WO2009054923A2 - Synthesis and crystalline forms of cb-1 antagonist/inverse agonist - Google Patents
Synthesis and crystalline forms of cb-1 antagonist/inverse agonist Download PDFInfo
- Publication number
- WO2009054923A2 WO2009054923A2 PCT/US2008/011927 US2008011927W WO2009054923A2 WO 2009054923 A2 WO2009054923 A2 WO 2009054923A2 US 2008011927 W US2008011927 W US 2008011927W WO 2009054923 A2 WO2009054923 A2 WO 2009054923A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- polymorphic form
- class
- hcl salt
- Prior art date
Links
- 0 CC(C)(C(CC(O*)=O)c1cc(Br)cc(F)c1)F Chemical compound CC(C)(C(CC(O*)=O)c1cc(Br)cc(F)c1)F 0.000 description 2
- SIODSZKEHGSAJF-YRNVUSSQSA-N CC(C)(/C(/c1cc(Br)cc(F)c1)=C/C(OC)=O)F Chemical compound CC(C)(/C(/c1cc(Br)cc(F)c1)=C/C(OC)=O)F SIODSZKEHGSAJF-YRNVUSSQSA-N 0.000 description 1
- UVLMTQALNTZYQQ-UHFFFAOYSA-N CC(C)(Cc1cc(Br)cc(F)c1)F Chemical compound CC(C)(Cc1cc(Br)cc(F)c1)F UVLMTQALNTZYQQ-UHFFFAOYSA-N 0.000 description 1
- WFYMIOTUYKCVRZ-FTJBHMTQSA-N CC(C)([C@@H](C(C1)CN1[C@@H](c(cc1)ccc1Cl)c1cccc(C(O2)=NNC2=O)c1)c1cc(C#N)cc(F)c1)F Chemical compound CC(C)([C@@H](C(C1)CN1[C@@H](c(cc1)ccc1Cl)c1cccc(C(O2)=NNC2=O)c1)c1cc(C#N)cc(F)c1)F WFYMIOTUYKCVRZ-FTJBHMTQSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/10—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles
- C07D271/113—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles with oxygen, sulfur or nitrogen atoms, directly attached to ring carbon atoms, the nitrogen atoms not forming part of a nitro radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/53—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
Definitions
- CB 1 modulators characterized as inverse agonists/antagonists useful as centrally acting drugs in the treatment of various diseases related to CB-I modulation, including, but not limited to, psychosis, memory deficits, cognitive disorders, Alzheimer's disease, Huntington's disease, migraine, neuropathy, neuro-inflammatory disorders including multiple sclerosis and Guillain- - Barre syndrome and the inflammatory sequelae of viral encephalitis, cerebral vascular accidents, and head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, movement disorders, and schizophrenia.
- diseases related to CB-I modulation including, but not limited to, psychosis, memory deficits, cognitive disorders, Alzheimer's disease, Huntington's disease, migraine, neuropathy, neuro-inflammatory disorders including multiple sclerosis and Guillain- - Barre syndrome and the inflammatory sequelae of viral encephalitis, cerebral vascular accidents, and head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, movement disorders, and schizophrenia.
- the compounds are also useful for the treatment of substance abuse disorders, the treatment of obesity or eating disorders, and complications associated therewith, including left ventricular hypertrophy, as well as the treatment of asthma, constipation, chronic intestinal pseudo-obstruction, and cirrhosis of the liver.
- the invention describes a novel and efficient process for the synthesis of the potent CB-I inverse agonist Compound I, which was previously prepared using a linear synthesis requiring the use of HF to install the fluorine group and requiring column chromatography to separate the diastereomers of Compound 1.
- the synthesis of the present invention is convergent, provides a higher yield of product and provides crystalline intermediates, which is an advantage of this invention with regard to isolation and purification without the use of chromatography. Reviews of Witting and Horner-Wadsworth-Emmons reactions are provided in Bonadies,
- Rhodium catalyzed additions of arylboronic acids to sulfinylimines is described in Weix, D. et al., J. Am. Chem . Soc. 127(4), 1092-1093 (2005); and Bolshan, Y. et al., Org. Lett. 7 (8), 1481-1484 (2005).
- Metal-halogen exchange reactions utilizing n-BuLi/n-Bu 2 Mg are described in Kitagawa, K. et al., Angew. Chem., Int. Ed. 39 (14), 2481-2483 (2000).
- the preparation of azetidines from diols via bis-alkylation is described in Hillier, M.C.; Chen, C-y., J. Org. Chem. 71, 7885-7887 (2006).
- This invention provides a novel and efficient process for producing 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile of structural formula I from benzhydrylamine II and cyanodiol III.
- This invention further provides eleven novel crystalline forms of 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile that have been identified are designated as 1) free base anhydrous polymorphic Form I of Compound I; 2) free base toluene/heptane solvate polymorphic Form I, Type B of Compound I; 3) free base isopropyl acetate/methyl cyclohexane solvate polymorphic Form I, Type A of Compound I; and 4) HCl salt anhydrous polymorphic Forms A, B, C, D, E, F, G and H of Compound I.
- the crystalline forms of these free base and hydrochloric acid salt polymorphs are new and may have advantages in the preparation of pharmaceutical compositions of Compound I, such as ease of processing, handling and dosing.
- the anhydrous crystalline free base Form I of Compound I has improved physiochemical properties, such as lipid based solubility; good pK exposure; chemical and physical stability; purity; ease of purification and isolation; and formulation due to desirable crystal size, crystal surface area, and the lack of crystal aggregation that render it particularly suitable for the manufacture of pharmaceutical dosage forms.
- the novel HCl salt anhydrous polymorphic Form G of Compound I is the most thermodynamically stable crystalline HCl salt form of Compound I, however, forms A, B, F and H are more kinetically favored.
- the present invention also relates to pharmaceutical formulations comprising the novel polymorphs and salts of compound I as active pharmaceutical ingredients, as well as methods for using them as CB-I inverse agonists/antagonists in the treatment of CB-I related disorders.
- FIG. 1 is the X-ray diffraction (XRPD) pattern for the anhydrous free base polymorphic Form I of Compound I.
- FIG. 2 is the Thermogravimetry analysis (TGA) curve for the anhydrous free base polymorphic Form I of Compound I.
- FIG. 3 is the Differential scanning calorimetry (DSC) curve for the anhydrous free base polymorphic Form I of Compound I.
- FIG. 4 is the X-ray diffraction (XRPD) pattern for anhydrous HCl salt polymorphic Form A of Compound I.
- FIG. 5 is the Differential scanning calorimetry (DSC) curve for anhydrous HCl salt polymorphic Form A of Compound I.
- FIG. 6 is the X-ray diffraction (XRPD) pattern for anhydrous HCl salt polymorphic Form
- FIG. 7 is the Differential scanning calorimetry (DSC) curve for anhydrous HCl salt polymorphic Form B of Compound I.
- FIG. 8 is the X-ray diffraction (XRPD) pattern for anhydrous HCl salt polymorphic Form G of Compound I.
- FIG. 9 is the Differential scanning calorimetry (DSC) curve for anhydrous HCl salt polymorphic Form G of Compound I.
- FIG. 10 is the X-ray diffraction (XRPD) pattern for the freebase isopropyl acetate/methylcyclohexane solvate polymorphic Form I Type A of Compound I.
- FIG. 11 is the Differential scanning calorimetry (DSC) curve for the freebase isopropyl acetate/methylcyclohexane solvate polymorphic Form I Type A of Compound I.
- FIG. 12 is the Thermogravimetry analysis (TGA) curve for the freebase isopropyl acetate/methylcyclohexane solvate polymorphic Form I Type A of Compound I.
- FIG. 13 is the X-ray diffraction (XRPD) pattern for the freebase toluene/heptane solvate of polymorphic Form I Type B of Compound I.
- FIG. 14 is the Differential scanning calorimetry (DSC) curve for the freebase toluene/heptane solvate of polymorphic Form I Type B of Compound I.
- FIG. 15 is the X-ray diffraction (XRPD) pattern for HCl salt polymorphic Form C of Compound I.
- FIG. 16 is the X-ray diffraction (XRPD) pattern for HCl salt polymorphic Form D of
- FIG. 17 is the Differential scanning calorimetry (DSC) curve for the HCl salt polymorphic Form D of Compound I.
- FIG. 18 is the Thermogravimetry analysis (TGA) curve for the HCl salt polymorphic Form D of Compound I.
- FIG. 19 is the X-ray diffraction (XRPD) pattern for anhydrous HCl salt polymorphic Form E of Compound I.
- FIG. 20 is the X-ray diffraction (XRPD) pattern for HCl salt polymorphic Form F hydrate of Compound I.
- FIG. 21 is the X-ray diffraction (XRPD) pattern for HCl salt polymorphic Form H of
- the present invention provides a process for the preparation of 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile of structural formula I
- Compound I can be prepared via the reaction of benzhydrylamine II with cyanodiol III to form the protected oxadiazole compound 20, followed by cleavage of the protecting group, P, to give compound I.
- the free base of compound I has three known crystalline forms or polymorphs denoted as anhydrous free base polymorphic Form I, free base isopropyl acetate/methyl cyclohexane solvate polymorphic Form I type A , and free base toluene/heptane solvate polymorphic Form I type B.
- the X-ray powder diffraction (XPRD) patterns for the three free base crystalline forms of Compound I are shown in Figure 1 (Form I), Figure 10 (freebase isopropyl acetate/methyl cyclohexane solvate Form I Type A) and Figure 13 (freebase toluene/heptane solvate Form I, Type B).
- TGA thermogravimetric analysis
- the DSC curve in Figure 3 for anhydrous free base polymorphic Form I of Compound I is characterized by one endotherms with an extrapolated onset temperature of 157.8 0 C, a peak temperature of 163.6 0 C, and an associated heat of 44.6 J/g.
- Figures 10, 11 and 12 show the X-ray diffraction pattern, TGA curve and DSC curve of the anhydrous freebase isopropyl acetate/methylcyclohexane solvate of Form I Type A of compound I.
- Figures 13 and 14 show the X-ray diffraction pattern and DSC curve of the freebase toluene/heptane solvate polymorphic Form I Type B of Compound I.
- Compound I may further be converted to a hydrochloric acid salt as described below.
- Figures 4 and 5 show the X-ray diffraction pattern and DSC curve of the hydrochloric acid salt Form A of Compound I.
- Figures 6 and 7 show the X-ray diffraction pattern and DSC curve of the hydrochloric acid salt Form B of Compound I.
- Figures 8 and 9 show the X-ray diffraction pattern and DSC curve of the hydrochloric acid salt From G of Compound I.
- Figure 15 shows the X-ray diffraction pattern for the hydrochloric acid salt Form C of Compound I.
- Figures 16, 17 and 18 show the X-ray diffraction pattern, TGA curve and DSC curve of the hydrochloric acid salt Form D of Compound I.
- Figure 19 shows the X-ray diffraction pattern for the hydrochloric acid salt Form E of Compound I.
- Figure 20 shows the X-ray diffraction pattern for the hydrochloric acid salt Form F of Compound I.
- Figure 21 shows the X-ray diffraction pattern for the hydrochloric acid salt Form H of Compound I.
- One embodiment of the present invention provides a process for preparing a compound of formula I, or a salt, hydrate or polymorph thereof,
- the protecting group P of Step A and B is selected from Boc and CBZ.
- the leaving groups of Step A are selected from Inflates, tosylates, nosylates, and mesylates.
- the leaving groups of Step A are triflates and compound III is treated with triflic anhydride to form a di- triflate intermediate
- the hindered amine base of Step A is selected from: diisopropyl ethyl amine, triethylamine, triisopropylamine and dicyclohexylamine.
- the hindered amine base of Step A is diisopropyl ethyl amine.
- the reaction of Step A is run in acetonitrile.
