WO2009114763A2 - Enzymatic substrates for multiple detection systems - Google Patents
Enzymatic substrates for multiple detection systems Download PDFInfo
- Publication number
- WO2009114763A2 WO2009114763A2 PCT/US2009/037087 US2009037087W WO2009114763A2 WO 2009114763 A2 WO2009114763 A2 WO 2009114763A2 US 2009037087 W US2009037087 W US 2009037087W WO 2009114763 A2 WO2009114763 A2 WO 2009114763A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- substrate
- heteroatom
- aryl
- ether
- Prior art date
Links
- 239000000758 substrate Substances 0.000 title claims abstract description 184
- 230000002255 enzymatic effect Effects 0.000 title claims abstract description 30
- 238000001514 detection method Methods 0.000 title abstract description 43
- 102000004190 Enzymes Human genes 0.000 claims abstract description 88
- 108090000790 Enzymes Proteins 0.000 claims abstract description 88
- 238000000034 method Methods 0.000 claims abstract description 44
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims abstract description 14
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 13
- 230000009471 action Effects 0.000 claims abstract description 4
- 229910052760 oxygen Inorganic materials 0.000 claims description 84
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 82
- 125000005842 heteroatom Chemical group 0.000 claims description 60
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 claims description 50
- 125000003118 aryl group Chemical group 0.000 claims description 47
- 229910052717 sulfur Inorganic materials 0.000 claims description 42
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 36
- 125000001424 substituent group Chemical group 0.000 claims description 34
- 125000000217 alkyl group Chemical group 0.000 claims description 33
- 125000000623 heterocyclic group Chemical group 0.000 claims description 32
- KDXKERNSBIXSRK-UHFFFAOYSA-N lysine Chemical group NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 27
- 229910052757 nitrogen Inorganic materials 0.000 claims description 19
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 18
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 18
- 239000001301 oxygen Substances 0.000 claims description 18
- 239000007787 solid Substances 0.000 claims description 18
- 239000011593 sulfur Substances 0.000 claims description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 14
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 13
- 239000004472 Lysine Substances 0.000 claims description 13
- 238000004458 analytical method Methods 0.000 claims description 10
- 125000006736 (C6-C20) aryl group Chemical group 0.000 claims description 8
- 230000000269 nucleophilic effect Effects 0.000 claims description 8
- 150000001413 amino acids Chemical group 0.000 claims description 7
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 7
- 150000002772 monosaccharides Chemical class 0.000 claims description 7
- 125000000747 amidyl group Chemical group [H][N-]* 0.000 claims description 6
- 150000002016 disaccharides Chemical class 0.000 claims description 5
- GZCGUPFRVQAUEE-UHFFFAOYSA-N 2,3,4,5,6-pentahydroxyhexanal Chemical compound OCC(O)C(O)C(O)C(O)C=O GZCGUPFRVQAUEE-UHFFFAOYSA-N 0.000 claims description 4
- 125000003342 alkenyl group Chemical group 0.000 claims description 3
- 125000006755 (C2-C20) alkyl group Chemical group 0.000 claims description 2
- AEMOLEFTQBMNLQ-UHFFFAOYSA-N 3,4,5,6-tetrahydroxyoxane-2-carboxylic acid Chemical compound OC1OC(C(O)=O)C(O)C(O)C1O AEMOLEFTQBMNLQ-UHFFFAOYSA-N 0.000 claims description 2
- WQZGKKKJIJFFOK-SVZMEOIVSA-N (+)-Galactose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-SVZMEOIVSA-N 0.000 claims 2
- 238000006243 chemical reaction Methods 0.000 abstract description 33
- 238000004949 mass spectrometry Methods 0.000 abstract description 20
- 235000000346 sugar Nutrition 0.000 abstract description 18
- 125000003277 amino group Chemical group 0.000 abstract description 11
- 125000001453 quaternary ammonium group Chemical group 0.000 abstract description 10
- 150000001875 compounds Chemical class 0.000 abstract description 9
- 125000005647 linker group Chemical group 0.000 abstract description 7
- 150000001735 carboxylic acids Chemical class 0.000 abstract description 5
- 230000035945 sensitivity Effects 0.000 abstract description 5
- 230000021615 conjugation Effects 0.000 abstract description 2
- 229940088598 enzyme Drugs 0.000 description 83
- 239000000047 product Substances 0.000 description 83
- 239000000523 sample Substances 0.000 description 54
- 238000003556 assay Methods 0.000 description 41
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- 239000002245 particle Substances 0.000 description 30
- 230000000694 effects Effects 0.000 description 28
- 210000004369 blood Anatomy 0.000 description 27
- 239000008280 blood Substances 0.000 description 27
- 239000000243 solution Substances 0.000 description 27
- 230000002132 lysosomal effect Effects 0.000 description 23
- 230000027455 binding Effects 0.000 description 19
- 238000009739 binding Methods 0.000 description 19
- 238000006911 enzymatic reaction Methods 0.000 description 18
- 208000015439 Lysosomal storage disease Diseases 0.000 description 16
- 238000003018 immunoassay Methods 0.000 description 15
- 239000003153 chemical reaction reagent Substances 0.000 description 14
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 102100033448 Lysosomal alpha-glucosidase Human genes 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- 238000004885 tandem mass spectrometry Methods 0.000 description 12
- 239000000872 buffer Substances 0.000 description 11
- 0 *C(C(*)C1O)C(CO)OC1[U]c(cc1)ccc1N Chemical compound *C(C(*)C1O)C(CO)OC1[U]c(cc1)ccc1N 0.000 description 10
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- 102000005840 alpha-Galactosidase Human genes 0.000 description 10
- 108010030291 alpha-Galactosidase Proteins 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 10
- 238000005406 washing Methods 0.000 description 10
- 208000015872 Gaucher disease Diseases 0.000 description 8
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 8
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 8
- 239000002953 phosphate buffered saline Substances 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 208000032007 Glycogen storage disease due to acid maltase deficiency Diseases 0.000 description 7
- 206010053185 Glycogen storage disease type II Diseases 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 239000008103 glucose Substances 0.000 description 7
- 201000004502 glycogen storage disease II Diseases 0.000 description 7
- 238000011534 incubation Methods 0.000 description 7
- 230000037361 pathway Effects 0.000 description 7
- -1 phenyl ester Chemical class 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 102000004547 Glucosylceramidase Human genes 0.000 description 6
- 108010017544 Glucosylceramidase Proteins 0.000 description 6
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 6
- 238000002820 assay format Methods 0.000 description 6
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 6
- 238000002372 labelling Methods 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 208000024720 Fabry Disease Diseases 0.000 description 5
- 208000010055 Globoid Cell Leukodystrophy Diseases 0.000 description 5
- 208000028226 Krabbe disease Diseases 0.000 description 5
- 102000003992 Peroxidases Human genes 0.000 description 5
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 5
- WQZGKKKJIJFFOK-DVKNGEFBSA-N alpha-D-glucose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-DVKNGEFBSA-N 0.000 description 5
- 108010028144 alpha-Glucosidases Proteins 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000002981 blocking agent Substances 0.000 description 5
- 239000003599 detergent Substances 0.000 description 5
- 238000007824 enzymatic assay Methods 0.000 description 5
- 239000003112 inhibitor Substances 0.000 description 5
- 239000004471 Glycine Substances 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- MEFKEPWMEQBLKI-AIRLBKTGSA-O S-adenosyl-L-methionine Chemical compound O[C@@H]1[C@H](O)[C@@H](C[S+](CC[C@H]([NH3+])C([O-])=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MEFKEPWMEQBLKI-AIRLBKTGSA-O 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 125000001931 aliphatic group Chemical class 0.000 description 4
- 239000011324 bead Substances 0.000 description 4
- 208000035475 disorder Diseases 0.000 description 4
- 238000001952 enzyme assay Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 125000006501 nitrophenyl group Chemical class 0.000 description 4
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 4
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- WQZGKKKJIJFFOK-FPRJBGLDSA-N beta-D-galactose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-FPRJBGLDSA-N 0.000 description 3
- 239000013060 biological fluid Substances 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical group OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 3
- 230000007812 deficiency Effects 0.000 description 3
- 229910052805 deuterium Inorganic materials 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000000835 fiber Substances 0.000 description 3
- SXXHYAMKYMNIHI-UHFFFAOYSA-N fluoroimino(sulfanylidene)methane Chemical compound FN=C=S SXXHYAMKYMNIHI-UHFFFAOYSA-N 0.000 description 3
- 229930182830 galactose Natural products 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 239000003550 marker Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 208000030159 metabolic disease Diseases 0.000 description 3
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 108040007629 peroxidase activity proteins Proteins 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 238000010846 tandem mass spectrometry analysis Methods 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- PKYCWFICOKSIHZ-UHFFFAOYSA-N 1-(3,7-dihydroxyphenoxazin-10-yl)ethanone Chemical compound OC1=CC=C2N(C(=O)C)C3=CC=C(O)C=C3OC2=C1 PKYCWFICOKSIHZ-UHFFFAOYSA-N 0.000 description 2
- KHNIALSGSRWBKQ-UHFFFAOYSA-N 3-hydroxy-3-[(trimethylazaniumyl)methyl]pentadecanoate Chemical compound C(CCCCCCCCCCC)C(O)(C[N+](C)(C)C)CC([O-])=O KHNIALSGSRWBKQ-UHFFFAOYSA-N 0.000 description 2
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 2
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 2
- 241000605059 Bacteroidetes Species 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000283074 Equus asinus Species 0.000 description 2
- 241000283073 Equus caballus Species 0.000 description 2
- 239000004366 Glucose oxidase Substances 0.000 description 2
- 108010015776 Glucose oxidase Proteins 0.000 description 2
- 229920002527 Glycogen Polymers 0.000 description 2
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 2
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- LEBBDRXHHNYZIA-LDUWYPJVSA-N [(2s,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] n-[(z)-1,3-dihydroxyoctadec-4-en-2-yl]carbamate Chemical compound CCCCCCCCCCCCC\C=C/C(O)C(CO)NC(=O)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O LEBBDRXHHNYZIA-LDUWYPJVSA-N 0.000 description 2
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 239000012062 aqueous buffer Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 210000000601 blood cell Anatomy 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 2
