WO2010065681A1 - Inhibitors of hcv ns5a - Google Patents
Inhibitors of hcv ns5a Download PDFInfo
- Publication number
- WO2010065681A1 WO2010065681A1 PCT/US2009/066467 US2009066467W WO2010065681A1 WO 2010065681 A1 WO2010065681 A1 WO 2010065681A1 US 2009066467 W US2009066467 W US 2009066467W WO 2010065681 A1 WO2010065681 A1 WO 2010065681A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- heteroaryl
- compound
- heterocycle
- independently
- Prior art date
Links
- 0 CC1CI*(*)CC1 Chemical compound CC1CI*(*)CC1 0.000 description 22
- LLPKQRMDOFYSGZ-UHFFFAOYSA-N Cc1cnc(C)[nH]1 Chemical compound Cc1cnc(C)[nH]1 LLPKQRMDOFYSGZ-UHFFFAOYSA-N 0.000 description 2
- GAMKNDOVVQFPJJ-UHFFFAOYSA-N Cc1n[nH]c(I)n1 Chemical compound Cc1n[nH]c(I)n1 GAMKNDOVVQFPJJ-UHFFFAOYSA-N 0.000 description 2
- ZVYGJRFJPXLXEA-CQSZACIVSA-N CC(C)(C)OC(N[C@@H](C(N1CCCC1)=O)c1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@@H](C(N1CCCC1)=O)c1ccccc1)=O ZVYGJRFJPXLXEA-CQSZACIVSA-N 0.000 description 1
- GAFZEKJMUQTIAF-QXLYETOESA-N CC(C)(C)OC(N[C@H](C(N(CCC1)C1C(N(CC1)CCC1c(cc1)ccc1-c1cnc(C(CCC2)N2C([C@@H](c2ccccc2)NC(OC(C)(C)C)=O)=O)[nH]1)=O)=O)c1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@H](C(N(CCC1)C1C(N(CC1)CCC1c(cc1)ccc1-c1cnc(C(CCC2)N2C([C@@H](c2ccccc2)NC(OC(C)(C)C)=O)=O)[nH]1)=O)=O)c1ccccc1)=O GAFZEKJMUQTIAF-QXLYETOESA-N 0.000 description 1
- ZKALWXKGVATMBC-UHFFFAOYSA-N CC(CC1)(CC2)CCC12N Chemical compound CC(CC1)(CC2)CCC12N ZKALWXKGVATMBC-UHFFFAOYSA-N 0.000 description 1
- FBXOMYIYXKYYDH-MWSTZMHHSA-N CC(NC=O)NC([C@H](CCC1)N1C([C@@H](c1ccccc1)NC(OC(C)(C)C)=O)=O)=N Chemical compound CC(NC=O)NC([C@H](CCC1)N1C([C@@H](c1ccccc1)NC(OC(C)(C)C)=O)=O)=N FBXOMYIYXKYYDH-MWSTZMHHSA-N 0.000 description 1
- GMZLBOXZHZKHIP-SVSMMVOBSA-N CC(NCC(CCC1)N1C([C@@H](c1ccccc1)NC(C1CC1)=O)=O)NC([C@H](CCC1)N1C([C@@H](c1ccccc1)NC(C1CC1)=O)=O)=N Chemical compound CC(NCC(CCC1)N1C([C@@H](c1ccccc1)NC(C1CC1)=O)=O)NC([C@H](CCC1)N1C([C@@H](c1ccccc1)NC(C1CC1)=O)=O)=N GMZLBOXZHZKHIP-SVSMMVOBSA-N 0.000 description 1
- UAEPNZWRGJTJPN-UHFFFAOYSA-N CC1CCCCC1 Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 1
- SHZRQFKYUKEHCK-UHFFFAOYSA-N Cc([nH]1)ccc1[IH]Cc1ncc(C)[nH]1 Chemical compound Cc([nH]1)ccc1[IH]Cc1ncc(C)[nH]1 SHZRQFKYUKEHCK-UHFFFAOYSA-N 0.000 description 1
- DMJJJHIFADRWEF-UHFFFAOYSA-N Cc([nH]1)nc2c1nc(C)cc2 Chemical compound Cc([nH]1)nc2c1nc(C)cc2 DMJJJHIFADRWEF-UHFFFAOYSA-N 0.000 description 1
- JKVQOVGFARCXAU-UHFFFAOYSA-N Cc([nH]1)nc2c1nc(C)cn2 Chemical compound Cc([nH]1)nc2c1nc(C)cn2 JKVQOVGFARCXAU-UHFFFAOYSA-N 0.000 description 1
- VWDPVZSFDURBLW-UHFFFAOYSA-N Cc([nH]1)nc2c1nc(C)nc2 Chemical compound Cc([nH]1)nc2c1nc(C)nc2 VWDPVZSFDURBLW-UHFFFAOYSA-N 0.000 description 1
- ZGZXJLBRWYRNNE-UHFFFAOYSA-N Cc([o]1)cnc1I Chemical compound Cc([o]1)cnc1I ZGZXJLBRWYRNNE-UHFFFAOYSA-N 0.000 description 1
- HUSIZGWEEAVFKT-UHFFFAOYSA-N Cc([s]1)cnc1I Chemical compound Cc([s]1)cnc1I HUSIZGWEEAVFKT-UHFFFAOYSA-N 0.000 description 1
- SOPCMQAOTNXTBK-UHFFFAOYSA-N Cc1n[nH]c(I)c1 Chemical compound Cc1n[nH]c(I)c1 SOPCMQAOTNXTBK-UHFFFAOYSA-N 0.000 description 1
- LWZFOIDXBIQJDH-UHFFFAOYSA-N Cc1n[nH]c([IH]Cc2nc(C)c[nH]2)c1 Chemical compound Cc1n[nH]c([IH]Cc2nc(C)c[nH]2)c1 LWZFOIDXBIQJDH-UHFFFAOYSA-N 0.000 description 1
- YZWJUWITJYOVMH-UHFFFAOYSA-N Cc1nc(CCc2c-3ccc(CCc4nc(ccc5c6ccc(C)c5)c6[nH]4)c2)c-3[nH]1 Chemical compound Cc1nc(CCc2c-3ccc(CCc4nc(ccc5c6ccc(C)c5)c6[nH]4)c2)c-3[nH]1 YZWJUWITJYOVMH-UHFFFAOYSA-N 0.000 description 1
- HIQPWBUJSYNVRL-UHFFFAOYSA-N Cc1nc(CN(c2c-3ccc(C)c2)N)c-3[nH]1 Chemical compound Cc1nc(CN(c2c-3ccc(C)c2)N)c-3[nH]1 HIQPWBUJSYNVRL-UHFFFAOYSA-N 0.000 description 1
- UWXXUKDLTRPMMF-UHFFFAOYSA-N Cc1nc(COc2c-3ccc(C)c2)c-3[nH]1 Chemical compound Cc1nc(COc2c-3ccc(C)c2)c-3[nH]1 UWXXUKDLTRPMMF-UHFFFAOYSA-N 0.000 description 1
- MVHOAOSHABGEFL-UHFFFAOYSA-N Cc1nc(ccc(C)c2)c2[nH]1 Chemical compound Cc1nc(ccc(C)c2)c2[nH]1 MVHOAOSHABGEFL-UHFFFAOYSA-N 0.000 description 1
- BHTLHVMHQVCJCE-UHFFFAOYSA-N Cc1nc(cnc(C)c2)c2[nH]1 Chemical compound Cc1nc(cnc(C)c2)c2[nH]1 BHTLHVMHQVCJCE-UHFFFAOYSA-N 0.000 description 1
- LZSXKOQVSYULPZ-UHFFFAOYSA-N Cc1nc(ncc(C)c2)c2[nH]1 Chemical compound Cc1nc(ncc(C)c2)c2[nH]1 LZSXKOQVSYULPZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the invention relates to compounds useful for inhibiting hepatitis C virus (“HCV”) replication, particularly functions of the non-structural 5 A (“NS5A”) protein of HCV.
- HCV hepatitis C virus
- HCV is a single-stranded RNA virus that is a member of the Flaviviridae family.
- the virus shows extensive genetic heterogeneity as there are currently seven identified genotypes and more than 50 identified subtypes.
- viral RNA is translated into a polyprotein that is cleaved into ten individual proteins.
- the core (C) protein and the envelope glycoproteins El and E2.
- p7 an integral membrane protein, follows El and E2.
- there are six nonstructural proteins, NS2, NS3, NS4A, NS4B, NS5A and NS5B which play a functional role in the HCV lifecycle. (see, for example, Lindenbach, B.D. and CM. Rice, Nature. 436:933- 938, 2005).
- HCV infection is a serious health issue. It is estimated that 170 million people worldwide are chronically infected with HCV. HCV infection can lead to chronic hepatitis, cirrhosis, liver failure and hepatocellular carcinoma. Chronic HCV infection is thus a major worldwide cause of liver-related premature mortality.
- a and A' are independently selected from the group consisting of a single bond, -(CR 2 ) n -C(O)-(CR 2 ) p - -(CR 2 ) n -O-(CR 2 ) p - -(CR 2 ) n -N(R N )-(CR 2 ) p - -(CR 2 ) n -S(O) k -N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-N(R N )-(CR 2 )p-, -(CR 2 ) n -N(R N )-C(O)-N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-O-(CR 2 ) p -, -(CR 2 ) n -N(R N )-C(O)-O-(CR
- heteroaryl group selected from the group consisting of
- X 1 is CH 2 , NH, O or S,
- Y 1 , Y 2 and Z 1 are each independently CH or N,
- X 2 is NH, O or S
- a and b are independently 0, 1, 2, or 3 with the proviso that a and b are not both 0, optionally includes 1 or 2 nitrogens as heteroatoms on the phenyl residue,
- the carbons of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- the nitrogens, if present, of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide,
- a and b are independently 1, 2, or 3.
- c and d are independently 1 or 2
- n and p are independently 0, 1, 2 or 3
- k 0, 1, or 2
- each R is independently selected from the group consisting of hydrogen, -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- each R N is independently selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide, and
- the A-B-A' can be any of:
- B is Q or Q Q , wherein each Q is independently selected from the group consisting of a cycloalkyl group, cycloalkenyl group, heterocycle, aryl group or heteroaryl group, with the proviso that only one Q is a six member aromatic ring when B is Q Q and with the proviso that if B is Q Q , any Q is that is poly cyclic is connected to the remainder of the molecule through only one cycle of the poly cycle;
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, aralkyl and a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl, wherein, each hetero atom, if present, is independently N, O or S, each of R c , R d , R e and R f may optionally be substituted by Ci to C 8 alkyl, Ci to C 8 heteroalkyl, aralkyl, or a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl and wherein each heteroatom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring, and R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring;
- Y and Y' are each independently carbon or nitrogen;
- Z and Z' are independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, 1-3 amino acids, -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 , -U-(CR 4 2 ),-R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 , wherein,
- U is selected from the group consisting of -C(O)-, -C(S)- and -S(O) 2 -,
- each R 4 , R 5 and R 7 is independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 8 is selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, -C(O)-R 81 , -C(S)-R 81 , -C(O)-O-R 81 , -C(O)-N-R 81 2, -S(O) 2 -R 81 and -S(O) 2 -N-R 81 2 , wherein each R 81 is independently chosen from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 7 and R 8 together form a 4-7 membered ring
- each t is independently 0, 1, 2, 3, or 4, and
- u 0, 1, or 2.
- each Q is independently optionally substituted with one or more substituents each independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino, and if Q is not aromatic, it is optionally substituted with oxo.
- substituents each independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulf
- each Q is independently optionally substituted with -CN, -OCF 3 , -OCHF 2 , -CF 3 or -F.
- B is selected from the group consisting of:
- each r and s is independently 0, 1, 2, 3, or 4;
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; and
- each R b is independently Ci-Ci 2 alkyl, hydroxyl, halogen, or oxo.
- each phenyl residue optionally includes 1 or 2 nitrogens as heteroatoms.
- each R a if present, -CN, -OCF 3 , -OCHF 2 , -CF 3 , or -F.
- a and A' are independently selected from the group consisting of a single bond, -(CR2) n -C(O)-(CR2) p -, -(CR2) n -O-(CR2) p -, -(CR 2 )n-N(R N )-(CR2)p-, -(CR2)n-C(O)-N(R N )-(CR 2 )p-, -(CR 2 ) n -N(R N )-C(O)-N(R N )-(CR 2 ) p - and -(CR 2 ) n -N(R N )-C(O)-O-(CR 2 ) p - and a
- heteroaryl group selected from the group consisting of - ⁇ X 1 ,
- a and A' are independently selected
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to Cg alkyl and Ci to Cg heteroalkyl, wherein, each hetero atom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle, and
- R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- R c and R d or R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- R c and R d are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R N is selected from the group consisting of hydrogen, -OH, Ci to C 12 alkyl, Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- R e and R f are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R N is selected from the group consisting of hydrogen, -OH, Ci to
- C 12 alkyl Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- A' is selected from the group consisting of single bond, H H
- R N is selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide;
- ⁇ / optionally includes 1 or 2 nitrogens as heteroatoms
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; and
- r is selected from the group consisting of 0, 1, 2, 3, or 4.
- A' is V H
- A' is selected from the group consisting of
- a and A' are independently selected from the group consisting of single bond, H
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino;
- r is O, 1, 2, 3 or 4;
- each R b is independently C1-C5 alkyl, hydroxyl, halogen, or oxo;
- s is O, 1, 2, 3, 4, 5, or 6;
- each of X 1 and X 2 are independently C or N.
- a and A' are each independently
- a and A' are independently selected from the group consisting of single bond, C(O)-N(R N )-(CR 2 ) P -, wherein R N is selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide;
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; and
- r is O, 1, 2, 3 or 4. 30]
- a and A' are each independently
- each - ⁇ ⁇ - --v- 1 iSs 1 iTndependently a divalent aryl or heteroaryl group which may be polycyclic with varying connective patterns; each r is independently 0, 1, 2, 3, or 4;
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino;
- a and A' are independently selected from the group consisting of a single bond, -(CR 2 ) n -C(O)-(CR 2 ) p - -(CR 2 ) n -O-(CR 2 ) p - -(CR 2 ) n -N(R N )-(CR 2 ) p - -(CR 2 ) n -S(O) k -N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-N(R N )-(CR 2 ) p -, -(CR 2 ) n -N(R N )-C(O)-N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-O-(CR 2 )p-, -(CR 2 ) n -N(R N )-C(O)-O-(CR
- X 1 is CH 2 , NH, O or S,
- Y 1 , Y 2 and Z 1 are each independently CH or N,
- X 2 is NH, O or S
- a and b are independently 0, 1, 2, or 3 with the proviso that a and b are not both 0, optionally includes 1 or 2 nitrogens as heteroatoms on the phenyl residue,
- the carbons of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- the nitrogens, if present, of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide,
- a and b are independently 1, 2, or 3.
- c and d are independently 1 or 2
- n and p are independently 0, 1, 2 or 3
- k 0, 1, or 2
- each R is independently selected from the group consisting of hydrogen, -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- each R N is independently selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide, and
- a or A' being the A-B-A' can be any of:
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to Cg alkyl, Ci to Cs heteroalkyl, aralkyl and a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl, wherein, each hetero atom, if present, is independently N, O or S, each of R c , R d , R e and R f may optionally be substituted by Ci to Cg alkyl, Ci to Cg heteroalkyl, aralkyl, or a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl and wherein each heteroatom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring, and R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring;
- Y and Y' are each independently carbon or nitrogen;
- Z and Z' are independently selected from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, 1-3 amino acids, -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ) t ] u -U-(CR 4 2 ) t -NR 7 -(CR 4 2 ) t -R 8 , -U-(CR 4 2 ) t -R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 , wherein,
- U is selected from the group consisting of -C(O)-, -C(S)- and -S(O) 2 -,
- each R 4 , R 5 and R 7 is independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 8 is selected from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, -C(O)-R 81 , -C(S)-R 81 , -C(O)-O-R 81 , -C(O)-N-R 81 2 , -S(O) 2 -R 81 and -S(O) 2 -N-R 81 2 , wherein each R 81 is independently chosen from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl, optionally, R 7 and R 8 together form a 4-7 membered ring, each t is independently 0, 1, 2, 3, or 4, and u is 0, 1, or 2.
- each - ⁇ - IS 1 ndependently
- each phenyl residue optionally includes 1 or 2 nitrogens as heteroatoms.
- a and A' are independently selected from the group consisting of a single bond, -(CR2) n -C(O)-(CR2) p -, -(CR2) n -O-(CR2) p -, -(CR 2 )n-N(R N )-(CR2)p-, -(CR2)n-C(O)-N(R N )-(CR 2 )p-, -(CR 2 )n-N(R N )-C(O)-N(R N )-(CR 2 )p- and -(CR 2 )n-N(R N )-C(O)-O-(CR 2 )p- and a
- a and A' are independently selected
- a and A' are each independently
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to Cs alkyl and Ci to Cs heteroalkyl, wherein, each hetero atom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle, and
- R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- one of R c and R d or R e and R f are joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- both of R c and R d and R e and R f are joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- R c and R d are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R N is selected from the group consisting of hydrogen, -OH, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- R e and R f are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R N is selected from the group consisting of hydrogen, -OH, Ci to
- C 12 alkyl Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- each R a if present in a compound of any of the second through sixth aspects, is independently -CN, -OCF3, -OCHF 2 , -CF 3 , or -F.
- one of Y and Y' is N.
- both Y and Y' are N.
- Z and Z' in any of the previous aspects are each 1-3 amino acids.
- the amino acids are in the D configuration.
- Z and Z' in any of the previous aspects are each independently selected from the group consisting of -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 , -U-(CR 4 2 ),-R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- Z and Z' are -U-(CR 4 2 ),-NR 5 -(CR 4 2 ),-U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -[C(O)-(CR 4 2 )t-NR 5 -(CR 4 2 )t] u -U-(CR 4 2 )t-NR 7 -(CR 4 2 )t-R 8 .
- Z and Z' are -C(O)-(CR 4 2 ),-NR 5 -(CR 4 2 ),-U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -[C(O)-(CR 4 2 )t-NR 5 -(CR 4 2 )t] u -C(O)-(CR 4 2 )t-NR 7 -(CR 4 2 )t-R 8 .
- Z and Z' are -C(O)-(CR 4 2 ),-NR 5 -(CR 4 2 ),-C(O)-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- Z and Z' are -C(O)-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -(CR 4 2 ) n -C(O)-R 81 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -C(O)-R 81 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -(CR 4 2 ) n -C(O)-O-R 81 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -C(O)-O-R 81 .
- one or both of Z and Z' are -U- (CR 4 2 ) t -R 8 .
- one or both of Z and Z' are - C(O)-(CR 4 2 ) t -R 8 .
- one or both of Z and Z' are -[U-(CR 4 2 )t-NR 5 -(CR 4 2 )t] u -U-(CR 4 2 )t-O-(CR 4 2 )t-R 8 .
- Z and Z' are -U-(CR 4 2 ),-NR 5 -(CR 4 2 ),-U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 )t-NR 5 -(CR 4 2 )t-C(O)-(CR 4 2 )t-O-(CR 4 2 ) t -R 8 .
- Z and Z' are -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 )t-O-(CR 4 2 )t-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -R 8 wherein R 7 and R 8 together form a 4-7 membered ring.
- r is from 0 to 4
- each R a is independently selected from the group consisting of -OH,
- Ci to Ci 2 alkyl Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino;
- a and A' are independently selected from the group consisting of
- a or A' is a heteroaryl group, it is optionally substituted with one or more of the substituents selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino, and
- n and p are independently 0, 1, or 2
- each R is independently selected from the group consisting of hydrogen, -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- R N is selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide, and
- B' is optionally substituted with between 1 and 4 substituents, each independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide, amino and oxo,
- X b is either C or N;
- Y and Y' are each independently carbon or nitrogen;
- Z and Z' are independently selected from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, 1-3 amino acids, -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 , -U-(CR 4 2 ),-R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 , wherein,
- U is selected from the group consisting of -C(O)-, -C(S)- and -S(O) 2 -,
- each R 4 , R 5 and R 7 is independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 8 is selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, -C(O)-R 81 , -C(S)-R 81 , -C(O)-O-R 81 , -C(O)-N-R 81 2 , -S(O) 2 -R 81 and -S(O) 2 -N-R 81 2 , wherein each R 81 is independently chosen from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 7 and R 8 together form a 4-7 membered ring
- each t is independently O, 1, 2, 3, or 4, and
- B' is selected from the group consisting of
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- B' is selected from the group consisting of ⁇ ⁇ V_v- s ⁇ ⁇ N — /- ,- ⁇ — / and
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- B' is selected from the group consisting of - ⁇ - - ⁇ - -
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- the sixteenth aspect of the invention provides a pharmaceutical composition comprising the compounds of the invention.
- the seventeenth aspect of the invention provides the use of the compounds of the invention in the manufacture of a medicament.
- the medicament is for the treatment of hepatitis C.
- the eighteenth aspect of the invention provides a method of treating hepatitis C comprising administering to a subject in need thereof, a therapeutically effective amount of a compound of the invention.
- alkanoyl as used herein contemplates a carbonyl group with a lower alkyl group as a substituent.
- alkenyl as used herein contemplates substituted or unsubstituted, straight and branched chain alkene radicals, including both the E- and Z-forms, containing from two to eight carbon atoms.
- the alkenyl group may be optionally substituted with one or more substituents selected from the group consisting of halogen, -CN, -NO 2 , CO 2 R, C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(O)R, -N(R N )S(O) 2 R, -SR, -C(O)N(R N ) 2 , -OC(O)R, -0C(0)N(R N ) 2 , S(O)R, SO 2 R, -SO 3 R, -S(O) 2 N(R N ) 2 , phosphate, phosphonate, cycloalkyl, cycloalkenyl
- alkoxy contemplates an oxygen with a lower alkyl group as a substituent and includes methoxy, ethoxy, butoxy, trifluromethoxy and the like. It also includes divalent substituents linked to two separated oxygen atoms such as, without limitation, -O-(CH 2 )i. 4 -O-, -0-CF 2 -O-, -0-(CH 2 )L 4 -O-(CH 2 CH 2 -O)L 4 - and
- alkoxycarbonyl as used herein contemplates a carbonyl group with an alkoxy group as a substituent.
- alkyl as used herein contemplates substituted or unsubstituted, straight and branched chain alkyl radicals containing from one to fifteen carbon atoms.
- lower alkyl as used herein contemplates both straight and branched chain alkyl radicals containing from one to six carbon atoms and includes methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert- butyl and the like.
- the alkyl group may be optionally substituted with one or more substituents selected from halogen, -CN, -NO 2 , -C(O) 2 R, -C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(0)R, -N(R N )S(O) 2 R, -SR, -C(0)N(R N ) 2 , -OC(O)R, -0C(0)N(R N ) 2 , -SOR, -SO 2 R, -SO3R, -S(O) 2 N(R N ) 2 , phosphate, phosphonate, cycloalkyl, cycloalkenyl, aryl and heteroaryl.
- substituents selected from halogen, -CN, -NO 2 , -C(O) 2 R, -C(O)R, -O-R, -N(R N ) 2 ,
- alkylene alkenylene and alkynylene as used herein refers to the groups “alkyl,” “alkenyl” and “alkynyl” respectively, when they are divalent, ie, attached to two atoms.
- alkylsulfonyl as used herein contemplates a sulfonyl group which has a lower alkyl group as a substituent.
- alkynyl as used herein contemplates substituted or unsubstituted, straight and branched carbon chain containing from two to eight carbon atoms and having at least one carbon-carbon triple bond.
- alkynyl includes, for example ethynyl, 1- propynyl, 2- propynyl, 1-butynyl, 3 -methyl- 1-butynyl and the like.
- the alkynyl group may be optionally substituted with one or more substituents selected from halo, -CN, NO 2 , CO 2 R, C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(0)R, -N(R N )S(O) 2 R, -SR, -C(0)N(R N ) 2 , -OC(O)R, -0C(0)N(R N ) 2 , -SOR, -SO 2 R, -SO3R, -S(O)2N(R N )2, phosphate, phosphonate, cycloalkyl, cycloalkenyl, aryl and heteroaryl.
- substituents selected from halo, -CN, NO 2 , CO 2 R, C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(0)R, -N(R N
- amino as used herein contemplates a group of the structure -NR N 2 .
- amino acid as used herein contemplates a group of the structure
- the present invention also includes, without limitation, D-configuration amino acids, beta-amino acids, amino acids having side chains as well as all non-natural amino acids known to one skilled in the art.
- aralkyl as used herein contemplates a lower alkyl group which has as a substituent an aromatic group, which aromatic group may be substituted or unsubstituted.
- the aralkyl group may be optionally substituted with one or more substituents selected from halogen, -CN, -NO 2 , -CO 2 R, -C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(O)R, -N(R N )S(O) 2 R, -SR, -
- aryl as used herein contemplates substituted or unsubstituted single-ring and multiple aromatic groups (for example, phenyl, pyridyl and pyrazole, etc.) and polycyclic ring systems (naphthyl and quinolinyl, etc.).
- the polycyclic rings may have two or more rings in which two atoms are common to two adjoining rings (the rings are "fused") wherein at least one of the rings is aromatic, e.g., the other rings can be cycloalkyls, cycloalkenyls, aryl, heterocycles and/or heteroaryls.
- the aryl group may be optionally substituted with one or more substituents selected from halogen, alkyl, -CN, -NO 2 , -CO 2 R, -C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(O)R, - N(R N )S(O) 2 R, -SR, -C(O)N(R N ) 2 ,
- arylsulfonyl contemplates a sulfonyl group which has as a substituent an aryl group. The term is meant to include, without limitation, monovalent as well as multiply valent aryls (eg, divalent aryls).
