WO2009080542A1 - Process for the preparation of a macrocycle - Google Patents
Process for the preparation of a macrocycle Download PDFInfo
- Publication number
- WO2009080542A1 WO2009080542A1 PCT/EP2008/067309 EP2008067309W WO2009080542A1 WO 2009080542 A1 WO2009080542 A1 WO 2009080542A1 EP 2008067309 W EP2008067309 W EP 2008067309W WO 2009080542 A1 WO2009080542 A1 WO 2009080542A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- halogen
- formula
- amino
- Prior art date
Links
- 0 **1C=CC(CN(C2)C(O[C@](C3)CN(C[C@](CCCCCC=C[C@](C4)[C@]4C(N(N)S(C4CC4)(=O)=O)=O)(C=O)N*)[C@@]3C(N)=O)=O)=C2C=C1 Chemical compound **1C=CC(CN(C2)C(O[C@](C3)CN(C[C@](CCCCCC=C[C@](C4)[C@]4C(N(N)S(C4CC4)(=O)=O)=O)(C=O)N*)[C@@]3C(N)=O)=O)=C2C=C1 0.000 description 4
- TXQAZWIBPGKHOX-UHFFFAOYSA-N Nc1c[nH]c2ccccc12 Chemical compound Nc1c[nH]c2ccccc12 TXQAZWIBPGKHOX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
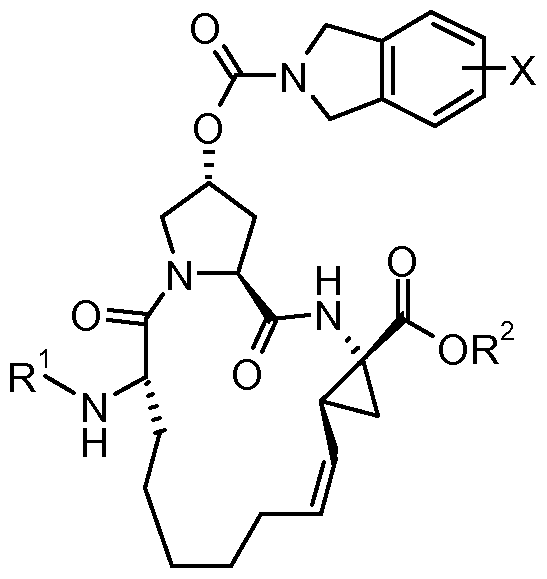
- the present invention relates to a new process for the preparation of macrocyclic HCV protease inhibitor compounds of the formula
- R 1 is an amino protecting group and X is halogen.
- Object of the present invention therefore was to find an improved process which is applicable on technical scale which is able to overcome the disadvantages known in the art.
- R is an amino protecting group and X is a halogen atom, comprises one or more of the steps a) subjecting a diene compound of formula
- R 1 is an amino protecting group
- R 2 is Ci_ 4 -alkyl
- X is halogen to ring closing metathesis reaction in the presence of pentacoordinated ruthenium (II) carbene complex catalyst
- R 1 is an amino protecting group
- R 2 is Ci_ 4 -alkyl and X is halogen
- R 1 is an amino protecting group and X is halogen
- R 1 is an amino protecting group and X is halogen by coupling the macrocyclic acid of formula XX with cyclopropyl sulfonamide and
- amino protecting group refers to any substituents conventionally used to hinder the reactivity of the amino group. Suitable amino protecting groups are described in Green T., "Protective Groups in Organic Synthesis", Chapter 7, John Wiley and Sons, Inc. ,1991, 309-385. Suitable amino protecting groups are Fmoc, Cbz, Moz, Boc, Troc, Teoc or Voc. Preferred amino protecting group, as defined for R 1 is Boc.
- halogen refers to fluorine, chlorine, bromine and iodine.
- the preferred halogen as a rule is chlorine, while the preferred halogen for X is fluorine.
- Ci_ 6 -alkyl refers to a branched or straight-chain monovalent saturated aliphatic hydrocarbon radical of one to six carbon atoms, preferably one to four carbon atoms. This term is further exemplified by radicals as methyl, ethyl, n-propyl, isopropyl, n-butyl, s-butyl, t-butyl and pentyl or hexyl and its isomers.
- Ci_ 4 -alkyl refers to a branched or straight-chain monovalent saturated aliphatic hydrocarbon radical of one to four carbon atoms such as methyl, ethyl, n-propyl, isopropyl, n-butyl, s-butyl, t-butyl, preferably to ethyl.
- C 2 - 6 -alkenyl refers to a branched or straight-chain monovalent unsaturated aliphatic hydrocarbon radical of two to six carbon atoms, preferably two to four carbon atoms. This term is further exemplified by radicals as vinyl, propenyl, butenyl, pentenyl and hexenyl and their isomers. Preferred alkenyl radical is vinyl.
- C 2 - 6 -alkynyl refers to a branched or straight-chain monovalent unsaturated aliphatic hydrocarbon radical of two to six carbon atoms, preferably two to four carbon atoms. This term is further exemplified by radicals as ethynyl, propynyl, butynyl, pentynyl or hexynyl their isomers.
- halogen-Ci_ 6 -alkyl refers to a halogen substituted Ci_ 6 -alkyl radical wherein halogen has the meaning as above.
- Preferred "halogen-Ci_6-alkyl” radicals are the fluorinated Ci_ 6-alkyl radicals such as CF 3 , CH 2 CF 3 , CH (CF 3 ) 2 , CH (CH 3 ) (CF 3 ), C 4 F 9 .
- Ci_6-alkoxy refers to a branched or straight-chain monovalent saturated aliphatic hydrocarbon radical of one to six carbon atoms, preferably 1 to 4 carbon atoms attached to an oxygen atom. Examples of “alkoxy” are methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy and hexyloxy. Preferred are the alkoxy groups specifically exemplified herein.
- the alkyl chain of the alkoxy group can optionally be substituted, particularly mono-, di- or tri-substituted by alkoxy groups as defined above, preferably methoxy, or ethoxy or by aryl groups, preferably phenyl.