- 125000000600 disaccharide group Chemical group 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 239000007850 fluorescent dye Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 150000002303 glucose derivatives Chemical class 0.000 description 2
- 229940116332 glucose oxidase Drugs 0.000 description 2
- 235000019420 glucose oxidase Nutrition 0.000 description 2
- 229940096919 glycogen Drugs 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 2
- 238000002542 parent ion scan Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 210000003296 saliva Anatomy 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- UQDQLMLHJRVMAS-JEDNCBNOSA-N (2s)-2,6-diaminohexanoic acid;2-nitrophenol Chemical compound NCCCC[C@H](N)C(O)=O.OC1=CC=CC=C1[N+]([O-])=O UQDQLMLHJRVMAS-JEDNCBNOSA-N 0.000 description 1
- QGKMIGUHVLGJBR-UHFFFAOYSA-M (4z)-1-(3-methylbutyl)-4-[[1-(3-methylbutyl)quinolin-1-ium-4-yl]methylidene]quinoline;iodide Chemical compound [I-].C12=CC=CC=C2N(CCC(C)C)C=CC1=CC1=CC=[N+](CCC(C)C)C2=CC=CC=C12 QGKMIGUHVLGJBR-UHFFFAOYSA-M 0.000 description 1
- PHIQHXFUZVPYII-ZCFIWIBFSA-O (R)-carnitinium Chemical compound C[N+](C)(C)C[C@H](O)CC(O)=O PHIQHXFUZVPYII-ZCFIWIBFSA-O 0.000 description 1
- IQUPABOKLQSFBK-UHFFFAOYSA-N 2-nitrophenol Chemical compound OC1=CC=CC=C1[N+]([O-])=O IQUPABOKLQSFBK-UHFFFAOYSA-N 0.000 description 1
- LXAHHHIGZXPRKQ-UHFFFAOYSA-N 5-fluoro-2-methylpyridine Chemical compound CC1=CC=C(F)C=N1 LXAHHHIGZXPRKQ-UHFFFAOYSA-N 0.000 description 1
- WRDABNWSWOHGMS-UHFFFAOYSA-N AEBSF hydrochloride Chemical compound Cl.NCCC1=CC=C(S(F)(=O)=O)C=C1 WRDABNWSWOHGMS-UHFFFAOYSA-N 0.000 description 1
- 108010087765 Antipain Proteins 0.000 description 1
- 108010039627 Aprotinin Proteins 0.000 description 1
- 241000238097 Callinectes sapidus Species 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- PHOQVHQSTUBQQK-SQOUGZDYSA-N D-glucono-1,5-lactone Chemical compound OC[C@H]1OC(=O)[C@H](O)[C@@H](O)[C@@H]1O PHOQVHQSTUBQQK-SQOUGZDYSA-N 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- POHLHDSBJAEFFB-UHFFFAOYSA-N OCC(CC1O)OCC1O Chemical compound OCC(CC1O)OCC1O POHLHDSBJAEFFB-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108700020962 Peroxidase Proteins 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 108010061312 Sphingomyelin Phosphodiesterase Proteins 0.000 description 1
- 102000010126 acid sphingomyelin phosphodiesterase activity proteins Human genes 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- SDNYTAYICBFYFH-TUFLPTIASA-N antipain Chemical compound NC(N)=NCCC[C@@H](C=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 SDNYTAYICBFYFH-TUFLPTIASA-N 0.000 description 1
- 229960004405 aprotinin Drugs 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 125000000188 beta-D-glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 230000008238 biochemical pathway Effects 0.000 description 1
- 230000009287 biochemical signal transduction Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229960004203 carnitine Drugs 0.000 description 1
- 239000003518 caustics Substances 0.000 description 1
- 239000011797 cavity material Substances 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 125000001295 dansyl group Chemical group [H]C1=C([H])C(N(C([H])([H])[H])C([H])([H])[H])=C2C([H])=C([H])C([H])=C(C2=C1[H])S(*)(=O)=O 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 239000012470 diluted sample Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000009088 enzymatic function Effects 0.000 description 1
- 238000002641 enzyme replacement therapy Methods 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- ZFKJVJIDPQDDFY-UHFFFAOYSA-N fluorescamine Chemical compound C12=CC=CC=C2C(=O)OC1(C1=O)OC=C1C1=CC=CC=C1 ZFKJVJIDPQDDFY-UHFFFAOYSA-N 0.000 description 1
- 238000001215 fluorescent labelling Methods 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 150000002305 glucosylceramides Chemical class 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 208000003532 hypothyroidism Diseases 0.000 description 1
- 230000002989 hypothyroidism Effects 0.000 description 1
- 238000007373 indentation Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 1
- 108010052968 leupeptin Proteins 0.000 description 1
- CIPMKIHUGVGQTG-VFFZMTJFSA-N leupeptin hemisulfate Chemical compound OS(O)(=O)=O.CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N.CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N CIPMKIHUGVGQTG-VFFZMTJFSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000000622 liquid--liquid extraction Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 150000002668 lysine derivatives Chemical class 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000002552 multiple reaction monitoring Methods 0.000 description 1
- 238000007837 multiplex assay Methods 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 239000000382 optic material Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 239000013610 patient sample Substances 0.000 description 1
- 108010091212 pepstatin Proteins 0.000 description 1
- FAXGPCHRFPCXOO-LXTPJMTPSA-N pepstatin A Chemical compound OC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)CC(C)C FAXGPCHRFPCXOO-LXTPJMTPSA-N 0.000 description 1
- FCJSHPDYVMKCHI-UHFFFAOYSA-N phenyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OC1=CC=CC=C1 FCJSHPDYVMKCHI-UHFFFAOYSA-N 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002541 precursor ion scan Methods 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 235000004252 protein component Nutrition 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000007430 reference method Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- HSSLDCABUXLXKM-UHFFFAOYSA-N resorufin Chemical compound C1=CC(=O)C=C2OC3=CC(O)=CC=C3N=C21 HSSLDCABUXLXKM-UHFFFAOYSA-N 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229960001866 silicon dioxide Drugs 0.000 description 1
- 239000007974 sodium acetate buffer Substances 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000011775 sodium fluoride Substances 0.000 description 1
- 235000013024 sodium fluoride Nutrition 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical class ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 229920002994 synthetic fiber Polymers 0.000 description 1
- 210000001138 tear Anatomy 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- LSGOVYNHVSXFFJ-UHFFFAOYSA-N vanadate(3-) Chemical compound [O-][V]([O-])([O-])=O LSGOVYNHVSXFFJ-UHFFFAOYSA-N 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 229910000166 zirconium phosphate Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/34—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving hydrolase
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/22—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/203—Monocyclic carbocyclic rings other than cyclohexane rings; Bicyclic carbocyclic ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K19/00—Hybrid peptides, i.e. peptides covalently bound to nucleic acids, or non-covalently bound protein-protein complexes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/90—Enzymes; Proenzymes
- G01N2333/914—Hydrolases (3)
- G01N2333/924—Hydrolases (3) acting on glycosyl compounds (3.2)
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2458/00—Labels used in chemical analysis of biological material
- G01N2458/15—Non-radioactive isotope labels, e.g. for detection by mass spectrometry
Definitions
- the present invention relates to analytical reagents for detecting enzymatic activity using detection methods such as mass spectrometry, immunoassay, and high performance liquid chromatography.
- the present invention relates to substrates for detecting lysosomal enzyme activity.
- Lysosomal storage disorders are a group of inherited disorders characterized by deficiencies in specific enzymes in the body, which results in the body' s inability to break down metabolic substances.
- Fabry disease is a lysosomal storage disorder seen in one out of every 40,000 people. It is caused by a deficiency in the enzyme alpha-galactosidase which results in the body's inability to break down specific fatty substances called globotriaosylceramides.
- Gaucher disease a lysosomal storage disorder caused by an inability to break down fatty substances or lipids called glucosylceramides (also called glucocerebrosides).
- glucocerebrosidase an enzyme needed to break down these fatty substances. These fatty substances then accumulate in cells of the liver, spleen, and bone marrow.
- a third example is Pompe disease, a lysosomal storage disorder caused by a deficiency in the enzyme acid alpha-glucosidase, which is needed to break down certain sugars called glycogen. When the enzyme acid alpha- glucosidase is missing, glycogen accumulates in various tissues and organs in the body. [0004] Lysosomal storage disorders are, for the most part, childhood disorders although some manifest in adulthood. In most of them, patients are normal at birth and have progressive neurological deterioration beginning at some later time.
- the clinical phenotype depends on the type and severity of the biochemical defect. Some of these lysosomal disorders, such as Pompe disease and Krabbe disease, manifest primarily in infancy. There have been ongoing efforts in developing methods to detect such disorders before the onset of clinical symptoms so that therapeutic interventions can be initiated. [0005] Over the past decade laboratories that test for metabolic disorders have introduced tandem mass spectrometry into their newborn screening programs. Tandem mass spectrometry continues to gain popularity in the clinic because this technology allows for assay of many metabolites in a single sample. For example, this technology has been implemented as a routine clinical practice for the detection of hereditary metabolic disorders in newborns using dry blood spot samples (Schulze A et al., Pediatrics 2003; 111:1399-406).
- a second commonly used clinical assay protocol is the enzyme linked immunosorbent assay (ELISA).
- ELISA enzyme linked immunosorbent assay
- biomarkers are presently used to detect and monitor Gaucher disease (see, for example Aerts, JM, and Hollack, CD, Bailliere's Clin. Haematol, 1997; 10:691-709; Deegan PB, et al., Blood Cells MoI Dis, 2005; 35:259-67; Beutler, E, et al., J Exp Med, 1976; 143:975-80).
- compositions and processes for detecting enzymatic reactions using detection systems such as mass spectrometry, immunoassay, and high performance liquid chromatography are provided according to embodiments of the present invention.
- the invention provides chemical compounds useful for assessing the level of lysosomal enzyme activity in a sample. Testing of lysosomal enzyme activity is useful, for example, when screening for metabolic disorders in newborns as well as when assessing an individual having a medical condition affecting enzyme activity or one undergoing a medical treatment such as enzyme replacement therapy, gene therapy, or bone marrow transplantation.
- the chemical compounds described herein include substrates for target enzymes and related molecules useful as controls or standards in enzyme assays.
