WO2008083248A2 - Cyclopamine analogs - Google Patents
Cyclopamine analogs Download PDFInfo
- Publication number
- WO2008083248A2 WO2008083248A2 PCT/US2007/088990 US2007088990W WO2008083248A2 WO 2008083248 A2 WO2008083248 A2 WO 2008083248A2 US 2007088990 W US2007088990 W US 2007088990W WO 2008083248 A2 WO2008083248 A2 WO 2008083248A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- independently
- aryl
- aralkyl
- Prior art date
Links
- QASFUMOKHFSJGL-LAFRSMQTSA-N Cyclopamine Chemical class C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H](CC2=C3C)[C@@H]1[C@@H]2CC[C@@]13O[C@@H]2C[C@H](C)CN[C@H]2[C@H]1C QASFUMOKHFSJGL-LAFRSMQTSA-N 0.000 title abstract description 11
- 150000001875 compounds Chemical class 0.000 claims description 135
- 125000000217 alkyl group Chemical group 0.000 claims description 117
- -1 -OR Chemical group 0.000 claims description 93
- 125000003118 aryl group Chemical group 0.000 claims description 77
- 125000003342 alkenyl group Chemical group 0.000 claims description 66
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 63
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 55
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 51
- 238000000034 method Methods 0.000 claims description 51
- 125000000304 alkynyl group Chemical group 0.000 claims description 50
- 125000001072 heteroaryl group Chemical group 0.000 claims description 43
- 125000004475 heteroaralkyl group Chemical group 0.000 claims description 40
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 38
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 35
- 230000008569 process Effects 0.000 claims description 35
- 150000003839 salts Chemical class 0.000 claims description 32
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 24
- 239000003795 chemical substances by application Substances 0.000 claims description 24
- 150000002148 esters Chemical class 0.000 claims description 23
- 150000004820 halides Chemical class 0.000 claims description 22
- 239000002253 acid Substances 0.000 claims description 20
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 18
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 18
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 17
- 235000000346 sugar Nutrition 0.000 claims description 16
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 15
- 229910019142 PO4 Inorganic materials 0.000 claims description 13
- 150000002825 nitriles Chemical class 0.000 claims description 13
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 13
- 239000010452 phosphate Substances 0.000 claims description 13
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 12
- 125000003368 amide group Chemical group 0.000 claims description 11
- 125000001188 haloalkyl group Chemical group 0.000 claims description 11
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 claims description 11
- 125000003545 alkoxy group Chemical group 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 9
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 9
- 125000005842 heteroatom Chemical group 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- 125000001424 substituent group Chemical group 0.000 claims description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 229910052760 oxygen Inorganic materials 0.000 claims description 8
- 229910052717 sulfur Inorganic materials 0.000 claims description 8
- 239000011701 zinc Substances 0.000 claims description 8
- 229910052725 zinc Inorganic materials 0.000 claims description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 6
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 6
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims description 6
- 125000004432 carbon atom Chemical group C* 0.000 claims description 6
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 6
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 claims description 5
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 claims description 5
- 125000005910 alkyl carbonate group Chemical group 0.000 claims description 5
- 150000001408 amides Chemical group 0.000 claims description 5
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 5
- 229910052751 metal Inorganic materials 0.000 claims description 5
- 239000002184 metal Substances 0.000 claims description 5
- 229910052698 phosphorus Inorganic materials 0.000 claims description 5
- 125000005547 pivalate group Chemical group 0.000 claims description 5
- 229940124530 sulfonamide Drugs 0.000 claims description 5
- 150000003456 sulfonamides Chemical class 0.000 claims description 5
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 claims description 5
- 150000003751 zinc Chemical class 0.000 claims description 5
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical group [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical group NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 claims description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 4
- 229910004749 OS(O)2 Inorganic materials 0.000 claims description 4
- 229940022663 acetate Drugs 0.000 claims description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 4
- PUJDIJCNWFYVJX-UHFFFAOYSA-N benzyl carbamate Chemical compound NC(=O)OCC1=CC=CC=C1 PUJDIJCNWFYVJX-UHFFFAOYSA-N 0.000 claims description 4
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 4
- PIZLBWGMERQCOC-UHFFFAOYSA-N dibenzyl carbonate Chemical compound C=1C=CC=CC=1COC(=O)OCC1=CC=CC=C1 PIZLBWGMERQCOC-UHFFFAOYSA-N 0.000 claims description 4
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical group I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 4
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 claims description 4
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 3
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 claims description 3
- 125000004849 alkoxymethyl group Chemical group 0.000 claims description 3
- 150000004832 aryl thioethers Chemical class 0.000 claims description 3
- 150000001558 benzoic acid derivatives Chemical class 0.000 claims description 3
- IEPBPSSCIZTJIF-UHFFFAOYSA-N bis(2,2,2-trichloroethyl) carbonate Chemical compound ClC(Cl)(Cl)COC(=O)OCC(Cl)(Cl)Cl IEPBPSSCIZTJIF-UHFFFAOYSA-N 0.000 claims description 3
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 claims description 3
- FOCAUTSVDIKZOP-UHFFFAOYSA-M chloroacetate Chemical compound [O-]C(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-M 0.000 claims description 3
- 229940089960 chloroacetate Drugs 0.000 claims description 3
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 claims description 3
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 claims description 3
- 229940120124 dichloroacetate Drugs 0.000 claims description 3
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 claims description 3
- 229940044170 formate Drugs 0.000 claims description 3
- 125000004665 trialkylsilyl group Chemical group 0.000 claims description 3
- 229940066528 trichloroacetate Drugs 0.000 claims description 3
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 claims description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 claims description 3
- 229910015900 BF3 Inorganic materials 0.000 claims description 2
- 229910001115 Zinc-copper couple Inorganic materials 0.000 claims description 2
- 125000002947 alkylene group Chemical group 0.000 claims description 2
- 125000004104 aryloxy group Chemical group 0.000 claims description 2
- TVZPLCNGKSPOJA-UHFFFAOYSA-N copper zinc Chemical compound [Cu].[Zn] TVZPLCNGKSPOJA-UHFFFAOYSA-N 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 125000005039 triarylmethyl group Chemical group 0.000 claims description 2
- 239000011592 zinc chloride Substances 0.000 claims description 2
- 235000005074 zinc chloride Nutrition 0.000 claims description 2
- MKRZFOIRSLOYCE-UHFFFAOYSA-L zinc;methanesulfonate Chemical compound [Zn+2].CS([O-])(=O)=O.CS([O-])(=O)=O MKRZFOIRSLOYCE-UHFFFAOYSA-L 0.000 claims description 2
- 150000001805 chlorine compounds Chemical group 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 165
- 238000006243 chemical reaction Methods 0.000 description 111
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 95
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 91
- 239000000243 solution Substances 0.000 description 87
- 239000000203 mixture Substances 0.000 description 77
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 72
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 65
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 61
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 57
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 57
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 50
- 239000012044 organic layer Substances 0.000 description 50
- 235000019439 ethyl acetate Nutrition 0.000 description 47
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 40
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 37
- 229910052938 sodium sulfate Inorganic materials 0.000 description 37
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 33
- 229910052757 nitrogen Inorganic materials 0.000 description 33
- 239000000047 product Substances 0.000 description 33
- 229910052739 hydrogen Inorganic materials 0.000 description 31
- 239000001257 hydrogen Substances 0.000 description 31
- 238000003818 flash chromatography Methods 0.000 description 30
- 239000000741 silica gel Substances 0.000 description 30
- 229910002027 silica gel Inorganic materials 0.000 description 30
- 206010028980 Neoplasm Diseases 0.000 description 29
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 29
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 29
- 239000011541 reaction mixture Substances 0.000 description 28
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 26
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 25
- 239000000463 material Substances 0.000 description 25
- 239000007787 solid Substances 0.000 description 24
- 239000010410 layer Substances 0.000 description 23
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 22
- 235000011152 sodium sulphate Nutrition 0.000 description 21
- 239000000725 suspension Substances 0.000 description 21
- 239000002904 solvent Substances 0.000 description 20
- 239000003921 oil Substances 0.000 description 19
- 235000019198 oils Nutrition 0.000 description 19
- 230000037361 pathway Effects 0.000 description 19
- 238000005888 cyclopropanation reaction Methods 0.000 description 18
- 244000060234 Gmelina philippensis Species 0.000 description 17
- 229960004592 isopropanol Drugs 0.000 description 17
- 238000003756 stirring Methods 0.000 description 17
- 239000007832 Na2SO4 Substances 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 239000012267 brine Substances 0.000 description 15
- 239000000706 filtrate Substances 0.000 description 15
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 15
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 14
- 239000012298 atmosphere Substances 0.000 description 14
- 239000013058 crude material Substances 0.000 description 14
- 230000003637 steroidlike Effects 0.000 description 14
- 238000001914 filtration Methods 0.000 description 13
- 125000000623 heterocyclic group Chemical group 0.000 description 13
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 13
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- 150000001412 amines Chemical class 0.000 description 12
- 239000012299 nitrogen atmosphere Substances 0.000 description 12
- 125000006239 protecting group Chemical group 0.000 description 12
- 229920006395 saturated elastomer Polymers 0.000 description 12
- 239000007858 starting material Substances 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 229930013930 alkaloid Natural products 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 235000017557 sodium bicarbonate Nutrition 0.000 description 11
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 11
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 10
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 10
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 10
- 150000003797 alkaloid derivatives Chemical class 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 229910052681 coesite Inorganic materials 0.000 description 10
- 229940125936 compound 42 Drugs 0.000 description 10
- 229910052906 cristobalite Inorganic materials 0.000 description 10
- 239000006260 foam Substances 0.000 description 10
- 150000002576 ketones Chemical class 0.000 description 10
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 10
- 239000000377 silicon dioxide Substances 0.000 description 10
- 229910052682 stishovite Inorganic materials 0.000 description 10
- 229910052905 tridymite Inorganic materials 0.000 description 10
- 201000011510 cancer Diseases 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 229940086542 triethylamine Drugs 0.000 description 9
- 239000003981 vehicle Substances 0.000 description 9
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- QASFUMOKHFSJGL-UHFFFAOYSA-N cyclopamine Natural products C1C=C2CC(O)CCC2(C)C(CC2=C3C)C1C2CCC13OC2CC(C)CNC2C1C QASFUMOKHFSJGL-UHFFFAOYSA-N 0.000 description 8
- 235000011007 phosphoric acid Nutrition 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 7
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 7
- 0 C**(C[C@](C[C@]1(OCC(*(C)=C)=C(C[C@]2(C)[C@](CCC(*)C3)(*=C)C3=CC3)[C@@](C)(CC4)C23C=C)I)*=C)C1(C=C)C4=N Chemical compound C**(C[C@](C[C@]1(OCC(*(C)=C)=C(C[C@]2(C)[C@](CCC(*)C3)(*=C)C3=CC3)[C@@](C)(CC4)C23C=C)I)*=C)C1(C=C)C4=N 0.000 description 7
- 239000007864 aqueous solution Substances 0.000 description 7
- 229940125851 compound 27 Drugs 0.000 description 7
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 7
- 238000006049 ring expansion reaction Methods 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 7
- 241000894007 species Species 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- 239000002841 Lewis acid Substances 0.000 description 6
- 208000000172 Medulloblastoma Diseases 0.000 description 6
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 6
- 239000004698 Polyethylene Substances 0.000 description 6
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 125000004414 alkyl thio group Chemical group 0.000 description 6
- HQWPLXHWEZZGKY-UHFFFAOYSA-N diethylzinc Chemical compound CC[Zn]CC HQWPLXHWEZZGKY-UHFFFAOYSA-N 0.000 description 6
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 6
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 6
- 229920001971 elastomer Polymers 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 239000012528 membrane Substances 0.000 description 6
- 229920000573 polyethylene Polymers 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 230000008707 rearrangement Effects 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 5
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 5
- 229920000742 Cotton Polymers 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- 229910017974 NH40H Inorganic materials 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 150000001299 aldehydes Chemical group 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 150000007517 lewis acids Chemical class 0.000 description 5
- KCRRYOSHXIMXSZ-UHFFFAOYSA-N n-(2-oxoethyl)acetamide Chemical compound CC(=O)NCC=O KCRRYOSHXIMXSZ-UHFFFAOYSA-N 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 239000002002 slurry Substances 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 4
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 4
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 238000002512 chemotherapy Methods 0.000 description 4
- 229940127204 compound 29 Drugs 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 125000000392 cycloalkenyl group Chemical group 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- 239000012065 filter cake Substances 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 229910052736 halogen Inorganic materials 0.000 description 4
- 150000002367 halogens Chemical group 0.000 description 4
- 150000002431 hydrogen Chemical class 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- 150000007522 mineralic acids Chemical class 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 239000007848 Bronsted acid Substances 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- KBHCPIJKJQNHPN-UHFFFAOYSA-N N=NP(O)=O Chemical group N=NP(O)=O KBHCPIJKJQNHPN-UHFFFAOYSA-N 0.000 description 3
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 3
- 150000001540 azides Chemical group 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- XSCHRSMBECNVNS-UHFFFAOYSA-N benzopyrazine Natural products N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 3
- 229960004562 carboplatin Drugs 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 229960005420 etoposide Drugs 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 239000000543 intermediate Substances 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 201000005202 lung cancer Diseases 0.000 description 3
- 208000020816 lung neoplasm Diseases 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 3
- 210000000963 osteoblast Anatomy 0.000 description 3
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical group [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 description 3
- 125000003367 polycyclic group Polymers 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000000967 suction filtration Methods 0.000 description 3
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 2
- ITQTTZVARXURQS-UHFFFAOYSA-N 3-methylpyridine Chemical compound CC1=CC=CN=C1 ITQTTZVARXURQS-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- 108700028369 Alleles Proteins 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 206010004146 Basal cell carcinoma Diseases 0.000 description 2
- 239000004342 Benzoyl peroxide Substances 0.000 description 2
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- KRAPECIPHSRAHW-UHFFFAOYSA-K CC[Zn+].CC[Zn+].CC[Zn+].[O-]P([O-])([O-])=O Chemical compound CC[Zn+].CC[Zn+].CC[Zn+].[O-]P([O-])([O-])=O KRAPECIPHSRAHW-UHFFFAOYSA-K 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 108090000031 Hedgehog Proteins Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 206010041067 Small cell lung cancer Diseases 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 2
- 150000001502 aryl halides Chemical class 0.000 description 2
- 235000019400 benzoyl peroxide Nutrition 0.000 description 2
- 229960003328 benzoyl peroxide Drugs 0.000 description 2
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 2
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 150000001720 carbohydrates Chemical group 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 150000001860 citric acid derivatives Chemical class 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 239000002274 desiccant Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 230000003828 downregulation Effects 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000012039 electrophile Substances 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 238000007306 functionalization reaction Methods 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 230000009459 hedgehog signaling Effects 0.000 description 2
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 238000009434 installation Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 2
- 239000012038 nucleophile Substances 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 150000002923 oximes Chemical class 0.000 description 2
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 238000007149 pericyclic reaction Methods 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- RDOWQLZANAYVLL-UHFFFAOYSA-N phenanthridine Chemical compound C1=CC=C2C3=CC=CC=C3C=NC2=C1 RDOWQLZANAYVLL-UHFFFAOYSA-N 0.000 description 2
- 229940074439 potassium sodium tartrate Drugs 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 2
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 2
- 238000007348 radical reaction Methods 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 229930192474 thiophene Natural products 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 2
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 2
- IPSRAFUHLHIWAR-UHFFFAOYSA-N zinc;ethane Chemical compound [Zn+2].[CH2-]C.[CH2-]C IPSRAFUHLHIWAR-UHFFFAOYSA-N 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- FLBAYUMRQUHISI-UHFFFAOYSA-N 1,8-naphthyridine Chemical compound N1=CC=CC2=CC=CN=C21 FLBAYUMRQUHISI-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical class CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- UXGVMFHEKMGWMA-UHFFFAOYSA-N 2-benzofuran Chemical compound C1=CC=CC2=COC=C21 UXGVMFHEKMGWMA-UHFFFAOYSA-N 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- HFQDRFAYVXFIKP-UHFFFAOYSA-N 4,5,6,7-tetrahydrofuro[3,2-b]pyridine Chemical group C1CCNC2=C1OC=C2 HFQDRFAYVXFIKP-UHFFFAOYSA-N 0.000 description 1
- XZKIHKMTEMTJQX-UHFFFAOYSA-N 4-Nitrophenyl Phosphate Chemical compound OP(O)(=O)OC1=CC=C([N+]([O-])=O)C=C1 XZKIHKMTEMTJQX-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- XQABVLBGNWBWIV-UHFFFAOYSA-N 4-methoxypyridine Chemical compound COC1=CC=NC=C1 XQABVLBGNWBWIV-UHFFFAOYSA-N 0.000 description 1
