WO2009111277A1 - Imdizo [4. 5-b] pyridine derivatives used as raf inhibitors - Google Patents
Imdizo [4. 5-b] pyridine derivatives used as raf inhibitors Download PDFInfo
- Publication number
- WO2009111277A1 WO2009111277A1 PCT/US2009/035379 US2009035379W WO2009111277A1 WO 2009111277 A1 WO2009111277 A1 WO 2009111277A1 US 2009035379 W US2009035379 W US 2009035379W WO 2009111277 A1 WO2009111277 A1 WO 2009111277A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- optionally substituted
- halogen
- imidazo
- Prior art date
Links
- 0 *c([n]1)nc2c1ncc(NC(c1c(*)c(*)cc(NS(*)(=O)=O)c1*)=O)c2 Chemical compound *c([n]1)nc2c1ncc(NC(c1c(*)c(*)cc(NS(*)(=O)=O)c1*)=O)c2 0.000 description 5
- NSYMCQJZIIQOHK-UHFFFAOYSA-N CCCCS(Nc(ccc(F)c1C(O)=O)c1F)(=O)=O Chemical compound CCCCS(Nc(ccc(F)c1C(O)=O)c1F)(=O)=O NSYMCQJZIIQOHK-UHFFFAOYSA-N 0.000 description 1
- KRJUPEKMRLMDNK-UHFFFAOYSA-N CCCS(N(c(cc1)c(C)c(C(O)=O)c1F)S(CCC)(=O)=O)(=O)=O Chemical compound CCCS(N(c(cc1)c(C)c(C(O)=O)c1F)S(CCC)(=O)=O)(=O)=O KRJUPEKMRLMDNK-UHFFFAOYSA-N 0.000 description 1
- FBFKEZIPKITEAW-UHFFFAOYSA-N CCCS(N(c(ccc(F)c1C(OC)=O)c1F)S(CCC)(=O)=O)(=O)=O Chemical compound CCCS(N(c(ccc(F)c1C(OC)=O)c1F)S(CCC)(=O)=O)(=O)=O FBFKEZIPKITEAW-UHFFFAOYSA-N 0.000 description 1
- RZBOEMRTZZTEMI-UHFFFAOYSA-N CCCS(Nc(cc1C(O)=O)ccc1Cl)(=O)=O Chemical compound CCCS(Nc(cc1C(O)=O)ccc1Cl)(=O)=O RZBOEMRTZZTEMI-UHFFFAOYSA-N 0.000 description 1
- FRVJLIJAEVYZMY-UHFFFAOYSA-N CCCS(Nc(ccc(F)c1C(Nc2cc(nc(-c(cc3C(F)(F)F)ccc3Cl)[nH]3)c3nc2)=O)c1F)(=O)=O Chemical compound CCCS(Nc(ccc(F)c1C(Nc2cc(nc(-c(cc3C(F)(F)F)ccc3Cl)[nH]3)c3nc2)=O)c1F)(=O)=O FRVJLIJAEVYZMY-UHFFFAOYSA-N 0.000 description 1
- RJMVTZVGTAANND-UHFFFAOYSA-N CCCS(Nc(ccc(F)c1C(Nc2cc(nc(-c3ccc(C)cc3)[nH]3)c3nc2)=O)c1F)(=O)=O Chemical compound CCCS(Nc(ccc(F)c1C(Nc2cc(nc(-c3ccc(C)cc3)[nH]3)c3nc2)=O)c1F)(=O)=O RJMVTZVGTAANND-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/48—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups having nitrogen atoms of sulfonamide groups further bound to another hetero atom
Definitions
- the present invention relates to novel compounds, to pharmaceutical compositions comprising the compounds, to a process for making the compounds and to the use of the compounds in therapy. More particularly, it relates to certain substituted 3H- imidazo[4,5-b]pyridine compounds useful for inhibiting Raf kinase and for treating disorders mediated thereby.
- Raf/MEK/ERK pathway is critical for cell survival, growth, proliferation and tumorigenesis.
- Li Nanxin, et al. "B-Raf kinase inhibitors for cancer treatment.”
- Raf kinases exist as three isoforms, A-Raf, B-Raf and C-Raf. Among the three isoforms, studies have shown that B-
- Raf functions as the primary MEK activator.
- B-Raf is one of the most frequently mutated genes in human cancers.
- B-Raf kinase represents an excellent target for anticancer therapy based on preclinical target validation, epidemiology and drugability.
- Raf inhibitors are also known, see for example, U.S. Patent Application Publication
- Patent Application Publication WO 2008/028617 and International Patent Application Publication WO 2009/012283 also disclose kinase inhibitors.
- the invention relates to compounds that are inhibitors of Raf kinases, particularly B-Raf inhibitors.
- Certain hyperproliferative disorders are characterized by the over activation of Raf kinase function, for example by mutations or over expression of the protein. Accordingly, the compounds of the invention are useful in the treatment of hyperproliferative disorders such as cancer.
- one aspect of the present invention provides compounds of
- Another aspect of the present invention provides methods of preventing or treating a disease or disorder modulated by B-Raf, comprising administering to a mammal in need of such treatment an effective amount of a compound of this invention or a stereoisomer or pharmaceutically acceptable salt thereof.
- diseases and disorders include, but are not limited to, hyperproliferative disorders (such as cancer, including melanoma and other cancers of the skin), neurodegeneration, cardiac hypertrophy, pain, migraine and neurotraumatic disease.
- Another aspect of the present invention provides methods of preventing or treating cancer, comprising administering to a mammal in need of such treatment an effective amount of a compound of this invention, or a stereoisomer or pharmaceutically acceptable salt thereof, alone or in combination with one or more additional compounds having anticancer properties.
- Another aspect of the present invention provides a method of treating a hyperproliferative disease in a mammal comprising administering a therapeutically effective amount of a compound of this invention to the mammal.
- Another aspect of the present invention provides methods of preventing or treating kidney disease, comprising administering to a mammal in need of such treatment an effective amount of a compound of this invention, or a stereoisomer or pharmaceutically acceptable salt thereof, alone or in combination with one or more additional compounds.
- Another aspect of the present invention provides methods of preventing or treating polycystic kidney disease, comprising administering to a mammal in need of such treatment an effective amount of a compound of this invention, or a stereoisomer or pharmaceutically acceptable salt thereof, alone or in combination with one or more additional compounds.
- Another aspect of the present invention provides the compounds of the present invention for use in therapy.
- Another aspect of the present invention provides the compounds of the present invention for use in the treatment of a hyperproliferative disease.
- the hyperproliferative disease may be cancer (or still further, a specific cancer as defined herein).
- kidney disease may be polycystic kidney disease.
- Another aspect of the present invention provides the use of a compound of this invention in the manufacture of a medicament for the treatment of a hyperproliferative disease.
- the hyperproliferative disease may be cancer (or still further, a specific cancer as defined herein).
- Another aspect of the present invention provides the use of a compound of this invention in the manufacture of a medicament for the treatment of a kidney disease.
- the kidney disease may be polycystic kidney disease.
- Another aspect of the present invention provides the use of a compound of the present invention in the manufacture of a medicament, for use as a B-Raf inhibitor in the treatment of a patient undergoing cancer therapy.
- Another aspect of the present invention provides the use of a compound of the present invention in the manufacture of a medicament, for use as a B-Raf inhibitor in the treatment of a patient undergoing polycystic kidney disease therapy.
- Another aspect of the present invention provides a pharmaceutical composition comprising a compound of the present invention for use in the treatment of a hyperproliferative disease.
- Another aspect of the present invention provides a pharmaceutical composition comprising a compound of the present invention for use in the treatment of cancer.
- Another aspect of the present invention provides a pharmaceutical composition comprising a compound of the present invention for use in the treatment of polycystic kidney disease.
- Another aspect of the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of this invention or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- Another aspect of the present invention includes methods of preparing, methods of separation, and methods of purification of the compounds of this invention.
- Another aspect of the present invention provides intermediates for preparing compounds of Formula I. Certain compounds of Formula I may be used as intermediates for other compounds of Formula I.
- alkyl includes linear or branched-chain radicals of carbon atoms.
- the alkyl radical is one to six carbon atoms (C 1 -C 6 ). In other examples, the alkyl radical is C 1 -C 5 , C 1 -C 4 or C 1 -C 3 .
- Some alkyl moieties have been abbreviated, for example, methyl ("Me”), ethyl (“Et”), propyl (“Pr”) and butyl ("Bu”), and further abbreviations are used to designate specific isomers of compounds, for example, 1 -propyl or n-propyl (“n-Pr"), 2-propyl or isopropyl (“i-Pr”), 1 -butyl or n-butyl (“n-Bu”), 2-methyl-l- propyl or isobutyl (“i-Bu”), 1-methylpropyl or s-butyl (“s-Bu”), 1,1-dimethylethyl or t-butyl (“t-Bu”) and the like.
- alkyl groups include 1-pentyl (n-pentyl, -CH 2 CH 2 CH 2 CH 2 CH 3 ), 2-pentyl (-CH(CH 3 )CH 2 CH 2 CH 3 ), 3-pentyl (-CH(CH 2 CH 3 ) 2 ), 2- methyl-2-butyl (-C(CH 3 ) 2 CH 2 CH 3 ), 3-methyl-2-butyl (-CH(CH 3 )CH(CH 3 ) 2 ), 3-methyl-l- butyl (-CH 2 CH 2 CH(CH 3 ) 2 ), 2-methyl-l -butyl (-CH 2 CH(CH 3 )CH 2 CH 3 ), 1-hexyl (-CH 2 CH 2 CH 2 CH 2 CH 2 CH 3 ), 2-hexyl (-CH(CH 3 )CH 2 CH 2 CH 2 CH 3 ), 3-hexyl
- alkenyl refers to linear or branched-chain monovalent hydrocarbon radical with at least one site of unsaturation, i.e., a carbon-carbon double bond, wherein the alkenyl radical may be optionally substituted independently with one or more substituents described herein, and includes radicals having "cis” and “trans” orientations, or alternatively, "E” and "Z” orientations.
- the alkenyl radical is two to six carbon atoms (C 2 - C 6 ). In other examples, the alkenyl radical is C 2 -C 3 .
- alkynyl refers to a linear or branched monovalent hydrocarbon radical with at least one site of unsaturation, i.e., a carbon-carbon, triple bond, wherein the alkynyl radical may be optionally substituted independently with one or more substituents described herein.
- the alkynyl radical is two to eighteen carbon atoms (C 2 - C 6 ). In other examples, the alkynyl radical is C 2 -C 3 .
- Examples include, but are not limited to, ethynyl (-C ⁇ CH), prop-1-ynyl (-C ⁇ CCH 3 ), prop-2-ynyl (propargyl, CH 2 C ⁇ CH), but-1-ynyl, but-2-ynyl and but-3-ynyl.
- alkenyl and alkynyl also include linear or branched-chain radicals of carbon atoms containing at least one unsaturated bond.
- Cycloalkyl refers to a non-aromatic, saturated or partially unsaturated hydrocarbon ring group wherein the cycloalkyl group may be optionally substituted independently with one or more substituents described herein.
- the cycloalkyl group is 3 to 6 carbon atoms (C 3 -C 6 ).
- cycloalkyl is C 3 -C 4 or C 3 -C 5 .
- the cycloalkyl group, as a monocycle is C 3 -C 6 or C 5 -C 6 .
- the cycloalkyl group, as a bicycle is C 7 -C 12 .
- Examples of monocyclic cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-l-enyl, l-cyclopent-2-enyl, l-cyclopent-3- enyl, cyclohexyl, 1-cyclohex-l-enyl, l-cyclohex-2-enyl, l-cyclohex-3-enyl, cyclohexadienyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl, and cyclododecyl.
- Exemplary arrangements of bicyclic cycloalkyls having 7 to 12 ring atoms include, but are not limited to, [4,4], [4,5], [5,5], [5,6] or [6,6] ring systems.
- Exemplary bridged bicyclic cycloalkyls include, but are not limited to, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, and bicyclo[3.2.2]nonane.
- heterocyclic or “heterocycle” or “heterocyclyl” refers to a saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) cyclic group in which at least one ring atom is a heteroatom independently selected from nitrogen, oxygen, and sulfur, the remaining ring atoms being carbon.
- heterocyclyl includes saturated or partially unsaturated 4-6 membered heterocyclyl groups.
- the heterocyclyl group may be optionally substituted with one or more substituents described herein.
- heterocyclyl groups include, but are not limited to, oxiranyl, aziridinyl, thiiranyl, azetidinyl, oxetanyl, thietanyl, 1,2-dithietanyl, 1,3-dithietanyl, pyrrolidinyl, piperidinyl, dihydropyridinyl, tetrahydropyridinyl, morpholinyl, thiomorpholinyl, thioxanyl, piperazinyl, homopiperazinyl, homopiperidinyl, azepanyl, oxepanyl, thiepanyl, 1,4-oxathianyl, 1,4-dioxepanyl, 1,4-oxathiepanyl, 1,4-oxazepanyl, 1,4- dithiepanyl, 1,4-thiazepanyl and 1,4-diazepane 1 ,
- heteroaryl refers to an aromatic cyclic group in which at least one ring atom is a heteroatom independently selected from nitrogen, oxygen and sulfur, the remaining ring atoms being carbon. Heteroaryl groups may be optionally substituted with one or more substituents described herein. In one example, heteroaryl includes 5-6 membered heteroaryl groups.
- heteroaryl groups include, but are not limited to, pyridinyl, imidazolyl, imidazopyridinyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, 1,2,3 -triazolyl, 1,3,4- triazolyl, l-oxa-2,3-diazolyl, l-oxa-2,4-
- treatment refers to therapeutic, prophylactic, palliative or preventative measures.
- treatment includes therapeutic and palliative treatment.
- beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- Those in need of treatment include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented.
- phrases "therapeutically effective amount” or “effective amount” mean an amount of a compound of the present invention that, when administered to a mammal in need of such treatment, sufficient to (i) treat or prevent the particular disease, condition, or disorder, (ii) attenuate, ameliorate, or eliminate one or more symptoms of the particular disease, condition, or disorder, or (iii) prevent or delay the onset of one or more symptoms of the particular disease, condition, or disorder described herein.
- the amount of a compound that will correspond to such an amount will vary depending upon factors such as the particular compound, disease condition and its severity, the identity (e.g., weight) of the mammal in need of treatment, but can nevertheless be routinely determined by one skilled in the art.
- cancer and “cancerous” refer to or describe the physiological condition in mammals that is typically characterized by abnormal or unregulated cell growth.
- a “tumor” comprises one or more cancerous cells. Examples of cancer include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies.
- cancers include squamous cell cancer (e.g., epithelial squamous cell cancer), lung cancer including small-cell lung cancer, non-small cell lung cancer ("NSCLC”), adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial or uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, as well as head and neck cancer.
- the term cancer may be used generically to include various types of cancer or specifically (as listed above).
- phrases "pharmaceutically acceptable” indicates that the substance or composition is compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- pharmaceutically acceptable salt refers to pharmaceutically acceptable organic or inorganic salts of a compound of the invention.
- the compounds of this invention also include other salts of such compounds which are not necessarily pharmaceutically acceptable salts, and which may be useful as intermediates for preparing and/or purifying compounds of this invention and/or for separating enantiomers of compounds of this invention.
