-yimethyl)carbamate (1 equiv) was stirred in a mixture of methyl isocyanate (1.1 equiv), and triethylamine (1.5 equiv) in dichloromethane at 0 °C until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction mixture was poured into saturated aqueous ammonium chloride, and then extracted twice with dichloromethane. The combined organics were dried over sodium sulfate, filtered, concentrated, and purified on silica gel to give the desired product. This material (1 equiv) and NBS (1.05 equiv) in acetonitriie was stirred at room temperature until the reaction was deemed complete by HPLC, LC- MS, or TLC analysis. The reaction mixture was concentrated, re-dissolved in dichloromethane, washed with saturated aqueous sodium thiosulfate, dried over sodium sulfate, filtered, concentrated, and purified on silica gel. The resulting material was stirred with the palladium catalysts (0.2 equiv each) and stannane (1.5 equiv) in dioxane at 160 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The reaction was allowed to cool to room temperature, diluted with ethyl acetate, washed with brine, dried over sodium sulfate, filtered, concentrated, and purified in silica gel. The resulting material was stirred in TFA and water at room temperature until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The mixture was concentrated, purified by prep-HPLC, and freeze-dried to afford compound 4-((7-amino-6-cyano-3-(6-fluoroquinolin-3-yl) pyrazolo [1 , 5-a] pyrimidin-5-yiamino) methyl)-N-methylpiperidine-1 -carboxamide.
Genera! Synthesis of 1-(4-((7-amino-6-bromo-3-(6-fluoroquinolin-3-v1)pyrazolori ,5- aipyrimidin-5-ylamino)alkvQpiperidin-1-yl)ethanone:

SCHEM E-49
Sodium cyanoborohydride ( 3.5 equiv) was added to a stirring mixture of 7-(bis((2- (tπmethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3-yl)pyrazolo[1 ,5-a]pyrimidine-- 5-carbaidehyde (1 equiv) and amine (2 equiv) in ethanol and acetic acid at room temperature. When the reaction was deemed complete by HPLC, LC-MS, or TLC analysis, the mixture was quenched with saturated aqueous sodium bicarbonate, and then extracted with dichloromethane (2x). The combined organics were dried over sodium sulfate, filtered, concentrated to dryness, and purified on siiica gel to afford the desired product, N5-({1-benzylpipeπdin-4-yl)methyl)-3-(6-fluoroquinolin-3-yl)-N7,N7- bis((2-(trimethylsilyl)ethoxy)methyl)pyrazolo[1 ,5-a]pyrimidine-5,7-diamine.
This material, (1 equiv), BoC2O (2 equiv), DMAP (0.2 equiv), and triethylamine (3 equiv) in dichloromethane was stirred at room temperature until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The mixture was diluted with additional dichloromethane, washed with saturated aqueous ammonium chloride, dried over sodium sulfate, filtered, concentrated, and purified on siiica gel to give the product, tert-butyl {1-benzylpiperidin-4-yl)methyl(7-(bis((2-
(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin- 5-yl)carbamate. This material was then stirred with Pd(OH)2/C (0.2 equiv) and ammonium formate (20 equiv) in ethanol at 80 °C until the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. The mixture was allowed to cool to room temperature, filtered through a pad of ceiite, concentrated and purified on silica gel to afford compound tert- butyl 7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl(piperidin-4-ylmethyl)carbamate.
This material was stirred with carboxyiic acid (1.5 equiv), HATU (1.5 equiv), and DIPEA (3 equiv) in DMF until the reaction was deemed complete by TLC, HPLC, or LC-MS analysis. The reaction mixture was concentrated, the resulting residue dissolved in ethyl acetate, washed with water, saturated aqueous sodium bicarbonate, and brine, dried over sodium sulfate, filtered, concentrated, and purified on silica gel to give the product. This material was stirred in a mixture of TFA, dichloromethane, and water, then concentrated to dryness when the reaction was deemed complete by HPLC, LC-MS, or TLC analysis. This material was then treated with NBS (1.05 equiv) in acetonitrile. When the reaction was deemed complete by HPLC, LC-MS, or TLC analysis, the mixture was concentrated to dryness, purified by prep-HPLC, and freeze- dried to afford compound 1 -(4-((7-amino-6-bromo-3-(6-fluoroquinoiin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-ylamino)methyl)piperidin-1 -yi)ethanone.
Synthesis of 1-(4-((7-amino-6-bromo-3-(6-fluoroαuinolin-3-yl)pyrazoioM ,5-aipyrimidin- 5-yl)methylamino)piperidin-1 -yl)-2-methoxypropan-1 -one:
Scheme-49

To a solution of tert-butyl (7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6- f!uoroquinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)methyl(piperidin-4-yl)carbamate (190 mg, 0.25 mmoi) in dichloromethane (3 mL) was added imidazolium salt (84 mg, 0.25mmol) {prepared according to the literature procedure:Grzyb, J.A. et. al. Tetrahedron, 2005, 61, 7153-7175) and Et3N (35 μl_, 0.25 mmol). The resulting mixture was stirred at 25 °C for 16 hours to give. The reaction mixture was diluted with CH2CI2 (50 mL) and was washed with 0.1 N HCI (3 mL), brine (10 mL), dried over Na2SO4. LCMS indicated that the crude product, crude tert-butyl (7-(bis((2- {trimethylsiIyl)ethoxy)methyl)amino)-3-(6-fluoroquinolin-3-yl)pyrazolo[1 ,5-a]pyrimidin- 5-yl)methyl(1-(2-methoxypropanoyl)piperidin-4-yl)carbamate was clean, This material was then converted to the final compound, by bromination with NBS and followed by treatment with 4NHCI, to give 1-(4-((7-amino-6-bromo-3-(6-fluoroquinotin-3- yl)pyrazolo[1 ,5-a]pyrimidin-5-yl)methy!amino)piperidin-1-yl)-2-methoxypropan-1 -one following a similar procedure as that described above schemes: MW: 556.13; MS MH+ m/z: 557; HPLC tR: 4.17. . LCMS: 2.67 mins, m/z =485.0 (MH+).
By essentially the same procedure given in Schemes 45 through 49, the compounds listed ϊn Table 14 can be prepared
TABLE-14









