WO2009014972A1 - Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein - Google Patents
Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein Download PDFInfo
- Publication number
- WO2009014972A1 WO2009014972A1 PCT/US2008/070254 US2008070254W WO2009014972A1 WO 2009014972 A1 WO2009014972 A1 WO 2009014972A1 US 2008070254 W US2008070254 W US 2008070254W WO 2009014972 A1 WO2009014972 A1 WO 2009014972A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- amino
- mmol
- aryl
- optionally substituted
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 179
- 238000000034 method Methods 0.000 title abstract description 71
- 230000030136 gastric emptying Effects 0.000 title abstract description 11
- 230000002496 gastric effect Effects 0.000 title description 14
- 230000005176 gastrointestinal motility Effects 0.000 claims abstract description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 90
- 239000001257 hydrogen Substances 0.000 claims description 90
- -1 alkyl-heterocycle Chemical group 0.000 claims description 83
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 81
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 57
- 125000003118 aryl group Chemical group 0.000 claims description 54
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 51
- 125000000623 heterocyclic group Chemical group 0.000 claims description 47
- 229910052736 halogen Inorganic materials 0.000 claims description 25
- 150000002367 halogens Chemical group 0.000 claims description 25
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 22
- 150000002431 hydrogen Chemical group 0.000 claims description 21
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 20
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 19
- 150000003839 salts Chemical class 0.000 claims description 14
- 125000003545 alkoxy group Chemical group 0.000 claims description 13
- 239000003814 drug Substances 0.000 claims description 13
- 229910052757 nitrogen Inorganic materials 0.000 claims description 12
- 125000003107 substituted aryl group Chemical group 0.000 claims description 12
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 10
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims description 10
- 229910052760 oxygen Inorganic materials 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 6
- 229910052717 sulfur Inorganic materials 0.000 claims description 6
- 239000012453 solvate Substances 0.000 claims description 5
- 125000002950 monocyclic group Chemical group 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 108010031944 Tryptophan Hydroxylase Proteins 0.000 abstract description 13
- 102000005506 Tryptophan Hydroxylase Human genes 0.000 abstract description 13
- 230000002401 inhibitory effect Effects 0.000 abstract description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 300
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 215
- 239000000203 mixture Substances 0.000 description 202
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 173
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 149
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 144
- 239000002904 solvent Substances 0.000 description 131
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical group CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 125
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 120
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 110
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 102
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 89
- 239000000243 solution Substances 0.000 description 89
- 238000006243 chemical reaction Methods 0.000 description 84
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 80
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 77
- 239000000047 product Substances 0.000 description 75
- 238000003786 synthesis reaction Methods 0.000 description 72
- 230000015572 biosynthetic process Effects 0.000 description 71
- 239000011541 reaction mixture Substances 0.000 description 71
- 238000005160 1H NMR spectroscopy Methods 0.000 description 69
- 235000019439 ethyl acetate Nutrition 0.000 description 63
- 229910000029 sodium carbonate Inorganic materials 0.000 description 52
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 50
- 229940093499 ethyl acetate Drugs 0.000 description 47
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 44
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 44
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 43
- 229910052938 sodium sulfate Inorganic materials 0.000 description 43
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 42
- 235000019260 propionic acid Nutrition 0.000 description 42
- 238000001816 cooling Methods 0.000 description 41
- 238000002953 preparative HPLC Methods 0.000 description 39
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 37
- NFIVJOSXJDORSP-QMMMGPOBSA-N (2s)-2-amino-3-(4-boronophenyl)propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(B(O)O)C=C1 NFIVJOSXJDORSP-QMMMGPOBSA-N 0.000 description 36
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 36
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 32
- 239000012043 crude product Substances 0.000 description 32
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 31
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 31
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 30
- 239000012267 brine Substances 0.000 description 30
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 29
- 201000010099 disease Diseases 0.000 description 26
- 235000011152 sodium sulphate Nutrition 0.000 description 26
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 25
- 239000012044 organic layer Substances 0.000 description 25
- 210000004027 cell Anatomy 0.000 description 22
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 22
- 238000000746 purification Methods 0.000 description 22
- 238000004128 high performance liquid chromatography Methods 0.000 description 21
- JPZOAVGMSDSWSW-UHFFFAOYSA-N 4,6-dichloropyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(Cl)=N1 JPZOAVGMSDSWSW-UHFFFAOYSA-N 0.000 description 20
- 239000000706 filtrate Substances 0.000 description 20
- 229910000104 sodium hydride Inorganic materials 0.000 description 20
- 230000002829 reductive effect Effects 0.000 description 19
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- 239000007832 Na2SO4 Substances 0.000 description 17
- 125000000217 alkyl group Chemical group 0.000 description 17
- 239000000460 chlorine Substances 0.000 description 17
- 230000000694 effects Effects 0.000 description 17
- 239000003112 inhibitor Substances 0.000 description 17
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 16
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 16
- 208000035475 disorder Diseases 0.000 description 16
- 229940076279 serotonin Drugs 0.000 description 16
- 239000007787 solid Substances 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 230000003389 potentiating effect Effects 0.000 description 15
- MWKJTNBSKNUMFN-UHFFFAOYSA-N trifluoromethyltrimethylsilane Chemical compound C[Si](C)(C)C(F)(F)F MWKJTNBSKNUMFN-UHFFFAOYSA-N 0.000 description 15
- 238000005481 NMR spectroscopy Methods 0.000 description 14
- 239000002253 acid Substances 0.000 description 14
- 235000019441 ethanol Nutrition 0.000 description 14
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 14
- 229940073584 methylene chloride Drugs 0.000 description 13
- 230000008569 process Effects 0.000 description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 12
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 12
- 239000000741 silica gel Substances 0.000 description 12
- 229910002027 silica gel Inorganic materials 0.000 description 12
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 11
- 241000700159 Rattus Species 0.000 description 11
- 239000002552 dosage form Substances 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 108010069013 Phenylalanine Hydroxylase Proteins 0.000 description 10
- 102100038223 Phenylalanine-4-hydroxylase Human genes 0.000 description 10
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 10
- 229910000024 caesium carbonate Inorganic materials 0.000 description 10
- 239000003960 organic solvent Substances 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 9
- 108091000117 Tyrosine 3-Monooxygenase Proteins 0.000 description 9
- 102000048218 Tyrosine 3-monooxygenases Human genes 0.000 description 9
- 238000004440 column chromatography Methods 0.000 description 9
- 239000002480 mineral oil Substances 0.000 description 9
- 235000010446 mineral oil Nutrition 0.000 description 9
- 230000002093 peripheral effect Effects 0.000 description 9
- 239000003981 vehicle Substances 0.000 description 9
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 8
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 8
- 230000014759 maintenance of location Effects 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- 239000000377 silicon dioxide Substances 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- 208000024891 symptom Diseases 0.000 description 8
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 8
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 7
- 238000013459 approach Methods 0.000 description 7
- 210000004556 brain Anatomy 0.000 description 7
- 125000004122 cyclic group Chemical group 0.000 description 7
- 239000005457 ice water Substances 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 6
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 6
- 238000006069 Suzuki reaction reaction Methods 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 229910052796 boron Inorganic materials 0.000 description 6
- 239000003610 charcoal Substances 0.000 description 6
- 238000004587 chromatography analysis Methods 0.000 description 6
- 229910052681 coesite Inorganic materials 0.000 description 6
- 229910052906 cristobalite Inorganic materials 0.000 description 6
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 6
- 125000001072 heteroaryl group Chemical group 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 239000006201 parenteral dosage form Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 229910052682 stishovite Inorganic materials 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 229910052905 tridymite Inorganic materials 0.000 description 6
- KOGJTURHFMAARC-ZDUSSCGKSA-N (2s)-3-[4-(2-amino-6-chloropyrimidin-4-yl)phenyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound C1=CC(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CC=C1C1=CC(Cl)=NC(N)=N1 KOGJTURHFMAARC-ZDUSSCGKSA-N 0.000 description 5
- ARSUABFPGZPNOY-FPOVZHCZSA-N (2s)-3-[4-[2-amino-6-[(1s)-1-(4-bromophenyl)-2,2,2-trifluoroethoxy]pyrimidin-4-yl]phenyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound C1=CC(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CC=C1C1=CC(O[C@@H](C=2C=CC(Br)=CC=2)C(F)(F)F)=NC(N)=N1 ARSUABFPGZPNOY-FPOVZHCZSA-N 0.000 description 5
- GIHOZZNRGGLUNZ-UHFFFAOYSA-N 2,2,2-trifluoro-1-[2-(3-methylphenyl)phenyl]ethanol Chemical compound CC1=CC=CC(C=2C(=CC=CC=2)C(O)C(F)(F)F)=C1 GIHOZZNRGGLUNZ-UHFFFAOYSA-N 0.000 description 5
- FZFWPURYSWKIRT-UHFFFAOYSA-N 3-cyclopentyloxy-4-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1OC1CCCC1 FZFWPURYSWKIRT-UHFFFAOYSA-N 0.000 description 5
- MCLXKFUCPVGZEN-UHFFFAOYSA-N 4,6-dichloro-1,3,5-triazin-2-amine Chemical compound NC1=NC(Cl)=NC(Cl)=N1 MCLXKFUCPVGZEN-UHFFFAOYSA-N 0.000 description 5
- KRRTXVSBTPCDOS-UHFFFAOYSA-N 5-bromopyrazin-2-amine Chemical compound NC1=CN=C(Br)C=N1 KRRTXVSBTPCDOS-UHFFFAOYSA-N 0.000 description 5
- 229910004373 HOAc Inorganic materials 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- 230000009471 action Effects 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 230000003197 catalytic effect Effects 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 235000012054 meals Nutrition 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 125000001624 naphthyl group Chemical group 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 239000008194 pharmaceutical composition Substances 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 230000005855 radiation Effects 0.000 description 5
- 230000002441 reversible effect Effects 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 239000012279 sodium borohydride Substances 0.000 description 5
- 229910000033 sodium borohydride Inorganic materials 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 238000011282 treatment Methods 0.000 description 5
- YBMVIGVXVXAKDM-LLVKDONJSA-N (1r)-1-[4-chloro-2-(3-methylpyrazol-1-yl)phenyl]-2,2,2-trifluoroethanol Chemical compound N1=C(C)C=CN1C1=CC(Cl)=CC=C1[C@@H](O)C(F)(F)F YBMVIGVXVXAKDM-LLVKDONJSA-N 0.000 description 4
- HNXQXTQTPAJEJL-UHFFFAOYSA-N 2-aminopteridin-4-ol Chemical compound C1=CN=C2NC(N)=NC(=O)C2=N1 HNXQXTQTPAJEJL-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 0 CC(C)(CC(C)(C)*(CCCC1)CCCC1(*)[C@](*=C*C(**)(*C1(*)CCCCCCC1)C(CCCC1)CCCC1C1=*)*1=C)[C@@](C(O*)=O)N* Chemical compound CC(C)(CC(C)(C)*(CCCC1)CCCC1(*)[C@](*=C*C(**)(*C1(*)CCCCCCC1)C(CCCC1)CCCC1C1=*)*1=C)[C@@](C(O*)=O)N* 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- NIGWMJHCCYYCSF-UHFFFAOYSA-N Fenclonine Chemical compound OC(=O)C(N)CC1=CC=C(Cl)C=C1 NIGWMJHCCYYCSF-UHFFFAOYSA-N 0.000 description 4
- 229920001213 Polysorbate 20 Polymers 0.000 description 4
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- 125000003342 alkenyl group Chemical group 0.000 description 4
- 125000000304 alkynyl group Chemical group 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- 210000003169 central nervous system Anatomy 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000008297 liquid dosage form Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 4
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 4
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 125000002577 pseudohalo group Chemical group 0.000 description 4
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 229930192474 thiophene Natural products 0.000 description 4
- 150000003852 triazoles Chemical class 0.000 description 4
- KHSYYLCXQKCYQX-SECBINFHSA-N (1r)-1-naphthalen-2-ylethanamine Chemical compound C1=CC=CC2=CC([C@H](N)C)=CC=C21 KHSYYLCXQKCYQX-SECBINFHSA-N 0.000 description 3
- XWIYUCRMWCHYJR-UHFFFAOYSA-N 1h-pyrrolo[3,2-b]pyridine Chemical compound C1=CC=C2NC=CC2=N1 XWIYUCRMWCHYJR-UHFFFAOYSA-N 0.000 description 3
- XJPZKYIHCLDXST-UHFFFAOYSA-N 4,6-dichloropyrimidine Chemical compound ClC1=CC(Cl)=NC=N1 XJPZKYIHCLDXST-UHFFFAOYSA-N 0.000 description 3
- DUUGKQCEGZLZNO-UHFFFAOYSA-N 5-hydroxyindoleacetic acid Chemical compound C1=C(O)C=C2C(CC(=O)O)=CNC2=C1 DUUGKQCEGZLZNO-UHFFFAOYSA-N 0.000 description 3
- LDCYZAJDBXYCGN-UHFFFAOYSA-N 5-hydroxytryptophan Chemical compound C1=C(O)C=C2C(CC(N)C(O)=O)=CNC2=C1 LDCYZAJDBXYCGN-UHFFFAOYSA-N 0.000 description 3
- XKVUYEYANWFIJX-UHFFFAOYSA-N 5-methyl-1h-pyrazole Chemical compound CC1=CC=NN1 XKVUYEYANWFIJX-UHFFFAOYSA-N 0.000 description 3
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 3
- 229930003347 Atropine Natural products 0.000 description 3
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 3
- 206010012735 Diarrhoea Diseases 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 3
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 3
- 229960000396 atropine Drugs 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000004305 biphenyl Substances 0.000 description 3
- 235000010290 biphenyl Nutrition 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 3
- QGBSISYHAICWAH-UHFFFAOYSA-N dicyandiamide Chemical compound NC(N)=NC#N QGBSISYHAICWAH-UHFFFAOYSA-N 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- IMBKASBLAKCLEM-UHFFFAOYSA-L ferrous ammonium sulfate (anhydrous) Chemical compound [NH4+].[NH4+].[Fe+2].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O IMBKASBLAKCLEM-UHFFFAOYSA-L 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 3
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- MQUPWTBHHPUUMC-UHFFFAOYSA-N isoindole Chemical compound C1=CC=C[C]2C=NC=C21 MQUPWTBHHPUUMC-UHFFFAOYSA-N 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 3
- 238000007726 management method Methods 0.000 description 3
- QUSNBJAOOMFDIB-UHFFFAOYSA-N monoethyl amine Natural products CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 3
- 239000002105 nanoparticle Substances 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000001543 one-way ANOVA Methods 0.000 description 3
- 239000006186 oral dosage form Substances 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 229960005190 phenylalanine Drugs 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- KNGKJEAJYVSAIV-SSDOTTSWSA-N (1r)-1-(2-bromo-4-chlorophenyl)-2,2,2-trifluoroethanol Chemical compound FC(F)(F)[C@H](O)C1=CC=C(Cl)C=C1Br KNGKJEAJYVSAIV-SSDOTTSWSA-N 0.000 description 2
- SRADOQOAOBUPOB-LLVKDONJSA-N (1r)-1-[5-chloro-2-(3-methylpyrazol-1-yl)phenyl]-2,2,2-trifluoroethanol Chemical compound N1=C(C)C=CN1C1=CC=C(Cl)C=C1[C@@H](O)C(F)(F)F SRADOQOAOBUPOB-LLVKDONJSA-N 0.000 description 2
