WO2009055557A1 - Process for the preparation of macrolide antibacterial agents - Google Patents
Process for the preparation of macrolide antibacterial agents Download PDFInfo
- Publication number
- WO2009055557A1 WO2009055557A1 PCT/US2008/080936 US2008080936W WO2009055557A1 WO 2009055557 A1 WO2009055557 A1 WO 2009055557A1 US 2008080936 W US2008080936 W US 2008080936W WO 2009055557 A1 WO2009055557 A1 WO 2009055557A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- acyl
- compound
- heteroaryl
- aryl
- Prior art date
Links
- 0 CC[C@]([C@](C)([C@]([C@@](C)C(C)C)N1*B[n]2nncc2)OC1=O)OC([C@](C)(C([C@](C)[C@]([C@](C)(CC(C)C)OC)O[C@@]([C@]1O)O[C@](C)C[C@@]1N(C)C)=O)F)=O Chemical compound CC[C@]([C@](C)([C@]([C@@](C)C(C)C)N1*B[n]2nncc2)OC1=O)OC([C@](C)(C([C@](C)[C@]([C@](C)(CC(C)C)OC)O[C@@]([C@]1O)O[C@](C)C[C@@]1N(C)C)=O)F)=O 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
Definitions
- the invention described herein relates to processes for preparing macrolide antibacterial agents.
- the invention relates to intermediates and processes for preparing ketolides and other macrolides that include a 1,2,3-triazole substituted side chain.
- Erythromycin was the first compound of this class to be introduced into clinical practice. Since then, additional macrolides, including ketolides have garnered much attention for their ability to treat a wide range of disease states. In particular, macrolides are an important component of therapies for treating bacterial, protozoal, and viral infections. In addition, macrolides are often used in patients allergic to penicillins.
- macrolide compounds have been found to be effective for the treatment and prevention of infections caused by a broad spectrum of bacterial and protozoal infections. They are also useful for infections of respiratory tract and soft tissue infections. Macrolide anitbiotics are found to be effective on beta-hemolytic streptococci, pneumococci, staphylococci and enterococci. They are also found to be effective against mycoplasma, mycobacteria, some rickettsia, and chlamydia.
- Macrolide compounds are characterized by the presence of a large lactone ring, which is generally a 14, 15, or 16-membered macrocyclic lactone, to which one or more saccharides, including deoxy sugars such as cladinose and desosamine, may be attached.
- erythromycin is a 14-membered macrolide that includes two sugar moieties.
- Spiramycin belongs to a second generation of macrolide compounds that include a 16- membered ring.
- Third generation macrolide compounds include for example semi-synthetic derivatives of erythromycin A, such as azithromycin and clarithromycin.
- ketolides represent a newer class of macro lide antibiotics that have received much attention recently due to their acid stability, and most importantly due to their excellent activity against organisms that are resistant to other macro lides.
- ketolides are 14-membered ring macro lide derivatives characterized by a keto group at the C-3 position (Curr. Med. Chem., "Anti-Infective Agents," 1 : 15-34 (2002)).
- telithromycin U.S. Patent No. 5,635,485
- telithromycin U.S. Patent No. 5,635,485
- Liang et al. describes compounds including those of formula (I):
- R 1 is a monosaccharide or polysaccharide
- A is -CH 2 -, -C(O)-, -C(O)O-, -C(O)NH-, -S(O)2-, -S(O)2NH-, -C(O)NHS(O)2-;
- B is -(CH 2 )D- where n is an integer ranging from 0-10, or B is an unsaturated carbon chain of 2-10 carbons, which may contain any alkenyl or alkynyl group;
- C represents 1 or 2 substituents independently selected in each instance from hydrogen, halogen, hydroxy, alkyl, aralkyl, alkylaryl, alkoxy, heteroalkyl, aryl, heteroaryl, heteroarylalkyl, aminoaryl, alkylaminoaryl, acyl, acyloxy, sulfonyl, ureyl, and carbamoyl, each of which is optionally substituted;
- W is hydrogen, F, Cl, Br, I, or OH
- R 1 is a monosaccharide or polysaccharide
- A is -CH 2 -, -C(O)-, -C(O)O-, -C(O)NH-, -S(O)2-, -S(0)2NH-, -C(0)NHS(0)2-;
- B is -(CH 2 ) n - where n is an integer ranging from 0-10, or B is an unsaturated carbon chain of 2-10 carbons, which may contain any alkenyl or alkynyl group;
- C represents 1 or 2 substituents independently selected in each instance from hydrogen, halogen, hydroxy, alkyl, aralkyl, alkylaryl, alkoxy, heteroalkyl, aryl, heteroaryl, heteroarylalkyl, aminoaryl, alkylaminoaryl, acyl, acyloxy, sulfonyl, ureyl, and carbamoyl, each of which is optionally substituted;
- W is hydrogen, F, Cl, Br, I, or OH
- R 1 is a monosaccharide that includes an optionally protected 2'-hydroxy group.
- R 1 is a monosaccharide that includes a protected 2'-hydroxy group, where the protecting group is a sterically hindered acyl group, such as a branched alkyl, aryl, heteroaryl, arylalkyl, arylalkyl, or heteroarylalkyl acyl group, each of which is optionally substituted.
- -A-B- is alkylene, cycloalkylene, or arylene; and C is optionally substituted aryl or heteroaryl.
- R 1 is desosamine; -A-B- is 1,4-butylene and C is 4-(3-aminophenyl).
- W is F.
- R 1 is desosamine that includes a protected 2'-hydroxyl group, where the protecting group is a sterically hindered acyl group.
- the sterically hindered acyl group is benzoyl or substituted benzoyl.
- R la is a sterically hindered acyl group, and A, B, C, and V are as described herein.
- -A-B- is alkylene, cycloalkylene, or arylene; and C is optionally substituted aryl or heteroaryl.
- R la is benzoyl; -A-B- is 1,4- butylene and C is 4-(3-aminophenyl).
- A, B, C, and V are as described herein.
- -A-B- is alkylene, cycloalkylene, or arylene; and C is optionally substituted aryl or heteroaryl.
- -A-B- is 1,4-butylene and C is 4-(3-aminophenyl).
- R 1 is a monosaccharide that includes a 2'-hydroxyl group, and V, W, X, and Y are as defined herein, with a sterically hindered acylating agent R la -L, wherein R la is a sterically hindered acyl group and L is a leaving or activating group, to form the corresponding 2'-acyl derivative.
- the process includes the step of (a) reacting compound (1) with a sterically hindered acylating agent to form the corresponding 2'-acyl or 2',4"-diacyl derivative, compound (2), as follows:
- W and R la are as defined herein.
- a process for preparing a compound of formula (I), (II), or (III) comprising the step of (b) reacting a compound of formula (IV) with a carbonylating reagent to form a compound of formula (V): where L is a leaving group, and R 1 , V, W, X, and Y are as defined herein.
- the process includes the step of (b) reacting compound (2) with carbonyldiimidazole to prepare compound (3):
- R la and W are as defined herein.
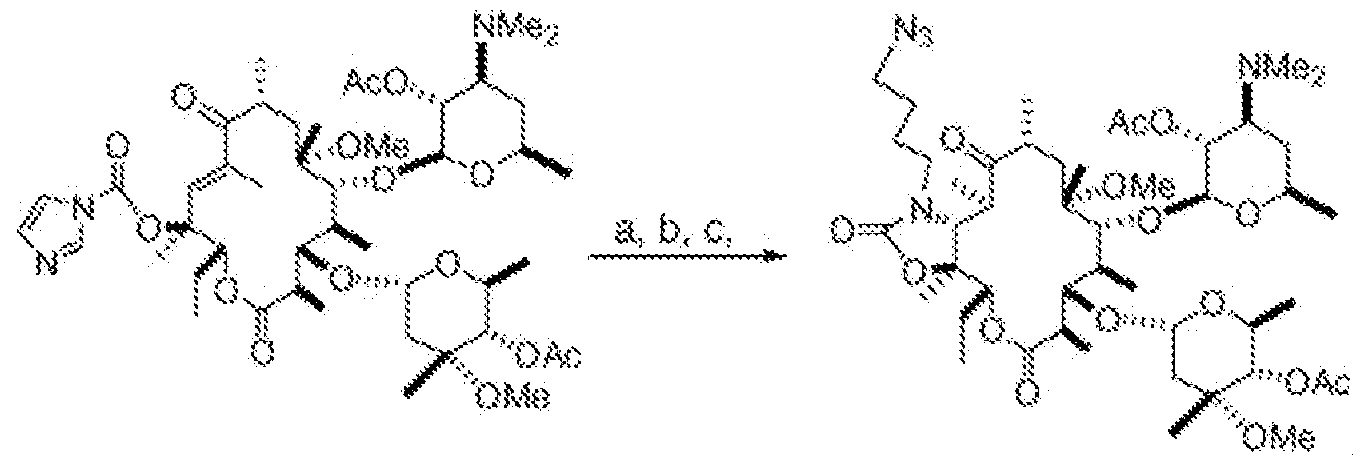
- a process for preparing a compound of formula (I), (II), or (III) comprising the step of (c) reacting a compound of formula (V) with a compound of formula N 3 -B-A-NH 2 to obtain a compound of formula (VI):
- R 1 , A, B, V, W, X, and Y are as described herein.
