WO2009154122A1 - D-キシロース利用機能が向上したコリネ型細菌形質転換体 - Google Patents
D-キシロース利用機能が向上したコリネ型細菌形質転換体 Download PDFInfo
- Publication number
- WO2009154122A1 WO2009154122A1 PCT/JP2009/060637 JP2009060637W WO2009154122A1 WO 2009154122 A1 WO2009154122 A1 WO 2009154122A1 JP 2009060637 W JP2009060637 W JP 2009060637W WO 2009154122 A1 WO2009154122 A1 WO 2009154122A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- xylose
- corynebacterium glutamicum
- gene
- arae
- coryneform bacterium
- Prior art date
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/74—Vectors or expression systems specially adapted for prokaryotic hosts other than E. coli, e.g. Lactobacillus, Micromonospora
- C12N15/77—Vectors or expression systems specially adapted for prokaryotic hosts other than E. coli, e.g. Lactobacillus, Micromonospora for Corynebacterium; for Brevibacterium
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/02—Preparation of oxygen-containing organic compounds containing a hydroxy group
- C12P7/04—Preparation of oxygen-containing organic compounds containing a hydroxy group acyclic
- C12P7/06—Ethanol, i.e. non-beverage
- C12P7/065—Ethanol, i.e. non-beverage with microorganisms other than yeasts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/02—Preparation of oxygen-containing organic compounds containing a hydroxy group
- C12P7/04—Preparation of oxygen-containing organic compounds containing a hydroxy group acyclic
- C12P7/06—Ethanol, i.e. non-beverage
- C12P7/08—Ethanol, i.e. non-beverage produced as by-product or from waste or cellulosic material substrate
- C12P7/12—Ethanol, i.e. non-beverage produced as by-product or from waste or cellulosic material substrate substrate containing sulfite waste liquor or citrus waste
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/40—Preparation of oxygen-containing organic compounds containing a carboxyl group including Peroxycarboxylic acids
- C12P7/44—Polycarboxylic acids
- C12P7/46—Dicarboxylic acids having four or less carbon atoms, e.g. fumaric acid, maleic acid
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E50/00—Technologies for the production of fuel of non-fossil origin
- Y02E50/10—Biofuels, e.g. bio-diesel
Definitions
- the present invention relates to D-xylose utilization technology. More specifically, the present invention relates to a coryneform bacterium transformant that has been subjected to a specific genetic manipulation in order to improve D-xylose utilization function, and a technique for producing a highly efficient organic compound thereby.
- Cellulosic biomass is useful as a raw material for producing various organic acid compounds, ethanol, and the like by biological methods.
- Cellulosic biomass unlike sugar-based biomass such as corn starch and sugar, can be obtained in a wide variety of inexpensive forms such as agricultural waste or wooden waste, and will be an obstacle to securing food resources in the future. This is because it is a raw material species that must not be released.
- Cellulosic biomass consists of approximately 35% to 45% cellulose, 30% to 40% hemicellulose, 10% lignin and 10% other components by weight.
- Cellulose is a polymer of glucose (hexose).
- pentoses such as xylose and arabinose are main components.
- D-xylose accounts for the majority of hemicelluloses of cellulosic biomass such as corn stover, wheat straw, rice straw, and bagasse. Therefore, in order to effectively use cellulosic biomass, it is indispensable to establish a highly efficient technique for using D-xylose by a biological method.
- Saccharomyces cerevisiae which cannot use D-xylose in the wild type, is derived from Pichia stipitis, xylose reductase gene and xylitol dehydrogenase gene, and Saccharomyces cerevisiae.
- a technique for imparting D-xylose utilization function by introducing three genes of the xylulokinase gene by plasmid and a technique for utilizing D-xylose by the resulting transformant Saccharomyces cerevisiae are disclosed.
- Non-Patent Document 2 as a technique using Saccharomyces cerevisiae, in addition to expressing the xylose isomerase gene derived from the mold Piromyces sp., Xylulokinase gene derived from Saccharomyces cerevisiae, Ribulose 5- A transformant, Saccharomyces cerevisiae, in which the aldose reductase gene is highly expressed and highly expressing the phosphate isomerase gene, ribulose ⁇ ⁇ 5-phosphate emiperase gene, transketolase gene, and transaldolase gene, was prepared, and further, Anaerobic xylose-restricted chemostat (anaerobic, ylxylose-limitedmostchimostat), followed by anaerobic automated continuous-batch culture (anaerobic automated sequencing-batch reactor) Thus, a technique for isolating a mutant strain with improved D-xylose uptake and utilization rate and
- Patent Document 2 Zymomonas mobilis that cannot use D-xylose in the wild type, Escherichia coli-derived xylose isomerase gene, xylulokinase gene, transketolase gene, and transaldolase gene
- a technique using D-xylose using a transformant Zymomonas mobilis obtained by introducing and expressing the above four genes by a plasmid is disclosed.
- Patent Document 3 is obtained by introducing and expressing four genes, a xylose isomerase gene derived from Escherichia coli, a xylulokinase gene, a transketolase gene, and a transaldolase gene, in Zymomonas mobilis.
- a technique for utilizing D-xylose by a transformant Zymomonas mobilis in which the introduced gene is more stable is disclosed.
- pentose simultaneous utilization to hexose has been achieved to some extent, the consumption rate (utilization rate) of D-xylose is insufficient compared with the consumption rate of D-glucose, and it is practically used. Further improvement is required as a technique for simultaneous use of hexose and pentose in a typical sense.
- Non-patent document 3 and non-patent document 4 indicate that a microorganism having the simultaneous utilization of D-glucose and D-xylose can be obtained by destroying the ptsG (glucose phosphotransferase system (PTS) transport) gene involved in glucose uptake.
- PTS glucose phosphotransferase system
- Corynebacterium glutamicum and its recombinant strains can produce organic compounds without growth in the bioconversion reaction from sugars to organic compounds such as organic acids under reducing conditions. It is a microorganism useful for effective use (Patent Document 4).
- Patent Document 4 since a reactor volume for growth is not required, a compact reactor design is possible.
- the present inventors introduced a pyruvate ⁇ ⁇ decarboxylase gene and an alcohol dehydrogenase gene derived from Zymomonas mobilis into Corynebacterium glutamicum, and ethanol is produced with high efficiency by the transformant expressing the transgene. The technology is disclosed (Non-Patent Document 5).
- Non-Patent Document 7 discloses a technique for imparting the availability of D-xylose by introducing and expressing xylose isomerase gene and xylulokinase gene derived from Escherichia coli in Corynebacterium glutamicum.
- Corynebacterium glutamicum R (FERM P-18976) strain that does not have L-arabinose utilization ability is added to Escherichia coli-derived arabinose isomerase gene, librokinase gene, and ribulose.
- a Corynebacterium glutamicum ATCC31831-derived proton symporter gene derived from Corynebacterium glutamicum ATCC31831 is further introduced into the Corynebacterium glutamicum transformant expressed by introducing the -5-phosphate 4-epimerase gene.
- the new transformant thus created can greatly improve the rate of L-arabinose utilization, and completely inhibits glucose inhibition against L-arabinose utilization in the presence of both D-glucose and L-arabinose sugars. It is released and D-glucose and L-arabinose can be used at the same time. However, as described above, glucose suppression on D-xylose utilization in the presence of both D-glucose and D-xylose is completely released, and D-xylose and D-glucose are efficiently used in parallel and simultaneously. A corynebacterium transformant that can be produced has not yet been developed.
- An object of the present invention is to provide a recombinant microorganism having an improved function of utilizing D-xylose necessary for effective utilization of cellulosic biomass resources, and a method for producing an organic compound using the microorganism.
- the present inventor has conducted research, and a transformation created by introducing a specific foreign gene into a coryneform bacterium, that is, a foreign gene encoding a protein having a sugar transporter function. It was found that the body efficiently produces organic compounds from D-xylose.
- the coryneform bacterium transformant can use “D-xylose and D-glucose (or D-glucose, D-xylose and L-arabinose) in parallel and simultaneously” or “ “With simultaneous use (ability)” means that the transformant is used in a medium containing a mixed sugar of D-xylose and D-glucose (or D-glucose, D-xylose and L-arabinose) as a carbon source. This means that when the organic compound is produced, the transformant can use these carbon sources in parallel and at the same rate of consumption.
- the present invention has been completed based on the above findings, and provides the following methods for producing microorganisms and organic compounds. Since the recombinant microorganism of the present invention has an excellent simultaneous use performance of D-glucose and D-xylose, it provides a technology for easy production process design and operation of useful organic compounds from cellulosic biomass raw materials. It will be a technology that leads to doing.
- a coryneform bacterial transformant wherein an exogenous gene encoding a protein having a sugar transporter function is introduced into a coryneform bacterium having D-xylose utilization ability.
- DNA comprising the base sequence represented by SEQ ID NO: 13, or DNA comprising a base sequence complementary to the base sequence represented by SEQ ID NO: 13 and a string
- the coryneform bacterium transformant according to (1) or (2) above which uses a DNA that encodes a polypeptide that hybridizes under a gentle condition and has a sugar transporter function.
- Corynebacterium glutamicum R (FERM P-18976), cellobiose-utilizing recombinant strain or mutant strain FERM P-18979, FERM P- 18977 and FERM P-18978, succinic acid-producing recombinant strains FERM P-19446 and FERM P-19477, and ethanol-producing recombinant strains FERM P-17887, FERM P-17888, FERM P-19361 and FERM P (1) to (3) above, which is a coryneform bacterium of any one strain selected from the group consisting of -19362 and is provided with the ability to use D-xylose The coryneform bacterium transformant according to any one of the above.
- coryneform bacterium transformant according to any one of (1) to (5), wherein D-glucose and D-xylose can be used in parallel and simultaneously.
- coryneform bacterium transformant (9) The coryneform bacterium transformant according to (1) above, wherein the coryneform bacterium transformant is Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD (Accession No. NITE BP-577). . (10) The coryneform bacterium transformant according to (1) above, wherein the coryneform bacterium transformant is Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh / pEthAra (Accession No. NITE BP-581). .
- D-xylose According to the present invention, efficient use of D-xylose becomes possible, and D-glucose and D-xylose can be used simultaneously in parallel and at the same rate of use. Effective use for various organic compounds can be realized by an efficient rational process.
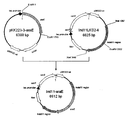
- FIG. 1 is a schematic diagram showing the vector pCRA811 prepared in Example 1 (1).
- FIG. 2 is a view showing an ara gene map in Corynebacterium glutamicum ATCC31831.
- FIG. 3 is a diagram showing a method for producing the vector Ind11-araE produced in Example 1, (3).
- FIG. 4 shows aerobic conditions under the conditions of Corynebacterium glutamicum R / pCRA811 (Fig. 4 (1)) and Corynebacterium glutamicum Ind-araE / pCRA811 (Fig. 4 (2)).
- FIG. 3 is a graph showing changes in D-xylose consumption and growth over time during culture in a BT medium containing D-xylose as a single carbon source.
- FIG. 1 is a schematic diagram showing the vector pCRA811 prepared in Example 1 (1).
- FIG. 2 is a view showing an ara gene map in Corynebacterium glutamicum ATCC31831.
- FIG. 3 is a diagram showing a
- FIG. 5 shows the conditions under the conditions of Corynebacterium®glutamicum R / pCRA811 (FIG. 5 (1)) and Corynebacterium®glutamicum Ind-araE / pCRA811 (FIG. 5 (2)). It is a figure which shows a time-dependent change of D-xylose consumption and lactic acid production
- FIG. 6 shows aerobic conditions under the conditions of Corynebacterium glutamicum R / pCRA811 (FIG. 6 (1)) and Corynebacterium glutamicum Ind-araE / pCRA811 (FIG. 6 (2)).
- FIG. 6 shows aerobic conditions under the conditions of Corynebacterium glutamicum R / pCRA811 (FIG. 6 (1)) and Corynebacterium glutamicum Ind-araE / pCRA811 (FIG. 6 (2)).
- FIG. 3 is a graph showing time-dependent changes in D-glucose and D-xylose consumption and growth during culturing in a BT medium containing D-glucose and D-xylose as carbon sources.
- FIG. 7 shows the conditions under the conditions of Corynebacterium®glutamicum R / pCRA811 (FIG. 7 (1)) and Corynebacterium®glutamicum Ind-araE / pCRA811 (FIG. 7 (2)). It is a figure which shows the time-dependent change of D-glucose and D-xylose consumption and lactic acid production in a mixed sugar having a content ratio of D-glucose and D-xylose of 2: 1.
- FIG. 7 shows the conditions under the conditions of Corynebacterium®glutamicum R / pCRA811 (FIG. 7 (1)) and Corynebacterium®glutamicum Ind-araE / pCRA811 (FIG. 7 (2)). It
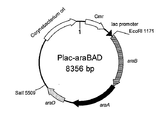
- FIG. 8 is a schematic diagram showing the vector Plac-araBAD prepared in Example 6, item (1).
- FIGS. 9 (1) and (2) show Corynebacterium glutamicum X5 / Plac-araBAD (FIG. 9 (1)) and Corynebacterium amicglutamicum X5-Ind-araE / Plac-araBAD ( FIG. 9 (2)) is a graph showing changes over time in sugar consumption and production of lactic acid in a mixed sugar having a D: glucose, D-xylose and L-arabinose content ratio of 5: 2.5: 1 under reducing conditions. It is.
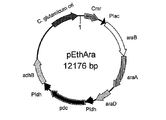
- FIG. 10 is a schematic diagram showing the vector pEthAra prepared in Example 9.
- FIG. 9 (1) and (2) show Corynebacterium glutamicum X5 / Plac-araBAD (FIG. 9 (1)) and Corynebacterium amicglutamicum X5-Ind-araE / Plac-araBAD ( FIG. 9 (2)) is a graph showing
- 11 shows the sugar consumption and the sugar consumption in a mixed sugar with a content ratio of D-glucose, D-xylose and L-arabinose of 5: 2.5: 1 by Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh / pEthAra. It is a figure which shows the time-dependent change of ethanol production.
- the coryneform bacterial transformant of the present invention has a foreign gene encoding a protein having a sugar transporter function and has D-xylose utilization ability. It was introduced into coryneform bacteria. By introducing a foreign gene encoding a protein having a sugar transporter function into a coryneform bacterium having D-xylose utilization ability, the D-xylose utilization ability of the coryneform bacterium is remarkably improved. Therefore, the coryneform bacterium transformant can use D-glucose and D-xylose in parallel and simultaneously.
- the coryneform bacterium having D-xylose utilization ability into which an exogenous gene encoding a protein having a sugar transporter function in the present invention is particularly limited as long as it is a coryneform bacterium imparted with D-xylose utilization ability. Is not to be done. Wild strains of coryneform bacteria cannot utilize pentoses such as D-xylose.
- the method for imparting D-xylose utilization ability to coryneform bacteria is not particularly limited, and examples thereof include a method of introducing a D-xylose metabolism-related gene derived from another species into the coryneform bacteria.
- Metabolism from D-xylose to D-xylulose-5-phosphate in prokaryotes and some molds is specifically: (a) xylose isomerase (hereinafter referred to as “catalyst”) that catalyzes the reaction from D-xylose to D-xylulose. (B) the xylulokinase that catalyzes the reaction from D-xylulose to D-xylulose-5-phosphate (hereinafter referred to as “XylB”, the gene It is carried out by a two-step reaction catalyzed by two enzymes (also referred to as “xylB”). In the present invention, the metabolic system can be used.
- xylose-kinase isomerase gene derived from Escherichia coli (xylA) and a xylulokinase gene (xylB) as D-xylose metabolism-related genes in coryneform bacteria.