- the protecting group P is CBZ. hi another class of this embodiment, the protecting group P is CBZ, and the CBZ protecting group in Step B is removed by hydrogenation.
- the protecting group P is Boc.
- the protecting group is Boc and the Boc protecting group in Step B is removed using an acid.
- the acid is selected from: HCl, H2SO4, H3PO4 and
- the acid is HCl. In another subclass of this subclass, the acid is HCl in isopropanol.
- Step B is run in a solvent selected from: isopropyl acetate, isopropanol, methylene chloride and THF. In a subclass of this class, the solvent of Step B is isopropyl acetate.
- the process further comprises isolating the compound of formula I. In a subclass of this class, the compound of formula I is isolated by recrystallizing from toluene/heptane.
- Another embodiment of the present invention provides a process for preparing a compound of formula I, or a salt, solvate, hydrate or polymorph thereof,
- the protecting group P of Step A and B is selected from
- the protecting group P is CBZ. In another class of this embodiment, the protecting group P is CBZ, and the CBZ protecting group in Step B is removed by hydrogenation. In another class of this embodiment, the protecting group P is Boc. In another class of this embodiment, the protecting group is Boc and the Boc protecting group in Step B is removed using an acid. In a subclass of this subclass, the acid is selected from: HCl, H2SO4, H3PO4 and
- Step B is run in a solvent selected from: isopropyl acetate, isopropanol, methylene chloride and THF. In a subclass of this class, the solvent of Step B is isopropyl acetate.
- the process further comprises isolating the compound of formula I.
- the compound of formula I is isolated by recrystallizing from toluene/heptane.
- Another embodiment of the present invention provides a process for preparing a compound of formula I, or a salt, solvate, hydrate or polymorph thereof,
- the Boc protecting group is removed using an acid.
- the acid is selected from: HCl, H2SO4, H3PO4 and TFA.
- the acid is HCl.
- the acid is HCl in isopropanol.
- the deprotection reaction is run in a solvent selected from: isopropyl acetate, isopropanol, methylene chloride and THF. In a subclass of this class, the solvent is isopropyl acetate.
- the process further comprises isolating the compound of formula I.
- the compound of formula I is isolated by recrystallizing from toluene/heptane.
- Another embodiment of the present invention provides a process for preparing a compound of formula 20, wherein P is a protecting group
- the protecting group P is selected from Boc and CBZ.
- the leaving groups are selected from triflates, tosylates, nosylates, and mesylates.
- the leaving groups are triflates and compound III is treated with triflic anhydride to form a di-triflate intermediate.
- the hindered amine base is selected from: diisopropyl ethyl amine, triethylamine, triisopropylamine and dicyclohexylamine.
- the hindered amine base is diisopropyl ethyl amine.
- the reaction is run in acetonitrile.
- the process further comprises isolating the compound of formula 20.
- the compound of formula 20 is isolated by recrystallizing from isopropyl acetate, dicholoromethane, acetonitrile, heptane, or a mixture thereof.
- the compound of formula 20 is isolated by recrystallizing from isopropyl acetate and heptane.
- Another embodiment of the present invention provides a process for preparing a compound of formula II wherein P is a protecting group, or a salt thereof,
- the base of Step A is selected from: DABCO, triethyl amine, and diisopropyl ethyl amine. In a subclass of this class, the base of Step A is DABCO. In another class of this embodiment, the reaction of Step A is run in methanol. In another class of this embodiment, the reaction of Step A is run between 50 to 55 0 C. In another class of this embodiment, the hydrazine of Step A is 64% hydrazine. In another class of this embodiment, the reaction of Step A is run with DABCO in methanol, followed by treatment with 64% hydrazine. In another class of this embodiment, the process further comprises isolating the hydrazide of formula 3. In a subclass of this class, the hydrazide of formula 3 is a solid. In yet another class of this embodiment, the process further comprises working up the reaction of Step A and using the hydrazide of formula 3 in a solution for Step B.
- the coupling agent of Step B is selected from CDI, triphosgene and phosgene, hi another class of this embodiment, the coupling agent of Step B is CDI.
- the reaction of Step B is run in an aprotic solvent.
- the aprotic solvent is THF, toluene and ether.
- the aprotic solvent is THF.
- the reaction of Step B is run at room temperature.
- the reaction of Step B is run with CDI in THF. In a subclass of this class, the reaction is run at room temperature.
- the process further comprises isolating the oxadiazole of formula 4 of Step B. hi a subclass of this class, the oxadiazole of formula 4 is a solid. In yet another class of this embodiment, the process further comprises working up the reaction of Step B and using the oxadiazole of formula 4 in a solution for Step C.
- the alkyl magnesium compound of Step C is selected from: dibutyl magnesium, dimethyl magnesium, diethyl magnesium, and dipropyl magnesium. In another class of this embodiment, the alkyl magnesium compound of Step C is di-w-butyl magnesium. In another class of this embodiment, the alkyl lithium compound of Step C is selected from: n-butyl lithium, sec-butyl lithium, tert-butyl lithium, and hexyl lithium. In another class of this embodiment, the alkyl lithium compound of Step C is n-butyl lithium.
- the reaction of Step C is run in an aprotic solvent, hi a subclass of this class, the aprotic solvent of Step C is selected from: THF, toluene, MTBE, and diethyl ether. In another subclass of this class, the aprotic solvent of Step C is THF. In another class of this embodiment, the reaction of Step C is run at a temperature between about -20 to -78 0 C. In a subclass of this class, the reaction of Step C is run at a temperature between about -40 to -50 0 C.
- the reaction of Step C is run with di-n-butyl magnesium and w-butyl lithium in THF at a temperature between about -20 to -78 0 C. In a subclass of this class, the temperature is between about -40 to -50 0 C.
- the reaction of Step C is worked up with acid, hi a subclass of this class, the acid of Step C is HCl or H2SO4. In a subclass of this class, the acid of Step C is HCl.
- the process further comprises isolating the aldehyde of formula 5. In a subclass of this class, the aldehyde of formula 5 is a solid. In yet another class of this embodiment, the process further comprises working up the reaction of Step C and using aldehyde of formula 5 in solution for Step D.
- the catalyst of Step D is selected from: PPTS, KHSO4, BF3-etherate, Ti(0Et)4, and TiCl4/triethyl amine.
- the catalyst of Step D is PPTS.
- the reaction of Step D is run in a solvent selected from: toluene, methylene chloride and THF.
- the reaction of Step D is run in toluene.
- the reaction of Step D is run with PPTS in toluene.
- the reaction of Step D is run at approximately 40 0 C.
- the process further comprises isolating the N-tert-butyl sulfinyl imine of formula 6 of Step D.
- the N- tert-buty ⁇ sulfinyl imine of formula 6 is a solid.
- the process further comprises working up the reaction of Step D and using the iV-ter/-butyl sulfinyl imine of formula 6 in solution for Step E.
- the protecting group P of the compound of formula 7 is a CBZ or Boc group.
- the compound of formula 7 is a N-CBZ protected oxadiazole wherein the protecting group P is CBZ.
- the protected oxadiazole compound of formula 7 is a iV-Boc protected oxadiazole wherein the protecting group P is Boc.
- the iV-Boc protected oxadiazole of formula 7 is prepared by treating the N-tert-buty ⁇ sulfinyl imine of formula 6 with boc anhydride in the presence of a base.
- the base is tertiary amine base. In another subclass of this subclass, the base is triethylamine.
- the TV-Boc protected oxadiazole is prepared by treating the N-tert-buty ⁇ sulfinyl imine of formula 6 with Boc anhydride in the presence of triethylamine in an aprotic solvent. In a subclass of this subclass, the aprotic solvent is THF. In another subclass of this subclass, the reaction of Step E is run at about 40 0 C.
- the compound of formula 7 is a N-Boc protected oxadiazole prepared by treating the N-tert-buty ⁇ sulfinyl imine of formula 6 with boc anhydride and triethyl amine in THF.
- the process further comprises isolating the protected oxadiazole compound of formula 7 of Step E.
- the protected oxadiazole compound of formula 7 is a solid.
- the process further comprises working up the reaction of Step E and using the protected oxadiazole compound of formula 7 in solution for Step F.
- the protecting group P of Step F is CBZ. In another class of this embodiment, the protecting group P of Step F is Boc. In another class of this embodiment, the rhodium catalyst of Step F is Rh(acac)(CH2CH2)2- In another class of this embodiment, the ligand is a phosphine ligand. In a subclass of this class, the phosphine ligand is selected from: l,2-bis(diphenyl phosphino)benzene and 1 ,2-bis(diphenyl phosphino)ethane. In another subclass of this class, the phosphine ligand is l,2-bis(diphenyl phosphino)benzene.
- the solvent is selected from: tert-amyl alcohol, tert-butanol, THF, and dioxane.
- the solvent is tert-amyl alcohol.
- the reaction of Step F is run at a temperature of about room temperature to about 45 0 C.
- the reaction of Step F, wherein the protecting group P is Boc is run in the presence of Rh(acac)(CH2CH2)2 and l,2-bis(diphenyl phosphino)benzene.
- the reaction of Step F is run in tert-amyl alcohol.
- the reaction of Step F is run at a temperature of about room temperature to about 45 0 C.
- the process further comprises isolating the 7V-tert-butyl sulfinyl amine of formula 8 of Step F.
- the N- tert-butyl sulfinyl amine of formula 8 is a solid.
- the process further comprises working up the reaction of Step F and using the jV-tert-butyl sulfinyl amine of formula 8 in solution for Step G.
- the protecting group P of Step G is CBZ.
- the protecting group P of Step G is Boc.
- the tert-butyl sulfoxide group of Step G is cleaved with an acid.
- the tert-butyl sulfoxide group is cleaved by treatment with an acid selected from the group consisting of: hydrochloric acid, sulfuric acid, phosphoric acid and trifluoroacetic acid.
- the cleavage of Step G is run in a halogenated solvent.
- the halogenated solvent is selected from: dichloromethane, chloroform and carbon tetrachloride.
- the halogenated solvent is dichloromethane.
- the cleavage of Step G is run at room temperature.
- the tert-butyl sulfoxide group of compound 8 in Step G is cleaved by treatment with hydrochloric acid.
- the process further comprises isolating the compound of formula II.
- the compound of formula II is a solid.
- the process further comprises working up the reaction of Step G and using the compound of formula Ha in solution for the coupling reaction to give compound 20.
- Another embodiment of the present invention provides a process for preparing a compound of formula III, or a salt thereof,
- R Ci -3 alkyl, by esterification of the compound of formula 16;
- R Ci -3 alkyl, by carboxylation of the compound of formula 17;
- R is -CH 3 . In another class of this embodiment, R is CH 2 CH 3 . In yet another class of this embodiment, R is -CH 2 CH 2 CH 3 or -CH(CH 3 ) 2 .
- the Grignard reagent of Step A is isopropyl magnesium chloride.
- the reaction Step A is run in the presence of one or more transition metal halide salt catalysts.
- the transition metal halide salt catalyst is selected from CuCl, ZnCl 2 and CoCl 2 .
- the transition metal halide salt catalysts are CuCl and ZnCl 2 .
- the Grignard reaction of Step A is run in an ether solvent. In a subclass of this class, the ether solvent is tetrahydrofuran.
- reaction of Step A is run in tetrahydrofuran with isopropyl magnesium chloride, in the presence of CuCl and ZnCl 2 .
- process further comprises isolating the compound of formula 12.
- process further comprises working up the reaction of Step A and using the compound of formula 12 in a toluene solution for Step B.
- the fluorine source of Step B is Select-FluorTM fluorinating agent.
- the base of Step B is an alkoxide base or sodium amylate.
- the alkoxide base is potassium tert-butoxide.
- the base of Step B is sodium amylate.
- the silyl halide and silyl triflate in Step B are selected from: ferf-butyldimethylsilyl chloride, trimethyl silyl chloride, and tert-butyldimethylsilyl triflate.
