WO2009135580A1 - Thienopyridone derivatives as amp-activated protein kinase (ampk) activators - Google Patents
Thienopyridone derivatives as amp-activated protein kinase (ampk) activators Download PDFInfo
- Publication number
- WO2009135580A1 WO2009135580A1 PCT/EP2009/002606 EP2009002606W WO2009135580A1 WO 2009135580 A1 WO2009135580 A1 WO 2009135580A1 EP 2009002606 W EP2009002606 W EP 2009002606W WO 2009135580 A1 WO2009135580 A1 WO 2009135580A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyridin
- hydroxy
- thieno
- dihydro
- phenyl
- Prior art date
Links
- 0 **c(c(C(O)=C(*)C(N1)=O)c1[s]1)c1I Chemical compound **c(c(C(O)=C(*)C(N1)=O)c1[s]1)c1I 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
Definitions
- the invention relates to thienopyridone derivatives that are activators of AMPK- activated protein kinase (AMPK) of formula (I).
- AMPK AMPK- activated protein kinase
- the invention also relates to the preparation and use of these thienopyridones in the treatment of disorders such as diabetes, metabolic syndrome, obesity, cancer, inflammation.
- the invention had the object of finding novel compounds having valuable properties, in particular those which can be used for the preparation of medicaments.
- the present invention relates to compounds that are useful in the treatment and/or prevention of diseases such as diabetes, metabolic syndrome, obesity, cancer, inflammation.
- the present invention therefore relates to compounds according to the invention as medicaments and/or medicament active ingredients in the treatment and/or prophylaxis of the said diseases and to the use of compounds according to the invention for the preparation of a pharmaceutical for the treatment and/or prophylaxis of the said diseases and also to a process for the treatment of the said diseases which comprises the administration of one or more compounds according to the invention to a patient in need of such an administration.
- thienopyridone derivatives activate AMPK; therefore, these compounds are especially suitable for the prevention and treatment of diabetes, metabolic syndrome, obesity, cancer, inflammation. It has been found that the compounds according to the invention and salts thereof have very valuable pharmacological properties while being well tolerated. In particular, they exhibit AMPK activating effects.
- the host or patient may belong to any mammal species, for example a primate species, particularly humans; rodents, including mice, rats and hamsters; rabbits; horses, cows, dogs, cats, etc.
- Animal models are of interest for experimental investigations, where they provide a model for the treatment of a human disease.
- AMPK is well established as a sensor and regulator of cellular energy homeostasis (Hardie D.G. and Hawley S.A; "AMP-activated protein kinase: the energy charge hypothesis revisited” Bioassays, 23, 1112, (2001), Kemp B. E. et al. "AMP-activated protein kinase, super metabolic regulator", Biochem; Soc. Transactions, 31 , 162 (2003)). Allosteric activation of this kinase due to rising AMP levels occurs in states of cellular energy depletion. The resulting serine/Threonine phosphorylation of target enzymes leads to an adaptation of cellular metabolism to low energy state. The net effect of AMPK activation induced changes is inhibition of ATP consuming processes and activation of
- AMPK substrates include acetyl-CoA carboxylase (ACC) and HMG-CoA - reductase (Carling D. et al., "A commun bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis", FEBS letters, 223, 217 (1987)).
- ACC acetyl-CoA carboxylase
- HMG-CoA - reductase Carling D. et al., "A commun bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis", FEBS letters, 223, 217 (1987)).
- Phosphorylation and therefore inhibition of ACC leads to a decrease in fatty acid synthesis (ATP-consuming) and at the same time to an increase in fatty acid oxidation (ATP-generating).
- Phosphorylation and resulting inhibition of HMG-CoA-reductase leads to a decrease in cholesterol synthesis.
- AMPK hormone sensitive lipase
- AMP-activated protein kinase a substrate of AMPK
- glycerol-3-phosphate acyltransferase glycerol-3-phosphate acyltransferase
- AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltranferase is a novel target", Biochem.
- AMPK is also implicated in the regulation of liver metabolism. Elevated glucose production by the liver is a major cause of fasting hyperglycemia in T2D (Saltiel et al., "new perspectives into the molecular pathogenesis and treatment of type 2 diabetes, cell 10, 517-529 (2001 )). Gluconeogenesis in the liver is regulated by multiple enzymes such as phosphoenolpyruvate carboxylase (PEPCK) and glucose-6-phosphatase -G6Pase).
- PEPCK phosphoenolpyruvate carboxylase
- G6Pase glucose-6-phosphatase
- AMPK Activation of AMPK suppresses the transcription of theses genes in hepatoma cells (Lochhead et al, "5- aminoimidazoie-4-carboxamide riboside, mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6- phosphatase, Diabetes, 49,896-903 (2000)).
- AMPK activation also down-regulates gluconeogenesis acting on some other genes expression. These effects may be due to its ability to down- regulate key transcription factors such as SREBP-Ic (Zhou G. et al., "Role of AMP-activated protein kinase in mechanism of metformin action", J. Clin. Invest., 108, 1167 (2001 )), ChREBP (Kawaguchi T. et al., "mechanism for fatty acids sparing effect on glucose induced transcription: regulation of carbohydrate response element binding protein by AMP-activated protein kinase" J. Biol. Chem. 277, 3829 involved in (Leclerc I.
- Hepatocyte nuclear facto r-4 D (2001 )) or HNF-4 type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase" Diabetes, 50, 1515 (2001 )) or by direct phosphorylation of transcriptional coactivators such as p300 (Yang W; et al., "Regulation of transcription by AMP-activated protein kinase; Phosphorylation of p300 blocks its interaction with nuclear receptors” J. Biol. Chem. 276, 38341 (2001 )) and TORC2.
- AMPK is considered as an attractive candidate for contraction-induced skeletal muscle glucose uptake because it is activated in parallel with elevation in AMP and a reduction in creatine phosphate energy stores (Hutber et al. "Electrical stimulation inactivates muscle acetyl - CoA carboxylase and increases AMP- activated protein kinase" Am. J. Physiol. Endocrinol. Metab. 272, E262-E66 (1997)). Furthermore, AICAR-induced activation of AMPK increases glucose uptake (Merrill et al. "AICA Riboside increases AMP-activated protein kinase, fatty acid oxidation and glucose uptake in rat muscle” Am. J. Physiol. Endocrinol.
- AS160 appeared to be a downstream target of AMPK in mediating glucose uptake in skeletal muscle. Taken together all these metabolic effects provide evidence that AMPK suppresses liver gluconeogenesis and lipid production, while decreasing hepatic lipid deposition via increased lipid oxidation, thus improving the glucose and lipid profile in T2D.
- adiponectin stimulates glucose utilization and fatty acid oxidation by activating AMP-activated protein kinase
- Nature Medicine, 8, 1288, (2002) Tomas E. et al., " Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: Acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation” PNAS, 99, 16309, (2002)).
- the activation of AMPK in these circumstances seems to be independent of increasing cellular AMP levels but rather due to phosphorylation by one or more yet to be identified upstream kinases.
- A-769662 a member of the Thienopyridone family in vivo induces a decrease in plasma glucose and triglycerides (Cool. B. et al., "Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome", cell Metab., 3, 403-416, (2006)).
- AMPK has a role in regulating the mTOR pathway.
- mTOR is a serine/threonine kinase and is a key regulator of protein synthesis.
- AMPK phosphorylates TSC2 at Thr-1227 and SeM 345 increasing the activity of the
- AMPK inhibits mTOR action by phosphorylation on Thr-2446.
- AMPK indirectly and directly inhibits the activity of mTOR to limit protein synthesis.
- AMPK may also be a therapeutic target for many cancers that have constitutive activation of the PI3K-Akt signalling pathway.
- Treatment of various cancer cell lines by AICAR attenuated the cell proliferation both in vitro and in vivo studies (Giri R; R.,”5- Aminoimidazole-4-carboxamide-1-beta-4- ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase (AMPK", J. Biol. Chem.
- AICAR as an AMPK activator exerts anti-inflammatory diseases. It has been observed that AICAR attenuates the production of proinflammatory cytokines and mediators (S. Giri et al. J. Neuroscience 2004, 24:479-487), AICAR in rat model and in vitro attenuates EAE progression by limiting infiltration of leucocytes across blood brain barrier (BBB) (N. Nath. Et al. J. of Immunology 2005, 175:566-574; R. Prasad et al. J. Neurosci Res.
- BBB blood brain barrier
- AMPK activating agents act as anti-inflammatory agents and can hold a therapeutic potential in Krabbe disease/twitcher disease (an inherited neurological disorder) (S.Giri et al. J. Neurochem. 2008, Mar 19).
- US 5,602,144 discloses thienopyridone derivatives for the treatment of cerebral ischemia or schizophrenia.
- US 7,119,205 discloses thienopyridones derivatives for the treatment useful for the treatment of diabetes, obesity as AMPK activators.
- WO2007019914 discloses thienopyridone derivatives for the treatment useful for the treatment of diabetes, obesity as AMPK activators.
