WO2009023402A2 - Heterocyclic compounds and uses as anticancer agents - Google Patents
Heterocyclic compounds and uses as anticancer agents Download PDFInfo
- Publication number
- WO2009023402A2 WO2009023402A2 PCT/US2008/070348 US2008070348W WO2009023402A2 WO 2009023402 A2 WO2009023402 A2 WO 2009023402A2 US 2008070348 W US2008070348 W US 2008070348W WO 2009023402 A2 WO2009023402 A2 WO 2009023402A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- ring
- alkyl
- pharmaceutically acceptable
- acceptable salt
- Prior art date
Links
- 0 CC(*)C(C)NC(C)C(*)*=C Chemical compound CC(*)C(C)NC(C)C(*)*=C 0.000 description 7
- WWLUBUQGCQZFBQ-UHFFFAOYSA-N CC1N=C2N=CC=CN2C1c1c[s]c(Nc(cc2)ccc2O)n1 Chemical compound CC1N=C2N=CC=CN2C1c1c[s]c(Nc(cc2)ccc2O)n1 WWLUBUQGCQZFBQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the field of the invention is heterocyclic compounds, pharmaceutical compositions and methods, and especially as they relate to compositions and methods for the treatment and prevention of cancer and related diseases.
- Cancer including over 200 diseases, is the second biggest cause of death in the developed countries. Therefore, cancer remains one of the most important unmet medical challenges to civilization.
- a number of options for treating tumors are available, including surgery, radiation, chemotherapy, or any combination of these approaches.
- chemotherapy is widely used for all types of cancers, in particular for those inoperable or with metastatic characteristics.
- chemotherapeutic compounds being used in clinics for improvement of survival rates of different human cancers, chemotherapy is generally not curative, but only delays disease progression.
- tumors and their metastasis become refractory to chemotherapy, as the tumor cells develop the ability of multi-drug resistance. In some cases, the tumors are inherently resistant to some classes of chemotherapeutic agents.
- chemotherapeutic agents In other cases, the acquired resistance against chemotherapeutic agents is developed during the chemotherapeutic intervention. Thus, there remain significant limitations to the efficacy of available chemotherapeutic compounds in treating different classes of tumors. Furthermore, many cytotoxic and cytostatic agents used for chemotherapeutic treatment of tumors have severe side effects, resulting in termination of the chemotherapy in some patients. Thus, there remains a need for new chemotherapeutic agents.
- the present invention is directed to various classes of heteroaryl-substituted bicyclic heteroaryl derivatives, pharmaceutical compositions, and methods of using thereof.
- Exemplary embodiments have a thiazole or oxazole or imidazole heterocyclic moiety that is further substituted with an optionally substituted aryl amino, arylthio, aryloxy, heterocyclic amino, heterocyclic thio, or heterocyclic oxy group.
- the compounds as described herein exhibit anti-tumor, anticancer, anti-inflammation, anti-infectious, and antiproliferation activity.
- the present invention also relates to the methods of making and formulating the described compounds.
- the present invention also relates to pharmaceutical compositions containing such compounds that may be used for treating tumors, cancer, infective and/or proliferative diseases.
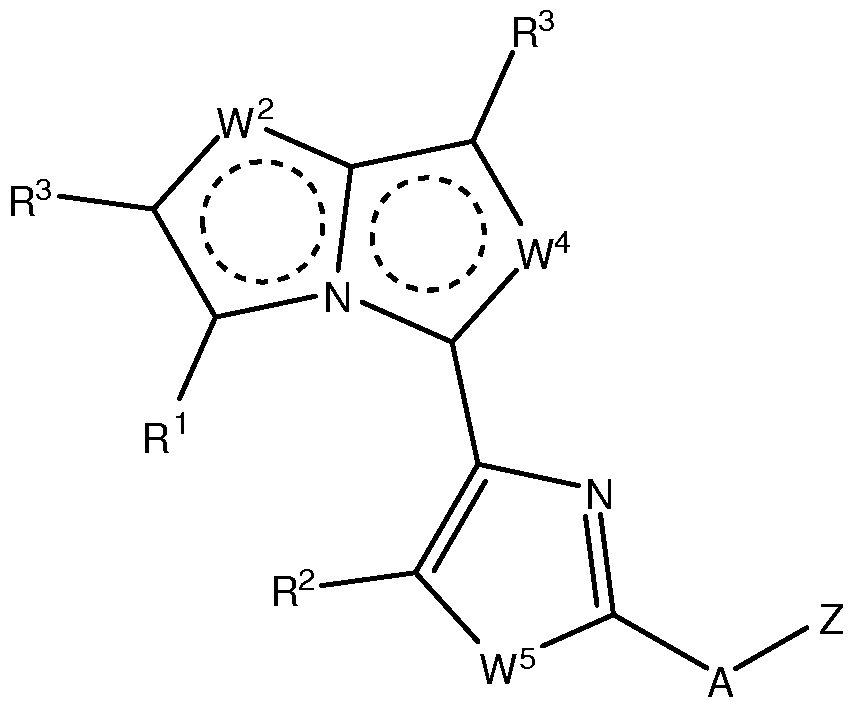
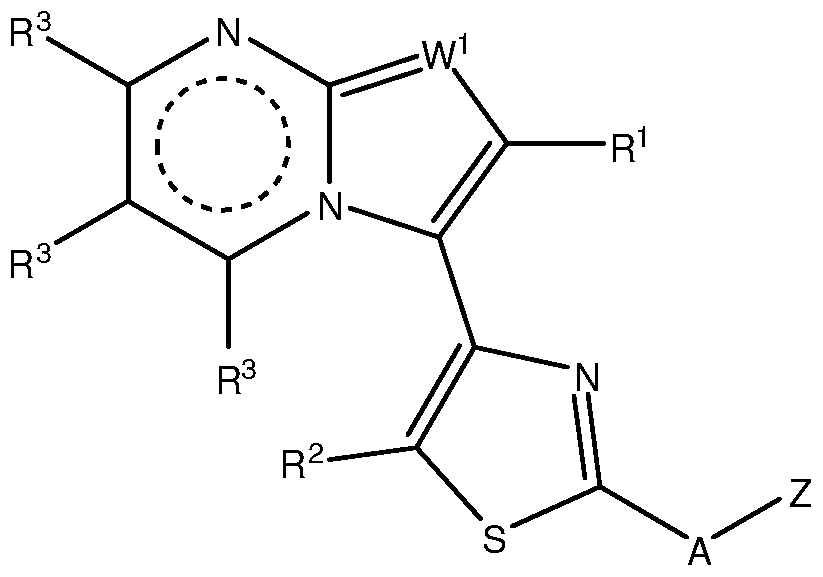
- contemplated heterocyclic compounds will generally have a structure according to Formula I and II:
- each W 1 , W 2 , W 3 , and W 4 is independently N, S, O, or CR 3 ;
- W 5 is S or O or CR 3 ;
- Z is sometimes substituted with up to three substituents.
- not more than one of W 2 , W 3 , W 4 and W 5 is a bond, and at least one of W 2 , W 3 , W 4 and W 5 is not CR 3 ; and not more than two of W 2 , W 3 , W 4 and W 5 represent N, but at least one of W 2 , W 3 , W 4 and W 5 is CR 3 .
- Z is not unsubstituted imidazopyridine, and when A is NAc, Z is not methoxy- substituted pyridinyl.
- W 1 is N
- R 1 is Me and A is NH
- Z is not -CH 2 -(2-furanyl), unsubstituted phenyl, unsubstituted benzyl, or phenyl substituted with -NO 2 , Br, - OH, -NHAc, SO 2 NH-heteroaryl, or COOH.
- R 1 is preferably not H when W 2 is CH.
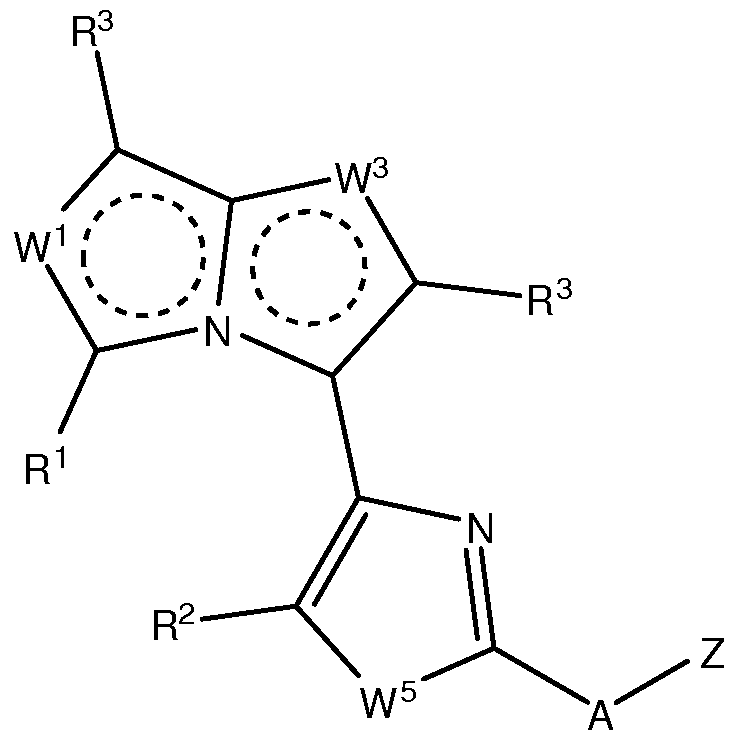
- the invention provides compounds having formula III,
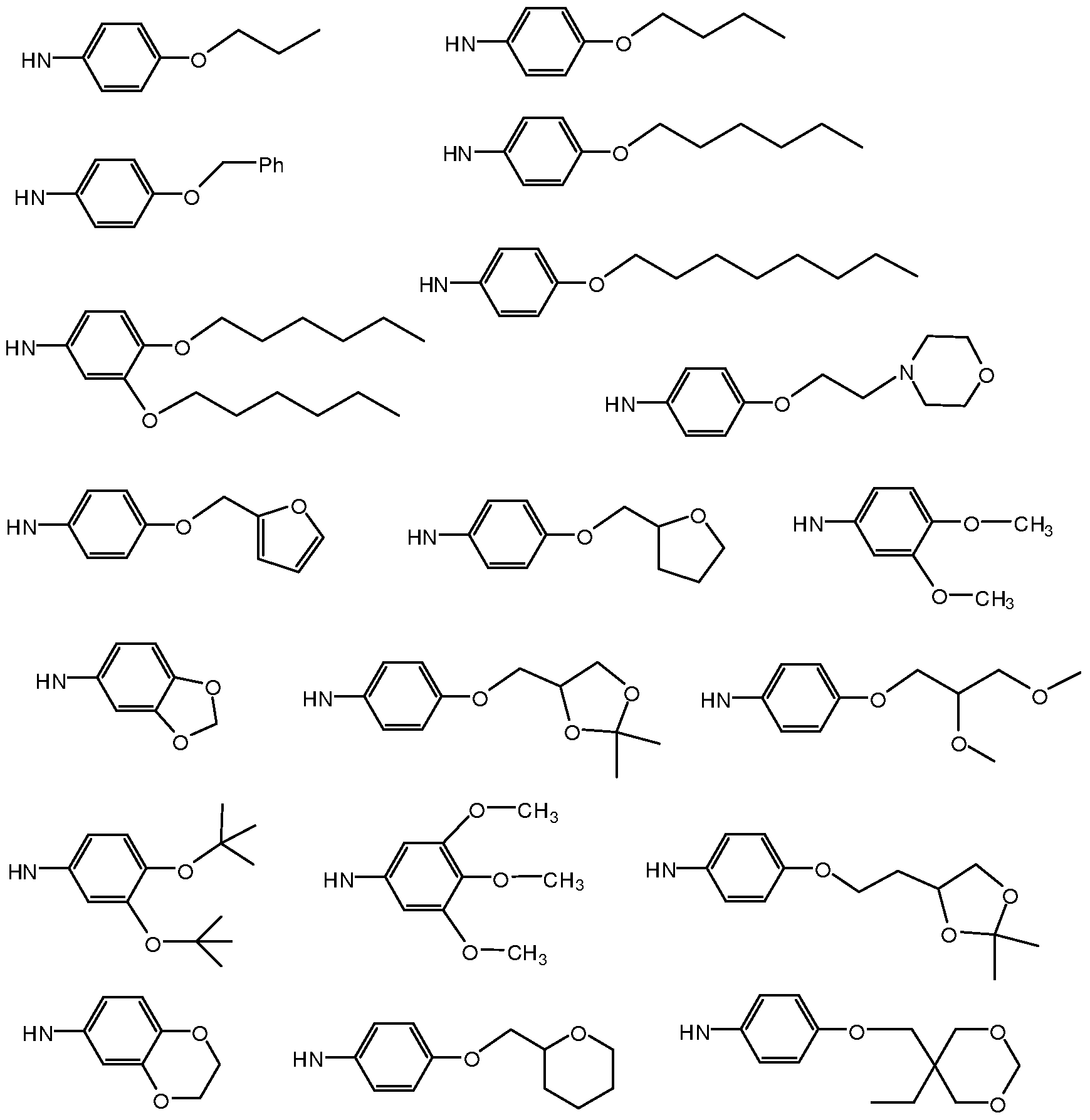
- Z may be an aromatic or heterocyclic moiety selected from the following structures:
- the aromatic ring(s) comprising Z can be substituted by up to five groups other than H, preferably up to four such groups, and in some embodiments with up to three groups other than H.
- Z is substituted with 1-3 non-hydrogen groups. These groups can be at any position of the aryl / heteroaryl ring of Z.
- Z is a 6-membered ring, in some embodiments at least one substituent other than H is present at the para position of the ring, or at the meta position of the ring.
- Z is selected from:
- each R group is as defined above.
- Some preferred groups that R can represent are listed in Table 1. Specific embodiments of the portion of these compounds of formula I- VII corresponding to -A-Z are set forth in Tables 3-7, and are preferred embodiments of -A-Z in each class of compounds related to the invention.
- Z is preferably not a benzyl group; a phenyl substituted with more than one Br or with SO 2 NRR; CH 2 -(2-furanyl); or methoxy- substituted pyridyl. In other embodiments, however, such as for methods of treating cancer, these limitations may not be applicable.
- Z may be mono-/di-/tri-substituted or unsubstituted benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine, oxazole, isoxazole, thiazole, isothiazole, oxadiazole, triazole, thiadiazole, pyrazole, imidazole, benzoxazole, pyrrole, furan, thiophene, indolizine, indole, isoindole, indoline, benzofuran, benzothiophene, indazole, benzimidazole, benzthiazole, purine, quinoxaline, quinoline, isoquinoline, cinnoline, phthalazine, quinazoline, naphthyridine, pteridine, acridine, phenazine, phenothiazine
- substituents include Cl-8 alkyl, C2-8 alkenyl, C2-8 alkynyl, C3-8 cyclic alkyl, C2-8 alkenyl, C2-8 alkynyl, an aryl, heteroaryl, a carbocyclic ring or a heterocyclic ring, each of which can also be substituted.
- W 5 is S.
- A is NR 4 , where R 4 is as defined above.
- Z is Ar, where Ar represents substituted or unsubstituted phenyl. In some embodiments, Z is -CH 2 -Ar, where Ar is substituted or unsubstituted phenyl.
- R is C1-C4 al
- the invention provides compounds of formula Ilia or Vila:
- R 1 is optionally substituted C1-C4 alkyl; each R 3 is independently H, halo, C1-C4 alkoxy, or C1-C4 alkyl; R 2 is H, halo, C1-C4 alkoxy, or C1-C4 alkyl; wherein Z is selected from the group consisting of:
- each R 1 is as defined above;
- R 5 is OR', SR', NR' 2 , OCHF 2 , OCF 3 , CF 3 , OCH 2 CF 3 , OCF 2 CF 3 , F, halo, (CF 2 ) 2 . 7 CF 3 , 0(CH 2 CH 2 )R', 0(CH 2 CH 2 O) 0 -OH, 0(CH 2 CH 2 )I -2 R',
- R 5 can also be selected from the following groups:
- R' is as defined above; and 1-5 such R 5 groups can be attached on the same benzene ring.
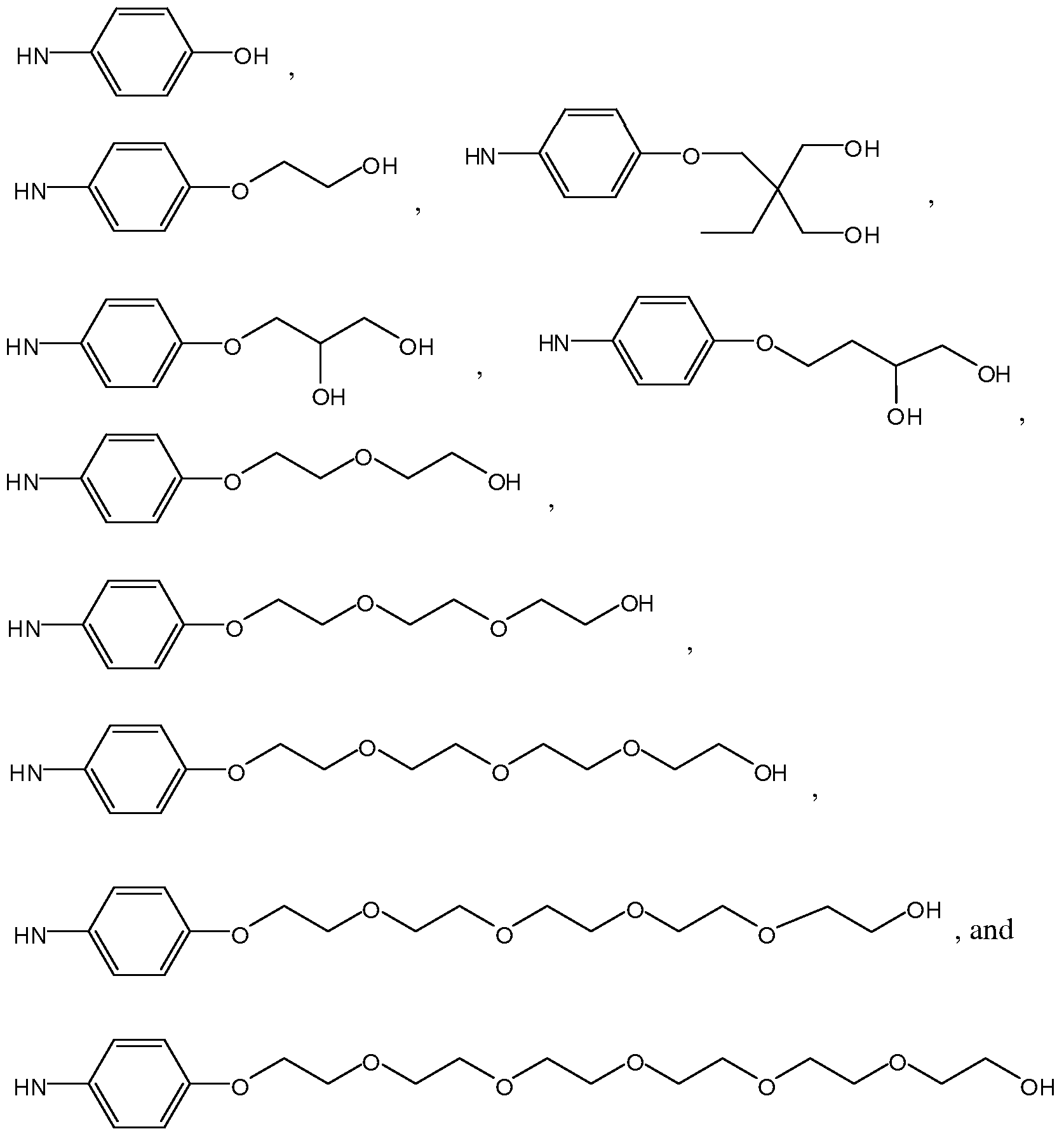
- the group corresponding to -NH-Z in formula Ilia or Vila is selected from the group consisting of:
- this group corresponding to -NH-Z in formula Ilia or Vila is selected from the group consisting of:
- R 1 is Methyl. In some of these compounds R 2 is H. In some of these compounds, R 3 is H.
- the invention provides compounds of formula (VI) as set forth above, and compositions comprising such compounds, and methods of using such compounds for treating various conditions described herein, including cancers.
- W 5 is S. In some embodiments of the compounds of formula (VI), W is S. In some embodiments of the compounds of formula (VI), W 3 is N.
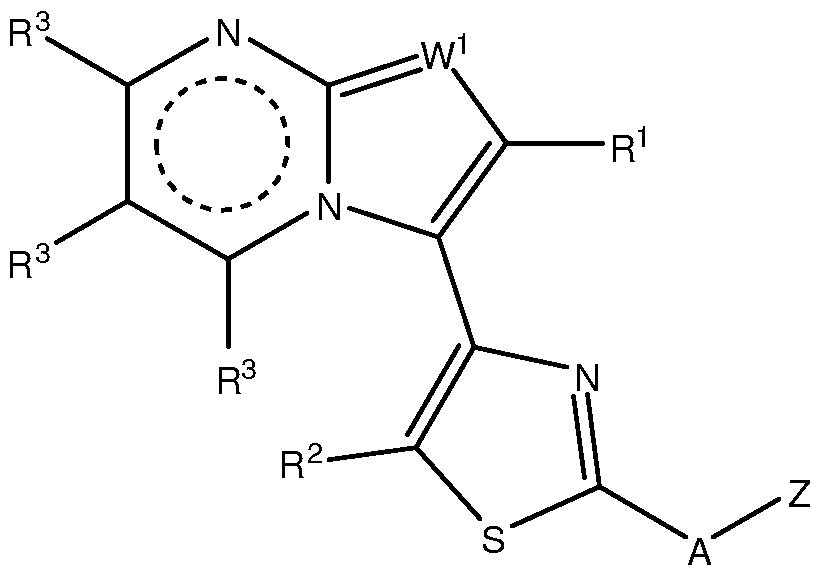
- the invention provides compounds, compositions and uses thereof wherein the compounds have the following structure:
- W 1 is CR 3 or N; each of W 2 , W 3 , W 4 and W 5 is CR 3 , N, O, or S or a bond, provided not more than one of W 2 , W 3 , W 4 and W 5 is a bond, and at least one of W 2 , W 3 , W 4 and W 5 is not CR 3 ; and not more than two of W 2 , W 3 , W 4 and W 5 represent N; and at least one of W 2 , W 3 , W 4 and W 5 is CR 3 ;
- Z is Ar or CH 2 Ar, where Ar is a 5-10 atom monocyclic or bicyclic aromatic group containing 0-4 heteroatoms selected from N, O and S as ring members and optionally substituted with up to four R 4 ; provided Z is not unsubstituted imidazopyridine, and when A is NAc, Z is not methoxy-substituted pyridinyl; each of R 1 , R 2 , R 3 , R 4 , R 5 is independently H, halo, OR, NRR', S(O) m R, COOR, SO 2 NRR' , NO 2 , CN, or a substituted or unsubstituted alkyl, alkenyl, alkynyl, aryl, fused aryl, heteroaryl, fused heterocycle, carbocyclic ring or heterocyclic ring, and two R 1 , R 2 , R 3 , R 4 , R 5 on the same or adjacent atoms can optionally be linked together to form
- Z is not unsubstituted imidazopyridine, and when A is NAc, Z is not methoxy-substituted pyridinyl; and when W 1 is N, R 1 is Me and A is NH, Z is not -CH 2 -(2-furanyl), unsubstituted phenyl, unsubstituted benzyl, or phenyl substituted with -NO 2 , Br, -OH, -NHAc, SOiNH-heteroaryl, or COOH.
- a and Z are as described above, so exemplary embodiments of Z are described in Tables 3-7, and A is sometimes NH.
- Z is selected from structures depicted in Tables 1-2.
- Z is substituted with at least one such group.
- W 1 is N or CH. Frequently, W 1 is N.
- R 1 is H, halo or optionally substituted C1-C4 alkyl.
- each R 3 is selected from H, halo, CN, optionally substituted C1-C4 alkyl, and C1-C4 alkoxy.
- R 2 is H, halo, CN, CONRR', COOR, or CF 3 , or an optionally substituted C1-C4 alkyl or alkoxy.
- A is NR 5 or O or S, where R 5 is as defined above.
- A is NH or NR 5 , where R 5 is optionally substituted C1-C4 alkyl or a C1-C4 acyl group.
- Z is a 5-membered aromatic or heteroaromatic ring or a 6-membered aromatic or heteroaromatic ring that is substituted with 0-3 substituents.
- Z is a substituted phenyl ring or a substituted or unsubstituted 2-pyridyl, 3-pyridyl or 4-pyridyl ring. Phenyl is sometimes preferred.
- the compounds in above formula I-X can be used as neutral compounds or as their pharmaceutically suitable salts with inorganic and organic anions.
- Their salts include, but are not limited to, halides (Cl “ , Br “ , I “ ), nitrate, mesylate, p-toluene sulfonate /tosulate, oxalate, citrate, malate, maleate, tartrate, fumarate, formate, acetate and the similar anions in the classes.
- heterocyclic compounds include the compounds themselves, as well as their salts and their prodrugs, if applicable.
- Such salts can be formed between a positively charged substitute group (e.g., an amino group on heterocyclic or aromatic rings) on a compound and a pharmaceutically suitable anion.
- Suitable anions include, but are not limited to, chloride, bromide, iodide, sulfate, nitrate, phosphate, citrate, methanesulfonate, trifluoroacetate, maleate, and acetate.
- a negatively charged substituted group e.g., carboxylate group on heterocyclic or aromatic rings
- a compound can form a salt with a cation.