- mammal means a warm-blooded animal that has or is at risk of developing a disease described herein and includes, but is not limited to, guinea pigs, dogs, cats, rats, mice, hamsters, and primates, including humans.
- the present invention provides compounds, and pharmaceutical formulations thereof, that are potentially useful in the treatment of diseases, conditions and/or disorders modulated by B-Raf.
- R 1 and R 2 are independently selected from hydrogen, halogen, CN, C 1 -C 3 alkyl, and C 1 -C 3 alkoxy;
- R 3 is hydrogen, halogen or C 1 -C 3 alkyl
- R 4 is C 3 -C 5 cycloalkyl, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, phenyl, a 5-6 membered heteroaryl, or NR h R', wherein the cycloalkyl, alkyl, alkenyl, alkynyl and phenyl are optionally substituted with OR 0 , halogen, phenyl, C 3 -C 4 cycloalkyl or C 1 -C 4 alkyl optionally substituted with halogen;
- R 5 is hydrogen, phenyl optionally substituted with one to three R a groups, -N(R 0 )- phenyl optionally substituted with R a , -CH 2 -phenyl optionally substituted with one to three R b groups, a 5-6 membered heteroaryl optionally substituted with one to three R e groups, saturated or partially unsaturated C 3 -C 6 cycloalkyl optionally substituted with halogen or C 1 - C 4 alkyl, a 5-6 membered heterocyclyl, or C 1 -C 6 alkyl optionally substituted with one or more R s groups; each R a is independently selected from halogen, CN, a 5-6 membered heterocyclyl, NR°R d , -S(O) 2 R f , -0(C 1 -C 4 alkyl), and C 1 -C 4 alkyl, wherein the alkyl or alkoxy are optionally substituted with halogen
- R e is selected from a 5-6 membered heterocyclyl or NR c R d ;
- R f is selected from C 1 -C 4 alkyl or NR c R d ; each R 8 is independently selected from halogen, CN, OR C , C 3 -C 6 cycloalkyl or NR c R d ; and
- R h and R 1 are independently selected from hydrogen and C 1 -C 5 alkyl optionally substituted with halogen, or
- R h and R 1 together with the nitrogen to which they are attached form a 4 to 6 membered heterocyclic ring.
- R 1 and R 2 are independently selected from hydrogen, halogen, CN, C 1 -C 3 alkyl, and C 1 -C 3 alkoxy;
- R 3 is hydrogen, halogen or C 1 -C 3 alkyl
- R 4 is C 3 -C 5 cycloalkyl, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, or C 2 -C 6 alkynyl, wherein the cycloalkyl, alkyl, alkenyl and alkynyl are optionally substituted with OR C , halogen or C 3 -C 4 cycloalkyl;
- R 5 is hydrogen, phenyl optionally substituted with one to three R a groups, -N(R 0 )- phenyl optionally substituted with R a , -CH 2 -phenyl optionally substituted with one to three R b groups, a 5-6 membered heteroaryl optionally substituted with one to three R e groups, saturated or partially unsaturated C 3 -C 6 cycloalkyl optionally substituted with halogen or C 1 - C 4 alkyl, a 5-6 membered heterocyclyl, or C 1 -C 6 alkyl optionally substituted with one or more R g groups; each R a is independently selected from halogen, CN, a 5-6 membered heterocyclyl, NR°R d , -S(O) 2 R f , -0(C 1 -C 4 alkyl), and C 1 -C 4 alkyl, wherein the alkyl or alkoxy are optionally substituted with halogen
- R e is selected from a 5-6 membered heterocyclyl or NR°R d ;
- R f is selected from C 1 -C 4 alkyl or NR c R d ; and each R 8 is independently selected from halogen, CN, OR C , C 3 -C 6 cycloalkyl or NR c R d .
- R 1 , R 2 and R 3 are independently selected from H, halogen or C 1 -C 3 alkyl
- R 4 is C 3 -C 4 cycloalkyl, or C 1 -C 6 alkyl optionally substituted with OH, halogen or C 3 - C 4 cycloalkyl;
- R 5 is hydrogen, phenyl optionally substituted with one to three R a groups, -NH- phenyl, -CH 2 -phenyl optionally substituted with one to three R b groups, a 5-6 membered heteroaryl optionally substituted with one to three R e groups, C 3 -C 6 cycloalkyl, a 5-6 membered heterocyclyl, or C 1 -C 6 alkyl; each R a is independently selected from halogen, CN, a 5-6 membered heterocyclyl, NR c R d , -S(O) 2 R 1 , -0(C 1 -C 4 alkyl) and C 1 -C 4 alkyl, wherein the alkyl or alkoxy are optionally substituted with halogen; each R b is independently selected from halogen, OH or OCH 3 ; each R c and R d are independently selected from hydrogen or C 1 -C 4 alkyl;
- R e is selected from a 5-6 membered heterocyclyl or NR e R d ;
- R f is selected from C 1 -C 4 alkyl or NR c R d .
- R 1 and R 2 are independently selected from hydrogen, halogen, CN, C 1 -C 3 alkyl or C 1 -C 3 alkoxy.
- R 1 , R 2 and R 3 are independently selected from hydrogen, halogen or C 1 -C 3 alkyl.
- R 1 , R 2 and R 3 are independently selected from hydrogen, F and Cl.
- R 1 is hydrogen, halogen, CN, C 1 -C 3 alkyl or C 1 -C 3 alkoxy.
- R 1 is hydrogen
- R 1 is halogen. In certain embodiments, R 1 is F or Cl.
- R 1 is C 1 -C 3 alkyl. In certain embodiments, R 1 is methyl.
- R 2 is hydrogen, halogen, CN, C 1 -C 3 alkyl or C 1 -C 3 alkoxy.
- R 2 is hydrogen
- R 2 is halogen. In certain embodiments, R 2 is F or Cl. [0059] In certain embodiments, R 2 is C 1 -C 3 alkyl. In certain embodiments, R 2 is methyl.
- R 2 is Cl
- R 2 is hydrogen
- R 3 is hydrogen, halogen or C 1 -C 3 alkyl.
- R 3 is hydrogen
- R 3 is halogen. In certain embodiments, R 3 is F or Cl.
- R 1 and R 2 are F and R 3 is hydrogen.
- R 1 is F and R 2 is Cl and R 3 is hydrogen.
- R 1 is Cl and R 2 is F and R 3 is hydrogen.
- R 1 is F and R 2 and R 3 are hydrogen.
- R and R are hydrogen and R is F.
- R 2 and R 3 are F and R 1 is hydrogen.
- R 1 is Cl and R 2 and R 3 are hydrogen.
- R , R and R are F.
- R 1 is F and R 2 is methyl and R 3 is hydrogen.
- R 1 is methyl and R 2 is F and R 3 is hydrogen.
- R 1 is F and R 2 and R 3 are hydrogen.
- R 1 is Cl and R 2 and R 3 are hydrogen.
- R 2 is F and R 1 and R 3 are hydrogen.
- R 4 is C 3 -C 5 cycloalkyl, C 1 -C 6 alkyl, C 2 -C 6 alkenyl,
- R c is independently selected from hydrogen, phenyl and C 1 -C 4 alkyl optionally substituted with oxo. In certain embodiments, R c is hydrogen. [0081] In certain embodiments, R 4 is C 3 -C 5 cycloalkyl, C 1 -C 6 alkyl, C 2 -C 6 alkenyl,
- R 4 is C 3 -C 5 cycloalkyl, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, or
- R 4 is cyclopropyl, ethyl, propyl, butyl, isobutyl,
- R 4 is cyclopropyl, ethyl, propyl, isobutyl,
- R 4 is propyl, butyl, isobutyl, -CH 2 CH 2 CH 2 F,
- R 4 is C 3 -C 5 cycloalkyl, or C 1 -C 6 alkyl optionally substituted with OH, halogen or C 3 -C 4 cycloalkyl.
- R 4 is C 3 -C 5 cycloalkyl. In certain embodiments, R 4 is
- R 4 is cyclopropyl or cyclobutyl. [0088] In certain embodiments, R 4 is C 3 -C 5 cycloalkyl. In certain embodiments, R 4 is
- R 4 is cyclopropyl
- R 4 is C 1 -C 6 alkyl. In certain embodiments, R 4 is ethyl, propyl, butyl or isobutyl.
- R 4 is C 1 -C 6 alkyl. In certain embodiments, R 4 is propyl, butyl or isobutyl.
- R 4 is C 1 -C 6 alkyl optionally substituted with OR C .
- R c is hydrogen.
- R 4 is C 1 -C 6 alkyl optionally substituted with OH.
- R 4 is -CH 2 CH 2 CH 2 OH.
- R 4 is C 1 -C 6 alkyl optionally substituted with halogen.
- R 4 is -CF 3 , -CH 2 Cl, -CH 2 CF 3 , -CH 2 CH 2 CH 2 F, -CH 2 CH 2 CF 3 , -CF 2 CF 3 or -CF 2 CF 2 CF 3 .
- R 4 is C 1 -C 6 alkyl optionally substituted with halogen.
- R 4 is -CF 3 , -CH 2 CF 3 , -CH 2 CH 2 CH 2 F, -CH 2 CH 2 CF 3 , -CF 2 CF 3 or -CF 2 CF 2 CF 3 . In certain embodiments, R 4 is -CH 2 CH 2 CH 2 F or -CH 2 CH 2 CF 3 . [0094] In certain embodiments, R 4 is C 1 -C 6 alkyl optionally substituted with OR C , halogen or C 3 -C 4 cycloalkyl. In certain embodiments, R 4 is C 1 -C 6 alkyl optionally substituted with OH, halogen or C 3 -C 4 cycloalkyl.
- R 4 is cyclopropylmethyl (- CH ⁇ cyclopropyl) or cyclobutylmethyl (-CHz-cyclobutyl). In certain embodiments, R 4 is cyclopropylmethyl (-CH ⁇ cyclopropyl).
- R 4 is C 1 -C 6 alkyl optionally substituted with phenyl.
- R 4 is phenylmethyl
- R 4 is phenyl optionally substituted with OR C , halogen,
- R 4 is phenyl optionally substituted with halogen. In certain embodiments, R 4 is phenyl optionally substituted with C 1 -C 4 alkyl optionally substituted with halogen. In certain embodiments, R 4 is phenyl optionally substituted with halogen and C 1 -C 4 alkyl optionally substituted with halogen, hi certain embodiments, R 4 is phenyl.
- R 4 is phenyl, 2-fluorophenyl, 3 -fluorophenyl, 4-fluorophenyl, 2,5-difluorophenyl or 4-chloro-3- trifluoromethylphenyl.
- R 4 is a 5-6 membered heteroaryl optionally substituted with OR C , halogen, C 3 -C 4 cycloalkyl or C 1 -C 4 alkyl optionally substituted with halogen. In certain embodiments, R 4 is a 5-6 membered heteroaryl optionally substituted with C 1 -C 4 alkyl. In certain embodiments, R 4 is a 5-6 membered heteroaryl optionally substituted with OR C , halogen, C 3 -C 4 cycloalkyl or C 1 -C 4 alkyl optionally substituted with halogen, wherein the heteroaryl contains one or two heteroatoms selected from the group consisting of oxygen, nitrogen and sulfur.
- R 4 is a 5-6 membered heteroaryl optionally substituted with OR 0 , halogen, C 3 -C 4 cycloalkyl or C 1 -C 4 alkyl optionally substituted with halogen, wherein the heteroaryl is imidazolyl, furanyl, pyridinyl or thiophenyl.
- R 4 is 1 -methyl- lH-imidazol-4-yl, furan-2-yl, pyridin- 2-yl, pyridin-3-yl or thiophen-2-yl.
- R 4 is NR h R ⁇
- R h and R 1 are independently selected from hydrogen and C 1 -C 5 alkyl optionally substituted with halogen.
- R 1 is hydrogen or methyl.
- R h is C 1 -C 5 alkyl optionally substituted with halogen.
- R h is methyl, ethyl, propyl, isopropyl, or 2,2-difluoroethyl.
- R 4 is selected from the group consisting of -NHCH 2 CH 3 , -NHCH 2 CH 2 CH 3 , -N(CH 3 )CH 2 CH 3 , -NHCH(CH 3 ) 2 , -NHCH 2 CHF 2 , and -N(CH 3 ) 2 .
- R 4 is NR 11 R 1 , wherein R h and R' together with the nitrogen to which they are attached form a 4 to 6 membered heterocyclic ring.
- R 4 is NR 11 R 1 , wherein R h and R 1 together with the nitrogen to which they are attached form a 4 to 6 membered heterocyclic ring, wherein the heterocyclic ring contains one or two heteroatoms selected from nitrogen and oxygen.
- R is NR 11 R 1 , wherein R h and R 1 together with the nitrogen to which they are attached form a 5 membered heterocyclic ring.
- R 4 is NR 11 R 1 , wherein R h and R 1 together with the nitrogen to which they are attached form a 5 membered heterocyclic ring, wherein the heterocyclic ring contains one nitrogen heteroatom.
- R 4 is pyrrolidin-1-yl.
- R 1 and R 2 are F, R 3 is hydrogen and R 4 is propyl, such that the compounds of Formula I, have the structure of Formula Ia:
- R 1 is Cl
- R 2 is F
- R 3 is hydrogen
- R 4 is propyl
- R 1 is F
- R 2 is Cl
- R 3 is hydrogen
- R 4 is propyl
- R 5 is hydrogen, phenyl optionally substituted with one to three R a groups, -N(R c )-phenyl optionally substituted with R a , -CH 2 -phenyl optionally substituted with one to three R b groups, a 5-6 membered heteroaryl optionally substituted with one to three R e groups, saturated or partially unsaturated C 3 -C 6 cycloalkyl optionally substituted with halogen or C 1 -C 4 alkyl, a 5-6 membered heterocyclyl, or C 1 -C 6 alkyl optionally substituted with one or more R s groups.
- R 5 is hydrogen, phenyl optionally substituted with one to three R a groups, -NH-phenyl, -CH 2 -phenyl optionally substituted with one to three R b groups, a 5-6 membered heteroaryl optionally substituted with one to three R e groups, C 3 -C 6 cycloalkyl, a 5-6 membered heterocyclyl, or C 1 -C 6 alkyl.
- R 5 is selected from hydrogen, phenyl, 2-fluorophenyl,
- R 5 is phenyl optionally substituted with one to three
- each R a is independently selected from halogen, CN, a 5- 6 membered heterocyclyl, NR c R d , -S(O) 2 R f , -0(C 1 -C 4 alkyl), and C 1 -C 4 alkyl, wherein the alkyl or alkoxy are optionally substituted with halogen.
- R a is a 5-6 membered heterocyclyl, wherein the heterocyclyl is morpholinyl.
- R 5 is selected from phenyl, 2-fluorophenyl, 2-chlorophenyl, 2-bromophenyl, 3 -fluorophenyl, 3- chlorophenyl, 3-bromophenyl, 3-trifluoromethylphenyl, 3-methoxyphenyl, 4-fluorophenyl, A- chlorophenyl, 4-bromophenyl, 4-cyanophenyl, 4-methylphenyl, 4-trifluoromethylphenyl, A- methoxyphenyl, 4-trifluoromethoxyphenyl, 4-dimethylaminophenyl, A-
- R 1 and R 2 are F
- R 3 is hydrogen
- R 4 is propyl
- R 5 is phenyl optionally substituted with one to three R a groups, such that the compounds of Formula I, have the structure of Formula Ib:
- R 5 is phenyl
- R 5 is phenyl optionally substituted with one R a group.