- xylA xylose-kinase isomerase gene
- xylB xylulokinase gene
- a coryneform bacterium to which D-xylose utilization ability produced by such a technique is imparted can be used.
- a transformant into which a gene derived from a species other than the above Escherichia coli is introduced may be used as a method for imparting D-xylose utilization ability.
- genes encoding the D-xylose metabolism-related enzymes of (a) and (b) above may be present on the same locus, or these two genes may be present on different loci. May be.
- An example in which two genes are present on the same locus is, for example, an operon formed by linking the genes (a) and (b) above.
- the genes (a) and (b) are usually held in microorganisms having the ability to metabolize D-xylose.
- Examples of the genes (a) and (b) in the present invention include Escherichia coli, Corynebacterium glutamicum (only xylB gene), Bacillus subtilis, Salmonella typhimurium (Salmonella). It is preferable to use an xylA gene derived from the same or different microorganism selected from the group consisting of typhimurium, Bacillus halodurans, Sinorhizobium meliloti, and Agrobacterium tumefaciens, and xylB gene. . Among these, in the present invention, the use of the xylA gene and xylB gene derived from Escherichia coli is most preferable.
- a DNA fragment synthesized according to the sequence can be used as the gene encoding the D-xylose metabolism-related enzyme. Even when the DNA sequence is unknown, it is possible to obtain a fragment by a hybridization method or a PCR method based on an amino acid sequence conserved between D-xylose metabolism-related enzyme proteins. Furthermore, fragments can be obtained by degenerate PCR using mixed primers designed based on other known D-xylose metabolism-related gene sequences.
- a gene encoding a D-xylose metabolism-related enzyme may have a part of its base sequence replaced with another base as long as the D-xylose metabolism activity of the polypeptide encoded by the gene is retained.
- the base sequence may be deleted, a base may be newly inserted, and a part of the base sequence may be rearranged. Any of the derivatives of genes encoding these D-xylose metabolism-related enzymes can be used in the present invention.
- the part of the base sequence may be, for example, 1 to several amino acids (usually 1 to 5, preferably 1 to 3, more preferably 1 to 2).
- the base sequence of a gene encoding a D-xylose metabolism-related enzyme encodes a D-xylose metabolism-related enzyme derived from prokaryotes, that is, having the same amino acid sequence as a D-xylose metabolism-related enzyme naturally present in prokaryotes. Preferably it is.
- the expression of prokaryotic D-xylose metabolism-related enzymes increases the likelihood that D-xylose metabolism-related enzymes will be expressed in active form in bacteria.
- the coryneform bacterium serving as a host is not particularly limited as long as it has a metabolic conversion function of saccharides. Depending on the type of saccharide and the type of target organic compound, it may be necessary to introduce a new function such as the uptake of saccharides that are not originally possessed by coryneform bacteria, the metabolic pathway, or the conversion pathway to the target organic compound. In such a case, these functions may be imparted by a recombination technique or a mutagenesis treatment.
- the coryneform bacterium used in the present invention is a group of microorganisms defined in Bargeys Manual Determinative Bacteriology, Vol. 8, 599 (1974), There is no particular limitation as long as it grows under aerobic conditions. Specific examples include Corynebacterium, Brevibacterium, Arthrobacter, Mycobacterium, Micrococcus, and the like.
- Corynebacterium genus Corynebacterium glutamicum R (FERMFERP-18976), ATCC13032, ATCC13058, ATCC13059, ATCC13060, ATCC13232, ATCC13286, ATCC13287, ATCC13655, ATCC13745, ATCC13746, ATCC13761, ATCC14020, ATCC31831 etc. are mentioned.
- Brevibacterium spp. include Brevibacterium lactofermentum ATCC 13869, Brevibacterium flavum MJ-233 (FERM BP-1497) or MJ-233AB-41 (FERM BP-1498), Another example is Brevibacterium ammoniagenes ATCC6872.
- Arthrobacter examples include Arthrobacter globiformis ATCC8010, ATCC4336, ATCC21056, ATCC31250, ATCC31738 or ATCC35698.
- Mycobacterium examples include Mycobacterium bovis ATCC19210 or ATCC27289.
- Micrococcus genus micrococcus freudenreichii (Micrococcus freudenreichii) NO. 239 (FERM P-13221), Micrococcus leuteus (Micrococcus leuteus) NO. 240 (FERM P-13222), Micrococcus ureae (Micrococcus ureae) IAM1010
- Micrococcus roseus IFO3764 and the like can be mentioned.
- the coryneform bacterium is a coryneform bacterium that can actively or passively transport D-xylose into cells.
- the transformed coryneform bacterium has a normally functioning glycolytic pathway and a pentose phosphate pathway.
- the coryneform bacterium to be used preferably has high resistance to ethanol; organic acids such as lactic acid, acetic acid, formic acid and the like; and glycolysis products such as furfural and hydroxymethylfurfural.
- the coryneform bacterium used in the present invention includes Corynebacterium glutamicum R (FERM P-18976, Japanese Patent Application No. 2002-252190 (Japanese Patent Laid-Open No. 2004-089029)) or Corynebacterium glutamicum ATCC31831. Etc. are preferred.
- coryneform bacteria are wild-type mutant strains that exist in nature (for example, cellobiose-utilizing mutant strains: FERM P-18977, FERM P-18978) (Japanese Patent Application No. 2002-252190 (Japanese Patent Application Laid-Open No. 2004-089029)).
- artificial genetically modified strains include, for example, ethanol-producing recombinant strains such as FERM P-17887 (FERM BP-7621), FERM P-17888 (FERM BP-7622) (PCT / JP01 / 04935 (International Publication WO01). / 096573)) and J. Mol. Microbiol.
- coryneform bacteria having the ability to use D-xylose are Corynebacterium glutamicum R (FERM P-18976), cellobiose-utilizing recombinant strains or mutant strains (FERM P-18979, FERM P -18977 and FERM P-18978), succinic acid-producing recombinant strains (FERM P-19446 and FERM P-19477), and ethanol-producing recombinant strains (FERM P-17887 (FERM BP-7621), FERM P-17888 ( FERM BP-7622) and a strain described in J. Mol. Microbiol.
- the strain is a coryneform bacterium and is provided with the ability to use D-xylose.
- a particularly preferred coryneform bacterium in the present invention is Corynebacterium glutamicum R (FERM P-18976) imparted with the ability to use D-xylose.
- a foreign gene encoding a protein having a sugar transporter function (also referred to as a sugar transporter gene) is added to a coryneform bacterium having D-xylose utilization ability. Is introduced. Thereby, the D-xylose utilization ability of the coryneform bacterium transformant can be further improved.
- the protein having a sugar transporter function may be a protein having a function of transporting hexose and pentose, but among them, an L-arabinose transport system proton symporter is preferable.
- a gene encoding a protein having a proton symporter function of L-arabinose transport system (also referred to as a proton symporter gene of L-arabinose transport system) ) Is preferably used.
- the proton symporter gene for the L-arabinose transport system is known and is called araE.
- araE gene sequences and enzyme properties have been reported in the following strains and the like. Bacillus subtilis (J.JBacteriol., Vol. 179, 7705-7711 (1997)), Klebsiella oxytoca 8017 (J. Bacteriol., Vol. 177, 5379-5380 (1995)), Escherichia coli (J. Biol. Chem., Vol. 263, 8003-8010 (1988)).
- exogenous gene encoding a protein having a sugar transporter function in the present invention examples include Corynebacterium glutamicum ATCC31831, Escherichia coli, Bacillus ⁇ subtilis, Bacillus liceniformis (Bacillus).
- licheniformis Bacillus coagulans, Lactobacillus sakei, Pediococcus Pentiococcus pentosaceus, Lactobacillus occus heyocto ), Leuconostoc mesenteroides, Lactobacillus plantarum, Lactobacillus fermentum, Lactobacillus brevis, Leuconostoc citreum, Enterococcus faecium, Klebsiella physium, Klebsiella physium An araE gene derived from a microorganism selected from the group can be used.
- exogenous genes encoding proteins with proton symporter activity of L-arabinose transport system suitable for the present invention include Corynebacterium glutamicum ATCC31831, Escherichia coli, Bacillus subtilis, Klebsiella oxytoca, and Salmonella typhim It is preferable to use an araE gene derived from a microorganism selected from the group consisting of lithium. Further, as a proton symporter gene of L-arabinose transport system, it hybridizes under stringent conditions with a DNA sequence represented by SEQ ID NO: 13 or a DNA sequence consisting of a base sequence complementary to the DNA sequence represented by SEQ ID NO: 13.
- a DNA encoding a polypeptide having a sugar transporter function for example, a proton symporter function of an L-arabinose transport system. That is, in the present invention, the foreign gene encoding a protein having a sugar transporter function is composed of DNA consisting of the base sequence represented by SEQ ID NO: 13 or a base sequence complementary to the base sequence represented by SEQ ID NO: 13. It is preferable to use DNA encoding a polypeptide that hybridizes with DNA under stringent conditions and has a sugar transporter function.
- the DNA having the nucleotide sequence of SEQ ID NO: 13 is a gene derived from Corynebacterium glutamicum ATCC31831 and the like, and is the nucleotide sequence of the araE gene DNA.
- Methods for regulating the appropriate expression level of the araE gene in D-xylose-utilizing strains are well known to those skilled in the art.
- the type, combination, order of introduction, and the like of microorganisms derived from the D-xylose metabolism-related enzyme gene and the sugar transporter gene are not particularly limited.
- stringent conditions mean that hybridization occurs when homology of 90% or more, preferably 95% or more, more preferably 97% or more, and even more preferably 98% or more exists between sequences. Means. Usually, the hybridization occurs at a temperature about 5 to 30 ° C., preferably about 10 to 25 ° C., more preferably about 15 to 20 ° C. lower than the melting temperature (Tm) of the complete hybrid.
- Tm melting temperature
- Stringent conditions are described in J. Sambrook et al., Molecular Cloning, A Laboratory Manual, Second edition, Cold Spring Harbor Laboratory Press (1989), especially section 11.45 “Conditions for Hybridization of Oligonucleotide Probes”. The described conditions can be used.
- the homology between the base sequences is calculated using the calculation software GENETYX (registered trademark) Ver. It is a value calculated using 8 (manufactured by Genetics).
- a DNA that hybridizes with a certain DNA under stringent conditions for example, a DNA that hybridizes with a DNA having a base sequence complementary to the base sequence represented by SEQ ID NO: 13 under stringent conditions, It preferably has about 90% or more sequence homology with the base sequence of SEQ ID NO: 13, more preferably about 95% or more sequence homology, and particularly preferably about 98% or more sequence homology. .
- the coryneform bacterium transformant of the present invention preferably uses D-glucose and D-xylose in parallel and simultaneously, and uses D-glucose, D-xylose and L-arabinose in parallel and simultaneously. More preferably.
- the coryneform bacterium transformant of the present invention can be prepared, for example, by introducing the above sugar transporter gene into a coryneform bacterium having D-xylose utilization ability and transforming it.
- Coryneform bacterial transformants that use D-glucose, D-xylose and L-arabinose in parallel and simultaneously include, for example, the above-mentioned D-xylose metabolism-related enzyme gene and sugar transporter gene, In addition, it can be prepared by introducing a gene related to L-arabinose metabolism.
- genes related to L-arabinose metabolism include, for example, a gene encoding Escherichia coli L-arabinose isomerase, a gene encoding L-librokinase, and L-ribulose-5-phosphate-4-epimerase. Suitable genes are suitable. There are no particular limitations on the types, combinations, and order of introduction of microorganisms derived from the D-xylose metabolism-related enzyme gene, sugar transporter gene, and L-arabinose metabolism-related gene.
- oligonucleotide primer set is prepared.
- examples of such a primer set include those represented by the nucleotide sequences of SEQ ID NOs: 14 and 15.
- a known PCR device such as a thermal cycler can be used. The PCR cycle may be performed according to a known technique.
- DNA isolated from a microorganism having a sugar transporter gene can be used to synthesize DNA cDNA encoding AraE by PCR.
- the gene obtained by the PCR method can be introduced into an appropriate cloning vector.
- Cloning methods include commercially available PCR cloning systems such as pGEM-T easy vector system (Promega), TOPO TA-cloning system (Invitrogen), Mighty Cloning Kit (Takara), etc. You can also In addition, one example of the method will be described in detail in the Examples.
- a DNA fragment containing this region is a high fragment using a synthetic primer appropriately designed based on the base sequence of DNA encoding known AraE as a template. It can also be obtained by a hybridization method.
- a cloning vector containing the gene obtained by the PCR method is introduced into a microorganism such as Escherichia coli JM109 strain, and the strain is transformed.
- the transformed strain is cultured in a medium containing an appropriate antibiotic (for example, ampicillin, chloramphenicol, etc.), and the cells are recovered from the culture.
- Plasmid DNA is extracted from the collected cells.
- Plasmid DNA can be extracted by a known technique, or can be easily extracted using a commercially available plasmid extraction kit. Examples of commercially available plasmid extraction kits include Qiaquick Plasmid Purification Kit (trade name: Qiaquick plasmid purification kit, manufactured by Qiagen).
- the gene sequence encoding AraE can be confirmed.
- the base sequence of DNA can be determined by a known method such as a dioxynucleotide enzyme method.
- the base sequence can be determined using a capillary electrophoresis system using a multi-fluorescence technique for detection.
- the base sequence can also be determined using a DNA sequencer such as ABI PRISM 3730xl DNA Analyzer (Applied Biosystems).
- Said method can be performed based on the conventional method of genetic engineering experiment.
- Introduction and expression methods of vector and the foreign gene of various microorganisms because they are described in many experimental books [e.g., Molecular Cloning: A Laboratory Manual ( 3 rd Edition) CSHL Press (2001), or Current protocols in molecular biology. Green Publishing and Wiley Interscience, New York (1987), etc.], vector selection, gene introduction and expression can be performed accordingly.
- the araE gene is expressed on the plasmid or chromosome in the aforementioned coryneform bacterium having the ability to use D-xylose.
- these genes are introduced under expressible regulatory sequences.
- “under the control sequence” means that these genes can be transcribed and translated by joint work with a promoter, an inducer, an operator, a ribosome binding site, a transcription terminator, and the like.
- any plasmid vector may be used as long as it contains a gene that controls the autonomous replication function in coryneform bacteria.
- pAM330 derived from Brevbacterium lactofermentum 2256 (Japanese Patent Laid-Open No. 58-67699, Agric. Biol. Chem., Vol. 48, 2901-2903 (1984) and Nucleic Acids Symp Ser., Vol. 16, 265-267 (1985)], Corynebacterium glutamicum ⁇ ATCC13058-derived pHM1519 [Agric. Biol. Chem., Vol. 48, 2901-2903 (1984)] and pCRY30 [Appl. Environ. Microbiol., Vol.
- the vector contains a multiple cloning site having various restriction enzyme sites therein or a single restriction enzyme site.
- promoters can be suitably used.
- Such a promoter can be obtained from many known sources including yeast, bacteria, and other cell sources, and can be any nucleotide sequence that has a function of initiating transcription of a target gene in coryneform bacteria. It may be.
- an efficient, non-glucose-repressed promoter is preferable.
- Preferable examples of such a promoter include, for example, the lac, trc, and tac promoters known as strong constitutive promoters in coryneform bacteria.
- the promoter used in the present invention can be modified as necessary to change its regulatory mechanism.
- the terminator under the control sequence arranged downstream of the target gene may be any nucleotide sequence as long as it has a function of terminating transcription of the gene in coryneform bacteria.