- Preferred substituted alkoxy group is the benzyloxy group.
- Ci_ 6 -alkyl carbonyl refers to Ci_ 6 -alkyl substituted carbonyl group, preferably to a Ci_4-alkycarbonyl group. It includes for example acetyl, propanoyl, butanoyl or pivaloyl. Preferred alkyl carbonyl group is acetyl.
- Ci_6-alkylthio refers to the group Ci_6-alkyl-S-, preferably Ci_4-alkyl e.g. methylthio or ethylthio. Preferred are the alkylthio groups specifically exemplified herein.
- arylthio refers to a group aryl-S-, preferably to phenylthio.
- Ci_ 6 -alkylsulfonyl refers to a Ci_ 6 -alkyl substituted sulfonyl group, preferably to methylsulfonyl.
- Ci_ 6 -alkylsulf ⁇ nyl refers to a Ci_ 6 -alkyl substituted sulf ⁇ nyl group, preferably to methylsulfinyl.
- SO 2 - aryl refers to a sulfonyl substituted aryl radical.
- Preferred SO 2 -aryl radical is SO 2 -phenyl.
- SO 2 -NR R' ' refers to a sulfonyl group substituted with an amino group NR R' ' wherein R' and R" independently of each other have the meaning of hydrogen or Ci_ 6 -alkyl or R' and R" together with the N atom form a carbocycle, eg. - (CH 2 ) 4 - or -(CH) 4 -.
- Preferred SO 2 - NR R" radical is SO 2 -N (CH 3 ) 2 .
- the term "mono- or di-Ci_6-alkyl-amino” refers to an amino group, which is mono- or disubstituted with Ci_6-alkyl, preferably Ci_4-alkyl.
- a mono-Ci_6-alkyl-amino group includes for example methylamino or ethylamino.
- the term "di-Ci_6-alkyl-amino” includes for example dimethylamino, diethylamino or ethylmethylamino. Preferred are the mono- or di-Ci_4- alkylamino groups specifically exemplified herein.
- di-Ci_6- alkyl-amino includes ring systems wherein the two alkyl groups together with the nitrogen atom to which they are attached form a 4 to 7 membered heterocycle which also may carry one further hetero atom selected from nitrogen, oxygen or sulfur.
- cycloalkyl denotes a "C 3 _ 7 -cycloalkyl” group containing from 3 to 7 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
- aryl relates to the phenyl or naphthyl group, preferably the phenyl group, which can optionally be mono-, di-, tri- or multiply-substituted by halogen, hydroxy, CN, halogen-Ci_6-alkyl, NO 2 , NH 2 , N(H,alkyl), N(alkyl) 2 , carboxy, aminocarbonyl, alkyl, alkoxy, alkylcarbonyl, Ci_ 6 -alkylsulfonyl, SO 2 -aryl, SO 3 H, SO 3 -alkyl, SO 2 -NR 5 R", aryl and/or aryloxy.
- Preferred aryl group is phenyl.
- aryloxy relates to an aryl radical attached to an oxygen atom.
- aryl has the meaning as defined above.
- Preferred aryloxy group is phenyloxy.
- arylalkyl relates to an aryl radical attached to an alkyl group.
- aryl has the meaning as defined above.
- Preferred arylalkyl group is benzyl.
- heteroaryl relates to a heterocyclic aryl radical containing 1 to 3 heteroatoms in the ring with the remainder being carbon atoms. Suitable heteroatoms include, without limitation, oxygen, sulfur, and nitrogen. Exemplary heteroaryl groups include furanyl, thienyl, pyridyl, pyrrolyl, N-alkyl pyrrolo, pyrimidyl, pyrazinyl, imidazolyl, benzo furanyl, quinolinyl, and indolyl.
- the heteroaryl group can optionally be mono-, di-, tri- or multiply- substituted by halogen, hydroxy, CN, NO 2 , NH 2 , N(H,alkyl), N(alkyl) 2 , carboxy, aminocarbonyl, alkyl, alkoxy, alkylcarbonyl, Ci_ 6 -alkylsulfonyl, SO 2 -aryl, SO 3 H, SO 3 -alkyl, SO 2 -NR 5 R", aryl and/or aryloxy.
- the diene starting compound of formula II can be prepared following the scheme below:
- vinylcyclopropancarboxylate X is treated with sulfuric acid to form XI, then coupled with Boc-(2S,4R)-hydroxyproline to form XII.
- Carbamate formation at the free OH group with 4-fluoroisoindoline leads to XIII and removal of the Boc-protecting group and addition of the (5)-2-tert-Butoxycarbonylamino-non-8-enoic acid side chain can then provide diene lib.
- Step a) requires the transformation of the dien compound of formula II via RCM reaction into the macrocyclic ester of formula I.