- GDRVFDDBLLKWRI-UHFFFAOYSA-N 4H-quinolizine Chemical compound C1=CC=CN2CC=CC=C21 GDRVFDDBLLKWRI-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- 238000008940 Alkaline Phosphatase assay kit Methods 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 238000009020 BCA Protein Assay Kit Methods 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- GVQVVAVJOMJVER-GZQFWVIOSA-N CC1([C@H]2N(CCN(C)C)CCC1)O[C@@](CC1)(CC(C)=C(C3)[C@@H]1C(CC1)[C@H]3[C@@](C)(CC3)[C@H]1CC3=O)C2=C Chemical compound CC1([C@H]2N(CCN(C)C)CCC1)O[C@@](CC1)(CC(C)=C(C3)[C@@H]1C(CC1)[C@H]3[C@@](C)(CC3)[C@H]1CC3=O)C2=C GVQVVAVJOMJVER-GZQFWVIOSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- MNTFKIPEVNIGPL-BFNIZCPRSA-N C[C@H]1C(CC2)(C(C)=C(C3)[C@@H]2C(CC2)[C@H]3C(C)(CC3)[C@H]2CC3=O)O[C@H](CC(C)C2)C1N2C(OCc1ccccc1)=O Chemical compound C[C@H]1C(CC2)(C(C)=C(C3)[C@@H]2C(CC2)[C@H]3C(C)(CC3)[C@H]2CC3=O)O[C@H](CC(C)C2)C1N2C(OCc1ccccc1)=O MNTFKIPEVNIGPL-BFNIZCPRSA-N 0.000 description 1
- XJTIRBYBKXIIDC-HEUPZQNYSA-N C[C@H]1C(CC2)(C(C)=C(C3)[C@@H]2C(CC2)[C@H]3C(C)(CC3)[C@H]2C[C@@H]3NS(C)(=O)=O)O[C@H]2C1NC[C@@H](C)C2 Chemical compound C[C@H]1C(CC2)(C(C)=C(C3)[C@@H]2C(CC2)[C@H]3C(C)(CC3)[C@H]2C[C@@H]3NS(C)(=O)=O)O[C@H]2C1NC[C@@H](C)C2 XJTIRBYBKXIIDC-HEUPZQNYSA-N 0.000 description 1
- VZIZZORINJKQOX-JHJHRAALSA-N C[C@H]1C(CC2)(C(C)=C(C3)[C@@H]2C(CC2)[C@H]3C(C)(CC3)[C@H]2C[C@H]3O)O[C@H](CC(C)C2)C1N2C(OCc1ccccc1)=O Chemical compound C[C@H]1C(CC2)(C(C)=C(C3)[C@@H]2C(CC2)[C@H]3C(C)(CC3)[C@H]2C[C@H]3O)O[C@H](CC(C)C2)C1N2C(OCc1ccccc1)=O VZIZZORINJKQOX-JHJHRAALSA-N 0.000 description 1
- MOJMSJLVDDRFQS-LUENTKBMSA-N C[C@H]1[C@](CC2)(CC(C)=C(C3)C2[C@H](CC2)[C@H]3C(C)(CC3)C2CC3=O)OC(CCC2)[C@H]1N2OC(c1ccccc1)=O Chemical compound C[C@H]1[C@](CC2)(CC(C)=C(C3)C2[C@H](CC2)[C@H]3C(C)(CC3)C2CC3=O)OC(CCC2)[C@H]1N2OC(c1ccccc1)=O MOJMSJLVDDRFQS-LUENTKBMSA-N 0.000 description 1
- HIRGSKHUPIRIOQ-PTKIWVSWSA-N C[C@H]1[C@](CC2)(CC(C)=C(C3)[C@@H]2[C@H](CC2)[C@H]3[C@@](C)(CC3)[C@H]2CC3=O)O[C@H](C[C@H](C)C2)[C@H]1N2C(OCc1ccccc1)=O Chemical compound C[C@H]1[C@](CC2)(CC(C)=C(C3)[C@@H]2[C@H](CC2)[C@H]3[C@@](C)(CC3)[C@H]2CC3=O)O[C@H](C[C@H](C)C2)[C@H]1N2C(OCc1ccccc1)=O HIRGSKHUPIRIOQ-PTKIWVSWSA-N 0.000 description 1
- FFWOTSFEBDWHRZ-PRJPMFTFSA-N C[C@H]1[C@](CC2)(CC(C)=C(C3)[C@@H]2[C@H](CC2)[C@H]3[C@@](C)(CC3)[C@H]2C[C@@H]3OC(C)=O)O[C@H]2[C@H]1NCCC2 Chemical compound C[C@H]1[C@](CC2)(CC(C)=C(C3)[C@@H]2[C@H](CC2)[C@H]3[C@@](C)(CC3)[C@H]2C[C@@H]3OC(C)=O)O[C@H]2[C@H]1NCCC2 FFWOTSFEBDWHRZ-PRJPMFTFSA-N 0.000 description 1
- ISJVNSARWLMBRK-PJHMHFDISA-N C[C@H]1[C@](CC2)(CC(C)=C(C3)[C@@H]2[C@H](CC2)[C@H]3[C@@](C)(CC3)[C@H]2C[C@H]3O)O[C@H](C[C@H](C)C2)[C@H]1N2C(OCc1ccccc1)=O Chemical compound C[C@H]1[C@](CC2)(CC(C)=C(C3)[C@@H]2[C@H](CC2)[C@H]3[C@@](C)(CC3)[C@H]2C[C@H]3O)O[C@H](C[C@H](C)C2)[C@H]1N2C(OCc1ccccc1)=O ISJVNSARWLMBRK-PJHMHFDISA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229940126639 Compound 33 Drugs 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 206010057648 Cyclopia Diseases 0.000 description 1
- 241001219085 Cyclopia Species 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-CBPJZXOFSA-N D-Gulose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@H](O)[C@H]1O WQZGKKKJIJFFOK-CBPJZXOFSA-N 0.000 description 1
- WQZGKKKJIJFFOK-WHZQZERISA-N D-aldose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-WHZQZERISA-N 0.000 description 1
- WQZGKKKJIJFFOK-IVMDWMLBSA-N D-allopyranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@H](O)[C@@H]1O WQZGKKKJIJFFOK-IVMDWMLBSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102000003693 Hedgehog Proteins Human genes 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- CLEXYFLHGFJONT-DNMILWOZSA-N Jervine Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H](C(=O)C2=C3C)[C@@H]1[C@@H]2CC[C@@]13O[C@@H]2C[C@H](C)CN[C@H]2[C@H]1C CLEXYFLHGFJONT-DNMILWOZSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VSOAQEOCSA-N L-altropyranose Chemical compound OC[C@@H]1OC(O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-VSOAQEOCSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- CJUMAFVKTCBCJK-UHFFFAOYSA-N N-benzyloxycarbonylglycine Chemical compound OC(=O)CNC(=O)OCC1=CC=CC=C1 CJUMAFVKTCBCJK-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 238000010240 RT-PCR analysis Methods 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- WBTCZXYOKNRFQX-UHFFFAOYSA-N S1(=O)(=O)NC1=O Chemical group S1(=O)(=O)NC1=O WBTCZXYOKNRFQX-UHFFFAOYSA-N 0.000 description 1
- 229910052772 Samarium Inorganic materials 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical class [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 241000489524 Veratrum californicum Species 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- SRBFZHDQGSBBOR-STGXQOJASA-N alpha-D-lyxopyranose Chemical compound O[C@@H]1CO[C@H](O)[C@@H](O)[C@H]1O SRBFZHDQGSBBOR-STGXQOJASA-N 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- MNFORVFSTILPAW-UHFFFAOYSA-N azetidin-2-one Chemical class O=C1CCN1 MNFORVFSTILPAW-UHFFFAOYSA-N 0.000 description 1
- 229940054066 benzamide antipsychotics Drugs 0.000 description 1
- 150000003936 benzamides Chemical class 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- QSNONXQMKXTNQH-UHFFFAOYSA-N benzyl n-(2-oxoethyl)carbamate Chemical compound O=CCNC(=O)OCC1=CC=CC=C1 QSNONXQMKXTNQH-UHFFFAOYSA-N 0.000 description 1
- ZBDOVHVJWVWSPQ-UHFFFAOYSA-N benzylpenilloaldehyde Chemical compound O=CCNC(=O)CC1=CC=CC=C1 ZBDOVHVJWVWSPQ-UHFFFAOYSA-N 0.000 description 1
- APOXBWCRUPJDAC-UHFFFAOYSA-N bis(2,6-dimethylphenyl) hydrogen phosphate Chemical compound CC1=CC=CC(C)=C1OP(O)(=O)OC1=C(C)C=CC=C1C APOXBWCRUPJDAC-UHFFFAOYSA-N 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 230000005880 cancer cell killing Effects 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- JJWKPURADFRFRB-UHFFFAOYSA-N carbonyl sulfide Chemical compound O=C=S JJWKPURADFRFRB-UHFFFAOYSA-N 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- WRJWRGBVPUUDLA-UHFFFAOYSA-N chlorosulfonyl isocyanate Chemical compound ClS(=O)(=O)N=C=O WRJWRGBVPUUDLA-UHFFFAOYSA-N 0.000 description 1
- QZHPTGXQGDFGEN-UHFFFAOYSA-N chromene Chemical compound C1=CC=C2C=C[CH]OC2=C1 QZHPTGXQGDFGEN-UHFFFAOYSA-N 0.000 description 1
- WCZVZNOTHYJIEI-UHFFFAOYSA-N cinnoline Chemical compound N1=NC=CC2=CC=CC=C21 WCZVZNOTHYJIEI-UHFFFAOYSA-N 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000002993 cycloalkylene group Chemical group 0.000 description 1
- 150000001942 cyclopropanes Chemical class 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000007976 developmental deficit Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- YVPJCJLMRRTDMQ-UHFFFAOYSA-N ethyl diazoacetate Chemical compound CCOC(=O)C=[N+]=[N-] YVPJCJLMRRTDMQ-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 150000002243 furanoses Chemical group 0.000 description 1
- JKFAIQOWCVVSKC-UHFFFAOYSA-N furazan Chemical compound C=1C=NON=1 JKFAIQOWCVVSKC-UHFFFAOYSA-N 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 150000002390 heteroarenes Chemical class 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- GPRLSGONYQIRFK-UHFFFAOYSA-N hydron Chemical compound [H+] GPRLSGONYQIRFK-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 230000006607 hypermethylation Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002951 idosyl group Chemical class C1([C@@H](O)[C@H](O)[C@@H](O)[C@H](O1)CO)* 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 150000002471 indium Chemical class 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- CLEXYFLHGFJONT-UHFFFAOYSA-N jervine Natural products C1C=C2CC(O)CCC2(C)C(C(=O)C2=C3C)C1C2CCC13OC2CC(C)CNC2C1C CLEXYFLHGFJONT-UHFFFAOYSA-N 0.000 description 1
- QRXOCOSLDOPPKH-UHFFFAOYSA-N jervine sulfate Natural products CC1CNC2C(C1)OC3(CCC4=C(C3C)C(=O)C5C4CC=C6CC(O)CCC56C)C2C QRXOCOSLDOPPKH-UHFFFAOYSA-N 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-M lactobionate Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-M 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 150000002603 lanthanum Chemical class 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- QDLAGTHXVHQKRE-UHFFFAOYSA-N lichenxanthone Natural products COC1=CC(O)=C2C(=O)C3=C(C)C=C(OC)C=C3OC2=C1 QDLAGTHXVHQKRE-UHFFFAOYSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 230000004777 loss-of-function mutation Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 230000036244 malformation Effects 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 238000010907 mechanical stirring Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 210000003716 mesoderm Anatomy 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 150000002780 morpholines Chemical class 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- DZVUYVAKBFPVIR-UHFFFAOYSA-N n-(2-oxoethyl)-3-phenylpropanamide Chemical compound O=CCNC(=O)CCC1=CC=CC=C1 DZVUYVAKBFPVIR-UHFFFAOYSA-N 0.000 description 1
- KMXMGVCPNMHREE-UHFFFAOYSA-N n-methyl-n-(2-oxoethyl)-3-phenylpropanamide Chemical compound O=CCN(C)C(=O)CCC1=CC=CC=C1 KMXMGVCPNMHREE-UHFFFAOYSA-N 0.000 description 1
- BZLWCXGFPPXARR-UHFFFAOYSA-N n-methyl-n-(2-oxoethyl)acetamide Chemical compound CC(=O)N(C)CC=O BZLWCXGFPPXARR-UHFFFAOYSA-N 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 125000005151 nonafluorobutanesulfonyl group Chemical group FC(C(C(S(=O)(=O)*)(F)F)(F)F)(C(F)(F)F)F 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-N palmitic acid group Chemical group C(CCCCCCCCCCCCCCC)(=O)O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- GJSGGHOYGKMUPT-UHFFFAOYSA-N phenoxathiine Chemical compound C1=CC=C2OC3=CC=CC=C3SC2=C1 GJSGGHOYGKMUPT-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical compound C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 208000029340 primitive neuroectodermal tumor Diseases 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- CPNGPNLZQNNVQM-UHFFFAOYSA-N pteridine Chemical compound N1=CN=CC2=NC=CN=C21 CPNGPNLZQNNVQM-UHFFFAOYSA-N 0.000 description 1
- 150000003214 pyranose derivatives Chemical class 0.000 description 1
- 150000004040 pyrrolidinones Chemical class 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- KZUNJOHGWZRPMI-UHFFFAOYSA-N samarium atom Chemical compound [Sm] KZUNJOHGWZRPMI-UHFFFAOYSA-N 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- NVBFHJWHLNUMCV-UHFFFAOYSA-N sulfamide Chemical compound NS(N)(=O)=O NVBFHJWHLNUMCV-UHFFFAOYSA-N 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- 150000008053 sultones Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- ACNRTYKOPZDRCO-UHFFFAOYSA-N tert-butyl n-(2-oxoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCC=O ACNRTYKOPZDRCO-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- GVIJJXMXTUZIOD-UHFFFAOYSA-N thianthrene Chemical compound C1=CC=C2SC3=CC=CC=C3SC2=C1 GVIJJXMXTUZIOD-UHFFFAOYSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000000464 thioxo group Chemical group S=* 0.000 description 1
- 150000003608 titanium Chemical class 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- KPZSTOVTJYRDIO-UHFFFAOYSA-K trichlorocerium;heptahydrate Chemical compound O.O.O.O.O.O.O.Cl[Ce](Cl)Cl KPZSTOVTJYRDIO-UHFFFAOYSA-K 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229910000165 zinc phosphate Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4355—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4747—Quinolines; Isoquinolines spiro-condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/17—Amides, e.g. hydroxamic acids having the group >N—C(O)—N< or >N—C(S)—N<, e.g. urea, thiourea, carmustine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/196—Carboxylic acids, e.g. valproic acid having an amino group the amino group being directly attached to a ring, e.g. anthranilic acid, mefenamic acid, diclofenac, chlorambucil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid or pantothenic acid
- A61K31/198—Alpha-amino acids, e.g. alanine or edetic acid [EDTA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4741—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having oxygen as a ring hetero atom, e.g. tubocuraran derivatives, noscapine, bicuculline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/58—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids containing heterocyclic rings, e.g. danazol, stanozolol, pancuronium or digitogenin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/664—Amides of phosphorus acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0014—Skin, i.e. galenical aspects of topical compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61N—ELECTROTHERAPY; MAGNETOTHERAPY; RADIATION THERAPY; ULTRASOUND THERAPY
- A61N5/00—Radiation therapy
- A61N5/10—X-ray therapy; Gamma-ray therapy; Particle-irradiation therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/94—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom spiro-condensed with carbocyclic rings or ring systems, e.g. griseofulvins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J69/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by contraction of only one ring by one atom and expansion of only one ring by one atom
Definitions
- the present invention generally relates to cyclopamine analogs and pharmaceutical compositions thereof, and methods for preparing such analogs and compositions. These compounds and compositions are useful for the treatment of disorders mediated by the hedgehog pathway, such as cancer.
- Inhibition of the hedgehog pathway in certain cancers has been shown to result in inhibition of tumor growth.
- anti-hedgehog antibodies have been shown to antagonize the function of the hedgehog pathway and inhibit the growth of tumors.
- Small molecule inhibition of hedgehog pathway activity has also been shown to result in cell death in a number of cancer types.
- PCT publication WO 2006/026430 published 9 March 2006 and assigned to the same assignee as the present application, discloses a wide variety of cyclopamine analogs, focusing on those with unsaturation in the A or B ring.
- the surprisingly potent analogs contain completely saturated A and B rings.
- the present invention relates to analogs of steroidal alkaloids, such as cyclopamine, pharmaceutical compositions containing these compounds, and methods for preparing these compounds.
- the present invention relates to a compound represented by the following structure: or a pharmaceutically acceptable salt thereof; wherein R 1 is H, alkyl, -OR, amino, sulfonamido, sulfamido, -OC(O)R 5 , - N(R 5 )C(O)R 5 , or a sugar;
- R 3 is H, alkyl, alkenyl, or alkynyl
- R 4 is H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, heteroaralkyl, haloalkyl, -OR 5 , -C(O)R 5 , -CO 2 R 5 , -SO 2 R 5 , -C(O)N(R 5 )(R 5 ), -[C(R) 2 ] q -R 5 , -[(W)-N(R)C(0)] q R 5 , -[(W)-C(0)] q R 5 , -[(W)-C(0)0] q R 5 , -[(W)-0C(0)] q R 5 , -[(W)-SO 2 ] q R 5 , -[(W)-N(R 5 )SO 2 ] q R 5 , -[(W)-C(O)N(R 5
- each R 5 is independently H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, heteroaralkyl or -[C(R) 2 ] P -R 6 ; wherein p is 0-6; or any two occurrences of R 5 can be taken together to form a 4-8 membered optionally substituted ring which contains 0-3 heteroatoms selected from N, O, S, and P; each R 6 is independently hydroxyl, -N(R)COR, -N(R)C(O)OR, -N(R)SO 2 (R), -C(O)N(R) 2 , -OC(O)N(R)(R), -SO 2 N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(O) 2 OR, -C(O)N(OH)(
- R 1 is H, hydroxyl, alkoxyl, aryloxy, or amino.
- R 3 is H and/or R 4 is H, alkyl, hydroxyl, aralkyl, -[C(R) 2 ] q -R 5 , -[(W)-N(R)C(0)] q R 5 , -[(W)-N(R)SO 2 ] q R 5 , -[(W)-C(0)N(R)] q R 5 , -[(W)-0] q R 5 , -[(W)-C(0)] q R 5 , or -[(W)-C(0)0] q R 5 .
- R 1 is H or -OR
- R 2 is H or alkyl
- R 4 is H.
- R 2 is H or alkyl
- R 3 is H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, or aralkyl
- R 4 is H, alkyl, aralkyl, -[(W)-N(R)C(0)] q R 5 , -[(W)-N(R)SO 2 ] q R 5 , -[(W)-C(0)N(R)] q R 5 , -[(W)-0] q R 5 , -[(W)-C(0)] q R 5 , or -[(W)-C(0)0] q R 5 .
- R 1 is sulfonamide
- the present invention relates to a compound selected from the group consisting of:
- the compounds mentioned above are isolated.
- the present invention relates to an isolated compound selected from the group consisting of:
- the present invention relates to a compound represented by the following structure: or a pharmaceutically acceptable salt thereof; wherein R 1 is H, alkyl, -OR, amino, sulfonamido, sulfamido, -OC(O)R 5 , - N(R 5 )C(O)R 5 , or a sugar;
- R 3 is H, alkyl, alkenyl, or alkynyl
- R 4 is H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, heteroaralkyl, haloalkyl, -OR 5 , -C(O)R 5 , -CO 2 R 5 , -SO 2 R 5 , -C(O)N(R 5 )(R 5 ), -[C(R) 2 ] q -R 5 , -[(W)-N(R)C(0)] q R 5 , -[(W)-C(0)] q R 5 , -[(W)-C(0)0] q R 5 , -[(W)-0C(0)] q R 5 , -[(W)-SO 2 ] q R 5 , -[(W)-N(R 5 )SO 2 ] q R 5 , -[(W)-C(O)N(R 5
- X " is a halide; each R is, independently, H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, heteroaralkyl or -[C(R) 2 ] P -R 6 ; wherein p is 0-6; or any two occurrences of R 5 can be taken together to form a 4-8 membered optionally substituted ring which contains 0-3 heteroatoms selected from N, O, S, and P; each R 6 is, independently, hydroxyl, -N(R)COR, -N(R)C(O)OR, -N(R)SO 2 (R), -C(O)N(R) 2 , -OC(O)N(R)(R), -SO 2 N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(O) 2
- R 8 and R 9 are H;
- the compound is epimerically pure and/or isolated.
- R 4 is alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, heteroaralkyl, haloalkyl, -OR 5 , -[C(R) 2 ] q -R 5 , -[(W)-N(R)C(0)] q R 5 , -[(W)-C(0)] q R 5 , -[(W)-C(0)0] q R 5 , -[(W)-0C(0)] q R 5 , -[(W)-SO 2 ] q R 5 , -[(W)-N(R 5 )SO 2 ] q R 5 , -[(W)-C(O)N(R 5 )] q R 5 , -[(W)-0] q R
- the present invention relates to a compound selected from the group consisting of:
- the above compounds are epimerically pure and/or isolated.
- the present invention relates to an epimerically pure compound selected from the group consisting of:
- Another aspect of the present invention relates to a pharmaceutical composition including any of the aforementioned compounds, and a pharmaceutically acceptable excipient.
- the present invention relates to a process for preparing cyclopropyl derivatives of cyclopamine and related analogs having the formula 136:
- Y is CR 7 R 8 ;
- R 1 is H, alkyl, amino, hydroxyl, carboxyl, carbamoyl, alkoxy, hydroxyl, sugar or a protected hydroxyl group;
- R ' 3 , R " 4 , a aniidu R K. 5 i iss,, i unidueeppeeniidueeniitLliyy,, H n,, a alik ⁇ yyli,, a alik ⁇ eeniiyyli,, a alik ⁇ y; nyl, aryl, cycloalk kyyll,, hheetteerrooccyyccllooaallkkyyll,, aarralkyl, heteroaryl, or heteroaralkyl; or R 3 and R 4 or R 4 and R 31 taken together form a bond; R is H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, heteroaralkyl,
- the process includes the steps of contacting a compound of formula 136a with a haloalkylzinc phosphate cyclopropanating agent to yield a compound of formula 136:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 are as defined in compound 136.
- the present invention provides methods for preparing a compound of formula 137:
- Y is CR 7 R 8 ;
- R 1 is H, alkyl, amino, hydroxyl, carboxyl, carbamoyl, alkoxy, hydroxyl, sugar or a protected hydroxyl group;
- R is H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, heteroaralkyl, haloalkyl, -OR 9 , -C(O)R 9 , -CO 2 R 9 , -SO 2 R 9 , -C(O)N(R 9 )(R 9 ), -[C(R 9 ) 2 ] q R 9 , -[(W)-N(R 9 )C(O)] q R 9 , -[(W)-C(0)] q R 9 , -[(W)-C(0)0] q R 9 , -[(W)-0C(0)] q R 9 , -[(W)-SO 2 ] q R 9 , -[(W)-N(R 9 )SO 2 ] q R 9 , -[(W)-C(O)N(
- R 1 , R 2 , R 3 , R 4 , R , R and Y are as defined in compound 137; and then contacting the compound of formula 137b with an acid to give a compound of formula 137.
- R 7 and R 8 can both be H; in other embodiments R 1 can be a protected hydroxyl; and/or R 6 is a nitrogen protecting group.
- the haloalkylzinc phosphate cyclopropanating agent is formed by combining a phosphoric acid of formula 141a, a dialkylzinc, and a dihaloalkylane of formula 141b:
- m is, independently for each occurrence, 0, 1, 2, 3, or 4; n is, independently for each occurrence, 0,1, or 2; and each of R 12 , R 13 , R 14 , R 15 , R 16 , R 17 and R 18 is, independently, alkyl, aryl, aralkyl, or halide.
- the present invention relates to a process for preparing a compound of formula 142:
- the process includes the steps of contacting a compound of formula 142a with a cyclopropanating agent to form a compound formula 142b;
- Y is CR R ; R is a protected hydroxyl group;
- R 2 is H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, or heteroaralkyl; each of R 3 , R 4 , and R 5 is, independently, H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, or heteroaralkyl; or
- R 6 is a nitrogen protecting group;
- each of R 7 and R 8 is, independently, H, alkyl, alkenyl, aryl, nitrile, amido, halide, or ester;
- each R 9 is, independently, H, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, or heteroaralkyl.
- R and R can both be H; in other embodiments the protected hydroxyl group can be an ester or a carbonate; the nitrogen protecting can be a
- R carbamate or an amide R and R can both be H and the nitrogen protecting can be a carbamate or an amide; R 2 and R 3 can be H and R 4 and R taken together can form a bond; and/or the cyclopropanating agent is generated from a dihaloalkane and a metal species (e.g., dialkyl zinc or a zinc copper couple).
- a metal species e.g., dialkyl zinc or a zinc copper couple
- the cyclopropanating agent is generated from a dihaloalkane species and a dialkyl zinc species, and a phosphoric acid species or a salt thereof.
- the phosphoric acid species can have a structure of formula 151:
- each of R 10 and R 11 is independently alkyl, alkenyl, aralkyl, aryl, heteroaryl, heteroaralkyl; or R 10 and R 11 taken together have the formula 151a, 151b, or 151c;
- n independently for each occurrence is 0,1, or 2; each of R 12 , R 13 , R 14 , R 15 , R 16 , R 17 and R 18 is, independently, alkyl, aryl, aralkyl, or halide.