- the construction of the plasmid vector used to create the coryneform bacterium transformant of the present invention is, for example, when using the araE gene of Corynebacterium ⁇ ⁇ ⁇ ⁇ glutamicum ATCC31831, the gene whose base sequence has been confirmed, After ligation with a control sequence such as a terminator, it can be carried out by inserting it into an appropriate restriction enzyme site of any of the plasmid vectors exemplified above. Details are described in the examples.
- an electric pulse method electropultation method
- an electric pulse method particularly limited as long as it is a method capable of target gene transfer into CaCl 2 method coryneform bacterium Is not to be done.
- Specific examples thereof include, for example, a known method [Agric. Biol. Chem., Vol. 54, 443-447 (1990)] or [Res. Microbiol., Vol. 144, 181-185 (1993). )] Can be used.
- a drug resistance gene or the like is incorporated into a plasmid containing the target gene, and the target gene introduction treatment is performed on a plate medium containing the drug at an appropriate concentration.
- the transformed coryneform bacterium can be selected. Specific examples of the method include those described in [Agric. Biol. Chem., Vol. 54, 443-447 (1990)] or [Res. Microbiol., Vol. 144, 181-185 (1993)]. The method etc. can be mentioned.
- Corynebacterium glutamicum Ind-araE / pCRA811 (Accession number NITE BP-576) and Corynebacterium glutamicum (Corynebacterium glutamicum) X5-Ind-araE / Plac-araBAD (Accession number: NITE BP-577) (both of which are the National Institute for Product Evaluation Technology Patent Biological Depositary Center (2-5 Kazusa Kamashika, Kisarazu City, Chiba, Japan) -8 (Postal Code 292-0818)) (Department date: May 28, 2008) and Corynebacterium glutenum X5-Ind-araE- ⁇ ldh / pEthAra (Accession Number NITE BP-581 ) (National Institute of Technology and Evaluation, Patent Biological Deposit Center (2-5-8 Kazusa Kamashi, Kisarazu City, Chiba Prefecture, Japan) 292-0818)) to deposit pre-
- the coryneform bacterium transformant created by the present invention increases the flow rate of the pentose phosphate pathway; increases resistance to ethanol, osmotic pressure or organic acids, and by-products (target production) to improve organic compound productivity. It may further comprise a genetic modification that produces one or more of the characteristics selected from the group consisting of reduced production (understood to mean carbon-containing molecules other than products). Such genetic modification can be specifically introduced by overexpression of a foreign gene and / or inactivation of an endogenous gene; classical mutagenesis; screening and / or selection of a target mutant. .
- the thus produced coryneform bacterium transformant of the present invention uses D-xylose as a raw material (in the case of producing a polycarboxylic acid present in the TCA pathway, further using carbonate ion or the like as a raw material), monocarboxylic acid, dicarboxylic acid, Various organic compounds such as acids, ketocarboxylic acids, hydroxycarboxylic acids, amino acids, monoalcohols, polyols and vitamins can increase the rate of saccharide metabolism or suppress a decrease in the rate of saccharide metabolism over time, while at the same time providing a high production rate. It can be produced in the reaction solution with a high accumulated concentration.
- the culture of the coryneform bacterium transformant can be performed using a normal nutrient medium containing a carbon source, a nitrogen source, an inorganic salt, and the like.
- a carbon source for example, glucose or waste molasses or the like as a carbon source; for example, ammonia, ammonium sulfate, ammonium chloride, ammonium nitrate or urea can be used alone or in combination of two or more as a nitrogen source.
- the inorganic salt for example, potassium monohydrogen phosphate, potassium dihydrogen phosphate, magnesium sulfate, or the like can be used.
- nutrients such as peptone, meat extract, yeast extract, corn steep liquor, casamino acid; various vitamins such as biotin and thiamine can be appropriately added to the medium as necessary.
- Cultivation can be performed at a temperature of about 15 to 45 ° C., preferably about 25 to 37 ° C. under aerobic conditions such as aeration stirring or shaking.
- the pH of the medium during culture is in the range of about 5 to 10, preferably about 6.5 to 8.5, and pH adjustment during culture can be performed by adding an acid or an alkali.
- the carbon source concentration of the medium at the start of the culture is preferably about 1 to 20% (W / V), more preferably about 2 to 5% (W / V).
- the culture period is usually about 1 to 7 days.
- the method for recovering and separating the cultured cells from the culture obtained as described above is not particularly limited, and known methods such as centrifugation and membrane separation can be used.
- the collected cultured microbial cells may be treated, and the resulting microbial cell processed product may be used in the next step.
- the cell-treated product may be any product obtained by applying some treatment to cultured cells, and examples thereof include immobilized cells obtained by immobilizing cells with acrylamide or carrageenan.
- a step of generating an organic compound (also referred to as an organic compound generation step) is performed using the cultured microbial cells of the coryneform bacterium transformant collected and separated from the culture obtained as described above or the processed microbial cells. .
- Organic Compound Generation Step an organic compound is generated using the coryneform bacterium transformant in a medium (reaction medium) containing D-xylose.
- a method for producing an organic compound including such steps is also one aspect of the present invention.
- the reaction medium contains D-xylose as a carbon source, but can also contain D-glucose in addition to D-xylose.
- the D-xylose or D-glucose may be D-xylose or D-glucose itself, for example, an oligomer or polymer of a carbohydrate comprising D-xylose or D-glucose units such as lignocellulose, arabinan, cellulose, starch and the like. But you can.
- an appropriate carbohydrate degrading enzyme xylanase, glucanase, amylase, etc.
- xylanase glucanase, amylase, etc.
- the coryneform bacterial transformant will be Enzymes may be produced.
- the reaction medium may contain an organic carbon source other than D-xylose and D-glucose.
- an organic carbon source include saccharides that can be used for biochemical reactions by the coryneform bacterium transformant of the present invention.
- the saccharide include monosaccharides such as (D- or L-) arabinose, galactose, fructose, or mannose; disaccharides such as cellobiose, sucrose, lactose, maltose; polysaccharides such as dextrin or soluble starch Is mentioned.
- monosaccharides such as C6 sugar and C5 sugar are preferable, and L-arabinose is particularly preferable.
- the composition of the reaction medium used for the organic compound production reaction is a component necessary for maintaining the metabolic function of the coryneform bacterium transformant or the processed product thereof, that is, a carbon source such as various sugars; Nitrogen sources necessary for synthesis; salts such as phosphorus, potassium or sodium; and trace metal salts such as iron, manganese or calcium.
- a carbon source such as various sugars
- Nitrogen sources necessary for synthesis nitrogen sources necessary for synthesis
- salts such as phosphorus, potassium or sodium
- trace metal salts such as iron, manganese or calcium.
- These addition amounts can be appropriately determined depending on the required reaction time, the type of the target organic compound product, the type of coryneform bacterium transformant used, and the like.
- Carbon sources, nitrogen sources, inorganic salts, vitamins, and trace metal salts may be known ones, for example, those exemplified for the growth culture step.
- the pH of the medium is preferably about 6-8.
- the reaction between the coryneform bacterium transformant or a treated product thereof and a carbon source containing D-xylose is performed under temperature conditions where the coryneform bacterium transformant of the present invention or the treated product thereof can act. Is preferable, and can be appropriately selected depending on the type of coryneform bacterium transformant or a treated product thereof.
- the temperature is usually about 25-35 ° C.
- carbon dioxide or inorganic carbonates such as various carbonates or hydrogen carbonates may be added to the reaction medium in addition to organic carbon sources such as sugars. Depending on the target organic compound, it may be effective.
- the concentration of the carbon source in the medium is not particularly limited, but is usually used at a concentration of about 0.1 to 30% by mass, preferably about 0.5 to 20% by mass.
- the (D-xylose / D-glucose) mixing ratio (mass ratio) of saccharides in the carbon source is usually about 1/3 to 2/3 although it depends on the saccharide-derived biomass raw material species.
- the mixing ratio of C6 sugars and C5 sugars is usually 1/4 to 3/4. Degree.
- the reaction medium is preferably in a reduced state. That is, the coryneform bacterium transformant or the treated product thereof is preferably subjected to a reaction for producing a target organic compound in a reaction medium in a reduced state.
- the organic compound generation method can be any of batch, fed-batch, and continuous methods.
- the coryneform bacterium of the present invention when an organic compound is produced using a coryneform bacterium transformant in a reaction medium in a reduced state, that is, an organic compound is produced by a biochemical reaction in a reduced state, the coryneform bacterium of the present invention
- the growth division of the transformant is completely suppressed, and a substantial complete suppression of secreted by-products accompanying the growth can be realized.
- the coryneform bacterium transformant recovered from the culture growth step or the treated product thereof is used in the reaction medium, the environmental state during aerobic growth inside and outside the coryneform bacterium cell is the reaction medium. It is preferred to use methods and conditions that do not result.
- the reaction medium is substantially free from a product that is generated during the growth culture process and is present outside and inside the cells. More specifically, secreted by-products generated during the growth culture process and released to the outside of the cells, and substances generated by the aerobic metabolic function in the cultured cells and remaining in the cells are substantially contained in the reaction medium. It is preferable that it is not present in Such a state is, for example, by performing a method such as centrifugation or membrane separation using a culture solution after growth and / or leaving the cultured cells in a reduced state for about 2 to 10 hours. It is realized by doing.
- reaction medium in a reduced state, but the reaction medium may have any shape such as solid, semi-solid, or liquid.
- the reduced state in the present invention is defined by the redox potential of the reaction system (reaction medium), and means that the redox potential of the reaction medium is a negative ( ⁇ ) value.
- the redox potential of the reaction medium in the reduced state is preferably about -200 mV to -500 mV, more preferably about -250 mV to -500 mV.
- the reduction state of the reaction medium can be estimated to some extent with a resazurin indicator (decolorization from blue to colorless in the reduction state) to some extent, but to be precise, use a redox potentiometer (eg ORP Electrodes, BROADLEY JAMES) Use to measure.
- a resazurin indicator decolorization from blue to colorless in the reduction state
- a redox potentiometer eg ORP Electrodes, BROADLEY JAMES
- the oxidation-reduction potential of the reaction medium is maintained at about -200 mV to -500 mV for about 50% or more of the reaction time, more preferably about 70% or more, and further preferably about 90% or more. preferable.
- the reduction state is realized by the method for preparing cultured cells after the culture, the method for preparing the reaction medium, the method for maintaining the reduction state during the reaction, or the like.
- a known method may be used as a method for preparing the reaction medium in a reduced state.
- a method for preparing an aqueous solution for a reaction medium is, for example, a method for preparing a culture solution for an anaerobic microorganism such as a sulfate-reducing microorganism (The ⁇ dissimilatory sulfate-reducing bacteria, In The Prokaryotes, A Handbook on Habitats, Isolation and Identification of Bacteria, Ed. By Starr, M. P. et. Al.
- a method for preparing the reaction medium more specifically, a method of removing dissolved gas by subjecting the reaction medium to heat treatment or reduced pressure treatment, and the like can be mentioned. More specifically, by treating the reaction medium for about 1 to 60 minutes, preferably about 5 to 40 minutes under reduced pressure of about 10 mmHg or less, preferably about 5 mmHg or less, more preferably about 3 mmHg or less.
- the dissolved gas, particularly dissolved oxygen can be removed, and a reaction medium under reducing conditions can be prepared.
- An appropriate reducing agent for example, thioglycolic acid, ascorbic acid, cysteine hydrochloride, mercaptoacetic acid, thiolacetic acid, glutathione, sodium sulfide, etc.
- an appropriate combination of these methods also provides a method for preparing an effective reduced reaction medium.
- the organic compound produced in the reaction medium as described above is collected.
- a known method used in a bioprocess can be used. Such known methods include salting out, recrystallization, organic solvent extraction, esterification distillation separation, chromatographic separation, or electrodialysis, etc. of the organic compound production liquid. Accordingly, the separation, purification and collection method can be appropriately determined.
- Examples of organic compounds that can be produced by the method of the present invention include organic acids, alcohols, amino acids, and vitamins.
- the organic acid include acetic acid, lactic acid, 3-hydroxypropionic acid, acrylic acid, succinic acid, fumaric acid, malic acid, oxaloacetic acid, citric acid, cisaconic acid, isocitric acid, itaconic acid, 2-oxoglutaric acid or shikimi An acid etc. are mentioned.
- Examples of the alcohol include ethanol, butanol, 1,3-propanediol, glycerol, xylitol, sorbitol, or 1,4-butanediol.
- amino acids examples include valine, leucine, alanine, aspartic acid, lysine, isoleucine or threonine.
- the method of the present invention is suitable for producing one or more organic compounds selected from the group consisting of ethanol, lactic acid, succinic acid, xylitol, acetic acid and amino acids.
- the present invention also provides a D-xylose-utilizing coryneform bacterium transformant having a markedly improved ability to convert D-xylose or a mixture of D-glucose and D-xylose into an organic compound by the reaction under the conditions described above. To do.
- Plasmid pCRA811 construction method a) Escherichia coli Extraction of chromosomal DNA from JM109 Escherichia coli JM109 is dissolved in L medium (tryptone 10 g, yeast extract 5 g and NaCl 5 g in distilled water 1 L. ) was inoculated using a platinum loop, and cultured at 37 ° C. with shaking until the logarithmic growth phase, and the cells were collected. Using a DNA genome extraction kit (trade name: GenomicPrep Cells and Tissue DNA Isolation Kit, manufactured by Amersham), chromosomal DNA was recovered from the collected cells according to the instruction manual.
- L medium tryptone 10 g, yeast extract 5 g and NaCl 5 g in distilled water 1 L.
- the former is added with an EcoRI site and the latter with a SmaI site.
- the template DNA the chromosomal DNA of Escherichia coli JM109 extracted in Example 1, (1), a) above was used.
- PCR was performed under the following conditions using a “DNA thermal cycler” manufactured by Perkin Elmer Citas Co., Ltd. and using a DNA polymerase (trade name: Takara LA Taq HS DNA polymerase, manufactured by Takara Shuzo Co., Ltd.) as a reaction reagent.
- Reaction solution The above was mixed, and this 100 ⁇ L reaction solution was subjected to PCR.
- PCR cycle Denaturation process: 94 ° C., 1 minute Annealing process: 55 ° C., 1 minute Extension process: 72 ° C., 2 minutes
- One cycle was performed for 30 cycles.
- electrophoresis was performed on a 1% (W / V) agarose gel using a part of the reaction solution generated above, DNA fragments of about 1.4 kb and about 1.6 kb containing the xylA gene and the xylB gene, respectively. was detected.
- the amplification product of about 1.6 kb treated with the restriction enzymes EcoRI and SmaI described above and the vector pTrc99A (Pharmacia) treated with restriction enzymes with EcoRI and SmaI were mixed, and ligation kit (trade name: Mighty Cloning) was mixed therewith. Kit, manufactured by Takara Shuzo Co., Ltd.) and then reacted according to the instruction manual.
- Escherichia coli JM109 was transformed by the calcium chloride method [Journal of Molecular Biology., Vol.
- a growing strain having a white color on this medium is subjected to liquid culture by a conventional method, and a plasmid into which a DNA fragment having a length of about 1.4 kb containing the xylA gene is inserted is extracted from the culture solution using a plasmid extraction kit (trade name: QIAprep Spin Extraction was performed using Miniprep Kit (manufactured by Qiagen).
- the plasmid containing the xylA gene was named pCRA801.
- the amplified product of about 1.6 kb treated with the restriction enzymes EcoRI and SmaI was ligated with the vector pTrc99A treated with the restriction enzymes EcoRI and SmaI in the same manner to construct a plasmid pCRA802 containing the xylB gene.
- a DNA fragment containing the xylB gene linked to the trc promoter (Ptrc) was used, and PCR was performed under the same conditions as described above except that the primer set shown below was used.