- the silyl halide of Step B is tert-butyldimethylsilyl chloride.
- the fluorination source of Step B is Select-FluorTM fluorinating agent
- the base is sodium amylate
- the silyl halide is tert-butyldimethylsilyl chloride.
- the process further comprises isolating the compound of formula 13.
- the process further comprises working up the reaction of Step B and using the compound of formula 13 in a toluene solution for Step C.
- the base of Step C is selected from: cesium carbonate, potassium carbonate, lithium carbonate, potassium terf-butoxide, lithium hydride, sodium hydride, and sodium amylate.
- the base of Step C is potassium carbonate.
- the trimethylphosphonoacetate of Step C is pretreated with base before addition to the compound of formula 13.
- the base in Step C is selected from: cesium carbonate, potassium carbonate, lithium carbonate, potassium tert-butoxide, lithium hydride, sodium hydride, and sodium amylate.
- the base of Step C is potassium carbonate.
- the reaction of Step C is run in a polar aprotic solvent.
- the polar aprotic solvent is selected from: dimethyl formamide, tetrahydrofuran or ether.
- the solvent of Step C is dimethyl formamide.
- the compound of formula 13 in Step C is reacted with trimethylphosphonoacetate which was pretreated with potassium carbonate.
- the reaction in Step C is run in dimethyl formamide.
- the process further comprises isolating the compound of formula 14 of Step C.
- the process further comprises working up the reaction of Step C and using compound of formula 14 in toluene for Step D.
- the hydrolysis of Step D is run using sodium hydroxide, lithium hydroxide or potassium hydroxide.
- the hydrolysis of Step D is run in using sodium hydroxide.
- the hydrolysis of Step D is run in an aqueous solvent.
- the aqueous solvent is methanol/water.
- the aqueous solvent is methanol/water/toluene.
- the hydrolysis of Step D is run using sodium hydroxide in methanol/water.
- the hydrolysis of Step D is run using sodium hydroxide in methanol/water/toluene.
- the process further comprises isolating the compound of formula 15 of Step D.
- the reduction of compound 15 of Step E is a hydrogenation in the presence of hydrogen and a ruthenium catalyst.
- the ruthenium catalyst has an axial chiral ligand.
- the axial chiral ligand is a JosiphosTM type ligand, a SolphosTM type ligand, a CH 3 O-BIPHEPTM type ligand, a BINAP type ligand, or a SegphosTM type ligand.
- the axial chiral ligand is (R)-C1,CH 3 O-BIPHEPTM, (S)-SolphosTM, (R)-Furyl-SolphosTM and SL-J212-1.
- the JosiphosTM type axial chiral ligand is SL-J212-1.
- the hydrogenation of Step E is run under pressure. In a subclass of this subclass, the hydrogenation of Step E is run at 200 psig. In another subclass of this class, the hydrogenation of Step E is run at about 40-50 0 C.
- the reduction of Step E is a hydrogenation in the presence of a ruthenium catalyst with a JosiphosTM type axial chiral ligand SL-J212-1. In a subclass of this class, the ruthenium catalyst is prepared by reacting [(cymene)RuCl] 2 with SL-J212-1. In another class of this embodiment, the process further comprises isolating the compound of formula 16 of Step E.
- the process further comprises isolating the compound of formula 16 of Step E as a solid and recrystallizing the compound of formula 16.
- the process further comprises working up the reaction of Step E and using the compound of formula 16 in solution for Step F.
- the process further comprises working up the reaction of Step E and using the compound of formula 16 in a methanolic solution for Step F.
- the esterification of Step F is run in the presence of an acid chloride in an alcohol solvent.
- the ester formed by the esterification in Step F is a methyl ester and the esterification is run in the presence of acetyl chloride in methanol.
- the esterification is run at room temperature.
- the process further comprises isolating the compound of formula 17 of Step F.
- the carboxylation of Step G is run by treating the compound of formula 17 with a base and in an ether or polar solvent, followed by the addition of CO 2 .
- the base is selected from: lithium hexamethyl disilazide, sodium hexamethyl disilazide, potassium hexamethyl disilazide and LDA.
- the ether or polar solvent is selected from one or more of: THF, THF, MTBE, DME, toluene, and DMPU.
- the carboxylation reaction was run by treating the compound of formula 17, in THF or toluene, with lithium hexamethyl disilazide, followed by the addition of CO 2 .
- the carboxylation reaction was run by treating the compound of formula 17, in THF or toluene and DMPU, with lithium hexamethyl disilazide, followed by the addition of CO 2 .
- the carboxylation reaction was run by treating the compound of formula 17, in DME/toluene and DMPU, with lithium hexamethyl disilazide, followed by the addition of CO 2 .
- the process further comprises isolating the compound of formula 18 of Step G.
- the reduction of Step H was run in the presence of a reducing agent.
- the reducing agent is sodium borohydride, sodium borohydride/I 2 , sodium borohydride/Br 2 , sodium borohydrideBFs/tetrahydrofuran complex, BFa/etherate complex, borane/tetrahydrofuran complex, and borane/dimethyl sulfide complex.
- the reducing agent is sodium borohydride/Br 2 .
- the reduction of Step H is run in a solvent selected from one or more of: toluene, DME, THF, DME/toluene, and dichloromethane.
- the reduction of Step H is run in a solvent selected from DME and toluene.
- the process further comprises isolating the compound of formula 19 of Step H.
- the cyanation of Step I is run in the presence of zinc, bromine, Zn(CN) 2 and a palladium catalyst.
- the palladium catalyst is a bidentate or monodentate palladium catalyst.
- the palladium phospine catalyst In another subclass of this class, the palladium phospine catalyst. In a subclass of this subclass, the palladium catalyst is palladium tetrakis triphenylphosphine. In another subclass of this class, the palladium catalyst is Pd(dppf) 2 . In another subclass of this class, the cyanation of Step I is run in DMF. In another subclass of this class, the cyanation of Step I is run in DMF at 80 0 C.
- the process further comprises isolating the compound of formula III of Step I.
- Another embodiment of the present invention provides for a method of preventing or treating a disease related to CB-I modulation comprising administering a therapeutically effective amount of a polymorph, hydrate or salt of Compound I to a subject in need thereof.
- Another embodiment of the present invention provides for the use of a therapeutically effective amount of a polymorph, solvate, hydrate or salt of Compound I for the manufacture of a medicament useful for the treatment, control, or prevention of a disease related to CB-I modulation in a subject in need of such treatment.
- Another embodiment of the present invention provides for a method of preventing or treating obesity, eating disorders, or an obesity related disorder comprising administering a therapeutically effective amount of a polymorph, hydrate or salt of Compound I to a subject in need thereof.
- Another embodiment of the present invention provides for the use of a therapeutically effective amount of a polymorph, solvate, hydrate or salt of Compound I for the manufacture of a medicament useful for the treatment, control, or prevention of obesity, eating disorders, or an obesity-related disorder in a subject in need of such treatment.
- Polymorphs are compounds having the same chemical composition but different crystal structures. Polymorphism is the ability of the same chemical substance to exist as different crystalline structures.
- the compound 3-[(liS)-l-(l- ⁇ (5)-(4-chlorophenyl)[3-(5-oxo-4,5-dihydro- 1 ,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2-fluoro-2-methylpropyl]-5- fluorobenzonitrile of structural formula I, and the HCl salt thereof, has been found it exist in at least eleven polymorphic or crystalline forms each of which can be formed by careful control of the crystallization conditions.
- hydrate is meant to include all full, multiple and partial hydrates of compound I, including, but not limited to, the mono hydrate, hemi-hydrate and bis hydrate.
- solvate is meant to include compound forms containing solvent molecules within the crystal structure of Compound I, or solvent molecules bound to or associated with Compound I, including but not limited to toluene, heptane, isopropyl acetate, ethyl acetate, methyl cyclohexane and water.
- amorphous refers to solid forms that have no long-range molecular order.
- Additional salts of compounds of formula I refer to the pharmaceutically acceptable and common salts, for example, base addition salt to carboxyl group when the compound has a carboxyl group, or acid addition salt to amino or basic heterocycle when the compound has an amino or basic heterocycle group, and the like.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- the base addition salts include salts with alkali metals (including, but not limited to, sodium, potassium); alkaline earth metals (including, but not limited to, calcium, magnesium); ammonium or organic amines (including, but not limited to, trimethylamine, triethylamine, dicyclohexylamine, ethanolamine, diethanolamine, triethanolamine, procaine, N,N'-dibenzylethylenediamine), and the like.
- alkali metals including, but not limited to, sodium, potassium
- alkaline earth metals including, but not limited to, calcium, magnesium
- ammonium or organic amines including, but not limited to, trimethylamine, triethylamine, dicyclohexylamine, ethanolamine, diethanolamine, triethanolamine, procaine, N,N'-dibenzylethylenediamine, and the like.
- the acid addition salts include salts with inorganic acids (including, but not limited to, hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, perchloric acid), organic acids (including, but not limited to, acetic acid, oxalic acid, maleic acid, fumaric acid, tartaric acid, citric acid, ascorbic acid, trifluoroacetic acid, acetic acid), sulfonic acids (including, but not limited to, methanesulfonic acid, isethionic acid, benzenesulfonic acid, /Moluenesulfonic acid, p-toluenesulfonic acid monohydrate, p- toluene sulfonic acid hydrate, camphor sulfonic acid), and the like.
- inorganic acids including, but not limited to, hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, perchloric acid
- organic acids including, but not limited to
- a pharmaceutical composition comprising 3-[(I 1 S)-I -(l- ⁇ (S)-(4-chlorophenyl)[3-(5-oxo-4,5-dihydro-l, 3,4- oxadiazol-2-yl)phenyl] -methyl ⁇ azetidin-3-yl)-2-fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as a free base, salt, hydrate or polymorph thereof.
- Compound I is in substantially pure form.
- Compound I is crystalline.
- Compound I is crystalline anhydrous free base.
- Compound I is a crystalline free base solvate. In another class of this embodiment, Compound I is a crystalline anhydrous salt. In another class of this embodiment, Compound I is a crystalline salt hydrate. In another class of this embodiment, Compound I is a crystalline anhydrous HCl salt, hi another class of this embodiment, Compound I is a crystalline HCl salt hydrate.
- the composition comprises 3-[(15)-l-(l- ⁇ (5)-(4-chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2- yl)phenyl] methyl ⁇ azetidin-3-yl)-2-fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the free base Form I of Compound I.
- the free base Form I of Compound I is in substantially pure form.
- free base Form I of Compound I is crystalline.
- free base Form I of Compound I is anhydrous.
- free base Form I of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the free base toluene/heptane solvate Form I, Type B of Compound I.
- the free base toluene/heptane solvate Form I, Type B of Compound I is in substantially pure form.
- the free base toluene/heptane solvate Form I, Type B of Compound I is crystalline.
- the free base toluene/heptane solvate Form I, Type B of Compound I is anhydrous.
- the free base toluene/heptane solvate Form I, Type B of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(l.S)-l-(l- ⁇ (S)-(4- chloro-phenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methyl-propyl]-5-fluorobenzonitrile (Compound I) as the free base isopropyl acetate/methyl cyclohexane solvate Form I, Type A of Compound I.
- the free base isopropyl acetate/methyl cyclohexane solvate Form I Type A of Compound I is in substantially pure form.
- the free base isopropyl acetate/methyl cyclohexane solvate Form I Type A of Compound I is crystalline.
- the free base isopropyl acetate/methyl cyclohexane solvate Form I Type A of Compound I is anhydrous.
- the free base isopropyl acetate/methyl cyclohexane solvate Form I Type A of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro- 1 ,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fiuorobenzonitrile (Compound I) as the HCl salt Form A of Compound I.
- the HCl salt Form A of Compound I is in substantially pure form.