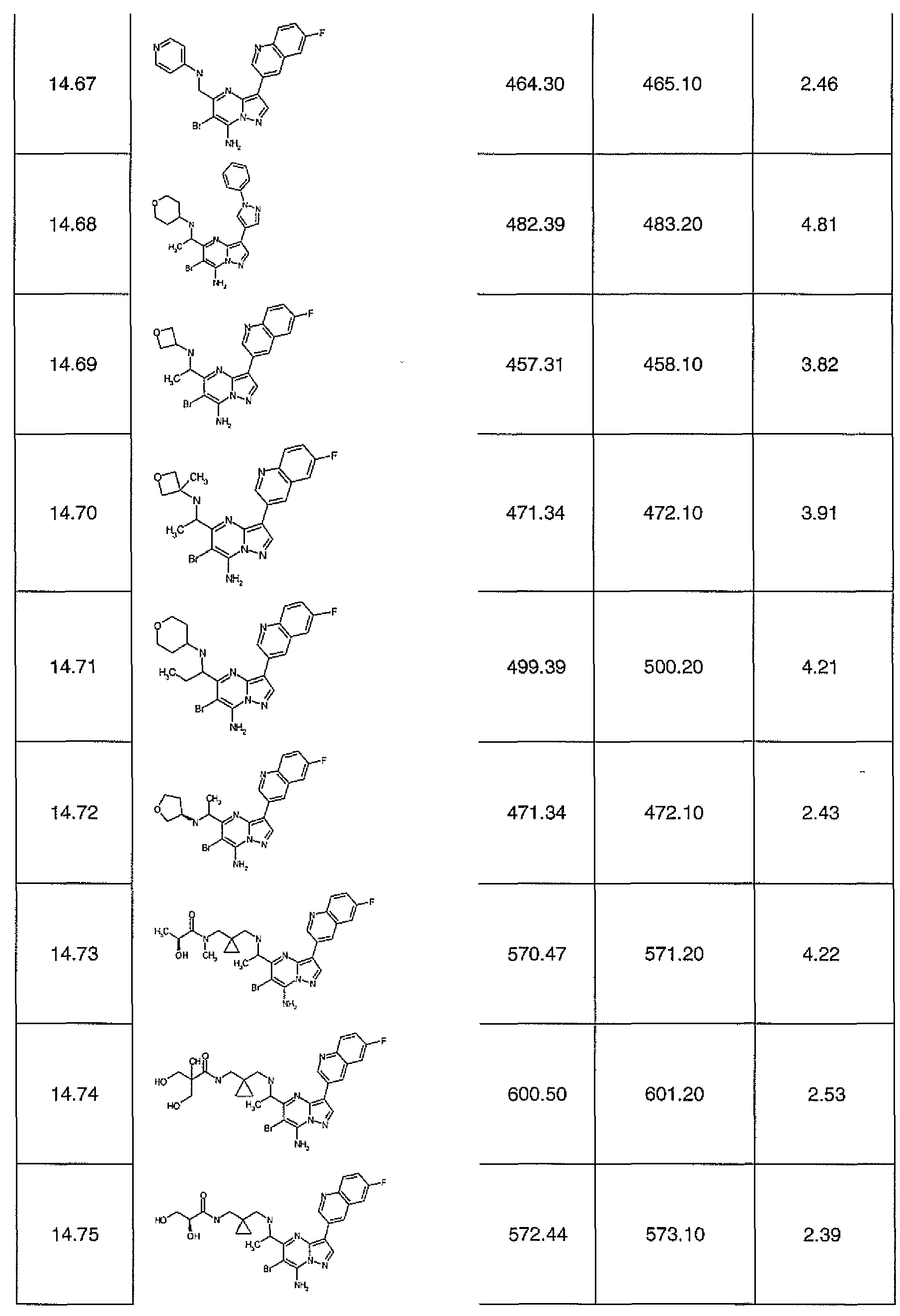







6-(7-amino<3-(quinoiin-3-vl)pvrazolof1 ,5-aipvrimidin-5-vl)hexanoic acid

Scheme 70
To a solution of methyl hex-5-enoate (12 mg, 0.1 mmol) in THF (0.5 m!) under argon at 0 °C was added 9-BBN (0.2 mmol, 0.4 mi of 0.5 M solution in THF). The mixture
was warmed to room temperature and stirred for 16 h. Potassium phosphate (3 M in H2O, 0.2 mmol) was added followed by the addition of 5-chloro-3-(quino!in-3-yl)-N,N- bis((2-(trimethylsilyi)ethoxy)methyl) pyrazolo[1 ,5-a]pyπmidin-7-amine (50 mg, 0.09 mmol) in DMF (0.5 ml) and Pd(dppf)Cl2-CH2Ci2 (5 mg, 0.005 mmol). The reaction mixture was heated at 90 °C for 16 h under argon. The solution was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in 2:1 THF: MeOH (3 ml_) and was treated with 2N NaOH<aq) (0.25 mL). The resulting solution was stirred at room temperature for 4 hours followed by the addition of 2N hydrochloride solution (1.0 mi). The solution was heated at 65 °C for 2 h, cooled to room temperature, concentrated in vacuo and purified by prep-LC to afford the title compound (8.2 mg): LC/MS RT = 2.74 min. Mass calculated for, M+H 376.17, observed 376.17.
By essentially the same procedure given in Scheme 50, the compounds listed in Table 15 can be prepared.
Table 15

Incorporation of deuterium at the 3-position
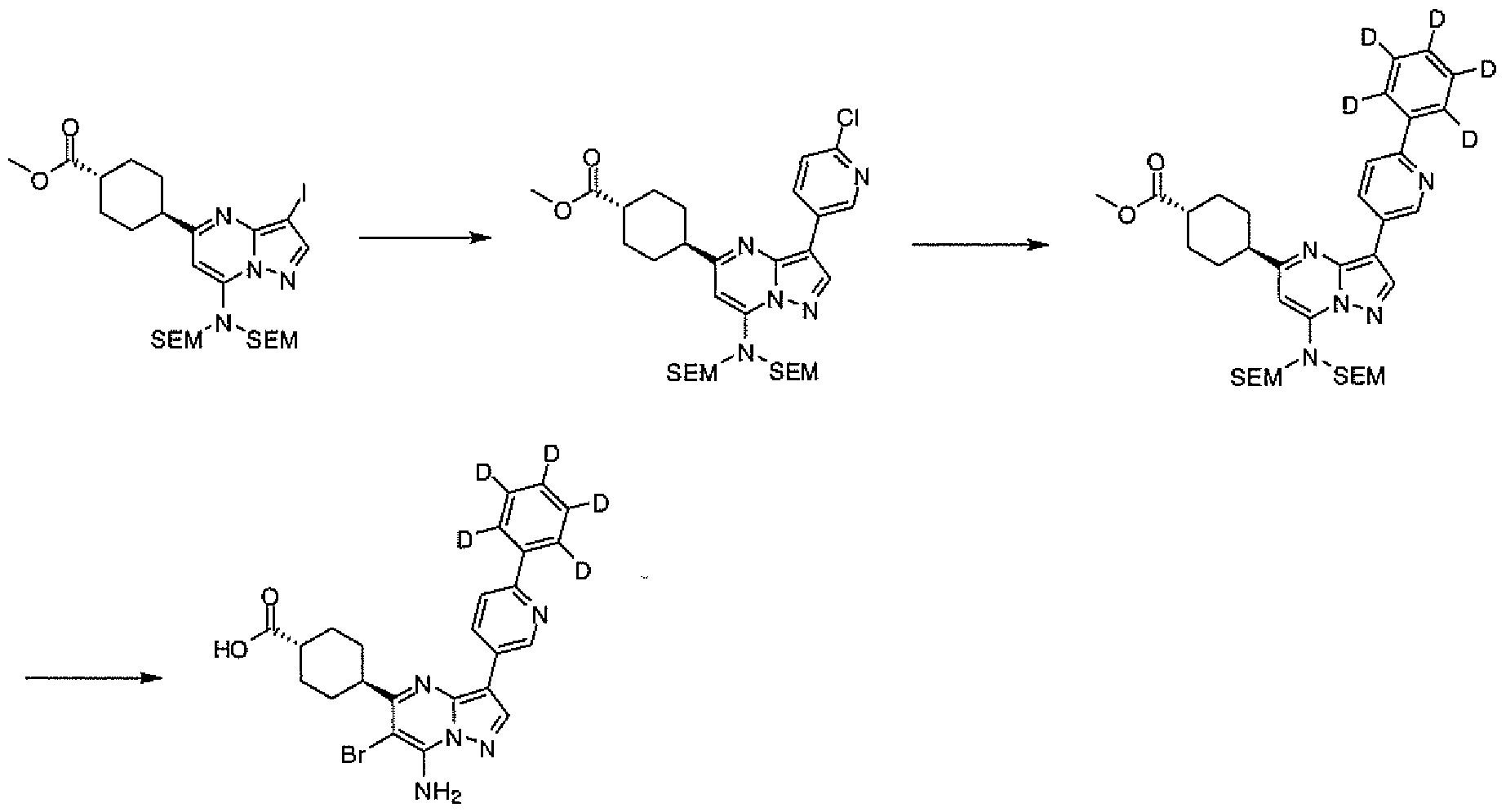
Scheme 51
Synthesis of Methyl 4-(7-(bis((2-(trimethylsiM)ethoxy)methyDamino)-3-(6- chloropyridin-3-yl)pyrazoloH ,5-alpyrimidin-5-yl)cvc!ohexanecarboxy[ate

2-Chioro-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridine (0.55 mmol, 131 rng), K3PO4 (1.36 mmol, 290 mg), and PdCI2{dppf) ■ CH2CI2 (0.045 mmol, 37 mg) is added to a solution of 4-{7-[Bis-(2-trimethylsilanyl-ethoxymethy!)-amino]-3-iodo-pyrazolo[1 ,5- a3pyrimidin-5-yl}-cyciohexanecarboxylic acid methyl ester (0.45 mmol, 300 mg) in dioxane (3.8 ml_). To this suspension is added distilled H2O (0.38 mL). The resulting solution is stirred at 90° C under argon for 18 hours. The reaction mixture is concentrated in vacuo and then purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield the title compound (220 mg, 75% yield) as yellow oil. LC-MS: 646 [M+H].
Synthesis of (1 r.4r)-methyl 4-(7-(bis((2-(trimethylsilyl)ethoxy)methyQamino)-3-(6- phenyl-d5-pyridin-3-yl)pyrazoloπ ^-alpyrimidin-5-yl)cvclohexanecarboxylate

Phenyl-d5-boronic acid (1.08 mmol, 137 mg), K3PO4 (1.63 mmol, 345 mg), and PdCI2(dppf) • CH2CI2 (0.054 mmol, 45 mg) is added to a solution of methyl 4-(7- (bis((2-(trimethylsilyl)ethoxy)methyl)amino)-3-(6-chloropyridin-3-yi)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexanecarboxylate (0.54 mmol, 350 mg) in dioxane (6.0 ml_). To this suspension is added distilled H2O (0.6 mL). The resulting solution is stirred at 100° C under argon for 18 hours. The reaction mixture is concentrated in vacuo and then purified via silica gel chromatography (0% to 60% ethyl acetate in hexanes gradient) to yield the title compound.
Synthesis of (1r,4r)-4-(7-amino-6-bromo-3-(6-phenyl-d5-pyridin-3-vQpyrazolori ,5- alpyrSmidin-5-yl)cvclohexanecarboxylic acid

N-bromosuccinimide (73 mg, 0.41 mmol) is added to a solution of <1 r,4r)-methyl 4-(7- (bis((2-(trimethyls!lyl)ethoxy)methyl)amino)-3-(6-phenyl-d5"pyπdin-3-yl)pyrazolo[1 ,5- a]pyrimidin-5-yl)cyclohexanecarboxyiate (284 mg, 0.41 mmoi) in acetonitrile (4 mL). The resulting solution is stirred at room temperature for 1 hour. The reaction mixture is concentrated in vacuo and dissolved in 3:3:2 MeOH:THF:H2O (8 mL), followed by the treatment with 3.75 N NaOH(aq) (0.6 mL). The resulting solution is stirred at room temperature for 18 hours. The reaction mixture is treated with 1 N hydrochloride
solution (4 ml) at 65 °C for 4 h. The reaction solution is concentrated and purified by prep-LC to afford the title compound.
incorporation of deuterium at the 5-position

Scheme 52
Synthesis of ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)methyl)amino)pyrazoloπ ,5- alpyrimidin-5-yl)cvclohexyl)acetate

To a 50 mL roundbottom flask is charged ethyl 2-(4-(7-(bis((2-(trimethylsilyl)ethoxy)- methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5-yl)cyclohexylidene)acetate (800 mg, 1.43 mmol) and ethyl acetate (15 mL). The flask is flushed with argon and 5% palladium on carbon (100 mg) is added. The flask is sealed and degassed under vacuum.
Deuterium gas is then added via balloon. The reaction is stirred under D2 atmosphere 18 hours. The reaction is then filtered through celite to yield deuterated ethyl 2-{4-(7- (bis((2-(trimethylsily!)ethoxy)methyl)amino)pyrazolo[1 ,5-a]pyrimidin-5- yl)cyclohexyl)acetate (761 mg, 1.35 mmol, 95% yield) as pale yellow oil.
Incorporate deuterium at both 3- and 5-positions

