WO2009095479A2 - Prodrug comprising a self-cleavable linker - Google Patents
Prodrug comprising a self-cleavable linker Download PDFInfo
- Publication number
- WO2009095479A2 WO2009095479A2 PCT/EP2009/051079 EP2009051079W WO2009095479A2 WO 2009095479 A2 WO2009095479 A2 WO 2009095479A2 EP 2009051079 W EP2009051079 W EP 2009051079W WO 2009095479 A2 WO2009095479 A2 WO 2009095479A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- prodrug
- group
- mmol
- poly
- alkyl
- Prior art date
Links
- 229940002612 prodrug Drugs 0.000 title claims abstract description 74
- 239000000651 prodrug Substances 0.000 title claims abstract description 74
- 239000003814 drug Substances 0.000 claims abstract description 49
- 125000005647 linker group Chemical group 0.000 claims abstract description 49
- 229940079593 drug Drugs 0.000 claims abstract description 44
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 29
- 150000003839 salts Chemical class 0.000 claims abstract description 20
- 239000001257 hydrogen Substances 0.000 claims abstract description 17
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 16
- 125000001424 substituent group Chemical group 0.000 claims abstract description 13
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 12
- 239000000126 substance Substances 0.000 claims abstract description 10
- 125000006850 spacer group Chemical group 0.000 claims abstract description 6
- -1 antitrypsins Proteins 0.000 claims description 116
- 125000000217 alkyl group Chemical group 0.000 claims description 55
- 238000000034 method Methods 0.000 claims description 31
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 25
- 125000003342 alkenyl group Chemical group 0.000 claims description 22
- 125000000304 alkynyl group Chemical group 0.000 claims description 22
- 125000000623 heterocyclic group Chemical group 0.000 claims description 21
- 229910052757 nitrogen Inorganic materials 0.000 claims description 21
- 229960001519 exenatide Drugs 0.000 claims description 20
- 108010011459 Exenatide Proteins 0.000 claims description 19
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical group C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 claims description 18
- 102000004169 proteins and genes Human genes 0.000 claims description 18
- 108090000623 proteins and genes Proteins 0.000 claims description 18
- 101001032756 Rattus norvegicus Granzyme-like protein 1 Proteins 0.000 claims description 14
- 239000000017 hydrogel Substances 0.000 claims description 14
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 13
- 239000012634 fragment Substances 0.000 claims description 11
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 11
- 229910052736 halogen Inorganic materials 0.000 claims description 10
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 8
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 7
- 102000004877 Insulin Human genes 0.000 claims description 7
- 108090001061 Insulin Proteins 0.000 claims description 7
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 claims description 7
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 claims description 7
- 229920001223 polyethylene glycol Polymers 0.000 claims description 7
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 7
- 150000003384 small molecules Chemical class 0.000 claims description 7
- 108010039627 Aprotinin Proteins 0.000 claims description 6
- 108010024976 Asparaginase Proteins 0.000 claims description 6
- 229920002472 Starch Polymers 0.000 claims description 6
- 108060008682 Tumor Necrosis Factor Proteins 0.000 claims description 6
- 229960002519 amoxapine Drugs 0.000 claims description 6
- 239000012867 bioactive agent Substances 0.000 claims description 6
- 229920001222 biopolymer Polymers 0.000 claims description 6
- 210000002381 plasma Anatomy 0.000 claims description 6
- 229920001184 polypeptide Polymers 0.000 claims description 6
- 239000008107 starch Substances 0.000 claims description 6
- 235000019698 starch Nutrition 0.000 claims description 6
- 102000003390 tumor necrosis factor Human genes 0.000 claims description 6
- 229960005486 vaccine Drugs 0.000 claims description 6
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 claims description 5
- 239000013543 active substance Substances 0.000 claims description 5
- 125000004429 atom Chemical group 0.000 claims description 5
- 239000003795 chemical substances by application Substances 0.000 claims description 5
- 150000002367 halogens Chemical group 0.000 claims description 5
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 claims description 5
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 claims description 5
- 125000001624 naphthyl group Chemical group 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- 239000002243 precursor Substances 0.000 claims description 5
- 108020003175 receptors Proteins 0.000 claims description 5
- 102000005962 receptors Human genes 0.000 claims description 5
- 125000005329 tetralinyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 claims description 5
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 4
- 102000018233 Fibroblast Growth Factor Human genes 0.000 claims description 4
- 108050007372 Fibroblast Growth Factor Proteins 0.000 claims description 4
- 102000003972 Fibroblast growth factor 7 Human genes 0.000 claims description 4
- 108090000385 Fibroblast growth factor 7 Proteins 0.000 claims description 4
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 claims description 4
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 claims description 4
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 claims description 4
- 102000002812 Heat-Shock Proteins Human genes 0.000 claims description 4
- 108010004889 Heat-Shock Proteins Proteins 0.000 claims description 4
- RPTUSVTUFVMDQK-UHFFFAOYSA-N Hidralazin Chemical compound C1=CC=C2C(NN)=NN=CC2=C1 RPTUSVTUFVMDQK-UHFFFAOYSA-N 0.000 claims description 4
- 108060003951 Immunoglobulin Proteins 0.000 claims description 4
- XUIIKFGFIJCVMT-LBPRGKRZSA-N L-thyroxine Chemical compound IC1=CC(C[C@H]([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-LBPRGKRZSA-N 0.000 claims description 4
- 102100031538 Phosphatidylcholine-sterol acyltransferase Human genes 0.000 claims description 4
- 102000019197 Superoxide Dismutase Human genes 0.000 claims description 4
- 108010012715 Superoxide dismutase Proteins 0.000 claims description 4
- 102000011923 Thyrotropin Human genes 0.000 claims description 4
- 108010061174 Thyrotropin Proteins 0.000 claims description 4
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 claims description 4
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 claims description 4
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 claims description 4
- 229940024142 alpha 1-antitrypsin Drugs 0.000 claims description 4
- QWGDMFLQWFTERH-UHFFFAOYSA-N amoxapine Chemical compound C12=CC(Cl)=CC=C2OC2=CC=CC=C2N=C1N1CCNCC1 QWGDMFLQWFTERH-UHFFFAOYSA-N 0.000 claims description 4
- BLFLLBZGZJTVJG-UHFFFAOYSA-N benzocaine Chemical compound CCOC(=O)C1=CC=C(N)C=C1 BLFLLBZGZJTVJG-UHFFFAOYSA-N 0.000 claims description 4
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 claims description 4
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 4
- WHBIGIKBNXZKFE-UHFFFAOYSA-N delavirdine Chemical compound CC(C)NC1=CC=CN=C1N1CCN(C(=O)C=2NC3=CC=C(NS(C)(=O)=O)C=C3C=2)CC1 WHBIGIKBNXZKFE-UHFFFAOYSA-N 0.000 claims description 4
- 229960001042 dexmethylphenidate Drugs 0.000 claims description 4
- DUGOZIWVEXMGBE-CHWSQXEVSA-N dexmethylphenidate Chemical compound C([C@@H]1[C@H](C(=O)OC)C=2C=CC=CC=2)CCCN1 DUGOZIWVEXMGBE-CHWSQXEVSA-N 0.000 claims description 4
- AEUTYOVWOVBAKS-UWVGGRQHSA-N ethambutol Chemical compound CC[C@@H](CO)NCCN[C@@H](CC)CO AEUTYOVWOVBAKS-UWVGGRQHSA-N 0.000 claims description 4
- 229940126864 fibroblast growth factor Drugs 0.000 claims description 4
- 108020001507 fusion proteins Proteins 0.000 claims description 4
- 102000037865 fusion proteins Human genes 0.000 claims description 4
- BGHSOEHUOOAYMY-JTZMCQEISA-N ghrelin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)CN)C1=CC=CC=C1 BGHSOEHUOOAYMY-JTZMCQEISA-N 0.000 claims description 4
- 108010085742 growth hormone-releasing peptide-2 Proteins 0.000 claims description 4
- 102000018358 immunoglobulin Human genes 0.000 claims description 4
- 229950008325 levothyroxine Drugs 0.000 claims description 4
- 239000003446 ligand Substances 0.000 claims description 4
- RHCSKNNOAZULRK-UHFFFAOYSA-N mescaline Chemical compound COC1=CC(CCN)=CC(OC)=C1OC RHCSKNNOAZULRK-UHFFFAOYSA-N 0.000 claims description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 4
- 229920000642 polymer Polymers 0.000 claims description 4
- 229940044601 receptor agonist Drugs 0.000 claims description 4
- 239000000018 receptor agonist Substances 0.000 claims description 4
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 claims description 4
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 claims description 4
- 229960002073 sertraline Drugs 0.000 claims description 4
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 claims description 4
- 229960000187 tissue plasminogen activator Drugs 0.000 claims description 4
- 108010010803 Gelatin Proteins 0.000 claims description 3
- 108060003199 Glucagon Proteins 0.000 claims description 3
- 108010088406 Glucagon-Like Peptides Proteins 0.000 claims description 3
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 claims description 3
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 claims description 3
- 108091034117 Oligonucleotide Proteins 0.000 claims description 3
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 claims description 3
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 claims description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims description 3
- 230000003197 catalytic effect Effects 0.000 claims description 3
- 229920002678 cellulose Polymers 0.000 claims description 3
- 239000001913 cellulose Substances 0.000 claims description 3
- 229920001577 copolymer Polymers 0.000 claims description 3
- 238000005516 engineering process Methods 0.000 claims description 3
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 claims description 3
- 239000008273 gelatin Substances 0.000 claims description 3
- 229920000159 gelatin Polymers 0.000 claims description 3
- 235000019322 gelatine Nutrition 0.000 claims description 3
- 235000011852 gelatine desserts Nutrition 0.000 claims description 3
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 claims description 3
- 229960004666 glucagon Drugs 0.000 claims description 3
- 239000000137 peptide hydrolase inhibitor Substances 0.000 claims description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 3
- 229960005356 urokinase Drugs 0.000 claims description 3
- MYPYJXKWCTUITO-UHFFFAOYSA-N vancomycin Natural products O1C(C(=C2)Cl)=CC=C2C(O)C(C(NC(C2=CC(O)=CC(O)=C2C=2C(O)=CC=C3C=2)C(O)=O)=O)NC(=O)C3NC(=O)C2NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CC(C)C)NC)C(O)C(C=C3Cl)=CC=C3OC3=CC2=CC1=C3OC1OC(CO)C(O)C(O)C1OC1CC(C)(N)C(O)C(C)O1 MYPYJXKWCTUITO-UHFFFAOYSA-N 0.000 claims description 3
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 claims description 2
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 claims description 2
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 claims description 2
- HMJIYCCIJYRONP-UHFFFAOYSA-N (+-)-Isradipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)C1C1=CC=CC2=NON=C12 HMJIYCCIJYRONP-UHFFFAOYSA-N 0.000 claims description 2
- XWTYSIMOBUGWOL-UHFFFAOYSA-N (+-)-Terbutaline Chemical compound CC(C)(C)NCC(O)C1=CC(O)=CC(O)=C1 XWTYSIMOBUGWOL-UHFFFAOYSA-N 0.000 claims description 2
- XEEQGYMUWCZPDN-DOMZBBRYSA-N (-)-(11S,2'R)-erythro-mefloquine Chemical compound C([C@@H]1[C@@H](O)C=2C3=CC=CC(=C3N=C(C=2)C(F)(F)F)C(F)(F)F)CCCN1 XEEQGYMUWCZPDN-DOMZBBRYSA-N 0.000 claims description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 claims description 2
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 claims description 2
- IGLYMJRIWWIQQE-QUOODJBBSA-N (1S,2R)-2-phenylcyclopropan-1-amine (1R,2S)-2-phenylcyclopropan-1-amine Chemical compound N[C@H]1C[C@@H]1C1=CC=CC=C1.N[C@@H]1C[C@H]1C1=CC=CC=C1 IGLYMJRIWWIQQE-QUOODJBBSA-N 0.000 claims description 2
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 claims description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 claims description 2
- YWPHCCPCQOJSGZ-LLVKDONJSA-N (2r)-2-[(2-ethoxyphenoxy)methyl]morpholine Chemical compound CCOC1=CC=CC=C1OC[C@@H]1OCCNC1 YWPHCCPCQOJSGZ-LLVKDONJSA-N 0.000 claims description 2
- BUJAGSGYPOAWEI-SECBINFHSA-N (2r)-2-amino-n-(2,6-dimethylphenyl)propanamide Chemical compound C[C@@H](N)C(=O)NC1=C(C)C=CC=C1C BUJAGSGYPOAWEI-SECBINFHSA-N 0.000 claims description 2
- YKFCISHFRZHKHY-NGQGLHOPSA-N (2s)-2-amino-3-(3,4-dihydroxyphenyl)-2-methylpropanoic acid;trihydrate Chemical compound O.O.O.OC(=O)[C@](N)(C)CC1=CC=C(O)C(O)=C1.OC(=O)[C@](N)(C)CC1=CC=C(O)C(O)=C1 YKFCISHFRZHKHY-NGQGLHOPSA-N 0.000 claims description 2
- OOBHFESNSZDWIU-GXSJLCMTSA-N (2s,3s)-3-methyl-2-phenylmorpholine Chemical compound C[C@@H]1NCCO[C@H]1C1=CC=CC=C1 OOBHFESNSZDWIU-GXSJLCMTSA-N 0.000 claims description 2
- DDYAPMZTJAYBOF-ZMYDTDHYSA-N (3S)-4-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-amino-1-[[(2S,3R)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-4-amino-1-[[(2S)-1-[[(2S)-4-amino-1-[[(2S)-4-amino-1-[[(2S,3S)-1-[[(1S)-1-carboxyethyl]amino]-3-methyl-1-oxopentan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-4-methylsulfanyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S,3R)-2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]-3-hydroxybutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]amino]-5-oxopentanoyl]amino]-4-oxobutanoic acid Chemical class [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O DDYAPMZTJAYBOF-ZMYDTDHYSA-N 0.000 claims description 2
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 claims description 2
- VQHRZZISQVWPLK-UIRGBLDSSA-N (7s,9s)-7-[(2r,4s,5s,6s)-5-[(2s,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-4-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@@H](O)C[C@H](O[C@@H]2C3=C(O)C=4C(=O)C5=CC=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)O[C@H]1C VQHRZZISQVWPLK-UIRGBLDSSA-N 0.000 claims description 2
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 claims description 2
- 229930182837 (R)-adrenaline Natural products 0.000 claims description 2
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 claims description 2
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 claims description 2
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 claims description 2
- XUBOMFCQGDBHNK-JTQLQIEISA-N (S)-gatifloxacin Chemical compound FC1=CC(C(C(C(O)=O)=CN2C3CC3)=O)=C2C(OC)=C1N1CCN[C@@H](C)C1 XUBOMFCQGDBHNK-JTQLQIEISA-N 0.000 claims description 2
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 claims description 2
- INGWEZCOABYORO-UHFFFAOYSA-N 2-(furan-2-yl)-7-methyl-1h-1,8-naphthyridin-4-one Chemical compound N=1C2=NC(C)=CC=C2C(O)=CC=1C1=CC=CO1 INGWEZCOABYORO-UHFFFAOYSA-N 0.000 claims description 2
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 claims description 2
- SHXWCVYOXRDMCX-UHFFFAOYSA-N 3,4-methylenedioxymethamphetamine Chemical compound CNC(C)CC1=CC=C2OCOC2=C1 SHXWCVYOXRDMCX-UHFFFAOYSA-N 0.000 claims description 2
- FERWCFYKULABCE-UHFFFAOYSA-N 3-(2-aminoethoxymethyl)-2,5,9-trimethylfuro[3,2-g]chromen-7-one Chemical compound O1C(=O)C=C(C)C2=C1C(C)=C1OC(C)=C(COCCN)C1=C2 FERWCFYKULABCE-UHFFFAOYSA-N 0.000 claims description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 claims description 2
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 claims description 2
- LSLYOANBFKQKPT-DIFFPNOSSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-hydroxyphenyl)propan-2-yl]amino]ethyl]benzene-1,3-diol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(O)C=C(O)C=1)C1=CC=C(O)C=C1 LSLYOANBFKQKPT-DIFFPNOSSA-N 0.000 claims description 2
- AWFDCTXCTHGORH-HGHGUNKESA-N 6-[4-[(6ar,9r,10ar)-5-bromo-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline-9-carbonyl]piperazin-1-yl]-1-methylpyridin-2-one Chemical compound O=C([C@H]1CN([C@H]2[C@@H](C=3C=CC=C4NC(Br)=C(C=34)C2)C1)C)N(CC1)CCN1C1=CC=CC(=O)N1C AWFDCTXCTHGORH-HGHGUNKESA-N 0.000 claims description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 claims description 2
- WUWFMDMBOJLQIV-UHFFFAOYSA-N 7-(3-aminopyrrolidin-1-yl)-1-(2,4-difluorophenyl)-6-fluoro-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid Chemical compound C1C(N)CCN1C(C(=C1)F)=NC2=C1C(=O)C(C(O)=O)=CN2C1=CC=C(F)C=C1F WUWFMDMBOJLQIV-UHFFFAOYSA-N 0.000 claims description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 2
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 claims description 2
- 102000055025 Adenosine deaminases Human genes 0.000 claims description 2
- 229920000936 Agarose Polymers 0.000 claims description 2
- 108010088751 Albumins Proteins 0.000 claims description 2
- 102000009027 Albumins Human genes 0.000 claims description 2
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 claims description 2
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 claims description 2
- 108010001779 Ancrod Proteins 0.000 claims description 2
- 108010058207 Anistreplase Proteins 0.000 claims description 2
- 102000004411 Antithrombin III Human genes 0.000 claims description 2
- 108090000935 Antithrombin III Proteins 0.000 claims description 2
- QTGIAADRBBLJGA-UHFFFAOYSA-N Articaine Chemical compound CCCNC(C)C(=O)NC=1C(C)=CSC=1C(=O)OC QTGIAADRBBLJGA-UHFFFAOYSA-N 0.000 claims description 2
- 102000015790 Asparaginase Human genes 0.000 claims description 2
- 101800001288 Atrial natriuretic factor Proteins 0.000 claims description 2
- LTKOVYBBGBGKTA-SFHVURJKSA-N Avizafone Chemical compound NCCCC[C@H](N)C(=O)NCC(=O)N(C)C1=CC=C(Cl)C=C1C(=O)C1=CC=CC=C1 LTKOVYBBGBGKTA-SFHVURJKSA-N 0.000 claims description 2
- KPYSYYIEGFHWSV-UHFFFAOYSA-N Baclofen Chemical compound OC(=O)CC(CN)C1=CC=C(Cl)C=C1 KPYSYYIEGFHWSV-UHFFFAOYSA-N 0.000 claims description 2
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 claims description 2
- 102100026189 Beta-galactosidase Human genes 0.000 claims description 2
- 108010006654 Bleomycin Proteins 0.000 claims description 2
- 102000004506 Blood Proteins Human genes 0.000 claims description 2
- 108010017384 Blood Proteins Proteins 0.000 claims description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 2
- YNXLOPYTAAFMTN-SBUIBGKBSA-N C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 Chemical compound C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 YNXLOPYTAAFMTN-SBUIBGKBSA-N 0.000 claims description 2
- ABFNTRQPWNXUHA-VEVJRHMJSA-N C([C@H]1C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CC=2C=CC=CC=2)C(=O)N(CC(N)=O)CCCC(=O)NCCCC(=O)N1)=O)[C@H](O)C)C1=CC=CC=C1 Chemical group C([C@H]1C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CC=2C=CC=CC=2)C(=O)N(CC(N)=O)CCCC(=O)NCCCC(=O)N1)=O)[C@H](O)C)C1=CC=CC=C1 ABFNTRQPWNXUHA-VEVJRHMJSA-N 0.000 claims description 2
- 102000000905 Cadherin Human genes 0.000 claims description 2
- 108050007957 Cadherin Proteins 0.000 claims description 2
- 229920001661 Chitosan Polymers 0.000 claims description 2
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 claims description 2
- 102100022641 Coagulation factor IX Human genes 0.000 claims description 2
- 102100023804 Coagulation factor VII Human genes 0.000 claims description 2
- 102000008186 Collagen Human genes 0.000 claims description 2
- 108010035532 Collagen Proteins 0.000 claims description 2
- 102000029816 Collagenase Human genes 0.000 claims description 2
- 108060005980 Collagenase Proteins 0.000 claims description 2
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 claims description 2
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 claims description 2
- 108010053770 Deoxyribonucleases Proteins 0.000 claims description 2
- 102000016911 Deoxyribonucleases Human genes 0.000 claims description 2
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 claims description 2
- 229920002307 Dextran Polymers 0.000 claims description 2
- 108010061435 Enalapril Proteins 0.000 claims description 2
- 108010049140 Endorphins Proteins 0.000 claims description 2
- 102000009025 Endorphins Human genes 0.000 claims description 2
- XRHVZWWRFMCBAZ-UHFFFAOYSA-L Endothal-disodium Chemical compound [Na+].[Na+].C1CC2C(C([O-])=O)C(C(=O)[O-])C1O2 XRHVZWWRFMCBAZ-UHFFFAOYSA-L 0.000 claims description 2
- 108010032976 Enfuvirtide Proteins 0.000 claims description 2
- 108010092674 Enkephalins Proteins 0.000 claims description 2
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 claims description 2
- 229940122601 Esterase inhibitor Drugs 0.000 claims description 2
- 108010076282 Factor IX Proteins 0.000 claims description 2
- 108010023321 Factor VII Proteins 0.000 claims description 2
- 108010054218 Factor VIII Proteins 0.000 claims description 2
- 102000001690 Factor VIII Human genes 0.000 claims description 2
- 108010088842 Fibrinolysin Proteins 0.000 claims description 2
- DJBNUMBKLMJRSA-UHFFFAOYSA-N Flecainide Chemical compound FC(F)(F)COC1=CC=C(OCC(F)(F)F)C(C(=O)NCC2NCCCC2)=C1 DJBNUMBKLMJRSA-UHFFFAOYSA-N 0.000 claims description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 claims description 2
- 102000002464 Galactosidases Human genes 0.000 claims description 2
- 108010093031 Galactosidases Proteins 0.000 claims description 2
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 claims description 2
- 229930182566 Gentamicin Natural products 0.000 claims description 2
- 101800001586 Ghrelin Proteins 0.000 claims description 2
- 102400000442 Ghrelin-28 Human genes 0.000 claims description 2
- 108010017544 Glucosylceramidase Proteins 0.000 claims description 2
- 102000004547 Glucosylceramidase Human genes 0.000 claims description 2
- 108010086677 Gonadotropins Proteins 0.000 claims description 2
- 102000006771 Gonadotropins Human genes 0.000 claims description 2
- 108010026389 Gramicidin Proteins 0.000 claims description 2
- AIJTTZAVMXIJGM-UHFFFAOYSA-N Grepafloxacin Chemical compound C1CNC(C)CN1C(C(=C1C)F)=CC2=C1C(=O)C(C(O)=O)=CN2C1CC1 AIJTTZAVMXIJGM-UHFFFAOYSA-N 0.000 claims description 2
- 102000018997 Growth Hormone Human genes 0.000 claims description 2
- 108010051696 Growth Hormone Proteins 0.000 claims description 2
- 102000001554 Hemoglobins Human genes 0.000 claims description 2
- 108010054147 Hemoglobins Proteins 0.000 claims description 2
- DKLKMKYDWHYZTD-UHFFFAOYSA-N Hexylcaine Chemical compound C=1C=CC=CC=1C(=O)OC(C)CNC1CCCCC1 DKLKMKYDWHYZTD-UHFFFAOYSA-N 0.000 claims description 2
- 108010007267 Hirudins Proteins 0.000 claims description 2
- 102000007625 Hirudins Human genes 0.000 claims description 2
- 102000001974 Hyaluronidases Human genes 0.000 claims description 2
- 108050009363 Hyaluronidases Proteins 0.000 claims description 2
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 claims description 2
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 claims description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 claims description 2
- 102000014150 Interferons Human genes 0.000 claims description 2
- 108010050904 Interferons Proteins 0.000 claims description 2
- 102000015696 Interleukins Human genes 0.000 claims description 2
- 108010063738 Interleukins Proteins 0.000 claims description 2
- JUZNIMUFDBIJCM-ANEDZVCMSA-N Invanz Chemical compound O=C([C@H]1NC[C@H](C1)SC=1[C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)NC1=CC=CC(C(O)=O)=C1 JUZNIMUFDBIJCM-ANEDZVCMSA-N 0.000 claims description 2
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 claims description 2
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 claims description 2
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 claims description 2
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 claims description 2
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 claims description 2
- XAGMUUZPGZWTRP-ZETCQYMHSA-N LSM-5745 Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1C1(N)CC1 XAGMUUZPGZWTRP-ZETCQYMHSA-N 0.000 claims description 2
- 108010059881 Lactase Proteins 0.000 claims description 2
- 108090001090 Lectins Proteins 0.000 claims description 2
- 102000004856 Lectins Human genes 0.000 claims description 2
- 108010092277 Leptin Proteins 0.000 claims description 2
- 102000016267 Leptin Human genes 0.000 claims description 2
- URLZCHNOLZSCCA-VABKMULXSA-N Leu-enkephalin Chemical class C([C@@H](C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=CC=C1 URLZCHNOLZSCCA-VABKMULXSA-N 0.000 claims description 2
- 108010000817 Leuprolide Proteins 0.000 claims description 2
- 108010007859 Lisinopril Proteins 0.000 claims description 2
- 102000009151 Luteinizing Hormone Human genes 0.000 claims description 2
- 108010073521 Luteinizing Hormone Proteins 0.000 claims description 2
- 229920000057 Mannan Polymers 0.000 claims description 2
- PKVZBNCYEICAQP-UHFFFAOYSA-N Mecamylamine hydrochloride Chemical compound Cl.C1CC2C(C)(C)C(NC)(C)C1C2 PKVZBNCYEICAQP-UHFFFAOYSA-N 0.000 claims description 2
- 101710151321 Melanostatin Proteins 0.000 claims description 2
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 claims description 2
- 108020001621 Natriuretic Peptide Proteins 0.000 claims description 2
- 102000004571 Natriuretic peptide Human genes 0.000 claims description 2
- 206010028980 Neoplasm Diseases 0.000 claims description 2
- 102400000064 Neuropeptide Y Human genes 0.000 claims description 2
- PHVGLTMQBUFIQQ-UHFFFAOYSA-N Nortryptiline Chemical compound C1CC2=CC=CC=C2C(=CCCNC)C2=CC=CC=C21 PHVGLTMQBUFIQQ-UHFFFAOYSA-N 0.000 claims description 2
- 108010016076 Octreotide Proteins 0.000 claims description 2
- 108010035766 P-Selectin Proteins 0.000 claims description 2
- 108700036316 PTR 3173 Proteins 0.000 claims description 2
- 102000018886 Pancreatic Polypeptide Human genes 0.000 claims description 2
- 108010067035 Pancrelipase Proteins 0.000 claims description 2
- 108090000526 Papain Proteins 0.000 claims description 2
- 102000003982 Parathyroid hormone Human genes 0.000 claims description 2
- 108090000445 Parathyroid hormone Proteins 0.000 claims description 2
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 claims description 2
- 102000057297 Pepsin A Human genes 0.000 claims description 2
- 108090000284 Pepsin A Proteins 0.000 claims description 2
- 108010088847 Peptide YY Proteins 0.000 claims description 2
- 102100029909 Peptide YY Human genes 0.000 claims description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 claims description 2
- RMUCZJUITONUFY-UHFFFAOYSA-N Phenelzine Chemical compound NNCCC1=CC=CC=C1 RMUCZJUITONUFY-UHFFFAOYSA-N 0.000 claims description 2
- 229920002732 Polyanhydride Polymers 0.000 claims description 2
- 229920001710 Polyorthoester Polymers 0.000 claims description 2
- 101710184309 Probable sucrose-6-phosphate hydrolase Chemical group 0.000 claims description 2
- 108010057464 Prolactin Proteins 0.000 claims description 2
- 102000003946 Prolactin Human genes 0.000 claims description 2
- 239000004365 Protease Substances 0.000 claims description 2
- 101800004937 Protein C Proteins 0.000 claims description 2
- 108020004511 Recombinant DNA Proteins 0.000 claims description 2
- 108010039491 Ricin Proteins 0.000 claims description 2
- 101800001700 Saposin-D Proteins 0.000 claims description 2
- 108010086019 Secretin Proteins 0.000 claims description 2
- 102100037505 Secretin Human genes 0.000 claims description 2
- 229940122055 Serine protease inhibitor Drugs 0.000 claims description 2
- 101710102218 Serine protease inhibitor Proteins 0.000 claims description 2
- 102000013275 Somatomedins Human genes 0.000 claims description 2
- 102000005157 Somatostatin Human genes 0.000 claims description 2
- 108010056088 Somatostatin Chemical group 0.000 claims description 2
- 108010023197 Streptokinase Chemical group 0.000 claims description 2
- 102400000472 Sucrase Human genes 0.000 claims description 2
- 101710112652 Sucrose-6-phosphate hydrolase Chemical group 0.000 claims description 2
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 claims description 2
- DRHKJLXJIQTDTD-OAHLLOKOSA-N Tamsulosine Chemical compound CCOC1=CC=CC=C1OCCN[C@H](C)CC1=CC=C(OC)C(S(N)(=O)=O)=C1 DRHKJLXJIQTDTD-OAHLLOKOSA-N 0.000 claims description 2
- 108010010056 Terlipressin Chemical group 0.000 claims description 2
- 108010055044 Tetanus Toxin Chemical group 0.000 claims description 2
- 108010078233 Thymalfasin Proteins 0.000 claims description 2
- 108010046075 Thymosin Proteins 0.000 claims description 2
- 102000007501 Thymosin Human genes 0.000 claims description 2
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 claims description 2
- 102000004338 Transferrin Human genes 0.000 claims description 2
- 108090000901 Transferrin Proteins 0.000 claims description 2
- 108010009583 Transforming Growth Factors Proteins 0.000 claims description 2
- 102000009618 Transforming Growth Factors Human genes 0.000 claims description 2
- 108010001957 Ularitide Proteins 0.000 claims description 2
- 108010092464 Urate Oxidase Proteins 0.000 claims description 2
- 102400001279 Urodilatin Human genes 0.000 claims description 2
- HDOVUKNUBWVHOX-QMMMGPOBSA-N Valacyclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCOC(=O)[C@@H](N)C(C)C)C=N2 HDOVUKNUBWVHOX-QMMMGPOBSA-N 0.000 claims description 2
- WPVFJKSGQUFQAP-GKAPJAKFSA-N Valcyte Chemical compound N1C(N)=NC(=O)C2=C1N(COC(CO)COC(=O)[C@@H](N)C(C)C)C=N2 WPVFJKSGQUFQAP-GKAPJAKFSA-N 0.000 claims description 2
- 108010059993 Vancomycin Proteins 0.000 claims description 2
