WO2009010789A2 - Pyrimidine derivatives 934 - Google Patents
Pyrimidine derivatives 934 Download PDFInfo
- Publication number
- WO2009010789A2 WO2009010789A2 PCT/GB2008/050562 GB2008050562W WO2009010789A2 WO 2009010789 A2 WO2009010789 A2 WO 2009010789A2 GB 2008050562 W GB2008050562 W GB 2008050562W WO 2009010789 A2 WO2009010789 A2 WO 2009010789A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- formula
- group
- hydrogen
- hydroxy
- Prior art date
Links
- 0 CC*C(C)(C=C1)C=C(CCBCC2)C2=C1N(*)c1ccnc(NC2=*C(*)=IC(*)=*2)n1 Chemical compound CC*C(C)(C=C1)C=C(CCBCC2)C2=C1N(*)c1ccnc(NC2=*C(*)=IC(*)=*2)n1 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Definitions
- the present invention relates to novel pyrimidine derivatives, to pharmaceutical compositions containing these derivatives and to their use in therapy, in particular in the prevention and treatment of solid tumour disease in a warm blooded animal such as man.
- a cell may become cancerous by virtue of the transformation of a portion of its DNA into an oncogene i.e. a gene which, on activation, leads to the formation of malignant tumour cells (Bradshaw, Mutagenesis 1986, 1, 91).
- oncogenes give rise to the production of peptides which are receptors for growth factors. Activation of the growth factor receptor complex subsequently leads to an increase in cell proliferation.
- oncogenes encode tyrosine kinase enzymes and that certain growth factor receptors are also tyrosine kinase enzymes (Yarden et ah, Ann. Rev. Biochem., 1988, 57, 443; Larsen et ah, Ann. Reports in Med. Chem.. 1989, Chpt. 13).
- Receptor tyrosine kinases are important in the transmission of biochemical signals which initiate a variety of cell responses including proliferation, survival and migration.
- EGF epidermal growth factor
- Various classes of receptor tyrosine kinases are known (Wilks, Advances in Cancer Research, 1993, 60 43-73) based on families of growth factors which bind to different receptor tyrosine kinases.
- the classification includes Class I receptor tyrosine kinases comprising the EGF family of receptor tyrosine kinases such as the EGF, TGF ⁇ , Neu and erbB receptors, Class II receptor tyrosine kinases comprising the insulin family of receptor tyrosine kinases such as the insulin and IGFl receptors and insulin-related receptor (IRR) and Class III receptor tyrosine kinases comprising the platelet-derived growth factor (PDGF) family of receptor tyrosine kinases such as the PDGF ⁇ , PDGF ⁇ and colony-stimulating factor 1 (CSFl) receptors.
- EGF EGF family of receptor tyrosine kinases
- TGF ⁇ TGF ⁇
- Neu and erbB receptors Class II receptor tyrosine kinases comprising the insulin family of receptor tyrosine kinases such as the insulin and IGFl receptors and insulin-related receptor (IRR)
- Eph family is the largest known family of receptor tyrosine kinses, with 14 receptors and 8 cognate ephrin ligands in mammals (Reviewed in Kullander and Klein, Nature Reviews Molecular Cell Biology, 2002, 3_, 475-486).
- the receptor family is further sub-divided into two sub-families defined largely by homology of extracellular domains and affinity towards ligand type.
- all Ephs contain an intracellular tyrosine kinase domain and an extracellular Ig-like domain with a cysteine- rich region with 19 conserved cysteines and two fibronectin type III domains.
- EphAl-8 The A-class of Ephs consists of 8 receptors termed EphAl-8, which generally bind to their cognate ephrinA class of ligands termed ephrinAl-5.
- EphBl-6 The B-class consistents of 6 receptors termed EphBl-6, which bind to their cognate ephrinB ligands termed ephrinBl-3.
- Eph receptor ligands are unusual and differ to most other receptor tyrosine kinase ligands in that they are also tethered to cells, via a glycosylphosphatidylinositol linker in ephrinA ligands or an integral transmembrane region in ephrinB ligands.
- Binding of ephrin ligand to the Eph partner induces a conformational change within the Eph intracellular domain that enables phosphorylation of tyrosine residues within an auto-inhibitory juxtamembrane region, which relieves this inhibition of catalytic site and enables additional phosphorylation to stabilise the active conformation and generate more docking sites for downstream signalling effectors.
- Eph/ ephrin signalling can regulate other cell responses such as proliferation and survival.
- Eph receptor signalling may contribute to tumourigenesis in a wide variety of human cancers, either on tumour cells directly or indirectly via modulation of vascularisation.
- Eph receptors are over- expressed in various tumour types (Reviewed in Surawska et al., Cytokine & Growth Factor Reviews, 2004, 1_5, 419-433, Nakamoto and Bergemann, Microscopy Res and Technique, 2002, 59, 58-67); EphA2 and other EphA receptor levels are elevated in diverse tumours such as leukemias, breast, liver, lung, ovarian and prostate.
- expression of EphB receptors including EphB4 is up-regulated in tumours such as neuroblastomas, leukemias, breast, liver, lung and colon.
- EphA2 and EphB4 have indicated that over-expression of Eph receptors on cancer cells is able to confer tumourigenic phenotypes such as proliferation and invasion, consistent with the speculated role in oncogenesis.
- EphA2 over-expression in MCF-IOA mammary epithelial cells is sufficient to cause tumourigenesis (Zelinski et ah, Cancer Res., 2001, 61_, 2301-2306).
- Inhibition of EphA2 function with therapeutic antibodies (Coffman et ah, Cancer Res., 2003, 63, 7907-7912) or interfering-RNA (Landen et ah, Cancer Res.. 2005, 15, 6910-6918) has been demonstrated to inhibit tumour growth in in vivo xenograft models.
- EphA2 and EphB4 may contribute to tumour vascularisation
- EphB4 (Reviewed in Brantley-Sieders et ah, Current Pharmaceutical Design. 2004, JJ), 3431- 3442, Cheng et ah, Cytokine and Growth Factor Reviews. 2002, 13, 75-85).
- EphA2 and EphB4 are expressed on endothelial cells.
- Transgenic studies have shown that disruption of EphB4 (Gerety et ah, Molecular Cell, 1999, 4, 403-414) or its ligand ephrinB2 (Wang et ah, CeU, 1998, 93, 741-753) causes embryonic lethality associated with vascular modelling defects consistent with a critical role in vessel development.
- EphB4 activation stimulates endothelial cell proliferation and migration in vitro (Steinle et ah, J. Biol. Chem.. 2002, 277, 43830-43835).
- EphB4 signalling using soluble extracellular-domains of EphB4 have been shown to inhibit tumour growth and anagiogenesis in in vivo xenograft studies (Martiny-Baron et al., Neoplasia, 2004, 6, 248-257, Kertesz et ah, Blood, 2005, Pre -published online).
- soluble EphA2 inhibited tumour vascularisation in a variety of in vivo models (Brantley et ah, Oncogene, 2002, 21_, 7011-7026, Cheng et al, Neoplasia. 2003, 5, 445-456).
- an inhibitor of Eph receptors should be of value as a selective inhibitor of the proliferation and survival of tumour cells either targeted at tumour cells directly or via effects on tumour vascularisation.
- such inhibitors should be valuable therapeutic agents for the containment and/or treatment of tumour disease.