- the acid is a Bronsted acid (e.g., acetic acid, trifluoromethanesulfonic acid, phosphoric acid, methanesulfonic acid or HCl).
- the acid is a Lewis acid (e.g., BF 3 , zinc chloride, zinc methanesulfonate, or a zinc salt).
- the present invention also relates to a process for preparing a compound of formula 156:
- the process includes the steps of: contacting a compound of formula 156a with a cyclopropanating agent to form a compound formula 156b;
- R 1 is an oxygen protecting group selected from the group consisting of formate, acetate, chloroacetate, dichloroacetate, trichloroacetate, pivaloate, benzoate, alkyl carbonate, alkenyl carbonate, aryl carbonates, aralkyl carbonate, 2,2,2-trichloroethyl carbonate, alkoxymethyl ether, aralkoxymethyl ether, alkylthiomethl ether, aralkylthio ether, arylthio ether, trialkylsilyl ether, alkylarylsilyl ether, benzyl ether, arylmethyl ether, allyl ether; and R 2 is a nitrogen protecting group selected from the group consisting of formyl, chloroacetyl, trichloroacetyl, trifluoroacetyl, phenyl acetyl, benzoyl
- n independently for each occurrence is 0,1, or 2; each of R 12 , R 13 , R 14 , R 15 , R 16 , R 17 and R 18 is, independently, alkyl, aryl, aralkyl, or halide.
- the oxygen protecting group can be, in some embodiments, selected from alkyl carbonates, aralkyl carbonates (e.g., benzylcarbonate), benzoates, pivaloate, or formate.
- the nitrogen protecting group can be selected from benzoyl, trichloroacetyl, trifluoroacetyl, formyl, alkyl carbamates, aralkyl carbamates (e.g., benzylcarbamate), aryl carbamates, diarylphosphinamides, dialkylphosphinamidates, or diarylphosphinamidates.
- each expression e.g., alkyl, m, n, etc., when it occurs more than once in any structure, is intended to be independent of its definition elsewhere in the same structure.
- acylamino refers to a moiety that may be represented by the general formula:
- R50 wherein R50 and R54 independently represent a hydrogen, an alkyl, an alkenyl or -(CH 2 ) m -R61, where R61 represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocycle or a polycycle; and m is zero or an integer in the range of 1 to 8.
- alkenyl and alkynyl refer to unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- alkoxyl refers to an alkyl group, as defined above, having an oxygen radical attached thereto.
- Representative alkoxyl groups include methoxy, ethoxy, propyloxy, tert-butoxy and the like.
- alkyl refers to the radical of saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups.
- a straight chain or branched chain alkyl has 30 or fewer carbon atoms in its backbone (e.g., C1-C30 for straight chain, C3-C30 for branched chain), 20 or fewer.
- certain cycloalkyls have from 3-10 carbon atoms in their ring structure, and others have 5, 6 or 7 carbons in the ring structure.
- alkylthio refers to an alkyl group, as defined above, having a sulfur radical attached thereto.
- the "alkylthio" moiety is represented by one of -S-alkyl, -S-alkenyl, -S-alkynyl, and -S-(CH 2 ) m -R61, wherein m and R61 are defined above.
- Representative alkylthio groups include methylthio, ethyl thio, and the like.
- amino is art recognized as an amino-substituted carbonyl and includes a moiety that may be represented by the general formula:
- R50 and R51 each independently represent a hydrogen, an alkyl, an alkenyl, -(CH 2 ) m -R61, R61 represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocycle or a polycycle; and m is zero or an integer in the range of 1 to 8, or R50 and R51, taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure. Certain embodiments of the amide in the present invention will not include imides which may be unstable.
- amine and “amino” are art-recognized and refer to both unsubstituted and substituted amines, e.g., a moiety that may be represented by the general formulas:
- R50 and R51 each independently represent a hydrogen, an alkyl, an alkenyl, or -(CH 2 ) m -R61, where R61 is defined as above.
- alkylamine includes an amine group, as defined above, having a substituted or unsubstituted alkyl attached thereto, i.e., at least one of R50 and R51 is an alkyl group.
- aralkyl refers to an alkyl group substituted with an aryl group (e.g., an aromatic or heteroaromatic group).
- aryl as used herein includes 5-, 6- and 7-membered single-ring aromatic groups that may include from zero to four heteroatoms, for example, benzene, anthracene, naphthalene, pyrene, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, triazole, pyrazole, pyridine, pyrazine, pyridazine and pyrimidine, and the like.
- aryl groups having heteroatoms in the ring structure may also be referred to as "aryl heterocycles" or “heteroaromatics.”
- the aromatic ring may be substituted at one or more ring positions with such substituents as described above, for example, halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, -CF3, -CN, or the like.
- aryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, e.g., the other cyclic rings may be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls.
- Random acid refers to any substance that can act as a hydrogen ion (proton) donor.
- carboxyl is defined to include esters, thiocarboxyl, aldehydes, ketones and the like and thus includes such moieties as may be represented by the general formulas:
- X50 is a bond or represents an oxygen or a sulfur
- each of R55 and R56 represents independently a hydrogen, an alkyl, an alkenyl, -(CH 2 ) m -R61, where m and R61 are defined above.
- dirtyical refers to any of a series of divalent groups from alkyl, alkenyl, alkynyl, aryl, cycloalkyl, heterocycloalkyl, aralkyl, heteroaryl, and heteroaralkyl
- alkyl heteroaralkyl diradical.
- Typical examples include alkylenes of general structure (CH 2 ) ⁇ where X is 1-6, and corresponding alkenylene and alkynylene linkers having 2-6 carbon atoms and one or more double or triple bonds; cycloalkylene groups having 3-8 ring members; and aralkyl groups wherein one open valence is on the aryl ring and one is
- haloalkyl refers to an alkyl group where anywhere from 1 to all hydgrogens have been replaced with a halide.
- a "perhaloalkyl” is where all of the hydrogens have been replaced with a halide.
- heteroatom as used herein means an atom of any element other than carbon or hydrogen.
- heteroatoms include boron, nitrogen, oxygen, phosphorus, sulfur and selenium.
- heterocyclyl or “heterocyclic group” refer to 3- to 10- membered ring structures, in some instances from 3- to 7-membered rings, whose ring structures include one to four heteroatoms. Heterocycles can also be polycycles.
- Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxathiin, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, o
- the heterocyclic ring may be substituted at one or more positions with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
- substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl,
- isolated in connection with a compound of the present invention means the compound is not in a cell or organism and the compound is separated from some or all of the components that typically accompany it in nature.
- Lower alkyl as used herein means an alkyl group, as defined above, but having from one to ten carbons, in some embodiments from one to six carbon atoms in its backbone structure. Likewise, “lower alkenyl” and “lower alkynyl” have similar chain lengths. Certain alkyl groups are lower alkyls. In some embodiments, a substituent designated herein as alkyl is a lower alkyl.
- nitro means -NO2; the term “halogen” designates -F, -Cl, -Br or -I; the term “sulfhydryl” means -SH; the term “hydroxyl” means -OH; and the term “sulfonyl” means -SO2-.
- polycyclyl or “polycyclic group” refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings". Rings that are joined through non-adjacent atoms are termed "bridged" rings.
- Each of the rings of the polycycle may be substituted with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
- substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl
- epimerically pure in connection with a compound of the present invention means that the compound is substantially free of stereoisomers of the compound wherein the configuration of the stereogenic center that R 3 is bonded to is inverted.
- an epimerically pure compound represented by the following formula:
- R 1 , R 2 , R 3 , R 4 , R 7 , R 7 , R 8 , and R 9 are as defined below, is substantially free of compounds represented by the following formula:
- R 1 , R 2 , R 3 , R 4 , R 7 , R 7 , R 8 , and R 9 are as defined below.
- Epimerically pure compounds contain less than about 20 % by mass, less than about 15% by mass, less than about 10% by mass, less than about 5% by mass, or less than about 3% by mass of stereoisomeric compounds wherein the configuration of the stereogenic center that R 3 is bonded to is inverted relative to the compound.
- protecting group means temporary substituents which protect a potentially reactive functional group from undesired chemical transformations.
- protecting groups include esters of carboxylic acids, silyl ethers of alcohols, and acetals and ketals of aldehydes and ketones, respectively.
- the field of protecting group chemistry has been reviewed (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2 nd ed.; Wiley: New York, 1991).
- the functional group being protected and the protecting group are together referred to as one moiety.
- the fragment shown below is sometimes referred to as a benzyl carbonate; i.e., the protected (underlined) O makes up part of the carbonate.
- saccharide refers to a natural or an unnatural monosaccharide, disaccharide or oligosaccharide comprising one or more pyranose or furanose rings.
- the sugar may be covalently bonded to the steroidal alkaloid of the present invention through an ether linkage or through an alkyl linkage.
- the saccharide moiety may be covalently bonded to a steroidal alkaloid of the present invention at an anomeric center of a saccharide ring.
- Sugars may include, but are not limited to ribose, arabinose, xylose, lyxose, allose, altrose, glucose, mannose, gulose, idose, galactose, talose, glucose, and trehalose.
- R50 O O wherein R50 and R56 are as defined above.
- triflyl refers to trifluoromethanesulfonyl, p-toluenesulfonyl, methanesulfonyl, and nonafluorobutanesulfonyl groups, respectively.
- triflate refers to trifluoromethanesulfonate ester, p-toluenesulfonate ester, methanesulfonate ester, and nonafluorobutanesulfonate ester functional groups and molecules that contain the groups, respectively.
- substitution or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc.
- Certain compounds of the present invention may exist in particular geometric or stereoisomeric forms.
- the present invention contemplates all such compounds, including CM- and trans -isomers, R- and 5-enantiomers, diastereomers, (D)- isomers, (L)-isomers, the racemic mixtures thereof, and other mixtures thereof, as falling within the scope of the invention.
- Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
- certain embodiments of the present compounds may contain a basic functional group, such as amino or alkylamino, and are, thus, capable of forming pharmaceutically-acceptable salts with pharmaceutically-acceptable acids.
- pharmaceutically-acceptable salts refers to the relatively non-toxic, inorganic and organic acid addition salts of compounds of the present invention. These salts can be prepared in situ in the administration vehicle or the dosage form manufacturing process, or by separately reacting a purified compound of the invention in its free base form with a suitable organic or inorganic acid, and isolating the salt thus formed during subsequent purification.
- Representative salts include the hydrobromide, hydrochloride, sulfate, bisulfate, phosphate, nitrate, acetate, valerate, oleate, palmitate, stearate, laurate, benzoate, lactate, phosphate, tosylate, citrate, maleate, fumarate, succinate, tartrate, napthylate, mesylate, glucoheptonate, lactobionate, and laurylsulphonate salts and the like.
- sulfate bisulfate
- phosphate nitrate
- acetate valerate
- oleate palmitate
- stearate laurate
- benzoate lactate
- phosphate tosylate
- citrate maleate
- fumarate succinate
- tartrate napthylate
- mesylate mesylate
- glucoheptonate lactobionate
- laurylsulphonate salts and the like See, for example, Berg
- the pharmaceutically acceptable salts of the compounds of the present invention include the conventional nontoxic salts or quaternary ammonium salts of the compounds, e.g., from non-toxic organic or inorganic acids.
- such conventional nontoxic salts include those derived from inorganic acids such as hydrochloride, hydrobromic, sulfuric, sulfamic, phosphoric, nitric, and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, palmitic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicyclic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isothionic, and the like.
- the compounds of the present invention may contain one or more acidic functional groups and, thus, are capable of forming pharmaceutically- acceptable salts with pharmaceutically-acceptable bases.
- pharmaceutically- acceptable salts refers to the relatively non-toxic, inorganic and organic base addition salts of compounds of the present invention. These salts can likewise be prepared in situ in the administration vehicle or the dosage form manufacturing process, or by separately reacting the purified compound in its free acid form with a suitable base, such as the hydroxide, carbonate or bicarbonate of a pharmaceutically-acceptable metal cation, with ammonia, or with a pharmaceutically- acceptable organic primary, secondary or tertiary amine.
- Representative alkali or alkaline earth salts include the lithium, sodium, potassium, calcium, magnesium, and aluminum salts and the like.
- Representative organic amines useful for the formation of base addition salts include ethylamine, diethylamine, ethylenediamine, ethanolamine, diethanolamine, piperazine and the like. (See, for example, Berge et al, supra)
- the ring expanded steroidal alkaloid derivatives described above can be prepared directly from naturally occurring steroidal alkaloids or synthetic analogs thereof.
- the steroidal alkaloid starting materials can be cyclopamine or jervine. These steroidal alkaloids can be purchased commercially or extracted from Veratrum Californicum.
- the process of the present invention comprises the steps of cyclopropanating suitable starting steroidal alkaloid derivatives followed by ring expansion rearrangement of the cyclopropyl derivatives. In some instances, it may be desirable to suitably protect or otherwise transform reactive functionalities present on the molecule prior to cyclopropanation.
- an alcohol present at R 1 and a secondary nitrogen present on the fused furano-piperidine ring can both be protected prior to cyclopropanation.
- protecting groups that are efficiently added and removed from the alkaloid, yield intermediates in the synthetic process with improved handling properties and which allow for the efficient purification of the synthetic intermediates formed may be preferred.
- oxygen protecting groups include, but are not limited to formate, acetate, chloroacetate, dichloroacetate, trichloroacetate, pivaloate, benzoates, alkyl carbonate, alkenyl carbonate, aryl carbonates, aralkyl carbonate (e.g., benzyl carbonate), 2,2,2-trichloroethyl carbonate, alkoxymethyl ether, aralkoxymethyl ether, alkylthiomethl ether, aralkylthio ether, arylthio ether, trialkylsilyl ether, alkylarylsilyl ether, benzyl ether, arylmethyl ether, and allyl ether.
- formate acetate, chloroacetate, dichloroacetate, trichloroacetate, pivaloate
- benzoates alkyl carbonate, alkenyl carbonate, aryl carbonates, aralkyl carbonate (e.g., benzyl
- nitrogen protecting groups include, but are not limited to formyl, chloroacetyl, trichloroacetyl, trifluoroacetyl, phenyl acetyl, benzoyls, benzamides, alkyl carbamates, aralkyl carbamates (e.g., benzyl carbamates), aryl carbamates, allyl, aralkyl, alkoxymethyl, aralkoxymethyl, N-2-cyanoethyl, diary lphosphinamides, dialkylphosphinamidates, diarylphosphinamidates, and trialkylsilyl.
- cyclopropanating agents can be used to cyclopropanate the steroidal alkaloid.
- 1 -haloalkylmetal complexes and reactive species referred to as carbenoids are commonly used to cyclopropanate olefins.
- These reagents are typically made using a diiodoalkane or diazoalkane and a metal or organometalic species such as Et 2 Zn, JBu 3 Al, samarium, copper, rhodium, or palladium.
- Et 2 Zn and diiodomethane are used to generate the 1,1 -haloalkylmetal species.
- the reactivity and the ease of handling of the 1,1 -haloalkylzinc complexes can be modified by the addition of certain reagents, such as acids. It is believed that the addition of an acid to the 1,1 -haloalkylzinc species generates an alkyl zinc mixed salt.
- a biarylphosphoric acid is combined with diiodomethane and diethylzinc to generate a putative haloalkyl zinc phosphate cyclopropanating agent.
- a variety of phosphoric acids can be used to generate the putative haloalkylzinc phosphate.
- cyclopropanation methods such as those utilizing sulfur ylides to react with an olefin conjugated to a carbonyl to add a CH 2 or CH-alkyl or CH- aryl group, and metal-catalyzed decomposition of diazoalkyl and ⁇ -diazo-carbonyl compounds, such as diazomethane and ethyl diazoacetate, can also be used: these methods readily provide cyclopropanes having alkyl, aryl, alkoxycarbonyl (-COOR), or acyl substituents. Additional cyclopropanating agents are described in Masalov et al., Organic Letters (2004) Vol. 6, pp. 2365-8 and Hansen et al., Chem. Comm. (2006) 4838- 40.
- the cyclopropyl ring may be substituted or unsubstituted. In cases where the cyclopropyl ring is substituted, the groups attached to the methylene of the cyclopropane will be installed onto the D ring after rearrangement and ring expansion. [0073] The cyclopropanation reactions may be conducted in an aprotic solvent.
- Suitable solvents include ethers, such as diethyl ether, 1,2-dimethoxyethane, diglyme, t- butyl methyl ether, tetrahydrofuran and the like; halogenated solvents, such as chloroform, dichlorome thane, dichloroethane, and the like; aliphatic or aromatic hydrocarbon solvents, such as benzene, xylene, toluene, hexane, pentane and the like; esters and ketones, such as ethyl acetate, acetone, and 2-butanone; polar aprotic solvents, such as acetonitrile, dimethylsulfoxide, dimethylformamide, and the like; or combinations of two or more solvents.
- dichlorome thane is the solvent used for the cyclopropanation when a dialkyl zinc and diiodomethane is used.
- a solution containing the cyclopropanating agent is prepared by first adding a solution of a phosphoric acid to a solution of diethylzinc, followed by addition of diiodomethane to the reaction solution.
- the cyclopropanation substrate is then added to this solution.
- the cyclopropanation agent can be prepared in the presence of the cyclopropanation substrate by changing the order of addition of the reagents.
- the cyclopropanation reaction is conducted by first adding the phosphoric acid to a solution of dialkylzinc, followed by the addition of the cyclopropanation substrate, and finally the dihaloalkane is added.
- cyclopropanating agent is generated under controlled conditions and immediately reacts with the cyclopropanation substrate.
- the cyclopropanation methods described herein can also be used to cyclopropanate other polycyclic compounds, for example, those with steroidal backbones.
- the compound may be derivatized using a variety of functionalization reactions known in the art.
- Representative examples include palladium coupling reactions to alkenylhalides or aryl halides, oxidations, reductions, reactions with nucleophiles, reactions with electrophiles, pericyclic reactions, radical reactions, installation of protecting groups, removal of protecting groups, and the like.
- the cyclopropyl analogs undergo a rearrangement and ring expansion to afford steroidal alkaloid analogs in which the D ring has been expanded by one carbon.
- the cyclopropanation and ring expansion can take place in a two-step one reaction vessel process or in a two-step two reaction vessel process.
- the acid used to initiate the ring expansion rearrangement is added after completion of the cyclopropanation reaction.
- the zinc salts that are generated in the course of cyclopropanating the steroidal alkaloid can themselves act as Lewis acids to catalyze the ring expansion rearrangement.
- the reactivity of the zinc salts generated after the cyclopropanation can be modified by the addition of acids to generate more active Lewis acids.
- the methanesulfonic acid is added to the cyclopropanation reaction vessel after completion of the cyclopropanation.
- suitable acids include, but are not limited to zinc salts, boron compounds, magnesium salts, titanium salts, indium salts, aluminum salts, tin salts, lanthanum salts, trifluoromethanesulfonic acid, diaryloxyphosporic acids, acetic acid, and HCl.
- the Lewis acid used is a zinc salt or BF 3 .
- ring expanded analogs may be further functionalized using a variety of functionalization reactions known in the art.
- Representative examples include palladium coupling reactions to alkenylhalides or aryl halides, oxidations, reductions, reactions with nucleophiles, reactions with electrophiles, pericyclic reactions, radical reactions, installation of protecting groups, removal of protecting groups, and the like.
