WO2008147518A1 - Pyridyl piperidine orexin receptor antagonists - Google Patents
Pyridyl piperidine orexin receptor antagonists Download PDFInfo
- Publication number
- WO2008147518A1 WO2008147518A1 PCT/US2008/006563 US2008006563W WO2008147518A1 WO 2008147518 A1 WO2008147518 A1 WO 2008147518A1 US 2008006563 W US2008006563 W US 2008006563W WO 2008147518 A1 WO2008147518 A1 WO 2008147518A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- methoxy
- pyridine
- piperidin
- methylpiperidin
- Prior art date
Links
- 0 C[C@@](CCCC1)N1C(C(CC1)CCC2(*)C3(*)C1(*)*23)=O Chemical compound C[C@@](CCCC1)N1C(C(CC1)CCC2(*)C3(*)C1(*)*23)=O 0.000 description 2
- CDGCGQSPMXFURV-MRXNPFEDSA-N CC(C)CC[C@H](CNC(c1cc(C(O)=O)ccc1-[n]1nccn1)=O)COc(cc1)ncc1F Chemical compound CC(C)CC[C@H](CNC(c1cc(C(O)=O)ccc1-[n]1nccn1)=O)COc(cc1)ncc1F CDGCGQSPMXFURV-MRXNPFEDSA-N 0.000 description 1
- LBBYKGSFSNUJCF-QDAZHVTCSA-N C[C@H](CCC(C1)(C2)C1Oc(cc1)ncc1F)N2C(c1cc(Br)ccc1-[n]1nccn1)=O Chemical compound C[C@H](CCC(C1)(C2)C1Oc(cc1)ncc1F)N2C(c1cc(Br)ccc1-[n]1nccn1)=O LBBYKGSFSNUJCF-QDAZHVTCSA-N 0.000 description 1
- FCAPCGVHMBXWCR-HZPDHXFCSA-N C[C@H](CC[C@@H](COc(nc1)ccc1F)C1)N1C(c1cc(C(OC)=O)ccc1-[n]1nccn1)=O Chemical compound C[C@H](CC[C@@H](COc(nc1)ccc1F)C1)N1C(c1cc(C(OC)=O)ccc1-[n]1nccn1)=O FCAPCGVHMBXWCR-HZPDHXFCSA-N 0.000 description 1
- DLRPPDYCDAXXAJ-GHMZBOCLSA-N C[C@H]1NC[C@H](COc(cc2)ccc2F)CC1 Chemical compound C[C@H]1NC[C@H](COc(cc2)ccc2F)CC1 DLRPPDYCDAXXAJ-GHMZBOCLSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the orexins (hypocretins) comprise two neuropeptides produced in the hypothalamus: the orexin A (OX-A) (a 33 amino acid peptide) and the orexin B (OX-B) (a 28 amino acid peptide) (Sakurai T. et al., Cell, 1998, 92, 573-585). Orexins are found to stimulate food consumption in rats suggesting a physiological role for these peptides as mediators in the central feedback mechanism that regulates feeding behaviour (Sakurai T. et al., Cell, 1998, 92, 573-585).
- Orexins regulate states of sleep and wakefulness opening potentially novel therapeutic approaches for narcoleptic or insomniac patients (Chemelli R.M. et al., Cell, 1999, 98, 437-451). Orexins have also been indicated as playing a role in arousal, reward, learning and memory (Harris, et al., Trends Neurosci., 2006, 29 (10), 571-577). Two orexin receptors have been cloned and characterized in mammals. They belong to the super family of G-protein coupled receptors (Sakurai T.
- the orexin-1 receptor (OX or OXlR) is selective for OX-A and the orexin-2 receptor (0X2 or 0X2R) is capable to bind OX-A as well as OX-B.
- OX 1 receptor the orexin-1 receptor
- OX 2 receptor the orexin-2 receptor (0X2 or 0X2R) is capable to bind OX-A as well as OX-B.
- the physiological actions in which orexins are presumed to participate are thought to be expressed via one or both of OX 1 receptor and OX 2 receptor as the two subtypes of orexin receptors.
- Orexin receptors are found in the mammalian brain and may have numerous implications in pathologies such as depression; anxiety; addictions; obsessive compulsive disorder; affective neurosis; depressive neurosis; anxiety neurosis; dysthymic disorder; behaviour disorder; mood disorder; sexual dysfunction; psychosexual dysfunction; sex disorder; schizophrenia; manic depression; delirium; dementia; severe mental retardation and dyskinesias such as Huntington's disease and Tourette syndrome; eating disorders such as anorexia, bulimia, cachexia, and obesity; addictive feeding behaviors; binge/purge feeding behaviors; cardiovascular diseases; diabetes; appetite/taste disorders; emesis, vomiting, nausea; asthma; cancer; Parkinson's disease; Cushing's syndrome/disease; basophile adenoma; prolactinoma; hyperprolactinemia; hypophysis tumour/adenoma; hypothalamic diseases; inflammatory bowel disease; gastric diskinesia; gastric ulcers; Froehlich'
- HIV post-chemotherapy pain
- post-stroke pain post-operative pain
- neuralgia emesis, nausea, vomiting; conditions associated with visceral pain such as irritable bowel syndrome, and angina
- migraine urinary bladder incontinence e.g. urge incontinence
- tolerance to narcotics or withdrawal from narcotics sleep disorders; sleep apnea; narcolepsy; insomnia; parasomnia; jet lag syndrome; and neurodegenerative disorders including nosological entities such as disinhibition-dementia-parkinsonism-amyotrophy complex; pallido-ponto-nigral degeneration; epilepsy; seizure disorders and other diseases related to general orexin system dysfunction.
- the present invention is directed to pyridyl piperidine compounds which are antagonists of orexin receptors, and which are useful in the treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved.
- the invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which orexin receptors are involved.
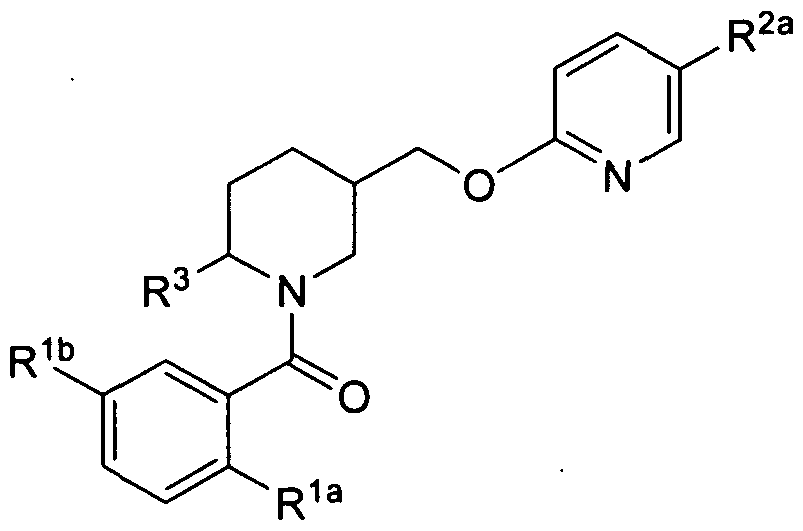
- the present invention is directed to compounds of the formula I:
- A is selected from the group consisting of phenyl, napthyl and heteroaryl
- R Ia 5 Rib and Rl c may be absent if the valency of Al does not permit such substitution and are independently selected from the group consisting of:
- R2a, R2b and R2c are independently selected from the group consisting of:
- R.3 is hydrogen, Ci_6alkyl or C3-6cycloalkyl, which is unsubstituted or substituted with one or more substituents selected from Rl 3;
- R4 and R5 are independently selected from hydrogen and Ci-6alkyl, which is unsubstituted or substituted with one or more substituents selected from Rl 3, or R4 and R5 may be joined together to form a C3_6cycloalkyl with the carbon atom to which they are attached, where the cycloalkyl is unsubstituted or substituted with one or more substituents selected from Rl 3;
- Rl 3 is selected from the group consisting of:
- Rl 4 is selected from the group consisting of: (1) hydroxyl,
- An embodiment of the present invention includes compounds of the formula Ia':
- R Ia 1 wherein A, R Ia, Rib, R Ic, R2a, R2b, R2C, R3, R4 and R5 are defined herein; or a pharmaceutically acceptable salt thereof.
- An embodiment of the present invention includes compounds of the formula Ia":
- R Ia 5 RIb 5 RIc 5 R2a, R2b 5 R2C and R3 are defined herein; or a pharmaceutically acceptable salt thereof.
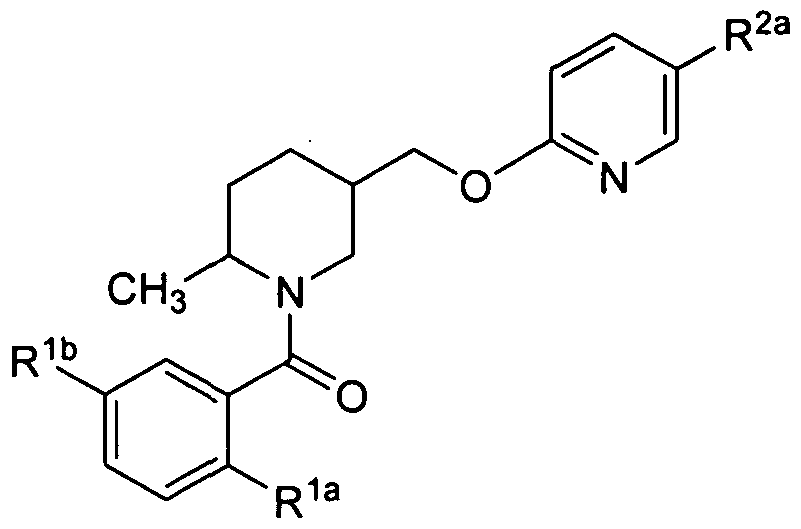
- An embodiment of the present invention includes compounds of the formula Id:
- Rl a , Rib, Rlc, R2a and R3 are defined herein; or a pharmaceutically acceptable salt thereof.
- An embodiment of the present invention includes compounds of the formula Ie:
- R Ia, Rib, R2a and R3 are defined herein; or a pharmaceutically acceptable salt thereof.
- An embodiment of the present invention includes compounds of the formula If:
- Rl a, Rib and R2a are defined herein; or a pharmaceutically acceptable salt thereof.
- An embodiment of the present invention includes compounds wherein Al is phenyl.
- An embodiment of the present invention includes compounds wherein Al is heteroaryl.
- An embodiment of the present invention includes compounds wherein Al is pyrazolyl.
- An embodiment of the present invention includes compounds wherein Al is thiazolyl.
- An embodiment of the present invention includes compounds wherein Rl a , Rib and Rl c are independently selected from the group consisting of: (1) hydrogen,
- Ci-6alkyl which is unsubstituted or substituted with halogen, hydroxyl, phenyl or napthyl,
- -O-Ci_6alkyl which is unsubstituted or substituted with halogen, hydroxyl or phenyl, (6) heteroaryl, wherein heteroaryl is selected from triazolyl, oxazolyl, pyrrolyl, imidazolyl, indolyl, pyridyl, and pyrimidinyl, which is unsubstituted or substituted with halogen, hydroxyl, Ci-6alkyl, -O-Ci-6alkyl or-NO2,
- Rl a , Rib and Rl c are independently selected from the group consisting of:
- Ci-6alkyl which is unsubstituted or substituted with halogen, hydroxyl or phenyl or napthyl,
- -O-Ci-6alkyl which is unsubstituted or substituted with halogen, hydroxyl or phenyl
- heteroaryl wherein heteroaryl is selected from triazolyl, oxazolyl and pyrimidinyl, which is unsubstituted or substituted with halogen, hydroxyl or Ci-6alkyl, and (7) phenyl, which is unsubstituted or substituted with halogen, hydroxyl or Ci-6alkyl.
- R Ia, Rib and Rl c are independently selected from the group consisting of: (1) hydrogen,
- R Ia, Rib and Rl c are independently selected from the group consisting of:
- An embodiment of the present invention includes compounds wherein R2a ? R2b and R2c are independently selected from the group consisting of: (1) hydrogen, (2) halogen,
- Ci-6alkyl which is unsubstituted or substituted with halogen, hydroxyl or phenyl or napthyl
- -O-C 1 - ⁇ alkyl which is unsubstituted or substituted with halogen, hydroxyl or phenyl
- heteroaryl wherein heteroaryl is selected from pyrrolyl, imidazolyl, indolyl, pyridyl, and pyrimidinyl, which is unsubstituted or substituted with halogen, hydroxyl, C].