- 108010003205 Vasoactive Intestinal Peptide Proteins 0.000 claims description 2
- GXBMIBRIOWHPDT-UHFFFAOYSA-N Vasopressin Natural products N1C(=O)C(CC=2C=C(O)C=CC=2)NC(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CCCN=C(N)N)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C1CC1=CC=CC=C1 GXBMIBRIOWHPDT-UHFFFAOYSA-N 0.000 claims description 2
- 108010004977 Vasopressins Proteins 0.000 claims description 2
- 102000002852 Vasopressins Human genes 0.000 claims description 2
- 108010015940 Viomycin Proteins 0.000 claims description 2
- OZKXLOZHHUHGNV-UHFFFAOYSA-N Viomycin Natural products NCCCC(N)CC(=O)NC1CNC(=O)C(=CNC(=O)N)NC(=O)C(CO)NC(=O)C(CO)NC(=O)C(NC1=O)C2CC(O)NC(=N)N2 OZKXLOZHHUHGNV-UHFFFAOYSA-N 0.000 claims description 2
- WREGKURFCTUGRC-POYBYMJQSA-N Zalcitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)CC1 WREGKURFCTUGRC-POYBYMJQSA-N 0.000 claims description 2
- FZSPJBYOKQPKCD-VIFPVBQESA-N [1-(4-chlorophenyl)-2-methylpropan-2-yl] (2s)-2-aminopropanoate Chemical compound C[C@H](N)C(=O)OC(C)(C)CC1=CC=C(Cl)C=C1 FZSPJBYOKQPKCD-VIFPVBQESA-N 0.000 claims description 2
- 229960002632 acarbose Drugs 0.000 claims description 2
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 claims description 2
- 229960003225 alaproclate Drugs 0.000 claims description 2
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 claims description 2
- 229940062527 alendronate Drugs 0.000 claims description 2
- 229940072056 alginate Drugs 0.000 claims description 2
- 229920000615 alginic acid Polymers 0.000 claims description 2
- 235000010443 alginic acid Nutrition 0.000 claims description 2
- 229960003318 alteplase Drugs 0.000 claims description 2
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 claims description 2
- 229960003805 amantadine Drugs 0.000 claims description 2
- 229960004821 amikacin Drugs 0.000 claims description 2
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 claims description 2
- 229960000959 amineptine Drugs 0.000 claims description 2
- VDPUXONTAVMIKZ-UHFFFAOYSA-N amineptine hydrochloride Chemical compound [Cl-].C1CC2=CC=CC=C2C([NH2+]CCCCCCC(=O)O)C2=CC=CC=C21 VDPUXONTAVMIKZ-UHFFFAOYSA-N 0.000 claims description 2
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 claims description 2
- 229960003437 aminoglutethimide Drugs 0.000 claims description 2
- NTJOBXMMWNYJFB-UHFFFAOYSA-N amisulpride Chemical compound CCN1CCCC1CNC(=O)C1=CC(S(=O)(=O)CC)=C(N)C=C1OC NTJOBXMMWNYJFB-UHFFFAOYSA-N 0.000 claims description 2
- 229960003036 amisulpride Drugs 0.000 claims description 2
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 claims description 2
- 229960000528 amlodipine Drugs 0.000 claims description 2
- 229950004267 amotosalen Drugs 0.000 claims description 2
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 claims description 2
- 229960003022 amoxicillin Drugs 0.000 claims description 2
- 229940025084 amphetamine Drugs 0.000 claims description 2
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 claims description 2
- 229960003942 amphotericin b Drugs 0.000 claims description 2
- 229960000723 ampicillin Drugs 0.000 claims description 2
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 claims description 2
- 229960001830 amprenavir Drugs 0.000 claims description 2
- YMARZQAQMVYCKC-OEMFJLHTSA-N amprenavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 YMARZQAQMVYCKC-OEMFJLHTSA-N 0.000 claims description 2
- RNLQIBCLLYYYFJ-UHFFFAOYSA-N amrinone Chemical compound N1C(=O)C(N)=CC(C=2C=CN=CC=2)=C1 RNLQIBCLLYYYFJ-UHFFFAOYSA-N 0.000 claims description 2
- 229960002105 amrinone Drugs 0.000 claims description 2
- 230000000202 analgesic effect Effects 0.000 claims description 2
- LKYQLAWMNBFNJT-UHFFFAOYSA-N anileridine Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCC1=CC=C(N)C=C1 LKYQLAWMNBFNJT-UHFFFAOYSA-N 0.000 claims description 2
- 229960002512 anileridine Drugs 0.000 claims description 2
- 229960000983 anistreplase Drugs 0.000 claims description 2
- 230000003266 anti-allergic effect Effects 0.000 claims description 2
- 230000000844 anti-bacterial effect Effects 0.000 claims description 2
- 230000003178 anti-diabetic effect Effects 0.000 claims description 2
- 230000000843 anti-fungal effect Effects 0.000 claims description 2
- 230000002924 anti-infective effect Effects 0.000 claims description 2
- 230000003110 anti-inflammatory effect Effects 0.000 claims description 2
- 230000000118 anti-neoplastic effect Effects 0.000 claims description 2
- 230000003579 anti-obesity Effects 0.000 claims description 2
- 230000001475 anti-trypsic effect Effects 0.000 claims description 2
- 239000003146 anticoagulant agent Substances 0.000 claims description 2
- 229940127219 anticoagulant drug Drugs 0.000 claims description 2
- 239000003472 antidiabetic agent Substances 0.000 claims description 2
- 102000025171 antigen binding proteins Human genes 0.000 claims description 2
- 108091000831 antigen binding proteins Proteins 0.000 claims description 2
- 229940034982 antineoplastic agent Drugs 0.000 claims description 2
- 229960005348 antithrombin iii Drugs 0.000 claims description 2
- 229960002610 apraclonidine Drugs 0.000 claims description 2
- IEJXVRYNEISIKR-UHFFFAOYSA-N apraclonidine Chemical compound ClC1=CC(N)=CC(Cl)=C1NC1=NCCN1 IEJXVRYNEISIKR-UHFFFAOYSA-N 0.000 claims description 2
- XZNUGFQTQHRASN-XQENGBIVSA-N apramycin Chemical compound O([C@H]1O[C@@H]2[C@H](O)[C@@H]([C@H](O[C@H]2C[C@H]1N)O[C@@H]1[C@@H]([C@@H](O)[C@H](N)[C@@H](CO)O1)O)NC)[C@@H]1[C@@H](N)C[C@@H](N)[C@H](O)[C@H]1O XZNUGFQTQHRASN-XQENGBIVSA-N 0.000 claims description 2
- 229950006334 apramycin Drugs 0.000 claims description 2
- 229960004405 aprotinin Drugs 0.000 claims description 2
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 claims description 2
- 229960003831 articaine Drugs 0.000 claims description 2
- 229960002274 atenolol Drugs 0.000 claims description 2
- 229960002430 atomoxetine Drugs 0.000 claims description 2
- LUCXVPAZUDVVBT-UNTBIKODSA-N atomoxetine hydrochloride Chemical compound Cl.O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=CC=C1C LUCXVPAZUDVVBT-UNTBIKODSA-N 0.000 claims description 2
- VWXRQYYUEIYXCZ-OBIMUBPZSA-N atosiban Chemical compound C1=CC(OCC)=CC=C1C[C@@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CCCN)C(=O)NCC(N)=O)CSSCCC(=O)N1 VWXRQYYUEIYXCZ-OBIMUBPZSA-N 0.000 claims description 2
- 108700007535 atosiban Proteins 0.000 claims description 2
- 229960002403 atosiban Drugs 0.000 claims description 2
- 229950009166 avizafone Drugs 0.000 claims description 2
- 229960000794 baclofen Drugs 0.000 claims description 2
- 229960004530 benazepril Drugs 0.000 claims description 2
- BNQDCRGUHNALGH-UHFFFAOYSA-N benserazide Chemical compound OCC(N)C(=O)NNCC1=CC=C(O)C(O)=C1O BNQDCRGUHNALGH-UHFFFAOYSA-N 0.000 claims description 2
- 229960000911 benserazide Drugs 0.000 claims description 2
- 229960005274 benzocaine Drugs 0.000 claims description 2
- 108010005774 beta-Galactosidase Proteins 0.000 claims description 2
- 229960004324 betaxolol Drugs 0.000 claims description 2
- CHDPSNLJFOQTRK-UHFFFAOYSA-N betaxolol hydrochloride Chemical compound [Cl-].C1=CC(OCC(O)C[NH2+]C(C)C)=CC=C1CCOCC1CC1 CHDPSNLJFOQTRK-UHFFFAOYSA-N 0.000 claims description 2
- 108010033394 biphalin Proteins 0.000 claims description 2
- DESSEGDLRYOPTJ-VRANXALZSA-N biphalin Chemical compound C([C@H](N)C(=O)N[C@H](C)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NNC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)CNC(=O)[C@@H](C)NC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 DESSEGDLRYOPTJ-VRANXALZSA-N 0.000 claims description 2
- 108010055460 bivalirudin Proteins 0.000 claims description 2
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 claims description 2
- 229960001500 bivalirudin Drugs 0.000 claims description 2
- 229960001561 bleomycin Drugs 0.000 claims description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 claims description 2
- WZXHSWVDAYOFPE-UHFFFAOYSA-N brofaromine Chemical compound C=1C2=CC(OC)=CC(Br)=C2OC=1C1CCNCC1 WZXHSWVDAYOFPE-UHFFFAOYSA-N 0.000 claims description 2
- 229950004068 brofaromine Drugs 0.000 claims description 2
- ZBPLOVFIXSTCRZ-UHFFFAOYSA-N bromfenac Chemical compound NC1=C(CC(O)=O)C=CC=C1C(=O)C1=CC=C(Br)C=C1 ZBPLOVFIXSTCRZ-UHFFFAOYSA-N 0.000 claims description 2
- 229960003655 bromfenac Drugs 0.000 claims description 2
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 claims description 2
- 229960003773 calcitonin (salmon synthetic) Drugs 0.000 claims description 2
- VDTNNGKXZGSZIP-UHFFFAOYSA-N carbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(N)C=C1 VDTNNGKXZGSZIP-UHFFFAOYSA-N 0.000 claims description 2
- 229960003362 carbutamide Drugs 0.000 claims description 2
- 239000002327 cardiovascular agent Substances 0.000 claims description 2
- 229940125692 cardiovascular agent Drugs 0.000 claims description 2
- 229920001525 carrageenan Polymers 0.000 claims description 2
- 239000000679 carrageenan Substances 0.000 claims description 2
- 235000010418 carrageenan Nutrition 0.000 claims description 2
- 229940113118 carrageenan Drugs 0.000 claims description 2
- 229960004195 carvedilol Drugs 0.000 claims description 2
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 claims description 2
- DLNKOYKMWOXYQA-IONNQARKSA-N cathine Chemical compound C[C@H](N)[C@@H](O)C1=CC=CC=C1 DLNKOYKMWOXYQA-IONNQARKSA-N 0.000 claims description 2
- 229960003609 cathine Drugs 0.000 claims description 2
- PUAQLLVFLMYYJJ-ZETCQYMHSA-N cathinone Chemical compound C[C@H](N)C(=O)C1=CC=CC=C1 PUAQLLVFLMYYJJ-ZETCQYMHSA-N 0.000 claims description 2
- 229950002698 cathinone Drugs 0.000 claims description 2
- 229940083181 centrally acting adntiadrenergic agent methyldopa Drugs 0.000 claims description 2
- ZAIPMKNFIOOWCQ-UEKVPHQBSA-N cephalexin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC=C1 ZAIPMKNFIOOWCQ-UEKVPHQBSA-N 0.000 claims description 2
- 229940045110 chitosan Drugs 0.000 claims description 2
- 150000001805 chlorine compounds Chemical group 0.000 claims description 2
- 229960003405 ciprofloxacin Drugs 0.000 claims description 2
- QGPKADBNRMWEQR-UHFFFAOYSA-N clinafloxacin Chemical compound C1C(N)CCN1C1=C(F)C=C2C(=O)C(C(O)=O)=CN(C3CC3)C2=C1Cl QGPKADBNRMWEQR-UHFFFAOYSA-N 0.000 claims description 2
- 229950001320 clinafloxacin Drugs 0.000 claims description 2
- 229920001436 collagen Polymers 0.000 claims description 2
- 229960002424 collagenase Drugs 0.000 claims description 2
- 230000000295 complement effect Effects 0.000 claims description 2
- 239000003433 contraceptive agent Substances 0.000 claims description 2
- 230000002254 contraceptive effect Effects 0.000 claims description 2
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 claims description 2
- 229960000258 corticotropin Drugs 0.000 claims description 2
- 229920006037 cross link polymer Polymers 0.000 claims description 2
- 102000003675 cytokine receptors Human genes 0.000 claims description 2
- 108010057085 cytokine receptors Proteins 0.000 claims description 2
- KWGRBVOPPLSCSI-UHFFFAOYSA-N d-ephedrine Natural products CNC(C)C(O)C1=CC=CC=C1 KWGRBVOPPLSCSI-UHFFFAOYSA-N 0.000 claims description 2
- 229960000860 dapsone Drugs 0.000 claims description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 2
- 229960000975 daunorubicin Drugs 0.000 claims description 2
- 229960000958 deferoxamine Drugs 0.000 claims description 2
- 229960005319 delavirdine Drugs 0.000 claims description 2
- 108010051768 des-n-octanoyl ghrelin Proteins 0.000 claims description 2
- 229960003914 desipramine Drugs 0.000 claims description 2
- 229960002086 dextran Drugs 0.000 claims description 2
- LBOJYSIDWZQNJS-CVEARBPZSA-N dizocilpine Chemical compound C12=CC=CC=C2[C@]2(C)C3=CC=CC=C3C[C@H]1N2 LBOJYSIDWZQNJS-CVEARBPZSA-N 0.000 claims description 2
- 229950004794 dizocilpine Drugs 0.000 claims description 2
- DLNKOYKMWOXYQA-UHFFFAOYSA-N dl-pseudophenylpropanolamine Natural products CC(N)C(O)C1=CC=CC=C1 DLNKOYKMWOXYQA-UHFFFAOYSA-N 0.000 claims description 2
- 229960003638 dopamine Drugs 0.000 claims description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 claims description 2
- IAVUPMFITXYVAF-XPUUQOCRSA-N dorzolamide Chemical compound CCN[C@H]1C[C@H](C)S(=O)(=O)C2=C1C=C(S(N)(=O)=O)S2 IAVUPMFITXYVAF-XPUUQOCRSA-N 0.000 claims description 2
- 229960003933 dorzolamide Drugs 0.000 claims description 2
- 229960004679 doxorubicin Drugs 0.000 claims description 2
- 229960002866 duloxetine Drugs 0.000 claims description 2
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 claims description 2
- 229960002759 eflornithine Drugs 0.000 claims description 2
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 claims description 2
- 229960000873 enalapril Drugs 0.000 claims description 2
- PEASPLKKXBYDKL-FXEVSJAOSA-N enfuvirtide Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(C)=O)[C@@H](C)O)[C@@H](C)CC)C1=CN=CN1 PEASPLKKXBYDKL-FXEVSJAOSA-N 0.000 claims description 2
- 229960002062 enfuvirtide Drugs 0.000 claims description 2
- 229960002549 enoxacin Drugs 0.000 claims description 2
- IDYZIJYBMGIQMJ-UHFFFAOYSA-N enoxacin Chemical compound N1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 IDYZIJYBMGIQMJ-UHFFFAOYSA-N 0.000 claims description 2
- 229960005139 epinephrine Drugs 0.000 claims description 2
- 229960001904 epirubicin Drugs 0.000 claims description 2
- 229960002770 ertapenem Drugs 0.000 claims description 2
- AQNDDEOPVVGCPG-UHFFFAOYSA-N esmolol Chemical compound COC(=O)CCC1=CC=C(OCC(O)CNC(C)C)C=C1 AQNDDEOPVVGCPG-UHFFFAOYSA-N 0.000 claims description 2
- 229960003745 esmolol Drugs 0.000 claims description 2
- 239000002329 esterase inhibitor Substances 0.000 claims description 2
- 229960000285 ethambutol Drugs 0.000 claims description 2
- 229960004222 factor ix Drugs 0.000 claims description 2
- 229940012413 factor vii Drugs 0.000 claims description 2
- 229960000301 factor viii Drugs 0.000 claims description 2
- 229960001582 fenfluramine Drugs 0.000 claims description 2
- TVURRHSHRRELCG-UHFFFAOYSA-N fenoldopam Chemical compound C1=CC(O)=CC=C1C1C2=CC(O)=C(O)C(Cl)=C2CCNC1 TVURRHSHRRELCG-UHFFFAOYSA-N 0.000 claims description 2
- 229960002724 fenoldopam Drugs 0.000 claims description 2
- 229960001022 fenoterol Drugs 0.000 claims description 2
- 229940001501 fibrinolysin Drugs 0.000 claims description 2
- 229960000449 flecainide Drugs 0.000 claims description 2
- CJOFXWAVKWHTFT-XSFVSMFZSA-N fluvoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 CJOFXWAVKWHTFT-XSFVSMFZSA-N 0.000 claims description 2
- 229960004038 fluvoxamine Drugs 0.000 claims description 2
- 229960003142 fosamprenavir Drugs 0.000 claims description 2
- MLBVMOWEQCZNCC-OEMFJLHTSA-N fosamprenavir Chemical compound C([C@@H]([C@H](OP(O)(O)=O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 MLBVMOWEQCZNCC-OEMFJLHTSA-N 0.000 claims description 2
- 229960003704 framycetin Drugs 0.000 claims description 2
- PGBHMTALBVVCIT-VCIWKGPPSA-N framycetin Chemical compound N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CN)O2)N)O[C@@H]1CO PGBHMTALBVVCIT-VCIWKGPPSA-N 0.000 claims description 2
- SIBNYOSJIXCDRI-SECBINFHSA-N frovatriptan Chemical compound C1=C(C(N)=O)[CH]C2=C(C[C@H](NC)CC3)C3=NC2=C1 SIBNYOSJIXCDRI-SECBINFHSA-N 0.000 claims description 2
- 229960002284 frovatriptan Drugs 0.000 claims description 2
- 229960003883 furosemide Drugs 0.000 claims description 2
- 230000004927 fusion Effects 0.000 claims description 2
- 229960002870 gabapentin Drugs 0.000 claims description 2
- 229960003923 gatifloxacin Drugs 0.000 claims description 2
- 229960002518 gentamicin Drugs 0.000 claims description 2
- 239000002622 gonadotropin Substances 0.000 claims description 2
- 229960004905 gramicidin Drugs 0.000 claims description 2
- ZWCXYZRRTRDGQE-SORVKSEFSA-N gramicidina Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](C(C)C)NC(=O)[C@H](C)NC(=O)[C@H](NC(=O)[C@H](C)NC(=O)CNC(=O)[C@@H](NC=O)C(C)C)CC(C)C)C(=O)NCCO)=CNC2=C1 ZWCXYZRRTRDGQE-SORVKSEFSA-N 0.000 claims description 2
- 229960000642 grepafloxacin Drugs 0.000 claims description 2
- 239000000122 growth hormone Substances 0.000 claims description 2
- 208000002672 hepatitis B Diseases 0.000 claims description 2
- 229960005388 hexylcaine Drugs 0.000 claims description 2
- WQPDUTSPKFMPDP-OUMQNGNKSA-N hirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(OS(O)(=O)=O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 WQPDUTSPKFMPDP-OUMQNGNKSA-N 0.000 claims description 2
- 229940006607 hirudin Drugs 0.000 claims description 2
- 229940088597 hormone Drugs 0.000 claims description 2
- 239000005556 hormone Substances 0.000 claims description 2
- 229920002674 hyaluronan Polymers 0.000 claims description 2
- 229960003160 hyaluronic acid Drugs 0.000 claims description 2
- 229960002474 hydralazine Drugs 0.000 claims description 2
- 229960002003 hydrochlorothiazide Drugs 0.000 claims description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 2
- 229950003053 icofungipen Drugs 0.000 claims description 2
- 229960000908 idarubicin Drugs 0.000 claims description 2
- 229960002751 imiquimod Drugs 0.000 claims description 2
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 claims description 2
- 230000002519 immonomodulatory effect Effects 0.000 claims description 2
- 229960003971 influenza vaccine Drugs 0.000 claims description 2
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 claims description 2
- 239000004026 insulin derivative Substances 0.000 claims description 2
- 102000028416 insulin-like growth factor binding Human genes 0.000 claims description 2
- 108091022911 insulin-like growth factor binding Proteins 0.000 claims description 2
- 229940047124 interferons Drugs 0.000 claims description 2
- 229940047122 interleukins Drugs 0.000 claims description 2
- 230000003834 intracellular effect Effects 0.000 claims description 2
- 235000011073 invertase Nutrition 0.000 claims description 2
- VBUWHHLIZKOSMS-RIWXPGAOSA-N invicorp Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)C(C)C)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 VBUWHHLIZKOSMS-RIWXPGAOSA-N 0.000 claims description 2
- 229940039009 isoproterenol Drugs 0.000 claims description 2
- 229960004427 isradipine Drugs 0.000 claims description 2
- 229960000318 kanamycin Drugs 0.000 claims description 2
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 claims description 2
- 229930182823 kanamycin A Natural products 0.000 claims description 2
- 229960001632 labetalol Drugs 0.000 claims description 2
- 229940116108 lactase Drugs 0.000 claims description 2
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 claims description 2
- 229960001627 lamivudine Drugs 0.000 claims description 2
- 239000002523 lectin Substances 0.000 claims description 2
- 229940039781 leptin Drugs 0.000 claims description 2
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 claims description 2
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 claims description 2
- 229960004338 leuprorelin Drugs 0.000 claims description 2
- IXHBTMCLRNMKHZ-LBPRGKRZSA-N levobunolol Chemical compound O=C1CCCC2=C1C=CC=C2OC[C@@H](O)CNC(C)(C)C IXHBTMCLRNMKHZ-LBPRGKRZSA-N 0.000 claims description 2
- 229960000831 levobunolol Drugs 0.000 claims description 2
- 229960004502 levodopa Drugs 0.000 claims description 2
- RLAWWYSOJDYHDC-BZSNNMDCSA-N lisinopril Chemical compound C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 RLAWWYSOJDYHDC-BZSNNMDCSA-N 0.000 claims description 2
- 229960002394 lisinopril Drugs 0.000 claims description 2
- ZEKZLJVOYLTDKK-UHFFFAOYSA-N lomefloxacin Chemical compound FC1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNC(C)C1 ZEKZLJVOYLTDKK-UHFFFAOYSA-N 0.000 claims description 2
- 229960002422 lomefloxacin Drugs 0.000 claims description 2
- 229960001977 loracarbef Drugs 0.000 claims description 2
- 229940040129 luteinizing hormone Drugs 0.000 claims description 2
- QSLMDECMDJKHMQ-GSXCWMCISA-N maprotiline Chemical compound C12=CC=CC=C2[C@@]2(CCCNC)C3=CC=CC=C3[C@@H]1CC2 QSLMDECMDJKHMQ-GSXCWMCISA-N 0.000 claims description 2
- 229960004090 maprotiline Drugs 0.000 claims description 2
- 229960001962 mefloquine Drugs 0.000 claims description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 claims description 2
- 229960001924 melphalan Drugs 0.000 claims description 2
- 229960004640 memantine Drugs 0.000 claims description 2
- LDDHMLJTFXJGPI-UHFFFAOYSA-N memantine hydrochloride Chemical compound Cl.C1C(C2)CC3(C)CC1(C)CC2(N)C3 LDDHMLJTFXJGPI-UHFFFAOYSA-N 0.000 claims description 2
- DMJNNHOOLUXYBV-PQTSNVLCSA-N meropenem Chemical compound C=1([C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)S[C@@H]1CN[C@H](C(=O)N(C)C)C1 DMJNNHOOLUXYBV-PQTSNVLCSA-N 0.000 claims description 2
- 229960002260 meropenem Drugs 0.000 claims description 2
- KBOPZPXVLCULAV-UHFFFAOYSA-N mesalamine Chemical compound NC1=CC=C(O)C(C(O)=O)=C1 KBOPZPXVLCULAV-UHFFFAOYSA-N 0.000 claims description 2
- 229960004963 mesalazine Drugs 0.000 claims description 2
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 claims description 2
- 229960002237 metoprolol Drugs 0.000 claims description 2
- 229960000600 milnacipran Drugs 0.000 claims description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 2
- 229960003702 moxifloxacin Drugs 0.000 claims description 2
- FABPRXSRWADJSP-MEDUHNTESA-N moxifloxacin Chemical compound COC1=C(N2C[C@H]3NCCC[C@H]3C2)C(F)=CC(C(C(C(O)=O)=C2)=O)=C1N2C1CC1 FABPRXSRWADJSP-MEDUHNTESA-N 0.000 claims description 2
- 239000000692 natriuretic peptide Substances 0.000 claims description 2
- 229960002748 norepinephrine Drugs 0.000 claims description 2
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 claims description 2
- OGJPXUAPXNRGGI-UHFFFAOYSA-N norfloxacin Chemical compound C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 OGJPXUAPXNRGGI-UHFFFAOYSA-N 0.000 claims description 2
- 229960001180 norfloxacin Drugs 0.000 claims description 2
- 229960001158 nortriptyline Drugs 0.000 claims description 2
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 claims description 2
- 229960000988 nystatin Drugs 0.000 claims description 2
- VQOXZBDYSJBXMA-NQTDYLQESA-N nystatin A1 Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/CC/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 VQOXZBDYSJBXMA-NQTDYLQESA-N 0.000 claims description 2
- 229960002700 octreotide Drugs 0.000 claims description 2
- 229960003752 oseltamivir Drugs 0.000 claims description 2
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 claims description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 claims description 2
- 229960003978 pamidronic acid Drugs 0.000 claims description 2
- 229940045258 pancrelipase Drugs 0.000 claims description 2
- 229940055729 papain Drugs 0.000 claims description 2
- 235000019834 papain Nutrition 0.000 claims description 2
- 239000000199 parathyroid hormone Substances 0.000 claims description 2
- 229960001319 parathyroid hormone Drugs 0.000 claims description 2
- 229960002296 paroxetine Drugs 0.000 claims description 2
- 229960002625 pazufloxacin Drugs 0.000 claims description 2
- RKOUGZGFAYMUIO-RITPCOANSA-N pdl 118 Chemical compound N[C@H]1CC(=C)C[C@H]1C(O)=O RKOUGZGFAYMUIO-RITPCOANSA-N 0.000 claims description 2
- 229960005079 pemetrexed Drugs 0.000 claims description 2
- 229940111202 pepsin Drugs 0.000 claims description 2
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 claims description 2
- 229960002582 perindopril Drugs 0.000 claims description 2
- 229960000964 phenelzine Drugs 0.000 claims description 2
- 229960003209 phenmetrazine Drugs 0.000 claims description 2
- 229920000765 poly(2-oxazolines) Polymers 0.000 claims description 2
- 229920001308 poly(aminoacid) Polymers 0.000 claims description 2
- 229920002401 polyacrylamide Polymers 0.000 claims description 2
- 229920000728 polyester Polymers 0.000 claims description 2
- 229920001451 polypropylene glycol Polymers 0.000 claims description 2
- 229920002635 polyurethane Polymers 0.000 claims description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 claims description 2
- 108010029667 pramlintide Proteins 0.000 claims description 2
- NRKVKVQDUCJPIZ-MKAGXXMWSA-N pramlintide acetate Chemical compound C([C@@H](C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CCCCN)[C@@H](C)O)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NRKVKVQDUCJPIZ-MKAGXXMWSA-N 0.000 claims description 2
- 229960001233 pregabalin Drugs 0.000 claims description 2
- AYXYPKUFHZROOJ-ZETCQYMHSA-N pregabalin Chemical compound CC(C)C[C@H](CN)CC(O)=O AYXYPKUFHZROOJ-ZETCQYMHSA-N 0.000 claims description 2
- 108010055741 pro-diazepam Proteins 0.000 claims description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 claims description 2
- 229960004919 procaine Drugs 0.000 claims description 2
- 229940097325 prolactin Drugs 0.000 claims description 2
- 229960000856 protein c Drugs 0.000 claims description 2
- BWPIARFWQZKAIA-UHFFFAOYSA-N protriptyline Chemical compound C1=CC2=CC=CC=C2C(CCCNC)C2=CC=CC=C21 BWPIARFWQZKAIA-UHFFFAOYSA-N 0.000 claims description 2
- 229960002601 protriptyline Drugs 0.000 claims description 2
- KWGRBVOPPLSCSI-WCBMZHEXSA-N pseudoephedrine Chemical compound CN[C@@H](C)[C@@H](O)C1=CC=CC=C1 KWGRBVOPPLSCSI-WCBMZHEXSA-N 0.000 claims description 2
- 229960003908 pseudoephedrine Drugs 0.000 claims description 2
- CBQGYUDMJHNJBX-RTBURBONSA-N reboxetine Chemical compound CCOC1=CC=CC=C1O[C@H](C=1C=CC=CC=1)[C@@H]1OCCNC1 CBQGYUDMJHNJBX-RTBURBONSA-N 0.000 claims description 2
- 229960003770 reboxetine Drugs 0.000 claims description 2
- 229940044551 receptor antagonist Drugs 0.000 claims description 2
- 239000002464 receptor antagonist Substances 0.000 claims description 2
- 229960001634 ritodrine Drugs 0.000 claims description 2
- IOVGROKTTNBUGK-SJCJKPOMSA-N ritodrine Chemical compound N([C@@H](C)[C@H](O)C=1C=CC(O)=CC=1)CCC1=CC=C(O)C=C1 IOVGROKTTNBUGK-SJCJKPOMSA-N 0.000 claims description 2
- 229950000615 sabarubicin Drugs 0.000 claims description 2
- 229960002052 salbutamol Drugs 0.000 claims description 2
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 claims description 2
- 229960002101 secretin Drugs 0.000 claims description 2
- OWMZNFCDEHGFEP-NFBCVYDUSA-N secretin human Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(N)=O)[C@@H](C)O)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)C1=CC=CC=C1 OWMZNFCDEHGFEP-NFBCVYDUSA-N 0.000 claims description 2
- 239000003001 serine protease inhibitor Substances 0.000 claims description 2
- WGWPRVFKDLAUQJ-MITYVQBRSA-N sermorelin Chemical compound C([C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(N)=O)C1=CC=C(O)C=C1 WGWPRVFKDLAUQJ-MITYVQBRSA-N 0.000 claims description 2
- 229960002758 sermorelin Drugs 0.000 claims description 2
- 229940076279 serotonin Drugs 0.000 claims description 2
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 claims description 2
- 229960004034 sitagliptin Drugs 0.000 claims description 2
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical group C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 claims description 2
- 229960000553 somatostatin Drugs 0.000 claims description 2
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 claims description 2
- 229960002370 sotalol Drugs 0.000 claims description 2
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 claims description 2
- 229960000268 spectinomycin Drugs 0.000 claims description 2
- 230000003637 steroidlike Effects 0.000 claims description 2
- 229960005202 streptokinase Drugs 0.000 claims description 2
- SEEPANYCNGTZFQ-UHFFFAOYSA-N sulfadiazine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)NC1=NC=CC=N1 SEEPANYCNGTZFQ-UHFFFAOYSA-N 0.000 claims description 2
- 229960004306 sulfadiazine Drugs 0.000 claims description 2
- QPPBRPIAZZHUNT-UHFFFAOYSA-N sulfamerazine Chemical compound CC1=CC=NC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 QPPBRPIAZZHUNT-UHFFFAOYSA-N 0.000 claims description 2
- 229960002597 sulfamerazine Drugs 0.000 claims description 2
- KXRZBTAEDBELFD-UHFFFAOYSA-N sulfamethopyrazine Chemical compound COC1=NC=CN=C1NS(=O)(=O)C1=CC=C(N)C=C1 KXRZBTAEDBELFD-UHFFFAOYSA-N 0.000 claims description 2
- 229960005404 sulfamethoxazole Drugs 0.000 claims description 2
- JLKIGFTWXXRPMT-UHFFFAOYSA-N sulphamethoxazole Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 JLKIGFTWXXRPMT-UHFFFAOYSA-N 0.000 claims description 2
- 229940099093 symlin Drugs 0.000 claims description 2
- 229960001685 tacrine Drugs 0.000 claims description 2
- YLJREFDVOIBQDA-UHFFFAOYSA-N tacrine Chemical compound C1=CC=C2C(N)=C(CCCC3)C3=NC2=C1 YLJREFDVOIBQDA-UHFFFAOYSA-N 0.000 claims description 2
- 229960002613 tamsulosin Drugs 0.000 claims description 2
- 229960000195 terbutaline Drugs 0.000 claims description 2
- BENFXAYNYRLAIU-QSVFAHTRSA-N terlipressin Chemical group NCCCC[C@@H](C(=O)NCC(N)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@H]1NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)CN)CSSC1 BENFXAYNYRLAIU-QSVFAHTRSA-N 0.000 claims description 2
- 229960003813 terlipressin Drugs 0.000 claims description 2
- IMCGHZIGRANKHV-AJNGGQMLSA-N tert-butyl (3s,5s)-2-oxo-5-[(2s,4s)-5-oxo-4-propan-2-yloxolan-2-yl]-3-propan-2-ylpyrrolidine-1-carboxylate Chemical compound O1C(=O)[C@H](C(C)C)C[C@H]1[C@H]1N(C(=O)OC(C)(C)C)C(=O)[C@H](C(C)C)C1 IMCGHZIGRANKHV-AJNGGQMLSA-N 0.000 claims description 2
- 229940118376 tetanus toxin Drugs 0.000 claims description 2
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 claims description 2
- 229960004231 thymalfasin Drugs 0.000 claims description 2