- Hedgehog signaling is essential in many stages of development, especially in formation of left-right symmetry. Loss or reduction of hedgehog signaling leads to multiple developmental deficits and malformations, one of the most striking of which is cyclopia.
- lung cancer (Watkins et al. (2003) Nature 422: 313-317), prostate cancer (Karhadkar et al (2004) Nature 431 : 707-12, Sheng et al. (2004) Molecular Cancer 3: 29-42, Fan et al. (2004) Endocrinology 145: 3961-70), breast cancer (Kubo et al. (2004) Cancer Research 64: 6071-74, Lewis et al. (2004) Journal of Mammary Gland Biology and Neoplasia 2: 165-181) and hepatocellular cancer (Sictician et al. (2005) ASCO conference, Mohini et al. (2005) AACR conference).
- small molecule inhibition of the hedgehog pathway has been shown to inhibit the growth of basal cell carcinoma (Williams, et al., 2003 PNAS 100: 4616-21), medulloblastoma (Berman et al., 2002 Science 297: 1559-61), pancreatic cancer (Berman et al., 2003 Nature 425: 846-51), gastrointestinal cancers (Berman et al., 2003 Nature 425: 846-51, published PCT application WO 05/013800), esophageal cancer (Berman et al., 2003 Nature 425: 846-51), lung cancer (Watkins et al., 2003. Nature 422: 313-7), and prostate cancer (Karhadkar et al., 2004. Nature 43J_: 707-12).
- the present invention provides pharmaceutically acceptable compositions which comprise a therapeutically-effective amount of one or more of the compounds described above, formulated together with one or more pharmaceutically acceptable carriers (additives) and/or diluents.
- the pharmaceutical compositions of the present invention may be specially formulated for administration in solid or liquid form, including those adapted for the following: (1) oral administration, for example, drenches (aqueous or non-aqueous solutions or suspensions), tablets, e.g., those targeted for buccal, sublingual, and systemic absorption, capsules, boluses, powders, granules, pastes for application to the tongue; (2) parenteral administration, for example, by subcutaneous, intramuscular, intravenous or epidural injection as, for example, a sterile solution or suspension, or sustained-release formulation; (3) topical application, for example, as a cream, ointment, or a controlled-release patch or spray applied to the skin; (4) intravaginally or intrarectally, for
- aqueous and nonaqueous carriers examples include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate.
- polyols such as glycerol, propylene glycol, polyethylene glycol, and the like
- vegetable oils such as olive oil
- injectable organic esters such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
- compositions may also contain adjuvants such as preservatives, wetting agents, emulsifying agents, dispersing agents, lubricants, and/or antioxidants.
- adjuvants such as preservatives, wetting agents, emulsifying agents, dispersing agents, lubricants, and/or antioxidants.
- Prevention of the action of microorganisms upon the compounds of the present invention may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like.
- isotonic agents such as sugars, sodium chloride, and the like into the compositions.
- prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents which delay absorption such as aluminum monostearate and gelatin.
- Methods of preparing these formulations or compositions include the step of bringing into association a compound of the present invention with the carrier and, optionally, one or more accessory ingredients.
- the formulations are prepared by uniformly and intimately bringing into association a compound of the present invention with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- the compounds of the present invention are administered as pharmaceuticals, to humans and animals, they can be given per se or as a pharmaceutical composition containing, for example, about 0.1 to 99%, or about 10 to 50%, or about 10 to 40%, or about 10 to 30, or about 10 to 20%, or about 10 to 15% of active ingredient in combination with a pharmaceutically acceptable carrier.
- compositions of the present invention may be varied so as to obtain an amount of the active ingredient which is effective to achieve the desired therapeutic response for a particular patient, composition, and mode of administration, without being toxic to the patient.
- the selected dosage level will depend upon a variety of factors including the activity of the particular compound of the present invention employed, or the ester, salt or amide thereof, the route of administration, the time of administration, the rate of excretion or metabolism of the particular compound being employed, the rate and extent of absorption, the duration of the treatment, other drugs, compounds and/or materials used in combination with the particular compound employed, the age, sex, weight, condition, general health and prior medical history of the patient being treated, and like factors well known in the medical arts.
- a suitable daily dose of a compound of the invention will be that amount of the compound which is the lowest dose effective to produce a therapeutic effect. Such an effective dose will generally depend upon the factors described above.
- oral, intravenous and subcutaneous doses of the compounds of the present invention for a patient when used for the indicated effects, will range from about 0.0001 to about 100 mg, or about 0.001 to about 100 mg, or about 0.01 to about 100 mg, or about 0.1 to about 100 mg per, or about 1 to about 50 mg per kilogram of body weight per day.
- the subject receiving this treatment is any animal in need, including primates, in particular humans, and other mammals such as equines, cattle, swine and sheep; and poultry and pets in general.
- Recrystallized cyclopamine 2 (14.1 g, 34.0 mmol, 1 eq) is dissolved in anhydrous DCM (70 mL) and anhydrous MeOH (29 mL). The clear solution is cooled, and triethylamine (10.4 g, 102.7 mmol, 3 eq) followed by benzyl chloroformate (6.20 g, 36.3 mmol, 1.1 eq) is added. After the addition is complete, the solution is stirred in the ice bath for 30 mm. Three portions of benzyl chloroformate (3 X 0.35 g, 3.46 mmol, 0.03 eq) are added over the 3 h.
- the reaction is slowly quenched with water (71 mL), while maintaining the temperature below 20 0 C.
- the mixture is stirred for 15 mm before the layers are settled and separated.
- the organic layer is dried over sodium sulfate and filtered.
- the combined filtrate is buffered with anhydrous pyridine (30 mL), concentrated, and solvent exchanged with additional anhydrous pyridine (43 mL) and concentrated.
- Bis(2,6-dimethyphenyl)phosphate (10.65 g, 34.8 mmol, 3.1 eq) is dried by concentration from anhydrous DCM (42 mL) and held under a nitrogen atmosphere. The phosphate is then redissolved in anhydrous DCM (110 mL). In a separate flask, a solution of neat diethylzinc (4.17 g, 33.8 mmol, 3.0 eq) in anhydrous DCM (35 mL) is prepared and cooled to -25 0 C. The phosphate solution is slowly transferred to the vessel containing the diethylzinc solution over 1 h, maintaining the temperature at or below -10 0 C.
- the solution is filtered under nitrogen, and the filtrate is washed with a 2 % (wt/wt) aqueous solution of ethylenediamine (125 mL) then water (130 mL), and then dried over sodium sulfate.
- the drying agent is removed by filtration and the filtrate is concentrated to dryness under vacuum.
- the solids that remained are chased with toluene (2 x 55 mL) on the rotary evaporator and the resulting material used without further purification in the next step
- a round bottom flask is sequentially charged with the homo-allylic alcohol (7.50 g, 17.6 mmol, 1 eq), aluminum tri-fe?t-butoxide (6.10 g, 24.8 mmol, 1.4 eq), anhydrous toluene ( 115 mL), and 2-butanone (90 g, 1.24 mol, 7 eq).
- the suspension is heated under a nitrogen atmosphere to 75 0 C for 16 h.
- the reaction temperature is then allowed to cool to 49 0 C.
- Aqueous 20 % (w/w) potassium sodium tartrate solution (226 g) is added to the stirred suspension.
- the suspension is stirred at rt for 3.5 h.
- the layers are separated.
- Cyclopamine 2 (5.02 g, 12.2 mmol, 1.0 eq) is dissolved in anhydrous pyridine (25 mL).
- DMAP 300 mg, 2.44 mmol, 0.2 eq.
- triethyl amine 5.5 mL, 39.1 mmol, 3.2 eq
- BtO-Cbz 10.5 g, 39.1 mmol, 3.2 eq
- the mixture is cooled to rt, treated with 30 mL water, heated to get a homogeneous solution and allowed to cool to room temp.
- a round bottom flask is sequentially charged with the homo-allylic alcohol 8 (7.50 g, 17.6 mmol, 1 eq), aluminum tri-fe?t-butoxide (6.10 g, 24.8 mmol, 1.4 eq), anhydrous toluene (115 mL), and 2-butanone (90 g, 1.24 mol, 7 eq).
- the suspension is heated under a nitrogen atmosphere to 75 0 C for 16 h.
- the reaction temperature is then allowed to cool to 49 0 C.
- Aqueous 20 % (w/w) potassium sodium tartrate solution (226 g) is added to the stirred suspension.
- the suspension is stirred at rt for 3.5 h.
- the layers are separated.
- ketone 6 85 mg, 0.199 mmol, 1 equiv. was charged and triethyleneglycol (2 mL) was added followed by hydrazine monohydrate (500 mg, 10 mmol, 50 equiv.) and potassium carbonate (138 mg, 1 mmol, 5 equiv.).
- the tube was sealed and the reaction was heated at 150 0 C for 16 h.
- the reaction was cooled to rt and water was added.
- the residue was extracted with chloroform (3X).
- the combined organic layers are washed with water, dried over Na 2 SO 4 , and concentrated to dryness.
- the colorless oil was purified using silica gel flash chromatography (DCM/MeOH 96:4).
- the reaction was cooled to rt and water was added.
- the residue was extracted with MTBE (3X).
- the organic layers were combined washed with brine, dried over Na 2 SO 4 , filtered, and concentrated to dryness.
- the white foam was purified using silica gel flash chromatography (DCM/MeOH 99:1) to give 206 mg of the N-isopropyl derivative as a white foam.
- the crude was purified using silica gel flash chromatography (hexane/EtOAc (9:1 to 4: 1)) to give the N-O derivative product (60 mg, 0.11 mmol) as a white foam.
- This foam was dissolved in 2 mL of MeOH followed by 2N aqueous KOH (0.4 mL). The reaction mixture was stirred for Ih. Most of the MeOH was evaporated under a stream of nitrogen and IN HCl (500 ⁇ L) was added. The material was extracted with DCM (3X). The combined organic layers were washed with water, dried over Na 2 SO 4 , filtered and concentrated under reduced pressure.
- Recrystallized cyclopamine (2.07 g) is charged to an appropriately sized reaction vessel and placed under an inert atmosphere.
- EtOAc (7.6 g), triethylamine (1.53 g), and DMAP (307 mg) are added sequentially.
- the suspension is warmed to 4O 0 C.
- Cbz-OBt is added in three portions over 90 minutes, keeping the internal temperature below 45 0 C.
- the reaction mixture is stirred at 4O 0 C for 90 minutes.
- the temperature is maintained while methanol (26.4 g) is slowly added to the reaction mixture.
- the resulting suspension is cooled to room temperature and stirred for at least 15 hours.
- the crude product is collected by filtration and rinsed with methanol (5 g).
- the white solid is dried under vacuum to a constant weight and recrystallized from heptane (30.3 g) and toluene (3.2 g) to afford Compound 24a (3.0 g).
- Johnson Matthey Pd/C catalyst A-305038-5 (890 mg) is charged to an appropriately sized reaction vessel, followed by 24b (2.24 g).
- the reaction vessel is purged with N 2 and toluene (21.8 g) and 2-propanol (6.7 g) are added sequentially.
- the system is degassed and placed under a nitrogen atmosphere, and the process is repeated with hydrogen.
- the system is stirred vigorously and the hydrogen blanket is maintained at one atmosphere for 4-5 hours.
- the reaction is monitor by either TLC or HPLC. If incomplete, the reaction is inerted, additional catalyst (145 mg) is charged, and the hydrogen atmosphere is returned for another hour. Ethylenediamine (12.9 mg) is charged and the mixture was stirred for 15 minutes.
- the catalyst is removed by filtration with a toluene:IPA (3:1) rinse.
- the filtrate and rinses are concentrated and solvent exchanged to toluene.
- the product is crystallized from toluene (19.0 g) and heptane (18.0 g) to afford 24c as a white crystalline solid (1.34 g, 98% yield).
- 24c (644 mg) is charged to an appropriately sized reaction vessel followed by aluminum t-butoxide (525 mg), toluene (8.34 g, 15 vol), and 2-butanone (7.83 g, 15 vol).
- the contents of the flask are degassed with evacuation/nitrogen purge cycles to remove oxygen and the reaction mixture is heated at 75°C with vigorous stirring for 16-18 hours.
- the reaction is quenched by the addition of aqueous Rochelle's salt (2.6 g in 10.3 g water) and the mixture vigorously stirred for one hour at 45 0 C.
- the aqueous and organic layers are separated.
- the aqueous layer is back extracted with a mixture of toluene (2.9 g) and EtOAc (2.9 g).
- the enone 24d (459 mg) and Johnson-Matthey 5% palladium on carbon (A503023-5, 101 mg) are charged to an appropriately sized multi neck reaction vessel.
- the vessel is purged with nitrogen and 3-picoline (2.2 g) is charged as the solvent. Stirring is started and the vessel is first degassed using nitrogen and then stirred under hydrogen at atmospheric pressure for 8 hours.
- the catalyst is removed by filtration through 0.2 micron media, rinsing with ACN (1.4 ml). The filtrate and rinse are combined in a clean reaction vessel equipped with mechanical stirring, an internal temperature probe, and a nitrogen atmosphere.
- citric acid (3.7 g) in water (9.2 ml) is charged to the reaction vessel at or below 3O 0 C, and IPI-335589 is allowed to slowly crystallize from solution as the citrate salt at 20 and then O 0 C.
- the crystalline product is recovered by suction filtration and washed with water (3.7 ml). After drying, the citrate salt, 24e, is isolated as a hydrate (3-5 wt% water) in 89.5% yield (622 mg) with a ⁇ : ⁇ ratio approaching 90:1.
- the reaction is quenched at low temperature with MeOH (0.33 g), then 3M NaOH (2.4 g) at -20 0 C and 15% hydrogen peroxide in water (1.04 g) at or below 5°C, then stirring overnight at ambient temperatures.
- the layers are split and the organic layer is washed sequentially with IM aqueous NaOH (2 ml), 0.5 M aqueous Na 2 SO 3 (2 ml), and water (2 ml) adjusted to a pH of 3 with HCl.
- the organic layer is dried over sodium sulfate (0.82 g), filtered and concentrated.
- the product 24g (0.457 g) is re-concentrated from DCM (0.9 g) and used in the next reaction.
- the layers are separated and the organic layer is washed with 2.5 wt% sodium bicarbonate (13.8 g) and then water (10.9 g). The organic layer is dried over of sodium sulfate (4 g), filtered, and concentrated.
- the product solution is solvent exchanged via charge and concentration to t-butyl methyl ether (10.9 ml) and then l,3-dimethyl-3,4,5,6-tetrahydro- 2(lH)-pyrimidinone (DMPU, 4.7 ml). The DMPU solution is used directly in the next reaction.
- 24j (1.09 g) is dissolved and transferred to an appropriately sized reaction vessel with anhydrous DCM (15.8 g) and placed under a nitrogen atmosphere. The solution is cooled to 0 0 C. N,N-diisopropylethylamine (357 mg) and neat methanesulfonyl chloride (0.165 ml) are charged sequentially while maintaining temperature between below 5 0 C. The reaction is monitored by HPLC. Incomplete reactions are driven to completion with additional methanesulfonyl chloride. The reaction is quenched with 0.4 M aqueous sodium bicarbonate (11.4 g) and warmed to ambient temperature. The layers are separated and the aqueous phase is back extracted with DCM (5.8 g). The combined organic layers are dried over magnesium sulfate (0.55 g), filtered and concentrated. The product 24k is dissolved and striped from 2- propanol (4.0 g) to remove residual DCM and used directly in the next reaction.
- the reaction is stirred under 1 atm of hydrogen for another 15 hours at ambient temperature.
- the reaction is monitored by HPLC. Incomplete reactions are treated with additional catalyst and hydrogen.
- the reaction is filtered, treated with steam activated carbon (200 mg), and filtered again.
- the solution is dried by partial concentration transferred to a reaction vessel and diluted with anhydrous 2-propanol to 0.09 M based on the theoretical yield.
- a 1.25 M HCl solution in 2- propanol (1.64 g) is charged over 20 minutes.
- the hydrochloride salt crystallizes slowly with gentle stirring and is isolated by filtration.
- the crystals are washed with 2-propanol (2.5 g) and vacuum dried to afford Compound 42 (916 mg, 80% yield) as a 1:1 IPA solvate.
- Step B The mixture was diluted with CH 2 CI 2 (30 mL) and washed with three portions (30 mL each) of water. The organic layer was dried over Na 2 SO 4 , filtered, and evaporated to dryness. The desired white solid 51 was taken to the next step without purification.
- the ketone 57 was azeotroped a couple times with CH 2 CI 2 and dried under vacuum for 1 h. Under nitrogen, the ketone 2 (693 mg, 1.27 mmol, 1 equiv.) was dissolved in anhydrous THF (20 mL) and the solution was cooled to -78C. A I M solution of K-selectride in THF (1.9 mL, 1.9 mmol, 1.5 equiv.) was added dropwise. After 1 h, the reaction was complete by TLC. The reaction was quenched by addition of 2.6 mL of 5 N NaOH followed by slow addition of 2.6 mL of 30% wt H 2 O 2 .
- Example 28 Inhibition of the Hedgehog pathway in cell culture
- Hedgehog pathway specific cancer cell killing effects may be ascertained using the following assay.
- C3H10T1/2 cells differentiate into osteoblasts when contacted with the sonic hedgehog peptide (Shh-N). Upon differentiation; these osteoblasts produce high levels of alkaline phosphatase (AP) which can be measured in an enzymatic assay (Nakamura et al., 1997 BBRC 237: 465).
- AP alkaline phosphatase
- Compounds that block the differentiation of C3H10T1/2 into osteoblasts can therefore be identified by a reduction in AP production (van der Horst et al., 2003 Bone 33: 899).
- the assay details are described below.
- the results approximate (EC 50 for inhibition) of the differentiation assay is shown below in Table 1.
- Mouse embryonic mesoderm fibroblasts C3H10T1/2 cells obtained from ATCC were cultured in Basal MEM Media (Gibco/Invitrogen) supplemented with 10% heat inactivated FBS (Hyclone), 50 units/ml penicillin and 50ug/ml streptomycin (Gibco/Invitrogen) at 37C with 5% CO2 in air atmosphere.
- C3H10T1/2 cells were plated in 96 wells with a density of 8xlO 3 cells/well. Cells were grown to confluence (72hrs). After sonic Hedgehog (250ng/ml), and/or compound treatment, the cells were lysed in 110 ⁇ L of lysis buffer (50 mM Tris pH 7.4, 0.1% TritonXlOO), plates were sonicated and lysates spun through 0.2 ⁇ m PVDF plates (Corning). 40 ⁇ L of lysates was assayed for AP activity in alkaline buffer solution (Sigma) containing lmg/ml p-Nitrophenyl Phosphate.
- lysis buffer 50 mM Tris pH 7.4, 0.1% TritonXlOO
- the activity of Compound 42 was also evaluated in a transgenic mouse model of medulloblastoma. Mice that are heterozygous for loss of function mutations in the tumor suppressors Patchedl (Ptchl) and Hypermethylated in Cancer (Hicl) develop spontaneous medulloblastoma. Similar to human medulloblastoma, these tumors demonstrate complete promoter hypermethylation of the remaining Hicl allele, as well as loss of expression of the wild type Ptchl allele. When passaged as subcutaneous allografts, these tumors grow aggressively and are Hedgehog pathway-dependent. This model was employed to evaluate the efficacy of orally administered Compound, and to correlate activity with drug exposure in plasma and tumors. Oral administration (PO) of a single dose of Compound 42 led to dose-dependent down-regulation of the HH pathway in subcutaneously implanted tumors, as measured by decreased GIi-I mRNA expression 8 hours post dose administration.
- Ptchl tumor suppressors Patchedl
- LX22 cells were implanted subcutaneously into the flank of the right leg.
- LX22 is primary xenograft model of SCLC derived from chemo -naive patients, which has been maintained by mouse to mouse passaging. This tumor responds to etoposide/carboplatin chemotherapy in way that closely resembles a clinical setting. LX22 regresses during chemotherapy treatment, goes through a period of remission, and then begins to recur.
- Compound single agent activity and its ability to modulate the chemoresistant recurrence was tested.
- mice were randomized into three dosing groups to receive Vehicle (30% HBPCD), Compound, or the chemotherapy combination of etoposide and carboplatin (E/P).
- Compound 42 was administered at a dose of 40mg/kg/day, and after 16 consecutive doses there was no measurable difference between the treated and vehicle groups.
- Etoposide was administered i.v at 12mg/kg on days 34, 35, 36, and 48, while Carboplatin was administered i.v. at 60mg/kg on days 34, 41, and 48, post tumor implant.
- the E/P treated mice were further randomized to receive either Vehicle (30%HPBCD) or Compound follow up treatment.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Oncology (AREA)
- Dermatology (AREA)
- Pathology (AREA)
- Radiology & Medical Imaging (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Steroid Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description