- Ptrc-xylB gene amplification primer set (Ptrc)
- a amplification product of about 1.7 kb treated with the restriction enzymes FbaI and BamHI was also subjected to restriction enzyme treatment with FbaI and BamHI in the same manner as above except that chloramphenicol was used instead of ampicillin in the selection medium.
- a plasmid pCRA811 (vector pCRA811) containing the Ptrc-xylB gene was constructed by ligation with pCRA810 (FIG. 1).
- the chromosomal DNA of Corynebacterium glutamicum 31831 extracted in the above Example 1, (2), a) was used.
- PCR was performed using “DNA Thermal Cycler” manufactured by Perkin Elmer Citas Co., Ltd., and using DNA polymerase (trade name: Takara LA Taq HS HS DNA Polymerase, manufactured by Takara Shuzo Co., Ltd.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ L of the reaction solution was subjected to PCR.
- PCR cycle Denaturation process: 94 ° C., 1 minute Annealing process: 37 ° C., 1 minute Extension process: 72 ° C., 1 minute One cycle was performed for 30 cycles.
- a plasmid in which a DNA fragment of about 390 bp in length containing the araA homologous region was inserted under the same conditions and method as in Example 1, (1), b) above was obtained.
- the DNA sequence was determined by sequencing the plasmid thus prepared. The sequence thus obtained showed very high homology with the base sequence of araA reported in other bacteria.
- the chromosomal DNA of Corynebacterium glutamicum ATCC31831 extracted in the above Example 1, (2), item a) was used.
- PCR was performed using “DNA thermal cycler” manufactured by Perkin Elmer Citas Co., Ltd. under the following conditions using DNA polymerase (trade name: Pyrobest DNA polymerase, manufactured by Takara Shuzo Co., Ltd.) as a reaction reagent.
- Reaction solution The above was mixed, and this 100 ⁇ L reaction solution was subjected to PCR.
- Escherichia coli JM109 was transformed by the calcium chloride method, and an L agar medium containing 50 ⁇ g / mL ampicillin [composition: containing 1.5% (W / V) agar] The same composition as that of the L medium was applied.
- a growing strain on this medium is subjected to liquid culture by a conventional method, and a plasmid into which a DNA fragment having a length of about 1.7 kb containing the araE gene is inserted from the culture solution using a QIAprep Spin Miniprep Kit (Qiagen). Extracted.
- the plasmid containing the araE gene was named pKK223-3-araE (FIG. 3).
- the chromosomal DNA of Corynebacterium glutamicum R extracted by the same method as in Example 1, (2), a) above was used.
- PCR was performed using Takara-LA-Taq-HS-DNA-polymerase, no DMSO in the reaction solution, and the PCR cycle was carried out under the following conditions (Examples 1, (2), b)) The same conditions were used.
- Example 1 Using this ligation solution, the same as in Example 1, (1), b) above, except that kanamycin 50 ⁇ g / mL was used instead of chloramphenicol as an antibiotic in the transformant selection medium.
- the plasmid containing the Indel11 region for araE gene introduction was named Ind11LKS2-4 (FIG. 3).
- Plasmid pKK223-3-araE was treated with EcoRI, separated by agarose electrophoresis, and a DNA fragment containing about 2.0 kb gene excised from the gel was gel extraction kit (trade name: Minelute Gel Extraction Kit, manufactured by Qiagen) Was collected using The resulting DNA was blunt-ended using a blunt end treatment kit (trade name: DNA : Blunting Kit, manufactured by Takara Shuzo Co., Ltd.), mixed with Ind11LKS2-4 treated with EcoRV and mixed with Mighty Cloning. After adding Kit (Takara Shuzo Co., Ltd.), the reaction was carried out according to the instruction manual.
- Escherichia coli JM109 was transformed by the calcium chloride method, L agar medium containing 50 ⁇ g / mL kanamycin [composition: containing 1.5% (W / V) agar] Except for the above, the composition was the same as that of the L medium.
- a growing strain on this medium is subjected to liquid culture by a conventional method, and a plasmid into which a DNA fragment having a length of about 1.7 kb containing the araE gene is inserted from the culture solution using a QIAprep Spin Miniprep Kit (Qiagen). Extracted.
- the araE gene introduction plasmid was named Ind11-araE (FIG. 3).
- the plasmid for introducing araE gene Ind11-araE is a plasmid that cannot replicate in Corynebacterium glutamicum R. Ind11-araE was prepared according to the method of electric pulse method [Agric. Biol. Chem., Vol. 54, 443-447 (1990) and Res. Microbiol., Vol. 144, 181-185 (1993)].
- a agar medium containing 50 ⁇ g / mL kanamycin introduced into Corynebacterium glutamicum R [Composition: Same composition as the above liquid medium except that 1.5% (W / V) agar is included] It was applied to.
- the strain obtained in the above medium was added to the BT agar medium shown in Table 2 containing 10% (W / V) of sucrose [composition: 1.5% (W / V) of agar was included. Except for the same composition as that of the BT liquid medium].
- the plasmid Ind11-araE undergoes one-point homologous recombination with a homologous region on the chromosome, it results from kanamycin resistance by expression of the kanamycin resistance gene on Ind11-araE and by expression of the sacR-sacB gene of Bacillus subtilis.
- sucrose lethality when two-point homologous recombination occurred, kanamycin sensitivity due to loss of the kanamycin resistance gene on Ind11-araE and growth in a sucrose-containing medium due to loss of the sacR-sacB gene Indicates.
- the target araE gene chromosome-introduced strain exhibits kanamycin sensitivity and growth of sucrose-containing medium.
- the strain that showed kanamycin sensitivity and growth of sucrose-containing medium was named Corynebacterium glutamicum Ind-araE.
- Corynebacterium glutamicum R and Corynebacterium glutamicum Ind11-araE an A agar medium containing 5 ⁇ g / mL chloramphenicol [Composition: 1.5% agar (W / V) The same composition as in the liquid medium A except that it is contained), and transformants Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 Got.
- Corynebacterium glutamicum (Ind-araE / pCRA811) is an independent administrative agency of the National Institute of Technology and Evaluation, Patent Biological Deposit, 2-5-8 Kazusa Kamashi, Kisarazu, Chiba, Japan Deposited at the center (date of deposit: May 28, 2008, deposit number: NITE BP-576).
- each of the two strains contains 5 ⁇ g / mL of chloramphenicol
- one platinum ear was inoculated into 10 mL of the same component composition medium as the above liquid medium and aerobic at 33 ° C. for 13 hours at 200 rpm. After culturing, the cells were washed twice with the above-described BT medium, and inoculated to 100 mL of BT medium containing 25 mM D-xylose as a single carbon source so that OD610 was 0.2. The D-xylose concentration was analyzed by liquid chromatography after sampling a small amount of the culture medium, and the change in consumption over time was examined. The results are shown in FIGS. 4 (1) and (2).
- FIG. 4 (1) is a graph showing changes in D-xylose consumption and growth over time by Corynebacterium glutamicum R / pCRA811 into which the araE gene has not been introduced.
- FIG. 4 (2) is a diagram showing time-dependent changes in D-xylose consumption and proliferation by Corynebacterium glutamicum Ind-araE / pCRA811 in which the araE gene is introduced into the chromosome.
- FIGS. 4 (1) and (2) under aerobic conditions, Corynebacterium glutamicum Ind-araE / pCRA811 (FIG.
- Example 3 Sugar Consumption and Lactic Acid Production Reaction of Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 using D-xylose as a single substrate (1) Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum (Corynebacterium glutamicum) prepared in Example 1 and (5) above, which are stored at ⁇ 80 ° C.
- Ind-araE / pCRA811 is a Agar medium containing 5 ⁇ g / mL of chloramphenicol, which is a medium for plate culture [Composition: except that 1.5% (W / V) of agar is included The same component composition as that of the liquid medium A] was allowed to stand at 33 ° C. for 12 hours in a dark place.
- Each of the two coryneform bacterial strains grown on the above plate was inoculated into a test tube containing 10 mL of chloramphenicol A liquid medium containing 5 ⁇ g / mL, and then plated at 33 ° C. for 12 hours.
- the cells were cultured aerobically at 200 rpm.
- Each of the two strains grown under the above conditions was transferred to a 1 L Erlenmeyer flask containing 500 mL of A liquid medium containing 5 ⁇ g / mL of chloramphenicol, which is an aerobic culture growth medium.
- the aerobic culture was performed at 200 rpm for 13 hours.
- the cells grown in this way were collected by centrifugation (4 ° C., 10 minutes, 5,000 ⁇ g) and subjected to the lactic acid production reaction under the following reducing conditions.
- the pH in the reaction vessel was constantly maintained at 7.5 using a 2.5 N (normal) concentration NH 4 OH aqueous solution (2.5 N ammonia aqueous solution).
- the D-xylose and lactic acid concentrations in the reaction vessel were analyzed by liquid chromatography by sequentially sampling a small amount of the medium, and the changes in consumption and production amount over time were examined.
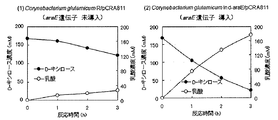
- FIG. 5 (1) is a diagram showing the time course of D-xylose consumption and lactic acid production by Corynebacterium glutamicum R / pCRA811 into which the araE gene has not been introduced.
- FIG. 5 (2) is a graph showing time-dependent changes in D-xylose consumption and lactic acid production by Corynebacterium glutamicum Ind-araE / pCRA811 in which the araE gene is introduced into the chromosome.
- black circles ( ⁇ ) indicate changes in the D-xylose concentration in the medium
- white circles ( ⁇ ) indicate changes in the lactic acid concentration in the medium.
- the D-xylose consumption rate and the lactic acid production rate are the consumption and production per unit time after 3 hours from the start of the reaction.
- Corynebacterium glutamicum R / pCRA811 Figure 5 (1)
- Corynebacterium glutamicum Ind-araE / pCRA811 Figure 5 (2)
- D-xylose consumption rate and lactic acid production rate Corynebacterium glutamicum R / pCRA811, D-xylose consumption rate 15.1 mM / h and lactic acid production rate 18.5 mM / h
- Corynebacterium glutamicum Ind-araE / pCRA811 The D-xylose consumption rate was 48.4 mM / h and the lactic acid production rate was 58.5 mM / h.
- Example 4 Aerobic culture of Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 using a medium containing D-glucose and D-xylose mixed sugar as a carbon source The reaction was carried out except that the carbon source in Example 2 was changed from D-xylose to a mixture of D-glucose and D-xylose (3.6 g / L and 3.6 g / L; abundance ratio 1: 1). Under the same conditions and method as in Example 2, aerobic culture growth and sugar consumption of Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 were examined.
- FIGS. 6 (1) and (2) The results are shown in FIGS. 6 (1) and (2).
- FIG. 6 (1) is a graph showing changes in D-glucose and D-xylose consumption and growth over time by Corynebacterium glutamicum R / pCRA811 into which the araE gene has not been introduced.
- FIG. 6 (2) is a graph showing changes in D-glucose and D-xylose consumption and growth over time by Corynebacterium glutamicum Ind-araE / pCRA811 in which the araE gene is introduced into the chromosome.
- 6A and 6B an asterisk (*) indicates a change in OD610.
- Black circles ( ⁇ ) indicate changes in D-glucose concentration in the medium.
- Open circles ( ⁇ ) indicate changes in D-xylose concentration in the medium.
- Corynebacterium glutamylum R / pCRA811 is observed to inhibit glucose in terms of D-xylose utilization in the presence of D-glucose and D-xylose, whereas corynebacteria Corynebacterium glutamicum Ind-araE / pCRA811 was able to use D-glucose and D-xylose in parallel and simultaneously.
- Example 5 Sugar consumption and lactic acid under reducing conditions of Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 using D-glucose and D-xylose mixed sugars as substrates
- Production reaction Example 3 except that the reaction was carried out by replacing the substrate in Example 3 above from D-xylose with a mixture of D-glucose and D-xylose (40 g / L and 20 g / L; abundance ratio 2: 1). After aerobic culture growth under the same conditions and method as 3, reaction under reducing conditions was carried out.
- FIGS. 7 (1) and (2) The results are shown in FIGS. 7 (1) and (2).
- FIG. 7 (1) is a graph showing changes in D-glucose and D-xylose consumption and lactic acid production over time by Corynebacterium glutamicum R / pCRA811 into which the araE gene has not been introduced.
- FIG. 7 (2) is a graph showing changes in D-glucose and D-xylose consumption and lactic acid production over time by Corynebacterium glutamicum Ind-araE / pCRA811 in which the araE gene is introduced into the chromosome.
- black circles ( ⁇ ) indicate changes in the xylose concentration in the medium.
- An asterisk (*) indicates a change in the D-glucose concentration in the medium.
- Open circles ( ⁇ ) indicate changes in lactic acid concentration in the medium.
- Corynebacterium glutamicum Ind-araE / pCRA811 was able to completely consume D-glucose and D-xylose within 8 hours, whereas Corynebacterium glutamicum (Corynebacterium glutamicum) ) With R / pCRA811, a significant amount of xylose remained in the same 8 hours.
- the lactic acid production concentrations produced by Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 after 8 hours are 339 mM and 431 mM, respectively, and the latter is about 1.3 times it was high.
- Corynebacterium glutamicum R / pCRA811 under reduced conditions shows that glucose suppression is observed for D-xylose utilization in the presence of D-glucose and D-xylose, whereas Corynebacterium glutamicum R / pCRA811 Corynebacterium glutamicum Ind-araE / pCRA811 was able to use D-glucose and D-xylose in parallel and simultaneously.
- araBAD gene amplification primer set The former has an EcoRI site and the latter has a SalI site added to the ends.
- Escherichia coli JM109 chromosomal DNA extracted in Example 1, (1), a) above was used.
- PCR was performed using “DNA thermal cycler” manufactured by Perkin Elmer Citas Co., Ltd. and using DNA polymerase (trade name: PrimeSTAR HS DNA polymerase, manufactured by Takara Shuzo Co., Ltd.) as a reaction reagent under the following conditions.
- PCR buffer 10 ⁇ L 2.5 mM dNTP mixture: 4 ⁇ L
- Template DNA 1 ⁇ L (DNA content 200 ng or less)
- primers 1 ⁇ L each (final concentration 0.2 ⁇ M)
- PrimeSTAR HS DNA polymerase 0.5 ⁇ L Sterile distilled water: 33.5 ⁇ L
- the above was mixed, and 50 ⁇ L of the reaction solution was subjected to PCR.
- PCR cycle Denaturation process: 98 ° C., 10 seconds Annealing process: 55 ° C., 10 seconds Extension process: 72 ° C., 5 minutes One cycle was performed for 30 cycles.
- Escherichia coli JM109 was transformed by the calcium chloride method [Journal of Molecular Biology., Vol. 53, 159 (1970)], and chloramphenicol 50 ⁇ g / mL, X-gal ( 5-Bromo-4-chloro-3-indoxyl-beta-D-galactopyranoside) 200 ⁇ g / mL and IPTG (isopropyl 1-thio-beta-d-galactoside) 100 ⁇ g / mL L agar medium [Composition: agar is 1. The composition was the same as that of the L medium except that 5% (W / V) was contained.
- a growing strain having a white color on this medium is subjected to liquid culture by a conventional method, and a plasmid into which a DNA fragment having a length of about 4.5 kb containing the araBAD gene is inserted from the culture solution is extracted with a plasmid extraction kit (trade name: QIAprep Spin Extraction was performed using Miniprep Kit (manufactured by Qiagen).
- the plasmid containing the araBAD gene was named Plac-araBAD (FIG. 8).