- ⁇ alkyl, -O-Cl-6alkyl or-NO2 (7) phenyl, which is unsubstituted or substituted with halogen, hydroxyl, Ci-6alkyl, -
- R.2a 5 R2b and R2c are independently selected from the group consisting of:
- R.2a ⁇ R2b and R2c are independently selected from the group consisting of:
- R.2a, R2b and R2c are independently selected from the group consisting of: (1) hydrogen,
- R.2a s R.2b and R2c are independently selected from the group consisting of:
- An embodiment of the present invention includes compounds wherein R3 is hydrogen, Ci-6alkyl or C3-6cycloalkyl. An embodiment of the present invention includes compounds wherein R.3 is other than hydrogen. An embodiment of the present invention includes compounds wherein R3 is Ci-6alkyl. An embodiment of the present invention includes compounds wherein R3 is C3_6cycloalkyl. An embodiment of the present invention includes compounds wherein R3 is methyl or ethyl. An embodiment of the present invention includes compounds wherein R3 is methyl. An embodiment of the present invention includes compounds wherein R3 is in the trans configuration on the piperidine ring relative to the pyridyloxymethyl substituent.
- An embodiment of the present invention includes compounds wherein R3 is in the cis configuration on the piperidine ring relative to the pyridyloxymethyl substituent.
- An embodiment of the present invention includes compounds wherein R3 is in the R configuration on the piperidine ring.
- An embodiment of the present invention includes compounds wherein the substituent at the 6-position of the piperidine ring is in the R configuration.
- An embodiment of the present invention includes compounds wherein pyridyloxymethyl group is in the R configuration on the piperidine ring.
- An embodiment of the present invention includes compounds wherein the substituent at the 3-position of the piperidine ring is in the R configuration.
- An embodiment of the present invention includes compounds wherein R4 is hydrogen or Ci-6alkyl.
- An embodiment of the present invention includes compounds wherein
- R4 is hydrogen or methyl.
- An embodiment of the present invention includes compounds wherein R4 is hydrogen.
- An embodiment of the present invention includes compounds wherein R5 is hydrogen or Ci-6alkyl.
- An embodiment of the present invention includes compounds wherein R 5 is hydrogen or methyl.
- An embodiment of the present invention includes compounds wherein R.5 is hydrogen.
- Specific embodiments of the present invention include a compound which is selected from the group consisting of the subject compounds of the Examples herein or a pharmaceutically acceptable salt thereof.
- the compounds of the present invention may contain one or more asymmetric centers and can thus occur as "stereoisomers” including racemates and racemic mixtures, enantiomeric mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers and it is intended that all of the possible optical isomers and diastereomers in mixtures and as pure or partially purified compounds are included within the scope of this invention. The present invention is meant to comprehend all such isomeric forms of these compounds.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diasteromeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- any enantiomer of a compound may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration by methods well known in the art.
- Ci-6alkyl is defined to identify the group as having 1 , 2, 3, 4, 5 or 6 carbons in a linear or branched arrangement, such that Ci-8alkyl specifically includes methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert- butyl, pentyl, and hexyl.
- a group which is designated as being independently substituted with substituents may be independently substituted with multiple numbers of such substituents.
- heterocycle includes both unsaturated and saturated heterocyclic moieties, wherein the unsaturated heterocyclic moieties (i.e. "heteroaryl”) include benzoimidazolyl, benzimidazolonyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzothiazolyl, benzotriazolyl, benzothiophenyl, benzoxazepin, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxazoline, isoxazoline, oxazoline, oxazoline,
- salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particular embodiments include the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylene-diamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p- toluenesulfonic acid, and the like.
- Specific compounds within the present invention include a compound which selected from the group consisting of the compounds disclosed in the following Examples and pharmaceutically acceptable salts thereof and individual enantiomers or diastereomers thereof.
- the subject compounds are useful in a method of antagonizing orexin receptor activity in a patient such as a mammal in need of such inhibition comprising the administration of an effective amount of the compound.
- the present invention is directed to the use of the compounds disclosed herein as antagonists of orexin receptor activity. In addition to primates, especially humans, a variety of other mammals can be treated according to the method of the present invention.
- the present invention is directed to a compound of the present invention or a pharmaceutically acceptable salt thereof for use in medicine.
- the present invention is further directed to a use of a compound of the present invention or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for antagonizing orexin receptor activity or treating the disorders and diseases noted herein in humans and animals.
- the subject treated in the present methods is generally a mammal, such as a human being, male or female.
- the term "therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- treatment and “treating” refer to all processes wherein there may be a slowing, interrupting, arresting, controlling, or stopping of the progression of the neurological and psychiatric disorders described herein, but does not necessarily indicate a total elimination of all disorder symptoms, as well as the prophylactic therapy of the mentioned conditions, particularly in a patient who is predisposed to such disease or disorder.
- composition as used herein is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- CHO cells expressing the rat orexin- 1 receptor or the human orexin-2 receptor, are grown in Iscove's modified DMEM containing 2 mM L-glutamine, 0.5 g/ml G418, 1% hypoxanthine-thymidine supplement, 100 U/ml penicillin, 100 ug/ml streptomycin and 10 % heat-inactivated fetal calf serum (FCS).
- FCS heat-inactivated fetal calf serum
- the cells are seeded at 20,000 cells / well into Becton-Dickinson black 384-well clear bottom sterile plates coated with poly-D- lysine. All reagents were from GIBCO-Invitrogen Corp.
- Ala-6,12 human orexin-A as the agonist is prepared as a 1 mM stock solution in 1% bovine serum albumin (BSA) and diluted in assay buffer (HBSS containing 20 mM HEPES, 0.1% BSA and 2.5mM probenecid, pH7.4) for use in the assay at a final concentration of 7OpM.
- Test compounds are prepared as 10 mM stock solution in DMSO, then diluted in 384-well plates, first in DMSO, then assay buffer.
- Fluorescence is measured for each well at 1 second intervals for 5 minutes and the height of each fluorescence peak is compared to the height of the fluorescence peak induced by 70 pM Ala-6,12 orexin-A with buffer in place of antagonist.
- IC50 value the concentration of compound needed to inhibit 50 % of the agonist response
- compound potency can be assessed by a radioligand binding assay (described in Bergman et. al. Bioorg. Med. Chem. Lett. 2008, 18, 1425 - 1430) in which the inhibition constant (Kj) is determined in membranes prepared from CHO cells expressing either the OXl or OX2 receptor.
- the intrinsic orexin receptor antagonist activity of a compound which may be used in the present invention may be determined by these assays.
- the compounds of the following examples had activity in antagonizing the rat orexin- 1 receptor and/or the human orexin-2 receptor in the aforementioned assays, generally with an IC50 of less than about 50 ⁇ M.
- Many of compounds within the present invention had activity in antagonizing the rat orexin- 1 receptor and/or the human orexin-2 receptor in the aforementioned assays with an IC50 of less than about 100 nM.
- Compounds of the present invention also have activity in the radioligand binding assay, generally with a Ki ⁇
- the present invention also includes compounds within the generic scope of the invention which possess activity as agonists of the orexin- 1 receptor and/or the orexin-2 receptor. With respect to other piperidine compounds, the present compounds exhibit unexpected properties, such as with respect to increased potency, oral bioavailability, metabolic stability, and/or selectivity.
- the present compounds wherein R3 is substituted such as with a Ci -6 alkyl or C3.6 cycloalkyl possess unexpectedly greater potency at the orexin- 1 receptor and/or the orexin-2 receptor.
- the orexin receptors have been implicated in a wide range of biological functions. This has suggested a potential role for these receptors in a variety of disease processes in humans or other species.
- the compounds of the present invention have utility in treating, preventing, ameliorating, controlling or reducing the risk of a variety of neurological and psychiatric disorders associated with orexin receptors, including one or more of the following conditions or diseases: sleep disorders, sleep disturbances, including enhancing sleep quality, improving sleep quality, increasing sleep efficiency, augmenting sleep maintenance; increasing the value which is calculated from the time that a subject sleeps divided by the time that a subject is attempting to sleep; improving sleep initiation; decreasing sleep latency or onset (the time it takes to fall asleep); decreasing difficulties in falling asleep; increasing sleep continuity; decreasing the number of awakenings during sleep; decreasing intermittent wakings during sleep; decreasing nocturnal arousals; decreasing the time spent awake following the initial onset of sleep; increasing the total amount of sleep; reducing the fragmentation of sleep; altering the timing, frequency or duration of REM sleep bout
- the present invention provides methods for: enhancing the quality of sleep; augmenting sleep maintenance; increasing REM sleep; increasing stage 2 sleep; decreasing fragmentation of sleep patterns; treating insomnia; enhancing cognition; increasing memory retention; treating or controlling obesity; treating or controlling depression; treating, controlling, ameliorating or reducing the risk of epilepsy, including absence epilepsy; treating or controlling pain, including neuropathic pain; treating or controlling Parkinson's disease; treating or controlling psychosis; or treating, controlling, ameliorating or'reducing the risk of schizophrenia, in a mammalian patient in need thereof which comprises administering to the patient a therapeutically effective amount of a compound of the present invention.
- the subject compounds are further useful in a method for the prevention, treatment, control, amelioration, or reducation of risk of the diseases, disorders and conditions noted herein.
- the dosage of active ingredient in the compositions of this invention may be varied, however, it is necessary that the amount of the active ingredient be such that a suitable dosage form is obtained.
- the active ingredient may be administered to patients (animals and human) in need of such treatment in dosages that will provide optimal pharmaceutical efficacy.
- the selected dosage depends upon the desired therapeutic effect, on the route of administration, and on the duration of the treatment.
- the dose will vary from patient to patient depending upon the nature and severity of disease, the patient's weight, special diets then being followed by a patient, concurrent medication, and other factors which those skilled in the art will recognize.
- dosage levels of between 0.0001 to 10 mg/kg. of body weight daily are administered to the patient, e.g., humans and elderly humans, to obtain effective antagonism of orexin receptors.
- the dosage range will generally be about 0.5 mg to 1.0 g. per patient per day which may be administered in single or multiple doses. In one embodiment, the dosage range will be about 0.5 mg to 500 mg per patient per day; in another embodiment about 0.5 mg to 200 mg per patient per day; and in yet another embodiment about 5 mg to 50 mg per patient per day.
- Pharmaceutical compositions of the present invention may be provided in a solid dosage formulation such as comprising about 0.5 mg to 500 mg active ingredient, or comprising about 1 mg to 250 mg active ingredient.
- the pharmaceutical composition may be provided in a solid dosage formulation comprising about 1 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg or 250 mg active ingredient.
- the compositions may be provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, such as 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, and 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, such as once or twice per day.
- the compounds of the present invention may be used in combination with one or more other drugs in the treatment, prevention, control, amelioration, or reduction of risk of diseases or conditions for which compounds of the present invention or the other drugs may have utility, where the combination of the drugs together are safer or more effective than either drug alone.
- Such other drug(s) may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of the present invention is contemplated.
- the combination therapy may also includes therapies in which the compound of the present invention and one or more other drugs are administered on different overlapping schedules.
- the compounds of the present invention and the other active ingredients may be used in lower doses than when each is used singly.
- the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of the present invention.
- the above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
- compounds of the present invention may be used in combination with other drugs that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present invention are useful.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is contemplated.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
- the weight ratio of the compound of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the present invention is combined with another agent, the weight ratio of the compound of the present invention to the other agent will generally range from about 1000:1 to about 1 :1000, such as about 200:1 to about 1 :200. Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations the compound of the present invention and other active agents may be administered separately or in conjunction. In addition, the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s).
- the compounds of the present invention may be administered in conbination with other compounds which are known in the art to be useful for enhancing sleep quality and preventing and treating sleep disorders and sleep disturbances, including e.g., sedatives, hypnotics, anxiolytics, antipsychotics, antianxiety agents, antihistamines, benzodiazepines, barbiturates, cyclopyrrolones, GABA agonists, 5HT-2 antagonists including 5HT-2A antagonists and 5HT-2A/2C antagonists, histamine antagonists including histamine H3 antagonists, histamine H3 inverse agonists, imidazopyridines, minor tranquilizers, melatonin agonists and antagonists, melatonergic agents, other orexin antagonists, orexin agonists, prokineticin agonists and antagonists, pyrazolopyrimidines, T-type calcium channel antagonists, triazolopyridines, and the like, such as: adinazolam, allobar
- the subject compound may be employed in combination with other compounds which are known in the art, either administered separately or in the same pharmaceutical compositions, include, but are not limited to: insulin sensitizers including (i) PPAR ⁇ antagonists such as glitazones (e.g.
