WO2010052011A1 - Novel process for the preparation of amino acid derivatives - Google Patents
Novel process for the preparation of amino acid derivatives Download PDFInfo
- Publication number
- WO2010052011A1 WO2010052011A1 PCT/EP2009/007962 EP2009007962W WO2010052011A1 WO 2010052011 A1 WO2010052011 A1 WO 2010052011A1 EP 2009007962 W EP2009007962 W EP 2009007962W WO 2010052011 A1 WO2010052011 A1 WO 2010052011A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- benzyl
- methoxypropion
- amide
- acetamido
- process according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- LGERAMWGVIFEPS-UHFFFAOYSA-N CC(COC)C(NCc1ccccc1)=O Chemical compound CC(COC)C(NCc1ccccc1)=O LGERAMWGVIFEPS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/16—Preparation of optical isomers
- C07C231/20—Preparation of optical isomers by separation of optical isomers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/16—Preparation of optical isomers
- C07C231/18—Preparation of optical isomers by stereospecific synthesis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/22—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
Definitions
- the present patent application relates to a novel process for the preparation of amino acid derivatives.
- the present application relates to an improved process for the manufacture of Lacosamide (LCM), (R)-2-acetamido-N-benzyl-3-methoxypropion-amide, which is useful as an anticonvulsive drug.
- LCM Lacosamide
- R -2-acetamido-N-benzyl-3-methoxypropion-amide
- LCM has demonstrated antiepileptic effectiveness in different rodent seizure models and antinociceptive potential in experimental animal models that reflect distinct types and symptoms of neuropathic as well as chronic inflammatory pain.
- US patent 5,378,729 describes the preparation of functionalized amino acids by reacting amines with acetylating derivates of a carboxylic acid under amide forming conditions. US patent 5,378,729 is however silent on the direct preparation of a single enantiomer of functionalized amino acids, such as Lacosamide.
- US patent 5,773,475 relates to methods of preparation of 'substantially optically pure' Lacosamide, as defined therein, starting from D-Serine.
- Said method of preparation involves the use of methyl iodide and silver (I) oxide as O-methylation agent which presents the disadvantages of being expensive and leads to partial racemization of the product undergoing the O-methylation. This is a main drawback in terms of industrial productivity of the process.
- 2006/037574 may lead to safety or environmental issues when producing Lacosamide on a large scale. Moreover the use of N-protection/N-deprotection steps of the amine moiety may lead to cost and productivity issues for the industrial production of the overall process.
- the present invention relates to a process of manufacture of optically enriched (R)-2-acetamido-N-benzyl-3-methoxypropion-amide (I) comprising resolution of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) as shown in following scheme 1.
- optically enriched when referring to a particular compound means that more than 50%, preferably more than 75%, more preferably more than 85%, most preferably more than 94% of the compound has the stereogenic center indicated by (*) in a given configuration (R) or (S).
- optically enriched (R)-2-acetamido-N-benzyl-3- methoxypropion-amide means that more than 50%, preferably more than 75%, more preferably more than 85%, most preferably more than 94% of the compound has the stereogenic center indicated by ( * ) in configuration (R).
- the present invention relates to a process of manufacture of substantially optically pure (R)-2-acetamido-N-benzyl-3-methoxypropion-amide (I) comprising resolution of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) as shown in scheme 1.
- substantially optically pure as used herein when referring to a particular compound means that at least 95%, preferably at least 96%, more preferably at least 97%, most preferably at least 98%, even most preferably at least 99% of the compound has the stereogenic center indicated by ( * ) in a given configuration (R) or (S). Therefore, the expression “substantially optically pure (R)-2-acetamido-N-benzyl-3- methoxypropion-amide” means that at least 95%, preferably at least 96%, more preferably at least 97%, most preferably at least 98%, even most preferably at least 99% of the compound has the stereogenic center indicated by (*) in configuration (R).
- the term "resolution” as used herein refers to the separation of a mixture of enantiomers into its corresponding individual enantiomers.
- the enantiomers may be present in the mixture in various ratios of enantiomer versus the other. Typical ratios of enantiomers according to the present invention range from about 3/97 to 97/3, preferably from about 5/95 to 95/5, more preferably from about 30/70 to 70/30, most preferably from about 40/60 to 60/40, even more preferably from about 45/55 to 55/45.
- the mixture is a racemic mixture.
- a racemic mixture as herein defined is a mixture comprising 50% of one enantiomer and 50% of the other enantiomer.
- Resolution can be achieved by various methods including conversion to diastereoisomers, differential absorption, chiral recognition, biochemical processes, mechanical separation, kinetic resolution and deracemization as detailed in Jerry March “Advanced Organic Chemistry", fourth edition, Chapter 4, pages 120-125.
- the resolution according to the present invention is achieved by the differential separation method, more preferably by chiral chromatographic separation using columns packed with a chiral stationary phase (CSP) and a mobile phase.
- Chiral chromatographic separation may be carried out in batch or by Multi Column Chromatography (MCC).
- MCC Multi Column Chromatography
- MCC Multi Column Chromatography
- SMB mode Simulated Moving Bed chromatography mode
- Varicol mode mode where the inlet and outlet ports are shifted asynchronously
- inlet and oulet flowrates and/or concentrations are changing in time during the switching period.
- resolution of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) is performed by chiral chromatographic separation.
- resolution of 2-acetamido- N-benzyl-3-methoxypropion-amide (II) is performed by MCC.
- the polymeric chiral selector may additionally be immobilized onto the silica backbone which provides to the column, among other advantages, a better resistance to solvents.
- chiral chromatographic separation of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) is performed using a column which comprises a polymeric chiral selector which is immobilized onto the silica backbone.
- the polymeric chiral selector according to the present invention generally comprises a polysaccharide, for example amylose or cellulose.
- polymeric chiral selector which may be used according to the present invention are cellulose tris(4-methylbenzoate), cellulose tribenzoate, amylose tris(3,5- dimethylphenylcarbamate) cellulose tris(3,5-dimethylphenylcarbamate), cellulose tris(4- methylphenylcarbamate) cellulose tris (3,5-dichlorophenylcarbamate), amylose tris [(S)- ⁇ - methylbenzylcarbamate] and cellulose tris(3-chloro-4-methylphenylcarbamate).
- chiral chromatographic separation of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) into its enantiomers is performed using cellulose tris (3,5-dichlorophenylcarbamate) immobilized onto a silica backbone as chiral stationary phase.
- Examples of mobile phase that may be used according to the present invention are alkanes, such as heptane, hexane, alcohols, such as methanol, ethanol, iso-propanol, n- propanol, acetonitrile, isopropyl acetate, ethyl acetate, dichloromethane, chloroform, ethers, such are methyl t-butyl ether (MTBE), or mixtures thereof. If mixtures of solvents are used, the ratio will depend upon the type of solvents constituting the mixture, upon the type of column which is used and upon the solubility in those mixtures of the compound to be separated.
- alkanes such as heptane, hexane
- alcohols such as methanol, ethanol, iso-propanol, n- propanol, acetonitrile, isopropyl acetate, ethyl acetate, dichloromethane, chloroform
- mixtures of solvents according to the present invention are mixtures of dichloromethane and an alcohol or mixtures comprising acetonitrile and an alcohol or mixtures comprising ethyl acetate and an alcohol.
- mixtures of dichloromethane and an alcohol comprise between 90% and 99% of dichloromethane.