- a asterisk indicates a primer for inverse PCR.
- the base sequence indicated by b underline indicates a restriction enzyme site prepared for cloning.
- Escherichia coli JM109 was transformed by the calcium chloride method, and an L agar medium containing 50 ⁇ g / mL kanamycin [composition: containing 1.5% (W / V) agar] The same composition as that of the L medium was applied.
- the growing strain on this medium is subjected to liquid culture by a conventional method, and a plasmid inserted with a DNA fragment of about 3.3 kb in length containing the xylA-xylB gene from the culture is designated as QIAprep Spin Miniprep Kit (manufactured by Qiagen). Extracted using.
- the araE gene introduction plasmid Ind11-araE prepared in Example 1 and (3) above was introduced into Corynebacterium glutenum X5 according to the same method as described above, and the strain obtained by introducing the araE gene introduction plasmid Ind11-araE into Corynebacterium It was named as Corynebacterium glutamicum X5-Ind-araE.
- each is introduced into each of Corynebacterium glutamicum X5 and Corynebacterium glutamicum X5-Ind-araE, and an A agar medium containing 5 ⁇ g / mL of chloramphenicol [ Composition: Same composition as in the above liquid medium except that 1.5% (W / V) agar is contained], and transformed strains Corynebacterium glutamicum X5 / Plac-araBAD and Coryne Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD was obtained.
- Corynebacterium glutamicum (Corynebacterium glutamicum) X5-Ind-araE / Plac-araBAD is an independent administrative agency product evaluation technology base of 2-5-8 Kazusa Kamashi, Kisarazu City, Chiba Prefecture, Japan (zip code 292-0818) Organization Deposited at the Patent Organism Depositary (Trust Date: May 28, 2008, Deposit Number: NITE BP-577).
- Example 7 Reduction conditions of Corynebacterium glutamicum X5 / Plac-araBAD and Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD using D-glucose and D-xylose mixed sugar as substrates The following sugar consumption and lactic acid production reaction Under the same conditions and method as in Example 3 above, Corynebacterium glutamicum X5 / Plac-araBAD and Corynebacterium glutamicum X5-Ind-araE / Each Plac-araBAD was grown in aerobic culture.
- Example 3 Thereafter, the substrate in Example 3 was changed from D-xylose to a mixture of D-glucose, D-xylose and L-arabinose (32 g / L, 16 g / L and 6.4 g / L, respectively; abundance ratio 5: 2.
- the reaction was carried out under reducing conditions under the same conditions and method as in Example 3 except that the reaction was performed instead of 5: 1).
- FIG. 9 (1) is a diagram showing the change over time in sugar consumption by Corynebacterium glutamicum X5 / Plac-araBAD into which the araE gene has not been introduced.
- FIG. 9 (2) is a diagram showing the time-dependent change in sugar consumption by Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD in which the araE gene is introduced into the chromosome.
- black circles ( ⁇ ) indicate changes in the L-arabinose concentration in the medium.
- Triangles ( ⁇ ) indicate changes in D-xylose concentration in the medium.
- An asterisk (*) indicates a change in the D-glucose concentration in the medium.
- Open circles ( ⁇ ) indicate changes in lactic acid concentration in the medium.
- Corynebacterium glutamicum (X) -Ind-araE / Plac-araBAD (FIG. The consumption rate of D-xylose and L-arabinose in 2)) was significantly faster than that of Corynebacterium glutamicum X5 / Plac-araBAD (Fig. 9 (1)) without the araE gene introduced. .
- Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD completely consumes the three sugars D-glucose, D-xylose and L-arabinose in the medium within 9 hours.
- Corynebacterium glutamicum X5 / Plac-araBAD showed D-xylose and L-arabinose remaining in the same 9 hours. After 9 hours, the concentrations of lactic acid produced by Corynebacterium glutamicum X5 / Plac-araBAD and Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD were 342 mM and 374 mM, respectively. .
- Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD uses three sugars, D-glucose, D-xylose and L-arabinose, in parallel and simultaneously. Was possible.
- Example 8 Production of succinic acid under reducing conditions using Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 mixed sugars of D-glucose and D-xylose as substrates Reaction Corynebacterium glutamicum R / pCRA811 and Corynebacterium glutamicum Ind-araE / pCRA811 prepared in Example 1, (5) above are the same as in Example 5 above. Under the conditions and method, succinic acid production reaction was performed under reducing conditions after aerobic culture growth.
- Escherichia coli JM109 was transformed by the calcium chloride method, and an L agar medium containing 50 ⁇ g / mL of chloramphenicol [Composition: 1.5% (W / V) agar was included. Except for the above, the same composition as that of the L medium was applied.
- the grown strain on this medium is subjected to liquid culture by a conventional method, and a plasmid inserted with a DNA fragment of about 4.0 kb in length containing the adhB-pdc gene from the culture solution is designated as QIAprep Spin Miniprep Kit (manufactured by Qiagen). Extracted using.
- the prepared plasmid was named pEthAra (FIG. 10).
- Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh was introduced into Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh, and an A agar medium containing 5 ⁇ g / mL of chloramphenicol [Composition: 1.5% agar (W / V) Transformant Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh / pEthAra was obtained by the same composition as that of the liquid medium A except that it was included.
- Corynebacterium glutamicum (Corynebacterium glutamicum) X5-Ind-araE- ⁇ ldh / pEthAra is an independent administrative agency product evaluation technology base of 2-5-8 Kazusa Kamashi, Kisarazu City, Chiba Prefecture, Japan (zip code 292-0818) Organization Deposited at the Patent Biological Deposit Center (Accession date: June 4, 2008, Accession number: NITE BP-581).
- Example 10 Sugar consumption and ethanol production reaction under reducing conditions of Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh / pEthAra using D-glucose, D-xylose and L-arabinose mixed sugar as substrates Under the same conditions and method as in Example 3, Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh / pEthAra was grown in aerobic culture, and then the substrate in Example 3 was converted from D-xylose to D-xylose.
- Example 3 The same conditions as in Example 3 except that the mixture was replaced with a mixture of glucose, D-xylose and L-arabinose (32 g / L, 16 g / L and 6.4 g / L; abundance ratio 5: 2.5: 1). And the reaction was carried out under reducing conditions.
- Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh / pEthAra is a mixture of 3 of D-glucose, D-xylose and L-arabinose in the medium.
- the seed sugar was consumed within 9 hours to produce ethanol.
- the consumption rates of the three kinds of sugars were almost the same as those of Corynebacterium glutamicum X5-Ind-araE / Plac-araBAD in Example 7.
- black circles ( ⁇ ) indicate changes in D-glucose concentration in the medium.
- Triangles ( ⁇ ) indicate changes in D-xylose concentration in the medium.
- Black squares ( ⁇ ) indicate changes in L-arabinose concentration in the medium.
- White squares ( ⁇ ) indicate changes in ethanol concentration in the medium.
- Corynebacterium glutamicum X5-Ind-araE- ⁇ ldh / pEthAra can use three types of sugars, D-glucose, D-xylose and L-arabinose, in parallel and simultaneously. It was possible.
- the present invention can be suitably used for the production of organic compounds from cellulosic biomass resources.
Abstract
Description
また、特許文献3では、同様に、ザイモモナス モビリスに、エシェリヒア コリ由来のキシロース イソメラーゼ遺伝子、キシルロキナーゼ遺伝子、トランスケトラーゼ遺伝子及びトランスアルドラーゼ遺伝子の4つの遺伝子を染色体に導入し、発現することにより得られる、導入した遺伝子がより安定な形質転換体ザイモモナス モビリスによるD-キシロース利用技術が開示されている。これらの公知技術においては、ヘキソースに対するペントースの同時利用性はある程度達成されてはいるものの、D-キシロースの消費速度(利用速度)はD-グルコースの消費速度に比し、不十分であり、実用的な意味でのヘキソース及びペントース同時利用性技術としては更なる改良が求められている。
しかしながら、上述したように、D-グルコース及びD-キシロースの両糖の存在下でのD-キシロース利用に対するグルコース抑制が完全に解除され、D-キシロース及びD-グルコースを並行的かつ同時に効率よく利用できるコリネバクテリウム形質転換体は、未だ開発されていない。
具体的には、上述したD-キシロースの利用能を付与したコリネバクテリウム グルタミカムに、コリネバクテリウム グルタミカム(Corynebacterium glutamicum) ATCC31831由来のL-アラビノース輸送系のプロトンシンポーター遺伝子を導入したところ、思いもかけず、基質特異性が発現することもなく、D-キシロース利用速度が大幅に向上すると共に、D-グルコース及びD-キシロースの両糖の存在下での、D-キシロース利用に対するグルコース抑制が完全に解除され、D-グルコース及びD-キシロースの並行的かつ同時利用が可能となることを見出した。
(2) シュガートランスポーター機能を有するタンパク質が、L-アラビノース輸送系のプロトンシンポーターである上記(1)記載のコリネ型細菌形質転換体。
(3) シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子として、配列番号13で示される塩基配列からなるDNA、又は配列番号13で示される塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつシュガートランスポーター機能を有するポリペプチドをコードするDNAを用いる上記(1)又は(2)記載のコリネ型細菌形質転換体。
(4) D-キシロース利用能を有するコリネ型細菌が、コリネバクテリウム グルタミカム(Corynebacterium glutamicum) R (FERM P-18976)、セロビオース利用性組換え株もしくは変異株であるFERM P-18979、FERM P-18977及びFERM P-18978、コハク酸生産組換え株であるFERM P-19446及びFERM P-19477、並びにエタノール生産組換え株であるFERM P-17887、FERM P-17888、FERM P-19361及びFERM P-19362からなる群より選択されるいずれか1種の菌株のコリネ型細菌であって、かつD-キシロース利用能が付与されているものであることを特徴とする上記(1)~(3)のいずれか一項に記載のコリネ型細菌形質転換体。
(5) シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子として、コリネバクテリウム グルタミカム (Corynebacterium glutamicum) ATCC31831、エシェリヒア コリ (Escherichia coli)、バチラス サブチリス(Bacillus subtilis)、バチラス リチェニフォルミス(Bacillus licheniformis)、バチラス コアグランス(Bacillus coagulans)、ラクトバチルス サケイ(Lactobacillus sakei)、ペディオコッカス ペントサセウス(Pediococcus pentosaceus)、ラクトバチルス レウテリ(Lactobacillus reuteri)、オーシャノバチルス イヘエニス(Oceanobacillus iheyensis)、ラクトコッカス ラクティス(Lactococcus lactis)、ロイコノストック メセンテロイデス(Leuconostoc mesenteroides)、ラクトバチルス プランタリューム(Lactobacillus plantarum)、ラクトバチルス ファーメンタム(Lactobacillus fermentum)、ラクトバチルス ブレビス(Lactobacillus brevis)、ロイコノストック シテリューム(Leuconostoc citreum)、エンテロコッカス フェシウム(Enterococcus faecium)、クレブシエラ オキシトカ(Klebsiella oxytoca)、及びサルモネラ ティフィムリウム(Salmonella typhimurium)からなる群より選ばれる微生物由来のaraE遺伝子を用いる上記(1)又は(2)に記載のコリネ型細菌形質転換体。
(6) D-グルコース及びD-キシロースを並行的かつ同時利用することができる上記(1)~(5)のいずれか一項に記載のコリネ型細菌形質転換体。
(7) D-グルコース、D-キシロース及びL-アラビノースを並行的かつ同時利用することができる上記(6)記載のコリネ型細菌形質転換体。
(8) コリネ型細菌形質転換体が、コリネバクテリウム グルタミカム (Corynebacterium glutamicum) Ind-araE/pCRA811 (受託番号 NITE BP-576)である上記(1)記載のコリネ型細菌形質転換体。
(9) コリネ型細菌形質転換体が、コリネバクテリウム グルタミカム (Corynebacterium glutamicum) X5-Ind-araE/Plac-araBAD (受託番号 NITE BP-577)である上記(1)記載のコリネ型細菌形質転換体。