- An inventive substrate has the general formula A — (B 1 — B 2 — B 3 — B 4 ) where A is a monosaccharide or a disaccharide and B 1 is a C 1 -C 20 alkyl, C 1 -C 20 having a substituted C 6 -C 2O aryl, C 6 -C 2O heterocyclic containing a heteroatom of N, O or S; B 2 is an amino acid; 2,6- diaminohexanoic acid; or
- R 1 ' is a C 1 -C 20 alkyl; C 4 -C 20 ether; C 1 -C 20 alkyl having a substituent of N, O, or S; heteroatom C 6 -C 2 O aryl, C1-C20 carbonyl, C1-C20 amidyl, C1-C20 ether, C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O, or S;
- X is a nullity, oxygen, sulfur, or nitrogen;
- R 2 ' is a nullity or C 1 -C 20 alkyl, C 4 -C 20 ether; C 1 -C 20 alkyl having a substituent of N, O, or S; C 1 - C 2 O ester; C 1 -C 2 0 alcohol; C 1 -C 2 0 alkenyl; heteroatom C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a
- R 1 is a C 1 -C 2 0 alkyl; C 4 -C 2 0 ether; C 1 -C 2 0 alkyl having a substituent of N, O, or S; heteroatom C 6 -C 2 O aryl, C1-C20 carbonyl, C1-C20 amidyl, C1-C20 ether, C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O, or S; R is independently in each occurrence a H, a C 1 -C 20 alkyl, a C 2 -C 2O alkyl having a substituent of C 1 -C 20 alkyl; X is independently in each occurrence a nullity, oxygen, sulfur, or nitrogen; R 3 is independently in each occurrence a nullity, C 1 -C 2 0 alkyl, C 1 -C 2 0 having a substituted C 6 -C 2 O aryl, C 6 -C 2
- R 1 ' is a C 1 -C 2 O alkyl; C4-C20 ether; C1-C20 alkyl having a substituent of N, O, or S; heteroatom C 6 -C 2 O aryl, C 1 -C 2 O carbonyl, C1-C20 amidyl, C1-C20 ether, C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O, or S;
- X is a nullity, oxygen, sulfur, or nitrogen;
- R 2 ' is a nullity or C 1 -C 20 alkyl, C 4 -C 20 ether; C 1 -C 20 alkyl having a substituent of N, O, or S; C 1 - C20 ester; C 1 -C 2 O alcohol; C 1 -C 2 O alkenyl; heteroatom C 6 -C 2 O aryl, C 6 -C
- R 1 is a C1-C20 alkyl; C4-C20 ether; C1-C20 alkyl having a substituent of N, O, or S; heteroatom C 6 -C 2 O aryl, C1-C20 carbonyl, C1-C20 amidyl, C1-C20 ether, C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O, or S; R is independently in each occurrence a H, a C1-C20 alkyl, a C 2 -C 2 O alkyl having a substituent of C1-C20 alkyl; X is independently in each occurrence a nullity, oxygen, sulfur, or nitrogen; R 3 is independently in each occurrence a nullity, C1-C20 alkyl, C1-C20 having a substituted C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O or S;
- Figure 1 is an exemplary substrate structure for detecting lysosomal storage disorders and illustrative method of synthesis.
- Figure 2 is an exemplary substrate structure for detecting lysosomal storage disorders of general composition and an illustrative method of synthesis.
- Figure 3 is an exemplary substrate structure highlighting structural moieties.
- Figure 4 is an exemplary substrate structure highlighting multiple functional centers amenable to multiple detection methods.
- Figure 5 is a generic enzymatic reaction scheme using an inventive substrate.
- Figure 6 is an alternative method of detecting enzymatic activities using double labeling of an inventive substrate.
- Figure 7 is an inventive method of detecting enzymatic reactions using mass spectrometry that is advantageous in comparison to a prior art reference method.
- the present invention has utility as an analytical reagent composition for detecting acid hydrolase enzyme activity, such as lysosomal enzyme activities associated with lysosomal storage disorders.
- acid hydrolase enzyme activity such as lysosomal enzyme activities associated with lysosomal storage disorders.
- enzyme substrates and related compounds useful as experimental controls or standards that are readily dissolvable in solutions adaptable for analytical methods such as mass spectrometry, HPLC and immunoassay, detecting enzyme activities associated with lysosomal storage diseases is more practical and less cumbersome.
- the present invention provides substrates specific for lysosomal enzymes illustratively including acid ⁇ -galactosidase A (GLA), acid ⁇ -glucocerebrosidase (ABG), galactocerebroside ⁇ -galactosidase (GALC) and acid ⁇ -glucosidase (GAA).
- GLA acid ⁇ -galactosidase A
- ABSG acid ⁇ -glucocerebrosidase
- GALC galactocerebroside ⁇ -galactosidase
- GAA acid ⁇ -glucosidase
- the action of these enzymes toward inventive substrates is used to measure the corresponding enzyme activities in a sample, and thus, these substrates can be used to detect lysosomal storage disorders including Fabry (GLA), Gaucher (ABG), Krabbe (GALC) and Pompe (GAA).
- An inventive substrate has the general formula of A — (B 1 — B 2 — B 3 — B 4 ) where A is a monosaccharide or a disaccharide and B 1 is a C 1 -C 20 alkyl, C 1 -C 20 having a substituted C 6 - C20 aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O or S; B 2 is an amino acid; 2,6- diaminohexanoic acid; or — R 1 ⁇ XR 2 XY) n I where R 1 ' is a C 1 -C 20 alkyl; C 4 -C 20 ether; C 1 -C 20 alkyl having a substituent of N, O, or S; heteroatom C 6 -C 2 O aryl, C 1 -C 2 O carbonyl, C 1 -C 2 O amidyl, C 1 -C 2 O ether, C 6 -C 20 aryl, C 6 -
- R 1 is a C 1 -C 20 alkyl; C 4 -C 20 ether; C 1 -C 20 alkyl having a substituent of N, O, or S; heteroatom C 6 -C 2 O aryl, C1-C20 carbonyl, C1-C20 amidyl, C1-C20 ether, C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O, or S; R is independently in each occurrence a H, a Ci-C 2O alkyl, a C 2 -C 2O alkyl having a substituent of Ci-C 2O alkyl; X is independently in each occurrence a nullity, oxygen, sulfur, or nitrogen; R is independently in each occurrence a nullity, Ci-C 2 O alkyl, Ci-C 2 O having a substituted C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O or
- sugar moiety A such as A being a monosaccharide or a disaccharide.
- sugar moieties include ⁇ -D-Glucose for detecting Pompe disease; ⁇ - D-Glucose for detecting Gaucher disease; ⁇ -D-Galactose for detecting Fabry disease; and ⁇ -D- Galactose for detecting Krabbe disease.
- B 1 is a linker moiety which functions to allow conjugation of the sugar moiety A to the remaining structure of the substrate.
- an inventive substrate of the general formula of A-B 1 - B 2 -B 3 -B 4 can be hydrophilic in a solvent such as pure methanol or pure ethanol.
- the B 1 -B 2 -B 3 -B 4 moiety in toto generally is hydrophilic.
- the inventive substrates can be soluble in aqueous buffer systems.
- B 2 provides a nucleophilic group(s) for interactions with a solid support or detectable tag, such as a fluorescent tag.
- B 2 is a derivative of an alkyl diamino acid, such as 2,6-diaminohexanoic acid.
- the 6-amino group optionally serves as a nucleophile for binding to a solid support or detectable tag.
- a quaternary ammonium group is a component of B 3 . Upon an enzymatic reaction, a cleaved product of B 1 -B 2 -B 3 -B 4 carries with it a permanent positive charge located on the B 3 .
- an inventive substrate is structurally terminated by a B 4 group.
- B 4 can be structurally tailored to provide different chain lengths. Such different chain lengths are useful for distinguishing different substrates, as well as enzymatic products thereof, from each other in enzyme assays.
- a substrate containing a 13 carbon atom chain has a different mass-to-charge ratio than a substrate containing a 14 carbon atom chain and as such, substrates containing 13 carbon atom or 14 carbon atom chains can be distinguished.
- substrates having differing chain lengths can be distinguished using antibodies selective for particular chemical moieties.
- the invention provides a reagent of formula A — B 1 — B 2 — B 3 - B 4 wherein A is a monosaccharide or a disaccharide and preferably an aldohexose or a ketohexose; B 1 is a phenol, a nitrophenol, or a phenyl ester such as a phenyl benzoate; B 2 contains an alkyl group such as the amino acid lysine; B contains a pendent quaternary ammonium cation extending from structure that prior to condensation to B 1 alone or also with a B 4 tail is carnitine.
- the present invention provides methods for detecting enzyme activity.
- the activity of a particular enzyme can be assessed by its capability or rate of acting on a cognate substrate to product enzymatic products.
- action of a target enzyme on substrate A-B 1 -B 2 -B 3 -B 4 results in generation of two products: A and B 1 -B 2 -B 3 -B 4 .
- the activity of the target enzyme can be determined.
- the invention provides compounds having the general formula of B 1 -B 2 -B 3 -B 4 .
- the activities of certain lysosomal enzymes in the blood of an individual can be used to test whether that individual has a lysosomal storage disorder. Therefore, the invention provides substrates for detecting medical conditions, in particular, lysosomal storage disorders such as Pompe disease, Gaucher disease, Fabry disease and Krabbe disease.
- an exemplary sugar moiety is ⁇ -D-glucose and an exemplary B 1 -B 2 -B 3 -B 4 portion is 4-aminophenyl-diaminohexanoyl-cartnitinyl-alkyl chain with B 4 of 10-20 carbons.
- an exemplary sugar moiety is ⁇ -D-glucose and an exemplary B 1 -B 2 -B 3 -B 4 portion is 4-aminophenyl-diaminohexanoyl-carnitinyl-alkyl with B 4 of 10-20 carbons in length.
- an exemplary sugar moiety is ⁇ -D-galactose and an exemplary B 1 -B 2 -B 3 -B 4 portion is 4-aminophenyl-diaminohexanoyl-carnitinyl-alkyl with B 4 of 10-20 carbons in length.
- an exemplary sugar moiety is ⁇ -D-galactose and an exemplary B 4 portion is 4-aminophenyl-diaminohexanoyl-carnitinyl-alkyl with B 4 of 10-20 carbons in length.
- a specific example of an inventive substrate specific for detecting Krabbe disease has a group A of ⁇ -D-Galactose, a group B 1 of a methyl, a group B 2 of an amidylaminoacyl group, a group B 3 of a amidyl terminating with a quaternary ammonium, and group B 4 of alkenyl alcohol with a carbon length of 12 to 20.