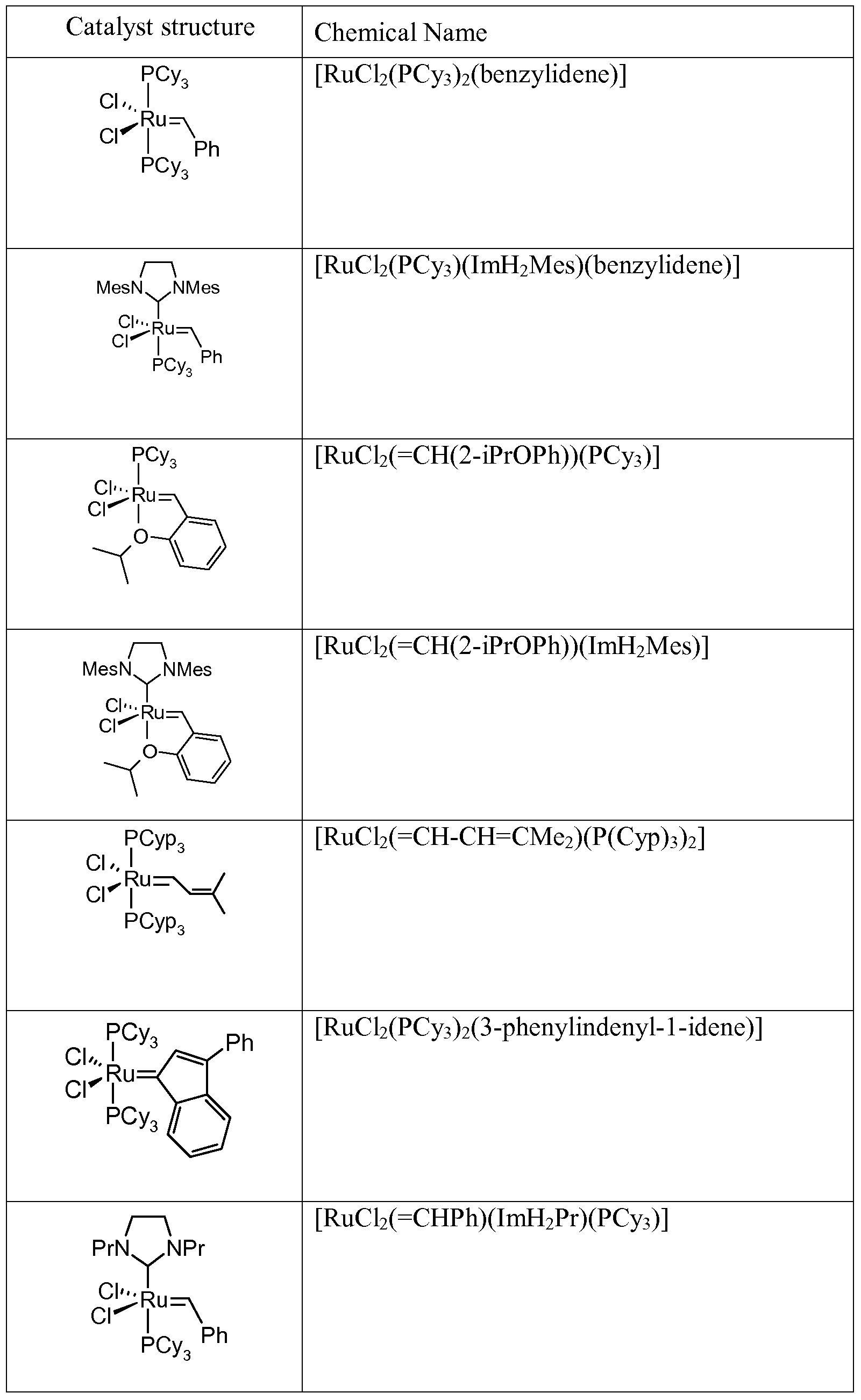
- the RCM reaction is as outlined above performed with a pentacoordinated ruthenium (II) carbene complex catalyst selected from compounds of the formula
- X and X independently of each other are anionic ligands;
- Y 1 and Y 2 independently of each other are hydrogen, Ci_ 6 -alkyl, C 2 - 6 -alkenyl, C 2-6 -alkynyl, Ci_6-alkylthio, aryl, arylthio, Ci_6-alkylsulfonyl, Ci_6-alkylsulf ⁇ nyl,
- G being hydrogen or aryl
- Y 3 is hydrogen, Ci_ 6 -alkyl, C 2 - 6 -alkenyl, C 2 - 6 -alkynyl, Ci_ 6 -alkylthio, aryl, arylthio, Ci_6-alkylsulfonyl, Ci_6-alkylsulf ⁇ nyl;
- R al , R a2 and R a3 independently of each other are Ci_ 6 -alkyl, C 3 _ 7 -cycloalkyl, aryl, heteroaryl or R al and R a2 or R a2 and R a3 or R al and R a3 form together a 1,5-bridged cyclooctyl group ;
- R b is Ci_ 6 -alkyl, C 2 - 6 -alkenyl, halogen- Ci_ 6 -alkyl, C 2 - 6 -alkynyl, aryl, Ci_ 6 -alkoxycarbonyl, Ci_ 6 -alkylcarbonyl, mono-Ci_ 6 -alkyl-or di-Ci_ 6 -alkylamino, Ci_ 6 -alkylaminocarbonyl, Ci_ 6 -alkylthiocarbonyl, Ci_ 6 -alkylsulfonyl, Ci_ 6 -alkylsulfinyl or arylalkyl;
- R 3 , R 4 , R 5 , R 6 , R 7 and R 8 independently of each other have the meaning of hydrogen, C 1-6 - alkyl, halogen-Ci_ 6 -alkyl, C 2 - 6 -alkenyl, C 2-6 -alkynyl, halogen-Ci_ 6 -alkyl, Ci_ 6 -alkoxy, C 2 -6-alkenyloxy, C 2 -6-alkynyloxy, Ci_6-alkylcarbonyl, aryl, hydroxy, aryloxy, nitro, Ci_ 6 -alkoxycarbonyl, amino, mono-Ci_ 6 -alkyl-or di-Ci_ 6 -alkylamino, halogen, thio, Ci_ 6 -alkylthio, arylthio, Ci_ 6 -alkylsulfonyl, Ci_ 6 -alkylsulfinyl, arylsulfonyl
- the ligand L is a neutral ligand preferably selected from — P(R a1 )(R a2 )(R a3 ) .
- R 10 and R 11 independently of each other are Ci_ 6 -alkyl, aryl, C 2-6 - alkenyl or 1-adamantyl and
- R 9a d are independently of each other hydrogen, Ci_ 6 -alkyl, C 2-6 - alkenyl or aryl, or R 9b and R 9c or R 9a and R 9d taken together form a-(CH 2 ) 4 -bridge;
- R al a3 are as outlined above, but preferably cyclohexyl or phenyl.
- R 10 and R 11 are Ci_ 6 -alkyl or a phenyl group which is mono-, di- or tri- substituted with Ci_ 6 -alkyl.
