WO2009079765A1 - Compounds with activity at the 5-ht2c receptor - Google Patents
Compounds with activity at the 5-ht2c receptor Download PDFInfo
- Publication number
- WO2009079765A1 WO2009079765A1 PCT/CA2008/002223 CA2008002223W WO2009079765A1 WO 2009079765 A1 WO2009079765 A1 WO 2009079765A1 CA 2008002223 W CA2008002223 W CA 2008002223W WO 2009079765 A1 WO2009079765 A1 WO 2009079765A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tetrahydro
- pyrido
- alkyl
- azepine
- compound
- Prior art date
Links
- 0 C*C(*)(C(*)(*)N*)c1c(C(*)(*)C(O*)=O)c(*)c(*)c(*)n1 Chemical compound C*C(*)(C(*)(*)N*)c1c(C(*)(*)C(O*)=O)c(*)c(*)c(*)n1 0.000 description 17
- YDNABXHXQHNWNL-UHFFFAOYSA-N CCC(CCc(c1n2)ccc2O)CC1S Chemical compound CCC(CCc(c1n2)ccc2O)CC1S YDNABXHXQHNWNL-UHFFFAOYSA-N 0.000 description 1
- QDJZUXHFLWKJOA-UHFFFAOYSA-N CCC(CN(CC1)C(OC(C)(C)C)=O)c2c1ccc(OS(C(F)(F)F)(=O)=O)n2 Chemical compound CCC(CN(CC1)C(OC(C)(C)C)=O)c2c1ccc(OS(C(F)(F)F)(=O)=O)n2 QDJZUXHFLWKJOA-UHFFFAOYSA-N 0.000 description 1
- SYGNXXICOWKEQP-UHFFFAOYSA-N COc1ccc(CC#N)c(CCOS(C)(=O)=O)n1 Chemical compound COc1ccc(CC#N)c(CCOS(C)(=O)=O)n1 SYGNXXICOWKEQP-UHFFFAOYSA-N 0.000 description 1
- OIPHVICVUVKZQC-ATNAJCNCSA-N C[C@@H](CN(CC1)C(OC(C)(C)C)=O)c2c1ccc(N(CC1)CCC1(F)[F]C(C1)[O]1=C)n2 Chemical compound C[C@@H](CN(CC1)C(OC(C)(C)C)=O)c2c1ccc(N(CC1)CCC1(F)[F]C(C1)[O]1=C)n2 OIPHVICVUVKZQC-ATNAJCNCSA-N 0.000 description 1
- GISOLDYLUSAQFV-CQSZACIVSA-N C[C@H](CN(CC1)C(OC(C)(C)C)=O)c2c1ccc(N(CC1)CCC1(F)F)n2 Chemical compound C[C@H](CN(CC1)C(OC(C)(C)C)=O)c2c1ccc(N(CC1)CCC1(F)F)n2 GISOLDYLUSAQFV-CQSZACIVSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- This invention relates to compounds which act at the 5-HT 2 c receptor and to the use of such compounds in the treatment of diseases.
- 5-Hydroxytryptamine 5-HT or serotonin
- PNS and CNS peripheral and central nervous system
- the diverse effects of this neurotransmitter are related to the extensive projections of serotonergic neurons throughout the brain and the large number of distinct serotonin receptor subtypes. At least 14 distinct serotonin receptor subtypes are expressed in the mammalian CNS. The contribution of these receptors to the action of serotonin has been difficult to ascertain owing to the paucity of selective pharmacological agents.
- the 5-HT 2 subfamily of serotonin receptors is composed of three subtypes; namely the 5-HT 2 A, 5-HT 2B and 5-HT 2 c receptors. All the members of this subfamily couple to the activation of the inositol phosphate and diacyl glycerol pathway via the G-protein,G q/ n.
- Other second messenger systems have been shown to be regulated by 5-HT 2 stimulation including mitogen activated protein kinase (MAP-kinase).
- MAP-kinase mitogen activated protein kinase
- the limited access to selective pharmacological tools amongst the 5-HT 2 subfamily of serotonin receptors has led to the use of gene targeting techniques to generate mouse lines that selectively lack functional receptor genes. This strategy has been applied to the study of 5-HT 2C receptor function.
- the 5-HT 2C receptor is expressed in many brain regions including the limbic system, extrapyramidal motor pathways, hypothalamus, thalamus and monoaminergic cell groups.
- 5-HT 2 c receptors have been implicated in the regulation of food intake and anxiety.
- the non-selective 5-HT 2 c receptor agonist, m- chlorophenylpiperazine 1 (mCPP) produces hypophagic and anxiogenic effects that were attenuated by 5-HT 2 c receptor antagonists.
- mCPP m- chlorophenylpiperazine 1
- the propensity of a 5-HT 2C receptor agonist to regulate food intake suggests a critical role for this receptor subtype in controlling obesity (Vickers, S.; Clifton, P.; Dourish, C; Tecott, L. Psychopharmacology (Berlin) 1999, 143:309; Nilsson, B. J. Med. Chem. 2006, 49:4023 ).
- the 5-HT 2 c receptor is the only seven-transmembrane spanning receptor whose messenger RNA (mRNA) undergoes RNA editing (i.e. adenosine to inosine editing) which change the coding for amino acids within the putative second intracellular domain (i2) of the receptor.
- mRNA messenger RNA
- RNA editing i.e. adenosine to inosine editing
- the non-edited receptor contains the amino acids isoleucine, asparagines, and isoleucine (INI) at positions 156, 158 and 160, respectively while the principle fully edited isoforms express valine, serine and valine (VSV) or valine, glycine and valine (VGV).
- Partially edited isoforms also exist where alteration of one or two amino acids within i2 occur giving isoforms such as the VNI isoforms.
- the 5-HT 2 c receptor produces profound changes in receptor function that differ depending upon whether the receptor is unoccupied or occupied by agonist.
- Many studies have shown that in addition to selectivity for receptor subtypes, agonists have selectivity for different signaling pathways coupled to a single receptor subtype; a process known as 'functional selectivity' (Berg KA et al. Drug Discov Today: Therap Strat. 2006, 4:421 ; Urban et al. J Pharmacol Exp Ther. 2007, 320:1 ).
- BMI body mass index
- m 2 body weight index
- Overweight is defined as a BMI in the range 25-30 kg/m 2
- obesity is a BMI greater than 30 kg/m 2 .
- body fat content is also be defined on the basis of body fat content: greater than 25% and 30% in males and females, respectively.
- Schizophrenia affects approximately 5 million people.
- the most prevalent treatments for schizophrenia are currently the 'atypical' antipsychotics, which combine dopamine (D 2 ) and serotonin (5-HT2A) receptor antagonism.
- D 2 dopamine
- 5-HT2A serotonin
- these compounds do not appear to adequately treat all the symptoms of schizophrenia and are accompanied by problematic side effects, such as weight gain (Allison, D. B., et. al., Am. J. Psychiatry 1999, 156:1686-1696; Masand, P. S., Exp. Opin. Pharmacother. 2000, 1:377-389; Whitaker, R., Spectrum Life Sciences. Decision Resources. 2000 2:1-9).
- Atypical antipsychotics also bind with high affinity to 5-HT 2 c receptors and function as 5-HT 2 c receptor antagonists or inverse agonists. Weight gain is a problematic side effect associated with atypical antipsychotics such as clozapine and olanzapine, and it has been suggested that 5-HT 2 c antagonism -A-
- 5-HT 2 c receptor agonism or partial agonism as a treatment for schizophrenia.
- 5-HT 2 c antagonist increase synaptic levels of dopamine and may be effective in animal models of Parkinson's disease (Di Matteo, V., et. al., Neuropharmacology 1998, 37:265-272; Fox, S. H., et. al., Experimental Neurology 1998, 151 :35-49).
- compounds with actions opposite to those of 5-HT 2C antagonists such as 5-HT 2C agonists and partial agonists, should reduce levels of synaptic dopamine.
- 5-HT 2C agonists decrease levels of dopamine in the prefrontal cortex and nucleus accumbens (Millan, M. J., et. al., Neuropharmacology 1998, 37:953-955; Di Matteo, V., et. al., Neuropharmacology 1999, 38:1195-1205; Di Giovanni, G., et. al., Synapse 2000, 35:53-61 ), brain regions that are thought to mediate critical antipsychotic effects of drugs like clozapine.
- 5-HT 2 c agonists do not decrease dopamine levels in the striatum, the brain region most closely associated with extrapyramidal side effects.
- 5-HT 2 c agonists decrease firing in the ventral tegmental area (VTA), but not in the substantia nigra.
- VTA ventral tegmental area
- 5-HT 2C agonists have limbic selectivity, and will be less likely to produce extrapyramidal side effects associated with typical antipsychotics.
- 5-HT 2 c receptors might also be involved in modulation of the rewarding properties of food, which is linked to increased mesolimbic dopamine levels in the nucleus accumbens of the brain in response to food ingestion.
- a number of studies have suggested that food and drug rewards may share some common neural substrates, specifically the nucleus accumbens (Saper, C. B.; Chou, T. C; Elmquist, J. K. Neuron 2002, 36:199- 211 ).
- 5-HT 2 c receptor agonists may decrease dopamine levels in the nucleus accumbens and that reward-related behaviors (e.g., cocaine or nicotine self-administration in rats) may be reduced by 5-HT 2 C receptor activation
- reward-related behaviors e.g., cocaine or nicotine self-administration in rats
- 5-HT 2C receptor agonists may reduce the rewarding properties of food should also be considered (Higgins, G. A.; Fletcher, P. J. Eur. J. Pharmacol. 2003, 480:151-162).
- Epilepsy a brain disorder manifested by recurrent seizures, refers to a complicated constellation of more than 40 distinct disorders.
- the seizure a sudden massive neuronal discharge, can be either partial or complete, depending on the area of brain involved or whether or not consciousness is impaired. Normally there is a balance between excitation and inhibition in the brain. When this balance is disrupted by increased excitation or decreased inhibition, a seizure may result.
- the neuronal discharges may stimulate muscles innervated by the nerves involved, resulting in involuntary muscle contractions, or convulsions (Lee, G. V.; Jones, E. J. Neurobiology of Diseases 2000, 7: 549-551 ).
- a sodium ion channel is a structure in the cell membrane that is selectively permeable to sodium ions and is opened by changes in voltage across the cell membrane.
- Other drugs affect calcium ion channels.
- the third category of drugs affects some aspect of inhibitory synapses that are activated by the neurotransmitter ⁇ -aminobutyric acid (GABA).
- mice lacking the 5-HT 2 c receptors were significantly more seizure susceptible than wild-type controls. Results indicate that mutants have lower focal seizure thresholds, increased focal seizure excitability, and facilitated propagation within the forebrain seizure system. Mutants also exhibit lower generalized seizure threshold for the expression of both generalized clonic and generalized tonic seizures.
- the 5-HT receptor antagonist, mesulergine (2 or 4 mg/kg) administered prior to electroshock testing, recapitulated the mutant phenotype in wild-type mice.
- the selective 5- HT 2C receptor antagonist, SB 242084 do not induce pro-convulsant effects in rats, which are characteristic of mutant mice lacking the 5-HT 2 c receptor. This failure to exhibit pro-convulsant properties in rats in contrast to the reported characteristics of mutant mice lacking 5-HT 2 c receptors might be accounted for by species differences (Di Matteo, V.; Di Giovanni, G.; Esposito, E. CNS Drug Rev. 2000, 6:195-205).
- SSRIs Selective serotonin reuptake inhibitors
- 5-HT serotonin
- OCD obsessive compulsive disorder
- SSRIs have become standard therapy for neuropsychiatric disorders such as obsessive compulsive disorder (OCD), depression, and panic anxiety.
- OCD obsessive compulsive disorder
- 5-HT 2 c receptor-mediated functions There is accumulating evidence for the involvement of 5-HT 2 c receptor-mediated functions in the therapeutic efficacy of SSRIs (Palvimaki, E. -P.; Roth, B. L.; Majasuo, H.; Laakso, A.; Kuoppamaki, M.; Syvalahti, E.; Hietala, J.
- Urinary incontinence (Ul), the involuntary release of urine, may be caused by physiologic, pharmacologic, pathologic, or psychological factors. It is a common condition and often constitutes an embarrassment which can lead to social isolation, depression, loss of quality of life and is a major cause of institutionalization in the elderly population
- Continence and micturition involve a balance between urethral closure and detrusor muscle activity.
- Urethral pressure normally exceeds bladder pressure, resulting in urine remaining in the bladder.
- the proximal urethra and bladder are both within the pelvis.
- Intraabdominal pressure increases (from coughing and sneezing) are transmitted to both urethra and bladder equally, leaving the pressure differential unchanged, resulting in continence.
- Urinary incontinence affects more than 10 million Americans according to the the American foundation of Urologic Diseases. Both men and woman suffer from urinary incontinence although women are disproportionately affected.
- SAI stress urinary incontinenence
- Ul urge incontinence
- mixed incontinence stress urinary incontinence
- SI urinary leakage (loss of small amounts of urine) that occurs during physical activity such as coughing, laughing, sneezing, exercising or other movements that increase intraabdominal pressure and thus increase pressure on the bladder.
- Physical changes resulting from pregnancy, childbirth, and menopause often cause stress incontinence, and in men it is a common problem following a prostatectomy. It is the most common form of incontinence in men.
- Urge incontinence (also known as bladder instability, neurogenic bladder, voiding dysfunction, hyperactive bladder, or detrusor overactivity) refers to the involuntary loss of urine occurring for no apparent reason while suddenly feeling the need or urge to urinate. The most common cause of urge incontinence is involuntary and inappropriate detrusor muscle contractions. Urge incontinence may also be called “reflex incontinence” if it results from overactive nerves controlling the bladder.
- Pain is both a sensory and emotional experience, and is generally associated with tissue damage or inflammation.
- pain is divided into two general categories - acute pain and chronic pain. Both differ in their etiology, pathophysiology, diagnosis, and most importantly treatment.
- Acute pain is short term, and is typically of readily identifiable cause. Patients suffering from acute pain typically respond well to medications. In contrast, chronic pain - medically defined as pain that lasts for 3-6 months or longer, is often not associated with an obvious injury; indeed, patients can suffer from protracted pain that persists for months or years after the initial insult. Whilst acute pain is generally favorably treated with medications, chronic pain is often much more difficult to treat, generally requiring expert care.
- neuropathic pain can, for instance, manifest itself as burning, stabbing, and shock-like sensations.
- neuropathic pain management is at best inconsistent, and often ineffective. This is in part due to the subjective nature of the pain, but also due to poor diagnosis, especially when the chronic pain is not clearly associated with a nerve injury or other insult.
- many, if any, ethical drugs have been prospectively developed for the treatment of chronic pain. Instead, the current medications used to treat chronic pain are "borrowed” from other diseases, most commonly antiepileptic drugs and antidepressants.
- Current first-line treatments for chronic pain include opioids, analgesics such as gabapentin, and tricyclic antidepressants. When opioids are administered over prolonged periods, undesirable side effects such as drug tolerance, chemical dependency and even physiological addiction can occur.
- opioids are administered over prolonged periods, undesirable side effects such as drug tolerance, chemical dependency and even physiological addiction can occur.
- Treatment remedies currently available for chronic pain at best approximately
- serotonin plays a major role in the inhibition of nociceptive transmission. Furthermore, it appears that the 5HT 2 c receptor have an inhibitory role in pain (Eur. J. Pharmacol. 2007, 567(1 -2):89-94; Eur. J. Pharmacol. 2004, 502(3):205-11 ; Pain. 2004108(1 -2): 163-9; Pol. J. Pharmacol. 1994, 46(5):423-8 and U.S. Patent Application Publication 2007/0225277).
- 5-HT 2 c receptors share substantial sequence homology with the 5-HT 2A and 5-HT 2 B receptor.
- the interaction of the compound with the target protein which in this case is the 5- HT 2 C receptor (particularly the more prevalent human edited isoforms VSV, VGV and VNI) and its pharmacokinetic profile, that is its ability to access the target protein in sufficient quantity for a sufficient duration of time to have the desired therapeutic effect.
- the target protein which in this case is the 5- HT 2 C receptor (particularly the more prevalent human edited isoforms VSV, VGV and VNI)
- the pharmacokinetic profile that is its ability to access the target protein in sufficient quantity for a sufficient duration of time to have the desired therapeutic effect.
- the present invention relates to 5-HT 2 c receptor agonists with improved drug- like characteristics such as reduced cytochrome P450 inhibition.
- R 1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing at least two rings, wherein said rings are fused rings, bridged fused rings and/or spiro rings;
- R 2 , R 3 and R 5 to R 12 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH 2 OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-
- R 4 is selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, alkylene- O-alkyl, alkylene-O-cycloalkyl, alkylene-O-alkylene-cycloalkyl;
- the compound of Formula I comprises a compound of Formula IA:
- R 1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing two bridged fused rings;
- R 3 and R 7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 1 is a substituted or unsubstituted N-heterocycloalkyl or a substituted or unsubstituted N-heterocycloalkyl containing two bridged fused rings; and R 3 and R 7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 1 is a substituted N-heterocycloalkyl containing two bridged fused rings substituted with alkyl, alkoxy, hydroxy, cyano, halo, haloalkyl, and/or haloalkyloxy
- R 7 is selected from H, halo, alkyl, haloalkyl, haloalkyloxy, cyano, alkoxy
- R 3 is selected from H, or lower alkyl, and/or a pharmaceutically- acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- the compound of Formula I comprises a compound of Formula IB, IC and/or ID:
- R 3 and R 7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH 2 OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalky
- R 13 and R 14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
- R 15 is selected from the group consisting of H and alkyl; n is an integer; and m is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 3 is selected from H, or lower alkyl
- R 7 is selected from H, halo
- X is O, S, SO, SO 2 , CR 13 R 14 ; NR 15 ; R 13 and R 14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl; R 15 is selected from the group consisting of H and alkyl of C1 to C6; n is either 0, 1 or 2; and m is 0, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 3 and R 7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH 2 OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalky
- R 15 is selected from the group consisting of H and alkyl; and n is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 3 is selected from H, or lower alkyl;
- R 7 is selected from H, halo;
- X is O, S, SO, SO 2 , CR 13 R 14 ;
- R 13 and R 14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
- R 15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either O, 1 or 2; and/or a pharmaceutically- acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- the compound of Formula Il comprises a compound of Formula HA and/or MB :
- R 3 is selected from H, or lower alkyl
- R 7 is selected from H, halo
- X is O, S, SO, SO 2 , CR 13 R 14 ; NR 15 ;
- R 13 and R 14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
- R 15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either O, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- composition comprising at least one of the compounds noted above and at least one pharmaceutically acceptable carrier and/or excipient.
- a method for making the compound of Formula IA to ID and Il comprising: reducing carbonyl groups of a cyclic imide:
- IA-ID, HA-IIB reacting a compound of Formula C under conditions (a), wherein said (a) comprises selective cyano reduction followed by cyclization of the compound of Formula C to provide Formula IA to ID and Il (including MA to MB), whereby R' is alkyl or cycloalkyl.
- IA-ID, MA-IIB reacting a compound of Formula D under conditions (a), wherein said (a) comprises cyclization of the compound of Formula D to provide Formula IA to ID and Il (including HA to HB), whereby R 1 is alkyl or cycloalkyl.
- a method for treating a 5-HT 2 c receptor- mediated disorder in a mammal comprising administering to the mammal a therapeutically effective amount of a compound or composition noted above; the compound or composition exhibiting reduced inhibition of a cytochrome
- the mammal is a human.
- the disorder is selected from urinary incontinence, obesity, schizophrenia, epilepsy, depression, panic anxiety, alcoholism or obsessive compulsive disorder, a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure.
- the disorder is selected from obesity, schizophrenia, epilepsy, a depressive disorder, panic attack, alcoholism, drug addiction or obsessive compulsive disorder.
- the compound or composition is administered orally and/or parenterally. In yet another aspect, the compound or composition is administered intravenously and/or intraperitoneally.
- the mammal is a human.
- the disorder is selected from urinary incontinence obesity, schizophrenia, epilepsy, depression, panic anxiety, alcoholism or obsessive compulsive disorder, a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure.
- the disorder is selected from obesity, schizophrenia, epilepsy, a depressive disorder, panic attack, alcoholism, drug addiction or obsessive compulsive disorder.
- the compound or composition is administrable orally and/or parenterally. In yet another aspect, the compound or composition is administrable intravenously and/or intraperitoneally.
- a method for decreasing food intake in a mammal comprising administering to the mammal a therapeutically effective amount of a compound or composition as noted above; the compound or composition exhibiting reduced inhibition of a cytochrome P450.
- a method of controlling weight gain in a mammal comprising administering to the mammal a therapeutically effective amount of a compound or composition as noted above; the compound or composition exhibiting reduced inhibition of a cytochrome P450.
- Figure 1 shows graphically the effect on feeding in adult male Sprague- Dawley rats of one exemplary compound (22.34) of the invention at various doses (mg/ml) or vehicle administered intraperitoneally (X-axis) on rat food consumption (Y-axis).
- * p ⁇ 0.05 vs. vehicle pretreatment # p ⁇ 0.05 vs. 204 0.6 mg/kg.
- the compounds of the present invention may have asymmetric centers, chiral axes, and chiral planes (e.g., as described in: E. L. Eliel and S. H. Wilen, Stereo-chemistry of Carbon Compounds, John Wiley & Sons, New York, 1994, pages 1119-1190), and occur as racemates, racemic mixtures, and as individual diastereomers, with all possible isomers and mixtures thereof, including optical isomers, being included in the present invention.
- alkyl as used herein means a straight- or branched-chain hydrocarbon radical; in one aspect, having from one to eight carbon atoms, and includes, for example, and without being limited thereto, methyl, ethyl, propyl, isopropyl, t-butyl and the like.
- alkyl encompasses substituted alkyl.
- Substituted alkyl includes, for example, and without being limited thereto, haloalkyl, hydroxyalkyl, cyanoalkyl, and the like. This is applied to any of the groups mentioned herein.
- Groups such as “alkenyl”, “alkynyl", “aryl”, etc. encompass substituted “alkenyl", "alkynyl", “aryl”, etc.
- alkenyl as used herein means a straight- or branched-chain alkenyl radical; in one aspect, having from two to eight carbon atoms, and includes, for example, and without being limited thereto, ethenyl, 1-propenyl, 1-butenyl and the like.
- alkenyl encompass radicals having "cis” and “trans” orientations, or alternatively, 11 E” and "Z” orientations.
- alkynyl as used herein means a straight- or branched-chain alkynyl radical; in one aspect, having from two to eight carbon atoms, and includes, for example, and without being limited thereto, 1-propynyl (propargyl), 1- butynyl and the like.
- cycloalkyl as used herein means a carbocyclic system (which may be unsaturated) containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused.
- the ring(s) may have from three to seven carbon atoms, and includes, for example, and without being limited thereto, cyclopropyl, cyclohexyl, cyclohexenyl and the like.
- heterocycloalkyl as used herein means a heterocyclic system (which may be unsaturated) having at least one heteroatom selected from N, S and/or O and containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused.
- the ring(s) may have a three- to seven-membered cyclic group and includes, for example, and without being limited thereto, piperidinyl, piperazinyl, pyrrolidinyl, tetrahydrofuranyl and the like.
- alkoxy as used herein means a straight- or branched-chain alkoxy radical; in one aspect, having from one to eight carbon atoms and includes, for example, and without being limited thereto, methoxy, ethoxy, propyloxy, isopropyloxy, f-butoxy and the like.
- halo as used herein means halogen and includes, for example, and without being limited thereto, fluoro, chloro, bromo, iodo and the like, in both radioactive and non-radioactive forms.
- alkylene as used herein means a difunctional branched or unbranched saturated hydrocarbon radical; in one aspect, having one to eight carbon atoms, and includes, for example, and without being limited thereto, methylene, ethylene, n-propylene, n-butylene and the like.
- alkenylene as used herein means a difunctional branched or unbranched hydrocarbon radical; in one aspect, having two to eight carbon atoms, and having at least one double bond, and includes, for example, and without being limited thereto, ethenylene, n-propenylene, n-butenylene and the like.
- alkynylene as used herein means a difunctional branched or unbranched hydrocarbon radical; in one aspect, having two to eight carbon atoms, and having at least one triple bond, and includes, for example, and without being limited thereto, ethynylene, n-propynylene, n-butynylene and the like.
- aryl alone or in combination, as used herein means a carbocyclic aromatic system containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused. In particular embodiments, aryl is one, two or three rings. In one aspect, the aryl has five to twelve ring atoms.
- aryl encompasses aromatic radicals such as phenyl, naphthyl, tetrahydronaphthyl, indanyl, biphenyl, phenanthryl, anthryl or acenaphthyl.
- the "aryl” group may have 1 to 4 substituents such as lower alkyl, hydroxyl, halo, haloalkyl, nitro, cyano, alkoxy, lower alkylamino and the like.
- heteroaryl alone or in combination, as used herein means an aromatic system having at least one heteroatom selected from N, S and/or O and containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused.
- heteroaryl is one, two or three rings. In one aspect, the heteroaryl has five to twelve ring atoms.
- heteroaryl encompasses heteroaromatic radicals such as pyridyl, indolyl, furyl, benzofuryl, thienyl, benzothienyl, quinolyl, oxazolyl and the like.
- the "heteroaryl” group may have 1 to 4 substituents such as lower alkyl, hydroxyl, halo, haloalkyl, nitro, cyano, alkoxy, lower alkylamino and the like.
- substituents and substitution patterns on the compounds of the invention may be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those methods set forth below. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, as long as a stable structure results.
- pharmaceutically acceptable salt means either an acid addition salt or a basic addition salt which is compatible with the treatment of patients.
- a "pharmaceutically acceptable acid addition salt” is any non-toxic organic or inorganic acid addition salt of the base compounds represented by Formula I or any of its intermediates.
- Illustrative inorganic acids which form suitable salts include, but are not limited thereto, hydrochloric, hydrobromic, sulfuric and phosphoric acid and acid metal salts such as sodium monohydrogen orthophosphate and potassium hydrogen sulfate.
- Illustrative organic acids which form suitable salts include the mono-, di- and tricarboxylic acids.
- lllustrative of such acids are, for example, acetic, glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, hydroxymaleic, benzoic, hydroxybenzoic, phenylacetic, cinnamic, salicylic, 2- phenoxybenzoic, p-toluenesulfonic acid and other sulfonic acids such as methanesulfonic acid and 2-hydroxyethanesulfonic acid.
- Either the mono- or di-acid salts can be formed, and such salts can exist in either a hydrated, solvated or substantially anhydrous form.
- the acid addition salts of these compounds are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms.
- the selection criteria for the appropriate salt will be known to one skilled in the art.
- Other non-pharmaceutically acceptable salts e.g. oxalates may be used for example in the isolation of compounds of Formula I for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
- a "pharmaceutically acceptable basic addition salt” is any non-toxic organic or inorganic base addition salt of the acid compounds represented by Formula I or any of its intermediates.
- Illustrative inorganic bases which form suitable salts include, but are not limited thereto, lithium, sodium, potassium, calcium, magnesium or barium hydroxides.
- Illustrative organic bases which form suitable salts include aliphatic, alicyclic or aromatic organic amines such as methylamine, trimethyl amine and picoline or ammonia. The selection of the appropriate salt may be important so that an ester functionality, if any, elsewhere in the molecule is not hydrolyzed. The selection criteria for the appropriate salt will be known to one skilled in the art.
- Solvate means a compound of Formula I or the pharmaceutically acceptable salt of a compound of Formula I wherein molecules of a suitable solvent are incorporated in a crystal lattice.
- a suitable solvent is physiologically tolerable at the dosage administered as the solvate.
- suitable solvents but are not limited thereto, are ethanol, water and the like. When water is the solvent, the molecule is referred to as a hydrate.
- stereoisomers is a general term for all isomers of the individual molecules that differ only in the orientation of their atoms in space. It includes mirror image isomers (enantiomers), geometric (cis/trans) isomers and isomers of compounds with more than one chiral centre that are not mirror images of one another (diastereomers).
- treat or “treating” means to alleviate symptoms, eliminate the causation of the symptoms either on a temporary or permanent basis, or to prevent or slow the appearance of symptoms of the named disorder or condition.
- terapéuticaally effective amount means an amount of the compound which is effective in treating the named disorder or condition.
- pharmaceutically acceptable carrier means a non-toxic solvent, dispersant, excipient, adjuvant or other material which is mixed with the active ingredient in order to permit the formation of a pharmaceutical composition, i.e., a dosage form capable of administration to the patient.
- a pharmaceutical composition i.e., a dosage form capable of administration to the patient.
- a pharmaceutically acceptable oil typically used for parenteral administration.
- a “5-HT 2 C receptor-mediated disorder”, as used herein, is a disorder in which there is believed to be involvement of the pathway controlled by the 5-HT 2 c receptor and which is ameliorated by treatment with an agonist of the 5-HT 2C receptor.
- 5-HT 2C receptor-mediated disorders include a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age- related memory disorder, personality disorder and raised intracranial pressure.
- R 1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing at least two rings, wherein said rings are fused rings, bridged fused rings and/or spiro rings;
- R 2 , R 3 and R 5 to R 12 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH 2 OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-
- R 1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing two bridged fused rings; and R 3 and R 7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 1 is a substituted or unsubstituted N-heterocycloalkyl containing two bridged fused rings; and R 3 and R 7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 1 is a substituted N-heterocycloalkyl containing two bridged fused rings substituted with alkyl, alkoxy, hydroxy, cyano, halo, haloalkyl, and/or haloalkyloxy;
- R 7 is selected from H, halo, alkyl, haloalkyl, haloalkyloxy, cyano, alkoxy;
- R 3 is selected from H, or lower alkyl, and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- the compound of Formula I can comprise a compound of Formula IB, IC and/or ID:
- R 3 and R 7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH 2 OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O- heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O- heterocycloalkyl, alkylene-O-alkylene-cycloalkyl, alkylene-O-alkylene- heterocycloalkyl, S-alkyl, S(O)-alkyl, S(O) 2 -alkyl, S-cycloalkyl, S(O)-
- R 3 is selected from H, or lower alkyl;
- R 7 is selected from H, halo;
- X is O, S, SO, SO 2 , CR 13 R 14 ;
- R 13 and R 14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
- R 15 is selected from the group consisting of H and alkyl of C1 to C6; n is either O, 1 or 2; and m is 0, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- the compound of Formula I comprises a compound of Formula II:
- R 3 and R 7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH 2 OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O- heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O- heterocycloalkyl, alkylene-O-alkylene-cycloalkyl, alkylene-O-alkylene- heterocycloalkyl, S-alkyl, S(O)-alkyl, S(O) 2 -alkyl, S-cycloalkyl, S(O)-
- R 3 is selected from H, or lower alkyl;
- R 7 is selected from H, halo;
- X is O 1 S, SO, SO 2 , CR 13 R 14 ;
- R 13 and R 14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
- R 15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either O, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- R 3 and R 7 are as outlined above.
- R 3 is selected from H, or lower alkyl
- R 7 is selected from H, halo
- X is O, S, SO, SO 2 , CR 13 R 14 ;
- NR 15 is each independently selected from the group consisting of H, alkyl, halo and haloalkyl
- R 15 is selected from the group consisting of H and alkyl of C1 to C6
- n is either O, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
- the compounds of the invention may exist in, and be isolated as, enantiomeric or diastereomeric forms, or as a racemic mixture.
- the present invention includes any possible enantiomers, diastereomers, racemates or mixtures thereof, of a compound of Formula I.
- the optically active forms of the compound of the invention may be prepared, for example, by chiral chromatographic separation of a racemate or chemical or enzymatic resolution methodology, by synthesis from optically active starting materials or by asymmetric synthesis based on the procedures described thereafter.
- salts of the compounds of Formula I and II are also salts of the compounds of Formula I and II.
- pharmaceutically acceptable salts of compounds of the present invention are obtained using standard procedures well known in the art, for example, by reacting a sufficiently basic compound, for example an alkyl amine with a suitable acid, for example, HCI or acetic acid, to afford a salt with a physiologically acceptable anion.
- alkali metal such as sodium, potassium, or lithium
- alkaline earth metal such as a calcium
- quaternary ammonium salts can be prepared by the addition of alkylating agents, for example, to neutral amines.
- the compound of Formula I and Il may be converted to a pharmaceutically acceptable salt or solvate thereof, particularly, an acid addition salt such as a hydrochloride, hydrobromide, phosphate, acetate, fumarate, maleate, tartrate, citrate, methanesulphonate or p-toluenesulphonate.
- an acid addition salt such as a hydrochloride, hydrobromide, phosphate, acetate, fumarate, maleate, tartrate, citrate, methanesulphonate or p-toluenesulphonate.
- R' can be alkyl or cycloalkyl and (a) comprises heat and base assisted cyclization of a compound of Formula A to provide a compound of Formula B and (b) comprises reduction of the carbonyl of the amide of the compound of Formula B.
- (a) comprises heating in DMF and (b) comprises reduction with LiAIH 4 ZAICb.
- Compounds of Formula I and IA to ID and Il may also be prepared as follows: or, more specific to Formula IA to ID and Il (including MA to HB),
- R' can be alkyl or cycloalkyl and (a) comprises heat and base assisted cyclization of a compound of Formula AA to provide a compound of Formula BB and (b) comprises reduction of the carbonyl of the amide of the compound of Formula B.
- (a) comprises heating in DMF and (b) comprises reduction with LiAIH 4 ZAICI 3 .
- R 11 and R 12 are H or, more specific to Formula IA to ID and Il (including HA to MB),
- R' can be alkyl or cycloalkyl.
- cyano reduction using LiAIH 4 ZAICI 3 and DIPEA in acetonitrile for cyclization.
- the resultant Formula I and IA to ID and Il can be converted to a salt addition of an acid, for example.
- R' can be alkyl or cycloalkyl.
- cyano reduction using LiAIH 4 ZAICI 3 and DIPEA in acetonitrile for cyclization.
- the resultant Formula I and IA to ID and Il can be converted to a salt addition of an acid, for example.
- (a) comprises cyclization of the compound of Formula D, whereby R' can be alkyl or cycloalkyl.
- R' can be alkyl or cycloalkyl.
- base can be used to initiate cyclization.
- (a) comprises cyclization of the compound of Formula DD, whereby R' can be alkyl or cycloalkyl.
- R' can be alkyl or cycloalkyl.
- base can be used to initiate cyclization.
- the intermediate V was prepared from 2-(2- chloroethyl)-3-(chloromethyl)-6-methoxypyridine [Feng, S.; He, X.; Yu, G.; Yu, X.; Bai, D. Org. Prep. Proced. Int. 2004, 36 (2); 129-134] via mono-cyanation and the Finkelstien chloro-iodo exchange.
- Acid addition salts of the compounds of Formula I and Il are most suitably formed from pharmaceutically acceptable acids, and include for example those formed with inorganic acids e.g. hydrochloric, sulphuric or phosphoric acids and organic acids e.g. succinic, maleic, acetic or fumaric acid.
- Other non- pharmaceutically acceptable salts e.g. oxalates may be used for example in the isolation of compounds of Formula I and Il for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
- base addition salts such as sodium, potassium and ammonium salts
- solvates and hydrates of compounds of the invention are also included within the scope of the invention. The conversion of a given compound salt to a desired compound salt is achieved by applying standard techniques, well known to one skilled in the art.
- the compounds of the invention have an EC 50 value for the human 5-HT 2 c receptor less than 1000 nM, or less than 500 nM, or less than 300 nM, or less than 10O nM.
- the compounds of the invention are therefore of interest for the treatment of 5-HT 2C receptor-mediated disorders, including a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, urinary incontinence (such as stress urinary incontinenence (SUI), urge incontinence (Ul) and mixed incontinence), pain, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure.
- a depressive disorder an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia
- bipolar disorder post-traumatic stress
- an eating disorder obesity
- Compounds of the invention have also been shown to be effective in inhibiting locomotor activity in a rodent model relevant to treatment of schizophrenia or other psychotic disorders. Compounds of the invention have also been shown to exhibit reduced cytochrome P450 inhibition thus minimizing potential drug-drug interactions.
- the compounds of the invention are, for instance, administered orally, sublingually, rectally, nasally, vaginally, topically (including the use of a patch or other transdermal delivery device), by pulmonary route by use of an aerosol, or parenterally, including, for example, intramuscularly, subcutaneously, intraperitoneally, intraarterially, intravenously or intrathecally. Administration can be by means of a pump for periodic or continuous delivery.
- the compounds of the invention are administered alone, or are combined with a pharmaceutically-acceptable carrier or excipient according to standard pharmaceutical practice.
- the compounds of the invention are used in the form of tablets, capsules, lozenges, chewing gum, troches, powders, syrups, elixirs, aqueous solutions and suspensions, and the like.
- carriers that are used include lactose, sodium citrate and salts of phosphoric acid.
- Various disintegrants such as starch, and lubricating agents such as magnesium stearate and talc, are commonly used in tablets.
- useful diluents are lactose and high molecular weight polyethylene glycols. If desired, certain sweetening and/or flavoring agents are added.
- sterile solutions of the compounds of the invention are usually prepared, and the pHs of the solutions are suitably adjusted and buffered.
- the total concentration of solutes should be controlled to render the preparation isotonic.
- ointments or droppable liquids may be delivered by ocular delivery systems known to the art such as applicators or eye droppers.
- Such compositions can include mucomimetics such as hyaluronic acid, chondroitin sulfate, hydroxypropyl methylcellulose or polyvinyl alcohol, preservatives such as sorbic acid, EDTA or benzylchromium chloride, and the usual quantities of diluents and/or carriers.
- diluents and/or carriers will be selected to be appropriate to allow the formation of an aerosol.
- Suppository forms of the compounds of the invention are useful for vaginal, urethral and rectal administrations. Such suppositories will generally be constructed of a mixture of substances that is solid at room temperature but melts at body temperature.
- the substances commonly used to create such vehicles include theobroma oil, glycerinated gelatin, hydrogenated vegetable oils, mixtures of polyethylene glycols of various molecular weight and fatty acid esters of polyethylene glycol.
- Analogous gels or creams can be used for vaginal, urethral and rectal administrations.
- Examples of pharmaceutically acceptable acid addition salts for use in the present invention include those derived from mineral acids, such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acids, and organic acids, such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic, p-toluenesulphonic and arylsulphonic acids, for example.
- Examples of pharmaceutically acceptable base addition salts for use in the present invention include those derived from non-toxic metals such as sodium or potassium, ammonium salts and organoamino salts such as triethylamine salts. Numerous appropriate such salts will be known to those of ordinary skill.
- the physician or other health care professional can select the appropriate dose and treatment regimen based on the subject's weight, age, and physical condition. Dosages will generally be selected to maintain a serum level of compounds of the invention between about 0.01 ⁇ g/cc and about 1000 ⁇ g/cc, preferably between about 0.1 ⁇ g/cc and about 100 ⁇ g/cc.
- an alternative measure of preferred amount is from about 0.001 mg/kg to about 10 mg/kg (alternatively, from about 0.01 mg/kg to about 10 mg/kg), more preferably from about 0.01 mg/kg to about 1 mg/kg (from about 0.1 mg/kg to about 1 mg/kg), will be administered.
- an alternative measure of preferred administration amount is from about 0.001 mg/kg to about 10 mg/kg (from about 0.1 mg/kg to about 10 mg/kg), more preferably from about 0.01 mg/kg to about 1 mg/kg (from about 0.1 mg/kg to about 1 mg/kg).
- an alternative measure of preferred administration amount is from about 0.1 mg/kg to about 10 mg/kg, more preferably from about 0.1 mg/kg to about 1 mg/kg.
- a transformation of a group or substituent into another group or substituent by chemical manipulation can be conducted on any intermediate or final product on the synthetic path toward the final product, in which the possible type of transformation is limited only by inherent incompatibility of other functionalities carried by the molecule at that stage to the conditions or reagents employed in the transformation.
- Such inherent incompatibilities, and ways to circumvent them by carrying out appropriate transformations and synthetic steps in a suitable order will be readily understood to the one skilled in the art of organic synthesis. Examples of transformations are given below, and it is to be understood that the described transformations are not limited only to the generic groups or substituents for which the transformations are exemplified.
- Example 17.1 The title compound from Example 17.1 (5.72 g, 17. 54 mmol) was suspended in dichloromethane (90 ml_) under a nitrogen atmosphere and cooled to 0 0 C. Diisopropylethyamine (7.93 g, 61.39 mmol) was added to the suspension with stirring. In a separate flask, di-tert-butyl dicarbonate (8.04 g, 36.83 mmol) was dissolved in dichloromethane (50 ml_) under a nitrogen atmosphere. This solution was added slowly to the main reaction vessel via cannula. The reaction was stirred at RT for 2 h.
- the salt was mixed with diisopropylethyamine (2.5 mL) and di-tert-butyl dicarbonate (570 mg, 2.5 mmol) in dichloromethane (20 mL) and water (10 mL) at 0 0 C and stirred for two h.
- the organic layer was separated and dried, concentrated to give tert-butyl 9-ethyl-2-hydroxy-5 ) 6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7- carboxylate.
- This intermediate was mixed with diisopropylethyalmine (1 mL) and triflic anhydride (420 ⁇ L, 2.5 mmol) in dichloromethane at -50 0 C and stirred overnight.