- LCJVIYPJPCBWKS-NXPQJCNCSA-N thymosin Chemical compound SC[C@@H](N)C(=O)N[C@H](CO)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CO)C(=O)N[C@H](CO)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@H]([C@H](C)O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@H](CCC(O)=O)C(O)=O LCJVIYPJPCBWKS-NXPQJCNCSA-N 0.000 claims description 2
- 229960000874 thyrotropin Drugs 0.000 claims description 2
- 230000001748 thyrotropin Effects 0.000 claims description 2
- 229960002203 tilactase Drugs 0.000 claims description 2
- 229960004605 timolol Drugs 0.000 claims description 2
- 229960000707 tobramycin Drugs 0.000 claims description 2
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 claims description 2
- 229960002872 tocainide Drugs 0.000 claims description 2
- 229950008187 tosufloxacin Drugs 0.000 claims description 2
- 229960002051 trandolapril Drugs 0.000 claims description 2
- GYDJEQRTZSCIOI-LJGSYFOKSA-N tranexamic acid Chemical compound NC[C@H]1CC[C@H](C(O)=O)CC1 GYDJEQRTZSCIOI-LJGSYFOKSA-N 0.000 claims description 2
- 229960000401 tranexamic acid Drugs 0.000 claims description 2
- 239000012581 transferrin Substances 0.000 claims description 2
- 229960003741 tranylcypromine Drugs 0.000 claims description 2
- 229960000497 trovafloxacin Drugs 0.000 claims description 2
- WVPSKSLAZQPAKQ-CDMJZVDBSA-N trovafloxacin Chemical compound C([C@H]1[C@@H]([C@H]1C1)N)N1C(C(=CC=1C(=O)C(C(O)=O)=C2)F)=NC=1N2C1=CC=C(F)C=C1F WVPSKSLAZQPAKQ-CDMJZVDBSA-N 0.000 claims description 2
- IUCCYQIEZNQWRS-DWWHXVEHSA-N ularitide Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@@H](N)[C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)=O)[C@@H](C)CC)C1=CC=CC=C1 IUCCYQIEZNQWRS-DWWHXVEHSA-N 0.000 claims description 2
- 229940005267 urate oxidase Drugs 0.000 claims description 2
- 229940093257 valacyclovir Drugs 0.000 claims description 2
- 229960002149 valganciclovir Drugs 0.000 claims description 2
- 229960003165 vancomycin Drugs 0.000 claims description 2
- 239000005526 vasoconstrictor agent Substances 0.000 claims description 2
- 230000000304 vasodilatating effect Effects 0.000 claims description 2
- 239000003071 vasodilator agent Substances 0.000 claims description 2
- 229960003726 vasopressin Drugs 0.000 claims description 2
- 229960001255 viloxazine Drugs 0.000 claims description 2
- GXFAIFRPOKBQRV-GHXCTMGLSA-N viomycin Chemical compound N1C(=O)\C(=C\NC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)C[C@@H](N)CCCN)CNC(=O)[C@@H]1[C@@H]1NC(=N)N[C@@H](O)C1 GXFAIFRPOKBQRV-GHXCTMGLSA-N 0.000 claims description 2
- 229950001272 viomycin Drugs 0.000 claims description 2
- 229920001221 xylan Polymers 0.000 claims description 2
- 150000004823 xylans Chemical class 0.000 claims description 2
- 229960000523 zalcitabine Drugs 0.000 claims description 2
- BPKIMPVREBSLAJ-QTBYCLKRSA-N ziconotide Chemical compound C([C@H]1C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]2C(=O)N[C@@H]3C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@H](C(N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CSSC2)C(N)=O)=O)CSSC[C@H](NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)CNC(=O)[C@H](CCCCN)NC(=O)CNC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CSSC3)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(N1)=O)CCSC)[C@@H](C)O)C1=CC=C(O)C=C1 BPKIMPVREBSLAJ-QTBYCLKRSA-N 0.000 claims description 2
- 229960002811 ziconotide Drugs 0.000 claims description 2
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 claims description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims 1
- 102400000321 Glucagon Human genes 0.000 claims 1
- 102000017975 Protein C Human genes 0.000 claims 1
- 102400000015 Vasoactive intestinal peptide Human genes 0.000 claims 1
- JAPHQRWPEGVNBT-UTUOFQBUSA-N loracarbef Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CC[C@@H]32)C([O-])=O)=O)[NH3+])=CC=CC=C1 JAPHQRWPEGVNBT-UTUOFQBUSA-N 0.000 claims 1
- VSZGPKBBMSAYNT-RRFJBIMHSA-N oseltamivir Chemical compound CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 VSZGPKBBMSAYNT-RRFJBIMHSA-N 0.000 claims 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 claims 1
- 229920000515 polycarbonate Polymers 0.000 claims 1
- 239000004417 polycarbonate Substances 0.000 claims 1
- MYPYJXKWCTUITO-LYRMYLQWSA-O vancomycin(1+) Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C([O-])=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)[NH2+]C)[C@H]1C[C@](C)([NH3+])[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-O 0.000 claims 1
- 150000001412 amines Chemical class 0.000 abstract description 12
- 229920005989 resin Polymers 0.000 description 105
- 239000011347 resin Substances 0.000 description 105
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 92
- 230000015572 biosynthetic process Effects 0.000 description 78
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 77
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 76
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 71
- 238000003786 synthesis reaction Methods 0.000 description 71
- 238000004007 reversed phase HPLC Methods 0.000 description 54
- 239000000203 mixture Substances 0.000 description 47
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 45
- 229910001868 water Inorganic materials 0.000 description 45
- 239000000243 solution Substances 0.000 description 39
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 34
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 31
- 238000003776 cleavage reaction Methods 0.000 description 29
- 230000007017 scission Effects 0.000 description 29
- 239000011734 sodium Substances 0.000 description 24
- 239000002904 solvent Substances 0.000 description 24
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 21
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 20
- 239000000047 product Substances 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 16
- 239000000463 material Substances 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 15
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 13
- 125000006239 protecting group Chemical group 0.000 description 13
- 239000011541 reaction mixture Substances 0.000 description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 150000001875 compounds Chemical class 0.000 description 12
- KSDTXRUIZMTBNV-INIZCTEOSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)butanedioic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CC(=O)O)C(O)=O)C3=CC=CC=C3C2=C1 KSDTXRUIZMTBNV-INIZCTEOSA-N 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 238000010168 coupling process Methods 0.000 description 11
- 238000010511 deprotection reaction Methods 0.000 description 11
- 238000004128 high performance liquid chromatography Methods 0.000 description 11
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 10
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 10
- 102100022831 Somatoliberin Human genes 0.000 description 10
- 101710142969 Somatoliberin Proteins 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 229910052799 carbon Inorganic materials 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- 238000005859 coupling reaction Methods 0.000 description 10
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 10
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 10
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 10
- 230000007062 hydrolysis Effects 0.000 description 10
- 238000006460 hydrolysis reaction Methods 0.000 description 10
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 10
- 230000004048 modification Effects 0.000 description 10
- 238000012986 modification Methods 0.000 description 10
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- 0 CC(*)(*N(*)C([C@@](CCCC1)[C@]1C(C)=O)=O)N(*)* Chemical compound CC(*)(*N(*)C([C@@](CCCC1)[C@]1C(C)=O)=O)N(*)* 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 235000011054 acetic acid Nutrition 0.000 description 9
- 230000008878 coupling Effects 0.000 description 9
- 238000000338 in vitro Methods 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- 239000008363 phosphate buffer Substances 0.000 description 9
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 8
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- 125000003277 amino group Chemical group 0.000 description 8
- 150000008064 anhydrides Chemical class 0.000 description 8
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 8
- 239000000872 buffer Substances 0.000 description 8
- 230000002255 enzymatic effect Effects 0.000 description 8
- 238000011534 incubation Methods 0.000 description 8
- JFLSOKIMYBSASW-UHFFFAOYSA-N 1-chloro-2-[chloro(diphenyl)methyl]benzene Chemical compound ClC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 JFLSOKIMYBSASW-UHFFFAOYSA-N 0.000 description 7
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 150000001408 amides Chemical class 0.000 description 7
- 239000007998 bicine buffer Substances 0.000 description 7
- 238000005277 cation exchange chromatography Methods 0.000 description 7
- 125000006413 ring segment Chemical group 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- 235000013877 carbamide Nutrition 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 239000008103 glucose Substances 0.000 description 6
- 238000011068 loading method Methods 0.000 description 6
- 239000012064 sodium phosphate buffer Substances 0.000 description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 239000007821 HATU Substances 0.000 description 5
- FSVCELGFZIQNCK-UHFFFAOYSA-N N,N-bis(2-hydroxyethyl)glycine Chemical compound OCCN(CCO)CC(O)=O FSVCELGFZIQNCK-UHFFFAOYSA-N 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 150000004985 diamines Chemical class 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- 235000019439 ethyl acetate Nutrition 0.000 description 5
- 125000005843 halogen group Chemical group 0.000 description 5
- 150000003840 hydrochlorides Chemical class 0.000 description 5
- 229940125396 insulin Drugs 0.000 description 5
- 239000012038 nucleophile Substances 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- AOCSUUGBCMTKJH-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCN AOCSUUGBCMTKJH-UHFFFAOYSA-N 0.000 description 5
- 239000003039 volatile agent Substances 0.000 description 5
- FODJWPHPWBKDON-IBGZPJMESA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-4-[(2-methylpropan-2-yl)oxy]-4-oxobutanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CC(=O)OC(C)(C)C)C(O)=O)C3=CC=CC=C3C2=C1 FODJWPHPWBKDON-IBGZPJMESA-N 0.000 description 4
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 description 4
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 4
- RDRBIXSNGAYLPT-UHFFFAOYSA-N CC1=CC=C(COC2=CC3=C(C=C2)C(NC(=O)OCC2C4=C(C=CC=C4)C4=C2C=CC=C4)C2=C(O3)C=CC=C2)C=C1 Chemical compound CC1=CC=C(COC2=CC3=C(C=C2)C(NC(=O)OCC2C4=C(C=CC=C4)C4=C2C=CC=C4)C2=C(O3)C=CC=C2)C=C1 RDRBIXSNGAYLPT-UHFFFAOYSA-N 0.000 description 4
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 4
- 239000004202 carbamide Substances 0.000 description 4
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 4
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 4
- 238000004108 freeze drying Methods 0.000 description 4
- DILRJUIACXKSQE-UHFFFAOYSA-N n',n'-dimethylethane-1,2-diamine Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 238000001542 size-exclusion chromatography Methods 0.000 description 4
- 239000001509 sodium citrate Substances 0.000 description 4
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 4
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- DYLAXLLCKLPULN-UHFFFAOYSA-N 2-bromo-n-(2-tritylsulfanylethyl)acetamide Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SCCNC(=O)CBr)C1=CC=CC=C1 DYLAXLLCKLPULN-UHFFFAOYSA-N 0.000 description 3
- IUTPJBLLJJNPAJ-UHFFFAOYSA-N 3-(2,5-dioxopyrrol-1-yl)propanoic acid Chemical compound OC(=O)CCN1C(=O)C=CC1=O IUTPJBLLJJNPAJ-UHFFFAOYSA-N 0.000 description 3
- ILAPAFBLFPIIBL-UHFFFAOYSA-N 6-tritylsulfanylhexanoic acid Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SCCCCCC(=O)O)C1=CC=CC=C1 ILAPAFBLFPIIBL-UHFFFAOYSA-N 0.000 description 3
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 3
- DYHSDKLCOJIUFX-UHFFFAOYSA-N Di-tert-butyl dicarbonate Substances CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- FSQQTNAZHBEJLS-UPHRSURJSA-N maleamic acid Chemical compound NC(=O)\C=C/C(O)=O FSQQTNAZHBEJLS-UPHRSURJSA-N 0.000 description 3
- 239000011859 microparticle Substances 0.000 description 3
- 229940124531 pharmaceutical excipient Drugs 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 230000028327 secretion Effects 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- UWYZHKAOTLEWKK-UHFFFAOYSA-N 1,2,3,4-tetrahydroisoquinoline Chemical compound C1=CC=C2CNCCC2=C1 UWYZHKAOTLEWKK-UHFFFAOYSA-N 0.000 description 2
- LBUJPTNKIBCYBY-UHFFFAOYSA-N 1,2,3,4-tetrahydroquinoline Chemical compound C1=CC=C2CCCNC2=C1 LBUJPTNKIBCYBY-UHFFFAOYSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- AECGEIVNZGQBJT-UHFFFAOYSA-N 3-tritylsulfanylpropanoic acid Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SCCC(=O)O)C1=CC=CC=C1 AECGEIVNZGQBJT-UHFFFAOYSA-N 0.000 description 2
- JLAKCHGEEBPDQI-UHFFFAOYSA-N 4-(4-fluorobenzyl)piperidine Chemical compound C1=CC(F)=CC=C1CC1CCNCC1 JLAKCHGEEBPDQI-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- FZTIWOBQQYPTCJ-UHFFFAOYSA-N 4-[4-(4-carboxyphenyl)phenyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=C(C=2C=CC(=CC=2)C(O)=O)C=C1 FZTIWOBQQYPTCJ-UHFFFAOYSA-N 0.000 description 2
- KIVCMHJKLQHCCI-UHFFFAOYSA-N 6-tritylsulfanylhexan-1-ol Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SCCCCCCO)C1=CC=CC=C1 KIVCMHJKLQHCCI-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 108090000531 Amidohydrolases Proteins 0.000 description 2
- 102000004092 Amidohydrolases Human genes 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 108090000371 Esterases Proteins 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102000051325 Glucagon Human genes 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000012062 aqueous buffer Substances 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000011033 desalting Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 238000005538 encapsulation Methods 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000004255 ion exchange chromatography Methods 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000006574 non-aromatic ring group Chemical group 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- KNCYXPMJDCCGSJ-UHFFFAOYSA-N piperidine-2,6-dione Chemical group O=C1CCCC(=O)N1 KNCYXPMJDCCGSJ-UHFFFAOYSA-N 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 230000000291 postprandial effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910000162 sodium phosphate Inorganic materials 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical group O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- PTMFUWGXPRYYMC-UHFFFAOYSA-N triethylazanium;formate Chemical compound OC=O.CCN(CC)CC PTMFUWGXPRYYMC-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 2
- 150000003672 ureas Chemical class 0.000 description 2
- KLBPUVPNPAJWHZ-UMSFTDKQSA-N (2r)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-tritylsulfanylpropanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)SC(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 KLBPUVPNPAJWHZ-UMSFTDKQSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- VZXQYACYLGRQJU-IBGZPJMESA-N (3s)-3-(9h-fluoren-9-ylmethoxycarbonylamino)-4-[(2-methylpropan-2-yl)oxy]-4-oxobutanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CC(O)=O)C(=O)OC(C)(C)C)C3=CC=CC=C3C2=C1 VZXQYACYLGRQJU-IBGZPJMESA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- WSEVKKHALHSUMB-RYVRVIGHSA-N (4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2R)-5-amino-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-carboxypropanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-5-oxopentanoyl]amino]-4-methylsulfanylbutanoyl]amino]-4-carboxybutanoyl]amino]-4-carboxybutanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-4-amino-1-[[2-[[2-[(2S)-2-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-1-[(2S)-2-[(2S)-2-[(2S)-2-[[(2S)-1-amino-3-hydroxy-1-oxopropan-2-yl]carbamoyl]pyrrolidine-1-carbonyl]pyrrolidine-1-carbonyl]pyrrolidin-1-yl]-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-2-oxoethyl]amino]-2-oxoethyl]amino]-1,4-dioxobutan-2-yl]amino]-1-oxohexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N1CCC[C@H]1C(=O)N1CCC[C@H]1C(=O)N[C@@H](CO)C(N)=O WSEVKKHALHSUMB-RYVRVIGHSA-N 0.000 description 1
- CSWRAOHDICNMPU-LLZJGCNPSA-N (4r)-n-tert-butyl-3-[(2s,3s)-3-[[2-(2,6-dimethylphenoxy)acetyl]amino]-2-hydroxy-4-phenylbutanoyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide Chemical compound CC1=CC=CC(C)=C1OCC(=O)N[C@H]([C@H](O)C(=O)N1[C@@H](C(C)(C)SC1)C(=O)NC(C)(C)C)CC1=CC=CC=C1 CSWRAOHDICNMPU-LLZJGCNPSA-N 0.000 description 1
- GPYKKBAAPVOCIW-HSASPSRMSA-N (6r,7s)-7-[[(2r)-2-amino-2-phenylacetyl]amino]-3-chloro-8-oxo-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid;hydrate Chemical compound O.C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CC[C@@H]32)C(O)=O)=O)N)=CC=CC=C1 GPYKKBAAPVOCIW-HSASPSRMSA-N 0.000 description 1
- 125000006555 (C3-C5) cycloalkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- BYEAHWXPCBROCE-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoropropan-2-ol Chemical compound FC(F)(F)C(O)C(F)(F)F BYEAHWXPCBROCE-UHFFFAOYSA-N 0.000 description 1
- NENLYAQPNATJSU-UHFFFAOYSA-N 1,2,3,4,4a,5,6,7,8,8a-decahydroisoquinoline Chemical compound C1NCCC2CCCCC21 NENLYAQPNATJSU-UHFFFAOYSA-N 0.000 description 1
- POTIYWUALSJREP-UHFFFAOYSA-N 1,2,3,4,4a,5,6,7,8,8a-decahydroquinoline Chemical compound N1CCCC2CCCCC21 POTIYWUALSJREP-UHFFFAOYSA-N 0.000 description 1
- UUZJJNBYJDFQHL-UHFFFAOYSA-N 1,2,3-triazolidine Chemical compound C1CNNN1 UUZJJNBYJDFQHL-UHFFFAOYSA-N 0.000 description 1
- CSNIZNHTOVFARY-UHFFFAOYSA-N 1,2-benzothiazole Chemical compound C1=CC=C2C=NSC2=C1 CSNIZNHTOVFARY-UHFFFAOYSA-N 0.000 description 1
- KTZQTRPPVKQPFO-UHFFFAOYSA-N 1,2-benzoxazole Chemical compound C1=CC=C2C=NOC2=C1 KTZQTRPPVKQPFO-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- IOEPOEDBBPRAEI-UHFFFAOYSA-N 1,2-dihydroisoquinoline Chemical compound C1=CC=C2CNC=CC2=C1 IOEPOEDBBPRAEI-UHFFFAOYSA-N 0.000 description 1
- IRFSXVIRXMYULF-UHFFFAOYSA-N 1,2-dihydroquinoline Chemical compound C1=CC=C2C=CCNC2=C1 IRFSXVIRXMYULF-UHFFFAOYSA-N 0.000 description 1
- CIISBYKBBMFLEZ-UHFFFAOYSA-N 1,2-oxazolidine Chemical compound C1CNOC1 CIISBYKBBMFLEZ-UHFFFAOYSA-N 0.000 description 1
- CZSRXHJVZUBEGW-UHFFFAOYSA-N 1,2-thiazolidine Chemical compound C1CNSC1 CZSRXHJVZUBEGW-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- FQUYSHZXSKYCSY-UHFFFAOYSA-N 1,4-diazepane Chemical compound C1CNCCNC1 FQUYSHZXSKYCSY-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- DQFQCHIDRBIESA-UHFFFAOYSA-N 1-benzazepine Chemical compound N1C=CC=CC2=CC=CC=C12 DQFQCHIDRBIESA-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- JIOMOSMOFRFEGZ-UHFFFAOYSA-N 1-chloro-3-[chloro(diphenyl)methyl]benzene Chemical compound ClC1=CC=CC(C(Cl)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 JIOMOSMOFRFEGZ-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 1
- OGNSCSPNOLGXSM-UHFFFAOYSA-N 2,4-diaminobutyric acid Chemical compound NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- DNITTWABAYHZQU-UHFFFAOYSA-N 2-(2,5-dioxopyrrol-1-yl)propanamide Chemical compound NC(=O)C(C)N1C(=O)C=CC1=O DNITTWABAYHZQU-UHFFFAOYSA-N 0.000 description 1
- OXQGTIUCKGYOAA-UHFFFAOYSA-N 2-Ethylbutanoic acid Chemical compound CCC(CC)C(O)=O OXQGTIUCKGYOAA-UHFFFAOYSA-N 0.000 description 1
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 description 1
- PHXRLXSFXHFCPA-UHFFFAOYSA-N 2-[4-(2-aminoethyl)piperazin-1-yl]acetic acid Chemical compound NCCN1CCN(CC(O)=O)CC1 PHXRLXSFXHFCPA-UHFFFAOYSA-N 0.000 description 1
- RGNOTKMIMZMNRX-XVFCMESISA-N 2-amino-1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-4-one Chemical compound NC1=NC(=O)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 RGNOTKMIMZMNRX-XVFCMESISA-N 0.000 description 1
- LSTRKXWIZZZYAS-UHFFFAOYSA-N 2-bromoacetyl bromide Chemical compound BrCC(Br)=O LSTRKXWIZZZYAS-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- XZOWICPSVWHCTC-UHFFFAOYSA-N 2-tritylsulfanylethanamine Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SCCN)C1=CC=CC=C1 XZOWICPSVWHCTC-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- JYNHRDJTWNEGJE-UHFFFAOYSA-N 3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(9h-fluoren-9-ylmethoxycarbonylamino)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoic acid Chemical compound C1=CC=C2C(COC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCC(=O)O)C3=CC=CC=C3C2=C1 JYNHRDJTWNEGJE-UHFFFAOYSA-N 0.000 description 1
- IHBVNSPHKMCPST-UHFFFAOYSA-N 3-bromopropanoyl chloride Chemical compound ClC(=O)CCBr IHBVNSPHKMCPST-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- WEQPBCSPRXFQQS-UHFFFAOYSA-N 4,5-dihydro-1,2-oxazole Chemical compound C1CC=NO1 WEQPBCSPRXFQQS-UHFFFAOYSA-N 0.000 description 1
- GUUULVAMQJLDSY-UHFFFAOYSA-N 4,5-dihydro-1,2-thiazole Chemical compound C1CC=NS1 GUUULVAMQJLDSY-UHFFFAOYSA-N 0.000 description 1
- WEDKTMOIKOKBSH-UHFFFAOYSA-N 4,5-dihydrothiadiazole Chemical compound C1CN=NS1 WEDKTMOIKOKBSH-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- ZLOIGESWDJYCTF-UHFFFAOYSA-N 4-Thiouridine Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=S)C=C1 ZLOIGESWDJYCTF-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- ZLOIGESWDJYCTF-XVFCMESISA-N 4-thiouridine Chemical class O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=S)C=C1 ZLOIGESWDJYCTF-XVFCMESISA-N 0.000 description 1
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 1
- FCMCSZXRVWDVAW-UHFFFAOYSA-N 6-bromo-1-hexanol Chemical compound OCCCCCCBr FCMCSZXRVWDVAW-UHFFFAOYSA-N 0.000 description 1
- HBPVGJGBRWIVSX-UHFFFAOYSA-N 6-bromohexanoyl chloride Chemical compound ClC(=O)CCCCCBr HBPVGJGBRWIVSX-UHFFFAOYSA-N 0.000 description 1
- DKCVJKZTTJDMMM-UHFFFAOYSA-N 6-tritylsulfanylhexanal Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SCCCCCC=O)C1=CC=CC=C1 DKCVJKZTTJDMMM-UHFFFAOYSA-N 0.000 description 1
- DGGKXQQCVPAUEA-UHFFFAOYSA-N 8-azabicyclo[3.2.1]octane Chemical compound C1CCC2CCC1N2 DGGKXQQCVPAUEA-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- ZXGVRMNJCJXPAV-WBLYCXFISA-N CCCCCCCCCCCC(NC(CCCCNC(CCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCNC(CCN(C(CC1SCCNC(CN2CCN(CCNC([C@H](CCCC3)[C@H]3C(N(CC3)CCN3C(c3c4)=Nc5ccccc5Oc3ccc4Cl)=O)=O)CC2)=O)=O)C1=O)=O)=O)C(N)=O)=O Chemical compound CCCCCCCCCCCC(NC(CCCCNC(CCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCNC(CCN(C(CC1SCCNC(CN2CCN(CCNC([C@H](CCCC3)[C@H]3C(N(CC3)CCN3C(c3c4)=Nc5ccccc5Oc3ccc4Cl)=O)=O)CC2)=O)=O)C1=O)=O)=O)C(N)=O)=O ZXGVRMNJCJXPAV-WBLYCXFISA-N 0.000 description 1
- 101100054570 Caenorhabditis elegans acn-1 gene Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- NTURVSFTOYPGON-UHFFFAOYSA-N Dihydroquinazoline Chemical compound C1=CC=C2C=NCNC2=C1 NTURVSFTOYPGON-UHFFFAOYSA-N 0.000 description 1
- 241000854350 Enicospilus group Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- 102400000322 Glucagon-like peptide 1 Human genes 0.000 description 1
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 1
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101000825742 Homo sapiens Somatoliberin Proteins 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- 208000022120 Jeavons syndrome Diseases 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical compound C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- WINXNKPZLFISPD-UHFFFAOYSA-M Saccharin sodium Chemical compound [Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 WINXNKPZLFISPD-UHFFFAOYSA-M 0.000 description 1
- 102400000827 Saposin-D Human genes 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 1
- 102000055135 Vasoactive Intestinal Peptide Human genes 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000002820 allylidene group Chemical group [H]C(=[*])C([H])=C([H])[H] 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000001195 anabolic effect Effects 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000002098 anti-diabetogenic effect Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- XYOVOXDWRFGKEX-UHFFFAOYSA-N azepine Chemical compound N1C=CC=CC=C1 XYOVOXDWRFGKEX-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- MXMZCLLIUQEKSN-UHFFFAOYSA-N benzimidazoline Chemical compound C1=CC=C2NCNC2=C1 MXMZCLLIUQEKSN-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000006065 biodegradation reaction Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- IOVVFSGCNWQFQT-UHFFFAOYSA-N bis(2,3,4,5,6-pentafluorophenyl) carbonate Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1OC(=O)OC1=C(F)C(F)=C(F)C(F)=C1F IOVVFSGCNWQFQT-UHFFFAOYSA-N 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 235000019577 caloric intake Nutrition 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 239000007857 degradation product Substances 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- QPMLSUSACCOBDK-UHFFFAOYSA-N diazepane Chemical compound C1CCNNCC1 QPMLSUSACCOBDK-UHFFFAOYSA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 230000009881 electrostatic interaction Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 108010024703 exendin (9-39) Proteins 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 235000013312 flour Nutrition 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 230000010030 glucose lowering effect Effects 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 150000003949 imides Chemical group 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 230000002473 insulinotropic effect Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 229920001427 mPEG Polymers 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 108091070501 miRNA Proteins 0.000 description 1
- 239000002679 microRNA Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- LLYKPZOWCPVRPD-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine;n,n-dimethylpyridin-4-amine Chemical compound CN(C)C1=CC=NC=C1.CN(C)C1=CC=CC=N1 LLYKPZOWCPVRPD-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical class C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 230000001019 normoglycemic effect Effects 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- NENPYTRHICXVCS-YNEHKIRRSA-N oseltamivir acid Chemical compound CCC(CC)O[C@@H]1C=C(C(O)=O)C[C@H](N)[C@H]1NC(C)=O NENPYTRHICXVCS-YNEHKIRRSA-N 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 150000002918 oxazolines Chemical class 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- WBXPDJSOTKVWSJ-ZDUSSCGKSA-N pemetrexed Chemical compound C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 WBXPDJSOTKVWSJ-ZDUSSCGKSA-N 0.000 description 1
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 1
- 239000000813 peptide hormone Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 150000003141 primary amines Chemical group 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 125000006238 prop-1-en-1-yl group Chemical group [H]\C(*)=C(/[H])C([H])([H])[H] 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- CPNGPNLZQNNVQM-UHFFFAOYSA-N pteridine Chemical compound N1=CN=CC2=NC=CN=C21 CPNGPNLZQNNVQM-UHFFFAOYSA-N 0.000 description 1
- USPWKWBDZOARPV-UHFFFAOYSA-N pyrazolidine Chemical compound C1CNNC1 USPWKWBDZOARPV-UHFFFAOYSA-N 0.000 description 1
- DNXIASIHZYFFRO-UHFFFAOYSA-N pyrazoline Chemical compound C1CN=NC1 DNXIASIHZYFFRO-UHFFFAOYSA-N 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- ZVJHJDDKYZXRJI-UHFFFAOYSA-N pyrroline Natural products C1CC=NC1 ZVJHJDDKYZXRJI-UHFFFAOYSA-N 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000005932 reductive alkylation reaction Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000000452 restraining effect Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000036186 satiety Effects 0.000 description 1
- 235000019627 satiety Nutrition 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid Chemical compound NS(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- QYJVBVKFXDHFPQ-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)-n-methylcarbamate Chemical compound NCCN(C)C(=O)OC(C)(C)C QYJVBVKFXDHFPQ-UHFFFAOYSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- ZYXWYDDFNXBTFO-UHFFFAOYSA-N tetrazolidine Chemical compound C1NNNN1 ZYXWYDDFNXBTFO-UHFFFAOYSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- RLTPJVKHGBFGQA-UHFFFAOYSA-N thiadiazolidine Chemical compound C1CSNN1 RLTPJVKHGBFGQA-UHFFFAOYSA-N 0.000 description 1
- CBDKQYKMCICBOF-UHFFFAOYSA-N thiazoline Chemical compound C1CN=CS1 CBDKQYKMCICBOF-UHFFFAOYSA-N 0.000 description 1
- XSROQCDVUIHRSI-UHFFFAOYSA-N thietane Chemical compound C1CSC1 XSROQCDVUIHRSI-UHFFFAOYSA-N 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- MYPYJXKWCTUITO-LYRMYLQWSA-N vancomycin Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C(O)=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)NC)[C@H]1C[C@](C)(N)[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-N 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/1703—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- A61K38/1709—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/18—Growth factors; Growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/26—Glucagons
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/542—Carboxylic acids, e.g. a fatty acid or an amino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/545—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
Definitions
- Prodrug comprising a drug linker conjugate
- the present invention relates to a prodrug or a pharmaceutically acceptable salt thereof comprising a drug linker conjugate D-L.