- R a is selected from halogen, CN, a 5-6 membered heterocyclyl, NR c R d , -S(O) 2 R f , -0(C 1 -C 4 alkyl), and C 1 -C 4 alkyl, wherein the alkyl or alkoxy are optionally substituted with halogen.
- R a is -0(C 1 -C 4 alkyl), wherein R a is methoxy.
- R a is -0(C 1 -C 4 alkyl) optionally substituted with halogen, wherein R a is trifluoromethoxy.
- R a is C 1 -C 4 alkyl optionally substituted with halogen, wherein R a is trifluoromethyl.
- R a is a 5-6 membered heterocyclyl, wherein the heterocyclyl is morpholinyl.
- R c and R d are C 1 -C 4 alkyl.
- R c and R d are methyl.
- R a is -S(O) 2 R f .
- R f is C 1 -C 4 alkyl.
- R 5 is selected from 2-fluorophenyl, 2-chlorophenyl, 2-bromophenyl, 3- fluorophenyl, 3-chlorophenyl, 3-bromophenyl, 3-trifluoromethylphenyl, 3-methoxyphenyl, 4- fluorophenyl, 4-chlorophenyl, 4-bromophenyl, 4-cyanophenyl, 4-methylphenyl, 4- trifluoromethylphenyl, 4-methoxyphenyl, 4-trifluoromethoxyphenyl, 4- dimethylaminophenyl, 4-(methylsulfonyl)phenyl and 4-morpholinophenyl.
- R 5 is phenyl optionally substituted with two R a groups.
- R a is selected from halogen and C 1 -C 4 alkyl, wherein the alkyl is optionally substituted with halogen.
- R a is C 1 -C 4 alkyl, wherein R a is methyl.
- R a is C 1 -C 4 alkyl optionally substituted with halogen, wherein R a is trifluoromethyl.
- R is selected from 3,4- dichlorophenyl, 3-trifluoromethyl-4-methylphenyl, 3-trifluoromethyl-4-chlorophenyl and 2- methyl-4-bromophenyl.
- R 5 is -N(R c )-phenyl optionally substituted with R a .
- R c is selected from hydrogen or C 1 -C 4 alkyl. In certain embodiments, R c is hydrogen. In certain embodiments, R 5 is -NH-phenyl optionally substituted with R a .
- R 5 is -NH-phenyl optionally substituted with R a . In certain embodiments, R 5 is -NH-phenyl.
- R 5 is -CH 2 -phenyl optionally substituted with one to three R b groups.
- each R b is independently selected from halogen, OH or OCH 3 .
- R 5 is selected from -CH 2 -4-chlorophenyl, -CH2-3,5- difluorophenyl, -CH 2 -4-methoxyphenyl, -CH 2 -4-bromophenyl and -CH 2 -3,4-difluorophenyl.
- R 5 is a 5-6 membered heteroaryl optionally substituted with one to three R e groups.
- R e is selected from a 5-6 membered heterocyclyl or NR c R d .
- R c and R d are independently selected from hydrogen and C 1 -C 4 alkyl.
- R 5 is a 5-6 membered heteroaryl optionally substituted with a 5-6 membered heterocyclyl, wherein the heteroaryl is pyridinyl.
- R 5 is a 5-6 membered heteroaryl optionally substituted with a 5-6 membered heterocyclyl, wherein the heterocyclyl is morpholinyl.
- R 5 is a 5-6 membered heteroaryl optionally substituted with a 5-6 membered heterocyclyl, wherein the heteroaryl is pyridinyl and the heterocyclyl is morpholinyl.
- R 5 is selected from pyridin-3-yl, pyridin-4-yl, 6-morpholinopyridin-3- yl and 6-(dimethylamino)pyridin-3-yl.
- R 5 is saturated or partially unsaturated C 3 -C 6 cycloalkyl optionally substituted with halogen or C 1 -C 4 alkyl.
- R 5 is saturated or partially unsaturated C 3 -C 6 cycloalkyl.
- R 5 is saturated C 3 -C 6 cycloalkyl.
- R 5 is C 3 -C 6 cycloalkyl, wherein the cycloalkyl is cyclobutyl.
- R 5 is cyclobutyl.
- R 5 is a 5-6 membered heterocyclyl. In certain embodiments, R 5 is a 5-6 membered heterocyclyl, wherein the heterocyclyl is piperdinyl. In certain embodiments, R 5 is selected from piperdin-3-yl and piperdin-4-yl. [00118] In certain embodiments, R 5 is C 1 -C 6 alkyl optionally substituted with one or more R g groups, hi certain embodiments, each R 8 is independently selected from halogen, CN, OR C , C 3 -C 6 cycloalkyl or NR c R d . [00119] In certain embodiments, R 5 is C 1 -C 6 alkyl. In certain embodiments, R 5 is selected from methyl, ethyl, isopropyl and tert-butyl.
- stereoisomers are contemplated and included as the compounds of the invention. Where stereochemistry is specified by a solid wedge or dashed line representing a particular configuration, then that stereoisomer is so specified and defined.
- compounds of Formula I include tautomeric forms. Tautomers are compounds that are interconvertible by tautomerization. This commonly occurs due to the migration of a hydrogen atom or proton, accompanied by the switch of a single bond and adjacent double bond.
- 3H-imidazo[4,5-b]pyridine is one tautomeric form
- 4H-imidazo[4,5-b]pyridine is another tautomeric form
- Other tautomers of Formula I may also form at other positions, including, but not limited to, the sulfonamide or R 5 position depending on the substitution.
- the compounds of Formula I are intended to include all tautomeric forms.
- the compounds of Formula I include the tautomer 4H-imidazo[4,5-b]pyridine, shown as Formula II:
- R 1 , R 2 , R 3 , R 4 and R 5 are as defined herein.
- R 20 is hydrogen, C 1 -C 6 alkyl, benzyl or phenyl and R 1 , R 2 , R 3 , R 4 are as defined herein.
- the compounds of the present invention may exist in unsolvated, as well as solvated forms with pharmaceutically acceptable solvents, such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms.
- Compounds of the present invention may be synthesized by synthetic routes that include processes analogous to those well-known in the chemical arts, particularly in light of the description contained herein.
- the starting materials are generally available from commercial sources such as Sigma-Aldrich (St. Louis, MO), Alfa Aesar (Ward Hill, MA), or
- TCI Portableland, OR
- TCI are readily prepared using methods well known to those skilled in the art (e.g., prepared by methods generally described in Louis F. Fieser and Mary Fieser,
- Schemes 1-4 show a general method for preparing the compounds of the present invention, as well as key intermediates. For a more detailed description of the individual reaction steps, see the Examples section below. Those skilled in the art will appreciate that other synthetic routes may be used to synthesize the inventive compounds. Although specific starting materials and reagents are depicted in the Schemes and discussed below, other starting materials and reagents can be easily substituted to provide a variety of derivatives and/or reaction conditions. In addition, many of the compounds prepared by the methods described below can be further modified in light of this disclosure using conventional chemistry well known to those skilled in the art.
- Scheme 1 shows a general scheme for the synthesis of imidazopyridine 7, wherein R 1 , R 2 , R 3 , R 4 , and R 5 are as defined herein.
- Dinitropyridine 1 can be treated with ammonium hydroxide to provide compound 2, which then can be selectively reduced to compound 3 using ammonium sulfide as the reducing agent.
- Imidazopyridine 4 may be prepared using carboxylic acids in a suitable solvent with various additives, such as triphenylphosphite, under microwave conditions or with a carboxylic acid and a dehydrating agent, such as POCl 3 .
- the nitro group may be reduced by hydrogenation with a suitable catalyst, such as palladium on carbon, tin (II) chloride dehydrate in refluxing methanol, or by zinc or iron in aqueous ammonium chloride, to give compound 5.
- a suitable catalyst such as palladium on carbon, tin (II) chloride dehydrate in refluxing methanol, or by zinc or iron in aqueous ammonium chloride, to give compound 5.
- Compound 5 may be coupled with benzoic acid 6 in the presence of a coupling reagent, such as 2-(1H- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (“HBTU”) or 1-ethyl- (3-dimethylaminopropyl)carbodiimide hydrochloride (“EDCl”), with additives, such as hydroxybenzotriazole monohydrate, in a suitable solvent, such as dichloromethane (“DCM”),
- Scheme 2 shows another preparation of imidazopyridine 7, wherein R , R , R ,
- Imidazopyridine 5 may be coupled with bis-sulfonylated benzoate 8 using Weinreb conditions (trimethylaluminum in toluene) to provide compound 10.
- a suitable base such as aqueous sodium hydroxide, potassium carbonate, or sodium carbonate, and deprotection provides compound 7.
- Scheme 3 illustrates yet another preparation of imidazopyridine 7, wherein R 1 ,
- Imidazopyridine 5 may be coupled with bis- sulfonylated benzoic acid 9 using standard amide coupling conditions, such as those described in Scheme 1, to provide compound 10. Hydrolysis with a suitable base, such as aqueous sodium hydroxide or sodium carbonate, provides compound 7.
- Scheme 4 shows a general method for preparing benzoate 8 and benzoic acid
- Benzoic acid 10 is esterified by standard methods, such as by Fischer esterification conditions.
- the nitro group may be reduced by hydrogenation with a suitable catalyst, such as palladium on carbon.
- Aniline 11 may be sulfonylated with a substituted sulfonyl chloride in the presence of a suitable base, such as triethylamine, to provide benzoate 8.
- Hydrolysis of benzoate 8 with a base, such as aqueous sodium hydroxide, in an optional solvent, such as an alcohol (e.g., methanol), tetrahydrofuran (“THF”)or a mixture thereof, provides benzoic acid 2.
- Another embodiment of the present invention provides a process for preparing compounds of Formula I, comprising: (a) coupling a compound of Formula 5:
- R 5 is as defined herein, with a compound of Formula 6:
- R 1 , R 2 , R 3 and R 4 are as defined herein, to provide a compound of Formula I; or (b) coupling a compound of Formula 5:
- Suitable amino-protecting groups include acetyl, trifluoroacetyl, t-butyloxycarbonyl ("Boc”), benzyloxycarbonyl ("CBz”) and 9- fluorenylmethyleneoxycarbonyl ("Fmoc”).
- Boc t-butyloxycarbonyl
- Fmoc 9- fluorenylmethyleneoxycarbonyl
- reaction products may be advantageous to separate reaction products from one another and/or from starting materials.
- the desired products of each step or series of steps is separated and/or purified (hereinafter separated) to the desired degree of homogeneity by the techniques common in the art.
- separations involve multiphase extraction, crystallization from a solvent or solvent mixture, distillation, sublimation, or chromatography.
- Chromatography can involve any number of methods including, for example: reverse-phase and normal phase; size exclusion; ion exchange; high, medium and low pressure liquid chromatography methods and apparatus; small scale analytical; simulated moving bed (SMB) and preparative thin or thick layer chromatography, as well as techniques of small scale thin layer and flash chromatography.
- SMB simulated moving bed
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods well known to those skilled in the art, such as by chromatography and/or fractional crystallization.
- Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereoisomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride
- Enantiomers can also be separated by use of a chiral HPLC column.
- a single stereoisomer e.g., an enantiomer, substantially free of its stereoisomer may be obtained by resolution of the racemic mixture using a method such as formation of diastereomers using optically active resolving agents (Eliel, E. and Wilen, S. Stereochemistry of Organic Compounds. New York: John Wiley & Sons, Inc., 1994; Lochmuller, C. H., et al. "Chromatographic resolution of enantiomers: Selective review.” J. Chromatogr.. 113(3) (1975): pp. 283-302).
- Racemic mixtures of chiral compounds of the invention can be separated and isolated by any suitable method, including: (1) formation of ionic, diastereomeric salts with chiral compounds and separation by fractional crystallization or other methods, (2) formation of diastereomeric compounds with chiral derivatizing reagents, separation of the diastereomers, and conversion to the pure stereoisomers, and (3) separation of the substantially pure or enriched stereoisomers directly under chiral conditions. See: Wainer, Irving W., Ed. Drug Stereochemistry: Analytical Methods and Pharmacology. New York: Marcel Dekker, Inc., 1993.
- diastereomeric salts can be formed by reaction of enantiomerically pure chiral bases such as brucine, quinine, ephedrine, strychnine, ⁇ -methyl- ⁇ -phenylethylamine (amphetamine), and the like with asymmetric compounds bearing acidic functionality, such as carboxylic acid and sulfonic acid.
- the diastereomeric salts may be induced to separate by fractional crystallization or ionic chromatography.
- the substrate to be resolved is reacted with one enantiomer of a chiral compound to form a diastereomeric pair (Eliel, E. and Wilen, S. Stereochemistry of Organic Compounds. New York: John Wiley & Sons, Inc., 1994, p. 322).
- Diastereomeric compounds can be formed by reacting asymmetric compounds with enantiomerically pure chiral derivatizing reagents, such as menthyl derivatives, followed by separation of the diastereomers and hydrolysis to yield the pure or enriched enantiomer.
- a method of determining optical purity involves making chiral esters, such as a menthyl ester, e.g., (-) menthyl chloroformate in the presence of base, or Mosher ester, ⁇ -methoxy- ⁇ - (trifluoromethyl)phenyl acetate (Jacob III, Peyton. "Resolution of ( ⁇ )-5-Bromonornicotine. Synthesis of (R)- and (S)-Nornicotine of High Enantiomeric Purity.” J. Qrg. Chem. Vol. 47, No. 21 (1982): pp.
- chiral esters such as a menthyl ester, e.g., (-) menthyl chloroformate in the presence of base, or Mosher ester, ⁇ -methoxy- ⁇ - (trifluoromethyl)phenyl acetate (Jacob III, Peyton. "Resolution of ( ⁇ )-5-Bromonornicotine. Synthesis of (R)-
- Stable diastereomers of atropisomeric compounds can be separated and isolated by normal- and reverse-phase chromatography following methods for separation of atropisomeric naphthyl-isoquinolines (WO 96/15111).
- a racemic mixture of two enantiomers can be separated by chromatography using a chiral stationary phase Lough, WJ., Ed. Chiral Liquid Chromatography. New York: Chapman and Hall, 1989; Okamoto, Yoshio, et al. "Optical resolution of dihydropyridine enantiomers by high-performance liquid chromatography using phenylcarbamates of polysaccharides as a chiral stationary phase.” J. of Chromatogr. Vol. 513 (1990): pp. 375-378).
- Enriched or purified enantiomers can be distinguished by methods used to distinguish other chiral molecules with asymmetric carbon atoms, such as optical rotation and circular dichroism. BIOLOGICAL EVALUATION
- B-Raf mutant protein 447-717 (V600E) was co-expressed with the chaperone protein Cdc37, complexed with Hsp90 (Roe, S. Mark, et al. "The Mechanism of Hsp90
- Activity of human recombinant B-Raf protein may be assessed in vitro by assay of the incorporation of radio labeled phosphate to recombinant MAP kinase (MEK), a known physiologic substrate of B-Raf, according to US 2004/0127496 and WO 03/022840.
- MEK MAP kinase
- the activity/inhibition of V600E full-length B-Raf was estimated by measuring the incorporation of radio labeled phosphate from [ ⁇ - 33 P]ATP into FSBA-modified wild-type MEK (see Example A).