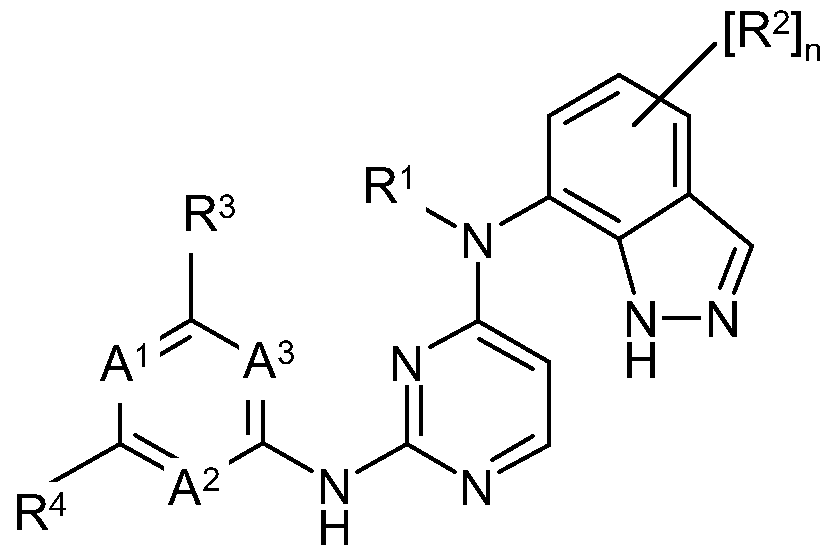
- R 1 is hydrogen or a (l-4C)alkyl group which is optionally substituted by one or more substituent groups selected from -OR 5 (wherein R 5 is selected from hydrogen or (l-2C)alkyl), cyano, halo, or -NR 6 R 7 (where R 6 and R 7 are independently selected from hydrogen, (l-2C)alkyl or (l-2C)alkanoyl);
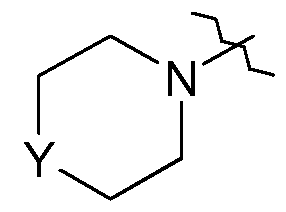
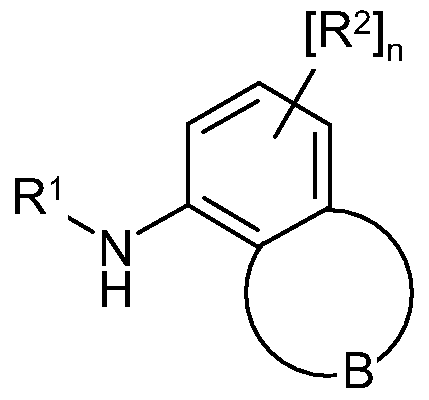
- ring B is a fused 5 or 6-membered carbocyclic or heterocyclic ring which is optionally substituted on a carbon atom by one or more halo groups or Ci_6alkyl groups, and where any nitrogen atoms in the ring are optionally substituted by a Ci_ 6 alkyl or Ci -6 alkylcarbonyl;
- n is 0, 1, 2 or 3 and each group R 2 is independently selected from halogeno, trifluoromethyl, cyano, nitro or a group of sub-formula (i) :
- X 1 is selected from a direct bond or O, S, SO, SO 2 , OSO 2 , NR 13 , CO, CH(OR 13 ), CONR 13 , N(R 13 )C0, SO 2 N(R 13 ), N(R 13 )SO 2 , C(R 13 ) 2 O, C(R 13 ) 2 S, C(R 13 ) 2 N(R 13 ) and N(R 13 )C(R 13 ) 2 , wherein R 13 is hydrogen or Ci -6 alkyl and
- R 11 is selected from hydrogen, Ci -6 alkyl, C 2-8 alkenyl, C 2-8 alkynyl, C 3-8 cycloalkyl, aryl or heterocyclyl, Ci -6 alkylC ⁇ .scycloalkyl, Ci -6 alkylaryl or Ci -6 alkylheterocyclyl, any of which may be optionally substituted with one or more groups selected from halogeno, trifluoromethyl, cyano, nitro, hydroxy, amino, carboxy, carbamoyl,
- R 3 is selected from: (i) hydrogen, halo, nitro, cyano, or hydroxy;
- W is selected from -0-, -S(0) p - (where p is O, 1 or 2),
- R b is selected from hydrogen or (l-2C)alkyl
- R 9 is selected from hydrogen or (l-4C)alkyl
- R 10 and R 1Oa are independently selected from hydrogen, or (l-2C)alkyl, or R 10 and R 1Oa are linked to form a 4, 5, 6 or 7 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 10 and R 1Oa are attached, one or two further heteroatoms selected from O, N or S, and wherein any S atoms that are present may be optionally oxidised to form an SO and SO 2 group, and wherein any carbon atom present in the ring is optionally substituted by oxo, halo,
- R 12 and R 12a are each independently selected from hydrogen or (l-6C)alkyl, or R 12 and R 12a are linked to form a 4, 5, 6 or 7-membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 12 and R 12a are attached, one or two further heteroatoms selected from O, N or S, and wherein any S atoms that are present may be optionally oxidised to form an SO and SO 2 group, and wherein any carbon atom present in the ring is optionally substituted by oxo, halo, hydroxy, cyano, (l-4C)alkyl, hydroxy(l-4C)alkyl, (l-4C)alkoxy, (l-2C)alkoxy-(l-4C)alkyl, (l-4C)alkanoyl, (l-4C)alkanesulfonyl, (l-4C)alkoxycarbonyl, (l-6
- X is selected from -O-, -S(O) P - (where p is 0, 1 or 2), -CO-, -NR C CO-, -CONR C -, -NR 0 COO-, and -NR 0 SO 2 -, where R° is selected hydrogen or (l-2C)alkyl; R 14 is a (l-4C)alkyl group which is optionally substituted by halo, hydroxy, cyano, (l-4C)alkoxy, or R 14 is
- R 15 and R 16 are independently selected from hydrogen, (l-2C)alkanoyl or (l-2C)alkyl, or R 15 and R 16 are linked to form a 4, 5, 6 or 7-membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 15 and R 16 are attached, one or two further heteroatoms selected from O, N or S, and wherein any S atoms that are present may be optionally oxidised to form an SO and SO 2 group, and wherein any carbon atom present in the ring is optionally substituted by oxo, halo, hydroxy, cyano, (l-4C)alkyl, hydroxy(l-4C)alkyl, (l-4C)alkoxy, (l-2C)alkoxy-(l-4C)alkyl, (l-4C)alkanoyl, (l-4C)alkanesulfonyl, (l-4C)alkoxycarbonyl, (l-6C)alkyla
- R 4 is a group -NR 17 R 18 , wherein R 17 and R 18 are linked to form a 4, 5, 6 or 7 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 17 and R 18 are attached, one or two further heteroatoms selected from O, N or S, and wherein any S atoms that are present may be optionally oxidised to form an SO or SO 2 group, and wherein any carbon atom present in the ring is optionally substituted by oxo, halo, hydroxy, cyano, (l-4C)alkyl, hydroxy(l-4C)alkyl, (l-4C)alkoxy,
- alkyl includes both straight-chain and branched-chain alkyl groups such as propyl, isopropyl and tert-butyl.
- references to individual alkyl groups such as "propyl” are specific for the straight-chain version only
- references to individual branched-chain alkyl groups such as “isopropyl” are specific for the branched-chain version only.
- (l-6C)alkoxy includes methoxy, ethoxy and isopropoxy
- (l-6C)alkylamino includes methylamino, isopropylamino and ethylamino
- di-[(l-6Calkyl]amino includes dimethylamino, diethylamino and N-methyl-N-isopropylamino
- alkenyl or alkynyl groups may be straight chain or branched.
- aryl refers to phenyl or naphthyl, particularly phenyl.
- halogen or halogeno includes fluoro, chloro, bromo, or iodo.
- heterocyclyl or “heterocyclic” refers to saturated, partially saturated or unsaturated, mono, bicyclic or tricyclic rings containing 3-15 atoms, of which at least one atom is chosen from nitrogen, sulphur or oxygen. These groups may, unless otherwise specified, be carbon or nitrogen linked. In addition, or a ring sulphur atom may be optionally oxidised to form the S-oxides. More particularly a “heterocyclyl” is a saturated, partially saturated or unsaturated, mono or bicyclic ring containing 3-12 atoms.
- Monocyclic rings suitably contain from 3-7 ring atoms, in particular 5 or 6 ring atoms.
- heterocyclyl examples and suitable values of the term "heterocyclyl” are thienyl, piperidinyl, morpholinyl, furyl, thiazolyl, pyridyl, imidazolyl, 1,2,4-triazolyl, thiomorpholinyl, coumarinyl, pyrimidinyl, phthalidyl, pyrazolyl, pyrazinyl, pyridazinyl, benzothienyl, benzimidazolyl, tetrahydrofuryl, [l,2,4]triazolo[4,3-a]pyrimidinyl, piperidinyl, indolyl, 1,3-benzodioxolyl and pyrrolidinyl, pyrrolyl, quinolinyl, isoquinolinyl, isoxazolyl, benzofuranyl, 1,2,3-thiadiazolyl, 1,2,5-thiadiazolyl, pyrimi
- Heterocyclyl groups may be non-aromatic or aromatic in nature. Aromatic heterocyclyl groups are referred to as heteroaryl. Heteroaryl groups are totally unsaturated, mono or bicyclic rings containing 3-12 atoms of which at least one atom is chosen from nitrogen, sulphur or oxygen, which may, unless otherwise specified, be carbon or nitrogen linked. Suitably “heteroaryl” refers to a totally unsaturated, monocyclic ring containing 5 or 6 atoms or a bicyclic ring containing 8 - 10 atoms of which at least one atom is chosen from nitrogen, sulphur or oxygen, which may, unless otherwise specified, be carbon or nitrogen linked.
- heteroaryl examples and suitable values of the term "heteroaryl” are thienyl, furyl, thiazolyl, pyrazolyl, isoxazolyl, imidazolyl, pyrrolyl, thiadiazolyl, isothiazolyl, triazolyl, pyranyl, indolyl, pyrimidyl, pyrazinyl, pyridazinyl, benzothienyl, pyridyl and quinolyl.
- novel compounds of the invention include, for example, compounds of Formula I, or pharmaceutically-acceptable salts thereof, wherein, unless otherwise stated, each of R 1 , n, R 2 , R 3 , R 4 , A 1 , A 2 , A 3 or B has any of the meanings defined hereinbefore or in paragraphs (1) to (45) hereinafter:-
- R 1 is (l-4C)alkyl
- R 1 is selected from hydrogen, methyl, ethyl, propyl, isopropyl, 2-methylpropyl or cyclopropylmethyl;
- R 1 is selected from hydrogen, methyl, ethyl, isopropyl or cyclopropylmethyl;
- R 1 is methyl
- R 1 is isopropyl
- R 1 is cyclopropylmethyl
- R 1 is ethyl; (8) R 1 is hydrogen;
- n 0, 1 or 2;
- n O or l
- n O
- n is i; (13) each R group present is independently selected from halogeno, trifluoromethyl, cyano, hydroxy, C2-salkenyl, C2-salkynyl and
- each R group present is independently selected from chloro, fiuoro, bromo, trifluoromethyl, cyano, hydroxy, methyl, ethyl, ethynyl, methoxy and ethoxy;
- each R 2 group present is halogeno
- each R group present is selected from bromo, chloro or fiuoro.
