WO2009056513A1 - Process for the preparation of a polyether polyol suitable for preparing rigid polyurethane foams - Google Patents
Process for the preparation of a polyether polyol suitable for preparing rigid polyurethane foams Download PDFInfo
- Publication number
- WO2009056513A1 WO2009056513A1 PCT/EP2008/064509 EP2008064509W WO2009056513A1 WO 2009056513 A1 WO2009056513 A1 WO 2009056513A1 EP 2008064509 W EP2008064509 W EP 2008064509W WO 2009056513 A1 WO2009056513 A1 WO 2009056513A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mixture
- polyhydric alcohols
- polyether polyol
- catalyst
- polyhydric alcohol
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
- C08G65/2603—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds the other compounds containing oxygen
- C08G65/2606—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds the other compounds containing oxygen containing hydroxyl groups
- C08G65/2609—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds the other compounds containing oxygen containing hydroxyl groups containing aliphatic hydroxyl groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4829—Polyethers containing at least three hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4833—Polyethers containing oxyethylene units
- C08G18/4837—Polyethers containing oxyethylene units and other oxyalkylene units
- C08G18/4845—Polyethers containing oxyethylene units and other oxyalkylene units containing oxypropylene or higher oxyalkylene end groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
- C08G65/2642—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds characterised by the catalyst used
- C08G65/2669—Non-metals or compounds thereof
- C08G65/2672—Nitrogen or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2110/00—Foam properties
- C08G2110/0025—Foam properties rigid
Definitions
- the present invention relates to a process for the preparation of a polyether polyol . More in particular, the present invention relates to a process for the preparation of a polyether polyol which is suitable for preparing rigid polyurethane foams therefrom. Further, the present invention relates to a polyether polyol obtainable by such process. Still further, the present invention relates to a process for preparing a polyurethane foam by foaming a composition comprising such polyether polyol and a polyisocyanate component, to a polyurethane foam obtainable by the latter process, and to a shaped article comprising such polyurethane foam.
- Rigid polyurethane foams are well known in the art and have numerous applications, particularly as an insulating material. Examples include insulation of refrigerators and freezers, insulation of pipes and tanks in industrial plants and use as insulating material in the construction industry.
- Polyether polyols which are suitable for preparing rigid polyurethane foams therefrom, are in this specification simply referred to as "rigid polyether polyols".
- polyether polyols are made by the polymerization reaction of an alkylene oxide with a polyhydric alcohol containing at least two active hydrogen atoms, the latter compound functioning as a starter or initiator, in the presence of a catalyst.
- Rigid polyether polyols are generally characterized as adducts of the alkylene oxide to polyols having 3 to 8 hydroxyl groups per molecule, the chain derived from one hydroxyl group being relatively short, around 0.5 to 2 alkylene oxide units.
- Polyether polyols which are suitable for preparing flexible polyurethane foams therefrom (hereinafter "flexible polyether polyols"), generally have longer chains attached to each hydroxyl group of the starter.
- the hydroxyl number of rigid poleyether polyols is relatively high, generally in the range of 300 to 800 mg KOH/g, as compared to that of flexible polyether polyols.
- the molecular weight of rigid polyether polyols is relatively low, generally 100 to 800 g/mole, as compared to that of flexible polyether polyols.
- tertiary amine catalysts generally may be left in the final product as they do not inhibit but in fact may catalyze such subsequent polyurethane forming reaction.
- tertiary amine catalysts have lower activity as compared to alkali metal hydroxide catalysts .
- the objective of the present invention is to provide a process for the preparation of a polyether polyol, more in particular a rigid polyether polyol, wherein the catalyst to be used is one which both has a high activity and does not have to be removed from the final polyol product before the latter can be used in the production of rigid polyurethane foams.
- the present invention relates to a process for the preparation of a polyether polyol, which comprises reacting an alkylene oxide with a polyhydric alcohol or a mixture of polyhydric alcohols, the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols containing at most ten active hydrogen atoms, in the presence of a catalyst comprising a guanidine of formula
- R 1 R 2 N-Cf NH)-NR 3 R 4 (I) wherein R 1 , R 2 , R 3 and R 4 may be the same or different and each of R 1 , R 2 , R 3 and R 4 is hydrogen or hydrocarbyl, at a reaction temperature in the range of from 85 to 170 0 C, in which process preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols is substantially absent.
- an alkylene oxide is reacted with a polyhydric alcohol or a mixture of polyhydric alcohols. It is required that the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols contains at most ten active hydrogen atoms, more preferably at most nine active hydrogen atoms, and most preferably at most eight active hydrogen atoms. Further, the polyhydric alcohol (s) should contain at least two active hydrogen atoms, more preferably at least three active hydrogen atoms. A wide range of polyhydric alcohols may be used in the present process, as long as they contain at least two and at most ten active hydrogen atoms. Polyhydric polyols containing more than ten active hydrogen atoms should not be used.
- the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols contains at least two and at most eight active hydrogen atoms.
- the polyhydric alcohol may be any alcohol or mixture of alcohols having a Fn (hydroxyl functionality) of from 2.0 to at most 10.0, preferably of from 2.0 to 8.0.
- Fn hydroxyl functionality
- examples include diols like diethylene glycol, monoethylene glycol, monopropylene glycol, dipropylene glycol and diethanol amine, and polyols like glycerol, trimethylol propane, sucrose, sorbitol, pentaerythritol and diglycerine.
- polyhydric alcohols having a relatively high Fn may be used, such as sucrose which has 8 OH groups and sorbitol which has 6 OH groups.
- sorbitol is used as a polyhydric alcohol, more preferably in combination with some water to keep the sorbitol sufficiently fluid which depends on the reaction temperature concerned, as further discussed below.
- Water is the preferred "solvent” as it will also act as an initiator.
- Other low molecular weight initiators e.g. diethanol amine, may also be used in admixture with the higher boiling polyhydric alcohols, in such amounts so that no additional solvent is needed.
- an inert solvent for the solid polyol starter, because the use of an inert solvent would disadvantageously lead to the need of recycling and a more complicated installation and loss of capacity.
- diethanol amine and glycerol are suitable polyhydric alcohol initiators . All of said polyol starters may be used alone or in admixture with each other .
- the amount of water used in the reaction mixture may be of from 0.5 to 5 wt.%, based on total amount of the polyhydric alcohol (s) and water.
- Diols in the final polyether polyol may have originated from the reaction of water with the alkylene oxide. An increase in the diols content leads to a decrease of the average Fn of the final polyether polyol. This may be undesired in some applications of rigid polyols.