- the invention also relates to pharmaceutical compositions comprising said prodrugs and their use as medicaments.
- Such drug can be conjugated with a carrier.
- carriers in drug delivery are either used in a non-covalent fashion, with the drug physicochemically formulated into a solvent-carrier mixture, or by covalent attachment of a carrier reagent to one of the drug's functional groups.
- the drugs may be conjugated to a carrier through covalent bonds.
- This approach is applied to various classes of molecules, from so-called small molecules, through natural products up to larger proteins.
- Covalent drug carrier conjugates can be divided into two groups. Firstly, conjugates, where the covalent bond between carrier and drug is mostly present during the action of the drug ("permanent covalent bond"), i.e. a derivative of the drug exhibits its pharmacological effects as it is known for the drug as such. Secondly, the covalent bond is mostly previously cleaved to release the drug as such, which can exhibit its known pharmacological effects. In the latter case the covalent drug carrier conjugate is called carrier linked prodrug or carrier prodrug. In order to ensure cleavage of the covalent bond between carrier and drug easy removal of said bond in vivo is required to release the drug (prodrug activation).
- Prodrug activation may occur by enzymatic or non-enzymatic cleavage of the bond between the carrier and the drug molecule, or a sequential combination of both, i.e. an enzymatic step followed by a non-enzymatic rearrangement.
- Enzymatically induced prodrug activation is characterized in that the cleavage in enzyme-free in-vitro environment such as an aqueous buffer solution, of, e.g., an ester or amide may occur, but the corresponding rate of hydrolysis may be much too slow and not therapeutically useful.
- enzyme-free in-vitro environment such as an aqueous buffer solution
- esterases or amidases are typically present and the esterases and amidases may cause significant catalytic acceleration of the kinetics of hydrolysis from twofold up to several orders of magnitude. Therefore, the cleavage is predominantly controlled by the enzymatic reaction.
- Enzyme levels may differ significantly between individuals resulting in biological variation of prodrug activation by the enzymatic cleavage.
- the enzyme levels may also vary depending on the site of administration. For instance it is known that in the case of subcutaneous injection, certain areas of the body yield more predictable therapeutic effects than others. To reduce this unpredictable effect, non-enzymatic cleavage or intramolecular catalysis is of particular interest.
- enzyme-independent autocatalytic cleavage of carrier and biologically active moiety is preferred. In most cases this is achieved by an appropriately designed linker moiety between the carrier and the biologically active moiety, which is directly attached to the functional group of a biologically active moiety via covalent bond.
- ester based prodrugs where the carrier is water-soluble and the biologically active moiety is derived from HIV-I protease inhibitor KNI-727.
- the linker used is attached to the biologically active moiety via ester group.
- the mechanism of this prodrug system is cyclization-activation by cyclic imide formation for the cleavage of ester bonds.
- ester groups may be less chemoselectively addressable for the conjugation of the carrier or linker and the drug.
- prodrug system based on N,N-bis-(2-hydroxyethyl)glycine amide (bicine) linker is described.
- two PEG carrier molecules are linked to a bicine molecule coupled to an amino group of the drug molecule.
- the first two steps in prodrug activation is the enzymatic cleavage of the first linkages connecting both PEG carrier molecules with the hydroxy groups of the bicine activating group.
- Different linkages between PEG and bicine are described resulting in different prodrug activation kinetics.
- the second step in prodrug activation is the cleavage of the second linkage connecting the bicine activating group to the amino group of the drug molecule.
- an object of the present invention is to provide such drug linker conjugates, where the linker is covalently attached via a cleavable bond to a biologically active moiety (representing the drug after release), and where the linker is further covalently attached via a permanent bond to a carrier directly or via a spacer to form the carrier- linked prodrug.
- -D is a nitrogen containing biologically active moiety
- -L is a non-bio logically active linker moiety -L 1 represented by formula (I),
- X is C(R 4 R 4a ); N(R 4 ); O; C(R 4 R 4a )-C(R 5 R 5a ); C(R 5 R 5a )-C(R 4 R 4a ); C(R 4 R 4a )-N(R 6 ); N(R 6 )- C(R 4 R 4a ); C(R 4 R 4a )-O; or O-C(R 4 R 4a );
- X I is C; or S(O);
- X 2 is C(R 7 , R 7a ); or C(R 7 , R 7a )-C(R 8 , R 8a );
- X 3 is O; S; or N-CN;
- R 1 , R la , R 2 , R 2a , R 3 , R 3a , R 4 , R 4a , R 5 , R 5a , R 6 , R 7 , R 7a , R 8 , R 8a are independently selected from the group consisting of H; and Ci_ 4 alkyl;
- one or more of the pairs R la /R 4a , R la /R 5a , R 4a /R 5a , R 7a /R 8a form a chemical bond;
- one or more of the pairs RVR la , R 2 /R 2a , R 4 /R 4a , R 5 /R 5a , R 7 /R 7a , R 8 /R 8a are joined together with the atom to which they are attached to form a C 3 _ 7 cycloalkyl; or 4 to 7 membered heterocyclyl;
- R 4 /R 6 are joined together with the atoms to which they are attached to form a saturated 4 to 7 membered heterocyclyl;
- one or more of the pairs RVR 4 , RVR 5 , RVR 6 , R 4 /R 5 , R 4 /R 6 , R 7 /R 8 , R 2 /R 3 are joined together with the atoms to which they are attached to form a ring A;
- R 3 /R 3a are joined together with the nitrogen atom to which they are attached to form a 4 to 7 membered heterocycle;
- A is selected from the group consisting of phenyl; naphthyl; indenyl; indanyl; tetralinyl; C 3-10 cycloalkyl; 4 to 7 membered heterocyclyl; and 9 to 11 membered heterobicyclyl; and
- L 1 is substituted with one to four groups L 2 -Z and optionally further substituted, provided that the hydrogen marked with the asterisk in formula (I) is not replaced by a substituent;
- L 2 is a single chemical bond or a spacer; and Z is a carrier group.
- Examples for such preferred cyclic cleavage products are substituted succinimide or glutarimide ring structures.
- Prerequisite for such cyclization activation is the presence of an amine-containing nucleophile in the linker structure and another amide bond which is not the amide prodrug bond but an amide bond substituted with a hydrogen atom.
- the amine-containing nucleophile serves as a neighbouring group to enhance the nucleophilicity of the nitrogen contained in the permanent amide bond which in turn attacks the prodrug amide carbonyl group and consequently induces intramolecular acylation of the permanent amide bond generating the cyclic imide ring.
- linker structures comprise a permanent linkage to a carrier, an amine- containing nucleophile, and a permanent amide bond with a hydrogen attached to the nitrogen of the amide bond.
- Corresponding carrier- linked prodrugs comprise a linker containing a permanent linkage to a carrier, an amine-containing nucleophile and said permanent amide bond, and a nitrogen containing biologically active moiety derived from the drug conjugated to the linker by means of a cleavable amide bond.
- Fig. 1 shows an example of the cleavage resulting in a cyclic imide.
- the nitrogen of the biologically active moiety is shown as hydrogen containing amine, which results in a drug having a primary amine functional group.
- a secondary amine may be part of the drug.
- the one to four mandatory substituents L 2 -Z including the carrier are not shown.
- Preferred properties of the prodrug are given by a half- life of hydrolysis in aqueous buffer at pH 7.4 and 37°C between 1 h and 3 months; similar rates of hydrolysis under physiological conditions in buffer and plasma.
- the prodrug according to the present invention may show excellent in vivo/in vitro correlation of linker cleavage, a high degree of enzyme independence and can be stored at lower pH (pH dependent cleavage).
- Bioly active moiety D means the part of the drug linker conjugate, which results after cleavage in a drug D-H of known biological activity.
- Non-active linker means a linker which does not show the pharmacological effects of the drug derived from the biologically active agent.
- Alkyl means a straight-chain or branched carbon chain. Each hydrogen of an alkyl carbon may be replaced by a substituent.
- Ci_ 4 alkyl means an alkyl chain having 1 - 4 carbon atoms, e.g. if present at the end of a molecule: methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl tert-butyl, or e.g. - CH 2 -, -CH 2 -CH 2 -, -CH(CH 3 )-, -CH 2 -CH 2 -CH 2 -, -CH(C 2 H 5 )-, -C(CH 3 ) 2 -, when two moieties of a molecule are linked by the alkyl group.
- Each hydrogen of a Ci_4 alkyl carbon may be replaced by a substituent.
- Ci_6 alkyl means an alkyl chain having 1 - 6 carbon atoms, e.g. if present at the end of a molecule: Ci_ 4 alkyl, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl; tert-butyl, n-pentyl, n-hexyl, or e.g.
- Ci_6 alkyl carbon when two moieties of a molecule are linked by the alkyl group.
- Each hydrogen of a Ci_6 alkyl carbon may be replaced by a substituent.
- C 1-18 alkyl means an alkyl chain having 1 to 18 carbon atoms and "C 8-18 alkyl” means an alkyl chain having 8 to 18 carbon atoms. Accordingly, “Ci_ 5 o alkyl” means an alkyl chain having 1 to 50 carbon atoms.
- Each hydrogen of a C 2 _ 5 o alkenyl carbon may be replaced by a substituent as further specified. Accordingly, the term "alkenyl” relates to a carbon chain with at least one carbon carbon double bond. Optionally, one or more triple bonds may occur.
- C 2 _ 5 o alkynyl means a branched or unbranched alkynyl chain having 2 to 50 carbon atoms, e.g. if present at the end of a molecule: -C ⁇ CH, -CH 2 -C ⁇ CH, CH 2 -CH 2 -C ⁇ CH, CH 2 -C ⁇ C- CH 3 , or e.g. -C ⁇ C- when two moieties of a molecule are linked by the alkynyl group.
- Each hydrogen of a C2-50 alkynyl carbon may be replaced by a substituent as further specified.
- alkynyl relates to a carbon chaim with at lest one carbon carbon triple bond. Optionally, one or more double bonds may occur.
- C 3 _ 7 cycloalkyl or “C 3 _ 7 cycloalkyl ring” means a cyclic alkyl chain having 3 to 7 carbon atoms, which may have carbon-carbon double bonds being at least partially saturated, e.g. cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, cycloheptyl. Each hydrogen of a cycloalkyl carbon may be replaced by a substituent.
- the term “C 3 _ 7 cycloalkyl” or “C 3 _ 7 cycloalkyl ring” also includes bridged bicycles like norbonane or norbonene. Accordingly, "C 3 _ 5 cycloalkyl” means a cycloalkyl having 3 to 5 carbon atoms.
- C3_io cycloalkyl means a cyclic alkyl having 3 to 10 carbon atoms, e.g. C3_7 cycloalkyl; cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl.
- C 3-10 cycloalkyl also includes at least partially saturated carbomono- and -bicycles.
- Halogen means fluoro, chloro, bromo or iodo. It is generally preferred that halogen is fluoro or chloro.
- Examples for a 4 to 7 membered heterocycles are azetidine, oxetane, thietane, furan, thiophene, pyrrole, pyrroline, imidazole, imidazoline, pyrazole, pyrazoline, oxazole, oxazoline, isoxazole, isoxazoline, thiazole, thiazoline, isothiazole, isothiazoline, thiadiazole, thiadiazoline, tetrahydro furan, tetrahydrothiophene, pyrrolidine, imidazolidine, pyrazolidine, oxazolidine, isoxazolidine, thiazolidine, isothiazolidine, thiadiazolidine, sulfolane, pyran, dihydropyran, tetrahydropyran, imidazolidine, pyridine, pyridazine, pyrazine, pyr
- Examples for a 9 to 11 membered heterobicycle are indole, indoline, benzofuran, benzothiophene, benzoxazole, benzisoxazole, benzothiazole, benzisothiazole, benzimidazole, benzimidazoline, quinoline, quinazoline, dihydroquinazoline, quinoline, dihydroquinoline, tetrahydroquinoline, decahydroquinoline, isoquinoline, decahydroisoquinoline, tetrahydroisoquinoline, dihydroisoquinoline, benzazepine, purine or pteridine.
- 9 to 11 membered heterobicycle also includes spiro structures of two rings like l,4-dioxa-8-azaspiro[4.5]decane or bridged heterocycles like 8-aza-bicyclo[3.2.1]octane.
- the invention also comprises their corresponding pharmaceutically or toxicologically acceptable salts, in particular their pharmaceutically utilizable salts.
- the compounds of the formula (I) which contain acidic groups can be used according to the invention, for example, as alkali metal salts, alkaline earth metal salts or as ammonium salts. More precise examples of such salts include sodium salts, potassium salts, calcium salts, magnesium salts or salts with ammonia or organic amines such as, for example, ethylamine, ethanolamine, triethanolamine or amino acids.
- Compounds of the formula (I) which contain one or more basic groups i.e.
- acids which can be protonated, can be present and can be used according to the invention in the form of their addition salts with inorganic or organic acids.
- suitable acids include hydrogen chloride, hydrogen bromide, phosphoric acid, sulfuric acid, nitric acid, methanesulfonic acid, p-toluenesulfonic acid, naphthalenedisulfonic acids, oxalic acid, acetic acid, tartaric acid, lactic acid, salicylic acid, benzoic acid, formic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid, pimelic acid, fumaric acid, maleic acid, malic acid, sulfaminic acid, phenylpropionic acid, gluconic acid, ascorbic acid, isonicotinic acid, citric acid, adipic acid, and other acids known to the person skilled in the art.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions).
- the respective salts according to the formula (I) can be obtained by customary methods which are known to the person skilled in the art like, for example by contacting these with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange with other salts.
- the present invention also includes all salts of the compounds of the formula (I) which, owing to low physiological compatibility, are not directly suitable for use in pharmaceuticals but which can be used, for example, as intermediates for chemical reactions or for the preparation of pharmaceutically acceptable salts.
- pharmaceutically acceptable means approved by a regulatory agency such as the EMEA (Europe) and/or the FDA (US) and/or any other national regulatory agency for use in animals, preferably in humans.
- “Pharmaceutical composition” means one or more active ingredients, and one or more inert ingredients, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable excipient (pharmaceutically acceptable carrier).
- excipient refers to a diluent, adjuvant, or vehicle with which the therapeutic is administered.
- Such pharmaceutical excipient can be sterile liquids, such as water and oils, including those of petroleum, animal, vegetable or synthetic origin, including but not limited to peanut oil, soybean oil, mineral oil, sesame oil and the like.
- Water is a preferred excipient when the pharmaceutical composition is administered orally.
- Saline and aqueous dextrose are preferred excipients when the pharmaceutical composition is administered intravenously.
- Saline solutions and aqueous dextrose and glycerol solutions are preferably employed as liquid excipients for injectable solutions.
- Suitable pharmaceutical excipients include starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol, propylene, glycol, water, ethanol and the like.
- the composition if desired, can also contain minor amounts of wetting or emulsifying agents, or pH buffering agents. These compositions can take the form of solutions, suspensions, emulsions, tablets, pills, capsules, powders, sustained-release formulations and the like.
- the composition can be formulated as a suppository, with traditional binders and excipients such as triglycerides.
- Oral formulation can include standard excipients such as pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate, etc. Examples of suitable pharmaceutical excipients are described in "Remington's Pharmaceutical Sciences” by E.W. Martin. Such compositions will contain a therapeutically effective amount of the therapeutic, preferably in purified form, together with a suitable amount of excipient so as to provide the form for proper administration to the patient. The formulation should suit the mode of administration.
- X 3 is O.
- X is N(R 4 ), X 1 is C and X 3 is O.
- X 2 is C(R 7 R 7a ).
- L 1 is selected from the group consisting of
- R is H; or Ci -4 alkyl
- Y is NH; O; or S
- R 1 , R la , R 2 , R 2a , R 3 , R 3a , R 4 , X, X 1 , X 2 have the meaning as indicated above.
- L 1 is selected from the group consisting of
- At least one (up to four) hydrogen is replaced by a group L 2 -Z.
- each L 2 and each Z can be selected independently.
- L 2 can be attached to L 1 at any position apart from the replacement of the hydrogen marked with an asterisk in formula (I).
- one to four of the hydrogen given by R, R 1 to R 8 directly or as hydrogen of the Ci_ 4 alkyl or further groups and rings given by the definition of R and R 1 to R 8 are replaced by L 2 -Z.
- L 1 may be optionally further substituted.
- any substituent may be used as far as the cleavage principle is not affected.
- one or more further optional substituents are independently selected from the group consisting of halogen; CN; COOR 9 ; OR 9 ; C(O)R 9 ; C(O)N(R 9 R 9a ); S(O) 2 N(R 9 R 9a ); S(O)N(R 9 R 9a ); S(O) 2 R 9 ; S(O)R 9 ; N(R 9 )S(O) 2 N(R 9a R 9b ); SR 9 ; N(R 9 R 9a ); NO 2 ; OC(O)R 9 ; N(R 9 )C(O)R 9a ; N(R 9 )S(O) 2 R 9a ; N(R 9 )S(O)R 9a ; N(R 9 )C(O)OR 9a ; N(R 9 )C(O)N(R 9a R 9b ); OC(O)N(R 9 R 9a ); T; Ci -50 alkyl; C
- T is selected from the group consisting of phenyl; naphthyl; indenyl; indanyl; tetralinyl; C 3-10 cycloalkyl; 4 to 7 membered heterocyclyl; or 9 to 11 membered heterobicyclyl, wherein T is optionally substituted with one or more R 10 , which are the same or different;
- R 11 , R l la , R 12 , R 12a , R 12b are independently selected from the group consisting of H; or Ci_6 alkyl, wherein Ci_6 alkyl is optionally substituted with one or more halogen, which are the same or different.
- interrupted means that between two carbons a group is inserted or at the end of the carbon chain between the carbon and hydrogen.
- L 2 is a single chemical bond or a spacer.
- L 2 is a spacer, it is preferably defined as the one or more optional substituents defined above, provided that L 2 is substituted with Z.
- L 2 -Z is COOR 9 ; OR 9 ; C(O)R 9 ; ( ); ( ) ; ( ) ( ) ; ( ) ( ) 2 ; ( ) ( ) ( ) ; ( ) ( ) ; alkenyl; or C 2 - 50 alkynyl, wherein T; Ci_5o alkyl; C2-50 alkenyl; and C2-50 alkynyl are optionally substituted with one or more R 10 , which are the same or different and wherein Ci .50 alkyl; C2-50 alkenyl; and C2-50 alkynyl are optionally interrupted by one or more groups selected from the group consisting of -T-, -
- R 9 , R 9a , R 9b are independently selected from the group consisting of H; Z; T; and Ci_so alkyl; C 2 _5o alkenyl; or C 2 _so alkynyl, wherein T; Ci_so alkyl; C 2 _so alkenyl; and C 2 _so alkynyl are optionally substituted with one or more R 10 , which are the same or different and wherein Ci_so alkyl; C 2 _so alkenyl; and C 2 _so alkynyl are optionally interrupted by one or more groups selected from the group consisting of ( ) ( ) ( ) ( 11 ) ( ) ( 11 ) - ;
- T is selected from the group consisting of phenyl; naphthyl; indenyl; indanyl; tetralinyl; C 3-10 cycloalkyl; 4 to 7 membered heterocyclyl; or 9 to 11 membered heterobicyclyl, wherein t is optionally substituted with one or more R 10 , which are the same or different;
- alkyl is optionally substituted with one or more halogen, which are the same or different;
- R 11 , R l la , R 12 , R 12a , R 12b are independently selected from the group consisting of H; Z; or Ci_ 6 alkyl, wherein Ci_6 alkyl is optionally substituted with one or more halogen, which are the same or different;
- R 9 , R 9a , R 9b , R 10 , R 11 , R l la , R 12 , R 12a , R 12b is Z.
- L 2 is a Ci_ 2 o alkyl chain, which is optionally interrupted by one or more groups independently selected from -0-; and C(0)N(R 3aa ); optionally substituted with one or more groups independently selected from OH; and C(O)N(R 3aa R 3aa ); and wherein R 3aa , R 3aaa are independently selected from the group consisting of H; and Ci_4 alkyl.
- L 2 has a molecular weight in the range of from 14 g/mol to 750 g/mol.
- L 2 is attached to Z via a terminal group selected from
- L 2 has such terminal group it is furthermore preferred that L 2 has a molecular weight in the range of from 14 g/mol to 500 g/mol calculated without such terminal group.
- L is represented by formula (Ia)
- R 4 , L 2 , and Z have the meaning as indicated above, and wherein R 3aa , R 3aaa are independently selected from the group consisting of H; and Ci_4 alkyl; or are joined together with the nitrogen atom to which they are attached to form a 4 to 7 membered heterocycle.
- R 4 is H; or methyl.
- L is represented by formula (Ib)
- R 1 , R la , R 4 , L 2 and Z have the meaning as indicated above, and wherein R 3aa is H; or Ci_4 alkyl.
- R 4 is H; or methyl.
- R 1 in formula (I) is L 2 -Z.
- R 3 in formula (I) is L 2 -Z.
- R 3 , R 3a in formula (I) are joined together with the nitrogen atom to which they are attached to form a 4 to 7 membered heterocycle, wherein the heterocycle is substituted with i ⁇ z.
- D-H is a small molecule bioactive agent or a biopolymer.
- D-H is a biopolymer selected from the group of biopolymers consisting of proteins, polypeptides, oligonucleotides, and peptide nucleic acids.
- Oligonucleotides means either DNA, RNA, single-stranded or double-stranded, siRNA, miRNA, aptamers, and any chemical modifications thereof with preferably 2 to 1000 nucleotides. Modifications include, but are not limited to, those which provide other chemical groups that incorporate additional charge, polarizability, hydrogen bonding, electrostatic interaction, and fluxionality to the nucleic acid ligand bases or to the nucleic acid ligand as a whole.
- Such modifications include, but are not limited to, 2'-position sugar modifications, 5- position pyrimidine modifications, 8-position purine modifications, modifications at exocyclic amines, substitution of 4-thiouridine, substitution of 5-bromo or 5 -io do -uracil; backbone modifications, methylations, unusual base- pairing combinations such as the isobases isocytidine and isoguanidine and the like. Modifications can also include 3' and 5' modifications such as capping and change of stereochemistry.
- D-H is a polypeptide selected from the group of polypeptides consisting of ACTH, adenosine deaminase, agalsidase, alfa-1 antitrypsin (AAT), alfa-1 proteinase inhibitor (API), alteplase, amylins (amylin, symlin), anistreplase, ancrod serine protease, antibodies (monoclonal or polyclonal, and fragments or fusions), antithrombin III, antitrypsins, aprotinin, asparaginases, atosiban, biphalin, bivalirudin, bone-morphogenic proteins, bovine pancreatic trypsin inhibitor (BPTI), cadherin fragments, calcitonin (salmon), collagenase, complement Cl esterase inhibitor, conotoxins, cytokine receptor fragments, DNase, dynorphine A, endorphins,
- ACTH
- D-H is a protein prepared by recombinant DNA technologies.
- D-H is a protein selected from the group of proteins consisting of antibody fragments, single chain antigen binding proteins, catalytic antibodies and fusion proteins.
- D-H is a small molecule bioactive agent selected from the group of agents consisting of central nervous system-active agents, anti-infective, anti-allergic, immunomodulating, anti-obesity, anticoagulants, antidiabetic, anti-neoplastic, antibacterial, anti-fungal, analgesic, contraceptive, anti-inflammatory, steroidal, vasodilating, vasoconstricting, and cardiovascular agents with at least one primary or secondary amino group.
- agents consisting of central nervous system-active agents, anti-infective, anti-allergic, immunomodulating, anti-obesity, anticoagulants, antidiabetic, anti-neoplastic, antibacterial, anti-fungal, analgesic, contraceptive, anti-inflammatory, steroidal, vasodilating, vasoconstricting, and cardiovascular agents with at least one primary or secondary amino group.
- D-H is a small molecule bioactive agent selected from the group of agents consisting of acarbose, alaproclate, alendronate, amantadine, amikacin, amineptine, aminoglutethimide, amisulpride, amlodipine, amotosalen, amoxapine, amoxicillin, amphetamine, amphotericin B, ampicillin, amprenavir, amrinone, anileridine, apraclonidine, apramycin, articaine, atenolol, atomoxetine, avizafone, baclofen, benazepril, benserazide, benzocaine, betaxolol, bleomycin, bromfenac, brofaromine, carvedilol, cathine, cathinone, carbutamid, cefalexine, clinafloxacin, ciprofloxacin, deferox
- Z is a polymer of at least 500 Da or a C 8-18 alkyl group.
- Z is selected from the group of optionally crosslinked polymers consisting of poly(propylene glycol), poly(ethylene glycol), dextran, chitosan, hyaluronic acid, alginate, xylan, mannan, carrageenan, agarose, cellulose, starch, hydroxyalkyl starch (HAS), poly( vinyl alcohols), poly (oxazo lines), poly(anhydrides), poly(ortho esters), poly(carbonates), poly(urethanes), poly(acrylic acids), poly(acrylamides), poly(acrylates), poly(methacrylates), poly(organophosphazenes), polyoxazoline, poly(siloxanes), poly(amides), poly(vinylpyrrolidone), poly(cyanoacrylates), poly(esters), poly(iminocarbonates), poly(amino acids), collagen, gelatin, hydrogel or a blood plasma protein, and copolymers thereof.
- Z is a protein.
- Z is a protein selected from the group consisting of albumin, transferrin, immunoglobulin.
- Z is a linear or branched poly( ethylene glycol) with a molecular weight from
- a prodrug of the present invention wherein D-H is a GLP-I receptor agonist; L is L 1 represented by formula (I) as indicated above; and Z is a hydrogel. Even more preferably, in formula (I) X is N(R 4 ), X 1 is C and X 3 is O. Even more preferably, L is represented by formula (Ia) as indicated above.
- GLP-I is one of the intestinal peptide hormones that are released into the circulatory system after food intake. It augments the postprandial release of insulin, when nutritions (especially carbohydrates) are absorbed and their level postprandially elevated. GLP-I associates with
- GLP-I receptor sites located on pancreatic ⁇ -cells and elevates endogenous cAMP levels in a dose dependent manner.
- GLP-I stimulates the release of insulin.
- a therapeutic potential for GLP-I in type 2 diabetes patients was suggested before, owing to the profound efficacy of this insulinotropic peptide to stimulate secretion of insulin when glucose levels are elevated and to cease doing so upon return to normoglycemia.
- Exendin-4 is reported to associate with GLP-I receptors located on pancreatic beta-cells with 2.5 times higher affinity than GLP-I. In isolated rat islets and beta-cells in presence of glucose, exendin enhances secretion of insulin in a dose-dependent fashion. Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide l-(7-36)-amide receptor of insulin-secreting beta-cells (see J. Biol. Chem. 268(26): 19650-19655). Studies in type 2 diabetic rodents revealed that exendin-4 is 5530-fold more potent than GLP-I in lowering blood glucose levels.
- exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers (see e.g. Am. J. Physiol. Endocrinol. Metab. 281(1):E155-61).
- the GLP-I receptor agonist is Exendin-4.
- Hydrogels to be used are known in the art. Suitable hydrogels may be used which are described in WO-A 2006/003014. Accordingly, a hydrogel may be defined as a three- dimensional, hydrophilic or amphiphilic polymeric network capable of taking up large quantities of water.
- the networks are composed of homopolymers or copolymers, are insoluble due to the presence of covalent chemical or physical (ionic, hydrophobic interactions, entanglements) crosslinks.
- the crosslinks provide the network structure and physical integrity.
- Hydrogels exhibit a thermodynamic compatibility with water which allow them to swell in aqueous media.
- the chains of the network are connected in such a fashion that pores exist and that a substantial fraction of these pores are of dimensions between 1 nm and 1000 nm.
- Another object of the present invention is a pharmaceutical composition
- a pharmaceutical composition comprising a prodrug of the present invention or a pharmaceutical salt thereof together with a pharmaceutically acceptable excipient.
- Yet another object of the present invention is a prodrug of the present invention or a pharmaceutical composition of the present invention for use as a medicament.
- Yet another object of the present invention is a method of treating, controlling, delaying or preventing in a mammalian patient in need of the treatment of one or more conditions comprising administering to said patient a therapeutically effective amount of a prodrug of the present invention or a pharmaceutical composition of the present invention or a pharmaceutically acceptable salt thereof.
- Another object of the present invention is a prodrug precursor of formula Act-L, wherein L has the meaning as indicated above and Act is a leaving group.
- Act is chloride, bromide, fluoride, nitrophenoxy, imidazolyl, N- hydroxysuccinimidyl, N-hydroxybenzotriazolyl, N-hydroxyazobenzotriazo IyI, pentafluorophenoxy, 2-thiooxo-thiazolidinyl, or N-hydroxysulfosuccinimidyl.
- 4OkDa methoxy poly(ethylene glycol) maleimido-propionamide (PEG40kDa-maleimide) was obtained from Chirotech Technology Ltd, Cambridge, UK.
- 2-Chlorotrityl chloride resin, Sieber amide resin and amino acids were from Merck Biosciences GmbH, Schwalbach/Ts, Germany, if not stated otherwise.
- Fmoc-D- Homocysteine(Trt)-OH and S-Trityl-3-mercaptopropionic acid (Trt-MPA) were obtained from Bachem AG, Bubendorf, Switzerland.
- O-(N-Fmoc-2-aminoethyl)-O'-(2-carboxyethyl)- undecaethyleneglycol (Fmoc-Pop-OH) was obtained from Polypure AS, Oslo, Norway.
- Fmoc-4-(2-aminoethyl)-l-carboxymethyl-piperazine (Fmoc-Acp-OH) was purchased from NeoMPS SA, France, France.
- cis-Cyclohexane-l,2-dicarboxylic anhydride was obtained from Alfa Aesar GmbH & Co KG, Düsseldorf, Germany.
- Solid phase synthesis was performed on 2-Chlorotrityl chloride resin with a loading of 1.3 mmol/g or Sieber amide resin with a loading of 0.55 mmol/g. Syringes equipped with polypropylene frits were used as reaction vessels. Loading of the first amino acid to resins was performed according to manufacturer's instructions.
- Fmoc deprotection For Fmoc protecting-group removal, the resin was agitated with 2/2/96 (v/v/v) piperidine/DBU/DMF (two times, 10 min each) and washed with DMF (ten times).
- the resin was agitated with 98/2 (v/v) DMF/hydrazine hydrate (3 times, 10 min each) and washed with DMF (ten times).
- N-terminus of a peptide was boc-protected by agitating the resin with 30 eq (boc)2 ⁇ and
- Coupling of 3-maleimido propionic acid to free amino groups on resin was achieved by agitating resin with 2 eq of acid, 2 eq DIC and 2 eq HOBt in relation to free amino groups in DMF at room temperature. After 30 min, resin was washed with DMF (10 times).
- Synthesis of ureas on resin was achieved by agitating resin with 2.5 eq of bis(pentafluorophenyl) carbonate, 5 eq DIEA, and 0.25 eq DMAP in relation to free amino groups in DCM/ACN 1/1 at room temperature. After 15 min resin was washed with DMF (10 times). 5 eq of amine was dissolved in DMF. Mixture was added to resin and agitated for 60 min at room temperature. Resin was washed with DMF (10 times).
- the resin was washed with DCM, dried in vacuo and treated with 2 ml of TFA cleavage cocktail (TFA/TES/Water/DTT 95/2/2/1) per 100 mg resin for 60 min at room temperature. Volatiles were removed under a nitrogen stream. Unpolar side products and protecting groups were removed by precipitating peptide from diethyl ether. Precipitate was dried in vacuo and dissolved in ACN/water 1/1 and purified by RP-HPLC.
- TFA cleavage cocktail TFA cleavage cocktail
- RP-HPLC was done on a 100x20 or a 100x40 mm Cl 8 ReproSil-Pur 300 ODS-3 5 ⁇ column (Dr. Maisch, Ammerbuch, Germany) connected to a Waters 600 HPLC System and Waters
- Electrospray ionization mass spectrometry was performed on a Waters ZQ 4000 ESI instrument and spectra were, if necessary, interpreted by Waters software MaxEnt.
- Size exclusion chromatography was performed using an Amersham Bioscience AEKTAbasic system equipped with a Superdex200 10/300 column (Amersham Bioscience/GE Healthcare), if not stated otherwise. 10 rnM sodium phosphate, 140 mM NaCl, pH 7.4, 3 mM EDTA was used as mobile phase
- Desalting was performed using an Amersham Bioscience AEKTAbasic system equipped with a HiPrep 26/10 Desalting column and 0.1% acetic acid in water as mobile phase.
- hydrogel conjugates For hydrogel conjugates, compounds were suspended in buffer A and incubated at 37°C. Samples were taken after centrifugation of the suspension and analyzed by RP-HPLC at 215 nm. UV-signals correlating to liberated drug molecule were integrated and plotted against incubation time.
- Curve-fitting software was applied to estimate the corresponding halftime of release.
- Linker reagent 2 was synthesized on 3-chlorotrityl chloride resin (300 mg, 0.39 mmol) by loading of resin with Fmoc-Cys(Trt)-OH, fmoc deprotection, and on-resin urea formation using N,N-dimethyl-ethylenediamine as amine, cleavage from resin as depicted above and described in "Materials and Methods".
- 0.01% HCl in water was used as solution A and 0.01% HCl in acetonitrile was used as solution B. Yield: 82 mg of HCl salt (0.16 mmol).
- Fmoc-Acp-OH • 2 HCl (100 mg, 0.21 mmol) was suspendend in 400 ⁇ l DMF/DMSO 1/1 (v/v).
- S-tritylcysteamine • HCl 75 mg, 0.21 mmol
- PyBOP 109 mg, 0.21 mmol
- DIEA 146 ⁇ l, 0.86 mmol
- Fmoc group was removed by adding 75 ⁇ l piperidine and 25 ⁇ l DBU. After 15 min mixture was hydro lyzed and acidified (AcOH) and compound was purified by RP-HPLC. After lyophilization 98 mg (0.14 mmol, double TFA salt) were obtained.
- 5b (7 mg, 0.010 mmol) was preactivated by incubating with PyBOP (12.5 mg, 0.024 mmol) and DIEA (5 ⁇ l, 0.03 mmol) in 200 ⁇ l of dry DMF for 45 min at RT.
- 5a (20 mg, 0.028 mmol) and DIEA (15 ⁇ l, 0.09 mmol) were added and mixture was incubated for further 60 min.
- Mixture was quenched with 0.5 ml of acetonitrile/acetic acid/water (1/1/1) and purified by RP-HPLC. After lyophilization 3 mg (0.0026 mmol, double TFA salt) of 5c were obtained.
- Fmoc-Asp(tBu)-OH (411 mg, 1 mmol), HOBt (153 mg, 1 mmol), and DIC (160 ⁇ l, 1 mmol) were dissolved in 2 ml of DMF and incubated for 10 min at RT. N,N-dimethyl ethylenediamine (160 ⁇ l, 1.5 mmol) was added and stirred at RT for 30 min. Acetic acid (300 ⁇ l) was added and Fmoc-Asp(tBu)-NH-(CH 2 )2-N(CH 3 )2 was purified by RP-HPLC.
- Fmoc-Asp(H)-NH-(CH 2 ) 2 -NH 2 (140 mg, 0.27 mmol, TFA salt) was dissolved in 1 ml of DMF and DIEA (140 ⁇ l, 0.81 mmol) and boc 2 ⁇ (100 mg, 0.46 mmol) added. The solution was stirred at RT for 15 min and then acidified with acetic acid (300 ⁇ l). 7a was purified by RP- HPLC. Yield 7a: 120 mg (0.24 mmol)
- Exendin-4 on resin (40 mg, 4 ⁇ mol) and incubated for 1 h at room temperature. Resin was washed ten times with DMF and then incubated for 5 min with 500 ⁇ l of 1/1/2 acetic anhydride/pyridine/DMF. Resin was washed 10 times with DMF and fmoc group was removed. Trt-mercaptopropionic acid was coupled and 8a was cleaved from resin and purified by RP-HPLC.
- 8b was synthesized as described above for 8a except for the use of 7b instead of 7a.
- 9a was purified by ion exchange chromatography using 10 mM sodium citrate pH 3 as solvent A and 10 mM sodium citrate pH 3 and 1 M NaCl as solvent B and a step-gradient (0 to 40% B). Fractions containing 9a were desalted and lyophilized:
- Trt-mercaptopropionic acid was coupled according to standard coupling method and resin was washed five times with DMF and ten times with DCM. Mmt protecting group of Lys27 was removed by incubation of resin five times in 2 ml of 9/1 (v/v) DCM/HFIP for 5 min. Resin was washed five times with DCM and five times with DMF and 5,6-carboxy-flourescein-NHS ester (20 mg, 42 ⁇ mol) and DIEA (20 ⁇ l, 115 ⁇ l) in 300 ⁇ l DMF were added to resin and incubated for 30 min. 12a was cleaved from resin and purified by RP-HPLC Yield: 12 mg
- Trt-mercaptopropionic acid was coupled according to standard coupling method to side-chain protected Exendin-4 on resin (120 mg, 12 ⁇ mol). Mmt protecting group removal of Lys27 and
- 12d was synthesized as described for 12c except for the use of GRF(I -29) on resin (120mg,
- N,N-dimethylethylenediamine (198 ⁇ L, 1.8 mmol) and NaCNBH 3 (58 mg, 0.9 mmol) were dissolved in methanol (5 mL) and brought to pH 5.5 by addition of AcOH (250 ⁇ L).
- a suspension of 2,4,6, -trimethoxybenzaldehyde (294 mg, 1.5 mmol) in EtOH (5 mL) was added and the reaction was stirred at RT for 1 h.
- 5 N HCl (0.5 mL) was added and the mixture was stirred for further 12 h.
- the solvent was removed under reduced pressure; the residue was dissolved in sat. NaHCO 3 and extracted 3x with DCM.
- the combined organic phases were dried over NaSO 4 and the solvent was evaporated under reduced pressure. Yield: 303 mg (1.13 mmol)
- Fmoc-Asp(OtBu)-N(Tmob)CH 2 CH 2 N(CH 3 ) 2 225 mg, 0.29 mmol was dissolved in a solution of piperidine (50 ⁇ L) and DBU (15 ⁇ L) in DMF (1.5 mL). The mixture was stirred at
- the TFA salt OfH-ASp(OtBu)-N(TmOb)CH 2 CH 2 N(CHs) 2 (114 mg, 0.21 mmol) was dissolved in sat. NaHCO 3 (10 mL) and extracted 3x with DCM (3x 10 mL). The combined organic layers were dried over NaSO 4 and the solvent was removed under reduced pressure. The residue was dissolved in DMF (1.0 mL), 6-tritylmercaptohexanoic acid (121 mg, 0.31 mmol), HATU (118 mg, 0.31 mmol) and DIEA (108 ⁇ L, 0.62 mmol) were added. The mixture was stirred for 30 min.
- TrtS(CH 2 ) 5 CONH-Asp(OtBu)- N(TmOb)CH 2 CH 2 N(CHs) 2 was purified by RP-HPLC. Yield: 95 mg (0.10 mmol, TFA salt)
- TrtS(CH 2 ) 5 CONH- ASp(OtBu)-N(TmOb)CH 2 CH 2 N(CHs) 2 (95 mg, 0.10 mmol) was dissolved in a 3:1 mixture of MeOH/H 2 O (1.0 mL), LiOH (7.4 mg, 0.31 mmol) was added and the mixture was stirred for 5 h at 60 0 C. AcOH was added (100 ⁇ L) and 17a was purified by RP- HPLC.
- 19b was synthesized as described for 19a except for the use of 18b instead of 18a.
- Fmoc-Asp(NH(CH 2 )2N(CH 3 )-boc)OtBu (91 mg, 0.13 mmol) was dissolved in DMF (1.0 mL), piperidine (50 ⁇ L) and DBU (15 ⁇ L) were added and the mixture was stirred for 45 min at RT. AcOH (100 ⁇ L ) was added and NH 2 -Asp(NH(CH 2 ) 2 N(CH 3 )-boc)OtBu was purified by RP-HPLC.
- 6-Tritylmercaptohexanoic acid 200 mg, 0.51 mmol
- (PIpO) 2 CO 202 mg, 0.51 mmol
- collidine 340 ⁇ L, 2.65 mmol
- the mixture was added to a solution of Fmoc-Lys-OH (170 mg, 0.46 mmol) in H 2 O/pyridine/tBuOH (3:3:1, 6 mL).
- the reaction was heated at 60 0 C for 2h, diluted with EtOAc, extracted 2x with 0.1M H 2 SO 4 , 2x with brine and dried over Na 2 SO 4 .
- the solvent was evaporated under reduced pressure and the residue was purified by RP-HPLC. Yield: 109 mg
- 25c was synthesized as described for 25a except for the use of 24c instead of 24a. Yield: 1.3 mg
- 26c was synthesized as described for 26a except for the use of 25c instead of 25a.
- H 25a (2.0 mg) was dissolved in 1 :1 H 2 0/MeCN containing 0.1 % TFA (200 ⁇ l).
- the solution was incubated at RT, after 5 min AcOH (20 ⁇ l) was added and 27a was purified by cation exchange chromatography, desalted and lyophilized.
- 27b was synthesized as described for 27a except for the use of 25b instead of 25a.
- 27c was synthesized as described for 27a except for the use of 25c instead of 25a.
- JV-Boc-ethylenediamine (81 ⁇ l, 0.51 mmol) was added to a solution of Br-(CH 2 )s-CONH-
- Boc-NH-(CH 2 ) 2 -N(CH 3 )-(CH 2 ) 5 -CONH-(CH 2 ) 2 -STrt (62.8 mg, 0.11 mmol) was dissolved in
- 31b 31b was synthesized as described for 31a except for the use of 30b instead of 30a. Yield: 16.8 mg
- JV-Alloc-ethylenediamine HCl-salt (43.5 mg, 0.24 mmol) and DIEA (38 ⁇ l, 0.22 mmol) were added to a solution of Br-(CH 2 ) 2 CONH-(CH 2 )-STrt (100 mg, 0.22 mmol) and Na 2 CO 3 (93 mg, 0.87 mmol) in DMF (1 ml). The reaction mixture was stirred for 10 h at 70 0 C.
- 33a was synthesized as described for 30a except for the use of 32 instead of 28.
- 34b was synthesized as described for 34a except for the use of 33b instead of 33a. Yield: 11.5 mg
- Linker-exendin conjugates were synthesized according to general synthesis method A, B, C, D, E or F.
- Diacid anhydride (0.2 mmol) and pyridine (0.2 mmol) were dissolved in 0.3 ml of dry DMF. Mixture was added to exendin-4 on resin (2 ⁇ mol) and agitated for 30 min at room temperature. Resin was washed with DMF (10 times). PyBOP (0.1 mmol), HOBt (0.1 mmol), and collidine (0.4 mmol) were dissolved in 0.3 ml of dry DMF. Mixture was added to resin and agitated for 30 min at room temperature. Resin was washed with DMF (10 times). Diamine (0.1 mmol) and DIEA (0.3 mmol) were dissolved in a mixture of 0.4 ml of DMF and 0.4 ml of EtOH.
- Exendin- linker conjugates were cleaved and purified by RP-HPLC as described in "Materials and Methods".
- maleimide-functionalized hydrogel microparticles were synthesized as described in EP 1 625 856 A1. 30 mg of maleimide-derivatized hydrogel microparticles (loading 40 ⁇ mol/g, 1.2 ⁇ mol) were reacted with 6 mg of compound 25a (1.32 ⁇ mol, 1.1 eq) in 600 ⁇ l 20/80 (v/v) acetonitrile/50 mM phosphate buffer (pH 7.4) for 10 min to give exendin- linker loaded hydrogel microparticles 39. The loaded hydrogel 39 was washed 5 times with 50/50 (v/v) acetonitrile/water and three times with water.
- 46 46 was synthesized as described for 42 except for the use of 45 instead of 41.
- Tritylsulf ⁇ de (247 mg, 0.89 mmol) was suspended in 1 ml DMSO.
- DBU 152 ⁇ l, 1.02 mmol
- 6-bromohexan-l-ol 173 mg, 0.96
- Reaction mixture was dissolved in 20 ml ethylacetate and washed with 1 N H 2 SO 4 (2x) and brine (3x). Organic layer was dried (Na 2 SO 4 ) and volatiles were removed in vacuo. Product was purified by flash chromatography on silica (heptane/ AcOEt 1/1).
- Fmoc- AIa-OH 250 mg, 0.8 mmol
- DIEA 170 ⁇ L, 1,0 mmol
- 2-chlorotrityl chloride resin 312 mg, 1.3 mmol/g
- Methanol 0.6 mL
- the resin was washed with DCM (1Ox) and DMF (1Ox). Fmoc-deprotection and urea formation was achieved according to general procedures (see Materials and Methods) by reaction with ethylene diamine.
- Release kinetics in vivo were determined by comparing the pharmacokinetics of 13a with the pharmacokinetics of 13c and 13b with 13d, respectively, after intravenous injection into rat. Animal studies were performed at Heidelberg Pharma AG, Heidelberg, Germany. 13a (27 mg) was dissolved in 3.5 ml PBS and 500 ⁇ l of the resulting solution were injected intravenously into six rats each. Male SD rats with approximately 270 g weight were used.
- Pharmacokinetics of 13c were determined as described for 13a.
- Pharmacokinetics of 13b and 13d were determined as described for 13a, except for the use of 20 mg 13 b and 13d each in 2.5 ml PBS and four rats.
- Linker hydrolysis half-life was calculated from the ratio of fluorescence of 13a compared to fluorescence of 13c and 13b compared to 13d, respectively, at the respective time points.
- Half-life of in vivo linker hydrolysis was determined to be 115 h and 160 h for 13a and 13b, respectively, which is in excellent correlation to the half-life of in vitro linker hydrolysis of 12O h and 16O h for 13a and 13b, respectively.
- Fig. 3 shows in vivo and in vitro linker cleavage data of 13b, wherein in vivo (triangles) and in vitro (diamonds) cleavage kinetics are shown by semilogarithmic representation.
Abstract
Description


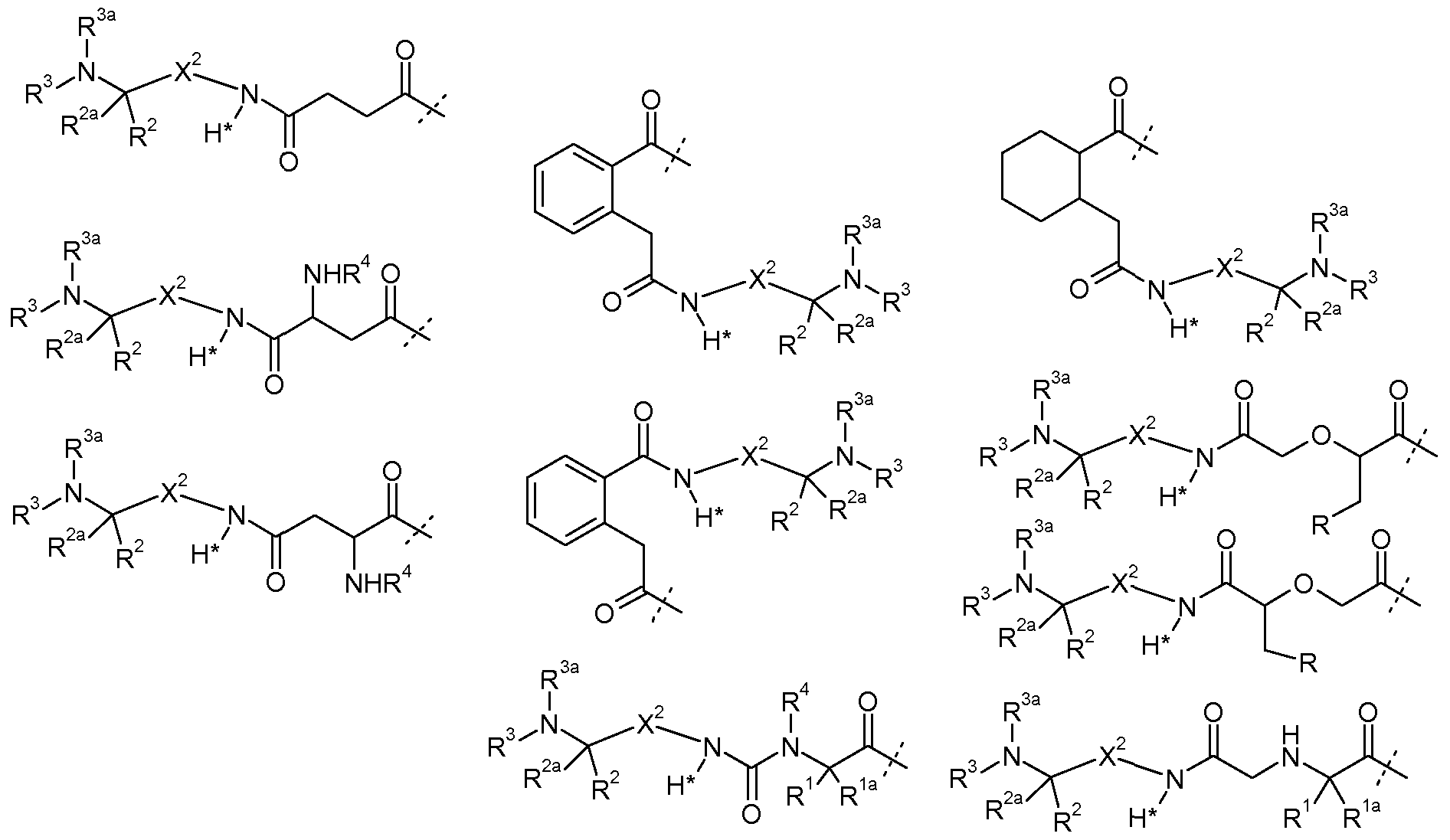

































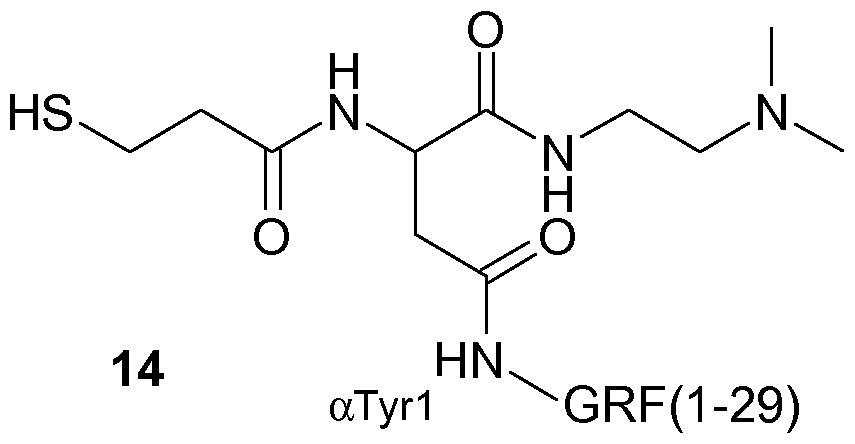







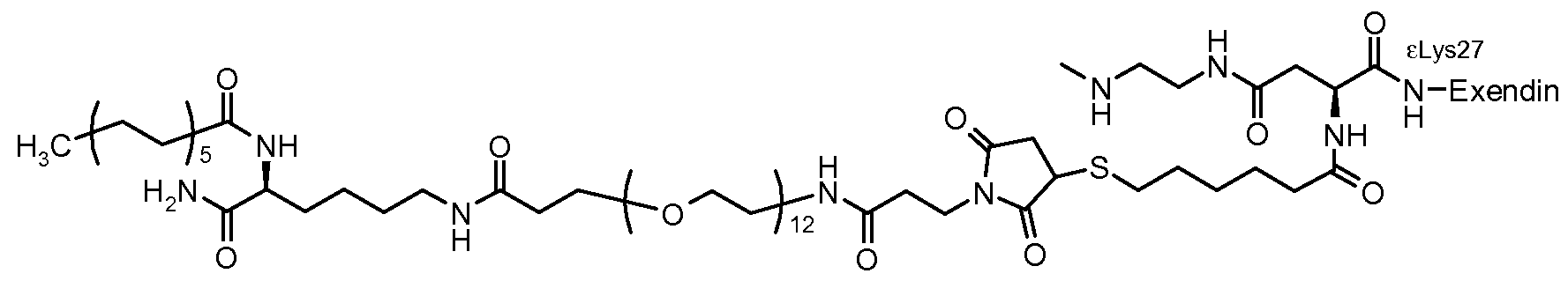
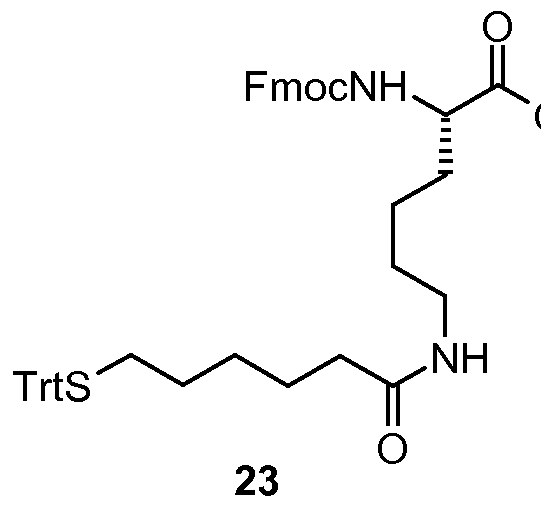

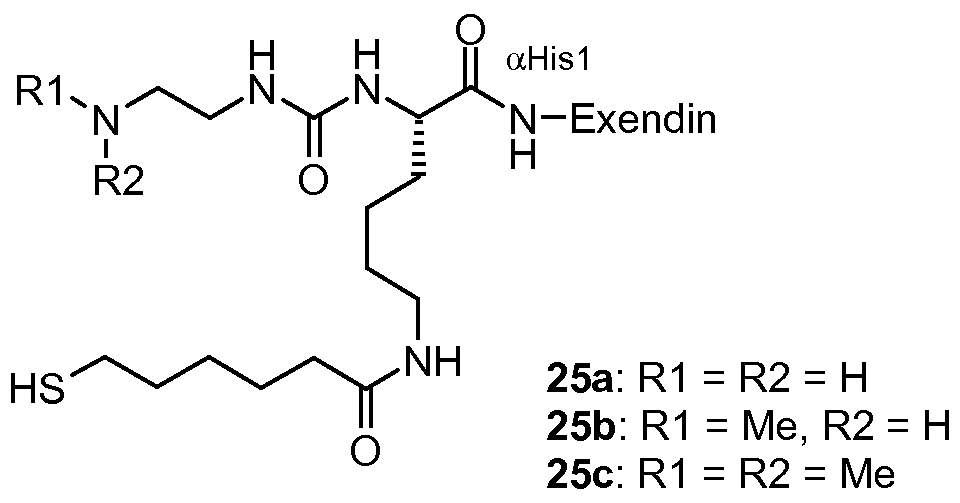



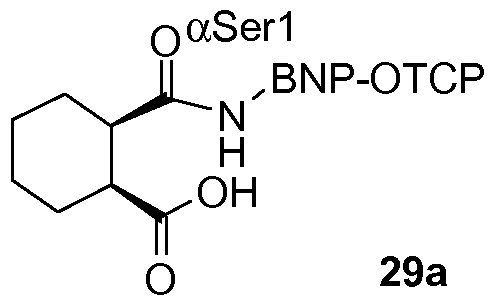



















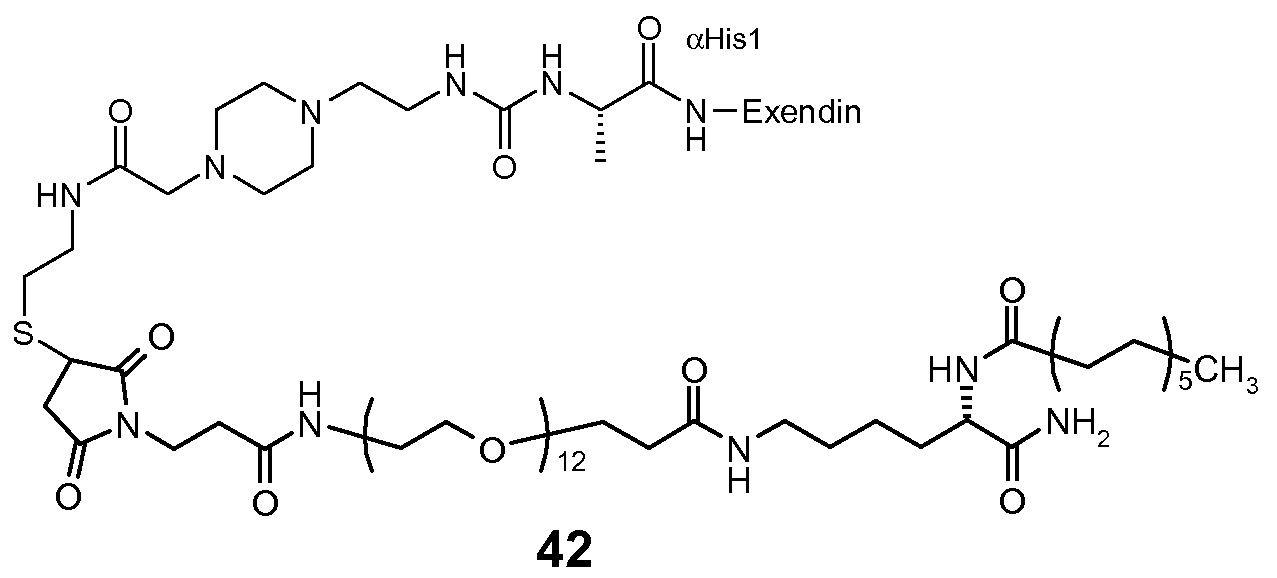

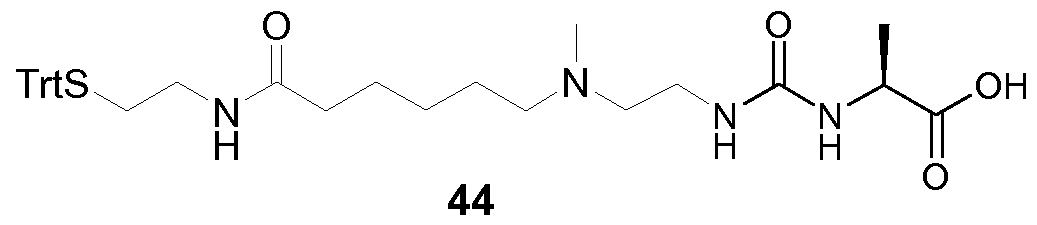





Claims









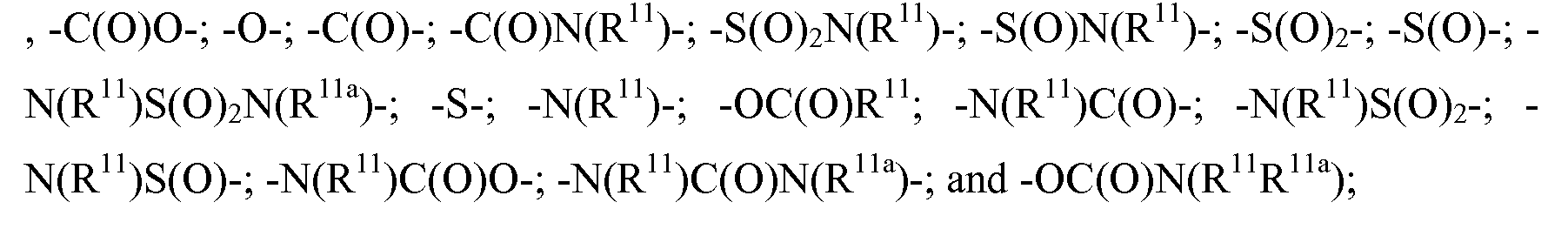

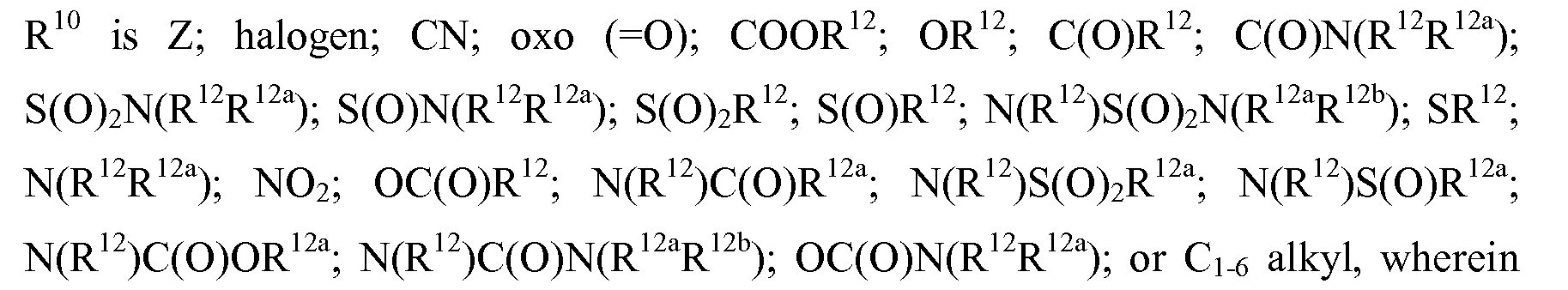

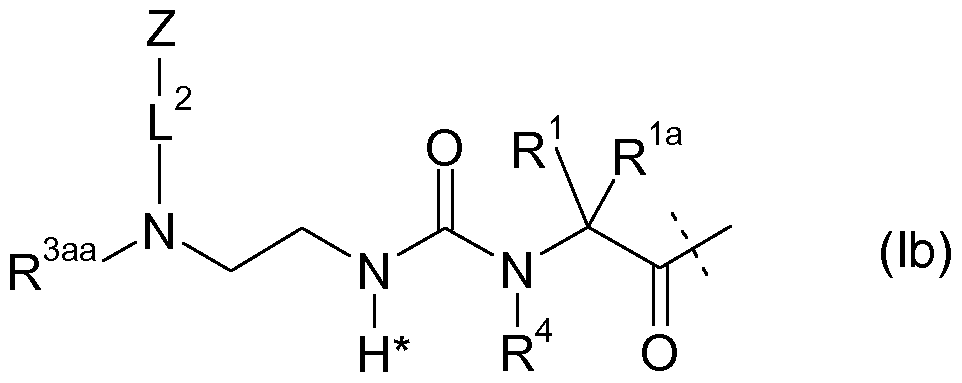
Priority Applications (26)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21202920.1A EP3981761A3 (en) | 2008-02-01 | 2009-01-30 | Intermediates for prodrugs |
| EP13156350.4A EP2596805B1 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a drug-linker conjugate |
| CN2009801110881A CN101980725B (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a drug linker conjugate |
| ES09706788T ES2733355T3 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
| AU2009209565A AU2009209565B2 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
| SI200931976T SI2237799T1 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
| US12/865,693 US8906847B2 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a drug linker conjugate |
| DK09706788.8T DK2237799T3 (en) | 2008-02-01 | 2009-01-30 | PRODRUG INCLUDING A SELF-SPLITABLE LINKS |
| MX2010008024A MX2010008024A (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker. |
| EP09706788.8A EP2237799B1 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
| RS20190789A RS58948B1 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
| PL09706788T PL2237799T3 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
| CA2712224A CA2712224C (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a drug linker conjugate |
| BRPI0906643A BRPI0906643A2 (en) | 2008-02-01 | 2009-01-30 | a prodrug comprising a drug-binder conjugate |
| LTEP09706788.8T LT2237799T (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
| JP2010544717A JP5588354B2 (en) | 2008-02-01 | 2009-01-30 | Prodrugs containing self-cleavable linkers |
| IL207150A IL207150A (en) | 2008-02-01 | 2010-07-22 | Prodrug comprising a drug linker conjugate |
| ZA2010/05413A ZA201005413B (en) | 2008-02-01 | 2010-07-29 | Prodrug comprising a self-cleavable linker |
| HK11106352.6A HK1152240A1 (en) | 2008-02-01 | 2011-06-21 | Prodrug comprising a self-cleavable linker |
| US14/528,362 US20150057221A1 (en) | 2008-02-01 | 2014-10-30 | Prodrug comprising a drug linker conjugate |
| US15/385,179 US20170100490A1 (en) | 2008-02-01 | 2016-12-20 | Prodrug Comprising a Drug Linker Conjugate |
| HRP20191052 HRP20191052T1 (en) | 2008-02-01 | 2019-06-12 | Prodrug comprising a self-cleavable linker |
| CY20191100707T CY1121961T1 (en) | 2008-02-01 | 2019-07-03 | PRODRUG CONTAINING A SELF-DISPERSIBLE LINKER |
| US16/654,667 US20200046725A1 (en) | 2008-02-01 | 2019-10-16 | Prodrug Comprising a Drug Linker Conjugate |
| US17/847,750 US20220331334A1 (en) | 2008-02-01 | 2022-06-23 | Prodrug Comprising a Drug-Linker Conjugate |
| US18/479,445 US20240041897A1 (en) | 2008-02-01 | 2023-10-02 | Prodrug comprising a drug linker conjugate |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08150973.9 | 2008-02-01 | ||
| EP08150973 | 2008-02-01 | ||
| EP08170872 | 2008-12-05 | ||
| EP08170872.9 | 2008-12-05 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/865,693 A-371-Of-International US8906847B2 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a drug linker conjugate |
| US14/528,362 Continuation US20150057221A1 (en) | 2008-02-01 | 2014-10-30 | Prodrug comprising a drug linker conjugate |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009095479A2 true WO2009095479A2 (en) | 2009-08-06 |
| WO2009095479A3 WO2009095479A3 (en) | 2009-10-01 |
Family
ID=40823379
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/051079 WO2009095479A2 (en) | 2008-02-01 | 2009-01-30 | Prodrug comprising a self-cleavable linker |
Country Status (23)
| Country | Link |
|---|---|
| US (6) | US8906847B2 (en) |
| EP (3) | EP2596805B1 (en) |
| JP (2) | JP5588354B2 (en) |
| CN (1) | CN101980725B (en) |
| AU (1) | AU2009209565B2 (en) |
| BR (1) | BRPI0906643A2 (en) |
| CA (1) | CA2712224C (en) |
| CY (1) | CY1121961T1 (en) |
| DK (2) | DK2237799T3 (en) |
| ES (2) | ES2904673T3 (en) |
| HK (1) | HK1152240A1 (en) |
| HR (1) | HRP20191052T1 (en) |
| HU (1) | HUE044174T2 (en) |
| IL (1) | IL207150A (en) |
| LT (1) | LT2237799T (en) |
| MX (1) | MX2010008024A (en) |
| PL (1) | PL2237799T3 (en) |
| PT (1) | PT2237799T (en) |
| RS (1) | RS58948B1 (en) |
| RU (2) | RU2010136023A (en) |
| SI (1) | SI2237799T1 (en) |
| WO (1) | WO2009095479A2 (en) |
| ZA (1) | ZA201005413B (en) |
Cited By (141)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010042596A2 (en) * | 2008-10-08 | 2010-04-15 | The Uab Research Foundation | Photo-activated protecting groups |
| WO2011012722A1 (en) * | 2009-07-31 | 2011-02-03 | Ascendis Pharma As | Prodrugs containing an aromatic amine connected by an amido bond to a linker |
| WO2011089216A1 (en) * | 2010-01-22 | 2011-07-28 | Ascendis Pharma As | Dipeptide-based prodrug linkers for aliphatic amine-containing drugs |
| US20120035320A1 (en) * | 2010-04-03 | 2012-02-09 | University Of Iowa Research Foundation | Polyacridine nucleic acid delivery peptide complexes |
| WO2012035139A1 (en) | 2010-09-17 | 2012-03-22 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an exendin linker conjugate |
| WO2011073234A3 (en) * | 2009-12-15 | 2012-04-19 | Ascendis Pharma As | Dry growth hormone composition transiently linked to a polymer carrier |
| US20120184489A1 (en) * | 2009-07-31 | 2012-07-19 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an insulin linker conjugate |
| US8338368B2 (en) | 2005-11-07 | 2012-12-25 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting physiological solubility and stability |
| WO2013010840A2 (en) | 2011-07-18 | 2013-01-24 | Arts Biologics A/S | Long acting biologically active luteinizing hormone (lh) compound |
| WO2013024048A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | Polymeric hyperbranched carrier-linked prodrugs |
| WO2013024047A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | High-loading water-soluble carrier-linked prodrugs |
| WO2013024049A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | Protein carrier-linked prodrugs |
| WO2013024053A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | Carrier-linked prodrugs having reversible carboxylic ester linkages |
| WO2013053856A1 (en) | 2011-10-12 | 2013-04-18 | Ascendis Pharma A/S | Prevention and treatment of ocular conditions |
| US8450270B2 (en) | 2008-06-17 | 2013-05-28 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting enhanced solubility and stability in physiological pH buffers |
| US8454971B2 (en) | 2007-02-15 | 2013-06-04 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| CN103179979A (en) * | 2010-06-24 | 2013-06-26 | 印第安纳大学研究及科技有限公司 | Amide based glucagon superfamily peptide prodrugs |
| US8481485B2 (en) | 2008-12-19 | 2013-07-09 | Indiana University Research And Technology Corporation | Insulin analogs |
| US8507428B2 (en) | 2010-12-22 | 2013-08-13 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting GIP receptor activity |
| US8546327B2 (en) | 2008-06-17 | 2013-10-01 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| WO2013160340A1 (en) | 2012-04-25 | 2013-10-31 | Ascendis Pharma A/S | Prodrugs of hydroxyl-comprising drugs |
| WO2013167750A2 (en) | 2012-05-11 | 2013-11-14 | Prorec Bio Ab | Method for diagnosis and treatment of prolactin associated disorders |
| US8669228B2 (en) | 2007-01-05 | 2014-03-11 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting enhanced solubility in physiological pH buffers |
| US8697632B2 (en) | 2008-12-19 | 2014-04-15 | Indiana University Research And Technology Corporation | Amide based insulin prodrugs |
| US8697838B2 (en) | 2008-01-30 | 2014-04-15 | Indiana University Research And Technology Corporation | Ester-based insulin prodrugs |
| WO2014056915A1 (en) | 2012-10-11 | 2014-04-17 | Ascendis Pharma A/S | Diagnosis, prevention and treatment of diseases of the joint |
| WO2014056926A1 (en) | 2012-10-11 | 2014-04-17 | Ascendis Pharma A/S | Hydrogel prodrugs |
| WO2014056923A1 (en) | 2012-10-11 | 2014-04-17 | Ascendis Pharma Ophthalmology Division A/S | Vegf neutralizing prodrugs for the treatment of ocular conditions |
| US8703701B2 (en) | 2009-12-18 | 2014-04-22 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US8729017B2 (en) | 2011-06-22 | 2014-05-20 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| WO2014080730A1 (en) | 2012-11-22 | 2014-05-30 | 株式会社糖鎖工学研究所 | Glycosylated linker, compound containing glycosylated linker moiety and physiologically active substance moiety or salt thereof, and methods for producing said compound or salt thereof |
| WO2014084110A1 (en) | 2012-11-30 | 2014-06-05 | 株式会社糖鎖工学研究所 | Sugar chain-attached linker, compound containing sugar chain-attached linker and physiologically active substance or salt thereof, and method for producing same |
| US8859491B2 (en) | 2011-11-17 | 2014-10-14 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhibiting glucocorticoid receptor activity |
| WO2014173759A1 (en) | 2013-04-22 | 2014-10-30 | Ascendis Pharma A/S | Hydrogel-linked prodrugs releasing modified drugs |
| US8940860B2 (en) | 2010-06-16 | 2015-01-27 | Indiana University Research And Technology Corporation | Single-chain insulin agonists exhibiting high activity at the insulin receptor |
| US8946147B2 (en) | 2010-06-24 | 2015-02-03 | Indiana University Research And Technology Corporation | Amide-based insulin prodrugs |
| US8969288B2 (en) | 2008-12-19 | 2015-03-03 | Indiana University Research And Technology Corporation | Amide based glucagon and superfamily peptide prodrugs |
| US8980830B2 (en) | 2007-10-30 | 2015-03-17 | Indiana University Research And Technology Corporation | Peptide compounds exhibiting glucagon antagonist and GLP-1 agonist activity |
| US8981047B2 (en) | 2007-10-30 | 2015-03-17 | Indiana University Research And Technology Corporation | Glucagon antagonists |
| WO2015052154A1 (en) * | 2013-10-08 | 2015-04-16 | Ascendis Pharma Osteoarthritis Division A/S | Hydrogel-linked il-1ra prodrug |
| WO2015067791A1 (en) | 2013-11-11 | 2015-05-14 | Ascendis Pharma Relaxin Division A/S | Relaxin prodrugs |
| EP2768856A4 (en) * | 2011-10-18 | 2015-05-27 | Prolynx Llc | Peg conjugates of exenatide |
| WO2015081891A1 (en) | 2013-12-06 | 2015-06-11 | Baikang (Suzhou) Co., Ltd | Bioreversable promoieties for nitrogen-containing and hydroxyl-containing drugs |
| US9062124B2 (en) | 2008-06-17 | 2015-06-23 | Indiana University Research And Technology Corporation | GIP-based mixed agonists for treatment of metabolic disorders and obesity |
| US9127088B2 (en) | 2010-05-13 | 2015-09-08 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhibiting nuclear hormone receptor activity |
| US9145451B2 (en) | 2010-05-13 | 2015-09-29 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhbiting G protein coupled receptor activity |
| US9156902B2 (en) | 2011-06-22 | 2015-10-13 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US9265723B2 (en) | 2009-07-31 | 2016-02-23 | Sanofi-Aventis Deutschland Gmbh | Long acting insulin composition |
| US9272048B2 (en) | 2008-04-29 | 2016-03-01 | Ascendis Pharma Growth Disorders Division A/S | PEGylated recombinant human growth hormone compounds |
| WO2016110577A1 (en) * | 2015-01-09 | 2016-07-14 | Ascendis Pharma A/S | Cnp prodrugs |
| US9487571B2 (en) | 2010-01-27 | 2016-11-08 | Indiana University Research And Technology Corporation | Glucagon antagonist-GIP agonist conjugates and compositions for the treatment of metabolic disorders and obesity |
| WO2016196124A2 (en) | 2015-05-29 | 2016-12-08 | Ascendis Pharma Inc. | Prodrugs comprising a pyroglutamate linker |
| WO2016193371A1 (en) | 2015-06-05 | 2016-12-08 | Sanofi | Prodrugs comprising an glp-1/glucagon dual agonist linker hyaluronic acid conjugate |
| US9533056B2 (en) | 2010-01-22 | 2017-01-03 | Ascendis Pharma As | Dipeptide-based prodrug linkers for aliphatic amine-containing drugs |
| US9573987B2 (en) | 2011-12-20 | 2017-02-21 | Indiana University Research And Technology Corporation | CTP-based insulin analogs for treatment of diabetes |
| US9593156B2 (en) | 2012-09-26 | 2017-03-14 | Indiana University Research And Technology Corporation | Insulin analog dimers |
| WO2017053807A2 (en) | 2015-09-23 | 2017-03-30 | Genentech, Inc. | Optimized variants of anti-vegf antibodies |
| US9670261B2 (en) | 2012-12-21 | 2017-06-06 | Sanofi | Functionalized exendin-4 derivatives |
| US9694053B2 (en) | 2013-12-13 | 2017-07-04 | Sanofi | Dual GLP-1/glucagon receptor agonists |
| WO2017118704A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Controlled-release cnp agonists with low npr-c binding |
| WO2017118693A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Cnp prodrugs with large carrier moieties |
| WO2017118703A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Controlled-release cnp agonists with low initial npr-b activity |
| WO2017118698A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Cnp prodrugs with carrier attachment at the ring moiety |
| US9751926B2 (en) | 2013-12-13 | 2017-09-05 | Sanofi | Dual GLP-1/GIP receptor agonists |
| US9750788B2 (en) | 2013-12-13 | 2017-09-05 | Sanofi | Non-acylated exendin-4 peptide analogues |
| WO2017148883A1 (en) | 2016-03-01 | 2017-09-08 | Ascendis Pharma Bone Diseases A/S | Pth prodrugs |
| US9758561B2 (en) | 2014-04-07 | 2017-09-12 | Sanofi | Dual GLP-1/glucagon receptor agonists derived from exendin-4 |
| US9771406B2 (en) | 2014-04-07 | 2017-09-26 | Sanofi | Peptidic dual GLP-1/glucagon receptor agonists derived from exendin-4 |
| US9775904B2 (en) | 2014-04-07 | 2017-10-03 | Sanofi | Exendin-4 derivatives as peptidic dual GLP-1/glucagon receptor agonists |
| US9789165B2 (en) | 2013-12-13 | 2017-10-17 | Sanofi | Exendin-4 peptide analogues as dual GLP-1/GIP receptor agonists |
| US9790263B2 (en) | 2009-06-16 | 2017-10-17 | Indiana University Research And Technology Corporation | GIP receptor-active glucagon compounds |
| WO2017186250A1 (en) | 2016-04-29 | 2017-11-02 | Defensin Therapeutics Aps | Treatment of liver, biliary tract and pancreatic disorders |
| WO2018011266A1 (en) | 2016-07-13 | 2018-01-18 | Ascendis Pharma A/S | Conjugation method for carrier-linked prodrugs |
| US9932381B2 (en) | 2014-06-18 | 2018-04-03 | Sanofi | Exendin-4 derivatives as selective glucagon receptor agonists |
| WO2018060314A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Growth Disorders A/S | Combination therapy with controlled-release cnp agonists |
| WO2018060311A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Bone Diseases A/S | Incremental dose finding in controlled-release pth compounds |
| WO2018060312A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Bone Diseases A/S | Pth compounds with low peak-to-trough ratios |
| WO2018060310A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Bone Diseases A/S | Dosage regimen for a controlled-release pth compound |
| US9982029B2 (en) | 2015-07-10 | 2018-05-29 | Sanofi | Exendin-4 derivatives as selective peptidic dual GLP-1/glucagon receptor agonists |
| WO2018100174A1 (en) | 2016-12-02 | 2018-06-07 | Sanofi | Conjugates comprising an glp-1/glucagon dual agonist, a linker and hyaluronic acid |
| WO2018108971A2 (en) | 2016-12-13 | 2018-06-21 | Defensin Therapeutics Aps | Methods for treating inflammatory conditions of the lungs |
| US10040850B2 (en) | 2013-10-08 | 2018-08-07 | Ascendis Pharma A/S | Protecting group comprising a purification tag |
| WO2018175788A1 (en) | 2017-03-22 | 2018-09-27 | Genentech, Inc. | Hydrogel cross-linked hyaluronic acid prodrug compositions and methods |
| US10232020B2 (en) | 2014-09-24 | 2019-03-19 | Indiana University Research And Technology Corporation | Incretin-insulin conjugates |
| WO2019076675A1 (en) * | 2017-10-06 | 2019-04-25 | Instituto De Medicina Molecular | Treatment of obesity-related conditions |
| WO2019092201A2 (en) | 2017-11-10 | 2019-05-16 | Defensin Therapeutics Aps | Maturation of mucosal defense and gut/lung function in the preterm infant |
| WO2019101773A2 (en) | 2017-11-24 | 2019-05-31 | Defensin Therapeutics Aps | Prevention and treatment of graft-versus-host-disease with defensins |
| US10385107B2 (en) | 2014-09-24 | 2019-08-20 | Indiana Univeresity Researc and Technology Corporation | Lipidated amide-based insulin prodrugs |
| WO2019185706A1 (en) | 2018-03-28 | 2019-10-03 | Ascendis Pharma A/S | Conjugates |
| WO2019185705A1 (en) | 2018-03-28 | 2019-10-03 | Ascendis Pharma A/S | Il-2 conjugates |
| WO2019219896A1 (en) | 2018-05-18 | 2019-11-21 | Ascendis Pharma Bone Diseases A/S | Starting dose of pth conjugates |
| WO2019229225A1 (en) | 2018-05-30 | 2019-12-05 | Sanofi | Conjugates comprising an glp-1/glucagon/gip triple receptor agonist, a linker and hyaluronic acid |
| WO2020064844A1 (en) | 2018-09-26 | 2020-04-02 | Ascendis Pharma A/S | Treatment of infections |
| WO2020064847A1 (en) | 2018-09-26 | 2020-04-02 | Ascendis Pharma A/S | Degradable hyaluronic acid hydrogels |
| WO2020064846A1 (en) | 2018-09-26 | 2020-04-02 | Ascendis Pharma A/S | Novel hydrogel conjugates |
| US10696726B2 (en) | 2013-03-14 | 2020-06-30 | Indiana University Research And Technology Corporation | Insulin-incretin conjugates |
| WO2020141225A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Minimization of systemic inflammation |
| WO2020141221A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Conjugates of pattern recognition receptor agonists |
| WO2020141223A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Induction of sustained local inflammation |
| WO2020141222A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Sustained local drug levels for innate immune agonists |
| WO2020165087A1 (en) | 2019-02-11 | 2020-08-20 | Ascendis Pharma Bone Diseases A/S | Liquid pharmaceutical formulations of pth conjugates |
| WO2020165081A1 (en) | 2019-02-11 | 2020-08-20 | Ascendis Pharma Growth Disorders A/S | Dry pharmaceutical formulations of cnp conjugates |
| US10751417B2 (en) | 2017-04-20 | 2020-08-25 | Novartis Ag | Sustained release delivery systems comprising traceless linkers |
| US10758592B2 (en) | 2012-10-09 | 2020-09-01 | Sanofi | Exendin-4 derivatives as dual GLP1/glucagon agonists |
| US10800738B2 (en) | 2017-12-05 | 2020-10-13 | Sunovion Pharmaceuticals Inc. | Crystal forms and production methods thereof |
| USRE48267E1 (en) | 2009-05-20 | 2020-10-20 | Biomarin Pharmaceutical Inc. | Variants of C-type natriuretic peptide |
| WO2020254617A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Anti-ctla4 compounds with localized pk properties |
| WO2020254607A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Anti-ctla4 compounds with localized pd properties |
| WO2020254606A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma A/S | Conjugates of heteroaromatic nitrogen-comprising compounds |
| WO2020254613A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Controlled-release tyrosine kinase inhibitor compounds with localized pk properties |
| WO2020254609A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Tyrosine kinase inhibitor conjugates |
| WO2020254611A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Anti-ctla4 conjugates |
| WO2020254603A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma A/S | CONJUGATES OF π-ELECTRON-PAIR-DONATING HETEROAROMATIC NITROGEN-COMPRISING COMPOUNDS |
| WO2020254612A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Controlled-release tyrosine kinase inhibitor compounds with localized pd properties |
| US10874639B2 (en) | 2017-12-05 | 2020-12-29 | Sunovion Pharmaceuticals Inc. | Nonracemic mixtures and uses thereof |
| WO2021136808A1 (en) | 2020-01-03 | 2021-07-08 | Ascendis Pharma A/S | Conjugates undergoing intramolecular rearrangements |
| WO2021144249A1 (en) | 2020-01-13 | 2021-07-22 | Ascendis Pharma Bone Diseases A/S | Hypoparathyroidism treatment |
| US11116816B2 (en) | 2014-10-22 | 2021-09-14 | Extend Biosciences, Inc. | Therapeutic vitamin d conjugates |
| US11160758B2 (en) | 2019-06-04 | 2021-11-02 | Sunovion Pharmaceuticals Inc. | Modified release formulations and uses thereof |
| WO2021224169A1 (en) | 2020-05-04 | 2021-11-11 | Ascendis Pharma A/S | Hydrogel irradiation |
| US11179467B2 (en) | 2015-09-25 | 2021-11-23 | Brightgene Bio-Medical Technology Co., Ltd. | Exenatide modifier and use thereof |
| WO2021245130A1 (en) | 2020-06-03 | 2021-12-09 | Ascendis Pharma Oncology Division A/S | Il-2 sequences and uses thereof |
| US11224661B2 (en) | 2016-01-08 | 2022-01-18 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with increased NEP stability |
| WO2022029178A1 (en) | 2020-08-05 | 2022-02-10 | Ascendis Pharma A/S | Conjugates comprising reversible linkers and uses thereof |
| WO2022043493A1 (en) | 2020-08-28 | 2022-03-03 | Ascendis Pharma Oncology Division A/S | Glycosylated il-2 proteins and uses thereof |
| WO2022053696A1 (en) * | 2020-09-11 | 2022-03-17 | Compass Pathfinder Limited | Novel safrylamine derivatives having prodrug properties |
| WO2022064035A1 (en) | 2020-09-28 | 2022-03-31 | Ascendis Pharma Bone Diseases A/S | Improvement of physical and mental well-being of patients with hypoparathyroidism |
| US11357828B2 (en) | 2018-09-12 | 2022-06-14 | Quiapeg Pharmaceuticals Ab | Releasable GLP-1 conjugates |
| US11389541B2 (en) | 2018-10-03 | 2022-07-19 | Novartis Ag | Sustained delivery of angiopoetin-like 3 polypeptides |
| US11389511B2 (en) | 2016-01-08 | 2022-07-19 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with reduced side-effects |
| WO2022207798A1 (en) | 2021-04-01 | 2022-10-06 | Ascendis Pharma A/S | Use of long-acting growth hormone for treating inflammation-induced diseases |
| US11559482B2 (en) | 2009-07-31 | 2023-01-24 | Ascendis Pharma A/S | Biodegradable polyethylene glycol based water-insoluble hydrogels |
| WO2023046732A1 (en) | 2021-09-22 | 2023-03-30 | Ascendis Pharma Bone Diseases A/S | Long-acting pth compound treatments |
| US11633487B2 (en) | 2014-08-06 | 2023-04-25 | Ascendis Pharma A/S | Prodrugs comprising an aminoalkyl glycine linker |
| WO2023110727A2 (en) | 2021-12-13 | 2023-06-22 | Ascendis Pharma Oncology Division A/S | Novel cancer treatments with tlr7/8 agonists |
| WO2023110758A1 (en) | 2021-12-13 | 2023-06-22 | Ascendis Pharma Growth Disorders A/S | Effective doses of cnp conjugates |
| US11786599B2 (en) | 2017-03-10 | 2023-10-17 | Quiapeg Pharmaceuticals Ab | Releasable conjugates |
| RU2806753C2 (en) * | 2018-05-18 | 2023-11-07 | Асцендис Фарма Боун Дизизис А/С | Method of treating or control of hypoparathyroidus using parathrooid hormone (pth) conjugate |
| WO2023227505A1 (en) | 2022-05-23 | 2023-11-30 | Ascendis Pharma Growth Disorders A/S | Liquid pharmaceutical formulations of cnp compounds |
| US11845736B2 (en) | 2021-10-01 | 2023-12-19 | Empathbio, Inc. | Prodrugs of MDMA, MDA, and derivatives thereof |
| US11912680B2 (en) | 2021-12-28 | 2024-02-27 | Empathbio, Inc. | Nitric oxide releasing prodrugs of MDA and MDMA |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2596805B1 (en) * | 2008-02-01 | 2021-10-27 | Ascendis Pharma A/S | Prodrug comprising a drug-linker conjugate |
| WO2010099200A1 (en) | 2009-02-24 | 2010-09-02 | Nektar Therapeutics | Oligomer-amino acid conjugates |
| EP2485712A1 (en) | 2009-10-06 | 2012-08-15 | Ascendis Pharma A/S | Subcutaneous paliperidone composition |
| MX2012004499A (en) | 2009-10-29 | 2012-05-29 | Ascendis Pharma As | Sterilization of biodegradable hydrogels. |
| US9561285B2 (en) | 2010-01-22 | 2017-02-07 | Ascendis Pharma As | Carrier-linked carbamate prodrug linkers |
| CN103857440B (en) | 2011-06-22 | 2018-09-25 | 维奥姆生物科学有限公司 | Antimycotic and antibacterium prodrug based on conjugate |
| RU2015101697A (en) | 2012-06-21 | 2016-08-10 | Индиана Юниверсити Рисерч Энд Текнолоджи Корпорейшн | GLUCAGON ANALOGUES WITH GIP RECEPTOR ACTIVITY |
| CA2883320A1 (en) * | 2012-08-30 | 2014-03-06 | The Scripps Research Institute | Small molecules targeting repeat r(cgg) sequences |
| WO2014187315A1 (en) * | 2013-05-21 | 2014-11-27 | 成都先导药物开发有限公司 | Compound administration precursor and medicament carrier preparation |
| JP6276390B2 (en) * | 2013-05-21 | 2018-02-07 | 成都先導薬物開発有限公司 | Method of cell membrane permeation of compounds |
| KR101768446B1 (en) | 2014-03-21 | 2017-08-17 | 애니젠 주식회사 | Novel Exenatide Analogs and Uses thereof |
| CN104945499B (en) * | 2014-03-31 | 2019-12-10 | 博瑞生物医药(苏州)股份有限公司 | GLP-1 analogue with modified structure and preparation method thereof |
| CN106456768B (en) | 2014-06-13 | 2019-12-03 | 腾博龙公司 | Conjugate containing anti-EGFR1 antibody |
| US10265413B2 (en) | 2014-11-05 | 2019-04-23 | University Of The Sciences In Philadelphia | High molecular weight biodegradable gelatin-doxorubicin conjugate |
| TWI660741B (en) | 2015-11-03 | 2019-06-01 | 財團法人工業技術研究院 | Antibody-drug conjugate (adc) and method for forming the same |
| WO2017127528A1 (en) * | 2016-01-19 | 2017-07-27 | University Of Florida Research Foundation, Inc. | Peptoid agonists of fibroblast growth receptors |
| MA45328A (en) | 2016-04-01 | 2019-02-06 | Avidity Biosciences Llc | NUCLEIC ACID-POLYPEPTIDE COMPOSITIONS AND USES THEREOF |
| EP3598866A1 (en) * | 2016-12-29 | 2020-01-29 | Development Center for Biotechnology | Klk6-mediated cns-specific antibody prodrug activation |
| GB2571696B (en) | 2017-10-09 | 2020-05-27 | Compass Pathways Ltd | Large scale method for the preparation of Psilocybin and formulations of Psilocybin so produced |
| CN109160883B (en) * | 2017-11-30 | 2021-10-08 | 银隆新能源股份有限公司 | Anthraquinone salt positive electrode material and synthetic method thereof |
| WO2019113393A1 (en) | 2017-12-06 | 2019-06-13 | Avidity Biosciences Llc | Compositions and methods of treating muscle atrophy and myotonic dystrophy |
| US10799138B2 (en) | 2018-04-05 | 2020-10-13 | University Of Maryland, Baltimore | Method of administering sotalol IV/switch |
| CN112543763A (en) | 2018-04-10 | 2021-03-23 | 赛诺菲-安万特德国有限公司 | Method for cleaving solid phase bound peptides from solid phase |
| WO2019197469A1 (en) | 2018-04-10 | 2019-10-17 | Sanofi-Aventis Deutschland Gmbh | Lixisenatide synthesis with capping |
| US11610660B1 (en) | 2021-08-20 | 2023-03-21 | AltaThera Pharmaceuticals LLC | Antiarrhythmic drug dosing methods, medical devices, and systems |
| US11344518B2 (en) | 2018-08-14 | 2022-05-31 | AltaThera Pharmaceuticals LLC | Method of converting atrial fibrillation to normal sinus rhythm and loading oral sotalol in a shortened time frame |
| US10512620B1 (en) | 2018-08-14 | 2019-12-24 | AltaThera Pharmaceuticals, LLC | Method of initiating and escalating sotalol hydrochloride dosing |
| US11696902B2 (en) | 2018-08-14 | 2023-07-11 | AltaThera Pharmaceuticals, LLC | Method of initiating and escalating sotalol hydrochloride dosing |
| CN109125707B (en) * | 2018-10-19 | 2022-01-04 | 艾伟伦 | GnRH analogue sustained-release composition and preparation method thereof |
| WO2020212948A1 (en) | 2019-04-17 | 2020-10-22 | Compass Pathfinder Limited | Methods of treating neurocognitive disorders, chronic pain and reducing inflammation |
| AU2021237465A1 (en) | 2020-03-19 | 2022-10-13 | Avidity Biosciences, Inc. | Compositions and methods of treating Facioscapulohumeral muscular dystrophy |
| CN113842379B (en) * | 2021-09-12 | 2024-04-23 | 东北农业大学 | New application of betaxolol as NDM-1 inhibitor or antibiotic protectant |
| US11912779B2 (en) | 2021-09-16 | 2024-02-27 | Avidity Biosciences, Inc. | Compositions and methods of treating facioscapulohumeral muscular dystrophy |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4179337A (en) * | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| JPS6253997A (en) * | 1985-09-03 | 1987-03-09 | Kowa Co | Novel amino glycoside and pharmaceutical preparation containing same |
| TW517067B (en) * | 1996-05-31 | 2003-01-11 | Hoffmann La Roche | Interferon conjugates |
| EP0986539A1 (en) * | 1997-04-06 | 2000-03-22 | California Institute Of Technology | Dna-binding pyrrole and imidazole polyamide derivatives |
| CA2623458A1 (en) | 1999-05-17 | 2000-11-23 | Conjuchem Biotechnologies Inc. | Protection of endogenous therapeutic peptides from peptidase activity through conjugation to blood components |
| US7087229B2 (en) * | 2003-05-30 | 2006-08-08 | Enzon Pharmaceuticals, Inc. | Releasable polymeric conjugates based on aliphatic biodegradable linkers |
| EP1764360A4 (en) * | 2004-07-02 | 2010-08-11 | Sankyo Co | Urea derivative |
| US7968085B2 (en) | 2004-07-05 | 2011-06-28 | Ascendis Pharma A/S | Hydrogel formulations |
| EP1625856A1 (en) | 2004-08-13 | 2006-02-15 | Complex Biosystems GmbH | Hydrogel polymeric conjugates of a prodrug |
| US20060115865A1 (en) * | 2004-10-25 | 2006-06-01 | Anlong Ouyang | Lamotrigine analogs |
| US7678551B2 (en) | 2004-10-25 | 2010-03-16 | Seradyn, Inc. | Immunoassays for lamotrigine |
| US7407117B2 (en) | 2004-10-28 | 2008-08-05 | Meadwestvaco Calmar, Inc. | Liquid sprayer assembly |
| EP1838317A4 (en) * | 2005-01-05 | 2012-02-29 | Univ Minnesota | Analgesic conjugates |
| GB2427360A (en) * | 2005-06-22 | 2006-12-27 | Complex Biosystems Gmbh | Aliphatic prodrug linker |
| US20090017004A1 (en) * | 2007-07-11 | 2009-01-15 | Enzon Pharmaceuticals, Inc. | Polymeric drug delivery systems containing an aromatic allylic acid |
| EP2596805B1 (en) * | 2008-02-01 | 2021-10-27 | Ascendis Pharma A/S | Prodrug comprising a drug-linker conjugate |
| EP2438930A1 (en) * | 2010-09-17 | 2012-04-11 | Sanofi-Aventis Deutschland GmbH | Prodrugs comprising an exendin linker conjugate |
-
2009
- 2009-01-30 EP EP13156350.4A patent/EP2596805B1/en active Active
- 2009-01-30 ES ES13156350T patent/ES2904673T3/en active Active
- 2009-01-30 CA CA2712224A patent/CA2712224C/en active Active
- 2009-01-30 RS RS20190789A patent/RS58948B1/en unknown
- 2009-01-30 DK DK09706788.8T patent/DK2237799T3/en active
- 2009-01-30 LT LTEP09706788.8T patent/LT2237799T/en unknown
- 2009-01-30 HU HUE09706788 patent/HUE044174T2/en unknown
- 2009-01-30 CN CN2009801110881A patent/CN101980725B/en active Active
- 2009-01-30 PL PL09706788T patent/PL2237799T3/en unknown
- 2009-01-30 DK DK13156350.4T patent/DK2596805T3/en active
- 2009-01-30 ES ES09706788T patent/ES2733355T3/en active Active
- 2009-01-30 EP EP09706788.8A patent/EP2237799B1/en active Active
- 2009-01-30 WO PCT/EP2009/051079 patent/WO2009095479A2/en active Application Filing
- 2009-01-30 BR BRPI0906643A patent/BRPI0906643A2/en not_active Application Discontinuation
- 2009-01-30 JP JP2010544717A patent/JP5588354B2/en active Active
- 2009-01-30 RU RU2010136023/15A patent/RU2010136023A/en unknown
- 2009-01-30 US US12/865,693 patent/US8906847B2/en active Active
- 2009-01-30 PT PT09706788T patent/PT2237799T/en unknown
- 2009-01-30 MX MX2010008024A patent/MX2010008024A/en active IP Right Grant
- 2009-01-30 EP EP21202920.1A patent/EP3981761A3/en active Pending
- 2009-01-30 SI SI200931976T patent/SI2237799T1/en unknown
- 2009-01-30 AU AU2009209565A patent/AU2009209565B2/en active Active
-
2010
- 2010-07-22 IL IL207150A patent/IL207150A/en active IP Right Grant
- 2010-07-29 ZA ZA2010/05413A patent/ZA201005413B/en unknown
-
2011
- 2011-06-21 HK HK11106352.6A patent/HK1152240A1/en unknown
-
2014
- 2014-05-30 RU RU2014122036A patent/RU2676324C2/en active
- 2014-06-04 JP JP2014115482A patent/JP2014193908A/en active Pending
- 2014-10-30 US US14/528,362 patent/US20150057221A1/en not_active Abandoned
-
2016
- 2016-12-20 US US15/385,179 patent/US20170100490A1/en not_active Abandoned
-
2019
- 2019-06-12 HR HRP20191052 patent/HRP20191052T1/en unknown
- 2019-07-03 CY CY20191100707T patent/CY1121961T1/en unknown
- 2019-10-16 US US16/654,667 patent/US20200046725A1/en not_active Abandoned
-
2022
- 2022-06-23 US US17/847,750 patent/US20220331334A1/en not_active Abandoned
-
2023
- 2023-10-02 US US18/479,445 patent/US20240041897A1/en active Pending
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (252)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8338368B2 (en) | 2005-11-07 | 2012-12-25 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting physiological solubility and stability |
| US9018164B2 (en) | 2005-11-07 | 2015-04-28 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting physiological solubility and stability |
| US8669228B2 (en) | 2007-01-05 | 2014-03-11 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting enhanced solubility in physiological pH buffers |
| US8454971B2 (en) | 2007-02-15 | 2013-06-04 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US8980830B2 (en) | 2007-10-30 | 2015-03-17 | Indiana University Research And Technology Corporation | Peptide compounds exhibiting glucagon antagonist and GLP-1 agonist activity |
| US8981047B2 (en) | 2007-10-30 | 2015-03-17 | Indiana University Research And Technology Corporation | Glucagon antagonists |
| US8697838B2 (en) | 2008-01-30 | 2014-04-15 | Indiana University Research And Technology Corporation | Ester-based insulin prodrugs |
| US9089539B2 (en) | 2008-01-30 | 2015-07-28 | Indiana University Research And Technology Corporation | Ester-based insulin prodrugs |
| US10098930B2 (en) | 2008-04-29 | 2018-10-16 | Ascendis Pharma Endocrinology Division A/S | PEGylated Recombinant Human Growth Hormone Compounds |
| US10960053B2 (en) | 2008-04-29 | 2021-03-30 | Ascendis Pharma Endocrinology Division A/S | PEGylated recombinant human growth hormone compounds |
| US9272048B2 (en) | 2008-04-29 | 2016-03-01 | Ascendis Pharma Growth Disorders Division A/S | PEGylated recombinant human growth hormone compounds |
| US9062124B2 (en) | 2008-06-17 | 2015-06-23 | Indiana University Research And Technology Corporation | GIP-based mixed agonists for treatment of metabolic disorders and obesity |
| US8546327B2 (en) | 2008-06-17 | 2013-10-01 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US8450270B2 (en) | 2008-06-17 | 2013-05-28 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting enhanced solubility and stability in physiological pH buffers |
| WO2010042596A2 (en) * | 2008-10-08 | 2010-04-15 | The Uab Research Foundation | Photo-activated protecting groups |
| WO2010042596A3 (en) * | 2008-10-08 | 2010-07-01 | The Uab Research Foundation | Photo-activated protecting groups |
| US8969288B2 (en) | 2008-12-19 | 2015-03-03 | Indiana University Research And Technology Corporation | Amide based glucagon and superfamily peptide prodrugs |
| US8481485B2 (en) | 2008-12-19 | 2013-07-09 | Indiana University Research And Technology Corporation | Insulin analogs |
| US8697632B2 (en) | 2008-12-19 | 2014-04-15 | Indiana University Research And Technology Corporation | Amide based insulin prodrugs |
| USRE48267E1 (en) | 2009-05-20 | 2020-10-20 | Biomarin Pharmaceutical Inc. | Variants of C-type natriuretic peptide |
| US9790263B2 (en) | 2009-06-16 | 2017-10-17 | Indiana University Research And Technology Corporation | GIP receptor-active glucagon compounds |
| US9457066B2 (en) | 2009-07-31 | 2016-10-04 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an insulin linker conjugate |
| US9265723B2 (en) | 2009-07-31 | 2016-02-23 | Sanofi-Aventis Deutschland Gmbh | Long acting insulin composition |
| WO2011012722A1 (en) * | 2009-07-31 | 2011-02-03 | Ascendis Pharma As | Prodrugs containing an aromatic amine connected by an amido bond to a linker |
| JP2013500951A (en) * | 2009-07-31 | 2013-01-10 | サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | Prodrugs containing insulin linker conjugates |
| US11559482B2 (en) | 2009-07-31 | 2023-01-24 | Ascendis Pharma A/S | Biodegradable polyethylene glycol based water-insoluble hydrogels |
| US20120184489A1 (en) * | 2009-07-31 | 2012-07-19 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an insulin linker conjugate |
| US9138462B2 (en) | 2009-07-31 | 2015-09-22 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an insulin linker conjugate |
| US9173953B2 (en) | 2009-07-31 | 2015-11-03 | Ascendis Pharma As | Prodrugs containing an aromatic amine connected by an amido bond to a linker |
| JP2013513647A (en) * | 2009-12-15 | 2013-04-22 | アセンディス ファーマ エー/エス | Dry growth hormone composition temporarily linked to a polymer carrier |
| US9511122B2 (en) | 2009-12-15 | 2016-12-06 | Ascendis Pharma Growth Disorders Division A/S | Dry growth hormone composition transiently linked to a polymer carrier |
| CN105125508A (en) * | 2009-12-15 | 2015-12-09 | 阿森迪斯药物生长障碍股份有限公司 | Growth hormone composition |
| US10682395B2 (en) | 2009-12-15 | 2020-06-16 | Ascendis Pharma Endocrinology Division A/S | Dry growth hormone composition transiently linked to a polymer carrier |
| WO2011073234A3 (en) * | 2009-12-15 | 2012-04-19 | Ascendis Pharma As | Dry growth hormone composition transiently linked to a polymer carrier |
| AU2010332958B2 (en) * | 2009-12-15 | 2014-09-18 | Ascendis Pharma Endocrinology Division A/S | Dry growth hormone composition transiently linked to a polymer carrier |
| CN102711733A (en) * | 2009-12-15 | 2012-10-03 | 阿森迪斯药物股份有限公司 | Growth hormone composition which is instantly connected with polymer carrier |
| US9919033B2 (en) | 2009-12-15 | 2018-03-20 | Ascendis Pharma Endocrinology Division A/S | Dry growth hormone composition transiently linked to a polymer carrier |
| US8703701B2 (en) | 2009-12-18 | 2014-04-22 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US9533056B2 (en) | 2010-01-22 | 2017-01-03 | Ascendis Pharma As | Dipeptide-based prodrug linkers for aliphatic amine-containing drugs |
| US9062094B2 (en) | 2010-01-22 | 2015-06-23 | Ascendis Pharma As | Dipeptide-based prodrug linkers for aliphatic amine-containing drugs |
| WO2011089216A1 (en) * | 2010-01-22 | 2011-07-28 | Ascendis Pharma As | Dipeptide-based prodrug linkers for aliphatic amine-containing drugs |
| US9487571B2 (en) | 2010-01-27 | 2016-11-08 | Indiana University Research And Technology Corporation | Glucagon antagonist-GIP agonist conjugates and compositions for the treatment of metabolic disorders and obesity |
| US20120035320A1 (en) * | 2010-04-03 | 2012-02-09 | University Of Iowa Research Foundation | Polyacridine nucleic acid delivery peptide complexes |
| US9783592B2 (en) | 2010-05-13 | 2017-10-10 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhibiting nuclear hormone receptor activity |
| US9145451B2 (en) | 2010-05-13 | 2015-09-29 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhbiting G protein coupled receptor activity |
| US9127088B2 (en) | 2010-05-13 | 2015-09-08 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhibiting nuclear hormone receptor activity |
| US9458220B2 (en) | 2010-06-16 | 2016-10-04 | Indiana University Research And Technology Corporation | Single-chain insulin agonists exhibiting high activity at the insulin receptor |
| US8940860B2 (en) | 2010-06-16 | 2015-01-27 | Indiana University Research And Technology Corporation | Single-chain insulin agonists exhibiting high activity at the insulin receptor |
| US10233225B2 (en) | 2010-06-16 | 2019-03-19 | Indiana University Research And Technology Corporation | Single chain insulin agonists exhibiting high activity at the insulin receptor |
| CN103179979A (en) * | 2010-06-24 | 2013-06-26 | 印第安纳大学研究及科技有限公司 | Amide based glucagon superfamily peptide prodrugs |
| RU2580317C2 (en) * | 2010-06-24 | 2016-04-10 | Индиана Юниверсити Рисерч Энд Текнолоджи Корпорейшн | Peptide pro-drugs of amide glucagon superfamily |
| US8946147B2 (en) | 2010-06-24 | 2015-02-03 | Indiana University Research And Technology Corporation | Amide-based insulin prodrugs |
| WO2011163012A3 (en) * | 2010-06-24 | 2013-07-11 | Indiana University Research And Technology Corporation | Amide based glucagon superfamily peptide prodrugs |
| US8778872B2 (en) | 2010-06-24 | 2014-07-15 | Indiana University Research And Technology Corporation | Amide based glucagon superfamily peptide prodrugs |
| US9133276B2 (en) | 2010-09-17 | 2015-09-15 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an exendin linker conjugate |
| AU2011303823B2 (en) * | 2010-09-17 | 2014-07-17 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an exendin linker conjugate |
| CN103237561B (en) * | 2010-09-17 | 2015-12-02 | 赛诺菲-安万特德国有限公司 | Comprise the prodrug of Exendin junctional complex conjugate |
| RU2593774C2 (en) * | 2010-09-17 | 2016-08-10 | Санофи-Авентис Дойчланд Гмбх | Prodrugs containing exendin conjugate-linker |
| WO2012035139A1 (en) | 2010-09-17 | 2012-03-22 | Sanofi-Aventis Deutschland Gmbh | Prodrugs comprising an exendin linker conjugate |
| CN103237561A (en) * | 2010-09-17 | 2013-08-07 | 赛诺菲-安万特德国有限公司 | Prodrugs comprising an exendin linker conjugate |
| EP2438930A1 (en) | 2010-09-17 | 2012-04-11 | Sanofi-Aventis Deutschland GmbH | Prodrugs comprising an exendin linker conjugate |
| JP2013538224A (en) * | 2010-09-17 | 2013-10-10 | サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | Prodrugs containing exendin linker conjugates |
| US8507428B2 (en) | 2010-12-22 | 2013-08-13 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting GIP receptor activity |
| US10730923B2 (en) | 2011-06-22 | 2020-08-04 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US8729017B2 (en) | 2011-06-22 | 2014-05-20 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US9156902B2 (en) | 2011-06-22 | 2015-10-13 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US10174093B2 (en) | 2011-06-22 | 2019-01-08 | Indiana University Research And Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| US9758562B2 (en) | 2011-06-22 | 2017-09-12 | Indiana University and Technology Corporation | Glucagon/GLP-1 receptor co-agonists |
| WO2013010840A2 (en) | 2011-07-18 | 2013-01-24 | Arts Biologics A/S | Long acting biologically active luteinizing hormone (lh) compound |
| US9353172B2 (en) | 2011-07-18 | 2016-05-31 | Arts Biologics A/S | Long acting biologically active luteinizing hormone (LH) compound |
| EP3643306A2 (en) | 2011-08-12 | 2020-04-29 | Ascendis Pharma A/S | Carrier-linked prodrugs having reversible carboxylic ester linkages |
| WO2013024053A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | Carrier-linked prodrugs having reversible carboxylic ester linkages |
| WO2013024049A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | Protein carrier-linked prodrugs |
| WO2013024048A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | Polymeric hyperbranched carrier-linked prodrugs |
| WO2013024047A1 (en) | 2011-08-12 | 2013-02-21 | Ascendis Pharma A/S | High-loading water-soluble carrier-linked prodrugs |
| US20140323402A1 (en) * | 2011-08-12 | 2014-10-30 | Ascendis Phama A/S | Protein Carrier-Linked Prodrugs |
| WO2013053856A1 (en) | 2011-10-12 | 2013-04-18 | Ascendis Pharma A/S | Prevention and treatment of ocular conditions |
| EP2768856A4 (en) * | 2011-10-18 | 2015-05-27 | Prolynx Llc | Peg conjugates of exenatide |
| US8859491B2 (en) | 2011-11-17 | 2014-10-14 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhibiting glucocorticoid receptor activity |
| US9573987B2 (en) | 2011-12-20 | 2017-02-21 | Indiana University Research And Technology Corporation | CTP-based insulin analogs for treatment of diabetes |
| AU2013254756B2 (en) * | 2012-04-25 | 2017-03-30 | Ascendis Pharma A/S | Prodrugs of hydroxyl-comprising drugs |
| WO2013160340A1 (en) | 2012-04-25 | 2013-10-31 | Ascendis Pharma A/S | Prodrugs of hydroxyl-comprising drugs |
| US20150087688A1 (en) * | 2012-04-25 | 2015-03-26 | Ascendis Pharma A/S | Prodrugs of hydroxyl-comprising drugs |
| WO2013167750A2 (en) | 2012-05-11 | 2013-11-14 | Prorec Bio Ab | Method for diagnosis and treatment of prolactin associated disorders |
| US9593156B2 (en) | 2012-09-26 | 2017-03-14 | Indiana University Research And Technology Corporation | Insulin analog dimers |
| US10758592B2 (en) | 2012-10-09 | 2020-09-01 | Sanofi | Exendin-4 derivatives as dual GLP1/glucagon agonists |
| RU2682676C2 (en) * | 2012-10-11 | 2019-03-20 | Асцендис Фарма Ас | Diagnosis, prevention and treatment of joint diseases |
| KR102235868B1 (en) * | 2012-10-11 | 2021-04-07 | 아센디스 파마 옵탈몰로지 디비젼 에이/에스 | Vegf neutralizing prodrugs for the treatment of ocular conditions |
| US10519226B2 (en) | 2012-10-11 | 2019-12-31 | Ascendis Pharma Opthalmology Division A/S | VEGF neutralizing prodrugs for the treatment of ocular conditions |
| US10980860B2 (en) | 2012-10-11 | 2021-04-20 | Ascendis Pharma A/S | Diagnosis, prevention and treatment of diseases of the joint |
| WO2014056923A1 (en) | 2012-10-11 | 2014-04-17 | Ascendis Pharma Ophthalmology Division A/S | Vegf neutralizing prodrugs for the treatment of ocular conditions |
