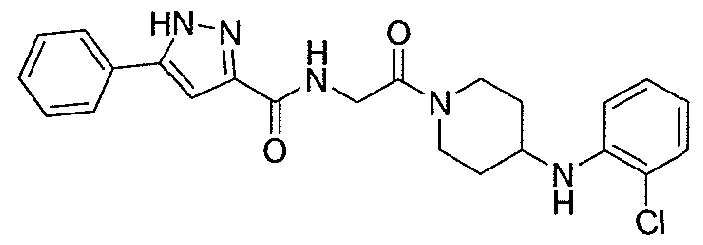
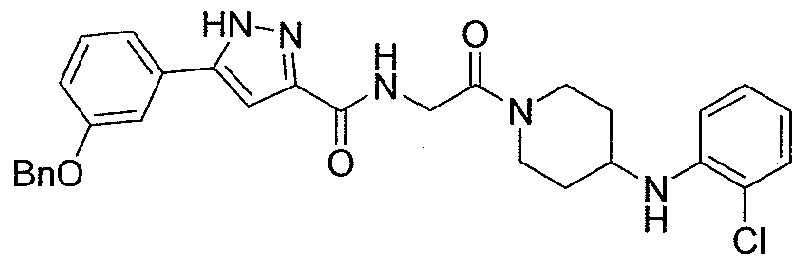
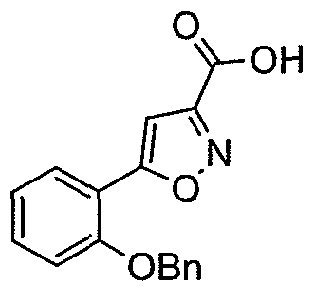
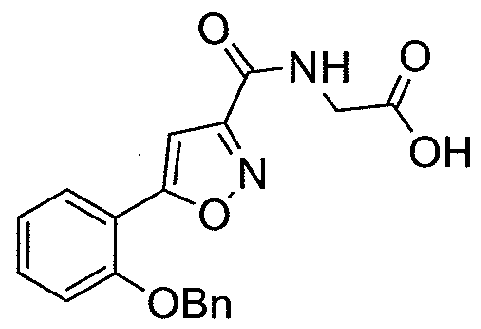
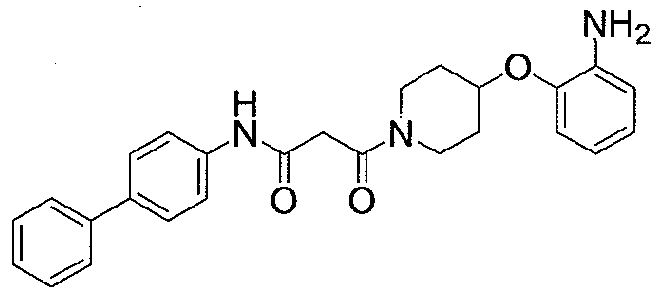
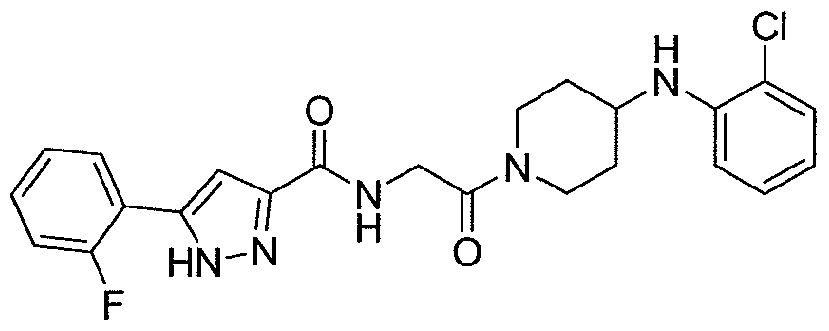
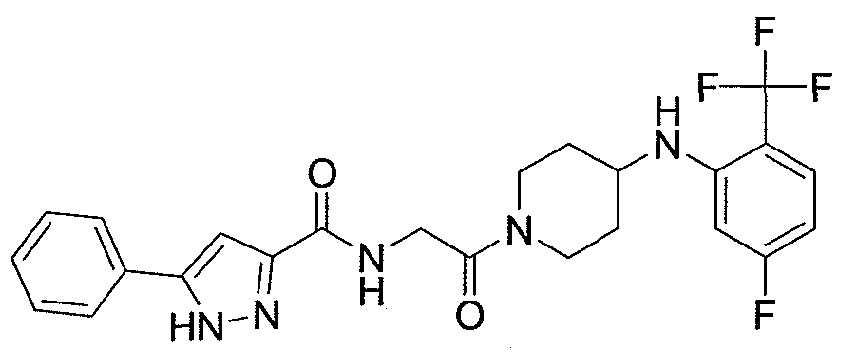
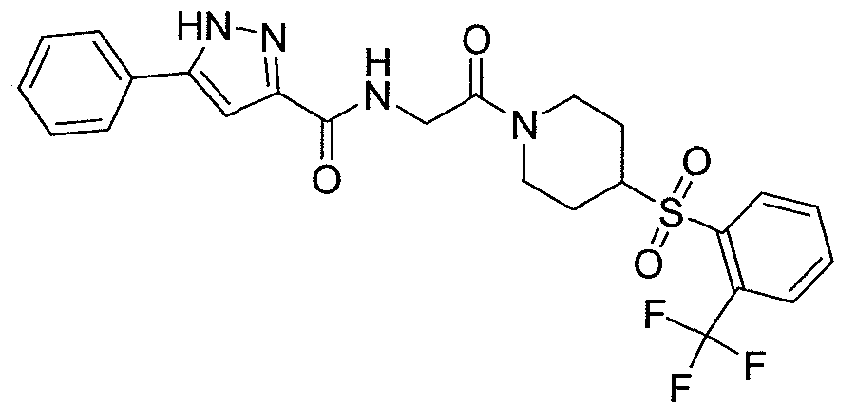
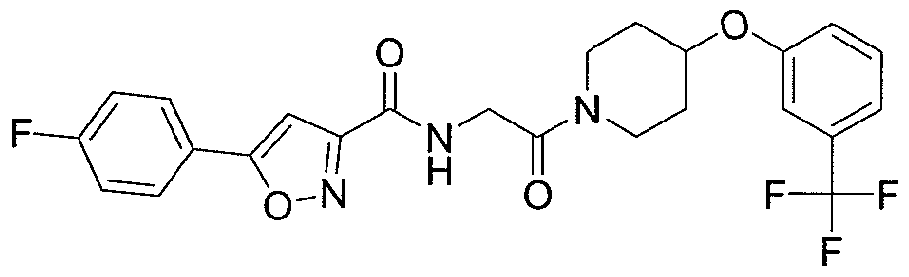
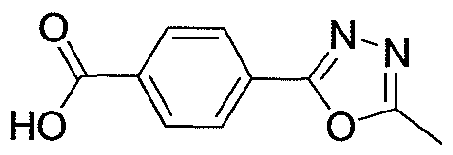
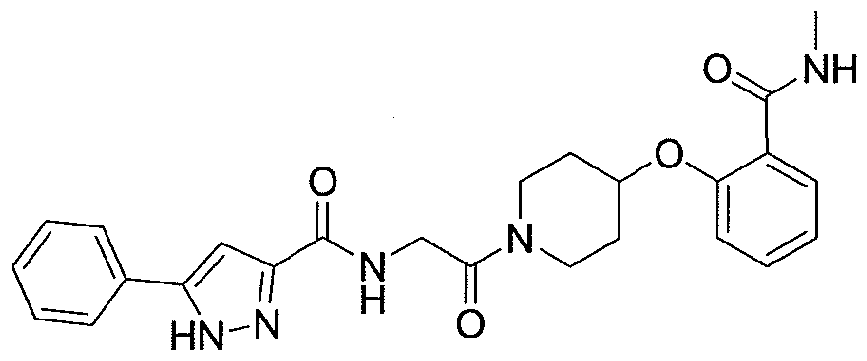
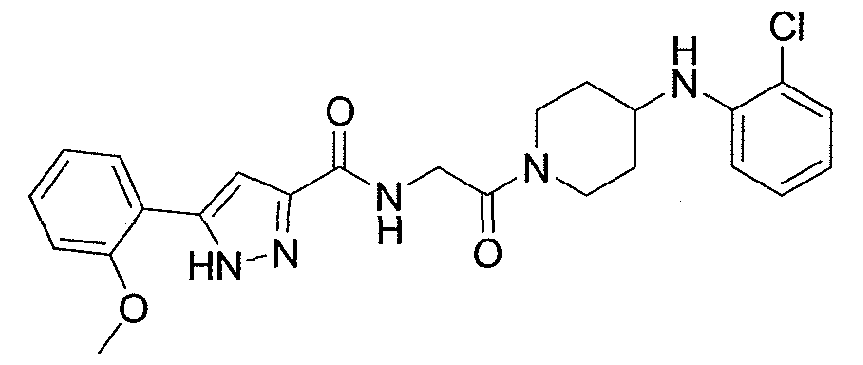
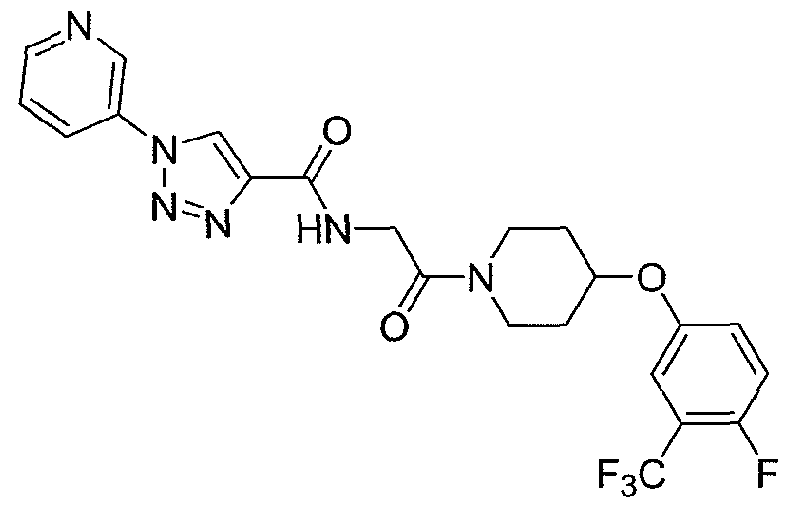
NOVEL PIPERIDINE DERIVATIVES AS INHIBITORS OF STEAROYL-COA DESATURASE
CROSS-REFERENCE TO RELATED APPLICATIONS This application claims priority to U.S. Provisional Patent Application 61/049,480, which was filed on May 1, 2008; and to Indian Patent Application 575/KOL/2008, which was filed on March 20, 2008.
FIELD OF THE INVENTION The present invention relates to piperidine derivatives that act as inhibitors of stearoyl-
CoA desaturase. The invention also relates to methods of preparing the compounds, compositions containing the compounds, and to methods of treatment using the compounds.
BACKGROUND OF THE INVENTION Metabolic syndrome has become one of the leading health problems in the world. As a component of metabolic syndrome, obesity also has causal roles in other components of the syndrome, including insulin resistance, dyslipidemia, and cardiovascular diseases. Effective treatments for metabolic syndrome in general and obesity in particular have been lacking. Effective therapies for the treatment of obesity, a key element of metabolic syndrome, are urgently needed. A number of mammalian stearoyl-coenzyme A desaturase (SCD) genes have been cloned.
For example, two genes have been cloned from rat (SCDl, SCD2) and four SCD genes have been isolated from mouse (SCDl, 2, 3, and 4). While the basic biochemical role of SCD has been known in rats and mice since the 1970's (see, e.g., Jeffcoat, R. et al, Elsevier Science , Vol. 4, pp. 85-112, 1984; de Antueno, R J, Lipids, Vol. 28, No. 4, pp. 285-290, 1993), it has only recently been directly implicated in human disease processes.
A single SCD gene, stearoyl-coenzyme A desaturase- 1 (SCDl) has been characterized in humans. SCDl is described in, e.g., International Publication No. application, WO 01/62954. A second human SCD isoform has recently been identified, and because it bears little sequence homology to alternate mouse or rat isoforms it has been named human SCD5 or hSCD5 (see, e.g., International Publication No. WO 02/26944).
SCD-I catalyzes conversion of saturated fatty acids, stearoyl-CoA and palmitoyl-CoA, to monounsaturated fatty acids, oleoyl-CoA and pamitoleoyl-CoA, respectively. These fatty acids are
components of membrane phospholipids, triglycerides, and cholesterol esters. Changes in SCD activity ultimately change membrane fluidity, lipoprotein metabolism, and adiposity. SCD-I inhibition can lead to decreased adiposity and thus be a potential therapy for metabolic syndrome. Since obesity is becoming increasingly prevalent worldwide, much effort is being devoted to understanding its pathogenesis and treatment. In recent years, several candidate genes have been proposed as therapeutic targets. However, stearoyl-CoA desaturase 1 is of special significance, because it is the major gene target of leptin — a central mediator of energy homeostasis. There is evidence that SCDl deficiency activates metabolic pathways that promote b-oxidation and decrease lipogenesis in liver and skeletal muscles. One mechanism is via increased activation of AMP-activated protein kinase. SCDl mutation results also in global changes in expression of genes involved in lipid metabolism. SCDl deficient mice have increased energy expenditure, reduced body adiposity, and are resistant to diet-induced obesity.
Thus, SCDl inhibition represents a new and important target for the treatment of various disorders such as obesity and related metabolic disorders. Accordingly, there is a need in the art for derivatives that act as inhibitors of stearoyl-CoA desaturase, such as SCDl .
SUMMARY OF THE INVENTION
The present invention relates to piperidine derivatives that act as inhibitors of stearoyl- CoA desaturase. The invention also relates to methods of preparing the compounds, compositions containing the compounds, and to methods of treatment using the compounds.
DETAILED DESCRIPTION OF THE INVENTION
In some embodiments, the present invention provides compounds of the formula:
R is aryl or heteroaryl;
R2 is aryl or heteroaryl ;
R3 and R4 are each independently hydrogen, halogen or alkyl; or
R and R , together with the carbon atom to which they are attached, form a cycloalkyl group;
R5 is hydrogen or alkyl; m and n are, independently, 1 or 2; X is -O-, -NR6-, -S-, -S(O) - or -S(O) 2- where R6 is hydrogen or alkyl; wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfinyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfinyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof; and pharmaceutically acceptable salts, solvates, hydrates, solvates of pharmaceutically acceptable salts thereof, or enantiomer or diasteromer thereof; with the proviso that said compound is not
4-[[(2R)-2,3-dihydro-2-methyl-6-nitroimidazo[2,l-b]oxazol-2-yl]methoxy]-N-[2-oxo-2-[4- [4-(trifluoromethoxy)phenoxy] - 1 -piperidinyl] ethyl] -benzamide,
N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2-oxoethyl]- M-methyl-benzamide,
4-amino-N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2- oxoethyl] -benzamide, or a pharmaceutically acceptable salt thereof.
In some embodiments, the present invention provides compounds of the formula:
R is heteroaryl;
R is aryl or heteroaryl ;
R and R are each independently hydrogen, halogen or alkyl; or
R3 and R4, together with the carbon atom to which they are attached, form a cycloalkyl group;
R5 is hydrogen or alkyl; m and n are, independently, 1 or 2;
X is -O-, -NR6-, -S-, -S(O) - or -S(O) 2- where R6 is hydrogen or alkyl; wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfmyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulflnyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof; and pharmaceutically acceptable salts, solvates, hydrates, solvates of pharmaceutically acceptable salts thereof, or enantiomer or diasteromer thereof.
In some embodiments, the present invention provides compounds of the formula:
R1 is aryl or heteroaryl;
R2 is aryl or heteroaryl ;
R3 and R4 are each independently hydrogen, halogen or alkyl; or
R3 and R4, together with the carbon atom to which they are attached, form a cycloalkyl group;
R5 is hydrogen or alkyl;
X is -O-, -NR6-, -S-, -S(O) - or -S(O) 2- where R6 is hydrogen or alkyl;
wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfinyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfϊnyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof; and pharmaceutically acceptable salts or solvates, hydrates, or solvates of pharmaceutically acceptable salts thereof; with the proviso that said compound is not
4-[[(2R)-2,3-dihydro-2-methyl-6-nitroimidazo[2,l-b]oxazol-2-yl]methoxy]-N-[2-oxo-2-[4- [4-(trifluoromethoxy)phenoxy]-l-piperidinyl]ethyl]-benzamide,
N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2-oxoethyl]- M-methyl-benzamide,
4-amino-N- [2- [4- [[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl] amino] - 1 -piperidinyl] -2- oxoethyl] -benzamide, or a pharmaceutically acceptable salt thereof.
In some embodiments, the present invention provides compounds of the formula:
R7 is aryl or heteroaryl; R is is aryl or heteroaryl; R9 and R10 are each independently hydrogen, halogen or alkyl; or
R9 and R10, together with the carbon atom to which they are attached, form a cycloalkyl group;
R11 is hydrogen or alkyl;
X is -O-, -NR12-, -S-, -S(O) - or -S(O) 2- where R12 is hydrogen or alkyl;
wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfinyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfinyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof.
In some embodiments, the present invention includes compounds of the formula:
R1 is aryl or heteroaryl;
R2 is aryl or heteroaryl ;
R3 and R4 are each independently hydrogen, halogen or alkyl; or R3 and R4, together with the carbon atom to which they are attached, form a cycloalkyl group;
R5 is hydrogen or alkyl; m and n are, independently, 1 or 2 (in some embodiments, m is 1 or 2; and n is 1 or 2; wherein the sum of m and n is between 2 and 4, such as, for example, wherein the difference between m and n is 0 or 1);
X is -O-, -NR6-, -S-, -S(O) - or -S(O) 2- where R6 is hydrogen or alkyl; wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfinyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfinyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof;
and pharmaceutically acceptable salts, solvates, hydrates, solvates of pharmaceutically acceptable salts thereof, or enantiomer or diasteromer thereof; such as, with the proviso that said compound is not
4-[[(2R)-2,3-dihydro-2-methyl-6-nitroimidazo[2,l-b]oxazol-2-yl]methoxy]-N-[2-oxo-2-[4- [4-(trifluoromethoxy)phenoxy]-l-piperidinyl]ethyl]-benzamide,
N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2-oxoethyl]- M-methyl-benzamide,
4-ammo-N-[2-[4-[[4-ammo-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2- oxoethyl] -benzamide,
In some embodiments, the present invention includes compounds of the formula:
R1 is aryl or heteroaryl; R2 is aryl or heteroaryl ;
R3 and R4 are each independently hydrogen, halogen or alkyl; or
R3 and R4, together with the carbon atom to which they are attached, form a cycloalkyl group;
R5 is hydrogen or alkyl; X is -O-, -NR6-, -S-, -S(O) - or -S(O) 2- where R6 is hydrogen or alkyl; wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfmyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfinyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof;
and pharmaceutically acceptable salts, solvates, hydrates, solvates of pharmaceutically acceptable salts thereof, or enantiomer or diasteromer thereof; such as, with the proviso that said compound is not
4-[[(2R)-2,3-dihydro-2-methyl-6-nitroimidazo[2,l-b]oxazol-2-yl]methoxy]-N-[2-oxo-2-[4- [4-(trifluoromethoxy)phenoxy]-l-piperidinyl]ethyl]-benzamide,
N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2-oxoethyl]- M-methyl-benzamide,
4-amino-N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2- oxoethyl] -benzamide, or a pharmaceutically acceptable salt thereof.
In some embodiments, the present invention includes compounds of the formula:
wherein R
1 is aryl or heteroaryl;
R2 is aryl or heteroaryl ;
R3 and R4 are each independently hydrogen, halogen or alkyl; or
R3 and R4, together with the carbon atom to which they are attached, form a cycloalkyl group; R5 is hydrogen or alkyl;
X is -O-, -NR6-, -S-, -S(O) - or -S(O) 2- where R6 is hydrogen or alkyl; wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfinyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfinyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof;
and pharmaceutically acceptable salts, solvates, hydrates, solvates of pharmaceutically acceptable salts thereof, or enantiomer or diasteromer thereof; such as, with the proviso that said compound is not
4-[[(2R)-2,3-dihydro-2-methyl-6-nitroimidazo[2,l-b]oxazol-2-yl]methoxy]-N-[2-oxo-2-[4- [4-(trifluoromethoxy)phenoxy]-l-piperidinyl]ethyl]-benzamide,
N-[2-[4-[[4-amino-5-(2,ό-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2-oxoethyl]- M-methyl-benzamide,
4-amino-N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2- oxoethyl]-benzamide, or a pharmaceutically acceptable salt thereof.
In some embodiments, the present invention includes compounds of the formula:
wherein R
1 is aryl or heteroaryl;
R2 is aryl or heteroaryl ;
R3 and R4 are each independently hydrogen, halogen or alkyl; or
R3 and R4, together with the carbon atom to which they are attached, form a cycloalkyl group; R5 is hydrogen or alkyl;
X is -O-, -NR6-, -S-, -S(O) - or -S(O) 2- where R6 is hydrogen or alkyl; wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfϊnyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfinyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof;
and pharmaceutically acceptable salts, solvates, hydrates, solvates of pharmaceutically acceptable salts thereof, or enantiomer or diasteromer thereof; such as, with the proviso that said compound is not
4-[[(2R)-2,3-dihydro-2-methyl-6-nitroimidazo[2,l-b]oxazol-2-yl]methoxy]-N-[2-oxo-2-[4- [4-(trifluoromethoxy)phenoxy] - 1 -piperidinyl] ethyl] -benzamide,
N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2-oxoethyl]- M-methyl-benzamide,
4-amino-N-[2-[4-[[4-amino-5-(2,6-difluorobenzoyl)-2-thiazolyl]amino]-l-piperidinyl]-2- oxoethyl] -benzamide, or a pharmaceutically acceptable salt thereof.
In another aspect, the present invention includes compounds of the formula:
R is aryl or heteroaryl; R8 is is aryl or heteroaryl;
R9 and R10 are each independently hydrogen, halogen or alkyl; or
R9 and R10, together with the carbon atom to which they are attached, form a cycloalkyl group;
R11 is hydrogen or alkyl; X is -O-, -NR12-, -S-, -S(O) - or -S(O) 2- where R12 is hydrogen or alkyl;
In some embodiments, R1 is aryl (e.g., phenyl) or heteroaryl (e.g., pyridinyl, oxazolyl, imidazolyl), R2 is aryl (e.g., phenyl), R3, R4 and R5 are each hydrogen, and X is -O-, -S-, or -NR6- where R6 is hydrogen or alkyl (e.g., methyl). In some embodiments, R1 is optionally substituted aryl (e.g., phenyl) or heteroaryl (e.g., pyridinyl, isoxazolyl, pyrazolyl). For example, R1 is aryl (e.g., phenyl) or heteroaryl (e.g., pyridinyl, isoxazolyl, pyrazolyl) optionally substituted by one or more aryl (e.g., phenyl, substituted phenyl (e.g., -hydroxyphenyl)), or arylamino (i.e., -NH-aryl, e.g., -NHC6Hs). For example, R1 may be biphenyl (e.g., 4-biphenyl), (phenyl)isoxazolyl (e.g., 5-phenyl-isoxazol-3-yl),
(phenylamino)phenyl (e.g., 4-phenylaminophenyl), (phenyl)pyrazolyl (e.g., 5 -phenyl- lH-pyrazol- 3-yl), (hydroxyphenyl)pyrazolyl (e.g., 5-(3-hydroxyphenyl)-lH-pyrazol-3-yl, 5-(4- hydroxyphenyl)-lH-pyrazol-3-yl), (phenyl)pyridinyl (e.g., 5-phenyl-pyridin-2-yl), (hydroxyphenyl)methylpyrazolyl (e.g., 5-(2-hydroxyphenyl)-l-methyl-lH-pyrazol-3-yl), (hydroxyphenyl)isoxazolyl (e.g., 5-(2-hydroxyphenyl)-isoxazol-3-yl, 5-(4-hydroxyphenyl)- isoxazol-3-yl), (phenylamino)pyridinyl (e.g., 6-phenylamino-pyridin-3-yl, 5 -phenyl amino-pyridin- 2-yl). In some embodiments, Rl is aryl that is substituted by one or more aryl groups. In some embodiments, Rl is heteroaryl and is substituted by one or more aryl or heteroaryl groups. In some embodiments, Rl is pyrazole, triazole, or isoxazole. In some embodiments, R2 is optionally substituted aryl (e.g., phenyl). For example, R2 is aryl (e.g., phenyl) optionally substituted by one or more halogen (e.g., F, Cl, Br), alkyl (e.g., methyl, t-butyl) nitro, amino or halogenated alkyl (e.g., CF3). For example, R2 is trifluoromethylphenyl (e.g., 2-trifluorornethylphenyl), bromophenyl (e.g., 2-bromophenyl), chlorophenyl (e.g., 2-chlorophenyl), trifluoromethylphenyl (e.g., 2-trifluoromethylphenyl), (chloro)flourophenyl (e.g., 2-chloro-5-fluorophenyl), nitrophenyl (e.g., 2-nitrophenyl), aminophenyl (e.g., 2-aminophenyl), methylphenyl (e.g., 2-methylphenyl), dimethylphenyl (e.g., 2,3-dimethylphenyl, 2,4-dimethylphenyl, 2,5-dimethylphenyl), t-butylphenyl (e.g., 2-t- butylphenyl) or difluorophenyl (e.g., 2,5-difluorophenyl).
In some embodiments, X is -O-, -S-, or -NR6- where R6 is hydrogen or alkyl (e.g., methyl). For example, X is -O-, -S-, -NH- or -N(CH3)-
In some embodiments, R3 and R4 are each independently hydrogen, halogen (e.g., F, Cl, Br) or alkyl (e.g., methyl). In other embodiments, R3 and R4 are hydrogen or alkyl (e.g., methyl).
In some embodiments, R3 and R4 are hydrogen or halogen (e.g., F). In one embodiment, R3 and R4 are hydrogen. In further embodiments, R3 and R4, together with the carbon atom to which they are attached, form a cycloalkyl group, such as a C3-C6 cycloalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl), e.g., R3 and R4, together with the carbon atom to which they are attached, form a cyclopropyl ring.
In some embodiments, R5 is hydrogen or methyl. For example R5 is hydrogen.
In some embodiments, R7 is aryl (e.g., phenyl, biphenyl) or heteroaryl (e.g., pyridinyl, thiazolyl, thiadiazolyl), R8 is aryl (e.g., phenyl), X is -O-, -S-, or -NH-, R11 is hydrogen, and R9 and R10 are hydrogen or together with the carbon atom to which they are attached R9 and R10 form a C3-C6 ring (e.g., cyclopropyl).
In some embodiments, R7 is optionally substituted aryl (e.g., phenyl) or heteroaryl (e.g., pyridinyl, thiazolyl, thiadiazolyl). For example, R7 is aryl (e.g., phenyl) or heteroaryl (e.g., pyridinyl, isoxazolyl, pyrazolyl) optionally substituted by one or more aryl (e.g., phenyl), or heteroaryl (e.g., oxadiazolyl). For example, R7 may be biphenyl (e.g., 4-biphenyl), (phenyl)pyridinyl (e.g., 6-phenyl- pyridin-3-yl, 5-phenyl-pyridin-2-yl), (phenyl)thiadiazolyl (e.g., 3-phenyl-[l,2,4]thiadiazol-5-yl), (oxadiazolyl)phenyl (e.g., 4-[l,2,4]oxadiazol-3-yl-phenyl) or (phenyl)thiazolyl (e.g., 5-phenyl- thiazol-2-yl).
In some embodiments, R is optionally substituted aryl (e.g., phenyl). For example, R is aryl (e.g., phenyl) optionally substituted by one or more halogen (e.g., F, Cl, Br), alkyl (e.g., methyl, t-butyl) nitro, amino or halogenated alkyl (e.g., CF3). For example, R8 is trifluoromethylphenyl (e.g., 2-trifluoromethylphenyl), bromophenyl (e.g., 2-bromophenyl), chlorophenyl (e.g., 2-chlorophenyl), (chloro)flourophenyl (e.g., 2-chloro-5-fluorophenyl), nitrophenyl (e.g., 2-nitrophenyl), aminophenyl (e.g., 2-aminophenyl), methylphenyl (e.g., 2- methylphenyl), dimethylphenyl (e.g., 2,3-dimethylphenyl, 2,4-dimethylphenyl, 2,5- dimethylphenyl), t-butylphenyl (e.g., 2-t-butylphenyl) or difluorophenyl (e.g., 2,5-difluorophenyl).
In certain embodiments, X is -O-, -S-, or -NR12- where R12 is hydrogen or alkyl (e.g., methyl). For example, X is -O-, -S- or -NH-.
In certain embodiments, R9 and R10 are each independently hydrogen, halogen (e.g., F, Cl, Br) or alkyl (e.g., methyl). In other embodiments, R9 and R10 are hydrogen or alkyl (e.g., methyl). In other embodiments, R9 and R10 are hydrogen or halogen (e.g., F). In one embodiment, R9 and R10 are hydrogen. In further embodiments, R9 and R10, together with the carbon atom to which they are attached, form a cycloalkyl group, such as a C3-C6 cyclalkyl (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl), e.g., R9 and R10, together with the carbon atom to which they are attached, form a cyclopropyl ring.
In some embodiments, R11 is hydrogen or methyl. For example R11 is hydrogen.
In some embodiments, R12 is hydrogen or methyl. For example R12 is hydrogen.
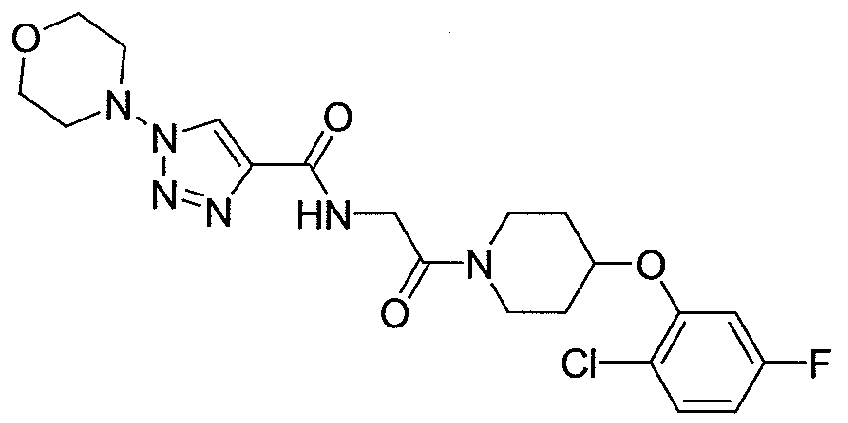
In certain embodiments, the compound is selected from: i) l-Cyclopentyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(5-chloro-pyridin-3- yloxy)-piperidin- 1 -yl]-2-oxo-ethyl} -amide;
ii) l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(5-cyano-2-methyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide; iii) l-Morpholin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide; iv) l-Morpholin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-5-fluoro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide; v) l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(2,5-difluoro-phenoxy)- pyrrolidin-l-yl]-2-oxo-ethyl}-amide; vi) 1 -Phenyl- lH-imidazole-4-carboxylic acid {2-[4-(2,5-difiuoro-phenoxy)-piperidin- l-yl]-2-oxo-ethyl}-amide; and vii) 1 -Phenyl- lH-imidazole-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-ρiperidin-l- yl]-2-oxo-ethyl}-amide, and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
In certain embodiments, the compound is selected from:
1. Biphenyl-4-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-phenoxy)-piperidin-l-yl]- ethyl} -amide
2. Biphenyl-4-carboxylic acid {2-[4-(2-bromo-phenoxy)-piperidin-l-yl]-2-oxo-ethyl} -amide 3. Biphenyl-4-carboxylic acid {2-[4-(2-bromo-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}- amide
4. Biphenyl-4-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}- amide
5. Biphenyl-4-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-phenylamino)-piperidin-l-yl]- ethyl}-amide
6. Biphenyl-4-carboxylic acid (2- {4-[methyl-(2-trifluoromethyl-phenyl)-amino]-piperidin- 1 - yl } -2-oxo-ethyl)-amide
7. Biphenyl-4-carboxylic acid (2-{4-[(2-chloro-phenyl)-methyl-amino]-piperidin-l-yl}-2- oxo-ethyl)-amide 8. Biphenyl-4-carboxylic acid (2-{4-[(2-bromo-phenyl)-methyl-amino]-piperidin-l-yl}-2- oxo-ethyl)-amide 9. S-Phenyl-isoxazole-S-carboxylic acid {2-[4-(2-bromo-phenoxy)-piperidin-l-yl]-2-oxo-
ethyl} -amide
10. S-Phenyl-isoxazole-S-carboxylic acid {2-[4-(2-bromo-phenylamino)-piperidin-l-yl]-2- oxo-ethyl } -amide
11. S-Phenyl-isoxazole-S-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo- ethyl} -amide
12. S-Phenyl-isoxazole^-carboxylic acid (2-{4-[(2-bromo-phenyl)-methyl-amino]-piperidin- l-yl}-2-oxo-ethyl)-amide
13. 5-Phenyl-isoxazole-3-carboxylic acid (2-oxo-2-[4-(2-trifluoromethyl-phenylamino)- piperidin-l-yl]-ethyl}-amide 14. S-Phenyl-isoxazole-S-carboxylic acid (2-{4-[methyl-(2-trifluoromethyl-phenyl)-amino]- piperidin- 1 -yl } -2-oxo-ethyl)-amide
15. N-{2-Oxo-2-[4-(2-trifluoromethyl-phenylamino)-piperidin-l-yl]-ethyl}-4-phenylamino- benzamide
16. N-{2-[4-(2-Chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-4-phenylamino-benzamide 17. N- {2-[4-(2-Bromo-phenylamino)-piperidin- 1 -yl]-2-oxo-ethyl} -4-phenylamino-benzamide
18. N-(2- {4-[Methyl-(2-trifluoromethyl-phenyl)-amino] -piperidin- 1 -yl } -2-oxo-ethyl)-4- phenylamino-benzamide
19. N-{2-[4-(2-Bromo-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-4-phenylamino-benzamide
20. 5 -Phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-bromo-phenylamino)-piperidin-l-yl]-2- oxo-ethyl} -amide
21. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2- oxo-ethyl } -amide
22. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-bromo-phenoxy)-piperidin-l-yl]-2-oxo- ethyl} -amide 23. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo- ethyl} -amide
24. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l- yl]-2-oxo-ethyl} -amide
25. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-phenylsulfanyl)- piperidin- 1-yl] -ethyl} -amide
26. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-henylsulfanyl)-piperidin-l-yl]-2- oxo-ethyl} -amide
27. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-mtro-phenoxy)-piperidin-l-yl]-2-oxo- ethyl} -amide
28. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-amino-phenoxy)-piperidin-l-yl]-2-oxo- ethyl} -amide 29. S-Phenyl-lH-pyrazole-S-carboxylic acid (2-[4-(2, 3-dimethyl-phenylammo)-piperidin-l- yl]-2-oxo-ethyl } -amide
30. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2, 4-dimethyl-phenylamino)-piperidin-l- yl] -2-oxo-ethyl} -amide, and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
In certain embodiments, the compound is selected from:
31. 5 -Phenyl- lH-pyrazole-S-carboxylic acid {2-[4-(2, 5-dimethyl-phenylarnino)-piperidin-l- yl] -2-oxo-ethyl } -amide 32. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-tert-butyl-phenylamino)-piperidin-l-yl]-
2-oxo-ethyl}-amide
33. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-2- oxo-ethyl} -amide
34. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-bromo-phenylsulfanyl)-piperidin-l-yl]- 2-oxo-ethyl}-amide
35. 5-Phenyl-lH-pyrazole-3-carboxylic acid [2-oxo-2-(4-o-tolylamino-piperidin-l-yl)-ethyl]- amide
36. 5-(3-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)- piperidin- 1 -yl]-2-oxo-ethyl} -amide 37. 5-Phenyl-pyridine-2-carboxylic acid {2-oxo-2-[4-(2-chloro-phenoxy)-piperidin-l-yl]- ethyl} -amide
38. 5-Phenyl-pyridine-2-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2- oxo-ethyl} -amide
39. 5-(4-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)- piperidin- 1 -yl] -2-oxo-ethyl } -amide
40. 5-(2 -Hydroxy-phenyl)- 1 -methyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro- phenylamino)-piperidin- 1 -yl] -2-oxo-ethyl } -amide
41. Synthesis of 5-(2-Hydroxy-phenyl)-isoxazole-3-carboxylic acid (2-[4-(2-chloro-phenoxy)- piperidin-l-yl]-2-oxo-ethyl} -amide
42. 5-(2-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide 43. 5-(2-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-ρhenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide
44. 5-(2-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- 1 -yl]-2-oxo-ethyl} -amide
45. N-{2-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-6-phenylamino-nicotinamide 46. N-{2-[4-(2-Chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-6-phenylamino- nicotinamide
47. 5-Phenylamino-pyridine-2-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]- 2-oxo-ethyl } -amide
48. 5-Phenylamino-pyridine-2-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]- 2-oxo-ethyl} -amide
49. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(5-bromo-2-methoxy-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide
50. 5 -Phenyl- lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-1 -yl]-ethyl}-amide 51. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-cyano-phenoxy)-piperidin-l-yl]-2-oxo- ethyl} -amide
52. 5-(2-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide
53. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2,4-difluoro-phenylamino)-piperidin-l-yl]- 2-oxo-ethyl}-amide
54. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(5-fluoro-2-trifluoromethyl-phenylamino)- piperidin-1-yl] -2-oxo-ethyl} -amide
55. 5-Phenyl-lH-pyrazole-3-carboxylic acid (2-[4-(4-fluoro-2-trifluoromethyl-phenylamino)- piperidin- 1 -yl]-2-oxo-ethyl } -amide 56. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-acetyl-ρhenoxy)-piperidin-l-yl]-2-oxo- ethyl} -amide 57. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(5-cyano-2-methyl-phenylamino)-
piperidin- 1 -yl]-2-oxo-ethyl}-amide
58. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-benzenesulfinyl)- piperidin- 1 -yl] -ethyl } -amide
59. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(pyridin-4-yloxy)-piperidin-l-yl]- ethyl} -amide
60. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]- 2-oxo-ethyl } -amide, and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
In certain embodiments, the compound is selected from:
61. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-hydroxy-phenoxy)-piperidin-l-yl]-2- oxo-ethyl} -amide
62. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-benzenesulfonyl)- piperidin- 1-yl] -ethyl} -amide
63. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(6-chloro-pyridin-2-yloxy)-piperidin-l-yl]- 2-oxo-ethyl } -amide
64. 4-Methyl-3-(l-{2-[(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetyl}-piperidin-4-yloxy)- benzoic acid methyl ester 65. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-fluoro-5-trifluoromethyl-phenoxy)- piperidin- 1 -yl]-2-oxo-ethyl } -amide
66. 5-(2-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l- yl] -2-oxo-ethyl } -amide
67. 5-(2-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1-yl] -ethyl } -amide
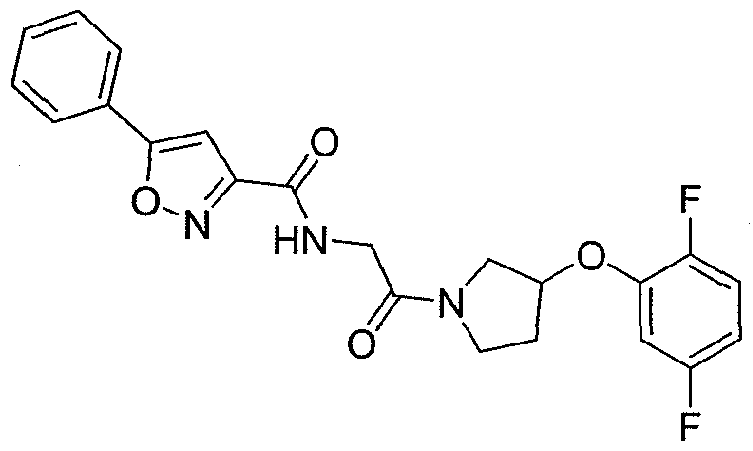
68. 5-(4-Trifluoromethyl-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)- piperidin- 1 -yl]-2-oxo-ethyl } -amide
69. 5-(3-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1-yl] -ethyl} -amide 70. 5-(3-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide 71. 5-(2-Trifluoromethyl-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-
trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethyl } -amide
72. 5-(4-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide
73. 5-(3-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide
74. 5-(4-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide
75. 5-(4-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-pyridin-3-yloxy)- piperidin-l-yl]-2-oxo-ethyl}-amide 76. 5-(4-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)- piperidin-l-yl]-2-oxo-ethyl}-amide
77. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(3-chlorophenoxy)-piperidin-l-yl]-2-oxo- ethyl} -amide
78. 3-(l-{2-[(5-Phenyl-lH-pyrazole-3-carbonyl)-amino]-acetyl}-piperidin-4-yloxy)-benzoic acid
79. 5-(3-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)- piperidin- 1 -yl] -2-oxo-ethyl } -amide
80. S-Phenyl-lH-pyrazole-S-carboxylic acid [2-oxo-2-(4-m-tolyloxy-piperidin-l-yl)-ethyl]- amide 81. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-methyl-pyridin-3-yloxy)-piperidm-l-yl]-
2-oxo-ethyl } -amide 82. 5-Pyridin-2-yl-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2- oxo-ethyl} -amide
83. 3-(5-{2-Oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethylcarbamoyl}-lH- pyrazol-3-yl)-benzoic acid
84. 5-Pyridin-3-yl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin- 1 -yl] -ethyl} -amide
85. 5-Pyridin-3-yl-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2- oxo-ethyl}-amide 86. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(4-methyl-pyridin-3-yloxy)-piperidin-l-yl]-
2-oxo-ethyl}-amide 87. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(5-trifluoromethyl-pyridin-3-yloxy)-
piperidin- 1 -yl]-ethyl} -amide
88. 5-(5-Chloro-thiophen-2-yl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide
89. 5-(5-Chloro-thiophen-2-yl)-lH-pyrazole-3-carboxylic acid {2-[4-(2,5-difluoro-phenoxy)- piperidin- 1 -yl] -2-oxo-ethyl } -amide
90. 5-(2-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifiuoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide, and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
In certain embodiments, the compound is selected from:
91. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-methanesulfonyl-phenoxy)-piperidin-l- yl]-2-oxo-ethyl } -amide
92. 5-(2-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin- l-yl]-2-oxo-ethyl}-amide
93. 5-Phenyl-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethyl } -amide
94. 5-(2-Fluoro-phenyl)-isoxazole-3-carboxylic acid (2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin- 1 -yl]-ethyl} -amide 95. 5-(2-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide
96. 5-(3-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide
97. 5-(4-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl] -ethyl} -amide
98. 5-(3-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin- 1 -yl]-ethyl } -amide
99. 5-(4-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-1 -yl]-ethyl}-amide 100. 5-(4-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)- piperidin-l-yl]-2-oxo-ethyl} -amide 101. 5-(4-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-[4-(2-chloro-pyridin-3-yloxy)-
piperidin- 1 -yl]-2-oxo-ethyl} -amide
102. 5-(3-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)- piperidin-l-yl]-2-oxo-ethyl}-amide
103. 1 -Phenyl- lH-pyrazole-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]- 2-oxo-ethyl} -amide
104. 2-[(Biphenyl-4-ylmethyl)-amino]-l-[4-(2-chloro-phenoxy)-piperidin-l-yl]- ethanone
105. N-{2-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-4-[l,3,4]oxadiazol-2-yl- benzamide 106. 4-Phenyl-pyrazole-l-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2- oxo-ethyl } -amide
107. 1 -Phenyl- IH-[1 ,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- 1 -yl] -2-oxo-ethyl} -amide
108. l-Phenyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- l-yl]-2-oxo-ethyl} -amide
109. l-(3-Fluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
110. l-CS-Fluoro-phenyO-lH-fl^^Jtriazole^-carboxylic acid {2-[4-(2-chloro-5-fluoro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide 111. l-m-Tolyl-lH-[l ,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifhioromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide
112. l-m-Tolyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)- piperidin- 1 -yl] -2-oxo-ethyl } -amide
113. l-(2-Cyano-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin- 1 -yl]-ethyl} -amide
114. l-(2-Cyano-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethyl} -amide
115. l-o-Tolyl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide 116. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid (2-[4-(2-chloro-phenoxy)- piperidin- 1 -yl]-2-oxo-ethyl } -amide 117. 1-Cyclopentyl- IH-[1, 2,3 ]triazole-4-carboxylic acid {2-[4-(5-chloro-pyridin-3-
yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
118. l-(5-Fluoro-pyridin-3-yl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
119. N-{2-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-4-(5-methyl- [ 1 ,3 ,4]oxadiazol-2-yl)-benzamide
120. 3'-Dimethylamino-biphenyl-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- 1 -yl]-2-oxo-ethyl} -amide, and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
In certain embodiments, the compound is selected from:
121. N-{2-Oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-4-(pyrrolidine- 1 -carbonyl)-benzamide
122. 9H-Carbazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethyl}-amide
123. 1 -Phenyl- 1 H-imidazole-4-carboxylic acid {2-oxo-2- [4-(3 -trifluoromethyl- phenoxy)-piperidin-l-yl]-ethyl}-amide
124. 1 -Phenyl- 1 H-imidazole-4-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)- piperidin-l-yl]-2-oxo-ethyl}-amide 125. 5-Phenyl-l H-pyrazole-3-carboxylic acid {2-[4-(2-formyl-phenoxy)-piperidin-l-yl]-
2-oxo-ethyl}-amide
126. 2-(l-{2-[(5-Phenyl-lH-pyrazole-3-carbonyl)-amino]-acetyl}-piperidin-4-yloxy)- benzoic acid
127. 5-Phenyl-l H-pyrazole-3-carboxylic acid {2-[4-(2-hydroxymethyl-phenoxy)- piperidin- 1 -yl] -2-oxo-ethyl } -amide
128. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(3,4,5-trifluoro-phenoxy)- piperidin- 1 -yl] -ethyl } -amide
129. 5-Phenyl- 1 H-pyrazole-3-carboxylic acid (2- {4-[2-(hydroxyimino-methyl)- phenoxy] -piperidin- 1 -yl } -2-oxo-ethyl)-amide 130. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(2-trifiuoromethyl-phenoxy)- piperidin- 1 -yl] -ethyl } -amide 131. 5-Phenyl-l H-pyrazole-3-carboxylic acid {2-[4-(3-cyano-phenoxy)-piperidin-l-yl]-
2-oxo-ethyl}-amide
132. S-Phenyl-lH-pyrazole-S-carboxylic acid (2-{4-[2-(methoxyimino-methyl)- phenoxy]-piperidin-l-yl}-2-oxo-ethyl)-amide
133. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-methylcarbamoyl-phenoxy)- piperidin- 1 -yl]-2-oxo-ethyl } -amide
134. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-carbamoyl-phenoxy)-piperidin-l- yl]-2-oxo-ethyl } -amide
135. 5-(2-Trifluoromethyl-phenyl)-lH-pyrazole-3-carboxylic acid (2-[4-(2-chloro- phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide 136. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3-cyano-phenylamino)-piperidin-l- yl]-2-oxo-ethyl} -amide
137. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(adamantan-2-ylamino)-piperidin-l- yl]-2-oxo-ethyl} -amide
138. 5-(2-Methoxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro- phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
139. l-Pyrrolidin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethyl} -amide
140. l-(l-Methyl-pyrrolidin-3-yl)-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin- 1 -yl]-ethyl} -amide 141. l-(3,5-Difluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro- phenoxy)-piperidin- 1 -yl] -2-oxo-ethyl } -amide
142. l-(3,5-Difluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(5-chloro- pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
143. l-Piperidin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl] -ethyl} -amide hydrochloride
144. l-(l-Methyl-piperidin-4-yl)-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl} -amide
145. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2,5-difluoro-phenoxy)- piperidin- 1 -yl] -2-oxo-ethyl } -amide 146. l-Pyridm-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(5-cyano-2-methyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide 147. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[3-(3-trifluoromethyl-phenoxy)-
pyrrolidin-l-yl]-ethyl}-amide
148. 4-(2-Oxo-pyrrolidin-l-yl)-N-{2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l- yl]-ethyl}-benzamide
149. l-Cyclopropyl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl} -amide
150. l-Morpholin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethyl } -amide, and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
In certain embodiments, the compound is selected from:
151. l-Phenyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(3-cyano-phenoxy)-piperidin- l-yl]-2-oxo-ethyl} -amide
152. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(2,5-difluoro-phenoxy)- pyrrolidin- 1 -yl] -2-oxo-ethyl } -amide
153. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[3-(3-trifluoromethyl-phenoxy)- azetidin- 1 -yl] -ethyl } -amide
154. 5-Pyridin-3-yl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[3-(3-trifluoromethyl- phenoxy)-azetidin-l-yl]-ethyl}-amide 155. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(2,5-difluoro-phenoxy)-pyrrolidin-
1 -yl]-2-oxo-ethyl} -amide
156. 1 -Phenyl- lH-imidazole-4-carboxylic acid {2-[4-(2,5-difiuoiO-phenoxy)-piperidin- 1 -yl] -2-oxo-ethyl} -amide
157. 1 -Phenyl- 1 H-imidazole-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- 1 - yl]-2-oxo-ethyl}-amide
158. 1 -Phenyl- 1 H-imidazole-4-carboxylic acid {2-[4-(5-cyano-2-methyl-ρhenoxy)- piperidin- 1 -yl] -2-oxo-ethyl } -amide
159. 1 -Phenyl- 1 H-imidazole-4-carboxylic acid {2-[3-(3-fluoro-5-trifluoromethyl- phenoxy)-azetidin-l -yl] -2-oxo-ethyl} -amide 160. 1 -Phenyl- 1 H-imidazole-4-carboxylic acid {2-[4-(3-fluoro-5-trifluoromethyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide 161. 1 -Phenyl- 1 H-imidazole-4-carboxylic acid {2-[4-(4-fluoro-3-trifluoromethyl-
phenoxy)-piperidin- 1 -yl] -2-oxo-ethyl } -amide
162. 1 -Phenyl- lH-imidazole-4-carboxylic acid {2-[3-(2-chloro-phenoxy)-azetidin-l-yl]- 2-oxo-ethyl} -amide
163. 1 -Phenyl- lH-imidazole-4-carboxylic acid {2-[3-(5-cyano-2-methyl-phenoxy)- azetidin- 1 -yl]-2-oxo-ethyl } -amide
164. 1 -Phenyl- lH-imidazole-4-carboxylic acid {2-[3-(2-chloro-phenoxy)-pyrrolidin-l- yl]-2-oxo-ethyl } -amide
165. S-Phenyl-isoxazole-S-carboxylic acid {2-[3-(2,5-difluoro-phenoxy)-pyrrolidin-l- yl]-2-oxo-ethyl} -amide 166. 2-Phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid {2-[4-(2-chloro-5-fluoro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
167. 6-Pyrazol-l-yl-imidazo[l,2-a]pyridine-2-carboxylic acid {2-[4-(2-chloro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
168. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(3-cyano-phenoxy)- piperidin-1 -yl]-2-oxo-ethyl}-amide
169. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(3-fluoro-5- trifluoromethyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
170. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(3-fluoro-5- trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide 171. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(2-chloro-phenoxy)- pyrrolidin-l-yl]-2-oxo-ethyl}-amide
172. l-Pyridin-3-yl-lH-[ 1 ,2,3]triazole-4-carboxylic acid {2-[4-(4-fluoro-3- trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
173. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(2-chloro-phenoxy)- azetidin- 1 -yl] -2-oxo-ethyl } -amide
174. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(5-cyano-2-methyl- phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
175. l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl- phenoxy)-piperidin- 1 -yl] -ethyl } -amide 176. 5-Phenyl-lH-pyrazole-3-carboxylic acid {2-[3-(3-fluoro-5-trifluoromethyl- phenoxy)-azetidin- 1 -yl]-2-oxo-ethyl } -amide 177. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3-fluoro-5-trifluoromethyl-
phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
178. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(4-fluoro-3-trifluoromethyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
179. S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(2-chloro-phenoxy)-azetidin-l-yl]- 2-oxo-ethyl } -amide
180. S-Phenyl-lH-pyrazole-S-carboxylic acid (2-[3-(5-cyano-2-methyl-phenoxy)- azetidin- 1 -yl] -2-oxo-ethyl} -amide, and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
In certain embodiments, the compound is selected from:
181. N-Biphenyl-4-yl-3 - [4-(2-bromo-phenoxy)-piperidin- 1 -yl] -3 -oxo-propionamide
182. N-Biphenyl-4-yl-3-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo- propionamide 183. N-Biphenyl-4-yl-3-[4-(2-bromo-phenylamino)-piperidin-l-yl]-3-oxo-propionamide
184. N-Biphenyl-4-yl-3-[4-(2-bromo-phenylsulfanyl)-piperidin-l-yl]-3-oxo- propionamide
185. N-Biphenyl-4-yl-3-oxo-3-(4-o-tolylamino-piperidm- 1 -yl)-propionamide
186. N-Biphenyl-4-yl-3 - [4-(2-nitro-phenoxy)-piperidin- 1 -yl] -3 -oxo-propionamide 187. 3-[4-(2-Amino-phenoxy)-piperidin-l-yl]-N-biphenyl-4-yl-3-oxo-propionamide
188. N-Biphenyl-4-yl-3-[4-(2,3-dimethyl-ρhenylamino)-piperidin-l-yl]-3-oxo- propionamide
189. N-Biphenyl-4-yl-3-[4-(2,4-dimethyl-phenylamino)-piperidin-l -yl]-3-oxo- propionamide 190. N-Biphenyl-4-yl-3-[4-(2, 5-dimethyl-phenylamino)-piperidin-l-yl]-3-oxo- propionamide
191. N-Biphenyl-4-yl-3-[4-(2-tert-butyl-phenylamino)-piperidin- 1 -yl] -3 -oxo- propionamide
192. N-Biphenyl-4-yl-3-[4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-3-oxo-propionamide 193. Synthesis of 3-[4-(2-Chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo-N-(6-phenyl- pyridin-3 -yl)-propionamide 194. 3-[4-(2-Chloro-5-fluoro-phenoxy)-piperidin-l -yl]-3-oxo-N-(5-phenyl-pyridin-2-
yl)-propionamide
195. 3 - [4-(2-Chloro-phenoxy)-piperidin- 1 -yl] -3 -oxo-N-(6-ρhenyl-pyridin-3 -yl)- propionamide
196. 3-[4-(2-Chloro-phenylamino)-piperidin- 1 -yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)- propionamide
197. 3-[4-(2-Bromo-phenylammo)-piperidin-l-yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)- propionamide
198. 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-[4-(2-trifluoromethyl-phenylamino)-piperidin- 1 -yl] -propionamide 199. 3-[4-(2-Chloro-phenylsulfanyl)-piperidin-l-yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)- propionamide
200. 3-[4-(2-Bromo-phenylsulfanyl)-piperidin-l-yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)- propionamide
201. 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-[4-(2-trifluoromethyl-ρhenylsulfanyl)- piperidin-l-yl]-propionamide
202. 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-[4-(2-trifluoromethyl-phenoxy)-piperidin-l- yl]-propionamide
203. 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-(4-o-tolylamino-piperidin-l-yl)-propionamide
204. 3-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-3-oxo-N-(3-phenyl-[l,2,4]thiadiazol-5- yl)-propionamide
205. 3-[4-(2-Chloro-phenoxy)-piperidin- 1 -yl]-N-(4-[ 1 ,2,4]oxadiazol-3-yl-phenyl)-3- oxo-propionamide
206. 3-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-3-oxo-N-(5-phenyl-thiazol-2-yl)- propionamide 207. 3-[4-(2-Chloro-phenylamino)-piperidin-l-yl]-3-oxo-N-(5-phenyl-thiazol-2-yl)- propionamide
208. l-[4-(2-Chloro-phenoxy)-piperidine-l-carbonyl]-cyclopropane carboxylic acid biphenyl-4-ylamide
209. N-Biphenyl-4-yl-3 -oxo-3 -[4-(3 ,4,5-trifluoro-phenoxy)-piperidin- 1 -yl] - propionamide
210. N-Biphenyl-4-yl-3-[4-(3-cyano-phenoxy)-piperidin-l-yl]-3-oxo-propionamide,
and pharmaceutically acceptable salts thereof, pharmaceutically acceptable solvates thereof, and solvates of pharmaceutically acceptable salts thereof.
wherein free base forms listed above can also be in the form of a pharmaceutically acceptable salt, wherein a compound listed above (in either a free base form or in the form of a pharmaceutically acceptable salt) can also be in the form of a solvate (such as a hydrate), wherein a compound listed above (in either a free base form or in the form of a pharmaceutically acceptable salt) can also be in the form of an N-oxide, wherein a compound listed above (in a free base form or solvate or N-oxide thereof, or in the form of a pharmaceutically acceptable salt or solvate thereof,) can also be in the form of a polymorph, and wherein if the compound exhibits chirality it can be in the form of a mixture of enantiomers such as a racemate or a mixture of diastereomers, or can be in the form of a single enantiomer or a single diastereomer. wherein, when present, an aryl, heteroaryl or heterocycle group may optionally be substituted by one or more halogen, hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, arylamino, diarylamino, amido, alkylamido, carboxyl, alkyl, halogenated alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, aroyl, acyl, alkoxy, aryloxy, heteroaryloxy, cycloalkyloxy, cycloalkylalkyloxy, arylalkyloxy, heteroarylalkyloxy, alkythio, arylthio, alkylsulfmyl, alkylsulfonyl, arylsulfinyl, arylsulfonyl, heteroarylsulfinyl, heteroarylsulfonyl alkoxycarbonyl, aryloxycarbonyl or heteroaryloxycarbonyl, and combinations thereof.
As used herein the term "halogen" means F, Cl, Br, and I. The term "alkyl" means a substituted or unsubstituted saturated hydrocarbon radical which may be straight-chain or branched-chain and may comprise about 1 to about 20 carbon atoms, for instance 1 to 12 carbon atoms, such as 1 to 8 carbon atoms, e.g., 1 to 4 carbon atoms. Suitable alkyl groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert- butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, and dodecyl. Other examples of suitable alkyl groups include, but are not limited to, 1-, 2- or 3-methylbutyl, 1,1-, 1,2- or 2,2- dimethylpropyl, 1-ethylpropyl, 1-, 2-, 3- or 4-methylpentyl, 1,1-, 1,2-, 1,3-, 2,2-, 2,3- or 3,3-
dimethylbutyl, 1 - or 2-ethylbutyl, ethylmethylpropyl, trimethylpropyl, methylhexyl, dimethylpentyl, ethylpentyl, ethylmethylbutyl, dimethylbutyl, and the like.
Substituted alkyl groups are alkyl groups as described above which are substituted in one or more positions by, e.g., halogen, hydroxyl, amino, carboxy, alkylamino, dialkylamino, aryl, heteroaryl, alkoxy, nitro and cyano, and combinations thereof.
The term "halogenated alkyl" means a saturated hydrocarbon radical which may be straight-chain or branched-chain and may comprise about 1 to about 20 carbon atoms, for instance 1 to 12 carbon atoms, such as 1 to 8 carbon atoms, e.g., 1 to 4 carbon atoms, that is substituted by one ore more halogens, such as, but not limited to, -CF3, CF2CF3, CHF2, CH2F, and the like. The use of the term "halogenated alkyl" should not be construed to mean that a "substituted alkyl" group may not be substituted by one or more halogens.
The term "alkenyl" means a substituted or unsubstituted hydrocarbon radical which may be straight-chain or branched-chain, which contains one or more carbon-carbon double bonds, and which may comprise about 1 to about 20 carbon atoms, such as 1 to 12 carbon atoms, for instance 1 to 6 carbon atoms. Suitable alkenyl groups include ethenyl, propenyl, butenyl, etc.
Substituted alkenyl groups are alkenyl groups as described above which are substituted in one or more positions by, e.g., halogen, hydroxyl, amino, carboxy, alkylamino, dialkylamino, aryl, heteroaryl, alkoxy, nitro and cyano, and combinations thereof.
The term "alkylene" means a linear saturated divalent hydrocarbon radical of one to six carbon atoms or a branched saturated divalent hydrocarbon radical of three to six carbon atoms unless otherwise stated e.g., methylene, ethylene, propylene, 1-methylpropylene, 2- methylpropylene, butylene, pentylene, and the like.
The term "alkynyl" means a substituted or unsubstituted aliphatic hydrocarbon radical which may be straight-chain or branched-chain and which contains one or more carbon-carbon triple bonds. Preferably the alkynyl group contains 2 to 15 carbon atoms, such as 2 to 12 carbon atoms, e.g., 2 to 8 carbon atoms. Suitable alkynyl groups include ethynyl, propynyl, butynyl, etc.
Substituted alkynyl groups are alkynyl groups as described above which are substituted in one or more positions by, e.g., halogen, hydroxyl, amino, carboxy, alkylamino, dialkylamino, aryl, heteroaryl, alkoxy, nitro and cyano, and combinations thereof. The term "amino" means -NH2.
The term "alkylamino" means -NH( alkyl), wherein alkyl is as described above.
The term "dialkylamino" means -N(alkyl)2, wherein alkyl is as described above.
The term "aryl" means a substituted or unsubstituted aromatic monocyclic or bicyclic ring system comprising about 5 to about 14 carbon atoms, e.g., about 6 to about 10 carbon atoms. Suitable aryl groups include, but are not limited to, phenyl, naphthyl, anthracenyl.
Substituted aryl groups include the above-described aryl groups which are substituted one or more times by, for example, but not limited to, halogen, hydroxyl, amino, carboxy, alkylamino, dialkylamino, aryl, heteroaryl, alkoxy, nitro and cyano, and combinations thereof.
The term "arylamino" means -NH(aryl), wherein aryl is as described above.
The term "diarylamino" means -N(aryl)2, wherein aryl is as described above.
The term "amido" means -CONH2. The term "arylalkyl" refers to an -(alkylene)-aryl group in which the aryl and alkylene portions are in accordance with the previous descriptions. Suitable examples include, but are not limited to, benzyl, 1-phenethyl, 2-phenethyl, phenpropyl, phenbutyl, phenpentyl, and napthylmethyl.
The term "carboxyl" means -C(O)OH. The term "cycloalkyl" means a monocyclic, bicyclic or tricyclic nonaromatic saturated hydrocarbon radical having 3 to 10 carbon atoms, such as 3 to 8 carbon atoms, for example, 3 to 6 carbon atoms. Suitable cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, norbornyl, 1-decalin, adamant- 1-yl, and adamant-2-yl. Other suitable cycloalkyl groups include, but are not limited to, spiropentyl, bicyclo[2.1.0]pentyl, bicyclo[3.1.0]hexyl, spiro[2.4]heptyl, spiro[2.5]octyl, bicyclo[5.1.0]octyl, spiro[2.6]nonyl, bicyclo[2.2.0]hexyl, spiro[3.3]heptyl, bicyclo[4.2.0]octyl, and spiro[3.5]nonyl. Preferred cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl. The cycloalkyl group can be substituted, for example, by one or more halogens and/or alkyl groups.
The term "cycloalkylalkyl" means a -(alkylene)-cycloalkyl in which the cycloalkyl group is as previsouly described; e.g., cyclopropylmethyl, cyclobutylmethyl, cyclopentylethyl, or cyclohexylmethyl, and the like.
The term "heteroaryl" means a substituted or unsubstituted aromatic monocyclic or multicyclic ring system comprising 5 to 14 ring atoms, preferably about 5 to about 10 ring atoms and most preferably 5 or 6 ring atoms, wherein at least one of the ring atoms is an N, O or S atom. Suitable heteroaryl groups include, but are not limited to furyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrimidinyl, benzimidazolyl, indazolyl, indolyl, quinolinyl, isoquinolinyl, naphthyridinyl and the like.
Substituted heteroaryl groups include the above-described heteroaryl groups which are substituted one or more times by, for example, but not limited to, halogen, hydroxyl, amino, carboxy, alkylamino, dialkylamino, aryl, heteroaryl, alkoxy, nitro and and combinations thereof.
The term "heteroarylalkyl" refers to a -(alkylene)-heteroaryl group wherein the heteroaryl and alkylene portions are in accordance with the previous discussions. Suitable examples include, but are not limited to, pyridylmethyl, thiazolylmethyl, thienylmethyl, pyrimidinylmethyl, pyrazinylmethyl, and isoquinolinylmethyl, and the like.
The term "heterocycle" means a substituted or unsubstituted non-aromatic mono- or multi cyclic ring system comprising 3 to 10 atoms, preferably 5 or 6, wherein at least one of the ring atoms is an N, O or S atom. Suitable heterocyle groups include, but are not limited to tetrahydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, pyrrolidinyl, piperidinyl, piperazinyl, thiomorpholinyl, morpholinyl, isoxazolinyl, and the like
Substituted heterocycle groups include the above-described heterocycle groups which are substituted one or more times by, for example, halogen, amino, alkyl, hydroxy, carboxy, and combinations thereof. Heterocycle groups may also be substituted by, e.g., aryl or heteroaryl.
The term "heterocyclealkyl" refers to a -(alkylene)-heterocycle group wherein the heterocycle and alkylene portions are in accordance with the previous discussions.
The term "aroyl" means an aryl-C(O)-, in which the aryl group is as previously described. Suitable aroyl groups include, but are not limited to, benzoyl and 1- naphthoyl. The term "acyl" means an HC(O)-, alkyl-C(O)-, cycloalkyl-C(O)-, aryl-C(O)-, or heteroalkyl-C(O)-, in which the various groups are as previously described, e.g., acetyl, propionyl, benzoyl, pyridinylcarbonyl, and the like.
The term "alkoxy" means alkyl-O- groups in which the alkyl portion is in accordance with the previous discussion. Suitable alkoxy groups include, but are not limited to, methoxy, ethoxy, n-propoxy, iso-propoxy, n-butoxy, t-butoxy, pentoxy, hexoxy, heptoxy, octoxy, and the like. For example, the alkoxy can be methoxy or ethoxy.
The term "aryloxy" means an aryl-O- group, in which the aryl group is as previously described.
The term "heteroaryloxy" means an heteroaryl-O- group, in which the heteroaryl group is as previously described.
The term "cycloalkylalkyloxy" means a -O-(alkylene)-cycloalkyl group, in which the cycloalkyl and alkylene groups are as previously described.
The term "alkylthio" means an alkyl-S- group, in which the alkyl group is as previously described.
The term "arylthio" means an aryl-S- group, in which the aryl group is as previously described. The term "alkylsulfinyl" means a -SOR radical where R is alkyl as defined above, e.g., methylsulfϊnyl, ethylsulfinyl, and the like.
The term "alkylsulfonyl" means a -SO2R radical where R is alkyl as defined above, e.g., methylsulfonyl, ethylsulfonyl, and the like.
The term "arylsulfinyl" means a -SOR radical where R is aryl as defined above, e.g., phenylsulfinyl, and the like.
The term "arylsulfonyl" means a -SO2R radical where R is aryl as defined above, e.g., phenylsulfonyl, and the like.
The term "heteroarylsulfinyl" means a -SOR radical where R is heteroaryl as defined above. The term "heteroarylsulfonyl" means a -SO2R radical where R is heteroaryl as defined above.
The term "alkoxycarbonyl" means an alkyl-O-C(O)- group, in which the alkyl group is as previously described.
The term "aryloxycarbonyl" means an aryl-O-C(O)- group, in which the aryl group is as previously described.
The term "heteroaryloxycarbonyl" means an heteroaryl-O-C(O)- group, in which the heteroaryl group is as previously described.
The term "cycloalkyloxy" means a -O-cycloalkyl group in which the cycloalkyl group is as previously described, e.g., cyclopropyloxy, cyclobutyloxy, cyclopentyloxy, cyclohexyloxy, and the like
The term "arylalkyloxy" means -O-(alkylene)-aryl group, in which the aryl and alkylene groups are as previously described.
The term "heteroarylalkyloxy" means -O-(alkylene)-heteroaryl group, in which the heteroaryl and alkylene groups are as previously described. One of ordinary skill in the art will recognize that compounds of the present invention can exist in different tautomeric and geometrical isomeric forms. All of these compounds, including cis isomers, trans isomers, diastereomic mixtures, racemates, nonracemic mixtures of
enantiomers, substantially pure, and pure enantiomers, are within the scope of the present invention. Substantially pure enantiomers contain no more than 5% w/w of the corresponding opposite enantiomer, preferably no more than 2%, most preferably no more than 1%.
The optical isomers can be obtained by resolution of the racemic mixtures according to conventional processes, for example, by the formation of diastereoisomeric salts using an optically active acid or base or formation of covalent diastereomers. Examples of appropriate acids are tartaric, diacetyltartaric, dibenzoyltartaric, ditoluoyltartaric and camphorsulfonic acid. Mixtures of diastereoisomers can be separated into their individual diastereomers on the basis of their physical and/or chemical differences by methods known to those skilled in the art, for example, by chromatography or fractional crystallization. The optically active bases or acids are then liberated from the separated diastereomeric salts. A different process for separation of optical isomers involves the use of chiral chromatography (e.g., chiral HPLC columns), with or without conventional derivation, optimally chosen to maximize the separation of the enantiomers. Suitable chiral HPLC columns are manufactured by Diacel, e.g., Chiracel OD and Chiracel OJ among many others, all routinely selectable. Enzymatic separations, with or without derivitization, are also useful. The optically active compounds of the invention can likewise be obtained by utilizing optically active starting materials in chiral synthesis processes under reaction conditions which do not cause racemization.
In addition, one of ordinary skill in the art will recognize that the compounds can be used in different enriched isotopic forms, e.g., enriched in the content of 2H, 3H, 11C, 13C and/or 14C. In one particular embodiment, the compounds are deuterated. Such deuterated forms can be made the procedure described in U.S. Patent Nos. 5,846,514 and 6,334,997. As described in U.S. Patent Nos. 5,846,514 and 6,334,997, deuteration can improve the efficacy and increase the duration of action of drugs. Deuterium substituted compounds can be synthesized using various methods such as described in: Dean, Dennis C; Editor. Recent Advances in the Synthesis and Applications of Radiolabeled Compounds for Drug Discovery and Development. [In: Curr., Pharm. Des., 2000; 6(10)] (2000), 110 pp. CAN 133:68895 AN 2000:473538 CAPLUS; Kabalka, George W.; Varma, Rajender S. The synthesis of radiolabeled compounds via organometallic intermediates. Tetrahedron (1989), 45(21), 6601-21, CODEN: TETRAB ISSN:0040-4020. CAN 112:20527 AN 1990:20527 CAPLUS; and Evans, E. Anthony. Synthesis of radiolabeled compounds, J.
Radioanal. Chem. (1981), 64(1-2), 9-32. CODEN: JRACBN ISSN:0022-4081, CAN 95:76229 AN 1981:476229 CAPLUS.
Where applicable, the present invention also relates to useful forms of the compounds as disclosed herein, such as base free forms, and pharmaceutically acceptable salts or prodrugs of all the compounds of the present invention for which salts or prodrugs can be prepared.
Pharmaceutically acceptable salts include those obtained by reacting the main compound, functioning as a base with an inorganic or organic acid to form a salt, for example, salts of hydrochloric acid, sulfuric acid, phosphoric acid, methane sulfonic acid, camphor sulfonic acid, oxalic acid, maleic acid, succinic acid, citric acid, formic acid, hydrobromic acid, benzoic acid, tartaric acid, fumaric acid, salicylic acid, mandelic acid, and carbonic acid. Pharmaceutically acceptable salts also include those in which the main compound functions as an acid and is reacted with an appropriate base to form, e.g., sodium, potassium, calcium, magnesium, ammonium, and choline salts. Those skilled in the art will further recognize that acid addition salts of the claimed compounds may be prepared by reaction of the compounds with the appropriate inorganic or organic acid via any of a number of known methods. Alternatively, alkali and alkaline earth metal salts can be prepared by reacting the compounds of the invention with the appropriate base via a variety of known methods.
The following are further examples of acid salts that can be obtained by reaction with inorganic or organic acids: acetates, aDIPEAtes, alginates, citrates, aspartates, benzoates, benzenesulfonates, bisulfates, butyrates, camphorates, digluconates, cyclopentanepropionates, dodecylsulfates, ethanesulfonates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, fumarates, hydrobromides, hydroiodides, 2-hydroxy-ethanesulfonates, lactates, maleates, methanesulfonates, nicotinates, 2-naphthalenesulfonates, oxalates, palmoates, pectinates, persulfates, 3-phenylpropionates, picrates, pivalates, propionates, succinates, tartrates, thiocyanates, tosylates, mesylates and undecanoates.
For example, the pharmaceutically acceptable salt can be a hydrochloride, a hydrobromide, a hydroformate, or a maleate.
Preferably, the salts formed are pharmaceutically acceptable for administration to mammals. However, pharmaceutically unacceptable salts of the compounds are suitable as intermediates, for example, for isolating the compound as a salt and then converting the salt back to the free base compound by treatment with an alkaline reagent. The free base can then, if desired, be converted to a pharmaceutically acceptable acid addition salt.
One of ordinary skill in the art will also recognize that some of the compounds of the present invention can exist in different polymorphic forms. As known in the art, polymorphism is an ability of a compound to crystallize as more than one distinct crystalline or "polymorphic" species. A polymorph is a solid crystalline phase of a compound with at least two different arrangements or polymorphic forms of that compound molecule in the solid state. Polymorphic forms of any given compound are defined by the same chemical formula or composition and are as distinct in chemical structure as crystalline structures of two different chemical compounds. One of ordinary skill in the art will further recognize that compounds of the present invention can exist in different solvate forms. Solvates of the compounds of the invention may also form when solvent molecules are incorporated into the crystalline lattice structure of the compound molecule during the crystallization process.
The present invention also includes prodrugs of compounds of the present invention. The term prodrug is intended to represent covalently bonded carriers, which are capable of releasing the active ingredient of the present invention when the prodrug is administered to a mammalian subject. Release of the active ingredient occurs in vivo. Prodrugs can be prepared by techniques known to one skilled in the art. These techniques generally modify appropriate functional groups in a given compound. These modified functional groups however regenerate original functional groups by routine manipulation or in vivo. Prodrugs of compounds of the present invention include compounds wherein a hydroxy, amino, carboxylic, or a similar group is modified. Examples of prodrugs include, but are not limited to esters (e.g., acetate, formate, and benzoate derivatives), carbamates (e.g., N,N-dimethylaminocarbonyl) of hydroxy or amino functional groups in compounds of the present invention), amides (e.g., trifluoroacetylamino, acetylamino, and the like), and the like. Prodrugs of compounds discussed herein are also within the scope of this invention. The present invention also provides processes for preparing the compounds discussed herein through methods described in the following General Scheme:

Coupling with (C) STEP-6
The starting materials for the above reaction scheme are commercially available or can be prepared according to methods known to one skilled in the art or by methods disclosed herein.
Compound (A) may be N-protected with a Boc or Cbz group via standard protection procedures and then activated as a mesylate or tosylate using procedures known in the art. The activated intermediate may then be reacted with an appropriately substitiuted ArOH or ArSH nucleophile or, alternatively, the alcohol intermediate can be reacted directly with a nucleophile under Mitsunobu conditions. Deprotection in a standard manner affords the desired amine compound (B) wherein X is O or S.
Compound (G), which is commercially available (for example, from Aldrich, St Louis, MS) may be protected with a Boc or Cbz group via standard protecting conditions known to the one skilled in the art and then treated with an appropriately substituted aryl amine under reductive
amination conditions to generate compound (B).
Carboxylic acid (C) may be reacted with an appropriately substituted amine (D) in the presence of a standard peptide coupling reagent (such as EDCI) to give the desired amide product, which undergoes standard hydrolysis procedure known to the one skilled in the art to generate the carboxylic acid (E). Coupling between compounds (B) and (E) under standard amide bond formation conditions known to the one skilled in the art affords a compound of the present invention.
Compound (N), a glycine derivative, may be reacted with compound (B) under standard amide bond formation conditions known to the one skilled in the art to afford compound (O). Following standard hydrolysis of the N-protection group, compound (O) may be reacted with compound (C) under standard amide bond formation conditions known to the one skilled in the art affords a compound of the present invention.
Compound (J) may be reacted with an appropriately substituted malonic acid mono-ethyl ester (K) (when W = OH) in the presence of a standard peptide coupling reagents kown to one skilled in the art, or alternatively compound (J) may be reacted with (K) as an acid chloride (when W = Cl) to give the desired amide product, which undergoes standard hydrolysis by procedures known to the one skilled in the art to generate the carboxylic acid (L). The coupling between compounds (L) and (B) under standard amide bond formation conditions known to the one skilled in the art affords a compound of the present invention. Compound (B) may be reacted with an appropriately substituted malonic acid mono-ethyl ester (K) (when W = OH) in the presence of a standard peptide coupling reagents known to one skilled in the art, or alternatively compound (B) may be reacted with (K) as an acid chloride (when W = Cl) to give the desired amide product, which undergoes standard hydrolysis procedure known to the one skilled in the art to generate the carboxylic acid (M). The coupling between compounds (M) and (B) under standard amide bond formation conditions known to the one skilled in the art affords the compound of the present invention.
The compounds of the invention can be administered alone or as an active ingredient of a formulation. Thus, the present invention also includes pharmaceutical compositions of compounds of the present invention, containing, for example, one or more pharmaceutically acceptable carriers.
Numerous standard references are available that describe procedures for preparing various formulations suitable for administering the compounds according to the invention. Examples of potential formulations and preparations are contained, for example, in the Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (current edition); Pharmaceutical Dosage Forms: Tablets (Lieberman, Lachman and Schwartz, editors) current edition, published by Marcel Dekker, Inc., as well as Remington's Pharmaceutical Sciences (Arthur Osol, editor), 1553-1593 (current edition).
Administration of the compounds of the present invention may be accomplished according to patient needs, for example, orally, nasally, parenterally (subcutaneously, intraveneously, intramuscularly, intrasternally and by infusion) by inhalation, rectally, vaginally, topically and by ocular administration.
Various solid oral dosage forms can be used for administering compounds of the invention including such solid forms as tablets, gelcaps, capsules, caplets, granules, lozenges and bulk powders. The compounds of the present invention can be administered alone or combined with various pharmaceutically acceptable carriers, diluents (such as sucrose, mannitol, lactose, starches) and excipients known in the art, including but not limited to suspending agents, solubilizers, buffering agents, binders, disintegrants, preservatives, colorants, flavorants, lubricants and the like. Time release capsules, tablets and gels are also advantageous in administering the compounds of the present invention. Various liquid oral dosage forms can also be used for administering compounds of the inventions, including aqueous and non-aqueous solutions, emulsions, suspensions, syrups, and elixirs. Such dosage forms can also contain suitable inert diluents known in the art such as water and suitable excipients known in the art such as preservatives, wetting agents, sweeteners, flavorants, as well as agents for emulsifying and/or suspending the compounds of the invention. The compounds of the present invention may be injected, for example, intravenously, in the form of an isotonic sterile solution. Other preparations are also possible.
Suppositories for rectal administration of the compounds of the present invention can be prepared by mixing the compound with a suitable excipient such as cocoa butter, salicylates and polyethylene glycols. Formulations for vaginal administration can be in the form of a pessary, tampon, cream, gel, past foam, or spray formula containing, in addition to the active ingredient, such suitable carriers as are known in the art.
For topical administration the pharmaceutical composition can be in the form of creams, ointments, liniments, lotions, emulsions, suspensions, gels, solutions, pastes, powders, sprays, and drops suitable for administration to the skin, eye, ear or nose. Topical administration may also involve transdermal administration via means such as transdermal patches. Aerosol formulations suitable for administering via inhalation also can be made.
For example, the compounds of the present invention can be administered by inhalation in the form of a powder (e.g., micronized) or in the form of atomized solutions or suspensions. The aerosol formulation can be placed into a pressurized acceptable propellant.
The compounds of the present invention may be useful as inhibitors of stearoyl- CoA desaturase (SCD) enzymes, for example, as inhibitors of SCD-I enzyme. Therefore, the compounds are useful in the treatment of conditions mediated by stearoyl-CoA desaturase (SCD) enzymes, e.g., SCD-I enzyme.
According to another embodiment, the present invention relates to a method of treating a disease or condition mediated by stearoyl-CoA desaturase (e.g., SCD-I) by administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention. An SCD-mediated disease or condition includes but is not limited to a disease or condition which is, or is related to, cardiovascular disease, dyslipidemias (including but not limited to disorders of serum levels of triglycerides, hypertriglyceridemia, VLDL, HDL, LDL, fatty acid Desaturation Index (e.g. the ratio of 18:1/18:0 fatty acids, or other fatty acids), cholesterol, and total cholesterol, hypercholesterolemia, as well as cholesterol disorders (including disorders characterized by defective reverse cholesterol transport), familial combined hyperlipidemia, coronary artery disease, atherosclerosis, heart disease, cerebrovascular disease (including, but not limited to stroke, ischemic stroke and transient ischemic attack (TIA)), peripheral vascular disease, and ischemic retinopathy. In an embodiment, compounds of the invention will, in a patient, increase HDL levels and/or decrease triglyceride levels and/or decrease LDL or non-HDL- cholesterol levels.
An SCD-mediated disease or condition also includes metabolic syndrome (including but not limited to dyslipidemia, obesity and insulin resistance, hypertension, microalbuminemia, hyperuricaemia, and hypercoagulability), Syndrome X, diabetes, insulin resistance, decreased glucose tolerance, non-insulin-dependent diabetes mellitus, Type II diabetes, Type I diabetes, diabetic complications, body weight disorders (including but not limited to obesity, overweight, cachexia and anorexia), weight loss, body mass index and leptin related diseases. In an
embodiment, the compounds of the present invention are useful in the treatment of diabetes mellitus and obesity. In another embodiment, the compounds of the present invention are useful in the treatment of obesity.
As used herein, the term "metabolic syndrome" is a recognized clinical term used to describe a condition comprising combinations of Type II diabetes, impaired glucose tolerance, insulin resistance, hypertension, obesity, increased abdominal girth, hypertriglyceridemia, low HDL, hyperuricaemia, hypercoagulability and/or microalbuniinemia.
An SCD-mediated disease or condition also includes fatty liver, hepatic steatosis, hepatitis, non-alcoholic hepatitis, non-alcoholic steatohepatitis (NASH), alcoholic hepatitis, acute fatty liver, fatty liver of pregnancy, drug-induced hepatitis, erythrohepatic protoporphyria, iron overload disorders, hereditary hemochromatosis, hepatic fibrosis, hepatic cirrhosis, hepatoma and conditions related thereto.
An SCD-mediated disease or condition also includes, but is not limited to, a disease or condition which is, or is related to primary hypertriglyceridemia, or hypertriglyceridemia secondary to another disorder or disease, such as hyperlipoproteinemias, familial histiocytic reticulosis, lipoprotein lipase deficiency, apolipoprotein deficiency (such as ApoCII deficiency or ApoE deficiency), and the like, or hypertriglyceridemia of unknown or unspecified etiology.
An SCD-mediated disease or condition also includes a disorder of polyunsaturated fatty acid (PUFA) disorder, or a skin disorder, including, but not limited to, eczema, acne, psoriasis, keloid scar formation or prevention, diseases related to production or secretions from mucous membranes, such as monounsaturated fatty acids, wax esters, and the like.
An SCD-mediated disease or condition also includes inflammation, sinusitis, asthma, pancreatitis, osteoarthritis, rheumatoid arthritis, cystic fibrosis, and pre-menstrual syndrome.
An SCD-mediated disease or condition also includes but is not limited to a disease or condition which is, or is related to cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like.
An SCD-mediated disease or condition also includes a condition where increasing lean body mass or lean muscle mass is desired, such as is desirable in enhancing performance through muscle building. Myopathies and lipid myopathies such as carnitine palmitoyltransferase deficiency (CPT I or CPT II) are also included herein. Such treatments are useful in humans and in animal husbandry, including for administration to bovine, porcine or avian domestic animals or
any other animal to reduce triglyceride production and/or provide leaner meat products and/or healthier animals.
An SCD-mediated disease or condition also includes a disease or condition which is, or is related to, neurological diseases, psychiatric disorders, multiple sclerosis, eye diseases, and immune disorders.
An SCD-mediated disease or condition also includes a disease or condition which is, or is related to, viral diseases or infections including but not limited to all positive strand RNA viruses, coronaviruses, SARS virus, SARS-associated coronavirus, Togaviruses, Picornaviruses, Coxsackievirus, Yellow Fever virus, Flaviviridae, ALPHA VIRUS (TOGA VIRID AE) including Rubella virus, Eastern equine encephalitis virus, Western equine encephalitis virus, Venezuelan equine encephalitis virus, Sindbis virus, Semliki forest virus, Chikungunya virus, O'nyong'nyong virus, Ross river virus, Mayaro virus, Alphaviruses; ASTRO VIRID AE including Astrovirus, Human Astroviruses; CALICIVIRIDAE including Vesicular exanthema of swine virus, Norwalk virus, Calicivirus, Bovine calicivirus, Pig calcivirus, Hepatitis E; CORONAVIRIDAE including Coronavirus, SARS virus, Avian infectious bronchitis virus, Bovine coronavirus, Canine coronavirus, Feline infectious peritonitis virus, Human coronavirus 299E, Human coronavirus OC43, Murine hepatitis virus, Porcine epidemic diarrhea virus, Porcine hemagglutinating encephalomyelitis virus, Porcine transmissible gastroenteritis virus, Rat coronavirus, Turkey coronavirus, Rabbit coronavirus, Berne virus, Breda virus; FLAVIVIRIDAE including Hepatitis C virus, West Nile virus, Yellow Fever virus, St. Louis encephalitis virus, Dengue Group, Hepatitis G virus, Japanese B encephalitis virus, Murray Valley encephalitis virus, Central European tick- borne encephalitis virus, Far Eastern tick-borne encephalitis virus, Kyasanur forest virus, Louping ill virus, Powassan virus, Omsk hemorrhagic fever virus, Kumilinge virus, Absetarov anzalova hypr virus, Ilheus virus, Rocio encephalitis virus, Langat virus, Pestivirus, Bovine viral diarrhea, Hog cholera virus, Rio Bravo Group, Tyuleniy Group, Ntaya Group, Uganda S Group, Modoc Group; PICORNA VIRID AE including Coxsackie A virus, Rhinovirus, Hepatitis A virus, Encephalomyocarditis virus, Mengovirus, ME virus, Human poliovirus 1, Coxsackie B; POTYVIRIDAE including Potyvirus, Rymovirus, Bymovirus. Additionally it can be a disease or infection caused by or linked to Hepatitis viruses, Hepatitis B virus, Hepatitis C virus, human immunodeficiency virus (HIV) and the like. Treatable viral infections include those where the virus employs an RNA intermediate as part of the replicative cycle (hepatitis or HIV); additionally
it can be a disease or infection caused by or linked to RNA negative strand viruses such as influenza and parainfluenza viruses.
In one embodiment, the compounds of the inventions are useful in the treatment of elevated levels of lipids, cardiovascular diseases, diabetes, obesity, and metabolic syndrome. The term "treating" means to relieve, alleviate, delay, reduce, reverse, improve or prevent at least one symptom of a condition in a subject. The term "treating" may also mean to arrest, delay the onset (i.e., the period prior to clinical manifestation of a disease) and/or reduce the risk of developing or worsening a condition.
An "effective amount" means the amount of a compound of the present invention that, when administered to a patient (e.g., a mammal) for treating a disease, is sufficient to effect such treatment for the disease to achieve the objectives of the invention. The "effective amount" will vary depending on the compound, the disease and its severity and the age, weight, etc., of the patient to be treated.
A subject or patient in whom administration of the therapeutic compound is an effective therapeutic regimen for a disease or disorder is preferably a human, but can be any animal, including a laboratory animal in the context of a clinical trial or screening or activity experiment. Thus, as can be readily appreciated by one of ordinary skill in the art, the methods, compounds and compositions of the present invention are particularly suited to administration to any animal, particularly a mammal, and including, but by no means limited to, humans, domestic animals, such as feline or canine subjects, farm animals, such as but not limited to bovine, equine, caprine, ovine, and porcine subjects, wild animals (whether in the wild or in a zoological garden), research animals, such as mice, rats, rabbits, goats, sheep, pigs, dogs, cats, etc., avian species, such as chickens, turkeys, songbirds, etc., i.e., for veterinary medical use.
In some embodiments, the compounds of the present invention are administered as a mono-therapy. In other embodiments, the compounds of the present invention are administered as part of a combination therapy. For example, a compound of the present invention may be used in combination with other drugs or therapies that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds discussed herein are useful. Such other drug(s) may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention. When a compound of the present invention is used contemporaneously with one or more other drags, a pharmaceutical unit dosage form containing such other drugs in addition to the compound of
formula I may be employed. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
The present invention also provides processes for preparing the compounds of the present invention through methods described in the following General Scheme:
Coupling with (C) STEP-6 (D)
The starting materials for the above reaction Scheme are commercially available or can be prepared according to methods known to one skilled in the art or by methods disclosed herein. Compound (A) may be N-protected with a Boc or Cbz group via standard protection procedures and then activated as a mesylate or tosylate using procedures known in the art. The activated intermediate may then be reacted with an appropriately substitiuted ArOH or ArSH
nucleophile or, alternatively, the alcohol intermediate can be reacted directly with a nucleophile under Mitsunobu conditions. Deprotection in a standard manner affords the desired amine compound (B) wherein X is O or S.
Compound (G), which is commercially available (for example, from Aldrich, St Louis, MS) may be protected with a Boc or Cbz group via standard protecting conditions known to the one skilled in the art and then treated with an appropriately substituted aryl amine under reductive amination conditions to generate compound (B).
Carboxylic acid (C) may be reacted with an appropriately substituted amine (D) in the presence of a standard peptide coupling reagent (such as EDCI) to give the desired amide product, which undergoes standard hydrolysis procedure known to the one skilled in the art to generate the carboxylic acid (E). Coupling between compounds (B) and (E) under standard amide bond formation conditions known to the one skilled in the art affords a compound of the present invention.
Compound (N), a glycine derivative, may be reacted with compound (B) under standard amide bond formation conditions known to the one skilled in the art to afford compound (O). Following standard hydrolysis of the N-protection group, compound (O) may be reacted with compound (C) under standard amide bond formation conditions known to the one skilled in the art affords a compound of the present invention.
Compound (J) may be reacted with an appropriately substituted malonic acid mono-ethyl ester (K) (when W = OH) in the presence of a standard peptide coupling reagents kown to one skilled in the art, or alternatively compound (J) may be reacted with (K) as an acid chloride (when W = Cl) to give the desired amide product, which undergoes standard hydrolysis by procedures known to the one skilled in the art to generate the carboxylic acid (L). The coupling between compounds (L) and (B) under standard amide bond formation conditions known to the one skilled in the art affords a compound of the present invention.
Compound (B) may be reacted with an appropriately substituted malonic acid mono-ethyl ester (K) (when W = OH) in the presence of a standard peptide coupling reagents known to one skilled in the art, or alternatively compound (B) may be reacted with (K) as an acid chloride (when W = Cl) to give the desired amide product, which undergoes standard hydrolysis procedure known to the one skilled in the art to generate the carboxylic acid (M). The coupling between compounds (M) and (B) under standard amide bond formation conditions known to the one skilled in the art affords the compound of the present invention.
EXAMPLES
The present invention will now be further described by way of the following non-limiting examples. In applying the disclosure of these examples, it should be kept clearly in mind that other and different embodiments of the methods and synthetic schemes disclosed according to the present invention will no doubt suggest themselves to those of skill in the relevant art.
In the foregoing and in the following examples, all temperatures are set forth uncorrected in degrees Celsius; and, unless otherwise indicated, all parts and percentages are by weight.
The folloing abbreviations are used herein: Ac (acetyl), BINAP (2,2'- bis(diphenylphosphino)-l,l '-binaphthyl), Bn (benzyl), DCM (dichloromethane), DMF (dimethylformamide), DIPEA/ DIEA (N,N-diisopropyl ethyl amine), EDCI (1 -ethyl-3-[3- dimethylaminopropyl]carbodiimide hydrochloride), Et (ethyl), HOBT (1-hydroxybenzotriazole), Me (methyl), TFA (trifluoroacetic acid), THF (tetrahydrofuran), EtOAc (ethyl acetate), MeOH (methanol), Pd(OAc)2 (palladium acetate), K2CO3 (potassium carbonate), HCOONH4 (ammonium formate), Pd/C (palladium on carbon), Boc (tert-butoxycarbonyl), Na2SO4 (sodium sulfate), NaHCO3 (sodium bicarbonate) HCl (hydrochloric acid), HBr (hydrogen bromide), NaCl (sodium chloride), brine (saturated aqueous sodium chloride solution), CHCI3 (chloroform), Cs2CO3 (caesium carbonate, cesium carbonate), NaClO2 (sodium chlorite), NH3SO3 [NH2.SO3H] (sulphamic acid), NaOH (sodium hydroxide), Cbz (benzyloxy carbonyl), DMAP (4- (dimethylamino)pyridine), celite (diatomaceous earth), TLC (thin layer chromatography), NMR (nuclear magnetic resonance), DMSO-d6 (deuterated dimethyl sulfoxide), CDCI3 (deuterated chloroform), LC-MS (LC-MS liquid chromatography-mass spectrometry), HPLC (high pressure liquid chromatography or high performance liquid chromatography),
Intermediate 1 - Synthesis of 4-Oxo-piperidine-l-carboxylic acid tert-butyl ester
To a stirred solution of sodium carbonate (4.76 g, 0.0567 mole) in water (5.4 mL), was added 4-piperidone hydrochloride monohydrate (7.27 g, 0.0473 mole) in water (24mL) followed by dropwise addition of di-tert-butyl dicarbonate (10.5 g, 0.048 mole) over a period of 30 minutes. The reaction mixture was warmed to 35 0C for 1 hour then heated to 50 0C for 2.5 hours. The
reaction mixture was cooled to 1O0C. The solid precipitate so obtained was filtered, washed with water and dried to afford 8.6 g (81%) of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester. 1H NMR (DMSOd6): δ 3.6 (t, 4H), 2.4 (t, 4H), 1.4 (s, 9H).
Intermediate 2 - Synthesis of (2-Bromo-phenyl)-piperidin-4-yl-amine dihydrochloride
To a stirred solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (0.5 g, 0.0025 mole) in dry 1 ,2-dichloroethane (5 niL) under an atmosphere of nitrogen was added 2- bromoaniline (0.474 g, 0.00276 mole), acetic acid (0.18 g, 0.00301 mole) and sodium triacetoxyborohydride (0.638 g, 0.00301 mole). Stirring was continued at ambient temperature for 14 hours. The reaction mixture was quenched in cold aqueous IN NaOH solution and the product extracted with ether. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford the crude product as transparent viscous liquid. Purification by column chromatography using silica gel 60-120 mesh (2% ethyl acetate in hexane) afforded 0.32O g (35%) of desired 4-(2-bromo-phenylamino)-piperidine- 1 -carboxylic acid tert- butyl ester. 1H NMR (DMSOd6): δ 7.44-7.38 (m, IH), 7.2-7.12 (m, IH), 6.84-6.78 (m, IH), 6.58- 6.48 (m, IH), 4.65 (d, IH), 3.9 (m, 2H), 3.5 (m, IH), 3.0-2.8 (m, 2H), 2.0-1.8 (m, 2H), 1.4 (s,9H). To a cooled solution (O0C) of 4-(2-bromo-phenylamino)-piperidine-l -carboxylic acid tert-butyl ester (0.31 g, 0.00087 mole) in dioxane (0.5 mL) was added dioxane.HCl (2.5 mL). The mixture was stirred at the same temperature for 10 minutes, then gradually brought to ambient temperature with continued stirring for a further 15 minutes. The reaction mixture was evaporated under reduced pressure to afford a residue which was washed with dry ether to afford 0.106 g (42%) of 2-bromo-phenyl)-piperidin-4-yl-amine dihydrochloride.
Intermediate 3 - Synthesis of Piperidin-4-yl-(2-trifluoromethyl-phenyl)-amine dihydrochloride
To a stirred solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (0.20 g, 0.0001 mole) in dry 1,2-dichloroethane (7 mL) under an atmosphere of nitrogen for 10 minutes, was added 2-trifluoromethylaniline (0.161 g, 0.0001 mole), acetic acid (0.06 g, 0.0001 mole) and sodium triacetoxyborohydride (1.05 g, 0.0005 mole). The stirring was continued at ambient temperature for 14 hours. The reaction mixture was then quenched in cold aqueous IN NaOH solution and the product extracted with ether. The ether layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to get the crude product as transparent viscous liquid. Purification by column chromatography using silica gel 60-120 mesh (2% ethyl acetate in hexane) afforded 0.106 g (31%) of 4-(2-trifluoromethyl-phenylamino)- piperidine-1-carboxylic acid tert-butyl ester.
1H NMR (DMSOd
6): δ 7.4 (t, 2H), 7.0 (d, IH), 6.7 (t, IH), 3.9 (m, 2H), 2.9 (m, 2H), 1.9 (m, 2H), 1.4 (s, 9H). To a solution of 4-(2-trifluoromethyl- phenylamino)-piperidine-l-carboxylic acid tert-butyl ester (0.106 g, 0.000307 mole) in dioxane (0.5 mL) which was cooled to O
0C, was added, dioxane.HCl (2.5 mL) and the misture was stirred at the same temperature for 10 minutes. The reaction mixture was gradually brought to ambient temperature and stirring was continued for 15 minutes. The reaction mixture was evaporated under reduced pressure and the resulting residue was washed with dry ether to afford 0.04 g (41%) of piperidin-4-yl-(2-trifluoromethyl-phenyl)-amine dihydrochloride.'H NMR (DMSOd
6): δ 7.41 (t, 2H), 7.0 (d, 2H), 6.75 (t, IH), 4.8 (d, IH), 3.7 (bs, IH), 3.3 (bs, IH), 3.0 (m, 2H), 2.1 (m, 2H), 1.7 (m, 2H).
Intermediate 4 - Synthesis of Piperidin-4-yl-o-tolyl-amine dihydrochloride
To a stirred solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (Ig, 0.00502 mole) in dry 1,2-dichloroethane (10 mL) under an atmosphere of nitrogen was added o-toluidine (0.699 g, 0.00652 mole), acetic acid (0.301 g, 0.005 mole) and sodium triacetoxyborohydride
(1.596 g, 0.00753 mole) portionwise. The resulting mixture was stirred at ambient temperature for 16 hours. The mixture was then basified with sodium bicarbonate solution and the product extracted with dichloromethane. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford a viscous liquid which was purified by column chromatography using neutral aluminium oxide (0.5% ethyl acetate in hexane)
to afford 1.2 g (82%) of 4-O-tolylamino-piperidine-l-carboxylic acid tert-butyl ester. LCMS: 291.2 (M+l)+,%, 1H NMR (CDCl3): δ 7.1 (dd, 2H), 6.6 (t, 2H), 4.0 (d, 2H), 3.5-3.2 (m, 2H), 3.0- 2.8 (t, 2H), 2.1 (m, 4H), 1.45 (s, 9H). A solution of 4-O-tolylamino-piperidine-l-carboxylic acid tert-butyl ester (1.2 g, 0.00413 mole) in ethyl acetate.HCl (10 mL) was stirred at ambient temperature for 2 hours. The reaction mixture was then concentrated under reduced pressure and the resulting residue was washed with ether to afford 0.9 g (96%) of piperidin-4-yl-o-tolyl-amine dihydrochloride. LCMS: 191.15 (M+l)+, 98%
1H NMR (DMSO-D6): δ 9.5 (d, IH), 9.0 (d, IH), 7.3 (m, 4H), 3.7 (m, IH), 3.3 (d, 2H), 2.9 (q, 2H), 2.4 (s, 3H), 2.1 (m, 4H).
Intermediate 5 - Synthesis of (2-Chloro-phenyl)-piperidin-4-yl-amine dihydrochloride
To a stirred solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (0.5 g, 0.0025 mole) in dry 1,2-dichloroethane (5 mL) (under an atmosphere of nitogen for 10 minutes) was added, 2-chloroaniline (0.352 g, 0.0027 mole), acetic acid (0.125 g, 0.00209 mole) and sodium triacetoxyborohydride (0.442 g, 0.00209 mole). The resulting mixture was stirred at ambient temperature for 16 hours. The reaction mixture was quenched in cold aqueous IN NaOH solution and the product was extracted with dichloromethane. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was stirred with hexane. The hexane was then decanted and the residue was dried to afford 0.615 g (79%) of 4-(2-chloro-phenylamino)-piperidine-l-carboxylic acid tert-butyl ester as a solid. 1H NMR (DMSO-d6): δ 7.6 (dd, IH), 7.1 (t, IH), 6.8 (d, IH), 6.54 (t, IH), 4.8 (d, IH), 3.9 (d, 3H), 3.5 (m, IH), 2.9 (bs, 2H), 1.8 (d, 2H), 1.4 (s, 9H), 1.3 (d,lH). To a stirred, cooled (O0C) solution of 4-(2-chloro-phenylamino)-piperidine-l-carboxylic acid tert-butyl ester (0.150 g, 0.00048 mole) in dioxane (0.5 mL) was added dioxane.HCl (1.5 mL). The stirring was continued at the same temperature for 10 minutes, then the mixture was gradually brought to ambient temperature with continued stirring for a further 15 minutes. The reaction mixture was evaporated under reduced pressure and the resulting residue was washed with dry ether and dried to afford 0.118 g (99%) of (2-chloro-phenyl)-piperidin-4-yl-amine dihydrochloride.
Intermediate 6 - Synthesis of (2, 3-Dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride
To a stirred solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (Ig, 0.00502 mole) in dry 1 ,2-dichloroethane (10 mL) (under an atmosphere of nitogen for 10 minutes) was added 2,3-dimethylaniline (0.73 g, 0.00602 mole), acetic acid (0.301 g, 0.005 mole) and sodium triacetoxyborohydride (1.596 g, 0.00753 mole) portionwise with stirring. The stirring was continued at ambient temperature for a further 16 hours. The reaction mixture was basified with sodium bicarbonate solution and the product extracted with dichloromethane. The dichloromethane layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to as afford a transparent viscous liquid which was purified by column chromatography using neutral aluminium oxide (1% ethyl acetate in hexane) to afford 1.5 g (98%) of 4-(2, 3-dimethyl-phenylamino)-piperidine-l-carboxylic acid tert-butyl ester. 1H NMR (CDCI3): δ 7.0 (t, IH), 6.6 (m, 2H), 4.1 (m, 2H), 3.5 (m, IH), 3.0-2.9 (t, 2H), 2.3 (s, 3H), 2.1 (m, 2H), 2.0 (s, 3H), 1.5 (s, 9H), 1.4 (m, 2H). A solution of 4-(2, 3-dimethyl-phenylamino)-piperidme- 1 - carboxylic acid tert-butyl ester (1.45 g, 0.00476 mole) in ethyl acetate.HCl (15 mL) was stirred at ambient temperature for 2 hours. The reaction mixture was concentrated under reduced pressure and the resulting residue was washed with ether and dried to afford l.lg (83) of (2, 3-dimethyl- phenyl)-piperidin-4-yl-arnine dihydrochloride. LCMS: 205.16 (M+l)+, 95.98%, 1H NMR (DMSO- d6): δ 9.4 (d, IH), 9.0 (d, IH), 7.3 (m, 3H), 3.7 (m, 2H), 3.3 (d, 2H), 3.1 (bs, IH), 2.9 (m, 2H), 2.3 (d, 6H), 2.1 (m, 4H).
Intermediate 7 - Synthesis of (2,4-Dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride
To a stirred solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (Ig, 0.00502 mole) in dry 1,2-dichloroethane (10 mL) (under an atmosphere of nitogen for 10 minutes) was added 2,4-dimethylaniline (0.73 g, 0.00602 mole), followed by acetic acid (0.301 g, 0.005 mole) and sodium triacetoxyborohydride (1.596 g, 0.00753 mole) portionwise. The resulting mixture
was stirred at ambient temperature for a further 16 hours. The reaction mixture was basified with sodium bicarbonate solution and the product was extracted with dichloromethane. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting transparent viscous liquid was purified by column chromatography using neutral aluminium oxide (1% ethyl acetate in hexane) to 1.5 g (82%) of 4-(2,4-dimethyl- phenylamino)-piperidine-l-carboxylic acid tert-butyl ester. 1H NMR (CDCI3): δ 6.9 (d, 2H), 6.6 (d, IH), 4.1 (bs, 2H), 3.4 (m, IH), 3.2 (bs, IH), 2.9 (t, 2H), 2.2 (s, 3H), 2.1 (s, 3H), 2.1 (s, 3H), 2.0 (s, 2H), 1.5 (s, 9H), 1.4-1.2 (m, 2H). A solution of 4-(2,4-dimethyl-phenylamino)-piperidine-l- carboxylic acid tert-butyl ester (1.26 g, 0.00413 mole) in ethyl acetate.HCl (10 mL) was stirred at ambient temperature for 2 hours. The reaction mixture was concentrated under reduced pressure and the resulting residue was washed with ether and dried to afford 1.12 g (97%) of (2,4-dimethyl- phenyl)-piperidin-4-yl-amine dihydrochloride. LCMS: 205.16(M+l)+, 95.9%. 1H NMR (DMSO- d6): δ 9.2(d, IH), 8.2(d, IH), 7.2(m, 3H), 3.7(m, 3H), 3.3(d, 3H), 3.0(q, 2H), 2.3(m, 5H), 2.1(d, 2H), 1.9(d, 2H).
Intermediate 8 - Synthesis of (2,5-Dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride
A solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (1 g, 0.00502 mole) in dry 1,2-dichloroethane (10 mL) was stirred under an atmosphere of nitrogen for 10 minutes. 2,5- dimethylaniline (0.73 g, 0.00602 mole), acetic acid (0.301 g, 0.005 mole) and sodium triacetoxyborohydride (1.596 g, 0.00753 mole) were then added portionwise and stirring was continued at ambient temperature for 16 hours. The reaction mixture was basified with sodium bicarbonate solution and the product was extracted with dichloromethane. The dichloromethane layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting transparent viscous liquid was purified by column chromatography using neutral aluminium oxide (1% ethyl acetate in hexane) to afford 1.5 g (82%) of 4-(2,5-dimethyl- phenylamino)-piperidine-l-carboxylic acid tert-butyl ester.
1H NMR (CDCI
3): δ 7.0 (t, IH), 6.6 (d, 2H), 4.2 (dd, 2H), 3.5 (m, IH), 2.9 (t, 2H), 2.3 (t, 3H), 2.15 (s, 3H), 2.05 (s, 3H), 1.5 (s, 9H), 1.3 (t, 2H). A solution of 4-(2,5-dimethyl-phenylamino)-piperidine-l-carboxylic acid tert-butyl ester
(1.5 g, 0.00492 mole) in ethyl acetate.HCl (7 mL) was stirred at ambient temperature for 2 hours. The reaction mixture was concentrated under reduced pressure and the residue was washed with ether and dried to afford l.lg (80%) of (2,5-dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride. LCMS: 205.16(M+l)
+, 92.74%,
1H NMR (DMSO-d
6): δ 9.4 (d, IH), 9.0 (d, IH), 7.2 (d, 2H), 7.1 (s, IH), 3.7 (m, IH), 3.6 (s, IH), 3.4 (d, 2H), 3.2 (bs, IH), 2.9 (m, 2H), 2.4 (s, 3H), 2.3 (s, 3H), 2.1 (m, 4H).
Intermediate 9 - Synthesis of (2-tert-Butyl-phenyl)-piperidin-4-yl-amine dihydrochloride
A solution of 4-oxo-piperidine-l-carboxylic acid tert-butyl ester (1 g, 0.00502 mole) in dry
1,2-dichloroethane (10 mL) was stirred under an atmosphere of nitrogen for 10 minutes. 2-Tert- butylaniline (0.974 g, 0.00652 mole), acetic acid (0.301 g, 0.005 mole) and sodium triacetoxyborohydride (1.596 g, 0.00753 mole) were then added portionwise and stirring was continued at ambient temperature for 16 hours. The reaction mixture was basifϊed with sodium bicarbonate solution and the product was extracted with dichloromethane. The dichloromethane layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting transparent viscous liquid was purified by column chromatography using neutral aluminium oxide (1-2% ethyl acetate in hexane) to afford 1.4 g (83%) of 4-(2-tert-butyl- phenylamino)-piperidine-l-carboxylic acid tert-butyl ester. 1H NMR (CDCl3): δ 7.25 (m, IH), 7.15 (t, IH), 6.8-6.6 (m, 2H), 4.0 (dd, 2H), 3.9-3.8 (dd, 2H), 3.6 (bs, IH), 3.1 (t, 2H), 2.2-2.2 (dd, 2H), 1.5-1.4 (d, 18H). A solution of 4-(2-tert-butyl-phenylamino)-piperidine-l-carboxylic acid tert-butyl ester (1.35 g, 0.00406 mole) in ethyl acetate.HCl (10 mL) was stirred at ambient temperature for 2 hours. The reaction mixture was concentrated under reduced pressure and the resulting residue was washed with ether and dried to afford 0.9 g (72%) of (2-tert-butyl-phenyl)- piperidin-4-yl-amine dihydrochloride. LCMS: 233.19(M+1)\ 86.32%, 1H NMR (DMSOd6): δ 9.1 (d, 2H), 7.2 (d, IH), 7.1 (s, 3H), 6.8 (d, IH), 6.6 (t, IH), 3.7 (m, IH), 3.4 (m, 3H), 3.0 (q, 3H), 2.6 (t, IH), 2.2 (d, 2H), 1.7 (m, 2H), 1.4 (s, 9H).
Intermediate 10 - Synthesis of (2-Bromo-phenyl)-methyl-piperidin-4-yI-amine
To a solution of 4-(2-bromophenylamino)-piperidine-l-carboxylic acid tert-butyl ester (2.0 g, 0.00562 mole), in DMF (10 mL) was added NaH (60% w/w dispersion in oil) (0.9 g, 0.02251 mole) and the resulting mixtured was stirred at ambient temperature for 10 minutes under an atmosphere of nitrogen. Methyl iodide (3.19 g, 0.0225 mole) was then added and stirring was continued for 30 minutes at ambient temperature. The reaction mixture was quenched with aqueous NH4CI solution and the product was extracted with ether. The ether layer with washed with aqueous NaHCO3 solution followed by brine solution. The ether layer collected was dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was washed with ether and dried to afford 0.893g (45%) of 4-[(2-bromo-phenyl)-methyl-amino]-piperidine-l- carboxylic acid tert-butyl ester. 1H NMR (CDCl3 ): δ 7.6-7.54 (m, IH), 7.28-7.2 (m, IH), 7.14- 7.08 (m, IH), 6.96-6.88 (m, IH), 4.2-4.0 (m, 2H),3.3-3.2 (m, IH), 2.8-2.7 (m, 2H), 2.7 (s,3H), 1.7-1.6 (m, 2H), 1.6 (m, IH), 1.45 (s, 9H). A solution of 4-[(2-bromo-phenyl)-methyl-amino]- piperidine-1-carboxylic acid tert-butyl ester (0.89 g, 0.0024 mole) in dioxane.HCl was stirred for 30 minutes. The reaction was concentrated under reduced pressure to afford 0.704 g (95%) of (2- bromo-phenyl)-methyl-piperidin-4-yl-amine. 1H NMR (DMSOd6): δ 9.0 (bd, 3H), 7.6 (m, IH), 7.4 (m, 2H), 7.0 (m, IH), 3.2 (m, 3H), 2.9 (m, 2H), 2.6 (s, 3H), 1.9 (m, 3H).
Intermediate 11 - Synthesis of (2-Chloro-phenyl)-methyl-piperidin-4-yl-amine hydrochloride
To a solution of 4-(2-chloro-phenylamino)-piperidine-l-carboxylic acid tert-butyl ester
(0.166 g, 0.00053 mole), in DMF (10 mL) was added, NaH (60% w/w dispersion in oil) (0.0512 g, 0.0021 mole) and the resulting mixture was stirred at ambient temperature for 10 minutes under an atmosphere of nitrogen. Methyl iodide (0.303 g, 0.0021 mole) was then added, and the stirring was continued for 30 minutes at ambient temperature. The reaction mixture was heated to 45 0C
for 30 minutes. The reaction mixture was then quenched with aqueous 10% NH4Cl solution and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with aqueous NaHCO3 solution followed by brine solution. The organic layer was collected, dried over sodium sulfate and concentrated under reduced pressure to afford 0.08g (46%) of 4-[(2-chloro-ρhenyl)- methyl-ammoj-piperidine-l-carboxylic acid tert-butyl ester.1!! NMR (CDCl3 ): δ 7.6-7.54 (m, IH), 7.28-7.2 (m, IH), 7.14-7.08 (m, IH), 6.96-6.88 (m, IH), 4.2-4.0 (m, 2H), 3.3-3.2 (m, IH), 2.8-2.7 (m, 2H), 2.7 (s,3H), 1.7-1.6 (m, 2H), 1.6 (m, IH), 1.45 (s, 9H). A solution of 4-(2-chloro- phenylamino)-piperidine-l-carboxylic acid tert-butyl ester (0.078 g, 0.00024 mole) in dioxane.HCl (1 mL) was stirred for 30 minutes. The reaction mixture was then concentrated under reduced pressure to afford 0.072 g (99%) of (2-chloro-phenyl)-methyl-piperidm-4-yl-amine hydrochloride which was used in the next step without further purification.
Intermediate 12 - Synthesis of Methyl-piperidin-4-yl-(2-trifluoromethyl-phenyl)- amine hydrochloride
To a solution of 4-(2-trifluoromethyl-phenylamino)-piperidine-l-carboxylic acid tert-butyl ester (0.5 g, 0.00053 mole), in DMF (5 mL) was added, NaH (60% w/w dispersion in oil) (0.0696 g, 0.0029 mole) and the resulting mixture was stirred at ambient temperature for 10 minutes under an atmosphere of nitrogen. Methyl iodide (0.617 g, 0.00435 mole) was then added, and stirring was continued for 30 minutes at ambient temperature. The reaction mixture was quenched with aqueous 10% NH4 Cl solution and the product was extracted with ether. The organic layer was washed with aqueous NaHCO3 solution followed by brine solution. The ether layer was then collected, dried over sodium sulfate and concentrated under reduced pressure to afford 0.5 g of 4- [methyl-(2-trifluoromethyl-phenyl)-amino]-piperidine-l-carboxylic acid tert-butyl ester that which was used in the next step without further purification. 4-[Methyl-(2-trifluoromethyl-phenyl)- amino]-piperidine-l-carboxylic acid tert-butyl ester obtained (0.5 g) was stirred in dioxane.HCl (5 mL) for 30 minutes. The reaction mixture was concentrated under reduced pressure to afford 0.36 g (88%) of methyl-piperidin-4-yl-(2-trifluoromethyl-phenyl)-amine hydrochloride which was used
in the next step without further purification.
Intermediate 13 - Synthesis of 4-Methanesulfonyloxy-piperidine-l-carboxylic acid tert-butyl ester
To a stirred solution of 4-hydroxy-piperidine hydrochloride monohydrate (10 g, 0.0988 mole) in THF (80 mL) was added triethylamine (11.98 g, 16.48 mL 0.1186 mole) and the resulting mixture was cooled to 1O0C. Di-tert-butyl dicarbonate (23.68 g, 0.1086 mole) was added dropwise and stirring was continued at ambient temperature overnight. The reaction mixture was concentrated under reduced pressure and the resulting residue was acidified with concentrated HCl and extracted with ethyl acetate. The ethyl acetate layer was collected, dried over sodium sulfate and concentrated under reduced pressure to afford 19.47 g (98%) of 4-hydroxy-piperidine- 1- carboxylic acid tert-butyl ester.1!! NMR (DMSOd6): δ 4.7 (d, IH), 3.7-3.5 (m, 3H), 3.0-2.8 (t, 2H), 1.7-1.6 (m, 2H), 1.4 (s, 9H), 1.3-1.2 (m, 2H). To a stirred solution of 4-hydroxy-piperidine- 1- carboxylic acid tert-butyl ester (10 g, 0.0497 mole) in THF (100 mL), was added triethylamine
(6.026 g, 8.29 mL, 0.0596 mole) and the resulting mixture was cooled to 0-5 0C. Methane sulfonyl chloride (6.76 g, 0.1086 mole) was then added dropwise over a period of 30 minutes and the mixture was maintained at 0-50C for 2hrs. The reaction mixture was then diluted with cold water and the product extracted with ethyl acetate. The ethyl acetate layer was washed with IN aqueous HCl solution, followed by saturated aqueous sodium bicarbonate solution and brine solution. The organic layer was collected, dried over sodium sulfate and concentrated under reduced pressure to afford 13.59 g (98%) of 4-methanesulfonyloxy-piperidine-l -carboxylic acid tert-butyl ester. 1H NMR (CDCl3): δ 5.0-4.9 (m, IH), 3.8-3.6 (m, 2H), 3.4-3.2 (m, 2H), 3.0 (s, 3H), 2.04-1.9 (m, 2H), 1.9-1.74 (m, 2H), 1.5-1.42 (s, 9H).
Intermediate 14 - Synthesis of 4-(2-Trifluoromethyl-phenoxy)-piperidine trifluoracetate
To a stirred solution of 2-trifluoromethyl-phenol (1 g, 0.00617 mole) in DMF (10 mL) was added cesium carbonate (4.01 g, 0.0123 mole), followed by 4-methanesulfonyloxy-piperidine-l- carboxylic acid tert-butyl ester (1.72 g, 0.00616 mole). The reaction mixture was heated at 60
0C overnight. The mixture was then diluted with water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (0.1% ethyl acetate in hexane) to afford 0.72 g (34%) of 4-(2-trifluoromethyl-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester. LCMS: 346.16 (M+l)
+, 94.42%. A solution of 4-(2-trifluoromethyl-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester (0.72 g, 0.00208 mole) in dichloromethane was stirred at 0-5
0C. To the cold solution was added TFA (1 mL) dropwise and stirring was continued at 10
0C for 2 hours. The reaction mixture was concentrated under reduced pressure and the resulting residue was washed with ether and dried to afford 0.22 g (99%) of 4-(2-trifluoromethyl-phenoxy)-piperidine trifluoracetate. LCMS: 360.1 (M+l)
+, 99.3%,
1H NMR (CDCl
3): δ 8.9-8.5 (bd, 2H), 7.62-7.6 (d, IH), 7.56-7.46 (t, IH), 7.1-7.02 (t, IH), 7.0-6.94 (d, IH), 5.0-4.8 (m, IH), 3.5-3.3 (s, 4H), 2.3-2.2 (S, 4H).
Intermediate 15 - Synthesis of 4-(2-ChIoro-phenoxy)-piperidine hydrochloride
To a stirred solution of 2-chlorophenol (20 g, 0.155 mole) in DMF (200 mL), was added cesium carbonate (101 g, 0.311 mole), followed by 4-methanesulfonyloxy-piperidine-l-carboxylic acid tert-butyl ester (43.45 g, 0.155 mole). The reaction mixture was heated at 65
0C for 7 hours. The mixture was then filtered and concentrated under reduced. The resulting residue was diluted with ice cold water and the solid obtained was filtered and washed with water. The solid was dissolved in ether, washed with 2.5N aqueous NaOH solution, dried over sodium sulfate and concentrated under reduced pressure to afford 24.24 g (50%) of 4-(2-chloro-phenoxy)-piperidine- 1-carboxylic acid tert-butyl ester.LCMS: 312.13 (M+l)
+, 98.35%. A solution of 4-(2-chloro- phenoxy)-piperidine-l-carboxylic acid tert-butyl ester (24.2 g, 0.0776 mole) in dioxane.HCl (30 mL), was stirred at ambient temperature for 2 hours. The reaction mixture was then concentrated under reduced pressure and the resulting residue was washed with hexane twice to afford 19.13g
(99.6%) of 4-(2-chloro-phenoxy)-piperidine hydrochloride. LCMS: 248.05 (M+l)
+, 92.79%.
1H NMR (DMSO-d
6): δ 9.3-8.9 (bs, 2H), 7.5-7.42 (d, IH), 7.38-7.24 (m, 2H), 7.04-6.94 (t, 2H), 4.8- 4.7 (m, IH), 3.3-3.0 (bd, 4H), 2.2-2.05 (bs, 2H), 2.0-1.8 (bs, 2H).
Intermediate 16 - Synthesis of 4-(2-Bromo-phenoxy)-piperidine trifluoroacetate
To a stirred solution of 2-bromophenol (0.5 g, 0.00289 mole) in DMF (4 mL) was added potassium carbonate (0.478 g, 0.003468 mole), followed by 4-methanesulfonyloxy-piperidine-l- carboxylic acid tert-butyl ester (0.888 g, 0.00318 mole). The reaction mixture was heated at 80 0C for 6 hours. The mixture was then diluted with water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (2% ethyl acetate in hexane) to afford 0.64g (68%) of 4-(2-bromo-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester. LCMS: 356.08 (M+l)+, 99.56%. A solution of 4-(2-bromo-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester (0.64 g, 0.0018 mole) in dichloromethane (6.4 mL) was stirred at 0-5 0C. To the cold solution was added TFA (3.2 mL) dropwise and stirring was continued at ambient temperature for 3 hours. The reaction mixture was concentrated under reduced pressure and the resulting residue was washed with hexane to afford 0.64 g (96%) of 4-(2-bromo-phenoxy)-piperidine trifluoroacetate. LCMS: 370.02 (M+l)+, 91.37%. 1H NMR (CDCl3-D2O): 5 7.6-7.54 (d, IH), 7.32-7.26 (d, IH), 6.94-6.86 (d, 2H), 4.8-4.7 (bs, IH), 3.5-3.4 (m, 2H), 3.3-3.2 (m, 2H), 2.2-2.1 (s, 4H).
Intermediate 17 - Synthesis of 4-(2,5-Difluoro-phenoxy)-piperidine hydrochloride
To a stirred solution of 2,5-difluorophenol (1.0 g, 0.0076 mole) in DMF (20 mL) was added cesium carbonate (12.4 g, 0.038 mole) followed by 4-methanesulfonyloxy-piperidine-l- carboxylic acid tert-butyl ester (2.1 g, 0.076 mole). The reaction mixture was heated at 80
0C overnight. The reaction mixture was then diluted with water and the product extracted with ethyl
acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate, and concentrated. The resulting residue was purified by column chromatography using silica gel 60- 120 mesh (10% ethyl acetate in hexane) to afford 1.5 g (63.0%) of 4-(2,5-Difluoro-phenoxy)- piperidine-1-carboxylic acid tert-butyl ester. LCMS: 356.08 (M+l)
+, 85%.
1H NMR (CDCl
3): δ 7.0 (m, IH), 6.6 (m, IH), 6.5 (m, IH), 4.5 (m, IH), 3.7 (m, 2H), 3.4 (m, 4H), 2 (m, 2H), 1.9 (m, 2H). 1.5 (s, 9H). A solution of 4-(2,5-difluoro-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester (1.5 g, 0.0047 mole) in dioxane.HCl (2 mL) was stirred at ambient temperature for 1 hour. Ether was then added, and the resulting precipitate was isolated by filtration and dried to afford 0.65 g (65%) of 4-(2,5-difluoro-phenoxy)-piperidine hydrochloride. LCMS: 214.1 (M+l)
+,100%.
1H NMR (CDCl
3): δ 9.2 (bs, 2H), 7.4 (m, 2H), 6.9 (m, IH), 4.8 (m, IH), 3.2 (m, 2), 3.0 (m, 2H), 2.2 (m, 2H), 1.9 (m, 2H).
Intermediate 18 - Synthesis of 4-(2-Chloro-5-fluoro-phenoxy)-piperidine hydrochloride
To a stirred solution of 2-chloro-5-fluorophenol (6 g, 0.0413 mole) in DMF (20 mL) was added cesium carbonate (26.89 g, 0.00827 mole) followed by 4-methanesulfonyloxy-piperidine-l- carboxylic acid tert-butyl ester (11.54 g, 0.04137 mole). The reaction mixture was heated at 80 0C for overnight. The mixture was then diluted with water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 230-400 mesh (2% ethyl acetate in hexane) to afford 8.9 g (65%) of4-(2-chloro-5-fluoro-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester. LCMS: 330.12 (M+ 1)+, 100%. A solution of 4-(2-chloro-5-fluoro-phenoxy)-piperidine-l-carboxylic acid tert- butyl ester (8.9 g, 0.027 mole) in ethyl acetate.HCl (10 mL) was stirred at ambient temperature for 2 hours. The reaction mixture was then concentrated under reduced pressure and the resulting residue was washed with ether to afford 6.54 g (92%) of . LCMS: 230.07 (M+l)+,100%, 1H NMR (DMSOd6): δ 9.6-9.1 (bs, 2H), 7.55-7.45 (t, IH), 7.35-7.2 (d, IH), 6.9-6.8 (t, IH), 4.9-4.7 (s, IH), 3.26-3.0 (s, 4H), 2.2-2.06 (s, 2H), 2.0-1.8 (s, 2H).
Intermediate 19 - Synthesis of 4-(2-Nitro-phenoxy)-piperidine hydrochloride
To a stirred solution of 2-nitrophenol (1.5 g, 0.010 lmole) in DMF (15 mL) was added cesium carbonate (16.4 g, 0.0505 mole) followed by 4-methanesulfonyloxy-piperidine-l- carboxylic acid tert-butyl ester (3.0 g, 0.0101 mole). The reaction mixture was heated at 800C overnight. The mixture was then diluted with water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate, and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (20% ethyl acetate in hexane) to afford 1.5 g (47%) of 4-(2-nitro-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester. LCMS: 323.15 (M+l)+, 98%,
1H NMR (CDCl3): δ 7.8 (m, IH), 7.6 (m, IH), 7.0 (m, 2H), 4.7 (m, IH), 3.6 (m, 4H), 1.9 (m, 4H), 1.5 (s, 9H). A solution of 4-(2-nitro-phenoxy)-piperidine-l-carboxylic acid tert-butyl ester (1.5 g, 0.00465 mole) in dioxane.HCl (1OmL) was stirred at ambient temperature for 1 hour. Ether was then added the resulting precipitate was isolated by filtration and dried to afford 1 g (99%) of 4-(2-nitro-phenoxy)-piperidine hydrochloride. LCMS: 223.1 (M+l)+, 55%, 1H NMR (DMSO-d6): δ 9.0 (bs, 2H), 7.9 (m, IH), 7.8 (m, IH), 7.4 (m, IH), 7.0 (m, IH), 5.0 (m, IH), 3.2 (m, 4H), 2.2 (m,2H), 2.0 (m, 2H).
Intermediate 20 - Synthesis of 4-(2-Bromo-phenylsulfanyl)-piperidine hydrochloride
To a stirred solution of 2-bromobenzenethiol (1.0 g, 0.00529 mole) in DMF (10 mL) was added cesium carbonate (2.063 g, 0.0063 mole) followed by 4-methanesulfonyloxy-piperidine-l- carboxylic acid tert-butyl ester (1.478 g, 0.00529 mole). The reaction mixture was heated at 800C overnight. The mixture was then diluted with water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was washed with hexane to afford 1.93 g (98%) of 4-(2-bromo-phenylsulfanyl)-piperidine-l-carboxylic acid tert-butyl ester. LCMS:
372.06 (M+l)+, 92.78%. A solution of 4-(2-bromo-phenylsulfanyl)-piperidine-l-carboxylic acid tert-butyl ester (1.93 g, 0.0052 mole) in dioxane.HCl (3 mL), was stirred at ambient temperature for 30 minutes. The reaction mixture was then concentrated under reduced pressure and the resulting resiude was washed with hexane twice to afford 1.38g (99%) of 4-(2-bromo- phenylsulfanyO-piperidine hydrochloride. 1H NMR (DMSOd6): δ 9.1-8.9 (bs, IH), 7.7-7.64 (d, IH), 7.56-7.52 (d, IH), 7.46-7.36 (t, IH), 7.24-7.16 (t, IH), 3.7-3.6 (m, IH), 3.34-3.2 (bd, 3H), 3.1-2.94 (q, 2H), 2.16-2.04 (d, 2H), 1.84-1.66 (q, 2H).
Intermediate 21 - Synthesis of 4-(2-Trifluoromethyl-phenylsulfanyl)-piperidine hydrochloride
To a stirred solution of 2-trifluoromethylbenzenethiol (0.637 g, 0.00358 mole) in DMF (5 mL) was added cesium carbonate (1.396 g, 0.00429 mole) followed by 4-methanesulfonyloxy- piperidine-1-carboxylic acid tert-butyl ester (1.0 g, 0.00358 mole). The reaction mixture was heated at 80 0C overnight. The mixture was then diluted with water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed brine solution, dried over sodium sulfate, filtered and concentrated. The resulting residue was washed with hexane to afford 0.8 Ig (99%) of 4-(2-trifluoromethyl-phenylsulfanyl)-piperidine-l-carboxylic acid tert-butyl ester. LCMS: 362.13 (M+l)+, 99%. A solution of 4-(2-trifluoromethyl-phenylsulfanyl)-piperidine-l-carboxylic acid tert-butyl ester (0.81 g, 0.00224 mole) in dioxane.HCl (2 mL) was stirred at ambient temperature for 30 minutes. The reaction mixture was then concentrated under reduced pressure and the resulting residue was washed with hexane twice to afford 0.604g (90%) of 4-(2-trifluoromethyl- phenylsulfanyl)-piperidine hydrochloride which was used in the next step without further purification. LCMS: 262.08 (M+l)+, 95.07%.
Intermediate 22 - Synthesis of 4-(2-Chloro-phenylsulfanyI)-piperidine hydrochloride
To a stirred solution of 2-chlorobenzenethiol (2.5 g, 0.017 mole) in DMF (20 mL) was
added cesium carbonate (6.77 g, 0.0208 mole), followed by 4-methanesulfonyloxy-piperidine-l- carboxylic acid tert-butyl ester (4.8g, 0.0173 mole). The reaction mixture was heated at 80
0C overnight. The mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 4.64 g (82%) of 4-(2-chloro-phenylsulfanyl)- piperidine-1-carboxylic acid tert-butyl ester. LCMS: 328.11 (M+l)
+, 95.12%. A solution of 4-(2- chloro-phenylsulfanyl)-piperidine-l-carboxylic acid tert-butyl ester (4 g, 0.0122 mole) in dioxane.HCl (7 mL) was stirred at ambient temperature for 30 minutes. The reaction mixture was then concentrated under reduced pressure and the resulting residue was washed with hexane twice to afford 3.75g (99%) of 4-(2-chloro-phenylsulfanyl)-piperidine hydrochloride. LCMS : 228.05 (M+l)
+, 92.02%.
Intermediate 23 - Synthesis of [(BiphenyM-carbonvO-aminoJ-acetic acid
To a stirred solution of biphenyl-4-carboxylic acid (10 g, 0.05044 mole) in DMF (50 mL), was added DIPEA (22.82 g, 0.176 mol), HOBt (7.496 g, 0.0554 mol) and EDCLHCl (17.4 g, 0.09 mol) at ambient temperature. After 2 minutes glycine ethyl ester hydrochloride (8.45 g, 0.06 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was diluted with cold water and the resulting precipitate was isolated by filtration and dried to afford 14.26 g (99.8%) of [(biρhenyl-4-carbonyl)-amino]-acetic acid ethyl ester.
1H NMR (DMSOd
6): δ9.0(t, IH), 8.0-7.9(d, 2H), 7.84-7.7(dd, 4H), 7.5(t, 2H), 7.46-7.36(m, IH), 4.2-4. l(q, 2H), 4.0(d, 2H), 1.25(t, 3H). To a stirred solution of [(biphenyl-4-carbonyl)-amino]-acetic acid ethyl ester (14.26 g, 0.05 mol) in a mixture of THF (60 mL), methanol (60 mL) and H
2O (30 mL) was added LiOH.H
2O (12.68 g, 0.3023 mol) and the resulting mixture was stirred at ambient temperature fro 1 hour. The volatiles were then evaporated and the residue was acidified with 10%aqueous HCl solution. The resulting precipitate was isolated by filtrationwashed with water followed by hexane and dried to afford 12.8g (99%) of [(biphenyl-4-carbonyl)-amino] -acetic acid.
1H NMR (DMSO- d
6): δ 8.0-7.85(m, 3H), 7.8-7.7(m, 4H), 7.55-7.45(t, 2H), 7.45-7.35(t, IH), 3.5(d, 2H).
EXAMPLE 1 - Synthesis of Biphenyl-4-carboxylic acid {2-oxo-2-[4-(2- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino]-acetic acid (0.09128 g, 0.00036 mol) in DMF (2 mL) was added DIPEA (0.126g, 0.00098 mol), HOBt (0.05266 g,
0.000390 lmol) and EDCI.HC1 (0.07457g, 0.00039 mol) at ambient temperature. After 2 minutes 4-(2-trifluoromethyl-phenoxy)-piperidine trifluoroactetate (0.1 Ig, 0.00033 mol) was added and the resulting mixture was stirred at the same temperature overnight. The reaction mixture was diluted with cold water and the resulting precipitate was isolated by filtration and dried to afford 0.072 g (46%) of biphenyl-4-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-phenoxy)-piperidin-l-yl]- ethyl}-amide. LCMS: 483.18 (M+l)+, 95.73%, 1H NMR (CDCl3): δ 7.9 (d, 2H), 7.64-7.54 (m, 4H), 7.46-7.3 (m, 4H), 7.02-6.9 (q, 2H), 4.75 (s, IH), 4.4-4.2 (dd, 2H), 4.2-4.0 (m, 2H), 3.65-3.35 (m, 4H), 2.05-1.8 (m, 4H).
EXAMPLE 2 - Synthesis of Biphenyl-4-carboxylic acid {2-[4-(2-bromo-phenoxy)- piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino]-acetic acid (0.08278 g, 0.00032 mol) in DMF (2mL) was added, DIPEA (0.104g, 0.00081 mol), HOBt (0.0437 g, 0.00032 mol) and EDCLHCl (0.0619g, 0.00032 mol) at ambient temperature. After 2 minutes 4-(2-bromo- phenoxy)-piperidine trifluoroacetate (0.1 g, 0.00027 mol) was added and the resulting mixture was stirred at the same temperature overnight. The reaction mixture was diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by preparative HPLC [(column-Zorbax XDB C^-21.2x150mm, mobile
phase-0.1%TFA in water (A)/acetonitrile(B), gradient: (Time): (%B)-0:50; 2:50; 5:80)]) to afford 0.033 g (22%) of biphenyl-4-carboxylic acid {2-[4-(2-bromo-phenoxy)-piperidin-l-yl]-2-oxo- ethyl}-amide. LCMS: 493.1 (M+l)+, 98.35%, 1H NMR (DMSOd6): δ 8.65 (t, IH), 8.0 (d, 2H), 7.8 (m, 4H), 7.6 (dd, IH), 7.52 (t, 2H), 7.4 (m, 2H), 7.24 (m, IH), 6.95 (m,lH), 4.8 (m, IH), 4.3 (m, 2H), 3.7 (m, 2H), 3.55 (m, 2H), 1.9 (m, 2H), 1.7 (m, 2H).
EXAMPLE 3 - Synthesis of Biphenyl-4-carboxylic acid {2-[4-(2-bromo-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino]-acetic acid (0.075 g, 0.00029 mol) in DMF (2 mL), was added DIPEA (0.1139 g, 0.00088 mol) HOBt (0.0398 g, 0.00029 mol) and EDCI.HC1 (0.06758 g, 0.00035 mol) at ambient temperature. After 2 minutes (2-bromo-phenyl)- piperidin-4-yl-amine dihydrochloride (0.09639 g, 0.00029 mol) was added and the resulting mixture was stirred at the same temperature for overnight. The reaction mixture was diluted with cold water and the resulting precipitate was isolated by filtration, washed with hexane followed by ether and dried to afford 0.067 g (46%) of biphenyl-4-carboxylic acid {2-[4-(2-bromo- phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide. LCMS: 492.0 (M+l)
+, 90.08%,
1H NMR (DMSOd
5): δ 8.62 (t, IH), 7.94 (d, 2H), 7.79 (m, 4H), 7.5 (t, 2H), 7.4 (t, 2H), 7.2 (t, IH), 6.84 (d, IH), 6.57 (d,lH), 4.6 (d, IH), 4.3 (d, IH), 4.2 (d, 2H), 3.9 (d, IH), 3.6 (d, IH), 2.7 (t, IH), 1.9 (bs, 2H), 1.5-1.2(m, 3H).
EXAMPLE 4 - Synthesis of Biphenyl-4-carboxylic acid {2-[4-(2-chloro-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino]-acetic acid (0.134 g, 0.00053 mol)
in DMF (2 mL) was added DIPEA (0.185 g, 0.00143 mol), HOBt (0.0646 g, 0.00048 mol) and EDCI.HC1 (0.1098 g, 0.00057 mol) at ambient temperature. After 2 minutes (2-chloro-phenyl)- piperidin-4-yl-amine dihydrochloride (0.118g, 0.00048 mol) was added and the resulting mixture was stirred at the same temperature overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by preparative TLC using 30% ethyl acetate in hexane as eluent afforded 0.105g (45%) of biphenyl-4-carboxylic acid {2-[4- (2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide. LCMS: 448 (M+l)
+, 97.78%,
1H NMR (CDCl
3): δ 7.94 (d, 2H), 7.74-7.6 (m, 4H), 7.5-7.38 (m, 2H), 7.2-7.1 (m, IH), 6.75-6.6 (m, 2H), 4.5 (m, IH), 4.3 (d, 2H), 4.25 (d,lH), 3.9 (m, IH), 3.6 (m, IH), 3.3 (m, 2H), 3.1 (m, IH), 2.2 (bt, 2H), 1.5 (m, 2H).
EXAMPLE 5 - Synthesis of Biphenyl-4-carboxylic acid {2-oxo-2-[4-(2- trifluoromethyl-phenylamino)-piperidin-l-yl]-ethyl}-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino] -acetic acid (0.03219 g, 0.00012 mol) in DMF (1 mL), was added, DIPEA (0.065 g, 0.0005 mol), HOBt (0.017 g, 0.00012 mol) and EDCLHCl (0.2689 g, 0.00013 mol) at ambient temperature. After 2 minutes piperidin-4-yl-(2- trifluoromethyl-phenyl)-amine dihydrochloride (0.04 g, 0.00012 mol) was and the resulting mixture was stirred at the same temperature overnight. The reaction mixture was diluted with cold water, and the resulting precipitate was isolated by filtration. The solid was recrystallized from a mixture of ethyl acetate and hexane to afford 0.042g (61%) of biphenyl-4-carboxylic acid {2-oxo- 2-[4-(2-trifluoromethyl-phenylamino)-piperidin-l-yl]-ethyl} -amide. LCMS: 482.1 (M+l)
+, 97.99%,
1H NMR (DMSO-d
6): δ 8.6 (t, IH), 8.0 (m, 2H), 7.78-7.5 (m, 4H), 7.6-7.4 (m, 5H), 7.0 (d,lH), 6.7 (t, IH), 4.7 (d, IH), 4.3 (m, IH), 4.2 (d, 2H), 4.0-3.7 (m, 2H), 3.2 (m, 2H), 2.8 (m, 2H), 2.0 (m, 3H), 1.6-1.2 (m, 4H).
EXAMPLE 6 - Synthesis of Biphenyl-4-carboxylic acid (2-{4-[methyl-(2- trifluoromethyl-phenyl)-amino]-piperidin-l-yl}-2-oxo-ethyl)-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino] -acetic acid (0.075 g, 0.00029 mol) in DMF (3 mL), was added DIPEA (0.1519 g, 0.00117 mol), HOBt (0.03981 g, 0.00029 mol) and EDCLHCl (0.06758 g, 0.00035 mol) at ambient temperature. After 2 minutes methyl-piperidin-4- yl-(2-trifluoromethyl-phenyl)-amine hydrochloride (0.06959 g, 0.00026 mol) was added and the resulting mixture was stirred at the same temperature overnight. The reaction mixture was diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed aqueous sodium bicarbonate solution followed by brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by preparative HPLC to afford 0.023 g ( 16%) of biphenyl-4-carboxylic acid (2- {4-[methyl-(2-trifluoromethyl- phenyl)-amino]-piperidm-l-yl}-2-oxo-ethyl)-amide. LCMS: 496.2 (M+l)+, 98.28%, 1H NMR (DMSOd6): δ 8.6 (t, IH), 7.56-7.3 (m, 4H), 7.8-7.6 (m, 7H), 7.98 (d, 2H), 4.4 (bs, IH), 4.15 (d, 2H), 3.9 (bs, IH), 3.2-2.9 (m, 2H), 1.7 (bs, 2H), 1.5-1.2 (m, 2H).
EXAMPLE 7 - Synthesis of Biphenyl-4-carboxylic acid (2-{4-[(2-chloro-phenyl)- methyl-amino]-piperidin-l-yl}-2-oxo-ethyl)-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino] -acetic acid (0.0659 g, 0.00025 mol) in DMF (1 mL), was added DIPEA (0.0911 g, 0.0007 mol), HOBt (0.0318 g, 0.00023 mol) and EDCI.HC1 (0.054 g, 0.00028 mol) at ambient temperature. After 2 minutes (2-chloro-phenyl)- methyl-piperidin-4-yl-amine hydrochloride (0.07g, 0.00023 mol) was added and the resulting mixture was stirred at the same temperature overnight. The reaction mixture was diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with aqueous sodium bicarbonate solution followed by brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue obtained was purified by column chromatography using basic aluminium oxide (10-30% ethyl acetate in hexane) to afford 0.028 g
(23%) of biphenyl-4-carboxylic acid (2-{4-[(2-chloro-phenyl)-methyl-amino]-piperidin-l-yl}-2- oxo-ethyl)-amide. LCMS: 462.1 (M+l)+, 96.19%,'H NMR (DMSO-d6): δ 8.6 (t, IH), 7.95 (d, 2H), 7.75 (m, 3H), 7.5 (t, 2H), 7.42 (m, 2H), 7.3-7.2 (m, 2H), 7.05 (m, IH), 4.4 (d, IH), 4.2 (m, 2H), 3.95 (d, IH), 3.05 (t,lH), 2.65 (s,3H), 1.6 (bs,3H), 1.6-1.4 (m,lH).
EXAMPLE 8 - Synthesis of Biphenyl-4-carboxylic acid (2-{4-[(2-bromo-phenyl)- methyl-amino] -piperidin- 1 -yl}-2-oxo-ethyl)-amide
To a stirred solution of [(biphenyl-4-carbonyl)-amino]-acetic acid (0.1 g, 0.00039 mol) in DMF (2 mL), was added DIPEA (0.2025 g, 0.00156 mol), HOBt (0.053g, 0.00039 mol) and EDCLHCl (0.0901 g, 0.00047 mol) at ambient temperature. After 2 minutes (2-bromo-phenyl)- methyl-piperidin-4-yl-amine hydrochloride (0.119g, 0.00039 mol) was added and the resulting mixture was stirred at the same temperature overnight. The reaction mixture was diluted with cold water, and the resulting precipitate was isolated by filtration. Purification by preparative TLC using 30% ethyl acetate in hexane afforded 0.072g (36%) of biphenyl-4-carboxylic acid (2-{4-[(2- bromo-phenyl)-methyl-amino]-piperidin-l-yl}-2-oxo-ethyl)-amide. LCMS: 506.1 (M+l)+, 97.07%,^ NMR (DMSOd6): δ 8.6 (t, IH), 8.0 (d, 2H), 7.8 (d, 2H), 7.75 (m, 4H), 7.6-7.25 (m, 6H), 7.0 (t, IH), 4.4 (m, IH), 4.2 (m,lH), 3.9 (m, IH), 3.05 (m,lH), 2.7 (m,lH), 2.6 (s,3H), 1.8- 1.5 (m,4H).
Intermediate 24 - Synthesis of 2,4-Dioxo-4-phenyl-butyric acid ethyl ester
A stirred solution of acetophenone (5 g, 0.0416 mole), diethyl oxalate (7.23 g, 0.0416 mole) in DMF (40 mL) was cooled to O0C for 10 minutes. NaH (60% w/w dispersion in oil) (2.0 g, 0.083 mole) was then added, and the resulting mixture was stirred at the same temperature for 30 minutes, then stirred at ambient temperature for 1 hour. The mixture was then heated at 5O0C for 30 minutes. The reaction mixture was quenched with iced water, acidified with aqueous 2.4N HCl
solution and the resulting precipitate was isolated by filtration and dried to afford 3.8 g (42%) of 2,4-dioxo-4-phenyl-butyric acid ethyl ester. LCMS: 221.07 (M+l)+, 85.2%.
Intermediate 25 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid
To a stirred solution of 2, 4-dioxo-4-phenyl-butyric acid ethyl ester (3.86 g, 0.01754 mole) in methanol (78 mL) was added hydroxylamine hydrochloride (3.657 g , 0.0526 mole) at ambient temperature. The resulting mixture was then heated to reflux overnight. Volatiles were then removed by evaporation and the resulting residue was diluted with water and the product extracted with chloroform. The chloroform layer was collected and washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120mesh (4% ethyl acetate in hexane) to afford 2.8 g (79%) of 5-phenyl-isoxazole-3-carboxylic acid methyl ester. LCMS: 204.06 (M+l)+, 97.58%. To a stirred solution of 5-phenyl-isoxazole-3-carboxylic acid methyl ester (2.8 g, 0.01379 mol) in a mixture of THF (10 mL), methanol (10 mL) and H2O (1OmL) was added LiOH-H2O (0.87 g,
0.02073 mol) and the resulting mixture was stirred at ambient temperature for 2.5 hours. Volatiles were then evaporated and the resulting residue was diluted with water, acidified with concentrated HCl and extracted with ethyl acetate. The ethyl acetate layer was collected and washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 2.2g (85%) of 5-phenyl-isoxazole-3-carboxylic acid. LCMS: 190.04 (M+l )+, 96.4%.
Intermediate 26 - Synthesis of [(S-Phenyl-isoxazole-S-carbony^-aminoJ-acetic acid
To a stirred solution of S-phenyl-isoxazole-S-carboxylic acid (1 g, 0.00529 mol) in DMF (5 mL) was added DIPEA (2.73 g, 0.0211 mol), HOBt (0.89 g, 0.006078 mol) and EDCLHCl
(1.266 g, 0.00661 mol) at ambient temperature. After 2 minutes glycine ethyl ester hydrochloride
(0.811 g, 0.00581 mol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water and the resulting precipitate was isolated by filtration and dried to afford 0.744 g (56%) of 5-phenyl-isoxazole-3-carbonyl)-amino]- acetic acid ethyl ester. LCMS: 275.1 (M+l)+, 94%. To a stirred solution of [(5-phenyl-isoxazole- 3-carbonyl)-amino]-acetic acid ethyl ester (0.74 g, 0.00270 mol) in a mixture of THF (5 mL), methanol (5 mL) and H2O (5 mL) was added LiOH-H2O (0.34g, 0.00809 mol) and the resulting mixture was stirred at ambient temperature for 1 hour. Volatiles were then evaporated and the resulting residue was diluted with water and acidified with concentrated HCl. The resulting precipitate was isolated by filtration and dried to afford 0.57 g (76%) of [(5-phenyl-isoxazole-3- carbonyl)-amino] -acetic acid . LCMS: 247.06 (M+l)+, 98.18%.
EXAMPLE 9 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid {2-[4-(2-bromo- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-isoxazole-3-carbonyl)-amino]-acetic acid (72.5 mg,
0.00026 mol) in DMF (1.5 mL) was added DIPEA (0.166 g, 0.00128 mol), HOBt (43.3 mg, 0.00032 mol) and EDCI.HC1 (61.5 mg, 0.00032 mol) at ambient temperature. After 2 minutes 4- (2bromo-phenoxy)-piperidine trifluoroacetate (0.1 g, 0.00027 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel, 60-120 mesh (40% ethyl acetate in hexane) to afford 0.044 g (35%) of 5-phenyl-isoxazole-3-carboxylic acid (2-[4-(2-bromo-phenoxy)- piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 484.08 (M+l)+, 77.84%, 1H NMR (DMSO-d6): δ 8.63 (t, IH), 7.98 (m, 2H), 7.6 (m, 4H), 7.4 (m, 2H), 7.2 (m, IH), 6.9 (t, IH), 4.8 (s, IH), 4.3 (m, 2H), 3.5 (m, 4H), 1.5 (m, 4H).
EXAMPLE 10 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid {2-[4-(2-bromo- phenylamino)-piperidin-l-yl]-2-oxo-ethyI}-amide
To a stirred solution of [(5-phenyl-isoxazole-3-carbonyl)-amino]-acetic acid (0.075 g, 0.00027 mol) in DMF (2 mL), was added DIPEA (171.5 mg, 0.00133 mol), HOBt (44.8 mg, 0.00033mol) and EDCLHCl (0.064 g, 0.00033 mol) at ambient temperature. After 2 minutes (2- brorno-phenyl)-piperidin-4-yl-amine dihydrochloride (0.078 g, 0.00026 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using basic aluminium oxide (35% ethyl acetate in hexane) to afford 0.028 g (22%) of S-phenyl-isoxazole^-carboxylic acid {2-[4-(2-bromo- phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 483.1 (M+l)+, 97.4%, 1H NMR (DMSOd6): δ 8.6 (t, IH), 7.85 (m, 2H), 7.6 (m. 3H), 7.4 (m, 2H), 7.2 (t, IH), 6.85 (d, IH), 6.55 (t, IH), 4.7 (d,lH), 4.3 (d, IH), 4.2 (d, 2H), 3.9 (d, IH), 3.6 (m, IH), 3.2 (m, IH), 2.8 (m, IH), 2.0 (t, 2H), 1.4 (m, 2H).
EXAMPLE 11 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid {2-[4-(2-chloro- phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-isoxazole-3-carbonyl)-amino]-acetic acid (0.075 g, 0.00027 mol) in DMF (2 mL) was added DIPEA (0.171 g, 0.00132 mol), HOBt (0.0448 g, 0.00033 mol) and EDCI.HC1 (0.064 g, 0.00033 mol) at ambient temperature. After 2 minutes piperidin-4-yl-(2-chloro-phenyl)-amine dihydrochloride (0.066g, 0.00027 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using basic aluminium oxide (25% ethyl acetate in hexane) to afford 0.066 g (57%) of 5-phenyl-isoxazole-3-carboxylic acid {2-[4-(2-chloro-
phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide.LCMS: 439.15 (M+l)
+, 97.26%,
1H NMR (DMSO-de): δ 8.62 (t, IH), 7.95 (m, 2H), 7.55 (m, 3H), 7.4 (s, IH), 7.25 (dd, IH), 7.2 (m, 2H), 6.85 (d, IH), 6.6 (t, IH), 4.9 (d, IH), 4.4 (d,lH), 4.2 (d, 2H), 3.9 (d, IH), 3.7 (m, IH), 3.2 (m, IH), 2.8 (m, 2H), 1.95 (t, 2H), 1.4 (m, 2H).
EXAMPLE 12 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid (2-{4-[(2-bromo- phenyl)-methyl-amino]-piperidin-l-yl}-2-oxo-ethyl)-amide
To a stirred solution of [(5-phenyl-isoxazole-3-carbonyl)-amino]-acetic acid (62.9 mg, 0.00025 mol) in DMF (2 mL) was added DIPEA (0.159 g, 0.00123 mol), HOBt (41.4 mg,
0.00031 mol) and EDCLHCl (58.8 mg, 0.0003' mol) at ambient temperature. After 2 minutes (2- bromo-phenyl)-methyl-piperidin-4-yl-amine hydrochloride (0.068 g, 0.00025 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using basic aluminium oxide (50-100% ethyl acetate in hexane) afforded 0.039 g (35%) of 5- phenyl-isoxazole-3-carboxylic acid (2-{4-[(2-bromo-phenyl)-methyl-amino]-piperidin-l-yl}-2- oxo-ethyl)-amide. LCMS: 497.11 (M+l)+, 97.87%, 1H NMR (DMSO-d6): δ 8.58 (t, IH), 7.93 (m, 2H), 7.52 (m, 4H), 7.4 (s, IH), 7.15 (m, 2H), 6.9 (m, IH), 4.3 (d, IH), 4.15 (t,2H), 3.8 (d, IH), 3.0 (m, IH), 2.65 (m, IH), 2.55 (s, 3H), 1.75 (m, 3H), 1.5(m, 2H).
EXAMPLE 13 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid {2-oxo-2-[4-(2- trifluoromethyl-phenylamino)-piperidin-l-yl]-ethyl}-amide
To a stirred solution of [(5-phenyl-isoxazole-3-carbonyl)-amino] -acetic acid (0.0685 g, 0.00024 mol) in DMF (2 mL) was added DIPEA (0.173 g, 0.00134 mol), HOBt (0.045 g, 0.00034 mol) and EDCLHCl (0.064g, 0.00034 mol) at ambient temperature. After 2 minutes piperidin-4- yl-(2-trifluoromethyl-phenyl)-amine dihydrochloride (0.068 g, 0.00024 mol) was added and the
resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was collected and washed with brine solution, dried over sodium sulfate and concentrated .The resulting residue was purified by column chromatography using neutral aluminium oxide (35% ethyl acetate in hexane) to afford 0.0684 g (60%) of S-phenyl-isoxazole-S-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl- phenylamino)-piperidin-l-yl]-ethyl}-amide. LCMS: 473.17 (M+l)+, 97.41%, 1H NMR (DMSO- d6): δ 8.62 (t, IH), 7.94 (m, 2H), 7.52 (m, 3H), 7.38 (m, 3H), 6.98 (d, IH), 6.7 (t, IH), 4.7 (d, IH), 4.3 (d,lH), 4.2 (d, 2H), 3.9 (d, IH), 3.7 (m, IH), 3.2 (m, IH), 2.8 (m, IH), 2.0 (m, 2H), 1.4 (m, 2H).
EXAMPLE 14 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid (2-{4-[methyl-(2- trifluoromethyl-phenyl)-amino]-piperidin-l-yl}-2-oxo-ethyl)-amide
To a stirred solution of [(5-phenyl-isoxazole-3-carbonyl)-amino]-acetic acid (0.0576 g, 0.00021 mol) in DMF (1 mL), was added DIPEA (0.1317 g, 0.0010 mol), HOBt (0.035 g, 0.00025 mol) and EDCI.HC1 (0.049g, 0.00025 mol) at ambient temperature. After 2 minutes methyl-piperidin-4-yl-(2-trifluoromethyl-phenyl)-amine hydrochloride (0.06 g, 0.00021 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product extracted with ethyl acetate. The ethyl acetate layer was collected and washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by preprative HPLC [(column-Zorbax SB Ci8-21.2x150mm, mobile phase-0.1%TFA in water (A)/acetonitrile (B), gradient: (Time): (%B)-0:30; 2:50; 8:80)]) to afford 0.015 g (15%) of S-phenyl-isoxazole-S-carboxylic acid (2-{4-[methyl-(2-trifluoromethyl- phenyl)-amino]-piperidin-l-yl}-2-oxo-ethyl)-amide. LCMS: 487.19 (M+l)+, 98.16%, 1H NMR (DMSO-d6):δ 8.58 (t, IH), 7.94 (m, 2H), 7.7 (m, 2H), 7.56 (m, 3H), 7.34 (m, 2H), 4.3 (m, IH), 4.15 (d, 2H), 3.8 (d,lH), 3.0 (m, 2H), 2.6 (s, 3H), 1.7 (m, 2H), 1.4 (m, IH), 1.3 (m, 2H).
Intermediate 27 - Synthesis of 4-Phenylamino-benzoic acid
A mixture of BINAP (1.6 g, 0.00257 mole), palladium acetate (0.23 g, 0.001 mole) and toluene (30 mL) was degassed with argon for 15 minutes. This mixture was then added to a mixture of aniline (5.0 g, 0.0536 mole), 4-bromobenzoic acid (12.9 g, 0.0644 mole) and cesium carbonate (52.47 g, 0.161 mole) in toluene (30 mL) (previously degassed with argon for 15 minutes). The resulting mixture was heated at reflux for 22 hours. The reaction mixture was then concentrated under reduced pressure. The resulting residue was acidified with 10% HCl and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography using silica gel 60-120 mesh (15% ethyl acetate in hexane) to afford 9.55 g (83%) of 4-phenylamino-benzoic acid.
Intermediate 28 - Synthesis of (4-Phenylamino-benzoylamino)-acetic acid
To a stirred solution of 4-phenylamino-benzoic acid (3.0 g, 0.01401 mol) in DMF (10 mL), was added DIPEA (5.4 g, 0.04205 mol), HOBt (2.27 g, 0.01682 mol) and EDCLHCl (6.69 g, 0.03504 mol) at ambient temperature. After 2 minutes amino-acetic acid ethyl ester hydrochloride (2.33 g, 0.01682 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration, washed with water followed by hexane and dried to afford 4 g (95%) of (4-phenylamino- benzoylamino)-acetic acid ethyl ester. To a stirred solution (4-phenylamino-benzoylamino)-acetic acid ethyl ester (4 g, 0.01337 mol) in a mixture of THF (15 mL), methanol (15 mL) and H
2O (8mL) was added LiOH. H
2O (2.24 g, 0.0535 mol). The resulting mixture was stirred at ambient temperature for 3 hours and then concentrated. The residue was diluted with water and acidified with 10% aqueous citric acid solution. The the resulting precipitate was isolated by filtration and dried to afford 3.3 g (91%) of (4-phenylamino-benzoylamino)-acetic acid.
EXAMPLE 15 - Synthesis of N-{2-Oxo-2-[4-(2-trifluoromethyl-phenyIamino)- piperidin-l-yl]-ethyl}-4-phenylamino-benzamide
To a stirred solution of (4-phenylamino-benzoylamino)-acetic acid (0.058 g, 0.00021 mol) in DMF (5 niL) was added DIPEA (0.069 g, 0.00054 mol), HOBt (0.028 g, 0.0002142 mol) and EDCI.HC1 (0.085 g, 0.00045 mol) at ambient temperature. After 2 minutes piperidin-4-yl-(2- trifluoromethyl-phenyl)-amine dihydrochloride (0.050 g, 0.00019 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was collected, washed with brine solution, dried over sodium sulfate and concentrated under reduced. The resulting residue was purified by column chromatography using silica gel 60-120mesh (60% ethyl acetate in hexane) to afford 0.06 g (68%) of N-{2-oxo-2-[4-(2-trifluoromethyl-phenylamino)-piperidin-l- yl]-ethyl}-4-phenylamino-benzamide. LCMS: 497.21 (M+l)+, 95.3%, 1H NMR (DMSOd5): δ 8.6 (s, IH), 8.2 (t, IH), 7.8 (d, 2H), 7.4 (m, 4H), 7.2 (d, 2H), 7.1 (m, 3H), 6.9 (t, IH), 6.7 (t,lH), 4.7 (d, IH), 4.3 (d, IH), 4.1 (s, 2H), 3.9 (d, IH), 3.7 (s, IH), 2.8 (t, IH), 2.0 (t, 2H), 1.5 (m,2H).
EXAMPLE 16 - Synthesis of N-{2-[4-(2-Chloro-phenylamino)-piperidin-l-yl]-2-oxo- ethyl}-4-phenyIamino-benzamide
To a stirred solution of (4-phenylamino-benzoylamino)-acetic acid (0.083 g, 0.00031 mol) in DMF (5 mL) was added DIPEA (0.099 g, 0.00077 mol), HOBt (0.041 g, 0.000307 mol) and EDCI.HC1 (0.073 g, 0.00038 mol) at ambient temperature. After 2 minutes (2-chloro-phenyl)- piperidin-4-yl-amine dihydrochloride (0.063 g, 0.00026 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated. The residue was purified by column chromatography using silica
gel 60-120mesh (60% ethyl acetate in hexane) to afford 0.04g (33%) of N-{2-[4-(2-chloro- phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-4-phenylamino-benzamide. LCMS: 463.17 (M+ 1)
+, 95.82%,
1H NMR (DMSOd
6): δ 8.6 (s, IH), 8.2 (t, IH), 7.8 (d, 2H), 7.5 (q, 3H), 7.2 (t, 3H), 7.1 (d, 2H), 6.9 (q, 2H), 6.6 (t, IH), 4.9 (d, IH), 4.3 (d, IH), 4.1 (d, 2H), 3.9 (d, IH), 3.6 (m, IH), 3.2 (m, IH), 2.8 (m, 2H), 2.0 (t,3H).
EXAMPLE 17 - Synthesis of N-{2-[4-(2-Bromo-phenylamino)-piperidin-l-yl]-2-oxo- ethyl}-4-phenylamino-benzamide
To a stirred solution of (4-phenylamino-benzoylamino)-acetic acid (0.074 g, 0.00027 mol) in DMF (5 mL) was added DIPEA (0.088 g, 0.0006827 mol), HOBt (0.036 g, 0.00027 mol) and EDCI.HC1 (0.065 g, 0.00034 mol) at ambient temperature. After 2 minutes (2-bromo-phenyl)- piperidin-4-yl-amine dihydrochloride (0.066 g, 0.00023 mol) and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120mesh (60% ethyl acetate in hexane) to afford 0.036 g (31%) ofN-{2-[4-(2-bromo-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-4-phenylamino- benzamide. LCMS: 507.13 (M+l)
+, 96.75%,
1H NMR (DMSOd
6): δ 8.6 (s, IH), 8.2 (t, IH), 7.8 (d, 2H), 7.4 (dd, IH), 7.3 (t, 2H), 7.2 (m, 3H), 7.1 (d, 2H), 6.9 (t, IH), 6.8 (d,lH), 6.6 (t, IH), 4.6 (d, IH), 4.3 (d, IH), 4.1 (d, 2H), 3.9 (d, IH), 3.6 (s, IH), 3.2 (m, 3H), 2.8 (m,2H), 2.0 (m,3H).
EXAMPLE 18 - Synthesis of N-(2-{4-[Methyl-(2-trifluoromethyl-phenyl)-amino]- piperidin-l-yl}-2-oxo-ethyl)-4-phenylamino-benzamide
To a stirred solution of (4-phenylamino-benzoylamino)-acetic acid (0.051 g, 0.00019 mol)
in DMF (5 mL) was added DIPEA (0.061 g, 0.00048 mol), HOBt (0.025 g, 0.00019 mol) and EDCI.HC1 (0.076 g, 0.00040 mol) at ambient temperature. After 2 minutes methyl-piperidin-4-yl- (2-trifluoromethyl-phenyl)-amine hydrochloride (0.047 g, 0.00016 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by preparative HPLC [(column-Zorbax SB Ci8-21.2xl50mm, mobile phase-0.1%TFA in water (A)/ Acetonitrile (B), gradient: (Time): (%B)-0:30; 2:50; 8:80)]) to afford 0.031 (38%) of N- (2- {4-[methyl-(2-trifluoromethyl-phenyl)-amino]-piperidin- 1 -yl } -2-oxo-ethyl)-4-phenylamino- benzamide. LCMS: 511.22 (M+l)+, 99.08%, 1H NMR (DMSOd6): δ 8.6 (s, IH), 8.2 (t, IH), 7.8 (d, 2H), 7.7 (m, 3H), 7.4 (m, 3H), 7.2 (d, 2H), 7.1 (d, 2H), 6.9 (t, IH), 4.4 (d,lH), 4.1 (d, 2H), 3.9 (d, IH), 3.1 (m, 3H), 2.6 (s, 4H), 1.8 (d,2H), 1.4 (m,lH).
EXAMPLE 19 - Synthesis of N-{2-[4-(2-Bromo-phenoxy)-piperidin-l-yl]-2-oxo- ethyl}-4-phenylamino-benzamide
To a stirred solution of (4-phenylamino-benzoylamino)-acetic acid (0.087 g, 0.00032 mol) in DMF (5 mL) was added DIPEA (0.104 g, 0.00081 mol), HOBt (0.043 g, 0.00032 mol) and EDCLHCl (0.061 g, 0.00032 mol) at ambient temperature. After 2 minutes 4-(2-bromo-phenoxy)- piperidine trifluoroacetate (0.1 g, 0.00027 mol) and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution and dried over sodium sulfate. The organic layer was collected and concentrated under reduced pressure to afford 0.045 g (32%) of N- {2-[4-(2-bromo-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-4-phenylamino-benzamide. LCMS: 508.12 (M+l)+, 96.85%, 1H NMR (DMSO-d6): δ 8.6 (s, IH), 8.3 (t, IH), 7.8 (d, 2H), 7.6 (dd, IH), 7.3 (m, 4H), 7.2 (d, 2H), 7.1 (d, 2H), 6.9 (q, 2H), 4.8 (m,lH), 4.2 (t, 2H), 3.7 (m, 2H), 3.5 (m, 2H), 2.0 (m, 2H), 1.7 (m,2H).
Intermediate 29 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid
A solution of 2,4-dioxo-4-phenyl -butyric acid ethyl ester (1.68 g, 0.0076 mole), acetic acid (10 niL) and hydrazine hydrate (0.42 g, 0.0083 mole) was heated to reflux for 5 hours. The reaction mixture was then poured into iced water, basified with saturated aqueous NaHCO3 solution and the product was extracted with ethyl acetate. The ethyl acetate layer was collected and washed with brine solution, dried over sodium and concentrated under reduced pressure. The resulting residue was purified by column chromatography using basic aluminium oxide (50% ethyl acetate in hexane) to afford 1.442 g (88%) of S-phenyl-lH-pyrazole-S-carboxylic acid ethyl ester. LCMS: 217.09 (M+l)+, 97.92%. To a stirred solution of 5-phenyl-lH-pyrazole-3-carboxylic acid ethyl ester (1.4 g, 0.00667 mol) in a mixture of THF (30 mL), methanol (15 mL) and H2O (10 mL) was added LiOH-H2O (0.839 g, 0.0199 mol) at ambient temperature. The resulting mixture was stirred at 45 0C overnight. Volatiles were evaporated and the residue was diluted with water, acidified with concentrated HCl solution and the product was extracted with ethyl acetate. The ethyl acetate layer was collected, dried over sodium sulfate and concentrated under reduced pressure to afford 1.26 g (86%) of 5-phenyl-lH-pyrazole-3-carboxylic acid. LCMS: 189.06 (M+l)+, 97.7%.
Intermediate 30 - Synthesis of [(S-Phenyl-lH-pyrazole-S-carbonyty-aminoJ-acetic acid
To a stirred solution of 5-phenyl-l H-pyrazole-3-carboxylic acid (1.25 g, 0.00665 mol) in
DMF (3 mL) was added DIPEA (4.296 g, 0.03324 mol), HOBt (1.123 g, 0.00831 mol) and EDCI.HC1 (1.593 g, 0.00831 mol) at ambient temperature. After 2 minutes glycine acid ethyl ester hydrochloride (0.928 g, 0.00665 mol) was and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was collected and washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.76 g (42% of [(5-phenyl-lH-pyrazole-3-
carbonyl)-amino] -acetic acid ethyl ester. LCMS: 274.11 (M+l)+, 96.7%. To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid ethyl ester (0.615 g, 0.0025 mol) in a mixture of THF (10 mL), methanol (5mL) and H2O (5 niL) was added LiORH2O (0.283 g, 0.00675 mol) at ambient temperature. The reaction mixture was stirred at the same temperature for 2 hours. Volatiles were evaporated and the residue was diluted with water, acidified with 10% HCl solution. The resulting precipitate was isolated by filtration, washed water followed by hexane and to afford 0.238 g (38%) of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid. LCMS: 246.08 (M+l)+, 95.60%
EXAMPLE 20 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-bromo- phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-arnino]-acetic acid (0.038 g, 0.00013 mol) in DMF (2 mL), was added DIPEA (0.0872 g, 0.00067 mol), HOBt (0.023 g, 0.00017 mol) and EDCLHCl (0.0325 g, 0.0001686 mol) at ambient temperature. After 2 minutes (2-bromo-phenyl)-piperidin-4-yl-amine dihydrochloride (0.0393 g, 0.00013 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.0375 g (58%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-bromo-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 482.11 (M+l)\ 91.09%, 1H NMR (DMSO-d6): δ 13.7 (s, IH), 8.0 (s, IH), 7.8 (d, 2H), 7.38 (m, 4H), 7.16 (t, IH), 7.06 (s, IH), 6.84 (d, IH), 6.5 (t, IH), 4.6 (d,lH), 4.3 (d, IH), 4.1 (d, 2H), 3.9 (m, IH), 3.6 (m, IH), 3.2 (m,lH), 2.8 (m, IH), 1.9 (m, 2H), 1.4(m, 2H).
EXAMPLE 21 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-chloro- phenylamino)-piperidin-l-yI]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino] -acetic acid (0.038 g,
0.00013 mol) in DMF (2 mL) was added DIPEA (0.0872 g, 0.00067 mol), HOBt (0.023 g, 0.00017 mol) and EDCLHCl (0.0325 g, 0.00017 mol) at ambient temperature. After 2 minutes (2- chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (0.0334 g, 0.00013 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.0382 g (65%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 438.16 (M+l)+, 98.29%, 1H NMR (DMSOd6): δ 13.8 (s, IH), 8.1 (s, IH), 7.78 (d, 2H), 7.44 (m, 2H), 7.38 (m, IH), 7.26 (d, 2H), 7.1 (m, 2H), 6.84 (d, IH), 6.6 (t, IH), 4.9 (d, IH), 4.3 (d, IH), 4.2 (d, 2H), 3.8 (d, IH), 3.6 (m, IH), 3.2 (m, IH), 2.8 (m, IH), 1.9 (t, 2H), 1.3 (m, 2H).
EXAMPLE 22 - Synthesis of S-Phenyl-lH-pyrazoIe-S-carboxylic acid {2-[4-(2-bromo- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.038 g, 0.000135 mol) in DMF (2 mL) was added DIPEA (0.0872 g, 0.00068 mol), HOBt (0.023 g,
0.00017 mol) and EDCI.HC1 (0.0325 g, 0.00017 mol) at ambient temperature. After 2 minutes 4- (2-bromophenoxy)-piperidine trifluoroacetate (0.05 g, 0.0001349 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.044 g (68%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-bromo-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide.
LCMS: 483.1 (M+l)+,99.71%, 1H NMR (DMSO-d6): δ 13.7 (s, IH), 8.02 (s, IH), 7.79 (d, IH), 7.6 (m, IH), 7.42 (m, 2H), 7.3 (m, 2H), 7.26 (d, IH), 7.0 (s, IH), 6.9 (t, IH), 4.8 (m, IH), 4.2 (d, 2H), 3.4 (m, 4H), 1.7 (m, 2H), 1.6 (m, 2H).
EXAMPLE 23 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-chloro- phenoxy)-piperidin-l-yl]-2-oxo-ethyI}-amide
To a stirred solution of [(S-phenyl-lH-pyrazole-S-carbonyty-aminoJ-acetic acid (0.038 g, 0.00013 mol) in DMF (2 mL) was added DIPEA (0.0872 g, 0.00067 mol), HOBt (0.023 g, 0.00017 mol) and EDCLHCl (0.0325 g, 0.00017 mol) at ambient temperature. After 2 minutes 4- (2-chlorophenoxy)-piperidine hydrochloride (0.03347 g, 0.00013 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.0517 g (87%) of 5-phenyl-lH- pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 439.15 (M+l)
+, 98.84%,
1H NMR (DMSO-d
6): δ 13.7 (s, IH), 8.1 (s, IH), 7.8 (d, 2H), 7.42 (m, 3H), 7.32 (m, IH), 7.24 (m, 2H), 7.1 (s, IH), 7.0 (t, IH), 4.7 (m, IH), 4.1 (d,2H), 3.7 (m, 2H), 3.4 (m, 2H), 1.9 (m, 2H), 1.6 (m, 2H).
EXAMPLE 24 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-chloro- 5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.038 g,
0.00013 mol) in DMF (2 mL) was added DIPEA (0.0872 g, 0.00067 mol), HOBt (0.023 g, 0.00017 mol) and EDCLHCl (0.0325 g, 0.00017 mol) at ambient temperature. After 2 minutes 4- (2-chloro-5-fluoro-phenoxy)-piperidine hydrochloride (0.036 g, 0.0001349 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.056 g (91%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}- amide. LCMS: 457.14 (M+l)+, 98.34%, 1H NMR (DMSO-d6): δ 13.7 (s, IH), 8.0 (s, IH), 7.8 (d, 2H), 7.44 (m, 2H), 7.38 (m, IH), 7.24 (m, IH), 7.1 (s, IH), 6.8 (m, IH), 4.8 (m, IH), 4.2 (d,2H), 3.6 (m, 2H), 3.4 (m, 2H), 1.9 (m, 2H), 1.7 (m, 2H).
EXAMPLE 25 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(2- trifluoromethyl-phenylsulfanyl)-piperidin-l-yl]-ethyl}-amide
To a stirred solution of [(5-Phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.05 g, 0.00018 mol) in DMF (1 mL) was added DIPEA (0.0689 g, 0.00053 mol), HOBt (0.028 g, 0.00021 mol) and EDCLHCl (0.0407 g, 0.00021 mol) at ambient temperature. After 2 minutes 4- (2-trifluoromethyl-phenylsulfanyl)-piperidine hydrochloride (0.063 g, 0.00021 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.068 g (78%) of 5- phenyl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(2-trifIuoromethyl-phenylsulfanyl)-piperidin- l-yl]-ethyl}-amide. LCMS: 489.15 (M+l)+, 94.5%, 1H NMR (DMSO-d6): δ 13.8(s, IH), 8.0 (s, IH), 7.8 (m, 4H), 7.65 (t, IH), 7.5-7.3 (m, 4H), 7.1 (s, IH), 4.3-4.05 (m, 3H), 3.9-3.6 (m, 2H), 3.2 (m, IH), 2.9 (m, IH), 1.9 (m, 2H), 1.6 (m, IH), 1.4 (m, IH).
EXAMPLE 26 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-chloro- henylsulfanyI)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.04 g, 0.00014 mol) in DMF (ImL) was added DIPEA (0.05517 g, 0.00043 mol), HOBt (0.023 g, 0.00017 mol) and EDCI.HC1 (0.0326 g, 0.00017 mol) at ambient temperature. After 2 minutes 4- (2-chloro-phenylsulfanyl)-piperidine hydrochloride (0.0449g, 0.00017 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.054 g (84%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-henylsulfanyl)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 455.12 (M+l)+, 83.96%, 1H NMR (DMSO-d6): δ 13.65 (s, IH), 8.1 (s, IH), 7.8 (d, 2H), 7.5 (m, 4H), 7.38 (t, 2H), 7.24 (t, 2H), 7.15 (s, IH), 4.2 (m, 3H), 3.85 (m, IH), 3.65 (m, IH), 3.25 (m, IH), 3.0 (m, IH), 2.0 (m, 2H), 1.56 (m, IH), 1.4 (m, IH).
EXAMPLE 27 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-nitro-
phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino] -acetic acid (0.15 g, 0.0006 mol) in DMF (3 niL) was added DIPEA (0.27 g, 0.0021 mol), HOBt (0.12 g, 0.0009 mol) and EDCLHCl (0.17 g, 0.0009 mol) at ambient temperature. After 2 minutes 4-(2-nitro-phenoxy)- piperidine hydrochloride (0.19 g, 0.00073 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The product was extracted with ethyl acetate, and the organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using neutral aluminium oxide (5% methanol in chloroform) to afford 0.1 g
(44%) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-nitro-phenoxy)-piperidin-l-yl]-2-oxo- ethyl}-amide. LCMS: 450.17 (M+l)+, 96.5%, 1H NMR (DMSOd6): δ 8.1 (bs, IH), 7.9 (m, 3H), 7.6 (m, IH), 7.5 (m, 4H), 7.1 (m, 2H), 5.0 (m, IH), 4.2 (m, 2H), 3.8 (m, 8H), 2.0 (m, 2H).
EXAMPLE 28 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-amino- phenoxy)-piperidin-l-yI]-2-oxo-ethyl}-amide
To a stirred solution of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-nitro-phenoxy)- piperidin-l-yl]-2-oxo-ethyl} -amide (0.07 g, 0.00016 mole) in a mixture of methanol (5 mL) and THF (5 mL) was added 10% Pd/C (0.01 g). The resulting mixture was stirred under an atmosphere of hydrogen for 30 minutes. The mixture was then filtered through celite, and the celite was washed with methanol. The combined organic layers were concentrated under reduced pressure. The resulting residue was re-crystalized from a hexane/chloroform mixture to afford 0.025 g (39%) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-amino-phenoxy)-piperidin-l-yl]-2- oxo-ethyl}-amide. LCMS: 420.2 (M+l)+, 96.9%, 1H NMR (DMSOd6): δ 8.1 (bs, IH), 7.8 (d, 2H), 7.5-7.3 (m, 3H), 7.1 (m, IH), 6.85 (d, IH), 6.7 (m, 2H), 6.5 (m, IH), 4.7 (s, 2H), 4.6 (s,lH), 4.2 (s, 2H), 3.9 (m, 2H), 2.0 (m, 2H), 1.8 (m, 2H), 1.4 (m, 2H).
EXAMPLE 29 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2, 3- dimethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.097 g,
0.00036 mol) in DMF (1 mL) was added DIPEA (0.233 g, 0.00180 mol), HOBt (0.053 g, 0.00036 mol) and EDCI.HC1 (0.138 g, 0.00072 mol) at ambient temperature. After 2 minutes (2,3- dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride (0.1 g, 0.00036 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.12 g (77%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2, 3-dimethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}- amide. LCMS: 432.23 (M+l)+ , 95.88%, 1H NMR (DMSOd6): δ 13.7 (s, IH), 8.1 (s, IH), 7.8 (t, 2H), 7.6 (m, 3H), 7.1 (s, IH), 6.9 (t, IH), 6.5 (d, IH), 6.4 (d, IH), 4.4 (m, 2H), 4.2 (s, 2H), 3.9 (d, IH), 3.5 (s, IH), 3.2 (m, IH), 2.8 (m, IH), 2.2 (s, 3H), 2.0 (s, 5H), 1.4 (m, 2H).
EXAMPLE 30 - Synthesis of 5-Phenyl-lH-pyrazole-3-carboxyUc acid {2-[4-(2, 4- dimethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.097 g, 0.00040 mol) in DMF (1 mL) was added DIPEA (0.233 g, 0.00180 mol), HOBt (0.053 g, 0.00040 mol) and EDCI.HC1 (0.138 g, 0.0007214 mol) at ambient temperature. After 2 minutes (2,4- dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride (O.lg, 0.00036 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.12 g (77%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2, 4-dimethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}- amide. LCMS: 432.23 (M+l)+, 97.64%, 1H NMR (DMSO-d6): δ 8.1 (s, IH), 7.8 (d, 2H), 7.5 (t, 2H), 7.4 (d, IH), 7.1 (s, IH), 6.6 (d, IH), 4.3 (m, 2H), 4.1 (t, 2H), 3.9 (d, IH), 3.5 (bs, IH), 3.2 (t,
IH), 2.8 (t, IH), 2.2 (s, 3H), 2.1 (s, 3H), 1.9 (t, 2H), 1.5 (m, IH), 1.3 (m, IH).
EXAMPLE 31 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2, 5- dimethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(S-phenyl-lH-pyrazole-S-carbony^-aminoJ-acetic acid (0.097 g, 0.00040 mol) in DMF (1 mL) was added DIPEA (0.233 g, 0.00180 mol), HOBt (0.053 g, 0.00040 mol) and EDCI.HC1 (0.138 g, 0.00072 mol) at ambient temperature. After 2 minutes (2,5- dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride (0.1 g, 0.00036 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.12 g (77%) of 5-phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(2, 5-dimethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}- amide. LCMS: 432.23 (M+l)+, 96.91%, 1H NMR (DMSO-d6): δ 8.1 (s, IH), 7.8 (d, 2H), 7.5 (t, 2H), 7.4 (t, IH), 7.1 (s, IH), 6.8 (d, IH), 6.5 (s, IH), 6.3 (d, IH), 4.4 (d, 2H), 4.2 (bs, 2H), 3.9 (d, IH), 3.6 (bs, IH), 3.2 (t, IH), 2.8 (t, IH), 2.2 (s, 3H), 2.0 (s, 3H), 1.9 (d, IH), 1.5 (d, IH), 1.3 (d, IH).
EXAMPLE 32 - Synthesis of S-Phenyl-lH-pyrazoIe-S-carboxylic acid {2-[4-(2-tert- butyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.088 g,
0.00036 mol) in DMF (2 mL) was added DIPEA (0.211 g, 0.0016 mol), HOBt (0.048 g, 0.00036 mol) and EDCLHCl (0.125 g, 0.000655mol) at ambient temperature. After 2 minutes (2-tert-butyl- phenyl)-piperidin-4-yl-amine dihydrochloride (0.1 g, 0.00033 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.07 g (49%) of 5-phenyl-lH- pyrazole-3-carboxylic acid {2-[4-(2-tert-butyl-phenylamino)-piperidin-l -yl]-2-oxo-ethyl} -amide.
LCMS: 460.26 (M+l)+, 89.75%, 1H NMR (DMSOd6): δ 13.8 (s, IH), 8.1 (s, IH), 7.8 (d, 2H), 7.6-7.4 (m, 3H), 7.2-7.0 (m, 3H), 6.8 (d, IH), 6.6 (t, IH), 4.3 (d, IH), 4.2 (d, 2H), 3.9 (m, 2H), 3.7 (s, IH), 3.2 (t, IH), 2.8 (t, IH), 2.0 (t, 3H), 1.4 (s, 9H), 1.0 (bs, IH).
EXAMPLE 33 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2,5- difluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino] -acetic acid (0.075 g, 0.0003 mol) in DMF (2 ml) was added DIPEA (0.135 g, 0.0012 mol), HOBt (0.061 g, 0.00045 mol) and EDCI.HC1 (0.086 g, 0.00045mol) at ambient temperature. After 2 minutes, 4-(2,5- difluoro-phenoxy)-piperidme hydrochloride (0.076 g, 0.0003 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was collected and washed with brine solution, dried over sodium sulfate and concentrated. The resulting residue was purified by column chromatography using neutral aluminum oxide (5% methanol in chloroform) to afford
0.065 g (49%) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2,5-difluoro-phenoxy)-piperidin- l-yl]-2-oxo-ethyl}-amide. LCMS: 441.17 (M+l)+, 91.07%, 1H NMR (DMSOd6): δ 13.8 (bs, IH), 8.0 (bs, IH), 7.8 (m, 2H), 7.4 (m, 5H), 7.2 (m, IH), 6.8 (m, IH), 4.6 (m, IH), 4.2 (m, 2H), 3.9 (m, IH), 3.8 (m,lH), 2.0 (m, 2H), 1.6 (m, 2H).
EXAMPLE 34 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-bromo- phenyIsulfanyl)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.15 g, 0.000533 mol) in DMF (3ml) was added DIPEA (0.206 g, 0.00160 mol), HOBt (0.0864 g,
0.00064 mol) and EDCI.HC1 (0.122 g, 0.00064 mol) at ambient temperature. After 2 minutes 4-(2- bromo-phenylsulfanyl)-piperidine hydrochloride (0.197 g, 0.00064 mol) was added and the
resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried. Purification by column chromatography using silica gel 60-120 mesh (40% ethyl acetate in hexane) afforded 0.097 g (36%) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-bromo-phenylsulfanyl)-piperidin-l- yl]-2-oxo-ethyl}-amide. LCMS: 499.07 (M+l)+, 95.7%, 1H NMR (DMSOd6): δ 13.8 (d, IH), 8.1 (t, IH), 7.8 (d, 2H), 7.6 (dd, IH), 7.58-7.3 (m, 6H), 7.22-7.06 (m, 2H), 4.3-4.1 (m, 3H), 3.9-3.6 (m, 2H), 3.0 (m, IH), 2.0 (m, 2H), 1.7-1.4 (m, 3H).
EXAMPLE 35 - Synthesis of δ-Phenyl-lH-pyrazole-S-carboxylic acid [2-oxo-2-(4-o- tolylamino-piperidin-l-yl)-ethyl]-amide
To a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (0.102 g, 0.00042 mol) in DMF (1 mL) was added DIPEA (0.245 g, 0.00190 mol), HOBt (0.056 g, 0.000418 mol) and EDCI.HC1 (0.145 g, 0.00076 mol) at ambient temperature. After 2 minutes piperidin-4-yl-o-tolyl-amine dihydrochloride (O.lg, 0.00038 mol) was added and the resulting mixture was stirred for 16 hours. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.15 g (94% of 5-phenyl-lH- pyrazole-3-carboxylic acid [2-oxo-2-(4-o-tolylamino-piperidin-l-yl)-ethyl]-amide.
LCMS: 418.22 (M+l)+, 92.96%, 1H NMR (CDCl3): δ 8.8 (bs, IH), 7.7 (d, 2H), 7.4 (m, 3H), 7.1 (t, 3H), 6.7 (q, 2H), 4.6-4.3 (m, 4H), 4.1 (d, IH), 3.6 (s, IH), 3.4 (t, 2H), 3.0 (t,lH), 2.2 (d, 2H), 2.1 (s, 3H), 1.5(q, 4H).
Intermediate 31 - Synthesis of 5-(3-Benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid
To a solution of l-(3-hydroxy-phenyl)-ethanone (5 g, 0.03672 mole) in DMF (75 mL) was added NaH (60% w/w dispersion in oil) (1.66 g, 0.0416 mole) and the resulting mixture was
stirred at ambient temperature for 15 minutes. Benzyl bromide (7.23 g, 0.0422 mole) was then added and stirring was continued for 4 hours at ambient temperature. The reaction mixture was then quenched with cold aqueous NH
4Cl solution. The product was extracted with ethyl acetate and the ethyl acetate layer was washed with water followed by brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 8.5 g (100%) of l-(3-benzyloxy- phenyl)-ethanone. A mixture of THF (150 mL) and NaH (60% w/w dispersion in oil) (4.4 g, 0.11 mole) was cooled to O
0C with stirring for 10 minutes. Diethyl oxalate (10.73 g, 0.0734 mole) was then added and the resulting mixture was heated to reflux for 10 minutes. The mixture was then cooled to ambient temperature and l-(3-benzyloxy-phenyl)-ethanone (8.3 g, 0.0367 mole) in THF (50 mL) was added dropwise over a period of 45 minutes. The resulting mixture was then heated to 50
0C for 30 minutes. The reaction mixture was quenched with cold aqueous NH
4CI solution and the product was extracted with ethyl acetate. The ethyl acetate layer was collected and washed with water followed by brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (4% ethyl acetate in hexane) to afford 10.9 g (92%) of 4-(3-benzyloxy-phenyl)-2, 4-dioxo- butyric acid ethyl ester. LCMS: 327.12 (M+l)
+, 86%. A solution of 4-(3-benzyloxy-phenyl)-2,4- dioxo-butyric acid ethyl ester (5 g, 0.0153 mole), acetic acid (25 mL) and hydrazine hydrate (0.8426 g, 0.0168 5mole) was heated to reflux for 3 hours. The reaction mixture was then poured into iced water, basified with saturated aqueous NaHCO
3 solution and the product was extracted with ethyl acetate. The ethyl acetate layer was collected, washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 4.876 g (99%) of 5-(3- benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester. LCMS: 323.13 (M+l)
+, 99%. To a stirred solution 5-(3-benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester (4 g, 0.0124 mol) in a mixture of THF (20 mL), methanol (10 mL) and H
2O (1OmL) was added LiOH-H
2O (1.56g, 0.0372 mol) at ambient temperature. The resulting mixture was then stirred for 6 hours. Volatiles were removed by evaporation and the resulting residue was diluted with water and acidified with 10%HCl. The resulting precipitate was isolated by filtration and dried to afford 3.78 g (92%) of 5-(3-benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid. LCMS: 295.1 (M+l)
+, 97.1%.
Intermediate 32 - Synthesis of {[5-(3-Benzyloxy-phenyl)-lH-pyrazole-3-carbonyl]- amino}-acetic acid
To a stirred solution of 5-(3-hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid (1 g, 0.00340 mol) in DMF (5 mL) was added DIPEA (1.976 g, 0.01529 mol), HOBt (0.574 g, 0.00425 mol) and EDCLHCl (0.814 g, 0.00425 mol) at ambient temperature. After 5 minutes glycine ethyl ester hydrochloride (0.498 g, 0.00357 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried. The crude product was recrystallized from ethyl acetate to afford 0.54 g (36%) of {[5-(3-benzyloxy-phenyl)-lH-pyrazole-3-carbonyl]-amino}-acetic acid ethyl ester. LCMS: 380.15 (M+l)+, 92.05%. To a stirred solution of {[5-(3-benzyloxy-phenyl)-lH-pyrazole-3- carbonyl]-amino}-acetic acid ethyl ester (0.54g, 0.00136 mmol) in a mixture of THF (50 mL), methanol (25 mL) and H2O (25 mL) was added LiORH2O (0.18 g, 0.0042 mol) at ambient temperature. The resulting mixture was stirredat ambient temperature overnight. Volatiles were removed by evaporation and the resulting residue was diluted with water, acidified with concentrated HCl solution. The resulting precipitate was isolated by filtration and dried afford 0.44 g (92%) of {[S-^-beiizyloxy-phenyty-lH-pyrazole-S-carbony^-aminoJ-acetic acid. LCMS: 352.12 (M+l)\ 97.85%,
Intermediate 33 - Synthesis of 5-(3-Benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of {[5-(3-benzyloxy-phenyl)-lH-pyrazole-3-carbonyl]-amino} -acetic acid (0.15 g, 0.00039 mol) in DMF (2 mL) was added DIPEA (0.224 g, 0.00174 mol) HOBt (0.065g, 0.000482 mol) and EDCLHCl (0.0925 g, 0.00048 mol) at ambient temperature. After 5 minutes (2-chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (0.1 g, 0.00041 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration. Purification by column chromatography
using basic aluminium oxide (60% ethyl acetate in hexane) afforded 0.19 g (91%) of 5-(3- benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]- 2-oxo-ethyl} -amide. LCMS: 544.2 (M+l)+, 96.18%.
EXAMPLE 36 - Synthesis of 5-(3-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2- [4-(2-chloro-phenylamino)-piperidin-l-yl)-2-oxo-ethyl}-amide
A solution of 5-(3-benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro- phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide (0.19 g, 0.00035 mole) in dichloromethane was cooled to -7O0C. 1 mL of a 1.6M solution OfBBr3 in dichloromethane was then added dropwise with vigorous stirring. The reaction mixture was slowly brought to ambient temperature and stirring was continued at the same temperature for 1 hour. The reaction mixture was then cooled to -7O0C and ice cold water was added dropwise with stirring and while the mixture was slowly brought to ambient temperature. The product was extracted with dichloromethane and the organic layers were dried over sodium sulfate. The organic layer was collected and concentrated under reduced pressure. The resulting residue was recrystallized from ethyl acetate to afford 0.071 g (45%) of 5-(3-hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 454.16 (M+l)+, 94.25%, 1H NMR (DMSOd6): δ 13.6 (s, IH), 9.6 (s, IH), 8.1 (s, IH), 7.0 (m, 5H), 6.84 (d, IH), 6.76 (d, IH), 6.6 (m, IH), 4.8 (s, IH), 4.3 (d, IH), 4.1 (d, 2H), 3.8 (d, IH), 3.7 (s, IH), 3.2 (m, IH), 2.8 (m, IH), 1.95 (m, 2H), 1.3 (m, 2H).
Intermediate 34 - Synthesis of S-Phenyl-pyridine^-carboxylic acid
OH
O A mixture of toluene (20 mL) and water (5 mL) was degassed with argon for 5 minutes.
Sodium carbonate (1.53 g, 0.01444 mole) was then added and the the mixture was degassed with argon for a further 5 minutes. Phenyl boronic acid (1.126 g, 0.00866 mole) and 5-chloro-pyridine- 2-carbonitrile (1 g, 0.00722 mole) were added and the resulting mixture was degassed with argon for a further 5 minutes. tetrakis(triphenylphosphine)palladium(0) (1.67 g, 0.00144 mole) was
added and the resulting mixture was degassed with argon for a further 5 minutes. The reaction mixture was then heated to reflux for 3 hours. The reaction mixture was diluted with ethyl acetate. The ethyl acetate layer was washed with water followed by brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (4% ethyl acetate in hexane) to afford 0.9 g (69%) of S-phenyl-pyridine^-carbonitrile. LCMS: 181.07 (M+l)+, 28.900Zo5 1H NMR (DMSOd6): δ 9.1 (s, IH), 8.4-8.3 (m, IH), 8.2-8.1 (m, IH), 7.9-7.78 (m, 2H), 7.6-7.4 (m, 3H). To a stirred solution of 5-phenyl-pyridine-2-carbonitrile (0.9 g, 0.005 mol) in a mixture of ethanol (10 mL) and water (5 mL) was added NaOH (1 g, 0.025 mol) and the resulting mixture was stirring overnight. The reaction mixture was then concentrated under reduced pressure. The residue was dissolved in water, washed with ether and the aqueous layer was acidified with citric acid. The acidified aqueous solution was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.75 g (75%) of 5-phenyl-pyridine-2-carboxylic acid. LCMS: 200.06 (M+l)+, 97.53%, 1H NMR (DMSO- d6): δ 13 (bs, IH), 9.1-9.0 (d, IH), 8.3-8.2 (dd, IH), 8.2-8.1 (d, IH), 7.8 (d, IH), 7.6-7.4 (m, 3H).
Intermediate 35 - Synthesis of [(5-Phenyl-pyridine-2-carbonyl)-amino]-acetic acid
To a stirred solution of 5-phenyl-pyridine-2-carboxylic acid (0.3 g, 0.00151 mol) in DMF (5 mL) was added DIPEA (0.580 g, 0.00452 mol), HOBt (0.243 g, 0.00180 7mol) and EDCLHCl (0.346 g, 0.001807 mol) at ambient temperature. After 2 minutes glycine ethyl ester hydrochloride (0.252 g, 0.00181 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.32 g (75%) of [(5 -phenyl-pyridine-2-carbonyl)-amino] -acetic acid ethyl ester. To a stirred solution of [(5-phenyl-pyridine-2-carbonyl)-amino]-acetic acid ethyl ester (0.32 g, 0.00113 mol) in a mixture of THF (3.2 mL), methanol (2mL) and H
2O (1 mL) was added LiOH.H
2O (0.142 g, 0.00338 mol) and the resulting mixture was stirred at ambient temperature overnight. Volatiles were removed by evaporation and the resulting residue was
diluted with water and acidified with 10% HCl. The product was extracted with ethyl acetate, which was dried over sodium sulfate and concentrated under reduced pressure to afford 0.24 g (83%) of [(5-phenyl-pyridine-2-carbonyl)-amino]-acetic acid.
EXAMPLE 37 - Synthesis of S-Phenyl-pyridine^-carboxylic acid {2-oxo-2-[4-(2- chloro-phenoxy)-piperidin-l-yl]-ethyl}-amide
To a stirred solution of [(5-phenyl-pyridine-2-carbonyl)-amino]-acetic acid (0.07 g, 0.00027 mol) in DMF (1.5 mL) was added DIPEA (0.105 g, 0.00082 mol), HOBt (0.044 g, 0.00033 mol) and EDCLHCl (0.062 g, 0.00033 mol) at ambient temperature. After 2 minutes 4-(2- chlorophenoxy)piperidine hydrochloride (0.08 Ig, 0.000327mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.0545 g (44%) of 5- phenyl-pyridine-2-carboxylic acid {2-oxo-2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-ethyl}-amide. LCMS: 484.18 (M+l)+, 95.40%, 1H NMR (DMSOd6): δ 9.0 (d, IH), 8.8 (t, IH), 8.3 (dd, IH), 8.1 (d, IH), 7.8 (d, IH), 7.6-7.4 (m, 2H), 7.3-7.2 (m, 2H), 7.0-6.9 (m, IH), 4.8-4.7 (m, IH), 4.3 (d, IH), 3.8-3.6 (m, 2H), 3.5 (m, 2H), 2.0-1.9 (m, 2H), 1.8-1.6 (m, 2H).
EXAMPLE 38 - Synthesis of 5-Phenyl-pyridine-2-carboxylic acid {2-[4-(2-chloro-5- fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenyl-pyridine-2-carbonyl)-amino]-acetic acid (0.07 g, 0.00027 mol) in DMF (1.5 mL) was added DIPEA (0.105 g, 0.000819 mol), HOBt (0.044 g, 0.00033 mol) and EDCI.HC1 (0.062g, 0.00033 mol) at ambient temperature. After 2 minutes 4-(2-
chloro-5-fluoro-phenoxy)-piperidine hydrochloride (0.087g, 0.00033 mol) wasand the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure afford 0.069 g (54%) of 5- phenyl-pyridine-2-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo- ethyl}-amide. LCMS: 468.14 (M+l)+, 82.99%, 1H NMR (DMSOd6): δ 9.0 (d, IH), 8.8 (t, IH), 8.3 (dd, IH), 8.1 (d, IH), 7.8 (d, 2H), 7.6-7.4 (m, 4H), 7.2 (dd, IH), 6.9-6.8 (m, IH), 4.9-4.8 (m, IH), 4.3 (d, 2H), 3.8-3.6 (m, 2H), 3.5 (m, 2H), 2.1-1.9 (m, 2H), 1.8-1.5 (m, 2H).
Intermediate 36 - Synthesis of 5-(4-Benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester
To a solution of l-(4-hydroxy-phenyl)-ethanone (10 g, 0.0735 mole) in DMF (50 mL) was added K2CO3 (20.2 g, 0.147 mole) at ambient temperature. Benzyl bromide (13.83 g, 0.0808 mole) was then added and the resulting mixture was stirred at ambient temperature for 5 hours. The reaction mixture was then quenched with iced water. The resulting precipitate was isolated by filtration and dried to afford 16.2 g (98%) of l-(4-benzyloxy-phenyl)-ethanone. LCMS: 227.1 (M+l)+, 98.38%. A mixture of THF (130 mL) and NaH (60% w/w dispersion in oil) (5.28g , 0.132 mole) was cooled to 00C with stirring for 5 minutes. Diethyl oxalate (12.87 g, 0.0886 mole) was added and the mixture was heated to reflux for 15 minutes.after cooling to ambient temperature 1- (4-benzyloxy-phenyl)-ethanone (10 g, 0.0442 mole) in THF (50 mL) was added dropwise over a period of 45 minutes. The resulting mixture was heated to 7O0C for 1 hour. The reaction mixture was then quenched with cold aqueous NH4Cl solution. The resulting precipitate was isolated by filtration and dried to afford 21.2 g of 4-(4-benzyloxy-phenyl)-2,4-dioxo-butyric acid ethyl ester. LCMS: 327.12 (M+l)+, 36.5%. A solution of 4-(4-benzyloxy-phenyl)-2,4-dioxo-butyric acid ethyl ester (10 g, 0.03064 mole), acetic acid (20 mL) and hydrazine hydrate (1.685 g, 0.0337 mole) was heated to reflux for 8 hours. The reaction mixture was then poured into iced water and basified with saturated aqueous NaHCO3 solution. The product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under
reduced pressure to afford 6.3 g (64%) of 5-(4-benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester. LCMS: 323.13 (M+l)+, 95.10%.
Intermediate 37 - Synthesis of 5-(4-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid
To a stirred solution of 5-(4-benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester
(5.8 g, 0.018 mole) in a mixture of methanol (50 mL) and THF (180 mL) was added 10% Pd/C (2 g) and the resulting mixture was stirred under an atmosphere of hydrogen for 5 hours. The reaction mixture was then filtered through celite, and the celit e was washed with methanol. The filtrate was collected and concentrated under reduced pressure afford 4.05 g (97%) of 5-(4-Benzyloxy- phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester. LCMS: 233.08 (M+l)+, 97%. To a stirred solution of 5-(4-hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester (4 g, 0.01724 mol) in a mixture of 1 ,4-dioxane (88 mL) and H2O (88mL) was added NaOH (1.57 g, 0.03793 mol) and the resuling mixture was stired at ambient temperature overnight. The reaction mixture was then acidified with concentrated HCl solution. The mixture was concentrated and the resulting precipitate was isolated by filtration and dried to afford 3.1 g (76%) of 5-(4-hydroxy-phenyl)-lH- pyrazole-3-carboxylic acid. LCMS: 205.05 (M+l)+, 91.6%.
Intermediate 38 - Synthesis of 2-Amino-l-[4-(2-chloro-phenylamino)-piperidin-l-yl]- ethanone hydrochloride
To a stirred mixture of glycine (30 g, 0.3996 mole) in IN aqueous NaOH (39.96 g, 0.999 mole) was added tert-butanol (270 mL). The mixture was cooled to O
0C and di-tert-butyl dicarbonate (96.42 g, 0.4395 mole) was added dropwise. The mixture was stirred at ambient temperature for 5 hours. The reaction mixture was then concentrated under reduced pressure and the resulting resiude was acidified with citric acid. The product was extracted with ethyl acetate, the organic layer was washed with brine solution, dried over sodium sulfate and concentrated
under reduced pressure to afford 49 g (70%) of tert-butoxycarbonylamino-acetic acid.
1H NMR (DMSOd
6): δ 12.8-12.0 (bs, IH), 7.1(t, IH), 3.6 (d, 2H), 1.4 (s 9H). To a stirred solution oftert- butoxycarbonylamino-acetic acid (0.2 g, 0.00114 mol) in DMF (5 mL) was added DIPEA (0.59 g, 0.00457 mol), HOBt (0.193 g, 0.001427 mol) and EDCLHCl (0.274 g, 0.00143 mol) at ambient temperature. After 5 minutes (2-chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (0.283 g, 0.00114 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and purified by column chromatography using basic aluminium oxide (60% ethyl acetate in hexane) to afford 0.41 g (98%) of {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-carbamic acid tert-butyl ester. LCMS: 368.18 (M+l)
+, 93.53%. A solution of {2-[4-(2-chloro-phenylamino)-piperidin-l- yl]-2-oxo-ethyl}-carbamic acid tert-butyl ester (0.41 g, 0.00111 mole) in methanol (10 mL) was added dioxane.HCl (5 mL) and the resulting mixture was stirred at ambient temperature for 10 minutes. The mixture was then concentrated under reduced and the resulting residue was washed with ether to afford 0.34 g (98%) of 2-ammo-l-[4-(2-chloro-phenylamino)-piperidin-l-yl]- ethanone hydrochloride. LCMS: 268.11 (M+l)
+, 84.63%.
EXAMPLE 39 - Synthesis of 5-(4-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2- [4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of 5-(4-hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid (0.1 g,
0.00049 mol) in DMF (5mL) was added DIPEA (0.253 g, 0.00196 mol), HOBt (0.0827 g, 0.00061 mol) and EDCI.HC1 (0.117 g, 0.00061 mol) at ambient temperature. After 5 minutes 2-amino-l- [4-(2-chloro-phenylamino)-piperidin-l-yl]-ethanone dihydrochloride (0.164 g, 0.00054 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and recrystallized from ethyl acetate to afford 0.092 g (41%) of 5-(4-hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro- phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 426.16 (M+l)+, 91.47%, 1H NMR (DMSO-d6): δ 13.5 (s, IH), 9.7 (s, IH), 8.0 (s, IH), 7.58 (d, 2H), 7.1 (m, 2H), 6.8 (m, 4H), 6.56 (m, IH), 4.9 (d, IH), 4.3 (d, IH), 4.1 (d, 2H), 3.8 (m, IH), 3.6 (m, IH), 3.1 (m, IH), 2.7 (m, IH),
1.9 (m, 2H), 1.3 (m, 2H)
Intermediate 39 - Synthesis of S-^-Benzyloxy-pheny^-lH-pyrazole-S-carboxylic acid ethyl ester
To a solution of l-(2-hydroxy-phenyl)-ethanone (5 g, 0.03672 mole) in DMF (75 mL) was added NaH (60% w/w dispersion in oil) (1.66 g, 0.0416 mole) and the resulting mixture was stirred at ambient temperature for 15 minutes. Benzyl bromide (7.23 g, 0.0422 mole) was then added and the mixture as stirred for an additional 4 hours. The reaction mixture was then quenched with cold aqueous NH4Cl solution and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with water, brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 8.4 g (100%) of l-(2-benzyloxy-phenyl)-ethanone. LCMS: 227.1 (M+l)+, 84.14%. To a mixture of THF (200 mL) and NaH (60% w/w dispersion in oil) (4.4 g, 0.11 mole) cooled to O0C was added diethyl oxalate (10.73 g, 0.07345 mole). The resulting mixture was heated to reflux for 15 minutes. After cooing to ambient temperature, l-(2- benzyloxy-phenyl)-ethanone (8.3 g, 0.0376 mole) was added dropwise over a period of 45 minutes. The resulting mixture was heated to 55 0C for 30 minutes. The reaction mixture was then quenched with cold aqueous NH4Cl solution and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with water, brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (4% ethyl acetate in hexane) to afford 8.7 g (71%) of 4-(2-benzyloxy-phenyl)-2,4-dioxo-butyric acid ethyl ester. LCMS: 327.12 (M+l)+, 78.6%. A solution of 4-(2-benzyloxy-phenyl)-2,4-dioxo-butyric acid ethyl ester (3.75 g, 0.01149 mole), acetic acid (20 mL) and hydrazine hydrate (0.632 g, 0.01264 mole) was heated to reflux for 3 hours. The reaction mixture was then poured into iced water basifϊed with saturated aqueous NaHCO3 solution and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 3.703 g (100%) of 5-(2-benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester. LCMS: 323.13 (M+l)+, 97.14%.
Intermediate 40 - Synthesis of 5-(2-Hydroxy-phenyl)-l-methyl-lH-pyrazole-3- carboxylic acid
To a solution of 5-(2-benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid ethyl ester
(0.25 g, 0.00078 mole) in DMF (5 mL) was added K2CO3 (0.225 g, 0.00163 mole) followed by methyl iodide (0.116 g, 0.00081 mole) and at the resuling mixture was stirred at ambient temperature for 1 hour. The mixture was then quenched with iced water and the resulting precipitate was isolated by filtration. The solid was recrystallized from ethyl acetate to afford 0.251 g (96%) of 5-(2-benzyloxy-phenyl)-l-methyl-lH-pyrazole-3-carboxylic acid ethyl ester. LCMS: 337.15 (M+l)+, 90.13%. To a stirred solution of 5-(2-benzyloxy-phenyl)-l -methyl- IH- pyrazole-3-carboxylic acid ethyl ester (0.251 g, 0.00075 mole) in methanol (40 mL) was added 10% Pd/C (0.05 g) and the resuling mixture was stirred under an atmosphere of hydrogen for 1 hour. The mixture was then filtered through celite and the celite was washed with methanol. The combined filtrate was concentrated under reduced pressure to afford 0.169 g (92%) of 5-(2- hydroxy-phenyl)-l -methyl- lH-pyrazole-3-carboxylic acid ethyl ester. LCMS: 247.1 (M+ 1)+, 99%. To a stirred solution of 5-(2-hydroxy-phenyl)-l -methyl- lH-pyrazole-3-carboxylic acid ethyl ester (0.169 g, 0.00069 mol) in a mixture of THF (10 mL), methanol (5 mL) and H2O (5mL), was added LiOH-H2O (0.087 g, 0.00206 mol) at ambient temperature and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and acidified with 10% HCl. The product was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to afford 0.13 g (87%) of 5-(2-hydroxy-phenyl)- l-methyl-lH-pyrazole-3-carboxylic acid. LCMS: 219.07 (M+l)+, 81.5%
EXAMPLE 40 - Synthesis of 5-(2-Hydroxy-phenyl)-l-methyl-lH-pyrazole-3- carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of 5-(2-hydroxy-phenyl)-l -methyl- lH-pyrazole-3-carboxylic acid (0.13g, 0.0006 mol) in DMF (2 mL) was added DIPEA (0.308 g, 0.00234 mol), HOBt (0.101 g, 0.00075 mol) and EDCLHCl (0.143 g, 0.00075 mol) at ambient temperature. After 5 minutes 2- amino-l-[4-(2-chloro-phenylamino)-piperidin-l-yl]-ethanone hydrochloride (0.181 g, 0.00060 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and recrystallized from ethyl acetate to afford 0.113 g (41%) of 5-(2-hydroxy-phenyl)-l -methyl- lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 468.18 (M+l)+, 96.98%. 1H NMR (DMSOd6): δ 10.2 (s, IH), 8.7 (t, IH), 7.7 (d, IH), 7.4 (s, IH), 7.1 (m, 3H), 6.8 (m, 3H), 6.6 (m, IH), 4.9 (m, IH), 4.3 (d, IH), 4.1 (m, 4H), 3.9 (m, IH), 3.6 (m, IH), 3.1 (m, IH), 2.7 (m, IH), 1.9 (m, 2H), 1.4 (m, 2H).
Intermediate 41 - Synthesis of 5-(2-Benzyloxy-phenyl)-isoxazoIe-3-carboxylic acid
To a stirred solution of 4-(2-benzyloxy-phenyl)-2,4-dioxo-butyric acid ethyl ester (3.75 g, 0.01149 mole) in acetic acid (20 mL) was added hydroxylamine hydrochloride (0.878 g, 0.0126 mole) and the resulting mixture was heated to reflux for 3 hours. Volatiles were removed and the resulting residue was diluted with water, basified with sodium bicarbonate solution and extracted with ethyl acetate, washed the ethyl acetate with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 3.42 g (92%) of 5-(2-benzyloxy-phenyl)-isoxazole- 3-carboxylic acid ethyl ester. LCMS: 324.12 (M+l)+, 78.58%. To a stirred solution of 5-(2- benzyloxy-phenyl)-isoxazole-3-carboxylic acid ethyl ester (1.75 g, 0.00542 mol) in a mixture of THF (30 mL), methanol (15mL) and H2O (15 mL) was added NaOH (0.651 g, 0.01628 mol) and the resulting mixture was stirred for 2 hours. The reaction mixture was then diluted with cold water. Volatiles were removed by evaporation and the residue was diluted with water acidified
with concentrated HCl. The resulting precipitate was isolated by filtration and dried to afford 1.57 g (98%) of 5-(2-benzyloxy-phenyl)-isoxazole-3-carboxylic acid. LCMS: 296.08 (M+l)+, 84.73%
Intermediate 42 - Synthesis of {[5-(2-Benzyloxy-phenyl)-isoxazole-3-carbonyl]- amino}-acetic acid
To a stirred solution of 5-(2-benzyloxy-phenyl)-isoxazole-3-carboxylic acid (0.7 g, 0.00237 mol) in DMF (4mL) was added DIPEA (1.226 g, 0.00948 mol), HOBt (0.4g, 0.0029 mol) and EDCLHCl (0.57g, 0.0029 mol) at ambient temperature. After 5 minutes glycine ethyl ester hydrochloride (0.347 g, 0.0024 8mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 60-120 mesh (65% ethyl acetate in hexane) afforded 0.66 g (73%) of {[5-(2-benzyloxy-phenyl)-isoxazole-3-carbonyl]-amino}- acetic acid ethyl ester. To a stirred solution of {[5-(2-benzyloxy-phenyl)-isoxazole-3-carbonyl]- amino}-acetic acid ethyl ester (0.65 g, 0.00171 mol) in a mixture of THF (1OmL), methanol (5mL) and H2O (5 mL) was added LiOH-H2O (0.215 g, 0.00512 mol) at ambient temperature. The resulting mixture was stirred overnight. Volatiles were removed by evaporation and the residue was diluted with water acidified with concentrated HCl. The resulting precipitate was isolated by filtration and dried to afford 0.56 g of {[5-(2-benzyloxy-phenyl)-isoxazole-3-carbonyl]-amino}- acetic acid. LCMS: 353.11 (M+l)+, 60.4%.
Intermediate 43 - Synthesis of 5-(2-Benzyloxy-phenyl)-isoxazole-3-carboxylic acid {2- [4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of {[5-(2-benzyloxy-phenyl)-isoxazole-3-carbonyl]-amino}-acetic
acid (0.15 g, 0.00039 mol) in DMF (2 niL) was added DIPEA (0.224 g, 0.00174 mol), HOBt (0.065g, 0.000482mol) and EDCI.HC1 (0.0925 g, 0.00048 mol) at ambient temperature. After 5 minutes 4-(2-chloro-phenoxy)-piperidine hydrochloride (0.1 g, 0.00041 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 230-400 mesh (70% ethyl acetate in hexane) afforded 0.153 g (73%) of 5-(2-benzyloxy- phenyl)-isoxazole-3 -carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- 1 -yl]-2-oxo-ethyl } - amide. LCMS: 546.17 (M+l)
+, 97.6%.
EXAMPLE 41 - Synthesis of 5-(2-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-[4-
(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of 5-(2-benzyloxy-phenyl)-isoxazole-3-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl} -amide (0.153 g, 0.00028 mole) in methanol (50 mL) was added 10% Pd/C (0.03 g) and the resulting mixture was stirred under an atmosphere of hydrogen for 3 hours. The mixture was then filtered through celite, the celite was washed with methanol and the organic layers were concentrated under reduced pressure. The resulting residue was purified by preparative HPLC to afford 0.055 g (43%) of 5-(2-hydroxy-phenyl)-isoxazole-3- carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 456.12 (M+l)+, 98.75%. 1H NMR (DMSOd6): δ 10.8 (s, IH), 8.7 (t, IH), 7.8 (d, IH), 7.4 (d, IH), 7.24 (m, 3H), 7.12 (s, IH), 7.04 (d, IH), 6.92 (t, 2H), 4.7 (m, IH), 4.2 (d, 2H), 3.7 (m, 2H), 3.4 (m, 2H), 1.9 (m, 2H), 1.6 (m, 2H).
Intermediate 44 - Synthesis of 5-(2-Benzyloxy-phenyl)-isoxazole-3-carboxylic acid {2- [4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of {[5-(2-benzyloxy-phenyl)-isoxazole-3-carbonyl] -amino} -acetic acid (0.15 g, 0.00039 mol) in DMF (2 mL) was added DIPEA (0.224 g, 0.00174 mol), HOBt (0.065 g, 0.000482mol) and EDCLHCl (0.0925 g, 0.00048 mol) at ambient temperature. After 5 minutes 4~(2-chloro-phenylamino)-piperidine dihydrochloride (0.1 g, 0.00041 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using basic alumina (50% ethyl acetate in hexane) afforded 0.142 g (78%) of 5-(2-benzyloxy- phenyl)-isoxazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}- amide.
EXAMPLE 42 - Synthesis of 5-(2-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-[4- (2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-ainide
To a stirred solution of 5-(2-benzyloxy-phenyl)-isoxazole-3-carboxylic acid {2-[4-(2- chloro-phenylamino)-piperidin- 1 -yl]-2-oxo-ethyl} -amide (0.142 g, 0.00026 mole) in a mixture of methanol (60 mL) and THF (10 mL) was added 10% Pd/C (0.05 g) and the resuling mixture was stirred under an atmosphere of hydrogen for 1 hour. The mixture was then filtered through celite, the celite was washed with methanol and the organic layers were concentrated under reduced pressure. The resulting residue was recrystallized from ethyl acetate to afford 0.02 g (13%) of 5- (2-hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-[4-(2-chloro-phenylarnino)-piperidin-l-yl]-2- oxo-ethyl} -amide. LCMS: 455.14 (M+l)+. 94.82%, 1H NMR (DMSOd6): δ 10.8 (s, IH), 8.7 (t, IH), 7.81 (d, IH), 7.38 (t, IH), 7.25 (d, IH), 7.0 (m, 4H), 6.8 (d, IH), 6.6 (t, IH), 4.9 (d, IH), 4.3 (d, IH), 4.1 (d, 2H), 3.9 (d, IH), 3.6 (s, IH), 3.2 (m, IH), 2.7 (m, IH), 2.2 (m, 2H), 1.5 (m, 2H).
Intermediate 45 - Synthesis of 5-(2-Benzyloxy-phenyl)-lH-pyrazoIe-3-carboxylic acid
{2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of {[5-(2-benzyloxy-phenyl)-lH-pyrazole-3-carbonyl]-amino}-acetic acid (0.15 g, 0.00039 mol) in DMF (2 mL) was added DIPEA (0.224 g, 0.00174 mol), HOBt (0.065 g, 0.00048 mol) and EDCI.HC1 (0.0925 g, 0.00048 mol) at ambient temperature. After 5 minutes (2-chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (O.lg, 0.00041 mol) was added to the reaction mixture continued stirring at the same temperature for overnight. The reaction mixture was diluted with cold water, filtered the solid precipitated and dried to afford 0.205 g (98%) of 5- (2-benzyloxy-phenyl)- 1 H-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin- 1 - yl]-2-oxo-ethyl}-amide. LCMS: 544.2 (M+l)+, 92.2%.
EXAMPLE 43 - Synthesis of 5-(2-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2- [4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of 5-(2-benzyloxy-phenyl)-l H-pyrazole-3-carboxylic acid {2-[4-(2- chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide (0.2 g, 0.00039 mole) in methanol
(6OmL), added 10% Pd/C (0.04 g) and the resulting mixture was stirred under an atmosphere of hydrogen for 2 hours. The mixture was then filtered through celite, the celite was washed with methanol and the organic layers were concentrated under reduced pressure. The residue was recrystallized from a mixture of ethyl acetate and methanol to afford 0.055 g (32%) of 5-(2- hydroxy-phenyl)- 1 H-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2- oxo-ethyl}-amide. LCMS: 454.16 (M+l)+, 94.65%. 1H NMR (DMSOd6): δ 13.2 (s, IH), 10.4 (s, IH), 8.0 (s, IH), 7.6 (d, IH), 7.2 (m, 4H), 7.0 (d, IH), 6.8 (m, 2H), 6.6 (t, IH), 4.9 (d, IH), 4.3 (d, IH), 4.2 (d, 2H), 3.8 (d, IH), 3.6 (m, IH), 3.2 (t, IH), 2.7 (t, IH), 1.9 (m, 2H), 1.3 (m, 2H).
Intermediate 46 - Synthesis of 5-(2-Benzyloxy-phenyl)-lH-pyrazole-3-carboxylic acid
{2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of {[5-(2-benzyloxy-phenyl)-lH-pyrazole-3-carbonyl] -amino} -acetic acid (0.15 g, 0.00039 mol) in DMF (2 mL) was added DIPEA (0.224 g, 0.001736mol), HOBt (0.065 g, 0.00048 mol) and EDCI.HC1 (0.0925g, 0.000482mol) at ambient temperature. After 5 minutes (2-chlorophenoxy)-piperidin-4-yl-amine hydrochloride (0.1 g, 0.00041 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration and dried to afford 0.08 g (33%) of 5- (2-benzyloxy-phenyl)- 1 H-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- 1 -yl]-2- oxo-ethyl} -amide. LCMS: 544.2 (M+l)+, 99.03%.
EXAMPLE 44 - Synthesis of 5-(2-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2- [4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a solution of 5-(2-benzyloxy-phenyl)-l H-pyrazole-3-carboxylic acid {2-[4-(2-chloro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl} -amide (0.075 g, 0.00014 mole), in methanol (50 mL) was added 10% Pd/C (0.015 g) and the resulting mixture was stirred under an atmosphere of hydrogen for 1.5 hours. The mixture was then filtered through celite, the celite was washed with methanol and the organic layers were concentrated under reduced pressure. The resulting residue was recrystallized from a mixture of ethyl acetate and methanol to afford 0.052 g (83%) of 5-(2- hydroxy-phenyl)- 1 H-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo- ethyl}-amide. LCMS: 455.16 (M+l)+, 90.34%, 1H NMR (DMSOd6): δ 13.2 (s, IH), 10.2 (s, IH), 8.0 (s, IH), 7.64 (m, IH), 7.42 (m, IH), 7.24 (m, 2H), 7.18 (m, IH), 7.06 (s, IH), 6.96 (m, 2H), 6.84 (t, IH), 4.7 (m, IH), 4.2 (d, 2H), 3.61 (m, 2H), 3.4 (m, 2H), 1.9 (m, 2H), 1.6 (m, 2H).
Intermediate 47 - Synthesis of 6-Phenylamino-nicotinic acid
A mixture of 6-chloronicotinic acid ethyl ester (0.2 g, 0.00108 mole) and aniline (0.119 g,
0.00129 mole) in ethoxyethanol (10 mL) was heated to reflux for overnight. The mixture was then concentrated under reduced pressure and the residue was dissolved in ethyl acetate. The oganic layer was washed with water, brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (5% ethyl acetate in hexane) to afford 0.23 g (88%) of 6-phenylamino-nicotinic acid ethyl ester. To a stirred solution 6-phenylamino-nicotinic acid ethyl ester (0.23 g, 0.00095 mol) in a mixture of THF (4 mL), methanol (4 mL) and H2O (2mL) was added LiOH-H2O (0.159 g, 0.0038 mol) at ambient temperature and the resulting mixture was stirred overnight. Volatiles were then evaporated and the resulting residue was diluted with water, acidified with 10% aqueous citric acid solution. The resulting precipitate was isolated by filtration and dried to afford 0.174 g (85%) of 6-Phenylamino-nicotimc acid.
Intermediate 48 - Synthesis of [(ό-Phenylamino-pyridine-S-carbonylJ-aminol-acetic acid
To a stirred solution of 6-phenylamino-nicotinic acid (0.1 g, 0.00047 mol) in DMF (2 mL), was added DIPEA (0.181 g, 0.001401 mol), HOBt (0.0756 g, 0.00056 mol) and EDCI.HC1 (0.107 g, 0.00056 mol) at ambient temperature. After 2 minutes glycine ethyl ester hydrochloride (0.0782 g, 0.00056 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and purified by column chromatography using silica gel 60-120 mesh (60% ethyl acetate in hexane) to afford 0.135 g (97%) of [(ό-phenylamino-pyridine-S-carbony^-aminoJ-acetic acid ethyl ester. LCMS: 299.13 (M+l)+, 99%. To a stirred solution of [(6-phenylamino-pyridine-3-carbonyl)-amino]-acetic acid ethyl ester (0.135 g, 0.00045 mol) in a mixture of THF (5 mL), methanol (7 mL) and H2O (2 mL) was added LiORH2O (0.0568 g, 0.00135 mol) at ambient temperature and the resulting
mixture was stirred for 2 hours. Volatiles were then evaporated and the resulting residue was diluted with water, acidified with 10% aqueous HCl solution. The resulting precipitate was isolated by filtration and dried to afford 0.078 g (64%) of [(6-phenylamino-pyridine-3-carbonyl)- amino] -acetic acid.
EXAMPLE 45 - Synthesis of N-{2-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-2-oxo- ethyl}-6-phenylamino-nicotinamide
To a stirred solution of [(6-phenylamino-pyridine-3-carbonyl)-amino] -acetic acid (0.035 g, 0.00013 mol) in DMF (1 mL) was added DIPEA (0.05 g, 0.00039 mol), HOBt (0.0209 g, 0.00015 mol) and EDCLHCl (0.029 g, 0.0001549 mol) at ambient temperature. After 2 minutes 4-(2- chloro-phenoxy)-piperidine hydrochloride (0.0382 g, 0.00015 mol) and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120mesh (70% ethyl acetate in hexane) to afford 0.037 g (62%) of N- {2- [4-(2-chloro-phenoxy)-piperidin- 1 -yl] -2-oxo-ethyl} -6-phenylamino-nicotinamide. LCMS: 465.16 (M+l)
+, 96.6%,
1H NMR (DMSOd
6): δ 9.4 (s, IH), 8.8 (s, IH), 8.4 (t, IH), 8.0 (dd, IH), 7.7 (d, 2H), 7.45 (d, IH), 7.4 (m, 4H), 7.0 (d,2H), 6.8 (d, IH), 4.7 (m, IH), 4.25 (m, 2H), 3.7 (m, 2H), 3.5 (m, 2H), 2.0 (m, 2H), 1.7 (q, 2H).
EXAMPLE 46 - Synthesis of N-{2-[4-(2-Chloro-phenylamino)-piperidin-l-yl]-2-oxo- ethyl}-6-phenylamino-nicotinamide
To a stirred solution of [(6-phenylamino-pyridine-3-carbonyl)-amino]-acetic acid (0.035 g,
0.00013 mol) in DMF (1 mL) was added DIPEA (0.05 g, 0.00039 mol), HOBt (0.0209 g, 0.00015 mol) and EDCLHCl (0.029 g, 0.00015 mol) at ambient temperature. After 2 minutes (2-chloro- phenyl)-piperidin-4-yl-amine dihydrochloride (0.0382 g, 0.00015 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.051 g (85%) of N-{2-[4- (2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-6-phenylamino-nicotinamide. LCMS: 464.18 (M+l)
+, 98.3%,
1H NMR (DMSOd
6): δ 9.4 (s, IH), 8.7 (d, IH), 8.4 (t, IH), 8.0 (d, IH), 7.7 (d, 2H), 7.3 (m, 3H), 7.15 (t, IH), 7.0 (t, IH), 6.85 (d,2H), 6.4 (t, IH), 4.9 (d, IH), 4.35 (d, IH), 4.1 (d, 2H), 3.9 (d, IH), 3.6 (s, IH), 3.2 (m, IH), 2.8 (m,lH), 2.0 (t, 2H), 1.5 (m, IH), 1.4 (m,lH).
Intermediate 49 - Synthesis of S-Phenylamino-pyridine-l-carboxylic acid
A mixture of BLNAP (0.167 g, 0.00027 mole), palladium acetate (0.06 g, 0.00027 mole) and toluene (10 mL) was degassed with argon for 15 minutes. This mixture was then added to a mixture of aniline (0.5 g, 0.00537 mole), 3-chloro-6-cyanopyridine (0.893 g, 0.00645 mole) and cesium carbonate (3.49 g, 0.01075 mole) in toluene (10 mL). The resulting mixture was heated to reflux for 22 hours. The reaction mixture was then concentrated under reduced pressure and the residue was extracted with ethyl acetate. The ethyl acetate layer was washed with water, brine solution, dried over sodium sulfate and concentrated under reduced pressure. Purification of the residue by column chromatography using silica gel 60-120 mesh (15% ethyl acetate in hexane) afforded 0.4 g (38%) of 5-phenylamino-pyridine-2-carbonitrile. To a stirred solution of 5- phenylamino-pyridine-2-carbonitrile (0.4 g, 0.00205 mol) in EtOH (15 mL) was added NaOH (0.246 g, 0.0061 mol) and water (10 mL) and the resulting mixture was heated to reflux for 4 hours. The mixture was then concentrated under reduced pressure and the residue was diluted with cold water. The resulting precipitate was isolated by filtration and dried to afford 0.385 g (87%) of 5-phenylamino-pyridine-2-carboxylic acid
Intermediate 50 - Synthesis of [(5-Phenylamino-pyridine-2-carbonyl)-amino]-acetic acid
To a stirred solution of 5-phenylamino-pyridine-2-carboxylic acid (0.26 g, 0.00121 mol) in DMF (2 mL) was added DIPEA (0.471 g, 0.00364 mol), HOBt (0.196 g, 0.00145 mol) and EDCI.HC1 (0.278 g, 0.00145 mol) at ambient temperature. After 2 minutes glycine ethyl ester hydrochloride (0.203 g, 0.00146 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water. The resulting precipitate was isolated by filtration and purified by column chromatography using silica gel 60-120 mesh (60% ethyl acetate in hexane) to afford 0.32 g (88%) of [(5-phenylamino-pyridine-2-carbonyl)-amino]-acetic acid ethyl ester. LCMS: 300.13 (M+ 1)+, 97%. To a stirred solution [(5-phenylamino-pyridine-2- carbonyl)-amino] -acetic acid ethyl ester (0.32 g, 0.00107 mol) in a mixture of THF (5 mL), methanol (10 mL) and H2O (2 mL) was added LiORH2O (0.134 g, 0.00321 mol) at ambient temperature and the resulting mixture was stirred for 2 hours. Volatiles were then evaporated and the resulting residue was diluted with water, acidified with 10% aqueous HCl solution. The resulting precipitate was isolated by filtration and dried to afford 0.26 g (90%) of [(5- phenylamino-pyridine-2-carbonyl)-amino] -acetic acid.
EXAMPLE 47 - Synthesis of S-Phenylamino-pyridine-^-carboxylic acid {2-[4-(2- chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenylamino-pyridine-2-carbonyl)-amino]-acetic acid (0.035 g,
0.00013 mol) in DMF (1 mL) was added DIPEA (0.05 g, 0.00039 mol), HOBt (0.0209 g, 0.00015 mol) and EDCLHCl (0.0296 g, 0.00015 mol) at ambient temperature. After 2 minutes piperidin-4- yl-(2-chloro-phenyl)-amine dihydrochloride (0.0382 g, 0.00015 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration and dried to afford 0.038 g (64%) of 5-phenylamino- pyridine-2-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 464.18 (M+l)+, 92.88%, 1H NMR (DMSO-d6): δ 8.9 (s, IH), 8.5 (t, IH), 8.3 (d, IH), 7.9
(d, IH), 7.55 (dd, IH), 7.35 (t, 2H), 7.2 (m, 4H), 7.0 (t,lH), 6.9 (d, IH), 6.6 (t, IH), 4.9 (d, IH), 4.35 (d, IH), 4.2 (d, 2H), 3.85 (d, IH), 3.55 (s, IH), 3.2 (m, 2H), 2.85 (m, IH), 1.95 (t,2H), 1.5 (m, IH), 1.35 (m, IH).
EXAMPLE 48 - Synthesis of 5-Phenylamino-pyridine-2-carboxylic acid {2-[4-(2- chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
To a stirred solution of [(5-phenylamino-pyridine-2-carbonyl)-amino]-acetic acid (0.035 g, 0.00013 mol) in DMF (1 mL) was added DIPEA (0.05 g, 0.00039 mol), HOBt (0.0209 g, 0.00015 mol) and EDCLHCl (0.0296 g, 0.00015 mol) at ambient temperature. After 2 minutes 4-(2-chloro- phenoxy)-piperidine hydrochloride (0.0382g, 0.00015 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration and dried to afford 0.032 g (53%) of 5-phenylamino-pyridine- 2-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LCMS: 465.16 (MH-I)+, 98.06%, 1H NMR (DMSOd6): δ 8.85 (s, IH), 8.5 (t, IH), 8.35 (d, IH), 7.85 (d, IH), 7.55 (dd, IH), 7.4 (d, IH), 7.3 (m, 4H), 7.2 (d, 2H), 7.0 (m, 2H)5 4.75 (m, IH), 4.2 (d, 2H), 3.7 (m, 2H), 3.5 (m, 2H), 1.95 (m, 3H), 1.7 (m, 2H).
Intermediate 51 - Synthesis of N-Biphenyl-4-yl-malonamic acid
To a stirred solution of monoethyl malonate (0.86 g, 0.00649 mole) in DMF (20 mL) was added HOBt (0.795 g, 0.0059 mole) and DMAP (0.790 g, 0.00649 mole). The mixture was cooled to 10 0C and EDCLHCl (1.7 g, 0.008 mole) followed by biphenyl-4-ylamine (1.0 g, 0.0059 mole) were added and the resulting mixture was stirred at the ambient temperature for overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration and dried to afford 1.76 g (94%) of N-biphenyl-4-yl-malonamic acid ethyl ester. To a solution of N-biphenyl-4-yl-malonamic acid ethyl ester (1.7 g, 0.0060 mole) in a mixture of
methanol (5 mL), THF (10 mL) and H2O (10 mL) was added LiOH.H2O (0.5 g, 0.012 mole). The reaction mixture was stirred for 2 hours at ambient temperature. The mixture was then concentrated and the residue was diluted with water acidified with cone. HCl. The resulting precipitate was isolated by filtration and dried to afford 1.5 g (98%) of N-biphenyl-4-yl- malonamic acid. LC-MS purity: 95.4%, 1H NMR (DMSOd6): δ 10.3 (s, IH), 7.7 (m, 6H), 7.4 (t, 2H), 7.32 (t, IH).
EXAMPLE 49 - Synthesis of N-Biphenyl-4-yl-3-[4-(2-bromo-phenoxy)-piperidin-l- yl] -3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.075 g, 0.00029 mole) in DMF
(2.0 mL) was added HOBt (0.042 g, 0.00031 mole) and DIPEA (0.083 g, 0.00065 mole). The mixture was cooled to 10 0C and EDCI.HC1 (0.060 g, 0.00031 mole) followed by 4-(2-bromo- phenoxy)-piperidin-l-ylamine trifluoroacetate (0.106g, 0.00029mole) were added. The mixture was stirred at the ambient temperature overnight. The mixture was then diluted with water and the product was extracted with ethyl acetate. The organic layer was washed with brine and concentrated. The resulting residue was purified by column chromatography using silica gel 60- 120 mesh (40% ethyl acetate in hexane) to afford 0.045 g (31%) of N-biphenyl-4-yl-3-[4-(2- bromo-phenoxy)-piperidin-l-yl]-3-oxo-propionamide. LCMS: 493 (M+l)+, 94.19%, 1H NMR (CDCl3): δ 10.0 (s, IH), 7.66 (d, 2H), 7.58 (m, 5H), 7.44 (t, 2H), 7.32 (m, IH), 6.9 (m, 2H), 4.52 (m, IH), 4.15 (m,lH), 3.56 (m, IH), 3.54 (m, 2H), 3.51 (d, 2H), 2.0 (m,2H), 1.8 (m, 2H).
EXAMPLE 50 - Synthesis of N-Biphenyl-4-yl-3-[4-(2-chloro-5-fluoro-phenoxy)- piperidin-l-yl]-3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.1 g, 0.00039 mole) in DMF (2
niL) was added HOBt (0.065 g, 0.00047 mole) and DIPEA (0.126 g, 0.00098 mole). The mixture was cooled to 10°C and EDCI.HC1 (0.090 g, 0.00047 mole) followed by 4-(2-chloro-5-fluoro- phenoxy)-piperidine hydrochloride (0.114g, 0.00043 mole) were added. The resulting mixture was stirred at the ambient temperature overnight. The mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 60-120 mesh (35% ethyl acetate in hexane) afforded 0.024 g (13%) of N-biphenyl-4-yl-3-[4- (2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo-propionamide. LCMS: 467 (M+l)+, 97.5%, 1H NMR (CDCl3): δ 10.0 (s, IH), 7.66 (d, 2H), 7.58 (m, 4H), 7.42 (t, 2H), 7.34 (m, 2H), 6.68 (m, 2H), 4.64 (m, IH), 4.3 (m, IH), 3.7 (m, 3H), 3.52 (s, 2H), 1.96 (m, 4H).
EXAMPLE 51 - Synthesis of N-Biphenyl-4-yl-3-[4-(2-bromo-phenylamino)-piperidin- l-yl]-3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.071 g, 0.00028 mole) in DMF (2 mL) was added DIPEA (0.089 g, 0.0007 mole), HOBt (0.046 g, 0.00034 mole) and EDCI.HC1 (0.065 g, 0.00034 mole). After 2 minutes (2-bromo-phenyl)-piperidin-4-yl-amine dihydrochloride (0.082g, 0.00028mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with aqueous sodium bicarbonate solution, brine and concentrated. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (70% ethyl acetate in hexane) to afford 0.025 g (17%) of N-biphenyl-4-yl-3-[4-(2-bromo-phenylamino)- piperidin-l-yl]-3-oxo-propionamide. LC-MS purity: 492 (M+l)+, 92.2%, 1H NMR (CDCl3): δ 10.0 (s, IH), 7.66 (d, 2H), 7.58 (m, 4H), 7.44 (t, 3H), 7.34 (t, IH), 7.18 (t, IH), 6.68 (d, IH), 6.6 (UH), 4.45 (d, IH), 4.0 (d, IH), 3.65 (bs, IH), 3.5 (s, 2H), 3.35 (UH), 3.1 (UH), 2.2 (m,2H), 1.5 (m, 2H).
EXAMPLE 52 - Synthesis of N-Biphenyl-4-yl-3-[4-(2-bromo-phenyIsulfanyl)- piperidin-l-yl]-3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.15 g, 0.00059 mole) in DMF (4 mL) was added DIPEA (0.226 g, 0.00176 mole), HOBt (0.095 g, 0.00071 mole) and EDCI.HC1 (0.134 g, 0.00071 mole). After 2 minutes 4-(2-bromo-phenylsulfanyl)-piperidine hydrochloride (0.21 g, 0.00071 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 60-120 mesh (60% ethyl acetate in hexane) afforded 0.153 g (51%) of N-biphenyl-4-yl-3-[4-(2-bromo-phenylsulfanyl)-piperidin-l- yl]-3-oxo-propionamide. LC-MS purity: 509.08 (M+l)+, 87.3%, 1H NMR (DMSOd6): δ 10.2 (s, IH), 7.82-7.6 (m, 6H), 7.48-7.36 (m, 3H), 7.36-7.28 (m, IH), 7.22-7.12 (m, IH), 5.6-5.2 (d, IH), 4.25 (d, IH), 3.7(m, IH), 3.55(s, 2H), 2.95 (m, IH), 2.0 (m, 2H), 1.7-1.5 (m, IH), 1.5-1.35 (m, IH).
EXAMPLE 53 - Synthesis of N-Biphenyl-4-yl-3-oxo-3-(4-o-toIylamino-piperidin-l-yI)- propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.1 g, 0.00038 mole) in DMF (1 mL) was added DIPEA (0.245 g, 0.0019 mole), HOBt (0.056 g, 0.00042 mole) and EDCLHCl (0.145 g, 0.00076 mole). After 2 minutes piperidin-4-yl-o-tolyl-amine dihydrochloride (0.106 g, 0.00042 mole) was added and the resulting mixture was stirred for 16 hours. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel (60-120 mesh) (1% methanol in chloroform) afforded 0.04 g (25%) of N-biphenyl-4-yl-3-oxo-3-(4-o-tolylamino-piperidin-l-yl)- propionamide. LC-MS purity: 428.23 (M+l)+, 93.11%, 1H NMR (DMSO-d6): δ 7.7 (m, 6H), 7.5 (t, 2H), 7.3 (t, IH), 7.2 (q, 2H), 6.65 (q, 2H), 4.5 (d, IH), 4.0 (d, IH), 3.6(m, IH), 3.5(s, 2H), 3.3 (s, IH), 3.0 (t, IH), 2.2 (m, 3H), 2.1 (s, 2H), 1.5(t, 2H).
EXAMPLE 54 - Synthesis of N-Biphenyl-4-yl-3-[4-(2-nitro-phenoxy)-piperidin-l-yl]-
3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.1 g, 0.00039 mole) in DMF
(3mL) was added DIPEA (0.176 g, 0.0014 mole), HOBt (0.078 g, 0.00058 mole) and EDCLHCl (0.11 g, 0.00058 mole). After 2 minutes (2-nitro-phenoxy)-piperidine hydrochloride (0.12 g, 0.00046 mole) and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layers were dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using neutral aluminium oxide (5% methanol in chloroform) to afford 0.130 g (72%) of N-biphenyl-4-yl-3-[4-(2-nitro-phenoxy)-piperidin-l-yl]-3-oxo- propionamide. LC-MS purity: 460.18 (M+l)+, 95%, 1H NMR (DMSO-d6): δ 7.9 (m, IH), 7.7 (m, 7H), 7.5 (m, 3H), 7.3 (m, IH), 7.1 (m, IH), 5.0(m, IH), 3.6 (m, 6H), 2.0 (m, 2H), 1.8 (m, 2H).
EXAMPLE 55 - Synthesis of 3-[4-(2-Amino-phenoxy)-piperidin-l-yl]-N-biphenyl-4- yl-3-oxo-propionamide
To a solution of N-biphenyl-4-yl-3-[4-(2-nitro-phenoxy)-piperidin-l-yl]-3-oxo- propionamide (0.09 g, 0.00019 mole) in methanol (10 mL) was added 10% Pd/C (0.015 g) and the resulting mixture was stirred under an atmosphere of hydrogen for 30 minutes. The mixture was then filtered through celite, the celite was washed with methanol and the organic layers were concentrated under reduced pressure. The resulting residue was recystallized from a mixture of hexane and chloroform to afford 0.025 g (29%) of 3-[4-(2-amino-phenoxy)-piperidin-l-yl]-N- biphenyl-4-yl-3-oxo-propionamide. LC-MS purity: 430.21 (M+l)+, 90.7%, 1H NMR (DMSOd6): δ 7.7 (m, 6H), 7.4 (m, 2H), 7.3 (m, IH), 6.8 (m, IH), 6.6 (m, 2H), 6.5 (m, IH), 4.6 (bs, 2H), 4.5
(bs, IH), 3.8 (m, 2H), 3.5 (m, 4H), 2.0 (m, 2H), 1.8 (m, 2H).
EXAMPLE 56 - Synthesis of N-Biphenyl-4-yl-3-[4-(2,3-dimethyl-phenylamino)- piperidin- 1-yl] -3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.101 g, 0.00039 mole) in DMF (3 mL) was added DIPEA (0.233 g, 0.0018mole), HOBt (0.053 g, 0.00039 mole) and EDCLHCl (0.138 g, 0.00072 mole). After 2 minutes (2,3-dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride (0.1 g, 0.00036 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 60-120 mesh (1 % methanol in chloroform) afforded 0.07 g (44%) of N-biphenyl-4-yl-3-[4-(2,3-dimethyl-phenylamino)- piperidin-l-yl]-3-oxo-propionamide. LC-MS purity: 442.24 (M+l)+, 95.5%, 1H NMR (DMSOd6): δ 10.2 (s, IH), 7.7 (m, 6H), 7.6 (t, 2H), 7.4 (d, IH), 6.8 (t, IH), 6.5 (d, IH), 6.4 (d, IH), 4.4 (t, 2H), 4.0 (d, IH), 3.6( bs,3 H), 3.2 (t, IH), 2.8 (t, IH), 2.2 (s, 3H), 2.0 (bs, 5H), 1.5 (d, IH), 1.4 (d, IH).
EXAMPLE 57 - Synthesis of N-Biphenyl-4-yl-3-[4-(2,4-diraethyl-phenylamino)- piperidin-l-yl]-3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.101 g, 0.00039 mole) in DMF
(3 mL) was added DIPEA (0.233 g, 0.0018 mole), HOBt (0.053 g, 0.00039 mole) and EDCI.HC1 (0.138 g, 0.000721 mole). After 2 minutes (2,4-dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride (0.1 g, 0.00036 mole was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 60-120 mesh (1% methanol in chloroform) afforded 0.07 g (44%) of N-biphenyl-4-yl-3-[4-(2,4-dimethyl-phenylamino)-
piperidin-l-yl]-3-oxo-propionamide.LC-MS purity: 442.24 (M+l)+, 98.89%, 1H NMR (DMSO- d6): δ 10.2 (s, IH), 7.8-7.6 (m, 6H), 7.5 (t, 2H), 7.4 (t, 3H), 6.8 (d, 2H), 6.6 (d, IH), 4.3 (dd, 2H), 3.9 (d, IH), 3.7 (s, 2H), 3.5 (bs, IH), 3.2 (t, IH), 2.7 (m, IH), 2.2 (s, 3H), 2.1 (s, 3H), 1.9 (t, 2H), 1.5 (t, IH), 1.3 (d, IH).
EXAMPLE 58 - Synthesis of N-Biphenyl-4-yl-3-[4-(2, 5-dimethyl-phenylamino)- piperidin-l-yl]-3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.101 g, 0.00039 mole) in DMF (1 mL) was added DIPEA (0.233 g, 0.0018 mole), HOBt (0.053 g, 0.00039 mole) and EDCLHCl (0.138 g, 0.00072 lmole). After 2 minutes (2,5-dimethyl-phenyl)-piperidin-4-yl-amine dihydrochloride (0.101 g, 0.00039 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 60-120 mesh (1% methanol in chloroform) afforded 0.025 g (16%) of N-biphenyl-4-yl-3-[4-(2,5-dimethyl- phenylamino)-piperidin-l-yl]-3-oxo-propionamide. LC-MS purity: 442.24 (M+l)+, 97.08%, 1H NMR (DMSO-d6): δ 10.1 (s, IH), 7.7-7.5 (m, 6H), 7.4 (t, 2H), 7.35 (d, IH), 6.9 (d, IH), 6.5 (t, 2H), 4.5 (d, IH), 4.0 (d, IH), 3.7 (s, IH), 3.5 (s, 2H), 3.4 (t, 2H), 3.1 (t, IH), 2.3 (s, 3H), 2.2 (d, 2H), 2.1 (s, 3H), 1.5 (t, 2H).
EXAMPLE 59 - Synthesis of N-Biphenyl-4-yl-3-[4-(2-tert-butyl-phenylamino)- piperidin-l-yl]-3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.091 g, 0.00036 mole) in DMF (1 mL) was added DIPEA (0.211 g, 0.0016 mole), HOBt (0.048 g, 0.00036 mole) and EDCI.HC1 (0.125 g, 0.00065 mole). After 2 minutes (2-tert-butyl-phenyl)-piperidin-4-yl-amine
dihydrochloride (0.1 g, 0.00033 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the resulting precipitate was isolated by filtration. Purification by column chromatography using silica gel 60-120 mesh (1% methanol in chloroform) afforded 0.062 g (40%) of N-biphenyl-4-yl-3-[4-(2-tert-butyl-phenylamino)- piperidin-l-yl]-3-oxo-propionamide. LC-MS purity: 470.27 (M+l)+, 96.38%, 1H NMR (DMSO- d6): δ 7.8-7.6 (m, 6H), 7.5 (t, 2H), 7.3 (d, IH), 7.1 (d, IH), 7.0 (t, IH), 6.8 (d, IH), 6.6 (t, IH), 4.3 (d, IH), 3.9 (d, 2H), 3.7 (s, IH), 3.6 (s, 2H), 3.2 (t, IH), 2.9 (t, IH), 2.0 (t, 2H), 1.5 (t, IH), 1.4 (s, 9H).
EXAMPLE 60 - Synthesis of N-Biphenyl-4-yl-3- [4-(2,5-difluoro-phenoxy)-piperidin-
1-yl] -3-oxo-propionamide
To a stirred solution of N-biphenyl-4-yl-malonamic acid (0.075 g, 0.0003 mole) in DMF (2mL) was added, HOBt (0.06 g, 0.00045 mole) and DIPEA (0.135g, 0.001 mole). The mixture was then cooled to 10 0C and EDCLHCl (0.086 g, 0.00045 mole) followed by 4-(2,5-difluoro- phenoxy)-piperidine hydrochloride (0.073 g, 0.0003 mole) were added. The resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using neutral aluminium oxide (5% methanol in chloroform) to afford 0.03 g (22%) of N-biphenyl-4-yl- 3-[4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-3-oxo-propionamide. LC-MS purity: 451.18 (M+l)+, 90.58%, 1H NMR (DMSO-d6): δ 10.2 (s, IH), 7.7 (m, 6H), 7.4 (m, 2H), 7.2 (m, 2H), 6.8 (m, IH), 4.7 (m, IH), 3.9 (m, IH), 3.8 (m, IH), 3.6 (s, 2H), 3.4 (m, IH), 2.0 (m, 2H), 1.7 (m, IH), 1.6 (m, IH).
Intermediate 52 - Synthesis of 6-Phenyl-pyridin-3-ylamine
A mixture of toluene (15 roL) and water (5 mL) was degassed with argon for 5 minutes. Sodium carbonate (0.802 g, 0.00454 mole) was then added and the mixture was degassed with argon for 5 minutes. Phenylboronic acid (0.587 g, 0.00454 mole) and 2-chloro-5-nitro-pyridine (0.6 g, 0.0037 8mole) were then added and the resulting mixture was degassed with argon for 5 minutes. Tetrakis(triphenylphosphine)palladium(0) (0.878 g, 0.00076 mole) was added and the mixture was degassed with argon for 5 minutes. The resulting mixture was then heated to reflux for 3 hours. The mixture was diluted with ethyl acetate, and the organic layer was washed with water followed by brine solution. The ethyl acetate layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography using silica gel 60- 120 mesh (5% ethyl acetate in hexane) afforded 0.5 g (66%) of 5-nitro-2-phenyl-pyridine. LCMS purity: 201 (M+l)
+, 98.2%,
1H NMR (DMSOd
6): δ 9.5 (s, IH), 8.55 (dd, IH), 8.1 (m, 2H), 7.9 (d, IH), 7.55 (m, 3H). To a stirred solution of 5-nitro-2-phenyl-pyridine (0.5 g, 0.0025 mole) in THF (10 mL) was added ammonium chloride (1.1 g, 0.020 mole) dissolved in water (15 mL). Methanol (5 mL) was then added, resulting in a clear solution. Zinc powder (1.3 g, 0.020 mole) was then added portionwise and the resulting mixture was stirred for 1 hour. The mixture was filtered through a bed of celite and the filtrate was concentrated under reduced pressure. The residue was extracted with ethyl acetate, and the organic layer was washed with brine solution, dried over Na
2SO
4, and evaporated under reduced pressure to afford 0.35 g (82%) of 6-phenyl-pyridin-3- ylamine. LCMS purity: 171.08 (M+l)
+, 87.9%,
1H NMR (DMSOd
6): δ 8.12 (d, IH), 8.0 (d, 2H), 7.72 (d, IH), 7.48 (t, 2H), 7.36 (t, IH), 7.1 (dd, IH), 5.6 (s, 2).
Intermediate 53 - Synthesis of 3-[4-(2-Chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3- oxo-propionic acid
To a stirred solution of malonic acid monoethyl ester (0.5 g, 0.00377 mole) in DMF (10 mL) was added DIPEA (1.18 g, 0.0094 mole), HOBt (0.608 g, 0.0045 mole) and EDCI.HC1 (0.865 g, 0.0045 mole). After 2 minutes 4-(2-chloro-5-fluoro-ρhenoxy)-piperidine hydrochloride (1 g, 0.003 7mole) and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layers were dried over sodium sulfate and concentrated under reduced pressure to get afford 0.9 g (70%) of 3-
[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo-propionic acid ethyl ester. LC-MS purity: 344.78 (M+l)
+, 82.56%. To a stirred solution of 3-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]- 3-oxo-propionic acid ethyl ester (0.9 g, 0.0026 mole) in a mixture of THF (10 mL), methanol (3 mL) and H
2O (2 mL) was added LiOH-H
2O (0.218 g, 0.0052 mole) at ambient temperature and the resulting mixture was stirred for 30 minutes. Volatiles were then evaporated and the resulting residue was diluted with water, acidified with 10% aqueous HCl solution. The product was extracted with ethyl acetate, and the organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.6 g (73%) of 3-[4-(2-chloro-5- fluoro-phenoxy)-piperidin-l-yl]-3-oxo-propionic acid. LC-MS purity: 316.72 (M+l)
+, 75.8%.
EXAMPLE 61 - Synthesis of 3-[4-(2-Chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo- N-(6-phenyl-pyridin-3-yl)-propionamide
To a stirred solution of 3-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo-propionic acid (0.1 g, 0.00032 mole) in DMF (5 mL) was added DMAP (0.059 g, 0.00048 mole), HOBt (0.051 g, 0.00038 mole) and EDCI.HC1 (0.073 g, 0.00038 mole). After 2 minutes 6-phenyl- pyridin-3-ylamine (0.06 g, 0.00035 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layers were dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (50% ethyl acetate in hexane) to afford 0.026 g (19%) of 3-[4-(2-chloro-5-fluoro- phenoxy)-piperidin-l-yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)-propionamide. LC-MS purity: 468 (M+l)+, 93.7%, 1H NMR (CDCl3): δ 10.5 (s, IH), 8.8 (s, IH), 8.3 (d, IH), 8.0 (d, 2H), 7.7 (d, IH), 7.4 (m, 4H), 6.7 (m, 2H), 4.7 (m, IH), 4.2 (m, IH), 3.8 (m, IH), 3.7 (m, 2H), 3.55 (s, 2H), 2.0 (m, 4H).
Intermediate 54 - Synthesis of 5-Phenyl-pyridin-2-ylamine
A mixture of toluene (80 mL) and water (2OmL) was degassed with argon for 5 minutes. Sodium carbonate (9.63 g, 0.0909 mole) was added and the mixture was degassed with argon for 5 minutes. Phenylboronic acid (6.65 g, 0.05454 mole) and 2-amino-5-iodo-pyridine (10.0 g, 0.04545 mole) were then added and the mixture was degassed with argon for 5 minutes.
Tetrakis(triphenylphosphine)palladium(0) (5.252 g, 0.00454 mole) was added was degassed with argon for 5 minutes. The resulting mixture was then heated to reflux for 3 hours. The mixture was diluted with ethyl acetate, and the organic layer was washed with water followed by brine solution. The ethyl acetate layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography using silica gel 60-120 mesh (1% methanol in chloroform) afforded 3.2 g (41%) of 5-phenyl-pyridin-2-ylamine. LC-MS purity: 171.08 (M+l)+, 87%.
EXAMPLE 62 - Synthesis of 3-[4-(2-Chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo- N-(5-phenyl-pyridin-2-yl)-propionamide
To a stirred solution of 5-phenyl-pyridin-2-ylamine (0.041 g, 0.00024 mole) in THF (3 mL) was added 3-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo-propionic acid (0.08 g, 0.00025 mole) followed by DIC (0.036 g, 0.00028 mole). The mixture was heated to reflux for 3 hours, and then concentrated under educed pressure. The resulting residue was stirred with cold ether. The organic layers were removed by filtration and concentrated under reduced pressure to afford 0.021 g (18%) of 3-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-3-oxo-N-(5-phenyl- pyridin-2-yl)-propionamide. LC-MS purity: 468.14 (M+l)+, 98%, 1H NMR (DMSOd6) δ 11.2 (bs, IH), 8.2 (t, 2H), 8.0 (d, IH), 7.4 (m, 4H), 7.1 (t, IH), 6.6 (d, 2H), 4.6 (s, IH), 4.0 (d, IH), 3.7 (m, IH), 3.6 (m, 3H), 2.1 (bs, 9H), 1.8 (t, 4H).
Intermediate 55 - Synthesis of 3-[4-(2-ChIoro-phenoxy)-piperidin-l-yl]-3-oxo-
propionic acid
To a stirred solution of malonic acid monoethyl ester (1.2 g, 0.0089 mole) in DMF (10 mL) was added DIPEA (2.5g, 0.02 mole), HOBt (1.3 g, 0.0097 mole) and EDCI.HC1 (1.9 g, 0.0097 mole). After 2 minutes 4-(2-chloro-phenoxy)-piperidine hydrochloride (2 g, 0.0081 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layers were dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography using silica gel 60-120 mesh (60% ethyl acetate in hexane) to afford 1.1 g (42%) of 3-[4-(2-chloro-phenoxy)-piperidm- 1 -yl]-3-oxo-propionic acid ethyl ester. LC-MS purity: 326.11 (M+l)+, 90.2%. To a stirred solution of 3-[4-(2-chloro-phenoxy)-piperidin-l-yl]-3- oxo-propionic acid ethyl ester (1.1 g, 0.0034 mole) in a mixture of THF (20 mL), methanol (4 mL) and H2O (4 mL) was added LiORH2O (0.286 g, 0.0068 mole) at ambient temperature. The resulting mixture was stirred for 30 minutes. Volatiles were then evaporated and the resulting residue was diluted with water, acidified with 10% aqueous HCl solution. The resulting precipitate was isolated by filtration and dried to afford 0.75 g (75%) of 3-[4-(2-chloro-phenoxy)-piperidin-l- yl]-3-oxo-propionic acid.
EXAMPLE 63 - Synthesis of 3-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-3-oxo-N-(6- phenyl-pyridin-3-yl)-propionamide
To a stirred solution of 3-[4-(2-chloro-phenoxy)-piperidin-l-yl]-3-oxo-propionic acid (0.08 g, 0.00027 mole) in DMF (3 mL) was added DMAP (0.049 g, 0.00040 mole), HOBt (0.043 g, 0.00032 mole) and EDCI.HC1 (0.06 g, 0.00032 mole). After 2 minutes 6-phenyl-pyridin-3- ylamine (0.055 g, 0.00032 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate
ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.0642 g (53%) of 3-[4-(2-chloro-phenoxy)- piperidin-l-yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)-propionamide. LC-MS purity: 450.15 (M+l)+, 99.25%, 1H NMR (DMSOd6): δ 10.4 (s, IH), 8.8 (s, IH), 8.2 (dd, IH), 8.1-8.0 (m, 2H), 8.0-7.9 (m, IH), 7.5-7.2 (m, 6H), 7.0-6.9 (m, IH), 4.8-4.7 (m, IH), 3.8-3.7 (m, 2H), 3.7-3.6 (s, 2H), 3.6- 3.5 (m, 2H), 2.1-1.5 (m, 4H).
Intermediate 56 - Synthesis of N-(6-PhenyI-pyridin-3-yl)-malonamic acid
To a stirred solution of 5-amino-2-phenyl-pyridine (4.0 g, 0.0235 mol) in dichloromethane
(40 mL) was added ethyl malonyl chloride (5.307 g, 0.035 mol) dropwise. The resulting mixture was stirred for 1 hour. The mixture was diluted with water and the product was extracted with dichloromethane. The organic layer was washed with saturated sodium bicarbonate solution, brine, dried over Na2SO4 and evaporated to afford 5.46 g (80%) of N-(6-phenyl-pyridin-3-yl)-malonamic acid ethyl ester. To a solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid ethyl ester (5.46 g, 0.019 mol) in the mixture of methanol (27.5 mL), THF (55 mL) and H2O (16.5 mL) was added LiOH-H2O (1.212 g, 0.0288 mol). The resulting reaction mixture was stirred for 1 hour at ambient temperature then concentrated. The resulting residue was diluted with water, acidified with concentrated HCl and the product was extracted with ethyl acetate. The ethyl acetate was washed with brine, dried over Na2SO4 and evaporated under reduced pressure to afford 4.0 g (82%) of N- (6-phenyl-pyridin-3 -yl)-malonamic acid.
EXAMPLE 64 - Synthesis of 3-[4-(2-Chloro-phenylamino)-piperidm-l-yl]-3-oxo-N-(6- phenyl-pyridin-3-yl)-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid (0.07 g, 0.00024 mole) in DMF (2 mL) was added, DIPEA (0.153 g, 0.00120 mole), HOBt (0.039 g, 0.00029 mole) and
EDCI.HC1 (0.055 g, 0.00029 mole). After 2 minutes (2-chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (0.08 Ig, 0.00029 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.082 g (77%) of 3-[4-(2-chloro-phenylamino)- piperidin-l-yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)-propionamide. LC-MS purity: 449.17 (M+l)+, 98.46%, 1H NMR (DMSOd6): δ 10.5 (s, IH), 8.8 (s, IH), 8.2 (dd, IH), 8.1 (d, 2H), 8.0 (d, IH), 7.5-7.2 (m, 2H), 7.3 (d, IH), 7.2 (t, IH), 6.9 (d, IH), 6.4 (t, IH), 4.9 (d, IH), 4.4 (d, IH), 4.0 (d, IH), 3.7-3.6 (m, 3H), 3.3-3.2 (m, IH), 2.9-2.7 (m, IH), 2.0 (t, 2H), 1.6-1.5 (m, IH), 1.4-1.3 (m, IH).
EXAMPLE 65 - Synthesis of 3-[4-(2-Bromo-phenylamino)-piperidin-l-yI]-3-oxo-N-(6- phenyl-pyridin-3-yl)-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid (0.07 g, 0.00024 mole) in DMF (2 mL) was added DIPEA (0.153 g, 0.00120 mole), HOBt (0.039 g, 0.00029 mole) and EDCI.HC1 (0.055 g, 0.00029 mole). After 2 minutes (2-bromo-phenyl)-piperidin-4-yl-amine dihydrochloride (0.093 g, 0.00029 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 77.9 mg (66%) of 3-[4-(2-bromo- phenylamino)-piperidin- 1 -yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)-propionamide. LC-MS purity: 493.12 (M+l)
+, 99.22%,
1H NMR (DMSO-d
6): δ 10.5 (s, IH), 8.8 (d, IH), 8.2 (dd, IH), 8.1 (d, IH), 8.0 (d, IH), 7.6-7.4 (m, 4H), 7.2 (t, IH), 6.9 (d, IH), 6.6 (t, IH), 4.7 (d, IH), 4.4 (d, IH), 4.0(d, IH), 3.7-3.6 (m, 3H), 3.3-3.2 (m, IH), 2.0 (t, 2H), 1.6-1.5 (m, IH), 1.4-1.3 (m, IH).
EXAMPLE 66 - Synthesis of 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-[4-(2- trifluoromethyl-phenylamino)-piperidm-l-yl]-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid (0.07 g, 0.00024 mole) in DMF (2 mL) was added DIPEA (0.153 g, 0.00120 mole), HOBt (0.039 g, 0.00029 mole) and EDCLHCl (0.055 g, 0.00029 mole) After 2 minutes (2-trifluoromethyl-phenyl)-piperidin-4-yl- amine dihydrochloride (0.075 g, 0.00029 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 73.7 mg (64%) of 3-oxo-N-(6-phenyl-pyridin- 3-yl)-3-[4-(2-trifluoromethyl-phenylamino)-piperidin-l-yl]-propionamide. LC-MS purity: 483.19 (M+l)+, 95.78%, 1H NMR (DMSOd6) δ 10.5 (s, IH), 8.8 (d, IH), 8.2 (dd, IH), 8.1 (d, 2H), 8.0 (d, IH), 7.5-7.4 (m, 4H), 7.0 (d, IH), 6.8 (t, IH), 4.7 (d, IH), 4.4 (d, IH), 4.0 (d, IH), 3.8-3.6 (m, 3H), 3.3-3.2 (m, IH), 2.9-2.8 (m, IH), 2.0 (m, IH), 1.6-1.5 (m, IH), 1.5-1.4 (m, IH).
EXAMPLE 67 - Synthesis of 3-[4-(2-Chloro-phenylsulfanyl)-piperidin-l-yl]-3-oxo-N- (6-phenyl-pyridin-3-yl)-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid (0.07 g, 0.00024 mole) in DMF (1 mL) was added DIPEA (0.153 g, 0.00120 mole), HOBt (0.039 g, 0.00029 mole) and EDCLHCl (0.055 g, 0.00029 mole). After 2 minutes (2-chloro-phenylsulfanyl)-piperidin-4-yl- amine hydrochloride (0.076 g, 0.00029 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.07 g (63%) of 3-[4-(2-chloro-phenylsulfanyl)- piperidin-l-yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)-propionamide. LC-MS purity: 466.13 (M+l)+, 93.65%, 1H NMR (DMSO-d6): δ 10.5 (s, IH), 8.8 (d, IH), 8.2 (d, IH), 8.1 (d, 2H), 8.0 (d, IH), 7.6-7.2 (m, 6H), 4.3 (d, IH), 3.9 (d, IH), 3.6-3.5 (m, 3H), ), 3.0 (t, IH), 2.0 (m, 2H), 1.7-1.6 (m,
IH), 1.5-1.4 (m, IH).
EXAMPLE 68 - Synthesis of 3-[4-(2-Bromo-phenylsulfanyl)-piperidm-l-yl]-3-oxo-N- (6-phenyl-pyridin-3-yl)-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid (0.07 g, 0.00024 mole) in DMF (1 mL) was added DIPEA (0.153 g, 0.00120 mole), HOBt (0.039 g, 0.00029 mole) and EDCI.HC1 (0.055 g, 0.00029 mole). After 2 minutes (2-bromo-phenylsulfanyl)-piperidin-4-yl- amine hydrochloride (0.088 g, 0.00028 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 73.2 mg (60%) of 3-[4-(2-bromo- phenylsulfanyl)-piperidin- 1 -yl]-3-oxo-N-(6-phenyl-pyridin-3-yl)-propionamide. LC-MS purity: 510.08 (M+l)+, 93.64%, 1H NMR (DMSO-d6): δ 10.5 (s, IH), 8.8 (d, IH), 8.2 (d, IH), 8.1 (d, 2H), 8.0 (d, IH), 7.84-7.7 (m, 2H), 7.7 (t, IH)3 7.54-7.36 (m, 4H), 4.3 (d, IH), 3.9 (d, IH), 3.8-3.7 (m, IH), 3.6 (s, 2H), 3.3-3.2 (m, IH), 3.0-2.9 (m, IH), 2.0 (t, 2H), 1.6-1.5 (m, IH), 1.5-1.4 (m, IH).
EXAMPLE 69 - Synthesis of 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-[4-(2- trifluoromethyl-phenylsulfanyl)-piperidin-l-yl]-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid (0.07 g, 0.00024 mole) in DMF (1 mL) was added DIPEA (0.153 g, 0.00120 mole), HOBt (0.039g, 0.000286 mole) and EDCLHCl (0.055 g, 0.00029 mole). After 2 minutes (2-trifluoromethyl-phenylsulfanyl)-piperidin- 4-yl-amine hydrochloride (0.085 g, 0.00029 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate
and concentrated under reduced pressure to afford 80.2 mg (67%) of 3-oxo-N-(6-phenyl-pyridin- 3-yl)-3-[4-(2-trifluoromethyl-phenylsulfanyl)-piperidin-l-yl]-propionamide. LC-MS purity: 501.15 (M+l)+, 95.38%, 1H NMR (DMSO-d6): δ 10.5(s, IH), 8.8 (d, IH), 8.2 (d, IH), 8.1 (d, 2H), 8.0 (d, IH), 7.7 (d, IH), 7.6-7.4 (m, 5H), 7.2 (t, IH), 4.3 (d, IH), 3.9 (d, IH), 3.7-3.6 (m, 3H), 3.3- 3.2 (m, IH), 3.0 (m, IH), 2.0 (m, 2H), 1.7-1.6 (m, IH), 1.5-1.4 (m, IH).
EXAMPLE 70 - Synthesis of 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-[4-(2- trifluoromethyl-phenoxy)-piperidin-l-yl]-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic acid (0.07 g, 0.00024 mole) in DMF (1 niL) was added DIPEA (0.153 g, 0.00120 mole), HOBt (0.039 g, 0.00029 mole) and EDCLHCl (0.055 g, 0.00029 mole). After 2 minutes (2-trifluoromethyl-phenoxy)-piperidin-4-yl- amine hydrochloride (0.080 g, 0.00029 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 57.5 mg (50%) of 3-oxo-N-(6-phenyl-pyridin- 3-yl)-3-[4-(2-trifluoromethyl-phenoxy)-piperidin-l-yl]-propionamide. LC-MS purity: 484.18 (M+l)
+, 98.83%,
1H NMR (DMSOd
6): δ 10.5 (s, IH), 8.8 (d, IH), 8.2 (d, IH), 8.1 (d, 2H), 8.0 (d, IH), 7.7-7.6 (m, 2H), 7.5-7.3 (m, 4H), 7.1 (t, IH), 4.9 (m, IH), 3.7-3.5 (m, 6H), 2.0-1.6 (m, 4H).
EXAMPLE 71 - Synthesis of 3-Oxo-N-(6-phenyl-pyridin-3-yl)-3-(4-o-toIylamino- piperidin-l-yl)-propionamide
To a stirred solution of N-(6-phenyl-pyridin-3-yl)-malonamic (0.07 g, 0.00024 mole) in DMF (1 mL) was added DIPEA (0.153 g, 0.00120 mole), HOBt (0.039 g, 0.00029 mole) and
EDCI.HC1 (0.055 g, 0.00029 mole). After 2 minutes piperidin-4-yl-o-tolyl-amine dihydrochloride (0.075 g, 0.00029 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The organic layer was washed with water followed by brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 0.075 g (74%) of 3-oxo-N-(6-phenyl-pyridin-3-yl)- 3-(4-o-tolylamino-piperidin-l-yl)-propionamide. LC-MS purity: 429.22 (M+l)+, 98.41%, 1H NMR (DMSOd6): δ 10.5 (s, IH), 8.8 (d, IH), 8.2 (d, IH), 8.1 (d, 2H), 8.0 (d, IH), 7.5-7.3 (m, 2H), 7.0 (t, 2H), 6.7 (d, IH), 6.5(t, IH), 4.5-4.3 (m, 2H), 4.0 (d, IH), 3.7-3.5 (m, 3H), 3.3-3.2 (m, IH), 2.9-2.8 (m, IH), 2.1 (s, 3H) 2.0-1.9 (m, 2H).
Intermediate 57 - Synthesis of N-(3-Phenyl-[l,2,4]thiadiazol-5-yl)-malonamic acid
To a stirred solution of 3-phenyl-[l,2,4]thiadiazol-5-ylamine (0.25 g, 0.00141 mol) in chloroform (2.5 mL) was added mono ethylmalonylchloride (0.233 g, 0.00155 mol) dropwise, the resulting mixture was stirred for 3 hours. The mixture was then concentrated and the residue was stirred in ethyl acetate. The filtrate was removed and concentrated to afford 0.26 g (63%) of N-(3- phenyl-[l,2,4]thiadiazol-5-yl)-malonamic acid ethyl ester. LC-MS purity: 292.07 (M+l)+, 67%. To a solution of N-(3-phenyl-[l,2,4]thiadiazol-5-yl)-malonamic acid ethyl ester (0.26 g, 0.00089 mol) in a mixture of methanol (3 mL) and H2O (1 mL) was added LiORH2O (0.112 g, 0.00267 mol). The reaction mixture was stirred for 4 hours at ambient temperature tehnconcentrated. The resulting residue was diluted with cold water, acidified with 2N HCl and the product was extracted with ethyl acetate. The ethyl acetate layer washed with brine, dried over Na2SO4 and evaporated under reduced pressure to afford 0.105 g (45%) of N-(3-phenyl-[l,2,4]thiadiazol-5-yl)-malonamic acid. LCMS: 264.27 (M+l)+: 97.64%.
EXAMPLE 72 - Synthesis of 3-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-3-oxo-N-(3- phenyl-[l,2,4]thiadiazol-5-yl)-propionamide
To a stirred solution of N-(3-phenyl-[l,2,4]thiadiazol-5-yl)-malonamic acid (0.07 g, 0.00027 mole) in THF ( 3mL) was added, 4-(2-chloro-phenoxy)-piperidine hydrochloride (0.072 g, 0.00029 mol) followed by DIC (0.04 g, 0.00031 mole). The resulting mixture was heated to reflux for 4 hours then concentrated. The resulting residue was purified by column chromatography using neutral aluminium oxide (0.5% methanol in dichloromethane) to afford 0.045 g (37%) of 3-[4-(2-chloro-phenoxy)-piperidin-l-yl]-3-oxo-N-(3-phenyl-[l,2,4]thiadiazol-5- yl)-propionamide. LC-MS purity: 457.1 (M+l)
+, 84.85%'
1H NMR (CDCl
3): δ 8.2 (s, 2H), 7.5 (m, 4H), 7.2 (m, IH), 7.0 (d, 2H), 4.7 (s, IH), 4.2 (d, IH), 3.8 (m, IH), 3.6 (m, 5H), 2.0 (dd, 4H), 2.6 (bs, 3H).
Intermediate 58 - Synthesis of 4-[l, 2, 4] Oxadiazol-3-yI-phenylamine
To a stirred solution of 4-nitrobenzonitrile (1 g, 0.0068 mol) in ethanol (20 mL) and water (8 mL) was added hydroxylamine hydrochloride (1.9 g, 0.0272 mol) followed by sodium carbonate (2.2 g, 0.0204 mol). The resulting mixture was heated to reflux at 85° C under an atmosphere of nitrogen for 2 hoursThe reaction was monitored by TLC (50% Ethyl acetate in Hexane). The volatiles were evaporated and the residue was extracted with ethyl acetate. The ethyl aetate was washed with brine solution, dried over Na2Sθ4 and concentrated under reduced pressure to get afford 1.2 g (98%) of N-hydroxy-4-nitro-benzamidine. 1H NMR (DMSOd6): δ 10.2 (s, IH), 8.24 (d, 2H), 7.97 (d, 2H), 6.03 (s, 2H). To a stirred solution of N-hydroxy-4-nitro- benzamidine (1.2 g, 0.0066 mol) in THF (15 mL) was added triethyl orthoformate (2.93 g, 0.0198mol). The resulting mixture was cooled to between 0 and 5 0C. Boron triflouride dimethyl ether (0.9 g, 0.0079 mol) was then added dropwise and the resulting mixture was stirred at ambient temperature for 3 hours. Volatiles were evaporated under reduced pressure and the resulting residue was washed with ether and dried to afford 0.65 g (55%) of 3-(4-nitro-phenyl)-[l, 2, 4]oxadiazole. To a stirred solution of 3-(4-nitro-phenyl)-[ 1 , 2, 4]oxadiazole (0.2 g, 0.001 mol) in THF (15 mL) was added ammonium chloride (0.214 g, 0.004 mol) in water (5 mL) followed by zinc powder (0.262 g, 0.004 mol) portionwise. The resulting mixture was stirred at ambient
temperature for 1 hr and then heated to 65 0C for 5 hours. The mixture was then filtered through celite and the organic layers were concentrated under reduced pressure. The residue was extracted with ethyl acetate. The organic layers were washed with brine solution, dried over Na2SO4 and evaporated under reduced pressure to afford 0.155 g (92%) of 4-[l, 2, 4]oxadiazol-3-yl- phenylamine. 1H NMR (DMSO-d6): δ 9.5 (s, IH), 7.7 (d, 2H), 6.7 (d, 2H), 5.8 (s, 2H).
EXAMPLE 73 - Synthesis of 3-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-N-(4- [l,2,4]oxadiazol-3-yl-phenyl)-3-oxo-propionamide
To a stirred solution of 3-[4-(2-chloro-phenoxy)-piperidin-l-yl]-3-oxo-propionic acid (0.1 g, 0.00034 mole) in DMF (2 mL) was added DMAP (0.063 g, 0.00051 mole), HOBt (0.055 g, 0.00041 mole) and EDCI.HC1 (0.079 g, 0.00041 mole). After 2 minutes 4-[l, 2, 4]-oxadiazol-3-yl- phenylamine (0.06 g, 0.00037 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was dried over sodium sulfate and concentrated under reduced pressure to afford 0.09 g (61%) of 3-[4-(2-chloro-phenoxy)-piperidin-l-yl]-N-(4-[l,2,4]oxadiazol- 3-yl-phenyl)-3-oxo-propionamide. LC-MS purity: 441 (M+l)
+, 94.4%,
1H NMR (CDCl
3): δ 10.5(s, IH), 8.8 (s, IH), 8.1 (d, 2H), 7.74 (d, 2H), 7.4 (d, IH), 7.2 (d, IH), 6.96 (t, 2H), 4.7 (q, IH), 4.1 (m, IH), 3.8 (m, IH), 3.6 (m, 2H), 3.55 (s, 2H), 2.0 (m, 5H).
Intermediate 59 - Synthesis of N-(5-Phenyl-thiazol-2-yl)-malonamic acid
To a stirred solution of 5-phenyl-thiazol-2-ylamine (0.4 g, 0.0022 mol) and DIEA (0.73 g,0.0056 mol) in chloroform (4 mL) was added mono-ethylmalonyl chloride (0.375 g, 0.0024 mol) dropwise at 0 0C and the resulting mixture was stirred at ambient temperature for 20 minutes. The reaction mixture was then diluted with cold water and the product was extracted with chloroform.
The chloroform was washed with saturated sodium bicarbonate solution and brine, dried over Na2SO4 and evaporated under reduced pressure to afford 0.3 g (46%) of N-(5-phenyl-thiazol-2- yl)-malonamic acid ethyl ester. To a solution of N-(5-phenyl-thiazol-2-yl)-malonamic acid ethyl ester (0.292 g, 0.001 mol) in the mixture of methanol (1 mL), THF (1.5 mL) and H2O (1 mL) was added LiOH-H2O (0.084 g, 0.002 mol) and the resulting mixture was stirred for 1 hour. The reaction mixture was then concentrated. The residue was diluted with water, acidified with concentrated HCl and the product was extracted with ethylacetate. The ethyl acetate was washed with brine, dried over Na2SO4 and evaporated under reduced pressure to afford 0.23 g (87%) of N- (5-phenyl-thiazol-2-yl)-malonamic acid.
EXAMPLE 74 - Synthesis of 3-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-3-oxo-N-(5- phenyl-thiazol-2-yl)-propionamide
To a stirred solution of N-(5-phenyl-thiazol-2-yl)-malonamic acid (0.075 g, 0.00028 mole) in DMF (2 mL) was added DIPEA (0.11 g, 0.00085 mole), HOBt (0.038 g, 0.00028 mole) and EDCI.HC1 (0.065 g, 0.00034 mole). After 2 minutes 4-(2-chloro-phenoxy)-piperidine hydrochloride (0.078 g, 0.00031 mole) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was dried over sodium sulfate and concentrated under reduced pressure to afford 0.031 g (24%) of 3-[4-(2-chloro-phenoxy)-piperidin-l -yl]-3-oxo-N-(5-phenyl- thiazol-2-yl)-propionamide. LC-MS purity: 456.11 (M+l)
+, 93.27%,
1H NMR (CDCl
3): δ 11.6 (s, IH), 7.9 (d, 2H), 7.44 (t, 3H), 7.3 (d, IH), 7.22 (d, IH), 7.14 (s, IH), 6.92 (d, 2H), 4.7 (s, IH), 4.1 (m, IH), 3.8 (m, IH), 3.7-3.5 (m, 4H), 2.0 (d, 4H).
EXAMPLE 75 - Synthesis of 3-[4-(2-Chloro-phenylamino)-piperidin-l-yl]-3-oxo-N-(5- phenyl-thiazol-2-yl)-propionamide
To a stirred solution of N-(5-phenyl-thiazol-2-yl)-malonamic acid (0.075 g, 0.00028 mole) in DMF (2 mL) was added DIPEA (0.11 g, 0.00085 mole), HOBt (0.038 g, 0.00028 mole) and EDCLHCl (0.065 g, 0.00034 mol). After 2 minutes (2-chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (0.077 g, 0.00031 mol) was added and the resulting mixture was stirred overnight. The reaction mixture was then diluted with cold water and the product was extracted with ethyl acetate. The ethyl acetate layer was dried over sodium sulfate and concentrated under reduced pressure. The residue was recrystallized from a mixture of ethyl acetate and hexane to afford 0.035 g (28%) of3-[4-(2-chloro-phenylamino)-piperidin-l-yl]-3-oxo-N-(5-phenyl-thiazol-2-yl)- propionamide. LC-MS purity: 455.11 (M+l)+, 98.03%, 1H NMR (CDCl3): δ 11.6 (s, IH), 7.9 (d, 2H), 7.4 (t, 3H), 7.3 (t, 2H), 7.2 (m, 2H), 7.1, 6.70 (m, 2H), 4.5 (d, IH), 4.2 (d, IH), 3.9 (d, IH), 3.6 (s, 3H), 3.3 (t, IH), 3.1 (t, IH), 2.2 (s, 2H), 1.5 (m, 2H).
Intermediate 60 - Synthesis of l-(Biphenyl-4-ylcarbamoyl)-cyclopropanecarboxylic acid
KOH (420 mg, 7.5 mmol) was added to a solution cyclopropane- 1,1-dicarboxylic acid diethyl ester (1 g, 6.3 mmol) in methanol (7 mL). The reaction mixture was stirred for 4 hours at ambient temperature then concentrated. The resulting residue was diluted with water, acidified with cone. HCl and the product was extracted with dichloromethane. The dichloromethane layer was dried over sodium sulfate and concentrated under reduced pressure to afford 680 mg (75%) of cyclopropane- 1,1-dicarboxylic acid methyl ester. HOBt (764 mg, 5.6 mmol), DMAP (1.72 g, 14.15 mmol) and EDCLHCl (1.08 g, 5.6 mmol) followed by biphenyl-4-ylamine (957 mg, 5.6 mmol) were added to a stirred solution of cyclopropane- 1,1-dicarboxylic acid methyl ester (680 mg, 4.7 mmol) in DMF (7 mL) and the resulting mixture was stirred at ambient temperature overnight. The mixture was then diluted with water and the product was extracted with ethyl
acetate .The ethylacetate layer was washed with brine solution and concentrated to afford 1.1 g (79%) of 1 -(biphenyl-4-ylcarbamoyl)-cyclopropanecarboxylic acid methyl ester. LiOHLH2O (234 mg, 5.5 mmol) was added to a solution of l-(biphenyl-4-ylcarbamoyl)-cyclopropanecarboxylic acid methyl ester (1.1 g, 3.7 mmol) in a mixture of methanol (5 mL), THF (11 mL) and H2O (3 mL) and the resulting mixture was stirred for 2 hours at ambient temperature. The reaction mixture was then concentrated and the residue was diluted with water, acidified with cone. HCl. The resulting precipitate was isolated by filtration and dried to afford 340mg (33%) of 1 -(biphenyl-4- ylcarbamoyl)-cyclopropane carboxylic acid.
EXAMPLE 76 - Synthesis of l-[4-(2-Chloro-phenoxy)-piperidine-l-carbonyl]- cyclopropane carboxylic acid biphenyl-4-ylamide
HOBt (57 mg, 0.42 mol) and DIPEA (137 mg, 1.06 mmole), EDCLHCl (82 mg, 0.42 mol) followed by 4-(2-chloro-phenoxy)-piperidine hydrochloride (105 mg, 0.42 mol) were added to a stirred solution of l-(biphenyl-4-ylcarbamoyl)-cyclopropanecarboxylic acid (100 mg, 0.35 mmol) in DMF (2.0 mL) and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was then diluted with water. The product was extracted with ethylacetate and the organic layers were washed with brine and concentrated to afford 78 mg (46%) of l-[4-(2-chloro- phenoxy)-piperidine-l-carbonyl]-cyclopropanecarboxylic acid biphenyl-4-ylamide. LCMS: 475.17 (M+l)+, 92.81%, 1H NMR (DMSO-d6): δ 9.8 (s, IH), 7.8-7.5 (d, 2H), 7.7-7.6 (t, 4H), 7.5- 7.4 (m, 3H), 7.4-7.2 (m, 3H), 7.0-6.9 (m, IH), 4.7 (m, IH), 3.8-3.6 (m, 2H), 3.5 (m, 2H), 2.0-1.8 (m, 2H), 1.7-1.6 (m, 2H), 1.4 (m,2H), 1.3-1.2 (m, 2H).
EXAMPLE 77 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(5-bromo- 2-methoxy-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (45mg, 0.35mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (26mg, O.lmmol) in DMF (ImL) followed by HOBt (20mg, 0.15mmol) and EDCLHCl (28mg, 0.15mmol). After 2 minutes of stirring, 2-amino-l-[4-(5- bromo-2-methoxy-phenylamino)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 2 and 5 of the General Scheme) (0.03g, O.OOOlmmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure Purification by column chromatography (using neutral alumina and 5% MeOH in CHCl
3 as eluent) to afford 60mg (35%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(5-bromo-2-methoxy- phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]
+: 512.4, 91
0Zo
5 1H NMR (300MHz, DMSO-d
6): δlθ.2 (m, IH), 7.9(m, IH), 7.7(m, 7H), 7.4(m, 2H), 7.2(m, IH), 3.6(m, 5H), 2.0(m, 2H), 1.8(m, 2H).
EXAMPLE 78 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (360mg, 2.8mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (200mg, 0.8mmol) in DMF (2mL) followed by HOBt (131mg, 0.97mmol) and EDCLHCl (187mg, 00.97mmol). After 2 minutes of stirring, 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (200mg, 0.8mmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography (using neutral alumina and 5% MeOH in CHCl3 as eluent) to afford 62mg (16.2%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 473.17, 91%, 1H NMR (300MHz, DMSO-d6): δ8.1(m, IH), 7.8(d, 2H), 7.5(m, 3H), 7.4(m, 4H), 7.1(s, IH), 4.8(m, IH), 4.2(d, 2H), 3.8(m, IH), 3.6(m, IH), 3.5(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 79 - Synthesis of S-Phenyl-lH-pyrazole-θ-carboxylic acid {2-[4-(2-cyano- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (1.3g, O.Olmmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (720mg, 2.9mmol) in DMF (4mL) followed by HOBt (459mg, 3.4mmol) and EDCLHCl (664mg, 3.4mmol). After 2 minutes of stirring, 2-[l-(2-amino-acetyl)- piperidin-4-yloxy]-benzonitrile hydrochloride (prepared by method used for the synthesis of Intermediate 15) (600mg, 2.9mmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography (using neutral alumina and 5%MeOH in CHCl3 as eluent) afforded 87mg (6%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-cyano-phenoxy)-piperidin-l-yl]-2- oxo-ethyl} -amide. LC/MS [M+H]+: 430.18, 96.4%/H NMR (300MHZ, DMSOd6): δ8.0(m, IH), 7.8(m, 3H), 7.2(m, IH), 7.4(m, 4H), 7.2(m, 2H), 4.9(m, IH), 4.2(bs, 2H), 3.7(m, 2H), 3.5(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 80 - Synthesis of 5-(2-Fluoro-phenyI)-lH-pyrazole-3-carboxylic acid {2- [4-(2-chIoro-phenylamino)-piperidin-l-yI]-2-oxo-ethyl}-amide
DIPEA (860mg, 0.66mmol) was added to a stirred solution of [(5-(2-fluoro-phenyl-lH- pyrazole-3-carbonyl)-amino] -acetic acid (prepared by the method used for the synthesis of Intermediate 30, starting from (2'-fluorophenyl)acetophenone) (50mg, 0.19mmol) in DMF (2mL) followed by HOBt (27mg, 0.19mmol) and EDCLHCl (39mg, 0.19mmol). After 2 minutes (2- chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (47mg, 0.189mmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold
water, the precipitate was collected to afford 44mg (50.86%Yield) of 5-(2-fluoro-phenyl)-lH- pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylammo)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]
+:456.1, 94.91%,
1H NMR (300MHz, DMSO-d
6): 513.7(d, IH), 7.9(m, 2H), 7.2(m, 4H), 7.0(m, 2H), 6.8(m, 2H), 6.6(m, IH), 4.9(d, IH), 4.3(d, IH), 4.1(d, 2H), 3.8(m,lH), 3.6(m, IH), 3.1(m, IH), 2.7(m, IH), 1.9(m, 2H), 1.3(m, 2H).
EXAMPLE 81 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2,4- difluoro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (232mg, 1.8mmol) was added to a stirred solution of [(5 -phenyl- 1 H-pyrazole-3- carbonyl)-amino]-acetic acid (148mg, O.όmmol) in DMF (2mL) followed by HOBt (95mg, 0.7mmol) and EDCLHCl (137mg, 0.7mmol. After 2 minutes (2,4-difluoro-phenyl)-piperidin-4-yl- amine dihydrochloride (prepared by the method used for the synthesis of Intermediate 3) (150mg, O.όmmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer thus collected was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 5% MeoH in CHCl
3 as eluent) to afford 43mg (16.5%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2,4- difluoro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]
+: 440.18, 94.52%.
]H NMR ( 300MHz, DMSO-d
6): δl3.8(s, IH), 8.1(m, IH), 7.8(m, 2H), 7.4(m, 4H), 7.0(m, IH),
6.9(m, 2H), 5.0(m, IH), 4.4(m, IH), 4.2(m, 2H), 3.5(m,lH), 3.2(m, IH), 2.8(m, IH), 2.0(m, 2H), 1.5(m, 2H).
EXAMPLE 82 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(5-fluoro- 2-trifluoromethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (232mg, 1.8mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (148mg, O.όmmol) in DMF (2mL) followed by HOBt (95mg, 0.7mmol) and EDCLHCl (137mg, 0.7mmol). After 2 minutes (5-fluoro-2-trifluoromethyl-phenyl)- piperidin-4-yl-amine dihydrochloride (prepared according to the method used for the synthesis of Intermediate 3) (150mg, O.όmmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 5%MeoH in CHCl
3 as eluent) afforded 129mg (44.4%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(5- fluoro-2-trifluoromethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]
+: 490.18, 95.08
0Zo-
1H NMR ( 300MHz, DMSOd
6): δl3.8(s, IH), 8.0(bs, IH), 7.8(d, 2H), 7.5(t, 4H), 7.1(bs, IH), 6.8(d, IH), 6.5(t, IH), 5.0(d, IH), 4.4(d, IH), 4.3(d, 2H), 3.8(m,2H), 3.7(m, 2H), 3.2(t, IH), 2.8(t, IH), 2.0(m, 2H), 1.5(m, 2H).
EXAMPLE 83 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(4-fluoro- 2-trifluoroniethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-ainide
DIPEA (232mg, 1.8mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (140mg, O.όmmol) in DMF (2mL) followed by HOBt (97mg,
0.7mmol) and EDCI.HC1 (137mg, 0.7mmol). After 2 minutes (4-fluoro-2-trifluoromethyl-phenyl)- piperidin-4-yl-amine dihydrochloride (prepared according to the method used for the synthesis of Intermediate 3) (150mg, O.όmmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure.
Purification by column chromatography (using 60-120 silica gel and 5%MeoH in CHCl3 as eluent) to affordl05mg (36%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(4-fluoro-2- trifluoromethyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 490.18, 95.36%.'H NMR (300MHZ, DMSO-d6): δl3.8(s, IH), 8.0(t, IH), 7.8(m, 2H), 7.4(m, 6H), 7.1(m,
2H), 4.6(d, IH), 4.4(m, IH), 4.2(m, 2H), 3.9(m,lH), 3.7(m, IH), 3.2(m, IH), 2.8(m, IH), 2.0(m, 2H), 1.5(m, 2H), 1.4(m, IH).
EXAMPLE 84 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-acetyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (232mg, 1.8mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (144mg, O.όmmol) in DMF (2mL) followed by HOBt (97mg, 0.7mmol) and EDCI.HC1 (137mg, OJrnmol). After 2 minutes l-[2-(piperidin-4-yloxy)-phenyl]- ethanone dihydrochloride (prepared according to Step 1 and 5 of the General Scheme) (150mg, O.όmmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 5%MeoH in CHCl3 as eluent) to afford 112mg (50.9%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-acetyl-phenoxy)-piperidin-l- yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+:447.2, 95.680Zc1H NMR (300MHz, DMSOd6): 813.8(s, IH), 8.2(bs, IH), 7.8(d, 2H), 7.5(m, 4H), 7.4(m, IH), 7.3(m, IH), 7.2(s, IH), 7.0(t, IH), 4.8 (m, IH), 4.4(m, 2H), 3.8(m, IH), 3.4(m,lH), 3.5(m, 2H), 2.5(s, 3H), 2.0(m, 2H), 1.7(m, 2H).
EXAMPLE 85 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(5-cyano-
2-methyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyI}-amide
DIPEA (426mg, 3.3mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (285mg, l.lmmol) in DMF (3mL) followed by HOBt (175mg, 1 ,3mmol) and EDCI.HC1 (252mg, 1.3mmol). After 2 minutes 4-methyl-3-(piperidin-4-ylamino)- benzonitrile dihydrochloride (250mg, l.lmmol) (prepared according to the method used for the
synthesis of Intermediate 3) was added and the stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 5%MeoH in CHCl3 as eluent) afforded lOOmg (22.7%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(5-cyano-2- methyl-phenylamino)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]+: 443.31, 91.960Zc1H NMR (300MHz, DMSOd6): δl3.8(s, IH), 8.1(t, IH), 7.8(d, 2H), 7.4(m, 4H), 7.2(m, 2H), 7.0(s, IH), 6.9(d, IH), 5.0(d, IH), 4.4 (m, IH), 4.2(m, 2H), 3.9(m, IH), 3.7(m,lH), 3.2(m, IH), 2.8(m, IH), 2.2(s, 3H), 2.0(m, 2H), 1.7(m, 2H).
EXAMPLE 86 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(2- trifluoromethyl-benzenesulfinyl)-piperidin-l-yl]-ethyl}-amide
m-Chloroperbenzoic acid (18.5mg, O.lmmol) was added to a cold (0-4
0C) solution of 5- phenyl- lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-phenylsulfanyl)-piperidin- 1-yl] -ethyl} -amide (50mg, O.lmmol) in DCM (1OmL) and stirring was continued for lhr. The reaction mixture was concentrated. Purification by column chromatography (using 60-120 silica gel and 0.6%MeOH in CHCl
3 as eluent) afforded 30mg (58.35% yield) of 5 -phenyl- lH-pyrazole- 3-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-benzenesulfinyl)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]
+: 505.15, 95.9%.
1H NMR (300MHz, DMSO-d6): δ 13.8(s, IH), 8.2-7.9 (m, 4H), 7.9-7.8 (m, 3H), 7.6-7.4(t, 2H), 7.4 (t, IH), 7.1 (s, IH), 4.5 (d, IH), 4.2 (d, 2H), 4.0(d, IH), 3.1 (m, 2H), 2.8-2.5 (m, 2H), 1.9(d, IH), 1.7 (dd, IH), δ 1.6-1.4 (m, 2H).
EXAMPLE 87 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4- (pyridin-4-yloxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (540mg, 4.2mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3-
carbonyl)-amino] -acetic acid (344mg, 1.4mmol) in DMF (3mL) followed by HOBt (229mg, 1.7mmol) and EDCI.HC1 (321mg, 1.7mmol). After 2 minutes 2-ammo-l-[4-(pyridin-4-yloxy)- piperidin-l-yl]-ethanone hydrochloride (250mg, 1.4mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 5% MeOH in CHCl
3 as eluent) afforded 198mg (% 34.9Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4- (pyridin-4-yloxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]
+: 406.18, 95.8"/0.
1H NMR (300MHz, DMSO-d
6): δl3.8(m, IH), 8.4(m, 2H), 8.1(m, IH), 7.8(m, 2H), 7.1(m, IH), 7.0(s, 2H), 4.8(m, IH), 4.2(m, 2H), 3.9 (m, IH), 3.8(m, IH), 3.4(m, IH), 3.3(m,lH), 2.0(m, 2H), 1.7(m, 2H).
EXAMPLE 88 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(5-chloro- pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (387mg, 3.0mmol) was added a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (247mg, l.Ommol) in DMF (2mL) followed by HOBt (162mg, 1.2mmol) and EDCLHCl (229mg, 1.2mmol). After 2 minutes 3-chloro-5-(piperidin-4-yloxy)- pyridine hydrochloride (prepared according to the method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 5%MeoH in CHCI3 as eluent) to afford 56mg (12.71%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(5-chloro- pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+:440.14, 95.7%. 1H NMR (300MHz, DMSO-d6): δl3.8(s, IH), 8.2(m, 2H), 8.0(m, IH), 7.8(m, 3H), 7.4(m, 4H), 7.1(m, IH), 4.8(m, IH), 4.2(m, 2H), 3.9(m, IH), 3.8(m, IH), 3.4(m, IH), 3.3(m,lH), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 89 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- hydroxy-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
10%Pd/C (10mg) was added to a stirred solution of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-benzyloxy-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide (prepared from Intermediate 30 and (2-benzyloxyoxy-phenyl)-piperidin-4-yl-amine hydrochloride which was prepared according to the method used for the synthesis of Intermediate 15) (lOOmg, 0.2mmol) in a mixture of MeOH:H2O (1:1, 1OmL) under inert atmosphere and stirring was continued under H2 gas atmosphere for 2hr. The reaction mixture was filtered through celite. The filtrate collected was concentrated under reduced pressure to afford the residue. The residue was purified by preparative HPLC to afford 60mg (71.5% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- hydroxy-phenoxy)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]+:421.8, 96.21%.]H NMR (300MHz, DMSOd6): δl3.8(b, IH), 8.9(b, IH), 8.2(b, IH), 7.8(m, 2H), 7.5(m, 3H), 7.2(s, IH), 7.0(d, IH), 6.8(m, 3H), 4.5(m, IH), 4.2(m, 2H), 3.8(m, 2H), 3.3(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 90 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(2- trifluoromethyl-benzenesulfonyl)-piperidin-l-yl]-ethyl}-amide
DIPEA (88mg, 0.68mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (36mg, 0.15mmol) in DMF (ImL) followed by HOBt (20mg, O.ISmmol) and EDCI.HC1 (39mg, 0.2mmol). After 2 minutes 4-(2-trifluoromethyl- benzenesulfonyl)-piperidine hydrochloride (250mg, l.Ommol) (prepared from 4-(2- trifluoromethyl-phenylsulfanyl)-piperidine-l-carboxylic acid tert-butyl ester by means of oxidation with hydrogen peroxide and subsequent hydrolysis of the N-protection group with hydrochloric acid) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, the precipitate was collected. The solid obtained was purified by column chromatography (using 60-120 silica gel and
5%MeoH in DCM as eluent) to afford 21mg (29.6%Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-benzenesulfonyl)-piperidin-l-yl]-ethyl} -amide. LC/MS [M+H]+: 521.14, 1H NMR (300MHz, DMSOd6): δl3.8(s, IH), 8.2(m, 5H), 7.7(d, 2H), 7.5-7.3(m, 3H), 7.1(s, IH), 4.5(d, IH), 4.2-4.0(m, 2H), 4.0(d, IH), 3.7-3.5(t, IH), 3.1(t, IH), 2.7(m,lH), 1.9- 1.7(m, 2H), 1.7(m, IH), 1.5(m, IH).
EXAMPLE 91 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(6-chloro- pyridin-2-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (271mg, 2.1mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (162mg, 0.7mmol) in DMF (2mL) followed by HOBt (94mg, 0.7mmol) and EDCLHCl (134mg, 0.7mmol). After 2 minutes 2-chloro-6-(piperidin-4-yloxy)- pyridine hydrochloride (prepared according to the method used for the synthesis of Intermediate 15) (140mg, 0.7mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 5%MeoH in CHCl
3 as eluent) afforded 53mg (18.27%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(6-chloro- pyridin-2-yloxy)-piperidm-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]
+: 440.14, 9O
0Zc
1H NMR (300MHz, DMSOd
6): δl3.8(s, IH), 8.2(b, IH), 7.8(m, 3H), 7.4(m, 3H), 7.1(m, IH), 6.8(m, IH), 5.2(m, IH), 4.2(m, 2H), 3.8(m, IH), 3.7(m, IH), 3.5(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 92 - Synthesis of 4-Methyl-3-(l-{2-[(5-phenyMH-pyrazole-3-carbonyl)- amino]-acetyl}-piperidin-4-yloxy)-benzoic acid methyl ester
DIPEA (423mg, 2.4mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (200mg, 0.81mmol) in DMF (3mL) followed by HOBt (130mg,
0.93mmol) and EDCLHCl (187mg, 0.93mmol). After 2 minutes 4-methyl-3-(piperidin-4-yloxy)- benzoic acid methyl ester hydrochloride (prepared according to the method used for the synthesis of Intermediate 15) (280mg, 0.97mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford 266mg (68.55%Yield) of 4-methyl-3-(l-{2-[(5-phenyl-lH- pyrazole-3-carbonyl)-amino]-acetyl}-piperidin-4-yloxy)-benzoic acid methyl ester. LC/MS [M+H]+: 477.21, 96.630Zo-1H NMR (300MHz, DMSOd6): 513.8(s, IH), 8.1-8.0(m, IH), 7.8(d, 2H), 7.54-7.28(m, 6H), 7.1(bs, IH), 4.8(m, IH), 4.2(d, 2H), 3.8(s, 3H), 3.8-3.6(m, 2H), 3.6-3.4(m, 2H), 2.2(s, 3H), 2.1-1.8(m, 2H), 1.8-1.5(m, 2H).
EXAMPLE 92 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2-fluoro- 5-trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (11 Omg, 0.85mmol) was added to a stirred solution of 5-phenyl-l H-pyrazole-3- carboxylic acid (50mg, 0.24mmol) in DMF (2mL) followed by HOBt (33mg, 0.24mmol) and EDCLHCl (49mg, 0.25mmol). After 2 minutes 2-amino-l-[4-(2-fluoro-5-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. The residue obtained was purified by recrystallisation using 25% EtOAc in hexane to afford 89mg (80.9%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-fluoro- 5-trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide.LC/MS [M+H]
+: 457, 90.38
0Zc
1H NMR (300MHz, DMSO-d
6): δl3.8(s, IH), 8.2(bs, IH), 7.9(t, IH), 7.1 (m, 7H), 4.61 (m, IH), 4.4(d, 2H), 3.7(m, 2H), 3.3(m, 2H), 1.9(m, 2H), 1.5(m, 2H).
EXAMPLE 94 - Synthesis of 5-(2-Fluoro-phenyI)-lH-pyrazole-3-carboxylic acid {2- [4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (1 lOmg, 0.85mmol) was added to a stirred solution of 5-(2-fluoro-phenyl)-lH- pyrazole-3-carboxylic acid (50mg, 0.24mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 2'-fiuoroacetophenone) in DMF (2mL) followed by HOBt (33mg, 0.24mmol) and EDCLHCl (49mg, 0.25mmol). After 2 minutes 2-amino-l-[4-(2-chloro-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (75mg, 0.24mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. Recrystallisation using using 25% EtOAc in hexane afforded 89mg (80.9%Yield) of 5-(2-fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)- piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 457, 90.380Zc1H NMR (300MHz, DMSO- d6): δl3.8(s, IH), 8.2(bs, IH), 7.9(t, IH), 7.1(m, 7H), 4.61(m, IH), 4.4(d, 2H), 3.7(m, 2H), 3.3(m, 2H), 1.9(m, 2H), 1.5(m, 2H).
EXAMPLE 95 - Synthesis of 5-(2-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (160mg, 1.3mmol) was added to a stirred solution of 5-(2-Fluoro-phenyl)-lH- pyrazole-3-carboxylic acid (77mg, 0.37mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 2'-fluoroacetophenone) in DMF (2mL) followed by HOBt (52mg, 0.39mmol) and EDCLHCl (74mg, 0.39mmol). After 2 minutes 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (125mg, 0.37mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated
under reduced pressure to afford the residue. Washing with ethyl acetate afforded 57mg (31.67% Yield) of 5-(2-fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 491,93.63%. 1H NMR (300MHz, DMSOd6): δl3.8(d, IH), 8.7(m, IH), 7.8(m, IH), 7.2(m, 6H), 6.9(m, 2H), 4.8(m, IH), 4.2(d, 2H), 3.7(m, 2H), 3.4(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 96 - Synthesis of 5-(4-Trifluoromethyl-phenyl)-lH-pyrazoIe-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (150mg, 1.15mmol) was added to a stirred solution of 5-(4-trifluoromethyl- phenyl)-lH-pyrazole-3-carboxylic acid (84mg, 0.33mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from (p-trifluoromethyl)acetophenone) in DMF (2mL) followed by HOBt (46mg, 0.34mmol) and EDCI.HC1 (66mg, 0.34mmol). After 2 minutes 2- amino-l-[4-(2-chloro-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (lOOmg, 0.33mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. The residue was purified by washing with mixture of ethyl acetate and methanol to afford 80mg (48.2%Yield) of 5-(4-trifluoromethyl- phenyl)- 1 H-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin- 1 -yl] -2-oxo-ethyl } - amide. LC/MS [M+H]+: 507, 92.02%. 1H NMR (300MHz, DMSOd6): 513.7(d, IH), 8.0(m, 3H), 7.8(m, 2H), 7.44(d, IH), 7.24(m, 3H), 6.96(m, IH), 4.7(m, IH), 4.2(d, 2H), 3.7(m, 2H), 3.4(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 97 - Synthesis of S-β-Fluoro-phenyty-lH-pyrazole-S-carboxylic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (165mg, 1.3mmol) was added to a stirred solution of 5-(3-fluoro-phenyl)-lH- pyrazole-3-carboxylic acid (75mg, 0.36mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 3'-fluoroacetophenone) in DMF (2mL) followed by HOBt (51mg, 0.38mmol) and EDCI.HC1 (73mg, 0.38mmol). After 2 minutes 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (120mg, 0.36mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. The residue was purified by dissolving in ethyl acetate and then reprecipitating with hexane to afford 103mg (57.86% Yield) of 5-(3-fluoro- phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]- ethyl}-amide. LC/MS [M+H]+: 491, 92.15%. 1B NMR (300MHz, DMSOd6): δl3.8(s, IH), 8.1(m, IH), 7.46(m, 4H), 7.1(m, 5H), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, IH), 3.3(m, IH), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 98 - Synthesis of S-β-Fluoro-phenyO-lH-pyrazole-S-carboxylic acid {2- [4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (165mg, 1.3mmol) was added to a stirred solution of 5-(3-fluoro-phenyl)-lH- pyrazole-3-carboxylic acid (75mg, 0.36mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 3'-fluoroacetophenone) in DMF (2mL) followed by HOBt (51mg, 0.38mmol) and EDCI.HC1 (73mg, 0.38mmol). After 2 minutes 2-amino-l-[4-(2-chloro- phenylamino)-piperidin-l-yl]-ethanone hydrochloride (120mg, 0.36mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate.
The organic layer was concentrated under reduced pressure to afford the residue. Washing with methanol afforded 76mg (43.1% Yield) of 5-(3-fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2- [4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]
+: 456, 100%.
1H NMR (300MHz, DMSO-d
6): δl3.8(s, IH), 8.06(t, IH), 7.44(m, 4H), 7.12(m, 4H), 6.84(d, IH), 6.58(m, IH), 4.9(d, IH), 4.3(d, IH), 4.2(m, 2H), 3.7(m, IH), 3.6(m, IH), 3.2(m, IH), 2.8(m, IH), 1.9(m, 2H), 1.4(m, 2H).
EXAMPLE 99 - Synthesis of 5-(2-Trifluoromethyl-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (76mg, 0.59mmol)was added to a stirred solution of 5-(2-trifluoromethyl-phenyl)- lH-pyrazole-3-carboxylic acid (43 mg, 0.167mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from (o-trifluoromethyl)acetophenone) in DMF (2mL) followed by HOBt (24mg, 0.176mmol) and EDCLHCl (34mg, 0.176mmol). After 2 minutes 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (0.057mg, 0.167mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. The residue was purified by washing with chloroform to afford 51mg (56% Yield) of 5-(2-trifluoromethyl-phenyl)-lH-pyrazole-3- carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 541, 87.52%. 1H NMR (300MHz, DMSOd6): δl3.8(d, IH), 8.1(m, IH), 7.6(m, 4H), 7.5(t, IH), 7.3(t, 3H), 6.76(s, IH), 4.8(m, IH), 4.1(d, 2H), 3.7(m, 2H), 3.4(m, IH), 3.2(m, IH), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 100 - Synthesis of 5-(4-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-ainide
DIPEA (150mg, 1.2mmol) was added to a stirred solution of 5-(4-hydroxy-phenyl)-lH- pyrazole-3-carboxylic acid (70mg, 0.34mmol) in DMF (2mL) followed by HOBt (48mg, 0.36mmol) and EDCLHCl (69mg, 0.36mmol). After 2 minutes 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (116mg, 0.34mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. The residue was purified by washing with 1% MeOH in EtOAc to afford 92mg (55%Yield) of 5-(4-hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 489, 96%. 1H NMR (300MHz, DMSO-d6): δl3.8(s, IH), 9.8(s, IH), 8.02(t, IH), 7.5(m, 3H), 7.28(t, 3H), 6.8(m, 3H), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.6(m, IH), 3.4(m, IH), 3.2(m, IH), 2.0(m, 2H), 1.7(m, 2H).
EXAMPLE 101 - Synthesis of 5-(3-Hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (133mg, 1.03mmol) was added to a stirred solution of 5-(3-hydroxy-phenyl)-lH- pyrazole-3-carboxylic acid (60mg, 0.294mmol) (prepared from 5-(3-benzyloxy-phenyl)-lH- pyrazole-3-carboxylic acid ethyl ester) in DMF (2mL) followed by HOBt (41.7mg, 0.308mmol) and EDCLHCl (59mg, 0.308mmol). After 2 minutes 2-amino-l-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (99mg, 0.294mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. Washing with ethyl acetate afforded 74mg (51.7% Yield) of 5-(3-
hydroxy-phenyl)- lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 489, 96%. 1H NMR (300MHz, DMSO-d6): δl3.6(s, IH), 9.6(s, IH), 8.0(t, IH), 7.5(t, IH), 7.1(m, 7H), 6.9(d, IH), 6.8(m, IH), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 102 - Synthesis of 5-(4-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (131mg, l.Ommol) was added to a stirred solution of 5-(4-fiuoro-phenyl)-lH- pyrazole-3-carboxylic acid (60mg, 0.29mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 4'-fhαoroacetophenone) in DMF (2mL) followed by HOBt (41mg, 0.305mmol) and EDCLHCl (59mg, 0.306mmol). After 2 minutes 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (98mg, 0.294mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by washing with l%MeOH in EtOAc to afford 97mg (68.3%Yield) of 5-(4-fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 491, 92.85%. 1H NMR (300MHz, DMSO-d6): δl3.8(s, IH), 8.1(t, IH), 7.9(m, 2H), 7.5(t, IH), 7.24(m, 5H), 7.1(d, IH), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 103 - Synthesis of 5-(4-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-
[4-(2-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (226mg, 1.75mmol) was added to a stirred solution of {[5-(4-fluoro-phenyl)-lH- pyrazole-3-carbonyl] -amino} -acetic acid (130mg, 0.5mmol) (prepared by the method used for the
synthesis of Intermediate 30, starting from (4-fluorophenyl)acetophenone) in DMF (2mL) followed by HOBt (71mg, 0.52mmol) and EDCI.HC1 (lOOmg, 0.52mmol). After 2 minutes 3- chloro-5-(piperidin-4-yloxy)-pyridine hydrochloride (125mg, 0.5mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by column chromatography (using 60-120 silicagel and 40% ethyl acetate in hexane as eluent) to afford 134mg (58.5% Yield) of 5-(4-fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 458, 93.27%. 1H NMR (300MHz, DMSOd6): δl3.8(s, IH), 8.0(m, 2H), 7.8(m, 2H), 7.7(d, IH), 7.4(m, IH), 7.25(m, 2H), 7.1(m, IH), 4.8(m, IH), 4.2(d, 2H), 3.7(m, 2H), 3.5(m, 2H), 1.9(m, 2H), 1.7(m, 2H).
EXAMPLE 104 - Synthesis of 5-(4-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2- [4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (200mg, l.όmmol) was added to a stirred solution of {[5-(4-fluoro-phenyl)-lH- pyrazole-3-carbonyl]-amino}-acetic acid (120mg, 0.45mmol) (prepared by the method used for the synthesis of Intermediate 30, starting from (4'-fluorophenyl)acetophenone) in DMF (2mL) followed by HOBt (65mg, 0.48mmol) and EDCI.HC1 (92mg, 0.48mmol). After 2 minutes 3- chloro-5-(piperidin-4-yloxy)-pyridine hydrochloride (0.114g, 0.00045mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by column chromatography (using 60-120 silicagel and 50% ethyl acetate in hexane as eluent) to afford 11 lmg (55.5% Yield) of 5-(4-fluoro-phenyl)-lH-pyrazole-3- carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+Hf: 458, 97.78%. 1H NMR (300MHz, DMSO-d6): δl3.8(s, IH), 8.3 (m, IH), 8.21 (m, IH), 8.04(m, IH), 7.8(m, 2H), 7.7(m, IH), 7.3(m, 2H), 7.1(s, IH), 4.8(m, IH), 4.2(d, 2H), 3.9(m, 2H), 3.7(m, IH), 3.4(m, IH), 3.2(m, IH), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 105 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3- chloro-phenoxy)-piperidin-l-yI]-2-oxo-ethyl}-amide
DIPEA (155mg, 1.2mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (99mg, 0.4mmol) in DMF (2mL) followed by HOBt (54mg, 0.4mmol) and EDCLHCl (84mg, 0.44mmol). After 2 minutes 4-(3-chloro-phenoxy)-piperidine hydrochloride (lOOmg, 0.4mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by washing with methanol to afford 67mg (39.4% Yield) S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3-chloro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]
+:439, 92.8%.
1H NMR (300MHz, DMSOd
6): δl3.8(m, IH), 8.0(m, IH), 7.8(m, 2H), 7.5(m, 2H), 7.4(m, IH), 7.3(m, 2H), 7.1(m, 2H), 7.0(m, 2H), 4.7(m, IH), 4.2(m, 2H), 4.0(m, IH), 3.7(m, IH), 3.4(m, IH), 3.2(m, IH), 2.0(m, 2H), 1.5(m, 2H).
EXAMPLE 106 - Synthesis of 3-(l-{2-[(5-Phenyl-lH-pyrazole-3-carbonyl)-amino]- acetyl}-piperidin-4-yIoxy)-benzoic acid
LiORH2O (44mg, l .Ommol) was added to a stirred mixture of 3-(l-{2-[(5-phenyl-lH- pyrazole-3-carbonyl)-amino]-acetyl}-piperidin-4-yloxy)-benzoic acid methyl ester (prepared by the method used to generate Example 92) (99mg, 0.2mmol) in MeOH: H2O (1:1, 4 mL) was added, and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was concentrated under reduced pressure. Cold water was then added and acidified it with 10% aqueous HCl and extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by washing with diethyl ether to afford 87 mg (97.7%Yield) 3-(l-{2-[(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetyl}-piperidin-4-yloxy)-
benzoic acid. LC/MS [M+H]+: 463, 96.9%. 1H NMR (300MHz, DMSOd6): δl3.8(b, IH), 13.6(b, IH), 8.1(b, IH), 7.8(q, 2H), 7.5(m, 6H), 7.3(m, IH), 7.1(b, IH), 4.8(m, IH), 4.2(m, 2H), 3.9(m, IH), 3.8(m, IH), 3.4(m, 2H)3 2.0(m, 2H), 1.7(m, IH).
EXAMPLE 107 - Synthesis of 5-(3-Fluoro-phenyl)-lH-pyrazole-3-carboxylic acid {2-
[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (253mg, 1.96mmol) was added to a stirred solution of 5-(3-fluoro-phenyl)-lH- pyrazole-3-carboxylic acid (81mg, 0.39mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 3'-fluoroacetophenone) in DMF (2mL) followed by HOBt (56mg, 0.41mmol) and EDCI.HC1 (79mg, 0.41mmol). After 2 minutes 2-amino-l-[4-(5-chloro-pyridin-3- yloxy)-piperidin-l-yl]-ethanone hydrochloride (120mg, 0.39mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by washing with ethyl acetate to afford 78mg (43.5%Yield) of 5-(3-fluoro-phenyl)-lH- pyrazole-3-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]+: 458, 90%. 1H NMR (300MHz, DMSO-d6): δ 13.8(s, IH), 8.3(d, IH), 8.22(d, IH), 8.04(m, IH), 7.6(m, 3H), 7.5(m, 2H), 7.18(m, 2H), 4.8(m, IH), 4.2(m, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, IH), 3.2(m, IH), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 108 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid [2-oxo-2-(4-m- tolyloxy-piperidin-l-yl)-ethyl]-amide
DIPEA (155mg, 1.2mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (lOOmg, 0.4mmol) in DMF (2mL) followed by HOBt (54mg, 0.4mmol) and EDCLHCl (84mg, 0.44mmol). After 2 minutes 4-m-tolyloxy-piperidine hydrochloride (107mg, 0.4mmol) (prepared by method used for the synthesis of Intermediate 15)
was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, the solid collected. The solid was purified by washing with 10% MeOH in CHCl
3 to afford 92 mg (56%Yield) 5-phenyl-lH-pyrazole-3- carboxylic acid [2-oxo-2-(4-m-tolyloxy-piperidin-l-yl)-ethyl] -amide. LC/MS [M+H]
+: 419, 95.4%.
1H NMR (300MHz, DMSOd
6): δl3.8(m, IH), 8.6(m, IH), 8.0(m, 2H), 7.8(m, 2H), 7.4(m, 4H), 7.2(m, 2H), 6.8(m, 3H), 4.6(m, IH), 4.2(m, 2H), 3.8(m, IH), 3.6(m, IH), 3.4(m, IH), 3.3(m, IH), 2.3(s, 3H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 109 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- methyl-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyI}-amide
DIPEA (193mg, 1.5mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (127mg, 0.5mmol) in DMF (2mL) followed by HOBt (67mg, 0.5mmol) and EDCI.HC1 (105mg, 0.55mmol). After 2 minutes 2-methyl-3-(piperidin-4-yloxy)- pyridine hydrochloride ( 1 OOmg, 0.5mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, and the solid was collected. Purification of the solid by washing with 10% MeOH in CHCl3 afforded 95mg (47.5%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-methyl-pyridin-3-yloxy)- piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 420, 96.7%. 1H NMR (300MHz, DMSO- d6): δ 8.3(m, IH), 8.2(b, IH), 8.0(m, IH), 7.8(m, 2H), 7.6(m, IH), 7.5(m, 2H), 7.4(m, IH), 7.2(b, IH), 4.9(m, IH), 4.2(m, 2H), 3.8(m, 2H), 3.5(m, 2H), 2.5(m, 3H), 2.0(m, 2H), 1.8(m, 2H).
EXAMPLE 110 - Synthesis of S-Pyridin^-yl-lH-pyrazole-S-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin-l-yl] -2-oxo-ethyl}-amide
DIPEA (287mg, 2.2mmol) was added to a stirred solution of 5-pyridin-2-yl-lH-pyrazole- 3-carboxylic acid hydrochloride (lOOmg, 0.44mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 2-acetylpyridine) in DMF (2mL) followed by HOBt (63mg, 0.46mmol) and EDCLHCl (89mg, 0.46mmol). After 2 minutes 2-amino- 1 -[4-(2-chloro- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (135mg, 0.44mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by filtering through 60-120 silicagel column and filtrate collected was concentrated under reduced pressure to afford 145mg (74.3%Yield) of 5-pyridin-2-yl-lH-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 440, 90%. 1H NMR (300MHz, DMSOd6): δ 13.9(s, IH), 8.6(d, IH), 8.1(bs, IH), 7.84(m, 2H), 7.24(m, 5H), 6.94(m, 2H), 4.75(m, IH), 4.15(d, 2H), 3.6(m, 2H), 3.4(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 111 - Synthesis of 3-(5-{2-Oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethylcarbamoyI}-lH-pyrazol-3-yl)-benzoic acid
LiOH-H2O (32mg, 0.76mmol) was added to a stirred mixture of 3-(5-{2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethylcarbamoyl}-lH-pyrazol-3-yl)-benzoic acid methyl ester (99mg, 0.2mmol) (prepared by the method used for the synthesis of Example 102, starting, alternatively from methyl 3-acetylbenzoate to generate the lH-pyrazole intermediate) in THF: MeOH: H2O (3:2:1, 38mL) was added, and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was concentrated under reduced pressure Cold water
was then added and the contents were acidified with 10% aqueous HCl, the precipitate was filtered. The solid obtained was purified by recrystallisation form methanol to afford 50mg (38.1%Yield) of3-(5-{2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethylcarbamoyl}- 2H-pyrazol-3-yl)-benzoic acid. LC/MS [M+H]+:517, 95.29%. 1H NMR (300MHz, DMSOd6): δl3.8(s, IH), 13.2(s, IH), 8.38(s, IH), 8.04(d, IH), 7.9(d, IH), 7.5(m, 2H), 7.26(t, 3H), 7.18(bs, IH), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 112 - Synthesis of S-Pyridin-S-yl-lH-pyrazole-S-carboxylic acid {2-oxo-2- [4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (290mg, 2.26mmol)was added to a stirred solution of 5-pyridin-3-yl-lH-pyrazole- 3-carboxylic acid hydrochloride (prepared by the method used for the synthesis of Intermediate 29, starting from 3-acetylpyridine) (lOOmg, 0.44mmol) in DMF (2mL) followed by HOBt (63mg, 0.46mmol) and EDCLHCl (90mg, 0.46mmol). After 5 minutes 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidm-l-yl]-ethanone hydrochloride (150mg, 0.44mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by filtering through 60-120 silicagel column and filtrate collected was concentrated under reduced pressure to afford 138mg (66%Yield) of 5-pyridm-3-yl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 474, 94.7%. 1H NMR (300MHz, DMSO-d6): δ 13.8(s, IH), 9.4(s, IH), 8.56(d, IH), 8.16(m, IH), 7.46(m, 2H), 7.22(m, 4H), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.44(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 113 - Synthesis of S-Pyridin-S-yl-lH-pyrazole-S-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (290mg, 2.26mmol) to a stirred solution of 5-pyridin-3-yl-lH-pyrazole-3- carboxylic acid hydrochloride (lOOmg, 0.44mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 3-acetylpyridine) in DMF (2mL) followed by HOBt (63mg, 0.46mmol) and EDCI.HC1 (90mg, 0.46mmol). After 2 minutes 2-amino-l-[4-(2-chloro-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (150mg, 0.44mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from 10% EtOAc in hexane afforded 1 OOmg (51.5 %Yield) of 5 -pyridin-3 -yl- 1 H- pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 440, 96.47%. 1H NMR (300MHz, DMSO-d6): δ 13.9(s, IH), 9.0(m, IH), 8.5(m, IH), 8.1(m, 2H), 7.4(m, 2H), 7.2(m, 3H), 6.9(m, IH), 4.75(m, IH), 4.2(m, 2H), 3.65(m, 2H), 3.4(m, 2H), 1.9(m, 2H), 1.7(m, 2H).
EXAMPLE 114 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(4- methyl-pyridin-3-yloxy)-piperidm-l-yl]-2-oxo-ethyl}-amide
DIPEA (154mg, 1.4mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (107mg, 0.4mmol) in DMF (2mL) followed by HOBt (54mg,
0.4mmol) and EDCI.HC1 (84mg, 0.44mmol). After 2 minutes 4-methyl-3-(piperidin-4-yloxy)- pyridine hydrochloride (lOOmg, 0.4mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient
temperature overnight. The reaction mixture was diluted with cold water, the solid collected. Purification of the solid by washing with 10% MeOH in CHC13 afforded 106mg (66%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(4-methyl-pyridm-3-yloxy)-piperidin-l-yl]-2-oxo- ethyl}-amide. LC/MS [M+H]+: 420, 98.9%. 1H NMR (300MHz, DMSOd6): δ 14.0(b, IH), 8.6(s, IH)5 8.4(d, IH), 8.2(b, IH), 7.8(m, 3H), 7.5(m, 2H), 7.4(m, IH), 7.2(s, IH), 4.9(m, IH), 4.2(m, 2H), 3.8(m, 2H), 3.5(m, 2H), 2.4(s, 3H), 2.0(m, 2H), 1.7(m, 2H).
EXAMPLE 115 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(5- trifluoromethyl-pyridin-3-yloxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (140mg, l.lmmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (87mg, 0.35mmol) in DMF (2mL) followed by HOBt (47mg, 0.35mmol) and EDCLHCl (73mg, 0.38mmol). After 2 minutes 3-(piperidin-4-yloxy)-5- trifluoromethyl-pyridine hydrochloride (lOOmg, 0.35mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight., The reaction mixture was diluted with cold water, the solid was collected. Purification of the solid by washing with 10% MeOH in CHCl3 afforded 76mg (47.5%Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(5-trifluoromethyl-pyridin- 3-yloxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 474, 99%. 1H NMR (300MHz, DMSO- d6): δ 13.8(m, IH), 8.68(m, IH), 8.58(s, IH), 8.1(m, IH), 7.9(m, IH), 7.8(m, 2H), 7.4(m, 4H), 7.1(m, IH), 5.0(m, IH), 4.2(m, 2H), 4.0(m, IH), 3.8(m, IH), 3.4(m, IH), 3.3(m, IH), 2.0(m, 2H), 1.7(m, 2H).
EXAMPLE 116 - Synthesis of 5-(5-Chloro-thiophen-2-yI)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyI-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (200mg, 1.5mmol) was added to a stirred solution of 5-(5-chloro-thiophen-2-yl)- lH-pyrazole-3-carboxylic acid (lOOmg, 0.44mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 2-acetyl-5-chlorothiophene) in DMF (2mL) followed by HOBt (63mg, 0.46mmol) and EDCI.HC1 (90mg, 0.46mmol). After 5 minutes 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (140mg, 0.44mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from 10% EtOAc in hexane afforded 146mg (65.1% Yield) of 5-(5-chloro-thiophen-2-yl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl] -ethyl} -amide. LC/MS [M+H]+: 513, 98.3%. 1H NMR (300MHz, DMSO-ds): δ 13.7(s, IH), 8.64(m, IH), 7.5(t, IH), 7.3(m, 3H), 7.2(m, 2H), 6.9(m, IH), 4.7(m, IH), 4.2(m, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, IH), 3.3(m, IH), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 117 - Synthesis of 5-(5-Chloro-thiophen-2-yl)-lH-pyrazole-3-carboxylic acid {2-[4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (120mg, 0.98mmol) was added to a stirred solution of {[5-(5-chloro-thiophen-2- yl)-lH-pyrazole-3-carbonyl]-amino}-acetic acid (80mg, 0.28mmol) (prepared by the method used for the synthesis of Intermediate 30, starting from 2-acetyl-5-chlorothiophene) in DMF (2mL) followed by HOBt (40mg, 0.29mmol) and EDCI.HC1 (56mg, 0.29mmol). After 5minutes 4-(2,5- difluoro-phenoxy)-piperidine hydrochloride (70mg, 0.28mmol) (prepared by method used for the
synthesis of Intermediate 15)was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from 10% EtOAc in hexane afforded 84mg (65.1%Yield) of 5-(5-chloro- thiophen-2-yl)-lH-pyrazole-3-carboxylic acid {2-[4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-2- oxo-ethyl}-amide. LC/MS [M+H]+: 481, 90.38%. 1H NMR (300MHz, DMSOd6): δ 13.6(s, IH), 8.6(m, IH), 7.18(m, 4H), 7.1(m, IH), 6.8(m, IH), 4.7(m, IH), 4.2(d, 2H), 3.8(m, IH), 3.7(m, IH), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 118 - Synthesis of S-φ-Hydroxy-phenylHH-pyrazole-S-carboxylic acid
{2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
10%Pd/C (50mg) was added to a stirred solution of 5-(2-benzyloxy-phenyl)-lH-pyrazole- 3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide (195mg, 0.34mmol) (prepared by method used for the synthesis of Intermediate 45) in methanol (3OmL) under inert atmosphere and stirred under H2 atmosphere with pressure for 3hrs. The reaction mixture was filtered through celite, the celite was washed with methanol and the filtrate was concnetrated under reduced pressure Washing with ethyl acetate afforded 58mg (35%Yield) of 5- (2-hydroxy-phenyl)-lH-pyrazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-1-yl] -ethyl} -amide. LC/MS [M+H]+: 489, 91.45%. 1H NMR (300MHz, DMSOd6): δ8.2(m, IH), 7.64(d, IH), 7.5(t, IH), 7.26(t, 2H), 7.1(m, 2H),6.94(m, IH), 6.84(t, IH), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
Synthesis of 4-(2-methanesulfonyl-phenoxy)-piperidine hydrochloride
m-Chloroperbenzoic acid (280mg, 1.62mmol) was added to a cold (0-4
0C) solution of 1- methoxy-2-methylsulfanyl-benzene (lOOmg, 0.64mmol) in DCM (3mL) and stirring was continued for lhr. The reaction mixture was diluted with water and extracted with
dichloromethane. The organic layer was washed with saturated brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 1 lOmg (91.6%Yield) of 1- methanesulfonyl-2-methoxy-benzene. Boron tribromide (370mg, 1.47mmol) was added to a cold solution (-70
0C ) solution of 1 -methanesulfonyl-2-methoxy-benzene (1 lOmg, 0.591mmol) in DCM (2mL) and stirring was continued for lOminutes. After which the reaction mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with water and extracted with dichloromethane. The organic layer was washed with saturated brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 65mg (64.35%Yield) of 2- methanesulfonyl-phenol. 2-Methylsulfonyl-phenol was converted to 4-(2-methanesulfonyl- phenoxy)-piperidine hydrochloride according to Step 1 of the General Scheme).
EXAMPLE 119 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- methanesulfonyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (63mg, 0.48mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (40mg, 0.163mmol) in DMF (ImL) followed by HOBt (26mg, 0.195mmol) and EDCLHCl (37mg, 0.195mmol). After 2 minutes 4-(2-methanesulfonyl-phenoxy)- piperidine hydrochloride (57mg, 0.195mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water and extracted with ethyl acetate. The ethyl acetate was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by preparative HPLC to afford 28mg (26.66%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2- methanesulfonyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]
+: 483, 98.59%.
1H NMR (300MHz, CDCl
3): δ 8.8(bs, IH), 8.0(m, IH), 7.7-7.6(d, 2H), 7.6-7.5(m, IH), 7.5-7.3(m, 3H),7.2-7.0(m, 3H), 5.1-4.9(m, 3H), 4.6-4.5(dd,lH), 4.2-4. l(m, 2H), 3.9-3.4(m, 3H), 3.2(s, 3H), 2.1-1.9(m, 4H).
EXAMPLE 120 - Synthesis of 5-(2-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-[4- (2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (328mg, 2.5mmol) to a stirred solution of 5-(2-fluoro-phenyl)-lH-pyrazole-3- carboxylic acid (150mg, 0.724mmol) (prepared by the method used for the synthesis of Intermediate 29, starting from 2'fluoroacetophenone) in DMF (5mL) followed by HOBt (lOOmg, 0.76mmol) and EDCI.HC1 (140mg, 0.76mmol). After 2 minutes 2-amino-l-[4-(2-chloro- phenylammo)-piperidin-l-yl]-ethanone dihydrochloride (220mg, 0.724mmol) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, the precipitate was collected. The solid was purified by recrystallisation from ethyl acetate The residue obtained was again purified by column chromatography (using 60-120 silica gel and 60% EtOAc in hexane as eluent) to afford 88mg (26.6% Yield) of 5-(2-fluoro-phenyl)-isoxazole-3-carboxylic acid (2-[4-(2-chloro-phenylamino)- piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+:457, 91.47%.1H NMR (300MHz, DMSO- d6): δ8.75(t, IH), 8.0(m, IH), 7.6(m, IH), 7.38(m, 2H), 7.1(m, 3H),6.82(d, IH), 6.6(m, IH), 4.8(m, IH), 4.3(d, IH), 4.1(d, 2H), 3.8(d, IH), 3.6(m, IH), 3.1(t, IH), 2.8(t, IH), 1.9(t, 2H), 1.45(m, 2H).
EXAMPLE 121 - Synthesis of S-Phenyl-isoxazole-S-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (96mg, 0.745mmol) was added to a stirred solution of [(5-phenyl-isoxazole-3- carbonyl)-amino]-acetic acid (67mg, 0.27mmol) in DMF (3mL) followed by HOBt (36mg, 0.273mmol) and EDCLHCl (62mg, 0.32mmol). After 2 minutes 4-(3-trifluoromethyl-phenoxy)- piperidine hydrochloride (70mg, 0.248mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was
purified by column chromatography (using 60-120 silica gel and 50%EtOAc in hexane as eluent) to afford 60mg (34%Yield) of 5-phenyl-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3- trifluorometliyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]
+: 474, 70.17%.
1H NMR (300MHz, DMSOd
6): δ8.7-8.6(t, IH), 8.0-7.9(m, 2H), 7.6-7.5(m, 4H), 7.4(s, IH), 7.3-7.26(t, 3H), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.5(m, IH), 3.3(m, IH), 2.0(m, 2H), 1.8-1.4(m, 2H).
EXAMPLE 122 - Synthesis of 5-(2-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-oxo- 2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (167mg, 1.3mmol) was added to a stirred solution of 5-(2-fluoro-phenyl)- isoxazole-3-carboxylic acid (76mg, 0.37mmol) in DMF (2mL) (prepared by the method used for the synthesis of Intermediate 25, starting from 2'-fluoroacetophenone) followed by HOBt (52mg, 0.38mmol) and EDCI.HC1 (74mg, 0.39mmol). After 2 minutes 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (125mg, 0.37mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by washing with methanol to afford the 40mg (34%Yield) of 5-(2-fluoro-phenyl)- isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}- amide. LC/MS [M+H]+: 492, 92.84%. 1H NMR (300MHz, DMSOd6): δ8.8(t, IH), 8.0(m, IH), 7.6(m, IH), 7.4(m, 3H), 7.25(m, 3H), 7.2(d, IH), 4.8(m, IH), 4.2(m, 2H), 3.7(m, 2H), 3.4(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 123 - Synthesis of 5-(2-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
10%Pd/C (50mg) was added to a stirred solution of 5-(2-benzyloxy-phenyl)-isoxazole-3- carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl} -amide (192mg, 0.33mmol) (prepared by the method used to generate Intermediate 43) in methanol (5OmL) and stirred under H
2 atmosphere with pressure for 3hrs. The reaction mixture was filtered through celite, the celite was washed with methanol and the filtrate was concnetrated under reduced pressure. Washing with ethyl acetate afforded the residue. The residue was purified by preparative HPLC to afford 31mg (19.1%Yield) of 5-(2-hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]
+: 490, 96.83%.
1H NMR (300MHz, DMSOd
6): δlθ.8(s, IH), 8.68(t, IH), 7.8(m, IH), 7.5(t, IH), 7.3(m, 4H), 7.18(s, IH), 7.04(d, IH), 6.94(t, IH), 4.8(m, IH), 4.2(d, 2H), 3.8(m, IH), 3.7(m, IH), 3.4(m, IH), 3.25(m, IH), 1.9(m, 2H), 1.65(m, 2H).
EXAMLE 124 - Synthesis of 5-(3-Hydroxy-phenyl)-isoxazole-3-carboxylic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-ainide
DIPEA (133mg, 1.03mmol) was added to a stirred solution of 5-(3-hydroxy-phenyl)- isoxazole-3-carboxylic acid (61mg, 0.294mmol) ) (prepared by the method used for the synthesis of Intermediate 25, starting from 2'-benzyloxyacetophenone) in DMF (2mL) followed by HOBt (41.7mg, 0.308mmol) and EDCLHCl (59mg, O.308mmol). After 2 minutes 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (99mg, 0.294mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure The residue was purified by washing with ethylacetate to afford 54mg (37.7% Yield) of 5-(3-hydroxy-phenyl)-isoxazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 490, 94.4%. 1H NMR(300MHz, DMSOd6): δ9.9(s, IH), 8.65(t, IH), 7.5(t, IH), 7.3(m, 7H), 6.95(m, IH), 4.8(m, IH), 4.2(d, 2H), 3.9(s, IH), 3.7(m, IH), 3.4(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 125 - Synthesis of 5-(4-Hydroxy-phenyl)-isoxazole-3-carboxyIic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (133mg, 1.03mmol) was added to a stirred solution of 5-(4-hydroxy-phenyl)- isoxazole-3-carboxylic acid (61 nig, 0.294mmol) (prepared by the method used for the synthesis of Intermediate 25, starting from 3'benzyloxyacetophenone) in DMF (2mL) followed by HOBt (41.7mg, 0.308mmol) and EDCLHCl (59mg, 0.308mmol). After 2 minutes 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (99mg, 0.294mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from 10%EtOAc in hexane afforded 74mg (51.3%Yield) of 5-(4-hydroxy-phenyl)-isoxazole-3-carboxylic acid (2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 490, 90.53%. 1H NMR (300MHz, DMSOd5): δlθ.2(s, IH), 8.6(s, IH), 7.78(d, 2H), 7.5(t, IH), 7.3(t, 3H), 7.16(s, IH), 6.9(m, 2H), 4.7(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 126 - Synthesis of 5-(3-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-oxo- 2-[4-(3-trifluoromethyI-phenoxy)-piperidin-l-yI]-ethyl}-amide
DIPEA (131mg, l.Ommol) was added to a stirred solution of 5-(3-fluoro-phenyl)- isoxazole-3-carboxylic acid (60mg, 0.29mmol) (prepared by the method used for the synthesis of Intermediate 25, starting from 3'-fluoroacetophenone) in DMF (2mL) followed by HOBt (41mg, OJmmol) and EDCLHCl (58mg, 0.3mmol). After 2 minutes 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (125mg, 0.37mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with
ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from methanol afforded 84mg (59.15%Yield) of 5-(3-fluoro-phenyl)-isoxazole-3- carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 492, 70.25%. 1H NMR (300MHz, DMSOd6): δ8.6(t, IH), 7.8(m, 2H), 7.5(m, 3H), 7.24(m, 4H), 4.7(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 127 - Synthesis of 5-(4-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-oxo- 2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (13 lmg, 1.Ommol) was added to a stirred solution of 5-(4-fluoro-phenyl)- isoxazole-3-carboxylic acid (60mg, 0.29mmol) (prepared by the method used for the synthesis of Intermediate 25, starting from 4'-fiuoroacetophenone) in DMF (2mL) followed by HOBt(41mg, 0.3mmol) and EDCLHCl (58mg, 0.3mmol). After 2 minutes 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (125mg, 0.37mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from chloroform afforded 77mg (54.2% Yield) of 5-(4-fiuoro-phenyl)-isoxazole- 3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]
+: 492, 96.7%.
1H NMR(300MHz, DMSOd
6): δ8.62(t, IH), 8.0(m, 2H), 7.5(t, IH), 7.38(m, 3H), 7.26(t, 3H), 4.8(m, IH), 4.2(d, 2H), 3.7(m, 4H), 3.4(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 128 - Synthesis of 5-(4-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2- [4- (5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (253mg, 1.96mmol) was added to a stirred solution of 5-(4-fluoro-phenyl)- isoxazole-3-carboxylic acid (81mg, 0.39mmol) (prepared by the method used for the synthesis of
Intermediate 25, starting from 4'-fluoroacetophenone) in DMF (2mL), followed by HOBt (56mg, 0.41mmol) and EDCI.HC1 (79mg, 0.41mmol). After 2 minutes 2-amino-l-[4-(5-chloro-pyiidin-3- yloxy)-piperidin-l-yl]-ethanone hydrochloride (120mg, 0.39mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from 10% ethyl acetate in hexane afforded 32mg (17.8%Yield) of 5-(4-fluoro- phenyl)-isoxazole-3-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo- ethyl}-amide. LC/MS [M+H]+: 459, 92.84%. 1H NMR (300MHz, CDCl3): δ8.2(s, 2H), 7.86(m, IH), 7.76(m, 2H), 7.14(m, 3H), 6.9(s, IH), 4.7(m, IH), 4.3(m, 2H), 3.7(m, 3H), 3.5(m, IH), 1.9(m, 4H).
EXAMPLE 129 - Synthesis of 5-(4-Fluoro-phenyl)-isoxazole-3-carboxylic acid {2-[4- (2-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (186mg, 1.43mmol) was added to a stirred solution of {[5-(4-fiuoro-phenyl)- isoxazole-3-carbonyl] -amino} -acetic acid (76mg, 0.29mmol) (prepared by the method used for the synthesis of Intermediate 26, starting from 4'-fluoroacetophenone) in DMF (2mL) followed by HOBt (41mg, 0.3mmol) and EDCI.HC1 (58mg, 0.3mmol). After 2 minutes 2-chloro-3-(piperidin- 4-yloxy)-pyridine hydrochloride (72mg, 0.29mmol) (prepared by method used for the synthesis of Intermediate 15) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from 10% ethyl acetate in hexane to afford 66mg (50% Yield ) of 5-(4-fluoro- phenyl)-isoxazole-3-carboxylic acid {2-[4-(2-chloro-pyridm-3-yloxy)-piperidin-l-yl]-2-oxo- ethyl}-amide. LC/MS [M+H]+: 459, 94.53%. 1H NMR(300MHz, DMSO-d6): δ 8.65(s, IH), 7.98(m, 3H), 7.72(m, IH), 7.38(m, 4H), 4.8(m, IH), 4.2(d, 2H), 3.7(m, 2H), 3.4(m, 2H), 1.9(m, 2H), 1.6(m, 2H).
EXAMPLE 130 - Synthesis of S-β-Fluoro-phenylHsoxazole-S-carboxylic acid {2-[4- (5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (253mg, 1.96mmol) was added to a stirred solution 5-(3-fluoro-phenyl)-isoxazole-
3-carboxylic acid (81mg, 0.39mmol) (prepared by the method used for the synthesis of Intermediate 25, starting from 3'-fiuoroacetophenone) in DMF (2mL) followed by HOBt(56mg, 0.41mmol) and EDCLHCl (79mg, 0.41mmol). After 2 minutes 2-amino-l-[4-(5-Chloro-pyridin-3- yloxy)-piperidin-l-yl]-ethanone hydrochloride (120mg, 0.38mmol) (prepared according to Step 1 and 5 of the General Scheme) was added to the reaction mixture and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from 1% methanol in ethyl acetate to afford 35mg (19.5% Yield) of 5-(3-fluoro- phenyl)-isoxazole-3-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)-piperidin-l -yl]-2-oxo- ethyl}-amide. LC/MS [M+H]+: 459, 100%. 1H NMR (300MHz, DMSO-d6): δ 8.7(t, IH), 8.32(d, IH), 8.22(s, IH), 7.78(t, 2H), 7.72(s, IH), 7.58(m, IH), 7.5(s, IH), 7.36(m, IH), 4.8(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, 2H), 3.4(m, IH), 3.2(m, IH), 2.0(m, 2H), 1.6(m, 2H).
Intermediate 61 - Synthesis of l-Phenyl-lH-pyrazoIe-4-carboxylic acid
1 -Phenyl- lH-pyrazole (250mg, 1.7mmol) was added to a cold (0-40C) solution of DMF
(1.5g, 1.6mL, 9.7mmol) and POCl3 (1.86g, l.lmL, 19.2mmol) and stirring continued for 10 minutes. The resulting mixture was heated at 1060C for 2.5hrs. The reaction mixture was cooled and quenched with ice cold water, basified with 20% aqueous NaOH solution, the solid was collected to afford 330mg (crude) of 1 -phenyl- lH-pyrazole-4-carbaldehyde. 1H NMR (300MHz,
CDCl3): δ 10.0(s, IH), 8.45 (s, IH), 8.2 (s, IH), 7.75 (d, 2H), 7.55 (t, 2H), 7.45 (t, IH). Sulphamic acid (253mg, 2.6mmol) in water (0.5 mL) was added at 00C to a mixture of phenyl- lH-pyrazole-4-carbaldehyde (0.5 g, 2.34 mmol) in acetone (3mL). After 2 minutes sodium chlorite (315 mg, 3.5 mmol) was added and the resulting mixture was stirred at 00C for 30mimites. Water was added and the solid obtained was isolated by filtration to afford 140mg (85%yield) of 1 -phenyl- lH-pyrazole-4-carboxylic acid.
EXAMPLE 131 - Synthesis of l-Phenyl-lH-pyrazole-4-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (223mg, 0.3mL, 1.72mmol) was added to a stirred solution of 1 -phenyl- IH- pyrazole-4-carboxylic acid (65mg, 0.34mmol) in DMF (5mL) followed by HOBt (51 mg, 0.38mmol) and EDCI (165mg, 0.86 mmol). After 2 minutes of stirring, 2-amino-l-[4-(2-chloro- phenoxy)-piperidin-l-yl]-ethanone hydrochloride salt (116 mg, 0.38mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the product was extracted with EtOAc and the organic layer was washed with brine. The organic phase was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (using 60-120 silica gel and 70% EtOAc in hexane as eluent) to afford 70 mg (46.35% yield) of 1- phenyl- lH-pyrazole-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}- amide. LC/MS [M+H]+: 439.15, 95.23%. 1H NMR (300MHz, CDCl3): δ 8.4 (s, IH), 8.06 (s, IH), 7.76(d, 2H), 7.54 (t, 2H), 7.42 (m, 2H), 7.2 (d, IH), 7.1 (t, IH), 7.0(t, 2H), 4.7 (t, IH), 4.4-4.2 (m, 2H), 4.1(m, IH), 3.8 - 3.5 (m, 2H), δ 3.5 (m, IH), 2.1-1.8 (m, 4H).
EXAMPLE 132 - Synthesis of 2-[(BiphenyI-4-ylmethyl)-amino]-l-[4-(2-chloro- phenoxy)-piperidin- 1-yl] -ethanone
A mixture of biphenyl-4-yl -methanol (250mg, 1.35mmol) and aqueous HBr (48%) (3mL) was stirred at 1000C for 3hrs. After completion of the reaction, the mixture was diluted with cold water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer collected was concentrated under reduced pressure to afford 330mg (98.5%Yield) 4-bromomethyl-biphenyl. 4- Bromomethyl-biphenyl (120mg, 0.485mmol) was added to a stirred mixture of 2-amino-l-[4-(2- chloro-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (134mg, 0.44mmol) (prepared according to Step 1 and 5 of the General Scheme), LiOH.H2O (40mg, 0.97mmol) and 4A molecular sieves (350mg) in DMF (4mL) and stirring was continued overnight. The reaction mixture was filtered and filtrate was diluted with cold water, extracted with ethyl acetate and organic layer was dried over sodium sulfate. The organic layer was concentrated under reduced pressure. Purification by column chromatography (using neutral alumina and 5% MeOH in DCM as eluent) to afford 22mg (11.4%Yield) of 2-[(biphenyl-4-ylmethyl)-amino]-l-[4-(2-chloro-phenoxy)-piperidin-l-yl]- ethanone. LC/MS [M+H]+: 435, 90.56%. 1H NMR (CDCl3): δ7.64-7.52(m, 4H), 7.5-7.3(m, 6H), 7.24-7.15(dt, IH), 7.0-6.86(m, 2H), 4.6(q, lH),4.0-3.8(m, 3H), 3.7-3.6(m, 3H), 3.5(s, 2H), 3.4- 3.3(m, IH), 2.0-1.8(m, 4H).
Intermediate 62 - Synthesis of 4-[l,3,4]Oxadiazol-2-yl-benzoic acid
Hydrazine hydrate was added to a solution of terephthalic acid monomethyl ester (Ig,
5.5mmol) in MeOH (1OmL) and stirring was continued for lhr. The reaction mixture was concentrated to afford 900mg (90.09%Yield) of 4-hydrazinocarbonyl-benzoic acid. p-Toluene sulfonic acid (48mg, 0.277mmol) was added to a solution of 4-hydrazinocarbonyl-benzoic acid (500mg, 2.77mmol) in triethylorthoformate (7.5mL, 44.0mmol) and stirring was continued with heating at 1000C for 3hrs. The reaction mixture was diluted with water, the solid was collected to
afford 200mg (37.9%Yield) of 4-[l,3,4]oxadiazol-2-yl-benzoic acid.
EXAMPLE 133 - Synthesis of N-{2-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-2-oxo- ethyI}-4-[l,3,4]oxadiazol-2-yl-benzamide
DIPEA (203mg, 0.27mL, 1.57mmol) was added to a stirred solution of 4-[l,3,4]oxadiazol- 2-yl-benzoic acid (lOOmg, 0.52mmol) in DMF (2mL) followed by HOBt (85mg, 0.63mmol) and EDCI (121mg, 0.63mmol). After 2 minutes of stirring, 2-amino-l-[4-(2-chloro-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (192mg, 0.63mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the product was extracted with EtOAc and the organic layer was washed with brine. The organic phase was dried over Na2SO4 and concentrated under reduced pressure to afford 88 mg (38.09% yield) of N-{2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2- oxo-ethyl}-4-[l,3,4]oxadiazol-2-yl-benzamide. LC/MS [M+H]+: 441.13, 95.23%. 1H NMR (300MHz, DMSO-d6): δ 9.4 (s, IH), 8.8 (t, IH), 8.1(q, 4H), 7.4 (m, IH), 7.3 (m, 2H), 7.0(m, IH), 4.8(m, IH), 4.2(m, 2H), 3.7 (m, 2H), 3.5 (m, 2H), 2.18(m, 2H), 1.8-1.5(m, 4H).
Intermediate 63 - Synthesis of 4-Phenyl-lH-pyrazole hydrochloride
Trityl chloride (1.58g, 5.67mmol) was added to a stirred cold (0-5° C) solution of 4-iodo pyrazole (Ig, 5.15mmol) and triethylamine (1.04g, 10.3mmol) in DCM (12mL). Stirring was continued at room temperature overnight. Cold water was then added and the product was extracted with DCM and the organic layer was washed with saturated sodium bicarbonate solution followed by brine. The organic phase collected was dried over Na
2SO
4 and concentrated under reduced pressure The residue was purified by column chromatography (using neutral alumina and 2% EtOAc in hexane as eluent) to afford 1.9g (84.4%Yield) of 4-iodo- 1 -trityl- lH-pyrazole.
Na
2CO
3 (727 mg, 6.86mmol) was added to a stirred solution of 4-iodo-l-trityl-lH-pyrazole (1.5g, 3.43mmol) in toluene: H
2O (4:1, 2OmL). Pd(PPh
3)
4 (790mg, 0.686mmol) and phenylboronic acid (838mg, 6.86mmol) were then added and the reaction mixture was heated to reflux for 2 hrs. The reaction mixture was then diluted with water and the product was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over sodium sulfate and concentrated to in vacuo. Purification by column chromatography (using neutral alumina and 5% EtOAc in hexane as eluent) afforded 790mg (59.4%Yield) of 4-phenyl-l-trityl-lH-pyrazole.
1H NMR (300MHz, CDCl
3): δ 7.96-7.94 (s, IH), 7.64-7.6(s, IH), 7.46-7.4 (d, 2H), 7.36 -7.0 (m, HH), 7.24-7.16(m, 7H). A solution of 4-phenyl-l-trityl-lH-pyrazole (785mg, 2.03mmol) in ether.HCl (15 mL) was stirred for lhr. The reaction mixture was then concentrated under reduced pressure and washed with hexane to afford 320mg (87.4%Yield) of 4-phenyl-lH-pyrazole hydrochloride.
1H NMR (300MHz, DMSO-d
6): δ 8.1-8.08 (s, 2H), 7.64(d, 2H), 7.38(t, 2H), 7.22 (t, IH).
EXAMPLE 134 - Synthesis of 4-Phenyl-pyrazoIe-l-carboxylic acid {2-[4-(2-chloro- phenoxy)-piperidin-l-yI]-2-oxo-ethyl}-amide
A mixture of 4-phenyl-lH-ρyrazole hydrochloride (70mg, 0.387mmol), DIPEA (lOOmg, 0.81mmol) and DCM (5mL) was added to a stirred solution of triphosgene (36 mg, 0.12 mmol) in DCM (2mL) at room temperature. After 30 minutes, to the above solution, a mixture of 2-amino- l-[4-(2-chloro-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared by method used for the synthesis of Intermediate 15) 118mg, 0.3875mmol), DIPEA (lOOmg, 0.81mmol) and DCM (5mL) was added and the resulting mixture was stirred at room temperature for lhr. Cold water was then added and the product was extracted with EtOAc and the organic layer was washed with brine. The organic phase was dried over Na2SO4 and concentrated under reduced pressure The residue was purified by preparative HPLC to afford 60mg (17.6%Yield) of 4-phenyl-pyrazole-l- carboxylic acid {2-[4-(2-chloro-phenoxy)-ρiperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 439.15, 93.19%. 1H NMR (300MHz, DMSO-d6): δ 8.8 (s, IH), 8.4 (t, IH), 8.35-8.3 (s, IH), 7.8- 7.72(d, 2H), 7.48-7.38 (m, 3H), 7.35 -7.24 (m, 3H), 7.0(t, IH), 4.8(m, IH), 4.2 (d, 2H), 3.8-3.6(m,
2H),3.6-3.4 (m, 2H), 2.1-1.8 (m, 2H), 1.8-1.6 (m, 2H).
Intermediate 64 - Synthesis of l-Phenyl-lH-[l,2,3]triazole-4-carboxylic acid
Oxalyl chloride (4.7g, 3.ImL, 37.0mmol) was added to a cold (0-4
0C) solution of DMF
(2.25g, 2.4mL, 30.8mmol) in CHCl3 (2OmL) and stirring was continued for 10 minutes. The reaction mixture was heated at 4O0C for 10 minutes and cooled to -1O0C. Diazoacetic acid ethyl ester (3.5g, 3.5mL, 30.6mmol) was then added and stirred at room temperature for lhrThe reaction mixture was concentrated ether was then added, the precipite was collected and dissolved in 10%aq Hac (1OmL) and stirring was continued for lhr. The reaction mixture was extracted with ether , washed with saturated sodium bicarbonate solution and brine. The organic phase was dried over Na2SO4 and concentrated under reduced pressure to afford 590mg (21% Yield) of 2-diazo-3- oxo-propionic acid ethyl ester. 1H NMR (300MHz, CDCl3): δ 9.7 (s, IH), 4.4 (q, 2H), 1.4 (t, 3H). Aniline (143mg, 1.5mmol) was added to a solution of 2-diazo-3-oxo-propionic acid ethyl ester (200mg, 1.4mmol) and HOAc (0.2mL) in EtOH (0.5mL) and stirring was at room temperature overnight. The reaction mixture was concentrated and cold water was then added, the solid was collected to afford 264mg (87.41% Yield) of 1 -phenyl- IH-[1, 2,3]triazole-4-carboxylic acid ethyl ester. 1H NMR (300MHz, CDCl3): δ 8.5 (s, IH), 7.8 (d, 2H), 7.6-7.48 (m, 3H), 4.5 (q, 2H), 1.4 (t, 3H). LiOH-H2O (80mg, 1.9mmol) was added to a stirred solution of 1 -phenyl- IH-[I, 2, 3] triazole-4-carboxylic acid ethyl ester (130mg, 0.6 mmol) in THFrH2O (1 :1, 4 mL), and the resulting mixture was stirred at room temperature for 45 min. The reaction mixture was concentrated under reduced pressure. Cold water was then added and it was acidified with 10% aqueous HCl, the solid was collected to afford 40 mg (35.4% yield) of l-phenyl-lH-[l,2,3] triazole-4-carboxylic acid. 1H NMR (300MHz, DMSO-d6): δ 13.4 (bs, IH), 9.4 (s, IH), 8.0 (d, 2H), 7.7 (t, 2H), 7.6 (t, IH).
EXAMPLE 135 - Synthesis of l-PhenyMH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (200mg, 0.27mL, 1.55mmol) was added to a stirred solution of 1 -phenyl- IH- [l,2,3]triazole-4-carboxylic acid (60mg, 0.32mmol) in DMF (5mL) followed by HOBt (47.6 mg, 0.35mmol) and EDCI (153 mg, 0.8 mmol). After 2 minutes of stirring, 2-amino-l -[4-(2-chloro- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (107 mg, O.35mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the precipitate formed was collected to afford 108mg (76.6% Yield) of l-phenyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 440.14, 95.71%. 1H NMR
(300MHz, DMSOd6): δ 9.36 (s, IH), 8.5 (t, IH), 8.02 (d, 2H), 7.68 (m, 3H), 7.48 (d, IH), 7.36- 7.22 (m, 2H), 7.0 (t, IH), 4.8 (m, IH), 4.3 (d, 2H), 3.8(t, 2H), 3.6 (s, 2H), 2.1 (d, 2H), 1.8 (d, 2H).
EXAMPLE 136 - Synthesis of l-Phenyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (137mg, l.Ommol) was added to stirred solution of l-phenyl-lH-[l,2,3] triazole-4- carboxylic acid (50mg, 0.26mmol) in DMF (3mL) followed by HOBT (39mg, 0.29mmol) and EDCI (lOOmg, 0.53mmol). After 2 minutes of stirring, 2-amino-l-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General
Scheme) (90mg, 0.26mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with cold water and extracted with ethyl acetate. The organic layer collected was dried over sodium sulfate and concentrated under reduced pressure afforded residue. The residue was purified by column chromatography (using 60-120 silica gel and 50%EtOAc in hexane as eluent) afforded 63mg (50%Yield) of 1 -phenyl- IH-[1, 2,3]triazole-4- carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 474.14, 94.PA1H NMR (300MHz, DMSO-d6): δ 9.38 (s, IH), 8.5 (t, IH), 8.0 (d, 2H), 7.7-7.6 (t,
2H), 7.6-7.5 (t, 2H), 7.36-7.24 (t, 3H), 4.8 (q, IH), 4.25 (d, 2H), 4.0-3.8(m, IH), 3.8-3.7 (m, IH), 3.5(m, 2H), 2.0 (m, 2H), 1.8-1.5 (m, 2H).
EXAMPLE 137 - Synthesis of l-(3-Fluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyI-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (168mg, 1.3mmol) was added to a stirred solution of l-(3-fluoro-phenyl)-lH- [1,2,3] triazole-4-carboxylic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-fluoroaniline) (60mg, 0.29mmol) in DMF (5mL) followed by, HOBt (43mg, 0.32mmol) and EDCI (139mg, 0.72mmol). After 2 minutes of stirring, 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride salt (prepared according to Step 1 and 5 of the General Scheme) (108mg, 0.32mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the precipitate formed was collected to affordl lOmg (77.46%Yield) of l-(3-fluoro-phenyl)- IH-[1, 2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 492.16, 90.71%. 1H NMR (300MHz, DMSO-d6): δ 9.4(s, IH), 8.56(t, IH), 8.0 (q, 2H), 7.74 (q, IH), 7.58 (t, IH), 7.46-7.24 (m, 4H), 4.9(m, IH), 4.3(d, 2H), 4.0 (bs, IH), 3.8(bs, IH), 3.5(bs, 2H), 2.1 (d, 2H), 1.8(d, 2H).
EXAMPLE 138 - Synthesis of l-(3-Fluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidύi-l-yl]-2-oxo-ethyl}-amide
DIPEA (168mg, 1.3mmol) was added to a stirred solution of l-(3-fiuoro-phenyl)-lH- [l,2,3]triazole-4-carboxylic acid (60mg, 0.29mmol) (prepared by the method used for the
synthesis of Intermediate 64, starting from 3-fluoroaniline) in DMF (5mL) followed by HOBt (43mg, 0.32mmol) and EDCI (139mg, 0.72mmol). After 2 minutes of stirring, 2-amino-l-[4-(2- chloro-5-fluoro-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (103mg, 0.32mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and the solid was collected to afford 128mg (92.75% Yield) of l-(3-fluoro-phenyl)-lH-
[ 1, 2,3 ]triazole-4-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}- amide. LC/MS [M+H]+: 476, 96.08%. 1H NMR (300MHz, DMSOd6): δ 9.4 (s, IH), 8.54 (t, IH), 8.0-7.96 (t, 2H), 7.72 (q, IH), 7.54-7.34 (m, 2H), 7.34 (d, IH), 6.9 (t, IH), 4.9(s, IH), 4.3(d, 2H), 3.8(bs, 2H), 3.55(bs, 2H), 2.05(d, 2H), 1.8(d, 2H).
EXAMPLE 139 - Synthesis of l-m-Tolyl-lH-[l,2,3]triazole-4-carboxyUc acid {2-oxo- 2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (171mg, 1.32mmol) was added to a stirred solution of 1-m-Tolyl-lH- [1 ,2,3]triazole-4-carboxylic acid (60mg, 0.29mmol) (prepared by the method used for the synthesis of Intermediate 64, starting from 3-methylaniline) in DMF (5mL) followed by HOBt (44mg, 0.32mmol) and EDCI (141mg, 0.73mmol). After 2 minutes of stirring, 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (1 lOmg, 0.32mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and the solid was collected to afford the crude solid. The crude solid obtained was purified by recrystallisation from a solvent system (3:7:0.2), EtOAc: hexane: MeOH to afford lOOmg (70.5%Yield) of 1-m-tolyl-lH- [l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}- amide. LC/MS [M+H]+: 488, 96.08%. 1H NMR (300MHz, DMSO-d6): δ 9.3 (s, IH), 8.5 (t, IH), 7.84 (s, IH), 7.8 (d, IH), 7.56-7.44 (m, 2H), 7.38-7.24 (m, 4H), 4.8(m, IH), 4.2(d, 2H), 3.9(s, IH), 3.8(s, IH), 3.5(s, IH), 3.3(s, IH), 2.4(s, 3H), 2.1(t, 2H), 1.8(d, 2H).
EXAMPLE 140 - Synthesis of l-m-Tolyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2- chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA ( 171 mg, 1.32mmol) was added to a stirred solution of 1 -m-tolyl- IH-
[l,2,3]triazole-4-carboxylic acid (60mg, 0.29mmol) (prepared by the method used for the synthesis of Intermediate 64, starting from 3-methylaniline) in DMF (5mL) followed by HOBt (44mg, 0.32mmol) and EDCI (141mg, 0.73mmol). After 2 minutes of stirring, 2-amino-l-[4-(2- chloro-5-fluoro-phenoxy)-piperidin-l-yl]-ethanone (105mg, 0.32mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and the precipitate was collected The solid obtained was purified by recrystallisation from a solvent system EtOAc:hexane:MeOH (2:8:0.2) to afford 103mg (74.1%Yield) of 1 -m-tolyl- 1 H- [l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}- amide. LC/MS [M+H]+: 472, 96.02%. 1H NMR (300MHz, DMSO-d6): δ 8.5 (t, IH), 7.86 (s, IH), 7.8 (d, IH), 7.54-7.44 (m, 2H), 7.38-7.24 (m, 4H), 6.88(t, IH), 4.9(m, IH), 4.3(d, 2H), 3.8(m, 2H), 3.5(s, 2H), 2.4(s, 3H), 2.1(d, 2H), 1.8(d, 2H).
EXAMPLE 141 - Synthesis of l-(2-Cyano-phenyl)-lH-[l,2,3]triazoIe-4-carboxylic acid {2-OXO-2- [4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl] -ethyl}-amide
DIPEA (163mg, 1.26mmol) was added to a stirred solution of l-(2-cyano-phenyl)-lH- [l,2,3]triazole-4-carboxylic acid (60mg, 0.28mmol) (prepared by the method used for the
synthesis of Intermediate 64, starting from 2-aminobenzonitrile) in DMF (5mL) followed by HOBt (41mg, 0.3mmol) and EDCI (134mg, 0.7mmol). After 2 minutes of stirring, 2-amino-l-[4- (3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (104mg, 0.3mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and the solid was collected. The solid obtained was purified by column chromatography (using 60-120 silica gel and 30-70% EtOAc in hexane as eluent) to afford 52mg (37.6%Yield) of l-(2-cyano-phenyl)-lH- [ 1, 2,3 ]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}- amide. LCMS [M+H]+: 499, 96.38%. 1H NMR (300MHz, DMSO-d6): δ 9.3 (s, IH), 8.64 (t, IH), 8.22 (d, IH), 8.02 (t, 2H), 7.84 (t, IH), 7.6 (t, IH), 7.4 (t, 3H), 4.9(m, IH), 4.3(d, 2H), 4.0(s, IH), 3.8(s, IH), 3.5(s, IH), 2.1(t, 2H), 1.8(d, 2H).
EXAMPLE 142 - Synthesis of l-(2-Cyano-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (61mg, 4.7mmol) was added to a stirred solution of l-(3-cyano-phenyl)-lH-
[l,2,3]triazole-4-carboxylic acid (202mg, 0.94mmol) (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminobenzonitrile) in DMF (5mL) followed by HOBt (14mg, 1.03mmol) and EDCI (452mg, 2.36mmol). After 2 minutes of stirring, 2-amino-l- [4-(3 -trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethanone hydrochloride (prepared according to
Step 1 and 5 of the General Scheme) (351mg, 1.03mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and the solid was collected. The solid obtained was purified by column chromatography (using 60-120 silica gel and 30-70% EtOAc in hexane as eluent) to afford 75mg (16% Yield) of l-(2-cyano-phenyl)-lH- [l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}- amide. LC/MS [M+H]+: 499, 99.08%. 1H NMR (300MHz, DMSO-d6): δ 9.48(s, IH), 8.58 (t, 2H),
8.4 (d, IH), 8.06 (d, IH), 7.9 (t, IH), 7.38(t, 3H), 4.9(m, IH), 4.3(d, 2H), 4.0(s, IH), 3.8(s, IH), 3.5(m, 2H), 2.1(t, 2H), 1.8(s, IH), 1.6(s, IH).
EXAMPLE 143 - Synthesis of l-o-Tolyl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2- [4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (171mg, 1.32mmol) was added to a stirred solution of l-o-tolyl-lH-[l,2,3]triazole- 4-carboxylic acid (60mg, 0.29mmol) (prepared by the method used for the synthesis of Intermediate 64, starting from 2-methylaniline) in DMF (5mL) followed by HOBt(44mg, 0.32mmol) and EDCI (141mg, 0.73mmol). After 2 minutes of stirring, 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (1 lOmg, 0.32mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and the solid was collected. The solid obtained was purified by recrystallisation from a solvent system (3:7:0.2), EtOAc: hexane: MeOH to afford 124mg (86.17%Yield) l-o-tolyl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2- [4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 488, 98.68%. 1H NMR (300MHz, DMSO-d6): δ 9.0 (s, IH), 8.5 (t, IH), 7.6-7.48 (m, 4H), 7.48 (m, IH), 7.36 (t, 3H), 4.9(m, IH), 4.3(d, 2H), 4.0 (s, IH), 3.8(s, IH), 3.6(m, 2H), 2.2(s, 3H), 2.1(t, 2H), 1.7(s, IH), 1.6(s, lH).
EXAMPLE 144 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2- [4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (215mg, 1.66mmol)was added o a stirred solution of l-pyridin-3-yl-lH- [l,2,3]triazole-4-carboxylic acid (70mg, 0.37mmol) (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine) in DMF (5mL) followed by HOBt (55mg, 0.41mmol) and EDCI (177mg, 0.92mmol) and stirring was continued at ambient temperature. After 2 minutes of stirring, 2-amino-l-[4-(2-chloro-phenoxy)-piperidin-l-yl]- ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (124mg, 0.4mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and filtered the precipitate was filtered to afford 147mg (90.74%Yield) of l-pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piρeridin-l-yl]-2- oxo-ethyl} -amide. LC/MS [M+H]+: 441, 98.31%. 1H NMR (300MHz, DMSO-d6): δ 9.5 (s, IH), 9.24 (d, IH), 8.76 (d, IH), 8.56 (t, IH), 8.46 (d, IH), 7.74 (d, IH), 7.48 (d, IH), 7.36 (q, 2H), 7.02 (t, IH), 4.8(m, IH), 4.4(d, 2H), 3.8(t, 2H), 3.6(t, 2H), 2.1(d, 2H), 1.8(d, 2H).
EXAMPLE 145 - Synthesis of l-Cyclopentyl-lH-[l,2,3]triazole-4-carboxylic acid {2-
[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (300mg, 2.3mmol) was added to a stirred solution of 1-Cyclopentyl-lH- [l,2,3]triazole-4-carboxylic acid (75mg, 0.41mmol (prepared by the method used for the synthesis of Intermediate 64, starting from cyclopentylamine) in DMF (5mL) followed by HOBt (61mg, 0.4mmol) and EDCI (200mg, lmmol). After 2 minutes of stirring, 2-amino-l-[4-(5-chloro- pyridin-3-yloxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (156mg, 0.5mmol) was added and the resulting mixture was stirred at
ambient temperature overnight. Cold water was then added and filtered the precipitate was filtered. The solid obtained was purified by recrystallisation from a solvent system EtOAc:hexane:MeOH (3:7:0.2) to afford 98mg (55%Yield) of l-cyclopentyl-lH-[l,2,3]triazole-4- carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide.LC/MS [M+H]+ 433, 98.59%. 1H NMR (300MHz, DMSO-dό): δ 8.7 (s, IH), 8.34 (m, 2H), 8.24 (s, IH), 7.74 (s, IH), 5.1 (m, IH), 4.9(m, IH), 4.2(d, 2H), 4.0(bs, IH), 3.8(bs, IH), 3.4(s, IH), 3.3 (m, IH), 2.3 (m, 2H), 2.1(m, 4H), 1.9-1.5(m, 6H).
EXAMPLE 146 - Synthesis of l-(5-Fluoro-pyridin-3-yl)-lH-[l,2,3]triazole-4- carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (195mg, 1.66mmol) was added to a stirred solution of l-(5-fluoro-pyridin-3-yl)- lH-[l,2,3]triazole-4-carboxylic acid (70mg, 0.33mmol) (prepared by the method used for the synthesis of Intermediate 64, starting from 3-amino-5-fluoropyridine) in DMF (5mL) followed by HOBt (55mg, 0.41mmol) and EDCI (177mg, 0.92mmol). After 2 minutes of stirring, 2-amino-l- [4-(2-chloro-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (124mg, 0.4mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was then added and the solid was collected to afford 147mg (90.74%Yield) of l-(5-fluoro-pyridin-3-yl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 441 (M+l), 98.31%. 1H NMR (300MHz, DMSO-d6): δ 9.5 (s, IH), 9.24 (d, IH), 8.76 (d, IH), 8.56 (t, IH), 8.46 (d, IH), 7.74 (d, IH), 7.48 (d, IH), 7.36 (q, 2H), 7.02 (t, IH), 4.8(m, IH), 4.4(d, 2H), 3.8(t, 2H), 3.6(t, 2H), 2.1(d, 2H), 1.8(d, 2H).
Intermediate 65 - Synthesis of 4-(5-Methyl-[l,3,4]oxadiazol-2-yl)-benzoic acid
Oxalyl chloride(1.05g,8.3mmol) was added to a stirred soluton of terephthalic acid monomethyl ester (Ig, 5.55mmol) in DCM (12mL) and stirring was continued at ambient temperature for 4hr.The reaction mixture was concentrated under redueced pressure to afford the residue. The residue was dissolved in DCM (4mL) and to the resulting solution was added, acetic acid hydrazide (490mg,6.66mmol), Et
3N (670mg, 6.66mmol) and stirring was continued at temperature overnight. The reaction mixture was diluted with water and extracted with ethylacetate, dried over sodium sulfate and concentrated under reduced pressute to afford Ig (76.39%Yield) of 4-(N'-acetyl-hydrazinocarbonyl)-benzoic acid methyl ester. A stirred soluton of 4-(N'-acetyl-hydrazinocarbonyl)-benzoic acid methyl ester (500mg, 5.55mmol) in POCl
3 (12mL) was heated at 100
0C for 3hr. The reaction mixture was concentrated under reduced pressure to afford the residue. The residue was diluted with cold water, extrated with ethyl acetate, washed the organic layer with sodiuim bicarbonate solution, saturated brine solution and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to 350mg (75.92%Yield) of afford 4-(5-methyl-[l,3,4]oxadiazol-2-yl)-benzoic acid methyl ester. LiOKH
2O (330mg, 8 mmol) was added to a solution of 4-(5-methyl-[l,3,4]oxadiazol-2-yl)-benzoic acid methyl ester (350mg, 1.6 mmol) in the mixture of methanol (4mL), THF (1OmL) and H
2O (4mL). The resulting reaction mixture was stirred at ambient temperature for 2hrs. The reaction mixture was concentrated The residue was diluted with water, acidified with aqueous citric acid solution, extracted with ethyl acetate and dreid over sodium sulfate. The organic layer was concentrated under reduced pressure to afford 280mg (87.46%Yield)of 4-(5-methyl-[l,3,4]oxadiazol-2-yl)-benzoic acid.
EXAMPLE 147 - Synthesis of N-{2-[4-(2-Chloro-phenoxy)-piperidin-l-yl]-2-oxo- ethyl}-4-(5-methyl-[l,3>4]oxadiazol-2-yl)-benzamide
DIPEA (180mg, 1.47mmol) was added to a stirred solution of 1 4-(5-methyl-
[l,3,4]oxadiazol-2-yl)-benzoic acid (lOOmg, 0.48mmol) in DMF (2mL) followed by HOBt (79mg, 0.58mmol) and EDCI (113mg, 0.587mmol). After 2 minutes of stirring, 2-amino-l-[4-(2-chloro- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (179mg, 0.587mmol) (prepared according to
Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the product was extracted with EtOAc and the organic layer was washed with brine. The organic phase was dried over Na2SO4 and concentrated under reduced pressure The residue was purified by preparative HPLC to afford 50mg (22.52%Yield) of N-{2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-4-(5-methyl- [l,3,4]oxadiazol-2-yl)-benzamide. LC/MS [M+H]+: 93.97%. 1H NMR (300MHz, DMSO-d6): δ 8.8 (s, IH), 8.2-7.9 (m, 4H), 7.5-7.2(m, 3H), 7.0 (bt, IH), 4.8 (bs, IH), 4.2 (bs, 2H), 3.7(bs, 2H), 3.5 (m, 2H), 2.7-2.5 (m, 3H), 2.1-1.5 (m, 4H).
EXAMPLE 148 - Synthesis of 3 '-Dimethylamino-biphenyl-4-carboxylic acid {2- [4-(2- chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (170mg, 1.3mmol) was added to a stirred solution of to a stirred solution of 3'- dimethylamino-biphenyl-4-carboxylic acid (80mg, O.33mmol) (generated from methyl 3'- amino(l , 1 !-biphenyl)-4-carboxylate) in DMF (2mL) followed by HOBt (49mg, 0.36mmol) and EDCI (126mg, 0.66mmol). After 2 minutes of stirring, 2-amino-l-[4-(2-chloro-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (1 lOmg, 0.36rnmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the product was extracted with EtOAc and the organic layer was washed with brine. The organic phase was dried over Na2SO4 and concentrated under reduced pressure The residue was purified by preparative HPLC to afford 36mg (22%Yield) of 3'- dimethylamino-biphenyl-4-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l -yl]-2-oxo- ethyl}-amide. LC/MS [M+H]+: 492, 86.69%. 1H NMR (300MHz, DMSO-d6): δ 8.6 (t, IH), 8.0- 7.9 (d, 2H), 7.8-7.7(d, 3H), 7.5-7.4 (d, IH), 7.36-7.24 (m, 3H), 7.1-7.0 (m, 2H), 7.0-6.94 (m, IH)5 6.88-6.8 (d, IH), 4.8 (q, IH), 4.2 (d, 2H), 4.0-3.7 (m, 4H), 3.5(m, 3H), 3.0 (s, 6H), 2.0-1.8 (m, 3H), 1.8-1.6 (m, 3H).
Intermediate 66 - Synthesis of 4-(Pyrrolidine-l-carbonyl)-benzoic acid
DIPEA (1.26g, 9.7mmol)was added to a stirred solution of terephthalic acid monomethyl ester (500mg, 2.77mmol) in DMF (3mL) followed by HOBt (410mg, 3.0mmol) and EDCI (1.32g, 7.0mmol). After 2 minutes of stirring, pyrrolidine (215mg, 3.0mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with cold water, the precipitate was collected to afford 53mg (81.9%Yield) of 4-(pyrrolidine-l-carbonyl)- benzoic acid methyl ester. LiOH. H2O (285mg, 6.8mmol) was added to a solution of 4- (pyrrolidine-l-carbonyl)-benzoic acid methyl ester (530mg, 2.27mmol) in a mixture of methanol (2mL), THF (6mL) and H2O (2mL). The reaction mixture was stirred at ambient temperature for 3hrs. The reaction mixture was concentrated, the residue was diluted with water, and acidified with cone. HCl. The solid was filtered and dried to afford 41mg (82.32%Yield).
EXAMPLE 149 - Synthesis of N-{2-Oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin- l-yl]-ethyI}-4-(pyrrolidine-l-carbonyl)-benzamide
DIPEA (200mg, 1.5mmol) was added to a stirred solution of 4-(ρyrrolidine-l-carbonyl)- benzoic acid (75g, 0.34mmol) in DMF (5mL) followed by HOBt (50mg, 0.37mmol) and EDCI (164mg, 0.85mmol). After 2 minutes of stirring, 2-amino-l-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (127mg, 0.37mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with cold water, the precipitate was collected to afford 130mg (75.58%Yield) of N- {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethyl } -4- (pyrrolidme-l-carbonyl)-benzamide. LC/MS [M+H]+: 504, 94.9%. 1H NMR (300MHz, DMSO- d6): δ 7.2(t, IH), 7.96 (d, 2H), 7.64-7.48(m, 3H), 7.36 (t, 3H), 4.9(b, IH), 4.2 (d, 2H), 4.0(d, 2H), 3.5(t, 2H), 3.4(t, 2H), 2.1-1.8 (m, 6H), 1.7 (m, 3H).
Intermediate 67 - Synthesis of 9H-Carbazole-3-carboxylic acid
A mixture of dicylcohexylphosphino-2', 3'-dimethoxy biphenyl (17mg, 0.04mmol), palladium(II) acetate (5mg, 0.02mmol) in toluene (2OmL) was degassed with argon gas for 15min. To the resulting mixture was added aniline (136mg, 1.4mmol), K3PO4 (636mg, 3.0mmol), and 4- bromobenzoic acid methyl ester (300mg, 1.4mmol) and stirring was continued at reflux temperature overnight. After completion of the reaction, the reaction mixture was filtered through celite and the filtrate collected was concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 15% ethyl acetate in hexane as eluent) to afford 200mg (63.4% Yield) of 4-phenylamino-benzoic acid methyl ester. A stirred solution of 4- phenylamino-benzoic acid methyl ester (98mg, 0.43mmol), Palladium acetate (193mg, 0.86mmol) in acetic acid (2mL) was heated at 1100C for 15hr. After completion of the reaction, the reaction mixture was diluted with water, extracted with ethyl acetate and washed with sodium bicarbonate solution. The organic layer collected was dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography (using 60-120 silica gel and 10% ethyl acetate in hexane as eluent) to afford 120mg (60.3%Yield) of 9H-carbazole-3-carboxylic acid methyl ester. LiOELH2O (680mg, 16.2 mmol) was added to a stirred solution of 9H-carbazole-3- carboxylic acid methyl ester (120mg, 0.53 mmol) in the mixture of methanol (ImL), THF (3mL) and H2O (ImL). The reaction mixture was stirred at 60 C for 3hrs. The reaction mixture was concentrated. The residue was diluted with water, acidified with aqueous 10% aqueous HCl solution, filtered the solid to afford 104mg (92.85%Yields) of 9H-carbazole-3-carboxylic acid.
EXAMPLE 150 - Synthesis of 9H-Carbazole-3-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (137mg, l.Ommol) was added to a stirred solution of to a stirred solution of 9H-
carbazole-3-carboxylic acid (50mg, 0.23mmol) in DMF (4mL) followed by HOBt (35mg, 0.26mmol) and EDCI (116mg, 0.59mmol). After 2minutes, 2-amino-l-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethanone hydrochloride (88mg, 0.26mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water and the precipitate was filtered. The solid obtained was purified by recrystallisation from a mixture of 10% ethyl acetate in hexane and 50% MeOH in H2O to afford 81mg (69.23%Yield) of 9H-carbazole-3-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 496, 91.03%. 1H NMR (SOOMHZ5 DMSO^6): δ 11.6(s, IH), 8.8(s, IH), 8.6(t, IH), 8.1(d, IH), 8.0(d, IH), 7.6(d, 2H), 7.46 (t, IH), 7.36(t, 2H), 7.24 (t, IH), 4.9(b, IH), 4.3 (d, 3H), 4.0(d, 2H), 3.5(d, 2H), 2.1 (t, 2H), 1.7 (d, 2H).
Intermediate 68 - Synthesis of l-Phenyl-lH-imidazole-4-carboxyIic acid
A stirred solution of lH-imidazole-4-carboxylic acid (0.5g, 0.00446mmol), concentrated sulfuric acid (0.5mL) and MeOH (2OmL) was heated to reflux overnight. The reaction mixture was concentrated under reduced pressure. The residue was diluted with cold water, extracted with ethyl acetate and washed the organic layer with sodium bicarbonate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to afford 325mg (58%Yield) of IH- imidazole-4-carboxylic acid methyl ester. A mixture of lH-imidazole-4-carboxylic acid methyl ester (225mg, l.δmmol), cuprous oxide (225mg, 1.8mmol), iodobenzene (736mg, 3.6mmol), 1,10- phenanthroline (320mg, 1.8mmol) and cesium carbonate (1.74g, 5.3mmol) in DMSO (2mL) in a seal tube was subjected to reaction in a microwave reactor (time:5min, temp: 90
0C, power: zero)The reaction mixture was filtered through celite and the filtrate collected was concentrated under reduced pressure. The residue was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure to afford the residue. Purification by column chromatography (using 60-120 silica gel and 50% ethyl acetate in hexane as eluent) to afford 200mg (55%Yield) of 1 -phenyl- lH-imidazole-4-carboxylic acid methyl ester.
LiOH.H
2O (143mg, 3.4mmol) was added to a stirred solution of 1 -phenyl- lH-imidazole-4- carboxylic acid methyl ester (231mg, l.lmmol) in a mixture of THF(3mL), MeOH(ImL), H
2O (ImL) and stirring was continued at ambient temperature for 3hrs. The reaction mixture was concentrated under reduced pressure. Cold water was then added and acidified it with 10% aqueous HCl, the solid was collected to afford 180mg (83.7%Yield) of 1 -phenyl- lH-imidazole- 4-carboxylic acid.
EXAMPLE 151 - l-Phenyl-lH-imidazole-4-carboxylic acid {2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (186mg, 1.4mmol) was added to a stirred solution of 1 -phenyl- lH-imidazole-4- carboxylic acid (60mg, 0.32mmol) in DMF (5mL) followed by HOBt (47mg, 0.35mmol) and EDCI (153mg, O.δmmol). After 2 minutes of stirring, 2-amino-l-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (119mg, 0.35mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water, the solid was collected to afford the 129mg (86%Yield) of 1 -phenyl- lH-imidazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: Purity: 473(M+1), 96.61%. 1H NMR (300MHz, DMSO-d6): δ 8.4(s, IH), 8.32(s, IH), 8.08(t, IH), 7.8(d, 2H), 7.6-7.48(t, 3H), 7.46 (t, IH), 7.36-7.24(t, 3H), 4.9(s, IH), 4.2 (d, 2H), 4.0(bs, IH), 3.8(bs, IH), 3.5(d, 2H), 2.1 (t, 2H), 1.8 (d, 2H).
EXAMPLE 152 - Synthesis of l-Phenyl-lH-imidazole-4-carboxylic acid {2-[4-(2- chϊoro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (186mg, 1.44mmol) was added to a stirred solution of 1 -phenyl- lH-imidazole-4- carboxylic acid (60mg, 0.32mmol) in DMF (5mL) followed by HOBt (47mg, 0.35mmol) and EDCI (153mg, O.δmmol). After 2 minutes of stirring, 2-amino-l-[4-(2-chloro-5-fiuoro-phenoxy)-
piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (119mg, 0.37mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Cold water was added and the precipitate formed was collected to afford the residue. The residue was purified by preparative HPLC to afford 80mg (55% Yield) of 1- phenyl- lH-imidazole-4-carboxylic acid {2-[4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo- ethylj-amide. LC/MS [M+H]+: 457.14, 96.27%. 1H NMR (300MHz, DMSOd6): δ 8.46(s, IH), 8.14(s, IH), 8.1 (t, 2H), 7.8 (d, 2H), 7.6-7.4 (m, 4H), 7.32 (d, IH), 6.9 (t, IH), 4.9(bs, IH), 4.2(d, 2H), 3.8(b, 2H), 3.55(b, 2H), 2.1(d, 2H), 1.8(d, 2H).
EXAMPLE 153 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- formyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (360mg, 2.8mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (190mg, 0.8mmol) in DMF (2.OmL) followed by HOBt (160mg, 1.2mmol) and EDCLHCl (230mg, 1.2mmol). After 2 minutes of stirring, 2-(piperidin-4-yloxy)- benzaldehyde hydrochloride (200mg, O.δmmol) (prepared according to Step 1 and 5 of the General Scheme) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and concentrated The residue was purified by column chromatography (using neutral alumina and 5% MeOH in CHCl3) to afford 150mg (44% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4- (2-formyl-phenoxy)-piρeridin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 433.18, 98.8%, 1H NMR (300MHz, DMSO-d6): δl3.8(m, IH), 10.3(s, IH), 8.2(m, IH), 7.8 (m, 2H), 7.7 (m, 2H), 7.5(m, 2H), 7.4(m, 2H), 7.1(m, 2H), 5.0(m,lH), 4.2(m, 2H), 3.7(m, 2H), 3.5(m, 2H), 2.0(m, 2H), 1.8(m, 2H).
EXAMPLE 154 - Synthesis of 2-(l-{2-[(5-Phenyl-lH-pyrazole-3-carbonyl)-amino]- acetyl}-piperidin-4-yloxy)-benzoic acid
Sulphamic acid (34mg, 0.3mmol) was added to stirred a mixture of 5-phenyl-lH-pyrazole- 3-carboxylic acid {2-[4-(2-formyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide (50mg, O.lmmol) in acetone (ImL). After 2 minutes, sodium chlorite (36 mg, 0.4mmol) was added and the resulting mixture was stirred at ambient temperature for 1 hour. Water was added and the solid obtained was isolated by filtration to afford 24mg (53%yield) of 2-(l-{2-[(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetyl}-piperidin-4-yloxy)-benzoic acid.LC/MS [M+H]+: 449.17, 90.6%, 1H NMR (300MHz, DMSOd6): δl3.8(b, IH), 12.8(b, IH), 8.1(m, IH), 7.8 (m, 2H), 7.6 (m, IH), 7.5(m, 3H), 7.4(m, IH), 7.2(m, IH), 7.1(m,lH), 7.0(m, IH), 4.8(m, IH), 4.2(m, 2H), 3.7(m, 3H), 3.5(m, IH), 2.0(m, IH), 1.7(m, 3H).
EXAMPLE 155 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- hydroxymethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
NaBH4 (5mg, 0.12mmol) was added to stirred a mixture of 5 -phenyl- lH-pyrazole-3- carboxylic acid {2-[4-(2-formyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide (50mg, O.lmmol) in MeOH (2mL) and stirring was continued for lhr. The reaction mixture was diluted with water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford the residue. The residue was purified by recrystallisation from CHCl3 in hexane mixture to afford 25mg (58%yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-hydroxymethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 435.2, 93.8%, 1H NMR (300MHz, DMSOd6): δl3.8(m, IH), 8.0(m, IH), 7.8(m, 2H), 7.5 (m, 2H), 7.4 (m, 2H), 7.2(m, IH), 7.1(m, IH), 7.0(m, IH), 6.9(m,lH), 5.0(m, IH), 4.7(m, IH), 4.5(m, 2H), 3.7(m, 3H), 3.5(m, 2H), 2.0(m, 2H), 1.7(m, 2H).
EXAMPLE 155 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-
(3,4,5-trifluoro-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (135mg, l.Ommol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (69mg, 0.3mmol) in DMF (2.OmL) followed by HOBt (61mg, 0.45mmol) and EDCLHCl (85mg, 0.45mmol). After 2 minutes of stirring, 4-(3,4,5-trifiuoro- phenoxy)-piperidine hydrochloride (75mg, 0.3mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and concentrated The residue was purified by column chromatography (using neutral alumina and 5% MeOH in CHCl3) to afford 26mg (19% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-oxo- 2-[4-(3,4,5-trifluoro-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 459.16, 97.98%, 1H NMR (300MHz, DMSO-d6): 613.8(m, IH), 8.1(m, IH), 7.8(m, 2H), 7.4 (m, 4H), 7.1 (m, IH), 4.7(m, IH), 4.2(m, 2H), 3.9(m, IH), 3.7(m,lH), 2.0(m, 3H), 1.7(m, 3H).
EXAMPLE 156 - Synthesis of δ-Phenyl-lH-pyrazole-S-carboxylic acid (2-{4-[2- (hydroxyimino-methyI)-phenoxy]-piperidin-l-yl}-2-oxo-ethyI)-amide
Hydroxylamine hydrochloride (33mg, 0.5mmol) was added to a stirred cold (0 C) mixture of 5-phenyl- 1 H-pyrazole-3 -carboxylic acid {2-[4-(2-formyl-phenoxy)-piperidin- 1 -yl]-2-oxo- ethyl}-amide (150mg, 0.4mmol) and sodium acetate (65mg, 0.8mmol) in MeOH (3mL) and stirring was continued at ambient temperature for 4hrs. The reaction mixture was diluted with water, extracted with ethyl acetate and dried over sodium sulfate. The organic layer was concentrated under reduced pressure to afford 25mg (58%yield) of 5-phenyl- lH-pyrazole-3- carboxylic acid {2-[4-(2-hydroxymethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 435.2, 93.8%, 1H NMR (300MHz, DMSO-d6): δ8.3(s, IH), 8.2(b, IH), 7.8(d, 2H), 7.7 (d, IH), 7.5 (t, 2H), 7.4(t, 2H), 7.2(d, 2H), 7.0(t, IH), 4.8(m,lH), 4.2(m, 2H), 3.8(m, 2H), 3.5(m, 2H),
2.0(m, 2H), 1.8(m, 2H).
EXAMPLE 157 - Synthesis of δ-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[4-(2- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (154mg, 1.2mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (lOOmg, 0.4mmol) in DMF (2.OmL) followed by HOBt (60mg, 0.49mmol) and EDCI.HC1 (93mg, 0.49mmol). After 2 minutes of stirring, 4-(2-trifluoromethyl- phenoxy)-ρiperidine trifluoroacetate (167mg, 0.49mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethylacetate, washed with brine and concentrated to afford 11 lmg (57.81% Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-oxo- 2-[4-(2-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 473.17, 97.38%, 1H NMR (300MHz, DMSOd6): δl3.8(m, IH), 8.1(bs, IH), 7.9-7.3(m, 8H), 7.2-7.0 (m, 2H), 4.9(bs, IH), 4.2(m, 2H), 3.8-2.9(m, 4H), 2.0-1.7(m, 3H).
EXAMPLE 158 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3-cyano- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (316mg, 2.5mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (18 lmg, 0.7mmol) in DMF (3mL) followed by HOBt (108mg, 0.8mmol) and EDCI.HC1 (160mg, 0.8mmol). After 2 minutes of stirring, 3-(piperidin-4-yloxy)- benzonitrile hydrochloride (150mg, 0.7mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and concentrated The residue was purified by column chromatography (using neutral alumina and 5% MeOH in CHCl
3) afford 44mg (14.3% Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(3-cyano-
phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]
+: 473.17, 99.16%,
1H NMR (300MHz, DMSOd
6): δ 8.1(m, IH), 7.8(m, 2H), 7.6(m, 4H), 7.4(m, 3H), 7.1(b, IH), 4.8(m, IH), 4.2(m, 2H), 4.0(m, IH), 3.8(m, IH), 2.0(m, 2H), 1.5(m, 2H).
EXAMPLE 159 - Synthesis of S-Phenyl-lH-pyrazole-θ-carboxylic acid (2-{4-[2- (methoxyimino-methyl)-phenoxy]-piperidin-l-yl}-2-oxo-ethyl)-amide
O-Methyl-hydroxylamine hydrochloride (23mg, 0.27mmol) was added to a stirred cold (O0C) mixture of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4-(2-formyl-phenoxy)-piperidin-l- yl]-2-oxo-ethyl}-amide (lOOmg, 0.23mmol) and sodium acetate (56mg, 0.7mmol) in MeOH (1OmL) and stirring was continued at ambient temperature for 4hrs. The reaction mixture was diluted with water, extracted with ethyl acetate, dried over sodium sulfate, and the organic layer was concentrated under reduced pressure. The residue was purified by preparative HPLC to afford 33mg (31.13%yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid (2-{4-[2-(methoxyimino- methyl)-phenoxy]-piperidin-l-yl}-2-oxo-ethyl)-amide. LC/MS [M+H]+: 462.2, 96.30Zc1H NMR (300MHz, DMSOd6): δl3.7(s, IH), 8.4(s, IH), 8.1(t, IH), 7.8 (t, 2H), 7.7 (d, IH), 7.5-7.34(m, 4H), 7.2(d, IH), 7.1(s, IH), 7.0(t, IH), 4.8(bs,lH), 4.2(b, 2H), 3.9(s, 3H), 3.8-3.6(bs, 2H), 3.5(b, 2H), 2.1-1.9(b, 2H), 1.8-1.6(b, 2H).
EXAMPLE 160 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- methylcarbamoyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (72mg, 0.56mmol) was added to a stirred solution of 2-(l-{2-[(5-phenyl-lH- pyrazole-3-carbonyl)-amino] -acetyl }-piperidin-4-yloxy)-benzoic acid (75mg, 0.16mmol) in DMF
(2mL) followed by HOBt (33mg, 0.24mmol) and EDCLHCl (46mg, 0.24mmol). After 2 minutes of stirring, methyl amine hydrochloride (1 lmg, O.lόmmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and concentrated The residue was purified by column chromatography (using neutral alumina and 5% MeOH in CHCl3) afford 23mg (31.5% Yield) of 5-phenyl- 1 H-pyrazole-3-carboxylic acid {2-[4-(2-methylcarbamoyl-phenoxy)-piperidin- 1 -yl]-2- oxo-ethyl} -amide. LC/MS [M+H]+: 462.2, 96.9%. 1H NMR (300MHz, DMSO-d6): δ 8.1(m, IH), 8.0(m, IH), 7.8(m, 2H), 7.6(m, IH), 7.4(m, 4H), 7.2(m, IH), 7.1(b, IH), 7.0(m, IH), 4.8(b, IH), 4.2(m, 2H), 3.6(m, 3H), 2.8(d, 3H), 2.0(m, 2H), 1.8(m, 2H).
EXAMPLE 161 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(2- carbamoyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
N-Methyl moφholine (28mg, 0.28 mmol) was added to a cold (-70
0C) solution of 2-(l-{2- [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetyl} -piρeridin-4-yloxy)-benzoic acid (100 mg, 0.2 mmol) in THF (2mL) followed by isobutyl chloroformate (41mg, 0.3mmol) and stirring was continued at the at -70
0C for 1 hr. After complete formation of anhydride, ammonia gas was bubbled through the reaction mixture at -70
0C for 10 minutes. The resulting mixture was then stirred at room temperature for lhr. Water (10 mL) was added, the precipitated was filtered and dried to afford 54 mg (60.6% yield) of 5-phenyl- 1 H-pyrazole-3-carboxylic acid {2-[4-(2- carbamoyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]
+: 448.18, 93.4%.
1H NMR (300MHz, DMSO-d
6): δ 7.9(m, IH), 7.8(m, 2H), 7.5(m, 3H), 7.4(m, IH), 7.3(m, IH), 7.1(m, 2H), 4.3(s, 2H), 4.0(m, IH), 3.8(m, IH), 3.5(m, 2H), 2.1(m, 3H), 1.9(m, 3H), l.l(m, IH).
EXAMPLE 162 - Synthesis of 5-(2-Trifluoromethyl-phenyl)-lH-pyrazole-3- carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (137mg, l.Oόmmol) was added to a stirred solution {[5-(2-trifluoromethyl-phenyl)- lH-pyrazole-3-carbonyl]-amino}-acetic acid (95mg, 0.3mmol) (prepared by the method used for the synthesis of Intermediate 30, starting from (2'-trifluoromethyl)acetophenone) in DMF (2.OmL) followed by HOBt (47mg, 0.35mmol) and EDCLHCl (67mg, 0.35mmol). After 2 minutes of stirring, (2-chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (75mg, 0.3mmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and concentrated under reduced pressure. The residue was purified washing with diethyl ether to afford 67mg (43.5% Yield) of 5-(2- trifiuoromethyl-phenyl)- 1 H-pyrazole-3-carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin- 1 - yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 506.12, 97.9%, 1H NMR (300MHz, DMSOd6): δl3.2(d, IH), 8.2(m, IH), 7.5(m, 5H), 7.2 (m, 2H), 6.85(m, IH), 6.5(m, 2H), 4.8(m,lH), 4.3(m, IH), 4.1(d, 2H), 4.0(m, IH), 3.6(m, IH), 3.2(m, IH), 2.7(m, IH), 1.9(m, 2H), 1.3(m, 2H).
EXAMPLE 163 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3-cyano- phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (135mg, l.Ommol) was added to a stirred of [(5-phenyl-lH-pyrazole-3-carbonyl)- amino]-acetic acid (150mg, 0.7mmol) in DMF (4mL) followed by HOBt (108mg, O.δmmol) and EDCLHCl (160mg, 0.8mmol). After 2 minutes of stirring3-(piperidin-4-ylamino)-benzonitrile dihydrochloride (prepared according to Step 2 and 5 of the General Scheme) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and concentrated. The residue was purified by column chromatography (using neutral alumina and 5% MeOH in CHCl3) to afford 91mg (42% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3-cyano-phenylamino)-piperidin-l-yl]- 2-oxo-ethyl}-amide. LC/MS [M+H]+: 429.2, 92.9%, 1H NMR (300MHz, DMSO-d6): δ 8.0(m,
IH), 7.8(m, 2H), 7.5 (m, 2H), 7.4(m, IH), 7.2(m, IH), 7.1(b, IH), 6.9(m, 2H), 6.2(m, IH), 4.3(m, IH), 4.2(m, IH), 3.8(m, IH), 3.6(m, IH), 3.4(m, IH), 2.8(m, IH), 2.0(m, 2H), 1.4(m, IH), 1.2(m,lH).
Intermediate 69 - Synthesis of 4-(Adamantan-2-ylamino)-piperidine dihydrochloride
Ammonium formate (300mg, 4.8mmol) was added to a stirred solution of 4-oxo- piperidine-1-carboxylic acid tert-butyl ester (250mg, 1.2mmol) in methanolic ammonia (2.5mL) followed by 10%Pd/C (50mg) and stirring was continued at room temperature overnight. The above mixture was filtered through celite, filtrate was collected, and concentrated under reduced pressure to furnish a crude residue. The residue was treated with 2N aqueous NaOH solution, extracted with EtOAc, dried over Na2SO4 and concentrated under reduced pressure to afford 200mg (83% yield) of (4-amino-piperidine-l-carboxylic acid tert-butyl ester. Titanium isopropoxide (756mg, 2.66mmol) was added to a stirred solution of 4-amino-piperidine-l- carboxylic acid tert-butyl ester (293mg, 1.46mmol) and adamantan-2-one (200mg, 1.33mmol) in EtOH (5mL) and stirring was continued at room temperature overnight. NaBH4 (lOOmg, 2.64mmol) was added in portionwise and the resulting mixture was stirred at room temperature for lOhrs. The reaction mixture was quenched with 2N aqueous NH3 solution and filtered. The filtrate was extracted with EtOAc, dried over Na2SO4 and concentrated under reduced pressure The residue was purified by column chromatography (using neutral alumina and 20% EtOAc in hexane as eluent) to afford 420mg (94.4% Yield) of 4-(adamantan-2-ylaniino)-piperidine-l -carboxylic acid tert-butyl ester. 1H NMR (300MHz, CDCl3): δ 4.0 (s, 2H), 2.9-2.4(m, 4H), 2.0-1.6(m, 9H), 1.7 (s, 4H), 1.6-1.5 (d, 4H), 1.45 (s, 9H), 1.35-1.2 (m, 3H). A mixture of 4-(adamantan-2- ylamino)-piperidine-l -carboxylic acid tert-butyl ester (420mg, 1.25mmol) in dioxane.HCl (2OmL) was stirred at ambient temperature for 2hrs. The reaction mixture was concentrated under reduced pressure to get the residue, which was washed with ether to afford 363mg (93.8%Yield) of 4- (adamantan-2-ylamino)-piperidine dihydrochloride. 1H NMR (300MHz, DMSO-d6): δ 9.2 (s, IH), 9.0 (s, 3H), 3.8(s, 4H), 3.5-3.3 (m, 4H), 3.0-2.8 (m, 2H), 2.4(d, 2H), 2.3(d, 3H), 2.0 (t, 2H), 1.9-1.8 (m, 5H), 1.7 (s, 2H), 1.6-1.5 (d, 2H).
EXAMPLE 164 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4- (adamantan-2-yIamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (150mg, 3.5mmol) was added to a stirred solution [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (150mg, 0.53mmol) in DMF (4mL) followed by HOBt (82mg, O.όlmmol) and EDCI.HC1 (270mg, 1.4mmol). After 2 minutes of stirring, adamantan-2-yl- piperidin-4-yl-amine dihydrochloride (150mg, 0.59mmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water and the precipitate was filtered to afford 200mg (81.63% Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[4- (adamantan-2-ylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 462.28, 91.77%.!H NMR (300MHz, DMSOd6): δ8.2-8.0(bs, IH), 7.82-7.76(d, 2H), 7.5-7.42(t, 2H), 7.4-7.34 (t, IH), 7.2-7. l(bs, IH), 4.3-4.1(m, 3H), 3.8(d, IH), 3.1(t, IH), 2.9-2.7(m, 3H), 2.1(d, 2H), 1.9-1.6(m, IH), 1.4-1.3(d, 3H), 1.2-l.l(m, 2H).
EXAMPLE 165 - Synthesis of N-Biphenyl-4-yl-3-oxo-3-[4-(3,4,5-trifluoro-phenoxy)- piperidin-l-yl]-propionamide
DIPEA (170mg, 1.3mmol) was added to a stirred solution of N-biphenyl-4-yl-malonamic acid (lOOmg, 0.4mmol) in DMF (2.OmL) followed by HOBt (81mg, 0.6mmol) and EDCI.HC1 (114mg, O.όmmol). After 2 minutes of stirring, 4-(3,4,5-trifTuoro-phenoxy)-piperidine hydrochloride (lOOmg, 0.4mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethy lacetate, washed with brine and concentrated. The remaining residue was purified by column chromatography (using neutral alumina and 5% MeOH in CHCl3) to afford 30mg (17% Yield) of N-biphenyl-4-yl-3-oxo-3-[4-(3,4,5-trifluoro-phenoxy)- piρeridin-l-yl]-propionamide. LC/MS [M+H]+: 469.17, 98.06%, 1H NMR (300MHz, DMSO-d6):
δ7.6(m, 6H), 7.4(m, 2H), 7.3(m, IH), 7.1 (m, 2H), 4.6(m, IH), 3.9(m, IH), 3.8(m,lH), 3.6(m, 2H), 3.4(m, IH), 3.2(m, IH), 2.0(m, 2H), 1.7(m, IH), 1.5(m, IH).
EXAMPLE 166 - Synthesis of N-Biphenyl-4-yI-3-[4-(3-cyano-phenoxy)-piperidin-l- yl]-3-oxo-propionamide
DIPEA (135mg, l.Ommol) was added to a stirred solution of N-biphenyl-4-yl-malonamic acid (64mg, 0.3mmol) in DMF (2.OmL) followed by HOBt (61mg, 0.45mmol) and EDCI.HC1 (85mg, 0.45mmol). After 2 minutes of stirring, 3-(piperidin-4-yloxy)-benzonitrile hydrochloride (60mg, 0.3mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethylacetate, washed with brine and concentrated The residue was purified by column chromatography (using neutral alumina and 5%MeOH in CHCI3) to afford 27mg (19.7% Yield) of N-biphenyl-4-yl-3-[4-(3-cyano-phenoxy)-piperidin-l -yl]-3-oxo-propionamide. LC/MS [M+H]+: 440.2, 98.14%, 1H NMR (300MHz, DMSOd6): δlθ.2(s, IH), 7.7(m, 5H),
7.5(m, 5H), 7.3 (m, 2H), 4.8(m, IH), 4.0(m, IH), 3.8(m,lH), 3.6(m, 2H), 3.3(m, 2H), 2.0(m, 2H), 1.7(m, IH), 1.6(m, IH).
EXAMPLE 167 - Synthesis of 5-(2-Methoxy-phenyl)-lH-pyrazoIe-3-carboxylic acid {2-[4-(2-chIoro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (99mg, 0.76mmol) was added to a stirred solution {[5-(2-methoxy-phenyl)-lH- pyrazole-3-carbonyl]-amino}-acetic acid (60mg, 0.22mmol) (prepared by the method used for the synthesis of Intermediate 30, starting from (2'-methoxy)acetophenone) in DMF (2.OmL) followed by HOBt (34mg, 0.25mmol) and EDCI.HC1 (49mg, 0.25mmol). After 2 minutes of stirring, (2-
chloro-phenyl)-piperidin-4-yl-amine dihydrochloride (54mg, 0.22mmol) was added and stirring was continued at ambient temperature overnight. The reaction mixture was diluted with water, extracted with ethyl acetate, washed with brine and concentrated. The residue was purified washing with diethyl ether to afford 67mg (43.5% Yield) of 5-(2-methoxy-phenyl)-lH-pyrazole-3- carboxylic acid {2-[4-(2-chloro-phenylamino)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS
[M+H]+: 468.2, 98.33%.Η NMR (300MHZ, DMSO-d6): δl3.6(S, IH), 8.05(m, IH), 7.7(m, IH), 7.35 (m, IH), 7.24(m, IH), 7.12(m, 2H), 7.0(m, 2H), 6.82(m, IH), 4.85(d,lH), 4.3(d, IH), 4.1(d, 2H), 3.9(s, 3H), 3.85(m, IH), 3.6(m, IH), 3.1(m, IH), 2.7(m, IH), 1.9(m, 2H), 1.4(m, 2H).
Intermediate 70 - Synthesis of 3-Hydroxy-4-methyl-benzonitrile
K2CO3 (18. Ig, 131.2mmol) was added to a stirred solution of 3-hydroxy-4-methyl-benzoic acid (5g, 33mmol) in DMF (5OmL) followed by benzyl bromide (11.8g, 69mmol) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was filtered, the fϊlterate was diluted with water and extracted with EtOAc. The organic layer was collected, dried over sodium sulfate and concentrated under reduced pressure to afford 11.3g (crude) of 3- benzyloxy-4-methyl-benzoic acid benzyl ester which was such taken for the next step without purification. NaOH (4.Og, 102mmol) was added to a stirred solution of 3-benzyloxy-4-methyl- benzoic acid benzyl ester (11.3g, 34mmol) in a mixture of MeOH (5OmL) and H2O (5OmL), stirring was continued at ambient temperature for 2hrs. The reaction mixture was concentrated under reduced pressure Cold water was then added and acidified it with 10% aqueous HCl, the precipitate was collected to afford 7.2 Ig (87.8%Yield) of 3-benzyloxy-4-methyl-benzoic acid. A mixture of 3-benzyloxy-4-methyl-benzoic acid (7.21g, 29.7mmol) in thionyl chloride (5OmL) was stirred at 790C for 4hrs. The reaction mixture was concentrated under reduced pressure to afford 7.75g (crude) of 3-benzyloxy-4-methyl -benzoyl chloride. A solution of 3-benzyloxy-4-methyl- benzoyl chloride (7.75g) in THF(IOmL) was poured to cold aqueous solution of ammonia (5OmL) with stirring, The precipitate was collected to afford 6.7g (94%Yield) of 3-benzyloxy-4-methyl- benzamide. A solution of 3-benzyloxy-4-methyl-benzamide (3.Og, 12.4mmol) in pyridine (4mL) was cooled to -300C. Imidazole (1.68g, 24.8mmol) followed by POCl3 (2.64g, 49.6mmol) were added to the cold solution with stirring and the reaction was continued at the same temperature for
30minutes. The reaction mixture was diluted with ethyl acetate. The organic layer was washed with water followed by 10%aqueous HCl solution. The organic layer separated was dried over sodium sulfate and concentrated under reduced pressure to afford 2.46g (89%Yield) of 3- benzyloxy-4-methyl-benzonitrile. Pd/C (476mg) was added to a stirred solution of 3-benzyloxy- 4-methyl-benzonitrile (2.46g, 11.Ommol) in MeOH (25mL) in an inert atmosphere and stirring was continued under H2 gas atmosphere at ambient temperature, overnight. The reaction mixture was filtered through celite. The filtrate collected was concentrated under reduced pressure to afford 1.367g (93.6% Yield) of 3-hydroxy-4-methyl-benzonitrile.
Intermediate 71 - Synthesis of 3-(3-TrifluoromethyI-phenoxy)-azetidine hydrochloride
Pd(OH)2 (lOOmg) was added to a stirred solution of l-benzhydryl-3-(3-trifluoromethyl- phenoxy)-azetidine (506mg, 1.32mmol) (prepared according to Step 1 and 5 of the General Scheme from l-(diphenylmethyl)-3-azetidinyl methanesulfonate) in EtOH (5OmL) in an inert atmosphere and shaken in a Parr aparatus under H2 atmosphere (60psi) for 7hrs. The reaction mixture was filtered through celite. The filtrate collected was concentrated under reduced pressure. The residue thus collected was stirred in EtOAcHCl for 5 minutes, the supernatant liquid was decanted, and it was dried to afford 160mg (47.9% Yield) of 3-(3-trifluoromethyl-phenoxy)- azetidine hydrochloride.
Intermediate 72 - Synthesis of 3-(3-TrifluoromethyI-phenoxy)-pyrrolidine
Pd(OH)2 (125mg) was added to a stirred solution of l-benzyl-3-(3-trifluoromethyl- phenoxy)-pyrrolidine (prepared according to Step 1 and 5 of the General Scheme from 1- benzylprrrolidin-3-ol) (125mg, 0.4mmol) in MeOH (15mL) in an inert atmosphere and stirring was continued under H2 gas atmosphere overnight. The reaction mixture was filtered through celite. The filtrate collected was concentrated under reduced pressure to afford 81 lmg (85.9%
Yield) of 3-(3-Trifluoromethyl-phenoxy)-pyrrolidine.
EXAMPLE 168 - Synthesis of l-Pyrrolidin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
Pd(OH)2 (80mg) was added to a stirred solution of l-(l-benzyl-pyrrolidin-3-yl)-lH- [ 1 ,2,3 ]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin- 1 -yl] -ethyl } - amide (prepared by the method used for the synthesis of Example 136, starting from l-benzyl-3- aminopyrrolidine) (407mg, 0.73mmol) in MeOH (4mL) in an inert atmosphere and stirring was continued under H2 gas atmosphere for 6hrs. The reaction mixture was filtered through celite. The filtrate collected was concentrated under reduced pressure to afford 260mg (80.2% Yield) of 1- pyrrolidin-3-yl- IH-[1 ,2,3 ]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+:467.19.
EXAMPLE 169 - Synthesis of l-(l-Methyl-pyrrolidin-3-yl)-lH-[l,2,3]triazole-4- carboxylic acid {2-oxo-2- [4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl] -ethyl}-amide
37% Aqueous formaldehyde (12mg, 0.4mmol) solution was added to a stirred solution of l-pyrrolidin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-1-yl] -ethyl} -amide (lOOmg, 0.15mmol) in a mixture of acetic acid (37.62mg, 0.62mmol) and H2O (0.5mL) and stirring was continued at ambient temperature for 5minutes. To the above mixture was added, Zinc powder (39.23mg, O.όmmol) and stirring was continued at ambient temperature for lhr. The reaction mixture was cooled, basifϊed with aqueous ammonia solution and extracted with DCM. The organic layer thus collected was dried over sodium sulfate and concentrated under reduced pressure to afford 88mg (88% Yield) of 1 -(I -methyl -pyrrolidin-3-yl)- IH-[1 ,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin- 1 -yl]- ethyl}-amide. LC/MS [M+H]+: 481.2. 1H NMR (300MHz, DMSO-d6): δ 11.2-11.0(b, IH), 8.85(s, IH), 8.4 (s, IH), 7.5 (t, IH), 7.3 (t, 3H), 5.51 (b, IH), 4.7(m, 2H), 4.4(d, 2H), 3.8(m, IH), 3.7(m,
2H), 3.4 (m, 2H), 2.9 (s, 3H), 2.5(m, 2H), 2.0 (m, 2H), 1.7-1.5 (m, 2H).
EXAMPLE 170 - Synthesis of l-(3,5-Difluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chIoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (160.7mg, 1.24mmol) was added to a stirred solution of l-(3,5-difluoro-phenyl)- lH-[l,2,3]triazole-4-carboxylic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3,5-difluoroaniline) (70mg, 0.31mmol) in DMF (2mL) followed by HOBt (46.2 mg, 0.34mmol) and EDCI (119mg, 0.62mmol). After 2 minutes of stirring, 2-amino- l-[4-(2-chloro-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (94.8mg, 0.31mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and the precipitate was collected. The crude solid was stirred in a mixture of 30% EtOAc in hexane and MeOH (0.5mL) for lhr and filtered to afford 95mg (64.19% Yield) of l-(3,5-difluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2- [4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 476.12, 98.71%. 1H NMR (300MHz, DMSO-d6): δ 9.5(s, IH), 8.5(t, IH), 7.9 (m, 2H), 7.6-7.4 (m, 2H), 7.4-7.2 (m, 2H), 7.0 (m, IH), 4.8 (m, IH), 4.2 (d, 2H), 3.8-3.6(m, 2H), 3.6-3.4 (m, 2H), 2.1-1.8 (m, 2H), 1.8- 1.5 (m, 2H).
EXAMPLE 171 - Synthesis of l-(3,5-Difluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(5-chloro-pyridin-3-yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (149.2mg, 1.15mmol) was added to a stirred solution of l-(3,5-difluoro-phenyl)- lH-[l,2,3]triazole-4-carboxylic acid (65mg, 0.28mmol) in DMF (2mL) followed by HOBt (43mg, 0.3 lmmol) and EDCI (1 lOmg, 0.57mmol). After 2 minutes of stirring, 2-amino-l-[4-(5-chloro- pyridin-3-yloxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (88.4mg, 0.28mmol) was added and the resulting mixture was stirred at room
temperature overnight. Cold water was added and the precipitate was collected. The crude solid was purified by recrystallisation from a mixture of DCM and hexane to afford 95mg (54.4% Yield) of l-(3,5-difluoro-phenyl)-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(5-chloro-pyridin-3- yloxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 477.12, 98.45%. 1H NMR (300MHz, DMSOd6): δ 9.5(s, IH), 8.5(t, IH), 8.3(d, IH), 8.2(d, IH), 7.9 (d, 2H), 7.75 (t, IH), 7.58-7.5 (m, IH), 4.8 (m, IH), 4.2 (d, 2H), 4.0-3.8(m, IH), 3.8-3.6(m, IH), 3.4(m, IH), 3.2(m, IH), 2.1-1.9(m, 2H), 1.9-1.5 (m, 2H).
EXAMPLE 172 - Synthesis of l-Piperidin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide hydrochloride
A mixture of 4-(4- {2-oxo-2- [4-(3 -trifluoromethyl-phenoxy)-piperidin- 1 -yl] - ethylcarbamoyl}-[l,2,3]triazol-l-yl)-piperidine-l-carboxylic acid tert-butyl ester (prepared by the method used for the synthesis of Example 136, starting from l-Boc-4-aminopiperidine) (380mg, 0.65mmol) in dioxane.HCl (5mL) was stirred at ambient temperature for 1 hr. The reaction mixture was concentrated under reduced pressure to get the crude solid which was purified by recrystallisation from a mixture of MeOH and diethyl ether to afford 252mg (73.9%Yield) of 1- piperidin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethyl}-amide hydrochloride.
EXAMPLE 173 - Synthesis of l-(l-MethyI-piperidin-4-yl)-lH-[l,2,3]triazole-4- carboxylic acid {2-oxo-2-[4-(3-trifIuoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
37% Aqueous formaldehyde (29.22mg, 0.97mmol) solution was added to a stirred solution of l-piperidin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethyl} -amide hydrochloride (252mg, 0.48mmol) in a mixture of acetic acid (240mg, 4.0mmol) and H2O (2mL) and stirring was continued at ambient temperature for 5 minutes. Zinc dust was added to the reaction mixture (94.1mg, 1.44mmol) and stirring was
continued at ambient temperature for lhr. The reaction mixture was cooled, basified with aqueous ammonia solution and extracted with DCM. The organic layer was collected and dried over sodium sulfate and concentrated under reduced pressure to afford 1 lOmg (45.6% Yield) of 1-(1- methyl-piperidin-4-yl)- IH-[1 ,2,3]triazole-4-carboxylic acid {2-oxo-2- [4-(3-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 495.2. 1H NMR (300MHz, DMSOd6): δ 8.7(s, IH), 8.3(t, IH), 7.5 (t, IH), 7.3 (t, 3H), 4.8 (m, IH), 4.5(m, IH), 4.2(d, 2H), 3.9(m, IH), 3.7(m, IH), 3.4 (m, 2H), 2.9 (m, 2H), 2.2(s, 3H), 2.1(m, 6H), 1.95 (m, 2H).
EXAMPLE 174 - Synthesis of l-Pyridin-3-yHH-[l,2,3]triazole-4-carboxylic acid {2- [4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (135.9mg, 1.05mmol) was added to a stirred solution of [(l-pyridin-3-yl-lH- [l,2,3]triazole-4-carbonyl)-amino]-acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (65mg, 0.26mmol) in DMF (2mL) followed by HOBt (39mg, 0.29mmol) and EDCI (lOlmg, 0.52mmol). After 2 minutes of stirring, 4-(2,5-difluoro-phenoxy)-piperidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (65.6mg, 0.26mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the resultant was extracted with DCM. The organic layer was collected and dried over sodium sulfate and concentrated under reduced pressure The residue obtained was purified by stirring in mixture of 30% EtOAc in hexane (1OmL), H2O (0.5mL) and MeOH (0.5mL) for 30minutes. Filtered the mixture to afford 54mg (46.5% Yield) of l-pyridin-3-yl-lH- [ 1, 2,3 ]triazole-4-carboxylic acid {2-[4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}- amide. LC/MS [M+H]+:443.16, 92.48%. 1H NMR (300MHz, DMSO-d6): δ 9.45(s, IH), 9.2(d,
IH), 8.75(m, IH), 8.5(t, IH), 8.4 (m, IH), 7.7(q, IH), 7.32-7.2(m, 2H), 6.7-6.4(m, IH), 4.8-4.7(m, IH), 4.25(d, 2H), 4.0-3.8(m, IH), 3.8-3.7(m, IH), 3.4(m, IH), 3.3(m, IH), 2.1-1.9(m, 2H), 1.8-1.5 (m, 2H).
EXAMPLE 175 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylk acid {2- [4-(5-cyano-2-methyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (94.1mg, 0.72mmol) was added to a stirred solution of [(l-pyridin-3-yl-lH-
[l,2,3]triazole-4-carbonyl)-amino]-acetic acid (45mg, 0.18mmol) (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) in DMF (2mL) followed by HOBt (27mg, 0.2mmol) and EDCI (69.8mg, 0.36mmol). After 2 minutes of stirring, 4-methyl-3-(piperidin-4-yloxy)-benzonitrile hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (50.6mg,
0.2mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and the precipitate was collected. The solid was purified by stirring in mixture of DCM in hexane to afford 40mg (49.2% Yield) of l-pyridin-3-yl- IH- [1,2,3 ]triazole-4-carboxylic acid {2-[4-(5-cyano-2-methyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 446.19, 96.84%. 1H NMR (300MHz, DMSOd6): δ 9.45(s, IH), 9.2(d, IH), 8.75(d, IH), 8.55- 8.45(t, IH), 8.4 (m, IH), 7.72-7.64(m, IH), 7.55(s, IH), 7.4-7.3(q, 2H), 4.8(m, IH), 4.25(d, 2H), 3.8-3.6(m, 2H), 3.5-3.4(m, 2H), 2.25(s, 3H), 2.1-1.8(m, 2H), 1.8-1.5 (m, 2H).
EXAMPLE 176 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[3-(3- trifluoromethyl-phenoxy)-pyrrolidin-l-yl]-ethyl}-amide
DIPEA (167.7mg, 1.29mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (106mg, 0.43mmol) in DMF (4mL) followed by HOBt (70.13mg, 0.52mmol) and EDCI (99.5mg, 0.52mmol). After 2 minutes of stirring, 3-(3-trifluoromethyl-
phenoxy)-pyrrolidine (lOOmg, 0.43mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and the precipitate was collected to afford 122mg (61.5% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[3-(3-trifluoromethyl- phenoxy)-pyrrolidin-l-yl]-ethyl} -amide. LC/MS [M+H]
+: 459.16, 96.52%.
1H NMR (SOOMHZ, DMSO-d
6): δ 13.8(s, IH), 8.05 (bs, IH), 7.8(db, 2H), 7.7-7.6(m, 7H), 7.05(bs, IH), 5.4(s, IH), 5.3(s, IH), 4.1(m, 2H), 3.9(m, IH), 3.8-3.6(m, 3H), 3.4(m, IH), 2.3-2.0(m, 2H).
Intermediate 73 - Synthesis of 4-(2-Oxo-pyrrolidin-l-yl)-benzoic acid
A mixture of pyrrolidin-2-one (500mg, 5.9mmol), 4-bromo benzoic acid methyl ester
(1.5g, 6.97mmol), Pd2(dba)3 (135mg, 0.14mmol), xantphos (256mg, 0.44mmol) and cesium carbonate (2.7g, 8.28mmol) in dioxane (2mL) in a sealed tube was subjected to reaction in a microwave reactor (time:20min, temp: 1050C, power: zero). The reaction mixture was filtered through celite and the filtrate collected was concentrated under reduced pressure. The residue was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure to afford the residue. The residue obtained was purified by column chromatography (using 60-120 silica gel and 50% ethyl acetate in hexane as eluent) to afford 485mg (37.89%Yield) of 4-(2-oxo-pyrrolidin-l-yl)-benzoic acid methyl ester. NaOH (295mg, 7.4mmol) was added to a stirred mixture of 4-(2-oxo-pyrrolidin-l-yl)-benzoic acid methyl ester (485mg, 2.2mmol) in a mixture of MeOH (5mL) and H2O (5mL), stirring was continued at 600C for lhr. After completion of the reaction, the reaction mixture was concentrated under reduced pressure to get the residue, which was acidified with 10% aqueous HCl solution, the precipitate was collected to afford 300mg (66%Yield) of 4-(2-oxo-pyrrolidin-l-yl)-benzoic acid.
EXAMPLE 177 - Synthesis of 4-(2-Oxo-pyrroIidin-l-yl)-N-{2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-benzamide
DIPEA (200mg, 1.55mmol) was added to a stirred solution of 4-(2-oxo-pyrrolidin-l-yl)- benzoic acid (70mg, 0.34mmol) in DMF (5mL) followed by HOBt (50mg, 0.37mmol) and EDCI (163mg, 0.85mmol). After 2 minutes of stirring, 2-amino-l-[4-(3-trifluoromethyl-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (127mg, 0.37mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the precipitate was filtered off. The solid obtained was purified by column chromatography (using 60-120 silica gel and 70% ethyl acetate in hexane as eluent) to afford 94mg (56.62% Yield) of 4-(2-oxo-pyrrolidin-l-yl)-N-{2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-benzamide. LC/MS [M+H]
+: 490.19, 98.95%.
1H NMR (300MHz, DMSOd
6): δ 8.6(t, IH), 7.94(d, 2H), 7.8(d, 2H), 7.6(t, IH), 7.38 (t, 3H), 4.8(m, IH), 4.2(d, IH), 4.0(t, 3H), 3.8(d, IH), 3.5(d, IH), 3.3(bs, IH), 2.5(bs, 2H), 2.1(m, 2H), 2.0(d, 2H), 1.7 (d, 2H).
EXAMPLE 178 - Synthesis of l-Cyclopropyl-lH-[l,2,3]triazole-4-carboxylic acid {2- oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (190mg, 1.47mmol) was added to a stirred solution of 1-cyclopropyl-lH- [l,2,3]triazole-4-carboxylic acid (prepared by the method used for the synthesis of Intermediate 64, starting from cyclopropylamine) (50mg, 0.32mmol) in DMF (5mL) followed by HOBt (48mg, 0.35mmol) and EDCI (156mg, 0.81mmol). After 2 minutes of stirring, 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (127mg, 0.37mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and the solid precipitate was filtered. The solid was purified by column chromatography (using 60-120 silica gel and 70% ethyl acetate in hexane as eluent) to afford 94mg (56.62% Yield) of 4-(2-oxo-pyrrolidin-l-yl)-N-{2-oxo-2-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl] -ethyl }-benzamide. LC/MS [M+H]+: 490.19, 98.95%. 1H NMR (300MHz, DMSO-d6): δ 8.6(t, IH), 7.94(d, 2H), 7.8(d, 2H), 7.6(t, IH), 7.38 (t, 3H), 4.8(m, IH), 4.2(d, IH), 4.0(t, 3H), 3.8(d, IH), 3.5(d, IH), 3.3(bs, IH), 2.5(bs, 2H), 2.1(m, 2H), 2.0(d,
2H), 1.7 (d, 2H).
EXAMPLE 179 - Synthesis of l-Morpholin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (200mg, 1.54mmol) was added to a stirred solution of l-morpholin-4-yl-lH- [ 1 ,2,3 ]triazole-4-carboxylic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 4-aminomorpholine) (70mg, 0.35mmol) in DMF (5mL) followed by HOBt (52mg, 0.38mmol) and EDCI (170mg, 0.88mmol). After 2 minutes of stirring, 2-amino-l-[4-(3- trifluoromethyl-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (120mg, 0.35mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the precipitate was collected out. The solid obtained was purified by recrystallisation from a mixture of 20% ethyl acetate in hexane and MeOH to afford 144mg (84.7% Yield) of l-morpholin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(3-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+:
483.19, 92.7%. 1H NMR (300MHz, DMSO-d6): δ 8.82(s, IH), 8.4 (t, IH), 7.6(t, IH), 7.36(t, 3H), 4.9(m, IH), 4.2(d, 2H), 4.0-3.6(m, 6H), 3.4(m, 2H), 3.2(m, 2H), 2.1(t, 2H), 1.7 (d, 2H).
Synthesis of l-Morpholin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2-chloro-5- fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (200mg, 1.54mmol) was added to a stirred solution of l-morpholin-4-yl-lH- [l,2,3]triazole-4-carboxylic acid (prepared by the method used for the synthesis of Intermediate
64, starting from 4-aminomorpholine) (70mg, 0.35mmol) in DMF (5mL) followed by HOBt (52mg, 0.38mmol) and EDCI (170mg, 0.88mmol). After 2 minutes of stirring, 2-amino-l-[4-(2- chloro-5-fluoro-phenoxy)-piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (125mg, 0.38mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and the precipitate was collected . The solid was purified by recrystallizing from a mixture of 20% ethyl acetate in hexane and MeOH to afford 116mg (70.3% Yield) of l-moφholin-4-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(2- chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+:467.15, 96.7%. 1H NMR (300MHz, DMSO-d6): δ 8.8(s, IH), 8.4 (t, IH), 7.5(t, IH), 7.3(d, IH), 6.8(t, IH), 4.9(m, IH), 4.2(d, 2H), 3.85(t, 4H), 3.7(d, 2H), 3.5(b, 2H), 3.3(bs, 4H), 2.0(d, 2H), 1.8(d, 2H).
EXAMPLE 180 - Synthesis of l-Phenyl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(3- cyano-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (167.9mg, 1.3mmol) was added to a stirred solution of [(1-phenyl-lH-
[l,2,3]triazole-4-carbonyl)-amino]-acetic acid (80mg, 0.32mmol) in DMF (3mL) followed by HOBt (48.2mg, 0.35mmol) and EDCI (124.5mg, 0.65mmol). After 2 minutes of stirring, 3- (piperidin-4-yloxy)-benzonitrile hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (77.5mg, 0.32mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and precipitate was collected. The solid was purified by column chromatography (using 70% ethyl acetate in hexane as eluent) to afford 45mg (32.6% Yield) of 1 -phenyl- IH-[1, 2,3 ]triazole-4-carboxylic acid {2-[4-(3-cyano-phenoxy)-piperidin-l-yl]- 2-oxo-ethyl} -amide. LC/MS [M+H]+: 431.18, 85.06%. 1H NMR (300MHz, DMSO-d6): δ 9.35(s, IH), 8.5 (t, IH), 8.0(d, 2H), 7.7-7.6(m, 2H), 7.58-7.45(m, 3H), 7.45-7.32(m, 2H), 4.75(m, IH), 4.2(d, 2H), 4.0-3.9(m, IH), 3.8-3.7(m, IH), 2.1-1.9(m, 2H), 1.75-1.5(m, 2H).
EXAMPLE 181 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2- [3-(2,5-difluoro-phenoxy)-pyrrolidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (156.8mg, 1.2mmol) was added to a stirred solution of [(l-pyridin-3-yl-lH- [l,2,3]triazole-4-carbonyl)-amino]-acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (75mg, OJmmol) in DMF (3mL) followed by HOBt (45mg, 0.33mmol) and EDCI (116mg, O.όmmol). After 2 minutes of stirring, 3-(2,5-difluoro-phenoxy)-pyrrolidine hydrochloride (prepared by the method used for the synthesis of Intermediate 72) (71.5mg, 0.3mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was then added and precipitate was collected to afford 48mg (37.2% Yield) of l-pyridin-3- yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(2,5-difluoro-phenoxy)-pyrrolidin-l-yl]-2-oxo- ethyl}-amide. LC/MS [M+H]+: 429.14, 92.32%. 1H NMR (300MHz, DMSOd6): δ 9.5(s, IH), 9.2 (d, IH), 8.75(d, IH), 8.6(m, IH), 8.4(m, IH), 7.7(q, IH), 7.3-7.2(m, 2H), 6.9-6.7(m, IH), 5.3- 5.1(d, IH), 4.25-4.1(m, 2H), 4.0-3.5(m, 4H), 2.3-2.2(m, IH), 2.2-2.05(m, IH).
EXAMPLE 182 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-oxo-2-[3-(3- trifluoromethyl-phenoxy)-azetidin-l-yl]-ethyl}-amide
DIPEA (158.2mg, 1.22mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino] -acetic acid (75mg, 0.30mmol) in DMF (2mL) followed by HOBt (43.4mg, 0.32mmol) and EDCI (61.6mg, 0.32mmol). After 2 minutes of stirring, 3-(3-trifluoromethyl- phenoxy)-azetidine hydrochloride (77.5mg, 0.3mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added followed by extraction with ethyl acetate. The organic phase was dried over sodium sulfate and concentrated under reduced pressure
to afford the residue. The residue was purified by column chromatography (using silica gel 60-and 100% ethyl acetate as eluent) to afford 69mg (51.1% Yield) of 5-phenyl-lH-pyrazole-3- carboxylic acid {2-oxo-2-[3-(3-trifluoromethyl-phenoxy)-azetidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 445.14, 90.72%. 1H NMR (300MHz, DMSOd6): δ 14.8(s, IH), 8.2 (t, IH), 7.78(t, 2H), 7.5(t, IH), 7.3(m, 5H), 7.16(d, 2H), 7.1(d, IH), 5.18(m, IH), 4.64(q, IH), 4.36(m, IH), 4.18(m, IH), 3.84(m, 3H).
EXAMPLE 183 - Synthesis of S-Pyridin-S-yl-lH-pyrazole-S-carboxylic acid {2-oxo-2- [3-(3-trifluoromethyl-phenoxy)-azetidin-l-yl]-ethyl}-amide
DIPEA (158.2mg, 1.22mmol) was added to a stirred solution of [(5-pyridin-3-yl-lH- pyrazole-3-carbonyl)-amino]-acetic acid (prepared by the method used for the synthesis of Intermediate 30, starting from 3-acetylpyridine) (76mg, 0.30mmol) in DMF (2mL) followed by HOBt (43.4mg, 0.32mmol) and EDCI (61.6mg, 0.32mmol). After 2 minutes of stirring, 3-(3- trifluoromethyl-phenoxy)-azetidine hydrochloride (77.5mg, 0.3mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure The residue obtained purified by column chromatography (using silica gel 60-and 100% ethyl acetate as eluent) to afford 43mg (32% Yield) of 5-pyridin-3-yl-lH-pyrazole-3-carboxylic acid {2-oxo-2-[3-(3-trifluoromethyl-phenoxy)-azetidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 446.14, 93.76%. 1H NMR (300MHz, DMSO-d6): δ 9.44(s, IH), 9.2 (d, IH), 8.72(m, 2H), 8.38(m, IH), 7.64(m, IH), 7.52(t, IH), 7.35(d, IH), 7.16(m, 2H), 5.2(m, IH), 4.7(m, IH), 4.3(m, IH), 4.2(m, IH), 3.95(d, 2H), 3.8(m, IH).
EXAMPLE 184 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(2,5- difluoro-phenoxy)-pyrrolidin-l-yI]-2-oxo-ethyl}-amide
DIPEA (166mg, 1.28mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3- carbonyl)-amino]-acetic acid (70mg, 0.28mmol) in DMF (5mL) followed by HOBt (42mg, 0.31mmol) and EDCI (136mg, 0.71mtnol). After 2 minutes of stirring, 3-(2,5-difluoro-phenoxy)- pyrrolidine hydrochloride (prepared by the method used for the synthesis of Intermediate 72)
(74mg, 0.3 lmmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and the precipitate was collected. The solid was purified by recrystallisation from a mixture of 20% ethyl acetate in hexane (15mL), water (0.5mL) and MeOH (0.03mL) to afford 92mg (76% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(2,5-difluoro- phenoxy)-pyrrolidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+:427.15. 1H NMR (300MHz, DMSO-d6): δ 13.8(s, IH), 8.1 (s, IH), 7.84(d, 2H), 7.54-7.12(m, 6H), 7.14(s, IH), 6.88(t, IH), 5.3(d, IH), 4.1(d, 2H), 3.9-3.5(m, 4H), 2.3(s, IH), 2.1(s, IH).
EXAMPLE 185 - Synthesis of l-Phenyl-lH-imidazole-4-carboxylic acid {2-[4-(2,5- difluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (170mg, 1.3mmol) was added to a stirred solution of 1 -phenyl- lH-imidazole-4- carboxylic acid (55mg, 0.29mmol) in DMF (5mL) followed by HOBt (43mg, 0.32mmol) and EDCI (140mg, 0.7mmol). After 2 minutes of stirring, 2-amino-l-[4-(2,5-difluoro-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (98.5mg, 0.32mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. The residue obtained
was purified by washing with ether to afford 63.5mg (49.6% Yield) of 1 -phenyl- lH-imidazole-4- carboxylic acid {2-[4-(2,5-difluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide.LC/MS [M+H]+: 441.17. 1H NMR (300MHz, DMSOd6): δ 8.4(s, IH), 8.3(s, IH), 8.08(t, IH), 7.8(d, 2H), 7.6(t, 2H), 7.44(t, IH), 7.3(m, IH), 6.84(m, IH), 4.8(m, IH), 4.2 (d, 2H), 4.0(bs, IH), 3.8(bs, IH), 3.7(bs, 2H), 3.5(bs, IH), 2.1 (t, 2H), 1.8 (d, 2H).
EXAMPLE 186 - Synthesis of l-PhenyI-lH-imidazole-4-carboxylic acid {2-[4-(2- chloro-phenoxy)-piperidin- 1 -yl] -2-oxo-ethyl}-amide
DIPEA (170mg, 1.3mmol) was added to a stirred solution of [(I -phenyl- lH-imidazole-4- carbonyl)-amino]-acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (55mg, 0.29mmol) in DMF (5mL) followed by HOBt (43mg, 0.32mmol) and EDCI (140mg, 0.7mmol). After 2 minutes of stirring, 4-(2-chloro-phenoxy)-piperidine hydrochloride (98mg, 0.32mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water and the precipitate was collected. The precipitate was purified by recrystallisation from a mixture of 20% EtOAc in hexane (15mL) and MeOH (O.lmL) to afford the 80mg (62.5%Yield) of 1 -phenyl- lH-imidazole-4-carboxylic acid {2- [4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]
+: 439.15, 98.75%.
1H NMR (300MHz, DMSO-d
6): δ 8.4(s, IH), 8.3(s, IH), 8.08(t, IH), 7.8(d, 2H), 7.6(t, 2H), 7.48(m, 2H), 7.34(m, 2H), 7.1(t, IH), 4.8(m, IH), 4.2 (d, 2H), 3.8(t, 2H), 3.6(t, 2H), 2.0(d, 2H), 1.8 (d, 2H).
Synthesis of l-Phenyl-lH-imidazole-4-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl- phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (236mg, 1.826mmol) was added to a stirred solution of [(1-Phenyl-lH-imidazole-
4-carbonyl)-amino] -acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (lOOmg, 0.4mmol) in DMF (5mL) followed by HOBt (60.5mg, 0.4mmol) and EDCI (195mg, 1.02mmol). After 2 minutes of stirring, 4-(2-trifluoromethyl-phenoxy)-piperidine trifluoroacetate (126mg, 0.45mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water, and the precipitate was filtered. The precipitae was purified by recrystallisation from a mixture of 20% EtOAc in hexane (15mL) and MeOH (O. ImL) to afford 113mg (58.2%Yield) of 1 -phenyl- 1 H-imidazole-4- carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide. LC/MS [M+H]+: 473, 96.85%. 1H NMR (300MHz, DMSO-d6): δ 8.4(s, IH), 8.3(s, IH), 8.08(t, IH), 7.8(d, 2H), 7.68-7.6(t, 2H), 7.6(t, 2H), 7.4-7.34(m, 2H), 7.14(t, IH), 5.0(m, IH), 4.2 (d, 2H), 3.7- 3.4(m, 4H), 2.1(d, 2H), 1.8 (d, 2H).
EXAMPLE 187 - Synthesis of l-Phenyl-lH-imidazole^-carboxylic acid {2-[4-(5- cyano-2-methyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (236mg, 1.826mmol) was added to a stirred solution of [(I -phenyl- lH-imidazole-
4-carbonyl)-amino]-acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (lOOmg, 0.4mmol) in DMF (5mL) followed by HOBt (60.5mg, 0.4mmol) and EDCI (195mg, 1.02mmol). After 2 minutes of stirring, 4-methyl-3-(piperidin-4-yloxy)-benzonitrile hydrochloride (113mg, 0.45mmol) (prepared by the method used for the synthesis of Intermediate 15) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water, the precipitate was collected and purified by recrystallisation from a mixture of diethyl ether (15mL) and MeOH (O.lmL) to afford 149mg (82.7%Yield) of 1 -phenyl- lH-imidazole-4-carboxylic acid {2-[4-(5-cyano-2-methyl-phenoxy)- piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]+: 443, 98.73%. 1H NMR (300MHz, DMSO- d6): δ 8.4(s, IH), 8.3(s, IH), 8.08(t, IH), 7.8(d, 2H), 7.6(t, 3H), 7.4-7.18(m, 3H), 4.85(m, IH), 4.2(d, 2H), 3.85-3.6(d, 2H), 3.5 (bs, 2H), 2.3(s, 3H), 2.1(d, 2H), 1.8 (d, 2H).
EXAMPLE 188 - Synthesis of l-Phenyl-lH-imidazole-4-carboxylic acid {2-[3-(3- fluoro-5-trifluoromethyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (143mg, l.lmmol) was added to a stirred solution of [(l-phenyl-lH-imidazole-4- carbonyl)-amino]-acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (68g, 0.27mmol) in DMF (2mL) followed by HOBt (39mg, 0.29mmol) and EDCI (56mg, 0.29mmol). After 2 minutes of stirring, 3-(3-fluoro-5-trifluoromethyl-ρhenoxy)-azetidine hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (75mg, 0.27mmol) was added and it was stirred overnight at ambient temperatue. Partitioning with cold water and ethyl acetate and concentration of the organic layer after drying over sodium sulfate afforded a residue which was purified by preparative HPLC to give 40mg (31.4%Yield) of 1 - phenyl- lH-imidazole-4-carboxylic acid {2-[3-(3-fluoro-5-trifluoromethyl-phenoxy)-azetidin-l- yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 463, 99.18%. 1H NMR (300MHz, DMSOd6): δ 8.44(d, IH), 8.34(d, IH), 8.2(t, IH), 7.8(d, 2H), 7.56(t, 2H), 7.44(m, IH), 7.34(d, IH), 7.12(m, 2H), 5.25(m, IH), 4.7(m, IH), 4.4(m, IH), 4.3 (m, IH), 3.9(m, 3H).
EXAMPLE 189 - Synthesis of l-Phenyl-lH-imidazole-4-carboxylic acid {2-[4-(3- fluoro-5-trifluoroniethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (97mg, 0.75mmol) was added to a stirred solution of [(I -phenyl- lH-imidazole-4- carbonyl)-amino] -acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (61mg, 0.25mmol) in DMF (2mL) followed by HOBt (35mg, 0.26mmol) and EDCI (50mg, 0.26mmol). After 2 minutes of stirring, 4-(3-fluoro-5-trifluoromethyl-phenoxy)-piperidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (75mg, 0.25mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water and extracted with ethyl acetate. The organic layer
was dried over sodium sulfate, concentrated under reduced pressure The residue obtained was purified by column chromatography (using silica gel 60-120 and 40%EtOAc in hexane as eluent) to afford 66mg (54%Yield) of 1 -phenyl- 1 H-imidazole-4-carboxylic acid {2-[4-(3-fluoro-5- trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]
+: 491, 96%.
1H NMR (300MHz, DMSOd
6): δ 8.4(m, IH), 8.3(m, IH), 8.03(m, IH), 7.78(m, 2H), 7.58(m, 2H), 7.4(m, IH), 7.3(m, IH), 7.2(m, 2H), 4.8(m, IH)
3 4.2(m, 2H), 3.9(m, IH), 3.7 (m, IH), 3.4(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 190 - Synthesis of l-Phenyl-lH-imidazole-4-carboxylic acid {2-[4-(4- fluoro-3-trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (97mg, 0.75mmol) was added to a stirred solution of [(I -phenyl- lH-imidazole-4- carbonyl)-amino]-acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (61mg, 0.25mmol) in DMF (2mL) followed by HOBt (35mg, 0.26mmol) and EDCI (50mg, 0.26mmol). After 2 minutes of stirring, 4-(4-fluoro-3-trifluoromethyl-phenoxy)-piperidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (75mg, 0.25mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water and extracted with ethyl acetate. The organic layer was dried over sodium sulfate, concentrated under reduced pressure. The residue obtained was purified by column chromatography (using silica gel 60-120 and 40%EtOAc in hexane as eluent) to afford 59.3mg (49%Yield) of 1 -phenyl- 1 H-imidazole-4-carboxylic acid {2-[4-(4-fluoro-3- trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 491, 96%. 1H NMR (300MHz, DMSOd6): δ 8.4(m, IH), 8.3(m, IH), 8.05(m, IH), 7.75(m, 2H), 7.55(m, 2H), 7.4(m, 4H), 4.8(m, IH), 4.2(m, 2H), 3.9(m, IH), 3.6 (m, IH), 3.4(b, IH), 3.3(m, IH), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 191 - Synthesis of l-Phenyl-lH-imidazole-4-carboxylic acid {2-[3-(2- chloro-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (141mg, 1.09mmol) was added to a stirred solution of [(I -phenyl- lH-imidazole-4- carbonyl)-amino] -acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (67mg, 0.27mmol) in DMF (3mL) followed by HOBt (39mg, 0.28mmol) and EDCI (56mg, 0.28mmol). After 2 minutes of stirring, 3-(2-chloro-phenoxy)-azetidine hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (60mg, 0.27mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Partitioning with cold water and ethyl acetate and concentration of the organic layer after drying over sodium sulfate afforded a residue which was purified by column chromatography (using silica gel 60-120 and 100% EtOAc as eluent) to afford 30mg (26.7%Yield) of 1 -phenyl- 1 H-imidazole-4-carboxylic acid {2-[3-(2-chloro-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 411, 98.45%. 1H NMR (300MHz, DMSO-d6): δ 8.34(d, IH), 8.28(d, IH), 8.12(t, IH), 7.72(d, IH), 7.44(m, 3H), 7.38(m, IH), 7.26(m, IH), 6.8(m, IH), 6.7(d, IH), 5.1(m, IH), 4.7(m, IH), 4.4(m, IH), 4.2 (m, IH), 3.8(m, 3H).
EXAMPLE 192 - Synthesis of l-Phenyl-lH-imidazole-^carboxylic acid {2-[3-(5- cyano-2-methyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (193.9mg, 1.5mmol) was added to a stirred solution of [(I -phenyl- lH-imidazole-4- carbonyl)-amino]-acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (92mg, 0.37mmol) in DMF (3mL) followed by HOBt (53.2mg, 0.39mmol) and EDCI (75.5mg, 0.39mmol). After 2 minutes of stirring, 3-(azetidin-3-yloxy)-4-methyl-benzonitrile hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (84mg, 0.37mmol) was added and the resulting mixture was stirred at ambient temperature overnight. Partitioning with cold water and ethyl acetate and concentration of the organic layer after drying over sodium sulfate afforded a residue which was purified by preparative HPLC to afford 29mg (18.7%Yield) of 1 -phenyl- 1 H-imidazole-4-carboxylic acid {2-[3-(5-cyano-2-methyl-phenoxy)-
azetidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 416, 98.6%. 1H NMR (300MHz, DMSOd6): δ 8.38(d, IH), 8.28(d, IH), 8.12(t, IH), 7.7(d, 2H), 7.5(t, 2H), 7.38(m, 3H), 7.16(s, IH), 5.2(m, IH), 4.7(t, IH), 4.4(m, IH), 4.2(m, IH), 3.8(m, 3H).
EXAMPLE 193 - Synthesis of l-Phenyl-lH-imidazole^-carboxylic acid {2-[3-(2- chloro-phenoxy)-pyrrolidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (167.9mg, 1.3mmol) was added to a stirred solution of [(I -phenyl- lH-imidazole-4- carbonyl)-amino] -acetic acid (prepared from Intermediate 68 by means of Step 3 of the General Scheme) (80mg, 0.32mmol) in DMF (2mL) followed by HOBt (48.3mg, 0.35mmol) and EDCI (124mg, 0.65mmol). After 2 minutes of stirring, 3-(2-chloro-phenoxy)-pyrrolidine hydrochloride (prepared by the method used for the synthesis of Intermediate 72) (76mg, 0.32mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate, and concentrated under reduced pressure. Purification by recrystallisation from a mixture of DCM in hexane afforded 48mg (34.7%Yield) of 1 -phenyl- lH-imidazole-4-carboxylic acid {2-[3-(2-chloro- phenoxy)-pyrrolidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 425, 93.15%. 1H NMR (300MHz, DMSOd6): δ 8.4(s, IH), 8.3(s, IH), 8.1-8.0(t, IH), 7.8-7.7(t, 2H), 7.5-7.4(m, 2H), 7.4- 7.2(m, 2H), 7.06-7.0(m, IH), 5.3-5.1(d, IH), 4.2-4.0(m, 2H), 4.0-3.5(m, 4H), 2.3-2.2(m, IH), 2.2- 2.0(m, IH).
EXAMPLE 194 - Synthesis of S-Phenyl-isoxazole-θ-carboxylic acid {2-[3-(2,5- difluoro-phenoxy)-pyrrolidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (153.5mg, 1.2mmol) followed by HOBt (44.1mg, 0.32mmol) and EDCI (113.8mg,
0.59mmol) were added to a stirred solution of [(5-phenyl-isoxazole-3-carbonyl)-amino]-acetic acid (73.4mg, 0.29mmol) in DMF (2mL). After 2 minutes of stirring, 3-(2,5-difluoro-phenoxy)- pyrrolidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (70mg, 0.29mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The organic layer was dried over sodium sulfate, concentrated under reduced pressure. Purification by column chromatography (using silica gel 60-120 and 50% EtOAc in hexane) afforded 45mg (35.4%Yield) of 5-phenyl-isoxazole-3-carboxylic acid {2-[3-(2,5-difluoro-phenoxy)-pyrrolidin-l-yl]-2-oxo- ethyl}-amide. LC/MS [M+H]
+: 428, 91.77%.
1H NMR (300MHz, DMSOd
6): δ 8.8-8.68(m, IH), 8.0-7.9(m, 2H), 7.6-7.5(m, 3H), 7.4(s, IH), 7.34-7.2(m, 2H), 6.9-6.75(m, IH), 5.3-5.0(m, 2H), 4.2-4.0(m, 2H), 3.9(m, IH), 3.8-3.7(m, IH), 3.7-3.5(m, 2H), 2.3-2.2(m, IH), 2.1(m, IH).
Intermediate 74 - Synthesis of 2-Phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid
Thionyl chloride (350mg, 3.0mmol) was added to cold (O
0C) solution of 5-bromo- thiophene-2-carboxylic acid (lOOmg, 0.4mmol) in MeOH (2mL) and stirred at ambient temperature overnight. The reaction mixture was concentrated under reduced pressure The residue was diluted with ethylacetate. The organic layer was washed with water followed by saturated brine solution, dried over sodium sulfate and concentrated under reduced pressure to afford 200mg of 5-bromo-thiophene-2-carboxylic acid methyl ester. Aqueous 2M Na
2CO
3 solution (7.35mL) was added to a stirred solution of 5-bromo-thiophene-2-carboxylic acid methyl ester (1.Og, 4.5mmol) and phenylboronic acid (660mg, 5.4mmol) in DME (15mL) purged with N
2 for 10 minutes. Pd (PPh
3^ (250mg, 0.22mmol) was added and the reaction mixture was heated to reflux for 2 hrs. The reaction mixture was diluted with water and the product was extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over sodium sulfate and concentrated. Purification by column chromatography (using silica gel 60-120 and 2% EtOAc in hexane as eluent) afforded 900mg (92%Yield) of 5-phenyl-thiophene-2-carboxylic acid methyl ester. A solution of 5-phenyl-thiophene-2-carboxylic acid methyl ester (800mg, 3.9mmol) in THF (3mL) was added dropwise to reaction flask containing LAH (299mg, 7.9mmol) at O
0C and stirring was continued at ambient temperature for 3 hrs. The reaction mixture was quenched with
aqueous NaOH solution and filtered through celite. The filtrated was collected was extracted with ethyl acetate, dried over sodium sulfate and concentrated to afford 710mg (91.l%Yield) of (5- phenyl-thiophen-2-yl)-methanol IBX (3.Og, 1 l.Ommol) was added to a stirred solution of (5- phenyl-thiophen-2-yl)-methanol (700mg, 3.6mmol) in THF (1OmL) and stirring was continued at ambient temperature overnight. The reaction mixture was filtered through celite, filtrated was collected, diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over sodium sulfate and concentrated. Purification by column chromatography (using silica gel 60-120 and 5% EtOAc in hexane as eluent) afforded 600mg (89.5%Yield) of 5-phenyl-thiophene-2-carbaldehyde. 5-phenyl-thiophene-2-carbaldehyde (500mg, 2.6mmol) and azido-acetic acid ethyl ester (1.3g, 10.4mmol) were added to a stirred solution of sodium metal (240mg, 10.4mmol) dissolved in EtOH (1OmL) and cooled to O
0C while stirring under N
2 atmosphere. Stirring was continued at 10
0C for 1.5hrs. The reaction mixture was quenched with saturated NH
4Cl solution and extracted with ethyl acetate. The organic layer was washed with saturated brine solution, dried over sodium sulfate and concentrated to afford 500mg (crude) of 2-azido-3-(5-phenyl-thiophen-2-yl)-acrylic acid ethyl ester. A mixture of 2-azido-3-(5- phenyl-thiophen-2-yl)-acrylic acid ethyl ester (350mg, crude) in xylene (5mL) was stirred at reflux temperature for 30minutes. The reaction mixture was concentrated. Purification by column chromatography (using silica gel 60-120 and 15% EtOAc in hexane as eluent) afforded 75mg of 2- phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid ethyl ester. LiOKH
2O (17mg, 0.4mmol) was added to a stirred solution of 2-phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid ethyl ester
(55mg, 0.2mmol) in a mixture of methanol (0.5mL), THF (1 ImL) and H2O (ImL). The reaction mixture was stirred at ambient temperature for 2hrs. The reaction mixture was concentrated. The residue was diluted with water, acidified with cone. HCl, and extracted with ethyl acetate. The organic layer collected was dried over sodium sulfate and concentrated under reduced pressure to afford 42mg (87.5%) of 2-phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid.
EXAMPLE 195 - Synthesis of 2-Phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid {2- [4-(2-chloro-5-fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (62mg, 0.48mmol) followed by HOBt (23 mg, O.lβmmol) and EDCI (32mg, O.lόmmol) were added to a stirred solution of 2-phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid (40mg, O.lόmmol) in DMF (2mL). After 2 minutes of stirring, 2-amino-l-[4-(2-chloro-phenoxy)- piperidin-l-yl]-ethanone hydrochloride (prepared according to Step 1 and 5 of the General Scheme) (51mg, 0.16 mmol) was added and the resulting mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate, and concentrated under reduced pressure. Purification by column chromatography (using silica gel60-120 and 40% EtOAc in hexane as eluent) afforded 18.5mg (23%Yield) of 2-phenyl-4H-thieno[3,2-b]pyrrole-5-carboxylic acid {2-[4-(2-chloro-5- fluoro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 512, 97.8%. 1H NMR (300MHz, DMSO-d6): δ 11.9(s, IH), 8.4(m, IH), 7.7(m, 2H), 7.4(m, 4H), 7.3(m, 2H), 7.17(m, IH), 6.8(m, IH), 4.8(m, IH), 4.2(m, 2H), 3.6(m, 2H), 3.5(m, 2H), 2.0(m, 2H), 1.7(m, 2H).
Intermediate 75 - Synthesis of 6-Pyrazol-l-yl-imidazo [l,2-a]pyridine-2-carboxylic acid
A mixture of sulfuric acid (0.5mL), acetic acid (12mL) and water (2mL) was added to a reaction flask containing 2-aminopyridine (2.Og, 21.26mmol) and the solution was stirred for 5 minutes. NaIO4 (1.8Ig, 8.5mmol) followed by I2 (2.16g, 8.5mmol) was added into the reaction flask and stirred at 800C for 4hrs. The reaction mixture was cooled to ambient temperature, diluted with cold water, basified with 20% aqueous KOH solution and extracted with DCM. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to obtain the crude residue. Purification by column chromatography (using silica gel 60- 120 and 8%EtOAc in hexane as eluent) and subsequent recrystallisation from ethanol afforded 53g (58.6%Yield) of 5-iodo-pyridin-2-ylamine. Ethyl bromopyruvate (4.89g, 25.11mmol) was added
to solution of 5-iodo-pyridin-2-ylaniine (5.Og, 22.83mmol) in DMF (25mL) and stirring was continued at 800C for 2hrs. The reaction mixture was diluted with cold water and extracted with ethyl acetate. The organic layer was washed with brine solution, dried over sodium sulfate and concentrated under reduced pressure to obtain the crude residue. The residue obtained was purified by recrystallisation from methanol to afford 4.2g (57.8%Yield) of 6-iodo-imidazo [l,2-a]ρyridine- 2-carboxylic acid ethyl ester. A mixture of imidazole (513mg, 7.5mmol), cuprous oxide (71mg, 0.5mmol), 6-iodo-imidazo [l,2-a]pyridine-2-carboxylic acid ethyl ester (1.6g, 5.02mmol), salox (137mg, l.Ommol) and cesium carbonate (3.27g, 10.05mmol) in ACN (2mL) in a seal tube were subjected to reaction in a microwave reactor (time:15min, temp: 850C, power: zero, pressure: zero). The reaction mixture was filtered through celite and the filtrate collected was concentrated under reduced pressure. The residue was diluted with cold water, extracted with ethyl acetate, dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (using neutral alumina and 0.2% MeOH in DCM as eluent) to afford 400mg (31%Yield) of 6-pyrazol-l-yl-imidazo [l,2-a]pyridine-2-carboxylic acid ethyl ester. A mixture of 6-pyrazol-l-yl-imidazo[l,2-a]pyridine-2-carboxylic acid ethyl ester (250mg,
0.97mmol) in 8N aqueous HCl (3mL) was stirred at 1000C for 2hrs. The reaction mixture was concentrated under reduced pressure to afford 130mg (58.5%Yield) of 6-pyrazol-l-yl-imidazo [l,2-a]pyridine-2-carboxylic acid.
EXAMPLE 196 - Synthesis and purification of 6-Pyrazol-l-yl-imidazo[l,2-a]pyridine-
2-carboxylic acid {2- [4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyI}-amide
DIPEA (169mg, 1.3mmol) followed by HOBt (39mg, 0.29mmol) and EDCI (75mg, 0.39mmol) was added to a stirred solution of 6-pyrazol-l-yl-imidazo[l,2-a]pyridine-2-carboxylic acid (60mg, 0.26mmol) in DMF (ImL). After 2 minutes of stirring, 2-amino-l-[4-(2-chloro- ρhenoxy)-piperidin-l-yl]-ethanone hydrochloride (80mg, 0.26mmol) (prepared according to Step 1 and 5 of the General Scheme) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and the reaction mixture was partitioned with ethyl
acetate, the organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by preparative HPLC afforded 45mg (33% Yield) of 6-pyrazol-l-yl-imidazo[l,2- a]pyridine-2-carboxylic acid {2-[4-(2-chloro-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 479, 97.25%. 1H NMR (300MHz, DMSOd6): δ 9.2(s, IH), 8.5(m, 2H), 8.4(t, IH), 8.0(dd, IH), 7.8(m, 2H), 7.4(dd, IH), 7.53(m, 2H), 7.0(m, IH), 6.6(t, IH), 4.8(m, IH), 4.0(bs, IH), 4.2(d, 2H), 3.7(m, 2H), 3.4(m, 2H), 2.0 (m, 2H), 1.7(m, 2H).
EXAMPLE 197 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2- [4-(3-cyano-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (156.8mg, 1.2mmol) followed by HOBt (45mg, 0.33mmol) and EDCI (116mg,
O.όmmol) were added to a stirred solution of [(l-pyridin-3-yl-lH-[l,2,3]triazole-4-carbonyl)- aminoj-acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (75mg, 0.3mmol) in DMF (3mL). After 2 minutes of stirring, 3-(piperidin-4-yloxy)-benzonitrile hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (72.4mg, 0.3mmol) was added and the resulting mixture was stirred at room temperature overnight. Cold water was added and formed precipitate was collected. Purification by preparative HPLC afforded 15mg (11.5% Yield) of l-pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(3-cyano- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 432.19, 99.3%. 1H NMR
(300MHz, DMSOd6): δ 9.5(s, IH), 9.22(d, IH), 8.8(d, IH), 8.58(t, IH), 8.44(d, IH), 7.7(q, IH), 7.58-7.44(d, 2H), 7.44(dd, 2H), 4.8(m, IH), 4.3(d, 2H), 4.0(bs, IH), 3.8(d, IH), 2.5(b, 2H), 2.1(t, 2H), 1.7 (bd, 2H).
EXAMPLE 198 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxyIic acid {2-
[3-(3-fluoro-5-trifluoromethyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (143mg, 1.lmmol) followed by HOBt (39mg, 0.29mmol) and EDCI (56mg, 0.29mmol) was added to a stirred solution of [(l-pyridin-3-yl-lH-[l,2,3]triazole-4-carbonyl)- amino] -acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (68.3mg, 0.27mmol) in DMF (2mL). After 2 minutes of stirring, 3-(3-fluoro-5-trifluoromethyl-phenoxy)- azetidine hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (75mg, 0.27mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate. The organic layer was, dried over sodium sulfate and concentrated under reduced pressure. Purification by preparative HPLC afforded 21mg (16.4% Yield) of l-pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3- (3-fluoro-5-trifluoromethyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 465, 93.03%. 1H NMR (300MHz, DMSOd6): δ 9.5(s, IH), 9.2(d, IH), 8.7(m, 2H), 8.4(m, IH), 8.3(s, IH), 7.65(m, IH), 7.3(d, IH), 7.1(m, 2H), 5.2(m, IH), 4.7(m, IH), 4.4(m, IH), 4.2(m, IH), 3.8(m, 3H).
EXAMPLE 199 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazoIe-4-carboxylic acid {2- [4-(3-fluoro-5-trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (97mg, 0.75mmol) followed by HOBt (35mg, 0.26mmol) and EDCI (50mg,
0.26mmol) were added to a stirred solution of [(l-pyridin-3-yl- IH-[1, 2,3]triazole-4-carbonyl)-
amino] -acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (62mg, 0.25mmol) in DMF (3mL). After 2 minutes of stirring, 4-(3-fluoro-5-trifluoromethyl-phenoxy)- piperidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (75mg, 0.25mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography (using silicagel 60-120 and 40% EtOAc in hexane as eluent) afforded 38.5mg (32% Yield) of l-pyridin-3-yl- IH-[1, 2,3]triazole-4-carboxylic acid {2-[4-(3-fluoro-5- trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 493, 98%. 1H NMR (300MHz, DMSOd6): δ 9.43(s, IH), 9.2(d, IH), 8.75(d, IH), 8.5(t, IH), 7.7(m, IH), 7.26(m, 3H), 4.85(m, IH), 4.3(m, 2H), 3.9(m, IH), 3.7(m, IH), 3.4(m, IH), 3.3(m, IH), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 200 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-
[3-(2-chloro-phenoxy)-pyrrolidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (167mg, 1.3mmol) followed by HOBt (48mg, 0.35mmol) and EDCI (124mg, 0.64mmol) were added to a stirred solution of [( l-pyridin-3-yl- IH- [1,2,3 ]triazole-4-carbonyl)- amino] -acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (80mg, 0.32mmol) in DMF (2mL). After 2 minutes of stirring, 3-(2-chloro-phenoxy)-pyrrolidine hydrochloride (prepared by the method used for the synthesis of Intermediate 72) (75.7mg, 0.32mniol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by recrystallisation from a mixture of DCM and hexane afforded 60mg (43.4% Yield) of 1-pyridin- 3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(2-chloro-phenoxy)-pyrrolidin-l-yl]-2-oxo-ethyl}-
amide. LC/MS [M+H]+: 427. 1H NMR (300MHz, DMSO-d6): δ 9.45(s, IH), 9.2(d, IH), 8.75(d, IH), 8.6(q, IH), 8.4(m, IH), 7.7(q, IH), 7.5-7.4(m, IH), 7.4-7.2(m, 2H), 7.06-6.96(m, IH), 5.3- 5.1(d, IH), 4.25-4.0(m, 2H), 4.0-3.6(m, 4H), 3.5-3.4(m, IH), 2.3-2.2(m, IH).
EXAMPLE 201 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-
[4-(4-fluoro-3-trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (97mg, 0.75mmol) was added to a stirred solution of [(l-ρyridin-3-yl-lH- [l,2,3]triazole-4-carbonyl)-amino]-acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (62mg, 0.25mmol) in DMF (2mL) followed by HOBt (35mg, 0.26mmol) and EDCI (50mg, 0.26mmol). After 2 minutes of stirring, 4-(4-fluoro-3-trifluoromethyl-phenoxy)- piperidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (75mg, 0.25mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography (using silicagel 60-120 and 40% EtOAc in hexane as eluent) afforded 24mg (20% Yield) of l-pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[4-(4-fluoro-3-trifluoromethyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 493, 95.48%. 1H NMR (300MHz, DMSO-d6): δ 9.43(s, IH), 9.2(m, IH), 8.75(m, IH), 8.5(m, IH), 8.4(m, IH), 7.7(m, IH), 7.4(m, 3H), 4.75(m, IH), 4.3(m, 2H), 3.9(m, IH), 3.7(m, IH), 3.5(m, 2H), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 202 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2- [3-(2-chloro-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (141mg, l.lmmol) followed by HOBt (39mg, 0.29mmol) and EDCI (56mg, 0.29mmol) were added to a stirred solution of [(l-pyridin-3-yl-lH-[l,2,3]triazole-4-carbonyl)- amino]-acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (67.5mg, 0.27mmol) in DMF (3mL). After 2 minutes of stirring, 3-(2-chloro-phenoxy)-azetidine hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (60mg, 0.27mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by preparative HPLC afforded 33mg (29.4% Yield) of l-pyridm-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3- (2-chloro-phenoxy)-azetidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]+: 413, 98.7%. 1H NMR (300MHz, DMSOd6): δ 9.42(s, IH), 9.2(d, IH), 8.7(m, 2H),-8.4(m, IH), 7.64(m, IH), 7.42(m, IH), 7.24(m, IH), 7.0(t, IH), 6.9(d, IH), 5.12(m, IH), 4.68(m, IH), 4.38(m, IH), 4.24(m, IH), 3.96(m, IH), 3.84(m, IH).
EXAMPLE 203 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxyIic acid {2- [3-(5-cyano-2-methyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (193.9mg, 1.5mmol) followed by HOBt (53mg, 0.29mmol) and EDCI (75mg,
0.39mmol) were added to a stirred solution of [(l-pyridin-3-yl- IH-[1, 2,3 ]triazole-4-carbonyl)- amino] -acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting
from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (93g, 0.37mmol) in DMF (3mL). After 2 minutes of stirring, 3-(azetidin-3-yloxy)-4-methyl-benzonitrile hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (84mg, 0.37mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by preparative HPLC afforded llmg (7%Yield) of l-pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2-[3-(5- cyano-2-methyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]+: 418, 96.08%. 1H NMR (300MHz, DMSOd6): δ 9.42(s, IH), 9.2(d, IH), 8.7(m, 2H), 8.38(m, IH), 7.62(m, IH), 7.4(s, IH), 7.16(s, IH), 5.1(m, IH), 4.7(m, IH), 4.4(m, IH), 4.2(m, IH), 4.0(d, 2H), 3.8(m, IH).
EXAMPLE 204 - Synthesis of l-Pyridin-3-yl-lH-[l,2,3]triazole-4-carboxylic acid {2- oxo-2-[4-(2-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}-amide
DIPEA (156.8mg, 1.5mmol) followed by HOBt (53mg, 0.29mmol) and EDCI (75mg,
0.39mmol) were added to a stirred solution of [(l-pyridin-3-yl-lH-[l,2,3]triazole-4-carbonyl)- amino] -acetic acid (prepared by the method used for the synthesis of Intermediate 64, starting from 3-aminopyridine, and subsequently, application of Step 3 of the General Scheme) (75g, 0.3mmol) in DMF (4mL). After 2 minutes of stirring, 4-(2-trifluoromethyl-phenoxy)-piperidine trifluoroacetate (85mg, 0.3mmol) was added and the resulting mixture was stirred at room temperature overnight. Addition of cold water lead to the formation of a precipitate which was collected and purified by preparative HPLC to afford 24mg (15.6%Yield) of l-pyridin-3-yl-lH- [l,2,3]triazole-4-carboxylic acid {2-oxo-2-[4-(2-trifluoromethyl-phenoxy)-piperidin-l-yl]-ethyl}- amide. LC/MS [M+H]+: 475, 99.07%. 1H NMR (300MHz, DMSOd6): δ 9.45(s, IH), 9.2(d, IH), 8.75(m, IH), 8.5(t, IH), 8.4(m, IH), 7.7-7.6(m, 3H), 7.4-7.35(d, IH), 7.1(t, IH), 5.0(q, IH), 4.25(d, 2H), 3.7-3.5(m, 4H), 2.0-1.8(m, 2H), 1.8-1.5(m, 2H).
EXAMPLE 205 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(3- fluoro-5-trifluoromethyI-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (143mg, 1. lmmol) followed by HOBt (39mg, 0.29mmol) and EDCI (56mg,
0.29mrαol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (68mg, 0.27mmol) in DMF (2mL) After 2 minutes of stirring, 3-(3-fiuoro-5-trifluoromethyl- phenoxy)-azetidine hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (75mg, 0.27mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by preparative HPLC afforded 31mg (24.4% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2- [3-(3-fluoro-5-trifluoromethyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 463, 81.67%. 1H NMR (300MHz, DMSOd6): δ 13.75(s, IH), 7.8(d, 2H), 7.42(t, 2H), 7.26(m, 2H), 7.1(d, IH), 7.04(s, IH), 5.2(m, IH), 4.7(m, IH), 4.4(m, IH), 4.2(m, 2H), 3.7(m, 2H).
EXAMPLE 206 - Synthesis of S-Phenyl-lH-pyrazoIe-S-carboxyiic acid {2-[4-(3- fluoro-5-trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (97mg, 0.75mmol) followed by HOBt (35mg, 0.26mmol) and EDCI (50mg,
0.26mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (62mg, 0.25mmol) in DMF (2mL). After 2 minutes of stirring, 3-(3-fluoro-5-trifluoromethyl-
phenoxy)-azetidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (75mg, 0.25mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography (using silicagel 60-120 and 40% EtOAc in hexane as eluent) afforded 54mg (45% Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(3-fluoro-5-trifluoromethyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 491, 94%. 1H NMR (300MHz, DMSOd6): δ 13.8(s, IH), 8.5(b, IH), 7.8(d, 2H), 7.4(m, 2H), 7.3(m, 2H), 7.2(m, 2H), 7.1(m, IH), 4.8(m, IH), 4.2(m, 2H), 3.8(m, IH), 3.6(m, IH), 3.4(m, IH), 3.3(m, IH), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 207 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[4-(4- fluoro-3-trifluoromethyl-phenoxy)-piperidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (97mg, 0.75mmol) followed by HOBt (35mg, 0.26mmol) and EDCI (50mg,
0.26mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (62mg, 0.25mmol) in DMF (2mL). After 2 minutes of stirring, 3-(3-fluoro-5-trifluoromethyl- phenoxy)-azetidine hydrochloride (prepared by the method used for the synthesis of Intermediate 15) (75mg, 0.25mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography (using silicagel 60-120 and 40% EtOAc in hexane as eluent) afforded 55mg (45% Yield) of 5 -phenyl- lH-pyrazole-3-carboxylic acid {2-[4-(4-fluoro-3-trifluoromethyl- phenoxy)-piperidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]+: 491, 99%. 1H NMR (300MHz, DMSO-d6): δ 13.6(m, IH), 8.0(m, IH), 7.8(m, 2H), 7.4(m, 6H), 7.3(m, IH), 4.7(m, IH), 4.2(m, 2H), 3.8(m, IH), 3.7(m, IH), 3.4(m, IH), 3.0(m, IH), 2.0(m, 2H), 1.6(m, 2H).
EXAMPLE 208 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(2- chloro-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (143mg, 1.lmmol) followed by HOBt (39mg, 0.29mmol) and EDCI (56mg, 0.29mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino] -acetic acid (67g, 0.27mmol) in DMF (2mL). After 2 minutes of stirring, 3-(2-chloro-phenoxy)- azetidinehydrochloride (prepared by the method used for the synthesis of Intermediate 71) (60mg, 0.27mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by preparative HPLC afforded 48mg (42.8%Yield) of 5-phenyl-lH-pyrazole-3-carboxylic acid {2-[3-(2-chloro- phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide. LC/MS [M+H]+: 411, 99.1%. 1H NMR (300MHz, DMSOd6): δ 13.7(s, IH)3 8.2(t, IH), 7.54(m, 2H), 7.28(m, 5H), 7.08(d, IH), 7.0(t, IH), 6.86(d, IH), 5.1(m, IH), 4.64(m, IH), 4.36(m, IH), 4.2(m, IH), 3.84(m, 3H).
EXAMPLE 209 - Synthesis of S-Phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(5-cyano- 2-methyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl}-amide
DIPEA (193.9mg, 1.5mmol) followed by HOBt (53mg, 0.29mmol) and EDCI (75mg, 0.39mmol) was added to a stirred solution of [(5-phenyl-lH-pyrazole-3-carbonyl)-amino]-acetic acid (92g, 0.37mmol) in DMF (3mL) After 2 minutes of stirring, 3-(azetidin-3-yloxy)-4-methyl- benzonitrile hydrochloride (prepared by the method used for the synthesis of Intermediate 71) (84mg, 0.37mmol) was added and the resulting mixture was stirred at room temperature overnight.
The reaction mixture was partitioned between cold water and ethyl acetate .The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification by preparative HPLC afforded 35mg (22.5%Yield) of S-phenyl-lH-pyrazole-S-carboxylic acid {2-[3-(5-cyano-2- methyl-phenoxy)-azetidin-l-yl]-2-oxo-ethyl} -amide. LC/MS [M+H]
+:416, 92.49%.
1H NMR (300MHz, DMSOd
6): δ 13.8(s, IH), 8.2(t, IH), 7.74(t, 2H), 7.42(m, 2H), 7.3(m, 3H), 7.16(s, IH), 7.08(s, IH), 5.2(m, IH), 4.7(m, IH), 4.4(m, IH), 4.2(m, IH), 3.8(m, 3H).
The entire disclosures of all applications, patents and publications, cited above and below, are hereby incorporated by reference. While the invention has been depicted and described by reference to exemplary embodiments of the invention, such a reference does not imply a limitation on the invention, and no such limitation is to be inferred. The invention is capable of considerable modification, alteration, and equivalents in form and function, as will occur to those ordinarily skilled in the pertinent arts having the benefit of this disclosure. The depicted and described embodiments of the invention are exemplary only, and are not exhaustive of the scope of the invention.
Consequently, the invention is intended to be limited only by the spirit and scope of the appended claims, giving full cognizance to equivalence in all respects.