- the amount of water used in the reaction mixture is of from 0.5 to 2.5 wt.%, more preferably 0.5 to 2.0 wt.%, most preferably 1 to 1.5 wt.%, based on total amount of the polyhydric alcohol (s) and water.
- preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols is substantially absent.
- the amount of such preformed reaction product is less than 5 wt . % based on the combined weight of the polyhydric alcohol or mixture of polyhydric alcohols and said preformed reaction product.
- the amount of such preformed reaction product is less than 4 wt.%, more preferably less than 3 wt.%, more preferably less than 2.5 wt.%, more preferably less than 2 wt.%, more preferably less than 1.5 wt.%, more preferably less than 1 wt.%, more preferably less than 0.7 wt.%, and most preferably less than 0.5 wt.%.
- the latter amounts are not sufficient so that the preformed reaction product could act as a solvent for the polyhydric alcohol (s) and/or as a reaction medium for the process.
- US3510471A discloses the use of tetramethyl guanidine in a process for preparing a polyether polyol .
- the process of US3510471A is characterized in that a mixture of starter polyols is used, wherein one of the starter polyols contains at least three monosaccharide units and should have at least eleven hydroxyl groups, whereas the other starter polyol has three to nine hydroxyl groups.
- the disadvantage of using such starter polyol which contains at least three monosaccharide units and has at least eleven hydroxyl groups is its relatively high viscosity, because of which it is difficult to handle in practice.
- US3346557A also discloses the use of tetramethyl guanidine in a process for preparing a polyether polyol.
- the process of US3346557A is characterized in that it is a multi-stage process. First an adduct of the polyol starter with alkylene oxide is formed, which is then used as a solvent in a next alkoxylation step. It is required in US3346557A that the amount of said adduct employed being sufficient so that it can act as the solvent and reaction medium.
- the catalyst to be used in the present invention is a guanidine of formula
- R 1 R 2 N-Cf NH)-NR 3 R 4 (I) wherein R 1 , R 2 , R 3 and R 4 may be the same or different and each of R 1 , R 2 , R 3 and R 4 is hydrogen or hydrocarbyl.
- R 1 , R 2 , R 3 and R 4 are the same.
- the hydrocarbyl group is an aryl group or a linear or cyclic alkyl group, preferably a linear or cyclic Ci to Ci 0 alkyl group, more preferably a linear or cyclic Ci to C 6 alkyl group, and most preferably methyl.
- the guanidine to be used in the present invention is tetramethyl guanidine or N,N,N',N'- tetramethyl guanidine or 1, 1, 3, 3-tetramethyl guanidine (TMG), i.e. a compound of formula (I) wherein all of R 1 , R 2 , R 3 and R 4 are methyl.
- TMG 1, 1, 3, 3-tetramethyl guanidine
- Alkylene oxides usually applied, and also useful for the present invention, are ethylene oxide, propylene oxide and butylene oxide or mixtures thereof.
- propylene oxide is, however, preferred to use propylene oxide as the sole alkylene oxide .
- the present process can be carried out in any way known to a skilled person as long as a guanidine catalyst is used, as defined above.
- preparing polyether polyols by alkoxylating a polyhydric alcohol i.e. reacting an alkylene oxide with a polyhydric alcohol
- One preferred way of carrying out the present process is one wherein in a first step the catalyst and the polyhydric alcohol (s) are charged into a reactor.
- the catalyst is suitably added to the reactor after all polyhydric alcohol (s) has (have) been added and before the alkylene oxide is added.
- said catalyst and polyhydric alcohol (s) are added all at once.
- the amount of catalyst used is in the range normally applied, i.e.
- the reactor After adding the polyhydric alcohol (s) and catalyst, the reactor is brought to the reaction conditions in terms of a nitrogen atmosphere, temperature and pressure.
- the reaction temperature is comprised in the range of from 85 to 170 0 C, more preferably 95 to 155 0 C, and most preferably 100 to 140 0 C.
- the pressure may be up to and including 10 bar.
- the addition of the alkylene oxide is started. Preferably, said alkylene oxide is added incrementally or gradually.
- the remaining alkylene oxide is allowed to react during a certain time period (so-called after-reaction or digestion time) .
- the volatile compounds e.g. alkylene oxide, such as propylene oxide, and water
- the reduced pressure during said stripping may be 1 to 200 mbara.
- Said stripping may be carried out ambient temperature, but preferably heating takes place during stripping, preferably at a temperature between 90 and 180 0 C, more preferably 110 and 160 0 C, and most preferably 120 and 140 0 C.
- the polyol product is treated with a carboxylic acid, such as for example formic acid or acetic acid, to at least partly neutralize any remaining amine containing compounds .
- the catalyst and/or catalyst derivatives do not have to be removed from the final polyether polyol product which may be used as such in preparing rigid polyurethane foam. Purification and/or filtration are not necessary. Since the polyether polyol that is directly obtained in the present process, still contains the guanidine catalyst and/or derivatives therefrom, it has another composition than prior art polyether polyols made in a similar process but using another catalyst. Therefore, the present invention also relates to a polyether polyol, preferably a polyether polyol that has not been filtered and/or purified, obtainable by the process of the present invention .
- the polyether polyols obtainable by the process of the present invention have hydroxyl values in the range of from 300 to 800, more preferably 300 to 700, and most preferably 400 to 650 mg KOH/g.
- the hydroxyl functionality (Fn) of said polyether polyols is preferably of from 2.0 to 6.0, more preferably 2.5 to 5.5.
- the molecular weight of said polyether polyols is preferably of from 100 to 800, more preferably 200 to 800, and most preferably 200 to 700 g/mole.
- the viscosity of said polyether polyols may vary widely, e.g. in the range of from 100 to 4,000 mm 2 /s (at 40 0 C) .
- the polyether polyol obtainable by the process of the present invention is very suitable for the preparation of polyurethane foams, especially rigid polyurethane foams, by reacting it with a suitable polyisocyanate in the presence of one or more suitable polyurethane catalysts, a suitable blowing agent, one or more surfactants, and optionally a cross-linking agent.
- This reaction is also commonly denoted as foaming.
- the present invention also relates to a process for preparing a polyurethane foam by foaming a composition comprising a polyether polyol obtainable by the process of the present invention and a polyisocyanate component.
- the polyether polyol is not purified prior to use in the foaming reaction.
- the present invention relates to a polyurethane foam obtainable by said foaming process. Still further, the present invention relates to a shaped article comprising said polyurethane foam.
- polyurethane catalysts are known in the art and include many different compounds.
- suitable catalysts include tin-based catalysts, such as tin salts and dialkyl tin salts of carboxylic acids. Specific examples are stannous octoate, stannous oleate, dibutyltin dilaureate, dibutyltin acetate and dibutyltin diacetate.