- the HCl salt Form A of Compound I is crystalline.
- the HCl salt Form A of Compound I is anhydrous, hi another subclass of this class, the HCl salt Form A of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the HCl salt Form B of Compound I.
- the HCl salt Form B of Compound I is in substantially pure form.
- the HCl salt Form B of Compound I is crystalline.
- the HCl salt Form B of Compound I is anhydrous.
- the HCl salt Form B of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(lS)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l ,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the HCl salt Form C of Compound I.
- the HCl salt Form C of Compound I is in substantially pure form.
- the HCl salt Form C of Compound I is crystalline.
- the HCl salt Form C of Compound I is anhydrous.
- the HCl salt Form C of Compound I is anhydrous and crystalline.
- the composition comprises 3-[( IS)-I -(I - ⁇ (S)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro- 1 ,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the HCl salt Form D of Compound I.
- the HCl salt Form D of Compound I is in substantially pure form.
- the HCl salt Form D of Compound I is crystalline.
- the HCl salt Form D of Compound I is anhydrous.
- the HCl salt Form D of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(lS)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the HCl salt Form E of Compound I.
- the HCl salt Form E of Compound I is in substantially pure form.
- the HCl salt Form E of Compound I is crystalline.
- the HCl salt Form E of Compound I is anhydrous.
- the HCl salt Form E of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(lS)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the HCl salt Form F of Compound I.
- the HCl salt Form F of Compound I is in substantially pure form.
- the HCl salt Form F of Compound I is crystalline.
- the HCl salt Form F of Compound I is a hydrate. In another subclass of this class, the HCl salt Form F of Compound I is a crystalline hydrate. In another class of this embodiment, the composition comprises 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the HCl salt Form G of Compound I.
- the HCl salt Form G of Compound I is in substantially pure form. In another subclass of this class, the HCl salt Form G of Compound I is crystalline. In another subclass of this class, the HCl salt Form G of Compound I is anhydrous. In another subclass of this class, the HCl salt Form G of Compound I is anhydrous and crystalline.
- the composition comprises 3-[(15)-l-(l- ⁇ (5)-(4- chlorophenyl)[3-(5-oxo-4,5-dihydro-l,3,4-oxadiazol-2-yl)phenyl]methyl ⁇ azetidin-3-yl)-2- fluoro-2-methylpropyl]-5-fluorobenzonitrile (Compound I) as the HCl salt Form H of Compound I.
- the HCl salt Form H of Compound I is in substantially pure form.
- the HCl salt Form H of Compound I is crystalline.
- the HCl salt Form H of Compound I is anhydrous.
- the HCl salt Form H of Compound I is anhydrous and crystalline.
- the compounds in the processes of the present invention include stereoisomers, such as optical isomers, diastereomers and geometerical isomers, or tautomers depending on the mode of substitution.
- the present invention is meant to comprehend all such isomeric forms of the compounds in the compositions of the present invention, and their mixtures. All hydrates, solvates and polymorphic crystalline forms of the above-described compounds and their use, including their use in the processes of the instant invention, are encompassed within scope of the instant invention.
- Neurokinin- 1 (NK-I) receptor antagonists may be favorably employed in combination with a compound of the present invention. NK-I receptor antagonists of use in the present invention are fully described in the art.
- Specific neurokinin- 1 receptor antagonists of use in the present invention include: ( ⁇ )-(2R3R,2S3S)-N- ⁇ [2-cyclopropoxy-5-(trifluoromethoxy)- phenyl]methyl ⁇ -2-phenylpiperidin-3-amine; 2-(R)-(l-(R)-(3,5- bis(trifluoromethyl)phenyl)ethoxy)-3-(S)-(4-fluorophenyl)-4-(3-(5-oxo-lH r ,4H-l,2,4- triazolo)methyl)morpholine; aperpitant; CJ17493; GW597599; GW679769; R673; RO67319; Rl 124; R1204; SSR146977; SSR240600; T-2328; and T2763.; or a pharmaceutically acceptable salts thereof.
- compositions which comprise a polymorph, hydrate or salt of Compound I and a pharmaceutically acceptable carrier.
- the pharmaceutical compositions of the present invention comprise a compound of Formula I as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- the compositions include compositions suitable for oral, rectal, topical, parenteral
- ocular ophthalmic
- pulmonary nasal or buccal inhalation
- nasal administration although the most suitable route in any given case will depend on the nature and severity of the conditions being treated and on the nature of the active ingredient. They may be conveniently presented in unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
- the polymorphs, hydrates and salts of Compound I can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations.
- oral liquid preparations such as, for example, suspensions, elixirs and solutions
- carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparation
- tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques. Such compositions and preparations should contain at least 0.1 percent of active compound. The percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2 percent to about 60 percent of the weight of the unit. The amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained.
- the active compounds can also be administered intranasally as, for example, liquid drops or spray.
- the tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- a liquid carrier such as a fatty oil.
- Various other materials may be present as coatings or to modify the physical form of the dosage unit. For instance, tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
- the polymorphs, hydrates and salts of Compound I may also be administered parenterally.
- Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxy-propylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- the present invention provides a method for the treatment and/or prevention of obesity and obesity-related disorders in a subject in need thereof comprising administering a therapeutically effective amount of a hydrate, salt or polymorph of Compound I to the subject in need thereof.
- the present invention also provides for the use of the hydrates, salts and polymorphs of Compound I for the manufacture of a medicament for the prevention and/or treatment of CB-I modulated disorders, such as psychosis, memory deficits, cognitive disorders, Alzheimer's disease, migraine, neuropathy, neuro-inflammatory disorders including multiple sclerosis and Guillain-Barre syndrome and the inflammatory sequelae of viral encephalitis, cerebral vascular accidents, and head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, movement disorders, and schizophrenia.
- CB-I modulated disorders such as psychosis, memory deficits, cognitive disorders, Alzheimer's disease, migraine, neuropathy, neuro-inflammatory disorders including multiple sclerosis and Guillain-Barre syndrome and the inflammatory sequelae of viral encepha
- the compounds are also useful for the treatment of substance abuse disorders, the treatment of obesity or eating disorders, obesity- related disorders and complications associated therewith, including left ventricular hypertrophy, as well as the treatment of asthma, constipation, chronic intestinal pseudo-obstruction, and cirrhosis of the liver.
- the obesity-related disorders herein are associated with, caused by, or result from obesity.
- obesity-related disorders include restenosis, atherosclerosis, arteriosclerosis, overeating and bulimia, hypertension, diabetes, elevated plasma insulin concentrations and insulin resistance, dyslipidemias, hyperlipidemia, endometrial, breast, prostate and colon cancer, osteoarthritis, obstructive sleep apnea, cholelithiasis, gallstones, heart disease, abnormal heart rhythms and arrythmias, myocardial infarction, congestive heart failure, coronary heart disease, sudden death, stroke, polycystic ovary disease, craniopharyngioma, the Prader-Willi Syndrome, Frohlich's syndrome, GH-def ⁇ cient subjects, normal variant short stature, Turner's syndrome, and other pathological conditions showing reduced metabolic activity or a decrease in resting energy expenditure as a percentage of total fat-free mass, e.g, children with acute lymphoblastic leukemia, metabolic syndrome, insulin resistance syndrome, reproductive hormone abnormalities, sexual and reproductive dysfunction,
- Treatment refers to the administration of the compounds or combinations of the present invention to reduce or maintain the body weight of an obese subject.
- prevention refers to the administration of the compounds or combinations of the present invention to reduce or maintain the body weight of a subject at risk of obesity.
- subject refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
- subject in need thereof refers to a subject who is in need of treatment or prophylaxis as determined by a researcher, veterinarian, medical doctor or other clinician.
- the subject in need of treatment is an obese mammal.
- the subject in need of treatment is an obese human with one or more obesity-related co-morbidities.
- the subject in need of treatment is an obese human without obesity-related comorbidities.
- terapéuticaally effective amount means the amount of the active compounds in the composition that will elicit the biological or medical response in a tissue, system, subject, or human that is being sought by the researcher, veterinarian, medical doctor or other clinician, which includes alleviation of the symptoms of the disorder being treated.
- the daily dose range of a salt, hydrate or polymorph of compound I is administered at a daily dosage of from about 0.0001 mg/kg to about 100 mg/kg, preferably from about 0.001 mg/kg to about 100 mg/kg, more preferably from about 0.001 mg/kg to about 10 mg/kg of body weight of a subject in single or divided doses two to six times a day, or in sustained release form.
- the compounds of this invention can be administered to humans in the dosage ranges specific for each compound.
- the compositions are preferably provided in the form of tablets containing from 0.01 mg to 1,000 mg, preferably 0.01, 0.05, 0.1, 0.2, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 7.5, 10, 15, 20, 25, 30, 40, 50, 60, 75, 80, 100, 125, 150, 175, 200, 225, 250, 500, 750, 850 and 1,000 milligrams of each active ingredient for the symptomatic adjustment of the dosage to the subject to be treated.
- This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the X-ray powder diffraction spectrum was recorded at ambient temperature.
- DSC data were acquired using a TA Instruments DSC-2910 differential scanning calorimeter at a heating rate of 10°C/min under N 2 flow.
- TA Instruments DSC 2910 or equivalent instrumentation Between 2 and 6 mg sample is weighed into an open aluminum pan. This pan is then crimped and placed at the sample position in the calorimeter cell. The sample is heated in a closed pan. An empty pan is placed at the reference position. The calorimeter cell is closed and a flow of nitrogen is passed through the cell.
- the heating program is set to heat the sample at a heating rate of 10 °C/min to a temperature of approximately 250 0 C. The heating program is started.
- the data are analyzed using the DSC analysis program contained in the system software.
- the melting endotherm is integrated between baseline temperature points that are above and below the temperature range over which the endotherm is observed.
- the data reported are the onset temperature, peak temperature and enthalpy.
- TGA data were acquired using Perkin Elmer TGA-7 thermogravimetric analyzer.
- the furnace is raised and a flow of nitrogen is passed over the sample.
- the heating program is set to heat the sample under a nitrogen flow at a heating rate of 10°C/min to a temperature of approximately 25O 0 C.
- the heating program is started.
- the data are analyzed using the delta Y function in the analysis program contained in the system software.
- the percent weight loss by the sample is calculated from the onset of the heating program to the melt/decomposition of the sample.
- acac is acetyl acetonate; aq is aqueous; /-AmOH is tert-amy ⁇ alcohol; BINAP is 2,2'-bis(diphenylphosphino)-l,r-binaphthyl; BuLi or n-BuLi is butyl lithium; Bu 2 Mg is dibutyl magnesium; Boc is ter/-butoxy carbonyl; Boc anhydride is tert-butoxy carbonyl anhydride; CBZ is carbobenzyloxy; CDI is l,l '-carbonyldiimidazole; DABCO is 1,4- diazabicyclo[2.2.2]-octane; DME is ethylene glycol dimethyl ether; DMF is dimethylformamide; DMPU is l,3-dimethyl-3,4,5,6-tetrahydro-2(lH)-
- the invention describes an efficient process for the synthesis of the potent CB-I inverse agonist Compound I.
- the fully functionalized chiral benzhydrylamine II is synthesized from the commercially available 3-bromobenzoyl chloride 1.
- 3-Bromobenzoyl chloride 1 was converted to hydrazide 3 by treatment with DABCO and hydrazine.
- Hydrazide 3 was treated with CDI to give oxadiazole 4, which was converted to aldehyde 5 via a metal- halogen exchange reaction.
- Aldehyde 5 was converted to the Ellman's Imine compound 6 by treatment with PPTS and (S)-sulfinamide.
- Scheme 2 illustrates the synthesis of the cyanodiol III intermediate.
- the synthesis of cyanodiol III was commenced from 1,3-dibromofluorobenzene 11 via Grignard formation with isopropylmagnesium chloride followed by CuCl/ZnCl 2 catalyzed addition to isobutyryl chloride which afforded ketone 12.
- Fluoro ketone 13 was treated with potassium carbonate and trimethylphosphonoacetate to give the Horner-Wadsworth- Emmons adduct, ⁇ , ⁇ -unsaturated ester 14; this compound was then hydrolyzed in situ with NaOH to give the ⁇ , ⁇ -unsaturated acid 15.
- Rhodium catalyzed asymmetric hydrogenation of ⁇ , ⁇ -unsaturated acid 15 gave saturated acid 16, which, following in situ esterification, provided saturated ester 17.
- Carboxylation of saturated ester 17 afforded ⁇ -carboxyester 18, which was reduced to bromodiol 19 via reduction with sodium borohydride-bromine in DME.
- the palladium catalyzed cyanation of bromo diol 19 yielded the requisite cyanodiol III.
- Compound I is completed via removal of the N-Boc protecing group from intermediate 20.
- AU of the intermediates in this route are crystalline, which is an advantage of this invention with regard to isolation and purification.
- Step A Preparation of Hydrazide 3.
- DABCO 2.81 kg, 25.06 mol
- MeOH 35 L
- 3-bromobenzoyl chloride 1 5.0 kg, 22.78 mol
- the mixture was stirred at room temperature for 10 - 20 minutes.
- Hydrazine (64%, 8.8 L, 182 mol) was added over 20 minutes, and the reaction mixture was heated at 50 to 55 0 C for 3 hours.
- Water (35 L) was added to crystallize the batch at room temperature over 1 hour. The resulting slurry was stirred at room temperature for 1 -2 hours and filtered.
- hydrazide 3 The wet cake was washed with water (3 x 15 L), and dried at room temperature under a vacuum/N 2 sweep to afford hydrazide 3 as white solid.
- Step B Preparation of Oxadiazole 4.
- 3- bromobenzoic hydrazide 3 3.5 kg, 16.3 mol
- THF 35 L
- the slurry was stirred at room temperature for 5 -10 min.
- CDI 3.17 kg, 19.5 mol
- the reaction mixture turned to a clear solution gradually, and the solution was stirred at room temperature for 2 - 3 h.
- IPAc 35 L
- water 35 L
- Step C Preparation of Aldehyde 5.
- oxadiazole 4 3.3 kg, 13.7 mol
- THF 33 L
- the solution was cooled to -50 0 C, and Bu 2 Mg (IMin heptane, 2N, 10.3 L, 10.3 mol) was added over 30 - 50 min at -40 to -45 0 C.
- a dry ice acetone bath was used to control the temperature.
- the batch was cooled to 0 0 C and the reaction was quenched by adding 2NHC1 (10 L) over 20 minutes keeping the batch temperature below 10 0 C.
- the batch was transferred to 160 L extractor, and EtOAc (33 L) and 2NHC1 (23 L) were added.
- EtOAc 33 L
- 2NHC1 23 L
- the resulting layers were separated and the aqueous layer was extracted with EtOAc (10 L).
- the combined organic layers was washed with water (33 L) and followed by brine (20 L), concentrated, and flushed with heptane (10 L).
- the slurry was stirred in 1 :2 EtOAc and heptane (12 L) at room temperature for 2 hours, and filtered.
- N-ter/-butane sulfinyl imine 6 (3.1 kg, 9.83 mol), THF (39 L), Et 3 N (2.5 kg, 24.7 mol) and followed by Boc anhydride (3.2 kg, 14.66 mol).
- the resulting mixture was heated at 40 0 C for 5 h and room temperature for 8 - 10 hours.
- the mixture was concentrated and flushed with IPAc (25 L), followed by heptane (10 L). During concentration, a solid formed and the mixture volume was then adjusted to about 22 L. The slurry was stirred at room temperature for 1 - 2 hours and filtered.
- Step F Preparation of boroxine 10.
- distillation unit batch concentrator
- 4-chlorophenylboronic acid 9 3.6 kg, 23 mol
- toluene 30 L
- the resulting slurry was heated to boil (95 — 110 0 C) and water was removed by azeotropic distillation.
- Fresh toluene (total about 30 - 35 L) was added during the distillation to maintain constant volume in the vessel. After about 30 L of solvent had distilled, the batch was cooled to room temperature and stirred for 0.5 -1 hour.
- Step G Preparation of Sulfinamide 8. To a 75 L round bottom flask under nitrogen was added N-Boc Imine 7 (3.1 kg, 7.88 mol), boroxine 10 (1.64 kg, 3.94 mol) and t-AmOH (62 L). The reaction mixture was heated to 45 0 C, and 1, 2-bis(diphenyl-phosphino)benzene (93 g, 0.208 mol) was added.
- Rh(acac)(CH 2 CH 2 ) 2 (45 g, 0.174 mol) was added.
- the reaction mixture was sparged with nitrogen gas at the same temperature for an additional 10 minutes, and then heated at 45 0 C for 3 - 5 hours.
- the batch was cooled to room temperature and diluted with EtOAc (25 L).
- the resulting thin slurry was transferred to a 160 L extractor containing EtOAc (20 L) and 0.5 Maq Na 2 CO 3 (22 L). After vigorous mixing for 10 minutes at room temperature, the layers were separated. The top organic layer was washed with 3% brine (3 x 20 L) and then brine (20 L).
- the organic solution was concentrated to 25 - 30 L, and flushed with heptane (20 L). During concentration, the product precipitated and then the batch volume was adjusted to about 60 L with heptane. The resulting slurry was stirred at room temperature for 1 - 2 hours, and then filtered. The resulting wet cake was washed with 1 :4 EtO Ac/heptane (2 x 20 L), and heptane
- the solution was concentrated and distilled azeotropically, with additional i-PrOH (6 L), at an internal temperature of ⁇ 45°C to give a thick slurry, and then solvent switched to dichloromethane by adding 18 L of dichloromethane and concentrating to ⁇ 8 L.
- the mixture was then heated to 35 0 C to completely dissolve all the solids, and i-PrOH (24 L) as an anti-solvent was slowly charged to the batch via addition funnel. After the addition of i-PrOH, the batch was slowly cooled to ambient temperature overnight. The temperature was then lowered to 0-5 0 C for 2 hours.
- HPLC retention time of benzyhydrylamine II 8.95 on Zorbax RX C8, 5.0 micron, 4.6 mm x 250 mm, P.N.:880967; temperature: 25°C, UV detection at 210 run, column flow is 1.0 ml/min, solvent A is acetonitrile, solvent B is H 2 O buffered with 0.1 % H 3 PO 4 ; gradient elution: 0 minutes: 10% A/90% B;l minutes: 90% A/10% B; and 15 minutes: 98% A/2% B.
- the reaction was diluted with toluene (11 L) and IN citric acid (10 L). The bi-phasic mixture was stirred for 10 minutes before separating the layers. The organic layer was then treated with IN citric acid (10 L) and stirred for an additional 10 minutes. The layers were separated and the organic layer was washed sequentially with IN K 2 HPO 4 (2 x 11 L) and water (11 L). The wet toluene batch was then held for 36 hours while a second batch of equal size was processed. The wet toluene batches containing isopropyl ketone 12 were then combined and concentrated to approximately 2-3 volumes and the KF ⁇ 200 ppm and were used directly in the next step.
- HPLC retention time of isopropyl ketone 12 8.1 min on 25 cm Zorbax SB C- 18, using MeCN / 0.1 H 3 PO 4 , 1 mL / min. gradient elution: 70% MeCN for 5 minutes then to 90% MeCN at 10 minutes; 210 nm; hold for 5 minutes.
- 1 H NMR 400 MHz, CDCl 3 ) ⁇ 7.85 (IH, br. s, Ar-H), 7.56 (IH, dd, 8.9 Hz, 1.0 Hz, Ar-H), 7.43 (IH, dd, 8 Hz, 1.0 Hz, Ar-H), 3.43 (IH, sep. 6.9 Hz, C-H), 1.21 (6H, d, 6.9 Hz, (CHj) 2 CH).
- Step B Synthesis of Fluoro Ketone 13.
- N 2 To a 100 L round bottomed flask fitted with a thermocouple, under an atmosphere of N 2 was added sequentially sodium amylate (3.0 kg, 27.3 mol) and DMF (11 L). The slurry was aged for 30 minutes until almost all of the base had dissolved. Then the solution was cooled to 10 0 C and a ⁇ 50 weight % solution of the isopropyl ketone 12 (10.8 kg, 50 weight %, 5.4 kg, 22 mol) was added over 55 minutes while maintaining the temperature between 15 - 20 0 C via the controlled addition of the isopropyl ketone.
- the solution was aged for 30 minutes, cooled to 10 0 C and treated with TBSCl (4.3 kg, 28.5 mol) over a 30 minute period while maintaining the temperature between 20 - 28 0 C via controlled TBSCl addition.
- the reaction was aged for 30 minutes, then Select-FluorTM (8.5 kg, 24 mol, Air Products) was then added over 1.5 hours while maintaining the reaction temperature 28 - 35 0 C and the slurry aged for 1 hour.
- water (18 L) was added, followed by toluene (12 L).
- the resulting biphasic mixture was transferred to a 100 L cylindrical vessel and aged for 10 minutes. The layers were separated and the organic phase was washed with additional water (9 L).
- the wet toluene solution containing fluoro ketone 13 was held for 36 hours while a second batch of equal size was processed. The combined toluene batches containing fluoro ketone 13 were then concentrated until the KF ⁇ 200 ppm and about 50 weight %. The resulting toluene solution of fluoro ketone 13 was used directly in the next step.
- HPLC retention time fluoro ketone 13 8.5 minutes on 25 cm Zorbax SB C-18, using MeCN / 0.1 H 3 PO 4 , 1 mL / min. gradient elution: 70% MeCN for 5 minutes, then to 90% MeCN at 10 minutes, hold for 5 minutes at 210 nm.
- Step C Synthesis of ⁇ . ⁇ -Unsaturated Ester 14.
- DMF sequentially DMF (10 L), potassium carbonate (5.3 kg, 38.4 mol) and trimethylphosphonoacetate (5.3 kg, 29.1 mol).
- the slurry was heated to 70 0 C and treated with the toluene solution of fluoro ketone 13 (9.9 kg of 50 weight % solution, 4.96 kg, 18.8 mol) over 46 minutes.
- the reaction was heated to 80 - 82 0 C and aged at this temperature for 10 hours. Then the reaction was cooled to room temperature and aged overnight for 10 hours.
- Step D Synthesis of ⁇ . ⁇ -Unsaturated Acid 15.
- the toluene solution containing ⁇ , ⁇ -unsaturated ester 14 of Step C was charged to a 50 L round bottomed flask fitted with a thermocouple, under a N 2 atmosphere. Next, methanol (17 L) and 5N NaOH (8 L) were added.
- the solid ⁇ , ⁇ -unsaturated acid 15 was collected by filtration, washed with 1 :1 methanol / water (15 L) and dried by pulling N 2 through the cake for 36 hours. A second batch on the same scale was processed to afford ⁇ , ⁇ -unsaturated acid 15 as an off white solid.
- HPLC retention time ⁇ , ⁇ -unsaturated acid 15 13.3 minutes on 25 cm Zorbax SB C-18, using MeCN / 0.1 H 3 PO 4 , 1 mL / min. gradient elution: 10% MeCN - 50% MeCN over 0-5 minutes, then 50 - 90% MeCN over 5 - 20 minutes, and hold at 90 % MeCN for additional 5 minutes at 210 nm.
- Step E Synthesis of ⁇ , ⁇ -Saturated Acid 16.
- the ⁇ , ⁇ -unsaturated acid 15 (2.2 kg, 7.2 mol) was weighed into a 4 L beaker and transferred into a poly jug. The beaker was then rinsed with MeOH (2 L) and the solution was transferred to the poly jug. Next, triethylamine (252 mL, 1.8 mol) was added and the funnel was rinsed down with the remaining MeOH (7 L). After rigorous mixing, the batch was transferred into a 5 gallon vessel followed by degassing of the solution.
- the catalyst was prepared as follows: the [Ru(cymene)Cl] 2 precursor (1.7 g, 0.0055 mol 0.076 mol %) and the (R)-l-[(S)-2-Di-2-furylphosphino)-ferrocenyl]ethyldi-/ert.-butyl-phosphine (or SL-J212-1) ligand (3.1 g, 0.006 mol, 0.083 mol%) were charged into a 100 mL round bottomed flask under a N 2 atmosphere. Next, the solids were dissolved in 1 :3 CH 2 Cl 2 /Me0H (21 mL) and stirred using a magnetic stir bar for 1 hour.
- Step F Preparation of ⁇ , ⁇ -Saturated Ester 17. To a 100 L round bottom flask was added a methanolic solution of the ⁇ , ⁇ -saturated acid 16 (4.50 kg, 14.6 mol).
- the reaction was then aged for 19 hours at room temperature, upon which time HPLC analysis indicated complete consumption of ⁇ -carboxy ester.
- the white heterogeneous mixture was inverse quenched into toluene (4 L) and 2N K 2 CO 3 (38.6 kg) at 5 0 C with cooling to control the exotherm. Additional toluene (7 L) was used to rinse the glassware.
- the combined toluene solutions were warmed up to 35 0 C for 2 hours, then allowed to cool to room temperature and aged for 15 hours.
- Step I Synthesis of cvano diol III.
- Crude bromo diol 19 (5 kg, 15.5 mol) in toluene ( ⁇ 20 weight %) was solvent switched into DMF (2.5 volumes, approximately 12.5 L), so that toluene was reduced to ⁇ 5 LCAP by HPLC.
- the filtrate was transferred to an extractor containing 10% NH 4 OH (60 L).
- the organic solution was washed once with 5% NaCl (30 L) and water (30 L).
- the organic layer was solvent switched to toluene ( ⁇ 9 L / kg cyano diol), at constant volume, at -40 0 C until IPAc was reduced to ⁇ 1 mol % (assay by GC).
- Heptane (5 L / kg cyanodiol) was added at ⁇ 45 0 C and the resulting slurry was allowed to cool to room temperature, and then to 0 0 C.
- the resulting azetidine 20 was obtained with a purity of 92 wt % and 97.8 LCAP.
- HPLC retention time of Azetidine 20 11.23 on Zorbax RX C8, Analytical 5.0 micron, 4.6 mm x 250 mm, P.N.: 880967-901; temperature: 25°C; detection at 210 nm; column flow is 1.0 ml/min; solvent A is acetonitrile; solvent B is H 2 O buffered with 0.1 % H 3 PO 4 , gradient elution: 0 minutes: 10% solvent A, 90% solvent B; 10 minutes: 90% solvent A, 10% solvent B; 15 minutes: 98% solvent A, 2% solvent B.
- Step B Preparation of Compound I
- azetidine 20 (1.85 assay kg, 2.91 mol)
- IPAc 5.6 L
- a solution of HCl in IPA 7.4 L, 4.55 M
- the reaction was complete by HPLC after aging overnight at room temperature.
- the reaction mixture was quenched by slowly transferring the batch to a 100 L extractor containing saturated NaHCO 3 (30.0 L) with a mild exotherm ( ⁇ 5°C).
- Additional saturated NaHCO 3 (5.0 L for a total of 35.0 L) was slowly added to adjust the pH to ⁇ 7.
- Additional IPAc (13.0 L) was added, then the layers were separated and the organic layer was washed with GMP water (1 x 9.3 L).
- the organic layer was transferred to a 50 L vessel through a 1 micron in-line filter.
- the organic layer was concentrated to ⁇ 10 L and the solvent was switched to IPAc using 18 L of IPAc, and concentrated to ⁇ 18 L.
- the mixture was then treated with Nuchar Aquaguard PowderTM (0.45 kg), heated to 5O 0 C for 1 hour and then cooled to room temperature overnight.
- the resulting slurry was filtered through a pad of Solka FloeTM (1.85 kg), and the cake was washed with IPAc (4 x 7.0 L).
- the organic layer was transferred to a 50 L vessel through a 1 micron in-line filter, concentrated to ⁇ 6 L and solvent switched to toluene using 28 L of toluene.
- the resulting solution was concentrated to ⁇ 9.0 L, slowly heated to 45 0 C and slowly treated with heptane (1.9 L over 1 hour while maintaining the internal temperature between 40-45 0 C.
- the batch was seeded (7 g) and aged for 1 hour at 45 0 C.
- HPLC retention time of Compound I 9.54 on Zorbax RX C8, Analytical 5.0 micron, 4.6 mm x 250 mm, P.N.: 880967-901; temperature: 25°C; detection at 210 nm; column flow: 1.0 ml/min; solvent A: acetonitrile; solvent B: H 2 O buffered with 0.1 % H 3 PO 4 , gradient elution: 0 minutes: 10% solvent A, 90% solvent B; 10 minutes: 90% solvent A, 10% solvent B; 15 minutes: 98% solvent A, 2% solvent B.
- anhydrous free base toluene/heptane solvate polymorphic Form I is characterized by the complete group of angle 2 theta values listed in Table 1, all the values are not required for such identification.
- the free base toluene/heptane solvate polymorphic Form I, Type B of Compound I can be identified by the angle theta value of 4.7°.
- the free base toluene/heptane solvate polymorphic Form I, Type B of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 4.7°; b) 4.7° and 7.1°; c) 4.7°, 7.1° and 8.4°; d) 4.7°, 7.1°, 8.4° and 8.7°; e) 4.7°, 7.1°, 8.4°, 8.7° and 9.5°; f) 4.7°, 7.1°, 8.4°, 8.7°, 9.5° and 11.6°; g) 4.7°, 7.1°, 8.4°, 8.7°, 9.5°, 11.6° and 14.3°; h) 4.7°, 7.1°, 8.4°, 8.7°, 9.5°, 11.6°, 14.3° and 15.4°; i) 4.7°, 7.1°, 8.4°, 8.7°, 9.5°, 11.6°, 14.3
- the free base toluene/heptane solvate polymorphic Form I, Type B of Compound I can also be identified by one or more reflections at d-spacings of: 18.800, 12.450, 10.526, 10.163, 9.309, 7.628, 6.194, 5.754 and 5.126 A from an x-ray powder diffraction pattern obtained using
- thermogravimetric (TG) analysis curve for the free base toluene/heptane solvate polymorphic Form I Type B of Compound I ( Figure 14) was obtained under a nitrogen flow at a heating rate of 10°C/min in a Perkin Elmer TGA-7 instrument.
- the anhydrous free base Form I of compound I was obtained by drying the heptane/toluene solvate, which was prepared as described in Example 4. After drying the filter pot overnight, the white solid was transferred to the trays and dried in a vacuum at 25 0 C / 75
- Compound I ( Figure 1) was generated on a Philips Analytical X'Pert PRO X-ray Diffraction System with PW3040/60 console. A PW3373/00 ceramic Cu LFF X-ray tube K-Alpha radiation was used as the source. The X-ray powder diffraction spectrum was recorded at ambient temperature (CuKa radiation, 2° to 40° (2 ⁇ ), steps of 0.0167°, 5.08 sec per step). Cu K- ⁇ of wavelength 1.54187A was used for d-spacing calculation.
- anhydrous free base polymorphic Form I of Compound I is characterized by the complete group of angle 2 theta values listed in Table 2, all the values are not required for such identification.
- the anhydrous free base polymorphic Form I of Compound I can be identified by the angle theta value of 5.2°.
- the anhydrous free base polymorphic Form I of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 5.2°; b) 5.2° and 15.4°; c) 5.2°, 15.4° and 9.3°; d) 5.2°, 15.4°, 9.3° and 17.4°; e) 5.2°, 15.4°, 9.3°, 17.4° and 7.0°; f) 5.2°, 15.4°, 9.3°, 17.4°, 7.0° and 15.7°; g) 5.2°, 15.4°, 9.3°, 17.4°, 7.0°, 15.7° and 22.5°; h) 5.2°, 15.4°, 9.3°, 17.4°, 7.0°, 15.7°, 22.5° and 11.8°; i) 5.2°, 15.4°, 9.3°, 17.4°, 7.0°, 15.7°, 22.5°, 11.8° and 16.4°.
- the anhydrous free base polymorphic Form I of Compound I can also be identified by one or more reflections at d-spacings of: 16.994, 5.754, 9.509, 5.096, 12.627, 5.644, 3.951, 7.499 and 5.405 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- Isopropyl Acetate /Methylcvclohexane Solvate Free Base Polymorphic Form I Type A A portion of 300 mg of the free base of Compound I was dissolved in 0.25 mL of isopropylacetate. The mixture was heated to 70 0 C, and then 0.25 mL of methylcyclohexane was added. The mixture was cooled to 40 0 C, then seeded with the free base of Compound I. Solid began to slowly precipitate. The solution was cooled to room temperature and stirred for 30 minutes. Then 0.1 mL of isopropyl acetate and 0.5 mL methylcyclohexane was added. The resulting solid was filtered to give the methylcyclohexane/isopropyl acetate solvate of free base polymorphic Form I of Compound I.
- Type A of Compound I is characterized by the complete group of angle 2 theta values listed in Table 3, all the values are not required for such identification.
- the isopropyl acetate/methylcyclohexane solvate free base polymorphic Form I Type A of Compound I can be identified by the angle theta value of 4.6°.
- the isopropyl acetate/methylcyclohexane solvate free base polymorphic Form I, Type A of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 4.6°; b) 4.6° and 7.5°; c) 4.6°, 7.5° and 9.1°; d) 4.6°, 7.5°, 9.1° and 9.7°; e) 4.6°, 7.5°, 9.1°, 9.7° and 14.5°; f) 4.6°, 7.5°, 9.1°, 9.7°, 14.5° and 15.1°; g) 4.6°, 7.5°, 9.1°, 9.7°, 14.5°, 15.1° and 16.8°; h) 4.6°, 7.5°, 9.1°, 9.7°, 14.5°, 15.1°, 16.8° and 17.8°; i) 4.6°, 7.5°, 9.1°, 9.7°, 14.5°,
- the isopropyl acetate/methylcyclohexane solvate free base polymorphic Form I can also be identified by one or more reflections at d-spacings of: 19.209, 11.787, 9.718, 9.118, 6.109, 5.867, 5.277, 4.983 and 4.874 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- the thermogravimetric analysis (TGA) curve for the isopropyl acetate/methylcyclohexane solvate free base polymorphic Form I Type A of Compound I ( Figure 12) was obtained under a nitrogen flow at a heating rate of 10°C/min in a Perkin Elmer TGA-7 instrument.
- the anhydrous HCl salt polymorphic Form A of Compound I may also be formed by a) crystallizing the HCl salt of Compound I from ethanol and drying; and b) drying the HCl salt polymorphic Form H of Compound I of Example 14.
- anhydrous HCl salt polymorphic Form A of Compound I is characterized by the complete group of angle 2 theta values listed in Table 4, all the values are not required for such identification.
- the anhydrous HCl salt polymorphic Form A of Compound I can be identified by the angle theta value of 13.0°.
- the anhydrous HCl salt Form A of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 13.0°; b) 13.0° and 19.3°; c) 13.0°, 19.3° and 15.8°; d) 13.0°, 19.3°, 15.8° and 16.1°; e) 13.0°, 19.3°, 15.8°, 16.1° and 17.3°; f) 13.0°, 19.3°, 15.8°, 16.1°, 17.3° and 13.6°; g) 13.0°, 19.3°, 15.8°, 16.1°, 17.3°, 13.6° and 9.3°; h) 13.0°, 19.3°, 15.8°, 16.1°, 17.3°, 13.6°, 9.3° and 16.6°; i) 13.0°, 19.3°, 15.8°, 16.1°, 17.3°, 13.6°, 9.3°, 16.6° and 9.6°.