- each R 2 group present is chloro
- R 3 is selected from:
- an optionally substituted (l-6C)alkyl group wherein the optional substituents are selected from cyano, halo, or a group of sub-formula: wherein W is selected from -0-, -S(0) p - (where p is 0, 1 or 2), -CO-, -NR b CO-, or -CONR b -; R b is selected from hydrogen or (l-2C)alkyl; and R 9 is selected from hydrogen or (l-4C)alkyl; or -NR 10 R 10a where R 10 and R 1Oa are independently selected from hydrogen, (l-2C)alkanoyl or (l-2C)alkyl, or R 10 and R 1Oa are linked to form a 5, or 6 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 10 and R 1Oa are attached, one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon
- (l-4C)alkanesulfonyl and any available nitrogen atom present in the ring is optionally substituted by (l-4C)alkyl or (l-4C)alkanoyl;
- a group -NR 12 R 12a wherein R 12 and R 12a are each independently selected from hydrogen or (l-6C)alkyl, or R 12 and R 12a are linked to form a 5, 6 or 7-membered heterocyclic ring which comprises, in addition to the nitrogen atom to which R 12 and R 12a are attached, one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, (l-4C)alkyl, or (l-4C)alkanesulfonyl, and any available nitrogen atom present in the ring is optionally substituted by
- X is selected from -O-, -S(O) P - (where p is 0, 1 or 2), -CO-, -NR C CO-, -CONR C -, or -NR 0 COO-, where R° is selected hydrogen or (l-2C)alkyl;
- R 14 is a (l-4C)alkyl group which is optionally substituted by halo, hydroxy, cyano, (l-4C)alkoxy, or R 14 is
- R 15 and R 16 are linked to form a 5, or 6-membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 15 and R 16 are attached, one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, (l-4C)alkyl, or (l-4C)alkanesulfonyl, and any available nitrogen atom present in the ring is optionally substituted by (l-4C)alkyl or (l-4C)alkanoyl; (19) R 3 is selected from:
- W is selected from -O-, -S(O) P - (where p is 0, 1 or 2), -CO-, -NR b CO-, or -CONR b -;
- R b is selected from hydrogen or (l-2C)alkyl and R 9 is selected from hydrogen or (l-4C)alkyl; or -NR 10 R 10a , where R 10 and R 1Oa are independently selected from hydrogen or (l-2C)alkyl, or R 10 and R 1Oa are linked to form a 5 or 6 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 10 and R 1Oa are attached, one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, or (l-4C)alkyl, and any available nitrogen atom present in the ring is optionally
- the ring optionally comprises one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, or (l-4C)alkyl, and any available nitrogen atom present in the ring is optionally substituted by (l-4C)alkyl; or (iv) a group of formula (II):
- X is selected from -O-, -S(O) P - (where p is 0, 1 or 2), or -CONR C -, where R c is selected hydrogen or (l-2C)alkyl;
- R 14 is a (l-4C)alkyl group which is optionally substituted by halo, hydroxy, cyano, (l-4C)alkoxy; (20) R 3 is selected from:
- W is selected from -O-, -S(O) P - (where p is 0, 1 or 2), -CO-, -NR b CO-, or -CONR b -;
- R b is selected from hydrogen or (l-2C)alkyl and R 9 is selected from hydrogen or (l-4C)alkyl; or -NR 10 R 10a , where R 10 and R 1Oa are independently selected from hydrogen or (l-2C)alkyl), or R 10 and R 11 are linked to form a 5 or 6 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 10 and R 11 are attached, one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, or (l-4C)alkyl, and any available nitrogen atom present in the ring is optionally substituted by (
- the ring optionally comprises one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, or (l-4C)alkyl, and any available nitrogen atom present in the ring is optionally substituted by (l-4C)alkyl; or (iv) a group of formula (II):
- X is selected from -O-, -S(O) P - (where p is 0, 1 or 2), or -CONR C -, where R c is selected hydrogen or (l-2C)alkyl;
- R 14 is a (l-4C)alkyl group which is optionally substituted by halo, hydroxy, cyano, (l-4C)alkoxy;
- R 3 is a group -NR 12 R 12a , wherein R 12 and R 12a are each independently selected from hydrogen or (l-6C)alkyl, or R 12 and R 12a are linked to form a 5, 6 or 7-membered heterocyclic ring, and wherein, in addition to the nitrogen atom to which R 12 and R 12a are attached, the ring optionally comprises one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, (l-4C)alkyl, or (l-4C)alkanesulfonyl, and any available nitrogen atom present in the ring is optionally substituted by (l-4C)alkyl or
- R 3 is a group — NR 12 R 12a where R 12 and R 12a are linked to form a 6 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 12 and R 12a are attached, one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, or (l-4C)alkyl, and any available nitrogen atom is optionally substituted by (l-4C)alkyl, hydroxy(l-4C)alkyl or (l-4C)alkanoyl;
- R 3 is a group of formula:
- R 3 is a group of formula: wherein Y' is selected from O, NR y , or CR Z , where R y is selected from hydrogen, (l-2C)alkyl, hydroxy(l-2C)alkyl, (l-2C)alkoxy(l-2C)alkyl, or (l-2C)alkanoyl, and R z is selected from hydrogen, hydroxy, (l-2C)alkyl, hydroxy(l-2C)alkyl, (l-2C)alkoxy(l-2C)alkyl, or (l-2C)alkanoyl;
- R 3 is a group of formula: wherein Y' is selected from O, NR y , or CR Z , where R y is selected from hydrogen or (l-2C)alkyl, and R z is selected from hydrogen or hydroxy;
- R 3 is selected from morpholin-4yl, 4-methylpiperazin-l-yl, or 4-hydroxypiperidin-l-yl;
- R 3 is morpholin-4-yl
- R 3 is halo such as chloro
- R 3 is a 4-7 membered heterocyclic group which is linked via a carbon atom;
- R 3 is a 5-6 membered heterocyclic group which is linked via a carbon atom; (30) R 3 is a 5-6 membered heteroaryl group which is linked via a carbon atom;
- R 3 is selected from carbon linked pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, oxazolidinyl, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, isoxazolyl, oxazolyl, oxadiazolyl, thiadiazolyl, triazolyl, tetrazolyl, pyridinyl, pyrazinyl, pyridazinyl or pyrimidinyl;
- R 4 is a group -NR 17 R 18 , wherein R 17 and R 18 are linked to form a 5 or 6 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 17 and R 18 are attached, one or two further heteroatoms selected from O, N or S, and wherein any S atoms that are present may be optionally oxidised to form an SO or SO 2 group, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, (l-4C)alkyl, or (l-4C)alkanesulfonyl, and any available nitrogen atom is optionally substituted by (l-4C)alkyl, hydroxy(l-4C)alkyl, or (l-4C)alkanoyl;
- R 4 is a group -NR 17 R 18 , wherein R 17 and R 18 are linked to form a 6 membered heterocyclic ring which optionally comprises, in addition to the nitrogen atom to which R 17 and R 18 are attached, one or two further heteroatoms selected from O, N or S, and wherein the ring is optionally substituted on any available carbon atom by one or two substituent groups selected from oxo, halo, hydroxy, cyano, or (l-4C)alkyl, and any available nitrogen atom is optionally substituted by (l-4C)alkyl, hydroxy(l-4C)alkyl or (l-4C)alkanoyl; (34) R 4 is a group of formula:
- R 4 is a group of formula:
- Y is selected from O, NR y , or CR Z , where R y is selected from hydrogen or (l-2C)alkyl, and R z is selected from hydrogen or hydroxy;
- R 4 is selected from morpholin-4yl, 4-methylpiperazin-l-yl, or 4-hydroxypiperidin- 1 -yl;
- R 4 is morpholin-4-yl
- a 1 or A 2 is nitrogen and A 3 is CH; (39) A 2 or A 3 is CH;
- a 1 is nitrogen and A 2 and A 3 are CH;
- a 1 and A 3 are both nitrogen and A 2 is CH;
- a 2 and A 3 are both nitrogen and A 1 is CH;
- n is O or 1.
- B groups are set out below, and include for example groups
- B' as defined below.
- R 2 or R 2a groups are groups selected from halogeno, trifluoromethyl, cyano, hydroxy, Ci_ 6 alkyl, C 2 -salkenyl, C 2- salkynyl and Ci -6 alkoxy.
- R 2 or R 2a may be selected from chloro, fiuoro, bromo, trifluoromethyl, cyano, hydroxy, methyl, ethyl, ethynyl, methoxy and ethoxy.
- R 2 or R 2a is halogeno, such as bromo, chloro or fiuoro, and in particular chloro.
- n 1 and R 2 or R 2a is halogeno such as chloro.
- ring B include those made up of a group of formula:
- a group B includes more than one group R 20 or R 22 , at least one such group is hydrogen.
- groups R 20 include hydrogen, methyl, ethyl or methylcarbonyl, in particular hydrogen.
- groups R 22 include hydrogen, chloro, fluoro, methyl or ethyl, in particular hydrogen.
- ring B is a fused five-membered ring.
- R 20 include hydrogen, methyl, and acetyl.
- R 20 is hydrogen.
- Ring B includes one nitrogen atom.
- Ring B may also include two nitrogen atoms.
- a substituent R 3 is suitably positioned on the available ortho- carbon atom of the ring, forming a compound of formula (IA)
- R 2a is a group R 2 as defined above, and in particular is halogeno, and m is O, 1 or 2.
- m is O.
- examples of compounds of formula (I) are compounds of formula (IB)
- the invention provides a compound of formula (IC)
- a 1 , A 2 A 3 , R 1 , R 2 , R 3 , R 4 , n and R 20 in formula (IC) are as set out above in relation to formula (I).
- n is 0.