- Other suitable catalysts are tertiary amines, such as, for instance, bis (2, 2 ' -dimethylamino) ethyl ether, trimethylamine, triethylamine, triethylenediamine and dimethylethanol- amine.
- tertiary amine catalysts examples include those sold under the tradenames NIAX, TEGOAMIN and DABCO (all trademarks) .
- the catalyst is typically used in an amount of from 0.01 to 2.0 parts by weight per hundred parts by weight of polyether polyol (php) .
- Preferred amounts of catalyst are from 0.05 to 1.0 php .
- Suitable blowing agents include water, acetone, (liquid) carbon dioxide, halogenated hydrocarbons, aliphatic alkanes and alicyclic alkanes. Due to the ozone depleting effect of the fully chlorinated, fluorinated alkanes (CFCs) the use of this type of blowing agents is generally not preferred, although it is possible to use them within the scope of the present invention.
- CFCs fluorinated alkanes
- the halogenated alkanes, wherein at least one hydrogen atom has not been substituted by a halogen atom have no or hardly any ozone depleting effect and therefore are the preferred halogenated hydrocarbons to be used in physically blown foams .
- a very suitable HCFC type blowing agent is 1-chloro-l, 1-dif luoroethane .
- the use of water as a (chemical) blowing agent is also well known. Water reacts with isocyanate groups according to the well known NCO/H2O reaction, thereby releasing carbon dioxide which causes the blowing to occur.
- the aliphatic and alicyclic alkanes were developed as alternative blowing agents for the CFC ' s . Examples of such alkanes are n-pentane and n-hexane (aliphatic) and cyclopentane and cyclohexane (alicyclic) .
- blowing agents may be used singly or in mixtures of two or more.
- the amounts wherein the blowing agents are to be used are those convention- ally applied, i.e. between 0.1 to 5 php in case of water and between about 0.1 and 20 php in case of halogenated hydrocarbons, aliphatic alkanes and alicyclic alkanes.
- Example 1 In addition, other well known auxiliaries, such as flame retardants and fillers, may also be used.
- auxiliaries such as flame retardants and fillers.
- the invention is further illustrated by the following Examples .
- the activity of the DMCHA catalyst was measured by determining the number of moles of converted PO per mole of catalyst per hour.
- the catalyst activity amounted to 433 mole/mole. h.
- Example 2 Upon comparing the results of Example 1 with those of Comparative Example 1, it can clearly be seen that the activity of the TMG catalyst is considerably higher than that of the DMCHA catalyst.
- Example 2 and Comparative Example 2
- Example 2 In Example 2 and Comparative Example 2, different catalysts were used for reacting sorbitol (and water) with propylene oxide (PO) .
- the catalyst amounts used were such that per kg of final polyol product 57 mmoles of catalyst (s) had been used, both in Example 2 and in Comparative Example 2.
- Example 2 0.65 wt.% of 1, 1, 3, 3-tetramethyl guanidine (TMG), based on total reaction mixture, was used as the catalyst.
- TMG 1, 1, 3, 3-tetramethyl guanidine
- Comparative Example 2 0.6 wt . % of a mixture of catalysts, based on total reaction mixture, was used, namely 0.3 wt.% of dimethylamino ethanol (DMAE) admixed with 0.3 wt.% of dimethyl cyclohexyl amine (DMCHA) .
- DMAE dimethylamino ethanol
- DMCHA dimethyl cyclohexyl amine
- Example 2 The polyol product that was obtained directly after reacting down, in Example 2 contained 524 mg PO/kg, whereas in Comparative Example 2 this was 1247 mg PO/kg. Therefore, the crude polyol product of Comparative Example 2 contained relatively more unconverted PO. Furthermore, as can be seen from Table 2, the total reaction time in Example 2 was considerably shorter than in Comparative Example 2. This also indicates that the activity of the TMG catalyst used in Example 2 was considerably higher than that of the mixture of DMAE and DMCHA catalysts used in Comparative Example 2.
- Example 2 After reacting down, the polyol product was subjected to a stripping procedure involving heating said product to a temperature of 130 to 135 0 C under a reduced pressure of 15 mbara. Said vacuum and temperature were maintained for 0.5 to 2 hours.
- the amount of condensate i.e. the amount of volatile compounds removed by the stripping procedure, was determined.
- the amount of condensate was 4.3 g whereas for Comparative Example 2, it was 26.5 g. This indicates that by using TMG rather than a mixture of DMAE and DMCHA as the catalyst, a relatively smaller amount of volatile byproducts is formed. Further, because when using TMG as the catalyst less condensate is formed upon stripping, the time needed for working up the crude reaction mixture is considerably shorter.
- the stripped product in both Example 2 and Comparative Example 2 was then cooled to a temperature of about 80 0 C, treated with 2.8 g of formic acid, and finally further cooled to a temperature of about 30 0 C.
- the colour properties of the polyols were determined by the ASTM D1209 method. In this method, the colour of a sample is compared with the colour of aqueous Pt/Co standard solutions, reported as mg/1 Pt/Co in water. As can be seen from Table 3, the polyol obtained in Example 2 contained far less colour impurities than the polyol obtained in Comparative Example 2. Therefore, the latter polyol may need to be subjected to a colour removal method before it even can be used in making polyurethane from it. Such additional step is cumbersome. Examples 3 and 4
- the diols content for the polyols obtained in Example 2 and Comparative Example 2 were 7 and 4 wt.%, respectively (see Table 3) .
- the water content was 3.0 wt.% based on total amount of sorbitol and water. It appeared that in Example 2 approximately twice the molar amount of water had reacted with PO, as compared with Comparative Example 2. Therefore, the TMG catalyst has a higher activity towards water than the DMAE and DMCHA catalyst mixture .
- Example 3 the procedure of Example 2 was repeated unless indicated otherwise below.
- the catalyst amount used was such that per kg of final polyol product 38 mmoles of catalyst had been used.
- Example 3 the water content was 3.0 wt.% based on total amount of sorbitol and water, which is the same as the water content used in Example 2 and Comparative Example 2. However, in Example 4, the water content was only 1.3 wt.% based on total amount of sorbitol and water
- Table 5 The addition times and reacting down times for Examples 3 and 4 are mentioned in Table 5.
- the final polyol products obtained after the stripping and acid treatment procedures had the properties as shown in Table 6.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyurethanes Or Polyureas (AREA)
- Polyethers (AREA)
Abstract
The invention relates to a process for the preparation of a polyether polyol, which comprises reacting an alkylene oxide with a polyhydric alcohol or a mixture of polyhydric alcohols, the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols containing at most ten active hydrogen atoms, in the presence of a catalyst comprising a guanidine of formula: R1R2N-C(=NH) -NR3R4 (I) wherein R1, R2, R3 and R4 may be the same or different and each of R1, R2, R3 and R4 is hydrogen or hydrocarbyl, at a reaction temperature in the range of from 85 to 170 °C, in which process preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols is substantially absent.