- R 10 and R 11 more preferably have the meaning of t-butyl, 1-adamantyl, isopropyl, 2-methylphenyl, 2, 6-diisopropylphenyl or 2, 4, 6-trimethylphenyl, most preferably 2, 4, 6-trimethylphenyl.
- R 9a and R 9c are methyl or phenyl and R 9b and R 9d are hydrogen, or R 9a and R 9c or R 9b and R 9d are taken together to form a -(CE ⁇ ) n - bridge with n having the meaning of 3 or 4. Its herby understood that if chiral carbon atoms are present, both the racemic and the enantiomerically pure form are comprised.
- R 9a d is hydrogen
- L is Vi la Vi l la
- the anionic ligands X 1 and X 2 are preferably selected from a halogenide or a pseudo halogenide such as cyanide, a rhodanide, a cyanate, an isocyanate, acetate or trifluoro acetate may be selected.
- Preferred anionic ligand for X 1 and X 2 is a halogenide, whereas chloro is the most preferred anionic ligand.
- Y preferably is hydrogen
- Y 1 and Y 2 are the same or different and preferably stand for hydrogen, Ci_ 6 -alkyl, C2-6-alkenyl, Ci_6-alkylthio, phenyl, phenylthio, or
- G being hydrogen or phenyl
- Y 3 preferably is hydrogen.
- R b is as outlined above, but preferably stands for Ci_6-alkyl and halogen-Ci_6-alkyl.
- the preferred meaning for c is hydrogen, halogen, nitro, Ci_ 6 -alkylcarbonyl amino, aryl carbonyl amino, aryl sulfonyl amino, alkyl sulfonyl amino, halogen-Ci_ 6 -alkyl sulfonyl amino, SO 2 -NR R" wherein R' and R" independently of each other have the meaning of hydrogen, Ci_ 6 -alkyl, aryl or R' and R" together with the N atom form a carbocycle.
- More preferred c means hydrogen, Cl, nitro, SO 2 -NR 5 R".
- the following catalysts represent preferred pentacoordinated ruthenium (II) carbene complex catalysts
- the RCM reaction is usually performed in an organic solvent, preferably in an aromatic organic solvent such as in benezene, toluene or mesitylene or in halogenated aromatic solvents such as in polyfluorinated benzenes or toluenes.
- aromatic organic solvent such as in benezene, toluene or mesitylene
- halogenated aromatic solvents such as in polyfluorinated benzenes or toluenes.
- halogenated hydrocarbons such as dichloromethane or dichloro ethane are suitable solvents.
- the solvents may be used as single solvent or as a mixture of different solvents.
- a co-solvent selected from an aliphatic hydrocarbon such as pentane, hexane or heptane may be used as well.
- the reaction temperature is as a rule selected in a range of 20 0 C to 140 0 C, preferably 40 to
- the molar substrate to catalyst ratio S/C is usually selected in a range of 20 to 10000, but preferably in a range of 200 to 4000.
- the macrocyclic ester of formula I can be isolated by applying methods known to the skilled in the art such as by column chromatography or by cristallisation.
- the metathesis reaction mixture can also, after a simple extractive work-up, be brought directly into the next step.
- ethylenediamine In order to remove most catalyst from the solution of the macrocyclic ester I it is convenient to treat the reaction mixture with a complexing agent such as ethylenediamine and to extract the resulting soluble ruthenium species into acidic water.
- a complexing agent such as ethylenediamine
- the amount of ethylenediamine is not critical; it can be used in a 1 :1 to 100: 1 molar ratio relative to the catalyst, preferentially in 20:1 to 70:1 molar ratio.
- Step b requires the hydrolysis of the macrocyclic ester of formula I into the macrocyclic acid of formula XX.
- the hydrolysis can usually be accomplished by treatment with an aqueous alkali hydroxide solution such as with an aqueous sodium hydroxide solution in solvents like methanol or ethanol at a temperature of 0 0 C to 40 0 C.
- an aqueous alkali hydroxide solution such as with an aqueous sodium hydroxide solution in solvents like methanol or ethanol at a temperature of 0 0 C to 40 0 C.
- the macrocyclic acid of formula XX can be isolated by way of extraction with a suitable solvent such as with dichloromethane. Crystallization in a suitable solvent, preferably in tetrahydrofuran leads to a crystalline product with a purity of over 98 %.
- Step c requires the coupling of the macrocyclic acid of formula XX with cyclopropyl sulfonamide to form the macrocyclic sulfonamide of formula XXI.
- a first step the macrocyclic acid of formula XX is reacted with acetic acid anhydride in the presence of an inorganic base, such as with an alkali carbonate like sodium carbonate and a suitable organic solvent such as with tetrahydrofuran into an azlacton intermediate of the formula
- R 1 is an amino protecting group and X is halogen.
- the reaction is expediently performed at a temperature of 10 0 C to 50 0 C.
- the azlacton intermediate will not be isolated but in situ further reacted with cyclopropyl sulfonamide in the presence of an inorganic base, such as with an alkali carbonate like potassium carbonate to the macrocyclic sulfonamide of formula XXI.
- reaction in this second step is expediently performed at a temperature of 50 0 C to 70 0 C.
- reaction mixture Upon completion of the reaction the reaction mixture can be treated with water. After separation and removal of the water phase the organic phase may further be diluted with a suitable organic solvent such as with ethyl acetate or toluene and washed e.g. with an aqueous sulphuric acid and water.
- a suitable organic solvent such as with ethyl acetate or toluene and washed e.g. with an aqueous sulphuric acid and water.
- Isolation of the macrocyclic sulfonamide of formula XXI can then be accomplished by a solvent switch to ethanol followed by addition of the ethanolic solution to water thereby causing precipitation of the desired product.
- the macrocyclic sulfonamide of formula XXI will not be isolated, but the organic phase which has been treated as hereinbefore described will be freed of residual water by way of a continuous azeotropic distillation.
- the mixture can then directly be used for subsequent step d).
- Step d requires the treatment of the macrocyclic sulfonamide of formula XXI with a sodium base to form the end product, i.e. the macrocyclic compound of formula VII.