- LSVGONLXDSVXAA-LLVKDONJSA-N (1r)-2,2,2-trifluoro-1-[5-fluoro-2-(3-methylpyrazol-1-yl)phenyl]ethanol Chemical compound N1=C(C)C=CN1C1=CC=C(F)C=C1[C@@H](O)C(F)(F)F LSVGONLXDSVXAA-LLVKDONJSA-N 0.000 description 2
- HXGZPMHPSBJMKB-UHFFFAOYSA-N (2-bromo-5-fluorophenyl)methanol Chemical compound OCC1=CC(F)=CC=C1Br HXGZPMHPSBJMKB-UHFFFAOYSA-N 0.000 description 2
- DGUWACLYDSWXRZ-UHFFFAOYSA-N (2-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC=C1C=O DGUWACLYDSWXRZ-UHFFFAOYSA-N 0.000 description 2
- BTANRVKWQNVYAZ-BYPYZUCNSA-N (2S)-butan-2-ol Chemical group CC[C@H](C)O BTANRVKWQNVYAZ-BYPYZUCNSA-N 0.000 description 2
- BVHDAPRBZWLVRX-DEOSSOPVSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-[4-[5-(naphthalen-2-ylmethylamino)pyrazin-2-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CC=C1C(N=C1)=CN=C1NCC1=CC=C(C=CC=C2)C2=C1 BVHDAPRBZWLVRX-DEOSSOPVSA-N 0.000 description 2
- ANXHGSTXRHZKHW-RXVVDRJESA-N (2s)-2-amino-3-[4-[2-amino-6-[(1s)-2,2,2-trifluoro-1-[4-(1,2-oxazol-4-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC(O[C@@H](C=2C=CC(=CC=2)C2=CON=C2)C(F)(F)F)=NC(N)=N1 ANXHGSTXRHZKHW-RXVVDRJESA-N 0.000 description 2
- IDABDDFSKOAXMD-NRFANRHFSA-N (2s)-2-amino-3-[4-[5-(naphthalen-2-ylmethylamino)pyrazin-2-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C(N=C1)=CN=C1NCC1=CC=C(C=CC=C2)C2=C1 IDABDDFSKOAXMD-NRFANRHFSA-N 0.000 description 2
- PZWGGRIVPTXYGU-NSHDSACASA-N (2s)-3-(4-azidophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CC1=CC=C(N=[N+]=[N-])C=C1 PZWGGRIVPTXYGU-NSHDSACASA-N 0.000 description 2
- BJQCPCFFYBKRLM-UHFFFAOYSA-N (3-methylphenyl)boronic acid Chemical compound CC1=CC=CC(B(O)O)=C1 BJQCPCFFYBKRLM-UHFFFAOYSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- DSCQFYSEGYGGBW-UHFFFAOYSA-N 1-cyano-1-ethylguanidine Chemical compound CCN(C#N)C(N)=N DSCQFYSEGYGGBW-UHFFFAOYSA-N 0.000 description 2
- WDWKMZMIJJTUES-UHFFFAOYSA-N 1-cyclohexyl-2,2,2-trifluoroethanol Chemical compound FC(F)(F)C(O)C1CCCCC1 WDWKMZMIJJTUES-UHFFFAOYSA-N 0.000 description 2
- FMAQCDSETDNKDW-UHFFFAOYSA-N 2,2,2-trifluoro-1-(2-pyrimidin-5-ylphenyl)ethanol Chemical compound FC(F)(F)C(O)C1=CC=CC=C1C1=CN=CN=C1 FMAQCDSETDNKDW-UHFFFAOYSA-N 0.000 description 2
- RYIFCXNFAICWRE-UHFFFAOYSA-N 2,2,2-trifluoro-1-[4-(3-fluorophenyl)phenyl]ethanol Chemical compound C1=CC(C(O)C(F)(F)F)=CC=C1C1=CC=CC(F)=C1 RYIFCXNFAICWRE-UHFFFAOYSA-N 0.000 description 2
- VNOMEAQPOMDWSR-UHFFFAOYSA-N 2,2,2-trifluoro-1-phenylethanol Chemical compound FC(F)(F)C(O)C1=CC=CC=C1 VNOMEAQPOMDWSR-UHFFFAOYSA-N 0.000 description 2
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 2
- PFOXNJMCQNJJES-UHFFFAOYSA-N 2-(1,3-thiazol-2-yl)benzaldehyde Chemical compound O=CC1=CC=CC=C1C1=NC=CS1 PFOXNJMCQNJJES-UHFFFAOYSA-N 0.000 description 2
- YFTHTJAPODJVSL-UHFFFAOYSA-N 2-(1-benzothiophen-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(SC=C2)C2=C1 YFTHTJAPODJVSL-UHFFFAOYSA-N 0.000 description 2
- DTKDANCMEMZESZ-UHFFFAOYSA-N 2-amino-3-[4-[6-[(3-cyclopentyloxy-4-methoxyphenyl)methylamino]pyrazin-2-yl]phenyl]propanoic acid Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC(N=1)=CN=CC=1C1=CC=C(CC(N)C(O)=O)C=C1 DTKDANCMEMZESZ-UHFFFAOYSA-N 0.000 description 2
- JPSJBEALHNYNBL-UHFFFAOYSA-N 2-pyrimidin-5-ylbenzaldehyde Chemical compound O=CC1=CC=CC=C1C1=CN=CN=C1 JPSJBEALHNYNBL-UHFFFAOYSA-N 0.000 description 2
- UBPQITZLYDWRFS-UHFFFAOYSA-N 3,4-dihydro-1H-triazine-2,4-diamine Chemical compound NC1NN(N)NC=C1 UBPQITZLYDWRFS-UHFFFAOYSA-N 0.000 description 2
- GRFNBEZIAWKNCO-UHFFFAOYSA-N 3-pyridinol Chemical compound OC1=CC=CN=C1 GRFNBEZIAWKNCO-UHFFFAOYSA-N 0.000 description 2
- RLKMANFYMOECQK-UHFFFAOYSA-N 4-benzylsulfanyl-6-chloropyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(SCC=2C=CC=CC=2)=N1 RLKMANFYMOECQK-UHFFFAOYSA-N 0.000 description 2
- AQQBAQRBIOYDMF-UHFFFAOYSA-N 4-bromo-n-[(3-cyclopentyloxy-4-methoxyphenyl)methyl]aniline Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC1=CC=C(Br)C=C1 AQQBAQRBIOYDMF-UHFFFAOYSA-N 0.000 description 2
- ATGTXNKADXUIMA-UHFFFAOYSA-N 4-chloro-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine Chemical compound C=1C=CC=CC=1C(C(F)(F)F)OC1=CC(Cl)=NC=N1 ATGTXNKADXUIMA-UHFFFAOYSA-N 0.000 description 2
- WQAGPHHDEHVVDO-UHFFFAOYSA-N 4-chloro-6-(2-fluorophenoxy)pyrimidine Chemical compound FC1=CC=CC=C1OC1=CC(Cl)=NC=N1 WQAGPHHDEHVVDO-UHFFFAOYSA-N 0.000 description 2
- XPHPDTOSZBVYRA-UHFFFAOYSA-N 4-chloro-6-(naphthalen-2-ylmethylsulfanyl)pyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(SCC=2C=C3C=CC=CC3=CC=2)=N1 XPHPDTOSZBVYRA-UHFFFAOYSA-N 0.000 description 2
- HTYYSQYFKUIVEH-UHFFFAOYSA-N 4-chloro-6-[1-(3,4-difluorophenyl)-2,2,2-trifluoroethoxy]pyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(OC(C=2C=C(F)C(F)=CC=2)C(F)(F)F)=N1 HTYYSQYFKUIVEH-UHFFFAOYSA-N 0.000 description 2
- WSSVMZRDIVEFAJ-UHFFFAOYSA-N 4-chloro-6-[2,2,2-trifluoro-1-[2-(3-methylphenyl)phenyl]ethoxy]pyrimidin-2-amine Chemical compound CC1=CC=CC(C=2C(=CC=CC=2)C(OC=2N=C(N)N=C(Cl)C=2)C(F)(F)F)=C1 WSSVMZRDIVEFAJ-UHFFFAOYSA-N 0.000 description 2
- PQVUOKKRKFNGNS-UHFFFAOYSA-N 4-chloro-6-[2,2,2-trifluoro-1-[4-(3-fluorophenyl)phenyl]ethoxy]pyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(OC(C=2C=CC(=CC=2)C=2C=C(F)C=CC=2)C(F)(F)F)=N1 PQVUOKKRKFNGNS-UHFFFAOYSA-N 0.000 description 2
- ISDBWOPVZKNQDW-UHFFFAOYSA-N 4-phenylbenzaldehyde Chemical compound C1=CC(C=O)=CC=C1C1=CC=CC=C1 ISDBWOPVZKNQDW-UHFFFAOYSA-N 0.000 description 2
- YLKJWJIGKPDADU-UHFFFAOYSA-N 5-bromo-n-(naphthalen-2-ylmethyl)pyrazin-2-amine Chemical compound C1=NC(Br)=CN=C1NCC1=CC=C(C=CC=C2)C2=C1 YLKJWJIGKPDADU-UHFFFAOYSA-N 0.000 description 2
- HKEIVRCTOYZHLY-UHFFFAOYSA-N 5-bromo-n-[(1,3-dimethylpyrazol-4-yl)methyl]pyrazin-2-amine Chemical compound CC1=NN(C)C=C1CNC1=CN=C(Br)C=N1 HKEIVRCTOYZHLY-UHFFFAOYSA-N 0.000 description 2
- TZNQDFXFXJYGJK-UHFFFAOYSA-N 5-bromo-n-[(3-cyclopentyloxy-4-methoxyphenyl)methyl]pyridin-3-amine Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC1=CN=CC(Br)=C1 TZNQDFXFXJYGJK-UHFFFAOYSA-N 0.000 description 2
- IOIZGHRWMLXSPW-UHFFFAOYSA-N 5-bromo-n-[(4-phenylphenyl)methyl]pyrazin-2-amine Chemical compound C1=NC(Br)=CN=C1NCC1=CC=C(C=2C=CC=CC=2)C=C1 IOIZGHRWMLXSPW-UHFFFAOYSA-N 0.000 description 2
- FCIQEWWWAWYLGA-UHFFFAOYSA-N 5-bromo-n-[[2-(4-methylphenyl)phenyl]methyl]pyrazin-2-amine Chemical compound C1=CC(C)=CC=C1C1=CC=CC=C1CNC1=CN=C(Br)C=N1 FCIQEWWWAWYLGA-UHFFFAOYSA-N 0.000 description 2
- CJJIWSJVIZMHOI-UHFFFAOYSA-N 5-chloro-n-(3,4-dimethoxyphenyl)pyrazine-2-carboxamide Chemical compound C1=C(OC)C(OC)=CC=C1NC(=O)C1=CN=C(Cl)C=N1 CJJIWSJVIZMHOI-UHFFFAOYSA-N 0.000 description 2
- LDCYZAJDBXYCGN-VIFPVBQESA-N 5-hydroxy-L-tryptophan Chemical compound C1=C(O)C=C2C(C[C@H](N)C(O)=O)=CNC2=C1 LDCYZAJDBXYCGN-VIFPVBQESA-N 0.000 description 2
- 229940000681 5-hydroxytryptophan Drugs 0.000 description 2
- HWOZEJJVUCALGB-UHFFFAOYSA-N 6-Methyltetrahydropterin Chemical compound N1C(N)=NC(=O)C2=C1NCC(C)N2 HWOZEJJVUCALGB-UHFFFAOYSA-N 0.000 description 2
- JWQWWWPMMDUJDH-UHFFFAOYSA-N 6-chloro-n-[(3-cyclopentyloxy-4-methoxyphenyl)methyl]pyrazin-2-amine Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC1=CN=CC(Cl)=N1 JWQWWWPMMDUJDH-UHFFFAOYSA-N 0.000 description 2
- UGSMFPYQTDZKSC-UHFFFAOYSA-N 6-chloro-n-[(3-cyclopentyloxy-4-methoxyphenyl)methyl]pyrimidin-4-amine Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC1=CC(Cl)=NC=N1 UGSMFPYQTDZKSC-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 208000019901 Anxiety disease Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 102100035882 Catalase Human genes 0.000 description 2
- 108010053835 Catalase Proteins 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- 102000003951 Erythropoietin Human genes 0.000 description 2
- 108090000394 Erythropoietin Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 102000010909 Monoamine Oxidase Human genes 0.000 description 2
- 108010062431 Monoamine oxidase Proteins 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- 102000003729 Neprilysin Human genes 0.000 description 2
- 108090000028 Neprilysin Proteins 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 239000008156 Ringer's lactate solution Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 229910001573 adamantine Inorganic materials 0.000 description 2
- 125000005213 alkyl heteroaryl group Chemical group 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 2
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 2
- 235000011130 ammonium sulphate Nutrition 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 229960002903 benzyl benzoate Drugs 0.000 description 2
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 2
- 229960004853 betadex Drugs 0.000 description 2
- 239000012888 bovine serum Substances 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- FUSUHKVFWTUUBE-UHFFFAOYSA-N buten-2-one Chemical compound CC(=O)C=C FUSUHKVFWTUUBE-UHFFFAOYSA-N 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 239000007894 caplet Substances 0.000 description 2
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 2
- 150000001722 carbon compounds Chemical class 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- CZZBXGOYISFHRY-UHFFFAOYSA-N copper;hydroiodide Chemical compound [Cu].I CZZBXGOYISFHRY-UHFFFAOYSA-N 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- KVFDZFBHBWTVID-UHFFFAOYSA-N cyclohexanecarbaldehyde Chemical compound O=CC1CCCCC1 KVFDZFBHBWTVID-UHFFFAOYSA-N 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- FXORZKOZOQWVMQ-UHFFFAOYSA-L dichloropalladium;triphenylphosphane Chemical compound Cl[Pd]Cl.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 FXORZKOZOQWVMQ-UHFFFAOYSA-L 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 229940105423 erythropoietin Drugs 0.000 description 2
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N ethenoxyethane Chemical compound CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 239000003365 glass fiber Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000001475 halogen functional group Chemical group 0.000 description 2
- 125000004404 heteroalkyl group Chemical group 0.000 description 2
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 2
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 2
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 2
- 229940011051 isopropyl acetate Drugs 0.000 description 2
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 2
- 238000011813 knockout mouse model Methods 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- BIFARHLBYAKSSN-UHFFFAOYSA-N methyl 2-bromo-4-chlorobenzoate Chemical compound COC(=O)C1=CC=C(Cl)C=C1Br BIFARHLBYAKSSN-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- IDSXLJLXYMLSJM-UHFFFAOYSA-N morpholine;propane-1-sulfonic acid Chemical compound C1COCCN1.CCCS(O)(=O)=O IDSXLJLXYMLSJM-UHFFFAOYSA-N 0.000 description 2
- 230000004899 motility Effects 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- FXLOVSHXALFLKQ-UHFFFAOYSA-N p-tolualdehyde Chemical compound CC1=CC=C(C=O)C=C1 FXLOVSHXALFLKQ-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- 208000019116 sleep disease Diseases 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 150000003871 sulfonates Chemical class 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000012353 t test Methods 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- SJSGRCXYBOKZNN-UHFFFAOYSA-N tert-butyl 2-(benzhydrylideneamino)-3-(4-bromo-2-fluorophenyl)propanoate Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)=NC(C(=O)OC(C)(C)C)CC1=CC=C(Br)C=C1F SJSGRCXYBOKZNN-UHFFFAOYSA-N 0.000 description 2
- YSHDPXQDVKNPKA-UHFFFAOYSA-N tert-butyl 2-(benzhydrylideneamino)acetate Chemical compound C=1C=CC=CC=1C(=NCC(=O)OC(C)(C)C)C1=CC=CC=C1 YSHDPXQDVKNPKA-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- GQIJQMFDUUUQCO-SSDOTTSWSA-N (1r)-1-(2-bromo-5-fluorophenyl)-2,2,2-trifluoroethanol Chemical compound FC(F)(F)[C@H](O)C1=CC(F)=CC=C1Br GQIJQMFDUUUQCO-SSDOTTSWSA-N 0.000 description 1
- WXGBZJJAGLSBPR-UHFFFAOYSA-N (2-fluoropyridin-4-yl)boronic acid Chemical compound OB(O)C1=CC=NC(F)=C1 WXGBZJJAGLSBPR-UHFFFAOYSA-N 0.000 description 1
- BTANRVKWQNVYAZ-SCSAIBSYSA-N (2R)-butan-2-ol Chemical compound CC[C@@H](C)O BTANRVKWQNVYAZ-SCSAIBSYSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- BVPJISSXTXZVDS-FQEVSTJZSA-N (2s)-2-amino-3-[4-[2-amino-6-(naphthalen-2-ylmethylsulfanyl)pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC(SCC=2C=C3C=CC=CC3=CC=2)=NC(N)=N1 BVPJISSXTXZVDS-FQEVSTJZSA-N 0.000 description 1
- DTBUCLLNKBWYSZ-REWPJTCUSA-N (2s)-2-amino-3-[4-[2-amino-6-[(1s)-2,2,2-trifluoro-1-(6-methoxynaphthalen-2-yl)ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound O([C@@H](C1=CC2=CC=C(C=C2C=C1)OC)C(F)(F)F)C(N=C(N)N=1)=CC=1C1=CC=C(C[C@H](N)C(O)=O)C=C1 DTBUCLLNKBWYSZ-REWPJTCUSA-N 0.000 description 1
- XWXBYPPPMHARHY-CVDCTZTESA-N (2s)-2-amino-3-[4-[2-amino-6-[(1s)-2,2,2-trifluoro-1-[4-(2-fluoropyridin-4-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC(O[C@@H](C=2C=CC(=CC=2)C=2C=C(F)N=CC=2)C(F)(F)F)=NC(N)=N1 XWXBYPPPMHARHY-CVDCTZTESA-N 0.000 description 1
- OWIQRMSSSNCLCT-URXFXBBRSA-N (2s)-2-amino-3-[4-[2-amino-6-[(1s)-2,2,2-trifluoro-1-[4-(5-methoxypyridin-3-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound COC1=CN=CC(C=2C=CC(=CC=2)[C@H](OC=2N=C(N)N=C(C=2)C=2C=CC(C[C@H](N)C(O)=O)=CC=2)C(F)(F)F)=C1 OWIQRMSSSNCLCT-URXFXBBRSA-N 0.000 description 1
- WTXPBHFTINZRTG-REWPJTCUSA-N (2s)-2-amino-3-[4-[2-amino-6-[(1s)-2,2,2-trifluoro-1-[4-[4-(trifluoromethyl)pyridin-3-yl]phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC(O[C@@H](C=2C=CC(=CC=2)C=2C(=CC=NC=2)C(F)(F)F)C(F)(F)F)=NC(N)=N1 WTXPBHFTINZRTG-REWPJTCUSA-N 0.000 description 1
- IYOGQIPGNBOBFJ-UCFFOFKASA-N (2s)-2-amino-3-[4-[2-amino-6-[2,2,2-trifluoro-1-(4-methylphenyl)ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(C)=CC=C1C(C(F)(F)F)OC1=CC(C=2C=CC(C[C@H](N)C(O)=O)=CC=2)=NC(N)=N1 IYOGQIPGNBOBFJ-UCFFOFKASA-N 0.000 description 1
- UVQUGMFVHPEDLT-XADRRFQNSA-N (2s)-2-amino-3-[4-[2-amino-6-[2,2,2-trifluoro-1-[2-(3-methylphenyl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound CC1=CC=CC(C=2C(=CC=CC=2)C(OC=2N=C(N)N=C(C=2)C=2C=CC(C[C@H](N)C(O)=O)=CC=2)C(F)(F)F)=C1 UVQUGMFVHPEDLT-XADRRFQNSA-N 0.000 description 1
- PRBVXMSGNUWOIE-XEGCMXMBSA-N (2s)-2-amino-3-[4-[2-amino-6-[2,2,2-trifluoro-1-[2-fluoro-4-(5-methoxypyridin-3-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound COC1=CN=CC(C=2C=C(F)C(C(OC=3N=C(N)N=C(C=3)C=3C=CC(C[C@H](N)C(O)=O)=CC=3)C(F)(F)F)=CC=2)=C1 PRBVXMSGNUWOIE-XEGCMXMBSA-N 0.000 description 1
- JZWUKILTKYJLCN-XEGCMXMBSA-N (2s)-2-amino-3-[4-[2-amino-6-[2,2,2-trifluoro-1-[4-(3-fluorophenyl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC(OC(C=2C=CC(=CC=2)C=2C=C(F)C=CC=2)C(F)(F)F)=NC(N)=N1 JZWUKILTKYJLCN-XEGCMXMBSA-N 0.000 description 1
- GZTQKJGGHFJLST-XGLRFROISA-N (2s)-2-amino-3-[4-[2-amino-6-[2,2,2-trifluoro-1-[4-(6-methoxypyridin-2-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound COC1=CC=CC(C=2C=CC(=CC=2)C(OC=2N=C(N)N=C(C=2)C=2C=CC(C[C@H](N)C(O)=O)=CC=2)C(F)(F)F)=N1 GZTQKJGGHFJLST-XGLRFROISA-N 0.000 description 1
- NOGGRGSSBDYZGI-ZHRRBRCNSA-N (2s)-2-amino-3-[4-[2-amino-6-[methyl-[(1r)-1-naphthalen-2-ylethyl]amino]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound CN([C@H](C)C=1C=C2C=CC=CC2=CC=1)C(N=C(N)N=1)=CC=1C1=CC=C(C[C@H](N)C(O)=O)C=C1 NOGGRGSSBDYZGI-ZHRRBRCNSA-N 0.000 description 1
- SSAHOVGFOLLZKM-SFHVURJKSA-N (2s)-2-amino-3-[4-[5-[4-(thiophene-2-carbonylamino)phenyl]triazol-1-yl]phenyl]propanoic acid Chemical group C1=CC(C[C@H](N)C(O)=O)=CC=C1N1C(C=2C=CC(NC(=O)C=3SC=CC=3)=CC=2)=CN=N1 SSAHOVGFOLLZKM-SFHVURJKSA-N 0.000 description 1
- YTAJLHDFOPMRSG-DEOSSOPVSA-N (2s)-2-amino-3-[4-[5-[[2-(4-methylphenyl)phenyl]methylamino]pyrazin-2-yl]phenyl]propanoic acid Chemical compound C1=CC(C)=CC=C1C1=CC=CC=C1CNC1=CN=C(C=2C=CC(C[C@H](N)C(O)=O)=CC=2)C=N1 YTAJLHDFOPMRSG-DEOSSOPVSA-N 0.000 description 1
- FNKPGBGZZHUTIY-NRFANRHFSA-N (2s)-2-amino-3-[4-[6-[(3-cyclopentyloxy-4-methoxyphenyl)methylamino]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC(N=CN=1)=CC=1C1=CC=C(C[C@H](N)C(O)=O)C=C1 FNKPGBGZZHUTIY-NRFANRHFSA-N 0.000 description 1
- JKMKDLFIAQBHOK-YDNXMHBPSA-N (2s)-2-amino-3-[4-[6-[2,2,2-trifluoro-1-(4-imidazol-1-ylphenyl)ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC(OC(C=2C=CC(=CC=2)N2C=NC=C2)C(F)(F)F)=NC=N1 JKMKDLFIAQBHOK-YDNXMHBPSA-N 0.000 description 1
- SSZJLFJRIFYICD-BDYUSTAISA-N (2s)-3-[4-[2-amino-6-[(1s)-2,2,2-trifluoro-1-[4-(5-methoxypyridin-3-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound COC1=CN=CC(C=2C=CC(=CC=2)[C@H](OC=2N=C(N)N=C(C=2)C=2C=CC(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CC=2)C(F)(F)F)=C1 SSZJLFJRIFYICD-BDYUSTAISA-N 0.000 description 1
- CYWKGNNLCLPBQV-AHWVRZQESA-N (2s)-3-[4-[2-amino-6-[(1s)-2,2,2-trifluoro-1-[4-[4-(trifluoromethyl)pyridin-3-yl]phenyl]ethoxy]pyrimidin-4-yl]phenyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound C1=CC(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CC=C1C1=CC(O[C@@H](C=2C=CC(=CC=2)C=2C(=CC=NC=2)C(F)(F)F)C(F)(F)F)=NC(N)=N1 CYWKGNNLCLPBQV-AHWVRZQESA-N 0.000 description 1
- HWJFQDKAZGIGSY-PVCWFJFTSA-N (2s)-3-[4-[2-amino-6-[2,2,2-trifluoro-1-[2-fluoro-4-(5-methoxypyridin-3-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound COC1=CN=CC(C=2C=C(F)C(C(OC=3N=C(N)N=C(C=3)C=3C=CC(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CC=3)C(F)(F)F)=CC=2)=C1 HWJFQDKAZGIGSY-PVCWFJFTSA-N 0.000 description 1
- JBNHJYGLRPSMFT-BXXZMZEQSA-N (2s)-3-[4-[2-amino-6-[2,2,2-trifluoro-1-[4-(6-methoxypyridin-2-yl)phenyl]ethoxy]pyrimidin-4-yl]phenyl]-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound COC1=CC=CC(C=2C=CC(=CC=2)C(OC=2N=C(N)N=C(C=2)C=2C=CC(C[C@H](NC(=O)OC(C)(C)C)C(O)=O)=CC=2)C(F)(F)F)=N1 JBNHJYGLRPSMFT-BXXZMZEQSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- OBFAPCIUSYHFIE-UHFFFAOYSA-N (5-hydroxyindol-3-yl)acetaldehyde Chemical compound OC1=CC=C2NC=C(CC=O)C2=C1 OBFAPCIUSYHFIE-UHFFFAOYSA-N 0.000 description 1
- RBSMKSPHBJFXCJ-UHFFFAOYSA-N (5-phenylthiophen-2-yl)boronic acid Chemical compound S1C(B(O)O)=CC=C1C1=CC=CC=C1 RBSMKSPHBJFXCJ-UHFFFAOYSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- IGJREDVLGVEPFI-UHFFFAOYSA-N 1,3-dimethylpyrazole-4-carbaldehyde Chemical compound CC1=NN(C)C=C1C=O IGJREDVLGVEPFI-UHFFFAOYSA-N 0.000 description 1
- FAKFWQATIYZKFW-UHFFFAOYSA-N 1-(2-bromo-4-chlorophenyl)-2,2,2-trifluoroethanone Chemical compound FC(F)(F)C(=O)C1=CC=C(Cl)C=C1Br FAKFWQATIYZKFW-UHFFFAOYSA-N 0.000 description 1
- IHGSAQHSAGRWNI-UHFFFAOYSA-N 1-(4-bromophenyl)-2,2,2-trifluoroethanone Chemical compound FC(F)(F)C(=O)C1=CC=C(Br)C=C1 IHGSAQHSAGRWNI-UHFFFAOYSA-N 0.000 description 1
- XZHBBIBTBQHRQY-UHFFFAOYSA-N 1-(5,7-difluoronaphthalen-2-yl)ethanone Chemical compound FC1=CC(F)=CC2=CC(C(=O)C)=CC=C21 XZHBBIBTBQHRQY-UHFFFAOYSA-N 0.000 description 1
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 1
- VQMJVTDTOGVWHB-UHFFFAOYSA-N 1-[3-(3-bromopyrazin-2-yl)-5-cyclopentyloxy-4-methoxyphenyl]-n-methylmethanamine Chemical compound COC=1C(C=2C(=NC=CN=2)Br)=CC(CNC)=CC=1OC1CCCC1 VQMJVTDTOGVWHB-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- 125000006039 1-hexenyl group Chemical group 0.000 description 1
- RUFPHBVGCFYCNW-UHFFFAOYSA-N 1-naphthylamine Chemical compound C1=CC=C2C(N)=CC=CC2=C1 RUFPHBVGCFYCNW-UHFFFAOYSA-N 0.000 description 1
- 125000006023 1-pentenyl group Chemical group 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- FZKKEKNWKXLQRG-UHFFFAOYSA-N 2,2,2-trifluoro-1-(6-methoxynaphthalen-2-yl)ethanol Chemical compound C1=C(C(O)C(F)(F)F)C=CC2=CC(OC)=CC=C21 FZKKEKNWKXLQRG-UHFFFAOYSA-N 0.000 description 1
- 125000004793 2,2,2-trifluoroethoxy group Chemical group FC(CO*)(F)F 0.000 description 1
- 125000006069 2,3-dimethyl-2-butenyl group Chemical group 0.000 description 1
- IPHDQSNAYMVQCP-UHFFFAOYSA-N 2,6-dichloro-1h-triazine Chemical compound ClN1NC(Cl)=CC=N1 IPHDQSNAYMVQCP-UHFFFAOYSA-N 0.000 description 1
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 description 1
- BNZMLIHMNWLHRL-UHFFFAOYSA-N 2-(4-methylphenyl)benzaldehyde Chemical compound C1=CC(C)=CC=C1C1=CC=CC=C1C=O BNZMLIHMNWLHRL-UHFFFAOYSA-N 0.000 description 1
- KKFDCBRMNNSAAW-UHFFFAOYSA-N 2-(morpholin-4-yl)ethanol Chemical compound OCCN1CCOCC1 KKFDCBRMNNSAAW-UHFFFAOYSA-N 0.000 description 1
- ZDVRPKUWYQVVDX-UHFFFAOYSA-N 2-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC=CC=C1C=O ZDVRPKUWYQVVDX-UHFFFAOYSA-N 0.000 description 1
- QVHJQCGUWFKTSE-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound OC(=O)C(C)NC(=O)OC(C)(C)C QVHJQCGUWFKTSE-UHFFFAOYSA-N 0.000 description 1
- OIQOAYVCKAHSEJ-UHFFFAOYSA-N 2-[2,3-bis(2-hydroxyethoxy)propoxy]ethanol;hexadecanoic acid;octadecanoic acid Chemical compound OCCOCC(OCCO)COCCO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O OIQOAYVCKAHSEJ-UHFFFAOYSA-N 0.000 description 1
- OSMNVPCUTGALDP-UHFFFAOYSA-N 2-[4-[2-amino-6-[2,2,2-trifluoro-1-(4-methylphenyl)ethoxy]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound NC1=NC(=CC(=N1)C1=CC=C(C=C1)C(C(=O)O)C)OC(C(F)(F)F)C1=CC=C(C=C1)C OSMNVPCUTGALDP-UHFFFAOYSA-N 0.000 description 1
- JMYNJBLTMDPNKZ-UHFFFAOYSA-N 2-amino-3-[4-[2-amino-6-(1-naphthalen-2-ylethylamino)pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C=1C=C2C=CC=CC2=CC=1C(C)NC(N=C(N)N=1)=CC=1C1=CC=C(CC(N)C(O)=O)C=C1 JMYNJBLTMDPNKZ-UHFFFAOYSA-N 0.000 description 1
- ZHWSZEIBIWNBPY-UHFFFAOYSA-N 2-amino-3-[4-[2-amino-6-[[4-(trifluoromethyl)phenyl]methylamino]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=CC(CC(N)C(O)=O)=CC=C1C1=CC(NCC=2C=CC(=CC=2)C(F)(F)F)=NC(N)=N1 ZHWSZEIBIWNBPY-UHFFFAOYSA-N 0.000 description 1
- NOGGRGSSBDYZGI-UHFFFAOYSA-N 2-amino-3-[4-[2-amino-6-[methyl(1-naphthalen-2-ylethyl)amino]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C=1C=C2C=CC=CC2=CC=1C(C)N(C)C(N=C(N)N=1)=CC=1C1=CC=C(CC(N)C(O)=O)C=C1 NOGGRGSSBDYZGI-UHFFFAOYSA-N 0.000 description 1
- FNKPGBGZZHUTIY-UHFFFAOYSA-N 2-amino-3-[4-[6-[(3-cyclopentyloxy-4-methoxyphenyl)methylamino]pyrimidin-4-yl]phenyl]propanoic acid Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC(N=CN=1)=CC=1C1=CC=C(CC(N)C(O)=O)C=C1 FNKPGBGZZHUTIY-UHFFFAOYSA-N 0.000 description 1
- WSPSMPPSYYEOSU-UHFFFAOYSA-N 2-amino-3-[5-(5-phenylthiophen-2-yl)-1h-indol-3-yl]propanoic acid Chemical compound C1=C2C(CC(N)C(O)=O)=CNC2=CC=C1C(S1)=CC=C1C1=CC=CC=C1 WSPSMPPSYYEOSU-UHFFFAOYSA-N 0.000 description 1
- DIJNYDVGZLDZAL-UHFFFAOYSA-N 2-amino-3-[5-[4-amino-6-(1-naphthalen-2-ylethylamino)-1,3,5-triazin-2-yl]pyridin-2-yl]propanoic acid Chemical compound C=1C=C2C=CC=CC2=CC=1C(C)NC(N=1)=NC(N)=NC=1C1=CC=C(CC(N)C(O)=O)N=C1 DIJNYDVGZLDZAL-UHFFFAOYSA-N 0.000 description 1
- JTPXVCKCLBROOJ-UHFFFAOYSA-N 2-amino-6-chloropyrazine Chemical compound NC1=CN=CC(Cl)=N1 JTPXVCKCLBROOJ-UHFFFAOYSA-N 0.000 description 1
- RXNZFHIEDZEUQM-UHFFFAOYSA-N 2-bromo-1,3-thiazole Chemical compound BrC1=NC=CS1 RXNZFHIEDZEUQM-UHFFFAOYSA-N 0.000 description 1
- USMQLFCVCDEXAK-UHFFFAOYSA-N 2-bromo-4-chlorobenzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1Br USMQLFCVCDEXAK-UHFFFAOYSA-N 0.000 description 1
- CJUCIKJLMFVWIS-UHFFFAOYSA-N 2-bromo-5-fluorobenzaldehyde Chemical compound FC1=CC=C(Br)C(C=O)=C1 CJUCIKJLMFVWIS-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- XCEZQXDBXQYHGE-UHFFFAOYSA-N 2-chloro-4-[3-(4-chlorophenyl)piperidin-1-yl]-1,3,5-triazine Chemical compound C1=CC(Cl)=CC=C1C1CN(C=2N=C(Cl)N=CN=2)CCC1 XCEZQXDBXQYHGE-UHFFFAOYSA-N 0.000 description 1
- HFHFGHLXUCOHLN-UHFFFAOYSA-N 2-fluorophenol Chemical compound OC1=CC=CC=C1F HFHFGHLXUCOHLN-UHFFFAOYSA-N 0.000 description 1
- 125000006040 2-hexenyl group Chemical group 0.000 description 1
- 125000006029 2-methyl-2-butenyl group Chemical group 0.000 description 1
- DEYDDEZRKCIBBM-UHFFFAOYSA-N 2-methyl-6-phenylbenzaldehyde Chemical compound CC1=CC=CC(C=2C=CC=CC=2)=C1C=O DEYDDEZRKCIBBM-UHFFFAOYSA-N 0.000 description 1
- 239000001431 2-methylbenzaldehyde Substances 0.000 description 1
- AFXKCBFBGDUFAM-UHFFFAOYSA-N 2-methylpropan-2-amine;hydrofluoride Chemical compound [F-].CC(C)(C)[NH3+] AFXKCBFBGDUFAM-UHFFFAOYSA-N 0.000 description 1
- IMEPJEKULYYELS-MHZLTWQESA-N 2-morpholin-4-ylethyl (2s)-2-amino-3-[4-[5-(naphthalen-2-ylmethylamino)pyrazin-2-yl]phenyl]propanoate Chemical compound O=C([C@H](CC=1C=CC(=CC=1)C=1N=CC(NCC=2C=C3C=CC=CC3=CC=2)=NC=1)N)OCCN1CCOCC1 IMEPJEKULYYELS-MHZLTWQESA-N 0.000 description 1
- PJKVFARRVXDXAD-UHFFFAOYSA-N 2-naphthaldehyde Chemical compound C1=CC=CC2=CC(C=O)=CC=C21 PJKVFARRVXDXAD-UHFFFAOYSA-N 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- JPHKMYXKNKLNDF-UHFFFAOYSA-N 3,4-difluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C=C1F JPHKMYXKNKLNDF-UHFFFAOYSA-N 0.000 description 1
- LGDHZCLREKIGKJ-UHFFFAOYSA-N 3,4-dimethoxyaniline Chemical compound COC1=CC=C(N)C=C1OC LGDHZCLREKIGKJ-UHFFFAOYSA-N 0.000 description 1
- GLXKIKPTKUQMAP-UHFFFAOYSA-N 3-(4-chlorophenyl)piperidine Chemical compound C1=CC(Cl)=CC=C1C1CNCCC1 GLXKIKPTKUQMAP-UHFFFAOYSA-N 0.000 description 1
- DVLFYONBTKHTER-UHFFFAOYSA-N 3-(N-morpholino)propanesulfonic acid Chemical compound OS(=O)(=O)CCCN1CCOCC1 DVLFYONBTKHTER-UHFFFAOYSA-N 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- UCFSYHMCKWNKAH-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound CC1(C)OBOC1(C)C UCFSYHMCKWNKAH-UHFFFAOYSA-N 0.000 description 1
- WEQPBCSPRXFQQS-UHFFFAOYSA-N 4,5-dihydro-1,2-oxazole Chemical compound C1CC=NO1 WEQPBCSPRXFQQS-UHFFFAOYSA-N 0.000 description 1
- VADZUJOCSAESJS-UHFFFAOYSA-N 4-(2-oxopyrrolidin-1-yl)benzaldehyde Chemical compound C1=CC(C=O)=CC=C1N1C(=O)CCC1 VADZUJOCSAESJS-UHFFFAOYSA-N 0.000 description 1
- OQYLQLAVBZSSDL-UHFFFAOYSA-N 4-(6-methoxypyridin-2-yl)benzaldehyde Chemical compound COC1=CC=CC(C=2C=CC(C=O)=CC=2)=N1 OQYLQLAVBZSSDL-UHFFFAOYSA-N 0.000 description 1
- BZYIRYKJNSACFP-UHFFFAOYSA-N 4-[2-(trifluoromethyl)phenyl]piperidine;hydrochloride Chemical compound Cl.FC(F)(F)C1=CC=CC=C1C1CCNCC1 BZYIRYKJNSACFP-UHFFFAOYSA-N 0.000 description 1
- UPCARQPLANFGQJ-UHFFFAOYSA-N 4-bromo-2-fluorobenzaldehyde Chemical compound FC1=CC(Br)=CC=C1C=O UPCARQPLANFGQJ-UHFFFAOYSA-N 0.000 description 1
- WDFQBORIUYODSI-UHFFFAOYSA-N 4-bromoaniline Chemical compound NC1=CC=C(Br)C=C1 WDFQBORIUYODSI-UHFFFAOYSA-N 0.000 description 1
- LCUQVQCGGNAGTI-UHFFFAOYSA-N 4-chloro-6-[2,2,2-trifluoro-1-(2-pyrimidin-5-ylphenyl)ethoxy]pyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(OC(C=2C(=CC=CC=2)C=2C=NC=NC=2)C(F)(F)F)=N1 LCUQVQCGGNAGTI-UHFFFAOYSA-N 0.000 description 1
- NXAJXYYLEYUNAT-UHFFFAOYSA-N 4-chloro-6-[2,2,2-trifluoro-1-(6-methoxynaphthalen-2-yl)ethoxy]pyrimidin-2-amine Chemical compound C1=CC2=CC(OC)=CC=C2C=C1C(C(F)(F)F)OC1=CC(Cl)=NC(N)=N1 NXAJXYYLEYUNAT-UHFFFAOYSA-N 0.000 description 1
- NQZZWULTCNKWBC-UHFFFAOYSA-N 4-chloro-6-[2,2,2-trifluoro-1-[2-(trifluoromethyl)phenyl]ethoxy]pyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(OC(C=2C(=CC=CC=2)C(F)(F)F)C(F)(F)F)=N1 NQZZWULTCNKWBC-UHFFFAOYSA-N 0.000 description 1
- JAVXNTVGPPPBEI-UHFFFAOYSA-N 4-chloro-6-[4-[2-(trifluoromethyl)phenyl]piperidin-1-yl]pyrimidin-2-amine Chemical compound NC1=NC(Cl)=CC(N2CCC(CC2)C=2C(=CC=CC=2)C(F)(F)F)=N1 JAVXNTVGPPPBEI-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- GCNTZFIIOFTKIY-UHFFFAOYSA-N 4-hydroxypyridine Chemical compound OC1=CC=NC=C1 GCNTZFIIOFTKIY-UHFFFAOYSA-N 0.000 description 1
- MDQXGHBCDCOOSM-UHFFFAOYSA-N 5-bromopyridin-3-amine Chemical compound NC1=CN=CC(Br)=C1 MDQXGHBCDCOOSM-UHFFFAOYSA-N 0.000 description 1
- GYCPLYCTMDTEPU-UHFFFAOYSA-N 5-bromopyrimidine Chemical compound BrC1=CN=CN=C1 GYCPLYCTMDTEPU-UHFFFAOYSA-N 0.000 description 1
- KZDNJQUJBMDHJW-UHFFFAOYSA-N 5-bromotryptophan Chemical compound C1=C(Br)C=C2C(CC(N)C(O)=O)=CNC2=C1 KZDNJQUJBMDHJW-UHFFFAOYSA-N 0.000 description 1
- CFEMQJVHMRAYNZ-UHFFFAOYSA-N 5-chloropyrazine-2-carbonyl chloride Chemical compound ClC(=O)C1=CN=C(Cl)C=N1 CFEMQJVHMRAYNZ-UHFFFAOYSA-N 0.000 description 1
- RVWZUOPFHTYIEO-UHFFFAOYSA-N 5-hydroxyindoleacetic acid Natural products C1=C(O)C=C2C(C(=O)O)=CNC2=C1 RVWZUOPFHTYIEO-UHFFFAOYSA-N 0.000 description 1
- 239000003310 5-hydroxyindoleacetic acid Substances 0.000 description 1
- 102000040125 5-hydroxytryptamine receptor family Human genes 0.000 description 1
- 108091032151 5-hydroxytryptamine receptor family Proteins 0.000 description 1
- IQZQZXRXGFLQML-UHFFFAOYSA-N 6-bromo-n-[(3-cyclopentyloxy-4-methoxyphenyl)methyl]pyrazin-2-amine Chemical compound C1=C(OC2CCCC2)C(OC)=CC=C1CNC1=CN=CC(Br)=N1 IQZQZXRXGFLQML-UHFFFAOYSA-N 0.000 description 1
- DUKKRSPKJMHASP-UHFFFAOYSA-N 6-chloropyrimidin-4-amine Chemical compound NC1=CC(Cl)=NC=N1 DUKKRSPKJMHASP-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 206010001497 Agitation Diseases 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- 102000005369 Aldehyde Dehydrogenase Human genes 0.000 description 1
- 108020002663 Aldehyde Dehydrogenase Proteins 0.000 description 1
- 229920001450 Alpha-Cyclodextrin Polymers 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 235000003276 Apios tuberosa Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000010744 Arachis villosulicarpa Nutrition 0.000 description 1
- CDSJHWQRPDMDMS-UHFFFAOYSA-N B1CCCOO1 Chemical compound B1CCCOO1 CDSJHWQRPDMDMS-UHFFFAOYSA-N 0.000 description 1
- 108010023063 Bacto-peptone Proteins 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- VYBSVCHAYATELV-QFIPXVFZSA-N CN(Cc(cc1)cc(OC2CCCC2)c1OC)c(nc1)cnc1-c1ccc(C[C@@H](C(O)=O)N)cc1 Chemical compound CN(Cc(cc1)cc(OC2CCCC2)c1OC)c(nc1)cnc1-c1ccc(C[C@@H](C(O)=O)N)cc1 VYBSVCHAYATELV-QFIPXVFZSA-N 0.000 description 1
- MHLQEGOEWFLOIA-MRXNPFEDSA-N C[C@H](c1cc(cccc2)c2cc1)N(C)c1cc(-c2ccc(CC(C(O)=O)=N)cc2)nc(N)n1 Chemical compound C[C@H](c1cc(cccc2)c2cc1)N(C)c1cc(-c2ccc(CC(C(O)=O)=N)cc2)nc(N)n1 MHLQEGOEWFLOIA-MRXNPFEDSA-N 0.000 description 1
- 101100296719 Caenorhabditis elegans pde-4 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- DXXKANYPIGARCL-PGRDOPGGSA-N Cc(cc1)n[n]1-c(c([C@H](C(F)(F)F)Oc1cc(-c2ccc(C[C@@H](C(O)=O)N)cc2)nc(N)n1)c1)ccc1Cl Chemical compound Cc(cc1)n[n]1-c(c([C@H](C(F)(F)F)Oc1cc(-c2ccc(C[C@@H](C(O)=O)N)cc2)nc(N)n1)c1)ccc1Cl DXXKANYPIGARCL-PGRDOPGGSA-N 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 102100022641 Coagulation factor IX Human genes 0.000 description 1
- 206010053567 Coagulopathies Diseases 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 206010010774 Constipation Diseases 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- 208000020401 Depressive disease Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- 206010013654 Drug abuse Diseases 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 239000001116 FEMA 4028 Substances 0.000 description 1
- 108010076282 Factor IX Proteins 0.000 description 1
- 108010074864 Factor XI Proteins 0.000 description 1
- 108010012088 Fibrinogen Receptors Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 101000606113 Homo sapiens Tyrosine 3-monooxygenase Proteins 0.000 description 1
- 102100022337 Integrin alpha-V Human genes 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 150000007945 N-acyl ureas Chemical class 0.000 description 1
- IGQBAVBZQAEKQE-FQEVSTJZSA-N N[C@@H](Cc(cc1)ccc1-c1nc(N)nc(/[O]=C(\C(F)(F)F)/c(cc2)ccc2-c2c(C(F)(F)F)ccnc2)c1)C(O)=O Chemical compound N[C@@H](Cc(cc1)ccc1-c1nc(N)nc(/[O]=C(\C(F)(F)F)/c(cc2)ccc2-c2c(C(F)(F)F)ccnc2)c1)C(O)=O IGQBAVBZQAEKQE-FQEVSTJZSA-N 0.000 description 1
- UPANRWLPMYJVDU-YDNXMHBPSA-N N[C@@H](Cc(cc1)ccc1-c1nc(N)nc(OC(C(F)(F)F)c(cccc2)c2-c2cncnc2)c1)C(O)=O Chemical compound N[C@@H](Cc(cc1)ccc1-c1nc(N)nc(OC(C(F)(F)F)c(cccc2)c2-c2cncnc2)c1)C(O)=O UPANRWLPMYJVDU-YDNXMHBPSA-N 0.000 description 1
- ULJBXNJTQMXJNJ-BUSXIPJBSA-N N[C@@H](Cc(cc1)ccc1-c1nc(N)nc(OC(C2CCCCC2)C(F)(F)F)c1)C(O)=O Chemical compound N[C@@H](Cc(cc1)ccc1-c1nc(N)nc(OC(C2CCCCC2)C(F)(F)F)c1)C(O)=O ULJBXNJTQMXJNJ-BUSXIPJBSA-N 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 238000011887 Necropsy Methods 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 1
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 1
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 1
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 1
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 229910006074 SO2NH2 Inorganic materials 0.000 description 1
- 229910006124 SOCl2 Inorganic materials 0.000 description 1
- 239000002262 Schiff base Substances 0.000 description 1
- 150000004753 Schiff bases Chemical class 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- 108010000134 Vascular Cell Adhesion Molecule-1 Proteins 0.000 description 1
- 102100023543 Vascular cell adhesion protein 1 Human genes 0.000 description 1
- 108010048673 Vitronectin Receptors Proteins 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- GPOLKFXETWUREM-UHFFFAOYSA-N [4-(trifluoromethyl)pyridin-3-yl]boronic acid Chemical compound OB(O)C1=CN=CC=C1C(F)(F)F GPOLKFXETWUREM-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 201000007930 alcohol dependence Diseases 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 1
- 235000008206 alpha-amino acids Nutrition 0.000 description 1
- HFHDHCJBZVLPGP-RWMJIURBSA-N alpha-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO HFHDHCJBZVLPGP-RWMJIURBSA-N 0.000 description 1
- 229940043377 alpha-cyclodextrin Drugs 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 239000012223 aqueous fraction Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 125000005140 aralkylsulfonyl group Chemical group 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 125000001691 aryl alkyl amino group Chemical group 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 238000010560 atom transfer radical polymerization reaction Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 150000003935 benzaldehydes Chemical class 0.000 description 1
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzenecarboxaldehyde Natural products O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- RROBIDXNTUAHFW-UHFFFAOYSA-N benzotriazol-1-yloxy-tris(dimethylamino)phosphanium Chemical compound C1=CC=C2N(O[P+](N(C)C)(N(C)C)N(C)C)N=NC2=C1 RROBIDXNTUAHFW-UHFFFAOYSA-N 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- UENWRTRMUIOCKN-UHFFFAOYSA-N benzyl thiol Chemical compound SCC1=CC=CC=C1 UENWRTRMUIOCKN-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 235000011175 beta-cyclodextrine Nutrition 0.000 description 1
- 230000003851 biochemical process Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- LRJRPHROCLHMHK-UHFFFAOYSA-N boron;n,n-dimethylmethanamine Chemical compound [B].CN(C)C LRJRPHROCLHMHK-UHFFFAOYSA-N 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical class [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- BPKIGYQJPYCAOW-FFJTTWKXSA-I calcium;potassium;disodium;(2s)-2-hydroxypropanoate;dichloride;dihydroxide;hydrate Chemical compound O.[OH-].[OH-].[Na+].[Na+].[Cl-].[Cl-].[K+].[Ca+2].C[C@H](O)C([O-])=O BPKIGYQJPYCAOW-FFJTTWKXSA-I 0.000 description 1
- BMLSTPRTEKLIPM-UHFFFAOYSA-I calcium;potassium;disodium;hydrogen carbonate;dichloride;dihydroxide;hydrate Chemical compound O.[OH-].[OH-].[Na+].[Na+].[Cl-].[Cl-].[K+].[Ca+2].OC([O-])=O BMLSTPRTEKLIPM-UHFFFAOYSA-I 0.000 description 1
- 229940041514 candida albicans extract Drugs 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- ZDQWVKDDJDIVAL-UHFFFAOYSA-N catecholborane Chemical compound C1=CC=C2O[B]OC2=C1 ZDQWVKDDJDIVAL-UHFFFAOYSA-N 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 230000035602 clotting Effects 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 201000007750 congenital bile acid synthesis defect Diseases 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate Chemical compound [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000003413 degradative effect Effects 0.000 description 1
- 238000009795 derivation Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000008356 dextrose and sodium chloride injection Substances 0.000 description 1
- 239000008355 dextrose injection Substances 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- ZHXTWWCDMUWMDI-UHFFFAOYSA-N dihydroxyboron Chemical compound O[B]O ZHXTWWCDMUWMDI-UHFFFAOYSA-N 0.000 description 1
- 125000000597 dioxinyl group Chemical group 0.000 description 1
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 229960004222 factor ix Drugs 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 235000020375 flavoured syrup Nutrition 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 230000010243 gut motility Effects 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 150000002373 hemiacetals Chemical class 0.000 description 1
- 230000023597 hemostasis Effects 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005468 isobutylenyl group Chemical group 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940074928 isopropyl myristate Drugs 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- XQNUHMQSOMLVGM-UHFFFAOYSA-M magnesium;1,3-difluorobenzene-5-ide;bromide Chemical compound [Mg+2].[Br-].FC1=C[C-]=CC(F)=C1 XQNUHMQSOMLVGM-UHFFFAOYSA-M 0.000 description 1
- DTKMKZYEOXZPQZ-UHFFFAOYSA-M magnesium;fluorobenzene;bromide Chemical compound [Mg+2].[Br-].FC1=CC=C[C-]=C1 DTKMKZYEOXZPQZ-UHFFFAOYSA-M 0.000 description 1
- 201000006512 mast cell neoplasm Diseases 0.000 description 1
- 208000006971 mastocytoma Diseases 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- FCMQMRAFVRTHCR-UHFFFAOYSA-N methyl 2-bromo-5-fluorobenzoate Chemical compound COC(=O)C1=CC(F)=CC=C1Br FCMQMRAFVRTHCR-UHFFFAOYSA-N 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 230000036651 mood Effects 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- VGVHJESBFMWXJF-UHFFFAOYSA-N n-(4-ethylphenyl)thiophene-2-carboxamide Chemical compound C1=CC(CC)=CC=C1NC(=O)C1=CC=CS1 VGVHJESBFMWXJF-UHFFFAOYSA-N 0.000 description 1
- NWJUMMHVDMNYMJ-UHFFFAOYSA-N n-benzyl-1,1,1-trifluoromethanamine Chemical compound FC(F)(F)NCC1=CC=CC=C1 NWJUMMHVDMNYMJ-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VRPIUODNLOVPNH-UHFFFAOYSA-N n-methyl-1-naphthalen-2-ylethanamine Chemical compound C1=CC=CC2=CC(C(C)NC)=CC=C21 VRPIUODNLOVPNH-UHFFFAOYSA-N 0.000 description 1
- VOVJEXLDAMDFBR-UHFFFAOYSA-N n-methyl-4-(4-methylphenyl)aniline Chemical compound C1=CC(NC)=CC=C1C1=CC=C(C)C=C1 VOVJEXLDAMDFBR-UHFFFAOYSA-N 0.000 description 1
- 239000006070 nanosuspension Substances 0.000 description 1
- XBCAHQUVHHVHHL-UHFFFAOYSA-N naphthalen-2-ylmethanamine Chemical compound C1=CC=CC2=CC(CN)=CC=C21 XBCAHQUVHHVHHL-UHFFFAOYSA-N 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 230000037125 natural defense Effects 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- YPJKMVATUPSWOH-UHFFFAOYSA-N nitrooxidanyl Chemical compound [O][N+]([O-])=O YPJKMVATUPSWOH-UHFFFAOYSA-N 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 229940055076 parasympathomimetics choline ester Drugs 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- FKRCODPIKNYEAC-UHFFFAOYSA-N propionic acid ethyl ester Natural products CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000004800 psychological effect Effects 0.000 description 1
- 150000003216 pyrazines Chemical class 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 150000003248 quinolines Chemical class 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000000862 serotonergic effect Effects 0.000 description 1
- 230000009329 sexual behaviour Effects 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 208000022925 sleep disturbance Diseases 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000008354 sodium chloride injection Substances 0.000 description 1
- GSVCNPRYQWGKBC-UHFFFAOYSA-N sodium dicyanide Chemical compound [Na+].[C-]#N.[C-]#N GSVCNPRYQWGKBC-UHFFFAOYSA-N 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000007905 soft elastic gelatin capsule Substances 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000012453 sprague-dawley rat model Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 150000003461 sulfonyl halides Chemical class 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 229940066765 systemic antihistamines substituted ethylene diamines Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- WGJGINMQCMATNP-UHFFFAOYSA-N tert-butyl 3-(4-hydroxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound CC(C)(C)OC(=O)NC(C(=O)OC(C)(C)C)CC1=CC=C(O)C=C1 WGJGINMQCMATNP-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- DHCDFWKWKRSZHF-UHFFFAOYSA-L thiosulfate(2-) Chemical compound [O-]S([S-])(=O)=O DHCDFWKWKRSZHF-UHFFFAOYSA-L 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- QYYZHXHYNLXWAW-UHFFFAOYSA-N trimethyl(2-phenylethynyl)stannane Chemical compound C[Sn](C)(C)C#CC1=CC=CC=C1 QYYZHXHYNLXWAW-UHFFFAOYSA-N 0.000 description 1
- 229940094989 trimethylsilane Drugs 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000006442 vascular tone Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000008136 water-miscible vehicle Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000012138 yeast extract Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- This invention relates to methods of affecting gastric transit and gastric emptying, and to compounds and compositions useful therein.
- the neurotransmitter serotonin [5-hydroxytryptamine (5-HT)] is involved in multiple central nervous facets of mood control and in regulating sleep, anxiety, alcoholism, drug abuse, food intake, and sexual behavior. It has also been implicated in the regulation of vascular tone, gut motility and cell-mediated immune responses. Walther, D. J., et al., Science 299:76 (2003). 5-HT also plays a role in clotting and hemostasis: platelets — which cannot themselves make 5-HT — take up large amounts of peripheral 5-HT. Goodman & Gilman's The Pharmacological Basis of Therapeutics, 10 th ed., p. 274-5 (McGraw-Hill, 2001).
- Serotonin is synthesized in two steps from the amino acid tryptophan. Goodman & Gilman's, p. 270. The first step is rate-limiting, and is catalyzed by the enzyme tryptophan hydroxylase (TPH), which has two known isoforms: TPHl, which is expressed in the periphery, and TPH2, which is expressed primarily in the brain. Walther, D. J., et al., Science 299:76 (2003).
- TPH tryptophan hydroxylase
- the principle route by which serotonin is removed from the body involves the enzyme monoamine oxidase (MAO), which converts the compound to 5-hydroxyindole acetaldehyde, which is then converted to 5-hydroxyindole acetic acid (5 -HIAA) by the enzyme aldehyde dehydrogenase.
- MAO monoamine oxidase
- 5 -HIAA 5-hydroxyindole acetic acid
- mice reportedly expressed normal amounts of serotonin in classical serotonergic brain regions, but largely lacked serotonin in the periphery. Id. In another, the knockout mice exhibited abnormal cardiac activity, which was attributed to a lack of peripheral serotonin. Cote, F., et al, PNAS 100(23): 13525-13530 (2003). Because serotonin is involved in so many biochemical processes, drugs that affect serotonin levels or affect serotonin receptors are often attended by adverse effects. For example, parenteral injection of the TPH inhibitor p-chlorophenylalanine (p-CPA) to rats reportedly decreased their gastrointestinal motility. Sailer, C. F., Strieker, E.
- p-CPA TPH inhibitor
- This invention is directed, in part, to methods of affecting gastrointestinal transit and gastric emptying, which comprise inhibiting peripheral tryptophan hydroxylase (TPH) in patients in need thereof, without substantially affecting their brain 5 -HT levels.
- TPH peripheral tryptophan hydroxylase
- the TPH is inhibited by administering to the patient an effective amount of a compound of formula I:
- A is optionally substituted cycloalkyl, aryl, or heterocycle
- Figure 1 shows the effect of oral administration of a potent TPHl inhibitor on the gastrointestinal (GI) motility of rats.
- the asterisk identifies data wherein p ⁇ 0.01 when compared with vehicle control using the t test or one-way ANOVA test.
- Figure 2 shows the effect of oral administration of a potent TPHl inhibitor on the gastric emptying of rats.
- the asterisk identifies data wherein p ⁇ 0.01 when compared with vehicle control using the t test or one-way ANOVA test.
- Figure 3 shows the effect of oral administration of a potent TPHl inhibitor on the blood and proximal colon levels of 5-HT of the rats for which data is presented in figures 1 and 2. In both cases, p ⁇ 0.0001 using one-way ANOVA.
- This invention is based, in part, on the discovery of compounds that are potent inhibitors of TPH (e.g., TPHl). When administered to mammals, preferred compounds of the invention reduce peripheral serotonin levels.
- TPH potent inhibitors of TPH
- alkenyl means a straight chain, branched and/or cyclic hydrocarbon having from 2 to 20 (e.g., 2 to 10 or 2 to 6) carbon atoms, and including at least one carbon-carbon double bond.
- alkenyl moieties include vinyl, allyl, 1-butenyl, 2-butenyl, isobutylenyl, 1-pentenyl, 2-pentenyl, 3-methyl-l-butenyl, 2-methyl-2-butenyl, 2,3-dimethyl-2-butenyl, 1-hexenyl, 2-hexenyl, 3-hexenyl, 1-heptenyl, 2- heptenyl, 3-heptenyl, 1-octenyl, 2-octenyl, 3-octenyl, 1-nonenyl, 2-nonenyl, 3-nonenyl, 1- decenyl, 2-decenyl and 3-decenyl.
- alkyl means a straight chain, branched and/or cyclic (“cycloalkyl”) hydrocarbon having from 1 to 20 (e.g., 1 to 10 or 1 to 4) carbon atoms. Alkyl moieties having from 1 to 4 carbons are referred to as "lower alkyl.” Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, n-butyl, t-butyl, isobutyl, pentyl, hexyl, isohexyl, heptyl, 4,4-dimethylpentyl, octyl, 2,2,4-trimethylpentyl, nonyl, decyl, undecyl and dodecyl.
- Cycloalkyl moieties may be monocyclic or multicyclic, and examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and adamantyl. Additional examples of alkyl moieties have linear, branched and/or cyclic portions (e.g., l-ethyl-4-methyl- cyclohexyl).
- alkyl includes saturated hydrocarbons as well as alkenyl and alkynyl moieties.
- alkoxy means an -O-alkyl group. Examples of alkoxy groups include -OCH 3 , -OCH 2 CH 3 , -O(CH 2 ) 2 CH 3 , -O(CH 2 ) 3 CH 3 , -O(CH 2 ) 4 CH 3 , and -O(CH 2 ) 5 CH 3 .
- alkylaryl or "alkyl-aryl” means an alkyl moiety bound to an aryl moiety.
- alkylheteroaryl or “alkyl-heteroaryl” means an alkyl moiety bound to a heteroaryl moiety.
- alkylheterocycle or “alkyl-heterocycle” means an alkyl moiety bound to a heterocycle moiety.
- alkynyl means a straight chain, branched or cyclic hydrocarbon having from 2 to 20 (e.g., 2 to 20 or 2 to 6) carbon atoms, and including at least one carbon-carbon triple bond.
- alkynyl moieties include acetylenyl, propynyl, 1-butynyl, 2-butynyl, 1-pentynyl, 2-pentynyl, 3-methyl-l-butynyl, 4-pentynyl, 1-hexynyl, 2-hexynyl, 5-hexynyl, 1-heptynyl, 2-heptynyl, 6-heptynyl, 1-octynyl, 2-octynyl, 7-octynyl, 1-nonynyl, 2-nonynyl, 8-nonynyl, 1-decynyl, 2-decynyl and 9-decynyl.
- aryl means an aromatic ring or an aromatic or partially aromatic ring system composed of carbon and hydrogen atoms.
- An aryl moiety may comprise multiple rings bound or fused together.
- aryl moieties include anthracenyl, azulenyl, biphenyl, fluorenyl, indan, indenyl, naphthyl, phenanthrenyl, phenyl, 1,2,3,4-tetrahydro-naphthalene, and to IyI.
- arylalkyl or “aryl-alkyl” means an aryl moiety bound to an alkyl moiety.
- biohydrolyzable amide means an amide, ester, carbamate, carbonate, ureido, or phosphate, respectively, of a compound that either: 1) does not interfere with the biological activity of the compound but can confer upon that compound advantageous properties in vivo, such as uptake, duration of action, or onset of action; or 2) is biologically inactive but is converted in vivo to the biologically active compound.
- biohydrolyzable esters include lower alkyl esters, alkoxyacyloxy esters, alkyl acylamino alkyl esters, and choline esters.
- biohydrolyzable amides include lower alkyl amides, ⁇ -amino acid amides, alkoxyacyl amides, and alkylaminoalkyl-carbonyl amides.
- biohydrolyzable carbamates include lower alkylamines, substituted ethylenediamines, aminoacids, hydroxy alkylamines, heterocyclic and heteroaromatic amines, and polyether amines.
- disease or disorder mediated by peripheral serotonin and “disease and disorder mediated by peripheral serotonin” mean a disease and/or disorder having one or more symptoms, the severity of which are affected by peripheral serotonin levels.
- heteroalkyl refers to an alkyl moiety (e.g., linear, branched or cyclic) in which at least one of its carbon atoms has been replaced with a heteroatom (e.g., N, O or S).
- heteroaryl means an aryl moiety wherein at least one of its carbon atoms has been replaced with a heteroatom (e.g., N, O or S).
- heteroatom e.g., N, O or S.
- examples include acridinyl, benzimidazolyl, benzofuranyl, benzoisothiazolyl, benzoisoxazolyl, benzoquinazolinyl, benzothiazolyl, benzoxazolyl, furyl, imidazolyl, indolyl, isothiazolyl, isoxazolyl, oxadiazolyl, oxazolyl, phthalazinyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrimidyl, pyrrolyl, quinazolinyl, quinolinyl, tetrazolyl, thiazolyl, and tri
- heterocycle refers to an aromatic, partially aromatic or non-aromatic monocyclic or polycyclic ring or ring system comprised of carbon, hydrogen and at least one heteroatom (e.g., N, O or S).
- a heterocycle may comprise multiple (i.e., two or more) rings fused or bound together.
- Heterocycles include heteroaryls.
- heterocyclealkyl refers to a heterocycle moiety bound to an alkyl moiety.
- heterocycloalkyl refers to a non-aromatic heterocycle.
- heterocycloalkylalkyl or “heterocycloalkyl- alkyl” refers to a heterocycloalkyl moiety bound to an alkyl moiety.
- the terms “manage,” “managing” and “management” encompass preventing the recurrence of the specified disease or disorder, or of one or more of its symptoms, in a patient who has already suffered from the disease or disorder, and/or lengthening the time that a patient who has suffered from the disease or disorder remains in remission.
- the terms encompass modulating the threshold, development and/or duration of the disease or disorder, or changing the way that a patient responds to the disease or disorder.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic acids or bases including inorganic acids and bases and organic acids and bases.
- suitable pharmaceutically acceptable base addition salts include metallic salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made from lysine, N 5 N'- dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine (N-methylglucamine) and procaine.
- Suitable non-toxic acids include inorganic and organic acids such as acetic, alginic, anthranilic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethenesulfonic, formic, fumaric, furoic, galacturonic, gluconic, glucuronic, glutamic, glycolic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phenylacetic, phosphoric, propionic, salicylic, stearic, succinic, sulfanilic, sulfuric, tartaric acid, and p-toluenesulfonic acid.
- inorganic and organic acids such as acetic, alginic, anthranilic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethe
- non-toxic acids include hydrochloric, hydrobromic, phosphoric, sulfuric, and methanesulfonic acids.
- specific salts thus include hydrochloride and mesylate salts.
- Others are well-known in the art. See, e.g., Remington' s Pharmaceutical Sciences, 18 ed. (Mack Publishing, Easton PA: 1990) and Remington: The Science and Practice of Pharmacy, 19 th ed. (Mack Publishing, Easton PA: 1995).
- the term "potent TPHl inhibitor” is a compound that has a TPH 1 IC 50 of less than about 10 ⁇ M.
- the terms “prevent,” “preventing” and “prevention” contemplate an action that occurs before a patient begins to suffer from the specified disease or disorder, which inhibits or reduces the severity of the disease or disorder, or of one or more of its symptoms.
- the terms encompass prophylaxis.
- prodrug encompasses pharmaceutically acceptable esters, carbonates, thiocarbonates, N-acyl derivatives, N-acyloxyalkyl derivatives, quaternary derivatives of tertiary amines, N-Mannich bases, Schiff bases, amino acid conjugates, phosphate esters, metal salts and sulfonate esters of compounds disclosed herein.
- prodrugs include compounds that comprise a biohydrolyzable moiety ⁇ e.g., a biohydrolyzable amide, biohydrolyzable carbamate, biohydrolyzable carbonate, biohydrolyzable ester, biohydrolyzable phosphate, or biohydrolyzable ureide analog).
- Prodrugs of compounds disclosed herein are readily envisioned and prepared by those of ordinary skill in the art. See, e.g., Design of Prodrugs, Bundgaard, A. Ed., Elseview, 1985; Bundgaard, H., “Design and Application of Prodrugs," A Textbook of Drug Design and Development, Krosgaard-Larsen and H. Bundgaard, Ed., 1991, Chapter 5, p. 113-191; and Bundgaard, H., Advanced Drug Delivery Review, 1992, 8, 1-38.
- a prophylactically effective amount of a compound is an amount sufficient to prevent a disease or condition, or one or more symptoms associated with the disease or condition, or prevent its recurrence.
- a prophylactically effective amount of a compound is an amount of therapeutic agent, alone or in combination with other agents, which provides a prophylactic benefit in the prevention of the disease.
- the term “prophylactically effective amount” can encompass an amount that improves overall prophylaxis or enhances the prophylactic efficacy of another prophylactic agent.
- protecting group when used to refer to part of a molecule subjected to a chemical reaction, means a chemical moiety that is not reactive under the conditions of that chemical reaction, and which may be removed to provide a moiety that is reactive under those conditions.
- Protecting groups are well known in the art. See, e.g., Greene, T.W. and Wuts, P.G.M., Protective Groups in Organic Synthesis (3 rd ed., John Wiley & Sons: 1999); Larock, R.C., Comprehensive Organic Transformations (2 nd ed., John Wiley & Sons: 1999). Some examples include benzyl, diphenylmethyl, trityl, Cbz, Boc, Fmoc, methoxycarbonyl, ethoxycarbonyl, and pthalimido.
- pseudohalogen refers to a polyatomic anion that resembles a halide ion in its acid-base, substitution, and redox chemistry, generally has low basicity, and forms a free radical under atom transfer radical polymerization conditions.
- pseudohalogens include azide ions, cyanide, cyanate, thiocyanate, thiosulfate, sulfonates, and sulfonyl halides.
- selective TPHl inhibitor is a compound that has a TPH2_IC 50 that is at least about 10 times greater than its TPH 1 IC 50 .
- the terms “serotonin-mediated disease,” “serotonin- mediated disorder” and “serotonin-mediated disease or disorder” refer to a disease or disorder having one or more symptoms that are attributable to increased levels of peripheral 5- hydroxytryptamine (5-HT).
- the term “stereomerically enriched composition of a compound refers to a mixture of the named compound and its stereoisomer(s) that contains more of the named compound than its stereoisomer(s).
- a stereoisomerically enriched composition of (S)-butan-2-ol encompasses mixtures of (S)-butan-2-ol and (R)- butan-2-ol in ratios of, e.g., about 60/40, 70/30, 80/20, 90/10, 95/5, and 98/2.
- stereomerically pure means a composition that comprises one stereoisomer of a compound and is substantially free of other stereoisomers of that compound.
- a stereomerically pure composition of a compound having one stereocenter will be substantially free of the opposite stereoisomer of the compound.
- a stereomerically pure composition of a compound having two stereocenters will be substantially free of other diastereomers of the compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers of the compound, greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound, or greater than about 99% by weight of one stereoisomer of the compound and less than about 1% by weight of the other stereoisomers of the compound.
- substituted when used to describe a chemical structure or moiety, refers to a derivative of that structure or moiety wherein one or more of its hydrogen atoms is substituted with an atom, chemical moiety or functional group such as, but not limited to, alcohol, aldehylde, alkoxy, alkanoyloxy, alkoxycarbonyl, alkenyl, alkyl (e.g.
- a “therapeutically effective amount” of a compound is an amount sufficient to provide a therapeutic benefit in the treatment or management of a disease or condition, or to delay or minimize one or more symptoms associated with the disease or condition.
- a therapeutically effective amount of a compound is an amount of therapeutic agent, alone or in combination with other therapies, which provides a therapeutic benefit in the treatment or management of the disease or condition.
- the term “therapeutically effective amount” can encompass an amount that improves overall therapy, reduces or avoids symptoms or causes of a disease or condition, or enhances the therapeutic efficacy of another therapeutic agent.
- TPH1_IC 5O is the IC50 of a compound for
- TPHl as determined using the in vitro inhibition assay described in the Examples, below.
- TPH2_IC 50 is the IC50 of a compound for TPH2 as determined using the in vitro inhibition assay described in the Examples, below.
- treat contemplate an action that occurs while a patient is suffering from the specified disease or disorder, which reduces the severity of the disease or disorder, or one or more of its symptoms, or retards or slows the progression of the disease or disorder.
- one or more adjectives immediately preceding a series of nouns is to be construed as applying to each of the nouns.
- the phrase "optionally substituted alky, aryl, or heteroaryl” has the same meaning as "optionally substituted alky, optionally substituted aryl, or optionally substituted heteroaryl.”
- a chemical moiety that forms part of a larger compound may be described herein using a name commonly accorded it when it exists as a single molecule or a name commonly accorded its radical.
- the terms "pyridine” and “pyridyl” are accorded the same meaning when used to describe a moiety attached to other chemical moieties.
- stereochemistry of a structure or a portion of a structure is not indicated with, for example, bold or dashed lines, the structure or the portion of the structure is to be interpreted as encompassing all stereoisomers of it.
- names of compounds having one or more chiral centers that do not specify the stereochemistry of those centers encompass pure stereoisomers and mixtures thereof.
- any atom shown in a drawing with unsatisfied valences is assumed to be attached to enough hydrogen atoms to satisfy the valences.
- chemical bonds depicted with one solid line parallel to one dashed line encompass both single and double (e.g. , aromatic) bonds, if valences permit.
- Particular methods of this invention comprise the use of potent TPHl inhibitors.
- potent TPHl inhibitors are disclosed herein and in U.S. patent application nos. 11/638,677 and 60/874,596, both filed December 12, 2006. These compounds are significantly more potent than p-chlorophenylalanine, which has a TPHl IC 5 O of about 93 ⁇ M.
- A is optionally substituted cycloalkyl, aryl, or heterocycle
- A is optionally substituted cycloalkyl, aryl, or heterocycle
- particular compounds include those wherein A is optionally substituted cycloalkyl (e.g. , 6-membered and 5-membered).
- A is optionally substituted aryl (e.g., phenyl or naphthyl).
- A is optionally substituted heterocycle (e.g., 6-membered and 5-membered). Examples of 6-membered heterocycles include pyridine, pyridazine, pyrimidine, pyrazine, and triazine.
- 5-membered heterocycles include pyrrole, imidazole, triazole, thiazole, thiophene, and furan.
- A is aromatic. In others, A is not aromatic.
- A is an optionally substituted bicyclic moiety (e.g., indole, iso-indole, pyrrolo-pyridine, or napthylene).
- each of Ai and A 2 is independently a monocyclic optionally substituted cycloalkyl, aryl, or heterocycle.
- Compounds encompassed by this formula include those wherein Ai and/or A 2 is optionally substituted cycloalkyl (e.g., 6-membered and 5-membered).
- Ai and/or A 2 is optionally substituted aryl (e.g., phenyl or naphthyl).
- Ai and/or A 2 is optionally substituted heterocycle (e.g., 6-membered and 5-membered).
- 6- membered heterocycles include pyridine, pyridazine, pyrimidine, pyrazine, and triazine.
- 5-membered heterocycles include pyrrole, imidazole, triazole, thiazole, thiophene, and furan.
- Ai and/or A 2 is aromatic. In others, Ai and/or A 2 is not aromatic.
- D is optionally substituted aryl (e.g., phenyl or naphthyl).
- D is optionally substituted heterocycle (e.g., 6-membered and 5-membered).
- 6-membered heterocycles include pyridine, pyridazine, pyrimidine, pyrazine, and triazine.
- 5- membered heterocycles include pyrrole, imidazole, triazole, thiazole, thiophene, and furan.
- D is aromatic.
- D is not aromatic.
- D is an optionally substituted bicyclic moiety (e.g. , indole, iso-indole, pyrrolo-pyridine, or napthylene).
- E is optionally substituted aryl (e.g., phenyl or naphthyl).
- E is optionally substituted heterocycle (e.g., 6-membered and 5-membered).
- 6- membered heterocycles include pyridine, pyridazine, pyrimidine, pyrazine, and triazine.
- 5-membered heterocycles include pyrrole, imidazole, triazole, thiazole, thiophene, and furan.
- E is aromatic.
- E is not aromatic.
- E is an optionally substituted bicyclic moiety (e.g. , indole, iso-indole, pyrrolo-pyridine, or napthylene).
- particular compounds include those wherein Ri is hydrogen or optionally substituted alkyl.
- R 2 is hydrogen or optionally substituted alkyl.
- n is 1 or 2.
- X is a bond or S.
- X is -O-, -C(R 3 R 4 )O-, or -OC(R 3 R 4 )-, and, for example, R 3 is hydrogen or optionally substituted alkyl, and R 4 is hydrogen or optionally substituted alkyl.
- R 3 is hydrogen and R 4 is trifluromethyl.
- X is -S(O 2 )-, -S(O 2 )N(R 5 )-, -N(R 5 )S(O 2 )-, -C(R 3 R 4 )S(O 2 )-, or -S(O 2 )C(R 3 R 4 )-, and, for example, R 3 is hydrogen or optionally substituted alkyl, R 4 is hydrogen or optionally substituted alkyl, and R 5 is hydrogen or optionally substituted alkyl.
- X is -N(R 5 )-, -N(R 5 )C(O)N(R 5 )-, -C(R 3 R 4 )N(R 5 )-, or -N(R 5 )C(R 3 R 4 )-, and, for example, R 3 is hydrogen or optionally substituted alkyl, R 4 is hydrogen or optionally substituted alkyl, and each R 5 is independently hydrogen or optionally substituted alkyl.
- R 3 is trifluoromethyl. Others are encompassed by the formula:
- R 3 is hydrogen
- each of Zi, Z 2 , Z 3 , and Z 4 is independently N or CR 6 ; each R 6 is independently hydrogen, cyano, halogen, OR 7 , NRgR ⁇ , amino, hydroxyl, or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R 7 is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each Rg is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R 9 is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; and m is 1-4. Certain such compounds are of the formula:
- R 3 is trifluoromethyl. Others are of the formula:
- R 3 is hydrogen
- some compounds are such that all of Zi, Z 2 , Z3, and Z 4 are N. In others, only three of Zi, Z 2 , Z3, and Z 4 are N. In others, only two of Zi, Z 2 , Z 3 , and Z 4 are N. In others, only one of Zi, Z 2 , Z 3 , and Z 4 is N. In others, none of Zi, Z 2 , Z 3 , and Z 4 are N.
- each of Z'i, Z' 2 , and Z' 3 is independently N, NH, S, O or CR 6 ; each R 6 is independently amino, cyano, halogen, hydrogen, OR 7 , SR 7 , NRgR 9 , or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R 7 is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each Rg is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R 9 is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; and p is 1-3. Certain such compounds are of the formula:
- R 3 is trifluoromethyl. Others are of the formula: wherein, for example, R 3 is hydrogen.
- some compounds are such that all of Z'i, Z' 2 , and Z' 3 are N or NH. In others, only two of Z' I , Z' 2 , and Z' 3 are N or NH. In others, only one of Z'i, Z' 2 , and Z' 3 is N or NH. In others, none of Z' h Z' 2 , and Z' 3 are N or NH.
- each of Z"i, Z" 2 , Z" 3 , and Z" 4 is independently N or CR10; each Ri 0 is independently amino, cyano, halogen, hydrogen, ORn, SRn, NR12R13, or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each Rn is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each Ri 2 is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; and each Ri 3 is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle. Certain such compounds are of the formula:
- R 3 is hydrogen
- some compounds are such that all of Z"i, Z" 2 , Z"3, and Z" 4 are N. In others, only three of Z" ls Z" 2 , Z' f 3, and Z" 4 are N. In others, only two of Z"i, Z" 2 , Z" 3 , and Z" 4 are N. In others, only one of Z"i, Z" 2 , Z" 3 , and Z" 4 is N. In others, none of Z" ls Z" 2 , Z" 3 , and Z" 4 are N.
- R 3 is trifluoromethyl. Others are of the formula:
- R 3 is hydrogen
- some compounds are such that all of Z" ls Z" 2 , Z" 3 , and Z" 4 are N. In others, only three of Z" ls Z" 2 , Z" 3 , and Z" 4 are N. In others, only two of Z"i, Z" 2 , Z" 3 , and Z" 4 are N. In others, only one of Z" ls Z" 2 , Z" 3 , and Z" 4 is N. In others, none of Z" ls Z" 2 , Z" 3 , and Z" 4 are N.
- each Ri 4 is independently amino, halogen, hydrogen, C(O)R A , OR A , NR B R C , S(O 2 )R A , or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R A is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each R B is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl-heterocycle; each Rc is independently hydrogen or optionally substituted alkyl, alkyl-aryl or alkyl- heterocycle; and m is 1-4.
- particular compounds include those wherein both A and E are optionally substituted phenyl and, for example, X is -O-, -C(RsR 4 )O-, or -OC(RsR 4 )- and, for example, R3 is hydrogen and R 4 is trifluoromethyl and, for example, n is 1.
- Stereoisomers may be asymmetrically synthesized or resolved using standard techniques such as chiral columns, chiral resolving agents, or enzymatic resolution. See, e.g., Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S. H., et al, Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw Hill, NY, 1962); and Wilen, S. H., Tables of Resolving Agents and Optical Resolutions, p. 268 (EX. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972).
- Particular compounds of the invention are potent TPHl inhibitors.
- Specific compounds have a TPHl IC 50 of less than about 10, 5, 2.5, 1, 0.75, 0.5, 0.4, 0.3, 0.2, 0.1, or 0.05 ⁇ M.
- Particular compounds are selective TPHl inhibitors.
- Specific compounds have a TPHl IC 50 that is about 10, 25, 50, 100, 250, 500, or 1000 times less than their TPH2_IC 50 .
- Particular compounds do not significantly inhibit human tyrosine hydroxylase (TH).
- specific compounds have an IC 50 for TH of greater than about 100, 250, 500 or 1000 ⁇ M.
- Particular compounds do not significantly inhibit human phenylalanine hydroxylase (PAH).
- PAH human phenylalanine hydroxylase
- specific compounds have an IC50 for PAH of greater than about 100, 250, 500 or 1000 ⁇ M.
- Particular compounds of the invention do not significantly bind (e.g., inhibit with an IC 50 of greater than about 10, 25, 50, 100, 250, 500, 750, or 1000 ⁇ M) to one or more of the following: angiotensin converting enzyme, erythropoietin (EPO) receptor, factor IX, factor XI, integrin (e.g., ⁇ 4), isoxazoline or isoxazole fibrinogen receptor, metalloprotease, neutral endopeptidase (NEP), phosphatase (e.g., tyrosine phosphatase), phosphodiesterase (e.g., PDE-4), polymerase, PPAR ⁇ , TNF- ⁇ , vascular cell adhesion molecule- 1 (VCAM-I), or the vitronectin receptor.
- angiotensin converting enzyme EPO
- factor IX factor IX
- factor XI factor XI
- integrin e.g., ⁇ 4
- certain compounds of the invention do not readily cross the blood/brain barrier (e.g., less than about 5, 2.5, 2, 1.5, 1, 0.5, or 0.01 percent of compound in the blood passes into the brain).
- the ability or inability of a compound to cross the blood/brain barrier can be determined by methods known in the art. See, e.g. , Riant, P. et al. , Journal of Neurochemistry 51 :421 -425 (1988); Kastin, A.J., Akerstrom, V., J. Pharmacol. Exp. Therapeutics 294:633-636 (2000); W. A. Banks, W. A., et al., J. Pharmacol. Exp. Therapeutics 302:1062-1069 (2002).
- A is optionally substituted phenyl, biphenyl or napthyl.
- Pi is Ri or a protecting group
- P 2 is a protecting group
- P3 is OR 2 or a protecting group
- X' is, for example, O or N
- Yi and Y 3 are halogen (e.g., Br, Cl) or an appropriate pseudohalide (e.g., triflate); and each R' is independently hydrogen or optionally substituted alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle, or are taken together with the oxygen atoms to which they are attached to provide a cyclic dioxaborolane (e.g., 4,4,5,5-tetramethyl- 1,3,2-dioxaborolane).
- a cyclic dioxaborolane e.g., 4,4,5,5-tetramethyl- 1,3,2-dioxaborolane.
- the groups A, R 1 , R 2 , R 3 , R 6 and m are defined elsewhere herein.
- the moieties Z"i, Z M 2, Z M 3, and Z M 4 are also defined herein, although it is to be understood that with regard to the scheme shown above, one of them is attached to the phenyl ring.
- Z"i and Z M 4 may be independently CRio (which is defined herein), while Z M 2 is N and Z" 3 is a carbon atom bound to the adjacent phenyl ring.
- the individual reactions shown above can be performed using conditions known in the art. For example, palladium catalysts and conditions suitable for the Suzuki coupling of the boron and halogen-containing moieties are well known, and examples are provided below.
- types and appropriate uses of protecting groups are well known, as are methods of their removal and replacement with moieties such as, but not limited to, hydrogen (e.g., hydrolysis under acidic or basic conditions).
- the A moiety can be bicyclic (e.g., optionally substituted biphenyl).
- the starting material containing A can be prepared as shown below:
- Y 2 is halogen or pseudohalogen, and each R is independently hydrogen or optionally substituted alkyl, alkyl-aryl, alkyl-heterocycle, aryl, or heterocycle, or are taken together with the oxygen atoms to which they are attached to provide a cyclic dioxaborolane (e.g., 4,4,5,5- tetramethyl-l,3,2-dioxaborolane).
- a cyclic dioxaborolane e.g., 4,4,5,5- tetramethyl-l,3,2-dioxaborolane.
- X is N, O or S
- cyclic moiety D can be any of a variety of structures, which are readily incorporated into compounds of the invention.
- D is oxazole
- Scheme 7 compounds wherein D is oxazole can be prepared as shown below in Scheme 7:
- This invention encompasses methods of affecting ⁇ e.g., slowing) gastrointestinal transit and gastric emptying, which comprise inhibiting peripheral tryptophan hydroxylase ⁇ e.g., TPHl) in patients in need thereof.
- Patients in need thereof include patients with diarrhea and patients susceptible to diarrhea ⁇ e.g., patients taking medications or undergoing therapies, such as chemotherapy, that can cause diarrhea).
- Preferred methods avoid measurably affecting serotonin levels in the central nervous system (CNS).
- CNS central nervous system
- One embodiment encompasses a method of slowing gastrointestinal transit in a patient, which comprises administering to the patient a sufficient amount of a potent TPHl inhibitor.
- Another embodiment encompasses a method of slowing gastric emptying in a patient, which comprises administering to the patient a sufficient amount of a potent TPHl inhibitor.
- the amount of active pharmaceutical ingredient e.g., a potent TPHl inhibitor
- amount of active pharmaceutical ingredient sufficient to achieve the desired pharmacological effect can be readily determined by those skilled in the art. For example, a patient can be administered a low dose of a compound, and then increasingly larger doses over time until the desired effect is achieved.
- Particular methods of the invention avoid adverse effects associated with alteration of CNS serotonin levels.
- adverse effects include agitation, anxiety disorders, depression, and sleep disorders (e.g., insomnia and sleep disturbance).
- compositions comprising one or more compounds of the invention.
- Certain pharmaceutical compositions are single unit dosage forms suitable for oral, mucosal (e.g., nasal, sublingual, vaginal, buccal, or rectal), parenteral (e.g., subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial), or transdermal administration to a patient.
- dosage forms include, but are not limited to: tablets; caplets; capsules, such as soft elastic gelatin capsules; cachets; troches; lozenges; dispersions; suppositories; ointments; cataplasms (poultices); pastes; powders; dressings; creams; plasters; solutions; patches; aerosols (e.g., nasal sprays or inhalers); gels; liquid dosage forms suitable for oral or mucosal administration to a patient, including suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions, or a water-in-oil liquid emulsions), solutions, and elixirs; liquid dosage forms suitable for parenteral administration to a patient; and sterile solids (e.g.
- crystalline or amorphous solids that can be reconstituted to provide liquid dosage forms suitable for parenteral administration to a patient.
- the formulation should suit the mode of administration.
- the oral administration of a compound susceptible to degradation in the stomach may be achieved using an enteric coating.
- a formulation may contain ingredients that facilitate delivery of the active ingredient(s) to the site of action.
- compounds may be administered in liposomal formulations in order to protect them from degradative enzymes, facilitate transport in circulatory system, and effect their delivery across cell membranes.
- poorly soluble compounds may be incorporated into liquid dosage forms (and dosage forms suitable for reconstitution) with the aid of solubilizing agents, emulsif ⁇ ers and surfactants such as, but not limited to, cyclodextrins (e.g., ⁇ -cyclodextrin, ⁇ -cyclodextrin, Captisol ® , and EncapsinTM (see, e.g., Davis and Brewster, Nat. Rev. Drug Disc.
- solubilizing agents emulsif ⁇ ers and surfactants
- cyclodextrins e.g., ⁇ -cyclodextrin, ⁇ -cyclodextrin, Captisol ®
- EncapsinTM see, e.g., Davis and Brewster, Nat. Rev. Drug Disc.
- Labrasol ® Labrafil ® , Labrafac ® , cremafor, and non-aqueous solvents, such as, but not limited to, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethyl formamide, dimethyl sulfoxide (DMSO), biocompatible oils (e.g., cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols, fatty acid esters of sorbitan, and mixtures thereof (e.g., DMSOxornoil).
- DMSO dimethyl formamide
- biocompatible oils e.g., cottonseed, groundnut, corn, germ, olive, castor, and sesame oils
- glycerol tetrahydr
- Nanoparticles of a compound may be suspended in a liquid to provide a nanosuspension (see, e.g., Rabinow, Nature Rev. Drug Disc. 3:785-796 (2004)).
- Nanoparticle forms of compounds described herein may be prepared by the methods described in U.S. Patent Publication Nos. 2004-0164194, 2004-0195413, 2004-0251332, 2005-0042177 Al, 2005-0031691 Al, and U.S. Patent Nos.
- the nanoparticle form comprises particles having an average particle size of less than about 2000 nm, less than about 1000 nm, or less than about 500 nm.
- the composition, shape, and type of a dosage form will typically vary depending with use. For example, a dosage form used in the acute treatment of a disease may contain larger amounts of one or more of the active ingredients it comprises than a dosage form used in the chronic treatment of the same disease.
- a parenteral dosage form may contain smaller amounts of one or more of the active ingredients it comprises than an oral dosage form used to treat the same disease. How to account for such differences will be apparent to those skilled in the art. See, e.g. , Remington 's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990).
- compositions of the invention suitable for oral administration can be presented as discrete dosage forms, such as, but are not limited to, tablets (e.g., chewable tablets), caplets, capsules, and liquids (e.g., flavored syrups).
- dosage forms contain predetermined amounts of active ingredients, and may be prepared by methods of pharmacy well known to those skilled in the art. See generally, Remington 's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990).
- Typical oral dosage forms are prepared by combining the active ingredient(s) in an intimate admixture with at least one excipient according to conventional pharmaceutical compounding techniques. Excipients can take a wide variety of forms depending on the form of preparation desired for administration.
- tablets and capsules represent the most advantageous oral dosage unit forms.
- tablets can be coated by standard aqueous or non-aqueous techniques.
- Such dosage forms can be prepared by conventional methods of pharmacy.
- pharmaceutical compositions and dosage forms are prepared by uniformly and intimately admixing the active ingredients with liquid carriers, finely divided solid carriers, or both, and then shaping the product into the desired presentation if necessary.
- Disintegrants may be incorporated in solid dosage forms to facility rapid dissolution. Lubricants may also be incorporated to facilitate the manufacture of dosage forms (e.g. , tablets).
- Parenteral dosage forms can be administered to patients by various routes including subcutaneous, intravenous (including bolus injection), intramuscular, and intraarterial. Because their administration typically bypasses patients' natural defenses against contaminants, parenteral dosage forms are specifically sterile or capable of being sterilized prior to administration to a patient. Examples of parenteral dosage forms include solutions ready for injection, dry products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection, suspensions ready for injection, and emulsions.
- Suitable vehicles that can be used to provide parenteral dosage forms of the invention are well known to those skilled in the art. Examples include: Water for Injection USP; aqueous vehicles such as Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection; water-miscible vehicles such as ethyl alcohol, polyethylene glycol, and polypropylene glycol; and nonaqueous vehicles such as corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate. 6.
- Water for Injection USP Water for Injection USP
- aqueous vehicles such as Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection
- water-miscible vehicles such as ethyl alcohol
- HPLC high performance liquid chromatography
- the organic layer was separated and washed with H 2 O (2x100ml), dried over Na 2 SO 4 , and concentrated in vacuo to give crude intermediate.
- the crude compound was dissolved in 5ml of MeCN and 5ml of H 2 O in a 20ml microwave reaction vial. To this solution were added L-/?-borono-phenylalanine (253mg, 1.21mmol), sodium carbonate (256mg, 2.42mmol) and catalytic amount of dichlorobis(triphenylphosphine)-palladium(II) (42.1mg, O.O ⁇ mmol). The mixture was sealed and stirred in the microwave reactor at 150 0 C for 5 minutes, followed by the filtration through celite.
- (R)-l-(l-(Napthalen-2-yl) ethyl) cyanoguanidine was prepared by forming a mixture of naphthalene amine (1 equivalent), sodium dicyanide (0.95 eq.) and followed by 5N HCl (1 eq.) in n-BuOH: H 2 O (1 : 1). The mixture was refluxed for 1 day in a sealed tube at 160 0 C, and progress of reaction was monitored by LCMS. After completion of reaction, solvent (n- BuOH) was removed under reduced pressure and IN HCl was added to adjust pH to 3-5 range. The aqueous solution was extracted with EtOAc (2x100) and combined organic phase was dried over Na 2 SO 4 .
- the crude intermediate was then dissolved in 1.5ml of MeCN and 1.5ml of H 2 O in a 5ml microwave vial. To this solution were added L-/?-borono- phenylalanine (126mg, 0.606mmol), sodium carbonate (128mg, 1.21mmol) and catalytic amount of dichlorobis(triphenylphosphine)-palladium(II) (21. lmg, 0.03mmol). The mixture was sealed and stirred in the microwave reactor at 150 0 C for 5 minutes followed by the filtration through celite. The filtrate was concentrated and dissolved in MeOH and H 2 O (1 :1) and purified by preparative HPLC using MeOH/H 2 O/TFA solvent system.
- Tetrabutylammonium fluoride (0.1 ml; 1.0 M solution in tetrahydrofuran) was added to a solution of 2-trifluoromethyl-benzaldehyde (1.74g, lOmmol) and trifluoromethyltrimethylsilane (TMSCF 3 ) (1.8ml, 12 mmol) in 10 ml THF at 0 0 C.
- TMSCF 3 trifluoromethyltrimethylsilane
- the formed mixture was warmed up to room temperature and stirred for 4 hours.
- the reaction mixture was then treated with 12 ml of IN HCl and stirred overnight.
- the product was extracted with ethyl acetate (3x20ml).
- the organic layer was separated and dried over sodium sulfate.
- the organic solvent was evaporated to give 2.2g of l-(2- trifluoromethylphenyl)-2,2,2-trifluoro-ethanol, yield 90%.
- Tetrabutylammonium fluoride (0.1 ml; 1.0 M solution in tetrahydrofuran) was added to a solution of 4-methyl-benzaldehyde (1.2g, lOmmol) and TMSCF3 (1.8ml, 12 mmol) in 10 ml THF at 0 0 C. The formed mixture was warmed up to room temperature and stirred for 4 hours. The reaction mixture was then treated with 12 ml of IN HCl and stirred overnight. The product was extracted with ethyl acetate (3x20ml). The organic layer was separated and dried over sodium sulfate. The organic solvent was evaporated to give 1.6g of l-(4- methylphenyl)-2,2,2-trifluoro-ethanol, yield 86%.
- a microwave vial was charged with 4-chloro-2-amino-6-[l-(4-methylphenyl)-2,2,2- trifluoro-ethoxy]-pyrimidine (33mg, 0. lmmol), 4-borono-L-phenylalanine (31mg, 0.15mmol) and 1 ml of acetonitrile, 0.7ml of water.
- Aqueous sodium carbonate (0.3 ml, IN) was added to above solution followed by 5 mol percent of dichlorobis(triphenylphosphine)- palladium(II).
- the reaction vessel was sealed and heated to 150 0 C for 5 minutes with microwave. After cooling, the reaction mixture was evaporated to dryness.
- Cyclohexanecarbaldehyde (0.9 g, 5mmol) was dissolved in 10ml aqueous 1,4- dioxane, to which 200mg (10 mmol) sodium borohydride was added. The reaction was run overnight at room temperature. After completion of the reaction, 5ml 10% HCl solution was added and the product was extracted with ethyl acetate. The organic layer was separated and dried over sodium sulfate. The organic solvent was evaporated to give 0.8g of 1-cyclohexyl- 2,2,2-trifluoro-ethanol, yield 88%.
- a microwave vial (2ml) was charged with 4-chloro-6-[2-fluorophenoxy]-pyrimidine, (33mg, 0. lmmol), 4-borono-L-phenylalanine(31mg, 0.15mmol) and 1 ml of actonitrile, 0.7ml of water, 0.3 ml of aqueous sodium carbonate (IM) was added to above solution followed by 5 mol % of dichlorobis(triphenylphosphine)-palladium(II). The reaction vessel was sealed and heated to 150 0 C for 5 minutes by microwave.
- 3-(4-Chlorophenyl)piperidine (232mg, lmmol) was added to a solution of 2,4- dichlorotriazine (149.97mg, lmmol), and 300mg diisopropylethyl amine in 10ml THF at 0 0 C.
- the formed mixture was warmed up to room temperature and stirred for 1 hour.
- the product was extracted with ethyl acetate (3x20ml).
- the organic layer was separated and dried over sodium sulfate.
- the organic solvent was evaporated to give 328mg of 2-chloro-4-[3-(4- chlorophenyl)-piperidin-l-yl]-[l, 3, 5] triazine.
- a microwave vial was charged with 2-chloro-4-[3-(4-chlorophenyl)-piperidin-l-yl]- [1, 3, 5]triazine (62mg, 0.2mmol), 4-borono-L-phenylalanine(60mg, 0.3mmol), 1 ml of acetonitrile, and 0.7ml of water.
- Aqueous sodium carbonate (0.6 ml; IM) was added to the solution, followed by 5 mol percent dichlorobis(triphenylphosphine)-palladium(II).
- the reaction vessel was sealed and heated to 150 0 C for 5 minutes with microwave. After cooling, the reaction mixture was evaporated to dryness.
- a microwave vial was charged with 4-chloro-6-[2,2,2-trifluoro-l-phenyl-ethoxy]- [l,3,5]triazine-2-ylamine (33mg, 0. lmmol), 4-borono-L-phenylalanine(31mg, 0.15mmol), 1ml of actonitrile, and 0.7ml of water.
- Aqueous sodium carbonate (0.3 ml, IM) was added to above solution followed by 5 mol percent dichlorobis(triphenylphosphine)-palladium(II).
- the reaction vessel was sealed and heated to 150 0 C for 5 minutes by microwave. After cooling, the reaction mixture was evaporated to dryness.
- a microwave vial was charged with 6-chloro-N-[l-naphthalen-2yl-ethyl]- [l,3,5]triazine-2,4-diamine (30mg, O.lmmol), 2-boc protected-amino-3- ⁇ 5-[4,4,5,5,- tetramethyl-[l,3,2]dioxaborolan-2-yl)-pyridin2-yl-]-propionic acid (50mg, 0.15mmol) 1 ml of acetonitrile, and 0.7ml of water.
- Aqueous sodium carbonate (0.3 ml; IN) was added to the solution, followed by 5 mol percent dichlorobis(triphenylphosphine)-palladium(II).
- the reaction vessel was sealed and heated to 150 0 C for 5 mintues by microwave. After cooling, the reaction mixture was evaporated to dryness. The residue was dissolved in 2.5 ml of methanol, and was then purified by Prep-LC to give 7 mg of boc protected 2-amino-3- ⁇ 5-[4- amino-6-( 1 -naphthalen-2-yl-ethylamino)- [ 1 ,3 ,5 ]triazin-2-yl] -pyridin-2-yl ⁇ proionic acid.
- reaction vessel was sealed and heated to 150 0 C for 5 minutes with microwave. After cooling, the reaction mixture was evaporated to dryness, the residue was dissolved in 2.5 ml of methanol, and then was purified with Prep- LC to give 6.8 mg of boc protected 2-amino-3- ⁇ 3-[4-amino-6-(l-naphthalen-2-yl- ethylamino)[ 1 ,3,5]triazin-2-yl]-pyrazol- 1 -yl ⁇ proionic acid.
- Aqueous sodium carbonate (2 ml, IM) was added to above solution followed by 5 mol percent of dichlorobis- (triphenylphosphine)-palladium(II).
- the reaction vessel was sealed and heated to 15O 0 C for 5 minutes with microwave. After cooling, the reaction mixture was evaporated to dryness. The residue was dissolved in 2.5 ml of methanol and purified with Prep-LC to give 5.3 mg of 2-amino-3- ⁇ 4-[6-(3-cyclopentyloxy-4-methoxy-benzylamino)-pyrimidin-4-yl]-phenyl ⁇ - propionic acid, yield 6%.
- Emrys process vial (2-5ml) for microwave was charged with (6-chloro-pyrazin-2- yl)-(3-cyclopentyloxy-4-methoxy-benzyl)-amine (50mg, 0.15mmol), 4-borono-L- phenylalanine (31mg, 0.15mmol) and 2 ml of acetonitrile.
- Aqueous sodium carbonate (2 ml, IM) was added to the solution followed by 5 mol percent of dichlorobis(triphenylphosphine)- palladium(II).
- the reaction vessel was sealed and heated to 15O 0 C for 5 minutes by microwave. After cooling, the reaction mixture was evaporated to dryness.
- An Emrys process vial (2-5ml) for microwave was charged with (5-bromo-pyrazin-2- yl)-(4'-methyl-biphenyl-2-ylmethyl)-amine (25mg, 0.071mmol), 4-borono-L-phenylalanine (22mg, 0.1 lmmol) and 1 ml of acetonitrile.
- Aqueous sodium carbonate (1 ml, IM) was added to the solution followed by 5 mol percent dichlorobis(triphenylphosphine)- palladium(II).
- the reaction vessel was sealed and heated to 15O 0 C for 5 mintues by microwave. After cooling, the reaction mixture was evaporated to dryness.
- An Emrys process vial (2-5ml) for microwave was charged with 4-chloro-6-(2,2,2- trifluoro-l-phenyl-ethoxy)-pyrimidine (30mg, O.l lmmol), 4-borono-L-phenylalanine (32mg, O.l ⁇ mmol), 1 ml of acetonitrile and 0.6 ml of water.
- Aqueous sodium carbonate (0.42 ml, IM) was added to above solution followed by 10 mol percent of POPd 2 (dihydrogen di- ⁇ - chlorodichlorobis(di-tert-butylphosphinito- ⁇ P) dipalladate.
- reaction vessel was sealed and heated to 12O 0 C for 30 minutes by microwave. After cooling, the reaction mixture was evaporated to dryness. The residue was dissolved in 2.5 ml of methanol, and the product was purified with Prep-LC to give 4.8mg of 2-amino-3- ⁇ 4-[6-(2,2,2-trifluoro-lphenyl-ethoxy)- pyrimidin-4-yl]-phenyl ⁇ -propionic acid, yield 11%.
- Tetrabutylammonium fluoride (TBAF: 0.1 ml, IM) in THF was added to a solution of 3,4-difluro-benzaldehyde (1.42g, lOmmol) and (trifluromethyl)trimethylsilane (1.7Og, 12mmol) in 10 ml THF at O 0 C.
- the mixture was warmed up to room temperature and stirred for 4 hours.
- the reaction mixture was treated with 12 ml of IM HCl and stirred overnight.
- the product was extracted with dicloromethane (3x20ml), the organic layer was combined and passed through a pad of silica gel. The organic solvent was evaporated to give 1.9g of 1- (3,4-difiuoro-phenyl)-2,2,2-trifluoro-ethanol, yield 90%.
- Aqueous sodium carbonate (0.3 ml, IM) was added to above solution followed by 5 mol % of dichlorobis(triphenylphosphine)-palladium(II).
- the reaction vessel was sealed and heated to 15O 0 C for 5 minutes by microwave. After cooling, the reaction mixture was evaporated to dryness. The residue was dissolved in 2.5 ml of methanol, then purified with Prep-LC to give 10 mg of 2-amino-3-(4- ⁇ 6-[l-(3,4-difluoro-phenyl)-2,2,2-trifluoro-ethoxy]-pyridin-4-yl ⁇ - phenyl)-propionic acid, yield 21%.
- reaction mixture was cooled, filtered through a syringe filter and then separated by a reverse phase preparative-HPLC using YMC-Pack ODS 100x30 mm ID column (MeOH/H 2 O/TFA solvent system). The pure fractions were concentrated in vacuum. The product was then suspended in 5 ml of water, frozen and lyophilized to give the title compound as a trifluoro salt (12 mg, 20 %).
- the above alcohol (290 mg, 1.151 mmol) was dissolved in anhydrous THF (10 ml).
- Sodium hydride 55 mg, 1.375 mmol was added all at once, and the mixture was stirred at room temperature for 30 minutes.
- the solution was then transferred into a flask that contained a suspension of 2-amino-4,6-dichloro-triazine (190 mg, 1.152 mmol) in THF (20 ml). The mixture was stirred at room temperature overnight.
- the reaction mixture was stirred at about -40 0 C for 0.5 hours, then the cold bath was removed and the temperature was allowed to rise slowly to room temperature.
- the solvent was evaporated and the residue was extracted with hexane (4x20 ml). The collected extractions were washed with cold 10% aqueous NaHCO 3 and dried over Na 2 SO 4 .
- the solvent was evaporated at reduced pressure to afford 3,5-difluorophenyl-l- trimethylsilyloxyalkene (2.03g, 7.929 mmol, 57% crude yield), which was used in the successive reaction without further purification.
- Powered calcium carbonate (3.806g, 38.06 mmol) and ethyl vinyl ether (2.184g, 30.329 mmol) were added to a solution of eerie ammonium nitrate (10.43Og, 19.033 mmol) in methanol (40 ml) under nitrogen atmosphere.
- the water layer was basified to pH « 10 with aqueous sodium hydroxide (IM), and was extracted with dichloromethane and the organic layers were combined, dried over magnesium sulfate and concentrated to afford 290 mg of l-(5,7-difluoro-naphthalen-2-yl)- ethylamine (38% yield).
- IM aqueous sodium hydroxide
- the fresh made amine (290mg, 1.401mmol) was added directly to a suspension of 2- amino-4,6-dichloro triazine (277mg, 1.678 mmol) in anhydrous 1,4-dioxane (60 ml), and followed by addition of N,N-diisopropylethylamine (1 ml, 5.732 mmol).
- the mixture was heated to mild reflux for about 3 hours.
- the reaction mixture was then cooled, and the solvent was removed under reduced pressure. To the residue was added water and the mixture was sonicated for 2-3 minutes.
- 5-Chloro-pyrazine-2 carboxylic acid (3,4-dimethoxy-phenyl)-amide (0.18 g, 0.61 mmol), L-p-borono phenylalanine (0.146 g, 0.70 mmol), CH 3 CN (2.5 ml), H 2 O (2.5 ml), Na 2 CO 3 (0.129 g, 1.22 mmol) were combined in a microwave vial. The mixture was sealed and kept at 150 0 C for 5 minutes. The mixture was filtered and concentrated.
- 2-Amino 4,6-dichloro pyrimidine 0.327 g, 2 mmol
- methyl-(l-naphthalen-2yl- ethyl)-amine (0.360 g, 2 mmol)
- cesium carbonate 0.717 g, 2.2 mmol
- the vial was sealed and stirred at 210 0 C for 20 minutes in a microwave reactor.
- N-(biphenyl-4-ylmethyl)-5-bromopyrazin-2-amine 60 mg, 0.176 mmol
- L-p- boronophenylalanine 37 mg, 0.176 mmol
- palladiumtriphenylphosphine dichloride 3.6 mg, 0.0052 mmol
- Na 2 CO 3 37 mg, 0.353 mmol
- acetonitrile 1.25 mis
- water (1.25 mis
- Benzylmercaptan (0.14g, 1.11 mmol) was treated with NaH (60% in mineral oil, 67 mg, 1.66 mmol) in dry THF (15 ml) for 30 minutes.
- 2-Amino-4,6-dichloropyrimidine (0.2 g, 1.22 mmol) was added and the mixture was stirred overnight.
- the mixture was diluted with methylenechloride, washed with water, then brine, dried over MgSO4, and concentrated to give 0.11 g of 4-(benzylthio)-6-chloropyrimidin-2-amine.
- 2-Mercaptonapthalene (0.2 g, 1.148) was treated with NaH (60% in Mineral oil, 92 mg, 2.30 mmol) in dry THF (10 ml) for 30 minutes.
- 2-Amino-4,6-dichloropyrimidine (0.21 g, 1.26 mmol) was added and the mixture was stirred overnight.
- the mixture was diluted with methylenechloride, washed with water, then brine, dried over MgSO4, and concentratred to give 0.18 g 4-chloro-6-(naphthalen-2-ylmethylthio)pyrimidin-2-amine.
- 3,5-Difluorophenyl-trifluoromethyl ketone was treated with NaBH 4 (0.18 g, 4.76 mmol) in THF (5 ml) for 2 hours. The mixture was quenched with water, extracted with methylene chloride (2x). The organics were combined, filtered through silica gel and concentrated to give 0.46g of l-(3,4-difluorophenyl)-2,2,2-trifluoroethanol. l-(3,4-Difluorophenyl)-2,2,2-trifluoroethanol (0.1 g, 0.471 mmol) was treated with NaH (60% in mineral oil, 38 mg, 0.943 mmol) in dry THF (3 ml) for 30 minutes.
- tetrabutylammoniumfluoride (TBAF 1.0 N in THF 13 uL, 3.3 mg, 0.013 mmol) was added to a mixture of 3-methyl-biphenyl-2-carboxaldehyde (0.25g, 1.27 mmol) and trifluoromethytrimethyl silane (0.25 g, 1.53 mmol), in THF (1.5 ml) at 0 0 C.
- the reaction was warmed to room temperature and stirred for 4 hours.
- HCl (3.0 N, 2.0 ml) was added, and the mixture was stirred for 3 hours.
- the mixture was concentrated, dissolved in methylene chloride, filtered through silica gel, and concentrated to give 0.15 g of 2,2,2- trifluoro-l-(3'-methylbiphenyl-2-yl)ethanol.
- reaction vessel was sealed and heated to 19O 0 C for 10 minutes with microwave. After cooling, the reaction mixture was evaporated to dryness. The residue was dissolved in 10 ml of THF, to which was added 5N.HC1 (5ml). The mixture was refluxed for 2 hours in order to deprotect the benzophone and tert-butyl groups. The resulting reaction mixture was concentrated and dissolved in methanol (8ml) and purified with Prep-LC to afford 15mg of 2-amino-3-(4(4-amino-6-((R)-l-(naphthalene-2-yl)ethylamino)-l,3,5-trizin-2- yl)phenyl)propanoic acid.
- Emrys process vial (20ml) for microwave was charged with tert-butyl 3-(4- bromo-2-fluorophenyl)-2-(diphenylmethylene-amino)propanoate (600mg, 1.24mmol), Pd(dba)2 (71mg, 0.124mmol), PCy3 (35mg, 0.124mmol), 4,4,4 > ,4 > ,5,5,5',5'-octamethyl-2,2 > - bi(l,3,2-dioxaborolane (346mg, l.leq. 1.36mmol) and KOAc (182mg, 1.5eq., 1.86mmol) 20ml of DMF.
- reaction vessel was sealed and heated to 16O 0 C for 20 minutes by microwave. After cooling, the reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in H 2 O (30ml), extracted with EtOAc (2x40ml), and purified with Prep-LC to give 220mg of tert-butyl 2-(diphenylmethyleneamino)-3-(2-fluoro- 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)propanoate.
- Aqueous sodium carbonate (2 ml, IM) was added to above solution followed by 10 mol percent dichlorobis(triphenylphosphine)-palladium(II).
- the reaction vessel was sealed and heated to 19O 0 C for 10 minutes by microwave. After cooling, the reaction mixture was evaporated to dryness. The residue was dissolved in 10 ml of THF, to which 5N.HC1 (2ml) was then added. The mixture was refluxed for 2 hours (deprotection of benzophone and tert-butyl groups).
- the crude intermediate (250mg, 0.83mmol) was then dissolved in 6.0ml of MeCN and 6ml of H 2 O in a 20ml microwave vial. To this solution were added L-p-borono- phenylalanine (173.6mg, 0.83mmol), sodium carbonate (173.6mg, 1.66mmol) and catalytic amount of dichlorobis(triphenylphosphine)-palladium(II) (11.6mg, 0.0166mmol). The reaction vial was then sealed and stirred in the microwave reactor at 150 0 C for 7 minutes.
- the crude intermediate (150mg, 0.497mmol) was then dissolved in 3.0ml of MeCN and 3ml of H 2 O in a 10 ml microwave vial.
- L-p-borono- phenylalanine (104mg, 0.497mmol)
- sodium carbonate 150mg, 0.994mmol
- catalytic amount of dichlorobis(triphenylphosphine)-palladium(II) (6.9mg, 0.00994mmol).
- the reaction vial was then sealed and stirred in the microwave reactor at 150 0 C for 5 minutes.
- Reaction mixture was cooled, filtered through a syringe filter and then separated by a reverse phase preparative-HPLC using YMC-Pack ODS 100x30 mm ID column (MeOH/H 2 O/TFA solvent system). The pure fractions were concentrated in vacuum. The product was then suspended in 5 ml of water, frozen and lyophilized to give 5 mg of pure product, 2-amino-3-[5-(5-phenyl-thiophen-2-yl)- lH-indol-3-yl]-propionic acid.
- Residue was purified by preparative HPLC using MeOH/H 2 O/TFA as solvent system to obtain (S)-2-amino-3-[4-(2- amino-6-phenylethynyl-pyrimidin-4-yl(-phenyl]-propionic acid as a TFA salt.
- 1 H-NMR 400 MHz, CD 3 OD: ⁇ (ppm) 3.20-3.42 (m, 2H), 4.31 (m, IH), 7.40-7.51 (m, 6H), 7.62 (d, 2H), 8.18 (d, 2H). 6.52.
- Step 1 Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone.
- thionyl chloride 29.2 ml, 400 mmol
- the ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added.
- the mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated.
- reaction mixture was cooled to -78°C (dry ice/acetone bath), and l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2h.
- the reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 h, and further stirred at -32°C for another 24h.
- 3N NaOH 250 ml was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes.
- Step 3 Synthesis of R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro- ethanol.
- R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65g, 54.06 mmol)
- 3- methylpyrazole (5.33 g, 65 mmol)
- K 2 CO 3 (15.7 g, 113.5 mmol)
- (lR,2R)-N,N'-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 130 0 C (oil bath temperature) for 12 h.
- reaction mixture was diluted with ethyl acetate and washed with H 2 O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-I- [4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %).
- H 3 PO 4 40 ml, 85 % in water
- water 300 ml
- 50 % NaOH in water 50 % NaOH in water to adjust pH to 6.15.
- the temperature was raised to 58°C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3.
- H 3 PO 4 40 ml, 85 % in water
- water 300 ml
- 50 % NaOH in water 50 % NaOH in water to adjust the pH to 6.15.
- the temperature was raised to 58°C, and the aqueous Li-salt of the compound was added dropwise into the buffer with simultaneous addition of 3N HCl so that the pH was maintained at 6.1 to 6.2.
- tetra-n-butyl ammonium fluoride (0.05 eq.) was added to a mixture of substituted benzaldehyde (1 eq.) and trifluoromethyl trimethylsilane (1.2 eq.) in THF at 0 0 C. The temperature was then allowed to warm to room temperature. The mixture was stirred at room temperature for 5h, then diluted with ethyl acetate, washed with water, brine and dried by MgSO 4 . The solvent was removed under reduced pressure to give the trifluoro- alcohol as crude product, which was used in next step without further purification.
- the above crude product (1 eq.) was added to a 5 ml microwave vial containing 4- borono-L-phenylalanine (1 eq.), Na 2 CO 3 (2 eq.), acetonitrile (2 ml), water (2 ml) and dichlorobis(triphenylphosphine)-palladium (0.05 eq.).
- the vial was capped, and the mixture was heated at 150 0 C for 5 min under microwave radiation.
- the mixture was cooled, filtered through a syringe filter, and then separated by a reverse phase preparative-HPLC using YMC-Pack ODS 100x30 mm ID column (MeOH/H 2 O/TFA solvent system). The pure fractions were combined and concentrated in vacuum.
- the product was then suspended in 5 ml of water, frozen and lyophilized to give the product as a trifluoro acetic acid (TFA) salt.
- TFA trifluoro acetic acid
- the monochloride (0.460 g, 1.104 mmol) made above was added to a 20 ml microwave vial, which contained 4-borono-L-phenylalanine (0.277 g, 1.325 mmol), Na 2 CO 3 (0.234 g, 2.208 mmol), acetonitrile (8 ml) / water (8 ml) and dichlorobis(triphenylphosphine)- palladium (0.039 g, 0.055 mmol).
- the vial was capped and the mixture stirred at 150 0 C for 10 minutes under microwave radiation.
- Emrys process vial (20 ml) for microwave was charged with l-(4-(2-amino-6- chloro-pyrimidin-4-yloxy)-2,2,2-trifluoro-ethyl)-phenyl)-pyrrolidine-2-one (200 mg, 0.51 mmol), 4-borono-L-phenylalanine (108 mg, 0.51 mmol) and 5 ml of acetonitrile. 5 ml of aqueous sodium carbonate (IM) was added to above solution followed by 5 mol % of dichlorobis(triphenylphosphine)-palladium (II). The reaction vessel was sealed and heated to 16O 0 C for 7min with microwave irradiation.
- IM aqueous sodium carbonate
- R-l-(2-Bromo-5-fluoro-phenyl)-2,2,2-trifluoro-ethanol (4.Og, 14.65 mmol), 3-methyl pyrazole (1.56 g, 19.04 mmol), CuI (0.557g, 2.93 mmol), K 2 CO 3 (4.25 g, 30.76 mmol), (lR,2R)-N,N'-dimethyl-cyclohexane-l,2-diamine (0.416 g, 2.93 mmol) and toluene (15 ml) were taken in 50 ml of sealed tube and the resulting mixture was heated at 130 0 C (oil bath temperature) for 2 days.
- the reaction vessel was sealed, and the mixture was heated at 16O 0 C for 10 minutes with microwave irradiation. After cooling, the reaction mixture was evaporated to dryness. The residue was dissolved in H 2 O (10 ml) and extracted with ether. The ethereal layer was discarded. Then most of the water in the aqueous phase was removed in vacuo followed by addition of 10 ml of methanol. The crude product was purified with Prep-HPLC to give 1.163 g (yield 75%) of product.
- Tetrabutylammonium fluoride (0.1 ml of IM in THF) was added to a solution of 4-(6-methoxy-pyridine-2-yl)-benzaldehyde (213 mg, 1 mmol) and trifluoromethyl trimethylsilane (0.2 ml, 1.2 mmol) in 10 ml THF at 0 0 C. The mixture was warmed up to room temperature and stirred for 4 hours. The reaction mixture was then treated with 12 ml of IM HCl and stirred overnight. The product was extracted with ethyl acetate (3x20ml). The organic layer was separated and dried over sodium sulfate. The organic solvent was evaporated to give 0.25g of l-[4-(6-methoxy-pyridine-2-yl)-phenyl]-2,2,2-trifluoro-ethanol which was directly used in next step without purification, yield: 90%.
- TBAF Tetrabutylammonium fluoride
- Tetrabutylammonium fluoride (TBAF, 0.1 ml of IM in THF) was added to a solution of 2-pyrimidin-5-yl-benzaldehyde (184 mg, 1 mmol) and trifluoromethyl trimethylsilane (TMSCF 3 , 0.2 ml, 1.2 mmol) in 10 ml THF at 0 0 C.
- TMSCF 3 trifluoromethyl trimethylsilane
- the product was extracted with ethyl acetate (3x20ml).
- the organic layer was separated and dried over sodium sulfate.
- the organic solvent was evaporated to give 0.21 g of 2,2,2-trifluoro-l-(2-pyrimidin-5-yl-phenyl)-ethanol (yield: 84%), which was directly used in next step without purification.
- Human TPHl, TPH2, tyrosine hydroxylase (TH) and phenylalanine hydroxylase (PH) were all generated using genes having the following accession numbers, respectively: X52836, AY098914, X05290, and U49897.
- the full-length coding sequence of human TPHl was cloned into the bacterial expression vector pET24 (Novagen, Madison, WI, USA).
- a single colony of BL21(DE3) cells harboring the expression vector was inoculated into 50 ml of L broth (LB)- kanamycin media and grown up at 37°C overnight with shaking.
- Expression of TPHl was induced with 15% D-lactose over a period of 10 hours at 25°C. The cells were spun down and washed once with phosphate buffered saline (PBS). TPHl was purified by affinity chromatography based on its binding to pterin.
- PBS phosphate buffered saline
- the cell pellet was resuspended in a lysis buffer (100 ml/20 g) containing 50 mM Tris-Cl, pH 7.6, 0.5 M NaCl, 0.1% Tween-20, 2 mM EDTA, 5 mM DTT, protease inhibitor mixture (Roche Applied Science, Indianapolis, IN, USA) and 1 mM phenylmethanesulfonyl fluoride (PMSF), and the cells were lyzed with a microfluidizer.
- a lysis buffer 100 ml/20 g
- PMSF phenylmethanesulfonyl fluoride
- the lysate was centrifuged and the supernatant was loaded onto a pterin-coupled sepharose 4B column that was equilibrated with a buffer containing 50 mM Tris, pH 8.0, 2 M NaCl, 0.1% Tween-20, 0.5 mM EDTA, and 2 mM DTT.
- the column was washed with 50 ml of this buffer and TPHl was eluded with a buffer containing 30 mM NaHCO 3 , pH 10.5, 0.5 M NaCl, 0.1% Tween-20, 0.5 mM EDTA, 2 mM DTT, and 10% glycerol.
- Eluted enzyme was immediately neutralized with 200 mM KH 2 PO 4 , pH 7.0, 0.5 M NaCl, 20 mM DTT, 0.5mM EDTA, and 10% glycerol, and stored at -80 0 C.
- Human tryptophan hydroxylase type II (TPH2), tyrosine hydroxylase (TH) and phenylalanine hydroxylase (PAH) were expressed and purified essentially in the same way, except the cells were supplemented with tyrosine for TH and phenylalanine for PAH during growth.
- TPHl and TPH2 activities were measured in a reaction mixture containing 50 mM 4- morpholinepropanesulfonic acid (MOPS), pH 7.0, 60 ⁇ M tryptophan, 100 mM ammonium sulfate, 100 ⁇ M ferrous ammonium sulfate, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), 0.3 mM 6-methyl tetrahydropterin, 0.05 mg/ml catalase, and 0.9 mM DTT.
- v is the initial velocity at a given compound concentration C
- b is the background signal
- D is the Hill slope which is approximately equal to 1
- I c so is the concentration of the compound that inhibits half of the maximum enzyme activity.
- Human TH and PAH activities were determined by measuring the amount of 3 H 2 O generated using L- [3, 4- H] -tyrosine and L- [4- H] -phenylalanine, respectively.
- the enzyme 100 nM was first incubated with its substrate at 0.1 mM for about 10 minutes, and added to a reaction mixture containing 50 mM MOPS, pH 7.2, 100 mM ammonium sulfate, 0.05% Tween-20, 1.5 mM TCEP, 100 ⁇ M ferrous ammonium sulfate, 0.1 mM tyrosine or phenylalanine, 0.2 mM 6-methyl tetrahydropterin, 0.05 mg/ml of catalase, and 2 mM DTT.
- RBL2H3 is a rat mastocytoma cell line, which contains TPHl and makes 5-hydroxytrypotamine (5HT) spontaneously
- BON is a human carcinoid cell line, which contains TPHl and makes 5-hydroxytryptophan (5HTP).
- the CBAs were performed in 96-well plate format.
- the mobile phase used in HPLC contained 97% of 100 mM sodium acetate, pH 3.5 and 3% acetonitrile.
- a Waters Cl 8 column (4.6 x 50 mm) was used with Waters HPLC (model 2795).
- a multi-channel fluorometer (model 2475) was used to monitor the flow through by setting at 280 nm as the excitation wavelength and 360 nm as the emission wavelength.
- RBL CBA Cells were grown in complete media (containing 5 % bovine serum) for 3-4 hours to allow cells to attach to plate wells (7K cell/well). Compounds were then added to each well in the concentration range of 0.016 ⁇ M to 11.36 ⁇ M. The controls were cells in complete media without any compound present. Cells were harvested after 3 days of incubation at 37°C. Cells were >95% confluent without compound present. Media were removed from plate and cells were lysed with equal volume of 0.1 N NaOH. A large portion of the cell lysate was treated by mixing with equal volume of IM TCA and then filtered through glass fiber. The filtrates were loaded on reverse phase HPLC for analyzing 5HT concentrations. A small portion of the cell lysate was also taken to measure protein concentration of the cells that reflects the cytotoxicity of the compounds at the concentration used. The protein concentration was measured by using BCA method.
- the average of 5HT level in cells without compound treated was used as the maximum value in the IC50 derivation according to the equation provided above.
- the minimum value of 5HT is either set at 0 or from cells that treated with the highest concentration of compound if a compound is not cytotoxic at that concentration.
- BON CBA Cells were grown in equal volume of DMEM and F12K with 5 % bovine serum for 3-4 hours (2OK cell/well) and compound was added at a concentration range of 0.07 ⁇ M to 50 ⁇ M. The cells were incubated at 37°C overnight. Fifty ⁇ M of the culture supernatant was then taken for 5HTP measurement. The supernatant was mixed with equal volume of IM TCA, then filtered through glass fiber. The filtrate was loaded on reverse phase HPLC for 5HTP concentration measurement. The cell viability was measured by treating the remaining cells with Promega Celltiter-Glo Luminescent Cell Viability Assay. The compound potency was then calculated in the same way as in the RBL CBA.
- GI gastrointestinal
- the effect of a potent TPHl inhibitor of the invention on gastrointestinal (GI) transit time and gastric emptying was determined in Sprague-Dawley rats.
- the compound was administred at doses of 50, 125 and 250 mpk, po, qd, for 14 days.
- Each dosing group utilized nine rats.
- Nine rats were also used as a negative control group (vehicle administration only), and another six were used as a positive control (Atropine).
- the rats were dosed compound or vehicle at 10 ml/kg.
- Atropine was given to the positive control group on day 14 only, whereas vehicle was given on days 1-14.
- Body weights and observations were taken through out study, and the rats were fasted overnight on day 13 prior to the charcoal meal.
- the potent TPHl inhibitor, Atropine or vehicle wereorally dosed 30 minutes prior to the charcoal meal.
- the charcoal meal (5% charcoal in vehicle) was orally dosed at 15 ml/kg.
- Necropsy was performed 25 minutes after the charcoal meal dose.
- GI transit times were determined by measuring the distance the charcoal meal traveled down the small intestine, and dividing that distance by the total length of the small intestine. Gastric emptying times were determined by weighing the stomachs of the rats.
- Brain 5 -HT levels were unaffected by the compound.
Landscapes
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description








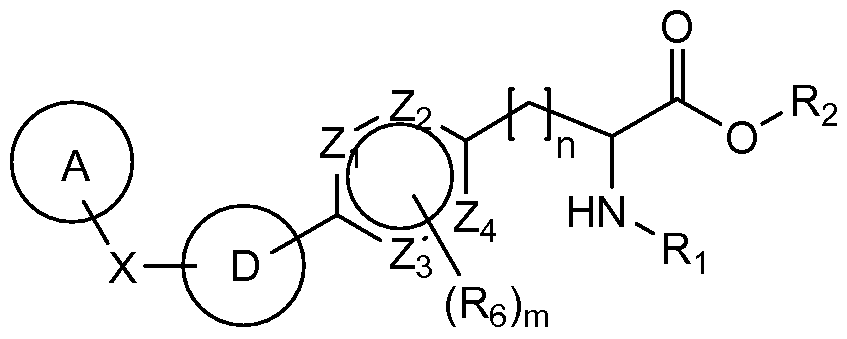


















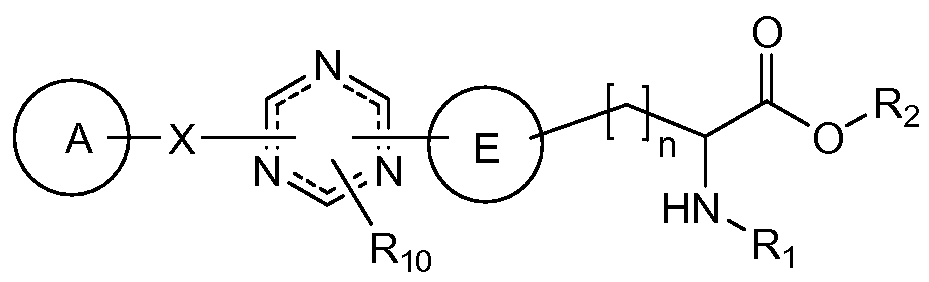















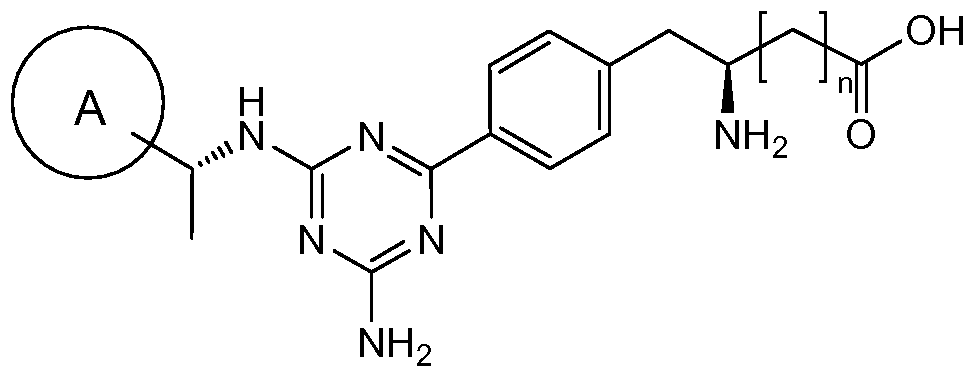


























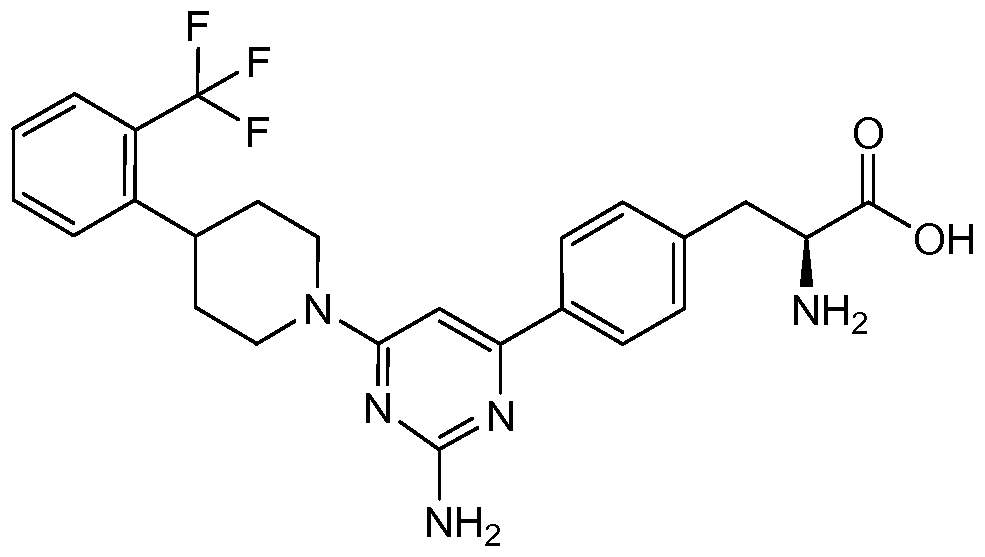


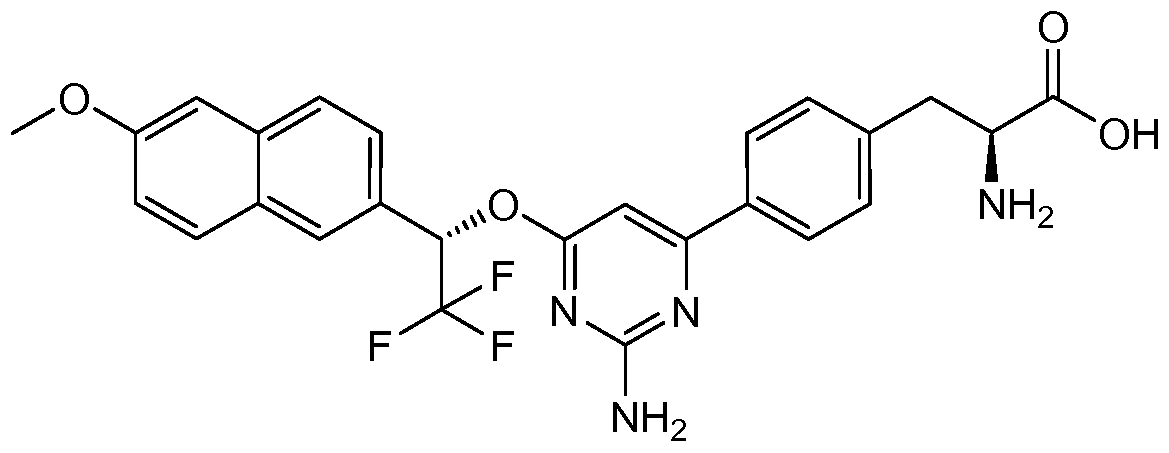





















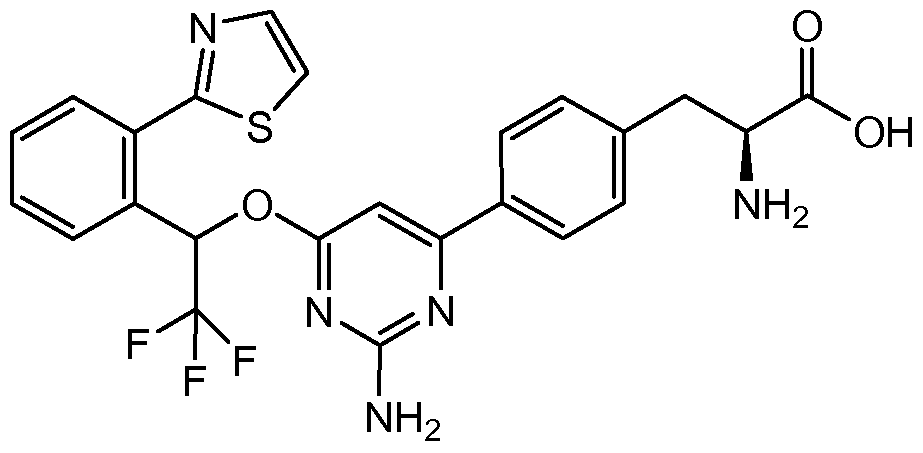













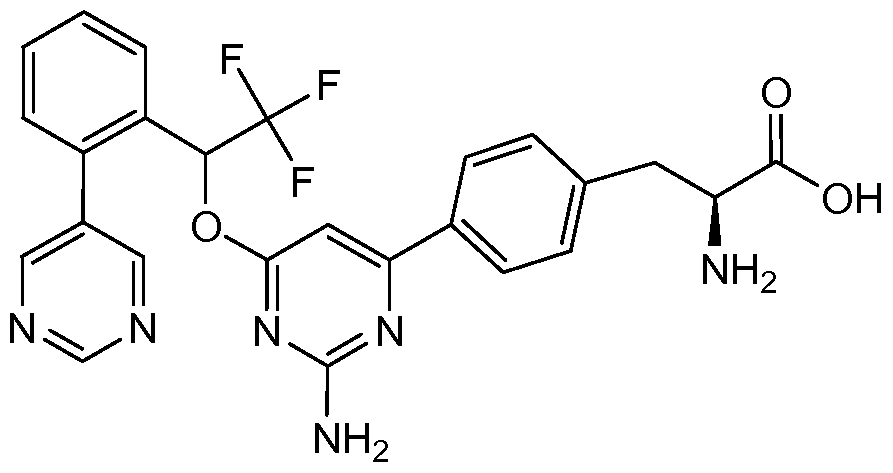













Claims




















Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2008279426A AU2008279426A1 (en) | 2007-07-26 | 2008-07-17 | Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein |
| EP08826540A EP2178536A1 (en) | 2007-07-26 | 2008-07-17 | Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein |
| CA2694443A CA2694443A1 (en) | 2007-07-26 | 2008-07-17 | Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein |
| BRPI0813835A BRPI0813835A2 (en) | 2007-07-26 | 2008-07-17 | methods of affecting gastrointestinal transit and gastric emptying, and useful compounds therein |
| JP2010518297A JP2010534662A (en) | 2007-07-26 | 2008-07-17 | Methods acting on gastrointestinal transit and gastric emptying, and compounds useful therefor |
| CN200880100490A CN101801385A (en) | 2007-07-26 | 2008-07-17 | Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95207107P | 2007-07-26 | 2007-07-26 | |
| US60/952,071 | 2007-07-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009014972A1 true WO2009014972A1 (en) | 2009-01-29 |
Family
ID=39745649
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/070254 WO2009014972A1 (en) | 2007-07-26 | 2008-07-17 | Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US20090029993A1 (en) |
| EP (1) | EP2178536A1 (en) |
| JP (1) | JP2010534662A (en) |
| KR (1) | KR20100055436A (en) |
| CN (1) | CN101801385A (en) |
| AU (1) | AU2008279426A1 (en) |
| BR (1) | BRPI0813835A2 (en) |
| CA (1) | CA2694443A1 (en) |
| RU (1) | RU2010107066A (en) |
| WO (1) | WO2009014972A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011100285A1 (en) * | 2010-02-10 | 2011-08-18 | Lexicon Pharmaceuticals, Inc. | Tryptophan hydroxylase inhibitors for the treatment of metastatic bone disease |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013148978A1 (en) * | 2012-03-30 | 2013-10-03 | Lexicon Pharmaceuticals, Inc. | Methods and compositions for the treatment of necrotizing enterocolitis |
| US8716281B2 (en) | 2010-05-11 | 2014-05-06 | Amgen Inc. | Pyrimidine compounds that inhibit anaplastic lymphoma kinase |
| US8759364B2 (en) | 2008-03-31 | 2014-06-24 | The Trustees Of Columbia University In The City Of New York | Methods of treating bone mass diseases |
| US8815883B2 (en) | 2009-11-02 | 2014-08-26 | The Trustees Of Columbia Unviersity In The City Of New York | Compounds and methods for inhibiting serotonin synthesis |
| US9199994B2 (en) | 2013-09-06 | 2015-12-01 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9611201B2 (en) | 2015-03-05 | 2017-04-04 | Karos Pharmaceuticals, Inc. | Processes for preparing (R)-1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethanol and 1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethanone |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0813867A2 (en) * | 2007-06-26 | 2015-01-06 | Lexicon Pharmaceuticals Inc | COMPOSITIONS UNDERSTANDING TRYPTOPHAN HYDROLASE INHIBITORS. |
| WO2009029499A1 (en) * | 2007-08-24 | 2009-03-05 | Lexicon Pharmaceuticals, Inc. | Methods of preparing 4-phenyl-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine-based compounds |
| TW200932729A (en) * | 2007-10-08 | 2009-08-01 | Lexicon Pharmaceuticals Inc | Solid forms of (S)-2-amino-3-(4-(2-amino-6-((R)-2,2,2-trifluoro-1-(3'-methoxybiphenyl-4-yl)ethoxy)pyrimidin-4-yl)phenyl)propanoic acid and methods of their use |
| US20110124667A1 (en) * | 2009-11-23 | 2011-05-26 | Philip Manton Brown | Methods for the treatment of irritable bowel syndrome |
| JP2013530180A (en) | 2010-06-16 | 2013-07-25 | パーデュー、ファーマ、リミテッド、パートナーシップ | Aryl substituted indoles and uses thereof |
| WO2018060949A1 (en) | 2016-09-30 | 2018-04-05 | Roivant Sciences Gmbh | Tryptophan hydroxylase inhibitors for use in the treatment of liver diseases |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007089335A2 (en) * | 2005-12-29 | 2007-08-09 | Lexicon Pharmaceutical Inc. | Multicyclic amino acid derivatives and methods of their use |
| WO2008073933A2 (en) * | 2006-12-12 | 2008-06-19 | Lexicon Pharmaceuticals, Inc. | 4-phenyl-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine-based compounds and methods of their use |
-
2008
- 2008-07-17 WO PCT/US2008/070254 patent/WO2009014972A1/en active Application Filing
- 2008-07-17 BR BRPI0813835A patent/BRPI0813835A2/en not_active IP Right Cessation
- 2008-07-17 CA CA2694443A patent/CA2694443A1/en not_active Abandoned
- 2008-07-17 RU RU2010107066/15A patent/RU2010107066A/en unknown
- 2008-07-17 US US12/174,741 patent/US20090029993A1/en not_active Abandoned
- 2008-07-17 JP JP2010518297A patent/JP2010534662A/en not_active Withdrawn
- 2008-07-17 AU AU2008279426A patent/AU2008279426A1/en not_active Abandoned
- 2008-07-17 EP EP08826540A patent/EP2178536A1/en not_active Withdrawn
- 2008-07-17 CN CN200880100490A patent/CN101801385A/en active Pending
- 2008-07-17 KR KR1020107004150A patent/KR20100055436A/en not_active Application Discontinuation
-
2010
- 2010-04-05 US US12/754,284 patent/US20100317664A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007089335A2 (en) * | 2005-12-29 | 2007-08-09 | Lexicon Pharmaceutical Inc. | Multicyclic amino acid derivatives and methods of their use |
| WO2008073933A2 (en) * | 2006-12-12 | 2008-06-19 | Lexicon Pharmaceuticals, Inc. | 4-phenyl-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine-based compounds and methods of their use |
Non-Patent Citations (3)
| Title |
|---|
| GERSHON M D: "REVIEW ARTICLE: SEROTONIN RECEPTORS AND TRANSPORTERS - ROLES IN NORMAL AND ABNORMAL GASTROINTESTINAL MOTILITY", ALIMENTARY PHARMACOLOGY & THERAPEUTICS, BLACKWELL SCIENTIFIC PUBLICATIONS LTD., CAMBRIDGE, GB, vol. 20, no. 7, 1 January 2004 (2004-01-01), pages 3 - 14, XP009075257, ISSN: 0269-2813 * |
| SAKOWSKI ET AL: "Differential tissue distribution of tryptophan hydroxylase isoforms 1 and 2 as revealed with monospecific antibodies", BRAIN RESEARCH, ELSEVIER, AMSTERDAM, NL, vol. 1085, no. 1, 26 April 2006 (2006-04-26), pages 11 - 18, XP005436826, ISSN: 0006-8993 * |
| SHI Z -C ET AL: "Modulation of peripheral serotonin levels by novel tryptophan hydroxylase inhibitors for the potential treatment of functional gastrointestinal disorders", JOURNAL OF MEDICINAL CHEMISTRY 20080710 US, vol. 51, no. 13, 10 July 2008 (2008-07-10), pages 3684 - 3687, XP002497512, ISSN: 0022-2623 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8759364B2 (en) | 2008-03-31 | 2014-06-24 | The Trustees Of Columbia University In The City Of New York | Methods of treating bone mass diseases |
| US8815883B2 (en) | 2009-11-02 | 2014-08-26 | The Trustees Of Columbia Unviersity In The City Of New York | Compounds and methods for inhibiting serotonin synthesis |
| WO2011100285A1 (en) * | 2010-02-10 | 2011-08-18 | Lexicon Pharmaceuticals, Inc. | Tryptophan hydroxylase inhibitors for the treatment of metastatic bone disease |
| US8716281B2 (en) | 2010-05-11 | 2014-05-06 | Amgen Inc. | Pyrimidine compounds that inhibit anaplastic lymphoma kinase |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013148978A1 (en) * | 2012-03-30 | 2013-10-03 | Lexicon Pharmaceuticals, Inc. | Methods and compositions for the treatment of necrotizing enterocolitis |
| US10660893B2 (en) | 2013-09-06 | 2020-05-26 | ROIVANT SCIENCES, GmbH | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9512122B2 (en) | 2013-09-06 | 2016-12-06 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9750740B2 (en) | 2013-09-06 | 2017-09-05 | Kanos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US10045988B2 (en) | 2013-09-06 | 2018-08-14 | Roivant Sciences Gmbh | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US10350208B2 (en) | 2013-09-06 | 2019-07-16 | Roivant Sciences Gmbh | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9199994B2 (en) | 2013-09-06 | 2015-12-01 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US10946018B2 (en) | 2013-09-06 | 2021-03-16 | Roivant Sciences Gmbh | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US11759462B2 (en) | 2013-09-06 | 2023-09-19 | Altavant Sciences Gmbh | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9611201B2 (en) | 2015-03-05 | 2017-04-04 | Karos Pharmaceuticals, Inc. | Processes for preparing (R)-1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethanol and 1-(5-chloro-[1,1′-biphenyl]-2-yl)-2,2,2-trifluoroethanone |
| US10710948B2 (en) | 2015-03-05 | 2020-07-14 | Roivant Sciences Gmbh | Processes for preparing (R)-1-(5-chloro-[1,1″-biphenyl] -2-yl)-2,2,2-trifluoroethanol and 1-(5-chloro-[1,1″-biphenyl]-2-yl)-2,2,2-trifluoroethanone |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2010107066A (en) | 2011-09-10 |
| US20090029993A1 (en) | 2009-01-29 |
| US20100317664A1 (en) | 2010-12-16 |
| AU2008279426A1 (en) | 2009-01-29 |
| CN101801385A (en) | 2010-08-11 |
| EP2178536A1 (en) | 2010-04-28 |
| BRPI0813835A2 (en) | 2017-06-06 |
| KR20100055436A (en) | 2010-05-26 |
| CA2694443A1 (en) | 2009-01-29 |
| JP2010534662A (en) | 2010-11-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2386547B1 (en) | Multicyclic amino acid derivatives and methods of their use | |
| AU2008275179B2 (en) | Methods and compositions for treating pulmonary hypertension and related diseases and disorders | |
| EP2178536A1 (en) | Methods of affecting gastrointestinal transit and gastric emptying, and compounds useful therein | |
| EP2170306A1 (en) | Methods of treating serotonin-mediated diseases and disorders | |
| AU2008268409B2 (en) | Compositions comprising tryptophan hydroxylase inhibitors | |
| WO2011100285A1 (en) | Tryptophan hydroxylase inhibitors for the treatment of metastatic bone disease | |
| CA2779681A1 (en) | Tryptophan hydroxylase inhibitors for the treatment of cancer | |
| WO2010062829A1 (en) | Tryptophan hydroxylase inhibitors for treating osteoporosis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880100490.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08826540 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 582535 Country of ref document: NZ Ref document number: 203263 Country of ref document: IL Ref document number: 2008279426 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12010500135 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/000847 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010518297 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2694443 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2008279426 Country of ref document: AU Date of ref document: 20080717 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008826540 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20107004150 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010107066 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0813835 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100126 |