WO2009012125A1 - Compounds and methods for modulating fxr - Google Patents
Compounds and methods for modulating fxr Download PDFInfo
- Publication number
- WO2009012125A1 WO2009012125A1 PCT/US2008/069719 US2008069719W WO2009012125A1 WO 2009012125 A1 WO2009012125 A1 WO 2009012125A1 US 2008069719 W US2008069719 W US 2008069719W WO 2009012125 A1 WO2009012125 A1 WO 2009012125A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- isoxazol
- dichloro
- cyclopropyl
- ylmethoxy
- Prior art date
Links
- 0 C[U]1NC(c2c(*)cccc2*)=CC(C*)=C1*=C Chemical compound C[U]1NC(c2c(*)cccc2*)=CC(C*)=C1*=C 0.000 description 5
- CRFSWDBNKHNGGA-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1)CCC1O)=O Chemical compound CC(C)(C)OC(N(CCC1)CCC1O)=O CRFSWDBNKHNGGA-UHFFFAOYSA-N 0.000 description 1
- SSWKGOFXGAQDQI-UHFFFAOYSA-N C[n]1c2cc(N(CCC3)CCC3OCc3c(C4CC4)[o]nc3-c(c(Cl)ccc3)c3Cl)ccc2c(C(OC)=O)c1 Chemical compound C[n]1c2cc(N(CCC3)CCC3OCc3c(C4CC4)[o]nc3-c(c(Cl)ccc3)c3Cl)ccc2c(C(OC)=O)c1 SSWKGOFXGAQDQI-UHFFFAOYSA-N 0.000 description 1
- PNZIYSWKRBYITO-UHFFFAOYSA-N Clc1c(-c2n[o]c(C3CC3)c2CBr)c(Cl)ccc1 Chemical compound Clc1c(-c2n[o]c(C3CC3)c2CBr)c(Cl)ccc1 PNZIYSWKRBYITO-UHFFFAOYSA-N 0.000 description 1
- WQOFPZFDXPHYLX-UHFFFAOYSA-N Clc1c(-c2n[o]c(C3CC3)c2COC2CCNCC2)c(Cl)ccc1 Chemical compound Clc1c(-c2n[o]c(C3CC3)c2COC2CCNCC2)c(Cl)ccc1 WQOFPZFDXPHYLX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the current invention relates to the fields of medicinal chemistry, pharmacology, and medicine. Specifically, the invention relates to novel compounds useful for the treatment dyslipidemia and diseases related to dyslipidemia.
- Dyslipidemia and diseases related to dyslipidemia e.g. atherosclerosis, coronary artery disease, stroke, etc., are major causes of death, morbidity, and economic loss.
- Plasma lipids, especially cholesterol fractions, are recognized as having a significant role in cardiovascular health.
- Favorable modulation of plasma lipids such as triglycerides, HDL cholesterol, and LDL cholesterol is desirable.
- patent application WO 2004/048349 Al discloses compounds purportedly useful as farnesoid X receptor (FXR) agonists.
- PCT international application WO2007/092751 A3 discloses isoxazole compounds useful for modulating FXR.
- PCT International application WO 2007/140174 A2 discloses aryltriazole derivatives as FXR modulators and their preparation, pharmaceutical compositions, and use in the treatment of dyslipidemia and related diseases.
- FXR agonists are ligands for a nuclear receptor that regulates the transcription of genes that control triglyceride, cholesterol, and carbohydrate metabolism.
- Such compounds would be useful for the treatment of disorders characterized by or resulting from an undesirable lipid profile including dyslipidemia and related diseases e.g., atherosclerosis.
- the present invention provides a compound of formula I
- U is O, N or C; provided that when U is O or N, R 3a is absent; and provided that when U is N or C, the UN bond is a double bond; and provided that when W is C, the WN bond is a double bond; W is C or N; X is C or N;
- R 1 is chloro, fluoro, or trifluoromethoxy
- R is hydrogen chloro, fluoro, or trifluoromethoxy
- R 3a is hydrogen, or absent
- R 3b is trifluoromethyl, cyclopropyl or isopropyl;
- Ar 1 is selected from the group consisting of 6- indolyl, 6-benzothienyl, 4-naphthyl, 4-phenyl, and 2-pyridinyl, each optionally substituted with one or two groups independently selected from the group consisting of methyl, ethyl, and phenyl; and
- R 5 is COOH; or a pharmaceutically acceptable salt, enantiomer, diastereomer or mixture thereof.
- the compounds of the present invention are agonists of FXRs.
- the compounds of present invention are useful for beneficially altering lipid profiles including but not limited to lowering total cholesterol levels, lowering LDL cholesterol levels, lowering VLDL cholesterol levels, raising HDL cholesterol levels, and /or lowering triglyceride levels.
- the present invention provides a method for treating FXR mediated conditions such as dyslipidemia and diseases related to dyslipidemia comprising administering a therapeutically effective amount of a compound of the present invention to a patient in need thereof.
- the present invention also provides a method for treating atherosclerosis comprising administering a therapeutically effective amount of a compound of the present invention to a patient in need thereof.
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the present invention and a pharmaceutically acceptable carrier.
- the present invention also relates to compounds of the present invention for use in therapy, in particular for use in treating dyslipidemia and related diseases.
- the present invention also relates to compounds of the present invention for use in treating atherosclerosis.
- the present invention also relates to the use of a compound of the present invention in the manufacture of a medicament for the treatment of dyslipidemia or related diseases.
- the present invention also relates to the use of a compound of the present invention in the manufacture of a medicament for the treatment of atherosclerosis.
- dyslipidemia refers to an abnormality in, or abnormal amounts of lipids and lipoproteins in the blood and the disease states resulting, caused by, exacerbated by, or adjunct to such abnormality (see Dorland's Illustrated Medical Dictionary, 29th edition, W.B Saunders publishing Company, New York, NY).
- Disease states encompassed within the definition of dyslipidemia as used herein include hyperlipidemia, hypertriglyceremia, low plasma HDL, high plasma LDL, high plasmaVLDL, liver cholestasis, and hypercholesterolemia.
- diabetes refers to diseases including but not limited to atherosclerosis, thrombosis, coronary artery disease, stroke, and hypertension.
- Diseases related to dyslipidemia also include metabolic diseases such as obesity, diabetes, insulin resistance, and complications thereof. Complications of diabetes include but are not limited diabetic retinopathy.
- terapéuticaally effective amount means an amount of a compound of the invention that is part of an approved therapeutic regimen, or is determined by a qualified prescriber to be sufficient taken as directed, for treating a condition herein described.
- Compounds of the invention may possess one or more chiral centers, and thus, may exist in optically active forms. All such optically active forms or stereoisomers are -A-
- R 1 is chloro or trifluoromethoxy.
- R 2 is chloro or H.
- R 3b is cyclopropyl or isopropyl.
- a preferred Ar 1 group is an optionally substituted 4-phenyl, 2-pyridinyl, 6- benzothienyl or 6-indolyl. More preferred Ar 1 is an optionally substituted 4-phenyl, 6- indolyl, or 6-benzothienyl. Most preferred Ar 1 is optionally substituted 4-phenyl or 6- indolyl. Preferably Ar 1 is optionally substituted with a group selected from the group consisting of methyl, ethyl and phenyl. A more preferred optional substituent is methyl.
- R 1 is chloro or trifluoromethoxy
- R 2 is hydrogen or chloro
- R 3a is hydrogen or absent
- R 3b is cyclopropyl or isopropyl
- Ar 1 group is 4-phenyl, 2-pyridinyl, 6-indolyl, or 6-benzothienyl each optionally substituted with a group selected from methyl or trifluoromethyl or phenyl.
- a compound of formula I wherein q is 1 ; R 1 is chloro or trifluoromethoxy; R 2 is hydrogen or chloro; R 3b is cyclopropyl and Ar 1 group is 4-phenyl, 2-pyridinyl, or 6-indolyl, each optionally substituted with methyl. Also preferred is a compound of formula I wherein q is 2; R 1 is chloro or trifluoromethoxy; R 2 is hydrogen or chloro; R 3b is cyclopropyl; X is N and Ar 1 group is A- phenyl, 2-pyridinyl, or 6-indolyl, each optionally substituted with methyl.
- U is oxygen, and W is carbon forming an isoxazole ring;
- R 1 is chloro or trifluoromethoxy;
- R 2 is hydrogen or chloro;
- R 3a is absent and
- R 3b is cyclopropyl and
- Ar 1 group is 4-phenyl, 2-pyridinyl, 6-indolyl or 6- benzothienyl each optionally substituted with methyl.
- a compound of formula I wherein: U and W are both nitrogen forming a triazole ring; R 1 is chloro or trifluoromethoxy; R 2 is hydrogen or chloro; R 3a is absent and R 3b is cyclopropyl or isopropyl and Ar 1 group is 4-phenyl, 6-indolyl or 6- benzothienyl, each optionally substituted with methyl or phenyl.
- U is carbon, W is nitrogen forming a pyrazole ring;
- R 1 is chloro or trifluoromethoxy;
- R 2 is hydrogen or chloro;
- R 3a is hydrogen and R 3b is cyclopropyl, or isopropyl and
- Ar 1 group is 4-phenyl, 6-indolyl or 6- benzothienyl, each optionally substituted with methyl or phenyl.
- a compound of formula I wherein q is 1 ; U is oxygen, and W is carbon forming an isoxazole ring; R 1 is chloro or trifluoromethoxy; R 2 is hydrogen or chloro; R 3a is absent and R 3b is cyclopropyl; X is C and Ar 1 group is 4-phenyl, 2- pyridinyl, 6-indolyl or 6-benzothienyl each optionally substituted with methyl.
- a compound of formula I wherein: q is 1 ; U and W are both nitrogen forming a triazole ring; R 1 is chloro or trifluoromethoxy; R 2 is hydrogen or chloro; R 3a is absent and R 3b is cyclopropyl or isopropyl; X is C and Ar 1 group is A- phenyl, 6-indolyl or 6-benzothienyl, each optionally substituted with methyl or phenyl.
- a compound of formula I wherein q is 1 ; U is carbon, W is nitrogen forming a pyrazole ring; R 1 is chloro or trifluoromethoxy; R 2 is hydrogen or chloro; R 3a is hydrogen and R 3b is cyclopropyl, or isopropyl; X is C and Ar 1 group is A- phenyl, 6-indolyl or 6-benzothienyl, each optionally substituted with methyl or phenyl.
- U is oxygen, and W is carbon forming an isoxazole ring;
- R 1 is chloro or trifluoromethoxy;
- R 2 is hydrogen or chloro;
- R 3a is absent and
- R 3b is cyclopropyl;
- X is N and
- Ar 1 group is 4-phenyl, 2-pyridinyl, 6-indolyl or 6-benzothienyl each optionally substituted with methyl.
- U and W are both nitrogen forming a triazole ring;
- R 1 is chloro or trifluoromethoxy;
- R 2 is hydrogen or chloro;
- R 3a is hydrogen and
- R 3b is cyclopropyl or isopropyl;
- X is N and
- Ar 1 group is 4-phenyl, 6-indolyl or 6-benzothienyl, each optionally substituted with methyl or phenyl.
- U is carbon, W is nitrogen forming a pyrazole ring;
- R 1 is chloro or trifluoromethoxy;
- R 2 is hydrogen or chloro;
- R 3a is hydrogen and
- R 3b is cyclopropyl, or isopropyl;
- X is N and
- Ar 1 group is 4-phenyl, 6- indolyl or 6-benzothienyl, each optionally substituted with methyl or phenyl.
- the present invention also provides a compound of formula Ia
- U is O, N or C; provided that when U is O or N, R 3a is absent; and provided that when U is N or C, the UN bond is a double bond; provided that U and W are not simultaneously
- WN are not simultaneously double bonds
- W is C or N
- X is C or N
- R 1 is chloro, fluoro, or trifluoromethoxy
- R 2 is hydrogen chloro, fluoro, or trifluoromethoxy
- R 3a is hydrogen, or absent
- R 3b is trifluoromethyl, cyclopropyl or isopropyl
- Ar 1 is selected from the group consisting of indolyl, benzothienyl, naphthyl, phenyl, benzoisothiazolyl, indazolyl, and pyridinyl, each optionally substituted with a group selected from the group consisting of methyl, ethyl, and phenyl; and
- R 5 is -COOH; or a pharmaceutically acceptable salt, or enantiomer thereof.
- U is C and W is N forming a pyrazole ring:
- the present invention further provides a compound of formula II
- R 1 is chloro, fluoro, or trifluoromethoxy
- R is hydrogen chloro, fluoro, or trifluoromethoxy
- R 3b is trifluoromethyl, cyclopropyl or isopropyl
- X is C or N, provided that when X is C, q is 1 ;
- Ar 1 is selected from the group consisting of indolyl, benzothienyl, naphthyl, phenyl, benzoisothiazolyl, indazolyl, and pyridinyl, each optionally substituted methyl or phenyl; or a pharmaceutically acceptable salt or enantiomer thereof.
- a preferred compound of the invention is a compound of formula I, Ia or II wherein R 1 is chloro or trifluoromethoxy and R 2 is hydrogen or chloro.
- R 1 and R 2 are both Chloro or R 1 is trifluoromethoxy and R 2 is hydrogen.
- a preferred compound of the invention is a compound of formula I, Ia or II wherein R 3b is cyclopropyl or isopropyl. Preferably R 3b is cyclopropyl.
- a preferred compound of the invention is a compound of formula Ia or II wherein Ar 1 is 6-benzoisothiazolyl, 5 -benzothienyl, 6-benzothienyl, 6-indazolyl, 5 -indolyl or 6- indolyl, 4-phenyl and 2-pyridinyl, each optionally substituted with methyl or phenyl.
- Ar 1 is 6-benzoisothiazolyl, 5 -benzothienyl, 6-benzothienyl, 6-indazolyl, 5- indolyl, 6-indolyl, or 4-phenyl, each optionally substituted with methyl.
- Ar 1 group is 5 -benzothienyl, 6-benzothienyl, 5 -indolyl, 6-indolyl or 4-phenyl, each optionally substituted with methyl.
- a preferred compound of the invention is a compound of formula I, Ia or II wherein q is 1 and X is N.
- a preferred compound of the invention is a compound of formula I, Ia or II wherein q is 1 and X is C.
- a preferred compound of the invention is a compound of formula I, Ia or II wherein q is 2 and X is N.
- the compounds of the invention may be prepared by the combination of a variety of stepwise procedures known in the art including those described below.
- the products of each step in the schemes below can be recovered by conventional methods including extraction, evaporation, precipitation, chromatography, filtration, trituration, crystallization, and the like.
- substituents unless otherwise indicated, are as previously defined and suitable reagents are well known and appreciated in the art.
- Scheme 1 depicts the reaction of an appropriate compound of formula (1) with an appropriate compound of formula (2) to give a compound of formula I or II where X is N.
- an appropriate compound of formula (1) in which R 1 , R 2 , R 3a , R 3b ,q, and R 5 are defined for formula I and Y is a leaving group and an appropriate compound of formula (2) is one in which R 5 , and Ar 1 are as defined in formula (I) or a group which gives rise to R 5 as defined in formula (I), for example, by formation of an ester, amide, sulfonamide, or acid are reacted to form the compound of formula (I) with appropriate protections and/or deprotections or other processing steps known to one of skill in the art or disclosed herein.
- Suitable leaving groups are well-known in the art and include halides, particularly chloro, bromo, and iodo; and sulfonate esters, such as brosyl, tosyl, methanesulfonyl, and trifluromethane sulfonyl.
- a compound of formula (1) is reacted with a compound of formula (2) in a suitable solvent, such as acetonitrile, dimethylformamide, dimethylsulfoxide, tetrahydrofuran, pyridine, methylethyl ketone and the like.
- a suitable solvent such as acetonitrile, dimethylformamide, dimethylsulfoxide, tetrahydrofuran, pyridine, methylethyl ketone and the like.
- a suitable base such as sodium hydride, potassium carbonate, potassium t- butoxide, sodium carbonate, cesium carbonate, sodium bicarbonate, triethylamine, diisopropyethylamine is usually used in the reaction.
- Such reactions generally are carried out at temperatures from about room temperature to about the reflux temperature of the chosen solvent and typically use from about 1 to 2 equivalents of the compound of formula (2).
- R 5 is an ester
- suitable solvents such as THF, acetonitrile, methanol, ethanol, water mixtures thereof at temperatures from about 25-100 0 C with suitable bases including for example, NaOH, LiOH, and KOH.
- a microwave apparatus may be used as an energy/heat source, especially when the ester is sterically hindered.
- a laboratory microwave utilizing the lowest power setting at about 125 0 C for about 20 minutes in solvent mixtures described above is useful.
- R 5 is a t-butyl ester the corresponding acid can be formed under acidic conditions well known to those skilled in the art.
- a pharmaceutically acceptable salt of a compound of formula (I) or (II) is formed.
- the formation of pharmaceutically acceptable salts is well known and appreciated in the art. See, e.g., P. Stahl, et ah, Handbook of Pharmaceutical Salts: Properties, Selection and Use, (VCHA/Wiley-VCH, 2002; S.M. Berge, et ah, "Pharmaceutical Salts, "Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977.
- compounds of formula (1) and (2) can be readily prepared by methods that are well-known and established in the art including methods and procedures similar to those described herein.
- the isoxazole esters may be reduced to the alcohol compounds of formula (1) with well known methods (e.g. DIBAL-H, LAH) and subsequently converted to a leaving group such as for example, a halide leaving group.
- a suitable base such as triethylamine or sodium methoxide
- Compounds of formula (2) are prepared by carbon-nitrogen bond formation/coupling reactions. For example, reaction of an appropriate piperidine derivative with a fluorobenzene under nucleophilic aromatic substitution conditions in suitable solvents such as acetonitrile using a base such as potassium carbonate at temperatures ranging from room temperature to the reflux temperature of the solvent affords a compound of formula 2 wherein X is N; q is 1 ; and Ar 1 is phenyl.
- appropriate piperidines can be reacted with aryl halides under palladium mediated coupling conditions in solvents such as dioxane at temperatures ranging from room temperature to 120 0 C to yield compounds of formula (2).
- appropriate piperidines can be reacted with aryl halides under copper mediated coupling conditions in solvents such as dimethylsulfoxide at temperatures ranging from 85 0 C to 120 0 C to yield compounds of formula (2).
- the steps required to prepare a compound of formula (I) can be carried out in any order including, for example, reaction of a partial compound of formula (2) with a compound of formula (1), such that the later carried out carbon-nitrogen bond formation/coupling reaction provide a compound of formula I.
- a compound of formula (3) can be reacted with a compound of formula (1) as described above to afford compounds of formula (4) which, following deprotection, can be converted to compounds of formula (I) via carbon-nitrogen bond forming reactions with compounds of formula (5) (Scheme T).
- Compounds of formula (5) may be purchased or prepared from purchased intermediates.
- the 6-bromoindole-3- carboxylic acid may be purchased and converted to the methyl or t-butyl ester derivative.
- the ⁇ -bromoindole-S-carboxylic acid, methyl ester derivative may be employed directly in the coupling reaction with the amine as described herein.
- the 6-bromoindole-3- carboxylic acid, t-butyl ester derivative may be used in the C-N coupling reaction described herein or preferably, it may be converted to the iodo derivative using for example, NaI and CuI in the presence of a suitable base and solvent using procedures known to one of skill in the art.
- the methyl ester derivative of 6- bromoindole-3-carboxylic acid may be used as the bromo.
- a person skilled in the art will recognize that different protecting groups and or different leaving groups may be better suited to particular Ar 1 substrates.
- carbon- nitrogen bond forming reactions including, for example, using copper (I) iodide and a suitable organic base, using a palladium catalyst such as Pd 2 (dba) 3 , in the presence of ligands such as S-Phos, X-Phos, or BINAP, using an inorganic base such as cesium carbonate, and a suitable solvent such as xylene to generate compounds of formula (I).
- a palladium catalyst such as Pd 2 (dba) 3
- ligands such as S-Phos, X-Phos, or BINAP
- an inorganic base such as cesium carbonate
- a suitable solvent such as xylene
- an appropriate compound of formula (1) in which R 1 , R 2 , R 3a , R 3 ,q, and R 5 are defined for formula (I) and Y is a leaving group and an appropriate compound of formula (6) is one in which R 5 , X is CH and Ar 1 are as defined in formula (I) or a group which gives rise to R 5 as defined in formula (I), for example, by formation of an ester or acid are reacted to form the compound of formula (I) with appropriate protections and/or deprotections or other processing steps known to one of skill in the art or disclosed herein.
- Suitable leaving groups are well-known in the art and include halides, particularly chloro, bromo, and iodo; and sulfonate esters, such as brosyl, tosyl, methanesulfonyl, and trifluromethanesulfonyl.
- a compound of formula (1) is reacted with a compound of formula (6) in a suitable solvent, such as acetonitrile, dimethylformamide, dimethylsulfoxide, tetrahydrofuran, pyridine, methylethyl ketone and the like.
- a suitable solvent such as acetonitrile, dimethylformamide, dimethylsulfoxide, tetrahydrofuran, pyridine, methylethyl ketone and the like.
- a suitable base is usually used in the reaction, including sodium hydride, potassium carbonate, potassium t-butoxide, sodium carbonate, cesium carbonate, sodium bicarbonate, triethylamine, diisopropyethylamine.
- Such reactions generally are carried out at temperatures of about room temperature to about the reflux temperature of the chosen solvent and typically use from about 1 to 2 equivalents of the compound of formula (1).
- compounds of formula (7) are converted to compounds of formula
- penultimate compounds of formula (I) wherein R 5 is an ester can be converted to compounds of formula (I) wherein R 5 is an acid or compounds of formula
- a pharmaceutically acceptable salt of a compound of formula (I) is formed.
- the formation of salts is well known and appreciated in the art.
- compounds of formula (6) can be prepared by methods that are well-known and established in the art including methods and procedures similar to those described herein.
- Compounds of formula (6) are prepared by carbon- carbon bond formation/coupling reactions.
- an appropriately substituted cyclohexanone alcohol can be converted to the vinyl triflate under well known conditions and subsequently converted to a vinylboronate.
- the boronate can then be reacted with arylbromides for example under Suzuki conditions to afford the requisite arylcyclohexenes that can be reduced with hydrogen and catalytic palladium on carbon to afford compounds of formula (6).
- Certain compounds of the invention exist as solid amorphous or crystalline forms.
- a compound of the invention may also exist in multiple crystalline forms wherein one or more of the crystalline forms are preferred over others on account of having more desirable properties such as, for example, improved solubility, improved bioavailability and/or improved stability. All such crystalline forms are within the ambit of the present invention
- LDL Low Density Lipoprotein
- HDL High Density Lipoprotein
- VLDL Very Low Density Lipoprotein
- LDLR-/- Low Density Lipoprotein receptor deficient
- DMEM Dulbecco's Modified Eagle's Medium
- GAPDH glyceraldehyde-3- phosphate dehydrogenase
- NaCMC sodium carboxymethylcellulose
- SLS sodium lauryl sulfate
- FPLC fast protein liquid chromatography
- PBS is phosphate buffered saline
- VLDL-C Very Low Density Lipoprotein-Cholesterol
- HDL-C High Density Lipoprotein-Cholesterol
- CMV cytomegalovirus.
- RT indicates room temperature.
- Human FXR/FXR-Response Element Luciferase Reporter Cotransfection Assay Compounds of the present invention are tested using the Human FXR/FXR- Response Element Luciferase Reporter Cotransfection Assay. The assay is performed essentially as described in J. Biological Chem. 2006, 281 (52), 39831-39838, except that 10 ⁇ g total DNA per million cells is used. One of skill in the art is able to perform this assay without undue experimentation.
- Exemplified compounds of the invention are found to be potent using this assay exhibiting ECsos in the range of about 75 to about 2590 nM.
- the compound of Example 7 showed an EC50 of 220 nM.
- Compounds are evaluated by an FXR-SRC-I Cofactor Recruitment assay using the Alpha (Amplified Luminescent Proximity Homogeneous Assay) Screen technology according to the manufacturer instructions (Perkin Elmer Corporation, Norwalk, CT, USA) at various concentrations. Mix purified 6-HIS-tagged human FXR ligand-binding domain (amino acids 242-472), purified GST-tagged human SRC-I nuclear receptor- interacting domain (amino acids 220-394), Nickel Chelate donor beads (Perkin Elmer Corporation) and Anti-GST antibody acceptor beads (Perkin Elmer Corporation) together and aliquot 12 ⁇ L per well into 384 well plates.
- mice Acclimate animals for two weeks prior to study initiation. Maintain mice on a 12: 12 hour light-dark cycle at 21 0 C. Provide deionized water ad libitum and maintain for two weeks on 'western diet' TD 88137 Diet (42 % fat, 0.15 % cholesterol, Harlan Teklad, Madison, WI, USA) ad libitum. Optimize groups of five ten-week-old male LDLR-/- mice based on serum triglyceride and cholesterol levels.
- Cholesterol reagent (available from Roche Diagnostics, Indianapolis, IN, USA) at 0.16 mL/min is mixed with the column effluent through a T connection; the mixture is then passed through a 15 m x 0.5 mm knitted tubing reactor (Aura Industries, New York, NY, USA) immersed in a 37 0 C water bath.
- the colored product produced in the presence of cholesterol is monitored in the flow stream at 505 nm, and the analog voltage from the monitor is converted to a digital signal for collection and analysis.
- the change in voltage corresponding to change in cholesterol concentration is plotted vs.
- the specific dose of a compound administered according to this invention will, of course, be determined by the particular circumstances surrounding the case including, for example, the compound administered, the route of administration, the state of being of the patient, and the pathological condition being treated.
- the compounds of the present invention are preferably formulated as pharmaceutical compositions administered by a variety of routes. Most preferably, such compositions are for oral administration.
- Pharmaceutical compositions and processes for preparing same are well known in the art. See, e.g., Remington: The Science and Practice of Pharmacy (A. Gennaro, et ah, eds., 20 l ed., Lippincott, Williams and Wilkins Publishers, 2003).
- the present invention is further illustrated by the examples and preparations disclosed herein. These examples and preparations are illustrative only and are not intended to limit the invention in any way.
- the terms used in the examples and preparations have their normal meanings unless otherwise designated. All chromatography is performed using silica gel, unless otherwise indicated.
- 2,6-Dichloro-benzaldehyde (7.0 g, 40 mmol) and hydroxylamine hydrochloride (2.16 g, 44 mmol) are added to 10 mL of water and 30 mL of methanol.
- Sodium hydroxide (4.0 g, 100 mmol) is dissolved in 8 mL of water slowly.
- the sodium hydroxide solution is added to the benzaldehyde solution.
- the reaction is stirred overnight.
- the reaction mixture is partitioned between ethyl acetate and water.
- the organic layer is washed with brine and dried over solid sodium sulfate.
- the organic layer is filtered and the solvent is removed under reduced pressure to yield the title compound.
- 2,6-Dichloro-benzaldehyde oxime (7.6 g, 40 mmol) is dissolved in 56 mL of DMF and N-chlorosuccinimide (5.9 g, 44.0 mmol) is added followed by a catalytic amount of HCl gas. The reaction mixture is stirred overnight. The reaction mixture is partitioned between ether and water. The layers are separated and the ether layer is washed with brine and is dried over sodium sulfate. The ether layer is filtered and the solvent is removed under reduced pressure to yield the crude product.
- reaction mixture is concentrated under reduced pressure to 51 g of crude material and purified via column chromatography using a gradient of 35-60% DCM in Hexanes to yield the title compound (28 g, 59%).
- a re-sealable tube is charged with 6-bromo-l -methyl- lH-indole-3-carboxylic acid methyl ester (850 mg, 1.00 equiv, 3.17 mmol), 4-hydroxypiperidine (491 mg, 1.5 equiv, 4.76 mmol), copper(I) iodide (60.379 mg, 0.1 equiv, 0.32 mmole), proline (73 mg, 0.2 equiv, 0.64 mmol) and potassium carbonate (885 mg, 2 equiv, 6.34 mmol) and purged with nitrogen. Anhydrous dimethyl sulfoxide (2 mL) is added. The tube is closed and the mixture is stirred at 110 0 C for 22 h.
- a flask containing a solution of trifluoro-methanesulfonic acid 4-(tert-butyl- dimethyl-silanyloxy)-cyclohex- 1 -enyl ester (3.82 g, 10.6 mmol) and bis(pinacolato)diboron (3.02 g; 11.7 mmol) in 1,4-dioxane (50 mL) is evacuated and refilled with N 2 three times.
- Oxalyl chloride (717.2 g, 5.65 mol, 3.5 eq) is added to a 0-5 0 C suspension of dichloromethane (3.44 L) and aluminum chloride (753.4 g, 5.65 mol, 3.5 eq). The resulting suspension is stirred for 30-60 minutes at 0-5 0 C and cooled -20 to -25 0 C.
- a solution of 6-bromobenzo[b]thiophene (344 g, 1.614 mol, 1 eq) in dichloromethane (1.72 L) is added over 1 h while maintaining the temperature at -20 to -25 0 C.
- the reaction mixture is stirred for 30 minutes at -20 to -25 0 C and warmed to 18 to 20 0 C using a warm water bath.
- the reaction mixture is stirred for 1.5 h at this temperature.
- the reaction mixture is filtered and the filter cake is washed with dichloromethane (3 x 300 mL).
- the combined filtrate is concentrated to yield a thick black oil in the flask (600 g). This residue is dissolved in dichloromethane (1 L) and added to ethanol (3.5 L) at -10 to 0 0 C in portions at such rate to maintain temperature at 10 to 20 0 C. Once the addition is complete, the reaction mixture is partially concentrated to remove the dichloromethane only and then the vacuum is released.
- the reaction mixture is heated to 60-70 0 C and stirred at this temperature for 1 h. Upon completion of the reaction, the solution is decanted from the resulting tars. The tars are discarded. The ethanol solution is evaporated to a residue. The residue is diluted with EtOAc (2 L).
- reaction mixture is combined with reaction mixture from another batch of the reaction for further work up (started with 330 g of 6- Bromobenzo[b]thiophene, 1.549 mol).
- the combined reaction mixture is poured into a stirred mixture of EtOAc (1 L) and brine solution (10 L). The layers are separated and the organic layer is washed with brine solution (2 L). The combined aqueous layer is extracted with EtOAc (4 L). The organic layer is washed with brine solution (1 L). The combined organic layers are dried over magnesium sulfate and charcoal, filtered, and concentrated under reduced pressure. The resulting oil is further concentrated in a vacuum oven for 15 h at room temperature to afford waxy solids after drying (750 g).
- the solids are suspended in heptane (5 L) with stirring and the suspension is heated to 70 0 C.
- Magnesium sulfate (300 g) is added and the resulting suspension is stirred for 10 minutes at 70 0 C.
- the suspension is filtered.
- the solids are suspended in heptane (5 L) and heated to 70 0 C.
- the suspension is stirred for 10-20 minutes at this temperature and filtered.
- the filter cake is washed with heptane (1 L).
- the heptane filtrates are collected and concentrated under reduced pressure to give light brown solids (550 g).
- the solids are dissolved in heptane (4 L) at 60 0 C.
- the resulting solution is cooled to 35 to 50 0 C.
- the solution is evenly loaded onto two plugs of silica gel (1.5 kg each) eluting with 0.5% EtOAc in heptane.
- the pure product fractions are combined and concentrated under reduced pressure.
- the impure product fractions are combined, concentrated, and purified as described above.
- the total purified product is isolated (500 g) and crystallized from heptane (1.2 L).
- the solids are collected by filtration, washed with cold heptane (200 mL, -20 0 C), and dried in a vacuum oven at room temperature for 15 h to afford the title compound (460 g, 51%).
- Example 14 involves the use of LiOH in place of NaOH for hydrolysis in step 2.
- Tetrahydrofuran (4 mL) is added at room temperature to a mixture of 18-crown-6 (306 mg, 1.14 mmole), tert-butyl alcohol, potassium derivative (134 mg, 1.14 mmole) and 6-(4-hydroxy-piperi din- 1-y I)-I -methyl- lH-indole-3 -carboxylic acid methyl ester (300 mg, 1.040 mmole).
- the mixture is stirred for 15 minutes and then 4-bromomethyl-5- cyclopropyl-3-(2,6-dichloro-phenyl)-isoxazole (361 mg, 1.04 mmole) is added.
- Anhydrous tetrahydrofuran (5 mL) is added at room temperature to a mixture of 4-(4-hydroxy-azepan-l-yl)-benzoic acid ethyl ester (364 mg, 1.38 mmol), 18-crown-6 (407 mg, 1.52 mmol) and tert-butyl alcohol, potassium derivative (178 mg, 1.52 mmol).
- the mixture is stirred for 5 minutes and then 4-bromomethyl-5-cyclopropyl-3-(2,6- dichloro-phenyl)-isoxazole (480 mg, 1.38 mmol) is added. After 1.5 h, the solvent is removed and water is added. The aqueous phase is extracted with ethyl acetate.
- a solution 2M of lithium hydroxide (283 ⁇ L, 566 ⁇ moles) is added to a solution of 4- ⁇ 4-[5-cyclopropyl-3-(2,6-dichloro-phenyl)-isoxazol-4-ylmethoxy]-azepan-l-yl ⁇ - benzoic acid ethyl ester (isomer A, 60 mg, 113 ⁇ moles) in 1,4-dioxane (2 mL) and the mixture is stirred at 90 0 C overnight. The organic solvent is removed and HCl (IM) is added until the solution reaches pH 3-4.
- IM HCl
- Example 27 listed in TABLE 3, is prepared essentially as described in the preparation of Example 26, 6- ⁇ 4-[5-cyclopropyl-3-(2,6-dichloro-phenyl)-isoxazol-4- ylmethoxy]-azepan-l-yl ⁇ -l -methyl- lH-indole-3-carboxylic acid using the appropriate starting materials.
- Examples 27A-27D in TABLE 3 follow the procedure essentially as described in Example 26, Step 1, except the reactions are completed in a sealed tube with heating to 110 0 C.
- Examples 27C-27D, Step 2 substitute approximately 10 eq LiOH for NaOH sufficient to drive reaction to completion and add water in equal (vol/vol) amount to tetrahydrofuran and methanol. Stir the reaction mixture overnight at room temperature, neutralize to p ⁇ 6 with 1 N HCl, and concentrate to dryness. Dilute the material with water, extract with ethyl acetate, dry with Na 2 SO 4 and concentrate to give the title compounds.
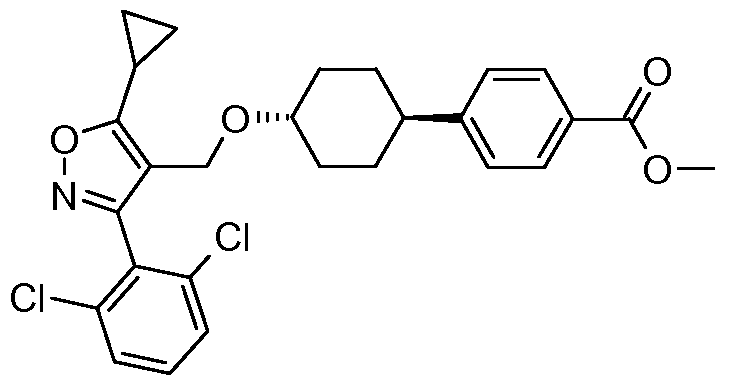
- the title compound as a cis/trans mixture is prepared essentially as described in the preparation oftrans-5-cyclopropyl-3-(2,6-dichloro-phenyl)-4- ⁇ 4-[4-(4-methoxy- benzyloxy)-phenyl]-cyclohexyloxymethyl ⁇ -isoxazole starting from 1 ,6-(4-hydroxy- cyclohexyl)-l -methyl- 1 H-indole-3 -carboxylic acid methyl ester (202 mg, 0.703 mmol) and 4-bromomethyl-5-cyclopropyl-3-(2-trifluoromethoxy-phenyl)-isoxazole (382 mg, 1.05 mmol).
- the isomers are separated by silica gel chromatography eluting with a gradient of 25-40% EtOAc/Hexanes to provide the cis isomer [62 mg,15.5%; MS m/z 569.0 (M+l)] and trans isomer [52 mg, 13%; MS m/z 569.0 (M+l)].
- N-Chlorosuccinimide (1162g, 8.53mol) in DMF (4.5L) is added dropwise over a solution of 2,6-dichloro-benzaldehyde oxime (1621.78g, 8.53mol) in DMF (5.3L) heated at 4O 0 C (addition is complete in about 6 hours). The mixture is stirred for Ih at that temperature. The reaction is cooled at room temperature, poured onto H2O (30L) at O 0 C, and extracted with MTBE (36L) and the aqueous phase was discarded. The organic layer is washed with brine, dried over Na 2 SO 4 , filtered and evaporated to dryness (at 3O 0 C). The crude, as a solid-oil, is triturated in IL of hexane and the solid formed is filtered and dried under vacuum to obtain the desired compound (1440.9g, 75% yield). MS (m/e): 224 (M+l).
- Triethylamine (1.82L, 12.84mol) is added to S-cyclopropyl-S-oxo-propionic acid methyl ester (913g, 6.42mol) and the mixture is stirred at room temperature for 30 minutes. Then, the mixture is cooled to about 1O 0 C and a suspension of 2,6-dichloro- benzaldehyde chloro-oxime (1440.9g, 6.42mol) in EtOH (3.2L) is added slowly (the internal temperature does not exceed 24 0 C). After the addition, the reaction is stirred overnight at room temperature. The reaction is diluted with EtOAc (5.3L) and washed with water (1.7L). The layers are separated and the aqueous layer is extracted with EtOAc (3L).
- Step 6 4- [5 -Cyclopropyl-3 -(2 ,6-dichloro-phenyiy isoxazol-4-ylmethoxyi -piperidine- 1 -carboxylic acid tert-butyl ester
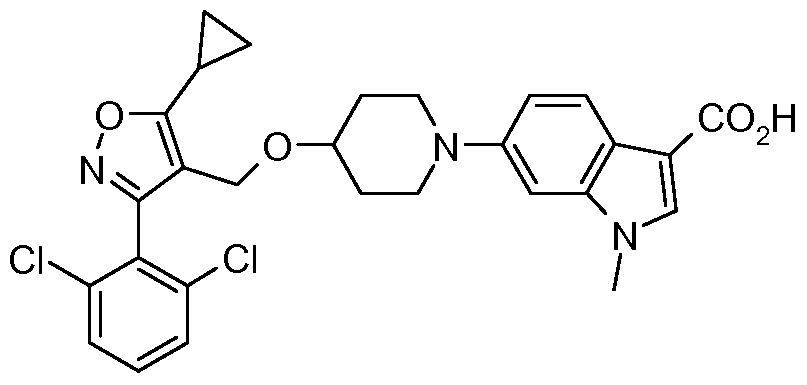
- Step 12 6- ⁇ 4-r5-Cyclopropyl-3-( ' 2,6-dichloro-phenyl)-isoxazol-4-ylmethoxy1-piperidin-l-yl ⁇ -l- methyl-lH-indole-3-carboxylic acid
- Step 1 IA fe/t-Butyl 6-(4- ⁇ r3-(2,6-dichlorophenyl)-5-cvclopropylisoxazol-4- yl1methoxy ⁇ piperidyl)-l-methylindole-3-carboxylate
- N-Chlorosuccinimide (1162g, 8.53mol) in DMF (4.5L) is added dropwise over a solution of 2,6-dichloro-benzaldehyde oxime (1621.78g, 8.53mol) in DMF (5.3L) heated at 4O 0 C (addition is complete in about 6 hours). The mixture is stirred for Ih at that temperature. The reaction is cooled at room temperature, poured onto H 2 O (30L) at O 0 C, and extracted with MTBE (36L). The organic layer is washed with brine, dried over Na 2 SO 4 , filtered and evaporated to dryness (at 3O 0 C). The crude, as a solid-oil, is triturated in IL of hexane and the solid formed is filtered and dried under vacuum to obtain the desired compound (1440.9g, 75% yield). MS (m/e): 224 (M+ 1)
- Triethylamine (1.82L, 12.84mol) is added to S-cyclopropyl-S-oxo-propionic acid methyl ester (913g, 6.42mol) and the mixture is stirred at room temperature for 30 minutes. Then, the mixture is cooled to about 1O 0 C and a suspension of 2,6-dichloro- benzaldehyde chloro-oxime (1440.9g, 6.42mol) in EtOH (3.2L) is added slowly (the internal temperature does not exceed 24 0 C). After the addition, the reaction is stirred overnight at room temperature. The reaction is diluted with EtOAc (5.3L) and washed with water (1.7L). The layers are separated and the aqueous layer is extracted with EtOAc (3L).
- the resulting mixture is stirred for 20 min at room temperature under nitrogen, followed by addition of a solution of 48.5g ( 139.75 mmol) of 4-Bromomethyl-5-cyclopropyl-3-(2,6- dichloro-phenyl)-isoxazole in 100 mL tetrahydrofuran over a 1 hour period.
- the resulting mixture is stirred at room temperature under nitrogen atmosphere for 12 hours.
- Water (20OmL) is added to the reaction mixture and the solvent is removed under reduced pressure.
- the resulting brown oil is diluted with EtOAc (2 x 25OmL), and washed with brine (2 x 150 mL).
- Step 10 4- ⁇ 4-r5-Cyclopropyl-3-( ' 2,6-dichloro-phenyl)-isoxazol-4-ylmethoxy1-azepan-l-yl ⁇ - benzoic acid ethyl ester
- racemic material is subjected to chiral purification (ChiralpakTM column, hex-DMEA (0.2%)/ IPA 6:4 as eluent) to afford 8.28g of the desired chiral isomer (isomer B). MS (m/e): 529 (M+ 1).
Abstract
Description








































































Claims




Priority Applications (20)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RS20120099A RS52216B (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
| ES08781651T ES2376176T3 (en) | 2007-07-16 | 2008-07-11 | COMPOUNDS AND PROCEDURES TO MODULATE FXR. |
| PL08781651T PL2178851T3 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
| EP08781651A EP2178851B1 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
| AU2008276236A AU2008276236B2 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating FXR |
| BRPI0814571-7A2A BRPI0814571A2 (en) | 2007-07-16 | 2008-07-11 | FXR MODULATING COMPOUNDS, THEIR USE AND COMPOSITION UNDERSTANDING THE SAME |
| CA2693406A CA2693406C (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
| KR1020107000896A KR101157334B1 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
| CN2008800247651A CN101743232B (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
| DK08781651.8T DK2178851T3 (en) | 2007-07-16 | 2008-07-11 | Compounds and Methods for Modulating FXR |
| EA201070148A EA016475B1 (en) | 2007-07-16 | 2008-07-11 | Azol compounds for use as fxr modulators |
| JP2010517082A JP5373788B2 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating FXR |
| AT08781651T ATE539065T1 (en) | 2007-07-16 | 2008-07-11 | COMPOUNDS AND METHODS FOR MODULATING FX RECEPTORS |
| SI200830514T SI2178851T1 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
| US12/600,879 US8153624B2 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating FXR |
| MX2010000502A MX2010000502A (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr. |
| IL202234A IL202234A0 (en) | 2007-07-16 | 2009-11-19 | Compounds and methods for modulating fxr |
| TNP2010000028A TN2010000028A1 (en) | 2007-07-16 | 2010-01-15 | Compounds and methods for modulating fxr |
| MA32615A MA31683B1 (en) | 2007-07-16 | 2010-02-11 | Fxr modulation components and methods |
| HR20120048T HRP20120048T1 (en) | 2007-07-16 | 2012-01-16 | Compounds and methods for modulating fxr |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US94997407P | 2007-07-16 | 2007-07-16 | |
| US60/949,974 | 2007-07-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009012125A1 true WO2009012125A1 (en) | 2009-01-22 |
Family
ID=39855171
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/069719 WO2009012125A1 (en) | 2007-07-16 | 2008-07-11 | Compounds and methods for modulating fxr |
Country Status (31)
| Country | Link |
|---|---|
| US (1) | US8153624B2 (en) |
| EP (1) | EP2178851B1 (en) |
| JP (1) | JP5373788B2 (en) |
| KR (1) | KR101157334B1 (en) |
| CN (1) | CN101743232B (en) |
| AR (1) | AR067540A1 (en) |
| AT (1) | ATE539065T1 (en) |
| AU (1) | AU2008276236B2 (en) |
| BR (1) | BRPI0814571A2 (en) |
| CA (1) | CA2693406C (en) |
| CL (1) | CL2008002051A1 (en) |
| CO (1) | CO6270212A2 (en) |
| CY (1) | CY1112298T1 (en) |
| DK (1) | DK2178851T3 (en) |
| DO (1) | DOP2010000018A (en) |
| EA (1) | EA016475B1 (en) |
| EC (1) | ECSP109879A (en) |
| ES (1) | ES2376176T3 (en) |
| HR (1) | HRP20120048T1 (en) |
| IL (1) | IL202234A0 (en) |
| MA (1) | MA31683B1 (en) |
| MX (1) | MX2010000502A (en) |
| PE (1) | PE20090809A1 (en) |
| PL (1) | PL2178851T3 (en) |
| PT (1) | PT2178851E (en) |
| RS (1) | RS52216B (en) |
| SI (1) | SI2178851T1 (en) |
| SV (1) | SV2010003458A (en) |
| TN (1) | TN2010000028A1 (en) |
| TW (1) | TW200906823A (en) |
| WO (1) | WO2009012125A1 (en) |
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011020615A1 (en) | 2009-08-19 | 2011-02-24 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4 ) binding and activity modulating compounds |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2012087519A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating fxr |
| WO2012087520A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| WO2012087521A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| EP2545964A1 (en) | 2011-07-13 | 2013-01-16 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| WO2013037482A1 (en) | 2011-09-15 | 2013-03-21 | Phenex Pharmaceuticals Ag | Farnesoid x receptor agonists for cancer treatment and prevention |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| WO2015069666A1 (en) * | 2013-11-05 | 2015-05-14 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| EP3006939A1 (en) | 2014-10-06 | 2016-04-13 | Gilead Sciences, Inc. | Histidine-rich Glycoprotein as a marker for hepatic Farnesoid X receptor activation |
| WO2016086218A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| EP3034501A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| EP3034499A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| US9394285B2 (en) | 2013-03-15 | 2016-07-19 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| WO2016127924A1 (en) | 2015-02-13 | 2016-08-18 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| WO2017218337A1 (en) | 2016-06-13 | 2017-12-21 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| WO2018085148A1 (en) * | 2016-11-04 | 2018-05-11 | Hepagene Therapeutics, Inc. | Nitrogen-containing heterocyclic compounds as fxr modulators |
| WO2018178260A1 (en) | 2017-03-30 | 2018-10-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for reducing persistence and expression of episomal viruses |
| KR20180115233A (en) * | 2017-04-12 | 2018-10-22 | 일동제약(주) | An isoxazole derivatives as nuclear receptor agonist and used thereof |
| CN109689050A (en) * | 2016-09-14 | 2019-04-26 | 诺华股份有限公司 | New departure of FXR agonist |
| WO2019089667A1 (en) | 2017-11-01 | 2019-05-09 | Bristol-Myers Squibb Company | Bridged bicyclic compounds as farnesoid x receptor modulators |
| WO2019149158A1 (en) | 2018-02-02 | 2019-08-08 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous tricyclic compounds and uses thereof in medicine |
| US10421730B2 (en) | 2016-06-13 | 2019-09-24 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2020042114A1 (en) * | 2018-08-30 | 2020-03-05 | Terns Pharmaceuticals, Inc. | Treating liver disorders |
| EP3523298A4 (en) * | 2016-10-04 | 2020-06-24 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as fxr agonists and methods of use thereof |
| EP3711762A1 (en) | 2013-09-11 | 2020-09-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A farnesoid x receptor agonsits foruse and pharmaceutical compositions for the treatment of chronic hepatitis b virus infection |
| WO2020231917A1 (en) * | 2019-05-13 | 2020-11-19 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2022082197A1 (en) * | 2020-10-15 | 2022-04-21 | Eli Lilly And Company | Polymorphs of an fxr agonist |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| US11419878B2 (en) | 2016-03-28 | 2022-08-23 | Intercept Pharmaceuticals, Inc. | Medicine obtained by combining FXR agonist and ARB |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| US11667629B2 (en) | 2017-12-22 | 2023-06-06 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Isoxazole derivative, preparation method therefor, and use thereof |
| US11718619B2 (en) | 2016-08-23 | 2023-08-08 | Ardelyx, Inc. | Isoxazolyl-carbonyloxy azabicyclo[3.2.1]octanyl compounds as FXR activators |
| US11753410B2 (en) | 2017-09-14 | 2023-09-12 | Ardelyx, Inc. | Hormone receptor modulators for treating metabolic mutagenic and fibrotic conditions and disorders |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
| US11958879B2 (en) | 2015-03-31 | 2024-04-16 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104045635A (en) * | 2014-06-23 | 2014-09-17 | 华东理工大学 | 3,4,5-tri-substituted isoxazole compounds and applications thereof |
| CR20170356A (en) | 2015-02-06 | 2018-02-28 | Intercept Pharmaceuticals Inc | PHARMACEUTICAL COMPOSITIONS FOR COMBINED THERAPY |
| TN2017000426A1 (en) | 2015-04-07 | 2019-04-12 | Intercept Pharmaceuticals Inc | Pharmaceutical compositions for combination therapy |
| CN106946867B (en) * | 2016-01-06 | 2019-11-12 | 广州市恒诺康医药科技有限公司 | FXR receptor modulators and its preparation method and application |
| CN107021958A (en) * | 2016-02-01 | 2017-08-08 | 山东轩竹医药科技有限公司 | FXR receptor stimulating agents |
| WO2017189652A1 (en) | 2016-04-26 | 2017-11-02 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| WO2017189651A1 (en) * | 2016-04-26 | 2017-11-02 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| US10080743B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2017201150A1 (en) | 2016-05-18 | 2017-11-23 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as fxr agonists and methods of use thereof |
| WO2017201155A1 (en) | 2016-05-18 | 2017-11-23 | Enanta Pharmaceuticals, Inc. | lSOXAZOLE DERIVATIVES AS FXR AGONISTS AND METHODS OF USE THEREOF |
| US10138228B2 (en) | 2016-05-18 | 2018-11-27 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use therof |
| TW201808283A (en) | 2016-08-05 | 2018-03-16 | 廣東東陽光藥業有限公司 | Nitrogen-containing tricyclic compounds and uses thereof in medicine |
| US10793568B2 (en) * | 2016-08-23 | 2020-10-06 | Ardelyx, Inc. | Hormone receptor modulators for treating metabolic conditions and disorders |
| US10597391B2 (en) | 2016-10-26 | 2020-03-24 | Enanta Pharmaceuticals, Inc. | Urea-containing isoxazole derivatives as FXR agonists and methods of use thereof |
| CN108218852A (en) * | 2016-12-15 | 2018-06-29 | 宁波百纳西药业有限公司 | A kind of spiro-compound, preparation method, composition and purposes |
| CN109265471B (en) * | 2017-06-30 | 2021-06-04 | 轩竹生物科技有限公司 | FXR receptor agonists |
| CN109320509B (en) * | 2017-07-31 | 2022-02-08 | 轩竹生物科技股份有限公司 | FXR receptor agonists |
| AU2018360575A1 (en) | 2017-11-01 | 2020-06-18 | Bristol-Myers Squibb Company | Alkene spirocyclic compounds as farnesoid X receptor modulators |
| JP7264906B2 (en) * | 2017-11-01 | 2023-04-25 | ブリストル-マイヤーズ スクイブ カンパニー | Alkene compounds as farnesoid X receptor modulators |
| SG11202003825TA (en) | 2017-11-01 | 2020-05-28 | Bristol Myers Squibb Co | Spirocyclic compounds as farnesoid x receptor modulators |
| US11370785B2 (en) | 2017-11-01 | 2022-06-28 | Bristol-Myers Squibb Company | Multicyclic compounds as farnesoid X receptor modulators |
| US10689391B2 (en) | 2017-12-12 | 2020-06-23 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| WO2019160813A1 (en) | 2018-02-14 | 2019-08-22 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| CN110452235B (en) * | 2018-05-08 | 2023-02-17 | 中国科学院上海药物研究所 | Fluorine-containing isoxazole compound and preparation method, pharmaceutical composition and application thereof |
| AR118050A1 (en) | 2019-02-15 | 2021-09-15 | Bristol Myers Squibb Co | BICYCLIC COMPOUNDS REPLACED AS MODULATORS OF THE FARNESOID X RECEIVER |
| JP2022536060A (en) | 2019-05-30 | 2022-08-12 | インターセプト ファーマシューティカルズ, インコーポレイテッド | Pharmaceutical composition comprising an FXR agonist and a fibrate for use in treating cholestatic liver disease |
| CN114667142A (en) * | 2019-11-08 | 2022-06-24 | 拓臻制药公司 | Treating liver disorders |
| US11820754B2 (en) | 2020-08-25 | 2023-11-21 | Eli Lilly And Company | Polymorphs of an SSAO inhibitor |
| MX2023002799A (en) * | 2020-09-11 | 2023-05-26 | Terns Pharmaceuticals Inc | Solid dispersion formulations of an fxr agonist. |
| US20230241071A1 (en) | 2021-11-11 | 2023-08-03 | Terns Pharmaceuticals, Inc. | Combination treatment of liver disorders |
| WO2023220404A1 (en) * | 2022-05-13 | 2023-11-16 | Terns Pharmaceuticals, Inc. | Treatment of non-alcoholic steatohepatitis |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004048349A1 (en) | 2002-11-22 | 2004-06-10 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2007092751A2 (en) | 2006-02-03 | 2007-08-16 | Eli Lilly And Company | Compounds and methods for modulating fx-receptors |
| WO2007140183A1 (en) * | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Fxr agonists |
| WO2007140174A2 (en) | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Compounds and methods for modulating fxr |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07138258A (en) | 1993-11-16 | 1995-05-30 | Taiho Yakuhin Kogyo Kk | Thiazolidinedione derivative or salt thereof |
| WO2000037077A1 (en) | 1998-12-23 | 2000-06-29 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| US6967212B2 (en) * | 2001-05-30 | 2005-11-22 | Bristol-Myers Squibb Company | Substituted azole acid derivatives useful as antidiabetic and antiobesity agents and method |
| US7105556B2 (en) | 2001-05-30 | 2006-09-12 | Bristol-Myers Squibb Company | Conformationally constrained analogs useful as antidiabetic and antiobesity agents and method |
| ATE381542T1 (en) | 2001-08-13 | 2008-01-15 | Phenex Pharmaceuticals Ag | NR1H4 CORE RECEPTOR BINDING COMPOUNDS |
| MXPA05000279A (en) | 2002-07-09 | 2005-03-31 | Squibb Bristol Myers Co | Substituted heterocyclic derivatives useful as antidiabetic and antiobesity agents and method. |
-
2008
- 2008-07-03 TW TW097125064A patent/TW200906823A/en unknown
- 2008-07-11 EA EA201070148A patent/EA016475B1/en not_active IP Right Cessation
- 2008-07-11 PE PE2008001168A patent/PE20090809A1/en not_active Application Discontinuation
- 2008-07-11 WO PCT/US2008/069719 patent/WO2009012125A1/en active Application Filing
- 2008-07-11 KR KR1020107000896A patent/KR101157334B1/en not_active IP Right Cessation
- 2008-07-11 MX MX2010000502A patent/MX2010000502A/en active IP Right Grant
- 2008-07-11 CA CA2693406A patent/CA2693406C/en not_active Expired - Fee Related
- 2008-07-11 AU AU2008276236A patent/AU2008276236B2/en not_active Ceased
- 2008-07-11 RS RS20120099A patent/RS52216B/en unknown
- 2008-07-11 PL PL08781651T patent/PL2178851T3/en unknown
- 2008-07-11 DK DK08781651.8T patent/DK2178851T3/en active
- 2008-07-11 SI SI200830514T patent/SI2178851T1/en unknown
- 2008-07-11 EP EP08781651A patent/EP2178851B1/en active Active
- 2008-07-11 ES ES08781651T patent/ES2376176T3/en active Active
- 2008-07-11 US US12/600,879 patent/US8153624B2/en active Active
- 2008-07-11 CN CN2008800247651A patent/CN101743232B/en active Active
- 2008-07-11 AT AT08781651T patent/ATE539065T1/en active
- 2008-07-11 JP JP2010517082A patent/JP5373788B2/en active Active
- 2008-07-11 PT PT08781651T patent/PT2178851E/en unknown
- 2008-07-11 BR BRPI0814571-7A2A patent/BRPI0814571A2/en not_active IP Right Cessation
- 2008-07-14 AR ARP080103026A patent/AR067540A1/en not_active Application Discontinuation
- 2008-07-14 CL CL2008002051A patent/CL2008002051A1/en unknown
-
2009
- 2009-11-19 IL IL202234A patent/IL202234A0/en unknown
-
2010
- 2010-01-12 DO DO2010000018A patent/DOP2010000018A/en unknown
- 2010-01-12 CO CO10002335A patent/CO6270212A2/en not_active Application Discontinuation
- 2010-01-15 EC EC2010009879A patent/ECSP109879A/en unknown
- 2010-01-15 TN TNP2010000028A patent/TN2010000028A1/en unknown
- 2010-01-15 SV SV2010003458A patent/SV2010003458A/en not_active Application Discontinuation
- 2010-02-11 MA MA32615A patent/MA31683B1/en unknown
-
2012
- 2012-01-16 HR HR20120048T patent/HRP20120048T1/en unknown
- 2012-01-27 CY CY20121100098T patent/CY1112298T1/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004048349A1 (en) | 2002-11-22 | 2004-06-10 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2007092751A2 (en) | 2006-02-03 | 2007-08-16 | Eli Lilly And Company | Compounds and methods for modulating fx-receptors |
| WO2007140183A1 (en) * | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Fxr agonists |
| WO2007140174A2 (en) | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | Compounds and methods for modulating fxr |
Non-Patent Citations (4)
| Title |
|---|
| "Dorland's Illustrated Medical Dictionary", W.B SAUNDERS PUBLISHING COMPANY |
| J. MED. CHEM., vol. 43, no. 16, 2000, pages 2971 - 2974 |
| P. STAHL ET AL.: "Handbook of Pharmaceutical Salts: Properties, Selection and Use", 2002, VCHA/WILEY-VCH |
| S.M. BERGE ET AL.: "Pharmaceutical Salts", JOURNAL OFPHARMACEUTICAL SCIENCES, vol. 66, no. 1, January 1977 (1977-01-01), XP002675560, DOI: doi:10.1002/jps.2600660104 |
Cited By (97)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102548974A (en) * | 2009-08-19 | 2012-07-04 | 菲尼克斯药品股份公司 | Novel FXR (NR1H4 ) binding and activity modulating compounds |
| EP2289883A1 (en) | 2009-08-19 | 2011-03-02 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| CN102548974B (en) * | 2009-08-19 | 2015-11-25 | 菲尼克斯药品股份公司 | Novel FXR (NR1H4) combines and activity modulating compounds |
| US8952042B2 (en) | 2009-08-19 | 2015-02-10 | Phenex Pharmaceuticals Ag | FXR (NR1H4) binding and activity modulating compounds |
| WO2011020615A1 (en) | 2009-08-19 | 2011-02-24 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4 ) binding and activity modulating compounds |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2012087520A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| EA025569B1 (en) * | 2010-12-20 | 2017-01-30 | Новартис Аг | Compositions and methods for modulating fxr |
| WO2012087521A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| KR101626046B1 (en) | 2010-12-20 | 2016-06-01 | 노파르티스 아게 | Compositions and methods for modulating fxr |
| US20130261108A1 (en) * | 2010-12-20 | 2013-10-03 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| KR20130109210A (en) * | 2010-12-20 | 2013-10-07 | 아이알엠 엘엘씨 | Compositions and methods for modulating fxr |
| CN103443099A (en) * | 2010-12-20 | 2013-12-11 | Irm责任有限公司 | Compositions and methods for modulating FXR |
| US20140039007A1 (en) * | 2010-12-20 | 2014-02-06 | David C. Tully | Compositions and methods for modulating farnesoid x receptors |
| CN103443099B (en) * | 2010-12-20 | 2016-03-23 | 诺华股份有限公司 | Regulate composition and the method for FXR |
| US9150568B2 (en) | 2010-12-20 | 2015-10-06 | Novartis Ag | Compositions and methods for modulating FXR |
| WO2012087519A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating fxr |
| AU2011345233B2 (en) * | 2010-12-20 | 2015-11-12 | Novartis Ag | Compositions and methods for modulating FXR |
| AP3414A (en) * | 2010-12-20 | 2015-09-30 | Irm Llc | Compositions and methods for modulating fxr |
| US10485795B2 (en) | 2011-07-13 | 2019-11-26 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US9139539B2 (en) | 2011-07-13 | 2015-09-22 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| EP2987532A1 (en) | 2011-07-13 | 2016-02-24 | Gilead Sciences, Inc. | Novel fxr (nr1h4) binding and activity modulating compounds |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| WO2013007387A1 (en) | 2011-07-13 | 2013-01-17 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4) binding and activity modulating compounds |
| EP3246070A1 (en) | 2011-07-13 | 2017-11-22 | Gilead Sciences, Inc. | Novel fxr (nr1h4) binding and activity modulating azoles |
| US9820979B2 (en) | 2011-07-13 | 2017-11-21 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| EP2545964A1 (en) | 2011-07-13 | 2013-01-16 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| US9539244B2 (en) | 2011-07-13 | 2017-01-10 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| WO2013037482A1 (en) | 2011-09-15 | 2013-03-21 | Phenex Pharmaceuticals Ag | Farnesoid x receptor agonists for cancer treatment and prevention |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| US9394285B2 (en) | 2013-03-15 | 2016-07-19 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| EP3711762A1 (en) | 2013-09-11 | 2020-09-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A farnesoid x receptor agonsits foruse and pharmaceutical compositions for the treatment of chronic hepatitis b virus infection |
| US10077240B2 (en) | 2013-11-05 | 2018-09-18 | Novartis Ag | Compositions and methods for modulating farnesoid X receptors |
| KR102350357B1 (en) | 2013-11-05 | 2022-01-14 | 노파르티스 아게 | Compositions and methods for modulating farnesoid x receptors |
| KR20160079091A (en) * | 2013-11-05 | 2016-07-05 | 노파르티스 아게 | Compositions and methods for modulating farnesoid x receptors |
| US10683271B2 (en) | 2013-11-05 | 2020-06-16 | Novartis Ag | Compositions and methods for modulating farnesoid X receptors |
| US9682939B2 (en) | 2013-11-05 | 2017-06-20 | Novartis Ag | Compositions and methods for modulating farnesoid X receptors |
| WO2015069666A1 (en) * | 2013-11-05 | 2015-05-14 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| US11021446B2 (en) | 2013-11-05 | 2021-06-01 | Novartis Ag | Compositions and methods for modulating farnesoid X receptors |
| EA030430B1 (en) * | 2013-11-05 | 2018-08-31 | Новартис Аг | Compositions and methods for modulating farnesoid x receptors |
| EP3006939A1 (en) | 2014-10-06 | 2016-04-13 | Gilead Sciences, Inc. | Histidine-rich Glycoprotein as a marker for hepatic Farnesoid X receptor activation |
| WO2016086218A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| US9751874B2 (en) | 2014-12-17 | 2017-09-05 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| CN107108595A (en) * | 2014-12-17 | 2017-08-29 | 吉利德科学公司 | New FXR (NR1H4) modulating compound |
| US9938278B2 (en) | 2014-12-17 | 2018-04-10 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| EP3034499A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| CN108064223A (en) * | 2014-12-17 | 2018-05-22 | 吉利德科学公司 | FXR (NR1H4) modulating compound of hydroxyl |
| WO2016096116A1 (en) * | 2014-12-17 | 2016-06-23 | Gilead Sciences, Inc. | Novel fxr (nr1h4) modulating compounds |
| EP3034501A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| EA033642B1 (en) * | 2014-12-17 | 2019-11-12 | Gilead Sciences Inc | Fxr (nr1h4) modulating compounds |
| WO2016096115A1 (en) * | 2014-12-17 | 2016-06-23 | Gilead Sciences, Inc. | Hydroxy containing fxr (nr1h4) modulating compounds |
| CN108064223B (en) * | 2014-12-17 | 2021-06-01 | 吉利德科学公司 | Fxr (nr1h4) modulating compounds containing hydroxy groups |
| KR101955840B1 (en) | 2014-12-17 | 2019-03-07 | 길리애드 사이언시즈, 인코포레이티드 | Novel fxr (nr1h4) modulating compounds |
| CN107108595B (en) * | 2014-12-17 | 2021-04-06 | 吉利德科学公司 | Novel FXR (NR1H4) modulating compounds |
| TWI697483B (en) * | 2014-12-17 | 2020-07-01 | 美商基利科學股份有限公司 | Hydroxy containing fxr (nr1h4) modulating compounds |
| KR20170095955A (en) * | 2014-12-17 | 2017-08-23 | 길리애드 사이언시즈, 인코포레이티드 | Hydroxy containing fxr (nr1h4) modulating compounds |
| KR20170093969A (en) * | 2014-12-17 | 2017-08-16 | 길리애드 사이언시즈, 인코포레이티드 | Novel fxr (nr1h4) modulating compounds |
| KR101998007B1 (en) | 2014-12-17 | 2019-07-08 | 길리애드 사이언시즈, 인코포레이티드 | Hydroxy containing fxr (nr1h4) modulating compounds |
| WO2016127924A1 (en) | 2015-02-13 | 2016-08-18 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| US11958879B2 (en) | 2015-03-31 | 2024-04-16 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| US11419878B2 (en) | 2016-03-28 | 2022-08-23 | Intercept Pharmaceuticals, Inc. | Medicine obtained by combining FXR agonist and ARB |
| US10329286B2 (en) | 2016-06-13 | 2019-06-25 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10981881B2 (en) | 2016-06-13 | 2021-04-20 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10421730B2 (en) | 2016-06-13 | 2019-09-24 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2017218337A1 (en) | 2016-06-13 | 2017-12-21 | Gilead Sciences, Inc. | Fxr (nr1h4) modulating compounds |
| US11739065B2 (en) | 2016-06-13 | 2023-08-29 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10774054B2 (en) | 2016-06-13 | 2020-09-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11247986B2 (en) | 2016-06-13 | 2022-02-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| EP3730487A1 (en) | 2016-06-13 | 2020-10-28 | Gilead Sciences, Inc. | Azetidine derivatives as fxr (nr1h4) modulators |
| US11718619B2 (en) | 2016-08-23 | 2023-08-08 | Ardelyx, Inc. | Isoxazolyl-carbonyloxy azabicyclo[3.2.1]octanyl compounds as FXR activators |
| CN109689050A (en) * | 2016-09-14 | 2019-04-26 | 诺华股份有限公司 | New departure of FXR agonist |
| US11034684B2 (en) | 2016-10-04 | 2021-06-15 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| EP3523298A4 (en) * | 2016-10-04 | 2020-06-24 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as fxr agonists and methods of use thereof |
| CN109963849B (en) * | 2016-11-04 | 2023-03-28 | 和博医药有限公司 | Nitrogen-containing heterocyclic compounds as FXR modulators |
| CN109963849A (en) * | 2016-11-04 | 2019-07-02 | 合帕吉恩治疗公司 | Nitrogen-containing heterocycle compound as FXR regulator |
| US10919903B2 (en) | 2016-11-04 | 2021-02-16 | Hepagene Therapeutics (HK) Limited | Nitrogen-containing heterocyclic compounds as FXR modulators |
| WO2018085148A1 (en) * | 2016-11-04 | 2018-05-11 | Hepagene Therapeutics, Inc. | Nitrogen-containing heterocyclic compounds as fxr modulators |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
| WO2018178260A1 (en) | 2017-03-30 | 2018-10-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for reducing persistence and expression of episomal viruses |
| KR102168543B1 (en) | 2017-04-12 | 2020-10-21 | 일동제약(주) | Isoxazole derivatives as nuclear receptor agonist and uses thereof |
| KR20180115233A (en) * | 2017-04-12 | 2018-10-22 | 일동제약(주) | An isoxazole derivatives as nuclear receptor agonist and used thereof |
| US10988449B2 (en) | 2017-04-12 | 2021-04-27 | Il Dong Pharmaceutical Co., Ltd. | Isoxazole derivatives as nuclear receptor agonists and uses thereof |
| US11753410B2 (en) | 2017-09-14 | 2023-09-12 | Ardelyx, Inc. | Hormone receptor modulators for treating metabolic mutagenic and fibrotic conditions and disorders |
| WO2019089667A1 (en) | 2017-11-01 | 2019-05-09 | Bristol-Myers Squibb Company | Bridged bicyclic compounds as farnesoid x receptor modulators |
| US11667629B2 (en) | 2017-12-22 | 2023-06-06 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Isoxazole derivative, preparation method therefor, and use thereof |
| WO2019149158A1 (en) | 2018-02-02 | 2019-08-08 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous tricyclic compounds and uses thereof in medicine |
| WO2020042114A1 (en) * | 2018-08-30 | 2020-03-05 | Terns Pharmaceuticals, Inc. | Treating liver disorders |
| CN112771026A (en) * | 2018-08-30 | 2021-05-07 | 拓臻制药公司 | Treating liver disorders |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| US11555032B2 (en) | 2019-05-13 | 2023-01-17 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| WO2020231917A1 (en) * | 2019-05-13 | 2020-11-19 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| WO2022082197A1 (en) * | 2020-10-15 | 2022-04-21 | Eli Lilly And Company | Polymorphs of an fxr agonist |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2693406C (en) | Compounds and methods for modulating fxr | |
| AU2007267692B2 (en) | Compounds and methods for modulating FXR | |
| TW480256B (en) | Angiogenesis inhibiting pyridazinamines | |
| ES2255294T3 (en) | ISOXAZOL DERIVATIVES REPLACED AS MODULATORS OF THE STROGEN RECEPTOR. | |
| CA2651378C (en) | Fxr agonists | |
| JP7398605B2 (en) | FXR small molecule agonists and their preparation methods and uses | |
| WO2005087710A1 (en) | Aminophenylpropanoic acid derivative | |
| CN101759645A (en) | P38 inhibitors and methods of use thereof | |
| SG176864A1 (en) | Compounds which selectively modulate the cb2 receptor | |
| JP2005539000A (en) | 2-Phenylpyridin-4-yl derivatives as ALK5 inhibitors | |
| WO2015024448A1 (en) | Benzo piperidine ring and benzo morpholine ring compounds, manufacturing method therefor, and medical applications thereof | |
| EP2638035B1 (en) | Triazole derivatives as ligands for Gaba receptors | |
| US4562187A (en) | (Isoxazol-3-yl)arylmethanones, compositions and pharmaceutical use | |
| JPH11509532A (en) | 1,6-disubstituted isochroman for the treatment of migraine | |
| KR20200074128A (en) | Method for producing benzothiophen-2-yl boronate | |
| JP2023545677A (en) | FXR small molecule agonists and their preparation methods and uses | |
| ES2342222T3 (en) | FXR AGONISTS. | |
| JPH0725854A (en) | Condensed benzeneoxyacetic acid derivative | |
| CN112424173A (en) | Isoxazoles as FXR receptor agonists | |
| HU211690A9 (en) | Phenoxy- and phenoxyalkyl-piperidines as antiviral agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880024765.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08781651 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12600879 Country of ref document: US Ref document number: 2009111697 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 581356 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008276236 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2252/MUMNP/2009 Country of ref document: IN Ref document number: 2008781651 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2008276236 Country of ref document: AU Date of ref document: 20080711 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2009000763 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010517082 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10002335 Country of ref document: CO Ref document number: MX/A/2010/000502 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2693406 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20107000896 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12010500108 Country of ref document: PH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201070148 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201000324 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PI 2010000156 Country of ref document: MY |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2012/0099 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: PI0814571 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100115 |