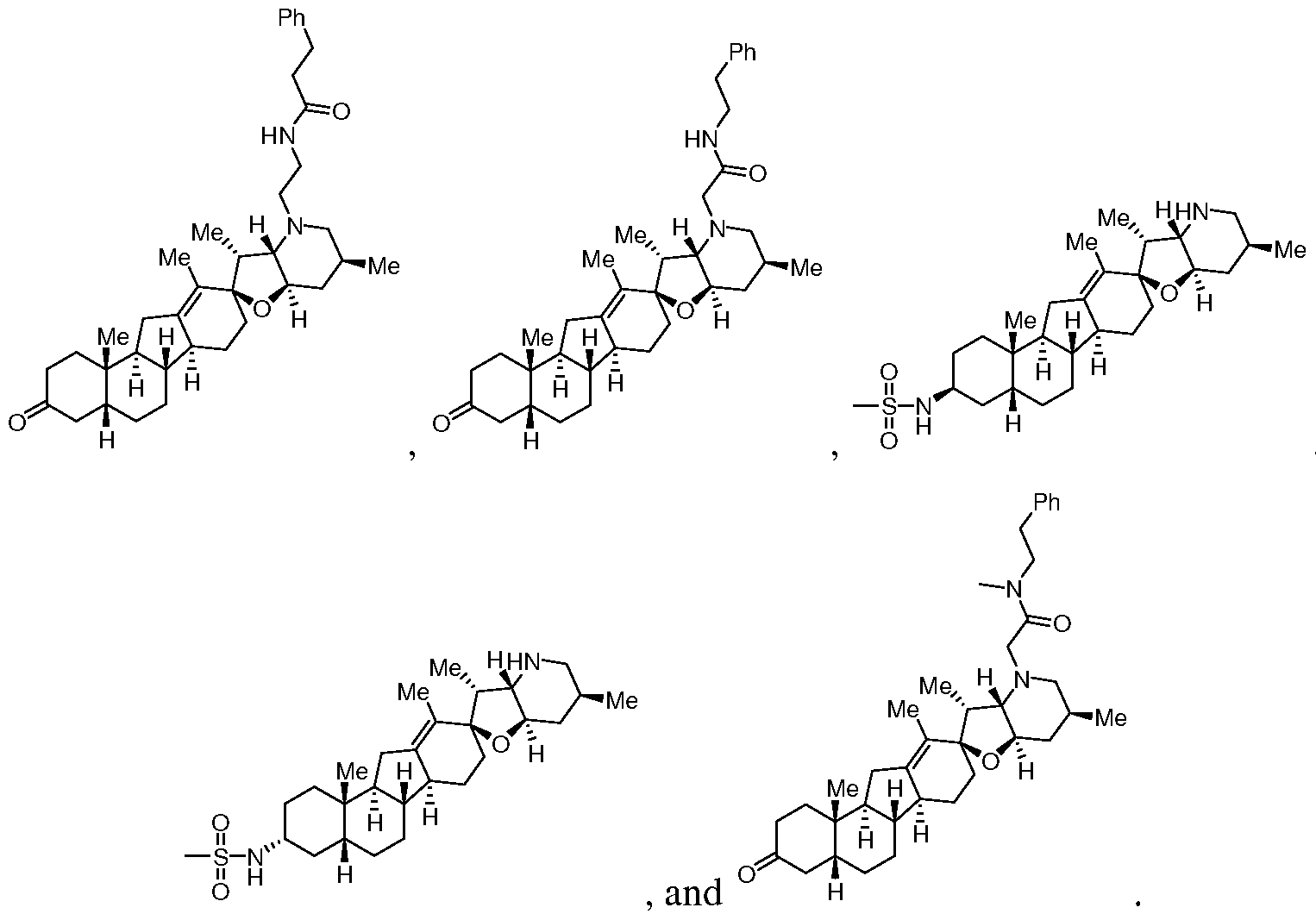
















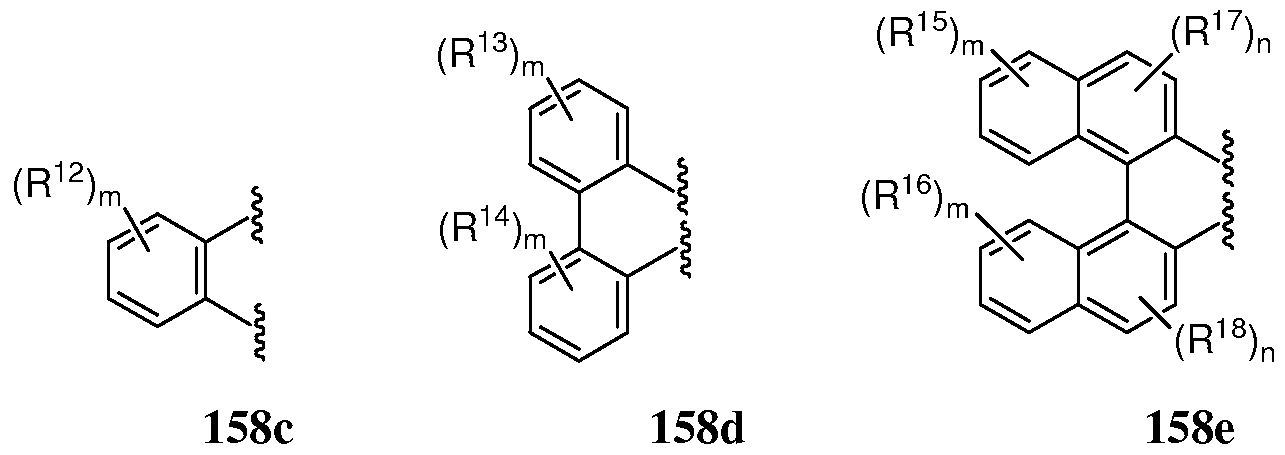































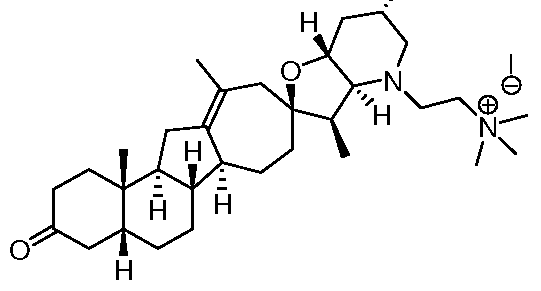
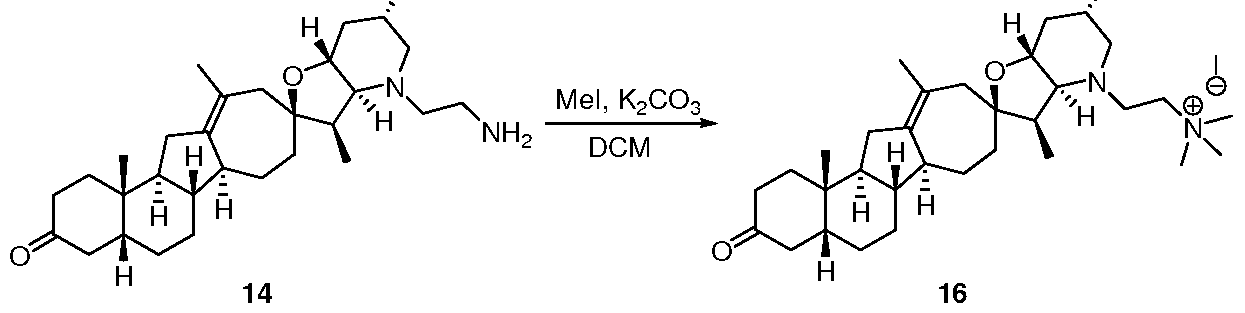