- Example 21.45 Oxidation of Example 21.45 to its sulfoxide and sulfones derivatives:
- Example 21.50 tert-butvl 2-(1.i-dioxidothiomorpholin ⁇ -yO- ⁇ . ⁇ . ⁇ . ⁇ - tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate
- Example 21.78 Tert-butvl 3-chloro-2-piperidin-1-yl 5,6,8,9-tetrahydro-7H- pyrido[2,3-d]azepine-7-carboxylate
- Example 21.84 (9R)- and (9S)-tert-butyl 9-methyl-2-piperidin-1-yl-5,6,8,9- tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate were separated from racemic were separated from racemic tert-butyl 2-[ethyl(methyl)amino]-9- methyl-5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate by Chiralcel OJ with 1 % ethanol in hexanes by Chiralcel OJ with 1% ethanol in hexanes.
- Demonstration of the activity of the compounds of the present invention may be accomplished through in vitro, ex vivo and in vivo assays that are well known in the art, including the assays described in the following examples.
- Gq coupled 5-HT 2 receptors stimulates phospholipase C activity and leads to formation of inositol trisphosphate (IP3) and the subsequent release of calcium from intracellular stores.
- Functional activity of Gq coupled receptors can be quantified in a FLIPR assay by measuring intracellular calcium levels with calcium sensitive dyes (using a fluorescence imaging plate reader, FLIPR) and in a Phosphatidyl Inositol Hydrolysis Assay (IP accumulation assay) which measures IPs derived from IP3. Both assays provide robust functional readouts of receptor activation.
- Stable cell lines expressing human 5-HT 2A , 5-HT 2B and 5-HT 2C (both INI and VSV isoforms) receptors were created in an MHEK cell background (an HEK293-based cell background which also expresses the
- Macrophage Scavenger Receptor 1 to increase the adherence of cells to tissue .culture plates).
- Recombinant cell lines were cultured in Growth Medium (High glucose DMEM (Hyclone) with 10% dialyzed fetal bovine serum (Hyclone), and L-glutamine (Gibco; 0.8mM for 5-HT 2 A and 5-HT 2 c. 2.0 mM for 5-HT 2 B), and grown under selection with 200 ⁇ g/ml Zeocin (Invitrogen), and either 200 ⁇ g/ml Hygromycin B (Invitrogen for 5-HT 2A and 5-HT 2 c) or 500 ug/ml Geneticin (Invitrogen) for 5-HT 2B .
- Growth Medium High glucose DMEM (Hyclone) with 10% dialyzed fetal bovine serum (Hyclone), and L-glutamine (Gibco; 0.8mM for 5-HT 2 A and 5-HT 2 c. 2.0 mM for 5-
- FLIPR assay methodology Cells that recombinantly expressed the 5-HT 2 receptors were enzymatically dissociated with Trypsin/EDTA 0.25% (Hyclone) 24 h prior to testing, and seeded at 60,000 cells per well in 100 ⁇ l Growth Medium in black sided, clear bottom 96 well plates (Greiner, BioExpress) at 37 0 C and 5% CO 2 .
- Phosphatidyl Inositol Hydrolysis Assay 24 h prior to testing, cells were plated in poly-D-Lysine-coated 96 well plates (VWR) at 100,000 cells/ well in 200 ⁇ l culture medium containing 10 ⁇ Ci/ml of [ 3 H]-myo-lnositol (Perkin Elmer). Cell monolayers were washed twice with HBSS (HEPES Buffered Saline solution: 20 mM HEPES 1 146 mM NaCI, 4.2 mM KCI, 0.5 mM MgCI 2 , 0.1 % Glucose, pH 7.4).
- HBSS HEPES Buffered Saline solution: 20 mM HEPES 1 146 mM NaCI, 4.2 mM KCI, 0.5 mM MgCI 2 , 0.1 % Glucose, pH 7.4
- the cell monolayers were pre-incubated for 5 min at 37°C in 100 ⁇ l/well HBSS containing 10 mM LiCI. Compounds were tested for agonist activity in duplicate at concentrations ranging from 3nM to 30 ⁇ M. Compounds were added (100 ⁇ l) at 2 times the required final concentration and incubated for 30 min at 37°C. Medium was aspirated and the soluble 3H- inositol phosphates were extracted from the cells by adding 100 ⁇ l/well of ice- cold 5% perchloroacetic acid solution. Plates were placed on ice for 1 hour, and extracts collected in a 2ml, 96 well, polypropylene, round bottom Uniplate (VWR).
- VWR Uniplate
- Total phosphatidyl inositols were eluted with 800 ⁇ l 30 mM ammonium formate, and the eluate was discarded.
- Total inositol phosphates were eluted with 600 ⁇ l (2X 300 ⁇ l) 700 mM ammonium formate / 100 mM formic acid and collected in a clean 2ml, 96 well, polypropylene, round bottom Uniplate.
- 75 ⁇ l eluate was transferred to a Hewlett Packard Optiplate and 150 ⁇ l Scint 40 was added to each well. The plate was sealed with a Topseal (Packard) and shaken for 1 minute on a platform plate shaker. Plates were counted in the Hewlett Packard Topcount to quantify the amount of radioactivity in each well.
- Topseal Packard
- Cytochrome P450 Assay In-vitro screening for drugs that inhibit cytochrome P450 enzymes is well established as a means for predicting potential metabolism related drug interactions in-vivo. Pooled human liver microsomes were acquired from a commercial supplier (Human Biologies International, Scottsdale, AZ, USA) and stored at -70 0 C until use. Exact assay conditions (e.g. substrate for the given CYP isoenzyme and positive control inhibitor) varied according to CYP isoenzyme under study (see summary table below). The metabolism reaction is started by the addition of substrate. Final concentration of substrate was measured by HPLC.
- the extent of metabolism in percentage of a given substrate for the given CYP assay is calculated from the extent of metabolism of the given substrate in the presence test compound compared to incubation of the substrate without test compound.
- Each assay uses as a positive control an inhibitor with known inhibitory activity for the metabolism of the given substrate in the particular CYP assay, which is used to determine the validity of the given assay.
- the data shown for the compounds of the present invention in Table 2 are results from assays in which the positive control gave appropriate results for the given CYP assay.
- Substrate S-Mephenytoin Inhibitor: Omeprazole
- Inhibitor Concentration 10 ⁇ M (omeprazole) or Buffer 890 the indicated for test compound (1 or 10 ⁇ M) ⁇ l
- Animals and housing Male, Sprague-Dawley rats or CD-1 mice were used for all studies. All animals were allowed ad-lib access to food and water except during experiment. Animals were housed within an animal vivarium maintained under a 12 h light:dark cycle (lights on: 07:00 h), and all experiments were conducted in the animals' light phase. For all experiments, animals were habituated to the vivarium for a minimum of 72 h before experimentation. The experimental procedures used in the present investigation were conducted under the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the Canadian Council on Animal Care (CCAC) guidelines.
- AALAC Association for Assessment and Accreditation of Laboratory Animal Care
- CCAC Canadian Council on Animal Care
- Test Compounds All compounds were dissolved in 5% Tween 80® in saline and injected in a dose volume of 5 ml/kg or 10 ml/kg (rat), and 10 ml/kg (mouse). Compounds were administered by either the oral or intraperitoneal route.
- Mouse hypolocomotion assay Selective 5-HT 2 c receptor agonists have been reported to produce hypolocomotion in rodent species by a relatively well defined CNS mechanism. A mouse locomotor assay was therefore used to screen compounds. Male, CD-1 mice were administered test compound 15 min before placement in a chamber where locomotor activity was measured through photocell beam breaks. Test compounds were administered either by the oral or intraperitoneal route.
- test compound or vehicle as control
- Test compound or vehicle was administered 10 to 15 min before the beginning of the 2 h food access period, and food intake over that period was measured as during the training period.
- Test compound or vehicle was administered on Tuesdays and Fridays, with drug free (washout) days in between.
- the animals received 3 doses of test compound and vehicle in a counterbalanced sequence.
- mice Male Sprague-Dawley rats (Charles River, St. Constant, Quebec, Canada) of approximate weight 180-20Og are pair housed on arrival in the animal facility (lights on 7:00-19:00h). After a 7 day acclimitisation period where the animals receive ad-libitum access to standard rodent lab chow (Harlan Teklad rodent maintenance diet, 2014; Harlan Teklad, Madison, Wl), the animals are trained to receive single 45mg food pellets under a fixed time interval of 60s over a 2h period within an operant chamber equipped with a water bottle. Thus during the 2h session, the rats can earn a maximum of 120 pellets. The total volume of water consumed by rats during this 2h period is recorded. Daily food allowance is supplemented by a 45min access period sometime between 15:00-18:00h.
- the rats may be dosed orally or parentally with vehicle or test compound.
- Test compound or vehicle is administered on Tuesdays and Fridays with drug free (washout) days in between.
- the animals will receive 3 doses of test compound .and vehicle in a counterbalanced sequence.
- a modification to the above procedure is to pre-treat rats with either vehicle or a selective 5-HT 2 c receptor antagonist, 6-chloro-5-methyl-N-(2-(2- methylpyridin-3-yl-oxy)pyridine-5-yl)aminocarbonyl)-2,3-dihydroindole (1 mg/kg in 8% HPCD, 25mM citric acid in saline) prior to the oral or parental dose of test compound.
- Antagonism of clonic-tonic seizures produced by chemical convulsants such as pentylenetetrazol have been widely utilized to identify novel anticonvulsants.
- mice Male, CD-1 mice (Charles River, St. Constant, Quebec, Canada) of approximate body weight 20-3Og are housed in groups of four on arrival at the facility. Food (Harlan Teklad rodent maintenance diet, 2014; Harlan Teklad, Madison, Wl) and water are available ad-libitum. After a minimum 3 day acclimatization period the animals would be tested in a s.c pentylenetetrazol assay - which is considered both a model of primary generalized convulsive seizures and non-convulsive absence (petit mal) seizures (Upton, N. (1994) Trends Pharmacol. Sci. 15: 456-463).
- the experiment is conducted within a single day with animals receiving a single pretreatment, i.e independent groups design. Following drug or vehicle control treatment by either oral, or parenteral route, the animals would receive pentylenetetrazol (85mg/kg mice) administered by the subcutaneous route. The dose of pentylenetetrazol is selected as it is of sufficient intensity to induce a clonic seizure in the majority of animals, i.e a CD97 dose. The animals are restrained by hand to deliver the chemical convulsant, following which the animals are released and transferred to a test cage to permit observation of the subsequent seizure throughout its course. The animal would receive a single pentylenetetrazol injection and would be terminated on reaching endpoint, i.e clonic seizure. If an animal displays no seizure activity after 60 min it is considered protected and the experiment completed as endpoint reached.
- pentylenetetrazol 85mg/kg mice
- the dose of pentylenetetrazol is selected as it is of sufficient
- a parallel tests of motor function using the rotorod would be undertaken to establish a therapeutic index (Tl), e.g ratio between the ED 50 dose required to block seizures, compared to ED 50 dose required to disrupt motor function in same species.
- Tl therapeutic index
- the rotorod test consists of placing the animal on a rotating treadmill (a rod) traveling at a constant speed of 16 r.p.m.
- the dependant measure is the time that the animal remains on the rod before falling. Up to three separate measures may be taken to get a meaningful measure of performance.
- a modification to the above procedure is to pretreat mice with either vehicle or a selective 5-HT 2 c receptor antagonist, 6-chloro-5-methyl-N-(2-(2- methylpyridin-3-yl-oxy)pyridine-5-yl)aminocarbonyl)-2,3-dihydroindole (1 mg/kg in 8% HPCD, 25mM citric acid in saline) prior to the oral or parental dose of test compound.
- Antagonism of increased locomotion produced by the psychostimulant amphetamine in rodents is a feature of many drugs with antipsychotic property in man. As such reversal of amphetamine hyperlocomotion is a widely used preclinical test to detect novel drugs for the treatment of schizophrenia.
- mice Male Sprague-Dawley rats (Charles River, St. Constant, Quebec, Canada) of approximate weight 200 g are pair housed on arrival in the animal facility (lights on 7:00-19:00 h). After a 7 day acclimitisation period where the animals receive ad-libitum access to standard rodent lab chow (Harlan Teklad rodent maintenance diet, 2014; Harlan Teklad, Madison, Wl) the animals may undergo behavioural testing.
- standard rodent lab chow Harlan Teklad rodent maintenance diet, 2014; Harlan Teklad, Madison, Wl
- Animals would be singly placed within the test apparatus (Perspex chamber of dimensions: rat 42 cm x 42 cm x 30 cm (L x W x H)) for a limited time period (approximately 30min) to habituate to the novel environment. After such habituation period has passed, animals will be treated with test article or vehicle control via the oral, or parental route, and then returned to the observation test chambers. After a predetermined period, the animals would be dosed with either saline vehicle or d-amphetamine (0.5mg/kg) by the intraperitoneal route and returned to the test chamber for 2h.
- the animal's activity will be monitored automatically by infrared sensors and/or manually by an experimenter for expression of 'normal' behaviors such as sniffing, grooming, rearing, and 'abnormal' behaviors such as 'circling'.
- 'normal' behaviors such as sniffing, grooming, rearing, and 'abnormal' behaviors such as 'circling'.
- the animals will be returned to their holding cages.
- a modification to the above procedure is to pretreat rats with either vehicle or a selective 5-HT 2 c receptor antagonist, 6-chloro-5-methyl-N-(2-(2- methylpyridin-3-yl-oxy)pyridine-5-yl)aminocarbonyl)-2,3-dihydroindole (1 mg/kg in 8% HPCD, 25mM citric acid in saline) prior to the oral or parental dose of test compound.
- a cystometry tube is implanted into the bladder of adult guinea-pigs (600-80Og).
- the cystometry tube is connected to an infusion pump and pressure transducer to enable fluid perfusion and the recording of intravesicular bladder pressure.
- Electromyographic (EMG) leads are inserted into the external urethral sphincter (EUS) to allow the measurement of sphincter tone.
- the bladder was filled at a rate of 150 ⁇ l/min with physiological saline (room temperature) until initiation of a micturition reflex. Following this reflex, the bladder was drained and the filling procedure repeated to establish a mean bladder threshold capacity for initiation of micturition reflex. Further, EUS EMG activity and intravesicular pressure was recorded throughout bladder filling.
- test drug or vehicle is administered i.v and bladder filling (150 ⁇ l/min) reinitiated until induction of micturition reflex. Bladder pressure and maximum EUS EMG activity is recorded.
- the formalin test is a chemically-induced tonic pain model in which injection of formalin into a hind paw elicits a biphasic nociceptive behavior.
- the second phase of formalin response is predominantly due to a central sensitization phenomenon.
- Most clinically used drugs against neuropathic pain are active on this second phase of formalin response.
- Formalin test is accepted as a valid model of persistent clinical pain.
- the test was done by pretreating the rats with the test compound and 30 min later (pretreatment time), 50 ⁇ l of 2.5% formalin was injected into the right hind paw of the animal. The number of paw licking and flinching episodes were scored for 60 min post-formalin injection.
- the compounds were administered either intraperitoneal ⁇ or orally. The compounds significantly inhibited the second phase of the formalin response.
- the compounds were tested for their efficacy in reducing mechanical allodynia and cold allodynia in the spared nerve injury (SNI) model of chronic neuropathic pain.
- SNI spared nerve injury
- the left sciatic nerve of the rat is exposed under anesthesia.
- Two of the branches of the sciatic nerve viz. the common peroneal and tibial nerve are ligated and sectioned.
- the third branch (sural nerve) is left intact.
- the animals were allowed a post-operative recovery period of 7 days before they were subjected to any test.
- the presence of mechanical allodynia was assessed using the Dynamic Plantar Aesthesiometer (Ugo Basile, Italy) which is a modified version of the Von Frey Hair test.
- a test filament is positioned below the animal's hind paw and the unit is activated which causes the filament to move up and touch the plantar surface of the hind paw. Increasing force is applied to the paw via the filament.
- the unit is inactivated automatically and the threshold force required to elicit the paw withdrawal is displayed.
- the cut-off force is set at 50 g.
- the tests were done on both the non-injured (control) and the injured (SNI) paw. Pilot studies showed the presence of mechanical allodynia 7 days after the surgery and lasted up to 4 weeks (end of the test period).
- Cold allodynia was assessed by using the acetone test. In this model, 25 ⁇ l of acetone is sprayed on to the plantar surface of the hind paw. Evaporation of acetone causes cooling of the skin. The cold stimulus sets up nociceptive responses from the injured paw as evidenced by paw lifting, paw licking and grooming. The duration of the nociceptive responses is noted. Similar stimulus to the uninjured (control) paw usually does not elicit nociceptive responses.
- test compound or the vehicle was administered (10 ml/kg) either intraperitoneally or orally. The readings were taken 30 min after the administration. Cold allodynia in neuropathic rats was measured as evidenced by the reduced nociceptive duration in these animals.
- FCA FCA-induced edema
- 100 ⁇ l of FCA is injected subcutaneously into the dorsal aspect of the left hind paw of the rats.
- the edema appears within 2 h reaches a peak by 6 h and lasts for about 7 days.
- the effects of compounds on mechanical allodynia and paw volume were studied 2 days after FCA administration.
- the effect of the compounds in alleviating mechanical allodynia was assessed in the FCA-treated rats using the modified Randall-Selitto method (Analgesy- Meter, Ugo Basile, Italy).
- the animal's paw is placed on a small plinth under a cone-shaped pusher. Increasing force is applied to the paw by depressing a pedal until vocalization or withdrawal of the paw occurs. The minimum force required to elicit this vocalization/withdrawal is the paw pressure threshold.
- the cut-off was set at 150 g.
- test compound/vehicle was administered orally in a volume of 10 ml/kg.
- the paw pressure thresholds were measured 30, 60 and 180 min post administration.
- the inhibition of mechanical allodynia was measured at 30 min by all compounds tested.
- Table 1 shows examples of selective 5-HT 2C receptor agonists in accordance with the invention, determined by the IP3 assay described above.
- Table 2 shows examples of compounds with reduced cytochrome P450 inhibition in accordance with the invention, determined by the cytochrome P450 assay described above. The data show the percent inhibition of the metabolism of the substrate in the assay resulting from incubation in the presence of the indicated compound. Compounds having less than 50% inhibition at 1 uM are considered to be a compound with reduced inhibition of cytochrome P450 for the present invention.
- Figure 1 shows the dose-related reduction in food intake in rats treated intraperitoneally with Example 22.34 of the invention.
Abstract
A compound of Formula (I): wherein R1 is a substituted or unsubstituted heterocycloalkyl containing at least two rings, wherein said rings are fused rings, bridged fused rings and/or spiro rings. A method for treating a 5-HT2C receptor-mediated disorder in a mammal using the same.
Description
Compounds with Activity at the 5-HTgc Receptor
Field of the Invention
This invention relates to compounds which act at the 5-HT2c receptor and to the use of such compounds in the treatment of diseases.
Background of the Invention
5-Hydroxytryptamine (5-HT or serotonin), a key neurotransmitter of the peripheral and central nervous system (PNS and CNS), has been implicated in a variety of sensory, motor and behavioral processes. The diverse effects of this neurotransmitter are related to the extensive projections of serotonergic neurons throughout the brain and the large number of distinct serotonin receptor subtypes. At least 14 distinct serotonin receptor subtypes are expressed in the mammalian CNS. The contribution of these receptors to the action of serotonin has been difficult to ascertain owing to the paucity of selective pharmacological agents.
The 5-HT2 subfamily of serotonin receptors is composed of three subtypes; namely the 5-HT2A, 5-HT2B and 5-HT2c receptors. All the members of this subfamily couple to the activation of the inositol phosphate and diacyl glycerol pathway via the G-protein,Gq/n. Recently, other second messenger systems have been shown to be regulated by 5-HT2 stimulation including mitogen activated protein kinase (MAP-kinase). The limited access to selective pharmacological tools amongst the 5-HT2 subfamily of serotonin receptors has led to the use of gene targeting techniques to generate mouse lines that selectively lack functional receptor genes. This strategy has been applied to the study of 5-HT2C receptor function. The 5-HT2C receptor is expressed in many brain regions including the limbic system, extrapyramidal motor pathways, hypothalamus, thalamus and monoaminergic cell groups. 5-HT2c receptors have been implicated in the regulation of food intake and anxiety. For example, the non-selective 5-HT2c receptor agonist, m- chlorophenylpiperazine 1 (mCPP) produces hypophagic and anxiogenic effects that were attenuated by 5-HT2c receptor antagonists. The propensity of a 5-HT2C receptor agonist to regulate food intake suggests a critical role for
this receptor subtype in controlling obesity (Vickers, S.; Clifton, P.; Dourish, C; Tecott, L. Psychopharmacology (Berlin) 1999, 143:309; Nilsson, B. J. Med. Chem. 2006, 49:4023 ).
Notably, the 5-HT2c receptor is is the only seven-transmembrane spanning receptor whose messenger RNA (mRNA) undergoes RNA editing (i.e. adenosine to inosine editing) which change the coding for amino acids within the putative second intracellular domain (i2) of the receptor. In human brain, the non-edited receptor contains the amino acids isoleucine, asparagines, and isoleucine (INI) at positions 156, 158 and 160, respectively while the principle fully edited isoforms express valine, serine and valine (VSV) or valine, glycine and valine (VGV). Partially edited isoforms also exist where alteration of one or two amino acids within i2 occur giving isoforms such as the VNI isoforms. As a result of mRNA editing, the 5-HT2c receptor produces profound changes in receptor function that differ depending upon whether the receptor is unoccupied or occupied by agonist. Many studies have shown that in addition to selectivity for receptor subtypes, agonists have selectivity for different signaling pathways coupled to a single receptor subtype; a process known as 'functional selectivity' (Berg KA et al. Drug Discov Today: Therap Strat. 2006, 4:421 ; Urban et al. J Pharmacol Exp Ther. 2007, 320:1 ). Consequently, changes in receptor function as a result of mRNA editing would be expected to alter serotonergic neurotransmission as well as responses to drugs that act via the 5-HT2c receptor system (Berg et al. J Pharmacol Exp Ther. 2007, in print).
It has been widely recognized that obesity is a disease process influenced by environmental factors in which the traditional weight loss methods of dieting and exercise need to be supplemented by therapeutic products (S. Parker, Obesity: Trends and Treatments", Scrip Reports, PJB Publications Ltd, 1969).
Whether someone is classified as overweight or obese is generally determined on the basis of their body mass index (BMI), which is calculated
by dividing body weight (kg) by height squared (m2). Thus, the units of BMI are kg/m2 and it is possible to calculate the BMI range associated with minimum mortality in each decade of life. Overweight is defined as a BMI in the range 25-30 kg/m2, and obesity as a BMI greater than 30 kg/m2. There are problems with this definition in that it does not take into account the proportion of body mass that is muscle in relation to fat (adipose tissue). To account for this, obesity can also be defined on the basis of body fat content: greater than 25% and 30% in males and females, respectively. As the BMI increases there is an increased risk of death from a variety of causes that in independent of other risk factors. The most common diseases with obesity are cardiovascular disease (particularly hypertension), diabetes (obesity aggravates the development of diabetes), gall bladder disease (particularly cancer) and diseases of reproduction. Research has shown that even a modest reduction in body weight can correspond to a significant reduction in the risk of developing coronary heart disease.
In addition to its growing role in the regulation of food intake and hence obesity, the 5-HT2c receptor has been implicated in the treatment of Schizophrenia. Schizophrenia affects approximately 5 million people. The most prevalent treatments for schizophrenia are currently the 'atypical' antipsychotics, which combine dopamine (D2) and serotonin (5-HT2A) receptor antagonism. Despite the reported improvements in efficacy and side-effect liability of atypical antipsychotics relative to typical antipsychotics, these compounds do not appear to adequately treat all the symptoms of schizophrenia and are accompanied by problematic side effects, such as weight gain (Allison, D. B., et. al., Am. J. Psychiatry 1999, 156:1686-1696; Masand, P. S., Exp. Opin. Pharmacother. 2000, 1:377-389; Whitaker, R., Spectrum Life Sciences. Decision Resources. 2000 2:1-9).
Atypical antipsychotics also bind with high affinity to 5-HT2c receptors and function as 5-HT2c receptor antagonists or inverse agonists. Weight gain is a problematic side effect associated with atypical antipsychotics such as clozapine and olanzapine, and it has been suggested that 5-HT2c antagonism
-A-
is responsible for the increased weight gain. Conversely, stimulation of the 5- HT2C receptor is known to result in decreased food intake and body weight (Walsh et. al., Psychopharmacology 1996, 124:57-73; Cowen, P. J., et. al., Human Psychopharmacology 1995, 10: 385-391 ; Rosenzweig-Lipson, S., et. al., ASPET abstract, 2000).
Several lines of evidence support a role for 5-HT2c receptor agonism or partial agonism as a treatment for schizophrenia. Studies suggest that 5-HT2c antagonist increase synaptic levels of dopamine and may be effective in animal models of Parkinson's disease (Di Matteo, V., et. al., Neuropharmacology 1998, 37:265-272; Fox, S. H., et. al., Experimental Neurology 1998, 151 :35-49). Since the positive symptoms of schizophrenia are associated with increased levels of dopamine, compounds with actions opposite to those of 5-HT2C antagonists, such as 5-HT2C agonists and partial agonists, should reduce levels of synaptic dopamine. Recent studies have demonstrated that 5-HT2C agonists decrease levels of dopamine in the prefrontal cortex and nucleus accumbens (Millan, M. J., et. al., Neuropharmacology 1998, 37:953-955; Di Matteo, V., et. al., Neuropharmacology 1999, 38:1195-1205; Di Giovanni, G., et. al., Synapse 2000, 35:53-61 ), brain regions that are thought to mediate critical antipsychotic effects of drugs like clozapine. However, 5-HT2c agonists do not decrease dopamine levels in the striatum, the brain region most closely associated with extrapyramidal side effects. In addition, a recent study demonstrates that 5-HT2c agonists decrease firing in the ventral tegmental area (VTA), but not in the substantia nigra. The differential effects of 5-HT2C agonists in the mesolimbic pathway relative to the nigrostriatal pathway suggest that 5-HT2c agonists have limbic selectivity, and will be less likely to produce extrapyramidal side effects associated with typical antipsychotics.
Additionally, 5-HT2c receptors might also be involved in modulation of the rewarding properties of food, which is linked to increased mesolimbic dopamine levels in the nucleus accumbens of the brain in response to food ingestion. A number of studies have suggested that food and drug rewards
may share some common neural substrates, specifically the nucleus accumbens (Saper, C. B.; Chou, T. C; Elmquist, J. K. Neuron 2002, 36:199- 211 ). Given that 5-HT2c receptor agonists may decrease dopamine levels in the nucleus accumbens and that reward-related behaviors (e.g., cocaine or nicotine self-administration in rats) may be reduced by 5-HT2C receptor activation, the possibility that 5-HT2C receptor agonists may reduce the rewarding properties of food should also be considered (Higgins, G. A.; Fletcher, P. J. Eur. J. Pharmacol. 2003, 480:151-162).
Another therapeutic area where the use of a 5-HT2C receptor agonist is considered of value is in the treatment of epilepsy. Epilepsy, a brain disorder manifested by recurrent seizures, refers to a complicated constellation of more than 40 distinct disorders. The seizure, a sudden massive neuronal discharge, can be either partial or complete, depending on the area of brain involved or whether or not consciousness is impaired. Normally there is a balance between excitation and inhibition in the brain. When this balance is disrupted by increased excitation or decreased inhibition, a seizure may result. The neuronal discharges may stimulate muscles innervated by the nerves involved, resulting in involuntary muscle contractions, or convulsions (Lee, G. V.; Jones, E. J. Neurobiology of Diseases 2000, 7: 549-551 ).
There are currently several drugs in clinical use to inhibit seizures, which fall into three different categories in terms of their target (Cosford, N. D. P.; McDonald, I. A.; Schweiger, E. J. Annu. Rep. Med. Chem. 1998, 33:61-70). Most common are the drugs that affect the flow of sodium into the cell via voltage-gated sodium ion channels. A sodium ion channel is a structure in the cell membrane that is selectively permeable to sodium ions and is opened by changes in voltage across the cell membrane. Other drugs affect calcium ion channels. The third category of drugs affects some aspect of inhibitory synapses that are activated by the neurotransmitter γ-aminobutyric acid (GABA). Despite the availability of these drugs, a large proportion of patients continue to have seizures. Furthermore, among those in whom seizures are effectively inhibited, substantial numbers experienced persistent and
undesirable effects from these drugs. In light of this, the current goal of researchers is to identify new classes of anti-seizure drugs that act on novel molecular targets and by novel mechanisms that may permit effective treatment of large numbers of individuals unsatisfactorily treated at present. The recently cloned 5-HT2c receptor has revealed a novel molecular target that provides just this opportunity for the development of novel antiepileptic drugs.
There is growing evidence that serotonergic neurotransmission modulates a wide variety of experimentally-induced seizures and involved in the enhanced seizure susceptibility observed in some genetically prone rodents Przegalinski, E.; Baran, L.; Siwanowicz, J. Epilepsia, 1994, 35:889-894; Wada, Y.; Shiraishi, J.; Nakamura, M.; Koshino, Y. Brain Res., 1997, 747:338- 342). Studies have shown that mice bearing a targeted disruption of the 5- HT2C receptor genes exhibit an epilepsy syndrome associated with sporadic spontaneous seizures that occasionally result in death. In all epileptic paradigms, mice lacking the 5-HT2c receptors were significantly more seizure susceptible than wild-type controls. Results indicate that mutants have lower focal seizure thresholds, increased focal seizure excitability, and facilitated propagation within the forebrain seizure system. Mutants also exhibit lower generalized seizure threshold for the expression of both generalized clonic and generalized tonic seizures. Importantly, the 5-HT receptor antagonist, mesulergine (2 or 4 mg/kg), administered prior to electroshock testing, recapitulated the mutant phenotype in wild-type mice. Together, these data strongly implicate a role for serotonin and the 5-HT2c receptors in the modulation of neuronal network excitability and seizure propagation throughout the CNS (Appelgate, C. D.; Tecott, L. H. Exp. Neurol. 1998, 154: 522-530; Heisler, L. K.; Chu, H. M.; Tecott, L. H. Ann. N. Y. Acad. Sci. 1998, 861 : 74-78; Rueter, L. E.; Tecott, L. H.; Blier, P. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000, 361 : 484-491 ). Furthermore, agents that elevate extracellular serotonin (5-HT) levels, such as 5-hydroxytryptophan and 5-HT reuptake blockers, inhibit both limbic and generalized seizures. Conversely,
depletion of brain 5-HT lowers the threshold to audiogenically, chemically and electrically evoked convulsion (Loscher, W. Methods Find Exp. Clin. Pharmacol., 1984, 6:531-547; Prendiville, S.; Gale, K. Epilepsia, 1993, 34:381-384; Yan, Q. S.; Jobe, P. C; Cheong, J. H.; Ko, K. H.; Daily, J. W. Naunyn-Schmiedeberg's Arch. Pharmacol. , 1994, 350: 149-152).
Reduction in seizure activity has been observed for the 5-HT2c receptor agonists mCPP and TFMPP when microinjected bilaterally into the rat substantia nigra. This indicates that the 5-HT2c receptors in the substantia nigra may contribute to seizure regulation (Gobert, A.; Rivet, J.; Lejeune, F.; et al. Synapse 2000, 36:205-221 ; Hutson, P. H.; Barton, C. L.; Jay, M.; et al. Neuropharmacology 2000, 39:2318-2328). Among the clinically effective anticonvulsants such as carbamazepine, dose-related anticonvulsant effects correlate with increased extracellular serotonin further implicating the role of serotonin and hence the 5-HT2C receptor agonist in epileptic seizures. Nevertheless, cross talk between the 5-HT2c and γ-amino butyric acid (GABA) receptors in the mediation of the observed anticonvulsant activity should not be overlooked (Huidobro-Toro, J. P.; Valenzuela, C. F.; Harris, R. A. Neuropharmacology 1996, 35:1355-1363).
Despite the fact that a large number of 5-HT receptors with different anatomical localization and function have been identified, there are only few studies investigating the role of 5-HT receptor subtypes in the modulation of seizure activity and the results are sometimes controversial depending on the experimental epilepsy model used (Jakus, R.; Graf, M.; Juhasz, G.; Geber, K.;
Evay, G.; Halasz, P.; Bagdy, G. Exp. Neurol. 2003, 184:964-972). In order to further delineate the role of the 5-HT20 receptors in seizure generation, the effects of the 5-HT2c preferring agonist, mCPP, were evaluated in a genetic absence epilepsy model. mCPP weakly elevated seizure threshold in mice
(but not in rats) electroshock test, however appreciable protection against pentylenetetrazol-induced myoclonic and/or tonic seizures in mice and rats were observed. This protection against pentylenetetrazol-induced myoclonic and/or tonic seizures in mice and rats was inhibited by the 5-HT2C/2B receptor
antagonist, SB 206533. The fact that the 5-HT2B agonist, BW-723C86, had no effect on animal seizure models supports the view that the 5-HT2c receptor mediated the mCPP-induced anticonvulsant effects (Upton, N.; Middlemiss, D.; Blackburn, T.; et al. Eur. J. Pharmacol. 1998, 359:33-40). The selective 5- HT2C receptor antagonist, SB 242084 do not induce pro-convulsant effects in rats, which are characteristic of mutant mice lacking the 5-HT2c receptor. This failure to exhibit pro-convulsant properties in rats in contrast to the reported characteristics of mutant mice lacking 5-HT2c receptors might be accounted for by species differences (Di Matteo, V.; Di Giovanni, G.; Esposito, E. CNS Drug Rev. 2000, 6:195-205).
Curiously, given the link between transferrin and the 5-HT2c receptor, it would be of interest to study whether other transport proteins synthesized in the choroid plexus, in particular transthyretin (formerly called prealbumin), also are modulated by 5-HT2c receptors. While speculative, this may be relevant for research on Alzheimer's disease (AD) because independent studies have indicated that both 5-HT2c receptor agonism and transthyretin may reduce the amyloidogenic cleavage of the amyloid precursor protein (APP), a cleavage that produces neurotoxic /3-amyloid protein, the principal proteinaceous component of brain amyloid plaques characteristic of AD (Arjona, A. A.; Pooler, A. M.; Lee, R. K.; Wurtman, R. J. Brain Res. 2002, 951 :135-140; Stein, T. D.; Anders, N. J.; DeCarli, C; Chan, S. L.; Mattson, M. P.; Johnson, J. A. J. Neurosci. 2004, 24:7707-7717).
In recent years, several case reports of the efficacy of psilocybin in the treatment of obsessive-compulsive disorder (OCD) have been published (Delgado, P. L.; Moreno, F. A. J. Psychoactive Drugs 1998, 30:359). As a result, an FDA-approved clinical trial for patients suffering from OCD is now underway (Sard, H.; Kumaran, G.; Morency, C; Roth, B. L.; Toth, B.; He, P.; Shuster, L. Bioorg. Med. Chem. Lett. 2005, 15:4555). The hallucinogenic activity of psilocybin and psilocin is believed to be largely due to activation of 5-HT2A receptors, while the anti-OCD activity is associated with agonist activity at 5-HT2c- Thus, it is believed that a selective 5-HT2C agonist would
have considerable potential for treatment of OCD (Roth, B. L.; Shapiro, D. Expert Opin. Ther. Targets 2001 , 5:685).
Selective serotonin reuptake inhibitors (SSRIs) increase extracellular levels of serotonin (5-HT) and thereby nonselective^ cause stimulation of all postsynaptic 5-HT receptor subtypes. SSRIs have become standard therapy for neuropsychiatric disorders such as obsessive compulsive disorder (OCD), depression, and panic anxiety. There is accumulating evidence for the involvement of 5-HT2c receptor-mediated functions in the therapeutic efficacy of SSRIs (Palvimaki, E. -P.; Roth, B. L.; Majasuo, H.; Laakso, A.; Kuoppamaki, M.; Syvalahti, E.; Hietala, J. Psychopharmacology 1996, 126:234-240; Jenck, F.; Moreau, J. -L.; Mutel, V.; Martin, J. R.; Haefely, W. E. Eur. J. Pharmacol. 1993, 231 :223-229). The increased 5-HT synaptic content resulting from the reuptake inhibition also allows 5-HT to act on the other 5- HT receptor subtypes, possibly explaining some of the side effects associated with SSRI treatment. Selective 5-HT2C receptor agonists, therefore, may represent a direct means to produce the beneficial therapeutic effects of SSRIs without concomitant side effects.
Urinary incontinence (Ul), the involuntary release of urine, may be caused by physiologic, pharmacologic, pathologic, or psychological factors. It is a common condition and often constitutes an embarrassment which can lead to social isolation, depression, loss of quality of life and is a major cause of institutionalization in the elderly population Continence and micturition involve a balance between urethral closure and detrusor muscle activity. Urethral pressure normally exceeds bladder pressure, resulting in urine remaining in the bladder. The proximal urethra and bladder are both within the pelvis. Intraabdominal pressure increases (from coughing and sneezing) are transmitted to both urethra and bladder equally, leaving the pressure differential unchanged, resulting in continence. Normal voiding is the result of changes in both of these pressure factors: urethral pressure falls and bladder pressure rises.
Urinary incontinence affects more than 10 million Americans according to the the American foundation of Urologic Diseases. Both men and woman suffer from urinary incontinence although women are disproportionately affected.
There are several different type of urinary incontinence with the major forms being stress urinary incontinenence (SUI), urge incontinence (Ul) and mixed incontinence. Stress urinary incontinence (SUI) refers to urinary leakage (loss of small amounts of urine) that occurs during physical activity such as coughing, laughing, sneezing, exercising or other movements that increase intraabdominal pressure and thus increase pressure on the bladder. Physical changes resulting from pregnancy, childbirth, and menopause often cause stress incontinence, and in men it is a common problem following a prostatectomy. It is the most common form of incontinence in men.
Urge incontinence (also known as bladder instability, neurogenic bladder, voiding dysfunction, hyperactive bladder, or detrusor overactivity) refers to the involuntary loss of urine occurring for no apparent reason while suddenly feeling the need or urge to urinate. The most common cause of urge incontinence is involuntary and inappropriate detrusor muscle contractions. Urge incontinence may also be called "reflex incontinence" if it results from overactive nerves controlling the bladder.
Mixed incontenance is the combination of stress urinary incontinenence (SUI) and urge incontinence (Ul).
The medical need for effective pharmacological treatment of urinary incontenance is high and one promising area of investigation centres around the finding that the neurotransmitter serotonin (5-HT) has a key role in mechanisms involved in micturition and continence. Recent evidence suggest that compounds with agonist activity at the 5-HT2C receptor may be beneficial in the treatment of conditions related to abnormal bladder activity
(International Patent Applications WO 2004/096169 and WO 2007/132841 and U.S. Patent Application Publication 2007/0225274).
In recent years pain management has become an area of increasing focus in the medical profession, partly due to the growing elderly population, issues surrounding quality of life and the growing numbers of patients reportedly suffering from pain. Pain is both a sensory and emotional experience, and is generally associated with tissue damage or inflammation. Typically, pain is divided into two general categories - acute pain and chronic pain. Both differ in their etiology, pathophysiology, diagnosis, and most importantly treatment.
Acute pain is short term, and is typically of readily identifiable cause. Patients suffering from acute pain typically respond well to medications. In contrast, chronic pain - medically defined as pain that lasts for 3-6 months or longer, is often not associated with an obvious injury; indeed, patients can suffer from protracted pain that persists for months or years after the initial insult. Whilst acute pain is generally favorably treated with medications, chronic pain is often much more difficult to treat, generally requiring expert care.
According to the American Chronic Pain Association, over 86 million Americans suffer from chronic pain, and the management of chronic pain has long been recognized as an unmet clinical need. Most chronic pain is neuropathic in nature (also referred to as neuralgia). Neuropathic pain can, for instance, manifest itself as burning, stabbing, and shock-like sensations.
Unfortunately, neuropathic pain management is at best inconsistent, and often ineffective. This is in part due to the subjective nature of the pain, but also due to poor diagnosis, especially when the chronic pain is not clearly associated with a nerve injury or other insult. Moreover, few, if any, ethical drugs have been prospectively developed for the treatment of chronic pain. Instead, the current medications used to treat chronic pain are "borrowed" from other diseases, most commonly antiepileptic drugs and antidepressants.
Current first-line treatments for chronic pain include opioids, analgesics such as gabapentin, and tricyclic antidepressants. When opioids are administered over prolonged periods, undesirable side effects such as drug tolerance, chemical dependency and even physiological addiction can occur. Of treatment remedies currently available for chronic pain, at best approximately
30% are effective in significantly diminishing the pain, and even these may lose their efficiency over time. Although numerous pharmacological agents are available for the treatment of neuropathic pain, a definitive therapy has remained elusive.
It is well known that serotonin (5-HT) plays a major role in the inhibition of nociceptive transmission. Furthermore, it appears that the the 5HT2c receptor have an inhibitory role in pain (Eur. J. Pharmacol. 2007, 567(1 -2):89-94; Eur. J. Pharmacol. 2004, 502(3):205-11 ; Pain. 2004108(1 -2): 163-9; Pol. J. Pharmacol. 1994, 46(5):423-8 and U.S. Patent Application Publication 2007/0225277).
Although these studies implicated the 5-HT2C receptors in the modulation of feeding (obesity), schizophrenia, epilepsy, OCD, urinary incontinence, pain and other related disorders, elucidation of the functional roles of these receptors has been hindered by a limited availability of selective agents. In addition, such paucity of selective agents can be attributed to the fact that the
5-HT2c receptors share substantial sequence homology with the 5-HT2A and 5-HT2B receptor.
In addition to its receptor selectivity profile, there are properties of a compound that determine its suitability as a drug product. For example, the interaction of the compound with the target protein, which in this case is the 5- HT2C receptor (particularly the more prevalent human edited isoforms VSV, VGV and VNI) and its pharmacokinetic profile, that is its ability to access the target protein in sufficient quantity for a sufficient duration of time to have the desired therapeutic effect.
Summary of the Invention
The present invention relates to 5-HT2c receptor agonists with improved drug- like characteristics such as reduced cytochrome P450 inhibition.
In one aspect, there is provided a compound of Formula I:

wherein:
R1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing at least two rings, wherein said rings are fused rings, bridged fused rings and/or spiro rings; R2, R3 and R5 to R12 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, S-alkyl, S(O)- alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S- heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2-heterocycloalkyl, O-aryl, O- heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O-heteroaryl, alkylene-O- alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S-heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl,
C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group; and
R4 is selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, alkylene- O-alkyl, alkylene-O-cycloalkyl, alkylene-O-alkylene-cycloalkyl;
and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In another aspect, the compound of Formula I comprises a compound of Formula IA:

R1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing two bridged fused rings; and
R3 and R7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further aspect, there is provided a compound of Formula IA wherein: R1 is a substituted or unsubstituted N-heterocycloalkyl or a substituted or unsubstituted N-heterocycloalkyl containing two bridged fused rings; and R3 and R7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In another aspect, there is provided a compound of Formula IA wherein: R1 is a substituted N-heterocycloalkyl containing two bridged fused rings substituted with alkyl, alkoxy, hydroxy, cyano, halo, haloalkyl, and/or haloalkyloxy; R7 is selected from H, halo, alkyl, haloalkyl, haloalkyloxy, cyano, alkoxy; and R3 is selected from H, or lower alkyl, and/or a pharmaceutically- acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further aspect, the compound of Formula I comprises a compound of Formula IB, IC and/or ID:

IB IC

ID wherein:
R3 and R7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, S-alkyl, S(O)- alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S-
heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2-heterocycloalkyl, O-aryl, O- heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O-heteroaryl, alkylene-O- alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S-heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl, C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group; X is O, S, SO, SO2, CR13R14; NR15;
R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
R15 is selected from the group consisting of H and alkyl; n is an integer; and m is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further aspect, there is provided a compound of Formula IB, IC and/or ID wherein: R3 is selected from H, or lower alkyl; R7 is selected from H, halo;
X is O, S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl; R15 is selected from the group consisting of H and alkyl of C1 to C6; n is either 0, 1 or 2; and m is 0, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In yet a further aspect, there is provided a compound of Formula II:

Il wherein:
R3 and R7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, S-alkyl, S(O)- alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S- heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2-heterocycloalkyl, O-aryl, O- heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O-heteroaryl, alkylene-O- alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S-heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl, C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group; X is O, S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
R15 is selected from the group consisting of H and alkyl; and n is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further aspect, the compound of Formula Il wherein: R3 is selected from H, or lower alkyl; R7 is selected from H, halo; X is O, S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl; R15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either O, 1 or 2; and/or a pharmaceutically- acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further aspect, the compound of Formula Il comprises a compound of Formula HA and/or MB :

HA "B wherein:
R3 is selected from H, or lower alkyl; R7 is selected from H, halo;
X is O, S, SO, SO2, CR13R14; NR15;
R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
R15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either O, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further aspect, there is provided a compound selected from: (9R)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H- pyrido[2,3d]azepine;
(gR^-i^fluoropiperidin-i-yO-θ-methyl-ej.δ.θ-tetrahydro-SH-pyrido^.S- d]azepine;
(9R)-9-methyl-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyridot2,3- d]azepine; (9R)-9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9R)-9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9S)-2-(4)4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9S)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7I8>9-tetrahydro-5H-pyrido[2,3- d]azepine;
(ΘSJ-θ-methyl^-CI ^-oxazepan^-yO-ej.δ.θ-tetrahydro-δH-pyrido^.S- d]azepine
(9S)-9-methyl-2-morpholin-4-yl-6,7,8)9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9S)-9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-(1 ,4-diazepan-1 -yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2)3-d]azepine;
2-(1-oxidothiomorpholin-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoroazepan-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoroazepan-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
2-(4,4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
2-(4-fluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4-methylpiperazin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(8-azabicyclo[3.2.1]oct-8-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-9-isopropyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-azepan-1-yl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-cyclopropyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-cyclopropyl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-piperazin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-pyrrolidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-thiomorpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-bromo-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-bromo-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-(4)4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
3-chloro-2-(4-fluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-ethyl-2-morpholin-4-yl-6>718,9-tetrahydro-5H-pyrido[2,3-d]azepine; 9-ethyl-2-piperidin-1 -yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-isopropyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-isopropyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; ethyl 4-(6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepin-2-yl)piperazine-1- carboxylate; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further aspect, there is provided a compound selected from:
(9R)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H- pyrido[2,3d]azepine;
(9R)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9R)-9-methyl-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(ΘRJ-θ-methyl^-morpholin-^yl-ej.δ.θ-tetrahydro-δH-pyriclo^.a-cllazepine;
(9R)-9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9S)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine; (9S)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(ΘSJ-θ-methyl^-O Aoxazepan^-yO-ej.δ.θ-tetrahydro-δH-pyridop.S- d]azepine
(ΘS^Θ-methyl^-moφholin^-yl-ej.δ.θ-tetrahydro-δH-pyridoβ.S-dlazepine; (9S)-9-methyl-2-piperidin-1 -yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-diazepan-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoroazepan-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-(4-fluoropiperidin-1 -yO-βJ.δ.θ-tetrahydro-δH-pyrido^.S-dJazepine;
2-azepan-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-9-methyl-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-cyclopropyl-9-methyl-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-morpholin-4-yl-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-piperidin-1-yl-6>7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-thiomorpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
S-chloro^-CI ^-oxazepan^-yO-βJ.δ.θ-tetrahydro-δH-pyrido^.S-dlazepine;
3-chloro-2-(4,4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine; 3-chloro-2-(4-fluoropiperidin-1-yl)-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-piperidin-1-yl-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-isopropyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 9-methyl-2-piperidin-1-yl-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In yet another aspect, there is provided a compound according to any one of the compounds noted above, wherein the compound has an EC50 for a human 5-HT2C receptor selected from less than 1000 nM, less than 500 nM, less than 300 nM, or less than 100 nM.
In another aspect, there is provided a Boc-protected precusor of the compound according to any one of the compounds noted above, or mixtures thereof.
In another aspect, there is provided a pharmaceutical composition comprising at least one of the compounds noted above and at least one pharmaceutically acceptable carrier and/or excipient.
In a further aspect, there is provided a method for making the compound of Formula I, wherein R11 and R12 are H, the method comprising:

B
reacting a compound of Formula A under conditions (a), wherein said (a) comprises heat and base assisted cyclization of the compound of Formula A to provide an amide of Formula B; and reducing the carbonyl of the amide of Formula B.
In a further aspect, there is provided a method for making the compound of Formula IA to ID and Il (including MA to HB), the method comprising:

reacting a compound of Formula A under conditions (a), wherein said (a) comprises heat and base assisted cyclization of the compound of Formula A to provide an amide of Formula B; and reducing the carbonyl of the amide of Formula B.
In a further aspect, there is provided another method for making the compound of Formula I, wherein R9 and R10 are H, the method comprising:
 BB reacting a compound of Formula AA under conditions (a), wherein said (a) comprises heat and base assisted cyclization of the compound of Formula AA to provide an amide of Formula BB; and reducing the carbonyl of the amide of Formula BB.
BB reacting a compound of Formula AA under conditions (a), wherein said (a) comprises heat and base assisted cyclization of the compound of Formula AA to provide an amide of Formula BB; and reducing the carbonyl of the amide of Formula BB.
In a further aspect, there is provided another method for making the compound of Formula IA to ID and Il (including MA to MB), the method comprising:

AA IA-ID IIA-IIB

BB reacting a compound of Formula AA under conditions (a), wherein said (a) comprises heat and base assisted cyclization of the compound of Formula AA to provide an amide of Formula BB; and reducing the carbonyl of the amide of Formula BB.
In a further aspect, there is provided a method for making the compound of Formula I, wherein R11 and R12 are H, the method comprising: reducing a carbonyl of an amide:

In a further aspect, there is provided a method for making the compound of Formula I, wherein R9 and R10 are H, the method comprising: reducing a carbonyl of an amide:

In a further aspect, there is provided a method for making the compound of Formula I, wherein R9, R10, R11 and R12 are H, the method comprising: reducing carbonyl groups of an imide:

In a yet further aspect, there is provided a method for making the compound of Formula I, wherein R11 and R12 are H, the method comprising:

In a yet further aspect, there is provided a method for making the compound of Formula I, wherein R9 and R10 are H, the method comprising:

CC
reacting a compound of Formula CC under conditions (a), wherein said (a) comprises selective cyano reduction followed by cyclization of the compound of Formula CC to provide Formula I, whereby R' is alkyl or cycloalkyl.
In a yet further aspect, there is provided a method for making the compound of Formula I, wherein the method comprises:

reacting a compound of Formula D under conditions (a), wherein said (a) comprises cyclization of the compound of Formula D to provide Formula I, whereby R' is alkyl or cycloalkyl.
In a yet further aspect, there is provided a method for making the compound of Formula I, wherein the method comprises:

DD reacting a compound of Formula DD under conditions (a), wherein said (a) comprises cyclization of the compound of Formula DD to provide Formula I1 whereby R' is alkyl or cycloalkyl.
In a further aspect, there is provided a method for making the compound of Formula IA to ID and Il (including MA to MB), the method comprising: reducing carbonyl groups of a cyclic imide:

IA-ID, HA-IlB
In a yet further aspect, there is provided a method for making the compound of Formula IA to ID and Il (including MA to MB), the method comprising:

IA-ID, HA-IIB
reacting a compound of Formula C under conditions (a), wherein said (a) comprises selective cyano reduction followed by cyclization of the compound of Formula C to provide Formula IA to ID and Il (including MA to MB), whereby R' is alkyl or cycloalkyl.
In a yet further aspect, there is provided a method for making the compound of Formula IA to ID and Il (including MA to HB), the method comprising:

IA-ID, MA-IIB
CC
reacting a compound of Formula CC under conditions (a), wherein said (a) comprises selective cyano reduction followed by cyclization of the compound of Formula CC to provide Formula IA to ID and Il (including HA to HB), whereby R' is alkyl or cycloalkyl.
In a yet further aspect, there is provided a method for making the compound of Formula IA to ID and Il (including HA to HB), wherein the method comprises:
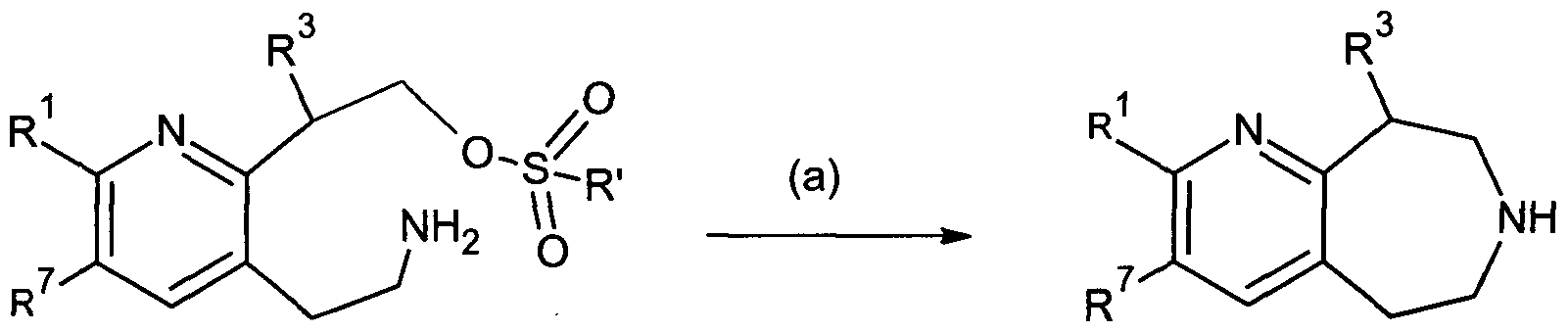
IA-ID, MA-IIB
reacting a compound of Formula D under conditions (a), wherein said (a) comprises cyclization of the compound of Formula D to provide Formula IA to ID and Il (including HA to HB), whereby R1 is alkyl or cycloalkyl.
In a yet further aspect, there is provided a method for making the compound of Formula IA to ID and Il (including HA to HB), wherein the method comprises:

IA-ID MA-IIB
DD
reacting a compound of Formula DD under conditions (a), wherein said (a) comprises cyclization of the compound of Formula DD to provide Formula IA to ID and Il (including HA to HB), whereby R' is alkyl or cycloalkyl.
In another aspect, there is provided a method for treating a 5-HT2c receptor- mediated disorder in a mammal, comprising administering to the mammal a therapeutically effective amount of a compound or composition noted above; the compound or composition exhibiting reduced inhibition of a cytochrome
P450. In a further aspect, the mammal is a human. In another aspect, the disorder is selected from urinary incontinence, obesity, schizophrenia, epilepsy, depression, panic anxiety, alcoholism or obsessive compulsive disorder, a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular
disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure. In a further aspect, the disorder is selected from obesity, schizophrenia, epilepsy, a depressive disorder, panic attack, alcoholism, drug addiction or obsessive compulsive disorder. In still a further aspect, the compound or composition is administered orally and/or parenterally. In yet another aspect, the compound or composition is administered intravenously and/or intraperitoneally.
In yet a further aspect, there is provided the use of the compound or composition noted above for the manufacture of a medicament for treatment of a 5-HT2c receptor-mediated disease in a mammal; the compound or composition exhibiting reduced inhibition of a cytochrome P450. In yet a further aspect, there is provided the use of the compound or composition noted above to treat a 5-HT2c receptor-mediated disease in a mammal the compound or composition exhibiting reduced inhibition of a cytochrome P450. In a further aspect, the mammal is a human. In another aspect, the disorder is selected from urinary incontinence obesity, schizophrenia, epilepsy, depression, panic anxiety, alcoholism or obsessive compulsive disorder, a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure. In a further aspect, the disorder is selected from obesity, schizophrenia, epilepsy, a depressive disorder, panic attack, alcoholism, drug addiction or obsessive compulsive disorder. In still a further aspect, the compound or composition is administrable orally and/or
parenterally. In yet another aspect, the compound or composition is administrable intravenously and/or intraperitoneally.
In another aspect, there is provided a method for decreasing food intake in a mammal comprising administering to the mammal a therapeutically effective amount of a compound or composition as noted above; the compound or composition exhibiting reduced inhibition of a cytochrome P450.
In another aspect, there is provided a method of controlling weight gain in a mammal comprising administering to the mammal a therapeutically effective amount of a compound or composition as noted above; the compound or composition exhibiting reduced inhibition of a cytochrome P450.
The novel features of the present invention will become apparent to those of skill in the art upon examination of the following detailed description of the invention. It should be understood, however, that the detailed description of the invention and the specific examples presented, while indicating certain embodiments of the present invention, are provided for illustration purposes only because various changes and modifications within the spirit and scope of the invention will become apparent to those of skill in the art from the detailed description of the invention and claims that follow.
Brief Description of the Drawings
Certain embodiments of the invention are described, reference being made to the accompanying drawings, wherein:
Figure 1 shows graphically the effect on feeding in adult male Sprague- Dawley rats of one exemplary compound (22.34) of the invention at various doses (mg/ml) or vehicle administered intraperitoneally (X-axis) on rat food consumption (Y-axis). The treatment groups are as follows: 1 = vehicle, 2 = 22.34 at 0.1 mg/kg ip, 3 = 22.34 at 0.3 mg/kg ip, 4 = 22.34 at 0.6 mg/kg ip, 5 =
22.34 at 1 mg/kg ip, 6 = 22.34 at 0.6 mg/kg ip + SB242084 at 1 mg/kg ip. * p<0.05 vs. vehicle pretreatment, # p<0.05 vs. 204 0.6 mg/kg.
Definitions Unless specified otherwise within this specification, the nomenclature used in this specification generally follows the examples and rules stated in Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F, and H, Pergamon Press, Oxford, 1979, which is incorporated by references herein for its exemplary chemical structure names and rules on naming chemical structures. Optionally, a name of a compound may be generated using a chemical naming program: ACD/ChemSketch, Version 5.09/September 2001 , Advanced Chemistry Development, Inc., Toronto, Canada.
The compounds of the present invention may have asymmetric centers, chiral axes, and chiral planes (e.g., as described in: E. L. Eliel and S. H. Wilen, Stereo-chemistry of Carbon Compounds, John Wiley & Sons, New York, 1994, pages 1119-1190), and occur as racemates, racemic mixtures, and as individual diastereomers, with all possible isomers and mixtures thereof, including optical isomers, being included in the present invention.
Generally, reference to a certain element such as hydrogen or H is meant to, if appropriate, include all isotopes of that element.
The following terms are meant to encompass unsubstituted and/or substituted.
The term "alkyl" as used herein means a straight- or branched-chain hydrocarbon radical; in one aspect, having from one to eight carbon atoms, and includes, for example, and without being limited thereto, methyl, ethyl, propyl, isopropyl, t-butyl and the like. As noted above, "alkyl" encompasses substituted alkyl. Substituted alkyl includes, for example, and without being limited thereto, haloalkyl, hydroxyalkyl, cyanoalkyl, and the like. This is
applied to any of the groups mentioned herein. Groups such as "alkenyl", "alkynyl", "aryl", etc. encompass substituted "alkenyl", "alkynyl", "aryl", etc.
The term "alkenyl" as used herein means a straight- or branched-chain alkenyl radical; in one aspect, having from two to eight carbon atoms, and includes, for example, and without being limited thereto, ethenyl, 1-propenyl, 1-butenyl and the like. The term "alkenyl" encompass radicals having "cis" and "trans" orientations, or alternatively, 11E" and "Z" orientations.
The term "alkynyl" as used herein means a straight- or branched-chain alkynyl radical; in one aspect, having from two to eight carbon atoms, and includes, for example, and without being limited thereto, 1-propynyl (propargyl), 1- butynyl and the like.
The term "cycloalkyl" as used herein means a carbocyclic system (which may be unsaturated) containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused. In one aspect, the ring(s) may have from three to seven carbon atoms, and includes, for example, and without being limited thereto, cyclopropyl, cyclohexyl, cyclohexenyl and the like.
The term "heterocycloalkyl" as used herein means a heterocyclic system (which may be unsaturated) having at least one heteroatom selected from N, S and/or O and containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused. In one aspect, the ring(s) may have a three- to seven-membered cyclic group and includes, for example, and without being limited thereto, piperidinyl, piperazinyl, pyrrolidinyl, tetrahydrofuranyl and the like.
The term "alkoxy" as used herein means a straight- or branched-chain alkoxy radical; in one aspect, having from one to eight carbon atoms and includes,
for example, and without being limited thereto, methoxy, ethoxy, propyloxy, isopropyloxy, f-butoxy and the like.
The term "halo" as used herein means halogen and includes, for example, and without being limited thereto, fluoro, chloro, bromo, iodo and the like, in both radioactive and non-radioactive forms.
The term "alkylene" as used herein means a difunctional branched or unbranched saturated hydrocarbon radical; in one aspect, having one to eight carbon atoms, and includes, for example, and without being limited thereto, methylene, ethylene, n-propylene, n-butylene and the like.
The term "alkenylene" as used herein means a difunctional branched or unbranched hydrocarbon radical; in one aspect, having two to eight carbon atoms, and having at least one double bond, and includes, for example, and without being limited thereto, ethenylene, n-propenylene, n-butenylene and the like.
The term "alkynylene" as used herein means a difunctional branched or unbranched hydrocarbon radical; in one aspect, having two to eight carbon atoms, and having at least one triple bond, and includes, for example, and without being limited thereto, ethynylene, n-propynylene, n-butynylene and the like.
The term "aryl" , alone or in combination, as used herein means a carbocyclic aromatic system containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused. In particular embodiments, aryl is one, two or three rings. In one aspect, the aryl has five to twelve ring atoms. The term "aryl" encompasses aromatic radicals such as phenyl, naphthyl, tetrahydronaphthyl, indanyl, biphenyl, phenanthryl, anthryl or acenaphthyl. The "aryl" group may have 1 to 4 substituents such as lower
alkyl, hydroxyl, halo, haloalkyl, nitro, cyano, alkoxy, lower alkylamino and the like.
The term "heteroaryl", alone or in combination, as used herein means an aromatic system having at least one heteroatom selected from N, S and/or O and containing one or more rings wherein such rings may be attached together in a pendent manner or may be fused. In particular embodiments, heteroaryl is one, two or three rings. In one aspect, the heteroaryl has five to twelve ring atoms. The term "heteroaryl" encompasses heteroaromatic radicals such as pyridyl, indolyl, furyl, benzofuryl, thienyl, benzothienyl, quinolyl, oxazolyl and the like. The "heteroaryl" group may have 1 to 4 substituents such as lower alkyl, hydroxyl, halo, haloalkyl, nitro, cyano, alkoxy, lower alkylamino and the like.
It is understood that substituents and substitution patterns on the compounds of the invention may be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those methods set forth below. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, as long as a stable structure results.
The term "pharmaceutically acceptable salt" means either an acid addition salt or a basic addition salt which is compatible with the treatment of patients.
A "pharmaceutically acceptable acid addition salt" is any non-toxic organic or inorganic acid addition salt of the base compounds represented by Formula I or any of its intermediates. Illustrative inorganic acids which form suitable salts include, but are not limited thereto, hydrochloric, hydrobromic, sulfuric and phosphoric acid and acid metal salts such as sodium monohydrogen orthophosphate and potassium hydrogen sulfate. Illustrative organic acids which form suitable salts include the mono-, di- and tricarboxylic acids.
lllustrative of such acids are, for example, acetic, glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, hydroxymaleic, benzoic, hydroxybenzoic, phenylacetic, cinnamic, salicylic, 2- phenoxybenzoic, p-toluenesulfonic acid and other sulfonic acids such as methanesulfonic acid and 2-hydroxyethanesulfonic acid. Either the mono- or di-acid salts can be formed, and such salts can exist in either a hydrated, solvated or substantially anhydrous form. In general, the acid addition salts of these compounds are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms. The selection criteria for the appropriate salt will be known to one skilled in the art. Other non-pharmaceutically acceptable salts e.g. oxalates may be used for example in the isolation of compounds of Formula I for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
A "pharmaceutically acceptable basic addition salt" is any non-toxic organic or inorganic base addition salt of the acid compounds represented by Formula I or any of its intermediates. Illustrative inorganic bases which form suitable salts include, but are not limited thereto, lithium, sodium, potassium, calcium, magnesium or barium hydroxides. Illustrative organic bases which form suitable salts include aliphatic, alicyclic or aromatic organic amines such as methylamine, trimethyl amine and picoline or ammonia. The selection of the appropriate salt may be important so that an ester functionality, if any, elsewhere in the molecule is not hydrolyzed. The selection criteria for the appropriate salt will be known to one skilled in the art.
"Solvate" means a compound of Formula I or the pharmaceutically acceptable salt of a compound of Formula I wherein molecules of a suitable solvent are incorporated in a crystal lattice. A suitable solvent is physiologically tolerable at the dosage administered as the solvate. Examples of suitable solvents, but are not limited thereto, are ethanol, water and the like. When water is the solvent, the molecule is referred to as a hydrate.
The term "stereoisomers" is a general term for all isomers of the individual molecules that differ only in the orientation of their atoms in space. It includes mirror image isomers (enantiomers), geometric (cis/trans) isomers and isomers of compounds with more than one chiral centre that are not mirror images of one another (diastereomers).
The term "treat" or "treating" means to alleviate symptoms, eliminate the causation of the symptoms either on a temporary or permanent basis, or to prevent or slow the appearance of symptoms of the named disorder or condition.
The term "therapeutically effective amount" means an amount of the compound which is effective in treating the named disorder or condition.
The term "pharmaceutically acceptable carrier" means a non-toxic solvent, dispersant, excipient, adjuvant or other material which is mixed with the active ingredient in order to permit the formation of a pharmaceutical composition, i.e., a dosage form capable of administration to the patient. One example of such a carrier is a pharmaceutically acceptable oil typically used for parenteral administration.
A "5-HT2C receptor-mediated disorder", as used herein, is a disorder in which there is believed to be involvement of the pathway controlled by the 5-HT2c receptor and which is ameliorated by treatment with an agonist of the 5-HT2C receptor. 5-HT2C receptor-mediated disorders include a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive
compulsive disorder, premenstrual tension, chronic fatigue syndrome, age- related memory disorder, personality disorder and raised intracranial pressure.
Detailed Description
Compounds of the invention conform generally to Formula I:
I wherein: R1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing at least two rings, wherein said rings are fused rings, bridged fused rings and/or spiro rings; R2, R3 and R5 to R12 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, S-alkyl, S(O)- alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S- heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2-heterocycloalkyl, O-aryl, O- heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O-heteroaryl, alkylene-O- alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S-heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl, C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or
S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group; and R4 is selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-alkylene- cycloalkyl; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In an embodiment, there is provided compound(s) of Formula I that comprise a compound of Formula IA:

IA wherein R1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing two bridged fused rings; and R3 and R7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof. In a further embodiment, R1 is a substituted or unsubstituted N-heterocycloalkyl containing two bridged fused rings; and R3 and R7 are as outlined above; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof. In still a further embodiment, R1 is a substituted N-heterocycloalkyl containing two bridged fused rings substituted with alkyl, alkoxy, hydroxy, cyano, halo, haloalkyl, and/or haloalkyloxy; R7 is selected from H, halo, alkyl, haloalkyl, haloalkyloxy, cyano, alkoxy; and R3 is selected from H, or lower alkyl, and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
The compound of Formula I can comprise a compound of Formula IB, IC and/or ID:

IB IC

ID wherein: R3 and R7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O- heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O- heterocycloalkyl, alkylene-O-alkylene-cycloalkyl, alkylene-O-alkylene- heterocycloalkyl, S-alkyl, S(O)-alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S-heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2- heterocycloalkyl, O-aryl, O-heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O- heteroaryl, alkylene-O-alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S- heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl, C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group; X is O, S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and
haloalkyl; R15 is selected from the group consisting of H and alkyl; n is an integer; and m is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof. In a further embodiment, R3 is selected from H, or lower alkyl; R7 is selected from H, halo; X is O, S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl; R15 is selected from the group consisting of H and alkyl of C1 to C6; n is either O, 1 or 2; and m is 0, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In another embodiment, the compound of Formula I comprises a compound of Formula II:

Il wherein: R3 and R7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O- heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O- heterocycloalkyl, alkylene-O-alkylene-cycloalkyl, alkylene-O-alkylene- heterocycloalkyl, S-alkyl, S(O)-alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S-heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2- heterocycloalkyl, O-aryl, O-heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O- heteroaryl, alkylene-O-alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S- heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl,
OC(O)alkyl, C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group; X is O, S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl; R15 is selected from the group consisting of H and alkyl; and n is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof. In a further embodiment, R3 is selected from H, or lower alkyl; R7 is selected from H, halo; X is O1 S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl; R15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either O, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
In a further embodiment of Formula II, n=1 (Formula MA) and n=2 (Formula MB):