| KR20150065891A (en) * | 2012-10-11 | 2015-06-15 | 아센디스 파마 옵탈몰로지 디비젼 에이/에스 | Vegf neutralizing prodrugs for the treatment of ocular conditions |
| WO2014056915A1 (en) | 2012-10-11 | 2014-04-17 | Ascendis Pharma A/S | Diagnosis, prevention and treatment of diseases of the joint |
| US9855340B2 (en) | 2012-10-11 | 2018-01-02 | Ascendis Pharma A/S | Hydrogel prodrugs |
| RU2673881C2 (en) * | 2012-10-11 | 2018-12-03 | Асцендис Фарма Офтальмолоджи Дивижн А/С | Vegf neutralising prodrugs for the treatment of ocular conditions |
| WO2014056926A1 (en) | 2012-10-11 | 2014-04-17 | Ascendis Pharma A/S | Hydrogel prodrugs |
| WO2014080730A1 (en) | 2012-11-22 | 2014-05-30 | 株式会社糖鎖工学研究所 | Glycosylated linker, compound containing glycosylated linker moiety and physiologically active substance moiety or salt thereof, and methods for producing said compound or salt thereof |
| US9708413B2 (en) | 2012-11-22 | 2017-07-18 | Glytech, Inc. | Glycosylated linker, compound containing glycosylated linker moiety and physiologically active substance moiety or salt thereof, and methods for producing said compound or salt thereof |
| KR102227918B1 (en) * | 2012-11-22 | 2021-03-15 | 가부시키가이샤 도우사 고가쿠 겐큐쇼 | Glycosylated linker, compound containing glycosylated linker moiety and physiologically active substance moiety or salt thereof, and methods for producing said compound or salt thereof |
| KR20150102003A (en) * | 2012-11-22 | 2015-09-04 | 가부시키가이샤 도우사 고가쿠 겐큐쇼 | Glycosylated linker, compound containing glycosylated linker moiety and physiologically active substance moiety or salt thereof, and methods for producing said compound or salt thereof |
| US10202469B2 (en) | 2012-11-30 | 2019-02-12 | Glytech, Inc. | Sugar chain-attached linker, compound containing sugar chain-attached linker and physiologically active substance or salt thereof, and method for producing same |
| WO2014084110A1 (en) | 2012-11-30 | 2014-06-05 | 株式会社糖鎖工学研究所 | Sugar chain-attached linker, compound containing sugar chain-attached linker and physiologically active substance or salt thereof, and method for producing same |
| US10253079B2 (en) | 2012-12-21 | 2019-04-09 | Sanofi | Functionalized Exendin-4 derivatives |
| US9670261B2 (en) | 2012-12-21 | 2017-06-06 | Sanofi | Functionalized exendin-4 derivatives |
| US9745360B2 (en) | 2012-12-21 | 2017-08-29 | Sanofi | Dual GLP1/GIP or trigonal GLP1/GIP/glucagon agonists |
| US10696726B2 (en) | 2013-03-14 | 2020-06-30 | Indiana University Research And Technology Corporation | Insulin-incretin conjugates |
| WO2014173759A1 (en) | 2013-04-22 | 2014-10-30 | Ascendis Pharma A/S | Hydrogel-linked prodrugs releasing modified drugs |
| US11116849B2 (en) | 2013-04-22 | 2021-09-14 | Ascendis Pharma A/S | Hydrogel-linked prodrugs releasing tagged drugs |
| WO2015052154A1 (en) * | 2013-10-08 | 2015-04-16 | Ascendis Pharma Osteoarthritis Division A/S | Hydrogel-linked il-1ra prodrug |
| AU2014333954C1 (en) * | 2013-10-08 | 2020-08-27 | Ascendis Pharma A/S | Protecting group comprising a purification tag |
| US10040850B2 (en) | 2013-10-08 | 2018-08-07 | Ascendis Pharma A/S | Protecting group comprising a purification tag |
| AU2014333954B2 (en) * | 2013-10-08 | 2020-03-05 | Ascendis Pharma A/S | Protecting group comprising a purification tag |
| WO2015067791A1 (en) | 2013-11-11 | 2015-05-14 | Ascendis Pharma Relaxin Division A/S | Relaxin prodrugs |
| US10626156B2 (en) | 2013-12-06 | 2020-04-21 | Jie Han | Bioreversable promoieties for nitrogen-containing and hydroxyl-containing drugs |
| WO2015081891A1 (en) | 2013-12-06 | 2015-06-11 | Baikang (Suzhou) Co., Ltd | Bioreversable promoieties for nitrogen-containing and hydroxyl-containing drugs |
| US9750788B2 (en) | 2013-12-13 | 2017-09-05 | Sanofi | Non-acylated exendin-4 peptide analogues |
| US9694053B2 (en) | 2013-12-13 | 2017-07-04 | Sanofi | Dual GLP-1/glucagon receptor agonists |
| US9789165B2 (en) | 2013-12-13 | 2017-10-17 | Sanofi | Exendin-4 peptide analogues as dual GLP-1/GIP receptor agonists |
| US9751926B2 (en) | 2013-12-13 | 2017-09-05 | Sanofi | Dual GLP-1/GIP receptor agonists |
| US9771406B2 (en) | 2014-04-07 | 2017-09-26 | Sanofi | Peptidic dual GLP-1/glucagon receptor agonists derived from exendin-4 |
| US9775904B2 (en) | 2014-04-07 | 2017-10-03 | Sanofi | Exendin-4 derivatives as peptidic dual GLP-1/glucagon receptor agonists |
| US9758561B2 (en) | 2014-04-07 | 2017-09-12 | Sanofi | Dual GLP-1/glucagon receptor agonists derived from exendin-4 |
| US9932381B2 (en) | 2014-06-18 | 2018-04-03 | Sanofi | Exendin-4 derivatives as selective glucagon receptor agonists |
| US11633487B2 (en) | 2014-08-06 | 2023-04-25 | Ascendis Pharma A/S | Prodrugs comprising an aminoalkyl glycine linker |
| US10232020B2 (en) | 2014-09-24 | 2019-03-19 | Indiana University Research And Technology Corporation | Incretin-insulin conjugates |
| US10385107B2 (en) | 2014-09-24 | 2019-08-20 | Indiana Univeresity Researc and Technology Corporation | Lipidated amide-based insulin prodrugs |
| US11116816B2 (en) | 2014-10-22 | 2021-09-14 | Extend Biosciences, Inc. | Therapeutic vitamin d conjugates |
| IL278970B2 (en) * | 2015-01-09 | 2024-03-01 | Ascendis Pharma Growth Disorders As | Cnp prodrugs |
| RU2728656C2 (en) * | 2015-01-09 | 2020-07-31 | Асцендис Фарма Гроус Дизордерс А/С | Cnp prodrugs |
| WO2016110577A1 (en) * | 2015-01-09 | 2016-07-14 | Ascendis Pharma A/S | Cnp prodrugs |
| IL278970B1 (en) * | 2015-01-09 | 2023-11-01 | Ascendis Pharma Growth Disorders As | Cnp prodrugs |
| US11298427B2 (en) | 2015-05-29 | 2022-04-12 | Ascendis Pharma A/S | Prodrugs comprising a pyroglutamate linker |
| WO2016196124A2 (en) | 2015-05-29 | 2016-12-08 | Ascendis Pharma Inc. | Prodrugs comprising a pyroglutamate linker |
| WO2016193371A1 (en) | 2015-06-05 | 2016-12-08 | Sanofi | Prodrugs comprising an glp-1/glucagon dual agonist linker hyaluronic acid conjugate |
| US20180289822A1 (en) * | 2015-06-05 | 2018-10-11 | Sanofi | Prodrugs comprising an glp-1/glucagon dual agonist linker hyaluronic acid conjugate |
| RU2719482C2 (en) * | 2015-06-05 | 2020-04-17 | Санофи | Prodrugs containing hyaluronic acid conjugate, linker and double agonist glp-1/glucagon |
| US10806797B2 (en) | 2015-06-05 | 2020-10-20 | Sanofi | Prodrugs comprising an GLP-1/glucagon dual agonist linker hyaluronic acid conjugate |
| US9982029B2 (en) | 2015-07-10 | 2018-05-29 | Sanofi | Exendin-4 derivatives as selective peptidic dual GLP-1/glucagon receptor agonists |
| WO2017053807A2 (en) | 2015-09-23 | 2017-03-30 | Genentech, Inc. | Optimized variants of anti-vegf antibodies |
| US11179467B2 (en) | 2015-09-25 | 2021-11-23 | Brightgene Bio-Medical Technology Co., Ltd. | Exenatide modifier and use thereof |
| IL259829B2 (en) * | 2016-01-08 | 2023-03-01 | Ascendis Pharma Growth Disorders As | Controlled-release cnp agonists with low npr-c binding |
| US11224661B2 (en) | 2016-01-08 | 2022-01-18 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with increased NEP stability |
| US11413351B2 (en) | 2016-01-08 | 2022-08-16 | Ascendis Pharma Growth Disorders A/S | CNP prodrugs with carrier attachment at the ring moiety |
| US11389511B2 (en) | 2016-01-08 | 2022-07-19 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with reduced side-effects |
| EP4162955A1 (en) * | 2016-01-08 | 2023-04-12 | Ascendis Pharma Growth Disorders A/S | Cnp prodrugs with carrier attachment at the ring moiety |
| AU2022202280B2 (en) * | 2016-01-08 | 2023-11-02 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low initial NPR-B activity |
| US11154593B2 (en) | 2016-01-08 | 2021-10-26 | Ascendis Pharma Growth Disorders A/S | CNP prodrugs with large carrier moieties |
| US10835578B2 (en) | 2016-01-08 | 2020-11-17 | Ascendis Pharma Growth Disorders A/S | CNP prodrugs with large carrier moieties |
| WO2017118704A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Controlled-release cnp agonists with low npr-c binding |
| US11389510B2 (en) | 2016-01-08 | 2022-07-19 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low initial NPR-B activity |
| AU2017205694B2 (en) * | 2016-01-08 | 2022-05-26 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low NPR-C binding |
| US11311604B2 (en) | 2016-01-08 | 2022-04-26 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low NPR-C binding |
| WO2017118693A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Cnp prodrugs with large carrier moieties |
| WO2017118703A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Controlled-release cnp agonists with low initial npr-b activity |
| WO2017118698A1 (en) * | 2016-01-08 | 2017-07-13 | Ascendis Pharma Growth Disorders A/S | Cnp prodrugs with carrier attachment at the ring moiety |
| AU2017205693B2 (en) * | 2016-01-08 | 2022-03-31 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low initial NPR-B activity |
| KR102373744B1 (en) | 2016-01-08 | 2022-03-15 | 아센디스 파마 그로우쓰 디스오더스 에이/에스 | Controlled release CNP agonists with low NPR-C binding |
| IL259829B (en) * | 2016-01-08 | 2022-11-01 | Ascendis Pharma Growth Disorders As | Controlled-release cnp agonists with low npr-c binding |
| KR20180100625A (en) * | 2016-01-08 | 2018-09-11 | 아센디스 파마 그로우쓰 디스오더스 에이/에스 | Controlled-release CNP agonists with low NPR-C binding |
| IL260756B1 (en) * | 2016-03-01 | 2023-05-01 | Ascendis Pharma Bone Diseases As | Pth prodrugs |
| AU2022256158A9 (en) * | 2016-03-01 | 2023-09-28 | Ascendis Pharma Bone Diseases A/S | PTH prodrugs |
| AU2022256158B2 (en) * | 2016-03-01 | 2023-09-28 | Ascendis Pharma Bone Diseases A/S | PTH prodrugs |
| KR20180118197A (en) * | 2016-03-01 | 2018-10-30 | 아센디스 파마 본 디지즈 에이/에스 | PTH prodrug |
| WO2017148883A1 (en) | 2016-03-01 | 2017-09-08 | Ascendis Pharma Bone Diseases A/S | Pth prodrugs |
| AU2017227962B2 (en) * | 2016-03-01 | 2020-11-12 | Ascendis Pharma Bone Diseases A/S | PTH prodrugs |
| IL260756B2 (en) * | 2016-03-01 | 2023-09-01 | Ascendis Pharma Bone Diseases As | Pth prodrugs |
| KR102524255B1 (en) * | 2016-03-01 | 2023-04-20 | 아센디스 파마 본 디지즈 에이/에스 | Pth prodrugs |
| KR102331820B1 (en) | 2016-03-01 | 2021-11-29 | 아센디스 파마 본 디지즈 에이/에스 | PTH prodrug |
| KR20210147085A (en) * | 2016-03-01 | 2021-12-06 | 아센디스 파마 본 디지즈 에이/에스 | Pth prodrugs |
| US11793861B2 (en) | 2016-03-01 | 2023-10-24 | Ascendis Pharma Bone Diseases A/S | PTH prodrugs |
| AU2017227962C1 (en) * | 2016-03-01 | 2021-05-13 | Ascendis Pharma Bone Diseases A/S | PTH prodrugs |
| WO2017186250A1 (en) | 2016-04-29 | 2017-11-02 | Defensin Therapeutics Aps | Treatment of liver, biliary tract and pancreatic disorders |
| AU2017295938B2 (en) * | 2016-07-13 | 2021-05-20 | Ascendis Pharma A/S | Conjugation method for carrier-linked prodrugs |
| US11896671B2 (en) | 2016-07-13 | 2024-02-13 | Ascendis Pharma A/S | Conjugation method for carrier-linked prodrugs |
| WO2018011266A1 (en) | 2016-07-13 | 2018-01-18 | Ascendis Pharma A/S | Conjugation method for carrier-linked prodrugs |
| AU2017295938C1 (en) * | 2016-07-13 | 2021-10-07 | Ascendis Pharma A/S | Conjugation method for carrier-linked prodrugs |
| CN109789221B (en) * | 2016-09-29 | 2022-11-01 | 阿森迪斯药物骨疾病股份有限公司 | Pharmaceutical composition for controlled release of PTH |
| WO2018060312A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Bone Diseases A/S | Pth compounds with low peak-to-trough ratios |
| US11918628B2 (en) | 2016-09-29 | 2024-03-05 | Ascendis Pharma Bone Diseases A/S | Controlled-release PTH compound |
| WO2018060314A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Growth Disorders A/S | Combination therapy with controlled-release cnp agonists |
| IL290054B2 (en) * | 2016-09-29 | 2024-03-01 | Ascendis Pharma Growth Disorders As | Combination therapy with controlled-release cnp agonists |
| WO2018060311A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Bone Diseases A/S | Incremental dose finding in controlled-release pth compounds |
| US11890326B2 (en) | 2016-09-29 | 2024-02-06 | Ascendis Pharma Bone Diseases A/S | Controlled-release PTH compound |
| US11857603B2 (en) | 2016-09-29 | 2024-01-02 | Ascendis Pharma Bone Diseases A/S | PTH compounds with low peak-to-trough ratios |
| EP4275677A2 (en) | 2016-09-29 | 2023-11-15 | Ascendis Pharma Bone Diseases A/S | Dosage regimen for a controlled-release pth compound |
| US11759504B2 (en) | 2016-09-29 | 2023-09-19 | Ascendis Pharma Bone Diseases A/S | PTH compounds with low peak-to-trough ratios |
| AU2017336250C1 (en) * | 2016-09-29 | 2023-08-03 | Ascendis Pharma Bone Diseases A/S | Incremental dose finding in controlled-release PTH compounds |
| WO2018060310A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Bone Diseases A/S | Dosage regimen for a controlled-release pth compound |
| IL290054B1 (en) * | 2016-09-29 | 2023-11-01 | Ascendis Pharma Growth Disorders As | Combination therapy with controlled-release cnp agonists |
| CN109789221A (en) * | 2016-09-29 | 2019-05-21 | 阿森迪斯药物骨疾病股份有限公司 | Ascending-dose discovery in controlled release PTH compound |
| US11564974B2 (en) | 2016-09-29 | 2023-01-31 | Ascendis Pharma Growth Disorders A/S | Combination therapy with controlled-release CNP agonists |
| US11590207B2 (en) | 2016-09-29 | 2023-02-28 | Ascendis Pharma Bone Diseases A/S | Dosage regimen for a controlled-release PTH compound |
| AU2017336250B2 (en) * | 2016-09-29 | 2023-04-20 | Ascendis Pharma Bone Diseases A/S | Incremental dose finding in controlled-release PTH compounds |
| US10792367B2 (en) | 2016-12-02 | 2020-10-06 | Sanofi | Conjugates comprising an GLP-1/glucagon dual agonist, a linker and hyaluronic acid |
| WO2018100174A1 (en) | 2016-12-02 | 2018-06-07 | Sanofi | Conjugates comprising an glp-1/glucagon dual agonist, a linker and hyaluronic acid |
| US11141489B2 (en) | 2016-12-02 | 2021-10-12 | Sanofi | Conjugates comprising an GLP-1/Glucagon dual agonist, a linker and hyaluronic acid |
| WO2018108971A2 (en) | 2016-12-13 | 2018-06-21 | Defensin Therapeutics Aps | Methods for treating inflammatory conditions of the lungs |
| US11786599B2 (en) | 2017-03-10 | 2023-10-17 | Quiapeg Pharmaceuticals Ab | Releasable conjugates |
| WO2018175788A1 (en) | 2017-03-22 | 2018-09-27 | Genentech, Inc. | Hydrogel cross-linked hyaluronic acid prodrug compositions and methods |
| US11642415B2 (en) | 2017-03-22 | 2023-05-09 | Ascendis Pharma A/S | Hydrogel cross-linked hyaluronic acid prodrug compositions and methods |
| US10751417B2 (en) | 2017-04-20 | 2020-08-25 | Novartis Ag | Sustained release delivery systems comprising traceless linkers |
| WO2019076675A1 (en) * | 2017-10-06 | 2019-04-25 | Instituto De Medicina Molecular | Treatment of obesity-related conditions |
| WO2019092201A2 (en) | 2017-11-10 | 2019-05-16 | Defensin Therapeutics Aps | Maturation of mucosal defense and gut/lung function in the preterm infant |
| WO2019101773A2 (en) | 2017-11-24 | 2019-05-31 | Defensin Therapeutics Aps | Prevention and treatment of graft-versus-host-disease with defensins |
| US11517558B2 (en) | 2017-12-05 | 2022-12-06 | Sunovion Pharmaceuticals Inc. | Nonracemic mixtures and uses thereof |
| US11370753B2 (en) | 2017-12-05 | 2022-06-28 | Sunovion Pharmaceuticals Inc. | Crystal forms and production methods thereof |
| US10800738B2 (en) | 2017-12-05 | 2020-10-13 | Sunovion Pharmaceuticals Inc. | Crystal forms and production methods thereof |
| US11767293B2 (en) | 2017-12-05 | 2023-09-26 | Sunovion Pharmaceuticals Inc. | Crystal forms and production methods thereof |
| US10874639B2 (en) | 2017-12-05 | 2020-12-29 | Sunovion Pharmaceuticals Inc. | Nonracemic mixtures and uses thereof |
| WO2019185706A1 (en) | 2018-03-28 | 2019-10-03 | Ascendis Pharma A/S | Conjugates |
| WO2019185705A1 (en) | 2018-03-28 | 2019-10-03 | Ascendis Pharma A/S | Il-2 conjugates |
| WO2019219896A1 (en) | 2018-05-18 | 2019-11-21 | Ascendis Pharma Bone Diseases A/S | Starting dose of pth conjugates |
| RU2806753C2 (en) * | 2018-05-18 | 2023-11-07 | Асцендис Фарма Боун Дизизис А/С | Method of treating or control of hypoparathyroidus using parathrooid hormone (pth) conjugate |
| WO2019229225A1 (en) | 2018-05-30 | 2019-12-05 | Sanofi | Conjugates comprising an glp-1/glucagon/gip triple receptor agonist, a linker and hyaluronic acid |
| US11957735B2 (en) | 2018-09-12 | 2024-04-16 | Quiapeg Pharmaceuticals Ab | Releasable GLP-1 conjugates |
| US11357828B2 (en) | 2018-09-12 | 2022-06-14 | Quiapeg Pharmaceuticals Ab | Releasable GLP-1 conjugates |
| WO2020064846A1 (en) | 2018-09-26 | 2020-04-02 | Ascendis Pharma A/S | Novel hydrogel conjugates |
| WO2020064847A1 (en) | 2018-09-26 | 2020-04-02 | Ascendis Pharma A/S | Degradable hyaluronic acid hydrogels |
| WO2020064844A1 (en) | 2018-09-26 | 2020-04-02 | Ascendis Pharma A/S | Treatment of infections |
| US11389541B2 (en) | 2018-10-03 | 2022-07-19 | Novartis Ag | Sustained delivery of angiopoetin-like 3 polypeptides |
| WO2020141225A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Minimization of systemic inflammation |
| WO2020141221A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Conjugates of pattern recognition receptor agonists |
| WO2020141223A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Induction of sustained local inflammation |
| WO2020141222A1 (en) | 2019-01-04 | 2020-07-09 | Ascendis Pharma A/S | Sustained local drug levels for innate immune agonists |
| WO2020165081A1 (en) | 2019-02-11 | 2020-08-20 | Ascendis Pharma Growth Disorders A/S | Dry pharmaceutical formulations of cnp conjugates |
| WO2020165087A1 (en) | 2019-02-11 | 2020-08-20 | Ascendis Pharma Bone Diseases A/S | Liquid pharmaceutical formulations of pth conjugates |
| US11654113B2 (en) | 2019-06-04 | 2023-05-23 | Sunovion Pharmaceuticals Inc. | Modified release formulations and uses thereof |
| US11160758B2 (en) | 2019-06-04 | 2021-11-02 | Sunovion Pharmaceuticals Inc. | Modified release formulations and uses thereof |
| WO2020254603A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma A/S | CONJUGATES OF π-ELECTRON-PAIR-DONATING HETEROAROMATIC NITROGEN-COMPRISING COMPOUNDS |
| WO2020254611A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Anti-ctla4 conjugates |
| WO2020254617A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Anti-ctla4 compounds with localized pk properties |
| WO2020254609A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Tyrosine kinase inhibitor conjugates |
| WO2020254607A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Anti-ctla4 compounds with localized pd properties |
| WO2020254606A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma A/S | Conjugates of heteroaromatic nitrogen-comprising compounds |
| WO2020254612A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Controlled-release tyrosine kinase inhibitor compounds with localized pd properties |
| WO2020254613A1 (en) | 2019-06-21 | 2020-12-24 | Ascendis Pharma Oncology Division A/S | Controlled-release tyrosine kinase inhibitor compounds with localized pk properties |
| WO2021136808A1 (en) | 2020-01-03 | 2021-07-08 | Ascendis Pharma A/S | Conjugates undergoing intramolecular rearrangements |
| WO2021144249A1 (en) | 2020-01-13 | 2021-07-22 | Ascendis Pharma Bone Diseases A/S | Hypoparathyroidism treatment |
| WO2021224169A1 (en) | 2020-05-04 | 2021-11-11 | Ascendis Pharma A/S | Hydrogel irradiation |
| US11879001B2 (en) | 2020-06-03 | 2024-01-23 | Ascendis Pharma Oncology Division A/S | Conjugate comprising an IL-2 moiety |
| WO2021245130A1 (en) | 2020-06-03 | 2021-12-09 | Ascendis Pharma Oncology Division A/S | Il-2 sequences and uses thereof |
| WO2022029178A1 (en) | 2020-08-05 | 2022-02-10 | Ascendis Pharma A/S | Conjugates comprising reversible linkers and uses thereof |
| WO2022043493A1 (en) | 2020-08-28 | 2022-03-03 | Ascendis Pharma Oncology Division A/S | Glycosylated il-2 proteins and uses thereof |
| WO2022053696A1 (en) * | 2020-09-11 | 2022-03-17 | Compass Pathfinder Limited | Novel safrylamine derivatives having prodrug properties |
| WO2022064035A1 (en) | 2020-09-28 | 2022-03-31 | Ascendis Pharma Bone Diseases A/S | Improvement of physical and mental well-being of patients with hypoparathyroidism |
| WO2022207798A1 (en) | 2021-04-01 | 2022-10-06 | Ascendis Pharma A/S | Use of long-acting growth hormone for treating inflammation-induced diseases |
| WO2023046732A1 (en) | 2021-09-22 | 2023-03-30 | Ascendis Pharma Bone Diseases A/S | Long-acting pth compound treatments |
| US11845736B2 (en) | 2021-10-01 | 2023-12-19 | Empathbio, Inc. | Prodrugs of MDMA, MDA, and derivatives thereof |
| WO2023110758A1 (en) | 2021-12-13 | 2023-06-22 | Ascendis Pharma Growth Disorders A/S | Effective doses of cnp conjugates |
| WO2023110727A2 (en) | 2021-12-13 | 2023-06-22 | Ascendis Pharma Oncology Division A/S | Novel cancer treatments with tlr7/8 agonists |
| US11912680B2 (en) | 2021-12-28 | 2024-02-27 | Empathbio, Inc. | Nitric oxide releasing prodrugs of MDA and MDMA |
| WO2023227505A1 (en) | 2022-05-23 | 2023-11-30 | Ascendis Pharma Growth Disorders A/S | Liquid pharmaceutical formulations of cnp compounds |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220331334A1 (en) | Prodrug Comprising a Drug-Linker Conjugate | |
| CA2811352C (en) | Prodrugs comprising an exendin linker conjugate | |
| EP2459171A1 (en) | Long acting insulin composition | |
| SG178193A1 (en) | Prodrugs comprising an insulin linker conjugate | |
| RU2798085C2 (en) | Prodrug containing a self-cleavable linker | |
| RU2798085C9 (en) | Prodrug containing a self-cleavable linker |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980111088.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09706788 Country of ref document: EP Kind code of ref document: A2 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2009209565 Country of ref document: AU Ref document number: 2712224 Country of ref document: CA Ref document number: 2009706788 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 207150 Country of ref document: IL Ref document number: MX/A/2010/008024 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4702/CHENP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010544717 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2009209565 Country of ref document: AU Date of ref document: 20090130 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010136023 Country of ref document: RU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12865693 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0906643 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100730 |