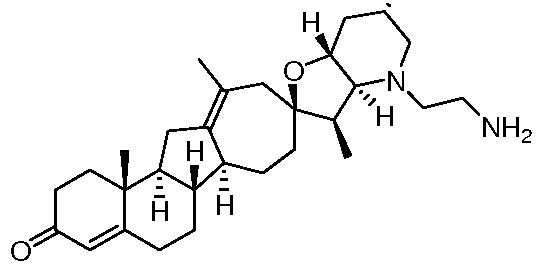

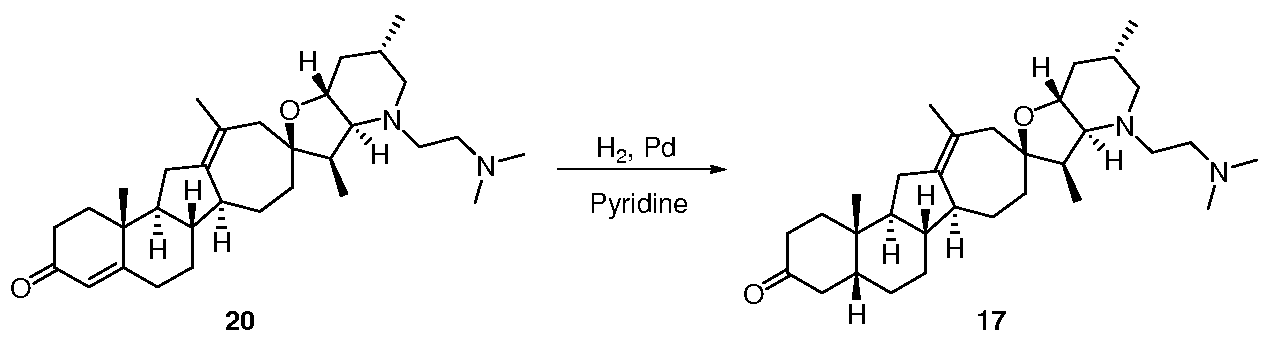












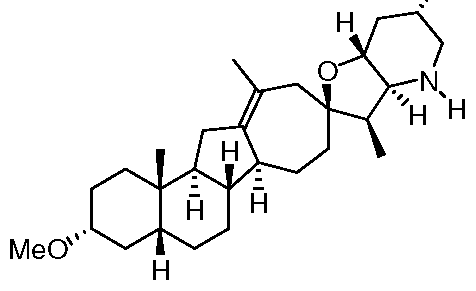
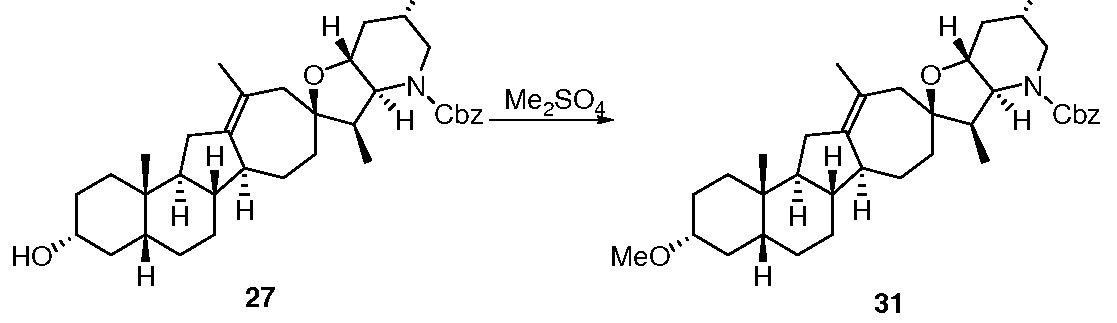
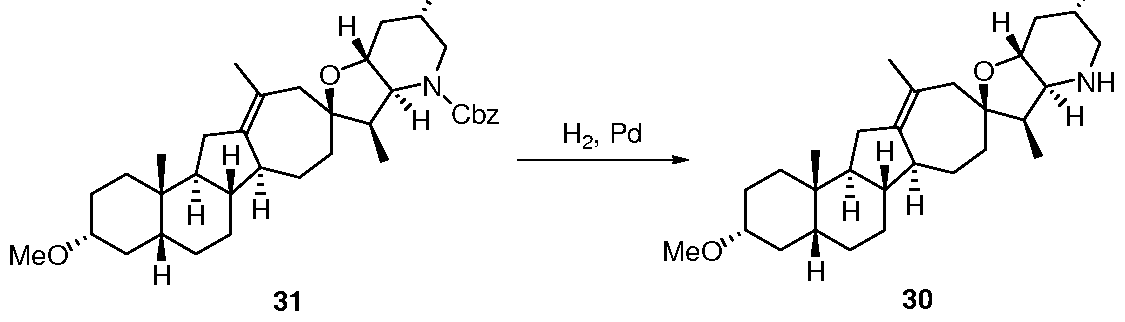