- Non-limiting examples of suitable cations are sodium ion, potassium ion, magnesium ion, calcium ion, and a organic ammonium ion such as teteramethylammonium ion, tetrabutylammonium ion, and other organic cations.
- Compounds of the invention may exist as isomers, including optical isomers, geometric isomers, tautomers, and rotational isomers.
- the invention includes each such isomer of the compounds of formula I-X, and mixtures thereof. Where a compound has a chiral center, for example, the invention includes each individual isomer as well as mixtures of both isomers in varying amounts, including a racemic mixture having equal amounts of both isomers. Because the compounds of the invention are biaryls, they can exist as rotational isomers about the biaryl linkage, also, and each isomer as well as mixtures of such isomers are included within the scope of the invention.
- the compounds and compositions comprising the compounds of the invention are useful to treat conditions characterized by undesired cell proliferation.
- the compounds are useful to treat sarcoma, epidermoid cancer, fibrosarcoma, cervical cancer, leukemia, lymphoma, lung cancer, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, breast cancer, head and neck cancers, pancreatic cancer, and other types of a proliferative disease.
- sarcoma epidermoid cancer, fibrosarcoma, cervical cancer, leukemia, lymphoma, lung cancer, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, breast cancer, head and neck cancers, pancreatic cancer, and other types of a proliferative disease.
- Figure 1 shows the dynamic response pattern of A549 cells (human non small cell lung cancer cell line) to different concentrations of classical anti-mitotic agents paclitaxel and vinblastin, as determined on Real-Time Electronic Sensing System.
- Figure 2 shows the dynamic response pattern of A549 cells (human non small cell lung cancer cell line) to different concentrations of Compound No. 28 (2-(4- bromophenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5-yl)thiazole, ACEA100161) in Table 8, as determined on Real-Time Cell Electronic Sensing System.
- Figure 3 shows the dynamic response pattern of A549 cell to different concentrations of paclitaxel and Compound 28 (ACEA100161) in Table 8, as determined on Real- Time Electronic Sensing System.
- A549 cell exhibited similar responsive patterns to the compound No. 28 (ACEA100161) in Table 8 and to paclitaxel.
- Figure 4 shows dose response curves of A549 cells to the treatment of the compound No. 28 (2-(4-bromophenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5-yl)thiazole, ACEA100161) in Table 8, at the treatment time of 24 hrs after treatment.
- Figure 5 shows the dynamic response pattern of A549 cells to different concentrations of compound No. 26 (2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin- 3-yl)thiazole, ACEA100160) in Table 8 as determined on Real-Time Cell Electronic Sensing System.
- Figure 6 shows dose response curves of A549 cells to the treatment of the compound No. 26 (2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin-3-yl)thiazole, ACEA 100160) in Table 8, at the treatment time of 24 hrs after treatment.
- Figure 7 shows the dynamic response pattern of A549 cell to different concentrations of paclitaxel and compound No. 26 (2-(4-ethoxyphenyl)amino-4-(6- methylimidazo[l,2-a]pyrimidin-3-yl)thiazole, ACEA100160) in Table 8, as determined on Real-Time Electronic Sensing System.
- A549 cell exhibited similar responsive patterns to ACEA 100160 in Table 8 and to paclitaxel.
- Figure 8 shows the dynamic response pattern of A549 cells to different concentrations of compound No. 27 (2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5- yl)thiazole, ACEA 100162) in Table 8, as determined on Real-Time Cell Electronic Sensing System.
- Figure 9 shows dose response curves of A549 cells to the treatment of the compound No. 27 (2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5-yl)thiazole, ACEA 100162) in Table 8 at the treatment time of 24 hrs after treatment.
- Figure 10 shows the time-dependent cell index for a number of cell lines prior to and after addition of ACEA100162 at various concentrations: (A) MCF7adr (human breast adenocarcinoma), (B) PC3 (human prostate cancer), (C) KB (human head-neck cancer), (D) KB200 (human oral epithelioma) and (E) Bcap37 (human breast adenocarcinoma) .
- MCF7adr human breast adenocarcinoma
- PC3 human prostate cancer
- C human head-neck cancer
- D KB200
- E Bcap37
- Figure 11 shows the time-dependent cell index for a number of cell lines prior to and after addition of ACEA100160 at various concentrations: (A) MCF7adr (human breast adenocarcinoma), (B) PC3 (human prostate cancer), (C) KB (human head-neck cancer), (D) KB200 (human oral epithelioma) and (E) Bcap37 (human breast adenocarcinoma) .
- Figure 12 shows Tumor growth suppression by ACEA100160 and ACEA100162 treated on mouse S180 carcinoma model. Animals were treated with ACEA100160 and ACEA100162 for 9 days.
- Figure 13 shows Tumor growth suppression by ACEA100160 and ACEA100162 treated on mouse lewis lung cancer model. Animals were treated with ACEA100160 and ACEA100162 for 12 days.
- alkyl refers to saturated hydrocarbon groups in a straight, branched, or cyclic configuration and particularly contemplated alkyl groups include lower alkyl groups (i.e., those having ten or less carbon atoms). Exemplary alkyl groups are methyl, ethyl, propyl, isopropyl, butyl, sec -butyl, tertiary butyl, pentyl, isopentyl, hexyl, etc.
- alkenyl refers to an alkyl as defined above and having at least one double bond.
- alkenyl groups include straight, branched, or cyclic alkenyl groups having two to ten carbon atoms (e.g., ethenyl, propenyl, butenyl, pentenyl, etc.).
- alkynyl refers to an alkyl or alkenyl as defined above and having at least one triple bond.
- alkynyls include straight, branched, or cyclic alkynes having two to ten total carbon atoms (e.g., ethynyl, propynyl, butynyl, etc.).
- cycloalkyl refers to a cyclic alkane (i.e., in which a chain of carbon atoms of a hydrocarbon forms a ring), preferably including three to eight carbon atoms.
- exemplary cycloalkanes include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- Cycloalkyls also include one or two double bonds, which form the "cycloalkenyl” groups. Cycloalkyl groups are also further substituted by alkyl, alkenyl, alkynyl, halo and other general groups.
- aryl or "aromatic moiety” as used herein refers to an aromatic ring system, which may further include one or more non-carbon atoms.
- contemplated aryl groups include (e.g., phenyl, naphthyl, etc.) and pyridyl.
- Further contemplated aryl groups may be fused (i.e., covalently bound with 2 atoms on the first aromatic ring) with one or two 5- or 6-membered aryl or heterocyclic group, and are thus termed “fused aryl” or “fused aromatic”.
- heterocycle cycloheteroalkyl
- heterocyclic moieties are used interchangeably herein and refer to any compound in which a plurality of atoms form a ring via a plurality of covalent bonds, wherein the ring includes at least one atom other than a carbon atom.
- Particularly contemplated heterocyclic bases include 5- and 6-membered rings with nitrogen, sulfur, or oxygen as the non-carbon atom (e.g., imidazole, pyrrole, triazole, dihydropyrimidine, indole, pyridine, thiazole, tetrazole etc.).
- heterocycles may be fused (i.e., covalently bound with two atoms on the first heterocyclic ring) to one or two ring or heterocycle, and are thus termed “fused heterocycle” or “fused heterocyclic base” or “fused heterocyclic moieties” as used herein.
- fused heterocycle or "fused heterocyclic base” or “fused heterocyclic moieties” as used herein.
- fused heterocycle or “fused heterocyclic base” or “fused heterocyclic moieties” as used herein.
- imidazooxazole herein refer to any compound in which the two designated heterocyclic rings are fused by any two adjacent atoms on the two heterocyclic rings.
- alkoxy refers to straight or branched alkyl connecting to an oxygen atom called alkoxides, wherein the hydrocarbon portion may have any number of carbon atoms, may further include a double or triple bond and may include one or two oxygen, sulfur or nitrogen atoms in the alkyl chains.
- suitable alkoxy groups include methoxy, ethoxy, propyloxy, isopropoxy, methoxyethoxy, etc.
- alkylthio refers to straight or branched chain alkylsulfides, wherein the hydrocarbon portion may have any number of carbon atoms, may further include a double or triple bond and may include one or two oxygen, sulfur or nitrogen atoms in the alkyl chains.
- contemplated alkylthio groups include methylthio, ethylthio, isopropylthio, methoxyethylthio, etc.
- alkylamino refers to straight or branched alkylamines, wherein the amino nitrogen “N” can be substituted by one or two alkyls and the hydrocarbon portion may have any number of carbon atoms and may further include a double or triple bond.
- the hydrogen of the alkylamino may be substituted with another alkyl group. Therefore, exemplary alkylamino groups include methylamino, dimethylamino, ethylamino, diethylamino, etc.
- aryloxy refers to an aryl group connecting to an oxygen atom, wherein the aryl group may be further substituted.
- suitable aryloxy groups include phenyloxy, etc.
- arylthio refers to an aryl group connecting to a sulfur atom, wherein the aryl group may be further substituted.
- suitable arylthio groups include phenylthio, etc.
- halogen refers to fluorine, chlorine, bromine and iodine.
- substituted refers to a replacement of an H atom with another atom or group.
- Alkyl, alkenyl and alkynyl groups are often substituted to the extent that such substitution makes sense chemically.
- Alkyl, alkenyl and alkynyl groups can also be substituted by C1-C8 acyl, C2-C8 heteroacyl, C6-C10 aryl or C5-C10 heteroaryl, each of which can be substituted by the substituents that are appropriate for the particular group.
- Heteroalkyl “heteroalkenyl”, and “hetero alkynyl” and the like are defined similarly to the corresponding hydrocarbyl (alkyl, alkenyl and alkynyl) groups, but the 'hetero' terms refer to groups that contain 1-3 O, S or N heteroatoms or combinations thereof within the backbone residue; thus at least one carbon atom of a corresponding alkyl, alkenyl, or alkynyl group is replaced by one of the specified heteroatoms to form a heteroalkyl, heteroalkenyl, or heteroalkynyl group.
- heteroforms of alkyl, alkenyl and alkynyl groups are generally the same as for the corresponding hydrocarbyl groups, and the substituents that may be present on the heteroforms are the same as those described above for the hydrocarbyl groups.
- substituents that may be present on the heteroforms are the same as those described above for the hydrocarbyl groups.
- such groups do not include more than two contiguous heteroatoms except where an oxo group is present on N or S as in a nitro or sulfonyl group.
- alkyl as used herein includes cycloalkyl and cycloalkylalkyl groups
- cycloalkyl may be used herein to describe a carbocyclic non-aromatic group that is connected via a ring carbon atom
- cycloalkylalkyl may be used to describe a carbocyclic non-aromatic group that is connected to the molecule through an alkyl linker.
- heterocyclyl may be used to describe a non-aromatic cyclic group that contains at least one heteroatom as a ring member and that is connected to the molecule via a ring atom, which may be C or N;
- heterocyclylalkyl may be used to describe such a group that is connected to another molecule through a linker.
- the sizes and substituents that are suitable for the cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl groups are the same as those described above for alkyl groups As used herein, these terms also include rings that contain a double bond or two, as long as the ring is not aromatic.
- acyl encompasses groups comprising an alkyl, alkenyl, alkynyl, aryl or arylalkyl radical attached at one of the two available valence positions of a carbonyl carbon atom
- heteroacyl refers to the corresponding groups wherein at least one carbon other than the carbonyl carbon has been replaced by a heteroatom chosen from N, O and S.
- Acyl and heteroacyl groups are bonded to any group or molecule to which they are attached through the open valence of the carbonyl carbon atom. Typically, they are C1-C8 acyl groups, which include formyl, acetyl, pivaloyl, and benzoyl, and C2-C8 heteroacyl groups, which include methoxyacetyl, ethoxycarbonyl, and A- pyridinoyl.
- the hydrocarbyl groups, aryl groups, and heteroforms of such groups that comprise an acyl or heteroacyl group can be substituted with the substituents described herein as generally suitable substituents for each of the corresponding component of the acyl or heteroacyl group.
- Aromaatic moiety or aryl moiety refers to a monocyclic or fused bicyclic moiety having the well-known characteristics of aromaticity; examples include phenyl and naphthyl.
- hetero aromatic and heteroaryl refer to such monocyclic or fused bicyclic ring systems which contain as ring members one or more heteroatoms selected from O, S and N. The inclusion of a heteroatom permits aromaticity in 5-membered rings as well as 6-membered rings.
- Typical heteroaromatic systems include monocyclic C5-C6 aromatic groups such as pyridyl, pyrimidyl, pyrazinyl, thienyl, furanyl, pyrrolyl, pyrazolyl, thiazolyl, oxazolyl, and imidazolyl and the fused bicyclic moieties formed by fusing one of these monocyclic groups with a phenyl ring or with any of the heteroaromatic monocyclic groups to form a C8-C10 bicyclic group such as indolyl, benzimidazolyl, indazolyl, benzotriazolyl, isoquinolyl, quinolyl, benzothiazolyl, benzofuranyl, pyrazolopyridyl, quinazolinyl, quinoxalinyl, cinnolinyl, and the like.
- monocyclic C5-C6 aromatic groups such as pyridyl, pyrimidy
- any monocyclic or fused ring bicyclic system which has the characteristics of aromaticity in terms of electron distribution throughout the ring system is included in this definition. It also includes bicyclic groups where at least the ring which is directly attached to the remainder of the molecule has the characteristics of aromaticity.
- the ring systems contain 5-12 ring member atoms.
- the monocyclic heteroaryls contain 5-6 ring members, and the bicyclic heteroaryls contain 8-10 ring members.
- Aryl and heteroaryl moieties may be substituted with a variety of substituents including C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, C3-C8 cycloalkyl, C5-C12 aryl, C1-C8 acyl, and heteroforms of these, each of which can itself be further substituted with appropriate substituents; other substituents for aryl and heteroaryl moieties include halo, O((CH2)pO)qR, where each p is independently 1-4 and q is 1- 6, OR, NR2, SR, SO2R, SO2NR2, NRSO2R, NRC0NR2, NRCOOR, NRCOR, CN, COOR, C0NR2, 0OCR, COR, and NO2, wherein each R is independently H, C1-C8 alkyl, C2-C8 heteroalkyl, C2-C8 alkenyl, C2-C8 heteroalkenyl, C2-C8 al
- substituent groups on an aryl or heteroaryl group may of course be further substituted with the groups described herein as suitable for each type of such substituents or for each component of the substituent; and such substituents can be substituted with one or more substituents selected from halo, CF 3 , Cl-4 alkyl and Cl-4 alkoxy.
- an arylalkyl substituent may be substituted on the aryl portion with substituents described herein as typical for aryl groups, and it may be further substituted on the alkyl portion with substituents described herein as typical or suitable for alkyl groups.
- arylalkyl and
- heteroarylalkyl refer to aromatic and heteroaromatic ring systems which are bonded to their attachment point through a linking group such as an alkylene, including substituted or unsubstituted, saturated or unsaturated, cyclic or acyclic linkers.
- linker is C1-C8 alkyl or a hetero form thereof.
- linkers may also include a carbonyl group, thus making them able to provide substituents as an acyl or heteroacyl moiety.
- An aryl or heteroaryl ring in an arylalkyl or heteroarylalkyl group may be substituted with the same substituents described above for aryl groups.
- an arylalkyl group includes a phenyl ring optionally substituted with the groups defined above for aryl groups and a C1-C4 alkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl groups or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane.
- a heteroarylalkyl group preferably includes a C5-C6 monocyclic heteroaryl group that is optionally substituted with the groups described above as substituents typical on aryl groups and a C1-C4 alkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl groups or heteroalkyl groups, or it includes an optionally substituted phenyl ring or C5-C6 monocyclic heteroaryl and a C1-C4 heteroalkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane.
- substituents may be on either the alkyl or heteroalkyl portion or on the aryl or heteroaryl portion of the group.
- the substituents optionally present on the alkyl or heteroalkyl portion are the same as those described above for alkyl groups generally; the substituents optionally present on the aryl or heteroaryl portion are the same as those described above for aryl groups generally.
- Arylalkyl groups as used herein are hydrocarbyl groups if they are unsubstituted, and are described by the total number of carbon atoms in the ring and alkylene or similar linker.
- a benzyl group is a C7-arylalkyl group
- phenylethyl is a C8-arylalkyl.
- Heteroarylalkyl refers to a moiety comprising an aryl group that is attached through a linking group, and differs from “arylalkyl” in that at least one ring atom of the aryl moiety or one atom in the linking group is a heteroatom selected from N, O and S.
- the heteroarylalkyl groups can be described herein according to the total number of atoms in the ring and linker combined, and they include aryl groups linked through a heteroalkyl linker; heteroaryl groups linked through a hydrocarbyl linker such as an alkylene; and heteroaryl groups linked through a heteroalkyl linker.
- C7-heteroarylalkyl would include pyridylmethyl, phenoxy, and N-pyrrolylmethoxy.
- groups can be otherwise described, e.g. as a C5-C6-aryl-Cl-C2-alkyl, which would refer to a 5-6 membered aryl ring connected to the base molecule through a C1-C2 linker.
- Alkylene refers to a divalent hydrocarbyl group; because it is divalent, it can link two other groups together. Typically it refers to -(CH 2 ) n - where n is 1-8 and preferably n is 1-4, though where specified, an alkylene can also be substituted by other groups, and can be of other lengths, and the open valences need not be at opposite ends of a chain. Thus -CH(Me)- and -C(Me) 2 - may also be referred to as alkylenes, as can a cyclic group such as cyclopropan-l,l-diyl. Where an alkylene group is substituted, the substituents include those typically present on alkyl groups as described herein.
- any alkyl, alkenyl, alkynyl, acyl, or aryl or arylalkyl group or any heteroform of one of these groups that is contained in a substituent may itself optionally be substituted by additional substituents.
- the nature of these substituents is similar to those recited with regard to the primary substituents themselves if the substituents are not otherwise described.
- R 7 is alkyl
- this alkyl may optionally be substituted by the remaining substituents listed as embodiments for R 7 where this makes chemical sense, and where this does not undermine the size limit provided for the alkyl per se; e.g., alkyl substituted by alkyl or by alkenyl would simply extend the upper limit of carbon atoms for these embodiments, and is not included.
- each such alkyl, alkenyl, alkynyl, acyl, or aryl group may be substituted with a number of substituents according to its available valences; in particular, any of these groups may be substituted with fluorine atoms at any or all of its available valences, for example.
- Particularly contemplated functional groups include nucleophilic groups (e.g., -NH 2 , -OH, -SH, -NC, etc.), electrophilic groups (e.g., C(O)OR, C(X)OH, etc.), polar groups (e.g., -OH), non-polar groups (e.g., heterocycle, aryl, alkyl, alkenyl, alkynyl, etc.), ionic groups (e.g., -NH 3 + ), and halogens (e.g., -F, -Cl), NHCOR, NHCONH 2 , OCH 2 COOH, OCH 2 CONH 2 , OCH 2 CONHR, NHCH 2 COOH, NHCH 2 CONH 2 , NHSO 2 R, OCH 2 -heterocycles, PO 3 H, SO 3 H, amino acids, and all chemically reasonable combinations thereof.
- nucleophilic groups e.g., -NH 2 , -OH
- substituted also includes multiple degrees of substitution, and where multiple substituents are disclosed or claimed, the substituted compound can be independently substituted by one or more of the disclosed or claimed substituent moieties.
- substituted compound can be independently substituted by one or more of the disclosed or claimed substituent moieties.
- mono-/di- /tri-/tetra- substituted used herein refers to one, or two, or three or four functional groups described above that substituted onto the aromatic or heterocyclic or fused aromatic or heterocyclic moiety, in which such multi-functional groups are substituted at the combination of any ortho- ox para- or meto-position of the aromatic or heterocyclic moiety.
- W 2 is S and W 3 is N.
- W 5 is S.
- A is NR 4 .
- Z is CH 2 -Phenyl or Phenyl, where the phenyl ring is optionally substituted.
- R H, F, Cl, Br, I, Me, Et, Propyl, Bu, CF 3 , OMe, OEt, OiPr, OCF 3 , COCH 3 ,
- R H, F, Cl, Br, I, Me, Et, Propyl, Bu, CF 3 , OMe, OEt, OiPr, OCF 3 , COCH 3 , NO2, NMe 2 , NEt 2 , NHCOMe, SO 2 NH 2 , SO 2 NHPh, SO 2 NH-thiazole, SO 2 NH-oxazole, SO 2 NH-pyridine, NH 2 , OH, SMe, SEt. Table 3. Representative fluoro-substituted derivatives
- 3-Chloro-2,5-pentanedione (2) (106 niL, 119 g, 887 mmol, 1.2 eq) was dissolved in 650 niL of anhydrous ethanol.
- the resulting mixture was refluxed for 40 h at an oil bath temperature of 100-105 0 C.
- the black reaction mixture was cooled and concentrated under reduced pressure.
- the residue was treated with saturated sodium bicarbonate solution (-500 mL) in portions, and swelled the flask to mix well.
- the mixture was extracted with dichloromethane (x 6).
- the crude product was taken up with acetone- ethanol (3:1) and stirred at room temperature for more than 6 h.
- the crude product was filtered, washed, and re-crystallized from methanol.
- the methanol solution was filtered while still hot to remove black dust, and then heated into solution.
- the crude product was filtered and washed. It was re-crystallized two more times methanol, and dried under vacuum to provide the desired product 15 as light yellow crystals yield 1.448 g (81%).
- TLC R/0.25 (dichloromethane-methanol: 20:1); R/0.28 (100% ethyl acetate, XZ).
- the neutral compound was prepared as described above.
- Other salts were prepared as described above.
- the compounds of the invention can be prepared by modifications of known synthetic methods from known or available starting materials. Some exemplary compounds are synthesized as illustrated in Schemes III and IV. Additional methods that may be used to make the products or precursors for making them are disclosed in, for example, Andreani, et al, J. Med. Chem. vol. 49, 7897-7901 (2006); Nafziger, et al., Cvtotechnology, vol. 6, 227-32 (1991); Andreani, et al., ARKIVOC 2004(v) 76- 84; Andreani, et al., Bioorg. Med. Chem., vol. 8, 2359-66 (2000); Saldabol, et al., Engl. Transl.
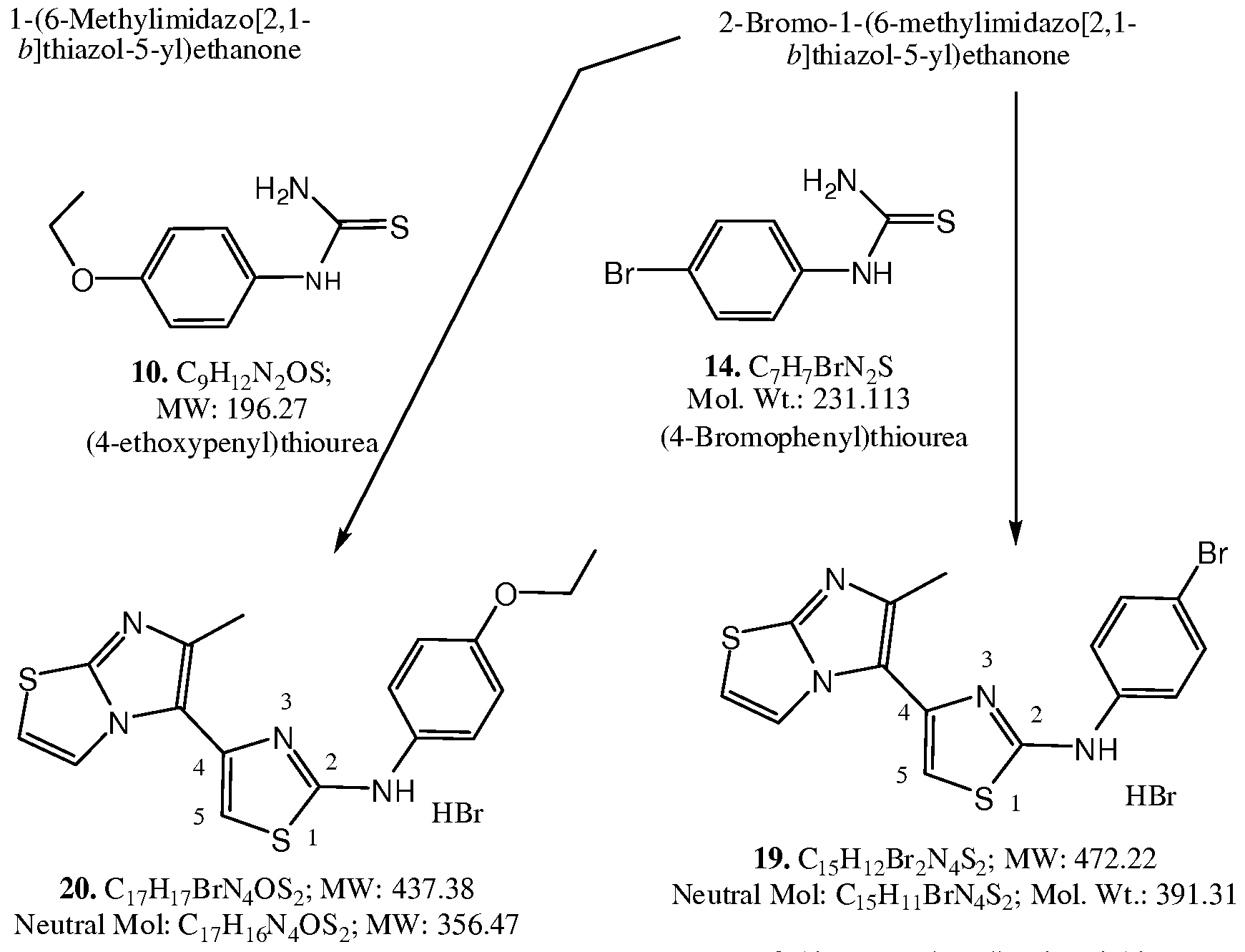
- the methanol solution was filtered while still hot to remove possible dust, and then heated into solution. It was re-crystallized two more times from methanol, and dried under vacuum to provide the desired product 20 as a white solid, yield 0.64 g (22.4%), HPLC purity 97.81%, mp >240 0 C.
- alkyloxy and substituted alkyloxy derivatized compounds 25 and 26 are also synthesized according to the Schemes IV and V utilizing the parallel last combinatorial approach.
- the intermediate bromide 5 is condensed with thiourea 21 utilizing the same procedure as the synthesis of 11 and 15 described above.
- the resulting compound 22 is reacted with reactive electrophiles, such as bromides, iodides, or tosylates, under basic conditions providing the desired products 23.
- the intermediate bromide 18 is condensed with thiourea 21 utilizing the same procedure as the synthesis of 19 and 20 described above.
- the resulting compound 24 is reacted with reactive electrophiles, such as bromides, iodides, or tosylates, under basic conditions providing the desired products 25.
- any suitable formulation of the compounds described herein can be prepared.
- administration of the compounds as salts may be appropriate.
- pharmaceutically acceptable salts are organic acid addition salts formed with acids that form a physiological acceptable anion, for example, tosylate, methanesulfonate, acetate, citrate, malonate, tartarate, succinate, benzoate, ascorbate, ⁇ -ketoglutarate, and ⁇ -glycerophosphate.
- Suitable inorganic salts may also be formed, including hydrochloride, sulfate, nitrate, bicarbonate, and carbonate salts.
- salts are obtained using standard procedures well known in the art, for example, by contacting a sufficiently basic compound such as an amine with a suitable acid, affording a physiologically acceptable salt.
- a sufficiently basic compound such as an amine
- suitable acid affording a physiologically acceptable salt.
- Alkali metal (e.g., sodium, potassium or lithium) or alkaline earth metal (e.g., calcium) salts of carboxylic acids also are included, and are prepared by conventional methods.
- the invention also includes pharmaceutical compositions comprising at least one compound of the invention admixed with at least one pharmaceutically acceptable excipient.
- at least one such excipient is an excipient other than water or a C1-C3 alcohol or a dimethyl sulfoxide.
- Compounds of the invention can be administered by conventional routes, including orally, topically, transdermally, or by inhalation or injection.
- the compounds of the invention can be formulated by those skilled in the art by reference to known methods, and the formulation can be tailored according to the intended route of administration. Suitable methods for formulating organic compounds are described, for example, in REMINGTON'S PHARMACEUTICAL SCIENCES, 18 th ed. (1990), which is incorporated herein by reference.
- contemplated compounds can be formulated in admixture with a pharmaceutically acceptable carrier.
- contemplated compounds can be administered orally as pharmacologically acceptable salts, or intravenously in a physiological saline solution.
- Conventional buffers such as phosphates, bicarbonates or citrates can be used for this purpose.
- contemplated compounds may be modified to render them more soluble in water or other vehicle, which for example, may be easily accomplished with minor modifications (salt formulation, esterification, etc.) that are well within the ordinary skill in the art.
- the compounds having formula I-X as described herein are generally soluble in organic solvents such as chloroform, dichloromethane, ethyl acetate, ethanol, methanol, isopropanol, acetonitrile, glycerol, iV,iV-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, etc.
- organic solvents such as chloroform, dichloromethane, ethyl acetate, ethanol, methanol, isopropanol, acetonitrile, glycerol, iV,iV-dimethylformamide, N,N- dimethylacetamide, dimethylsulfoxide, etc.
- the present invention provides formulations prepared by mixing a compound having formula I-X with a pharmaceutically acceptable carrier.
- the formulation may be prepared using a method comprising: a) dissolving a described compound in a water-soluble organic solvent, a non-ionic solvent, a water-soluble lipid, a cyclodextrin, a vitamin such as tocopherol, a fatty acid, a fatty acid ester, a phospholipid, or a combination thereof, to provide a solution; and b) adding saline or a buffer containing 1-10% carbohydrate solution.
- the carbohydrate comprises dextrose.
- water soluble organic solvents for use in the present methods include and are not limited to polyethylene glycol (PEG), alcohols, acetonitrile, iV-methyl-2-pyrrolidone, iV,iV-dimethylformamide, N,N- dimethylacetamide, dimethyl sulfoxide, or a combination thereof.
- suitable alcohols include but are not limited to methanol, ethanol, isopropanol, glycerol, or propylene glycol.
- Illustrative examples of water soluble non-ionic surfactants for use in the present methods include and are not limited to CREMOPHOR ® EL, polyethylene glycol modified CREMOPHOR ® (polyoxyethyleneglyceroltriricinoleat 35), hydrogenated CREMOPHOR ® RH40, hydrogenated CREMOPHOR ® RH60, PEG- succinate, polysorbate 20, polysorbate 80, SOLUTOL ® HS (polyethylene glycol 660 12-hydroxystearate), sorbitan monooleate, poloxamer, LABRAFIL ® (ethoxylated persic oil), LABRASOL ® (capryl-caproyl macrogol-8-glyceride), GELUCIRE ® (glycerol ester), SOFTIGEN ® (PEG 6 caprylic glyceride), glycerin, glycol- polysorbate, or a combination thereof.
- CREMOPHOR ® EL polyethylene glycol modified CREMOP
- lipid oils include but are not limited to castor oil, polyoxyl castor oil, corn oil, olive oil, cottonseed oil, peanut oil, peppermint oil, safflower oil, sesame oil, soybean oil, hydrogenated vegetable oil, hydrogenated soybean oil, a triglyceride of coconut oil, palm seed oil, and hydrogenated forms thereof, or a combination thereof.
- fatty acids and fatty acid esters for use in the present methods include but are not limited to oleic acid, monoglycerides, diglycerides, a mono- or di-fatty acid ester of PEG, or a combination thereof.
- cyclodextrins for use in the present methods include but are not limited to alpha-cyclodextrin, beta-cyclodextrin, hydroxypropyl-beta- cyclodextrin, or sulfobutyl ether-beta-cyclodextrin.
- Illustrative examples of phospholipids for use in the present methods include but are not limited to soy phosphatidylcholine, or distearoyl phosphatidylglycerol, and hydrogenated forms thereof, or a combination thereof.
- One of ordinary skill in the art may modify the formulations within the teachings of the specification to provide numerous formulations for a particular route of administration.
- the compounds may be modified to render them more soluble in water or other vehicle. It is also well within the ordinary skill of the art to modify the route of administration and dosage regimen of a particular compound in order to manage the pharmacokinetics of the present compounds for maximum beneficial effect in a patient.
- the compounds of the present invention can be used as cytotoxic and/or cytostatic agents in treating cancers or other types of proliferative disease. These compounds may function through any type of action mechanisms.
- the compounds may inhibit molecules and/or signal transduction pathways leading to arrest of the cell cycle at G2/M phase, which might eventually induce apoptosis in tumor cells (see, e.g., Weung et al. (1997) Biochim. Biophys. Res. Comm., vol: 263, pp 398-404).
- the compounds may disturb tubulin assembly/disassembly, which may inhibit the cell mitosis and induce the cell apoptosis (see, e.g., Panda et al., (1997) Proc. Natl.
- the compounds may also inhibit endothelial cell proliferation and angiogenesis effect (see, e.g., Witte et al., 1998, Cancer Metastasis Rev, vol. 17: 155- 161).
- the present invention is directed to a method of treatment of cancers of all tissue or organ origin including but not limited to sarcoma, epidermoid cancer, fibrosarcoma, cervical cancer, leukemia, lymphoma, lung cancer, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, breast cancer, head and neck cancers, pancreatic cancer and other types of proliferative disease in a mammal comprising administering a therapeutically effective amount of compound having Formula I-X as a cytotoxic and/or cytostatic agent to said subject in need of such treatment, in at least one treatment.
- the present invention is directed to a method for manufacturing a pharmaceutical preparation for the treatment of cancers of all tissue or organ origin including but not limited to leukemia, lymphoma, lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer or breast cancer, and other types of a proliferative disease, comprising admixing a therapeutically effective amount of a compound having Formula I-X with a pharmaceutically acceptable carrier.
- compounds having formula I- X and pharmaceutical compositions thereof may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally, via an implanted reservoir, or other drug administration methods.
- parenteral as used herein includes subcutaneous, intracutaneous, intravenous, intramuscular, intraarticular, intraarterial, intrasynovial, intrasternal, intrathecal, intralesional and intracranial injection or infusion techniques.
- the compounds of the invention are delivered by injection, i.e., parenterally.
- the preferred route of administration is by intravenous or intraperitoneal injection.
- a sterile injectable composition such as a sterile injectable aqueous or oleaginous suspension, may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent.
- acceptable vehicles and solvents include mannitol, water, Ringer' s solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium (e.g., synthetic mono- or diglycerides).
- Fatty acids such as oleic acid and its glyceride derivatives
- injectables are useful in the preparation of injectables, as are pharmaceutically acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- oils such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions can also contain a long-chain alcohol diluent or dispersant, or carboxymethyl cellulose or similar dispersing agents.
- Various emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms can also be used for the purpose of formulation.
- a composition for oral administration may be any orally acceptable dosage form including, but not limited to, tablets, capsules, emulsions and aqueous suspensions, dispersions and solutions.
- commonly used carriers include lactose and corn starch.
- Lubricating agents such as magnesium stearate, can also be added.
- useful diluents include lactose and dried corn starch.
- a nasal aerosol or inhalation compositions can be prepared according to techniques well-known in the art of pharmaceutical formulation and can be prepared as solutions in, for example saline, employing suitable preservatives (for example, benzyl alcohol), absorption promoters to enhance bioavailability, and/or other solubilizing or dispersing agents known in the art.
- suitable preservatives for example, benzyl alcohol
- absorption promoters to enhance bioavailability
- an effective amount of a compound of the invention can be determined by routine experimentation as is known in the art. Typically, this involves administration of an amount shown to be well tolerated, and gradually increasing the dosage until a desired effect is achieved, such as reduction in symptoms, reduction n tumor size, or cessation of tumor growth.
- a starting dosage of about 5-10 mg/kg is used, and the dosage is increased incrementally once per week by about 50% each time until a desired effect is noted or tolerance problems are observed.
- a suitable dosage is between about 5 and 250 mg/kg; or between about 10 and 150 mg/kg. Dosages between 10 and 100 mg/kg are sometimes preferred. Dosing can be done once, once weekly, once daily or more than once daily. In some embodiments, 1-4 doses are delivered per day to a subject in need of treatment.
- Combination therapies according to the present invention comprise the administration of at least one compound of the present invention or a functional derivative thereof and at least one other pharmaceutically active ingredient.
- the active ingredient(s) and pharmaceutically active agents may be administered separately or together.
- the amounts of the active ingredient(s) and pharmaceutically active agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
- the present invention is directed to a method of treatment of restenosis after coronary stenting for patients with coronary artery diseases with a compound having formula I-X.
- the main cause of restenosis after coronary stenting for patients with coronary artery disease is neointimal hyperplasia resulting from the proliferation and migration of smooth-muscle cells and extracellular matrix productions (see, for example, "Pathology of acute and chronic coronary stenting in humans", by Farb, A., Sangiorgi, G., Certer, AJ., et al, in Circulation, vol. 99, pp 44-52, 1999).
- Compounds that have anti-proliferation capability may have an effect in reducing the risk of clinical and angiographic restenosis when such compounds are delivered with a suitable means (see, for example, "A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease", by Stone, G.W., Ellis, S. G., Cox, D. A, et al, in New England Journal of Medicine, vol. 350: pp 221-231, 2004).
- compounds having formula I- IX in treating tumor they may be also useful in inhibiting proliferation of the cells involved in neointimal hyperplasia and thus reducing the incidence of neointimal hyperplasia and restenosis.
- compositions comprising above-described compounds having formula I-X can be administered orally, parenterally, or via an implanted reservoir.
- approaches described in the following papers may also be used: "A polymer- based, paclitaxel-eluting stent in patients with coronary artery disease", by Stone, G.W., Ellis, S. G., Cox, D.A. et al, in New England Journal of Medicine, vol. 350: pp 221-231, 2004; "A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization", by Morice, M.
- RT-CES® Real-Time Cell Electronic Sensing
- the RT-CES system utilizes cell- substrate impedance technology to monitor cellular behavior inside tissue culture wells in a microtiter plate format.
- the technology features in the integration of molecular and cell biology with microelectronics and is based on the electronic detection of biological assay process.
- RT-CES technology is further disclosed in United States provisional application number 60/519,567, filed on November 12, 2003, and United States provisional application number 60/542,927, filed on February 9, 2004.
- microelectrodes having appropriate geometries are fabricated onto the bottom surfaces of microtiter plate or similar device, facing into the wells. Cells are introduced into the wells of the devices, and make contact to and attach to the electrode surfaces. The presence, absence or change of properties of cells affects the electronic and ionic passage on the electrode sensor surfaces. Measuring the impedance between or among electrodes provides important information about biological status of cells present on the sensors.
- analogue electronic readout signals are measured automatically and in real time, and are converted to digital signals for processing and for analysis.
- a cell index (arbitrary representation of change in impedance) is automatically derived and provided based on measured electrode impedance values.
- the cell index obtained for a given well reflects: 1) how many cells are attached to the electrode surfaces in this well; 2) how well cells are attached to the electrode surfaces in this well.
- the RT-CES system comprises three components, an electronic sensor analyzer, a device station and 16X or 96X microtiter devices.
- Microelectrode sensor array was fabricated on glass slides with lithographical microfabrication methods and the electrode-containing slides are assembled to plastic trays to form electrode- containing wells.
- the device station receives the 16X or 96X microtiter plate devices and is capable of electronically switching any one of the wells to the sensor analyzer for impedance measurement.
- the devices with cells cultured in the wells are placed into a device station that is located inside an incubator. Electrical cables connect the device station to the sensor analyzer.
- the sensor analyzer Under the RT-CES software control, the sensor analyzer can automatically select wells to be measured and continuously conduct impedance measurements.
- the impedance data from the analyzer is transferred to a computer, analyzed and processed by the integrated software.
- Impedance measured between electrodes in an individual well depends on electrode geometry, ionic concentration in the well and whether there are cells attached to the electrodes. In the absence of the cells, electrode impedance is mainly determined by the ion environment both at the electrode/solution interface and in the bulk solution. In the presence of the cells, cells attached to the electrode sensor surfaces will alter the local ionic environment at the electrode/solution interface, leading to an increase in the impedance. The more cells there are on the electrodes, the larger the increase in cell-electrode impedance. Furthermore, the impedance change also depends on cell morphology and the extent to which cells attach to the electrodes.
- Cell Index is a quantitative measure of the status of the cells in an electrode-containing well. Under the same physiological conditions, more cells attached on to the electrodes leads to larger cell-electrode resistance value, leading to a larger value for Cell Index. Furthermore, for the same number of cells present in the well, a change in the cell status such as morphology will lead to a change in the Cell Index. For example, an increase in cell adhesion or cell spreading leads to larger cell-electrode contact area which will lead to an increase in cell-electrode resistance and thus a larger value for Cell Index.
- the interaction of biologically active compounds with cells growing inside the wells of the E-Plates results in unique activity patterns (i.e., unique cell impedance curves or cell index curves in response to a compound treatment) that is dependent on the biological mechanism of the compound itself, the concentration, length of incubation and the cell type.
- the "signature" cell responsive patterns to each compound correlates with specific biological phenomenon such as cell cycle arrest, morphology change and cell death.
- Cell response profiling on the RT-CES system has proven effective and we have shown that compounds with similar mechanism of action displays similar patterns.
- the similarity in the cell responsive patterns to compound treatment may indicate similarity in mechanism of action, mode of resistance and possibly molecular targets.
- Figure 1 shows specific profile of A549 lung cancer cells treated with different concentrations of well know anti-mitotic agents paclitaxel and vinblastine. As shown by Figure 1, the cell responsive patterns to paclitaxel and vinblastine compounds are very similar even though the potency may vary between the two compounds.
- Table 8 shows some representative compounds of the present invention whose in vitro and in vivo against tumor activities were studied.
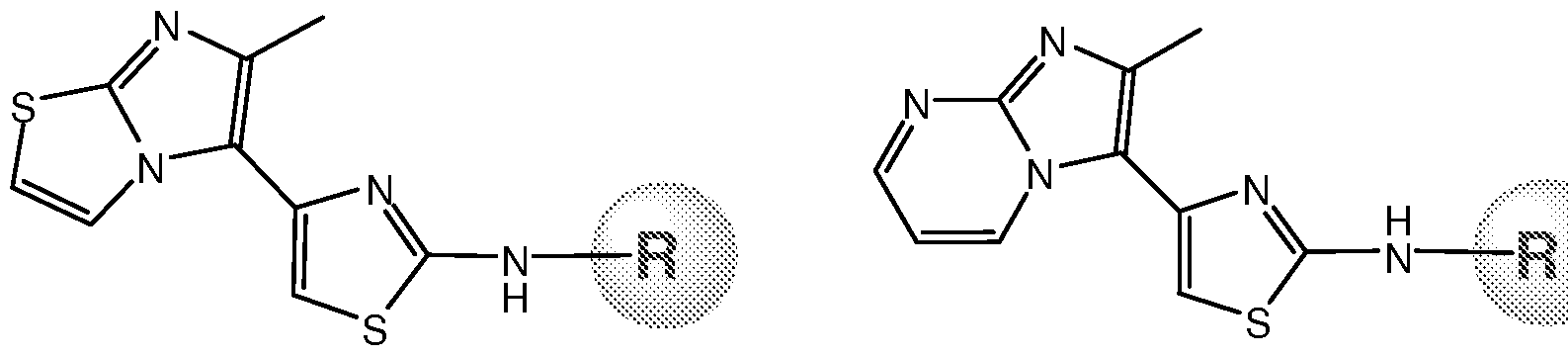
- these compounds are numbered as No. 26, 2-(4-ethoxyphenyl)amino-4-(6- methylimidazo[l,2-a]pyrimidin-3-yl)thiazole (ACEA100160); No. 27, (2-(4- ethoxyphenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5-yl)thiazole (ACEA100162); and No. 28, 2-(4-bromophenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5-yl)thiazole (ACEA100161).
- Compound 26 is 2-(4- ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin-3-yl)thiazole (ACEA100160).
- Compound 27 is 2-(4-ethoxyphenyl)amino-4-(6- methylimidazo[2,l-b]thiazol-5-yl)thiazole (ACEA100162).
- Compound 28 is 2-(4- bromophenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5-yl)thiazole (ACEA100161).
- Figure 2 shows the time-dependent cell index for A549 cells prior to and after addition of different concentrations of compound No. 28 (2-(4- bromophenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5-yl)thiazole, or
- ACEA100161 in Table 8, as determined on Real-Time Cell Electronic Sensing System.
- compound No. 28 ACEA100161
- ACEA100161 exhibited inhibitory ability against the proliferation of A549 cells at various concentrations studied.
- the figure indicate that after compound addition (compound No. 28, or ACEA100161) at concentration of 0.14 uM or above, the cell indices for A549 cells first decreased with time and then increased, showing that A549 cells had complex kinetic responses to compound No. 28 (ACEAlOOl 61).
- Figure 3 shows the time-dependent cell index for A549 cells prior to and after addition of different concentrations of paclitaxel and compound No. 28 (ACEA00161) in Table 8, as determined on Real-Time Electronic Sensing System.
- response pattern of A549 cells to 560 nM of compound No. 28 (ACEA100161) is somewhat similar to that of A549 cells to 25 nM of paclitaxel.
- response pattern of A549 cells to 140 nM of compound No. 28 (ACEA100161) is somewhat similar to that of A549 cells to 12.5 nM of paclitaxel.
- the compound ACEA100161 may have mechanisms of anticancer action similar to those of paclitaxel.
- the compound ACEAlOOl 61 may act on cancer cells through other mechanisms of action, different from those of paclitaxel, even though the time- dependent, cell responsive patterns of compound ACEA100161 are similar to those of paclitaxel. It is also possible that the compound No. 28 (ACEA100161) act on cancer cells through multiple mechanisms of action, including the mechanism of action similar to those of paclitaxel.
- Figure 4 shows dose response curves of A549 cells to the treatment of the compound No. 28 (ACEA100161) in Table 8, at 24 hrs after treatment. The dose- response curve is obtained by plotting the normalized Cell Index value at 24 hours after compound treatment as a function of the compound concentration, based on dose-dependent response profiles shown Figure 2.
- IC50 values i.e. the concentration of the compound at which cell proliferation has been inhibited by 50% due to compound treatment for a specified length of time
- the calculated IC 50 value for compound No. 28 is 86.4 nM.
- Figure 5 shows the time-dependent cell index for A549 cells prior to and after addition of different concentrations of compound No. 26 (2-(4- ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin-3-yl)thiazole, ACEA 100160) in Table 8, as determined on Real-Time Cell Electronic Sensing System.
- ACEA100160 exhibited inhibitory ability against the proliferation of A549 cells at various concentrations studied. Furthermore, the figure indicate that after compound addition (ACEA 100160) at concentration of 4.38 nM or above, the cell indices for A549 cells first decreased with time and then increased, showing that A549 cells had complex kinetic responses to ACEA100160.
- Figure 6 shows dose response curves of A549 cells to the treatment of the compound No. 26 (2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin-3- yl)thiazole, ACEA100160) in Table 8, at 24 hrs after treatment.
- the dose-response curve is obtained by plotting the normalized Cell Index value at 24 hours after compound treatment as a function of the compound concentration, based on dose- dependent response profiles shown Figure 5. From the dose response curve in Figure 6, one can calculate the IC50 values (i.e. the concentration of the compound at which cell proliferation has been inhibited by 50% due to compound treatment for a specified length of time) at 24 hours after compound treatment.
- the calculated IC 50 value at 24 hr after treatment with ACEA 100160 is 23.7 nM.
- Figure 7 shows the time-dependent cell index for A549 cells prior to and after addition of different concentrations of paclitaxel and compound No. 26 (2-(4- ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin-3-yl)thiazole, ACEA100160) in Table 8, as determined on Real-Time Electronic Sensing System.
- response pattern of A549 cells to 35 nM of ACEA100160 is somewhat similar to that of A549 cells to 25 nM of paclitaxel.
- response pattern of A549 cells to 140 nM of ACEA 100160 is somewhat similar to that of A549 cells to 50 nM of paclitaxel.
- the compound ACEA100160 may have mechanisms of anticancer action similar to those of paclitaxel.
- ACEA100160 may act on cancer cells through other mechanisms of action, different from those of paclitaxel, even though the time- dependent, cell responsive patterns of ACEA 100160 are similar to those of paclitaxel.
- ACEA100160 act on cancer cells through multiple mechanisms of action, including the mechanism of action similar to those of paclitaxel.
- Figure 8 shows the time-dependent cell index for
- ACEA100162 exhibited inhibitory ability against the proliferation of A549 cells at various concentrations studied.
- the figure indicate that after compound addition (ACEA 100162) at concentration of 62.5 nM or above, the cell indices for A549 cells first decreased with time and then increased, showing that A549 cells had complex kinetic responses to ACEA100162.
- Figure 9 shows dose response curves of A549 cells to the treatment of the compound No. 27 (2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[2,l-b]thiazol-5- yl)thiazole, ACEA100162) in Table 8, at 24 hrs after treatment.
- the dose-response curve is obtained by plotting the normalized Cell Index value at 24 hours after compound treatment as a function of the compound concentration, based on dose- dependent response profiles shown Figure 8. From the dose response curve in Figure 9, one can calculate the IC50 values (i.e. the concentration of the compound at which cell proliferation has been inhibited by 50% due to compound treatment for a specified length of time) at 24 hours after compound treatment.
- the calculated IC 50 value at 24 hr after treatment with ACEA100162 is 57.4 nM.
- NCI-H460 non-small cell lung cancer cells
- MCF7 breast cancer cells
- SKO V3 Ovarian cancer cells
- Jurkat Leukemia
- PC3 prostate cancer cells
- Panc-1 Pancreatic Carcinoma
- SH-SY5Y Neroblastoma
- HepG2 human hepatosarcoma
- GTL16 Gastric carcinoma
- B16- Iuc Melanoma
- KB Head-Neck cancer cells
- HeLa Cervical carcinoma
- HT1080 fibrosarcoma cancer cells
- MDCK Kidney cells
- HT29 Cold cancer cells
- A549 non-small cell lung cancer cells
- other cell lines see Table 9
- the cells were allowed to grow for about 20 hours prior to the addition of compound 27 (2-(4-ethoxyphenyl)amino-4-(6- methylimidazo[2,l-b]thiazol-5-yl)thiazole, ACEA100162)) dissolved in DMSO solution (final DMSO concentration: 0.2%; final ACEA100162 concentration: between 3.13 nM and 200 nM).
- DMSO solution final DMSO concentration: 0.2%
- final ACEA100162 concentration between 3.13 nM and 200 nM
- Neuroblastmoa (Human) SY5Y 13.4 nM 6.7 nM Hepatosarcoma (Human) HepG2 >200 nM >200 nM Gastric carcinoma (Human) GTL16 138.5 nM 2.1 nM Melanoma (Rat) B16-luc 705.3 nM 643.2 nM Head-Neck (Human) KB 22.2 nM 18.1 nM Cervical carcinoma (Human) HeIa 5.85 nM 6.95 nM Fibrosarcoma cancer (Human) HT1080 2.39 nM 2.69 nM Kidney tumor (Monkey) MDCK 929.6 nM 1 .87 uM Colon cancer (Human) HT29 53.6 nM 58.8 nM fibroblast (Mouse) 3T3 20.6 nM 51.5 nM Ovary adenocarcinoma (Human) ST30 38.8 nM 42.4 nM Oral epit
- FIG. 10 shows the time-dependent cell index for a number of cell lines prior to and after addition of compound 27 (ACEA 100162) at various concentrations.
- ACEA 100162 exhibited inhibitory effect on the proliferation of a number of cancer cell lines.
- the susceptibility to ACEA 100162 differs among the cancer cell types. For some cancer cell types, a low dosage of ACEA100162 is sufficient to significantly inhibit cancer cell proliferation, whilst for other cancer cell types, a higher dosage is needed to achieve similar inhibition degree.
- NCI-H460 non-small cell lung cancer cells
- MCF7 breast cancer cells
- SKO V3 Ovarian cancer cells
- Jurkat Leukemia
- PC3 prostate cancer cells
- Panc-1 Pancreatic Carcinoma
- SH-SY5Y Neroblastoma
- HepG2 human hepatosarcoma
- GTL16 Gastric carcinoma
- B16- Iuc Melanoma
- KB Head-Neck cancer cells
- HeLa Cervical carcinoma
- HT1080 fibrosarcoma cancer cells
- MDCK Kidney cells
- HT29 Cold cancer cells
- A549 non-small cell lung cancer cells
- A549 non-small cell lung cancer cells
- other cell lines see Table 10
- the cells were allowed to grow for about 20 hours prior to the addition of (2-(4-ethoxyphenyl)amino-4-(6- methylimidazo[l,2-a]pyrimidin-3-yl)thiazole, ACEA100160) dissolved in DMSO solution (final DMSO concentration: 0.2%; final ACEA100160 concentration: between ⁇ 2.nM and ⁇ 2 uM.
- DMSO solution final DMSO concentration: 0.2%
- final ACEA100160 concentration between ⁇ 2.nM and ⁇ 2 uM.
- the cell-electrode impedance was continuously measured and the corresponding, time dependent, dose-dependent cell-index values were derived and recorded.
- dose-response curves similar to those in Figure 4 and Figure 6 were plotted at selected time points of 24 hrs and 48 hrs after compound treatment. IC50 values were then calculated based on such dose response curves and are summarized in Table 10.
- Neuroblastmoa Human SY5Y Hepatosarcoma (Human) HepG2 >2uM >2uM Gastric carcinoma (Human) GTL16 147.6 nM 0.4 nM Melanoma (Rat) B16-IUC 3.88 uM 10O nM Head-Neck (Human) KB 12.5nM 83.4nM Cervical carcinoma (Human) HeIa 31 .7nM 56.7nM (42hr) Fibrosarcoma cancer (Human) HT1080 17.7nM 25.5nM Kidney tumor (Monkey) MDCK 2.O uM 5.09 uM Colon cancer (Human) HT29 61.
- FIG. 11 shows the time-dependent cell index for a number of cell lines prior to and after addition of ACEA 100160 at various concentrations.
- ACEA 100160 exhibited inhibitory effect on the proliferation of a number of cancer cell lines.
- the susceptibility to ACEA 100160 differs among the cancer cell types. For some cancer cell types, a low dosage of ACEA100160 is sufficient to significantly inhibit cancer cell proliferation, whilst for other cancer cell types, a higher dosage is needed to achieve similar inhibition degree.
- the RT-CES System for such a study includes a RT-CES analyzer, an Mult-E-Plate Station, E-Plate device and RT-CES software.
- A549 cells human lung cancer cells
- the E-Plates containing the cells were placed onto an Mult-E-Plate station inside the CO 2 incubator.
- the cell-electrode resistance for each well of the E-Plate devices is continuously monitored by RT-CES analyzer under the control of RT-CES software every 30 minutes .
- Figure 2 shows the normalized cell index as a function of time prior to and after compound addition for different concentrations of Compound 28 (ACEAlOOl 61).
- the cell index data was normalized at the time point immediately prior to compound addition (about 20 hrs after cell seeding).
- Acute oxidative stress we plotted the normalized cell-index values at a time point of 24 hrs after compound addition as a function of the compound concentration in Figure 4, based on dose-dependent cell response profiles shown in Figure 2. From the dose response curve in Figure 4, one can calculate the IC50 values (i.e. the concentration of the compound at which cell proliferation has been inhibited by 50% due to compound treatment for a specified length of time) at 24 hours after compound treatment. The calculated
- the RT-CES System for such a study includes a RT-CES analyzer, an Mult-E-Plate Station, E-Plate device and RT-CES software.
- A549 cells human lung cancer cells
- the E-Plates containing the cells were placed onto an Mult-E-Plate station inside the CO 2 incubator.
- the cell-electrode resistance for each well of the E-Plate devices is continuously monitored by RT-CES analyzer under the control of RT-CES software every 30 minutes . After about 20 hours, the measurement on RT-CES system was paused; the E-Plate was removed from the Mult-E-Plate station for compound addition.
- Figure 5 shows the normalized cell index as a function of time prior to and after compound addition for different concentrations of ACEA100160.
- the cell index data was normalized at the time point immediately prior to compound addition (about 20 hrs after cell seeding).
- the normalized cell-index values at a time point of 24 hrs after compound addition as a function of the compound concentration in Figure 6, based on dose-dependent cell response profiles shown in Figure 5. From the dose response curve in Figure 6, one can calculate the IC50 values (i.e. the concentration of the compound at which cell proliferation has been inhibited by 50% due to compound treatment for a specified length of time) at 24 hours after compound treatment.
- the calculated IC 50 value for ACEA100160 is 23.7 nM.
- the RT-CES System for such a study includes a RT-CES analyzer, a Mult-E-Plate Station, E-Plate device and RT-CES software.
- A549 cells human lung cancer cells
- the E- Plates containing the cells were placed onto a Mult- E- Plate station inside the CO 2 incubator.
- the cell-electrode resistance for each well of the E- Plate devices is continuously monitored by RT-CES analyzer under the control of RT-CES software every 30 minutes . After about 20 hours, the measurement on
- ACEA 100162 The cell index data was normalized at the time point immediately prior to compound addition (about 20 hrs after cell seeding).
- the calculated IC 50 value for ACEA 100162 is 57.4 nM.
- Example 1 The anti-tumor activity of ACEA100160 and ACEA100162 against S 180 Bearing Mouse Model A. Materials and Methods A.I Compound and solution
- ACEA100160 2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin-3- yl)thiazole
- ACEA100162 2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[2,l- b]thiazol-5-yl)thiazole
- HP-beta-CD solution is used as a vehicle negative control.
- S 180 sarcoma murine cancer cell line S 180.
- the malignant ascites model of ICR mouse was induced by S 180 cells.
- ICR murine S 180 bearing mice Under a sterilized condition, sacrifice the ICR murine S 180 bearing mice, take out of the ascites and dilute with NS to certain cell numbers. Inject 0.2 ml cell suspension (10 6 cells) subcutaneously into the left flank on ICR mice. On the first treatment day before iv or ip, implanted animals are randomly divided into ten groups as following. The drugs were administrated in the schedule of Table 11 respectively. The solvent HP-beta-CD solution is used as a vehicle negative control. Observe clinical signs at least daily.
- Tumor weight was recorded.
- ACEA 100160 40 mg/kg (qd, i,p) and 40 mg/kg (qd, i,v) groups inhibit S 180 tumor growth, with the inhibitory rate value of 52.7% and 52.2% respectively.
- ACEA100162 40 mg/kg (qd, i,p), ACEA100162 80 mg/kg (qd, i,p) and ACEA100162 40 mg/kg (qd, i,v) groups showed anti-tumor activities0 with the inhibitory rate value of 59.3%, 77.2%, and 60.7% respectively.
- Table 12 Effects of ACEA100160 and ACEA100162 on ICR mice body weight and tumor weight at pre-dose and post-dose. ICR mice bearing S 180 cancer were treated with ACEA100160 and ACEA100162 for 9 days ( ⁇ ⁇ SD).
- Example 2 The Anti-tumor activity of ACEA100160 and
- ACEA100160 2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[l,2-a]pyrimidin-3- yl)thiazole
- ACEA100162 2-(4-ethoxyphenyl)amino-4-(6-methylimidazo[2,l- b]thiazol-5-yl)thiazole
- HP-beta-CD solution is used as a vehicle negative control.
- LLC murine Lewis lung cancer cell line.
- Tumor weight was recorded.
- ACEA 100160 40 mg/kg (qd, i,p) group exhibited anti-tumor activities, with the inhibitory rate value of 27.13%, respectively.
- ACEA100162 40 mg/kg (qd, i,p) and ACEA100162 40 mg/kg (qd, i,v) groups 5 showed anti-tumor activities, with the inhibitory rate value of 39.15%, and 25.5%, respectively (Table 14 and Figure 13).
- Table 14 Effects of ACEA100160 and ACEA100162 on C57 BL/6 mice body weight and tumor weight at pre-dose and post-dose. C57 BL/6 mice bearing LLC cancer were treated with ACEA100160, ACEA100162 and vehicle negative control for 12 days ( x ⁇ SD).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description






























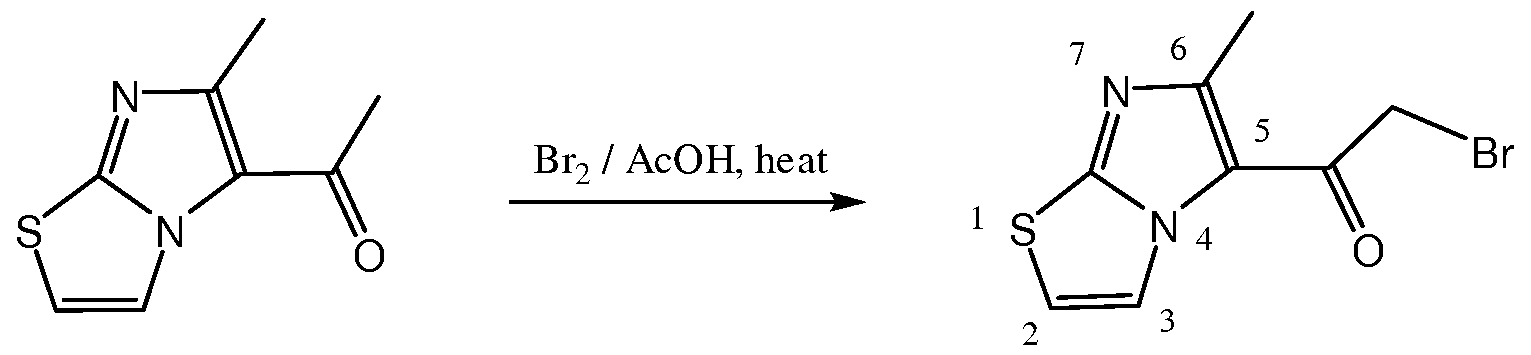





Claims















Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010517163A JP5457344B2 (en) | 2007-07-17 | 2008-07-17 | Use as heterocyclic compounds and anticancer agents |
| EP08827232.3A EP2188271B1 (en) | 2007-07-17 | 2008-07-17 | Heterocyclic compounds and uses as anticancer agents |
| ES08827232.3T ES2617305T3 (en) | 2007-07-17 | 2008-07-17 | Heterocyclic compounds and uses as anticancer agents |
| CN2008801074039A CN101801942B (en) | 2007-07-17 | 2008-07-17 | Heterocyclic compounds and uses as anticancer agents |
| CA2693162A CA2693162C (en) | 2007-07-17 | 2008-07-17 | Heterocyclic compounds and uses as anticancer agents |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95019107P | 2007-07-17 | 2007-07-17 | |
| US95019707P | 2007-07-17 | 2007-07-17 | |
| US60/950,191 | 2007-07-17 | ||
| US60/950,197 | 2007-07-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009023402A2 true WO2009023402A2 (en) | 2009-02-19 |
| WO2009023402A3 WO2009023402A3 (en) | 2009-04-23 |
Family
ID=40351402
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/070348 WO2009023402A2 (en) | 2007-07-17 | 2008-07-17 | Heterocyclic compounds and uses as anticancer agents |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8252822B2 (en) |
| EP (2) | EP2188271B1 (en) |
| JP (1) | JP5457344B2 (en) |
| CN (1) | CN101801942B (en) |
| CA (1) | CA2693162C (en) |
| ES (1) | ES2617305T3 (en) |
| WO (1) | WO2009023402A2 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2306836A1 (en) * | 2008-07-01 | 2011-04-13 | PTC Therapeutics, Inc. | Bmi-1 protein expression modulators |
| US20110207751A1 (en) * | 2010-02-19 | 2011-08-25 | Long Mao | Heterocyclic compounds and uses as anticancer agents |
| WO2011124190A2 (en) | 2010-04-06 | 2011-10-13 | Zentiva, K.S. | Method of producing 4-(2-(substituted)-1-(1-hydroxycyclohexyl)ethyl)phenols by o- demethylation of their methylethers by means of inodorous aromatic thiols |
| US8252822B2 (en) | 2007-07-17 | 2012-08-28 | Acea Biosciences, Inc. | Heterocyclic compounds and uses as anticancer agents |
| WO2012162468A1 (en) * | 2011-05-25 | 2012-11-29 | Janssen Pharmaceutica Nv | Thiazol derivatives as pro -matrix metalloproteinase inhibitors |
| US9505728B2 (en) | 2012-03-09 | 2016-11-29 | Inception 2, Inc. | Triazolone compounds and uses thereof |
| US9676754B2 (en) | 2012-12-20 | 2017-06-13 | Inception 2, Inc. | Triazolone compounds and uses thereof |
| US9776976B2 (en) | 2013-09-06 | 2017-10-03 | Inception 2, Inc. | Triazolone compounds and uses thereof |
| KR20220020651A (en) * | 2020-08-12 | 2022-02-21 | 광주과학기술원 | Composition for prevention or treatment of triple-negative breast cancer through blocking microtubule acetylation |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103204862B (en) * | 2012-01-12 | 2014-12-17 | 清华大学深圳研究生院 | 6-phenylimidazol[2, 1-b]thiazole-3-amide derivative, its preparation method and application |
| MX2017002610A (en) | 2014-08-29 | 2017-10-11 | Tes Pharma S R L | INHIBITORS OF A-AMINO-ß-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE. |
| PE20180529A1 (en) | 2015-05-20 | 2018-03-19 | Amgen Inc | TRIAZOLE AGONISTS OF THE APJ RECEPTOR |
| EP3452466B1 (en) | 2016-05-03 | 2020-08-12 | Amgen Inc. | Heterocyclic triazole compounds as agonists of the apj receptor |
| US11046680B1 (en) | 2016-11-16 | 2021-06-29 | Amgen Inc. | Heteroaryl-substituted triazoles as APJ receptor agonists |
| MA46827A (en) | 2016-11-16 | 2019-09-25 | Amgen Inc | CYCLOALKYL SUBSTITUTE TRIAZOLE COMPOUNDS AS APJ RECEPTOR AGONISTS |
| EP3541810B1 (en) | 2016-11-16 | 2020-12-23 | Amgen Inc. | Triazole phenyl compounds as agonists of the apj receptor |
| EP3541803B1 (en) | 2016-11-16 | 2020-12-23 | Amgen Inc. | Triazole pyridyl compounds as agonists of the apj receptor |
| EP3541802A1 (en) | 2016-11-16 | 2019-09-25 | Amgen Inc. | Alkyl substituted triazole compounds as agonists of the apj receptor |
| EP3541792B1 (en) | 2016-11-16 | 2020-12-23 | Amgen Inc. | Triazole furan compounds as agonists of the apj receptor |
| WO2019089335A1 (en) | 2017-11-03 | 2019-05-09 | Amgen Inc. | Fused triazole agonists of the apj receptor |
| MA52487A (en) | 2018-05-01 | 2021-03-10 | Amgen Inc | PYRIMIDINONES SUBSTITUTED AS APJ RECEPTOR AGONISTS |
| JP7002436B2 (en) | 2018-11-13 | 2022-01-20 | 三菱電機ビルテクノサービス株式会社 | Electric blinds |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003313176A (en) | 2002-04-24 | 2003-11-06 | Sankyo Co Ltd | Aminoazole derivative |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5391485A (en) * | 1985-08-06 | 1995-02-21 | Immunex Corporation | DNAs encoding analog GM-CSF molecules displaying resistance to proteases which cleave at adjacent dibasic residues |
| JPS63500636A (en) * | 1985-08-23 | 1988-03-10 | 麒麟麦酒株式会社 | DNA encoding multipotent granulocyte colony stimulating factor |
| US4810643A (en) * | 1985-08-23 | 1989-03-07 | Kirin- Amgen Inc. | Production of pluripotent granulocyte colony-stimulating factor |
| AU3740101A (en) * | 2000-03-01 | 2001-09-12 | Janssen Pharmaceutica Nv | 2,4-disubstituted thiazolyl derivatives |
| KR101034489B1 (en) * | 2001-08-13 | 2011-05-17 | 얀센 파마슈티카 엔.브이. | 2-amino-4,5-trisubstituted thiazolyl derivatives |
| US7158675B2 (en) * | 2002-05-14 | 2007-01-02 | Microsoft Corporation | Interfacing with ink |
| JP2005537498A (en) * | 2002-07-20 | 2005-12-08 | アセア バイオサイエンシーズ,インク. | Measuring device and method using impedance |
| US7468255B2 (en) * | 2002-12-20 | 2008-12-23 | Acea Biosciences | Method for assaying for natural killer, cytotoxic T-lymphocyte and neutrophil-mediated killing of target cells using real-time microelectronic cell sensing technology |
| US7470533B2 (en) * | 2002-12-20 | 2008-12-30 | Acea Biosciences | Impedance based devices and methods for use in assays |
| US20050227989A1 (en) * | 2004-04-13 | 2005-10-13 | Icagen, Inc. | Polycyclic thiazoles as potassium ion channel modulators |
| JP2008540537A (en) | 2005-05-09 | 2008-11-20 | アキリオン ファーマシューティカルズ,インコーポレーテッド | Thiazole compounds and methods of use |
| AU2006290814B2 (en) * | 2005-09-13 | 2012-06-07 | Janssen Pharmaceutica N.V. | 2-aniline-4-aryl substituted thiazole derivatives |
| ES2617305T3 (en) | 2007-07-17 | 2017-06-16 | Acea Biosciences, Inc. | Heterocyclic compounds and uses as anticancer agents |
| EP2306836B1 (en) | 2008-07-01 | 2016-09-07 | PTC Therapeutics, Inc. | Bmi-1 protein expression modulators |
-
2008
- 2008-07-17 ES ES08827232.3T patent/ES2617305T3/en active Active
- 2008-07-17 US US12/175,154 patent/US8252822B2/en active Active
- 2008-07-17 CN CN2008801074039A patent/CN101801942B/en active Active
- 2008-07-17 JP JP2010517163A patent/JP5457344B2/en active Active
- 2008-07-17 CA CA2693162A patent/CA2693162C/en active Active
- 2008-07-17 EP EP08827232.3A patent/EP2188271B1/en active Active
- 2008-07-17 EP EP17150851.8A patent/EP3173414A1/en not_active Withdrawn
- 2008-07-17 WO PCT/US2008/070348 patent/WO2009023402A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003313176A (en) | 2002-04-24 | 2003-11-06 | Sankyo Co Ltd | Aminoazole derivative |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2188271A4 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8252822B2 (en) | 2007-07-17 | 2012-08-28 | Acea Biosciences, Inc. | Heterocyclic compounds and uses as anticancer agents |
| US8680113B2 (en) | 2008-07-01 | 2014-03-25 | Ptc Therapeutics, Inc. | BMI-1 protein expression modulators |
| EP2306836A4 (en) * | 2008-07-01 | 2011-12-21 | Ptc Therapeutics Inc | Bmi-1 protein expression modulators |
| EP2306836A1 (en) * | 2008-07-01 | 2011-04-13 | PTC Therapeutics, Inc. | Bmi-1 protein expression modulators |
| CN102822173A (en) * | 2010-02-19 | 2012-12-12 | 美国艾森生物科学公司 | Heterocyclic compounds and uses as anticancer agents |
| US20110207751A1 (en) * | 2010-02-19 | 2011-08-25 | Long Mao | Heterocyclic compounds and uses as anticancer agents |
| WO2011103509A1 (en) | 2010-02-19 | 2011-08-25 | Acea Biosciences Inc. | Heterocyclic compounds and uses as anticancer agents |
| WO2011124190A2 (en) | 2010-04-06 | 2011-10-13 | Zentiva, K.S. | Method of producing 4-(2-(substituted)-1-(1-hydroxycyclohexyl)ethyl)phenols by o- demethylation of their methylethers by means of inodorous aromatic thiols |
| WO2012162468A1 (en) * | 2011-05-25 | 2012-11-29 | Janssen Pharmaceutica Nv | Thiazol derivatives as pro -matrix metalloproteinase inhibitors |
| US9505728B2 (en) | 2012-03-09 | 2016-11-29 | Inception 2, Inc. | Triazolone compounds and uses thereof |
| US9676754B2 (en) | 2012-12-20 | 2017-06-13 | Inception 2, Inc. | Triazolone compounds and uses thereof |
| US10568871B2 (en) | 2012-12-20 | 2020-02-25 | Tempest Therapeutics, Inc. | Triazolone compounds and uses thereof |
| US11666557B2 (en) | 2012-12-20 | 2023-06-06 | Tempest Therapeutics, Inc. | Triazolone compounds and uses thereof |
| US9776976B2 (en) | 2013-09-06 | 2017-10-03 | Inception 2, Inc. | Triazolone compounds and uses thereof |
| KR20220020651A (en) * | 2020-08-12 | 2022-02-21 | 광주과학기술원 | Composition for prevention or treatment of triple-negative breast cancer through blocking microtubule acetylation |
| KR102397817B1 (en) * | 2020-08-12 | 2022-05-13 | 광주과학기술원 | Composition for prevention or treatment of triple-negative breast cancer through blocking microtubule acetylation |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101801942B (en) | 2013-03-27 |
| CN101801942A (en) | 2010-08-11 |
| EP3173414A1 (en) | 2017-05-31 |
| ES2617305T3 (en) | 2017-06-16 |
| JP5457344B2 (en) | 2014-04-02 |
| US8252822B2 (en) | 2012-08-28 |
| US20090048271A1 (en) | 2009-02-19 |
| CA2693162A1 (en) | 2009-02-19 |
| CA2693162C (en) | 2017-05-16 |
| JP2011507798A (en) | 2011-03-10 |
| WO2009023402A3 (en) | 2009-04-23 |
| EP2188271B1 (en) | 2017-01-11 |
| EP2188271A4 (en) | 2010-12-08 |
| EP2188271A2 (en) | 2010-05-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2188271B1 (en) | Heterocyclic compounds and uses as anticancer agents | |
| Wang et al. | Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships | |
| US20210100802A1 (en) | Heterocyclic compounds and uses as anticancer agents | |
| CN107266453B (en) | Pyrrolopyrimidines as protein kinase inhibitors | |
| WO2015006754A2 (en) | Heterocyclic compounds and uses thereof | |
| EP2361254A1 (en) | Raf inhibitors and their uses | |
| Zou et al. | Synthesis and evaluation of N-heteroaromatic ring-based analogs of piperlongumine as potent anticancer agents | |
| CN111247152B (en) | Cyclic iminopyrimidine derivatives as kinase inhibitors | |
| Spanò et al. | [1, 2] Oxazolo [5, 4-e] isoindoles as promising tubulin polymerization inhibitors | |
| CN117940436A (en) | 7- (Naphthalene-1-yl) pyrido [4,3-d ] pyrimidine derivative and preparation and application thereof | |
| JP2011162562A (en) | Substituted organosulfur compound and method of using the same | |
| KR101754664B1 (en) | Substituted pyrido[2,3-d]pyrimidin-7(8h)-ones and therapeutic uses thereof | |
| Ouellette et al. | 4-(3-Alkyl-2-oxoimidazolidin-1-yl)-N-phenylbenzenesulfonamide salts: Novel hydrosoluble prodrugs of antimitotics selectively bioactivated by the cytochrome P450 1A1 in breast cancer cells | |
| CA2653298A1 (en) | Crystalline form of 5(s)-(2'-hydroxyethoxy)-20(s)-camptothecin | |
| WO2012097196A1 (en) | Pyrazolopyrimidine derivatives and uses as anticancer agents | |
| CA2789249A1 (en) | Heterocyclic compounds and uses as anticancer agents | |
| CN116249703A (en) | Macrocyclic K-RAS G12C inhibitor, preparation method and application thereof | |
| TWI640518B (en) | Fused tricyclic compounds as raf kinase inhibitors | |
| PL227918B1 (en) | 10H-1,8-diazaphenothiazine, its application and method for obtaining, 10-substituted 1,8-diazaphenothiazines, their applications and method for obtaining and the pharmaceutical compositions containing 10H-dipyridothiazine with the structure of 10H-1,8-diazaphenothiazine and/or new 10-substituted 1,8-diazaphenothiazines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880107403.9 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08827232 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2693162 Country of ref document: CA Ref document number: 2010517163 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 561/KOLNP/2010 Country of ref document: IN |
|
| REEP | Request for entry into the european phase |
Ref document number: 2008827232 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008827232 Country of ref document: EP |