WO2009132978A1 - 4-dimethylaminobutyric acid derivatives - Google Patents
4-dimethylaminobutyric acid derivatives Download PDFInfo
- Publication number
- WO2009132978A1 WO2009132978A1 PCT/EP2009/054635 EP2009054635W WO2009132978A1 WO 2009132978 A1 WO2009132978 A1 WO 2009132978A1 EP 2009054635 W EP2009054635 W EP 2009054635W WO 2009132978 A1 WO2009132978 A1 WO 2009132978A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dimethylamino
- butyric acid
- phenyl
- formula
- nonanoylamino
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
- C07C237/12—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/04—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms
- C07C275/06—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton
- C07C275/16—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton being further substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/24—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
Definitions
- This invention is concerned with novel 4-dimethylaminobutyric acid derivatives, a process for the manufacture of these compounds, pharmaceutical preparations which contain such compounds as well as the use of these compounds for the production of medicaments.
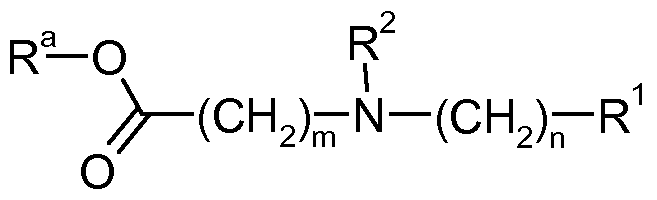
- the invention relates to compounds of the formula
- a 1 is NH or a bond
- a 2 is selected from the group consisting of a bond, O, O(CH 2 )2 ⁇ , S, SO 2 , CF 2 and NR 2 , wherein R 2 is hydrogen or lower alkyl,
- n 2 + 3, 4, 5, 6, 7, 8, 9, 10 and 11
- n is selected from 0, 1, 2, 3, 4 and 5
- R 1 is aryl selected from phenyl and naphthyl, said aryl being unsubstituted or substituted by one, two, three, four or five groups selected from the group consisting of lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl, or
- heteroaryl selected from the group consisting of pyridyl, thienyl and thiazolyl, said heteroaryl being unsubstituted or substituted by one, two or three groups selected from lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl,
- FFAs free fatty acids
- ATP energy
- NADH reducing force
- CPTs membrane-bound carnitine-dependent palmitoyltransferases
- Liver (L- CPTl) and muscle (M-CPTl) CPTl iso forms are encoded by two different genes and inhibited by malonyl-CoA.
- the N-terminal domain of L-CPTl confers its lower sensitivity to malonyl CoA.
- CPT2 the inner mitochondrial membrane enzyme, reconverts long-chain acylcarnitines into long-chain acyl CoA esters.
- Long-chain acyl-CoAs are then ⁇ -oxidized to acetyl-CoA, which activates the pyruvate carboxylase and gluconeogenesis.
- pharmaceutically active substances which inhibit transport of long chain FFA though the inhibition of CPTs, reduce liver ⁇ -oxidation, consequently inhibit gluconeogenesis and therefore counteract hyperglycemia.
- the present invention relates to novel compounds which inhibit carnitine palmitoyl transferase(CPT) activity, in particular/preferentially CPT2 activity.
- the compounds of the present invention can be used as pharmaceutically active agents, which are useful in the prevention and/or treatment of diseases which are modulated by CPT inhibitors, in particular/preferentially CPT2 inhibitors, particularly diseases which are related to hyperglycemia and/or glucose tolerance disorders.
- diseases include e.g. diabetes and associated pathologies, non insulin dependent diabetes mellitus (also referred to as diabetes type II), obesity, hypertension, insulin resistance syndrome, metabolic syndrome, hyperlipidemia, hypercholesterolemia, fatty liver disease, atherosclerosis, congestive heart failure and renal failure.
- novel compounds of the present invention exceed the compounds known in the art, inasmuch as they inhibit in particular or preferentially CPT2 activity. They are therefore expected to have an enhanced therapeutic potential compared to the compounds already known in the art. Unless otherwise indicated, the following definitions are set forth to illustrate and define the meaning and scope of the various terms used to describe the invention herein.
- lower is used to mean a group consisting of one to seven, preferably of one to four carbon atom(s).
- halogen refers to fluorine, chlorine, bromine and iodine, with fluorine, chlorine and bromine being preferred.
- alkyl refers to a branched or straight-chain monovalent saturated aliphatic hydrocarbon radical of one to twenty carbon atoms, preferably one to sixteen carbon atoms, more preferably one to ten carbon atoms.
- C 1- l o-alkyl refers to a branched or straight-chain monovalent saturated aliphatic hydrocarbon radical of one to ten carbon atoms, such as e.g. methyl, ethyl, n-propyl, isopropyl, n-butyl, s- butyl, t-butyl, pentyl, 1,1,3,3-tetramethyl-butyl and the like.
- Lower alkyl groups as described below also are preferred alkyl groups.
- lower alkyl or "Ci-Cy-alkyl”, alone or in combination with other groups, refers to a branched or straight-chain monovalent alkyl radical of one to seven carbon atoms, preferably one to four carbon atoms. This term is further exemplified by such radicals as methyl, ethyl, n- propyl, isopropyl, n-butyl, s-butyl, t-butyl and the like.
- lower halogenalkyl or “halogen-Ci-Cy-alkyl” refers to lower alkyl groups as defined above wherein at least one of the hydrogen atoms of the lower alkyl group is replaced by a halogen atom, preferably fluoro or chloro, most preferably fluoro.
- halogen atom preferably fluoro or chloro, most preferably fluoro.
- preferred lower halogenalkyl groups are trifluoromethyl, difluoromethyl, trifluoro ethyl, 2,2-difluoroethyl, fluoromethyl and chloromethyl, with trifluoromethyl being especially preferred.
- alkoxy refers to the group R'-O-, wherein R' is lower alkyl and the term “lower alkyl” has the previously given significance.
- lower alkoxy groups are e.g. methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy and tert-butoxy, preferably methoxy and ethoxy and most preferred methoxy.
- Compounds of formula (I) can form pharmaceutically acceptable salts.
- Compounds of formula (I) can form salts with bases. Examples of such salts are alkaline, earth-alkaline and ammonium salts such as e.g. Na-, K-, Ca- and trimethylammonium salt.
- Compounds of formula I can also form pharmaceutically acceptable acid addition salts.
- salts of compounds of formula (I) are salts of compounds of formula (I) with physiologically compatible mineral acids, such as hydrochloric acid, sulphuric acid or phosphoric acid; or with organic acids, such as methanesulphonic acid, p-toluenesulphonic acid, acetic acid, lactic acid, citric acid, fumaric acid, maleic acid, tartaric acid, succinic acid or salicylic acid.
- physiologically compatible mineral acids such as hydrochloric acid, sulphuric acid or phosphoric acid
- organic acids such as methanesulphonic acid, p-toluenesulphonic acid, acetic acid, lactic acid, citric acid, fumaric acid, maleic acid, tartaric acid, succinic acid or salicylic acid.
- pharmaceutically acceptable salts refers to all these salts.
- the present invention relates to compounds of the formula
- a 1 is NH or a bond
- a 2 is selected from the group consisting of a bond, O, O(CH2)2 ⁇ , S, SO 2 , CF 2 and NR 2 , wherein R 2 is hydrogen or lower alkyl,
- n 2 + 3, 4, 5, 6, 7, 8, 9, 10 and 11
- n is selected from 0, 1, 2, 3, 4 and 5
- R 1 is aryl selected from phenyl and naphthyl, said aryl being unsubstituted or substituted by one, two, three, four or five groups selected from the group consisting of lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl, or
- heteroaryl selected from the group consisting of pyridyl, thienyl and thiazolyl, said heteroaryl being unsubstituted or substituted by one, two or three groups selected from lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl,
- a 2 2 is selected from the group consisting of a bond, O, O(CH 2 )2 ⁇ , S, SO 2 and NR 2 , wherein R 2 is hydrogen or lower alkyl. More preferably, A 2 is selected from the group consisting of a bond, O and O(CH2)2 ⁇ .
- a group of more preferred compounds of formula I are those, wherein A 2 is O or O(CH 2 ) 2 ⁇ , with those compounds of formula I being especially preferred, wherein A 2 is O (oxygen).
- Another group of preferred compounds of formula I are those, wherein A 2 is a bond.
- n is selected from 0, 1, 2 and 3, with those compounds being more preferred, wherein n is selected from 0 or 1 , and those being most preferred wherein n is 1.
- a group of preferred compounds of formula (I) according to the invention are further those, wherein R 1 is aryl selected from phenyl and naphthyl, said aryl being unsubstituted or substituted by one, two, three, four or five groups selected from the group consisting of lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl.
- R 1 is phenyl substituted by one, two, three, four or five groups selected from the group consisting of lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl. More preferred are those compounds of formula I, wherein R 1 is phenyl substituted by one, two, three, four or five groups selected from the group consisting of lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl provided that at least one of the substituents is halogen or lower halogenalkyl. Especially preferred R 1 is phenyl substituted by one, two, three, four or five groups selected from halogen and lower halogenalkyl.
- R 1 is heteroaryl selected from the group consisting of pyridyl, thienyl and thiazolyl, said heteroaryl being unsubstituted or substituted by one, two or three groups selected from lower alkyl, halogen, lower halogenalkyl, lower alkoxy and phenyl.
- Preferred compounds of formula I are those selected from the group consisting of: (R)-3-[8-(3,4-difluoro-benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid, (R)-3-[8-(2,5-difluoro-benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid, (R)-3-[8-(2,4-difluoro-benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid, (R)-4-dimethylamino-3-[8-(2,3,4-trifluoro-benzyloxy)-octanoylamino]-butyric acid, (R)-4-dimethylamino-3-(8-pentafluorophenylmethoxy-octanoylamino)-butyric acid, (
- Particularly preferred compounds of formula I are those selected from the group consisting of:
- a compound of formula I which is (R)-4-dimethylamino-3-(10-phenyl-decanoylamino)-butyric acid.
- the compounds of general formula I in this invention may be derivatised at functional groups to provide derivatives which are capable of conversion back to the parent compound in vivo.
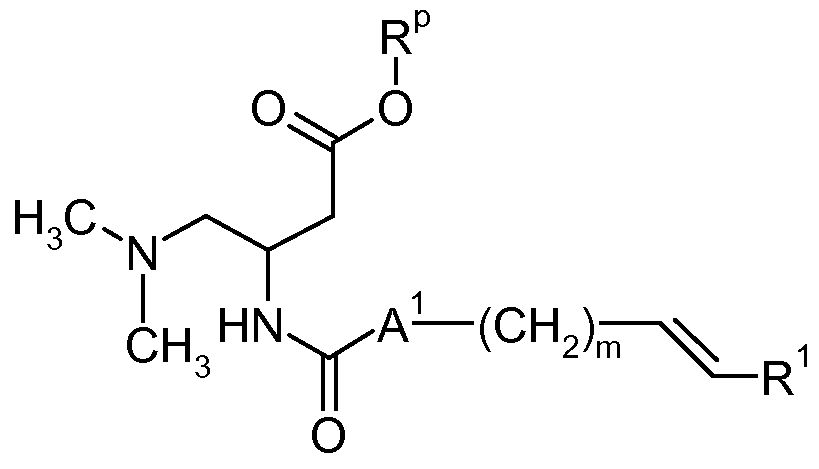
- the invention also relates to a process for the preparation of compounds of formula I as defined above, which process comprises a) condensating an amine of formula
- R p is methyl, ethyl or benzyl, with a carboxylic acid of the formula
- a 1 is a bond and A 2 , m, n and R 1 are as defined herein before, in the presence of a base and a condensing agent to obtain a compound of the formula
- R p is benzyl, with a carboxylic acid of the formula
- Ester hydrolysis means a base-catalyzed hydrolysis using reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide, in solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between O 0 C and 100 0 C.
- reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide, in solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between O 0 C and 100 0 C.
- Hydrogenation is normally carried out at a hydrogen pressure of 1 to 10 bar, using a suitable catalyst such as palladium on activated charcoal, in a solvent such as methanol or ethanol, at a temperature between 0 0 C and 50 0 C, but hydrogenation can also mean reduction of a double bond using triethysilane and trifluoro acetic acid in an inert solvent such as toluene or dichloromethane followed by ester hydrolysis as described hereinbefore.
- a suitable catalyst such as palladium on activated charcoal
- solvent such as methanol or ethanol
- the present invention also relates to compounds of formula I as defined above, when prepared by a process as described above.
- R p benzyl
- esters 1 can be transformed into compounds of formula I by base-catalyzed hydrolysis, using reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide, in solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between 0 0 C and 100 0 C.
- reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide
- solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between 0 0 C and 100 0 C.
- compounds of formula I wherein A 2 is a bond and n is 2, can also be synthesized from ester 2 (in the case where R p is benzyl) by hydrogenation as described above, whereby a double bond eventually adjacent to R 1 as a result of the synthetic protocol used (see below) is also reduced.
- R p is methyl, ethyl, or benzyl
- the transformation of 2 into a compound of formula I can also be accomplished in two steps as follows: In a first step the aforementioned double bond is reduced using triethysilane and trifluoro acetic acid in an inert solvent such as toluene or dichloromethane.
- ester group is hydrolyzed, using reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide, in solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between 0 0 C and 100 0 C.
- reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide, in solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between 0 0 C and 100 0 C.
- the reaction is preferably accomplished in an aprotic solvent such as dichloromethane or tetrahydrofuran, optionally in the presence of a base, e. g., triethylamine or 4-methylmorpholine.
- Such reactions can conveniently be carried out for example by mixing carboxylic acid 5 with amine 3 in aprotic solvents such as dichloromethane, tetrahydrofuran, N,N-dimethylformamide, N- methylpyrrolidinone and mixtures thereof at temperatures between 0 0 C and 60 0 C in the presence or absence of a base such as triethylamine or N,N-diisopropylethylamine, and a condensing agent.
- aprotic solvents such as dichloromethane, tetrahydrofuran, N,N-dimethylformamide, N- methylpyrrolidinone and mixtures thereof at temperatures between 0 0 C and 60 0 C in the presence or absence of a base such as triethylamine or N,N-diisopropylethylamine, and a condensing agent.
- Appropriate condensing agents can be for example O-(7-benzotriazol-l-yl)- N,N,N',N'-tetramethyluronium-tetrafluoroborate (TBTU), O-(7-azabenzotriazol- 1 -yl)-N,N,N',N'- tetramethyluronium-hexaflurophophate (HATU), N,N'-dicyclohexylcarbodiimide, l-(3- dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, O-(benzotriazol- 1 -yl)-N,N,N',N'- tetramethyluronium hexafluoro-phosphate, bromo-tris-pyrrolidino-phosphonium hexafluorophosphate or others well known to the person skilled in the art.
- TBTU O-(7-benzotri
- Such reactions can be performed in two steps involving first formation of the acyl halide derivative of 5 and subsequent coupling reaction with amine 3 in the presence of a base.
- reagents for the formation of the acyl chloride are thionyl chloride, phosphorus pentachloride, oxalyl chloride or cyanuric chloride, and the reaction is generally conducted in the absence of a solvent or in the presence of an aprotic solvent like dichloromethane, toluene or acetone.
- a base can optionally be added, like for example pyridine, triethylamine, N,N-diisopropylethylamine or 4-methylmorpholine.
- acyl chloride can be isolated or reacted as such with an amine 3 in an aprotic solvent, like dichloromethane, tetrahydrofuran or acetone, in the presence of a base.
- aprotic solvent like dichloromethane, tetrahydrofuran or acetone
- Typical bases are triethylamine, A- methylmorpholine, pyridine, N,N-diisopropylethyl-amine or dimethylaminopyridine or mixtures thereof.
- Such reactions can be performed in two steps involving first formation of a mixed anhydride derivative of 5 obtained by reaction with a reagent such as ethyl chloro formate, isobutyl chloroformate, or acetic anhydride, and subsequent reaction with amine 3 as described above.
- a reagent such as ethyl chloro formate, isobutyl chloroformate, or acetic anhydride
- step a carboxylic acid 7 is reacted with dimethylamine to the N 5 N- dimethylamide derivative 8, using reagents and methods as described for the reaction of carboxylic acid 5 with amine 3.
- step b scheme I 5 N,N-dimethylamide 8 is converted to dimethylamine derivative 3 by reduction and subsequent removal of the tert-butoxycarbonyl protective group.
- Preferred reagents for the reduction are borane-tetrahydrofuran complex or borane-dimethylsulfide complex, in an aprotic solvent such as tetrahydrofuran, at temperature between -20 0 C and 80 0 C.
- Removal of the tert-butoxycarbonyl group is accomplished in an acidic environment, using hydrochloric acid or sulfuric acid, in solvents such as ethanol, methanol, water or mixtures thereof, at temperatures between 0 0 C and 20 0 C.
- Isocyanate 4 is synthesized from carboxylic acid 5 as highlighted in scheme 2. This conversion is accomplished by methods well known in the art, e. g., Curtius rearrangement. A typical procedure starts the transformation of 5 to its acyl halide derivative. Typically employed reagents for the formation of the acyl chloride are thionyl chloride, phosphorus pentachloride, oxalyl chloride, ethyl chloroformate, isobutyl chloroformate, or cyanuric chloride, and the reaction is generally conducted in the absence of a solvent or in the presence of an aprotic solvent like dichloromethane or toluene.
- a base can optionally be added, like for example pyridine, triethylamine, diisopropyl ethyl amine or 4-methylmorpholine.
- the obtained acyl chloride can be isolated or reacted as such with sodium azide, leading to the acyl azide derivative of 5, which is not isolated but heated to >60 0 C, whereupon it rearranges to isocyanate 4 under elimination of nitrogen gas.
- the conversion of 5 to 4 can be accomplished in a single step, using diphenylphosphoryl azide as azide source, optionally in the presence of a base, e. g., triethylamine, at temperatures between 0 0 C and 110 0 C, preferably in toluene.
- a base e. g., triethylamine
- Carboxylic acids 5 are either commercially available or can be produced as outlined in schemes 3 to 10.
- the carboxylic acids 5 can be produced as described in scheme 3, where X is a leaving group such as bromine, iodine, or methanesulfonyloxy and PG is an optional protective group, e. g., tetrahydropyran-2-yl.
- step a scheme 3
- compound 10 is alkylated with optionally protected ⁇ -halo or ⁇ - sulfonyloxy alcohol 9, leading to 11.
- the reaction is performed in a solvent such as ethanol, acetonitrile, or N, ⁇ /-dimethylformamide, in the presence of a base, e. g., potassium carbonate, sodium hydroxide, potassium tert-butylate, or sodium hydride, at temperatures between 0 0 C and 100 0 C.
- a base e. g., potassium carbonate, sodium hydroxide, potassium tert-butylate, or sodium hydride
- step b i. e., in the case where PG ⁇ H
- the protective group of 11 is removed, leading to alcohol 12.
- this reaction is accomplished using an acid catalyst such as hydrochloric acid, toluene-4-sulfonic acid, or pyridinium toluene- 4-sulfonate, in a solvent such as water, methanol, or ethanol, at temperatures between 0 0 C and 100 0 C.
- step c scheme 3, alcohol 12 is oxidized to carboxylic acid 5.
- reagents and conditions for the oxidation of alcohol 12 include pyridinium dichromate, chromium(VI)oxide, or potassium permanganate. This oxidation of 12 to 5 is also possible for alcohols 12 in which A 2 is a bond.
- alcohol 12 can be synthesized as outlined in scheme 4.
- a 2 is oxygen, sulfur, or SO 2 , R 1 , m and n are as defined above.
- diol 13 and compound 14 are reacted under Mitsunobu conditions using a phosphine, e. g., triphenylphosphine, and an azodicarboxylic acid diester, e. g., diethyl azodicarboxylate or diisopropyl azodicarboxylate, in a solvent such as tetrahydrofuran, dichloromethane, or toluene, at temperatures between 0 0 C and 50 0 C, leading to 12.
- a phosphine e. g., triphenylphosphine
- an azodicarboxylic acid diester e. g., diethyl azodicarboxylate or diisopropyl azodicarboxylate
- alcohol 12 can be synthesized as outlined in scheme 5.
- a 2 is O, S or SO 2 , R 1 , m and n are as defined above and X is a leaving group such as bromine, iodine, or methanesulfonyloxy.
- compound 15 is alkylated with halide or sulfonate 16.
- the reaction is performed in a solvent such as ethanol, acetonitrile, or N, ⁇ /-dimethylformamide, in the presence of a base, e. g., potassium carbonate, sodium hydroxide, potassium tert-butylate, or sodium hydride, at temperatures between 0 0 C and 100 0 C.
- a base e. g., potassium carbonate, sodium hydroxide, potassium tert-butylate, or sodium hydride
- Acid 5 can also be synthesized as outlined in scheme 6.
- a 2 is O, S or SO 2 , R 1 , m and n are as defined above, X is a leaving group such as bromine, iodine, or methanesulfonyloxy.
- the alkylation of carboxylic acid 17 with halide or sulfonate 16 is performed in an analogous fashion to that of 16 with 17 (scheme 5).
- Acid 5 in which A 2 is N(R 2 ), is represented as a compound of formula 18.
- the compound of formula 18 can be synthesized as outlined in scheme 7. R 1 , R 2 , m and n are as defined above.
- step a dicarboxylic acid monoester 19 is reduced to ⁇ -hydroxyester 20, using reagents known in the art, e. g., borane-tetrahydrofuran complex, in a solvent such as tetrahydrofuran, at temperatures between 0 0 C and 50 0 C.
- reagents known in the art e. g., borane-tetrahydrofuran complex, in a solvent such as tetrahydrofuran, at temperatures between 0 0 C and 50 0 C.
- step b scheme 7, the hydroxy group of 20 is oxidized to a formyl group, leading to 21.
- Suitable reagents are e. g., sodium hypochlorite, in the presence of potassium bromide, 2,2,6,6- tetramethylpiperidin-1-oxyl, and sodium hydrogencarbonate, in a biphasic mixture of water and dichloromethane, at around 0 0 C.
- dimethyl sulfoxide-based reagents can be employed, such as dimethyl sulfoxide - oxalyl chloride, or dimethyl sulfoxide - trifluoro acetic anhydride, in a solvent such as dichloromethane, at temperatures below 0 0 C, typically at -78 0 C.
- aldehyde 21 is reacted with amine 22 in the presence of a reducing agent to give aminoester 23.
- a reducing agent typically used reagents are sodium boro hydride (optionally in the presence of titanium(IV)isopropoxide), sodium cyanoborohydride, or sodium triacetoxyborohydride, in solvents such as methanol, acetic acid, tetrahydrofuran, 1,2- dichloroethane, or mixtures thereof, at temperatures between 0 0 C and 100 0 C.
- step d aminoester 23 is converted to acid 18 by base-catalyzed hydrolysis, using reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide, in solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between 0 0 C and 100 0 C.
- reagents such as lithium hydroxide, sodium hydroxide, potassium hydroxide, in solvents such as water, methanol, ethanol, tetrahydrofuran, or mixtures thereof, at temperatures between 0 0 C and 100 0 C.
- aminoester 23 can be accessed as outlined in scheme 8.
- R 1 , R 2 and m are as defined above, R a is lower alkyl, e. g., methyl or ethyl.
- step a scheme 8
- amide 24 is obtained from acid 19 by treatment with amine 22, using reagents and methods as described for the reaction of carboxylic acid 5 with amine 3.
- aminoester 23 is obtained by reduction of amide 24, using reagents such as diborane, borane-dimethylsulf ⁇ de complex or borane-tetrahydrofuran complex in solvents such as tetrahydrofuran at temperatures between 0 0 C and 100 0 C.
- reagents such as diborane, borane-dimethylsulf ⁇ de complex or borane-tetrahydrofuran complex in solvents such as tetrahydrofuran at temperatures between 0 0 C and 100 0 C.
- Unsaturated acids of general formula 6 can be synthesized as outlined in scheme 9.
- R 1 and m are as defined above, R a is lower alkyl, e. g., methyl or ethyl.
- step a unsaturated ester 25 is reacted with styrene derivative 26 in an alkene cross-metathesis reaction, leading to 27.
- This reaction is carried out in an inert solvent, such as dichloromethane or toluene and requires a suitable catalyst, e. g., dichloro(l,3-dimesityl-4,5- dihydroimidazol-2-ylidene)(phenylmethylene)(tricyclohexyl-phosphine)ruthenium, at temperatures between 20 0 C and 100 0 C.
- step b scheme 9, ester 27 is converted to acid 6 by base-catalyzed hydrolysis, in analogy to scheme 7, step d.
- unsaturated acids of formula 6 can be synthesized as outlined in scheme 10.
- R 1 and m are as defined above.
- step a scheme 10
- ⁇ -bromoacid 28 is reacted with triphenylphosphine, leading to phosphonium salt 29.
- This reaction is carried out in an inert solvent such as toluene, at temperatures between 20 0 C and 110 0 C.
- step b scheme 10, phosphonium salt 29 is reacted with aldehyde 30, leading to 6.
- This reaction is carried out in the presence of a base, e. g., sodium hydride, n-butyllithium, or potassium tert-butylate, in a solvent such as diethyl ether or tetrahydrofuran, at temperatures between -20 0 C and 50 0 C.
- a base e. g., sodium hydride, n-butyllithium, or potassium tert-butylate
- the novel compounds of formula I of the present invention have been found to inhibit carnitine palmitoyl transferase 2 (CPT2) activity.
- CPT2 carnitine palmitoyl transferase 2
- the compounds of the present invention can therefore be used in the treatment and/or prophylaxis of diseases that are modulated by CPT2 inhibitors, particularly diseases that are related to hyperglycemia and/or glucose tolerance disorders.
- diseases include e.g. diabetes and associated pathologies, non insulin dependent diabetes mellitus, obesity, hypertension, insulin resistance syndrome, metabolic syndrome, hyperlipidemia, hypercholesterolemia, fatty liver disease, atherosclerosis, congestive heart failure and renal failure.
- the invention therefore also relates to pharmaceutical compositions comprising a compound of formula I as defined above and a pharmaceutically acceptable carrier and/or adjuvant.
- the invention also embraces compounds of formula I as described above for use as therapeutically active substances, especially as therapeutically active substances for the treatment and/or prophylaxis of diseases which are modulated by CPT2 inhibitors, particularly for use as therapeutically active substances for the treatment and/or prophylaxis of hyperglycemia, glucose tolerance disorders, diabetes and associated pathologies, non insulin dependent diabetes mellitus, obesity, hypertension, insulin resistance syndrome, metabolic syndrome, hyperlipidemia, hypercholesterolemia, fatty liver disease, atherosclerosis, congestive heart failure and renal failure.
- CPT2 inhibitors particularly for use as therapeutically active substances for the treatment and/or prophylaxis of hyperglycemia, glucose tolerance disorders, diabetes and associated pathologies, non insulin dependent diabetes mellitus, obesity, hypertension, insulin resistance syndrome, metabolic syndrome, hyperlipidemia, hypercholesterolemia, fatty liver disease, atherosclerosis, congestive heart failure and renal failure.
- the invention relates to a method for the therapeutic and/or prophylactic treatment of diseases which are modulated by CPT2 inhibitors, particularly for the therapeutic and/or prophylactic treatment of hyperglycemia, glucose tolerance disorders, diabetes and associated pathologies, non insulin dependent diabetes mellitus, obesity, hypertension, insulin resistance syndrome, metabolic syndrome, hyperlipidemia, hypercholesterolemia, fatty liver disease, atherosclerosis, congestive heart failure and renal failure, which method comprises administering a compound of formula I as defined above to a human being or animal.
- the invention also relates to the use of compounds of formula I as described above for the preparation of medicaments for the therapeutic and/or prophylactic treatment of diseases which are modulated by CPT2 inhibitors, particularly for the therapeutic and/or prophylactic treatment of hyperglycemia, glucose tolerance disorders, diabetes and associated pathologies, non insulin dependent diabetes mellitus, obesity, hypertension, insulin resistance syndrome, metabolic syndrome, hyperlipidemia, hypercholesterolemia, fatty liver disease, atherosclerosis, congestive heart failure and renal failure.
- Such medicaments comprise a compound of formula I as described above.
- Prevention and/or treatment of hyperglycemia and non insulin dependent diabetes mellitus is the preferred use.
- the compounds according to formula I preferably have an IC50 value (CPT2) below 10 ⁇ M, preferably 1 nM to 10 ⁇ M, more preferably 1 nM to 1 ⁇ M.
- CPT2 IC50 value
- the compounds of formula I and/or their pharmaceutically acceptable salts can be used as medicaments, e.g. in the form of pharmaceutical preparations for enteral, parenteral or topical administration. They can be administered, for example, perorally, e.g. in the form of tablets, coated tablets, dragees, hard and soft gelatine capsules, solutions, emulsions or suspensions, rectally, e.g. in the form of suppositories, parenterally, e.g. in the form of injection solutions or suspensions or infusion solutions, or topically, e.g. in the form of ointments, creams or oils. Oral administration is preferred.
- the production of the pharmaceutical preparations can be effected in a manner which will be familiar to any person skilled in the art by bringing the described compounds of formula I and/or their pharmaceutically acceptable salts, optionally in combination with other therapeutically valuable substances, into a galenical administration form together with suitable, non-toxic, inert, therapeutically compatible solid or liquid carrier materials and, if desired, usual pharmaceutical adjuvants.
- Suitable carrier materials are not only inorganic carrier materials, but also organic carrier materials.
- lactose, corn starch or derivatives thereof, talc, stearic acid or its salts can be used as carrier materials for tablets, coated tablets, dragees and hard gelatine capsules.
- Suitable carrier materials for soft gelatine capsules are, for example, vegetable oils, waxes, fats and semi-solid and liquid polyols (depending on the nature of the active ingredient no carriers might, however, be required in the case of soft gelatine capsules).
- Suitable carrier materials for the production of solutions and syrups are, for example, water, polyols, sucrose, invert sugar and the like.
- Suitable carrier materials for injection solutions are, for example, water, alcohols, polyols, glycerol and vegetable oils.
- Suitable carrier materials for suppositories are, for example, natural or hardened oils, waxes, fats and semi-liquid or liquid polyols.
- Suitable carrier materials for topical preparations are glycerides, semi- synthetic and synthetic glycerides, hydrogenated oils, liquid waxes, liquid paraffins, liquid fatty alcohols, sterols, polyethylene glycols and cellulose derivatives.
- Usual stabilizers preservatives, wetting and emulsifying agents, consistency-improving agents, flavour-improving agents, salts for varying the osmotic pressure, buffer substances, solubilizers, colorants and masking agents and antioxidants come into consideration as pharmaceutical adjuvants.
- the dosage of the compounds of formula I can vary within wide limits depending on the disease to be controlled, the age and the individual condition of the patient and the mode of administration, and will, of course, be fitted to the individual requirements in each particular case. For adult patients a daily dosage of about 1 to 2000 mg, especially about 1 to 500 mg, comes into consideration. Depending on the severity of the disease and the precise pharmacokinetic profile the compound could be administered with one or several daily dosage units, e.g. in 1 to 3 dosage units.
- the pharmaceutical preparations conveniently contain about 1 to 500 mg, preferably 1 to 200 mg, of a compound of formula I.
- HPLC high pressure liquid chromatography
- m/e mass to charge ratio as measured by mass spectrometry (MS).
- Step 1 A solution of 1,8-octanediol (300 mg, 2.05 mmol) in tetrahydrofuran/iV,iV- dimethylformamide 2:1 (3 mL) was added dropwise at 0 0 C to a suspension of sodium hydride (60% dispersion in mineral oil, 90 mg, 2.3 mmol) in N, ⁇ /-dimethylformamide (1.5 mL), then after 2 h 3,4-difluorobenzyl bromide (445 mg, 2.15 mmol) was added. After 4 h the reaction mixture was partitioned between water and ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and evaporated.
- Step 2 Pyridinium dichromate (1.23 g, 3.27 mmol) was added at 0 0 C to a solution of 8-
- Step 3 A solution of 8-(3,4-difluoro-benzyloxy)-octanoic acid (153 mg, 0.53 mmol), N,N- diisopropylethylamine (414 mg, 3.21 mmol), and O-(7-azabenzotriazol-l-yl)-N,N,N',N'- tetramethyluronium-hexaflurophophate (244 mg, 0,64 mmol) in N, ⁇ /-dimethylformamide was stirred for 1 h at room temperature, then a solution of (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride (198 mg, 0.64 mmol) in N, ⁇ /-dimethylformamide (1 mL) was added.
- Triethylamine (2.8 mL, 20.0 mmol) was added at 0 0 C to a solution of Boc-D-aspartic acid 4-benzyl ester (5.00 g, 15.0 mmol) in dichloromethane, then ethyl chloroformate (1.91 mL, 20.0 mmol) was added dropwise.
- the reaction mixture was stirred for 1 h at 0 0 C, then a solution of dimethylamine hydrochloride (2.65 g, 32.0 mmol) and triethylamine (4.53 mL, 32.0 mmol) in dichloromethane (100 mL) were added dropwise.
- 1,8-octanediol was alkylated in step 1 with 2,5-difluorobenzyl bromide, leading to 8-(2,5-difluoro-benzyloxy)-octan-l-ol, which was oxidized in step 2 to 8-(2,5-difluoro- benzyloxy)-octanoic acid.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(2,5-difluoro-benzyloxy)-octanoylamino]- 4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(2,4-difluoro-benzyloxy)-octanoylamino]- 4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(2,3,4-trifluoro- benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(pentafluoro-benzyloxy)-octanoylamino]- 4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3- [8-(3-fluoro-4-trifluoromethyl-benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydro genated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(4-methoxy-benzyloxy)-octanoylamino]-4- dimethylamino -butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino- butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(biphenyl-4-ylmethoxy)- octanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- 1,8-octanediol was alkylated in step 1 with 2-fluoro-4-trifluoromethyl-benzyl bromide, leading to 8-(2-fluoro-4-trifluoromethyl-benzyloxy)-octan-l-ol, which was oxidized in step 2 to 8-(2-fluoro-4-trifluoromethyl-benzyloxy)-octanoic acid.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3- [8-(2-fluoro-4-trifluoromethyl-benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydro genated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)- 3-[8-(2,3,5,6-tetrafluoro-4-methoxy-benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino- butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(naphthalen-l-ylmethoxy)- octanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(3-fluoro-benzyloxy)-octanoylamino]-4- dimethylamino -butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(4-fluoro-benzyloxy)-octanoylamino]-4- dimethylamino -butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-3-[8-(2,3-difluoro-benzyloxy)-octanoylamino]- 4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)- 3-[8-benzyloxy)-octanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydro genated in step 4.
- Step 1 Potassium carbonate (2.59 g, 18.7 mmol) and 9-bromo-l-nonanol (1.39 g, 6.24 mmol) were added at room temperature to a solution of 2-fluorophenol (700 mg, 6.24 mmol) in N, ⁇ /-dimethylformamide (20 mL), then after 40 h insoluble material was removed by filtration. The filtrate was evaporated and the residue taken up in dichloromethane, washed with 1 M aq. sodium hydroxide solution, dried over sodium sulfate, filtered, and evaporated, to afford 9-(2- fluoro-phenoxy)-nonan-l-ol (1.7 g), which was directly used in the next step.
- Step 2 Oxidation of 9-(2-fluoro-phenoxy)-nonan-l-ol in analogy with example 1, step 2 gave 9-(2-fluoro-phenoxy)-nonanoic acid.
- Step 3 Amide coupling of 9-(2-fluoro-phenoxy)-nonanoic acid with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydro chloride in analogy with example 1, step 3 led to (R)-4-dimethylamino-3-[9-(2-fluoro-phenoxy)-nonanoylamino]-butyric acid benzyl ester.
- Step 4 Hydrogenation of (R)-4-dimethylamino-3-[9-(2-fluoro-phenoxy)-nonanoylamino]- butyric acid benzyl ester in analogy with example 1, step 4 produced (R)-4-dimethylamino-3-[9- (2-fluoro-phenoxy)-nonanoylamino]-butyric acid.
- White solid, m/e 395.5 ([M-H] " ).
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(3-fluoro-phenoxy)-nonanoylamino]- butyric acid benzyl ester, which was hydro genated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(4-fluoro-phenoxy)-nonanoylamino]- butyric acid benzyl ester, which was hydro genated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(2,3-difluoro-phenoxy)- nonanoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(2,4-difluoro-phenoxy)- nonanoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(2,3,4-trifluoro- phenoxy)-nonanoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(biphenyl-4-yloxy)-nonanoylamino]- butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9- (3,4-dimethoxy-phenoxy)-nonanoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9- (4-trifluoromethyl-phenoxy)-nonanoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(4-methoxy-phenoxy)- nonanoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- naphthalen-1-ol was alkylated in step 1 with 9-bromo-l-nonanol, leading to 9- (naphthalen-l-yloxy)-nonan-l-ol, which was oxidized in step 2 to 9-(naphthalen-l-yloxy)- nonanoic acid.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4-dimethylamino-3-[9-(naphthalen-l-yloxy)- nonanoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- Step 2 Sodium hydride (60% dispersion in mineral oil; 0.65 g, 16 mmol) was added portionwise at room temperature to a stirred solution of (8-carboxy-octyl)-triphenyl- phosphonium bromide (2.8 g, 5.6 mmol) in tetrahydrofuran (30 mL) at room temperature, then after 1 hour 4-fluorobenzaldehyde (700 mg, 5.64 mmol) was added portionwise to the reaction mixture and the solution stirred for 2 days at room temperature. On reaction completion, water (10ml) was added and the solution acidified to pH 3 with concentrated hydrochloric acid.
- Step 3 A solution of 10-(4-fluoro-phenyl)-dec-9-enoic acid (230 mg, 0.87 mmol), oxalyl chloride (0.11 mL, 1.3 mmol), and N, ⁇ /-dimethylformamide (one drop) in dichloromethane (3 mL) was stirred at room temperature for 2 hours, then volatile material was removed by distillation to afford 10-(4-fluoro-phenyl)-dec-9-enoyl chloride.
- Step 1 To a solution of 9-decenoic acid ethyl ester (Tetrahedron 2003, 59, 7973; 500 mg, 2.53 mmol) and 2-fluorostyrene (617 mg, 5.05 mmol) in dichloromethane (12.5 mL) was added dichloro(l,3-dimesityl-4,5-dihydroimidazol-2-ylidene)(phenylmethylene)
- Step 2 To a solution of 10-(2-fluoro-phenyl)-dec-9-enoic acid ethyl ester (420 mg, 1.44 mmol) in tetrahydrofuran (2 mL) was added 2 M aq. lithium hydroxide solution (2 mL, 4 mmol). The reaction mixture was stirred at room temperature for 16 h, then partitioned between 1 M aq. hydrochloric acid solution and ethyl acetate. The organic layer was dried over magnesium sulfate, filtered, and evaporated, to afford 10-(2-fluoro-phenyl)-dec-9-enoic acid (250 mg, 66%).
- Step 3 In analogy with example 34, step 3, 10-(2-fluoro-phenyl)-dec-9-enoic acid was converted to 10-(2-fluoro-phenyl)-dec-9-enoyl chloride, then reacted with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10- (2-fluoro-phenyl)-dec-9-enoylamino]-butyric acid benzyl ester.
- step 3 this was converted to 10-(2,5-dimethyl- phenyl)-dec-9-enoyl chloride, then reacted with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10-(2,5-dimethyl-phenyl)-dec- 9-enoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 this was converted to 10-(2,6-dimethyl-phenyl)-dec-9-enoyl chloride, then reacted with (R)-3- amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4- dimethylamino-3-[10-(2,6-dimethyl-phenyl)-dec-9-enoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- Example 38
- step 3 this was converted to 10-(4-methoxy- phenyl)-dec-9-enoyl chloride, then reacted with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10-(4-methoxy-phenyl)-dec-9- enoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 this was converted to l ⁇ -(naphthalene-l-yl)- dec-9-enoyl chloride, then reacted with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10-(naphthalene-l-yl)-dec-9-enoylamino]- butyric acid benzyl ester, which was hydrogenated in step 4.
- step 3 this was converted to 10-(4- trifluoromethyl-phenyl)-dec-9-enoyl chloride, then reacted with (R)-3-amino-4-dimethylamino- butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10-(4- trifluoromethyl-phenyl)-dec-9-enoylamino]-butyric acid benzyl ester, which was hydrogenated in step 4.
- Example 41
- step 3 this was converted to 10-(3-fluoro-phenyl)-dec-9- enoyl chloride, then reacted with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10-(3-fluoro-phenyl)-dec-9-enoylamino]- butyric acid benzyl ester, which was hydro genated in step 4.
- step 3 this was converted to 10-(2,3-difluoro-phenyl)-dec-9-enoyl chloride, then reacted with (R)-3- amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4- dimethylamino-3-[10-(2,3-difluoro-phenyl)-dec-9-enoylamino]-butyric acid benzyl ester, which was hydro genated in step 4.
- Step 1 Reaction of 3-thiophenecarboxaldehyde with (8-carboxy-octyl)-triphenyl- phosphonium bromide, in analogy with example 34, step 2, produced 10-(thiophen-3-yl)-dec-9- enoic acid.
- Step 2 In analogy with example 34, step 3, 10-(thiophen-3-yl)-dec-9-enoic acid was converted to 10-(thiophen-3-yl)-dec-9-enoyl chloride, then reacted with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10- (thiophen-3-yl)-dec-9-enoylamino]-butyric acid benzyl ester.
- Step 3 Triethylsilane (0.23 mL, 1.43 mmol) and trifluoro acetic acid (0.21 mL, 2.9 mmol) were added to a solution of 4-dimethylamino-3-(10-thiophen-3-yl-dec-9-enoylamino)-butyric acid benzyl ester (67 mg, 0.14 mmol) in toluene (8 mL). The reaction mixture was stirred at room temperature for 12 hours after which time the solution was added to cold saturated aq. sodium bicarbonate solution. The aqueous phase was separated and extracted twice with dichloromethane.
- step 2 this was converted to 10-( thiazol-5-yl)-dec-9-enoyl chloride, then reacted with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[10- (2,3-difluoro-phenyl)-dec-9-enoylamino]-butyric acid benzyl ester.
- step 3 reaction of (R)-4- dimethylamino-3-[10-(2,3-difluoro-phenyl)-dec-9-enoylamino]-butyric acid benzyl ester with triethylsilane-trifluoroacetic acid produced 4-dimethylamino-3-(10-thiazol-5-yl- decanoylamino)-butyric acid benzyl ester, which was hydro lyzed in step 4.
- step 3 commercially available 8-phenyloctanoic acid was coupled in step 3 with (R)-3- amino-4-dimethylamino-butyric acid benzyl ester dihydro chloride to produce (R)-4- dimethylamino-3-(8-phenyl-octanoylamino)-butyric acid benzyl ester, which was hydrogenated in step 4.
- Step 2 Amide coupling of 4-(5-phenyl-pentyloxy)-butyric acid with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydro chloride in analogy with example 1, step 3 led to (R)-4-dimethylamino-3-[4-(5-phenyl-pentyloxy)-butyrylamino]-butyric acid benzyl ester.
- Step 3 Hydrogenation of (R)-4-dimethylamino-3-[4-(5-phenyl-pentyloxy)-butyrylamino]- butyric acid benzyl ester in analogy with example 1, step 4 produced (R)-4-dimethylamino-3-[9- (2-fluoro-phenoxy)-nonanoylamino]-butyric acid.
- White solid, m/e 379.5 ([M+H] + ).
- Step 1 A solution of 9-bromo-l-nonanol (2.00 g, 8.96 mmol), 3,4-dihydro-2H-pyran (792 mg, 9.41 mmol), and pyridinium toluene-4-sulfonate (1.08 g, 4.30 mmol) in dichloromethane (34 mL) was stirred at room temperature for 16 h, then washed with 2 M aq. sodium carbonate solution and brine, dried over sodium sulfate, filtered, and evaporated.
- Step 2 Sodium hydride (60% dispersion in mineral oil, 264 mg, 6.6 mmol) was added at 0 0 C to a solution of 2-phenylethanol (807 mg, 6.61 mmol) in tetrahydrofuran (8.6 mL) and N,N- dimethylformamide (3.4 mL), then after 1 h a solution of 2-(9-bromo-nonyloxy)-tetrahydro- pyran (2.44 g, 7.93 mmol) in N, ⁇ /-dimethylformamide (1.5 mL) was added dropwise. The solution was allowed to reach room temperature over 16 h, then partitioned between water and ethyl acetate.
- Step 4 Oxidation of 9-phenethyloxy-nonan-l-ol in analogy with example 1, step 2 gave 9- phenethyloxy-nonanoic acid.
- White solid, m/e 277.4 ([M-H] " ).
- Step 5 Amide coupling of 9-phenethyloxy-nonanoic acid with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydro chloride in analogy with example 1, step 3 led to (R)-4-dimethylamino-3-(9-phenethyloxy-nonanoylamino)-butyric acid benzyl ester.
- Light yellow oil, m/e 497.5 ([M+H] + ).
- Step 6 Hydrogenation of (R)-4-dimethylamino-3-(9-phenethyloxy-nonanoylamino)- butyric acid benzyl ester in analogy with example 1, step 4 produced (R)-4-dimethylamino-3-[9- (2-fluoro-phenoxy)-nonanoylamino]-butyric acid.
- White solid, m/e 405.6 ([M-H] " ).
- step 4 This was oxidized in step 4 to 8-(3-phenyl-propoxy)-octanoic acid, which was coupled in step 5 with with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[8-(3-phenyl-propoxy)-octanoylamino]- butyric acid benzyl ester, which was hydro genated in step 6.
- step 4 This was oxidized in step 4 to 8-(2-phenoxy-ethoxy)-octanoic acid, which was coupled in step 5 with with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-4-dimethylamino-3-[8-(2-phenoxy-ethoxy)-octanoylamino]- butyric acid benzyl ester, which was hydrogenated in step 6.
- Step 1 2-Phenylethanethiol was alkylated with 7-bromo-l-heptanol in analogy with example 18, step 1, affording 7-phenethylsulfanyl-heptan-l-ol.
- Step 2 Oxone®(3.3 g, 5.5 mmol) was added to a solution of 7-phenethylsulfanyl-heptan-l- ol (930 mg, 3.68 mmol) in methanol (40 mL), then after 16 h insoluble material was removed by filtration and the filtrate evaporated, affording 7-(2-phenyl-ethanesulfonyl)-heptan-l-ol (1.6 g), which was directly used in the next step.
- Step 3 Oxidation of 7-(2-phenyl-ethanesulfonyl)-heptan-l-ol in analogy with example 1, step 2 gave 7-(2-phenyl-ethanesulfonyl)-heptanoic acid.
- Step 4 Amide coupling of 7-(2-phenyl-ethanesulfonyl)-heptanoic acid with (R)-3-amino- 4-dimethylamino-butyric acid benzyl ester dihydro chloride in analogy with example 1, step 3 led to (R)-4-dimethylamino-3-[7-(2-phenyl-ethanesulfonyl)-heptanoylamino]-butyric acid benzyl ester.
- Step 5 Hydrogenation of (R)-4-dimethylamino-3-[7-(2-phenyl-ethanesulfonyl)- heptanoylamino]-butyric acid benzyl ester in analogy with example 1, step 4 produced (R)-4- dimethylamino-3-[7-(2-phenyl-ethanesulfonyl)-heptanoylamino]-butyric acid.
- White solid, m/e 427.3 ([M+H] + ).
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino- butyric acid benzyl ester dihydro chloride to produce (R)-3-(9-benzenesulfonyl-nonanoylamino)- 4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 4.
- step 4 This was oxidized in step 4 to 7-[2-(2,3-difluoro-phenyl)-ethoxy]-heptanoic acid, which was coupled in step 5 with with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-3- ⁇ 7-[2-(2,3-difluoro-phenyl)-ethoxy]-heptanoylamino ⁇ -4-dimethylamino-butyric acid benzyl ester, which was hydrogenated in step 6.
- Step 1 Ethyl chloroformate (414 mg, 3.82 mmol) was added at 0 0 C to a solution of 7-[2- (2,3-difluoro-phenyl)-ethoxy]-heptanoic acid (800 mg, 2.94 mmol) and triethylamine (387 mg, 3.82 mmol) in dichloromethane (15 mL), then after 2 h volatile material was removed by distillation, producing 7-[2-(2,3-difluoro-phenyl)-ethoxy]-heptanoyl chloride (855 mg).
- Step 2 A solution of [6-(2,3-difluoro-benzyloxy)-hexyl]-isocyanate (224 mg, 0.83 mmol) in dichloromethane (1 mL) was added at 0 0 C to a suspension of (R)-3-amino-4-dimethylamino butyric acid benzyl ester dihydro chloride (255 mg, 0.83 mmol) in dichloromethane (4 mL), then a solution of triethylamine (167 mg, 1.65 mmol) in dichloromethane (1 mL) was added dropwise over 25 min. The reaction mixture was kept at ⁇ 5 0 C for 1 h, then allowed to reach room temperature over 1 h.
- Step 2 9-(Methyl-phenethyl-amino)-nonanoic acid methyl ester was hydrolyzed in analogy with example 35, step 2, leading to 9-(methyl-phenethyl-amino)-nonanoic acid.
- Step 3 Amide coupling of 9-(methyl-phenethyl-amino)-nonanoic acid with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydrochloride in analogy with example 1, step 3, led to (R)-4-dimethylamino-3-[9-(methyl-phenethyl-amino)-nonanoylamino]-butyric acid benzyl ester.
- step 3 This was coupled in step 3 with (R)-3-amino-4-dimethylamino-butyric acid benzyl ester dihydrochloride, leading to (R)-3-[9-(benzyl-phenethyl-amino)-nonanoylamino]-4-dimethylamino-butyric acid benzyl ester, which was hydro genated in step 4.
- Step 1 Azelaic acid monomethyl ester was coupled with N-methylaniline in analogy with example 1, step 3, leading to 8-(methyl-phenyl-carbamoyl)-octanoic acid methyl ester.
- Step 2 Borane-tetrahydrofuran complex (1 M in tetrahydrofuran, 3.09 mL, 3.09 mmol) was added at 0 0 C to a solution of 8-(methyl-phenyl-carbamoyl)-octanoic acid methyl ester (300 mg, 1.03 mmol) in tetrahydrofuran (20 mL) The reaction mixture was stirred at room temperature overnight. A further aliquot of borane-tetrahydrofuran complex (2.1 mL, 2.1 mmol) was added and the reaction mixture was stirred at room temperature for 3.5 hours. Methanol (60 mL) was added followed by concentrated sulfuric acid.
- the reaction mixture was refluxed at 80 0 C for 2 hours. After cooling to room temperature, saturated sodium carbonate was added and the reaction mixture was extracted 3 times with dichloromethane . The combined organic layers were dried over sodium sulfphate, filtered and the solvent was removed in vacuo. The crude product was purified by chromatography (SiO 2 ; heptane-ethyl acetate gradient) to afford 9- (methyl-phenyl-amino)-nonanoic acid methyl ester (89 mg, 31%).
- Step 3 9-(Methyl-phenyl-amino)-nonanoic acid methyl ester was hydrolyzed in analogy with example 35, step 2, leading to 9-(methyl-phenyl-amino)-nonanoic acid.
- Step 4 Amide coupling of 9-(methyl-phenyl-amino)-nonanoic acid with (R)-3-amino-4- dimethylamino -butyric acid benzyl ester dihydro chloride in analogy with example 1, step 3 led to (R)-4-dimethylamino-3-[9-(methyl-phenyl-amino)-nonanoylamino]-butyric acid benzyl ester.
- Step 5 Hydro genation of (R)-4-dimethylamino-3-[9-(methyl-phenyl-amino)- nonanoylamino]-butyric acid benzyl ester in analogy with example 1, step 4 produced (R)-4- dimethylamino-3-[9-(methyl-phenethyl-amino)-nonanoylamino]-butyric acid.
- White solid, m/e 392.3 ([M+H] + ).
- Film coated tablets containing the following ingredients can be manufactured in a conventional manner:
- the active ingredient is sieved and mixed with micro crystalline cellulose and the mixture is granulated with a solution of polyvinylpyrrolidon in water.
- the granulate is mixed with sodium starch glycolate and magesiumstearate and compressed to yield kernels of 120 or 350 mg respectively.
- the kernels are lacquered with an aqueous solution / suspension of the above mentioned film coat.
- Capsules containing the following ingredients can be manufactured in a conventional manner:
- the components are sieved and mixed and filled into capsules of size 2.
- Injection solutions can have the following composition:
- the active ingredient is dissolved in a mixture of polyethylene glycol 400 and water for injection (part).
- the pH is adjusted to 5.0 with acetic acid.
- the volume is adjusted to 1.0 ml by addition of the residual amount of water.
- the solution is filtered, filled into vials using an appropriate overage and sterilized.
- Soft gelatin capsules containing the following ingredients can be manufactured in a conventional manner:
- Soya bean oil HO.O mg
- Example E The active ingredient is dissolved in a warm melting of the other ingredients and the mixture is filled into soft gelatin capsules of appropriate size.
- the filled soft gelatin capsules are treated according to the usual procedures.
- Sachets containing the following ingredients can be manufactured in a conventional manner:
- Microcrystalline cellulose (AVICEL PH 102) 1400.0 mg
- Flavoring additives 1.0 mg
- the active ingredient is mixed with lactose, microcrystalline cellulose and sodium carboxymethyl cellulose and granulated with a mixture of polyvinylpyrrolidone in water.
- the granulate is mixed with magnesiumstearate and the flavouring additives and filled into sachets.
Abstract
Description























Claims








Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020107024228A KR101225223B1 (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derivatives |
| JP2011505469A JP5474047B2 (en) | 2008-04-29 | 2009-04-20 | 4-Dimethylaminobutyric acid derivative |
| AU2009242240A AU2009242240B2 (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derivatives |
| EP09738022.4A EP2268606B1 (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derivatives |
| CN200980105617.7A CN101952243B (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derivatives |
| ES09738022.4T ES2507089T3 (en) | 2008-04-29 | 2009-04-20 | Derivatives of 4-dimethylaminobutyric acid |
| RU2010148532/04A RU2474573C2 (en) | 2008-04-29 | 2009-04-20 | 4-dimethyl aminobutyric acid derivatives |
| CA2722087A CA2722087C (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derivatives |
| MX2010008196A MX2010008196A (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derivatives. |
| BRPI0911159A BRPI0911159A2 (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derived compounds, process for their preparation, pharmaceutical compositions comprising them, method for the therapeutic or prophylactic treatment of diseases that are modulated by cpt2 inhibitors and use of these compounds |
| IL206524A IL206524A0 (en) | 2008-04-29 | 2010-06-21 | 4-dimethylminobutyric acid derivatives |
| ZA2010/05321A ZA201005321B (en) | 2008-04-29 | 2010-07-26 | 4-dimethylaminobutyric acid derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08155323.2 | 2008-04-29 | ||
| EP08155323 | 2008-04-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009132978A1 true WO2009132978A1 (en) | 2009-11-05 |
Family
ID=40897585
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/054635 WO2009132978A1 (en) | 2008-04-29 | 2009-04-20 | 4-dimethylaminobutyric acid derivatives |
Country Status (18)
| Country | Link |
|---|---|
| US (2) | US8344027B2 (en) |
| EP (1) | EP2268606B1 (en) |
| JP (1) | JP5474047B2 (en) |
| KR (1) | KR101225223B1 (en) |
| CN (1) | CN101952243B (en) |
| AR (1) | AR073842A1 (en) |
| AU (1) | AU2009242240B2 (en) |
| BR (1) | BRPI0911159A2 (en) |
| CA (1) | CA2722087C (en) |
| CL (1) | CL2009001006A1 (en) |
| ES (1) | ES2507089T3 (en) |
| IL (1) | IL206524A0 (en) |
| MX (1) | MX2010008196A (en) |
| PE (1) | PE20091735A1 (en) |
| RU (1) | RU2474573C2 (en) |
| TW (1) | TWI369344B (en) |
| WO (1) | WO2009132978A1 (en) |
| ZA (1) | ZA201005321B (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8344027B2 (en) | 2008-04-29 | 2013-01-01 | Hoffmann-La Roche Inc. | 4-dimethylaminobutyric acid derivatives |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5373888B2 (en) * | 2008-04-29 | 2013-12-18 | エフ.ホフマン−ラ ロシュ アーゲー | 4-Trimethylammonio-butyrates as CPT2 inhibitors |
| FR2951448B1 (en) * | 2009-10-15 | 2012-09-07 | Centre Nat Rech Scient | POLYURETHANE SYNTHESIS BY AUTOCONDENSATION |
| CN105859658A (en) * | 2016-05-12 | 2016-08-17 | 青岛大学 | (R)-2-hydroxy-N-((2S,3S)-2-((R,E)-1-hydroxy pentadecane-4-alkene-1-yl)oxygen heterocycle-3-yl)lignoceric acid amino and preparation method thereof |
| CN115304569A (en) * | 2022-09-14 | 2022-11-08 | 浙江世佳科技股份有限公司 | Method for protecting alcohol-terminated hydroxyl group by using 4-pyridinium methyl benzenesulfonate as catalyst |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4632925A (en) | 1985-10-07 | 1986-12-30 | Hoffmann-La Roche Inc. | N-substituted diphenylpiperidines and antiobesity use thereof |
| WO1999059957A1 (en) | 1998-05-15 | 1999-11-25 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Compounds having reversible inhibiting activity of carnitine palmitoyl-transferase |
| WO2006092204A1 (en) | 2005-03-02 | 2006-09-08 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Derivatives of aminobutanoic acid inhibiting cpt |
| WO2008015081A1 (en) | 2006-08-02 | 2008-02-07 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Derivatives of 4-trimethylammonium-3-aminobutyrate and 4-trimethylphosphonium-3-aminobutyrate as cpt-inhibitors |
| WO2008109991A1 (en) * | 2007-03-09 | 2008-09-18 | University Health Network | Inhibitors of carnitine palmitoyltransferase and treating cancer |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0127098B1 (en) | 1983-05-25 | 1989-02-08 | Takeda Chemical Industries, Ltd. | Derivatives of beta-amino-gamma-trimethylammonio-butyrate, their production and use |
| ITMI20010395A1 (en) * | 2001-02-27 | 2002-08-27 | Dompe Spa | OMEGA-AMINO ALKYLAMIDS OF R-2-ARYL-PROPIONIC ACIDS AS INHIBITORS OF CHEMOTAXIS OF POLYMORPHONUCLEATED AND MONONUCLEATE CELLS |
| ITMI20040230A1 (en) * | 2004-02-12 | 2004-05-12 | Defiante Farmaceutica Lda | ANTI-CANCER ACTIVITY COMPOUNDS |
| RU2309144C2 (en) * | 2005-03-25 | 2007-10-27 | Общество С Ограниченной Ответственностью "Фарминтерпрайсез" | Phenyl-containing n-acylamine derivatives, method for production thereof, pharmaceutical composition and uses thereof as anti-inflammatory and analgesic agents |
| ES2507089T3 (en) | 2008-04-29 | 2014-10-14 | F. Hoffmann-La Roche Ag | Derivatives of 4-dimethylaminobutyric acid |
-
2009
- 2009-04-20 ES ES09738022.4T patent/ES2507089T3/en active Active
- 2009-04-20 RU RU2010148532/04A patent/RU2474573C2/en not_active IP Right Cessation
- 2009-04-20 AU AU2009242240A patent/AU2009242240B2/en not_active Ceased
- 2009-04-20 KR KR1020107024228A patent/KR101225223B1/en not_active IP Right Cessation
- 2009-04-20 EP EP09738022.4A patent/EP2268606B1/en not_active Not-in-force
- 2009-04-20 MX MX2010008196A patent/MX2010008196A/en active IP Right Grant
- 2009-04-20 JP JP2011505469A patent/JP5474047B2/en not_active Expired - Fee Related
- 2009-04-20 CN CN200980105617.7A patent/CN101952243B/en not_active Expired - Fee Related
- 2009-04-20 CA CA2722087A patent/CA2722087C/en not_active Expired - Fee Related
- 2009-04-20 WO PCT/EP2009/054635 patent/WO2009132978A1/en active Application Filing
- 2009-04-20 BR BRPI0911159A patent/BRPI0911159A2/en active Search and Examination
- 2009-04-27 PE PE2009000570A patent/PE20091735A1/en not_active Application Discontinuation
- 2009-04-27 AR ARP090101488A patent/AR073842A1/en not_active Application Discontinuation
- 2009-04-27 TW TW098113900A patent/TWI369344B/en not_active IP Right Cessation
- 2009-04-28 CL CL2009001006A patent/CL2009001006A1/en unknown
- 2009-04-28 US US12/430,934 patent/US8344027B2/en not_active Expired - Fee Related
-
2010
- 2010-06-21 IL IL206524A patent/IL206524A0/en unknown
- 2010-07-26 ZA ZA2010/05321A patent/ZA201005321B/en unknown
-
2012
- 2012-07-24 US US13/556,250 patent/US20120289554A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4632925A (en) | 1985-10-07 | 1986-12-30 | Hoffmann-La Roche Inc. | N-substituted diphenylpiperidines and antiobesity use thereof |
| WO1999059957A1 (en) | 1998-05-15 | 1999-11-25 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Compounds having reversible inhibiting activity of carnitine palmitoyl-transferase |
| WO2006092204A1 (en) | 2005-03-02 | 2006-09-08 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Derivatives of aminobutanoic acid inhibiting cpt |
| WO2008015081A1 (en) | 2006-08-02 | 2008-02-07 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Derivatives of 4-trimethylammonium-3-aminobutyrate and 4-trimethylphosphonium-3-aminobutyrate as cpt-inhibitors |
| WO2008109991A1 (en) * | 2007-03-09 | 2008-09-18 | University Health Network | Inhibitors of carnitine palmitoyltransferase and treating cancer |
Non-Patent Citations (7)
| Title |
|---|
| DATABASE CA [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; GIANNESSI, FABIO ET AL: "Discovery of a Long-Chain Carbamoyl Aminocarnitine Derivative, a Reversible Carnitine Palmitoyltransferase Inhibitor with Antiketotic and Antidiabetic Activity", XP002539907, retrieved from STN Database accession no. 2002:954524 * |
| GIANNESSI ET AL., J. MED. CHEM, vol. 46, no. 2, 2003, pages 303 - 309 |
| J. ORG. CHEM., vol. 72, 2007, pages 9471 |
| JACKSON ET AL., BIOCHEM. J., vol. 341, 1999, pages 483 - 489 |
| JACKSON ET AL., J. BIOL. CHEM., vol. 275, 2000, pages 19560 - 19566 |
| JOURNAL OF MEDICINAL CHEMISTRY , 46(2), 303-309 CODEN: JMCMAR; ISSN: 0022-2623, 2003 * |
| TETRAHEDRON, vol. 59, 2003, pages 7973 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8344027B2 (en) | 2008-04-29 | 2013-01-01 | Hoffmann-La Roche Inc. | 4-dimethylaminobutyric acid derivatives |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101952243A (en) | 2011-01-19 |
| MX2010008196A (en) | 2010-08-11 |
| AR073842A1 (en) | 2010-12-09 |
| AU2009242240B2 (en) | 2011-09-22 |
| RU2474573C2 (en) | 2013-02-10 |
| CA2722087A1 (en) | 2009-11-05 |
| AU2009242240A1 (en) | 2009-11-05 |
| US20120289554A1 (en) | 2012-11-15 |
| US20090270505A1 (en) | 2009-10-29 |
| EP2268606B1 (en) | 2014-08-06 |
| JP5474047B2 (en) | 2014-04-16 |
| KR20100127306A (en) | 2010-12-03 |
| TW200946106A (en) | 2009-11-16 |
| EP2268606A1 (en) | 2011-01-05 |
| CN101952243B (en) | 2014-07-16 |
| CA2722087C (en) | 2013-07-09 |
| US8344027B2 (en) | 2013-01-01 |
| ES2507089T3 (en) | 2014-10-14 |
| ZA201005321B (en) | 2011-04-28 |
| RU2010148532A (en) | 2012-06-10 |
| KR101225223B1 (en) | 2013-01-22 |
| PE20091735A1 (en) | 2009-11-13 |
| IL206524A0 (en) | 2010-12-30 |
| CL2009001006A1 (en) | 2010-06-04 |
| JP2011518205A (en) | 2011-06-23 |
| BRPI0911159A2 (en) | 2015-10-06 |
| TWI369344B (en) | 2012-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20120289554A1 (en) | 4-dimethylaminobutyric acid derivatives | |
| EP1881958B1 (en) | Diacylglycerol acyltransferase inhibitors | |
| AU734967B2 (en) | Certain cyclic thio substituted acylaminoacid amide derivatives | |
| EP2280935B1 (en) | 4-trimethylammonio-butyrates as cpt2 inhibitors | |
| US7375120B2 (en) | Peptide deformylase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980105617.7 Country of ref document: CN |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09738022 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009738022 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009242240 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2009242240 Country of ref document: AU Date of ref document: 20090420 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/008196 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5749/DELNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12010502313 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2722087 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011505469 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 20107024228 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010148532 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0911159 Country of ref document: BR Kind code of ref document: A2 Effective date: 20101019 |