- the invention provides a compound of formula (ID)
- a 1 , A 2 A 3 , R 1 , R 2 , R 3 , R 4 n and R 20 in formula (ID) are as set out above in relation to formula (I).
- n is 0.
- the invention provides a compound of formula (IE), or a pharmaceutically acceptable salt thereof where:
- a 1 is N and A 2 and A 3 are both CH; A 1 and A 2 are both N and A 3 is CH; or A 2 is N and A 1 and A 3 are both CH;
- R 1 is hydrogen or a (l-4C)alkyl group;
- R is halogeno; and
- n is 0 or 1.
- the invention provides a compound of formula (IE) as defined above where A 1 is N and A 2 and A 3 are both CH.
- the invention provides a compound of formula (IE) as defined above where R 1 is a (l-4C)alkyl group, and in one particular embodiment R 1 is methyl.
- the invention provides a compound of formula (IE) as defined above where R 2 is chloro, and in one particular embodiment n is 1 and R 2 is chloro positioned on the available carbon atom in the ortho position relative to the amine linkage. In a further embodiment, the invention provides a compound of formula (IE) as defined above where n is 0.
- At least one of A 2 or A 3 is -CH-.
- a 1 is N and A 2 and A 3 are CH.
- a suitable pharmaceutically acceptable salt of a compound of the invention is, for example, an acid-addition salt of a compound of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulphuric, phosphoric, trifiuoroacetic, citric or maleic acid.
- a suitable pharmaceutically acceptable salt of a compound of the invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium or magnesium salt, an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation, for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine.
- an alkali metal salt for example a sodium or potassium salt
- an alkaline earth metal salt for example a calcium or magnesium salt
- an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation
- a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxye
- the invention relates to any and all tautomeric forms of the compounds of the formula (I) that possess EphB4 or EphA2 inhibitory activity.
- optically active forms may be carried out by standard techniques of organic chemistry well known in the art, for example by synthesis from optically active starting materials or by resolution of a racemic form.
- Suitable leaving groups L are halogeno such as chloro.
- the reaction is suitably carried out in an organic solvent such as a Ci_6alkanol, for instance, n-butanol, isopropanol or 2-pentanol, dimethylacetamide (DMA), or N-methylpyrrolidine (NMP) or mixtures thereof.
- An acid, and in particular an inorganic acid such as hydrochloric acid is suitably added to the reaction mixture.
- the reaction is suitably conducted at elevated temperatures for example at from 80-150 0 C, conveniently at the reflux temperature of the solvent.
- the reaction between (II) and (III) may be catalysed by transition metals complexes, such as palladium catalysts.
- Suitable palladium catalysts include Pd2(dba)3 (tris(dibenzylideneacetone)dipalladium), Pd(PPh 3 ) 4 and Pd(OAc) 2 .
- This palladium catalysed reaction conveniently carried out in the presence of a suitable base, such as potassium carbonate, cesium carbonate, potassium phosphate, sodium tert-butoxide, or l,8-diazabicyclo[5.4.0]undec-7-ene (DBU).
- Suitable solvents for such a reaction include toluene, dioxane or ethylene glycol dimethylether (DME).
- Suitable ligands for use in such a reaction include Xantphos (4,5-bis(diphenylphosphino)-9,9- -dimethylxanthene), BINAP (2,2'-bis(diphenylphosphino)-l,l '-binaphtyl) or DPPF (l,l '-bis(diphenylphosphino)ferrocene).
- the reaction is conveniently carried out at an elevated temperature, generally at the reflux temperature of the particular solvent used. A temperature of 90-140 0 C would be typical.
- a 1 , A 2 , A 3 , R 3 and R 4 are as defined in relation to formula I, with a suitable halogenating agent such as phosphorus oxychloride.
- a suitable halogenating agent such as phosphorus oxychloride.
- the reaction is conducted under reactions conditions appropriate to the halogenating agent employed. For instance, it may be conducted at elevated temperatures, for example of from 50-100 0 C, in an organic solvent such as acetonitrile or dichloromethane (DCM).
- the reaction is suitably effected in an organic solvent such as diglyme, again at elevated temperatures, for example from 120-180 0 C, and conveniently at the reflux temperature of the solvent.
- organic solvent such as diglyme
- compounds of formula I may be prepared by reacting a compound of formula (VII)
- any protecting groups can be removed using conventional methods, and if required, the compound of formula I can be converted to a different compound of formula I or a salt, again using conventional chemical methods.
- L 1 and L 2 are leaving groups such as halogen, and in particular chloro.
- the reaction is suitably effected in the presence of a base for example, an organic base such as triethylamine or N.N-diisopropylethylamine.
- a base for example, an organic base such as triethylamine or N.N-diisopropylethylamine.
- the reaction is also suitably carried out at an elevated temperature, for example between 80 and 120 0 C in a suitable organic solvent such as a C 1-6 alkanol, for instance, ethanol.
- the reaction can also be performed in presence of a strong base such as sodium hydride, in an organic solvent such as DMA.
- depressed temperatures for example from -20 0 C to 20 0 C, conveniently at about 0 0 C are suitably employed.
- L is a leaving group as defined hereinbefore and B and R and n are as defined in relation to Formula I with a compound
- R r -X where X is a suitable leaving group such as halogen and R 1 is as defined above in relation to Formula I but is other than hydrogen.
- R r -X where X is a suitable leaving group such as halogen and R 1 is as defined above in relation to Formula I but is other than hydrogen, and P is a suitable protecting group for this reaction, for example a 4-methoxybenzyl group.
- This reaction is conveniently performed using a strong base such as sodium hydride in a suitable solvent, for example dimethylformamide.
- a strong base such as sodium hydride in a suitable solvent, for example dimethylformamide.
- Such a reaction forms an example of a reaction in which a compound of formula (I) is converted to a different compound of formula (I), but there may be many other examples of suitable conversion reactions as would be apparent to a chemist.
- Another method for preparing compounds of formula I is to react a compound of formula (XI)
- This reaction is suitably carried out in the presence of a suitable catalyst such as a palladium catalyst.
- a suitable catalyst such as a palladium catalyst.
- suitable palladium catalysts include Pd2(dba)3 (tris(dibenzylideneacetone)dipalladium), Pd(PPh 3 ⁇ and Pd(OAc) 2 .
- This palladium catalysed reaction conveniently carried out in the presence of a suitable base, such as potassium carbonate, cesium carbonate, potassium phosphate, sodium tert-butoxide, or l,8-diazabicyclo[5.4.0]undec-7-ene (DBU).
- Suitable solvents for such a reaction include toluene, dioxane or ethylene glycol dimethylether (DME).
- Suitable ligands for use in such a reaction include Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene), BINAP (2,2'-bis(diphenylphosphino)-l,l '-binaphtyl) or DPPF (l,l '-bis(diphenylphosphino) ferrocene).
- the reaction is conveniently carried out at an elevated temperature, generally at the reflux temperature of the particular solvent used. A temperature of 90-140 0 C would be typical.
- Compounds of formula (VI) are also either known compounds or they can be prepared from known compounds using routine methods.
- aromatic substitution reactions include the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halogeno group.
- nucleophilic substitution reactions include the introduction of an alkoxy group or of a monoalkylamino group, a dialkyamino group or a N-containing heterocycle using standard conditions.
- reduction reactions include the reduction of a carbonyl group to a hydroxy group with sodium borohydride or of a nitro group to an amino group by catalytic hydrogenation with a nickel catalyst or by treatment with iron in the presence of hydrochloric acid with heating.
- a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl.
- the deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an acyl group such as a t-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulphuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate).
- a suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example dimethylaminopropylamine, or with hydrazine.
- a suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl.
- the deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a base such as sodium hydroxide
- a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- the protecting groups may be removed at any convenient stage in the synthesis using conventional techniques well known in the chemical art.
- Examples of the types of conversion reactions that may be used to convert a compound of formula (I) to a different compound of formula (I) include introduction of a substituent by means of an aromatic substitution reaction or of a nucleophilic substitution reaction, reduction of substituents, alkylation of substituents and oxidation of substituents.
- the reagents and reaction conditions for such procedures are well known in the chemical art.
- aromatic substitution reactions include the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under
- nucleophilic substitution reactions include the introduction of an alkoxy group or of a monoalkylamino group, a dialkyamino group or a N-containing heterocycle using standard conditions.
- reduction reactions include the reduction of a carbonyl group to a hydroxy group with sodium borohydride or of a nitro group to an amino group by catalytic hydrogenation with a nickel catalyst or by treatment with iron in the presence of hydrochloric acid with heating.
- a pharmaceutical composition which comprises a compound of the formula (I) and in particular a compound of formula (IA), (IB), (IC) or (IE), or a pharmaceutically acceptable salt or pro-drug thereof, as defined hereinbefore in association with a pharmaceutically-acceptable diluent or carrier.
- the composition may be in a form suitable for oral administration, for example as a tablet or capsule, for parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion) as a sterile solution, suspension or emulsion, for topical administration as an ointment or cream or for rectal administration as a suppository.
- parenteral injection including intravenous, subcutaneous, intramuscular, intravascular or infusion
- a sterile solution, suspension or emulsion for topical administration as an ointment or cream or for rectal administration as a suppository.
- the above compositions may be prepared in a conventional manner using conventional excipients.
- the compound of formula (I) will normally be administered to a warm-blooded animal at a unit dose within the range 5-5000 mg/m 2 body area of the animal, i.e. approximately 0.1-100 mg/kg, and this normally provides a therapeutically-effective dose.
- a unit dose form such as a tablet or capsule will usually contain, for example 1-250 mg of active ingredient.