Description
PROCESS FOR THE PREPARATION OF A POLYETHER POLYOL SUITABLE FOR PREPARING RIGID POLYURETHANE FOAMS
The present invention relates to a process for the preparation of a polyether polyol . More in particular, the present invention relates to a process for the preparation of a polyether polyol which is suitable for preparing rigid polyurethane foams therefrom. Further, the present invention relates to a polyether polyol obtainable by such process. Still further, the present invention relates to a process for preparing a polyurethane foam by foaming a composition comprising such polyether polyol and a polyisocyanate component, to a polyurethane foam obtainable by the latter process, and to a shaped article comprising such polyurethane foam.
Rigid polyurethane foams are well known in the art and have numerous applications, particularly as an insulating material. Examples include insulation of refrigerators and freezers, insulation of pipes and tanks in industrial plants and use as insulating material in the construction industry. Polyether polyols which are suitable for preparing rigid polyurethane foams therefrom, are in this specification simply referred to as "rigid polyether polyols".
In general, polyether polyols are made by the polymerization reaction of an alkylene oxide with a polyhydric alcohol containing at least two active hydrogen atoms, the latter compound functioning as a starter or initiator, in the presence of a catalyst. Rigid polyether polyols are generally characterized as adducts of the alkylene oxide to polyols having 3 to 8 hydroxyl groups per molecule, the chain derived from one
hydroxyl group being relatively short, around 0.5 to 2 alkylene oxide units. Polyether polyols, which are suitable for preparing flexible polyurethane foams therefrom (hereinafter "flexible polyether polyols"), generally have longer chains attached to each hydroxyl group of the starter. Further, the hydroxyl number of rigid poleyether polyols is relatively high, generally in the range of 300 to 800 mg KOH/g, as compared to that of flexible polyether polyols. On the other hand, the molecular weight of rigid polyether polyols is relatively low, generally 100 to 800 g/mole, as compared to that of flexible polyether polyols.
As mentioned in Chapter 13 ("Polyether Polyols for Rigid Polyurethane Foams") of "Chemistry and Technology of Polyols for Polyurethanes " , Rapra Technology Ltd., 2005, the most important catalysts used in industrial practice for the preparation of rigid polyether polyol are basic catalysts, such as alkali metal hydroxides, such as potassium hydroxide (KOH) , and tertiary amines, such as dimethylamino ethanol (DMAE) and dimethyl cyclohexyl amine (DMCHA) . Generally, where an alkali metal hydroxide has been used it must be removed from the final product by neutralisation and further purification in order to prevent it from interfering in the subsequent polyurethane forming reaction. On the other hand, tertiary amine catalysts generally may be left in the final product as they do not inhibit but in fact may catalyze such subsequent polyurethane forming reaction. However, unfortunately, tertiary amine catalysts have lower activity as compared to alkali metal hydroxide catalysts .
Although the prior art rigid polyol preparation processes perform satisfactory in many respects, there is still room for improvement, as outlined above.
The objective of the present invention is to provide a process for the preparation of a polyether polyol, more in particular a rigid polyether polyol, wherein the catalyst to be used is one which both has a high activity and does not have to be removed from the final polyol product before the latter can be used in the production of rigid polyurethane foams.
The catalyst found by the present inventors to meet the above requirements was a guanidine catalyst. More in particular, the present invention relates to a process for the preparation of a polyether polyol, which comprises reacting an alkylene oxide with a polyhydric alcohol or a mixture of polyhydric alcohols, the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols containing at most ten active hydrogen atoms, in the presence of a catalyst comprising a guanidine of formula
R1R2N-Cf=NH)-NR3R4 (I) wherein R1, R2, R3 and R4 may be the same or different and each of R1, R2, R3 and R4 is hydrogen or hydrocarbyl, at a reaction temperature in the range of from 85 to 170 0C, in which process preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols is substantially absent.
In the process of the present invention, an alkylene oxide is reacted with a polyhydric alcohol or a mixture of polyhydric alcohols. It is required that the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols contains at most ten active hydrogen atoms, more preferably at most nine
active hydrogen atoms, and most preferably at most eight active hydrogen atoms. Further, the polyhydric alcohol (s) should contain at least two active hydrogen atoms, more preferably at least three active hydrogen atoms. A wide range of polyhydric alcohols may be used in the present process, as long as they contain at least two and at most ten active hydrogen atoms. Polyhydric polyols containing more than ten active hydrogen atoms should not be used. Preferably, the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols contains at least two and at most eight active hydrogen atoms. Thus, the polyhydric alcohol may be any alcohol or mixture of alcohols having a Fn (hydroxyl functionality) of from 2.0 to at most 10.0, preferably of from 2.0 to 8.0. Examples include diols like diethylene glycol, monoethylene glycol, monopropylene glycol, dipropylene glycol and diethanol amine, and polyols like glycerol, trimethylol propane, sucrose, sorbitol, pentaerythritol and diglycerine. Thus, for example, polyhydric alcohols having a relatively high Fn may be used, such as sucrose which has 8 OH groups and sorbitol which has 6 OH groups. Preferably, sorbitol is used as a polyhydric alcohol, more preferably in combination with some water to keep the sorbitol sufficiently fluid which depends on the reaction temperature concerned, as further discussed below. Water is the preferred "solvent" as it will also act as an initiator. Other low molecular weight initiators, e.g. diethanol amine, may also be used in admixture with the higher boiling polyhydric alcohols, in such amounts so that no additional solvent is needed. In general, it is not preferred to use an inert solvent for the solid polyol starter, because the use of an inert solvent would disadvantageously lead to the need of
recycling and a more complicated installation and loss of capacity. Further, diethanol amine and glycerol are suitable polyhydric alcohol initiators . All of said polyol starters may be used alone or in admixture with each other .
Generally, in a case where water is used in addition to the polyhydric alcohol (s), the amount of water used in the reaction mixture may be of from 0.5 to 5 wt.%, based on total amount of the polyhydric alcohol (s) and water. Diols in the final polyether polyol may have originated from the reaction of water with the alkylene oxide. An increase in the diols content leads to a decrease of the average Fn of the final polyether polyol. This may be undesired in some applications of rigid polyols. Therefore, preferably, when performing the present process with water as an initiator in addition to the polyhydric alcohol (s), especially sorbitol, the amount of water used in the reaction mixture is of from 0.5 to 2.5 wt.%, more preferably 0.5 to 2.0 wt.%, most preferably 1 to 1.5 wt.%, based on total amount of the polyhydric alcohol (s) and water. Surprisingly, it has been found, as demonstrated in the Examples below, that by using such relatively low water content in the present process, a diols content in the final polyether polyol can be achieved which is the same as that in a case where a catalyst other than a catalyst comprising a guanidine of above formula (I) is used.