- the water free mixture obtained from step c) is treated with a sodium base sodium hydroxide, preferably an aqueous solution thereof, sodium methylate or sodium ethoxide, preferably with sodium methylate in the presence of methanol at a temperature of 0 0 C and 50 0 C.
- a sodium base sodium hydroxide preferably an aqueous solution thereof, sodium methylate or sodium ethoxide, preferably with sodium methylate in the presence of methanol at a temperature of 0 0 C and 50 0 C.
- reaction mixture Upon completion of the reaction the reaction mixture can be treated with a mixture of a suitable organic solvent such as ethyl acetate and water where after the crystals of the sodium compound of formula VII, preferably the compound of formula VIII can be collected in good purity and yield.
- a suitable organic solvent such as ethyl acetate and water
- ImMes l,3-bis-(2,4,6-trimethylphenyl)-2-imidazolylidene
- ImH 2 Pr l,3-bis-(2,6-diisopropylphenyl)-2-imidazolidinylidene
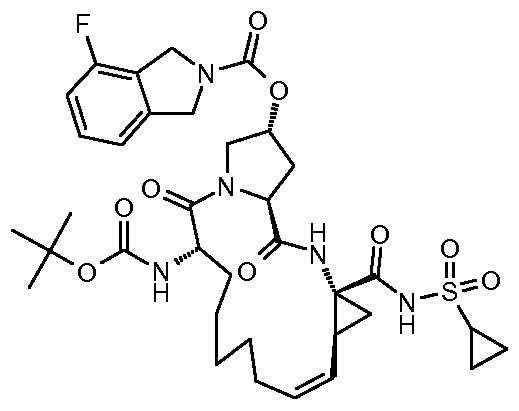
- Diene lib 4-Fluoro- 1 ,3-dihydro-isoindole-2-carboxylic acid (3R,5S)- 1 -((5)-2-tert-butoxy- carbonylamino-non-8-enoyl)-5-((li?,25)-l-ethoxycarbonyl-2-vinyl-cyclopropylcarbamoyl)- pyrrolidin-3-yl ester of the formula
- RCM-Ester Ib (2R,6S,12Z,13aS,14aR,16aS)-Cyclopropa[e]pyrrolo[l,2-a][l,4]diazacyclo- pentadecine-14a(5H)-carboxylic acid, 6-[[(tert-butoxy)carbonyl]amino]-2-[[(4-fluoro-l,3- dihydro-2H-isoindol-2-yl)carbonyl]oxy]- 1 ,2,3,6,7,8,9, 10, 11 , 13a, 14, 15, 16, 16a-hexadecahydro- 5,16-dioxo-, ethyl ester
- the reaction mixture was stirred for 2.5 h at 0 0 C. Salts were filtered off and the filtrate was treated with 20 ml of aqueous HCl (0.5 N). The solvents were removed at 50 0 C under reduced pressure using a rotary evaporator and the residue was extracted twice with 50 ml of ethyl acetate. The extract was washed with 40 ml of water and 40 ml of aqueous sodium carbonate solution (10 % w/w), and dried over sodium sulfate.
- the resulting suspension was heated to 52°C bath temperature. After stirring for 3 h at this temperature the reaction mixture was cooled with an ice bath. 70 ml of aqueous HCl (IM) were added. The mixture was extracted with 50 ml of toluene. The separated aqueous layer was extracted twice with 50 ml toluene. The combined toluene extracts were washed with 30 ml of water and 30 ml of an aqueous solution of sodium carbonate (5 % w/w). The toluene extract was dried with sodium sulfate, filtered, and the solvent was completely removed.
- IM aqueous HCl
- the phases were separated and the aqueous layer was extracted with toluene (3 x 3 ml).
- the combined organic layers were washed with 2 ml of water, 5 ml of aqueous sodium carbonate (5 % w/w) and dried over sodium sulfate.
- the solvent was removed at 50 0 C under reduced pressure using a rotary evaporator.
- %y. % yield determined by HPLC with internal standard; a%: HPLC area%; n.d.: not determined. Reactions 4e-g: after 4 h, additional 0.005 mmol of catalyst were added, total reaction time was 6 h.
- %y. % yield determined by HPLC with internal standard; a%: HPLC area%; n.d.: not determined.
- %y. % yield determined by HPLC with internal standard; a%: HPLC area% Reaction Nr. 7d and 7e have been carried out at S/C of 600. $) Catalyst was added in one portion at beginning of reaction.
- Example 10 was carried out in analogy to example 8, but 2.3 mg of catalyst 5008 were added during 1 h. After 2 h of total reaction time, work-up as in example 8 with final charcoal treatment afforded after evaporation of the solvent RCM-ester Ib as an off-white solid (3.48 g) with 78% purity (83.1% yield)
- the mixture was concentrated to a residual volume of 200 ml and then treated with 200 g of water.
- the biphasic mixture was stirred for 15 minutes and the layers were then allowed to separate.
- the lower aqueous phase was removed.
- the organic phase was diluted with 90 g of ethyl acetate and washed with 3% sulfuric acid (1x140 g) and water (3x130 g).
- the organic layer was concentrated to dryness and then diluted with 400 ml of ethyl acetate. Residual amounts of water were removed by a continuous azeotropic distillation with ethyl acetate.
- the mixture was then treated at 10 0 C with 20 ml of methanol, followed by 10.0 g of sodium methylate (30% in methanol). From the resulting mixture approx. 300 ml of ethyl acetate/methanol were then distilled off. The mixture was then treated at 34°C within one hour with 300 ml of ethyl acetate and 5 g of water. The resulting mixture was allowed to cool to ambient temperature within 4 hours. The crystals were filtered off, washed with 80 ml of ethyl acetate and dried at 80°C/ ⁇ 30 mbar for 20 hours to afford 30.4 g (87% corrected yield) of the title compound as white crystals with an assay of 92.7 %(m/m).
