WO2009010871A2 - Pyrazole derivatives as antagonists of adenosine a3 receptor - Google Patents
Pyrazole derivatives as antagonists of adenosine a3 receptor Download PDFInfo
- Publication number
- WO2009010871A2 WO2009010871A2 PCT/IB2008/002243 IB2008002243W WO2009010871A2 WO 2009010871 A2 WO2009010871 A2 WO 2009010871A2 IB 2008002243 W IB2008002243 W IB 2008002243W WO 2009010871 A2 WO2009010871 A2 WO 2009010871A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- optionally substituted
- cycloalkyl
- pyrazol
- Prior art date
Links
- 239000005557 antagonist Substances 0.000 title claims abstract description 39
- 102000008161 Adenosine A3 Receptor Human genes 0.000 title 1
- 108010060261 Adenosine A3 Receptor Proteins 0.000 title 1
- 150000003217 pyrazoles Chemical class 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 170
- 239000003814 drug Substances 0.000 claims abstract description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 11
- 230000002265 prevention Effects 0.000 claims abstract description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 7
- 201000010099 disease Diseases 0.000 claims abstract description 5
- 238000004519 manufacturing process Methods 0.000 claims abstract description 4
- 125000000623 heterocyclic group Chemical group 0.000 claims description 162
- 238000000034 method Methods 0.000 claims description 134
- 125000001072 heteroaryl group Chemical group 0.000 claims description 123
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 120
- 239000001257 hydrogen Substances 0.000 claims description 117
- 229910052739 hydrogen Inorganic materials 0.000 claims description 117
- 125000003118 aryl group Chemical group 0.000 claims description 113
- -1 imidazopyridazinyl ring Chemical group 0.000 claims description 108
- 125000000217 alkyl group Chemical group 0.000 claims description 69
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 60
- 229960005305 adenosine Drugs 0.000 claims description 60
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 56
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 54
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 53
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 claims description 52
- 239000000203 mixture Substances 0.000 claims description 52
- 241000282414 Homo sapiens Species 0.000 claims description 43
- 229910052736 halogen Inorganic materials 0.000 claims description 40
- 150000002367 halogens Chemical class 0.000 claims description 40
- 150000002431 hydrogen Chemical class 0.000 claims description 39
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 35
- 125000002837 carbocyclic group Chemical group 0.000 claims description 35
- 229910052799 carbon Inorganic materials 0.000 claims description 32
- 125000005213 alkyl heteroaryl group Chemical group 0.000 claims description 28
- 125000003107 substituted aryl group Chemical group 0.000 claims description 26
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 25
- 125000004076 pyridyl group Chemical group 0.000 claims description 25
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 24
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 23
- 150000003839 salts Chemical class 0.000 claims description 23
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 21
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 19
- 239000002253 acid Substances 0.000 claims description 19
- 230000000694 effects Effects 0.000 claims description 19
- 125000002883 imidazolyl group Chemical group 0.000 claims description 19
- 125000001425 triazolyl group Chemical group 0.000 claims description 19
- 229910052757 nitrogen Inorganic materials 0.000 claims description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 16
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 claims description 13
- 208000006673 asthma Diseases 0.000 claims description 13
- 125000000590 4-methylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 claims description 11
- 229910052760 oxygen Inorganic materials 0.000 claims description 11
- 206010028980 Neoplasm Diseases 0.000 claims description 10
- 229910052717 sulfur Inorganic materials 0.000 claims description 10
- 208000010412 Glaucoma Diseases 0.000 claims description 9
- 230000000302 ischemic effect Effects 0.000 claims description 9
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 8
- 210000002268 wool Anatomy 0.000 claims description 8
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 7
- 208000028867 ischemia Diseases 0.000 claims description 7
- VJHTZTZXOKVQRN-UHFFFAOYSA-N 1,2,4-thiadiazol-5-amine Chemical compound NC1=NC=NS1 VJHTZTZXOKVQRN-UHFFFAOYSA-N 0.000 claims description 6
- 230000004054 inflammatory process Effects 0.000 claims description 6
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 5
- 206010061218 Inflammation Diseases 0.000 claims description 5
- 241000124008 Mammalia Species 0.000 claims description 5
- 239000002580 adenosine A3 receptor antagonist Substances 0.000 claims description 5
- 208000035475 disorder Diseases 0.000 claims description 5
- 208000031225 myocardial ischemia Diseases 0.000 claims description 5
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 5
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 4
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims description 4
- 208000019901 Anxiety disease Diseases 0.000 claims description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 4
- 208000015114 central nervous system disease Diseases 0.000 claims description 4
- 230000002757 inflammatory effect Effects 0.000 claims description 4
- 230000004770 neurodegeneration Effects 0.000 claims description 4
- 235000005152 nicotinamide Nutrition 0.000 claims description 4
- 239000011570 nicotinamide Substances 0.000 claims description 4
- 229960003966 nicotinamide Drugs 0.000 claims description 4
- 239000001301 oxygen Substances 0.000 claims description 4
- 208000019022 Mood disease Diseases 0.000 claims description 3
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 claims description 3
- 206010030043 Ocular hypertension Diseases 0.000 claims description 3
- 150000001721 carbon Chemical group 0.000 claims description 3
- 230000000973 chemotherapeutic effect Effects 0.000 claims description 3
- 210000003979 eosinophil Anatomy 0.000 claims description 3
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 3
- 230000003287 optical effect Effects 0.000 claims description 3
- 208000028173 post-traumatic stress disease Diseases 0.000 claims description 3
- PVGHNTXQMCYYGF-UHFFFAOYSA-N thiadiazol-5-amine Chemical compound NC1=CN=NS1 PVGHNTXQMCYYGF-UHFFFAOYSA-N 0.000 claims description 3
- KYSBRECADCKEHL-UHFFFAOYSA-N 3-(3-methyl-1-propylpyrazol-4-yl)-n-(6-morpholin-4-ylpyridin-2-yl)-1,2,4-thiadiazol-5-amine Chemical compound CC1=NN(CCC)C=C1C1=NSC(NC=2N=C(C=CC=2)N2CCOCC2)=N1 KYSBRECADCKEHL-UHFFFAOYSA-N 0.000 claims description 2
- HGCWHLIVTQZQNF-UHFFFAOYSA-N 3-[1-(cyclopropylmethyl)pyrazol-4-yl]-n-pyrazin-2-yl-1,2,4-thiadiazol-5-amine Chemical compound C1=C(C=2N=C(NC=3N=CC=NC=3)SN=2)C=NN1CC1CC1 HGCWHLIVTQZQNF-UHFFFAOYSA-N 0.000 claims description 2
- BRAMQQVWNGOEIR-UHFFFAOYSA-N 4-(1-methylpyrazol-4-yl)-n-pyridin-2-yl-1,3-thiazol-2-amine Chemical compound C1=NN(C)C=C1C1=CSC(NC=2N=CC=CC=2)=N1 BRAMQQVWNGOEIR-UHFFFAOYSA-N 0.000 claims description 2
- IIUXKPGDUNEOES-UHFFFAOYSA-N 4-(1-propylpyrazol-4-yl)-n-pyridin-2-yl-1,3-thiazol-2-amine Chemical compound C1=NN(CCC)C=C1C1=CSC(NC=2N=CC=CC=2)=N1 IIUXKPGDUNEOES-UHFFFAOYSA-N 0.000 claims description 2
- 208000000103 Anorexia Nervosa Diseases 0.000 claims description 2
- 208000006096 Attention Deficit Disorder with Hyperactivity Diseases 0.000 claims description 2
- 208000036864 Attention deficit/hyperactivity disease Diseases 0.000 claims description 2
- 206010003805 Autism Diseases 0.000 claims description 2
- 208000020706 Autistic disease Diseases 0.000 claims description 2
- 206010006550 Bulimia nervosa Diseases 0.000 claims description 2
- OCNDPHQZSWOQFG-UHFFFAOYSA-N CC1=NN(C=C1C=1N=C(SC1)NC1=NC=CC=C1)CCC.FC1=C(C=CC=C1)NC=1SC=C(N1)C=1C=NN(C1)CCC Chemical compound CC1=NN(C=C1C=1N=C(SC1)NC1=NC=CC=C1)CCC.FC1=C(C=CC=C1)NC=1SC=C(N1)C=1C=NN(C1)CCC OCNDPHQZSWOQFG-UHFFFAOYSA-N 0.000 claims description 2
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 claims description 2
- 208000032131 Diabetic Neuropathies Diseases 0.000 claims description 2
- 208000019695 Migraine disease Diseases 0.000 claims description 2
- VZICTMSBZZTYHV-UHFFFAOYSA-N N-(5-chloropyridin-2-yl)-4-(3-methyl-1-propylpyrazol-4-yl)-1,3-thiazol-2-amine 4-(3-methyl-1-propylpyrazol-4-yl)-N-(6-methylpyridin-2-yl)-1,3-thiazol-2-amine Chemical compound ClC=1C=CC(=NC1)NC=1SC=C(N1)C=1C(=NN(C1)CCC)C.CC1=NN(C=C1C=1N=C(SC1)NC1=NC(=CC=C1)C)CCC VZICTMSBZZTYHV-UHFFFAOYSA-N 0.000 claims description 2
- 208000008589 Obesity Diseases 0.000 claims description 2
- 208000001647 Renal Insufficiency Diseases 0.000 claims description 2
- 208000000323 Tourette Syndrome Diseases 0.000 claims description 2
- 208000016620 Tourette disease Diseases 0.000 claims description 2
- 206010003246 arthritis Diseases 0.000 claims description 2
- 208000030963 borderline personality disease Diseases 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 208000019622 heart disease Diseases 0.000 claims description 2
- 230000008595 infiltration Effects 0.000 claims description 2
- 238000001764 infiltration Methods 0.000 claims description 2
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 2
- 201000006370 kidney failure Diseases 0.000 claims description 2
- 206010027599 migraine Diseases 0.000 claims description 2
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 claims description 2
- YHORQGSODLJLKL-UHFFFAOYSA-N n-(2-methoxypyridin-3-yl)-3-[1-propyl-3-(trifluoromethyl)pyrazol-4-yl]-1,2,4-thiadiazol-5-amine Chemical compound FC(F)(F)C1=NN(CCC)C=C1C1=NSC(NC=2C(=NC=CC=2)OC)=N1 YHORQGSODLJLKL-UHFFFAOYSA-N 0.000 claims description 2
- BEXJXDPVIRHSJB-UHFFFAOYSA-N n-(6-cyclobutyloxypyridin-2-yl)-3-[1-(cyclopropylmethyl)pyrazol-4-yl]-1,2,4-thiadiazol-5-amine Chemical compound C1=C(C=2N=C(NC=3N=C(OC4CCC4)C=CC=3)SN=2)C=NN1CC1CC1 BEXJXDPVIRHSJB-UHFFFAOYSA-N 0.000 claims description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 2
- 235000020824 obesity Nutrition 0.000 claims description 2
- 208000019906 panic disease Diseases 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 208000019899 phobic disease Diseases 0.000 claims description 2
- 208000023504 respiratory system disease Diseases 0.000 claims description 2
- 208000012201 sexual and gender identity disease Diseases 0.000 claims description 2
- 208000015891 sexual disease Diseases 0.000 claims description 2
- 208000019116 sleep disease Diseases 0.000 claims description 2
- 125000004434 sulfur atom Chemical group 0.000 claims description 2
- 208000002381 Brain Hypoxia Diseases 0.000 claims 2
- 206010008111 Cerebral haemorrhage Diseases 0.000 claims 2
- 206010014950 Eosinophilia Diseases 0.000 claims 2
- 208000011688 Generalised anxiety disease Diseases 0.000 claims 2
- 208000016988 Hemorrhagic Stroke Diseases 0.000 claims 2
- 206010059491 Intracranial haematoma Diseases 0.000 claims 2
- 208000006011 Stroke Diseases 0.000 claims 2
- 208000030886 Traumatic Brain injury Diseases 0.000 claims 2
- 208000029364 generalized anxiety disease Diseases 0.000 claims 2
- 208000020658 intracerebral hemorrhage Diseases 0.000 claims 2
- 208000020431 spinal cord injury Diseases 0.000 claims 2
- 230000009529 traumatic brain injury Effects 0.000 claims 2
- BSAXWMSAVVOOHO-UHFFFAOYSA-N 2,2-dioxo-1,5-dihydroimidazo[4,5-c][1,2,6]thiadiazin-4-one Chemical compound O=C1NS(=O)(=O)NC2=C1NC=N2 BSAXWMSAVVOOHO-UHFFFAOYSA-N 0.000 claims 1
- CIMHHWUUPKDZAV-UHFFFAOYSA-N 3-(1-ethylpyrazol-4-yl)-n-(6-morpholin-4-ylpyridin-2-yl)-1,2,4-thiadiazol-5-amine Chemical compound C1=NN(CC)C=C1C1=NSC(NC=2N=C(C=CC=2)N2CCOCC2)=N1 CIMHHWUUPKDZAV-UHFFFAOYSA-N 0.000 claims 1
- MLTFMMQBMRXJEY-UHFFFAOYSA-N 3-[1-(2-morpholin-4-ylethyl)pyrazol-4-yl]-n-pyridin-2-yl-1,2,4-thiadiazol-5-amine Chemical compound C1COCCN1CCN(N=C1)C=C1C(N=1)=NSC=1NC1=CC=CC=N1 MLTFMMQBMRXJEY-UHFFFAOYSA-N 0.000 claims 1
- YJPVSEACXYIGGD-UHFFFAOYSA-N 3-[1-(cyclopropylmethyl)-3,5-dimethylpyrazol-4-yl]-n-(6-methylpyridin-2-yl)-1,2,4-thiadiazol-5-amine Chemical compound CC1=C(C=2N=C(NC=3N=C(C)C=CC=3)SN=2)C(C)=NN1CC1CC1 YJPVSEACXYIGGD-UHFFFAOYSA-N 0.000 claims 1
- XEHGINAJEBNDNG-UHFFFAOYSA-N 4-[1-propyl-3-(trifluoromethyl)pyrazol-4-yl]-n-pyridin-2-yl-1,3-thiazol-2-amine Chemical compound FC(F)(F)C1=NN(CCC)C=C1C1=CSC(NC=2N=CC=CC=2)=N1 XEHGINAJEBNDNG-UHFFFAOYSA-N 0.000 claims 1
- 206010056508 Acquired epidermolysis bullosa Diseases 0.000 claims 1
- 208000008811 Agoraphobia Diseases 0.000 claims 1
- 206010027654 Allergic conditions Diseases 0.000 claims 1
- 208000033116 Asbestos intoxication Diseases 0.000 claims 1
- 206010003645 Atopy Diseases 0.000 claims 1
- 208000035143 Bacterial infection Diseases 0.000 claims 1
- 208000020925 Bipolar disease Diseases 0.000 claims 1
- 208000032841 Bulimia Diseases 0.000 claims 1
- 208000007596 Byssinosis Diseases 0.000 claims 1
- STGNPKYDXDTPQB-UHFFFAOYSA-N C(C)N1N=CC(=C1)C=1N=C(SC1)NC1=NC=CC=C1.C(C)(C)N1N=CC(=C1)C=1N=C(SC1)NC1=NC=CC=C1.C(C1=CC=CC=C1)N1N=CC(=C1)C=1N=C(SC1)NC1=CC=CC=C1 Chemical compound C(C)N1N=CC(=C1)C=1N=C(SC1)NC1=NC=CC=C1.C(C)(C)N1N=CC(=C1)C=1N=C(SC1)NC1=NC=CC=C1.C(C1=CC=CC=C1)N1N=CC(=C1)C=1N=C(SC1)NC1=CC=CC=C1 STGNPKYDXDTPQB-UHFFFAOYSA-N 0.000 claims 1
- QLFKWZOSUFLSAE-UHFFFAOYSA-N COC1=CC=C(CN2N=CC(=C2C)C=2N=C(SC2)NC2=CC=CC=C2)C=C1.COC1=CC=C(CN2N=C(C(=C2)C=2N=C(SC2)NC2=CC=CC=C2)C)C=C1 Chemical compound COC1=CC=C(CN2N=CC(=C2C)C=2N=C(SC2)NC2=CC=CC=C2)C=C1.COC1=CC=C(CN2N=C(C(=C2)C=2N=C(SC2)NC2=CC=CC=C2)C)C=C1 QLFKWZOSUFLSAE-UHFFFAOYSA-N 0.000 claims 1
- 206010010741 Conjunctivitis Diseases 0.000 claims 1
- 206010010744 Conjunctivitis allergic Diseases 0.000 claims 1
- 206010012335 Dependence Diseases 0.000 claims 1
- 206010012442 Dermatitis contact Diseases 0.000 claims 1
- 208000003556 Dry Eye Syndromes Diseases 0.000 claims 1
- 206010013774 Dry eye Diseases 0.000 claims 1
- 208000030814 Eating disease Diseases 0.000 claims 1
- 206010064212 Eosinophilic oesophagitis Diseases 0.000 claims 1
- 206010015150 Erythema Diseases 0.000 claims 1
- 208000019454 Feeding and Eating disease Diseases 0.000 claims 1
- 206010019663 Hepatic failure Diseases 0.000 claims 1
- 208000009319 Keratoconjunctivitis Sicca Diseases 0.000 claims 1
- 208000009668 Neurobehavioral Manifestations Diseases 0.000 claims 1
- 206010067013 Normal tension glaucoma Diseases 0.000 claims 1
- 208000022873 Ocular disease Diseases 0.000 claims 1
- 201000004681 Psoriasis Diseases 0.000 claims 1
- 208000032430 Retinal dystrophy Diseases 0.000 claims 1
- 206010039710 Scleroderma Diseases 0.000 claims 1
- 201000010001 Silicosis Diseases 0.000 claims 1
- 206010041250 Social phobia Diseases 0.000 claims 1
- 208000011962 Substance-induced mood disease Diseases 0.000 claims 1
- 231100000395 Substance-induced mood disorder Toxicity 0.000 claims 1
- 208000024780 Urticaria Diseases 0.000 claims 1
- 206010064930 age-related macular degeneration Diseases 0.000 claims 1
- 208000037883 airway inflammation Diseases 0.000 claims 1
- 201000009961 allergic asthma Diseases 0.000 claims 1
- 208000002205 allergic conjunctivitis Diseases 0.000 claims 1
- 208000004631 alopecia areata Diseases 0.000 claims 1
- 208000028462 aluminosis Diseases 0.000 claims 1
- 206010003441 asbestosis Diseases 0.000 claims 1
- 208000022362 bacterial infectious disease Diseases 0.000 claims 1
- 206010006451 bronchitis Diseases 0.000 claims 1
- 239000003183 carcinogenic agent Substances 0.000 claims 1
- 208000010247 contact dermatitis Diseases 0.000 claims 1
- 208000026725 cyclothymic disease Diseases 0.000 claims 1
- 238000007598 dipping method Methods 0.000 claims 1
- 235000014632 disordered eating Nutrition 0.000 claims 1
- 208000024732 dysthymic disease Diseases 0.000 claims 1
- 230000002708 enhancing effect Effects 0.000 claims 1
- 201000011114 epidermolysis bullosa acquisita Diseases 0.000 claims 1
- 231100000321 erythema Toxicity 0.000 claims 1
- 238000003384 imaging method Methods 0.000 claims 1
- 201000010659 intrinsic asthma Diseases 0.000 claims 1
- 208000007903 liver failure Diseases 0.000 claims 1
- 231100000835 liver failure Toxicity 0.000 claims 1
- 201000002978 low tension glaucoma Diseases 0.000 claims 1
- 206010025135 lupus erythematosus Diseases 0.000 claims 1
- 208000002780 macular degeneration Diseases 0.000 claims 1
- 208000024714 major depressive disease Diseases 0.000 claims 1
- 208000010125 myocardial infarction Diseases 0.000 claims 1
- WOMSKBWRPCXIFM-UHFFFAOYSA-N n-(6-methoxypyridin-3-yl)-4-(3-methyl-1-propylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=NN(CCC)C=C1C1=CSC(NC=2C=NC(OC)=CC=2)=N1 WOMSKBWRPCXIFM-UHFFFAOYSA-N 0.000 claims 1
- 230000000414 obstructive effect Effects 0.000 claims 1
- 208000007892 occupational asthma Diseases 0.000 claims 1
- 210000001328 optic nerve Anatomy 0.000 claims 1
- 230000003071 parasitic effect Effects 0.000 claims 1
- 208000022821 personality disease Diseases 0.000 claims 1
- 239000000700 radioactive tracer Substances 0.000 claims 1
- 210000001525 retina Anatomy 0.000 claims 1
- 230000001953 sensory effect Effects 0.000 claims 1
- 208000004003 siderosis Diseases 0.000 claims 1
- 201000005539 vernal conjunctivitis Diseases 0.000 claims 1
- 102000005962 receptors Human genes 0.000 abstract description 70
- 108020003175 receptors Proteins 0.000 abstract description 70
- 102000009346 Adenosine receptors Human genes 0.000 abstract description 25
- 108050000203 Adenosine receptors Proteins 0.000 abstract description 25
- 150000003254 radicals Chemical class 0.000 description 130
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 81
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 60
- 239000011541 reaction mixture Substances 0.000 description 57
- 239000000243 solution Substances 0.000 description 51
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 43
- 210000004027 cell Anatomy 0.000 description 34
- 239000002904 solvent Substances 0.000 description 32
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 31
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 30
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 29
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 27
- 239000007787 solid Substances 0.000 description 27
- 239000012043 crude product Substances 0.000 description 26
- 239000000741 silica gel Substances 0.000 description 23
- 229910002027 silica gel Inorganic materials 0.000 description 23
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 239000012074 organic phase Substances 0.000 description 20
- 238000001704 evaporation Methods 0.000 description 19
- 230000008020 evaporation Effects 0.000 description 19
- 230000002829 reductive effect Effects 0.000 description 19
- 239000007832 Na2SO4 Substances 0.000 description 18
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 18
- 239000012267 brine Substances 0.000 description 18
- 229910052938 sodium sulfate Inorganic materials 0.000 description 18
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 18
- 239000003480 eluent Substances 0.000 description 17
- 238000003818 flash chromatography Methods 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 239000012071 phase Substances 0.000 description 15
- 238000005160 1H NMR spectroscopy Methods 0.000 description 14
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 14
- 239000000556 agonist Substances 0.000 description 14
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 14
- 238000009472 formulation Methods 0.000 description 14
- 230000004913 activation Effects 0.000 description 13
- 210000002216 heart Anatomy 0.000 description 13
- 238000000746 purification Methods 0.000 description 13
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 12
- 230000005764 inhibitory process Effects 0.000 description 12
- 230000004410 intraocular pressure Effects 0.000 description 12
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 12
- 241000699670 Mus sp. Species 0.000 description 11
- 239000004480 active ingredient Substances 0.000 description 11
- 239000008346 aqueous phase Substances 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- 239000003643 water by type Substances 0.000 description 11
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 230000001886 ciliary effect Effects 0.000 description 9
- 238000011156 evaluation Methods 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 210000001519 tissue Anatomy 0.000 description 9
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 239000003208 petroleum Substances 0.000 description 8
- 238000003653 radioligand binding assay Methods 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 7
- 235000019270 ammonium chloride Nutrition 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000012299 nitrogen atmosphere Substances 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- 150000001409 amidines Chemical class 0.000 description 6
- 125000004122 cyclic group Chemical group 0.000 description 6
- 230000007423 decrease Effects 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 230000004224 protection Effects 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 238000004007 reversed phase HPLC Methods 0.000 description 6
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- HUJXGQILHAUCCV-MOROJQBDSA-N 3-iodobenzyl-5'-N-methylcarboxamidoadenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NC)O[C@H]1N1C2=NC=NC(NCC=3C=C(I)C=CC=3)=C2N=C1 HUJXGQILHAUCCV-MOROJQBDSA-N 0.000 description 5
- 0 CC(C)C(CC*1[C@]2*3(C)*CC2)*(*2CC2)C13N=C Chemical compound CC(C)C(CC*1[C@]2*3(C)*CC2)*(*2CC2)C13N=C 0.000 description 5
- 208000026935 allergic disease Diseases 0.000 description 5
- 230000006907 apoptotic process Effects 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 201000011510 cancer Diseases 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 230000000875 corresponding effect Effects 0.000 description 5
- 210000002919 epithelial cell Anatomy 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000007789 gas Substances 0.000 description 5
- 210000003630 histaminocyte Anatomy 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 230000000144 pharmacologic effect Effects 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 239000000523 sample Substances 0.000 description 5
- 238000010561 standard procedure Methods 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- QKSQWQOAUQFORH-UHFFFAOYSA-N tert-butyl n-[(2-methylpropan-2-yl)oxycarbonylimino]carbamate Chemical compound CC(C)(C)OC(=O)N=NC(=O)OC(C)(C)C QKSQWQOAUQFORH-UHFFFAOYSA-N 0.000 description 5
- 150000003557 thiazoles Chemical class 0.000 description 5
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 5
- IPSYPUKKXMNCNQ-PFHKOEEOSA-N (2s,3s,4r,5r)-5-[2-chloro-6-[(3-iodophenyl)methylamino]purin-9-yl]-3,4-dihydroxy-n-methyloxolane-2-carboxamide Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NC)O[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(I)C=CC=3)=C2N=C1 IPSYPUKKXMNCNQ-PFHKOEEOSA-N 0.000 description 4
- TVOHAUVCHFSBEO-UHFFFAOYSA-N 2-chloro-1-[1-(2,2,2-trifluoroethyl)pyrazol-4-yl]ethanone Chemical compound FC(F)(F)CN1C=C(C(=O)CCl)C=N1 TVOHAUVCHFSBEO-UHFFFAOYSA-N 0.000 description 4
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 4
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- 206010020751 Hypersensitivity Diseases 0.000 description 4
- JADDQZYHOWSFJD-FLNNQWSLSA-N N-ethyl-5'-carboxamidoadenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 JADDQZYHOWSFJD-FLNNQWSLSA-N 0.000 description 4
- 102100028874 Sodium-dependent serotonin transporter Human genes 0.000 description 4
- 230000007815 allergy Effects 0.000 description 4
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 210000001742 aqueous humor Anatomy 0.000 description 4
- 230000010261 cell growth Effects 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000006196 drop Substances 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 230000014509 gene expression Effects 0.000 description 4
- 239000003365 glass fiber Substances 0.000 description 4
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 4
- 150000002540 isothiocyanates Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 229910001629 magnesium chloride Inorganic materials 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 230000009871 nonspecific binding Effects 0.000 description 4
- 230000036542 oxidative stress Effects 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 125000003226 pyrazolyl group Chemical group 0.000 description 4
- 125000000168 pyrrolyl group Chemical group 0.000 description 4
- 239000002287 radioligand Substances 0.000 description 4
- 239000000018 receptor agonist Substances 0.000 description 4
- 229940044601 receptor agonist Drugs 0.000 description 4
- 229940044551 receptor antagonist Drugs 0.000 description 4
- 239000002464 receptor antagonist Substances 0.000 description 4
- 238000007363 ring formation reaction Methods 0.000 description 4
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 4
- 230000009870 specific binding Effects 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 4
- 230000000699 topical effect Effects 0.000 description 4
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 4
- 230000003827 upregulation Effects 0.000 description 4
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- TYFASWZBYOQUGO-UHFFFAOYSA-N 4-(1-propan-2-ylpyrazol-4-yl)-n-pyridin-2-yl-1,3-thiazol-2-amine Chemical compound C1=NN(C(C)C)C=C1C1=CSC(NC=2N=CC=CC=2)=N1 TYFASWZBYOQUGO-UHFFFAOYSA-N 0.000 description 3
- JRDTYLLRMGJYJV-UHFFFAOYSA-N 5-methyl-1-phenylpyrazole-4-carboximidamide Chemical compound CC1=C(C(N)=N)C=NN1C1=CC=CC=C1 JRDTYLLRMGJYJV-UHFFFAOYSA-N 0.000 description 3
- ACYSKLGOADDKAI-UHFFFAOYSA-N 5-morpholin-4-ylpyridin-2-amine Chemical compound C1=NC(N)=CC=C1N1CCOCC1 ACYSKLGOADDKAI-UHFFFAOYSA-N 0.000 description 3
- LEZFYYWHNXICNC-UHFFFAOYSA-N 6-morpholin-4-ylpyridin-2-amine Chemical compound NC1=CC=CC(N2CCOCC2)=N1 LEZFYYWHNXICNC-UHFFFAOYSA-N 0.000 description 3
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 3
- 102100036664 Adenosine deaminase Human genes 0.000 description 3
- 108091006101 Gi proteins Proteins 0.000 description 3
- 102000034354 Gi proteins Human genes 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 206010021143 Hypoxia Diseases 0.000 description 3
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 3
- 101710114597 Sodium-dependent serotonin transporter Proteins 0.000 description 3
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 3
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000008485 antagonism Effects 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 210000000981 epithelium Anatomy 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 125000002541 furyl group Chemical group 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 230000007954 hypoxia Effects 0.000 description 3
- 230000001976 improved effect Effects 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 208000037906 ischaemic injury Diseases 0.000 description 3
- 125000001786 isothiazolyl group Chemical group 0.000 description 3
- 125000000842 isoxazolyl group Chemical group 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 238000012417 linear regression Methods 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000004112 neuroprotection Effects 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 229940054534 ophthalmic solution Drugs 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 230000002018 overexpression Effects 0.000 description 3
- 125000001715 oxadiazolyl group Chemical group 0.000 description 3
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 3
- 125000002971 oxazolyl group Chemical group 0.000 description 3
- 230000007170 pathology Effects 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 230000002633 protecting effect Effects 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- 125000002098 pyridazinyl group Chemical group 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 239000012047 saturated solution Substances 0.000 description 3
- 230000028327 secretion Effects 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 208000011580 syndromic disease Diseases 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 125000003831 tetrazolyl group Chemical group 0.000 description 3
- 125000001113 thiadiazolyl group Chemical group 0.000 description 3
- 125000000335 thiazolyl group Chemical group 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- ZXAQFYZQHPGMMN-BZSJEYESSA-N (3R)-3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-phenylcyclohexane-1-carboxamide Chemical compound C1C[C@H](CC(C1)C(=O)NC2=CC=CC=C2)OC3=CC(=CC(=N3)C(F)(F)F)CN ZXAQFYZQHPGMMN-BZSJEYESSA-N 0.000 description 2
- YNGDWRXWKFWCJY-UHFFFAOYSA-N 1,4-Dihydropyridine Chemical class C1C=CNC=C1 YNGDWRXWKFWCJY-UHFFFAOYSA-N 0.000 description 2
- YMTASZPNDJAOKN-UHFFFAOYSA-N 1-(2,2,2-trifluoroethyl)pyrazole-4-carbonyl chloride Chemical compound FC(F)(F)CN1C=C(C(Cl)=O)C=N1 YMTASZPNDJAOKN-UHFFFAOYSA-N 0.000 description 2
- SFDOGOGLQYZMTP-UHFFFAOYSA-N 1-ethyl-3-methylpyrazole-4-carboximidamide;hydrochloride Chemical compound Cl.CCN1C=C(C(N)=N)C(C)=N1 SFDOGOGLQYZMTP-UHFFFAOYSA-N 0.000 description 2
- VLQMAVVBCYNBQC-UHFFFAOYSA-N 1-propylpyrazole-4-carboxylic acid Chemical compound CCCN1C=C(C(O)=O)C=N1 VLQMAVVBCYNBQC-UHFFFAOYSA-N 0.000 description 2
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 2
- QMPLSDOUROSVQS-UHFFFAOYSA-N 2-bromo-1-(1-propan-2-ylpyrazol-4-yl)ethanone Chemical compound CC(C)N1C=C(C(=O)CBr)C=N1 QMPLSDOUROSVQS-UHFFFAOYSA-N 0.000 description 2
- KCTSDEASOUHSGB-UHFFFAOYSA-N 2-ethyl-6-isothiocyanatopyridine Chemical compound CCC1=CC=CC(N=C=S)=N1 KCTSDEASOUHSGB-UHFFFAOYSA-N 0.000 description 2
- WFJPRCMYUOOTNS-UHFFFAOYSA-N 2-isothiocyanatopyridine Chemical compound S=C=NC1=CC=CC=N1 WFJPRCMYUOOTNS-UHFFFAOYSA-N 0.000 description 2
- SONNQRNOTIAJDS-GFCCVEGCSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-[(2R)-2,3-dihydroxypropyl]benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NC[C@H](CO)O)C=CC=1 SONNQRNOTIAJDS-GFCCVEGCSA-N 0.000 description 2
- GZPHSAQLYPIAIN-UHFFFAOYSA-N 3-pyridinecarbonitrile Chemical compound N#CC1=CC=CN=C1 GZPHSAQLYPIAIN-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- JAJUAAWSTSRHAD-UHFFFAOYSA-N 4-(6-nitropyridin-3-yl)morpholine Chemical compound C1=NC([N+](=O)[O-])=CC=C1N1CCOCC1 JAJUAAWSTSRHAD-UHFFFAOYSA-N 0.000 description 2
- MSHFRERJPWKJFX-UHFFFAOYSA-N 4-Methoxybenzyl alcohol Chemical compound COC1=CC=C(CO)C=C1 MSHFRERJPWKJFX-UHFFFAOYSA-N 0.000 description 2
- QEIVWSRXBYOTAZ-UHFFFAOYSA-N 4-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-phenylpiperidine-1-carboxamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC1CCN(CC1)C(=O)NC1=CC=CC=C1 QEIVWSRXBYOTAZ-UHFFFAOYSA-N 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- BKLJUYPLUWUEOQ-UHFFFAOYSA-N 6-bromopyridin-2-amine Chemical compound NC1=CC=CC(Br)=N1 BKLJUYPLUWUEOQ-UHFFFAOYSA-N 0.000 description 2
- SXMORUZBQAJDEL-UHFFFAOYSA-N 6-cyclobutyloxypyridin-2-amine Chemical compound NC1=CC=CC(OC2CCC2)=N1 SXMORUZBQAJDEL-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical class NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 108010062745 Chloride Channels Proteins 0.000 description 2
- 102000011045 Chloride Channels Human genes 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FFBDFADSZUINTG-UHFFFAOYSA-N DPCPX Chemical compound N1C=2C(=O)N(CCC)C(=O)N(CCC)C=2N=C1C1CCCC1 FFBDFADSZUINTG-UHFFFAOYSA-N 0.000 description 2
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 208000001953 Hypotension Diseases 0.000 description 2
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 2
- 229930010555 Inosine Natural products 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 102000043136 MAP kinase family Human genes 0.000 description 2
- 108091054455 MAP kinase family Proteins 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 108010012996 Serotonin Plasma Membrane Transport Proteins Proteins 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 102000014384 Type C Phospholipases Human genes 0.000 description 2
- 108010079194 Type C Phospholipases Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000013521 Visual disease Diseases 0.000 description 2
- BYWBCSRCPLBDFU-CYBMUJFWSA-N [3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxyphenyl]-[(3R)-3-aminopyrrolidin-1-yl]methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=CC=1)C(=O)N1C[C@@H](CC1)N BYWBCSRCPLBDFU-CYBMUJFWSA-N 0.000 description 2
- LJHFUFVRZNYVMK-ZDUSSCGKSA-N [3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxyphenyl]-[(3S)-3-hydroxypyrrolidin-1-yl]methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=CC=1)C(=O)N1C[C@H](CC1)O LJHFUFVRZNYVMK-ZDUSSCGKSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- 229940121359 adenosine receptor antagonist Drugs 0.000 description 2
- 150000003838 adenosines Chemical class 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- WXBLLCUINBKULX-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1.OC(=O)C1=CC=CC=C1 WXBLLCUINBKULX-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 230000017531 blood circulation Effects 0.000 description 2
- 230000006931 brain damage Effects 0.000 description 2
- 231100000874 brain damage Toxicity 0.000 description 2
- 208000029028 brain injury Diseases 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 2
- 230000001275 ca(2+)-mobilization Effects 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 150000001723 carbon free-radicals Chemical class 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 230000005779 cell damage Effects 0.000 description 2
- 208000037887 cell injury Diseases 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229940044683 chemotherapy drug Drugs 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 2
- 230000001120 cytoprotective effect Effects 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 125000005283 haloketone group Chemical group 0.000 description 2
- 210000002064 heart cell Anatomy 0.000 description 2
- 150000002391 heterocyclic compounds Chemical class 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 125000002632 imidazolidinyl group Chemical group 0.000 description 2
- 125000002636 imidazolinyl group Chemical group 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 229960003786 inosine Drugs 0.000 description 2
- 208000023589 ischemic disease Diseases 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 238000002483 medication Methods 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- MZNZAHMGUKUSBB-UHFFFAOYSA-N methyl 6-isothiocyanatopyridine-3-carboxylate Chemical compound COC(=O)C1=CC=C(N=C=S)N=C1 MZNZAHMGUKUSBB-UHFFFAOYSA-N 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- LCSFROYYNFLPTJ-UHFFFAOYSA-N n-pyridin-2-yl-4-[1-(2,2,2-trifluoroethyl)pyrazol-4-yl]-1,3-thiazol-2-amine Chemical compound C1=NN(CC(F)(F)F)C=C1C1=CSC(NC=2N=CC=CC=2)=N1 LCSFROYYNFLPTJ-UHFFFAOYSA-N 0.000 description 2
- 235000001968 nicotinic acid Nutrition 0.000 description 2
- 239000011664 nicotinic acid Substances 0.000 description 2
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 2
- 239000002997 ophthalmic solution Substances 0.000 description 2
- 150000002923 oximes Chemical class 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 2
- RYFZYYUIAZYQLC-UHFFFAOYSA-N perchloromethyl mercaptan Chemical compound ClSC(Cl)(Cl)Cl RYFZYYUIAZYQLC-UHFFFAOYSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000004928 piperidonyl group Chemical group 0.000 description 2
- 125000005936 piperidyl group Chemical group 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 239000000296 purinergic P1 receptor antagonist Substances 0.000 description 2
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 2
- 125000002755 pyrazolinyl group Chemical group 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 125000001422 pyrrolinyl group Chemical group 0.000 description 2
- 150000003246 quinazolines Chemical class 0.000 description 2
- 230000002207 retinal effect Effects 0.000 description 2
- 229940076279 serotonin Drugs 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- ZWZVWGITAAIFPS-UHFFFAOYSA-N thiophosgene Chemical compound ClC(Cl)=S ZWZVWGITAAIFPS-UHFFFAOYSA-N 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 208000029257 vision disease Diseases 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 229940075420 xanthine Drugs 0.000 description 2
- SDTRVGPIZUJPIE-JTQLQIEISA-N (2s)-2-amino-n-[(3-pyrazol-1-ylphenyl)methyl]propanamide Chemical class C[C@H](N)C(=O)NCC1=CC=CC(N2N=CC=C2)=C1 SDTRVGPIZUJPIE-JTQLQIEISA-N 0.000 description 1
- JEXZMSAMBNBAHM-ZDUSSCGKSA-N (2s)-2-amino-n-[[3-[5-(5-anilino-1,3,4-oxadiazol-2-yl)-3-(trifluoromethyl)pyrazol-1-yl]phenyl]methyl]propanamide Chemical compound C[C@H](N)C(=O)NCC1=CC=CC(N2C(=CC(=N2)C(F)(F)F)C=2OC(NC=3C=CC=CC=3)=NN=2)=C1 JEXZMSAMBNBAHM-ZDUSSCGKSA-N 0.000 description 1
- LOGOEBMHHXYBID-WBKNRDRNSA-N (2s,3s,4r,5r)-5-[6-[(4-amino-3-iodanylphenyl)methylamino]purin-9-yl]-3,4-dihydroxy-n-methyloxolane-2-carboxamide Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NC)O[C@H]1N1C2=NC=NC(NCC=3C=C([125I])C(N)=CC=3)=C2N=C1 LOGOEBMHHXYBID-WBKNRDRNSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- QUKGLNCXGVWCJX-UHFFFAOYSA-N 1,3,4-thiadiazol-2-amine Chemical compound NC1=NN=CS1 QUKGLNCXGVWCJX-UHFFFAOYSA-N 0.000 description 1
- VPAMEBUGVMETCV-UHFFFAOYSA-N 1-(2,2,2-trifluoroethyl)pyrazole-4-carboxylic acid Chemical compound OC(=O)C=1C=NN(CC(F)(F)F)C=1 VPAMEBUGVMETCV-UHFFFAOYSA-N 0.000 description 1
- BLBHNZSGKOYIRE-UHFFFAOYSA-N 1-(2-phenylmethoxyethyl)pyrazole-4-carboximidamide;hydrochloride Chemical compound Cl.C1=C(C(=N)N)C=NN1CCOCC1=CC=CC=C1 BLBHNZSGKOYIRE-UHFFFAOYSA-N 0.000 description 1
- PEULPBJQURLVEH-UHFFFAOYSA-N 1-[(4-methoxyphenyl)methyl]-3-methylpyrazole-4-carbonitrile Chemical compound C1=CC(OC)=CC=C1CN1N=C(C)C(C#N)=C1 PEULPBJQURLVEH-UHFFFAOYSA-N 0.000 description 1
- ZEIWXTWEIKKCFR-UHFFFAOYSA-N 1-[(4-methoxyphenyl)methyl]-3-methylpyrazole-4-carboximidamide Chemical compound C1=CC(OC)=CC=C1CN1N=C(C)C(C(N)=N)=C1 ZEIWXTWEIKKCFR-UHFFFAOYSA-N 0.000 description 1
- GELSZGAZSBNNLY-UHFFFAOYSA-N 1-[(4-methoxyphenyl)methyl]-5-methylpyrazole-4-carbonitrile Chemical compound C1=CC(OC)=CC=C1CN1C(C)=C(C#N)C=N1 GELSZGAZSBNNLY-UHFFFAOYSA-N 0.000 description 1
- KXRWYWQIRSZKRR-UHFFFAOYSA-N 1-[4-[5-(pyridin-2-ylamino)-1,2,4-thiadiazol-3-yl]pyrazol-1-yl]ethanol Chemical compound N1=C(C=CC=C1)NC1=NC(=NS1)C=1C=NN(C1)C(C)O KXRWYWQIRSZKRR-UHFFFAOYSA-N 0.000 description 1
- 125000004343 1-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C([H])([H])[H] 0.000 description 1
- UIXCWJIYHCTUEK-UHFFFAOYSA-N 1-propan-2-ylpyrazole-4-carbonyl chloride Chemical compound CC(C)N1C=C(C(Cl)=O)C=N1 UIXCWJIYHCTUEK-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- FRWUWOYTOIZGLT-UHFFFAOYSA-N 2-[4-[5-(pyridin-2-ylamino)-1,2,4-thiadiazol-3-yl]pyrazol-1-yl]ethanol Chemical compound C1=NN(CCO)C=C1C1=NSC(NC=2N=CC=CC=2)=N1 FRWUWOYTOIZGLT-UHFFFAOYSA-N 0.000 description 1
- DQHMUVAHWRHEDM-UHFFFAOYSA-N 2-bromo-1-[1-(2,2,2-trifluoroethyl)pyrazol-4-yl]ethanone Chemical compound FC(F)(F)CN1C=C(C(=O)CBr)C=N1 DQHMUVAHWRHEDM-UHFFFAOYSA-N 0.000 description 1
- FWOHDAGPWDEWIB-UHFFFAOYSA-N 2-bromoethoxymethylbenzene Chemical compound BrCCOCC1=CC=CC=C1 FWOHDAGPWDEWIB-UHFFFAOYSA-N 0.000 description 1
- STVKTHJPLIROOR-UHFFFAOYSA-N 2-isothiocyanato-6-methylpyridine Chemical compound CC1=CC=CC(N=C=S)=N1 STVKTHJPLIROOR-UHFFFAOYSA-N 0.000 description 1
- 125000004204 2-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000006479 2-pyridyl methyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- WOOIBJWFHSHPJN-UHFFFAOYSA-N 2h-triazolo[4,5-h]quinazoline Chemical compound C1=CC2=NNN=C2C2=NC=NC=C21 WOOIBJWFHSHPJN-UHFFFAOYSA-N 0.000 description 1
- SPINKLQLZIGOIS-UHFFFAOYSA-N 3-(1-ethyl-3-methylpyrazol-4-yl)-n-pyridin-2-yl-1,2,4-thiadiazol-5-amine Chemical compound CC1=NN(CC)C=C1C1=NSC(NC=2N=CC=CC=2)=N1 SPINKLQLZIGOIS-UHFFFAOYSA-N 0.000 description 1
- ZYAHTLKCOSUAEU-UHFFFAOYSA-N 3-(3,5-dimethyl-1-propylpyrazol-4-yl)-n-pyridin-2-yl-1,2,4-thiadiazol-5-amine Chemical compound CCCN1N=C(C)C(C=2N=C(NC=3N=CC=CC=3)SN=2)=C1C ZYAHTLKCOSUAEU-UHFFFAOYSA-N 0.000 description 1
- SRAYVDYKPZOEDB-UHFFFAOYSA-N 3-[1-(cyclopropylmethyl)pyrazol-4-yl]-n-(5-fluoropyridin-2-yl)-1,2,4-thiadiazol-5-amine Chemical compound N1=CC(F)=CC=C1NC1=NC(C2=CN(CC3CC3)N=C2)=NS1 SRAYVDYKPZOEDB-UHFFFAOYSA-N 0.000 description 1
- LGDQGHKDJVMPDB-UHFFFAOYSA-N 3-[1-(cyclopropylmethyl)pyrazol-4-yl]-n-(5-morpholin-4-ylpyridin-2-yl)-1,2,4-thiadiazol-5-amine Chemical compound C1=C(C=2N=C(NC=3N=CC(=CC=3)N3CCOCC3)SN=2)C=NN1CC1CC1 LGDQGHKDJVMPDB-UHFFFAOYSA-N 0.000 description 1
- FJVXDJBPNQCTPE-UHFFFAOYSA-N 3-[1-(cyclopropylmethyl)pyrazol-4-yl]-n-(6-methoxypyridin-2-yl)-1,2,4-thiadiazol-5-amine Chemical compound COC1=CC=CC(NC=2SN=C(N=2)C2=CN(CC3CC3)N=C2)=N1 FJVXDJBPNQCTPE-UHFFFAOYSA-N 0.000 description 1
- HSIXAZNSVOVYML-UHFFFAOYSA-N 3-[1-(cyclopropylmethyl)pyrazol-4-yl]-n-(6-methylpyridin-2-yl)-1,2,4-thiadiazol-5-amine Chemical compound CC1=CC=CC(NC=2SN=C(N=2)C2=CN(CC3CC3)N=C2)=N1 HSIXAZNSVOVYML-UHFFFAOYSA-N 0.000 description 1
- ORWLZBHASYGJOX-UHFFFAOYSA-N 3-[1-[(3-fluorophenyl)methyl]pyrazol-4-yl]-n-phenyl-1,2,4-thiadiazol-5-amine Chemical compound FC1=CC=CC(CN2N=CC(=C2)C=2N=C(NC=3C=CC=CC=3)SN=2)=C1 ORWLZBHASYGJOX-UHFFFAOYSA-N 0.000 description 1
- YNVUBNBCGRPWKB-UHFFFAOYSA-N 3-[1-propyl-3-(trifluoromethyl)pyrazol-4-yl]-2-quinolin-2-yl-3H-1,2,4-thiadiazole Chemical compound C(CC)N1N=C(C(=C1)C1N(SC=N1)C1=NC2=CC=CC=C2C=C1)C(F)(F)F YNVUBNBCGRPWKB-UHFFFAOYSA-N 0.000 description 1
- ZUNFPBNHELLPPP-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-[2-(dimethylamino)ethyl]benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCCN(C)C)C=CC=1 ZUNFPBNHELLPPP-UHFFFAOYSA-N 0.000 description 1
- BVJIRXMMXVSGIB-UHFFFAOYSA-N 3-[5-imidazol-1-yl-2-[(6-methylpyridin-2-yl)amino]-1,3-thiazol-4-yl]benzonitrile Chemical compound CC1=CC=CC(NC=2SC(=C(N=2)C=2C=C(C=CC=2)C#N)N2C=NC=C2)=N1 BVJIRXMMXVSGIB-UHFFFAOYSA-N 0.000 description 1
- XTUAFLZKZLLDNU-UHFFFAOYSA-N 3-isothiocyanato-2-methoxypyridine Chemical compound COC1=NC=CC=C1N=C=S XTUAFLZKZLLDNU-UHFFFAOYSA-N 0.000 description 1
- 125000006201 3-phenylpropyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- BKVALPQRYBWLNN-UHFFFAOYSA-N 4-(1-ethylpyrazol-4-yl)-n-pyridin-2-yl-1,3-thiazol-2-amine Chemical compound C1=NN(CC)C=C1C1=CSC(NC=2N=CC=CC=2)=N1 BKVALPQRYBWLNN-UHFFFAOYSA-N 0.000 description 1
- JALRQYRNZNTPIM-UHFFFAOYSA-N 4-(1h-indazol-3-yl)-n-phenyl-1,3-thiazol-2-amine Chemical compound N=1C(C=2C3=CC=CC=C3NN=2)=CSC=1NC1=CC=CC=C1 JALRQYRNZNTPIM-UHFFFAOYSA-N 0.000 description 1
- SXEBBMKZIMTECE-UHFFFAOYSA-N 4-(4-methoxyphenyl)-n,5-dipyridin-3-yl-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1C1=C(C=2C=NC=CC=2)SC(NC=2C=NC=CC=2)=N1 SXEBBMKZIMTECE-UHFFFAOYSA-N 0.000 description 1
- DBRYOGUMMWHDHS-UHFFFAOYSA-N 4-(4-methoxyphenyl)-n-phenyl-5-pyridin-3-yl-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1C1=C(C=2C=NC=CC=2)SC(NC=2C=CC=CC=2)=N1 DBRYOGUMMWHDHS-UHFFFAOYSA-N 0.000 description 1
- UMHSENIDASHEIK-UHFFFAOYSA-N 4-(5-methyl-1-phenylpyrazol-4-yl)-n-(2,4,5-trimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C(=CC(C)=C(C)C=3)C)SC=2)C=NN1C1=CC=CC=C1 UMHSENIDASHEIK-UHFFFAOYSA-N 0.000 description 1
- DPEMZDMNALJGCV-UHFFFAOYSA-N 4-(5-methyl-1-phenylpyrazol-4-yl)-n-(4-phenoxyphenyl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=CC(OC=4C=CC=CC=4)=CC=3)SC=2)C=NN1C1=CC=CC=C1 DPEMZDMNALJGCV-UHFFFAOYSA-N 0.000 description 1
- XXUZKUKKIGFFSA-UHFFFAOYSA-N 4-(5-methyl-1-phenylpyrazol-4-yl)-n-naphthalen-1-yl-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C4=CC=CC=C4C=CC=3)SC=2)C=NN1C1=CC=CC=C1 XXUZKUKKIGFFSA-UHFFFAOYSA-N 0.000 description 1
- WDPPDJRFTFUEMP-UHFFFAOYSA-N 4-(5-methyl-1-phenylpyrazol-4-yl)-n-phenyl-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=CC=CC=3)SC=2)C=NN1C1=CC=CC=C1 WDPPDJRFTFUEMP-UHFFFAOYSA-N 0.000 description 1
- ACRGAKFVEOSIPC-UHFFFAOYSA-N 4-[1-(cyclopropylmethyl)pyrazol-4-yl]-N-pyridin-2-yl-1,3-thiazol-2-amine 4-[1-propyl-3-(trifluoromethyl)pyrazol-4-yl]-N-pyridin-2-yl-1,3-thiazol-2-amine Chemical compound C1(CC1)CN1N=CC(=C1)C=1N=C(SC1)NC1=NC=CC=C1.C(CC)N1N=C(C(=C1)C=1N=C(SC1)NC1=NC=CC=C1)C(F)(F)F ACRGAKFVEOSIPC-UHFFFAOYSA-N 0.000 description 1
- NUAURNBNOUEEQX-UHFFFAOYSA-N 4-[1-[(4-methoxyphenyl)methyl]-3-methylpyrazol-4-yl]-n-phenyl-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1CN1N=C(C)C(C=2N=C(NC=3C=CC=CC=3)SC=2)=C1 NUAURNBNOUEEQX-UHFFFAOYSA-N 0.000 description 1
- YKGIENBTXSMKCV-UHFFFAOYSA-N 4-[1-[(4-methoxyphenyl)methyl]-5-methylpyrazol-4-yl]-n-phenyl-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1CN1C(C)=C(C=2N=C(NC=3C=CC=CC=3)SC=2)C=N1 YKGIENBTXSMKCV-UHFFFAOYSA-N 0.000 description 1
- LGUWZBBPEISDIC-UHFFFAOYSA-N 4-[2-[4-octoxy-3-(trifluoromethyl)anilino]-1,3-thiazol-4-yl]-1h-pyrazole-5-carbonitrile Chemical compound C1=C(C(F)(F)F)C(OCCCCCCCC)=CC=C1NC1=NC(C=2C(=NNC=2)C#N)=CS1 LGUWZBBPEISDIC-UHFFFAOYSA-N 0.000 description 1
- ONBUBUACLYDURG-UHFFFAOYSA-N 4-[[4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-yl]amino]phenol Chemical compound CC1=C(C=2N=C(NC=3C=CC(O)=CC=3)SC=2)C=NN1C1=CC=CC=C1 ONBUBUACLYDURG-UHFFFAOYSA-N 0.000 description 1
- DWNXGZBXFDNKOR-UHFFFAOYSA-N 4-bromo-2-fluoro-1-methoxybenzene Chemical compound COC1=CC=C(Br)C=C1F DWNXGZBXFDNKOR-UHFFFAOYSA-N 0.000 description 1
- 125000001572 5'-adenylyl group Chemical group C=12N=C([H])N=C(N([H])[H])C=1N=C([H])N2[C@@]1([H])[C@@](O[H])([H])[C@@](O[H])([H])[C@](C(OP(=O)(O[H])[*])([H])[H])([H])O1 0.000 description 1
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 description 1
- OOXNYFKPOPJIOT-UHFFFAOYSA-N 5-(3-bromophenyl)-7-(6-morpholin-4-ylpyridin-3-yl)pyrido[2,3-d]pyrimidin-4-amine;dihydrochloride Chemical compound Cl.Cl.C=12C(N)=NC=NC2=NC(C=2C=NC(=CC=2)N2CCOCC2)=CC=1C1=CC=CC(Br)=C1 OOXNYFKPOPJIOT-UHFFFAOYSA-N 0.000 description 1
- ATXXLNCPVSUCNK-UHFFFAOYSA-N 5-bromo-2-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Br)C=N1 ATXXLNCPVSUCNK-UHFFFAOYSA-N 0.000 description 1
- LXQAAVBTKBNTFA-UHFFFAOYSA-N 5-chloro-3-[1-(cyclopropylmethyl)pyrazol-4-yl]-1,2,4-thiadiazole Chemical compound S1C(Cl)=NC(C2=CN(CC3CC3)N=C2)=N1 LXQAAVBTKBNTFA-UHFFFAOYSA-N 0.000 description 1
- SBHYNBAKHDSUKT-UHFFFAOYSA-N 5-methyl-1h-pyrazole-4-carbonitrile Chemical compound CC=1NN=CC=1C#N SBHYNBAKHDSUKT-UHFFFAOYSA-N 0.000 description 1
- VNDXVYQZJKXWKP-UHFFFAOYSA-N 6-[[3-[1-(cyclopropylmethyl)pyrazol-4-yl]-1,2,4-thiadiazol-5-yl]amino]pyridine-3-carboxamide Chemical compound N1=CC(C(=O)N)=CC=C1NC1=NC(C2=CN(CC3CC3)N=C2)=NS1 VNDXVYQZJKXWKP-UHFFFAOYSA-N 0.000 description 1
- KDVBYUUGYXUXNL-UHFFFAOYSA-N 6-aminopyridine-3-carbonitrile Chemical compound NC1=CC=C(C#N)C=N1 KDVBYUUGYXUXNL-UHFFFAOYSA-N 0.000 description 1
- JXKAUUVMXZIJNZ-UHFFFAOYSA-N 6-ethylpyridin-2-amine Chemical compound CCC1=CC=CC(N)=N1 JXKAUUVMXZIJNZ-UHFFFAOYSA-N 0.000 description 1
- QUXLCYFNVNNRBE-UHFFFAOYSA-N 6-methylpyridin-2-amine Chemical compound CC1=CC=CC(N)=N1 QUXLCYFNVNNRBE-UHFFFAOYSA-N 0.000 description 1
- FFBDFADSZUINTG-LEZITTIZSA-N 8-cyclopentyl-1,3-bis(1,3-ditritiopropyl)-7h-purine-2,6-dione Chemical compound N1C=2C(=O)N(C([3H])CC[3H])C(=O)N(C([3H])CC[3H])C=2N=C1C1CCCC1 FFBDFADSZUINTG-LEZITTIZSA-N 0.000 description 1
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 1
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 1
- 208000016557 Acute basophilic leukemia Diseases 0.000 description 1
- 229940123786 Adenosine A3 receptor antagonist Drugs 0.000 description 1
- 102000055025 Adenosine deaminases Human genes 0.000 description 1
- 102100032534 Adenosine kinase Human genes 0.000 description 1
- 108010076278 Adenosine kinase Proteins 0.000 description 1
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 1
- 108010000239 Aequorin Proteins 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 102000013455 Amyloid beta-Peptides Human genes 0.000 description 1
- 108010090849 Amyloid beta-Peptides Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 201000006474 Brain Ischemia Diseases 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- KCHZUILKPICGHE-UHFFFAOYSA-N CC1(Cc2ccccc2)CCCC1 Chemical compound CC1(Cc2ccccc2)CCCC1 KCHZUILKPICGHE-UHFFFAOYSA-N 0.000 description 1
- PAOANWZGLPPROA-RQXXJAGISA-N CGS-21680 Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NCC)O[C@H]1N1C2=NC(NCCC=3C=CC(CCC(O)=O)=CC=3)=NC(N)=C2N=C1 PAOANWZGLPPROA-RQXXJAGISA-N 0.000 description 1
- PSXPFHYPXHLGCE-UHFFFAOYSA-N COC1=NC=CC=C1NC=1SC=C(N1)C=1C(=NN(C1)CCC)C.COC1=CC=C(C=N1)NC=1SC=C(N1)C=1C(=NN(C1)CCC)C Chemical compound COC1=NC=CC=C1NC=1SC=C(N1)C=1C(=NN(C1)CCC)C.COC1=CC=C(C=N1)NC=1SC=C(N1)C=1C(=NN(C1)CCC)C PSXPFHYPXHLGCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102100035882 Catalase Human genes 0.000 description 1
- 108010053835 Catalase Proteins 0.000 description 1
- LDZBDQQSUMBVBK-UHFFFAOYSA-N Cc1c(C)[n](-c2ccccc2)nc1 Chemical compound Cc1c(C)[n](-c2ccccc2)nc1 LDZBDQQSUMBVBK-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 206010008120 Cerebral ischaemia Diseases 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 244000124209 Crocus sativus Species 0.000 description 1
- 101710095468 Cyclase Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- 208000020401 Depressive disease Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 239000003109 Disodium ethylene diamine tetraacetate Substances 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- SNVFDPHQAOXWJZ-UHFFFAOYSA-N Furcelleran Chemical compound CCOC(=O)C1=C(C)NC(C=2C=CC=CC=2)=C(C(=O)OCC=2C=CC=CC=2)C1C#CC1=CC=CC=C1 SNVFDPHQAOXWJZ-UHFFFAOYSA-N 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108010063907 Glutathione Reductase Proteins 0.000 description 1
- 102000006587 Glutathione peroxidase Human genes 0.000 description 1
- 108700016172 Glutathione peroxidases Proteins 0.000 description 1
- 102100036442 Glutathione reductase, mitochondrial Human genes 0.000 description 1
- 108091006065 Gs proteins Proteins 0.000 description 1
- 102000002268 Hexosaminidases Human genes 0.000 description 1
- 108010000540 Hexosaminidases Proteins 0.000 description 1
- 101000836540 Homo sapiens Aldo-keto reductase family 1 member B1 Proteins 0.000 description 1
- 101000809450 Homo sapiens Amphiregulin Proteins 0.000 description 1
- 101000928259 Homo sapiens NADPH:adrenodoxin oxidoreductase, mitochondrial Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 206010020565 Hyperaemia Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 102000013462 Interleukin-12 Human genes 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 102000016924 KATP Channels Human genes 0.000 description 1
- 108010053914 KATP Channels Proteins 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 1
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 1
- WSMYVTOQOOLQHP-UHFFFAOYSA-N Malondialdehyde Chemical compound O=CCC=O WSMYVTOQOOLQHP-UHFFFAOYSA-N 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 206010028116 Mucosal inflammation Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- ZVDKLHJCVMVOKV-UHFFFAOYSA-N N-(2,5-difluorophenyl)-3-[1-propyl-3-(trifluoromethyl)pyrazol-4-yl]-1,2,4-thiadiazol-5-amine Chemical compound FC1=C(C=C(C=C1)F)NC1=NC(=NS1)C=1C(=NN(C1)CCC)C(F)(F)F ZVDKLHJCVMVOKV-UHFFFAOYSA-N 0.000 description 1
- AJBBEYXFRYFVNM-UHFFFAOYSA-N N-(4-cyanophenyl)-2-[4-(2,6-dioxo-1,3-dipropyl-7H-purin-8-yl)phenoxy]acetamide Chemical compound N1C=2C(=O)N(CCC)C(=O)N(CCC)C=2N=C1C(C=C1)=CC=C1OCC(=O)NC1=CC=C(C#N)C=C1 AJBBEYXFRYFVNM-UHFFFAOYSA-N 0.000 description 1
- YPMSWCFGSNSECI-UHFFFAOYSA-N N-(4-morpholin-4-ylphenyl)-3-[1-propyl-3-(trifluoromethyl)pyrazol-4-yl]-1,2,4-thiadiazol-5-amine Chemical compound O1CCN(CC1)C1=CC=C(C=C1)NC1=NC(=NS1)C=1C(=NN(C1)CCC)C(F)(F)F YPMSWCFGSNSECI-UHFFFAOYSA-N 0.000 description 1
- GIYLOBDVOMZXPA-UHFFFAOYSA-N N-(6-methoxypyridin-3-yl)-3-[1-propyl-3-(trifluoromethyl)pyrazol-4-yl]-1,2,4-thiadiazol-5-amine Chemical compound COC1=CC=C(C=N1)NC1=NC(=NS1)C=1C(=NN(C1)CCC)C(F)(F)F GIYLOBDVOMZXPA-UHFFFAOYSA-N 0.000 description 1
- 229910019065 NaOH 1 M Inorganic materials 0.000 description 1
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 1
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 1
- 208000027771 Obstructive airways disease Diseases 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 206010030348 Open-Angle Glaucoma Diseases 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000015433 Prostaglandin Receptors Human genes 0.000 description 1
- 108010050183 Prostaglandin Receptors Proteins 0.000 description 1
- 108091008611 Protein Kinase B Proteins 0.000 description 1
- 102000003708 Protein arginine N-methyltransferase Human genes 0.000 description 1
- 108020000912 Protein arginine N-methyltransferase Proteins 0.000 description 1
- 102000005765 Proto-Oncogene Proteins c-akt Human genes 0.000 description 1
- 102000007466 Purinergic P2 Receptors Human genes 0.000 description 1
- 108010085249 Purinergic P2 Receptors Proteins 0.000 description 1
- KAEGGIFPLJZUOZ-UHFFFAOYSA-N Renilla luciferin Chemical compound C1=CC(O)=CC=C1C(N1)=CN2C(=O)C(CC=3C=CC=CC=3)=NC2=C1CC1=CC=CC=C1 KAEGGIFPLJZUOZ-UHFFFAOYSA-N 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102000005038 SLC6A4 Human genes 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 102000019197 Superoxide Dismutase Human genes 0.000 description 1
- 108010012715 Superoxide dismutase Proteins 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000009270 Tumour necrosis factor alpha Human genes 0.000 description 1
- 108050000101 Tumour necrosis factor alpha Proteins 0.000 description 1
- 206010047141 Vasodilatation Diseases 0.000 description 1
- 230000004156 Wnt signaling pathway Effects 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- YQYBUJYBXOVWQW-UHFFFAOYSA-N [3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxyphenyl]-(3,4-dihydro-1H-isoquinolin-2-yl)methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=CC=1)C(=O)N1CC2=CC=CC=C2CC1 YQYBUJYBXOVWQW-UHFFFAOYSA-N 0.000 description 1
- DGYIJVNZSDYBOE-UHFFFAOYSA-N [CH2]C1=CC=NC=C1 Chemical group [CH2]C1=CC=NC=C1 DGYIJVNZSDYBOE-UHFFFAOYSA-N 0.000 description 1
- LRIUKPUCKCECPT-UHFFFAOYSA-N [hydroxy(phenyl)-$l^{3}-iodanyl] 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OI(O)C1=CC=CC=C1 LRIUKPUCKCECPT-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 108010018878 adenosine transporter Proteins 0.000 description 1
- 108060000200 adenylate cyclase Proteins 0.000 description 1
- 102000030621 adenylate cyclase Human genes 0.000 description 1
- 239000000695 adrenergic alpha-agonist Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 239000013566 allergen Substances 0.000 description 1
- 230000002009 allergenic effect Effects 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000009285 allergic inflammation Effects 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229950003476 aminothiazole Drugs 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 210000002159 anterior chamber Anatomy 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000002253 anti-ischaemic effect Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 229940006133 antiglaucoma drug and miotics carbonic anhydrase inhibitors Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 230000006851 antioxidant defense Effects 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 108010041089 apoaequorin Proteins 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 208000015802 attention deficit-hyperactivity disease Diseases 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 230000004378 blood-retinal barrier Effects 0.000 description 1
- 210000004155 blood-retinal barrier Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- 239000004044 bronchoconstricting agent Substances 0.000 description 1
- 230000003435 bronchoconstrictive effect Effects 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 239000003489 carbonate dehydratase inhibitor Substances 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- 125000005392 carboxamide group Chemical group NC(=O)* 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 210000004413 cardiac myocyte Anatomy 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000004700 cellular uptake Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- SFZULDYEOVSIKM-UHFFFAOYSA-N chembl321317 Chemical compound C1=CC(C(=N)NO)=CC=C1C1=CC=C(C=2C=CC(=CC=2)C(=N)NO)O1 SFZULDYEOVSIKM-UHFFFAOYSA-N 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 239000007958 cherry flavor Substances 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 238000010568 chiral column chromatography Methods 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- KTHXBEHDVMTNOH-UHFFFAOYSA-N cyclobutanol Chemical compound OC1CCC1 KTHXBEHDVMTNOH-UHFFFAOYSA-N 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000009795 derivation Methods 0.000 description 1
- XXTZHYXQVWRADW-UHFFFAOYSA-N diazomethanone Chemical compound [N]N=C=O XXTZHYXQVWRADW-UHFFFAOYSA-N 0.000 description 1
- 229940061607 dibasic sodium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000006001 difluoroethyl group Chemical group 0.000 description 1
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 235000019301 disodium ethylene diamine tetraacetate Nutrition 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 229940125542 dual agonist Drugs 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 229940124274 edetate disodium Drugs 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 230000004406 elevated intraocular pressure Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 230000013764 eosinophil chemotaxis Effects 0.000 description 1
- 230000002327 eosinophilic effect Effects 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- UNPPYAXHMYQLEC-UHFFFAOYSA-N ethyl 1-(2,2,2-trifluoroethyl)pyrazole-4-carboxylate Chemical compound CCOC(=O)C=1C=NN(CC(F)(F)F)C=1 UNPPYAXHMYQLEC-UHFFFAOYSA-N 0.000 description 1
- LZWKNSZXNNHBOJ-UHFFFAOYSA-N ethyl 1-(2-phenylmethoxyethyl)pyrazole-4-carboxylate Chemical compound C1=C(C(=O)OCC)C=NN1CCOCC1=CC=CC=C1 LZWKNSZXNNHBOJ-UHFFFAOYSA-N 0.000 description 1
- MKCNXJIANOZKRI-UHFFFAOYSA-N ethyl 1-(cyclopropylmethyl)pyrazole-4-carboxylate Chemical compound C1=C(C(=O)OCC)C=NN1CC1CC1 MKCNXJIANOZKRI-UHFFFAOYSA-N 0.000 description 1
- CUWKKMRPTUKJNT-UHFFFAOYSA-N ethyl 1-propylpyrazole-4-carboxylate Chemical compound CCCN1C=C(C(=O)OCC)C=N1 CUWKKMRPTUKJNT-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 208000030533 eye disease Diseases 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 210000000744 eyelid Anatomy 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 229930003935 flavonoid Natural products 0.000 description 1
- 150000002215 flavonoids Chemical class 0.000 description 1
- 235000017173 flavonoids Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000006005 fluoroethoxy group Chemical group 0.000 description 1
- 125000003784 fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 102000046818 human AR Human genes 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 230000037417 hyperactivation Effects 0.000 description 1
- 230000001631 hypertensive effect Effects 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 208000021822 hypotensive Diseases 0.000 description 1
- 230000001077 hypotensive effect Effects 0.000 description 1
- UVNXNSUKKOLFBM-UHFFFAOYSA-N imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=CSC2=NC=CN21 UVNXNSUKKOLFBM-UHFFFAOYSA-N 0.000 description 1
- 150000005235 imidazopyrazines Chemical class 0.000 description 1
- 125000005945 imidazopyridyl group Chemical group 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 229940117681 interleukin-12 Drugs 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 230000037427 ion transport Effects 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 239000002085 irritant Substances 0.000 description 1
- 231100000021 irritant Toxicity 0.000 description 1
- 230000002530 ischemic preconditioning effect Effects 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001810 isothiocyanato group Chemical class *N=C=S 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 230000003859 lipid peroxidation Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229920006008 lipopolysaccharide Polymers 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940118019 malondialdehyde Drugs 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000013160 medical therapy Methods 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000006609 metabolic stress Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- VNSGGDXXYZQRKM-UHFFFAOYSA-N methyl 1-ethyl-3-methylpyrazole-4-carboxylate Chemical compound CCN1C=C(C(=O)OC)C(C)=N1 VNSGGDXXYZQRKM-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- RMCDUNHIVVEEDD-UHFFFAOYSA-N methylcyclopropane Chemical compound [CH2]C1CC1 RMCDUNHIVVEEDD-UHFFFAOYSA-N 0.000 description 1
- 239000003697 methyltransferase inhibitor Substances 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 125000006203 morpholinoethyl group Chemical group [H]C([H])(*)C([H])([H])N1C([H])([H])C([H])([H])OC([H])([H])C1([H])[H] 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- XNOLEAFNGXIOIO-UHFFFAOYSA-N n-(1-methylindazol-4-yl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=4C=NN(C)C=4C=CC=3)SC=2)C=NN1C1=CC=CC=C1 XNOLEAFNGXIOIO-UHFFFAOYSA-N 0.000 description 1
- AYHHVSBHXLPOFI-UHFFFAOYSA-N n-(1h-indazol-3-yl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C4=CC=CC=C4NN=3)SC=2)C=NN1C1=CC=CC=C1 AYHHVSBHXLPOFI-UHFFFAOYSA-N 0.000 description 1
- HDLHPZCZQRDNSL-UHFFFAOYSA-N n-(1h-indazol-4-yl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=4C=NNC=4C=CC=3)SC=2)C=NN1C1=CC=CC=C1 HDLHPZCZQRDNSL-UHFFFAOYSA-N 0.000 description 1
- XHOSNDTTYLMVJE-UHFFFAOYSA-N n-(2,4-dimethoxyphenyl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound COC1=CC(OC)=CC=C1NC1=NC(C2=C(N(N=C2)C=2C=CC=CC=2)C)=CS1 XHOSNDTTYLMVJE-UHFFFAOYSA-N 0.000 description 1
- QLTITRMEXCXCFH-UHFFFAOYSA-N n-(3,5-dimethylphenyl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=C(C)C=C(C)C=3)SC=2)C=NN1C1=CC=CC=C1 QLTITRMEXCXCFH-UHFFFAOYSA-N 0.000 description 1
- UFMIQMPQVHEWCN-UHFFFAOYSA-N n-(3-bromophenyl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=C(Br)C=CC=3)SC=2)C=NN1C1=CC=CC=C1 UFMIQMPQVHEWCN-UHFFFAOYSA-N 0.000 description 1
- MOXVKUBNWSXVFV-UHFFFAOYSA-N n-(3-fluorophenyl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=C(F)C=CC=3)SC=2)C=NN1C1=CC=CC=C1 MOXVKUBNWSXVFV-UHFFFAOYSA-N 0.000 description 1
- HCEZRJCSTJQCGL-UHFFFAOYSA-N n-(4-chlorophenyl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=CC(Cl)=CC=3)SC=2)C=NN1C1=CC=CC=C1 HCEZRJCSTJQCGL-UHFFFAOYSA-N 0.000 description 1
- GGYVAZCXZMFFQI-UHFFFAOYSA-N n-(4-methoxyphenyl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1NC1=NC(C2=C(N(N=C2)C=2C=CC=CC=2)C)=CS1 GGYVAZCXZMFFQI-UHFFFAOYSA-N 0.000 description 1
- GKMXCMVIHGICGO-UHFFFAOYSA-N n-(4-methylphenyl)-4-(5-methyl-1-phenylpyrazol-4-yl)-1,3-thiazol-2-amine Chemical compound CC1=C(C=2N=C(NC=3C=CC(C)=CC=3)SC=2)C=NN1C1=CC=CC=C1 GKMXCMVIHGICGO-UHFFFAOYSA-N 0.000 description 1
- KGHQFJPAIPAWOK-UHFFFAOYSA-N n-(6-fluoropyridin-2-yl)-3-(3-methyl-1-propylpyrazol-4-yl)-1,2,4-thiadiazol-5-amine Chemical compound CC1=NN(CCC)C=C1C1=NSC(NC=2N=C(F)C=CC=2)=N1 KGHQFJPAIPAWOK-UHFFFAOYSA-N 0.000 description 1
- VDJVLRLNAOFIPW-UHFFFAOYSA-N n-[5-pyridin-4-yl-4-(3,4,5-trimethoxyphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound COC1=C(OC)C(OC)=CC(C2=C(SC(NC(C)=O)=N2)C=2C=CN=CC=2)=C1 VDJVLRLNAOFIPW-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VXZQKWHYMVRZMO-UHFFFAOYSA-N n-phenyl-4-(1h-pyrazol-5-yl)-1,3-thiazol-2-amine;hydrobromide Chemical compound Br.C=1C=CC=CC=1NC(SC=1)=NC=1C=1C=CNN=1 VXZQKWHYMVRZMO-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- MRWXACSTFXYYMV-FDDDBJFASA-N nebularine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC=C2N=C1 MRWXACSTFXYYMV-FDDDBJFASA-N 0.000 description 1
- 230000031990 negative regulation of inflammatory response Effects 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000003227 neuromodulating effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 108010028584 nucleotidase Proteins 0.000 description 1
- 229940100655 ophthalmic gel Drugs 0.000 description 1
- 229940100654 ophthalmic suspension Drugs 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000004792 oxidative damage Effects 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000004963 pathophysiological condition Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- QKFJKGMPGYROCL-UHFFFAOYSA-N phenyl isothiocyanate Chemical compound S=C=NC1=CC=CC=C1 QKFJKGMPGYROCL-UHFFFAOYSA-N 0.000 description 1
- 150000003905 phosphatidylinositols Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 108091008695 photoreceptors Proteins 0.000 description 1
- 125000002265 phtalazinyl group Chemical group 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000003518 presynaptic effect Effects 0.000 description 1
- 230000001686 pro-survival effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- UUSHFEVEROROSP-UHFFFAOYSA-N propyl 6-ethyl-5-ethylsulfanylcarbonyl-2-phenyl-4-propylpyridine-3-carboxylate Chemical compound CCCOC(=O)C1=C(CCC)C(C(=O)SCC)=C(CC)N=C1C1=CC=CC=C1 UUSHFEVEROROSP-UHFFFAOYSA-N 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 210000001747 pupil Anatomy 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 239000002212 purine nucleoside Substances 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical class O1C(C=CC=C1)* 0.000 description 1
- SLUHLANJIVXTRQ-UHFFFAOYSA-N pyridin-2-ylthiourea Chemical compound NC(=S)NC1=CC=CC=N1 SLUHLANJIVXTRQ-UHFFFAOYSA-N 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 125000005494 pyridonyl group Chemical group 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 102000009929 raf Kinases Human genes 0.000 description 1
- 108010077182 raf Kinases Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 230000027425 release of sequestered calcium ion into cytosol Effects 0.000 description 1
- 230000008085 renal dysfunction Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 208000032253 retinal ischemia Diseases 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 238000006748 scratching Methods 0.000 description 1
- 230000002393 scratching effect Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical class [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000001117 sulphuric acid Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000000946 synaptic effect Effects 0.000 description 1
- 230000005062 synaptic transmission Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 150000004867 thiadiazoles Chemical class 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 150000007979 thiazole derivatives Chemical class 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 210000001585 trabecular meshwork Anatomy 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 1
- 230000008728 vascular permeability Effects 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- QDLHCMPXEPAAMD-QAIWCSMKSA-N wortmannin Chemical compound C1([C@]2(C)C3=C(C4=O)OC=C3C(=O)O[C@@H]2COC)=C4[C@@H]2CCC(=O)[C@@]2(C)C[C@H]1OC(C)=O QDLHCMPXEPAAMD-QAIWCSMKSA-N 0.000 description 1
- QDLHCMPXEPAAMD-UHFFFAOYSA-N wortmannin Natural products COCC1OC(=O)C2=COC(C3=O)=C2C1(C)C1=C3C2CCC(=O)C2(C)CC1OC(C)=O QDLHCMPXEPAAMD-UHFFFAOYSA-N 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to novel compounds of Formula (I), wherein X 1 , X 2 , X 3 , X 4 , Y 1 , Y 2 , Y 3 , Y 4 , M 1 , M 2 , M 3 , A m and B n are defined as in Formula (I); invention compounds are antagonists of adenosine receptors — subtype 3 (A 3 ) which are useful for the treatment or prevention of disorders modulated by A 3 receptors.
- the invention is also directed to pharmaceutical compositions and the use of such compounds in the manufacture of medicaments, as well as to the use of such compounds for the prevention and treatment of such diseases in which A 3 receptor is involved.
- the extracellular purine nucleoside adenosine is present in all tissues and body fluids and is known to function as a modulator of a variety of physiological processes.
- adenosine One of the primary roles of adenosine is cytoprotection against ischemia-induced cell damage, mainly in tissues such as the heart, brain and kidney, which are especially prone to ischemic injury (Mubagwa and Flameng (2001) Cardiovasc. Res. 52:25-39).
- the effects of adenosine on tissue protection and repair include increasing the ratio of oxygen supply to demand, protecting against ischemic damage by cell conditioning, triggering anti-inflammatory responses and promoting angiogenesis.
- Other actions of adenosine include the regulation of cellular growth and differentiation, vasodilatation
- AR action also might be modulated by inhibition of the metabolism of extracellular adenosine (Parkinson et al (2005) Neurol. Res. 2:153-160) or its cellular uptake by adenosine transporter (McGaraughty et al (2005) Curr. Top.Med. Chem. 5:43-58).
- a 1 , A 2A , A 2B , and A 3 pharmacologically and biochemically distinct adenosine receptors
- a 1 , A 2A , A 2B , and A 3 pharmacologically and biochemically distinct adenosine receptors
- a 1 and A 3 receptor subtypes couple to Gi-protein, mediating the inhibition of adenylyl
- a 1 and A 3 ARs (49% sequence similarity) and the A 2A and A 2B ARs (59% similarity).
- degree of homology is somewhat low, it has been very difficult to develop highly selective or even specific adenosine receptor subtype agonists and antagonists.
- a 3 receptors are more variable.
- adenosine receptors may be useful for therapeutic intervention due to their distribution in several types of tissue throughout the body.
- Many pathological conditions such as renal failure, cardiac and cerebral ischemia, central nervous system disorders, neurodegenerative diseases and inflammatory pathologies may be treated with selective modulators of the different sub-types of adenosine receptors (for a review see Gao and Jacobson (2006) Nat Rev Drug Discov. 5:247-64).
- the adenosine A 3 receptor (A 3 AR) is the most recently discovered adenosine receptor (Zhou et al (1992) Proc Natl Acad Sci U S A. 89:7432-6; Salvatore et al (1993) Proc Natl Acad Sci U S A. 90:10365-9).
- the A 3 AR couples to second-messenger pathways resulting in stimulation of phospholipase C (PLC) (Abbracchio et al. (1995) MoL Pharmacol. 48:1038-1045) and calcium mobilization via a Gi/o-dependent pathway (Shneyvays et al (2005) Am. J. Physiol. Heart Circ. Physiol.
- the WNT signaling pathway is involved in A 3 AR agonist- mediated suppression of melanoma cells (Fishman et al (2002) Oncogene 21:4060- 4064.).
- the A 3 AR couples to MAPK, which suggests a possible role in cell growth, survival, death and differentiation (Schulte & Fredholm (2002) MoI. Pharmacol. 62:1137-1146; Schulte & Fredholm (2003) Cell Signal. 15:813-827).
- An A 3 AR agonist inhibits proliferation in A375 human melanoma cells via the phosphatidylinositol 3-kinase-protein kinase B-ERKl/2 pathway (Merighi et al. (2005) J. Biol. Chem. 280:19516-19526). It is suggested that the adenosine A 3 receptor activates ERK1/2 in human fetal astrocytes (Neary et al (1998) Neurosci Lett.; 242:159-62) and in CHO cells (Schulte and Fredholm (2000) MoI Pharmacol. 58:477- 82).
- Cl-IB-MECA and IB-MECA have been reported to potently inhibit and less potently to activate apoptosis in various cells (Abbracchio et al (1997) Ann NY Acad Sci 825:11-22).
- Cl-IB-MECA potently blocks UV irradiation-induced apoptosis by a process correlated with protein kinase B phosphorylation which is blocked by pertussis toxin and wortmannin (Gao et al (2001) MoI Pharmacol 59:76-82).
- a 3 AR selective agonists IB-MECA and Cl-IB-MECA have been used extensively as pharmacological probes in the elucidation of the physiological roles of this receptor (Jacobson (1998) Trends Pharmacol. Sci. 19:184-191).
- a 3 AR antagonists available including, among many, xanthine, adenine derivatives, imidazo[2,l-i]purin-5-ones, quinazolines and derivatives, 1,4-dihydropyridines and pyrans, pyrimidines derivatives (Zhou (1992) Proc. Natl Acad. Sci. USA 89:7432-7436; Yang (2005) Curr.Eye Res. 30:747-754;
- the conformationally constrained nucleoside MRS 1292 which is a selective A 3 AR antagonist, in both rat and human (Gao et al. (2002) J. Med. Chem. 45:4471-4484; Yang et al (2005) Curr. Eye Res. 30:747-754) is currently used in vitro as a reference antagonist.
- Adenosine A 3 receptors are found mostly in brain, lung, liver, heart, kidney and testis (Gao and Jacobson (2006) Nat Rev Drug Discov. 5:247-64) and have been implicated in cell cycle progression and cell growth (Brambilla et al. (2000) Naunyn
- adenosine A 3 receptors have functional effects that are dependent on the degree of receptor activation. Specifically, when activated moderately, A 3 ARs have a cytoprotective role for example reducing damage to heart cells from lack of oxygen or protecting cells from apoptosis.
- a 3 receptor is known to activate the cellular antioxidant defense system by increasing the activities of superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase, along with a reduction in malondialdehyde, a marker of lipid peroxidation (Maggirwar et al (1994) Biochem Biophys Res Comniun 201 :508-512).
- Such a mechanism may provide a mechanism by which adenosine exerts a cytoprotective action in ischemic conditions.
- high levels of A 3 AR stimulation can actually result in cell death.
- a role for the A 3 AR in mediating control of the cell cycle has been reported (Neary et al (1998) Neurosci. Lett 242:159-162). For example, there is a significant over- expression of A 3 ARs in several types of tumor cells (Madi et al (2004) Clin. Cancer Res. 10:4472-4479; Gessi et al (2004) Clin. Cancer Res. 10:5895-5901).
- a 3 AR antagonists might sensitize tumor cells to chemotherapeutic drugs as it is known that A3 receptor subtype activation plays a role in the prosurvival and in the antiapoptotic effect of adenosine (Merighi et al (2003) Biochem. Pharmacol. 66:739-748; Baraldi et
- a 3 AR antagonists might sensitize tumors cells to chemotherapeutic drugs and therefore could inhibit tumor growth and metastasis in patient. Possible indication therefore for selective adenosine A 3 antagonists is the use of such compounds class in synergistically improving chemotherapeutic treatment of cancers expressing A 3 ARs and cancers expressing P-glycoprotein or MRP in combination with other anti-tumor agents such as antiangiogenic agents and/or cytostatic agents.
- the adenosine A 3 receptor was initially implicated as the receptor subtype that triggers the degranulation of rat RBL 2H3 mast-like cells (Ramkumar et al., 1993) and perivascular mast cells of the hamster cheek pouch (Jin et al (1997) J Clin Invest. 100:2849-57) and therefore A 3 AR has been implicated in mediating allergic responses: A 3 receptor agonists induce mast cell degranulation and consequent release of allergic mediators, such as histamine, when administered to rats or mice (Ramkumar et al (1993) J. Biol. Chem. 268:16887-16890; Tilley et al (2000) J Clin Invest.lO5:361-7).
- a 3 AR receptor antagonists have the potential for treating diseases and disorders resulting from or including a component of inflammation. This could be extended to the treatment of asthma and others respiratory diseases involving inflammation and or allergenic responses. In the asthmatic lung, adenosine acts as an irritant and bronchoconstrictor, suggesting that a synthetic A 3 AR antagonist, could have therapeutic potential in asthma
- Glaucoma is characterized by elevated intraocular pressure (IOP) and is a leading cause of irreversible blindness.
- IOP intraocular pressure
- Molecular and pharmacological studies have provided evidence that all adenosine receptor subtypes are expressed in ocular tissues (Blazynski et al (1992) J Neurochem. 58:761- 767; Wax et al (1993) Exp Eye Res. 57:89 -95 ; Wax et al (1994) Invest Ophthalmol Vis Sci. ;35:3057-3063 ; Kvanta et al (1997) Exp Eye Res.
- adenosine and its receptors have been implicated in many ocular and systemic ischemic diseases such as retinal ischemia and in conditions with oxidative stress in rodents (Roth S et al (1997) Exp Eye Res. 65:771-779; Lutty and Mc Leod (2003) Prog Retin Eye Res. 22:95-111, Larsen and Osbourne (1996) Invest Ophthalmol Vis Sci. 37: 2603-2611).
- Civan and co-workers have found that the A 3 adenosine receptors regulate Cl(-) channels of non-pigmented ciliary epithelial cells (Von Arnini et al (2000) Neuroreport. 11:1223-1226).
- a 3 agonists have been shown to activate chloride channels in non pigmented ciliary epithelial cells in vitro, leading to the hypothesis that A 3 receptor agonists would increase aqueous humor secretion and thereby IOP in vivo (Mitchell et al (1999) Am J Physiol. 276:C659-C666).
- a 3 AR antagonist MRS 1292 was recently found to reduce mouse intraocular pressure but also inhibited adenosine-triggered human non-pigmented ciliary epithelial cell fluid release (Yang, H. et al (2005) Curr. Eye Res. 30: 747-754).
- OT-7999 a potent and selective A(3) receptor antagonist administered via topical eye-
- the A 3 receptor subtype has been immunolocalized to the basolateral surface infoldings of non-pigmented ciliary epithelial cells, which is consistent with the receptor's functional role in aqueous humor secretion.
- the mean aqueous adenosine levels were significantly elevated when compared to normotensive subjects and correlated with IOP levels (Daines et al (2003) J Ocul Pharmacol Ther. 19:113-119).
- parenteral infusion of adenosine induced a small but significant decrease in IOP (Polska et al (2003) Invest Ophthalmol Vis Sci. 44:3110-3114).
- PEX pseudoexfoliation
- a recent study further provided evidence of a selective and significant upregulation of the A 3 adenosine receptor on both the mRNA and protein levels, in the non-pigmented ciliary epithelium of all patients eyes with PEX syndrome confirming a previous study showing a 30-fold overexpression of A 3 adenosine receptor mRNA in the ciliary processes of PEX eyes compared with control eyes (Schl ⁇ tzer-Schrehardt et al (2004) IOVS 45: ARVO E-Abstract 3535).
- hypoxia and/or oxidative stress typical of all eyes with PEX syndrome/glaucoma (Ritch and Schl ⁇ tzer-Schrehardt (2001) Surv Ophthalmol. 45:265-315; Helbig et al (1994) German J Ophthalmol. 3:148 -153) promotes a selective upregulation of A 3 adenosine receptors in non-pigmented ciliary epithelium, which may confer protection against ischemic or oxidative damage to sustain prolonged periods of chronic hypoxia or oxidative stress.
- the known adenosine receptors promotes a selective upregulation of A 3 adenosine receptors in non-pigmented ciliary epithelium, which may confer protection against ischemic or oxidative damage to sustain prolonged periods of chronic hypoxia or oxidative stress.
- a 3 receptor has been mainly implicated in ischemic disease, such as ischemic brain damage or cardiac ischemia (Baraldi et al (2003) Eur. J. Med. Chem. 38:367-382).
- Adenosine is released in large amounts during myocardial ischemia, resulting in effective preconditioning in cardiomyocytes through the activation of A 1 and A 3 ARs (Shneyvays et al (2004) Cell Calcium 36:387-396; Tracey et al (1998) Cardiovasc. Res. 40:138-145 ; Mozzicato et al (2004) FASEB J. 18: 406-408; Auchampach (1997) Circ. Res. 80: 800-809).
- AR agonist to activate either or both of these receptors might therefore be beneficial to the survival of the ischemic heart.
- Various lines of evidence indicate that the A 3 AR has a role in protecting the heart (Auchampach (1997) Circ. Res. 80: 800-809; Tracey et al (2003) Am. J. Physiol. Heart Circ. Physiol. 285:H2780- H2787).
- Overexpression of A 3 ARs decreases heart rate, preserves energetics and protects ischemic heart (Cross et al (2002) Am. J. Physiol. Heart Circ. Physiol.
- synaptic serotonin (5-hydiOxytryptamine, 5-HT) is primarily through reuptake by the presynaptic, 5-HT transporter (SERT, SLC6A4)
- a 3 AR antagonists could be interesting in treating pathologies resulting from a low level of serotonin (including affective disorders including depression and depressive disorders, anxiety disorders including obsessive-compulsive disorder, post-traumatic stress disorder, panic disorder and phobias, borderline personality disorder, anorexia nervosa, bulimia nervosa, autism, attention deficit hyperactivity disorder, Tourette's syndrome, sexual disorders, migraine, diabetic neuropathy, obesity, drug or alcohol addiction, sleep disorders, arthritis, chronic fatigue syndrome or irritable bowel syndrome) because such compounds will decrease the SERT surface expression and/or catalytic rates.
- TNF- ⁇ Tumor Necrosis Factor- Alpha
- IL-12 Interleukin-12
- Hassan, N.M. et al (1997) J. Chem. Res., Syn. (10): 350-351 described a synthetic route to obtain 4-((l-aryl-4-carbonitrile-5-phenyl)-lH-pyrazolyl)-2- arninophenyl thiazoles;
- arylpyridinyl thiazoles such as N-(5-(pyridin-4-yl)-4-(3 5 4,5-trimethoxyphenyl)thiazol- 2-yl)acetamide, having adenosine A 3 receptor antagonism properties, useful as pharmacological tools;
- European patent publication EP1027050 and US patent publication US6620825 describe 1,3-thiazoles as adenosine A 3 receptor antagonist for the treatment of allergy, asthma and diabetes, such as compounds substituted at the 4- or 5 -position, or both, by a pyridyl, such as 4-(4-methoxyphenyl)-N-phenyl-5-(pyridin-3-yl)thiazol-2-amine or 4-
- Japan patent application JP2003313176 describes 2-aminothiazole derivatives having cell proliferation inhibitory activity for preventing and treating cancer.
- the present invention relates to a method of treating or preventing a condition in a mammal, including a human, the treatment or prevention of which is affected or facilitated by the neuromodulatory effect of Adenosine A3 receptor antagonists.
- the invention relates to compounds having A 3 receptor antagonist activity.
- the present invention provides a compound according to Formula (I),
- n is an integer ranging from 1 to 3;
- a m radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(Ci-C 6 )alkyl, - (Ci-C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(Ci-C 6 )alkylcyano 5 -(Ci-C 6 )alkylheteroaryl, - (Ci-C 6 )alkylaryl, aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 1 , ⁇ O-(C 2 -C 6 )alkyl- OR 1 , -NR 1 (C 2 -C 6 )alkyl-OR 2 , -(C 3 -C 7 )cycloalkyl-(C
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen or an optionally substituted radical selected from the group of -(CrC ⁇ alkylhalo, -(CrC ⁇ alkyl, -(C t -C ⁇ alkylcyano, -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and
- R Any two radicals of R (R 1 , R 2 , R 3 or R 4 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- Y 1 , Y 2 , Y 3 and Y 4 are each independently selected from the group of C and N representing 5 membered heteroaryl ring which may further be substituted by 1 to 3 radicals B n ;
- n is an integer ranging from 1 to 3;
- B n radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(Q-C ⁇ alkyl, - (Ci-C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(Ci-C 6 )alkylcyano, -(CrC ⁇ alkylheteroaryl, - (Ci-C f Oalkylaryl, aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 5 , -O-(C 2 -C 6 )alkyl- OR 5 , -NR 5 (C 2 -C 6 )alkyl-OR 6 , -(Ca-C ⁇ cycloalkyl-CCrC ⁇ alkyl, -O-(C 3 -C 7 )cycloalkyl- (Ci-C 6 )
- R 5 , R 6 , R 7 and R 8 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylhalo, -(Ci-C 6 )alkyl, -(C 1 -C 6 )alkylcyano, -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, aryl, heterocycle and -(Q-C ⁇ alkylaryl;
- R Any two radicals of R (R 5 , R 6 , R 7 or R 8 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- M 1 is selected from an optionally substituted 3 to 10 membered ring selected from the group of aryl, heteroaryl, heterocyclic and cycloalkyl;
- R 9 and R 10 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C 1 -C 6 )alkylhalo, -(CrC ⁇ alkyl, -(CrC ⁇ alkylcyano, ⁇ (C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(C t -C ⁇ alkylheteroaryl, aryl, heterocycle and -(Cj . -C 6 )alkylaryl;
- M 3 is an optionally substituted radical selected from the group of -(Co-C 6 )alkyl-R , - (d-C ⁇ alkylhalo, -(C 2 -C 6 )alkyl-NR ⁇ R 12 , -(C 2 -C 6 )alkyl-OR n and -(C 2 -C 6 )alkyl-SR U ; and
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylhalo, -(CrC ⁇ alkyl, -(CrC ⁇ alkylcyano, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(Ci-C ⁇ alkylheteroaryl, aryl, heterocycle and -(Q-C ⁇ alkylaryl; provided that according to proviso (i): when M 3 is -(Co)-R 11 (that is when M 3 is -R 11 ), then R 11 is not H; and provided that according to proviso (ii):
- a 1 and A 2 radicals are not linked to form an imidazopyridazinyl ring; and provided that according to proviso (v):
- M 1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl 5 lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M 3 is 4-methylphenyl, then B n can not be a phenyl; and provided that according to proviso (vii): when M 1 M 2 N is linked to X 1 , and X 1 is C 5 X 2 is S 5 X 3 is C 5 X 4 is C, to provide a thiazole ring, n is 1 , then A 1 is not a pyridyl; and provided that according to proviso (viii)
- the compounds are 3-[5-[(4-chlorophenyl)azo]-2-(phenylamino)-4-thiazolyl]-l-(4- methyl ⁇ henyl)-5 -phenyl- lH-pyrazole-4-carbonitrile [198840-15-2], l-(4- methylphenyl)-3 - [5 - [(4-methylphenyl)azo] -2-(phenylamino)-4-thiazolyl] -5 -phenyl- 1 H- pyrazole-4-carbonitrile [198840-14-1], l-(4-methylphenyl)-5-phenyl-3-[2-
- the invention provides a compound according to Formula (II),
- KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008 m is an integer ranging from 1 to 3;
- a m radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of - (CrC ⁇ alkylhalo, -(C 3 -C 7 )cycloalkyl, -(Ci-C 6 )alkylcyano, -(Ci-C 6 )alkylheteroaryl, - (C r C 6 )alkylaryl, aryl, heteroaryl, heterocycle, -(Co-C ⁇ alkyl-OR 1 , -NR 1 (C 2 -C 6 )alkyl- OR 2 , -(C 3 -C 7 )cycloalkyl-(Ci-C 6 )alkyl, -O-(C 3 -C 7 )cycloalkyl-(Ci-C 6 )alkyl, -NR ⁇ (C 3 - C 7 )cycloalkyl-(Ci-C 6
- Any two radicals of A m may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen or an optionally substituted radical selected from the group of -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and -(Ci-C ⁇ alkylaryl;
- R Any two radicals of R (R 1 , R 2 , R 3 or R 4 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- n is an integer ranging from 1 to 2;
- B n radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(d-C ⁇ alkyl, -
- R 5 , R 6 , R 7 and R 8 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C ⁇ alkylhalo, -(CrC ⁇ alkyl, -(C 1 -C 6 )alkylcyano, -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, aryl, heterocycle and -(Ci-C 6 )alkylaryl;
- R 5 , R 6 , R 7 or R 8 Any two radicals of R (R 5 , R 6 , R 7 or R 8 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- M 1 is selected from an optionally substituted 3 to 10 membered ring selected from the group of aryl, heteroaryl, heterocyclic and cycloalkyl;
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(Q-C ⁇ alkyl-R 11 , -(CrC ⁇ alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR ⁇ and -(C 2 -C 6 )alkyl-SR ⁇ ; and
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C ⁇ alkylhalo, -(CrC ⁇ alkyl, -(Q-C ⁇ alkylcyano, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(Ci-C ⁇ alkylheteroaryl, aryl, heterocycle and -(CrC ⁇ alkylaryl; provided that according to proviso (i): when M 3 is -(Co)-R 11 (that is when M 3 is -R 11 ), then R 11 is not H; and provided that according to proviso (ii):
- a 1 is H, then M 1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M 3 is 4-methylphenyl, then B n can not be a phenyl; and provided that according to proviso (vii): when X 2 is S, X 3 is C, X 4 is C, n is 1, then A 1 when linked to either X 3 or X 4 is not a pyridyl; and provided that according to proviso (viii): when X 2 is S, X 3 is C, X 4 is C, to provide a thiazole ring, n is 1, then A 1 is not an optionally substituted imidazolyl or triazolyl ring.
- X is a nitrogen, an oxygen, or a sulfur atom
- X is a carbon atom or a nitrogen atom
- X 4 is a carbon or a nitrogen atom, representing a 5 membered heteroaryl, which may further be substituted by 1 to 2 radicals A m
- a m radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(Ci-C 6 )alkyl, - (Ci-C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(Ci-Q)alkylcyano, -(Ci-C 6 )alkylheteroaiyl, - (Ci-C 6 )alkylaryl, aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 1 ,
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen or an optionally substituted radical selected from the group of -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(C t -C ⁇ alkylheteroaryl, aryl, heterocycle and -(Ci-C ⁇ alkylaryl;
- R Any two radicals of R (R 1 , R 2 , R 3 or R 4 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- n is an integer ranging from 1 to 2;
- B n radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(C 1 -C 6 )alkyl, - (CrC f Oalkylhalo, -(C 3 -C 7 )cycloalkyl, -(d-C ⁇ alkylcyano, -(C 1 -C 6 )alkylheteroaryl, - aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 5 , -O-(C 2 -C 6 )alkyl- OR 5 , -NR 5 (C 2 -C 6 )alkyl-OR 6 , -(C 3 -C 7 )cycloalkyl-(C 1 -C 6 )alkyl, -O-(C 3 -C 7 )cycl
- R 5 , R 6 , R 7 and R 8 each independently hydrogen or an optionally substituted radical selected from the group of -(d-C ⁇ alkylhalo, -(C t -C ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 - C 10 )alkylcycloalkyl and heterocycle;
- R 5 , R 6 , R 7 or R 8 Any two radicals of of R (R 5 , R 6 , R 7 or R 8 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- M 1 is selected from an optionally substituted aryl and heteroaryl; M is a hydrogen or an optionally substituted -(Ci-C ⁇ alkyl-R ; R 9 is a hydrogen;
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(Ci-C 6 )alkyl-R ⁇ , -(CrC ⁇ alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR U and and
- R and R are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(CrC ⁇ alkylhalo, -(CrC ⁇ alkyl, -(Q-C ⁇ alkylcyano, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, aryl, heterocycle and -(C 1 -C 6 )alkylaryl; provided that according to proviso (i): when M 3 is -(Co)-R 11 (that is when M 3 is -R 11 ), then R 11 is not H;
- a 1 and A 2 radicals are not linked to form an imidazopyridazinyl ring; and provided that according to proviso (v):
- a 1 is H, then M 1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M 3 is 4-methylphenyl, then B n can not be a phenyl; and provided that according to proviso (vii): when X 2 is S 5 X 3 is C, X 4 is C 5 n is I 5 then A 1 when linked to either X 3 or X 4 is not a pyridyl; and provided that according to proviso (viii): when X 2 is S 5 X 3 is C 5 X 4 is C 5 to provide a thiazole ring, n is 1, then A 1 is not an optionally substituted imidazolyl or triazolyl ring.
- X 3 is selected from C or N which may further be substituted by A 1 ;
- a 1 radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(C 1 -C 6 ⁇ IkVl, - (C] . -C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(CrC ⁇ alkylcyano, -(CrC ⁇ alkylheteroaryl, - (Ci-C 6 )alkylaiyl, heterocycle, -(Co-C ⁇ alkyl-OR 1 , -NR 1 (C 2 -C 6 )alkyl-OR 2 , -(C 3 - C 7 )cycloalkyl-(C 1 -C 6 )alkyl, -O-(C 3 -C 7 )cycloalkyl-(C 1 -C 6 )alkyl, -NR ⁇ (C 3 -
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen or an optionally substituted radical selected from the group of -(C 1 -C 6 )alkylhalo, -(CrC ⁇ alkyl, -(Ci-C 6 )alkylcyano, -(C 3 -
- R Any two radicals of R (R 1 , R 2 , R 3 or R 4 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
- B n radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(d-C ⁇ alkyl, - (CrC ⁇ alkylhalo, -(C 3 -C 7 )cycloalkyl, -(CrC ⁇ alkylcyano, -(d-C ⁇ alkylheteroaryl, - (CrC f Oalkylaryl, aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 5 , -O-(C 2 -C 6 )alkyl- OR 5 , -NR 5 (C 2 -C 6 )alkyl-OR 6 , -(Cs-C ⁇ cycloalkyHd-C ⁇ alkyl, -O-(C 3 -C 7 )cycloalkyl- (d-C 6 )alkyl, -NR 5
- R Any two radicals of R (R 5 , R 6 , R 7 or R 8 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- M 1 is selected from an optionally substituted aryl and heteroaryl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR U and -(C 2 -C 6 )alkyl-SR ⁇ ; and
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylhalo, -(Q-C ⁇ alkyl, -(Q-C ⁇ alkylcyano, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and -(Q-C ⁇ alkylaryl; and provided that according to proviso (v):
- a 1 radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(Ci-C ⁇ alkyl, -
- R 1 , R 2 and R 3 are each independently hydrogen or an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, -(C 4 - C 10 )alkylcycloalkyl and heterocycle; Any two radicals of R (R 1 , R 2 or R 3 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- B n radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(Q-C ⁇ alkyl, - (d-C ⁇ alkylhalo, -(C 3 -C 7 )cycloalkyl, -(Ci-C 6 )alkylcyano, -(Ci-C 6 )alkylheteroaryl 5 - (C 1 -C 6 )alkylaryl, aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 5 , -O-(C 2 -C 6 )alkyl- OR 5 , -NR 5 (C 2 -C 6 )alkyl-OR 6 , -(C 3 -C 7 )cycloalkyl-(C 1 -C 6 )alkyl, -O-(C 3 -C 7 )cycloalkyl
- R 5 , R 6 , R 7 and R 8 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C ⁇ alkylhalo, -(CrC ⁇ alkyl, -(C 1 -C 6 )alkylcyano, -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and -(Ci-C 6 )alkylaryl;
- R 5 , R 6 , R 7 and R 8 Any two radicals of R (R 5 , R 6 , R 7 and R 8 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- M 1 is selected from an optionally substituted aryl and heteroaryl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC ⁇ alkyl-R 11 , -(Ci-C 6 )alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR ⁇ and -(C 2 -C 6 )alkyl-SR n ; and
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C ⁇ alkylhalo, -(Ci-C ⁇ alkyl, -(CrC ⁇ alkylcyano, -(C 3 -C 7 )cycloalkyl 5 -(C 4 -C 10 )alkylcycloalkyl 5 heteroaryl, -(d-C ⁇ alkylheteroaryl, aryl, heterocycle and -(Cj . -C 6 )alkylaryl; provided that according to proviso (v):
- the invention provides a compound according to Formula (IIIA),
- a 1 radical is selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(Ci-C 6 )alkyl, -(C 1 -
- R 1 , R 2 and R 3 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylcyano, -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )aUiylcycloalkyl, heteroaryl, aryl, heterocycle and -(C t -C ⁇ alkylaryl;
- R Any two radicals of R (R 1 , R 2 and R 3 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
- KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008 B n radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of -(Ci-C 6 )alkyl, - (CrC ⁇ alkylhalo, -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 5 , - O-(C 2 -C 6 )alkyl-OR 5 , -NR 5 (C 2 -C 6 )alkyl-OR 6 , -(Cs-C ⁇ cycloalkyl-Cd-C ⁇ alkyl, -0-(C 3 - OR 5 , -(d-C ⁇ alkylhalo-NR ⁇ 6 , -(C 0 -C 6 )alkyl-NR
- R 5 , R 6 and R 7 are each independently hydrogen or an optionally substituted radical selected from the group of -(Q-C ⁇ alkylhalo, -(CrC ⁇ alkyl, -(CrC ⁇ alkylcyano, -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(d-C ⁇ alkylheteroaryl, aryl, heterocycle and -(Ci-C ⁇ alkylaryl; Any two radicals of R (R 5 , R 6 , or R 7 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- M 1 is selected from an optionally substituted aryl and heteroaryl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(C 1 -C 6 )alkyl-R 11 , -(d-C ⁇ alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR ⁇ and -(C 2 -C 6 )alkyl-SR n ; and
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(d-C ⁇ alkylhalo, -(Ct-C ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(d-C ⁇ alkylheteroaryl, aryl, heterocycle and - (d-C ⁇ alkylaryl; provided that according to proviso (v):
- a 1 radical is selected from the group of hydrogen, halogen, -CF 3 , and an optionally substituted radical selected from the group of -(Q-C ⁇ alkyl, -(Q-C ⁇ alkylhalo, -(C 3 - C 7 )cycloalkyl and heterocycle; n is an integer ranging from 1 to 2, and either;
- n is 1 and B 1 radical is selected from the group of hydrogen, halogen, -CF 3 , and an optionally substituted radical selected from the group of -(CrC ⁇ alkyl, -(C 1 - C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(C 0 -C 6 )alkyl-OR 5 , and -(C 3 -C 7 )CyClOaIlCyI-(C 1 - C 6 )alkyl; or
- R 5 is selected from the group of hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylhalo, -(Ci-C 6 )alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 - C 10 )alkylcycloalkyl and heterocycle;
- M 1 is an optionally substituted aryl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(C 1 -C 6 )alkyl-R 11 , -(Ci-C 6 )alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR n and -(C 2 -C 6 )alkyl-SR ⁇ ;
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C] . -C 6 )alkylhalo, -(Ci-C 6 )alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and - (Ci-C 6 )alkylaryl; provided that according to proviso (v):
- a 1 radical is selected from the group of hydrogen, halogen, -CF 3 , and an optionally substituted radical selected from the group of -(Ci-C 6 )alkyl, -(C t -C ⁇ alkylhalo, -(C 3 - C 7 )cycloalkyl and heterocycle; n is an integer ranging from 1 to2, and either;
- n 1 and B 1 radical is selected from the group of hydrogen, halogen, -CF 3 , and an optionally substituted radical selected from the group of -(Ci-C 6 )alkyl, -(C 1 - C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(C 0 -C 6 )alkyl-OR 5 , and -(C 3 -C 7 )cycloalkyl-(Ci- C 6 )alkyl; or (b) n is 2, and B 1 and B 2 radicals are each independently selected from the group of - CF 3 and an optionally substituted radical selected from the group of -(Q-C ⁇ alkyl, -(C 1 - C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, and -(
- R 5 is selected from the group hydrogen or an optionally substituted radical selected from the group of -(CrC ⁇ alkylhalo, -(Q-C ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 - C 10 )alkylcycloalkyl and heterocycle;
- M 1 is an optionally substituted heteroaryl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC ⁇ alkyl-R 11 , -(CrC ⁇ alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR ⁇ and -(C 2 -C 6 )alkyl-SR n ;
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(CrC ⁇ alkylhalo, -(d-C ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and - (Q-COal
- M 1 can not be 1-methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii):
- a 1 is not a pyridyl; and provided that according to proviso (viii):
- a 1 radical is hydrogen, n is an integer ranging from 1 to2, and either;
- n is 1 and B 1 radical is selected from the group of hydrogen, -CF 3 , -(Q-C ⁇ alkyl and -(d-COalkylhalo; or
- M 1 is an optionally substituted pyridyl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(Ci-C 6 )alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR ⁇ and -(C 2 -C 6 )alkyl-SR ⁇ ; and
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylhalo, -(Ci-C ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, aryl, heterocycle and - (Ci-C 6 )alkylaryl.
- the invention provides a compound according to Formula (IIIB),
- n is an integer ranging from 1 to 2;
- B n radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF 3 , and an optionally substituted radical selected from the group of - (CrC 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(C 0 -C 6 )alkyl-OR 5 , - O-(C 2 -C 6 )alkyl-OR 5 , -NR 5 (C 2 -C 6 )alkyl-OR 6 , -(C 3 -C 7 )cycloalkyl-(C 1 -C 6 )alkyl, -0-(C 3 - C ⁇ cycloalkyKd-C ⁇ alkyl, -NR 5 -(C 3 -C 7 )cycloalkyl-(C 1 -C 6 )alkyl, -(d-C ⁇ alkylhalo- OR 5 , -(d
- R 5 , R 6 and R 7 are each independently hydrogen or an optionally substituted radical selected from the group of -(d-C ⁇ alkylhalo, -(C 1 -C 6 )alkyl, -(C 1 -C 6 )alkylcyano, -(C 3 - C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and -(C 1 -C 6 )alkylaryl; Any two radicals of R (R 5 , R 6 , or R 7 ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
- M 1 is selected from an optionally substituted aryl and heteroaryl
- KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008 M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )CyClOaHCyI, aryl, heteroaryl, heterocycle, -(Ci-C 6 )alkyl-R n , -(Ci-C 6 )alkylhalo. -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR n and -(C 2 -C 6 )alkyl-SR ⁇ ; and
- R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylhalo, -(CrC ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, aryl, heterocycle and - (Ci-C 6 )alkylaryl.
- n is an integer ranging from 1 to 2, and either;
- n 1 and B 1 radical is selected from the group of hydrogen, halogen, -CF 3 , and an optionally substituted radical selected from the group of -(CrC ⁇ alkyl, -(C 1 - C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(C 0 -C 6 )alkyl-OR S , aryl, heteroaryl, and -(C 3 - C 7 )cycloalkyl-(C 1 -C 6 )alkyl; or (b) n is 2, and B 1 and B 2 radicals are each independently selected from the group of - CF 3 and an optionally substituted radical selected from the group of -(C t -C ⁇ alkyl, -(C 1 - C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, and -(C 3 -C 7 )cycloalkyl-(Ci
- R 5 is selected from the group hydrogen or an optionally substituted radical selected from the group of -(CrC 6 )alkylhalo, -(CrC ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 - C 10 )alkylcycloalkyl and heterocycle;
- M 1 is an optionally substituted aryl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(d-C ⁇ alkyl-R 11 , -(CrC ⁇ alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(Ca-C ⁇ alkyl-OR 11 and -(C 2 -C 6 )alkyl-SR ⁇ ; and R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl,
- n is an integer ranging from 1 to 2, and either;
- n is 1 and B 1 radical is selected from the group of hydrogen, halogen, -CF 3 , and an optionally substituted radical selected from the group of -(C 1 -C 6 )alkyl, -(C 1 - C 6 )alkylhalo, -(C 3 -C 7 )cycloalkyl, -(C 0 -C 6 )alkyl-OR 5 , aryl, heteroaryl, and -(C 3 - C 7 )cycloalkyl-(Ci-C 6 )alkyl; or (b) n is 2, and B 1 and B 2 radicals are each independently selected from the group of - CF 3 and an optionally substituted radical selected from the group -(C 1 - C 6 )alkylhalo, ⁇ (C 3 -C 7 )cycloalkyl, and -(C 3 -C 7 )cycloalkyl-(C 1 -C 6 )alkyl;
- R 5 is selected from the group hydrogen or an optionally substituted radical selected from the group of -(Ci-C 6 )alkylhalo, -(Ci-C 6 )alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 - C 10 )alkylcycloalkyl and heterocycle;
- M 1 is an optionally substituted heteroaryl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, aryl, heteroaryl, heterocycle, -(Q-C ⁇ alkyl-R 11 , -(Ci-C 6 )alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(C 2 -C 6 )alkyl-OR ⁇ and and R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C ⁇ alkylhalo, -(Q-C ⁇ alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(Q-C ⁇ alkylheteroaryl, aryl, heterocycle and - (Ci-C f Oalkylaryl.
- n is an integer ranging from 1 to 2, and either;
- n 1 and B 1 radical is selected from the group of hydrogen, -CF 3 , -(Q-C ⁇ alkyl and -(Ci-C 6 )alkylhalo; or
- n 2 ⁇ n + 2 ⁇ n + 2 ⁇ n + 2 ⁇ n
- M 1 is an optionally substituted pyridyl
- M 3 is an optionally substituted radical selected from the group of -(C 3 -C 7 )cycloalkyl, atyl, heteroaryl, heterocycle, -(Ci-C 6 )alkyl-R u s -(CrC ⁇ alkylhalo, -(C 2 -C 6 )alkyl- NR 11 R 12 , -(d-C ⁇ alkyl-OR 11 and -(C 2 -C 6 )alkyl-SR ⁇ ; and R 11 and R 12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C 1 -C 6 )alkymalo, -(Ci-C 6 )alkyl, -(C 3 -C 7 )cycloalkyl, -(C 4 -C 10 )alkylcycloalkyl, heteroaryl, -(CrC ⁇ alkylheteroaryl, aryl, heterocycle and - (
- Specific compounds of the invention according to Formula (I) to (III) are compounds as mentioned in the following list A and list B as well as a pharmaceutically acceptable acid or base addition salt thereof, a stereochemical ⁇ isomeric form thereof and an N- oxide form thereof:
- (C 1 -C 6 ) means a carbon radical having 1, 2, 3, 4, 5 or 6 carbon atoms.
- (Co-C 6 ) means a carbon radical having 0, 1, 2, 3, 4, 5 or 6 carbon atoms.
- C means a carbon atom
- N means a nitrogen atom
- O means an oxygen atom
- S means a sulphur atom.
- bonds refers to a saturated covalent bond.
- bonds When two or more bonds are adjacent to one another, they are assumed to be equal to one bond.
- alkyl includes both straight and branched chain alkyl radicals and may be methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, s-butyl, t-butyl, n-pentyl, i-pentyl, t-pentyl, neo-pentyl, n-hexyl, i-hexyl or t- hexyl.
- (C 0 -C 3 )alkyl refers to an alkyl radical having 0, 1, 2 or 3 carbon atoms and may be methyl, ethyl, n-propyl and i-propyl.
- cycloalkyl refers to an optionally substituted carbocycle containing no heteroatoms, including mono-, bi-, and tricyclic saturated carbocycles, as well as fused ring systems.
- fused ring systems can include one ring that is partially or fully unsaturated such as a benzene ring to form fused ring systems such as benzo- fused carbocycles.
- Cycloalkyl includes such fused ring systems as spirofused ring systems.
- cycloalkyl examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, decahydronaphthalene, adamantane, indanyl, fluorenyl and 1,2,3,4-tetrahydronaphthalene and the like.
- (C 3 -C 7 )cycloalkyT may be cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- alkenyl includes both straight and branched chain alkenyl radicals.
- (C 2 -C 6 )alkenyl refers to an alkenyl radical having 2 to 6 carbon atoms and one or two double bonds, and may be, but is not limited to vinyl, allyl, propenyl, i-propenyl, butenyl, i-butenyl, crotyl, pentenyl, i-
- alkynyl includes both straight and branched chain alkynyl radicals.
- aryl refers to an optionally substituted monocyclic or bicyclic hydrocarbon ring system containing at least one unsaturated aromatic ring.
- suitable values of the term “aryl” are phenyl, naphthyl, 1,2,3,4-tetrahydronaphthyl, indyl, indenyl, benzo[d][l,3]dioxolyl and the like.
- heteroaryl refers to an optionally substituted monocyclic or bicyclic unsaturated, aromatic ring system containing at least one heteroatom selected independently from N, O or S.
- heteroaryl may be, but are not limited to thienyl, pyridyl, thiazolyl, isothiazolyl, furyl, pyrrolyl, triazolyl, imidazolyl, oxadiazolyl, oxazolyl, isoxazolyl, pyrazolyl, imidazolonyl, oxazolonyl, thiazolonyl, tetrazolyl, thiadiazolyl, benzoimidazolyl, benzoxazolyl, benzothiazolyl, tetrahydrotriazolopyridyl, tetrahydrotriazolopyrimidinyl, benzofuryl, benzothiophenyl,
- Examples of pyrazolyl may be, but not limited to, IH- pyrazol-4-yl.
- pyridinyl may be, but not limited to, pyridin-2-yl, pyridin-3- yl and pyridin-4-yl.
- alkylaryl refers respectively to a substituent that is attached via the alkyl radical to an aryl, heteroaryl or cycloalkyl radical, respectively.
- (C 1 - C 6 )alkylaryl includes aryl-Ci-C 6 -alkyl radicals such as benzyl, 1-phenylethyl, 2- phenylethyl, 1-phenylpropyl, 2-phenylpropyl, 3-phenylpropyl, 1-naphthylmethyl and 2- naphthylmethyl.
- (Ci-C 6 )alkyheteroaryl includes heteroaryl-CrC ⁇ -alkyl radicals, wherein examples of heteroaryl are the same as those illustrated in the above definition, such as 2-furylmethyl, 3-furylmethyl, 2-thienylmethyl, 3-thienylmethyl, 1- imidazolylmethyl, 2-imidazolylmethyl, 3-imidazolyhnethyl, 2-oxazolylmethyl, 3- oxazolylmethyl, 2-thiazolylmethyl, 3-thiazolylmethyl, 2-pyridylmethyl, 3- pyridylmethyl, 4-pyridylmethyl, 1-quinolylmethyl or the like.
- heterocycle refers to an optionally substituted, monocyclic or bicyclic saturated, partially saturated or unsaturated ring system (containing at least one heteroatom selected independently from N, O and S.
- the said “heterocycle” refers respectively to a group linked either via the carbon or the nitrogen.
- heterocycle includes morpholine, thiomorpholine, tetrahydrofuran, tetrahydropyran radicals or the like.
- alkylheterocycle refers respectively to a substituent that is attached via the alkyl radical to a heterocycle radical, respectively.
- (Q-C ⁇ alkyl heterocycle” includes heterocycle-Ci-C 6 - alkyl radicals such as (tetrahydro-2H-pyran-4-yl)methyl, (tetrahydrofuran-2-yl)methyl or morpholinoethyl.
- a 5- or 6-membered ring containing one or more atoms independently selected from C, N, O and S includes aromatic and heteroaromatic rings as well as carbocyclic and heterocyclic rings which may be saturated or unsaturated.
- Such rings may be, but are not limited to, furyl, isoxazolyl, isothiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidyl,
- KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008 pyrrolyl, thiazolyl, thienyl, imidazolyl, imidazolidinyl, imidazolinyl, triazolyl, morpholinyl, piperazinyl, piperidyl, piperidonyl, pyrazolidinyl, pyrazolinyl, pyrrolidinyl, pyrrolinyl, tetxahydropyranyl, tetrahydrothiopyranyl, oxazolidinonyl, thiomorpholinyl, oxadiazolyl, thiadiazolyl, tetrazolyl, phenyl, cyclohexyl, cyclopentyl, cyclohexenyl and cyclopentenyl.
- a 3- to 10-nienibered ring containing one or more atoms independently selected from C, N, O and S includes aromatic and heteroaromatic rings as well as carbocyclic and heterocyclic rings which may be saturated or unsaturated.
- rings may be, but are not limited to imidazolidinyl, imidazolinyl, morpholinyl, piperazinyl, piperidyl, piperidonyl, pyrazolidinyl, pyrazolinyl, pyrrolidinyl, pyrrolinyl, tetrahydropyranyl, thiomorpholinyl, tetrahydrothiopyranyl, furyl, pyrrolyl, isoxazolyl, isothiazolyl, oxazolyl, oxazolidinonyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidyl, pyrrolyl, thiazolyl, thienyl, imidazolyl, triazolyl, phenyl, cyclopropyl, aziridinyl, cyclobutyl, azetidinyl, oxadia
- alkylhalo means an alkyl radical as defined above, substituted with one or more halo radicals.
- (C 1 - C 6 )alkylhalo may include, but is not limited to, fluoromethyl, difluoromethyl, trifluoromethyl, fluoroethyl, difluoroethyl and trifluoroethyl.
- 0-C 1 -C 6 - alkylhalo may include, but is not limited to, fluoromethoxy, difluoromethoxy, trifluoromethoxy and fluoroethoxy.
- alkylcyano means an alkyl radical as defined above, substituted with one or more cyano.
- the term “optionally substituted” refers to radicals further bearing one or more substituents which may be, but are not limited to, (Ci-C 6 )alkyl, (C 3 -C 7 )cycloalkyl, hydroxy, (CrC 6 )alkyloxy, mercapto, aryl, heterocycle, halogen, trifluoromethyl, pentafluoroethyl, cyano, cyanomethyl, nitro, amino, amido, amidinyl, carboxyl, carboxamide, (CrC ⁇ alkyloxycarbonyl, carbamate, sulfonamide, ester and sulfonyl.
- an optionally substituted (Q-C ⁇ alkyl radical, such as a methyl group, by a (C 3 -C 7 )cycloalkyl, such as cyclopropyl refers to a cyclopropylmethyl radical.
- pyridyl radicals substituted in the 3- position carboxyl, cyano and carboxamide group may be called nicotinic, nicotinotrile and nicotinamide radicals.
- solvate refers to a complex of variable stoichiometry formed by a solute (e.g. a compound of Formula (I)) and a solvent.
- the solvent is a pharmaceutically acceptable solvent as preferably water; such solvent may not interfere with the biological activity of the solute.
- the term “antagonists of A 3 refers also to a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV ⁇ oxide form thereof.
- Antagonists of A 3 described herein, and the pharmaceutically acceptable salts, solvates and hydrates thereof can be used in pharmaceutical preparations in combination with a pharmaceutically acceptable carrier or diluent.
- Suitable pharmaceutically acceptable carrier or diluent Suitable pharmaceutically acceptable
- KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008 carriers include inert solid fillers or diluents and sterile aqueous or organic solutions.
- the antagonists of A 3 will be present in such pharmaceutical compositions in amounts sufficient to provide the desired dosage amount in the range described herein. Techniques for formulation and administration of the compounds of the instant invention can be found in Remington: the Science and Practice of Pharmacy, 19 th edition, Mack Publishing Co., Easton, PA (1995).
- the amount of antagonists of A 3 receptor, administered to the subject will depend on the type and severity of the disease or condition and on the characteristics of the subject, such as general health, age, sex, body weight and tolerance to drugs. The skilled artisan will be able to determine appropriate dosages depending on these and other factors. Effective dosages for commonly used CNS drugs are well known to the skilled person.
- the total daily dose usually ranges from about 0.05 - 2000 mg.
- compositions which provide from about 0.01 to 1000 mg of the active ingredient per unit dose.
- the compositions may be administered by any suitable route.
- parenterally in the form of solutions for injection topically in the form of unguents or lotions, ocularly in the form of eye-drops, rectally in the form of suppositories, intranasally or transcutaneously in the form of delivery system like patches.
- the antagonists of A 3 receptor thereof can be combined with a suitable solid or liquid carrier or diluent to form capsules, tablets, pills, powders, syrups, solutions, suspensions and the like.
- the tablets, pills, capsules, and the like contain from about 0.01 to about 99 weight percent of the active ingredient and a binder such as gum tragacanth, acacias, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid, a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008 Various other materials may be present as coatings or to modify the physical form of the dosage unit. For instance, tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
- the disclosed antagonists of A 3 can be combined with sterile aqueous or organic media to form injectable solutions or suspensions.
- injectable solutions or suspensions for example, solutions in sesame or peanut oil, aqueous propylene glycol and the like can be used, as well as aqueous solutions of water-soluble pharmaceutically-acceptable salts of the compounds.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the compounds may also be formulated as a depot preparation.
- Such long acting formulations may be administered by implantation, for example, subcutaneously or intramuscularly or by intramuscular injection.
- implantation for example, subcutaneously or intramuscularly or by intramuscular injection.
- sparingly soluble derivatives for example, as sparingly soluble salts.
- the antagonists of A 3 receptor described herein, and their pharmaceutically acceptable salts can be incorporated into various types of ophthalmic formulations for delivery to the eye (e.g., topically, intracamerally, or via an implant). Such compounds are preferably incorporated into topical ophthalmic formulations for delivery to the eye.
- the compounds may be combined with ophthalmologically acceptable preservatives, surfactants, viscosity enhancers, penetration enhancers, buffers, sodium chloride, and water to form an aqueous, sterile ophthalmic suspension or solution.
- Ophthahnic solution formulations may be prepared by dissolving a compound in a physiologically acceptable isotonic aqueous buffer. Further, the ophthalmic solution may include an ophthalmologically acceptable surfactant to assist in dissolving the compound.
- the ophthalmic solution may contain an agent to increase viscosity, such as, hydroxyniethylcellulose, methylcellulose, polyvinylpyrrolidone, and the like, to improve the retention of the formulation in the conjunctival sac.
- Gelling agents can also be used, such as xanthan gum.
- the active ingredient is combined with a preservative in an appropriate vehicle, such as, mineral oil or liquid lanolin.
- Sterile ophthalmic gel formulations may be prepared by suspending the compound in a hydrophilic base, according to the published formulations for analogous ophthalmic preparations. Some preservatives and tonicity agents can be incorporated.
- the antagonists of A 3 receptor described herein can be formulated as topical ophthalmic suspensions or solutions, with a pH of about 4 to 8. Such compounds will normally be contained in these formulations in an amount 0.01% to 5% by weight.
- the dosage form may be a solution, suspension, or microemulsion.
- 1 to 2 drops of these formulations would be delivered to the surface of the eye 1 to 4 times per day according to the discretion of a skilled clinician.
- Preferably disclosed antagonists of A 3 or pharmaceutical formulations containing these compounds are in unit dosage form for administration to a mammal.
- the unit dosage form can be any unit dosage form known in the art including, for example, a capsule, an IV bag, a tablet, or a vial.
- the quantity of active ingredient in a unit dose of composition is an effective amount and may be varied according to the particular treatment involved. It may be appreciated that it may be necessary to make routine variations to the dosage depending on the age and condition of the patient.
- the dosage will also depend on the route of administration which may be by a variety of routes including oral, aerosol, rectal, transdermal, subcutaneous, intravenous, intramuscular, intraperitoneal, intraocular, eye drop and intranasal.
- the compounds according to the invention may be prepared by methods known in the art of organic synthesis as set forth in part by the following synthesis schemes. In all of the schemes described below, it is well understood that protecting groups for sensitive or reactive groups are employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (Green T.W. and Wuts P.G.M. (1991) Protecting Groups in Organic Synthesis, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art.
- the selection of process as well as the reaction conditions and order of their execution shall be consistent with the preparation of compounds of Formula (I), (II), (III), (IIIA) and (IIIB).
- the compounds according to the invention may be represented as a mixture of enantiomers, which may be resolved into the individual pure R- or S'-enantiomers. If for instance, a particular enantiomer is required, it may be prepared by asymmetric synthesis or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers.
- this resolution may be conveniently performed by fractional crystallization from various solvents as the salts of an optical active acid or by other methods known in the literature (e.g. chiral column chromatography). Resolution of the final product, an intermediate or a starting material may be performed by any suitable method known in the art (Eliel E.L., Wilen S. H. and Mander L.N. (1984) Stereochemistry of Organic Compounds, Wiley-Interscience).
- heterocyclic compounds of the invention can be prepared using synthetic routes well known in the art (Katrizky A.R. and. Rees CW. (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
- the product from the reaction can be isolated and purified employing standard techniques, such as extraction, chromatography, crystallization and distillation.
- the compounds of the invention may be prepared by general route of synthesis as disclosed in the following methods or according to any method known from the man skilled in the art.
- compounds of Formula (III) and (HIB) may be prepared according to the synthetic sequences illustrated in Scheme 1.
- Pyrazole gl can be substituted using Mitsunobu conditions.
- amidine can be synthesized either from ester treated with aluminium chloride in the presence of ammonium chloride or from nitrile by synthesis of amidoxime g3 followed by hydrogenation, in the presence of Pd/C and anhydride acetic.
- the cyclization between the amidine g4 and the isothiocyanate g5 may be promoted by di-tert-butylazodicarboxylate and a base such as DBU.
- the compounds of Formula (III) and (IIIA) may be prepared according to the synthetic sequences illustrated in Scheme 2.
- Compound g7 may be hydrolyzed by standard procedures followed by reaction with oxalyl chloride to yield compound g9.
- cyclization reaction may be performed between the halo-ketone glO and the thiourea gll to yield the aminothiazole gl2.
- Thioureas gll can be prepared form the corresponding isothiocyanates g5 by reaction with methanolic ammonia.
- isothiocyanate g5 was not commercially available or known in the literature, it was prepared from the corresponding amine by treatment either with TDCI (M.P.Gauthier at al (2006) Biorg. Med. Chem. 14: H918-H927) or with thiophosgene (R. D.Haugwitz et al (1985) J. Med. Chem. 9: H1234-H1241) as described in literature.
- the compounds of Formula (II) may be prepared according to the synthetic sequences illustrated in Scheme 3.
- Aminothiadiazole gl3 can be alkylated into gl4 in the presence of a base such as NaH and a solvent such as THF.
- the compounds of Formula (III) and (IIIB) may be prepared according to the synthetic sequences illustrated in Scheme 4.
- Pyrazole gl5 can be alkylated using the proper alkyl halide, such as bromide and iodide, or triflate in presence of an inorganic base, such as potassium carbonate, in a suitable organic solvent.
- Alternativerly pyrazole gl5 can be arylated using the proper aryl halide in the presence of an inorganic base, such as potassium carbonate, a copper catalyst, such as copper iodide, a proper ligand, such as 1,10-phenanthroline, in a suitable organic solvent, such as dioxane (Y-M.
- Compound gl6 can be alternatively obtained by esterification of the corresponding commercially available acids gl7 using standard procedures. Then amidine gl8 is synthesized from the corresponding ester by reaction with aluminium chloride in the presence of ammonium chloride in a suitable solvent, such as toluene. The cyclization between the amidine gl8 and the isothiocyanate gl9 is promoted by di- tert-butylazodicarboxylate and a base such as DBU, to provide compound g6.
- the compounds of Formula (III) and (IIIB) may be prepared according to the synthetic sequences illustrated in Scheme 5.
- Compound gl8, prepared as described in Scheme 4 can be converted into compound g20 by cyclization using trichloromethanesulfenyl chloride.in presence of an inorganic base, such as sodium hydroxide, in water.
- Compound g20 can be converted to compound g6 by substitution with the desired amine in presence of a suitable base, such as potassium fer/-butoxide, in a suitable solvent (dioxane) under reflux.
- a suitable base such as potassium fer/-butoxide
- Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7 ⁇ m, 50x2. lmm) from Waters, with a flow rate of 0.5 ml/min.
- 0-0.30min A: 92%, B: 8%), 0.30-1.50 min (A: 0%, B: 100%), 1.50- 2.00 min (A: 0%, B: 100%), 2.00-2.40 min (A: 95%, B: 5%).
- Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7 ⁇ m, 50x2. lmm) from Waters, with a flow rate of 0.5 ml/min.
- 0-0.30min A: 95%, B: 5%
- 0.30-3.30 min A: 0%, B: 100%
- 3.30- 3.90 min A: 0%, B: 100%
- 3.90-4.40 min A: 95%, B: 5%).
- Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7 ⁇ m, 50x2. lmm) from Waters, with a flow rate of 0.5 ml/min.
- 0-O.lOmin A: 95%, B: 5%
- 0.10-1.40 min A: 0%, B: 100%
- 1.40- 1.90 min A: 0%, B: 100%
- 1.90-2.40 min A: 95%, B: 5%).
- Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7 ⁇ m, 50x2. lmm) from Waters, with a flow rate of 0.6 ml/min.
- 0-0.25min A: 95%, B: 5%
- 0.25-3.30 min A: 0%, B: 100%
- 3.30- 4.00 min A: 0%, B: 100%
- 4.00-4.10 min A: 95%, B: 5%
- 4.10-5.00 min A: 95%, B: 5%).
- ES MS detector was used, acquiring both in positive and negative ionization modes. Cone voltages were 30 V (Method LC-A), 26V (Methods LC-B, LC-C and LC-D) and 25V (Methods LC-E and LC-F) for both positive and negative ionization modes. All mass spectra were taken under electrospray ionisation (ESI) methods (see Table 3).
- Table 1 Compounds prepared according to the Examples.
- Step 1 of the general methodology Triphenylphosphine (11 mmol, 2.9 g), (4-methoxyphenyl)methanol (10 mmol, 1.4 g) and di-tert- butylazodicarboxylate (11 mmol, 2.6 g) were added to a solution of 3 -methyl- IH- pyrazole-4-carbonitrile (9.3 mmol, 1.0 g), in DCM (40 mL) at O 0 C. The reaction mixture was stirred at room temperature overnight. The organic phase was washed with a saturated solution of NH 4 OH and brine. Then the organic phase was dried over MgSO 4 , was filtered and was concentrated under reduced pressure.
- the resulting crude product was purified by flash chromatography over silica gel using cyclohexane/ AcOEt (90:10) as eluent to yield l-(4-methoxybenzyl)-3-methyl-lH-pyrazole-4-carbonitrile and l-(4-methoxybenzyl)-5-methyl-lH-pyrazole-4-carbonitrile (9.3 mmol, 2.1g, 100%).
- Step 2 of the general methodology Method A: A mixture of 1- (4-methoxybenzyl)-3 -methyl- 1 H-pyrazole-4-carbonitrile and 1 -(4-methoxybenzyl)-5 - methyl-lH-pyrazole-4-carbonitrile (9.90 mmol, 2.25 g), hydroxylamine 50% in water (19.8 mmol, 1.21 mL) and EtOH (10 mL) was heated at 80 0 C for 12 hours.
- Step 3 of the general methodology DBU (0.39 mmol, 60.0 mg) was added to a solution of l-fluoro-2-isothiocyanatobenzene (0.39 mmol, 48 ⁇ l) and 1- (4-methoxybenzyl)-3 -methyl- lH-pyrazole-4-carboximidamide and l-(4- methoxybenzyl)-5 -methyl- lH-pyrazole-4-carboximidamide (0.39 mmol, 200 mg) in DMF (7 mL) under nitrogen. The reaction mixture was stirred at room temperature until total consumption of the amidine.
- di-fert-butylazodicarboxylate (0.43 mmol, 100 mg) was added dropwise and the reaction mixture was stirred for 5 minutes. After evaporation of the EtOH, water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with a solution of HCl 1 M, water and brine, was dried over Na 2 SO 4 , was filtered and was concentrated under reduced pressure.
- Step 3 of the general methodology A solution of TMSdiazomethane (6.0 mmol, 3.0 mL) was added to a solution of 1-isopropyl-lH- pyrazole-4-carbonyl chloride (1.95 mmol, 0.33 g) in acetonitrile (5 mL) at 0°C. The reaction mixture was stirred at room temperature overnight. HBr (8.0 mmol, 0.9 mL, 48%) was added at 0 0 C to the reaction mixture. The reaction mixture was stirred at room temperature for one hour.
- Step 4 of the general methodology A solution of 2-bromo-l- (l-isopropyl-lH-pyrazol-4-yl)ethanone (0.43 mmol, 0.10 g) and of l-(pyridin-2- yl)thiourea (0.35 mmol, 53 mg) in acetone (5 mL) was stirred under reflux for one hour. After evaporation of the solvent, DCM was added and the organic phase was washed with a saturated solution OfNaHCO 3 , water and brine. The organic phase was dried over Na 2 SO 4 , was filtered and was concentrated under reduced pressure.
- EXAMPLE 5 SKl-CCyclopropylmethy ⁇ -lH-pyrazol ⁇ -y ⁇ -N ⁇ pyridin-l-yl)-! ⁇ - thiadiazol-5-amine 1 (Final compound 1-22) and 3 ⁇ (l-(Cyclopropylmethyl)-lH- pyrazol-4-yl)-N-(pyridin ⁇ 2-yl)-l,2,4-thiadiazol-5 ⁇ amine monochlohydrate salt (Final compound 1.22a)
- Step Ia of the general methodology K 2 CO 3 (5.35 mmol, 740 mg) and cyclopropylmethylbromide (7.12 mmol, 962 mg) were added to a solution of ethyl l-H-pyrazole-4-carboxylate (3.57 mmol, 500 mg) in acetone (5 mL). The reaction mixture was stirred at reflux for 8 hours. After cooling to room temperature, inorganics were filtered off and the filtrated was concentrated to dryness affording ethyl 1- (cyclopropylmethyl)-lH-pyrazole-4-carboxylate (3.40 mmol, 660 mg, 95%) as colorless oil.
- 6-Cyclobutoxypyridin-2-amine (c3) Sodium hydride (3.80 mmol, 150 mg) was added portionwise to a solution of cyclobutanol (3.46mmmol, 0.27 mL) in acetonitrile and the reaction mixture was stirred 1 hour at room temperature. 6-Bromopyridine-2-amine (1.73 mmol, 0.30 g) was added and the reaction mixture was heated at 130°C for 3 hours under microwave irradiation. After evaporation of the solvent, water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with water, brine and dried over Na 2 SO 4 . The organic solvent was filtered and concentrated under reduced pressure.
- reaction mixture was concentrated and purified by flash chromatography over silica gel using petroleum ether/ AcOEt (90:10 to 80:20) as eluent affording 2-isothiocyanato-6-methylpyridine (1.21 mmol, 0.18 mg, 33 %) as pale yellow oil;
- Step Ib of the general methodology a solution of l-ethyl-3- methyl-lH-pyrazole-4-carboxylic acid (3.24 mmol, 0.50 g) and sulphuric acid 96% (0.5 mL) in methanol (5 mL) was heated at 70°C for 48 hours. After evaporation of the solvent, AcOEt was added and the organic phase was washed with a saturated solution of NaHCO 35 water and brine, dried over Na 2 SO 4 , filtered and concentrated under
- EXAMPLE 8 6-(3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazol-5- ylamino)-nicotinonitrile (Final compound 1-33)
- Step 2 of the general methodology a mixture of 5-chloro-3-(l- (cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazole (0.22 mmol, 54 mg), 6- aminonicotinonitrile (0.44 mmol, 53 mg) and sodium tert-butoxide (0.33 mmol, 32 mg) in dioxane (3 mL) was heated under reflux overnight. Sodium tert-butoxide (0.11 mmol, 12 mg) was added and the reaction mixture was refluxed for other 5 hours. After cooling at room temperature the solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography over silica gel using DCM/MeOH
- EXAMPLE 10 6-(3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazol-S- ylamino)nicotinamide (Final compound 1-35)
- Step 4b of the general methodology methyl 6-(3-(l- (cyclopropylmethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4-thiadiazol-5-ylamino) nicotinate (0.17 mmol, 60 mg) was suspended in 32% NH 4 OH solution and the mixture was heated in a closed vessel at 100°C under microwaves irradiation for Ih. After cooling, a precipitate was formed.
- Step 1 of the general methodology ethyl 1 -(2,2,2- trifluoroethyl)-lH-pyrazole-4-carboxylate (1.03 mmol, 230 mg) was suspended in a mixture of HCl 37% aqueous solution (2 mL) and dioxane (3 mL) and the mixture was heated overnight at reflux. The reaction mixture was concentrated to dryness affording l-(2,2,2-trifluoroethyl)-lH-pyrazole-4-carboxylic acid (1.03 mmol, 200 mg, 100%) as an off white solid. The crude product was used without further purification.
- Step 2 of the general methodology A solution of 1 -(2,2,2- trifluoroethyl)-lH-pyrazole-4-carboxylic acid (1.03 mmol, 200 mg), oxalyl chloride (2.58 mmol, 0.22 mL) and two drops of DMF in DCM (5 mL) was stirred for 4 hours at room temperature. After evaporation of the solvent, 1 -(2,2,2 -trifluoroethyl)- IH- pyrazole-4-carbonyl chloride (1.03 mmol, 219 mg) was obtained as a brown oil. The crude product was used without further purification.
- Step 3 of the general methodology a solution of TMSdiazomethane (3.90 mmol, 1.95 mL) was added to a solution of 1 -(2,2,2- trifluoroethyl)-lH-pyrazole-4-carbonyl chloride ⁇ (1.03 mmol, 219 mg) in acetonitrile (5 mL) at 0°C. The reaction mixture was stirred at room temperature overnight. After cooling to O 0 C, HBr (4.16 mmol, 0.7 mL, 48%) was added to the reaction mixture. The reaction mixture was stirred at room temperature for two hour. AcOEt and water were added to the reaction mixture and the organic layer was separated.
- Step 4 of the general methodology a solution of 2-bromo-l-(l- (2,2,2-trifluoroethyl)-lH- ⁇ yrazol-4-yl)ethanone and 2-chloro-l-(l-(2,2,2- trifluoroethyl)-lH-pyrazol-4-yl)ethanone (0.38 mmol, 105 mg) and of l-(pyridin-2-
- the compounds provided in the present invention are highly selective antagonists of the human adenosine A 3 receptor.
- the compounds of Formula I to III block the activation of adenosine A 3 receptor induced by an agonist of the receptor, while they have little or no effect against other subtypes of adenosine receptors including human A 1 , human A 2 A and human A 2B receptors.
- Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A 3 receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described in Salvatore et al. ( (1993) Proc. Natl. Acad. ScL USA, 90: 10365).
- cell membrane homogenates (40 ⁇ g protein) were incubated for 120 min at 22°C with 0.15 nM [ 125 I]AB-MECA in the absence or presence of the test compound in a buffer containing 50 niM Tris-HCl (pH 7.4), 5 mM MgCl 2 , 1 niM EDTA and 2 units/ml ADA.
- Nonspecific binding was determined in the presence of 1 ⁇ M IB- MECA.
- the samples were filtered rapidly under vacuum through glass fiber filters (GF/B, Packard) presoaked with 0.3% PEI and rinsed several times with ice-cold 50 niM Tris-HCl using a 96-sample cell harvester (Unifilter, Packard). The filters were dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard).
- the results are expressed as a percent inhibition of the control radioligand specific binding.
- the standard reference compound is IB-MECA, which is tested in each experiment at several concentrations to obtain a competition curve from which its IC 5O and Ki values are calculated.
- the inhibition curves were generated using the Prism GraphPad program (Graph Pad Software Inc, San Diego, USA). IC 50 and Ki determinations were extrapolated from data obtained from 3- to 8-point-concentration response curves using a non linear
- Compounds Nr 1-8, 1-9, 1-10, 1-12, 1-13, 1-14, 1-15, 1-19, 1-21, 1-22, 1-35, 1-42, 1- 50, 1-63, 1-64 and 1-74 of the prevent invention have a KIi value on human adenosine A 3 receptors of less than 1 ⁇ M.
- Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A 1 receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described by Townsend-Nicholson and Schofield (1994), J. Biol. Chem. 269: 2373.
- cell membrane homogenates (20 ⁇ g protein) were incubated for 60 min at 22°C with 1 nM [ 3 H]DPCPX in the absence or presence of the test compound in a buffer containing 50 mM Tris-HCl (pH 7.4), 5 mM MgCl 2 , 1 mM EDTA/Tris and 2 UI/ml ADA.
- the samples were filtered rapidly under vacuum through glass fiber filters (GF/B, Packard) presoaked with 0.3% PEI and rinsed several times with ice-cold 50 mM Tris-HCl using a 96-sample cell harvester (Unifilter, Packard).
- the filters are dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). Nonspecific binding was determined in the presence of 1 ⁇ M DPCPX.
- the results are expressed as a percent inhibition of the control radioligand specific binding.
- the standard reference compound is DPCPX, which is tested in each experiment at several concentrations to obtain a competition curve from which its IC 50 is calculated.
- Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A 2A receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described by Luthin et al ((1995) MoI. Pharmacol., 47: 307).
- cell membrane homogenates 50 ⁇ g protein were incubated for 120 min at 22°C with 6 nM [ 3 H]CGS 21680 in the absence or presence of the test compound in a buffer containing 50 mM Tris-HCl (pH 7.4), 10 mM MgCl 2 and 2 Ul/ml ADA.
- the samples are filtered rapidly under vacuum through glass fiber filters (GF/B, Packard) presoaked with 0.3% PEI and rinsed several times with ice-cold
- the inhibition curves were generated using the Prism GraphPad program (Graph Pad Software Inc, San Diego, USA). IC 50 and Ki determinations were extrapolated from data obtained from 3- to 8-point-concentration response curves using a non linear regression analysis. The mean of IC 5O and Ki obtained from at least three independent experiments of selected molecules performed in duplicate were calculated.
- Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A 2B receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described in Stehle et al ((1992) MoI. Endocrinol. 6:384-393). .
- cell membrane homogenates of HEK-293 cells (200 ⁇ g protein) were incubated for 120 min at 22°C with 0.5 nM [ 3 H]MRS1754 in the absence or presence of the test compound in a buffer containing 10 niM Hepes/Tris (pH 7.4), 1 mM MgCl 2 and 1 mM EDTA. Following incubation, the samples are filtered rapidly under vacuum
- the results are expressed as a percent inhibition of the control radioligand specific binding.
- the standard reference compound is NECA, which is tested in each experiment at several concentrations to obtain a competition curve from which its IC 50 is calculated.
- the inhibition curves were generated using the Prism GraphPad program (Graph Pad Software Inc, San Diego, USA). IC 50 and ELi determinations were extrapolated from data obtained from 3- to 8-point-concentration response curves using a non linear regression analysis. The mean of IC 5O and Ki obtained from at least three independent experiments of selected molecules performed in duplicate were calculated.
- the functional properties of the compounds of the present invention were assessed using a cell-based Ca 2+ -mobilization assay in which the luminescence properties of the photoprotein aequorin was directly proportional to the intracellular Ca 2+ released within
- the reference agonist IB-MECA was injected at a concentration corresponding to 80 % of the maximal agonist concentration (EC 8 o) in the wells containing the cells and test compounds and the light emission was recorded over 60 s using a FDSS 6000 luminometer (Hamamatsu). Results are expressed as relative light units (RLU).
- Compounds Nr 1-8, 1-9, 1-14, 1-15, 1-22, 1-35, 1-50 and 1-64 of the prevent invention have an IC 50 value on human adenosine A 3 receptors less than 1 ⁇ M.
- the selective antagonists of human A 3 receptor are expected to block the effectiveness of adenosine or A 3 AR agonists at human A 3 receptor. Therefore, these selective adenosine A 3 antagonists are expected to be useful in human for the treatment of various conditions associated with dysfunction of adenosine system when adenosine A 3 receptors are overstimulated due to presence of an excess of adenosine or metabolite or endogenous ligand with human A 3 AR agonist property or due to a sustained presence of these agonists in the vicinity of human A 3 ARs resulting in an hyperactivation of adenosinergic system.
- Potato starch ad 200 mg active ingredient can be replaced by the same amount of any of the compounds according to the present invention, in particular by the same amount of any of the exemplified compounds.
- An aqueous suspension is prepared for oral administration so that each 1 milliliter contains 1 to 5 mg of one of the active compounds, 50 mg of sodium carboxymethyl cellulose, 1 mg of sodium benzoate, 500 mg of sorbitol and water ad 1 ml.
- a parenteral composition is prepared by stirring 1.5 % by weight of active ingredient of the invention in 10% by volume propylene glycol and water.
- Disodium EDTA (Edetate disodium) 0.01%
- active ingredient can be replaced with the same amount of any of the compounds according to the present invention, in particular by the same amount of any of the exemplified compounds.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Ophthalmology & Optometry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to novel compounds of Formula (I), wherein X1, X2, X3, X4, Y1, Y2, Y3, Y4, M1, M2, M3, Am and Bn are defined as in Formula (I); invention compounds are antagonists of adenosine receptors - subtype 3 (A3) which are useful for the treatment or disorders modulated by A3 receptors. The invention is also directed to pharmaceutical compositions and the use of such compounds in the manufacture of medicaments, as well as to the use of such compounds for the prevention and treatment of such diseases in which A3 is involved.
Description
NOVEL HETEROARYL DERIVATIVES AS ANTAGONISTS OF ADENOSINE
A3 RECEPTOR
SUMMARY OF THE INVENTION

The present invention relates to novel compounds of Formula (I), wherein X1, X2, X3, X4, Y1, Y2, Y3, Y4, M1, M2, M3, Am and Bn are defined as in Formula (I); invention compounds are antagonists of adenosine receptors — subtype 3 (A3) which are useful for the treatment or prevention of disorders modulated by A3 receptors. The invention is also directed to pharmaceutical compositions and the use of such compounds in the manufacture of medicaments, as well as to the use of such compounds for the prevention and treatment of such diseases in which A3 receptor is involved.
BACKGROUND O~F THE INVENTION
The extracellular purine nucleoside adenosine is present in all tissues and body fluids and is known to function as a modulator of a variety of physiological processes.
One of the primary roles of adenosine is cytoprotection against ischemia-induced cell damage, mainly in tissues such as the heart, brain and kidney, which are especially prone to ischemic injury (Mubagwa and Flameng (2001) Cardiovasc. Res. 52:25-39). The effects of adenosine on tissue protection and repair include increasing the ratio of oxygen supply to demand, protecting against ischemic damage by cell conditioning, triggering anti-inflammatory responses and promoting angiogenesis. Other actions of adenosine include the regulation of cellular growth and differentiation, vasodilatation
KAS/ClientDocs/Addex/SS^.WOOl.FinalSpec.lO.O? 2008
miOOHIOOll
and blood flow control, inflammatory responses, central and peripheral neural function, neuroprotection and apoptosis (see in Bruns (1990) Ann NY Acad Sci. 603:211-225; for a recent review see Gao and Jacobson (2006) Nat Rev Drug Discov. 5:247-64).
Adenosine levels in tissues change with cellular activity and energy demand and the sources of adenosine are either release through an equilibrative transporter or as a result of cell damage (McGaraughty et al (2005) Curr. Top.Med. Chem. 5, 43-58), or nucleotidase-mediated hydrolysis of extracellular adenine nucleotides (Zimmermann (2000) Naunyn Schmiedebergs Arch. Pharmacol. 362:299-309), which have their own signalling properties that are mediated by purinergic P2 receptors. Adenosine itself is rapidly metabolized by adenosine kinase (Parkinson et al (2005) Neurol. Res. 2:153— 160) and, to a lesser extent, adenosine deaminase to AMP and inosine, respectively, both of which are less active than adenosine on adenosine receptors (ARs). AR action also might be modulated by inhibition of the metabolism of extracellular adenosine (Parkinson et al (2005) Neurol. Res. 2:153-160) or its cellular uptake by adenosine transporter (McGaraughty et al (2005) Curr. Top.Med. Chem. 5:43-58). Under metabolic stress conditions extracellular concentrations of adenosine and its metabolites inosine, hypoxanthine, and xanthine increase dramatically, mainly through breakdown of adenosine triphosphate (Roth et al (1997) Exp. Eye Res. 65:771-779; Von Arnim et al (2000) Neuroreport. 11:1223-1226; Ramkumar et al (2001) Jpn J Pharmacol. 86:265-274).
The protective effects are mediated by activation of four pharmacologically and biochemically distinct adenosine receptors, named A1, A2A, A2B, and A3, which belong to the family of G-protein-coupled receptors and that have been cloned from several mammalian and non-mammalian species, including man (see Nomenclature and Classification of adenosine receptors from the International Union of Basic and Clinical Pharmacology (IUPHAR): Fredholm et al (2001) Pharmacol Rev 53:527-552). The A1 and A3 receptor subtypes couple to Gi-protein, mediating the inhibition of adenylyl
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
cyclase and a decrease in cAMP levels, whereas the A2A and A2B receptors activate adenylyl cyclase and increase cAMP levels via stimulatory Gs-protein.
Among the human ARs, the most similar are the A1 and A3 ARs (49% sequence similarity) and the A2A and A2B ARs (59% similarity). Although the degree of homology is somewhat low, it has been very difficult to develop highly selective or even specific adenosine receptor subtype agonists and antagonists. There is high sequence similarity between species for the A1, A2A and A2B receptors, whereas A3 receptors are more variable.
The modulation of adenosine receptors may be useful for therapeutic intervention due to their distribution in several types of tissue throughout the body. Many pathological conditions such as renal failure, cardiac and cerebral ischemia, central nervous system disorders, neurodegenerative diseases and inflammatory pathologies may be treated with selective modulators of the different sub-types of adenosine receptors (for a review see Gao and Jacobson (2006) Nat Rev Drug Discov. 5:247-64).
The adenosine A3 receptor (A3AR) is the most recently discovered adenosine receptor (Zhou et al (1992) Proc Natl Acad Sci U S A. 89:7432-6; Salvatore et al (1993) Proc Natl Acad Sci U S A. 90:10365-9). In addition to Gi protein, the A3AR couples to second-messenger pathways resulting in stimulation of phospholipase C (PLC) (Abbracchio et al. (1995) MoL Pharmacol. 48:1038-1045) and calcium mobilization via a Gi/o-dependent pathway (Shneyvays et al (2005) Am. J. Physiol. Heart Circ. Physiol. 288 :H2792-H2801; Englert et al (2002) Biochem. Pharmacol. 64:61-65; Fosserta et al (2003) MoI. Pharmacol. 63:342-350; Shneyvays et al (2004) Cell Calcium 36:387-396). In cardiac cells, A3AR agonists induce protection through the activation of KATP channels (30). RhoA-phospholipase Dl signaling has been demonstrated to mediate the anti-ischemic effect of A3ARs (Mozzicato et al (2004)
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
FASEB J. 18:406-408.). The WNT signaling pathway is involved in A3AR agonist- mediated suppression of melanoma cells (Fishman et al (2002) Oncogene 21:4060- 4064.). In addition, like other ARs, the A3AR couples to MAPK, which suggests a possible role in cell growth, survival, death and differentiation (Schulte & Fredholm (2002) MoI. Pharmacol. 62:1137-1146; Schulte & Fredholm (2003) Cell Signal. 15:813-827). An A3AR agonist inhibits proliferation in A375 human melanoma cells via the phosphatidylinositol 3-kinase-protein kinase B-ERKl/2 pathway (Merighi et al. (2005) J. Biol. Chem. 280:19516-19526). It is suggested that the adenosine A3 receptor activates ERK1/2 in human fetal astrocytes (Neary et al (1998) Neurosci Lett.; 242:159-62) and in CHO cells (Schulte and Fredholm (2000) MoI Pharmacol. 58:477- 82). The A3 receptor agonists Cl-IB-MECA and IB-MECA have been reported to potently inhibit and less potently to activate apoptosis in various cells (Abbracchio et al (1997) Ann NY Acad Sci 825:11-22). In RBL-2H3 mast-like cells, Cl-IB-MECA potently blocks UV irradiation-induced apoptosis by a process correlated with protein kinase B phosphorylation which is blocked by pertussis toxin and wortmannin (Gao et al (2001) MoI Pharmacol 59:76-82).
The use of more selective pharmacological tools for the A3AR has led to a better understanding of the functions of the receptor. The A3AR selective agonists IB-MECA and Cl-IB-MECA have been used extensively as pharmacological probes in the elucidation of the physiological roles of this receptor (Jacobson (1998) Trends Pharmacol. Sci. 19:184-191). However, much less is known about the effects of antagonists OfA3ARs in part because the classical xanthine class antagonists OfA1, A2A and A2B ARs (e.g., caffeine and theophylline) have low binding affinities for the rat A3AR (Zhou (1992) Proc. Natl Acad. Sci. USA 89:7432-7436). In general, there is a higher affinity for the human A3 receptor as compared to the rat receptor for the different class of purine or non-purine like A3AR antagonists available including, among many, xanthine, adenine derivatives, imidazo[2,l-i]purin-5-ones, quinazolines and derivatives, 1,4-dihydropyridines and pyrans, pyrimidines derivatives (Zhou (1992) Proc. Natl Acad. Sci. USA 89:7432-7436; Yang (2005) Curr.Eye Res. 30:747-754;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
Mϋller, C. et al. (2001) Mini Reviews in Medicinal Chemistry 1:339-348). Therefore, the search for A3AR antagonists turned towards more novel heterocyclic systems (Jacobson (1998) Trends Pharmacol. Sci. 19:184-191) and screening of diverse chemical libraries resulted in the identification of new high-affinity non-xanthine compounds for the human A3AR, including 1,4-dihydropyridine, flavonoids, pyridines, thiazoles, triazoloquinazoline, isoquinoline and quinazoline, isoquinoline and quinazoline derivatives among others (Baraldi et al (2000) Med Res Rev. 20:103-128; Gao and Jacobson (2006) Nat Rev Drug Discov. 5:247-64). Although potency and selectivity towards human adenosine A3ARs have been improved in these novel heterocyclic compounds, the overall profile has not improved sufficiently to consider development in human beings. Some high-affinity derivatives of adenosine with added substituents or rigidified derivates of adenosine improve the affinity at the target but in both cases the overall profile of ligands is far from optimal, as pharmacokinetic properties remain poor. The conformationally constrained nucleoside MRS 1292, which is a selective A3AR antagonist, in both rat and human (Gao et al. (2002) J. Med. Chem. 45:4471-4484; Yang et al (2005) Curr. Eye Res. 30:747-754) is currently used in vitro as a reference antagonist.
Adenosine A3 receptors are found mostly in brain, lung, liver, heart, kidney and testis (Gao and Jacobson (2006) Nat Rev Drug Discov. 5:247-64) and have been implicated in cell cycle progression and cell growth (Brambilla et al. (2000) Naunyn
Schmiedebergs Arch Pharmacol. 361:225-34), modulation of apoptosis (Abbracchio et al. (1997) Biochem Biophys Res Commun. 241:297-304), cancer (Baraldi et al (2005)
Curr Med Chem. 12:1319-29), mast cell degranulation (Jin et al. (1997) J Clin Invest. 100:2849-57), cardiac ischemia and ischemic pre-conditioning in the heart (Strickler et al., (1996) J Clin Invest. 98:1773-9), neuroprotection (see in Fredholm (1997) Int Rev
Neurobiol. 40:259-80), pro- and anti-inflammatory modulation (Salvatore et al. (2000)
J. Biol. Chem. 275:4429-4434), asthma (Jacobson et al (1998) Drug Dev. Res. 45:113), neurodegeneration (Jacobson et al (1995) Drugs Future 20:689 ; Kohno et al (1996) Blood 88: 3569), ischemic brain damage (Von Lubitz et al (1994) Eur. J. Pharmacol.
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008
263:59; Von Lubitz et al (1999) Eur. J. Pharmacol. 367:157) and hypotension (Harmon et al (1995) Br. J. Pharmacol. 115:945). In addition, adenosine A3 receptors have functional effects that are dependent on the degree of receptor activation. Specifically, when activated moderately, A3ARs have a cytoprotective role for example reducing damage to heart cells from lack of oxygen or protecting cells from apoptosis. Indeed moderate activation of the A3 receptor is known to activate the cellular antioxidant defense system by increasing the activities of superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase, along with a reduction in malondialdehyde, a marker of lipid peroxidation (Maggirwar et al (1994) Biochem Biophys Res Comniun 201 :508-512). Such a mechanism may provide a mechanism by which adenosine exerts a cytoprotective action in ischemic conditions. However, high levels of A3AR stimulation can actually result in cell death. This could be linked to the observation that there is a subsequent upregulation of selective A3 receptor observed, for instance, by RBL-2H3 cells in vitro after exposure to oxidative stress (Ramkumar et al (2001) Jpn J Pharmacol 86:265-274) and after preconditioning of central nervous tissue, whereas expression OfA1, A2A and A2B ARs remained unchanged (Von Arnim et al (2000) Neuroreport 11:1223-1226). Excessive A3AR activation is toxic for the cells. Indeed A3ARs modulators are being tested for therapeutic potential, for example, treatment of cancer, heart conditions, neurological conditions, pain, asthma, inflammation, glaucoma and other immune implications. In particular, there are literature data strongly suggesting that a A3R antagonist may be used specifically in the treatment of several conditions as listed below:
A role for the A3AR in mediating control of the cell cycle has been reported (Neary et al (1998) Neurosci. Lett 242:159-162). For example, there is a significant over- expression of A3ARs in several types of tumor cells (Madi et al (2004) Clin. Cancer Res. 10:4472-4479; Gessi et al (2004) Clin. Cancer Res. 10:5895-5901). A3AR antagonists might sensitize tumor cells to chemotherapeutic drugs as it is known that A3 receptor subtype activation plays a role in the prosurvival and in the antiapoptotic effect of adenosine (Merighi et al (2003) Biochem. Pharmacol. 66:739-748; Baraldi et
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
al (2005) Curr. Med Chem 12:1319-1329). An opportunity would be then that A3AR antagonists might sensitize tumors cells to chemotherapeutic drugs and therefore could inhibit tumor growth and metastasis in patient. Possible indication therefore for selective adenosine A3 antagonists is the use of such compounds class in synergistically improving chemotherapeutic treatment of cancers expressing A3ARs and cancers expressing P-glycoprotein or MRP in combination with other anti-tumor agents such as antiangiogenic agents and/or cytostatic agents.
The adenosine A3 receptor was initially implicated as the receptor subtype that triggers the degranulation of rat RBL 2H3 mast-like cells (Ramkumar et al., 1993) and perivascular mast cells of the hamster cheek pouch (Jin et al (1997) J Clin Invest. 100:2849-57) and therefore A3AR has been implicated in mediating allergic responses: A3 receptor agonists induce mast cell degranulation and consequent release of allergic mediators, such as histamine, when administered to rats or mice (Ramkumar et al (1993) J. Biol. Chem. 268:16887-16890; Tilley et al (2000) J Clin Invest.lO5:361-7). Systemic infusion in mice of IB-MECA causes scratching that is prevented by coadministration of histamine antagonists. In contrast, in mice lacking A3ARs (Salvatore et al (2000) J. Biol. Chem 275:4429-4434), the potentiation by Cl-IB-MECA of antigen-dependent degranulation of mast cells, as measured by hexosaminidase release, was lost and lipopolysaccharide-induced tumour-necrosis factor-α (TNFα) production was lower than in control mice (Salvatore et al (2000) J. Biol. Chem 275:4429-4434). The effect of adenosine analogs on mast cell degranulation (Salvatore et al (2000) J. Biol. Chem 275:4429-4434) and the consequent decrease in vascular permeability (Tilley et al (2000) J Clin Invest.105:361-7) is decreased in mice with a targeted disruption of the A3AR receptor. Taken together, these observations suggest that A3AR receptor antagonists have the potential for treating diseases and disorders resulting from or including a component of inflammation. This could be extended to the treatment of asthma and others respiratory diseases involving inflammation and or allergenic responses. In the asthmatic lung, adenosine acts as an irritant and bronchoconstrictor, suggesting that a synthetic A3AR antagonist, could have therapeutic potential in asthma
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
treatment (Holgate (2005) Br. J. Pharmacol 145:1009-1015). In patients with asthma, relatively high density of functionally active A3 receptors are expressed in human eosinophils (Kohno et al (1996) Blood 88:3569-74) and appear to be involved in the inhibition of eosinophil chemotaxis when stimulated (Walker et al (1997) Am J Respir Cell MoI Biol 16:531-7). Since inflammation in allergic rhinitis is characterized by eosinophilic infiltration of the airways, it is possible that the elevated adenosine concentrations associated with allergic inflammation would contribute to inhibition of mucosal inflammation through stimulation of eosinophils-expressed A3 receptors (Rimmer et al (2007) Clin Exp Allergy; 37:8-14). A recent clinical trial has shown that a novel dual agonist Of A2ARs and antagonist Of A3ARs appears to have some clinical benefit in both the early-phase and the late-phase response to intranasal allergen challenge. Notably, there was a reduction of some pro-inflammatory mediators suggesting that comparable, more selective compounds of A2AR or A3ARs may have additional benefits (Rimmer et al (2007) Clin Exp Allergy 37:8-14).
A3ARs antagonists may be useful for the acute and chronic treatment of glaucoma and other visual disorders in general. Glaucoma is characterized by elevated intraocular pressure (IOP) and is a leading cause of irreversible blindness. Molecular and pharmacological studies have provided evidence that all adenosine receptor subtypes are expressed in ocular tissues (Blazynski et al (1992) J Neurochem. 58:761- 767; Wax et al (1993) Exp Eye Res. 57:89 -95 ; Wax et al (1994) Invest Ophthalmol Vis Sci. ;35:3057-3063 ; Kvanta et al (1997) Exp Eye Res. 65:595-602 ; Fleischhauer et al (2003) J Membr Biol. 193:121-136.) and that activation of these receptors has been shown to regulate retinal neurotransmission and neuroprotection (Blazynski et al (1992) J Neurochem. 58:761- 767; Macaluso et al (2003) Doc Ophthalmol. 106: 51- 59) retinal and choroidal blood flow (Braunagel et al (1988) J Ocul Pharmacol. 4:61- 73), photoreceptor phagocytosis (Gregory et al (1994) Invest Ophthalmol Vis Sci. 35:819-825) and integrity of the blood-retinal barrier (Sen et al (1989) Arch Ophthalmol. 107:1364-1367; Campochiaro et al (1989) Arch Ophthalmol. 107:412- 416). The adenosine system has also been shown to regulate ion transport in corneal
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008
. , n
PCT/IBzυ U D / u u i 2 U
(Riley et al (1996) Invest Ophthalmol Vis Sci. 37:1-10.) and ciliary epithelia (Carre et al (1997) Am J Physiol. 273:C1354-C1361; Mitchell et al (1999) Am J Physiol. 276:C659-C666) and to modulate aqueous humor in- and outflow (Crosson (1995) J Pharmacol Exp Ther. 273:320-326; Crosson and Gray (1996) Invest Ophthalmol Vis Sci. 37:1833-1839; Tian et al (1997) Exp Eye Res. 64:979-989).
In pathophysiological conditions, adenosine and its receptors have been implicated in many ocular and systemic ischemic diseases such as retinal ischemia and in conditions with oxidative stress in rodents (Roth S et al (1997) Exp Eye Res. 65:771-779; Lutty and Mc Leod (2003) Prog Retin Eye Res. 22:95-111, Larsen and Osbourne (1996) Invest Ophthalmol Vis Sci. 37: 2603-2611). Civan and co-workers have found that the A3 adenosine receptors regulate Cl(-) channels of non-pigmented ciliary epithelial cells (Von Arnini et al (2000) Neuroreport. 11:1223-1226). Furthermore, the knock-out of A3ARs gene in the mice or their pharmacological blockage with selective antagonists in normal mice with MRS 1191, MRS 1097, and MRS 1523 lowered IOP (Ramkumar et al (2001) Jpn J Pharmacol. 86:265-274; Safran et al (2001) MoI Cell Biochem. 217:143-152). These results suggest that antagonists Of A3ARs may provide a novel approach for the treatment of glaucoma (Fredholm et al (2001) Pharm Rev. 53:527— 552). This has extended in vitro observations implicating A3 receptors in tissues controlling aqueous humour physiology (Fredholm et al (2001) Pharm Rev. 53:527— 552). In contrast, A3 agonists have been shown to activate chloride channels in non pigmented ciliary epithelial cells in vitro, leading to the hypothesis that A3 receptor agonists would increase aqueous humor secretion and thereby IOP in vivo (Mitchell et al (1999) Am J Physiol. 276:C659-C666).
The cross-species A3AR antagonist MRS 1292 was recently found to reduce mouse intraocular pressure but also inhibited adenosine-triggered human non-pigmented ciliary epithelial cell fluid release (Yang, H. et al (2005) Curr. Eye Res. 30: 747-754). OT-7999, a potent and selective A(3) receptor antagonist administered via topical eye-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
drops was found to significantly decrease intraocular pressure in monkey without ophthalmologic side effects, such as appearance of eyelid closure, hyperemia of the external and anterior ocular segments or abnormality of the pupil (Okamura et al (2004) Bioorg. Med. Chem. Lett. 14: 3775-3779).
In human, the A3 receptor subtype has been immunolocalized to the basolateral surface infoldings of non-pigmented ciliary epithelial cells, which is consistent with the receptor's functional role in aqueous humor secretion. In ocular hypertensive individuals, the mean aqueous adenosine levels were significantly elevated when compared to normotensive subjects and correlated with IOP levels (Daines et al (2003) J Ocul Pharmacol Ther. 19:113-119). However, in the eyes of healthy subjects, parenteral infusion of adenosine induced a small but significant decrease in IOP (Polska et al (2003) Invest Ophthalmol Vis Sci. 44:3110-3114). An involvement of the adenosine system in ischemia/hypoxia and IOP elevation in pseudoexfoliation (PEX) eye syndrome, a common age-related extracellular matrix disorder that often leads to the development of ocular hypertension and secondary open-angle glaucoma, may therefore be hypothesized. A recent study further provided evidence of a selective and significant upregulation of the A3 adenosine receptor on both the mRNA and protein levels, in the non-pigmented ciliary epithelium of all patients eyes with PEX syndrome confirming a previous study showing a 30-fold overexpression of A3 adenosine receptor mRNA in the ciliary processes of PEX eyes compared with control eyes (Schlδtzer-Schrehardt et al (2004) IOVS 45: ARVO E-Abstract 3535).
This finding suggested that hypoxia and/or oxidative stress, typical of all eyes with PEX syndrome/glaucoma (Ritch and Schlδtzer-Schrehardt (2001) Surv Ophthalmol. 45:265-315; Helbig et al (1994) German J Ophthalmol. 3:148 -153) promotes a selective upregulation of A3 adenosine receptors in non-pigmented ciliary epithelium, which may confer protection against ischemic or oxidative damage to sustain prolonged periods of chronic hypoxia or oxidative stress. Considering, however, the known
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
-
properties of the A3 receptor in activating chloride transport in epithelial cells, its upregulation in the nonpigmented ciliary epithelium may have an additional influence on aqueous humor secretion and hence on IOP levels in PEX eyes. It has been previously reported that IOP elevation in PEX patients results from an increased outflow resistance in the trabecular meshwork, whereas the rate of aqueous flow through the anterior chamber was not different or even slightly lower in PEX eyes than in control eyes (Johnson and Brubaker (1982) Am. J. Ophthalmol. 93:629-634).
These results above mentioned suggest that antagonists OfA3ARs may be useful for the acute and chronic treatment of glaucoma and other visual disorders in general. International patent publication WO 00/03741 describes a method for decreasing intraocular pressure by administering an A3 adenosine receptor antagonist.
All of these finding could be of clinical and therapeutic significance and the reduction of chloride channel activity with A3 receptor antagonists if confirmed in human beings may be an alternative specific approach for treating ocular hypertension in patients, for example, with PEX who are refractory to standard medical therapy. Patients with glaucoma may require long-term administration of IOP -lowering medications. These medications belong to several classes of molecules including beta-adrenergic blockers, prostaglandin receptor inhibitors, cholinergic agents, alpha-adrenergic agonists, carbonic anhydrase inhibitors, and ocular hypotensive lipids and are associated with mild and ocular side-effects. However, several of them are associated with systemic risks as well as serious ocular effects, especially following chronic use (Roth et al (1997) Exp. Eye Res. 65:771-779).
Recent evidence suggested that both A1AR agonists and A3AR antagonists protect the kidney. Precisely, mice lacking A3ARs or wild-type mice in which the A3AR was blocked pharmacologically had significant renal protection (Lee et al (2003) Am. J.
KAS/ClientDocs/Addex/53195.WOO 1.FinalSpec.10.072008
Physiol. Renal. Physiol. 284: F267-F273), suggesting that A3AR antagonists might have general renal-protective properties. Ligands possessing dual acting and opposite properties at these AR subtypes could therefore be effective therapeutic agents for renal protection.
A3 receptor has been mainly implicated in ischemic disease, such as ischemic brain damage or cardiac ischemia (Baraldi et al (2003) Eur. J. Med. Chem. 38:367-382). Adenosine is released in large amounts during myocardial ischemia, resulting in effective preconditioning in cardiomyocytes through the activation of A1 and A3 ARs (Shneyvays et al (2004) Cell Calcium 36:387-396; Tracey et al (1998) Cardiovasc. Res. 40:138-145 ; Mozzicato et al (2004) FASEB J. 18: 406-408; Auchampach (1997) Circ. Res. 80: 800-809). AR agonist to activate either or both of these receptors might therefore be beneficial to the survival of the ischemic heart. Various lines of evidence indicate that the A3AR has a role in protecting the heart (Auchampach (1997) Circ. Res. 80: 800-809; Tracey et al (2003) Am. J. Physiol. Heart Circ. Physiol. 285:H2780- H2787). Overexpression of A3ARs decreases heart rate, preserves energetics and protects ischemic heart (Cross et al (2002) Am. J. Physiol. Heart Circ. Physiol. 283:H1562-H1568) and low-level expression OfA3ARs in the heart provides effective protection against ischemic injury without detectable adverse effects, although higher levels of A3AR expression lead to the development of a dilated cardiomyopathy (Black et al (2002) Circ. Res. 91:165-172). As said earlier, this could be not good during too long activation. And paradoxically, global deletion of the A3AR in mice also confers resistance to myocardial ischaemic injury and does not prevent early preconditioning (Guo et al (2001) J. MoI. Cell Cardiol. 33:825-830). In an isovolumic Langendorff perfusion model, A3AR-knockout mice also had improved functional recovery and tissue viability during reperfusion after ischemia when compared with control mice (Tracey et al (2003) Am. J. Physiol. Heart Circ. Physiol. 285:H2780-H2787).
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
Using rat basophilic leukemia 2H3 cell line (RBL-2H3), it has been established that activation OfA3ARs stimulates SERT activity via both PKG and ρ38 MAPK (Zhu et al (2004) MoI Pharmacol. 65:1462-74). Therefore, as it is known that the inactivation of synaptic serotonin (5-hydiOxytryptamine, 5-HT) is primarily through reuptake by the presynaptic, 5-HT transporter (SERT, SLC6A4), it is likely that A3AR antagonists could be interesting in treating pathologies resulting from a low level of serotonin (including affective disorders including depression and depressive disorders, anxiety disorders including obsessive-compulsive disorder, post-traumatic stress disorder, panic disorder and phobias, borderline personality disorder, anorexia nervosa, bulimia nervosa, autism, attention deficit hyperactivity disorder, Tourette's syndrome, sexual disorders, migraine, diabetic neuropathy, obesity, drug or alcohol addiction, sleep disorders, arthritis, chronic fatigue syndrome or irritable bowel syndrome) because such compounds will decrease the SERT surface expression and/or catalytic rates.
In conclusion, and considering hypotheses based on the known distribution Of A3ARs. its in vitro functional role and the knowledge of its role in physiological and pathological conditions from preclinical and clinical data, together with the use of subtype selective A3ARs antagonists, it is foreseen that the blockade OfA3ARs could be a valuable approach to treat eye disorders including glaucoma and related conditions, inflammatory processes including allergy, asthma and airway pathologies, ischemic brain and cardiac diseases and related, renal dysfunction, tumor and abnormal cell growth, and depression, anxiety and related neuropsychiatric and affective disorders.
The following compounds are known: (i) International patent publication WO2001/64674, as well as US patent publications US2003/0203897 and US7105550, describe 2,4-disubstituted thiazolyl derivatives, such as N-phenyl-4-(lH-pyrazol-3-yl)thiazol-2-amine hydrobromide [358779-21-2], for the prevention or the treatment of disease throught cytokines, in
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
particular, Tumor Necrosis Factor- Alpha (TNF-α) and Interleukin-12 (IL- 12) or throught activation of Adenosine A3 receptor;
International patent publication WO2006/122011 describes thiazole derivatives, such as 4-(2-(4-(octyloxy)-3-(trifluoromethyl)phenylamino)thiazol-4-yl)-lH-pyrazole- 3-carbonitrile [914668-55-6] as Hepatite C Virus replication inhibitors;
(ii) International patent publication WO2006/069155 describes (S)-2-amino-N-(3- (lH-pyrazol-l-yl)benzyl)propanamide derivatives, such as (2S)-2-amino-N-(3-(5-(5- (phenylamino)- 1 ,3 ,4-oxadiazol-2-yl)-3 -(trifluoromethyl)- 1 H-pyrazol- 1 -yl)benzyl)- propanamide [895524-13-7], as protein arginine methyl transferase inhibitors; (iii) International patent publication WO2005/112923 describes 5-anilino-4- heteroarylpyrazole derivatives, such as 5-methoxy-2-([l-(2-methoxyphenyl)-l',3- dimethyl-lH,l'H-4,4'-bipyrazol-5-yl]amino)benzoic acid [870188-47-9], useful for the treatment of diabetes or related disorders;
(iv) International patent publication WO2005/125101 describes imidazopyrazine derivatives such as (Z)-5-(3-(l-methyl-lH-pyrazol-4-yl)imidazo[l,2-a]pyrazin-2- ylamino)-2,3-dihydro-lH-inden-l-one oxime [915705-05-4] as RAF kinase inhibitors;
(v) Some (5 -methyl- 1 -phenyl- lH-pyrazol-4-yl)thiazol-2-amino derivatives have been reported: Singh, S.P. et al. (1997) J: Indian Chem. Soc. (74, 11-12): 940-942 described a synthetic route from 4-acylpyrazole, using [hydroxyl(tosyloxy)iodo]benzene; US patent publication US7244739 describes compounds as Amyloid-beta (Aβ) modulators useful for the treatment of neurodegenerative disorders;
(vi) Hassan, N.M. et al (1997) J. Chem. Res., Syn. (10): 350-351 described a synthetic route to obtain 4-((l-aryl-4-carbonitrile-5-phenyl)-lH-pyrazolyl)-2- arninophenyl thiazoles;
(vii) International patent publication WO 99/64418 describes arylpyridinyl thiazoles as adenosine receptor antagonists, such as 4-(4-methoxyphenyl)-N-(pyridin-2-yl)-5-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
(pyridin-4-yl)thiazol-2-amme or N-(5-(pyridin-4-yl)-4-(3 ,4,5-trimethoxyphenyl)thiazol- 2-yl)acetamide, having activity at the A3 and/or A2B receptors;
Press, NJ. et al (2004) Curr. Topics Med. Chem. (4): 863-870 described arylpyridinyl thiazoles, such as N-(5-(pyridin-4-yl)-4-(354,5-trimethoxyphenyl)thiazol- 2-yl)acetamide, having adenosine A3 receptor antagonism properties, useful as pharmacological tools;
European patent publication EP1027050 and US patent publication US6620825 describe 1,3-thiazoles as adenosine A3 receptor antagonist for the treatment of allergy, asthma and diabetes, such as compounds substituted at the 4- or 5 -position, or both, by a pyridyl, such as 4-(4-methoxyphenyl)-N-phenyl-5-(pyridin-3-yl)thiazol-2-amine or 4-
(4-methoxyphenyl)-N,5-di(pyridin-3-yl)thiazol-2-amine;
(viii) International patent publication WO 02/42298 as well as Press, NJ. et al (2005) Bioorg. Med. Chem. Lett. (15): 3081-3085 described aminothiazoles, such as 3-(5-(1H- imidazol- 1 -yl)-2-(6-methylpyridin-2-ylamino)thiazol-4-yl)benzonitrile, having adenosine receptor antagonism properties, particularly at the A3 and A2B receptors, and useful for the treatment of inflammatory or obstructive airways diseases;
(ix) Borghini, A. et al. (2005) Bioorg. Med. Chem. Lett. (13): 5330-5337 described thiazole and thiadiazole derivatives, such as 4-hydroxy-N-(3-phenyl-l,2,4-thiadiazol-5- yl)benzamide or N-(3-(4-methoxyphenyl)-l,2,4-thiadiazol-5-yl)acetamide, having adenosine Ai or A3 receptors antagonism properties.
(x) International patent publication WO2007/031440 describes 2-aniline-4-aryl substituted thiazole as positive modulators of nicotinic acetylcholine receptors;
(xi) Japan patent application JP2003313176 describes 2-aminothiazole derivatives having cell proliferation inhibitory activity for preventing and treating cancer.
It has now surprisingly been found that the compounds of general formula (I), (II), (III), (HIA) and (IIIB) show potent activity and subtype-selectivity on Adenosine A3 receptor. The compounds of the invention demonstrate advantageous properties over
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
compounds of the prior art, noteworthy selectivity versus the other adenosine receptor subtypes such as A1 receptor, A2A receptor and A2B receptor.
The present invention relates to a method of treating or preventing a condition in a mammal, including a human, the treatment or prevention of which is affected or facilitated by the neuromodulatory effect of Adenosine A3 receptor antagonists.
DETAILED DESCRIPTION OF THE INVENTION
The invention relates to compounds having A3 receptor antagonist activity. In its most general compound aspect, the present invention provides a compound according to Formula (I),