Further, it is required that in the process of the present invention, preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols is substantially absent. In the present specification, this means first of all, that during the present process no such preformed reaction
product is added, and secondly, that in a case where during the present process some of such preformed reaction product is present, the amount thereof is not sufficient so that it could act as a solvent for the polyhydric alcohol (s) and/or as a reaction medium for the process .
It is envisaged that some preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols originating from a previous batch reaction in the same reactor vessel, sometimes also referred to as "heel", is present during the present process. However, the amount of such preformed reaction product is less than 5 wt . % based on the combined weight of the polyhydric alcohol or mixture of polyhydric alcohols and said preformed reaction product. Preferably, the amount of such preformed reaction product is less than 4 wt.%, more preferably less than 3 wt.%, more preferably less than 2.5 wt.%, more preferably less than 2 wt.%, more preferably less than 1.5 wt.%, more preferably less than 1 wt.%, more preferably less than 0.7 wt.%, and most preferably less than 0.5 wt.%. The latter amounts are not sufficient so that the preformed reaction product could act as a solvent for the polyhydric alcohol (s) and/or as a reaction medium for the process.
US3510471A discloses the use of tetramethyl guanidine in a process for preparing a polyether polyol . The process of US3510471A is characterized in that a mixture of starter polyols is used, wherein one of the starter polyols contains at least three monosaccharide units and should have at least eleven hydroxyl groups, whereas the other starter polyol has three to nine hydroxyl groups. The disadvantage of using such starter
polyol which contains at least three monosaccharide units and has at least eleven hydroxyl groups, is its relatively high viscosity, because of which it is difficult to handle in practice. This is different from the present invention because in a case where a mixture of polyhydric alcohols is used as a starter in the present process, each of the polyhydric alcohols in such mixture may contain at most ten active hydrogen atoms . US3346557A also discloses the use of tetramethyl guanidine in a process for preparing a polyether polyol. The process of US3346557A is characterized in that it is a multi-stage process. First an adduct of the polyol starter with alkylene oxide is formed, which is then used as a solvent in a next alkoxylation step. It is required in US3346557A that the amount of said adduct employed being sufficient so that it can act as the solvent and reaction medium. This is different from the present invention because for the present process it is required that preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols is substantially absent, as defined above. This greatly simplifies the present process, in that no prepolymerisation is required and the final product is thus produced faster. Further, the use of a relatively large amount of heel leads to a considerable loss of capacity and leads to an increase in the molecular weight of the product and thus to variation in product performance which is highly undesired. These disadvantages are avoided with the present invention. In addition, the polyether polyol prepared in accordance with the process of the present invention is substantially free of undesired by-products .
Thus, the catalyst to be used in the present invention, is a guanidine of formula
R1R2N-Cf=NH)-NR3R4 (I) wherein R1, R2, R3 and R4 may be the same or different and each of R1, R2, R3 and R4 is hydrogen or hydrocarbyl. Preferably, R1, R2, R3 and R4 are the same. Further, preferably, the hydrocarbyl group is an aryl group or a linear or cyclic alkyl group, preferably a linear or cyclic Ci to Ci0 alkyl group, more preferably a linear or cyclic Ci to C6 alkyl group, and most preferably methyl. Most preferably, the guanidine to be used in the present invention is tetramethyl guanidine or N,N,N',N'- tetramethyl guanidine or 1, 1, 3, 3-tetramethyl guanidine (TMG), i.e. a compound of formula (I) wherein all of R1, R2, R3 and R4 are methyl.
Alkylene oxides usually applied, and also useful for the present invention, are ethylene oxide, propylene oxide and butylene oxide or mixtures thereof. For preparing rigid polyether polyols it is, however, preferred to use propylene oxide as the sole alkylene oxide .
The present process can be carried out in any way known to a skilled person as long as a guanidine catalyst is used, as defined above. In general, preparing polyether polyols by alkoxylating a polyhydric alcohol, i.e. reacting an alkylene oxide with a polyhydric alcohol, is well known in the art. One preferred way of carrying out the present process is one wherein in a first step the catalyst and the polyhydric alcohol (s) are charged into a reactor. The catalyst is suitably added to the reactor after all polyhydric alcohol (s) has (have) been added and before the alkylene oxide is added. Preferably, said catalyst and polyhydric alcohol (s) are
added all at once. The amount of catalyst used is in the range normally applied, i.e. from 0.05 to 2% by weight on final product. After adding the polyhydric alcohol (s) and catalyst, the reactor is brought to the reaction conditions in terms of a nitrogen atmosphere, temperature and pressure. In the present process, the reaction temperature is comprised in the range of from 85 to 170 0C, more preferably 95 to 155 0C, and most preferably 100 to 140 0C. The pressure may be up to and including 10 bar. In a second step, the addition of the alkylene oxide is started. Preferably, said alkylene oxide is added incrementally or gradually. In a third step, after completion of the alkylene oxide addition, the remaining alkylene oxide is allowed to react during a certain time period (so-called after-reaction or digestion time) . If the reaction of the alkylene oxide is completed, then in a fourth step the volatile compounds (e.g. alkylene oxide, such as propylene oxide, and water) present are removed from the product under reduced pressure, which is called "stripping". The reduced pressure during said stripping may be 1 to 200 mbara. Said stripping may be carried out ambient temperature, but preferably heating takes place during stripping, preferably at a temperature between 90 and 180 0C, more preferably 110 and 160 0C, and most preferably 120 and 140 0C. Preferably, after said stripping, the polyol product is treated with a carboxylic acid, such as for example formic acid or acetic acid, to at least partly neutralize any remaining amine containing compounds . The catalyst and/or catalyst derivatives do not have to be removed from the final polyether polyol product which may be used as such in preparing rigid polyurethane foam. Purification and/or filtration are not necessary.
Since the polyether polyol that is directly obtained in the present process, still contains the guanidine catalyst and/or derivatives therefrom, it has another composition than prior art polyether polyols made in a similar process but using another catalyst. Therefore, the present invention also relates to a polyether polyol, preferably a polyether polyol that has not been filtered and/or purified, obtainable by the process of the present invention . Preferably the polyether polyols obtainable by the process of the present invention have hydroxyl values in the range of from 300 to 800, more preferably 300 to 700, and most preferably 400 to 650 mg KOH/g. Further, the hydroxyl functionality (Fn) of said polyether polyols is preferably of from 2.0 to 6.0, more preferably 2.5 to 5.5. Still further, the molecular weight of said polyether polyols is preferably of from 100 to 800, more preferably 200 to 800, and most preferably 200 to 700 g/mole. The viscosity of said polyether polyols may vary widely, e.g. in the range of from 100 to 4,000 mm2/s (at 40 0C) .