- mixtures of acetonitrile and an alcohol comprise between 90% and 99% of acetonitrile.
- mixtures of ethyl acetate and alcohol comprise between 90% and 99% of ethylacetate.
- Preferred solvents according to the present invention are ethanol, methanol n- propanol, iso-propanol, acetonitrile, dichloromethane and ethyl acetate
- a mixture of ethyl acetate and methanol is used as mobile phase.
- a mixture of ethyl acetate and methanol in a ratio of 90/10 v:v is used as mobile phase.
- acetonitrile is used as mobile phase.
- a productivity of the chiral chromatographic separation greater than 1 Kg of racemic mixture separated per Kg of Chiral Stationary Phase per day is achieved.
- a productivity of the chiral chromatographic resolution greater than 2 Kg of racemic mixture separated per Kg of Chiral Stationary Phase per day is achieved.
- the present invention relates to a process of manufacture of substantially optically pure (R)-2-acetamido-N-benzyl-3- methoxypropion-amide (I) comprising: (a) resolution of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) into
- Steps (a) and (c) are generally performed by chiral chromatographic separation, preferably by MCC.
- (R)-2-acetamido-N-benzyl-3-methoxypropion-amide (I) obtained in steps (a) and (c) may be optically enriched or substantially optically pure.
- (R)-2-acetamido-N- benzyl-3-methoxypropion-amide (I) obtained in steps (a) and (c) is substantially optically pure. This is particularly advantageous, as it avoids the use of iterative purification steps, such as crystallization which would impact on the productivity of the overall process.
- step (S)-2-acetamido-N-benzyl-3-methoxypropion-amide (III) obtained in step (a) may be optically enriched or substantially optically pure.
- racemisation refers to the transformation of an optically enriched enantiomer or a substantially optically pure enantiomer into a mixture consisting of said enantiomer and of the other enantiomer, up to a racemic mixture.
- Step (b) may be typically achieved by reacting optically enriched or substantially optically pure (S)-2-acetamido-N-benzyl-3-methoxypropion-amide (III) with a base, with or without acidification of the media.
- bases that can be used according to the present invention are sodium methoxide, potassium hydroxide, sodium hydroxide, potassium carbonate, sodium carbonate, tertiary amines, such as triethylamine, 1 ,8-Diazabicyclo[5.4.0]undec-7-ene and strong or weakly basic anion-exchange resins, such as AMBERLYSTTM A21 , AMBERLITETM IRA400 or IRA410, and the like.
- Preferred bases according to the present invention are sodium methoxide, potassium hydroxide, sodium hydroxide, potassium carbonate andsodium carbonate.
- acidification of the media is made, it is preferably performed using a stoechiometric amount of acid with respect to the base to avoid the formation of any degradation products.
- Said overall process is particularly advantageous as it avoids any loss of productivity which may sometime occur when using a resolution step. Indeed, often the undesired enantiomer is a by-product of the process which needs to be eliminated from the reaction media. By recycling the undesired enantiomer and performing an additional resolution, for example via chiral chromatography separation, overall yield of process and productivity are increased.
- Steps (b) and (c) may be iterated to further increase overall yield of the process.
- sodium methoxide is used a the base.
- the racemisation is generally performed in a solvent at a temperature comprised between 20°C and 80 0 C, preferably at a temperature comprised between 4O 0 C and 60 0 C.
- the reaction is performed at a temperature lower than 60°C in order to avoid formation of degradation products.
- solvents examples include alcohol, such as methanol, ethanol, ethers, such as tetrahydrofuran, 2-methyl-tetrahydrofuran or acetonitrile.
- the solvent is methanol.
- 2-acetamido-N-benzyl-3-methoxypropion-amide (II) may be prepared by acetylation of 2-amino-N-benzyl-3-methoxypropion-amide (IV) according to methods known to the man skilled in the art, as shown in following scheme 3.
- the acetylation may be performed using an acetylation agent such as acetic anhydride or acetyl chloride.
- an acetylation agent such as acetic anhydride or acetyl chloride.
- a particularly preferred acetylation agent according to the present invention is acetic anhydride.
- the acetylation is generally performed in a solvent at a temperature comprised between 20 0 C and 7O 0 C.
- solvents that may be used for the acetylation are dichloromethane, tetrahydrofuran, ethyl acetate, isopropyl-acetate and isobutyl-acetate.
- Preferred solvents are isopropyl-acetate and isobutyl-acetate.
- Acetylation is preferably performed at a temperature comprised between 5O 0 C and 70°C. More preferably, the acetylation is performed at a temperature of about 60 0 C.
- 2-amino-N-benzyl-3-methoxypropion-amide (IV) may be prepared according to the method described in scheme 1 of US patent 6,048,899, incorporated herein as reference, starting from racemic serine, or according to any other method known to the person skilled in the art
- 2-amino-N-benzyl-3-methoxypropion-amide (IV) may be prepared by ammonolysis of compound (V), wherein X is a leaving group, according to following scheme 4.
- Examples of a leaving group according to the present invention are halogen or sulfonate groups.
- sulfonate group represents a group of formula -O-SO2- R a wherein R a is an alkyl or an aryl.
- Preferred sulfonate groups are methanesulfonate, para-toluenesulfonate group or trifluoromethanesulfonate.
- the leaving group X in compound of formula (V) is a halogen, more preferably bromine or chlorine. Most preferably, X is bromine.
- the ammonolysis reaction may be performed according to methods known in the art.
- the ammonolysis may be performed according to the method described in international patent application published under WO 03/014080.
- the ammonolysis according to the present invention is preferably performed with aqueous ammonia in the presence of methanol.
- an excess of ammonia with respect to compound (V) is used in order to avoid the formation of secondary amine impurities.
- 20 to 25 molar equivalents of ammonia with respect to compound (V) are used.
- 2-amino-N-benzyl-3-methoxypropion-amide (IV) is typically extracted from the reaction media with a solvent.
- solvents that may be used for said extraction are isobutylacetate, isopropylacetate, propylacetate, ethyl acetate, 2-methyl-tetrahydrofuran, dichloromethane and toluene.
- isobutylacetate is used as solvent for the extraction of 2-amino-N- benzyl-3-methoxypropion-amide (IV).
- the extraction with isobutylacetate is preferably performed at a pH higher than 10 and lower than 12, more preferably at a pH between 11 and 12 in order to increase yield of isolation of 2-amino-N-benzyl-3-methoxypropion-amide
- 2-amino-N-benzyl-3- methoxypropion-amide (IV) may be obtained by performing Gabriel synthesis on compound (V), wherein the leaving group is a halogen.
- Typical reaction conditions according to this embodiment comprise reacting compound of formula (V) with potassium phtalimide and further reacting the intermediate thereby formed with hydrazine in ethanol as shown in following scheme 5.
- Compound of formula (V) may be synthetized according to various methods known in the art.
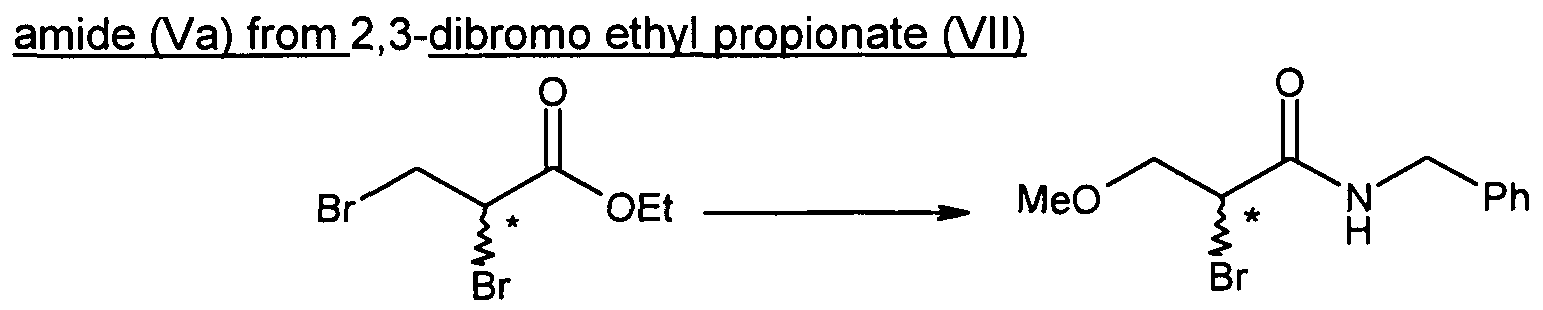
- 2-bromo-N-benzyl-3-methoxypropion-amide (Va) may be obtained by applying the method described in Bioorganic and Medicinal Chemistry 2004, 3079.
- compounds of formula (V) according to the present invention may be prepared by reacting compound of formula (Vl), wherein X is a leaving group as hereabove defined and Y is hydroxy or C- ⁇ io alkoxy, with benzylamine, according to following scheme 6.
- hydroxy represents a group of formula -OH.
- alkoxy represents a group of formula -ORb wherein R D is C- ⁇ io alkyl as defined above.
- alkyl is a group which represents saturated, monovalent hydrocarbon radicals having straight (unbranched), branched or cyclic moieties, or combinations thereof. Reaction conditions depend on the nature of the X and Y groups.
- Compound of formula (Vl), wherein X is a halogen and Y is a hydroxy group, herein after referred to as compound of formula (Via), may be converted in situ to a mixed anhydride (Villa) (Method A), wherein R is C-
- compound of formula (Via), wherein X is chlorine or bromine is reacted with an alkyl halo formate, for example ethyl chloroformate or isobutylchloroformate, at a temperature comprised between -10 c C and 10 0 C, followed by addition of a base, to afford the corresponding mixed anhydride (Villa) which is not isolated from the reaction media.
- the mixed anhydride (Villa) is reacted with benzylamine in a solvent at a temperature comprised between -10 0 C and 10 0 C.
- said temperature is comprised between -5°C and 0°C.
- solvent examples include dichloromethane, ethylacetate, isobutyl acetate, tetrahydrofuran, toluene, propylacetate, isopropylacetate.
- isobutylacetate or toluene is used as a solvent.
- Suitable bases are triethylamine, pyridine, N-methylmorpholine, h ⁇ ning base. In a preferred embodiment of the invention, the base is N-methylmorpholine.
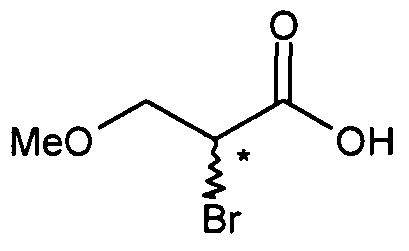
- Compound of formula (Via) that may be used as starting material in Method A are 2-chloro-3-methoxy-propionic acid (VIc) or 2-bromo-3-methoxy-propionic acid (VId).
- 2-bromo-3-methoxy propionic acid (VId) is easily obtainable by reaction of 2,3- bromo propionic acid, or corresponding alkyl esters, with sodium methoxide in methanol, in yields of more than 85 %.
- a method to produce 2-bromo-3-methoxy propionic acid (VId) is for example described by L.L. Wood & V. du Vigneaud, J. biol. Chemistry, 1940, 134, 413.
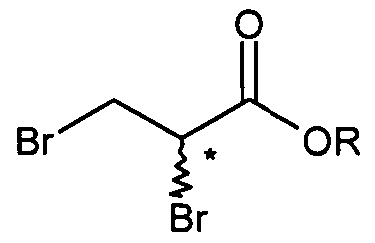
- 2-bromo-3-methoxy propionic acid may be obtained starting from commercially available 2,3-dibromo ethyl propionate (Vila) or 2,3-dibromo methyl propionate (VIIb).
- 2,3-dibromo ethyl propionate (Vila) or 2,3-dibromo methyl propionate (VIIb) is reacted with sodium methoxide in an organic solvent, preferably methanol, at a temperature lower than 10 0 C, preferably lower than 0 0 C, affording 2-bromo-3-methoxy methyl propionate (VIe) which is further reacted in situ with sodium hydroxide, at a temperature comprised between O 0 C and 25°C, preferably at a temperature comprised between 20°C and 25 0 C, to afford 2- bromo-3-methoxy propionic acid (VId), after acidification of the mixture with aqueous chlorhydric acid, in a yield comprised between 80% and 90%, according to following scheme 8.
- an organic solvent preferably methanol
- compounds of formula (Via), wherein X is chlorine or bromine may be converted into their corresponding acid chloride according to standard methods known to the person skilled in the art, or by reaction with a compound selected from the group consisting of thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride, phosphorus oxychloride, phenylphosphonic dichloride and N- chlorosuccinimide, preferably with thionylchloride.
- Said Method B may be performed in a solvent selected from dichloromethane, toluene, isobutylacetate or isopropylacetate at a temperature comprises between between 20 0 C and 60 0 C.
- Example of compound (Via) that may be used for Method B is 2-chloro-3-methoxy propionic acid (VIc).
- Compound of formula (Vl), wherein X is a halogen and Y is a C-J .-I Q alkoxy group, herein after referred to as compound of formula (VIb), may be either converted into corresponding compound of formula (V) by reaction with benzylamine (Method C), either saponified into the corresponding acid (Via) which can then undergo any of the transformations (Methods A & B) mentioned in scheme 7 above, as shown here below in scheme 9.
- 2-acetamido-N-benzyl-3-methoxypropion-amide (II) may be manufactured by a process comprising the following steps:
- X is a leaving group, with an alkylhaloformiate in the presence of a base and benzylamine;
- step (ii) performing ammonolysis of compound (V) resulting from step (i), wherein X is as defined in compound (Via);
- Step (i) is preferably performed by reacting compound (Via) with ethylchloroformate or isobutylchloroformate, followed by addition of N-Methyl-Morpholine, which results in the in situ formation of compound (Villa).
- _io alkyl is formed in situ, as shown in scheme 7 above.
- Reaction of step (i) is performed at a temperature comprised between -1O 0 C and 10 0 C.
- a temperature comprised between -5 0 C and 0°C.
- Step (ii) is preferably performed by treating compound (V) with a molar excess of aqueous ammonia in the presence of methanol. More preferably, 20 to 25 molar equivalents of ammonia with respect to compound (V) are used.
- Step (iii) is preferably performed using acetic anhydride as acetylating agent and is at a temperature comprised between 50 0 C and 70 0 C. More preferably, the acetylation is performed at a temperature of about 60°C.
- compound of formula (V) may be synthesized by reacting compound of formula (Vila) or (VIIb) under the conditions mentioned hereabove for the conversion of (Vila) or (VIIb) into (VId), followed by reaction with benzylamine, without isolation of the intermediate compound (Vl).
- Examples of compounds of formula (V) according to the present invention are 2-bromo-N-benzyl-3-methoxypropion-amide (Va) and 2-chloro-N-benzyl-3- methoxypropion-amide (Vb).
- Examples of compounds of formula (Vl) according to the present invention are 2- bromo-3-methoxy propionic acid (VId), 2-bromo-3-methoxy propionic acid methyl ester (VIe), 2-bromo-3-methoxy propionic acid ethyl ester, 2-chloro-3-methoxy propionic acid (VIc), 2-chloro-3-methoxy propionic acid methyl ester, 2-chloro-3-methoxy propionic acid ethyl ester and 2-bromo-3-methoxy propionyl chloride.
- compounds (V), (IV) and (II) are respectively isolated from the reaction media before undergoing any further chemical transformation.
- compound of formula (V) is isolated by crystallization in a mixture of solvents selected from heptane, toluene, isobutylacetate, propylacetate, methyl t-butyl ether.
- said mixture comprises heptane.
- Compound of formula (Vl) may be isolated from the reaction media or not.
- compound of formula (II) is isolated by crystallization in a mixture of solvents selected from isobutylacetate, isopropylacetate, propylacetate, 2-Me- tetrahydrofuran and acetonitrile.
- said mixture comprises isobutylacetate or ethyl acetate.
- 2-acetamido-N-benzyl-3-methoxypropion-amide (II) is manufactured by a process comprising the following steps:
- 2-bromo-3-methoxy methyl propionic acid is prepared from commercially available 2,3-dibromo ethyl propionate (Vila) or 2,3-dibromo methyl propionate (VIIb) as shown in scheme 8 of the present application.
- 2,3-dibromo ethyl propionate (Vila) may be transformed into 2-bromo-N-benzyl-3- methoxypropion-amide (Va) without isolation of 2-bromo-3-methoxy methyl propionic acid
- the present invention relates to a process of manufacture of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) comprising the following steps: (iii) reacting 2,3-dibromo ethyl propionate (Vila) or 2,3-dibromo methyl propionate (VIIb)
- 2-acetamido-N-benzyl- 3-methoxypropion-amide (II) is manufactured by reacting 2-acetamido-3-methoxypropionic acid (IX) with an alkyl chloroformiate, preferably ethyl- or isobutylchloroformiate, thereby forming a mixed anhydride which is then reacted with benzylamine.
- an alkyl chloroformiate preferably ethyl- or isobutylchloroformiate
- 2-acetamido-3-methoxypropionic acid may be obtained by acetylation of commercially available O-Methyl-D, L-serine (X) according to following scheme 10.
- the present invention also relates to a process for the manufacture of
- Step (i) is generally performed using acetic anhydride as acetylating agent in acetic acid, toluene, tetrahydrofuran, 2-methyl-tetrahydrofuran, isobutylacetate, dichloromethane or water, or mixtures thereof.
- said step is achieved in a mixture of tetrahydrofuran and water.
- Step (ii) is generally performed in the presence of ethylchloroformiate or isobutylchloroformiate and N-methylmorpholine or triethylamine in tetrahydrofuran, 2- methyl-tetrahydrofuran, toluene, ethyl acetate or dichloromethane.
- Step (ii) can be performed in the presence of a catalyst selected from the group consisting of boric acid, phenylboronic acid, 3,4,5-trifluorophenylboronic acid, 2- (N,N-di-isopropylaminomethyl)phenylboronic acid and 2-(N 1 N- dimethylaminomethyl)phenylboronic acid byrefluxing of a solvent selected from the group consisting of toluene, N-methylpyrrolidone and mixtures thereof, tetrahydrofuran, 2-methyl- tetrahydrofuran, cyclopentylmethyl ether, di n-butylether, fluorobenzene using a Dean- Stark apparatus, molecular sieves or sodium sulfate to continuously remove water.
- a catalyst selected from the group consisting of boric acid, phenylboronic acid, 3,4,5-trifluorophenylboronic acid, 2- (N,N-di-isopropylamino
- the catalyst according to the present invention may either be soluble in the reaction media or may be solid supported.
- a mixture of toluene and N-methylpyrrolidone is used as a solvent.
- the ratio of the volume of toluene with respect to N-methylpyrrolidone is for example 80/20 or 99/1.
- benzylation of compound of formula (IX) to afford compound of formula (II) may be performed with benzylamine in the presence of di-tert- butyl-dicarbonate (BoC 2 O) in the presence of pyridine, triethylamine or Hunig's base in a solvent selected from the group consisting of tetrahydrofuran, 2-methyl-tetrahydrofuran, ethyl acetate and dichloromethane.
- benzylation of compound of formula (iX) to afford compound of formula (Ii) may be performed with benzylamine in the presence of n- propanephosphonic acid anhydride (T3P®) in the presence of triethylamine or Hunig's base in a solvent selected from ethyl acetate, tetrahydrofuran, dichloromethane and 2- methyl-tetrahydrofuran.
- T3P® propanephosphonic acid anhydride
- benzylation of compound of formula (IX) to afford compound of formula (II) may be performed with benzylamine in the presence of dicyclohexyl- (DCC) or diisopropylcarbodiimide (DIC) in a solvent selected from the group consisting of tetrahydrofuran, ethyl acetate and dichloromethane.
- benzylation of compound of formula (IX) to afford compound of formula (II) may be performed in neat benzylamine in the presence of hexamethyldisalazane (HMDS).
- HMDS hexamethyldisalazane
- benzylation of compound of formula (IX) to afford compound of formula (II) may be performed by heating a solid 1 :1 mixture of (IX) with benzylamine above 130 0 C.
- Catalysts used for the benzylation step according to the present embodiment of the invention may be either soluble in the reaction or supported on a solid.
- the present invention relates to a process for the preparation of Lacosamide comprising the following steps:
- X is a leaving group, with an alkylhaloformiate in the presence of a base and benzylamine;
- step (ii) performing ammonolysis of compound (V) resulting from step (i), wherein X is as defined in compound (Via);
- the present invention relates to a process of manufacture of (R)-2-acetamido-N-benzyl-3-methoxypropion-amide (I) comprising the following steps:
- X is a leaving group, with an alkylhaloformiate in the presence of a base and benzylamine;
- step (ii) performing ammonolysis of compound (V) resulting from step (i), wherein X is as defined in compound (Via);
- Step (vi) racemisation of (S)-2-acetamido-N-benzyl-3-methoxypropion-amide (III) thereby obtained; and (vii) further resolution of the resulting 2-acetamido-N-benzyl-3-methoxypropion- amide (II).
- Step (iv) is preferably performed by crystallization in a solvent selected from the group consisting of toluene, ethyl acetate, isobutylacetate, isopropylacetate, acetonitrile, 2- methyl-tetrahydrofuran and mixtures thereof.
- Compound (II) is thereby preferably obtained with a purity of at least about 98% measured by HPLC, more preferably with a purity of at least about 99%, most preferably with a purity of at least about 99.5%
- Step (v) in the two embodiments detailed here above is preferably performed by chiral chromatographic separation.
- the chiral chromatographic separation is performed by MCC.
- Said separation is preferably performed using a CSP which comprises a polysaccharide chiral selector coated or immobilized on a silica backbone according to techniques well-known in the art and a mobile phase, as detailed here above in the specification.
- a CSP which comprises a polysaccharide chiral selector coated or immobilized on a silica backbone according to techniques well-known in the art and a mobile phase, as detailed here above in the specification.
- the polymeric chiral selector is selected from cellulose tris(4-methylbenzoate), cellulose tribenzoate, amylose tris(3,5- dimethylphenylcarbamate) cellulose tris(3,5-dimethylphenylcarbamate), cellulose tris(4- methylphenylcarbamate) cellulose tris (3,5-dichlorophenylcarbamate), amylose tris [(S)- ⁇ - methylbenzylcarbamate] and cellulose tris(3-chloro-4-methylphenylcarbamate) and the solvent is selected from alkanes, such as heptane, hexane, alcohols, such as methanol, ethanol, iso-propanol, n-propanol, acetonitrile, isopropyl acetate, ethyl acetate, dichloromethane, chloroform, ethers, such are methyl t-butyl
- the separation in step (v) is performed using cellulose tris (3,5-dichlorophenylcarbamate) immobilized on a silica backbone as polymeric chiral selector and a mixture of ethyl acetate and methanol in a 90/10 v:v ratio was used as mobile phase.
- Step (vi) is preferably performed by reacting compound (III) with sodium methoxide, followed by steochiometric acidification.
- the temperature of the racemisation is preferably lower than 60 0 C.
- the substantially optically pure (R)-2- acetamido-N-benzyl-3-methoxypropion-amide (I) obtained after the resolution step may be further crystallized for purification purposes. This crystallization is preferably performed in ethyl acetate.
- the crystallization is initiated by seeding the crystallization media with substantially optically pure (R)-2- acetamido-N-benzyl-3-methoxypropion-arnide (I).
- the seeding is be performed at a temperature comprises between 60 0 C and 80°C, preferably at a temperature comprised between 65°C and 75°C.
- the process according to the present invention is particularly advantageous over known processes of manufacture of Lacosamide because: - the starting materials are readily available; it does not require the use of protecting agent for amine functions present in synthetic intermediates which generates additional protection-deprotection process steps and thus increases costs of production; It does not use reagents which are detrimental to the environment; - the undesired enantiomer (III) may ultimately be recycled into Lacosamide, thereby increasing the overall productivity of the process.
- the present invention relates to a process of manufacture of substantially optically pure (R)-2-acetamido-N-benzyl-3-methoxypropion-amide (I) comprising the following steps:
- resolution is preferably performed by diastereosomeric salt formation by reacting compound of formula (IV) with an acid selected from the group consisting of R)-(-)-mandelic acid, (S)-(+)-mandelic acid, (D)-(+)-malic acid, (L)-(-)-malic acid, (+)-O,O'-dibenzoyl tartaric acid, (L)-N-acetyl-alanine and (D)-N-acetyl-leucine, i.e. 'Conversion to diastereoisomers" method described in Jerry March in "Advanced Organic Chemistry", fourth edition, Chapter 4, pages 120-125.
- an acid selected from the group consisting of R)-(-)-mandelic acid, (S)-(+)-mandelic acid, (D)-(+)-malic acid, (L)-(-)-malic acid, (+)-O,O'-dibenzoyl tartaric acid, (L)
- Resolution is preferably performed in a solvent selected from the group consisting of acetone, methanol, ethanol, 1-propanol, methyl-tert-butyl-ether, heptane, cyclohexane, methylethylketone, isopropylacetate and mixtures thereof.
- Resolution is preferably performed at a temperature comprised between 20 0 C and 6O 0 C followed by cooling at a temperature comprised between 0°C and 20 0 C.
- (R)-2-acetamido-N-benzyl-3-methoxypropion-amide is further crystallized to afford substantially optically pure (R)-2-acetamido-N-benzyl-3-methoxypropion-amide. Said crystallization is iterated until (R)-2-acetamido-N-benzyl-3-methoxypropion-amide is obtained in the desired optical purity.
- the resolution step may be performed by chiral chromatographic separation using diffentia absorption methods, more preferably using chiral chromatographic separation carried out in batch or in MCC (Multi Column Chromatography) including SMB (simulated moving bed) mode or mode where the inlet and outlet ports are shifted asynchronously or mode in which inlet and oulet flowrates and/or concentrations are changing in time during the switching period, as detailed here above in the specification for 2-acetamido-N-benzyl-3-methoxypropion-amide (II).
- 2-amino-N-benzyl-3-methoxypropion-amide (IV) used in the present embodiment may be prepared according to any of the methods described here above in the specification.
- All process steps mentioned here above, and particularly processes of manufacture of compound (II), including synthesis and extraction of the materials may be individually or collectively performed in batch mode or according to a continuous process, using for example micro-reactors.
- NMR spectra are recorded on a Bruker 400 MHz spectrometer as solutions in deuterated chloroform (CDCI3). Chemical shifts are expressed in parts per million (ppm, ⁇ ) downfield from tetramethylsilane and are referenced to the deuterated solvent (CDCI3).
- GC Gas chromatography
- MS Mass spectroscopy
- Benzylamine (1.1 equiv) is added so as to obtain a mass temperature between -5°C and 0 0 C.
- the addition funnel is rinsed with 0.25 volume isobutylacetate.
- the reaction mixture is allowed to warm up to 25-30 °C and post-stirred for about 1 hour (HPLC area of (VId) lower than 0,2%).
- 1 volume of water with respect to the initial solution of (VId) is added and the mixture stirred for 15 min.
- the aqueous layer is separated and the organic layer washed with water (0.5 volume).
- the solution is concentrated to ca.
- reaction can be monitiored by HPLC.
- LC-MS (+Cl) (rel intensity) 210 (12), 209 (M + +1 , 100).
- the combined organic layers in the preceding step are adjusted to 7 volumes with respect to the initial amount of 2-bromo-N-benzyl-3-methoxypropion-amide (Va) used in the preceding step and the temperature of the solution is adjusted to 60 0 C. 1 equivalent (with respect to the initial amount of 2-bromo-N-benzyl-3-methoxypropion-amide (Va) used in the preceding step) of acetic anhydride is added dropwise while the temperature is maintained below 70 0 C.
- a feed solution of 12 kg of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) in Ethyl acetate-MeOH (90/10) is prepared and stirred under nitrogen until complete dissolution is achieved.
- the solution is continuously injected in an SMB system which is equipped with six identical columns of 12.4 cm length and 4.8 cm internal diameter, in a 1- 2-2-1 configuration.
- Each column contains 125 g of a Chiral stationary phase comprising cellulose tris(3,5-dichlorophenylcarbamate) immobilized onto the silica backbone and the enantiomers are separated using Ethyl acetate-MeOH (90/10) as the mobile phase.
- Substantially optically pure (R)-2-acetamido-N-benzyl-3-methoxypropion-amide (I) is extracted from the stream and obtained with an enantiomeric excess greater than 99%.
- the mixture is quenched with a 0.05 equivalent of an aqueous solution of HCI while maintaining the mass temperature at about 20°C.
- the methanol is distilled under atmospheric pressure until about 1 volume of solvent remains.
- An azeotropic distillation with 9 volume of isopropylacetate is then performed.
- the distillation is carried out by continuous addition of isopropylacetate in order to maintain a total of 10 volumes (about 5 volumes are distilled to achieve a residual level of MeOH ⁇ 0.1% by GC).
- the mixture is cooled down to 60 0 C and is washed with water.
- the residual water in the organic layer is removed by azeotropic distillation with isopropylacetate according to the same method as mentioned here above.
- the solution is cooled down to 0 0 C for crystallization.
- the suspension is filtered and the cake is washed with isopropylacetate.
- the solid is dried under reduced pressure at 40 0 C.
- the suspension is extracted four times with: 10 ml Water 10 ml 5 % Sodiumhydrogene carbonate
- Example 3 Preparation of Lacosamide from 2-amino-N-benzyl-3-methoxy- propionamide (IV)
- Example 3.1 Diastereoisomeric resolution of 2-amino-N-benzyl-3-methoxy- propionamide (IV) with salts
- N-acetyl-O-methyl-D,L-serine (IX) is obtained as a white solid in 80% yield with a chemical purity of greater than 99% measured by HPLC.
- the NMR spectrum obtained is consistent with characteristics described for compound (IX) in the literature (S .V. Andurkar et al., Tetrahedron: Asymmetry 9, 3841- 3854).
- Example 4.2. Preparation of 2-acetamido-N-benzyl-3-methoxypropion-amide (II) from N-acetyl-O-methyl-D.L-serine (IX)
- Example 4.2.1 Use of isobutylchloroformiate and N-methylmorpholine
- a reaction vessel equipped with a mechanical stirrer were charged 1.0 eq. of N- acetyl-O-methyl-D,L-serine (IX) and 10 volumes of anhydrous THF.
- IX N- acetyl-O-methyl-D,L-serine
- 10 volumes of anhydrous THF 10 volumes of anhydrous THF.
- 1.15 eq. of isobutylchloroformiate and then 1.15 eq. of N-methylmorpholine were successively added dropwise maintaining the temperature below -15°C.
- the reaction mixture was stirred further 15 min. at -20°C and then 1.15 eq. of benzylamine were added dropwise maintaining the mass temperature below -15°C.
- 2-acetamido-N-benzyl-3-methoxypropion-amide (II) is obtained in a yield of 65% with a chemical purity greater than 99% measured by HPLC.
- the NMR spectra is consistent with the one obtained with a reference sample of Lacosamide.
- the homogenous mixture was cooled to 6O 0 C and seeded with 1% w:w of (II). The crystallization was allowed to develop at this T° and the mixture was filtered at 0 0 C, rinsed with a mixture of fresh toluene/ethyl acetate (70:30) and dried under vacuum for 24 hours.
- Compound (II) is obtained in 70% yield and a chemical purity greater than 94.5%.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Biomedical Technology (AREA)
- Public Health (AREA)
- Analytical Chemistry (AREA)
- Pain & Pain Management (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description

































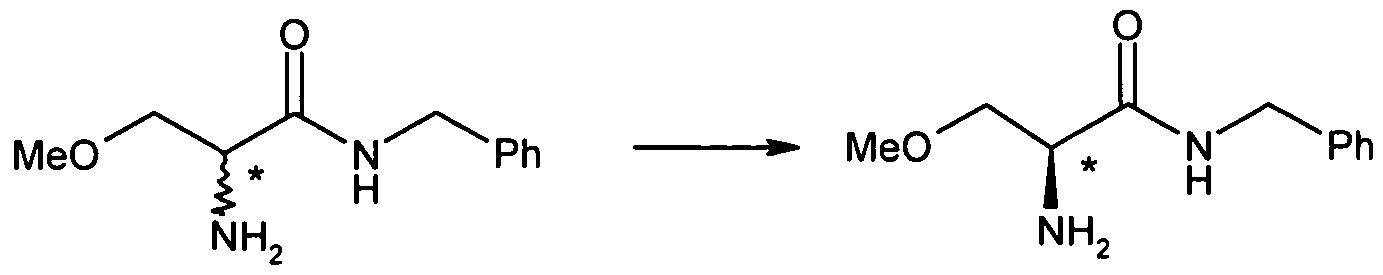

Claims









Priority Applications (19)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EA201100727A EA019040B1 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| BRPI0921408-9 BRPI0921408B8 (en) | 2008-11-07 | 2009-11-06 | process for the manufacture of optically pure (r)-2-acetamido-n-benzyl-3-methoxypropion-amide (i) |
| SI200930598T SI2352721T1 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| KR1020117012580A KR101755548B1 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| US13/127,555 US8946476B2 (en) | 2008-11-07 | 2009-11-06 | Process for the preparation of amino acid derivatives |
| ES09799015T ES2408208T3 (en) | 2008-11-07 | 2009-11-06 | New procedure to prepare amino acid derivatives. |
| RS20130253A RS52820B (en) | 2008-11-07 | 2009-11-06 | A NEW PROCESS TO OBTAIN AMINO ACID DERIVATIVES |
| MEP-2013-71A ME01618B (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| EP09799015A EP2352721B1 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| PL09799015T PL2352721T3 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| JP2011533631A JP5986380B2 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| MX2011004324A MX2011004324A (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives. |
| AU2009313037A AU2009313037B2 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| DK09799015.4T DK2352721T3 (en) | 2008-11-07 | 2009-11-06 | Hitherto unknown method of preparing amino acid derivatives |
| CN200980144201.6A CN102209707B (en) | 2008-11-07 | 2009-11-06 | A New Method to Prepare Amino Acid Derivatives |
| CA2739741A CA2739741C (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| HRP20130456AT HRP20130456T1 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| HK11113149.0A HK1158617B (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
| IL211987A IL211987A (en) | 2008-11-07 | 2011-03-29 | Process for the preparation of amino acid derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08105749.9 | 2008-11-07 | ||
| EP08105749 | 2008-11-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010052011A1 true WO2010052011A1 (en) | 2010-05-14 |
Family
ID=40627087
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/007962 Ceased WO2010052011A1 (en) | 2008-11-07 | 2009-11-06 | Novel process for the preparation of amino acid derivatives |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US8946476B2 (en) |
| EP (1) | EP2352721B1 (en) |
| JP (3) | JP5986380B2 (en) |
| KR (1) | KR101755548B1 (en) |
| CN (2) | CN104058991B (en) |
| AU (1) | AU2009313037B2 (en) |
| BR (1) | BRPI0921408B8 (en) |
| CA (1) | CA2739741C (en) |
| CY (1) | CY1114053T1 (en) |
| DK (1) | DK2352721T3 (en) |
| EA (1) | EA019040B1 (en) |
| ES (1) | ES2408208T3 (en) |
| HR (1) | HRP20130456T1 (en) |
| IL (1) | IL211987A (en) |
| ME (1) | ME01618B (en) |
| MX (1) | MX2011004324A (en) |
| PL (1) | PL2352721T3 (en) |
| PT (1) | PT2352721E (en) |
| SI (1) | SI2352721T1 (en) |
| WO (1) | WO2010052011A1 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITMI20100127A1 (en) * | 2010-01-29 | 2011-07-30 | Archimica Srl | PROCESS FOR THE PREPARATION OF LACOSAMIDE. |
| WO2011158194A1 (en) | 2010-06-15 | 2011-12-22 | Medichem, S.A. | Enzymatic resolution of racemic (2r,s)-2-(acetylamino)-3-methoxy-n-(phenylmethyl)propanamide |
| EP2399901A1 (en) | 2010-06-23 | 2011-12-28 | Archimica GmbH | Intermediate for producing lacosamide and a process for its preparation and conversion to lacosamide |
| WO2012014226A1 (en) * | 2010-07-27 | 2012-02-02 | Indoco Remedies Limited | Process for preparation of lacosamide and some n-benzyl-propanamide intermediate derivatives |
| WO2012041986A1 (en) | 2010-10-01 | 2012-04-05 | Ucb Pharma Gmbh | Process for the preparation of amino acid derivatives |
| WO2012046245A1 (en) * | 2010-10-05 | 2012-04-12 | Hetero Research Foundation | Novel polymorph of lacosamide |
| EP2444390A1 (en) | 2010-10-19 | 2012-04-25 | Archimica GmbH | Process for producing Lacosamide |
| WO2012065891A1 (en) | 2010-11-17 | 2012-05-24 | Ucb Pharma Gmbh | Process for preparing lacosamide |
| WO2012069855A1 (en) | 2010-11-25 | 2012-05-31 | Cambrex Karlskoga Ab | Process for the preparation of lacosamide |
| US8440861B2 (en) | 2009-08-06 | 2013-05-14 | Medichem, S.A. | Solid forms of an N-(phenylmethyl)propanamide derivative and processes of preparation |
| WO2014068333A3 (en) * | 2012-11-01 | 2014-06-26 | Cambrex Karlskoga Ab | New process |
| JP2014520074A (en) * | 2011-05-10 | 2014-08-21 | ディーエスエム アイピー アセッツ ビー.ブイ. | Separation of chiral isomers of chroman compounds and their derivatives and precursors |
| CN104030943A (en) * | 2014-03-12 | 2014-09-10 | 重庆福安药业(集团)股份有限公司 | A preparing process of lacosamide |
| EP2990399A1 (en) | 2014-08-28 | 2016-03-02 | Rao, Davuluri Ramamohan | Improved process for the preparation of lacosamide and its novel intermediate |
| WO2018159028A1 (en) | 2017-03-01 | 2018-09-07 | 株式会社エーピーアイ コーポレーション | N-benzyl-2-bromo-3-methoxypropionamide and method for producing intermediate thereof |
| EP3659997A1 (en) | 2015-11-13 | 2020-06-03 | API Corporation | Method for producing lacosamide and intermediate thereof |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101466390B (en) * | 2006-06-15 | 2014-03-12 | 优时比制药有限公司 | Peptide compounds for the treatment of refractory status epilepticus |
| WO2013072936A2 (en) | 2011-11-10 | 2013-05-23 | Ramamohan Rao Davuluri | A novel process for the preparation of (r)-n-benzyl-2 acetamido-3-methoxypropionamide |
| US8779355B2 (en) * | 2011-12-28 | 2014-07-15 | Quest Diagnostics Investments, Inc. | Methods for detecting lacosamide by mass spectrometry |
| KR101578979B1 (en) * | 2013-11-08 | 2015-12-18 | 에스티팜 주식회사 | A Method for preparing Lacosamide |
| WO2016021711A1 (en) * | 2014-08-07 | 2016-02-11 | 株式会社エーピーアイ コーポレーション | Method for producing amino acid derivative |
| CN104892461B (en) * | 2015-06-24 | 2017-04-19 | 上海上药第一生化药业有限公司 | Lacosamide analogue and preparation method thereof |
| CN106811492B (en) * | 2017-01-18 | 2019-11-01 | 长兴制药股份有限公司 | A kind of preparation method of scheme for lacosamide |
| EP3725769A1 (en) * | 2019-04-17 | 2020-10-21 | Newron Pharmaceuticals S.p.A. | Process for the production of substituted 2-[2-(phenyl) ethylamino]alkaneamide derivatives |
| CN112574058B (en) * | 2020-12-31 | 2023-04-28 | 珠海润都制药股份有限公司 | Synthetic route of lacosamide |
| JP7667998B2 (en) * | 2021-03-03 | 2025-04-24 | 国立大学法人東海国立大学機構 | Methods for Producing Amides and Peptides |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5773475A (en) * | 1997-03-17 | 1998-06-30 | Research Corporation Technologies, Inc. | Anticonvulsant enantiomeric amino acid derivatives |
| EP1642889A1 (en) * | 2004-10-02 | 2006-04-05 | Schwarz Pharma Ag | Improved synthesis scheme for lacosamide |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS61197530A (en) * | 1985-02-25 | 1986-09-01 | Mitsubishi Gas Chem Co Inc | Racemization method |
| US6048899A (en) * | 1997-03-17 | 2000-04-11 | Research Corporation Tech., Inc. | Anticonvulsant enantiomeric amino acid derivatives |
| JP2001288153A (en) * | 2000-04-06 | 2001-10-16 | Mitsubishi Rayon Co Ltd | Racemization of optically active amino acid amides |
| GB0325055D0 (en) * | 2003-10-27 | 2003-12-03 | Smithkline Beecham Corp | New process |
| DE602006011952D1 (en) * | 2005-12-20 | 2010-03-11 | Nps Pharma Inc | FLUORINATED CONNECTIONS |
| CN101130504B (en) * | 2006-08-25 | 2010-07-28 | 苏州雅本化学股份有限公司 | Synthesis, split and racemization method for preparing chirality medicament levetiracetam midbody (S)-(+)-2-amido butyramide hydrochlorate |
| CN100491365C (en) * | 2006-12-08 | 2009-05-27 | 浙江大学 | Process of racemizing 6-fluoro-3,4-dihydro-2H-1-benzopyran-2-formic acid |
-
2009
- 2009-11-06 HR HRP20130456AT patent/HRP20130456T1/en unknown
- 2009-11-06 DK DK09799015.4T patent/DK2352721T3/en active
- 2009-11-06 PL PL09799015T patent/PL2352721T3/en unknown
- 2009-11-06 SI SI200930598T patent/SI2352721T1/en unknown
- 2009-11-06 ME MEP-2013-71A patent/ME01618B/en unknown
- 2009-11-06 BR BRPI0921408-9 patent/BRPI0921408B8/en not_active IP Right Cessation
- 2009-11-06 PT PT97990154T patent/PT2352721E/en unknown
- 2009-11-06 US US13/127,555 patent/US8946476B2/en active Active
- 2009-11-06 JP JP2011533631A patent/JP5986380B2/en active Active
- 2009-11-06 CN CN201410247216.8A patent/CN104058991B/en active Active
- 2009-11-06 CN CN200980144201.6A patent/CN102209707B/en active Active
- 2009-11-06 ES ES09799015T patent/ES2408208T3/en active Active
- 2009-11-06 MX MX2011004324A patent/MX2011004324A/en active IP Right Grant
- 2009-11-06 WO PCT/EP2009/007962 patent/WO2010052011A1/en not_active Ceased
- 2009-11-06 CA CA2739741A patent/CA2739741C/en not_active Expired - Fee Related
- 2009-11-06 KR KR1020117012580A patent/KR101755548B1/en not_active Expired - Fee Related
- 2009-11-06 EP EP09799015A patent/EP2352721B1/en active Active
- 2009-11-06 AU AU2009313037A patent/AU2009313037B2/en not_active Ceased
- 2009-11-06 EA EA201100727A patent/EA019040B1/en not_active IP Right Cessation
-
2011
- 2011-03-29 IL IL211987A patent/IL211987A/en active IP Right Grant
-
2013
- 2013-06-12 CY CY20131100470T patent/CY1114053T1/en unknown
-
2015
- 2015-02-10 JP JP2015023801A patent/JP6272250B2/en active Active
-
2016
- 2016-07-29 JP JP2016149961A patent/JP6272409B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5773475A (en) * | 1997-03-17 | 1998-06-30 | Research Corporation Technologies, Inc. | Anticonvulsant enantiomeric amino acid derivatives |
| EP1642889A1 (en) * | 2004-10-02 | 2006-04-05 | Schwarz Pharma Ag | Improved synthesis scheme for lacosamide |
Non-Patent Citations (1)
| Title |
|---|