- ADMINISTRATION AND PHARMACEUTICAL FORMULATIONS [00143]
- the compounds of the invention may be administered by any convenient route appropriate to the condition to be treated.
- Suitable routes include oral, parenteral (including subcutaneous, intramuscular, intravenous, intraarterial, intradermal, intrathecal and epidural), transdermal, rectal, nasal, topical (including buccal and sublingual), vaginal, intraperitoneal, intrapulmonary and intranasal.
- the compounds may be administered in any convenient administrative form, e.g., tablets, powders, capsules, solutions, dispersions, suspensions, syrups, sprays, suppositories, gels, emulsions, patches, etc.
- Such compositions may contain components conventional in pharmaceutical preparations, e.g. diluents, carriers, pH modifiers, sweeteners, bulking agents, and further active agents. If parenteral administration is desired, the compositions will be sterile and in a solution or suspension form suitable for injection or infusion.
- a typical formulation is prepared by mixing a compound of the present invention and a carrier or excipient.
- Suitable carriers and excipients are well known to those skilled in the art and are described in detail in, e.g., Ansel, Howard C, et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; and Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005.
- the formulations may also include one or more buffers, stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents, diluents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
- buffers stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents, diluents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing
- One embodiment of the present invention includes a pharmaceutical composition comprising a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof.
- the present invention provides a pharmaceutical composition comprising a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof, together with a pharmaceutically acceptable carrier or excipient.
- Another embodiment of the present invention provides a pharmaceutical composition comprising a compound of Formula I for use in the treatment of a hyperproliferative disease.
- Another embodiment of the present invention provides a pharmaceutical composition comprising a compound of Formula I for use in the treatment of cancer.
- METHODS OF TREATMENT WITH COMPOUNDS OF THE INVENTION The invention includes methods of treating or preventing disease or condition by administering one or more compounds of this invention, or a stereoisomer or pharmaceutically acceptable salt thereof.
- a human patient is treated with a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, adjuvant, or vehicle in an amount to detectably inhibit B-Raf activity.
- a human patient is treated with a compound of
- Formula I or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, adjuvant, or vehicle in an amount to detectably inhibit B- Raf activity.
- a method of treating a hyperproliferative disease in a mammal comprising administering a therapeutically effective amount of the compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, to the mammal is provided.
- a method of treating a hyperproliferative disease in a mammal comprising administering a therapeutically effective amount of the compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof, to the mammal is provided.
- kidney disease in another embodiment, a method of treating kidney disease in a mammal comprising administering a therapeutically effective amount of the compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, to the mammal is provided.
- the kidney disease is polycystic kidney disease.
- a method of treating or preventing cancer in a mammal in need of such treatment comprises administering to said mammal a therapeutically effective amount of a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof.
- the cancer is selected from breast, ovary, cervix, prostate, testis, genitourinary tract, esophagus, larynx, glioblastoma, neuroblastoma, stomach, skin, keratoacanthoma, lung, epidermoid carcinoma, large cell carcinoma, NSCLC, small cell carcinoma, lung adenocarcinoma, bone, colon, adenoma, pancreas, adenocarcinoma, thyroid, follicular carcinoma, undifferentiated carcinoma, papillary carcinoma, seminoma, melanoma, sarcoma, bladder carcinoma, liver carcinoma and biliary passages, kidney carcinoma, myeloid disorders, lymphoid disorders, hairy cells, buccal cavity and pharynx (oral), lip, tongue, mouth, pharynx, small intestine, colon-rectum, large intestine, rectum, brain and central nervous system, Hodgkin's and leukemia.
- a method of treating or preventing cancer in a mammal in need of such treatment comprises administering to said mammal a therapeutically effective amount of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof.
- Another embodiment of the present invention provides the use of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of cancer.
- Another embodiment of the present invention provides the use of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of kidney disease.
- the kidney disease is polycystic kidney disease.
- a method of preventing or treating cancer comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, alone or in combination with one or more additional compounds having anti-cancer properties.
- a method of preventing or treating cancer comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof, alone or in combination with one or more additional compounds having anti-cancer properties.
- the cancer is a sarcoma.
- the cancer is a carcinoma.
- the carcinoma is squamous cell carcinoma.
- the carcinoma is an adenoma or adenocarcinoma.
- a method of treating or preventing a disease or disorder modulated by B-Raf comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof.
- diseases and disorders include, but are not limited to, cancer.
- the cancer is selected from breast, ovary, cervix, prostate, testis, genitourinary tract, esophagus, larynx, glioblastoma, neuroblastoma, stomach, skin, keratoacanthoma, lung, epidermoid carcinoma, large cell carcinoma, NSCLC, small cell carcinoma, lung adenocarcinoma, bone, colon, adenoma, pancreas, adenocarcinoma, thyroid, follicular carcinoma, undifferentiated carcinoma, papillary carcinoma, seminoma, melanoma, sarcoma, bladder carcinoma, liver carcinoma and biliary passages, kidney carcinoma, myeloid disorders, lymphoid disorders, hairy cells, buccal cavity and pharynx (oral), lip, tongue, mouth, pharynx, small intestine, colon-rectum, large intestine, rectum, brain and central nervous system, Hodgkin's and leukemia.
- a method of treating or preventing a disease or disorder modulated by B-Raf comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof.
- a method of preventing or treating kidney disease comprising administering to a mammal in need of such treatment an effective amount of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, alone or in combination with one or more additional compounds.
- a method of preventing or treating polycystic kidney disease comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, alone or in combination with one or more additional compounds.
- Another embodiment of the present invention provides the use of a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of cancer.
- the cancer is selected from breast, ovary, cervix, prostate, testis, genitourinary tract, esophagus, larynx, glioblastoma, neuroblastoma, stomach, skin, keratoacanthoma, lung, epidermoid carcinoma, large cell carcinoma, NSCLC, small cell carcinoma, lung adenocarcinoma, bone, colon, adenoma, pancreas, adenocarcinoma, thyroid, follicular carcinoma, undifferentiated carcinoma, papillary carcinoma, seminoma, melanoma, sarcoma, bladder carcinoma, liver carcinoma and biliary passages, kidney carcinoma, myeloid disorders, lymphoid disorders, hairy cells, buccal cavity and pharynx (oral), lip, tongue, mouth,
- Another embodiment of the present invention provides the use of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of cancer.
- Another embodiment of the present invention provides the use of a compound of Formula I, or a stereoisomer, tautomer or pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of polycystic kidney disease.
- the kidney disease is polycystic kidney disease.
- Another embodiment of the present invention provides the compounds of
- Another embodiment of the present invention provides the compounds of
- the hyperproliferative disease is cancer (as further defined and may be individually selected from those above).
- Another embodiment of the present invention provides the compounds of
- kidney disease is polycystic kidney disease.
- the compounds of this invention and stereoisomers and pharmaceutically acceptable salts thereof may be employed alone or in combination with other therapeutic agents for treatment.
- the compounds of the present invention can be used in combination with one or more additional drugs, for example an anti-hyperproliferative, anti-cancer or chemotherapeutic agent.
- the second compound of the pharmaceutical combination formulation or dosing regimen preferably has complementary activities to the compound of this invention such that they do not adversely affect each other.
- agents are suitably present in combination in amounts that are effective for the purpose intended.
- the compounds may be administered together in a unitary pharmaceutical composition or separately and, when administered separately this may occur simultaneously or sequentially in any order. Such sequential administration may be close in time or remote in time.
- a "chemotherapeutic agent” is a chemical compound useful in the treatment of cancer, regardless of mechanism of action.
- Chemotherapeutic agents include compounds used in "targeted therapy” and conventional chemotherapy.
- a number of suitable chemotherapeutic agents to be used as combination therapeutics are contemplated for use in the methods of the present invention.
- the present invention contemplates, but is not limited to, administration of numerous anticancer agents, such as: agents that induce apoptosis; polynucleotides (e.g., ribozymes); polypeptides (e.g., enzymes); drugs; biological mimetics; alkaloids; alkylating agents; antitumor antibiotics; antimetabolites; hormones; platinum compounds; monoclonal antibodies conjugated with anticancer drugs, toxins, and/or radionuclides; biological response modifiers (e.g., interferons [e.g., IFN-a, etc.] and interleukins [e.g., IL-2, etc.], etc.); adoptive immunotherapy agents; hematopoietic growth factors; agents that induce tumor cell differentiation (e.g., all-trans-retinoic acid, etc.); gene therapy reagents; antisense therapy reagents and nucleotides; tumor vaccines; inhibitors of angiogenesis, and the like.
- chemotherapeutic agents include Erlotinib (T ARCEV A®,
- dynemicin including dynemicin A; bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, carminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6- diazo-5-oxo-L-norleucine, ADRIAMYCIN® (doxorubicin), morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, e
- chemotherapeutic agent also included in the definition of "chemotherapeutic agent” are: (i) anti- hormonal agents that act to regulate or inhibit hormone action on tumors such as anti- estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen (including NOLVADEX®; tamoxifen citrate), raloxifene, droloxifene, 4- hydroxytamoxifen, trioxifene, keoxifene, LYl 17018, onapristone, and FARESTON® (toremifine citrate); (ii) aromatase inhibitors that inhibit the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)-imidazoles, aminoglutethimide, MEGASE® (megestrol acetate), AROMASIN® (exemestane; Pfizer), formestanie, fadrozole, RIVISOR® (vorozole), FEMARA®
- chemotherapeutic agent therapeutic antibodies such as alemtuzumab (Campath), bevacizumab (AVASTIN®, Genentech); cetuximab (ERBITUX®, Imclone); panitumumab (VECTIBIX®, Amgen), rituximab (RITUXAN®, Genentech/Biogen pie), pertuzumab (OMNITARG®, 2C4, Genentech), trastuzumab (HERCEPTIN®, Genentech), tositumomab (Bexxar, Corixia), and the antibody drug conjugate, gemtuzumab ozogamicin (MYLOTARG®, Wyeth).
- therapeutic antibodies such as alemtuzumab (Campath), bevacizumab (AVASTIN®, Genentech); cetuximab (ERBITUX®, Imclone); panitumumab (VECTIBIX®, Amgen), rituximab (RIT
- Humanized monoclonal antibodies with therapeutic potential as chemotherapeutic agents in combination with the Raf inhibitors of the invention include: alemtuzumab, apolizumab, aselizumab, atlizumab, bapineuzumab, bevacizumab, bivatuzumab mertansine, cantuzumab mertansine, cedelizumab, certolizumab pegol, cidfusituzumab, cidtuzumab, daclizumab, eculizumab, efalizumab, epratuzumab, erlizumab, felvizumab, fontolizumab, gemtuzumab ozogamicin, inotuzumab ozogamicin, ipilimumab, labetuzumab, lintuzumab, matuzumab, mepolizumab, motavizumab, moto
- Activity of human recombinant B-Raf protein may be assessed in vitro by assay of the incorporation of radio labeled phosphate to recombinant MAP kinase (MEK), a known physiologic substrate of B-Raf, according to US 2004/0127496 and WO 03/022840.
- Catalytically active human recombinant B-Raf protein is obtained by purification from sf9 insect cells infected with a human B-Raf recombinant baculovirus expression vector.
- V600E full-length B-Raf The activity/inhibition of V600E full-length B-Raf was estimated by measuring the incorporation of radio labeled phosphate from [ ⁇ - 33 P]ATP into FSBA-modified wild-type MEK.
- the 30- ⁇ L assay mixtures contained 25mM Na Pipes, pH 7.2, 10OmM KCl, 1OmM MgCl 2 , 5mM ⁇ -glycerophosphate, lOO ⁇ M Na Vanadate, 4 ⁇ M ATP, 500 nCi [ ⁇ - 33 P]ATP, l ⁇ M FSBA-MEK and 2OnM V600E full-length B-Raf. Incubations were carried out at 22°C in a Costar 3365 plate (Corning).
- the B-Raf and FSBA-MEK were preincubated together in assay buffer at 1.5x (20 ⁇ L of 3OnM and 1.5 ⁇ M, respectively) for 15 minutes, and the assay was initiated by the addition of 10 ⁇ L of lO ⁇ M ATP.
- the assay mixtures were quenched by the addition of 100 ⁇ L of 25% TCA, the plate was mixed on a rotary shaker for 1 minute, and the product was captured on a Perkin-Elmer GF/B filter plate using a Tomtec Mach III Harvester. After sealing the bottom of the plate, 35 ⁇ L of Bio-Safe II (Research Products International) scintillation cocktail were added to each well and the plate was top-sealed and counted in a TopcountNXT (Packard).
- Step A A 1 L flask was charged with 2,6-difluoro-3-nitrobenzoic acid (17.0 g, 83.7 mmol) and MeOH (170 mL, 0.5M). The flask was placed in a cold water bath, and an addition funnel charged with a 2M solution of trimethylsilyl (“TMS”) diazomethane in hexanes (209 mL, 419 mmol) was attached to the flask. The TMS diazomethane solution was added slowly to the reaction flask over the course of 2 hours.
- TMS trimethylsilyl
- Step B 10% (wt.) Pd on activated carbon (4.46 g, 4.19 mmol) was added to a
- Step C Propane- 1-sulfonyl chloride (23.46 niL, 209.3 mmol) was slowly added to a solution of methyl 3-amino-2,6-difluorobenzoate (15.66 g, 83.7 mmol) and triethylamine (35.00 mL, 251.1 mmol) in CH 2 Cl 2 (175 mL, 0.5M) maintained in a cool water bath. The reaction mixture was stirred for 1 hour at room temperature.
- the mixture was then partitioned between saturated NaHCO 3 (100 mL) and ethyl acetate (75 mL).
- the aqueous layer was washed with ethyl acetate (50 mL) and then acidified with concentrated HCl to a pH of about 1.
- the acidified aqueous layer was extracted with ethyl acetate (2 X 50 mL), and the combined ethyl acetate extracts were dried (Na 2 SO 4 ), filtered and concentrated.
- 6-fluoro-2-methyl-3- ⁇ S[-(propylsulfonyl)propylsulfonamido)benzoic acid [00190] 6-Fluoro-2-methyl-3-(N-(propylsulfonyl)propylsulfonamido)benzoic acid
- Step A 2-Chloro-6-fluorobenzoic acid (2.00 g, 11.5 mmol) was dissolved in sulfuric acid (20 mL) and cooled to 0°C. Nitric acid (0.529 mL, 12.6 mmol) was added, and the reaction mixture was warmed to room temperature for one hour. The reaction mixture was diluted with water, and the aqueous portion was extracted with DCM (3 X), dried over Na 2 SO 4 , concentrated to a solid, 2-chloro-6-fluoro-3-nitrobenzoic acid (97%), which was used directly in the next step without further purification.
- Step B 2-Chloro-6-fluoro-3-nitrobenzoic acid (0.100 g, 0.455 mmol) and Zn dust (0.298 g, 4.55 mmol) were taken up in tetrahydrofuran (4 mL) and saturated aqueous NH 4 Cl (2 mL) and stirred at room temperature overnight. The reaction mixture was filtered through Celite, concentrated to a solid, and dissolved in water. The pH was adjusted to 2 with IN HCl, and the aqueous portion was extracted with DCM (3 X). The organic portion was dried over Na 2 SO 4 and concentrated to a solid, 3-amino-2-chloro-6-fluorobenzoic acid (49%), which was used directly in the next step without further purification.