The polyether polyol obtainable by the process of the present invention is very suitable for the preparation of polyurethane foams, especially rigid polyurethane foams, by reacting it with a suitable polyisocyanate in the presence of one or more suitable polyurethane catalysts, a suitable blowing agent, one or more surfactants, and optionally a cross-linking agent. This reaction is also commonly denoted as foaming. Therefore, the present invention also relates to a process for preparing a polyurethane foam by foaming a composition comprising a polyether polyol obtainable by the process of the present invention and a polyisocyanate
component. Preferably, the polyether polyol is not purified prior to use in the foaming reaction.
Further, the present invention relates to a polyurethane foam obtainable by said foaming process. Still further, the present invention relates to a shaped article comprising said polyurethane foam.
Polyurethane catalysts are known in the art and include many different compounds. For the purpose of the present invention, suitable catalysts include tin-based catalysts, such as tin salts and dialkyl tin salts of carboxylic acids. Specific examples are stannous octoate, stannous oleate, dibutyltin dilaureate, dibutyltin acetate and dibutyltin diacetate. Other suitable catalysts are tertiary amines, such as, for instance, bis (2, 2 ' -dimethylamino) ethyl ether, trimethylamine, triethylamine, triethylenediamine and dimethylethanol- amine. Examples of commercially available tertiary amine catalysts are those sold under the tradenames NIAX, TEGOAMIN and DABCO (all trademarks) . The catalyst is typically used in an amount of from 0.01 to 2.0 parts by weight per hundred parts by weight of polyether polyol (php) . Preferred amounts of catalyst are from 0.05 to 1.0 php .
Suitable blowing agents include water, acetone, (liquid) carbon dioxide, halogenated hydrocarbons, aliphatic alkanes and alicyclic alkanes. Due to the ozone depleting effect of the fully chlorinated, fluorinated alkanes (CFCs) the use of this type of blowing agents is generally not preferred, although it is possible to use them within the scope of the present invention. The halogenated alkanes, wherein at least one hydrogen atom has not been substituted by a halogen atom (the so-called HCFCs) have no or hardly any ozone depleting effect and
therefore are the preferred halogenated hydrocarbons to be used in physically blown foams . A very suitable HCFC type blowing agent is 1-chloro-l, 1-dif luoroethane . The use of water as a (chemical) blowing agent is also well known. Water reacts with isocyanate groups according to the well known NCO/H2O reaction, thereby releasing carbon dioxide which causes the blowing to occur. The aliphatic and alicyclic alkanes, finally, were developed as alternative blowing agents for the CFC ' s . Examples of such alkanes are n-pentane and n-hexane (aliphatic) and cyclopentane and cyclohexane (alicyclic) . It will be understood that the above blowing agents may be used singly or in mixtures of two or more. The amounts wherein the blowing agents are to be used are those convention- ally applied, i.e. between 0.1 to 5 php in case of water and between about 0.1 and 20 php in case of halogenated hydrocarbons, aliphatic alkanes and alicyclic alkanes.
In addition, other well known auxiliaries, such as flame retardants and fillers, may also be used. The invention is further illustrated by the following Examples . Example 1
50 g of glycerol (0.54 mole) and 0.10 ml of 1,1,3,3- tetramethyl guanidine (0.8 mmole; TMG catalyst) were charged into an autoclave. The temperature inside the autoclave was raised to 130 0C and the autoclave was flushed with nitrogen. Propylene oxide (PO) was then added to the autoclave at such a rate that the pressure was maintained at 4 bar . After the consumption of PO stopped, the activity of the TMG catalyst was measured by determining the number of moles of converted PO per mole of catalyst per hour. The catalyst activity amounted to 2,500 mole/mole. h.
Comparative Example 1
50 g of glycerol (0.54 mole) and 0.12 ml of dimethyl cyclohexyl amine (0.8 mmole; DMCHA catalyst) were charged into an autoclave. The temperature inside the autoclave was raised to 130 0C and the autoclave was flushed with nitrogen. Propylene oxide (PO) was then added to the autoclave at such a rate that the pressure was maintained at 4 bar .
After the consumption of PO stopped, the activity of the DMCHA catalyst was measured by determining the number of moles of converted PO per mole of catalyst per hour. The catalyst activity amounted to 433 mole/mole. h.
Upon comparing the results of Example 1 with those of Comparative Example 1, it can clearly be seen that the activity of the TMG catalyst is considerably higher than that of the DMCHA catalyst. Example 2 and Comparative Example 2
In Example 2 and Comparative Example 2, different catalysts were used for reacting sorbitol (and water) with propylene oxide (PO) . The catalyst amounts used were such that per kg of final polyol product 57 mmoles of catalyst (s) had been used, both in Example 2 and in Comparative Example 2.
In Example 2, 0.65 wt.% of 1, 1, 3, 3-tetramethyl guanidine (TMG), based on total reaction mixture, was used as the catalyst. In Comparative Example 2, 0.6 wt . % of a mixture of catalysts, based on total reaction mixture, was used, namely 0.3 wt.% of dimethylamino ethanol (DMAE) admixed with 0.3 wt.% of dimethyl cyclohexyl amine (DMCHA) .
The amounts of sorbitol, water, catalysts and PO used in Example 2 and Comparative Example 2 are mentioned in Table 1.
Table 1

The experiments were carried out as follows. Sorbitol was added to the reactor as a syrup containing about 30 wt .% of water. Under nitrogen the reactor content was heated to a temperature of 130 0C. Under vacuum the water was removed. Then the reactor content was cooled to a temperature of 110 0C, and water and the catalyst (s) in the amounts indicated in Table 1 were added to the reactor. Propylene oxide (PO) was then added to the reactor at such a rate that the pressure remained below 6 bara.
After the total amount of PO indicated in Table 1 had been added during a certain period of time ("addition time"), any unconverted PO was allowed to react at a temperature of 110 0C during a certain period of time ("reacting down time" or "after-reaction time") . During the reacting down time, the pressure in the reactor decreased to a constant value. Said addition times and reacting down times for Example 2 and Comparative Example 2 are mentioned in Table 2.