- a and B are taken together to form alkylene, cycloalkylene, including spirocycloalkylene, or arylene, each of which is optionally substituted.
- the process includes the step of (c) reacting compound (3) with N 3 -B-A-NH 2 to obtain compound (4):
- R la , A, B, and W are as described herein.
- a process for preparing a compound of formula (I), (II), or (III) comprising the step of (d) reacting a compound of formula (I), where X is hydrogen and Y is OR 7 ; where R 7 is a monosaccharide or disaccharide with an acid to prepare the corresponding compound of formula (I) where R 7 is hydrogen.
- the process includes the step of (d) reacting compound (4) with an acid to prepare compound (5):
- R la , A, B, and W are as described herein.
- the process includes the step of (e) oxidizing compound (5) with an oxidizing agent to prepare compound (6): where R , 1a a , A, B, and W are as described herein.
- a process for preparing a compound of formula (I), (II), or (III) comprising the step of (f) reacting a compound of formula (I), where W is hydrogen, with a fluorinating agent to prepare the corresponding compound of formula (I) where W is F.
- the process includes the step of (f) reacting compound (6) with a fluorinating agent to prepare compound (7):
- R , 1a a , A, and B are as described herein.
- a process for preparing a compound of formula (I), (II), or (III) comprising the step of converting the azide group on a compound of formula (VI) into the corresponding compound of formula (I) having a 1,2,3- triazole group.
- a process is described for preparing a compound of formula (I), (II), or (III) comprising the step of (g) reacting a compound of formula (VI) with an R 4 ,R 5 - substituted alkyne to obtain a compound of formula (VII):
- R 4 and R 5 are each independently selected from the group consisting of hydrogen, alkyl, heteroalkyl, aryl, and heteroaryl, each of which is optionally substituted, and R 1 , A, B, V, W, X, and Y are as described herein. In one aspect, both R 4 and R 5 are not hydrogen. In another aspect, at least one of R 4 and R 5 is hydrogen. In one variation, A and B are taken together to form alkylene, cycloalkylene, including spirocycloalkylene, or arylene, each of which is optionally substituted.
- the process includes the step of (g) performing a Huisgen cyclization in the presence of a copper catalyst and base on compound (7) to prepare compound (8):
- R , 1a a , A, and B are as described herein.
- a process for preparing a compound of formula (I) comprising the step (h) of reacting a compound of formula (I), where R 1 is a monosaccharide or polysaccharide having a acyl protecting group, with an alcohol to prepare the corresponding deprotected compound of formula (I).
- a process is described for preparing a compound of formula (III) comprising the step of reacting a compound of formula (II) with an alcohol.
- the process includes the step of (h) reacting compound (8) with an alcohol to prepare compound (9):
- the processes described herein may be advantageously performed simply and cost-effectively. It is further appreciated that the processes described herein may be scaled to large production batches. It is further appreciated that the processes described herein are performed in fewer steps than conventional processes. It is further appreciated that the processes described herein are performed in more convergent steps and fewer linear steps than conventional processes. It is further appreciated that the processes described herein may concomitantly produce fewer or different side products than known processes. It is further appreciated that the processes described herein may yield compounds described herein in higher purity than known processes.
- R 1 is a monosaccharide or polysaccharide.
- the monosaccharide is an aminosugar or a derivative thereof, such as a mycaminose derivatized at the C-4' position, desosamine, a 4-deoxy-3-amino-glucose derivatized at the C-6' position, chloramphenicol, clindamycin, and the like, or an analog or derivative of the foregoing.
- the polysaccharide is a disaccharide, such as a mycaminose derivatized at the C-4' position with another sugar or a 4-deoxy-3-amino-glucose derivatized at the C-6' position with another sugar, a trisaccharide, such as an aminosugar or halosugar, or an analog or derivative of the foregoing.
- R 1 is desosamine, or an analog or derivative thereof. It is to be understood that in this and other embodiments, derivatives include protected forms of the monosaccharide or polysaccharide.
- R 1 is a monosaccharide that includes a 2'-hydroxyl group
- V, W, X, and Y are as defined herein, with a sterically hindered acylating agent R la -L, wherein R la is a sterically hindered acyl group and L is a leaving or activating group, to form the corresponding 2'-acyl derivative.
- R la is a sterically hindered acylating agent
- L is a leaving or activating group
- the process includes the step of (a) reacting compound (1) with a sterically hindered acylating agent to form the corresponding 2'-acyl or 2',4"-diacyl derivative, compound (2), as follows: wherein W and R , 1a a are as defined herein.
- W is F.
- R la is an optionally substituted benzoyl group
- step (a) includes benzoic anhydride, or an equivalent activated benzoylating reagent capable of forming the benzoyl ester at the 2' or both the 2' and 4' positions of a compound of formula (IV), or alternatively compound (1).
- Illustrative bases include but are not limited to inorganic bases, such as sodium and potassium bicarbonates and carbonates, sodium and potassium hydroxides, and the like, and mixtures thereof; and amine bases, such as pyridine, dimethylaminopyridine (DMAP), triethylamine (TEA), diisopropylethylamine (DIPEA, H ⁇ nigs base), l,4-diazabicyclo[2.2.2]octane (DABCO), and the like, and mixtures thereof.
- the reaction may be performed at a variety of temperatures, such as in the range from about O 0 C to about 60 0 C, and illustratively at about 10 0 C to about 30 0 C.
- a process for preparing a compound of formula (I), (II), or (III) comprising the step of (b) reacting a compound of formula (IV) with a carbonylating reagent to form a compound of formula (V):
- Step (d) is generally performed in a solvent such as water, a polar organic solvent, including alcohols such as methanol, ethanol, isopropanol, n-propanol, tert-butanol, n-butanol, and the like, and mixtures thereof.
- Step (d) may be performed at a wide variety of temperatures, including temperatures in the range from about 0 0 C to about 70 0 C, and illustratively in the range from about 20 0 C to about 6O 0 C.
- the oxidizing agent is selected from Swern conditions such as DMSO/EDAC ⁇ Cl/pyridine-TFA, Dess-Martin conditions, Corey-Kim conditions, such as dimethylsulfide/N-chlorosuccinimide, Jones reagent and other chromium oxidizing agents, permanganate and other manganese oxidizing agents, Ni(Ac)2/hypochlorite, and others.
- the oxidation is carried out using the Dess-Martin periodinane in methylene chloride at a temperature from about 5°C to about 30 0 C utilizing a mole-equivalent ratio of Dess-Martin periodinane to compound (5) of from about 3.3 to 1 to about 1.3 to 1.
- the oxidation is carried out using the Dess-Martin periodinane in methylene chloride at a temperature from about 5°C to about 30 0 C utilizing a mole-equivalent ratio of Dess-Martin periodinane to compound (5) of about 1.3 to 1.
- the process includes the step of (f) reacting compound (6) with a fluorinating agent, such as (PhSC ⁇ N-F (NFSI or N- fluorosulfonimide), F-TEDA, F-TEDA-BF 4 , l-fluoro-4-hydroxy-l,4- diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate), and the like, in the presence of solvent and base, such as t-BuOK, to prepare compound (7):
- a fluorinating agent such as (PhSC ⁇ N-F (NFSI or N- fluorosulfonimide), F-TEDA, F-TEDA-BF 4 , l-fluoro-4-hydroxy-l,4- diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate), and the like
- Illustrative substituted alkynes include alkynes substituted with aromatic groups, substituted aromatic groups, heterocyclic groups, substituted heterocyclic groups, alkyl groups, branched alkyl groups, substituted alkyl groups, such as alkyl groups substituted with amino groups, including primary, secondary, and tertiary amino groups, one or more halogens, hydroxyls, ethers, including alkyl and aromatic ethers, ketones, thioethers, esters, carboxylic acids, cyanos, epoxides, and the like.
- R 1 is a monosaccharide that includes a 2'-hydroxyl group acylated with a sterically hindered acylating agent R la -L, wherein R la is a sterically hindered acyl group and L is a leaving or activating group; and A, B, and W are as defined herein.
- the steps (a), (b), (c), (d), (e), (f), (g), and (h) are performed as described herein.
- the processes described herein are useful for preparing compounds of formulae (I), (II), and (III) in higher yields and/or purity than conventional processes.
- the processes described herein allow for the direct introduction of an azide side chain onto the macro lide without requiring the prior activation of a side chain hydroxyl group, such as by using tosyl chloride or an equivalent activating group, and subsequent conversion into the corresponding side chain azide group.
- the direct introduction of the azide side chain as described herein reduces the overall number of synthetic steps that must be performed in preparing compounds of formulae (I), (II), and (III).
- Conventional syntheses disclose the introduction of a side chain containing an alcohol group that must be converted into the azide in a linear sequence in at least two steps.
- the adsorbent solid is selected from a reverse-phase adsorbent, silica gel, alumina, magnesia-silica gel, or the like
- the elutant is selected from ethyl acetate, isopropyl acetate, methylene chloride, heptane, cyclohexane, toluene, acetonitrile, methanol, isopropanol, ethanol, THF, water or the like, or combinations thereof.
- the solid adsorbent is magnesia-silica gel.
- the processes described herein improve the purity of the compounds of formulae (I), (II), and (III) described herein, and/or improve the purification of the compounds described herein.
- the processes described herein include the use of a sterically hindered acyl group that functions both to protect a hydroxyl group on the saccharide moieties of the macro lide and also functions to provide more effective purification of the compounds. For example, it has been discovered that performing the Huisgen cyclization leads to a mixture of triazole compound of formula (VII) and unreacted ethyne compound.
- heteroaryl refers to optionally substituted aromatic ring systems having one or more heteroatoms such as, for example, oxygen, nitrogen, sulfur, selenium and phosphorus.
- heteroaryl may include five- or six-membered heterocyclic rings, poly cyclic heteroaromatic ring systems and polyheteroaromatic ring systems.
- Example 2 Preparation of 10,11 -anhydro-2',4"-di-O-benzoyl- 12-0- imidazolylcarbonyl-6-O-methylerythromycin A.
- Dimethylformamide (DMF, 100 mL) was added to 2',4"-di-O-benzoyl-6-O-methylerythromycin A at 25-35°C, then 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU 6.4 g) was added to the reaction mixture and stirred at ambient temperature.
- l,l'-Carbonyldiimidazole (CDI, 17 g) was added to the reaction and it was stirred until completion at ambient temperature.
- the title compound is isolated by addition of water, and collecting the resulting precipitate.
- the solid was treated with dichloromethane followed by extraction and removal of solvent to give the title compound.
- the mole-equivalent ratio of 4-azido butyl amine to 10,1 l-anhydro-2',4"-di-O-benzoyl-12-O- imidazolylcarbonyl-6-O-methylerythromycin A is optionally selected to be from about 4 to 1 to about 3 to 1.
- the molar ratio of DBU to 10,1 l-anhydro-2',4"-di-O-benzoyl- 12-0- imidazolylcarbonyl-6-O-methylerythromycin A is optionally selected to be from about 1 to 1 to about 0.75 to 1.
- Example 5 Preparation of 1 l-N-(4-Azidobutyl)-5-(2'-benzoyldesosaminyl)-3- oxo-6-O-methylerythronolide A 11,12-cyclic carbamate.
- Dichloromethane 50 mL was added to N-chlorosuccinimide (2 g) under nitrogen at room temperature cooled to 0 0 C.
- Dimethylsulfide (1.8 mL) was added slowly to the reaction mixture at 0 0 C under stirring.
- the reaction mixture was quenched with 5 % aqueous sodium hydroxide solution.
- the organic layer was washed with water and sat. solution of sodium chloride.
- the solvent was removed by distillation of the organic layer and the product was isolated from a mixture of diisopropyl ether and hexane.
- the separated solid was filtered and dried under vacuum at 30-35 0 C to give the title compound.
- the mole-equivalent ratio of Dess-Martin periodinane to 1 l-N-(4-azidobutyl)- 5-(2'-benzoyldesosaminyl)-3-hydroxy-6-O-methylerythronolide A 11,12-cyclic carbamate is optionally from about 3.3 to 1 to about 1.3 to 1.
- the material is converted to a salt by addition of an acid followed by precipitation of the salt.
- Analysis of the material indicated the title compound with >98% purity. Examples 1-8 were repeated to prepare a 5 kg sample of the title compound of Example 8. It was determined that the large sample contained less than about 0.1% aminophenylethynes, or about 0.07% aminophenylethynes.
- Example 9 Purification of 1 l-N-(3-amino-phenyl-l-ylmethyl-[l,2,3]-triazole-l- yl]butyl)-5-desosaminyl-3-oxo-2-fluoro-erythronolide A, 11,12-cyclic carbamate. Florisil (21 kg) was loaded into a column containing 63 L of ethyl acetate.
- the filtrate is slowly added to a solution of aqueous ammonia (0.79 L ammonia in 28 L water) at 10-25 0 C.
- aqueous ammonia (0.79 L ammonia in 28 L water) at 10-25 0 C.
- the resulting mixture is stirred for 30 minutes and the solid is collected by centrifugation.
- the solid was dried at 45-50 0 C until the moisture content was not more than 1.5%.
- Example 9A 1 l-N-(3-amino-phenyl-l-ylmethyl-[l,2,3]-triazole-l-yl]butyl)-5- desosaminyl-3-oxo-2-fluoro-erythronolide A, 11,12-cyclic carbamate is optionally purified by dissolving material in a minimum amount of a solvent and adding an acid to the mixture to form a solid that precipitates from the solvent or precipitates after addition of a second solvent to the acidified mixture.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Oncology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Communicable Diseases (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Saccharide Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description































Claims












Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08841217A EP2214484A4 (en) | 2007-10-25 | 2008-10-23 | Process for the preparation of macrolide antibacterial agents |
| CN200880123646.1A CN101917850B (en) | 2007-10-25 | 2008-10-23 | The preparation method of macrolide antibacterial agents |
| AU2008316830A AU2008316830B2 (en) | 2007-10-25 | 2008-10-23 | Process for the preparation of macrolide antibacterial agents |
| JP2010531238A JP5698979B2 (en) | 2007-10-25 | 2008-10-23 | Process for preparing macrolide antibacterial agents |
| US12/739,652 US9453042B2 (en) | 2007-10-25 | 2008-10-23 | Process for the preparation of macrolide antibacterial agents |
| CA2703475A CA2703475A1 (en) | 2007-10-25 | 2008-10-23 | Process for the preparation of macrolide antibacterial agents |
| IL205254A IL205254A (en) | 2007-10-25 | 2010-04-22 | Process for the preparation of macrolide antibacterial agents |
| US15/262,277 US10131684B2 (en) | 2007-10-25 | 2016-09-12 | Process for the preparation of macrolide antibacterial agents |
| US16/155,939 US20190241602A1 (en) | 2007-10-25 | 2018-10-10 | Process for the preparation of macrolide antibacterial agents |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US98244607P | 2007-10-25 | 2007-10-25 | |
| US60/982,446 | 2007-10-25 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/739,652 A-371-Of-International US9453042B2 (en) | 2007-10-25 | 2008-10-23 | Process for the preparation of macrolide antibacterial agents |
| US15/262,277 Continuation US10131684B2 (en) | 2007-10-25 | 2016-09-12 | Process for the preparation of macrolide antibacterial agents |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009055557A1 true WO2009055557A1 (en) | 2009-04-30 |
Family
ID=40580003
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/080936 WO2009055557A1 (en) | 2007-10-25 | 2008-10-23 | Process for the preparation of macrolide antibacterial agents |