- a daily dose in the range of 1-50 mg/kg is employed.
- the daily dose will necessarily be varied depending upon the host treated, the particular route of administration, and the severity of the illness being treated. Accordingly the optimum dosage may be determined by the practitioner who is treating any particular patient.
- the compounds of the present invention are expected to be useful in the treatment of diseases or medical conditions mediated alone or in part by EphB4 or EphA2, i.e. the compounds may be used to produce an EphB4 or EphA2 inhibitory effect in a warm-blooded animal in need of such treatment.
- the compounds of the present invention provide a method for treating the proliferation of malignant cells characterised by inhibition of EphB4 or EphA2, i.e. the compounds may be used to produce an anti-proliferative effect mediated alone or in part by the inhibition of EphB4 or EphA2.
- a method for producing an EphB4 or EphA2 inhibitory effect in a warm-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of the formula (I), or a pharmaceutically acceptable salt or pro-drug thereof, as defined hereinbefore.
- a method for producing an anti-angiogenic effect in a warm-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of the formula (I), (IA), (IB), (IC), (ID) or (IE), or a pharmaceutically acceptable salt or pro-drug thereof, as defined hereinbefore.
- a method of treating cancer in a warm-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of the formula (I), (IA), (IB), (IC), (ID) or (IE), or a pharmaceutically acceptable salt or pro-drug thereof, as defined hereinbefore.
- a method of treating neuroblastomas, breast, liver, lung and colon cancer or leukemias in a warm-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of the formula (I), or a pharmaceutically acceptable salt or pro-drug thereof, as defined hereinbefore.
- EphB4 or EphA2 inhibitory activity defined hereinbefore may be applied as a sole therapy or may involve, in addition to a compound of the invention, one or more other substances and/or treatments.
- Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate administration of the individual components of the treatment.
- the other component(s) of such conjoint treatment in addition to the anti-angiogenic treatment defined hereinbefore may be: surgery, radiotherapy or chemotherapy.
- Such chemotherapy may include one or more of the following categories of anti-tumour agents: (i) antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as alkylating agents (for example cis-platin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan and nitrosoureas); antimetabolites (for example antifolates such as fluoropyrimidines like 5-fiuorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside and hydroxyurea; antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine
- cytostatic agents such as antioestrogens (for example tamoxifen, toremifene, raloxifene, droloxifene and iodoxyfene), oestrogen receptor down regulators (for example fulvestrant), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and buserelin), progestogens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5 ⁇ -reductase such as finasteride;
- antioestrogens for example tamoxifen, toremifene, raloxifene, droloxifene and iodoxyfene
- Agents which inhibit cancer cell invasion for example metalloproteinase inhibitors like marimastat and inhibitors of urokinase plasminogen activator receptor function);
- inhibitors of growth factor function include growth factor antibodies, growth factor receptor antibodies (for example the anti-erbb2 antibody trastuzumab [HerceptinTM] and the anti-erbbl antibody cetuximab [C225]) , farnesyl transferase inhibitors, MEK inhibitors, tyrosine kinase inhibitors and serine/threonine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as 7V-(3-chloro-4-fluorophenyl)-7- methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib,), 7V-(3-ethynylphenyl)- 6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6-acrylamido-7V-
- growth factor antibodies for example the anti
- antiangiogenic agents such as those which inhibit the effects of vascular endothelial growth factor, (for example the anti-vascular endothelial cell growth factor antibody bevacizumab [AvastinTM], compounds such as those disclosed in International Patent Applications WO 97/22596, WO 97/30035, WO 97/32856 and WO 98/13354) and compounds that work by other mechanisms (for example linomide, inhibitors of integrin ⁇ v ⁇ 3 function and angiostatin); (vi) vascular damaging agents such as Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO00/40529, WO 00/41669, WO01/92224, WO02/04434 and WO02/08213;
- antisense therapies for example those which are directed to the targets listed above, such as ISIS 2503, an anti-ras antisense;
- gene therapy approaches including for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCAl or BRCA2, GDEPT (gene-directed enzyme pro-drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multi-drug resistance gene therapy;
- GDEPT gene-directed enzyme pro-drug therapy
- immunotherapy approaches including for example ex-vivo and in-vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interleukin 2, interleukin 4 or granulocyte -macrophage colony stimulating factor, approaches to decrease T-cell anergy, approaches using transfected immune cells such as cytokine-transfected dendritic cells, approaches using cytokine-transfected tumour cell lines and approaches using anti-idiotypic antibodies;
- cytokines such as interleukin 2, interleukin 4 or granulocyte -macrophage colony stimulating factor
- Cell cycle inhibitors including for example CDK inhibitiors (eg flavopiridol) and other inhibitors of cell cycle checkpoints (eg checkpoint kinase); inhibitors of aurora kinase and other kinases involved in mitosis and cytokinesis regulation (eg mitotic kinesins); and other histone deacetylase inhibitors; and
- a pharmaceutical composition comprising a compound of the formula (I) as defined hereinbefore and an additional anti-tumour substance as defined hereinbefore for the conjoint treatment of cancer.
- a pharmaceutical composition comprising a compound of the formula (I) as defined hereinbefore and an additional anti-tumour substance as defined hereinbefore for the conjoint treatment of cancer.
- a unit dose in the range for example, 1-100 mg/kg, preferably 1-50 mg/kg is envisaged.
- the compounds of formula (I), (IA), (IB), (IC), (ID) or (IE), and their pharmaceutically acceptable salts thereof are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of inhibitors of anti-angiogenic activity in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutic agents.
- Preparative HPLC was performed on Cl 8 reversed-phase silica, on a Phenomenex "Gemini" preparative reversed-phase column (5 microns silica, HOA, 21.1 mm diameter, 100 mm length) using decreasingly polar mixtures as eluent, for example decreasingly polar mixtures of water (containing 0.1% formic acid or 0.1% ammonia) as solvent A and acetonitrile as solvent B; either of the following preparative HPLC methods were used:
- Method A a solvent gradient over 9.5 minutes, at 25 mis per minute, from a 85:15 mixture of solvents A and B respectively to a 5:95 mixture of solvents A and B.
- Method B a solvent gradient over 9.5 minutes, at 25 mis per minute, from a 60:40 mixture of solvents A and B respectively to a 5:95 mixture of solvents A and B.
- 2,6-Dimorpholinopyridin-4-amine 140 mg, 0.53 mmol
- 2-chloro-N-(5- chlorobenzo[d][l,3]dioxol-4-yl)pyrimidin-4-amine 150 mg, 0.53 mmol
- 1,8- diazabicyclo-[5.4.0]-undec-7-ene (0.158 mL, 1.06 mmol)
- bis(dibenzylideneacetone)palladium(0) (45.5 mg, 0.08 mmol)
- 9,9-dimethyl-4,5- bis(diphenylphosphino)xanthene 92 mg, 0.16 mmol
- dioxane 3 mL
- the reaction was degassed, purged with nitrogen and heated at 120 0 C overnight.
- the reaction mixture was filtered off and washed thoroughly with dichloromethane.
- the filtrate was concentrated to dryness, diluted with dichloromethane (15 ml), washed with water (30 ml) and brine (15 ml), dried over magnesium sulfate and concentrated.
- the crude product was purified by flash chromatography on silica gel eluting with 0 to 50% ethyl acetate in dichloromethane.
- N4-(5-chlorobenzo[d] [1,3] dioxol-4-yl)-N2- (2,6-dimorpholinopyridin-4-yl)pyrimidine-2,4-diamine 80 mg, 29.6 %) as a pale beige foam.
- the 2,6-dimorpholinopyridin-4-amine used as starting material was made as follows: A mixture of 4-amino-2,6-dichloro-pyridine (900 mg, 5.52 mmol), morpholine (4.8 ml, 55.2 mmol) and DMA (1.0 ml, 11 mmol) was heated in a Personal Chemistry EMRYSTM Optimizer EXP microwave synthesisor at 240 0 C for 30 minutes. After cooling, morpholine was removed in vacuo, the residue was treated with 30% aqueous ammonium hydroxide and the resulting mixture was extracted with methylene chloride.
- 2,6-Dimorpholinopyridin-4-amine 160 mg, 0.60 mmol
- 2-chloro-N-(5- chlorobenzo[d][l,3]dioxol-4-yl)-N-methylpyrimidin-4-amine 180 mg, 0.60 mmol
- N4-(5-chlorobenzo[d][l,3]dioxol- 4-yl)-N2-(2,6-dimorpholinopyridin-4-yl)-N4-methylpyrimidine-2,4-diamine 80 mg, 25.2 %) as a pale yellow foam.
- N-(2-chloropyrimidin-4-yl)-l-(4-methoxybenzyl)-lH-indazol-4-amine 400 mg, 1.09 mmol
- 2,6-dimorpholinopyridin-4-amine (289 mg, 1.09 mmol) were reacted according to procedure of Example 1.
- TFA 8.2 mL
- anisole 0.594 mL, 5.47 mmol
- the reaction was heated to 130 0 C over a period of 30 minutes in a Personal Chemistry EMRYSTM Optimizer EXP microwave synthesisor.
- the reaction mixture was concentrated to dryness, diluted with dichloromethane (20 ml), washed with water (20 ml), brine (20 ml), dried over magnesium sulfate and concentrated.
- the crude product was purified by flash chromatography on silica gel eluting with 0 to 5% methanol in dichloromethane. The solvent was evaporated to dryness.