- Step C 2-Chloro-6-fluoro-3-(propylsulfonamido)benzoic acid (13%) was prepared according to the general procedure for Example H, substituting 3-amino-2-chloro-6- fluorobenzoic acid for 5-amino-2-fluorobenzoic acid.
- Step A A flame dried flask equipped with a stir bar and rubber septum was charged with 4-chloro-2-fluoroaniline (5.00 g, 34.35 mmol) and dry THF (170 mL). This solution was chilled to -78°C, and n-BuLi (14.7 mL, 1.07 eq. of 2.5M solution in hexanes) was then added over a 15 minute period.
- Benzyl chloroformate (7.40 g, 1.2 eq.) was then added slowly, and the mixture was stirred at -78°C for one hour. The cooling bath was then removed. The mixture was allowed to warm for 30 minutes and then quenched with water (70 mL) and concentrated HCl (25 mL). The mixture was allowed to continue to warm to room temperature. The mixture was then extracted with EtOAc. The extracts were washed twice with a saturated NaHCO 3 solution, once with water, dried over sodium sulfate and concentrated.
- Step B Benzyl 3-amino-6-chloro-2-fluorobenzoate (4.3 g, 15.37 mmol) was dissolved in dry dichloromethane (270 mL). Triethylamine (5.36 mL, 2.5 eq.) was added, and the mixture was chilled to 0°C. Propane- 1-sulfonyl chloride (3.63 mL, 32.3 mmol, 2.1 eq.) was then added via syringe, and a precipitate resulted. Once the addition was complete, the mixture was allowed to warm to room temperature, and the starting material was consumed as determined by TLC (3:1 hexane:ethyl acetate).
- Step A Cyclopropylmethanesulfonyl chloride (1.27 g, 8.20 mmol) was added to a mixture of 3-amino-2,6-difluorobenzoic acid (0.430 g, 2.48 mmol), triethylamine (1.52 mL, 10.9 mmol) and CH 2 Cl 2 (12 mL, 0.2M) cooled to 0 0 C. The reaction mixture was allowed to warm to room temperature and stirred for 1 hour. The mixture was then partitioned between saturated NaHCO 3 (75 mL) and ethyl acetate (50 mL).
- the aqueous layer was washed with ethyl acetate (50 mL) and then acidified to a pH of 1 with concentrated HCl.
- the acidified aqueous layer was extracted twice with ethyl acetate (2 X 50 mL), and the combined ethyl acetate extracts were dried (Na 2 SO 4 ), filtered and concentrated to provide crude 3-(l-cyclopropyl-N-(cyclopropylmethylsulfonyl)methylsulfonamido)-2,6- difluorobenzoic acid (380 mg, 37%).
- Step B A solution of IN NaOH (2.78 mL, 2.78 mmol) was added to a solution of crude 3-(l-cyclopropyl-N-(cyclopropylmethylsulfonyl)methylsulfonamido)-2,6- difluorobenzoic acid (380 mg, 0.928 mmol) in 4:1 THF/MeOH (5 niL, 0.2M). The reaction mixture was stirred at room temperature for 1 hour, after which most of the organic solvents were removed. IN HCl (3 mL) was slowly added to the mixture to acidify to a pH of 1. The acidified aqueous layer was extracted with ethyl acetate (75 mL). The ethyl acetate extract was washed with water (2 X 20 mL), brine (20 mL), dried (Na 2 SO 4 ), filtered and concentrated. Trituration of the residue with Et 2 O afforded 3-
- 2,6-Difluoro-3-(3-fluoropropylsulfonamido)benzoic acid was prepared according to the general procedure for Example C, substituting methyl 2,6-difluoro-3-(N-(3- fluoropropylsulfonyl)-3-fluoropropylsulfonamido)benzoate for methyl 2,6-difluoro-3-(N- (propylsulfonyl)-propylsulfonamido)benzoate.
- 2,6-Difluoro-3-(2-methylpropylsulfonamido)benzoic acid was prepared according to the general procedure for Example C, substituting methyl-2,6-difluoro-3-(N-(2- methylpropylsulfonyl)-2-methylpropylsulfonamido)benzoate for methyl 2,6-difluoro-3-(N- (propylsulfonyl)-propylsulfonamido)benzoate.
- Benzyl 6-chloro-2-fluoro-3-( ' 3-fluoro-N-(3-fluoropropylsulfonyl)propylsulfonamido)benzoate [00209] Benzyl 6-chloro-2-fluoro-3 -(3 -fluoro-N-(3 -fluoropropylsulfonyl)propylsulfon- amido)benzoate (92%) was prepared according to the general procedure for Example K, Step B substituting 3-fluoropropane- 1-sulfonyl chloride for propane- 1-sulfonyl chloride.
- 6-Chloro-2-fluoro-3-(3-fluoropropylsulfonamido)benzoic acid (71%) was prepared according to the general procedure for Example L substituting benzyl 6-chloro-2- fluoro-3-(3-fluoro-N-(3-fluoropropylsulfonyl)propylsulfonamido)benzoate for benzyl 6- chloro-2-fluoro-3-(N-(propylsulfonyl)propylsulfonamido)benzoate.
- 2-Fluoro-3-(3-fluoropropylsulfonamido)benzoic acid (81%) was prepared according to the general procedure for Example M substituting 6-chloro-2-fluoro-3-(3- fluoropropylsulfonamido)benzoic acid for 6-chloro-2-fluoro-3-(propylsulfonamido)benzoic acid.
- Step A 2,5-Difluorobenzoic acid (2.01 g, 9.90 mmol, 31.3% yield) was dissolved in concentrated sulfuric acid (25 mL) and cooled to 0°C. Nitric Acid (1.46 mL, 34.8 mmol) was added, and the reaction mixture was stirred at room temperature for one hour. The solution was extracted with DCM (3 X), and the combined organic layers were dried over sodium sulfate and concentrated. The residue was purified by column chromatography (1:1 hexanes:l%HCOOH/EtOAc) giving 2,5-difluoro-3-nitrobenzoic acid (2.01 g, 31.3%) as a solid.
- Step B 2,5-Difluoro-3-nitrobenzoic acid (2.00 g, 9.847 mmol) was dissolved in MeOH (60 mL). TMSCl (6.220 mL, 49.24 mmol) was added, and the reaction mixture was stirred at reflux for 4 hours. The reaction mixture was concentrated to about 20 mL, and the crystals produced were filtered and dried under high vacuum providing methyl 2,5- difluoro-3-nitrobenzoate (1.55 g, 72.4%) as a crystalline solid.
- Step C Methyl 3-amino-2,5-difluorobenzoate (96.5%) was prepared according to the general procedure for Example B, Step B, substituting methyl 2,5-difluoro- 3-nitrobenzoate for methyl 2,6-difluoro-3-nitrobenzoate.
- Step D Methyl 2,5-difluoro-3-(N-(propylsulfonyl)propylsulfon- amido)benzoate was prepared according to the general procedure for Example B, Step C, substituting methyl 3-amino-2,5-difluorobenzoate for methyl 3-amino-2,6-difluorobenzoate.
- Step E 2,5-Difluoro-3-(propylsulfonamido)benzoic acid (83.8%, two steps) was prepared according to the general procedure for Example C substituting methyl 2,5- difluoro-3-(N-(propylsulfonyl)propylsulfonamido)benzoate for methyl 2,6-difluoro-3-(N- (propylsulfonyl)propylsulfonamido)benzoate.
- Step A 2,2,2-Trifluoroethyl-sulfonyl chloride (459 mL, 4.15 mmol) was slowly added to a solution of methyl 3-amino-2,6-difluorobenzoate (311 g, 1.66 mmol) and pyridine (0.806 mL, 9.97 mmol) in dichloromethane (8.92 mL, 139 mmol), while applying external cooling using an acetone dry ice bath. The reaction mixture was stirred for 45 minutes, and the dry ice bath was removed. The reaction mixture was kept stirring for another hour.
- Step B 2,6-Difluoro-3-(2-trifluoroethylsulfonamido)benzoic acid was prepared according to the general procedure for Example C, substituting methyl 2,6-difluoro- 3-(2-trifluoroethylsulfonamido) benzoate for methyl 2,6-difluoro-3-(N-(propylsulfonyl)- propylsulfonamido)benzoate.
- Step A Methyl 2,6-difluoro-3-(N-(3,3,3-trifluoropropylsulfonyl)-3,3,3- trifluoropropyl-sulfonamido) benzoate was made according to the general procedure for Example B, substituting 3,3,3-trifluoropropyl sulfonyl chloride for propane- 1-sulfonyl chloride.
- Step B 2,6-Difluoro-3-(3,3,3-trifluoropropylsulfonamido)benzoic acid was prepared according to the general procedure for Example C, substituting methyl 2,6-difluoro- 3-(N-(3 ,3 ,3 -trifluoropropylsulfonyl)-3 ,3 ,3 -trifluoropropylsulfonamido)benzoate for methyl 2,6-difluoro-3-(N-(propylsulfonyl)-propylsulfonamido)benzoate.
- Step A Methyl 2,6-difluoro-3-(N-(2-chloromethylsulfonyl)-2-chloromethyl- sulfonamido) benzoate was made according to the general procedure for Example B, substituting 2-chloromethyl sulfonyl chloride for propane- 1-sulfonyl chloride.
- 1 H NMR 400 MHz, d 6 -DMSO
- ⁇ 8.08-7.97 m, IH
- 7.45 t, IH
- 4.55 (s, 2H) 4.02(s, 3H).
- Step B 2,6-Difluoro-3-(2-chloromethylsulfonamido)benzoic acid was prepared according to the general procedure for Example C, substituting methyl 2,6-difluoro- 3-(N-(2-chloromethylsulfonyl)-2-chloromethylsulfonamido)benzoate for methyl 2,6-difluoro- 3-(N-(propylsulfonyl)-propylsulfonamido)benzoate.
- Step A Benzyl 3-amino-2-chloro-6-fluorobenzoate (56%) was prepared according to the general procedure for Example K, substituting 2-chloro-4-fluoroaniline for 4-chloro-2-fluoroaniline.
- Step B Benzyl 3-amino-2-chloro-6-fluorobenzoate (330 mg, 1.2 mmol) was dissolved in dry dichloromethane (11.8 mL). Triethylamine (0.494 mL, 3.54 mmol) was added, and the mixture was chilled to 0°C. Propane- 1-sulfonyl chloride (0.332 mL, 2.95 mmol) was then added via syringe. Once the addition was complete, the mixture was allowed to warm to ambient temperature and stir for 16 hours.
- Step A Benzyl 2-chloro-6-fluoro-3-(N-(propylsulfonyl)propylsulfon- amido)benzoate (413.2 mg, 0.840 mmol) was dissolved in THF (8.4 mL) and 2.0M aqueous LiOH (1.26 mL). The mixture was refluxed for 16 hours and then allowed to cool to ambient temperature. The mixture was acidified to a pH of 0 with l.OM HCl (5.0 mL) and then adjusted to a pH of 4 using saturated sodium bicarbonate. The mixture was extracted with EtOAc (2 X).
- Step A 2,6-Dichloro-3-nitrobenzoic acid (2.13 g, 9.03 mmol) was dissolved in 2:1 THF:saturated aqueous NH 4 Cl and cooled to 0°C. The mixture was treated with zinc (11.8 g, 181 mmol). The reaction mixture was allowed to warm to ambient temperature and stir for 24 hours. The reaction mixture was filtered through GF/F paper while rinsing with THF. The mixture was acidified to a pH of 1 using 1.0M HCl and extracted with 15% 2- propanol:DCM (3 X).
- Step B 3-Amino-2,6-dichlorobenzoic acid (1.40 g, 6.82 mmol) was dissolved in dry dichloromethane (66.7 mL). Triethylamine (4.09 mL, 29.4 mmol) was added, and the mixture was chilled to 0°C. Propane- 1-sulfonyl chloride (2.48 mL, 22 mmol) was then added via syringe. Once the addition was complete, the mixture was allowed to warm to ambient temperature and stir for 1 hour. The mixture was concentrated in vacuo and diluted with diethyl ether.
- Step A 2-Chloro-3,5-dinitropyridine (6.8 g, 33.41 mmol) was taken up in ethanol (200 mL), and ammonium hydroxide solution (19.5 mL, 167 mmol, 5 eq.) was then dripped in slowly, which resulted in a mixture/precipitate. An exotherm was observed, so the reaction mixture was placed in an ice bath for 10 minutes and then removed. After stirring for about 20 minutes, the solids were collected by filtration and dried under vacuum.
- Step C 5-Nitropyridine-2,3-diamine (0.050 g, 0.32 mmol) was dissolved in formic acid (10 mL) and heated to 100°C for 6 hours. The reaction was diluted with water (10 mL) and brought to a pH of 7 with 3N NaOH. The aqueous portion was extracted with 25% isopropyl alcohol (“IPA”)/DCM (6 X), dried over Na 2 SO 4 and concentrated to give 6- nitro-3H-imidazo[4,5-b]pyridine (52 mg, 98%) as a solid.
- IPA isopropyl alcohol
- 6 X isopropyl alcohol
- Step D 6-Nitro-3H-imidazo[4,5-b]pyridine (0.026 g, 0.158 mmol) was taken up as a slurry in EtOH. 10% PdVC (0.00843 g, 0.00792 mmol) was added, and hydrogen gas was bubbled through for 10 minutes. The reaction was stirred under a balloon of hydrogen at room temperature overnight. The reaction was filtered through celite, and the filtrate was concentrated to give 3H-imidazo[4,5-b]pyridin-6-amine as a solid that was used without further purification. [00235] Step E: 3H-Imidazo[4,5-b]pyridin-6-amine (29 mg, 0.22 mmol), 2,6-difluoro-
- Step A 5-Nitropyridine-2,3-diamine (100 mg, 0.65 mmol, see Example 1) and benzoic acid (87 mg, 0.71 mmol, 1.1 eq.) were combined in POCl 3 (5 mL) and heated to reflux for 16 hours. After cooling to room temperature, the mixture was concentrated under reduced pressure, and the resulting residue was taken up in saturated sodium bicarbonate solution, and extracted with 25% IPA/DCM (2 X).
- Step B 2-Phenyl-3H-imidazo[4,5-b]pyridin-6-amine (71%) was prepared according to the general procedure in Example 1, step D, substituting 6-nitro-2-phenyl-3H- imidazo[4,5-b]pyridine for 6-nitro-3H-imidazo[4,5-b]pyridine, and was carried forward to the next step without further purification.
- Step C 2,6-Difluoro-N-(2-phenyl-3H-imidazo[4,5-b]pyridin-6-yl)-3-
- Step D Alternative formation of 2,6-difluoro-N-(2-phenyl-3H-imidazo[4,5- b]pyridin-6-yl)-3-(propylsulfonamido)benzamide: A reaction vial containing 2-phenyl-3H- imidazo[4,5-b]pyridin-6-amine (5.3 mg, 0.026 mmol) was taken up in dry toluene (0.25 mL). Trimethyl aluminum (36 mL, 0.075 mmol, 2M in toluene) was then added by syringe, and the mixture was stirred at room temperature for 20 minutes.