HA "B wherein R3 and R7 are as outlined above. In a specific embodiment, R3 is selected from H, or lower alkyl; R7 is selected from H, halo; X is O, S, SO, SO2, CR13R14; NR15; R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl; R15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either O, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
It will be understood by those of skill in the art that when compounds of the present invention contain one or more chiral centers, the compounds of the invention may exist in, and be isolated as, enantiomeric or diastereomeric forms, or as a racemic mixture. The present invention includes any possible enantiomers, diastereomers, racemates or mixtures thereof, of a compound of Formula I. The optically active forms of the compound of the invention may be prepared, for example, by chiral chromatographic separation of a racemate or chemical or enzymatic resolution methodology, by synthesis from optically active starting materials or by asymmetric synthesis based on the procedures described thereafter.
It will also be appreciated by those of skill in the art that certain compounds of the present invention may exist as geometrical isomers, for example E and Z isomers of alkenes. The present invention includes any geometrical isomer of a compound of Formula I and II. It will further be understood that the present invention encompasses tautomers of the compounds of Formula I and II.
It will also be understood by those of skill in the art that certain compounds of the present invention may exist in solvated, for example hydrated, as well as unsolvated forms. It will further be understood that the present invention encompasses all such solvated forms of the compounds of Formula I and II.
Within the scope of the invention are also salts of the compounds of Formula I and II. Generally, pharmaceutically acceptable salts of compounds of the present invention are obtained using standard procedures well known in the art, for example, by reacting a sufficiently basic compound, for example an alkyl amine with a suitable acid, for example, HCI or acetic acid, to afford a salt with a physiologically acceptable anion. It is also possible to make a corresponding alkali metal (such as sodium, potassium, or lithium) or an alkaline earth metal (such as a calcium) salt by treating a compound of the present invention having a suitably acidic proton, such as a carboxylic acid or
a phenol, with one equivalent of an alkali metal or alkaline earth metal hydroxide or alkoxide (such as the ethoxide or methoxide), or a suitably basic organic amine (such as choline or meglumine) in an aqueous medium, followed by conventional purification techniques. Additionally, quaternary ammonium salts can be prepared by the addition of alkylating agents, for example, to neutral amines.
In one embodiment of the present invention, the compound of Formula I and Il may be converted to a pharmaceutically acceptable salt or solvate thereof, particularly, an acid addition salt such as a hydrochloride, hydrobromide, phosphate, acetate, fumarate, maleate, tartrate, citrate, methanesulphonate or p-toluenesulphonate.
Specific examples of the present invention include the following compounds, their pharmaceutically acceptable salts, hydrates, solvates, optical isomers, and combinations thereof:
(9R)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H- pyrido[2,3d]azepine; (9R)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9R)-9-methyl-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9R)-9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; (9R)-9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9S)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9S)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine; (ΘS^Θ-methyl^^i ^-oxazepan^-yO-ej.δ.θ-tetrahydro-δH-pyrido^.S- d]azepine
(9S)-9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9S)-9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-diazepan-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1-oxidothiomorpholin-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-(4,4-difluoroazepan-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2>3-d]azepine;
2-(4,4-difluoroazepan-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
2-(4,4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4J4-difluoropiperidin-1-yl)-9-methyl-6,7)8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
2-(4-fluoropiperidin-1-yl)-6,7)8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4-methylpiperazin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(8-azabicyclo[3.2.1]oct-8-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-(trifluoromethyl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-9-isopropyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-cyclopropyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-cyclopropyl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-piperazin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-pyrrolidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-thiomorpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-bromo-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-bromo-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-(4,4-difluoropiperidin-1-yl)-6,7,8I9-tetrahydro-5H-pyrido[2,3- d]azepine;
3-chloro-2-(4-fluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2^iperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3<l]azepine;
9-ethyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-ethyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-isopropyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-isopropyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; ethyl 4-(6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepin-2-yl)piperazine-1- carboxylate;
Several methods for preparing compounds of this invention are illustrated in the following, non-limiting, Schemes and Examples. Starting materials and the requisite intermediates are in some cases commercially available, or can be prepared according to literature procedures or as illustrated herein.
Compounds of Formula I and IA to ID and Il (including MA to HB), may be prepared as follows:

B
(wherein R11 and R12 are H) or, more specific to Formula IA to ID and Il (including HA to HB),

whereby R' can be alkyl or cycloalkyl and (a) comprises heat and base assisted cyclization of a compound of Formula A to provide a compound of Formula B and (b) comprises reduction of the carbonyl of the amide of the compound of Formula B. For example, (a) comprises heating in DMF and (b) comprises reduction with LiAIH4ZAICb.
Compounds of Formula I and IA to ID and Il (including MA to MB), may also be prepared as follows:
 or, more specific to Formula IA to ID and Il (including MA to HB),
or, more specific to Formula IA to ID and Il (including MA to HB),

AA IA-ID HA-IIB

BB
whereby R' can be alkyl or cycloalkyl and (a) comprises heat and base assisted cyclization of a compound of Formula AA to provide a compound of
Formula BB and (b) comprises reduction of the carbonyl of the amide of the compound of Formula B. For example, (a) comprises heating in DMF and (b) comprises reduction with LiAIH4ZAICI3.
Compounds of Formula I may alternatively be prepared via reduction of the carbonyl groups of the following amide/cyclic imide:

(R11 and R12 are H) or

10
(Rs and R1U are H)
or


IA-ID IIA-IIB
Again, reduction of the carbonyl group(s) can occur, for example, using LiAIH4ZAICI3.
Compounds of Formula I and IA to ID and Il (including MA to MB), may be prepared as follows:

(R11 and R12 are H) or, more specific to Formula IA to ID and Il (including HA to MB),

IA-ID HA-IIB
whereby (a) comprises selective cyano reduction followed by cyclization of the compound of Formula C. R' can be alkyl or cycloalkyl. For example, cyano reduction using LiAIH4ZAICI3 and DIPEA in acetonitrile for cyclization. The
resultant Formula I and IA to ID and Il (including MA to HB) can be converted to a salt addition of an acid, for example.
Compounds of Formula I, may be prepared as follows:
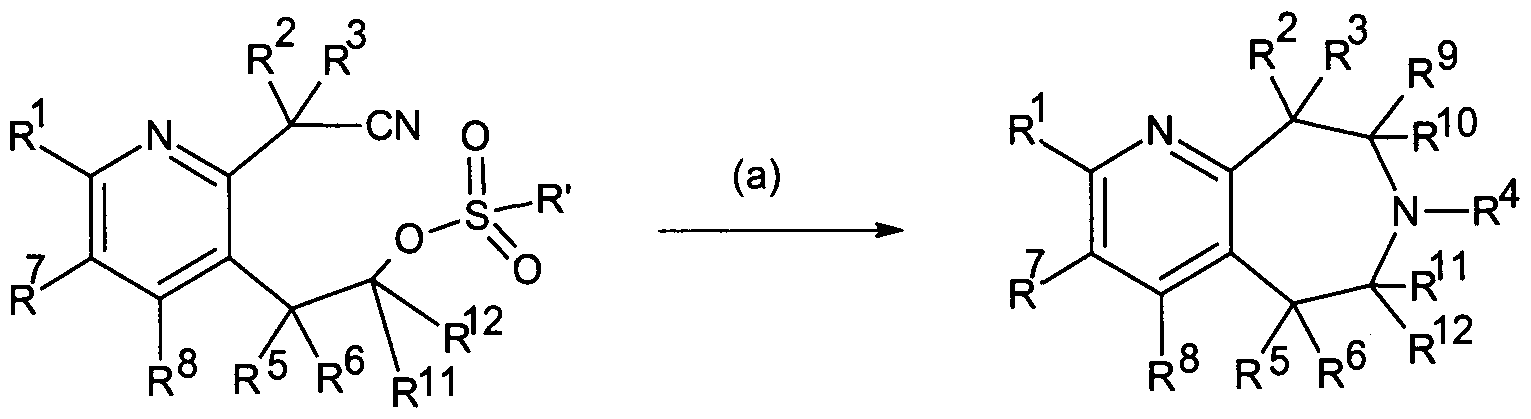
CC
(wherein R s9a and j C R-.110U are H) or, more specific to Formula IA to ID and Il (including MA to MB),

IA-ID MA-IIB
CC
whereby (a) comprises selective cyano reduction followed by cyclization of the compound of Formula CC. R' can be alkyl or cycloalkyl. For example, cyano reduction using LiAIH4ZAICI3 and DIPEA in acetonitrile for cyclization. The resultant Formula I and IA to ID and Il (including MA to MB) can be converted to a salt addition of an acid, for example.
Compounds of Formula I may also be prepared as follows:

or, more specific to Formula IA to ID and Il (including MA to MB),

IA-ID HA-IIB
whereby (a) comprises cyclization of the compound of Formula D, whereby R' can be alkyl or cycloalkyl. For example, base can be used to initiate cyclization.
Compounds of Formula I may also be prepared as follows:

DD or, more specific to Formula IA to ID and Il (including MA to MB),

DD IA-ID MA-IIB
whereby (a) comprises cyclization of the compound of Formula DD, whereby R' can be alkyl or cycloalkyl. For example, base can be used to initiate cyclization.
Compounds of Formula IA and Il where R3 and R7 are H and R1 is methoxy may be prepared according to Scheme 1 , below, from nitrile intermediate V by alkylation with benzylamine, followed by hydrolysis of the nitrile function to afford the amino ester VII. Microwave-assisted cyclization to azepinone VIII and reduction with UAIH4/AICI3 gave the benzyl-protected azepine IX. Subsequent hydrogenolysis of the protecting group provided the intermediate compound Example 13.1 . The intermediate V was prepared from 2-(2- chloroethyl)-3-(chloromethyl)-6-methoxypyridine [Feng, S.; He, X.; Yu, G.; Yu, X.; Bai, D. Org. Prep. Proced. Int. 2004, 36 (2); 129-134] via mono-cyanation and the Finkelstien chloro-iodo exchange.
H2NCH2Ph HCI in MeOH DMF, MW BuOH, 90° C reflux 200» C, 4h




Vl VII VIII
LAH, AICI3
Pd1 H2 NH
O N
THF, rt -CCH, MeOH
IX Example 13.1
Scheme 1
For another synthetic strategy to prepare the intermediate precursor to compounds of Formula IA and Il is outlined in Scheme 2. Condensation of the β-ketodiester B with the acetylinic amide A gives the functionalized pyridone C which on treatment with Ag2CO3 and methyl iodide gives the methoxypyridine intermediate D. The intermediate D can be treated with a number of alkylating groups (e.g. R5 = H or Methyl is shown) to introduce functionality onto the azepine ring. Treatment of the diester D with a reducing agent (e.g. LiAIH4) provides diol intermediate E. Treatment of diol E with mesyl chloride gives the chloro-mesylate F which on mild cyanation with NaCN in DMSO gives the cyano-mesylate G. Selective cyano reduction (e.g. with alane generated in situ from AICI3 and LiAIH4) followed by cyclization gives the pyridyl-fused azepine intermediate H. O-Deprotection with HBr in acetic acid to I followed by N- protection with for example (BOC^O affords intermediate J. Triflation of J to the versatile intermediate K followed by coupling with the various amines give the Boc-protected precusor L of the compounds of Formula III. Treatment of L with HCI gives M, the HCI salts of the compounds of this invention.

NaOetHMEl C F^H
TH= ^ R=Me Et2CTHF -780C 300C



Scheme 2
Acid addition salts of the compounds of Formula I and Il are most suitably formed from pharmaceutically acceptable acids, and include for example those formed with inorganic acids e.g. hydrochloric, sulphuric or phosphoric acids and organic acids e.g. succinic, maleic, acetic or fumaric acid. Other non- pharmaceutically acceptable salts e.g. oxalates may be used for example in the isolation of compounds of Formula I and Il for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt. Also included within the scope of the invention are base addition salts (such as sodium, potassium and ammonium salts), solvates and hydrates of compounds of the invention.
The conversion of a given compound salt to a desired compound salt is achieved by applying standard techniques, well known to one skilled in the art.
Compounds in accordance with the invention have been shown to be potent agonists or partial agonists of the 5-HT2c receptor and have good specificity for the 5-HT2C receptor compared to the 5-HT2A and 5-HT2B receptors.
In embodiments of the invention, the compounds of the invention have an EC50 value for the human 5-HT2c receptor less than 1000 nM, or less than 500 nM, or less than 300 nM, or less than 10O nM.
The compounds of the invention are therefore of interest for the treatment of 5-HT2C receptor-mediated disorders, including a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, urinary incontinence (such as stress urinary incontinenence (SUI), urge incontinence (Ul) and mixed incontinence), pain, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure.
The studies described herein of the effect of compounds of the invention in a feeding assay which is an accepted in vivo rodent model for studies on overeating and control of food intake confirms the potential of these compounds for treatment of obesity.
Compounds of the invention have also been shown to be effective in inhibiting locomotor activity in a rodent model relevant to treatment of schizophrenia or other psychotic disorders.
Compounds of the invention have also been shown to exhibit reduced cytochrome P450 inhibition thus minimizing potential drug-drug interactions.
For pharmaceutical use, the compounds of the invention are, for instance, administered orally, sublingually, rectally, nasally, vaginally, topically (including the use of a patch or other transdermal delivery device), by pulmonary route by use of an aerosol, or parenterally, including, for example, intramuscularly, subcutaneously, intraperitoneally, intraarterially, intravenously or intrathecally. Administration can be by means of a pump for periodic or continuous delivery. The compounds of the invention are administered alone, or are combined with a pharmaceutically-acceptable carrier or excipient according to standard pharmaceutical practice. For the oral mode of administration, the compounds of the invention are used in the form of tablets, capsules, lozenges, chewing gum, troches, powders, syrups, elixirs, aqueous solutions and suspensions, and the like. In the case of tablets, carriers that are used include lactose, sodium citrate and salts of phosphoric acid. Various disintegrants such as starch, and lubricating agents such as magnesium stearate and talc, are commonly used in tablets. For oral administration in capsule form, useful diluents are lactose and high molecular weight polyethylene glycols. If desired, certain sweetening and/or flavoring agents are added. For parenteral administration, sterile solutions of the compounds of the invention are usually prepared, and the pHs of the solutions are suitably adjusted and buffered. For intravenous use, the total concentration of solutes should be controlled to render the preparation isotonic. For ocular administration, ointments or droppable liquids may be delivered by ocular delivery systems known to the art such as applicators or eye droppers. Such compositions can include mucomimetics such as hyaluronic acid, chondroitin sulfate, hydroxypropyl methylcellulose or polyvinyl alcohol, preservatives such as sorbic acid, EDTA or benzylchromium chloride, and the usual quantities of diluents and/or carriers. For pulmonary administration, diluents and/or carriers will be selected to be appropriate to allow the formation of an aerosol.
Suppository forms of the compounds of the invention are useful for vaginal, urethral and rectal administrations. Such suppositories will generally be constructed of a mixture of substances that is solid at room temperature but melts at body temperature. The substances commonly used to create such vehicles include theobroma oil, glycerinated gelatin, hydrogenated vegetable oils, mixtures of polyethylene glycols of various molecular weight and fatty acid esters of polyethylene glycol. For example, see Remington's Pharmaceutical Sciences, 16th Ed., Mack Publishing, Easton, PA, 1980, pp. 1530-1533 for further discussion of suppository dosage forms. Analogous gels or creams can be used for vaginal, urethral and rectal administrations.
Numerous administration vehicles will be apparent to those of ordinary skill in the art, including without limitation slow release formulations, liposomal formulations and polymeric matrices.
Examples of pharmaceutically acceptable acid addition salts for use in the present invention include those derived from mineral acids, such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acids, and organic acids, such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic, p-toluenesulphonic and arylsulphonic acids, for example. Examples of pharmaceutically acceptable base addition salts for use in the present invention include those derived from non-toxic metals such as sodium or potassium, ammonium salts and organoamino salts such as triethylamine salts. Numerous appropriate such salts will be known to those of ordinary skill.
The physician or other health care professional can select the appropriate dose and treatment regimen based on the subject's weight, age, and physical condition. Dosages will generally be selected to maintain a serum level of compounds of the invention between about 0.01 μg/cc and about 1000 μg/cc, preferably between about 0.1 μg/cc and about 100 μg/cc. For parenteral
administration, an alternative measure of preferred amount is from about 0.001 mg/kg to about 10 mg/kg (alternatively, from about 0.01 mg/kg to about 10 mg/kg), more preferably from about 0.01 mg/kg to about 1 mg/kg (from about 0.1 mg/kg to about 1 mg/kg), will be administered. For oral administrations, an alternative measure of preferred administration amount is from about 0.001 mg/kg to about 10 mg/kg (from about 0.1 mg/kg to about 10 mg/kg), more preferably from about 0.01 mg/kg to about 1 mg/kg (from about 0.1 mg/kg to about 1 mg/kg). For administrations in suppository form, an alternative measure of preferred administration amount is from about 0.1 mg/kg to about 10 mg/kg, more preferably from about 0.1 mg/kg to about 1 mg/kg.
When introducing elements disclosed herein, the articles "a", "an", "the", and "said" are intended to mean that there are one or more of the elements. The terms "comprising", "having", "including" are intended to be open-ended and mean that there may be additional elements other than the listed elements.
The above disclosure generally describes the present invention. A more complete understanding can be obtained by reference to the following specific Examples. These Examples are described solely for purposes of illustration and are not intended to limit the scope of the invention. Changes in form and substitution of equivalents are contemplated as circumstances may suggest or render expedient. Although specific terms have been employed herein, such terms are intended in a descriptive sense and not for purposes of limitation.
Examples:
Methods of Preparation
Processes for the preparation of specific compounds of the present invention are described herein.
Throughout the following description of such processes it is to be understood that, where appropriate, suitable protecting groups will be added to, and subsequently removed from, the various reactants and intermediates in a manner that will be readily understood by one skilled in the art of organic synthesis. Conventional procedures for using such protecting groups as well as examples of suitable protecting groups are described, for example, in "Protective Groups in Organic Synthesis", T.W. Green, P. G. M. Wuts, Wiley- Interscience, New York, (1999). It is also to be understood that a transformation of a group or substituent into another group or substituent by chemical manipulation can be conducted on any intermediate or final product on the synthetic path toward the final product, in which the possible type of transformation is limited only by inherent incompatibility of other functionalities carried by the molecule at that stage to the conditions or reagents employed in the transformation. Such inherent incompatibilities, and ways to circumvent them by carrying out appropriate transformations and synthetic steps in a suitable order, will be readily understood to the one skilled in the art of organic synthesis. Examples of transformations are given below, and it is to be understood that the described transformations are not limited only to the generic groups or substituents for which the transformations are exemplified. References and descriptions on other suitable transformations are given in "Comprehensive Organic Transformations - A Guide to Functional Group Preparations" R. C. Larock, VHC Publishers, Inc. (1989). References and descriptions of other suitable reactions are described in textbooks of organic chemistry, for example, "Advanced Organic Chemistry", March, 4th ed. McGraw Hill (1992) or, "Organic Synthesis", Smith, McGraw Hill, (1994). Techniques for purification of intermediates and final products include for
example, straight and reversed phase chromatography on column or rotating plate, recrystallisation, distillation and liquid-liquid or solid-liquid extraction, which will be readily understood by the one skilled in the art. The definitions of substituents and groups are as in Formula I except where defined differently. The term "room temperature" and "ambient temperature" shall mean, unless otherwise specified, a temperature between 16 and 25 0C.
The term "reflux" shall mean, unless otherwise stated, in reference to an employed solvent a temperature at or above the boiling point of named solvent.
Abbreviations
atm Atmosphere
aq. Aqueous
BINAP 2,2'-bis(diphenylphosphino)-1 ,1 '-binaphthyl
Boc terf-butoxycarbonyl
CDI N.N'-Carbonyldiimidazole
DCC N.N-Dicyclohexylcarbodiimide
DCM Dichloromethane
DBU Diaza(1 ,3)bicyclo[5.4.0]undecane
DEA N,N-Diisopropyl ethylamine
DIBAL-H Diisobutylaluminium hydride
DIC N,N'-Diisopropylcarbodiimide
DMAP N,N-Dimethyl-4-aminopyridine
DMF Dimethylformamide
DMSO Dimethylsulfoxide
DPPF Diphenylphosphinoferrocene
EA Ethyl acetate
EDCI N-[3-(dimethylamino)propyl]-N'-ethylcarbodiimide hydrochloride
EDC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
EDTA Ethylenediaminetetraacetic acid
Et2O Diethyl ether
EtOAc Ethyl acetate
EtOH Ethanol
EtI lodoethane
Et Ethyl
Fmoc 9-fluorenylmethyloxycarbonyl
h hour(s)
HetAr Heteroaryl
HOBt N-Hydroxybenzotriazole
HBTU O-(Benzotriazol-1 -yO-N.N.N'.N'-tetramethyluronium hexafluorophosphate
HPLC High performance liquid chromatography
LAH Lithium aluminium hydride
LCMS HPLC mass spec
MCPBA m-Chloroperbenzoic acid
MeCN Acetonitrile
MeOH Methanol mm Min
MeI lodomethane MeMgCI Methyl magnesium chloride
Me Methyl n-BuLi 1-Butyllithium
NaOAc Sodium acetate
NMR Nuclear magnetic resonance
NMP N-Methyl pyrrolidinone nBuLi 1 -Butyl lithium o.n. Over night
RT Room temperature
TEA Triethylamine
THF Tetrahydrofuran nBu normal Butyl
OMs Mesylate or methane sulfonate ester
OTs Tosylate, toluene sulfonate or 4-methylbenzene sulfonate ester
PCC Pyridinium chlorochromate
PPTS Pyridinium p-toluenesulfonate
TBAF Tetrabutylammonium fluoride
pTsOH p-Toluenesulfonic acid
Second(s)
SEM Trimethyllsilylethoxymethyl
SPE Solid phase extraction (usually containing silica gel for mini- chromatography)
sat. Saturated
Example 1.1: Propiolamide

Methyl propioate (5 mL, 55 mmol) was added to ammonium hydroxide (15 ml_) at -50 to -60° C over 20 min. The reaction mixture was stirred at this temperature for 1 hour. The solvent was evaporated and the residue dried under vacuum to give the product (3.7 g, 92%). 1H NMR (300 MHz, CDCI3): δ (ppm) 6.35 (bs, 1H), 5.97 (bs, 1H), 2.88 (s, 1H).
Example 2.1: Ethyl 2-(2-ethoxy-2-oxoethyl)-6-oxo-1,6-dihydropyridine-3- carboxylate

Propiolamide (20.0 g, 289.6 mmol), diethyl 3-oxopentanedioate (87.8 g, 434.4 mmol) and sodium carbonate (24.6 g, 231.7 mmol) were mixed in water (800
ml_) at 0 °C and then warmed to RT over 4 h. The reaction was allowed to continue to stir at RT for 3 days. The reaction was neutralized with aqueous hydrochloric acid (5 M) at 0 °C with vigorous stirring. A solid precipitate was collected by filtration and washed with diethyl ether/hexanes (2:1) to yield a first batch of the title compound (43 g). The filtrate was further extracted with ethyl acetate and the combined organic phases were dried over sodium sulphate and purified by column chromatography (10-80% ethyl acetate/hexanes) to yield another batch of the title compound (7 g), in total gave 50 g (68%). 1H NMR (300 MHz, CDCI3): δ (ppm) 13.12 (br s, 1 H), 8.08 (d, 1 H), 6.52 (d, 1 H), 4.3 (q, 2H), 4.21 (q, 2H), 4.13 (s, 2H), 1.29 (m, 6H).
Example 3.1: Ethyl 2-(2-ethoxy-2-oxoethyl)-6-methoxynicotinate

The title compound from Example 2.1 (20 g, 79.05 mmol) was stirred with silver carbonate (23.7 g, 105.3 mmol) and iodomethane (40.7 g, 286.6 mmol) in chloroform (180 ml_) at 5O0C overnight. The reaction mixture was filtered and the filtrate was concentrated to give the crude title compound (22 g, quantitative). 1H NMR (300 MHz, CDCI3): δ (ppm) 8.21 (d, 1 H), 6.69 (d, 1H), 4.32 (q, 2H), 4.17 (m, 4H), 3.97 (s, 3H), 1.37 (t, 3H), 1.29 (t, 3H).
Example 3.2: Ethyl 2-(2-ethoxy-1-methyl-2-oxoethyl)-6- methoxynicotinate

To a suspension of sodium tert-butoxide (7.98 g, 82.3 mmol) in tetrahydrofuran (250 ml_) was added a solution of ethyl 2-(2-ethoxy-2- oxoethyl)-6-methoxynicotinate (20.0 g, 74.8 mmol) (the title compound from Example 3.1) in tetrahydrofuran (100 mL) at 0 0C over 15 min. After being stirred for 15 min, iodomethane (53.1 g, 374 mmol) was added at 0 0C. The reaction was allowed to warm up to RT over 2 h. Acetic acid (1 mL) was added at 0 °C and the mixture was stirred for 30 min. The reaction was diluted with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate, filtered and concentrated to give the title compound (20.16 g, 96%). 1H NMR (300 MHz, CDCI3): δ (ppm) 8.16 (d, 1 H), 6.64 (d, 1 H), 4.87 (q, 1 H), 4.33 (m, 2H), 4.13 (m, 2H), 3.94 (3H), 1.54 (d, 3H), 1.36 (m, 3H), 1.18 (m, 3H).
Example 4.1 : 2-(3-Hydroxymethyl-6-methoxy-pyridin-2-yl)-ethanol

To a suspension of lithium aluminum hydride (2.66 g, 70 mmol) in THF (120 mL) at 0° C was added 2-ethoxycarbonylmethyl-6-methoxy-nicotinic acid ethyl ester (5.26 g, 22 mmol). The reaction mixture was stirred at room temperature for 10 min and then at reflux for 80 min. The reaction mixture was cooled to 0° C and to it was successively added water (3 mL), aqueous
sodium hydroxide (15%, 3 ml_) and water (9 ml_). The resulting mixture was filtered and concentrated to give the product (3.68 g, 91.3%). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.57 (d, 1H), 6.63 (d, 1H), 4.63 (s, 2H), 4.07 (t, 2H), 3.92 (s, 3H), 3.02 (t, 2H).
Using the above general procedure, the following compound was synthesized:

Example 5.1: 2-(2-Chloro-ethyl)-3-chloromethyl-6-methoxy-pyridine

A mixture of 2-(3-hydroxymethyl-6-methoxy-pyridin-2-yl)-ethanol (3.68 g, 20 mmol), thionyl chloride (16 ml_) and chloroform (80 ml_) was stirred at room temperature overnight and then heated at reflux for 1 h. The reaction mixture was concentrated, diluted with ethyl acetate and washed with aqueous sodium carbonate containing ice. The organic layer was dried over sodium sulfate and concentrated. The residue was purified on silica gel using hexanes:ethyl acetate (10:0 to 9.5:0.5) to give the product (3.23 g, 73%). 1H
NMR (300 MHz, CDCI3): δ (ppm) 7.52 (d, 1 H), 6.62 (d, 1 H), 4.61 (s, 2H), 4.03 (t, 2H), 3.93 (s, 3H), 3.28 (t, 2H).
Example 6.1 : [2-(2-Chloro-ethyl)-6-methoxy-pyridin-3-yl]-acetonitrile

To a suspension of sodium cyanide (808 mg, 16.5 mmol) in N1N- dimethylformamide (20 mL) was added 2-(2-chloro-ethyl)-3-chloromethyl-6- methoxy-pyridine (2.88 g, 15 mmol). The reaction mixture was stirred at 25° C for 3 h. To the reaction mixture was added water and ethyl acetate. The organic layer was separated and washed further with water. The organic layer was dried over sodium sulfate and concentrated. The residue was purified on silica gel using hexanes: ethyl acetate (10:0 to 8:2) to give the product (2.4 g, 88%). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.56 (d, 1H), 6.67 (d, 1 H), 4.04 (t, 2H), 3.93 (s, 3H), 3.71 (s, 2H), 3.16 (t, 2H).
Example 7.1 : [2-(2-lodo-ethyl)-6-methoxy-pyridin-3-yl]-acetonitrile

To the crude mixture of [2-(2-chloro-ethyl)-6-methoxy-pyridin-3-yl]-acetonitrile (875 mg, 4.8 mmol), sodium iodide (5.0 g, 33.3 mmol) and acetone (10 mL) was heated at 58° C for 2 days. The reaction mixture was concentrated, diluted with dichloromethane, filtered and concentrated once again to give the product (1.30 g). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.55 (d, 1 H), 6.66 (d, 1 H), 3.93 (S, 3H), 3.68 (s, 2H), 3.63 (t, 2H), 3.3 (t, 2H).
Example 8.1 : [2-(2-Benzylamino-ethyl)-6-methoxy-pyridin-3-yl]- acetonitrile

A solution of [2-(2-iodo-ethyl)-6-methoxy-pyridin-3-yl]-acetonitrile (7.9 g, 29 mmol) in butanol (40 ml_) was added to a mixture of benzylamine (11 ml_, 0.1 mol) in butanol (80 ml_) at 90° C. The reaction mixture was stirred at 100° C for 1 hour. The reaction mixture was concentrated, diluted with ethyl acetate and washed with aqueous sodium carbonate. The organic layer was dried over sodium sulfate, concentrated and purified on silica gel using dichloromethane:2M NH3 in methanol (10:0 to 9.5:0.5) to give the product (5.2 g, 90% pure). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.54 (d, 1 H), 6.62 (d, 1 H), 3.86 (m, 5H), 3.66 (s, 2H), 3.13 (t, 2H), 2.91 (t, 2H).
Example 9.1: [2-(2-Benzylamino-ethyl)-6-methoxy-pyridin-3-yl]-acetic acid methyl ester

A mixture of [2-(2-benzylamino-ethyl)-6-methoxy-pyridin-3-yl]-acetonitrile (5.2 g) and a solution of hydrochloric acid in methanol (12%, 150 ml_) was heated at reflux overnight. The reaction mixture was cooled to room temperature and sodium bicarbonate was then added. After stirring for 30 min, methanol was evaporated. To the residue was added ethyl acetate and water. The aqueous layer was separated and further extracted with ethyl acetate. The
combined organic layer was dried over sodium sulfate and concentrated to give the product (4.85 g). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.27 (m, 6H), 6.56 (d, 1 H), 3.86 (m, 5H), 3.67 (m, 3H), 3.59 (s, 2H), 3.09 (t, 2H), 2.94 (t, 2H).
Example 10.1 : 7-Benzyl-2-methoxy-5,7,8,9-tetrahydro-pyrido[2,3- d]azepin-6-one

A mixture of [2-(2-benzylamino-ethyl)-6-methoxy-pyridin-3-yl]-acetic acid methyl ester (1.6 g, 5.4 mmol) in N,N-dimethylformamide (18 ml_) was heated at 200° C in a microwave for 4 h. The above reaction was repeated using 3.2 g of substrate. The two reaction mixtures were combined, diluted with ethyl acetate and washed with water. The organic layer was dried over sodium sulfate, concentrated and purified on silica gel using hexanes:ethyl acetate (9:1 to 1 :1 ) to give the product (2.0 g, 47%). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.32 (m, 6H), 6.54 (d, 1 H), 4.67 (s, 2H), 3.86 (s, 5H), 3.7 (t, 2H), 2.99 (t, 2H).
Example 11.1 : 7-Benzyl-2-methoxy-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine

To a suspension of lithium aluminum hydride (1.9 g, 50 mmol) in tetrahydrofuran (60 ml_) at -78° C, was added to aluminum chloride (2.4 g, 18
mmol) followed by a solution of 7-benzyl-2-methoxy-5,7,8,9-tetrahydro- pyrido[2,3-d]azepin-6-one (1.6 g, 6 mmol) in tetrahydrofuran (40 mL). The reaction mixture was stirred at room temperature for 2 days. To the reaction mixture at 0° C was slowly and successively added water (2 mL), aqueous sodium hydroxide (1 N, 2 mL) and water (4 mL). The resulting mixture was filtered through Celite® and concentrated. The above reaction was repeated using 422 mg of substrate. The combined residue was purified on silica gel using dichloromethane:2M NH3 in methanol (10:0 to 9.5:0.5) to give the product (1.8 g, 95%). The product was treated with hydrochloric acid in diethyl ether to give the hydrochloric acid salt (2 salt equivalents). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.31 (m, 6H), 6.48 (d, 1 H), 3.9 (s, 3H), 3.65 (s, 2H), 3.07 (m, 2H), 2.82 (m, 2H), 2.68 (m, 2H), 2.61 (m, 2H).
Example 12.1: 2-[3-(Chloromethyl)-6-methoxypyridin-2-yl]ethyl methanesulfonate

The title compound from Example 3 (24.70 g, 134.8 mmol) was dissolved in dichloromethane (500 mL) under a nitrogen atmosphere and cooled to -30 0C. Then triethylamine (30.01 g, 296.6 mmol) and methyl sulfonyl chloride (33.98 g, 296.6 mmol) were added. The reaction was allowed to warm to RT and was stirred overnight. The reaction mixture was diluted with hexanes (400 mL) and filtered to remove solids. The filtrate was concentrated and redissolved in ethyl acetate, washing with saturated sodium bicarbonate solution and brine. The organic layer was dried over magnesium sulfate,
filtered and concentrated to give the title compound as pale yellow oil (36.38 g, 96%). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.51 (d, 1 H), 6.60 (d, 1 H)1 4.75 (t, 2H), 4.57 (s, 2H), 3.90 (s, 3H), 3.25 (t, 2H), 2.92 (s, 3H).
Using the above general procedure, the following compound was synthesized:

Example 13.1 : 2-[3-(Cyanomethyl)-6-methoxypyridln-2-yl]ethyl methanesulfonate

The title compound from Example 4 (36.38 g, 130.05 mmol) was dissolved in dimethylformamide (400 ml_) and cooled to 0 0C. Sodium cyanide (6.69 g, 135.55 mmol) was added and the reaction mixture was allowed to warm up to RT overnight. The reaction mixture was filtered through celite filtering agent.
The filtrate was washed with 25% saturated sodium bicarbonate solution and extracted with portions of ethyl acetate. The organic extracts were dried over sodium sulphate, filtered and concentrated. The product was purified by column chromatography (30-50% ethyl acetate in hexanes) to give the title compound (21.13 g, 60%). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.48 (d, 1 H), 6.57 (d, 1 H), 4.65 (t, 2H), 3.84 (s, 3H), 3.62 (s, 2H), 3.07 (t, 2H), 2.90 (s, 3H).
Using the above general procedure, the following compound was synthesized:

Example 15.1: 2-Methoxy-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine
Method A
A mixture of 7-benzyl-2-methoxy-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine (1.05 g, 4.2 mmol of the compound from Example 11.1), palladium hydroxide (20% on carbon, 180 mg) and methanol (50 ml_) was stirred under hydrogen (36 psi) for 6 h. The reaction mixture was filtered through Celite® and concentrated to give the product (565 mg). The product was treated with hydrochloric acid in diethyl ether to give the hydrochloric acid salt (2 salt
equivalents). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.29 (d, 1 H), 6.48 (d, 1 H), 3.91 (s, 3H), 3.08 (m, 2H), 2.98 (m, 4H), 2.82 (m, 2H).
Method B To a mixture of aluminum chloride (1.8 g, 13.3 mmol) and lithium aluminum hydride (1.0g, 26.6 mmol) at -78 0C, dry diethyl ether (30 ml_) was added carefully. After being stirred at RT for 30 min, the reaction mixture was cooled down to -78 0C again. Then a solution of 2-[3-(cyanomethyl)-6- methoxypyridin-2-yl]ethyl methanesulfonate (3.6 g, 13.3 mmol of the compound from Example 13.1 ) in tetrahydrofuran (30 ml_) was added slowly. The reaction mixture was stirred at 0 0C for an hour and a half. Water (200 ml_) and saturated sodium carbonate (10 ml_) were used to quench the reaction. The reaction mixture was extracted with ethyl acetate, dried with sodium sulfate, concentrated. The residue was mixed with diisopropylethylamine (3.36 g, 26 mmol) in acetonitrile (45 ml) and stirred at 3O0C for 24 h, concentrated again, saturated sodium carbonate (15 ml_) was added. The mixture was extracted with ethyl acetate, dried, purified by chromatography to give the title compound (1.25g, 52.7%).
Using the Method B general procedure, the following compound was synthesized:


To a solution of the title compound Example 13.1 (1.07 g, 6 mmol) and diisopropylethylamine(1.5 mL) in dichlromethane (30 ml.) at -5O0C, di-tert- butyl dicarbonate (1.6 g, 7.2 mmol) was added. The reaction mixture was stirred at room temperature for two h, then quenched with water and extracted with dichloromethane. The product was purified by column chromatograph with 10% ethyl acetate in hexanes to give the title compound 1.188g (71%).
Example 16.2: tert-butyl 3-bromo-2-methoxy-5,6,8,9-tetrahydro-7H- pyrido[2,3-d]azepine-7-carboxylate

tert-Butyl 2-methoxy-5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate (140 mg, 0.5 mmol) was mixed with sodium acetate (50 mg, 0.6 mmol) in dichloromethane at O0C. Then bromine (96 mg, 0.6 mmol) was added, the reaction mixture was filtered and concentrated to dry to give the title compound 200 mg which can be carried on for the next step reaction without further purification. . 1H NMR (300 MHz, CDCI3): δ (ppm) 7.55 (s, 1 H), 3.99 (S, 3H), 3.58 (m, 4H), 3.04 (m, 2H), 2.78 (m, 2H), 1.49 (s, 9H).
Example 17.1 : 6,7,8,9-Tetrahydro-5H-pyrido[2,3-d]azepin-2-ol dihydrobromide

A mixture of of tert-Butyl 2-methoxy-5,6,8,9-tetrahydro-7H-pyrido[2,3- d]azepine-7-carboxylate, example 16.1 , (549 mg, 3.3 mmol), hydrobromic acid in ethanol (16 ml_) and acetic acid (16 ml_) was heated at 880C for 1 day. The reaction mixture was concentrated and the residue was triturated with hexanes to give the product (879 mg).
Using the above, the following compounds were synthesized:
Example Structure Name Yield
Λ 9-Methyl-6,7,8,9- Not taken
N H-Br tetrahydro-5H-pyri
Example HO-Ηr do[2,3-d]azepin-2-
17 O ol; hydrobromide
NMR 1H NMR (300 MHz, CDCI3) : δ (ppm) not characterized
Λ 3-bromo-6,7,8,9- Not taken
NH H-Br tetrahydro-5H-
Example pyrido[2,3-
17.3 d]azepin-2-ol; hydrobromide
NMR 1H NMR (300 MHz, CDCI3) : δ (ppm) not characterized
Example 19.1 : tert-Butyl 2-hydroxy-5,6,8,9-tetrahydro-7H-pyrido[2,3- d]azepine-7-carboxylate

The title compound from Example 17.1 (5.72 g, 17. 54 mmol) was suspended in dichloromethane (90 ml_) under a nitrogen atmosphere and cooled to 00C. Diisopropylethyamine (7.93 g, 61.39 mmol) was added to the suspension with stirring. In a separate flask, di-tert-butyl dicarbonate (8.04 g, 36.83 mmol) was dissolved in dichloromethane (50 ml_) under a nitrogen atmosphere. This solution was added slowly to the main reaction vessel via cannula. The reaction was stirred at RT for 2 h. The reaction mixture was washed with a 50% saturated solution of ammonium chloride and the aqueous phase was extracted with portions of dichloromethane. The combined organic extracts were washed with brine, dried over manesium sulfate, filtered and concentrated to give the title compound as a plae brown solid (4.64 g, quant.). 1H NMR (300 MHz, CDCI3): δ (ppm) 13.53 (br s, 1 H), 7.25 (d, 1 H), 6.38 (d, 1 H), 3.55 (m, 4H), 2.94 (m, 2H), 2.68 (m, 2H), 1.46 (s, 9H).
Using the above general procedure, the following compounds were synthesized:


Example 20.1 : tert-Butyl 2-{[(tιϊfluoromethyl)sulfonyl]oxy}-5,6,8,9- tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate

The title compound from Example 19.1 (4.64 g, 17.54 mmol) was dissolved in dichloromethane (150 ml_) and cooled to 0 °C under nitrogen. To this solution was added diisopropylethylamine (2.83 g, 21.93 mmol) and triflic anhydride (6.19 g, 21.93 g) and the reaction was allowed to warm to RT over 2 h. The reaction mixture was concentrated and partitioned between ethyl acetate and water. The organic phase was washed with 50% sat. ammonium chloride and brine. The organic phase was neutralized with sat. sodium bicarbonate and concentrated to give the title compound (1.94 g, 28%). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.59 (d, 1 H)1 6.94 (d, 1 H), 3.61 (br d, 4H), 3.12 (t, 2H), 2.93 (t, 2H), 1.49 (s, 9H).

Using the above general procedure, the following compounds were synthesized:
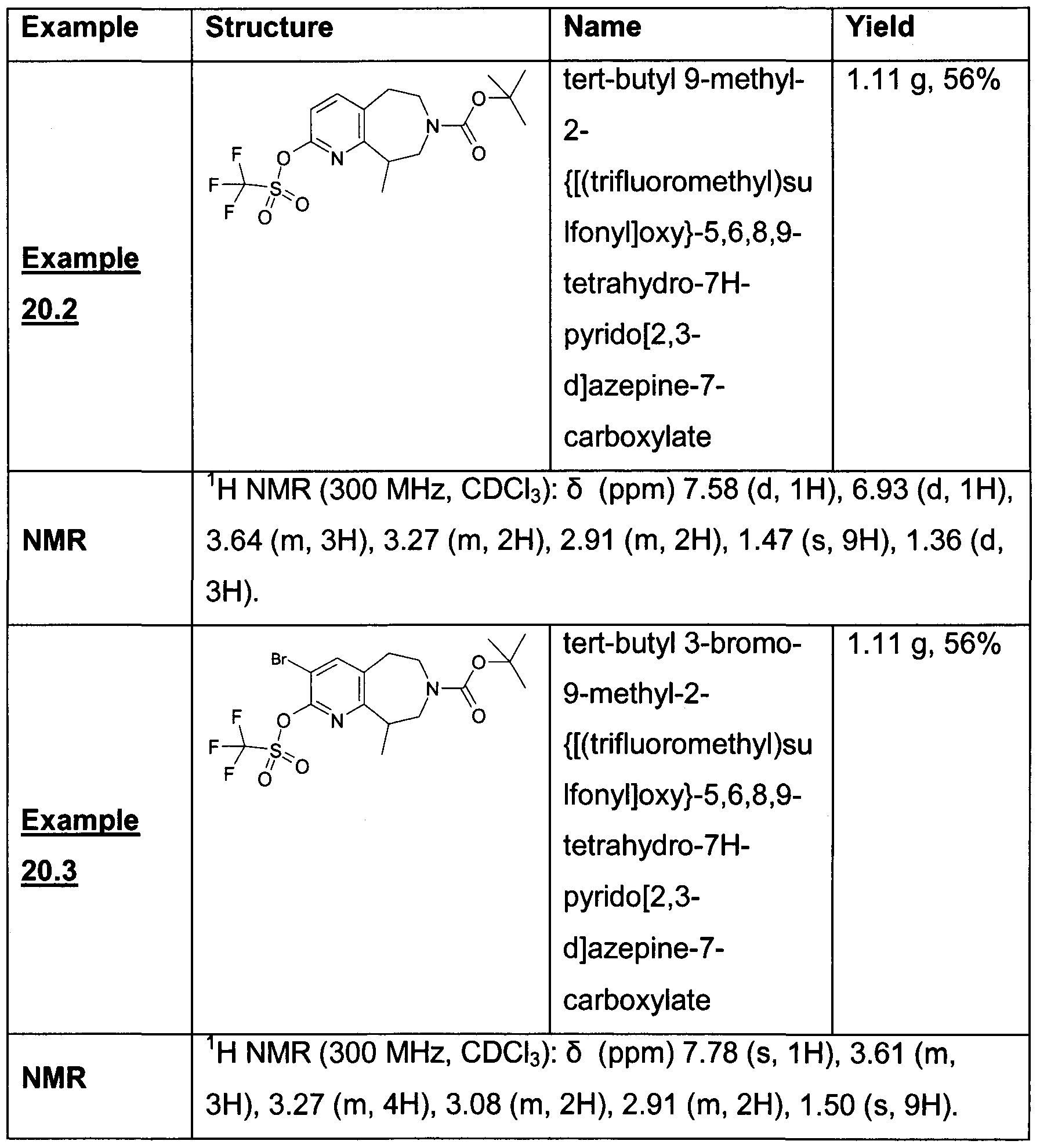

To a solution of 7-benzyl-2-methoxy-6>7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine (500 mg, 1.86 mmol) in toluene (8 mL) at -580C, n-BuLi (1.5 mL of 2.5 M in hexanes, 3.75 mmol) was aaded. After the mixture was stirred at O0C for two h, iodoethane (650 μl_, 8 mmol) was added and the reaction mixture was stirred at room temperature overnight. The reaction mixture was quenched with saturated ammonium chloride and extracted with ethyl acetate, dried and concentrated by Rotavapor. The residue was mixed with 20% Pd(OH)2 (400 mg) and methanol (30 mL) and stirred under H2 for a week. The reaction mixture was filtered and the filtrate was concentrated again to give 9- ethyl-2-methoxy-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine. This compound was mixed with 20%wt/v HBr in ethanol (16 mL) and acetic acid (16 mL) and heated at 90 0C overnight and then concentrated to give 9-ethyl-6, 7,8,9- tetrahydro-5H-pyrido[2,3-d]azepin-2-ol dihydrobromide salt. The salt was mixed with diisopropylethyamine (2.5 mL) and di-tert-butyl dicarbonate (570 mg, 2.5 mmol) in dichloromethane (20 mL) and water (10 mL) at 0 0C and stirred for two h. The organic layer was separated and dried, concentrated to give tert-butyl 9-ethyl-2-hydroxy-5)6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7- carboxylate. This intermediate was mixed with diisopropylethyalmine (1 mL) and triflic anhydride (420 μL, 2.5 mmol) in dichloromethane at -50 0C and stirred overnight. After work-up, the compound was purified by column chromatography with 20% ethyl acetate in hexanes to give the title compound
284 mg (37% in total). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.58 (d, 1 H), 6.93 (d, 1 H), 2.6-3.9 (m, 7H), 1.95 (m, 2H), 1.49 (s, 9H), 1.37 (m, 3H).
Using the above general procedure, the following compounds were synthesized:

Coupling of amines to triflates:
The above title triflates (1 equivalent) and excess of amines (at least 2 equivalents) dissolved in dimethyl sulfoxide were heated to 120-15O0C in a microwave for 30 min. The reaction mixtures were diluted with water and extracted with ethyl acetate, dried and concentrated. The coupling product was purified by column chromatography to give the title compounds.
Using the above general procedure, the following compounds were synthesized:










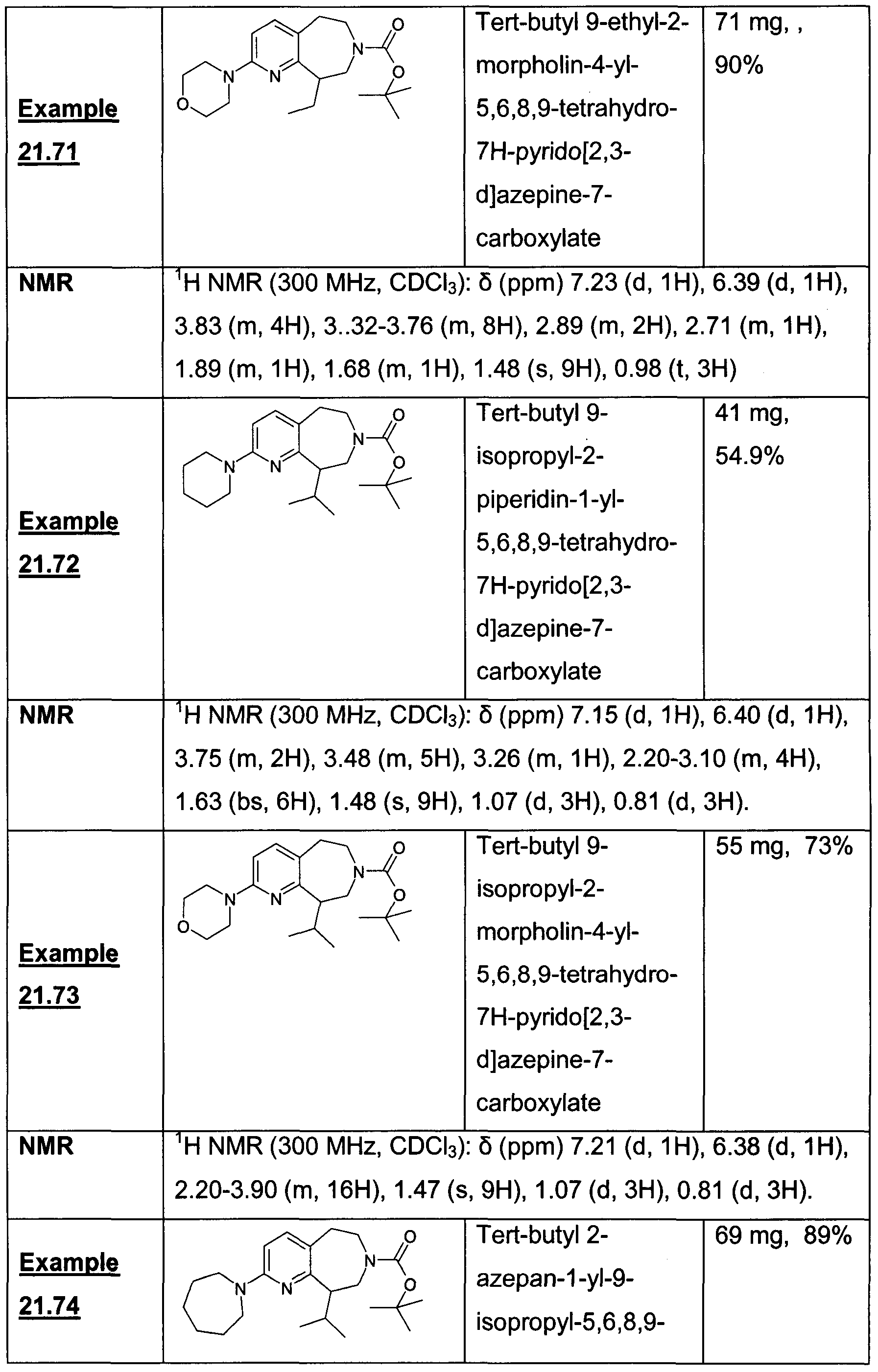
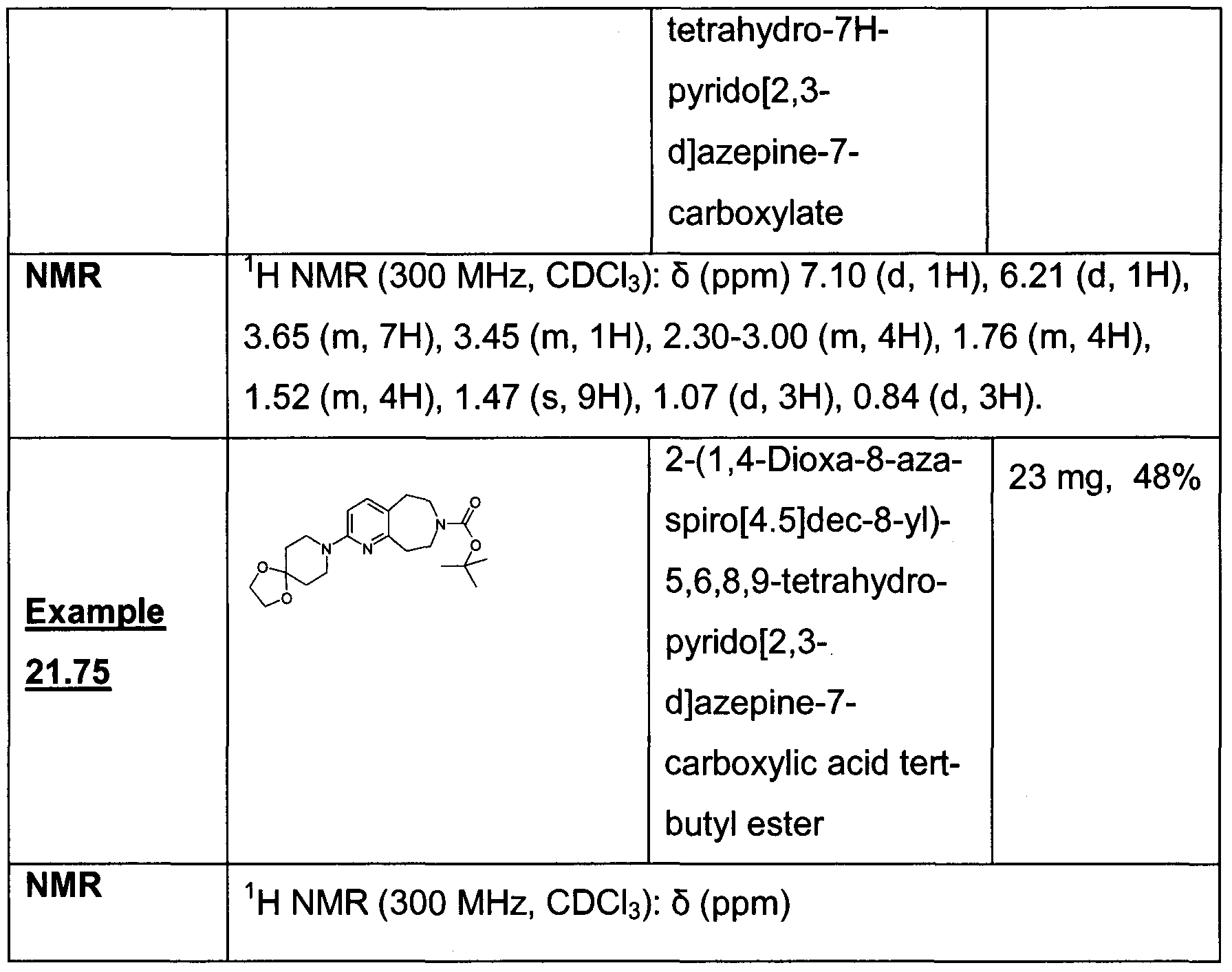
Oxidation of Example 21.45 to its sulfoxide and sulfones derivatives:
Example 21.77: tert-Butvl 2-(1 -oxidothiomorpholin-4-yl)-5,6,8,9-tetrahydro- 7H-pyrido[2,3-d]azepine-7-carboxylate

To a solution of NaIO4 (132.6 mg, 0.62 mmol) in water (2mL) at 0 0C, tert-butyl 2-(thiomorpholin-4-yl)-5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7- carboxylate (143.3 mg, 0.41 mmol) in methanol (4ml_) and DMF (4ml_) was added, the reaction mixture was stirred at 0 0C for 24 h and then filtered. The filtrate was extracted with dichloromethane and concentrated. The residue was purified by column chromatography to give the title compound 116.9 mg (78%). 1H NMR (300 MHz, CDCI3): δ (ppm) 7.23 (d, 1 H), 6.46 (d, 1 H), 4.10 (m, 4H), 3.52 (m, 4H), 2.96 (m, 2H), 2.75 (m, 6H), 1.45 (s, 9H).
Example 21.50: tert-butvl 2-(1.i-dioxidothiomorpholin^-yO-δ.β.δ.θ- tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate

From the same above protocol, 10 mg (21%) was obtained. 1H NMR (300 MHz, CDCI3): δ (ppm) 7.29(d, 1 H), 6.62 (d, 1 H), 4.14 (m, 4H), 3.57 (m, 4H), 3.03 (m, 6H), 2.78 (m, 2H), 1.50 (s, 9H).
Chlorination with N-Chlorosuccinimide:
Example 21.78: Tert-butvl 3-chloro-2-piperidin-1-yl 5,6,8,9-tetrahydro-7H- pyrido[2,3-d]azepine-7-carboxylate

In a similar manner the following compounds were synthesized:

1H NMR (300 MHz, CDCI3): δ (ppm) 7.32 (s, 1 H), 4.82 (dm, 1 H),
NMR 3.47 (m, 6H), 3.25 (m, 2H), 3.02 (m, 2H), 2.76 (m, 2H), 1.96 (m,
4H), 1.49 (s, 9H).
Tert-butyl 3- 17.9 chloro-2-(1 ,4- mg,
PT oxazepan-4- 60% yl)-5,6,8,9-
Example o T y \ tetrahydro-
21.82 7H-pyrido[2,3- d]azepine-7- carboxylate
1H NMR (300 MHz, CDCI3): δ (ppm) 7.27 (s, 1 H)1 3.86 (m, 4H),
NMR 3.70 (m, 4H), 3.55 (m, 4H), 2.97 (m, 2H), 2.73 (m, 2H), 2.05 (m,
2H), 1.49 (s, 9H).
Chiral separation:
Example 21.84: (9R)- and (9S)-tert-butyl 9-methyl-2-piperidin-1-yl-5,6,8,9- tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate were separated from racemic were separated from racemic tert-butyl 2-[ethyl(methyl)amino]-9- methyl-5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate by Chiralcel OJ with 1 % ethanol in hexanes by Chiralcel OJ with 1% ethanol in hexanes.