a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV-oxide form thereof, wherein:
X1, X2, X3 and X4 are each independently selected from the group of C, N3 O, S and C=C representing a 5 or 6 membered heteroaryl ring which may further be substituted by 1 to 3 radicals Am;
m is an integer ranging from 1 to 3;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
Am radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, - (Ci-C6)alkylhalo, -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano5 -(Ci-C6)alkylheteroaryl, - (Ci-C6)alkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR1, ~O-(C2-C6)alkyl- OR1 , -NR1(C2-C6)alkyl-OR2, -(C3-C7)cycloalkyl-(Ci-C6)alkyl, -O-(C3-C7)cycloalkyl- (Ci-C6)alkyl, -NR1-(C3-C7)cycloalkyl-(C1-C6)alkyl,
 -(C1- C(OaUCyIlIaIo-NR1R2, -(Co-C^alkyl-S-R1, -O-^-COalkyl-S-R1, -NR^Ca-C^alkyl-S- R2, -(Co-C6)alkyl-S(=0)-R1, -O-(Ci-C6)alkyl-S(=O)-R1, -NR^Q-CfOalkyl-S^OVR2, - (C0-C6)alkyl-S(=O)2-R1, -O-(Ci-C6)alkyl-S(=O)2-R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, - (Co-C^alkyl-NR^2, -O-(C2-C6)alkyl-NR1R2, -NR^Cz-C^alkyl-NR^3, -(C0-C6)alkyl- S(^O)2NR1R2, -O-(C1-C6)alkyl-S(=O)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3, -(C0- C6)alkyl-NR1-S(=O)2R2, -O-(C2-C6)alkyl-NR1-S(=O)2R2, -NR1-(C2-C6)alkyl-NR2- S(=O)2R3, -(C0-C6)alkyl-C(=O)-NR1R2, -O-(C1-C6)alkyl-C(=O)-NR1R2, -NR^(Ci- C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl-NR1C(=O)-R2, -O-(C2-C6)alkyl-NR1C(=O)-R2, - NR1-(C2-C6)alkyl-NR2C(=O)-R3, -O-(C2-C6)alkyl-OC(=O)-R1, -NR1-(C2-C6)alkyl- OC(=O)-R2, -(Co-C6)alkyl-C(=0)-OR1, -O-(C1-C6)alkyl-C(=O)-OR1,
-(C1- C(OaUCyIlIaIo-NR1R2, -(Co-C^alkyl-S-R1, -O-^-COalkyl-S-R1, -NR^Ca-C^alkyl-S- R2, -(Co-C6)alkyl-S(=0)-R1, -O-(Ci-C6)alkyl-S(=O)-R1, -NR^Q-CfOalkyl-S^OVR2, - (C0-C6)alkyl-S(=O)2-R1, -O-(Ci-C6)alkyl-S(=O)2-R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, - (Co-C^alkyl-NR^2, -O-(C2-C6)alkyl-NR1R2, -NR^Cz-C^alkyl-NR^3, -(C0-C6)alkyl- S(^O)2NR1R2, -O-(C1-C6)alkyl-S(=O)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3, -(C0- C6)alkyl-NR1-S(=O)2R2, -O-(C2-C6)alkyl-NR1-S(=O)2R2, -NR1-(C2-C6)alkyl-NR2- S(=O)2R3, -(C0-C6)alkyl-C(=O)-NR1R2, -O-(C1-C6)alkyl-C(=O)-NR1R2, -NR^(Ci- C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl-NR1C(=O)-R2, -O-(C2-C6)alkyl-NR1C(=O)-R2, - NR1-(C2-C6)alkyl-NR2C(=O)-R3, -O-(C2-C6)alkyl-OC(=O)-R1, -NR1-(C2-C6)alkyl- OC(=O)-R2, -(Co-C6)alkyl-C(=0)-OR1, -O-(C1-C6)alkyl-C(=O)-OR1,
 C6)alkyl-C(=O)-OR2, -(Co-C^alkyl-C^O)-^, -O-(C1-C6)alkyl-C(=O)-R1, -NR^(C1- C6)alkyl-C(=O)-R2, -(C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-C6)alkyl-NR1-C(=O)-NR2R3, -O-(C2-C6)alkyl-NR1-C(=O)-NR2R3, and -NR1-(C2-C6)alkyl-NR2-C(=O)-NR3R4; Any two radicals of Am (A1 and A2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
C6)alkyl-C(=O)-OR2, -(Co-C^alkyl-C^O)-^, -O-(C1-C6)alkyl-C(=O)-R1, -NR^(C1- C6)alkyl-C(=O)-R2, -(C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-C6)alkyl-NR1-C(=O)-NR2R3, -O-(C2-C6)alkyl-NR1-C(=O)-NR2R3, and -NR1-(C2-C6)alkyl-NR2-C(=O)-NR3R4; Any two radicals of Am (A1 and A2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(CrC^alkyl, -(Ct-C^alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and

Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
Y1, Y2, Y3 and Y4 are each independently selected from the group of C and N representing 5 membered heteroaryl ring which may further be substituted by 1 to 3 radicals Bn;
n is an integer ranging from 1 to 3;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Q-C^alkyl, - (Ci-C6)alkylhalo, -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano, -(CrC^alkylheteroaryl, - (Ci-CfOalkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, -O-(C2-C6)alkyl- OR5, -NR5(C2-C6)alkyl-OR6, -(Ca-C^cycloalkyl-CCrC^alkyl, -O-(C3-C7)cycloalkyl- (Ci-C6)alkyl5 -NR5-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(Ci-C^alkylhalo-OR5, -(C1- C6)alkylhalo-NR5R6, -(C0-C6)alkyl-S-R5, -O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S- R6, -(Co-C6)alkyl-S(=0)-R5, -O-(d-C6)alkyl-S(=O)-R5, -NR^d-CfOalkyl-S^CO-R6, - (C0-C6)alkyl-S(=O)2-R5, -O-(C1-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, - (C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl- S(=O)2NR5R6 5 -O-(C1-C6)alkyl-S(=O)2NR5R6, -NR5-(C1-C6)alkyl-S(=O)2NR6R7, -(C0- C6)alkyl-NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, -NR5-(Cr C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl-NR5C(=O)-R6, - NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, -NR5-(C2-C6)alkyl- OC(=O)-R6, -(C0-C6)alkyl-C(=O)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5 5 -NR5-(Cr C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(C1-C6)alkyl-C(=O)-R5, -NR^(C1- C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R5, R6, R7 and R8 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Ci-C6)alkyl, -(C1-C6)alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl,
 aryl, heterocycle and -(Q-C^alkylaryl;
aryl, heterocycle and -(Q-C^alkylaryl;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
Any two radicals of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted 3 to 10 membered ring selected from the group of aryl, heteroaryl, heterocyclic and cycloalkyl;
M2 is selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C0-C6)alkyl-R9, -(CrC6)alkylhalo, -(C2-C6)alkyl-NR9R10, - (C2-C6)alkyl-OR9, -(C2-C6)alkyl-SR9, -(C0-C6)alkyl-C(=O)-R9, -(C2-C6)alkyl-S(O)-R9, - (Co-C6)alkyl-C(=0)NR9R10 and -(C0-C6)alkyl-S(O)2-R9;
R9 and R10 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C1-C6)alkylhalo, -(CrC^alkyl, -(CrC^alkylcyano, ~(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Ct-C^alkylheteroaryl, aryl, heterocycle and -(Cj.-C6)alkylaryl;
M3 is an optionally substituted radical selected from the group of -(Co-C6)alkyl-R , - (d-C^alkylhalo, -(C2-C6)alkyl-NRπR12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRU; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(CrC^alkyl, -(CrC^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Ci-C^alkylheteroaryl, aryl, heterocycle and -(Q-C^alkylaryl; provided that according to proviso (i): when M3 is -(Co)-R11 (that is when M3 is -R11), then R11 is not H; and provided that according to proviso (ii):
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
 and provided that according to proviso (iii):
and provided that according to proviso (iii):
when M1 is aryl, M2 is H5 X1 is C, X2 is C5 X3 is C, X4 is N5 then
 is not linked to X2; and provided that according to proviso (iv):
is not linked to X2; and provided that according to proviso (iv):
A1 and A2 radicals are not linked to form an imidazopyridazinyl ring; and provided that according to proviso (v):
when
 is and linked to X4 , X1 is C, X2 is S, X3 is C, X4 is C, n is I5 A1 is H5 Y1, Y2, Y3 are C5 Y4 is N5 then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl5 lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when M1M2N is linked to X1, and X1 is C5 X2 is S5 X3 is C5 X4 is C, to provide a thiazole ring, n is 1 , then A1 is not a pyridyl; and provided that according to proviso (viii): when M1M2N is linked to X1, and X1 is C5 X2 is S, X3 is C5 X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
is and linked to X4 , X1 is C, X2 is S, X3 is C, X4 is C, n is I5 A1 is H5 Y1, Y2, Y3 are C5 Y4 is N5 then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl5 lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when M1M2N is linked to X1, and X1 is C5 X2 is S5 X3 is C5 X4 is C, to provide a thiazole ring, n is 1 , then A1 is not a pyridyl; and provided that according to proviso (viii): when M1M2N is linked to X1, and X1 is C5 X2 is S, X3 is C5 X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
The compounds N-phenyl-4-(lH-pyrazol-3-yl)-2-thiazolamine hydrobromide (1:1) [358779-21-2], 4-(lH-mdazol-3-yl)-N-(4-methoxyphenyl)-2-thiazolamine hydrobromide (1:1) [358779-27-8], 4-(lH-indazol-3-yl)-N-phenyl-2-thiazolamine
KAS/ClieniDocs/Addex/53195.WO01.FinalSpec.l0.07200δ
hydrobromide (1:1) [358779-28-9] known as such from international patent publication WO2001/64674 are excluded from the present invention by virtue of proviso (i);
The compounds 4-(2-(4-(octyloxy)-3 -(trifiuoromethyl)phenylammo)thiazol-4-yl)- IH- pyrazole-3-carbonitrile [914668-55-6] known as such from International patent publication WO2006/122011 are excluded from the present invention by virtue of proviso (i);
The compounds 2-amino-N-[[3-[5-[5-(phenylamino)-l ,3,4-oxadiazol-2-yl]-3- (trifluoromethyl)-lH-pyrazol-l-yl]phenyl]methyl]-(2S)-propanamide [895524-13-7], 2- amino-N- [[3 - [5 - [5 -[(2-methoxyphenyl)amino]- 1 ,3 ,4-oxadiazol-2-yl] -3 - (trifluoromethyl)-lH-pyrazol-l-yl]ρhenyl]methyl]-(2S)-propanamide [895524-14-8], 2- amino-N-[[3-[5-[5-[(2-methoxy-5-methylphenyl)amino]-l,3,4-oxadiazol-2-yl]-3- (trifiuoromethyl)-lH-pyrazol-l-yl]ρhenyl]methyl]-(2S)-proρanamide [895524-15-9], 2- amino-N-[[3-[5-[5-[(2,4-dimethoxyphenyl)arnino]-l,3,4-oxadiazol-2-yl]-3- (trifluoromethyl)- 1 H-pyrazol- 1 -yl]ρhenyl]methyl]-(2S)-proρanamide [895524- 16-0] , 2- amino-N-[[3-[5-[5-[(2,5-dimethoxyphenyl)amino]-l ,3,4-oxadiazol-2-yl]-3-
(trifiuoromethyl)-lH-ρyrazol-l-yl]ρhenyl]methyl]-(2S)-propanamide [895524-17-1], 2- amino-N-[[3-[5-[5-[(5-chloro-2-methoxyphenyl)amino]-l,3,4-oxadiazol-2-yl]-3- (trifluoromethyl)-lH-ρyrazol-l-yl]phenyl]methyl]-(2S)-propanamide [895524-18-2], 2- amino-N-[[3-[5-[5-[[2-(difluoromethoxy)phenyl]amino]-l,3,4-oxadiazol-2-yl]-3- (trifluoromethyl)-lH-ρyrazol-l-yl]ρhenyl]methyl]-(2S)-propanamide [895524-19-3], 2- amino-N- [[3 - [5 -[5 - [[2-(trifluoromethoxy)phenyl] amino] - 1 ,3 ,4-oxadiazol-2-yl] -3 - (trifluoromethyl)-l H-ρyrazol- l-yl]ρhenyl]methyl]-(2S)-propanamide [895524-20-6], 2- amino-N-[[3-[5-[5-[(4-methoxy[l,r-biphenyl]-3-yl)amino]-l,3,4-oxadiazol-2-yl]-3- (trifiuoromethyl)-lH-pyrazol-l-yl]phenyl]methyl]-(2S)-propanamide [895524-21-7], 2- amino-N-[[3-[5-[5-[(2-methoxy-5-nitrophenyl)amino]-l,3,4-oxadiazol-2-yl]-3-
(trifluoromethyl)-lH-pyrazol-l-yl]ρhenyl]methyl]-(2S)-propanamide [895524-22-8], 2- amino-N-[[3-[5-[5-(7-benzothiazolylamino)-l,3,4-oxadiazol-2-yl]-3-(trifluoromethyl)- lH-pyrazol-l-yl]phenyl]methyl]-(2S)-propanamide [895524-23-9], [(lS)-l-methyl-2- oxo-2-[[[3-[5-[5-(phenylamino)-l,3,4-oxadiazol-2-yl]-3-(trifluoromethyl)-lH-ρyrazol- l-yl]phenyl]methyl]amino]ethyl]-, 1,1-dimethylethyl ester carbamic acid [895524-52-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
4P] known as such from international patent publication WO2006/069155 are excluded from the present invention by virtue of proviso (ii);
The compound 5-methoxy-2-([l-(2-methoxyphenyl)-r,3-dimethyl-lH,rH-434'- bipyrazol-5-yl]amino)benzoic acid [870188-47-9] known as such from international patent publication WO2005/112923 is excluded from the present invention by virtue of proviso (iii).
The compounds (Z)-5-(3-(l-methyl-lH-pyrazol-4-yl)imidazo[l,2-a]pyrazin-2- ylamino)-2,3-dihydro-lH-inden-l-one oxime [915705-05-4] known as such from international patent publication WO2005/125101 is excluded from the present invention by virtue of proviso (iv) ;
The compounds N-(4-methylphenyl)-4-(5-methyl-l-phenyl-lH-pyrazol-4-yl) 2- Thiazolamine [209117-29-3], N-(4-methoxyphenyl)-4-(5-methyl-l-phenyl-lH-pyrazol- 4-yl)-2-thiazolamine [209117-30-6], 4-(5-methyl-l-phenyl-lH-pyrazol-4-yl)-N-phenyl- 2-thiazolamine [209117-27-1], N-(4-chlorophenyl)-4-(5-methyl-l -phenyl- lH-pyrazol- 4-yl)-2-thiazolamine [209117-32-8], known as such from Singh, S.P. et al. (1997) J: Indian Chem. Soc. (74, ll-12):940-942, are excluded from the present invention by virtue of proviso (v);
The compounds 4-(5-methyl-l-phenyl-lH-pyrazol-4-yl)-N-(3-
(methylthio)phenyl)thiazol-2-amine, N1 -isopropyl-N4-(4-(5 -methyl- 1 -phenyl- 1 H- pyrazol^-yOthiazol^-yO-N^phenylbenzene-l^-diamine, N4,N4-diethyl-2-methyl-N1- (4-(5-methyl-l-phenyl-lH-pyrazol-4-yl)thiazol-2-yl)benzene-l,4-diamine; N-(IH- indazol-4-yl)-4-(5-methyl-l-phenyl-lH-pyrazol-4-yl)thiazol-2-amine; N-(2,4- dimethoxyphenyl)-4-(5 -methyl- 1 -phenyl- 1 H-pyrazol-4-yl)thiazol-2-amine; 4-(4-(5- methyl- 1 -phenyl- 1 H-pyrazol-4-yl)thiazol-2-ylamino)phenol; 4-(5 -methyl- 1 -phenyl- 1 H- pyrazol-4-yl)-N-(4-phenoxyphenyl)thiazol-2-amine; 4-(5 -methyl- 1 -phenyl- lH-pyrazol- 4-yl)-N-(l-methyl-lH-indazol-4-yl)thiazol-2-amine; N-(lH-indazol-3-yl)-4-(5-methyl- 1 -phenyl- lH-pyrazol-4-yl)thiazol-2-amine; N-(3, 5 -dimethylphenyl)-4-(5 -methyl- 1- phenyl- 1 H-pyrazol-4-yl)thiazol-2-amine; 4-(5 -methyl- 1 -phenyl- 1 H-pyrazol-4-yl)-N- (naphthalen-l-yl)thiazol-2-amine; 4-(5-methyl-l-phenyl-lH-pyrazol-4-yl)-N-(2,4,5- trimethylphenyl)thiazol-2-amine; 4-(5-methyl-l -phenyl-lH-pyrazol-4-yl)-N-(3-
KAS/CUenωocs/Addex/53195.WO01.FinalSpec.l0.072008
(trifluoromethyl)phenyl)thiazol-2-amine; N-(3 -bromophenyl)-4-(5 -methyl- 1 -phenyl- lH-pyrazol-4-yl)thiazol-2-amine; 3 -(4-(5 -methyl- 1 -phenyl- lH-pyrazol-4-yl)thiazol-2- ylaraino)benzoyl chloride; 4-(5 -methyl- 1 -phenyl- lH-pyrazol-4-y I)-N- w-tolylthiazol-2- amine; N-(3 -fluorophenyl)-4-(5 -methyl- 1 -phenyl-1 H-pyrazol-4-yl)thiazol-2-amine; N- (2-methoxyρhenyl)-4-(5-methyl-l-phenyl-lH-pyrazol-4-yl)thiazol-2-amine, known as such from US patent publication US7244739 are excluded from the present invention by virtue of proviso (v).
The compounds are 3-[5-[(4-chlorophenyl)azo]-2-(phenylamino)-4-thiazolyl]-l-(4- methylρhenyl)-5 -phenyl- lH-pyrazole-4-carbonitrile [198840-15-2], l-(4- methylphenyl)-3 - [5 - [(4-methylphenyl)azo] -2-(phenylamino)-4-thiazolyl] -5 -phenyl- 1 H- pyrazole-4-carbonitrile [198840-14-1], l-(4-methylphenyl)-5-phenyl-3-[2-
(phenylamino)-5 -(phenylazo)-4-thiazolyl] - 1 H-pyrazole-4-carbonitrile [198840-13-0], l-(4-methylphenyl)-5-phenyl-3-[2-(phenylamino)-4-thiazolyl]-lH-pyrazole-4- carbonitrile [198840-12-9], known as such from Hassan, N.M. et al (1997) J. Chem. Res., Syn. (10): 350-351, are excluded from the present invention by virtue of proviso (vi).
In a preferred aspect of Formula (I), the invention provides a compound according to Formula (II),

a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV-oxide form thereof, wherein:
X2, X3 and X4 are each independently selected from the group of C, N, O, S and C=C representing a 5 or 6 membered heteroaryl ring which may further be substituted by 1 to 3 radicals Am;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
m is an integer ranging from 1 to 3;
Am radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of
 - (CrC^alkylhalo, -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano, -(Ci-C6)alkylheteroaryl, - (CrC6)alkylaryl, aryl, heteroaryl, heterocycle, -(Co-C^alkyl-OR1, -NR1(C2-C6)alkyl- OR2, -(C3-C7)cycloalkyl-(Ci-C6)alkyl, -O-(C3-C7)cycloalkyl-(Ci-C6)alkyl, -NR^(C3- C7)cycloalkyl-(Ci-C6)alkyl, -(d-C^alkylhalo-OR1, -(d-C^alkylhalo-N^R2, -NR1- (C2-C6)alkyl-S-R2, -(C0-C6)alkyl-S(=O)-R1, -NR1-(C1-C6)alkyl-S(=O)-R2, -(C0-
- (CrC^alkylhalo, -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano, -(Ci-C6)alkylheteroaryl, - (CrC6)alkylaryl, aryl, heteroaryl, heterocycle, -(Co-C^alkyl-OR1, -NR1(C2-C6)alkyl- OR2, -(C3-C7)cycloalkyl-(Ci-C6)alkyl, -O-(C3-C7)cycloalkyl-(Ci-C6)alkyl, -NR^(C3- C7)cycloalkyl-(Ci-C6)alkyl, -(d-C^alkylhalo-OR1, -(d-C^alkylhalo-N^R2, -NR1- (C2-C6)alkyl-S-R2, -(C0-C6)alkyl-S(=O)-R1, -NR1-(C1-C6)alkyl-S(=O)-R2, -(C0-
 C6)alkyl-NR2R3, -(C0-C6)alkyl-S(=O)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3, -(C0- C6)alkyl-NR1-S(=O)2R2, -NR1-(C2-C6)alkyl-NR2-S(=O)2R3, -(C0-C6)alkyl-C(=O)- NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(Co-C^alkyl-NR^^O)^2, -NR^(C2- C6)alkyl-NR2C(=O)-R3, -NR^^-C^alkyl-OC^O)^2, -(C0-C6)alkyl-C(=O)-OR1, - NR1-(C1-C6)alkyl-C(=O)-OR2, -(C0-C6)alkyl-C(=O)-R1, -NR1-(C1-C6)alkyl-C(=O)-R2, - (C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-C6)alkyl-NR1-C(=O)-NR2R3, and -NR^(C2- C6)alkyl-NR2-C(=O)-NR3R4;
C6)alkyl-NR2R3, -(C0-C6)alkyl-S(=O)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3, -(C0- C6)alkyl-NR1-S(=O)2R2, -NR1-(C2-C6)alkyl-NR2-S(=O)2R3, -(C0-C6)alkyl-C(=O)- NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(Co-C^alkyl-NR^^O)^2, -NR^(C2- C6)alkyl-NR2C(=O)-R3, -NR^^-C^alkyl-OC^O)^2, -(C0-C6)alkyl-C(=O)-OR1, - NR1-(C1-C6)alkyl-C(=O)-OR2, -(C0-C6)alkyl-C(=O)-R1, -NR1-(C1-C6)alkyl-C(=O)-R2, - (C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-C6)alkyl-NR1-C(=O)-NR2R3, and -NR^(C2- C6)alkyl-NR2-C(=O)-NR3R4;
Any two radicals of Am (A1 and A2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of
 -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl;
-(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl;
Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
n is an integer ranging from 1 to 2;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(d-C^alkyl, -
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
(Ci-C6)alkylhalo, -(C3-C7)cycloalkyl5 -(C^C^alkylcyano, -(d-C^alkylheteroaryl, - (CrC^alkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, -O-(C2-C6)alkyl- OR5, -NR5(C2-C6)alkyl-OR6 5 -(C3-C7)cycloalkyl-(CrC6)alkyls -O-(C3-C7)cycloalkyl- (Ci-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(Ci-C6)alkylhalo-OR5 5 -(C1- C6)alkylhalo-NR5R6, -(C0-C6)alkyl-S-R5, -O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S- R6, -(C0-C6)alkyl-S(=O)-R5, -O-(C1-C6)alkyl-S(=O)-R5, -NR^CrC^alkyl-S^-R6, - (C0-C6)alkyl-S(=O)2-R5, ~O-(CrC6)alkyl-S(=O)2-R5, -NR5-(d-C6)alkyl-S(=O)2-R6, - (C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl- S(=O)2NR5R6, -O-(Ci-C6)alkyl-S(=O)2NR5R6, -NR5-(C1-C6)alkyl-S(=O)2NR6R7 5 -(C0- C6)alkyl-NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(Co-C6)alkyl-C(=0)-NR5R6, -O-(C1-C6)alkyl-CC=O)-NR5R6, -NR5-(Cr C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl-NR5C(=O)-R6, - NR5-(C2-C6)allcyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, -NR5-(C2-C6)alkyl-
 C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(Ci-C6)alkyl-C(K))-R5, -NR5-(Ci- C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(Ci-C6)alkyl-C(K))-R5, -NR5-(Ci- C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R5, R6, R7 and R8 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C^alkylhalo, -(CrC^alkyl, -(C1-C6)alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl,
 aryl, heterocycle and -(Ci-C6)alkylaryl;
aryl, heterocycle and -(Ci-C6)alkylaryl;
Any two radicals of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted 3 to 10 membered ring selected from the group of aryl, heteroaryl, heterocyclic and cycloalkyl;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
M2 is selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C0-C6)alkyl-R9 5 -(Ci-C6)alkylhalo, -(C2-C6)alkyl-NR9R10, - (C2-C6)alkyl-OR9, -(C2-C6)alkyl-SR9, -(C0-C6)alkyl-C(=O)-R9, -(C2~C6)alkyl-S(O)-R9, - (C0-C6)alkyl-C(=O)NR9R10 and -(C0-C6)alkyl-S(O)2-R9; R9 and R10 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo,
 -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(CrC^alkylaryl;
-(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(CrC^alkylaryl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Q-C^alkyl-R11, -(CrC^alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORπ and -(C2-C6)alkyl-SRπ; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(CrC^alkyl, -(Q-C^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C 10)alkylcycloalkyl, heteroaryl, -(Ci-C^alkylheteroaryl, aryl, heterocycle and -(CrC^alkylaryl; provided that according to proviso (i): when M3 is -(Co)-R11 (that is when M3 is -R11), then R11 is not H; and provided that according to proviso (ii):

when M1 is aryl, M2 is H, X2 is C, X3 is C5 X4 is N, then
 is not linked to x2; and provided that according to proviso (iv):
is not linked to x2; and provided that according to proviso (iv):
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
A1 and A2 radicals are not linked to form an imidazopyridazinyl ring; and provided that according to proviso (v):
when
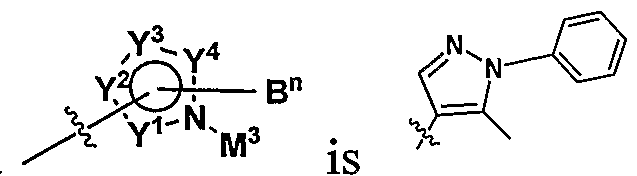 is and linked to X4, X2 is S, X3 is C, X4 is C, n is 1,
is and linked to X4, X2 is S, X3 is C, X4 is C, n is 1,
A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when X2 is S, X3 is C, X4 is C, n is 1, then A1 when linked to either X3 or X4 is not a pyridyl; and provided that according to proviso (viii): when X2 is S, X3 is C, X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
In a specific aspect of Formula (II), the invention provides a compound wherein:
X is a nitrogen, an oxygen, or a sulfur atom, X is a carbon atom or a nitrogen atom, X4 is a carbon or a nitrogen atom, representing a 5 membered heteroaryl, which may further be substituted by 1 to 2 radicals Am; m is an integer ranging from 1 to 2; Am radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, - (Ci-C6)alkylhalo, -(C3-C7)cycloalkyl, -(Ci-Q)alkylcyano, -(Ci-C6)alkylheteroaiyl, - (Ci-C6)alkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR1, -NR1(C2-C6)alkyl- OR2, -(C3-C7)cycloalkyl-(C1-C6)alkyl, -O-(C3-C7)cycloalkyl-(Ci-C6)alkyl, -NR^(C3- C7)cycloalkyl-(C1-C6)alkyl, -(Q-C^alkylhalo-OR1, -(d-C^alkylhalo-NR^2, -NR1-
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008
(C2-C6)alkyl-S-R2, -(C0-C6)alkyl-S(=O)-R1, -NR1-(C1-C6)alkyl-S(=O)-R2, -(C0- C6)alkyl-S(=O)2-R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, -(C0-C(OaIlCyI-NR1R2, -NR^(C2- C6)alkyl-NR2R3, -(C0-C6)alkyl-S(=O)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3 5 -(C0- C6)alkyl-NR1-S(=O)2R2, -NR1-(C2-C6)alkyl-NR2-S(=O)2R3, -(C0-C6)alkyl-C(=O)- NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl-NR1C(=O)-R2, -NR^(C2- C6)alkyl-NR2C(=O)-R3, -NR1-(C2-C6)alkyl-OC(=O)-R2 5 -(C0-C6)alkyl-C(=O)-OR1 5 - NR1-(C1-C6)alkyl-C(=O)-OR2, -(Co-CfOalkyl-C^CO-R1, -NR1-(C1-C6)alkyl-C(=O)-R2, - (C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-C6)alkyl-NR1-C(=O)-NR2R3, and -NR^(C2- C6)alkyl-NR2-C(=O)-NR3R4; Any two radicals of Am (A1 and A2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of
 -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Ct-C^alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl;
-(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Ct-C^alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl;
Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
n is an integer ranging from 1 to 2; Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(C1-C6)alkyl, - (CrCfOalkylhalo, -(C3-C7)cycloalkyl, -(d-C^alkylcyano, -(C1-C6)alkylheteroaryl, -
 aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, -O-(C2-C6)alkyl- OR5 , -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(C1-C6)alkyl, -O-(C3-C7)cycloalkyl- (C1-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(CrC^alkylhalo-OR5, -(C1- C6)alkylhalo-NR5R6, -(C0-C6)alkyl-S-R5, -O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S- R6, -(Co-C6)alkyl-S(=0)-R5, -O-(C1-C6)alkyl-S(=O)-R5, -NR5-(Ci-C6)alkyl-S(=O)-R6, - (C0-C6)alkyl-S(=O)2-R5, -O-(Ci-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, -
aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, -O-(C2-C6)alkyl- OR5 , -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(C1-C6)alkyl, -O-(C3-C7)cycloalkyl- (C1-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(CrC^alkylhalo-OR5, -(C1- C6)alkylhalo-NR5R6, -(C0-C6)alkyl-S-R5, -O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S- R6, -(Co-C6)alkyl-S(=0)-R5, -O-(C1-C6)alkyl-S(=O)-R5, -NR5-(Ci-C6)alkyl-S(=O)-R6, - (C0-C6)alkyl-S(=O)2-R5, -O-(Ci-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, -
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
(C0-C6)alkyl-NR5R6 5 -O-(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl- S(=O)2NR5R6, -O-(C1-C6)alkyl-S(=O)2NR5R6, -NR5-(C1-C6)alkyl-S(=O)2NR6R7 3 -(C0- C6)alkyl-NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(Ci-C6)alkyl-C(=O)-NR5R6, -NR5-(Cr C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl-NR5C(=O)-R6, - NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, -NR5-(C2-C6)alkyl- OC(=O)-R6, -(Co-C6)alkyl-C(=0)-OR5, -O-(d-C6)alkyl-C(=O)-OR5, -NR5-(d- C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(C1-C6)alkyl-C(=O)-R5, -NR5-(Cr C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-OR6 5 -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R5, R6, R7 and R8 each independently hydrogen or an optionally substituted radical selected from the group of -(d-C^alkylhalo, -(Ct-C^alkyl, -(C3-C7)cycloalkyl, -(C4- C10)alkylcycloalkyl and heterocycle;
Any two radicals of of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl; M is a hydrogen or an optionally substituted -(Ci-C^alkyl-R ; R9 is a hydrogen;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Ci-C6)alkyl-Rπ, -(CrC^alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORU and
 and
and
11 10
R and R are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(CrC^alkyl, -(Q-C^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl,
 aryl, heterocycle and -(C1-C6)alkylaryl; provided that according to proviso (i): when M3 is -(Co)-R11 (that is when M3 is -R11), then R11 is not H;
aryl, heterocycle and -(C1-C6)alkylaryl; provided that according to proviso (i): when M3 is -(Co)-R11 (that is when M3 is -R11), then R11 is not H;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
and provided that according to proviso (ii):

when M1 is aryl, X2 is C5 X3 is C, X4 is N, then
 is not linked to X2; and provided that according to proviso (iv):
is not linked to X2; and provided that according to proviso (iv):
A1 and A2 radicals are not linked to form an imidazopyridazinyl ring; and provided that according to proviso (v):
when
 is and linked to X4, X2 is S, X3 is C, X4 is C, n is 1,
is and linked to X4, X2 is S, X3 is C, X4 is C, n is 1,
A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when X2 is S5 X3 is C, X4 is C5 n is I5 then A1 when linked to either X3 or X4 is not a pyridyl; and provided that according to proviso (viii): when X2 is S5 X3 is C5 X4 is C5 to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
In a more preferred aspect of Formula (II), the invention provides a compound according to Formula (III),

a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV-oxide form thereof, wherein:
X3 is selected from C or N which may further be substituted by A1;
A1 radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(C1-C6^IkVl, - (C].-C6)alkylhalo, -(C3-C7)cycloalkyl, -(CrC^alkylcyano, -(CrC^alkylheteroaryl, - (Ci-C6)alkylaiyl, heterocycle, -(Co-C^alkyl-OR1, -NR1(C2-C6)alkyl-OR2, -(C3- C7)cycloalkyl-(C1-C6)alkyl, -O-(C3-C7)cycloalkyl-(C1-C6)alkyl, -NR^(C3-
C7)cycloalkyl-(Ci-C6)alkyl, -(CrC^alkylhalo-OR1, -(d-C^alkylhalo-NR^2, -NR1- (C2-C6)alkyl-S-R2, -(C0-C6)alkyl-S(=O)-R1, -NR1-(C1-C6)alkyl-S(=O)-R2, -(Qr C6)alkyl-S(=O)2-R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, -(Co-C^alkyl-NR^2, -NR^(C2- C6)alkyl-NR2R3, -(C0-C6)alkyl-S(=O)2NR1R2, -NR1-(Ci-C6)alkyl-S(=O)2NR2R3, -(Q,- C6)alkyl-NR1-S(=O)2R2, -NR1-(C2-C6)alkyl-NR2-S(=O)2R3, -(C0-C6)alkyl-C(=O)- NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(Co-C^alkyl-NR^CO-R2, -NR^(C2- C6)alkyl-NR2C(=O)-R3, -NR1-(C2-C6)alkyl-OC(=O)-R2, -(C0-C6)alkyl-C(-O)-OR1 5 - NR1-(C1-C6)alkyl-C(=O)-OR2, -(C0-C6)alkyl-C(=O)-R1, -NR1-(C1-C6)alkyl-C(=O)-R2, - (C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-C6)alkyl-NR1-C(=O)-NR2R3, and -NR^(C2- C6)alkyl-NR2-C(=O)-NR3R4;
R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of -(C1-C6)alkylhalo, -(CrC^alkyl, -(Ci-C6)alkylcyano, -(C3-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
43
C7)cycloalkyl, -(C4-C1o)alkylcycloalkyl, heteroaryl, -(d-C^alkylheteroaryl, aryl, heterocycle and -(CrC^alkylaryl;
Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(d-C^alkyl, - (CrC^alkylhalo, -(C3-C7)cycloalkyl, -(CrC^alkylcyano, -(d-C^alkylheteroaryl, - (CrCfOalkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, -O-(C2-C6)alkyl- OR5 , -NR5(C2-C6)alkyl-OR6, -(Cs-C^cycloalkyHd-C^alkyl, -O-(C3-C7)cycloalkyl- (d-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(Ci-C6)alkyl, -(d-C6)alkylhalo-OR5 5 -(Ci- C6)alkylhalo-NR5R6, -(C0-C6)alkyl-S-R5, -O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S- R6, -(C0-C6)alkyl-S(=O)-R5, -O-(C1-C6)alkyl-S(=O)-R5, -NR5-(C!-C6)alkyl-S(=O)-R6, - (C0-C6)alkyl-S(=O)2-R5 5 -O-(Ci-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, - (C0-C6)alkyl-NR5R6 5 -O-(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl- S(^O)2NR5R6, -O-(C1-C6)alkyl-S(=O)2NR5R6, -NR5-(Ci-C6)alkyl-S(=O)2NR6R7, -(C0- C6)alkyl-NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(Co-C6)alkyl-C(=0)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, -NR5-(d- C6)alkyl-C(=O)-NR6R7 5 -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl-NR5C(=O)-R6, - NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, -NR5-(C2-C6)alkyl- 0C(=0)-R6, -(C0-C6)alkyl-C(==O)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5, -NR5-(Ci- C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(Ci-C6)alkyl-C(=O)-R5, -NR5-(Ci- C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7 5 -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8; R5, R6, R7 and R8 are each independently hydrogen or an optionally substituted radical selected from the group of -(d-C^alkylhalo, -(Ci-C^alkyl, -(Ci-C^alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)aUcylcycloalkyl, heteroaryl, -(Ci-C6)alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
Any two radicals of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle,
 -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORU and -(C2-C6)alkyl-SRπ; and
-(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORU and -(C2-C6)alkyl-SRπ; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Q-C^alkyl, -(Q-C^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(Q-C^alkylaryl; and provided that according to proviso (v):
when
 , X is C, n is 1, A is H, then M can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol- 4-yl; and provided that according to proviso (vi) : when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when A1 is different from a pyridyl; and provided that according to proviso (viii): when X3 is C, to provide a thiazole ring, n is I5 then A1 is not an optionally substituted imidazolyl or triazolyl ring.
, X is C, n is 1, A is H, then M can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol- 4-yl; and provided that according to proviso (vi) : when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when A1 is different from a pyridyl; and provided that according to proviso (viii): when X3 is C, to provide a thiazole ring, n is I5 then A1 is not an optionally substituted imidazolyl or triazolyl ring.
In a specific aspect of Formula (III), the invention provides a compound wherein:
A1 radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Ci-C^alkyl, -
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
OOll
(Ci-CfOalkylhalo, -(C3-C7)cycloalkyl, -(C1-C6)alkylcyano, heterocycle, -(C0-C6)alkyl- OR1, -(Cs-C^cycloalkyl-CCi-C^alkyl, -NR^Cs-C^cycloalkyKCrC^alkyl, -(C1- COalkylhalo-OR1, -(CrC^alkylhalo-NR^2, (Co-C^alkyl-NR^2, -NR^CrC^alkyl- NR2R3, -(C0-C6)alkyl-C(=O)-NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3 5 -(C0-C6)alkyl- NR1CC=O)-R2, -NR1-(C2-C6)alkyl-NR2C(=O)-R3, -(Co-C^alkyl-C^-R1, -NR1^C1- C6)alkyl-C(=O)-R2;
R1, R2 and R3 are each independently hydrogen or an optionally substituted radical selected from the group of
 -(C3-C7)cycloalkyl, -(C4- C10)alkylcycloalkyl and heterocycle; Any two radicals of R (R1, R2 or R3) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
-(C3-C7)cycloalkyl, -(C4- C10)alkylcycloalkyl and heterocycle; Any two radicals of R (R1, R2 or R3) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Q-C^alkyl, - (d-C^alkylhalo, -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano, -(Ci-C6)alkylheteroaryl5 - (C1-C6)alkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, -O-(C2-C6)alkyl- OR5 , -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(C1-C6)alkyl, -O-(C3-C7)cycloalkyl- (C!-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(CrC6)alkylhalo-OR5, -(C1- C6)alkylhalo-NR5R6, -(C0-C6)alkyl-S-R5, -O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S- R6, -(C0-C6)alkyl-S(=O)-R5, -©-(d-C^alkyl-S^O)^5, -NR5-(C1-C6)alkyl-S(=O)-R6, - (C0-C6)alkyl-S(=O)2-R5, -O-(C1-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, - (C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl- S(^O)2NR5R6, -O-(C1-C6)alkyl-S(=O)2NR5R6, -NR5-(C1-C6)alkyl-S(=O)2NR6R7, -(C0- C6)alkyl-NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(Ci-C6)alkyl-C(=O)-NR5R6, -NR5-(C1- C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl-NR5C(=O)-R6, - NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, -NR5-(C2-C6)alkyl- OC(=O)-R6 3 -(C0-C6)alkyl-C(=O)-OR5, -O-(CrC6)alkyl-C(=O)-OR5, -NR5-(Ci- C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -©-(CrC^alkyl-C^O)^5, -NR5-(Cr
KAS/Clienωocs/Addex/53195.WO0LFinalSpec.l0.072008
C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R5, R6, R7 and R8 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C^alkylhalo, -(CrC^alkyl, -(C1-C6)alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(Ci-C6)alkylaryl;
Any two radicals of R (R5, R6, R7 and R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl; M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC^alkyl-R11, -(Ci-C6)alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORπ and -(C2-C6)alkyl-SRn; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Ci-C^alkyl, -(CrC^alkylcyano, -(C3-C7)cycloalkyl5 -(C4-C10)alkylcycloalkyl5 heteroaryl, -(d-C^alkylheteroaryl, aryl, heterocycle and -(Cj.-C6)alkylaryl; provided that according to proviso (v):
when
 is , X3 is C, n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol- 4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): A1 is not a pyridyl; and provided that according to proviso (viii):
is , X3 is C, n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol- 4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): A1 is not a pyridyl; and provided that according to proviso (viii):
KAS/ClientDocs/Addex/SSlQS.WOOLFinalSpec.lO.OV ZOOδ
when X3 is C to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
In a specific aspect of Formula (III), the invention provides a compound according to Formula (IIIA),

a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV-oxide form thereof, wherein:
A1 radical is selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, -(C1-
C6)alkylhalo, -(C3-C7)cycloalkyl, -(C1-C6)alkylcyano, heterocycle, -(Co-C^alkyl-OR1, -
(C3-C7)cycloalkyl-(C1-C6)alkyl, -NR^Cs-C^cycloalkyKCi-C^alkyl, -(C1-
C^alkylhalo-OR1, -(Q-C^alkylhalo-NR^R2, (Co-C^alkyl-NR^2, -NR1-(C2-C6)alkyl-
NR2R3, -(C0-C6)alkyl-C(=O)-NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl- NR!C(=O)-R2, -NR1-(C2-C6)alkyl-NR2C(=O)-R3, -(C0-C6)alkyl-C(=O)-R1, and -NR1-
(d-C^alkyl-C^O)^2;
R1, R2 and R3 are each independently hydrogen or an optionally substituted radical selected from the group of
 -(Ci-C6)alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)aUiylcycloalkyl, heteroaryl,
-(Ci-C6)alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)aUiylcycloalkyl, heteroaryl,
 aryl, heterocycle and -(Ct-C^alkylaryl;
aryl, heterocycle and -(Ct-C^alkylaryl;
Any two radicals of R (R1, R2 and R3) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, - (CrC^alkylhalo, -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, - O-(C2-C6)alkyl-OR5 , -NR5(C2-C6)alkyl-OR6, -(Cs-C^cycloalkyl-Cd-C^alkyl, -0-(C3-
 OR5, -(d-C^alkylhalo-NR^6, -(C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6 5 -NR5- (C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, - (C1-C6)alkyl-OC(=O)-R5, -(C0-C6)alkyl-C(=O)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5, - NR5-(C1-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(C1-C6)alkyl-C(=O)-Rs and -NR5-(CrC6)alkyl-C(=O)-R6;
OR5, -(d-C^alkylhalo-NR^6, -(C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6 5 -NR5- (C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, - (C1-C6)alkyl-OC(=O)-R5, -(C0-C6)alkyl-C(=O)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5, - NR5-(C1-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(C1-C6)alkyl-C(=O)-Rs and -NR5-(CrC6)alkyl-C(=O)-R6;
R5, R6 and R7 are each independently hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(CrC^alkyl, -(CrC^alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(d-C^alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl; Any two radicals of R (R5, R6, or R7) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C1-C6)alkyl-R11, -(d-C^alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORπ and -(C2-C6)alkyl-SRn; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(d-C^alkylhalo, -(Ct-C^alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(d-C^alkylheteroaryl, aryl, heterocycle and - (d-C^alkylaryl; provided that according to proviso (v):
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
when B" is
 , X3 is C5 n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-
, X3 is C5 n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-
4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
In a specific embodiment of the Formula (IIIA),
A1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Q-C^alkyl, -(Q-C^alkylhalo, -(C3- C7)cycloalkyl and heterocycle; n is an integer ranging from 1 to 2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(CrC^alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5, and -(C3-C7)CyClOaIlCyI-(C1- C6)alkyl; or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of - CF3 and an optionally substituted radical selected from the group of -(C1-Ce^UCyI5 -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(C1-C6)alkyl;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
R5 is selected from the group of hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Ci-C6)alkyl, -(C3-C7)cycloalkyl, -(C4- C10)alkylcycloalkyl and heterocycle;
M1 is an optionally substituted aryl; M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C1-C6)alkyl-R11, -(Ci-C6)alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRπ;
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C].-C6)alkylhalo, -(Ci-C6)alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and - (Ci-C6)alkylaryl; provided that according to proviso (v):
when B" is
 , X3 is C, n is 1, A1 is H, then M1 can not be 1-methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii) : A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C, X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
, X3 is C, n is 1, A1 is H, then M1 can not be 1-methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii) : A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C, X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
In another specific embodiment of the Formula (IIIA),
A1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, -(Ct-C^alkylhalo, -(C3- C7)cycloalkyl and heterocycle; n is an integer ranging from 1 to2, and either;
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.07 2008
(a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5, and -(C3-C7)cycloalkyl-(Ci- C6)alkyl; or (b) n is 2, and B1 and B2 radicals are each independently selected from the group of - CF3 and an optionally substituted radical selected from the group of -(Q-C^alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(CrC6)alkyl;
R5 is selected from the group hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(Q-C^alkyl, -(C3-C7)cycloalkyl, -(C4- C10)alkylcycloalkyl and heterocycle;
M1 is an optionally substituted heteroaryl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC^alkyl-R11, -(CrC^alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORπ and -(C2-C6)alkyl-SRn; R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(d-C^alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and - (Q-COalkylaryl; and provided that according to proviso (v):
when B" {s
 χ3 is C, n is 1, A1 is H, then M1 can not be 1-methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii):
χ3 is C, n is 1, A1 is H, then M1 can not be 1-methyl- lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii):
A1 is not a pyridyl; and provided that according to proviso (viii):
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
when X3 is C to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
In a more specific embodiment of the Formula (IIIA), A1 radical is hydrogen, n is an integer ranging from 1 to2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, -CF3, -(Q-C^alkyl and -(d-COalkylhalo; or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of - CF3 and an optionally substituted radical selected from the group
 and -
and -
(Ci-C6)alkylhalo;
M1 is an optionally substituted pyridyl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle,
 -(Ci-C6)alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORπ and -(C2-C6)alkyl-SRπ; and
-(Ci-C6)alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORπ and -(C2-C6)alkyl-SRπ; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Ci-C^alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl,
 aryl, heterocycle and - (Ci-C6)alkylaryl.
aryl, heterocycle and - (Ci-C6)alkylaryl.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
IB2008/002243
In a more specific aspect of Formula (III), the invention provides a compound according to Formula (IIIB),

a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV-oxide form thereof, wherein: n is an integer ranging from 1 to 2;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of
 - (CrC6)alkylhalo, -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, - O-(C2-C6)alkyl-OR5 , -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(C1-C6)alkyl, -0-(C3- C^cycloalkyKd-C^alkyl, -NR5-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(d-C^alkylhalo- OR5, -(d-C6)alkylhalo-NR5R6, -(C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6, -NR5- (C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, - (C1-C6)alkyl-OC(=O)-R5, -(C0-C6)alkyl-C(=O)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5, - NR5-(C1-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-^rC^alkyl-C^O)^5 and -NR5-(C1-C6)alkyl-C(=O)-R6;
- (CrC6)alkylhalo, -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-OR5, - O-(C2-C6)alkyl-OR5 , -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(C1-C6)alkyl, -0-(C3- C^cycloalkyKd-C^alkyl, -NR5-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(d-C^alkylhalo- OR5, -(d-C6)alkylhalo-NR5R6, -(C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6, -NR5- (C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, - (C1-C6)alkyl-OC(=O)-R5, -(C0-C6)alkyl-C(=O)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5, - NR5-(C1-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-^rC^alkyl-C^O)^5 and -NR5-(C1-C6)alkyl-C(=O)-R6;
R5, R6 and R7 are each independently hydrogen or an optionally substituted radical selected from the group of -(d-C^alkylhalo, -(C1-C6)alkyl, -(C1-C6)alkylcyano, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl; Any two radicals of R (R5, R6, or R7) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
M3 is an optionally substituted radical selected from the group of -(C3-C7)CyClOaHCyI, aryl, heteroaryl, heterocycle, -(Ci-C6)alkyl-Rn, -(Ci-C6)alkylhalo. -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRπ; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(CrC^alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl,
 aryl, heterocycle and - (Ci-C6)alkylaryl.
aryl, heterocycle and - (Ci-C6)alkylaryl.
In a specific embodiment of the Formula (IIIB), n is an integer ranging from 1 to 2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(CrC^alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-ORS, aryl, heteroaryl, and -(C3- C7)cycloalkyl-(C1-C6)alkyl; or (b) n is 2, and B1 and B2 radicals are each independently selected from the group of - CF3 and an optionally substituted radical selected from the group of -(Ct-C^alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(Ci-C6)alkyl;
R5 is selected from the group hydrogen or an optionally substituted radical selected from the group of -(CrC6)alkylhalo, -(CrC^alkyl, -(C3-C7)cycloalkyl, -(C4- C10)alkylcycloalkyl and heterocycle;
M1 is an optionally substituted aryl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(d-C^alkyl-R11, -(CrC^alkylhalo, -(C2-C6)alkyl- NR11R12, -(Ca-C^alkyl-OR11 and -(C2-C6)alkyl-SRπ; and R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of
 -(C3-C7)cycloalkyl,
-(C3-C7)cycloalkyl,
KAS/ClientDocs/Addex/SSm.WOOl.FinalSpec.lO.Oy ZOOδ
-(C4-C10)alkylcycloalkyl, heteroaryl, -(d-C^alkylheteroaryl, aryl, heterocycle and - (Ci-C6)alkylaryl.
In another specific embodiment of the Formula (IIIB), n is an integer ranging from 1 to 2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(C1-C6)alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5, aryl, heteroaryl, and -(C3- C7)cycloalkyl-(Ci-C6)alkyl; or (b) n is 2, and B1 and B2 radicals are each independently selected from the group of - CF3 and an optionally substituted radical selected from the group
 -(C1- C6)alkylhalo, ~(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(C1-C6)alkyl;
-(C1- C6)alkylhalo, ~(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(C1-C6)alkyl;
R5 is selected from the group hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Ci-C6)alkyl, -(C3-C7)cycloalkyl, -(C4- C10)alkylcycloalkyl and heterocycle;
M1 is an optionally substituted heteroaryl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Q-C^alkyl-R11, -(Ci-C6)alkylhalo, -(C2-C6)alkyl- NR11R12, -(C2-C6)alkyl-ORπ and
 and R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Q-C^alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Q-C^alkylheteroaryl, aryl, heterocycle and - (Ci-CfOalkylaryl.
and R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Q-C^alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Q-C^alkylheteroaryl, aryl, heterocycle and - (Ci-CfOalkylaryl.
In a more specific embodiment of the Formula (IIIB), n is an integer ranging from 1 to 2, and either;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
(a) n is 1 and B1 radical is selected from the group of hydrogen, -CF3, -(Q-C^alkyl and -(Ci-C6)alkylhalo; or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of - CF3 and an optionally substituted radical selected from the group of -(Ct-C^alkyl and - (Ci-C6)alkylhalo;
M1 is an optionally substituted pyridyl;
M3 is an optionally substituted radical selected from the group of -(C3-C7)cycloalkyl, atyl, heteroaryl, heterocycle, -(Ci-C6)alkyl-Ru s -(CrC^alkylhalo, -(C2-C6)alkyl- NR11R12, -(d-C^alkyl-OR11 and -(C2-C6)alkyl-SRπ; and R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C1-C6)alkymalo, -(Ci-C6)alkyl, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and - (CrC^alkylaryl.
Specific compounds of the invention according to Formula (I) to (III) are compounds as mentioned in the following list A and list B as well as a pharmaceutically acceptable acid or base addition salt thereof, a stereochemical^ isomeric form thereof and an N- oxide form thereof:
List A:
3-(l,3-Dimethyl-lH-pyrazol-4-yl)-N-methyl-N-phenyl-l,2,4-thiadiazol-5-amine
3-(l,5-Dimethyl-lH-pyrazol-4-yl)-N-methyl-iV-phenyl-l,254-thiadiazol-5-amine
N-(2-Fluorophenyl)-3-(l-(4-methoxybenzyl)-3-methyl-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5 -amine
N-(2-Fluorophenyl)-3-(l-(4-methoxybenzyl)-5-methyl-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5 -amine
3 -(I -(4-Methoxybenzyl)-3 -methyl- 1 H-ρyrazol-4-yl)-N-(pyridin-2-yl)- 1 ,2,4- thiadiazol-5 -amine
3-(l-(4-Methoxybenzyl)-5-methyl-lH-pyrazol-4-yl)-N-(ρyridin-2-yl)-l,2,4-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
thiadiazol-5 -amine
3-(l-(4-Methoxybenzyl)-lH-pyrazol-4-yl)-N-phenyl-l,2,4-tMadiazol-5-ainine
4-(l-(4-Methoxybenzyl)-3-methyl-lH-pyrazol-4-yl)-N-phenylthiazol-2-amine
4-(l-(4-Methoxybenzyl)-5-methyl-lH-pyrazol-4-yl)-N-phenylthiazol-2-amine
3 -(5 -Methyl- 1 -phenyl- 1 H-pyrazol-4-yl)-iV-phenyl- 1 ,2,4-thiadiazol-5 -amine
3-(3-Methyl-l-propyl-lf/-pyrazol-4-yl)-iV-(pyridin-2-yl)-l52,4-thiadiazol-5-amine
4-(l-Ben2yl-lH-pyrazol-4-yl)-N-phenylthiazol-2-amine
4-(l-Isopropyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine
4-( 1 -Ethyl- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine and
4-(l-Methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine.
List B:
4-(l -Propyl- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine N-(Benzo[d][l,3]dioxol-5-yl)-4-(l-propyl-lH-pyrazol-4-yl)thiazol-2-amine N-(2-Fluorophenyl)-4-(l-propyl-lH-pyrazol-4-yl)thiazol-2-amine 4-(3-Methyl-l-propyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine 4-(3 -Methyl- 1 -propyl- 1 H-pyrazol-4-yl)-N-(6-methylpyridin-2-yl)thiazol-2-amine N-(5-Chloropyridin-2-yl)-4-(3-methyl-l-propyl-lH-pyrazol-4-yl)thiazol-2-amine N-(6-Methoxypyridin-3 -yl)-4-(3 -methyl- 1 -propyl- 1 H-pyrazol-4-yl)thiazol-2-amine N-(2-Methoxypyridin-3 -yl)-4-(3 -methyl- 1 -propyl- lH-pyrazol-4-yl)thiazol-2-amine 4-(l-Propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine 4-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine N-(Pyridin-2-yl)-4-(l-(2,2,2-trifluoroethyl)-lH-ρyrazol-4-yl)thiazol-2-amine 3-(l-Ethyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-tMadiazol-5-amine 3-(l-Ethyl-lH-pyrazol-4-yl)-N-(6-methoxypyridin-2-yl)-l,2,4-thiadiazol-5-amine 3-(l-Ethyl-lH-pyrazol-4-yl)-N-(6-morpholinopyridin-2-yl)-l52,4-thiadiazol-5-amine 3-(l-Ethyl-3-methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l52,4-thiadiazol-5-amine 3-(l-Propyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5-amine 3-(l-(3-Fluorobenzyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,254-thiadiazol-5-amine 3-(l -(3 -Fluorobenzyl)- 1 H-pyrazol-4-yl)-N-phenyl- 1 ,2,4-thiadiazol-5-amine
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
N-(Pyridin-2-yl)-3-(l-((tetrahydro-2H-pyran-4-yl)methyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(pyridine-2-yl)-l,2,4-thiadiazol-5- amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine hydrochloride
3-(l -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(6-methylpyridin-2-yl)- 1 ,2,4- thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(6-ethylpyridin-2-yl)-l,2,4-thiadiazol-
5 -amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(6-fluoropyridin-2-yl)-l,2,4- thiadiazol-5-amine
3-(l -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(5-fluoropyridin-2-yl)- 1 ,2,4- thiadiazol-5 -amine
3 -( 1 -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(6-methoxypyridin-2-yl)- 1 ,2,4- thiadiazol-5 -amine
N-(6-Cyclobutoxypyridin-2-yl)-3 -(I -(cyclopropylmethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4- thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(6-morpholinopyridin-2-yl)-l,2,4- thiadiazol-5 -amine
3 -(I -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(5-morpholinopyridin-2-yl)- 1 ,2,4- thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(6-(trifluoromethyl)pyridin-2-yl)-
1 ,2,4-thiadiazol-5-amine
N-(5-Chloropyridin-2-yl)-3-(l-(cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5 -amine
6-(3 -(I -(Cyclopropylmethyl)- 1 H-ρyrazol-4-yl)- 1 ,2,4-thiadiazol-5- ylamino)nicotinonitrile
3 -(I -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(pyrazin-2-yl)- 1 ,2,4-thiadiazol-5 - amine
KASZClientDocsZAddexZ53195.WO01.FinalSpec.l0.072008
6-(3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-l:>2,4-thiadiazol-5- ylamino)nicotinamide
3-(l-(Cyclobutylmethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5-amine
3 -(I -(2-Morpholinoethyl)- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)- 1 ,254-thiadiazol-5-amine
N-(Pyridin-2-yl)-3-(l-((tetrahydrofuran-2-yl)methyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5 -amine
N-(Pyridin-2-yl)-3-(l-(2,2,2-trifluoroethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazol-5- amine
3-(l-(2-Methoxyethyl)-lH-pyrazol-4-yl)-N-(jpyridin-2-yl)-l,2,4-thiadiazol-5-aniine
2-(4-(5-(Pyridin-2-ylamino)- 1 ,2,4-thiadiazol-3-yl)- 1 H-pyrazol- 1 -yl)ethanol
3-(l-Cyclobutyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5-amine
3-(l-(3-Fluoro-4-niethoxyphenyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-ls2,4-thiadiazol-
5 -amine
3-(l-(4-Fluorophenyl)-5-methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-
5 -amine
3-(l-(4-Fluorophenyl)-5-methyl-lH-pyrazol-4-yl)-N-phenyl-l,2,4-thiadiazol-5-amine
3-(l-Propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine
N-(6-Methylρyridin-2-yl)-3 -(I -proρyl-3 -(trifluoromethyl)- 1 H-ρyrazol-4-yl)- 1 ,2,4- thiadiazol-5-amine
N-(6-Methoxypyridin-3 -yl)-3 -(I -propyl-3 -(trifluoromethyl)- lH-pyrazol-4-yl)- 1 ,2,4- thiadiazol-5 -amine
N-(2-Methoxypyridin-3 -yl)-3 -( 1 -propyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4- thiadiazol-5 -amine
N-(5-Chloroρyridin-2-yl)-3-(l-ρropyl-3-(trifluoiOmethyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5 -amine
3-(l-Propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-N-(4-(trifluoromethyl)pyridin-2- yl)- 132,4-thiadiazol-5 -amine
N-(355-Difluoropyridin-2-yl)-3-(l-ρroρyl-3-(trifluoromethyl)-lH-ρyrazol-4-yl)-l,254- thiadiazol-5 -amine
KASZClientDocsZAddexZ53195.WO01.FinalSpec.l0.07 2008
3 -(I -Propyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)-N-(quinolin-2-yl)- 1 ,2,4-thiadiazol-
5 -amine
N-(2-Methylpyridin-4-yl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5-amine
N-(2-Fluorophenyl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-l,234- thiadiazol-5-amine
N-(2,5-Difluorophenyl)-3 -(I -propyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4- thiadiazol-5 -amine
N-(Benzo[d][l,3]dioxol-5-yl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5-amine
N-(4-Morpholinophenyl)-3 -(I -propyl-3-(trifluoromethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4- thiadiazol-5-amine
N-(3-Methoxyphenyl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-l52,4- thiadiazol-5 -amine
3 -(I -Propyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)-N-(3 -(trifluoromethyl)phenyl)-
1 ,2,4-thiadiazol-5-amine
N-(4-Chloroρhenyl)-3-(l-proρyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5 -amine
3-(l-(Cyclopropylmethyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-
1 ,2,4-thiadiazol-5-amine
N-(Pyridin-2-yl)-3-(l-(2,2,2-trifluoroethyl)-3-(trifluoromethyl)-lH-pyrazol-4-yl)-
1 ,2,4-thiadiazol-5-amine
3 -(3 ,5-Dimethyl- 1 -propyl- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)- 1 ,2,4-thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-3,5-dimethyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4- thiadiazol-5 -amine
3-(l-(Cyclopropylmethyl)-3,5-dimethyl-lH-pyrazol-4-yl)-N-(6-methylpyridin-2-yl)-
1 ,2,4-thiadiazol-5 -amine
3-(3-Methyl-l-propyl-lH-pyrazol-4-yl)-N-(6-methylpyridin-2-yl)-l,2,4-thiadiazol-5- amine
N-(6-Ethylpyridin-2-yl)-3-(3-methyl-l-proρyl-lH-pyrazol-4-yl)-l,2,4-thiadiazol-5-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
amine
3-(3-Methyl-l-propyl-lH-pyrazol-4-yl)-N-(6-(trifluoromethyl)pyridin-2-yl)-l,2,4- thiadiazol-5-amine
3-(3-Methyl-l-propyl-lH-pyrazol-4-yl)-N-(3-(trifluoromethyl)pyridin-2-yl)-l,2,4- thiadiazol-5 -amine
N-(5-Chloropyridin-2-yl)-3-(3-methyl-l-propyl-lH-pyrazol-4-yl)-l,2,4-thiadiazol-5- amine
N-(5-Fluoropyridin-2-yl)-3-(3-methyl-l-propyl-lH-pyrazol-4-yl)-l52,4-thiadiazol-5- amine
6-(3-(3 -Methyl-1 -propyl- lH-pyrazol-4-yl)-l ,2,4-thiadiazol-5-ylamino)nicotinonitrile
3 -(3 -Methyl- 1 -propyl- 1 H-pyrazol-4-yl)-N-(6-morpholinopyridin-2-yl)- 1,2,4- thiadiazol-5 -amine
N-(6-Methoxypyridin-2-yl)-3-(3-methyl-l-propyl-lH-pyrazol-4-yl)-l,2,4-thiadiazol-
5 -amine
N-(6-Fluoropyridin-2-yl)-3-(3 -methyl- 1 -propyl- 1 H-pyrazol-4-yl)- 1 ,2,4-thiadiazol-5- amine and
3-(3-methyl-l-propyl-lH-pyrazol-4-yl)-N-(pyridin-3-yl)-l,2,4-thiadiazol-5-amine.
DEFINITION OF TERMS
Listed below are definitions of various terms used in the specification and claims to describe the present invention.
For the avoidance of doubt it is to be understood that in this specification "(C1-C6)" means a carbon radical having 1, 2, 3, 4, 5 or 6 carbon atoms. "(Co-C6)" means a carbon radical having 0, 1, 2, 3, 4, 5 or 6 carbon atoms. In this specification "C" means a carbon atom, "N" means a nitrogen atom, "O" means an oxygen atom and "S" means a sulphur atom.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
In the case where a subscript is the integer 0 (zero) the radical to which the subscript refers, indicates that the radical is absent, i.e. there is a direct bond between the radicals.
In this specification, unless stated otherwise, the term "bond" refers to a saturated covalent bond. When two or more bonds are adjacent to one another, they are assumed to be equal to one bond. For example, a radical -A-B-, wherein both A and B may be a bond, the radical is depicting a single bond.
In this specification, unless stated otherwise, the term "alkyl" includes both straight and branched chain alkyl radicals and may be methyl, ethyl, n-propyl, i-propyl, n-butyl, i- butyl, s-butyl, t-butyl, n-pentyl, i-pentyl, t-pentyl, neo-pentyl, n-hexyl, i-hexyl or t- hexyl. The term "(C0-C3)alkyl" refers to an alkyl radical having 0, 1, 2 or 3 carbon atoms and may be methyl, ethyl, n-propyl and i-propyl.
In this specification, unless stated otherwise, the term "cycloalkyl" refers to an optionally substituted carbocycle containing no heteroatoms, including mono-, bi-, and tricyclic saturated carbocycles, as well as fused ring systems. Such fused ring systems can include one ring that is partially or fully unsaturated such as a benzene ring to form fused ring systems such as benzo- fused carbocycles. Cycloalkyl includes such fused ring systems as spirofused ring systems. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, decahydronaphthalene, adamantane, indanyl, fluorenyl and 1,2,3,4-tetrahydronaphthalene and the like. The term "(C3-C7)cycloalkyT may be cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like. In this specification, unless stated otherwise, the term "alkenyl" includes both straight and branched chain alkenyl radicals. The term "(C2-C6)alkenyl" refers to an alkenyl radical having 2 to 6 carbon atoms and one or two double bonds, and may be, but is not limited to vinyl, allyl, propenyl, i-propenyl, butenyl, i-butenyl, crotyl, pentenyl, i-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
pentenyl and hexenyl.
In this specification, unless stated otherwise, the term "alkynyl" includes both straight and branched chain alkynyl radicals. The term (C2-C6)alkynyl having 2 to 6 carbon atoms and one or two triple bonds, and may be, but is not limited to ethynyl, propargyl, butynyl, i-butynyl, pentynyl, i-pentynyl and hexynyl.
The term "aryl" refers to an optionally substituted monocyclic or bicyclic hydrocarbon ring system containing at least one unsaturated aromatic ring. Examples and suitable values of the term "aryl" are phenyl, naphthyl, 1,2,3,4-tetrahydronaphthyl, indyl, indenyl, benzo[d][l,3]dioxolyl and the like.
In this specification, unless stated otherwise, the term "heteroaryl" refers to an optionally substituted monocyclic or bicyclic unsaturated, aromatic ring system containing at least one heteroatom selected independently from N, O or S. Examples of "heteroaryl" may be, but are not limited to thienyl, pyridyl, thiazolyl, isothiazolyl, furyl, pyrrolyl, triazolyl, imidazolyl, oxadiazolyl, oxazolyl, isoxazolyl, pyrazolyl, imidazolonyl, oxazolonyl, thiazolonyl, tetrazolyl, thiadiazolyl, benzoimidazolyl, benzoxazolyl, benzothiazolyl, tetrahydrotriazolopyridyl, tetrahydrotriazolopyrimidinyl, benzofuryl, benzothiophenyl, thionaphthyl, indolyl, isoindolyl, pyridonyl, pyridazinyl, pyrazinyl, pyrimidinyl, quinolyl, quinolinyl, phtalazinyl, naphthyridinyl, quinoxalinyl, quinazolyl, imidazopyridyl, oxazolopyridyl, thiazolopyridyl, imidazopyridazinyl, oxazolopyridazinyl, thiazolopyridazinyl, cynnolyl, pteridinyl, furazanyl, benzotriazolyl, pyrazolopyridinyl and purinyl. Examples of pyrazolyl may be, but not limited to, IH- pyrazol-4-yl. Examples of pyridinyl may be, but not limited to, pyridin-2-yl, pyridin-3- yl and pyridin-4-yl.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
In this specification, unless stated otherwise, the term "alkylaryl", "alkylheteroaryl" and "alkylcycloalkyl" refers respectively to a substituent that is attached via the alkyl radical to an aryl, heteroaryl or cycloalkyl radical, respectively. The term "(C1- C6)alkylaryl" includes aryl-Ci-C6-alkyl radicals such as benzyl, 1-phenylethyl, 2- phenylethyl, 1-phenylpropyl, 2-phenylpropyl, 3-phenylpropyl, 1-naphthylmethyl and 2- naphthylmethyl. The term "(Ci-C6)alkyheteroaryl" includes heteroaryl-CrCδ-alkyl radicals, wherein examples of heteroaryl are the same as those illustrated in the above definition, such as 2-furylmethyl, 3-furylmethyl, 2-thienylmethyl, 3-thienylmethyl, 1- imidazolylmethyl, 2-imidazolylmethyl, 3-imidazolyhnethyl, 2-oxazolylmethyl, 3- oxazolylmethyl, 2-thiazolylmethyl, 3-thiazolylmethyl, 2-pyridylmethyl, 3- pyridylmethyl, 4-pyridylmethyl, 1-quinolylmethyl or the like.
In this specification, unless stated otherwise, the term "heterocycle" refers to an optionally substituted, monocyclic or bicyclic saturated, partially saturated or unsaturated ring system (containing at least one heteroatom selected independently from N, O and S. The said "heterocycle" refers respectively to a group linked either via the carbon or the nitrogen. The term "heterocycle" includes morpholine, thiomorpholine, tetrahydrofuran, tetrahydropyran radicals or the like.
In this specification, unless stated otherwise, the term "alkylheterocycle" refers respectively to a substituent that is attached via the alkyl radical to a heterocycle radical, respectively. The term "(Q-C^alkyl heterocycle" includes heterocycle-Ci-C6- alkyl radicals such as (tetrahydro-2H-pyran-4-yl)methyl, (tetrahydrofuran-2-yl)methyl or morpholinoethyl. In this specification, unless stated otherwise, a 5- or 6-membered ring containing one or more atoms independently selected from C, N, O and S, includes aromatic and heteroaromatic rings as well as carbocyclic and heterocyclic rings which may be saturated or unsaturated. Examples of such rings may be, but are not limited to, furyl, isoxazolyl, isothiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidyl,
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
pyrrolyl, thiazolyl, thienyl, imidazolyl, imidazolidinyl, imidazolinyl, triazolyl, morpholinyl, piperazinyl, piperidyl, piperidonyl, pyrazolidinyl, pyrazolinyl, pyrrolidinyl, pyrrolinyl, tetxahydropyranyl, tetrahydrothiopyranyl, oxazolidinonyl, thiomorpholinyl, oxadiazolyl, thiadiazolyl, tetrazolyl, phenyl, cyclohexyl, cyclopentyl, cyclohexenyl and cyclopentenyl.
In this specification, unless stated otherwise, a 3- to 10-nienibered ring containing one or more atoms independently selected from C, N, O and S, includes aromatic and heteroaromatic rings as well as carbocyclic and heterocyclic rings which may be saturated or unsaturated. Examples of such rings may be, but are not limited to imidazolidinyl, imidazolinyl, morpholinyl, piperazinyl, piperidyl, piperidonyl, pyrazolidinyl, pyrazolinyl, pyrrolidinyl, pyrrolinyl, tetrahydropyranyl, thiomorpholinyl, tetrahydrothiopyranyl, furyl, pyrrolyl, isoxazolyl, isothiazolyl, oxazolyl, oxazolidinonyl, pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidyl, pyrrolyl, thiazolyl, thienyl, imidazolyl, triazolyl, phenyl, cyclopropyl, aziridinyl, cyclobutyl, azetidinyl, oxadiazolyl, thiadiazolyl, tetrazolyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, cycloheptenyl, cyclooctyl and cyclooctenyl.
In this specification, unless stated otherwise, the term "halo" or "halogen" may be fluoro, chloro, bromo or iodo.
In this specification, unless stated otherwise, the term "alkylhalo" means an alkyl radical as defined above, substituted with one or more halo radicals. The term "(C1- C6)alkylhalo" may include, but is not limited to, fluoromethyl, difluoromethyl, trifluoromethyl, fluoroethyl, difluoroethyl and trifluoroethyl. The term "0-C1-C6- alkylhalo" may include, but is not limited to, fluoromethoxy, difluoromethoxy, trifluoromethoxy and fluoroethoxy.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
In this specification, unless stated otherwise, the term "alkylcyano" means an alkyl radical as defined above, substituted with one or more cyano.
In this specification, unless stated otherwise, the term "optionally substituted" refers to radicals further bearing one or more substituents which may be, but are not limited to, (Ci-C6)alkyl, (C3-C7)cycloalkyl, hydroxy, (CrC6)alkyloxy, mercapto, aryl, heterocycle, halogen, trifluoromethyl, pentafluoroethyl, cyano, cyanomethyl, nitro, amino, amido, amidinyl, carboxyl, carboxamide, (CrC^alkyloxycarbonyl, carbamate, sulfonamide, ester and sulfonyl. As an example, an optionally substituted (Q-C^alkyl radical, such as a methyl group, by a (C3-C7)cycloalkyl, such as cyclopropyl, refers to a cyclopropylmethyl radical. In some examples, pyridyl radicals substituted in the 3- position carboxyl, cyano and carboxamide group may be called nicotinic, nicotinotrile and nicotinamide radicals.
In this specification, unless stated otherwise, the term "solvate" refers to a complex of variable stoichiometry formed by a solute (e.g. a compound of Formula (I)) and a solvent. The solvent is a pharmaceutically acceptable solvent as preferably water; such solvent may not interfere with the biological activity of the solute.
In this specification, unless stated otherwise, the term "antagonists of A3 refers also to a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV~oxide form thereof.
PHARMACEUTICAL COMPOSITIONS
Antagonists of A3 described herein, and the pharmaceutically acceptable salts, solvates and hydrates thereof can be used in pharmaceutical preparations in combination with a pharmaceutically acceptable carrier or diluent. Suitable pharmaceutically acceptable
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
carriers include inert solid fillers or diluents and sterile aqueous or organic solutions. The antagonists of A3 will be present in such pharmaceutical compositions in amounts sufficient to provide the desired dosage amount in the range described herein. Techniques for formulation and administration of the compounds of the instant invention can be found in Remington: the Science and Practice of Pharmacy, 19th edition, Mack Publishing Co., Easton, PA (1995).
The amount of antagonists of A3 receptor, administered to the subject will depend on the type and severity of the disease or condition and on the characteristics of the subject, such as general health, age, sex, body weight and tolerance to drugs. The skilled artisan will be able to determine appropriate dosages depending on these and other factors. Effective dosages for commonly used CNS drugs are well known to the skilled person. The total daily dose usually ranges from about 0.05 - 2000 mg.
The present invention relates to pharmaceutical compositions which provide from about 0.01 to 1000 mg of the active ingredient per unit dose. The compositions may be administered by any suitable route. For example orally in the form of capsules, etc., parenterally in the form of solutions for injection, topically in the form of unguents or lotions, ocularly in the form of eye-drops, rectally in the form of suppositories, intranasally or transcutaneously in the form of delivery system like patches.
For oral administration, the antagonists of A3 receptor thereof can be combined with a suitable solid or liquid carrier or diluent to form capsules, tablets, pills, powders, syrups, solutions, suspensions and the like.
The tablets, pills, capsules, and the like contain from about 0.01 to about 99 weight percent of the active ingredient and a binder such as gum tragacanth, acacias, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid, a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin. When a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
Various other materials may be present as coatings or to modify the physical form of the dosage unit. For instance, tablets may be coated with shellac, sugar or both. A syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
For parenteral administration the disclosed antagonists of A3 can be combined with sterile aqueous or organic media to form injectable solutions or suspensions. For example, solutions in sesame or peanut oil, aqueous propylene glycol and the like can be used, as well as aqueous solutions of water-soluble pharmaceutically-acceptable salts of the compounds. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
In addition, to the formulations described previously, the compounds may also be formulated as a depot preparation. Such long acting formulations may be administered by implantation, for example, subcutaneously or intramuscularly or by intramuscular injection. Thus, for example, as an emulsion in acceptable oil, or ion exchange resins, or as sparingly soluble derivatives, for example, as sparingly soluble salts.
The antagonists of A3 receptor described herein, and their pharmaceutically acceptable salts, can be incorporated into various types of ophthalmic formulations for delivery to the eye (e.g., topically, intracamerally, or via an implant). Such compounds are preferably incorporated into topical ophthalmic formulations for delivery to the eye. The compounds may be combined with ophthalmologically acceptable preservatives, surfactants, viscosity enhancers, penetration enhancers, buffers, sodium chloride, and water to form an aqueous, sterile ophthalmic suspension or solution. Ophthahnic solution formulations may be prepared by dissolving a compound in a physiologically acceptable isotonic aqueous buffer. Further, the ophthalmic solution may include an ophthalmologically acceptable surfactant to assist in dissolving the compound. In
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
addition, the ophthalmic solution may contain an agent to increase viscosity, such as, hydroxyniethylcellulose, methylcellulose, polyvinylpyrrolidone, and the like, to improve the retention of the formulation in the conjunctival sac. Gelling agents can also be used, such as xanthan gum. In order to prepare sterile ophthamic ointment formulations, the active ingredient is combined with a preservative in an appropriate vehicle, such as, mineral oil or liquid lanolin. Sterile ophthalmic gel formulations may be prepared by suspending the compound in a hydrophilic base, according to the published formulations for analogous ophthalmic preparations. Some preservatives and tonicity agents can be incorporated.
The antagonists of A3 receptor described herein can be formulated as topical ophthalmic suspensions or solutions, with a pH of about 4 to 8. Such compounds will normally be contained in these formulations in an amount 0.01% to 5% by weight. The dosage form may be a solution, suspension, or microemulsion. For topical presentation 1 to 2 drops of these formulations would be delivered to the surface of the eye 1 to 4 times per day according to the discretion of a skilled clinician.
Preferably disclosed antagonists of A3 or pharmaceutical formulations containing these compounds are in unit dosage form for administration to a mammal. The unit dosage form can be any unit dosage form known in the art including, for example, a capsule, an IV bag, a tablet, or a vial. The quantity of active ingredient in a unit dose of composition is an effective amount and may be varied according to the particular treatment involved. It may be appreciated that it may be necessary to make routine variations to the dosage depending on the age and condition of the patient. The dosage will also depend on the route of administration which may be by a variety of routes including oral, aerosol, rectal, transdermal, subcutaneous, intravenous, intramuscular, intraperitoneal, intraocular, eye drop and intranasal.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
METHODS OF SYNTHESIS
The compounds according to the invention, in particular the compounds according to the Formula (I), (II), (III), (IIIA) and (IIIB) may be prepared by methods known in the art of organic synthesis as set forth in part by the following synthesis schemes. In all of the schemes described below, it is well understood that protecting groups for sensitive or reactive groups are employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (Green T.W. and Wuts P.G.M. (1991) Protecting Groups in Organic Synthesis, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection of process as well as the reaction conditions and order of their execution shall be consistent with the preparation of compounds of Formula (I), (II), (III), (IIIA) and (IIIB). The compounds according to the invention may be represented as a mixture of enantiomers, which may be resolved into the individual pure R- or S'-enantiomers. If for instance, a particular enantiomer is required, it may be prepared by asymmetric synthesis or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers. Alternatively, where the molecule contains a basic functional group such as an amino or an acidic functional group such as carboxyl, this resolution may be conveniently performed by fractional crystallization from various solvents as the salts of an optical active acid or by other methods known in the literature (e.g. chiral column chromatography). Resolution of the final product, an intermediate or a starting material may be performed by any suitable method known in the art (Eliel E.L., Wilen S. H. and Mander L.N. (1984) Stereochemistry of Organic Compounds, Wiley-Interscience).
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
Many of the heterocyclic compounds of the invention can be prepared using synthetic routes well known in the art (Katrizky A.R. and. Rees CW. (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
The product from the reaction can be isolated and purified employing standard techniques, such as extraction, chromatography, crystallization and distillation.
The compounds of the invention may be prepared by general route of synthesis as disclosed in the following methods or according to any method known from the man skilled in the art.
In one embodiment of the present invention compounds of Formula (III) and (HIB) may be prepared according to the synthetic sequences illustrated in Scheme 1. Pyrazole gl can be substituted using Mitsunobu conditions. Then amidine can be synthesized either from ester treated with aluminium chloride in the presence of ammonium chloride or from nitrile by synthesis of amidoxime g3 followed by hydrogenation, in the presence of Pd/C and anhydride acetic. Finally, the cyclization between the amidine g4 and the isothiocyanate g5 may be promoted by di-tert-butylazodicarboxylate and a base such as DBU.