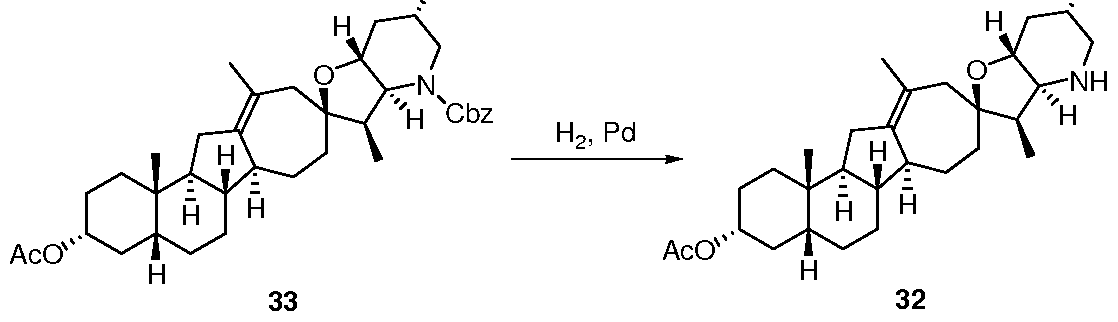



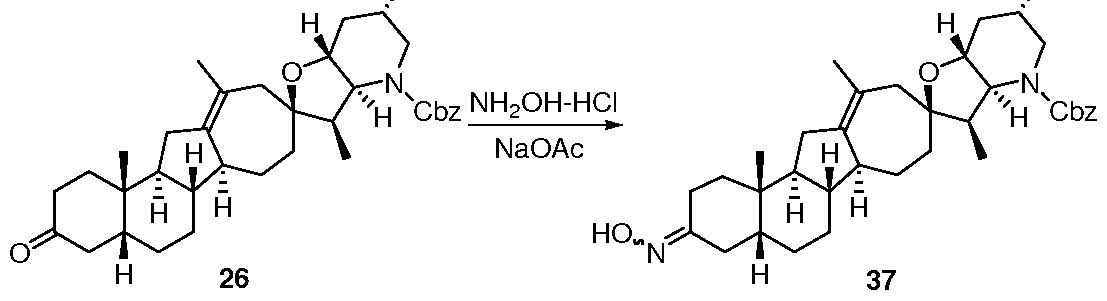






















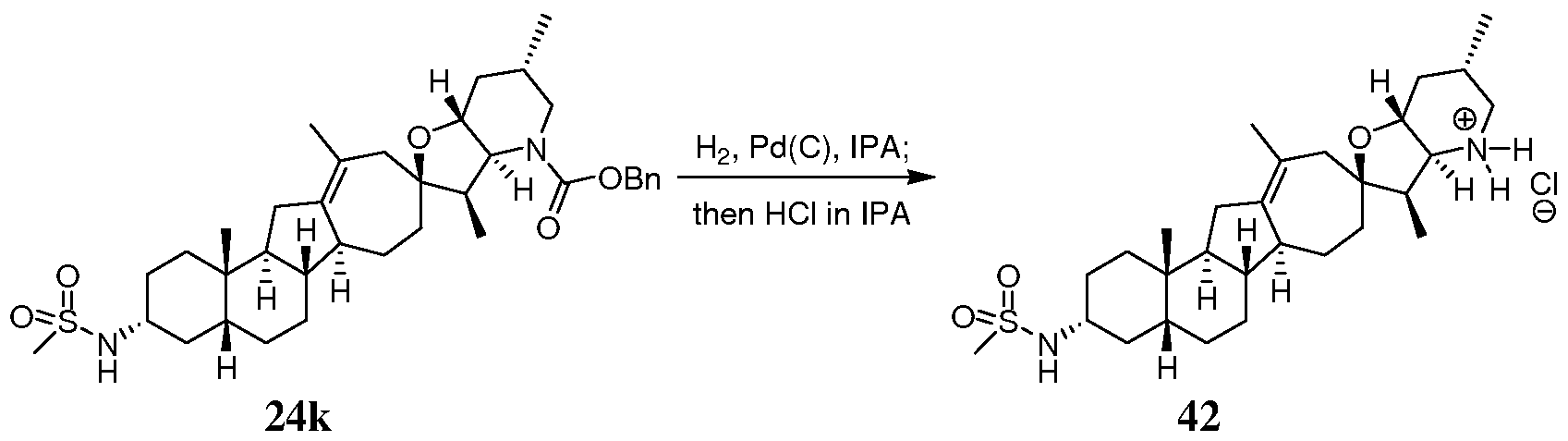








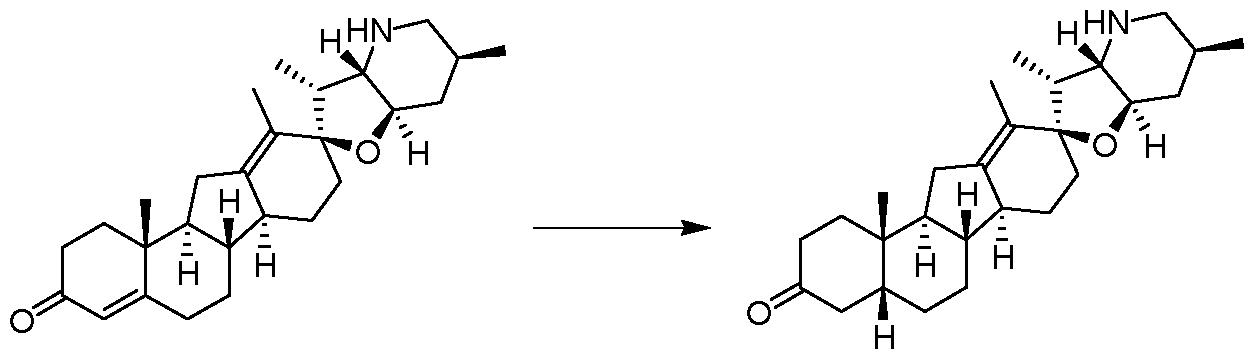







Claims












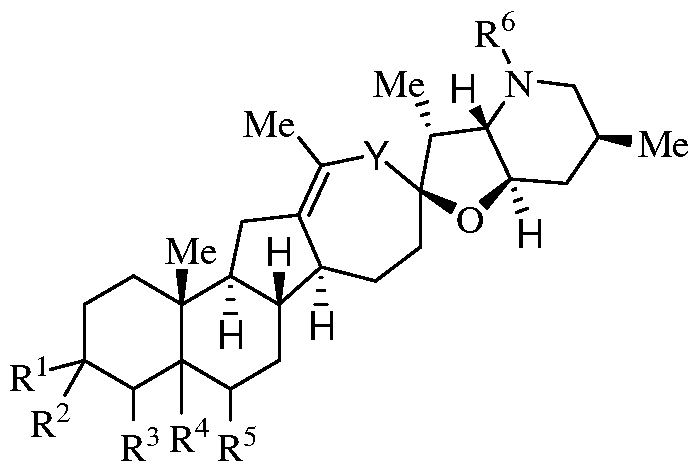




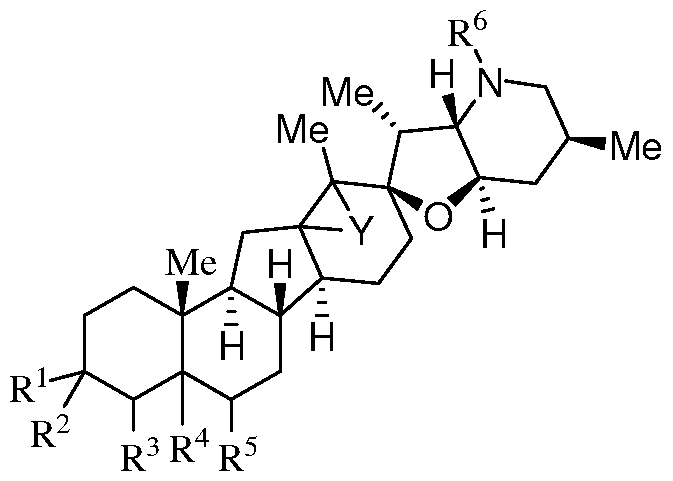

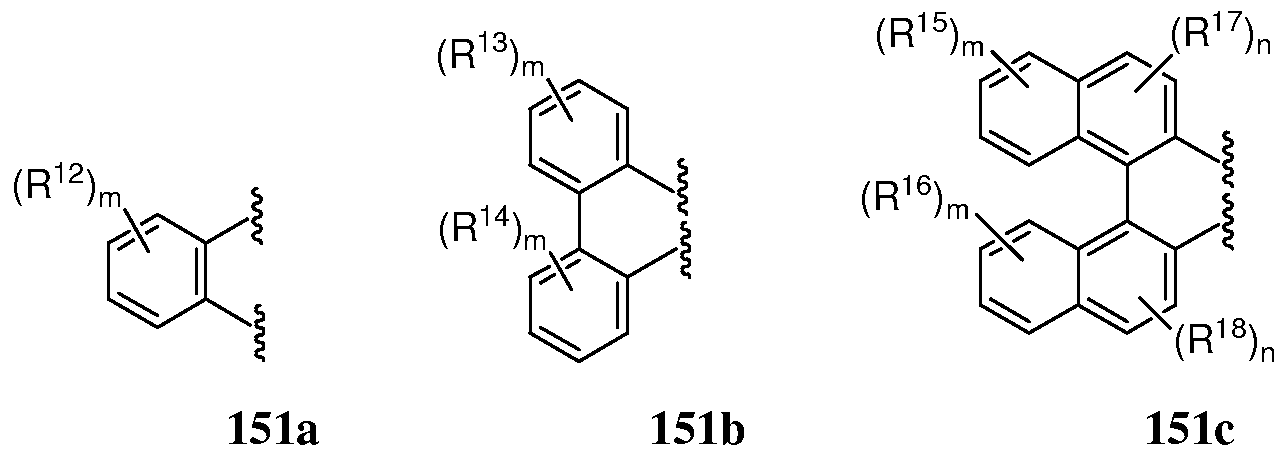


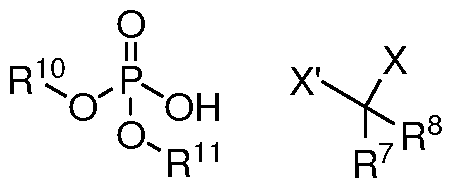

Priority Applications (19)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES07870001.0T ES2488466T3 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogues |
| EP07870001.0A EP2096917B1 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
| CA2673995A CA2673995C (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
| AU2007339782A AU2007339782B2 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
| JP2009544276A JP5412292B2 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analog |
| UAA200907837A UA97130C2 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine derivatives |
| NZ577916A NZ577916A (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
| RU2009128997/04A RU2486194C2 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogues |
| CN200780051114.7A CN101631463B (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
| KR1020097015913A KR101489394B1 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
| NO20190059A NO347310B1 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogues |
| MX2009007077A MX2009007077A (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs. |
| DK07870001.0T DK2096917T3 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine-ANALOGS |
| BRPI0720972A BRPI0720972B8 (en) | 2006-12-28 | 2007-12-27 | cyclopamine analog compounds and pharmaceutical compositions comprising said compounds |
| MX2012007856A MX342683B (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs. |
| PL07870001T PL2096917T3 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
| ZA2009/04445A ZA200904445B (en) | 2006-12-28 | 2009-06-25 | Cyclopamine analogs |
| IL199554A IL199554B (en) | 2006-12-28 | 2009-06-25 | Cyclopamine analogs and their use for preparing anti-cell-proliferation medicaments |
| NO20092710A NO343748B1 (en) | 2006-12-28 | 2009-07-20 | Cyclopamine analogues and pharmaceutical compositions comprising such |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US87801806P | 2006-12-28 | 2006-12-28 | |
| US60/878,018 | 2006-12-28 | ||
| US94159607P | 2007-06-01 | 2007-06-01 | |
| US60/941,596 | 2007-06-01 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008083248A2 true WO2008083248A2 (en) | 2008-07-10 |
| WO2008083248A3 WO2008083248A3 (en) | 2008-10-16 |
Family
ID=39589207
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/088995 WO2008083252A2 (en) | 2006-12-28 | 2007-12-27 | Methods of use for cyclopamine analogs |
| PCT/US2007/088990 WO2008083248A2 (en) | 2006-12-28 | 2007-12-27 | Cyclopamine analogs |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/088995 WO2008083252A2 (en) | 2006-12-28 | 2007-12-27 | Methods of use for cyclopamine analogs |
Country Status (26)
| Country | Link |
|---|---|
| US (18) | US8017648B2 (en) |
| EP (4) | EP2101580B1 (en) |
| JP (2) | JP5412292B2 (en) |
| KR (2) | KR101489394B1 (en) |
| CN (2) | CN101677551B (en) |
| AR (1) | AR081107A1 (en) |
| AU (2) | AU2007339786B2 (en) |
| BR (2) | BRPI0720993B8 (en) |
| CA (2) | CA2674302C (en) |
| CL (1) | CL2007003854A1 (en) |
| DK (1) | DK2096917T3 (en) |
| ES (2) | ES2488466T3 (en) |
| HK (1) | HK1158989A1 (en) |
| IL (1) | IL199554B (en) |
| JO (1) | JO2947B1 (en) |
| MX (3) | MX2009007075A (en) |
| NO (3) | NO347310B1 (en) |
| NZ (2) | NZ577916A (en) |
| PE (1) | PE20090042A1 (en) |
| PL (1) | PL2096917T3 (en) |
| PT (1) | PT2096917E (en) |
| RU (2) | RU2486194C2 (en) |
| SG (2) | SG177930A1 (en) |
| TW (1) | TWI433674B (en) |
| WO (2) | WO2008083252A2 (en) |
| ZA (2) | ZA200904445B (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7648994B2 (en) | 2007-03-07 | 2010-01-19 | Infinity Discovery, Inc. | Heterocyclic cyclopamine analogs and methods of use thereof |
| EP2225254A1 (en) * | 2007-12-27 | 2010-09-08 | Infinity Pharmaceuticals, Inc. | Therapeutic cancer treatments |
| EP2224807A1 (en) * | 2007-12-27 | 2010-09-08 | Infinity Pharmaceuticals, Inc. | Methods for stereoselective reduction |
| US7812164B2 (en) | 2006-12-28 | 2010-10-12 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogs |
| US7875628B2 (en) | 2004-08-27 | 2011-01-25 | Infinity Discovery, Inc. | Cyclopamine analogues and methods of use thereof |
| US20110135739A1 (en) * | 2009-11-06 | 2011-06-09 | Bennett Carter | Oral Formulations of a Hedgehog Pathway Inhibitor |
| US7964590B2 (en) | 2007-03-07 | 2011-06-21 | Infinity Discovery, Inc. | Cyclopamine lactam analogs and methods of use thereof |
| EP2389068A1 (en) * | 2009-01-23 | 2011-11-30 | Cancer Research Technology Limited | Hedgehog pathway inhibitors |
| WO2013191144A1 (en) | 2012-06-21 | 2013-12-27 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Novel indanesulfamide derivative |
| WO2015093515A1 (en) | 2013-12-19 | 2015-06-25 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Therapeutic and/or preventive agent comprising 1-indansulfamide derivative for pain |
| US9376447B2 (en) | 2010-09-14 | 2016-06-28 | Infinity Pharmaceuticals, Inc. | Transfer hydrogenation of cyclopamine analogs |
| WO2017137003A1 (en) * | 2016-02-14 | 2017-08-17 | 山东大学 | Compound for treating respiratory syncytial virus infection and preparation method and use thereof |
| WO2017139497A1 (en) * | 2016-02-11 | 2017-08-17 | PellePharm, Inc. | Hedgehog inhibitor for use in relief of and treatment of pruritus or itching |
| US9879293B2 (en) | 2009-08-05 | 2018-01-30 | Infinity Pharmaceuticals, Inc. | Enzymatic transamination of cyclopamine analogs |
| US10369147B2 (en) | 2015-06-04 | 2019-08-06 | PellePharm, Inc. | Topical formulations for delivery of hedgehog inhibitor compounds and use thereof |
Families Citing this family (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100222287A1 (en) * | 2007-12-27 | 2010-09-02 | Mcgovern Karen J | Therapeutic Cancer Treatments |
| US20100297118A1 (en) * | 2007-12-27 | 2010-11-25 | Macdougall John | Therapeutic Cancer Treatments |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| WO2009099625A2 (en) * | 2008-02-08 | 2009-08-13 | The Board Of Regents Of The University Of Texas System | Cyclopamine tartrate salt and uses thereof |
| WO2010080817A2 (en) * | 2009-01-06 | 2010-07-15 | Utah State University | Carbohydrate-cyclopamine conjugates as anticancer agents |
| US20110065645A1 (en) * | 2009-09-10 | 2011-03-17 | The Regents Of The University Of California | Compositions and Methods for Modulating Neuron Degeneration and Neuron Guidance |
| AU2010321773A1 (en) * | 2009-11-20 | 2012-06-14 | Infinity Pharmaceuticals, Inc. | Methods and compositions for treating hedgehog-associated cancers |
| WO2011088404A1 (en) * | 2010-01-15 | 2011-07-21 | Infinity Pharmaceuticals , Inc | Treatment of fibrotic conditions using hedgehog inhibitors |
| ES2593256T3 (en) | 2010-05-21 | 2016-12-07 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulations |
| EP2637669A4 (en) | 2010-11-10 | 2014-04-02 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof |
| ES2637113T3 (en) | 2011-01-10 | 2017-10-10 | Infinity Pharmaceuticals, Inc. | Procedures for preparing isoquinolinones and solid forms of isoquinolinones |
| TWI565709B (en) | 2011-07-19 | 2017-01-11 | 英菲尼提製藥股份有限公司 | Heterocyclic compounds and uses thereof |
| CN103930422A (en) | 2011-07-19 | 2014-07-16 | 无限药品股份有限公司 | Heterocyclic compounds and uses thereof |
| WO2013032591A1 (en) | 2011-08-29 | 2013-03-07 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| CA2752008A1 (en) * | 2011-09-13 | 2013-03-13 | Universite De Montreal | Combination therapy using ribavirin as elf4e inhibitor |
| US9630979B2 (en) | 2011-09-29 | 2017-04-25 | Infinity Pharmaceuticals, Inc. | Inhibitors of monoacylglycerol lipase and methods of their use |
| HUE055562T2 (en) | 2011-11-23 | 2021-11-29 | Therapeuticsmd Inc | Natural combination hormone replacement formulations and therapies |
| US9301920B2 (en) | 2012-06-18 | 2016-04-05 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| BR112014022103B1 (en) * | 2012-03-06 | 2022-04-19 | The Board Of Trustees Of The University Of Illinois | COMPOSITIONS AND THEIR USES |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US20150196640A1 (en) | 2012-06-18 | 2015-07-16 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable pk profile |
| US10806697B2 (en) | 2012-12-21 | 2020-10-20 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US20130338122A1 (en) | 2012-06-18 | 2013-12-19 | Therapeuticsmd, Inc. | Transdermal hormone replacement therapies |
| US10806740B2 (en) | 2012-06-18 | 2020-10-20 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| MX2020009849A (en) | 2012-11-01 | 2021-09-13 | Infinity Pharmaceuticals Inc | Treatment of cancers using pi3 kinase isoform modulators. |
| EP2925363A4 (en) | 2012-11-29 | 2016-11-09 | Strasspharma Llc | Methods of modulating follicle stimulating hormone activity |
| US9180091B2 (en) | 2012-12-21 | 2015-11-10 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US11266661B2 (en) | 2012-12-21 | 2022-03-08 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10471072B2 (en) | 2012-12-21 | 2019-11-12 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10568891B2 (en) | 2012-12-21 | 2020-02-25 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10537581B2 (en) | 2012-12-21 | 2020-01-21 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11246875B2 (en) | 2012-12-21 | 2022-02-15 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| EP2970194A1 (en) | 2013-03-15 | 2016-01-20 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9192609B2 (en) | 2013-04-17 | 2015-11-24 | Hedgepath Pharmaceuticals, Inc. | Treatment and prognostic monitoring of proliferation disorders using hedgehog pathway inhibitors |
| US20140377258A1 (en) | 2013-05-30 | 2014-12-25 | Infinity Pharmaceuticals, Inc. | Treatment Of Cancers Using PI3 Kinase Isoform Modulators |
| ES2900806T3 (en) | 2013-10-04 | 2022-03-18 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US20160244452A1 (en) | 2013-10-21 | 2016-08-25 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| JP6701088B2 (en) | 2014-03-19 | 2020-05-27 | インフィニティー ファーマシューティカルズ, インコーポレイテッド | Heterocyclic compounds for use in the treatment of PI3K-gamma mediated disorders |
| WO2015160986A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| WO2015168079A1 (en) | 2014-04-29 | 2015-11-05 | Infinity Pharmaceuticals, Inc. | Pyrimidine or pyridine derivatives useful as pi3k inhibitors |
| RU2016143081A (en) | 2014-05-22 | 2018-06-26 | Терапьютиксмд, Инк. | NATURAL COMBINED HORMONE SUBSTITUTION COMPOSITIONS AND THERAPIES |
| CN105232562B (en) * | 2014-07-07 | 2019-06-28 | 中国科学院上海巴斯德研究所 | A kind of inhibitor and antiviral drugs design target spot of human respiratory syncytial virus |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US10328087B2 (en) | 2015-07-23 | 2019-06-25 | Therapeuticsmd, Inc. | Formulations for solubilizing hormones |
| US10722484B2 (en) | 2016-03-09 | 2020-07-28 | K-Gen, Inc. | Methods of cancer treatment |
| US10286077B2 (en) | 2016-04-01 | 2019-05-14 | Therapeuticsmd, Inc. | Steroid hormone compositions in medium chain oils |
| KR20180126582A (en) | 2016-04-01 | 2018-11-27 | 쎄러퓨틱스엠디, 인코퍼레이티드 | Steroid hormone pharmaceutical composition |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| EP3474856B1 (en) | 2016-06-24 | 2022-09-14 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| MX2019002835A (en) | 2016-09-13 | 2019-09-04 | Allergan Inc | Stabilized non-protein clostridial toxin compositions. |
| MX2020004991A (en) | 2017-11-17 | 2022-02-10 | Univ Illinois | Cancer therapy by degrading dual mek signaling. |
| AU2019354771A1 (en) | 2018-10-05 | 2021-04-01 | The Board Of Trustees Of The University Of Illinois | Combination therapy for the treatment of uveal melanoma |
| US11633405B2 (en) | 2020-02-07 | 2023-04-25 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical formulations |
| CN113461771B (en) * | 2020-03-31 | 2022-09-30 | 南方科技大学 | Preparation method of sodium-potassium ATPase inhibitor |
Family Cites Families (146)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6333341A (en) | 1986-07-28 | 1988-02-13 | Seitetsu Kagaku Co Ltd | Treatment of glycoside |
| US4843071A (en) | 1986-12-05 | 1989-06-27 | Serotonin Industries Of Charleston | Method and composition for treating obesity, drug abuse, and narcolepsy |
| JP2851054B2 (en) | 1989-03-15 | 1999-01-27 | サントリー株式会社 | Benzoxepin derivative |
| US5169780A (en) | 1989-06-22 | 1992-12-08 | Celgene Corporation | Enantiomeric enrichment and stereoselective synthesis of chiral amines |
| FR2656310B1 (en) | 1989-12-22 | 1992-05-07 | Roussel Uclaf | NEW STEROUID PRODUCTS COMPRISING IN POSITION 10, A SUBSTITUTED THIOETHYL RADICAL, THEIR PREPARATION METHOD AND THE INTERMEDIATES THEREOF, THEIR APPLICATION AS MEDICAMENTS AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| US5378475A (en) | 1991-02-21 | 1995-01-03 | University Of Kentucky Research Foundation | Sustained release drug delivery devices |
| ES2123133T3 (en) | 1993-03-10 | 1999-01-01 | Magainin Pharma | STEROIDIAL DERIVATIVES, PHARMACEUTICAL COMPOSITIONS CONTAINING THESE STEROIDIAL DERIVATIVES AND USE OF THESE STEROIDIAL AS ANTIBIOTICS OR DISINFECTANTS. |
| IT1265001B1 (en) | 1993-12-16 | 1996-10-17 | Zambon Spa | PHARMACEUTICAL COMPOSITION FOR TOPICAL USE CONTAINING (S) -2- (4- ISOBUTYLPHENYL) PROPIONIC ACID |
| AU704178B2 (en) | 1993-12-30 | 1999-04-15 | Imperial Cancer Research Technology Ltd | Vertebrate embryonic pattern-inducing hedgehog-like proteins |
| US6281332B1 (en) | 1994-12-02 | 2001-08-28 | The Johns Hopkins University School Of Medicine | Hedgehog-derived polypeptides |
| JPH11514999A (en) | 1995-10-10 | 1999-12-21 | ストルーブ,マリリン | Treatment of pruritus with vitamin D and its derivatives |
| US6887820B1 (en) | 1995-12-06 | 2005-05-03 | Japan Science And Technology Corporation | Method for producing optically active compounds |
| DE69635874T2 (en) | 1995-12-06 | 2006-12-14 | Japan Science And Technology Agency, Kawaguchi | Process for the preparation of optically active alcohols |
| GB9706321D0 (en) | 1997-03-26 | 1997-05-14 | Zeneca Ltd | Catalytic hydrogenation |
| US7741298B2 (en) | 1997-06-20 | 2010-06-22 | New York University | Method and compositions for inhibiting tumorigenesis |
| US7709454B2 (en) | 1997-06-20 | 2010-05-04 | New York University | Methods and compositions for inhibiting tumorigenesis |
| US6238876B1 (en) | 1997-06-20 | 2001-05-29 | New York University | Methods and materials for the diagnosis and treatment of sporadic basal cell carcinoma |
| US7361336B1 (en) | 1997-09-18 | 2008-04-22 | Ivan Bergstein | Methods of cancer therapy targeted against a cancer stem line |
| US6231875B1 (en) | 1998-03-31 | 2001-05-15 | Johnson & Johnson Consumer Companies, Inc. | Acidified composition for topical treatment of nail and skin conditions |
| US6867216B1 (en) | 1998-04-09 | 2005-03-15 | Johns Hopkins University School Of Medicine | Inhibitors of hedgehog signal pathways, compositions and uses related thereto |
| US7291626B1 (en) | 1998-04-09 | 2007-11-06 | John Hopkins University School Of Medicine | Inhibitors of hedgehog signaling pathways, compositions and uses related thereto |
| US6432970B2 (en) | 1998-04-09 | 2002-08-13 | Johns Hopkins University School Of Medicine | Inhibitors of hedgehog signaling pathways, compositions and uses related thereto |
| JP2002522505A (en) * | 1998-08-13 | 2002-07-23 | ユニヴァースティ オブ サザーン カリフォルニア | How to increase blood flow to ischemic tissue |
| GB9821067D0 (en) | 1998-09-29 | 1998-11-18 | Zeneca Ltd | Transfer hydrogenation process |
| US6291516B1 (en) * | 1999-01-13 | 2001-09-18 | Curis, Inc. | Regulators of the hedgehog pathway, compositions and uses related thereto |
| FR2796938B1 (en) | 1999-07-29 | 2004-04-02 | Ppg Sipsy | NOVEL N-SUBSTITUTED NORPHEDRIN CHIRAL DERIVATIVES, THEIR PREPARATION AND THEIR USE FOR THE SYNTHESIS OF OPTICALLY ACTIVE FUNCTIONALIZED COMPOUNDS BY HYDROGEN TRANSFER |
| US20070021493A1 (en) | 1999-09-16 | 2007-01-25 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| AU780846B2 (en) | 1999-09-16 | 2005-04-21 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| US6993456B2 (en) | 1999-09-30 | 2006-01-31 | Rockwell Automation Technologies, Inc. | Mechanical-electrical template based method and apparatus |
| IL149069A0 (en) | 1999-10-13 | 2002-11-10 | Univ Johns Hopkins Med | Regulators of the hedgehog pathway, compositions and uses related thereto |
| US6552016B1 (en) | 1999-10-14 | 2003-04-22 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| IL148972A0 (en) | 1999-10-14 | 2002-11-10 | Curis Inc | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| US6893637B1 (en) | 1999-10-21 | 2005-05-17 | Zymogenetics, Inc. | Method of treating fibrosis |
| IL133809A0 (en) | 1999-12-30 | 2001-04-30 | Yeda Res & Dev | Steroidal alkaloids and pharmaceutical compositions comprising them |
| KR20010077591A (en) | 2000-02-03 | 2001-08-20 | 복성해 | A novel metalloprotease and a gene thereof derived from Aranicola proteolyticus |
| US6613798B1 (en) | 2000-03-30 | 2003-09-02 | Curis, Inc. | Small organic molecule regulators of cell proliferation |
| ES2250377T5 (en) | 2000-03-30 | 2010-04-06 | Curis, Inc. | SMALL ORGANIC MOLECULES REGULATING THE CELL PROLIFERATION. |
| AU2001264595A1 (en) | 2000-05-19 | 2001-12-03 | Guilford Pharmaceuticals Inc. | Sulfonamide and carbamide derivatives of 6(5h)phenanthridinones and their uses |
| DK1387674T3 (en) | 2000-10-13 | 2017-04-10 | Curis Inc | Hedgehog antagonists, methods and uses related thereto |