- cycloalkyl as used herein contemplates substituted or unsubstituted cyclic alkyl radicals containing from three to twelve carbon atoms and includes cyclopropyl, cyclopentyl, cyclohexyl and the like.
- cycloalkyl also includes polycyclic systems having two rings in which two or more atoms are common to two adjoining rings (the rings are "fused").
- the cycloalkyl group may be optionally substituted with one or more substituents selected from halo, -CN, -NO 2 , -CO 2 R, -C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(O)R, -N(R N )S(O) 2 R, -SR, -C(O)N(R N ) 2 , -OC(O)R, -OC(O)N(R N ) 2 , -SOR, -SO 2 R, -S(O) 2 N(R N ) 2 , phosphate, phosphonate, alkyl, cycloalkenyl, aryl and heteroaryl.
- substituents selected from halo, -CN, -NO 2 , -CO 2 R, -C(O)R, -O-R, -N(R N ) 2 , -N(R N
- cycloalkenyl as used herein contemplates substituted or unsubstituted cyclic alkenyl radicals containing from four to twelve carbon atoms in which there is at least one double bond between two of the ring carbons and includes cyclopentenyl, cyclohexenyl and the like.
- cycloalkenyl also includes polycyclic systems having two rings in which two or more atoms are common to two adjoining rings (the rings are "fused").
- the cycloalkenyl group may be optionally substituted with one or more substituents selected from halo, -CN, -NO 2 , -CO 2 R, -C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(O)R, -N(R N )S(O) 2 R, -SR, -C(O)N(R N ) 2 , -OC(O)R, -OC(O)N(R N ) 2 , -SOR, -SO 2 R, -S(O) 2 N(R N ) 2 , phosphate, phosphonate, alkyl, cycloalkenyl, aryl and heteroaryl.
- halo or "halogen” as used herein includes fluorine, chlorine, bromine and iodine.
- heteroalkyl as used herein contemplates an alkyl with one or more heteroatoms.
- heteroatom particularly within a ring system, refers to N, O and S.
- heterocyclic group contemplates substituted or unsubstituted aromatic and non-aromatic cyclic radicals having at least one heteroatom as a ring member.
- Preferred heterocyclic groups are those containing five or six ring atoms which includes at least one hetero atom and includes cyclic amines such as morpholino, piperidino, pyrrolidino and the like and cyclic ethers, such as tetrahydrofuran, tetrahydropyran and the like.
- Aromatic heterocyclic groups also termed "heteroaryl” groups, contemplates single-ring hetero-aromatic groups that may include from one to three heteroatoms, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, triazole, pyrazole, oxodiazole, thiadiazole, pyridine, pyrazine, pyridazine, pyrimidine and the like.
- heteroaryl also includes polycyclic hetero-aromatic systems having two or more rings in which two or more atoms are common to two adjoining rings (the rings are "fused") wherein at least one of the rings is a heteroaryl, e.g., the other rings can be cycloalkyls, cycloalkenyls, aryl, heterocycles and/or heteroaryls.
- polycyclic heteroaromatic systems examples include quinoline, isoquinoline, cinnoline, tetrahydroisoquinoline, quinoxaline, quinazoline, benzimidazole, benzofuran, benzothiophene, benzoxazole, benzothiazole, indazole, purine, benzotriazole, pyrrolepyridine, pyrrazolopyridine and the like.
- the heterocyclic group may be optionally substituted with one or more substituents selected from the group consisting of halo, alkyl, -CN, -NO 2 , -CO 2 R, -C(O)R, -O-R, -N(R N ) 2 , -N(R N )C(O)R, -N(R N )S(O) 2 R, -SR, -C(O)N(R N ) 2 , -OC(O)R, -OC(O)N(R N ) 2 , -SOR, -SO 2 R, -SO 3 R, -S(O) 2 N(R N ) 2 , -SiR 3 , -P(O)R, phosphate, phosphonate, cycloalkyl, cycloalkenyl, aryl and heteroaryl.
- substituents selected from the group consisting of halo, alkyl, -CN, -NO 2
- oxo as used herein contemplates an oxygen atom attached with a double bond.
- pharmaceutically acceptable or “pharmacologically acceptable” is meant a material which is not biologically or otherwise undesirable, i.e., the material may be administered to an individual without causing any undesirable biological effects or interacting in a deleterious manner with any of the components of the composition in which it is contained.
- “Pharmaceutically acceptable salt” refers to a salt of a compound of the invention which is made with counterions understood in the art to be generally acceptable for pharmaceutical uses and which possesses the desired pharmacological activity of the parent compound.
- Such salts include: (1) acid addition salts, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like; or formed with organic acids such as acetic acid, propionic acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, 3-(4-hydroxybenzoyl) benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 1 ,2-ethane-disulfonic acid,
- salts of amino acids such as arginates and the like, and salts of organic acids like glucurmic or galactunoric acids and the like (see, e.g., Berge et al, 1977, J. Pharm. Sci. 66:1-19).
- salts and “hydrates” refers to the hydrated forms of the compound that would favorably affect the physical or pharmacokinetic properties of the compound, such as solubility, palatability, absorption, distribution, metabolism and excretion.
- Other factors, more practical in nature, which those skilled in the art may take into account in the selection include the cost of the raw materials, ease of crystallization, yield, stability, solubility, hygroscopicity, flowability and manufacturability of the resulting bulk drug.
- sulfonamide as used herein contemplates a group having the structure
- R s is selected from the group consisting of hydrogen, C 1 -C 10 alkyl,
- alkylsulfonyl including, but not limited to alkylsulfonyl and arylsulfonyl.
- thiocarbonyl means a carbonyl wherein an oxygen atom has been replaced with a sulfur.
- Each R is independently selected from hydrogen, -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to C 12 heteroalkyl, alkenyl, alkynyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide, amino, and oxo.
- Each R N is independently selected from the group consisting of hydrogen, -OH, Ci to C 12 alkyl, Ci to C 12 heteroalkyl, alkenyl, alkynyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- Two R N may be taken together with C, O, N or S to which they are attached to form a five to seven membered ring which may optionally contain a further heteroatom.
- the compounds of the present invention may be used to inhibit or reduce the activity of HCV, particularly HCVs NS5A protein.
- inhibition and reduction of activity of the NS5A protein refers to a lower level of the measured activity relative to a control experiment in which the cells or the subjects are not treated with the test compound.
- the inhibition or reduction in the measured activity is at least a 10% reduction or inhibition.
- reduction or inhibition of the measured activity of at least 20%, 50%, 75%, 90% or 100%, or any number in between, may be preferred for particular applications.
- a and A' are independently selected from the group consisting of a single bond, -(CR 2 ) n -C(O)-(CR 2 ) p - -(CR 2 ) n -O-(CR 2 ) p - -(CR 2 ) n -N(R N )-(CR 2 ) p - -(CR 2 ) n -S(O) k -N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-N(R N )-(CR 2 ) p -, -(CR 2 ) n -N(R N )-C(O)-N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-O-(CR 2 )p-, -(CR 2 ) n -N(R N )-C(O)-O-(CR
- X 1 is CH 2 , NH, O or S,
- Y 1 , Y 2 and Z 1 are each independently CH or N,
- X 2 is NH, O or S
- the carbons of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- the nitrogens, if present, of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide,
- a and b are independently 1, 2, or 3.
- c and d are independently 1 or 2
- n and p are independently 0, 1, 2 or 3
- each R is independently selected from the group consisting of hydrogen, -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- each R N is independently selected from the group consisting of hydrogen, -OH, Ci to C 12 alkyl, Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide, and
- the A-B-A' can be any of:
- B is Q or Q Q , wherein each Q is independently selected from the group consisting of a cycloalkyl group, cycloalkenyl group, heterocycle, aryl group or heteroaryl group, with the proviso that only one Q is a six member aromatic ring when B is Q Q and with the proviso that if B is Q Q , any Q is that is poly cyclic is connected to the remainder of the molecule through only one cycle of the poly cycle;
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to C 8 alkyl, Ci to Cs heteroalkyl, aralkyl and a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl, wherein,
- each hetero atom if present, is independently N, O or S,
- each of R c , R d , R e and R f may optionally be substituted by Ci to C 8 alkyl, Ci to C 8 heteroalkyl, aralkyl, or a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl and wherein each heteroatom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring, and R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring;
- Y and Y' are each independently carbon or nitrogen;
- Z and Z' are independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, 1-3 amino acids, -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 , -U-(CR 4 2 ),-R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 , wherein,
- U is selected from the group consisting of -C(O)-, -C(S)- and -S(O) 2 -,
- each R 4 , R 5 and R 7 is independently selected from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 8 is selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, -C(O)-R 81 , -C(S)-R 81 , -C(O)-O-R 81 , -C(O)-N-R 81 2, -S(O) 2 -R 81 and -S(O) 2 -N-R 81 2 , wherein each R 81 is independently chosen from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 7 and R 8 together form a 4-7 membered ring
- each t is independently O, 1, 2, 3, or 4, and
- u 0, 1, or 2.
- the compounds of the present invention include pharmaceutically acceptable salts of I as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- each Q is independently optionally substituted with one or more substituents each independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino, and if Q is not aromatic, it is optionally substituted with oxo.
- each Q is independently optionally substituted with -CN, -OCF 3 , -OCHF 2 , -CF 3 , or -F.
- B is selected from the group consisting of
- each r and s is independently 0, 1, 2, 3, or 4;
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; and
- each R b is independently C 1 -C 12 alkyl, hydroxyl, halogen or oxo.
- each phenyl residue optionally includes 1 or 2 nitrogens as heteroatoms.
- each R a is independently -CN, -OCF 3 , -OCHF 2 , -CF 3 , or -F.
- a and A' are independently selected from the group consisting of a single bond, -(CR 2 ) n -C(O)-(CR 2 )p-, -(CR 2 ) n -O-(CR 2 )p-, -(CR 2 ) n -N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-N(R N )-(CR 2 )p-, -(CR 2 ) n -N(R N )-C(O)-N(R N )-(CR 2 ) P - and -(CR 2 ) n -N(R N )-C(O)-O-(CR 2 ) p - and a
- heteroaryl group selected from the group consisting of ⁇ X 1
- a and A' are independently selected
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to Cg alkyl and Ci to Cg heteroalkyl, wherein, each hetero atom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle, and
- R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- R c and R d or R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- R c and R d are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R N is selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- R e and R f are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R 1N is selected from the group consisting of hydrogen, -OH, Ci to
- A' is selected from the group consisting of single bond, H , H
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; and
- r is selected from the group consisting of 0, 1, 2, 3, or 4.
- the compounds of the present invention include pharmaceutically acceptable salts of III as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- A' is H , H , -(CR 2 ) n -C(O)-(CR 2 ) p - -(CR 2 ) n -C(O)N(R N )-(CR 2 ) p - or -(CR 2 ) n -N(R N )C(O)-(CR 2 ) p -.
- A' is V H
- the compounds of the present invention include pharmaceutically acceptable salts of Ilia as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof. [0133] In a fourth embodiment of the second aspect, compounds have formula III
- A' is selected from the group consisting of
- the compounds of the present invention include pharmaceutically acceptable salts of IHb as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof. 6] In a third aspect of the invention, compounds have formula IV:
- a and A' are independently selected from the group consisting of single bond, H
- R N is selected from the group consisting of hydrogen, - OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide;
- ⁇ / optionally includes 1 or 2 nitrogens as heteroatoms
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino;
- r is O, 1, 2, 3 or 4;
- each R b is independently C 1 -C 5 alkyl, hydroxyl, halogen, or oxo;
- s is O, 1, 2, 3, 4, 5, or 6;
- each of X 1 and X 2 are independently C or N.
- the compounds of the present invention include pharmaceutically acceptable salts of IV as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- a and A' are each independently ⁇ y H ⁇ or -(CR 2 ) n -C(O)N(R N )-(CR 2 ) p -.
- the compounds of the present invention include pharmaceutically acceptable salts of IVa as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- the compounds of the present invention include pharmaceutically acceptable salts of IVb as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- a and A' are independently selected from the group consisting of single bond,
- R N is selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide;
- ⁇ / optionally includes 1 or 2 nitrogens as heteroatoms
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; and
- r is O, 1, 2, 3 or 4.
- the compounds of the present invention include pharmaceutically acceptable salts of V as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- a and A' are each independently [0146]
- compounds have formula Va:
- the compounds of the present invention include pharmaceutically acceptable salts of Va as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- the compounds of the present invention include pharmaceutically acceptable salts of Vb as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- each -O- IS l independently a divalent aryl or heteroaryl group which may be polycyclic with varying connective patterns
- each r is independently 0, 1, 2, 3, or 4;
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino;
- a and A' are independently selected from the group consisting of a single bond, -(CR 2 ) n -C(O)-(CR 2 ) p - -(CR 2 ) n -O-(CR 2 ) p - -(CR 2 ) n -N(R N )-(CR 2 ) p - -(CR 2 ) n -S(O) k -N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-N(R N )-(CR 2 )p-, -(CR 2 ) n -N(R N )-C(O)-N(R N )-(CR 2 ) p -, -(CR 2 ) n -C(O)-O-(CR 2 )p-, -(CR 2 ) n -N(R N )-S(O) k -N(R
- X 1 is CH 2 , NH, O or S,
- Y 1 , Y 2 and Z 1 are each independently CH or N,
- X 2 is NH, O or S
- the carbons of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- the nitrogens, if present, of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide,
- a and b are independently 1, 2, or 3.
- c and d are independently 1 or 2
- n and p are independently 0, 1, 2 or 3
- k 0, 1, or 2
- each R is independently selected from the group consisting of hydrogen, -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- each R N is independently selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide, and
- a and A' may be attached to either side of A and A' so that in the example of A or A' being the A-B-A' can be any of:
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to Cs alkyl, Ci to Cs heteroalkyl, aralkyl and a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl, wherein,
- each hetero atom if present, is independently N, O or S,
- each of R c , R d , R e and R f may optionally be substituted by Ci to Cs alkyl, Ci to Cs heteroalkyl, aralkyl, or a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl and wherein each heteroatom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring, and
- R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring;
- Y and Y' are each independently carbon or nitrogen;
- Z and Z' are independently selected from the group consisting of hydrogen, Ci to Cs alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, 1-3 amino acids, -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 , -U-(CR 4 2 ),-R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 , wherein,
- U is selected from the group consisting of -C(O)-, -C(S)- and -S(O) 2 -,
- each R 4 , R 5 and R 7 is independently selected from the group consisting of hydrogen, Ci to Cs alkyl, Ci to Cs heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 8 is selected from the group consisting of hydrogen, Ci to Cs alkyl, Ci to Cs heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, -C(O)-R 81 , -C(S)-R 81 , -C(O)-O-R 81 , -C(O)-N-R 81 2, -S(O) 2 -R 81 and -S(O) 2 -N-R 81 2 , wherein each R 81 is independently chosen from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 7 and R 8 together form a 4-7 membered ring
- each t is independently 0, 1, 2, 3, or 4, and
- u 0, 1, or 2.
- the compounds of the present invention include pharmaceutically acceptable salts of VI as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- each - ⁇ - IS 1 ndependently
- each phenyl residue optionally includes 1 or 2 nitrogens as heteroatoms.
- a and A' are independently selected from the group consisting of a single bond, -(CR 2 )n-O-(CR 2 )p-, -(CR 2 )n-N(R N )-(CR 2 )p-, -(CR 2 ) n -C(O)-N(R N )-(CR 2 ) p - -(CR 2 ) n -N(R N )-C(O)-N(R N )-(CR 2 ) p - and -(CR 2 )n-N(R N )-C(O)-O-(CR 2 )p- and a heteroaryl group selected from the group consisting
- a and A' are independently selected
- a and A' are each independently ⁇ y H ⁇ or -(CR 2 ) n -C(O)N(R N )-(CR 2 ) p -.
- R c , R d , R e and R f are each independently selected from the group consisting of: hydrogen, Ci to Cg alkyl and Ci to Cg heteroalkyl, wherein, each hetero atom, if present, is independently N, O or S, R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle, and
- R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- one of R c and R d or R e and R f are joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- both of R c and R d and R e and R f are joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle.
- R c and R d are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R N is selected from the group consisting of hydrogen, -OH, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- R e and R f are joined and form a heterocyclic fused ring system selected from the group consisting of:
- R N is selected from the group consisting of hydrogen, -OH, Ci to
- C 12 alkyl Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide.
- each R a if present in a compound of any of the second through sixth aspects, is independently -CN, -OCF 3 , -OCHF 2 , -CF 3 , or -F.
- one of Y and Y' is N.
- both Y and Y' are N.
- Z and Z' in any of the previous aspects are each 1-3 amino acids.
- the amino acids are in the D configuration.
- Z and Z' in any of the previous aspects are each independently selected from the group consisting of -[U-(CR 4 2 ) t -NR 5 -(CR 4 2 ) t ] u -U-(CR 4 2 ) t -NR 7 -(CR 4 2 ) t -R 8 , -U-(CR 4 2 ),-R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -U-(CR 4 2 ),-NR 5 -(CR 4 2 ) t -U-(CR 4 2 ) t -NR 7 -(CR 4 2 ) t -R 8 .
- one or both of Z and Z' are -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -[C(O)-(CR 4 2 ) t -NR 5 -(CR 4 2 ) t ] u -U-(CR 4 2 ) t -NR 7 -(CR 4 2 ) t -R 8 .
- Z and Z' are -C(O)-(CR 4 2 ),-NR 5 -(CR 4 2 ),-U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -[C(O)-(CR 4 2 ) t -NR 5 -(CR 4 2 ) t ] u -C(O)-(CR 4 2 ) t -NR 7 -(CR 4 2 ) t -R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ),-NR 5 -(CR 4 2 ),-C(O)-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- Z and Z' are -C(O)-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -(CR 4 2 ) n -C(O)-R 81 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -C(O)-R 81 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -(CR 4 2 ) n -C(O)-O-R 81 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -C(O)-O-R 81 .
- one or both of Z and Z' are -U- (CR 4 2 ) t -R 8 .
- one or both of Z and Z' are - C(O)-(CR 4 2 ) t -R 8 .
- one or both of Z and Z' are -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- Z and Z' are -U-(CR 4 2 ),-NR 5 -(CR 4 2 ),-U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 )t-NR 5 -(CR 4 2 )t-C(O)-(CR 4 2 )t-O-(CR 4 2 ) t -R 8 .
- Z and Z' are -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ),-O-(CR 4 2 ),-R 8 .
- one or both of Z and Z' are -C(O)-(CR 4 2 ) n -NR 7 -R 8 wherein R 7 and R 8 together form a 4-7 membered ring.
- r is from 0 to 4
- each R a is independently selected from the group consisting of -OH,
- Ci to Ci 2 alkyl Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino;
- a and A' are independently selected from the group consisting of H
- a or A' is a heteroaryl group, it is optionally substituted with one or more of the substituents selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino, and
- n and p are independently O, 1, or 2
- each R is independently selected from the group consisting of hydrogen, -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- R N is selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide, and
- B' is optionally substituted with between 1 and 4 substituents, each independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to C 12 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide, amino and oxo,
- Y and Y' are each independently carbon or nitrogen;
- Z and Z' are independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, 1-3 amino acids, -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 , -U-(CR 4 2 ),-R 8 and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 , wherein,
- U is selected from the group consisting of -C(O)-, -C(S)- and -S(O) 2 -,
- each R 4 , R 5 and R 7 is independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 8 is selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, -C(O)-R 81 , -C(S)-R 81 , -C(O)-O-R 81 , -C(O)-N-R 81 2, -S(O) 2 -R 81 and -S(O) 2 -N-R 81 2 , wherein each R 81 is independently chosen from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 7 and R 8 together form a 4-7 membered ring
- each t is independently O, 1, 2, 3, or 4, and
- u is O, 1, or 2.
- the compounds of the present invention include pharmaceutically acceptable salts of VII as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- the compounds of the present invention include pharmaceutically acceptable salts of IX as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- B' is selected from the group consisting of - ⁇ - - ⁇ - -
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- the compounds of the present invention include pharmaceutically acceptable salts of IX as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- compounds have formula X:
- B' is selected from the group consisting of - ⁇ - - ⁇ - -
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- each R 81 is independently chosen from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl; and each R 4 is independently selected from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl.
- each R 4 is independently selected from the group consisting of hydrogen, Ci to Cg alkyl, Ci to Cg heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl.
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; r is O, 1, 2, 3 or 4;
- the compounds of the present invention include pharmaceutically acceptable salts of XI as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof. 7] In an additional aspect of the invention, compounds of formula XII are provided:
- a and A' are independently selected from the group consisting of a single bond
- X 1 is CH 2 , NH, O or S,
- Y 1 , Y 2 and Z 1 are each independently CH or N,
- X 2 is NH, O or S
- a and b are independently 0, 1, 2, or 3 with the proviso that a and b are not both 0, optionally includes 1 or 2 nitrogens as heteroatoms on the phenyl residue,
- the carbons of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to C12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- the nitrogens, if present, of the heteroaryl group are each independently optionally substituted with a substituent selected from the group consisting of -OH, Ci to C 12 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide,
- a and b are independently 1, 2, or 3
- c and d are independently 1 or 2
- n and p are independently 0, 1, 2 or 3
- k 0, 1, or 2
- each R is independently selected from the group consisting of hydrogen, -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino,
- each R N is independently selected from the group consisting of hydrogen, -OH, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate and sulfonamide, and
- the A-B-A' can be any of: B is selected f rom the group consisting of v ' s ,
- - ⁇ - i s a divalent aryl or heteroaryl group which may be polycyclic with varying connective patterns
- X 1 is CH 2 , NH, O or S,
- each Y 1 is independently CH or N
- each r and s is independently 0, 1, 2, 3 or 4;
- each R a is independently selected from the group consisting of -OH, -CN, -NO 2 , halogen, Ci to Ci 2 alkyl, Ci to Ci 2 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, alkoxy, alkoxycarbonyl, alkanoyl, carbamoyl, substituted sulfonyl, sulfonate, sulfonamide and amino; and
- each R b is independently selected from the group consisting of Ci-Ci 2 alkyl, hydroxyl, halogen and oxo;
- R c , R d , R e , and R f are each independently selected from the group consisting of: hydrogen, Ci to C 8 alkyl, Ci to Cs heteroalkyl, aralkyl and a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl, wherein, each hetero atom, if present, is independently N, O or S, each of R c , R d , R e , and R f may optionally be substituted by Ci to C 8 alkyl, Ci to C 8 heteroalkyl, aralkyl, or a 4- to 8- membered ring which may be cycloalkyl, heterocycle, heteroaryl or aryl and wherein each heteroatom, if present, is independently N, O or S,
- R c and R d are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring, and R e and R f are optionally joined to form a 4- to 8-membered heterocycle which is optionally fused to another 3- to 6- membered heterocycle or heteroaryl ring;
- Y and Y' are each independently carbon or nitrogen;
- Z and Z' are independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, 1-3 amino acids, -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-NR 7 -(CR 4 2 ),-R 8 , -U-(CR 4 2 ),-R 8 , and -[U-(CR 4 2 ),-NR 5 -(CR 4 2 ),] U -U-(CR 4 2 ),-O-(CR 4 2 ),-R 8 , wherein,
- U is selected from the group consisting of -C(O)-, -C(S)-, and -S(O) 2 -,
- each R 4 , R 5 and R 7 is independently selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 8 is selected from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl, aralkyl, -C(O)-R 81 , -C(S)-R 81 , -C(O)-O-R 81 , -C(O)-N-R 81 2 , -S(O) 2 -R 81 , and -S(O) 2 -N-R 81 2 , wherein each R 81 is independently chosen from the group consisting of hydrogen, Ci to C 8 alkyl, Ci to C 8 heteroalkyl, cycloalkyl, heterocycle, aryl, heteroaryl and aralkyl,
- R 7 and R 8 together form a 4-7 membered ring, each t is independently 0, 1, 2, 3, or 4, and
- u 0, 1, or 2.
- the compounds of the present invention include pharmaceutically acceptable salts of XII as well as an optically pure enantiomer, racemate or diastereomeric mixtures thereof.
- the compounds of the invention are prepared by synthetic routes and procedures that are illustrated in the various synthetic schemes below.
- the synthetic routes shown in schemes below are disclosed for the purpose of illustration and are not meant to be interpreted as limiting the processes to make the embodied compounds by any other methods. It will be appreciated by those skilled in the art that a number of methods are available for the preparation of the compounds of the present invention.
- These compounds may be prepared by processes which include processes known in the chemical art for the production of structurally analogous compounds or by a novel process described herein.
- One of the general strategies includes building the central scaffold followed by gradual functional group transformations of the two ends either simultaneously. In order to differentially functionalize the two ends, some orthogonal functional group protection and deprotection strategies are used (T.W. Greene & P. G.
- IC 50 The concentration of an inhibitor that causes a 50 % reduction in a measured activity
- Schemes 1-1 and 1-2 illustrate the various methods of preparing aryl-carbocyclic central scaffolds (Bs) and how the two ends of the molecule can be differentially constructed in order to allow for selective functionalization of either end of the molecule.
- Scheme 1-3 outlines the preparation of core structures connected through a carbon-nitrogen bond.
- the nitrogen arylation is achieved using one of the several methods.
- the acetophenone moiety and the V group in A-17 can be sequentially converted to a bromoketone as in A-18 for further introduction of the A-19 moiety.
- the coupling between A-15 and A-16 to A-17 represents a nucleophilic aromatic substitution method.
- Other methodologies employed include Buchwald or Ma couplings (J. Org. Chem. 2005, 70, 164), such as the coupling of A-15 with A-20 to B-6, A-15 with A-22 to B-7, A-21 with A-22 to B-8.