Country Status (9)
| Country | Link |
|---|---|
| US (3) | US9453042B2 (en) |
| EP (1) | EP2214484A4 (en) |
| JP (4) | JP5698979B2 (en) |
| CN (2) | CN105732745A (en) |
| AU (2) | AU2008316830B2 (en) |
| CA (1) | CA2703475A1 (en) |
| HK (1) | HK1226411A1 (en) |
| IL (1) | IL205254A (en) |
| WO (1) | WO2009055557A1 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010048599A1 (en) | 2008-10-24 | 2010-04-29 | Cempra Pharmaceuticals, Inc. | Methods for treating gastrointestinal diseases |
| WO2011146829A1 (en) | 2010-05-20 | 2011-11-24 | Cempra Pharmaceuticals, Inc. | Processes for preparing macrolides and ketolides and intermediates therefor |
| EP2550286A1 (en) * | 2010-03-22 | 2013-01-30 | Cempra Pharmaceuticals, Inc. | Crystalline forms of a macrolide, and uses therefor |
| US9200026B2 (en) | 2003-03-10 | 2015-12-01 | Merck Sharp & Dohme Corp. | Antibacterial agents |
| US9453042B2 (en) | 2007-10-25 | 2016-09-27 | Cempra Pharmaceuticals, Inc. | Process for the preparation of macrolide antibacterial agents |
| US9480679B2 (en) | 2009-09-10 | 2016-11-01 | Cempra Pharmaceuticals, Inc. | Methods for treating malaria, tuberculosis and MAC diseases |
| CN106518939A (en) * | 2015-09-14 | 2017-03-22 | 江苏奥赛康药业股份有限公司 | Method for preparing Solithromycin compound |
| EP3190122A1 (en) | 2016-01-08 | 2017-07-12 | LEK Pharmaceuticals d.d. | A novel synthetic pathway towards solithromycin and purification thereof |
| US9751908B2 (en) | 2013-03-15 | 2017-09-05 | Cempra Pharmaceuticals, Inc. | Convergent processes for preparing macrolide antibacterial agents |
| US9815863B2 (en) | 2010-09-10 | 2017-11-14 | Cempra Pharmaceuticals, Inc. | Hydrogen bond forming fluoro ketolides for treating diseases |
| US9861616B2 (en) | 2013-03-14 | 2018-01-09 | Cempra Pharmaceuticals, Inc. | Methods for treating respiratory diseases and formulations therefor |
| US9937194B1 (en) | 2009-06-12 | 2018-04-10 | Cempra Pharmaceuticals, Inc. | Compounds and methods for treating inflammatory diseases |
| US9982005B2 (en) | 2013-04-04 | 2018-05-29 | President And Fellows Of Harvard College | Macrolides and methods of their preparation and use |
| US10188674B2 (en) | 2012-03-27 | 2019-01-29 | Cempra Pharmaceuticals, Inc. | Parenteral formulations for administering macrolide antibiotics |
| US10633407B2 (en) | 2014-10-08 | 2020-04-28 | President And Fellows Of Harvard College | 14-membered ketolides and methods of their preparation and use |
| US10640528B2 (en) | 2015-03-25 | 2020-05-05 | President And Fellows Of Havard College | Macrolides with modified desosamine sugars and uses thereof |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104151382B (en) * | 2014-08-08 | 2016-09-14 | 广东东阳光药业有限公司 | A kind of crystal formation of solid-state macrolide |
| CN104311611B (en) * | 2014-08-13 | 2017-01-18 | 广东东阳光药业有限公司 | Preparing method of solid-state macrolide |
| CN107207418A (en) * | 2015-02-06 | 2017-09-26 | 森普拉制药公司 | 4 azido butylamine and preparation method |
| EP3265100A4 (en) * | 2015-03-06 | 2019-01-09 | Cempra Pharmaceuticals Inc. | Processes for preparing fluoroketolides |
| CN106554381A (en) * | 2015-09-25 | 2017-04-05 | 苏州鹏旭医药科技有限公司 | Ketolide antibiotics intermediate and its preparation method and application |
| CN105348341B (en) * | 2015-12-04 | 2018-01-09 | 浙江京新药业股份有限公司 | A kind of method for preparing rope Citropten |
| CN105503976B (en) * | 2015-12-04 | 2018-02-06 | 浙江京新药业股份有限公司 | A kind of rope Citropten intermediate |
| WO2018045294A1 (en) * | 2016-09-02 | 2018-03-08 | Cempra Pharmaceuticals, Inc. | Processes for preparing fluoroketolides |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0248279A2 (en) | 1986-06-04 | 1987-12-09 | Abbott Laboratories | Semisynthetic erythromycin antibiotics |
| US5635485A (en) | 1994-05-03 | 1997-06-03 | Roussel Uclaf | Erythromycin compounds |
| WO1998030574A1 (en) | 1997-01-07 | 1998-07-16 | Abbott Laboratories | Tricyclic erythromycin derivatives |
| US20060100164A1 (en) | 2003-03-10 | 2006-05-11 | Chang-Hsing Liang | Novel antibacterial agents |
Family Cites Families (147)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1354753A (en) | 1919-02-11 | 1920-10-05 | E M Lannes | Portable building |
| US2180006A (en) | 1936-09-09 | 1939-11-14 | Eastman Kodak Co | Process for the separation and refining of amines |
| GB891817A (en) | 1959-04-07 | 1962-03-21 | Upjohn Co | Improvements in or relating to injectable tetracyclic preparations |
| US3843787A (en) | 1969-01-15 | 1974-10-22 | Pierrel Spa | Water soluble derivative of erythromycin |
| US3668282A (en) | 1970-05-08 | 1972-06-06 | Stauffer Chemical Co | Stabilized mixture employing n-(beta-0,0-dialkyldithiophosphoryl) aryl sulfonamides |
| EP0012521B1 (en) | 1978-12-11 | 1983-03-23 | Bexford Limited | Vesicular recording materials and process for their production |
| SE458505B (en) | 1979-07-10 | 1989-04-10 | Lepetit Spa | APPLICATION OF RIFAMYCIN SV AND SALTS THEREOF PREPARING A PREPARATION FOR TREATMENT OF REUMATOID ARTHRITIS AND CERTAIN SALTS AND THEIR PREPARATION |
| US4331803A (en) | 1980-06-04 | 1982-05-25 | Taisho Pharmaceutical Co., Ltd. | Novel erythromycin compounds |
| US4474768A (en) | 1982-07-19 | 1984-10-02 | Pfizer Inc. | N-Methyl 11-aza-10-deoxo-10-dihydro-erytromycin A, intermediates therefor |
| JPS59104326A (en) | 1982-12-04 | 1984-06-16 | Toyo Jozo Co Ltd | Stable oral preparation of macrolide antibiotic substance, and method for stabilizing the same |
| JPS59175414A (en) | 1983-03-23 | 1984-10-04 | Toyo Jozo Co Ltd | Stable oral preparation of macrolide antibiotic substance and method for stabilizing the same |
| KR960000434B1 (en) | 1986-12-17 | 1996-01-06 | 다이쇼 세이야꾸 가부시끼가이샤 | Eryhromycin a derivatives and process of method for the same |
| DE3860503D1 (en) | 1987-09-03 | 1990-10-04 | Pliva Pharm & Chem Works | 10-DIHYDRO-10-DEOXO-11-AZAERYTHRONOLID-A COMPOUNDS, METHODS AND INTERMEDIATE PRODUCTS FOR THE PRODUCTION AND THEIR USE IN MEDICINAL PRODUCTS AND THE PRODUCTION THEREOF. |
| BE1001869A3 (en) | 1988-10-12 | 1990-04-03 | Franz Legros | METHOD OF PACKAGING liposomal AMINOGLUCOSIDIQUES ANTIBIOTICS IN PARTICULAR THE GENTAMYCIN. |
| IL114589A (en) | 1990-11-21 | 1999-12-22 | Roussel Uclaf | Intermediates for the preparation of erythromycin derivatives |
| US5985844A (en) | 1992-03-26 | 1999-11-16 | Merck & Co., Inc. | Homoerythromycin A derivatives modified at the 4"-and 8A-positions |
| TW271400B (en) | 1992-07-30 | 1996-03-01 | Pfizer | |
| US5527780A (en) | 1992-11-05 | 1996-06-18 | Roussel Uclaf | Erythromycin derivatives |
| FR2697524B1 (en) | 1992-11-05 | 1994-12-23 | Roussel Uclaf | New erythromycin derivatives, their preparation process and their use as drugs. |
| JPH07126172A (en) | 1993-11-05 | 1995-05-16 | Genichiro Soma | Lps-containing anti-mrsa agent and anti-mrsa agent for animal |
| FR2718450B1 (en) | 1994-04-08 | 1997-01-10 | Roussel Uclaf | New erythromycin derivatives, their preparation process and their use as drugs. |
| US5760010A (en) | 1995-01-01 | 1998-06-02 | Klein; Ira | Method of treating liver disorders with a macrolide antibiotic |
| US5834428A (en) | 1995-04-14 | 1998-11-10 | 1149336 Ontario Inc. | Glucagon-like peptide-2 and its therapeutic use |
| FR2739620B1 (en) | 1995-10-09 | 1997-12-19 | Roussel Uclaf | NOVEL DERIVATIVES OF 5-0-DESOSAMINYL 6-O-METHYL ERYTHRONOLIDE A, THEIR PREPARATION PROCESS AND THEIR APPLICATION TO THE PREPARATION OF BIOLOGICALLY ACTIVE PRODUCTS |
| FR2742757B1 (en) | 1995-12-22 | 1998-01-30 | Roussel Uclaf | NOVEL ERYTHROMYCIN DERIVATIVES, THEIR PREPARATION PROCESS AND THEIR APPLICATION AS MEDICAMENTS |
| FR2745290B1 (en) | 1996-02-28 | 1998-04-03 | Roussel Uclaf | NOVEL ERYTHROMYCIN DERIVATIVES, THEIR PREPARATION PROCESS AND THEIR APPLICATION AS MEDICAMENTS |
| US5719272A (en) | 1996-04-02 | 1998-02-17 | Abbott Laboratories | 2'-protected 3'-dimethylamine, 9-etheroxime erythromycin A derivatives |
| KR100523679B1 (en) | 1996-09-04 | 2005-10-26 | 아보트 러보러터리즈 | 6-0-substituted ketolides having antibacterial activity |
| BR9714394A (en) | 1996-12-13 | 2000-05-16 | Lilly Co Eli | Inhibitors of psa enzyme activity |
| US6407074B1 (en) | 1997-06-11 | 2002-06-18 | Pfizer Inc | C-4″-substituted macrolide derivatives |
| HN1998000074A (en) | 1997-06-11 | 1999-01-08 | Pfizer Prod Inc | DERIVATIVES FROM MACROLIDES C-4 SUBSTITUTED |
| EA199901016A1 (en) | 1997-06-11 | 2000-06-26 | Пфайзер Продактс Инк. | 9-OXYM DERIVATIVES OF ERYTHROMYCIN |
| HN1998000086A (en) | 1997-06-11 | 1999-03-08 | Pfizer Prod Inc | DERIVATIVES OF 9 - DESOFO - 9 AZA - 9A - HOMOERITROMICINA A - C - 4 SUBSTITUTED. |
| HN1998000159A (en) | 1997-10-29 | 1999-02-09 | Monsanto Co | DERIVATIVES OF 9- AMINO - 3 CETO ERITROMICINA |
| IL135792A0 (en) | 1997-12-01 | 2001-05-20 | Abbott Lab | 6-o-alkyl derivatives of erythronolide b |
| WO1999049863A1 (en) | 1998-03-26 | 1999-10-07 | Fujisawa Pharmaceutical Co., Ltd. | Sustained release preparations |
| FR2777282B1 (en) | 1998-04-08 | 2001-04-20 | Hoechst Marion Roussel Inc | NEW DERIVATIVES OF 2-FLUORO 3-DE ((2,6-DIDEOXY 3-C-METHYL 3-0-METHYL-ALPHA-L-RIBOHEXOPYRANOSYL) OXYL) 6-O-METHYL 3-OXO ERYTHROMYCIN, THEIR PREPARATION PROCESS AND THEIR APPLICATION TO THE SYNTHESIS OF ACTIVE INGREDIENTS OF MEDICINES |
| US6020521A (en) | 1998-08-26 | 2000-02-01 | Abbott Laboratories | Macrolide LHRH antagonists |
| US6387885B1 (en) | 1998-08-26 | 2002-05-14 | Abbott Laboratories | 3′,3′-N-bis-desmethyl-3′-N-cycloalkyl erythromycin derivatives as LHRH antagonists |
| FR2785612A1 (en) | 1998-11-10 | 2000-05-12 | Hoechst Marion Roussel Inc | NOVEL DERIVATIVES OF ERYTHROMYCIN, PROCESS FOR PREPARING THEM AND THEIR APPLICATION AS MEDICAMENTS |
| FR2786188B1 (en) | 1998-11-24 | 2002-10-31 | Hoechst Marion Roussel Inc | NOVEL ERYTHROMYCIN DERIVATIVES, THEIR PREPARATION PROCESS AND THEIR APPLICATION AS MEDICAMENTS |
| KR100317907B1 (en) | 1998-11-24 | 2001-12-24 | 김 완 주 | Novel Intermediates, process for preparing macrolide antibiotic agent therefrom |
| ES2273964T3 (en) | 1998-12-10 | 2007-05-16 | Pfizer Products Inc. | CARBAMATE AND CETOLID CARBAZATE ANTIBIOTICS. |
| ID30233A (en) | 1999-01-27 | 2001-11-15 | Pfizer Prod Inc | ANTIBIOTICS OF TETOLIDA |
| CA2411293A1 (en) | 1999-01-28 | 2000-07-28 | Pfizer Products Inc. | Novel azalides and methods of making same |
| FR2789392B1 (en) | 1999-02-04 | 2001-10-05 | Hoechst Marion Roussel Inc | NOVEL ERYTHROMYCIN DERIVATIVES, THEIR PREPARATION PROCESS AND THEIR APPLICATION AS MEDICAMENTS |
| US7056893B2 (en) | 1999-03-31 | 2006-06-06 | Insite Vision, Inc. | Topical treatment for prevention of ocular infections |
| AR023264A1 (en) | 1999-04-08 | 2002-09-04 | Hokuriku Pharmaceutical | ERYTHROMYCIN DERIVATIVES |
| JP2000351794A (en) | 1999-04-08 | 2000-12-19 | Hokuriku Seiyaku Co Ltd | Erythromycin derivative |
| ATE340183T1 (en) | 1999-04-16 | 2006-10-15 | Kosan Biosciences Inc | ANTINIFECTIVE MACROLIDE DERIVATIVES |
| WO2000062783A2 (en) | 1999-04-16 | 2000-10-26 | Ortho-Mcneil Pharmaceutical, Inc. | Ketolide antibacterials |
| US6395300B1 (en) | 1999-05-27 | 2002-05-28 | Acusphere, Inc. | Porous drug matrices and methods of manufacture thereof |
| US6420535B1 (en) | 1999-06-07 | 2002-07-16 | Abbott Laboratories | 6-O-carbamate ketolide derivatives |
| US6437106B1 (en) | 1999-06-24 | 2002-08-20 | Abbott Laboratories | Process for preparing 6-o-substituted erythromycin derivatives |
| AU6183700A (en) | 1999-08-06 | 2001-03-05 | Taisho Pharmaceutical Co., Ltd. | Erythromycin a derivatives |
| WO2001010787A1 (en) | 1999-08-09 | 2001-02-15 | Eric Au | An apparatus for treating wastewater |
| US6096922A (en) | 1999-11-01 | 2000-08-01 | Air Products And Chemicals, Inc. | Process for the synthesis of dialkyl, diaryl, and arylalkyl aminosulfur trifluorides |
| KR100336447B1 (en) | 1999-11-24 | 2002-05-15 | 민경윤 | Improved method of preparing clarithromycin |
| US20020009507A1 (en) | 2000-01-19 | 2002-01-24 | Alcon Universal Ltd. | Use of polyethoxylated castor oil for the treatment of dry eye |
| JP2001261694A (en) | 2000-03-06 | 2001-09-26 | Pfizer Prod Inc | Ketolide antibiotic |
| DE60122791T2 (en) | 2000-03-15 | 2007-10-04 | Hanmi Pharm. Co., Ltd. | Process for the preparation of non-pharmaceutical grade clarythromycin |
| NZ523693A (en) | 2000-07-10 | 2004-08-27 | Chiron Corp | Macrolide formulations for inhalation and methods of treatment of endobronchial infections |
| US20020115621A1 (en) | 2000-08-07 | 2002-08-22 | Wei-Gu Su | Macrolide antibiotics |
| GB0031312D0 (en) | 2000-12-21 | 2001-02-07 | Glaxo Group Ltd | Macrolides |
| FR2821747B1 (en) | 2001-03-09 | 2004-07-02 | Ethypharm Lab Prod Ethiques | SUSPENSION OF TELITHROMYCIN WITH A MASK TASTE |
| CA2447600C (en) | 2001-05-18 | 2015-10-20 | Chiron Corporation | Methods and unit dose formulations for the inhalation administration of aminoglycoside antibiotics |
| FR2826274B1 (en) | 2001-06-21 | 2003-09-26 | Aventis Pharma Sa | PHARMACEUTICAL FORMULATION FOR MASK TASTE AND METHOD FOR PREPARING THE SAME |
| US6756359B2 (en) | 2001-07-03 | 2004-06-29 | Chiron Corporation | C12 modified erythromycin macrolides and ketolides having antibacterial activity |
| US20030176327A1 (en) | 2001-10-25 | 2003-09-18 | Cassell Gail Houston | Antibiotics for treating biohazardous bacterial agents |
| US20040019012A1 (en) | 2002-02-22 | 2004-01-29 | Singh Satish K. | Ophthalmic antibiotic drug formulations containing a cyclodextrin compound and cetyl pyridinium chloride |
| AU2003240482B2 (en) | 2002-05-30 | 2009-03-12 | The Scripps Research Institute | Copper-catalysed ligation of azides and acetylenes |
| EP1553954A4 (en) | 2002-06-17 | 2009-12-23 | Epigenesis Pharmaceuticals Llc | Dihydrate dehydroepiandrosterone and methods of treating asthma or chronic obstructive pulmonary disease using compostions thereof |
| CA2489402A1 (en) | 2002-07-08 | 2004-01-15 | Pliva-Istrazivacki Institut D.O.O. | New compounds, compositions and methods for treatment of inflammatory diseases and conditions |
| US20040013737A1 (en) | 2002-07-19 | 2004-01-22 | Philippe Becourt | Taste masked oral composition of telithromycin |
| US7091196B2 (en) | 2002-09-26 | 2006-08-15 | Rib-X Pharmaceuticals, Inc. | Bifunctional heterocyclic compounds and methods of making and using same |
| ITMI20022292A1 (en) | 2002-10-29 | 2004-04-30 | Zambon Spa | 9A-AZALIDS WITH ANTI-INFLAMMATORY ACTIVITY. |
| US7163924B2 (en) | 2003-04-25 | 2007-01-16 | Chiron Corporation | Ketolide derivatives |
| US7332476B2 (en) * | 2003-04-25 | 2008-02-19 | Novartis Ag | Pyridyl substituted ketolide antibiotics |
| US20070293472A1 (en) | 2003-05-13 | 2007-12-20 | Glaxo Group Limited | Novel 14 and 15 Membered Ring Compounds |
| WO2005007143A2 (en) | 2003-07-14 | 2005-01-27 | The Board Of Trustees Of The University Of Illinois | Use of makrolides and ketolides for the treatment of tuberculosis |
| US7457520B2 (en) | 2003-07-24 | 2008-11-25 | Time Warner Cable, Inc. | Technique for providing a virtual digital video recorder service through a communications network |
| GB0402578D0 (en) | 2004-02-05 | 2004-03-10 | Cambridge Theranostics Ltd | Methods of treatment of atherosclerosis |
| US7468428B2 (en) | 2004-03-17 | 2008-12-23 | App Pharmaceuticals, Llc | Lyophilized azithromycin formulation |
| US20060116336A1 (en) | 2004-03-17 | 2006-06-01 | American Pharmaceutical Partners, Inc. | Lyophilized azithromycin formulation |
| AU2005238313A1 (en) | 2004-04-28 | 2005-11-10 | Alembic Limited | Process for the preparation of telithromycin |
| WO2005108412A1 (en) | 2004-05-06 | 2005-11-17 | Glaxosmithkline Istrazivacki Centar Zagreb D.O.O. | Ester linked macrolides useful for the treatment of microbial infections |
| BRPI0513903A (en) | 2004-07-28 | 2008-05-20 | Ranbaxy Lab Ltd | ketolide derivatives useful as antimicrobial agents, their preparation processes and pharmaceutical compositions comprising the same |
| US20060076536A1 (en) | 2004-09-29 | 2006-04-13 | Barshied Scott R | Oxygen scavenging pharmaceutical package and methods for making same |
| GB0424958D0 (en) | 2004-11-11 | 2004-12-15 | Glaxo Group Ltd | Novel compounds |
| GB0424959D0 (en) | 2004-11-11 | 2004-12-15 | Glaxo Group Ltd | Novel compounds |
| AU2005317735B2 (en) | 2004-12-21 | 2009-06-04 | Pfizer Products Inc. | Macrolides |
| EP1841437B1 (en) | 2005-01-14 | 2009-04-29 | GlaxoSmithKline istrazivacki centar Zagreb d.o.o. | 9a-carbamoyl and thiocarbamoyl azalides with antimalarial activity |
| ATE523519T1 (en) | 2005-01-14 | 2011-09-15 | Glaxo Group Ltd | MACROLIDE COMPOUNDS WITH BIOTIN AND PHOTO AFFINITY GROUP FOR IDENTIFYING A MACROLIDE TARGET |
| EP1835921B1 (en) | 2005-01-14 | 2010-08-18 | GlaxoSmithKline istrazivacki centar Zagreb d.o.o. | 9a-carbamoyl-y-aminopropyl- and 9a-thiocarbamoyl-y-aminopropyl-azalides with antimalarial activity |
| AU2006227199A1 (en) | 2005-03-22 | 2006-09-28 | Azevan Pharmaceuticals, Inc. | Beta-lactamylalkanoic acids for treating premenstrual disorders |
| CA2609549A1 (en) | 2005-05-25 | 2006-11-30 | Sirtris Pharmaceuticals, Inc. | Treatment of eye disorders with sirtuin modulators |
| US20070015719A1 (en) | 2005-07-07 | 2007-01-18 | Elan Pharma International Limited | Nanoparticulate clarithromycin formulations |
| US8048874B2 (en) | 2005-07-19 | 2011-11-01 | Azevan Pharmaceuticals, Inc. | Beta-lactamyl phenylalanine, cysteine, and serine vasopressin antagonists |
| WO2007039914A2 (en) | 2005-10-06 | 2007-04-12 | Alembic Limited | Novel polymorphs of telithromycin |
| BRPI0618081A2 (en) | 2005-10-31 | 2012-02-22 | Leo Pharma As | compound, mixture or composition of crystalline forms of fusidic acid, mixture or formulation, use of a compound, method for preparing a compound, and pharmaceutical composition |
| GB0522715D0 (en) | 2005-11-08 | 2005-12-14 | Helperby Therapeutics Ltd | New use |
| WO2007059307A2 (en) | 2005-11-15 | 2007-05-24 | Teva Pharmaceutical Industries Ltd. | Crystalline and amorphous forms of telithromycin |
| WO2007060627A2 (en) | 2005-11-23 | 2007-05-31 | Ranbaxy Laboratories Limited | Use of macrolide derivatives for treating acne |
| US20090005325A1 (en) | 2005-11-23 | 2009-01-01 | Biswajit Bas | Ketolide Derivatives as Antibacterial Agents |
| DOP2006000268A (en) | 2005-12-22 | 2007-07-31 | Pfizer Prod Inc | ANTIBACTERIAL AGENTS |
| FR2897154B1 (en) | 2006-02-08 | 2008-03-07 | Airbus France Sas | DEVICE FOR BUILDING AND SECURING A LOW ALTITUDE FLIGHT PATH TO BE FOLLOWED BY AN AIRCRAFT. |
| CN101045063B (en) | 2006-03-28 | 2011-01-26 | 广州朗圣药业有限公司 | Clarithromycin water soluble preparation for injection use |
| WO2007129646A1 (en) | 2006-05-01 | 2007-11-15 | Taisho Pharmaceutical Co., Ltd. | Macrolide derivative |
| WO2007143507A2 (en) | 2006-06-05 | 2007-12-13 | Auspex Pharmaceuticals, Inc. | Preparation and utility of substituted erythromycin analogs |
| CN101129383B (en) | 2006-08-25 | 2014-04-02 | 天津和美生物技术有限公司 | Antibiotic compound containing aminoglycoside antibiotic |
| EP2078034A2 (en) | 2006-09-18 | 2009-07-15 | The University of Manitoba | Synthesis of carbohydrate-templated amino acids and methods of using same |
| US8609148B2 (en) | 2006-10-25 | 2013-12-17 | Revalesio Corporation | Methods of therapeutic treatment of eyes |
| RU2469034C2 (en) | 2006-12-04 | 2012-12-10 | Астразенека Аб | Chemical compounds |
| JP5025249B2 (en) | 2006-12-15 | 2012-09-12 | Nskワーナー株式会社 | Starting clutch |
| EP2214484A4 (en) | 2007-10-25 | 2013-01-02 | Cempra Pharmaceuticals Inc | Process for the preparation of macrolide antibacterial agents |
| EP2220104A1 (en) | 2007-10-25 | 2010-08-25 | Sandoz AG | Process for the production of telithromycin |
| US7795316B1 (en) | 2007-12-19 | 2010-09-14 | Alcon Research, Ltd. | Topical ophthalmic compositions containing tobramycin and dexamethasone |
| US20090209547A1 (en) | 2008-02-15 | 2009-08-20 | In Jong Kim | C-8 halogenated macrolides |
| US8796232B2 (en) | 2008-10-24 | 2014-08-05 | Cempra Pharmaceuticals, Inc. | Methods for treating resistant diseases using triazole containing macrolides |
| WO2010077730A2 (en) | 2008-12-09 | 2010-07-08 | Auspex Pharmaceutical, Inc | Indanone inhibitors of acetylcholinesterase |
| US9814657B2 (en) | 2009-04-27 | 2017-11-14 | Premier Dental Products Company | Buffered microencapsulated compositions and methods |
| JP5202763B2 (en) | 2009-06-03 | 2013-06-05 | アルギファルマ エーエス | Treatment of Acinetobacter infection with alginate oligomers and antibiotics |
| WO2011008193A1 (en) | 2009-07-13 | 2011-01-20 | Cempra Pharmaceuticals Inc. | Fusidic acid dosing regimens for treatment of bacterial infections |
| AU2010292010B2 (en) | 2009-09-10 | 2016-01-07 | Cempra Pharmaceuticals, Inc. | Methods for treating malaria, tuberculosis and MAC diseases |
| US20110119604A1 (en) | 2009-11-19 | 2011-05-19 | Clevest Solutions Inc. | System and method for a configurable and extensible allocation and scheduling tool |
| JP5890785B2 (en) | 2010-03-10 | 2016-03-22 | センプラ ファーマシューティカルズ,インコーポレイテッド | Parenteral preparations of macrolide antibiotics |
| CN103080122A (en) | 2010-03-22 | 2013-05-01 | 森普拉制药公司 | Crystalline forms of a macrolide, and uses therefor |
| US9051346B2 (en) | 2010-05-20 | 2015-06-09 | Cempra Pharmaceuticals, Inc. | Process for preparing triazole-containing ketolide antibiotics |
| US8247394B2 (en) | 2010-06-02 | 2012-08-21 | Cempra Pharmaceuticals Inc. | Methods of treating urethritis and related infections using fusidic acid |
| US9260473B2 (en) | 2010-07-19 | 2016-02-16 | Virginia Commonwealth University | Bivalent multifunctional ligands targeting Aβ oligomers as treatment for Alzheimer's disease |
| US20130164351A1 (en) | 2010-08-30 | 2013-06-27 | Cempra Pharmaceuticals Inc. | Methods of treating bacterial infections through pulmonary delivery of fusidic acid |
| JP6042334B2 (en) | 2010-09-10 | 2016-12-14 | センプラ ファーマシューティカルズ,インコーポレイテッド | Hydrogen bond forming fluoroketolides for disease treatment |
| WO2012042534A2 (en) | 2010-09-28 | 2012-04-05 | Glenmark Generics Limited | Processes for the preparation of r-sitagliptin and intermediates thereof |
| JP5718488B2 (en) | 2011-03-01 | 2015-05-13 | ウォックハート リミテッド | Method for preparing ketolide intermediate |
| JP6072778B2 (en) | 2011-05-23 | 2017-02-01 | シーイーエム—102 ファーマシューティカルズ,インコーポレイテッド | Compositions containing fusidic acid and packages therefor |
| DK2748165T3 (en) | 2011-08-27 | 2016-12-19 | Wockhardt Ltd | 1,6-diazabicyclo [3,2,1] octane-7-ON DERIVATIVES AND THEIR USE IN THE TREATMENT OF BACTERIAL INFECTIONS |
| WO2013032591A1 (en) | 2011-08-29 | 2013-03-07 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| US8461188B2 (en) | 2011-10-20 | 2013-06-11 | Trius Therapeutics, Inc. | Therapeutic combination of daptomycin and protein synthesis inhibitor antibiotic, and methods of use |
| CN104470527B (en) | 2012-03-27 | 2019-05-28 | 森普拉制药公司 | For applying the parenteral administration of macrolide antibiotic |
| WO2014152326A1 (en) | 2013-03-14 | 2014-09-25 | Cempra Pharmaceuticals, Inc. | Methods for treating respiratory diseases and formulations therefor |
| US9751908B2 (en) | 2013-03-15 | 2017-09-05 | Cempra Pharmaceuticals, Inc. | Convergent processes for preparing macrolide antibacterial agents |
| CA3130061A1 (en) | 2013-04-04 | 2014-10-09 | President And Fellows Of Harvard College | Macrolides and methods of their preparation and use |
| WO2015123256A1 (en) | 2014-02-14 | 2015-08-20 | Cempra Pharmaceuticals, Inc. | Compositions and methods for treating diabetes and liver diseases |
| US20170101365A1 (en) | 2014-05-27 | 2017-04-13 | Dipharma Francis S.R.L. | Azidoalkylamine salts and their use as intermediates |
| AU2015301154A1 (en) | 2014-08-05 | 2017-02-23 | Cempra Pharmaceuticals, Inc. | Powder oral suspension formulations of antibacterial agents |
| EP3265100A4 (en) | 2015-03-06 | 2019-01-09 | Cempra Pharmaceuticals Inc. | Processes for preparing fluoroketolides |
| WO2018045294A1 (en) | 2016-09-02 | 2018-03-08 | Cempra Pharmaceuticals, Inc. | Processes for preparing fluoroketolides |
-
2008
- 2008-10-23 EP EP08841217A patent/EP2214484A4/en not_active Withdrawn
- 2008-10-23 AU AU2008316830A patent/AU2008316830B2/en not_active Ceased
- 2008-10-23 CA CA2703475A patent/CA2703475A1/en not_active Abandoned
- 2008-10-23 CN CN201610086292.4A patent/CN105732745A/en active Pending
- 2008-10-23 US US12/739,652 patent/US9453042B2/en active Active
- 2008-10-23 CN CN200880123646.1A patent/CN101917850B/en not_active Expired - Fee Related
- 2008-10-23 WO PCT/US2008/080936 patent/WO2009055557A1/en active Application Filing
- 2008-10-23 JP JP2010531238A patent/JP5698979B2/en active Active
-
2010
- 2010-04-22 IL IL205254A patent/IL205254A/en active IP Right Grant
-
2014
- 2014-11-10 JP JP2014227753A patent/JP6167095B2/en active Active
-
2016
- 2016-06-01 AU AU2016203649A patent/AU2016203649A1/en not_active Abandoned
- 2016-09-12 US US15/262,277 patent/US10131684B2/en active Active
- 2016-12-23 HK HK16114635A patent/HK1226411A1/en unknown
-
2017
- 2017-06-26 JP JP2017124117A patent/JP6845099B2/en active Active
-
2018
- 2018-10-10 US US16/155,939 patent/US20190241602A1/en not_active Abandoned
-
2019
- 2019-05-15 JP JP2019092242A patent/JP2019147827A/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0248279A2 (en) | 1986-06-04 | 1987-12-09 | Abbott Laboratories | Semisynthetic erythromycin antibiotics |
| US5635485A (en) | 1994-05-03 | 1997-06-03 | Roussel Uclaf | Erythromycin compounds |
| WO1998030574A1 (en) | 1997-01-07 | 1998-07-16 | Abbott Laboratories | Tricyclic erythromycin derivatives |
| US20060100164A1 (en) | 2003-03-10 | 2006-05-11 | Chang-Hsing Liang | Novel antibacterial agents |
Non-Patent Citations (4)
| Title |
|---|
| "Anti-Infective Agents", CURR. MED. CHEM., vol. 1, 2002, pages 15 - 34 |
| LIANG C. H. ET AL.: "Bioorganic & Medicinal Letters", vol. 15, PERGAMON, ELSEVIER SCIENCE, pages: 1307 - 1310 |
| ROMERO A. ET AL.: "Tetrahedron Letters", vol. 46, ELSEVIER, pages: 1483 - 1487 |
| See also references of EP2214484A4 |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9200026B2 (en) | 2003-03-10 | 2015-12-01 | Merck Sharp & Dohme Corp. | Antibacterial agents |
| US9453042B2 (en) | 2007-10-25 | 2016-09-27 | Cempra Pharmaceuticals, Inc. | Process for the preparation of macrolide antibacterial agents |
| US10131684B2 (en) | 2007-10-25 | 2018-11-20 | Cempra Pharmaceuticals, Inc. | Process for the preparation of macrolide antibacterial agents |
| US9669046B2 (en) | 2008-10-24 | 2017-06-06 | Cempra Pharmaceuticals, Inc. | Biodefenses using triazole-containing macrolides |
| US9901592B2 (en) | 2008-10-24 | 2018-02-27 | Cempra Pharmaceuticals, Inc. | Methods for treating resistant diseases using triazole containing macrolides |
| US9072759B2 (en) | 2008-10-24 | 2015-07-07 | Cempra Pharmaceuticals, Inc. | Biodefenses using triazole-containing macrolides |
| WO2010048599A1 (en) | 2008-10-24 | 2010-04-29 | Cempra Pharmaceuticals, Inc. | Methods for treating gastrointestinal diseases |
| US8791080B2 (en) | 2008-10-24 | 2014-07-29 | Cempra Pharmaceuticals, Inc. | Methods for treating gastrointestinal diseases |
| US8796232B2 (en) | 2008-10-24 | 2014-08-05 | Cempra Pharmaceuticals, Inc. | Methods for treating resistant diseases using triazole containing macrolides |
| US9439918B2 (en) | 2008-10-24 | 2016-09-13 | Cempra Pharmaceuticals, Inc. | Methods for treating gastrointestinal diseases |
| EP3031460A1 (en) | 2008-10-24 | 2016-06-15 | Cempra Pharmaceuticals Inc. | Biodefenses using triazole-containing macrolides |
| WO2010048600A1 (en) | 2008-10-24 | 2010-04-29 | Cempra Pharmaceuticals, Inc. | Methods for treating resistant diseases using triazole containing macrolides |
| US9937194B1 (en) | 2009-06-12 | 2018-04-10 | Cempra Pharmaceuticals, Inc. | Compounds and methods for treating inflammatory diseases |
| US9480679B2 (en) | 2009-09-10 | 2016-11-01 | Cempra Pharmaceuticals, Inc. | Methods for treating malaria, tuberculosis and MAC diseases |
| CN108570083A (en) * | 2010-03-22 | 2018-09-25 | 森普拉制药公司 | Crystal form of macrocyclic lactone and application thereof |
| US20190040096A1 (en) * | 2010-03-22 | 2019-02-07 | Cempra Pharmaceuticals, Inc. | Crystalline forms of a macrolide, and uses therefor |
| JP2020002168A (en) * | 2010-03-22 | 2020-01-09 | センプラ ファーマシューティカルズ,インコーポレイテッド | Crystalline forms of macrolide, and uses thereof |
| EP2550286B1 (en) | 2010-03-22 | 2015-12-09 | Cempra Pharmaceuticals, Inc. | Crystalline forms of a macrolide, and uses therefor |
| EP3009442A1 (en) | 2010-03-22 | 2016-04-20 | Cempra Pharmaceuticals Inc. | Crystalline forms of a macrolide, and uses therefor |
| JP2015063536A (en) * | 2010-03-22 | 2015-04-09 | センプラ ファーマシューティカルズ,インコーポレイテッド | Crystalline form of macrolide, and use therefor |
| KR102006200B1 (en) * | 2010-03-22 | 2019-08-01 | 셈프라 파마슈티컬스, 인크. | Crystalline forms of a macrolide, and uses therefor |
| US8975386B2 (en) | 2010-03-22 | 2015-03-10 | Cempra Pharmaceuticals, Inc. | Crystalline forms of a macrolide, and uses therefor |
| US8759500B2 (en) | 2010-03-22 | 2014-06-24 | Cempra Pharmaceuticals, Inc. | Crystalline forms of a macrolide, and uses therefor |
| AU2016203986B2 (en) * | 2010-03-22 | 2016-10-06 | Cempra Pharmaceuticals, Inc. | Crystalline forms of a macrolide, and uses therefor |
| EP2550286A4 (en) * | 2010-03-22 | 2013-10-02 | Cempra Pharmaceuticals Inc | Crystalline forms of a macrolide, and uses therefor |
| EP2550286A1 (en) * | 2010-03-22 | 2013-01-30 | Cempra Pharmaceuticals, Inc. | Crystalline forms of a macrolide, and uses therefor |
| RU2650883C2 (en) * | 2010-03-22 | 2018-04-18 | Семпра Фармасьютикалз, Инк. | Crystalline forms of macrolides and their application |
| CN103080122A (en) * | 2010-03-22 | 2013-05-01 | 森普拉制药公司 | Crystalline forms of a macrolide, and uses therefor |
| JP2017081953A (en) * | 2010-03-22 | 2017-05-18 | センプラ ファーマシューティカルズ,インコーポレイテッド | Crystalline forms of a macrolide, and uses therefor |
| JP2013522367A (en) * | 2010-03-22 | 2013-06-13 | センプラ ファーマシューティカルズ,インコーポレイテッド | Macrolide crystal forms and their use |
| KR20130056226A (en) * | 2010-03-22 | 2013-05-29 | 셈프라 파마슈티컬스, 인크. | Crystalline forms of a macrolide, and uses therefor |
| WO2011146829A1 (en) | 2010-05-20 | 2011-11-24 | Cempra Pharmaceuticals, Inc. | Processes for preparing macrolides and ketolides and intermediates therefor |
| US20200087335A1 (en) * | 2010-05-20 | 2020-03-19 | Têtard, Inc. | Processes for preparing macrolides and ketolides and intermediates therefor |
| US9051346B2 (en) | 2010-05-20 | 2015-06-09 | Cempra Pharmaceuticals, Inc. | Process for preparing triazole-containing ketolide antibiotics |
| AU2011255464B2 (en) * | 2010-05-20 | 2016-06-16 | Cempra Pharmaceuticals, Inc. | Processes for preparing macrolides and ketolides and intermediates therefor |
| KR101945324B1 (en) | 2010-05-20 | 2019-02-07 | 셈프라 파마슈티컬스, 인크. | Processes for preparing macrolides and ketolides and intermediates therefor |
| AU2011255464C1 (en) * | 2010-05-20 | 2017-02-23 | Cempra Pharmaceuticals, Inc. | Processes for preparing macrolides and ketolides and intermediates therefor |
| US20150232500A1 (en) * | 2010-05-20 | 2015-08-20 | Cempra Pharmaceuticals, Inc. | Processes for preparing macrolides and ketolides and intermediates therefor |
| RU2608390C2 (en) * | 2010-05-20 | 2017-01-18 | Семпра Фармасьютикалз, Инк. | Processes for preparing macrolides and ketolides and intermediates therefor |
| US9815863B2 (en) | 2010-09-10 | 2017-11-14 | Cempra Pharmaceuticals, Inc. | Hydrogen bond forming fluoro ketolides for treating diseases |
| US10188674B2 (en) | 2012-03-27 | 2019-01-29 | Cempra Pharmaceuticals, Inc. | Parenteral formulations for administering macrolide antibiotics |
| US9861616B2 (en) | 2013-03-14 | 2018-01-09 | Cempra Pharmaceuticals, Inc. | Methods for treating respiratory diseases and formulations therefor |
| US9751908B2 (en) | 2013-03-15 | 2017-09-05 | Cempra Pharmaceuticals, Inc. | Convergent processes for preparing macrolide antibacterial agents |
| US10913764B2 (en) | 2013-04-04 | 2021-02-09 | President And Fellows Of Harvard College | Macrolides and methods of their preparation and use |
| US9982005B2 (en) | 2013-04-04 | 2018-05-29 | President And Fellows Of Harvard College | Macrolides and methods of their preparation and use |
| US11634449B2 (en) | 2013-04-04 | 2023-04-25 | President And Fellows Of Harvard College | Macrolides and methods of their preparation and use |
| US10633407B2 (en) | 2014-10-08 | 2020-04-28 | President And Fellows Of Harvard College | 14-membered ketolides and methods of their preparation and use |
| US11466046B2 (en) | 2014-10-08 | 2022-10-11 | President And Fellows Of Harvard College | 14-membered ketolides and methods of their preparation and use |
| US10640528B2 (en) | 2015-03-25 | 2020-05-05 | President And Fellows Of Havard College | Macrolides with modified desosamine sugars and uses thereof |
| US11535643B2 (en) | 2015-03-25 | 2022-12-27 | President And Fellows Of Harvard College | Macrolides with modified desosamine sugars and uses thereof |
| CN106518939B (en) * | 2015-09-14 | 2019-12-31 | 江苏奥赛康药业有限公司 | Method for preparing Solithromycin compound |
| CN106518939A (en) * | 2015-09-14 | 2017-03-22 | 江苏奥赛康药业股份有限公司 | Method for preparing Solithromycin compound |
| EP3190122A1 (en) | 2016-01-08 | 2017-07-12 | LEK Pharmaceuticals d.d. | A novel synthetic pathway towards solithromycin and purification thereof |
| WO2017118690A1 (en) | 2016-01-08 | 2017-07-13 | Lek Pharmaceuticals D.D. | A novel synthetic pathway towards solithromycin and purification thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017200943A (en) | 2017-11-09 |
| EP2214484A1 (en) | 2010-08-11 |
| US9453042B2 (en) | 2016-09-27 |
| IL205254A0 (en) | 2010-12-30 |
| US20170096445A1 (en) | 2017-04-06 |
| JP6845099B2 (en) | 2021-03-17 |
| AU2008316830B2 (en) | 2016-03-17 |
| HK1226411A1 (en) | 2017-09-29 |
| IL205254A (en) | 2017-09-28 |
| AU2016203649A1 (en) | 2016-06-16 |
| CN101917850B (en) | 2016-01-13 |
| JP2019147827A (en) | 2019-09-05 |
| CN101917850A (en) | 2010-12-15 |
| CN105732745A (en) | 2016-07-06 |
| JP2011500834A (en) | 2011-01-06 |
| US10131684B2 (en) | 2018-11-20 |
| CA2703475A1 (en) | 2009-04-30 |
| JP5698979B2 (en) | 2015-04-08 |
| JP6167095B2 (en) | 2017-07-19 |
| JP2015051994A (en) | 2015-03-19 |
| AU2008316830A1 (en) | 2009-04-30 |
| EP2214484A4 (en) | 2013-01-02 |
| US20190241602A1 (en) | 2019-08-08 |
| US20100216731A1 (en) | 2010-08-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10131684B2 (en) | Process for the preparation of macrolide antibacterial agents | |
| AU2011255464C1 (en) | Processes for preparing macrolides and ketolides and intermediates therefor | |
| AU2014233240B2 (en) | Convergent processes for preparing macrolide antibacterial agents | |
| CZ303474B6 (en) | 6-O-substituted erythromycin ketolide derivative, process for its preparation, its use and pharmaceutical composition containing thereof | |
| BG64600B1 (en) | 15-membered lactam class ketolides with antibacterial activity | |
| KR20010023756A (en) | 6,9-Bridged erythromycin derivatives | |
| WO2004016634A1 (en) | Novel 11,12-Substituted Lactone Ketolide Derivatives Having Antibacterial Activity | |
| CN108610388B (en) | Preparation method of macrolide | |
| EP1633764B1 (en) | Regioselective process for the preparation of o-alkyl macrolide and azalide derivatives | |
| CZ20003704A3 (en) | 15-membered lactam ketolides exhibiting antibacterial activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880123646.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08841217 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 205254 Country of ref document: IL Ref document number: 2703475 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12739652 Country of ref document: US Ref document number: 2010531238 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008316830 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1722/KOLNP/2010 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2008316830 Country of ref document: AU Date of ref document: 20081023 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008841217 Country of ref document: EP |