HEPATITIS C INHIBITOR COMPOUNDS
RELATED APPLICATIONS
This application claims benefit of U.S. Serial No. 61/099292, filed September 23, 2008, and U.S. Serial No. 61/186632, filed June 12, 2009, which are herein incorporated by reference.
FIELD OF THE INVENTION
The present invention relates to compounds, processes for their synthesis, compositions and methods for the treatment of hepatitis C virus (HCV) infection. In particular, the present invention provides novel peptide analogs, pharmaceutical compositions containing such analogs and methods for using these analogs in the treatment of HCV infection.
BACKGROUND OF THE INVENTION
It is estimated that at least 170 million persons worldwide are infected with the hepatitis C virus (HCV). Acute HCV infection progresses to chronic infection in a high number of cases, and, in some infected individuals, chronic infection leads to serious liver diseases such as cirrhosis and hepatocellular carcinoma.
Currently, standard treatment of chronic hepatitis C infection involves administration of pegylated interferon-alpha in combination with ribavirin. However, this therapy is not effective in reducing HCV RNA to undetectable levels in many infected patients and is associated with often intolerable side effects such as fever and other influenza-like symptoms, depression, thrombocytopenia and hemolytic anemia. Furthermore, some HCV-infected patients have co-existing conditions which contraindicate this treatment.
Therefore, a need exists for alternative treatments for hepatitis C viral infection. One possible strategy to address this need is the development of effective antiviral agents which inactivate viral or host cell factors which are essential for viral replication.
HCV is an enveloped positive strand RNA virus in the genus Hepacivirus in the
Fiavivmαae family The single stranα HCV RNA genome is approximately 9500 nucleotides in length and has a single open reading frame (ORF), fιanκed by 5' ana 3' non-translated regions The HCV 5' non-translated region is 341 nucleotides in iength and functions as an internal πbosome entry site for cap-independent translation initiation The open reading frame encodes a single large polyprotein of about 3000 ammo acids which is cleaved at multiple sites by cellular and viral proteases to produce the mature structural and non-structural (NS2 NS3 NS4A, NS4B NS5A, and NS5B) proteins The viral NS2/3 protease cleaves at the NS2- NS3 junction while the viral NS3 protease mediates the cleavages downstream of !MS3, at the NS3-NS4A, NS4A-NS4B, NS4B-NS5A and N85A-NS5B cleavage sites The NS3 protein aiso exhibits nucleoside tnphospnatase and RNA helicase activities The NS4A protem acts as a cofactor for the NS3 protease and may also assist in the membrane localization of NS3 and other viral replicase components Although NS4B and the NS5A phosphoprotein are also likely components oi the rephcase, their specific roie≤ are unκnown The NS5B protein is the elongation subunit of the HCV rephcase possessing RNA-αependent RNA polymerase (RdRp] activity
The first evidence of the clinical antiviral activity of HCV NS3 protease inhibitors is provided by the results of a two day dinical trial, which indicate that the HCV NS3 protease inhibitor BiLN 2081 is effective m rapidly reducing viral loads m patients infected with the hepatitis C virus (Gastioenterology (2004) 127(5) 1347-1355) More recently, in 14- and 28-day dinical trials with the HCV NS3 protease inhibitor VX-95Q, alone (Gastroenterology (2006) 131 (4) 997-1002) or in combination with pegylated interferon with or witnout ribavirin, viral load for most HCV patients rapidly decreased to undetectable levels dunng treatment (Hepatology i 2006) 44(4 s1 ) 532A and 614A)
in WO 00/09543, compounds of the formula
wherein R
2 may be O-R
2Q and R
2Q nay be a Het, either unsubstituted or mono-, dι- or tri-substituted, are described as hepatitis C viral NS3 protease inhibitors, an enzyme essential for the replication of the hepatitis C virus
Oral administration is one of the most commonly used drug dosing route. In vitro approaches evaluating absorption distribution, metabolism and excretion have been developed to speed up characterization of the increased number of compounds synthesized in drug discovery programs These experiments are designed to identify candidates that are most likely to have adequate PK profile (Prediction of pharmacokinetic properties using experimental approaches during early drug discovery, Pravin R Chalurvedi*, Caroline J Decker and Aieksandrs Odinecs Current Opinion in Chemical Biology, 2001 , 5 452-463.) but do not yet replace in vivo methods (Pharmacokinetics and metabolism in early drug discovery, Dennis A Smith and Han van de WateΦeemd. Current Opinion in Chemical Biology, 1999. 3, 373-378) herein incorporated by reference.
The rat is among the most commonly used animal in preclinical PK studies and the fraction of oral dose absorbed in rats can be correlated to that observed in humans for many drugs (Lmear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Win L. Chiou and Abhyit Barve Pharmaceutical Research,Mo\. 15 No. 1 1 , 1792-1795,1998), herein incorporated by reference.
Accordingly, there is a need to provide novel compounds for drug development that exhibit good cell-based potency against HCV and favorable pharmacokinetic properties
SUMMARY OF THE SMVE^TΪOM
The present invention provides a novel series of compounds having at least one of
the following surprising advantages:
• unexpectedly good cell-based potency; and/or
• unexpectedly good rat plasma levels after oral administration.
Further objects of this invention arise for the one skilled in the art from the following description and the examples.
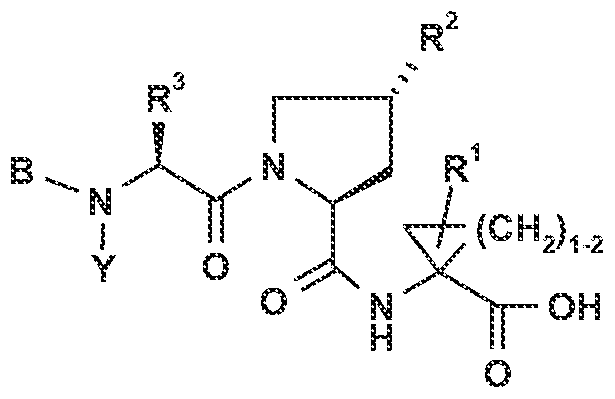
One aspect of the invention provides a racemate, diastereoisomer, or optical isomer of a compound of Formula (I):
R3 is (C2-8)alkyl, (C^cycloalkyl or (C1-3)alkyl-(C3-7)cycloalkyl, wherein each of said alkyl, cycloalkyl, and alkyl-cycloalkyl groups may be mono-, di- or tri- substituted with (C1-4)alkyl;
L0 is halogen, (C1-4)alkyl, -OH, -O-(C1-4)alkyl, -NH2, -NH(C1-4)alkyl or -N((C1-4)alkyl)2;
L1 is halogen, (C1-4)alkyl, -O-(Ci^)alkyl, -S-(C1-4)alkyl, -SO-(d^)alkyl, or -SO2- (Ci-4)alkyl, wherein each of said alkyl groups is optionally substituted with from one to three halogen atoms;
R2 is -NR22COR20, -NR22COOR20, -NR22R21 or -NR22CONR21R23, wherein
R20 is (C1-β)alkyl, (C^cycloalkyl or (C1.4)alkyl-(C3-7)cycloalkyl, wherein said alkyl, cycloalkyl or alkyl-cycloalkyl may be mono-, di- or tri-substituted with
(C1-3)alkyl or -O(C^)alkyl; R21 is H or R20 as defined above, R22 and R23 are independently H or methyl,
R1 is (C1-4)alkyl, (Cjwjalkenyl or (C^Jcycloalkyl;
Rc is hydroxy or NHSO2R8 wherein Rs is (C1-β)alkyl, (C^Jcycloalkyl, (C1-6)alkyl- (C3-7)CyClOaI kyl, aryl or Het; each of which optionally being mono-, di- or tri- substituted with substituents selected from halogen, hydroxy, cyano, (C1-4)alkyl, O-(C1-6)alkyl, -CO-NH2, -CO-NHfd^alkyl, -CO-Ntfd-^alkylk, -NH2, -NH(C1-4)alkyl and -N((C1-4)alkyl)2, wherein (C1-4)alkyl and O-(C1-6)alkyl are optionally substituted with one to three halogen atoms;
B is C(=0)-R4; wherein R4 is (C1-β)alkyl, (C^cycloalkyl, (C1-4)alkyl-(Ci.
7)cycloalkyl, (C^Jcycloalkenyl, (d^alkyHC^cycloalkenyl, Het, aryl, (C1- 4)alkyl-Het, or (C1-4)alkyl-aryl; all of which being optionally substituted 1 to 3 times with (C1-4)alkyl, hydroxy, O-(C1-4)alkyl or halogen; or B is aryl or Het, optionally mono-, di- or tri-substituted with halogen, hydroxy, (C^)alkyl, O-fC^alkyl, O-aryl, O-Hβt, S-(C1-6)alkyl, -CO-(C1^aIkVl, -CO-NH2, -CO-NH(C1-4)BIkVl, -CO-N((C1-4)alkyl)2, -NH2, -NH(C1-4)alkyl and -N((C1.4)alkyl)2, wherein said alkyl and O-alkyl groups may be optionally substituted with 1 to 3 halogen atoms; and wherein Het is a 4- to 7-membered saturated, unsaturated or aromatic heterocycle having 1 to 4 heteroatoms each independently selected from O1 N and S1 or a 7- to 14-membered saturated, unsaturated or aromatic heteropolycycle having wherever possible 1 to 5 heteroatoms, each independently selected from O, N and S, wherein each N heteroatom may, independently and where possible, exist in an oxidized state such that it is further bonded to an oxygen atom to form an N-oxide group and wherein each S heteroatom may, independently and where possible, exist in an oxidized state such that it is further bonded to one or two oxygen atoms to form the groups SO or SO2;
or a pharmaceutically acceptable salt or ester thereof.
Furthermore, compounds according to this invention exhibit one or more of the following surprising advantages:
« unexpectedly good cell-based potency; and/or * unexpectedly good DMPK profile.
Another aspect of this invention provides compounds of Formula (!) showing at least one of the following surprising advantages:
« unexpectedly good activity in a cell-based assay; and/or
« unexpectedly good rat plasma levels after oral administration.
Another aspect of this invention provides a compound of formula (I), or a pharmaceutically acceptable salt or ester thereof, as a medicament.
Included within the scope of this invention is a pharmaceutical composition comprising an anti-hepatitis C viraliy effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt or ester thereof, in admixture with at least one pharmaceutically acceptable carrier medium or auxiliary agent.
According to a further aspect of this embodiment the pharmaceutical composition according to this invention further comprises a therapeutically effective amount of at least one other antiviral agent.
The invention also provides the use of a pharmaceutical composition as described hereinabove for the treatment of a hepatitis C viral infection in a mammal having or at risk of having the infection.
Another important aspect of the invention involves a method of treating or preventing a hepatitis C viral infection m a mammal by administering to the mammal an anti-hepatitis C viraily effective amount of a compound of Formula (!}, a pharmaceutically acceptable salt or ester thereof, or a composition as described above, alone or in combination with at least one other antiviral agent, administered together or separately.
Also within the scope of this invention is the use of a compound of Formula (I), or a pharmaceutically acceptable salt or ester thereof, as described herein, for the manufacture of a medicament for the treatment or prevention of hepatitis C viral
infection in mammal
An additional aspecl oi this invention refers to an article o1 manι_1aclure comprising a composition effective to treat a hepatitis C virai infection, and packaging material comprising a labei which indicates that the composition can be used to treat infection by the hepatitis C virus, wherein the composition comprises a compound of formula (i) according to this invention or a pharmaceutically acceptable salt or ester thereof
Still another aspect of this invention reiates to a method of inhibiting the replication of hepatitis C virus comprising exposing the virus to an effective amount of the compound of formula (I) or a salt or ester thereof, under conditions where replication of hepatitis C virus is inhibited
Further included in the scope o! the invention is the use of a compound of formula (I), or a salt or ester thereof, to inhibit the replication of hepatitis C virus
Yet another aspect of this invention provides a method of inhibiting HCV NS3 protease activity in a mammal by administering a compound of Formula (I), including a pharmaceutically acceptable salt or ester thereof
Another aspect of this invention provides a method of decreasing the NS3 protease activity of the hepatitis C virus infecting a mammal by administering a compound of Formula (!), including a pharmaceutically acceptable salt or ester thereof
DETAILED DESCRIPTION OF PREFERRED EMBODIMEMTS
As used herein, the following definitions apply unless otherwise noted Witn reference to the instances where (R) or (S) is used to designate the absolute configuration of a substituent or asymmetric center of a compound of Formula (I), the designation is done in the context of the whole compound and not in the context o? the substituent or asymmetric center alone
The designation 'P1 , P2, and P3' as used herein reier to the position of the amino acid residues starting from the C-terminus end of the peptice analogs and extending towards the N-terminus (i e P1 refers to position 1 from the C-terminus, P2 seconc position from the C-terminus. etc ) (see Berger A & Schechter I , Transactions of the Royal Society London series B257. 2^9-284 (1970)), herein incorporated by reference
As used herein the term 'vinyl-AGCA" refers to a compound of formula
nameiy, ( IR, 2S) i-amιno-2-ethenylcyclopropanecarboxylιc acid Herein also refered to as vinyl-ACCA
As used herein the term "ethyl-ACCA" refers to a compound of formula
nameiy, (1R, 2R) i-amιno-2-ethylcyclopropanecarboxylιc acid Herein also reiered to as ethyl-ACCA
As used herein the term "cyclopropyl-ACCA" refers to a comoound of formula
nameiy, (1R, 2S) 1-amιno-2-cyclopropy!cyc!opropanecarboxylιc acιd Herein aiso refered to as cyclopropyi-ACCA
The term ' (C1 )a!kγi" as used herein, either alone or in combination with another
suDstiluent, means acyclic, straight or branched chain aikyi suDstiluents containing from 1 to n carbon atoms "(C &)alkyl" includes, but is not iimiteα to, methyl, ethyl, n- propyi n-bulyl, 1-melhy!ethyl (i-propyl), 1-methylpropyl 2-methylpropyl, 1 ,1- dimethylethyl (te/f-butyl), penty! and hexyi The abbreviations Me and Pr denote a methyl group and n-propy! respectively
The term "[C0 -jcycloalkyi" as used herein, either aione or in combination with another substituent, πeans a cycloalkyl substituent containing from 3 to n carbon atoms and includes, but is not limited to cyciopropyl cyciobutyi cyciopentyl, cyclohexyi and cycloheptyi
The term "(C3 )cyc!oa!kenyl" as used herein, either alone or in combination with another substituent, means an unsaturated cychc radicai containing from 3 to n carbon atoms and includes, but is not limited to, cyclopropenyl, cyclobutenyi, cyclopentenyi, cyciohexenyl and cycloheptenyl
The term ' (C1 -)a!kyi-(Cu P)cycloalkyr as used nerein means an alkylene radical containing 1 to n carbon atoms to which a cycioaikyi radical containing from 3 to n carbon atoms is directly linked, and includes, but is not limited to, cyclopropyimethyi, cyclobutylmethyl, cyclopentylmetnyi, 1-cycioρentyiethyi. 2-cydopentyiethy!, cyciohexyimethyl, 1-cyciohexyiethyi, 2-cyclohexyiethyi and cycloheptyi propyl
The term ' (Ci n)aikyi-(C3 r)cycloalkeny!" as used herein means an aikylene radical containing 1 to n carbon atoms to which a cycloalkenyl radicai containing from 3 to n carbon atoms is directly linked and includes, but is not limited to, cyclopropenylmethyl, cyclobutenyimethyl, cyclopentenylmethyi, 1- cyclopenlenyielhy!, 2-cyclopentenylethyi, cyclohexenylmethyl, 1-cyclohexenylethyl, 2-cycιonexenylethyι and cycloheptenylpropyl
The term O-(C. n)aikyi ' or "(C1 n)a!koxy' as used nerein, eitner alone or in combination with another radical, means the radical -O-(Ci n)aikyl wherein aikyi is as defined above containing from 1 to n carbon atoms, and includes methoxy, ethoxy. propoxy, 1~methyiethoxy, butoxy and 1 ,1-dιmethylethoxy The latter radical is known commonly as ?erf-butoxy
The term "(C^ ^alkenyl" as used herein, wherein n is an integer, either aione or in combination with another radical, is intended to mean an unsaturated, acychc straight or branched chain radical containing two to n carbon atoms, at least two of which are bonded to each other by a double bond Examples of such radicals include, but are not limited to, ethenyi (vinyl), 1-ρropenyi. 2-propeπyl, and 1-butenyi Unless specified otherwise, the term "(C2 -,)aikeny!' is understood to encompass individual stereoisomers where possible, including but not limited to (E) and (Z) isomers, and mixtures thereof When a (C2 n)alkeny! group is substituted it is understood to be substituted on any carbon atom thereof which would otherwise bear a hydrogen atom, unless specified otherwise, such that the substitution would give rise to a chemically stable compound, such as are recognized by those skilled in the art
The term ' aryl" as usec herein, either aione or in combination with another radicai, is intended to mean a carbocyclic aromatic monocyclic group containing 8 carbon atoms which may be further fused to a second 5- or 6-membered carbocyciic group which may be aromatic, saturated or unsaturated Aryl includes, but is not limited to, phenyl, mdaπyl, indenyl, 1-naphthyl, 2-naρhthyi, tetrahydronaphthyl and dihydronaphthyl
The term "cyano" or "CN" as used herein is intended to mean a nitrogen atom attached to a carbon atom by a triple bond (C≡N)
The term "haio" or "haiogen" as used herein means a halogen substituent selected from fluoro, chioro bromo or iodo
The term "carbocyde" or "caroocyclic" as usec herein, either aione or in combination with another radical, is intended to mean a cyclic compound, either aromatic or non- aromatic, saturated or unsaturated, in which all of the ring members are carbon atoms The earboeyde group may contain 5 or 6 carbon atoms and may be further fused to a second 5- or 6-membered carbocyclic group which may be aromatic, saturated or unsaturated The carbocyde may be substituted When the carbocycie is substituted, it is understood that substituents may be attached to any carbon atom
whicn would otherwise bear a hydrogen atom, unless specified otherwise, such that the substitution would give rise to a chemically stable compound, such as are recognized by tnose skilled in the art
The term "Net" as used herein, either alone or in combination with another radical, is intended to mean a 4- to 7-membered saturated, unsaturated or aromatic heterocycle having 1 to 4 heteroatoms each independently selected from O, N and S, or a 7- to 14-membered saturated unsaturated or aromatic heteropoiycycle having wherever possible 1 to 5 heteroatoms, each independently selected from O, H and S, wherein each N heteroatom may, independently and where possibie exist in an oxidized state such that it is further bonded to an oxygen atom to form an N- oxide group and wherein each S heteroatom may, independently and where possible, exist in an oxidized state such that it is further bonded to one or two oxygen atoms to form the groups SO or SO2, unless specified otherwise When a Net group is substituted, it is understood that substituents may De attached to any- carbon atom or heteroatom tnereof which would otherwise bear a hydrogen atom, unless specified otherwise, such that the substitution would give rise to a chemically stable compound, such as are recognized by those skilled in the art
The term ' (C1 -)a!kyi-Het" as used herein and unless specified otherwise, wherein n is an integer, either alone or in combination with another radical, is intended to mean an alky! radical having 1 to n carbon atoms as defined above which is itself substituted with a Net substituent as defined above Examples of (C1 ,)a!ky!-Het include, but are not limited to, thienylmethyl, furyimethyi, pipendinyiethyi 2- pyndinyimethyi, 3-pyrιdιnylmethyl, 4-pyrιdιnyimethyl, quinolinylpropyl, and the hke When an (C- n)alkyl-Het group is substituted, it is understood that substituents may be attached to either the Net or the alky! portion tnereof or both, unless specified otherwise, sucn that tne substitution would give rise to a chemically stable compound, sucn as are recognized by those skilled in the art
The term "heteroatom' as used herein is intended to mean O. S or N
The term "heterocyde" as used herein and unless specified otherwise, either alone or in combination with another radical, is intended to mean a 3- to 7-membered
saturated, unsaturated or aromatic neterocyde containing from 1 to 4 neteroatoms each independently selected from O, N ana S, or a monovalent radical denveα by- removal of a hyαrogen atom lhereirom Examples o! sucn heterocycie≤ include but are not limited to. azetichπe, pyrrolidine, tetrahydrofuran, tetrahydrothiopheπe, thiazohdine, oxazohdme, pyrrole, tniopnene, furan, pyrazoie, imidazole, isoxazole, oxazoie, isothiazoie, th'azoie, tπazole, tetrazoie, pipendine, piperazine, azepine, diazepine, pyran, 1 ,4-dιoxane 4-morphohne, 4-thιomorphohne, pyridine, pyridine-N-oxide, pyπdazine, pyrazine and pynmidine, and saturated, unsaturated and aromatic derivatives thereof
The terms "-S-(Gi η)alky!" or "(Ci n)alkylthιo" as used herein interchangeably, wherem n is an integer, either alone or in combination with another radical, is intended to mean an sulfur atom further bonded to an alky! radical having 1 to n carbon atoms as defmeα above Examples o1 -S-(C- r )aιkyi include but are not limited to methylthio (CH3S-), ethyllhiQ (CH3CH2S-), propyithio (CHJDH2CH2S-), 1-methyietnyithιo
(/sopropyithio, (CH3)2CH-S-) and 1 ,1-dιmelhylelhylthιo (terf-biiiylthio- (CHJ^C-S-) When -S-(C1 -)a!kyi radical, or an oxidized derivative thereof, such as an -SO-(C' p)aikyl radical or an -SO2-(Ci n)alkyl radical, is substituted, each is understood to be substituted on the (C1 r)aikyi portion thereof, such that the substitution would give rise to a chemically stable compound, such as are recognized by those skilled in the art
The term "pharmaceutically acceptable ester' as used herein, either alone or in combination with another substituent, means esters of the compound of Formula (I) m which any of the carboxyl functions of the molecule, but preferably the carboxy terminus, is replaced by an alkoxycarbonyi function
in which the R moiety of the ester is selected from alkyl (including, but not limited to, methyl, ethyl, n-propyi, t-butyl, n-butyl). aikoxyaikyl (including, but not limited to methoxymethyi), alkoxyacyl (including, but not limited to acetoxymethyi), alkyl-aryi (including, but not limited to benzyl), aryloxyaikyl (including, but not limited to phenoxymethyi), ary! (including, but not limited to phenyl), optionally substituted with halogen (C-
4)aikyi or (C
1 4)alkoxy Other suitable prodrug esters can be found in
Design o1 prodrugs Bundgaard, H Ed Elseviβr (1985), herein incorporated by reierence Sucn pharmaceutically acceptable esters are usually hydroiyzed in vivo when iniected in a mammal anc transformed into the acid form of the compound oi Formula (!) With regard to the esters described above, unless otherwise specified. any alky! moiety present advantageously contains 1 to 16 carbon atoms, particularly 1 to 8 carbon atoms Any aryl moiety present in such esters advantageously comprises a phenyl group In particular the esters may be a C< <
6 a'kyi ester an unsubstituted benzyl ester or a benzyl ester substituted with at least one halogen, C
1 6 alkyi, C,
R aikoxy, nitro or tnfluoromethyl
The term "pnarmaceuticaϋy acceptable salt" means a salt of a compound of formula (!) which is, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response, ana the hke, commensurate with a reasonable benefit/risk ratio generally water or oil-soluble or cispersible, and effective for their intended use The term includes pnarmaceuticaily-acceptabie acid addition salts and pharmaceuticaliy-acceptable base addition salts Usts of suitable salts are found in, e g , S M Berge et ai , J Pharm Sc* , 1977, 66, pp 1-19, herein incorporated by reference
The term ' pharmaceuticaily-acceptabie acd addition salt' means those salts which retam the biological effectiveness and properties of the free bases and which are not biologically or otherwise undesirable formed with inorgamc acids such as hydrochloric acid hyαrobromic acid sulfuric a&α, sulfamic acid, nitnc acid, phosphoric aciCi, and the like, and organic aciαs such as acetic acid, tnfluoroacetic acid, adipic acid ascorbic acid, aspartic acid, benzenesuifomc acid, benzoic acid, butyric acid, camphoric aαα, eamphorsuilonic acic, cinnamic acid, citric aod, digiuconic acid, ethanesulfonic aod, glutamic acid, glycolic acic, giycerophosphoric acid, nemisuliic acid, hexanoic acid, 1oπτ>ιc acid, fumaric acid, 2-hydroxyethane- suiionic acid (iselhionic acid), lactic acid, hydroxymaieic acid, malic acid malonic acid, mandeiic acid, mesitylenesuifonic acid, methaπesulfonic acid, naphthalenesulfonic acid, nicotinic acid, 2-naphthaienesulfonιc acid, oxalic ac*d, Damoic acid, pectinic acid, phenylacetic acid. 3-phenylρropιonιc aod, Divalic acid, propionic acid, pyruvic acid, sahcyhc acid, stearic acid, succinic acid, sulfamic acid,
tartaric aod, p-loiuenesulionic a&α, undecanoic acid, and the lικe
The term "pharmaceuticaily-acceptaDle base aαdilion salt" means tnose salts which retain the biological effectiveness and properties of the free acids and which are not biologically or otherwise undesirable, formed with inorganic bases such as ammonia or hydroxide, carbonate, or bicarbonate of ammonium or a metal cation such as sodium, potassium lithium calcium, magnesium, iron, zinc, copper, manganese, aluminum and the like Particularly preferred are the ammonium, potassium, sodium, calcium, and magnesium salts. Salts derived from pharmaceuticaliy- acceptable organic nontoxic bases include salts of primary, secondary, and tertiary amines, quaternary amine compounds, substituted amines including naturally occurring substituted amines cyclic amines and basic ion-exchange resins, such as methylamine dimethylamine, tnmethylamme, ethylamme, diethylamine, triethylamine, isopropylamine tripropylamine, tπbutylamine, ethanoiamine, diethanoiamine, 2-dιmethylamιnoethanol, 2-dιetnyiamιnoethanoι, dicyciohexylamine, lysine, arginme, histidine, caffeine, hydrabamine, cnohne, betaine, ethylenediamine, glucosamine, methyiglucamme, theobromine, purines, piperazine, piperidine, N- ethylpipeπdme, tetramethylaTimoniiim compounds, tetraethylammonium compounds, pyridine. N,N-dιmethylanιiιne, N-methyloioendine, N-methylmorphohne, dicyciohexylamine, dibenzylamine, N,N-dιbenzy!phenethylamιne, 1-ephenamιπe, N, N -dibenzylethyienediamine. poiyamme resins, and the like Particularly preferred organic nontoxic bases are isopropylamine. diethylamine, ethanoiamine, tπmethylamme, dicyciohexylamine, choline, and caffeine.
The term "mammal" as it is used herem is meant to encompass humans, as well as non-human mammals which are susceptible to infection by hepatitis C virus inducing domestic animals, such as cows, pigs, horses, dogs and cats, and non- domestic animals
The term "antiviral agent" as used herein means an agent (compound or Dioiogical) that is effective to inhibit the formation and/or replication of a virus in a mammal This includes agents that interfere with either host or viral mechanisms necessary for the formation and/or replication of a virus in a mammal. Such agents can be selected from another anti-HCV agent, HiV inhibitor. HAV inhibitor and HBV
inhibitor Antiviral agents include, for example ribavirin, amantadine, VX-497 (meπmepodib, Vertex Pharmaceuticals), VX-498 (Vertex Pharmaceuticals), Levoviπn, Viramiαine, Cepiene (maxarnine), XTL-001 and XTL-002 (XTL Biopnarmaceuticals)
The term "other anti-HCV agent" as used herein means those agents that are effective for diminishing or preventing the progression of hepatitis C related symptoms of disease Such agents can be selected from immunomodulatory agents inhibitors of HCV NS3 protease, inhibitors of HCV polymerase or inhibitors of another target in the HCV life cycle
The term "immunomodulatory agent" as used herein includes those agents (compounds or biologicais) that are effective to enhance or potentiate the immune system response in a mammal Immunomodulatory agents include, but are not limited to, inosine monophosphate dehydrogenase inhibitors sucn as VX-497 (menmepodib, Vertex Pharmaceuticals), dass I interferons, dass M interferons, consensus interferons, asialo-interferons pegylated interferons and conjugated interferons, including but not limited to interferons conjugated with other proteins including but not limited to human albumin Class I interferons are a group of interferons that all bind to receptor type I, including both naturally and synthetically produced class I interferons, while class M interferons ail bind to receptor type M. Examples of class i interferons include, but are not limited to, α-, β~, δ-. to-, and τ-mterferons, while examples of class Il interferons include, but are not limited to, ->~ interferons
The term "inhibitor of HCV NS3 protease ' as used herein means an agent (compound or biological) that is effective to inhibit the function of HCV NS3 protease in a mammal Inhibitors of HCV NS3 protease include, for example, those compounds described in VVO 99/07733, WO 99/07734. WO 00/09558, WO 00/09543. WO 00/59929, WO 03/064416, WO 03/064455, WO 03/064456, WO 2004/030670, WO 2004/037855, WO 2004/039833, WO 2004/101602, WO 2004/101605, WO 2004/103996, WO 2005/028501 , WO 2005/070955, WO 2006/000085, WO 2006/007700, WO 2006/007708, WO 2007/009227 (all by Boehringer Ingelheim), WO 02/080926 WO 03/053349 WO 03/099274, WO
03/099316, WO 2004/032827, WO 2004/043339, WO 2004/094452, WO 2005/046712, WO 2005/051410, WO 2005/054430, WO 2006/122188, WO 2007/056120, WO 2007/044933, WO 2007/008657, WO 2008/008776, WO 2008/064066, WO 2008/064057, WO 2008/060927, WO 2008/057871 , WO 2008/057873, WO 2008/057875 (ail by BMS), WO 200^/0722^3, WO 200^/093798, WO 2004/1 13365, WO 2005/010029 (all by Enanta), WO 2005/037214 (Intermune). WO 01/771 13, WO 01/81325, WO 02/08187 WO 02/08198, WO 02/08244, WO 02/08256 WO 02/48172, WO 03/062228, WO 03/062265, WO 2005/021584, WO 2005/030796, WO 2005/058821 , WO 2005/051980, WO 2005/085197, WO 2005/085242, WO 2005/085275, WO 2005/087721 , WO 2005/087725, WO
2005/087730, WO 2005/087731 , WO 2005/107745 and WO 2005/113581 (ah by Scheπngj, WO 2006/119061 , WO 2007/016441 , WO 2007/015855, WO 2007/015787, WO 2008/057208, WO 2008/051514 (all by Merck}, WO 2006/043145 (Pfizer), WO 2008/059046 (Tibotec), WO 2008/057209, WO 2008/051477, WO 2008/051475 (all by MercK & IRBM), ali of which are herein incorporated by reference, and the candidates VX-950, SCH-503034, iTMN-191 , TMC 435350. and MK7009
The term ' inhibitor of HCV polymerase" as used herein means an agent (compound or biological) that is effective to inhibit the function of an HCV polymerase in a mammal This includes, for example, inhibitors of HCV NS5B polymerase Inhibitors of HCV polymerase inducle for example those compounds described in WO 03/007945, WO 03/010140, WO 03/0101^1 , US 6,448, 281 , WO 02/04425, WO 2008/019477, WO 2007/087717, WO 2006/007693, WO 2005/080388, WO 2004/099241 , WO 2004/065367, WO 2004/064925 (all by Boehringer Ingelheimj, WO 2006/093801 , US 2005/0107364 WO 2005/019191 US 2004/0167123 WO 2004/041818, WO 2008/011337 (all by Abbolt Laboratories], WO 01/32153 (Biochem Pharma lnc }, WO 01/60315 (Biochem Pharma lnc ), US 2004/0138170, WO 2004/106350, WO 2006/050161 , WO 2006/104945, WO 2006/002231 , US 2005/080053, US 2004/0242599, US 2004/0229839, WO 03/087298, WO
02/069903 (ah by Biocryst Pharmaceuticals, lnc ), WO 2006/0943^7 (Biota, lnc ). WO 2005/021568 (Biota, lnc ), WO 2008/051637, WO 2007/150001 , WO 2006/066079 (all by Anadys Pharmaceuticals), WO 2007/033032. WO 2007/033175, WO 03/026587 WO 2007/143521 WO 2007/140109 WO
2007/140200, VVO 2007/140254, WO 2007/136982, WO 2007/092000, WO 2007/092888, WO 2006/020082, US 2005/01 19318, WO 2005/034850 (all by Bristol-Myers Squibb), VVO 2007/034127 (Arrow Therapeutics Limited), WO 2005/063734 (Bayer), VVO 03/093290, WO 2005/012288, WO 2008/01 1521 , VVO 2008/008907, WO 2008/008912, WO 2007/084157, WO 2007/019397, WO 2006/138744, WO 2006/121468, WO 2006/1 16557, WO 2006/102594, WO 2006/076529, WO 2006/075993, US 2006/01 1131 1 , WO 2005/054268, WO 2005/042556, US 2005/0090463, WO 2004/108687, WO 2004/028481 , WO 2006/093986, WO 2006/093987 (all by Genelabs Technologies), VVO 2006/1 17306, WO 2004/046159, WO 2007/113159, WO 2007/093541 , WO 2007/068615, WO 2007/065829, WO 2007/020193, VVO 2006/021341 , VVO 2006/021340, VVO 02/100415. WO 02/094289, WO 02/18404 (ail by F. Hoffmann-La Roche). WO 2007/039142, WO 2007/039145, VVO 2007/039144, VVO 2006/045613, VVO 2006/045615, WO 2005/103045, VVO 2005/092863, VVO 2005/079799, VVO 2004/096774, WO 2004/096210, VVO 2004/076415, VVO 2004/060889, VVO
2004/037818, WO 2004/009543, VVO 03/097646, WO 03/037893, WO 03/037894, WO 03/037895, WO 03/000713 (ail by Glaxo Group), WO 2007/144686, WO 2006/000922, WO 2004/046331 , WO 2004/002422, WO 2004/002999, WO 2004/003000, WO 2005/009418, WO 03/026675, WO 03/026589, WO 2007/025043 (all by Idemx), US 03/050320, WO 2007/1 19889, WO 2006/052013, WO
2005/080399, WO 2005/049622, WO 2005/014543, EP 1 162 196. WO 01/47883 (all by Japan Tobacco), WO 2007/095269, WO 2007/054741 , WO 03/06221 11 WO 00/06529, WO 99/64442, WO 2006/1 19975, WO 2006/046030, WO 2006/046039, WO 2005/034941 , VVO 2005/023819, VVO 2004/110442, VVO 2004/087714, VVO 2007/065883, WO 02/06246, WO 2007/129119, WO 2007/029029, WO
2007/028789, WO 2006/029912, WO 2006/027628, WO 2006/008556 (all by lstituto Di Richerche Di Bioiogia Molecolare P. Angelelti SPA), WO 2008/005542, WO 2006/091905, WO 2005/063751 , WO 2004/005286 (all by Gilead Sciences), VVO 2008/043704 (Medivir), WO 2005/123087, WO 2007/021610, WO 2006/065335, WO 2006/012078, VVO 2004/003138, VVO 2004/000858, VVO 03/105770, WO 03/020222. WO 2005/084192, WO 2004/009020, WO 2004/007512, WO 02/057425. WO 02/057287, WO 2007/022073. US 2004/0229840 (all by Merck and Co.), WO 2006/018725, WO 2004/073599. WO 2004/074270. WO 03/095441 , WO 03/082848 (ail by Pfizer). US 2005/00154056, WO 2004/002977. WO 2004/002944.
WO 2004/002940 (all by Pharmacia & Upjohn Company), WO 00/04141 (Ribozyme). WO 2006/050035 (Scheπng), WO 2006/050034 (Scheπngj, US 2003/0203948 (Shionogi). WO 02/20497 (Shionogi), WO 2005/121132 (Shionogi) EP 1321463 (Shire Biochem). WO 02/100851 (Shire Biochem), WO 02/100846 (Shire Biocheπ), WO 03/061385, WO 03/062256, WO 03/062255, US 6,906.190, WO 2004/080466 (all by Ribapharm), WO 2007/026024 (Tibotec), WO 2006/065590 (XTL Biopharmaceuticals), WO 2008/051244, WO 2007/092558, WO 2006/034337, WO 03/099275, WO 03/099824 (ail by Wyeth). WO 03/059356, WO 01/85172, WO 01/85720. WO 03/037262, WO 2008/059042, WO 2008/043791 , WO 2008/017688, WO 2007/147794, WO 2007/088148, WO 2007/071434, WO 2007/039146, WO 2006/100106, WO 2004/058150, WO 2004/052312, WO 2004/052313, WO 03/099801 WO 02/098424 (a!! by Smithkline Beecham), WO 2007/027248 (Vaieant), WO 2008/058393, WO 2006/1 19646, WO 2004/052879, WO 2004/052885, WO 00/18231 WO 00/13708, WO 00/10573, WO 2004/041201 , WO 03/090674 (ah by Viropharma), a!! oi which are herein incorporated by reference Specie exampies of inhibitors of an HCV polymerase, induce R-1626 and R-7128 (Roche), GL60667 (Geneiabs/Novartis), VCH-759 and VCH-916 (Virochem), GS9190 (Gilead). MK-3281 (Merck) and PF868554 (Pfizer), ABT-333 (Abbott), .
The term ' inhibitor of another target in the HCV life cycle" as used herein means an agent (compound or biologicai) that is effective to inhibit the formation and/or replication of HCV in a mammal other than by inhibiting the function of the HCV NS3 protease. This includes agents that interfere with either host or HCV viral targets necessary for the HCV life cycle or agents which specifically inhibit in HGV cell culture assays through an undefined or incompletely defined mechanism Inhibitors of another target in the HCV life cycle include, for example, agents that inhibit viral targets such as Core E1 , E2, p7, NS2/3 protease, NS3 nehcase, NS4A NS5A, NS5B polymerase, and internal πbo≤ome entry site (IRES), or host targets such as cyclophilin B, phosphatidylmositoi 4-kιnase IHu, CD81 , SR-B1 Ciaudin 1 VAP-A, VAP-B Specific examples of inhibitors of another target in the HCV life cycle include !SIS-14803 (ISIS Pharmaceuticals), GS9190 (G'lead), GS9132 (Gϋead), A-831 (AstraZeneca) NM-811 (Novartis), and DEBIO-025 (Debio Pharma)
The term "HIV inhibitor" as used herein means an agent (compound or biological)
that is effective to inhibit the formation and/or replication of HIV in a mamma! This induces agents that interfere with either host or viral mechanisms necessary for the formation and/or replication of HIV in a mammal HIV inhibitors include, for example nucleoside inhibitors, non-πiideoside inhibitors, protease inhibitors, fusion inhibitors and integrase inhibitors
The term "HAV inhibitor' as used herein means an agent (compound or bioiogica!) that is effective to inhibit the formation and/or replication of HAV in a mammal This includes agents that interfere with either host or viral mechanisms necessary for the formation and/or replication of HAV in a mammal HAV inhibitors include Hepatitis A vaccines, for example, HaVnX* (GlaxoSmithKlme), VAQT A* (Merck) and Avaxim^ (Aventis Pasteur)
The term "HBV inhibitor' as used herein means an agent (compound or bioiogical) that is effective to inhibit the formation and/or replication of HBV in a mammal Tlrs includes agents that interfere with either host or viral mechanisms necessary for the formation and/or replication of HBV in a mammal HBV inhibitors include, for example, agents that inhibit HBV viral DNA polymerase or HBV vaccines Specific examples of HBV inhibitors include Lamivudiπe (Epivir-HBV3), Adefovir Dipivoxil, Entecavir. FTC (Coviracil'5'), DAPD (DXG), L-FMAU (Cievudine*) AM365 (Amrad), Ldt (Teibivudine), monovai-LdC (Valtorcitabme) ACH-126,443 (L-Fd4C) (Achilhon). MCC478 (Eh Lilly), Racivir (RCV) Fluoro-L and D nucleosides Robustallavone, ICN 2001-3 (ICN). Bam 205 (Novels), XTL-001 (XTL), Immo-Sugars (Nonyi-DNJ) (Synergy), HepBzyme. and immunomoduiator products such as interferon alpha 2b, HE2000 (HoHis-Eden), Theradigm (Epimmune). EHT899 (Enzo Biochem),
Thymosin alpha- 1 (Zadaxin*), HBV DNA vaccine (PowderJect), HBV DNA vaccme (Jefferon Center), HBV antigen (OraGen), BayHep B's (Bayer). Nabi-HB^ (Nabi) and Anti-hepatitis B (Cangene); and HBV vaccme products such as the following Engeπx B, Recombivax HB, GenHevac B, Hepacare, Bio-Hep B, TwinRix, Comvax, Hexavac
Specific preferred examples of some of these agents are listed below
81 antiviral agents, ribavirin or amantadine,
81 immunomodulatory agents class I interferons, class Ii interferons or pegylated
forms tnereαf,
51 HCV polymerase inhibitors nucleoside analogs or non-nucleosides, 51 inhibitor of another target in the HCV hie cycie that inhibits a target selected from NS3 hehcase, NS2/3 protease, interna* πbosome entry site (IRES), NS^A, NS5A, NS5B polymerase, or host targets such as cyclophilin A or B,
81 HIV inhibitors nucleoside inhibitors, non-nυcleosidic inhibitors, protease inhibitors, fusion inhibitors or mtegrase inhibitors, or * HBV inhibitors agents that inhibit virai DMA polymerase or is an HBV vaccine
As discussed above, combination therapy is contemplated wnerem a compound of formula (!}, or a pharmaceutically acceptable salt thereof is co-ad ministered witn at least one additional agent selected from an antiviral agent, an immunomodulatory agent, another inhibitor of HCV NS3 protease, an inhibitor of HCV polymerase an innibitor of another target in the HCV life cycie, an HIV mniDilor, an HAV inhibitor and an HBV inhibitor Examples of such agents are provided in the Definitions section above These additional agents may be combined witn the compounds of this invention to create a single pharmaceutical dosage form Alternatively these additional agents may be separately administered to the patient as part of a multiple dosage form, for example, using a kit Such additional agents may be administered to the patient prior to, concurrently with, or following the administration of a compound of formula (i), or a pharmaceutically acceptable salt thereof
As used herein, the term 'treatment" means the administration of a compound or composition according to the present invention to alleviate or eliminate symptoms of the hepatitis C disease and/or to reduce viral load in a patient
As used herein, the term "prevention' means tne administration of a compound or composition according to the present invention post-exposure of the individual to the virus Dut before the appearance ot symptoms oi the disease and/or prior to the detection o1 the virus in the blood, to prevent the appearance of symptoms ot the disease
The term "therapeutically effective amount' means an amount of a compound according to the invention which when administered to a patient in need thereof, is
sufficient to effect treatment for disease-states, conditions, or disorders for which the compounds have utility. Such an amount would be sufficient to elicit the biological or medical response of a tissue system, or patient that is sought by a researcher or clinician. The amount of a compound according to the invention which constitutes a therapeutically effective amount will vary depending on such factors as the compound and its biological activity, the composition used for administration, the time of administration, the route of administration, the rate of excretion of the compound, the duration of the treatment, the type of disease-state or disorder being treated and its severity, drugs used in combination with or coincidentally with the compounds of the invention, and the age, body weight, general health, sex and diet of the patient. Such a therapeutically effective amount can be determined routinely by one of ordinary skill in the art having regard to their own knowledge, the state of the art, and this disclosure.
Preferred embodiments
In the following preferred embodiments, groups and substituents of the compounds of formula (I):
according to this invention are described in detail.
JL
B-A: In one embodiment, B is C(=O)-R4; wherein R4 is (Ci-β)alkyl, (C^cycloalkyl, (C1^)alkyl-(C3-7)cycloalkyl, (C3-7)cycloalkenyl, (C1^)alkyl-(C3-7)cycloalkenyl, Het, aryl, (C1-4)alkyl-Het, or (C^alkyl-aryl; all of which being optionally substituted 1 to 3 times with (Ci^alkyl, hydroxy, O-(Ci^)alkyl or halogen; or
B is aryl or Het, optionally mono-, di- or tri-substituted with halogen, hydroxy, (d-ejalkyl, O-tC^alkyl, O-aryl, O-Het, S-(C1-6)alkyl, -CO-(C^)alkyl, -CO-NH2, -CO-NH(C^)alkyl, -CO-N((C1-4)alkyl)2, -NH2, -NHfC^Jalkyl and
-N((C1_4)alkyl)2l wherein said alky! and O-alkyl groups may be optionally substituted with 1 to 3 halogen atoms; and wherein Het is defined as a 4- to 7-membered saturated, unsaturated or aromatic heterocycle having 1 to 4 heteroatoms each independently selected from O, N and S, or a 7- to 14-membered saturated, unsaturated or aromatic heteropolycycle having wherever possible 1 to 5 heteroatoms, each independently selected from O, N and S, wherein each N heteroatom may, independently and where possible, exist in an oxidized state such that it is further bonded to an oxygen atom to form an N-oxide group and wherein each S heteroatom may, independently and where possible, exist in an oxidized state such that it is further bonded to one or two oxygen atoms to form the groups SO or SO2. B-B: In another embodiment, B is C(=0)-R4; wherein R4 is (C1-β)alkyl, (C3- 7)cycloalkyl, (C1-4)alkyl-(C3-7)cycloalkyl, (Q^cycloalkenyl, (Ci-4JaIk^-(C3- 7)cycloalkenyl, aryl or (C1-4)alkyl-aryl; all of which being optionally substituted
1 to 3 times with (C^Jalkyl, hydroxy, O-(C1-4)alkyl or halogen; or
B is aryl or Het, optionally mono-, di- or tri-substituted with halogen, hydroxy, (C^alkyl, O-(C^)alkyl, O-aryl, O-Hβt, S-(C1-6)alkyl, -CO-(C^)alkyl, -NH2, -NH(C1-4)alkyl and -N((C^)alkyl)2, wherein said alkyl and O-alkyl groups may be optionally substituted with 1 to 3 halogen atoms; and wherein Het is defined as a 4- to 7-membered saturated, unsaturated or aromatic heterocycle having 1 to 3 heteroatoms each independently selected from O1 N and S, or a 7- to 14-membered saturated, unsaturated or aromatic heteropolycycle having wherever possible 1 to 3 heteroatoms, each independently selected from O, N and S.
B-C: In another embodiment, B is C(=O)-R4; wherein R4 is (C1-6)alkyl, (C3- 7)cycloalkyl, (d-^alkyKC^cycloalkyl, (Ci^)alkyl-(C3-7)cycloalkenyl or C6- aryl; all of which being optionally substituted 1 to 3 times with (C^alkyl, O- (C^alkyl or halogen; or B is aryl or Het, optionally mono-, di- or tri-substituted with halogen,
(C1-6)alkyl, O-(C1-β)alkyl, O-aryl, O-Het or S-(C1-6)alkyl, wherein said alkyl and O-alkyl groups may be optionally substituted with 1 to 3 halogen atoms; and wherein Het is a 4- to 6-membered saturated, unsaturated or aromatic heterocycle having 1 to 3 heteroatoms each independently selected
from O, N and S, or a 7- to 10-membered saturated, unsaturated or aromatic heteropolycycle having wherever possible 1 to 3 heteroatoms, each independently selected from O, N and S. -D: in another embodiment, B is C(=O)-R4; wherein R4 is (Ci 6)alkyl. (C3.
7)cydoaikyi, (Ci 4)aikyl-(C3.7)cycloalky!, (C|.i)alky!-(C3 7)cyc!oa!keπyl or C6- aryi; ail of which being optionally substituted 1 to 3 times with (Ci.,_)alkyl; or
B is aryl or Net, optionally mono-, di~ or tri-substituted with halogen, (Ci_6)a!kyi, O-(Ci-6)alkyl, O-aryl, O-Het or S-(Ci_6)a!kyi. wherein said aikyl and O-alkyl groups may be optionally substituted with 1 to 3 halogen atoms; and wherein the Net group is defined as:
-E: i n another embodiment, B is C(^Q)-R
4: wherein R
4 is (Chalky!, (C
3. 5)cycloalkyi, (Ci.?)a!kyl-(C3.5)cycloalky!, (Ci.2)alky!-(C
3^)cyc!oaikenyl or G
6- aryl; ail of which being optionally substituted 1 to 3 times with (Ci^)alkyl; or B is aryi or Het, optionally mono-, di- or tn-substituted with halogen, hydroxy, (Ci.
4)aikyl, O-(C
1.
4)a!kyi, O-phenyl, O-tetrahydropyranyl, S-(C
1.
4)a!kyi, wherein said aikyl and Oaikyl groups may be optionally substituted with 1 to 3 halogen atoms; and wherein the Het group is defined as:
B-F: In another embodiment, B is C(^Q)-R
4 wherein R
4 is (Chalky!, (C
3
5)cydoaikyi, (Ci.2)aikyl-(C3.5)cycloalkyi, (Ci.2)alky!-(C3.5)cycioaikenyl or C6- aryi; all of which being optionally mono-substituted with (C-^jalkyl: or
B is aryi or Het, optionally mono- or di-substituted with halogen, (Chalky!, O-(Ci_6)alkyl, O-phenyi, O-tetrahydropyranyl, S-(Ci_6)a!kyl, wherein said aikyl and O-alkyl groups may be optionally substituted with 1 to
3 halogen atoms; and wherein the Heϊ group is defined as
B-G: in another embodiment, B is C(=O)-R wherein R is:
B-H In another embodiment, B is C(=O)-R4, wherein R4 is (C1-β)alkyl, (C3.5)cycloalkyl, (C1.2)alkyl-(C3.5)cycloalkylI (C1.2)alkyl-(C3.5)cycloalkenyl or C6-aryl; all of which being optionally mono- substituted with (Ci_2)alkyl. B-I In another embodiment, B is aryl or Het, optionally mono-, di- or tri-substituted with halogen, hydroxy, (C^alkyl, O- (C1-4)alkyl, O-phenyl, O-tetrahydropyranyl, S-(C1^)alkyl, wherein said alkyl
and O-aikyi groups may be optionally substituted with 1 to 3 halogen atoms' and wherein tne Het group is αeimed as
Any and each individual definition of B as set out herein nay be combined with any and each individual definition of R!, R2. R3, Rc, L8 and L1 as set out herein
R3-A- in one embodiment, R3 is (C? e)alky! (C , ,.)cyc!oa!kyi or (C1 ,)alky!-(C3 ,')cye!oaiky!, wherein each of said alkyl, cycloalky!, and alkyl-cydoaikyl groups may De mono-, di- or tn-suDstiluted with (C1 4)a!kyi R3-B- In another emboαiment, R3 is (C2 6)alkyl (C3 ,-)cycloalkyι or (C1 3)alky!-(C ,')cycloaikyι, wherein each of said alkyl, cycloalky!, and alkyl-cydoaikyl groups may De mono- or di-suDsMuted with (C- 3)aιkyl
3 p I n another embodiment, R3 is ethyl, propyl, butyl, cyclopropyl, cyclobutyl, cyciopentyi or eydohexyl, each of which optionaliy being substituted with 1 or 2 substituents selected from methyl, ethyl and propyl
R3-D in another embodiment, R3 is 1-methylethyl, 1 1-dιmethylethyl, 1-methyipropyl, 2-methyl propyl, 1 ,1-dιmethyipropyl, 1.2-dιmethylpropyi
2,2-dιmethyiproρyl, butyl, cyclopropyl, cyclobutyl, cyciopentyi, cyciohexyl, 1- methylcyclopropyl, 1-methylcyclobutyl, 1-methyicyc!opentyi or 1- methylcyclohexyi
RJ-E I n another embodiment, R3 is
Any and each individual definition of RJ as set out herein may be combined with any and each individual definition of R1 R2, Rc, B, Ls and L1 as set out herein
L0
L°-A: In one embodiment, L0 is halogen, (C1-4)alkyl, -OH, -O-(C1-4)alkyl, "NH 2. -
NH(C1-4)alkyl or -N((C^)alkyl)2.
L°-B: In another embodiment, L0 is halogen, (C1-4)alkyl or -0-(C1 ^JaI kyl.
L°-C: In another embodiment, L0 is (C1-4)alkyl or -O-(C1^)alkyl. L°-D: In another embodiment, L0 is -O-tC^alkyl.
L°-E: In another embodiment, L0 is -O-(Ci-2)alkyl.
L°-F: In another embodiment, L0 is -OCH3.
Any and each individual definition of L0 as set out herein may be combined with any and each individual definition of R1, R2, R3, Rc, B and L1 as set out herein.
JL2I
L1-A: In one embodiment, L1 is halogen, (C1-4)alkyl, -O-(C1-4)alkyl, -S-(C1^JaI kyl, -
SO-(Ci-4)alkyl, or -SO2-(Ci-4)alkyl, wherein each of said alkyl groups is optionally substituted with from one to three halogen atoms.
L1-B: In another embodiment, L1 is halogen, (C1-4)alkyl or -O-(C1-4)alkyl.
L1-C: In another embodiment, L1 is -CH3, -C2H5, -C3H7, -F, -Cl, -Br1 -OCH3, -OC2H5 or -OC3H7.
L1-D: In another embodiment, L1 is -CH3, -C2H5, -Cl or -Br. L1-E: In another embodiment, L1 is CH3, -Cl or -Br.
Any and each individual definition of L1 as set out herein may be combined with any and each individual definition of R1, R2, Rc, R3, B and L0 as set out herein.
R2:
R2-A: In one embodiment, R2 is -NR22COR20, -NR22COOR20, -NR22R21 or -
NR22CONR21R23, wherein
R20 is (C1-β)alkyl, (C^Jcycloalkyl or (C1^)alkyl-(C3-7)cycloalkyl, wherein said alkyl, cycloalkyl or alkyl-cycloalkyl may be mono-, di- or tri-substituted with (C1-3)alkyl or -O(C1-3)alkyl;
R21 is H or R20 as defined above,
R22 and R23 are independently H or methyl. R2-B: In another embodiment, R2 is -NR22COR20, -NR22COOR20 or -NR22R21, wherein
R20 is (C1 6)aikyi, (C3.7)cycloalky! or (C,.,:)aikyl~(C3 7)cydoa!kyl, wherein said alkyi, cycloalkyl or alkyl-cycioaikyl may be mono-, di- or tri-subslituled With (C1 3)alkyl or -O(C1 3)alkyl; R21 is H or R20 as defined above, R22 is H or methyl.
R2-C: in another embodiment, R2 is -N(H)COR28, -N(H)COOR20 or -N(H)R21, wherein
R20 is (Ci_6)alkyl or (C3-7)cycioaikyl, wherein said alkyi or cydoalky! may be mono- or di-substituted with (Ci.3)alkyl or -O(Ci.3)alkyl; R21 is R20 as defined above.
R2-D: in another embodiment, R2 is -N(H)COR20, -N(H)COOR20 or -N(H)R21, wherein
R20 is is (C-M)aikyi or (C3-5)cyc!oa!kyi, wherein said aikyi or cycioaikyi nay be mono- or di-substituted with (Ci-3)alkyl or -O(Ci-3)alkyl; and R21 is R20 as defined above.
R?-E: In another embodiment, R2 is -N(H)CQR?0, -N(H)COOR20 or -N(H)R21, wherein
R20 is (Ci.i)alkyl or (C3.5)cycioaikyl, wherein said aikyi may be mono- or di- substituted with (C1 3)a!ky! or -0(C1 3)a!ky!: and R21 is R28 as defined above.
R2-F: in stϋl another embodiment, R2 is
Any and each individual definition of R as set out herein may be combined with any and each individual definition of R1, R3. Rc. L0. L1 and B as set out herein.
Rc-A: In one embodiment, Rc is hydroxy or NHSO2R8 wherein Rs is (C^Jalkyl,
((-W)CyClOaI kyl, (C14)alkyl-(C3.7)cycloalkyl, aryl or Het; each of which optionally being mono-, di- or tri-substituted with substituents selected from halogen, hydroxy, cyano, (C^Jalkyl, O-(Ci^)alkyl, -CO-NH2, -CO-NH(C1- 4)alkyl, -CO-N((C^)alkyl)2, -NH2, -NH(C14)alkyl and -NKd^alkylfc, wherein
(C1-4)alkyl and O-(C1-6)alkyl are optionally substituted with one to three halogen atoms. Rc-B: In another embodiment, Rc is hydroxy or NHSO2R8 wherein Rs is (C1-β)alkyl,
(C3-7)CyClOaI kyl or (C1-β)alky 1-(C3-7)CyClOaI ky I; each of which optionally being mono-, di- or tri-substituted with substituents selected from (Ci-^alkyl, O-(Ci_ β)alkyl, wherein (C^alkyl and O-(Ci-β)alkyl are optionally substituted with one to three halogen atoms. Rc-C: In another embodiment, Rc is hydroxy or NHSO2R8 wherein R8 is (C1-e)alkyl,
(C3-7)CyClOaI kyl or (Ci-β)alkyl-(C3-7)cycloal kyl; each of which optionally being mono-, di- or tri-substituted with substituents selected from (C1-4)alkyl and O-
(C1-β)alkyl. Rc-D: In another embodiment, Rc is hydroxy or NHSO2R8 wherein R8 is (Ci-β)alkyl,
(C3-7)cycloalkyl, each of which optionally being mono- or di-substituted with substituents selected from (C^alkyl and O-(C14)alkyl. Rc-E: In another embodiment, Rc is hydroxy or NHSO2R8 wherein R8 is (C1-6)alkyl,
(C3-7)CyClOaI kyl or (C1«)alkyl-(C3-7)cycloalkyl, each of which optionally being mono-substituted with substituents selected from (d^alkyl. Rc-F: In another embodiment, Rc is hydroxy or NHSO2R8 wherein R8 is
(C3-7)CyClOaI kyl optionally being mono-substituted with substituents selected from (Ci^)alkyl.
Rc-G: In another embodiment, Rc is hydroxy or NHSO2R8 wherein R8 is cyclopropyl optionally being mono-substituted with (CM)alkyl. Rc-H: In another embodiment, Rc is hydroxy or NHSO2R8 wherein R8 is
R
c-I: In another embodiment, R
c is hydroxy or NHSO
2R
8 wherein R
8 cyclopropyl. R
c-J: In another embodiment, R
c is hydroxy.
Any and each individual αeiinition of R
c as sel out herein may be combined witn any ana each individual definition o1 R
1, R
2, R
3 L
0 L
1 and B as sel out herein
Bl= R1-A. In one embodiment, R1 is (C- i)alkyl, (C24)aiκenyi or (C3 7)cycloalkyi.
R1-B. In another embodiment, R1 is (C2 i)alkyl, (C2 4)alκeny! or (C3 5)cycloalκyi. R1 -C in another embodiment, R1 is ethyl, vinyi or cyciopropyl
Any and each individual definition of R1 as set out herein may be combined with any and each individual definition of R2, R3 Rc L0 L1 and B as set out herein
in the moiety P1 the substituent R1 and the carbonyi take a syn orientation Therefore in the case R1 is ethyl the asymmetric carbon atoms in the cyciopropy! group take the R,R configuration according to the subiormula
in the case R1 is vinyl, the asymmetric carbon atoms in the cyciopropyl group take the R, S configuration according to the subformula
In the case R1 is cyciopropyl, the asymmetric carbon atoms in the cyciopropy! group take the R, S configuration according to the subformula'
Examples o! preferred suogenenc embodiments of the present invention are set fortn in the following table, wherein eacn substituent group o! each embodiment is defined according to the definitions set forth above
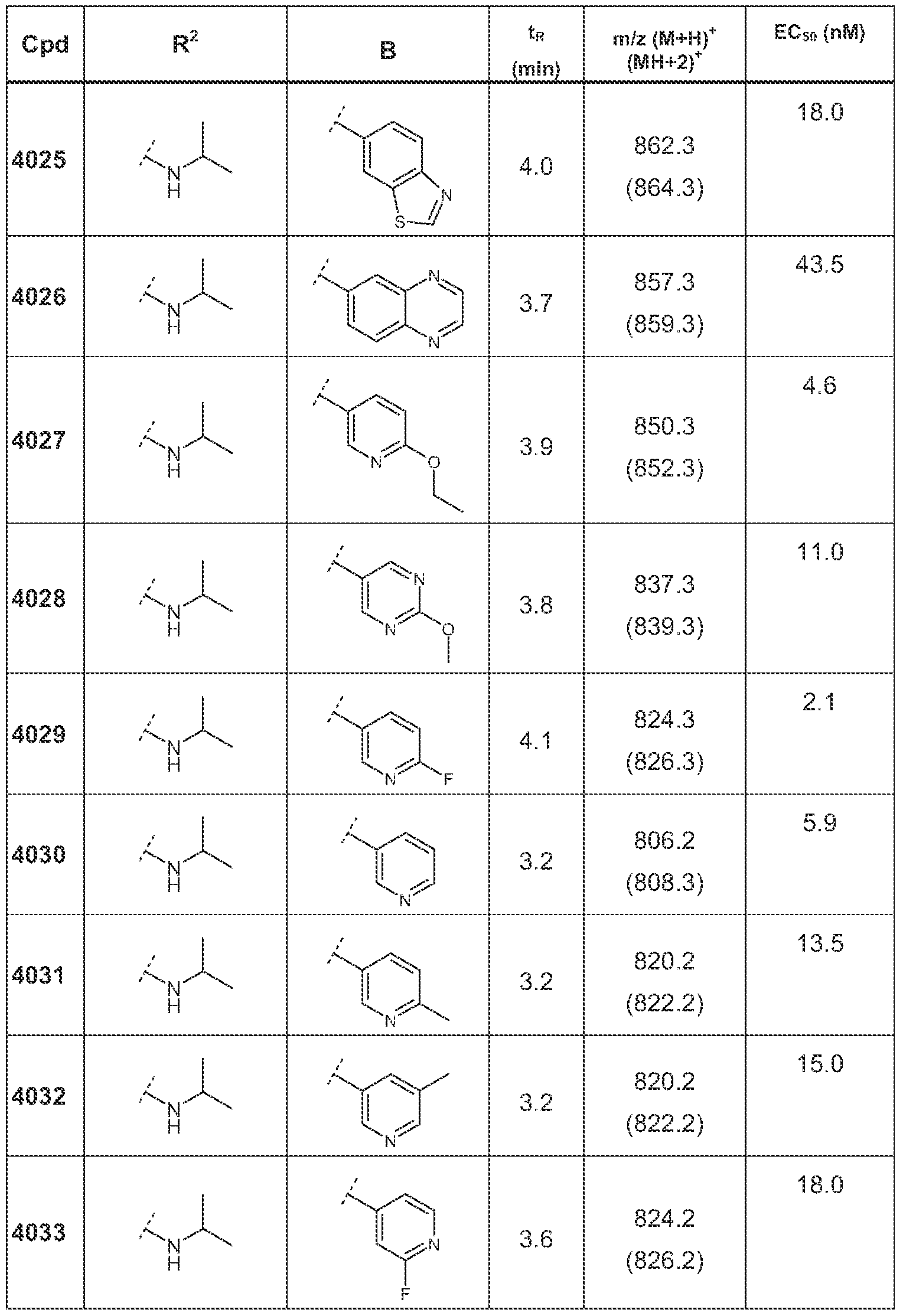
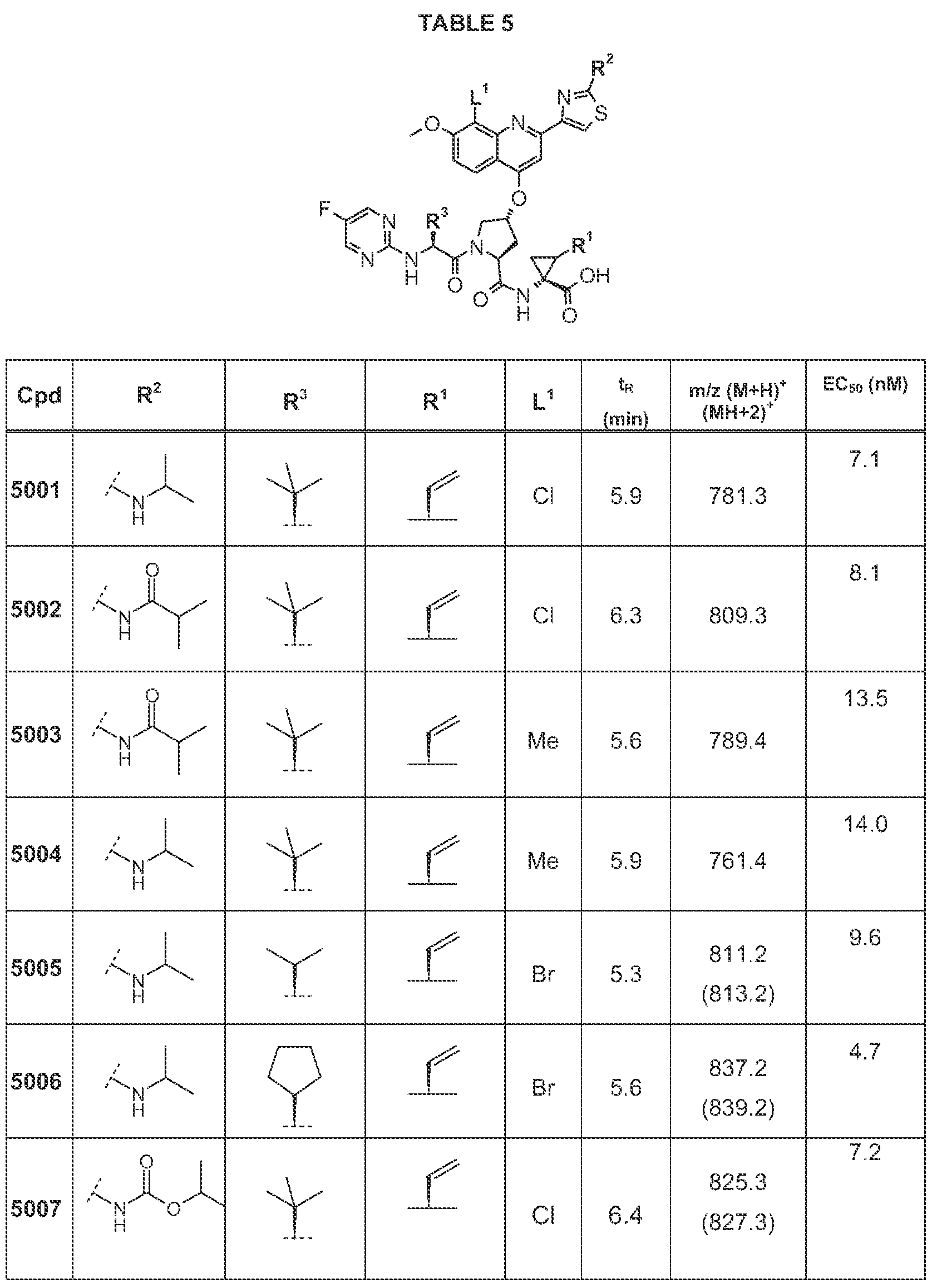
Examples of most preferred compounds according to this invention are each single
compound hsted in the following Tables 1 to 8
According to an alternate embodiment, the pharmaceutical composition o! this invention nay additionally compπse at least one other anti-HCV agent. Examples of anti-HCV agents include, α- (alpha), β- (beta), δ- (delta), y- (gamma), ω- (omega) or τ~ (tau) interferon, pegylated α~'nterferon, ribavirin and amantadine
According to another alternate embodiment, the pharmaceutical composition of this invention may additionally comprise at least one otner inhibitor of HCV NS3 protease
According to another alternate embodiment, the pharmaceutical composition of this invention may additionally comprise at least one inhibitor of HCV polymerase
According to yet another alternate embodiment, the pharmaceutical composition of this invention may additionally comprise at least one inhibitor of other targets in the HCV life cycle, including but not limited to, hehcase NS2/3 protease or internal πbosome entry site (IRES).
The pharmaceutical composition of this invention may be administered orally parenteraliy or via an implanted reservoir Orai administration or administration by iniection is preferred The pharmaceutical composition of this invention may contain any conventional non-toxic pharmaceiilicaliy-acceptable carriers, adjuvants or venicles In some cases, the pH of the formulation may be adjusted with pharmaceutically acceptable acids, bases or buffers to enhance tne stability of the formulated compound c its delivery form. The term parenteral as used herein ^eludes subcutaneous, intracutaneous, intravenous, intramuscular, intra-articuiar, intrasynovial, intrasternal, intrathecal, and intralesional injection or infusion techniques
The pharmaceutical composition may be in the form of a stenle injectable preparation, for example, as a sterile injectable aqueous or oleaginous suspension This suspension may be formulated according to techniques known m the art using suitable dispersing or wettmg agents (such as, for example Tween 80) and
suspending agents
The pharmaceutical composition of this invention may be orally administered in any oraily acceptable dosage form including, but not limited to, aqueous suspensions and solutions, capsules, powders, syrups, elixirs or tablets In the case of tablets for orai use, earners which are commonly used include iactose and corn starch Lubricating agents such as magnesium stearate are aiso typically added For orai administration !P a capsule form, useful diluents include lactose and dπed corn stanch When aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents If desired certain sweetening and/or flavoring and/or coloring agents may be adαed For systemic administration, including but not limited to administration Dy subcutaneous intracutaneous, intravenous, intramuscular, intra-articiiiar, intrasynovial, intrasternal intrathecal and intralesional injection or miusion techniques, it is preferred to use a solution of the compound, or a pharmaceutically acceptable salt or ester thereof , in a pharmaceutically acceptable steπle aqueous venicle
Pharmaceutically acceptable earners, adjuvants, diluents, vehicles, excipients and additives as well as methods of formulating pharmaceutical compositions for various modes of administration are well-known to those of skill in the art and are described m pharmaceutical texts such as Remington The Science and Practice of Pharmacy 21st Edition, Lippincott Wilhams & Wiikms, 2005 and L V Allen, N G Popovish and H C Ansel, Pharmaceutical Dosage Forms and Drug Delivery Systems, 8th ed , Lippincott Williams & Wiikms, 2004, herein incorporated by reference
Dosage levels of between about 0 01 and about 100 mg/kg body weight per day preferably between about 0 1 and about 50 mg/kg body weight per day of the protease inhibitor compound described herein are useful in a monotherapy or in combination therapy 1or the prevention and treatment oi HCV mediated disease Typically, the pharmaceutical composition of this invention will be administered 1rom about 1 to about 5 times per day or alternatively, as a continuous infusion Such administration can be used as a chronic or acute therapy The amount of active ingredient that may be combined with the earner materials to produce a single dosage form will vary depending upon the host treated and the particular node of
administration A typical preparation will contain from about 5% to aoout 95% active compound (w/w) Preferably, such preparations contain from about 20% to about 80% active compound
As the skilled artisan will appreciate, lower or higher doses than those recited above may be required. Specific dosage and treatment regimens for any particular patient wil! depend upon a variety of factors, including the activity of the specific compound employed the age, body weight, genera! health status, sex, diet, time of administration rate of excretion, drug combination, the severity and course of the infection, the patient's disposition to the infection and the judgment of the treating physician Generally treatment is initiated with smaϋ dosages substantially less than the optimum dose of the peptide Thereafter, the dosage is increased by smail increments until the optimum effect under the circumstances is reached in general, the compound is most desirably administered at a concentration level that will generally afford antiviraliy elective results witnout causing any harmful or deleterious side effects
When the composition of this invention comprises a combination of a compound of Formula (!) and one or more additional therapeutic or prophylactic agent, both the compound and the additional agent should be present at dosage levels of between about 10 to 100%, and more preferably between about 10 and 80% of the dosage normally administered in a monotherapy regimen
When these compounds, including their pharmaceutically acceptable salts and esters are formulated together with a pharmaceutically acceptable earner, the resulting composition may be administered in vivo to mammals, such as man to inhibit HCV NS3 protease or to treat or prevent HCV virus infection Such treatment may also be achieved using a compound of this invention in combination with another antiviral agent Preferred other antiviral agents are described within the Delinitions section and the section of preferred pharmaceutical compositions according to this invention and include but are not limited to α~, β-, ό-, ro~, y- or τ- mterferon ribavirin, amantadine, other inhibitors of HCV NS3 protease, inhibitors of HCV polymerase, inhibitors of other targets in the HCV life cycle, which include but are not limited to, heiicase, NS2/3 protease or interna! πbosome entry site (IRES).
or combinations thereof The additional agents may be combined with compounds of this invention to create a single dosage form Alternatively tnese additional agents may De separately administered to a mammal as part of a multiple cosage form
Accordingly, another embodiment of this invention provides a method of inhibiting HCV NS3 protease activity in a mammal by administering a compound of the Formula (I) including a pharmaceutically acceptable sait or ester thereof
in a preferred embodiment this metnod is useful in decreasing the NS3 protease activity of the nepatitis C virus infecting a mammal
As discussed above, combination therapy is contemplated wherein a compound of formula (I), or a pharmaceutically acceptable sait or ester thereof, is co-administered with at ieast one additional antiviral agent Preferred antiviral agents are described nereinbefore ana examples of such agents are provided in the Definitions section These additional agents may be combined with the compounds of this invention to create a single pharmaceutical dosage form Alternatively these additional agents may be separately administered to the patient as part of a multiple dosage form, for example, using a Kit Such additional agents may be administered to the patient prior to, concurrently with, or following the administration of a compound of formula (i), or a pharmaceutically acceptable salt or ester thereof
A compound of formula (!), or a pharmaceutically acceptable sait or ester thereof, set forth herein may also be used as a laboratory reagent Furthermore a compounα of this invention, including a pharmaceutically acceptable salt or ester tnereof, may also be used to treat or prevent viral contamination of materials and therefore reduce the risk of viral infection o1 laboratory or medical personnel or patients who come in contact witn such materials (e g biood, tissue, surgical instruments and garments, laboratory instruments and garments, ana biood collection apparatuses and materials)
A compound of formula (J), including a pharmaceutically acceptable sait or ester thereof set forth herein may also be used as a research reagent A compound of
formula (Ij, including a pharmaceutically acceptable salt or ester thereof, may aiso be used as positive control to validate surrogate cell-based assays or in vitro or in vivo viral replication assays
METHODOLOGY
The synthesis of compounds of Formula (!) according to this invention is conveniently accomplished following the general procedure outlined in the schemes below wherein R1, R2, R3, Rc. Rs, L0, L1 and B are as defined herein. Further instruction is provided to one skiϋed in the art by the specific examples set out herein below Other specific ways of synthesis or resolution of the compounds of this invention can be found in WO 00/09543- WO 00/09558, WO 00/59929, WO 99/07733 and WO2004/103998, all of which are hereby incorporated by reference
The following schemes illustrate a convenient process using known methods for preparing the compounds of Formula (!) when R1 is vinyl or ethyl and Rc is OH
The synthesis of dipeptide 1 is carried out by coupling the P1 residue to the properly protected frans-hydroxy proline under standard conditions. The stereochemistry of the hydroxyl group is inverted by the well known Mitsunobu reaction using para- nitrobenzoic acid. Coupling of the dipeptide with the P3 moiety (2a-h) (obtained from
commercial sources) yields tπpeplide 3 Introduction of tne quinohne moiety to the hydroxyl group of the tripeptide 3 with inversion of stereochemistry can be earned out using either a Mitsonobu reaction or by converting tne free hydroxy! group into a good leaving group (such as a brosyiate) and displacing it with the hydroxys quinohne derivative 5 For the synthesis of the 2-{2~amino~4-thiazo!yi) derivatives, the quiπoiine used contains a 2-carbomethoxy group as shown in 5 Conversion of the carboxyiate group to the aminothiazoie derivative !s carried out by well known synthetic methodology and is described and exemplified in VVO 00/09543 and VVO 00/09558 herein incorporated by reference Removal of the P3 boc protecting group followed by a peptide coupling reaction with an appropriate carboxyhc acid proviαes the desired amides Alternatively, removal of the P3 boc protecting group followed by an SNAr reaction with an appropriately substituted 2-pynmιdιne with a leaving group at the 2-ρosιtιon provides the corresponding 2-pyπmιdyi compounds For the introduction of other Het, see Scheme 3 The C-termina! ester is hydrolyzed under basic aqueous conditions to provide compounds oi formula (!) in which R1 is vinyl Tne vinyl group can De reduces using hydrazine monohydrale as a source of diιiτ»de to provide compounds of formula (I) in which R1 is ethyl
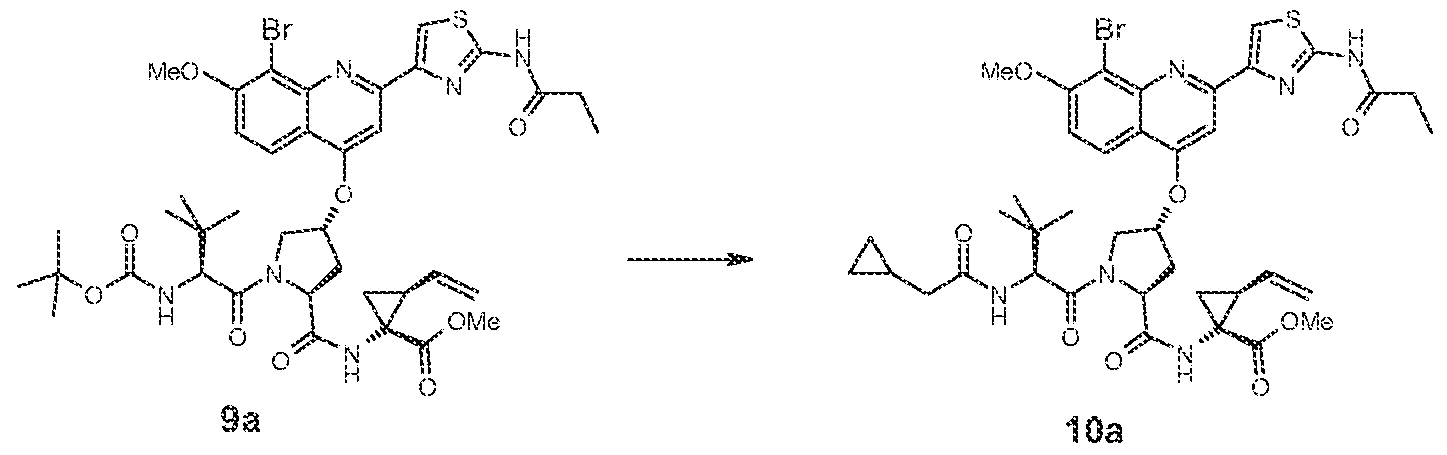
Scheme 2 describes another reaction sequence for making compounds o1 Formula {I} In this case the quinohne moiety is introduced to the dipeptide in a similar way as descπbed in Scheme 1 The PZ moiety (2a-h) is coupled under standard conditions with the dipeptide 17 to provide the corresponding tπpeptide analogs 9 Conversion of the resulting tπpeotides (9) to the desired inhibitors (11 and 12) of formula (i) is earned out as described in Scheme 1
Scheme 3
Scheme 3 demonstrates the reaction sequence used to prepare intermediates 21 and 22 To prepare P3 fragments 19a-h. the commercially available ammo acids (18a-h) were heated in the presence of commercially available aryi halides using copper catalysis and the N-arylated amino acids 19a-h are obtained The N-arylated ammo acids 1§a-h can undergo a peptide coupling reaction with the deprotected dipeptide 17 to provide the N-arylated tripeptides 20 Basic aqueous hydrolysis of
these intermediates provides compounds (21) o1 lormula (!) where R1 is vinyl arse B is aryi Reαuction of the vinyl group in using hydrazine monohyαrate as a source oi diimiαe provides compounαs (22) oi Formula (I) where R1 is ethyl and B is aryl For compounds of formula (!) where B is Het (Met being other than 2-pyπmιdyi described in Scheme 1 ), same methodology applies
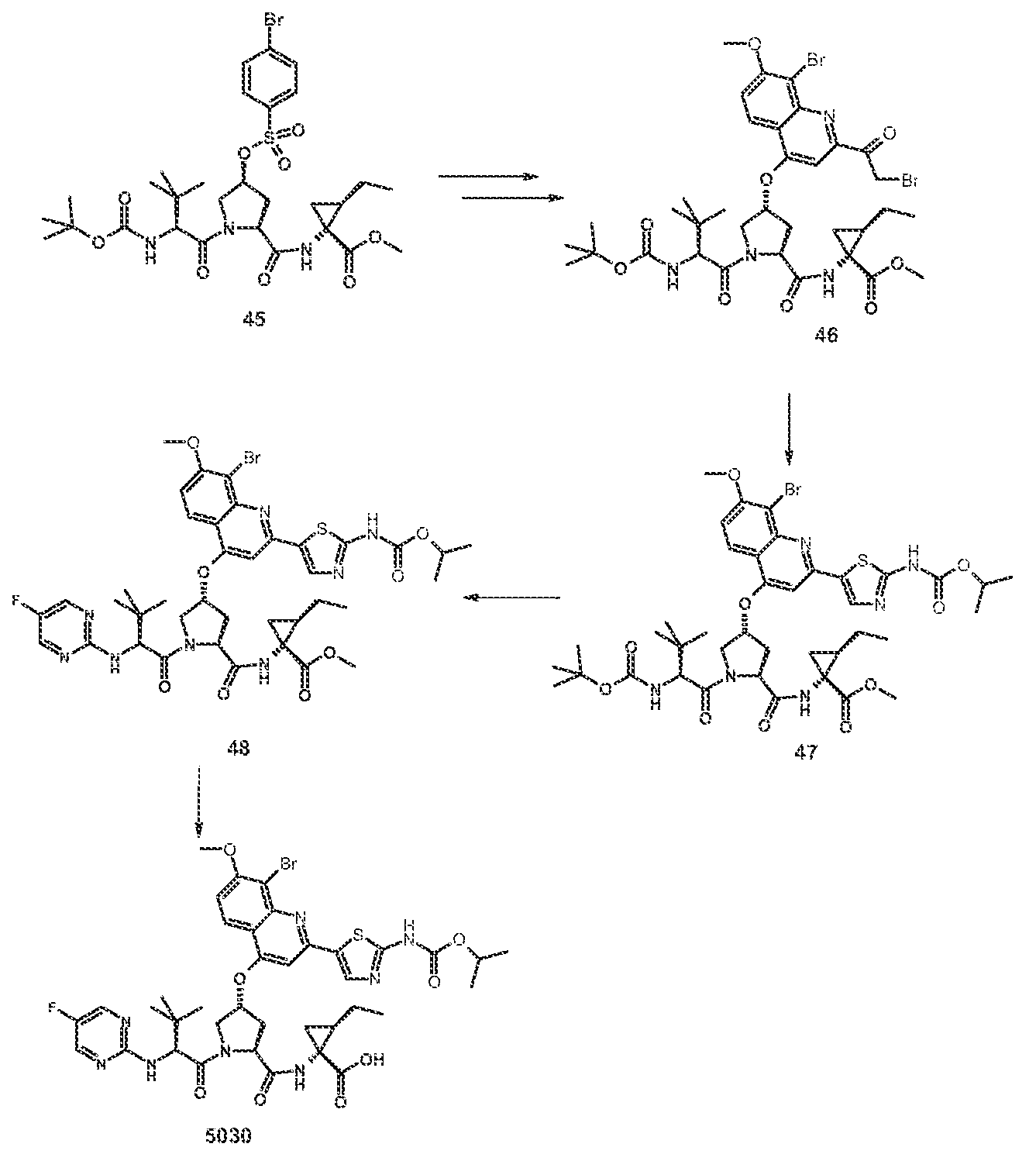
Scheme A describes an alternative synthetic route for the synthesis of compounds of formula (Ij in which R1 is ethyl The vinyl cyiopropane derivative 23 is reduced using Rh/C under 45 ps* of hydrogen gas to provide the ethyl-cyclopropane derivative 24 This compound is then sequentially coupled with protected amino acids 25 and 2a-fo to provide the tπ peptide analog 28 The tπ peptide 28 is converted to the quinohne aminothiazoie derivative 30 using similar transformations to those described in Scheme 1 for the conversion of 3 to 9. 30 can be converted to compounds of formula (i) where R1 is ethyl using similar transformations to those described in Scheme 1 for the conversion of 9 to 10,
Scheme 5
Scheme 5 described a reaction sequence for making compounds of formula (I) where R 1 is cyclopropyi. The vinyl cyclopropane derivative 17 is cyclopropanated using palladium acetate and diazomethane to give intermediate 33 Peptide coupling between the deprotected dipeptide 33 and one of the P3 fragments 2a-h or 19a-h provides tripeptide derivatives which can be converted to compounds of formula (I) using reaction sequences already described m Schemes 1 to 4
Scheme 6
Scheme 6 describes a reaction sequence which permits the conversion of carboxylic acids of formula (I) to the corresponding acyl sulfonamides. The acid is activated by a peptide coupling agent or a chloroformate and is thereby converted to the corresponding aza-lactone. The azalactone is opened by the nucleophilic addition of a sulfonamide to obtain the corresponding acyl sulfonamide analogs.
Scheme 7
Scheme 7 describes a reaction sequence for preparing sulfonamide 42-C used in Scheme 6. Compound 42-A is first reacted with sulfur dioxide, then reacted with N- chlorosuccinimide (NCS) in a suitable solvent at a suitable reaction temperature to provide compound 42-B, which is then reacted with ammonia in a suitable solvent, at a suitable reaction temperature, to provide sulfonamide 42-C. Rs is selected from (C1-β)alkyl, (C^cycloalkyl, (C1-β)alkyl-(C3-7)cycloalkyl, aryl or Het; each of which optionally being mono-, di- or tri-substituted with substituents selected from halogen, hydroxy, cyano, (C1-4JaIkVl1 O-(C1^)alkyl, -CO-NH2, -CO- NH(C1-4)alkyl, -CO-N((CM)alkyl)2, -NH2, -NH(C^)alkyl and -N((CM)alkyl)2, wherein (C1_4)alkyl and O-(C1^)alkyl are optionally substituted with one to three halogen atoms. In another embodiment, Rs is a C3-7cycloakyl group which is substituted by a group selected from C1-1(ralkyl, C3.12-cycloalkyl, aryl, aryl-C1-10-alkyl or heteroaryl.
Scheme 8
Scheme 8 shows an alternative method for preparing sulfonamide 42-C by the reaction of compound 42-A with sulfuryl dichloride, in a suitable solvent, at a suitable reaction temperature to provide compound 42-B, which is then reacted with ammonia in a suitable solvent, at a suitable reaction temperature, to provide sulfonamide 42-C.
Scheme 9
Scheme 9 describes an alternative method for preparing sulfonamide 42-C via an N -substituted sulfonamide intermediate. The compound 42-B is reacted with a substituted amine compound NHR1R2 to provide compound 42-D wherein R1 and R2 are, independently, hydrogen or C1-10-alkyl with the proviso that R1 and R2 are not both hydrogen. Compound 42-D is then reacted with an acid to provide the sulfonamide 42-C.
Scheme 10
Scheme 10 describes a reaction sequence for preparing the certain substituted cycloalkyl sulfonamides 42-F wherein Rs is a C^cycloakyl group which is substituted by an R6 group selected from C1-10-alkyl, C3-12-CyClOaIkVl1 aryl, aryl-C1-10- alkyl or heteroaryl. Compound 42-D is reacted with a base, followed by a halide R6X
wherein X is chioro bromo or iodide to provide compound 42-E, which is then reacted with an acid to provide 42-F sulfonamide
Synthesis of P1 Fragments
P1 moieties of compounds of Formula (!) are prepared using the protocols outlined in VVO 00/59929, published October 12, 2000, and WO 00/09543, published on February 24, 2000 herein incorporated by reference in particular reference is made to pages 33-35, Example 1 of WO00/59929 and Pages 58-69 Examples 9 to 20 of WO00/09543 for the preparation of i-ammocyclopropanecarboxyhc acid P1 moieties
Compounds of formula 5 can be synthesized from commercially available materials using the techniques describee in International Patent Applications WO 00/59929, WO 00/09543, WO 00/09558, WO2004/103996 and U S Patent 6,323,180, ah of which are herein incorporated by reference
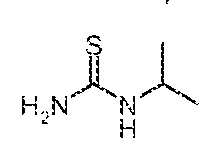
Synthesis of Thioureas 8a~k
Compounds of formula 8a-k can be synthesized from commercially available materials using the techniques described in International Patent Application WO2004/ 103996, nerem incorporated by reference
Synthesis of P3 residues:
Amino acids 2a-h and 18a-h are available commercially from various vendors and were used as received
P3 residues 19a-h were prepared according to Scheme 3 which is described in detail in Example 17, step 3.
[EXAMPLES Temperatures are given in degrees Celsius. Solution percentages express a weight to volume relationship, and solution ratios express a volume to volume relationship, unless stated otherwise. Flash chromatography is carried out on silica gel (Siθ2) according to Still's flash chromatography technique (W.C. Still et a/., J. Org. Chem., (1978), 43, 2923). Mass spectral analyses are recorded using flow injection analysis mass spectrometry. Analytical HPLC is carried out under standard conditions using a SunFire™ C18 3.5μM reverse phase column, 4.6 x 30 mm and a linear gradient (0 to 100% over 5 or 8 min with 2.5 mL/min) employing 0.1%TFA/acetonitrile and 0.1%TFA/water as solvents.
Abbreviations used in the examples include:
Ac: acetyl; ACCA : 1-Aminocyclopropyl-carboxylic acid; BOC or Boc: tert- butyloxycarbonyl; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; DCM: dichloromethane; DIAD: diisopropylazodicarboxylate; DIEA: diisopropylethylamine; DIPEA:
diisopropylethylarninβ' DMF N, Λ/-αιmetnyiformamιde, DMAP 4- (αιmetnyiamιno)pyrιdιne, DMSO dimethylsu!foxide- equiv equivalent, EtOAc ethyl acetate, HATLJ- [O-7-azabenzotrιazo!-1-yl)-1 ,1 ,3,3-telrametny!uronιι_m hexafluorophosphate], hex hexanes, HPLC high performance liquid chromatography. MS mass spectrometry (FiA MS- flow injection analysis mass spectrometry), Me methyl, MeOH methanol, mmoi. rrplhmole, NCS. N- chiorosuccinimide, Ph. phenyl, RT room temperature (18 to 220C), sat saturated, SNAr Nucleophilic aromatic substitution, tert-butyl or t~buty! 1 ,1-dimethyiethyi. Tbg. terf-buty! glycine terf-leucine, TBTU 2-(1 H-benzotrιazole-1-yi)-1 ,1 l3.3-tetramethyi uromum tetrafluoroboratβ" TEA' triethyiamme, TFA tnfiuoroacetic acid, and THF tetrahydrofuran
A mixture of Boc-hydroxyprohne P2 (50 0 g, 216 mmoi), vinyl-ACCA methyl ester P1 (23) (42 25 g, 238 mmoi), TBTU (76 36 g, 238 mmoi) and DiPEA (113 rrsL, 649 mmoi) in DMF (800 iτsL) is stirred at RT under a nitrogen atmosphere After about 3.5 h the solvent is evaporated and the residue is extracted with EtOAc. The extract is washed with HCI (10%) saturated sodium bicarbonate and brine. The organic phase is then dried over MgSO
4, filtered and evaporated to afford an oil After drying overnight under high vacuum, dipeptide 36 is obtained (72 0 g, 94% yield, punty >95% by HPLC)
Dipeptide 36 (72.0 g, 203 mmoi), tripnenylohosphine (63 9^ g, 2^3 8 mmoi) and 4-
nitrobenzene acid (41 08 g, 245 8 mmo!) are dissolved in dry THF (1 4 L) The stirred solution is cooled to 0cC under a nitrogen atmosphere Detn/I azodicarboxylate (38 4 ml_, 244 mmo!) is then added dropwise over about 45 mm and the reaction is allowed to warm to RT After about 4 h, the solvent is evaporated The residue is divided into four portions Each of these is purified by chromatography over tine silica gel (10-40 urn mesh, column diameter 12 cm, column length 16 cm) usmg a gradient of 2 1 hexanes/EtOAc to 1 1 hexanes/EtOAc to pure EtOAc Tne Boc-dipeptide ester 13 is obtained as an amorpnous white solid after evaporation of the solvents ana drying of the residues under nign vacuum at 700C for about 1 h (108 1 g quantitative yield; A solution of 4 N HC! in αioxane is adαed to the Boc-dipeptice ester 13 (108 g 243 rnmoi; Tne solution is stirred at RT for aoout 1 n The solvent is evaporated and the residue placed under nign vacuum for aoout 3 n to afford the hydrochloride salt o1 compound 1 (quantitative yieiα)
EXAMPLE 3: Synthesis of trϊpeptide 3a
Carbamate 2a (121 4 g, 525 mmo!) ana HATU (228 2 g, 2 6 moi; are suspended in DCM (1 L) and the suspension is stirred rapidly DIPEA (91 7 mL, 525 mmo!) is added at RT and after about 10 mm, the reaction is nearly homogeneous A solution of dipeptide 1 (220 0 g, 500 mmol) in anhydrous DCM (3 L) containing DIPEA (87 mL, 500 mmol) is then poured into the reaction The resulting yellow solution is allowed to stir for about 15 h As the reaction is not compiete (as indicated by HPLC), an additional amount of HATU (22 8 g, 260 mmo!), DIPEA (18 mL, 100 πmoi) and carbamate 2a (12 1 g, 52 5 mmo!) are added and the reaction is stirred for about an aααitiona! 5 h The solvent is then evaporated to yieid a yeiiow syrup wnich is extracted with EtOAc (300 + 150 mL; and washed with 0 05 N HCI (2 x 200 mL), saturated Na,CO_.. (300 mL) and bnne (150 mL) The combined extracts are dried over MgSO4 and evaporated to yield the tπpeptiαe 3a (529 g quantitative yield)
EXAMPLE 4: Synthesis of tripeptide 4a
The cruce tπpeptiαe 3a (529 g, 0 79 moi) is dissolved in THF (3L) and water (800 rnL) is added The resulting solution is cooled to 0 'C ana a solution of lithium hydroxide monohyαrate (1 11 1 g, 0 99 mol) is aαded over about 3 mm with vigorous stirring After approximately 3 h at 0°C, the excess base is neutralized with 1 N HCi (final pH -6) and the THF is evaporated, resulting in an aqueous suspension (yellow gum). The mixture is extracted with EtOAc (2 x 200 mϋ and is washed with saturated NaHCO3 (2 x 300 mϋ The combined extracts are dried over MgSO4 and are evaporated to yield a pale yellow foam Flash chromatography of the foam over silica gel using 6 4 EtOAc-Hex to 8 2 EtOAc-Hex as the eluent affords 4a (180 g, 43% yield based on crude mass of SM)
Tnethyiamme (187 m!_, 1.34 mol) is added dropwise to a cooled solution (O0C) of the tπpeptide 4a (18Og, 0 385 moi), 4-broπo-benzenesulfonyl chloride (208 6 g. 0 809 mol) and dimethyiaminopyridine (4 7 g. 38 5 mmoi) dissolved in DCM (2 L) The yeϋow solution is stirred for about 1 h at 00C before slowly warming to RT. The solution is then stirred for about 60 h at RT. The reaction mixture is concentrated to dryness, diluted with EtOAc, washed with saturated sodium bicarbonate solution water and brine, dried over MgSO4, filtered and evaporated to dryness to obtain the crude product (50 g) The crude material is purified by flash column chromatography with hexanes EtOAc, 40 80 to 20 80 to provide the pure product 37a (170 g, 84% yield)
EXAMPLE 6: Synthesis of compound 1005
Introduction of the quinoline moiety onto the tripeptides
The brosylate 37a (2 O g 2 91 mmoi), bromoquinohne 5a (1 1 g 3 58 mmoi} and ground cesium caroonate (1 9 g, 5 83 mmo!) are dissolved in 1-methyl-2- pyrrohαmone (37 mL) Tn e solution is neated to 7ϋ°C and stirred for about 7 h, then stirring is continued at RT overnignt The reaction mixture is poured into EtOAc, washed with H^O (3xj, saturates NaHCO^ (2x) H2O (2x), bnne (1x), dπed over MgSOA, filtered and concentrated to afiord the crude proαuct (2 1 g) Purification by Hash column chromatography (1 30 sihca gel, 5 5 EtOAc/hexanesj afioms pure product 8a (84% yeid, 1 86 g)
Selective monohydrolysis of ester 6a
Tπpeptide 6a (1 88 g 2 44 mmoi) in 30 mL of a 2 1 1 mixture of THF-MeOH- H
2O, >s cooied to O 'C anc a 1 N NaOH aqueous solution (2 8 mL, 2 58 mmoi) is added The resulting solution is stirred for about 15 mm at 0
0C, then stirring is continued for about 1 5 h at RT As the reaction is found to be incomplete (as indicated by HPLC), an additional 1 N NaOH (0 1 mL, 0 1 mmoi) is added and the reaction is stirred for about an additional 1 5 h. The mixture is evaporated to near dryness diluted with water, frozen and iyophihzed to provide the acid 38 (crude material used for next step. 1 84 g, 2 39 mmoi)
StejD_3 Synthesis of diazoketone 39
Sodium salt 38 (1.84 g, 2 39 mmoi) is dissolved m THF (40 mL). tπethylamine (467 uL, 3 35 mmoi) is added and the solution is cooied to 0"C isobutylchloroformate (434 μL 3 35 mmoi) is added dropwise and the white suspension is stirred at 0=C for about 1 5 n, followed by the addition of a solution of diazoπethane (0.87 M in diethyl ether, 22 mL, 14 70 mπoi) The reaction mixture is stirred for about 60 mm at O11C, then the mixture is warmed to RT and stimng is continued for about 1 h. The reaction mixture is evaporated to provide a thick suspension This suspension is dissolved in EtOAc, washed with saturated NaHCO3 (2x) brine (1x) dried over MgSO^, filtered and evaporated to give the crude diazoketone product 39 (2.1 g) Purification by flash column chromatography (1 30 silica gel, 4 6 EtOAc/hexanes) affords pure product 39 (87% yield, 1 60 g)
Synthesis of bromoketone 7a
At 00C, to a solution of chazoketone 39 (1 60 g, 2 07 mmo!) m THF (38 mL) is adαed dropwise an HBr solution (48% aqueous, 1 5 mL) and the mixture is stirred for about 1 25 h The mixture is quenched with a saturated NaHCO3 solution, then the THF is evaporated The residue is diluted with EtOAc, washed with a saturated NaHCO3 solution (2xj, bnπe (1x), dried over MgSO^, filtered and evaporated to provide the crude bromoketoπe 7a (1 72 g, 100% y!e!d)
Synthesis of thiazoiyl tπpeptide 9a
α-Bromoketone 7a (700 mg, 0 85 mmoij and thiourea 8h ( 134 mg, 1 02 mmol) are dissolved in isopropanoi (5 mLj and the yellow solution is heated at 75°C lor about 1 n Tne solution is allowed to cooi Io RT and evaporated to dryness Tms crude material 9a is used as such for next step
Deboc and Couphng of 9a
The crude 9a (107 mg, 0 125 mmol) is dissolved in 4 N HCI/dioxanes (2 mL) and
stirred at RT for about 1 h tnen evaporated to dryness under reduced pressure The materia! is co-evaporated twice with DCM The materia! is then reαissoived in DCM (3 ml), DiPEA (0 087 ml_. 0 5 mmoi), cyclopropyl acetic acid (0 013 mL 0 14 mmoi) and HATU (57 mg, 0 15 mmoi) are added and the reaction is stirred for about 18 n at RT The reaction mixture is diluted with EtOAc, washed with NaHCO3 (saturated), H2O, and brine then dried over MgSO4, ήitered and concentrated to dryness to obtain 10a ( 103 mg, 99% yieid) that is used as such in the subsequent reaction
. Hydro!ysιs of ester
To a solution of metnyi ester 10a (104 mg, 0 124 mmoi), in a 3 0 mL mixture of THF H2O (2.1 ), is added 1 N NaOH (1 24 mL, 1 2& mπo!) 1 πL of MeOH is required to obtain an homogeneous solution The resulting reaction is stirred at RT overnight The organic soiution is concentrated to provide a brown oii. The crude materia! is purified by preparatory HPLC (YMC Combiscreen ODS-AQ, 50 x 20 mm ID S-5 micron, 120 A, λ- 220 nm) using a linear gradient and 0.08% TFA CH3CN / H2O The pure fractions are combined, concentrated and lyophiiized to provide the product 1005 as the TF salt (44 mg, 38% yield)
EXAMPLE 7: Synthesis of compound 2008
Compound 1003 is prepared using the protocol descπoed in Example 8, sleps 1 to 7
and using
as the thiourea in Step 5
Step 8 Reduction of Vmyi ACCA
To a solution of compound 1003 (35 mg 0 04 mmol) in MeOH (0 4 m!_) is added nydrazine monohydrate (0 1 ml_, 2 08 mmo!) The reaction mixture is heated at 6QCC while air is oubbied lhrougn the reaction mixture The mixture is stirred at 60cC 1or about 4 5 n, then cooied to RT and concentrated to dryness in vacuo The materia! is redissolved in 1 1 DMSO MeCN, two drops ot AcOH are aαded and tne product is purified by preparative HPLC (YMC Combiscreen ODS-AQ, 50 x 20 mm ID S-5 micron, 120 A, λ= 220 nm, 30 to 45% MeCN, 0 1 % TFA) The appropriate fractions are combined, frozen and lyophylised to give compound 2008 (17 mg ^9% yield)
Compound 15b is prepared using the protocol described in Example 6, steps 1 to 5
using
(8b) as the thiourea in Step 5 and by using P3 fragment
(2b) during the synthesis of the tripeptide according to the procedure in example 3.
Steps 6-7:
15b is converted to 2001 using protocol described in Example 7, steps 6 and 7, using cyclobutyl acetic acid instead of cyclopropyl acetic acid.
Compound 2001 is converted to compound 2008 using a procedure analogous to that described in Example 7, Step 8.
EXAMPLE 9: Synthesis of compound 1008
Compound 1008 is prepared using the protocol described in Example 6, steps 1 to 7
and using
(8b) as the thiourea in Step 5 and by using
in place of cyclopropyl acetic acid in Step 6.
EXAMPLE 10: Synthesis of compound 1034
Compound 1034 is prepared using the protocol described in Example 6, steps 1 to 7
and using
(8b) as the thiourea in Step 5 and using
in place of cyclopropyl acetic acid in Step 6.
EXAMPLE 11 : Synthesis of acyl sulfonamide Analogs in table 3
EXAMPLE 11a: Synthesis of compounds 3002 and 3004
Step 1: Hydrolysis and Synthesis of Aza-lactone
Ester 40 was prepares according to tne syntnesis described in Example 6 sleps 1 through 5 using the C8 methyl analog ot 5a in step 1 anc tniourea 8b in step 5 Further details describing the synthesis of tne C8 methyl analogs o1 5a can be found in WO2004/103996 and U S patent 6,323,180. aϋ of which are herein incorporated by reference
Compound 40 (800 mg, 0 77 mmol) is dissolved in THF (4 0 mL), MeOH (2 0 mU and NaOH (7 7 mL of a 1 0 N solution 7 7 mmol) are added and the reaction is stirred for about 8 h at RT The reaction mixture is concentrated almost to dryness in vacuo The residue is taken up in EtOAc and acidified to about pH 5 5-8 0 with 1 0 N HCi The mixture is transferred to a separatory funnel and the aqueous phase is extracted with EtOAc (3xj The combined organics are washed with H?O and bnne and then dned over MgSO^, filtered and concentrated in vacuo to give a foam (507 5 mg, 86% yield) The foam (507 5 mg, 0 66 mmol) from above is re-dissolved in DCM (5 mL) and Et,N (0 31 mL, 2 2 mmol) and tne solution is cooked to O0C in an ice batn
IsoDutylchioroformate (0 129 mL, 1 0 mmol) is added dropwise anc tne reaction is stirred at O0C for about 1 h and then at RT for about 16 h The reaction mixture is evaporated to dryness to obtain azalactone 41 as a solid (0 715 mg, quantitative)
St§j)_2: CycloproDyisulfoπamide 42 is commercially avauabie, or can be prepared according to step 2A below
Step 2A Synthesis of cyciopropyisulfonamide 42
To a solution of cyclopropylmagnesium bromide (0 5 M 20 mL, 10 0 mmol) in anhydrous THF (10 mL) at about -100C is added a solution of SO; in THF (~ 16 wt%, 4 8 mL, 12 mmo!) slowiy over 10 mm at -10 to -5°C Tne reaction mixture is warmed to ambient temperature over 0 5h, and then NCS (2 O g 15 mmo!) is added at about -5 to 0°C The reaction mixture is warmed up to ambient temperature and diluted with 50 mL oi methyl terl-buty! ether To tne reaction mixture ι≤ added 50 mL water and the mixture is stirred for 5 mm The organic layer is washed with 50 mL of bnne The organic layer is concentrated and the resultant cyclopropylsuifony! chloride is dissolved in CH2Ci2 (total volume was about 50 mL) and ammonia gas is bubbled in at about O'C for about 5 mm and the mixture is slowly warmed up to ambient temperature and stored at that temperature for 2 h The mixture is filtered
through Cehte to remove the solid (NH4C!), and the filtrate is concentrated to obtain a crude cyclopropyisulfonamide solid (~ 1 2g) Re-cryslailization of the crude product from EtOAc/hexane produces cyclopropyisuifonarnide 42 in 80% overall yield.
Step 2B. Synthesis of Acyi Sulfonamide
Sulfonamide 42 is dissolved in anhydrous THF (8 5 ml.) in an oven dried flask and cooled to -150C. LiHMDS (0 66 mL of a 1.0 M solution in THF, 0.86 mmoi) is added all at once and the reaction is stirred for about 5 mm, warmed to RT for about 20 mm and then re-cooled to -150C The crude aza-lactone 41 (0 33 mmoi) is dissolved in anhydrous THF (3 mL) and added dropwise to the above solution over about 10 mm The cold bath is removed and the reaction is allowed to warm to RT and stirred for about 16 h The reaction is quenched by the addition of a few drops of giacia! HOAc and the reaction is stirred for about 5 mm and then concentrated to dryness in vacuo The residue is redissoived in THF (3 mL), MeOH (1 5 mL) and 1 N NaOH (2 mL, 2 mmo!) are added and the reaction is stirred for about 4 h at RT The reaction is concentrated in vacuo and the residue is taken up in EtOAc 1 0 N HCI is added to give approximately a pH of 3 and the aqueous phase is extracted with EtOAc (3x) The combined organics are washed with H2O, bπne and then dried over MgSO4. filtered and concentrated in vacuo to give a solid 43 (296.8 mg, quantitative yield).
StegJL Deboc and coupling of Amide fragment
The soϋd 43 (297 mg. 0 33 mmoi) is re-dιsso!ved in 4 N HCI m dioxanes (2 mL) and the reaction is stirred for about 30 mm at RT and then concentrated to dryness in vacuo Half of this materia! (0 17 mmoi) is re-dissolved in DCM (2 mL), DIPEA
(0 144 mL, 0.83 mmo!), cyciopropy! acetic acid (0 018 mL, 0 183 mmoi) and HATU (76 mg 0 199 mmo!) are added and the resulting reaction is stirred for about 16 h at RT The reaction mixture is diluted with EtOAc, washed with 10% citric acid (2x), H2O (2x), bπne (1x), dπed over MgSO4, filtered and concentrated in vacuo to obtain a sohd (153 mg, 0.17 mmoi). HaH oi this materia! (0.085 mmoi) is purified by prep HPLC, frozen and iyophilized to give 3002 as a white fluffy solid (42%)
Steρ_4_ Reduction of vinyl-ACCA
3002 is reduced using the hydrazine monohydrate procedure described in Example
6, step 8 to provide compound 3004 in 20% yield.
EXAMPLE 11b: Synthesis of compound 3001
Compound 3001 is prepared using the protocol described in Example 6, steps 1 to 7
and using
(8c) as the thiourea in Step 5 followed by steps 1 to 3 of Example 11a.
EXAMPLE 11c: Synthesis of compound 3003 Compound 3003 is prepared using the protocol described in Example 6, steps 1 to 7
and using
(8b) as the thiourea in Step 5 followed by steps 1 to 3 of
Example 11a. In step 3
\
S use{j j
n p|
ace of cyclopropyl acetic acid.
EXAMPLE 11d: Synthesis of compound 3006 Compound 3006 is prepared using the protocol described in Example 6, steps 1 to 7
and using
(8b) as the thiourea in Step 5 followed by steps 1 to 3 of
Example 11a. In step 3
is used in place of cyclopropyl acetic acid.
EXAMPLE 11e: Synthesis of compound 3005 Compound 3005 is obtained by reducing 3003 using the protocol described in Example 6, step 8.
EXAMPLE 11f: Synthesis of compound 3007
Compound 3006 is reduced to provide 3007 using the protocol described in Example 6, step 8.
EXAMPLE 12: Synthesis of compounds in which B is a pyrimidine using an
SNAr reaction
EXAMPLE 12a: Synthesis of compound 4001
Steps 1-5-
Compound 9b is prepared according the protocol in Example 6, Steps 1 to 5 using
SteβjL Deboc and Coupling of 9b
The crude 9b (1 2 g, 1.35 mmo!) >s dissolved in 4N HCI/dioxanes (20 nil) and stirred at RT for about 1 h then evaporated to dryness under reduced pressure The materia! is co-evaporated twice with DCM The material is then redissolved in EtOAc, and washed twice with saturated NaHCO3, dried over MgSθ4 filtered and concentrated in vacuo to give 1 O g of the neutralized amine 2-ch!oro-5-fluoropyπmιdme (0 664 mL, 5 38 mmo!) is dissolved in DMSO (16 5 mL), K3PO4 (0 343 g, 1 61 mmol) and TBAF (3 36 mL oi 1 0 M solution m THF 3 36 mmol) are added and the mixture is heated to 70 C for about 2 h The amine from above (1 0 g, 1 35 mmo!) is dissolved in DMSO (18 5 mL) and added dropwise to the pynmidine solution over about 25 mm The reaction is aged for about 65 h, then cooled to RT, diluted with EtOAc, washed with saturated NaHCO3 (2x), H2O (2x) and brine (1x). then dπed over MgSO4. filtered and concentrated in vacuo The residue is purified by fiash chromatography using hexanes/EtOAc (4/1 ) to give compound 44 (556 mg, 49% yield) as a pale yellow solid
Stej3_7_Hyd roiysis of ester
To a solution of methyl ester 44 (1.5 g, 1.79 mmol), in a mixture of THF (10 ml_), H2O (5 ml.) and MeOH (5 mL), is added 1 N NaOH (17.9 ml_, 17.9 mmol). The resulting reaction is stirred at RT overnight. The organic solution is concentrated in vacuo, redissolved in EtOAc, acidified with 1.0 N HCI to approximately pH 5.5-6.0, and the aqueous phase is extracted with EtOAc (3x). The combined organ ics are washed with H2O1 brine, then dried over MgSO4, filtered and concentrated in vacuo. The crude material is purified by flash chromatography using hexanes/EtOAc (8/2) as the eluent to give 4001 (1.13 g, 77% yield).
EΞXAMPLE 12b: Synthesis of compounds 4003, 4012, 4014 and 4017 These compounds are prepared from 9b using the protocol described in Example 12 steps 6 and 7, but replacing the 5-fluoro-2-chloropyrimidine with the appropriately substituted 2-chloro or 2-bromo pyrimidine.
EXAMPLE 12c: Synthesis of compounds 4002, 4004, 4005, 4006, 4007, 4008, 4009, 4010, 4011, 4013, 4015, 4016 and 4018
These compounds are prepared using the protocol described in Example 12a, Steps 6 and 7. The starting materials (9a-k analogs) for these compounds are obtained using the protocol described in Example 6, Steps 1 to 5 by using the appropriate thiourea (8a-k) in Step 5.
EXAMPLE 13: Reduction of vinyl-ACCA EXAMPLE 13a: Synthesis of compound 5009 Step 1: Reduction of Vinyl ACCA:
To a solution of compound 4001 (400 mg, 0.48 mmol) in MeOH (25 mL) is added hydrazine monohydrate (1.76 mL, 36.3 mmol). The reaction mixture is heated at 600C while air is bubbled through the reaction mixture. The mixture is stirred at 600C for about 5 h, then cooled to RT and concentrated to dryness in vacuo. The material is purified by flash chromatography using 7/3 hexanes/EtOAc as the eluent to yield 5009 (196 mg, 49% yield).
[EXAMPLE 13b: Synthesis of compounds 5010, 5011, 5015, 5016, 5017, 5021, 5022, 5023, 5025, 5028, 5029 and 5030
These compounds are also prepared by reducing the appropriate vinyl-ACCA derivative using the protocol described in Example 13a for the reduction of 4001. The reduction procedure can also be carried out on the ester (ie compound 44) followed by alkaline hydrolysis to give the carboxylic acid inhibitors although the yields are lower for this route.
EXAMPLE 14: Alternative synthesis of compounds containing ethyl-ACCA, illustrated by synthesis of compound 5030
Rather than performing a reduction at the end of the synthesis, the reduction of the vinyl cyclopropane to the ethyl-cyclopropane can be carried out early in the synthesis.
Step 1 : Reduction of vinyl cyclopropane amino ester
The tosyiate salt of vinyl-ACCA methyl ester 23 (5 0 g) and Rh/C (6 mole %) is dissolved in 50 mL of MeOH letrahydrofuran (1 3) and tne reaction is placed unαer a hydrogen atmosphere at 45 psi lor 3 5 hours under mechanical stirring (450 rpm) The reaction is complete (by hydrogen consumption monitoring) after 3 hours Alter nitrogen purge the reaction vessel is depressuπzed and the suspension is filtered through a piug of CehteΘ and the plug is washed with 100 mL methanol The combined filtrates are stnpped of solvent under reduced pressure and the residue is slurried with 20 mL EtOAc and the solid is ήitered off After drymg under vacuum, 4 5 g (90%) of white powdery solid (compound 24) is isolated.
Compound 45 can be prepared in a variety of ways One route involves utilizing the procedure for tne synthesis of 37a described in Examples 1 througn 5 and usmg
ethyl-ACCA analog 24 instead oi the vinyi-ACCA analog 23 for the preparation of the αipeptide described in Example 1
Compound 47 is prepared from 45 using the protocoi describee in Example 6, Steps
to 5 and using
(8k) as the thiourea in Step 5
Steβ_2 Deboc neutralization and Pyπmidme SNA'- (Synthesis of Compound 48)
Compound 47 (7 01g 6 05 mmoi) is dissolved in CHCi, (10 mL) ana 4N HCI in dioxanes (35 mL) is addeα The reaction is stirred for three hours at RT then concentrated in vacuo Io give the hydrochloride salt as a yellow solid Tnis solid is suspended in EtOAc (300 mL) and sat NaHCO0 (150 mL) is added and the mixture is stirred for 15 minutes until the solid is dissolved Tne mixture is transierred to a separatory funnel and the organic phase is washed once with NaHCOj. once with H2Q and once with brine, then dπed over MgSQ/j, filtered and concentrated in vacuo to give the neutralized amine as a tan gum in a separate flask 2-chioro-5-fiuoro pyπmidine (2 99 ml, 24 2 mmo!) is dissolved in DMSO (78 mL), K0PO4 (1 54 g 7 26 mmol) and TBAF (15 12 mL 1 0 M solution in THF, 15 12 mmol) are added The mixture is heated to 75LC and stirred for one hour
The gum from above is ^dissolved in DMSO (78 mL) and added dropwise to the above mixture over 25 minutes The reaction vessel is protected from hght and stirred at 750G for 42 hours The reaction is cooled to RT, diluted with EtOAc, washed twice with H2G, twice with 10% αtπc aciα, twice with sat NaHCO<, twice with Drine, tnen dried over MgSO1, filtered and concentrated in vacuo to give 5 7 g of compound 48 as a tan foam
Step 3 Hydrolysis
Compound 48 is dissolved in THF (55 mL) and MeOH (26 mL), then 1 0 N NaOH (80 5 mL, 60 5 mmoi) is added and the reaction is stirred at RT for 18 hours The reaction mixture is concentrated in vacuo then dissolved m H2O and then acidified to pH ~4 with 1 0 N HC! The materia! is then extracted with EtOAc (3x) The combined orgarpes are washed with H-O, brme and then dried over MgSO-., filtered and concentrated in vacuo
The yellow orange solid is purified by flash chromatography 7/3 hexanes/EtOAc, followed by trituration from Et2O to give 1.30 g (25 % yield) of the final compound 5030 as a pale yellow solid.
EXAMPLE 15: Synthesis of compound 5011
Compound 5011 is prepared using the protocol in Example 14a, steps 1 to 3 using
(8f) as the thiourea in the aminothiazole forming step (ie. conversion of 46 to 47).
EXAMPLE 16: Synthesis of compound 5023
Step 1: Cyclopropanation
Compound 49 (200 mg, 0.274 mmol) is dissolved in CH2CI2 (1 mL), diazomethane (4.1 mL of a 0.67 M solution in Et2O) is added and the mixture is cooled to -2O0C. Pd(OAc)2 (6.15 mg, 0.027 mmol) is added portionwise over 5 min. The reaction is
stirred for 4 hours at -2O0C and allowed to warm to RT and stir two hours at RT. The reaction is concentrated in vacuo and then purified by flash chromatography using 40% hexanes / EtOAc. The product 50 is obtained as a yellow foam (149 mg, 73% yield).
Step 2: Coupling with P3
Compound 50 (149 mg, 0.20 mmol) is dissolved in 4N HCI/dioxanes (3 ml_) and stirred for one hour at RT1 then concentrated in vacuo. The residue is redissolved in CH2CI2 and added to a mixture of Boocyclopenyl-L-Glycine 2b (54 mg, 0.22 mmol) in CH2CI2 (2 mL), HATU (91 mg, 0.24 mmol) and DIPEA (0.173 ml_, 1 mmol). The reaction mixture is diluted with EtOAc, washed with NaHCO3 (2x), water (2x), brine, dried over MgSO4, filtered and concentrated in vacuo to provide 180 mg of crude material that was used as such in step 3.
Step 3: Deboc. neutralization and SNAr with 2-chloro-5-fluoro pyrimidine These steps are carried out according to the protocol described for Step 2 in Example 14a to provide compound 51.
Step 4: Hydrolysis This step is carried out according to the protocol described for Step 3 in Example 14 to provide 5023.
EXAMPLE 17: Alternative synthesis of compounds in which B is phenyl or heterocyclic
EXAMPLE 17a: Synthesis of compound 6006
Step 1: Hydrolysis and P1-P2 coupling
Ester 52 (5 Og, 10 77 mmoij is dissolved in THF (100 mL) and MeOH (30 mL) 1 0 N NaOH (30 mL 30 mmo!) is added and the reaction is stirred for three hours at RT The reaction mixture is diluted with H
2O and concentrateα to remove the THF and MeOH The remaining aqueous solution is acidified with 1 0 N HCi and then extracted twice with EtOAc, the combined organics are dπed over MgSO
4, filtered and concentrated in vacuo
The resulting acid is redissolved in CH3CN ( 10 mL), Et3N (6 0 mL ^3 1 mmol) and HATU (4 1g 10 77 mmol) are added and the mixture is stirred for five rmn The amine 24, (3 4 g, 10 8 mmo*) is added and the reaction is stirred overnight at RT The mixture is concentrated in vacuo, the residue taken up in EtOAc, washed twice with 1 ON HCI, twice with 1 0 N NaOH dπeα over MgSO4, filtered and concentrated in vacuo Tn e crude material (6 3 g, quantitative yield) is useα as such in subsequent reactions
Step 2 Brosylate Displacement
Brosylate 53, (3 5g, 6 1 mmol) >s dissolved in NMP (10 0 mL) and Cs2CO3 (1 98 g, 6 1 mmol) is added The quinolone 54 (6 1 mmol) is added and the reaction is heated to 85~C for 16 hours The reaction mixture is cooled to ambient temperature then poured into EtOAc, washed with with 1 0 N HCI (2x), 1 0 N NaOH (2x), dπed over MgSO^, filtered and concentrated in vacuo Purification by fash chromatography using 30 - 100% EtOAc /hexanes as the eiuent provides compound 55 as a pale yeϋow soϋd (2 6 g 56 % yield)
StefjjB Preparation of N-Ary! Derivatives
iodo-benzene (4 0 mL 35 9 mmol) L-tert Leucine (4 71 g, 35 9 mmoi), CuI (2 05 g, 10 8 mmol) and Cs^CQ5 (17 55 g, 53 9 mmo!) are added to a rounα bottom 1iask containing DMA (35 ml) and the reaction is heated to 12CT C and vigorously stirred for 18 hours Tne reaction is cooled to RT and diluted with H2O (100 mL) the resulting solution is filtered through cehteΘ to remove any insolubles, the cehte pad is πnsed with another 50 mL of H2O The resulting aqueous solution is neutralized to pH ~ 6 with 1 0 N HCI The precipitated mateπa! is extracted with EtOAc (2 x) and the combined organics are washed with H2O (3x), dried over MgSO*, filtered and concentrated in vacuo The material is dried under high vacuum and the green sohd (6 4g, 86% yield) is used as such in subsequent reactions
Stej3_4 Deboc, P1 P2 - P3 coupling and hydrolysis
The boc-protected amine 55 (1 5g, 1 97 mmol) is dissolved in 4N HCI/dioxanes (30 mL) and stirred for two hours at RT The reaction is concentrated in vacuo and dried under high vacuum to give the amine hydrochloride as a bright yeliow solid In a separate flask the acid 56 (0 49 g, 2 37 mmoi) is dissolved in acetoπitπle (20 mL). Ef3N (1 1 mL, 7 89 mmoO and HATU (0 90 g, 2 37 mmol) are added and the reaction is stored for 10 minutes The hydrochloride salt is added as a solution in CH3CN and the resulting couphng reaction is stirred for three hours The acetonitrle is removed in vacuo and the residue is taken up in EtOAc, washed withi 0 N HC! (3xj, 1 0 N NaOH (2xj, dned over MgSθ4, filtered and concentrated in vacuo The
resulting yellow solid is triturated from Et2O (100 mL) to give a very fine solid which is collected by suction filtration (quite slow since the product is a very fine sohα) and is wasned with nexane≤ then dried unαer high vacuum to give 1 3 g of a pale yellow soiid (> 98% homogeneous) The pale yellow solid is redissolved in THF/MeOH, 1 0 N NaOH is added and the solution is stιτed overnight at RT The soiution is concentrated in vacuo, redissolved in H:O and the mixture is neutralized to pH ~5 with 1 0 N HCI The solid which precipitated out is taken up in EtOAc, washed with H?O (2x), dned over MgSO4 filtered and concentrated in vacuo The residue is triturated with Et7O to give a very fine solid again This solid is couected by suction filtration (slow) and washed with hexanes and dried under high vacuum The solid is taken up in CH3CN, HpQ is added and the mixture is frozen and lyophyhzed to give compound 6008 as a pale yeϋow very fluffy solid (1 Ig1 60% yield)
EXAMPLE 17b: Synthesis of compoursd 4021 to 4048 and 6001 to 8008
These compounds are prepared using a protocol described in Example 17a, steps 1 to 4 The appropriate commercially available aryi or heteroaryl halide is used in step 3 to prepare the P3 fragments 19a~h. For the preparation of compounds 4021 to 4048, compound 23 is used in step 1 instead of compound 24 and the aminotrnazole is installed through the bromoketone as described m Example 6. steps 1 to 5. For
step 5
(8b) is used as the thiourea
EXAMPLE 18: CeH~based iirøferase reporter HCV RMA Replication Assay CeH culture
Hiιh-7 cells with a stable subgenomic HCV rephcon that encodes a modified iuciferase reporter gene (expressed as a iuc!ferase-FMDV2A-neomycιn phosphotransferase fusion gene) are established as previously described (Lohman et a!., 1999. Science 285 1 10-1 13. Vroljik et al . 2003 J Virol Methods 110 201-209. all of which are herein incorporated by reference ), with the exception that rephcon ceϋs were selected with 0 25 mg/mL G4 \ 8 The amount of iuciferase expressed by selected ceils directly correlates with the level of HCV replication These cells designated as MP- 1 ceils, are maintained m Dulbecco's Modified Earie Medium
(DMEM) supplemented with 10% FBS and 0 25 mg/mL G418 (standard medium) The ceils are passaged Dy trypsimzation and frozen in 90% FBS/10% DMSO Duπng the assay DMEM medium supplemented witn 10% FBS, containing 0 5% DMSO and lacking G418, was used (Assay medium) The day of the assay, MP- 1 ceils are trypsinized and diluted to 15 000 celis/70 uL in assay medium 70 μL is distributed into each we*! of a black 96-weli ViewPiate M (Packard) The plate is then incubated at 37°C until the compound is added
Preparation of test compound
The test compound in 100% DMSO is first diluted in assay medium to a final DMSO concentration of 0 5% The solution is somcateα for 15 mm Into coiumn 3 of a Polypropylene Deep-Well Titer Plate, the appropriate volume is transferred into assay medium to obtain the starting concentration (2x) to be tested In columns 4 to 11 add 400 μL of assay medium (containing 0 5% DMSO) Senal dilutions (1/3) are prepared by transferring 200 μL from column 3 to coiumn 4, then from column 4 to coiumn 5. serially through to column 1 1 Column 12 is the no inhibition control
Addition of test compound to cells
A volume of 70μL from each well of the compound dilution plate is transferred to a corresponding well of the Cell Plate (Three columns wil* be used as the "No
inhibition control", nine columns are used for the dose response) The ceil culture plate is mcubatec at 37°C with 5% CO2 for 28 hours
Following the 28 n incubation period, the medium is aspirated from the 96-well assay piate and a volume of 50 μL of 1X Gio Lysis Buffer (Promega) previously warmed to RT was added to each well. The plate is incubated at RT for 10 mm with occasional shaking A black tape was put at the bottom of the plate 50 μL of Bright- Gio luciferase substrate (Promega) previously warmed to RT is added to each well followed by gentle mixing The luminescence is determined on a Packard Topcount instrument using the Data Mode Luminescence (CPS) with a count delay of 1 mm and a count time of 2 sec
The luminescence determination (CPS) in each well of the culture plate is a measure of the amount of HCV RNA replication m the presence of various concentrations of inhibitor The % inhibition is calculated with the following equation % inhibition = 100- [CPS (inhibitor) / CPS (control) x 100]
A non-hnear curve fit with the Hn! model is applied to the inhibition-concentration data, and the 50% effective concentration (EC5o) is calculated by the use of SAS software (Statistical Software, SAS institute, inc Gary, N. C )
The compounds of Tables 1 to 8 were found to have EC50 values of 50 nM or less when tested in the cell-based assay described above The results are shown in tables 1 to 8, wherein the listed EC50 values are rounded to one significant digit after the decimal pomt
EXAMPLE 19: Pharmacokinetic profiling in rats
Oral vβhseϋβ arsd compound preparation;
Compounds are dosed at 5 mg/kg using a suspension consisting of 1 %N~Methyi-2~ Pyrrαhdone, 0 5% aqueous methyiceliuiose and 0.3% of polyαxyethylene (20) sorbitan monooleafe (Tween~80). The dosing volume is 10mL/κg via oral gavage.
Dosing and plasma sarnpHrsg:
Male Sprague-Dawley rats are used for ai! experiments. Their right carotid artery is cannulated at least 16 hr prior to dosing. Animals are fasted overnight, with access to 10% dextrose in water. Biood samples are collected at 0. 0 25. 0.5, 1. 1.5, 2, 3. 4, 6 and 8 hours post-dosing, and pooled from 3 rats at each time point Plasma is separated by centnfugation and frozen at -20"C prior to analysis
Compound extraction arsd analysis:
Plasma samples are thawed and standard curves ranging kom 0 01 to 20 μM are prepared from blank plasma spiked with compound. All samples are extracted by solid phase extraction on Oasis HLB 30 mg/1cc cartridges according to the following method.
Condition Cartridges' 1 mL 5% NH4OH in acetonitπie
1 mL acetonitπle 1 mL miili-Q water
Load 5μL 20% H3PO^ in water, 50 μL plasma, 150 μL miϋi-Q water,
50 μL acetomtπie (vortex prior to loading)
Wash Cartridges 1 mL miϋi-Q water
1 mL 5% methanol in water 1 mL 2% CH3GOOH in methanol: acetonitrile: water (1 :1 8)
EiLite 2 x 500μL 5% NH4OH in acetomtπie
Evaporate to dryness under nitrogen flow using a TurboVap LV at 45C'C
Residue is reconstituted in 500 μL of 0 2% NH4OH in acetonitnie-water (1.1 )
Samples are analyzed by LC/MS-MS (Thermo Finnigan TSQ AM Quantum Ultra) and plasma concentrations are estimated based on the standard curves
When assayed in the preceding screen the compounds below are found to be present at significant levels in the plasma of rats Based on the observed plasma
concentration vs time profile, tne pharmacokinetic parameters (Cmax and AUC) are caicυated using ToxKin V3 3 and are reported in the following taoie, wherein the values are rounded to one significant digit after tne αecima! point
Representative compounds according to this invention are listed in Tabies 1 to 8 wherein Me defines methyl All of the compounds in Tables 1 to 8 are synthesized analogously to the Examples described above A person skilled in the art will recognize that obvious moddications to the Examples are required to generate tne compounds in Tables 1 to 8
TABLE 1
Each reference, including ai! patents, patent applications, ana publications cited in the present application is incorporated herein by reference in its entirety, as if each of them is individually incorporated Further, it would be appreciated that, in the above teaching of invention, the skilled in tne art could rτiaκe certain changes or modifications to the invention, and these equivalents would still be within the scope of the invention defined by the appended claims of the application