- V is a carboxylate, it can be extended via an amide linkage.
- the carboxylate can undergo a Curtis rearrangement to an amine, which in turn can be transformed to an amide (B-6 to B-9).
- the carboxylate can be hydrolyzed and converted to B-IO when reacts with a protected form of pyrrolidin-2- ylmethanamine.
- the methodologies outlined in this scheme are further depicted in other schemes described herein.
- Scheme 1-4 illustrates some of the ways for further functional group transformations using scaffold B-I as an example.
- the nitrogen protecting groups P and P' can be removed simultaneously to give B-Ia.
- B-Ia is treated together a suitably protected amino acid under standard peptide coupling conditions, such as HATU and a tertiary amine base, e.g. H ⁇ nig's base, the doubly coupled product B-Ib is obtained.
- P refers to a protecting group such as Boc-, Cbz-, Troc-, etc.
- P can also be other alkyl, acyl, alkoxylcarbonyl, alkylaminocarbonyl groups that are intended for this position.
- the newly introduced amino acid in B-If can be the same as the residue on the left-hand side of the molecule or can be a different one. From B- If, a variety of compounds (with a general formula of B-Ig) are prepared with differentially functionalized end pieces.
- the imidazole moiety is preferably protected with SEM or other protecting groups.
- the protection process does generate a mixture of regioisomers of the protecting group. However, such a mixture does not usually affect the reactivity of the intermediates toward further reaction and will become one compound upon the removal of the protecting group.
- the iodo- or bromo-imidazole intermediate A-27 is used converted to is the corresponding borate A-28 under the conditions shown, or using conditions that are known to promote similar transformations.
- the acetylene compound A-28 is obtained after subsequent treatment with base.
- Scheme 1-1 The use of intermediate as an alternative method of synthesizing arylimidazole intermediates such as A-I and B-3 is illustrated in Scheme 1-1. These versatile building blocks are used in many other manners as will be shown in the schemes to follow.
- the aryl-imidazole and benzoimidazole building blocks are synthesized using the route described in Scheme 1-6 (Ref. J. Med. Chem. 2008, 51, 5109; 2006, 49, ZIlA; and 2002, 45, 5556).
- Those in the art shall recognize that the phenyl, the proline and the protecting group on nitrogen may be replaced in order to achieve the desired functionality at a given position.
- V for example can be an acetyl group or a masked form
- OTf V can be a protected form of N
- Scheme 1-7 [0207]
- the building blocks used in Scheme 1-1 for the synthesis of scaffolds such as B-I to B-4 and others are synthesized using routes described in Scheme 1-7.
- the preparation of A-Ia starts with the dimethyl bicyclo[2.2.2]octane-l,4-dicarboxylate, one of the methyl ester group is selectively hydro lyzed to the acid, which in turn is converted to a bromo group to give methyl 4-bromobicyclo[2.2.2]octane-l-carboxylate.
- a Friedel- Crafts reaction between methyl 4-bromobicyclo[2.2.2]octane-l-carboxylate and a substituted benzene yields A-Ia (Ref. J. Org. Chem. 1970, 55, 917).
- Compound A-Ib is prepared similarly.
- Another method to prepare the cyclohexyl-containing compounds such as A-Ib and A-Ib' is through a cross coupling reaction between vinyltrifolate and a phenylboronate (or a boronic acid).
- the letter V represents a carboxylate group, such as an ethoxylcarbonyl, or a protected amino group, or can be another functional group that can be further functionalized.
- the resulted styrenyl group can be preserved (A- Ib') or can be saturated under hydrogenation condition to give A-Ib.
- the cyclopentyl analog A-Ic may be made by similar route.
- the four-carbon analog is made in sequence as shown.
- Reagents and solvents used below can be obtained from commercial sources such as Aldrich Chemical Co. (Milwaukee, Wisconsin, USA).
- 1 H-NMR spectra were recorded on a Bruker 400 MHz or 500 MHz NMR spectrometer. Significant peaks are tabulated in the order: multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br s, broad singlet), coupling constant(s) in Hertz (Hz) and number of protons.
- Electrospray spray ionization (ESI) mass spectrometry analysis was conducted on a Hewlett-Packard 1100 MSD electrospray mass spectrometer using the HPl 100 HPLC for sample delivery.
- Mass spectrometry results are reported as the ratio of mass over charge, followed by the relative abundance of each ion (in parentheses) or a single m/z value for the M+H (or, as noted, M-H) ion containing the most common atomic isotopes. Isotope patterns correspond to the expected formula in all cases. Normally the analyte was dissolved in methanol at 0.1 mg/mL and 5 microliter was infused with the delivery solvent into the mass spectrometer, which scanned from 100 to 1500 daltons. All compounds could be analyzed in the positive ESI mode, using an acetonitrile/water gradient (10%-90%) acetonitrile in water with 0.1% formic acid as delivery solvent.
- the compounds provided below could also be analyzed in the negative ESI mode, using 2 mM NH 4 OAc in acetonitrile/water as delivery solvent. Enantiomeric purity was determined using a Hewlett-Packard Series 1050 system equipped with a chiral HLPC column (ChiralPak AD, 4.6 mm x 150mm) and isocratic elution using 5:95 isopropanol-hexane as a mobile phase.
- Step a Referring to Scheme 2-3, compound 1 was prepared following procedures described in J. Med. Chem. 2007, 50, 6706.
- Step b A sample of compound 1 (1.Og, 2.82 mmol) in dichloromethane was treated with excess 4N HCl in dioxane. At the completion of removal of Boc group as indicated by LCMS, solvents were removed and the residue was dried under vaccum. This material was taken up in acetonitrile (6 mL) and treated with tert-butyi (lH-pyrazol-1- yl)methylenedicarbamate (2) (1.01 g, 3.2 mmol) and DIPEA (0.60 g, 4.65 mmol) at rt overnight. The solvents were evaporated off and the crude product was purified by silica gel column chromatography with a gradient eluent consisting various ratio of EtOAc and hexanes to give compound 3 (0.59 g, 71% yield).
- Step c A solution of 3 (1.34 g, 2.69 mmol, from combined runs) in THF (5 mL) was treated with excess 4N HCl in dioxane at rt overnight. Solvents were removed by evaporation, and residue was further dried under high vacuum. A portion of this de-Boc material (0.3 g, 1.0 mmol) was taken up in THF (4 mL) and water (0.34 mL). To this mixture was added K2CO3 (0.27 g, 2.02 mmol) followed by bromoketone compound 4 (0.33 g, 1.01 mmol, prepared following published procedures). The entire mixture was heated at reflux overnight.
- Step d A mixture of compound 5 (0.2 g, 0.38 mmol), bis(pinacolato)diboron (0.12 g, 0.46 mmol), potassium acetate (0.11 g, 1.1 mmol), and Pd(dppf)Cl 2 -CH 2 Cl2 (30 mg, 0.038 mmol) in dioxane (5 mL) was stirred at 80 0 C for 17 h under an atmosphere of Ar. Subsequently, the reaction mixture was filtered. The filtered cake was washed with EtOAc (5 mL x 3). The filtrate was washed with H 2 O and dried with anhydrous Na 2 SO 4 . The solvent was removed and the residue was purified by silica gel column chromatography to give compound 6 (0.14 g).
- Step e A solution of compound 6 (0.640 g, 1.1 mmol) in dioxane (24 mL) and water (2.4 mL) was sequentially added ⁇ S)-tert-hvXy ⁇ 2-(5-iodo-l-((2-(trimethylsilyl) ethoxy)methyl)-lH-imidazol-2-yl)pyrrolidine-l-carboxylate (1.10 g, 2.22 mmol), NaHCO 3 (0.37 g, 3.66 mmol), and Pd(dppf)Cl 2 -CH 2 Cl 2 (90 mg, 0.11 mmol) at rt under an atmosphere of Ar.
- Step f A sample of compound 8 (34 mg) was treated 4N HCl in dioxane (0.5 mL) at rt overnight to remove both the Boc group and the SEM group. The Cbz group was removed under hydrogeno lysis conditions (Pd/C, H 2 ).
- Step g Compound 9 (29 mg, 0.05 mmol) was treated with N-Moc-D-Phg-OH (20.6 mg, 0.11 mmol), DMTMM (42 mg, 0.15 mmol) and DIPEA (52 mg, 0.42 mmol) in THF/DMF (0.5 mL /0.5 mL) to give compound 10.
- Step a Referring to Scheme 2-4, to a stirred solution of 1 (1 eq., 0.2 M) in CH 2 Cl 2 was added DIPEA (1.2 eq.). The reaction flask was cooled with a water-ice bath, and TMSOTf (1.1 eq.) was slowly added over a 10 min period. The reaction was stirred at 0 0 C for 10 min, then at rt until silylation was complete, as determined by TLC (1.5 h). PTT (IM in THF, 1.05 eq) was added over 10 min. The reaction was stirred at rt until bromination was complete, as judged by TLC (30 min).
- the reaction was partitioned between CH 2 Cl 2 and NaHCO 3 with a 1/1 (v/v) ratio.
- the aqueous phase was extracted with CH 2 Cl 2 (2x).
- the combined organic layers were dried over Na 2 SO 4 , filtered, and concentrated.
- the crude product 2 was used without further purification.
- Step d A stream of nitrogen was bubbled through a solution of 5 (1 eq., 0.3M) in o-xylene in a sealable reaction tube for 10 min. NH 4 OAc (10 eq.) was added to the solution. The reaction tube was sealed and placed in a 150 0 C oil bath. The reaction was stirred at this temperature for 1 h then cooled to rt. The reaction tube was carefully opened, and its contents partitioned between aqueous Na 2 CO 3 and CH 2 Cl 2 /Me0H (10/1 (v/v)) with a 1 : 1 (v/v) ratio..
- Step e To a solution of 6 (1 eq.) in EtOH/AcOH (1/1 , v/v) was added Pd(OH) 2 on carbon (10%). After sealing the reaction flask with a rubber septum, the system was evacuated and backfilled with 1 atm H 2 (2x). The vigorously stirred reaction was stirred at rt under H 2 (1 atm) for 16 h. The mixture was filtered through a pad of CELITETM545 (pre- washed with EtOH), and the separated solids were washed with EtOH (5x). The filtrate was concentrated to provide crude 7.
- Residual AcOH was removed by dissolving the crude product in H 2 O then adjusting the pH to ⁇ 13 with 2N aqueous NaOH. The product was extracted with CH 2 Cl 2 until the extracts were free of 7. The crude product was used without further purification.
- Step g A stream of nitrogen was bubbled through a solution of 8 (1 eq, 0.3M) in o-xylene in a sealable reaction tube for 10 min. NH 4 OAc (10 eq) was added to the solution. The reaction tube was sealed and placed in a 150 0 C oil bath. The reaction was stirred at this temperature for 1 h then cooled to rt. The reaction tube was carefully opened, and its contents were partitioned between aqueous Na 2 CO 3 and CH 2 Cl 2 /Me0H (10/1 (v/v)) with a 1 : 1 (v/v) ratio.
- Step h To a solution of 9 (1 eq) in absolute EtOH was added Pd on carbon (10%, 50% H 2 O). After sealing the reaction flask with a rubber septum, the system was evacuated and backfilled with 1 atm H 2 (2x). The vigorously stirred reaction was stirred at rt under H 2 (1 atm) for 4 h. The mixture was filtered through a pad of CELITETM545 (pre -washed with EtOH), and the separated solids were washed with EtOH (5x). The filtrate was concentrated to provide crude 10 that was used without further purification.
- Step i To a stirred solution of 10 (1 eq, 0. IM) and N-Moc-D-Phg-OH(1 eq.) in DMF, DMTMM (1 eq.) and DIPEA (1 eq.) were sequentially added. The reaction was stirred at rt for 1 h and partitioned between aqueous Na 2 CO 3 and CH 2 Cl 2 /Me0H (10/1) with a 1/1 (v/v) ratio. The layers were separated, and the aqueous phase was extracted with CH 2 Cl 2 ZMeOH (10/1) until the extracts were free of any uv-active component.
- Step j Compound 11 (1 eq.) was treated with THF (7 mL/mmol 11) followed by slow addition of 4N HCl in dioxane (14 mL/1 mmol of 11). The reaction was stirred at rt for
- Step k To a stirred solution of 12 (1 eq, 0. IM) and N-Boc-D-Phg-OH (1 eq.) in DMF were sequentially added DMTMM (1 eq.) and DIPEA (1 eq.). The reaction was stirred at rt for 1 h. The crude reaction mixture was purified on a C18-Luna preparative HPLC column (H 2 O-MeCN, 0.1% HCO 2 H) to give compound 13 as a white solid.
- Step 1 Compound 13 (1 eq.) was treated with THF (7 mL/mmol) followed by slow addition of 4N HCl in dioxane (14 mL/1 mmol of 13). The reaction was stirred at rt for
- Step m To a stirred solution of 14 (1 eq, 0.1M) and cyclopropane carboxylic acid (1 eq.) in DMF were sequentially added DMTMM (1 eq.) and DIPEA (1 eq.). The reaction was stirred at rt until complete, as determined by LC-MS. The crude reaction mixture was purified on a C18-Luna preparative HPLC column (H 2 O-MeCN, 0.1% HCO 2 H) to give compound 15 as a white solid.
- Step b To a suspension OfAlCl 3 (5.9 g, 54 mmol) in DCM (200 mL) at 0 0 C was added l-(4-phenylcyclohexyl)ethanone (9 g in DCM), and acetyl chloride (10.4 mL, 146 mmol) dropwise over 30 min. The mixture was stirred to rt for 15 min. and heated at 45 0 C for 4h. After being cooled to rt, the mixture was poured it into ice-HCl (IN). The organic layer was collected, and the aqueous layer was extracted with ethyl acetate (3x).
- Step c To a solution of l-(4-(4-acetylcyclohexyl)phenyl)ethanone (A2) (900 mg, 3.7 mmol) in MeOH (20 mL) at O 0 C was added bromine (993 ⁇ L, 8.1 mmol) drop-wise and the reaction mixture was stirred below 5 0 C for 3 h. The reaction was quenched by adding saturated aqueous NaHCO 3 solution and the mixture was extracted with ethyl acetate (3x). The extracts were combined and concentrated.
- A2 l-(4-(4-acetylcyclohexyl)phenyl)ethanone
- Step d To a solution of N-Cbz-L-Pro-OH (1.36 g, 5.47 mmol) in acetonitrile (10 mL) was added triethyl amine (762 ⁇ L, 5.47 mmol) and 2-bromo-l-(4-(4-(2- bromoacetyl)cyclo hexyl)phenyl)ethanone (A3) (1 g, 2.49 mmol) in acetonitrile. The reaction mixture was stirred at rt overnight. The solvent was removed and product was diluted with ethyl acetate (3x), washed with NaHCO 3 (100 mL) and brine, and dried with Na 2 SO 4 .
- Step f (5)-benzyl 2-(4-(4-(4-(4-(4-(4-(4-((S)- 1 -(benzyloxycarbonyl)pyrrolidin-2-yl)- 1 H- imi-dazol-2-yl)phenyl)cyclohexyl)- 1 H-imidazol-2-yl)pyrrolidine- 1 -carboxylate (A5) (200 mg) and Pd/C (20 mg) in MeOH (20 mL) was purged with H 2 . The reaction was stirred under hydrogen balloon for 48 h, filtered on CELITETMand concentrated.
- Step g To a solution of 2-((5)-pyrrolidin-2-yl)-4-(4-(4-(4-((5)-pyrrolidin-2-yl)- lH-imidazol-2-yl) phenyl)cyclohexyl)-lH-imidazole (A6) (300 mg, 0.715 mmol) in DMF (20 mL) was added N-Boc-L- VaI-OH (432 mg, 1.72 mmol), HATU (654 mg, 1.72 mmol) and diisopropylethylamine (590 ⁇ L, 3.58 mmol). The reaction was stirred at rt overnight. The solvent was removed under reduced pressure.
- Step a Referring to Scheme 2-7 to a suspension of AICI3 (1.58 g, 12 mmol) in DCM (30 niL) at -78 0 C was added l-(4-phenylcyclohexyl)ethanone (Al') (1 g in DCM, 5.0 mmol), and bromoacetyl bromide (0.65 mL, 7.5 mmol) dropwise over 10 min. The mixture was stirred to rt for 15 min and heated at 40-50 0 C for 3 h, cooled to rt and poured into a mixture of ice and HCl (IN).
- Al' l-(4-phenylcyclohexyl)ethanone
- Step c To a solution of A3' (20.0 g, 40.7 mmol) and diethylisopropylamine (20 mL) in DCM (200 mL) in a round bottom flask was added trimethylsilyl trifluoromethanesulfonate (TMSOTf, 20 mL, 122 mmol) at -78 0 C. The reaction was stirred at rt overnight. To the solution was added a solution of phenyltrimethylammonium tribromide (PTT, 16.8 g, 45 mmol) in THF (50 mL). The reaction was stirred at rt 2 h and quenched with saturated NaHCO 3 solution.
- TMSOTf trimethylsilyl trifluoromethanesulfonate
- Step e Following the deprotection and amide formation operations described for similar systems and repeated the process twice, the differentially functionalized compound A8' was obtained.
- Step a Referring to Scheme 3-1, methyl S-phenylcyclobutanecarboxylate A2 was obtained by treating a solution of 3-phenylcyclobutanecarboxylic acid (5.3 g, 30 mmol, Reference: JOC, 1962, 27, 1647) in methanol (50 mL) and thionyl chloride (2.9 mL, 39 mmol) first at 0 0 C for 0.5 h and then at rt overnight. The solvent was removed, the residue was diluted with ethyl acetate (100 mL) and washed with brine and H 2 O, dried with Na 2 SO 4 .
- 3-phenylcyclobutanecarboxylic acid 5.3 g, 30 mmol, Reference: JOC, 1962, 27, 1647
- thionyl chloride 2.9 mL, 39 mmol
- Step b To a solution of methyl S-phenylcyclobutanecarboxylate (A2) (5.41 g, 28 mmol) and AICI3 (9.5 g, 71 mmol) in methylene chloride (150 mL) at 0 0 C was added acetyl chloride (4 mL) dropwise and the reaction mixture was stirred at rt overnight. The reaction mixture was diluted with IN HCl (100 mL) and extracted with CH 2 Cl 2 , and the organic layer was dried with Na 2 SO 4 , filtered and concentrated.
- A2 methyl S-phenylcyclobutanecarboxylate
- AICI3 9 g, 71 mmol
- Step c To a solution of methyl 3-(4-acetylphenyl)cyclobutanecarboxylate (A3) (6.56 g, 28.2 mmol) in methylene chloride (250 mL) at O 0 C was added PTT (10.6 g, 28.2 mmol) and the reaction mixture was stirred at rt overnight. The reaction was quenched with NaHCO 3 (100 mL), washed with brine, and dried with Na 2 SO 4 . After removal of the solvent, crude product A4 (7.2 g, 82% yield) was directly used for the next step.
- A3 methyl 3-(4-acetylphenyl)cyclobutanecarboxylate
- Step d A solution of crude methyl 3-(4-(2-bromoacetyl)phenyl) cyclobutanecarboxylate (A4) (8.8 g, 28.3 mmol), N-Boc-L-Pro-OH (6.7 g, 31.2 mmol) and triethylamine (4.34 mL) in CH 3 CN was stirred for 2 h. At the completion of reaction, the volatile components were removed in vacuo, and the residue was partitioned between H 2 O and CH 2 Cl 2 . The organic phase was dried, filtered and concentrated in vacuo.
- Step e A mixture of ketoester (S)-l-tert-butyl 2-(2-(4-(3-(methoxycarbonyl) cyclobutyl) phenyl)-2-oxoethyl) pyrrolidine- 1 ,2-dicarboxylate (A5) (3.7 g, 8.76 mmol) and NH 4 OAc (6.75 g, 87.6 mmol) in o-xylenes (10 niL) was heated in a sealed tube at 140 0 C for 1 h. The volatile component was removed in vacuo, and the residue was partitioned between H 2 Oand CH 2 Cl 2 . Te organic phase was dried, filtered and concentrated in vacuo.
- Step f To a solution of the compound (S)-tert-bv ⁇ y ⁇ 2-(4-(4-(3- (methoxycarbonyl)cyclobutyl) phenyl)- 1 H-imidazol-2-yl)pyrrolidine- 1 -carboxylate (A6) (1.76 g, 41.5 mmol) in THF (10 mL) and MeOH (10 mL), LiOH (IM, 10.0 mL) was added at 0 0 C.
- Step g The mixture of (5)-3-(4-(2-(l-(tert-butoxycarbonyl)pyrrolidin-2-yl)-lH- imidazol-4-yl) phenyl)cyclobutanecarboxylic acid (A7) (822 mg, 2 mmol), DPPA (0.53 mL, 2.4 mmol), and triethylamine (0.34 mL) in benzyl alcohol (10 mL) was heated at 95 0 C overnight. The volatile component was removed in vacuo, the residue was partitioned between H 2 O and CH 2 Cl 2 . The organic phase was dried, filtered and concentrated in vacuo.
- Step h To a solution of (S)-tert-butyl 2-(4-(4-(3-(benzyloxycarbonylamino) cyclobutyl)phenyl)- lH-imidazol-2-yl)pyrrolidine-l -carboxylate (A8) (449 mg, 0.87 mmol) in methanol (10 mL) was added 10% Pd/C (50 mg) and the mixture was stirred overnight under H 2 atmosphere in a balloon.
- Step i To a solution of (S)-tert-bv ⁇ yl 2-(4-(4-(3-aminocyclobutyl)phenyl)-7H- imidazol-2-yl)pyrrolidine-l -carboxylate (A9) (330 mg, 0.86mmol) in dichloromethane (60 mL), N-Boc-L-Pro-O ⁇ (204 mg, 0.95 mmol), and DIC (0.2 mL) were added at rt. After stirring overnight, the reaction mixture was diluted with methylene chloride (50 mL), and washed with saturated aqueous NaHCCh, dried with Na 2 SO 4 .
- Step j To a solution of (S)-tert-butyl 2-(3-(4-(2-((S)-l-(tert- butoxycarbonyl)pyrrolidin- 2-yl)-7H-imidazol-4-yl)phenyl)cyclobutylcarbamoyl)pyrrolidine- 1-carboxylate (AlO) (100 mg, 0.173 mmol) in T ⁇ F (8 mL), 4N HCl in dioxane (2 mL) was added slowly at rt.
- AlO 1-carboxylate
- Step k To a solution of (5)-N-(3-(4-(2-((5)-pyrrolidin-2-yl)-lH-imidazol-4- yl)phenyl) cyclobutyl)pyrrolidine-2-carboxamide (All) (100 mg, 0.98 mmol) in dichloromethane (20 mL), N-Boc-D-Phg-OH (87mg, 0.35 mmol), DIPEA (144 ⁇ L, 0.34 mmol) and HATU (134 mg, 0.35 mmol) were added at rt. After stirring overnight, the reaction mixture was washed with brine and H 2 O, dried with Na 2 SO 4 .
- Step a A sample of the product from Scheme 3-1, tert-butyl (R)-2-((S)-2-(3-(4- (2-((S)- 1 -((R)-2-( te/t-butoxycarbonyl amino)-2-phenylacetyl)pyrrolidin-2-yl)- 1 H-imidazol- 4-yl)phenyl)cyclobutylcarbamoyl)pyrrolidin- 1 -yl)-2-oxo- 1 -phenylethylcarbamate (A13) (222 mg, 0.262 mmol) in THF (10 niL) was treated with 4N HCl in dioxane (5 rnL) at rt for 8 h.
- Step b To a solution of (5)-l-((i?)-2-amino-2-phenylacetyl)-N-(3-(4-(2-((5)-l- ((i?)-2-amino -2-phenylacetyl)pyrrolidin-2-yl)- 1 H-imidazol-4-yl)phenyl)cyclobutyl) pyrrolidine-2-carboxamide (80 mg, 0.124 mmol) in dichloromethane (20 mL), cyclopropyl carboxylic acid (22 mg, 0.26 mmol), DIPEA (0.5 mL) and HATU (94 mg, 0.26 mmol) were added at rt.
- Step a Referring to Scheme 4-1, the starting building blocks, Al, Al, A3, and A4 can be prepared following published conditions in the literature. References: 1). Chang, H.; Fiesman, W. F. and Petter, R. C. Syn. Comm. 2007, 37, 1267. 2). Champans, N. B.; Sotheeswar, S. and Tonyne, K. J. J. Org. Chem. 1970, 35, 917. 3). Delia, E. W. and Tsanaktisidis, J. Aust. J. Chem. 1985, 38, 1705. [0260] Step b.
- Step d To a solution of A3 (17.8 g) in dibromomethane (1200 mL) was added mercury(II) oxide at rt. The reaction was warmed up to 80 0 C and bromine (6.0 mL) was added dropwise during 25 min. After additional 3 h stirring, the reaction was cooled down to rt and filtered through a pad of CELITETM, washed with dichloromethane, concentrated to afford A4 (19.8 g). The crude product A4 was used without further purification.
- Step e To a solution of A4 (19.8 g) in dichloromethane (500 mL) at -20 0 C was added aluminum chloride (43 g) in several portions. The reaction was then warmed up to O 0 C. After overnight stirring, the reaction was poured into a mixture of ethyl acetate (800 mL), IM HCl (800 mL) along with some ice. After separating the ethyl acetate layer, the aqueous layer was extracted with ethyl acetate (3 x 300 mL). The combined organic layers were washed with brine, H 2 O, dried over Na 2 SO 4 , filtered, and concentrated to afford A5 (19.2 g), which was used without further purification.
- Step f To a solution of A5 (19.2 g) in dichloromethane (500 mL) at -20 0 C was added acetyl chloride (16.8 mL), followed by aluminum chloride (43 g). The reaction was allowed to warm up to rt. After overnight stirring, the reaction was poured into a mixture of ethyl acetate (800 mL), IM HCl (800 mL) along with some ice. After separating the organic layer, the aqueous layer was extracted with ethyl acetate (3x300 mL). The combined organic layers were washed with brine, H 2 O, dried over Na 2 SO 4 , filtered, and concentrated to afford A6 (24 g), which was used for next reaction without further purification.
- Step g To a solution of A6 (21 g) in MeOH (500 mL) and THF (500 mL) was added aqueous lithium hydroxide (IM, 365 mL) at rt. After overnight stirring, the reaction was concentrated. The residue was diluted with water (1400 mL) and extracted with diethyl ether (3 x 500 mL), ethyl acetate (2 x 400 mL). The combined organic layers were extracted with lithium hydroxide (IM, 500 mL). All aqueous basic layers were combined and acidified to pH 2 by concentrated HCl, extracted with ethyl acetate (3 x 800 mL). The combined organic layers were dried over Na 2 SO 4 , filtered and concentrated to provide A7 (14.8 g).
- IM lithium hydroxide
- Step h To a solution of A7 (14.8 g) in dichloromethane (800 mL) was added oxalyl chloride (5.1 mL) at rt. DMF (209 ⁇ L) was added in three portions over 4 h. The reaction was concentrated and re-dissolved in dichloromethane (500 mL). The fresh made diazomethane (0.3M, 500 mL) was added at O 0 C. The reaction was slowly warmed up to rt. After overnight stirring, the reaction was concentrated to obtain the crude A8 (17.8 g).
- Step j To a stirred solution of A9 (18.5 g) and Cbz-Pro-OH (14.5 g) in MeCN was added Et 3 N (8.1 mL). The reaction was stirred at rt overnight and concentrated. The resulting crude product was diluted by ethyl acetate (1000 mL) and washed with saturated sodium carbonate, brine, H 2 O, dried over sodium sulfate and concentrated to provide AlO (27 g)- [0269] Step k. To a stirred solution of AlO (10 g) in CH 2 Cl 2 (400 mL) was added DIPEA (3.4 mL).
- Step 1 To a stirred solution of All (8.7 g) and N-Cbz-L-Pro-OH (3.45g) in MeCN was added Et 3 N (2.24 mL). The reaction was stirred at rt overnight and concentrated. The resulting crude product was diluted by ethyl acetate (500 mL) and washed with saturated sodium carbonate, brine, water, dried over sodium sulfate and concentrated to provide A12 (10.2 g).
- Step n To a stirred solution of A13 (500 mg) in dichloromethane (50 mL) was added trifluoroacetic acid (5 mL). After 3 h, the reaction was concentrated to dryness to give a TFA salt, which was dissolved in DMF (20 mL). To the solution were added DIEA (490 ⁇ L), N-Moc-D-Phg-OH (98 mg) and 4-(4,6-dimethoxy-l,3,5-triazin-2-yl)-4- methylmorpholinium chloride (DMTMM, 156 mg) subsequently. After one h stirring, the reaction was diluted with H 2 O. The suspension was filtered through a pad of CELITETM545 and washed with H 2 O. The filtrate cake was rinsed with dichloromethane and concentrated to provide A14 (380 mg) without further purification.
- Step 13 To a stirred solution of A14 (8 mg) in methanol (3 mL) was added 10% Pd/C (4 mg) and one drop of concentrated HCl. The reaction was in a Parr shaker at rt and under 60 psi of hydrogen for 12 hours. The reaction was filtered through a pad of CELITETM545 and washed with methanol. The filtrate was concentrated to dryness to provide a free amine, which was dissolved in DMF (1 mL). Subsequently, the mixture was added DIEA (6 ⁇ L), N-Moc- VaI-OH (1.8 mg) and DMTMM (2.8 mg). After stirring for one h, the reaction was quenched by adding water.
- the suspension was filtered through CELITETM545 and washed with water.
- the filtrate cake was rinsed by dichloromethane and concentrated.
- the residue was purified by prep-HPLC (Phenomenex, C18-Luna column, H 2 O-MeCN, 0.1% HCO 2 H) to provide A15 (3.0 mg, 98% purity).
- Step a Referring to Scheme 5-4, a mixture of Inflate 2 (0.43 g, 2.34 mmol), borate 3 from above (1.01 g, 2.3 mmol) and Pd(PPh 3 ) 2 Cl 2 (160 mg, 0.23 mmol) in THF (12 mL) and aqueous Na 2 CO 3 (2 M, 2.4 mL) was degassed and filled with nitrogen. The mixture heated at 8O 0 C overnight. The reaction mixture was purified by column chromatography eluting with 1% methanol in ethyl acetate to give compound 4 (0.95 g, 70% yield).
- Step b To a solution of 4 (0.95 g, 1.6 mmol) in DCM (32 mL) at O 0 C was add 4M HCl in dioxane. The mixture was stirred at O 0 C for 3 h and warmed to rt for 1 h. Solvent was removed to give a white solid 5.
- Step c A mixture of 5 obtained above, DMTMM (0.89g, 3.2 mmol), triethylamine (1.12 mL, 8.0 mmol) and N-Boc-D-Phg-OH (0.8 g, 3.2 mmol) in THF (16 mL) was stirred at rt for 14 h. The reaction was diluted with H 2 O (50 mL), extracted with ethyl acetate (3 x 20 mL). The combined organic layers were washed with brine, dried over Na 2 SO 4 , and then concentrated to give a residue.
- Step d A mixture of 5 (400 mg, 1.0 mmol) and Pd/C ( 100 mg) in methanol ( 12 mL) was degassed, and filled with hydrogen (60 psi). The mixture was shaken for 16 h. The solution was filtered through a pad of CELITETM545. The filtrate was concentrated to give a pale yellow solid 7.
- Step e A mixture of 7 (384 mg, 0.98 mmol), DMTMM (0.51g, 1.86 mmol), triethylamine (0.54 mL, 3.9 mmol) and Boc-D-Phg-OH (466 mg, 1.86 mmol) in THF (10 mL) was stirred at rt for 14 h. The reaction was diluted with H 2 O and extracted with ethyl acetate. The combined organic layers were washed with brine and dried over Na 2 SO 4 , concentrated to give a residue.
- Step a Referring to Scheme 5-5, to a solution of 8 (0.86 g, 1.0 mmol) in DCM (10 mL) at O 0 C was add TFA (2 mL). The mixture was stirred at rt for 3 h. Solvent was removed to give a white solid 9.
- Step b To a solution of 9 (90 mg, 0.14 mmol) and triethylamine (95 ⁇ L, 0.68 mmol) in THF/DMF (2 mL, 1/1 (v/v)) at -78 0 C was added morpholinyl chloroformate (30 ⁇ L, 0.26 mmol). The solution was stirred at rt overnight, diluted with H 2 Oand ethyl acetate. The organic layer was washed with H 2 O and brine, dried over sodium sulfate, and then concentrated to give a residue. The residue was purified with silica gel chromatography eluting with 5% methanol in DCM to afford 10 (40 mg).
- Step a Referring to Scheme 5-6, to prepare compound 1, to a solution of quinoline-6-carboxylic acid methyl ester (5.61 g, 30 mmol) in 100 mL of dichloromethane was added mCPBA (8.1 g, 36 mmol, 77% maximum purity) in four portions in 5 min at 0 0 C. The resulting mixture was warmed to rt and stirred for 2 h. The reaction mixture was diluted with 300 mL of dichloromethane and washed with aqueous Na 2 S 2 O 3 solution, saturated NaHCO 3 solution, H 2 Oand brine.
- Step b To the solution of quinoline iV-oxide (6.7 g, 33 mmol) in dichloromethane (60 mL) was added POCl 3 (30 mL) dropwise at rt. The reaction mixture was stirred for 16 h at 50 0 C in sealed vessel before it was concentrated to dryness. The residue was dissolved in dichloromethane and washed with saturated NaHCO 3 , H 2 Oand brine. The organic layer was dried over Na 2 SO 4 .
- Step c The mixture of 1 (1.8 g, 8.1 mmol), methyl isonipecotate 2 (1.75 g, 12.2 mmol) and K 2 HPO 4 (5.64 g, 32.4 mmol) in 15 mL of DMSO was stirred at 80 0 C for 20 hours. After cooling down, the resulting mixture was partitioned between 500 mL of EtOAc and 500 mL of H 2 O. The organic layer was washed with H 2 O and brine, and dried (Na 2 SO 4 ).
- Step d To the solution of 3 (1.81 g, 5.5 mmol) and chloroiodomethane (2.81 mL, 38.5 mmol) in THF (40 mL) was added LDA (pre-cooled to -78 0 C, freshly made from 10.9 mL of diisoproylamine and 28.6 mL of 2.5 M n-BuLi in hexanes in 40 mL of THF) at -78 0 C via cannula over 20 minutes. The reaction mixture was stirred for two hours at -78 0 C before it was quenched by dropwise addition of 60 niL AcOH/THF (v/v, 1/1).
- Step e A solution of 4 (1.2 g, 3.3 mmol), N-Boc-L-Pro-OH (2.1 g, 9.9 mmol), DIPEA (2.96 mL, 19.8 mmol) and potassium iodide (1.66 g, 9.9 mmol) in MeCN (50 mL) was stirred at 40 0 C for 14 h. After removing all solvent in vacuo, the residue was partitioned in EtOAc and saturated NaHCO 3 . The organic layer was washed with H 2 O and brine and dried over Na 2 SO 4 .
- Step f A mixture of 5 (723 mg, 1.0 mmol), ammonium acetate (2.31 g, 30 mmol) and Et 3 N (4.18 mL, 30 mmol) in xylene (30 mL) was stirred in sealed vessel at 140 0 C for 1.5 h. After removing all solvent in vacuo, the residue was partitioned in 5% MeOH in dichloromethane and water. The organic layer was washed with H 2 Oand brine. The aqueous layers were extracted with 5% MeOH in dichloromethane twice. The combined organic layers were dried over Na 2 SO 4 .
- Step g The solution of 6 (320 mg) in 6 mL of dichloromethane and TFA (5 mL) was stirred at rt for 2 h. After removing solvent in vacuo, the residue was purified by reverse- phase preparative HPLC to afford compound 7 (120 mg) as a yellow solid.
- Step h To a solution of N-Moc-L- VaI-OH (8) (77 mg, 0.44 mmol) and HATU (167 mg, 0.44 mmol) in 2 mL of DMF was added DIPEA (0.15 mL, 1.0 mmol). The resulting solution was stirred at rt for 20 min before 7 (97 mg, ca. 0.2 mmol) and DIPEA (0.15 mL, 1.0 mmol) in 3 mL of DMF was added. The reaction mixture was stirred at rt for another 2 h with determination of the completion of the reaction by LC-MS. The solution was partitioned between EtOAc and water and the organic layer was washed with H 2 Oand brine.
- Step a Referring to Scheme 5-7, to a suspension of quinoline-6-carboxylic acid (13.2 g, 76.2 mmol) in MeOH (120 mL) was added thionyl chloride (16.6 mL, 229 mmol) dropwise at 0 0 C over 10 minutes. The resulting mixture was refluxed for 3 h, then concentrated to remove all solvents in vacuo. The residue was partitioned between 300 mL of EtOAc and 500 mL of saturated NaHCCh. The organic layer was washed with H 2 O followed by brine and dried with Na 2 SO 4 .
- Step b To a solution of 4-hydroxy-cyclohexanecarboxylic acid (11.5 g, 80 mmol) in methanol (120 mL) was added thionyl chloride (16.5 mL, 224 mmol) dropwise at 0 0 C. The resulting mixture was then warmed to refluxing for 3 hours. All solvent was removed in vacuo and the residue was dissolved in EtOAc. The solution was washed with saturated aqueous NaHCO 3 solution, H 2 O and brine. Column chromatography (EtOAc/Hexanes, v/v, 1/1 to 2/1) gave 8.5 g methyl ester as colorless oil.
- Step c To a solution of compound 1 (3.45 g, 18.4 mmol) in absolute ethanol (100 mL) was added TFA (1.42 mL, 18.4 mmol) dropwise at rt, followed by compound 2 (3.18 g, 15.3 mmol). The reaction mixture was heated to reflux. (TMS) 3 SiH (5.7 mL, 18.4 mmol) and AIBN (2.52 g, 15.3 mmol) were added into the mixture in four portions over a period of 6 h. After refluxing for 8 h, the resulting mixture was cooled down and all solvents were removed.
- TMS TMS 3 SiH
- AIBN (2.52 g, 15.3 mmol
- Step d To a solution of Intermediate 3 (470 g, 1.5 mmol) in methanol (20 mL) was added thionyl chloride (0.55 mL, 7.5 mmol) dropwise at O 0 C. The resulting mixture was then heated to refluxing for 3 h. All solvent was removed in vacuo and the residue was dissolved in EtOAc. The solution was washed with saturated aqueous NaHCO 3 solution, water and brine, respectively. Column chromatography (EtOAc/Hexanes, v/v, 1/1) gave compound 4 (310 mg). LC-MS (ESI): m/z 328 (M+l) + . [0292] Step e.
- Step f A solution of Intermediate 5 (330 mg, 0.91 mmol), N-Boc-L-Pro-OH (0.58 g, 2.73 mmol), DIPEA (0.81 mL, 5.46 mmol) and potassium iodide (0.45 g, 2.7 mmol) in MeCN (20 mL) was stirred at 40 0 C for 14 h. After removing all solvent in vacuo, the residue was partitioned in EtOAc and saturated NaHCO3. The organic layer was washed with H 2 O and brine and dried over Na 2 SO 4 .
- Step g A mixture of Intermediate 6 (450 mg, 0.62 mmol), ammonium acetate (1.44 g, 18.7 mmol) and Et 3 N (2.6 mL, 18.7 mmol) in xylene (20 mL) was stirred in sealed vessel at 140 0 C for 1.5 h. After removing all solvent in vacuo, the residue was partitioned between 5% MeOH in dichloromethane and H 2 O. The organic layer was washed with H 2 O and brine. The aqueous layers were extracted with 5% MeOH in dichloromethane twice. The combined organic layers were dried over Na 2 SO 4 .
- Step h A solution of Intermediate 6 (110 mg) in dichloromethane (3 mL) and TFA (2 mL) was stirred at rt for 2 h. After removing all solvent in vacuo, compound 8 (122 mg) was obtained as a yellow TFA salt.
- Step i To a solution of N-Moc-D-Phg-OH (25 mg) and compound 8 (-40 mg) in 0.75 mL of THF and 1.5 mL of DMF was added DIPEA (0.01 mL). The resulting solution was stirred at rt for 20 min before DMTMM (33 mg) was added. The reaction mixture was stirred at rt for another 2 h as LC-MS indicating the completion of reaction. The solution was partitioned in EtOAc and water, the organic layer was washed with water and brine. The aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na 2 SO 4 before it was concentrated in vacuo.
- Step a Referring to Scheme 5-8, to a suspension of quinoline-6-carboxylic acid (13.2 g, 76.2 mmol) in 120 mL MeOH was added thionyl chloride (16.6 mL, 229 mmol) dropwise at O 0 C in 10 min. After being heated at reflux for 3 h, the volatile solvents were removed by a rotary evaporator in vacuo. The residue was taken up in 300 mL EtOAc and was washed with 500 mL saturated NaHCO 3 , H 2 Oand brine sequentially. The organic layer was dried over anhydrous Na 2 SO 4 .
- reaction mixture was kept at refluxing for another 12 h. After cooling down, the mixture was partitioned between EtOAc and water. The organic layer was washed with brine and dried over anhydrous Na 2 SO 4 . After filtration and concentration, the residue was purified by the flash column chromatography (silica, hexanes/ethyl acetate, v/v, 4/1 to 2/1) to afford compound C4 (4.6 g, 64 % yield) as a white solid. The product was further purified by recrystallization from a mixture of EtOAc and hexanes.
- Step c 2-Chloro-l- ⁇ 2-[4-(2-chloro-acetyl)-bicyclo[2.2.2]oct-l-yl]-quinolin-6-yl ⁇ - ethanone.
- C4 610 mg, 1.68 mmol
- chloroiodomethane (0.74 mL, 10.1 mmol)
- THF 10 mL
- LDA pre-cooled to -78 0 C, freshly made from 3 mL of diisoproylamine and 8.06 mL of 2.5 M n-BuLi in hexanes in 10 mL of THF
- Step e The mixture of C6 (700 mg, 0.94 mmol), ammonium acetate (2.16 g, 28 mmol) and Et 3 N (3.9 mL, 28 mmol) in xylene (30 mL) was stirred in sealed vessel at 140 0 C for 1.5 h. After removing all solvent in vacuo, the residue was partitioned in 5% MeOH in dichloromethane and water. The organic layer was washed with H 2 O and brine. The aqueous layers were extracted with 5% MeOH in dichloromethane twice. The combined organic layers were dried over Na 2 SO 4 .
- Step a Referring to Scheme 5-9, a mixture of acetanilide 1 (3.28g, 24 mmol), 4- bromo-bicyclo[2.2.2]octane-l-carboxylic acid methyl ester (2) (5 g, 20 mmol), ZnCl 2 (13.1 g, 96 mmol) in 1 ,2-dichloroethane (2 mL) in a sealed Parr bottle was heated at 120 0 C for 16 h. The reaction mixture was dissolved in ethyl acetate (100 mL) and washed with H 2 O (50 mL) and brine (50 mL). The organic phase was dried over Na 2 SO 4 and concentrated under reduced pressure to give a mixture of ester 3 and acid 3'.
- Step b The resulting mixture of ester 3 and acid 3' was dissolved in methanol (60 mL) and treated with aqueous lithium hydroxide (10 g in 20 mL of water, 240 mmol). The reaction mixture was refluxed for 4 h. Methanol was evaporated under reduced pressure and the resulting aqueous solution was washed with ethyl acetate (20 mL). The aqueous solution was adjusted to pH 2 by the addition of 1 N HCl (aq) and extracted with ethyl acetate (2 x 100 mL). The combined organic phases were dried over Na 2 SO 4 and concentrated in vacuo to give 4 (5.3 g, 92% yield) as a yellow solid.
- aqueous lithium hydroxide 10 g in 20 mL of water, 240 mmol
- Step c A nitrating mixture [ 1.17 mL, prepared from 67% HNO 3 (3.52 mmol) and H 2 SO 4 ] was added dropwise to a cooled mixture of acid 4 (1.01 g, 3.52 mmol) in glacial acetic acid (8 mL) and acetic anhydride (4 mL). The mixture was stirred at rt for 12 h and poured into iced H 2 O. The precipitate was filtered off, washed with H 2 OtO neutral, and dried in vacuo to give 5 (0.72 g, 70% yield) as a yellow solid.
- Step f A solution of N-Cbz-L-Pro-OH (2 g, 8 mmol), pentafluorophenol (1.48 g, 8 mmol) and DCC (1.82 g, 8.8 mmol) in ethyl acetate (20 mL) was stirred at rt for 2 h. The reaction solution was filtered and the filtrate was concentrated in vacuo to give compound 8 (3.2 g, 96% yield) as a white solid. The mixture of compound 7 (0.44 g, 1.61 mmol) and activated ester 8 (0.8 g, 1.93 mmol) in 2,2-dimethoxypropane (30 mL) was stirred at rt overnight. The reaction solution was concentrated to give crude compound 9.
- Step g Compound 9 was dissolved in acetic acid (2 mL) and heated at 60 0 C for 5 h. The reaction solution was neutralized by saturated NaHCO 3 and extracted with ethyl acetate (2x20 mL). The combined organic phase was concentrated and the resulting crude product was purified by silica gel column chromatography (ethyl acetate/hexane, 65/35, v/v) to give compound 10 (0.4 g, 52% yield) as a yellow oil.
- Step h To a solution of compound 10 (0.8 g, 1.68 mmol) in methanol (3 mL) was added aqueous lithium hydroxide (0.13 g in 1 mL of water, 3.4 mmol). The reaction mixture was then refluxed for 8 h. Methanol was evaporated and the resulting aqueous solution was washed by ethyl acetate and neutralized by 1 N HCl solution.
- Step i A solution of acid obtained (0.57 g, 1.2 mmol) and oxalyl chloride (1.05 mL, 12 mmol) in dichloromethane (5 mL) was stirred at rt overnight. After concentrated under reduced pressure, the remaining residue was dissolved in ether (10 mL) followed by addition of diazomethane (20 mL, 0.33 N in ether, 6.8 mmol) at 0 0 C. The reaction mixture was stirred at O 0 C for 30 min.
- Step k A solution of compounds 12 and 12' (0.37 g, 0.68 mmol), N-Boc-L-Pro-
- Step m A mixture of compoundsl4 and 14' (83.3 mg, 0.13 mmol), Pd/C (53.4 mg, 5% on carbon, 0.025 mmol) and one drop concentrated HCl in ethanol (3 mL) under an atmosphere of hydrogen (15 psi) was stirred at rt for 6 h. The reaction solution was then concentrated in vacuo to give compounds 15 and 15' (75 mg, 90% yield). To a solution of the amines obtained above (75 mg, 0.14 mmol) in THF (4 mL) was added 4N HCl in dioxane (1 mL) at rt. The reaction mixture was stirred at rt for 2 h. The solvent was evaporated and the residue was dried in vacuo to give compounds 16 and 16' (50 mg, 83% yield) as a yellow solid.
- Step n To a solution of compounds 16 and 16' (44 mg, 0.1 mmol), N-Moc-L-Val- OH (43 mg, 0.25 mmol) and DMTMM (68 mg, 0.25 mmol) in a solvent mixture of DMF- THF (2 mL, 1 :3) was added DIPEA (0.17 mL, 1.0 mmol) at rt. The reaction mixture was stirred at rt for 2 h. THF was evaporated and the remaining residue was purified by prep- HPLC to provide compounds 17 and 17' (4.6 mg, 5% yield).
- Step a Referring to Scheme 6-1, to a solution of N-Cbz-L-Pro-OH (7.52 g, 30.0 mmol) and DIPEA (5.50 g, 54.0 mmol) in THF (200 niL) was added HATU (11.5 g, 30.0 mmol) at rt. After stirring for 10 min, 4-bromobenzene-l,2-diamine (1) (5.1O g, 27.0 mmol) was added and the reaction mixture was stirred at rt for 2 h. The mixture was concentrated and the residue was diluted with EtOAc (250 mL) and water (50 mL). The organic phase was washed with brine and dried with anhydrous Na 2 SO 4 . The solvent was removed and the residue was dried in vacuo to give crude compounds 2 and 2 ' (10.0 g), which were used for the next step without further purification. LC-MS (ESI) m/z 418.1 (M + H) + .
- Step c To a mixture of compound 3 (5.05 g, 12.5 mmol), bis(pinacolato)diboron (7.10 g, 26.3 mmol), and KOAc (3.20 g, 32.5 mmol) in 1,4-dioxane (100 mL) was added Pd(dppf)Cl 2 (400 mg, 0.5 mmol) at rt under an atmosphere of N 2 . After stirring at 80 0 C for 3h under an atmosphere of N 2 , the reaction mixture was filtered through CELITETM545 and the filtered cake was washed with EtOAc (100 mL x 3). The filtrate was washed with brine and dried with anhydrous Na 2 SO 4 .
- Step d To a mixture of compound 4 (1.04 g, 2.20 mmol), 1 ,4-diiodobenzene (360 mg, 1.1 mmol), and NaHCO 3 (650 mg, 7.7 mmol) in 1 ,2-dimethoxyethane (36 mL) and water (12 mL) was added Pd(dppf)Cl 2 (80.0 mg, 0.10 mmol) at rt under an atmosphere of N 2 . After stirring at 80 0 C overnight under an atmosphere of N 2 , the reaction mixture was concentrated and the residue was diluted with EtOAc (100 mL) and water (25 mL). The organic phase was washed with brine and dried with anhydrous Na 2 SO 4 .
- Step e To a solution of compound 5 (200 mg, 0.28 mmol) in CHCl 3 (5 mL) was added TMSI (168 mg, 0.84 mmol) at rt. After stirring at rt overnight, the reaction was quenched by adding MeOH (3.0 mL), followed by 4N HCl in dioxane (2.0 mL). The resulting mixture was stirred at rt for 30 min and concentrated. The residue was washed with DCM (10 mL) and dried in vacuo to give crude compound 6 (220 mg), which was used for the next step without further purification. LC-MS (ESI) m/z 449.2 (M + H) + .
- Step f To a solution of crude compound 6 (150 mg, 0.20 mmol) in DMF (2 mL) was added Et 3 N (0.22 mL, 2.10 mmol), followed by N-Moc-L-Val-OH (87.0 mg, 0.50 mmol) and HATU (190 mg, 0.50 mmol). After stirring at rt for 1 h, the reaction mixture was concentrated and the residue was diluted with DCM (50 mL) and water (10 mL). The organic phase was washed with brine and dried with anhydrous Na 2 SO 4 .
- Step a Referring to Scheme 6-2, to a solution of veratrol (8) (40.Og, 0.29 mol) in anhydrous THF (100 mL) and TMEDA (40 mL) was added /?-BuLi (2.5 M in hexanes, 128 rnL, 0.32 mol) dropwise at rt under an atmosphere of N 2 . After stirring at rt for 28 h under an atmosphere of N 2 , the reaction mixture was cooled to -78 0 C, followed by adding TMSCl (45 mL). The reaction mixture was stirred at rt for 5 h and then added water (20 mL).
- Step b To a solution of compound 9 (40 g, 0.20 mol) in anhydrous TMEDA (40 mL) was added n-BuLi in hexanes (2.5 M, 120 mL, 0.24 mol) dropwise at 0 0 C under an atmosphere of N 2 . After stirring at rt for 25 h, the reaction mixture was cooled to -78 0 C, followed by adding TMSCl (40 mL). The reaction mixture was stirred at rt for 5h and then added water (50 mL). The mixture was concentrated and the residue was extracted with hexanes (100 mL x 3). The extracts were combined, washed with brine, and dried with anhydrous Na 2 SO 4 . The solvent was removed and the residue was purified by silica gel column chromatography to give compound 10 (52 g, 85%) as colorless oil. LC-MS: (ESI) m/z 283.1 (M+H) + .
- Step d To a solution of compound 11 (3.89g, 10.0 mmol) in CH 2 Cl 2 (20 mL) was added BBr 3 (2.5 M in DCM, 15 mL, 60 mmol) at -78 0 C. After stirring at rt for 14 h, the reaction mixture was poured into water and the aqueous layer was extracted with CH 2 Cl 2 (5OmL x 2). The organic phase was washed with brine and dried with anhydrous Na 2 SO 4 . The solvent was removed and the residue was purified by silica gel column chromatography to give compound 12 as a white solid (3.1 g, 85%).
- Step e A mixture of compound 12 (1.5 g, 4.2 mmol), K 2 CO 3 (4.60 g, 33.2 mmol), and l-bromo-2-methoxyethane (2.30 g, 16.6 mmol) in acetone (100 mL) was refluxed overnight. The reaction mixture was concentrated and the residue was diluted with H 2 O (50 mL) and EtOAc (150 mL).
- Step f To a solution of compound 13 (200 mg, 0.42 mmol) in 40 mL DME/H 2 O
- Step g A mixture of compound 14 (150 mg, 0.21 mmol) in dioxane (2 mL) was added 4N HCl in dioxane (2 mL) at rt. After stirring at rt for 2 h, the reaction mixture was concentrated and the residue was dried in vacuo to give an HCl salt, which was used for the next step without further purification.
- Step h Subsequently, the HCl salt was dissolved in DMF (2 mL) and the resulting solution was respectively added DIPEA (0.34 mL, 2.1 mmol), N-Boc-L- VaI-OH (90 mg, 0.51 mmol), and HATU (192 mg, 0.210 mmol). After stirring at rt for 30 min, the reaction mixture was poured into ice H 2 O. The solid was collected by filtration and purified by preparative HPLC to give compound 15. LC-MS (ESI): m/z 911.5 (M+H) + .
- Step a To prepare compound 2 of Scheme 7-1, tert-Butyi 4- oxocyclohexylcarbamate (9.0 g, 42.5 mmol) was treated with HCl (4N in 1,4-dioxane, 60 mL) at rt. After 4 h stirring, the reaction was concentrated to obtain the crude product (6.3 g), which was dissolved in anhydrous DMF (200 mL). N-Boc-L-Pro-OH (9.1 g, 42.3 mmol)), HATU (17.7 g, 46.6 mmol) and DIEA (22.8 mL, 131 mmol) were added at rt.
- Step b To a solution of ⁇ S)-tert-hvXy ⁇ 2-(4-oxocyclohexylcarbamoyl)pyrrolidine- 1-carboxylate (4.3 g, 13.87 mmol) in dichloromethane (350 mL) at -78 0 C was slowly added LHMDS (29.1 mL, IM in THF, 29.1 mmol). After stirring for 2 h, 1,1,1-trifluoro-N-phenyl- N-(trifluoromethylsulfonyl)methanesulfonamide (10.4 g, 29.1 mmol) was added as solid at - 78 0 C.
- Step c To a solution of (2S)-tert-bv ⁇ y ⁇ 2-(4-(trifluoromethylsulfonyloxy) cyclohex-3-enyl carbamoyl)pyrrolidine-l-carboxylate (2) (1.65 g, 3.73 mmol) and (5)-tert- butyl 2-(5-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-lH-imidazol-2- yl)pyrrolidine-l-carboxylate (3) (1.95 g, 4.44 mmol) in THF (19 mL), Pd(PPh 3 ) 2 Cl 2 (130 mg, 0.185 mmol) and aqueous Na 2 CO 3 (2M, 7.5 mL, 15 mmol) were added under N 2 protection at rt.
- Step d To a solution of (2S)-tert-buiyl 2-(4-(4-(2-((S)- 1 -(tert-butoxycarbonyl) pyrrolidin- 2-yl)- lH-imidazol-S-y ⁇ pheny ⁇ cyclohex-S-enylcarbamoy ⁇ pyrrolidine- 1 - carboxylate (50 mg, 0.082 mmol) in DCM (15 mL), was added TFA (1 mL) at rt. After 1.5 h stirring, the reaction was concentrated to give a TFA salt (5) (75 mg), which was dissolved in anhydrous T ⁇ F (5 mL).
- Step e To a solution of the compound 6 (210 mg, 0.24 mmol) in DCM (10 mL) was added TFA (1 mL) at rt. After 1.5 h stirring, the reaction was concentrated to give the crude product, which was dissolved in anhydrous THF (20 mL). 4-Morpholinecarbonyl chloride (62 ⁇ L, 0.54 mmol) and Et 3 N (206 ⁇ L, 1.48 mmol) were added at rt. After overnight stirring, the reaction was quenched by water, extracted by ethyl acetate.
- Step f A mixture of compound 7 (20 mg, 0.022mmol) and 10% Pd/C (5 mg) in MeOH (2 mL) was hydrogenated (60 psi) for 2 days. The reaction was filtered through a CELITETM pad and washed with MeOH.
- Biological activity of the compounds of the invention was determined using an HCV replicon assay.
- the lb_Huh-Luc/Neo-ET cell line persistently expressing a bicistronic genotype Ib replicon in Huh 7 cells was obtained from ReBLikon GMBH. This cell line was used to test compound inhibition using luciferase enzyme activity readout as a measurement of compound inhibition of replicon levels.
- each compound is added in triplicate to the cells. Plates incubated for 72 h prior to running the luciferase assay. Enzyme activity was measured using a Bright-Glo Kit (cat. number E2620) manufactured by Promega Corporation. The following equation was used to generate a percent control value for each compound.
- Example compounds of the disclosed invention are illustrated in Tables 1-9 attached as appendices.
- the tables show inhibitory activity of many of the example compounds with respect to HCV Ib.
- the biological activity is indicated as being *, ** , ***, or ****, which corresponds to EC50 ranges of > 1000 nM, 999 nM to 10 nM, 9.9 nM to 1 nM, or ⁇ 1 nM respectively.
- the tables further provide mass spectrometry results for the synthesized example compounds.
- compositions [0341] The sixteenth aspect of the invention provides a pharmaceutical composition comprising the compounds of the invention
- the pharmaceutical composition further comprises one or more pharmaceutically acceptable excipients or vehicles, and optionally other therapeutic and/or prophylactic ingredients.
- excipients are known to those of skill in the art.
- the compounds of the present invention include, without limitation, basic compounds such as free bases. A thorough discussion of pharmaceutically acceptable excipients and salts is available in Remington's Pharmaceutical Sciences, 18th Edition (Easton, Pennsylvania: Mack Publishing Company, 1990)..
- the pharmaceutical compositions may be in the form of solid, semi-solid or liquid dosage forms, such as, for example, tablets, suppositories, pills, capsules, powders, liquids, suspensions, creams, ointments, lotions or the like, preferably in unit dosage form suitable for single administration of a precise dosage.
- the compositions will include an effective amount of the selected drug in combination with a pharmaceutically acceptable carrier and, in addition, may include other pharmaceutical agents, adjuvants, diluents, buffers, etc.
- the invention includes a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the present invention including isomers, racemic or non-racemic mixtures of isomers, or pharmaceutically acceptable salts or solvates thereof together with one or more pharmaceutically acceptable carriers and optionally other therapeutic and/or prophylactic ingredients.
- conventional nontoxic solid carriers include, for example, pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharin, talc, cellulose, glucose, sucrose, magnesium carbonate and the like.
- the composition will generally take the form of a tablet, capsule, a softgel capsule nonaqueous solution, suspension or syrup. Tablets and capsules are preferred oral administration forms. Tablets and capsules for oral use will generally include one or more commonly used carriers such as lactose and corn starch. Lubricating agents, such as magnesium stearate, are also typically added. When liquid suspensions are used, the active agent may be combined with emulsifying and suspending agents. If desired, flavoring, coloring and/or sweetening agents may be added as well. Other optional components for incorporation into an oral formulation herein include, but are not limited to, preservatives, suspending agents, thickening agents and the like. [0347] The seventeenth aspect of the invention provides the use of the compounds of the invention in the manufacture of a medicament.
- the medicament is for the treatment of hepatitis C.
- the eighteenth aspect of the invention provides a method of treating hepatitis C comprising administering to a subject in need thereof, a therapeutically effective amount of a compound of the invention, optionally in a pharmaceutical composition.
- a pharmaceutically or therapeutically effective amount of the composition will be delivered to the subject.
- the precise effective amount will vary from subject to subject and will depend upon the species, age, the subject's size and health, the nature and extent of the condition being treated, recommendations of the treating physician, and the therapeutics or combination of therapeutics selected for administration. Thus, the effective amount for a given situation can be determined by routine experimentation.
- the subject may be administered as many doses as is required to reduce and/or alleviate the signs, symptoms or causes of the disorder in question, or bring about any other desired alteration of a biological system.
- One of ordinary skill in the art of treating such diseases will be able, without undue experimentation and in reliance upon personal knowledge and the disclosure of this application, to ascertain a therapeutically effective amount of the compounds of this invention for a given disease.
- the compounds of the present invention and their isomeric forms and pharmaceutically acceptable salts thereof are useful in treating and preventing HCV infection alone or when used in combination with other compounds targeting viral or cellular elements or functions involved in the HCV lifecycle.
- Classes of compounds useful in the invention may include, without limitation, all classes of HCV antivirals.
- mechanistic classes of agents that may be useful when combined with the compounds of the present invention include, for example, nucleoside and non-nucleoside inhibitors of the HCV polymerase, protease inhibitors, helicase inhibitors, NS4B inhibitors and medicinal agents that functionally inhibit the internal ribosomal entry site (IRES) and other medicaments that inhibit HCV cell attachment or virus entry, HCV RNA translation, HCV RNA transcription, replication or HCV maturation, assembly or virus release.
- IRS internal ribosomal entry site
- telaprevir VX-950
- boceprevir SCH-503034
- narlaprevir SCH-9005178
- ITMN-191 R-7227
- TMC-435350 a.k.a.
- Nucleosidic HCV polymerase (replicase) inhibitors useful in the invention include, but are not limited to, R7128, PSI-7851, IDX-184, IDX-102, R1479, UNX-08189, PSI-6130, PSI-938 and PSI-879 and various other nucleoside and nucleotide analogs and HCV inhibitors including (but not limited to) those derived as 2'-C-methyl modified nucleos(t)ides, 4'-aza modified nucleos(t)ides, and 7'-deaza modified nucleos(t)
- Non-nuclosidic HCV polymerase (replicase) inhibitors useful in the invention include, but are not limited to , HCV-796, HCV-371, VCH-759, VCH-916, VCH- 222, ANA-598, MK-3281, ABT-333, ABT-072, PF-00868554, BI-207127, GS-9190, A- 837093, JKT-109, GL-59728 and GL-60667.
- NS5A inhibitors of the present invention may be used in combination with cyclophyllin and immunophyllin antagonists (eg, without limitation, DEBIO compounds, NM-811 as well as cyclosporine and its derivatives), kinase inhibitors, , inhibitors of heat shock proteins (e.g., HSP90 and HSP70), other immunomodulatory agents that may include, without limitation, interferons (-alpha, -beta, -omega, -gamma, -lambda or synthetic) such as Intron ATM, Roferon-ATM, Canferon-A300TM, AdvaferonTM, InfergenTM, HumoferonTM, Sumiferon MPTM, AlfaferoneTM, IFN- ⁇ TM, FeronTM and the like; polyethylene glycol derivatized (pegylated) interferon compounds, such as PEG interferon- ⁇ -2a (PegasysTM), PEG interferon- ⁇ -2
- ITCA-638 omega-interferon delivered by the DUROS TM subcutaneous delivery system
- compounds that stimulate the synthesis of interferon in cells such as resiquimod and the like
- interleukins compounds that enhance the development of type 1 helper T cell response, such as SCV-07 and the like
- TOLL-like receptor agonists such as CpG-IOlOl (actilon), isotorabine, ANA773 and the like; thymosin ⁇ -1; ANA-245 and ANA-246; histamine dihydrochloride; propagermanium; tetrachlorodecaoxide; ampligen; IMP-321; KRN-7000; antibodies, such as civacir, XTL-6865 and the like and prophylactic and therapeutic vaccines such as InnoVac C, HCV E1E2/MF59 and the like.
- any of the above-described methods involving administering an NS5A inhibitor, a Type I interferon receptor agonist (e.g., an IFN- ⁇ ) and a Type II interferon receptor agonist (e.g., an IFN- ⁇ ) can be augmented by administration of an effective amount of a TNF- ⁇ antagonist.
- a Type I interferon receptor agonist e.g., an IFN- ⁇
- a Type II interferon receptor agonist e.g., an IFN- ⁇
- TNF- ⁇ antagonists that are suitable for use in such combination therapies include ENBREL TM, REMIC ADETM and HUMIRA TM.
- NS5A inhibitors of the present invention may be used in combination with antiprotozoans and other antivirals thought to be effective in the treatment of HCV infection, such as, without limitation, the prodrug nitazoxanide.
- Nitazoxanide can be used as an agent in combination the compounds disclosed in this invention as well as in combination with other agents useful in treating HCV infection such as peginterferon alfa-2a and ribavarin (see, for example,_Rossignol, JF and Keeffe, EB, Future Microbiol. 3:539-545, 2008).
- NS5A inhibitors of the present invention may also be used with alternative forms of interferons and pegylated interferons, ribavirin or its analogs (e.g., tarabavarin, levoviron), microRNA, small interfering RNA compounds (e.g., SIRPLEX-140-N and the like), nucleotide or nucleoside analogs, immunoglobulins, hepatoprotectants, anti-inflammatory agents and other inhibitors of NS5A.
- interferons and pegylated interferons e.g., tarabavarin, levoviron
- microRNA e.g., small interfering RNA compounds
- nucleotide or nucleoside analogs e.g., immunoglobulins, hepatoprotectants, anti-inflammatory agents and other inhibitors of NS5A.
- Inhibitors of other targets in the HCV lifecycle include NS3 helicase inhibitors; NS4A co-factor inhibitors; antisense oligonucleotide inhibitors, such as ISIS- 14803, AVI-4065 and the like; vector-encoded short hairpin RNA (shRNA); HCV specific ribozymes such as heptazyme, RPI, 13919 and the like; entry inhibitors such as HepeX-C, HuMax-HepC and the like; alpha glucosidase inhibitors such as celgosivir, UT- 23 IB and the like; KPE-02003002 and BIVN 401 and IMPDH inhibitors.
- NS3 helicase inhibitors such as ISIS- 14803, AVI-4065 and the like
- antisense oligonucleotide inhibitors such as ISIS- 14803, AVI-4065 and the like
- HCV inhibitor compounds include those disclosed in the following publications: U.S. Pat. No. 5,807,876; U.S. Pat. No. 6,498,178; U.S. Pat. No. 6,344,465; U.S. Pat. No.
- combinations of, for example, ribavirin and interferon may be administered as multiple combination therapy with at least one of the compounds of the present invention.
- the present invention is not limited to the aforementioned classes or compounds and contemplates known and new compounds and combinations of biologically active agents (see, Strader, D.B., Wright, T., Thomas, DX. and Seeff, L. B., AASLD Practice Guidelines. 1-22, 2009 and Manns, M.P., Foster, G. R., Rockstroh, J.K., Zeuzem, S., Zoulim, F. and Houghton, M., Nature Reviews Drug Discovery. 6:991-1000, 2007, Pawlotsky, J-M., Chevaliez, S.
- combination therapies of the present invention include any chemically compatible combination of a compound of this inventive group with other compounds of the inventive group or other compounds outside of the inventive group, as long as the combination does not eliminate the anti-viral activity of the compound of this inventive group or the anti-viral activity of the pharmaceutical composition itself.
- Combination therapy can be sequential, that is treatment with one agent first and then a second agent (for example, where each treatment comprises a different compound of the invention or where one treatment comprises a compound of the invention and the other comprises one or more biologically active agents) or it can be treatment with both agents at the same time (concurrently).
- Sequential therapy can include a reasonable time after the completion of the first therapy before beginning the second therapy. Treatment with both agents at the same time can be in the same daily dose or in separate doses.
- Combination therapy need not be limited to two agents and may include three or more agents. The dosages for both concurrent and sequential combination therapy will depend on absorption, distribution, metabolism and excretion rates of the components of the combination therapy as well as other factors known to one of skill in the art.
- Dosage values will also vary with the severity of the condition to be alleviated. It is to be further understood that for any particular subject, specific dosage regimens and schedules may be adjusted over time according to the individual's need and the professional judgment of the person administering or supervising the administration of the combination therapy. [0356] All publications and patent applications cited in this specification are herein incorporated by reference as if each individual publication or patent application were specifically and individually indicated to be incorporated by reference.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description












































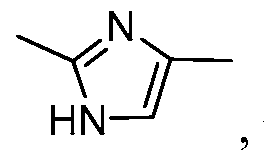













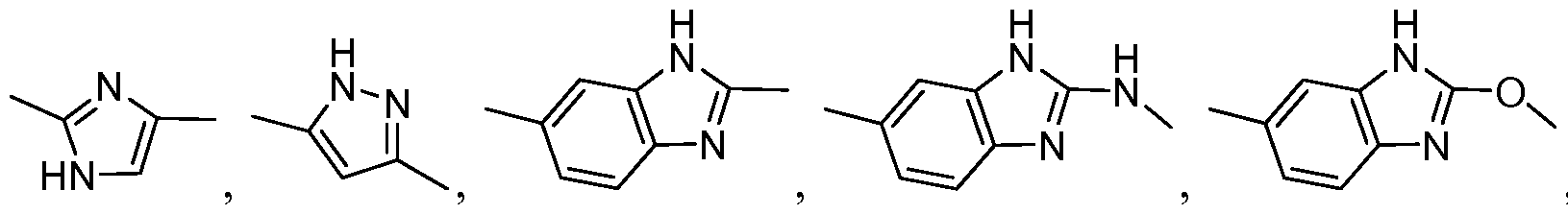

























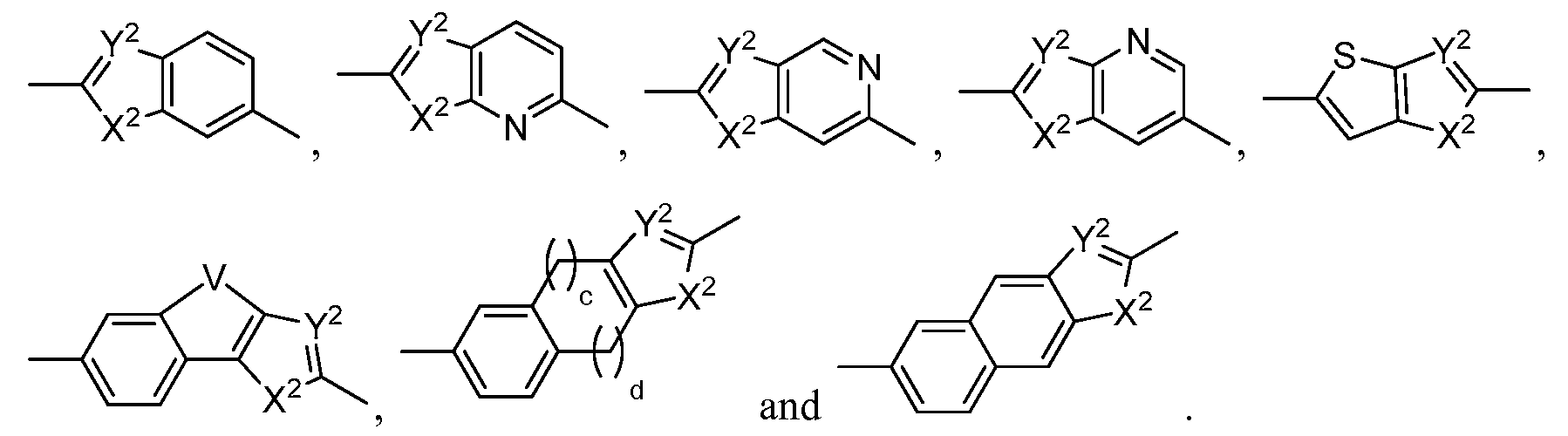










































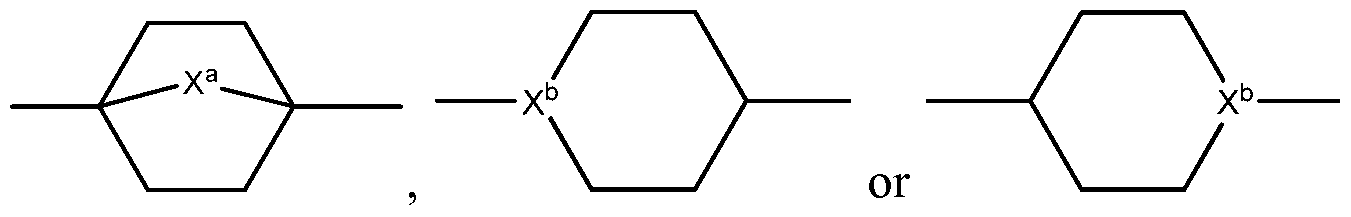
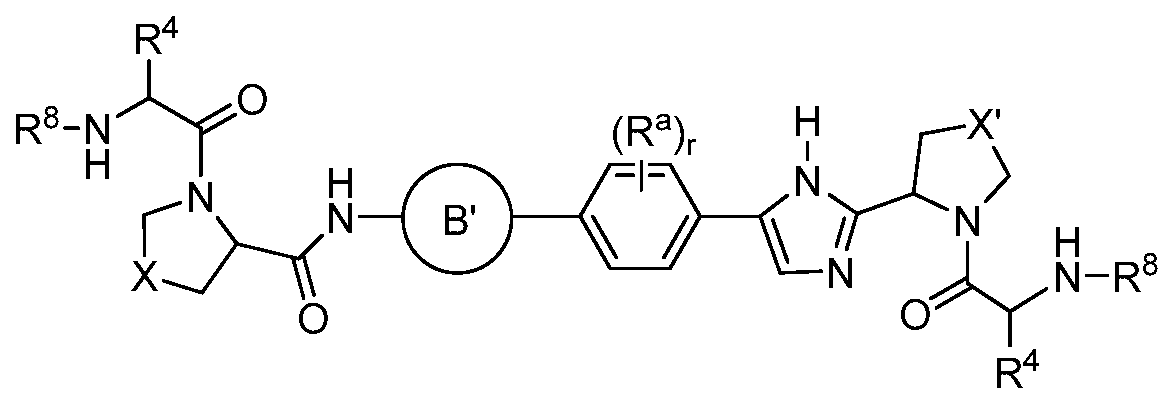





































































































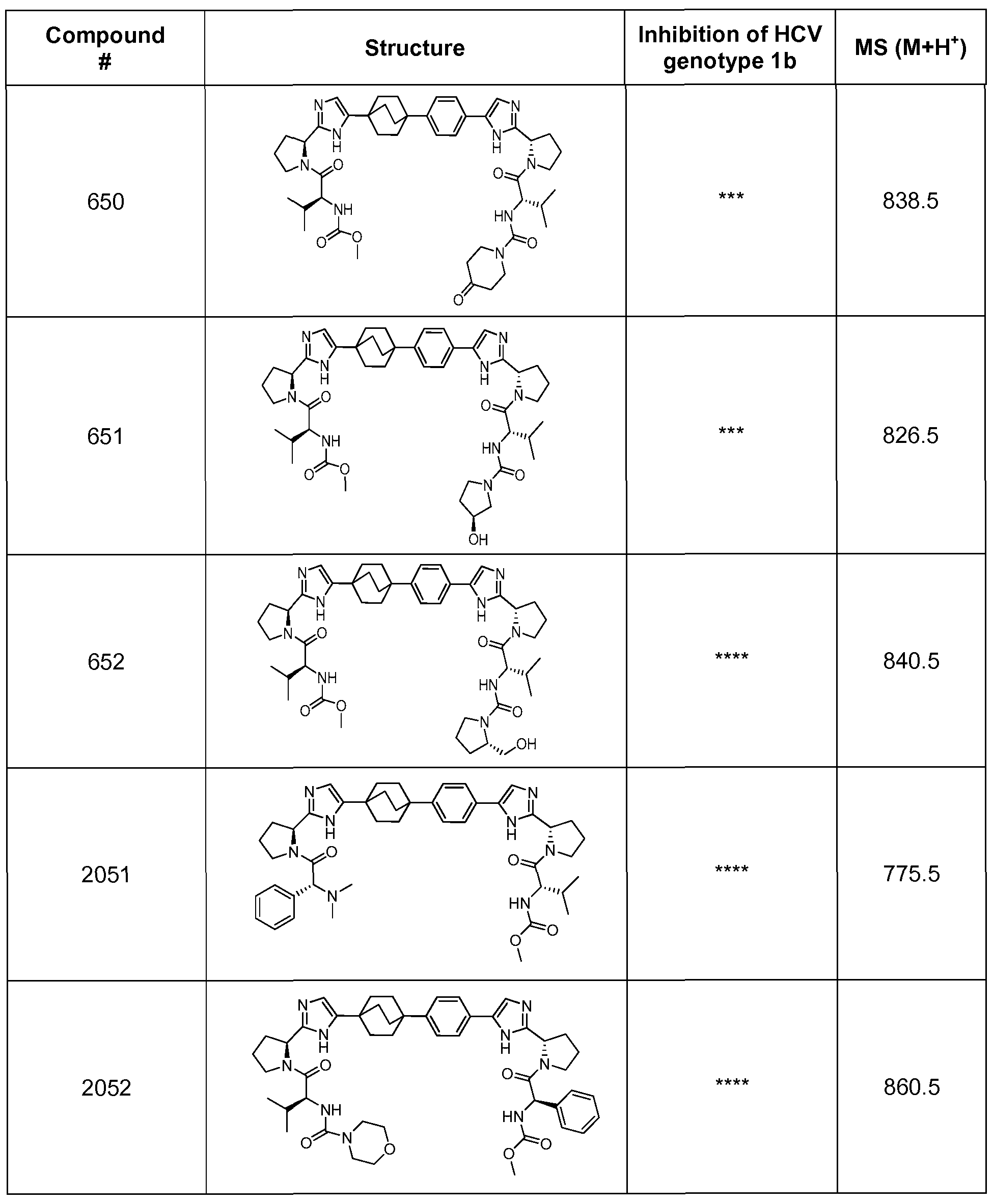









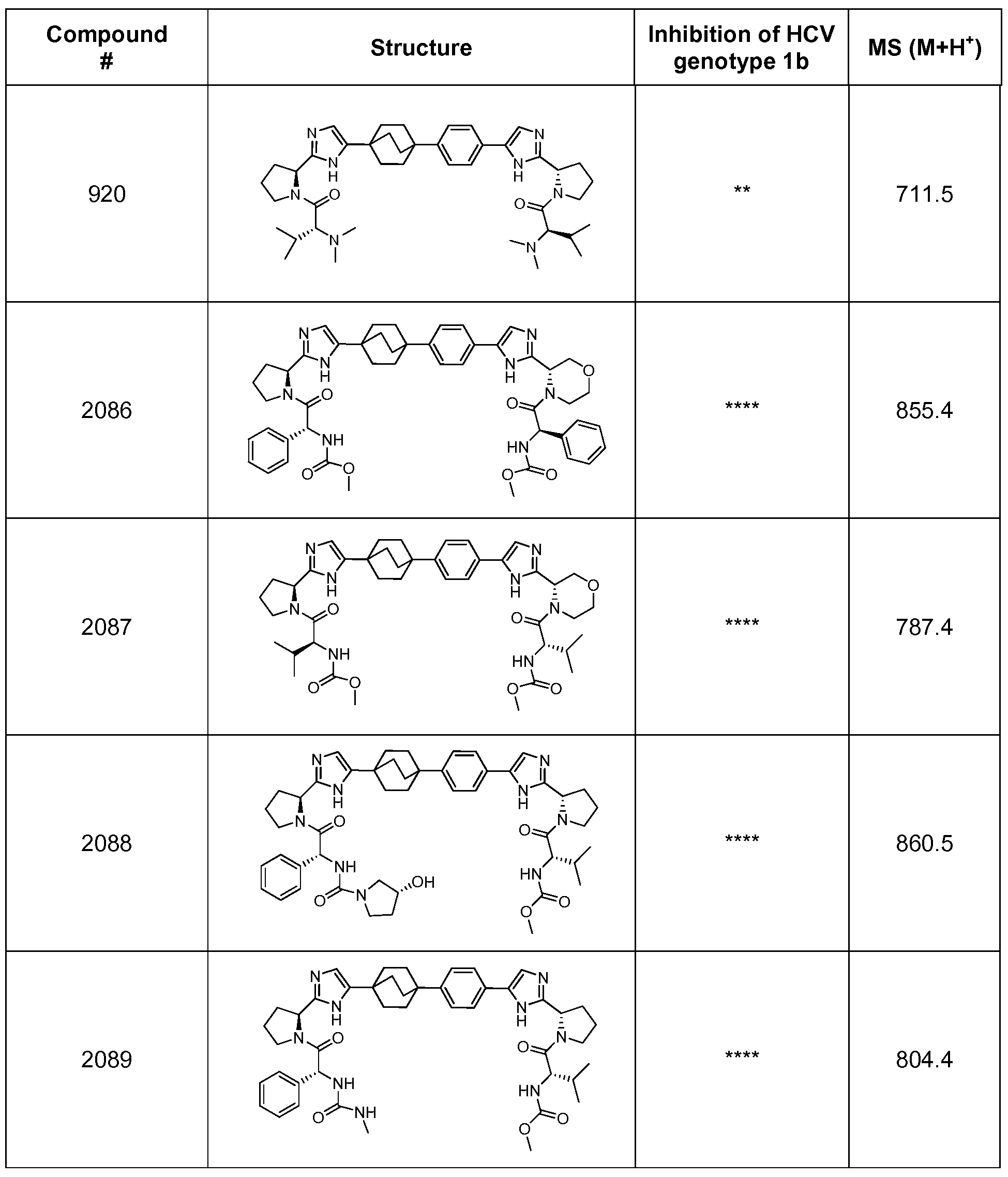


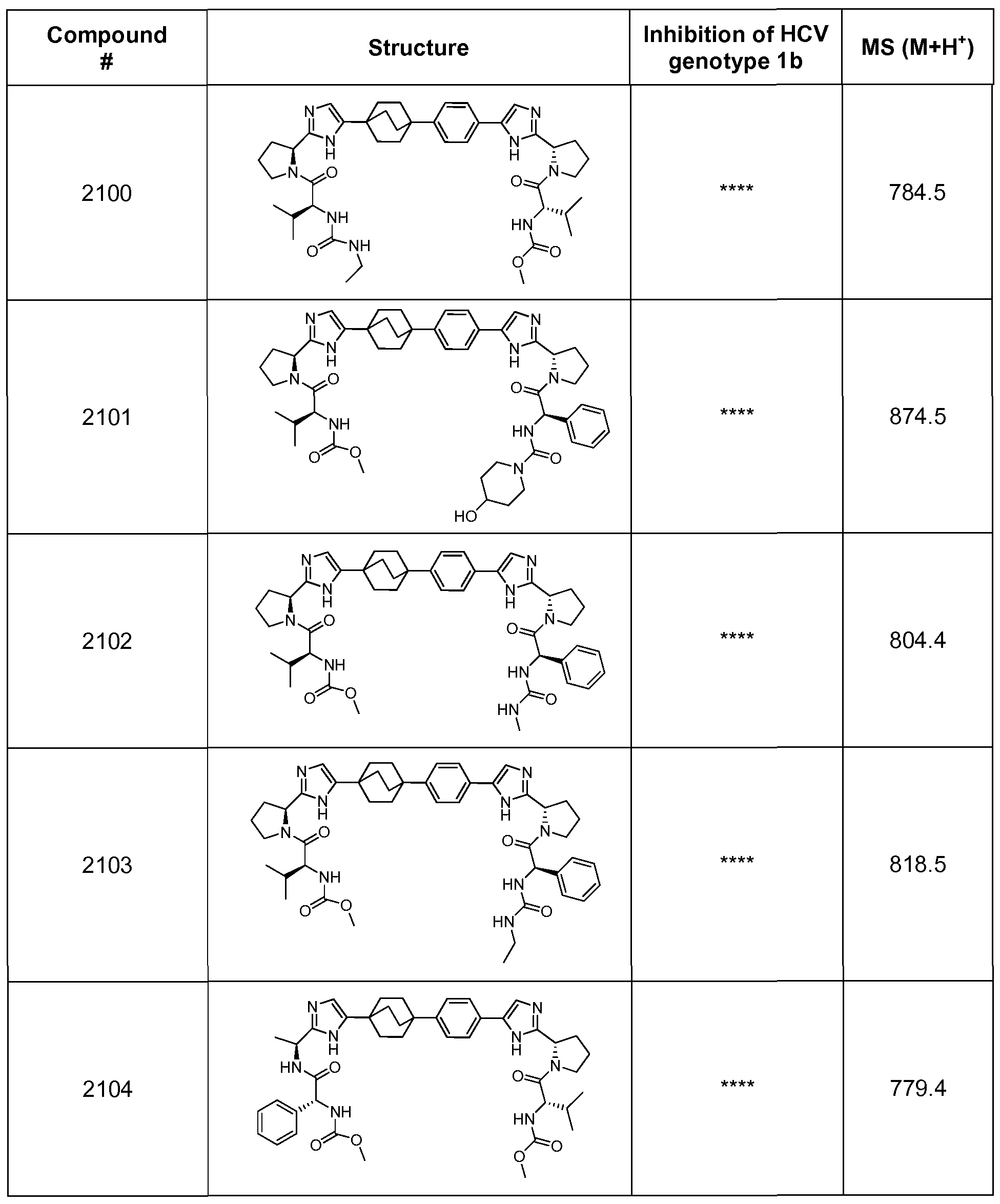





Claims





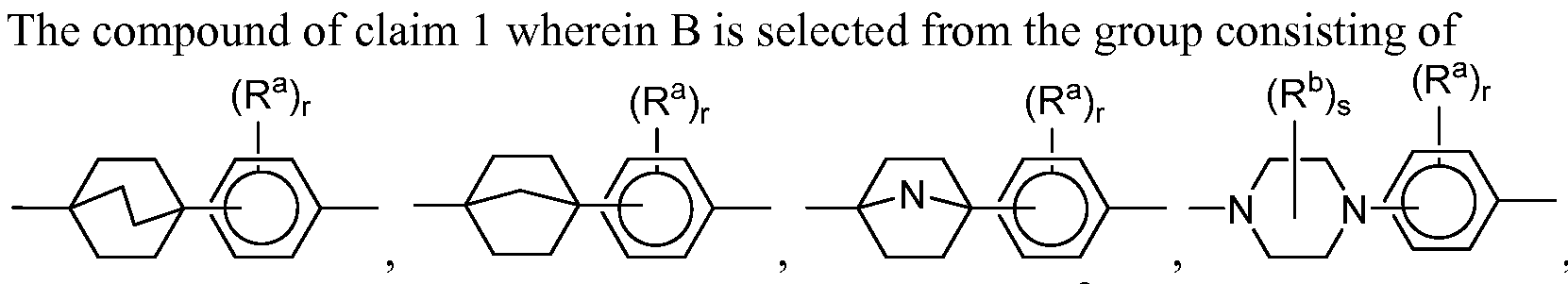
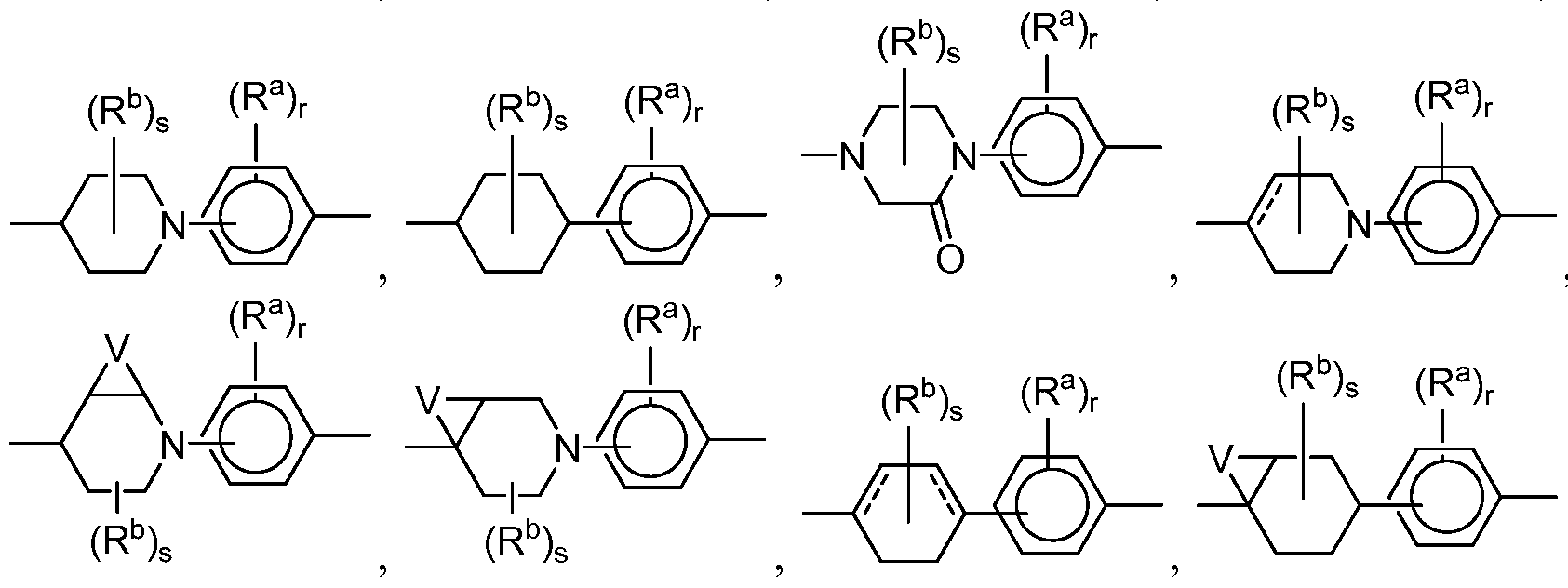




































Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2009322400A AU2009322400A1 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of HCV NS5A |
| EP09831084A EP2373168A4 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of hcv ns5a |
| CN200980155944.3A CN102300462B (en) | 2008-12-03 | 2009-12-02 | Inhibitors of HCV NS5A |
| NZ593808A NZ593808A (en) | 2008-12-03 | 2009-12-02 | Inhibitors of hcv ns5a |
| JP2011539670A JP2012510525A (en) | 2008-12-03 | 2009-12-02 | Inhibitor of HCVNS5A |
| US13/132,606 US9120779B2 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of HCV NS5A |
| CA2750577A CA2750577A1 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of hcv ns5a |
| SG2011039781A SG171890A1 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of hcv ns5a |
| ARP090104678A AR074474A1 (en) | 2008-12-03 | 2009-12-03 | HC5 NS5A INHIBITORS |
| IL213279A IL213279A0 (en) | 2008-12-03 | 2011-05-31 | Inhibitors of hcv ns5a |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11972308P | 2008-12-03 | 2008-12-03 | |
| US61/119,723 | 2008-12-03 | ||
| US17359009P | 2009-04-28 | 2009-04-28 | |
| US61/173,590 | 2009-04-28 | ||
| US18295209P | 2009-06-01 | 2009-06-01 | |
| US61/182,952 | 2009-06-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010065681A1 true WO2010065681A1 (en) | 2010-06-10 |
Family
ID=42233598
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/066451 WO2010065668A1 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of hcv ns5a |
| PCT/US2009/066467 WO2010065681A1 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of hcv ns5a |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/066451 WO2010065668A1 (en) | 2008-12-03 | 2009-12-02 | Inhibitors of hcv ns5a |
Country Status (20)
| Country | Link |
|---|---|
| US (3) | US9120779B2 (en) |
| EP (3) | EP2774927A1 (en) |
| JP (2) | JP2012510525A (en) |
| KR (1) | KR20110098779A (en) |
| CN (2) | CN102300462B (en) |
| AR (2) | AR080264A1 (en) |
| AU (2) | AU2009322387A1 (en) |
| BR (1) | BRPI0922364A2 (en) |
| CA (2) | CA2745119A1 (en) |
| CL (1) | CL2011001332A1 (en) |
| CO (1) | CO6390076A2 (en) |
| EA (1) | EA201190007A1 (en) |
| IL (2) | IL213278A0 (en) |
| MX (1) | MX2011005896A (en) |
| NZ (2) | NZ593808A (en) |
| PE (1) | PE20120207A1 (en) |
| SG (2) | SG171891A1 (en) |
| TW (2) | TWI472526B (en) |
| WO (2) | WO2010065668A1 (en) |
| ZA (1) | ZA201104911B (en) |
Cited By (123)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010117704A1 (en) * | 2009-03-30 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2010132601A1 (en) * | 2009-05-13 | 2010-11-18 | Gilead Sciences, Inc. | Antiviral compounds |
| WO2011015658A1 (en) | 2009-08-07 | 2011-02-10 | Tibotec Pharmaceuticals | Bis-benzimidazole derivatives as hepatitis c virus inhibitors |
| WO2011026920A1 (en) | 2009-09-03 | 2011-03-10 | Tibotec Pharmaceuticals | Bis-benzimidazole derivatives |
| WO2011054834A1 (en) | 2009-11-04 | 2011-05-12 | Tibotec Pharmaceuticals | Benzimidazole-imidazole derivatives |
| WO2011075607A1 (en) * | 2009-12-18 | 2011-06-23 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| WO2011081918A1 (en) | 2009-12-14 | 2011-07-07 | Enanta Pharmaceuticals, Inc | Hepatitis c virus inhibitors |
| WO2011087740A1 (en) | 2009-12-22 | 2011-07-21 | Schering Corporation | Fused tricyclic compounds and methods of use thereof for the treatment of viral diseases |
| WO2011112429A1 (en) | 2010-03-09 | 2011-09-15 | Schering Corporation | Fused tricyclic silyl compounds and methods of use thereof for the treatment of viral diseases |
| WO2012006055A2 (en) | 2010-06-28 | 2012-01-12 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flavivirus infections |
| WO2012006060A1 (en) | 2010-06-28 | 2012-01-12 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flavivirus infections |
| US8101643B2 (en) | 2009-02-27 | 2012-01-24 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| WO2012013643A1 (en) | 2010-07-26 | 2012-02-02 | Tibotec Pharmaceuticals | Hetero-bicyclic derivatives as hcv inhibitors |
| WO2012018534A2 (en) | 2010-07-26 | 2012-02-09 | Schering Corporation | Substituted biphenylene compounds and methods of use thereof for the treatment of viral diseases |
| WO2012024363A2 (en) | 2010-08-17 | 2012-02-23 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
| US8138215B2 (en) | 2009-05-29 | 2012-03-20 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8143414B2 (en) | 2009-04-13 | 2012-03-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8143301B2 (en) | 2009-04-09 | 2012-03-27 | Bristol Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2012040389A2 (en) * | 2010-09-22 | 2012-03-29 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic hcv inhibitors |
| EP2435421A1 (en) * | 2009-05-29 | 2012-04-04 | Schering Corporation | Antiviral compounds composed of three aligned aryl moieties to treat diseases such as hepatitis c |
| WO2012041014A1 (en) * | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives for treating hepatitis c virus infection |
| WO2012051361A1 (en) | 2010-10-13 | 2012-04-19 | Abbott Laboratories | Anti-viral compounds |
| WO2012050848A1 (en) | 2010-09-29 | 2012-04-19 | Schering Corporation | Fused tetracycle derivatives and methods of use thereof for the treatment of viral diseases |
| WO2012058125A1 (en) | 2010-10-26 | 2012-05-03 | Presidio Pharmaceuticals, Inc. | Inhibitors of hepatitis c virus |
| US8178531B2 (en) | 2010-02-23 | 2012-05-15 | Enanta Pharmaceuticals, Inc. | Antiviral agents |
| US8188132B2 (en) | 2009-02-17 | 2012-05-29 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| WO2012074437A2 (en) | 2010-11-30 | 2012-06-07 | Алла Хем, Ллс | Substituted azoles, anti-viral active ingredient, pharmaceutical composition, method for the production and use thereof |
| WO2012083170A1 (en) * | 2010-12-16 | 2012-06-21 | Abbott Laboratories | Anti-viral compounds |
| WO2012087976A2 (en) * | 2010-12-21 | 2012-06-28 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| US8211928B2 (en) | 2009-05-29 | 2012-07-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8221737B2 (en) | 2009-06-16 | 2012-07-17 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| WO2012083048A3 (en) * | 2010-12-15 | 2012-08-02 | Abbott Laboratories | Anti-viral compounds |
| US8242156B2 (en) | 2009-02-17 | 2012-08-14 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| JP2012519185A (en) * | 2009-02-27 | 2012-08-23 | エナンタ ファーマシューティカルズ インコーポレイテッド | Hepatitis C virus inhibitor |
| US8314135B2 (en) | 2009-02-09 | 2012-11-20 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole antivirals |
| US8344155B2 (en) | 2009-09-04 | 2013-01-01 | Glaxosmith Kline Llc | Chemical compounds |
| WO2012068234A3 (en) * | 2010-11-17 | 2013-01-17 | 12Gilead Sciences, Inc. | Antiviral compounds |
| US8362020B2 (en) | 2009-12-30 | 2013-01-29 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2013016491A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Thiophene compounds |
| WO2013016499A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Methods for preparation of thiophene compounds |
| US8377980B2 (en) | 2009-12-16 | 2013-02-19 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8394968B2 (en) | 2009-02-17 | 2013-03-12 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2013039876A1 (en) | 2011-09-14 | 2013-03-21 | Merck Sharp & Dohme Corp. | Silyl-containing heterocyclic compounds and methods of use thereof for the treatment of viral diseases |
| US8420686B2 (en) | 2009-02-17 | 2013-04-16 | Enanta Pharmaceuticals, Inc. | Linked diimidazole antivirals |
| US8426458B2 (en) | 2009-02-27 | 2013-04-23 | Enanta Pharmaceuticals, Inc. | Hepatitis C Virus inhibitors |
| WO2013098313A1 (en) | 2011-12-28 | 2013-07-04 | Janssen R&D Ireland | Hetero-bicyclic derivatives as hcv inhibitors |
| WO2013098320A1 (en) | 2011-12-28 | 2013-07-04 | Janssen R&D Ireland | Quinazolinone derivatives as hcv inhibitors |
| US8507522B2 (en) | 2009-03-06 | 2013-08-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8541424B2 (en) | 2008-12-23 | 2013-09-24 | Abbott Laboratories | Anti-viral compounds |
| US8546405B2 (en) | 2008-12-23 | 2013-10-01 | Abbott Laboratories | Anti-viral compounds |
| US8552047B2 (en) | 2011-02-07 | 2013-10-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8575135B2 (en) | 2011-11-16 | 2013-11-05 | Gilead Sciences, Inc. | Antiviral compounds |
| US8609648B2 (en) | 2009-07-02 | 2013-12-17 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| CN103459399A (en) * | 2010-09-29 | 2013-12-18 | 默沙东公司 | Tetracyclic indole derivatives for treating hepatitis c virus infection |
| US8618153B2 (en) | 2009-11-12 | 2013-12-31 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8623814B2 (en) | 2010-02-23 | 2014-01-07 | Enanta Pharmaceuticals, Inc. | Antiviral agents |
| US8637561B2 (en) | 2009-02-17 | 2014-01-28 | Enanta Pharmaceuticals, Inc. | Linked diimidazole derivatives |
| US8673954B2 (en) | 2009-02-27 | 2014-03-18 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US8686026B2 (en) | 2010-06-10 | 2014-04-01 | Abbvie Inc. | Solid compositions |
| US8691938B2 (en) | 2009-06-11 | 2014-04-08 | Abbvie Inc. | Anti-viral compounds |
| US8697704B2 (en) | 2010-08-12 | 2014-04-15 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8703938B2 (en) | 2009-09-11 | 2014-04-22 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8709999B2 (en) | 2009-03-27 | 2014-04-29 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic HCV inhibitors |
| US8716454B2 (en) | 2009-06-11 | 2014-05-06 | Abbvie Inc. | Solid compositions |
| US8759332B2 (en) | 2009-09-11 | 2014-06-24 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| CN103880823A (en) * | 2012-12-21 | 2014-06-25 | 广东东阳光药业有限公司 | Spiro compound serving as hepatitis c inhibitor and application thereof in medicine |
| US8765731B2 (en) | 2009-07-16 | 2014-07-01 | Vertex Pharmaceuticals Incorporated | Benzimidazole analogues for the treatment or prevention of flavivirus infections |
| US8779156B2 (en) | 2010-03-24 | 2014-07-15 | Vertex Pharmaceuticals Incorporated | Analogues for the treatment or prevention of flavivirus infections |
| US8778938B2 (en) | 2010-06-04 | 2014-07-15 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8785487B2 (en) | 2010-01-25 | 2014-07-22 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8796466B2 (en) | 2009-03-30 | 2014-08-05 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CN103987723A (en) * | 2011-12-16 | 2014-08-13 | 弗·哈夫曼-拉罗切有限公司 | Inhibitors of HCV NS5A |
| WO2014123456A2 (en) | 2013-02-07 | 2014-08-14 | ИВАЩЕНКО, Андрей Александрович | Alkyl [(s)-1-((s)-2-{5-[4-(4-{2-[(s)-1-((s)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidine-2-yl]-3h-imidazole-4-yl}-buta-1,3-dienyl)-phenyl]-1h-imidazole-2-yl}-pyrrolidine-1-carbonyl)-2-methyl-propyl]-carbamate naphthalene-1,5-disulfonate, pharmaceutical composition, medicinal agent and method for treatment of viral diseases |
| US8809548B2 (en) | 2009-02-17 | 2014-08-19 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8815928B2 (en) | 2009-09-11 | 2014-08-26 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8822700B2 (en) | 2009-09-11 | 2014-09-02 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| WO2014134251A1 (en) | 2013-02-28 | 2014-09-04 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions |
| US8927739B2 (en) | 2011-05-18 | 2015-01-06 | Enanta Pharmaceuticals, Inc. | Processes for the preparation of 5-azaspiro[2.4]heptane-6-carboxylic acid and its derivatives |
| US8927709B2 (en) | 2009-09-11 | 2015-01-06 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8933110B2 (en) | 2010-01-25 | 2015-01-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| JP2015044866A (en) * | 2009-11-11 | 2015-03-12 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Hepatitis c virus inhibitors |
| US8980920B2 (en) | 2009-05-29 | 2015-03-17 | Merck Sharp & Dohme Corp. | Antiviral compounds of three linked aryl moieties to treat diseases such as hepatitis C |
| US8999967B2 (en) | 2010-09-29 | 2015-04-07 | Presidio Pharmaceuticals, Inc. | Tricyclic fused ring inhibitors of hepatitis C |
| JP2015510512A (en) * | 2012-02-10 | 2015-04-09 | ルピン・リミテッドLupin Limited | Antiviral compounds having a heterotricyclic moiety |
| US9012427B2 (en) | 2012-03-22 | 2015-04-21 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| US9034832B2 (en) | 2011-12-29 | 2015-05-19 | Abbvie Inc. | Solid compositions |
| US9060971B2 (en) | 2010-03-04 | 2015-06-23 | Enanta Pharmaceuticals, Inc. | Combination pharmaceutical agents as inhibitors of HCV replication |
| WO2015094998A1 (en) | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Fused tetracyclic heterocyclic compounds and methods of use thereof for the treatment of viral diseases |
| US9127021B2 (en) | 2010-04-09 | 2015-09-08 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US9139569B2 (en) | 2009-05-12 | 2015-09-22 | Merck Sharp & Dohme Corp. | Fused tricyclic aryl compounds useful for the treatment of viral diseases |
| US9156818B2 (en) | 2009-09-11 | 2015-10-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US9187496B2 (en) | 2009-12-18 | 2015-11-17 | Idenix Pharmaceuticals Llc | 5,5-fused arylene or heteroarylene hepatitis C virus inhibitors |
| US9278922B2 (en) | 2009-04-15 | 2016-03-08 | Abbvie Inc. | Anti-viral compounds |
| US9326973B2 (en) | 2012-01-13 | 2016-05-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9333204B2 (en) | 2014-01-03 | 2016-05-10 | Abbvie Inc. | Solid antiviral dosage forms |
| JP2016106075A (en) * | 2010-10-13 | 2016-06-16 | アッヴィ・バハマズ・リミテッド | Anti-virus compound |
| AU2014203655B2 (en) * | 2010-10-13 | 2016-07-07 | Abbvie Ireland Unlimited Company | Anti-viral compounds |
| US9393256B2 (en) | 2011-09-16 | 2016-07-19 | Gilead Pharmasset Llc | Methods for treating HCV |
| US9394279B2 (en) | 2009-06-11 | 2016-07-19 | Abbvie Inc. | Anti-viral compounds |
| US9546160B2 (en) | 2011-05-12 | 2017-01-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9717712B2 (en) | 2013-07-02 | 2017-08-01 | Bristol-Myers Squibb Company | Combinations comprising tricyclohexadecahexaene derivatives for use in the treatment of hepatitis C virus |
| US9732080B2 (en) | 2006-11-03 | 2017-08-15 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| US9765087B2 (en) | 2009-02-27 | 2017-09-19 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US9770439B2 (en) | 2013-07-02 | 2017-09-26 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9775831B2 (en) | 2013-07-17 | 2017-10-03 | Bristol-Myers Squibb Company | Combinations comprising biphenyl derivatives for use in the treatment of HCV |
| US9776981B2 (en) | 2009-11-11 | 2017-10-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10039779B2 (en) | 2013-01-31 | 2018-08-07 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US10071979B2 (en) | 2010-04-22 | 2018-09-11 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| US10081621B2 (en) | 2010-03-25 | 2018-09-25 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US10086011B2 (en) | 2013-08-27 | 2018-10-02 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US10118920B2 (en) | 2015-04-20 | 2018-11-06 | Cellcentric Ltd | Isoxazolyl substituted benzimidazoles |
| US10167298B2 (en) | 2013-10-30 | 2019-01-01 | Merck Sharp & Dohme Corp. | Pseudopolymorphs of an HCV NS5A inhibitor and uses thereof |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| US10206877B2 (en) | 2014-04-15 | 2019-02-19 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US10428065B2 (en) | 2015-04-20 | 2019-10-01 | Cellcentric Ltd | Isoxazolyl substituted imidazopyridines |
| US10617675B2 (en) | 2015-08-06 | 2020-04-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2020117849A1 (en) | 2018-12-04 | 2020-06-11 | Bristol-Myers Squibb Company | Methods of analysis using in-sample calibration curve by multiple isotopologue reaction monitoring |
| US10800789B2 (en) | 2012-05-16 | 2020-10-13 | Gilead Pharmasset Llc | Antiviral compounds |
| US11203599B2 (en) | 2014-06-11 | 2021-12-21 | Gilead Pharmasset Llc | Solid forms of an antiviral compound |
| US11484534B2 (en) | 2013-03-14 | 2022-11-01 | Abbvie Inc. | Methods for treating HCV |
| US11760764B2 (en) | 2020-05-22 | 2023-09-19 | Aligos Therapeutics, Inc. | Methods and compositions for targeting PD-L1 |
| US12037340B2 (en) | 2021-05-21 | 2024-07-16 | Gilead Sciences, Inc. | Pentacyclic derivatives as Zika virus inhibitors |
| US12071428B2 (en) | 2020-12-30 | 2024-08-27 | Tyra Biosciences, Inc. | Indazole compounds as kinase inhibitors |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT2373172E (en) * | 2008-12-03 | 2013-10-21 | Presidio Pharmaceuticals Inc | Inhibitors of hcv ns5a |
| AU2013204195B2 (en) * | 2009-02-27 | 2016-09-22 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| CN102471327A (en) | 2009-07-21 | 2012-05-23 | 吉里德科学公司 | Inhibitors of flaviviridae viruses |
| CA2768637A1 (en) | 2009-08-07 | 2011-02-10 | Tibotec Pharmaceuticals | Phenyl ethynyl derivatives as hepatitis c virus inhibitors |
| CA2771124C (en) | 2009-09-09 | 2019-01-15 | Gilead Sciences, Inc. | Inhibitors of flaviviridae viruses |
| WO2011031934A1 (en) * | 2009-09-11 | 2011-03-17 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| US20130072523A1 (en) * | 2009-12-24 | 2013-03-21 | Vertex Pharmaceuticals Incorporated | Analogues for the treatment or prevention of flavivirus infections |
| EP3219713A1 (en) | 2010-01-15 | 2017-09-20 | Gilead Sciences, Inc. | Inhibitors of flaviviridae viruses |
| EP2523951B1 (en) | 2010-01-15 | 2015-04-22 | Gilead Sciences, Inc. | Inhibitors of flaviviridae viruses |
| TW201141857A (en) * | 2010-03-24 | 2011-12-01 | Vertex Pharma | Analogues for the treatment or prevention of flavivirus infections |
| CA2800530A1 (en) * | 2010-05-28 | 2011-12-01 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2011156543A2 (en) * | 2010-06-09 | 2011-12-15 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a protein |
| CA2807305A1 (en) * | 2010-08-04 | 2012-02-09 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| EP2963034A1 (en) * | 2010-08-26 | 2016-01-06 | RFS Pharma, LLC. | Potent and selective inhibitors of hepatitis c virus |
| EP2619195A1 (en) * | 2010-09-24 | 2013-07-31 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| CN103189371B (en) | 2010-11-04 | 2015-04-01 | 施万生物制药研发Ip有限责任公司 | Novel inhibitors of hepatitis C virus |
| US20140364616A1 (en) * | 2010-12-15 | 2014-12-11 | Abbvie Inc. | Anti-viral compounds |
| US20150031884A1 (en) * | 2010-12-15 | 2015-01-29 | Abbvie Inc. | Anti-viral compounds |
| WO2012083058A2 (en) * | 2010-12-15 | 2012-06-21 | Abbott Laboratories | Anti-viral compounds |
| WO2012091115A1 (en) * | 2010-12-29 | 2012-07-05 | キッセイ薬品工業株式会社 | Acetylene derivative and pharmaceutical application therefor |
| CA2840445A1 (en) | 2011-07-13 | 2013-01-17 | Gilead Sciences, Inc. | Thiophen-2-carboxylic acid derivatives useful as inhibitors of flaviviridae viruses |
| WO2013030750A1 (en) | 2011-09-01 | 2013-03-07 | Lupin Limited | Antiviral compounds |
| CA2870264C (en) | 2012-04-17 | 2016-11-08 | Fujifilm Corporation | Nitrogen-containing heterocyclic compound or salt thereof |
| TWI610916B (en) | 2012-08-03 | 2018-01-11 | 廣東東陽光藥業有限公司 | Bridged ring compounds as hepatitis c virus (hcv) inhibitors and pharmaceuticals applications thereof |
| US8759544B2 (en) | 2012-08-17 | 2014-06-24 | Gilead Sciences, Inc. | Synthesis of an antiviral compound |
| US8927741B2 (en) | 2012-08-17 | 2015-01-06 | Gilead Sciences, Inc. | Synthesis of an antiviral compound |
| US8841340B2 (en) | 2012-08-17 | 2014-09-23 | Gilead Sciences, Inc. | Solid forms of an antiviral compound |
| US9802949B2 (en) | 2012-11-29 | 2017-10-31 | Sunshine Lake Pharma Co., Ltd. | Fused ring compounds as hepatitis C virus inhibitors, pharmaceutical compositions and uses thereof |
| CN103848821B (en) | 2012-11-29 | 2016-10-12 | 广东东阳光药业有限公司 | Spiro-compound, pharmaceutical composition and their purposes as hepatitis c inhibitor |
| CN103664581B (en) * | 2013-12-20 | 2016-07-06 | 中节能万润股份有限公司 | A kind of anti-, trans-4, the preparation method of 4 '-dicyclohexyl dioctyl phthalate |
| WO2015110048A1 (en) | 2014-01-23 | 2015-07-30 | Sunshine Lake Pharma Co., Ltd. | Bridged ring compounds as hepatitis c virus inhibitors, pharmaceutical compositions and uses thereof |
| CN105175408B (en) * | 2014-06-04 | 2018-07-17 | 中国人民解放军第二军医大学 | Benzothiazole compound and its purposes as drug |
| CN106279117A (en) * | 2016-08-16 | 2017-01-04 | 上海同昌生物医药科技有限公司 | A kind of synthetic method of Lei Dipawei intermediate and products thereof |
| CN108727345B (en) * | 2017-04-25 | 2023-06-27 | 广东东阳光药业有限公司 | Preparation method of imidazole ring intermediate |
| KR20240035395A (en) | 2021-06-14 | 2024-03-15 | 스코르피온 테라퓨틱스, 인코퍼레이티드 | Urea derivatives that can be used in cancer treatment |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007014922A1 (en) * | 2005-07-29 | 2007-02-08 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis c virus |
Family Cites Families (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT1244507B (en) * | 1991-04-11 | 1994-07-15 | Sigma Tau Ind Farmaceuti | PYROGLUTAMIC ACID DERIVATIVES AS ACTIVATORS OF LEARNING PROCESSES AND MEMORY AND PHARMACEUTICAL COMPOSITIONS INCLUDING SUCH COMPOUNDS |
| DE4234295A1 (en) | 1992-10-12 | 1994-04-14 | Thomae Gmbh Dr K | Carboxylic acid derivatives, medicaments containing these compounds and process for their preparation |
| US5807876A (en) | 1996-04-23 | 1998-09-15 | Vertex Pharmaceuticals Incorporated | Inhibitors of IMPDH enzyme |
| US6054472A (en) | 1996-04-23 | 2000-04-25 | Vertex Pharmaceuticals, Incorporated | Inhibitors of IMPDH enzyme |
| JPH09227417A (en) * | 1996-02-26 | 1997-09-02 | Dainippon Ink & Chem Inc | Alkenyltolan derivative and its production |
| NZ332405A (en) | 1996-04-23 | 2000-06-23 | Vertex Pharma | oxazolyl, thiazolyl or phenyl urea derivatives as inhibitors of inosine monophosphate dehydrogenase enzyme |
| JP3865084B2 (en) * | 1996-07-04 | 2007-01-10 | 大日本インキ化学工業株式会社 | Dialkenyltolane derivatives |
| EP2409985A3 (en) | 1996-10-18 | 2013-05-01 | Vertex Pharmaceuticals Incorporated | Inhibitors de serine proteases, especially of the NS3 protease of the hepatitis C virus |
| GB9623908D0 (en) | 1996-11-18 | 1997-01-08 | Hoffmann La Roche | Amino acid derivatives |
| ES2201452T3 (en) | 1997-03-14 | 2004-03-16 | Vertex Pharmaceuticals Incorporated | INHIBITORS OF THE IMPDH ENZYME. |
| US6010848A (en) | 1997-07-02 | 2000-01-04 | Smithkline Beecham Corporation | Screening methods using an atpase protein from hepatitis C virus |
| ES2234144T3 (en) | 1997-08-11 | 2005-06-16 | Boehringer Ingelheim (Canada) Ltd. | ANALOGS OF INHIBITING PEPTIDES OF HEPATITIS C. |
| DE69925918T2 (en) | 1998-07-27 | 2006-05-11 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | DIKETIC ACID DERIVATIVES AS POLYMERASES INHIBITORS |
| US6323180B1 (en) | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| BR9913157A (en) | 1998-08-21 | 2001-05-15 | Viropharma Inc | Processes for treating or preventing infection caused by at least one virus of the flaviviridae and diseases associated with said infection and infection caused by at least one virus of the genus hepacivirus of flavivirity and diseases associated with said infection, pharmaceutical composition to treat or prevent viral infections , it's composed |
| AU751201B2 (en) | 1998-09-04 | 2002-08-08 | Viropharma Incorporated | Methods for treating or preventing viral infections and associated diseases |
| JP2002525295A (en) | 1998-09-25 | 2002-08-13 | バイロファーマ・インコーポレイテッド | How to treat or prevent viral infections and related diseases |
| TR200103428T2 (en) | 1999-03-19 | 2002-04-22 | Vertex Pharmaceuticals Incorporated | IMPDH enzyme inhibitors. |
| WO2000066578A1 (en) * | 1999-04-30 | 2000-11-09 | Pfizer Products Inc. | Compounds for the treatment of obesity |
| CA2389745C (en) | 1999-11-04 | 2010-03-23 | Shire Biochem Inc. | Method for the treatment or prevention of flaviviridae viral infection using nucleoside analogues |
| JP2001294541A (en) * | 2000-04-14 | 2001-10-23 | Dainippon Ink & Chem Inc | Method for producing tolane derivative |
| WO2001085172A1 (en) | 2000-05-10 | 2001-11-15 | Smithkline Beecham Corporation | Novel anti-infectives |
| US6448281B1 (en) | 2000-07-06 | 2002-09-10 | Boehringer Ingelheim (Canada) Ltd. | Viral polymerase inhibitors |
| SV2003000617A (en) | 2000-08-31 | 2003-01-13 | Lilly Co Eli | INHIBITORS OF PROTEASA PEPTIDOMIMETICA REF. X-14912M |
| EP1256628A3 (en) | 2001-05-10 | 2003-03-19 | Agouron Pharmaceuticals, Inc. | Hepatitis c virus (hcv) ns5b rna polymerase and mutants thereof |
| JP4544857B2 (en) | 2001-06-11 | 2010-09-15 | ヴァイロケム ファーマ インコーポレイテッド | Compounds and methods for the treatment or prevention of FLAVIRIRUS infection |
| WO2002100846A1 (en) | 2001-06-11 | 2002-12-19 | Shire Biochem Inc. | Compounds and methods for the treatment or prevention of flavivirus infections |
| AR035543A1 (en) | 2001-06-26 | 2004-06-16 | Japan Tobacco Inc | THERAPEUTIC AGENT FOR HEPATITIS C THAT INCLUDES A CONDENSED RING COMPOUND, CONDENSED RING COMPOUND, PHARMACEUTICAL COMPOSITION THAT UNDERSTANDS, BENZIMIDAZOL, THIAZOL AND BIFENYL COMPOUNDS USED AS INTERMEDIARY COMPARTMENTS OF COMPARTMENTS |
| WO2003007959A1 (en) * | 2001-07-16 | 2003-01-30 | Fujisawa Pharmaceutical Co., Ltd. | Quinoxaline derivatives which have parp inhibitory action |
| US6841566B2 (en) | 2001-07-20 | 2005-01-11 | Boehringer Ingelheim, Ltd. | Viral polymerase inhibitors |
| EP2335700A1 (en) | 2001-07-25 | 2011-06-22 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C virus polymerase inhibitors with a heterobicylic structure |
| EP1440069B1 (en) | 2001-11-02 | 2007-08-15 | Glaxo Group Limited | 4-(6-membered)-heteroaryl acyl pyrrolidine derivatives as hcv inhibitors |
| JP2005511572A (en) | 2001-11-02 | 2005-04-28 | グラクソ グループ リミテッド | Acyl dihydropyrrole derivatives as HCV inhibitors |
| JP2005511573A (en) | 2001-11-02 | 2005-04-28 | グラクソ グループ リミテッド | 4- (5-membered) -heteroarylacylpyrrolidine derivatives as HCV inhibitors |
| JP2006505570A (en) * | 2002-10-17 | 2006-02-16 | アムジエン・インコーポレーテツド | Benzimidazole derivatives and their use as vanilloid receptor ligands |
| JP4206382B2 (en) | 2002-11-19 | 2009-01-07 | アキリオン ファーマシューティカルズ,インコーポレーテッド | Substituted arylthioureas and related compounds; inhibitors of viral replication |
| AU2003294443A1 (en) * | 2002-11-22 | 2004-06-18 | Bristol-Myers Squibb Company | 3-(pyridinyl-piperazin-1-yl)-phenylethyl amides as potassium channel openers |
| JP4584909B2 (en) | 2003-05-09 | 2010-11-24 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Hepatitis C virus NS5B polymerase inhibitor binding pocket |
| JP2007534624A (en) * | 2003-07-16 | 2007-11-29 | ニューロジェン・コーポレーション | Biaryl piperazinyl-pyridine analogues |
| WO2005073195A2 (en) | 2004-01-30 | 2005-08-11 | Medivir Ab | Hcv ns-3 serine protease inhibitors |
| KR20070083484A (en) | 2004-07-14 | 2007-08-24 | 피티씨 테라퓨틱스, 인크. | Methods for treating hepatitis c |
| UA90477C2 (en) | 2004-07-14 | 2010-05-11 | ПиТиСи ТЕРАПЬЮТИКС, ИНК. | Methods for treating hepatitis c (variants) |
| US8143288B2 (en) * | 2005-06-06 | 2012-03-27 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| TW200813015A (en) | 2006-03-15 | 2008-03-16 | Mitsubishi Pharma Corp | 2-(cyclic amino)-pyrimidone derivatives |
| US8329159B2 (en) | 2006-08-11 | 2012-12-11 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7659270B2 (en) * | 2006-08-11 | 2010-02-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7759495B2 (en) * | 2006-08-11 | 2010-07-20 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| KR20090086081A (en) | 2006-11-15 | 2009-08-10 | 바이로켐 파마 인코포레이티드 | Thiophene analogues for the treatment or prevention of flavivirus infections |
| WO2008073461A2 (en) * | 2006-12-11 | 2008-06-19 | Wyeth | Ion channel modulators |
| US7723343B2 (en) * | 2007-03-30 | 2010-05-25 | King Pharmaceuticals Research And Development, Inc. | Adenosine A2A receptor antagonists |
| CN101280196B (en) * | 2008-06-03 | 2011-01-26 | 西安近代化学研究所 | Four-ring liquid crystal compounds |
| US7906655B2 (en) * | 2008-08-07 | 2011-03-15 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8383094B2 (en) | 2008-10-01 | 2013-02-26 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| PT2373172E (en) * | 2008-12-03 | 2013-10-21 | Presidio Pharmaceuticals Inc | Inhibitors of hcv ns5a |
| EP2393359A4 (en) | 2009-02-09 | 2012-10-03 | Enanta Pharm Inc | Linked dibenzimidazole derivatives |
| WO2010099527A1 (en) * | 2009-02-27 | 2010-09-02 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| NZ706236A (en) * | 2009-05-13 | 2016-07-29 | Gilead Pharmasset Llc | Antiviral compounds |
-
2009
- 2009-12-02 WO PCT/US2009/066451 patent/WO2010065668A1/en active Application Filing
- 2009-12-02 US US13/132,606 patent/US9120779B2/en not_active Expired - Fee Related
- 2009-12-02 AU AU2009322387A patent/AU2009322387A1/en not_active Abandoned
- 2009-12-02 EP EP13189259.8A patent/EP2774927A1/en not_active Withdrawn
- 2009-12-02 NZ NZ593808A patent/NZ593808A/en not_active IP Right Cessation
- 2009-12-02 WO PCT/US2009/066467 patent/WO2010065681A1/en active Application Filing
- 2009-12-02 JP JP2011539670A patent/JP2012510525A/en active Pending
- 2009-12-02 US US13/132,604 patent/US8865756B2/en not_active Expired - Fee Related
- 2009-12-02 CN CN200980155944.3A patent/CN102300462B/en not_active Expired - Fee Related
- 2009-12-02 CN CN200980155930.1A patent/CN102300461B/en not_active Expired - Fee Related
- 2009-12-02 EP EP09831084A patent/EP2373168A4/en not_active Withdrawn
- 2009-12-02 SG SG2011039799A patent/SG171891A1/en unknown
- 2009-12-02 BR BRPI0922364A patent/BRPI0922364A2/en not_active IP Right Cessation
- 2009-12-02 CA CA2745119A patent/CA2745119A1/en not_active Abandoned
- 2009-12-02 NZ NZ593806A patent/NZ593806A/en not_active IP Right Cessation
- 2009-12-02 EA EA201190007A patent/EA201190007A1/en unknown
- 2009-12-02 AU AU2009322400A patent/AU2009322400A1/en not_active Abandoned
- 2009-12-02 PE PE2011001155A patent/PE20120207A1/en not_active Application Discontinuation
- 2009-12-02 CA CA2750577A patent/CA2750577A1/en not_active Abandoned
- 2009-12-02 SG SG2011039781A patent/SG171890A1/en unknown
- 2009-12-02 MX MX2011005896A patent/MX2011005896A/en not_active Application Discontinuation
- 2009-12-02 KR KR1020117015471A patent/KR20110098779A/en not_active Application Discontinuation
- 2009-12-02 JP JP2011539662A patent/JP2012510523A/en active Pending
- 2009-12-02 EP EP20090831071 patent/EP2373167A4/en not_active Withdrawn
- 2009-12-03 TW TW98141406A patent/TWI472526B/en not_active IP Right Cessation
- 2009-12-03 AR ARP090104677A patent/AR080264A1/en unknown
- 2009-12-03 TW TW098141404A patent/TWI494309B/en not_active IP Right Cessation
- 2009-12-03 AR ARP090104678A patent/AR074474A1/en unknown
-
2011
- 2011-05-31 IL IL213278A patent/IL213278A0/en unknown
- 2011-05-31 IL IL213279A patent/IL213279A0/en unknown
- 2011-06-02 CL CL2011001332A patent/CL2011001332A1/en unknown
- 2011-06-30 ZA ZA2011/04911A patent/ZA201104911B/en unknown
- 2011-07-01 CO CO11082846A patent/CO6390076A2/en not_active Application Discontinuation
-
2014
- 2014-10-20 US US14/518,940 patent/US20150038501A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007014922A1 (en) * | 2005-07-29 | 2007-02-08 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis c virus |
Non-Patent Citations (2)
| Title |
|---|
| BARBATO ET AL.: "Inhibitor binding induces active site stabilization of the HCV NS3 protein serine protease domain.", THE EMBO JOURNAL, vol. 19, no. 6, 2000, pages 1195 - 1206 * |
| See also references of EP2373168A4 * |
Cited By (206)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9732080B2 (en) | 2006-11-03 | 2017-08-15 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| US9249138B2 (en) | 2008-12-23 | 2016-02-02 | Abbvie Inc. | Anti-viral compounds |
| US8546405B2 (en) | 2008-12-23 | 2013-10-01 | Abbott Laboratories | Anti-viral compounds |
| US8541424B2 (en) | 2008-12-23 | 2013-09-24 | Abbott Laboratories | Anti-viral compounds |
| US9163017B2 (en) | 2008-12-23 | 2015-10-20 | Abbvie Inc. | Anti-viral compounds |
| US8314135B2 (en) | 2009-02-09 | 2012-11-20 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole antivirals |
| US8637561B2 (en) | 2009-02-17 | 2014-01-28 | Enanta Pharmaceuticals, Inc. | Linked diimidazole derivatives |
| US8809548B2 (en) | 2009-02-17 | 2014-08-19 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8420686B2 (en) | 2009-02-17 | 2013-04-16 | Enanta Pharmaceuticals, Inc. | Linked diimidazole antivirals |
| US8394968B2 (en) | 2009-02-17 | 2013-03-12 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8242156B2 (en) | 2009-02-17 | 2012-08-14 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| US8188132B2 (en) | 2009-02-17 | 2012-05-29 | Enanta Pharmaceuticals, Inc. | Linked dibenzimidazole derivatives |
| US8426458B2 (en) | 2009-02-27 | 2013-04-23 | Enanta Pharmaceuticals, Inc. | Hepatitis C Virus inhibitors |
| US8673954B2 (en) | 2009-02-27 | 2014-03-18 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| JP2012519185A (en) * | 2009-02-27 | 2012-08-23 | エナンタ ファーマシューティカルズ インコーポレイテッド | Hepatitis C virus inhibitor |
| US9765087B2 (en) | 2009-02-27 | 2017-09-19 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US8101643B2 (en) | 2009-02-27 | 2012-01-24 | Enanta Pharmaceuticals, Inc. | Benzimidazole derivatives |
| US8507522B2 (en) | 2009-03-06 | 2013-08-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8709999B2 (en) | 2009-03-27 | 2014-04-29 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic HCV inhibitors |
| US8796466B2 (en) | 2009-03-30 | 2014-08-05 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2010117704A1 (en) * | 2009-03-30 | 2010-10-14 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US8822444B2 (en) * | 2009-03-30 | 2014-09-02 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20120277266A1 (en) * | 2009-03-30 | 2012-11-01 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| JP2012522056A (en) * | 2009-03-30 | 2012-09-20 | ブリストル−マイヤーズ スクイブ カンパニー | Hepatitis C virus inhibitor |
| EA021260B1 (en) * | 2009-03-30 | 2015-05-29 | Бристол-Маерс Сквибб Компани | Hepatitis c virus inhibitors |
| US8143301B2 (en) | 2009-04-09 | 2012-03-27 | Bristol Myers Squibb Company | Hepatitis C virus inhibitors |
| US8143414B2 (en) | 2009-04-13 | 2012-03-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9278922B2 (en) | 2009-04-15 | 2016-03-08 | Abbvie Inc. | Anti-viral compounds |
| US9139569B2 (en) | 2009-05-12 | 2015-09-22 | Merck Sharp & Dohme Corp. | Fused tricyclic aryl compounds useful for the treatment of viral diseases |
| US8273341B2 (en) | 2009-05-13 | 2012-09-25 | Gilead Sciences, Inc. | Antiviral compounds |
| US8822430B2 (en) | 2009-05-13 | 2014-09-02 | Gilead Pharmasset Llc | Antiviral compounds |
| US8841278B2 (en) | 2009-05-13 | 2014-09-23 | Gilead Pharmasset Llc | Antiviral compounds |
| JP2014169331A (en) * | 2009-05-13 | 2014-09-18 | Gilead Sciences Inc | Antiviral compound |
| EA026536B1 (en) * | 2009-05-13 | 2017-04-28 | Джилид Фармассет Ллс | Antiviral compounds |
| US8088368B2 (en) | 2009-05-13 | 2012-01-03 | Gilead Sciences, Inc. | Antiviral compounds |
| US9511056B2 (en) | 2009-05-13 | 2016-12-06 | Gilead Pharmasset Llc | Antiviral compounds |
| WO2010132601A1 (en) * | 2009-05-13 | 2010-11-18 | Gilead Sciences, Inc. | Antiviral compounds |
| EA021974B1 (en) * | 2009-05-13 | 2015-10-30 | Джилид Фармассет Ллс | Antiviral compound |
| US9981955B2 (en) | 2009-05-13 | 2018-05-29 | Gilead Pharmasset Llc | Antiviral compounds |
| EA027493B1 (en) * | 2009-05-13 | 2017-07-31 | Джилид Фармассет Ллс | Intermediates for preparing an antiviral compound |
| US8669234B2 (en) | 2009-05-13 | 2014-03-11 | Gilead Sciences, Inc. | Antiviral compounds |
| EP2857394A1 (en) * | 2009-05-13 | 2015-04-08 | Gilead Pharmasset LLC | Antiviral compounds |
| US8138215B2 (en) | 2009-05-29 | 2012-03-20 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8980920B2 (en) | 2009-05-29 | 2015-03-17 | Merck Sharp & Dohme Corp. | Antiviral compounds of three linked aryl moieties to treat diseases such as hepatitis C |
| EP2435421A1 (en) * | 2009-05-29 | 2012-04-04 | Schering Corporation | Antiviral compounds composed of three aligned aryl moieties to treat diseases such as hepatitis c |
| US8772505B2 (en) | 2009-05-29 | 2014-07-08 | Merck Sharp & Dohme Corp. | Antiviral compounds composed of three aligned aryl moieties to treat diseases such as hepatitis C |
| US8211928B2 (en) | 2009-05-29 | 2012-07-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10039754B2 (en) | 2009-06-11 | 2018-08-07 | Abbvie Inc. | Anti-viral compounds |
| US8716454B2 (en) | 2009-06-11 | 2014-05-06 | Abbvie Inc. | Solid compositions |
| US8921514B2 (en) | 2009-06-11 | 2014-12-30 | Abbvie Inc. | Anti-viral compounds |
| US9586978B2 (en) | 2009-06-11 | 2017-03-07 | Abbvie Inc. | Anti-viral compounds |
| US8937150B2 (en) | 2009-06-11 | 2015-01-20 | Abbvie Inc. | Anti-viral compounds |
| US9394279B2 (en) | 2009-06-11 | 2016-07-19 | Abbvie Inc. | Anti-viral compounds |
| US8691938B2 (en) | 2009-06-11 | 2014-04-08 | Abbvie Inc. | Anti-viral compounds |
| US10028937B2 (en) | 2009-06-11 | 2018-07-24 | Abbvie Inc. | Anti-viral compounds |
| US8221737B2 (en) | 2009-06-16 | 2012-07-17 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8609648B2 (en) | 2009-07-02 | 2013-12-17 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8765731B2 (en) | 2009-07-16 | 2014-07-01 | Vertex Pharmaceuticals Incorporated | Benzimidazole analogues for the treatment or prevention of flavivirus infections |
| CN102471324A (en) * | 2009-08-07 | 2012-05-23 | 泰博特克药品公司 | Bis-benzimidazole derivatives as hepatitis c virus inhibitors |
| AU2010280712B2 (en) * | 2009-08-07 | 2015-10-22 | Janssen Sciences Ireland Uc | Bis-benzimidazole derivatives as hepatitis C virus inhibitors |
| WO2011015658A1 (en) | 2009-08-07 | 2011-02-10 | Tibotec Pharmaceuticals | Bis-benzimidazole derivatives as hepatitis c virus inhibitors |
| US8623899B2 (en) | 2009-08-07 | 2014-01-07 | Janssen Research & Development Ireland | Bis-benzimidazole derivatives as hepatitis C virus inhibitors |
| WO2011026920A1 (en) | 2009-09-03 | 2011-03-10 | Tibotec Pharmaceuticals | Bis-benzimidazole derivatives |
| US9150587B2 (en) | 2009-09-04 | 2015-10-06 | Janssen Pharmaceuticals, Inc. | Chemical compounds |
| US9814699B2 (en) | 2009-09-04 | 2017-11-14 | Janssen Pharmaceuticals, Inc. | Chemical compounds |
| US8492554B2 (en) | 2009-09-04 | 2013-07-23 | Glaxosmithkline Llc | Chemical compounds |
| US8853416B2 (en) | 2009-09-04 | 2014-10-07 | Janssen Pharmaceuticals, Inc. | Chemical compounds |
| US8344155B2 (en) | 2009-09-04 | 2013-01-01 | Glaxosmith Kline Llc | Chemical compounds |
| US9156818B2 (en) | 2009-09-11 | 2015-10-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8703938B2 (en) | 2009-09-11 | 2014-04-22 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8815928B2 (en) | 2009-09-11 | 2014-08-26 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8759332B2 (en) | 2009-09-11 | 2014-06-24 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8822700B2 (en) | 2009-09-11 | 2014-09-02 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8927709B2 (en) | 2009-09-11 | 2015-01-06 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US9427428B2 (en) | 2009-11-04 | 2016-08-30 | Janssen Sciences Ireland Uc | Benzimidazole-imidazole derivatives |
| JP2013510119A (en) * | 2009-11-04 | 2013-03-21 | ヤンセン・アールアンドデイ・アイルランド | Benzimidazole-imidazole derivatives |
| WO2011054834A1 (en) | 2009-11-04 | 2011-05-12 | Tibotec Pharmaceuticals | Benzimidazole-imidazole derivatives |
| US9433609B2 (en) | 2009-11-04 | 2016-09-06 | Janssen Sciences Ireland Uc | Benzimidazole-imidazole derivatives |
| US9776981B2 (en) | 2009-11-11 | 2017-10-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9006455B2 (en) | 2009-11-11 | 2015-04-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| JP2015044866A (en) * | 2009-11-11 | 2015-03-12 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Hepatitis c virus inhibitors |
| US8618153B2 (en) | 2009-11-12 | 2013-12-31 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8653070B2 (en) | 2009-12-14 | 2014-02-18 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| WO2011081918A1 (en) | 2009-12-14 | 2011-07-07 | Enanta Pharmaceuticals, Inc | Hepatitis c virus inhibitors |
| US8377980B2 (en) | 2009-12-16 | 2013-02-19 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9187496B2 (en) | 2009-12-18 | 2015-11-17 | Idenix Pharmaceuticals Llc | 5,5-fused arylene or heteroarylene hepatitis C virus inhibitors |
| WO2011075607A1 (en) * | 2009-12-18 | 2011-06-23 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| US9796705B2 (en) | 2009-12-22 | 2017-10-24 | Merck Sharp & Dohme Corp. | Fused tricyclic compounds and methods of use thereof for the treatment of viral diseases |
| WO2011087740A1 (en) | 2009-12-22 | 2011-07-21 | Schering Corporation | Fused tricyclic compounds and methods of use thereof for the treatment of viral diseases |
| US8735398B2 (en) | 2009-12-30 | 2014-05-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8362020B2 (en) | 2009-12-30 | 2013-01-29 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8933110B2 (en) | 2010-01-25 | 2015-01-13 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8785487B2 (en) | 2010-01-25 | 2014-07-22 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8623814B2 (en) | 2010-02-23 | 2014-01-07 | Enanta Pharmaceuticals, Inc. | Antiviral agents |
| US8178531B2 (en) | 2010-02-23 | 2012-05-15 | Enanta Pharmaceuticals, Inc. | Antiviral agents |
| US9060971B2 (en) | 2010-03-04 | 2015-06-23 | Enanta Pharmaceuticals, Inc. | Combination pharmaceutical agents as inhibitors of HCV replication |
| WO2011112429A1 (en) | 2010-03-09 | 2011-09-15 | Schering Corporation | Fused tricyclic silyl compounds and methods of use thereof for the treatment of viral diseases |
| US8609635B2 (en) | 2010-03-09 | 2013-12-17 | Merck Sharp & Dohme Corp. | Fused tricyclic silyl compounds and methods of use thereof for the treatment of viral diseases |
| US8779156B2 (en) | 2010-03-24 | 2014-07-15 | Vertex Pharmaceuticals Incorporated | Analogues for the treatment or prevention of flavivirus infections |
| US10081621B2 (en) | 2010-03-25 | 2018-09-25 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US9127021B2 (en) | 2010-04-09 | 2015-09-08 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US10071979B2 (en) | 2010-04-22 | 2018-09-11 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| US8778938B2 (en) | 2010-06-04 | 2014-07-15 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| US8686026B2 (en) | 2010-06-10 | 2014-04-01 | Abbvie Inc. | Solid compositions |
| WO2012006055A2 (en) | 2010-06-28 | 2012-01-12 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flavivirus infections |
| WO2012006060A1 (en) | 2010-06-28 | 2012-01-12 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flavivirus infections |
| WO2012013643A1 (en) | 2010-07-26 | 2012-02-02 | Tibotec Pharmaceuticals | Hetero-bicyclic derivatives as hcv inhibitors |
| WO2012018534A2 (en) | 2010-07-26 | 2012-02-09 | Schering Corporation | Substituted biphenylene compounds and methods of use thereof for the treatment of viral diseases |
| US8815849B2 (en) | 2010-07-26 | 2014-08-26 | Janssen R&D Ireland | Hetero-bicyclic derivatives as HCV inhibitors |
| US8697704B2 (en) | 2010-08-12 | 2014-04-15 | Enanta Pharmaceuticals, Inc. | Hepatitis C virus inhibitors |
| WO2012024363A2 (en) | 2010-08-17 | 2012-02-23 | Vertex Pharmaceuticals Incorporated | Compounds and methods for the treatment or prevention of flaviviridae viral infections |
| WO2012040389A3 (en) * | 2010-09-22 | 2012-05-10 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic hcv inhibitors |
| WO2012040389A2 (en) * | 2010-09-22 | 2012-03-29 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic hcv inhibitors |
| US8822520B2 (en) | 2010-09-22 | 2014-09-02 | Presidio Pharmaceuticals, Inc. | Substituted bicyclic HCV inhibitors |
| WO2012041014A1 (en) * | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives for treating hepatitis c virus infection |
| WO2012050848A1 (en) | 2010-09-29 | 2012-04-19 | Schering Corporation | Fused tetracycle derivatives and methods of use thereof for the treatment of viral diseases |
| US8999967B2 (en) | 2010-09-29 | 2015-04-07 | Presidio Pharmaceuticals, Inc. | Tricyclic fused ring inhibitors of hepatitis C |
| CN103459399A (en) * | 2010-09-29 | 2013-12-18 | 默沙东公司 | Tetracyclic indole derivatives for treating hepatitis c virus infection |
| JP2020059696A (en) * | 2010-10-13 | 2020-04-16 | アッヴィ・アイルランド・アンリミテッド・カンパニー | Anti-viral compounds |
| JP2018065828A (en) * | 2010-10-13 | 2018-04-26 | アッヴィ・アイルランド・アンリミテッド・カンパニー | Anti-virus compound |
| WO2012051361A1 (en) | 2010-10-13 | 2012-04-19 | Abbott Laboratories | Anti-viral compounds |
| EP2692726A1 (en) | 2010-10-13 | 2014-02-05 | Abbvie Inc. | Anti-viral compounds |
| JP2021035964A (en) * | 2010-10-13 | 2021-03-04 | アッヴィ・アイルランド・アンリミテッド・カンパニー | Anti-viral compounds |
| EP3438106A1 (en) | 2010-10-13 | 2019-02-06 | AbbVie Ireland Unlimited Company | Anti-viral compounds |
| EP2692346A1 (en) | 2010-10-13 | 2014-02-05 | Abbvie Inc. | An antiviral 1-phenyl-2,5-dibenzimidazol-5-yl-pyrrolidine derivative |
| AU2014203655B2 (en) * | 2010-10-13 | 2016-07-07 | Abbvie Ireland Unlimited Company | Anti-viral compounds |
| JP2016106075A (en) * | 2010-10-13 | 2016-06-16 | アッヴィ・バハマズ・リミテッド | Anti-virus compound |
| WO2012058125A1 (en) | 2010-10-26 | 2012-05-03 | Presidio Pharmaceuticals, Inc. | Inhibitors of hepatitis c virus |
| KR101835474B1 (en) | 2010-11-17 | 2018-03-08 | 길리애드 파마셋 엘엘씨 | Antiviral compounds |
| WO2012068234A3 (en) * | 2010-11-17 | 2013-01-17 | 12Gilead Sciences, Inc. | Antiviral compounds |
| US10344019B2 (en) | 2010-11-17 | 2019-07-09 | Gilead Pharmasset Llc | Antiviral compounds |
| US9156823B2 (en) | 2010-11-17 | 2015-10-13 | Gilead Pharmasset Llc | Antiviral compounds |
| AU2011328980B2 (en) * | 2010-11-17 | 2015-07-30 | Gilead Sciences, Inc. | Antiviral compounds |
| KR101771093B1 (en) * | 2010-11-30 | 2017-08-24 | 이바센코 앤드리 알렌산드로비치 | Substituted azoles, anti-viral active ingredient, pharmaceutical composition, method for the production and use thereof |
| JP2013544281A (en) * | 2010-11-30 | 2013-12-12 | アラ・ケム・エルエルシー | Substituted azoles, antiviral active ingredients, pharmaceutical compositions, methods for their preparation and use. |
| EP2808325A1 (en) | 2010-11-30 | 2014-12-03 | Alla Chem, LLC. | Substituted azoles, anti-viral active ingredient, pharmaceutical composition, method for the production and use thereof |
| WO2012074437A2 (en) | 2010-11-30 | 2012-06-07 | Алла Хем, Ллс | Substituted azoles, anti-viral active ingredient, pharmaceutical composition, method for the production and use thereof |
| EP2651920A4 (en) * | 2010-12-15 | 2014-12-17 | Abbvie Inc | Anti-viral compounds |
| EP2651920A2 (en) * | 2010-12-15 | 2013-10-23 | Abbvie Inc. | Anti-viral compounds |
| WO2012083048A3 (en) * | 2010-12-15 | 2012-08-02 | Abbott Laboratories | Anti-viral compounds |
| WO2012083170A1 (en) * | 2010-12-16 | 2012-06-21 | Abbott Laboratories | Anti-viral compounds |
| CN103354808B (en) * | 2010-12-16 | 2016-08-10 | Abbvie公司 | Antiviral compound |
| JP2014504296A (en) * | 2010-12-16 | 2014-02-20 | アッヴィ・インコーポレイテッド | Antiviral compounds |
| CN103354808A (en) * | 2010-12-16 | 2013-10-16 | Abbvie公司 | Anti-viral compounds |
| WO2012087976A3 (en) * | 2010-12-21 | 2012-11-29 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| WO2012087976A2 (en) * | 2010-12-21 | 2012-06-28 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| US9340520B2 (en) | 2011-02-07 | 2016-05-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8552047B2 (en) | 2011-02-07 | 2013-10-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9546160B2 (en) | 2011-05-12 | 2017-01-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10201541B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| US8927739B2 (en) | 2011-05-18 | 2015-01-06 | Enanta Pharmaceuticals, Inc. | Processes for the preparation of 5-azaspiro[2.4]heptane-6-carboxylic acid and its derivatives |
| WO2013016499A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Methods for preparation of thiophene compounds |
| WO2013016492A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Thiophene compounds |
| WO2013016490A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Thiophene compounds |
| WO2013016501A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Formulations of thiophene compounds |
| WO2013016491A1 (en) | 2011-07-26 | 2013-01-31 | Vertex Pharmaceuticals Incorporated | Thiophene compounds |
| WO2013039876A1 (en) | 2011-09-14 | 2013-03-21 | Merck Sharp & Dohme Corp. | Silyl-containing heterocyclic compounds and methods of use thereof for the treatment of viral diseases |
| US9393256B2 (en) | 2011-09-16 | 2016-07-19 | Gilead Pharmasset Llc | Methods for treating HCV |
| US10456414B2 (en) | 2011-09-16 | 2019-10-29 | Gilead Pharmasset Llc | Methods for treating HCV |
| US10807990B2 (en) | 2011-11-16 | 2020-10-20 | Gilead Pharmasset Llc | Antiviral compounds |
| US9809600B2 (en) | 2011-11-16 | 2017-11-07 | Gilead Pharmasset Llc | Antiviral compounds |
| US9051340B2 (en) | 2011-11-16 | 2015-06-09 | Gilead Pharmasset Llc | Antiviral compounds |
| US8921341B2 (en) | 2011-11-16 | 2014-12-30 | Gilead Pharmasset Llc | Antiviral compounds |
| US9221833B2 (en) | 2011-11-16 | 2015-12-29 | Gilead Pharmasset Llc | Antiviral compounds |
| US8575135B2 (en) | 2011-11-16 | 2013-11-05 | Gilead Sciences, Inc. | Antiviral compounds |
| US9868745B2 (en) | 2011-11-16 | 2018-01-16 | Gilead Pharmasset Llc | Antiviral compounds |
| US8940718B2 (en) | 2011-11-16 | 2015-01-27 | Gilead Pharmasset Llc | Antiviral compounds |
| CN103987723B (en) * | 2011-12-16 | 2017-03-01 | 弗·哈夫曼-拉罗切有限公司 | Inhibitors of HCV NS5A |
| CN103987723A (en) * | 2011-12-16 | 2014-08-13 | 弗·哈夫曼-拉罗切有限公司 | Inhibitors of HCV NS5A |
| WO2013098313A1 (en) | 2011-12-28 | 2013-07-04 | Janssen R&D Ireland | Hetero-bicyclic derivatives as hcv inhibitors |
| US9126986B2 (en) | 2011-12-28 | 2015-09-08 | Janssen Sciences Ireland Uc | Hetero-bicyclic derivatives as HCV inhibitors |
| US9382261B2 (en) | 2011-12-28 | 2016-07-05 | Janssen Sciences Ireland Uc | Substituted quinazolinones as HCV inhibitors |
| WO2013098320A1 (en) | 2011-12-28 | 2013-07-04 | Janssen R&D Ireland | Quinazolinone derivatives as hcv inhibitors |
| US9034832B2 (en) | 2011-12-29 | 2015-05-19 | Abbvie Inc. | Solid compositions |
| US9326973B2 (en) | 2012-01-13 | 2016-05-03 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| JP2015510512A (en) * | 2012-02-10 | 2015-04-09 | ルピン・リミテッドLupin Limited | Antiviral compounds having a heterotricyclic moiety |
| US9012427B2 (en) | 2012-03-22 | 2015-04-21 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| US10800789B2 (en) | 2012-05-16 | 2020-10-13 | Gilead Pharmasset Llc | Antiviral compounds |
| CN103880823B (en) * | 2012-12-21 | 2017-12-05 | 广东东阳光药业有限公司 | Application as the spiro-compound of hepatitis c inhibitor and its in medicine |
| CN103880823A (en) * | 2012-12-21 | 2014-06-25 | 广东东阳光药业有限公司 | Spiro compound serving as hepatitis c inhibitor and application thereof in medicine |
| US10039779B2 (en) | 2013-01-31 | 2018-08-07 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| WO2014123456A2 (en) | 2013-02-07 | 2014-08-14 | ИВАЩЕНКО, Андрей Александрович | Alkyl [(s)-1-((s)-2-{5-[4-(4-{2-[(s)-1-((s)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidine-2-yl]-3h-imidazole-4-yl}-buta-1,3-dienyl)-phenyl]-1h-imidazole-2-yl}-pyrrolidine-1-carbonyl)-2-methyl-propyl]-carbamate naphthalene-1,5-disulfonate, pharmaceutical composition, medicinal agent and method for treatment of viral diseases |
| WO2014134251A1 (en) | 2013-02-28 | 2014-09-04 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions |
| US11484534B2 (en) | 2013-03-14 | 2022-11-01 | Abbvie Inc. | Methods for treating HCV |
| US9770439B2 (en) | 2013-07-02 | 2017-09-26 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9717712B2 (en) | 2013-07-02 | 2017-08-01 | Bristol-Myers Squibb Company | Combinations comprising tricyclohexadecahexaene derivatives for use in the treatment of hepatitis C virus |
| US9775831B2 (en) | 2013-07-17 | 2017-10-03 | Bristol-Myers Squibb Company | Combinations comprising biphenyl derivatives for use in the treatment of HCV |
| US11707479B2 (en) | 2013-08-27 | 2023-07-25 | Gilead Sciences, Inc. | Combination formulation of two antiviral compounds |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US10086011B2 (en) | 2013-08-27 | 2018-10-02 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US10167298B2 (en) | 2013-10-30 | 2019-01-01 | Merck Sharp & Dohme Corp. | Pseudopolymorphs of an HCV NS5A inhibitor and uses thereof |
| US9828365B2 (en) | 2013-12-20 | 2017-11-28 | Merck Sharp & Dohme Corp. | Fused tetracyclic heterocyclic compounds and methods of use thereof for the treatment of viral diseases |
| WO2015094998A1 (en) | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Fused tetracyclic heterocyclic compounds and methods of use thereof for the treatment of viral diseases |
| US9333204B2 (en) | 2014-01-03 | 2016-05-10 | Abbvie Inc. | Solid antiviral dosage forms |
| US9744170B2 (en) | 2014-01-03 | 2017-08-29 | Abbvie Inc. | Solid antiviral dosage forms |
| US10105365B2 (en) | 2014-01-03 | 2018-10-23 | Abbvie Inc. | Solid antiviral dosage forms |
| US10206877B2 (en) | 2014-04-15 | 2019-02-19 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US11203599B2 (en) | 2014-06-11 | 2021-12-21 | Gilead Pharmasset Llc | Solid forms of an antiviral compound |
| US10428065B2 (en) | 2015-04-20 | 2019-10-01 | Cellcentric Ltd | Isoxazolyl substituted imidazopyridines |
| US10118920B2 (en) | 2015-04-20 | 2018-11-06 | Cellcentric Ltd | Isoxazolyl substituted benzimidazoles |
| US10617675B2 (en) | 2015-08-06 | 2020-04-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2020117849A1 (en) | 2018-12-04 | 2020-06-11 | Bristol-Myers Squibb Company | Methods of analysis using in-sample calibration curve by multiple isotopologue reaction monitoring |
| US11760764B2 (en) | 2020-05-22 | 2023-09-19 | Aligos Therapeutics, Inc. | Methods and compositions for targeting PD-L1 |
| US12071428B2 (en) | 2020-12-30 | 2024-08-27 | Tyra Biosciences, Inc. | Indazole compounds as kinase inhibitors |
| US12037340B2 (en) | 2021-05-21 | 2024-07-16 | Gilead Sciences, Inc. | Pentacyclic derivatives as Zika virus inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9120779B2 (en) | Inhibitors of HCV NS5A | |
| EP2373172B1 (en) | Inhibitors of hcv ns5a | |
| US8877707B2 (en) | Inhibitors of HCV NS5A | |
| US20120040977A1 (en) | Inhibitors of hcv ns5a | |
| WO2011150243A1 (en) | Inhibitors of hcv ns5a | |
| WO2011156543A2 (en) | Inhibitors of hcv ns5a protein | |
| WO2010111673A1 (en) | Substituted bicyclic hcv inhibitors | |
| WO2012040389A2 (en) | Substituted bicyclic hcv inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980155944.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09831084 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2750577 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 213279 Country of ref document: IL Ref document number: 2009831084 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011539670 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13132606 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 593808 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2009322400 Country of ref document: AU Date of ref document: 20091202 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |