WO2009024550A1 - N-benzyl, n' -arylcarbonylpiperazine derivatives as lxr modulators - Google Patents
N-benzyl, n' -arylcarbonylpiperazine derivatives as lxr modulators Download PDFInfo
- Publication number
- WO2009024550A1 WO2009024550A1 PCT/EP2008/060788 EP2008060788W WO2009024550A1 WO 2009024550 A1 WO2009024550 A1 WO 2009024550A1 EP 2008060788 W EP2008060788 W EP 2008060788W WO 2009024550 A1 WO2009024550 A1 WO 2009024550A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tert
- piperazin
- butyl
- methyl
- benzamide
- Prior art date
Links
- 0 C*C(NC(C=CC(C(N(*)CC(C)(C)CNC(*)C1=CC(C([N+])=O)=CC(C)(C)C=C1)=O)=C1)=CC1(C)C(C)=*)=O Chemical compound C*C(NC(C=CC(C(N(*)CC(C)(C)CNC(*)C1=CC(C([N+])=O)=CC(C)(C)C=C1)=O)=C1)=CC1(C)C(C)=*)=O 0.000 description 1
- CWWXLBTUUOHODD-UHFFFAOYSA-N CC(C)(C(F)(F)F)NC(c1cc(CN(CC2)CCN2C(c(cc2F)ccc2NC(NCC2CC2)=O)=O)ccc1)=O Chemical compound CC(C)(C(F)(F)F)NC(c1cc(CN(CC2)CCN2C(c(cc2F)ccc2NC(NCC2CC2)=O)=O)ccc1)=O CWWXLBTUUOHODD-UHFFFAOYSA-N 0.000 description 1
- VOIHATWGCYFERM-UHFFFAOYSA-N CC(C)(C)CNC(Nc(c(F)c1)ccc1C(N1CCN(Cc2cccc(C(NC(C)(C)C(F)(F)F)=O)c2)CC1)=O)=O Chemical compound CC(C)(C)CNC(Nc(c(F)c1)ccc1C(N1CCN(Cc2cccc(C(NC(C)(C)C(F)(F)F)=O)c2)CC1)=O)=O VOIHATWGCYFERM-UHFFFAOYSA-N 0.000 description 1
- SSALVIWIQCAOEK-UHFFFAOYSA-N CC(C)(C)CNC(Nc(cc1)ccc1C(N1CCN(Cc(cccc2C(NC(C)(C)C)=O)c2F)CC1)=O)=O Chemical compound CC(C)(C)CNC(Nc(cc1)ccc1C(N1CCN(Cc(cccc2C(NC(C)(C)C)=O)c2F)CC1)=O)=O SSALVIWIQCAOEK-UHFFFAOYSA-N 0.000 description 1
- VVMPXOXYQONBIH-UHFFFAOYSA-N CC(C)(C)NC(c(cccc1CN(CC2)CCN2C(c(cc2)ccc2N)=O)c1F)=O Chemical compound CC(C)(C)NC(c(cccc1CN(CC2)CCN2C(c(cc2)ccc2N)=O)c1F)=O VVMPXOXYQONBIH-UHFFFAOYSA-N 0.000 description 1
- KSQFNNSNTGADLI-UHFFFAOYSA-N CC(C)(C)NC(c1cc(CN(CC2)C(CF)CN2C(c(cc2F)ccc2I)=O)ccc1)=O Chemical compound CC(C)(C)NC(c1cc(CN(CC2)C(CF)CN2C(c(cc2F)ccc2I)=O)ccc1)=O KSQFNNSNTGADLI-UHFFFAOYSA-N 0.000 description 1
- NQLJJOHKIYJKHS-UHFFFAOYSA-N CC(C)(C)NC(c1cc(CN(CC2)CCN2C(c(cc2)cc(C)c2N)=O)ccc1)=O Chemical compound CC(C)(C)NC(c1cc(CN(CC2)CCN2C(c(cc2)cc(C)c2N)=O)ccc1)=O NQLJJOHKIYJKHS-UHFFFAOYSA-N 0.000 description 1
- KOOHWGOYZFRXFW-UHFFFAOYSA-N CC(C)(C)NC(c1cc(CN(CC2)CCN2C(c(cc2F)ccc2NC(NCCC2N(C)CCC2)=O)=O)ccc1)=O Chemical compound CC(C)(C)NC(c1cc(CN(CC2)CCN2C(c(cc2F)ccc2NC(NCCC2N(C)CCC2)=O)=O)ccc1)=O KOOHWGOYZFRXFW-UHFFFAOYSA-N 0.000 description 1
- BYDFSLRGMHABSM-UHFFFAOYSA-N CC(C)(C)NC(c1cc(CN(CC2)CCN2C(c(ccc([N+]([O-])=O)c2)c2F)=O)ccc1)=O Chemical compound CC(C)(C)NC(c1cc(CN(CC2)CCN2C(c(ccc([N+]([O-])=O)c2)c2F)=O)ccc1)=O BYDFSLRGMHABSM-UHFFFAOYSA-N 0.000 description 1
- JGWBIQCLXCHAQU-UHFFFAOYSA-N CC(C)(C)NC(c1cccc(CN(CC2)CCN2C(c(cc2)ccc2NC(c2ccccc2)=O)=O)c1)=O Chemical compound CC(C)(C)NC(c1cccc(CN(CC2)CCN2C(c(cc2)ccc2NC(c2ccccc2)=O)=O)c1)=O JGWBIQCLXCHAQU-UHFFFAOYSA-N 0.000 description 1
- NNHJSJCKQOITIA-UHFFFAOYSA-N CCCCNC(Nc(cc1)ccc1C(N1CCN(Cc2cccc(C(NC(C)(C)C(F)(F)F)=O)c2)CC1)=O)=O Chemical compound CCCCNC(Nc(cc1)ccc1C(N1CCN(Cc2cccc(C(NC(C)(C)C(F)(F)F)=O)c2)CC1)=O)=O NNHJSJCKQOITIA-UHFFFAOYSA-N 0.000 description 1
- AUQOFWNRYLWFFA-MRXNPFEDSA-N C[C@H](C1)N(Cc2cccc(C(NC(C)(C)C)=O)c2)CCN1C(c(cc1F)ccc1I)=O Chemical compound C[C@H](C1)N(Cc2cccc(C(NC(C)(C)C)=O)c2)CCN1C(c(cc1F)ccc1I)=O AUQOFWNRYLWFFA-MRXNPFEDSA-N 0.000 description 1
- BHDFPYLPLOVBSR-LJQANCHMSA-N C[C@H](C1)N(Cc2cccc(C(NC(C)(C)C)=O)c2)CCN1C(c(cc1F)ccc1NC(NCC1CC1)=O)=O Chemical compound C[C@H](C1)N(Cc2cccc(C(NC(C)(C)C)=O)c2)CCN1C(c(cc1F)ccc1NC(NCC1CC1)=O)=O BHDFPYLPLOVBSR-LJQANCHMSA-N 0.000 description 1
- ILMBIMITNLUWLR-UHFFFAOYSA-N Nc(c(F)c1)ccc1C(N1CCN(Cc2cccc(C(NC3CCC3)=O)c2)CC1)=O Chemical compound Nc(c(F)c1)ccc1C(N1CCN(Cc2cccc(C(NC3CCC3)=O)c2)CC1)=O ILMBIMITNLUWLR-UHFFFAOYSA-N 0.000 description 1
- QWFUQIODOXXZJE-CEBUJLNPSA-N O=C(c1cccc(CN(CC2)CCN2C(c(cc2)cc(F)c2NC(NCC2CC2)=O)=O)c1)NC(C1)[C@@H]1c1ccccc1 Chemical compound O=C(c1cccc(CN(CC2)CCN2C(c(cc2)cc(F)c2NC(NCC2CC2)=O)=O)c1)NC(C1)[C@@H]1c1ccccc1 QWFUQIODOXXZJE-CEBUJLNPSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/192—Radicals derived from carboxylic acids from aromatic carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
- C07D207/09—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/18—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D207/22—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/24—Oxygen or sulfur atoms
- C07D207/26—2-Pyrrolidones
- C07D207/263—2-Pyrrolidones with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms
- C07D207/27—2-Pyrrolidones with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms with substituted hydrocarbon radicals directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/28—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/30—Oxygen or sulfur atoms
- C07D233/32—One oxygen atom
- C07D233/36—One oxygen atom with hydrocarbon radicals, substituted by nitrogen atoms, attached to ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D241/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with nitrogen atoms directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D241/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with nitrogen atoms directly attached to ring carbon atoms
- C07D241/28—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with nitrogen atoms directly attached to ring carbon atoms in which said hetero-bound carbon atoms have double bonds to oxygen, sulfur or nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/08—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D261/14—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D261/18—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/28—Radicals substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/48—Acylated amino or imino radicals by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof, e.g. carbonylguanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D285/00—Heterocyclic compounds containing rings having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by groups C07D275/00 - C07D283/00
- C07D285/01—Five-membered rings
- C07D285/02—Thiadiazoles; Hydrogenated thiadiazoles
- C07D285/04—Thiadiazoles; Hydrogenated thiadiazoles not condensed with other rings
- C07D285/12—1,3,4-Thiadiazoles; Hydrogenated 1,3,4-thiadiazoles
- C07D285/125—1,3,4-Thiadiazoles; Hydrogenated 1,3,4-thiadiazoles with oxygen, sulfur or nitrogen atoms, directly attached to ring carbon atoms, the nitrogen atoms not forming part of a nitro radical
- C07D285/135—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/155—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/20—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof
- C07D295/205—Radicals derived from carbonic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/20—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof
- C07D295/215—Radicals derived from nitrogen analogues of carbonic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/10—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/14—Radicals substituted by nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/52—Radicals substituted by nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/56—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/66—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/58—Radicals substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/14—Radicals substituted by singly bound hetero atoms other than halogen
- C07D333/20—Radicals substituted by singly bound hetero atoms other than halogen by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/46—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings substituted on the ring sulfur atom
- C07D333/48—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings substituted on the ring sulfur atom by oxygen atoms
Definitions
- the present invention relates to N-benzyl,N'arylcarbonylpiperazine derivatives, to pharmaceutical compositions comprising the same and to the use of these N- benzyl, N'arylcarbonylpiperazine derivatives in the treatment of atherosclerosis.
- LXRs The Liver X Receptors
- ⁇ and ⁇ Two subtypes of LXR ( ⁇ and ⁇ ) have been identified and exhibit 77% homology of both their ligand- and DNA-binding domains. Both subtypes are highly conserved between humans and rodents however their tissue expression patterns differ significantly.
- the expression of LXR ⁇ is restricted to tissues involved in lipid metabolism with highest expression in the liver; there are also significant levels in kidney, spleen, small intestine and adipose tissue.
- LXR ⁇ is very widely distributed and has been found in virtually every tissue examined, including liver and brain. Both LXR ⁇ and LXR ⁇ are expressed in macrophages. See Costet et al., J. Biol. Chem 275:28240-28245 (2000).
- LXR ATP-binding cassette transporter A1
- LXR activation Costet et al., supra.
- LXR agonists have also been shown to promote cholesterol efflux. See Claudel et al., Proc. Natl. Acad. Sci. U S A. 98:2610-2615 (2001).
- LXR receptors therefore play a critical role in cholesterol homeostasis in macrophages, and suppression within the local environment of the advanced atherosclerotic plaque may be a key feature of the pathology of the disease.
- LXR agonists in the treatment of atherosclerosis has been increasingly documented over the last few years. See for example Levin et al.,
- Atherosclerosis is a disease of the arteries that exists for many years without causing symptoms. Advanced atherosclerotic plaques can however become vulnerable to rupture, promoting acute thrombosis and clinical events such as myocardial infarction (Ml) and stroke.
- Ml myocardial infarction
- the primary mechanism for achieving efficacy in atherosclerosis with an LXR agonist is expected to occur by lowering the cholesterol burden of arteries (via upregulation of ABCA1 ), to generate more stable lesions and thus reduce the clinical events.
- LXR agonists may increase circulating HDL levels due to the role of ABCA 1 in generation of nascent HDL by the liver. There is potential for further anti- atherosclerotic effects of LXR agonists due to suppression of inflammation (Joseph et al., Nat.Med. 9:213-219 (2003)) and effects on glucose metabolism. See Latiffe et al., Proc. Natl. Acad. Sci. U S A. 100:5419-24 (2003).
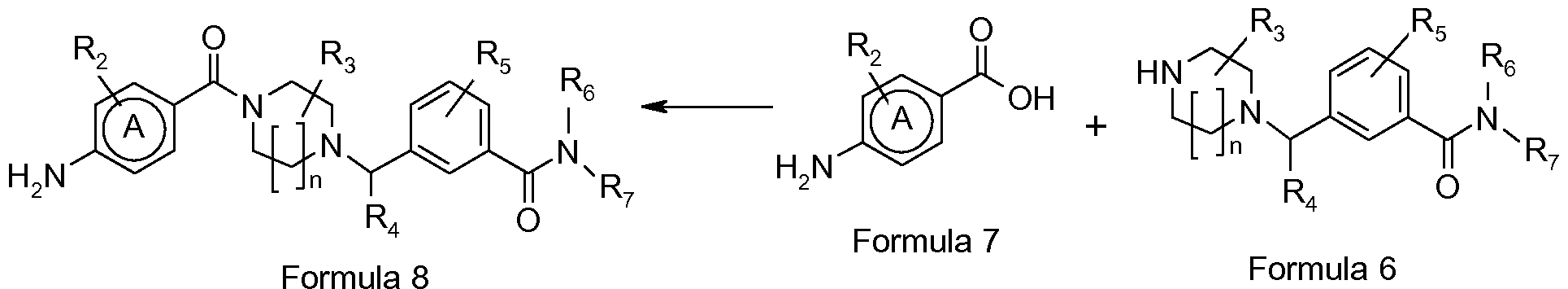
- N-benzyl,N'-arylcarbonylpiperazine derivatives having the general formula I
- n 1 or 2;
- A is a 6-membered aromatic ring optionally containing 1 or 2 nitrogen atoms;
- X is NR 8 , O or a bond;
- R 1 is H, (d- 8 )alkyl [optionally substituted with halogen, (d ⁇ )alkyloxy, (C ⁇ alkyloxy- carbonyl, (C 3 -8)cycloalkyl, OH, CF 3 , cyano or NR 9 R 10 ], (C 2 -6)alkenyl, (C 2 -6)alkynyl, (C 3 -S)- cycloalkyl, phenyl [optionally substituted with (C 1 ⁇ )alkyl, (C 1 _ 4 )alkyloxy, halogen, CN, CF 3 , OCF 3 or NO 2 ], a 5- or 6-membered saturated heterocyclic group containing 1 or 2 heteroatoms selected from O and S, or a 5- or 6-membered heteroaryl group containing 1-3 heteroatoms selected from O, S and N [optionally substituted with (C- ⁇ )alkyl, (C 1 ⁇ )- alkyloxy, halogen or CF
- R 3 optionally represents 1-4 substituents independently selected from (C- ⁇ )alkyl and (C 1 - 4 )alkyl substituted by OH or 1 or more halogens;
- R 4 is H or (C 1 _ 6 )alkyl;
- R 5 optionally represents 1-3 substituents independently selected from (C ⁇ alkyl, (C ⁇ alkyloxy and halogen;
- R 6 is H, (C ⁇ alkyl [optionally substituted with halogen, CF 3 or CN], (C 3 _ 6 )cycloalkyl [optionally containing a heteroatom selected from O and S, and optionally substituted by CN or phenyl], (C 3 _ 6 )cycloalkyl(C 1 ⁇ )alkyl, or a 5- or 6-membered (hetero)aryl group, optionally linked through a methylene group, and optionally substituted with (C- ⁇ )alkyl, (C 1 .
- R 7 is H or (Ci- 3 )alkyl
- R 8 when present, is H or (C ⁇ alkyl; or R 8 together with R 1 and the N to which they are bonded form a 4-8 membered ring optionally containing a further heteroatom selected from O and S [and which ring is optionally substituted with OH, (d_ 3 )alkyl, (d_ 3 )alkyloxy or halogen]
- R 9 and Ri 0 are independently selected from H, (Ci- 3 )alkyl or (Ci- 3 )alkylcarbonyl; or R 9 and R 10 together with the N to which they are bonded form a 4-8 membered ring optionally containing a further heteroatom selected from O and S; or a pharmaceutically acceptable salt thereof.
- (C ⁇ alkyl as used in the definition of Formula I means a branched or unbranched alkyl group having 1-8 carbon atoms, like octyl, hexyl, pentyl, isopentyl, butyl, isobutyl, tertiary butyl, propyl, isopropyl, ethyl and methyl.
- (d ⁇ )alkyl as used in the definition of Formula I means a branched or unbranched alkyl group having 1-4 carbon atoms, like butyl, isobutyl, tertiary butyl, propyl, isopropyl, ethyl and methyl.
- (d_ 3 )alkyl used in the definition of Formula I means a branched or unbranched alkyl group having 1-3 carbon atoms, like propyl, isopropyl, ethyl and methyl.
- (C 2 - 6 )alkenyl means a branched or unbranched alkenyl group having 2-6 carbon atomes, such as ethenyl, propen-2-yl, 2-methyl-propenyl, penten-4-yl and the like.
- (C 2 -6)alkynyl means a branched or unbranched alkynyl group having 2-6 carbon atomes, such as ethynyl, propyn-2-yl, pentyn-4-yl and the like.
- (C 3 _ 8 )cycloalkyl means a cycloalkyl group having 3-8 carbon atoms, like cycloheptyl, cyclohexyl, cyclopentyl, cyclobutyl and cyclopropyl.
- the (C 3 -8)cycloalkyl group may contain a heteroatom selected from O and S to form a saturated heteroromatic ring such as tetrahydrofuranyl, tetrahydrothienyl, tetrahydropyranyl and tetrahydrothiopyranyl.
- a 5- or 6-membered heteroaryl group containing 1 -3 heteroatoms selected from O, S and N is exemplified by oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, furanyl, pyrrolyl, furazanyl, pyrazolyl, pyrazinyl, pyridinyl, pyrimidinyl, imidazolyl, thiazolyl, thiadiazolyl, thienyl and the like.
- a 5- 6-membered saturated heterocyclic group containing 1 or 2 heteroatoms selected from O and S is exemplified by tetrahydrofuranyl, tetrahydrothienyl, tetrahydropyranyl and tetrahydrothiopyranyl.
- a 5- 6-membered saturated heterocyclic group containing 1 or 2 heteroatoms selected from O, S and N is exemplified by tetrahydrofuranyl, tetrahydrothienyl, tetra- hydropyranyl, tetrahydrothiopyranyl, morpholin-1-yl, thiomorpholin-1-yl, piperidinyl, pyrrolidinyl, imidazolidinyl and the like.
- halogen means F, Cl, Br or I.
- N-benzyl,N'-arylcarbonylpiperazine derivatives of formula wherein X is NH and n is 1.
- N-benzyl,N'-arylcarbonylpiperazine derivatives of formula I wherein in addition A is phenyl.
- R 1 is (C ⁇ alkyl [optionally substituted with (C 3 _ 8 )cycloalkyl], (C 2 - 6 )alkenyl, (C 3 _ 8 )cycloalkyl or phenyl [optionally substituted with halogen].
- N-benzyl,N'-arylcarbonylpiperazine derivatives of formula I wherein R 3 is absent or represents methyl, and wherein R 5 is absent or represents halogen.
- R 6 is (Ci_ 6 )alkyl optionally substituted with CF 3 , and R 7 is H.
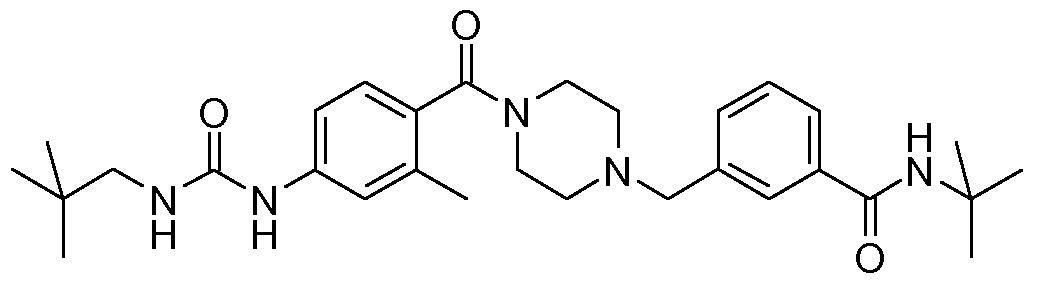
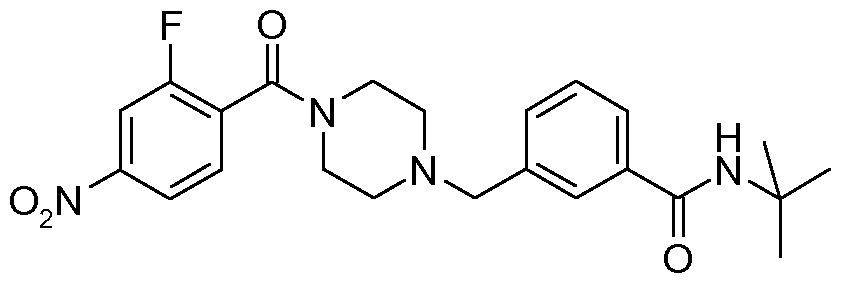
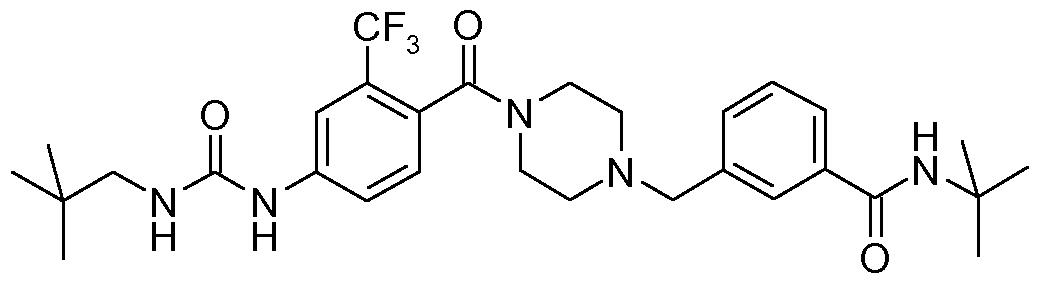
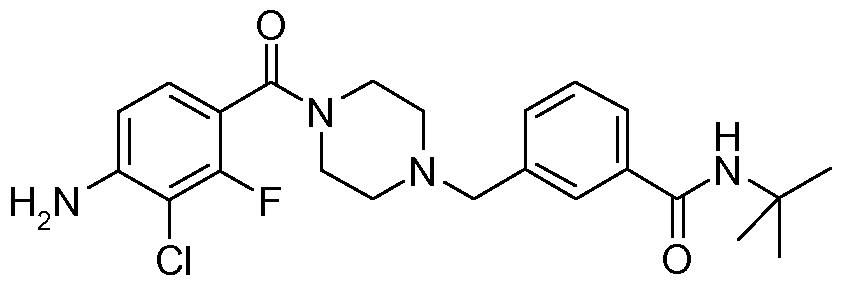
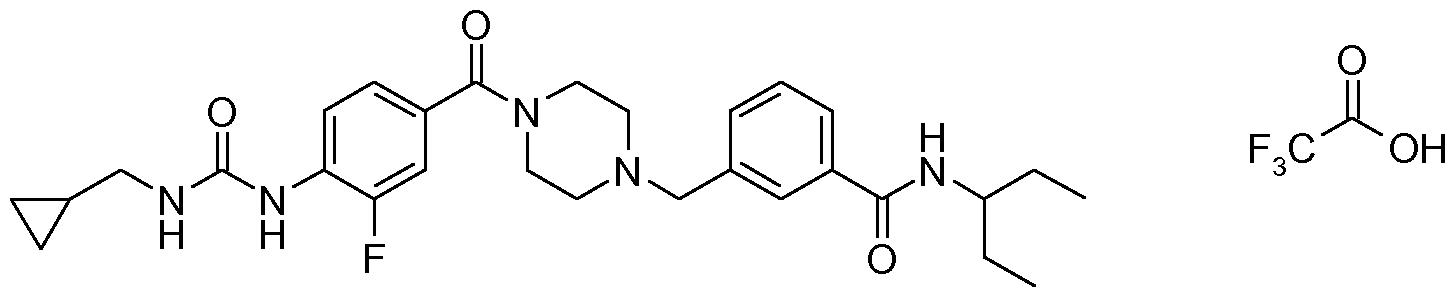
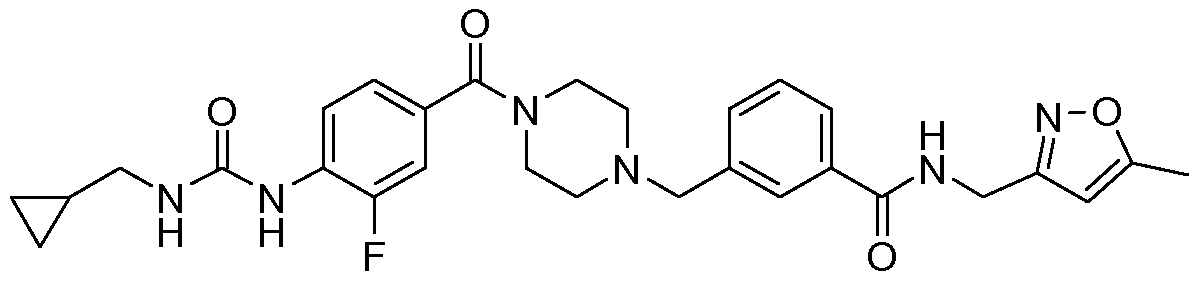
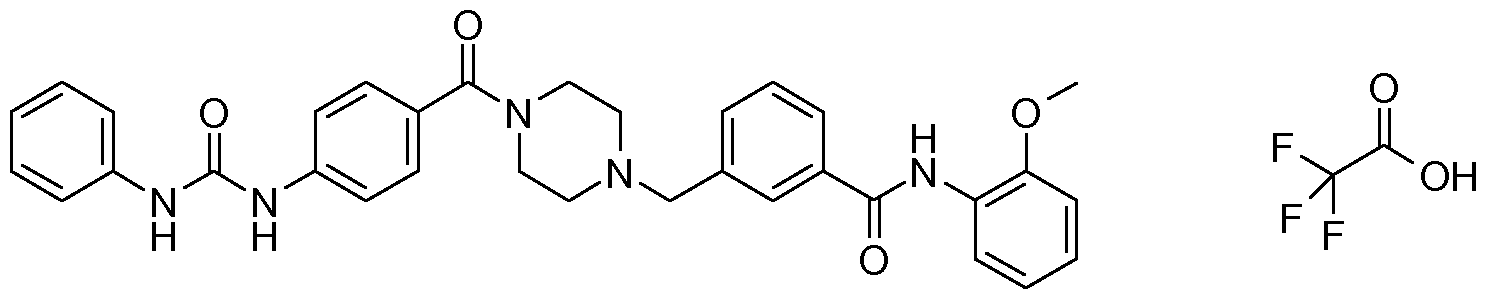
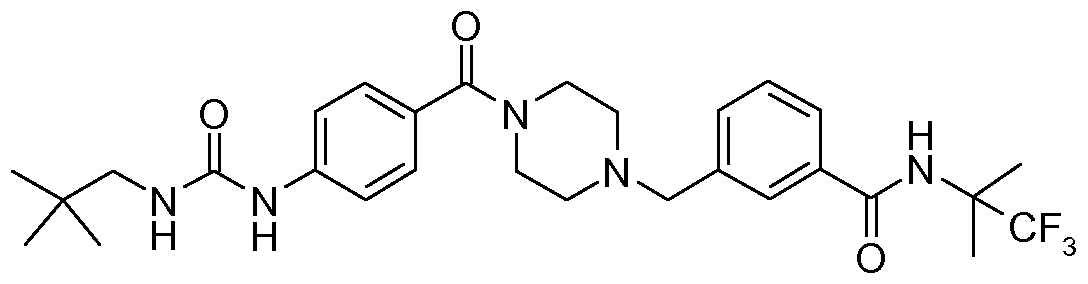
- N-benzyl,N'-arylcarbonylpiperazine derivatives of the invention are:
- N-benzyl,N'arylcarbonylpiperazine derivatives of the invention can be prepared using general synthetic methods known in the art of organic synthesis, for instance by using synthetic routes depicted in Scheme 1.
- a (homo)piperazine intermediate of Formula 2 wherein Y represents an amino protecting group, such as for example a tert-butyloxycarbonyl group (t-Boc), and R 3 has the meaning as previously defined, is alkylated with a benzamide derivative of Formula 3, wherein R 4 , R 5 , R 6 and R 7 have the previously defined meaning and wherein L represents a leaving group such as chloro or bromo, in a solvent e.g. dichloromethane or acetonitrile at room or elevated temperature using an organic base e.g. triethylamine or inorganic base e.g. potassium carbonate to give an intermediate N-benzyl(hom)piperazine derivative of Formula 5.
- Scheme 1
- the intermediates of Formula 5 can be obtained from a reductive amination reactiion between the (homo)piperazine intermediate of Formula 2 with the intermediate benzamide derivative of Formula 4, wherein R 4 , R 5 ,R 6 and R 7 have the meaning as previously defined, which reaction can be carried out in a solvent e.g. dichloromethane using a reducing agent e.g. sodium triacetoxyborohydride.
- a solvent e.g. dichloromethane
- a reducing agent e.g. sodium triacetoxyborohydride.
- Compounds of the invention according to Formula I wherein X is NR 8 , and wherein R 1 and R 8 have the meaning as previously defined, can be prepared from the intermediates of Formula 8 by reaction with 4-nitrophenylchoroformate or with (bis(trichloromethyl) carbonate (triphosgene) in a solvent e.g. dichloromethane at room or elevated temperature, followed by addition of the desired amine of Formula R 1 R 8 NH, wherin R 1 and R 8 have the meaning as previously defined, in the presence of a base e.g. triethylamine.
- a base e.g. triethylamine
- Compounds of the invention wherein X is NH can also be prepared by reacting a N- benzyl.N'-benzoylpiperazine intermediate of Formula 8 with an isocyanate of formula R 1 -NCO, wherein R 1 has the meaning as previously defined, in solvent e.g. dichloromethane.
- Compounds of Formula I wherein X is O can be prepared by reaction from an intermediate of Formula 8 with 4-nitrophenylchoroformate or triphosgene in a solvent e.g. dichloromethane at room or elevated temperature, followed by addition of an excess of an alcohol of formula R 1 -OH, wherein R 1 has the meaning as previously defined.
- Compounds of the invention wherein X is a bond can be prepared by reacting a N- benzyl.N'-benzoylpiperazine intermediate of Formula 8 with an acid chloride of formula R 1 CO 2 CI, wherein R 1 has the meaning as previously defined, in a solvent e.g. dichloromethane in the presence of an organic base e.g. triethylamine at room or elevated temperatures, or alternatively, by reaction with an acid derivative of formula R 1 COOH, wherein R 1 has the meaning as previously defined, in a solvent e.g. dichloromethane using a coupling reagent e.g. N-(3-dimethylaminopropyl)-N'- ethylcarbodiimide hydrochloride or 1-propanephosphonic acid cyclic anhydride and base e.g. triethylamine.
- a coupling reagent e.g. N-(3-dimethylaminopropyl)-N'
- the intermediate piperazine derivative of Formula 2, the benzamide derivatives of Formula 3 and Formula 4, as well as the 4-aminobenzoic acid derivatives of formula 7 are compounds that can be prepared using methods well known in the art from commercially available intermediates.
- ⁇ /-protecting group means a group commonly used for the protection of an amino group, like the alloxycarbonyl (Alloc) group, the tert- butyloxycarbonyl (Boc) group, the benzyloxycarbonyl (Z) group or the 9- fluorenylmethyloxycarbonyl (Fmoc) group. Removal of these and other protecting groups can take place in different ways, depending on the nature of those protecting groups. An overview of protecting groups and methods for their removal is given in T. W. Greene and P.G.M. Wuts, " Protective Groups in Organic Synthesis", 2 nd edition, 1991 , John Wiley & Sons, Inc.
- the N-benzyl,N'arylcarbonylpiperazine derivatives of Formula I and their salts may contain at least one centre of chirality, and exist therefore as stereoisomers, including enantiomers and diastereomers.
- the present invention includes the aforementioned stereoisomers within its scope and each of the individual R and S enantiomers of the compounds of formula I and their salts, substantially free, i.e. associated with less than 5%, preferably less than 2%, in particular less than 1% of the other enantiomer, and mixtures of such enantiomers in any proportions including the racemic mixtures containing substantially equal amounts of the two enantiomers.
- Pharmaceutically acceptable salts may be obtained by treating a free base of a compound of formula I with a mineral acid such as hydrochloric acid, hydrobromic acid, phosphoric acid and sulfuric acid, or an organic acid such as for example ascorbic acid, citric acid, tartaric acid, lactic acid, maleic acid, malonic acid, fumaric acid, glycolic acid, succinic acid, propionic acid, acetic acid, methane sulfonic acid, and the like.
- a mineral acid such as hydrochloric acid, hydrobromic acid, phosphoric acid and sulfuric acid
- an organic acid such as for example ascorbic acid, citric acid, tartaric acid, lactic acid, maleic acid, malonic acid, fumaric acid, glycolic acid, succinic acid, propionic acid, acetic acid, methane sulfonic acid, and the like.
- the compounds of the invention may exist in unsolvated as well as in solvated forms with pharmaceutically acceptable solvents such as water, ethanol and the like.
- the solvated forms are considered equivalent to the unsolvated forms for the purpose of the invention.
- compositions comprising a N- benzyl.N'arylcarbonylpiperazine derivative having the general Formula I, or a pharmaceutically acceptable salt thereof, in admixture with pharmaceutically acceptable auxiliaries, and optionally other therapeutic agents.
- acceptable means being compatible with the other ingredients of the composition and not deleterious to the recipients thereof.
- Compositions include e.g. those suitable for oral, sublingual, subcutaneous, intravenous, epidural, intrathecal, intramuscular, transdermal, pulmonary, local, or rectal administration, and the like, all in unit dosage forms for administration.
- the active ingredient may be presented as discrete units, such as tablets, capsules, powders, granulates, solutions, suspensions, and the like.
- the pharmaceutical composition of the invention may be presented in unit-dose or multi-dose containers, e.g. injection liquids in predetermined amounts, for example in sealed vials and ampoules, and may also be stored in a freeze dried (lyophilized) condition requiring only the addition of sterile liquid carrier, e.g. water, prior to use.
- sterile liquid carrier e.g. water
- the active agent may be compressed into solid dosage units, such as pills, tablets, or be processed into capsules, suppositories or patches.
- the active agent can be applied as a fluid composition, e.g. as an injection preparation, in the form of a solution, suspension, emulsion, or as a spray, e.g. a nasal spray.
- solid dosage units For making solid dosage units, the use of conventional additives such as fillers, colorants, polymeric binders and the like is contemplated. In general any pharmaceutically acceptable additive which does not interfere with the function of the active compounds can be used. Suitable carriers with which the active agent of the invention can be administered as solid compositions include lactose, starch, cellulose derivatives and the like, or mixtures thereof, used in suitable amounts. For parenteral administration, aqueous suspensions, isotonic saline solutions and sterile injectable solutions may be used, containing pharmaceutically acceptable dispersing agents and/or wetting agents, such as propylene glycol or butylene glycol.
- the invention further includes a pharmaceutical composition, as hereinbefore described, in combination with packaging material suitable for said composition, said packaging material including instructions for the use of the composition for the use as hereinbefore described.
- the N-benzyl,N'arylcarbonylpiperazine derivatives of the present invention were found to be modulators of LXR ⁇ , especially having agonistic activity thereon, and are as such useful in preventing and reducing the risk of atherosclerosis and related disorders associated with cholesterol and bile acids transport and metabolism, such as hypercholesterolemia (e.g. coronary heart disease), cholesterol gallstones, lipid storage diseases, diabetes and obesity.
- the compounds of the invention are potentially also useful in further indications such as:
- Ligand activation of LXR has been shown to inhibit a number of inflammatory pathways e.g. Interleukin1- ⁇ , lnterleukin-6, cyclooxygenase-2 and most recently shown to directly inhibit C-reactive protein expression. See Blaschke et al., Circ. Res. 99: 88-99. (2006).
- Compounds of the invention may have therapeutic utility in suppression of inflammation in inflammatory diseases such as contact dermititis (see Fowler et al., J. Invest.
- the LXR ligand T0901317 has been shown to inhibit vascular smooth muscle cell proliferation and neointima formation following balloon injury in vitro and in vivo. Compounds of the invention may therefore have therapeutic utility in proliferative vascular diseases. See Blaschke et al., Circ. Res. 95:110-123 (2004).
- the LXR agonist T0901317 delayed progression of tumours in an animal model of prostate cancer.
- Compounds of the invention may be potentially useful for treatment of prostate cancer. See Chuu et al.,Cancer.Res. 66:6482-6486 (2006).
- LXR agonists can reduce the deposition of ⁇ -amyloid in the brain.
- T0901317 has been shown to lower deposition of ⁇ - amyloid but also improve memory. See Riddell et al., MoI. Cell. Neurosci. 34: 621-628 (2007).
- the agonist derivatives of the present invention may therefore have therapeutic utility in neurodegenerative diseases such as Alzheimers disease.
- Combination therapies The compounds of the invention may be combined with another therapeutic agent that is useful in the treatment of other metabolic disorders such as; hypertension, hyperlipidaemias, dyslipidaemias, diabetes, chronic inflammatory disorders, obesity and in any condition where enhancement of reverse cholesterol transport and/or improvement of LDLHDL ratios would be of potential clinical benefit.
- therapies are: inhibitors of 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG CoA reductase) (e.g. atorvastatin, pravastatin, simvastatin, lovastatin, rosuvastatin and others), cholesterol absorption inhibitors (e.g. ezetimibe), bile sequestrants (e.g.
- cholestyramine microsomal triglyceride transfer protien (MTP) inhibitors
- MTP microsomal triglyceride transfer protien
- peroxisome proliferator-activated receptor modulators e.g.muraglitazar, rosiglitazone, fibrates and others
- cholesterol ester transfer protien inhibitiors e.g. nicotinic acid derivatives
- Acyl coenzyme A cholesterol acyl transferase (ACAT) inhibitors (e.g. eflucimibe), farnesoid X receptor modulators, therapies used for the treatment of metabollic syndrome or type 2 diabetes e.g. metformin.
- ACAT cholesterol acyl transferase
- Compounds of the invention may be combined with anti-inflammatory therapies (e.g. aspirin) and with treatments for neurodegenerative diseases (e.g Aricept®, Exelon®, Reminyl® and Ebixa®).
- the compounds of the invention may be administered for humans in a sufficient amount and for a sufficient amount of time to alleviate the symptoms.
- daily dosage levels for humans can be in the range of 0.001-50 mg per kg body weight, preferably in a daily dosage of 0.01-20 mg per kg body weight.
- HPLC purification is used within this experimental section and refers to High Performance Liquid Chromatography.
- Some examples of general methods that may be used to purify compounds are: acidic reverse phase HPLC (water / acetonitrile / 0.1 % trifluoroacetic acid) using a standard gradient of 5% acetonitrile / 95% water to 100% acetonitrile or basic reverse phase HPLC ( water / acetonitrile / 0.1 % ammonia solution) using a standard gradient of 10% acetonitrile / 90% water to 100% acetontrile.
- UV detection e.g. 254nM is used for the collection of fractions from HPLC. This description gives general methods and variations in types of equipment, columns, mobile phase, detection wavelength, solvent gradient and run time may also be used to purify compounds.
- Boc terf-butoxycarbonyl
- CDCI 3 chloroform-d
- CD 3 OD methanol-d4
- HPLC high performance liquid chromatography
- HOBt 1 H-benzo[d][1 ,2,3]triazol-1-ol
- HATU O-(7- azabenzotriazol-1-yl)-N,N,N,N-tetramethyl uronium hexafluorophosphate
- EDCI N-(3- dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride.
- N-tert-butyl-3-(chloromethyl)benzamide was placed under an argon atmosphere and treated with a solution of tert-butyl 1-piperazine-carboxylate (2.96g, 15.9mmol) and N-ethyl-N-isopropylpropan-2-amine (5.53ml_, 31.7mmol) in dimethyl sulfoxide (15ml_) at room temperature.
- the reaction mixture was stirred overnight, diluted with ethyl acetate (10OmL) and washed with saturated aqueous sodium chloride (x5).
- the organic phase was dried on magnesium sulfate, filtered and concentrated under vacuum to give a crude oil.
- N-tert-Butyl-3-(piperazin-1-ylmethyl)benzamide dihydrochloride (0.5Og, 1.44mmol) was suspended in stirred dichloromethane (2OmL) at O 0 C (ice-bath) under an argon atmosphere.
- N-ethyl-N-isopropylpropan-2-amine (1.OmL, 5.75mmol) was added slowly.
- the resulting solution was treated with a suspension of 4-nitrobenzoyl chloride (0.267g, 1.44mmol) in dichloromethane (1 OmL).
- the ice-bath was removed and stirring continued for 1 hour.
- the solution was diluted with dichloromethane (7OmL) and washed with water (x4).
- N-tert-Butyl-3-((4-(4-nitrobenzoyl)piperazin-1-yl)methyl)benzamide (0.56g, 1.31 mmol) was dissolved in methanol (1OmL) at room temperature.
- Raney 2800 nickel ⁇ 1 ml_ of an aqueous suspension
- a balloon of hydrogen was attached to the flask via a three-way stopcock and hydrogen was allowed to slowly flow through the system for 1 minute.
- the flask was sealed under 1 atmosphere of hydrogen and vigorous stirring continued for 1 hour.
- the balloon was removed and the flask was flushed with argon.
- the suspension was gravity-filtered over fluted paper and washed with methanol.
- N-tert-Butyl-3-(chloromethyl)benzamide 14.71g, 65.2mmol
- tert-butyl piperazine-1-carboxylate (12.14g, 65.2mmol)
- sodium iodide (1.95g, 13.0mmol
- triethylamine (27.3mL, 195.5mmol)
- tetrahydrofuran 125mL
- the reaction was then concentrated under vacuum and the residue dissolved in ethyl acetate.
- the organic mixture was washed consecutively with saturated aqueous sodium hydrogen carbonate solution, water (x2) and brine then concentrated under vacuum.
- Step 1 To a mixture of ethyl 1-cyanocyclopropanecarboxylate (35.9mmol, 5g), dimethoxyethane (10OmL) and methanol (1OmL) was added sodium borohydride (287mmol, 10.87g) slowly and the mixture stirred at room temperature for 18 hours. The solution was diluted with saturated sodium hydrogen carbonate slowly and then extracted with 10% methanol / dichloromethane (x3). The organic layers were combined, dried over sodium sulphate and concentrated under vacuum to give the intermediate 1-(hydroxymethyl)cyclopropanecarbonitrile (2.36g).
- Step 2 A stirred mixture of 1-(hydroxymethyl)cyclopropanecarbonitrile (24.30mmol, 2.36g) in dichloromethane (3OmL) was treated with triethylamine (48.6mmol, 6.83mL, 4.92g) and portionwise with methanesulfonyl chloride (31.6mmol, 2.445mL, 3.62g) keeping the reaction mixture at O 0 C.
- Step 3 A stirred mixture of (i-cyanocyclopropyl)methyl methanesulfonate (21.52mmol, 3.77g) and sodium azide (43.0mmol, 2.8Og) in N,N-diemethyl formamide (4OmL) was heated to 120 0 C for -18 hours. The mixture was allowed to cool and was diluted with water and ethyl acetate. The organic layer was separated, dried over sodium sulphate and concentrated under reduced pressure to give an oil.
- N-tert-Butyl-3-((4-(2-chloro-4-nitrobenzoyl)piperazin-1-yl)methyl)benzamide (1.9483g, 4.2mmol) was added to a suspension of reduced iron powder (2.3623g, 42mmol) and 1 M hydrochloric acid (6ml_) in isopropanol. The reaction was stirred at room temperature for 2 hours. The reaction was concentrated under reduced pressure and the remaining residue was taken up in dichloromethane and washed with water. The organic phase was dried over magnesium sulphate and concentrated under vacuum to afford the title compound (1.57g).
- Stepi To a stirred solution of N-tert-butyl-3-(piperazin-1-ylmethyl)benzamide (3.99mmol, 1.1g), 4-nitro-2-(trifluoromethyl)benzoic acid (3.83mmol, 0.9g) and triethylamine (28.7mmol, 4mL, 2.9Og) in dichloromethane (2OmL) was added 1 - propanephosphonic acid cyclic anhydride (8.94mmol, 5.3mL, 5.69g, 50% solution in ethyl acetate) dropwise. After 1 hour stirring, ethyl acetate was added and the organic mixture was washed with saturated sodium hydrogen carbonate (x2), water (x2) and finally brine.
- Step 2 To a stirred mixture of N-tert-butyl-3-((4-(4-nitro-2-(trifluoromethyl)benzoyl)- piperazin-1-yl)methyl)benzamide (2.234mmol, 1.1g) and iron powder (22.38mmol, 1.25g) in isopropanol (1OmL) was added 1 M hydrochloric acid (3.00mmol, 3mL). The reaction was allowed to stir for 12 hours and was then concentrated under vacuum. Dichloromethane was added and the organic mixture was washed with water, dried with sodium sulfate, filtered and concentrated under vacuum to yield the title compound (0.86g). MS (ESI) m/z 463.5 [M+H] +
- Methyl 6-(3-phenylureido)nicotinate (200mg, 0.43mmol) was dissolved in methanol (2ml_) at room temperature. Lithium hydroxide (100mg, 4.17mmol) was added, followed by water (10.5ml_). The mixture was heated to 5O 0 C and stirred overnight. The mixture was brought to pH 4-5 by addition of concentrated hydrochloric acid. The solid that had formed was then filtered and dried under vacuum to give the title compound (69mg). MS (ESI) m/z 258.1 [M+H] +
- N-tert-butyl-3-(piperazin-1-ylmethyl)benzamide dihydrochloride 16mg, 46 ⁇ mol
- N-ethyl-N-isopropylpropan-2-amine (20.4 ⁇ l, 117 ⁇ mol)
- N- diemthylformamide (3ml_)
- 6-(3-phenylureido)nicotinic acid 10mg, 39 ⁇ mol
- O-(7-azabenzotriazol-1-yl)-N,N,N,N-tetramethyl uronium hexafluorophosphate 18mg, 47.3 ⁇ mol, HATU
- tert-Butyl 4-(3-(tert-butylcarbamoyl)benzyl)-1 ,4-diazepane-1-carboxylate (0.53g, 1.36mmol) was treated with an ethanolic solution of hydrochloric acid (3ml_, 14.5 wt% hydrochloric acid in ethanol). The resultant mixture was stirred at room temperature for 2 hours. The mixture was concentrated under vacuum, azeotroped with dichloromethane (x2) and dried under vacuum to afford the title compound (0.394g) as a white foamy solid.
- N-tert-butyl-3-(((2S,6R)-2,6-dimethylpiperazin-1- yl)methyl)benzamide hydrochloride (0.025g, 0.083mmol) in N,N-dimethylformamide (2ml_) was added O-(7-azabenzotriazol-1-yl)-N,N,N,N-tetramethyl uronium hexafluorophosphate (0.032g, 0.083mmol, HATU), 4-(3-cyclobutylureido)benzoic acid (0.019g, 0.083mmol) and N-ethyl-N-isopropylpropan-2-amine (0.036mL, 0.21 mmol).
- Step 2 To a stirred mixture of tert-butyl 4-(4-amino-3-fluorobenzoyl)-2,2- dimethylpiperazine-1-carboxylate (0.569mmol, 200mg) in dichloromethane (2ml_) was added trifluoroacetic acid (10. lOmmol, 0.75ml_, 1151 mg). After 1 hour stirring, the reaction was concentrated under reduced pressure. The residue was taken up in dichloromethane/methanol and loaded on to strong cation exchange column (5g) which was washed with dichloromethane/methanol. The product was eluted from the column with 2M ammonia in methanol. The organic phase was concentrated under vacuum to yield the title compound (131mg).
- Step 1 A solution of methanesulfonyl chloride (165mg, 1.44mmol) in dichloromethane (2.OmL) was added to a stirred solution of N-tert-butyl-3-(1-hydroxy-2- methylpropyl)benzamide (300mg, 1.20mmol) in dichloromethane (8.OmL). After 1.5 hours stirring, the mixture was partitioned between dichloromethane and aqueous sodium hydrogen carbonate solution.
- Step 2 A solution of 1-(3-(tert-butylcarbamoyl)phenyl)propyl methanesulfonate, tert-butyl piperazine-1 -carboxylate (447mg, 2.40mmol) and N-ethyl-N-isopropylpropan-2 -amine (0.623mL, 3.60mmol) in N,N-dimethylformamide (8.OmL) was stirred at 70 0 C for 4 hours, then subjected to microwave irradiation at 160°C for 10 minutes.
- Methyl 4-methoxy-3-((4-(4-(3-phenylureido)benzoyl)piperazin-1-yl)methyl)benzoate (216mg, 0.43mmol) was dissolved in methanol (2OmL) at room temperature. Lithium hydroxide (51.5mg, 2.15mmol) was added, followed by water (1 mL). The mixture was stirred at 45 0 C overnight and concentrated under vacuum. The residue was dissolved in water (15mL) and brought to pH ⁇ 3 with 1 M aqueous hydrochloric acid. The mixture was extracted with ethyl acetate (x2) and with dichloromethane (x2).
- N-tert-butyl-2-fluoro-3-(piperazin-1-ylmethyl)benzamide (0.648mmol, 190mg), 4-amino- benzoic acid (0.648mmol, 89mg) and triethylamine (2.59mmol, 0.361 mL, 262mg) were dissolved in dichloromethane (6.476ml_).
- 1 -Propanephosphonic acid cyclic anhydride (1.295mmol, 0.767mL, 824mg, 50% solution in ethyl acetate) was added and the reaction mixture was stirred for 2 hours.
- tert-Butyl 4-(4-(3-cyclobutylureido)benzoyl)piperazine-1-carboxylate (48.7mmol, 19.6g) was dissolved in a mixture of dichloromethane (125ml_) and acetonitrile (1OmL). After complete dissolution, trifluoroacetic acid (269mmol, 2OmL, 30.7g) was added over 10 minutes. The reaction was stirred until no trace of starting material remained. The reaction was neutralised with sodium hydrogen carbonate, filtered and concentrated under vacuum to generate a white solid. The desired product was isolated by continuous extraction of this material using soxhlet apparatus with dichloromethane as elutant.
- N-(3,3-Difluorocyclobutyl)-3-(piperazin-1-ylmethyl)benzamide (5.14mmol, 1.59g)
- 4-amino-3-fluorobenzoic acid (5.14mmol, 0.797g)
- triethylamine (15.42mmol, 2.143mL, 1.56Og) were stirred in dichloromethane (3OmL) at room temperature.
- 1 - Propanephosphonic acid cyclic anhydride (7.71 mmol, 4.59mL, 4.91g, 50% solution in ethyl acetate) was added dropwise and the reaction stirred for 1 hour.
- the reaction mixture was concentrated under vacuum and the residue taken up in ethyl acetate.
- This assay is used to evaluate the potency of compounds in their ability to compete with the binding of the agonist radioligand [ 3 H] T0901317.
- This assay utilises purified ligand binding domain (LBD) of Liver X Receptor alpha (LXR ⁇ ) fused to glutathione-S-transferase (GST) tagged protein (LXR ⁇ -LB D-GST) and scintillation proximity assay (SpA) technology to determine binding affinities (pKi) of compounds at the ligand binding domain (LBD) of the human nuclear hormone receptor LXR ⁇ . Protocol for expression of human LXR ⁇ -LBD-GST:
- LBD ligand binding domain of Liver X Receptor alpha
- LXR ⁇ from the pGEX-4T-1 plasmid in E. CoIi resulted in the production of a recombinant glutathione-S-transferase (GST) LXR ⁇ -LBD fusion protein.
- GST glutathione-S-transferase
- E.coli containing the plasmid were propagated, induced, and harvested by centrifugation. The bacterial pellet was resuspended in lysis buffer (5OmM tris(hydroxy- methyl)aminomethane(TRIS)-pH 8.0, 10OmM sodium chloride (NaCI), 1mM ethylene- diaminetetraacetic acid (EDTA), one tablet of proteinase inhibitor cocktail complete/ EDTA free (Roche) (per 50ml of buffer).
- lysis buffer 5OmM tris(hydroxy- methyl)aminomethane(TRIS)-pH 8.0, 10OmM sodium chloride (NaCI), 1mM
- the mixture was sonicated on ice with a Branson sonifier.
- the suspension was centrifuged and dithiothreitol (DTT) added to the supernatant to obtain a final concentration of 25mM.
- DTT dithiothreitol
- Recombinant human LXR ⁇ -LBD- GST protein was purified from the resulting supernatant by affinity chromatography on Glutathione-Sepharose Fast flow (Amersham), and receptor was eluted with buffer containing glutathione (5OmM tris pH 8.0, 2mM DTT, 1OmM glutathione).
- T0901317 (50 Ci/mmol), + test compound was added and incubated at 15 ° C on a plate shaker for 2hr. After incubation, the assay plates are read on a Packard TopCount.
- the pKi value for T0901317 in this assay is: pKi 7.8 ⁇ 0.2.
- T0901317 at a concentration of 1 ⁇ M was used as the maximum binding control. Active compounds show pKi values > 5.5 and preferred compounds show pKi values of >7 in this assay.
- Intracellular agonist activity was measured in vitro using recombinant Chinese hamster ovary K1 (CHO. K1 ) cells stably expressing human Estrogen receptor ⁇ / Liver X receptor ⁇ chimeric receptor protein from a eukaryotic expression construct and a natural estrogen responsive element (ERE)-containing luciferase reporter construct.
- the ER ⁇ /LXR ⁇ chimeric receptor protein contains the human LXR ⁇ receptor ligand binding domain (LBD) fused to the human ERa receptor DNA binding domain (DBD).
- LBD human LXR ⁇ receptor ligand binding domain
- DBD human ERa receptor DNA binding domain
- the ERa DBD can induce ERE-mediated luciferase expression via a natural estrogen responsive element present in the rat oxytocin promotor luciferase construct.
- a LXR agonist-induced luciferase assay was generated using T0901317 as the agonist control.
- Expression constructs were prepared by inserting the ligand binding domain (LBD) of human LXR ⁇ of human LXR ⁇ cDNA adjacent to the human ERa transcription factor DNA binding domain (DBD) to create pNGVLER ⁇ DBD-LXR ⁇ LBD.
- the pNGV1 mammalian expression construct (EMBL nucleotide sequence database file ASPNGV1 , ace. # X99274) carries a selection marker for Neomycin (G418).
- the ERa responsive rat oxytocin (RO) was used to generate the promoter construct, pROLUC, and contains several copies of the ERa response element (ERE) placed adjacent to the luciferase reporter gene.
- the promoter construct was based on the rat oxytocin gene regulatory region (position -363/+16) as a Hindi I/Mbol restriction enzyme fragment linked to the firefly luciferase encoding sequence. See Ivell and Richter., Proc Natl Acad Sci U S A. 7: 2006-2010 (1984).
- a stable CHO. K1 cell line expressing both pNGVLER ⁇ DBD-LXR ⁇ LBD and pROLUC was generated following transfection and selection of positive expressing clones using Neomycin.
- the best cell line (CHO. K1 LXR ⁇ ) was selected on the basis of agonist window in response to 3 ⁇ M T0901317 and stability of response up to 20 passages. Assay of agonist activity of test compounds in LXR ⁇ transactivation assay.
- K1 LXR ⁇ cells were seeded at a density of 25000 cells/well in 96 well plates in 200 ⁇ l of Dulbecco's Modified Eagle Medium (phenol red free) containing 5% charcoal treated bovine calf serum at 37 ° C in a humidified atmosphere of 5% CO 2 . After 6 h, compounds were characterised by incubation with cells for 16 h across a concentration range. T0901317 at a concentration of 3 ⁇ M was used as the maximum agonist control. Luciferase activity was determined using a Luciferase assay kit (Perkin Elmer) by addition of lysis buffer to each well and light emission measured using a Packard Topcount reader.
- Luciferase activity was determined using a Luciferase assay kit (Perkin Elmer) by addition of lysis buffer to each well and light emission measured using a Packard Topcount reader.
- Agonist activities of test compounds were compared against the maximum agonist control.
- Preferred compounds of the invention were shown to have LXR ⁇ agonist activity using this assay protocol.
Abstract
Description





































































































































































Claims

Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT08787281T ATE495161T1 (en) | 2007-08-20 | 2008-08-18 | N-BENZYL-N'-ARYLCARBONYLPIPERAZINE DERIVATIVES AS LXR MODULATORS |
| CA2694604A CA2694604C (en) | 2007-08-20 | 2008-08-18 | N-benzyl, n' -arylcarbonylpiperazine derivatives as lxr modulators |
| JP2010521405A JP5444547B2 (en) | 2007-08-20 | 2008-08-18 | N-benzyl, N'-arylcarbonylpiperazine derivatives as LXR modulators |
| MX2010001847A MX2010001847A (en) | 2007-08-20 | 2008-08-18 | N-benzyl, n' -arylcarbonylpiperazine derivatives as lxr modulators. |
| DE602008004562T DE602008004562D1 (en) | 2007-08-20 | 2008-08-18 | N-BENZYL-N'-ARYLCARBONYLPIPERAZINE DERIVATIVES AS LXR MODULATORS |
| EP08787281A EP2190827B1 (en) | 2007-08-20 | 2008-08-18 | N-benzyl-n'-arylcarbonylpiperazine derivatives as lxr modulators |
| CN200880101573.6A CN101848899B (en) | 2007-08-20 | 2008-08-18 | As the N-benzyl of LXR conditioning agent, N '-aryl carbonyl bridged piperazine derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07114602 | 2007-08-20 | ||
| EP07114602.1 | 2007-08-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009024550A1 true WO2009024550A1 (en) | 2009-02-26 |
Family
ID=40032504
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/060788 WO2009024550A1 (en) | 2007-08-20 | 2008-08-18 | N-benzyl, n' -arylcarbonylpiperazine derivatives as lxr modulators |
Country Status (12)
| Country | Link |
|---|---|
| EP (1) | EP2190827B1 (en) |
| JP (1) | JP5444547B2 (en) |
| CN (1) | CN101848899B (en) |
| AR (1) | AR067975A1 (en) |
| AT (1) | ATE495161T1 (en) |
| CA (1) | CA2694604C (en) |
| CL (1) | CL2008002429A1 (en) |
| DE (1) | DE602008004562D1 (en) |
| MX (1) | MX2010001847A (en) |
| PE (1) | PE20091241A1 (en) |
| TW (1) | TW200922582A (en) |
| WO (1) | WO2009024550A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010025179A1 (en) * | 2008-08-28 | 2010-03-04 | N.V. Organon | 1-(4-ureidobenzoyl)piperazine derivatives |
| WO2011023773A1 (en) | 2009-08-28 | 2011-03-03 | Novartis Ag | Compounds and compositions as protein kinase inhibitors |
| WO2012033353A2 (en) | 2010-09-07 | 2012-03-15 | 서울대학교 산학협력단 | Sesterterpene compounds and use thereof |
| US8314091B2 (en) | 2007-08-20 | 2012-11-20 | Msd Oss B.V. | N-benzyl,N'-arylcarbonylpiperazine derivatives |
| CN103159687A (en) * | 2013-04-12 | 2013-06-19 | 苏州科捷生物医药有限公司 | Synthesis method of 2-aminopyrazinyl-5-formic acid |
| WO2014028461A2 (en) | 2012-08-13 | 2014-02-20 | The Rockefeller University | Treatment and diagnosis of melanoma |
| JP5774984B2 (en) * | 2009-04-29 | 2015-09-09 | 興和株式会社 | Carbinol compounds having a heterocyclic linker |
| WO2017123568A2 (en) | 2016-01-11 | 2017-07-20 | The Rockefeller University | Methods for the treatment of myeloid derived suppressor cells related disorders |
| US11174220B2 (en) | 2019-12-13 | 2021-11-16 | Inspirna, Inc. | Metal salts and uses thereof |
| US11214536B2 (en) | 2017-11-21 | 2022-01-04 | Inspirna, Inc. | Polymorphs and uses thereof |
| WO2023240376A1 (en) * | 2022-06-12 | 2023-12-21 | 中国医学科学院药物研究所 | Use of intestinal epithelial cell nuclear receptor repressor ncor as target in drug screening |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0742208A1 (en) * | 1995-05-05 | 1996-11-13 | Grelan Pharmaceutical Co., Ltd. | 2-Ureido-benzamide derivatives |
| WO2003015774A1 (en) * | 2001-08-17 | 2003-02-27 | Astrazeneca Ab | Compounds effecting glucokinase |
| WO2007051920A1 (en) * | 2005-11-02 | 2007-05-10 | Clinigenetics | Methanone compounds and the use thereof in pharmaceutical compositions, in particular for treating cardiovascular diseases |
| US20070142369A1 (en) * | 2005-12-21 | 2007-06-21 | Margaret Van Heek | Combination of an H3 antagonist/inverse agonist and an appetite suppressant |
| WO2007092065A2 (en) * | 2005-11-14 | 2007-08-16 | Irm Llc | Compounds and compositions as lxr modulators |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2448080A1 (en) * | 2001-05-22 | 2002-11-28 | Neurogen Corporation | Melanin concentrating hormone receptor ligands: substituted 1-benzyl-4-aryl piperazine analogues |
-
2008
- 2008-08-04 TW TW097129557A patent/TW200922582A/en unknown
- 2008-08-18 CN CN200880101573.6A patent/CN101848899B/en not_active Expired - Fee Related
- 2008-08-18 JP JP2010521405A patent/JP5444547B2/en not_active Expired - Fee Related
- 2008-08-18 MX MX2010001847A patent/MX2010001847A/en active IP Right Grant
- 2008-08-18 CL CL2008002429A patent/CL2008002429A1/en unknown
- 2008-08-18 EP EP08787281A patent/EP2190827B1/en active Active
- 2008-08-18 AT AT08787281T patent/ATE495161T1/en not_active IP Right Cessation
- 2008-08-18 CA CA2694604A patent/CA2694604C/en not_active Expired - Fee Related
- 2008-08-18 PE PE2008001401A patent/PE20091241A1/en not_active Application Discontinuation
- 2008-08-18 DE DE602008004562T patent/DE602008004562D1/en active Active
- 2008-08-18 WO PCT/EP2008/060788 patent/WO2009024550A1/en active Application Filing
- 2008-08-19 AR ARP080103603A patent/AR067975A1/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0742208A1 (en) * | 1995-05-05 | 1996-11-13 | Grelan Pharmaceutical Co., Ltd. | 2-Ureido-benzamide derivatives |
| WO2003015774A1 (en) * | 2001-08-17 | 2003-02-27 | Astrazeneca Ab | Compounds effecting glucokinase |
| WO2007051920A1 (en) * | 2005-11-02 | 2007-05-10 | Clinigenetics | Methanone compounds and the use thereof in pharmaceutical compositions, in particular for treating cardiovascular diseases |
| WO2007092065A2 (en) * | 2005-11-14 | 2007-08-16 | Irm Llc | Compounds and compositions as lxr modulators |
| US20070142369A1 (en) * | 2005-12-21 | 2007-06-21 | Margaret Van Heek | Combination of an H3 antagonist/inverse agonist and an appetite suppressant |
Non-Patent Citations (1)
| Title |
|---|
| D. J. BENNETT ET AL.: "An update on non-steroidal liver X receptor agonists and their potential use in the treatment of atherosclerosis", EXPERT. OPIN. THER. PATENTS, vol. 16, no. 12, 2006, pages 1673 - 1699, XP002500308 * |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8314091B2 (en) | 2007-08-20 | 2012-11-20 | Msd Oss B.V. | N-benzyl,N'-arylcarbonylpiperazine derivatives |
| WO2010025179A1 (en) * | 2008-08-28 | 2010-03-04 | N.V. Organon | 1-(4-ureidobenzoyl)piperazine derivatives |
| AU2009285823B2 (en) * | 2008-08-28 | 2014-04-17 | Merck Sharp & Dohme B.V. | 1-(4-ureidobenzoyl)piperazine derivatives |
| US8759352B2 (en) | 2008-08-28 | 2014-06-24 | Merck Sharp & Dohme B.V. | 1-(4-ureidobenzoyl)piperazine derivatives |
| JP5774984B2 (en) * | 2009-04-29 | 2015-09-09 | 興和株式会社 | Carbinol compounds having a heterocyclic linker |
| WO2011023773A1 (en) | 2009-08-28 | 2011-03-03 | Novartis Ag | Compounds and compositions as protein kinase inhibitors |
| WO2012033353A2 (en) | 2010-09-07 | 2012-03-15 | 서울대학교 산학협력단 | Sesterterpene compounds and use thereof |
| EP3626309A1 (en) | 2012-08-13 | 2020-03-25 | The Rockefeller University | Lxrbeta agonist for the treatment of cancer |
| WO2014028461A2 (en) | 2012-08-13 | 2014-02-20 | The Rockefeller University | Treatment and diagnosis of melanoma |
| EP4218935A1 (en) | 2012-08-13 | 2023-08-02 | The Rockefeller University | Lxrbeta agonist for the treatment of cancer |
| CN103159687A (en) * | 2013-04-12 | 2013-06-19 | 苏州科捷生物医药有限公司 | Synthesis method of 2-aminopyrazinyl-5-formic acid |
| WO2017123568A2 (en) | 2016-01-11 | 2017-07-20 | The Rockefeller University | Methods for the treatment of myeloid derived suppressor cells related disorders |
| US11214536B2 (en) | 2017-11-21 | 2022-01-04 | Inspirna, Inc. | Polymorphs and uses thereof |
| US11174220B2 (en) | 2019-12-13 | 2021-11-16 | Inspirna, Inc. | Metal salts and uses thereof |
| US11459292B2 (en) | 2019-12-13 | 2022-10-04 | Inspirna, Inc. | Metal salts and uses thereof |
| US11878956B2 (en) | 2019-12-13 | 2024-01-23 | Inspirna, Inc. | Metal salts and uses thereof |
| WO2023240376A1 (en) * | 2022-06-12 | 2023-12-21 | 中国医学科学院药物研究所 | Use of intestinal epithelial cell nuclear receptor repressor ncor as target in drug screening |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101848899A (en) | 2010-09-29 |
| CN101848899B (en) | 2016-03-02 |
| JP2010536821A (en) | 2010-12-02 |
| CA2694604A1 (en) | 2009-02-26 |
| TW200922582A (en) | 2009-06-01 |
| EP2190827A1 (en) | 2010-06-02 |
| ATE495161T1 (en) | 2011-01-15 |
| DE602008004562D1 (en) | 2011-02-24 |
| CL2008002429A1 (en) | 2009-01-02 |
| MX2010001847A (en) | 2010-03-11 |
| EP2190827B1 (en) | 2011-01-12 |
| AR067975A1 (en) | 2009-10-28 |
| JP5444547B2 (en) | 2014-03-19 |
| PE20091241A1 (en) | 2009-07-16 |
| CA2694604C (en) | 2016-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2694604C (en) | N-benzyl, n' -arylcarbonylpiperazine derivatives as lxr modulators | |
| JP5756747B2 (en) | N- (1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl) benzyl-N'-arylcarbonylpiperazine derivative | |
| KR101321728B1 (en) | Adamantyl-acetamide derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme | |
| CA2733661C (en) | 1-(4-ureidobenzoyl)piperazine derivatives | |
| US8183239B2 (en) | Substituted piperazines and piperidines as modulators of the neuropeptide Y2 receptor | |
| ZA200103680B (en) | Benzamide derivatives and their use as APOB-100 secretion inhibitors. | |
| AU2007330575B2 (en) | New benzamide derivatives as bradykinin antagonists | |
| WO2011051282A1 (en) | (1,1, 1,3, 3, 3-hexafluoro-2-hydroxypropan-2-yl) phenyl derivatives, pharmaceutical compositions thereof and their use for the treatment of atherosclerosis | |
| US20040009988A1 (en) | Bioisosteric bensamide derivatives and their use as apob-100 secretion inhibitors | |
| US8372841B2 (en) | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme | |
| US8314091B2 (en) | N-benzyl,N'-arylcarbonylpiperazine derivatives | |
| AU2011202889A1 (en) | Adamantyl-acetamide derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880101573.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08787281 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008787281 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2694604 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/001847 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2010521405 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |