WO2009037463A1 - Thienopyrimidine compounds - Google Patents
Thienopyrimidine compounds Download PDFInfo
- Publication number
- WO2009037463A1 WO2009037463A1 PCT/GB2008/003173 GB2008003173W WO2009037463A1 WO 2009037463 A1 WO2009037463 A1 WO 2009037463A1 GB 2008003173 W GB2008003173 W GB 2008003173W WO 2009037463 A1 WO2009037463 A1 WO 2009037463A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- compound
- alkyl
- methyl
- ring
- Prior art date
Links
- RBNBDIMXFJYDLQ-UHFFFAOYSA-N thieno[3,2-d]pyrimidine Chemical class C1=NC=C2SC=CC2=N1 RBNBDIMXFJYDLQ-UHFFFAOYSA-N 0.000 title description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 90
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 46
- -1 benzimidazol-2-yl-methyl Chemical group 0.000 claims abstract description 42
- 239000001257 hydrogen Substances 0.000 claims abstract description 18
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 17
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 13
- 125000002911 monocyclic heterocycle group Chemical group 0.000 claims abstract description 8
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 8
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 7
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims abstract description 7
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 7
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims abstract description 6
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims abstract description 5
- 125000003107 substituted aryl group Chemical group 0.000 claims abstract description 5
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims abstract description 4
- 229920006395 saturated elastomer Polymers 0.000 claims abstract description 4
- 238000000034 method Methods 0.000 claims description 27
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 18
- 150000003839 salts Chemical class 0.000 claims description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 12
- 238000011282 treatment Methods 0.000 claims description 12
- 208000035475 disorder Diseases 0.000 claims description 11
- 125000000217 alkyl group Chemical group 0.000 claims description 10
- 239000003814 drug Substances 0.000 claims description 10
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 9
- 229960005305 adenosine Drugs 0.000 claims description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 9
- 125000001072 heteroaryl group Chemical group 0.000 claims description 8
- 230000001404 mediated effect Effects 0.000 claims description 8
- 206010028980 Neoplasm Diseases 0.000 claims description 7
- 125000002947 alkylene group Chemical group 0.000 claims description 7
- 208000006673 asthma Diseases 0.000 claims description 7
- 201000011510 cancer Diseases 0.000 claims description 7
- 206010012601 diabetes mellitus Diseases 0.000 claims description 7
- 230000000694 effects Effects 0.000 claims description 7
- 230000020341 sensory perception of pain Effects 0.000 claims description 7
- 239000012453 solvate Substances 0.000 claims description 7
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 6
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 6
- 208000027866 inflammatory disease Diseases 0.000 claims description 6
- 125000001153 fluoro group Chemical group F* 0.000 claims description 5
- 125000002950 monocyclic group Chemical group 0.000 claims description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 5
- 125000001544 thienyl group Chemical group 0.000 claims description 5
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 3
- 230000008485 antagonism Effects 0.000 claims description 3
- 239000008194 pharmaceutical composition Substances 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 claims description 2
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 claims 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims 3
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims 1
- 125000003282 alkyl amino group Chemical group 0.000 claims 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 claims 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 abstract description 4
- 101150078577 Adora2b gene Proteins 0.000 abstract 1
- 239000002464 receptor antagonist Substances 0.000 abstract 1
- 229940044551 receptor antagonist Drugs 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 32
- 239000000243 solution Substances 0.000 description 27
- 102000005962 receptors Human genes 0.000 description 20
- 108020003175 receptors Proteins 0.000 description 20
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- 239000000203 mixture Substances 0.000 description 13
- 238000005160 1H NMR spectroscopy Methods 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 235000019439 ethyl acetate Nutrition 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 229940093499 ethyl acetate Drugs 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- 125000001424 substituent group Chemical group 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000000651 prodrug Substances 0.000 description 9
- 229940002612 prodrug Drugs 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 7
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 235000019253 formic acid Nutrition 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 239000003643 water by type Substances 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 5
- 239000005557 antagonist Substances 0.000 description 5
- 229940113088 dimethylacetamide Drugs 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 229960001866 silicon dioxide Drugs 0.000 description 5
- 102000009346 Adenosine receptors Human genes 0.000 description 4
- 108050000203 Adenosine receptors Proteins 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 125000001309 chloro group Chemical group Cl* 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 235000011121 sodium hydroxide Nutrition 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 229910014455 Ca-Cb Inorganic materials 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- 206010020772 Hypertension Diseases 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- 125000004566 azetidin-1-yl group Chemical group N1(CCC1)* 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 235000019441 ethanol Nutrition 0.000 description 3
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 3
- 125000000842 isoxazolyl group Chemical group 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 3
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 230000003185 calcium uptake Effects 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 239000008177 pharmaceutical agent Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 230000036515 potency Effects 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- HDOUGSFASVGDCS-UHFFFAOYSA-N pyridin-3-ylmethanamine Chemical compound NCC1=CC=CN=C1 HDOUGSFASVGDCS-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical class [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 2
- 239000001117 sulphuric acid Substances 0.000 description 2
- 235000011149 sulphuric acid Nutrition 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 238000000825 ultraviolet detection Methods 0.000 description 2
- NXQKWQMXPKUSTQ-UHFFFAOYSA-N (2,6-dichlorothieno[3,2-d]pyrimidin-4-yl)-thiophen-2-ylmethanone Chemical compound C=12SC(Cl)=CC2=NC(Cl)=NC=1C(=O)C1=CC=CS1 NXQKWQMXPKUSTQ-UHFFFAOYSA-N 0.000 description 1
- DIVNUTGTTIRPQA-UHFFFAOYSA-N (3,4-dimethoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1OC DIVNUTGTTIRPQA-UHFFFAOYSA-N 0.000 description 1
- DAXJNUBSBFUTRP-RTQNCGMRSA-N (8r,9s,10r,13s,14s)-6-(hydroxymethyl)-10,13-dimethyl-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthrene-3,17-dione Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(CO)C2=C1 DAXJNUBSBFUTRP-RTQNCGMRSA-N 0.000 description 1
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- NHCGQHAZAQDUDS-UHFFFAOYSA-N 1,2-dimethylimidazole;hydroiodide Chemical compound [I-].C[NH+]1C=CN=C1C NHCGQHAZAQDUDS-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- QAFVXBQPQCSSLI-UHFFFAOYSA-N 1h-thieno[3,2-d]pyrimidine-2,4-dione Chemical compound O=C1NC(=O)NC2=C1SC=C2 QAFVXBQPQCSSLI-UHFFFAOYSA-N 0.000 description 1
- YTBCURLKGMTHAH-UHFFFAOYSA-N 2,4,6-trichlorothieno[3,2-d]pyrimidine Chemical compound ClC1=NC(Cl)=C2SC(Cl)=CC2=N1 YTBCURLKGMTHAH-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- AXLGXWLKXBJBJY-UHFFFAOYSA-N 6-nitro-1h-thieno[3,2-d]pyrimidine-2,4-dione Chemical compound N1C(=O)NC(=O)C2=C1C=C([N+](=O)[O-])S2 AXLGXWLKXBJBJY-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- 239000004150 EU approved colour Substances 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical class [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 229910006074 SO2NH2 Inorganic materials 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 239000004141 Sodium laurylsulphate Substances 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102100026383 Vasopressin-neurophysin 2-copeptin Human genes 0.000 description 1
- BXEPZMXZIXWQED-UHFFFAOYSA-N [2,6-bis(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(NCC=2C=NC=CC=2)=NC=2C=C(NCC=3C=NC=CC=3)SC=2C=1C(=O)C1=CC=CS1 BXEPZMXZIXWQED-UHFFFAOYSA-N 0.000 description 1
- VGLSCMPCDPYQTM-UHFFFAOYSA-N [6-(dimethylamino)-2-(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(C(=O)C=2SC=CC=2)=C2SC(N(C)C)=CC2=NC=1NCC1=CC=CN=C1 VGLSCMPCDPYQTM-UHFFFAOYSA-N 0.000 description 1
- GQSJGNQPKLGYJZ-UHFFFAOYSA-N [6-(ethylamino)-2-(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(C(=O)C=2SC=CC=2)=C2SC(NCC)=CC2=NC=1NCC1=CC=CN=C1 GQSJGNQPKLGYJZ-UHFFFAOYSA-N 0.000 description 1
- RMJSDWIAVCAAMH-UHFFFAOYSA-N [6-(methylamino)-2-(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(C(=O)C=2SC=CC=2)=C2SC(NC)=CC2=NC=1NCC1=CC=CN=C1 RMJSDWIAVCAAMH-UHFFFAOYSA-N 0.000 description 1
- TWNISLQLWKPWDI-UHFFFAOYSA-N [6-amino-2-(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(C(=O)C=2SC=CC=2)=C2SC(N)=CC2=NC=1NCC1=CC=CN=C1 TWNISLQLWKPWDI-UHFFFAOYSA-N 0.000 description 1
- HWFLOIWTEDBAFA-UHFFFAOYSA-N [6-chloro-2-(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(C(=O)C=2SC=CC=2)=C2SC(Cl)=CC2=NC=1NCC1=CC=CN=C1 HWFLOIWTEDBAFA-UHFFFAOYSA-N 0.000 description 1
- STQYXQGAZSYWDR-UHFFFAOYSA-N [6-ethenyl-2-(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(C(=O)C=2SC=CC=2)=C2SC(C=C)=CC2=NC=1NCC1=CC=CN=C1 STQYXQGAZSYWDR-UHFFFAOYSA-N 0.000 description 1
- FARDIRBBHGVIGJ-UHFFFAOYSA-N [6-ethyl-2-(pyridin-3-ylmethylamino)thieno[3,2-d]pyrimidin-4-yl]-thiophen-2-ylmethanone Chemical compound N=1C(C(=O)C=2SC=CC=2)=C2SC(CC)=CC2=NC=1NCC1=CC=CN=C1 FARDIRBBHGVIGJ-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 102000030621 adenylate cyclase Human genes 0.000 description 1
- 108060000200 adenylate cyclase Proteins 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 230000003444 anaesthetic effect Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 125000005129 aryl carbonyl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000003684 cardiac depression Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 201000010064 diabetes insipidus Diseases 0.000 description 1
- IBDMRHDXAQZJAP-UHFFFAOYSA-N dichlorophosphorylbenzene Chemical compound ClP(Cl)(=O)C1=CC=CC=C1 IBDMRHDXAQZJAP-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- CNUDBTRUORMMPA-UHFFFAOYSA-N formylthiophene Chemical compound O=CC1=CC=CS1 CNUDBTRUORMMPA-UHFFFAOYSA-N 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- JUQAECQBUNODQP-UHFFFAOYSA-N furo[3,2-d]pyrimidine Chemical class C1=NC=C2OC=CC2=N1 JUQAECQBUNODQP-UHFFFAOYSA-N 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 239000003193 general anesthetic agent Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003871 intestinal function Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 238000013332 literature search Methods 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical class [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 235000012254 magnesium hydroxide Nutrition 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- MRWXACSTFXYYMV-FDDDBJFASA-N nebularine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC=C2N=C1 MRWXACSTFXYYMV-FDDDBJFASA-N 0.000 description 1
- 230000003957 neurotransmitter release Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 229940006093 opthalmologic coloring agent diagnostic Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 230000001991 pathophysiological effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000011118 potassium hydroxide Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000002212 purine nucleoside Substances 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000002278 tabletting lubricant Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ZGNPLWZYVAFUNZ-UHFFFAOYSA-N tert-butylphosphane Chemical compound CC(C)(C)P ZGNPLWZYVAFUNZ-UHFFFAOYSA-N 0.000 description 1
- 125000001302 tertiary amino group Chemical group 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical class CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 230000006442 vascular tone Effects 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D495/14—Ortho-condensed systems
Definitions
- This invention relates to novel thienopyrimidine derivatives having A 2B receptor antagonistic activity, to the use of such compounds in medicine, in relation to the treatment of disorders which are responsive to antagonism of the A 2B receptor such as nociception, asthma, COPD, inflammatory disorders, diabetes, diabetic retinopathy and cancer, and to pharmaceutical compositions containing such compounds.
- Adenosine is a naturally occurring purine nucleoside, the effects of which include stimulation of nociception afferents, bronchconstriction, immunosupression, vasodilation, inhibition of platelet aggregation, cardiac depression and inhibition of neurotransmitter release.
- Adenosine produces a wide range of pharmacological effects mediated by activation of specific cell surface receptors, which are members of the G- protein coupled receptor family.
- Four subtypes of adenosine receptors have been identified, designated A 1 , A 2A , A 2B and A 3 .
- the A 26 adenosine receptor subtype is coupled to the G 3 G-protein and stimulates adenylyl cyclase activity.
- PCT/G B00/02517 is concerned with a class of thieno- and furopyrimidine derivatives which are antagonists of the adenosine A 2 A receptor.
- This invention relates to a subset of compounds within the PCT/GB00/02517 class, but which are not specifically disclosed therein.
- the present invention relates to a class of substituted thienopyrimidine compounds useful as selective A 2B antagonists, for example, for the treatment of nociception, asthma, COPD, inflammatory disorders, diabetes, diabetic retinopathy and cancer.
- a core thieno-pyrimidine bicyclic ring, with substitution on the thieno portion by an amino group, and substitution on the pyrimidine portion by a (hetero)aryl-carbonyl group in addition to an amino group, are principle characterising features of the compounds with which the invention is concerned.
- Ri is optionally substituted aryl or an optionally substituted 5- or 6-membered heteroaryl ring
- R 2 and R 3 are independently selected from hydrogen, C 1 -C 6 alkyl, C 3 -C 8 cycloalkyl, C 3 -C 8 cycloalkyl-(CrC 6 )-alkyl, aryl-(CrC 6 )-alkyl optionally substituted in the ring part thereof, a 5- or 6-membered monocyclic heterocyclic group optionally linked via a Ci-C 6 alkylene chain and optionally substituted in the ring part thereof, benzimidazol-2-yl-methyl, pyrid-3-yl- carbonyl, or (1-methyl-piperidin-4-yl)-carbonyl-methyl;
- R 2 and R 3 taken together with the nitrogen atom to which they are attached form an optionally substituted 5- or 6-membered ring;
- R 4 is CrC 3 alkyl, C 2 -C 3 alkenyl, -N(-R 5 )-R 6 , or optionally substituted heteroarylmethylamino;
- R 5 and R 6 are independently selected from hydrogen or C r C 3 alkyl
- R 5 and R 6 taken together with the nitrogen atom to which they are attached form an optionally substituted 4- to 6-membered saturated ring.
- the active compounds of formula (I) are selective antagonists of the A 2B receptor and are useful for the treatment, prevention and suppression of disorders mediated by the A 28 receptor.
- disorders include nociception; asthma; chronic obstructive pulmonary disease (COPD); inflammatory diseases such as rheumatoid arthritis, multiple sclerosis, lupus, psoriasis and inflammatory bowel disease; diabetes mellitus or diabetes insipidus; diabetic retinopathy and cancer.
- a compound of formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, or prodrug thereof in the manufacture of a medicament for the treatment of disorders mediated by the adenosine A 2B receptor.
- a method of treatment of a disorder mediated by the A 2B receptor comprising administration to a subject in need of such treatment an effective dose of the compound of formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, or prodrug thereof.
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, or prodrug thereof, and a pharmaceutically acceptable carrier.
- (C a -C b )alkyl wherein a and b are integers refers to a straight or branched chain alkyl radical having from a to b carbon atoms.
- a 1 and b is 6, for example, the term includes methyl, ethyl, n- propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, n-pentyl and n-hexyl.
- divalent (C a 7C b )alkylene radical wherein a and b are integers refers to a saturated hydrocarbon chain having from a to b carbon atoms and two unsatisfied valences.
- (C a -C b )alkenyl wherein a and b are integers refers to a straight or branched chain alkenyl moiety having from a to b carbon atoms having at least one double bond of either E or Z stereochemistry where applicable.
- the term includes, for example, vinyl, allyl, 1- and 2-butenyl and 2-methyl-2-propenyl.
- divalent (C a -C b )alkenylene radical refers to a hydrocarbon chain having from a to b carbon atoms, at least one double bond, and two unsatisfied valences.
- cycloalkyl refers to a saturated carbocyclic radical having from 3-8 carbon atoms and includes, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- cycloalkenyl refers to a carbocyclic radical having from 3-8 carbon atoms containing at least one double bond, and includes, for example, cyclopentenyl, cyclohexenyl, cycloheptenyl and cyclooctenyl.
- Carbocyclic refers to a mono- or bi-cyclic radical whose ring atoms are all carbon, and includes monocyclic aryl, cycloalkyl, and cycloalkenyl radicals, provided that no single ring present has more than 8 ring members.
- a "carbocyclic” group includes a mono-bridged or multiply- bridged cyclic alkyl group.
- aryl refers to a mono-, bi- or tri-cyclic carbocyclic aromatic radical. Illustrative of such radicals are phenyl, biphenyl and napthyl.
- heteroaryl refers to a mono-, bi- or tri-cyclic aromatic radical containing one or more heteroatoms selected from S, N and
- radicals are thienyl, benzthienyl, furyl, benzfuryl, pyrrolyl, imidazolyl, benzimidazolyl, thiazolyl, benzthiazolyl, isothiazolyl, benzisothiazolyl, pyrazolyl, oxazolyl, benzoxazolyl, isoxazolyl, benzisoxazolyl, isothiazolyl, triazolyl, benztriazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, indolyl and indazolyl.
- heterocyclyl or “heterocyclic” includes “heteroaryl” as defined above, and in particular refers to a mono-, bi- or tricyclic non-aromatic radical containing one or more heteroatoms selected from S, N and O, to groups consisting of a monocyclic non-aromatic radical containing one or more such heteroatoms which is covalently linked to another such radical or to a monocyclic carbocyclic radical, and to a mono-, bi- or tri-cyclic non-aromatic radical containing one or more heteroatoms selected from S, N and O which is mono-bridged or multiply-bridged.
- radicals are pyrrolyl, furanyl, thienyl, piperidinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, pyrazolyl, pyridinyl, pyrrolidinyl, pyrimidinyl, morpholinyl, piperazinyl, indolyl, morpholinyl, benzfuranyl, pyranyl, isoxazolyl, benzimidazolyl, methylenedioxyphenyl, ethylenedioxyphenyl, maleimido and succinimido groups.
- substituted as applied to any moiety herein means substituted with at least one substituent, for example selected from (Ci-C ⁇ jalkyl, (d-C ⁇ alkoxy, hydroxy, hydroxy(CrC 6 )alkyl, mercapto, mercapto(Ci-C 6 )alkyl, (C 1 - C 6 )alkylthio, halo (including fluoro and chloro), trifluoromethyl, trifluoromethoxy, nitro, nitrile (-CN), oxo, phenyl, -COOH, -COOR A , -COR A , -SO 2 R A , -CONH 2 , -SO 2 NH 2 , -CONHR A , -SO 2 NHR A , -CONR A R B , -SO 2 NR A R B , -NH 2 , -NHR A , -
- R A and R B are independently a (CrC 6 )alkyl group, or R A and R B when attached to the same nitrogen may form a cyclic amino ring such as a morpholinyl, piperidinyl or piperazinyl ring.
- An "optional substituent” or “susbtituent” may be one of the foregoing substituent groups.
- salt includes base addition, acid addition and quaternary salts.
- Compounds of the invention which are acidic can form salts, including pharmaceutically or veterinarily acceptable salts, with bases such as alkali metal hydroxides, e.g. sodium and potassium hydroxides; alkaline earth metal hydroxides e.g. calcium, barium and magnesium hydroxides; with organic bases e.g. N-ethyl piperidine, dibenzylamine and the like.
- bases such as alkali metal hydroxides, e.g. sodium and potassium hydroxides; alkaline earth metal hydroxides e.g. calcium, barium and magnesium hydroxides; with organic bases e.g. N-ethyl piperidine, dibenzylamine and the like.
- Those compounds (I) which are basic can form salts, including pharmaceutically or veterinarily acceptable salts with inorganic acids, e.g.
- hydrohalic acids such as hydrochloric or hydrobromic acids, sulphuric acid, nitric acid or phosphoric acid and the like
- organic acids e.g. with acetic, tartaric, succinic, fumaric, maleic, malic, salicylic, citric, methanesulphonic and p- toluene sulphonic acids and the like.
- 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and a stoichiometric amount of one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- solvent molecules for example, ethanol.
- 'hydrate' is employed when said solvent is water.
- Compounds with which the invention is concerned which may exist in one or more stereoisomeric form, because of the presence of asymmetric atoms or rotational restrictions, can exist as a number of stereoisomers with R or S stereochemistry at each chiral centre or as atropisomeres with R or S stereochemistry at each chiral axis.
- the invention includes all such enantiomers and diastereoisomers and mixtures thereof.
- So-called 'pro-drugs' of the compounds of formula (I) are also within the scope of the invention.
- certain derivatives of compounds of formula (I) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) having the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as 'prodrugs'.
- Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association).
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
- metabolites of compounds of formula (I) that is, compounds formed in vivo upon administration of the drug.
- Some examples of metabolites include (i) where the compound of formula (I) contains a methyl group, an hydroxymethyl derivative thereof (-CH 3 -> -CH 2 OH):
- R 1 is optionally substituted aryl or an optionally substituted 5- or 6-membered heteroaryl ring.
- Ri is optionally substituted phenyl.
- the phenyl ring preferably has one substituent, selected from methyl, methoxy, fluoro, chloro, or cyano.
- R 1 is an optionally substituted 5- or 6-membered heteroaryl ring.
- the heteroaryl ring may be, for example, furan, thiophene, pyrrole, oxazole, thiazole, imidazole, or pyridine.
- Preferred substituents include methyl, ethyl, chloro, or bromo.
- Ri is optionally substituted thienyl, particularly thien-2-yl.
- R 2 and R 3 are independently selected from hydrogen, C 1 -C 6 alkyl, C 3 -C 8 cycloalkyl, C 3 -C 8 cycloalkyl-(Ci-C 6 )-alkyl, aryl-(C- ⁇ -C 6 )-alkyl optionally substituted in the ring part thereof, a 5- or 6-membered monocyclic heterocyclic group optionally linked via a C 1 -C 6 alkylene chain and optionally substituted in the ring part thereof, benzimidazol-2-yl-methyl, pyrid-3-yl-carbonyl, or (1-methyl-piperidin-4-yl)- carbonyl-methyl.
- R 2 is hydrogen and R 3 is a 5- or 6-membered monocyclic heterocyclic group optionally linked via a CrC 6 alkylene chain and optionally substituted in the ring part thereof.
- the heterocyclic ring may be, for example, pyran, piperidine, morpholine, imidazole, pyridine, pyrimidine, pyrazine, or tetrazole.
- methylene or ethylene is preferred for the C 1 -C 6 alkylene chain.
- R 2 is hydrogen and R 3 is aryl-(C- ⁇ -C 6 )-alkyl optionally substituted in the ring part thereof.
- Phenyl is preferred for aryl, and when substituted, the phenyl ring preferably has one substituent, selected from methyl, ethyl, methoxy, or chloro. Methyl or ethyl is preferred for C 1 -C 6 alkyl.
- R 2 and R 3 taken together with the nitrogen atom to which they are attached form an optionally substituted 5- or 6-membered ring.
- R 2 is hydrogen and R 3 is pyrid-3-ylmethyl.
- R 4 is C- 1 -C 3 alkyl, C 2 -C 3 alkenyl, -N(-R 5 )-R 6 , or optionally substituted heteroarylmethylamino.
- R 4 is C 1 -C 3 alkyl, preferably ethyl.
- R 4 is C 2 -C 3 alkenyl, preferably ethenyl.
- R 4 is optionally substituted heteroarylmethylamino.
- heteroaryl represents a 5- or 6-membered monocyclic heteroaryl ring, with pyridyl preferred, particularly pyrid-3-yl.
- R 4 is amino, mono-(CrC 3 -alkyl)amino, or di-(C- ⁇ -C 3 -alkyl)amino.
- R 4 is -N(-R 5 )-R 6 wherein R 5 and R 6 taken together with the nitrogen atom to which they are attached form an optionally substituted 4- to 6-membered saturated ring.
- -N(-Rs)-R 6 includes azetidin-1-yl, pyrrolidin-1-yl and piperidin-1-yl, with azetidin-1-yl and pyrrolidin-1-yl preferred, particularly azetidin-1-yl.
- R 4 is amino, methylamino, ethylamino, dimethylamino, ethyl, ethenyl, or pyrid-3-ylmethylamino.
- R 4 is amino or methylamino.
- Specific compounds with which the invention is concerned include those of the Examples.
- the present invention may be employed in respect of a human or animal subject, more preferably a mammal, more preferably a human subject.
- treatment includes prophylactic treatment.
- the compound of formula (I) may be used in combination with one or more additional drugs useful in the treatment of the disorders mentioned above, the components being in the same formulation or in separate formulations for administration simultaneously or sequentially.
- a suitable dose for orally administrable formulations will usually be in the range of 0.1 to 3000 mg, once, twice or three times per day, or the equivalent daily amount administered by infusion or other routes.
- optimum dose levels and frequency of dosing will be determined by clinical trials as is conventional in the art.
- the compounds with which the invention is concerned may be prepared for administration by any route consistent with their pharmacokinetic properties.
- the orally administrable compositions may be in the form of tablets, capsules, powders, granules, lozenges, liquid or gel preparations, such as oral, topical, or sterile parenteral solutions or suspensions.
- Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinyl-pyrrolidone; fillers for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricant, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants for example potato starch, or acceptable wetting agents such as sodium lauryl sulphate.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, glucose syrup, gelatin hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and if desired conventional flavouring or colouring agents.
- suspending agents for example sorbitol, syrup, methyl cellulose, glucose syrup, gelatin hydrogenated edible fats
- emulsifying agents for example lecithin, sorbitan monooleate, or acacia
- non-aqueous vehicles which may include edible oils
- almond oil fractionated coconut oil
- oily esters such as glycerine, propylene
- the drug may be made up into a cream, lotion or ointment.
- Cream or ointment formulations which may be used for the drug are conventional formulations well known in the art, for example as described in standard textbooks of pharmaceutics such as the British Pharmacopoeia.
- the active ingredient may also be administered parenterally in a sterile medium.
- the drug can either be suspended or dissolved in the vehicle.
- adjuvants such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle.
- Scheme 1 represents a method known in the art of organic chemistry in general, by which the compounds of the present invention may be prepared:
- Example 2 A suspension of Example 2 (7Og, 330mmol) in phenylphosphonic dichloride (320ml, 2310mmol) was heated, with stirring, to 180 0 C for 4h. The mixture was cooled to 100 0 C and transferred slowly onto vigorously stirred ice/water (2500ml). After 2h stirring at room temperature, a tan solid was filtered and dried in vacuo at 40 0 C. The solid was dissolved in the minimum volume of tetrahydrofuran and passed over a short pad of silica using ethyl acetate as an eluent.
- Example 3 A stirred solution of Example 3 (6.86g, 29mmol) in tetrahydrofuran (200ml) was treated with dimethylimidazolium iodide (2.13g, 9.6mmol), thiophene-2- carbaldehyde (3.19ml, 35mmol) and sodium hydride (1.51g, 38mmol). The mixture was first stirred at room temperature for 25min and then heated, with stirring, to 7O 0 C for 3h. The reaction mixture was cooled to room temperature and reduced in vacuo. The residue was partitioned between water and dichloromethane. The organic fraction was separated, dried over sodium sulfate and reduced in vacuo.
- Example 4 A stirred solution of Example 4 (5g,16mmol) in n-butanol (120ml) was treated with 3-picolylamine (8.1 ml, 80mmol) and heated to 100 0 C for 2h. The reaction mixture was cooled to room temperature, reduced in vacuo and the residue purified by silica gel (100g) column chromatography (3% MeOH/DCM) affording the title compound in 38% yield, >95% purity.
- Example 5 A stirred solution of Example 5 (0.055g, 0.143mmol) in dimethylacetamide (2ml) was treated with 3,4-dimethoxybenzylamine (0.22ml, 1.43mmol). The solution was heated to 170 0 C for 30min in a microwave reactor. The cooled solution was reduced in vacuo and taken up in neat trifluoroacetic acid (3ml). The mixture was stirred at 70 0 C for 48h, cooled and reduced in vacuo. The residue was taken up in ethylacetate (25ml), washed with saturated sodium bicarbonate solution (15ml), dried over sodium sulfate and reduced in vacuo.
- Example 5 A stirred solution of Example 5 (0.05g, 0.13mmol) in tetrahydrofuran (3ml) at room temperature was treated with tris[di(benzylidene)acetone]palladium (0) (0.006g, 0.0065mmol), tri-te/t-butylphosphine (0.01 ml, 0.04mmol), tributylvinyltin (0.06ml, 0.0.195mmol), and caesium carbonate (0.046g, 0.14mmol). The mixture was heated to 140 0 C for 20min in a microwave reactor.
- Example 10 A stirred solution of Example 10 (0.025g, 0.066mmol) in ethanol (5ml) was treated with palladium/activated carbon (10%, 0.0025g) and HCI (1.25M in MeOH, 0.19mmol). The Flask was thoroughly evacuated and placed under 1 atmosphere of hydrogen gas. The mixture was stirred under hydrogen overnight at room temperature, evacuated and filtered through celite. The filtrate was reduced in vacuo to give the title compound as a yellow solid in 70% yield, >95% purity.
- the compounds of the present invention were characterized by liquid chromatography-mass spectroscopy (LC-MS) using the following methods.
- UV detection from 220 to 400nm
- Some compounds of the invention were purified by preparative HPLC. These were performed on a Waters FractionLynx MS autopurification system, with a Gemini ® 5 ⁇ m C18(2), 100 mm * 20 mm i.d. column from Phenomenex, running at a flow rate of 20 cm 3 min ⁇ 1 with UV diode array detection (210—400 nm) and mass-directed collection. Gradients used for each compound are shown in Table 1.
- solvent A 10 mM ammonium acetate in HPLC grade water + 0.08% v/v formic acid.
- Solvent B 95% v/v HPLC grade acetonitrile + 5% v/v solvent A + 0.08% v/v formic acid.
- solvent A 10 mM ammonium acetate in HPLC grade water + 0.08% v/v ammonia solution.
- Solvent B 95% v/v HPLC grade acetonitrile + 5% v/v solvent A + 0.08% v/v ammonia solution.
- the mass spectrometer was a Waters Micromass ZQ2000 spectrometer, operating in positive or negative ion electrospray ionisation modes, with a molecular weight scan range of 150 to 1000.
- Fluoromethc Imaging Plate Reader FLIPR
- FLIPR Fluoromethc Imaging Plate Reader
Abstract
Compounds of formula (I) are A2B receptor antagonists, wherein R1 is optionally substituted aryl or an optionally substituted 5- or 6- membered heteroaryl ring; R2 and R3 are independently selected from hydrogen, C1-C6 alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkyl-(C1-C6)-alkyl, aryl- (C1-C6)-alkyl optionally substituted in the ring part thereof, a 5- or 6-membered monocyclic heterocyclic group optionally linked via a C1-C6 alkylene chain and optionally substituted in the ring part thereof, benzimidazol-2-yl-methyl, pyrid- 3-yl-carbonyl, or (1-methyl-piperidin-4-yl)-carbonyl-methyl; or R2 and R3 taken together with the nitrogen atom to which they are attached form an optionally substituted 5- or 6-membered ring; R4 is C1-C3 alkyl, C2-C3 alkenyl, -N(-R5)- R6, or optionally substituted heteroarylmethylamino; and R5 and R6 are independently selected from hydrogen or C1-C3 alkyl; or R5 and R6 taken together with the nitrogen atonrto which they are attached form an optionally substituted 4- to 6-membered saturated ring.
Description
THIENOPYRIMIDINE COMPOUNDS
This invention relates to novel thienopyrimidine derivatives having A2B receptor antagonistic activity, to the use of such compounds in medicine, in relation to the treatment of disorders which are responsive to antagonism of the A2B receptor such as nociception, asthma, COPD, inflammatory disorders, diabetes, diabetic retinopathy and cancer, and to pharmaceutical compositions containing such compounds.
Background to the invention
Adenosine is a naturally occurring purine nucleoside, the effects of which include stimulation of nociception afferents, bronchconstriction, immunosupression, vasodilation, inhibition of platelet aggregation, cardiac depression and inhibition of neurotransmitter release.
Adenosine produces a wide range of pharmacological effects mediated by activation of specific cell surface receptors, which are members of the G- protein coupled receptor family. Four subtypes of adenosine receptors have been identified, designated A1, A2A, A2B and A3.
The A26 adenosine receptor subtype is coupled to the G3 G-protein and stimulates adenylyl cyclase activity. Although significant advancement has been made in the understanding of the molecular pharmacology and physiology of A2B adenosine receptors, due to the lack of highly potent and selective ligands for this receptor subtype, many questions about the pathophysiological role of A2B receptors are yet to be resolved (Feoktistov and Biaggioni, Pharmacological Reviews (1997), 49(4), 381-402).
A2B receptors have been implicated in:
(i) the regulation of mast cell secretion (Feoktistov and Biaggioni., Journal of Clinical Investigation (1995), 96(4), 1979-86).
(ii) pain (Abo-Salem et al., Journal of Pharmacology and Experimental
Therapeutics (2004), 308(1), 358-366.). (iii) inflammation (Yang et al., Journal of Clinical Investigation (2006),
116(7), 1913-1923). (iv) cancer (Zeng et al., Drug Development Research (2003), 58(4),
405-411 ). (v) diabetes (Harada et al., Journal of Medicinal Chemistry (2001),
44(2), 170-179).
(vi) gene expression (Boyle et al., Arthritis & Rheumatism (1996), 39(6), 923-930).
(vii) cell growth (Dubey et al., Hypertension (1996), 27(3 Pt 2), 786-93
Hypertension (1996), 27(3 Pt 2), 786-93, Dubey et al.,
Hypertension (1998), 31(1 Pt 2), 516-21 ).
(viii) intestinal functions (Murthy et al., Journal of Neurochemistry (1995), 64(1), 77-84).
(ix) neurosecretion (Mateo et al., 1995).
(x) vascular tone (Haynes et al., American Journal of Physiology
(1995), 268(5, Pt. 2), H1862-H1868).
(xi) asthma (Feoktistov et al., Trends in pharmacological sciences (1998), 19(4), 148-153; Holgate, British Journal of Pharmacology
(2005), 145(8), 1009-1015). (xii) COPD (Van den Berge et al., Drugs in R&D (2007), 8(1 ), 13-23).
Thus, there remains a medical need for low molecular weight selective antagonists of the A2B receptor with pharmacokinetic and pharmacodynamic properties making them suitable for use as pharmaceutical agents. There also remains a medical need for new treatments of disorders mediated by the A2B receptor, by selective antagonism of the A2B receptor, particularly the treatment of nociception, asthma, COPD, inflammatory disorders, diabetes, diabetic retinopathy and cancer. The object of the present invention is to provide such pharmaceutical agents and treatments.
It has now been found that certain thienopyrimidine derivatives show efficacy as selective A2B antagonists.
Brief description of the invention
Our co-pending international patent application no. PCT/G B00/02517 is concerned with a class of thieno- and furopyrimidine derivatives which are antagonists of the adenosine A2A receptor. This invention relates to a subset of compounds within the PCT/GB00/02517 class, but which are not specifically disclosed therein.
The present invention relates to a class of substituted thienopyrimidine compounds useful as selective A2B antagonists, for example, for the treatment of nociception, asthma, COPD, inflammatory disorders, diabetes, diabetic retinopathy and cancer. A core thieno-pyrimidine bicyclic ring, with substitution on the thieno portion by an amino group, and substitution on the pyrimidine portion by a (hetero)aryl-carbonyl group in addition to an amino group, are principle characterising features of the compounds with which the invention is concerned.
Detailed description of the invention
According to the present invention, there is provided a compound of formula (I) or a pharmaceutically acceptable salt, hydrate or solvate thereof:

wherein
Ri is optionally substituted aryl or an optionally substituted 5- or 6-membered heteroaryl ring;
R2 and R3 are independently selected from hydrogen, C1-C6 alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkyl-(CrC6)-alkyl, aryl-(CrC6)-alkyl optionally substituted in the ring part thereof, a 5- or 6-membered monocyclic heterocyclic group optionally linked via a Ci-C6 alkylene chain and optionally substituted in the ring part thereof, benzimidazol-2-yl-methyl, pyrid-3-yl- carbonyl, or (1-methyl-piperidin-4-yl)-carbonyl-methyl;
or R2 and R3 taken together with the nitrogen atom to which they are attached form an optionally substituted 5- or 6-membered ring;
R4 is CrC3 alkyl, C2-C3 alkenyl, -N(-R5)-R6, or optionally substituted heteroarylmethylamino; and
R5 and R6 are independently selected from hydrogen or CrC3 alkyl;
or R5 and R6 taken together with the nitrogen atom to which they are attached form an optionally substituted 4- to 6-membered saturated ring.
The active compounds of formula (I) are selective antagonists of the A2B receptor and are useful for the treatment, prevention and suppression of disorders mediated by the A28 receptor. Such disorders include nociception; asthma; chronic obstructive pulmonary disease (COPD); inflammatory diseases such as rheumatoid arthritis, multiple sclerosis, lupus, psoriasis and inflammatory bowel disease; diabetes mellitus or diabetes insipidus; diabetic retinopathy and cancer.
According to a further embodiment of the present invention there is provided the use of a compound of formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, or prodrug thereof, in the manufacture of a medicament for the treatment of disorders mediated by the adenosine A2B receptor.
According to a further embodiment of the present invention there is provided a method of treatment of a disorder mediated by the A2B receptor comprising administration to a subject in need of such treatment an effective dose of the compound of formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, or prodrug thereof.
According to a further embodiment of the present invention there is provided a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt, hydrate, solvate, or prodrug thereof, and a pharmaceutically acceptable carrier.
As used herein, the term "(Ca-Cb)alkyl" wherein a and b are integers refers to a straight or branched chain alkyl radical having from a to b carbon atoms. Thus when a is 1 and b is 6, for example, the term includes methyl, ethyl, n- propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, n-pentyl and n-hexyl.
As used herein the term "divalent (Ca7Cb)alkylene radical" wherein a and b are integers refers to a saturated hydrocarbon chain having from a to b carbon atoms and two unsatisfied valences.
As used herein the term "(Ca-Cb)alkenyl" wherein a and b are integers refers to a straight or branched chain alkenyl moiety having from a to b carbon atoms having at least one double bond of either E or Z stereochemistry where applicable. The term includes, for example, vinyl, allyl, 1- and 2-butenyl and 2-methyl-2-propenyl.
As used herein the term "divalent (Ca-Cb)alkenylene radical" refers to a hydrocarbon chain having from a to b carbon atoms, at least one double bond, and two unsatisfied valences.
As used herein the term "cycloalkyl" refers to a saturated carbocyclic radical having from 3-8 carbon atoms and includes, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
As used herein the term "cycloalkenyl" refers to a carbocyclic radical having from 3-8 carbon atoms containing at least one double bond, and includes, for example, cyclopentenyl, cyclohexenyl, cycloheptenyl and cyclooctenyl.
As used herein the term "carbocyclic" refers to a mono- or bi-cyclic radical whose ring atoms are all carbon, and includes monocyclic aryl, cycloalkyl, and cycloalkenyl radicals, provided that no single ring present has more than 8 ring members. A "carbocyclic" group includes a mono-bridged or multiply- bridged cyclic alkyl group.
As used herein the term "aryl" refers to a mono-, bi- or tri-cyclic carbocyclic aromatic radical. Illustrative of such radicals are phenyl, biphenyl and napthyl.
As used herein the term "heteroaryl" refers to a mono-, bi- or tri-cyclic aromatic radical containing one or more heteroatoms selected from S, N and
O. Illustrative of such radicals are thienyl, benzthienyl, furyl, benzfuryl, pyrrolyl, imidazolyl, benzimidazolyl, thiazolyl, benzthiazolyl, isothiazolyl, benzisothiazolyl, pyrazolyl, oxazolyl, benzoxazolyl, isoxazolyl, benzisoxazolyl, isothiazolyl, triazolyl, benztriazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, indolyl and indazolyl.
As used herein the unqualified term "heterocyclyl" or "heterocyclic" includes "heteroaryl" as defined above, and in particular refers to a mono-, bi- or tricyclic non-aromatic radical containing one or more heteroatoms selected from S, N and O, to groups consisting of a monocyclic non-aromatic radical containing one or more such heteroatoms which is covalently linked to another such radical or to a monocyclic carbocyclic radical, and to a mono-, bi- or tri-cyclic non-aromatic radical containing one or more heteroatoms selected from S, N and O which is mono-bridged or multiply-bridged. Illustrative of such radicals are pyrrolyl, furanyl, thienyl, piperidinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, pyrazolyl, pyridinyl, pyrrolidinyl, pyrimidinyl, morpholinyl, piperazinyl, indolyl, morpholinyl, benzfuranyl, pyranyl, isoxazolyl, benzimidazolyl, methylenedioxyphenyl, ethylenedioxyphenyl, maleimido and succinimido groups.
Unless otherwise specified in the context in which it occurs, the term "substituted" as applied to any moiety herein means substituted with at least one substituent, for example selected from (Ci-Cβjalkyl, (d-C^alkoxy, hydroxy, hydroxy(CrC6)alkyl, mercapto, mercapto(Ci-C6)alkyl, (C1- C6)alkylthio, halo (including fluoro and chloro), trifluoromethyl, trifluoromethoxy, nitro, nitrile (-CN), oxo, phenyl, -COOH, -COORA, -CORA, -SO2RA, -CONH2, -SO2NH2, -CONHRA, -SO2NHRA, -CONRARB, -SO2NRARB, -NH2, -NHRA, -NRARB, -OCONH2, -OCONHRA , -OCONRARB, -NHCORA, -NHCOORA, -NRBCOORA, -NHSO2ORA, -NRBSO2ORA, -NHCONH2,
-NRACONH2, -NHCONHR8 , -NRACONHRB, -NHCONRARB , or -NRACONRARB wherein RA and RB are independently a (CrC6)alkyl group, or RA and RB when attached to the same nitrogen may form a cyclic amino ring such as a morpholinyl, piperidinyl or piperazinyl ring. An "optional substituent" or "susbtituent" may be one of the foregoing substituent groups.