- the resulting solid was triturated with diethyl ether / petroleum ether (1/1), collected by filtration and dried under vacuum to give N2-(2,6-dimorpholinopyridin-4-yl)-N4-(lH-indazol-4- yl)pyrimidine-2,4-diamine (178 mg, 34.4 %) as a pale beige solid.
- N-(2-chloropyrimidin-4-yl)-l-(4-methoxybenzyl)-lH-indazol-4-amine used as starting material was made as follows: To a suspension of 4-nitro-lH-indazole (10 g, 61.30 mmol) and potassium carbonate (9.32 g, 67.43 mmol) in DMF (100 mL) at 25°C was added 4-methoxybenzyl chloride (9.14 mL, 67.43 mmol). The resulting mixture was stirred at 110 0 C for 2 hours. The reaction mixture was cooled and diluted with water. The aqueous layer was extracted with DCM (2 x 100 mL).
- N-(2-Chloropyrimidin-4-yl)-l-(4-methoxybenzyl)-N-methyl-lH-indazol-4-amine (304 mg, 0.80 mmol) the corresponding aminoheteroaryl (0.80 mmol), l,8-diazabicyclo-[5.4.0]- undec-7-ene (0.239 mL, 1.60 mmol), bis(dibenzylideneacetone)palladium(0) (69 mg, 0.12 mmol) and 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene (139 mg, 0.24 mmol) were suspended in dioxane (5 mL) and sealed into a microwave tube.
- the reaction mixture was degassed, purged with nitrogen and heated at 120 0 C overnight in an oil bath.
- the reaction mixture was allowed to cool to room temperature, filtered off and washed with dichloromethane.
- the filtrate was concentrated to dryness.
- the residue was dissolved in a solution of 20% water in trifluoroacetic acid (5 mL) and the reaction was heated to 75°C for 24 hours.
- the mixture was was concentrated to dryness, taken up in methanol (2.5 mL) - toluene (2.5 mL) and concentrated to dryness.
- N-(2-Chloropyrimidin-4-yl)- 1 -(4-methoxybenzyl)-N-methyl- 1 H-indazol-4-amine used as starting material was made as follows: Methyl iodide (1.021 mL, 16.40 mmol) was added dropwise to a stirred suspension of N- (2-chloropyrimidin-4-yl)-l-(4-methoxybenzyl)-l H-indazol-4-amine (4 g, 10.93 mmol, Example 3 starting material) and potassium carbonate (2.267 g, 16.40 mmol) in DMF (40 mL) at 0 0 C under nitrogen.
- the resulting suspension was stirred at 0 0 C for 15 minutes and was allowed to warm to room temperature.
- the reaction mixture was stirred at room temperature overnight, filtered off and washed with ethyl acetate.
- the filtrate was concentrated to dryness, diluted with dichloromethane (40 ml), washed with water (40 ml), brine (40 ml), dried over magnesium sulfate and concentrated.
- This compound was prepared from 4,6-dimorpholinopyrimidin-2-amine following the same procedure as Example 4, using potassium carbonate (20 eq.) instead of 1,8- diazabicyclo-[5.4.0]-undec-7-ene and toluene as the solvent.
- 4,6-dimorpholinopyrimidin-2-amine was prepared using the following procedure: 2-amino-4,6-dichloropyrimidine (1 g, 6.10 mmol) and morpholine (5.33 ml, 60.9 mmol) were dissolved in DMA (1.1 ml) and sealed into a microwave tube. The mixture was heated to 150 0 C over a period of 20 minutes in the microwave reactor. The reaction mixture was diluted with AcOEt, filtered and the filtrate concentrated to give an oil. The crude product was purified by flash chromatography on silica gel eluting with 0 to 4% methanol in dichlorome thane. The solvent was evaporated to dryness to afford 4,6- dimorpholinopyrimidin-2-amine (1.15 g, 71 %) as a white foam.
- 2-Methyl-6-(4-methylpiperazin-l-yl)pyrimidin-4-amine (71 mg), palladium acetate (1 mg), Xantphos (24 mg), caesium carbonate (166 mg) and 2-chloro-N-(5-chloro-l,3- benzodioxol-4-yl)pyrimidin-4-amine (117 mg) were dissolved in dioxane (4 ml) under nitrogen and heated in a microwave reactor at 15O 0 C for 60 minutes. The reaction was cooled and partitioned between ethyl acetate and water.
- the 2-methyl-6-(4-methylpiperazin-l-yl)pyrimidin-4-amine used as starting material was prepared as follows: 6-Amino-2-methylpyrimidin-4-ol (5 g) was suspended in phosphorous oxychloride (50 ml) and heated at 8O 0 C for 1 hour. A few drops of DMF was added and the reaction heated to 105 0 C for 3 hours to give an orange solution. The reaction was cooled, concentrated in vacuo and azeotroped with toluene. The residue was cautiously partitioned between ethyl 5 acetate and ice-cold saturated aqueous sodium bicarbonate solution.
- This assay detects inhibitors of EphB4-mediated phosphorylation of a polypeptide substrate using AlphascreenTM luminescence detection technology. Briefly, recombinant EphB4 was incubated with a biotinylated-polypeptide substrate (biotin-poly-GAT) in presence of magnesium-ATP. The reaction was stopped by addition of EDTA, together5 with streptavidin-coated donor beads which bind the biotin-substrate containing any phosphorylated tyrosine residues. Anti-phosphotyrosine antibodies present on acceptor beads bind to phosphorylated substrate, thus bringing the donor & acceptor beads into close proximity.
- biotinylated-polypeptide substrate biotin-poly-GAT
- streptavidin-coated donor beads which bind the biotin-substrate containing any phosphorylated tyrosine residues.
- Anti-phosphotyrosine antibodies present on acceptor beads bind to phosphorylated substrate
- Test compounds were prepared as 1OmM stock solutions in DMSO (Sigma- Aldrich Company Ltd, Gillingham, Dorset SP8 4XT Catalogue No.154938) and serially diluted with 5% DMSO to give a range of test concentrations at 6x the required final concentration. A 2 ⁇ l aliquot of each compound dilution was transferred to appropriate wells of low volume white 384-well assay plates (Greiner, Stroudwater Business Park, Stonehouse, Gloucestershire, GLlO 3SX, Cat No. 784075) in duplicate.
- Each plate also contained control wells: maximum signal was created using wells containing 2 ⁇ l of 5% DMSO, and minimum signal corresponding to 100% inhibition were created using wells containing 2 ⁇ l of 0.5M EDTA (Sigma- Aldrich Company Ltd, Catalogue No. E7889).
- Test compounds were prepared in 100% DMSO and dispensed in multiples of 2.5nl droplets into the target wells of the assay plate using a Labcyte Echo550 (Sunnyvale, California 94089, USA). To ensure that each well contained a total of 120nl DMSO the wells were all backfilled as required. Maximum control wells contained DMSO, minimum control wells contained 120nl of a compound at a concentration sufficient to completely inhibit enzyme activity. The test range of compounds was 10Ox the required final concentration. For the assay using aqueous prepared compounds, in addition to the compound or control, each well of the assay plate contained; lO ⁇ l of assay mix containing final buffer
- the assay mix was adjusted such that the final assay volume of 12ul contained the same concentration of reagent as lOul of assay mix used when aqueous compounds were tested.
- the reaction was stopped by addition of 5 ⁇ l/well stop buffer (1OmM Tris, 495mM EDTA, lmg/ml BSA) containing 0.25ng each of AlphaScreen anti-phosphoTyrosine-100 acceptor beads and streptavidin- coated donor beads (Perkin Elmer, Catalogue No 6760620M).
- the plates were sealed under natural lighting conditions, wrapped in aluminium foil and incubated in the dark for a further 20 hours.
- the resulting assay signal was determined on the Perkin Elmer EnVision plate reader. The minimum value was subtracted from all values, and the signal plotted against compound concentration to generate IC 50 data. The method used to generate the compound dilutions was recorded with the IC50 value in the database. Data from compounds prepared using acoustic dispensing were marked “Echo” and the remaining results were marked “Genesis”. Compounds of the invention were tested in the in vitro EphB4 enzyme assay and the IC50 values so obtained are presented in Table A below.
- the assay identifies inhibitors of cellular EphB4 by measuring a decrease in phosphorylation of EphB4 following treatment of cells with compound.
- the endpoint assay used a sandwich ELISA to detect EphB4 phosphorylation status. Briefly, Myc- tagged EphB4 from treated cell lysate was captured on the ELISA plate via an anti-c-Myc antibody. The phosphorylation status of captured EphB4 was then measured using a generic phosphotyrosine antibody conjugated to HRP via a colourimetric output catalysed by HRP, with level of EphB4 phosphorylation directly proportional to the colour intensity. Absorbance was measured spectrophotometrically at 450nm.
- Full length human EphB4 (Swiss-Prot Ace. No. P54760) was cloned using standard techniques from cDNA prepared from HUVEC using RT-PCR. The cDNA fragment was then sub-cloned into a pcDNA3.1 expression vector containing a Myc-His epitope tag to generate full-length EphB4 containing a Myc-His tag at the C-terminus (Invitrogen Ltd. Paisley, UK). CHO-Kl cells (LGC Promochem, Teddington, Middlesex, UK, Catalogue No. CCL-61) were maintained in HAM's F12 medium (Sigma-Aldrich Company Ltd, Gillingham, Dorset SP8 4XT, Catalogue No.
- EphB4-CHO CHO- Kl cells were engineered to stably express the EphB4-Myc-His construct using standard stable transfection techniques, to generate cells hereafter termed EphB4-CHO.