Abstract
Description





































Claims











Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010538595A JP5439384B2 (en) | 2007-12-21 | 2008-12-11 | Manufacturing method for large rings |
| CN2008801220355A CN101903391B (en) | 2007-12-21 | 2008-12-11 | Process for the preparation of a macrocycle |
| AU2008340430A AU2008340430B2 (en) | 2007-12-21 | 2008-12-11 | Process for the preparation of a macrocycle |
| EP08864619.5A EP2225249B1 (en) | 2007-12-21 | 2008-12-11 | Process for the preparation of a macrocycle |
| CA2709032A CA2709032C (en) | 2007-12-21 | 2008-12-11 | Process for the preparation of a macrocycle |
| MX2010006659A MX2010006659A (en) | 2007-12-21 | 2008-12-11 | Process for the preparation of a macrocycle. |
| BRPI0820733-0A BRPI0820733A2 (en) | 2007-12-21 | 2008-12-11 | Process for macrocycle preparation |
| ES08864619.5T ES2599927T3 (en) | 2007-12-21 | 2008-12-11 | Process for the preparation of a macrocycle |
| IL206100A IL206100A (en) | 2007-12-21 | 2010-05-31 | Process for the preparation of sodium ((2r,6s,13as,14ar,16as,z)-6-(amino)-2-(4-haloisoindoline-2-carbonyloxy)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,15,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carbonyl)(cyclpropylsulfonyl)amide derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07150287 | 2007-12-21 | ||
| EP07150287.6 | 2007-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009080542A1 true WO2009080542A1 (en) | 2009-07-02 |
Family
ID=40456983
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/067309 WO2009080542A1 (en) | 2007-12-21 | 2008-12-11 | Process for the preparation of a macrocycle |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US7910728B2 (en) |
| EP (1) | EP2225249B1 (en) |
| JP (1) | JP5439384B2 (en) |
| KR (1) | KR101629523B1 (en) |
| CN (1) | CN101903391B (en) |
| AU (1) | AU2008340430B2 (en) |
| BR (1) | BRPI0820733A2 (en) |
| CA (1) | CA2709032C (en) |
| ES (1) | ES2599927T3 (en) |
| IL (1) | IL206100A (en) |
| MX (1) | MX2010006659A (en) |
| WO (1) | WO2009080542A1 (en) |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010015545A1 (en) * | 2008-08-07 | 2010-02-11 | F. Hoffmann-La Roche Ag | Process for the preparation of a macrocycle |
| WO2011017389A1 (en) | 2009-08-05 | 2011-02-10 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors useful against viral infections, particularly hcv |
| WO2011075615A1 (en) | 2009-12-18 | 2011-06-23 | Idenix Pharmaceuticals, Inc. | 5,5-fused arylene or heteroarylene hepatitis c virus inhibitors |
| US8232246B2 (en) | 2009-06-30 | 2012-07-31 | Abbott Laboratories | Anti-viral compounds |
| EP2483273A1 (en) * | 2009-09-28 | 2012-08-08 | F. Hoffmann-La Roche Ltd | Novel macrocyclic inhibitors of hepatitis c virus replication |
| WO2012109398A1 (en) | 2011-02-10 | 2012-08-16 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors, pharmaceutical compositions thereof, and their use for treating hcv infections |
| WO2012135581A1 (en) | 2011-03-31 | 2012-10-04 | Idenix Pharmaceuticals, Inc. | Methods for treating drug-resistant hepatitis c virus infection with a 5,5-fused arylene or heteroarylene hepatitis c virus inhibitor |
| US8420596B2 (en) | 2008-09-11 | 2013-04-16 | Abbott Laboratories | Macrocyclic hepatitis C serine protease inhibitors |
| US8937041B2 (en) | 2010-12-30 | 2015-01-20 | Abbvie, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US8951964B2 (en) | 2010-12-30 | 2015-02-10 | Abbvie Inc. | Phenanthridine macrocyclic hepatitis C serine protease inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2015042375A1 (en) | 2013-09-20 | 2015-03-26 | Idenix Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| US8993595B2 (en) | 2009-04-08 | 2015-03-31 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| WO2015134561A1 (en) | 2014-03-05 | 2015-09-11 | Idenix Pharmaceuticals, Inc. | Pharmaceutical compositions comprising a 5,5-fused heteroarylene flaviviridae inhibitor and their use for treating or preventing flaviviridae infection |
| WO2015134560A1 (en) | 2014-03-05 | 2015-09-11 | Idenix Pharmaceuticals, Inc. | Solid forms of a flaviviridae virus inhibitor compound and salts thereof |
| CN105001302A (en) * | 2009-09-28 | 2015-10-28 | 英特穆恩公司 | Cyclic peptide inhibitors of hepatitis C virus replication |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9333204B2 (en) | 2014-01-03 | 2016-05-10 | Abbvie Inc. | Solid antiviral dosage forms |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AP2010005416A0 (en) | 2008-04-15 | 2010-10-31 | Intermune Inc | Novel macrocyclic inhibitors of hepatitis c virus replication. |
| AR075584A1 (en) * | 2009-02-27 | 2011-04-20 | Intermune Inc | THERAPEUTIC COMPOSITIONS THAT INCLUDE beta-D-2'-DESOXI-2'-FLUORO-2'-C-METHYLYCTIDINE AND A CARDIEX ISOINDOL ACID DERIVATIVE AND ITS USES. COMPOUND. |
| ES2613608T3 (en) * | 2010-03-10 | 2017-05-24 | Abbvie Ireland Unlimited Company | Solid compositions |
| US8466159B2 (en) | 2011-10-21 | 2013-06-18 | Abbvie Inc. | Methods for treating HCV |
| TW201600087A (en) | 2011-10-21 | 2016-01-01 | 艾伯維有限公司 | Methods for treating HCV |
| DK2583677T1 (en) | 2011-10-21 | 2015-01-19 | Abbvie Inc | Methods for treatment of HCV comprising at least two direct-acting antiviral agents ribavirin, interferon but not |
| US8492386B2 (en) | 2011-10-21 | 2013-07-23 | Abbvie Inc. | Methods for treating HCV |