(10) コリネ型細菌形質転換体が、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araE-Δldh/pEthAra (受託番号 NITE BP-581)である上記(1)記載のコリネ型細菌形質転換体。
(11) D-キシロースを含有する培地中で上記(1)~(10)のいずれか一項に記載のコリネ型細菌形質転換体を用いて有機化合物を生成させる工程と、同培地より有機化合物を回収する工程とを含む有機化合物の製造方法。
(12) 有機化合物が、エタノール、乳酸、コハク酸、キシリトール、酢酸及びアミノ酸からなる群より選ばれる1種以上であることを特徴とする上記(11)記載の有機化合物の製造方法。
本発明のコリネ型細菌形質転換体は、シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子を、D-キシロース利用能を有するコリネ型細菌に導入したものである。
D-キシロース利用能を有するコリネ型細菌に、シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子を導入することにより、該コリネ型細菌のD-キシロース利用能が顕著に向上する。このため、コリネ型細菌形質転換体はD-グルコース及びD-キシロースを並行的かつ同時利用することができるものとなる。
中でも本発明では、エシェリヒア コリ(Escherichia coli)に由来するxylA遺伝子、及びxylB遺伝子の使用が最も好ましい。
マイクロコッカス属菌としては、マイクロコッカス フロイデンライヒ(Micrococcus freudenreichii)NO. 239(FERM P-13221)、マイクロコッカス ルテウス(Micrococcus leuteus)NO. 240(FERM P-13222)、マイクロコッカス ウレアエ(Micrococcus ureae)IAM1010又はマイクロコッカス ロゼウス(Micrococcus roseus)IFO3764等が挙げられる。
本発明において特に好ましいコリネ型細菌は、D-キシロース利用能が付与されたコリネバクテリウム グルタミカムR(FERM P-18976)である。
本発明では、D-キシロース利用能を有するコリネ型細菌に、シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子(シュガートランスポーター遺伝子ともいう)を導入する。これにより、コリネ型細菌形質転換体のD-キシロース利用能をより向上させることができる。シュガートランスポーター機能を有するタンパク質としては、ヘキソース及びペントースを輸送する機能を有するタンパク質であればよいが、中でも、L-アラビノース輸送系のプロトンシンポーターが好ましい。
本発明において、塩基配列間の相同性は、計算ソフト GENETYX(登録商標) Ver.8(ジェネティックス社製)を用いて計算した値である。
まず、上記のシュガートランスポーター遺伝子、例えば、L-アラビノース輸送系のプロトンシンポーターをコードするDNA(araE、タンパク質をAraEともいう)の配列をPCR (polymerase chain reaction) 法により増幅するためのオリゴヌクレオチドプライマーセットを作製する。このようなプライマーセットとしては、例えば配列番号14、15の塩基配列で示されるものなどが挙げられる。PCR法は、公知のPCR装置、例えばサーマルサイクラーなどを利用することができる。PCRのサイクルは、公知の技術にしたがって行なわれてよく、例えば、変性、アニーリング、伸張を1サイクルとし、通常10~100サイクル、好ましくは、約20~50サイクルである。PCRの鋳型としては、シュガートランスポーター遺伝子を有する微生物から単離したDNAを用いて、PCR法によりAraEをコードするDNAのcDNAを合成することができる。PCR法によって得られた遺伝子は、適当なクローニングベクターに導入することができる。クローニング法としては、pGEM-T easy vector system(Promega社製)、TOPO TA-cloning system(Invitrogen社製)、Mighty Cloning Kit(Takara社製)などの商業的に入手可能なPCRクローニングシステムなどを使用することもできる。また、方法の1つの例を実施例で詳記するが、該領域を含むDNA断片を、既知のAraEをコードするDNAの塩基配列に基づいて適当に設計された合成プライマーを鋳型として用いたハイブリダイゼーション法により取得することもできる。
目的遺伝子を含むプラスミドベクターのコリネ型細菌への導入方法としては、電気パルス法(エレクトロポレーション法)、CaC12法等コリネ型細菌への目的遺伝子導入が可能な方法であれば特に限定されるものではない。その具体例として、例えば電気パルス法としては、公知の方法〔Agric. Biol. Chem.、Vol. 54、443-447(1990)〕又は〔Res. Microbiol.、Vol. 144、181-185(1993)〕を用いることができる。
増殖培養工程
本発明のコリネ型細菌形質転換体を用いて有機化合物を製造する場合は、まず上述したコリネ型細菌形質転換体を好気条件下で増殖培養することが好ましい。
上記の如くして得られる培養物から回収分離されたコリネ型細菌形質転換体の培養菌体又はその菌体処理物を用いて、有機化合物を生成させる工程(有機化合物生成工程ともいう)を行う。
有機化合物生成工程においては、D-キシロースを含有する培地(反応培地)中で、上記コリネ型細菌形質転換体を用いて有機化合物を生成させる。このような工程を含む有機化合物の製造方法も、本発明の1つである。
具体的には、糖類としては、(D-又はL-)アラビノース、ガラクトース、フルクトース又はマンノースなどの単糖類;セロビオース、ショ糖、ラクトース、マルトースなどの二糖類;デキストリン又は可溶性澱粉などの多糖類などが挙げられる。特に、C6糖やC5糖などの単糖類が好ましく、中でも、L-アラビノースが好ましい。
培地のpHは、約6~8が好ましい。
本発明における還元状態とは、反応系(反応培地)の酸化還元電位で規定され、反応培地の酸化還元電位がマイナス(-)の値であることを意味する。還元状態下にある反応培地の酸化還元電位は、好ましくは約-200mV~-500mV程度、より好ましくは約-250mV~-500mV程度である。
(1) プラスミドpCRA811構築方法
a) エシェリヒア コリ(Escherichia coli) JM109からの染色体DNAの抽出
エシェリヒア コリ(Escherichia coli) JM109を、L培地(トリプトン 10g、イーストエキストラクト 5g及びNaCl 5gを蒸留水1Lに溶解)に、白金耳を用いて植菌後、対数増殖期まで37℃で振盪培養し、菌体を集めた。DNAゲノム抽出キット(商品名:GenomicPrep Cells and Tissue DNA Isolation Kit、アマシャム社製)を用いて、取扱説明書に従い、集めた菌体から染色体DNAを回収した。
エシェリヒア コリ(Escherichia coli)のキシロースイソメラーゼ遺伝子(以下、xylAと記す)(配列番号1)、及びキシルロキナーゼ遺伝子(以下、xylBと記す)(配列番号2)の2つの遺伝子を含むDNA断片を以下のPCR法により増幅した。

PCRは、パーキンエルマーシータス社製の「DNAサーマルサイクラー」を用い、反応試薬として、DNAポリメラーゼ(商品名:Takara LA Taq HS DNA polymerase、宝酒造株式会社製)を用いて下記の条件で行なった。

デナチュレーション過程: 94℃、1分
アニーリング過程: 55℃、1分
エクステンション過程: 72℃、2分
以上を1サイクルとし、30サイクル行なった。
上記で生成した反応液の一部を用いて1%(W/V)アガロースゲルにより電気泳動を行なったところ、それぞれxylA遺伝子及びxylB遺伝子を含む、約1.4kb及び約1.6kbのDNA断片が検出できた。
該xylA遺伝子を含むプラスミドをpCRA801と命名した。
Ptrc-xylA遺伝子増幅用プライマーセット

Ptrc-xylB遺伝子増幅用プライマーセット

a) コリネバクテリウム グルタミカム(Corynebacterium glutamicum)ATCC31831からの染色体DNAの抽出
コリネバクテリウム グルタミカム(Corynebacterium glutamicum)ATCC31831からの染色体DNA抽出は、培地として表1に示すA液体培地を使用することと、培養温度が30℃であること以外は、上記のエシェリヒア コリ(Escherichia coli)JM109と同様の条件で行なった。

L-アラビノースを単一炭素源として良好な増殖を示したコリネバクテリウム グルタミカム(Corynebacterium glutamicum)ATCC31831よりL-アラビノース代謝に関与する遺伝子のクローニングを試みた。データベースのホモロジー検索の結果より、3種のL-アラビノース代謝酵素L-アラビノースイソメラーゼ、L-リブロキナーゼ、L-リブロース-5-フォスフェイト-4-エピメラーゼをコードする各遺伝子araA、araB、araDの内、araAが最も異種間での保存性が高かった。そこで、このaraAにおける保存領域の配列から以下の一対のプライマーセットを、アプライド・バイオシステムズ社製「394 DNA/RNAシンセサイザー」を用いて合成し、PCRに使用した。


デナチュレーション過程: 94℃、1分
アニーリング過程: 37℃、1分
エクステンション過程: 72℃、1分
以上を1サイクルとし、30サイクル行なった。
PCR増幅産物は、pGEM-T easy vector system(Promega社製)を用いて取扱説明書に従い反応させた。
コリネバクテリウム グルタミカム(Corynebacterium glutamicum)ATCC31831のaraE遺伝子を含むDNA断片を以下のPCR法により増幅した。
araE遺伝子増幅用プライマーセット


デナチュレーション過程: 98℃、10秒
アニーリング過程: 55℃、30秒
エクステンション過程: 72℃、2分
以上を1サイクルとし、30サイクル行なった。
上述のEcoRI制限酵素で処理した増幅産物と、EcoRIで制限酵素処理をしたpKK223-3(ファルマシア社製)とを混合し、これにMighty Cloning Kit(宝酒造株式会社製)を添加した後、取扱説明書に従い反応させた。
この培地上の生育株を常法により液体培養し、培養液よりaraE遺伝子を含有する長さ約1.7kbのDNA断片が挿入されたプラスミドを、QIAprep Spin Miniprep Kit(キアゲン社製)を用いて抽出した。
該araE遺伝子を含むプラスミドをpKK223-3-araE(図3)と命名した。
次に、araE遺伝子をコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Rにマーカーレスで染色体に導入するために必要なDNA領域を、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)Rの生育に必須でないと報告されている配列〔Appl. Environ. Microbiol.、Vol. 71、3369-3372(2005)〕(SSI領域)を基に決定した。このDNA領域(Indel11領域)を以下のPCR法により増幅した。
araE遺伝子導入用Indel11領域増幅用プライマーセット

デナチュレーション過程: 94℃、30秒
アニーリング過程: 55℃、30秒
エクステンション過程: 72℃、3分
以上を1サイクルとし、30サイクル行なった。
上述の制限酵素XbaIで処理した増幅産物と、XbaIで制限酵素処理をしたマーカーレス遺伝子破壊用プラスミドpCRA725〔J. Mol. Microbiol. Biotechnol.、 Vol. 8、243-254(2004)、(特開2006-124440)〕とを混合し、これにMighty Cloning Kit(宝酒造株式会社製)を添加した後、取扱説明書に従い反応させた。
該araE遺伝子導入用Indel11領域を含むプラスミドをInd11LKS2-4(図3)と命名した。
該araE遺伝子導入用プラスミドをInd11-araE(図3)と命名した。
araE遺伝子導入用プラスミドInd11-araEは、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)R内で複製不可能なプラスミドである。Ind11-araEを、電気パルス法〔Agric. Biol. Chem.、Vol. 54、443-447(1990)及びRes. Microbiol.、Vol. 144、181-185(1993)〕の方法に従って、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)Rへ導入し、カナマイシン50μg/mLを含むA寒天培地〔組成:寒天が1.5%(W/V)含まれていることを除けば上記A液体培地と同一成分組成〕に塗布した。

カナマイシン感受性及びスクロース含有培地生育性を示した株をコリネバクテリウム グルタミカム(Corynebacterium glutamicum) Ind-araEと命名した。
プラスミドpCRA811を上記の実施例1、(4)項に示した電気パルス法に従って、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)Rとコリネバクテリウム グルタミカム(Corynebacterium glutamicum) Ind11-araEへ導入し、クロラムフェニコール5μg/mLを含むA寒天培地〔組成:寒天が1.5%(W/V)含まれていることを除けば上記A液体培地と同一成分組成〕により、形質転換株コリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811とコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811とを得た。尚、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811は、日本国千葉県木更津市かずさ鎌足2-5-8(郵便番号292-0818)の独立行政法人製品評価技術基盤機構 特許生物寄託センターに寄託した(受託日:2008年5月28日、受託番号:NITE BP-576)。
上記の実施例1、(5)項で作製したコリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811とコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811をそれぞれ用いて、D-キシロースを唯一の炭素源として含有するBT液体培地におけるD-キシロースの減少と菌の増殖とを検討した。該2菌株をそれぞれ、クロラムフェニコール5μg/mLを含むことを除けば上記A液体培地と同一成分組成培地10mLに一白金耳植菌して33℃、13時間、200rpmの条件で好気的に培養後、上記で示したBT培地で2回洗浄し、単一炭素源として25mM D-キシロースを含有する100mLのBT培地にOD610が0.2になるように植菌した。D-キシロース濃度は、逐次培地の少量をサンプリングして液体クロマトグラフィーによる分析を行い、経時的な消費量の変動を調べた。結果を、図4(1)及び(2)に示す。図4(1)及び(2)において、白丸(○)は、OD610の変化を、黒丸(●)は、培地中のD-キシロース濃度の変化を、それぞれ示す。図4(1)は、araE遺伝子を導入していないコリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811による、D-キシロース消費及び増殖の経時変化を示す図である。図4(2)は、araE遺伝子を染色体に導入したコリネバクテリウム グルタミカム(Corynebacterium glutamicum) Ind-araE/pCRA811による、D-キシロース消費及び増殖の経時変化を示す図である。結果、図4(1)及び(2)に示すように、好気条件下において、araE遺伝子を染色体に導入したコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811(図4(2))のD-キシロース消費速度と増殖速度は、araE遺伝子を導入していないコリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811(図4(1))と比較して、著しく速くなった。
(1)好気培養増殖
冷凍庫にて-80℃で保存してある上記の実施例1、(5)項で作製したコリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811とコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811を、それぞれプレート培養用培地であるクロラムフェニコール5μg/mLを含むA寒天培地〔組成:寒天が1.5%(W/V)含まれていることを除けば上記A液体培地と同一成分組成〕に塗布し、33℃、12時間暗所に静置した。
上記の実施例3、(1)項の好気培養増殖工程により回収された該2種の菌株それぞれの湿潤菌体 4g(Dry cell換算約0.8g)を、BT-U培地(組成:尿素が含まれていないことを除けば上記BT液体培地と同一成分組成)80mLに懸濁後、100mL容のバイアルに入れ、D-キシロース100mMを利用炭素源として加え、炭酸水素ナトリウム100mMを添加後、密閉した状態で33℃にてゆるく撹拌し、還元条件下の乳酸生成反応を行なった。
反応槽内のD-キシロース及び乳酸濃度は、逐次培地少量をサンプリングして液体クロマトグラフィーによる分析を行い、経時的な消費量及び生成量の変動をそれぞれ調べた。
この結果より、還元条件下においても、araE遺伝子を染色体に導入したコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811のD-キシロース消費速度及び乳酸生成速度はいずれも、araE遺伝子を導入していないコリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811と比較して、著しく速くなったことが分かった。
上記の実施例2における炭素源をD-キシロースから、D-グルコース及びD-キシロースの混合物(3.6g/L及び3.6g/L;存在比率1:1)に代えて反応を行なう以外は、実施例2と同様の条件及び方法にて、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811及びコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811の好気培養増殖及び糖消費を調べた。
上記の実施例3における基質をD-キシロースから、D-グルコース及びD-キシロースの混合物(40g/L及び20g/L;存在比率2:1)に代えて反応を行なう以外は、実施例3と同様の条件及び方法にて好気培養増殖後、還元条件下での反応を行なった。
(1) Plac-araBADの構築方法
エシェリヒア コリ(Escherichia coli)のL-アラビノースイソメラーゼ(以下araAと記す)、L-リブロキナーゼ(araBと記す)、L-リブロース-5-フォスフェイト-4-エピメラーゼ(araDと記す)の3つの遺伝子はaraB、araA、araDが順に連結したオペロン(araBAD)を形成している(配列番号18)〔Gene、Vol. 47、231-244(1986)〕。araBAD遺伝子を含むDNA断片を以下のPCR法により増幅した。
araBAD遺伝子増幅用プライマーセット

PCRは、パーキンエルマーシータス社製の「DNAサーマルサイクラー」を用い、反応試薬として、DNAポリメラーゼ(商品名:PrimeSTAR HS DNA polymerase、宝酒造株式会社製)を用いて下記の条件で行なった。
(5×)PCR緩衝液: 10μL
2.5mM dNTP混合液: 4μL
鋳型DNA: 1μL (DNA含有量200ng以下)
上記の2種のプライマー: 各々 1μL(最終濃度0.2μM)
PrimeSTAR HS DNA polymerase: 0.5μL
滅菌蒸留水: 33.5μL
以上を混合し、この50μLの反応液をPCRにかけた。
デナチュレーション過程: 98℃、10秒
アニーリング過程: 55℃、10秒
エクステンション過程: 72℃、5分
以上を1サイクルとし、30サイクル行なった。
上述の制限酵素EcoRI及びSal Iで処理した増幅産物と、EcoRI及びSal Iで制限酵素処理をしたコリネ型細菌-大腸菌シャトルベクターpCRB1(特開2006-124440)とを混合し、これにライゲーションキット(商品名:Mighty Cloning Kit、宝酒造株式会社製)を添加後、取扱説明書に従い反応させた。
該araBAD遺伝子を含むプラスミドをPlac-araBAD(図8)と命名した。
a) コリネバクテリウム グルタミカム(Corynebacterium glutamicum)RからのSSI領域のクローニング
コリネバクテリウム グルタミカム(Corynebacterium glutamicum)RのSSI領域にD-キシロース代謝関連遺伝子xylA及びxylB遺伝子の導入を行なうため、これらのDNA領域を、表3に示したプライマー及びコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Rの染色体DNAを用いたPCR反応により増幅させた。PCRは、Takara LA Taq HS DNA polymeraseを使用し、上記の実施例1、(3)項と同条件で行なった
上記で生成した反応液を用いて1%(W/V)アガロースゲルにより電気泳動を行なったところ、約2.0から3.0kbのDNA断片が検出できた。

表1において、b アンダーラインで示した塩基配列は、クローニング用に作製した制限酵素サイトを示している。
上記の実施例1、(1)項で作製したプラスミドpCRA811をXbaI及びXhoIで処理後、アガロース電気泳動で分離し、ゲルから切り出した約3.3kbのxylA-xylB遺伝子を含むDNA断片をゲル抽出キット(商品名:Minelute Gel Extraction Kit、キアゲン社製)を用いて回収した。得られたDNAを、XbaI及びXhoIで制限酵素処理をした上記実施例6、(2)、a)項で作製したプラスミドと混合し、これにMighty Cloning Kit(宝酒造株式会社製)を添加した後、取扱説明書に従い反応させた。
上記の実施例6、(2)、b)項で作製したxylA-xylB遺伝子導入用プラスミドを上記の実施例1、(3)の方法に従って、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)RのSSI領域にXyl3、7、8、4、1、11、6の順番に導入した。最終的に得られた菌株をコリネバクテリウム グルタミカム(Corynebacterium glutamicum) X5と命名した。