| CECILE BEGUIN ETAL: "N-Substituted amino acid N-benzylamides:synthesis, anticonvulsant and metabolic activities", BIOORGANIC & MEDICINAL CHEMISTRY., vol. 12, 2004, GBELSEVIER SCIENCE LTD., pages 3079 - 3096, XP002567548, ISSN: 0968-0896 * |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8440861B2 (en) | 2009-08-06 | 2013-05-14 | Medichem, S.A. | Solid forms of an N-(phenylmethyl)propanamide derivative and processes of preparation |
| US8946477B2 (en) | 2009-08-06 | 2015-02-03 | Medichem, S.A. | Solid forms of an N-(phenylmethyl) propanamide derivative and processes of preparation |
| RU2585762C2 (en) * | 2010-01-29 | 2016-06-10 | Эутикальс Спа | Method of producing lakosamide |
| ITMI20100127A1 (en) * | 2010-01-29 | 2011-07-30 | Archimica Srl | PROCESS FOR THE PREPARATION OF LACOSAMIDE. |
| WO2011092559A1 (en) * | 2010-01-29 | 2011-08-04 | Archimica Srl | Process for the synthesis of lacosamide |
| US8796488B2 (en) | 2010-01-29 | 2014-08-05 | Euticals S.P.A. | Process for the preparation of lacosamide |
| WO2011158194A1 (en) | 2010-06-15 | 2011-12-22 | Medichem, S.A. | Enzymatic resolution of racemic (2r,s)-2-(acetylamino)-3-methoxy-n-(phenylmethyl)propanamide |
| EP2399901A1 (en) | 2010-06-23 | 2011-12-28 | Archimica GmbH | Intermediate for producing lacosamide and a process for its preparation and conversion to lacosamide |
| WO2012014226A1 (en) * | 2010-07-27 | 2012-02-02 | Indoco Remedies Limited | Process for preparation of lacosamide and some n-benzyl-propanamide intermediate derivatives |
| US8957252B2 (en) | 2010-07-27 | 2015-02-17 | Indoco Remedies Limited | Process for preparation of lacosamide and some N-benzyl-propanamide intermediate derivatives |
| WO2012041986A1 (en) | 2010-10-01 | 2012-04-05 | Ucb Pharma Gmbh | Process for the preparation of amino acid derivatives |
| US8969620B2 (en) | 2010-10-01 | 2015-03-03 | Ucb Pharma Gmbh | Process for the preparation of amino acid derivatives |
| WO2012046245A1 (en) * | 2010-10-05 | 2012-04-12 | Hetero Research Foundation | Novel polymorph of lacosamide |
| US8598386B2 (en) | 2010-10-19 | 2013-12-03 | Euticals Gmbh | Process for producing lacosamide |
| EP2444390A1 (en) | 2010-10-19 | 2012-04-25 | Archimica GmbH | Process for producing Lacosamide |
| EP2487152A1 (en) | 2010-11-17 | 2012-08-15 | UCB Pharma GmbH | Process for the preparation of Lacosamide including resolution of O-methyl-DL-serine |
| WO2012065891A1 (en) | 2010-11-17 | 2012-05-24 | Ucb Pharma Gmbh | Process for preparing lacosamide |
| WO2012069855A1 (en) | 2010-11-25 | 2012-05-31 | Cambrex Karlskoga Ab | Process for the preparation of lacosamide |
| JP2014520074A (en) * | 2011-05-10 | 2014-08-21 | ディーエスエム アイピー アセッツ ビー.ブイ. | Separation of chiral isomers of chroman compounds and their derivatives and precursors |
| WO2014068333A3 (en) * | 2012-11-01 | 2014-06-26 | Cambrex Karlskoga Ab | New process |
| US9771317B2 (en) | 2012-11-01 | 2017-09-26 | Cambrex Karlskoga Ab | Process for preparing lacosamide and related compounds |
| CN104030943A (en) * | 2014-03-12 | 2014-09-10 | 重庆福安药业(集团)股份有限公司 | A preparing process of lacosamide |
| EP2990399A1 (en) | 2014-08-28 | 2016-03-02 | Rao, Davuluri Ramamohan | Improved process for the preparation of lacosamide and its novel intermediate |
| EP3659997A1 (en) | 2015-11-13 | 2020-06-03 | API Corporation | Method for producing lacosamide and intermediate thereof |
| US10975117B2 (en) | 2015-11-13 | 2021-04-13 | Api Corporation | Method for producing lacosamide and intermediate thereof |
| US11623943B2 (en) | 2015-11-13 | 2023-04-11 | Api Corporation | Method for producing lacosamide and intermediate thereof |
| WO2018159028A1 (en) | 2017-03-01 | 2018-09-07 | 株式会社エーピーアイ コーポレーション | N-benzyl-2-bromo-3-methoxypropionamide and method for producing intermediate thereof |
| US11136287B2 (en) | 2017-03-01 | 2021-10-05 | Api Corporation | Method for producing n-benzyl-2-bromo-3-methoxypropionamide and intermediates thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2352721B1 (en) | Novel process for the preparation of amino acid derivatives | |
| AU2019420442B2 (en) | L-glufosinate intermediate and L-glufosinate preparation method | |
| KR101699095B1 (en) | Process for manufacture and resolution of 2-acylamino-3-diphenylpropanoic acid | |
| CN117886708A (en) | A kind of synthetic method of milobalin besylate | |
| CN112272665B (en) | Method for preparing lifalast | |
| US8969620B2 (en) | Process for the preparation of amino acid derivatives | |
| HK1158617B (en) | Novel process for the preparation of amino acid derivatives | |
| RS52820B (en) | A NEW PROCESS TO OBTAIN AMINO ACID DERIVATIVES | |
| KR101088488B1 (en) | Method for preparing amlodipine having optical activity | |
| CN111333552B (en) | Synthesis method of beta-benzo amino acid compound | |
| KR20220048951A (en) | Method of preparing intermediate for synthesizing sphingosine-1-phosphate receptor agonist | |
| WO2013187406A1 (en) | Method for producing 4,4,7-trifluoro-1,2,3,4-tetrahydro-5h-1-benzazepine compound and intermediate for synthesis thereof | |
| ITMI20071722A1 (en) | PROCEDURE FOR THE PREPARATION OF ACID (S) (+) - 3- (AMINOMETHYL) -5-METHYLESANOIC | |
| JPH0822855B2 (en) | Optically active histidine derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980144201.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09799015 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2739741 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2011/004324 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011533631 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009313037 Country of ref document: AU Ref document number: 2009799015 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3373/DELNP/2011 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2009313037 Country of ref document: AU Date of ref document: 20091106 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20117012580 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201100727 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13127555 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2013/0253 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: PI0921408 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110506 |