- a specific example of an inventive substrate for detecting Gaucher disease has a group A of ⁇ -D-Glucose, a group B 1 of a methyl, a group B 2 of an amidylaminoacyl group, a group B 3 of a amidyl terminating with a quaternary ammonium, and group B 4 of alkenyl alcohol with a carbon length of 12 to 20.
- An inventive substrate can be tailored for assaying a variety of enzymes, in particular, enzymes associated with a disease state or birth detect, or one otherwise useful for medical purposes. Such tailoring is possible because a variety of monosaccharide and disaccharide groups can be present at A of the general formula A-B 1 -B 2 -B 3 -B 4 .
- an inventive substrate can be readily prepared using guidance provided herein.
- enzymes which can be assayed using an inventive substrate as described herein include acid ⁇ -galactosidase A, acid ⁇ - glucocerebrosidase, acid galactocerebroside ⁇ -galactosidase, acid sphingomyelinase, and acid ⁇ -glucosidase [0034]
- This inventive system provides for optional multiplex assays where two or more lysosomal enzymes are analyzed in the same sample or sample receptacle using structurally similar yet enzyme specific substrates.
- the present invention provides compounds that function as experimental controls or standards useful for assessing the amount of enzymatic product in a sample or sample receptacle.
- an internal standard corresponding to a particular inventive substrate is structurally identical to its enzymatic product, except that the internal standard differs in mass-to-charge (m/z) ratio.
- the internal standards of the present invention include modified forms of enzymatic products, for example, stable isotope- labeled analogs of enzymatic products in which one or more atoms are replaced by corresponding atomic isotopes so as to create a shift in the mass.
- the resulting spectrum reveals a spatial separation of the internal standard and enzymatic product, each represented by its own peak.
- the known amount of internal standard is reflected by peak magnitude at its known m/z ratio.
- the amount of enzymatic product can be assessed by comparison of peak magnitude at its known m/z, relative to the peak magnitude of the internal standard.
- An example of isotopic labeling to produce an internal standard is the replacement of 1 H on an acyl group of B 4 with 2 D. As a result, a "heavier" internal standard molecule with the substituted 2 D has a different m/z from the enzymatic product, as detected on a mass spectrum.
- an internal standard is labeled with deuterium to cause a mass change of 3 to 9 Daltons from the corresponding cleaved product.
- a combined B 2 -B 3 -B 4 subgroup is a lysine-acylcarnitine with a positively charged quaternary ammonium moiety and the acyl tail is of carbon length from 12 to 18.
- the inventive substrates are labeled with a detectable tag. Many fluorescent probes are recognized in the art as useful for labeling reactive amines. A particularly sensitive target for specific labeling of biomolecules is the side chain amino group of lysine.
- a preferred embodiment of the inventive substrates includes a lysine residue at position B 2 that possess this active terminal amino group.
- detectable tags suitable for labeling the inventive substrates include fluorophores such as isothiocyanates, dansyl and other sulfonyl chlorides, 7-nitrobenz-2-oxa-l,3-diazole derivatives, fluorescamine, and the like.
- An inventive substrate can be used in a variety of physical formats, for example, in solution as well as linked or immobilized to solid supports.
- a solid support can be composed of a natural or synthetic material, an organic or inorganic material, such as a polymer, resin, metal or glass, and combinations thereof.
- a suitable solid support can have a variety of physical formats, which can include for example, a membrane; column; a hollow, solid, semi-solid, pore or cavity-containing particle such as a bead; a gel; a fiber, including a fiber optic material; a matrix and sample receptacle.
- sample receptacles include sample wells, tubes, capillaries, vials and any other vessel, groove or indentation capable of holding a sample.
- a sample receptacle can be contained on a multi-sample platform, such as a microplate, slide, microfluidics device, and the like.
- suitable particles are known in the art and illustratively include Luminex®-type encoded particles, encoded fiber optic particles, magnetic particles, and glass particles. Covalent interaction of an inventive substrate and/or enzymatic cleavage product thereof with a solid support is useful for retaining the substrate and/or product during washing procedures performed in some assay formats, thus, producing a robust and accurate signal of enzymatic activity.
- the presence of the exemplary lysine group B 2 can be used, for example, for covalent bonding to high-binding solid supports.
- High binding solid supports are surfaces having exposed moieties that are chemically active or otherwise capable of covalent or high affinity binding to an inventive substrate or internal standard.
- Corning Life Sciences produces high-binding microwell plates that are irradiated to break the benzene ring and produce exposed carboxylic acids. These carboxylic acids are amenable to nucleophilic attack such as by the terminal amino group on the lysine derivative component of a preferred embodiment substrate. This reaction is rapid and produces a tight interaction between the substrate/product and the high-binding surface.
- the methods described herein can be performed in a multiplexed format such that a plurality of samples are assayed simultaneously.
- An illustrative multiplexed format involves using physically and/or chemically coded particles.
- Use of coded particles in multiplexed formats has been described, for example, in US 6,649,414 and US 6,939,720. Because the codes allow particles to be distinguished from each other, a plurality of distinct particles can be present in a single reaction mixture, allowing a plurality of different samples or different enzymes to be assayed simultaneously. Codes on particles can correspond, for example, to sample origins, particular enzymes to be assayed, particular substrates present, and the like, depending on the experimental goal of the user.
- a sample useful in the methods of the invention contains or is suspected of containing one or more target enzymes.
- Target enzymes can be contained in samples obtained from an individual, as well as from laboratory materials, such as cell lines, and synthetic protein sources.
- Exemplary sample sources include tissue homogenates; cell culture lysates; biological fluids including urine, blood in liquid or dry form, tears, saliva, and cerebrospinal fluid.
- a sample can be further fractionated, if desired, to a fraction containing particular cell types.
- a blood sample can be fractionated into serum or into fractions containing particular types of blood cells such as red blood cells or white blood cells (leukocytes).
- a sample can be a combination of samples from a subject such as a combination of a tissue and fluid sample, and the like.
- the sample is blood, which can be, for example, whole blood or a blood fraction thereof, or reconstituted from a dry blood sample.
- Methods for obtaining samples that preserve the activity or integrity of molecules in the sample are well known to those skilled in the art. Such methods include the use of appropriate buffers and/or inhibitors, including nuclease, protease and phosphatase inhibitors, which preserve or minimize changes in the molecules in the sample.
- Such inhibitors include, for example, chelators such as ethylenediamine tetraacetic acid (EDTA), ethylene glycol bis(P- aminoethyl ether)N,N,Nl,Nl-tetraacetic acid (EGTA), protease inhibitors such as phenylmethylsulfonyl fluoride (PMSF), aprotinin, leupeptin, antipain and the like, and phosphatase inhibitors such as phosphate, sodium fluoride, vanadate and the like.
- chelators such as ethylenediamine tetraacetic acid (EDTA), ethylene glycol bis(P- aminoethyl ether)N,N,Nl,Nl-tetraacetic acid (EGTA), protease inhibitors such as phenylmethylsulfonyl fluoride (PMSF), aprotinin, leupeptin, antipain and the like, and phosphatase inhibitors such as phosphate,
- Appropriate buffers and conditions for isolating molecules are well known to those skilled in the art and can be varied depending, for example, on the type of molecule in the sample to be characterized (see, for example, Ausubel et al. Current Protocols in Molecular Biology (Supplement 47), John Wiley & Sons, New York (1999); Harlow and Lane, Antibodies: A Laboratory Manual (Cold Spring Harbor Laboratory Press (1988); Harlow and Lane, Using Antibodies: A Laboratory Manual, Cold Spring Harbor Press (1999); Tietz Textbook of Clinical Chemistry, 3rd ed. Burtis and Ashwood, eds. W.B. Saunders, Philadelphia, (1999)).
- a sample also can be processed to eliminate or minimize the presence of interfering substances.
- Samples in the form of a dry blood spot are commonly used when screening blood from newborns and children patients.
- blood is collected and retained on a filter paper.
- the dried blood is eluted from the filter paper into an aqueous solution, which generally contains a buffer such as phosphate buffer saline and a protease inhibitor.
- protease inhibitor conditions include for example, include one or more of the following: AEBSF hydrochloride in a final concentration of 50 to 400 ⁇ g/ml, EDTA disodium dehydrate in a final concentration of 0.2 to 25 mg/ml, leupeptin hemisulfate in a final concentration of 0.5 to 1 ⁇ g/ml, and pepstatin A in a final concentration of 0.5 to 1 ⁇ g/ml.
- AEBSF hydrochloride in a final concentration of 50 to 400 ⁇ g/ml
- EDTA disodium dehydrate in a final concentration of 0.2 to 25 mg/ml
- leupeptin hemisulfate in a final concentration of 0.5 to 1 ⁇ g/ml
- pepstatin A in a final concentration of 0.5 to 1 ⁇ g/ml.
- a single extraction of a dry sample avoids the need to obtain several sample punches from the same sample, or to collect aliquots of other sample sources and accordingly reduces variation caused by inhomogeneous distribution of blood on the filter paper and errors in sample transfer.
- extraction efficiency may vary with the different enzymes being analyzed.
- the target enzymes may have different levels of activities when contained in different assay solutions.
- Composition of the inventive universal assay solution is optionally chosen such that each enzyme to be tested is active.
- the substrate can be detected in an assay when it is desired to observe substrate consumption during an enzymatic assay, while the product can be detected in the assay when it is desired to observe its formation during an enzymatic assay. Both substrate and product can be detected when it is desired to observe the enzymatic reaction from both perspectives, for example, to confirm that the amount of product produced correlates with the amount of substrate consumed.
- the amount of substrate A-B 1 -B 2 -B 3 -B 4 or product B 1 -B 2 -B 3 -B 4 can be detected using established tandem mass spectrometry procedures.
- An exemplary enzyme assay employing mass spectrometry can be performed as follows.
- a sample is incubated with an inventive substrate for a time period that allows formation of an enzymatic product.
- the substrate is cleaved by a target enzyme present in a blood sample to form a respective B1-B2-B3-B4 product.
- the reaction is then quenched by adding a reagent that precipitates protein components.
- exemplary reagents include alcohol, acetonitrile and dilute trifluoro acetic acid.
- a portion of the incubation mixture is then transferred to a new assay vessel.
- a dilution reagent such as methanol, acetonitrile, water-methanol mixtures or water- acetonitrile can be added to dilute the transferred portion.