As used herein the term "salt" includes base addition, acid addition and quaternary salts. Compounds of the invention which are acidic can form salts, including pharmaceutically or veterinarily acceptable salts, with bases such as alkali metal hydroxides, e.g. sodium and potassium hydroxides; alkaline earth metal hydroxides e.g. calcium, barium and magnesium hydroxides; with organic bases e.g. N-ethyl piperidine, dibenzylamine and the like. Those compounds (I) which are basic can form salts, including pharmaceutically or veterinarily acceptable salts with inorganic acids, e.g. with hydrohalic acids such as hydrochloric or hydrobromic acids, sulphuric acid, nitric acid or phosphoric acid and the like, and with organic acids e.g. with acetic, tartaric, succinic, fumaric, maleic, malic, salicylic, citric, methanesulphonic and p- toluene sulphonic acids and the like.
For a review on suitable salts, see Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
The term 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and a stoichiometric amount of one or more pharmaceutically acceptable solvent molecules, for example, ethanol. The term 'hydrate' is employed when said solvent is water.
Compounds with which the invention is concerned which may exist in one or more stereoisomeric form, because of the presence of asymmetric atoms or rotational restrictions, can exist as a number of stereoisomers with R or S stereochemistry at each chiral centre or as atropisomeres with R or S stereochemistry at each chiral axis. The invention includes all such enantiomers and diastereoisomers and mixtures thereof.
So-called 'pro-drugs' of the compounds of formula (I) are also within the scope of the invention. Thus certain derivatives of compounds of formula (I) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as 'prodrugs'. Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association).
Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
Also included within the scope of the invention are metabolites of compounds of formula (I), that is, compounds formed in vivo upon administration of the drug. Some examples of metabolites include
(i) where the compound of formula (I) contains a methyl group, an hydroxymethyl derivative thereof (-CH3 -> -CH2OH):
(ii) where the compound of formula (I) contains an alkoxy group, an hydroxy derivative thereof (-OR -> -OH);
(iii) where the compound of formula (I) contains a tertiary amino group, a secondary amino derivative thereof (-NR1R2 -> -NHR1 or -NHR2);
(iv) where the compound of formula (I) contains a secondary amino group, a primary derivative thereof (-NHR1 -> -NH2);
(v) where the compound of formula (I) contains a phenyl moiety, a phenol derivative thereof (-Ph -> -PhOH); and
(vi) where the compound of formula (I) contains an amide group, a carboxylic acid derivative thereof (-CONH2 -> COOH).
Variable substituents present in compounds (I) will now be further defined. It is to be inferred in the further description that any disclosed substituent or substituent class may be present in any combination with any of the other disclosed substituent classes.
The group Ri
In the compounds in accordance with the invention, R1 is optionally substituted aryl or an optionally substituted 5- or 6-membered heteroaryl ring.
In a subclass of compounds with which the invention is concerned, Ri is optionally substituted phenyl. When substituted, the phenyl ring preferably has one substituent, selected from methyl, methoxy, fluoro, chloro, or cyano.
In another subclass of compounds with which the invention is concerned, R1 is an optionally substituted 5- or 6-membered heteroaryl ring. In such cases,
the heteroaryl ring may be, for example, furan, thiophene, pyrrole, oxazole, thiazole, imidazole, or pyridine. Preferred substituents include methyl, ethyl, chloro, or bromo.
Presently, it is preferred that Ri is optionally substituted thienyl, particularly thien-2-yl.
The group -N(R2)-R3
In the compounds in accordance with the invention, R2 and R3 are independently selected from hydrogen, C1-C6 alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkyl-(Ci-C6)-alkyl, aryl-(C-ι-C6)-alkyl optionally substituted in the ring part thereof, a 5- or 6-membered monocyclic heterocyclic group optionally linked via a C1-C6 alkylene chain and optionally substituted in the ring part thereof, benzimidazol-2-yl-methyl, pyrid-3-yl-carbonyl, or (1-methyl-piperidin-4-yl)- carbonyl-methyl.
In a subclass of compounds with which the invention is concerned, R2 is hydrogen and R3 is a 5- or 6-membered monocyclic heterocyclic group optionally linked via a CrC6 alkylene chain and optionally substituted in the ring part thereof. In such cases, the heterocyclic ring may be, for example, pyran, piperidine, morpholine, imidazole, pyridine, pyrimidine, pyrazine, or tetrazole. When present, methylene or ethylene is preferred for the C1-C6 alkylene chain.
In another subclass of compounds with which the invention is concerned, R2 is hydrogen and R3 is aryl-(C-ι-C6)-alkyl optionally substituted in the ring part thereof. Phenyl is preferred for aryl, and when substituted, the phenyl ring preferably has one substituent, selected from methyl, ethyl, methoxy, or chloro. Methyl or ethyl is preferred for C1-C6 alkyl.
In a further subclass of compounds with which the invention is concerned, R2 and R3 taken together with the nitrogen atom to which they are attached form an optionally substituted 5- or 6-membered ring.
Presently, it is preferred that R2 is hydrogen and R3 is pyrid-3-ylmethyl.
The group R4
In the compounds in accordance with the invention, R4 is C-1-C3 alkyl, C2-C3 alkenyl, -N(-R5)-R6, or optionally substituted heteroarylmethylamino.
In a subclass of compounds with which the invention is concerned, R4 is C1-C3 alkyl, preferably ethyl.
In another subclass of compounds with which the invention is concerned, R4 is C2-C3 alkenyl, preferably ethenyl.
In a further subclass of compounds with which the invention is concerned, R4 is optionally substituted heteroarylmethylamino. In such cases, heteroaryl represents a 5- or 6-membered monocyclic heteroaryl ring, with pyridyl preferred, particularly pyrid-3-yl.
In yet another subclass of compounds with which the invention is concerned, R4 is amino, mono-(CrC3-alkyl)amino, or di-(C-ι-C3-alkyl)amino.
In a further subclass of compounds with which the invention is concerned, R4 is -N(-R5)-R6 wherein R5 and R6 taken together with the nitrogen atom to which they are attached form an optionally substituted 4- to 6-membered saturated ring. In such cases, -N(-Rs)-R6 includes azetidin-1-yl, pyrrolidin-1-yl and piperidin-1-yl, with azetidin-1-yl and pyrrolidin-1-yl preferred, particularly azetidin-1-yl.
Presently, it is preferred that R4 is amino, methylamino, ethylamino, dimethylamino, ethyl, ethenyl, or pyrid-3-ylmethylamino.
It is particularly preferred that R4 is amino or methylamino.
Specific compounds with which the invention is concerned include those of the Examples.
The present invention may be employed in respect of a human or animal subject, more preferably a mammal, more preferably a human subject.
As used herein, the term "treatment" as used herein includes prophylactic treatment.
The compound of formula (I) may be used in combination with one or more additional drugs useful in the treatment of the disorders mentioned above, the components being in the same formulation or in separate formulations for administration simultaneously or sequentially.
It will be understood that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, rate of excretion, drug combination and the causative mechanism and severity of the particular disease undergoing therapy. In general, a suitable dose for orally administrable formulations will usually be in the range of 0.1 to 3000 mg, once, twice or three times per day, or the equivalent daily amount administered by infusion or other routes. However, optimum dose levels and frequency of dosing will be determined by clinical trials as is conventional in the art.
The compounds with which the invention is concerned may be prepared for administration by any route consistent with their pharmacokinetic properties. The orally administrable compositions may be in the form of tablets, capsules, powders, granules, lozenges, liquid or gel preparations, such as oral, topical, or sterile parenteral solutions or suspensions. Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinyl-pyrrolidone; fillers for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting
lubricant, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants for example potato starch, or acceptable wetting agents such as sodium lauryl sulphate. The tablets may be coated according to methods well known in normal pharmaceutical practice. Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, glucose syrup, gelatin hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and if desired conventional flavouring or colouring agents.
For topical application to the skin, the drug may be made up into a cream, lotion or ointment. Cream or ointment formulations which may be used for the drug are conventional formulations well known in the art, for example as described in standard textbooks of pharmaceutics such as the British Pharmacopoeia.
The active ingredient may also be administered parenterally in a sterile medium. Depending on the vehicle and concentration used, the drug can either be suspended or dissolved in the vehicle. Advantageously, adjuvants such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle.
There are multiple synthetic strategies for the synthesis of the compounds (I) with which the present invention is concerned, but all rely on known chemistry, known to the synthetic organic chemist. Thus, compounds according to formula (I) can be synthesised according to procedures described in the standard literature and are well-known to the one skilled in the art. Typical literature sources are "Advanced organic chemistry", 4th Edition (Wiley), J March, "Comprehensive Organic Transformation", 2nd Edition (Wiley), R. C.
Larock , "Handbook of Heterocyclic Chemistry", 2nd Edition (Pergamon), A.R. Katritzky), review articles such as found in "Synthesis", "Ace. Chem. Res." , "Chem. Rev", or primary literature sources identified by standard literature searches online or from secondary sources such as "Chemical Abstracts" or "Beilstein". Such literature methods include those of the preparative Examples herein, and methods analogous thereto.
Scheme 1 represents a method known in the art of organic chemistry in general, by which the compounds of the present invention may be prepared:
Scheme 1

EXAMPLES
The following examples illustrate the preparation of specific compounds of the invention and are not intended to be limiting of the full scope of the invention.
Examples 1 to 5 relate to the method indicated in Scheme 1.
Preparative Example 1 1H-Thieno[3,2-d]pyrimidine-2,4-dione
A solid mixture of methyl-S-aminothiophene^-carboxylate (76g, 480mmol) and urea (189g, 3140mmol) was heated, with stirring, to 18O0C for 5h. The mixture was cooled to 900C and water (1000ml) added. After stirring at room temperature for 16h, a cream coloured solid was filtered and washed twice with further water. The solid was dried in vacuo at 40 0C to give the title compound in 94% yield, >95% purity. LC-MS m/z=169.0 [M+H]+; RT=1.29min; LC-MS method 2. 1H NMR: δH (400MHz, D6-DMSO) 6.90 (1 H, d, J 5.5Hz),
8.04 (1 H, d, J 5.5Hz), 11.10-11.80 (2H,b).
Preparative Example 2 6-Nitro-1H-thieno[3,2-d]pyrimidine-2,4-dione A stirred mixture of concentrated sulphuric acid (98%, 270ml) and fuming nitric acid (270ml) at 0 0C was treated portionwise with Example 1 (9Og, 530mmol). Upon complete dissolution, stirring was continued at room temperature for a further 20min. The solution was added slowly to vigorously stirred ice/water (2000ml). After 30min stirring at room temperature, a yellow solid was filtered, washed with water and dried in vacuo at 40 0C to give the title compound in 61 % yield, >95% purity. LC-MS m/z=251.0 [M+H]+; RT=2.92; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 7.66 (1 H, s), 11.71 (1 H, b), 11.79 (1 H, b).
Preparative Example 3
2,4,6-Trichloro-thieno[3,2-d]pyrimidine
A suspension of Example 2 (7Og, 330mmol) in phenylphosphonic dichloride (320ml, 2310mmol) was heated, with stirring, to 180 0C for 4h. The mixture was cooled to 100 0C and transferred slowly onto vigorously stirred ice/water (2500ml). After 2h stirring at room temperature, a tan solid was filtered and dried in vacuo at 40 0C. The solid was dissolved in the minimum volume of tetrahydrofuran and passed over a short pad of silica using ethyl acetate as an eluent. The filtrate was reduced in vacuo and the residue re-crystallised from iso-hexane:ethyl acetate (10:1 ) to give the title compound in 45% yield,
>95% purity. LC-MS m/z=239.0 [M+H]+; RT=3.39min; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 7.94 (1 H, s)
Preparative Example 4 (2,6-Dichloro-thieno[3,2-d]pyrimidin-4-yl)-thiophen- 2-yl-methan-one
A stirred solution of Example 3 (6.86g, 29mmol) in tetrahydrofuran (200ml) was treated with dimethylimidazolium iodide (2.13g, 9.6mmol), thiophene-2- carbaldehyde (3.19ml, 35mmol) and sodium hydride (1.51g, 38mmol). The mixture was first stirred at room temperature for 25min and then heated, with stirring, to 7O 0C for 3h. The reaction mixture was cooled to room temperature and reduced in vacuo. The residue was partitioned between water and dichloromethane. The organic fraction was separated, dried over sodium sulfate and reduced in vacuo. Trituration with methanol gave the title compound in 72% yield, 95% purity. LC-MS m/z=279.1 [M+H]+; RT=3.77min; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 7.40-7.42 (1H, m), 7.95 (1 H, s), 8.31 (1 H, d, J 4.5Hz), 8.61 (1 H, d, J 4.5Hz).
Preparative Example 5
{6-Chloro-2-[(pyridin-3-ylmethyl)-amino]-thieno[3,2-d]pyrimidin-4-yl}- thiophen-2-yl-methanone
A stirred solution of Example 4 (5g,16mmol) in n-butanol (120ml) was treated with 3-picolylamine (8.1 ml, 80mmol) and heated to 1000C for 2h. The reaction mixture was cooled to room temperature, reduced in vacuo and the residue purified by silica gel (100g) column chromatography (3% MeOH/DCM) affording the title compound in 38% yield, >95% purity. LC-MS m/z=387.0 [M+H]+; RT=3.30min; LC-MS method 1.
1H NMR: δH (400MHz, D6-DMSO) 4.73 (2H, b), 7.31-7.36 (2H, m), 7.47 (1 H, s), 7.80 (1 H1 d, J 6.0Hz), 8.20-8.28 (2H, m), 8.44 (1 H, d,J 6.0Hz), 8.64 (1 H, bd).
Example 6
{6-Methylamino-2-[(pyridin-3-ylmethyl)-amino]-thieno[3,2-d]pyrimidin-4- yl}-thiophen-2-yl-methanone
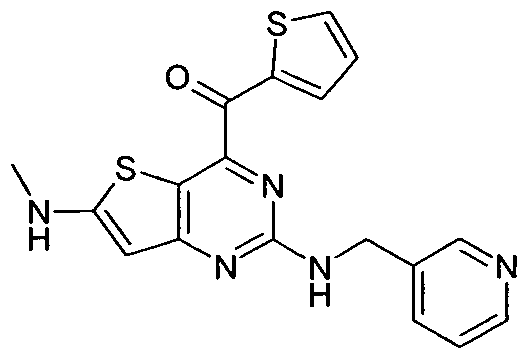
A stirred solution of Example 5 (0.4g, 1 mmol) in dimethyl acetamide (10ml) was treated with methylamine (2.0M in MeOH, 4.96ml,10mmol). The solution was heated to 15O 0C in a sealed tube for 1h. The solution was cooled to room temperature and slowly poured onto stirred iced water (100ml). After 15min stirring, a light orange solid was filtered. Recrystallisation from hot ethyl acetate gave the title compound in 58% yield, >95% purity. LC-MS /77/z=382.0 [M+H]+; RT=2.73; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 2.89 (3H ,d, J 5.0Hz), 4.67 (2H, d ,J 6.0Hz), 5.81 (1 H,s), 7.23 (1 H.W 4.5Hz), 7.77 (1 H.d.J 8.0Hz), 7.96 (1 H,b), 8.09 (1 H1 d, J 5.0Hz), 8.41 (1 H, d, J 5.0Hz), 8.36- 8.50 (1 H, b), 8.61 (1 H, bd)
Example 7 {6-dimethylamino-2-[(pyridine-3-ylmethyl)-amino]-thieno[3,2-d]pyrimidin- 4-yl}-thiophen-2-yl-methanone

A stirred solution of Example 5 (0.02g, 0.052mmol) in dimethylacetamide (1.5ml) was treated with dimethylamine (2.0M in THF, 0.52mmol). The solution was heated to 170 0C for 30min in a microwave reactor. The cooled solution was poured onto stirred iced water (20ml). After 15min stirring, an orange solid was filtered. Recrystallisation from hot ethyl acetate gave the title compound in 42% yield, >95% purity. LC-MS m/z=396.0 [M+H]+; RT=2.97;
LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 3.13 (6H, s), 4.68 (2H, bd), 5.91 (1 H, s), 7.26-8.61 (8H, m).
Example 8 {6-Ethylamino-2-[(pyridin-3-ylmethyl)-amino]-thieno[3,2-d]pyrimidin-4- yl}-thiophen-2-yl-methanone

A stirred solution of Example 5 (0.025g, 0.065mmol) in dimethyl acetamide (1.5ml) was treated with ethylamine (2.0M in THF, 0.65mmol). The solution was heated to 170 0C for 30min in a microwave reactor. The cooled solution was poured on to aqueous HCI solution (2.5M,25ml). The aqueous solution was washed with ethylacetate (50ml) and basified (pH 9) using aqueous sodium hydroxide solution (5M). The title compound was filtered as an orange solid in 40% yield, >90% purity. LC-MS m/z=396.0 [M+H]+; RT=2.78; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 1.21 (3H ,t, J 7.0Hz), 3.22-3.33 (2H1 m), 4.68 (2H, bd), 5.82 (1 H, s), 7.23-8.63 (11 H, m).
Example 9
{6-Amino-2-[(pyridin-3-ylmethyl)-amino]-thieno[3,2-d]pyrimidin-4-yl}- thiophen-2-yl-methanone

Example 10 {2-[(Pyridin-3-ylmethyl)-amino]-6-vinyl-thieno[3,2-d]pyrimidin-4-yl}- thiophen-2-yl-methanone

A stirred solution of Example 5 (0.05g, 0.13mmol) in tetrahydrofuran (3ml) at room temperature was treated with tris[di(benzylidene)acetone]palladium (0) (0.006g, 0.0065mmol), tri-te/t-butylphosphine (0.01 ml, 0.04mmol), tributylvinyltin (0.06ml, 0.0.195mmol), and caesium carbonate (0.046g, 0.14mmol). The mixture was heated to 140 0C for 20min in a microwave reactor. The cooled reaction mixture was poured onto aqueous HCI (2.5M, 25ml), washed with ethylacetate, and basified (pH9) using aqueous sodium hydroxide solution (5M). The aqueous phase was extracted with ethyl acetate (2X30ml). Combined extracts were dried over sodium sulfate and reduced in vacuo. Silica gel (10g) column chromatography (ethyl acetate) afforded the title compound as a yellow solid in 51% yield, >95% purity. LC-MS m/z=379.0
[M+H]+; RT=3.15; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 4.73 (2H1 b), 5.57 (1 H, d, J 10.5Hz), 6.00 (1 H, d, J 17.5Hz), 7.06-7.14 (1 H, m), 7.29-7.35 (3H, m), 7.79 (1 H, d, J 8.0Hz), 8.11-8.19 (2H, m), 8.43 (1 H, d, J 4.5Hz), 8.64 (1 H, bd), 8.43-8.64 (1 H, b)
Example 11
{6-Ethyl-2-[(pyridin-3-ylmethyl)-amino]-thieno[3,2-d]pyrimidin-4-yl}- thiophen-2-yl-methanone

A stirred solution of Example 10 (0.025g, 0.066mmol) in ethanol (5ml) was treated with palladium/activated carbon (10%, 0.0025g) and HCI (1.25M in MeOH, 0.19mmol). The Flask was thoroughly evacuated and placed under 1 atmosphere of hydrogen gas. The mixture was stirred under hydrogen overnight at room temperature, evacuated and filtered through celite. The filtrate was reduced in vacuo to give the title compound as a yellow solid in 70% yield, >95% purity. LC-MS m/z=381.0 [M+H]+; RT=3.19; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 1.33 (3H, t, J 7.5Hz), 2.95 (2H, q, J 7.5Hz), 4.71 (2H, b), 7.08 (1 H, s), 7.28-7.35 (2H, m), 7.78 (1 H, d, J 8.0Hz), 8.06 (1 H, bt), 8.17 (1 H, d, J 4.5Hz), 8.43 (1 H, d, J 4.5Hz), 8.63 (1 H, bd), 8.40- 8.65 (1 H, b).
Example 12 {6-(3-picolylamino)-2-[(pyridin-3-ylmethyl)-amino]-thieno[3,2-d]pyrimidin- 4-yl}-thiophen-2-yl-methanone

A stirred solution of Example 5 (0.02g, 0.052mmol) in dimethylacetamide (1.5ml) was treated with 3-picolylamine (0.105ml, 1.04mmol). The solution was heated to 170 0C for 30min in a microwave reactor. The cooled solution was poured on to aqueous HCI solution (2.5M, 25ml). The aqueous solution was washed with ethylacetate (50ml) and basified (pH 9) using aqueous sodium hydroxide solution (5M). The title compound was filtered as an orange solid in 59% yield, >95% purity. LC-MS m/z=459.0 [M+H]+; RT=2.67; LC-MS method 1. 1H NMR: δH (400MHz, D6-DMSO) 4.51 (2H, d ,J 5.5Hz), 4.66 (2H, bd), 5.93 (1 H, s), 7.23-7.41 (3H, m), 7.55 (1 H, bt), 7.75-7.81 (2H, m), 8.10 (1 H, d, J 5.0Hz), 8.40-8.63 (6H, m).
General Procedures
All reagents obtained from commercial sources were used without further purification. Anhydrous solvents were obtained from commercial sources and used without further drying. Flash chromatography was performed with prepacked silica-gel cartridges (Strata Si-1 ; 61 A, Phenomenex, Cheshire, UK or IST Flash II, 54 A, Argonaut, Hengoed, UK). Thin layer chromatography was conducted with 5 x 10 cm plates coated with Merck Type 60 F2S4 silica-gel. Microwave heating was performed with a Biotage Initiator™ 2.0 instrument.
The compounds of the present invention were characterized by liquid chromatography-mass spectroscopy (LC-MS) using the following methods.
LC-MS Method 1
Instrument: Waters 2695 pump and 2700 sample manager Waters ZQ2000, M/z range 100 to 900 amu
Column: Gemini 5μm, C18 110A, 30 mm x 2mm from Phenomenex. Pt no 00A-4435-B0
Temperature: Ambient Mobile Phase: A - Water + 10 mMol / ammonium formate + 0.04% (v/v) formic acid at pH ca 3.5 B - 100% Acetonitrile + 0.04% (v/v) formic acid
Injection Volume 1OuL Gradient:
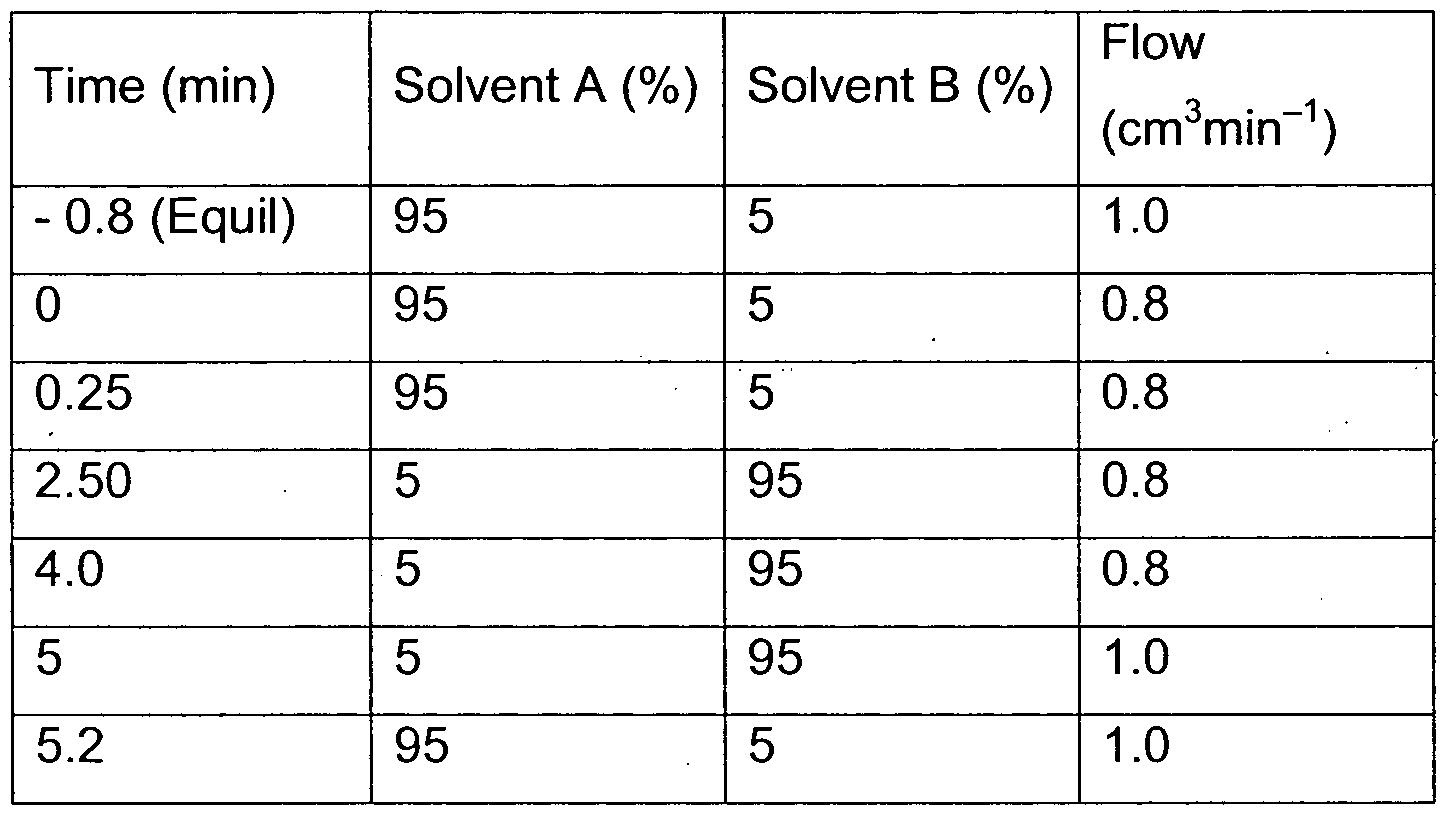
Detection: UV detection from 220 to 400nm (1 :3 split MS to UV)
LC-MS Method 2
Instrument: Waters 2695 pump and 2700 sample manager Waters ZQ2000, M/z range 100 to 900 amu
Column: Gemini 5μm, C18 110A, 30 mm x 2mm from Phenomenex. Pt no 00A-4435-B0 Temperature: Ambient Mobile Phase: A - Water + 10 mMol / ammonium formate + 0.04% (v/v) formic acid at pH ca 3.5 B - 100% Acetonitrile + 0.04% (v/v) formic acid
Injection Volume 5 uL
Gradient:

Detection: UV detection from 220 to 400nm
Nuclear magnetic resonance (NMR) analysis was performed with a Bruker DPX400 spectrometer and proton NMR spectra were measured at 400 MHz. The spectral reference was the known chemical shift of the solvent. Proton NMR data is reported as follows: chemical shift (δ) in ppm, followed by the integration, the multiplicity (where s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, dd = doublet of doublets and br = broad), and the coupling constant rounded to the nearest 0.1 Hz.
Some compounds of the invention were purified by preparative HPLC. These were performed on a Waters FractionLynx MS autopurification system, with a Gemini® 5 μm C18(2), 100 mm * 20 mm i.d. column from Phenomenex, running at a flow rate of 20 cm3min~1 with UV diode array detection (210—400 nm) and mass-directed collection. Gradients used for each compound are shown in Table 1.
At pH 4: solvent A = 10 mM ammonium acetate in HPLC grade water + 0.08% v/v formic acid. Solvent B = 95% v/v HPLC grade acetonitrile + 5% v/v solvent A + 0.08% v/v formic acid.
At pH 9: solvent A = 10 mM ammonium acetate in HPLC grade water + 0.08% v/v ammonia solution. Solvent B = 95% v/v HPLC grade acetonitrile + 5% v/v solvent A + 0.08% v/v ammonia solution.
The mass spectrometer was a Waters Micromass ZQ2000 spectrometer, operating in positive or negative ion electrospray ionisation modes, with a molecular weight scan range of 150 to 1000.

IUPAC chemical names were generated using AutoNom Standard.
Assay Description
The use of a Fluoromethc Imaging Plate Reader (FLIPR) to measure calcium flux in Adenosine-receptor expressing cells is a well-established technique. In this assay calcium flux is triggered by receptor activation and measured through the fluorescence of an incorporated calcium-sensitive dye. The potencies shown were determined using expressed human adenosine A2B receptors in mammalian cell lines. Selectivity values were obtained by using mammalian cell lines expressing the human adenosine A1, A2A and A3 receptors. Compound potency was determined from dose response curves and is reported as an IC50 value.
All examples were tested for activity in the functional assay described above. The resulting experimental data for each example are given in Table 2 below.
All examples demonstrate unexpected selectivity for binding at the A2B receptor versus the A2A receptor. The binding affinity of the examples is of magnitude 12 to 198-fold higher for the A2B receptor versus the A2A receptor, whilst also retaining selectivity over the Ai and A3 sub-types.

Claims
1. A compound of formula (I) or a pharmaceutically acceptable salt, hydrate or solvate thereof:

wherein
R1 is optionally substituted aryl or an optionally substituted 5- or 6-membered heteroaryl ring;
R2 and R3 are independently selected from hydrogen, Ci-C6 alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkyl-(C-ι-C6)-alkyl, aryl-(C-ι-C6)-alkyl optionally substituted in the ring part thereof, a 5- or 6-membered monocyclic heterocyclic group optionally linked via a CrC6 alkylene chain and optionally substituted in the ring part thereof, benzimidazol-2-yl-methyl, pyrid-3-yl- carbonyl, or (1-methyl-piperidin-4-yl)-carbonyl-methyl;
or R2 and R3 taken together with the nitrogen atom to which they are attached form an optionally substituted 5- or 6-membered ring;
R4 is CrC3 alkyl, C2-C3 alkenyl, -N(-R5)-R6, or optionally substituted heteroarylmethylamino; and
R5 and R6 are independently selected from hydrogen or CrC3 alkyl; or R5 and R6 taken together with the nitrogen atom to which they are attached form an optionally substituted 4- to 6-membered saturated ring.
2. A compound as claimed in claim 1 wherein R1 is an optionally substituted 5- or 6-membered heteroaryl ring.
3. A compound as claimed in claim 1 or claim 2 wherein R1 is an optionally substituted 5-membered heteroaryl ring.
4. A compound as claimed in any of the preceding claims wherein R1 is optionally substituted thienyl.
5. A compound as claimed in claim 4 wherein R1 is thienyl optionally substituted by fluoro, chloro, bromo, cyano, methyl, trifluoromethyl, ethyl, hydroxyl, hydroxymethyl, or hydroxyethyl.
6. A compound as claimed in any of the preceding claims wherein R1 is thien-2-yl.
7. A compound as claimed in any of the preceding claims wherein R2 is hydrogen, or a 5- or 6-membered monocyclic heterocyclic group optionally linked via a C1-Ce alkylene chain and optionally substituted in the ring part thereof.
8. A compound as claimed in claim 7 wherein R2 is hydrogen.
9. A compound as claimed in claim 7 wherein R2 is an optionally substituted 5- or 6-membered monocyclic heteroaryl group linked via a C1-C6 alkylene chain.