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
In another embodiment of the present invention, the compounds of Formula (III) and (IIIA) may be prepared according to the synthetic sequences illustrated in Scheme 2. Compound g7 may be hydrolyzed by standard procedures followed by reaction with oxalyl chloride to yield compound g9. Subsequently, the acid chloride can be transformed in halo-ketone glO (sometime present as a mixture of chloro (X=Cl) and bromo (X=Br) derivative) via the formation of diazoketone. Finally the cyclization reaction may be performed between the halo-ketone glO and the thiourea gll to yield the aminothiazole gl2. Thioureas gll can be prepared form the corresponding isothiocyanates g5 by reaction with methanolic ammonia. When isothiocyanate g5 was not commercially available or known in the literature, it was prepared from the corresponding amine by treatment either with TDCI (M.P.Gauthier at al (2006) Biorg. Med. Chem. 14: H918-H927) or with thiophosgene (R. D.Haugwitz et al (1985) J. Med. Chem. 9: H1234-H1241) as described in literature.

Scheme 2
In one embodiment of the present invention, the compounds of Formula (II) may be prepared according to the synthetic sequences illustrated in Scheme 3. Aminothiadiazole gl3 can be alkylated into gl4 in the presence of a base such as NaH and a solvent such as THF.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008

Scheme 3
In another embodiment of the present invention, the compounds of Formula (III) and (IIIB) may be prepared according to the synthetic sequences illustrated in Scheme 4. Pyrazole gl5 can be alkylated using the proper alkyl halide, such as bromide and iodide, or triflate in presence of an inorganic base, such as potassium carbonate, in a suitable organic solvent. Alternativerly pyrazole gl5 can be arylated using the proper aryl halide in the presence of an inorganic base, such as potassium carbonate, a copper catalyst, such as copper iodide, a proper ligand, such as 1,10-phenanthroline, in a suitable organic solvent, such as dioxane (Y-M. Zhu at al (2007) Tetrahedron Lett. 48: H6262-H6266).Compound gl6 can be alternatively obtained by esterification of the corresponding commercially available acids gl7 using standard procedures. Then amidine gl8 is synthesized from the corresponding ester by reaction with aluminium chloride in the presence of ammonium chloride in a suitable solvent, such as toluene. The cyclization between the amidine gl8 and the isothiocyanate gl9 is promoted by di- tert-butylazodicarboxylate and a base such as DBU, to provide compound g6.
When in Ml is present an additional functional group, such as an ester, it could be converted into a different related residue such as acide or amide by standard procedures known by those skilled in the art, to provide g6\
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

Scheme 4
In another embodiment of the present invention, the compounds of Formula (III) and (IIIB) may be prepared according to the synthetic sequences illustrated in Scheme 5. Compound gl8, prepared as described in Scheme 4, can be converted into compound g20 by cyclization using trichloromethanesulfenyl chloride.in presence of an inorganic base, such as sodium hydroxide, in water. Compound g20 can be converted to compound g6 by substitution with the desired amine in presence of a suitable base, such as potassium fer/-butoxide, in a suitable solvent (dioxane) under reflux.

EXPERIMENTAL
Unless otherwise noted, all starting materials were obtained from commercial suppliers and used without further purification.
Specifically, the following abbreviations may be used in the examples and throughout the specification.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

All references to brine refer to a saturated aqueous solution of NaCl. Unless otherwise indicated, all temperatures are expressed in °C (degrees Centigrade). All reactions are conducted not under an inert atmosphere at room temperature unless otherwise noted. Most of the reaction were monitored by thin-layer chromatography on 0.25mm Merck silica gel plates (60F-254), visualized with UV light. Flash column chromatography was performed on silica gel ( 40-63 μM, Merck) or on prepacked silica gel cartridges (15-40 μM, Merck or 40-63 μM, Biotage).
Melting point determination was performed on a Buchi B-540 apparatus. 1H NMR spectra were recorded on a Brucker 300MHz (see Table 4). Chemical shifts are expressed in parts of million (ppm, δ units). Coupling constants are in units of herts
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
(Hz) Splitting patterns describe apparent multiplicities and are designated as s (singlet), d (doublet), t (triplet), q (quadruplet), quint (quintuplet), m (multiplet), br (broad). Mobile protons are omitted for some products.
Physico-Chemical Data
LCMS chromatograms were recorded under the following conditions: Method LC-A:
Waters Micromass ZQ 2996 system Reversed phase HPLC was carried out on an Zorbax SB-C18 cartridge (1.8 μm, 4.6 x 30 mm) from Agilent, with a flow rate of 1.5 ml/min. The gradient conditions used are: 90 % A (water + 0.05 % of formic acid), 10% B (acetonitrile + 0.05 % of formic acid) to 100 % B at 3.5 minutes, kept till 3.7 minutes and equilibrated to initial conditions at 3.8 minutes until 4.5 minutes. Injection volume 5-20 μL.
Method LC-B:
Waters Acquity UPLC Micromass ZQ 2000 Single quadrupole
Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7μm, 50x2. lmm) from Waters, with a flow rate of 0.5 ml/min. The gradient conditions used are: Mobile phase: A phase= water/CH3CN 95/5 + 0.1% TFA; B phase= water/CH3CN 5/95 + 0.1% TFA. 0-0.30min (A: 92%, B: 8%), 0.30-1.50 min (A: 0%, B: 100%), 1.50- 2.00 min (A: 0%, B: 100%), 2.00-2.40 min (A: 95%, B: 5%). Injection volume 2μl
Method LC-C Waters Acquity UPLC Micromass ZQ 2000 Single quadrupole
Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7μm, 50x2. lmm) from Waters, with a flow rate of 0.5 ml/min. The gradient conditions used
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
are: Mobile phase: A phase= water/CH3CN 95/5 + 0.1% TFA; B ρhase= water/CH3CN 5/95 + 0.1% TFA. 0-0.30min (A: 95%, B: 5%), 0.30-3.30 min (A: 0%, B: 100%), 3.30- 3.90 min (A: 0%, B: 100%), 3.90-4.40 min (A: 95%, B: 5%). Injection volume 2μl
Method LC-D
LCMS were recorded on a Waters Acquity UPLC Micromass ZQ 2000 Single quadrupole system by the following conditions:
Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7μm, 50x2. lmm) from Waters, with a flow rate of 0.5 ml/min. The gradient conditions used are: Mobile phase: A phase= water/CH3CN 95/5 + 0.1% TFA; B phase= water/CH3CN 5/95 + 0.1% TFA. 0-O.lOmin (A: 95%, B: 5%), 0.10-1.40 min (A: 0%, B: 100%), 1.40- 1.90 min (A: 0%, B: 100%), 1.90-2.40 min (A: 95%, B: 5%). Injection volume 2μl
Method LC-E Waters Acquity UPLC Micromass ZQ 2000 Single quadrupole
Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7μm, 50x2. lmm) from Waters, with a flow rate of 0.6 ml/min. The gradient conditions used are: Mobile phase: A ρhase= water/CH3CN 95/5 + 0.1% TFA; B phase= water/CH3CN 5/95 + 0.1% TFA. 0-0.25min (A: 95%, B: 5%), 0.25-3.30 min (A: 0%, B: 100%), 3.30- 4.00 min (A: 0%, B: 100%), 4.00-4.10 min (A: 95%, B: 5%), 4.10-5.00 min (A: 95%, B: 5%). Injection volume 2μl
Method LC-F
Waters Acquity UPLC Micromass ZQ 2000 Single quadrupole Reversed phase HPLC was carried out on a Acquity UPLC-BEH Cl 8 cartridge (1.7μm, 50x2. lmm) from Waters, with a flow rate of 0.6 ml/min. The gradient conditions used are: Mobile phase: A phase= water/CH3CN 95/5 + 0.1% TFA; B phase= water/CH3CN 5/95 + 0.1% TFA. 0-0.50min (A: 95%, B: 5%), 0.50-6.00 min (A: 0%, B: 100%), 6.00-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
7.00 min (A: 0%, B: 100%), 7.00-7.10 min (A: 95%, B: 5%); 7.10-8.50 min (A: 95%, B: 5%); Injection volume: 2μl
ES MS detector was used, acquiring both in positive and negative ionization modes. Cone voltages were 30 V (Method LC-A), 26V (Methods LC-B, LC-C and LC-D) and 25V (Methods LC-E and LC-F) for both positive and negative ionization modes.All mass spectra were taken under electrospray ionisation (ESI) methods (see Table 3).
The compounds (Cpd) in the following Tables have been synthezised according to the same methods as previous examples 1 to 11, as denoted in the column denoted as "Exp. nr". The compounds denoted with the asterisk have been exemplified in the Examples.
Table 1: Compounds prepared according to the Examples.


KAS/ClientDocs/Addex/SS^S.WOOl.FinalSpec.lO.OT ZOOδ

KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008


KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008


Table 2: Compounds prepared according to the Examples.


EXAMPLES
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008
EXAMPLE 1 : iV-(2-Fluorophenyl)-3-(l-(4-methoxybenzyl)-3-methyl-lJH-pyrazol- 4-yl)-l,2,4-thiadiazol-5-amine and iV-(2-fluorophenyl)-3-(l-(4-methoxybenzyl)-5- methyl-lif-pyrazol-4-yl)-l,2,4-thiadiazoI-5-amine (Final Compounds 1-1 and 1-2)
1.1: l-(4-Methoxybenzyl)-3 -methyl- lH-pyrazole-4-carbonitrile and l-(4- methoxybenzyl)-5-methyl-lH-pyrazole-4-carbonitrile
According to Scheme 1 Step 1 of the general methodology: Triphenylphosphine (11 mmol, 2.9 g), (4-methoxyphenyl)methanol (10 mmol, 1.4 g) and di-tert- butylazodicarboxylate (11 mmol, 2.6 g) were added to a solution of 3 -methyl- IH- pyrazole-4-carbonitrile (9.3 mmol, 1.0 g), in DCM (40 mL) at O0C. The reaction mixture was stirred at room temperature overnight. The organic phase was washed with a saturated solution of NH4OH and brine. Then the organic phase was dried over MgSO4, was filtered and was concentrated under reduced pressure. The resulting crude product was purified by flash chromatography over silica gel using cyclohexane/ AcOEt (90:10) as eluent to yield l-(4-methoxybenzyl)-3-methyl-lH-pyrazole-4-carbonitrile and l-(4-methoxybenzyl)-5-methyl-lH-pyrazole-4-carbonitrile (9.3 mmol, 2.1g, 100%).
1.2: l-(4-Methoxybenzyl)-3 -methyl- lΗ-pyrazole-4-carboximidamide and l-(4- methoxybenzyl)-5-methyl-lH-pyrazole-4-carboximidamide
According to Scheme 1 Step 2 of the general methodology, Method A: A mixture of 1- (4-methoxybenzyl)-3 -methyl- 1 H-pyrazole-4-carbonitrile and 1 -(4-methoxybenzyl)-5 - methyl-lH-pyrazole-4-carbonitrile (9.90 mmol, 2.25 g), hydroxylamine 50% in water (19.8 mmol, 1.21 mL) and EtOH (10 mL) was heated at 800C for 12 hours. After evaporation of the solvent, 2.42 g (9.30 mmol, 94%) of iV-hydroxy-l-(4- methoxybenzyl)-3 -methyl- lH-ρyrazole-4-carboximidamide and JV-hydroxy- 1 -(4- methoxybenzyl)-5 -methyl- lH-pyrazole-4-carboximidamide were obtained. The crude product was used in the next step without purification.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
LC (Method LC-A): RT = 0.86 min; MS m/z ES+= 261.
A mixture of iV'-hydroxy-l-(4-methoxybenzyl)-3-methyl-lH-pyrazole-4- carboximidamide and iV-hydroxy-l-(4-methoxybenzyl)-5-methyl-lH-pyrazole-4- carboximidarnide (9.30 mmol, 2.42 g), Pd/C (200 mg) and anhydride acetic (9.30 nimol, 0.88 mL) in MeOH (30 mL) was stirred at room temperature for 13 hours under hydrogen atmosphere. After filtration and evaporation of the solvent, the crude product was triturated with Et2O and dried to yield l-(4-methoxybenzyl)-3 -methyl- IH- pyrazole-4-carboximidamide and l-(4-methoxybenzyl)-5-methyl-lH-pyrazole-4- carboximidamide (1.55 g, 5.11 mmol, 55%) as a white solid. LC (Method LC-A): RT = 0.91 min; MS m/z ES+= 245.
According to Scheme 1 Step 3 of the general methodology, DBU (0.39 mmol, 60.0 mg) was added to a solution of l-fluoro-2-isothiocyanatobenzene (0.39 mmol, 48 μl) and 1- (4-methoxybenzyl)-3 -methyl- lH-pyrazole-4-carboximidamide and l-(4- methoxybenzyl)-5 -methyl- lH-pyrazole-4-carboximidamide (0.39 mmol, 200 mg) in DMF (7 mL) under nitrogen. The reaction mixture was stirred at room temperature until total consumption of the amidine. Then, di-fert-butylazodicarboxylate (0.43 mmol, 100 mg) was added dropwise and the reaction mixture was stirred for 5 minutes. After evaporation of the EtOH, water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with a solution of HCl 1 M, water and brine, was dried over Na2SO4, was filtered and was concentrated under reduced pressure. The crude product was purified by flash chromatography over silica gel using cyclohexane/AcOEt (80:20) as eluent to afford iV-(2-fluorophenyl)-3-(l-(4- methoxybenzyl)-3-methyl-lH-pyrazol-4-yl)-l,2,4-thiadiazol-5-amine and N-Q.- fluorophenyl)-3-(l-(4-methoxybenzyl)-5-methyl-lH-pyrazol-4-yl)-l,2,4-thiadiazol-5- amine (0.13 mmol, 50 mg, 32%) as a white solid.
LC (Method LC-A): RT = 2.80 min; MS m/z ES+= 396.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
EXAMPLE 2: S-CS-Methyl-l-phenyl-liϊ-pyrazol^-y^-iV-phenyl-l^^-thiadiazol- 5-amine (Final Compound 1-8)
2.1: 5 -Methyl- 1 -phenyl- 1 H-pyrazole-4-carboximidamide According to Scheme 1 Step 2 of the general methodology, Method B: Trimethylaluminium, 2M solution in heptane (21.7 mmol, 10.9 mL), was added dropwise to a suspension of ammonium chloride (21.7 mmol, 1.16 g) in dry toluene (20 mL), under an argon atmosphere, at 0°C. The reaction mixture was stirred at room temperature until no more evolution of gas was observed. After addition of 5 -methyl- 1- phenyl- li7-pyrazole-4-carboxylic acid ethyl ester (2.17 mmol, 500 mg), the reaction mixture was stirred at 80°C overnight. The reaction mixture was then cooled down to 0°C and MeOH was added with consequent stirring for 1 hour at room temperature. The reaction mixture was added to a mixture of DCM and silica, then filtered on an empty cartridge. The cartridge was first eluted with DCM and then with DCM/MeOH (80:20) as eluent. After filtration and evaporation, 5-methyl-l-phenyl-lH-pyrazole-4- carboximidamide (1.55 mmol, 310 mg, 71%) was obtained as a white solid.
LC (Method LC-A): RT = 0.91 min; MS m/z ES+= 201.
2.2: 3-(5-Methyl-l-phenyl-lH-pyrazol-4-yl)-N-phenyl-l,2,4-thiadiazol-5-amine According to Scheme 1 Step 3 of the general methodology: DBU (0.42 mmol, 64 mg) was added to a solution of 5-methyl-l-phenyl-lH-pyrazole-4-carboximidamide (0.42 mmol, 100 mg) and isothiocyanatobenzene (0.42 mmol, 57 mg) in dry DMF (5 mL), under argon. The reaction mixture was stirred at room temperature until total consumption of the amidine. Then, di-/ert-butylazodicarboxylate (0.42 mmol, 97 mg) was added dropwise and was stirred for 5 minutes. The reaction mixture was quenched with water and was extracted with AcOEt. The organic phase was washed with brine, was dried over Na2SO4, was filtered and was concentrated under reduced pressure. The crude product was purified by flash chromatography over silica gel using
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
cyclohexane/ AcOEt (80:20) as eluent to afford 3-(5-methyl-l-phenyl-lH-pyrazol-4-yl)- JV-phenyl-l,2,4-thiadiazol-5 -amine (0.22 mmol, 75 mg, 53%) as a white powder.
LC (Method LC-A): RT = 2.85 min; MS m/z ES"= 332.
EXAMPLE 3: 4-(l-Isopropyl-ljH-pyrazol-4-yl)-iV-(pyridiii-2-yl)thiazol-2-amine (Final Compound 1-11)
3.1: l-Isopropyl-lH-pyrazole-4-carboxylic acid
According to Scheme 2 Step 1 of the general methodology: A solution of ethyl 1- isopropyl-li7-pyrazole-4-carboxylate (2.28 mmol, 417 mg) and LiOH (23.4 mmol, 1.00 g) in water/MeOH (1:1, 10 mL) was heated at 90°C overnight. After evaporation of the solvent, the aqueous phase was extracted with DCM then acidified with a solution of HCl 1 M until pH = 1-2 and extracted with DCM. The organic phase was washed with brine, was dried over Na2SO4, was filtered and was concentrated under reduced pressure to yield l-isopropyl-lH-pyrazole-4-carboxylic acid (1.95 mmol, 300 mg, 85%) as a white solid. The crude product was used without purification.
LC (Method LC-A): RT = 1.07 min; MS m/z ES"= 155.
3.2: l-Isopropyl-lΗ-pyrazole-4-carbonyl chloride
According to Scheme 2 Step 2: of the general methodology A solution of 1-isoρropyl- lH-pyrazole-4-carboxylic acid (1.95 mmol, 300 mg), oxalyl chloride (3.42 mmol, 0.30 mL) and three drops of DMF in DCM (5 mL) was stirred for 2 hours at room temperature. After evaporation of the solvent, the crude residue was treated with toluene and was coevaporated to dryness to yield l-isopropyl-lif-pyrazole-4-carbonyl chloride (1.95 mmol, 0.33 g). The crude product was used without purification. LC (Method LC-A): RT = 1.58 min.
3.3 : 2-Bromo- 1 -(I -isopropyl- 1 H-pyrazol-4-yl)ethanone
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
According to Scheme 2 Step 3 of the general methodology: A solution of TMSdiazomethane (6.0 mmol, 3.0 mL) was added to a solution of 1-isopropyl-lH- pyrazole-4-carbonyl chloride (1.95 mmol, 0.33 g) in acetonitrile (5 mL) at 0°C. The reaction mixture was stirred at room temperature overnight. HBr (8.0 mmol, 0.9 mL, 48%) was added at 00C to the reaction mixture. The reaction mixture was stirred at room temperature for one hour. After evaporation of the solvent, AcOEt was added and the aqueous phase was neutralized with a solution of NaOH 1 M. The aqueous phase was extracted with AcOEt. The organic phase was washed with brine, was dried over Na2SO4, was filtered and was concentrated under reduced pressure to yield 2-bromo-l- (l-isopropyl-lH"-pyrazol-4-yl)ethanone (1.95 mmol, 0.45 g, 100%) as an orange oil. The crude product was used without purification.
3.4: 4-(l -Isopropyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine
According to Scheme 2 Step 4 of the general methodology: A solution of 2-bromo-l- (l-isopropyl-lH-pyrazol-4-yl)ethanone (0.43 mmol, 0.10 g) and of l-(pyridin-2- yl)thiourea (0.35 mmol, 53 mg) in acetone (5 mL) was stirred under reflux for one hour. After evaporation of the solvent, DCM was added and the organic phase was washed with a saturated solution OfNaHCO3, water and brine. The organic phase was dried over Na2SO4, was filtered and was concentrated under reduced pressure. The crude product was purified by flash chromatography over silica gel using DCM/ AcOEt (90:10) as eluent to yield 4-(l-isopropyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2- amine (0.32 mmol, 92 mg) as a white solid.
LC (Method LC-A): RT - 1.91 min; MS m/z ES ; + = 286.
EXAMPLE 4: 3-(l,3-Dimethyl-lJΪ-pyrazol-4-yl)-iV-methyl-iV-phenyl-l,2,4- thiadiazol-5-amine and 3-(l,5-dimethyl-lH-pyrazol-4-yl)-JV-methyl-iV-phenyl- l,2,4-thiadiazol-5-amine (Final Compounds 2-1 and 2-2)
According to Scheme 3 of the general methodology: NaH (0.97 mmol, 42 mg) was added to a solution of 3-(3-methyl-lH"-pyrazol-4-yl)-iV-phenyl-l,2,4-thiadiazol-5-amine
KAS/ClientDocs/Addex/SSlPS.WOOLFinalSpec.lO.OV lOOδ
(0.39 mmol, 0.10 mg) in THF (10 mL). After 30 minutes, methyl iodide (1.2 mmol, 73 μL) was added and the reaction mixture was stirred at room temperature for one hour. The reaction mixture was quenched with water at 0°C. The aqueous phase was extracted twice with DCM. The organic phase was washed with a sodium bisulfite solution, water and brine. Then the organic phase was dried over Na2SO4, was filtered and was concentrated under reduced pressure. The crude product was purified by flash chromatography over silica gel using cyclohexane/AcOEt (85:15) as eluent to yield 3- (153-dimethyl-li7-pyrazol-4-yl)-Λr-methyl-N-phenyl-l ,2,4-thiadiazol-5-amine and 3- (l,5-dimethyl-lH-pyrazol-4-yl)-iV-methyl-iV-phenyl-l,2,4-thiadiazol-5-amme (0.32 mmol, 90 mg, 81%) as a colorless oil.
LC (Method LC-A): RT = 4.21 and 4.31min; MS m/z ES+= 286.
EXAMPLE 5: SKl-CCyclopropylmethy^-lH-pyrazol^-y^-N^pyridin-l-yl)-!^^- thiadiazol-5-amine 1 (Final compound 1-22) and 3~(l-(Cyclopropylmethyl)-lH- pyrazol-4-yl)-N-(pyridinτ2-yl)-l,2,4-thiadiazol-5~amine monochlohydrate salt (Final compound 1.22a)
5.1: Ethyl l-(cyclopropylmethyl)-lH-ρyrazole-4-carboxylate (al)
According to Scheme 4, Step Ia of the general methodology, K2CO3 (5.35 mmol, 740 mg) and cyclopropylmethylbromide (7.12 mmol, 962 mg) were added to a solution of ethyl l-H-pyrazole-4-carboxylate (3.57 mmol, 500 mg) in acetone (5 mL). The reaction mixture was stirred at reflux for 8 hours. After cooling to room temperature, inorganics were filtered off and the filtrated was concentrated to dryness affording ethyl 1- (cyclopropylmethyl)-lH-pyrazole-4-carboxylate (3.40 mmol, 660 mg, 95%) as colorless oil.
LC (Method LC-B): RT = 1.26 min; MS m/z ES+= 195
1H NMR (300 MHz, CDCl3) δ ppm: 8.01 (s, 1 H) 7.91 (s, 1 H) 4.31 (q, 2 H) 4.00 (d, 2
H) 1.36 (t, 3 H) 1.19 - 1.32 (m, 1 H) 0.63 - 0.76 (m, 2 H) 0.32 - 0.45 (m, 2 H).
KAS/Clienωocs/Addex/SSlθS.WOOl.FinalSpec.lO.O? 2008
The following compounds are synthetized according to the same method:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

5.2 l-(Cyclopropylmethyl)-lH-pyrazole-4-carboximidamide hydrochloride (bl) According to Scheme 4 Step 2 of the general methodology, trimethylaluminium, 2M solution in hexane (32.99 mmol, 16.5 mL), was added dropwise to a suspension of ammonium chloride (32.99 mmol, 1.75 g) in dry toluene (17 mL), under a nitrogen atmosphere, at O0C. The reaction mixture was stirred at room temperature until no more evolution of gas was observed. After addition of ethyl l-(cyclopropylmethyl)-lH- pyrazole-4-carboxylate (3.29 mmol, 640 mg), the reaction mixture was stirred at 95°C overnight. It was then cooled down to 00C and MeOH was added under stirring. The solvent was removed and the residue was triturated with MeOH (100 mL) and filtered. The filtrate was concentrated and the crude was purified by silica gel flash chromatography (eluent: DCM/MeOH = 80:20) to yield l-(cyclopropylmethyl)-lH- pyrazole-4-carboximidamide hydrochloride (3.26 mmol, 652 mg, 98%) as a white solid.
LC (Method LC-B): RT = 0.64 min; MS m/z ES+= 165.
1H NMR (300 MHz, DMSO-d6) δ ppm: 8.92 (br. s., 3 H) 8.72 (s, 1 H) 8.24 (d, 1 H) 4.06 (d, 2 H) 1.14 - 1.34 (m, 1 H) 0.53 - 0.65 (m, 2 H) 0.34 - 0.44 (m, 2 H).
The following compounds are synthetized according to the same method:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

5.3 Synthesis of isothiocyanato derivatives
5.3.1 6-Morpholinopyridin-2 -amine (cl)
A mixture of 6-bromopyridine-2-amine (5.20 mmol, 0.90 g) and morpholine (57.39 irunol, 5 mL) was heated at 150°C for 4 hours under microwave irradiation. AcOEt was added and the organic phase was washed with NaHCO3, water and brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure to yield 6-morpholinopyridin-2-amine (5.20 mmol, 1.06 g, 100%) as a light brown solid. The crude product was used in the next step without further purification. LC (Method LC-B): RT = 0.29 min; MS m/z ES+= 180.
5.3.2 5-Morpholinopyridin-2-amine (c2)
A mixture of 5-bromo-2-nitropyridine (0.49 mmol, 100 mg) and morpholine (11.48 mmol, 1 mL) was heated at 100°C for 30 minutes under microwave irradiation. After evaporation of the solvent, the crude mixture was purified on a silica gel cartridge (2g) eluted a first time with petroleum ether/AcOEt (90:10), then with DCM to afford 4-(6- nitropyridin-3-yl)morpholine (0.43 mmol, 89 mg, 88%) as a yellow solid. LC (Method LC-C): RT = 1.25 min; MS m/z ES+= 210. 4-(6-Nitropyridin-3-yl)morpholine (0.40 mmol, 83 mg) was dissolved in AcOEt (30 mL) and MeOH (2 mL). Palladium on activated charcoal 10% (20 mg) was added and the reaction mixture was hydrogenated on a Parr apparatus (15 psi) for 1.5 hours at
KAS/ClientDocs/Addex/SSlθS.WOOl.FinalSpec.lO.OV lOOS
room temperature. The reaction mixture was filtered to remove the catalyst and after evaporation of the solvent 5-morpholinopyridin-2-amine (0.40 mmol, 72 mg, 100%) was recovered as a pale brown solid. The crude product was used in the next step without purification. LC (Method LC-D): RT = 0.43 min; MS m/z ES+= 180.
1H NMR (300 MHz, DMSO-d6) δ ppm: 7.60 (d, 1 H) 7.16 (dd, 1 H) 6.41 (d, 1 H) 5.37 (S5 2 H) 3.64 - 3.77 (m, 4 H) 2.84 - 2.95 (m, 4 H).
5.3.3 6-Cyclobutoxypyridin-2-amine (c3) Sodium hydride (3.80 mmol, 150 mg) was added portionwise to a solution of cyclobutanol (3.46mmmol, 0.27 mL) in acetonitrile and the reaction mixture was stirred 1 hour at room temperature. 6-Bromopyridine-2-amine (1.73 mmol, 0.30 g) was added and the reaction mixture was heated at 130°C for 3 hours under microwave irradiation. After evaporation of the solvent, water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with water, brine and dried over Na2SO4. The organic solvent was filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography over silica gel using petroleum ether/AcOEt (80:20) as eluent affording 6-cyclobutoxypyridm-2-amine (0.85 mmol, 0.14 g, 49%) as a yellow oil. LC (Method LC-B): RT = 0.83 min; MS m/z ES+= 165.
5.3.4 2-Isothiocyanatopyridine (or its dimer 2-(pyridin-2-yl)-4-(pyridin-2-ylimino)-l,2- thiazetidine-3-thione) dl (Method A) l,r-Thiocarbonyldimidazole solution (5.84 mmol, 1.04 g) in dichloromethane (10 mL) was added dropwise to 2-amino-pyridine (5.31 mmol, 0.5Og) in dichloromethane (10 mL) at 0°C under nitrogen. The reaction mixture was warmed to room temperature and stirred for 5 hours. The reaction mixture was concentrated and purified on a silica gel cartridge (Isolute SPE column 1OG 25mL; eluent: petroleum ether/ AcOEt = 100:0 to 90:10) affording 2-isothiocyanatopyridine (1.76 mmol, 0.24 g, 33 %) as yellow oil; LC (Method LC-D): RT = 1.17 min; MS m/z ES+= 137.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
Upon standing in vacuo 48 hours the product converted quantitatively into 2-(pyridin-
2-yl)-4-(pyridin-2-ylimino)-l,2-thiazetidine-3-thione appearing as an orange solid.
LC (Method LC-D): RT = 0.92 min; MS m/z ES+= 273.
1H NMR (300 MHz5 DMSOd6) δ ppm: 9.16 - 9.32 (m, 1 H) 8.51 - 8.63 (m, 1 H) 8.03 -
8.12 (m, 1 H) 7.95 - 8.03 (m, 1 H) 7.41 - 7.49 (m, 2 H) 7.33 - 7.40 (m, 1 H) 7.25 (td, 1
H)
5.3.5 l-Isothiocyanato-β-methylpyridine d2 (Method A) l,r-Thiocarbonyldimidazole solution (3.70 mmol, 0.66 g) in DCM (5 niL) was added dropwise to 2-amino-6-methylpyridine (3.70 mmol, 0.40 g) in DCM (8 mL) at 00C under a nitrogen atmosphere. The reaction mixture was warmed to room temperature and stirred for 1 hour. The reaction mixture was concentrated and purified by flash chromatography over silica gel using petroleum ether/ AcOEt (90:10 to 80:20) as eluent affording 2-isothiocyanato-6-methylpyridine (1.21 mmol, 0.18 mg, 33 %) as pale yellow oil;
LC (Method LC-B): RT =1.40 min; MS m/z ES+= 151.
1H NMR (300 MHz, CDCl3) δ ppm: 7.60 (dd, 1 H) 7.07 (d, 1 H) 6.96 (d, 1 H) 2.53 (s, 3
H).
The following compounds are synthetized according to the same method:

KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.07 2008
5.3.6 2-Isothiocyanato-6-ethylpyridine d6 (Method B) TEA (1.50 mmol, 0.21 ml) and thiophosgene (0.52 mmol, 0.04 mL) were added to a solution of 2-amino-6- ethylpyridine (0.50 mmol, 60 mg) in THF (5 mL). The reaction mixture was stirred 1 hour at room temperature under a nitrogen atmosphere. After evaporation of the solvent 2-isothiocyanato-6-ethylpyridine was recovered as a brown solid and used in the next step without further purification. LC (Method LC-B): RT =1.62 min; MS m/z ES+= 165.
The following compounds are synthetized according to the same method:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

5.4 3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine 1 (Final compound 1-22) and 3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N- (pyridin-2-yl)-l,2,4-thiadiazol-5 -amine monochlohydrate salt (Final compound 1.22a)
According to Scheme 4 Step 3 of the general methodology, 2-(pyridin-2-yi)-4-(pyridin- 2-ylimino)-l,2-thiazetidine-3-thione (1.74 mmol, 475 mg) and DBU (5.237 mmol, 797 mg) were added to a solution of l-(cyclopropylmethyl)-lH-pyrazole-4- carboximidamide hydrochloride (1.74 mmol, 350 mg) in dry DMF (2.5 mL). The reaction mixture was stirred at room temperature for 15 minutes. Oi-tert- butylazodicarboxylate (1.92 mmol, 442 mg) was added and the reaction mixture was stirred at room temperature for 15 minutes. Water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with water and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was triturated twice in acetonitrile affording 3-(l-(cyclopropylmethyl)-lH-pyrazol-4-yl)-N- (pyridin-2-yl)-l,2,4-thiadiazol-5-amine (0.705 mmol, 210 mg, 40%) as a light brown powder.
LC (Method LC-E): RT = 1.89 min; MS m/z ES+= 299. 1HNMR (300 MHz, DMSO-d6) δ ppm: 12.05 (br. s., 1 H) 8.42 (ddd, 1 H) 8.25 (d, 1 H) 7.89 (d, 1 H) 7.77 - 7.86 (m, 1 H) 7.12 - 7.19 (m, 1 H) 7.02 - 7.09 (m, 1 H) 4.04 (d, 2 H) 1.20 - 1.38 (m, 1 H) 0.51 - 0.61 (m, 2 H) 0.34 - 0.45 (m, 2 H).
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
Melting point: 237-238 0C.
Preparation of 5.4 3-(l-(Cydopropylmethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4- thiadiaκol-5-amine 1 (Final compound l-22a) To a solution of 3-(l-(cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4- thiadiazol-5 -amine (0.134 mmol, 40 mg) in dioxane (8 mL), a solution of HCl in dioxane (4N, 0.4 mmol, 0.1 mL) was added and the reaction mixture was stirred 30 minutes at room temperature. After evaporation of the solvent, the residue was triturated in AcOEt affording 3-(l-(cyclopropylmemyl)-lH-pyrazol-4-yl)-N-(pyridin-2- yl)-l,2,4-thiadiazol-5-amine hydrochloride (0.107 mmol, 36 mg, 80%) as a pale yellow powder.
LC (Method LC-E): RT = 1.80 min; MS m/z ES+= 299.
1H NMR (300 MHz, DMSOd6) δ ppm 12.22 (s, 1 H)5 8.39 - 8.45 (m, 1 H), 8.25 (d, 1 H), 7.89 (d, 1 H), 7.77 - 7.86 (m, 1 H), 7.12 - 7.19 (m, 1 H), 7.02 - 7.10 (m, 1 H), 4.04 (d, 2 H), 1.19 - 1.39 (m, 1 H)3 0.51 - 0.60 (m, 2 H), 0.35 - 0.44 (m, 2 H). Melting point: 222-224 °C. Elemental analysis confirm formation of monochlorhydrate salt.
The following compounds are synthetized according to the same method used for compound 1.22:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/Clien1Docs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

EXAMPLE 6: 3-(l-Ethyl-3-methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4- thiadiazol-5-amine (Final compound 1-17)
6.1: Methyl 1 -ethyl-3 -methyl- 1 H-pyrazole-4-carboxylate al 7
According to Scheme 4 Step Ib of the general methodology, a solution of l-ethyl-3- methyl-lH-pyrazole-4-carboxylic acid (3.24 mmol, 0.50 g) and sulphuric acid 96% (0.5 mL) in methanol (5 mL) was heated at 70°C for 48 hours. After evaporation of the solvent, AcOEt was added and the organic phase was washed with a saturated solution of NaHCO35 water and brine, dried over Na2SO4, filtered and concentrated under
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
reduced pressure affording methyl l-ethyl-3 -methyl- lH-pyrazole-4-carboxylate (1.78 mmol, 0.30 g, 55%) as a pale yellow oil.
The crude product was used without further purification.
LC (Method LC-B): RT = 1.07 min; MS m/z ES+= 169.
The following compounds are synthesized according to the same method (for compound al9 ethanol was used instead of methanol):

6.2 l-Ethyl-3 -methyl- lH-pyrazole-4-carboximidamide hydrochloride b!7 According to Scheme 4 Step 2 of the general methodology, trimethylaluminium, 2M solution in toluene (17.83 mmol, 8.90 mL), was added dropwise to a suspension of ammonium chloride (17.83 mmol, 0.954 g) in dry toluene (15 mL), under a nitrogen atmosphere, at 0°C. The reaction mixture was stirred at room temperature until no more evolution of gas was observed. After addition of methyl l-ethyl-3 -methyl- lH-pyrazole- 4-carboxylate (1.783 mmol, 0.30 g), the reaction mixture was stirred at 90°C overnight. The reaction mixture was then cooled down to O0C and MeOH was added under stirring. The solvent was removed and the residue was tritured with MeOH (50 mL) and filtered. The filtrate was concentrated and the crude was purified by flash chromatography over silica gel, using DCM/MeOH (80:20) as eluent, to yield 1-ethyl- 3 -methyl- lH-pyrazole-4-carboximidamide hydrochloride (1.43 mmol, 270 mg, 80%) as a white solid. LC (Method LC-B): RT = 0.40 min; MS m/z ES+= 153.
The following compounds were synthesized according to the same method:
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

6.3 3 -(I -Ethyl-3 -methyl- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)- 1 ,2,4-thiadiazol-5-amine (Final compound 1-17)
According to Scheme 4 Step 3 of the general methodology, 2-(pyridin-2-yl)-4-(pyridin- 2-ylimino)-l,2-thiazetidine-3-thione (0.53 mmol, 145 mg) and DBU (1.59 mmol, 242 mg) were added to a solution of 1 -ethyl-3 -methyl- lH-pyrazole-4-carboximidamide hydrochloride (0.53 mmol, 100 mg) in dry DMF (1 rnL) and the reaction mixture was stirred at room temperature for 15 minutes. Di-tert-butyl diazene-l,2-dicarboxylate (0.583 mmol, 135 mg) was added and the reaction mixture was stirred at room temperature for 30 minutes. Water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with water and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography over silica gel using DCM/MeOH/NH4OH (32%) (95:5:0.5) as eluent and then the solid was triturated in di-isopropyl ether affording 3 -(I -ethyl-3 - methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5-amine (0.157 mmol, 45 mg, 30%) as a light brown powder. LC (Method LC-E): RT = 1.70 min; MS m/z ES+= 287.
1H NMR (300 MHz, CDCl3)δ ppm: 9.93 (s, 1 H) 8.46 (ddd, 1 H) 7.92 (s, 1 H) 7.60 (ddd, 1 H) 6.93 - 7.03 (m, 1 H) 6.71 (dt, 1 H) 4.11 (q, 2 H) 2.66 (s, 3 H) 1.45 (t, 3 H). Melting point: 203-205 °C.
The following compounds are synthesized according to the same method:
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/Clienωocs/Addex/53195.WO01.FinalSpec.10.072008

EXAMPLE 7: 3-(l-(3-Fluoro-4-methoxyphenyl)-lH-pyrazol-4-yl)-N-(pyridin-2- yl)-l,2,4-thiadiazol-5-amine (Final compound 1-43)
7.1: Ethyl l-(3-fluoro-4-methoxyphenyl)-lH-pyrazole-4-carboxylate a20
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
According to Scheme 4 Step Ia of the general methodology, copper iodide (0.214 rnmol, 41 mg), 1-10-phenanthroline (0.428 mmol, 77 mg) and K2CO3 (10.71 mmol, 1.48 g) were added to a solution of ethyl lH-pyrazole-4-carboxylate (4.28 mmol, 600 mg) and 4-bromo-2-fluoro-l-methoxybenzene (6.43 mmol, 0.83 mL) in dry dioxane (9 mL). The reaction mixture was heated at 150°C for 5h under microwave irradiation. The mixture was filtered through a Celite® pad. After evaporation of the solvent the crude product was purified on a silica gel cartridge eluted with petroleum ether/ AcOEt (100:0 to 90:10) as solvent affording ethyl l-(3-fluoro-4-methoxyphenyl)-lH-pyrazole- 4-carboxylate (0.728 rnmol, 191 mg, 17%) as a white solid. LC (Method LC-C): RT = 2.31 min; MS m/z ES+= 265.
7.2: l-(3-Fluoro-4-methoxyphenyl)-lH-pyrazole-4-carboximidamidehydrochloride b20
According to Scheme 4 Step 2 of the general methodology, trimemylaluminium, 2M solution in toluene (7.23 mmol, 3.62 mL), was added dropwise to a suspension of ammonium chloride (7.23 mmol, 383 mg) in dry toluene (10 mL), under a nitrogen atmosphere, at 0°C. The reaction mixture was stirred at room temperature until no more evolution of gas was observed. After addition of ethyl l-(3-fluoro-4-methoxyphenyl)- lH-pyrazole-4-carboxylate (0.723 mmol, 191 mg), the reaction mixture was stirred at 9O0C for 5 hours. The reaction mixture was then cooled down to 0°C and MeOH was added under stirring. The solvent was removed and the residue was tritured with MeOH
(50 mL) and filtered. The filtrate was concentrated and the crude obtained was purified on a silica gel cartridge eluted with DCM/MeOH (90:10 to 80:20) affording l-(3- fluoro-4-methoxyphenyl)-lH-pyrazole-4-carboximidamide hydrochloride (0.646 mmol, 175 mg, 90%) as a white solid.
LC (Method LC-C): RT = 1.22 min; MS m/z ES+= 235
7.3: 3-(l-(3-Fluoro-4-methoxyphenyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4- thiadiazol-5-amine (Final compound 1-43)
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008
According to Scheme 4 Step 3 of the general methodology, 2-(pyridin-2-yi)-4-(pyridin- 2-ylimino)-l,2-thiazetidme-3-thione (0.648 mmol, 176 mg) and DBU (1.296 mmol, 194 μL) were added to a solution of l-(3-fmoro-4-methoxyphenyl)-lH-pyrazole-4- carboximidamide hydrochloride b20 (0.648 mmol, 175 mg) in dry DCM (10 mL) and the reaction mixture was stirred at room temperature for 15 minutes. Di-tert- butylazodicarboxylate (0.713 mmol, 164 mg) was added and the reaction mixture was stirred at room temperature for 1 hour. After evaporation of the solvent, water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with water and brine, dried over Na2SO4, filtered and was concentrated under reduced pressure. The crude product was triturated in acetonitrile, isopropanol and then in methanol affording 3-(l-(3-fluoro-4-methoxyphenyl)-lH-pyrazol-4-yl)-N-(pyridin- 2-yl)-l,2,4-thiadiazol-5-amine (0.312 mmol, 115 mg, 48%) as apale pink powder. LC (Method LC-E): RT = 2.17 min; MS m/z ES+= 369. 1H NMR (300 MHz, DMSO-d6) δ ppm:12.15 (br. s., 1 H) 8.91 (s, 1 H) 8.44 (ddd, 1 H) 8.17 (s, 1 H) 7.80 - 7.93 (m, 2 H) 7.68 - 7.78 (m, 1 H) 7.32 (dd, 1 H) 7.15 - 7.23 (m, 1 H) 7.00 - 7.12 (m, 1 H) 3.90 (s, 3 H).
EXAMPLE 8: 6-(3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazol-5- ylamino)-nicotinonitrile (Final compound 1-33)
8.1: 5-Chloro-3-(l-(cyclopropyhnethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazole el from bl_: According to Scheme 5 Step 1 of the general methodology, trichloromethanesulfenyl chloride (2.00 mmol, 220 μL) was added under vigorous stirring to a solution of 1- (cyclopropyhnethyl)-lH-pyrazole-4-carboximidamide hydrochloride (2.00 mmol, 400 mg) in water (12 mL) cooled at 0°C. A solution of NaOH (9.40 mmol, 376 mg) in water (8 mL) was added and the reaction mixture was stirred 1 hour at room temperature. DCM was added and the organic phase was washed with water and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified on a silica gel cartridge eluted with petroleum ether/ AcOEt (100:0
KASZClientDocs/AddexZ53195.WO01.FinalSpec.l0.072008
to 90:10) as solvent affording 5-chloro-3-(l-(cyclopropylmethyl)-lH-pyrazol-4-yl)- 1,2,4-thiadiazole (0.436 mmol5 105 mg, 22%). LC (Method LC-D): RT = 1.31 min; MS m/z ES+= 241.
1H NMR (300 MHz, DMSOd6) δ ppm: 8.45 (d, 1 H) 8.00 (d, 1 H) 4.04 (d, 2 H) 1.21 - 1.37 (m, 1 H) 0.47 - 0.60 (m, 2 H) 0.35 - 0.47 (m, 2 H).
8.2: 6-(3-(l-(Cycloproρylmethyl)-lH-pyrazol-4-yl)-l,2,4-tliiadiazol-5-ylamino)- nicotinonitrile (Final compound 1-33)
According to Scheme 5 Step 2 of the general methodology, a mixture of 5-chloro-3-(l- (cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazole (0.22 mmol, 54 mg), 6- aminonicotinonitrile (0.44 mmol, 53 mg) and sodium tert-butoxide (0.33 mmol, 32 mg) in dioxane (3 mL) was heated under reflux overnight. Sodium tert-butoxide (0.11 mmol, 12 mg) was added and the reaction mixture was refluxed for other 5 hours. After cooling at room temperature the solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography over silica gel using DCM/MeOH
(95:5) as eluent. The resulting solid was further purified by trituration in MeOH affording 6-(3 -(I -(cyclopropylmethyl)- 1 H-pyrazol-4-yl)- 1 ,234-thiadiazol-5-ylamino)- nicotinonitrile (0.043 mmol, 14 mg, 20%).
LC (Method LC-E): RT = 1.89 min; MS m/z ES+= 324. 1HNMR (300 MHz, DMSOd6) δ ppm: 12.77 (br. s., 1 H) 8.91 (dd, 1 H) 8.27 (s, 1 H)
8.20 (dd, 1 H) 7.91 (s, 1 H) 7.23 (d, 1 H) 4.04 (d, 2 H) 1.19 - 1.37 (m, 1 H) 0.50 - 0.60
(m, 2 H) 0.35 - 0.44 (m, 2 H).
The following compound is prepared according to the same method:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

EXAMPLE 9: 2-(4-(5-(Pyridin-2-ylamino)-l,2,4-thiadiazol-3-yl)-lH-pyrazol-l- yl)ethanol (Final compound 1-41)
9.1: Ethyl 1 -(2-(benzyloxy)ethyl)- 1 H-pyrazole-4-carboxylate a21 According. to Scheme 4 Step Ia of the general methodology, a mixture of ethyl IH- pyrazole-4-carboxylate (3.57 mmol, 500 mg), ((2-bromoethoxy)methyl)benzene (5.31 mmol, 1.14 g) and K2CO3 (5.355 mmol, 739 mg) in acetone (20 mL) was heated under reflux for 24 hours. After cooling to room temperature the mixture was filtered. After evaporation of the solvent under reduced pressure, the crude was purified by flash chromatography over silica gel using petroleum ether/ AcOEt (90:10 to 80:20) as eluent affording ethyl l-(2-(benzyloxy)ethyl)-lH-pyrazole-4-carboxylate (2.916 mmol, 800 mg, 82%) as a colourless oil. LC (Method LC-B): RT = 1.40 min; MS m/z ES+= 275.
9.2: l-(2-(Benzyloxy)ethyl)-lH-pyrazole-4-carboximidamide hydrochloride b21 According to Scheme 4 Step 2 of the general methodology, trimethylaluminium, 2M solution in toluene (18.20 mmol, 9.15 mL), was added dropwise to a suspension of ammonium chloride (18.20 mmol, 975 mg) in dry toluene (15 mL), under a nitrogen atmosphere, at 0°C. The reaction mixture was stirred at room temperature until no more evolution of gas was observed. After addition of ethyl l-(2-(benzyloxy)ethyl)-lH- pyrazole-4-carboxylate (1.82 mmol, 500 mg), the reaction mixture was stirred at 95°C overnight. The reaction mixture was then cooled down to O0C and MeOH was added under stirring. The solvent was removed and the residue was tritured with MeOH (100 mL) and filtered. The filtrate was concentrated and the crude obtained was purified by
KAS/Clienfflocs/Addex/SS^.WOOLFinalSpec.lO.Oy 2008
flash chromatography over silica gel using DCM/MeOH (80:20) as eluent affording 1- (2-(benzyloxy)ethyl)-lH-pyrazole-4-carboximidamide hydrochloride (1.57 mmol, 440mg, 86%) as a pale yellow oil. LC (Method LC-B): RT = 0.87 min; MS m/z ES+= 245.
9.3: 3-(l-(2-(Benzyloxy)ethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine fl
According to Scheme 4 Step 3 of the general methodology, 2-(pyridin-2-yl)-4-(pyridin-
2-ylimino)-l,2-thiazetidine-3-thione (0.28 mmol, 75 mg) and DBU (0.57 mmol, 85 μL) were added to a solution of l-(2-(benzyloxy)ethyl)-lH-pyrazole-4-carboximidamide hydrochloride (0.28 mmol, 80 mg) in dry DMF (5 mL) and the reaction mixture was stirred at room temperature for 20 minutes. Di-tert-butylazodicarboxylate (0.31 mmol, 70 mg) was added and the reaction mixture was stirred at room temperature for 20 minutes. Water was added and the aqueous phase was extracted with AcOEt. The organic phase was washed with water and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was tritured in acetonitrile affording 3-(l-(2-(benzyloxy)ethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4- thiadiazol-5 -amine (0.172 mmol, 65 mg, 61%) as a pale brown solid. LC (Method LC-B): RT = 1.43 min; MS m/z ES+= 379.
9.4: 2-(4-(5-(Pyridin-2-ylamino)-l,2,4-thiadiazol-3-yl)-lH-pyrazol-l-yl)ethanol (Final compound 1-41)
According to Scheme 4 Step 4a of the general methodology, 3-(l-(2(benzyloxy)ethyl)- lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5-amine (0.052 mmol, 20mg) was dissolved in DCM (4 mL) and TMSBr (0.068 mmol, 9 μl) was added in a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 72 hours with regular addition of TMSBr in portions of 9 μL until the total amount of (0.68 mmol, 90 μl) was reached. The reaction mixture was diluted with DCM and washed with water. The organic layer was separated, dried over Na2SO4 and concentrated. The crude was purified by silica gel flash chromatography (eluent: DCM/MeOH = 95:5) to yield 1- 2-
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.07 2008
(4-(5 -(pyridin-2-ylamino)- 1 ,2,4-thiadiazol-3 -yl)- 1 H-pyrazol- 1 -yl)ethanol (0.035 mmol, 10 nig, 67%) as a beige solid.
LC (Method LC-E): RT = 1.40 min; MS m/z ES+= 289.
1H NMR (300 MHz5 DMSOd6) δ ppm: 12.19 (s, 1 H), 8.35 - 8.48 (m, 1 H), 8.19 (s, 1 H), 7.89 (s, 1 H), 7.76 - 7.86 (m, 1 H), 7.10 - 7.20 (m, 1 H), 6.98 - 7.10 (m, 1 H)5 4.21 (t, 2 H), 3.71 - 3.82 (m, 3 H).
EXAMPLE 10: 6-(3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4-thiadiazol-S- ylamino)nicotinamide (Final compound 1-35)
According to Scheme 4 Step 3 of the general methodology, methyl 6-(3-(l- (cyclopropylmethyl)-lH-pyrazol-4-yl)-l ,2,4-thiadiazol-5-ylamino) nicotinate was prepared following the procedure described in Example 5 starting from (cyclopropyhnethyl)-lH-pyrazole-4-carboximidamide hydrochloride (bl) and methyl 6-isothiocyanatonicotinate (dl9) (LC (Method LC-D): RT = 1.25 min; MS m/z ES+= 357.
According to Scheme 4 Step 4b of the general methodology, methyl 6-(3-(l- (cyclopropylmethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4-thiadiazol-5-ylamino) nicotinate (0.17 mmol, 60 mg) was suspended in 32% NH4OH solution and the mixture was heated in a closed vessel at 100°C under microwaves irradiation for Ih. After cooling, a precipitate was formed. The solid was recovered by filtration and washed with little MeOH affording 6-(3 -(I -(cyclopropylmethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4-thiadiazol-5-ylamino)- nicotinamide (0.047 mmol, 16 mg, 28%) as a white solid. LC (Method LC-E): RT = 1.42 min; MS m/z ES+= 342. 1H NMR (300 MHz, DMSO-d6) δ ppm: 12.49 (br. s., 1 H), 8.92 (d, 1 H), 8.25 (s, 1 H), 8.23 (dd, 1 H), 7.99 (br. s., 1 H)5 7.90 (s, 1 H), 7.41 (br. s.5 1 H)5 7.17 (d, 1 H)5 4.04 (d, 2 H)5 1.14 - 1.43 (m, 1 H), 0.51 - 0.62 (m, 2 H)5 0.33 - 0.44 (m, 2 H).
EXAMPLE 11 : N-(Pyridin-2-yl)-4-(l-(2,2,2-trifluoroethyl)-lH-pyrazol-4- yl)thiazol-2-amine (Final compound 1-87)
KAS/ClientDocs/Addex/53195.WO01.FmalSpec.l0.072008'
11.1 : l-(2,2,2-Trifluoroethyl)-lH-ρyrazole-4-carboxylic acid (hi)
According to Scheme 2 Step 1 of the general methodology, ethyl 1 -(2,2,2- trifluoroethyl)-lH-pyrazole-4-carboxylate (1.03 mmol, 230 mg) was suspended in a mixture of HCl 37% aqueous solution (2 mL) and dioxane (3 mL) and the mixture was heated overnight at reflux. The reaction mixture was concentrated to dryness affording l-(2,2,2-trifluoroethyl)-lH-pyrazole-4-carboxylic acid (1.03 mmol, 200 mg, 100%) as an off white solid. The crude product was used without further purification.
LC (Method LC-D): RT = 0.79 min; MS m/z ES"= 195.
11.2: l-(2,2,2-Trifluoroethyl)-lH-pyrazole-4-carbonyl chloride (il)
According to Scheme 2 Step 2 of the general methodology: A solution of 1 -(2,2,2- trifluoroethyl)-lH-pyrazole-4-carboxylic acid (1.03 mmol, 200 mg), oxalyl chloride (2.58 mmol, 0.22 mL) and two drops of DMF in DCM (5 mL) was stirred for 4 hours at room temperature. After evaporation of the solvent, 1 -(2,2,2 -trifluoroethyl)- IH- pyrazole-4-carbonyl chloride (1.03 mmol, 219 mg) was obtained as a brown oil. The crude product was used without further purification.
LC (Method LC-B): RT = 1.11 min; MS m/z ES"= 209 (methyl ester).
The following compounds are synthesized according to the same method starting from commercially available acid (h2) or from the freshly prepared acid described below :

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

The carboxylic acids h3, h4, and h5, used for the synthesis of intermediate compounds i3, i4 and i5, are prepared according to the following procedure:
1 -Propyl- lH-pyrazole-4-carboxylic acid (h3J
According to Scheme 2 Step 1 of the general methodology, ethyl 1 -Propyl- IH- pyrazole-4-carboxylate (4.39 mmol, 801 mg) was dissolved in a 1:1 mixture of THF and MeOH (15 mL). A solution of LiOH (2N, 10.54 mmol, 5.27 mL) was added and the mixture was heated at reflux for 96 hours. The reaction mixture was concentrated and pardoned between NaOH IN and AcOEt. The aqueous layer was separated, acidified with HCl 12N (pH=l) and extracted with DCM. The organic layer was separated, dried over Na2SO4 and concentrated to dryness affording 1-propyl-lH- pyrazole-4-carboxylic acid (3.85 mmol, 594 mg, 88 %) as an off white solid. The crude product was used without further purification.
LC (Method LC-D): RT = 0.71 min; MS m/z ES"= 155.
The following compounds are synthesized according to the same method using NaOH instead of LiOH:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
11.3: 2-Bromo-l-(l-(252,2-trifluoroethyl)-lH-pyrazol-4-yl)ethanone Ql) and 2-chloro- 1 -( 1 -(2,2,2-trifluoroethyl)- 1 H-pyrazol-4-yl)ethanone (kl)
According to Scheme 2 Step 3 of the general methodology, a solution of TMSdiazomethane (3.90 mmol, 1.95 mL) was added to a solution of 1 -(2,2,2- trifluoroethyl)-lH-pyrazole-4-carbonyl chloride ϋ (1.03 mmol, 219 mg) in acetonitrile (5 mL) at 0°C. The reaction mixture was stirred at room temperature overnight. After cooling to O0C, HBr (4.16 mmol, 0.7 mL, 48%) was added to the reaction mixture. The reaction mixture was stirred at room temperature for two hour. AcOEt and water were added to the reaction mixture and the organic layer was separated. The aqueous phase was neutralized with IM NaOH solution and extracted with AcOEt. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure to yield 2-bromo-l-(l-(2,2,2-trifluoroethyl)-lH-pyrazol-4- yl)ethanone and 2-chloro-l-(l-(2,2,2-trifluoroethyl)-lH-pyrazol-4-yl)ethanone in a ratio (54/34 by LC-MS peaks integration at 254nm) (105 mg) as a brown gummy solid. The crude product was used without further purification.
LC (Method LC-B): RT = 1.18; 1.13 min; MS m/z ES'= 271.1; 227.
The following compounds are synthesized according to the same method:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

11.4 1 -(2-Methoxypyridin-3 -ypthiourea (ml)
3-Isothiocyanato-2-methoxypyridine (1.20 mmol, 200 mg) was dissolved in 2M methanolic ammonia and the reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated to dryness affording l-(2- methoxypyridin-3-yl)thiourea (218 mg) as and off white solid.
LC (Method LC-D): RT = 0.57 min; MS m/z ES"= 184.
The following compounds are synthesized according to the same method

11.5: N-(pyridin-2-yl)-4-(l-(2,2,2-trifiuoroethyl)-lH-pyrazol-4-yl)thiazol-2-amine (Final compound 1-87)
According to Scheme 2 Step 4 of the general methodology, a solution of 2-bromo-l-(l- (2,2,2-trifluoroethyl)-lH-ρyrazol-4-yl)ethanone and 2-chloro-l-(l-(2,2,2- trifluoroethyl)-lH-pyrazol-4-yl)ethanone (0.38 mmol, 105 mg) and of l-(pyridin-2-
KAS/ClientDocs/Addex/SSlPS.WOOLFinalSpec.lO.OV aOOS
yl)thiourea (CAS 14294-11-2, 0.26 mmol, 40 mg) in absolute ethanol (5 mL) was stirred under reflux for 20 minutes. After evaporation of the solvent, AcOEt was added and the organic phase was washed with a IM NaOH solution and brine. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography over silica gel using DCM/MeOH/32% NH4OH (99:1:0.1) as eluent to yield N-(pyridin-2-yl)-4-(l -(2,2,2- trifluoroethyl)-lH-pyrazol-4-yl)thiazol-2-amine (0.06 mmol, 20 mg, 16%) as a white solid.
LC (Method LC-E): RT = 1.50 min; MS m/z ES+= 326. 1H NMR (300 MHz, DMSO-d6) δ ppm: 11.33 (br. s., 1 H) 8.25 - 8.32 (m, 1 H) 8.09 (s, 1 H) 7.93 (d, 1 H) 7.64 - 7.74 (m, 1 H) 7.04 - 7.11 (m, 2 H) 6.89 - 6.95 (m, 1 H) 5.17 (q, 2 H).
The following compounds are synthesized according to the same method:

KAS/ClientDocs/Addex/53195.WO01.FinalSρec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008

Table 3 : Physico-chemical data for some compounds (nd = not determined).

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

Table 4 ; 1H-NMR spectra for some compounds:

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008.

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195. WOO 1.FinalSpec.10.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008

KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008

PHARMACOLOGY
The compounds provided in the present invention are highly selective antagonists of the human adenosine A3 receptor. As such, the compounds of Formula I to III block the activation of adenosine A3 receptor induced by an agonist of the receptor, while they have little or no effect against other subtypes of adenosine receptors including human A1, human A2A and human A2B receptors.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
Material and methods
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A3 receptors:
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A3 receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described in Salvatore et al. ( (1993) Proc. Natl. Acad. ScL USA, 90: 10365).
Briefly, cell membrane homogenates (40 μg protein) were incubated for 120 min at 22°C with 0.15 nM [125I]AB-MECA in the absence or presence of the test compound in a buffer containing 50 niM Tris-HCl (pH 7.4), 5 mM MgCl2, 1 niM EDTA and 2 units/ml ADA. Nonspecific binding was determined in the presence of 1 μM IB- MECA. Following incubation, the samples were filtered rapidly under vacuum through glass fiber filters (GF/B, Packard) presoaked with 0.3% PEI and rinsed several times with ice-cold 50 niM Tris-HCl using a 96-sample cell harvester (Unifilter, Packard). The filters were dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard).
The results are expressed as a percent inhibition of the control radioligand specific binding. The standard reference compound is IB-MECA, which is tested in each experiment at several concentrations to obtain a competition curve from which its IC5O and Ki values are calculated.
The inhibition curves were generated using the Prism GraphPad program (Graph Pad Software Inc, San Diego, USA). IC50 and Ki determinations were extrapolated from data obtained from 3- to 8-point-concentration response curves using a non linear
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.10.07 2008
regression analysis. The mean of IC50 and Ki obtained from at least three independent experiments of selected molecules performed in duplicate were calculated.
Compounds Nr 1-8, 1-9, 1-10, 1-12, 1-13, 1-14, 1-15, 1-19, 1-21, 1-22, 1-35, 1-42, 1- 50, 1-63, 1-64 and 1-74 of the prevent invention have a KIi value on human adenosine A3 receptors of less than 1 μM.
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine Ai receptors:
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A1 receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described by Townsend-Nicholson and Schofield (1994), J. Biol. Chem. 269: 2373.
Briefly, cell membrane homogenates (20 μg protein) were incubated for 60 min at 22°C with 1 nM [3H]DPCPX in the absence or presence of the test compound in a buffer containing 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 1 mM EDTA/Tris and 2 UI/ml ADA. Following incubation, the samples were filtered rapidly under vacuum through glass fiber filters (GF/B, Packard) presoaked with 0.3% PEI and rinsed several times with ice-cold 50 mM Tris-HCl using a 96-sample cell harvester (Unifilter, Packard). The filters are dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). Nonspecific binding was determined in the presence of 1 μM DPCPX.
The results are expressed as a percent inhibition of the control radioligand specific binding. The standard reference compound is DPCPX, which is tested in each experiment at several concentrations to obtain a competition curve from which its IC50 is calculated.
KAS/ClientDocs/Addex/SSlθS.WOOLFinalSpec.lO.O? 2008
The inhibition curves were generated using the Prism GraphPad program (Graph Pad Software Inc, San Diego, USA). IC50 and Ki determinations were extrapolated from data obtained from 3- to 8-point-concentration response curves using a non linear regression analysis. The mean of IC50 and Ki obtained from at least three independent experiments of selected molecules performed in duplicate were calculated.
Compounds Nr 1-8, 1-9, 1-10, 1-12, 1-13, 1-14, 1-15, 1-19, 1-21, 1-22, 1-35, 1-42, 1- 50, 1-63, 1-64 and 1-74 of the prevent invention have a Ki value on human adenosine A1 receptors greater than 1 μM.
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A2A receptors:
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A2A receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described by Luthin et al ((1995) MoI. Pharmacol., 47: 307).
Briefly, cell membrane homogenates (50 μg protein) were incubated for 120 min at 22°C with 6 nM [3H]CGS 21680 in the absence or presence of the test compound in a buffer containing 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2 and 2 Ul/ml ADA.
Following incubation, the samples are filtered rapidly under vacuum through glass fiber filters (GF/B, Packard) presoaked with 0.3% PEI and rinsed several times with ice-cold
50 mM Tris-HCl using a 96-sample cell harvester (Unifilter, Packard). The filters are dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). Nonspecific binding was determined in the presence of 10 μM NECA.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
The results are expressed as a percent inhibition of the control radioligand specific binding. The standard reference compound is NECA, which is ' tested in each experiment at several concentrations to obtain a competition curve from which its IC50 is calculated.
The inhibition curves were generated using the Prism GraphPad program (Graph Pad Software Inc, San Diego, USA). IC50 and Ki determinations were extrapolated from data obtained from 3- to 8-point-concentration response curves using a non linear regression analysis. The mean of IC5O and Ki obtained from at least three independent experiments of selected molecules performed in duplicate were calculated.
Compounds Nr 1-8, 1-9, 1-10, 1-12, 1-13, 1-14, 1-15, 1-19, 1-21, 1-22, 1-35, 1-42, 1- 50, 1-63, 1-64 and 1-74 of the prevent invention have a Ki value on human adenosine A2A receptors greater than 1 μM.
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A2B receptors:
Radioligand binding assay for the evaluation of the affinity of compounds for the human adenosine A2B receptors expressed in transfected HEK-293 cells has been performed in 96-well plate and following the experimental conditions described in Stehle et al ((1992) MoI. Endocrinol. 6:384-393). .
Briefly, cell membrane homogenates of HEK-293 cells (200 μg protein) were incubated for 120 min at 22°C with 0.5 nM [3H]MRS1754 in the absence or presence of the test compound in a buffer containing 10 niM Hepes/Tris (pH 7.4), 1 mM MgCl2 and 1 mM EDTA. Following incubation, the samples are filtered rapidly under vacuum
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008 .
through glass fiber filters (GF/C, Whatman) presoaked with 0.3% PEI and rinsed several times with ice-cold 50 mM Tris-HCl using a 48-sample cell harvester (Brandel). The filters are dried then counted for radioactivity in a scintillation counter (LS series, Beckman) using a scintillation cocktail (Formula 989, Packard). Nonspecific binding is determined in the presence of 100 μM NECA.
The results are expressed as a percent inhibition of the control radioligand specific binding. The standard reference compound is NECA, which is tested in each experiment at several concentrations to obtain a competition curve from which its IC50 is calculated.
The inhibition curves were generated using the Prism GraphPad program (Graph Pad Software Inc, San Diego, USA). IC50 and ELi determinations were extrapolated from data obtained from 3- to 8-point-concentration response curves using a non linear regression analysis. The mean of IC5O and Ki obtained from at least three independent experiments of selected molecules performed in duplicate were calculated.
Compounds Nr 1-8, 1-9, 1-10, 1-12, 1-13, 1-14, 1-15, 1-19, 1-21, 1-22, 1-35, 1-42, 1- 50, 1-63, 1-64 and 1-74 of the prevent invention have a Ki value on human adenosine A2B receptors greater than 1 μM.
Luminescence-based Ca2+mobilization assay for the evaluation of the antagonist properties of compounds acting at human adenosine A3 receptors:
The functional properties of the compounds of the present invention were assessed using a cell-based Ca2+-mobilization assay in which the luminescence properties of the photoprotein aequorin was directly proportional to the intracellular Ca2+ released within
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
the cell cytoplasm as it is described in literature (Detheux et al (2000) J. Exp. Med. 192:1501-1508). A CHO-Kl cell line expressing adenosine A3AR, Ga16 and mitochondrial apoaequorin were established.
In brief, cells were collected from plates with PBS containing 5 mM EDTA, pelleted, and resuspended at 7.5 x 105 cells/ml in DMEM-F 12 medium and incubated with 5mM coelenterazine H (Molecular Probes) overnight at room temperature. Cells were then washed in DMEM-F 12 medium and resuspended at a concentration of 105 cells/ml. For measuring an agonist activity, cells were injected to the test compounds at 8 concentrations already distributed in microtiter plate, and the light emission was recorded over 60 s using a FDSS 6000 luminometer (Hamamatsu). Then, following an incubation of 3 min, the reference agonist IB-MECA was injected at a concentration corresponding to 80 % of the maximal agonist concentration (EC8o) in the wells containing the cells and test compounds and the light emission was recorded over 60 s using a FDSS 6000 luminometer (Hamamatsu). Results are expressed as relative light units (RLU).
The concentration-response curves of representative compounds of the present invention were generated using the Prism GraphPad software (Graph Pad Inc, San Diego, USA). The curves were fitted to a four-parameter logistic equation: (Y=Bottom + (Top-Bottom)/(l+10Λ((LogEC50-X)*Hill Slope) allowing determination OfIC50 values.
Compounds Nr 1-8, 1-9, 1-14, 1-15, 1-22, 1-35, 1-50 and 1-64 of the prevent invention have an IC50 value on human adenosine A3 receptors less than 1 μM.
These results demonstrate that the compounds described in the present invention have a higher affinity for human adenosine A3 receptor as compared to others human
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
adenosine receptors A1, A2A and A2B. Furthermore, these results show that these compounds functionally behave as antagonist of human Adenosine A3 receptors. These compounds do not show an activity by their own but they rather block the functional activation of human adenosine A3 receptors induced by adenosine or human A3 receptor agonist.
Thus, the selective antagonists of human A3 receptor provided in the present invention are expected to block the effectiveness of adenosine or A3AR agonists at human A3 receptor. Therefore, these selective adenosine A3 antagonists are expected to be useful in human for the treatment of various conditions associated with dysfunction of adenosine system when adenosine A3 receptors are overstimulated due to presence of an excess of adenosine or metabolite or endogenous ligand with human A3AR agonist property or due to a sustained presence of these agonists in the vicinity of human A3ARs resulting in an hyperactivation of adenosinergic system.
FORMULATION EXAMPLES
Typical examples of recipes for the formulation of the invention are as follows:
1. Tablets
Active ingredient 5 to 50 mg Di-calcium phosphate 20 mg
Lactose 30 mg
Talcum 10 mg
Magnesium stearate 5 mg
Potato starch ad 200 mg In this Example, active ingredient can be replaced by the same amount of any of the compounds according to the present invention, in particular by the same amount of any of the exemplified compounds.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
2. Suspension
An aqueous suspension is prepared for oral administration so that each 1 milliliter contains 1 to 5 mg of one of the active compounds, 50 mg of sodium carboxymethyl cellulose, 1 mg of sodium benzoate, 500 mg of sorbitol and water ad 1 ml.
3. Injectable
A parenteral composition is prepared by stirring 1.5 % by weight of active ingredient of the invention in 10% by volume propylene glycol and water.
4. Ointment
Active ingredient 5 to 1000 mg Stearyl alcohol 3 g Lanoline 5 g White petroleum 15 g Water ad 100 g
5. Opthalmic solution
Ingredients: Amount (wt %):
Active ingredient 0.01-2% Hydroxypropyl methylcellulose 0.5%
Dibasic sodium phosphate (anhydrous) 0.2%
Sodium chloride 0.5%
Disodium EDTA (Edetate disodium) 0.01%
Polysorbate 80 0.05% Benzalkonium chloride 0.01%
Sodium hydroxide/Hydrochloric acid For adjusting pH to 7.3-7.4
Purified water q.s. to 100%
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
In this Example, active ingredient can be replaced with the same amount of any of the compounds according to the present invention, in particular by the same amount of any of the exemplified compounds.
Reasonable variations are not to be regarded as a departure from the scope of the invention. It will be obvious that the thus described invention may be varied in many ways by those skilled in the art.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
Claims
1. A compound according to the general Formula (I),

X1, X2, X3 and X4 are each independently selected from the group of C, N5 O, S and C=C representing a 5 or 6 membered heteroaryl ring which may further be substituted by 1 to 3 radicals Am; m is an integer ranging from 1 to 3;
Am radicals are each independently selected from the group of hydrogen,
. halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, -(d-C^alkylhalo, -(C3-C7)cycloalkyl, -(C1-C6)alkylcyano, -(C1-
C6)alkylheteroaryl, -(C1-C6)alkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl- OR1, -O-(C2-C6)alkyl-OR\ -NR1(C2-C6)alkyl-OR2, -(C3-C7)cycloalkyl-(C1-
C6)alkyl, -O-(C3-C7)cycloalkyl-(Ci-C6)alkyl, -NR1 -(C3-C7)CyClOaUCyI-(C1- C6)alkyl, -(CrC^alkylhalo-OR1, -(CrC^alkylhalo-N^R2, -(Co-QOalkyl-S-R1, - O-(C2-C6)alkyl-S-R1, -NR1-(C2-C6)alkyl-S-R2, -(C0-C6)alkyl-S(=O)-R1, -0-(C1- C6)alkyl-S(=O)-R1, -NR1-(C1-C6)alkyl-S(=O)-R2, -(C0-C6)alkyl- S(^O)2-R1, -O- (C1-C6)alkyl-S(=O)2-R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, -(Co-C^alkyl-N^R2, -O-
 -NR1-(C2-C6)alkyl-NR2R3, -(C0-C6)alkyl-S(=O)2NR1R2, -O- (C1-C6)alkyl-S(=O)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3, -(C0-C6)alkyl- NR^S^O^R2, -O-(C2-C6)alkyl-NR1-S(=O)2R2, ^^-(Cs-C^alkyl-NR2- S(=O)2R3, -(Co-C6)alkyl-C(=0)-NR1R2, -O-(C1-C6)alkyl-C(=O)-NR1R2 5 -NR1-
-NR1-(C2-C6)alkyl-NR2R3, -(C0-C6)alkyl-S(=O)2NR1R2, -O- (C1-C6)alkyl-S(=O)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3, -(C0-C6)alkyl- NR^S^O^R2, -O-(C2-C6)alkyl-NR1-S(=O)2R2, ^^-(Cs-C^alkyl-NR2- S(=O)2R3, -(Co-C6)alkyl-C(=0)-NR1R2, -O-(C1-C6)alkyl-C(=O)-NR1R2 5 -NR1-
KAS/ClientDocs/Addex/SSlPS.WOOl.FinalSpec.lO.O? 2008
™2» 22«
(Ci-C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl-NR1C(=O)-R2, -O-(C2-C6)alkyl- NR1C(^O)-R2, -NR1-(C2-C6)alkyl-NR2C(=O)-R3, -O-(C2-C6)alkyl-OC(=O)-R1, - NR1-(C2-C6)alkyl-OC(=O)-R2, -(C0-C6)alkyl-C(=O)-OR1 5 -O-td-C^alkyl- CC=O)-OR1, -NR1-(C1-C6)alkyl-C(=O)-OR2, -(Co-C^alkyl-C^O)^1, -0-(Ci-
OR2, -(C0-C6)alkyl-NR1-C(=O)-NR2R3, -O-(C2-C6)alkyl-NR1-C(=O)-NR2R3, and -NR1-(C2-C6)alkyl-NR2-C(=O)-NR3R4;
Any two radicals of Am (A1 and A2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(Q-C^alkyl, -(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C1o)alkylcycloaUcyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(CrC^alkylaryl;
Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
Y1, Y2, Y3 and Y4 are each independently selected from the group of C and N representing 5 membered heteroaryl ring which may further be substituted by 1 to 3 radicals Bn;
n is an integer ranging from 1 to 3;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(CrC6)alkylhalo, -(C3-C7)cycloalkyl, -(CrC^alkylcyano, -(C1-
C6)alkylheteroaryl, -(Ci-C6)alkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl- OR5, -O-(C2-C6)alkyl-OR5, -NR5(C2-C6)alkyl-OR6, -(Cs-C^cycloalkyHCr C6)alkyl, -O-(C3-C7)cycloalkyl-(C1-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(C1-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
pc™ 2 o 2 3
C6)alkyl, -(Q-C^alkylhalo-OR5, -(CrC^alkylhalo-NR5^, -(C0-C6)alkyl-S-R5, - O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S-R6, -(C0-C6)alkyl-S(=O)-R5, -0-(C1- C6)alkyl-S(=O)-R5, -NR^CrC^alkyl-SC^-R6, -(C0-C6)alkyl-S(=O)2-R5, -O- (Ci-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6 5 -(C0-C6)alkyl-NR5R6, -O- (C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-S(=O)2NR5R6, -O-
(C1-C6)alkyl-S(=O)2NR5R6, -NR5-(C1-C6)alkyl-S(=O)2NR6R7, -(C0-C6)alkyl- NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(Co-C6)alkyl-C(=0)-NR5R6, -0-(C1-C6)alkyl-C(=0)-NR5R6, -NR5- (C1-C6)alkyl-C(-O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl- NR5C(=O)-R6, -NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, -
NR5-(C2-C6)alkyl-OC(=O)-R6, -(C0-C6)alkyl-C(=O)-OR5 5 -O-CCrC^alkyl- C(=0)-0R5, -NR5-(C1-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5 5 -0-(C1- C6)alkyl-C(=O)-R5, -NR5-(C!-C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)- OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
Any two radicals of Bn (B1 and B2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
R5, R6, R7 and R8 are each independently hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(C1-C6)alkyl, -(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C ^alkylcy cloalkyl, heteroaryl, -(C1-
C6)alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl;
Any two radicals of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted 3 to 10 membered ring selected from the group of aryl, heteroaryl, heterocyclic and cycloalkyl;
M2 is selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C0-C6)alkyl-R9, -(Ci-C6)alkylhalo, -(C2-C6)alkyl-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
NR9R10, -(C2-C6)alkyl-OR9, -(C2-C6)alkyl-SR9, -(C0-C6)alkyl-C(=O)-R9 3 -(C2- C6)alkyl-S(O)-R9, -(C0-C6)alkyl-C(=O)NR9R10 and -(C0-C6)alkyl-S(O)2-R9;
R9 and R10 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(Ci-C^alkyl, -(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1-
C6)alkylheteroaryl, aryl, heterocycle and -(CrC^alkylaryl;
M3 is an optionally substituted radical selected from the group of -(C0-C6)alkyl- R11, -(C1-C6)alkylhalo, -(C2-C6)alkyl-NRπR12, -(C2-C6)alkyl-ORn and -(C2- C6)alkyl-SRU; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C1-C6)alkylhalo, -(C1-C6)alkyl, -
 -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl5 heteroaryl, - (CrC^alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl; provided that according to proviso (i) : when M3 is -(Co)-R11 (that is when M3 is -R11), then R11 is not H; and provided that according to proviso (ii):
-(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl5 heteroaryl, - (CrC^alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl; provided that according to proviso (i) : when M3 is -(Co)-R11 (that is when M3 is -R11), then R11 is not H; and provided that according to proviso (ii):

when M1 is aryl, M2 is H, X1 is C, X2 is C, X3 is C, X4 is N, then
 is not linked to X2; and provided that according to proviso (iv):
is not linked to X2; and provided that according to proviso (iv):
A1 and A2 radicals are not linked to form an imidazopyridazinyl ring;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
and provided that according to proviso (v):
.when
 is and linked to X4 X1 is C, X2 is S5 X3 is C5
is and linked to X4 X1 is C, X2 is S5 X3 is C5
X4 is C5 n is I5 A1 is H5 Y1, Y2, Y3 are C, Y4 is N5 then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and IH- indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl5 then Bn can not be a phenyl; and provided that according to proviso (vii): when M1M2N is linked to X1, and X1 is C, X2 is S5 X3 is C, X4 is C5 to provide a thiazole ring, n is 1 , then A1 is not a pyridyl; and provided that according to proviso (viii): when M1M2N is linked to X1, and X1 is C5 X2 is S, X3 is C, X4 is C5 to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
2. A compound according to claim 1 having the Formula (II),

X2, X3 and X4 are each independently selected from the group of C5 N5 O5 S and C=C representing a 5 or 6 membered heteroaryl ring which may further be substituted by 1 to 3 radicals Am; m is an integer ranging from 1 to 3;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
Am radicals are each independently selected from the group of hydrogen, halogen, -CN5 -CF3, and an optionally substituted radical selected from the group of -(Ci-QOalkyl, -(Ci-C6)alkylhalo, -(C3-C7)cycloalkyl, -(C3-C8)cycloalkenyl, -
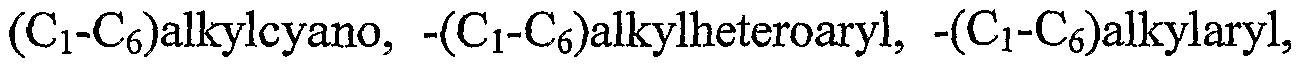 aryl, heteroaryl, heterocycle, -(Co-C6)alkyl-OR\ -NR1(C2-C6)alkyl-OR2, -(C3-C7)cycloalkyl-(Ci-
aryl, heteroaryl, heterocycle, -(Co-C6)alkyl-OR\ -NR1(C2-C6)alkyl-OR2, -(C3-C7)cycloalkyl-(Ci-
C6)alkyl, -O-(C3-C7)cycloalkyl-(Ci-C6)alkyl,
 C6)alkyl, -(Ci-C^alkylhalo-OR1, -(CrC^alkylhalo-NE^R2, -(C0-C6)alkyl-S-R1, - NR^Q-C^alkyl-S-R2, -(C0-C6)alkyl-S(=O)-R1, -NR1-(C1-C6)alkyl-S(=O)-R2, - (Co-C6)alkyl-S(=0)2-R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, -(Co-C^alkyl-N^R2, - NR^Cs-CeOalkyl-NR^3, -(C0-C6)alkyl-S(=O)2NR1R2 3 -NR^Ci-C^alkyl-
C6)alkyl, -(Ci-C^alkylhalo-OR1, -(CrC^alkylhalo-NE^R2, -(C0-C6)alkyl-S-R1, - NR^Q-C^alkyl-S-R2, -(C0-C6)alkyl-S(=O)-R1, -NR1-(C1-C6)alkyl-S(=O)-R2, - (Co-C6)alkyl-S(=0)2-R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, -(Co-C^alkyl-N^R2, - NR^Cs-CeOalkyl-NR^3, -(C0-C6)alkyl-S(=O)2NR1R2 3 -NR^Ci-C^alkyl-
S(O)2NR2R3, -(Co-C^alkyl-NR^S^O)^2, -NR1-(C2-C6)alkyl-NR2-S(=O)2R3, - (C0-C6)alkyl-C(=O)-NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl- NR1C(^O)-R2, -NR1-(C2-C6)alkyl-NR2C(=O)-R3, -NR1-(C2-C6)alkyl-OC(=O)- R2, -(Co-C6)alkyl-C(=0)-OR1, -NR1-(C1-C6)alkyl-C(=O)-OR2, -(C0-C6)alkyl-
 -(C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-
-(C0-C6)alkyl-NR1-C(=O)-OR2, -(C0-
C6)alkyl-NR1-C(=O)-NR2R3, and -NR1-(C2-C6)alkyl-NR2-C(=O)-NR3R4;
Any two radicals of Am (A1 and A2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(Ci-C6)alkyl, -(C1-
C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(Ci-C6)alkylaryl;
Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN5 -CF3, and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(d-C^alkylhalo, -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano, -(C1- C6)alkylheteroaryl5 -(Ci-C^alkylaryl, aryl, heteroaryl, heterocycle, -(Co-C6)alkyl-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
OR5, -O-(C2-C6)alkyl-OR5 , -NR5(C2-C6)alkyl-OR6 5 -(Cs-C^cycloalkyHQ- C6)alkyl, -O-(C3-C7)cycloalkyl-(Ci-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(Cr C6)alkyl, -(d-C^alkylhalo-OR5, -(Q-C^alkylhalo-NR^6, -(C0-C6)alkyl-S-R5, - O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S-R6, -(C0-C6)alkyl-S(=O)-R5, -0-(C1- C6)alkyl-S(=O)-R5, -NR5-(C1-C6)alkyl-S(=O)-R6 5 -(C0-C6)alkyl-S(-O)2-R5, -0-
(Ci-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, -(C0-C6)alkyl-NR5R6 5 -O- (C2-C6)alkyl-NR5R6, -NR5~(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-S(=O)2NR5R6, -O-
 -(C0-C6)alkyl- NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6 5 -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, -NR5-
-(C0-C6)alkyl- NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6 5 -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, -NR5-
(C1-C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl- NR5C(=O)-R6, -NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5 5 - NR5-(C2-C6)alkyl-OC(=O)-R6, -(C0-C6)alkyl-C(=O)-OR5, -O-CCrC^alkyl- C(K))-0R5, -NR5-(C!-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -0-(C1- C6)alkyl-C(=O)-R5, -NR5-(C1-C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-
OR6, -(Co-C6)alkyl-NR5-C(=0)-NR6R7 5 -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R5, R6, R7 and R8 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(CrC^alkyl, -(C1-
C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C 1o)alkylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and

Any two radicals of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; M1 is selected from an optionally substituted 3 to 10 membered ring selected from the group of aryl, heteroaryl, heterocyclic and cycloalkyl;
M2 is selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C0-C6)alkyl-R9, -(C1-C6)alkylhalo, -(C2-C6)alkyl-
KAS/ClientDocs/Addex/SSlPS.WOOLFinalSpec.lO.O? 2008
NR9R10, -(C2-C6)alkyl-OR9, -(C2-C6)alkyl-SR9, -(C0-C6)alkyl-C(=O)-R9, -(C2- C6)alkyl-S(O)-R9, -(C0-C6)alkyl-C(=O)NR9R10 and -(C0-C6)alkyl-S(O)2-R9;
R9 and R10 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Q-C^alkyl, -(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1-
C6)alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Ct-C^alkyl-R11, -(CrC^alkylhalo, -(C2-C6)alkyl-NR11R12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRπ; and R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Q-C^alkyl, - (CrC^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, - (CrC^alkylheteroaryl, aryl, heterocycle and -(Ci-C6)alkylaryl; provided that according to proviso (i): when M3 is -(Co)-R11 (that is when M3 is -R11), then R11 is not H; and provided that according to proviso (ii):

when M1 is aryl, M2 is H, X2 is C, X3 is C, X4 is N, then
 is not linked to X2; and provided that according to proviso (iv):
is not linked to X2; and provided that according to proviso (iv):
A1 and A2 radicals are not linked to form an imidazopyridazinyl ring; and provided that according to proviso (v):
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008 . .
when
 and linked to X4, X2 is S5 X3 is C, X4 is C, n is 1, A1 is H5 then M1 can not be an optionally substituted aryl, 1 -methyl- IH- indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when X2 is S, X3 is C5 X4 is C5 n is I5 then A1 when linked to either X3 or X4 is not a pyridyl; and provided that according to proviso (viii): when X2 is S5 X3 is C5 X4 is C5 to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
and linked to X4, X2 is S5 X3 is C, X4 is C, n is 1, A1 is H5 then M1 can not be an optionally substituted aryl, 1 -methyl- IH- indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): when X2 is S, X3 is C5 X4 is C5 n is I5 then A1 when linked to either X3 or X4 is not a pyridyl; and provided that according to proviso (viii): when X2 is S5 X3 is C5 X4 is C5 to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
3. A compound according to claim 2 having the Formula (III),

a pharmaceutically acceptable acid or base addition salt thereof, a stereochemical^ isomeric form thereof and an JV-oxide form thereof, wherein:
X3 is selected from C or N which may further be substituted by A1;
A1 radicals are each independently selected from the group of hydrogen, halogen,
-CN5 -CF3, and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(CrC6)alkylhalo5 -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano5 -(C1-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
C6)alkylheteroaryl, -(C1-C6)alkylaryl, heterocycle, -(Co-C^alkyl-OR1, -NR1CC2- C6)alkyl-OR2, -(C3-C7)cycloalkyl-(CrC6)alkyl, -0-(C3-C7)CyClOaIlCyI-(C1- C6)alkyl, -NR1-(C3-C7)cycloalkyl-(C1-C6)alkyl, -(Ci-C^alkylhalo-OR1, -(C1- C^alkylhalo-N^R2, -NR1-(C2-C6)alkyl-S-R2, -(Co-C^alkyl-S^O)^1, -NR1- (C1-C6)alkyl-S(=O)-R2, -(Co-C6)alkyl-S(=0)2-R1, -NR1-(C1-C6)alkyl-S(=0)2-R2,
-(Co-C^alkyl-N^R2, -NR1-(C2-C6)alkyl-NR2R3, -(C0-C6)alkyl-S(=O)2NR1R2, - NR1-(Ci-C6)alkyl-S(=O)2NR2R3, -(C0-C6)alkyl-NR1-S(=O)2R2, -NR^(C2- C6)alkyl-NR2-S(=O)2R3, -(C0-C6)alkyl-C(=O)-NR1R2, -NR^^i-C^alkyl-C^O)- NR2R3, -(Co-CfOalkyl-NR^CJhR2, -NR1-(C2-C6)alkyl-NR2C(=O)-R3, -NR1- (C2-C6)alkyl-OC(=O)-R2, -(C0-C6)alkyl-C(=O)-OR1, -NR1-(C1-C6)alkyl-C(=O)-
OR2, -(Co-C6)alkyl-C(=0)-R1, -NR1-(C1-C6)alkyl-C(=O)-R2, -(Co-C^alkyl-NR1- C(=O)-OR2, -(Co-C6)alkyl-NR1-C(=0)-NR2R3, and -NR^Q-C^alkyl-NR2- C(=O)-NR3R4;
R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C^alkylhalo, -(Ci-C^alkyl, -(C1-
C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)aUcylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(Ci-C6)alkylaryl;
Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
B" radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group Of -(C1- C6)alkyl, -(CrC^alkylhalo, -(C3-C7)cycloalkyl, -(Q-C^alkylcyano, -(C1- C6)alkylheteroaryl, -(C1-C6)alkylaryl, aryl, heteroaryl, heterocycle, -(Co-C6)alkyl- OR5, -O-(C2-C6)alkyl-OR5, -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(Ci-
C6)alkyl, -O-(C3-C7)cycloalkyl-(C1-C6)alkyl,
 C6)alkyl, -(d-C^alkylhalo-OR5, -(CrC^alkylhalo-NR^6, -(C0-C6)alkyl-S-R5, - O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S-R6, -(C0-C6)alkyl-S(=O)-R5, -0-(C1- C6)alkyl-S(=O)-R5,
C6)alkyl, -(d-C^alkylhalo-OR5, -(CrC^alkylhalo-NR^6, -(C0-C6)alkyl-S-R5, - O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S-R6, -(C0-C6)alkyl-S(=O)-R5, -0-(C1- C6)alkyl-S(=O)-R5,
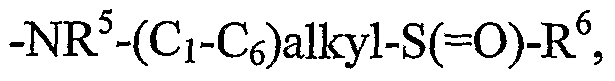 -(C0-C6)alkyl-S(=O)2-R5, -O-
-(C0-C6)alkyl-S(=O)2-R5, -O-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
Pc oo
(C1-C6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, -(C0-C6)alkyl-NR5R6, -O- (C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7 5 -(C0-C6)alkyl-S(=O)2NR5R6, -O- (C1-C6)alkyl-S(=O)2NR5R6, -NR5-(C1-C6)alkyl-S(=O)2NR6R7, -(C0-C6)alkyl- NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(Ci-C6)alkyl-C(=O)-NR5R6, -NR5-
(Ci-C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl- NR5C(=O)-R6, -NR5-(C2-C6)alkyl~NR6C(=O)-R7, -O-(C2-C6)alkyl-OCC=O)-R5, - NR5-(C2-C6)alkyl-OC(=O)-R6, -(C0-C6)alkyl-C(=O)-OR5, -0-(Ci-C6)alkyl- C(=0)-0R5, -NR5-(C1-C6)alkyl-C(=O)-OR6, -CC0-C6)alkyl-C(=O)-R5, -0-(C1- C6)alkyl-C(=O)-R5, -NR5-(C1-C6)alkyl-CC=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-
OR6, -(Co-C6)alkyl-NR5-C(=0)-NR6R7 5 -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R5, R6, R7 and R8 are each independently hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(Ci-C6)alkyl, -(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(Q-C^alkylcycloalkyl, heteroaryl, -(C1-
C6)alkylheteroaryl, aryl, heterocycle and

Any two radicals of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl; M3 is an optionally substituted radical selected from the group of -(C3-
C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C1-C6)alkyl-R11, -(d-C^alkylhalo, -(C2-C6)alkyl-NRπR12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRn; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of
 - (Q-C^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -
- (Q-C^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -
(C1-C6)alkylheteroaryl, aryl, heterocycle and -(Ci-C6)alkylaryl; and provided that according to proviso (v):
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
when
 , X3 is C, n is I5 A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and IH- indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): A1 is different from a pyridyl; and provided that according to proviso (viii): when X3 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
, X3 is C, n is I5 A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and IH- indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): A1 is different from a pyridyl; and provided that according to proviso (viii): when X3 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
4. A compound according to claim 3 having the Formula (IIIA),

a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an JV-oxide form thereof, wherein:
A1 radical is selected from the group of hydrogen, halogen, -CN5 -CF3, and an optionally substituted radical selected from the group of -(Q-C^alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl, -(CrC^alkylcyano, heterocycle, -(Co-C6)alkyl- OR1, -(C3-C7)cycloalkyl-(Ci-C6)alkyl, -NR1-(C3-C7)cycloalkyl-(C1-C6)alkyl, - (CrCøalkylhalo-OR1, -(C1-C6)alkylhalo-NR1R2, (Co-C^alkyl-NR^2, -NR1^C2-
KAS/ClientDocs/Addex/SSlθS.WOOLFinalSpec.lO.OV 2008
C6)alkyl-NR2R3, -(C0-C6)alkyl-C(=O)-NR1R2 5 -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl-NR1C(=O)-R2, -NR1-(C2-C6)alkyl-NR2C(=O)-R3, -(C0-C6)alkyl- CC=O)-R1, and -NR1-(C1-C6)alicyl-C(=O)-R2;
R1, R2 and R3 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ct-C^alkylhalo, -(Q-C^alkyl, -(C1-
C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(Ct-G^alkylaryl;
Any two radicals of R (R1, R2 or R3) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(CrC^alkylhalo, -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, - (Co-C6)alkyl-OR5, -O-(C2-C6)alkyl-OR5 , -NR5(C2-C6)alkyl-OR6, -(C3- C7)cycloalkyl-(C1-C6)alkyl, -O-^s-C^cycloalkyl-^rC^alkyl, -NR5-(C3-
C^cycloalkyKCrC^alkyl, -(Ci-C6)alkylhalo-OR5, -(Ci-C6)alkylhalo-NR5R6, - (C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0- C6)alkyl-C(=O)-NR5R6, -O-(Ci-C6)alkyl-C(=O)-NR5R6, -(Ci-C6)alkyl-OC(=O)- R5, -(Co-C6)alkyl-C(=0)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5, -NR^^i-C^alkyl- C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(C1-C6)alkyl-C(=O)-R5 and -NR5-(Cr
C6)alkyl-C(=O)-R6;
R5, R6 and R7 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo,
 -(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)ahcylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(Ci-C6)alkylaryl;
-(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)ahcylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(Ci-C6)alkylaryl;
Any two radicals of R (R5, R6, or R7) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C1-C6)alkyl-R11, -(Ci-C6)alkylhalo, -(C2-C6)BIlCyI-NR11R12, -(C2-C6)alkyl-ORU and -(C2-C6)alkyl-SRπ;
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Ci-C6)alkyl, - (C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Ci-C^alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl; provided that according to proviso (v):
 when B" is , X is C5 n is 1, A is H, then M can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and IH- indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
when B" is , X is C5 n is 1, A is H, then M can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and IH- indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bn can not be a phenyl; and provided that according to proviso (vii): A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
5. A compound according to claim 3 having the Formula (IIIB),
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
pCT/IB2008/002243

a pharmaceutically acceptable acid or base addition salt thereof, stereochemically isomeric form thereof and an JV-oxide form thereof, wherein:
n is an integer ranging from 1 to 2; Bn radicals are each independently selected from the group of hydrogen, halogen,
-CN, -CF3, and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(C1-C6)alkylhalo, -(C3-C7)cycloalkyl, aryl, heteroaryl, heterocycle, - (C0-C6)alkyl-OR5, -O-(C2-C6)alkyl-ORS , -NR5(C2-C6)alkyl-OR6, -(C3- C7)cycloalkyl-(Ci-C6)alkyl, -O-(C3-C7)cycloalkyl-(C1-C6)alkyl, -NR5-(C3- C7)cycloalkyl-(Ci-C6)alkyl, -(C1-C6)alkylhalo-OR5, -(Ci-C6)alkylhalo-NR5R6, -
(C0-C6)alkyl-NR5R6, -O-(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0- C6)alkyl-C(=O)-NR5R6, -O-(CrC6)alkyl-C(=O)-NR5R6 5 -(C1-C6)alkyl-OC(=O)- R5, -(C0-C6)alkyl-C(=O)-OR5, -O-(C1-C6)alkyl-C(=O)-OR5, -NR5-(Ci-C6)alkyl- C(=O)-OR6, -(Co-C6)alkyl-C(=0)-R5, -O-(CrC6)alkyl-C(=O)-R5 and -NR5-(Cr C6)alkyl-C(-O)-R6;
R5, R6 and R7 are each independently hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(Ci-C6)alkyl, -(C1- C6)alkylcyano, ~(C3-C7)cycloalkyl, -(C4-C1o)aIkylcycloaUiyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl; Any two radicals of R (R5, R6, or R7) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl;
KAS/ClientDocs/Addex/SSigS.WOOLFmalSpec.lO.OT lOOS
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC6)alkyl-Rn, -(CrC^alkylhalo, -(C2-C6)alkyl-NRπR12 5 -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRn; and
R11 and R are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(C1-C6)alkylhalo,
 -
-
(C3-C7)cycloalkyl, -(C4-C1o)alkylcycloalkyl5 heteroaryl,
 aryl, heterocycle and -(C1-C6)alkylaryl.
aryl, heterocycle and -(C1-C6)alkylaryl.
6. A compound according to claim 2 having the Formula (II), wherein: X2 is a nitrogen, an oxygen, or a sulfur atom, X3 is a carbon atom or a nitrogen atom, X4 is a carbon or a nitrogen atom, representing a 5 membered heteroaryl, which may further be substituted by 1 to 2 radicals Am; m is an integer ranging from 1 to 2;
Am radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group
 -(C1-
-(C1-
C6)alkylheteroaryl, -(CrC^alkylaryl, aryl, heteroaryl, heterocycle, -(C0-C6)alkyl-
OR1, -0-(C2-C(OaUCyI-OR1, -NR1(C2-C6)alkyl-OR2, -(Cs-C^cycloalkyHCr
C6)alkyl, -O-tCa-C^cycloalkyHCrC^alkyl, -NR1-(C3-C7)cycloalkyl-(C1- C6)alkyl, -(Q-C^alkylhalo-OR1, -(Q-C^alkylhalo-NR^2, -NR1-(C2-C6)alkyl-S-
R2, -(Co-CfOalkyl-S^-R1, -NR1-(Ci-C6)alkyl-S(=O)-R2, -(C0-C6)alkyl-S(=O)2-
R1, -NR1-(C1-C6)alkyl-S(=O)2-R2, -(Co-C^alkyl-N^R2, -NR^^-C^alkyl-
NR2R3, -(Co-C6)alkyl-S(=0)2NR1R2, -NR1-(C1-C6)alkyl-S(=O)2NR2R3, -(C0-
C6)alkyl-NR1-S(=O)2R2, -NR1-(C2-C6)alkyl-NR2-S(=O)2R3, -(C0-C6)alkyl- C(^O)-NR1R2, -NR1-(C1-C6)alkyl-C(=O)-NR2R3, -(C0-C6)alkyl-NR1C(=O)-R2, -
NR1-(C2-C6)alkyl-NR2C(=O)-R3, -NR^^a-C^alkyl-OC^O)^2, -(C0-C6)alkyl-
C(=O)-OR1, -NR1-(Ci-C6)alkyl-C(=O)-OR2, -(C0-C6)alkyl-C(=O)-R1, -NR^(C1-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
C6)alkyl-C(=O)-R2 5 -(C0-C6)alkyl-NR1-C(=O)-OR2, -(Co-C6)alkyl-NR1-C(=0> NR2R3, and -NR1-(C2-C6)alkyl-NR2-C(=O)-NR3R4;
Any two radicals of Am (A1 and A2) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; R1, R2, R3 and R4 are each independently hydrogen or an optionally substituted radical selected from the group of
 -(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(Ci.-C6)alkylaryl;
-(C1- C6)alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1- C6)alkylheteroaryl, aryl, heterocycle and -(Ci.-C6)alkylaryl;
Any two radicals of R (R1, R2, R3 or R4) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(CrC6)alkylhalo, -(C3-C7)cycloalkyl, -(CrC^alkylcyano, -(C1- C6)alkylheteroaryl, -(C^C^alkylaryl, aryl, heteroaryl, heterocycle, -(Co-C6)alkyl-
OR5, -O-(C2-C6)alkyl-OR5, -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(Cr C6)alkyl, -O-(C3-C7)cycloalkyl-(C1-C6)alkyl, -NR5-(C3-C7)cycloalkyl-(C1- C6)alkyl, -(Ci-C6)alkylhalo-OR5, -(Q-C^alkylhalo-NR^6, ~(C0-C6)alkyl-S-R5, - O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S-R6, -(C0-C6)alkyl-S(=O)-R5, -O-(Cr C6)alkyl-S(=O)-R5, -NR5-(C1-C6)alkyl-S(=O)-R6, -(C0-C6)alkyl-S(=O)2-R5, -O-
(C1-C6)alkyl-S(=O)2-R5, -NR5-(Ci-C6)alkyl-S(=O)2-R6, -(C0-C6)alkyl-NR5R6, -O- (C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-S(=O)2NR5R6, -O- (C!-C6)alkyl-S(=O)2NR5R6, -NR5-(Ci-C6)alkyl-S(=O)2NR6R7, -(C0-C6)alkyl- NR5-S(=O)2R6, -O-(C2-C6)alkyl-NR5-S(=O)2R6, -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(C1-C6)alkyl-C(=O)-NR5R6, -NR5-
(C1-C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl- NR5C(=O)-R6, -NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, - NR5-(C2-C6)alkyl-OC(=O)-R6, -(C0-C6)alkyl-C(=O)-OR5, -O-(d-C6)alkyl- C(=O)-OR5, -NR5-(C1-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -O-(Cr
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
C6)alkyl-C(=O)-R5, -NR^Q-C^alkyl-C^CO-R6, -(C0-C6)alkyl-NR5-C(=O)- OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NR5-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R 5 R 5 R and R each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo5 -(Ci-C^alkyl, -(C3-
C7)cycloalkyl, -(C4-C1o)alkylcycloalkyl and heterocycle;
Any two radicals of of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl; M2 is a hydrogen or an optionally substituted -(C1-Ce)alkyl-R9;
R9 is a hydrogen;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Q-C^alkyl-R11, -(C1-C6)alkylhalo, -(C2-C6)alkyl-NRnR12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRn; and R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of
 - (CrC^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C 10)alkylcycloalkyl, heteroaryl, -
- (CrC^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C 10)alkylcycloalkyl, heteroaryl, -
 provided that according to proviso (i): when M3 is -(C0)-Rn (that is when M3 is -R11), then R11 is not H; and provided that according to proviso (ii):
provided that according to proviso (i): when M3 is -(C0)-Rn (that is when M3 is -R11), then R11 is not H; and provided that according to proviso (ii):
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
pCT/IB2008/002243
when M1 is aryl, X2 is C, X3 is C5 X4 is N, then
 is not linked to X2; and provided that according to proviso (iv):
is not linked to X2; and provided that according to proviso (iv):
1 0
A and A radicals are not linked to form an imidazopyridazinyl ring; and provided that according to proviso (v):
when
 is and linked to X4 , X2 is S, X3 is C, X4 is C, n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- IH- indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bπ can not be a phenyl; and provided that according to proviso (vii): when X2 is S, X3 is C, X4 is C, n is 1, then A1 when linked to either X3 or X4 is not pyridyl; and provided that according to proviso (viii): when X2 is S5 X3 is C, X4 is C, to provide a thiazole ring, n is I5 then A1 is not an optionally substituted imidazolyl or triazolyl ring.
is and linked to X4 , X2 is S, X3 is C, X4 is C, n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- IH- indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then Bπ can not be a phenyl; and provided that according to proviso (vii): when X2 is S, X3 is C, X4 is C, n is 1, then A1 when linked to either X3 or X4 is not pyridyl; and provided that according to proviso (viii): when X2 is S5 X3 is C, X4 is C, to provide a thiazole ring, n is I5 then A1 is not an optionally substituted imidazolyl or triazolyl ring.
7. A compound according to claim 3 having the Formula (III), wherein: m is an integer ranging from 1 to 2;
Am radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, -(CrC6)alkylhalo5 -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano, heterocycle, -(Co-C^alkyl-OR1, -(C3-C7)cycloalkyl-(Ci-C6)alkyl, -NR^(C3- C^cycloalkyl-CCrCfOalkyl, -(Ci-C^alkylhalo-OR1, -(Q-CeOalkylhalo-NR^2,
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
(Co-C^alkyl-NR1^, -NR^Cs-CfOalkyl-NR^3, -(C0-C6)alkyl-C(=O)-NR1R2, -
 C6)alkyl-NR2C(=O)-R3, -(C0-C6)alkyl-C(=O)-R1 5 and
C6)alkyl-NR2C(=O)-R3, -(C0-C6)alkyl-C(=O)-R1 5 and
 R2; R1, R2 and R3 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Q-C^alkyl, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl and heterocycle,
R2; R1, R2 and R3 are each independently hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Q-C^alkyl, -(C3- C7)cycloalkyl, -(C4-C10)alkylcycloalkyl and heterocycle,
1 O 1X
Any two radicals of R (R , R or R ) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring; n is an integer ranging from 1 to 2;
Bn radicals are each independently selected from the group of hydrogen, halogen, -CN, -CF3, and an optionally substituted radical selected from the group Of -(C1- C6)alkyl, -(CrC6)alkylhalo, -(C3-C7)cycloalkyl, -(Ci-C6)alkylcyano, -(C1- C6)alkylheteroaryl, -(C1-C6)alkylaryl, aryl, heteroaryl, heterocycle, -(Co-C6)alkyl- OR5, -O-(C2-C6)alkyl-OR5, -NR5(C2-C6)alkyl-OR6, -(C3-C7)cycloalkyl-(Ci-
C6)alkyl, -O-(C3-C7)cycloalkyl-(C1-C6)alkyl,
 C6)alkyl, -(CrC^alkylhalo-OR5, -(CrC^alkylhalo-NR^6, -(C0-C6)alkyl-S-R5, - O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S-R6, -(C0-C6)alkyl-S(=O)-R5, -0-(C1- C6)alkyl-S(=O)-R5, -NR5-(Ci-C6)alkyl-S(=O)-R6, -(C0-C6)alkyl-S(=O)2-R5, -O- (CrC6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, -(C0-C6)alkyl-NR5R6, -O-
C6)alkyl, -(CrC^alkylhalo-OR5, -(CrC^alkylhalo-NR^6, -(C0-C6)alkyl-S-R5, - O-(C2-C6)alkyl-S-R5, -NR5-(C2-C6)alkyl-S-R6, -(C0-C6)alkyl-S(=O)-R5, -0-(C1- C6)alkyl-S(=O)-R5, -NR5-(Ci-C6)alkyl-S(=O)-R6, -(C0-C6)alkyl-S(=O)2-R5, -O- (CrC6)alkyl-S(=O)2-R5, -NR5-(C1-C6)alkyl-S(=O)2-R6, -(C0-C6)alkyl-NR5R6, -O-
(C2-C6)alkyl-NR5R6, -NR5-(C2-C6)alkyl-NR6R7, -(C0-C6)alkyl-S(=O)2NR5R6, -O- (C1-C6)alkyl-S(=O)2NR5R6, -NR5-(C1-C6)alkyl-S(=O)2NR6R7, -(C0-C6)alkyl- NR5-S(=O)2R6, -O-(C2-C6)atkyl-NR5-S(=O)2R6 5 -NR5-(C2-C6)alkyl-NR6- S(=O)2R7, -(C0-C6)alkyl-C(=O)-NR5R6, -O-(CrC6)alkyl-C(=O)-NR5R6, -NR5- (C1-C6)alkyl-C(=O)-NR6R7, -(C0-C6)alkyl-NR5C(=O)-R6, -O-(C2-C6)alkyl-
NR5C(=O)-R6, -NR5-(C2-C6)alkyl-NR6C(=O)-R7, -O-(C2-C6)alkyl-OC(=O)-R5, - NR5-(C2-C6)alkyl-OC(=O)-R6, -(C0-C6)alkyl-C(=O)-OR5, -O-(d-C6)alkyl- C(=O)-OR5, -NR5-(Ci-C6)alkyl-C(=O)-OR6, -(C0-C6)alkyl-C(=O)-R5, -0-(Ci- C6)alkyl-C(=O)-R5, -NR5-(Ci-C6)alkyl-C(=O)-R6, -(C0-C6)alkyl-NR5-C(=O)-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
OR6, -(C0-C6)alkyl-NR5-C(=O)-NR6R7, -O-(C2-C6)alkyl-NRs-C(=O)-NR6R7 and -NR5-(C2-C6)alkyl-NR6-C(=O)-NR7R8;
R 5 R 5 R and R each independently hydrogen or an optionally substituted radical selected from the group of -(CrC^alkylhalo, -(Ci-C6)alkyl5 -(C3- C7)cycloalkyl5 -(C4-C10)alkylcycloalkyl and heterocycle;
Any two radicals of of R (R5, R6, R7 or R8) may be taken together to form an optionally substituted 3 to 10 membered carbocyclic or heterocyclic ring;
M1 is selected from an optionally substituted aryl and heteroaryl;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Ci-C6)alkyl-Rπ 5 -(CrC6)alkylhalo,
-(C2-C6)alkyl-NRπR12 5 -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRπ;
R l and R1 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Q-C^alkyl, - (Ci-C^alkylcyano, -(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, - (C1-C6)alkylheteroaryl, aryl, heterocycle and -(C1-Ce)alkylaryl; provided that according to proviso (v):
when
 is , X3 is C, n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and IH- indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then B" can not be a phenyl; and provided that according to proviso (vii):
is , X3 is C, n is 1, A1 is H, then M1 can not be an optionally substituted aryl, 1 -methyl- lH-indazol-4-yl, lH-indazol-3-yl and IH- indazol-4-yl; and provided that according to proviso (vi): when M3 is 4-methylphenyl, then B" can not be a phenyl; and provided that according to proviso (vii):
A1 is not a pyridyl; and provided that according to proviso (viii):
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
when X3 is C to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
8. A compound according to claim 4 having the Formula (IIIA), wherein: A1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, -(C1- C6)alkylhalo, -(C3-C7)cycloalkyl and heterocycle; n is an integer ranging from 1 to 2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Ci-C^alkyl, -
(C1-C6)alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5, and -(C3-C7)cycloalkyl- (CrC6)alkyl; or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of -CF3 and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(CrC^alkylhalo, -(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(Ci-
C6)alkyl;
R5 is selected from the group of hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(CrC^alkyl, -(C3-C7)cycloalkyl, - (C4-Cio)alkylcycloalkyl and heterocycle; M1 is an optionally substituted aryl;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC^alkyl-R11, -(Ci-C6)alkylhalo, -(C2-C6)alkyl-NRπR12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRπ;
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of
 -
-
(C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(Q-C^alkylaryl;
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
provided that according to proviso (v):
when
 , X3 is C, n is 1, A1 is H, then M1 can not be 1- methyl-lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii): A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C, X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
, X3 is C, n is 1, A1 is H, then M1 can not be 1- methyl-lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii): A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C, X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
9. A compound according to claim 4 having the Formula (IIIA), wherein:
A1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Ci-C6)alkyl, -(Ci- C6)alkylhalo, -(C3-C7)cycloalkyl and heterocycle; n is an integer ranging from 1 to 2, and either; (a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Q-C^alkyl, - (Ci-C6)alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5, and -(C3-C7)cycloalkyl- (Ci-C6)alkyl; or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of -CF3 and an optionally substituted radical selected from the group of -(C1-
C6)alkyl, -(d-C^alkylhalo, -(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(Ci- C6)alkyl;
R5 is selected from the group of hydrogen or an optionally substituted radical selected from the group of -(C1-C6)alkylhalo, -(C1-C6)alkyl, -(C3-C7)cycloalkyl, - (C4-C ^alkylcy cloalkyl and heterocycle;
KAS/ClientDocs/Addex/SSlθSΛVOOl.FiiialSpec.lO.O? 2008
M1 is an optionally substituted heteroaryl;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC^alkyl-R11, -(Ct-C^alkylhalo, -(C2-C6)alkyl-NRnR12, -(C2-C6)alkyl-ORπ and -(C2-C6)alkyl-SRn;
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Ct-C^alkylhalo, -(Ci-C6)alkyl, - (C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(CrC^alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl; provided that according to proviso (v):
when
 is , X3 is C, n is 1, A1 is H, then M1 can not be 1- methyl-lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii):
is , X3 is C, n is 1, A1 is H, then M1 can not be 1- methyl-lH-indazol-4-yl, lH-indazol-3-yl and lH-indazol-4-yl; and provided that according to proviso (vii):
A1 is not a pyridyl; and provided that according to proviso (viii): when X3 is C, X4 is C, to provide a thiazole ring, n is 1, then A1 is not an optionally substituted imidazolyl or triazolyl ring.
10. A compound according to claim 4 having the Formula (IIIA), wherein:
A1 radical is hydrogen, n is an integer ranging from 1 to 2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, -CF3, -(C1- C6)alkyl and -(Ci-C^alkylhalo, or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of -CF3 and an optionally substituted radical selected from the group of -(C1- C6)alkyl and -(Ci-C6)alkylhalo;
KAS/ClientDocs/Addex/SS^.WOOl.FinalSpec.lO.O? 2008
M1 is an optionally substituted pyridyl;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Q-C^alkyl-R11, -(CrC^alkylhalo, -(C2-C6)alkyl-NR11R12 J -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRn; and R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of ~(Ci-C6)alkylhalo, -(Ct-C^alkyl, - (C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl5 heteroaryl, -(Ci-C^alkylheteroaryl, aryl, heterocycle and -(CrC^alkylaryl.
11. A compound according to claim 5 having the Formula (IIIB), wherein: n is an integer ranging from 1 to 2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of
 - (Ci-C^alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5 5 aryl, heteroaryl, and - (Cs-C^cycloalkyl-CCrC^alkyl; or
- (Ci-C^alkylhalo, -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5 5 aryl, heteroaryl, and - (Cs-C^cycloalkyl-CCrC^alkyl; or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of -CF3 and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(CrC^alkylhalo, -(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(Ci- C6)alkyl; R5 is selected from the group of hydrogen or an optionally substituted radical selected from the group of -(Ci-C6)alkylhalo, -(Q-C^alkyl, -(C3-C7)cycloalkyl, - (C4-C10)alkylcycloalkyl and heterocycle;
M1 is an optionally substituted aryl;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(Q-C^alkyl-R11, -(CrC^alkylhalo,
-(C2-C6)alkyl-NRπR12, -(C2-C6)alkyl-ORn and -(C2-C6)alkyl-SRu; and
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(d-C6)alkylhalo, -(Ci-C6)alkyL - (C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1-C6)alkylheteroaryl, aryl, heterocycle and -(Ci-C^alkylaryl.
12. A compound according to claim 5 having the Formula (IIIB), whereintn n is an integer ranging from 1 to 2, and either;
(a) n is 1 and B1 radical is selected from the group of hydrogen, halogen, -CF3, and an optionally substituted radical selected from the group of -(Q-C^alkyl, - (CrC6)alkylhalo5 -(C3-C7)cycloalkyl, -(C0-C6)alkyl-OR5, aryl, heteroaryl, and -
(C3-C7)cycloalkyl-(Ci-C6)alkyl; or
(b) n is 2, and B1 and B2 radicals are each independently selected from the group of -CF3 and an optionally substituted radical selected from the group of -(C1- C6)alkyl, -(Ci-C6)alkylhalo, -(C3-C7)cycloalkyl, and -(C3-C7)cycloalkyl-(Ci- C6)alkyl;
R5 is selected from the group of hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Ci~C6)alkyl, -(C3-C7)cycloalkyl, - (C4-C 10)alkylcycloalkyl and heterocycle;
M1 is an optionally substituted heteroaryl; M is an optionally substituted radical selected from the group of -(C3-
C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(C1-C6)alkyl-R11, -(Q-C^alkylhalo, -(C2-C6)alkyl-NRπR12 5 -(C2-C6)alkyl-ORn and -(Q-QOalkyl-SR11; and
R and R are selected from the group of a hydrogen or an optionally substituted radical selected from the group of -(Q-C^alkylhalo, -(Ci-C6)alkyl, - (C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(C1-C6)alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
13. A compound according to claim 5 having the Formula (IIIB), wherein: n is an integer ranging from 1 to 2, and either;
(a) n. 'is 1, and B1 radical is selected from the group of hydrogen, -CF3, -(C1- C6)alkyl and -(C1-C6)alkylhalo; or (b) n is 2, and B1 and B2 radicals are each independently selected from the group of -CF3 and an optionally substituted radical selected from the group of -(C1- C6)alkyl and -(Q-C^alkylhalo;
M1 is an optionally substituted pyridyl;
M3 is an optionally substituted radical selected from the group of -(C3- C7)cycloalkyl, aryl, heteroaryl, heterocycle, -(CrC6)alkyl-Rn, -(CrC^alkylhalo,
-(CrC^alkyl-NR11!^2, -(C2-C6)alkyl-OR11 and -(C2-C6)alkyl-SRU; and
R11 and R12 are selected from the group of a hydrogen or an optionally substituted radical selected from the group of
 -(Ci-C6)alkyl, - (C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Ci-C^alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl.
-(Ci-C6)alkyl, - (C3-C7)cycloalkyl, -(C4-C10)alkylcycloalkyl, heteroaryl, -(Ci-C^alkylheteroaryl, aryl, heterocycle and -(C1-C6)alkylaryl.
14. A compound according to claims 1 to 13, which can exist as optical isomers, wherein said compound is either the racemic mixture or one or both of the individual optical isomers.
15. A compound according to claims 1 to 2, wherein said compound is selected from:
3-(l,3-Dimethyl-lH-pyrazol-4-yl)-iV-methyl-N-phenyl-l,2,4-thiadiazol-5-amine 3-(l ,5-Dimethyl-lH-pyrazol-4-yl)-N-methyl-iV-phenyl-l ,2,4-thiadiazol-5-amine and a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an JV-oxide form thereof.
KAS/ClientDocs/Addex/SSlθS.WOOl.FinalSpec.lO.O? 2008
16. A compound according to claims 1 to 3, wherein said compound is selected from:
. N-(2-Fluoroρhenyl)-3-(l-(4-methoxybenzyl)^3-methyl-lHrpyrazol-4-yl)-l,2,4- thiadiazol-5-amine
N-(2-Fluorophenyl)-3-(l-(4-methoxybenzyl)-5-methyl-lH-pyrazol-4-yl)-l52,4- thiadiazol-5-amine
3 -(I -(4-Methoxybenzyl)-3 -methyl-1 H-pyrazol-4-yl)-N-(pyridin-2-yl)- 1 ,2,4- thiadiazol-5-amine
3 -(I -(4-Methoxybenzyl)-5 -methyl- 1 H-pyrazol-4-yl)-N-(ρyridin-2-yl)-l ,2,4- thiadiazol-5-amine
3-(l-(4-Methoxybenzyl)-li?-pyrazol-4-yl)-N-phenyl-l,2,4-thiadiazol-5-arnine 4-(l-(4-Methoxybenzyl)-3-methyl-lH-pyrazol-4-yl)-N-phenylthiazol-2-amine 4-(l -(4-Methoxybenzyl)-5 -methyl- 1 H-pyrazol-4-yl)-N-phenylthiazol-2-amine 3-(5-Methyl-l-phenyl-lH-pyrazol-4-yl)-N-phenyl-l,2,4-thiadiazol-5-amine 3-(3-Methyl-l-propyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine
4-(l -Benzyl- 1 H-pyrazol-4-yl)-N-phenylthiazol-2-amine 4-(l-Isopropyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine 4-(l -Ethyl- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine 4-(l-Methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine and a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV-oxide form thereof.
17. A compound according to claim 4, wherein said compound is selected from:
4-(l-Propyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine
N-(Benzo[d][l,3]dioxol-5-yl)-4-(l-propyl-lH-pyrazol-4-yl)thiazol-2-amine
N-(2-Fluorophenyl)-4-(l-propyl-lH-pyrazol-4-yl)thiazol-2-amine
4-(3 -Methyl- 1 -propyl- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-amine
4-(3-Methyl-l-propyl-lH-pyrazol-4-yl)-N-(6-methylpyridin-2-yl)thiazol-2- amine
N-(5-Chloropyridin-2-yl)-4-(3 -methyl- 1 -propyl- 1 H-pyrazol-4-yl)thiazol-2-amine
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.07 2008
N-(6-Methoxypyridin-3-yl)-4-(3-methyl-l-propyl-lH-pyrazol-4-yl)thiazol-2- amine
N-(2-Methoxyρyridin-3 -yl)-4-(3 -methyl- 1 -propyl- 1 H-pyrazol-4-yl)thiazol-2- amine
4-(l-Propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2- amine
4-(l -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(pyridin-2-yl)thiazol-2-aniine
N-(Pyridin-2-yl)-4-(l-(2,2}2-trifluoroethyl)-lH-pyrazol-4-yl)thiazol-2-amine and a pharmaceutically acceptable acid or base addition salt thereof, a stereochemical^ isomeric form thereof and an iV-oxide form thereof.
18. A compound according to claim 5, wherein said compound is selected from:
3-(l-Ethyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5-amine
3-(l-Ethyl-lH-pyrazol-4-yl)-N-(6-methoxypyridin-2-yl)-l,254-thiadiazol-5- amine
3 -( 1 -Ethyl- 1 H-pyrazol-4-y l)-N-(6-morpholinopyridin-2-yl)- 1 ,2,4-thiadiazol-5 - amine
3-(l-Ethyl-3-methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5-amine
3-(l-Proρyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,254-thiadiazol-5-amine
3-(l-(3-Fluorobenzyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine
3 -( 1 -(3 -Fluorobenzyl)- 1 H-pyrazol-4-yi)-N-phenyl- 1 ,2,4-thiadiazol-5 -amine
N-(Pyridin-2-yl)-3-(l-((tetrahydro-2H-ρyran-4-yl)methyl)-lH-ρyrazol-4-yl)-
1 ,2,4-thiadiazol-5-amine
3 -(I -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(pyridine-2-yl)- 1 ,2,4-thiadiazol-
5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine hydrochloride
3-(l-(Cycloproρylmethyl)-lH-pyrazol-4-yl)-N-(6-methylpyridin-2-yl)-l,2,4-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.10.072008
thiadiazol-5 -amine
3 -(I -(Cyclopropylmethyl)- lH-pyrazol-4-yl)-N-(6-ethylρyridin-2-yl)- 1 ,2,4- thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(6-fluoropyridin-2-yl)-l,2,4- thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(5-fluoropyridin-2-yl)-l,2,4- thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(6-methoxypyridin-2-yl)-l,2,4- thiadiazol-5-aniine
N-(6-Cyclobutoxypyridin-2-yl)-3-(l-(cyclopropylmethyl)-lH-pyrazol-4-yl)-
1 ,2,4-thiadiazol-5 -amine
3-(l-(Cyclopropylmethyl)-lH-pyrazol-4-yl)-N-(6-morpholinopyridin-2-yl)-
1 ,2,4-thiadiazol-5-amine
3-(l-(Cyclopropylmethyl)-lH-ρyrazol-4-yl)-N-(5-morpholinopyridin-2-yl)-
1 ,2,4-thiadiazol-5-amine
3 -(I -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(6-(trifluoromethyl)pyridin-2-yl)-
1 ,2,4-thiadiazol-5-amine
N-(5-Chloropyridin-2-yl)-3-(l-(cyclopropylmethyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5-amine
6-(3 -(I -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4-thiadiazol-5- ylamino)nicotinonitrile
3 -( 1 -(Cyclopropylmethyl)- 1 H-pyrazol-4-yl)-N-(pyrazin-2-yl)- 1 ,2,4-thiadiazol-5 - amine
6-(3-(l -(Cyclopropylmethyl)-lH-pyrazol-4-yl)-l ,2,4-thiadiazol-5- ylamino)nicotinamide
3-(l-(Cyclobutymiethyl)-lH-ρyrazol-4-yl)-N-(pyridin-2-yl)-l52,4-thiadiazol-5- amine
3 -(I -(2-Morpholinoethyl)- lH-pyrazol-4-yl)-N-(pyridin-2-yl)- 1 ,2,4-thiadiazol-5 - amine
N-(Pyridin-2-yl)-3-(l-((tetrahydrofuran-2-yl)methyl)-lH-pyrazol-4-yl)-l,2,4-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.072008
pcτ^oo oo 43
thiadiazol-5-amine
N-(Pyridin-2-yl)-3-(l-(2,2,2-trifluoroethyl)-lH-ρyrazol-4-yl)-l,254-thiadiazol-5- amine
3-(l-(2-Methoxyethyl)-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,2,4-thiadiazol-5- amine
2-(4-(5-(Pyridin-2-ylamino)-l,2s4-thiadiazol-3-yl)-lH-pyrazol-l-yl)ethanol
3-(l-Cyclobutyl-lH-pyrazol-4-yl)-TSf-(pyridin-2-yl)-l,2,4-tMadiazol-5-amine
3 -(I -(3 -Fluoro-4-methoxyphenyl)- lH-ρyrazol-4-yl)-N-(pyridin-2-yl)- 1 ,2,4- thiadiazol-5 -amine
3-(l-(4-Fluorophenyl)-5-methyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-l,254- thiadiazol-5-amine
3.(l.(4-Fluorophenyl)-5-methyl-lH-pyrazol-4-yl)-N-phenyl-l,2,4-thiadiazol-5- amine
3 -(I -Propyl-3 -(trifluoromethyl)- lH-pyrazol-4-yl)-N-(ρyridin-2-yl)- 1 ,2,4- thiadiazol-5-amine
N-(6-Methylpyridin-2-yl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-
1 ,2,4-thiadiazol-5 -amine
N-(6-Methoxypyridin-3 -yl)-3 -(I -propyl-3 -(trifluoromethyl)- lH-pyrazol-4-yl)-
152,4-thiadiazol-5-amine
N-(2-Methoxypyridin-3 -yl)-3 -(I -propyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)-
1 ,2,4-thiadiazol-5 -amine
N-(5-Chloropyridin-2-yl)-3 -(I -ρropyl-3-(trifluoromethyl)- 1 H-pyrazol-4-yl)-
1 ,2,4-thiadiazol-5 -amine
3-(l-Propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-N-(4-(trifluoromethyl)pyridin-
2-yl)-l ,2,4-thiadiazol-5-amine
N-(3 ,5 -Difluoropyridin-2-yl)-3-(l -propyl-3 -(trifluoromethyl)- lH-pyrazol-4-yl)-
1 ,2,4-thiadiazol-5 -amine
3 -(I -Proρyl-3 -(trifluoromethyl)- lH-pyrazol-4-yl)-N-(quinolin-2-yl)- 1 ,2,4- thiadiazol-5-amine
N-(2-Methylpyridin-4-yl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
1 ,2,4-thiadiazol-5-amine
N-(2-Fluorophenyl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-l52,4- thiadiazol-5 -amine
N-(255-Difluorophenyl)-3-(l-propyl-3-(trifluoroniethyl)-lH-pyrazol-4-yl)-l,2.4- thiadiazol-5 -amine
N-(Benzo[d][l,3]dioxol-5-yl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-
1 ;2,4-thiadiazol-5-amine
N-(4-Morpholinoρhenyl)-3 -(I -propyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)-
152,4-thiadiazol-5-aniine
N-(3 -Methoxyphenyl)-3 -( 1 -ρropyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)- 1 ,2,4- thiadiazol-5-amine
3 -( 1 -Propyl-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)-N-(3 -(trifluoromethyl)phenyl)-
1 ,2,4-thiadiazol-5 -amine
N-(4-Chlorophenyl)-3-(l-propyl-3-(trifluoromethyl)-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5-amine
3 -( 1 -(Cyclopropylmethyl)-3 -(trifluoromethyl)- 1 H-pyrazol-4-yl)-N~(pyridin-2- yl)- 1 ,2,4-thiadiazol-5-amine
N-(Pyridin-2-yl)-3 -(I -(2,2,2-trifluoroethyl)-3 -(trifluoromethyl)- 1 H-pyrazol-4- yl)- 1 ,2,4-thiadiazol-5-amine
3-(3,5-Dimethyl-l-propyl-lH-pyrazol-4-yl)-N-(ρyridin-2-yl)-l,254-thiadiazol-5- amine
3-(l-(Cyclopropylmethyl)-3,5-dimethyl-lH-pyrazol-4-yl)-N-(pyridin-2-yl)-
1 ,2,4-thiadiazol-5-amine
3 -( 1 -(Cyclopropylmethyl)-3 , 5 -dimethyl- 1 H-pyrazol-4-yl)-N-(6-methylpyridin-2- yl)- 1 ,2,4-thiadiazol-5-amine
3-(3-Methyl-l-propyl-lH-pyrazol-4-yl)-N-(6-methylpyridin-2-yl)-l,2,4- thiadiazol-5-amine
N-(6-Ethylpyridin-2-y l)-3 -(3 -methyl- 1 -propyl- 1 H-pyrazol-4-yl)- 1 ,2,4-thiadiazol-
5 -amine
3 -(3 -Methyl- 1 -propyl- 1 H-pyrazol-4-yl)-N-(6-(trifluoromethyl)pyridin-2-yl)-
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008
1 ,2,4-thiadiazol-5-amine
3 -(3 -Methyl- 1 -propyl- 1 H-pyrazol-4-yl)-N-(3 -(trifluoromethyl)pyridin-2-yl)-
1 ,2,4-thiadiazol-5-amine
N-(5-Chloroρyridin-2-yl)-3-(3-niethyl-l-proρyl-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5-amine
N-(5-Fluoropyridin-2-yl)-3 -(3-niethyl- 1 -propyl- 1 H-pyrazol-4-yl)- 1 ,2,4- thiadiazol-5-amine
6-(3 -(3 -Methyl-1 -propyl- 1 H-pyrazol-4-yl)- 1 ,2,4-thiadiazol-5- ylamino)nicotinonitrile
3 -(3 -Methyl- 1 -propyl- 1 H-pyrazol-4-yl)-N-(6-morpholinopyridin-2-yl)- 1 ,2,4- thiadiazol-5 -amine
N-(6-Methoxypyridin-2-yl)-3-(3-methyl-l-propyl-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5 -amine
N-(6-Fluoropyridin-2-yl)-3-(3-methyl-l-propyl-lH-pyrazol-4-yl)-l,2,4- thiadiazol-5-amine
3 -(3 -methyl- 1 -propyl- 1 H-pyrazol-4-y l)-N-(pyridin-3 -yl)- 1 ,2,4-thiadiazol-5 - amine and a pharmaceutically acceptable acid or base addition salt thereof, a stereochemically isomeric form thereof and an iV-oxide form thereof.
19. A pharmaceutical composition comprising a therapeutically effective amount of a compound according to claims 1 to 18 and a pharmaceutically acceptable carrier and/or excipient.
20. A method of treating or preventing a condition in a mammal, including a human, the treatment or prevention of which is affected or facilitated by the effect of A3 antagonists, comprising administering to a mammal in need of such treatment or prevention, an effective amount of a compound/composition according to claims 1 to 19.
KAS/ClientDocs/Addex/53195.WO01.FinalSpec.l0.07 2008
21. A method useful for treating or preventing ocular disorders such as ocular hypertension, glaucoma, normal tension glaucoma, neurodegenerative disease conditions of the retina and the optic nerve, retinal dystrophies, age-related Macular degeneration, and conditions of the eye such as conjunctivitis, keratoconjunctivitis sicca, and vernal conjunctivitis, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
22. A method useful for treating or preventing inflammatory or obstructive airway diseases, airway inflammation-related bronchial hyperractivity, asthma of whatever type, including non-allergic and allergic asthma (mild to severe), bronchitic asthma, exercice-induced asthma, occupational asthma and asthma induced following bacterial infection, morning dipping, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
23. A method useful for treating or preventing bronchitis of whatever type; aluminosis, antracosis, asbestosis, chalicosis, ptilosis, siderosis, silicosis, tabacosis and byssinosis, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
24. A method useful for treating or preventing eosinophil related disorders such as hypereosinophilia with eosinophil infiltration in the airways, eosinophilic oesophagitis, parasitic eosinophilia, inflammatory or allergic conditions of the skin, for example psoriasis, contact dermatitis, alopecia areata, erythema multiforma, scleroderma, atopic dermatis, urticaria, lupus erythematosus or epidermolysis bullosa acquisita, comprising administering to a mammalian
KAS/ClientDocs/Addex/53195. WOO 1.FinalSpec.10.072008
patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
25. A method useful for synergistically enhancing the chemotherapeutic treatment of tumors expressing adenosine A3 receptors comprising administering to a mammal in need thereof an effective amount of a high affinity adenosine A3 receptor antagonists either prior to, during or subsequent to administration of a chemotherapeutic cancer agent, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
26. A method useful for treating or preventing central nervous system disorders selected from the group consisting of anxiety disorders: Agoraphobia, Generalized Anxiety Disorder (GAD), Obsessive-Compulsive Disorder (OCD), Panic Disorder, Posttraumatic Stress Disorder (PTSD), Social Phobia, Other
Phobias, Substance-Induced Anxiety Disorder, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
27. A method useful for treating or preventing central nervous system disorders selected from the group consisting of mood disorders: Bipolar Disorders (I & II), Cyclothymic Disorder, Depression, Dysthymic Disorder, Major Depressive Disorder, Substance-Induced Mood Disorder, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
28. A method useful for treating or preventing central nervous system disorders selected from the group consisting of eating disorders including anorexia nervosa, bulimia, comprising administering to a mammalian patient in need of such
KAS/ClientDocs/Addex/53195. WOO 1.FinalSpec.10.072008
treatment an effective amount of a compound/composition according to claims 1 to 19.
29. A method useful for treating or preventing disorders including personality disorders such a borderline personality disorders; autism; ADHD; Tourette's syndrome; sexual disorders; migraine; diabetic neuropathy; obesity; addiction; sleep disorders; arthritis; chronic fatigue syndrome and irritable bowel syndrome, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
30. A method useful for treating or preventing inflammation and/or neurodegeneration, resulting from traumatic brain injury, stroke, hemorrhagic stroke, ischemia, spinal cord injury, cerebral hypoxia, cerebral haemorrhage or intracranial hematoma, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
31. A method useful for treating or preventing sensory, motor or cognitive symptoms resulting from traumatic brain injury, stroke, hemorrhagic stroke, ischemia, spinal cord injury, cerebral hypoxia, cerebral haemorrhage or intracranial haematoma, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
32. A method useful for treating or preventing ischemic cardiac diseases, including myocardial infarction, ischemic heart disease and related disease, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
33. A method useful for treating or preventing renal failure and consequences due to ischemia, comprising administering to a mammalian patient in need of such
KAS/Clienωocs/Addex/53195.WO01.FinalSpec.l0.072008
treatment an effective amount of a compound/composition according to claims 1 to 19.
34. A method useful for treating or preventing hepatic failure and consequences due to ischemia, comprising administering to a mammalian patient in need of such treatment an effective amount of a compound/composition according to claims 1 to 19.
35. Use of a compound according to claims 1 to 18 to prepare a tracer for imaging an adenosine A3 receptor.
36. Use of a compound according to claims 1 to 18 in the manufacture of a medicament for a treatment or prevention as defined in any of claims 20 to 34.
KAS/ClientDocs/Addex/53195. WOO 1.FinalSpec.10.072008
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0713687.2 | 2007-07-13 | ||
| GB0713687A GB0713687D0 (en) | 2007-07-13 | 2007-07-13 | New compounds 1 |
| GB0723344A GB0723344D0 (en) | 2007-11-28 | 2007-11-28 | Novel compounds 3 |
| GB0723344.8 | 2007-11-28 | ||
| GB0803103.1 | 2008-02-20 | ||
| GB0803103A GB0803103D0 (en) | 2008-02-20 | 2008-02-20 | Novel compounds 3 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009010871A2 true WO2009010871A2 (en) | 2009-01-22 |
| WO2009010871A3 WO2009010871A3 (en) | 2009-03-12 |
Family
ID=40089928
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2008/002243 WO2009010871A2 (en) | 2007-07-13 | 2008-07-11 | Pyrazole derivatives as antagonists of adenosine a3 receptor |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009010871A2 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7994185B2 (en) | 2008-05-06 | 2011-08-09 | Glaxo Smith Kline LLC | Benzene sulfonamide thiazole and oxazole compounds |
| WO2012068196A1 (en) * | 2010-11-18 | 2012-05-24 | Janssen Pharmaceutica Nv | Bis heteroaryl inhibitors of pro-matrix metalloproteinase activation |
| WO2012131031A1 (en) | 2011-04-01 | 2012-10-04 | H. Lundbeck A/S | New positive allosteric modulators of nicotinic acetylcholine receptor |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| CN103351339A (en) * | 2013-07-12 | 2013-10-16 | 雅本化学股份有限公司 | 3-fluorinated alkyl-1-substituted pyrazole-4-carboxylic acid and preparation method thereof |
| US8604033B2 (en) | 2010-01-15 | 2013-12-10 | Addex Pharma S.A. | Preparation of 4-amino-thiazoles and 3-amino-1,2,4-thiadiazoles and their use as allosteric modulators of metabotropic glutamate receptors |
| US9073907B2 (en) | 2009-01-12 | 2015-07-07 | Addex Pharma S.A. | Thiazoles derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors |
| US9334286B2 (en) | 2012-09-07 | 2016-05-10 | Cancer Research Technology Limited | Pharmacologically active compounds |
| US9371319B2 (en) | 2011-03-14 | 2016-06-21 | Cancer Research Technology Limited | Pyrrolopyridineamino derivatives as MPS1 inhibitors |
| WO2017054112A1 (en) * | 2015-09-28 | 2017-04-06 | 常州市卜弋科研化工有限公司 | Method of preparing 3-fluoroalkyl-1-methylpyrazole-4-carboxylic acid |
| WO2017194517A1 (en) * | 2016-05-10 | 2017-11-16 | Solvay Sa | Composition comprising 3-(haloalkyl or formyl)-1h-pyrazole-4-carboxylic acids or esters, its manufacture and its use for the preparation of carboxamides |
| CN108017584A (en) * | 2017-06-20 | 2018-05-11 | 南开大学 | A3The small molecular antagonists of adenosine receptor |
| WO2018180943A1 (en) * | 2017-03-27 | 2018-10-04 | Agc株式会社 | Method for producing halogen-containing pyrazol carboxylic acid and intermediate thereof |
| US10159712B2 (en) | 2013-08-13 | 2018-12-25 | Ostara Biomedical Ltd. | Embryo implantation |
| US10293029B2 (en) | 2015-01-27 | 2019-05-21 | Ostara Biomedical Ltd. | Embryo implantation |
| WO2019157317A1 (en) * | 2018-02-09 | 2019-08-15 | Astrocyte Pharmaceuticals, Inc. | Compounds and methods for treating addiction and related disorders |
| CN110198939A (en) * | 2017-01-20 | 2019-09-03 | 帕罗生物制药有限公司 | The regulator of adenosine A 3 receptor |
| US10588944B2 (en) | 2015-10-05 | 2020-03-17 | Ostara Biomedical Ltd. | Methods and compositions for managing reproduction |
| WO2020106773A1 (en) * | 2018-11-19 | 2020-05-28 | Saint Louis University | Treatment of irritable bowel syndrome |
| US11484545B2 (en) | 2016-04-21 | 2022-11-01 | Astrocyte Pharmaceuticals, Inc. | Compounds and methods for treating neurological and cardiovascular conditions |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11158073A (en) * | 1997-09-26 | 1999-06-15 | Takeda Chem Ind Ltd | Adenosine a3 antagonist |
| WO2001064674A1 (en) * | 2000-03-01 | 2001-09-07 | Janssen Pharmaceutica N.V. | 2,4-disubstituted thiazolyl derivatives |
| WO2007071348A1 (en) * | 2005-12-19 | 2007-06-28 | F. Hoffmann-La Roche Ag | Isoquinoline aminopyrazole derivatives, their manufacture and use as pharmaceutical agents for the treatment of cancer |
| WO2007076473A2 (en) * | 2005-12-23 | 2007-07-05 | Kalypsys, Inc. | Substituted pyrimidinyloxy ureas useful as inhibitors of protein kinases |
-
2008
- 2008-07-11 WO PCT/IB2008/002243 patent/WO2009010871A2/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11158073A (en) * | 1997-09-26 | 1999-06-15 | Takeda Chem Ind Ltd | Adenosine a3 antagonist |
| WO2001064674A1 (en) * | 2000-03-01 | 2001-09-07 | Janssen Pharmaceutica N.V. | 2,4-disubstituted thiazolyl derivatives |
| WO2007071348A1 (en) * | 2005-12-19 | 2007-06-28 | F. Hoffmann-La Roche Ag | Isoquinoline aminopyrazole derivatives, their manufacture and use as pharmaceutical agents for the treatment of cancer |
| WO2007076473A2 (en) * | 2005-12-23 | 2007-07-05 | Kalypsys, Inc. | Substituted pyrimidinyloxy ureas useful as inhibitors of protein kinases |
Non-Patent Citations (6)
| Title |
|---|
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; SHELKE, S. N. ET AL: "Synthesis of pyrazolyltriazoles and thiadiazoles by conventional and non-conventional methods" XP002507912 retrieved from STN Database accession no. 2006:1282971 & ORIENTAL JOURNAL OF CHEMISTRY , 22(2), 369-374 CODEN: OJCHEG; ISSN: 0970-020X, 2006, * |
| DATABASE REGISTRY [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 11 February 2000 (2000-02-11), XP002507907 retrieved from STN Database accession no. 255828-23-0 * |
| DATABASE REGISTRY [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 11 February 2000 (2000-02-11), XP002507908 retrieved from STN Database accession no. 255828-22-9 * |
| DATABASE REGISTRY [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 11 February 2000 (2000-02-11), XP002507909 retrieved from STN Database accession no. 255828-21-8 * |
| DATABASE REGISTRY [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 11 February 2000 (2000-02-11), XP002507910 retrieved from STN Database accession no. 255828-20-7 * |
| DATABASE REGISTRY [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 11 February 2000 (2000-02-11), XP002507911 retrieved from STN Database accession no. 255828-19-4 * |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8415345B2 (en) | 2008-05-06 | 2013-04-09 | Glaxo SmithKline LLC | Benzene sulfonamide thiazole and oxazole compounds |
| US7994185B2 (en) | 2008-05-06 | 2011-08-09 | Glaxo Smith Kline LLC | Benzene sulfonamide thiazole and oxazole compounds |
| US8642759B2 (en) | 2008-05-06 | 2014-02-04 | Glaxosmithkline Llc | Benzene sulfonamide thiazole and oxazole compounds |
| US9073907B2 (en) | 2009-01-12 | 2015-07-07 | Addex Pharma S.A. | Thiazoles derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors |
| US8604033B2 (en) | 2010-01-15 | 2013-12-10 | Addex Pharma S.A. | Preparation of 4-amino-thiazoles and 3-amino-1,2,4-thiadiazoles and their use as allosteric modulators of metabotropic glutamate receptors |
| WO2012068196A1 (en) * | 2010-11-18 | 2012-05-24 | Janssen Pharmaceutica Nv | Bis heteroaryl inhibitors of pro-matrix metalloproteinase activation |
| US9371319B2 (en) | 2011-03-14 | 2016-06-21 | Cancer Research Technology Limited | Pyrrolopyridineamino derivatives as MPS1 inhibitors |
| WO2012131031A1 (en) | 2011-04-01 | 2012-10-04 | H. Lundbeck A/S | New positive allosteric modulators of nicotinic acetylcholine receptor |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9895364B2 (en) | 2012-09-07 | 2018-02-20 | Cancer Research Technology Limited | Pharmacologically active compounds |
| US9334286B2 (en) | 2012-09-07 | 2016-05-10 | Cancer Research Technology Limited | Pharmacologically active compounds |
| US10188642B2 (en) | 2012-09-07 | 2019-01-29 | Cancer Research Technology Limited | Pharmacologically active compounds |
| CN103351339A (en) * | 2013-07-12 | 2013-10-16 | 雅本化学股份有限公司 | 3-fluorinated alkyl-1-substituted pyrazole-4-carboxylic acid and preparation method thereof |
| US10159712B2 (en) | 2013-08-13 | 2018-12-25 | Ostara Biomedical Ltd. | Embryo implantation |
| US10293029B2 (en) | 2015-01-27 | 2019-05-21 | Ostara Biomedical Ltd. | Embryo implantation |
| US10987406B2 (en) | 2015-01-27 | 2021-04-27 | Ostara Biomedical Ltd. | Embryo implantation |
| WO2017054112A1 (en) * | 2015-09-28 | 2017-04-06 | 常州市卜弋科研化工有限公司 | Method of preparing 3-fluoroalkyl-1-methylpyrazole-4-carboxylic acid |
| US10588944B2 (en) | 2015-10-05 | 2020-03-17 | Ostara Biomedical Ltd. | Methods and compositions for managing reproduction |
| US11484545B2 (en) | 2016-04-21 | 2022-11-01 | Astrocyte Pharmaceuticals, Inc. | Compounds and methods for treating neurological and cardiovascular conditions |
| WO2017194517A1 (en) * | 2016-05-10 | 2017-11-16 | Solvay Sa | Composition comprising 3-(haloalkyl or formyl)-1h-pyrazole-4-carboxylic acids or esters, its manufacture and its use for the preparation of carboxamides |
| CN110198939B (en) * | 2017-01-20 | 2023-03-28 | 帕罗生物制药有限公司 | Modulators of adenosine A3 receptors |
| CN110198939A (en) * | 2017-01-20 | 2019-09-03 | 帕罗生物制药有限公司 | The regulator of adenosine A 3 receptor |
| CN110461822A (en) * | 2017-03-27 | 2019-11-15 | Agc株式会社 | The manufacturing method of pyrazole carboxylic acid containing halogen and its intermediate |
| JPWO2018180943A1 (en) * | 2017-03-27 | 2020-02-06 | Agc株式会社 | Process for producing halogen-containing pyrazolecarboxylic acid and intermediates thereof |
| WO2018180943A1 (en) * | 2017-03-27 | 2018-10-04 | Agc株式会社 | Method for producing halogen-containing pyrazol carboxylic acid and intermediate thereof |
| CN108017584B (en) * | 2017-06-20 | 2021-03-23 | 南开大学 | A3Small molecule antagonists of adenosine receptors |
| CN108017584A (en) * | 2017-06-20 | 2018-05-11 | 南开大学 | A3The small molecular antagonists of adenosine receptor |
| WO2019157317A1 (en) * | 2018-02-09 | 2019-08-15 | Astrocyte Pharmaceuticals, Inc. | Compounds and methods for treating addiction and related disorders |
| US11839615B2 (en) | 2018-02-09 | 2023-12-12 | Astrocyte Pharmaceuticals, Inc. | Compounds and methods for treating addiction and related disorders |
| WO2020106773A1 (en) * | 2018-11-19 | 2020-05-28 | Saint Louis University | Treatment of irritable bowel syndrome |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009010871A3 (en) | 2009-03-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009010871A2 (en) | Pyrazole derivatives as antagonists of adenosine a3 receptor | |
| AU2006290814B2 (en) | 2-aniline-4-aryl substituted thiazole derivatives | |
| EP2730564B1 (en) | Heterocycle amines and uses thereof | |
| AU2003298693B2 (en) | Novel chemical compounds | |
| JP5063700B2 (en) | Anilinopiperazine derivatives and methods using anilinopiperazine derivatives | |
| CA2565599C (en) | Substituted 2-quinolyl-oxazoles useful as pde4 inhibitors | |
| JP4968860B2 (en) | Anilinopiperazine derivatives and methods using anilinopiperazine derivatives | |
| EP2590950B1 (en) | N-cyclyl-3-(cyclylcarbonylaminomethyl)benzamide derivatives as rho kinase inhibitors | |
| EP2181110A2 (en) | Novel heteroaromatic derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors | |
| KR102530580B1 (en) | Therapeutic compounds as inhibitors of the orexin-1 receptor | |
| US20070213338A1 (en) | Triazolo-Pyridazine Compounds and Derivatives Thereof Useful in the Treatment of Neuropathic Pain | |
| JP5205276B2 (en) | Enzyme inhibitor | |
| US20070249599A1 (en) | Novel Chemical Compounds | |
| SG172676A1 (en) | Antibacterial compositions | |
| KR20060030914A (en) | 3,5 disubstituted indazole compounds, pharmaceutical compositions, and methods for mediating or inhibiting cell proliferation | |
| JP2010013459A (en) | Use of oxy-indole derivative in treatment of dementia-associated disease, alzheimer disease and glycogen synthase kinase-3-associated symptom | |
| NZ526783A (en) | (Diazolo-pyridinyl)-pyrimidines for use in treatment of CNS disorders and diabetes | |
| CA3181415A1 (en) | Azalactam compounds as hpk1 inhibitors | |
| KR20240041987A (en) | Substituted pyridine derivatives as SARM1 inhibitors | |
| JP2008540442A (en) | New chemical compounds | |
| WO1996030350A1 (en) | Amidine derivatives | |
| US20090203692A1 (en) | Novel chemical compounds | |
| US20080261974A1 (en) | Novel Chemical Compounds | |
| WO2017051294A1 (en) | N-[2-(3-amino-2,5-dimethyl-1,1-dioxido-5,6-dihydro-2h-1,2,4-thiadiazin-5-yl)-1,3-thiazol-4-yl] amides useful as bace inhibitors | |
| EP4422753A1 (en) | Imidazolone derivatives as inhibitors of protein kinases in particular dyrk1a, clk1 and/or clk4 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08806945 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08806945 Country of ref document: EP Kind code of ref document: A2 |