- Step E Alternative formation of 2,6-difluoro-N-(2-phenyl-3H-imidazo[4,5- b]pyridin-6-yl)-3-(propylsulfonamido)benzamide: 2,6-Difluoro-N-(2-phenyl-3H- imidazo[4,5-b]pyridin-6-yl)-3-(N-(propylsulfonyl)propylsulfonamido)benzamide was prepared according to the general procedure in Example 1, Step E, substituting 2-phenyl-3H- imidazo[4,5-b]pyridin-6-amine for 3H-imidazo[4,5-b]pyridin-6-amine and 2,6-difluoro-3-(N- (propylsulfonyl)propylsulfonamido)benzoic acid for 2,6-difluoro-3-
- Step A N,N-Dimethyl-4-(6-nitro-3H-imidazo[4,5-b]pyridin-2-yl)aniline
- Step C N-(2-(4-(Dimethylamino)phenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-
- Step A A scintillation vial was charged with 5-nitropyridine-2,3-diamine (44 mg, 0.285 mmol), phenyl isothiocyanate (250 ⁇ L, 2.09 mmol) and THF (5 mL). The reaction vessel was sealed and heated to 70°C for 30 minutes. The vessel was then cooled to room temperature, and PS-carbodiimide (1.25 mmol/g; 1.20 g, 1.50 mmol) was added. The vessel was re-sealed and heated to 70°C for 24 hours. The reaction mixture was filtered through GF/F paper, and the solids were rinsed with excess CH 2 Cl 2 and MeOH alternately. The filtrate was concentrated in vacuo, and the residue was triturated with 5% MeOH/CH 2 Cl 2 to afford 6-nitro-N-phenyl-3H-imidazo[4,5-b]pyridin-2-amine.
- Step B N2-Phenyl-3H-imidazo[4,5-b]pyridine-2,6-diamine was prepared according to Example 1, Step D, substituting 6-nitro-N-phenyl-3H-imidazo[4,5-b]pyridin-2- amine for 6-nitro-3H-imidazo[4,5-b]pyridine.
- Step C 2,6-Difluoro-N-(2-(phenylamino)-3H-imidazo[4,5-b]pyridin-6-yl)-3-
- Step A A 10-20 mL Biotage Microwave reaction vial was charged with 5- nitropyridine-2,3 -diamine (85 mg, 0.55 mmol, Example 1, Step B) and acetic acid (5 mL). This mixture was subjected to microwave irradiation at 175°C for 75 minutes and then poured into a saturated sodium bicarbonate solution (200 mL). The solution was extracted with 25% IPA/DCM (2 X), and the extracts were dried over sodium sulfate.
- Step B 2-Methyl-3H-imidazo[4,5-b]pyridin-6-amine (87%) was prepared according to the general procedure Example 1, Step D, substituting 2-methyl-6-nitro-3H- imidazo[4,5-b]pyridine for 6-nitro-3H-imidazo[4,5-b]pyridine.
- Step C 2,6-Difluoro-N-(2-methyl-3H-imidazo[4,5-b]pyridin-6-yl)-3-
- Step A A 2-5 mL Biotage Microwave reaction vial was charged with 5- nitropyridine-2,3 -diamine (100 mg, 0.65 mmol), isonicotinic acid (80 mg, 0.65 mmol, 1 eq.), and triphenyl phosphite (204 ⁇ L, 0.78 mmol, 1.2 eq.) in pyridine (3 mL). This mixture was heated in the microwave at 200°C for 15 minutes and then concentrated under reduced pressure.
- Step B 2-(Pyridin-4-yl)-3H-imidazo[4,5-b]pyridin-6-amine (50%) was prepared according to the general procedure in Example 1, Step D (using 1 weight equivalent of 20% Pd(OH) 2 as the catalyst), substituting 6-nitro-2-(pyridin-4-yl)-3H-imidazo[4,5- b]pyridine for 6-Nitro-3H-imidazo[4,5-b]pyridine.
- Step C 2,6-Difluoro-3-(propylsulfonamido)-N-(2-(pyridin-4-yl)-3H- imidazo[4,5-b]pyridin-6-yl)benzamide (35%) was prepared according to the general procedure in Example 1, Step E, substituting 2-(pyridin-4-yl)-3H-imidazo[4,5-b]pyridin-6- amine for 3H-imidazo[4,5-b]pyridin-6-amine.
- Step A 6-Nitro-2-(pyridin-3-yl)-3H-imidazo[4,5-b]pyridine (20%) was prepared according to Example 6, Step A, substituting nicotinic acid for isonicotinic acid.
- Step B 2-(Pyridin-3-yl)-3H-imidazo[4,5-b]pyridin-6-amine (61%) was prepared according to Example 6, Step B, substituting 6-nitro-2-(pyridin-3-yl)-3H- imidazo[4,5-b]pyridine for 6-nitro-2-(pyridin-4-yl)-3H-imidazo[4,5-b]pyridine.
- Step C 2,6-Difluoro-3-(propylsulfonamido)-N-(2-(pyridin-3-yl)-3H- imidazo[4,5-b]pyridin-6-yl)benzarnide (42%) was prepared according to the general procedure in Example 1, Step E, substituting 2-(pyridin-3-yl)-3H-imidazo[4,5-b]pyridin-6- amine for 3H-imidazo[4,5-b]pyridin-6-amine.
- Step A Benzyl 4-(6-nitro-3H-imidazo[4,5-b]pyridin-2-yl)piperidine-l- carboxylate (34%) was prepared according to the general procedure in Example 6, Step 1, substituting l-(benzyloxycarbonyl)piperidine-4-carboxylic acid for isonicotinic acid.
- Step B Benzyl 4-(6-nitro-3H-imidazo[4,5-b]pyridin-2-yl)piperidine-l- carboxylate (80 mg, 0.21 mmol) was dissolved in methanol (2 mL). Tin (II) chloride di- hydrate (237 mg, 1.05 mmol, 5 eq.) was added, and the mixture was warmed to 70°C for 2 hours. The mixture was then allowed to cool to room temperature. The reaction mixture was diluted with EtOAc and washed once with saturated sodium bicarbonate solution (an emulsion formed, which was filtered through GF/F filter paper). The organics were isolated, dried and concentrated under reduced pressure.
- Step C Benzyl 4-(6-(2,6-difluoro-3-(propylsulfonamido)benzamido)-3H- imidazo[4,5-b]pyridin-2-yl)piperidine-l-carboxylate (46%) was prepared according to the general procedure in Example 1, Step E, substituting benzyl 4-(6-amino-3H-imidazo[4,5- b]pyridin-2-yl)piperidine-l-carboxylate for 3H-imidazo[4,5-b]pyridin-6-amine.
- Step D Benzyl 4-(6-(2,6-difluoro-3-(propylsulfonamido)benzamido)-3H- imidazo[4,5-b]pyridin-2-yl)piperidine-l-carboxylate (29 mg, 0.047 mmol) was dissolved in methanol (approximately 1 mL), and 20% palladium hydroxide (30 mg, 1 eq.) was added. The mixture was hydrogenated under a balloon of hydrogen for 1.5 hours.
- Step A Benzyl 3-(6-nitro-3H-imidazo[4,5-b]pyridin-2-yl)piperidine-l- carboxylate (27%) was prepared according to the general procedure in Example 6, Step 1, substituting l-(benzyloxycarbonyl)piperidine-3-carboxylic acid for isonicotinic acid.
- Step B Benzyl 3-(6-amino-3H-imidazo[4,5-b]pyridin-2-yl)piperidine-l- carboxylate (75%) was prepared according to the general procedure in Example 8, Step B, substituting benzyl 3-(6-nitro-3H-imidazo[4,5-b]pyridin-2-yl)piperidine-l-carboxylate for benzyl 4-(6-nitro-3H-imidazo[4,5-b]pyridin-2-yl)piperidine-l-carboxylate.
- Step C Benzyl 3-(6-(2,6-difluoro-3-(propylsulfonamido)benzamido)-3H- imidazo[4,5-b]pyridin-2-yl)piperidine-l-carboxylate (25%) was prepared according to the general procedure in Example 1, Step E, substituting 3-(6-amino-3H-imidazo[4,5-b]pyridin- 2-yl)piperidine-l-carboxylate for 3H-imidazo[4,5-b]pyridin-6-amine.
- m/z (APCI-neg) M-I 611.2
- (APCI-pos) M+l 613.1.
- Step D 2,6-Difluoro-N-(2-(piperidin-3-yl)-3H-imidazo[4,5-b]pyridin-6-yl)-3-
- Step A 2-(4-Chlorophenyl)-6-nitro-3H-imidazo[4,5-b]pyridine (49%) was prepared as in the general procedure in Example 2, Step A, substituting 4-chlorobenzoic acid for benzoic acid.
- Step B 2-(4-Chlorophenyl)-6-nitro-3H-imidazo[4,5-b]pyridine (50 mg, 0.182 mmol) was dissolved in THF (2 mL), and a saturated ammonium chloride solution (2 mL) was added followed by zinc powder (119 mg, 1.82 mmol, 10 eq.). This mixture was vigorously stirred at room temperature for 15 minutes, when TLC indicated complete consumption of starting material. The reaction mixture was then filtered through GF/F filter paper. The filtrate was diluted with water and extracted with EtOAc (with a little methanol, 2 X). The extracts were dried over sodium sulfate and concentrated to 2-(4-chlorophenyl)-3H- imidazo[4,5-b]pyridin-6-amine (41 mg, 92%) as a solid.
- Step C N-(2-(4-Chlorophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step A 2-(4-Methoxyphenyl)-6-nitro-3H-imidazo[4,5-b]pyridine (64%) was prepared as in the general procedure in Example 2, Step A, substituting 4-methoxybenzoic acid for benzoic acid.
- 1 H NMR 400 MHz, DMSOd 6 ) ⁇ 9.19-9.22 (m, IH), 8.72 (br s, IH), 8.21-8.26 (m, 2H), 7.16-7.20 (m, 2H), 3.87 (s, 3H); m/z (APCI-neg) M-I - 269.3.
- Step B 2-(4-Methoxyphenyl)-3H-imidazo[4,5-b]pyridin-6-amine (96%) was prepared as in the general procedure in Example 10, Step B, substituting 2-(4- methoxyphenyl)-6-nitro-3H-imidazo[4,5-b]pyridine for 2-(4-chlorophenyl)-6-nitro-3H- imidazo[4,5-b]pyridine.
- Step C 2,6-Difluoro-N-(2-(4-methoxyphenyl)-3H-imidazo[4,5-b]pyridin-6- yl)-3-(propylsulfonamido)benzamide (22%) was prepared according to the general procedure in Example 1, Step E, substituting 2-(4-methoxyphenyl)-3H-imidazo[4,5-b]pyridin-6-amine for 3H-imidazo[4,5-b]pyridin-6-amine.
- Step A 2-(4-Fluorophenyl)-6-nitro-3H-imidazo[4,5-b]pyridine (30%) was prepared according to the general procedure in Example 2, Step A, substituting 4- fluorobenzoic acid for benzoic acid.
- Step B 2-(4-Fluorophenyl)-3H-imidazo[4,5-b]pyridin-6-amine (59%) was prepared according to Example 10, Step B, substituting 2-(4-fluorophenyl)-6-nitro-3H- imidazo[4,5-b]pyridine for 2-(4-chlorophenyl)-6-nitro-3H-imidazo[4,5-b]pyridine.
- Step C 2,6-Difluoro-N-(2-(4-fluorophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-
- Step B 2-p-Tolyl-3H-imidazo[4,5-b]pyridin-6-amine was prepared according to the general procedure in Example 3, Step B, substituting 6-nitro-2-p-tolyl-3H-imidazo[4,5- b]pyridine for N,N-dimethyl-4-(6-nitro-3H-imidazo[4,5-b]pyridin-2-yl)aniline.
- Step C N-(2-(4-Bromophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step C N-(2-(3-Chlorophenyl)-3H-imidazo[4,5-b]pyridm-6-yl)-2,6-difluoro-
- Step A 6-Nitro-2-(3-(trifluoromethoxy)phenyl)-3H-imidazo[4,5-b]pyridine
- Step C 2,6-Difluoro-3-(propylsulfonamido)-N-(2-(4-)
- Step A 6-Nitro-2-(4-(trifluoromethyl)phenyl)-3H-imidazo[4,5-b]pyridine
- Step C 2,6-Difluoro-3-(propylsulfonamido)-N-(2-(4-)
- Step A 6-Nitro-2-(3-(trifluoromethyl)phenyl)-3H-imidazo[4,5-b]pyridine
- Step B 2-(3-(Trifiuoromethyl)phenyl)-3H-imidazo[4,5-b]pyridin-6-amine was prepared according to the general procedure in Example 3, Step B, substituting 6-nitro-2- (3 -(trifluoromethyl)phenyl)-3 H-imidazo [4,5 -bjpyridine for N,N-dimethyl-4-(6-nitro-3 H- imidazo[4,5-b]pyridin-2-yl)aniline.
- Step C 2,6-Difluoro-N-(2-(2-fluorophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-
- Step C N-(2-(3-Bromophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step C N-(2-(4-Cyanophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step C N-(2-(2-Chlorophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step C N-(2-(4-Chlorobenzyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step C N-(2-(2-Bromophenyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step C N-(2-(4-Bromobenzyl)-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-
- Step C N-(2-tert-Butyl-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-3-
- Step C 2,6-Difluoro-N-(2-isopropyl-3H-imidazo[4,5-b]pyridin-6-yl)-3-
- Step C N-(2-Ethyl-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-3-
- Step C N-(2-Cyclopropyl-3H-imidazo[4,5-b]pyridin-6-yl)-2,6-difluoro-3-
- Step A 4-(4-(6-Nitro-3H-imidazo[4,5-b]pyridin-2-yl)phenyl)morpholine
- Step C 2,6-Difluoro-N-(2-(4-morpholinophenyl)-3H-imidazo[4,5-b]pyridin-
- Step A 2-(4-Bromo-2-methylphenyl)-6-nitro-3H-imidazo[4,5-b]pyridine
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description






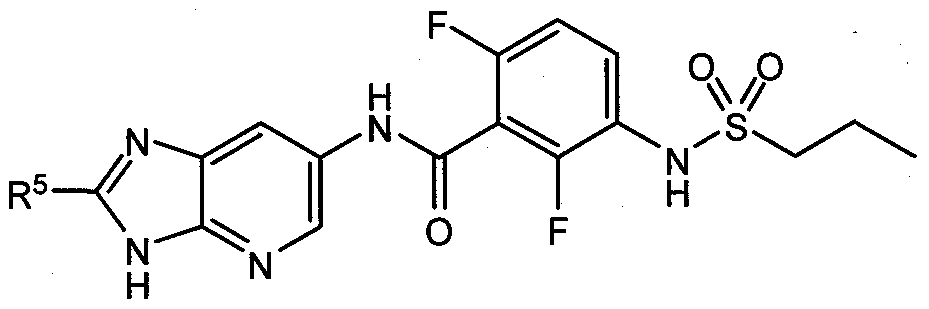













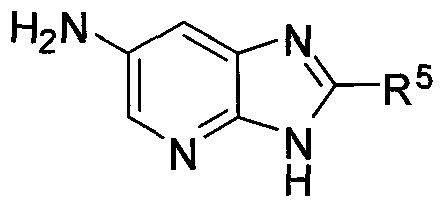






























































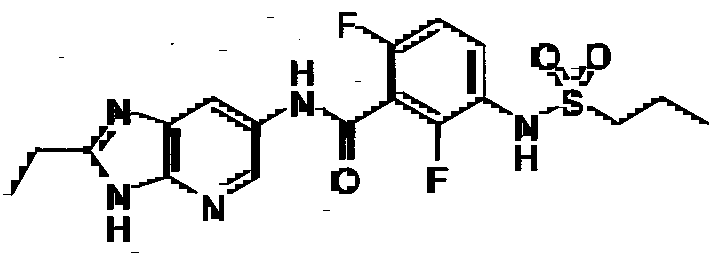
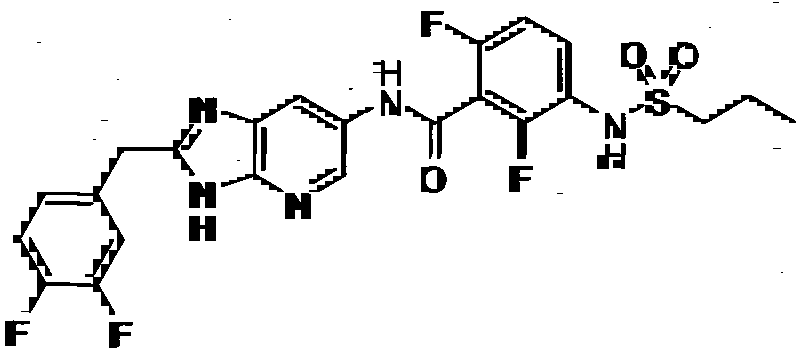

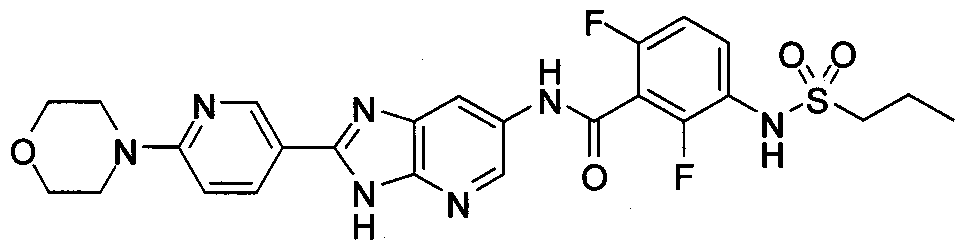





Claims











Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/919,981 US20110003809A1 (en) | 2008-02-29 | 2009-02-27 | Imidazo [4,5-b] pyridine derivatives used as raf inhibitors |
| JP2010548884A JP2011513329A (en) | 2008-02-29 | 2009-02-27 | Imidazo [4,5-b] pyridine derivatives used as RAF inhibitory compounds |
| CA2716947A CA2716947A1 (en) | 2008-02-29 | 2009-02-27 | Imidazo [4,5-b] pyridine derivatives used as raf inhibitors |
| ES09716688T ES2392482T3 (en) | 2008-02-29 | 2009-02-27 | Imidazo [4,5-b] pyridine derivatives used as RAF inhibitors |
| EP09716688A EP2265609B1 (en) | 2008-02-29 | 2009-02-27 | Imdizo [4. 5-b] pyridine derivatives used as raf inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3280108P | 2008-02-29 | 2008-02-29 | |
| US61/032,801 | 2008-02-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009111277A1 true WO2009111277A1 (en) | 2009-09-11 |
| WO2009111277A9 WO2009111277A9 (en) | 2009-12-30 |
Family
ID=40673985
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/035379 WO2009111277A1 (en) | 2008-02-29 | 2009-02-27 | Imdizo [4. 5-b] pyridine derivatives used as raf inhibitors |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20110003809A1 (en) |
| EP (1) | EP2265609B1 (en) |
| JP (1) | JP2011513329A (en) |
| CA (1) | CA2716947A1 (en) |
| ES (1) | ES2392482T3 (en) |
| WO (1) | WO2009111277A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012118492A1 (en) * | 2011-03-01 | 2012-09-07 | Array Biopharma Inc. | Heterocyclic sulfonamides as raf inhibitors |
| CN102659778A (en) * | 2012-03-22 | 2012-09-12 | 盛世泰科生物医药技术(苏州)有限公司 | Novel synthesis technology of 6-amino-2-methyl-3H-imidazo [4, 5-b] pyridine |
| CN102746241A (en) * | 2012-07-02 | 2012-10-24 | 西安交通大学 | 2, 3, 5-trisubstituted benzamide compound, and preparation method and application thereof |
| WO2013043715A1 (en) | 2011-09-19 | 2013-03-28 | Genentech, Inc. | Combination treatments comprising c-met antagonists and b-raf antagonists |
| US20140134133A1 (en) * | 2012-11-14 | 2014-05-15 | Sunshine Lake Pharma Co., Ltd. | Heteroaromatic compounds as pi3 kinase modulators and methods of use |
| US8741920B2 (en) | 2009-08-03 | 2014-06-03 | Hoffmann-La Roche, Inc. | Process for the manufacture of pharmaceutically active compounds |
| WO2014128235A1 (en) | 2013-02-22 | 2014-08-28 | F. Hoffmann-La Roche Ag | Methods of treating cancer and preventing drug resistance |
| WO2014144850A1 (en) | 2013-03-15 | 2014-09-18 | Genentech, Inc. | Methods of treating cancer and preventing cancer drug resistance |
| US9150570B2 (en) | 2012-05-31 | 2015-10-06 | Plexxikon Inc. | Synthesis of heterocyclic compounds |
| US9169250B2 (en) | 2006-11-22 | 2015-10-27 | Plexxikon Inc. | Compounds modulating c-fms and/or c-kit activity and uses therefor |
| WO2015191986A1 (en) | 2014-06-13 | 2015-12-17 | Genentech, Inc. | Methods of treating and preventing cancer drug resistance |
| WO2016011160A1 (en) | 2014-07-15 | 2016-01-21 | Genentech, Inc. | Compositions for treating cancer using pd-1 axis binding antagonists and mek inhibitors |
| US9447089B2 (en) | 2009-04-03 | 2016-09-20 | Plexxikon Inc. | Compositions and uses thereof |
| US9624213B2 (en) | 2011-02-07 | 2017-04-18 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2017087851A1 (en) | 2015-11-19 | 2017-05-26 | Genentech, Inc. | Methods of treating cancer using b-raf inhibitors and immune checkpoint inhibitors |
| US9844539B2 (en) | 2007-07-17 | 2017-12-19 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2018011138A1 (en) | 2016-07-11 | 2018-01-18 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ror1 activity |
| WO2018146253A1 (en) | 2017-02-10 | 2018-08-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of cancers associated with activation of the mapk pathway |
| CN108602821A (en) * | 2015-12-14 | 2018-09-28 | 齐尼思表观遗传学有限公司 | 1H- imidazos [4,5-B] pyridyl group and 2- oxos -2,3- dihydro -1H- imidazos [4,5-B] pyridinyl heterocycle BET bromine structural domain inhibitor |
| WO2019133810A1 (en) | 2017-12-28 | 2019-07-04 | Tract Pharmaceuticals, Inc. | Stem cell culture systems for columnar epithelial stem cells, and uses related thereto |
| US10550113B2 (en) | 2015-02-02 | 2020-02-04 | Kancera Ab | 2-phenyl-3H-imidazo[4,5-B]pyridine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| US10646567B2 (en) | 2011-08-01 | 2020-05-12 | Genentech, Inc. | Methods of treating cancer using PD-1 axis binding antagonists and MEK inhibitors |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| US11087354B2 (en) | 2012-08-17 | 2021-08-10 | Genentech, Inc. | Combination therapies |
| US11660303B2 (en) | 2016-07-11 | 2023-05-30 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| US12123019B2 (en) | 2020-10-15 | 2024-10-22 | Whitehead Institute For Biomedical Research | Uses of kinase inhibitors for inducing and maintaining pluripotency |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2716949A1 (en) * | 2008-02-29 | 2009-09-11 | Array Biopharma Inc. | N- (6-aminopyridin-3-yl) -3- (sulfonamido) benzamide derivatives as b-raf inhibitors for the treatment of cancer |
| AR072657A1 (en) * | 2008-02-29 | 2010-09-15 | Genentech Inc | RAF INHIBITING COMPOUNDS AND METHODS FOR USE |
| PE20091561A1 (en) * | 2008-02-29 | 2009-10-30 | Array Biopharma Inc | RAF INHIBITING COMPOUNDS AND METHODS FOR THEIR USE |
| WO2011057022A1 (en) | 2009-11-06 | 2011-05-12 | Plexxikon, Inc. | Compounds and methods for kinase modulation, and indications therefor |
| TWI558702B (en) | 2011-02-21 | 2016-11-21 | 普雷辛肯公司 | Solid forms of a pharmaceutically active substance |
| MX2014000452A (en) * | 2011-07-15 | 2014-03-21 | Shionogi & Co | Azabenzimidazole derivative having ampk-activating activity. |
| CN103772317B (en) * | 2014-01-06 | 2016-04-06 | 西安山川医药科技有限公司 | 2-methoxyl group-3-replaces sulfonamido-5-(2-acetylaminohydroxyphenylarsonic acid 6-benzothiazolyl) benzamide compound and preparation method thereof and purposes |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4328155A (en) * | 1977-01-27 | 1982-05-04 | Shionogi & Co., Ltd. | Meta-sulfonamido-benzamides |
| WO2006040039A1 (en) * | 2004-10-13 | 2006-04-20 | Merck Patent Gmbh | N,n'-dithenylurea derivatives used in the form of kinase inhibitors |
| WO2006042599A1 (en) * | 2004-10-13 | 2006-04-27 | Merck Patent Gmbh | Phenylurea derivatives used as inhibitors of tyrosinkinase for treating tumors |
| WO2006066913A2 (en) * | 2004-12-23 | 2006-06-29 | F. Hoffmann-La Roche Ag | Benzamide substituted imidazo- and pyrolo-pyridines as protein kinase inhibitors |
| WO2007017143A1 (en) * | 2005-07-29 | 2007-02-15 | F. Hoffmann-La Roche Ag | Azabenzimidazole derivatives, their manufacture and use as anti-cancer agents |
| WO2009012283A1 (en) * | 2007-07-17 | 2009-01-22 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
Family Cites Families (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7928239B2 (en) * | 1999-01-13 | 2011-04-19 | Bayer Healthcare Llc | Inhibition of RAF kinase using quinolyl, isoquinolyl or pyridyl ureas |
| CA2359244C (en) * | 1999-01-13 | 2013-10-08 | Bayer Corporation | .omega.-carboxy aryl substituted diphenyl ureas as p38 kinase inhibitors |
| US8124630B2 (en) * | 1999-01-13 | 2012-02-28 | Bayer Healthcare Llc | ω-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| EP1140840B1 (en) * | 1999-01-13 | 2006-03-22 | Bayer Pharmaceuticals Corp. | -g(v)-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| ME00275B (en) * | 1999-01-13 | 2011-02-10 | Bayer Corp | ?-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US7235576B1 (en) * | 2001-01-12 | 2007-06-26 | Bayer Pharmaceuticals Corporation | Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US20030207914A1 (en) * | 2001-04-20 | 2003-11-06 | Bayer Corporation | Inhibition of raf kinase using quinolyl, isoquinolyl or pyridyl ureas |
| US7071216B2 (en) * | 2002-03-29 | 2006-07-04 | Chiron Corporation | Substituted benz-azoles and methods of their use as inhibitors of Raf kinase |
| US7135575B2 (en) * | 2003-03-03 | 2006-11-14 | Array Biopharma, Inc. | P38 inhibitors and methods of use thereof |
| MXPA05013349A (en) * | 2003-06-13 | 2006-03-09 | Novartis Ag | 2-aminopyrimidine derivatives as raf kinase inhibitors. |
| US7691870B2 (en) * | 2003-07-11 | 2010-04-06 | Merck Patent Gmbh | Benzimidazole carboxamides as raf kinase inhibitors |
| ATE538787T1 (en) * | 2003-07-11 | 2012-01-15 | Merck Patent Gmbh | BENZIMIDAZOLE DERIVATIVES AS RAF-KINASE INHIBITORS |
| DE10337942A1 (en) * | 2003-08-18 | 2005-03-17 | Merck Patent Gmbh | aminobenzimidazole derivatives |
| DE10344223A1 (en) * | 2003-09-24 | 2005-04-21 | Merck Patent Gmbh | New 2-anilino-1,3-benzoxazole derivatives, are inhibitors of kinases, especially tyrosine- or Raf-kinases, useful e.g. for treating solid tumors, angiogenesis, diabetic retinopathy, inflammation or psoriasis |
| JP4758349B2 (en) * | 2003-10-08 | 2011-08-24 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as protein kinase inhibitors |
| US7691866B2 (en) * | 2003-10-16 | 2010-04-06 | Novartis Vaccines And Diagnostics, Inc. | 2,6-disubstituted quinazolines, quinoxalines, quinolines and isoquinolines and methods of their use as inhibitors of Raf kinase |
| ATE435015T1 (en) * | 2003-10-16 | 2009-07-15 | Novartis Vaccines & Diagnostic | SUBSTITUTED BENZAZOLES AND THEIR USE AS RAF-KINASE INHIBITORS |
| EP1696920B8 (en) * | 2003-12-19 | 2015-05-06 | Plexxikon Inc. | Compounds and methods for development of ret modulators |
| DE102004009238A1 (en) * | 2004-02-26 | 2005-09-08 | Merck Patent Gmbh | New aryl amide compounds are kinase inhibitors useful for the treatment and/or prophylaxis of e.g. tumors, psoriasis, rheumatoid arthritis, contact dermatitis, inflammations, endometriosis, scar and benign prostatic hyperplasia |
| TW200616974A (en) * | 2004-07-01 | 2006-06-01 | Astrazeneca Ab | Chemical compounds |
| BRPI0514691A (en) * | 2004-08-31 | 2008-06-17 | Astrazeneca Ab | compound or a pharmaceutically acceptable salt thereof, process for preparing it, pharmaceutical composition, and use of a compound or pharmaceutically acceptable salt thereof |
| CN101010303A (en) * | 2004-09-01 | 2007-08-01 | 阿斯利康(瑞典)有限公司 | Quinazolinone derivatives and their use as B-RAF inhibitors |
| US20070054916A1 (en) * | 2004-10-01 | 2007-03-08 | Amgen Inc. | Aryl nitrogen-containing bicyclic compounds and methods of use |
| AU2005293384A1 (en) * | 2004-10-15 | 2006-04-20 | Astrazeneca Ab | Quinoxalines as B Raf inhibitors |
| GB0423554D0 (en) * | 2004-10-22 | 2004-11-24 | Cancer Rec Tech Ltd | Therapeutic compounds |
| JP5197016B2 (en) * | 2004-12-23 | 2013-05-15 | デシファラ ファーマスーティカルズ, エルエルシー | Enzyme modulators and therapy |
| WO2006079791A1 (en) * | 2005-01-25 | 2006-08-03 | Astrazeneca Ab | Chemical compounds |
| US20090105250A1 (en) * | 2005-01-26 | 2009-04-23 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| TW200639163A (en) * | 2005-02-04 | 2006-11-16 | Genentech Inc | RAF inhibitor compounds and methods |
| BRPI0606281A2 (en) * | 2005-03-17 | 2009-06-09 | Novartis Ag | organic compounds |
| RU2411242C2 (en) * | 2005-05-13 | 2011-02-10 | Айрм, Ллк. | Compounds and compositions as proteinkinase inhibitors |
| NZ563444A (en) * | 2005-05-17 | 2011-04-29 | Plexxikon Inc | Pyrrolo(2,3-b)pyridine derivatives as protein kinase inhibitors |
| EP1902056A2 (en) * | 2005-05-20 | 2008-03-26 | Array Biopharma, Inc. | Raf inhibitor compounds and methods of use thereof |
| RU2008108898A (en) * | 2005-08-09 | 2009-09-20 | Айрм Ллк (Bm) | COMPOUNDS AND COMPOSITIONS AS PROTEINKINASE INHIBITORS |
| WO2007027855A2 (en) * | 2005-09-01 | 2007-03-08 | Array Biopharma Inc. | Raf inhibitor compounds and methods of use thereof |
| JP4908511B2 (en) * | 2005-09-15 | 2012-04-04 | エフ.ホフマン−ラ ロシュ アーゲー | 4-Amino-thieno [3,2-c] pyridine-7-carboxylic acid derivative |
| WO2007056625A2 (en) * | 2005-11-04 | 2007-05-18 | Smithkline Beecham Corporation | Thienopyridine b-raf kinase inhibitors |
| RU2008127486A (en) * | 2005-12-08 | 2010-01-20 | Милленниум Фармасьютикалз, Инк. (Us) | BICYCLIC COMPOUNDS WITH INHIBITOR ACTIVITY AGAINST KINASE |
| EP1966159A2 (en) * | 2005-12-22 | 2008-09-10 | AstraZeneca AB | Chemical compounds |
| TW200804349A (en) * | 2005-12-23 | 2008-01-16 | Kalypsys Inc | Novel substituted pyrimidinyloxy ureas as inhibitors of protein kinases |
| WO2007076474A1 (en) * | 2005-12-23 | 2007-07-05 | Kalypsys, Inc. | Novel substituted pyridinyloxy and pyrimidinyloxy amides useful as inhibitors of protein kinases |
| CN101415689A (en) * | 2006-04-05 | 2009-04-22 | 阿斯利康(瑞典)有限公司 | Substituted quinazolines with anti-cancer activity |
| WO2007113558A2 (en) * | 2006-04-05 | 2007-10-11 | Astrazeneca Ab | Quinazolinone derivatives having b-raf inhibitory activity |
| US20090149484A1 (en) * | 2006-04-18 | 2009-06-11 | Astrazeneca Ab | Quinazolin-4-one derivatives, process for their preparation and pharmaceutical compositions containing them |
| WO2008028617A1 (en) * | 2006-09-06 | 2008-03-13 | F. Hoffmann-La Roche Ag | Heteroaryl derivatives as protein kinase inhibitors |
| US7897762B2 (en) * | 2006-09-14 | 2011-03-01 | Deciphera Pharmaceuticals, Llc | Kinase inhibitors useful for the treatment of proliferative diseases |
| US8188113B2 (en) * | 2006-09-14 | 2012-05-29 | Deciphera Pharmaceuticals, Inc. | Dihydropyridopyrimidinyl, dihydronaphthyidinyl and related compounds useful as kinase inhibitors for the treatment of proliferative diseases |
| MX2009003456A (en) * | 2006-10-02 | 2009-04-14 | Irm Llc | Compounds and compositions as protein kinase inhibitors. |
| EP1914234A1 (en) * | 2006-10-16 | 2008-04-23 | GPC Biotech Inc. | Pyrido[2,3-d]pyrimidines and their use as kinase inhibitors |
| WO2008079909A1 (en) * | 2006-12-21 | 2008-07-03 | Plexxikon, Inc. | Pyrrolo [2,3-b] pyridines as kinase modulators |
| PE20121126A1 (en) * | 2006-12-21 | 2012-08-24 | Plexxikon Inc | PIRROLO [2,3-B] PYRIDINES COMPOUNDS AS KINASE MODULATORS |
| US7872018B2 (en) * | 2006-12-21 | 2011-01-18 | Plexxikon, Inc. | Compounds and methods for kinase modulation, and indications therefor |
| CN101707863A (en) * | 2007-06-15 | 2010-05-12 | Irm责任有限公司 | protein kinase inhibitors and methods for using thereof |
| WO2008157575A1 (en) * | 2007-06-21 | 2008-12-24 | Irm Llc | Protein kinase inhibitors and methods for using thereof |
| WO2009025358A1 (en) * | 2007-08-23 | 2009-02-26 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| EP2184285B1 (en) * | 2007-08-29 | 2015-11-04 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| CA2716949A1 (en) * | 2008-02-29 | 2009-09-11 | Array Biopharma Inc. | N- (6-aminopyridin-3-yl) -3- (sulfonamido) benzamide derivatives as b-raf inhibitors for the treatment of cancer |
| AR072657A1 (en) * | 2008-02-29 | 2010-09-15 | Genentech Inc | RAF INHIBITING COMPOUNDS AND METHODS FOR USE |
| PE20091561A1 (en) * | 2008-02-29 | 2009-10-30 | Array Biopharma Inc | RAF INHIBITING COMPOUNDS AND METHODS FOR THEIR USE |
| HUE032873T2 (en) * | 2008-03-17 | 2017-11-28 | Ambit Biosciences Corp | 1-(3-(6,7-dimethoxyquinazolin-4-yloxy)phenyl)-3-(5-(1,1,1-trifluoro-2-methylpropan-2-yl)isoxazol-3-yl)urea as raf kinase modulator in the treatment of cancer diseases |
| GB0807609D0 (en) * | 2008-04-25 | 2008-06-04 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| UA103319C2 (en) * | 2008-05-06 | 2013-10-10 | Глаксосмитклайн Ллк | Thiazole- and oxazole-benzene sulfonamide compounds |
| WO2009143018A2 (en) * | 2008-05-19 | 2009-11-26 | Plexxikon, Inc. | Compounds and methods for kinase modulation, and indications therefor |
| PE20091846A1 (en) * | 2008-05-19 | 2009-12-16 | Plexxikon Inc | PIRROLO [2,3-d] -PYRIMIDINE DERIVATIVES AS KINE MODULATORS |
| US8110576B2 (en) * | 2008-06-10 | 2012-02-07 | Plexxikon Inc. | Substituted pyrrolo[2,3b]pyrazines and methods for treatment of raf protein kinase-mediated indications |
| CN102112478A (en) * | 2008-06-10 | 2011-06-29 | 普莱希科公司 | 5H-pyrr0l0 [2, 3-B] pyrazine derivatives for kinase modulation, and indications therefor |
| KR101061599B1 (en) * | 2008-12-05 | 2011-09-02 | 한국과학기술연구원 | Novel indazole derivatives that are protein kinase inhibitors for the treatment of abnormal cell growth diseases, pharmaceutically acceptable salts thereof, and pharmaceutical compositions containing the same as active ingredients |
-
2009
- 2009-02-27 US US12/919,981 patent/US20110003809A1/en not_active Abandoned
- 2009-02-27 JP JP2010548884A patent/JP2011513329A/en not_active Withdrawn
- 2009-02-27 WO PCT/US2009/035379 patent/WO2009111277A1/en active Application Filing
- 2009-02-27 ES ES09716688T patent/ES2392482T3/en active Active
- 2009-02-27 CA CA2716947A patent/CA2716947A1/en not_active Abandoned
- 2009-02-27 EP EP09716688A patent/EP2265609B1/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4328155A (en) * | 1977-01-27 | 1982-05-04 | Shionogi & Co., Ltd. | Meta-sulfonamido-benzamides |
| WO2006040039A1 (en) * | 2004-10-13 | 2006-04-20 | Merck Patent Gmbh | N,n'-dithenylurea derivatives used in the form of kinase inhibitors |
| WO2006042599A1 (en) * | 2004-10-13 | 2006-04-27 | Merck Patent Gmbh | Phenylurea derivatives used as inhibitors of tyrosinkinase for treating tumors |
| WO2006066913A2 (en) * | 2004-12-23 | 2006-06-29 | F. Hoffmann-La Roche Ag | Benzamide substituted imidazo- and pyrolo-pyridines as protein kinase inhibitors |
| WO2007017143A1 (en) * | 2005-07-29 | 2007-02-15 | F. Hoffmann-La Roche Ag | Azabenzimidazole derivatives, their manufacture and use as anti-cancer agents |
| WO2009012283A1 (en) * | 2007-07-17 | 2009-01-22 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9169250B2 (en) | 2006-11-22 | 2015-10-27 | Plexxikon Inc. | Compounds modulating c-fms and/or c-kit activity and uses therefor |
| US10426760B2 (en) | 2007-07-17 | 2019-10-01 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| US9844539B2 (en) | 2007-07-17 | 2017-12-19 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| US9663517B2 (en) | 2009-04-03 | 2017-05-30 | Plexxikon Inc. | Compositions and uses thereof |
| US9447089B2 (en) | 2009-04-03 | 2016-09-20 | Plexxikon Inc. | Compositions and uses thereof |
| US8741920B2 (en) | 2009-08-03 | 2014-06-03 | Hoffmann-La Roche, Inc. | Process for the manufacture of pharmaceutically active compounds |
| US12076322B2 (en) | 2011-02-07 | 2024-09-03 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| US11337976B2 (en) | 2011-02-07 | 2022-05-24 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| US9624213B2 (en) | 2011-02-07 | 2017-04-18 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2012118492A1 (en) * | 2011-03-01 | 2012-09-07 | Array Biopharma Inc. | Heterocyclic sulfonamides as raf inhibitors |
| US10646567B2 (en) | 2011-08-01 | 2020-05-12 | Genentech, Inc. | Methods of treating cancer using PD-1 axis binding antagonists and MEK inhibitors |
| WO2013043715A1 (en) | 2011-09-19 | 2013-03-28 | Genentech, Inc. | Combination treatments comprising c-met antagonists and b-raf antagonists |
| CN102659778A (en) * | 2012-03-22 | 2012-09-12 | 盛世泰科生物医药技术(苏州)有限公司 | Novel synthesis technology of 6-amino-2-methyl-3H-imidazo [4, 5-b] pyridine |
| US9695169B2 (en) | 2012-05-31 | 2017-07-04 | Plexxikon Inc. | Synthesis of heterocyclic compounds |
| US9150570B2 (en) | 2012-05-31 | 2015-10-06 | Plexxikon Inc. | Synthesis of heterocyclic compounds |
| CN102746241A (en) * | 2012-07-02 | 2012-10-24 | 西安交通大学 | 2, 3, 5-trisubstituted benzamide compound, and preparation method and application thereof |
| US11087354B2 (en) | 2012-08-17 | 2021-08-10 | Genentech, Inc. | Combination therapies |
| US11783366B2 (en) | 2012-08-17 | 2023-10-10 | Genentech, Inc. | Combination therapies |
| US20140134133A1 (en) * | 2012-11-14 | 2014-05-15 | Sunshine Lake Pharma Co., Ltd. | Heteroaromatic compounds as pi3 kinase modulators and methods of use |
| US9926324B2 (en) * | 2012-11-14 | 2018-03-27 | Calitor Sciences, Llc | Heteroaromatic compounds as PI3 kinase modulators and methods of use |
| WO2014128235A1 (en) | 2013-02-22 | 2014-08-28 | F. Hoffmann-La Roche Ag | Methods of treating cancer and preventing drug resistance |
| WO2014144850A1 (en) | 2013-03-15 | 2014-09-18 | Genentech, Inc. | Methods of treating cancer and preventing cancer drug resistance |
| WO2015191986A1 (en) | 2014-06-13 | 2015-12-17 | Genentech, Inc. | Methods of treating and preventing cancer drug resistance |
| US10946093B2 (en) | 2014-07-15 | 2021-03-16 | Genentech, Inc. | Methods of treating cancer using PD-1 axis binding antagonists and MEK inhibitors |
| EP3563870A1 (en) | 2014-07-15 | 2019-11-06 | F. Hoffmann-La Roche AG | Methods of treating cancer using pd-1 axis binding antagonists and mek inhibitors |
| WO2016011160A1 (en) | 2014-07-15 | 2016-01-21 | Genentech, Inc. | Compositions for treating cancer using pd-1 axis binding antagonists and mek inhibitors |
| US10550113B2 (en) | 2015-02-02 | 2020-02-04 | Kancera Ab | 2-phenyl-3H-imidazo[4,5-B]pyridine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| WO2017087851A1 (en) | 2015-11-19 | 2017-05-26 | Genentech, Inc. | Methods of treating cancer using b-raf inhibitors and immune checkpoint inhibitors |
| CN108602821A (en) * | 2015-12-14 | 2018-09-28 | 齐尼思表观遗传学有限公司 | 1H- imidazos [4,5-B] pyridyl group and 2- oxos -2,3- dihydro -1H- imidazos [4,5-B] pyridinyl heterocycle BET bromine structural domain inhibitor |
| CN108602821B (en) * | 2015-12-14 | 2021-06-29 | 恒元生物医药科技(苏州)有限公司 | 1H-imidazo [4,5-B ] pyridinyl BET bromodomain inhibitors |
| WO2018011138A1 (en) | 2016-07-11 | 2018-01-18 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ror1 activity |
| US11008318B2 (en) | 2016-07-11 | 2021-05-18 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| US11660303B2 (en) | 2016-07-11 | 2023-05-30 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| WO2018146253A1 (en) | 2017-02-10 | 2018-08-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of cancers associated with activation of the mapk pathway |
| WO2019133810A1 (en) | 2017-12-28 | 2019-07-04 | Tract Pharmaceuticals, Inc. | Stem cell culture systems for columnar epithelial stem cells, and uses related thereto |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| US12123019B2 (en) | 2020-10-15 | 2024-10-22 | Whitehead Institute For Biomedical Research | Uses of kinase inhibitors for inducing and maintaining pluripotency |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009111277A9 (en) | 2009-12-30 |
| EP2265609B1 (en) | 2012-09-05 |
| ES2392482T3 (en) | 2012-12-11 |
| JP2011513329A (en) | 2011-04-28 |
| EP2265609A1 (en) | 2010-12-29 |
| US20110003809A1 (en) | 2011-01-06 |
| CA2716947A1 (en) | 2009-09-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2265609B1 (en) | Imdizo [4. 5-b] pyridine derivatives used as raf inhibitors | |
| EP2265610B1 (en) | Pyrazole [3, 4-b]pyridine raf inhibitors | |
| US8338452B2 (en) | Raf inhibitor compounds and methods of use thereof | |
| US20110003859A1 (en) | N- (6-aminopyridin-3-yl) -3- (sulfonamido) benzamide derivatives as b-raf inhibitors for the treatment of cancer | |
| US20130018033A1 (en) | Raf inhibitor compounds and methods of use thereof | |
| US20120157451A1 (en) | Raf inhibitor compounds and methods of use thereof | |
| US20120157439A1 (en) | Raf inhibitor compounds and methods of use thereof | |
| WO2011025968A1 (en) | 1h-pyrazolo [ 3, 4-b] pyridine compounds for inhibiting raf kinase | |
| US20120214811A1 (en) | Raf inhibitor compounds and methods of use thereof | |
| US20120157453A1 (en) | Raf inhibitor compounds and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09716688 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2716947 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12919981 Country of ref document: US Ref document number: 2010548884 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 6727/DELNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009716688 Country of ref document: EP |