Scheme 53 Synthesis of ethyl 2-(4-(7-amino-6-bromo-3-(6-phenylpyridin-3-yl)pyrazolori ,5- aipyrimidin-5-yl)cvclohexyl)acetate

ASSAYS: mTOR Kinase Assay
The nπTOR assay buffer contains 10 mM hepes (pH 7.4), 50 mM NaCI, 100 μg/ml BSA, 50 mM B-glycerophosphate, 10 mM MnCI2 and 0.5 mM DTT. 20 ng of mTOR enzyme is preincubated with the compound for 10 minutes. 5 μM ATP and 0.1 μM GSTS6K is added. The reaction is incubated for one hour at 30 °C. Anti phospho P70S6K (about 1.7 ng/well) and anti GSTXL665 (1 :1 Ratio with the substrate GSTS6K) are added after incubating. The plates are read at least 2 hours after adding the anti phospho p70S6K and the anti GSTXL665. ICm DETERMINATIONS: Dose-response curves were plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds. Concentration of compound was plotted against % kinase activity, calculated by CPM of treated samples divided by CPM of untreated samples. To generate IC5O values, the dose-response curves were then fitted to a standard sigmoidal curve and IC50 values were derived by nonlinear regression analysis.
CHK1 in yltro Kinase Assay
This in vitro assay utilizes recombinant His-CHK1 expressed in the baculovirus expression system as an enzyme source and a biotinyiated peptide based on CDC25C as substrate (biotin-RSGLYRSPSMPENLNRPR). Materials and Reagents:
1 ) CDC25C Ser 216 C-term Biotinyiated peptide substrate (25 mg), stored at -20° C1 Custom Synthesis by Research Genetics: biotin-RSGLYRSPSMPENLNRPR 2595.4 MW 2) His-CHK1 In House lot P976, 235 μg/mL, stored at -80° C.
3) D-PBS (without CaCI and MgCI): GIBCO, Cat.# 14190-144
4) SPA beads: Amersham, Cat.# SPQ0032: 500 mg/vial
Add 10 mL of D-PBS to 500 mg of SPA beads to make a working concentration of 50 mg/mL. Store at 4 °C. Use within 2 week after hydration. 5) 96-WeII White Micropiate with Bonded GF/B filter: Packard, Cat.# 6005177
6) Top seai-A 96 well Adhesive Film: Perkin Elmer, Cat.# 6005185
7) 96-weil Non-Binding White Polystyrene Plate: Corning, Cat. # 6005177
8) MgCl2: Sigma, Cat.# M-8266
9) DTT: Promega, Cat.# V3155 10) ATP, stored at 4 ^C: Sigma, Cat.# A-5394
11 ) γ33p-ATP, 1000-3000 Ci/mMol: Amersham, Cat.# AH9968 12} NaCI: Fisher Scientific, Cat.# BP358-212
13) H3PO4 85% Fisher, Cat.#A242-500
14) Tris-HCL pH 8.0: Bio-Whittaker, Cat. # 16-015V 15) Staurosporine, 100 μg: CALBIOCHEM, Cat. # 569397
16) Hypure Cell Culture Grade Water, 500 mL: HyClone, Cat.# SH30529.02 Reaction Mixtures:
1) Kinase Buffer: 50 mM Tris pH 8.0; 10 mM MgCl2; 1 mM DTT
2) His-CHK1 , In House Lot P976, MW -3OKDa1 stored at -80° C. 6 nM is required to yield positive controls of -5,000 CPM. For 1 piate (100 reaction): dilute 8 μi_ of 235 μg/mL (7.83 μM) stock in 2 mL Kinase Buffer. This makes a 31 nM mixture. Add 20 μL/well. This makes a final reaction concentration of 6 nM.
3) CDC25C Biotinylated peptide.
Dilute CDC25C to 1 mg/mL (385 μM) stock and store at -20 °C. For 1 plate (100 reactions): dilute 10 μL of 1 mg/mL peptide stock in 2 mL Kinase Buffer. This gives a 1.925 μM mix. Add 20 μL/reaction. This makes a final reaction concentration of 385 nM.
4) ATP Mix.
For 1 plate (100 reactions): dilute 10 μL of 1 mM ATP (cold) stock and 2 μL fresh P33-ATP (20 μCi) in 5 mL Kinase Buffer. This gives a 2 μM ATP (cold) solution; add 50 μL/well to start the reaction. Final volume is 100 μL/reaction so the final reaction concentrations will be 1 μM ATP (cold) and 0.2 μCi/reaction.
5) Stop Solution:
For 1 plate add: To 10 mL Wash Buffer 2 (2M NaCI 1 % H3PO4) : 1 mL SPA bead slurry (50 mg); Add 100 μL/well
6) Wash buffer 1 : 2 M NaCl
7) Wash buffer 2: 2 M NaCl, 1% H3PO4 Assay Procedure:

* Total reaction volume for assay.** Final reaction volume at termination of reaction (after addition of stop solution).
1) Dilute test compounds to desired concentrations in water/10% DMSO - this will give a final DMSO concentration of 1 % in the reaction. Dispense 10 μL/reaction to appropriate weils. Add 10 μl_ 10% DMSO to positive (CHK1 +CDC25C+ATP) and negative (CHK1+ATP only) control wells. 2) Thaw enzyme on ice -- dilute enzyme to proper concentration in kinase buffer (see
Reaction Mixtures) and dispense 20 μL to each well.
3) Thaw the Biotinylated substrate on ice and dilute in kinase buffer (see Reaction
Mixtures). Add 20 μL/well except to negative control wells. Instead, add 20 μL Kinase
Buffer to these wells. 4) Dilute ATP (cold) and P33-ATP in kinase buffer (see Reaction Mixtures). Add 50 μL/well to start the reaction.
5) Allow the reaction to run for 2 hours at room temperature.
6) Stop reaction by adding 100 μL of the SPA beads/stop solution (see Reaction Mixtures) and leave to incubate for 15 minutes before harvest 7) Place a blank Packard GF/B filter plate into the vacuum filter device (Packard plate harvester) and aspirate 200 mL water through to wet the system. 8) Take out the blank and put in the Packard GF/B filter plate.
9) Aspirate the reaction through the filter plate.
10) Wash: 200 ml_ each wash; 1 X with 2M NaCI; 1 X with 2M NaCi/ 1 % H3PO4
1 1 ) Allow filter plate to dry 15 minutes.
12) Put TopSeal-A adhesive on top of fitter plate. 13) Run filter plate in Top Count
Settings: Data mode: CPM
Radio nuclide: Manual SPA:P33 Scintillator: Liq/plast Energy Range: Low ICgn DETERMINATIONS: Dose-response curves were plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds. Concentration of compound was plotted against % kinase activity, calculated by CPM of treated samples divided by CPM of untreated samples. To generate IC50 values, the dose-response curves were then fitted to a standard sigmoida! curve and IC50 values were derived by nonlinear regression analysis.
Selected Thiazole Derivatives of the present invention were tested using this assay and provided IC50 values ranging from about 1 nM to about 5500 nM.
CDK2 Kinase Assay BACULQVIRUS CONSTRUCTIONS: Cyclin E was cloned into pVL1393
(Pharmingen, La JoIIa, California) by PCR, with the addition of 5 histidine residues at the amino-terminal end to allow purification on nickel resin. The expressed protein was approximately 45kDa. CDK2 was cloned into pVL1393 by PCR, with the addition of a haemaglutinin epitope tag at the carboxy-terminal end (YDVPDYAS). The expressed protein was approximately 34kDa in size.
ENZYME PRODUCTION: Recombinant bacuioviruses expressing cyclin E and CDK2 were co-infected into SF9 celis at an equal multiplicity of infection (MOi=5), for 48 hrs. Cells were harvested by centrifugation at 1000 RPM for 10 minutes, then pellets lysed on ice for 30 minutes in five times the peiiet volume of lysis buffer containing 5OmM Tris pH 8.0, 15OmM NaCl, 1% NP40, 1 mM DTT and protease inhibitors (Roche Diagnostics GmbH, Mannheim, Germany). Lysates were spun down at 15000 RPM for 10 minutes and the supernatant retained. 5mL of nickel beads (for
one liter of SF9 cells) were washed three times in lysis buffer (Qiagen GmbH, Germany). Imidazole was added to the baculovirus supernatant to a final concentration of 2OmM, then incubated with the nickel beads for 45 minutes at 4° C. Proteins were eluted with lysis buffer containing 25OmM imidazole. Eluate was dialyzed about 15 hours in 2 liters of kinase buffer containing 5OmM Tris pH 8.0, 1 mM DTT, 1OmM MgCI≥, 100μM sodium orthovanadate and 20% glycerol. Enzyme was stored in aliquots at -7O°C.
In Vitro Cyclin E/CDK2 Kinase Assays Cyclin E/CDK2 kinase assays can be performed as described below in low protein binding 96-we!S plates {Corning Inc, Corning, New York).
Enzyme is diluted to a final concentration of 50 μg/mL in kinase buffer containing 5OmM Tris pH 8.0, 10 mM MgCI2J mM DTT, and 0.1 mM sodium orthovanadate. The substrate used in these reactions is a biotinylated peptide derived from Histone H1 (from Amersham, UK). The substrate is thawed on ice and diluted to 2 μM in kinase buffer. Test compounds are diluted in 10% DMSO to desirable concentrations. For each kinase reaction, 20 μl_ of the 50 μg/mL enzyme solution (1 μg of enzyme) and 20 μl of the 2 μM substrate solution are mixed, then combined with 10 μL of diluted compound in each well for testing. The kinase reaction is initiated by addition of 50 μL of 2 μM ATP and 0.1 μCi of 33P-ATP (from Amersham, UK). The reaction iss allowed to run for 1 hour at room temperature, then is stopped by adding 200 μL of stop buffer containing 0.1 % Triton X-100, 1 mM ATP, 5mM EDTA, and 5 mg/mL streptavidine coated SPA beads (from Amersham, UK) for 15 minutes. The SPA beads are then captured onto a 96-well GF/B filter plate (Packard/Perkin Elmer Life Sciences) using a Filtermate universal harvester (Packard/Perkin Elmer Life Sciences.). Non-specific signals are eliminated by washing the beads twice with 2M NaCI then twice with 2 M NaCI with 1% phosphoric acid. The radioactive signal can then be measured using, for example, a TopCount 96 well liquid scintillation counter (from Packard/Perkin Elmer Life Sciences). ICgi DETERMINATIONS: Dose-response curves are plotted from inhibition data generated, each in duplicate, from 8 point serial dilutions of inhibitory compounds. Concentration of compound is plotted against % kinase activity,
calculated by CPM of treated samples divided by CPM of untreated samples. To generate IC50 values, the dose-response curves are then fitted to a standard stgmoidal curve and IC50 values can be derived using nonlinear regression analysis.
Compounds of the present invention exhibit mTOR IC50 values of about 1 nM to about 5500 nM, CHK1 IC50 values of about 100 nM to about 55000 nM, and CDK2 IC50 values of about 800 nM to about 30000 nM. In all cases, the compounds are much more selective for mTOR over CHK1 and CDK2. Table 15 shows the activity data for an illustrative list of compounds of the invention.
Table 15