- the anhydrous HCl salt polymorphic Form A of Compound I can also be identified by one or more reflections at d-spacings of: 6.810, 4.599, 9.509, 5.609, 5.505, 5.126, 9.213, 6.511 and 5.340 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- the anhydrous HCl salt polymorphic Form A of Compound I is also characterized by differential scanning calorimetry (DSC).
- DSC curve of the anhydrous HCl salt polymorphic Form A of Compound I ( Figure 5) was obtained on a TA Instruments DSC-2910 differential scanning calorimeter at a heating rate of 10°C/min under N 2 flow. The sample was heated in a closed pan. A single endotherm with onset at 213.3C is observed.
- the anhydrous HCl salt polymorphic Form B of Compound I may also be formed by: a) slurrying the HCl salt polymorphic Form C of Compound I of Example 9 in isopropyl acetate and drying; and b) slurrying the HCl salt polymorphic Form D of Compound I of Example 10 in ethanol with HCl salt polymorphic Form B Compound I seed and drying; c) slurrying the HCl salt polymorphic Form A of Compound I of Example 7 in isopropyl acetate and drying; and d) heating the HCl salt polymorphic Form A of Compound I to a temperature greater than 215 0 C.
- the X-ray powder diffraction spectra for the anhydrous HCl salt polymorphic Form B of Compound I ( Figure 6) was generated on a Philips Analytical X'Pert PRO X-ray Diffraction System with PW3040/60 console. A PW3373/00 ceramic Cu LFF X-ray tube K-Alpha radiation was used as the source.
- the X-ray powder diffraction spectrum was recorded at ambient temperature (CuKa radiation, 2° to 40° (2 ⁇ ), steps of 0.0167°, 5.08 sec per step). Cu K- ⁇ of wavelength 1.54178A was used for d-spacing calculation.
- anhydrous HCl salt polymorphic Form B of Compound I is characterized by the complete group of angle 2 theta values listed in Table 5, all the values are not required for such identification.
- the anhydrous HCl salt polymorphic Form B of Compound I can be identified by the angle theta value of 17.9°.
- the anhydrous HCl salt polymorphic Form B of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 17.9°; b) 17.9° and 15.3°; c) 17.9°, 15.3° and 19.4°; d) 17.9°, 15.3°, 19.4 °and 24.9°; e) 17.9°, 15.3°, 19.4°, 24.9° and 6.1°; f) 17.9°, 15.3°, 19.4°, 24.9°, 6.1° and 8.1°; g) 17.9°, 15.3°, 19.4°, 24.9°, 6.1°, 8.1° and 21.5°; h) 17.9°, 15.3°, 19.4°, 24.9°, 6.1°, 8.1°, 21.5° and 14.2°; i) 17.9°, 15.3°, 19.4°, 24.9°, 6.1°, 8.1°, 21.5°, 14.2° and 10.9°.
- the anhydrous HCl salt polymorphic Form B of Compound I can also be identified by one or more reflections at d-spacings of: 4.955, 14.488, 5.791, 10.915, 8.117, 6.237, 4.575, 4.133 and 3.576 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- the anhydrous HCl salt polymorphic Form B of Compound I is also characterized by differential scanning calorimetry (DSC).
- DSC differential scanning calorimetry
- Figure 7 The DSC curve for the anhydrous HCl salt polymorphic Form B of Compound I ( Figure 7) was obtained on a TA Instruments DSC-2910 differential scanning calorimeter at a heating rate of 10°C/min under N 2 flow. The sample was heated in a closed pan. A single endotherm with onset at 249.9C is observed.
- HCl salt polymorphic Form C of Compound I is characterized by the complete group of angle 2 theta values listed in Table 6, all the values are not required for such identification.
- the HCl salt polymorphic Form C of Compound I can be identified by the angle theta value of 6.5°.
- the HCl salt Form C of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: j) 6.5°; k) 6.5° and 8.2°; 1) 6.5°, 8.2° and 9.1°; m) 6.5°, 8.2°, 9.1° and 10°; n) 6.5°, 8.2°, 9.1°, 10° and 13.8°; o) 6.5°, 8.2°, 9.1°, 10°, 13.8° and 14.2°; p) 6.5°, 8.2°, 9.1°, 10°, 13.8°, 14.2° and 14.9°; q) 6.5°, 8.2°, 9.1°, 10°, 13.8°, 14.2°, 14.9° and 15.8°; r) 6.5°, 8.2°, 9.1 °, 10°, 13.8°, 14.2°, 14.9°, 15.8° and 18.1°.
- the HCl salt polymorphic Form C of Compound I can also be identified by one or more reflections at d-spacings of: 13.598, 10.782, 9.718, 8.845, 6.417, 6.237, 5.945, 5.609 and 4.901 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- the HCl salt polymorphic Form D of Compound I was prepared by drying crystalline HCl salt polymorphic Form F (hydrate) in a vacuum oven at 35 0 C.
- HCl salt polymorphic Form D of Compound I is characterized by the complete group of angle 2 theta values listed in Table 7, all the values are not required for such identification.
- the HCl salt polymorphic Form D of Compound I can be identified by the angle theta value of 7.2°.
- the HCl salt Form D of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 7.2°; b) 7.2° and 12.3°; c) 7.2°, 12.3° and 6.2°; d) 7.2°, 12.3°, 6.2° and 11.1°; e) 7.2°, 12.3°, 6.2°, 11.1° and 13.8°; f) 7.2°, 12.3°, 6.2°, 11.1°, 13.8° and 18°; g) 7.2°, 12.3°, 6.2°, 11.1°, 13.8°, 18° and 8.8°; h) 7.2°, 12.3°, 6.2°, 11.1°, 13.8°, 18°, 8.8° and 22°; i) 7.2°, 12.3°, 6.2°, 11.1°, 13.8°, 18°, 8.8°, 22° and 24.2°.
- the HCl salt polymorphic Form D of Compound I can also be identified by one or more reflections at d-spacings of: 12.277, 7.196, 14.255, 7.971, 6.417, 4.928, 10.048, 4.040 and 3.678 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- thermogravimetric (TG) analysis curve for the HCl salt polymorphic Form D of Compound I ( Figure 18) was obtained under a nitrogen flow at a heating rate of 10°C/min in a Perkin Elmer TGA-7 instrument.
- the DSC curve for the HCl salt polymorphic Form D of Compound I ( Figure 17) was obtained on a TA Instruments DSC-2910 differential scanning calorimeter at a heating rate of 10°C/min under N 2 flow.
- the anhydrous HCl salt polymorphic Form E was prepared by drying HCl salt polymorphic Form D to a temperature > 185 0 C.
- the X-ray powder diffraction spectra for the anhydrous HCl salt polymorphic Form E of Compound I ( Figure 19) was generated on a Philips Analytical X'Pert PRO X-ray Diffraction System with PW3040/60 console. A PW3373/00 ceramic Cu LFF X-ray tube K-Alpha radiation was used as the source. The X-ray powder diffraction spectrum was recorded at ambient temperature (CuKa radiation, 2° to 40° (2 ⁇ ), steps of 0.0167°, 5.08 sec per step). Cu K- ⁇ of wavelength 1.54178A was used for d-spacing calculation.
- anhydrous HCl salt polymorphic Form E of Compound I is characterized by the complete group of angle 2 theta values listed in Table 8, all the values are not required for such identification.
- the anhydrous HCl salt polymorphic Form E of Compound I can be identified by the angle theta value of 7.4°.
- the anhydrous HCl salt Form E of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 7.4°; b) 7.4° and 8.2°; c) 7.4°, 8.2° and 8.9°; d) 7.4°, 8.2°, 8.9° and 10.2°; e) 7.4°, 8.2°, 8.9°, 10.2° and 12.8°; f) 7.4°, 8.2°, 8.9°, 10.2°, 12.8° and 14.3°; g) 7.4°, 8.2°, 8.9°, 10.2°, 12.8°, 14.3° and 14.8°; h) 7.4°, 8.2°, 8.9°, 10.2°, 12.8°, 14.3°, 14.8° and 15.4°; i) 7.4°, 8.2°, 8.9 °, 10.2°, 12.8°, 14.3°, 14.8°, 15.4° and 19.5°.
- the anhydrous HCl salt polymorphic Form E of Compound I can also be identified by one or more reflections at d-spacings of: 11.946, 10.782, 9.936, 8.672, 6.916, 6.194, 5.985, 5.754 and 4.552 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- Compound I ( Figure 20) was generated on a Philips Analytical X'Pert PRO X-ray Diffraction System with PW3040/60 console. A PW3373/00 ceramic Cu LFF X-ray tube K-Alpha radiation was used as the source. The X-ray powder diffraction spectrum was recorded at ambient temperature (CuKa radiation, 2° to 40° (2 ⁇ ), steps of 0.0167°, 5.08 sec per step). Cu K- ⁇ of wavelength 1.54178A was used for d-spacing calculation.
- HCl salt polymorphic Form F hydrate of Compound I is characterized by the complete group of angle 2 theta values listed in Table 9, all the values are not required for such identification.
- the HCl salt polymorphic Form F hydrate of Compound I can be identified by the angle theta value of 6.1°.
- the HCl salt Form F hydrate of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 6.1°; b) 6.1° and 6.4°; c) 6.1°, 6.4° and 7.3°; d) 6.1°, 6.4°, 7.3° and 11°; e) 6.1°, 6.4°, 7.3°, 11° and 11.5°; f) 6.1°, 6.4°, 7.3°, 11°, 11.5° and 12°; g) 6.1°, 6.4°, 7.3°, 11°, 11.5°, 12° and 16.2°; h) 6.1°, 6.4°, 7.3°, 11°, 11.5°, 12°, 16.2° and 18°; i) 6.1°, 6.4°, 7.3 °, 11°, 11.5°, 12°, 16.2°, 18° and 19°.
- the HCl salt polymorphic Form F hydrate of Compound I can also be identified by one or more reflections at d-spacings of: 14.488, 13.810, 12.109, 8.043, 7.694, 7.375, 5.471, 4.928 and 4.671 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- the anhydrous HCl salt polymorphic Form G of Compound I may also be formed by: a) slurrying the HCl salt polymorphic Form D of Compound I of Example 10 with HCl salt polymorphic Form G of Compound I; b) slurrying the HCl salt polymorphic Form B of Compound I of Example 8 in ethanol with HCl salt polymorphic Form G Compound I seed; c) slurrying the HCl salt polymorphic Form B of Compound I of Example 8 in 25% water/ethanol; and d) slurrying the HCl salt polymorphic Form A of Compound I of Example 7 in ethanol with HCl salt polymorphic Form G of Compound I.
- the X-ray powder diffraction spectra for the anhydrous HCl salt polymorphic Form G of Compound I ( Figure 8) was generated on a Philips Analytical X'Pert PRO X-ray Diffraction System with PW3040/60 console. A PW3373/00 ceramic Cu LFF X-ray tube K-Alpha radiation was used as the source. The X-ray powder diffraction spectrum was recorded at ambient temperature (CuKa radiation, 2° to 40° (2 ⁇ ), steps of 0.0167°, 5.08 sec per step). Cu K- ⁇ of wavelength 1.54178A was used for d-spacing calculation. Table 10. Powder X-ray diffraction: anhydrous HCl salt polymorphic Form G of Compound I
- anhydrous HCl salt polymorphic Form G of Compound I is characterized by the complete group of angle 2 theta values listed in Table 10, all the values are not required for such identification.
- the anhydrous HCl salt polymorphic Form G of Compound I can be identified by the angle theta value of 8.2°.