プラスミPlac-araBADを上記の実施例1、(4)項に示した電気パルス法に従って、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5及びコリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araEそれぞれへ導入し、クロラムフェニコール5μg/mLを含むA寒天培地〔組成:寒天が1.5%(W/V)含まれていることを除けば上記A液体培地と同一成分組成〕により、形質転換株コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5/Plac-araBAD及びコリネバクテリウム グルタミカム(Corynebacterium glutamicum) X5-Ind-araE/Plac-araBADを得た。尚、コリネバクテリウム グルタミカム(Corynebacterium glutamicum) X5-Ind-araE/Plac-araBADは、日本国千葉県木更津市かずさ鎌足2-5-8(郵便番号292-0818)の独立行政法人製品評価技術基盤機構 特許生物寄託センターに寄託した(受託日:2008年5月28日、受託番号:NITE BP-577)。
上記の実施例3と同様の条件及び方法にて、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5/Plac-araBAD及びコリネバクテリウム グルタミカム(Corynebacterium glutamicum) X5-Ind-araE/Plac-araBADそれぞれを好気培養増殖させた。その後、上記の実施例3における基質をD-キシロースから、D-グルコース、D-キシロース及びL-アラビノースの混合物(それぞれ32g/L、16g/L及び6.4g/L;存在比率5:2.5:1)に代えて行なう以外は、上記の実施例3と同様の条件及び方法にて、還元条件下での反応を行なった。
上記の実施例1、(5)項で作製したコリネバクテリウム グルタミカム(Corynebacterium glutamicum)R/pCRA811及びコリネバクテリウム グルタミカム(Corynebacterium glutamicum)Ind-araE/pCRA811それぞれを、上記の実施例5と同様の条件及び方法にて、好気培養増殖後、還元条件下、コハク酸生成反応を行った。
(1) プラスミドpEthAra構築方法
エタノール生産に必要なザイモモナス モビリス(Zymomonas mobilis)のアルデヒドデヒドロゲナーゼ(以下adhBと記す)及びピルベートデカルボキシラーゼ(pdcと記す)の2つの遺伝子〔Journal Mol Microbiol Biotechnol、Vol. 8、4(2004)〕を含むプラスミドpCRA723〔J. Mol. Microbiol. Biotechnol.、 Vol. 8、243-254(2004)、(特開2006-124440)〕を制限酵素BamHI及びBgl IIで処理後、アガロース電気泳動で分離した。ゲルから切り出した約4.0kbのadhB-pdc遺伝子を含むDNA断片をゲル抽出キット(商品名:Minelute Gel Extraction Kit、キアゲン社製)を用いて回収した。得られたDNAを、平滑末端処理キット(商品名:DNA Blunting Kit、宝酒造株式会社製)を用いて平滑末端処理を行ない、Sal Iで制限酵素処理後、同じく平滑末端処理をした上記の実施例6、(1)項で作製したプラスミドPlac-araBADを混合し、これにMighty Cloning Kit(宝酒造株式会社製)を添加した後、取扱説明書に従い反応させた。
乳酸デヒドロゲナーゼ(ldh)遺伝子をマーカーレス破壊するために、マーカーレスldh遺伝子破壊用プラスミドpCRA728〔J. Mol. Microbiol. Biotechnol.、Vol. 8、243-254(2004)〕を上記の実施例1、(3)の方法に従って、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind11-araEに導入した。最終的に得られた菌株をコリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araE-Δldhと命名した。
上記の実施例9、(1)項で作製したプラスミドpEthAraを、上記の実施例1、(4)項に示した電気パルス法に従って、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araE-Δldhへ導入し、クロラムフェニコール5μg/mLを含むA寒天培地〔組成:寒天が1.5%(W/V)含まれていることを除けば上記A液体培地と同一成分組成〕により、形質転換株コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araE-Δldh/pEthAraを得た。尚、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araE-Δldh/pEthAraは、日本国千葉県木更津市かずさ鎌足2-5-8(郵便番号292-0818)の独立行政法人製品評価技術基盤機構 特許生物寄託センターに寄託した(受託日:2008年6月4日、受託番号:NITE BP-581)。
上記の実施例3と同様の条件及び方法にて、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araE-Δldh/pEthAraを好気培養増殖後、上記の実施例3における基質をD-キシロースからD-グルコース、D-キシロース及びL-アラビノースの混合物(32g/L、16g/L及び6.4g/L;存在比率5:2.5:1)に代えて行なう以外は、実施例3と同様の条件及び方法にて還元条件下での反応を行なった。
Claims (12)
- シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子を、D-キシロース利用能を有するコリネ型細菌に導入したことを特徴とする、コリネ型細菌形質転換体。
- シュガートランスポーター機能を有するタンパク質が、L-アラビノース輸送系のプロトンシンポーターである請求項1記載のコリネ型細菌形質転換体。
- シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子として、配列番号13で示される塩基配列からなるDNA、又は配列番号13で示される塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつシュガートランスポーター機能を有するポリペプチドをコードするDNAを用いる請求項1又は2記載のコリネ型細菌形質転換体。
- D-キシロース利用能を有するコリネ型細菌が、コリネバクテリウム グルタミカム(Corynebacterium glutamicum) R (FERM P-18976)、セロビオース利用性組換え株もしくは変異株であるFERM P-18979、FERM P-18977及びFERM P-18978、コハク酸生産組換え株であるFERM P-19446及びFERM P-19477、並びにエタノール生産組換え株であるFERM P-17887、FERM P-17888、FERM P-19361及びFERM P-19362からなる群より選択されるいずれか1種の菌株のコリネ型細菌であって、かつD-キシロース利用能が付与されているものであることを特徴とする請求項1~3のいずれか一項に記載のコリネ型細菌形質転換体。
- シュガートランスポーター機能を有するタンパク質をコードする外来性遺伝子として、コリネバクテリウム グルタミカム (Corynebacterium glutamicum) ATCC31831、エシェリヒア コリ (Escherichia coli)、バチラス サブチリス(Bacillus subtilis)、バチラス リチェニフォルミス(Bacillus licheniformis)、バチラス コアグランス(Bacillus coagulans)、ラクトバチルス サケイ(Lactobacillus sakei)、ペディオコッカス ペントサセウス(Pediococcus pentosaceus)、ラクトバチルス レウテリ(Lactobacillus reuteri)、オーシャノバチルス イヘエニス(Oceanobacillus iheyensis)、ラクトコッカス ラクティス(Lactococcus lactis)、ロイコノストック メセンテロイデス(Leuconostoc mesenteroides)、ラクトバチルス プランタリューム(Lactobacillus plantarum)、ラクトバチルス ファーメンタム(Lactobacillus fermentum)、ラクトバチルス ブレビス(Lactobacillus brevis)、ロイコノストック シテリューム(Leuconostoc citreum)、エンテロコッカス フェシウム(Enterococcus faecium)、クレブシエラ オキシトカ(Klebsiella oxytoca)、及びサルモネラ ティフィムリウム(Salmonella typhimurium)からなる群より選ばれる微生物由来のaraE遺伝子を用いる請求項1又は2に記載のコリネ型細菌形質転換体。
- D-グルコース及びD-キシロースを並行的かつ同時利用することができる請求項1~5のいずれか一項に記載のコリネ型細菌形質転換体。
- D-グルコース、D-キシロース及びL-アラビノースを並行的かつ同時利用することができる請求項6記載のコリネ型細菌形質転換体。
- コリネ型細菌形質転換体が、コリネバクテリウム グルタミカム (Corynebacterium glutamicum) Ind-araE/pCRA811 (受託番号 NITE BP-576)である請求項1記載のコリネ型細菌形質転換体。
- コリネ型細菌形質転換体が、コリネバクテリウム グルタミカム (Corynebacterium glutamicum) X5-Ind-araE/Plac-araBAD (受託番号 NITE BP-577)である請求項1記載のコリネ型細菌形質転換体。
- コリネ型細菌形質転換体が、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)X5-Ind-araE-Δldh/pEthAra (受託番号 NITE BP-581)である請求項1記載のコリネ型細菌形質転換体。
- D-キシロースを含有する培地中で請求項1~10のいずれか一項に記載のコリネ型細菌形質転換体を用いて有機化合物を生成させる工程と、同培地より有機化合物を回収する工程とを含む有機化合物の製造方法。
- 有機化合物が、エタノール、乳酸、コハク酸、キシリトール、酢酸及びアミノ酸からなる群より選ばれる1種以上であることを特徴とする請求項11記載の有機化合物の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/995,774 US8685703B2 (en) | 2008-06-17 | 2009-06-10 | Coryneform bacterium transformant having improved D-xylose-utilizing ability |
| CN2009801231392A CN102066553B (zh) | 2008-06-17 | 2009-06-10 | 具有改进的d-木糖利用能力的棒状杆菌转化体 |
| EP09766570.7A EP2287287B1 (en) | 2008-06-17 | 2009-06-10 | Coryneform bacterium transformant having improved d-xylose-utilizing function |
| JP2010517869A JP5564423B2 (ja) | 2008-06-17 | 2009-06-10 | D−キシロース利用機能が向上したコリネ型細菌形質転換体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008157409 | 2008-06-17 | ||
| JP2008-157409 | 2008-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009154122A1 true WO2009154122A1 (ja) | 2009-12-23 |
Family
ID=41434038
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/060637 WO2009154122A1 (ja) | 2008-06-17 | 2009-06-10 | D-キシロース利用機能が向上したコリネ型細菌形質転換体 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8685703B2 (ja) |
| EP (1) | EP2287287B1 (ja) |
| JP (1) | JP5564423B2 (ja) |
| KR (1) | KR20110027780A (ja) |
| CN (1) | CN102066553B (ja) |
| WO (1) | WO2009154122A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012067174A1 (ja) * | 2010-11-18 | 2012-05-24 | グリーンフェノール・高機能フェノール樹脂製造技術研究組合 | コリネ型細菌形質転換体及びそれを用いるフェノールの製造方法 |
| CN103097530A (zh) * | 2010-09-08 | 2013-05-08 | 绿色苯酚·高机能苯酚树脂制造技术研究组合 | 棒状细菌转化体及使用该转化体的苯酚的制造方法 |
| CN103228784A (zh) * | 2010-11-10 | 2013-07-31 | 绿色苯酚·高机能苯酚树脂制造技术研究组合 | 棒状细菌转化体及使用该转化体的苯酚的制造方法 |
| JP2014512818A (ja) * | 2011-04-22 | 2014-05-29 | ディーエスエム アイピー アセッツ ビー.ブイ. | アラビノース及びキシロースを含む糖類の変換能を有する酵母細胞 |
| JP2015173628A (ja) * | 2014-03-14 | 2015-10-05 | 公益財団法人地球環境産業技術研究機構 | キシロオリゴ糖利用能を付与したコリネ型細菌形質転換体 |
| CN111607601A (zh) * | 2020-04-24 | 2020-09-01 | 天津大学 | 谷氨酸棒杆菌转录调控因子IpsA突变体及应用 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8530203B2 (en) | 2008-09-01 | 2013-09-10 | Shinshu University | Process for producing useful substance |
| RU2584593C2 (ru) * | 2012-01-10 | 2016-05-20 | СиДжей ЧеилДжеданг Корпорейшн | МИКРООРГАНИЗМЫ Corynebacterium, СПОСОБНЫЕ УТИЛИЗИРОВАТЬ КСИЛОЗУ, И СПОСОБ ПОЛУЧЕНИЯ L-ЛИЗИНА С ПРИМЕНЕНИЕМ ТАКИХ МИКРООРГАНИЗМОВ |
| CN102559527B (zh) * | 2012-01-11 | 2013-12-11 | 山东大学 | 一株木糖转运能力高的酿酒酵母菌株及其应用 |
| KR101487810B1 (ko) * | 2013-07-29 | 2015-01-30 | 한국과학기술연구원 | 바이오 화학물질 발효균주의 제조방법 |
| CN104844698B (zh) * | 2014-02-16 | 2020-09-25 | 中国科学院天津工业生物技术研究所 | 一种促进微生物细胞转运葡萄糖、木糖和阿拉伯糖的方法及其在生物基产品发酵中的应用 |
| MX2018009634A (es) | 2016-02-19 | 2018-12-17 | Intercontinental Great Brands Llc | Procesos para crear multiples flujos de valores a partir de fuentes de biomasa. |
| CN107012161A (zh) * | 2017-04-03 | 2017-08-04 | 天津大学 | 利用秸秆水解液高产琥珀酸的谷氨酸棒杆菌及构建及应用 |
| CN110551648B (zh) * | 2018-05-30 | 2022-03-15 | 天津大学 | 发酵木糖生产琥珀酸的谷氨酸棒杆菌及用途 |
| CN112458108A (zh) * | 2020-11-24 | 2021-03-09 | 华东理工大学 | 一种在谷氨酸棒状杆菌中利用木糖生成谷氨酸的合成路径的构建方法 |
Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57183799A (en) | 1981-04-17 | 1982-11-12 | Kyowa Hakko Kogyo Co Ltd | Novel plasmid |
| JPS5867699A (ja) | 1981-10-16 | 1983-04-22 | Ajinomoto Co Inc | プラスミド |
| JPS62166890A (ja) | 1986-01-20 | 1987-07-23 | Asahi Chem Ind Co Ltd | イソクエン酸デヒドロゲナ−ゼ産生遺伝子を含むdna断片 |
| JPS644935A (en) | 1987-06-29 | 1989-01-10 | Toshiba Corp | Information recording medium |
| US5789210A (en) | 1993-11-08 | 1998-08-04 | Purdue Research Foundation | Recombinant yeasts for effective fermentation of glucose and xylose |
| WO2001096573A1 (fr) | 2000-06-16 | 2001-12-20 | Research Institute Of Innovative Technology For The Earth | Procede de production d'ethanol au moyen d'une bacterie coryneforme de recombinaison |
| WO2002038740A1 (en) | 2000-11-01 | 2002-05-16 | Midwest Research Institute | Recombinant zymomonas mobilis with improved xylose utilization |
| JP2002252190A (ja) | 2001-02-22 | 2002-09-06 | Toshiba Corp | 半導体基板の研磨方法および半導体基板の研磨装置 |
| JP2002338596A (ja) * | 1998-12-18 | 2002-11-27 | Ajinomoto Co Inc | 糖トランスポーター及びそれをコードする遺伝子 |
| JP2003512024A (ja) * | 1999-07-01 | 2003-04-02 | ビーエーエスエフ アクチェンゲゼルシャフト | ホスホエノールピルビン酸:糖ホスホトランスフェラーゼ系タンパク質をコードするコリネバクテリウム−グルタミカム遺伝子 |
| JP2004089029A (ja) | 2002-08-29 | 2004-03-25 | Research Institute Of Innovative Technology For The Earth | 新規なセロビオース資化性微生物 |
| WO2005010182A1 (ja) | 2003-07-29 | 2005-02-03 | Research Institute Of Innovative Technology For The Earth | コリネ型細菌形質転換体及びそれを用いるジカルボン酸の製造方法 |
| JP2006124440A (ja) | 2004-10-27 | 2006-05-18 | Kohjin Co Ltd | 非結晶性ポリエステル樹脂の製造方法 |
| JP2007222439A (ja) | 2006-02-24 | 2007-09-06 | Yamaha Livingtec Corp | 蒸気発生装置 |
| US7354755B2 (en) | 2000-05-01 | 2008-04-08 | Midwest Research Institute | Stable zymomonas mobilis xylose and arabinose fermenting strains |
| JP2008517583A (ja) * | 2004-10-22 | 2008-05-29 | 味の素株式会社 | 腸内細菌科の細菌を用いたl−アミノ酸の製造方法 |
| JP2009050236A (ja) | 2007-08-29 | 2009-03-12 | Research Institute Of Innovative Technology For The Earth | L−アラビノース利用機能を有するコリネ型細菌形質転換体 |
| JP4294373B2 (ja) | 2003-05-23 | 2009-07-08 | 財団法人地球環境産業技術研究機構 | エタノールの新規製造方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002338956A (ja) * | 2001-05-16 | 2002-11-27 | Yamagata Public Corp For The Development Of Industry | 紫外線吸収能を有する蛍光体 |
-
2009
- 2009-06-10 US US12/995,774 patent/US8685703B2/en active Active
- 2009-06-10 JP JP2010517869A patent/JP5564423B2/ja active Active
- 2009-06-10 EP EP09766570.7A patent/EP2287287B1/en active Active
- 2009-06-10 CN CN2009801231392A patent/CN102066553B/zh active Active
- 2009-06-10 KR KR1020117001106A patent/KR20110027780A/ko not_active Application Discontinuation
- 2009-06-10 WO PCT/JP2009/060637 patent/WO2009154122A1/ja active Application Filing
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57183799A (en) | 1981-04-17 | 1982-11-12 | Kyowa Hakko Kogyo Co Ltd | Novel plasmid |