Table 16 also lists compounds of the invention with activity data whereby the IC50 values are rated "A", "B1" "C," "D," "E" or T." The IC50 values are rated "A" for IC50 values less than 100 nM, "B" for ICs0 values in the range from 101 nM to 200 nM, "C" for IC50 values in the range from 201 nM to 500 nM, "D" for IC50 values in the range of about 501 nM to 1000 nM, Ε" for IC50 values in the range from 1001 nM to 5000 nM and "F" for IC50 values greater than about 5000 nM.
Table 16






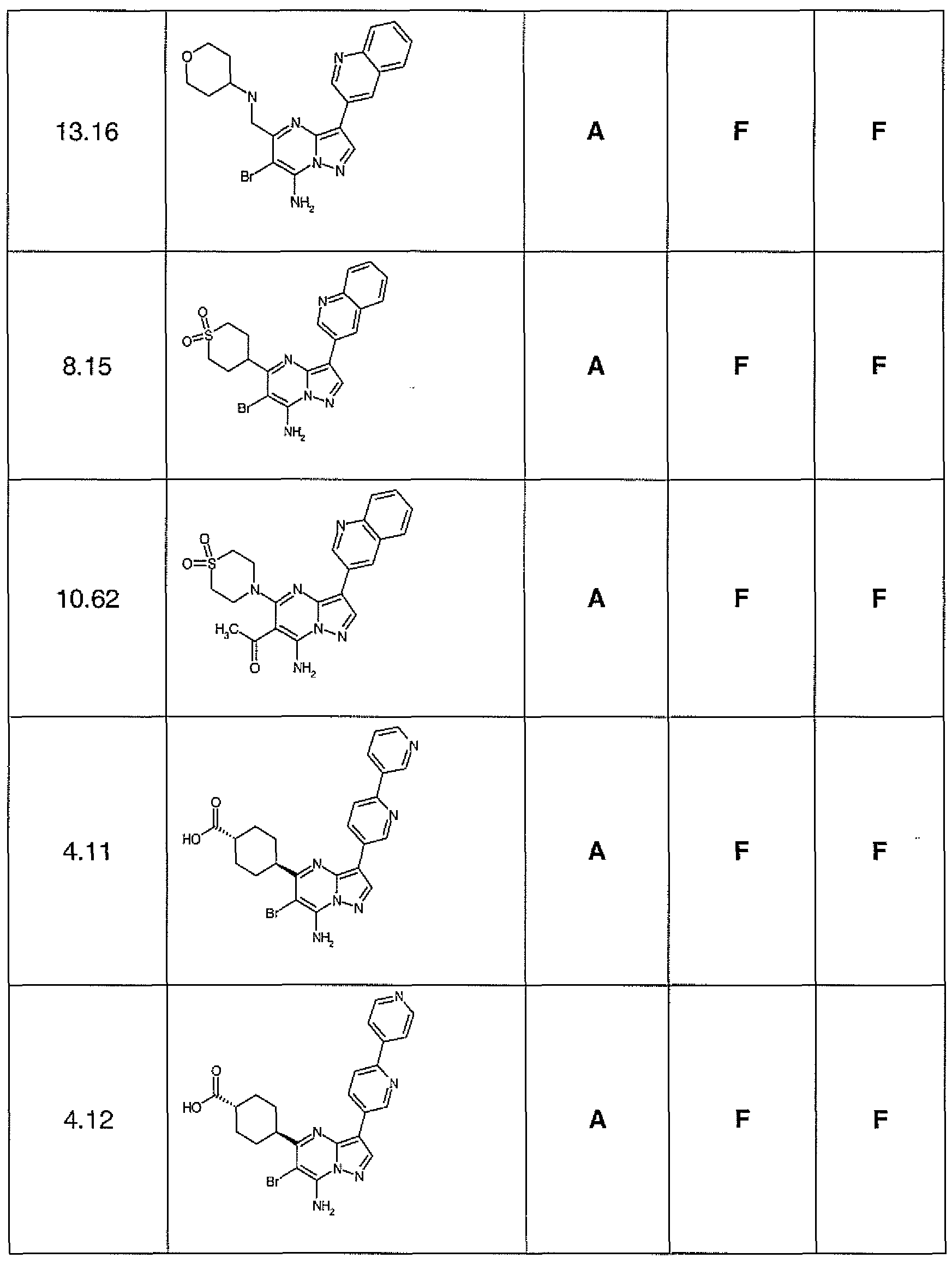


Whiie the present invention has been described in conjunction with the specific embodiments set forth above, many alternatives, modifications and other variations thereof will be apparent to those of ordinary skill in the art. All such alternatives, modifications and variations are intended to fall within the spirit and scope of the present invention.
Claims
What is claimed is: 1. A compound of the formuia:

