Example 21.85: (9R)- and (9S)-tert-butyl 9-methyl-2-morpholin-4-yl-5,6,8>9- tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate
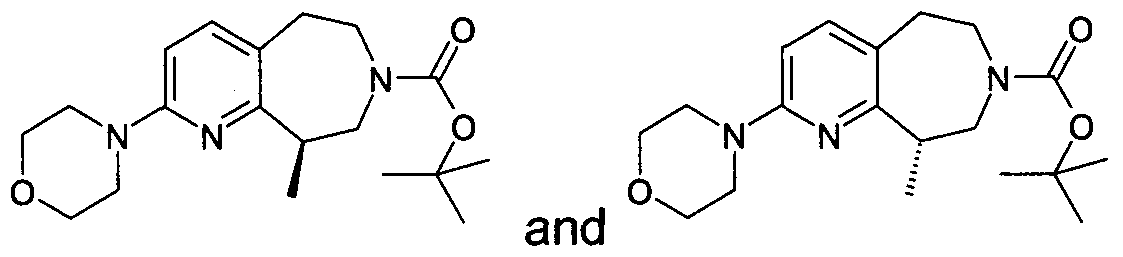
Example 21.86: (9R)- and (9S)-2-(4-fluoropiperidin-1-yl)-9-methyl-5,6,8,9- tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate

The (9R)-2-(4-fluoropiperidin-1-yl)-9-methyl-5)6,8,9-tetrahydro-7H-pyrido[2,3- d]azepine-7-carboxylate and the (S)-2-(4-fluoropiperidin-1-yl)-9-methyl- 5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate (respectively, Rt= 7.51 min, 11 mg; Rt=9.68 min, 8.7mg) were separated from racemic 2-(4- fluoropiperidin-1-yl)-9-methyl-5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7- carboxylate by CH I RALPACK AD-H with 2.5% EtOH in Hexanes.
Example 21.87: (9R)- and (9S)-2-(4-fluoropiperidin-1-yl)-9-methyl-5,6,8,9- tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate

The (9R)-2-(4-fluoropiperidin-1-yl)-9-methyl-5,6,8,9-tetrahydro-7H-pyrido[2l3- d]azepine-7-carboxylate and the (S)-2-(4-fluoropiperidin-1-yl)-9-methyl- 5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate (respectively, Rt= 5.12 min, 12.8 mg; Rt=6.62 min, 13 mg) were separated from 2-(4- fluoropiperidin-1-yl)-9-methyl-5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7- carboxylate by CHIRALPACK AD-H with EtOH in Hexanes.
Example 21.88: (9R)- and (9S)-tert-butyl 9-methyl-2-(1 ,4-oxazepan-4-yl)- 5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate

(9R)-tert-butyl 9-methyl-2-(1 )4-oxazepan-4-yl)-5,6,8,9-tetrahydro-7H- pyrido[2,3-d]azepine-7-carboxylate and (9S)-tert-butyl 9-methyl-2-(1 ,4- oxazepan-4-yl)-5,6,8,9-tetrahydro-7H-pyrido[2,3-d]azepine-7-carboxylate (respectively, Rt=10.2 min, 10 mg; Rt= 14.4 min, 10 mg) were separated from racemic tert-butyl 9-methyl-2-(1 ,4-oxazepan-4-yl)-5,6,8,9-tetrahydro-7H- pyrido[2,3-d]azepine-7-carboxylate by CHIRALPACK AD-H with 2.5% EtOH in Hexanes.
Cleavage of Boc protecting group
Method A: (TFA-DCM)
To a solution of the Boc protected amine intermediates in dichloromethane (1 ml_) at O0C, was added trifluoroacetic acid (0.5 ml_). The reaction mixture was stirred at O0C for 2 h. The reaction mixture was concentrated, diluted with ethyl acetate and washed with aqueous sodium carbonate. The organic layer was dried over sodium sulfate and concentrated to give the product. The product was treated with hydrochloric acid in diethyl ether to give the hydrochloric acid salt (2 salt equivalents).
Method B: (HCI in Et2O) A solution of the Boc protected amine intermediates in dichloromethane (2 ml_) and 2M hydrochloric acid in diethyl ether (3 mL) for 6 h. The reaction was concentrated to give the title compound.
In a similar manner the following compounds were synthesized:
Example Structure Name Method Yield
Example 2-pyrrolidin-1-yl- 42 22.27 6,7,8,9-tetrahydro- mg,
 5H-pyrido[2,3- 65% d]azepine
5H-pyrido[2,3- 65% d]azepine
NMR Η NMR (300 MHz, CDCI3): δ (ppm) 7.16 (d, 1H), 6.10 (d, 1H), 3.43 (m, 6H), 2.50-3.30 (m, 7H), 1.97 (m, 4H).
Example 2-piperidin-1-yl- 13 22.28 6,7,8,9-tetrahydro- mg, 5H-pyrido[2,3- 19%
 d]azepine
d]azepine
NMR 1H NMR (300 MHz, CDCI3): δ (ppm) 7.21 (d, 1 H), 6.45 (d, 1 H), 3.50 (m, 4H), 3.27 (m, 7H), 3.00 (m, 2H), 1.64 (br, 6H).
Example 9-methyl-2- B 61 22.29 piperidin-1-yl- mg,
6,7,8,9-tetrahydro- 96%
 5H-pyrido[2,3- d]azepine dihydrochloride
5H-pyrido[2,3- d]azepine dihydrochloride
NMR 1H NMR (300 MHz, MeOD): δ (ppm) 7.83 (d, 1 H), 7.25 (d, 1 H), 3.78 (m, 5H), 3.50 (m, 3H), 3.20 (m, 3H), 1.79 (br, 6H), 1.55 (d, 3H).
Example (9R)-9-methyl-2- B 42.5 22.30 piperidin-1-yl- mg
6,7,8,9-tetrahydro-
 5H-pyrido[2,3- d]azepine dihydrochloride
5H-pyrido[2,3- d]azepine dihydrochloride



Example 9-methyl-2- B 59m
22.42 i NH morpholin-4-yl- g
6,7,8,9-tetrahydro-
H-Cl 5H-pyrido[2,3- 93% H-Cl d]azepine dihydrochloride
NMR 1H NMR (300 MHz1 MeOD): δ (ppm) 7.90 (d, 1 H) , 7.25 (d, 1 H),
3.87 (m, 4H), 3.74 (m, 4H), 3.53 (m, 3H), 3.30 (m , 4H), 1.56 (d,
3H).
Example (9R)-9-methyl-2- B 47.5
22.43 morpholin-4-yl- mg
6,7,8,9-tetrahydro-
 5H-pyrido[2,3- 93% H-Cl d]azepine dihydrochloride
5H-pyrido[2,3- 93% H-Cl d]azepine dihydrochloride
Example (9S)-9-methyl-2- B 48.2
22.44 morpholin-4-yl- mg
6,7,8,9-tetrahydro-
 5H-pyrido[2,3- 93% H-Cl d]azepine dihydrochloride
5H-pyrido[2,3- 93% H-Cl d]azepine dihydrochloride
Example 9-ethyl-2- B 55
22.45 morpholin-4-yl- mg
6,7,8,9-tetrahydro-
 5H-pyrido[2,3- 82%
5H-pyrido[2,3- 82%
H Pl d]azepine dihydrochloride
NMR 1H NMR (300 MHz1 MeOD): δ (ppm) 7.93 (d, 1 H) 7.27 (d, 1 H),
3.87 (m, 4H), 3.05-3.80 (m, 11 H), 1.96 (m, 2H), 1 .08 (t, 3H).












Evaluation of Biological activity
Demonstration of the activity of the compounds of the present invention may be accomplished through in vitro, ex vivo and in vivo assays that are well known in the art, including the assays described in the following examples.
MATERIALS AND METHODS
Activation of the Gq coupled 5-HT2 receptors stimulates phospholipase C activity and leads to formation of inositol trisphosphate (IP3) and the subsequent release of calcium from intracellular stores. Functional activity of Gq coupled receptors can be quantified in a FLIPR assay by measuring intracellular calcium levels with calcium sensitive dyes (using a fluorescence imaging plate reader, FLIPR) and in a Phosphatidyl Inositol Hydrolysis Assay (IP accumulation assay) which measures IPs derived from IP3. Both assays provide robust functional readouts of receptor activation.
Cell Culture: Stable cell lines expressing human 5-HT2A, 5-HT2B and 5-HT2C (both INI and VSV isoforms) receptors were created in an MHEK cell background (an HEK293-based cell background which also expresses the
Macrophage Scavenger Receptor 1 , to increase the adherence of cells to
tissue .culture plates). Recombinant cell lines were cultured in Growth Medium (High glucose DMEM (Hyclone) with 10% dialyzed fetal bovine serum (Hyclone), and L-glutamine (Gibco; 0.8mM for 5-HT2A and 5-HT2c. 2.0 mM for 5-HT2B), and grown under selection with 200 μg/ml Zeocin (Invitrogen), and either 200 μg/ml Hygromycin B (Invitrogen for 5-HT2A and 5-HT2c) or 500 ug/ml Geneticin (Invitrogen) for 5-HT2B.
FLIPR assay methodology: Cells that recombinantly expressed the 5-HT2 receptors were enzymatically dissociated with Trypsin/EDTA 0.25% (Hyclone) 24 h prior to testing, and seeded at 60,000 cells per well in 100 μl Growth Medium in black sided, clear bottom 96 well plates (Greiner, BioExpress) at 370C and 5% CO2. On the day of the assay, Growth Medium was removed by aspiration, and 80 μl of Assay Buffer (2OmM HEPES, 146 mM Sodium Chloride, 5 mM Potassium Chloride, 1 mM Magnesium Chloride, 1 mg/mL BSA, 1 mg/mL Glucose, 1 mM CaCI2, pH 7.4, supplied by Amresco), containing 6 μM Fluo-3 AM and 0.01% pluronic acid (Biotium Inc., Hawyard, CA) was added. Cells were incubated in the Fluo-3 solution for 60 min in the dark at room temperature. The Fluo-3 solution was then removed by aspiration and cells were washed twice with assay buffer leaving 160 μl in each well.
All compounds were prepared at 5 times their final concentration prior to the online addition (40 μl) in the FLIPR (MDS Inc., Sunnyvale, CA). The fluorescent intensity was measured at 1 second intervals for 10 seconds prior to the compound addition and 65 seconds after the compound addition. All responses were measured as the peak height of the fluorescent response over baseline, within the sample period. Non-linear regression of the relative fluorescence unit (RFU) change was used to determine agonist potency. Antagonist activity was measured after pre-incubation of cells with compound for 30 min at RT, followed by the online addition of agonist (5-HT, EC80) in the FLIPR. Antagonist activity was determined by normalizing the response to the maximal 5-HT response in the absence of test compound.
Phosphatidyl Inositol Hydrolysis Assay: 24 h prior to testing, cells were plated in poly-D-Lysine-coated 96 well plates (VWR) at 100,000 cells/ well in 200 μl culture medium containing 10 μCi/ml of [3H]-myo-lnositol (Perkin Elmer). Cell monolayers were washed twice with HBSS (HEPES Buffered Saline solution: 20 mM HEPES1 146 mM NaCI, 4.2 mM KCI, 0.5 mM MgCI2, 0.1 % Glucose, pH 7.4). The cell monolayers were pre-incubated for 5 min at 37°C in 100 μl/well HBSS containing 10 mM LiCI. Compounds were tested for agonist activity in duplicate at concentrations ranging from 3nM to 30 μM. Compounds were added (100 μl) at 2 times the required final concentration and incubated for 30 min at 37°C. Medium was aspirated and the soluble 3H- inositol phosphates were extracted from the cells by adding 100 μl/well of ice- cold 5% perchloroacetic acid solution. Plates were placed on ice for 1 hour, and extracts collected in a 2ml, 96 well, polypropylene, round bottom Uniplate (VWR). Cell extracts were neutralized with 150-170μl HEPES / KOH (0.375 / 0.75 M) containing a pH indicator until all solutions turned pale green. 600μl HEPES / EDTA (2.5/0.5 mM, pH 7.4) was then added to all tubes, and contents were transferred to a 96 well PALL Filter Plate (VWR) loaded with 600 μl/well of Dowex resin (Dowex AG-1X8 formate form, 200-400 mesh, Bio- Rad, equilibrated in HEPES / EDTA (2.5/0.5 mM, pH 7.4). The Filter Plate was then placed in a vacuum manifold and a gentle vacuum was applied. Total phosphatidyl inositols were eluted with 800 μl 30 mM ammonium formate, and the eluate was discarded. Total inositol phosphates were eluted with 600 μl (2X 300 μl) 700 mM ammonium formate / 100 mM formic acid and collected in a clean 2ml, 96 well, polypropylene, round bottom Uniplate. 75μl eluate was transferred to a Hewlett Packard Optiplate and 150 μl Scint 40 was added to each well. The plate was sealed with a Topseal (Packard) and shaken for 1 minute on a platform plate shaker. Plates were counted in the Hewlett Packard Topcount to quantify the amount of radioactivity in each well.
Cytochrome P450 Assay:
In-vitro screening for drugs that inhibit cytochrome P450 enzymes is well established as a means for predicting potential metabolism related drug interactions in-vivo. Pooled human liver microsomes were acquired from a commercial supplier (Human Biologies International, Scottsdale, AZ, USA) and stored at -700C until use. Exact assay conditions (e.g. substrate for the given CYP isoenzyme and positive control inhibitor) varied according to CYP isoenzyme under study (see summary table below). The metabolism reaction is started by the addition of substrate. Final concentration of substrate was measured by HPLC. The extent of metabolism in percentage of a given substrate for the given CYP assay is calculated from the extent of metabolism of the given substrate in the presence test compound compared to incubation of the substrate without test compound. Each assay uses as a positive control an inhibitor with known inhibitory activity for the metabolism of the given substrate in the particular CYP assay, which is used to determine the validity of the given assay. The data shown for the compounds of the present invention in Table 2 are results from assays in which the positive control gave appropriate results for the given CYP assay.

2C19 Inhibition assay
Substrate: S-Mephenytoin Inhibitor: Omeprazole
Reaction conditions: Incubation conditions:
Time: 35 min Human Microsomal protein 50 μl
Temperature: 37°C Substrate (0.1 mg/ml in Acetonitrile) 10 μl
Protein Concentration: 500 μg Inhibitor or test compound 10 μl
Incubation Volume: 2 ml NADPH regenerating system 50 μl
Inhibitor Concentration: 10 μM (omeprazole) or Buffer 890 the indicated for test compound (1 or 10 μM) μl
Extraction procedure:
Quench with 5 ml ethyl acetate
Spike samples with internal standard (tolbutamide 5 μg/ml, 100 μl)
Shake on Vibrax shaker for 10 min
Centrifuge at 3000 rpm, 4°C for 10 min
Transfer 4 ml of upper layer to fresh tubes and evaporate to dryness under stream of nitrogen
(waterbath at 500C) for about 25 minutes
Reconstitute samples in 200 μl mobile phase and inject onto HPLC column.

2D6 Inhibition assay
Substrate: Dextromethorphan Inhibitor: Quinidine
Reaction conditions: Incubation conditions:
Time: 35 min Human Microsomal protein 25 μl
Temperature: 37°C Substrate (0.1 mg/ml in Acetonitrile) 10 μl
Protein Concentration: 500 μg Inhibitor or test compound 10 μl
Incubation Volume: 2 ml NADPH regenerating system 50 μl
Inhibitor Concentration: 1 μM (quinidine or the Buffer 915 indicated for test compound (1 or 10 μM) μl
Extraction procedure:
Quench with 75 μl full-strength perchloric acid
Spike samples with internal standard (5μg/ml OH-Bufurolol, 100 μl) and then vortex
Centrifuge at 3000 rpm, 4°C for 10 min
Pipette -200 μl from upper layer and inject onto HPLC column
Animals and housing: Male, Sprague-Dawley rats or CD-1 mice were used for all studies. All animals were allowed ad-lib access to food and water except during experiment. Animals were housed within an animal vivarium maintained under a 12 h light:dark cycle (lights on: 07:00 h), and all experiments were conducted in the animals' light phase. For all experiments, animals were habituated to the vivarium for a minimum of 72 h before experimentation. The experimental procedures used in the present investigation were conducted under the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the Canadian Council on Animal Care (CCAC) guidelines.
Test Compounds: All compounds were dissolved in 5% Tween 80® in saline and injected in a dose volume of 5 ml/kg or 10 ml/kg (rat), and 10 ml/kg (mouse). Compounds were administered by either the oral or intraperitoneal route.
Mouse hypolocomotion assay: Selective 5-HT2c receptor agonists have been reported to produce hypolocomotion in rodent species by a relatively well defined CNS mechanism. A mouse locomotor assay was therefore used to screen compounds. Male, CD-1 mice were administered test compound 15 min before placement in a chamber where locomotor activity was measured through photocell beam breaks. Test compounds were administered either by the oral or intraperitoneal route.
Deprivation-induced feeding in the rat: Male Sprague-Dawley rats (Charles River, St. Constant, Quebec, Canada) of approximate weight 180-20Og were pair housed on arrival in the animal facility (lights on 7:00-19:00h). After a 7 day acclimitisation period where the animals received ad-libitum access to standard rodent lab chow (Harlan Teklad rodent maintenance diet, 2014;
Harlan Teklad, Madison, Wl), the animals were trained to receive a daily ration of lab chow in distinct chambers over a 2h period. Animals were singly housed for the duration of this period and food intakes over the 2h access period were measured by weighing food containers before and after feeding periods, with correction for spillage. After the daily 2h food access period, the animals were returned to their holding cage with no further daily food allowance. Water was available ad-libitum. Body weights were recorded daily.
Once the daily food intake had stabilized (after one to two weeks training), a dose of a test compound (or vehicle as control) was administered 10 to 15 min before the beginning of the 2 h food access period, and food intake over that period was measured as during the training period.
Test compound or vehicle was administered on Tuesdays and Fridays, with drug free (washout) days in between. Typically the animals received 3 doses of test compound and vehicle in a counterbalanced sequence.
Schedule-induced polydipsia
Food deprived rats exposed to intermittent, uncontrollable presentations of food will drink quantities of water that are far in excess of their normal daily intake and in excess of their intake when given food at one time (FaIk JL (1961) Science 133: 195-196). This excessive behaviour is persistent and has been proposed as a model of obsessive-compulsive disorder based on pharmacological validation and symptomatic similarities (Woods, A. et al. (1993) Selective serotonin re-uptake inhibitors decrease schedule-induced polydipsia in rats: a potential model for obsessive compulsive disorder. Psychopharmacology 112: 195-198).
Male Sprague-Dawley rats (Charles River, St. Constant, Quebec, Canada) of approximate weight 180-20Og are pair housed on arrival in the animal facility (lights on 7:00-19:00h). After a 7 day acclimitisation period where the animals receive ad-libitum access to standard rodent lab chow (Harlan Teklad rodent maintenance diet, 2014; Harlan Teklad, Madison, Wl), the animals are trained to receive single 45mg food pellets under a fixed time interval of 60s over a 2h period within an operant chamber equipped with a water bottle. Thus during the 2h session, the rats can earn a maximum of 120 pellets. The total volume of water consumed by rats during this 2h period is recorded. Daily food allowance is supplemented by a 45min access period sometime between 15:00-18:00h.
Once daily fluid intakes within the 2 h test session become stable over days (approximately +15%), the rats may be dosed orally or parentally with vehicle or test compound. Test compound or vehicle is administered on Tuesdays and Fridays with drug free (washout) days in between. Typically the animals
will receive 3 doses of test compound .and vehicle in a counterbalanced sequence.
A modification to the above procedure is to pre-treat rats with either vehicle or a selective 5-HT2c receptor antagonist, 6-chloro-5-methyl-N-(2-(2- methylpyridin-3-yl-oxy)pyridine-5-yl)aminocarbonyl)-2,3-dihydroindole (1 mg/kg in 8% HPCD, 25mM citric acid in saline) prior to the oral or parental dose of test compound.
s.c Pentylenetetrazol assay
Antagonism of clonic-tonic seizures produced by chemical convulsants such as pentylenetetrazol have been widely utilized to identify novel anticonvulsants.
Male, CD-1 mice (Charles River, St. Constant, Quebec, Canada) of approximate body weight 20-3Og are housed in groups of four on arrival at the facility. Food (Harlan Teklad rodent maintenance diet, 2014; Harlan Teklad, Madison, Wl) and water are available ad-libitum. After a minimum 3 day acclimatization period the animals would be tested in a s.c pentylenetetrazol assay - which is considered both a model of primary generalized convulsive seizures and non-convulsive absence (petit mal) seizures (Upton, N. (1994) Trends Pharmacol. Sci. 15: 456-463).
The experiment is conducted within a single day with animals receiving a single pretreatment, i.e independent groups design. Following drug or vehicle control treatment by either oral, or parenteral route, the animals would receive pentylenetetrazol (85mg/kg mice) administered by the subcutaneous route. The dose of pentylenetetrazol is selected as it is of sufficient intensity to induce a clonic seizure in the majority of animals, i.e a CD97 dose. The animals are restrained by hand to deliver the chemical convulsant, following which the animals are released and transferred to a test cage to permit
observation of the subsequent seizure throughout its course. The animal would receive a single pentylenetetrazol injection and would be terminated on reaching endpoint, i.e clonic seizure. If an animal displays no seizure activity after 60 min it is considered protected and the experiment completed as endpoint reached.
Approximately 10-20 min prior to the PTZ test, a parallel tests of motor function using the rotorod would be undertaken to establish a therapeutic index (Tl), e.g ratio between the ED50 dose required to block seizures, compared to ED50 dose required to disrupt motor function in same species. The rotorod test consists of placing the animal on a rotating treadmill (a rod) traveling at a constant speed of 16 r.p.m. The dependant measure is the time that the animal remains on the rod before falling. Up to three separate measures may be taken to get a meaningful measure of performance.
A modification to the above procedure is to pretreat mice with either vehicle or a selective 5-HT2c receptor antagonist, 6-chloro-5-methyl-N-(2-(2- methylpyridin-3-yl-oxy)pyridine-5-yl)aminocarbonyl)-2,3-dihydroindole (1 mg/kg in 8% HPCD, 25mM citric acid in saline) prior to the oral or parental dose of test compound.
Amphetamine-induced hyperlocomotion
Antagonism of increased locomotion produced by the psychostimulant amphetamine in rodents is a feature of many drugs with antipsychotic property in man. As such reversal of amphetamine hyperlocomotion is a widely used preclinical test to detect novel drugs for the treatment of schizophrenia.
Male Sprague-Dawley rats (Charles River, St. Constant, Quebec, Canada) of approximate weight 200 g are pair housed on arrival in the animal facility (lights on 7:00-19:00 h). After a 7 day acclimitisation period where the animals
receive ad-libitum access to standard rodent lab chow (Harlan Teklad rodent maintenance diet, 2014; Harlan Teklad, Madison, Wl) the animals may undergo behavioural testing.
Animals would be singly placed within the test apparatus (Perspex chamber of dimensions: rat 42 cm x 42 cm x 30 cm (L x W x H)) for a limited time period (approximately 30min) to habituate to the novel environment. After such habituation period has passed, animals will be treated with test article or vehicle control via the oral, or parental route, and then returned to the observation test chambers. After a predetermined period, the animals would be dosed with either saline vehicle or d-amphetamine (0.5mg/kg) by the intraperitoneal route and returned to the test chamber for 2h. While in the test chamber, the animal's activity will be monitored automatically by infrared sensors and/or manually by an experimenter for expression of 'normal' behaviors such as sniffing, grooming, rearing, and 'abnormal' behaviors such as 'circling'. At the completion of such test, the animals will be returned to their holding cages.
A modification to the above procedure is to pretreat rats with either vehicle or a selective 5-HT2c receptor antagonist, 6-chloro-5-methyl-N-(2-(2- methylpyridin-3-yl-oxy)pyridine-5-yl)aminocarbonyl)-2,3-dihydroindole (1 mg/kg in 8% HPCD, 25mM citric acid in saline) prior to the oral or parental dose of test compound.
Model of Urinary Incontinence : Urethral function in Guinea pigs
Test compounds evaluated in a guinea-pig model designed to measure urethral incontinence and leak pressure point pressure using a paradigm to measure the response of urethral musculature to bladder filling to maintain continence, as measured by changes in sphincter tone.
Under anaesthesia, a cystometry tube is implanted into the bladder of adult guinea-pigs (600-80Og). The cystometry tube is connected to an infusion pump and pressure transducer to enable fluid perfusion and the recording of intravesicular bladder pressure. Electromyographic (EMG) leads are inserted into the external urethral sphincter (EUS) to allow the measurement of sphincter tone. Following a post-surgery stabilization period the bladder was filled at a rate of 150 μl/min with physiological saline (room temperature) until initiation of a micturition reflex. Following this reflex, the bladder was drained and the filling procedure repeated to establish a mean bladder threshold capacity for initiation of micturition reflex. Further, EUS EMG activity and intravesicular pressure was recorded throughout bladder filling.
At this stage the test drug or vehicle is administered i.v and bladder filling (150 μl/min) reinitiated until induction of micturition reflex. Bladder pressure and maximum EUS EMG activity is recorded.
Pain Assays
Persistent Inflammatory Pain (Formalin Model)
The formalin test is a chemically-induced tonic pain model in which injection of formalin into a hind paw elicits a biphasic nociceptive behavior. The second phase of formalin response is predominantly due to a central sensitization phenomenon. Most clinically used drugs against neuropathic pain are active on this second phase of formalin response. Formalin test is accepted as a valid model of persistent clinical pain.
The test was done by pretreating the rats with the test compound and 30 min later (pretreatment time), 50 μl of 2.5% formalin was injected into the right hind paw of the animal. The number of paw licking and flinching episodes were scored for 60 min post-formalin injection.
The compounds were administered either intraperitoneal^ or orally. The compounds significantly inhibited the second phase of the formalin response.
Chronic Neuropathic Pain Model (Spared Nerve Injury or SNI Model)
The compounds were tested for their efficacy in reducing mechanical allodynia and cold allodynia in the spared nerve injury (SNI) model of chronic neuropathic pain. In this model, the left sciatic nerve of the rat is exposed under anesthesia. Two of the branches of the sciatic nerve viz. the common peroneal and tibial nerve are ligated and sectioned. The third branch (sural nerve) is left intact. The animals were allowed a post-operative recovery period of 7 days before they were subjected to any test.
Mechanical Allodynia
The presence of mechanical allodynia was assessed using the Dynamic Plantar Aesthesiometer (Ugo Basile, Italy) which is a modified version of the Von Frey Hair test. In this, a test filament is positioned below the animal's hind paw and the unit is activated which causes the filament to move up and touch the plantar surface of the hind paw. Increasing force is applied to the paw via the filament. When the animal withdraws its paw, the unit is inactivated automatically and the threshold force required to elicit the paw withdrawal is displayed. The cut-off force is set at 50 g. The tests were done on both the non-injured (control) and the injured (SNI) paw. Pilot studies showed the presence of mechanical allodynia 7 days after the surgery and lasted up to 4 weeks (end of the test period).
Testing of compounds was done after 21 days post-operatively. After initial basal readings were taken, the test compound or the vehicle was administered (10 ml/kg) either intraperitoneally or orally. The readings were taken again 30, 60 and 180 min after the compound/vehicle administration. Mechanical allodynia in neuropathic rats was significantly inhibited by the compounds up to 180 min after the intraperitoneal administration as evidenced by the increase in the withdrawal threshold.
CoId Allodynia
Cold allodynia was assessed by using the acetone test. In this model, 25 μl of acetone is sprayed on to the plantar surface of the hind paw. Evaporation of acetone causes cooling of the skin. The cold stimulus sets up nociceptive responses from the injured paw as evidenced by paw lifting, paw licking and grooming. The duration of the nociceptive responses is noted. Similar stimulus to the uninjured (control) paw usually does not elicit nociceptive responses.
Testing of compounds was done after 21 days post-operatively. After initial basal readings were taken, the test compound or the vehicle was administered (10 ml/kg) either intraperitoneally or orally. The readings were taken 30 min after the administration. Cold allodynia in neuropathic rats was measured as evidenced by the reduced nociceptive duration in these animals.
Chronic Inflammatory Pain Model (Freund's Complete Adjuvant or FCA Model)
Administration of FCA to the hind paw of an animal induces the formation of a localized edema and has been widely used as a model of chronic inflammatory pain. 100 μl of FCA is injected subcutaneously into the dorsal aspect of the left hind paw of the rats. The edema appears within 2 h reaches a peak by 6 h and lasts for about 7 days. The effects of compounds on mechanical allodynia and paw volume were studied 2 days after FCA administration.
Mechanical Allodynia
The effect of the compounds in alleviating mechanical allodynia was assessed in the FCA-treated rats using the modified Randall-Selitto method (Analgesy- Meter, Ugo Basile, Italy). The animal's paw is placed on a small plinth under a cone-shaped pusher. Increasing force is applied to the paw by depressing a pedal until vocalization or withdrawal of the paw occurs. The minimum force
required to elicit this vocalization/withdrawal is the paw pressure threshold. The cut-off was set at 150 g.
After the initial basal (control) readings were taken, the test compound/vehicle was administered orally in a volume of 10 ml/kg. The paw pressure thresholds were measured 30, 60 and 180 min post administration. The inhibition of mechanical allodynia was measured at 30 min by all compounds tested.
Selective 5-HT?r Agonist Activity
Table 1 shows examples of selective 5-HT2C receptor agonists in accordance with the invention, determined by the IP3 assay described above.
Cytochrome P450 Activity
Table 2 shows examples of compounds with reduced cytochrome P450 inhibition in accordance with the invention, determined by the cytochrome P450 assay described above. The data show the percent inhibition of the metabolism of the substrate in the assay resulting from incubation in the presence of the indicated compound. Compounds having less than 50% inhibition at 1 uM are considered to be a compound with reduced inhibition of cytochrome P450 for the present invention.
Rat Deprivation Induced Feeding Assay
Figure 1 shows the dose-related reduction in food intake in rats treated intraperitoneally with Example 22.34 of the invention.
Pre-treatment of rats with the selective 5-HT2c antagonist SB 242084 blocked the effect of the agonist compounds, as shown by the hatched bars.
The description as set forth is not intended to be exhaustive or to limit the scope of the invention. Many modifications and variations are possible in light of the above teaching without departing from the spirit and scope of the following claims. It is intended that the scope of the present invention be defined by the claims appended hereto, giving full cognizance to equivalents in all respects.
Table 1
Functional selectivity of selected compounds at h5-HT? receptor subtypes in the IP3 cell-based assay. Data presented are ECsn values and Emax compared to 5-HT (5-HT = 1.0).

Activity of compounds in a cytochrome P450 assay.

Claims
1. A compound of Formula I:

I wherein:
R1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing at least two rings, wherein said rings are fused rings, bridged fused rings and/or spiro rings; R2, R3 and R5 to R12 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O- heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O- heterocycloalkyl, alkylene-O-alkylene-cycloalkyl, alkylene-O-alkylene- heterocycloalkyl, S-alkyl, S(O)-alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S-heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2- heterocycloalkyl, O-aryl, O-heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O- heteroaryl, alkylene-O-alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S- heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl, C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group; and R4 is selected from H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, alkylene- O-alkyl, alkylene-O-cycloalkyl, alkylene-O-alkylene-cycloalkyl;
and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
2. A compound according to Claim 1 , wherein R1 is a substituted or unsubstituted heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing two bridged fused rings.
3. A compound according to Claim 1 or Claim 2, wherein R1 is a substituted or unsubstituted N-heterocycloalkyl or a substituted or unsubstituted N-heterocycloalkyl containing two bridged fused rings
4. A compound according to Claim 3, wherein R1 is a substituted N- heterocycloalkyl or a substituted or unsubstituted heterocycloalkyl containing two bridged fused rings substituted with alkyl, alkoxy, hydroxy, cyano, halo, haloalkyl, and/or haloalkyloxy; R7 is selected from H, halo, alkyl, haloalkyl, haloalkyloxy, cyano, alkoxy; and R3 is selected from H, or lower alkyl
5. A compound according to Claim 1 , wherein Formula I comprises a compound of Formula IB, IC and/or ID:

IB IC

ID wherein:
R3 and R7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene-
O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, S-alkyl, S(O)- alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S- heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2-heterocycloalkyl, O-aryl, O- heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl,
N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O-heteroaryl, alkylene-O- alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S-heteroaryl, S(O)-aryl,
S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl,
C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or
S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group;
X is O, S, SO, SO2, CR13R14; NR15;
R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
R15 is selected from the group consisting of H and alkyl; n is an integer; and m is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
6. A compound according to Claim 5, wherein R3 is selected from H, or lower alkyl; R7 is selected from H, halo; R15 is selected from the group consisting of H and alkyl of C1 to C6.
7. A compound of Formula II:

Il wherein:
R3 and R7 are each independently selected from H, halo, hydroxy, cyano, nitro, alkyl, alkoxy, CH2OH, haloalkyl, O-haloalkyl, hydroxyalkyl, cyanoalkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, alkylaryl, alkylheteroaryl, O-cycloalkyl, O-heterocycloalkyl, alkylene-O-alkyl, alkylene-O-cycloalkyl, alkylene-O-heterocycloalkyl, alkylene- O-alkylene-cycloalkyl, alkylene-O-alkylene-heterocycloalkyl, S-alkyl, S(O)- alkyl, S(O)2-alkyl, S-cycloalkyl, S(O)-cycloalkyl, S(O)2-cycloalkyl, S- heterocycloalkyl, S(O)-heterocycloalkyl, S(O)2-heterocycloalkyl, O-aryl, O- heteroaryl, N(H)alkyl, N(alkyl)alkyl, N(H)-aryl, N(alkyl)-aryl, N(H)-heteroaryl, N(alkyl)-heteroaryl, alkylene-O-aryl, alkylene-O-heteroaryl, alkylene-O- alkylene-aryl, alkylene-O-alkylene-heteroaryl, S-aryl, S-heteroaryl, S(O)-aryl, S(O)-heteroaryl, S(O)2-aryl, S(O)2-heteroaryl, C(O)alkyl, OC(O)alkyl, C(O)Oalkyl, C(O)N(H)alkyl, C(O)N(alkyl)alkyl, S(O)2N(H)alkyl or S(O)2N(alkyl)alkyl; R2 and R3, R5 and R6, R9 and R10, and/or R11 and R12, together with the carbon atom to which they are attached, form a cycloalkyl group;
X is O, S1 SO, SO2, CR13R14; NR15;
R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
R15 is selected from the group consisting of H and alkyl; and n is an integer; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
8. A compound according to Claim 7, wherein R3 is selected from H, or lower alkyl; R7 is selected from H, halo; and R15 is selected from the group consisting of H and alkyl of C1 to C6.
9. A compound according to Claim 7, wherein Formula Il comprises a compound of Formula NA and/or MB:

HA HB wherein :
R3 is selected from H, or lower alkyl;
R7 is selected from H, halo;
X is O, S, SO, SO2, CR13R14; NR15;
R13 and R14 are each independently selected from the group consisting of H, alkyl, halo and haloalkyl;
R15 is selected from the group consisting of H and alkyl of C1 to C6; and n is either 0, 1 or 2; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
10. A compound according to any one of Claims 1 to 9, wherein the compound is a pharmaceutically-acceptable salt, optical isomer, or combination thereof.
11. A compound according to any one of Claims 1 to 10, wherein the pharmaceutically-acceptable salt comprises an acid addition salt or a basic addition salt.
12. A compound according to Claim 11 , wherein the acid addition salt is formed from hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, acid metal salt, monocarboxylic acids, dicarboxylic acids, or tricarboxylic acids.
13. A compound selected from:
(9R)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7I8)9-tetrahydro-5H- pyrido[2,3d]azepine; (9R)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9R)-9-methyl-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9R)-9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; (ΘRJ-θ-methyl^-piperidin-i-yl-ey.δ.θ-tetrahydro-δH-pyridop.S-dJazepine;
(9S)-2-(4I4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(9S)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine; (9S)-9-methyl-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- djazepine
(9S)-9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; (9S)-9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-diazepan-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1-oxidothiomorpholin-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-(4,4-difluoroazepan-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoroazepan-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
2-(4,4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
2-(4-fluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4-methylpiperazin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(8-azabicyclo[3.2.1]oct-8-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-azepan-1 -yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-9-isopropyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-cyclopropyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-cyclopropyl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-piperazin-1-yl-6>7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-pyrrolidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-thiomorpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 3-bromo-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-bromo-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-(4,4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine; 3-chloro-2-(4-fluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 9-ethyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 9-ethyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]a2epine; 9-isopropyl-2-morpholin-4-yl-6J,δ,9-tetrahydro-δH-pyrido[2,3-d]azepine; 9-isopropyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; θ-methyl^i ^-oxazepan^-yO-βJ.δ.θ-tetrahydro-δH-pyrido^.S-dlazepine; 9-methyl-2-morpholin-4-yl-67,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine; 9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; ethyl 4-(6, 7,8, 9-tetrahydro-5H-pyrido[2,3-d]azepin-2-yl)piperazine-1- carboxylate; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
14. A compound selected from:
(9R)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H- pyrido[2,3d]azepine;
(9R)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(ΘR^Θ-methyl^-O Aoxazepan^-yO-ΘJ.δ.θ-tetrahydro-δH-pyridoβ.S- d]azepine; (9R)-9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9R)-9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(9S)-2-(4,4-difluoropiperidin-1-yl)-9-methyl-6I7,8,9-tetrahydro-5H-pyrido[2)3- d]azepine;
(9S)-2-(4-fluoropiperidin-1-yl)-9-methyl-6,7,δ,9-tetrahydro-5H-pyrido[2,3- d]azepine;
(ΘS^Θ-methyl^i ^-oxazepan^-yO-ey.δ.θ-tetrahydro-δH-pyrido^.S- d]azepine
(9S)-9-methyl-2-morpholin-4-yl-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine;
(ΘS^g-methyl^-piperidin-i-yl-ej.δ.θ-tetrahydro-δH-pyrido^.S-dlazepine; 2-(1 ,4-diazepan-1-yl)-6,7)δ,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(1 ,4-oxazepan-4-yl)-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4,4-difluoroazepan-1-yl)-6,7,δ,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-(4,4-difluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-(4-fluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-azepan-1-yl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 2-cyclopropyl-9-methyl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-piperidin-1-yl-6,7,8>9-tetrahydro-5H-pyrido[2,3-d]azepine;
2-thiomorpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-(1 ,4-oxazepan-4-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 3-chloro-2-(4,4-difluoropiperidin-1 -yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3- d]azepine;
3-chloro-2-(4-fluoropiperidin-1-yl)-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
3-chloro-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; 9-isopropyl-2-piperidin-1 -yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-morpholin-4-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine;
9-methyl-2-piperidin-1-yl-6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine; and/or a pharmaceutically-acceptable salt, hydrate, solvate, isoform, tautomer, optical isomer, or combination thereof.
15. A compound according to any one of claims 1 to 14, wherein the compound has an EC50 for a human 5-HT2c receptor selected from less than
1000 nM, less than 500 nM, less than 300 nM, or less than 100 nM.
16. A pharmaceutical composition comprising a compound according to any one of Claims 1 to 15 and at least one pharmaceutically acceptable carrier and/or excipient.
17. A Boc-protected precusor of the compound according to any one of Claims 1 to 15 or mixtures thereof.
18. A method for making a compound of Claim 1 , wherein R11 and R12 are H, the method comprising:

reacting a compound of Formula A under conditions (a), wherein said (a) comprises heat and base assisted cyclization of the compound of Formula A to provide an amide of Formula B; and reducing the carbonyl of the amide of Formula B, whereby R' is alkyl or cycloalkyl.
19. A method for making a compound of Claim 1 , wherein R9 and R10 are H, the method comprising: 
BB reacting a compound of Formula AA under conditions (a), wherein said (a) comprises heat and base assisted cyclization of the compound of Formula AA to provide an amide of Formula BB; and reducing the carbonyl of the amide of Formula BB, whereby R' is alkyl or cycloalkyl.
20. A method for making a compound of Claim 1 , wherein R11 and R12 are H, the method comprising: reducing a carbonyl of an amide:

21. A method for making a compound of Claim 1 , wherein R9 and R10 are H1 the method comprising: reducing a carbonyl of an amide:

22. A method for making a compound of Claim 1 , wherein R9, R10, R11 and R12 are H, the method comprising: reducing carbonyl groups of a cyclic imide:

23. A method for making a compound of Claim 1 , wherein R11 and R12 are H, the method comprising:

24. A method for making a compound of Claim 1 , wherein R9 and R10 are H, the method comprising:

CC
reacting a compound of Formula CC under conditions (a), wherein said (a) comprises selective cyano reduction followed by cyclization of the compound of Formula CC to provide Formula I, whereby R' is alkyl or cycloalkyl.
25. A method for making a compound of Claim 1 , wherein the method comprises:

reacting a compound of Formula D under conditions (a), wherein said (a) comprises cyclization of the compound of Formula D to provide Formula I, whereby R' is alkyl or cycloalkyl.
26. A method for making a compound of Claim 1 , wherein the method comprises:

DD reacting a compound of Formula DD under conditions (a), wherein said (a) comprises cyclization of the compound of Formula DD to provide Formula I, whereby R' is alkyl or cycloalkyl.
27. A method for treating a 5-HT2c receptor-mediated disorder in a mammal, comprising administering to the mammal a therapeutically effective amount of a compound according to any one of Claims 1 to 15, said compound exhibiting reduced inhibition of a cytochrome P450.
28. A method for treating a 5-HT2c receptor-mediated disorder in a mammal, comprising administering to the mammal a therapeutically effective amount of a composition according to Claim 16, said composition exhibiting reduced inhibition of a cytochrome P450.
29. The method according to Claim 27 or 28, wherein the mammal is a human.
30. The method according to any one of Claims 27 to 29, wherein the disorder is selected from the group consisting of urinary incontinence, a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure.
31. The method according to Claim 30, wherein the urinary incontinence is selected from the group consisting of stress urinary incontinenence (SUI), urge incontinence (Ul) and mixed incontinence.
32. The method according to Claim 30, wherein the disorder is selected from the group consisting of obesity, schizophrenia, epilepsy, a depressive disorder, panic attack, alcoholism, drug addiction or obsessive compulsive disorder.
33. The method according to Claim 27, wherein the compound is administered orally and/or parenterally.
34. The method according to Claim 28, wherein the composition is administered orally and/or parenterally.
35. The method according to Claim 33, wherein the compound is administered intravenously and/or intraperitoneally.
36. The method according to Claim 34, wherein the compound is administered intravenously and/or intraperitoneally.
37. Use of a compound according to any one of Claims 1 to 15 for the manufacture of a medicament for treatment of a 5-HT2c receptor-mediated disorder in a mammal, said compound exhibiting reduced inhibition of a cytochrome P450.
38. Use of a composition according to Claim 16 for the manufacture of a medicament for treatment of a 5-HT2C receptor-mediated disorder in a mammal, said composition exhibiting reduced inhibition of a cytochrome P450.
39. Use of a compound according to any one of Claims 1 to 15 to treat a 5- HT2C receptor-mediated disorder in a mammal, said compound exhibiting reduced inhibition of a cytochrome P450.
40. Use of a composition according to Claim 16 to treat a 5-HT2c receptor- mediated disorder in a mammal, said composition exhibiting reduced inhibition of a cytochrome P450.
41. The use according to any one of Claims 37 to 40, wherein the mammal is a human.
42. The use according to any one of Claims 37 to 41 , wherein the disorder is selected from the group consisting of urinary incontinence, a depressive disorder, an anxiety disorder, including panic attack, agoraphobia, and specific or social phobia, bipolar disorder, post-traumatic stress, an eating disorder, obesity, a gastro-intestinal disorder, alcoholism, drug addiction, schizophrenia, a psychotic disorder, a sleep disorder, including sleep apnea, migraine, sexual dysfunction, a central nervous system disorder, including trauma, stroke and spinal cord injury, a cardio-vascular disorder, diabetes insipidus, obsessive compulsive disorder, premenstrual tension, chronic fatigue syndrome, age-related memory disorder, personality disorder and raised intracranial pressure.
43. The use according to Claim 42, wherein the urinary incontinence is selected from the group consisting of stress urinary incontinenence (SUI), urge incontinence (Ul) and mixed incontinence.
44. The use according to claim 42, wherein the disorder is selected from the group consisting of obesity, schizophrenia, epilepsy, a depressive disorder, panic attack, alcoholism, drug addiction or obsessive compulsive disorder,
45. The use according to Claim 37 or 39, wherein the compound is administrable orally and/or parenterally.
46. The use according to Claim 38 or 40, wherein the composition is administrable orally and/or parenterally.
47. The use according to Claim 45, wherein the compound is administrable intravenously and/or intraperitoneal^.
48. The use according to Claim 46, wherein the composition is administrable intravenously and/or intraperitoneal^.
49. A method for decreasing food intake in a mammal comprising administering to the mammal a therapeutically effective amount of a compound according to any one of Claims 1 to 15, said compound exhibiting reduced inhibition of a cytochrome P450.
50. A method for decreasing food intake in a mammal comprising administering to the mammal a therapeutically effective amount of a composition according to Claim 16, said composition exhibiting reduced inhibition of a cytochrome P450.
51. A method of controlling weight gain in a mammal comprising administering to the mammal a therapeutically effective amount of a compound according to any one of Claims 1 to 15, said compound exhibiting reduced inhibition of a cytochrome P450.
52. A method of controlling weight gain in a mammal comprising administering to the mammal a therapeutically effective amount of a composition according to Claim 16, said composition exhibiting reduced inhibition of a cytochrome P450.
53. Use of a compound according to any one of Claims 1 to 15 for the manufacture of a medicament for decreasing food intake or controlling weight gain in a mammal, said compound exhibiting reduced inhibition of a cytochrome P450.
54. Use of a composition according to Claim 16 for the manufacture of a medicament for decreasing food intake or controlling weight gain in a mammal, said composition exhibiting reduced inhibition of a cytochrome P450.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US1599207P | 2007-12-21 | 2007-12-21 | |
| US61/015,992 | 2007-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009079765A1 true WO2009079765A1 (en) | 2009-07-02 |
Family
ID=40800608
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2008/002223 WO2009079765A1 (en) | 2007-12-21 | 2008-12-18 | Compounds with activity at the 5-ht2c receptor |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009079765A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011071136A1 (en) | 2009-12-11 | 2011-06-16 | アステラス製薬株式会社 | Therapeutic agent for fibromyalgia |
| WO2012030953A1 (en) | 2010-09-01 | 2012-03-08 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists in the treatment of disorders ameliorated by reduction of norepinephrine level |
| CN102887856A (en) * | 2012-10-17 | 2013-01-23 | 河北师范大学 | Method for synthesizing Blonanserin |
| CN103130799A (en) * | 2011-12-01 | 2013-06-05 | 上海药明康德新药开发有限公司 | Preparing method of 2-hydroxy pyridino-6,7,8,9-tetrahydro-5H- pyridine[2,3-d] azepine derivative |
| WO2015066344A1 (en) | 2013-11-01 | 2015-05-07 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists and compositions and methods of use |
| CN109608451A (en) * | 2018-12-17 | 2019-04-12 | 上海药明康德新药开发有限公司 | A kind of 2- oxygen subunit-pyrido [2,3-d] azatropylidene -7 (2H)-t-butyl formate synthetic method |
| US10947257B2 (en) | 2017-10-09 | 2021-03-16 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11564935B2 (en) | 2019-04-17 | 2023-01-31 | Compass Pathfinder Limited | Method for treating anxiety disorders, headache disorders, and eating disorders with psilocybin |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5827849A (en) * | 1993-02-22 | 1998-10-27 | Karl Thomae Gmbh | Cyclic dericatives, pharmaceutical compositions containing these compounds and processes for preparing them |
| CA2294530A1 (en) * | 1997-07-02 | 1999-01-14 | Soong-Hoon Kim | Inhibitors of farnesyl protein transferase |
| CA2412632A1 (en) * | 2000-06-15 | 2001-12-20 | Bang-Chi Chen | Hmg-coa reductase inhibitors and method |
| WO2008009125A1 (en) * | 2006-07-20 | 2008-01-24 | Cascade Therapeutics Inc. | Tetrahydro-5h-pyrido[2,3-d]azepines as 5-ht2c ligands |
-
2008
- 2008-12-18 WO PCT/CA2008/002223 patent/WO2009079765A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5827849A (en) * | 1993-02-22 | 1998-10-27 | Karl Thomae Gmbh | Cyclic dericatives, pharmaceutical compositions containing these compounds and processes for preparing them |
| CA2294530A1 (en) * | 1997-07-02 | 1999-01-14 | Soong-Hoon Kim | Inhibitors of farnesyl protein transferase |
| CA2412632A1 (en) * | 2000-06-15 | 2001-12-20 | Bang-Chi Chen | Hmg-coa reductase inhibitors and method |
| WO2008009125A1 (en) * | 2006-07-20 | 2008-01-24 | Cascade Therapeutics Inc. | Tetrahydro-5h-pyrido[2,3-d]azepines as 5-ht2c ligands |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011071136A1 (en) | 2009-12-11 | 2011-06-16 | アステラス製薬株式会社 | Therapeutic agent for fibromyalgia |
| WO2012030953A1 (en) | 2010-09-01 | 2012-03-08 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists in the treatment of disorders ameliorated by reduction of norepinephrine level |
| CN103130799A (en) * | 2011-12-01 | 2013-06-05 | 上海药明康德新药开发有限公司 | Preparing method of 2-hydroxy pyridino-6,7,8,9-tetrahydro-5H- pyridine[2,3-d] azepine derivative |
| CN102887856A (en) * | 2012-10-17 | 2013-01-23 | 河北师范大学 | Method for synthesizing Blonanserin |
| CN102887856B (en) * | 2012-10-17 | 2014-05-28 | 河北师范大学 | Method for synthesizing Blonanserin |
| WO2015066344A1 (en) | 2013-11-01 | 2015-05-07 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists and compositions and methods of use |
| US10954259B1 (en) | 2017-10-09 | 2021-03-23 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US10947257B2 (en) | 2017-10-09 | 2021-03-16 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11149044B2 (en) | 2017-10-09 | 2021-10-19 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11180517B2 (en) | 2017-10-09 | 2021-11-23 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11447510B2 (en) | 2017-10-09 | 2022-09-20 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11505564B2 (en) | 2017-10-09 | 2022-11-22 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11629159B2 (en) | 2017-10-09 | 2023-04-18 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11851451B2 (en) | 2017-10-09 | 2023-12-26 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| US11939346B2 (en) | 2017-10-09 | 2024-03-26 | Compass Pathfinder Limited | Preparation of psilocybin, different polymorphic forms, intermediates, formulations and their use |
| CN109608451A (en) * | 2018-12-17 | 2019-04-12 | 上海药明康德新药开发有限公司 | A kind of 2- oxygen subunit-pyrido [2,3-d] azatropylidene -7 (2H)-t-butyl formate synthetic method |
| US11564935B2 (en) | 2019-04-17 | 2023-01-31 | Compass Pathfinder Limited | Method for treating anxiety disorders, headache disorders, and eating disorders with psilocybin |
| US11738035B2 (en) | 2019-04-17 | 2023-08-29 | Compass Pathfinder Limited | Method for treating anxiety disorders, headache disorders, and eating disorders with psilocybin |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009079765A1 (en) | Compounds with activity at the 5-ht2c receptor | |
| US20090312306A1 (en) | Tetrahydro-5h-pyrido[2,3-d]azepines as 5-ht2c ligands | |
| EP1109813B1 (en) | Pyrroloindoles, pyridoindoles and azepinoindoles as 5-ht2c agonists | |
| US6800627B1 (en) | Pirazino indole derivatives | |
| US5610174A (en) | Use of α1A -selective adrenoceptor agonists for the treatment of urinary incontinence | |
| US8815891B2 (en) | Tricyclic derivative or pharmaceutically acceptable salts thereof, preparation method thereof, and pharmaceutical composition containing the same | |
| JP2011504892A (en) | Aryl and heteroaryl fused imidazo (1,5-a) pyrazines as inhibitors of phosphodiesterase 10 | |
| EP1109809B1 (en) | Pyrroloquinolines for treatment of obesity | |
| US7763636B2 (en) | N-(arylalkyl)-1H-pyrrolopyridine-2-carboxamide derivatives, preparation and therapeutic use thereof | |
| US20080221129A1 (en) | New compounds | |
| JP2011505366A (en) | Aryl and heteroaryl fused imidazo [1,5-a] pyrazines as inhibitors of phosphoesterase 10 | |
| WO2006068164A1 (en) | Tricyclic compound and use thereof | |
| EP3344248B1 (en) | 6-membered aza-heterocyclic containing delta-opioid receptor modulating compounds, methods of using and making the same | |
| EP3582779B1 (en) | 5-membered aza-heterocyclic containing delta-opioid receptor modulating compounds, methods of using and making the same | |
| EP2521714B1 (en) | Aromatic sulfone compounds useful in the treatment of central nervous disorders | |
| CN104428286B (en) | Indole carboxamides derivant | |
| CA2792918A1 (en) | Benzazepine compound | |
| EP3582783B1 (en) | 7-membered aza-heterocyclic containing delta-opioid receptor modulating compounds, methods of using and making the same | |
| JP5898815B2 (en) | Novel pyrazolopyrimidine | |
| KR100705179B1 (en) | Thienopyranecarboxamide derivatives | |
| Kanayama | TECHNICAL FIELD |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08864225 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08864225 Country of ref document: EP Kind code of ref document: A1 |