- EphB4-CHO cells were seeded into each well of Costar 96- well tissue-culture plate (Fisher Scientific UK, Loughborough, Leicestershire, UK., Catalogue No. 3598) and cultured overnight in full media. On day 2, the cells were incubated overnight in 90 ⁇ l/ well of media containing 0.1% Hyclone stripped-serum (Fisher Scientific UK, Catalogue No. SH30068.02). Test compounds were prepared as 1OmM stock solutions in DMSO (Sigma-Aldrich Company Ltd, Gillingham, Dorset SP8 4XT Catalogue No.154938) and serially diluted with serum-free media to give a range of test concentrations at 1Ox the required final concentration.
- Oxon OX14 3NB UK, Catalogue No. 496-EB a Fc-tagged form of the cognate ligand for EphB4
- cells were stimulated with clustered ephrin-B2 at a final concentration of 1 ⁇ g/ml for 20 minutes at 37°C to induce EphB4 phosphorylation.
- lysis buffer 25mM Tris HCl, 3mM EDTA, 3mM EGTA, 5OmM NaF, 2mM orthovanadate, 0.27M Sucrose, 1OmM ⁇ -glycerophosphate, 5mM sodium pyrophosphate, 2% Triton X-100, pH 7.4.
- ELISA plates were washed twice with PBS/0.05% Tween-20 and incubated with lOO ⁇ l/well cell lysate overnight at 4°C.
- ELISA plates were washed four times with PBS/0.05% Tween-20 and incubated for 1 hour at room temperature with lOO ⁇ l/well HRP-conjugated 4G10 anti- phosphotyrosine antibody (Upstate, Dundee Technology Park, Dundee, UK, DD2 ISW, Catalogue No. 16-105) diluted 1:6000 in 3% Top Block.
- ELISA plates were washed four times with PBS/0.05% Tween-20 and developed with lOO ⁇ l/well TMB substrate (Sigma- Aldrich Company Ltd, Catalogue No. T0440).
- This assay detects inhibitors of EphA2 -mediated phosphorylation of a polypeptide substrate using AlphascreenTM luminescence detection technology. Briefly, recombinant EphA2 was incubated with a biotinylated-polypeptide substrate (biotin-poly-GAT) in presence of magnesium-ATP. The reaction was stopped by addition of EDTA, together with streptavidin-coated donor beads which bind the biotin-substrate containing any phosphorylated tyrosine residues. Anti-phosphotyrosine antibodies present on acceptor beads bind to phosphorylated substrate, thus bringing the donor & acceptor beads into close proximity.
- biotinylated-polypeptide substrate biotin-poly-GAT
- streptavidin-coated donor beads which bind the biotin-substrate containing any phosphorylated tyrosine residues.
- Anti-phosphotyrosine antibodies present on acceptor beads bind to phosphorylated substrate
- Test compounds were prepared as 1OmM stock solutions in DMSO (Sigma- Aldrich Company Ltd, Gillingham, Dorset SP8 4XT Catalogue No.154938) and serially diluted with 5% DMSO to give a range of test concentrations at 6x the required final concentration. A 2 ⁇ l aliquot of each compound dilution was transferred to appropriate wells of low volume white 384-well assay plates (Greiner, Stroudwater Business Park, Stonehouse, Gloucestershire, GLlO 3SX, Cat No. 784075) in duplicate.
- Each plate also contained control wells: maximum signal was created using wells containing 2 ⁇ l of 5% DMSO, and minimum signal corresponding to 100% inhibition were created using wells containing 2 ⁇ l of 0.5M EDTA (Sigma- Aldrich Company Ltd, Catalogue No. E7889).
- each well of the assay plate contained; lO ⁇ l of assay mix containing final buffer (1OmM Tris, lOO ⁇ M EGTA, 1OmM magnesium acetate, 4 ⁇ M ATP, 500 ⁇ M DTT, lmg/ml BSA), 0.5ng of recombinant active EphA2 (amino acids 591-976; Swiss-Prot Ace. No. P29317) (ProQinase GmbH, Breisacher Str.
- the reaction was stopped by addition of 5 ⁇ l/well stop buffer (1OmM Tris, 495mM EDTA, lmg/ml BSA) containing 0.25ng each of AlphaScreen anti-phosphoTyrosine-100 acceptor beads and streptavidin-coated donor beads (Perkin Elmer, Catalogue No 6760620M).
- the plates were sealed under natural lighting conditions, wrapped in aluminium foil and incubated in the dark for a further 20 hours.
- the resulting assay signal was determined on the Perkin Elmer EnVision plate reader. The minimum value was subtracted from all values, and the signal plotted against compound concentration to generate IC50 data. Examples 9 and 10 of the invention were tested in the in vitro EphA2 enzyme assay and the IC50 values so obtained were 0.131 and 0.566 ⁇ M respectively.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description



































Claims









Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010516595A JP2010533700A (en) | 2007-07-16 | 2008-07-14 | Pyrimidine derivatives |
| MX2010000658A MX2010000658A (en) | 2007-07-16 | 2008-07-14 | Pyrimidine derivatives 934. |
| BRPI0814821-0A2A BRPI0814821A2 (en) | 2007-07-16 | 2008-07-14 | COMPOUND, PHARMACEUTICAL COMPOSITION, AND, PROCESS TO PREPARE A COMPOUND |
| EP08776197A EP2183242A2 (en) | 2007-07-16 | 2008-07-14 | Pyrimidine derivatives 934 |
| CN200880105690A CN101796046A (en) | 2007-07-16 | 2008-07-14 | Pyrimidine derivatives 934 |
| AU2008277446A AU2008277446A1 (en) | 2007-07-16 | 2008-07-14 | Pyrimidine derivatives 934 |
| EA201000101A EA201000101A1 (en) | 2007-07-16 | 2008-07-14 | DERIVATIVES OF PYRIMIDINE 934 |
| CA2693880A CA2693880A1 (en) | 2007-07-16 | 2008-07-14 | Pyrimidine derivatives 934 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07301236.1 | 2007-07-16 | ||
| EP07301236 | 2007-07-16 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009010789A2 true WO2009010789A2 (en) | 2009-01-22 |
| WO2009010789A3 WO2009010789A3 (en) | 2009-05-07 |
Family
ID=40260140
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2008/050562 WO2009010789A2 (en) | 2007-07-16 | 2008-07-14 | Pyrimidine derivatives 934 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US7718653B2 (en) |
| EP (1) | EP2183242A2 (en) |
| JP (1) | JP2010533700A (en) |
| KR (1) | KR20100042272A (en) |
| CN (1) | CN101796046A (en) |
| AU (1) | AU2008277446A1 (en) |
| BR (1) | BRPI0814821A2 (en) |
| CA (1) | CA2693880A1 (en) |
| CO (1) | CO6260075A2 (en) |
| CR (1) | CR11220A (en) |
| DO (1) | DOP2010000022A (en) |
| EA (1) | EA201000101A1 (en) |
| EC (1) | ECSP109958A (en) |
| MX (1) | MX2010000658A (en) |
| NI (1) | NI201000011A (en) |
| SV (1) | SV2010003459A (en) |
| WO (1) | WO2009010789A2 (en) |
| ZA (1) | ZA201000107B (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9986898B2 (en) | 2013-04-18 | 2018-06-05 | Ankon Technologies Co., Ltd | Apparatus and method for controlling movement of a capsule endoscope in digestive tract of a human body |
| US10226461B2 (en) | 2014-01-30 | 2019-03-12 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103003264B (en) | 2010-05-21 | 2014-08-06 | 切米利亚股份公司 | Novel pyrimidine derivatives |
| EP2688883B1 (en) | 2011-03-24 | 2016-05-18 | Noviga Research AB | Pyrimidine derivatives |
| CN111499580A (en) | 2011-04-22 | 2020-08-07 | 西格诺药品有限公司 | Substituted diaminocarboxamides and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| WO2012150293A1 (en) | 2011-05-03 | 2012-11-08 | Scholz Peter-Dominik | Arrangement and method for contactless energy transmission with a coupling-minimized matrix of planar transmission coils |
| WO2016100308A1 (en) | 2014-12-16 | 2016-06-23 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-jun n-terminal kinase in skin |
| EP3233809B1 (en) | 2014-12-16 | 2021-04-21 | Signal Pharmaceuticals, LLC | Formulations of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methycyclohexylamino)-pyrimidine-5-carboxamide |
| EP3250557A4 (en) | 2015-01-29 | 2018-06-20 | Signal Pharmaceuticals, LLC | Isotopologues of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10252981B2 (en) | 2015-07-24 | 2019-04-09 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| CN106543089A (en) * | 2016-11-04 | 2017-03-29 | 山东铂源药业有限公司 | A kind of synthetic method of Dasatinib intermediate |
| AU2021280113B2 (en) * | 2020-05-29 | 2023-12-21 | Nanjing Chia Tai Tianqing Pharmaceutical Co., Ltd. | Pyrimidine compound as AXL inhibitor |
| CN116178434A (en) * | 2021-11-26 | 2023-05-30 | 南京正大天晴制药有限公司 | Mono-p-toluene sulfonate and crystal forms of AXL kinase inhibitor |
| CN116178433A (en) * | 2021-11-26 | 2023-05-30 | 南京正大天晴制药有限公司 | Salts of AXL kinase inhibitors, methods of preparation and use thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004080980A1 (en) * | 2003-03-14 | 2004-09-23 | Novartis Ag | 2, 4- di (phenylamino) pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2005016894A1 (en) * | 2003-08-15 | 2005-02-24 | Novartis Ag | 2, 4-pyrimidinediamines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2006021454A2 (en) * | 2004-08-27 | 2006-03-02 | Novartis Ag | Pyrimidine derivatives |
| WO2006129100A1 (en) * | 2005-06-03 | 2006-12-07 | Glaxo Group Limited | Novel compounds |
| WO2007028445A1 (en) * | 2005-07-15 | 2007-03-15 | Glaxo Group Limited | 6-indolyl-4-yl-amino-5-halogeno-2-pyrimidinyl-amino derivatives |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU3704101A (en) * | 2000-02-17 | 2001-08-27 | Amgen Inc | Kinase inhibitors |
| JP4495465B2 (en) | 2002-03-05 | 2010-07-07 | アストラゼネカ・アクチエボラーグ | Alkylammonium salts of omeprazole and esomeprazole |
| US7521457B2 (en) | 2004-08-20 | 2009-04-21 | Boehringer Ingelheim International Gmbh | Pyrimidines as PLK inhibitors |
| CN101155799A (en) | 2005-03-16 | 2008-04-02 | 塔格根公司 | Pyrimidine inhibitors of kinases |
| PE20070362A1 (en) | 2005-07-15 | 2007-04-23 | Glaxo Group Ltd | COMPOUNDS DERIVED FROM INDAZOLE-4-IL-2,4-PYRIMIDINDIAMINE AS INHIBITORS OF TYROSINE KINASE (KINASE Syk) |
| US20090149467A1 (en) | 2005-09-15 | 2009-06-11 | Merck & Co., Inc. | Tyrosine Kinase Inhibitors |
| BR122021011787B1 (en) * | 2005-11-01 | 2022-01-25 | Impact Biomedicines, Inc | Biaryl metapyrimidine kinase inhibitors, pharmaceutical composition and process for preparing a pharmaceutical composition |
| TW200736232A (en) | 2006-01-26 | 2007-10-01 | Astrazeneca Ab | Pyrimidine derivatives |
| WO2007085540A1 (en) | 2006-01-27 | 2007-08-02 | Glaxo Group Limited | 1h-indaz0l-4-yl-2 , 4-pyrimidinediamine derivatives |
| TW200840581A (en) * | 2007-02-28 | 2008-10-16 | Astrazeneca Ab | Novel pyrimidine derivatives |
| US7947698B2 (en) * | 2007-03-23 | 2011-05-24 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| WO2008132505A1 (en) | 2007-04-27 | 2008-11-06 | Astrazeneca Ab | N' - (phenyl) -n- (morpholin-4-yl-pyridin-2-yl) -pyrimidine-2, 4-diamine derivatives as ephb4 kinase inhibitors for the treatment of proliferative conditions |
-
2008
- 2008-07-14 BR BRPI0814821-0A2A patent/BRPI0814821A2/en not_active IP Right Cessation
- 2008-07-14 CN CN200880105690A patent/CN101796046A/en active Pending
- 2008-07-14 AU AU2008277446A patent/AU2008277446A1/en not_active Abandoned
- 2008-07-14 KR KR1020107002514A patent/KR20100042272A/en not_active Application Discontinuation
- 2008-07-14 JP JP2010516595A patent/JP2010533700A/en active Pending
- 2008-07-14 MX MX2010000658A patent/MX2010000658A/en active IP Right Grant
- 2008-07-14 CA CA2693880A patent/CA2693880A1/en not_active Abandoned
- 2008-07-14 EA EA201000101A patent/EA201000101A1/en unknown
- 2008-07-14 WO PCT/GB2008/050562 patent/WO2009010789A2/en active Application Filing
- 2008-07-14 EP EP08776197A patent/EP2183242A2/en not_active Withdrawn
- 2008-07-16 US US12/174,339 patent/US7718653B2/en not_active Expired - Fee Related
-
2010
- 2010-01-06 ZA ZA201000107A patent/ZA201000107B/en unknown
- 2010-01-15 DO DO2010000022A patent/DOP2010000022A/en unknown
- 2010-01-15 CR CR11220A patent/CR11220A/en not_active Application Discontinuation
- 2010-01-15 NI NI201000011A patent/NI201000011A/en unknown
- 2010-01-15 SV SV2010003459A patent/SV2010003459A/en not_active Application Discontinuation
- 2010-02-11 EC EC2010009958A patent/ECSP109958A/en unknown
- 2010-02-16 CO CO10017544A patent/CO6260075A2/en not_active Application Discontinuation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004080980A1 (en) * | 2003-03-14 | 2004-09-23 | Novartis Ag | 2, 4- di (phenylamino) pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2005016894A1 (en) * | 2003-08-15 | 2005-02-24 | Novartis Ag | 2, 4-pyrimidinediamines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2006021454A2 (en) * | 2004-08-27 | 2006-03-02 | Novartis Ag | Pyrimidine derivatives |
| WO2006129100A1 (en) * | 2005-06-03 | 2006-12-07 | Glaxo Group Limited | Novel compounds |
| WO2007028445A1 (en) * | 2005-07-15 | 2007-03-15 | Glaxo Group Limited | 6-indolyl-4-yl-amino-5-halogeno-2-pyrimidinyl-amino derivatives |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9986898B2 (en) | 2013-04-18 | 2018-06-05 | Ankon Technologies Co., Ltd | Apparatus and method for controlling movement of a capsule endoscope in digestive tract of a human body |
| US10226461B2 (en) | 2014-01-30 | 2019-03-12 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US10517873B2 (en) | 2014-01-30 | 2019-12-31 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US11241430B2 (en) | 2014-01-30 | 2022-02-08 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2693880A1 (en) | 2009-01-22 |
| CN101796046A (en) | 2010-08-04 |
| CR11220A (en) | 2010-05-20 |
| EP2183242A2 (en) | 2010-05-12 |
| AU2008277446A1 (en) | 2009-01-22 |
| US7718653B2 (en) | 2010-05-18 |
| US20090023719A1 (en) | 2009-01-22 |
| ZA201000107B (en) | 2010-09-29 |
| BRPI0814821A2 (en) | 2015-02-03 |
| EA201000101A1 (en) | 2010-08-30 |
| DOP2010000022A (en) | 2010-02-15 |
| ECSP109958A (en) | 2010-03-31 |
| MX2010000658A (en) | 2010-03-26 |
| SV2010003459A (en) | 2010-04-30 |
| NI201000011A (en) | 2010-12-07 |
| CO6260075A2 (en) | 2011-03-22 |
| JP2010533700A (en) | 2010-10-28 |
| KR20100042272A (en) | 2010-04-23 |
| WO2009010789A3 (en) | 2009-05-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7718653B2 (en) | Pyrimidine derivatives for inhibiting Eph receptors | |
| DK2970218T3 (en) | DNA-PK inhibitors | |
| AU2005254982B2 (en) | Compounds and compositions as protein kinase inhibitors | |
| ES2477878T3 (en) | Compounds and compositions of 5- (4- (aloalkoxy) phenyl) pyrimidin-2-amine as kinase inhibitors | |
| JP5303557B2 (en) | 2-Heteroarylamino-pyrimidine derivatives which are kinase inhibitors | |
| EP3746072B1 (en) | 2h-indazole derivatives as cdk4 and cdk6 inhibitors and therapeutic uses thereof | |
| CA2954298A1 (en) | 2-h-indazole derivatives as cyclin-dependent kinase (cdk) inhibitors and therapeutic uses thereof | |
| US20080242663A1 (en) | Novel pyrimidine derivatives 698 | |
| US20090036440A1 (en) | Novel pyrimidine derivatives - 816 | |
| WO2007085833A2 (en) | Pyrimidine derivatives | |
| CN101163694A (en) | Imidaz0l0-5-yl-2-anil0-pyrimidines as agents for the inhibition of cell proliferation | |
| WO2008144253A1 (en) | Protein kinase inhibitors and methods for using thereof | |
| AU2014250836A1 (en) | Quinazolines and azaquinazolines as dual inhibitors of RAS/RAF/MEK/ERK and PI3K/AKT/PTEN/mTOR pathways | |
| KR20200011977A (en) | Aminopyrimidine Compounds, Methods for Making the Same, and Uses | |
| US20090054428A1 (en) | Novel pyrimidine derivatives 965 | |
| KR20220114643A (en) | Cyano-substituted pyridine and cyano-substituted pyrimidine compounds, preparation method thereof, and application thereof | |
| CN101668749A (en) | Novel pyrimidine derivatives 698 | |
| WO2017211216A1 (en) | Fused pyrimidinopiperidine derivative, and manufacturing method and application thereof | |
| CN118221696A (en) | Pyrimidine heterocyclic compound containing N-methylpiperazine structure, and preparation method and application thereof | |
| CN101472916A (en) | Chemical compounds | |
| MX2008009676A (en) | Pyrimidine derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880105690.X Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2411/MUMNP/2009 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008277446 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2693880 Country of ref document: CA Ref document number: 2010010064 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010516595 Country of ref document: JP Ref document number: 201011220 Country of ref document: CR Ref document number: D2010009 Country of ref document: CU Ref document number: CR2010-011220 Country of ref document: CR Ref document number: 12010500105 Country of ref document: PH Ref document number: MX/A/2010/000658 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008776197 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2008277446 Country of ref document: AU Date of ref document: 20080714 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201000101 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20107002514 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 583315 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201001433 Country of ref document: UA Ref document number: 10017544 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PI 2010000189 Country of ref document: MY |
|
| ENP | Entry into the national phase |
Ref document number: PI0814821 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100114 |