| JP2015533124A (en) | 2012-10-08 | 2015-11-19 | アッヴィ・インコーポレイテッド | Compounds useful for making HCV protease inhibitors |
| JP7129703B2 (en) | 2016-04-28 | 2022-09-02 | エモリー ユニバーシティー | Alkyne-Containing Nucleotide and Nucleoside Therapeutic Compositions and Uses Associated Therewith |
| CN109280074A (en) * | 2017-07-20 | 2019-01-29 | 歌礼药业(浙江)有限公司 | Dan Nuoruiwei sodium crystal and preparation method thereof |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002083742A2 (en) * | 2001-04-16 | 2002-10-24 | California Institute Of Technology | Group 8 transition metal carbene complexes as enantioselective olefin metathesis catalysts |
| WO2005016944A1 (en) * | 2003-08-02 | 2005-02-24 | Boehringer Ingelheim International Gmbh | Novel metathesis catalysts |
| WO2005037214A2 (en) * | 2003-10-14 | 2005-04-28 | Intermune, Inc. | Macrocyclic carboxylic acids and acylsulfonamides as inhibitors of hcv replication |
| WO2006043145A1 (en) * | 2004-10-21 | 2006-04-27 | Pfizer Inc. | Inhibitors of hepatitis c virus protease, and compositions and treatments using the same |
| WO2007015824A2 (en) * | 2005-07-25 | 2007-02-08 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| EP1825913A1 (en) * | 2006-02-22 | 2007-08-29 | Lanxess Deutschland GmbH & Co.KG | New catalyst systems and their application for metathesis reactions |
| WO2007140954A1 (en) * | 2006-06-07 | 2007-12-13 | Umicore Ag & Co. Kg | Complex of ruthenium and osmium, method of production thereof and use thereof as (pre)catalysts of the metathesis reaction |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5831108A (en) * | 1995-08-03 | 1998-11-03 | California Institute Of Technology | High metathesis activity ruthenium and osmium metal carbene complexes |
| EA200900969A1 (en) | 2007-01-08 | 2010-02-26 | Феномикс Корпорейшн | MACROCYCLIC INHIBITORS OF HEPATITIS C PROTEASE |
-
2008
- 2008-12-11 CN CN2008801220355A patent/CN101903391B/en active Active
- 2008-12-11 MX MX2010006659A patent/MX2010006659A/en active IP Right Grant
- 2008-12-11 WO PCT/EP2008/067309 patent/WO2009080542A1/en active Application Filing
- 2008-12-11 CA CA2709032A patent/CA2709032C/en not_active Expired - Fee Related
- 2008-12-11 JP JP2010538595A patent/JP5439384B2/en not_active Expired - Fee Related
- 2008-12-11 AU AU2008340430A patent/AU2008340430B2/en not_active Ceased
- 2008-12-11 EP EP08864619.5A patent/EP2225249B1/en not_active Not-in-force
- 2008-12-11 BR BRPI0820733-0A patent/BRPI0820733A2/en not_active IP Right Cessation
- 2008-12-11 KR KR1020107013641A patent/KR101629523B1/en active IP Right Grant
- 2008-12-11 ES ES08864619.5T patent/ES2599927T3/en active Active
- 2008-12-19 US US12/317,116 patent/US7910728B2/en not_active Expired - Fee Related
-
2010
- 2010-05-31 IL IL206100A patent/IL206100A/en not_active IP Right Cessation
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002083742A2 (en) * | 2001-04-16 | 2002-10-24 | California Institute Of Technology | Group 8 transition metal carbene complexes as enantioselective olefin metathesis catalysts |
| WO2005016944A1 (en) * | 2003-08-02 | 2005-02-24 | Boehringer Ingelheim International Gmbh | Novel metathesis catalysts |
| WO2005037214A2 (en) * | 2003-10-14 | 2005-04-28 | Intermune, Inc. | Macrocyclic carboxylic acids and acylsulfonamides as inhibitors of hcv replication |
| WO2006043145A1 (en) * | 2004-10-21 | 2006-04-27 | Pfizer Inc. | Inhibitors of hepatitis c virus protease, and compositions and treatments using the same |
| WO2007015824A2 (en) * | 2005-07-25 | 2007-02-08 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| EP1825913A1 (en) * | 2006-02-22 | 2007-08-29 | Lanxess Deutschland GmbH & Co.KG | New catalyst systems and their application for metathesis reactions |
| WO2007140954A1 (en) * | 2006-06-07 | 2007-12-13 | Umicore Ag & Co. Kg | Complex of ruthenium and osmium, method of production thereof and use thereof as (pre)catalysts of the metathesis reaction |
Non-Patent Citations (3)
| Title |
|---|
| FURSTNER, ALOIS: "Olefin metathesis and beyond", ANGEWANDTE CHEMIE, INTERNATIONAL EDITION, vol. 39, no. 17, 2000, pages 3012 - 3043, XP002522005 * |
| TSANTRIZOS, YOULA S. ET AL: "Olefin ring-closing metathesis as a powerful tool in drug discovery and development - potent macrocyclic inhibitors of the hepatitis C virus NS3 protease", JOURNAL OF ORGANOMETALLIC CHEMISTRY, vol. 691, no. 24-25, 2006, pages 5163 - 5171, XP002522003 * |
| YEE, NATHAN K. ET AL: "Efficient Large-Scale Synthesis of BILN 2061, a Potent HCV Protease Inhibitor, by a Convergent Approach Based on Ring-Closing Metathesis", JOURNAL OF ORGANIC CHEMISTRY, vol. 71, no. 19, 2006, pages 7133 - 7145, XP002522004 * |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102112442A (en) * | 2008-08-07 | 2011-06-29 | 弗·哈夫曼-拉罗切有限公司 | Process for preparation of macrocycle |
| WO2010015545A1 (en) * | 2008-08-07 | 2010-02-11 | F. Hoffmann-La Roche Ag | Process for the preparation of a macrocycle |
| US8106187B2 (en) | 2008-08-07 | 2012-01-31 | Roche Palo Alto Llc | Process for the preparation of a macrocycle |
| US9309279B2 (en) | 2008-09-11 | 2016-04-12 | Abbvie Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US8420596B2 (en) | 2008-09-11 | 2013-04-16 | Abbott Laboratories | Macrocyclic hepatitis C serine protease inhibitors |