- the anhydrous HCl salt polymorphic Form G of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 8.2°; b) 8.2° and 20.8°; c) 8.2°, 20.8° and 16.8°; d) 8.2°, 20.8°, 16.8° and 21.6°; e) 8.2°, 20.8°, 16.8°, 21.6° and 12.6°; f) 8.2°, 20.8°, 16.8°, 21.6°, 12.6° and 10.4°; g) 8.2°, 20.8°, 16.8°, 21.6°, 12.6°, 10.4° and 12.1°; h) 8.2°, 20.8°, 16.8°, 21.6°, 12.6°, 10.4°, 12.1° and 14.1°; i) 8.2°, 20.8°, 16.8°, 21.6°, 12.6°, 10.4°, 12.1°, 14.1° and 13.3°.
- the anhydrous HCl salt polymorphic Form G of Compound I can also be identified by one or more reflections at d-spacings of: 10.782, 4.270, 8.506, 7.025, 7.314, 6.657, 6.281, 5.277 and 4.114 A from an x-ray powder diffraction pattern obtained using Cu radiation.
- the anhydrous HCl salt polymorphic Form G of Compound I is also characterized by differential scanning calorimetry (DSC).
- DSC differential scanning calorimetry
- Figure 9 The DSC curve for the anhydrous HCl salt polymorphic Form G of Compound I ( Figure 9) was obtained on a TA Instruments DSC-2910 differential scanning calorimeter at a heating rate of 10°C/min under N 2 flow. The sample was heated in a closed pan. A single endotherm with onset at 267.3C is observed.
- HCl salt polymorphic Form H of Compound I is characterized by the complete group of angle 2 theta values listed in Table 11, all the values are not required for such identification.
- the HCl salt polymorphic Form H of Compound I can be identified by the angle theta value of 9.1 °.
- the HCl salt Form H of Compound I can be identified by any one of the following angle theta values, or any one of the following groups of angle theta values: a) 9.1°; b) 9.1° and 9.4°; c) 9.1°, 9.4° and 12.8°; d) 9.1°, 9.4°, 12.8° and 13.4°; e) 9.1°, 9.4°, 12.8°, 13.4° and 14.3°; f) 9.1°, 9.4°, 12.8°, 13.4°, 14.3° and 14.9°; g) 9.1°, 9.4°, 12.8°, 13.4°, 14.3°, 14.9° and 15.3°; h) 9.1°, 9.4°, 12.8°, 13.4°, 14.3°, 14.9°, 15.3° and 16.9°; i) 9.1°, 9.4°, 12.8°, 13.4°, 14.3°, 14.9°, 15.3 °, 16.9° and 18.2°.
- the HCl salt polymorphic Form C of Compound I can also be identified by one or more reflections at d-spacings of: 9.718, 9.408, 6.916, 6.607, 6.194, 5.945, 5.791, 5.246 and 4.874 A from an x-ray powder diffraction pattern obtained using Cu radiation.
Abstract
Description













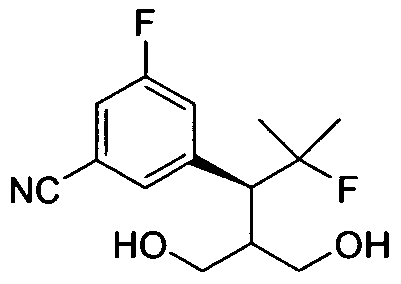
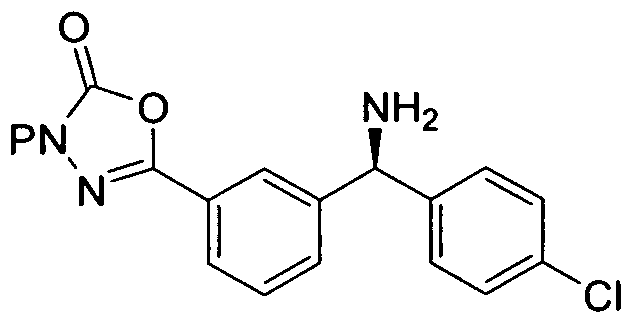









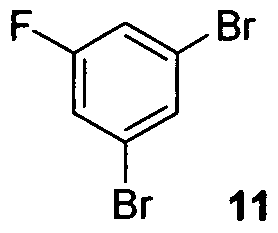















Claims












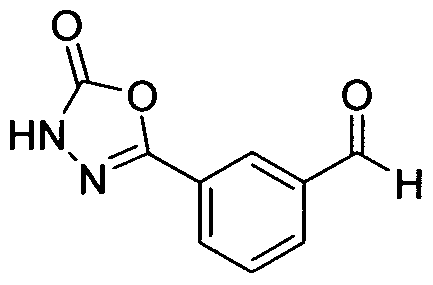

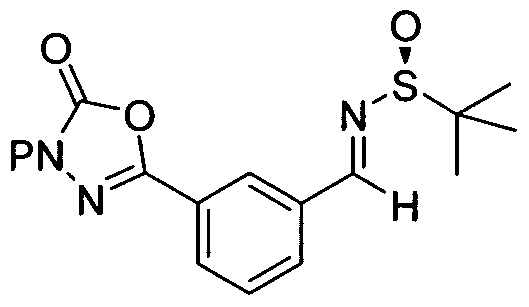



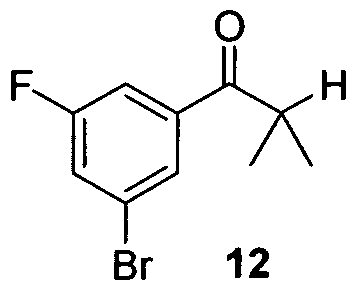







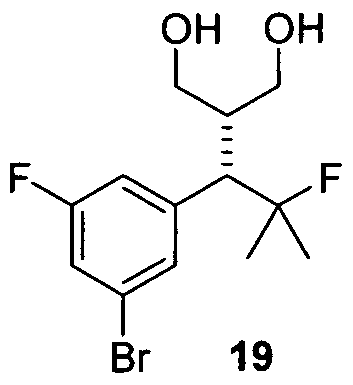

Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2008317482A AU2008317482A1 (en) | 2007-10-24 | 2008-10-20 | Synthesis and crystalline forms of CB-1 antagonist/inverse agonist |
| JP2010531013A JP2011502119A (en) | 2007-10-24 | 2008-10-20 | Synthesis and crystals of CB-1 antagonist / inverse agonist |
| CA2703465A CA2703465A1 (en) | 2007-10-24 | 2008-10-20 | Synthesis and crystalline forms of cb-1 antagonist/inverse agonist |
| US12/681,797 US20100292282A1 (en) | 2007-10-24 | 2008-10-20 | Synthesis and crystalline forms of cb-1 antagonist/inverse agonist |
| EP08841788A EP2212313A2 (en) | 2007-10-24 | 2008-10-20 | Synthesis and crystalline forms of cb-1 antagonist/inverse agonist |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15107P | 2007-10-24 | 2007-10-24 | |
| US61/000,151 | 2007-10-24 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009054923A2 true WO2009054923A2 (en) | 2009-04-30 |
| WO2009054923A3 WO2009054923A3 (en) | 2009-06-18 |
Family
ID=40229804
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/011927 WO2009054923A2 (en) | 2007-10-24 | 2008-10-20 | Synthesis and crystalline forms of cb-1 antagonist/inverse agonist |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20100292282A1 (en) |
| EP (1) | EP2212313A2 (en) |
| JP (1) | JP2011502119A (en) |
| AU (1) | AU2008317482A1 (en) |
| CA (1) | CA2703465A1 (en) |
| WO (1) | WO2009054923A2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111087305A (en) * | 2019-12-13 | 2020-05-01 | 南京工业大学 | β -fluoroalkyl cinnamate compound and preparation method thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104844643B (en) * | 2015-06-12 | 2017-03-22 | 沧州普瑞东方科技有限公司 | Method for preparing carboxyl phenylboronic acid |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2006100298A (en) * | 2003-06-11 | 2006-05-10 | Мерк энд Ко., Инк. (US) | SUBSTITUTED 3-ALKYL-AND 3-ALKENYLASETIDINE DERIVATIVES |
| AR058199A1 (en) * | 2005-11-28 | 2008-01-23 | Merck & Co Inc | DERIVATIVES OF 3- RENTED ALQUILAZETIDINA WITH HETEROCICLOS |
| US7906652B2 (en) * | 2005-11-28 | 2011-03-15 | Merck Sharp & Dohme Corp. | Heterocycle-substituted 3-alkyl azetidine derivatives |
-
2008
- 2008-10-20 AU AU2008317482A patent/AU2008317482A1/en not_active Abandoned
- 2008-10-20 WO PCT/US2008/011927 patent/WO2009054923A2/en active Application Filing
- 2008-10-20 US US12/681,797 patent/US20100292282A1/en not_active Abandoned
- 2008-10-20 JP JP2010531013A patent/JP2011502119A/en not_active Withdrawn
- 2008-10-20 CA CA2703465A patent/CA2703465A1/en not_active Abandoned
- 2008-10-20 EP EP08841788A patent/EP2212313A2/en not_active Withdrawn
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111087305A (en) * | 2019-12-13 | 2020-05-01 | 南京工业大学 | β -fluoroalkyl cinnamate compound and preparation method thereof |
| CN111087305B (en) * | 2019-12-13 | 2021-08-27 | 南京工业大学 | Beta-fluoroalkyl cinnamate compound and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2008317482A1 (en) | 2009-04-30 |
| US20100292282A1 (en) | 2010-11-18 |
| WO2009054923A3 (en) | 2009-06-18 |
| EP2212313A2 (en) | 2010-08-04 |
| CA2703465A1 (en) | 2009-04-30 |
| JP2011502119A (en) | 2011-01-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| USRE49860E1 (en) | Process for preparing (4S)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1,6-naphthyridine-3-carboxamide and purification thereof for use as pharmaceutical active ingredient | |
| US11352327B2 (en) | Process for the preparation of a PDE4 inhibitor | |
| TW201932465A (en) | Processes for preparing intermediates of substituted 5-fluoro-1H-pyrazolopyridines | |
| AU2016312904A1 (en) | Method for the preparation of (4S)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1-6-naphthyridine-3-carboxamide and the purification thereof for use as an active pharmaceutical ingredient | |
| KR20180030582A (en) | CGRP receptor antagonist | |
| WO2009054923A2 (en) | Synthesis and crystalline forms of cb-1 antagonist/inverse agonist | |
| US20220411382A1 (en) | 3-((r)-2-(amino-2-phenylethyl)-1-(2-fluoro-6 trifluoromethyl benzyl)-5-iodo-6-methyl-1h-pyrimidine-2,4-dione or a salt thereof, process for its preparation, and its use in the synthesis of elagolix | |
| EP1904465B1 (en) | Method of synthesis of imidazole-amino acid derivatives and related compounds | |
| JPH11193271A (en) | Arylacetamide derivative or its salt and medicine containing the same | |
| WO2023187833A1 (en) | A novel salt of bempedoic acid | |
| KR100377578B1 (en) | Process for the preparation of ondansetron and pharmaceutically acceptable salts thereof | |
| TW202408997A (en) | Processes and intermediates for the preparation of 2-(2,6-dichlorophenyl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(3-hydroxy-3-methylbutyl)-1-methyl-3,4-dihydroisoquinolin-2(1h)-yl]ethanone | |
| JP2000505427A (en) | Method for producing substituted pyridine derivative | |
| CZ20001012A3 (en) | Imidazopyridine derivatives and process of their preparation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08841788 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008317482 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2703465 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010531013 Country of ref document: JP Ref document number: 2008841788 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2008317482 Country of ref document: AU Date of ref document: 20081020 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12681797 Country of ref document: US |