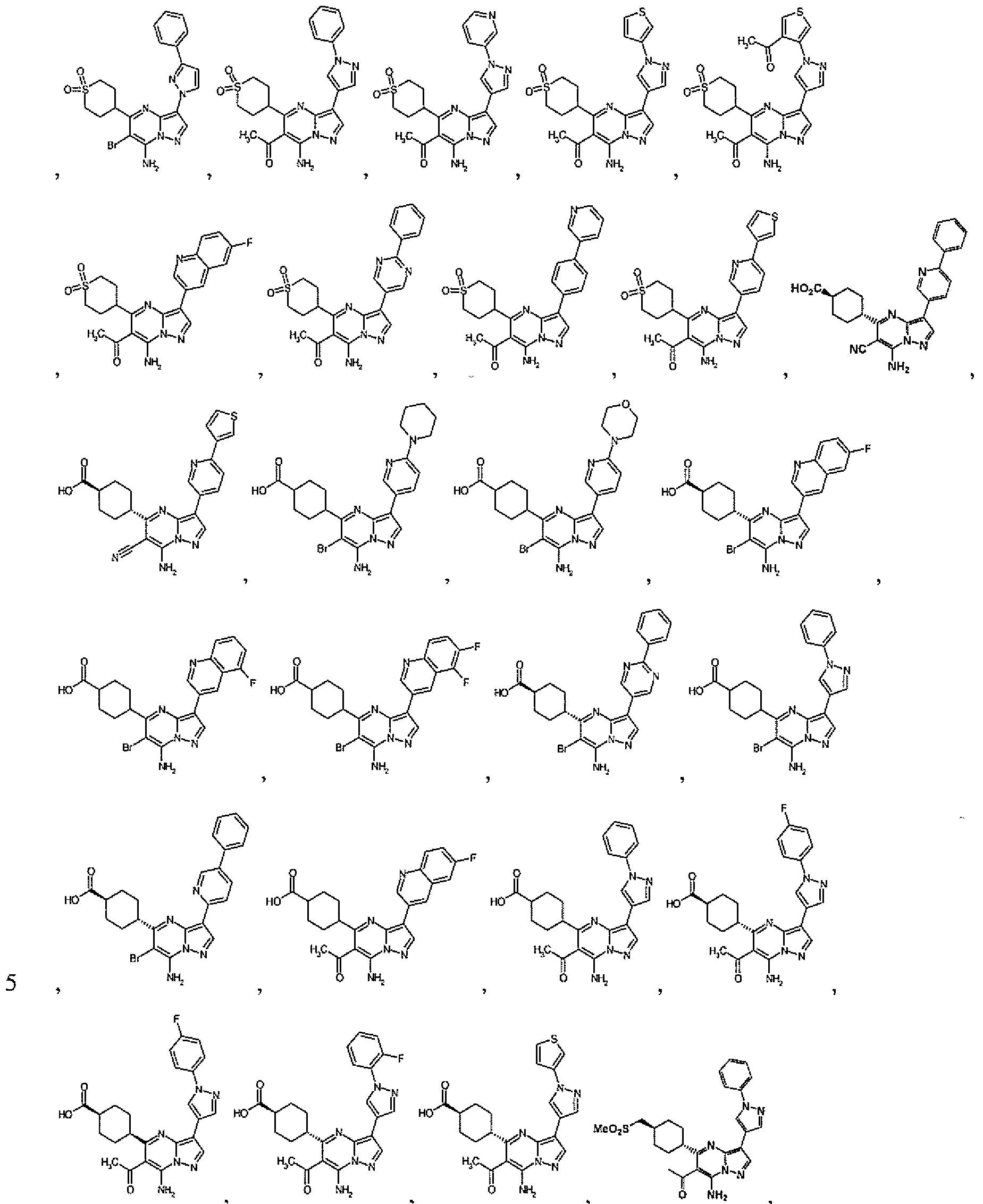









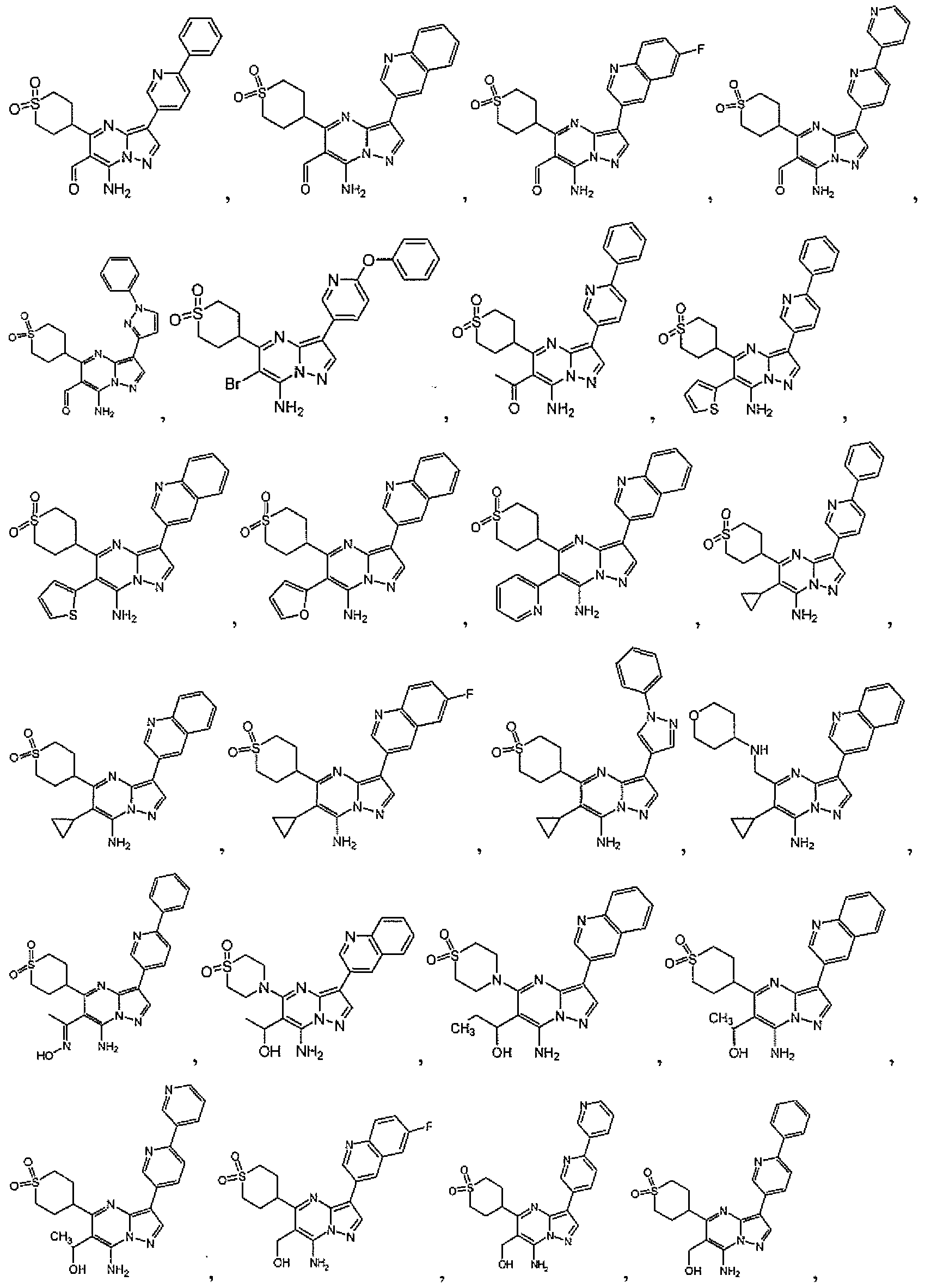


















or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
2. A pharmaceutical composition comprising a therapeutically effective amount of at least one compound of claim 1 or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, in combination with at least one pharmaceutically acceptable - carrier.
3. The pharmaceutical composition according to claim 2, further comprising one or more anti-cancer agents different from the compounds of claim 1.
4. The pharmaceutical composition according to claim 3, wherein the one or more anti-cancer agents are selected from the group consisting of Adriamycin, Altretamine, Amidox, Aminoglutethimide, Amsacrine, Anastrazole, Antibodies to EGFR, 3-AP, Aphidicolon, Ara-C, Arsenic trioxide, L-Asparaginase, Bevacizumab, Bleomycin, BMS 214662, Bortezomib, Busulfan, Campath, Camptostar, Capecitabine, Carboplatin, Carmustine, Centrosome associated protein E ("CENP-E") inhibitors, Cetuximab, Cladribine, Chlorambucil, Chlormethine, Chlorotrianisene, Cisplatin, Clofarabine, cyclophosphamide, Cytarabine, a Cytostatic agent, Cytoxan, Dacarbazine, Dactinomycin, Daunorubicin, Dasatinib, Deforolimus, Deoxycoformycin, Didox,
Diethylstilbestrol, Docetaxel, Doxorubicin, Dromostanolone, Droloxafine, Epirubicin, Epothilones, ERK inhibitors, Erlotinib, Etoposide, 17α-Ethinylestradiol, Estramustine, Exemestane, Floxuridine, Fludarabine, Fludarabine phosphate, 5-Fluorouracii, Fluoxymesterone, Flutamide, Fulvestrant, Gefitinib, Gemcitabine, Gemtuzumab ozogamcicin, Gosereliπ, GSK-923295, Hexamethylmelamine, Hydroxyprogesterone, Hydroxyurea, lbritumomab Tiuxetan, [darubicin, Ifosfamide, lmatinib mesylate, Intron, Irinotecan, ispinesib, KSP inhibitors, L778.123, Lapatinib, Leucovirin, Leuprolide, Lerozole, Letrazole, Levamisole, Liposomal Doxorubicin, Liposomal, Lomustine, lonafornib, Medroxyprogesteroneacetate, Megestrolacetate, Melphalan, 6-Mercaptopurine, Methoxtrexate, Methylprednisoione, Methyltestosterone, Mithramycin, Mitomycin-C, Mitotane, Mitoxantrone, Navelbene, Niiotinib, Oxaliplatin, Paclitaxel, Panitubimab, Pentostatin, Pipobroman, Porfimer, Prednisolone, Prednisone propionate, Procarbazine, Reioxafine, Rituximab, Satriplatin, SB-743921, SmII , Sorafinib, Streptozocin, Sunitinib, Tamoxifen, Taxotere, Taxol, Temozolomide, Teniposide, Testolactone, Testosterone, Tezacitabine, 6-Thioguanine, Thiotepa, Tipifarnib, Topotecan, Toremifene, Tositumomab, Trastuzumab, Triamcinolone, Triapine, Triethylenemelamine, Triethylenethiophosphoramine, Trimidox, Uracil mustard, Vinblastine, Vincristine, Vindesine, and Vinoreibine.
5. A method of treating, or slowing the progression of, a disease by inhibiting mammalian Target Of Rapamycin in a patient, said method comprising administering a therapeutically effective amount of at least one compound of claim 1 , or a pharmaceuticaliy acceptable salt, solvate, ester or prodrug of the compound, to a patient in need thereof.
6. A method of treatment of a disease selected from the group consisting of proliferative inflammatory diseases, allergic diseases, obstructive airways diseases, diseases related to transplant rejection and diseases that respond to inhibition of mTOR, comprising administering a therapeutically effective amount of compound of claim 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof to a patient in need of such treatment.
7. A method of treating a disease by inhibiting a mTOR, comprising administering to a patient in need of such treatment an amount of a first compound of claim 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, said second compound being an anti-cancer agent; wherein the amounts of the first compound and said second compound result in a therapeutic effect.
8. The method according to claim 7, wherein the one or more anti-cancer agents are selected from the group consisting of Adriamycin, Altretamine, Amidox, AminogSutethimide, Amsacrine, Anastrazole, Antibodies to EGFR, 3-AP, Aphidicolon, Ara-C, Arsenic trioxide, L-Asparaginase, Bevacizumab, Bleomycin, BMS 214662, Bortezomib, Busuifan, Campath, Camptostar, Capecitabine, Carboplatin, Carmustine, Centrosome associated protein E ("CENP-E") inhibitors, Cetuximab, Cladribine, Chlorambucil, Chiormethine, Chlorotrianisene, Cisplatin, Clofarabine, cyclophosphamide, Cytarabine, a Cytostatic agent, Cytoxan, Dacarbazine, Dactinomycin, Daunorubicin, Dasatinib, Deforolimus, Deoxycoformycin, Didox, Diethylstilbestroi, Docetaxel, Doxorubicin, Dromostanolone, Droloxafine, Epirubicin, Epothilones, ERK inhibitors, Erlotinib, Etoposide, 17α-Ethiny!estradiol, Estramustine, Exemestane, Fioxuridine, Fludarabine, Fludarabine phosphate, 5-F!uorouracil, Fluoxymesterone, Flutamide, Fulvestrant, Gefitinib, Gemcitabine, Gemtuzumab ozogamcicin, Goserelin, GSK-923295, Hexamethylmelamine, Hydroxyprogesterone, Hydroxyurea, lbritumomab Tiuxetan, Idarubicin, Ifosfamide, lmatinib mesylate, Intron, Irinotecan, ispinesib, KSP inhibitors, L778,123, Lapatinib, Leucovirin, Leuprolide, Lerozole, Letrazole, Levamisole, Liposomal Doxorubicin, Liposomal, Lomustine, Lonafarnib, Medroxyprogesteroneacetate, Megestrolacetate, Melphalan, 6-Mercaptopurine, Methoxtrexate, Methylprednisolone, Methyltestosterone, Mithramycin, Mitomycin-C, Mitotane, Mitoxantrone, Navelbene, Nilotinib, Oxaliplatin, Paclitaxel, Panitubimab, Pentostatin, Pipobroman, Porfimer, Prednisolone, Prednisone propionate, Procarbazine, Reloxafine, Rituximab, Satriplatin, SB-743921 , SmH , Sorafinib, Streptozocin, Sunitinib, Tamoxifen, Taxotere, Taxol, Temozoiomide, Teniposide, Testolactone, Testosterone, Tezacitabine, 6-Thioguanine, Thiotepa, Tipifarnib, Topotecan, Toremifene, Tositumomab, Trastuzumab, Triamcinolone, Triapine, Triethylenemeiamine, Triethylenethiophosphoramine, Trimidox, Uracil mustard, Vinblastine, Vincristine, Vindesine, and Vinorelbine.
9. A method of treating a cancer comprising administering a therapeutically effective amount of at least one compound of claim 1.
10. The method of claim 9, wherein said cancer is selected from the group consisting of: cancer of the bladder, breast, colon, kidney, liver, lung, small cell lung cancer, non-small ceil lung cancer, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, including squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma and Burkett's lymphoma; acute and chronic myelogenous leukemia, myelodysplastic syndrome and promyelocyte leukemia; fibrosarcoma, rhabdomyosarcoma; mantle ceil lymphoma, myeloma; astrocytoma, neuroblastoma, glioma and schwannomas; melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderma pigmentosum, keratoctanthoma, thyroid follicular cancer, endometrial cancer, gastrointestinal tract cancer and Kaposi's sarcoma.
1 1. A method of inhibiting a mammalian Target Of Rapamycin, comprising administering a therapeutically effective amount of a compound of claim 1.
12. A method of treating a disease by inhibiting a mTOR, comprising administering to a patient in need of such treatment an amount of a first compound represented by the structural Formula I:

R is independently selected from the group consisting of halo, hydroxyl, amino, -CN, H, -(CrC6)alkyl, alkoxy, -C(=O)alkyi, heteroaryl and aryl, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl or halo; R1 is independently selected from the group consisting of heterocycloalkyl, heterocycioalkylalkyl, spiroheterocycloalkyl, heterocyclenyl, -NR3R4, cycloaikyl, heteroaryl, aryl, alkynyi, heterocyclenylalkyi, cycloalkylalkyl, heteroarylaikyl heteroarylalkynyl, spiroheterocycloalkylalkyl, -N-heteroaryl, and arylalkyl, wherein each of said heterocycloalkyl, heterocycloalkyfaJkyl, spiroheterocycloaikyl, heterocycienyl, cycloaikyl, heteroaryl, aryl, alkynyi, heterocycienylalky!, cycloalkylaikyl, heteroarylaikyl, heteroarylalkynyl , -N-heteroaryi and arylalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloaikyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -a]kyl(CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2l -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyl, halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylaikyl, -C(O)-heteroaryl, -C(O)-alkyl-O-alkyl, - aikyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S(O)2-CF3, -N-aikyl, -SOa-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2l -alkyl-NS(O)2-alkyl, alkyl(CO)NS(O)2- cycloalkyl, -CO-CO2H1 -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C(O)-heteroaryl can be unsubstituted or substituted with one or more alkyl; R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3)2CN, trifiuromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl, and arylalkyl; R3 is cycloaikyl or heteroaryl, wherein each of said cycloaikyl or heteroaryl can be unsubstituted or substituted with one or more moieties independently selected from the group consisting of X; and
R4 is H; and an amount of at least one second compound, said second compound being an anti-cancer agent; wherein the amounts of the first compound and said second compound result in a therapeutic effect.
13. The method according to claim 12, wherein the one or more anti-cancer agents are selected from the group consisting of Adriamycin, Altretamine, Amidox, Aminoglutethimide, Amsacrine, Anastrazole, Antibodies to EGFR, 3-AP, Aphidicolon, Ara-C, Arsenic trioxide, L-Asparaginase, Bevacizumab, Bleomycin, BMS 214662, Bortezomib, Busulfan, Campath, Camptostar, Capecitabine, Carboplatin, Carmustine, Centrosome associated protein E ("CENP-E") inhibitors, Cetuximab, Cladribine, Chlorambucil, Chlormethine, Chlorotrianisene, Cisplatin, C10farabine, cyclophosphamide, Cytarabine, a Cytostatic agent, Cytoxan, Dacarbazine, Dactinomycin, Daunorubicin, Dasatinib, Deforolimus, Deoxycoformycin, Didox, Diethylstilbestrol, Docetaxel, Doxorubicin, Dromostanoione, Droloxafine, Epirubicin, Epothilone, ERK inhibitors, Erlotinib, Etoposide, 17tx-Ethinylestradiol, Estramustine, Exemestane, Fioxuridine, Fludarabine, Fludarabine phosphate, 5-Fluorouracil, Fluoxymesterone, Flutamide, Fulvestrant, Gefitinib, Gemcitabine, Gemtuzumab ozogamcicin, Goserelin, GSK-923295, Hexamethylmelamine, Hydroxyprogesterone, Hydroxyurea, lbritumomab Tiuxetan, idarubicin, Ifosfamide, lmatinib mesylate, Intron, Irinotecan, ispinesib, KSP inhibitors, L778,123, Lapatinib, Leucovirin, Leuprolide, Lerozoie, Letrazole, Levamisole, Liposomal Doxorubicin, Liposomal, Lomustine, Lonafarnib, Medroxyprogesteroneacetate, Megestrolacetate, Melphatan, 6-Mercaptopurine, Methoxtrexate, Methylprednisolone, Methyltestosterone, Mithramycin, Mitomycin-C, Mitotane, Mitoxantrone, Navelbene, Nilotinib, Oxaliplatin, Paclitaxel, Panitubimab, Pentostatin, Pipobroman, Porfimer, Prednisolone, Prednisone propionate, Procarbazine, Reloxafine, Rituximab, Satriplatin, SB-743921 , SmM , Sorafinib, Streptozocin, Sunitinib, Tamoxifen, Taxotere, Taxol, Temozolomide, Teniposide, Testolactone, Testosterone, Tezacitabine, 6-Thioguanine, Thiotepa, Tipifarnib, Topotecan, Toremifene, Tositumomab, Trastuzumab, Triamcinolone, Triapine, Triethylenemelamine, Triethylenethiophosphoramine, Trimidox, Uracil mustard, Vinblastine, Vincristine, Vindesine, and Vinorelbine.
14. A method of treating a cancer comprising administering a therapeutically effective amount of at least one compound represented by the structural Formula I: 
Formula I or a pharmaceutically acceptable salt, solvate, ester, or prodrug thereof, wherein: R is independently selected from the group consisting of halo, hydroxyl, amino,
-CN, H, -(CrCβ)alkyΙ, alkoxy, -C(=O)a!kyl, heteroaryl and aryi, wherein each of said heteroaryl and aryl can be unsubstituted or substituted with one or more alkyl or halo;
R1 is independently selected from the group consisting of heterocycloalkyl, heterocycloalkylalkyl, spiroheterocycloalkyl, heterocyclenyl, -NR3R4, cycloalky!, heteroaryl, aryl, alkynyl, heterocyclenylaikyl, cycloalky I alkyl, heteroarylalky^ heteroarylalkynyl, spiroheterocycloalkylalkyi, -N-heteroaryl, and arylalkyl, wherein each of said heterocycloalkyl, heterocycloalkylalkyl, spiroheterocycloalkyl, heterocyclenyl, cycioaikyl, heteroaryl, aryl, alkynyl, heterocyclenylaikyl , cycloalkyialkyl, heteroarylalkyl, heteroaryialkynyl, -N-heteroaryl and arylalkyl can be unsubstituted or substituted with one or moieties independently selected from the group X;
X is alkoxyl, alkyl, -C(O)alkyl, -C(O)-hydroxyalkyl, -C(O)2alkyl, -C(O)2H, hydroxyalkyl, -S(O)2alkyl, hydroxyl, heterocycloalkyl, -NH-heterocycloalkyl, - trihaloalkyl, -dihaloalkyl, -monohaloalkyl, -N-S(O)2-alkyl, -C(O)-heteroaryl, -alkyl- C(O)2H, -alkyl{CO)N(CH3)-O-CH3, -alkyl(CO)-heteroaryl, -C(O)2-alkyl, -alkyl-C(O)-NH2j -NH2, heteroaryl, -alkyl-CN, -C(O)2-arylalkyi( halo, carboxyesteralkyl, -C(O)-NH2, - alkyl-C(O)2alkyl, heteroarylalkyl, -C(O)-heteroaryl, -C(O)-alkyi-O-alkyl, - alkyl(CO)NS(O)2-cycloalkyl, -alkyl(CO)N-S{O)2-CF3, -N-alkyl. -SO2-cycloalkyl, - alkyl(CO)NS(O)2-alkyl, -alkyl-C(O)-N(alkyl)2, -alkyl-NS<O)2-alkyl, alky!(CO)NS(O)2- cycloalkyl, -CO-CO2H, -C(O)2-alkyl-aryl, -SO2-CF3 or -C(O)H, wherein each of said heterocycloalkyl, heteroaryl or -C (O)- heteroaryl can be unsubstituted or substituted with one or more alkyl;
R2 is heteroaryl or aryl, wherein each of said heteroaryl or aryl can be unsubstituted or independently substituted with one or more moieties independently selected from the group consisting of alkyl, alkoxyl, -CN, aryloxyl, aryl, halo, hydroxyl, -C(CH3^CN, trifluromethyl, difluromethyl, monofluromethyl,_heterocycloalkyl and arylalkyl;
R3 is cycloalkyl or heteroaryi, wherein each of said cycloalkyl or heteroaryl can be unsubstituted or substituted with one or more moieties independently selected from the group consisting of X; and R4 is H.
15. The method of claim 14, wherein said cancer is selected from the group consisting of: cancer of the bladder, breast, colon, kidney, liver, lung, small cell lung cancer, non-smail cell lung cancer, head and neck, esophagus, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, including squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma and Burkett's lymphoma; acute and chronic myelogenous leukemia, myeiodysplastic syndrome and promyelocytic leukemia; fibrosarcoma, rhabdomyosarcoma; mantle cell lymphoma, myeloma; astrocytoma, neuroblastoma, glioma and schwannomas; melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderma pigmentosum, keratoctanthoma, thyroid follicular cancer, endometrial cancer, gastrointestinal tract cancer and Kaposi's sarcoma.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10715010.4A EP2417138B1 (en) | 2009-04-09 | 2010-04-08 | Pyrazolo[1, 5-a]pyrimidine derivatives as mtor inhibitors |
| US13/263,193 US8591943B2 (en) | 2009-04-09 | 2010-04-08 | Pyrazolo[1,5-a]pyrimidine derivatives as mTOR inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16809309P | 2009-04-09 | 2009-04-09 | |
| US61/168,093 | 2009-04-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010118207A1 true WO2010118207A1 (en) | 2010-10-14 |
Family
ID=42936570
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/030350 WO2010118207A1 (en) | 2009-04-09 | 2010-04-08 | Pyrazolo [1, 5-a] pyrimidine derivatives as mtor inhibitors |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8591943B2 (en) |
| EP (1) | EP2417138B1 (en) |
| TW (1) | TW201102392A (en) |
| WO (1) | WO2010118207A1 (en) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013016164A1 (en) * | 2011-07-26 | 2013-01-31 | Merck Sharp & Dohme Corp. | FUSED TRICYCLIC COMPOUNDS AS mTOR INHIBITORS |
| US20130150362A1 (en) * | 2010-08-23 | 2013-06-13 | Merck Sharp & Dohme Corp. | NOVEL PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVES AS mTOR INHIBITORS |
| WO2013184876A1 (en) | 2012-06-06 | 2013-12-12 | Constellation Pharmaceuticals, Inc. | Benzo [c] isoxazoloazepine bromodomain inhibitors and uses thereof |
| US8871754B2 (en) | 2012-11-19 | 2014-10-28 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2015037716A1 (en) | 2013-09-12 | 2015-03-19 | 住友化学株式会社 | Nitrogen-containing saturated heterocyclic compound |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2016142508A1 (en) * | 2015-03-11 | 2016-09-15 | Centre Léon-Bérard | Composition for treating pancreatic neuroendocrine tumours |
| US9556169B2 (en) | 2012-11-19 | 2017-01-31 | Novartis Ag | Compounds and compositions for the treatment of parasitic diseases |
| JP2017507980A (en) * | 2014-03-17 | 2017-03-23 | レクシコン ファーマシューティカルズ インコーポレイテッド | Inhibitor of adapter-related kinase 1, composition containing the same, and method of use thereof |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| JP2019502678A (en) * | 2015-12-10 | 2019-01-31 | ユーシービー バイオファルマ エスピーアールエル | Hexahydropyrazinotriazinone derivatives as kinase inhibitors |
| WO2019054427A1 (en) | 2017-09-14 | 2019-03-21 | 第一三共株式会社 | Compound having cyclic structure |
| US20200206188A1 (en) * | 2012-03-23 | 2020-07-02 | Dennis M. Brown | Compositions and methods to improve the therapeutic benefit of indirubin and analogs thereof, including meisoindigo |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| CN112010860A (en) * | 2020-08-05 | 2020-12-01 | 南京纳丁菲医药科技有限公司 | Benzyloxypyrazolopyrimidine compounds, pharmaceutical compositions and uses thereof |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| JP2021506954A (en) * | 2017-12-15 | 2021-02-22 | ピラミッド バイオサイエンシズ インコーポレイテッド | 5- (2- (2,5-difluorophenyl) pyrrolidine-1-yl) -3- (1H-pyrazole-1-yl) pyrazolo [1,5] as a TRK kinase inhibitor for the treatment of cancer -A] Pyrimidine derivatives and related compounds |
| WO2021122745A1 (en) | 2019-12-16 | 2021-06-24 | Carrick Therapeutics Limited | 4-[[(7-aminopyrazolo[1,5-a]pyrimidin-5-yl)amino]methyl]piperidin-3-ol compounds and their therapeutic use |
| WO2021216454A1 (en) * | 2020-04-21 | 2021-10-28 | Lexicon Pharmaceuticals, Inc. | 4-(3-(pyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl)piperazine for use in the the treatment of cov-229e or cov-oc43 coronaviruses infections |
| CN114423756A (en) * | 2019-06-28 | 2022-04-29 | 上海瑛派药业有限公司 | Substituted fused heteroaromatic bicyclic compounds as kinase inhibitors and uses thereof |
| WO2023134692A1 (en) * | 2022-01-13 | 2023-07-20 | 浙江同源康医药股份有限公司 | Polycyclic compound and use thereof |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011090935A1 (en) | 2010-01-19 | 2011-07-28 | Merck Sharp & Dohme Corp. | PYRAZOLO[1,5-a]PYRIMIDINE COMPOUNDS AS mTOR INHIBITORS |
| CA2898294C (en) * | 2013-02-21 | 2020-06-09 | Calitor Sciences, Llc | Heteroaromatic compounds as pi3 kinase modulators |
| US9944648B2 (en) | 2014-01-09 | 2018-04-17 | Intra-Cellular Therapies, Inc. | Organic compounds |
| TWI820146B (en) * | 2018-06-15 | 2023-11-01 | 瑞典商阿斯特捷利康公司 | Purinone compounds and their use in treating cancer |
| CA3111980A1 (en) | 2018-09-10 | 2020-03-19 | Mirati Therapeutics, Inc. | Combination therapies |
| CA3134814A1 (en) | 2019-03-26 | 2020-10-01 | Ventyx Biosciences, Inc. | Tyk2 pseudokinase ligands |
| AU2020378345A1 (en) | 2019-11-08 | 2022-06-02 | Ventyx Biosciences, Inc. | TYK2 pseudokinase ligands |
| CN113372531B (en) * | 2021-05-13 | 2022-11-25 | 眉山尤博瑞新材料有限公司 | Polyurethane with increased modulus after water absorption and preparation method and application thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006090169A1 (en) * | 2005-02-25 | 2006-08-31 | Kudos Pharmaceuticals Limited | 2,4-diamino-pyridopyrimidine derivatives and their use as mtor inhibitors |
| US20070082900A1 (en) * | 2005-10-06 | 2007-04-12 | Schering Corporation | Methods for inhibiting protein kinases |
| WO2007044449A2 (en) * | 2005-10-06 | 2007-04-19 | Schering Corporation | Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors |
| WO2007061737A2 (en) * | 2005-11-17 | 2007-05-31 | Osi Pharmaceuticals, Inc. | FUSED BICYCLIC mTOR INHIBITORS |
| WO2007087395A2 (en) * | 2006-01-25 | 2007-08-02 | Osi Pharmaceuticals, Inc. | UNSATURATED mTOR INHIBITORS |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HU229024B1 (en) | 1996-07-24 | 2013-07-29 | Bristol Myers Squibb Pharma Co | Azolo-pyridimidines, pharmaceutical compositions containing the same and use thereof |
| US20020041880A1 (en) | 2000-07-05 | 2002-04-11 | Defeo-Jones Deborah | Method of treating cancer |
| WO2003091256A1 (en) | 2002-04-23 | 2003-11-06 | Shionogi & Co., Ltd. | PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVE AND NAD(P)H OXIDASE INHIBITOR CONTAINING THE SAME |
| EP1534710B1 (en) | 2002-09-04 | 2007-10-24 | Schering Corporation | Pyrazolopyrimidines as cyclin dependent kinase inhibitors |
| US7119200B2 (en) | 2002-09-04 | 2006-10-10 | Schering Corporation | Pyrazolopyrimidines as cyclin dependent kinase inhibitors |
| EP1537116B1 (en) | 2002-09-04 | 2010-06-02 | Schering Corporation | Pyrazolopyrimidines suitable for the treatment of cancer diseases |
| CA2527136A1 (en) | 2003-06-03 | 2004-12-09 | Basf Aktiengesellschaft | Substituted pyrazolopyrimidines, methods for the production thereof, use of the same for controlling pathogenic fungi, and agents containing said compounds |
| US20050222171A1 (en) | 2004-01-22 | 2005-10-06 | Guido Bold | Organic compounds |
| PE20051089A1 (en) | 2004-01-22 | 2006-01-25 | Novartis Ag | PYRAZOLE [1,5-A] PYRIMIDIN-7-IL-AMINE DERIVATIVES AS PROTEIN KINASE INHIBITORS |
| GB0515026D0 (en) | 2005-07-21 | 2005-08-31 | Novartis Ag | Organic compounds |
| EP1940839B1 (en) | 2005-10-07 | 2013-07-31 | Exelixis, Inc. | PYRIDOPYRIMIDINONE INHIBITORS OF PI3Kalpha |
| GB0525080D0 (en) | 2005-12-09 | 2006-01-18 | Astrazeneca Ab | Pyrimidine derivatives |
| CA2657200A1 (en) | 2006-07-28 | 2008-01-31 | Novartis Ag | 2,4-substituted quinazolines as lipid kinase inhibitors |
| WO2011002887A1 (en) * | 2009-07-02 | 2011-01-06 | Schering Corporation | FUSED TRICYCLIC COMPOUNDS AS NOVEL mTOR INHIBITORS |
-
2010
- 2010-04-08 US US13/263,193 patent/US8591943B2/en active Active
- 2010-04-08 WO PCT/US2010/030350 patent/WO2010118207A1/en active Application Filing
- 2010-04-08 EP EP10715010.4A patent/EP2417138B1/en active Active
- 2010-04-09 TW TW099111183A patent/TW201102392A/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006090169A1 (en) * | 2005-02-25 | 2006-08-31 | Kudos Pharmaceuticals Limited | 2,4-diamino-pyridopyrimidine derivatives and their use as mtor inhibitors |
| US20070082900A1 (en) * | 2005-10-06 | 2007-04-12 | Schering Corporation | Methods for inhibiting protein kinases |
| WO2007044449A2 (en) * | 2005-10-06 | 2007-04-19 | Schering Corporation | Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors |
| WO2007061737A2 (en) * | 2005-11-17 | 2007-05-31 | Osi Pharmaceuticals, Inc. | FUSED BICYCLIC mTOR INHIBITORS |
| WO2007087395A2 (en) * | 2006-01-25 | 2007-08-02 | Osi Pharmaceuticals, Inc. | UNSATURATED mTOR INHIBITORS |
Non-Patent Citations (9)
| Title |
|---|
| A. L. BINGHAM ET AL., CHEM. COMMUN., 2001, pages 603 - 604 |
| CANCER RESEARCH, vol. 57, 1997, pages 3375 |
| J. CELL SCI., vol. 108, 1995, pages 2897 |
| M. CAIRA ET AL., J. PHARMACEUTICAL SCI., vol. 93, no. 3, 2004, pages 601 - 611 |
| P. GOULD, INTERNATIONAL J. OF PHARMACEUTICS, vol. 33, 1986, pages 201 - 217 |
| S. BERGE ET AL., JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 66, no. 1, 1977, pages 1 - 19 |
| T. HIGUCHI; V. STELLA: "Pro-drugs as Novel Delivery Systems", A.C.S. SYMPOSIUM SERIES, 1987 |
| T. HIGUCHI; W. STELLA: "Pro-drugs as Novel Delivery Systems", A.C.S. SYMPOSIUM SERIES, vol. 14 |
| TONDER ET AL., AAPS PHARMSCITECH., vol. 12, 2004 |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130150362A1 (en) * | 2010-08-23 | 2013-06-13 | Merck Sharp & Dohme Corp. | NOVEL PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVES AS mTOR INHIBITORS |
| EP2608669A1 (en) * | 2010-08-23 | 2013-07-03 | Merck Sharp & Dohme Corp. | NOVEL PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVES AS mTOR INHIBITORS |
| EP2608669A4 (en) * | 2010-08-23 | 2014-01-22 | Merck Sharp & Dohme | NOVEL PYRAZOLO[1,5-a]PYRIMIDINE DERIVATIVES AS mTOR INHIBITORS |
| US8883801B2 (en) * | 2010-08-23 | 2014-11-11 | Merck Sharp & Dohme Corp. | Substituted pyrazolo[1,5-a]pyrimidines as mTOR inhibitors |
| US8901142B2 (en) | 2011-07-26 | 2014-12-02 | Merck Sharp & Dohme Corp. | Fused tricyclic compounds as mTOR inhibitors |
| WO2013016164A1 (en) * | 2011-07-26 | 2013-01-31 | Merck Sharp & Dohme Corp. | FUSED TRICYCLIC COMPOUNDS AS mTOR INHIBITORS |
| US20200206188A1 (en) * | 2012-03-23 | 2020-07-02 | Dennis M. Brown | Compositions and methods to improve the therapeutic benefit of indirubin and analogs thereof, including meisoindigo |
| US9255108B2 (en) | 2012-04-10 | 2016-02-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2013184876A1 (en) | 2012-06-06 | 2013-12-12 | Constellation Pharmaceuticals, Inc. | Benzo [c] isoxazoloazepine bromodomain inhibitors and uses thereof |
| US8871754B2 (en) | 2012-11-19 | 2014-10-28 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| US9556169B2 (en) | 2012-11-19 | 2017-01-31 | Novartis Ag | Compounds and compositions for the treatment of parasitic diseases |
| US9926314B2 (en) | 2012-11-19 | 2018-03-27 | Novartis Ag | Compounds and compositions for the treatment of parasitic diseases |
| WO2015037716A1 (en) | 2013-09-12 | 2015-03-19 | 住友化学株式会社 | Nitrogen-containing saturated heterocyclic compound |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10329299B2 (en) | 2013-10-04 | 2019-06-25 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9828377B2 (en) | 2013-10-04 | 2017-11-28 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| JP2017507980A (en) * | 2014-03-17 | 2017-03-23 | レクシコン ファーマシューティカルズ インコーポレイテッド | Inhibitor of adapter-related kinase 1, composition containing the same, and method of use thereof |
| US11541059B2 (en) | 2014-03-19 | 2023-01-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10675286B2 (en) | 2014-03-19 | 2020-06-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10941162B2 (en) | 2014-10-03 | 2021-03-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10253047B2 (en) | 2014-10-03 | 2019-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| FR3033499A1 (en) * | 2015-03-11 | 2016-09-16 | Centre Leon-Berard | COMPOSITION FOR THE TREATMENT OF PANCREATIC NEUROENDOCRINE TUMORS |
| WO2016142508A1 (en) * | 2015-03-11 | 2016-09-15 | Centre Léon-Bérard | Composition for treating pancreatic neuroendocrine tumours |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US11939333B2 (en) | 2015-09-14 | 2024-03-26 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US11247995B2 (en) | 2015-09-14 | 2022-02-15 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| JP2019502678A (en) * | 2015-12-10 | 2019-01-31 | ユーシービー バイオファルマ エスピーアールエル | Hexahydropyrazinotriazinone derivatives as kinase inhibitors |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2019054427A1 (en) | 2017-09-14 | 2019-03-21 | 第一三共株式会社 | Compound having cyclic structure |
| JP2021506954A (en) * | 2017-12-15 | 2021-02-22 | ピラミッド バイオサイエンシズ インコーポレイテッド | 5- (2- (2,5-difluorophenyl) pyrrolidine-1-yl) -3- (1H-pyrazole-1-yl) pyrazolo [1,5] as a TRK kinase inhibitor for the treatment of cancer -A] Pyrimidine derivatives and related compounds |
| JP7321183B2 (en) | 2017-12-15 | 2023-08-04 | ピラミッド バイオサイエンシズ インコーポレイテッド | 5-(2-(2,5-difluorophenyl)pyrrolidin-1-yl)-3-(1H-pyrazol-1-yl)pyrazolo[1,5] as TRK kinase inhibitors for treating cancer -A] pyrimidine derivatives and related compounds |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| EP3997087A4 (en) * | 2019-06-28 | 2023-05-31 | Impact Therapeutics (Shanghai), Inc | Substituted fused heteroaromatic bicyclic compounds as kinase inhibitors and the use thereof |
| CN114423756A (en) * | 2019-06-28 | 2022-04-29 | 上海瑛派药业有限公司 | Substituted fused heteroaromatic bicyclic compounds as kinase inhibitors and uses thereof |
| WO2021122745A1 (en) | 2019-12-16 | 2021-06-24 | Carrick Therapeutics Limited | 4-[[(7-aminopyrazolo[1,5-a]pyrimidin-5-yl)amino]methyl]piperidin-3-ol compounds and their therapeutic use |
| WO2021216454A1 (en) * | 2020-04-21 | 2021-10-28 | Lexicon Pharmaceuticals, Inc. | 4-(3-(pyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl)piperazine for use in the the treatment of cov-229e or cov-oc43 coronaviruses infections |
| CN112010860B (en) * | 2020-08-05 | 2023-03-10 | 南京纳丁菲医药科技有限公司 | Benzyloxypyrazolopyrimidine compounds, pharmaceutical compositions and uses thereof |
| CN112010860A (en) * | 2020-08-05 | 2020-12-01 | 南京纳丁菲医药科技有限公司 | Benzyloxypyrazolopyrimidine compounds, pharmaceutical compositions and uses thereof |
| WO2023134692A1 (en) * | 2022-01-13 | 2023-07-20 | 浙江同源康医药股份有限公司 | Polycyclic compound and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US8591943B2 (en) | 2013-11-26 |
| EP2417138B1 (en) | 2019-11-27 |
| TW201102392A (en) | 2011-01-16 |
| US20120114739A1 (en) | 2012-05-10 |
| EP2417138A1 (en) | 2012-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2417138A1 (en) | Pyrazolo [1, 5-a] pyrimidine derivatives as mtor inhibitors | |
| JP7229257B2 (en) | GCN2 inhibitors and uses thereof | |
| KR101659193B1 (en) | Heteroaryl pyridone and aza-pyridone compounds as inhibitors of btk activity | |
| AU2009276339B2 (en) | Pyrimidine compounds, compositions and methods of use | |
| JP6159028B2 (en) | Syk inhibitor | |
| JP5152922B2 (en) | Use of pyrazolo [1,5-A] pyrimidine derivatives to inhibit protein kinases and methods for inhibiting protein kinases | |
| JP5109109B2 (en) | A novel imidazopyrazine as a cyclin-dependent kinase inhibitor | |
| WO2008156614A2 (en) | Imidazopyrazines as protein kinase inhibitors | |
| KR20190018645A (en) | Positive allosteric modulator of muscarinic acetylcholine receptor M4 | |
| CA2628455A1 (en) | Imidazopyrazines as protein kinase inhibitors | |
| CA2665127A1 (en) | Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors | |
| EP2424859A1 (en) | Inhibitors of pi3 kinase and / or mtor | |
| AU2008279776A2 (en) | Heterocyclic amide compounds as protein kinase inhibitors | |
| CN113683629B (en) | Substituted heteroaryl compounds, compositions and uses thereof | |
| EP2448942B1 (en) | FUSED TRICYCLIC COMPOUNDS AS mTOR INHIBITORS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10715010 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010715010 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13263193 Country of ref document: US |