- ciglitazone darglitazone; englitazone; isaglitazone (MCC-555); pioglitazone; rosiglitazone; troglitazone; tularik; BRL49653; CLX-0921; 5-BTZD), GW-0207, LG- 100641, and LY-300512, and the like);
- biguanides such as metformin and phenformin
- insulin or insulin mimetics such as biota, LP-100, novarapid, insulin detemir, insulin lispro, insulin glargine, insulin zinc suspension (lente and ultralente); Lys-Pro insulin, GLP-I (73-7) (insulintropin); and GLP-I (7-36)-NH2)
- sulfonylureas such as acetohexamide; chlorpropamide; diabinese; glibenclamide; glipizide; gly
- CNTF Central neurotrophic factors
- GI-181771 Gaxo-SmithKline
- SR146131 Sanofi Synthelabo
- butabindide PD170,292, and PD 149164 (Pfizer)
- CNTF derivatives such as axokine (Regeneron)
- monoamine reuptake inhibitors such as sibutramine
- UCP-I uncoupling protein- 1
- activators such as phytanic acid, 4-[(E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-napthalenyl)-l- propenyl] benzoic acid (TTNPB), retinoic acid
- thyroid hormone ⁇ agonists such as KB- 2611 (KaroBioBMS
- FAS fatty acid synthase inhibitors, such as Cerulen
- dipeptidyl peptidase IV (DP-IV) inhibitors such as isoleucine thiazolidide, valine pyrrolidide, NVP-DPP728, LAF237, MK-431 , P93/01, TSL 225, TMC-2A/2B/2C, FE 999011, P9310/K364, VIP 0177, SDZ 274-444; (46) dicarboxylate transporter inhibitors; (47) glucose transporter inhibitors; (48) phosphate transporter inhibitors; (49) Metformin (Glucophage®); and (50) Topiramate (Topimax®); and (50) peptide YY, PYY 3-36, peptide YY analogs, derivatives, and fragments such as BIM- 43073D, BIM-43004C (Olitvak, D.A. et al., Dig. Dis. Sci. 44(3):643-
- Neuropeptide Y2 (NPY2) receptor agonists such NPY3-36, N acetyl [Leu(28,31)] NPY 24-36, TASP-V, and cyclo-(28/32)-Ac-[Lys28-Glu32]-(25-36)-pNPY;
- Neuropeptide Y4 (NPY4) agonists such as pancreatic peptide (PP), and other Y4 agonists such as 1229U91;
- cyclooxygenase-2 inhibitors such as etoricoxib, celecoxib, valdecoxib, parecoxib, lumiracoxib, BMS347070, tiracoxib or JTE522, ABT963, CS502 and GW406381, and pharmaceutically acceptable salts thereof;
- Neuropeptide Yl (NPYl) antagonists such as BIBP3226, J-1 15814, BIBO 3304, LY-357897, CP-671906,
- the subject compound may be employed in combination with an anti-depressant or anti-anxiety agent, including norepinephrine reuptake inhibitors (including tertiary amine tricyclics and secondary amine tricyclics), selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors (MAOIs), reversible inhibitors of monoamine oxidase (RIMAs), serotonin and noradrenaline reuptake inhibitors (SNRIs), corticotropin releasing factor (CRF) antagonists, ⁇ -adrenoreceptor antagonists, neurokinin- 1 receptor antagonists, atypical anti-depressants, benzodiazepines, 5-HTi A agonists or antagonists, especially 5-HTj A partial agonists, and corticotropin releasing factor (CRF) antagonists.
- norepinephrine reuptake inhibitors including tertiary amine tricyclics and secondary amine tricyclics
- Specific agents include: amitriptyline, clomipramine, doxepin, imipramine and trimipramine; amoxapine, desipramine, maprotiline, nortriptyline and protriptyline; fluoxetine, fluvoxamine, paroxetine and sertraline; isocarboxazid, phenelzine, tranylcypromine and selegiline; moclobemide: venlafaxine; aprepitant; bupropion, lithium, nefazodone, trazodone and viloxazine; alprazolam, chlordiazepoxide, clonazepam, chlorazepate, diazepam, halazepam, lorazepam, oxazepam and prazepam; buspirone, flesinoxan, gepirone and ipsapirone, and pharmaceutically acceptable salts thereof.
- the subject compound may be employed in combination with anti-Alzheimer's agents; beta-secretase inhibitors; gamma-secretase inhibitors; growth hormone secretagogues; recombinant growth hormone; HMG-CoA reductase inhibitors; NSAID's including ibuprofen; vitamin E; anti-amyloid antibodies; CB-I receptor antagonists or CB-I receptor inverse agonists; antibiotics such as doxycycline and rifampin; N-methyl-D- aspartate (NMDA) receptor antagonists, such as memantine; cholinesterase inhibitors such as galantamine, rivastigmine, donepezil, and tacrine; growth hormone secretagogues such as ibutamoren, ibutamoren mesylate, and caproniorelin; histamine H3 antagonists; AMPA agonists; PDE IV inhibitors; GABAA inverse agonists; or neuronal nicotinic
- the subject compound may be employed in combination with sedatives, hypnotics, anxiolytics, antipsychotics, antianxiety agents, cyclopyrrolones, imidazopyridines, pyrazolopyrimidines, minor tranquilizers, melatonin agonists and antagonists, melatonergic agents, benzodiazepines, barbiturates, 5HT-2 antagonists, and the like, such as: adinazolam, allobarbital, alonimid, alprazolam, amitriptyline, amobarbital, amoxapine, bentazepam, benzoctamine, brotizolam, bupropion, busprione, butabarbital, butalbital, capuride, carbocloral, chloral betaine, chloral hydrate, chlordiazepoxide, clomipramine, clonazepam, cloperidone, clorazepate, clorethate
- the subject compound may be employed in combination with levodopa (with or without a selective extracerebral decarboxylase inhibitor such as carbidopa or benserazide), anticholinergics such as biperiden (optionally as its hydrochloride or lactate salt) and trihexyphenidyl (benzhexol) hydrochloride, COMT inhibitors such as entacapone, MOA-B inhibitors, antioxidants, A2a adenosine receptor antagonists, cholinergic agonists, NMDA receptor antagonists, serotonin receptor antagonists and dopamine receptor agonists such as alentemol, bromocriptine, fenoldopam, lisuride, naxagolide, pergolide and pramipexole.
- levodopa with or without a selective extracerebral decarboxylase inhibitor such as carbidopa or benserazide
- anticholinergics such as biperi
- the dopamine agonist may be in the form of a pharmaceutically acceptable salt, for example, alentemol hydrobromide, bromocriptine mesylate, fenoldopam mesylate, naxagolide hydrochloride and pergolide mesylate.
- a pharmaceutically acceptable salt for example, alentemol hydrobromide, bromocriptine mesylate, fenoldopam mesylate, naxagolide hydrochloride and pergolide mesylate.
- Lisuride and pramipexol are commonly used in a non-salt form.
- the subject compound may be employed in combination with acetophenazine, alentemol, benzhexol, bromocriptine, biperiden, chlorpromazine, chlorprothixene, clozapine, diazepam, fenoldopam, fluphenazine, haloperidol, levodopa, levodopa with benserazide, levodopa with carbidopa, lisuride, loxapine, mesoridazine, molindolone, naxagolide, olanzapine, pergolide, perphenazine, pimozide, pramipexole, risperidone, sulpiride, tetrabenazine, trihexyphenidyl, thioridazine, thiothixene or trifluoperazine.
- the subject compound may be employed in combination with a compound from the phenothiazine, thioxanthene, heterocyclic dibenzazepine, butyrophenone, diphenylbutylpiperidine and indolone classes of neuroleptic agent.
- phenothiazines include chlorpromazine, mesoridazine, thioridazine, acetophenazine, fluphenazine, perphenazine and trifluoperazine.
- Suitable examples of thioxanthenes include chlorprothixene and thiothixene.
- An example of a dibenzazepine is clozapine.
- An example of a butyrophenone is haloperidol.
- An example of a diphenylbutylpiperidine is pimozide.
- An example of an indolone is molindolone.
- Other neuroleptic agents include loxapine, sulpiride and risperidone.
- the neuroleptic agents when used in combination with thesubject compound may be in the form of a pharmaceutically acceptable salt, for example, chlorpromazine hydrochloride, mesoridazine besylate, thioridazine hydrochloride, acetophenazine maleate, fluphenazine hydrochloride, flurphenazine enathate, fluphenazine decanoate, trifluoperazine hydrochloride, thiothixene hydrochloride, haloperidol decanoate, loxapine succinate and molindone hydrochloride.
- Perphenazine, chlorprothixene, clozapine, haloperidol, pimozide and risperidone are commonly used in a non-salt form.
- the subject compound may be employed in combination with an anoretic agent such as aminorex, amphechloral, amphetamine, benzphetamine, chlorphentermine, clobenzorex, cloforex, clominorex, clortermine, cyclexedrine, dexfenfluramine, dextroamphetamine, diethylpropion, diphemethoxidine, N-ethylamphetamine, fenbutrazate, fenfluramine, fenisorex, fenproporex, fiudorex, fluminorex, furfurylmethylamphetamine, levamfetamine, levophacetoperane, mazindol, mefenorex, metamfepramone, methamphetamine, norpseudoephedrine, pentorex, phendimetrazine, phenmetrazine, phentermine, phenylpropanolamine,
- the subject compound may be employed in combination with an opiate agonist, a lipoxygenase inhibitor, such as an inhibitor of 5-lipoxygenase, a cyclooxygenase inhibitor, such as a cyclooxygenase-2 inhibitor, an interleukin inhibitor, such as an interleukin- 1 inhibitor, an NMDA antagonist, an inhibitor of nitric oxide or an inhibitor of the synthesis of nitric oxide, a non-steroidal antiinflammatory agent, or a cytokine-suppressing antiinflammatory agent, for example with a compound such as acetaminophen, asprin, codiene, fentanyl, ibuprofen, indomethacin, ketorolac, morphine, naproxen, phenacetin, piroxicam, a steroidal analgesic, sufentanyl, sunlindac, tenidap, and the like.
- a lipoxygenase inhibitor such as an inhibitor of 5-lip
- the subject compound may be administered with a pain reliever; a potentiator such as caffeine, an H2-antagonist, simethicone, aluminum or magnesium hydroxide; a decongestant such as phenylephrine, phenylpropanolamine, pseudophedrine, oxymetazoline, epinephrine, naphazoline, xylometazoline, propylhexedrine, or levo-desoxy-ephedrine; an antiitussive such as codeine, hydrocodone, caramiphen, carbetapentane, or dextramethorphan; a diuretic; and a sedating or non-sedating antihistamine.
- a pain reliever such as caffeine, an H2-antagonist, simethicone, aluminum or magnesium hydroxide
- a decongestant such as phenylephrine, phenylpropanolamine, pseudophedrine, oxymetazoline, epinep
- the compounds of the present invention may be administered by oral, parenteral (e.g., intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or infusion, subcutaneous injection, or implant), by inhalation spray, nasal, vaginal, rectal, sublingual, or topical routes of administration and may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- parenteral e.g., intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or infusion, subcutaneous injection, or implant
- inhalation spray nasal, vaginal, rectal, sublingual, or topical routes of administration
- nasal, vaginal, rectal, sublingual, or topical routes of administration may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- the compounds of the invention are effective for
- compositions for the administration of the compounds of this invention may conveniently be presented in dosage unit form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing the active ingredient into association with the carrier which constitutes one or more accessory ingredients.
- the pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- Oily suspensions may be formulated by suspending the active ingredient in a suitable oil. Oil-in- water emulsions may also be employed.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- Pharmaceutical compositions of the present compounds may be in the form of a sterile injectable aqueous or oleagenous suspension.
- the compounds of the present invention may also be administered in the form of suppositories for rectal administration.
- creams, ointments, jellies, solutions or suspensions, etc., containing the compounds of the present invention may be employed.
- the compounds of the present invention may also be formulated for administered by inhalation.
- the compounds of the present invention may also be administered by a transdermal patch by methods known in the art.
- Several methods for preparing the compounds of this invention are illustrated in the following Schemes and Examples. Starting materials are made according to procedures known in the art or as illustrated herein. The following abbreviations are used herein: Me: methyl; Et: ethyl; t-Bu: /ert-butyl; Ar: aryl; Ph: phenyl; Bn: benzyl; Ac: acetyl; THF: tetrahydrofuran; DEAD: diethylazodicarboxylate; DIPEA: N,N-diisopropylethylamine; DMSO: dimethylsulfoxide; EDC: N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide; HOBT: hydroxybenzotriazole hydrate; Boc: tert-butyloxy carbonyl; Et 3 N: trie
- the final product may be further modified, for example, by manipulation of substituents.
- substituents may include, but are not limited to, reduction, oxidation, alkylation, acylation, and hydrolysis reactions which are commonly known to those skilled in the art.
- the order of carrying out the foregoing reaction schemes and examples n may be varied to facilitate the reaction or to avoid unwanted reaction products.
- the following examples are provided so that the invention might be more fully understood. These examples are illustrative only and should not be construed as limiting the invention in any way.
- a solution of the A-2 (23 g, 152 mmol) in EtOH (200 ml) was treated with 5 mol% platinum oxide (1.728 g, 7.61 mmol) and acetic acid (8.71 ml, 152 mmol).
- the Parr bottle was evacuated and backfilled with H 2 (g) three times and stirred under a H 2 (g) atmosphere (45 psi, recharged 4 times) at 22 0 C for 3h.
- the mixture was filtered though Celite and the filter cake was washed with MeOH.
- the filtrate was concentrated to yield product with a -3.5:1 cis:trans diastereomer ratio.
- the trans material was separated away from the cis diastereomers and into its enantiomers on a 5 cm OD chiral column by isocratic elution (93:7 Hexane:EtOH; 75 mL/min; 1 inj) with detection at 215 nm to yield 120 mg of A-5, peak 1 (colorless oil, 100% ee) and 1 14 mg of peak 2. (colorless oil, contaminated with 30% cis, 90% ee). Data for A-5: LRMS m/z (M+H): 264. Similarly, the cis diastereomer can be separated into its enantiomers and utilized in the following procedures to prepare (R 5 S) and (S,R) compounds.
- a solution of A-7 (67 mg, 0.299 mmol) in DMF (1 ml) was treated with acid A-8 (60.7 mg, 0.299 mmol), EDC (68.7 mg, 0.358 mmol), ⁇ OBT (54.9 mg, 0.358 mmol), and triethylamine (0.167 ml, 1.195 mmol). After stirring at 22 0 C overnight, the mixture was diluted with EtOAc and washed with water three times. The organic phase was dried over Na 2 SO 4 , filtered and concentrated. The crude material was purified by gradient elution on silica gel (0 to 75% EtOAc in Hex) to yield impure material.
- the pH was adjusted to 7.4 with NaOH and then 450 g ATA-117 transaminase, 9 g Lactate Dehydrogenase, and 45 g glucose dehydrogenase were added and rinsed into the vessel with 2.5 L water. After all enzymes were in solution, the rotavaped solution of D-I was added, followed by a final 2.5 L water. pH control utilizing 5 N NaOH was initiated. The reaction was allowed to stir for 42 hours; reaction was complete at 31 hours. To the reaction vessel was added 19.4 kg NaCl and 6.0 L 5N HCl to adjust the pH to 3.5. 20 L of acetonitrile was added and allowed to stir for 10 min. The agitator was turned off and the reaction mixture allowed to settle for 1 hr.
- the mixture was aged 45min at 20 0 C, then MTBE (10 mL/g, 30 L) was added over 45min. The mixture was aged for 45 min, then cooled to 2 0 C over 45min. The mixture was aged at this temperature for a period of 30min, then filtered.
- the salt was rinsed 2 x 6 mL/g (2 x 18 L) with THF/MTBE 1/1, then 1 x 6 mL/g (1 x 18 L) MTBE, and was dried on the frit under a nitrogen atmosphere for a period of 16hrs to provide 4.46 Kg (52%) of D-5 as a white solid.
- the diastereoselectivity of the salt was 40-50:1.
- Methyl 2-iodo-5-methylbenzoate (E-I) A visually clean 100 L flask equipped with a mechanical stirrer thermocouple and water chilled condenser was charged with MeOH (50 L). 2-iodo-5-methylbenzoic acid (5.85 kg, 22.32 mol) was then added while stirring. Concentrated sulfuric acid (0.595 L, 11.16 mol) was then added portion- wise which caused an increase in temperature from 17 0 C to 22 0 C. This mixture was gradually brought to an internal temperature of 64.6 0 C an aged overnight ( ⁇ 18h). The next morning the reaction had reached >98% conversion by HPLC.
- the flask was cooled to 16 0 C by placing in an ice bath and 850ml of ION NaOH (0.98 equiv.) was added slowly (over 10 minutes) while monitoring the pH. After the addition the pH was 5-6 (Caution: bringing pH over 9 can result in saponification during the work-up).
- the solution was then concentrated to about 16L and this suspension was transferred to a 100 L extractor.
- the flask was rinsed with 8L of IPAc and 4L of water which were also transferred to the extractor. 32L IPAc along with 1OL of 5w% NaHCO 3 and ⁇ 10L of 15w% Brine.
- the layers were cut and the aqueous layers were back- extracted with 2OL of IPAc.
- the organic layers were then combined and washed with 1OL of 15w% Brine.
- the organic layers were collected to provide E-I (6.055 kg, 21.93 mol, 98 % yield) in 98.3% purity.
- the internal temperature was set to 74 0 C and aged for 16 h. An aliquot was taken for HPLC analysis and revealed near complete consumption of the starting boronate (>97% conv.).
- the reaction was cooled to room temperature, and 12 L of water and 24 L of MTBE were added while maintaining stirring for 10 minutes. This solution was filtered on Solka floe and transferred to a 100 L extractor. The flask was further rinsed with 4L of both MTBE and water (x2) and then another 4 L of MTBE. The layers were cut and the aqueous layers were back-extracted with 21.5 L of MTBE. Assay of the organic layers showed the biaryl ester (2.76 kg, 12.09 mol, 76 % yield).
- the aqueous layer was then re-introduced into the reactor (100 L) through an in-line filter for the acidification.
- This precipitate was filtered.
- the beige filter cake was washed twice with 3 mL/g of cold water. Then the cake was washed with 3 mL/g of cold 15% MTBE/Heptane and 15% PhMe/Heptane. Finally it was washed with 1.5 mL/g of room temperature MTBE and twice with room temperature 3 mL/g Heptane.
- the reaction was heated to 44 0 C (small exotherm at 42°C, which causes the temperature to rise to 46.7 °C and maintain that temperature for 30 min). The reaction was aged at this temperature overnight. After 17 h the reaction was not complete and T3P (1.1 L, 1.870 mol) was added to accelerate conversion. The next day (42 h) the reaction was deemed complete by HPLC and was cooled in an ice bath to 4 °C. 20 L of water was added (slowly for the first 1.5 L then pretty fast.) keeping the reaction temperature under 17 0 C. This mixture was stirred at room temperature for 30 minutes. Then the mixture was transferred into a 50 L extractor charged with 20 L of MTBE.
- the flask was rinsed with an additional 2 L of water and 4 L of MTBE.
- the layers were cut and the organics are washed with 20 L IN NaOH and then 10 L of IN NaOH. Finally, the organics were washed twice with 10 L of brine 15%.
- the organic fractions (quantitative HPLC assay at 1.65 kg) are then treated with ⁇ 50w% of Darco KB (75Og) for 1.75 h, filtered on Solka floe and rinsed with 10 mL/g of MTBE (1.559 kg, 94.5% recovery).
- Carbon monoxide was bubbled through a solution of 500 mg (1.03 mmol) of H-2, 23.1 mg (0.10 mmol) palladium(II) acetate, 43 mg (0.10 mmol) 1 ,3-bis(diphenylphosphino)- propane, and 0.57 mL (4.1 mmol) triethylamine in 15 mL of methanol and 7.5 ml of DMSO at 80 0 C for 10 minutes. The reaction was then placed under a balloon of carbon monoxide and stirred at 80 0 C for 2.5 hours.
- TFA as a modifier
- fractions containing the product could be basified with NaHCO 3 and extracted with EtOAc, dried over Na 2 SO 4 , and concentrated to provide the free-base.
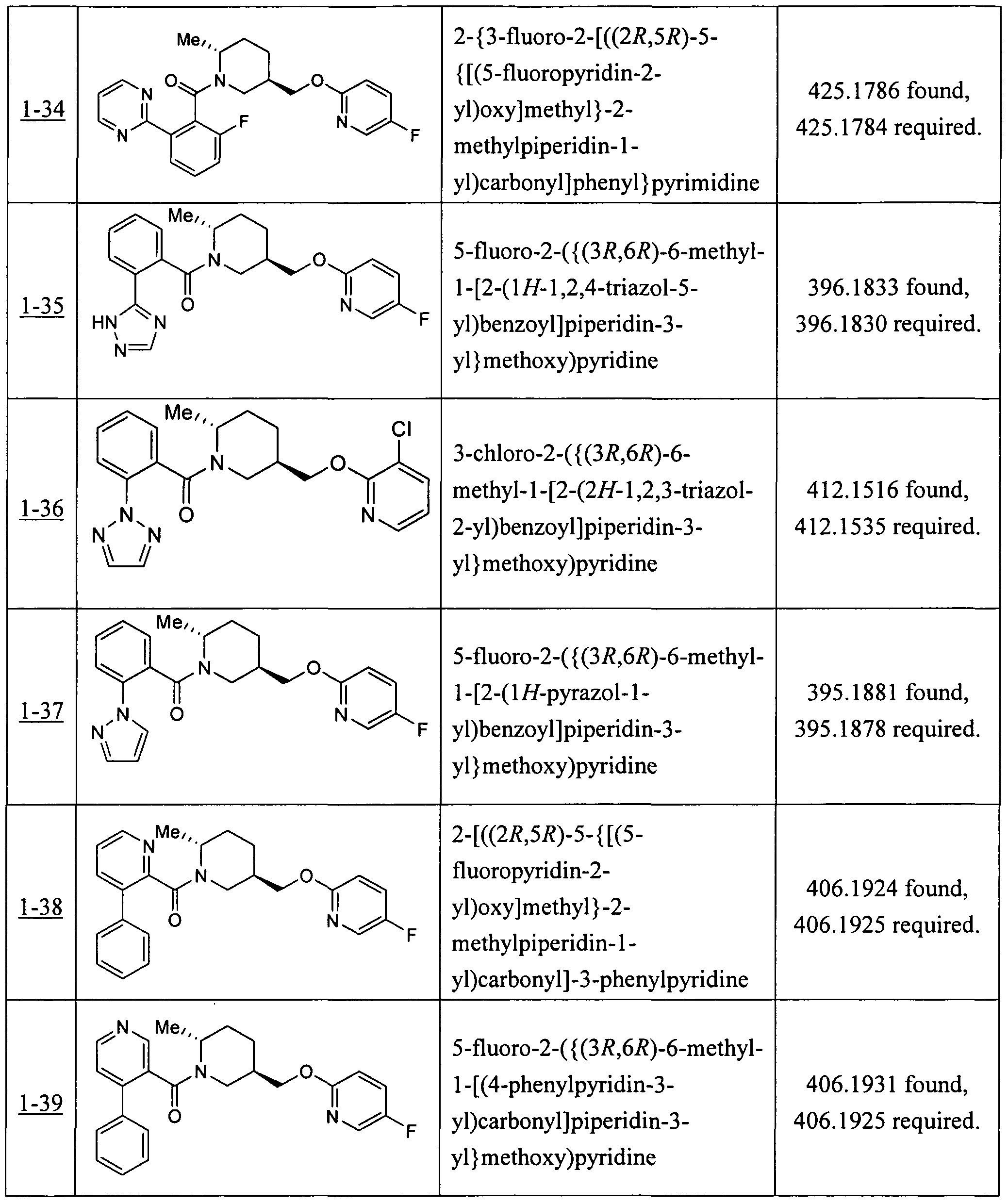
- Table 2 shows representative data for the compounds of the Examples as orexin receptor OXlR and/or OX2R antagonists as determined by the foregoing assays.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Addiction (AREA)
- Psychiatry (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Rheumatology (AREA)
- Obesity (AREA)
- Immunology (AREA)
- Hospice & Palliative Care (AREA)
- Urology & Nephrology (AREA)
- Epidemiology (AREA)
- Reproductive Health (AREA)
- Psychology (AREA)
- Emergency Medicine (AREA)
- Oncology (AREA)
- Ophthalmology & Optometry (AREA)
- Vascular Medicine (AREA)
Abstract
Description




























Claims






Priority Applications (21)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RS20120111A RS52200B (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| BRPI0811842-6A2A BRPI0811842A2 (en) | 2007-05-23 | 2008-05-22 | COMPOUND, PHARMACEUTICAL COMPOSITION, USE OF A COMPOUND, AND METHODS TO INCREASE SLEEP QUALITY IN A MAMMALIAN PATIENT, TO TREAT INSOMNIA IN A MAMMALIAN PATIENT, AND TO TREAT OR CONTROL OBESITY IN A MAMMALIAN PATIENT |
| JP2010509380A JP4881476B2 (en) | 2007-05-23 | 2008-05-22 | Pyridylpiperidine orexin receptor antagonist |
| AU2008257411A AU2008257411B2 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| AT08754659T ATE540944T1 (en) | 2007-05-23 | 2008-05-22 | PYRIDYLPIPERIDINOREXINE RECEPTOR ANTAGONISTS |
| CN2008800169258A CN101679366B (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| NZ580887A NZ580887A (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| SI200830593T SI2152690T1 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| KR1020097024362A KR101480279B1 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| DK08754659.4T DK2152690T3 (en) | 2007-05-23 | 2008-05-22 | Pyridylpiperidinorexin receptor antagonists |
| EP08754659A EP2152690B1 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| MX2009012579A MX2009012579A (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists. |
| CA2687321A CA2687321C (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| PL08754659T PL2152690T3 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| RU2009147733/04A RU2470021C2 (en) | 2007-05-23 | 2008-05-22 | Pyridyl-piperidine antagonists of orexin receptors |
| US12/600,388 US8242121B2 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| ES08754659T ES2379744T3 (en) | 2007-05-23 | 2008-05-22 | Pyridyl-piperidine antagonists of orexin receptors |
| IL201790A IL201790A0 (en) | 2007-05-23 | 2009-10-27 | Pyridyl piperidine orexin receptor antagonists |
| HK10101749.0A HK1134084A1 (en) | 2007-05-23 | 2010-02-18 | Pyridyl piperidine orexin receptor antagonists |
| HR20120240T HRP20120240T1 (en) | 2007-05-23 | 2012-03-15 | Pyridyl piperidine orexin receptor antagonists |
| US13/568,242 US8569311B2 (en) | 2007-05-23 | 2012-08-07 | Pyridyl piperidine orexin receptor antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US93145807P | 2007-05-23 | 2007-05-23 | |
| US60/931,458 | 2007-05-23 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/600,388 A-371-Of-International US8242121B2 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
| US13/568,242 Continuation US8569311B2 (en) | 2007-05-23 | 2012-08-07 | Pyridyl piperidine orexin receptor antagonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008147518A1 true WO2008147518A1 (en) | 2008-12-04 |
Family
ID=39708485
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/006563 WO2008147518A1 (en) | 2007-05-23 | 2008-05-22 | Pyridyl piperidine orexin receptor antagonists |
Country Status (34)
| Country | Link |
|---|---|
| US (2) | US8242121B2 (en) |
| EP (1) | EP2152690B1 (en) |
| JP (2) | JP4881476B2 (en) |
| KR (1) | KR101480279B1 (en) |
| CN (1) | CN101679366B (en) |
| AT (1) | ATE540944T1 (en) |
| AU (1) | AU2008257411B2 (en) |
| BR (1) | BRPI0811842A2 (en) |
| CA (1) | CA2687321C (en) |
| CO (1) | CO6251266A2 (en) |
| CR (1) | CR11146A (en) |
| CY (1) | CY1112969T1 (en) |
| DK (1) | DK2152690T3 (en) |
| DO (1) | DOP2009000263A (en) |
| EC (1) | ECSP099749A (en) |
| ES (1) | ES2379744T3 (en) |
| GT (1) | GT200900300A (en) |
| HK (1) | HK1134084A1 (en) |
| HR (1) | HRP20120240T1 (en) |
| IL (1) | IL201790A0 (en) |
| MA (1) | MA31451B1 (en) |
| MX (1) | MX2009012579A (en) |
| MY (1) | MY148544A (en) |
| NI (1) | NI200900193A (en) |
| NZ (1) | NZ580887A (en) |
| PL (1) | PL2152690T3 (en) |
| PT (1) | PT2152690E (en) |
| RS (1) | RS52200B (en) |
| RU (1) | RU2470021C2 (en) |
| SI (1) | SI2152690T1 (en) |
| SV (1) | SV2009003417A (en) |
| UA (1) | UA99620C2 (en) |
| WO (1) | WO2008147518A1 (en) |
| ZA (1) | ZA200907495B (en) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009124956A1 (en) * | 2008-04-10 | 2009-10-15 | Glaxo Group Limited | Pyridine derivatives used to treat orexin related disorders |
| WO2009143033A1 (en) * | 2008-05-22 | 2009-11-26 | Merck & Co., Inc. | Process for the preparation of an orexin receptor antagonist |
| WO2011006960A1 (en) | 2009-07-15 | 2011-01-20 | Rottapharm S.P.A. | Spiro amino compounds suitable for the treatment of inter alia sleep disorders and drug addiction |
| WO2011023578A1 (en) * | 2009-08-24 | 2011-03-03 | Glaxo Group Limited | 5-methyl-piperidine derivatives as orexin receptor antagonists for the treatment of sleep disorder |
| EP2484674A1 (en) | 2011-02-02 | 2012-08-08 | Rottapharm S.P.A. | Spiro aminic compounds with NK1 antagonist activity |
| US8242121B2 (en) | 2007-05-23 | 2012-08-14 | Merck Sharp & Dohme Corp. | Pyridyl piperidine orexin receptor antagonists |
| WO2012114252A1 (en) * | 2011-02-21 | 2012-08-30 | Actelion Pharmaceuticals Ltd | Novel indole and pyrrolopyridine amides |
| WO2013005755A1 (en) | 2011-07-05 | 2013-01-10 | 大正製薬株式会社 | Methylpiperidine derivative |
| WO2013059222A1 (en) | 2011-10-19 | 2013-04-25 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-nitrile orexin receptor antagonists |
| WO2013092893A1 (en) | 2011-12-21 | 2013-06-27 | Rottapharm Spa | Chemical compounds |
| EP2632253A1 (en) * | 2010-10-29 | 2013-09-04 | Merck Sharp & Dohme Corp. | Process for the preparation of an orexin receptor antagonist |
| WO2013127913A1 (en) | 2012-03-01 | 2013-09-06 | Rottapharm S.P.A. | 4,4-difluoro-piperidine-compounds |
| WO2014091876A1 (en) * | 2012-12-13 | 2014-06-19 | 大正製薬株式会社 | Fluorine-substituted piperidine compound |
| EP2768305A1 (en) * | 2011-10-21 | 2014-08-27 | Merck Sharp & Dohme Corp. | 2,5-disubstituted thiomorpholine orexin receptor antagonists |
| WO2015131773A1 (en) * | 2014-03-06 | 2015-09-11 | 上海海雁医药科技有限公司 | Piperidine derivatives as orexin receptor antagonist |
| US9174977B2 (en) | 2012-03-19 | 2015-11-03 | Rottapharm Biotech S.R.L. | 2-azabicyclo[4.1.0]heptane derivatives as orexin receptor antagonists for the treatment of certain disorders |
| WO2016025669A1 (en) | 2014-08-13 | 2016-02-18 | Eolas Therapeutics, Inc. | Difluoropyrrolidines as orexin receptor modulators |
| WO2016065585A1 (en) * | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Piperidine isoxazole and isothiazole orexin receptor antagonists |
| WO2016100161A1 (en) * | 2014-12-19 | 2016-06-23 | Merck Sharp & Dohme Corp. | Ethyldiamine orexin receptor antagonists |
| US9440982B2 (en) | 2012-02-07 | 2016-09-13 | Eolas Therapeutics, Inc. | Substituted prolines/piperidines as orexin receptor antagonists |
| US9458110B2 (en) | 2013-02-28 | 2016-10-04 | Bristol-Myers Squibb Company | Phenylpyrazole derivatives as potent ROCK1 and ROCK2 inhibitors |
| US9499517B2 (en) | 2012-02-07 | 2016-11-22 | Eolas Therapeutics, Inc. | Substituted prolines / piperidines as orexin receptor antagonists |
| WO2017028732A1 (en) * | 2015-08-14 | 2017-02-23 | 上海海雁医药科技有限公司 | Crystal form of orexin receptor antagonist compound, and preparation method and application thereof |
| EP3189840A1 (en) * | 2011-07-28 | 2017-07-12 | Kempharm, Inc. | Methylphenidate-prodrugs, processes of making and using the same |
| WO2017139603A1 (en) | 2016-02-12 | 2017-08-17 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| WO2017194548A1 (en) | 2016-05-10 | 2017-11-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of autoimmune inflammatory diseases |
| US9828345B2 (en) | 2013-02-28 | 2017-11-28 | Bristol-Myers Squibb Company | Phenylpyrazole derivatives as potent ROCK1 and ROCK2 inhibitors |
| US10308712B2 (en) | 2014-03-27 | 2019-06-04 | Bird Rock Bio, Inc. | Antibodies that bind human cannabinoid 1 (CB1) receptor |
| US10584113B2 (en) | 2016-12-11 | 2020-03-10 | Kempharm, Inc. | Methylphenidate-prodrugs, processes of making and using the same |
| US11421026B2 (en) | 2015-09-30 | 2022-08-23 | Bird Rock Bio, Inc. | Antibodies that bind human cannabinoid 1 (CB1) receptor |
Families Citing this family (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010504957A (en) * | 2006-09-29 | 2010-02-18 | アクテリオン ファーマシューティカルズ リミテッド | 3-Aza-bicyclo [3.1.0] hexane derivatives |
| JP2010511038A (en) | 2006-12-01 | 2010-04-08 | アクテリオン ファーマシューティカルズ リミテッド | 3-Heteroaryl (amino or amide) -1- (biphenyl or phenylthiazolyl) carbonylpiperidine derivatives as orexin receptor inhibitors |
| TW200833328A (en) | 2006-12-28 | 2008-08-16 | Actelion Pharmaceuticals Ltd | 2-aza-bicyclo[3.1.0]hexane derivatives |
| CL2008000836A1 (en) * | 2007-03-26 | 2008-11-07 | Actelion Pharmaceuticals Ltd | Thiazolidine derivative compounds, orexin receptor antagonists; pharmaceutical composition that includes them; and its use in the treatment of emotional neurosis, severe depression, psychotic disorders, Alzheimer's, parkinson's, pain, among others. |
| JP2010534647A (en) * | 2007-07-27 | 2010-11-11 | アクテリオン ファーマシューティカルズ リミテッド | 2-Aza-bicyclo [3.3.0] octane derivatives |
| WO2009040730A2 (en) * | 2007-09-24 | 2009-04-02 | Actelion Pharmaceuticals Ltd | Pyrrolidines and piperidines as orexin receptor antagonists |
| AU2009215243A1 (en) * | 2008-02-21 | 2009-08-27 | Actelion Pharmaceuticals Ltd. | 2-aza-bicyclo[2.2.1]heptane derivatives |
| EP2318367B1 (en) * | 2008-04-30 | 2013-03-20 | Actelion Pharmaceuticals Ltd. | Piperidine and pyrrolidine compounds |
| US8546576B2 (en) * | 2008-06-06 | 2013-10-01 | Sk Biopharmaceuticals Co., Ltd. | 3 or 4-substituted piperidine compounds |
| CA2739915A1 (en) * | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | 2,5-disubstituted piperidine orexin receptor antagonists |
| EP2348856B1 (en) * | 2008-10-21 | 2013-08-14 | Merck Sharp & Dohme Corp. | 2,4-disubstituted pyrrolidine orexin receptor antagonists |
| US8357700B2 (en) | 2008-10-21 | 2013-01-22 | Merck Sharp & Dohme Corp. | 2,3-disubstituted piperidine orexin receptor antagonists |
| WO2010048012A1 (en) * | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | 2,5-disubstituted piperidine orexin receptor antagonists |
| CA2739917A1 (en) | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | 2,5-disubstituted morpholine orexin receptor antagonists |
| CA2739927A1 (en) * | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | Disubstituted azepan orexin receptor antagonists |
| RU2617696C2 (en) * | 2012-02-17 | 2017-04-26 | ЭЙСАЙ Ар ЭНД Ди МЕНЕДЖМЕНТ КО., ЛТД | Methods and compounds that can be used to synthesize orexin-2 receptors antagonists |
| ES2613663T3 (en) * | 2012-06-15 | 2017-05-25 | Taisho Pharmaceutical Co., Ltd. | 1,3-oxazolidine or 1,3-oxazinan compounds as orexin receptor antagonists |
| US9273033B2 (en) | 2012-11-20 | 2016-03-01 | Merck Sharp & Dohme Corp. | Substituted pyridone derivatives as PDE10 inhibitors |
| EP2934527A4 (en) | 2012-12-20 | 2016-07-13 | Merck Sharp & Dohme | 2-pyridyloxy-4-ester orexin receptor antagonists |
| WO2014099698A1 (en) * | 2012-12-20 | 2014-06-26 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-3-nitrile-4-substituted orexin receptor antagonists |
| EP2934516A4 (en) | 2012-12-20 | 2016-07-20 | Merck Sharp & Dohme | 3-ester-4-substituted orexin receptor antagonists |
| WO2014113303A1 (en) | 2013-01-16 | 2014-07-24 | Merck Sharp & Dohme Corp. | 4-fluoropiperidine orexin receptor antagonists |
| US9745284B2 (en) | 2013-03-08 | 2017-08-29 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-ether orexin receptor antagonists |
| EP2988746A1 (en) * | 2013-04-23 | 2016-03-02 | Merck Sharp & Dohme Corp. | Halo and trifluoromethyl substituted orexin receptor antagonists |
| WO2014176142A1 (en) * | 2013-04-23 | 2014-10-30 | Merck Sharp & Dohme Corp. | Hydroxy-substituted orexin receptor antagonists |
| WO2015018029A1 (en) | 2013-08-08 | 2015-02-12 | Merck Sharp & Dohme Corp. | Oxazole orexin receptor antagonists |
| WO2015018027A1 (en) | 2013-08-08 | 2015-02-12 | Merck Sharp & Dohme Corp. | Thiazole orexin receptor antagonists |
| WO2015095441A1 (en) * | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | 2-amino-3-ester-pyridyl orexin receptor antagonists |
| WO2016065587A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Pyrazole orexin receptor antagonists |
| WO2016065586A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Pyrazole, triazole and tetrazole orexin receptor antagonists |
| WO2016065584A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Piperidine oxadiazole and thiadiazole orexin receptor antagonists |
| WO2016065583A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Oxazole orexin receptor antagonists |
| US9994570B2 (en) | 2014-11-26 | 2018-06-12 | Merck Sharp & Dohme Corp. | Bridged diazepane orexin receptor antagonists |
| WO2016085784A1 (en) | 2014-11-26 | 2016-06-02 | Merck Sharp & Dohme Corp. | Methyl diazepane orexin receptor antagonists |
| WO2016086358A1 (en) | 2014-12-02 | 2016-06-09 | Merck Sharp & Dohme Corp. | Hydroxymethyl piperidine orexin receptor antagonists |
| WO2016086357A1 (en) | 2014-12-02 | 2016-06-09 | Merck Sharp & Dohme Corp. | Methyl oxazole orexin receptor antagonists |
| WO2016100157A2 (en) | 2014-12-19 | 2016-06-23 | Merck Sharp & Dohme Corp. | 6,5-bicyclic octahydropyrrolopyridine orexin receptor antagonists |
| WO2016095204A1 (en) | 2014-12-19 | 2016-06-23 | Merck Sharp & Dohme Corp. | Pyrrolidine orexin receptor antagonists |
| US9987255B2 (en) | 2014-12-19 | 2018-06-05 | Merck Sharp & Dohme Corp. | 5,5-bicyclic oxazole orexin receptor antagonists |
| WO2016095205A1 (en) | 2014-12-19 | 2016-06-23 | Merck Sharp & Dohme Corp. | Heteroaryl orexin receptor antagonists |
| WO2016101118A1 (en) | 2014-12-23 | 2016-06-30 | Merck Sharp & Dohme Corp. | Amidoethyl azole orexin receptor antagonists |
| WO2016101119A1 (en) | 2014-12-23 | 2016-06-30 | Merck Sharp & Dohme Corp. | Fused heteroaryl derivatives as orexin receptor antagonists |
| WO2017012502A1 (en) | 2015-07-17 | 2017-01-26 | Sunshine Lake Pharma Co., Ltd. | Substituted quinazoline compounds and preparation and uses thereof |
| CN108025040A (en) * | 2015-08-25 | 2018-05-11 | 阿尔莫生物科技股份有限公司 | The method that disease and illness are treated using interleukin-10 |
| US11655241B2 (en) | 2018-06-29 | 2023-05-23 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US11098029B2 (en) | 2019-02-13 | 2021-08-24 | Merck Sharp & Dohme Corp. | 5-alkyl pyrrolidine orexin receptor agonists |
Citations (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999009024A1 (en) | 1997-08-14 | 1999-02-25 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as hfgan72 antagonists |
| WO1999058533A1 (en) | 1998-05-08 | 1999-11-18 | Smithkline Beecham Plc | Phenylurea and phenylthio urea derivatives |
| WO2000047580A2 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives |
| WO2000047577A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as orexin receptor antagonists |
| WO2000047576A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Cinnamide derivatives as orexin-1 receptors antagonists |
| WO2001068609A1 (en) | 2000-03-14 | 2001-09-20 | Actelion Pharmaceuticals Ltd. | 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2001085693A1 (en) | 2000-05-11 | 2001-11-15 | Banyu Pharmaceutical Co., Ltd. | N-acyltetrahydroisoquinoline derivatives |
| WO2001096302A1 (en) | 2000-06-16 | 2001-12-20 | Smithkline Beecham P.L.C. | Piperidines for use as orexin receptor antagonists |
| WO2002044172A1 (en) | 2000-11-28 | 2002-06-06 | Smithkline Beecham P.L.C. | Morpholine derivatives as antagonists of orexin receptors |
| WO2002051232A2 (en) | 2000-12-27 | 2002-07-04 | Actelion Pharmaceuticals Ltd. | Novel benzazepines and related heterocyclic derivatives |
| WO2002089800A2 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2002090355A1 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amines |
| WO2003002559A2 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | Piperidine compounds for use as orexin receptor antagonist |
| WO2003002561A1 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2003032991A1 (en) | 2001-10-11 | 2003-04-24 | Smithkline Beecham Plc | N-aroyl piperazine derivatives as orexin receptor antagonists |
| WO2003037847A1 (en) | 2001-11-01 | 2003-05-08 | Smithkline Beecham P.L.C. | Benzamide derivatives as antagonists of orexin receptors |
| WO2003041711A1 (en) | 2001-11-10 | 2003-05-22 | Smithkline Beecham P.L.C. | Piperazine bis-amide derivatives and their use as antagonists of the orexin receptor |
| WO2003051872A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham P.L.C. | Ethylene diamine derivatives and their use as orexin-receptor antagonists |
| WO2003051368A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham Plc | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2003051873A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham Plc | Piperazine compounds and their phamaceutical use |
| WO2004004733A1 (en) | 2002-07-09 | 2004-01-15 | Actelion Pharmaceuticals Ltd. | 7,8,9,10-tetrahydro-6h-azepino, 6,7,8,9-tetrahydro-pyrido and 2,3-dihydro-2h-pyrrolo[2,1-b]-quinazolinone derivatives |
| WO2004026866A1 (en) | 2002-09-18 | 2004-04-01 | Glaxo Group Limited | N-aroyl cyclic amines as orexin receptor antagonists |
| WO2004033418A2 (en) | 2002-10-11 | 2004-04-22 | Actelion Pharmaceuticals Ltd. | Sulfonylamino-acetic derivatives and their use as orexin receptor antagonists |
| WO2004041816A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Azacyclic compounds as orexin receptor antagonist |
| WO2004041807A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Novel compounds |
| WO2004052876A1 (en) | 2002-12-12 | 2004-06-24 | Janssen Pharmaceutica, N.V. | Substituted 4-phenyl-[1,3]-dioxanes |
| WO2004083218A1 (en) | 2003-03-20 | 2004-09-30 | Actelion Pharmaceuticals Ltd | Guanidine derivatives and use thereof as neuropeptide ff receptor antagonists |
| WO2004085403A1 (en) | 2003-03-26 | 2004-10-07 | Actelion Pharmaceuticals Ltd | Tetrahydroisoquinolyl acetamide derivatives for use as orexin receptor antagonists |
| WO2004096780A1 (en) | 2003-04-28 | 2004-11-11 | Actelion Pharmaceuticals Ltd | Quinoxalinone-3- one derivatives as orexin receptor antagonists |
| WO2005060959A1 (en) | 2003-12-22 | 2005-07-07 | Sanofi-Aventis | Pyrazole derivatives and use thereof as orexin receptor antagonists |
| WO2005075458A1 (en) | 2004-02-10 | 2005-08-18 | Sanofi-Aventis | Pyrimidine derivatives as orexin receptors antagonists |
| WO2005118548A1 (en) | 2004-03-01 | 2005-12-15 | Actelion Pharmaceuticals Ltd | Substituted 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2006067224A2 (en) | 2004-12-23 | 2006-06-29 | Biovitrum Ab (Publ) | Spiro-benzodioxole and spiro-benzodioxane compounds as orexin receptor antagonists |
| WO2006110626A1 (en) | 2005-04-12 | 2006-10-19 | Merck & Co., Inc. | Amidopropoxyphenyl orexin receptor antagonists |
| WO2006117669A1 (en) | 2005-05-03 | 2006-11-09 | Pfizer Inc. | Amide resorcinol compounds |
| WO2006127550A1 (en) | 2005-05-23 | 2006-11-30 | Merck & Co., Inc. | Proline bis-amide orexin receptor antagonists |
| WO2007019234A2 (en) | 2005-08-04 | 2007-02-15 | Merck & Co., Inc. | Aminoethane sulfonamide orexin receptor antagonists |
| WO2007025069A2 (en) | 2005-08-26 | 2007-03-01 | Merck & Co., Inc. | Diazaspirodecane orexin receptor antagonists |
| WO2007061763A2 (en) | 2005-11-22 | 2007-05-31 | Merck & Co., Inc. | Indole orexin receptor antagonists |
| WO2007116374A1 (en) | 2006-04-11 | 2007-10-18 | Actelion Pharmaceuticals Ltd | Novel sulfonamide compounds |
| WO2007122591A2 (en) | 2006-04-26 | 2007-11-01 | Actelion Pharmaceuticals Ltd | Pyrazolo-tetrahydro pyridine derivatives as orexin receptor antagonists |
| WO2007126935A2 (en) | 2006-03-29 | 2007-11-08 | Merck & Co., Inc. | Diazepan orexin receptor antagonists |
| WO2007126934A2 (en) | 2006-03-29 | 2007-11-08 | Merck & Co., Inc. | Amidoethylthioether orexin receptor antagonists |
| WO2008008551A2 (en) | 2006-07-14 | 2008-01-17 | Merck & Co., Inc. | 2-substituted proline bis-amide orexin receptor antagonists |
| WO2008008517A2 (en) | 2006-07-14 | 2008-01-17 | Merck & Co., Inc. | Bridged diazepan orexin receptor antagonists |
| WO2008008518A1 (en) | 2006-07-14 | 2008-01-17 | Merck & Co., Inc. | Substituted diazepan orexin receptor antagonists |
| WO2008020405A2 (en) | 2006-08-15 | 2008-02-21 | Actelion Pharmaceuticals Ltd | Azetidine compounds as orexin receptor antagonists |
| WO2008026149A1 (en) | 2006-08-28 | 2008-03-06 | Actelion Pharmaceuticals Ltd | 1,4,5,6, 7,8-hexahydro-i^1s-triaza-azulene derivatives as orexin receptor antagonists |
| WO2008038251A2 (en) | 2006-09-29 | 2008-04-03 | Actelion Pharmaceuticals Ltd | 3-aza-bicyclo[3.1.0]hexane derivatives |
| WO2008065626A2 (en) | 2006-12-01 | 2008-06-05 | Actelion Pharmaceuticals Ltd | 3-heteroaryl (amino or amido)-1- (biphenyl or phenylthiazolyl) carbonylpiperidine derivativesas orexin receptor inhibitors |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA848275B (en) | 1983-12-28 | 1985-08-28 | Degussa | New piridine-2-ethers or pyridine-2-thioethers having a nitrogen-containing cycloaliphatic ring |
| US6784192B2 (en) | 2000-01-20 | 2004-08-31 | Eisai Co., Ltd. | Piperidine compound and pharmaceutical composition thereof |
| GB0130388D0 (en) | 2001-12-19 | 2002-02-06 | Smithkline Beecham Plc | Compounds |
| RU2294927C2 (en) * | 2002-05-29 | 2007-03-10 | Танабе Сейяку Ко., Лтд. | Derivatives of piperidine, methods for their preparing, pharmaceutical composition based on thereof, their using and treatment method |
| TWI283241B (en) | 2002-05-29 | 2007-07-01 | Tanabe Seiyaku Co | Novel piperidine compound |
| ITMI20021273A1 (en) * | 2002-06-11 | 2003-12-11 | Milano Politecnico | SYSTEM AND METHOD FOR THE AUTOMATIC DETECTION OF THE EXPIRATORY FLOW LIMITATION |
| WO2004033445A1 (en) | 2002-10-08 | 2004-04-22 | Merck Frosst Canada & Co. | 4-amino-azepan-3-one compounds as cathepsin k inhibitors useful in the treatment of osteoporosis |
| GB0225944D0 (en) | 2002-11-06 | 2002-12-11 | Glaxo Group Ltd | Novel compounds |
| JO2355B1 (en) | 2003-04-15 | 2006-12-12 | ميرك شارب اند دوم كوربوريشن | CGRP receptor antagonists |
| DE102004062544A1 (en) | 2004-12-24 | 2006-07-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel substituted pyrrolidinones, their preparation and their use as pharmaceuticals |
| PE20081229A1 (en) | 2006-12-01 | 2008-08-28 | Merck & Co Inc | DIAZEPAM OREXIN RECEPTOR ANTAGONISTS REPLACED |
| PL2152690T3 (en) | 2007-05-23 | 2012-06-29 | Merck Sharp & Dohme | Pyridyl piperidine orexin receptor antagonists |
| GB0806536D0 (en) | 2008-04-10 | 2008-05-14 | Glaxo Group Ltd | Novel compounds |
| MX2010012631A (en) | 2008-05-22 | 2010-12-14 | Merck Sharp & Dohme | Process for the preparation of an orexin receptor antagonist. |
| WO2010048012A1 (en) | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | 2,5-disubstituted piperidine orexin receptor antagonists |
| US8357700B2 (en) | 2008-10-21 | 2013-01-22 | Merck Sharp & Dohme Corp. | 2,3-disubstituted piperidine orexin receptor antagonists |
| CA2739917A1 (en) | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | 2,5-disubstituted morpholine orexin receptor antagonists |
| CA2739927A1 (en) | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | Disubstituted azepan orexin receptor antagonists |
| CA2739915A1 (en) | 2008-10-21 | 2010-04-29 | Merck Sharp & Dohme Corp. | 2,5-disubstituted piperidine orexin receptor antagonists |
| EP2348856B1 (en) | 2008-10-21 | 2013-08-14 | Merck Sharp & Dohme Corp. | 2,4-disubstituted pyrrolidine orexin receptor antagonists |
| GB0823467D0 (en) | 2008-12-23 | 2009-01-28 | Glaxo Group Ltd | Novel Compounds |
| EP2470523A1 (en) | 2009-08-24 | 2012-07-04 | Glaxo Group Limited | 5-methyl-piperidine derivatives as orexin receptor antagonists for the treatment of sleep disorder |
-
2008
- 2008-05-22 PL PL08754659T patent/PL2152690T3/en unknown
- 2008-05-22 DK DK08754659.4T patent/DK2152690T3/en active
- 2008-05-22 CN CN2008800169258A patent/CN101679366B/en not_active Expired - Fee Related
- 2008-05-22 BR BRPI0811842-6A2A patent/BRPI0811842A2/en active Search and Examination
- 2008-05-22 EP EP08754659A patent/EP2152690B1/en active Active
- 2008-05-22 MX MX2009012579A patent/MX2009012579A/en active IP Right Grant
- 2008-05-22 SI SI200830593T patent/SI2152690T1/en unknown
- 2008-05-22 CA CA2687321A patent/CA2687321C/en not_active Expired - Fee Related
- 2008-05-22 KR KR1020097024362A patent/KR101480279B1/en active IP Right Grant
- 2008-05-22 JP JP2010509380A patent/JP4881476B2/en not_active Expired - Fee Related
- 2008-05-22 MY MYPI20094689A patent/MY148544A/en unknown
- 2008-05-22 US US12/600,388 patent/US8242121B2/en active Active
- 2008-05-22 NZ NZ580887A patent/NZ580887A/en not_active IP Right Cessation
- 2008-05-22 RS RS20120111A patent/RS52200B/en unknown
- 2008-05-22 AU AU2008257411A patent/AU2008257411B2/en not_active Ceased
- 2008-05-22 PT PT08754659T patent/PT2152690E/en unknown
- 2008-05-22 AT AT08754659T patent/ATE540944T1/en active
- 2008-05-22 UA UAA200913366A patent/UA99620C2/en unknown
- 2008-05-22 WO PCT/US2008/006563 patent/WO2008147518A1/en active Application Filing
- 2008-05-22 ES ES08754659T patent/ES2379744T3/en active Active
- 2008-05-22 RU RU2009147733/04A patent/RU2470021C2/en not_active IP Right Cessation
-
2009
- 2009-10-26 ZA ZA200907495A patent/ZA200907495B/en unknown
- 2009-10-27 IL IL201790A patent/IL201790A0/en unknown
- 2009-10-30 NI NI200900193A patent/NI200900193A/en unknown
- 2009-11-18 GT GT200900300A patent/GT200900300A/en unknown
- 2009-11-18 SV SV2009003417A patent/SV2009003417A/en not_active Application Discontinuation
- 2009-11-20 DO DO2009000263A patent/DOP2009000263A/en unknown
- 2009-11-20 EC EC2009009749A patent/ECSP099749A/en unknown
- 2009-11-23 CO CO09133050A patent/CO6251266A2/en active IP Right Grant
- 2009-12-04 CR CR11146A patent/CR11146A/en unknown
- 2009-12-17 MA MA32427A patent/MA31451B1/en unknown
-
2010
- 2010-02-18 HK HK10101749.0A patent/HK1134084A1/en not_active IP Right Cessation
-
2011
- 2011-10-19 JP JP2011229710A patent/JP5639564B2/en not_active Expired - Fee Related
-
2012
- 2012-03-15 HR HR20120240T patent/HRP20120240T1/en unknown
- 2012-04-10 CY CY20121100354T patent/CY1112969T1/en unknown
- 2012-08-07 US US13/568,242 patent/US8569311B2/en active Active
Patent Citations (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999009024A1 (en) | 1997-08-14 | 1999-02-25 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as hfgan72 antagonists |
| WO1999058533A1 (en) | 1998-05-08 | 1999-11-18 | Smithkline Beecham Plc | Phenylurea and phenylthio urea derivatives |
| WO2000047580A2 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives |
| WO2000047577A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as orexin receptor antagonists |
| WO2000047576A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Cinnamide derivatives as orexin-1 receptors antagonists |
| WO2001068609A1 (en) | 2000-03-14 | 2001-09-20 | Actelion Pharmaceuticals Ltd. | 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2001085693A1 (en) | 2000-05-11 | 2001-11-15 | Banyu Pharmaceutical Co., Ltd. | N-acyltetrahydroisoquinoline derivatives |
| WO2001096302A1 (en) | 2000-06-16 | 2001-12-20 | Smithkline Beecham P.L.C. | Piperidines for use as orexin receptor antagonists |
| WO2002044172A1 (en) | 2000-11-28 | 2002-06-06 | Smithkline Beecham P.L.C. | Morpholine derivatives as antagonists of orexin receptors |
| WO2002051232A2 (en) | 2000-12-27 | 2002-07-04 | Actelion Pharmaceuticals Ltd. | Novel benzazepines and related heterocyclic derivatives |
| WO2002051838A1 (en) | 2000-12-27 | 2002-07-04 | Actelion Pharmaceuticals Ltd. | Novel benzazepines and related heterocyclic derivatives which are useful as orexin receptor antagonists |
| WO2002089800A2 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2002090355A1 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amines |
| WO2003002559A2 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | Piperidine compounds for use as orexin receptor antagonist |
| WO2003002561A1 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2003032991A1 (en) | 2001-10-11 | 2003-04-24 | Smithkline Beecham Plc | N-aroyl piperazine derivatives as orexin receptor antagonists |
| WO2003037847A1 (en) | 2001-11-01 | 2003-05-08 | Smithkline Beecham P.L.C. | Benzamide derivatives as antagonists of orexin receptors |
| WO2003041711A1 (en) | 2001-11-10 | 2003-05-22 | Smithkline Beecham P.L.C. | Piperazine bis-amide derivatives and their use as antagonists of the orexin receptor |
| WO2003051872A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham P.L.C. | Ethylene diamine derivatives and their use as orexin-receptor antagonists |
| WO2003051368A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham Plc | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2003051873A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham Plc | Piperazine compounds and their phamaceutical use |
| WO2004004733A1 (en) | 2002-07-09 | 2004-01-15 | Actelion Pharmaceuticals Ltd. | 7,8,9,10-tetrahydro-6h-azepino, 6,7,8,9-tetrahydro-pyrido and 2,3-dihydro-2h-pyrrolo[2,1-b]-quinazolinone derivatives |
| WO2004026866A1 (en) | 2002-09-18 | 2004-04-01 | Glaxo Group Limited | N-aroyl cyclic amines as orexin receptor antagonists |
| WO2004033418A2 (en) | 2002-10-11 | 2004-04-22 | Actelion Pharmaceuticals Ltd. | Sulfonylamino-acetic derivatives and their use as orexin receptor antagonists |
| WO2004041816A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Azacyclic compounds as orexin receptor antagonist |
| WO2004041807A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Novel compounds |
| WO2004052876A1 (en) | 2002-12-12 | 2004-06-24 | Janssen Pharmaceutica, N.V. | Substituted 4-phenyl-[1,3]-dioxanes |
| WO2004083218A1 (en) | 2003-03-20 | 2004-09-30 | Actelion Pharmaceuticals Ltd | Guanidine derivatives and use thereof as neuropeptide ff receptor antagonists |
| WO2004085403A1 (en) | 2003-03-26 | 2004-10-07 | Actelion Pharmaceuticals Ltd | Tetrahydroisoquinolyl acetamide derivatives for use as orexin receptor antagonists |
| WO2004096780A1 (en) | 2003-04-28 | 2004-11-11 | Actelion Pharmaceuticals Ltd | Quinoxalinone-3- one derivatives as orexin receptor antagonists |
| WO2005060959A1 (en) | 2003-12-22 | 2005-07-07 | Sanofi-Aventis | Pyrazole derivatives and use thereof as orexin receptor antagonists |
| WO2005075458A1 (en) | 2004-02-10 | 2005-08-18 | Sanofi-Aventis | Pyrimidine derivatives as orexin receptors antagonists |
| WO2005118548A1 (en) | 2004-03-01 | 2005-12-15 | Actelion Pharmaceuticals Ltd | Substituted 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2006067224A2 (en) | 2004-12-23 | 2006-06-29 | Biovitrum Ab (Publ) | Spiro-benzodioxole and spiro-benzodioxane compounds as orexin receptor antagonists |
| WO2006110626A1 (en) | 2005-04-12 | 2006-10-19 | Merck & Co., Inc. | Amidopropoxyphenyl orexin receptor antagonists |
| WO2006117669A1 (en) | 2005-05-03 | 2006-11-09 | Pfizer Inc. | Amide resorcinol compounds |
| WO2006127550A1 (en) | 2005-05-23 | 2006-11-30 | Merck & Co., Inc. | Proline bis-amide orexin receptor antagonists |
| WO2007019234A2 (en) | 2005-08-04 | 2007-02-15 | Merck & Co., Inc. | Aminoethane sulfonamide orexin receptor antagonists |
| WO2007025069A2 (en) | 2005-08-26 | 2007-03-01 | Merck & Co., Inc. | Diazaspirodecane orexin receptor antagonists |
| WO2007061763A2 (en) | 2005-11-22 | 2007-05-31 | Merck & Co., Inc. | Indole orexin receptor antagonists |
| WO2007126935A2 (en) | 2006-03-29 | 2007-11-08 | Merck & Co., Inc. | Diazepan orexin receptor antagonists |
| WO2007126934A2 (en) | 2006-03-29 | 2007-11-08 | Merck & Co., Inc. | Amidoethylthioether orexin receptor antagonists |
| WO2007116374A1 (en) | 2006-04-11 | 2007-10-18 | Actelion Pharmaceuticals Ltd | Novel sulfonamide compounds |
| WO2007122591A2 (en) | 2006-04-26 | 2007-11-01 | Actelion Pharmaceuticals Ltd | Pyrazolo-tetrahydro pyridine derivatives as orexin receptor antagonists |
| WO2008008551A2 (en) | 2006-07-14 | 2008-01-17 | Merck & Co., Inc. | 2-substituted proline bis-amide orexin receptor antagonists |
| WO2008008517A2 (en) | 2006-07-14 | 2008-01-17 | Merck & Co., Inc. | Bridged diazepan orexin receptor antagonists |
| WO2008008518A1 (en) | 2006-07-14 | 2008-01-17 | Merck & Co., Inc. | Substituted diazepan orexin receptor antagonists |
| WO2008020405A2 (en) | 2006-08-15 | 2008-02-21 | Actelion Pharmaceuticals Ltd | Azetidine compounds as orexin receptor antagonists |
| WO2008026149A1 (en) | 2006-08-28 | 2008-03-06 | Actelion Pharmaceuticals Ltd | 1,4,5,6, 7,8-hexahydro-i^1s-triaza-azulene derivatives as orexin receptor antagonists |
| WO2008038251A2 (en) | 2006-09-29 | 2008-04-03 | Actelion Pharmaceuticals Ltd | 3-aza-bicyclo[3.1.0]hexane derivatives |
| WO2008065626A2 (en) | 2006-12-01 | 2008-06-05 | Actelion Pharmaceuticals Ltd | 3-heteroaryl (amino or amido)-1- (biphenyl or phenylthiazolyl) carbonylpiperidine derivativesas orexin receptor inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| CAI J ET AL: "Antagonists of the orexin receptors", EXPERT OPINION ON THERAPEUTIC PATENTS, ASHLEY PUBLICATIONS, GB, vol. 16, no. 5, 1 May 2006 (2006-05-01), pages 631 - 646, XP002458093, ISSN: 1354-3776 * |
| EXPERT OPIN. THER. PATENTS, vol. 1, no. 5, 2006, pages 631 - 646 |
Cited By (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8569311B2 (en) | 2007-05-23 | 2013-10-29 | Merch Sharp & Dohme Corp. | Pyridyl piperidine orexin receptor antagonists |
| US8242121B2 (en) | 2007-05-23 | 2012-08-14 | Merck Sharp & Dohme Corp. | Pyridyl piperidine orexin receptor antagonists |
| WO2009124956A1 (en) * | 2008-04-10 | 2009-10-15 | Glaxo Group Limited | Pyridine derivatives used to treat orexin related disorders |
| WO2009143033A1 (en) * | 2008-05-22 | 2009-11-26 | Merck & Co., Inc. | Process for the preparation of an orexin receptor antagonist |
| AU2009249306B8 (en) * | 2008-05-22 | 2013-07-25 | Merck Canada Inc. | Process for the preparation of an orexin receptor antagonist |
| US8674093B2 (en) | 2008-05-22 | 2014-03-18 | Merck Canada Inc. | Process for the preparation of an orexin receptor antagonist |
| AU2009249306B2 (en) * | 2008-05-22 | 2013-03-28 | Merck Canada Inc. | Process for the preparation of an orexin receptor antagonist |
| WO2011006960A1 (en) | 2009-07-15 | 2011-01-20 | Rottapharm S.P.A. | Spiro amino compounds suitable for the treatment of inter alia sleep disorders and drug addiction |
| WO2011023578A1 (en) * | 2009-08-24 | 2011-03-03 | Glaxo Group Limited | 5-methyl-piperidine derivatives as orexin receptor antagonists for the treatment of sleep disorder |
| EP2632253A4 (en) * | 2010-10-29 | 2014-05-14 | Merck Sharp & Dohme | Process for the preparation of an orexin receptor antagonist |
| EP2632253A1 (en) * | 2010-10-29 | 2013-09-04 | Merck Sharp & Dohme Corp. | Process for the preparation of an orexin receptor antagonist |
| EP2484674A1 (en) | 2011-02-02 | 2012-08-08 | Rottapharm S.P.A. | Spiro aminic compounds with NK1 antagonist activity |
| WO2012114252A1 (en) * | 2011-02-21 | 2012-08-30 | Actelion Pharmaceuticals Ltd | Novel indole and pyrrolopyridine amides |
| WO2013005755A1 (en) | 2011-07-05 | 2013-01-10 | 大正製薬株式会社 | Methylpiperidine derivative |
| EP3189840A1 (en) * | 2011-07-28 | 2017-07-12 | Kempharm, Inc. | Methylphenidate-prodrugs, processes of making and using the same |
| US10231960B2 (en) | 2011-07-28 | 2019-03-19 | Kempharm Inc. | Methylphenidate-prodrugs, processes of making and using the same |
| US9156819B2 (en) | 2011-10-19 | 2015-10-13 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-nitrile orexin receptor antagonists |
| WO2013059222A1 (en) | 2011-10-19 | 2013-04-25 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-nitrile orexin receptor antagonists |
| EP2768305A1 (en) * | 2011-10-21 | 2014-08-27 | Merck Sharp & Dohme Corp. | 2,5-disubstituted thiomorpholine orexin receptor antagonists |
| EP2768305A4 (en) * | 2011-10-21 | 2015-04-29 | Merck Sharp & Dohme | 2,5-disubstituted thiomorpholine orexin receptor antagonists |
| WO2013092893A1 (en) | 2011-12-21 | 2013-06-27 | Rottapharm Spa | Chemical compounds |
| US9896452B2 (en) | 2012-02-07 | 2018-02-20 | Eolas Therapeutics, Inc. | Substituted prolines/piperidines as orexin receptor antagonists |
| US9440982B2 (en) | 2012-02-07 | 2016-09-13 | Eolas Therapeutics, Inc. | Substituted prolines/piperidines as orexin receptor antagonists |
| US9499517B2 (en) | 2012-02-07 | 2016-11-22 | Eolas Therapeutics, Inc. | Substituted prolines / piperidines as orexin receptor antagonists |
| WO2013127913A1 (en) | 2012-03-01 | 2013-09-06 | Rottapharm S.P.A. | 4,4-difluoro-piperidine-compounds |
| US9174977B2 (en) | 2012-03-19 | 2015-11-03 | Rottapharm Biotech S.R.L. | 2-azabicyclo[4.1.0]heptane derivatives as orexin receptor antagonists for the treatment of certain disorders |
| WO2014091876A1 (en) * | 2012-12-13 | 2014-06-19 | 大正製薬株式会社 | Fluorine-substituted piperidine compound |
| US9828345B2 (en) | 2013-02-28 | 2017-11-28 | Bristol-Myers Squibb Company | Phenylpyrazole derivatives as potent ROCK1 and ROCK2 inhibitors |
| US9458110B2 (en) | 2013-02-28 | 2016-10-04 | Bristol-Myers Squibb Company | Phenylpyrazole derivatives as potent ROCK1 and ROCK2 inhibitors |
| AU2015226679B2 (en) * | 2014-03-06 | 2017-05-25 | Shanghai Haiyan Pharmaceutical Technology Co. Ltd | Piperidine derivatives as orexin receptor antagonist |
| US10100047B2 (en) * | 2014-03-06 | 2018-10-16 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Piperidine derivatives as orexin receptor antagonist |
| CN106414439A (en) * | 2014-03-06 | 2017-02-15 | 上海海雁医药科技有限公司 | Piperidine derivatives as orexin receptor antagonist |
| EP3115362A4 (en) * | 2014-03-06 | 2017-08-16 | Shanghai Haiyan Pharmaceutical Technology Co. Ltd. | Piperidine derivatives as orexin receptor antagonist |
| US20170233385A1 (en) * | 2014-03-06 | 2017-08-17 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Piperidine derivatives as orexin receptor antagonist |
| WO2015131773A1 (en) * | 2014-03-06 | 2015-09-11 | 上海海雁医药科技有限公司 | Piperidine derivatives as orexin receptor antagonist |
| US11566069B2 (en) | 2014-03-27 | 2023-01-31 | Bird Rock Bio, Inc. | Treatment of disease responsive to modulation of cannabanoid 1(CB1) receptor signaling |
| US10308712B2 (en) | 2014-03-27 | 2019-06-04 | Bird Rock Bio, Inc. | Antibodies that bind human cannabinoid 1 (CB1) receptor |
| US10221170B2 (en) | 2014-08-13 | 2019-03-05 | Eolas Therapeutics, Inc. | Difluoropyrrolidines as orexin receptor modulators |
| WO2016025669A1 (en) | 2014-08-13 | 2016-02-18 | Eolas Therapeutics, Inc. | Difluoropyrrolidines as orexin receptor modulators |
| WO2016065585A1 (en) * | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Piperidine isoxazole and isothiazole orexin receptor antagonists |
| US10017504B2 (en) | 2014-10-30 | 2018-07-10 | Merck Sharp & Dohme Corp. | Piperidine isoxazole and isothiazole orexin receptor antagonists |
| US10011595B2 (en) | 2014-12-19 | 2018-07-03 | Merck Sharp & Dohme Corp. | Ethyldiamine orexin receptor antagonists |
| WO2016100161A1 (en) * | 2014-12-19 | 2016-06-23 | Merck Sharp & Dohme Corp. | Ethyldiamine orexin receptor antagonists |
| WO2017028732A1 (en) * | 2015-08-14 | 2017-02-23 | 上海海雁医药科技有限公司 | Crystal form of orexin receptor antagonist compound, and preparation method and application thereof |
| US10874674B2 (en) | 2015-08-14 | 2020-12-29 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Crystal form of orexin receptor antagonist compound, and preparation method and application thereof |
| CN111499625B (en) * | 2015-08-14 | 2022-09-02 | 上海海雁医药科技有限公司 | Preparation method of orexin receptor antagonist receptor compound and intermediate and crystal form thereof |
| US10912775B2 (en) | 2015-08-14 | 2021-02-09 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Crystal form of orexin receptor antagonist compound, and preparation method and application thereof |
| US10898486B2 (en) | 2015-08-14 | 2021-01-26 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Crystal form of orexin receptor antagonist compound, and preparation method and application thereof |
| CN107709318A (en) * | 2015-08-14 | 2018-02-16 | 上海海雁医药科技有限公司 | Crystal formation of orexin receptor antagonists compound and its preparation method and application |
| US10668066B2 (en) | 2015-08-14 | 2020-06-02 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Crystal form of orexin receptor antagonist compound, and preparation method and application thereof |
| CN111499625A (en) * | 2015-08-14 | 2020-08-07 | 上海海雁医药科技有限公司 | Preparation method of orexin receptor antagonist receptor compound and intermediate and crystal form thereof |
| TWI631122B (en) * | 2015-08-14 | 2018-08-01 | 上海海雁醫藥科技有限公司 | Preparation method of orexin receptor resisting agent receptor compound, intermediate and crystal form thereof |
| US11421026B2 (en) | 2015-09-30 | 2022-08-23 | Bird Rock Bio, Inc. | Antibodies that bind human cannabinoid 1 (CB1) receptor |
| US11434236B2 (en) | 2016-02-12 | 2022-09-06 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| US10894789B2 (en) | 2016-02-12 | 2021-01-19 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| US12084437B2 (en) | 2016-02-12 | 2024-09-10 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| WO2017139603A1 (en) | 2016-02-12 | 2017-08-17 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| WO2017194548A1 (en) | 2016-05-10 | 2017-11-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of autoimmune inflammatory diseases |
| US10584112B2 (en) | 2016-12-11 | 2020-03-10 | Kempharm, Inc. | Methylphenidate-prodrugs, processes of making and using the same |
| US11021460B2 (en) | 2016-12-11 | 2021-06-01 | Kempharm, Inc. | Compositions comprising methylphenidate-prodrugs, processes of making and using the same |
| US11021459B2 (en) | 2016-12-11 | 2021-06-01 | Kempharm, Inc. | Compositions comprising methylphenidate-prodrugs, processes of making and using the same |
| US10954213B2 (en) | 2016-12-11 | 2021-03-23 | Kempharm, Inc. | Compositions comprising methylphenidate-prodrugs, processes of making and using the same |
| US10954212B2 (en) | 2016-12-11 | 2021-03-23 | Kempharm, Inc. | Compositions comprising methylphenidate-prodrugs, processes of making and using the same |
| US10858341B2 (en) | 2016-12-11 | 2020-12-08 | Kempharm, Inc. | Compositions comprising methylphenidate-prodrugs, processes of making and using the same |
| US11505537B2 (en) | 2016-12-11 | 2022-11-22 | Kempharm, Inc. | Compositions comprising methylphenidate-prodrugs, processes of making and using the same |
| US10759778B2 (en) | 2016-12-11 | 2020-09-01 | Kempharm, Inc. | Methylphenidate-prodrugs, processes of making and using the same |
| US10584113B2 (en) | 2016-12-11 | 2020-03-10 | Kempharm, Inc. | Methylphenidate-prodrugs, processes of making and using the same |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2152690B1 (en) | Pyridyl piperidine orexin receptor antagonists | |
| EP2348856B1 (en) | 2,4-disubstituted pyrrolidine orexin receptor antagonists | |
| EP2350066B1 (en) | 2,5-disubstituted piperidine orexin receptor antagonists | |
| EP2349270B1 (en) | 2,5-disubstituted morpholine orexin receptor antagonists | |
| EP2348846B1 (en) | Disubstituted azepan orexin receptor antagonists | |
| EP2350061B1 (en) | 2,3-disubstituted piperidine orexin receptor antagonists | |
| WO2010048012A1 (en) | 2,5-disubstituted piperidine orexin receptor antagonists | |
| WO2008008551A2 (en) | 2-substituted proline bis-amide orexin receptor antagonists | |
| WO2007061763A2 (en) | Indole orexin receptor antagonists | |
| WO2015095111A1 (en) | Diazepane orexin receptor antagonists | |
| WO2007126934A2 (en) | Amidoethylthioether orexin receptor antagonists | |
| WO2009011775A1 (en) | Amidoethyl alkylamino orexin receptor antagonists | |
| AU2015201015B2 (en) | Pyridyl piperidine orexin receptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880016925.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08754659 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201790 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2009000636 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008257411 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 580887 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2687321 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12600388 Country of ref document: US Ref document number: 7428/DELNP/2009 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12009502198 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010509380 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2009/012579 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009111711 Country of ref document: EG |
|
| ENP | Entry into the national phase |
Ref document number: 20097024362 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09133050 Country of ref document: CO |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008754659 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2008257411 Country of ref document: AU Date of ref document: 20080522 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2009-011146 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009147733 Country of ref document: RU Ref document number: A200913366 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2012/0111 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: PI0811842 Country of ref document: BR Kind code of ref document: A2 Effective date: 20091119 |