| JPS5867699A (ja) | 1981-10-16 | 1983-04-22 | Ajinomoto Co Inc | プラスミド |
| JPS62166890A (ja) | 1986-01-20 | 1987-07-23 | Asahi Chem Ind Co Ltd | イソクエン酸デヒドロゲナ−ゼ産生遺伝子を含むdna断片 |
| JPS644935A (en) | 1987-06-29 | 1989-01-10 | Toshiba Corp | Information recording medium |
| US5789210A (en) | 1993-11-08 | 1998-08-04 | Purdue Research Foundation | Recombinant yeasts for effective fermentation of glucose and xylose |
| JP2002338596A (ja) * | 1998-12-18 | 2002-11-27 | Ajinomoto Co Inc | 糖トランスポーター及びそれをコードする遺伝子 |
| JP2003512024A (ja) * | 1999-07-01 | 2003-04-02 | ビーエーエスエフ アクチェンゲゼルシャフト | ホスホエノールピルビン酸:糖ホスホトランスフェラーゼ系タンパク質をコードするコリネバクテリウム−グルタミカム遺伝子 |
| US7354755B2 (en) | 2000-05-01 | 2008-04-08 | Midwest Research Institute | Stable zymomonas mobilis xylose and arabinose fermenting strains |
| WO2001096573A1 (fr) | 2000-06-16 | 2001-12-20 | Research Institute Of Innovative Technology For The Earth | Procede de production d'ethanol au moyen d'une bacterie coryneforme de recombinaison |
| WO2002038740A1 (en) | 2000-11-01 | 2002-05-16 | Midwest Research Institute | Recombinant zymomonas mobilis with improved xylose utilization |
| JP2002252190A (ja) | 2001-02-22 | 2002-09-06 | Toshiba Corp | 半導体基板の研磨方法および半導体基板の研磨装置 |
| JP2004089029A (ja) | 2002-08-29 | 2004-03-25 | Research Institute Of Innovative Technology For The Earth | 新規なセロビオース資化性微生物 |
| JP4294373B2 (ja) | 2003-05-23 | 2009-07-08 | 財団法人地球環境産業技術研究機構 | エタノールの新規製造方法 |
| WO2005010182A1 (ja) | 2003-07-29 | 2005-02-03 | Research Institute Of Innovative Technology For The Earth | コリネ型細菌形質転換体及びそれを用いるジカルボン酸の製造方法 |
| JP2008517583A (ja) * | 2004-10-22 | 2008-05-29 | 味の素株式会社 | 腸内細菌科の細菌を用いたl−アミノ酸の製造方法 |
| JP2006124440A (ja) | 2004-10-27 | 2006-05-18 | Kohjin Co Ltd | 非結晶性ポリエステル樹脂の製造方法 |
| JP2007222439A (ja) | 2006-02-24 | 2007-09-06 | Yamaha Livingtec Corp | 蒸気発生装置 |
| JP2009050236A (ja) | 2007-08-29 | 2009-03-12 | Research Institute Of Innovative Technology For The Earth | L−アラビノース利用機能を有するコリネ型細菌形質転換体 |
Non-Patent Citations (37)
| Title |
|---|
| "Current protocols in molecular biology", 1987, GREEN PUBLISHING AND WILEY INTERSCIENCE |
| "Molecular Cloning: A Laboratory Manual", 2001, CSHL PRESS |
| "The Prokaryotes, A Handbook on Habitats, Isolation and Identification of Bacteria", 1981, SPRINGER VERLAG, article "The dissimilatory sulfate-reducing bacteria", pages: 926 - 940 |
| AGRIC. BIOL. CHEM., vol. 48, 1984, pages 2901 - 2903 |
| AGRIC. BIOL. CHEM., vol. 54, 1990, pages 443 - 447 |
| AGRIC., BIOL., CHEM., vol. 48, 1984, pages 2901 - 2903 |
| APPL. ENVIRON. MICROBIOL., vol. 57, 1991, pages 759 - 764 |
| APPL. ENVIRON. MICROBIOL., vol. 71, 2005, pages 3369 - 3372 |
| APPLIED AND ENVIRONMENTAL MICROBIOLOGY, vol. 64, 1998, pages 1852 - 1859 |
| APPLIED AND ENVIRONMENTAL MICROBIOLOGY, vol. 72, 2006, pages 3418 - 3428 |
| APPLIED AND ENVIRONMENTAL MICROBIOLOGY, vol. 73, 2007, pages 2349 - 2353 |
| APPLIED MICROBIOLOGY AND BIOTECHNOLOGY, vol. 56, 2001, pages 120 - 125 |
| APPLIED MICROBIOLOGY AND BIOTECHNOLOGY, vol. 57, 2001, pages 186 - 191 |
| BARGEYS MANUAL OF DETERMINATIVE BACTERIOLOGY, vol. 8, 1974, pages 599 |
| FEMS YEAST RESEARCH, vol. 5, 2005, pages 925 - 934 |
| GENE, vol. 102, 1991, pages 93 - 98 |
| GENE, vol. 47, 1986, pages 231 - 244 |
| HERNANDEZ-MONTALVO V. ET AL.: "Characterization of sugar mixtures utilization by an Escherichia coli mutant devoid of the phosphotransferase system.", APPL.MICROBIOL.BIOTECHNOL., vol. 57, no. 1-2, 2001, pages 186 - 191, XP002354251 * |
| HIDEO KAWAGUCHI ET AL.: "Coryne-gata Saikin ni Okeru Arabinose Taisha Keiro no Kochiku", NIPPON NOGEI KAGAKUKAI TAIKAI KOEN YOSHISHU, 5 March 2007 (2007-03-05), pages 43, XP008135489 * |
| HIDEO KAWAGUCHI ET AL.: "Coryne-gata Saikin ni Okeru xylose Taisha Keiro no Kochiku", NIPPON NOGEI KAGAKUKAI TAIKAI KOEN YOSHISHU, 5 March 2006 (2006-03-05), pages 262, XP008135491 * |
| J. BACTERIOL., vol. 159, 1984, pages 306 - 311 |
| J. BACTERIOL., vol. 177, 1995, pages 5379 - 5380 |
| J. BACTERIOL., vol. 179, 1997, pages 7705 - 7711 |
| J. BIOL. CHEM., vol. 263, 1988, pages 8003 - 8010 |
| J. MOL. MICROBIOL. BIOTECHNOL., vol. 8, 2004, pages 243 - 254 |
| J. SAMBROOK ET AL.: "Molecular Cloning, A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY PRESS |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 53, 1970, pages 159 |
| JOURNAL OF MOLECULAR MICROBIOLOGY AND BIOTECHNOLOGY, vol. 8, 2004, pages 243 - 254 |
| KAWAGUCHI H. ET AL.: "Engineering of a xylose metabolic pathway in Corynebacterium glutamicum.", APPL.ENVIRON.MICROBIOL., vol. 72, no. 5, 2006, pages 3418 - 3428, XP008135484 * |
| KYOTO DAIGAKU NOGAKUBU NOGEIKAGAKU KYOSHITU, NOGEIKAGAKU JIKKENSHO, vol. 3, 1990 |
| MIHO SASAKI ET AL.: "Kumikae Coryne-gata Saikin ni yoru Soft Biomass Yurai Kongoto no Doji Riyo", NIPPON NOGEI KAGAKUKAI TAIKAI KOEN YOSHISHU, 5 March 2008 (2008-03-05), pages 267, XP008135488 * |
| NICHOLS N.N. ET AL.: "Use of catabolite repression mutants for fermentation of sugar mixtures to ethanol.", APPL.MICROBIOL. BIOTECHNOL., vol. 56, no. 1-2, 2001, pages 120 - 125, XP008135486 * |
| NUCLEIC ACIDS SYMP. SER., vol. 16, 1985, pages 265 - 267 |
| RES. MICROBIOL., vol. 144, 1993, pages 181 - 185 |
| SA-NOGUEIRA I. ET AL.: "Cloning, functional analysis, and transcriptional regulation of the Bacillus subtilis araE gene involved in L- arabinose utilization.", J.BACTERIOL., vol. 179, no. 24, 1997, pages 7705 - 7711, XP008135485 * |
| See also references of EP2287287A4 |
| SHINSUKE SAKAI ET AL.: "Coryne-gata Saikin o Mochiita Kongo Torui kara no Bioethanol Seisan Process no Kochiku", NIPPON NOGEI KAGAKUKAI TAIKAI KOEN YOSHISHU, 5 March 2007 (2007-03-05), pages 42, XP008135490 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9328361B2 (en) | 2010-09-08 | 2016-05-03 | Green Phenol Development Co., Ltd. | Coryneform bacterium transformant and process for producing phenol using the same |
| CN103097530A (zh) * | 2010-09-08 | 2013-05-08 | 绿色苯酚·高机能苯酚树脂制造技术研究组合 | 棒状细菌转化体及使用该转化体的苯酚的制造方法 |
| JP5932649B2 (ja) * | 2010-09-08 | 2016-06-08 | グリーンフェノール開発株式会社 | コリネ型細菌形質転換体及びそれを用いるフェノールの製造方法 |
| US9090900B2 (en) | 2010-11-10 | 2015-07-28 | Green Phenol Development Co., Ltd. | Coryneform bacterium transformant and process for producing phenol using the same |
| CN103228784A (zh) * | 2010-11-10 | 2013-07-31 | 绿色苯酚·高机能苯酚树脂制造技术研究组合 | 棒状细菌转化体及使用该转化体的苯酚的制造方法 |
| CN103384721A (zh) * | 2010-11-18 | 2013-11-06 | 绿色苯酚·高机能苯酚树脂制造技术研究组合 | 棒状细菌转化体及使用该转化体的苯酚的制造方法 |
| EP2641964A4 (en) * | 2010-11-18 | 2014-05-07 | Green Phenol Technology Res Ass | CORNEFORMED BACTERIUM TRANSFORMER, AND PHENOL MANUFACTURING METHOD |
| US8846367B2 (en) | 2010-11-18 | 2014-09-30 | Green Phenol Development Co., Ltd. | Coryneform bacterium transformant and process for producing phenol using the same |
| WO2012067174A1 (ja) * | 2010-11-18 | 2012-05-24 | グリーンフェノール・高機能フェノール樹脂製造技術研究組合 | コリネ型細菌形質転換体及びそれを用いるフェノールの製造方法 |
| JP5887277B2 (ja) * | 2010-11-18 | 2016-03-16 | グリーンフェノール開発株式会社 | コリネ型細菌形質転換体及びそれを用いるフェノールの製造方法 |
| CN103384721B (zh) * | 2010-11-18 | 2016-03-23 | 绿色苯酚开发株式会社 | 棒状细菌转化体及使用该转化体的苯酚的制造方法 |
| EP2641964A1 (en) * | 2010-11-18 | 2013-09-25 | Green Phenol Technology Research Association | Coryneform bacterium transformant and method for producing phenol using same |
| JP2014512818A (ja) * | 2011-04-22 | 2014-05-29 | ディーエスエム アイピー アセッツ ビー.ブイ. | アラビノース及びキシロースを含む糖類の変換能を有する酵母細胞 |
| JP2015173628A (ja) * | 2014-03-14 | 2015-10-05 | 公益財団法人地球環境産業技術研究機構 | キシロオリゴ糖利用能を付与したコリネ型細菌形質転換体 |
| CN111607601A (zh) * | 2020-04-24 | 2020-09-01 | 天津大学 | 谷氨酸棒杆菌转录调控因子IpsA突变体及应用 |
| CN111607601B (zh) * | 2020-04-24 | 2022-10-18 | 天津大学 | 谷氨酸棒杆菌转录调控因子IpsA突变体及应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5564423B2 (ja) | 2014-07-30 |
| CN102066553A (zh) | 2011-05-18 |
| US20110117612A1 (en) | 2011-05-19 |
| JPWO2009154122A1 (ja) | 2011-12-01 |
| EP2287287A1 (en) | 2011-02-23 |
| US8685703B2 (en) | 2014-04-01 |
| EP2287287B1 (en) | 2015-04-08 |
| EP2287287A4 (en) | 2012-07-11 |
| CN102066553B (zh) | 2013-08-07 |
| KR20110027780A (ko) | 2011-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5564423B2 (ja) | D−キシロース利用機能が向上したコリネ型細菌形質転換体 | |
| Sasaki et al. | Xylitol production by recombinant Corynebacterium glutamicum under oxygen deprivation | |
| US10066217B2 (en) | Thermophilic organisms for conversion of lignocellulosic biomass to ethanol | |
| WO2008041840A1 (en) | Metabolic engineering of arabinose- fermenting yeast cells | |
| EP2545178A1 (en) | Acid production by fermentation | |
| Gopinath et al. | Corynebacterium glutamicum as a potent biocatalyst for the bioconversion of pentose sugars to value-added products | |
| KR102323473B1 (ko) | 코리네형 세균 형질 전환체 및 이를 이용하는 4-히드록시벤조산 또는 그 염의 제조 방법 | |
| US20110230682A1 (en) | Microorganisms with inactivated lactate dehydrogenase gene (ldh) for chemical production | |
| US20180030482A1 (en) | Use of acetaldehyde in the fermentative production of ethanol | |
| JP5074131B2 (ja) | L−アラビノース利用機能を有するコリネ型細菌形質転換体 | |
| Tamakawa et al. | Construction of a Candida utilis strain with ratio-optimized expression of xylose-metabolizing enzyme genes by cocktail multicopy integration method | |
| CA3049415A1 (en) | Obtaining high-performance yeast strains for metabolizing arabinose | |
| EP2643466B1 (en) | Yeast cells and process for converting acetaldehyde to ethanol | |
| JP2007295809A (ja) | コリネ型細菌形質転換体による高効率な有機化合物の製造方法 | |
| CN110760536A (zh) | 甲醇生物转化菌株的构建方法以及构建的菌株和应用 | |
| EP2397556A1 (en) | Thermophilic organisms for conversion of lignocellulosic biomass to ethanol | |
| JP6434704B2 (ja) | キシロオリゴ糖利用能を付与したコリネ型細菌形質転換体 | |
| SI24955A (sl) | Modificirana 6-fosfofrukto-1-kinaza, ki rekombinantnim celicam kvasovke Saccharomyces cerevisiae omogoča fermentativno uporabo pentoznih sladkorjev | |
| Yao | Metabolic engineering of ethanol production in Thermoanaerobacter mathranii |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980123139.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09766570 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010517869 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009766570 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4661/KOLNP/2010 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20117001106 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12995774 Country of ref document: US |