Table 2

The polyol product that was obtained directly after reacting down, in Example 2 contained 524 mg PO/kg, whereas in Comparative Example 2 this was 1247 mg PO/kg. Therefore, the crude polyol product of Comparative Example 2 contained relatively more unconverted PO. Furthermore, as can be seen from Table 2, the total reaction time in Example 2 was considerably shorter than in Comparative Example 2. This also indicates that the activity of the TMG catalyst used in Example 2 was considerably higher than that of the mixture of DMAE and DMCHA catalysts used in Comparative Example 2.
After reacting down, the polyol product was subjected to a stripping procedure involving heating said product to a temperature of 130 to 135 0C under a reduced pressure of 15 mbara. Said vacuum and temperature were maintained for 0.5 to 2 hours. The amount of condensate, i.e. the amount of volatile compounds removed by the stripping procedure, was determined. For Example 2, the amount of condensate was 4.3 g whereas for Comparative Example 2, it was 26.5 g. This indicates that by using TMG rather than a mixture of DMAE and DMCHA as the catalyst, a relatively smaller amount of volatile byproducts is formed. Further, because when using TMG as the catalyst less condensate is formed upon stripping, the time needed for working up the crude reaction mixture is considerably shorter.
The stripped product in both Example 2 and Comparative Example 2 was then cooled to a temperature of about 800C, treated with 2.8 g of formic acid, and finally further cooled to a temperature of about 300C.
The final polyol products obtained after the stripping and acid treatment procedures had the properties as shown in Table 3.
Table 3
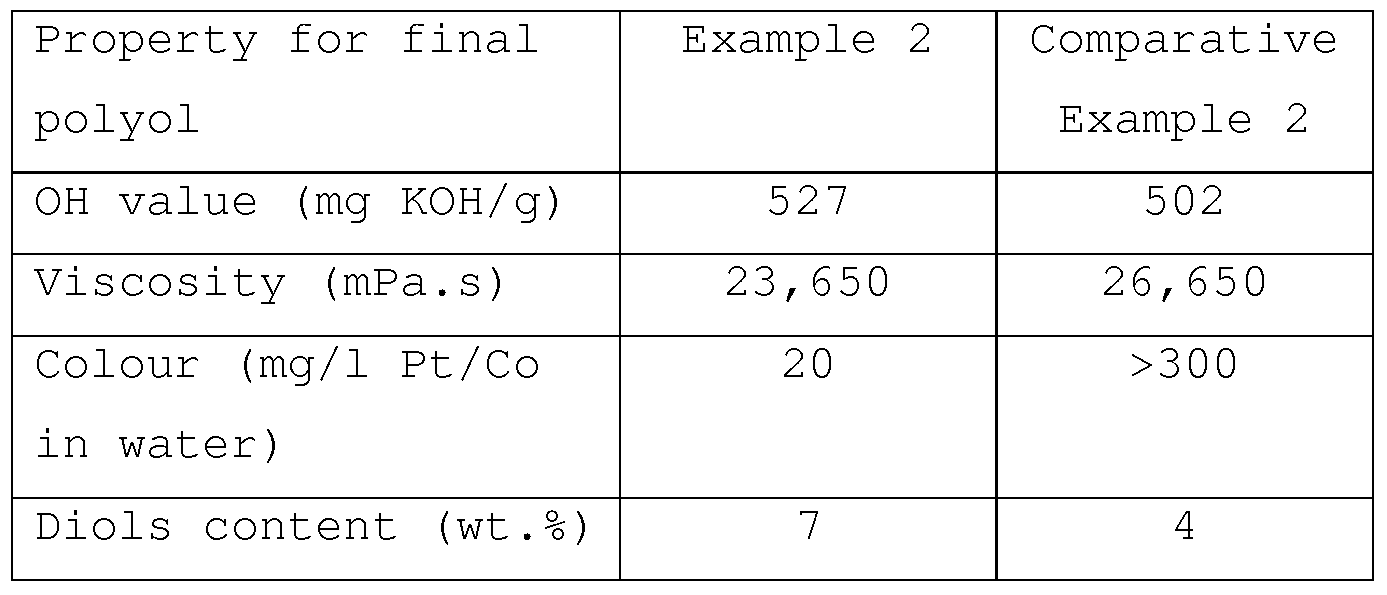
The hydroxyl values of the polyols obtained in Example 2 and Comparative Example 2 were comparable . However, the viscosity of the polyol obtained in Comparative Example 2 was unsatisfactorily higher.
Further, the colour properties of the polyols were determined by the ASTM D1209 method. In this method, the colour of a sample is compared with the colour of aqueous Pt/Co standard solutions, reported as mg/1 Pt/Co in water. As can be seen from Table 3, the polyol obtained in Example 2 contained far less colour impurities than the polyol obtained in Comparative Example 2. Therefore, the latter polyol may need to be subjected to a colour removal method before it even can be used in making polyurethane from it. Such additional step is cumbersome.
Examples 3 and 4
The diols content for the polyols obtained in Example 2 and Comparative Example 2, were 7 and 4 wt.%, respectively (see Table 3) . In both Example 2 and Comparative Example 2, the water content was 3.0 wt.% based on total amount of sorbitol and water. It appeared that in Example 2 approximately twice the molar amount of water had reacted with PO, as compared with Comparative Example 2. Therefore, the TMG catalyst has a higher activity towards water than the DMAE and DMCHA catalyst mixture .
In Examples 3 and 4, the procedure of Example 2 was repeated unless indicated otherwise below. The catalyst amount used was such that per kg of final polyol product 38 mmoles of catalyst had been used. In Examples 3 and 4, 0.43 wt.% of TMG, based on total reaction mixture, was used as the catalyst.
The amounts of sorbitol, water, catalyst and PO used in Examples 3 and 4 are mentioned in Table 4.
Table 4

In Example 3, the water content was 3.0 wt.% based on total amount of sorbitol and water, which is the same as the water content used in Example 2 and Comparative Example 2. However, in Example 4, the water content was only 1.3 wt.% based on total amount of sorbitol and water
The addition times and reacting down times for Examples 3 and 4 are mentioned in Table 5.
Table 5

Further, the amounts of formic acid used in the work-up procedure for Examples 3 and 4, were 4.2 and 4.0 g, respectively.
The final polyol products obtained after the stripping and acid treatment procedures had the properties as shown in Table 6.
Table 6

From Table 6 it appears that the diols content of the polyol obtained in Example 4 was the same as the diols content of the polyol obtained in Comparative Example 2. Further, the viscosity of the polyol from Example 4 was still lower than in Comparative Example 2 Finally, also the colour properties were still much better .
Claims
1. Process for the preparation of a polyether polyol, which comprises reacting an alkylene oxide with a polyhydric alcohol or a mixture of polyhydric alcohols, the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols containing at most ten active hydrogen atoms, in the presence of a catalyst comprising a guanidine of formula
R1R2N-Cf=NH)-NR3R4 (I) wherein R1, R2, R3 and R4 may be the same or different and each of R1, R2, R3 and R4 is hydrogen or hydrocarbyl, at a reaction temperature in the range of from 85 to 170 0C, in which process preformed reaction product of the alkylene oxide with the polyhydric alcohol or mixture of polyhydric alcohols is substantially absent.
2. Process according to claim 1, wherein R1, R2, R3 and R4 in formula (I) for the guanidine are the same and are a linear or cyclic Ci to Ci0 alkyl group.
3. Process according to claim 2, wherein R1, R2, R3 and R4 are methyl.
4. Process according to any one of the preceding claims, wherein the polyhydric alcohol and each of the polyhydric alcohols in the mixture of polyhydric alcohols contains at least two and at most eight active hydrogen atoms .
5. Process according to any one of the preceding claims, wherein the alkylene oxide is propylene oxide.
6. Process according to any one of the preceding claims, wherein the amount of preformed reaction product is less than 5 wt . % based on the combined weight of the polyhydric alcohol or mixture of polyhydric alcohols and preformed reaction product.
7. Polyether polyol obtainable by the process according to any one of claims 1 to 6.
8. Process for preparing a polyurethane foam by foaming a composition comprising a polyether polyol obtainable by the process according to any one of claims 1 to 6 and a polyisocyanate component.
9. Process according to claim 8, wherein the polyether polyol is not purified prior to use.
10. Polyurethane foam obtainable by the process according to claim 8 or 9.
11. Shaped article comprising the polyurethane foam according to claim 10.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07119571.3 | 2007-10-30 | ||
| EP07119571 | 2007-10-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009056513A1 true WO2009056513A1 (en) | 2009-05-07 |
Family
ID=38693356
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/064509 WO2009056513A1 (en) | 2007-10-30 | 2008-10-27 | Process for the preparation of a polyether polyol suitable for preparing rigid polyurethane foams |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009056513A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2551289A1 (en) | 2011-07-26 | 2013-01-30 | Basf Se | Process for the continuous production of polyetherols |
| WO2013149860A1 (en) | 2012-04-03 | 2013-10-10 | Basf Se | Process for the continuous production of polyetherols |
| CN111393631A (en) * | 2020-05-29 | 2020-07-10 | 上海多纶化工有限公司 | Synthesis method of secondary alcohol polyoxyethylene ether |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1082672A (en) * | 1964-06-10 | 1967-09-06 | Pfizer Ltd | Polyethers |
| US3346557A (en) * | 1965-06-04 | 1967-10-10 | Wyandotte Chemicals Corp | Process for oxyalkylating solid polyols |
| US4332936A (en) * | 1978-10-16 | 1982-06-01 | Mobay Chemical Corporation | Method of making polyether polyols from solid hydroxyl containing initiators |
| WO2004020506A2 (en) * | 2002-08-30 | 2004-03-11 | Huntsman Petrochemical Corporation | Polyether polyamine agents and mixtures therefor |
-
2008
- 2008-10-27 WO PCT/EP2008/064509 patent/WO2009056513A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1082672A (en) * | 1964-06-10 | 1967-09-06 | Pfizer Ltd | Polyethers |
| US3346557A (en) * | 1965-06-04 | 1967-10-10 | Wyandotte Chemicals Corp | Process for oxyalkylating solid polyols |
| US4332936A (en) * | 1978-10-16 | 1982-06-01 | Mobay Chemical Corporation | Method of making polyether polyols from solid hydroxyl containing initiators |
| WO2004020506A2 (en) * | 2002-08-30 | 2004-03-11 | Huntsman Petrochemical Corporation | Polyether polyamine agents and mixtures therefor |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2551289A1 (en) | 2011-07-26 | 2013-01-30 | Basf Se | Process for the continuous production of polyetherols |
| WO2013014153A1 (en) | 2011-07-26 | 2013-01-31 | Basf Se | Process for the continuous production of polyetherols |
| WO2013149860A1 (en) | 2012-04-03 | 2013-10-10 | Basf Se | Process for the continuous production of polyetherols |
| CN111393631A (en) * | 2020-05-29 | 2020-07-10 | 上海多纶化工有限公司 | Synthesis method of secondary alcohol polyoxyethylene ether |
| CN111393631B (en) * | 2020-05-29 | 2022-09-09 | 上海多纶化工有限公司 | Synthesis method of secondary alcohol polyoxyethylene ether |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101077759B1 (en) | A process for the production of rigid foams from alkaline polyether polyols | |
| JP3703263B2 (en) | Process for producing polyoxyalkylene polyol | |
| US5010187A (en) | Production of polyether polyols with reduced unsaturation | |
| CN106103520B (en) | Formulated isocyanate-reactive blends including olefin-based blowing agents | |
| US5840781A (en) | Polyether polyols, polyol formulation containing them and their use in the production of hard polyurethane foams | |
| JP5254719B2 (en) | Autocatalytic polyol | |
| RU2585290C2 (en) | Method of producing solid polyurethane foam materials | |
| JP5823496B2 (en) | Method for producing rigid polyurethane foam | |
| KR101475959B1 (en) | Method for the production of rigid polyurethane foams | |
| KR20130052563A (en) | Method for producing polyether alcohols | |
| US20110218259A1 (en) | Preparing polyurethanes | |
| SG192822A1 (en) | Producing pu rigid foams materials | |
| KR20130004587A (en) | Method for producing polyurethanes | |
| US5070125A (en) | Production of polyether polyols with reduced unsaturation | |
| EP0269271A2 (en) | Urea catalyst for preparation of sucrose polyols useful for rigid polyurethane foams | |
| JP5393146B2 (en) | Method for producing polyether alcohol | |
| WO2009056513A1 (en) | Process for the preparation of a polyether polyol suitable for preparing rigid polyurethane foams | |
| CA3183168A1 (en) | A process for recycling a polyurethane material | |
| JPH115833A (en) | Polyether-ester polyol and production of polyurethane resin using the same | |
| WO2002050161A2 (en) | Co-initiated polyols useful for the production of rigid polyurethane foams | |
| JP4282044B2 (en) | Polyester polyol for rigid polyurethane foam, method for producing the same, and method for producing rigid polyurethane foam | |
| JPH10158388A (en) | Modified polyetherpolyol and production of polyurethane resin | |
| KR101832380B1 (en) | Novel tertiary amine-based polyol and application of the same | |
| JP3724928B2 (en) | Method for producing polyoxyalkylene polyamine and method for producing polyurethane urea resin using the same | |
| JP2000017040A (en) | Preparation of polyurethane resin and rigid polyurethane foam |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08844874 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08844874 Country of ref document: EP Kind code of ref document: A1 |