| US8642538B2 (en) | 2008-09-11 | 2014-02-04 | Abbvie, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US8993595B2 (en) | 2009-04-08 | 2015-03-31 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US8232246B2 (en) | 2009-06-30 | 2012-07-31 | Abbott Laboratories | Anti-viral compounds |
| WO2011017389A1 (en) | 2009-08-05 | 2011-02-10 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors useful against viral infections, particularly hcv |
| US9284307B2 (en) | 2009-08-05 | 2016-03-15 | Idenix Pharmaceuticals Llc | Macrocyclic serine protease inhibitors |
| EP2483273A1 (en) * | 2009-09-28 | 2012-08-08 | F. Hoffmann-La Roche Ltd | Novel macrocyclic inhibitors of hepatitis c virus replication |
| EP2483273A4 (en) * | 2009-09-28 | 2013-05-01 | Hoffmann La Roche | Novel macrocyclic inhibitors of hepatitis c virus replication |
| CN105001302A (en) * | 2009-09-28 | 2015-10-28 | 英特穆恩公司 | Cyclic peptide inhibitors of hepatitis C virus replication |
| WO2011075615A1 (en) | 2009-12-18 | 2011-06-23 | Idenix Pharmaceuticals, Inc. | 5,5-fused arylene or heteroarylene hepatitis c virus inhibitors |
| US8937041B2 (en) | 2010-12-30 | 2015-01-20 | Abbvie, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US8951964B2 (en) | 2010-12-30 | 2015-02-10 | Abbvie Inc. | Phenanthridine macrocyclic hepatitis C serine protease inhibitors |
| US9353100B2 (en) | 2011-02-10 | 2016-05-31 | Idenix Pharmaceuticals Llc | Macrocyclic serine protease inhibitors, pharmaceutical compositions thereof, and their use for treating HCV infections |
| WO2012109398A1 (en) | 2011-02-10 | 2012-08-16 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors, pharmaceutical compositions thereof, and their use for treating hcv infections |
| WO2012135581A1 (en) | 2011-03-31 | 2012-10-04 | Idenix Pharmaceuticals, Inc. | Methods for treating drug-resistant hepatitis c virus infection with a 5,5-fused arylene or heteroarylene hepatitis c virus inhibitor |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9527885B2 (en) | 2011-05-05 | 2016-12-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10201541B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2015042375A1 (en) | 2013-09-20 | 2015-03-26 | Idenix Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| US9333204B2 (en) | 2014-01-03 | 2016-05-10 | Abbvie Inc. | Solid antiviral dosage forms |
| US9744170B2 (en) | 2014-01-03 | 2017-08-29 | Abbvie Inc. | Solid antiviral dosage forms |
| US10105365B2 (en) | 2014-01-03 | 2018-10-23 | Abbvie Inc. | Solid antiviral dosage forms |
| WO2015134561A1 (en) | 2014-03-05 | 2015-09-11 | Idenix Pharmaceuticals, Inc. | Pharmaceutical compositions comprising a 5,5-fused heteroarylene flaviviridae inhibitor and their use for treating or preventing flaviviridae infection |
| WO2015134560A1 (en) | 2014-03-05 | 2015-09-11 | Idenix Pharmaceuticals, Inc. | Solid forms of a flaviviridae virus inhibitor compound and salts thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| IL206100A0 (en) | 2010-11-30 |
| CN101903391B (en) | 2013-04-03 |
| ES2599927T3 (en) | 2017-02-06 |
| IL206100A (en) | 2015-09-24 |
| EP2225249A1 (en) | 2010-09-08 |
| JP5439384B2 (en) | 2014-03-12 |
| KR101629523B1 (en) | 2016-06-10 |
| CA2709032C (en) | 2016-02-23 |
| US7910728B2 (en) | 2011-03-22 |
| CN101903391A (en) | 2010-12-01 |
| JP2011508729A (en) | 2011-03-17 |
| CA2709032A1 (en) | 2009-07-02 |
| AU2008340430B2 (en) | 2013-01-24 |
| AU2008340430A1 (en) | 2009-07-02 |
| BRPI0820733A2 (en) | 2015-06-16 |
| KR20100106411A (en) | 2010-10-01 |
| EP2225249B1 (en) | 2016-08-03 |
| US20090163706A1 (en) | 2009-06-25 |
| MX2010006659A (en) | 2010-07-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2225249A1 (en) | Process for the preparation of a macrocycle | |
| JP5728752B2 (en) | Process for producing macrocyclic compounds | |
| KR101216900B1 (en) | new ruthenium complexes as catalysts for metathesis reactions | |
| EP2150548B1 (en) | Process for hcv protease inhibitor intermediate | |
| WO2007017698A1 (en) | Preparation of diazapentalene derivatives via epoxydation of dihydropyrroles | |
| Gütschow et al. | A new dimerization reaction producing 2‐amino‐9‐oxopyrrolo‐[2, 1‐b] quinazoline‐1‐carbonitriles and analogous pyrrolo‐[1, 2‐a] thieno [3, 2‐d] pyrimidinecar bonitriles | |
| CN113773323B (en) | Preparation method of 3R-amino substituted butyramide derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880122035.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08864619 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| REEP | Request for entry into the european phase |
Ref document number: 2008864619 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008864619 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 206100 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008340430 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2709032 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/006659 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3767/CHENP/2010 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20107013641 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010538595 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2008340430 Country of ref document: AU Date of ref document: 20081211 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0820733 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100618 |