- the invention relates to compounds of the formula (I)
- R 1 denotes H, A 1 OA, OH, Hal, NO 2 , COOA, COOH, CHO, COA, CONH 2 ,
- B 1 denotes Ar — diyl or Het-diyl
- B 2 denotes Ar or Het
- Ar denotes phenyl, naphthyl, each of which is unsubstituted or mono-, di-, tri-, tetra- or pentasubstituted by A, Hal, OA, OH, CHO, COA, NH 2 , NHA, NA 2 , NO 2 , COOA, COOH, CONH 2 , CONA, CONA 2 , SO 2 A, CN,
- Het denotes a mono- or bicyclic unsaturated or aromatic heterocycle having
- N, O and/or S atoms which may be mono-, di- or trisubstituted by Hal, A, OA, OH, CHO, COA, COOH, COOA, CN, NO 2 , NH 2 , NHA, NA 2 ,
- CONH 2 CONHA and/or CONA 2
- A denotes unbranched or branched alkyl having 1-10 C atoms, in which
- H atoms may be replaced by OH, F, Cl and/or Br, or denotes cycloalkyl having 3-7 C atoms, Hal denotes F, Cl, Br or I, and pharmaceutically usable derivatives, salts, solvates and stereoisomers thereof, including mixtures thereof in all ratios,
- Some preferred compounds of formula (I) are the following : 3-biphenyl-4-yl- 4-hydroxy-5-phenyl-6,7-dihydro-thieno[2,3-b]pyridin-6-one,
- the invention relates to the compounds of the formula (I) and salts thereof and to a process for the preparation of compounds of the formula (I) according to Claims 1-11 and pharmaceutically usable derivatives, solvates, salts and stereoisomers thereof, characterised in that a compound of the formula (II)
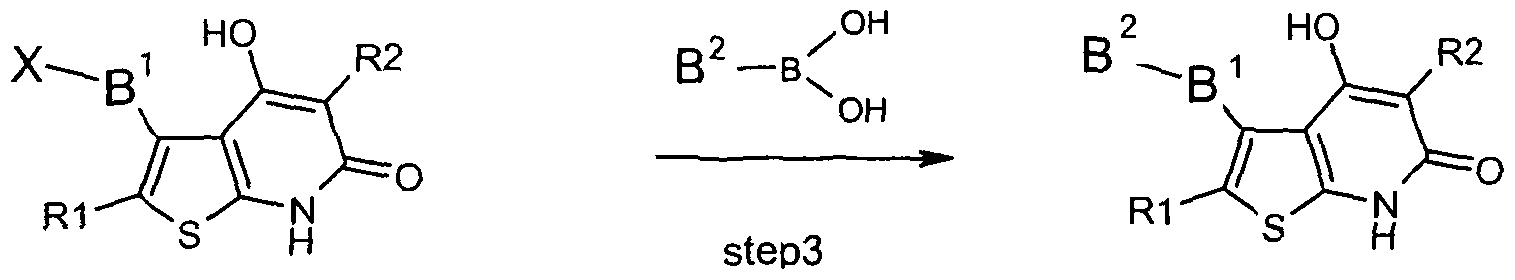
- B 2 is as defined in claim 1 , under Suzuki reaction conditions (Journal of organometallic chemistry, 1999, 576(1-2), 147-168 / Applied Homogeneous Catalysis with Organometallic Compounds (2nd Edition) (2002), 1 , 591-598), and/or a base or acid of the formula I is converted into one of its salts.
- the 2-aminothiophene starting compound (IV) is commercially available (chemos Gmbh, Fluorochem, Acros, Interchim) or easily prepared by a person skilled in the Art by a Gewald reaction described in Journal Heterocycle Chemistry, vol. 36, page 333, 1999.
- Y is preferably Cl, Br 1 1 or a free or reactively modified OH group, such as, for example, an activated ester, an imidazolide or alkylsulfonyloxy having 1-6 carbon atoms (preferably methylsulfonyloxy or trifluoromethylsulfonyloxy) or arylsulfonyloxy having 6-10 carbon atoms (preferably phenyl- or p-tolylsulfonyloxy), using of coupling agent as carbodiimidazole (CDI), Dicyclohexylcarbodiimine (DCC) preferably DCC.
- the coupling agent is a carbodiimine, the preferred derivative is described in reference internet link (http://chemicalland21.com/lifescience/phar/HBTU.htm).
- - is Y an halogene atome, preferably Cl, it is reacted in an inert solvent as tetrahydrofurane, dioxane, preferably dioxane from zero to 100 degree for 5 minutes to 24 hour to prepare compounds of formula (III).
- - Is Y is an OH, it is reacted in an non protic solvent as tetrahydrofurane, dioxane, preferably tetrahydrofurane with a condension agent as carbodiimidazole, diclohexylcarbodiimine (DCC) preferably DCC at zero to solvent reflux temperature for 15 minutes to 24 h preferably at room temperature at solvent reflux overnight.
- a condension agent as carbodiimidazole, diclohexylcarbodiimine (DCC) preferably DCC at zero to solvent reflux temperature for 15 minutes to 24 h preferably at room temperature at solvent reflux overnight.
- Suitable inert solvents are, for example, hydrocarbons, such as hexane, petroleum ether, benzene, toluene or xylene; chlorinated hydrocarbons, such as trichloroethylene, 1 ,2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; alcohols, such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; ethers, such as diethyl ether, diisopropyl ether, tetrahydrofuran (THF) or dioxane; glycol ethers, such as ethylene glycol monomethyl or monoethyl ether, ethylene glycol dimethyl ether (diglyme); ketones, such as acetone or butanone; amides, such as acetamide, dimethylacetamide or dimethylformamide (DMF); nitrile
- Compound of formula (III) is then cyclised under basic condition to synthezise compound of formula (II) by a cyclisation reaction using a base as for example hexamethyldisilylazane, a potassium- or sodium salt, e.g. sodium- or potassium tertioamylate, sodium ethylate with preferably use of hexamethyldisylazide in an inert solvent, preferably in tetrahydrofurane, dioxane, toluene at 20 0 C to 150 0 C preferably at room temprature for 30 minutes to 24 hours and more preferably from 30 minutes to 1 hour.
- a base as for example hexamethyldisilylazane, a potassium- or sodium salt, e.g. sodium- or potassium tertioamylate, sodium ethylate with preferably use of hexamethyldisylazide in an inert solvent, preferably in tetrahydrofurane,
- Suitable inert solvents are, for example, hydrocarbons, such as hexane, petroleum ether, benzene, toluene or xylene; chlorinated hydrocarbons, such as trichloroethylene, 1 ,2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; alcohols, such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; ethers, such as diethyl ether, diisopropyl ether, tetrahydrofuran (THF) or dioxane; glycol ethers, such as ethylene glycol monomethyl or monoethyl ether, ethylene glycol dimethyl ether (diglyme); ketones, such as acetone or butanone; amides, such as acetamide, dimethylacetamide or dimethylformamide (DMF); nitrile
- Compound of formula (I) is manufactured by a Suzuki reaction using a boronic acid derivative as for example an aryl boronic acid in presence of base preferably a carbonate salt and more preferably a cesium carbonate in presence of palladium catalyst such as palladium tetrakis (triphenyl phosphine) under inert atmosphere in a mixture of solvants such as polar aprotic solvent / inert solvent / protic solvent / water.
- the combination of solvent is preferably dimethylformamide / toluene / ethanol / water with 10/1/6/3 ratio or 25/2.6/15/7.5 at 20 degree to solvent reflux temperature for one hour to 48 hours, preferably 6 h to 24 h.
- the invention also relates to the racemic forms, tautomeric forms, enantiomers, diastereoisomers, epimers and organic or mineral salts of the compounds of the general formula (I), as well as their crystalline forms, including their polymorphic forms and the polymorphic forms of the compounds of formula (I).
- the present invention is directed not only to racemic mixtures of these compounds, but also to individual stereoisomers and/or diastereoisomers thereof as well or as mixtures of these in all proportions.
- the invention also relates to the stereoisomers (including E, Z isomers) and the hydrates and solvates of these compounds.
- Solvates of the compounds are taken to mean adductions of inert solvent molecules onto the compounds which form owing to their mutual attractive force. Solvates are, for example, mono- or dihydrates or alcoholates.
- compositions is taken to mean, for example, the salts of the compounds according to the invention and also so-called prodrug compounds.
- Prodrug derivatives is taken to mean compounds of the formula I which have been modified, with, for example, alkyl or acyl groups, sugars or oligopeptides and which are rapidly cleaved in the organism to form the active compounds according to the invention.
- These also include biodegradable polymer derivatives of the compounds according to the invention, as is described, for example, in Int. J. Pharm. 115, 61-67 (1995).
- prodrug refers to any compound that when administered to a biological system generates the "drug” substance (a biologically active compound) as a result of spontaneous chemical reaction(s), enzyme catalyzed chemical reaction(s), and/or metabolic chemical reaction(s).
- an effective amount means the amount of a medicament or pharmaceutical active ingredient which causes a biological or medical response which is sought or aimed at, for example by a researcher or physician, in a tissue, system, animal or human.
- the expression "therapeutically effective amount” means an amount which, compared with a corresponding subject who has not received this amount, has the following consequence: improved treatment, healing, prevention or elimination of a disease, syndrome, condition, complaint, disorder or prevention of side effects or also the reduction in the progress of a disease, condition, disorder or side effects or also the reduction in the progress of a disease, condition or disorder.
- the expression “therapeutically effective amount” also encompasses the amounts which are effective for increasing normal physiological function.
- the invention also relates to mixtures of the compounds of the formula I according to the invention, for example mixtures of two diastereomers, for example in the ratio 1 :1 , 1 :2, 1:3, 1:4, 1:5, 1 :10, 1 :100 or 1 :1000.
- mixtures of the compounds of the formula I according to the invention for example mixtures of two diastereomers, for example in the ratio 1 :1 , 1 :2, 1:3, 1:4, 1:5, 1 :10, 1 :100 or 1 :1000.
- These are particularly preferably mixtures of stereoisomeric compounds.
- A denotes alkyl, is unbranched (linear) or branched, and has 1 , 2, 3, 4, 5, 6, 7, 8, 9 or 10 C atoms.
- A preferably denotes methyl, furthermore ethyl, propyl, iso- propyl, butyl, isobutyl, sec-butyl or tert-butyl, furthermore also pentyl, 1-, 2- or 3-methylbutyl, 1 ,1- , 1 ,2- or 2,2-dimethylpropyl, 1-ethylpropyl, hexyl, 1- , 2- , 3- or 4-methylpentyl, 1 ,1- , 1 ,2- , 1 ,3- , 2,2- , 2,3- or 3,3-dimethylbutyl, 1- or
- 2-ethylbutyl 1-ethyl-1-methylpropyl, i-ethyl-2-methylpropyl, 1 ,1 ,2- or 1 ,2, 2-tri- methylpropyl, further preferably, for example, trifluoromethyl.
- A preferably denotes unbranched or branched alkyl having 1-10 C atoms, in which 1-7 H atoms may be replaced by OH, F and/or Cl.
- Cycloalkyl preferably denotes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
- R 1 preferably denotes H, A, Hal, COOA, COOH, CONH 2 , CONHA, CONA 2 , CN, SO 2 A, SO 2 NH 2 or phenyl.
- R 2 preferably denotes phenyl, naphthyl, each of which is unsubstituted or mono-, di-, tri-, tetra- or pentasubstituted by A, Hal, OA, OH, COOA, COOH, CONH 2 , CONA, CONA 2 and/or SO 2 A or denotes Het.
- B 1 is Ar-diyl, denotes 1 ,2 phenylene or 1 ,3 phenylene or 1 ,4 phenylene group which is unsubstituted or mono-, di-, tri-, tetrasubsituted by A, Hal, OA, OH, , COOA, COOH 1 CN, CONH 21 CONA, CONA 2 and/or SO 2 A.
- B 1 denotes 1 ,4 phenylene which is unsubstituted or mono-, di-, tri-, tetrasubstituted by A, Hal, OA, OH, , COOA, COOH.CN, CONH 2 ,CONA, CONA 2 and/or SO 2 A ;
- B 1 also denotes Het-diyl which preferably means unsubstituted Het-diyl most preferably 2,5 pyridyl.
- B 2 preferably denotes preferably denotes phenyl, naphthyl, each of which is unsubstituted or mono-, di-, tri-, tetra- or pentasubstituted by A, Hal, OA, OH, , COOA, COOH 1 CN, CONH 2 , CONA, CONA 2 and/or SO 2 A or denotes Het.
- Ar denotes, for example, phenyl, o-, m- or p-tolyl, o-, m- or p-ethylphenyl, o-, m- or p-propylphenyl, 0-, m- or p-isopropylphenyl, o-, m- or p-tert-butylphenyl, o-, m- or p-hydroxyphenyl, o-, m- or p-nitrophenyl, o-, m- or p-aminophenyl, o-, m- or p-(N-methylamino)phenyl, o-, m- or p-(N-methylaminocarbonyl)phenyl, o-, m- or p-acetamidophenyl, o-, m- or p-methoxyphenyl, 0-, m- or p-eth
- Het denotes, for example, 2- or 3-furyl, 2- or 3-thienyl, 1-, 2- or 3-pyrrolyl, 1-, 2, 4- or 5-imidazolyl, 1-, 3-, 4- or 5-pyrazolyl, 2-, 4- or 5-oxazolyl, 3-, 4- or 5-isoxazolyl, 2-, 4- or 5-thiazolyl, 3-, 4- or 5-isothiaz- olyl, 2-, 3- or 4-pyridyl, 2-, 4-, 5- or 6-pyrimidinyl, furthermore preferably 1 ,2,3- triazol-1-, -4- or -5-yl, 1 ,2,4-triazoM-, -3- or 5-yl, 1- or 5-tetrazolyl, 1 ,2,3-oxa- diazol-4- or -5-yl, 1 ,2,4-oxadiazol-3- or -5-yl, 1 ,3,4-thiadiazol-2- or -5-yl,
- heterocyclic radicals can also be partially or fully hydrogenated.
- Het can thus also denote, for example, 2,3-dihydro-2-, -3-, -A- or -5-furyl, 2,5- dihydro-2-, -3-, -A- or 5-furyl, tetrahydro-2- or -3-furyl, 1 ,3-dioxolan-4-yl, tetrahydro-2- or -3-thienyl, 2,3-dihydro-1-, -2-, -3-, -A- or -5-pyrrolyl, 2,5-dihydro- 1-, -2-, -3-, -A- or -5-pyrrolyl, 1-, 2- or 3-pyrrolidinyl, tetrahydro-1-, -2- or -A- imidazolyl, 2,3-dihydro-1-, -2-, -3-, -4- or -5-pyrazolyl, tetrahydro-1-, -3- or -4- pyr
- Het preferably denotes a mono- or bicyclic aromatic heterocycle having 1 to 4 N, O and/or S atoms,
- Het denotes pyridyl, pyrimidinyl, furanyl, isoxazolyl, imidazolyl, pyrazolyl, oxazolyl, pyrrolyl, thiazolyl, isothiazolyl, thienyl, triazolyl, tetrazolyl, indolyl, benzimidazolyl or indazolyl.
- the invention relates, in particular, to the compounds of the formula (I) in which at least one of the said radicals has one of the preferred meanings indicated above.
- Some preferred groups of compounds may be expressed by the following sub-formulae Ia to Ih, which conform to the formula I and in which the radicals not designated in greater detail have the meaning indicated for the formula I, but in which
- R 1 denotes H, A, Hal, COOA, COOH, CONH 2 , CONHA, CONA 2 , CN, SO 2 A, SO 2 NH 2 or phenyl;
- R 2 denotes phenyl, naphthyl, each of which is unsubstituted or mono-, di-, tri-, tetra- or pentasubstituted by A, Hal, OA, OH, COOA, COOH, CONH 2 , CONA, CONA 2 and/or SO 2 A;
- B 1 denotes Ar -diyl which said compound is 1 ,2 phenylene or 1 ,3 phenylene or 1 ,4 phenylene group, which is unsubstituted or mono-, di-, tri-, tetrasubstituted by A, Hal, OA, OH, COOA, COOH 1 CN, CONH 21 CONA, CONA 2 and/or SO 2 A;
- Id B 2 denotes phenyl, naphthyl, each of which is unsubstituted or mono-, di-, tri-, tetra- or pentasubstituted by A, Hal, OA, OH,
- Het denotes a mono- or bicyclic aromatic heterocycle having 1 to 4 N 1 O and/or S atoms;
- Het denotes pyridyl, pyrimidinyl, furanyl, isoxazolyl, imidazolyl, pyrazolyl, oxazolyl, pyrrolyl, thiazolyl, isothiazolyl, thienyl, triazolyl, tetrazolyl, indolyl, benzimidazolyl or indazolyl;
- Ig A denotes unbranched or branched alkyl having 1-10 C atoms, in which 1-7 H atoms may be replaced by OH, F, Cl and/or Br;
- Ih R 1 denotes H, A, Hal, COOA, COOH, CONH 2 , CONHA, CONA 2 ,
- R 2 denotes phenyl, naphthyl each of which is unsubstituted or mono-, di-, tri-, tetra- or pentasubstituted by A, Hal, OA, OH, ,
- COOA 1 and pharmaceutically usable derivatives, solvates, salts and stereoisomers thereof, including mixtures thereof in all ratios.
- the invention relates, in particular, to the compounds of the formula I in which at least one of the said radicals has one of the preferred meanings indicated above.
- Some preferred groups of compounds may be expressed by the following sub-formulae Ia to Ih, which conform to the formula I and in which the radicals not designated in greater detail have the meaning indicated for the formula I, but in which
- the compounds of the present invention may be prepared in a number of methods well known to those skilled in the art, including, but not limited to those described below, or through modifications of these methods by applying standard techniques known to those skilled in the art of organic synthesis. All processes disclosed in association with the present invention are contemplated to be practiced on any scale, including milligram, gram, multigram, kilogram, multikilogram or commercial industrial scale.
- the compounds of the present invention may contain one or more asymmetrically substituted carbon atoms, and may be isolated in optically active or racemic forms.
- optically active or racemic forms all chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated.
- optically active forms mixtures of stereoisomers may be separated by standard techniques including, but not limited to, resolution of racemic forms, normal, reverse-phase, and chiral chromatography, preferential salt formation, recrystallization, and the like, or by chiral synthesis either from active starting materials or by deliberate chiral synthesis of target centers.
- bases include: sodium hydroxide, potassium carbonate, potassium tertiobutylate.sodium tertioamylate.triethylamine, potassium hexamethyldisilazide .alkali metal hydrides, such as sodium hydride and potassium hydride; alkyllithium compounds, such as methyllithium and butyllithium; and alkali metal alkoxides, such as sodium methoxide and sodium ethoxide.
- reaction are carried out in a suitable solvent.
- solvents may be used, provided that it has no adverse effect on the reaction or on the reagents involved.
- suitable solvents include: hydrocarbons, which may be aromatic, aliphatic or cycloaliphatic hydrocarbons, such as hexane, cyclohexane, benzene, toluene and xylene; amides, such as dimethylformamide; alcohols such as ethanol and methanol and ethers, such as diethyl ether, dioxane and tetrahydrofuran.
- the reactions can take place over a wide range of temperatures.
- reaction in general, we find it convenient to carry out the reaction at a temperature of from 0 0 C to 150 0 C (more preferably from about room temperature to 100 0 C).
- the time required for the reaction may also vary widely, depending on many factors, notably the reaction temperature and the nature of the reagents. However, provided that the reaction is effected under the preferred conditions outlined above, a period of from 3 hours to 20 hours is preferred.
- the compound thus prepared may be recovered from the reaction mixture by conventional means.
- the compounds may be recovered by distilling off the solvent from the reaction mixture or, if necessary, after distilling off the solvent from the reaction mixture, pouring the residue into water followed by extraction with a water-immiscible organic solvent and distilling off the solvent from the extract.
- the product can, if desired, be further purified by various well-known techniques, such as recrystallization, reprecipitation or the various chromatography techniques, notably column chromatography or preparative thin layer chromatography.
- the said compounds according to the invention can be used in their final non- salt form.
- the present invention also encompasses the use of these compounds in the form of their pharmaceutically acceptable salts, which can be derived from various organic and inorganic acids and bases by procedures known in the art.
- Pharmaceutically acceptable salt forms of the compounds of the formula I are for the most part prepared by conventional methods. If the compound of the formula I contains a carboxyl group, one of its suitable salts can be formed by reacting the compound with a suitable base to give the corresponding base-addition salt.
- Such bases are, for example, alkali metal hydroxides, including potassium hydroxide, sodium hydroxide and lithium hydroxide; alkaline earth metal hydroxides, such as barium hydroxide and calcium hydroxide; alkali metal alkoxides, for example potassium ethoxide and sodium propoxide; and various organic bases, such as piperidine, diethanolamine and N-methylglutamine.
- alkali metal hydroxides including potassium hydroxide, sodium hydroxide and lithium hydroxide
- alkaline earth metal hydroxides such as barium hydroxide and calcium hydroxide
- alkali metal alkoxides for example potassium ethoxide and sodium propoxide
- organic bases such as piperidine, diethanolamine and N-methylglutamine.
- the aluminium salts of the compounds of the formula I are likewise included.
- acid-addition salts can be formed by treating these compounds with pharmaceutically acceptable organic and inorganic acids, for example hydrogen halides, such as hydrogen chloride, hydrogen bromide or hydrogen iodide, other mineral acids and corresponding salts thereof, such as sulfate, nitrate or phosphate and the like, and alkyl- and monoarylsulfonates, such as ethanesulfonate, toluenesulfonate and benzenesulfonate, and other organic acids and corresponding salts thereof, such as acetate, trifluoroacetate, tartrate, maleate, succinate, citrate, benzoate, salicylate, ascorbate and the like.
- organic and inorganic acids for example hydrogen halides, such as hydrogen chloride, hydrogen bromide or hydrogen iodide, other mineral acids and corresponding salts thereof, such as sulfate, nitrate or phosphate and the like, and alkyl- and monoarylsul
- pharmaceutically acceptable acid-addition salts of the compounds of the formula I include the following: acetate, adipate, alginate, arginate, aspartate, benzoate, benzenesulfonate (besylate), bisulfate, bisulfite, bromide, butyrate, camphorate, camphorsulfonate, caprylate, chloride, chlorobenzoate, citrate, cyclopentanepropionate, digluconate, dihydrogenphosphate, dinitro- benzoate, dodecylsulfate, ethanesulfonate, fumarate, galacterate (from mucic acid), galacturonate, glucoheptanoate, gluconate, glutamate, glycerophosphate, hemisuccinate, hemisulfate, heptanoate, hexanoate, hippurate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanethan
- the base salts of the compounds according to the invention include aluminium, ammonium, calcium, copper, iron(lll), iron(ll), lithium, magnesium, manganese(lll), manganese(ll), potassium, sodium and zinc salts, but this is not intended to represent a restriction.
- Salts of the compounds of the formula I which are derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary and tertiary amines, substituted amines, also including naturally occurring substituted amines, cyclic amines, and basic ion exchanger resins, for example arginine, betaine, caffeine, chloroprocaine, choline, N,N'-dibenzylethylenediamine (benzathine), dicyclohexylamine, diethanolamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropyl- amine, lidocaine, lysine, meglumine, N-methyl-D-glucamine, morpholine, piperazine, piperidine, polyamine resins, pro
- Compounds of the present invention which contain basic nitrogen-containing groups can be quatemised using agents such as (Ci-C 4 )alkyl halides, for example methyl, ethyl, isopropyl and tert-butyl chloride, bromide and iodide; di(C- ⁇ -C 4 )alkyl sulfates, for example dimethyl, diethyl and diamyl sulfate; (C 10 - Cis)alkyl halides, for example decyl, dodecyl, lauryl, myristyl and stearyl chloride, bromide and iodide; and aryl(CrC 4 )alkyl halides, for example benzyl chloride and phenethyl bromide. Both water- and oil-soluble compounds according to the invention can be prepared using such salts.
- (Ci-C 4 )alkyl halides for example methyl, ethyl,
- the above-mentioned pharmaceutical salts which are preferred include acetate, trifluoroacetate, besylate, citrate, fumarate, gluconate, hemisuccinate, hippurate, hydrochloride, hydrobromide, isethionate, mandelate, meglumine, nitrate, oleate, phosphonate, pivalate, sodium phosphate, stearate, sulfate, subsalicylate, tartrate, thiomalate, tosylate and tromethamine, but this is not intended to represent a restriction.
- the acid-addition salts of basic compounds of the formula I are prepared by bringing the free base form into contact with a sufficient amount of the desired acid, causing the formation of the salt in a conventional manner.
- the free base can be regenerated by bringing the salt form into contact with a base and isolating the free base in a conventional manner.
- the free base forms differ in a certain respect from the corresponding salt forms thereof with respect to certain physical properties, such as solubility in polar solvents; for the purposes of the invention, however, the salts otherwise correspond to the respective free base forms thereof.
- the pharmaceutically acceptable base-addition salts of the compounds of the formula I are formed with metals or amines, such as alkali metals and alkaline earth metals or organic amines.
- metals are sodium, potassium, magnesium and calcium.
- Preferred organic amines are N,N'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methyl-D-glucamine and procaine.
- the base-addition salts of acidic compounds according to the invention are prepared by bringing the free acid form into contact with a sufficient amount of the desired base, causing the formation of the salt in a conventional manner.
- the free acid can be regenerated by bringing the salt form into contact with an acid and isolating the free acid in a conventional manner.
- the free acid forms differ in a certain respect from the corresponding salt forms thereof with respect to certain physical properties, such as solubility in polar solvents; for the purposes of the invention, however, the salts otherwise correspond to the respective free acid forms thereof.
- a compound according to the invention contains more than one group which is capable of forming pharmaceutically acceptable salts of this type, the invention also encompasses multiple salts.
- Typical multiple salt forms include, for example, bitartrate, diacetate, difumarate, dimeglumine, diphosphate, disodium and trihydrochloride, but this is not intended to represent a restriction.
- the expression "pharmaceutically acceptable salt” in the present connection is taken to mean an active ingredient which comprises a compound of the formula I in the form of one of its salts, in particular if this salt form imparts improved pharmacokinetic properties on the active ingredient compared with the free form of the active ingredient or any other salt form of the active ingredient used earlier.
- the pharmaceutically acceptable salt form of the active ingredient can also provide this active ingredient for the first time with a desired pharmacokinetic property which it did not have earlier and can even have a positive influence on the pharmacodynamics of this active ingredient with respect to its therapeutic efficacy in the body.
- Compounds of the formula I according to the invention may be chiral owing to their molecular structure and may accordingly occur in various enantiomeric forms. They can therefore exist in racemic or in optically active form.
- the pharmaceutical activity of the racemates or stereoisomers of the compounds according to the invention may differ, it may be desirable to use the enantiomers.
- the end product or even the intermediates can be separated into enantiomeric compounds by chemical or physical measures known to the person skilled in the art or even employed as such in the synthesis.
- diastereoisomers are formed from the mixture by reaction with an optically active resolving agent.
- optically active acids such as the R and S forms of tartaric acid, diacetyltartaric acid, dibenzoyltartaric acid, mandelic acid, malic acid, lactic acid, suitably N-protected amino acids (for example N-benzoylproline or N-benzenesulfonylproline), or the various optically active camphorsulfonic acids.
- chromatographic enantiomer resolution with the aid of an optically active resolving agent (for example dinitrobenzoylphenylglycine, cellulose triacetate or other derivatives of carbohydrates or chirally derivatised methacrylate polymers immobilised on silica gel).
- optically active resolving agent for example dinitrobenzoylphenylglycine, cellulose triacetate or other derivatives of carbohydrates or chirally derivatised methacrylate polymers immobilised on silica gel.
- Suitable eluents for this purpose are aqueous or alcoholic solvent mixtures, such as, for example, hexane/isopropanol/ acetonitrile, for example in the ratio 82:15:3.
- (+)-D-di-O-benzoyltartaric acid (+)-D-di-O-benzoyltartaric acid, (-)-L-di-O-benzoyltartaric acid, (-)-L-di-O,O'-p-toluyl-L- tartaric acid, (+)-D- di-O,O'-p-toluyl-L-tartaric acid, (+)-(+)-malic acid, (S)-(-)-malic acid, (+)- camphoric acid, (-)-camphoric acid, R-(-)1 ,1'-binaphtalen-2,2'-diyl hydrogenophosphonic, (+)-camphanic acid, (-)-camphanic acid, (S)-(+)-2- phenylpropionic acid, (R)-(+)-2-phenylpropionic acid, D-(
- the following chiral amines can be used: quinine, brucine, (S)-1- (benzyloxymethyl)propylarnine (III), (-)-ephedrine, (4S,5R)-(+)-1 ,2,2,3,4- tetramethyl-5-phenyl-1 ,3-oxazolidine, (R)-1-phenyl-2-p-toly!ethylamine, (S)- phenylglycinol, (-)-N-methylephedrine, (+)-(2S,3R)-4-dimethylamino-3-methyl- 1 ,2-diphenyl-2-butanol, (S)-phenylglycinol, (S)- ⁇ -methylbenzylamine or any mixture of them.
- the invention furthermore relates to the use of the compounds and/or physiologically acceptable salts thereof for the preparation of a medicament (pharmaceutical composition), in particular by non-chemical methods. They can be converted into a suitable dosage form here together with at least one solid, liquid and/or semi-liquid excipient or adjuvant and, if desired, in combination with one or more further active ingredients.
- the invention furthermore relates to medicaments comprising at least one compound according to the invention and/or pharmaceutically usable derivatives, solvates and stereoisomers thereof, including mixtures thereof in all ratios, and optionally excipients and/or adjuvants.
- compositions can be administered in the form of dosage units which comprise a predetermined amount of active ingredient per dosage unit.
- a unit can comprise, for example, 0.5 mg to 1 g, preferably 1 mg to 700 mg, particularly preferably 5 mg to 100 mg, of a compound according to the invention, depending on the disease condition treated, the method of administration and the age, weight and condition of the patient, or pharmaceutical formulations can be administered in the form of dosage units which comprise a predetermined amount of active ingredient per dosage unit.
- Preferred dosage unit formulations are those which comprise a daily dose or part-dose, as indicated above, or a corresponding fraction thereof of an active ingredient.
- pharmaceutical formulations of this type can be prepared using a process which is generally known in the pharmaceutical art.
- compositions can be adapted for administration via any desired suitable method, for example by oral (including buccal or sublingual), rectal, nasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal) methods.
- oral including buccal or sublingual
- rectal nasal
- topical including buccal, sublingual or transdermal
- vaginal or parenteral including subcutaneous, intramuscular, intravenous or intradermal
- parenteral including subcutaneous, intramuscular, intravenous or intradermal
- compositions adapted for oral administration can be administered as separate units, such as, for example, capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or foam foods; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- the active-ingredient component in the case of oral administration in the form of a tablet or capsule, can be combined with an oral, nontoxic and pharmaceutically acceptable inert excipient, such as, for example, ethanol, glycerol, water and the like.
- an oral, nontoxic and pharmaceutically acceptable inert excipient such as, for example, ethanol, glycerol, water and the like.
- Powders are prepared by comminuting the compound to a suitable fine size and mixing it with a pharmaceutical excipient comminuted in a similar manner, such as, for example, an edible carbohydrate, such as, for example, starch or mannitol.
- a flavour, preservative, dispersant and dye may likewise be present.
- Capsules are produced by preparing a powder mixture as described above and filling shaped gelatine shells therewith.
- Glidants and lubricants such as, for example, highly disperse silicic acid, talc, magnesium stearate, calcium stearate or polyethylene glycol in solid form, can be added to the powder mixture before the filling operation.
- a disintegrant or solubiliser such as, for example, agar- agar, calcium carbonate or sodium carbonate, may likewise be added in order to improve the availability of the medicament after the capsule has been taken.
- suitable binders include starch, gelatine, natural sugars, such as, for example, glucose or beta-lactose, sweeteners made from maize, natural and synthetic rubber, such as, for example, acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes, and the like.
- the lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
- the disintegrants include, without being restricted thereto, starch, methylcellulose, agar, bentonite, xanthan gum and the like.
- the tablets are formulated by, for example, preparing a powder mixture, granulating or dry- pressing the mixture, adding a lubricant and a disintegrant and pressing the entire mixture to give tablets.
- a powder mixture is prepared by mixing the compound comminuted in a suitable manner with a diluent or a base, as described above, and optionally with a binder, such as, for example, carboxymethylcellulose, an alginate, gelatine or polyvinylpyrrolidone, a dissolution retardant, such as, for example, paraffin, an absorption accelerator, such as, for example, a quaternary salt, and/or an absorbent, such as, for example, bentonite, kaolin or dicalcium phosphate.
- a binder such as, for example, carboxymethylcellulose, an alginate, gelatine or polyvinylpyrrolidone
- a dissolution retardant such as, for example, paraffin
- an absorption accelerator such as, for example, a quaternary salt
- an absorbent such as, for example, bentonite, kaolin or dicalcium phosphate.
- the powder mixture can be granulated by wetting it with a binder, such as, for example, syrup, starch paste, acadia mucilage or solutions of cellulose or polymer materials and pressing it through a sieve.
- a binder such as, for example, syrup, starch paste, acadia mucilage or solutions of cellulose or polymer materials
- the powder mixture can be run through a tableting machine, giving lumps of non-uniform shape which are broken up to form granules.
- the granules can be lubricated by addition of stearic acid, a stearate salt, talc or mineral oil in order to prevent sticking to the tablet casting moulds. The lubricated mixture is then pressed to give tablets.
- the compounds according to the invention can also be combined with a free- flowing inert excipient and then pressed directly to give tablets without carrying out the granulation or dry-pressing steps.
- a transparent or opaque protective layer consisting of a shellac sealing layer, a layer of sugar or polymer material and a gloss layer of wax may be present. Dyes can be added to these coatings in order to be able to differentiate between different dosage units.
- Oral liquids such as, for example, solution, syrups and elixirs, can be prepared in the form of dosage units so that a given quantity comprises a prespecified amount of the compounds.
- Syrups can be prepared by dissolving the compound in an aqueous solution with a suitable flavour, while elixirs are prepared using a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersion of the compound in a non-toxic vehicle.
- Solubilisers and emulsifiers such as, for example, ethoxylated isostearyl alcohols and polyoxyethylene sorbitol ethers, preservatives, flavour additives, such as, for example, peppermint oil or natural sweeteners or saccharin, or other artificial sweeteners and the like, can likewise be added.
- the dosage unit formulations for oral administration can, if desired, be en- capsulated in microcapsules.
- the formulation can also be prepared in such a way that the release is extended or retarded, such as, for example, by coating or embedding of particulate material in polymers, wax and the like.
- the compounds according to the invention and salts, solvates and physio- logically functional derivatives thereof can also be administered in the form of liposome delivery systems, such as, for example, small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- liposomes can be formed from various phospholipids, such as, for example, cholesterol, stearylamine or phosphatidylcholines.
- the compounds according to the invention and the salts, solvates and physio- logically functional derivatives thereof can also be delivered using monoclonal antibodies as individual carriers to which the compound molecules are coupled.
- the compounds can also be coupled to soluble polymers as targeted medicament carriers.
- Such polymers may encompass polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamidophenol, polyhydroxy- ethylaspartamidophenol or polyethylene oxide polylysine, substituted by palmitoyl radicals.
- the compounds may furthermore be coupled to a class of biodegradable polymers which are suitable for achieving controlled release of a medicament, for example polylactic acid, poly-epsilon-caprolactone, polyhydroxybutyric acid, polyorthoesters, polyacetals, polydihydroxypyrans, polycyanoacrylates and crosslinked or amphipathic block copolymers of hydrogels.
- a class of biodegradable polymers which are suitable for achieving controlled release of a medicament, for example polylactic acid, poly-epsilon-caprolactone, polyhydroxybutyric acid, polyorthoesters, polyacetals, polydihydroxypyrans, polycyanoacrylates and crosslinked or amphipathic block copolymers of hydrogels.
- compositions adapted for transdermal administration can be administered as independent plasters for extended, close contact with the epidermis of the recipient.
- the active ingredient can be delivered from the plaster by iontophoresis, as described in general terms in Pharmaceutical Research, 3(6), 318 (1986).
- Pharmaceutical compounds adapted for topical administration can be for- mulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- the formulations are preferably applied as topical ointment or cream.
- the active ingredient can be employed either with a paraffinic or a water-miscible cream base.
- the active ingredient can be formulated to give a cream with an oil-in-water cream base or a water-in-oil base.
- compositions adapted for topical application to the eye include eye drops, in which the active ingredient is dissolved or suspended in a suitable carrier, in particular an aqueous solvent.
- compositions adapted for topical application in the mouth encompass lozenges, pastilles and mouthwashes.
- compositions adapted for rectal administration can be administered in the form of suppositories or enemas.
- compositions adapted for nasal administration in which the carrier substance is a solid comprise a coarse powder having a particle size, for example, in the range 20-500 microns, which is administered in the manner in which snuff is taken, i.e. by rapid inhalation via the nasal passages from a container containing the powder held close to the nose.
- suitable formulations for administration as nasal spray or nose drops with a liquid as carrier substance encompass active-ingredient solutions in water or oil.
- compositions adapted for administration by inhalation encompass finely particulate dusts or mists, which can be generated by various types of pressurised dispensers with aerosols, nebulisers or insufflators.
- compositions adapted for vaginal administration can be administered as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
- compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions comprising antioxidants, buffers, bacteriostatics and solutes, by means of which the formulation is rendered isotonic with the blood of the recipient to be treated; and aqueous and non-aqueous sterile suspensions, which may comprise suspension media and thickeners.
- the formulations can be administered in single-dose or multidose containers, for example sealed ampoules and vials, and stored in freeze-dried (lyophilised) state, so that only the addition of the sterile carrier liquid, for example water for injection purposes, immediately before use is necessary.
- Injection solutions and suspensions prepared in accordance with the recipe can be prepared from sterile powders, granules and tablets.
- formulations may also comprise other agents usual in the art with respect to the particular type of formulation; thus, for example, formulations which are suitable for oral administration may comprise flavours.
- a therapeutically effective amount of a compound of the present invention depends on a number of factors, including, for example, the age and weight of the human or animal, the precise disease condition which requires treatment, and its severity, the nature of the formulation and the method of administration, and is ultimately determined by the treating doctor or vet.
- an effective amount of a compound according to the invention is generally in the range from 0.1 to 100 mg/kg of body weight of the recipient (mammal) per day and particularly typically in the range from 1 to 10 mg/kg of body weight per day.
- the actual amount per day for an adult mammal weighing 70 kg is usually between 70 and 700 mg, where this amount can be administered as an individual dose per day or usually in a series of part-doses (such as, for example, two, three, four, five or six) per day, so that the total daily dose is the same.
- An effective amount of a salt or solvate or of a physiologically functional derivative thereof can be determined as the fraction of the effective amount of the compound according to the invention per se. It can be assumed that similar doses are suitable for the treatment of other conditions mentioned above.
- the compounds were characterised especially via the following analytical techniques.
- the NMR spectra were acquired using a Bruker Avance DPX 300 MHz NMR spectrometer.
- the masses were determined by HPLC coupled to an Agilent Series 1100 mass detector.
- the melting points (m.p.) were measured on a Stuart Scientific block.
- Step 1 To a solution of ethyl 2-amino-4-(4-bromophenyl)thiophene-3- carboxylate (12g, 36.8 mmol) in dioxanne (100 mL) was added dropwise a solution of phenylacetyl chloride (5.84 mL) in dioxanne (100 mL). After 15 minutes at room temperature, the reaction mixture was heated to 70 0 C for 1 hour. The solvent was removed under reduced pressure and the yellowish remaining solid taken up into a minimum of ethyl acetate. Petroleum ether was added and an off-white solid (14.3g) was recovered.
- Step 2 To a solution of previous compound (14.3g) in tetrahydrofurane (630 mL) was added potassium hexamethyldisilazane (256 mL, 0.5M in toluene). After 30 minutes at room temperature, the reaction mixtured was quenshed with hydrochloric solution (4M) and extracted with ethyl acetate. The organic phase was dried over sodium sulphate and the solvent removed under reduced pressure. The remaining yellow oil was taken up into a minimum of ethyl acetate. Petroleum ether was added and the off-white solid (11g) formed was recovered.
- Step 1 To a solution of ethyl 2-amino-4-(4-bromophenyl)thiophene-3- carboxylate (10g, 30.7 mmol), 3-pyridyl acetic acid hydrochloride (12.8g) in tetrahydrofurane (200 mL) was added dicyclohexylcarbodiimide (15g). The solution was heated to reflux overnight then filtered. The organic phase was taken up into ethyl acetate and washed with sodium bicarbonate solution. The organic solution was dried over sodium sulfate and the solvent removed under reduced pression. The crude solid obtained was washed with a mixture petroleum ether/minimum ethyl acetate. An off-white solid (9.8g) was recovered.
- Step 2 To the previous compound (5g, 11.2 mmol) in tetrahydrofurane (250 mL) was added dropwise potassium hexamethyldisilazane (90 mL, 0.5M solution in toluene). After 1 hour, the reaction mixture was concentrated to dryness and taken up into ethyl acetate / water mixture. The aquous phase was partially concentred until a solid precipitated. This one was filtered (3.46g) and washed with ethyl acetate; MS: 399.0 (M+1 );
- Step 1 A solution of intermediate 1 step 1 (2g, 4.50 mmol), 2-methoxyphenyl boronic acid (1.37g), cesium carbonate (4.4Og) and palladium tetrakis(triphenylphosphine) (468 mg) under argon in a mixture of toluene (55 ml_) / ethanol (65 mL) / water (32 ml_) was heated overnight at 80 0 C.
- Step 2 A solution of previous compound (1.54g, 3.27 mmol), N- chlorosuccinimide (0.48g) in chloroform (20 mL) was heated overnight at 50 0 C. The solvent was evaporated and the crude taken up into ethyl acetate. This solution was washed with water then dried over sodium sulphate. The solvent was removed under reduced pressure and the remaining oil was crystallisided with a mixture of isopropyl ether / minimum ethyl acetate. The formed solid (1.18g) was filtered.
- Step 3 To a solution of potassium hexamethyldisilazane (18.6 mL, 0.5 M in toluene) was added dropwise previuous compound (1.18g, 2.32 mmol, in 45 mL of tetrahydrofurane). After 1 hour, the solvants were removed under reduced pressure and a solution of hydrochloric acid (4N) was added to the crude solid.
- Example 4 4-hydroxy-3-(3'-hydroxymethylbiphenyl-4-yl)-5-(pyridine-3-yl)-6,7-dihydro- thieno[2,3-b]pyridin-6-one
- Step 1 A solution of 5-acetyl-2-chloro-pyridine (4.45 g, 28.6 mmol), 2- methoxyphenyl boronic acid (8.96 g), cesium carbonate (27.9 g) and palladium tetrakis(triphenylphosphine) (2.97 g) under argon in a mixture of toluene (10 ml_) / ethanol (12 mL) / water (6 ml_) was heated overnight at 8O 0 C. The solution was filtered over a pad of celite® and concentred under reduced pressure. The crude was purified over silica (dichloromethane then diisopropyl ether); MS: 228.1 (M+1 ).
- Step 2 A solution of previous compound (5.73 g, 25 mmol), sulphur (6.47 g), ethyl cyanoacetate (3.28 mL), morpholine (4.4 mL) and absolute ethanol (100 mL) was heated to 50 0 C overnight. The reaction mixture was filtered and the solvent evaporated. The remaining oil was taken up in a mixture of water / ethyl acetate. The aquous phase was extracted with ethyl acetate and the whole organic phase washed with brine, dried over sodium sulphate and concentred under reduced pressure. The crude oil was puridfied over silica (petroleum ether 80 / ethyl acetate 20) to afford the desired compound (2.38 g); MS: 355.1 (M+1 ).
- Step 3 To previous compound (2.38 g) in dioxane (15 mL) was added phenylaceyl chloride (1.15 mL in 5 mL of dioxane) dropwise. After 2 hours, the reaction mixture was evaporated to dryness. To the remaining oil was added iced water and diisopropyl ether. The precipited solid (2.4 g) was filtered and washed with water and diisopropylether.To this solid in tetrahydrofurane (40 ml_) was added potassium hexamethyldisilazane (4OL, 0.5 M in toluene) and the reaction mixture is heated at 70 0 C overnight. The solvants were removed under reduced pressure. To the remaining oil was added water and acetic acid (until pH 4). The precipated solid (1.77 g) was filtered and washed with water; MS: 427.1 (M+1 );
- AMPK enzyme activities were assayed by using A Delfia technology. AMPK enzyme activities were carried out in microtiter plates (50 mM Hepes buffer, pH 7.4 with 125 ⁇ M ATP respectively) in the presence of a synthetic peptide substrate (AMARAASAAALARRR, the "AMARA" peptide) and activators in serial dilutions. Reactions were initiated by the addition of AMPK (50-100 ng). Following mixing, the plates were incubated for 30 min at room temperature.
- Enzyme activity was assayed by using an anti-phosphoserine antibody to measure the quantity of phosphate incorporated into the AMARAA.
- Compounds of formula (I) in the table Il are considered as direct activator of AMPK if the ratio is 90% or higher.
- Skeletal muscle is the major site of insulin-stimulated glucose disposal and insulin resistance in this target tissue has long been viewed as a contribution factor in the pathogenesis of type 2 diabetes (T2D). Therefore alternative pathways that stimulate skeletal muscle glucose uptake independently of insulin signaling could potentially improve glycemic control in T2D subjects. Although exercise stimulates glucose uptake in skeletal muscle independently of the insulin pathway, the underlying molecular mechanisms remains largely elusive.
- AMPK is considered as an attractive candidate for contraction-induced skeletal muscle glucose uptake because it is activated in parallel with elevation in AMP and a reduction in creatine phosphate energy stores (Hubter CA. , Am. J. Physiol. Endocrinol. Metab. 272:E262-E266 ; 1997). Furthermore, AICAR-induced activation of AMPK increases glucose uptake (Merrill G. F. and al.., Am. J. Physiol. Endocrinol. Metab. 273:E1107-E1112 ; 1997).
- H-2Kb cells derived from heterozygous H-2Kb tsA58 transgenic mouse were grown in 24-well in plates coated with matrigel and were cultured at 33 0 C for 4 days under permissive conditions, as described previously by Fryer et al.
- cells were switched to non-permissive culture conditions (37 0 C in the absence of interferon- ⁇ ). After 3 days, cells were incubated for 4 hours in DMEM 1g/l glucose culture medium containing different concentrations of the tested molecules. Then glucose uptake was measured by incubating the cells for 10 min with radiolabeled 2-deoxy-D-[1 , 2 3 H] glucose. Glucose uptake was terminated by rapidly washing the plates 2 times with ice-cold NaCI 0,9%. The cells were then solubilized in 0.1 N NaOH for 30 min. Radioactivity was determined by liquid scintillation counting.
- N 0 Number of the molecule
- Activity tablelll Concentration of compound (I) for a glucose uptake equal or higher to glucose uptake induced by insulin (17OnM)
- Compounds of the invention are able to increase glucose uptake in a muscular cell line named H-2Kb independently of insulin. These data resulting of an enzymatic test followed by a cellular test shows that thienopyridone derivatives as defined in formula (I) are direct AMPK activators and these compounds are able to increase glucose uptake preferably in muscular cells.
Abstract
Description














Claims




Priority Applications (20)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09741799A EP2280952B1 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| AU2009243811A AU2009243811B2 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as AMP-activated protein kinase (AMPK) activators |
| NZ589690A NZ589690A (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as AMP-activated protein kinase (AMPK) activators |
| JP2011507805A JP5536757B2 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as protein kinase (AMPK) activators activated by AMP |
| US12/991,028 US8604202B2 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as AMP-activated protein kinase (AMPK) activators |
| KR1020167007434A KR20160038065A (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| ES09741799T ES2388485T3 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as activators of AMP-activated protein kinase (AMPK) |
| EA201001733A EA020773B1 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| CN200980116120.5A CN102015676B (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as AMP-activated protein kinase (AMPK) activators |
| UAA201014384A UA103617C2 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| SI200930319T SI2280952T1 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| PL09741799T PL2280952T3 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| MX2010011916A MX2010011916A (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators. |
| DK09741799.2T DK2280952T3 (en) | 2008-05-05 | 2009-04-08 | THIENOPYRIDINO DERIVATIVES AS AMP-ACTIVATED PROTEIN KINASE (AMPK) ACTIVATORS |
| BRPI0910832A BRPI0910832B8 (en) | 2008-05-05 | 2009-04-08 | thienopyridone derivatives as amp-activated protein kinase (ampk) activators, their uses, their preparation process and their intermediates, and drugs. |
| CA2723429A CA2723429C (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| IL208988A IL208988A (en) | 2008-05-05 | 2010-10-28 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| ZA2010/08722A ZA201008722B (en) | 2008-05-05 | 2010-12-03 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| HK11110571.3A HK1156313A1 (en) | 2008-05-05 | 2011-10-06 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators amp-(ampk) |
| HRP20120549AT HRP20120549T1 (en) | 2008-05-05 | 2012-07-05 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08290423 | 2008-05-05 | ||
| EP08290423.6 | 2008-05-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009135580A1 true WO2009135580A1 (en) | 2009-11-12 |
Family
ID=40756916
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/002606 WO2009135580A1 (en) | 2008-05-05 | 2009-04-08 | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US8604202B2 (en) |
| EP (1) | EP2280952B1 (en) |
| JP (1) | JP5536757B2 (en) |
| KR (2) | KR101648593B1 (en) |
| CN (1) | CN102015676B (en) |
| AR (1) | AR071530A1 (en) |
| AU (1) | AU2009243811B2 (en) |
| BR (1) | BRPI0910832B8 (en) |
| CA (1) | CA2723429C (en) |
| CO (1) | CO6331304A2 (en) |
| CY (1) | CY1113112T1 (en) |
| DK (1) | DK2280952T3 (en) |
| EA (1) | EA020773B1 (en) |
| EC (1) | ECSP10010654A (en) |
| ES (1) | ES2388485T3 (en) |
| HK (1) | HK1156313A1 (en) |
| HR (1) | HRP20120549T1 (en) |
| IL (1) | IL208988A (en) |
| MX (1) | MX2010011916A (en) |
| MY (1) | MY160357A (en) |
| NZ (1) | NZ589690A (en) |
| PL (1) | PL2280952T3 (en) |
| PT (1) | PT2280952E (en) |
| SI (1) | SI2280952T1 (en) |
| UA (1) | UA103617C2 (en) |
| WO (1) | WO2009135580A1 (en) |
| ZA (1) | ZA201008722B (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011121109A1 (en) | 2010-04-02 | 2011-10-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and compositions comprising ampk activator (metformin/troglitazone) for the treatment of myotonic dystrophy type 1 (dm1) |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| JP2013536818A (en) * | 2010-09-01 | 2013-09-26 | バイエル・インテレクチユアル・プロパテイー・ゲー・エム・ベー・ハー | Ketosultams and diketopyridines with herbicidal activity |
| EP2679591A1 (en) | 2012-06-29 | 2014-01-01 | Poxel | Thienopyridone derivatives useful as activators of AMPK |
| CN103517896A (en) * | 2011-03-07 | 2014-01-15 | 葛兰素史密斯克莱有限责任公司 | Quinolinone derivatives |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| US9394285B2 (en) | 2013-03-15 | 2016-07-19 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| WO2016205534A1 (en) * | 2015-06-16 | 2016-12-22 | Lazo John S | Inhibitors of ptp4a3 for the treatment of cancer |
| WO2017055925A2 (en) | 2015-09-30 | 2017-04-06 | Instituto De Medicina Molecular | Methods for attenuating parasite virulence |
| WO2019106087A1 (en) * | 2017-11-29 | 2019-06-06 | Syddansk Universitet | Ampk inhibitors |
| US10501474B2 (en) | 2015-06-25 | 2019-12-10 | University Health Network | HPK1 inhibitors and methods of using same |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| WO2022106892A1 (en) | 2020-11-17 | 2022-05-27 | Instituto De Medicina Molecular | Anti-malarial compounds |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10143703B2 (en) | 2014-01-02 | 2018-12-04 | Massachusetts Eye And Ear Infirmary | Treating ocular neovascularization |
| WO2015114663A1 (en) | 2014-01-30 | 2015-08-06 | Council Of Scientific & Industrial Research | Novel thieno [2,3-d]pyrimidin-4(3h)-one compounds with antimycobacterial properties |
| WO2019067442A1 (en) * | 2017-09-26 | 2019-04-04 | Ideaya Biosciences, Inc. | DIHYDROTHIENO[3,2-b]PYRIDINE COMPOUNDS |
| JP2023544026A (en) | 2020-09-30 | 2023-10-19 | バイオベラティブ セラピューティクス インコーポレイテッド | AMPK activator and its use |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0717044A1 (en) * | 1994-12-15 | 1996-06-19 | MERCK PATENT GmbH | Thienopyridone derivatives, their preparation and their use as NMDA-receptor antagonists |
| WO2003101979A1 (en) | 2002-05-31 | 2003-12-11 | Pharmacia & Upjohn Company Llc | Anthelmintic and insecticidal thiophene derivatives |
| US20050038068A1 (en) * | 2003-05-16 | 2005-02-17 | Iyengar Rajesh R. | Thienopyridones as AMPK activators for the treatment of diabetes and obesity |
| WO2007019914A1 (en) * | 2005-08-18 | 2007-02-22 | Merck Patent Gmbh | Use of thienopyridone derivatives as ampk activators and pharmaceutical compositions containing them |
| WO2007072094A1 (en) | 2005-12-20 | 2007-06-28 | Richter Gedeon Nyrt. | New compounds |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8627698D0 (en) * | 1986-11-20 | 1986-12-17 | Boots Co Plc | Therapeutic agents |
| WO2006093518A2 (en) * | 2004-06-25 | 2006-09-08 | Apath, Llc | Thienyl compounds for treating virus-related conditions |
| US7563813B2 (en) * | 2005-05-13 | 2009-07-21 | Wyeth | Iminothiazolidinone derivatives as SFRP-1 antagonists |
| PT2262500T (en) * | 2008-04-11 | 2016-08-16 | Merck Patent Gmbh | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
-
2009
- 2009-04-08 UA UAA201014384A patent/UA103617C2/en unknown
- 2009-04-08 CN CN200980116120.5A patent/CN102015676B/en active Active
- 2009-04-08 EP EP09741799A patent/EP2280952B1/en active Active
- 2009-04-08 WO PCT/EP2009/002606 patent/WO2009135580A1/en active Application Filing
- 2009-04-08 SI SI200930319T patent/SI2280952T1/en unknown
- 2009-04-08 KR KR1020107027208A patent/KR101648593B1/en active IP Right Grant
- 2009-04-08 KR KR1020167007434A patent/KR20160038065A/en not_active Application Discontinuation
- 2009-04-08 JP JP2011507805A patent/JP5536757B2/en active Active
- 2009-04-08 ES ES09741799T patent/ES2388485T3/en active Active
- 2009-04-08 CA CA2723429A patent/CA2723429C/en active Active
- 2009-04-08 EA EA201001733A patent/EA020773B1/en not_active IP Right Cessation
- 2009-04-08 PL PL09741799T patent/PL2280952T3/en unknown
- 2009-04-08 NZ NZ589690A patent/NZ589690A/en unknown
- 2009-04-08 MY MYPI2010005160A patent/MY160357A/en unknown
- 2009-04-08 AU AU2009243811A patent/AU2009243811B2/en active Active
- 2009-04-08 DK DK09741799.2T patent/DK2280952T3/en active
- 2009-04-08 MX MX2010011916A patent/MX2010011916A/en active IP Right Grant
- 2009-04-08 US US12/991,028 patent/US8604202B2/en active Active
- 2009-04-08 PT PT09741799T patent/PT2280952E/en unknown
- 2009-04-08 BR BRPI0910832A patent/BRPI0910832B8/en active IP Right Grant
- 2009-04-30 AR ARP090101562A patent/AR071530A1/en active IP Right Grant
-
2010
- 2010-10-28 CO CO10134194A patent/CO6331304A2/en active IP Right Grant
- 2010-10-28 IL IL208988A patent/IL208988A/en active IP Right Grant
- 2010-12-02 EC EC2010010654A patent/ECSP10010654A/en unknown
- 2010-12-03 ZA ZA2010/08722A patent/ZA201008722B/en unknown
-
2011
- 2011-10-06 HK HK11110571.3A patent/HK1156313A1/en unknown
-
2012
- 2012-07-05 HR HRP20120549AT patent/HRP20120549T1/en unknown
- 2012-09-13 CY CY20121100841T patent/CY1113112T1/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0717044A1 (en) * | 1994-12-15 | 1996-06-19 | MERCK PATENT GmbH | Thienopyridone derivatives, their preparation and their use as NMDA-receptor antagonists |
| WO2003101979A1 (en) | 2002-05-31 | 2003-12-11 | Pharmacia & Upjohn Company Llc | Anthelmintic and insecticidal thiophene derivatives |
| US20050038068A1 (en) * | 2003-05-16 | 2005-02-17 | Iyengar Rajesh R. | Thienopyridones as AMPK activators for the treatment of diabetes and obesity |
| WO2007019914A1 (en) * | 2005-08-18 | 2007-02-22 | Merck Patent Gmbh | Use of thienopyridone derivatives as ampk activators and pharmaceutical compositions containing them |
| WO2007072094A1 (en) | 2005-12-20 | 2007-06-28 | Richter Gedeon Nyrt. | New compounds |
Non-Patent Citations (4)
| Title |
|---|
| BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 17, 2007, pages 3254 - 3257 |
| BUCHSTALLER H-P ET AL: "THIENO[2,3-B]PYRIDINONES AS ANTAGONISTS ON THE GLYCINE SITE OF THE N-METHYL-D-ASPARTATE RECEPTOR-BINDING STUDIES, MOLECULAR MODELING AND STRUCTURE-ACTIVITY-RELATIONSHIPS", SCIENTIA PHARMACEUTICA, WIEN, vol. 68, 1 January 2000 (2000-01-01), pages 3 - 14, XP008059055, ISSN: 0036-8709 * |
| SCIENTIA PHARMAZEUTICA, vol. 68, 2000, pages 3 - 14 |
| ZHAO ET AL: "Discovery and SAR development of thienopyridones: A class of small molecule AMPK activators", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 17, no. 12, 15 June 2007 (2007-06-15), pages 3254 - 3257, XP022097763, ISSN: 0960-894X * |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011121109A1 (en) | 2010-04-02 | 2011-10-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and compositions comprising ampk activator (metformin/troglitazone) for the treatment of myotonic dystrophy type 1 (dm1) |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| JP2013536818A (en) * | 2010-09-01 | 2013-09-26 | バイエル・インテレクチユアル・プロパテイー・ゲー・エム・ベー・ハー | Ketosultams and diketopyridines with herbicidal activity |
| US8686000B2 (en) | 2010-09-01 | 2014-04-01 | Bayer Cropscience Ag | Herbicidally active ketosultams and diketopyridines |
| JP2017048211A (en) * | 2011-03-07 | 2017-03-09 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニーGlaxoSmithKline LLC | Quinolinone Derivative |
| CN103517896B (en) * | 2011-03-07 | 2016-09-21 | 葛兰素史密斯克莱有限责任公司 | (E)-3-(3-Acetyl-4-hydroxy-5-methoxy-phenyl)-N-(4-hydroxy-1-methyl-3-octyloxy-2-oxo-1,2-dihydro-quinolin-7-yl)-acrylamide |
| CN103517896A (en) * | 2011-03-07 | 2014-01-15 | 葛兰素史密斯克莱有限责任公司 | Quinolinone derivatives |
| JP2014507452A (en) * | 2011-03-07 | 2014-03-27 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | Quinoline derivatives |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| EP2679591A1 (en) | 2012-06-29 | 2014-01-01 | Poxel | Thienopyridone derivatives useful as activators of AMPK |
| WO2014001554A1 (en) | 2012-06-29 | 2014-01-03 | Poxel | Thienopyridone derivatives useful as activators of ampk |
| KR20150033709A (en) | 2012-06-29 | 2015-04-01 | 뽁셀 에스아에스 | Thienopyridone derivatives useful as activators of ampk |
| US9394285B2 (en) | 2013-03-15 | 2016-07-19 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| US10308663B2 (en) | 2015-06-16 | 2019-06-04 | University Of Virginia Patent Foundation | Inhibitors of PTP4A3 for the treatment of cancer |
| WO2016205534A1 (en) * | 2015-06-16 | 2016-12-22 | Lazo John S | Inhibitors of ptp4a3 for the treatment of cancer |
| US10501474B2 (en) | 2015-06-25 | 2019-12-10 | University Health Network | HPK1 inhibitors and methods of using same |
| US11059832B2 (en) | 2015-06-25 | 2021-07-13 | University Health Network | HPK1 inhibitors and methods of using same |
| TWI733679B (en) * | 2015-06-25 | 2021-07-21 | 加拿大健康網路大學 | Hpk1 inhibitors and methods of using same |
| WO2017055925A2 (en) | 2015-09-30 | 2017-04-06 | Instituto De Medicina Molecular | Methods for attenuating parasite virulence |
| WO2019106087A1 (en) * | 2017-11-29 | 2019-06-06 | Syddansk Universitet | Ampk inhibitors |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11851429B2 (en) | 2020-05-19 | 2023-12-26 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
| WO2022106892A1 (en) | 2020-11-17 | 2022-05-27 | Instituto De Medicina Molecular | Anti-malarial compounds |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2723429C (en) | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators | |
| US8563729B2 (en) | Thienopyridone derivatives as AMP-activated protein kinase (AMPK) activators | |
| EP2285786B1 (en) | Quinoxalinedione derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980116120.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09741799 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009741799 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10134194 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/011916 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2723429 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12991028 Country of ref document: US Ref document number: 2011507805 Country of ref document: JP Ref document number: 12010502474 Country of ref document: PH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009243811 Country of ref document: AU Ref document number: 201001733 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 589690 Country of ref document: NZ Ref document number: 4597/KOLNP/2010 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20107027208 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2009243811 Country of ref document: AU Date of ref document: 20090408 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0910832 Country of ref document: BR Kind code of ref document: A2 Effective date: 20101028 |