10. A compound as claimed in claim 7 wherein R2 is a 5- or 6-membered monocyclic heteroaryl group linked via a C1-C6 alkylene chain and optionally substituted in the ring part thereof by fluoro, chloro, bromo, cyano, methyl, trifluoromethyl, ethyl, hydroxyl, hydroxymethyl, or hydroxyethyl.
11. A compound as claimed in any of the preceding claims wherein R3 is hydrogen, or a 5- or 6-membered monocyclic heterocyclic group optionally linked via a CrC6 alkylene chain and optionally substituted in the ring part thereof.
12. A compound as claimed in claim 11 wherein R3 is an optionally substituted 5- or 6-membered monocyclic heteroaryl group linked via a C1-Ce alkylene chain.
13. A compound as claimed in claim 11 wherein R3 is a 5- or 6-membered monocyclic heteroaryl group linked via a CrC6 alkylene chain and optionally substituted in the ring part thereof by fluoro, chloro, bromo, cyano, methyl, thfluoromethyl, ethyl, hydroxyl, hydroxymethyl, or hydroxyethyl.
14. A compound as claimed in any of the preceding claims wherein R4 is amino.
15. A compound as claimed in any of claims 1 to 13 wherein R4 is mono CrC3 alkylamino.
16. A compound as claimed in claim 15 wherein R4 is methylamino.
17. A compound as claimed in claim 15 wherein R4 is ethylamino.
18. A compound as claimed in any of claims 1 to 13 wherein R4 is Ci-C3 alkyl.
19. A compound as claimed in claim 18 wherein R4 is ethyl.
20. A compound of formula (II) or a pharmaceutically acceptable salt, hydrate or solvate thereof: 
wherein
Ri is optionally substituted aryl or an optionally substituted 5- or 6-membered heteroaryl ring;
R2 and R3 are independently selected from hydrogen, C1-C6 alkyl, C3-C8 cycloalkyl, C3-Cs cycloalkyl-(CrC6)-alkyl, aryl-(CrC6)-alkyl optionally substituted in the ring part thereof, a 5- or 6-membered monocyclic heterocyclic group optionally linked via a C1-C6 alkylene chain and optionally substituted in the ring part thereof, benzimidazol-2-yl-methyl, pyrid-3-yl- carbonyl, or (1-methyl-piperidin-4-yl)-carbonyl-methyl;
or R2 and R3 taken together with the nitrogen atom to which they are attached form an optionally substituted 5- or 6-membered ring; and
R4 is hydrogen or C1-C3 alkyl.
21. A compound as claimed in claim 20 wherein R1 is thien-2-yl.
22. A compound as claimed in claim 20 or claim 21 wherein R2 is hydrogen and R3 is pyrid-3-ylmethyl.
23. A compound as claimed in any of claims 20 to 22 wherein R4 is hydrogen or methyl.
24. A pharmaceutical composition comprising a compound as claimed in any of the preceding claims and a pharmaceutically acceptable carrier.
25. The use of a compound as claimed in any of claims 1 to 23 in the manufacture of a medicament for the treatment of disorders mediated by the adenosine A2B receptor.
26. A method of treating a disorder mediated by the adenosine A2B receptor comprising the administration to a subject suffering such a disorder an effective amount of a compound as claimed in any of claims 1 to 23.
27. The use as claimed in claim 25, or a method as claimed in claim 26 wherein the disorder mediated by the adenosine A2B receptor is nociception, asthma, COPD, an inflammatory disorder, diabetes, diabetic retinopathy, or cancer.
28. A use or method as claimed in claim 27 for nociception, asthma, COPD, inflammatory disorders, diabetes, diabetic retinopathy, or cancer, by selective antagonism of A2B activity over A2A activity.
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/678,378 US9120807B2 (en) | 2007-09-21 | 2008-09-19 | Thienopyrimidine compounds |
| JP2010525422A JP2010540422A (en) | 2007-09-21 | 2008-09-19 | Thienopyrimidine compounds |
| EP20080806328 EP2205607A1 (en) | 2007-09-21 | 2008-09-19 | Thienopyrimidine compounds |
| US14/803,214 US9610290B2 (en) | 2007-09-21 | 2015-07-20 | Thienopyrimidine compounds |
| US15/436,988 US9920066B2 (en) | 2007-09-21 | 2017-02-20 | Thienopyrimidine compounds |
| US15/891,618 US10556911B2 (en) | 2007-09-21 | 2018-02-08 | Thienopyrimidine compounds |
| US16/726,308 US11028097B2 (en) | 2007-09-21 | 2019-12-24 | Thienopyrimidine compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0718434.4 | 2007-09-21 | ||
| GB0718434A GB0718434D0 (en) | 2007-09-21 | 2007-09-21 | New chemical compounds |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/678,378 A-371-Of-International US9120807B2 (en) | 2007-09-21 | 2008-09-19 | Thienopyrimidine compounds |
| US14/803,214 Continuation US9610290B2 (en) | 2007-09-21 | 2015-07-20 | Thienopyrimidine compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009037463A1 true WO2009037463A1 (en) | 2009-03-26 |
Family
ID=38670289
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2008/003173 WO2009037463A1 (en) | 2007-09-21 | 2008-09-19 | Thienopyrimidine compounds |
Country Status (5)
| Country | Link |
|---|---|
| US (5) | US9120807B2 (en) |
| EP (1) | EP2205607A1 (en) |
| JP (1) | JP2010540422A (en) |
| GB (1) | GB0718434D0 (en) |
| WO (1) | WO2009037463A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2023201267A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3481869A4 (en) | 2016-07-11 | 2020-02-26 | Corvus Pharmaceuticals, Inc. | Anti-cd73 antibodies |
| CN111629728A (en) | 2017-08-31 | 2020-09-04 | 科尔沃斯制药股份有限公司 | Compounds and methods for modulating adenosine A2B receptor and adenosine A2A receptor |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001002409A1 (en) * | 1999-07-01 | 2001-01-11 | Vernalis Research Limited | Thieno- and furopyrimidine derivatives as a2a-receptor antagonists |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6117878A (en) | 1998-02-24 | 2000-09-12 | University Of Virginia | 8-phenyl- or 8-cycloalkyl xanthine antagonists of A2B human adenosine receptors |
| EP1467995B1 (en) * | 2001-12-20 | 2010-05-19 | OSI Pharmaceuticals, Inc. | Pyrrolopyrimidine a2b selective antagonist compounds, their synthesis and use |
| KR20040080939A (en) | 2002-02-01 | 2004-09-20 | 킹 파머슈티칼스 리서치 앤드 디벨로프먼트 아이엔씨 | 8-Heteroaryl Xanthine Adenosine A2B Receptor Antagonists |
| AU2004285013A1 (en) * | 2003-10-31 | 2005-05-12 | Cv Therapeutics, Inc. | A2B adenosine receptor antagonists |
| US20070208040A1 (en) * | 2006-03-02 | 2007-09-06 | Elfatih Elzein | A2a adenosine receptor antagonists |
-
2007
- 2007-09-21 GB GB0718434A patent/GB0718434D0/en not_active Ceased
-
2008
- 2008-09-19 JP JP2010525422A patent/JP2010540422A/en active Pending
- 2008-09-19 US US12/678,378 patent/US9120807B2/en active Active
- 2008-09-19 WO PCT/GB2008/003173 patent/WO2009037463A1/en active Application Filing
- 2008-09-19 EP EP20080806328 patent/EP2205607A1/en not_active Withdrawn
-
2015
- 2015-07-20 US US14/803,214 patent/US9610290B2/en active Active
-
2017
- 2017-02-20 US US15/436,988 patent/US9920066B2/en active Active
-
2018
- 2018-02-08 US US15/891,618 patent/US10556911B2/en active Active
-
2019
- 2019-12-24 US US16/726,308 patent/US11028097B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001002409A1 (en) * | 1999-07-01 | 2001-01-11 | Vernalis Research Limited | Thieno- and furopyrimidine derivatives as a2a-receptor antagonists |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2023201267A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0718434D0 (en) | 2007-10-31 |
| US20170291907A1 (en) | 2017-10-12 |
| US9610290B2 (en) | 2017-04-04 |
| US9920066B2 (en) | 2018-03-20 |
| US20150342953A1 (en) | 2015-12-03 |
| US11028097B2 (en) | 2021-06-08 |
| US10556911B2 (en) | 2020-02-11 |
| US20180162872A1 (en) | 2018-06-14 |
| EP2205607A1 (en) | 2010-07-14 |
| JP2010540422A (en) | 2010-12-24 |
| US20200140455A1 (en) | 2020-05-07 |
| US9120807B2 (en) | 2015-09-01 |
| US20110118284A1 (en) | 2011-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2207780B1 (en) | Thienopyrimidine compounds and compositions | |
| CN115448923B (en) | Pyrimidine-fused ring compound, preparation method and application thereof | |
| US11028097B2 (en) | Thienopyrimidine compounds | |
| ES2607125T3 (en) | Tetrazolo [1,5-a] pyrazine compounds as histamine receptor inhibitors | |
| AU2009311756B2 (en) | Modulators of amyloid beta. | |
| ES2291628T3 (en) | THYROPHENE BASED THYMPHENE COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THAT INCLUDE SUCH COMPOUNDS. | |
| EP2324020A2 (en) | Piperidine derivatives as jak3 inhibitors | |
| MX2011001938A (en) | Thienopyrimidines for pharmaceutical compositions. | |
| US10239873B2 (en) | 7-azaindole or 4,7-diazaindole derivatives as IKKϵ epsilon and TBK1 inhibitor and pharmaceutical composition comprising same | |
| CN111925379B (en) | Nitrogen-containing heteroaryl substituted pyrimidinediones and uses thereof | |
| WO2012003829A1 (en) | Novel homopiperazine derivatives as protein tyrosine kinase inhibitors and pharmaceutical use thereof | |
| BR112021011147A2 (en) | PYRAZOLYL-AMINO-PYRIMIDINIL DERIVATIVES BENZAMIDES AND COMPOSITIONS AND METHODS THEREOF | |
| KR20210031925A (en) | P2X3 receptor antagonists | |
| EP2203452A1 (en) | Pyrrolopyrimidine compounds | |
| CN111793078B (en) | Bicyclic nitrogen-containing heteroaryl substituted pyrimidinediones and application thereof | |
| CN112142757B (en) | Five-membered nitrogen-containing heteroaryl substituted pyrimidinedione compound and application thereof | |
| CN112047957B (en) | Substituted pyrimidinediones and their use | |
| CN111909168B (en) | Nitrogenous heterocyclic group substituted pyrimidinediones and uses thereof | |
| KR20240021923A (en) | PARP7 inhibitors and their uses |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08806328 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010525422 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008806328 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12678378 Country of ref document: US |