- the sample so diluted reduces the amount of endogenous competing material so as to relatively increase the sensitivity of the tandem mass spectrometry analysis.
- Other types of reagents are selected by those skilled in the art to be compatible with tandem mass spectrometry analysis.
- the diluted sample is directly injected into the tandem mass spectrometer either manually or automatically with the aid of autosamplers and liquid handlers. If desired, the sample can be derivatized prior to analysis.
- Reagents are selected to be non-hostile to the MS/MS system. For example, suitable solvents lack detergents and corrosive agents, such as chloroform. Pure ethanol and pure methanol are often used simply because they easily vaporized upon mechanical drying processes.
- the tandem mass spectrometer can be set to simultaneously detect the added substrate, the corresponding resulting enzymatic product and the corresponding internal standards. Such detection is accomplished by means of parent ion scans, precursor ion scans or multiple reaction monitoring scans.
- the amount of substrate A-B ⁇ B ⁇ B ⁇ B 4 or product B ⁇ B ⁇ B ⁇ B 4 formed during an enzymatic assay also can be detected using antibodies and other target- specific binding molecules.
- an antibody can be used to detect the substrate, product or both.
- Antibodies useful in such methods can be specific, such that they recognize individual substrates, or non-specific, such that they recognize many or all substrates.
- An illustrative example is an antibody generated to the combination B1-B2 nitrophenol-lysine.
- the antibody is illustratively produced in animals including mouse, rat, rabbit, horse, donkey, or other suitable animal used for the production of antibodies.
- an antibody with a detectable tag, such as a fluorescent tag.
- detection can be performed by using a secondary antibody that is specific for the species IgG of the primary antibody is labeled illustratively with a fluorescent marker such as rhodamine.
- a fluorescent marker such as rhodamine.
- other antibody detection systems are similarly operable in the instant invention such as horseradish peroxidase labeled antibodies, or alkaline phosphatase labeled antibodies.
- samples containing target enzymes are contacted with substrates linked to particles in an assay solution.
- each particle is linked to a particular substrate, and there are multiple particles representing each substrate.
- the target enzymes act on the substrates to produce products A and B 1 -B 2 -B 3 -B 4 .
- the B 1 -B 2 -B 3 -B 4 product remains bound to the particle, while the A product is released into solution.
- Antibodies that recognize specific B 1 -B 2 -B 3 -B 4 products are then contacted with the assay solution.
- the antibodies will bind to the products, if produced during the enzymatic assay, to produce particles having bound antibodies.
- antibodies having different product specificities can have different detectable moieties, such as different fluorescent tags.
- antibodies that recognize substrate A-B 1 -B 2 -B 3 -B 4 can be used to detect substrate remaining on the beads after incubation with enzymes. In this situation, either product A or B 1 -B 2 -B 3 -B 4 would remain attached to the bead, if an enzymatic reaction occurred. In either case the selected substrate specific antibody would not significantly cross-react with product attached to the bead.
- samples containing target enzymes are contacted with substrates linked to encoded particles in an assay solution.
- the encoded particles have a feature, such as a bar code or optical profile, which allows them to be distinguished from each other.
- encoded particles can have different bar codes corresponding to different target enzyme substrates.
- the target enzymes act on the substrates to produce products A and B ⁇ B ⁇ B ⁇ B 4 .
- the B ⁇ B ⁇ B ⁇ B 4 would remain bound to the particle, while the A product would be released into solution, or visa versa.
- Antibodies that recognize specific products are then contacted with the assay solution.
- antibodies need not be specific for particular products, and thus one type of antibody can be used to detect products derived from multiple different substrates. Such non-specific antibodies will bind to the products, if produced during the enzymatic assay, to produce particles having bound antibodies. Particles having bound antibodies are then distinguished from those without antibodies, for example, by detecting a tag on the antibodies or physical behavior of the particles. The different products contained on the antibody-bound particles can be determined based on the encoding of each particle.
- antibodies directed to particular substrates are generated. Following quenching of an enzymatic reaction, the reaction solution is transferred to a high-binding microtiter plate whereby the reactive B 2 moiety covalently attaches to the plate via a terminal amino group. The enzyme and assay solution components are removed by washing. The specific primarily antibody is then incubated in each assay well followed by subsequent washing to remove unbound antibody. A secondary antibody is optionally used for detection and quantitation. The more product formed per unit time of initial reaction the greater the activity of the measured enzyme.
- an antibody specific for the B ⁇ B 2 subgroup is optionally used as a capture antibody on the surface of the microtiter plate in a standard sandwich ELISA assay.
- a primary antibody with a unique epitope on the product such as one directed to the B 4 moiety (or the B 4 moiety is modified with a specific binding pair member such as biotin) is used for detection.
- a labeled secondary antibody is optionally used for detection as described above.
- an exemplary antibody reacts with the OC-D- glucose A group bound to the inventive hydrophenol B 1 moiety.
- the substrate is attached to a solid support using a lysine B 2 moiety.
- the substrate is provided in solution, the reaction is transferred to a sample receptacle, in which Following quenching of an exemplary enzymatic reaction, the reaction solution is transferred to a high-binding microtiter plate whereby the reactive B 2 moiety covalently attaches to the receptacle via a terminal amino group.
- a capture antibody specific for an alternate epitope on the inventive product/substrate is employed. The unreacted enzyme and buffer components are removed by washing.
- the antibody specific to the A-B ⁇ B 2 moiety is then incubated in each assay well for detection and quantitation of remaining substrate. The greater the substrate remaining after the initial enzyme reaction, the lower the activity of the enzyme.
- the antibody is illustratively unlabeled and produced in animals including mouse, rat, rabbit, horse, donkey, or other suitable animal used for the production of antibodies.
- a secondary antibody that is specific for the species IgG of the primary antibody is labeled illustratively with a fluorescent marker such as rhodamine and subsequently used for detection of remaining substrate. It is appreciated in the art that other antibody detection systems are similarly operable in the instant invention such as horseradish peroxidase labeled antibodies, or alkaline phosphatase labeled antibodies.
- monoclonal mouse antibody specific for the exemplary the ⁇ -D-glucose A group bound to the inventive hydrophenol B 1 moiety and lysine B moiety is itself labeled illustratively by a fluorescent marker.
- multiple lysosomal enzymes are optionally simultaneously analyzed for activity toward a variety of specific substrates.
- An illustrative example includes a two enzyme system wherein inventive substrate are employed, one specific for GLA and another specific for GAA. Each substrate is simultaneously added to the reaction with the biological sample. As each substrate optionally contains a lysine B 2 group, both will similarly bind to the high-binding microtiter plate.
- Each antibody is illustratively labeled with a different fluorophore such as rhodamine or cyanine. As such the binding of each antibody is detected and quantitated without interference from the other, and the amount of each enzyme activity is detectable in the same well of the microtiter plate from the same sample.
- Another illustrative assay format is performed using mass spectrometry.
- An assay for target enzymes is performed by first obtaining a sample illustratively including serum, plasma, whole blood, urea, saliva, other biological fluids or tissue lysates, recombinant or native purified enzyme in solution, or chemically or functionally modified enzyme in biological fluid or liquid medium.
- a portion of the filter paper sample is then excised and deposited in a non-binding assay tube or micro titer plate well to which an assay solution is added.
- the assay solution comprises aqueous buffers, a substrate, a standard, as well as protease inhibitors.
- the sample mixture is then incubated for a determined period of time in the range of 30 minutes to 20 hours at a particular temperature ranging from 30 to 41 0 C.
- a stopping solution is illustratively 0.4 M glycine/NaOH pH 10.4 added at 6X reaction volume.
- a stopping solution is illustratively 0.4 M glycine/NaOH pH 10.4 added at 6X reaction volume.
- the amount of product formation is determined by transferring a known volume of sample to a high-binding assay tube or microtiter plate and incubated for 5 minutes to 2 hours. The unbound material is removed by washing.
- Detection of the intact substrates or products is illustratively performed using a coupled peroxidase enzyme approach.
- the level of released glucose or galactose product is measured in real time by a coupled enzyme approach.
- a non-limiting example involves the release of glucose from an inventive substrate specific for alpha-glucosidase in diagnosis of Pompe disease.
- glucose is reacted with glucose oxidase producing glucolactone and releasing hydrogen peroxide.
- the released hydrogen peroxide is detected by reaction with peroxidase to produce a fluorescent molecule that is measured on a standard fluorometer.
- suitable peroxidases are horseradish peroxidase or any other peroxidase known in the art.
- the hydrogen peroxide released by glucose oxidase interacts with a detector substrate molecule.
- the peroxidase catalyzes conversion of this substrate to a fluorescent product.
- a detector molecule suitable for use with the inventive substrates includes Amplex Red that is oxidized in to produce the fluorescent product resorufin. Amplex Red and kits for detecting free glucose are available from Invitrogen Corp. The increase in red fluorescent product is detected on a fluorometer set with an excitation wavelength at 571 and an emission wavelength at 585 with the band pass set at 5 nm. The greater amount of glycosidase activity the more rapidly the red fluorescent product is produced.
- multiple substrates for different lysosomal enzymes are generated with unique B ⁇ B 4 structure. This prevents product inhibition of one enzyme that is particularly important should the catalytic activity of one enzyme toward one inventive substrate be much greater than the catalytic activity of the other enzyme for its corresponding inventive substrate. This is additionally important in conditions where a single mutant glycosidase is being screened in a panel of substrates for 6 or more lysosomal enzymes.
- the product formed by the other lysosomal enzymes may inhibit the function of the lower activity enzyme such that its activity is not accurately measured.
- the specificity of the substrate and the product for each enzyme is appreciated to be optionally distinct.
- the substrates may differ not only in the type of sugar moiety which confers enzyme specificity, but also in the length of the B 4 tail moiety. This is particularly important with the use of MS/MS as a detection tool since the differentiated inventive substrate molecules having corresponding differentiated mass index correspond to various enzymes being examined.
- the approach described for assaying enzymes using substrate reagents, immunoassay, HPLC, and mass spectrometry according to the present invention may be broadly applied.
- the multiplex techniques may be expanded to assay dozens or more enzymes simultaneously in a single reaction, obviating the need for multiple assays to assist in confirming diagnoses of rare disorders.
- the method may be used to measure several enzymes simultaneously when evaluating the rate of chemical flux through a specific biochemical pathway or for monitoring biochemical signaling pathways. Because of the high sensitivity of the ESI-MS detection employed, which requires only sub-microgram quantities of the substrate reagents per assay, the synthesis of several hundred substrate reagents on a low-gram scale becomes practical and economical. [0061] In a preferred embodiment two lysosomal enzymes are simultaneously measured for activity by the use of inventive substrates. In an alternative embodiment 2-6 lysosomal enzymes are simultaneously measured.
- the substrates can be labeled with the same fluorophore, but possess significant mass or charge characteristics that differentiate one from the other.
- the amount of product produced following an enzymatic cleavage reaction is detected by reversed phase high performance liquid choromatography (HPLC). Reactions are quenched by the addition of alcohol, acetonitrile or dilute trifluoro acetic acid. A portion of the incubation mixture is transferred to a new assay vessel to which is added a neat solution such as methanol, acetonitrile, water-methanol mixtures or water- acetonitrile.
- reaction products and unreacted substrate are separated on a 5 ⁇ m particle size Ci 8 HPLC column and detected by a fluorescent detector or set of detectors.
- the amount of product is calculated based on a standard curve generated using increasing amounts of the relevant product.
- multiple substrates for multiple enzymes are optionally simultaneously detected by the HPLC method. If substrates with sufficiently different mass or retention characteristics are used each product is resolvable in the HPLC column and can be quantified in a single assay. Alternatively, each substrate is labeled with a different fluorophore that has different or the same excitation or emission properties. Detection may be by a family of fluorescent detectors that can simultaneously quantify individual products from each other and their corresponding labeled substrate. Other methods of detection are similarly suitable and are known in the art.
- Figure 1 shows ⁇ -D-glucose, modified with a 4-aminophenyl group is reacted with a di-protected lysine group.
- Methods for di-protecting lysine are known in the art and illustrative examples are described in WO 01/27074.
- the intermediate A-B ⁇ B 2 is formed which is specifically deprotected at the alpha-amino group to provide a suitable reactive site for subsequent synthetic steps. Methods of site-specific deprotection are known in the art.
- the alkyl chain is then added.
- An exemplary alkyl chain is provided by dodecyl-carnitine (B 3 -B 4 ) which is synthetically added to the A-B ⁇ B 2 intermediate to form a full length substrate molecule (A-B 1 -B 2 -B 3 -B 4 ).
- This substrate molecule is active as a GAA specific substrate and is optionally used in analyses involving mass spectrometry or other suitable assay and detection method.
- the 6-amino group on B 2 is deprotected so as to produce a similarly reactive substrate. It is appreciated in the art that synthesis of numerous derivatives of the above illustrated substrate are similarly achieved such as by varying the carbon chain on the acylcarnitine, altering the sugar A group or the B 1 group. Also, numerous derivatives of lysine (B 2 ) or other alkyl groups are similarly suitable.
- Figure 2 depicts a synthetic pathway similar to Figure 1 except for the protective groups and the choice of acylcarnitine whereby n is a value between 1 and 30. Preferably, n is between 1 and 20.
- Figure 3 depicts an exemplary substrate structure for detecting lysosomal storage diseases.
- the structure is composed of a sugar (A) in the form of a glucose or a galactose and an aliphatic group B.
- Group B is further composed of a linker arm (B 1 ) in the form of a nitrophenyl, a B 2 subgroup of a lysine, a B 3 subgroup of a carnitinyl, and a B 4 subgroup in the form of an alkyl with carbon length in the range of 10 to 30.
- a quaternary ammonium cation located on the B 3 subgroup avoids the need of further ionization otherwise needed for mass spectrometry detection.
- Figure 4 depicts the multiple functional groups that provide multiple assay format detection of enzymatic activity.
- the terminal amino group on B 2 is capable of forming covalent bonds with carboxylic acids on solid supports.
- the presence of the quaternary ammonium on B 3 provides a permanently charged moiety such that subsequent ionization is not required for analysis by MS/MS providing a robust and easily detectable and quantifiable detection of lysosomal enzyme activity.
- Figure 5 demonstrates a generic enzymatic reaction using an inventive substrate.
- the substrate Upon specific affinity binding and enzymatic reaction, the substrate is cleaved into two groups, a sugar moiety A and an aliphatic group B.
- the group B is optionally composed of a nitrophenyl, a lysine, a carnitinyl, and long-chain alkyl moieties. Both groups are optionally then analyzed by MS/MS.
- An internal standard is also concurrently subject to the MS/MS analysis. The internal standard is an isotopically labeled analog of B with deuterium to replace hydrogen atom(s) on a methyl group.
- the deprotected terminal amino group on B 2 is reactive with a solid support such that the presence or absence of product is optionally detected using immunoassay methods.
- the deprotected terminal amine can also be derivatized with a fluorescent reagent for HPLC, or other suitable detection method.
- Figure 6 illustrates exemplary inventive methods depicting detection of enzymatic reactions using mass spectrometry that are advantageous in comparison to prior art methods.
- lysosomal enzyme substrates each require a unique synthetic pathway.
- the inventive substrates can be synthesized using a common synthetic pathway. Having a common synthetic pathway for two or more of the inventive substrates means significant savings in production environments due to shorter and less complex production processes and the use of common raw materials.
- the inventive substrates also can be rapidly synthesized relative to the prior art. Synthesis is accomplished in as little as three steps. Synthesis of a preferred inventive substrate with specificity for the lysosomal enzyme GAA (Pompe disease) is illustrated in Figure 1.
- the compound B 1 -B 2 -B 3 -B 4 is also functional as an antagonist, an analytical control, or for clinical treatment of disease such as hypothyroidism, diabetes, and HIV.
- the B ⁇ B ⁇ B ⁇ B 4 molecule is: B 1 is a Ci-C 2 O alkyl, Ci-C 20 having a substituted C 6 -C 2 O aryl, C 6 -C 2 O heterocyclic containing a heteroatom of N, O or S; B 2 is an amino acid; 2,6-diaminohexanoic acid; or
- R 1 ' is a Ci-C 2 O alkyl; C 4 -C 2 O ether; Ci-C 2 O alkyl having a substituent of N, O, or S; heteroatom C 6 -C 20 aryl, C 1 -C 2 O carbonyl, C 1 -C 2 O amidyl, C 1 -C 2 O ether, C 6 -C 20 aryl, C 6 -C 20 heterocyclic containing a heteroatom of N, O, or S;
- X is a nullity, oxygen, sulfur, or nitrogen;
- R 2 ' is a nullity or Ci-C 20 alkyl, C 4 -C 20 ether; Ci-C 20 alkyl having a substituent of N, O, or S; C 1 - C 20 ester; Ci-C 20 alcohol; Ci-C 20 alkenyl; heteroatom C 6 -C 20 aryl, C 6 -C 20 heterocyclic containing a heteroatom of N, O or S;
- Y
- R 1 is a Ci-C 20 alkyl; C 4 -C 20 ether; Ci-C 20 alkyl having a substituent of N, O, or S; heteroatom C 6 -C 20 aryl, Ci-C 20 carbonyl, Ci-C 20 amidyl, Ci-C 20 ether, C 6 -C 20 aryl, C 6 -C 20 heterocyclic containing a heteroatom of N, O, or S;
- R is independently in each occurrence a H, a Ci-C 20 alkyl, a C 2 -C 20 alkyl having a substituent of Ci-C 20 alkyl;
- X is independently in each occurrence a nullity, oxygen, sulfur, or nitrogen;
- R is independently in each occurrence a nullity, Ci-C 20 alkyl, Ci-C 20 having a substituted C 6 -C 20 aryl, C 6 -C 20 heterocyclic containing a heteroatom of N, O or S;
- n is an integer between
- inventive substrates A-B 1 -B 2 -B 3 -B 4 are optionally synthesized with a non-hydrolyzable link between A and B 1 .
- Figure 1 shows ⁇ -D-glucose, modified with a 4-aminophenyl group is reacted with a differentially protected lysine group. Methods for differentially protecting lysine are known in the art and illustrative examples are described in WO 01/27074.
- the intermediate A-B ⁇ B 2 is formed which is specifically deprotected at the alpha-amino group to provide a suitable reactive site for subsequent synthetic steps. Methods of site-specific deprotection are known in the art.
- the alkyl chain is then added.
- An exemplary alkyl chain is provided by dodecyl-carnitine (B 3 - B 4 ) which is synthetically added to the A-B ⁇ B 2 intermediate to form a full length substrate molecule (A-B 1 -B 2 -B 3 -B 4 ).
- This substrate molecule is active as a GAA specific substrate and is optionally used in analyses involving mass spectrometry or other suitable assay and detection method.
- the 6-amino group on B 2 is deprotected so as to produce a similarly reactive substrate. It is appreciated in the art that synthesis of numerous derivatives of the above illustrated substrate are similarly achieved such as by varying the carbon chain on the acylcarnitine, altering the sugar A group or the B 1 group. Also, numerous derivatives of lysine (B 2 ) or other alkyl groups are similarly suitable. [0075] It is appreciated in the art that similar synthetic pathways are suitable for production of the inventive substrates. Figure 2 depicts a synthetic pathway similar to Figure 1 except for the protective groups and the choice of acylcarnitine whereby n is a value between 1 and 30. Preferably, n is between 1 and 20.
- Figure 3 depicts an exemplary substrate structure for detecting lysosomal storage diseases.
- the structure is composed of a sugar (A) in the form of a glucose or a galactose and an aliphatic group B.
- Group B is further composed of a linker arm (B 1 ) in the form of a nitrophenyl, a B 2 subgroup of a lysine, a B 3 subgroup of a carnitinyl, and a B 4 subgroup in the form of an alkyl with carbon length in the range of 10 to 30.
- a quaternary ammonium cation located on the B 3 subgroup avoids the need of further ionization otherwise needed for mass spectrometry detection.
- Figure 4 depicts the multiple functional groups that provide multiple assay format detection of enzymatic activity.
- the terminal amino group on B 2 is capable of forming covalent bonds with carboxylic acids on solid supports.
- the presence of the quaternary ammonium on B 3 provides a permanently charged moiety such that subsequent ionization is not required for analysis by MS/MS providing a robust and easily detectable and quantifiable detection of lysosomal enzyme activity.
- Figure 5 demonstrates a generic enzymatic reaction using an inventive substrate.
- the substrate Upon specific affinity binding and enzymatic reaction, the substrate is cleaved into two groups, a sugar moiety A and an aliphatic group B.
- the group B is optionally composed of a nitrophenyl, a lysine, a carnitinyl, and long-chain alkyl moieties. Both groups are optionally then analyzed by MS/MS.
- An internal standard is also concurrently subject to the MS/MS analysis. The internal standard is an isotopically labeled analog of B with deuterium to replace hydrogen atom(s) on a methyl group.
- the deprotected terminal amino group on B 2 is reactive with a solid support such that the presence or absence of product is optionally detected using immunoassay methods.
- the deprotected terminal amine can also be derivatized with a fluorescent reagent for HPLC, or other suitable detection method.
- Figure 6 illustrates exemplary inventive methods depicting detection of enzymatic reactions using mass spectrometry that are advantageous in comparison to prior art methods. The inventive method eliminates many steps inherent with the use of non-polar substrates and their relative internal standards. More specifically, the inventive method no longer needs steps to clean up non-polar solvent and or detergents the involvement of which is detrimental to mass spectrometry machinery.
- Example 1 For each sample, a disk of 3 mm diameter is punched from the areas of dried blood on a filter paper into a well of a 96-well microtiter plate. The blood disk is then incubated directly with an assay solution containing substrates directed to the Fabry disease at a final concentration of 5 ⁇ mol/L and internal standards at a final concentration of 0.1 ⁇ mol/L. To the assay solution, a final concentration of 0.5 mol/L sodium acetate buffer is also added. The assay mixture containing the blood disk is incubated for 15 to 24 hours at 37° Celsius with orbital shaking (150 rpm) in a thermostatic air shaker.
- Example 2 In an alternative embodiment the product of the reaction with the inventive substrates is quantified by immunoassay. Blood spotted on filter paper is reconstituted in buffer to liberate the active components.
- One or an array of inventive substrates are added to the reaction chamber and the reaction allowed to proceed overnight (-14 hours).
- the reaction is quenched by the addition of 6X volume glycine/NaOH pH 10.4.
- a sample of each reaction is added to the wells of a high-binding irradiated microtiter plate and incubated overnight to allow sufficient binding of the reaction product to the wells of the plate.
- a standard curve of product in similar buffer/sample is also added to the plate to serve as a basis for quantitation.
- the wells are washed twice with phosphate buffered saline (PBS) by the use of a squirt bottle, plate washer, or any other automated or non-automated plate washing system.
- PBS phosphate buffered saline
- any additional sites for protein binding are subsequently blocked by the addition of a blocking agent illustratively including 3% bovine serum albumin in PBS or any other synthetic or natural blocking agent known in the art.
- the blocking agent is incubated for two hours at room temperature.
- the wells are washed 3X with PBS.
- the primary antibody(s) is then added to the wells to recognize and bind the remaining substrate, or the product.
- the antibody(s) is incubated in the wells for at least 2 hours.
- the plate is washed four times to remove unbound antibody. If the primary antibody is labeled the plate is used for detection.
- Example 3 In an alternative embodiment the product of the reaction with the inventive substrates is quantified by immunoassay. Blood spotted on filter paper is reconstituted in buffer to liberate the active components. One or an array of inventive substrates immobilized to encoded particles are added to the reaction chamber, preferably a microplate well, and the reaction allowed to proceed overnight (-14 hours). A standard curve of enzyme in similar buffer/sample is also added to separate sets of encoded particles to serve as a basis for quantitation.
- the reaction is quenched by the addition of 6X volume glycine/NaOH pH 10.4.
- the primary antibody(s) is then added to the wells to recognize and bind the remaining substrate, or the product.
- the antibody(s) is incubated for at least 30 minutes. If the primary antibody is labeled the assay is ready for detection.
- a labeled secondary antibody is placed in the plate wells and allowed to incubate for an additional 30 minutes. Detection is accomplished a flow cytometer.
- Example 4 The active amino group on the B 2 of a preferred embodiment is amenable to numerous labeling procedures.
- fluorescent labeling offers one of the most powerful methods of detection as it provides excellent sensitivity, ability to quantitate relative to a standard, and can be combined with other substrates with other fluorophores in a panel assay for multiple lysosomal enzymes.
- the B 2 side chain amine is specifically labeled with fluoroisothiocyanate (FITC).
- FITC fluoroisothiocyanate
- the purified/lyophilized substrate is resuspended in 0.1 M sodium bicarbonate buffer, pH 9.0 at a concentration of 5 mg/ml.
- Immediately prior to reaction with substrate dissolve 5mg of FITC dye in 0.5 ml of DMSO in the dark. With gentle vortexing, add 0.1 ml of dye solution of the substrate solution and incubate for 1 hour at room temperature in the dark.
- the free unreacted dye is removed by gel filtration on a 10 x 300 mm Sephadex G column pre- equilibrated in phosphate buffered saline. Concentration of the final product is determined by mass spectrometry or other method known in the art.
- the labeled substrate is optionally concentrated and aliquoted for storage at -2O 0 C until further use.
- the labeled substrate is used in a reaction for the detection of glucocerebrosidase activity and the detection of Gaucher' s disease.
- a disk of 3 mm diameter is punched from the areas of dried blood on a filter paper into a micro-centrifuge tube or a well of a 96-well microtiter plate.
- the blood disk is then incubated directly with an assay solution containing labeled inventive substrate at a final concentration of 5 ⁇ mol/L and internal standards at a final concentration of 0.1 ⁇ mol/L.
- the assay mixture containing the blood disk is incubated for 15 to 24 hours at 37 0 C with orbital shaking (150 rpm) in a thermostatic air shaker.
- Fluorescence intensity is continuously monitored using a fluorescence detector (model L-7480; Hitachi, Naperville, IL) at a medium gain sensitivity.
- the amount of labeled product in the sample is determined by comparing the area of the peak to that of an external standard comprised of labeled product at a known concentration.
- the concentration of product in the reaction is readily determined and the activity of glucocerebrosidase determined by dividing the moles product/per unit of reaction time.
- Example 5 In an alternative embodiment the product of the reaction with the inventive substrates is quantified by immunoassay wherein a specific antibody targeted to the cleaved inventive substrate is employed. Blood spotted on filter paper is resolubilized in buffer to liberate the active components. One or an array of inventive substrates are added to the reaction chamber and the reaction allowed to proceed overnight (-14 hours). The reaction is quenched by the addition of 6X volume glycine/NaOH pH 10.4. A microtiter plate is prepared in advance by coating with an antibody generated in a mouse that is specific for the nitrophenyl-aminoacyl B ⁇ B 2 moiety.
- any additional sites for protein binding are subsequently blocked by the addition of a blocking agent illustratively including 3% bovine serum albumin in PBS or any other synthetic or natural blocking agent known in the art.
- a blocking agent illustratively including 3% bovine serum albumin in PBS or any other synthetic or natural blocking agent known in the art.
- a sample of each reaction is added to the wells of a microtiter plate and incubated for two hours to overnight to allow sufficient binding of the reaction product to the wells of the plate.
- a standard curve of control product in similar buffer/sample is also added to the plate to serve as a basis for quantitation.
- the wells are washed twice with phosphate buffered saline (PBS) by the use of a squirt bottle, plate washer, or any other automated or non-automated plate washing system.
- PBS phosphate buffered saline
- the primary antibody(s) is then added to the wells to recognize and bind the product at an alternative epitope.
- the antibody(s) is incubated in the wells for at least 2 hours.
- the plate is washed four times to remove unbound antibody. If the primary antibody is labeled the plate is used for detection.
- a labeled secondary antibody is placed in the plate wells and allowed to incubate for an additional 2 hours followed by washing 4 times and detection by the appropriate method such as by a fluorescent or optical plate reader.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Medicinal Chemistry (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Other Investigation Or Analysis Of Materials By Electrical Means (AREA)
Abstract
Description




Claims
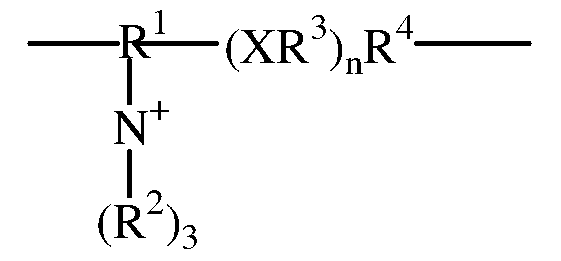

Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09720998A EP2265645A4 (en) | 2008-03-13 | 2009-03-13 | Enzymatic substrates for multiple detection systems |
| BRPI0906090A BRPI0906090A2 (en) | 2008-03-13 | 2009-03-13 | substrate for analysis of an enzyme, molecule and method for detecting enzymatic activity. |
| JP2010550887A JP2011514809A (en) | 2008-03-13 | 2009-03-13 | Enzyme substrates for multiplex detection systems |
| CN2009801167273A CN102014929A (en) | 2008-03-13 | 2009-03-13 | Enzymatic substrates for multiple detection systems |
| AU2009223222A AU2009223222A1 (en) | 2008-03-13 | 2009-03-13 | Enzymatic substrates for multiple detection systems |
| CA2718276A CA2718276A1 (en) | 2008-03-13 | 2009-03-13 | Enzymatic substrates for multiple detection systems |
| IL208521A IL208521A0 (en) | 2008-03-13 | 2010-10-06 | Enzymatic substrates for multiple detection systems |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3621108P | 2008-03-13 | 2008-03-13 | |
| US61/036,211 | 2008-03-13 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009114763A2 true WO2009114763A2 (en) | 2009-09-17 |
| WO2009114763A3 WO2009114763A3 (en) | 2009-11-19 |
Family
ID=41065853
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/037087 WO2009114763A2 (en) | 2008-03-13 | 2009-03-13 | Enzymatic substrates for multiple detection systems |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US8173784B2 (en) |
| EP (2) | EP2706121A1 (en) |
| JP (1) | JP2011514809A (en) |
| CN (1) | CN102014929A (en) |
| AU (1) | AU2009223222A1 (en) |
| BR (1) | BRPI0906090A2 (en) |
| CA (1) | CA2718276A1 (en) |
| IL (1) | IL208521A0 (en) |
| WO (1) | WO2009114763A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11591544B2 (en) | 2020-11-25 | 2023-02-28 | Akagera Medicines, Inc. | Ionizable cationic lipids |
| US12064479B2 (en) | 2022-05-25 | 2024-08-20 | Akagera Medicines, Inc. | Lipid nanoparticles for delivery of nucleic acids and methods of use thereof |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201017875D0 (en) * | 2010-10-22 | 2010-12-01 | Johnson Matthey Plc | Method of identifying a material |
| CA2906839A1 (en) * | 2013-03-15 | 2014-09-18 | Perkinelmer Health Sciences, Inc. | Compounds and methods relating to testing for lysosomal storage disorders |
| JP6624482B2 (en) * | 2014-07-29 | 2019-12-25 | 俊 保坂 | Micro accelerator and micro mass spectrometer |
| US9575064B2 (en) | 2014-09-02 | 2017-02-21 | Perkinelmer Health Sciences, Inc. | Methods relating to testing for lysosomal storage disorders |
| WO2019056251A1 (en) * | 2017-09-21 | 2019-03-28 | 大连理工大学 | Group of isotope labeling reagents for amino/phenolic hydroxyl labeling, and synthesis method therefor |
| CN111269280B (en) * | 2020-03-27 | 2021-07-13 | 江苏豪思睦可生物科技有限公司 | Compound, corresponding internal standard and kit for detecting enzyme related to lysosomal storage disease |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001027074A1 (en) | 1999-10-08 | 2001-04-19 | Abbott Laboratories | Preparation of amino-protected lysine derivatives |
| US6649414B1 (en) | 1999-08-17 | 2003-11-18 | Luminex Corporation | Microparticles with multiple fluorescent signals and methods of using same |
| US6939720B2 (en) | 1995-10-11 | 2005-09-06 | Luminex Corporation | Multiplexed analysis of clinical specimens apparatus and method |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2931833A (en) * | 1954-12-29 | 1960-04-05 | Armour & Co | Benzyldialkyl-2-(1-hydroxy-alkyl) alkyl ammonium chloride compounds |
| US3636114A (en) * | 1968-07-16 | 1972-01-18 | Union Carbide Corp | Novel quaternary ammonium compounds and method for preparation thereof |
| IT1116037B (en) * | 1979-04-23 | 1986-02-10 | Sigma Tau Ind Farmaceuti | ACIL CARNITINE ESTERS AND AMIDS THEIR PREPARATION PROCEDURES AND THERAPEUTIC USE |
| US4968785A (en) * | 1988-05-17 | 1990-11-06 | Henkel Kommanditgesellschaft Auf Aktien | Substituted glycoside compositions |
| JPH0243560A (en) * | 1988-08-04 | 1990-02-14 | Canon Inc | Electrophotographic sensitive body |
| US5138043A (en) * | 1989-12-07 | 1992-08-11 | Union Carbide Chemicals & Plastics Technology Corporation | Alkoxylated alkyl glucoside ether quaternaries useful in personal care |
| IT1261984B (en) * | 1993-06-22 | 1996-06-11 | Avantgarde Spa | USE OF L-CARNITINE ESTERS OR ACIL L-CARNITINE WITH HYDROXIACID TO PRODUCE PHARMACEUTICAL COMPOSITIONS FOR THE TREATMENT OF SKIN DISEASES. |
| EP0710107A4 (en) * | 1993-07-23 | 1997-05-28 | Biomembrane Inst | N,n,n-trimethylsphingosine derivatives |
| US5627171A (en) * | 1994-04-11 | 1997-05-06 | Oncomembrane, Inc. | Sphingosine-1-phosphate/trimethylsphingosine composition |
| FR2767134B1 (en) * | 1997-08-08 | 1999-09-24 | Ceca Sa | N-(DIALKYLAMINO)ALKYL GLYCOSYLAMINES AND DERIVATIVES, PROCESSES FOR THE PREPARATION, COMPOSITION CONTAINING THEM AND USES, PARTICULARLY IN THE FIELD OF COSMETICS. |
| WO2000011208A1 (en) | 1998-08-25 | 2000-03-02 | University Of Washington | Rapid quantitative analysis of proteins or protein function in complex mixtures |
| IT1306129B1 (en) * | 1999-04-13 | 2001-05-30 | Sigma Tau Ind Farmaceuti | ESTERS OF L-CARNITINE OR ALCANOYL L-CARNITINE USABLE CATIONIC COMELIPIDS FOR INTRACELLULAR PLACING OF COMPOUNDS |
| JP2003530553A (en) | 2000-04-10 | 2003-10-14 | エムシーダブリユー リサーチ フオンデーシヨン インコーポレーテツド | Fluorescent HPLC assay of 20-HETE and other P-450 metabolites of arachidonic acid |
| US7393640B2 (en) | 2003-02-05 | 2008-07-01 | Ge Healthcare Bio-Sciences Corp. | Terminal-phosphate-labeled nucleotides with new linkers |
| JP4861308B2 (en) * | 2004-04-07 | 2012-01-25 | ジェネンテック, インコーポレイテッド | Mass spectrometry of antibody conjugates |
| WO2007013601A1 (en) * | 2005-07-28 | 2007-02-01 | Shinichiro Isobe | Labeling dye for biomolecule, labeling kit, and method for detecting biomolecule |
| JP4364197B2 (en) | 2005-12-27 | 2009-11-11 | 日本リライアンス株式会社 | AC power supply apparatus and arc suppression method in the apparatus |
| CN101426926A (en) * | 2006-03-13 | 2009-05-06 | 珀金埃尔默Las公司 | Substrates and internal standards for mass spectroscopy detection |
| EP2155776A2 (en) * | 2007-05-11 | 2010-02-24 | Mpex Pharmaceuticals, Inc. | Quaternary alkyl ammonium bacterial efflux pump inhibitors and therapeutic uses thereof |
-
2009
- 2009-03-13 EP EP13184562.0A patent/EP2706121A1/en not_active Withdrawn
- 2009-03-13 US US12/403,790 patent/US8173784B2/en active Active
- 2009-03-13 AU AU2009223222A patent/AU2009223222A1/en not_active Abandoned
- 2009-03-13 CA CA2718276A patent/CA2718276A1/en not_active Abandoned
- 2009-03-13 CN CN2009801167273A patent/CN102014929A/en active Pending
- 2009-03-13 EP EP09720998A patent/EP2265645A4/en not_active Withdrawn
- 2009-03-13 JP JP2010550887A patent/JP2011514809A/en active Pending
- 2009-03-13 WO PCT/US2009/037087 patent/WO2009114763A2/en active Application Filing
- 2009-03-13 BR BRPI0906090A patent/BRPI0906090A2/en not_active IP Right Cessation
-
2010
- 2010-10-06 IL IL208521A patent/IL208521A0/en unknown
-
2012
- 2012-04-03 US US13/438,029 patent/US8791246B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6939720B2 (en) | 1995-10-11 | 2005-09-06 | Luminex Corporation | Multiplexed analysis of clinical specimens apparatus and method |
| US6649414B1 (en) | 1999-08-17 | 2003-11-18 | Luminex Corporation | Microparticles with multiple fluorescent signals and methods of using same |
| WO2001027074A1 (en) | 1999-10-08 | 2001-04-19 | Abbott Laboratories | Preparation of amino-protected lysine derivatives |
Non-Patent Citations (14)
| Title |
|---|
| AERTS, JM; HOLLACK, CD, BAILLIERE'S CLIN. HAEMATOL, vol. 10, 1997, pages 691 - 709 |
| AUSUBEL ET AL.: "Current Protocols in Molecular Biology", 1999, JOHN WILEY & SONS |
| BEUTLER, E ET AL., J EXP MED, vol. 143, 1976, pages 975 - 80 |
| BOOT, RG ET AL., J. BIOL. CHEM., vol. 282, 2006, pages 1305 - 12 |
| DEEGAN PB ET AL., BLOOD CELLS MOL DIS, vol. 35, 2005, pages 259 - 67 |
| GELB MH ET AL., CLINICAL CHEMISTRY, vol. 50, no. 10, 2004, pages 1785 - 1796 |
| HARLOW; LANE: "Antibodies: A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY PRESS |
| HARLOW; LANE: "Using Antibodies: A Laboratory Manual", 1999, COLD SPRING HARBOR PRESS |
| LEONARD R ET AL., J. BIOL. CHEM., vol. 281, 2006, pages 4867 - 75 |
| RICHARDS SM ET AL., BLOOD, vol. 82, 1993, pages 1402 |
| ROSENBERG, M. ET AL., BLOOD, vol. 93, 1999, pages 2081 - 2088 |
| SCHULZE A ET AL., PEDIATRICS, vol. 111, 2003, pages 1399 - 406 |
| See also references of EP2265645A4 |
| TIETZ: "Textbook of Clinical Chemistry", 1999, W.B. SAUNDERS |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11591544B2 (en) | 2020-11-25 | 2023-02-28 | Akagera Medicines, Inc. | Ionizable cationic lipids |
| US12077725B2 (en) | 2020-11-25 | 2024-09-03 | Akagera Medicines, Inc. | Ionizable cationic lipids |
| US12064479B2 (en) | 2022-05-25 | 2024-08-20 | Akagera Medicines, Inc. | Lipid nanoparticles for delivery of nucleic acids and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2718276A1 (en) | 2009-09-17 |
| US20090253117A1 (en) | 2009-10-08 |
| AU2009223222A1 (en) | 2009-09-17 |
| BRPI0906090A2 (en) | 2016-05-03 |
| CN102014929A (en) | 2011-04-13 |
| EP2265645A2 (en) | 2010-12-29 |
| EP2706121A1 (en) | 2014-03-12 |
| US8173784B2 (en) | 2012-05-08 |
| WO2009114763A3 (en) | 2009-11-19 |
| EP2265645A4 (en) | 2011-03-23 |
| US20120190043A1 (en) | 2012-07-26 |
| US8791246B2 (en) | 2014-07-29 |
| JP2011514809A (en) | 2011-05-12 |
| IL208521A0 (en) | 2011-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8791246B2 (en) | Enzymatic substrates for multiple detection systems | |
| AU2007226582B2 (en) | Substrates and internal standards for mass spectroscopy detection | |
| US20220010354A1 (en) | Compounds and methods relating to lysosomal storage disorders | |
| JP7361650B2 (en) | Methods related to testing for lysosomal storage disorders | |
| JP2016522677A5 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980116727.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09720998 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2718276 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010550887 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009223222 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009720998 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2009223222 Country of ref document: AU Date of ref document: 20090313 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0906090 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100913 |