| US7708998B2 (en) | 2000-10-13 | 2010-05-04 | Curis, Inc. | Methods of inhibiting unwanted cell proliferation using hedgehog antagonists |
| GB0029356D0 (en) | 2000-12-01 | 2001-01-17 | Avecia Ltd | Transfer hydrogenation |
| US6456928B1 (en) | 2000-12-29 | 2002-09-24 | Honeywell International Inc. | Prognostics monitor for systems that are subject to failure |
| CA2440884C (en) | 2001-03-16 | 2012-05-22 | Alza Corporation | Transdermal patch for administering fentanyl |
| EP1401469A2 (en) | 2001-04-09 | 2004-03-31 | Lorantis Limited | Therapeutic use and identification of modulators of a hedgehog signalling pathway or one of its target pathways |
| AUPR602401A0 (en) | 2001-06-29 | 2001-07-26 | Smart Drug Systems Inc | Sustained release delivery system |
| RU2308948C2 (en) * | 2001-07-02 | 2007-10-27 | ТАШ Шинан | Using cyclopamine in treatment of basal cell epithelioma (carcinoma) and other tumors |
| ES2250434T3 (en) | 2001-07-02 | 2006-04-16 | Tas, Sinan | USE OF CYCLOPAMINE IN THE TREATMENT OF CARCINOMA OF BASIC CELLS AND OTHER TUMORS. |
| CA2452152A1 (en) | 2001-07-02 | 2002-10-10 | Sinan Tas | Use of cyclopamine in the treatment of psoriasis |
| NZ530580A (en) | 2001-07-27 | 2007-02-23 | Curis Inc | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| JP2003192919A (en) * | 2001-10-17 | 2003-07-09 | Asahi Denka Kogyo Kk | Flame-retardant synthetic resin composition |
| US7112690B2 (en) | 2001-11-09 | 2006-09-26 | National Research Council Of Canada | Volatile noble metal organometallic complexes |
| US7105637B2 (en) | 2002-02-15 | 2006-09-12 | William Marsh Rice University | Dehydro-estriol (8-DHE3) and dehydro-pregnanetriol (7-DHPT), methods of their synthesis |
| EP1490031A1 (en) * | 2002-03-07 | 2004-12-29 | Vectura Limited | Fast melt multiparticulate formulations for oral delivery |
| CA2482022A1 (en) | 2002-04-19 | 2003-10-30 | Smithkline Beecham Corporation | Novel compounds |
| WO2003088970A2 (en) | 2002-04-22 | 2003-10-30 | Johns Hopkins University School Of Medicine | Modulators of hedgehog signaling pathways, compositions and uses related thereto |
| GB0210234D0 (en) | 2002-05-03 | 2002-06-12 | Novartis Ag | Process for the manufacture of organic compounds |
| NZ537686A (en) | 2002-06-20 | 2007-01-26 | Nippon Suisan Kaisha Ltd | A beta2 agonist prodrug having reduced side effects |
| WO2004020599A2 (en) | 2002-08-29 | 2004-03-11 | Curis, Inc. | Hedgehog antagonists, methods and uses related thereto |
| GB0221539D0 (en) | 2002-09-17 | 2002-10-23 | Medical Res Council | Methods of treatment |
| GB0230217D0 (en) | 2002-12-27 | 2003-02-05 | Biotica Tech Ltd | Borrelidin-producing polyketide synthase and its uses |
| FR2850022B1 (en) | 2003-01-22 | 2006-09-08 | Centre Nat Rech Scient | NOVEL USE OF MIFEPRISTONE AND ITS DERIVATIVES AS MODULATORS OF THE HEDGEHOG PROTEIN SIGNALING PATH AND ITS APPLICATIONS |
| EP1639097B1 (en) | 2003-06-25 | 2013-08-07 | Ottawa Health Research Institute | Methods and compositions for modulating stem cell growth and differentiation |
| US20080118493A1 (en) | 2003-07-15 | 2008-05-22 | Beachy Philip A | Elevated Hedgehog Pathway Activity In Digestive System Tumors, And Methods Of Treating Digestive Sytem Tumors Having Elevated Hedgehog Pathway Activity |
| WO2005033288A2 (en) | 2003-09-29 | 2005-04-14 | The Johns Hopkins University | Hedgehog pathway antagonists |
| US20070231828A1 (en) | 2003-10-01 | 2007-10-04 | Johns Hopkins University | Methods of predicting behavior of cancers |
| US20080095761A1 (en) | 2003-10-01 | 2008-04-24 | The Johns Hopkins University | Hedgehog Signaling in Prostate Regeneration Neoplasia and Metastasis |
| WO2005033286A2 (en) | 2003-10-01 | 2005-04-14 | Invitrogen Corporation | Compositions and methods for synthesizing, purifying and detecting biomolecules |
| US20080057071A1 (en) | 2003-10-20 | 2008-03-06 | Watkins David N | Use Of Hedgehog Pathway Inhibitors In Small-Cell Lung Cancer |
| US20070219250A1 (en) | 2003-11-28 | 2007-09-20 | Romi Singh | Pharmaceutical Compositions of Nateglinide |
| PL1745041T3 (en) | 2004-04-30 | 2012-11-30 | Genentech Inc | Quinoxaline inhibitors of the hedgehog signalling pathway |
| BRPI0514444A (en) * | 2004-08-27 | 2008-06-10 | Infinity Pharmaceuticals Inc | cyclopamine analogues and methods of use of these |
| KR101366414B1 (en) | 2004-09-02 | 2014-03-18 | 쿠리스 인코퍼레이션 | Pyridyl inhibitors of hedgehog signalling |
| WO2006039569A1 (en) | 2004-09-30 | 2006-04-13 | The University Of Chicago | Combination therapy of hedgehog inhibitors, radiation and chemotherapeutic agents |
| BRPI0517253A (en) | 2004-10-28 | 2008-10-07 | Irm Llc | compounds and compositions as hedgehog trajectory modulators |
| US20090208579A1 (en) * | 2004-12-27 | 2009-08-20 | Eisai R & D Management Co., Ltd. | Matrix Type Sustained-Release Preparation Containing Basic Drug or Salt Thereof, and Method for Manufacturing the Same |
| US20060252073A1 (en) | 2005-04-18 | 2006-11-09 | Regents Of The University Of Michigan | Compositions and methods for the treatment of cancer |
| CA2608236A1 (en) | 2005-05-12 | 2006-11-23 | Introgen Therapeutics, Inc. | P53 vaccines for the treatment of cancers |
| AU2006283040B2 (en) | 2005-08-22 | 2011-08-25 | The Johns Hopkins University | Hedgehog pathway antagonists to treat disease |
| JP4712524B2 (en) | 2005-10-28 | 2011-06-29 | 富士通コンポーネント株式会社 | Input device and electronic equipment |
| US20070112017A1 (en) | 2005-10-31 | 2007-05-17 | Braincells, Inc. | Gaba receptor mediated modulation of neurogenesis |
| CN101299921A (en) | 2005-11-04 | 2008-11-05 | 默克公司 | Methods of treating cancers with saha, carboplatin |
| US20090156611A1 (en) | 2005-11-11 | 2009-06-18 | Licentia Ltd. | Mammalian hedgehog signaling modulators |
| ES2604539T3 (en) | 2005-11-14 | 2017-03-07 | Genentech, Inc. | Bisamide type inhibitors of hedgehog signaling |
| WO2007076294A2 (en) | 2005-12-15 | 2007-07-05 | Wei Chen | Method of screening the activity of the smoothened receptor to identify theraputic modulation agents or diagnose disease |
| US20070179091A1 (en) | 2005-12-27 | 2007-08-02 | Genentech, Inc. | Hedgehog Kinases and Their Use in Modulating Hedgehog Signaling |
| EP1818411A1 (en) | 2006-02-13 | 2007-08-15 | Lonza AG | Process for the preparation of optically active chiral amines |
| US20080019961A1 (en) | 2006-02-21 | 2008-01-24 | Regents Of The University Of Michigan | Hedgehog signaling pathway antagonist cancer treatment |
| US20110034498A1 (en) | 2006-03-24 | 2011-02-10 | Mcgovern Karen J | Dosing regimens for the treatment of cancer |
| JP2009536156A (en) | 2006-04-14 | 2009-10-08 | ノバルティス アクチエンゲゼルシャフト | Use of biaryl carboxamides in the treatment of hedgehog pathway related disorders |
| UA93548C2 (en) | 2006-05-05 | 2011-02-25 | Айерем Елелсі | Compounds and compositions as hedgehog pathway modulators |
| DE102006031358A1 (en) * | 2006-07-05 | 2008-01-17 | Schott Ag | Method for housing optical or optoelectronic components, as well as according to the method manufacturable optical or optoelectronic housing element |
| WO2008011071A2 (en) | 2006-07-19 | 2008-01-24 | The Regents Of The University Of California | Interactions of hedgehog and liver x receptor signaling pathways |
| PE20080948A1 (en) | 2006-07-25 | 2008-09-10 | Irm Llc | IMIDAZOLE DERIVATIVES AS MODULATORS OF THE HEDGEHOG PATH |
| US20090305338A1 (en) | 2006-09-29 | 2009-12-10 | Anneli Ritala-Nurmi | Plant cell lines established from the medicinal plant veratrum californicum |
| MX2009003680A (en) | 2006-10-05 | 2009-07-17 | Univ Johns Hopkins | Water-dispersible oral, parenteral, and topical formulations for poorly water soluble drugs using smart polymeric nanoparticles. |
| US8198402B2 (en) | 2006-10-31 | 2012-06-12 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Smoothened polypeptides and methods of use |
| US20080103116A1 (en) | 2006-11-01 | 2008-05-01 | Jennings-Spring Barbara L | Method of treatment and compositions of D-chiro inositol and phosphates thereof |
| WO2008057497A2 (en) | 2006-11-02 | 2008-05-15 | Curis, Inc. | Small organic molecule regulators of cell proliferation |
| CN101610673A (en) | 2006-11-20 | 2009-12-23 | 萨托里医药公司 | Can be used for treating the compound of neurodegenerative disorders |
| WO2008063166A1 (en) | 2006-11-20 | 2008-05-29 | Thomson Licensing | Method for eliminating redundant connections |
| TWI433674B (en) | 2006-12-28 | 2014-04-11 | Infinity Discovery Inc | Cyclopamine analogs |
| WO2008089123A2 (en) | 2007-01-12 | 2008-07-24 | Infinity Discovery, Inc. | Methods for analysis of hedgehog pathway inhibitors |
| FI123481B (en) | 2007-02-05 | 2013-05-31 | Upm Kymmene Corp | Process for producing printing paper and blend composition |
| CN101687877B (en) * | 2007-03-07 | 2014-07-23 | 无限发现股份有限公司 | Heterocyclic cyclopamine analogs and methods of use thereof |
| EP2134719B1 (en) | 2007-03-07 | 2013-07-17 | Infinity Discovery, Inc. | Cyclopamine lactam analogs and methods of use thereof |
| WO2008112913A1 (en) | 2007-03-14 | 2008-09-18 | Exelixis, Inc. | Inhibitors of the hedgehog pathway |
| UA100684C2 (en) | 2007-03-15 | 2013-01-25 | Новартіс Аг | Normal;heading 1;heading 2;heading 3;BENZYL AND PYRIDINYL DERIVATIVES AS MODULATORS OF THE HEDGEHOG SIGNALING PATHWAY |
| US9095589B2 (en) | 2007-04-05 | 2015-08-04 | Johns Hopkins University | Chirally pure isomers of itraconazole for use as angiogenesis inhibitors |
| TW200901987A (en) | 2007-04-18 | 2009-01-16 | Merck & Co Inc | Triazole derivatives which are Smo antagonists |
| WO2008131354A2 (en) | 2007-04-20 | 2008-10-30 | The Curators Of The University Of Missouri | Phytoestrogens as regulators of hedgehog signaling and methods of their use in cancer treatment |
| CN101835374B (en) | 2007-07-09 | 2014-08-27 | 东弗吉尼亚医学院 | Substituted nucleoside derivatives with antiviral and antimicrobial properties |
| US8636982B2 (en) | 2007-08-07 | 2014-01-28 | Foamix Ltd. | Wax foamable vehicle and pharmaceutical compositions thereof |
| WO2009049258A1 (en) * | 2007-10-12 | 2009-04-16 | The Johns Hopkins University | Compounds for hedgehog pathway blockade in proliferative disorders, including hematopoietic malignancies |
| US8592463B2 (en) | 2007-12-13 | 2013-11-26 | Siena Biotech S.P.A. | Hedgehog pathway antagonists and therapeutic applications thereof |
| EP3190121B1 (en) | 2007-12-27 | 2019-02-20 | Infinity Pharmaceuticals, Inc. | Methods for stereoselective reduction of cyclopamine 4-en-3-one derivative |
| JP2011522773A (en) | 2007-12-27 | 2011-08-04 | インフィニティ ファーマスーティカルズ、インク. | Methods of treating cancer with therapeutic agents |
| US20100222287A1 (en) | 2007-12-27 | 2010-09-02 | Mcgovern Karen J | Therapeutic Cancer Treatments |
| US20100297118A1 (en) | 2007-12-27 | 2010-11-25 | Macdougall John | Therapeutic Cancer Treatments |
| WO2009099625A2 (en) | 2008-02-08 | 2009-08-13 | The Board Of Regents Of The University Of Texas System | Cyclopamine tartrate salt and uses thereof |
| US20090246841A1 (en) | 2008-03-26 | 2009-10-01 | Jamieson Andrew C | Methods and compositions for production of acetaldehyde |
| WO2009126840A1 (en) | 2008-04-11 | 2009-10-15 | Galaxy Biotech, Llc | Combination of hgf inhibitor and hedgehog inhibitor to treat cancer |
| CL2009001479A1 (en) | 2008-07-02 | 2010-01-04 | Infinity Pharmaceuticals Inc | A method of isolating a deglycosylated veratrum alkaloid comprising providing a veratrum plant material, contacting an aqueous solution, and extracting the veratrum plant material with a solvent to provide an extract comprising said alkaloid. |
| CA2636807A1 (en) | 2008-07-04 | 2010-01-04 | Steven Splinter | Methods for obtaining cyclopamine |
| US20100041663A1 (en) | 2008-07-18 | 2010-02-18 | Novartis Ag | Organic Compounds as Smo Inhibitors |
| SI2342198T1 (en) | 2008-09-17 | 2013-07-31 | Novartis Ag | A diphosphate salt of n-s6-cis-2,6-dimethylmorpholin-4yl)pyridine-3ylc-2-methyl-4'-(trifluoromethoxy)s1,1'-biphenylc-3-carboxamide |
| EP2342221B1 (en) | 2008-09-22 | 2018-11-07 | Aileron Therapeutics, Inc. | Methods for preparing purified polypeptide compositions |
| US20100081637A1 (en) | 2008-10-01 | 2010-04-01 | Innovia Skincare Corp. | Eczema treatment with vitamin D and analogs thereof method, composition and cream |
| EP2389068A4 (en) | 2009-01-23 | 2012-07-18 | Cancer Rec Tech Ltd | Hedgehog pathway inhibitors |
| AR077490A1 (en) | 2009-07-21 | 2011-08-31 | Novartis Ag | TOPICAL PHARMACEUTICAL COMPOSITIONS FOR THE TREATMENT OF A HYPERPROLIFERATIVE SKIN CONDITION |
| CN102574791A (en) | 2009-08-05 | 2012-07-11 | 无限药品股份有限公司 | Enzymatic transamination of cyclopamine analogs |
| WO2011057222A1 (en) | 2009-11-06 | 2011-05-12 | Infinity Pharmaceuticals, Inc. | Oral formulations of a hedgehog pathway inhibitor |
| AU2010321773A1 (en) | 2009-11-20 | 2012-06-14 | Infinity Pharmaceuticals, Inc. | Methods and compositions for treating hedgehog-associated cancers |
| WO2011088404A1 (en) | 2010-01-15 | 2011-07-21 | Infinity Pharmaceuticals , Inc | Treatment of fibrotic conditions using hedgehog inhibitors |
| GB201010954D0 (en) | 2010-06-29 | 2010-08-11 | Edko Pazarlama Tanitim Ticaret | Compositions |
| US20120010230A1 (en) | 2010-07-08 | 2012-01-12 | Macdougall John R | Methods and compositions for identification, assessment and treatment of cancers associated with hedgehog signaling |
| WO2012006584A2 (en) | 2010-07-08 | 2012-01-12 | Infinity Pharmaceuticals, Inc. | Therapeutic regimens for hedgehog-associated cancers |
| WO2012037217A1 (en) | 2010-09-14 | 2012-03-22 | Infinity Pharmaceuticals, Inc. | Transfer hydrogenation of cyclopamine analogs |
| CN102085176A (en) | 2010-12-31 | 2011-06-08 | 江苏中丹制药有限公司 | Nanometer itraconazole external preparation and preparation method and use thereof |
| US8865765B2 (en) | 2011-01-12 | 2014-10-21 | The William M. Yarbrough Foundation | Method for treating eczema |
| AU2012304407A1 (en) | 2011-09-09 | 2014-04-24 | Children's Hospital & Research Center Oakland | Topical itraconazole formulations and uses thereof |
| US9630979B2 (en) | 2011-09-29 | 2017-04-25 | Infinity Pharmaceuticals, Inc. | Inhibitors of monoacylglycerol lipase and methods of their use |
| NZ708371A (en) | 2012-12-14 | 2020-05-29 | Trevi Therapeutics Inc | Methods for treating pruritus |
| US9452139B2 (en) | 2013-03-14 | 2016-09-27 | Novartis Ag | Respirable agglomerates of porous carrier particles and micronized drug |
| NZ724691A (en) | 2014-03-24 | 2018-02-23 | Guangdong Zhongsheng Pharmaceutical Co Ltd | Quinoline derivatives as smo inhibitors |
| BR112017026103B1 (en) | 2015-06-04 | 2023-10-03 | Sol-Gel Technologies Ltd | TOPICAL COMPOSITIONS WITH HEDGEHOG INHIBITOR COMPOUND, TOPICAL DELIVERY SYSTEM AND THEIR USES |
| WO2017139497A1 (en) | 2016-02-11 | 2017-08-17 | PellePharm, Inc. | Hedgehog inhibitor for use in relief of and treatment of pruritus or itching |
-
2007
- 2007-12-24 TW TW096149773A patent/TWI433674B/en active
- 2007-12-27 AU AU2007339786A patent/AU2007339786B2/en active Active
- 2007-12-27 US US11/965,675 patent/US8017648B2/en active Active
- 2007-12-27 AU AU2007339782A patent/AU2007339782B2/en active Active
- 2007-12-27 WO PCT/US2007/088995 patent/WO2008083252A2/en active Application Filing
- 2007-12-27 NO NO20190059A patent/NO347310B1/en unknown
- 2007-12-27 CA CA2674302A patent/CA2674302C/en active Active
- 2007-12-27 KR KR1020097015913A patent/KR101489394B1/en active IP Right Grant
- 2007-12-27 MX MX2009007075A patent/MX2009007075A/en active IP Right Grant
- 2007-12-27 NZ NZ577916A patent/NZ577916A/en unknown
- 2007-12-27 WO PCT/US2007/088990 patent/WO2008083248A2/en active Application Filing
- 2007-12-27 NZ NZ578053A patent/NZ578053A/en unknown
- 2007-12-27 EP EP07870006.9A patent/EP2101580B1/en active Active
- 2007-12-27 EP EP11165825.8A patent/EP2368551B1/en active Active
- 2007-12-27 RU RU2009128997/04A patent/RU2486194C2/en active
- 2007-12-27 PT PT78700010T patent/PT2096917E/en unknown
- 2007-12-27 JO JO2007564A patent/JO2947B1/en active
- 2007-12-27 SG SG2011096674A patent/SG177930A1/en unknown
- 2007-12-27 CN CN200780051112.8A patent/CN101677551B/en active Active
- 2007-12-27 US US11/965,688 patent/US7812164B2/en active Active
- 2007-12-27 JP JP2009544276A patent/JP5412292B2/en active Active
- 2007-12-27 SG SG2011096666A patent/SG177929A1/en unknown
- 2007-12-27 KR KR1020097015914A patent/KR101482693B1/en active IP Right Grant
- 2007-12-27 RU RU2009129000/15A patent/RU2492859C2/en active
- 2007-12-27 CN CN200780051114.7A patent/CN101631463B/en active Active
- 2007-12-27 ES ES07870001.0T patent/ES2488466T3/en active Active
- 2007-12-27 JP JP2009544277A patent/JP5542448B2/en active Active
- 2007-12-27 CA CA2673995A patent/CA2673995C/en active Active
- 2007-12-27 MX MX2012007856A patent/MX342683B/en unknown
- 2007-12-27 BR BRPI0720993A patent/BRPI0720993B8/en active IP Right Grant
- 2007-12-27 DK DK07870001.0T patent/DK2096917T3/en active
- 2007-12-27 MX MX2009007077A patent/MX2009007077A/en active IP Right Grant
- 2007-12-27 EP EP11189894A patent/EP2443926A3/en not_active Withdrawn
- 2007-12-27 BR BRPI0720972A patent/BRPI0720972B8/en active IP Right Grant
- 2007-12-27 PL PL07870001T patent/PL2096917T3/en unknown
- 2007-12-27 ES ES07870006.9T patent/ES2612746T3/en active Active
- 2007-12-27 EP EP07870001.0A patent/EP2096917B1/en active Active
- 2007-12-28 CL CL200703854A patent/CL2007003854A1/en unknown
- 2007-12-28 AR ARP070105973A patent/AR081107A1/en active IP Right Grant
-
2008
- 2008-01-02 PE PE2008000074A patent/PE20090042A1/en active IP Right Grant
- 2008-08-27 US US12/199,689 patent/US8785635B2/en active Active
- 2008-08-27 US US12/199,722 patent/US8669365B2/en active Active
-
2009
- 2009-06-25 ZA ZA2009/04445A patent/ZA200904445B/en unknown
- 2009-06-25 IL IL199554A patent/IL199554B/en active IP Right Grant
- 2009-06-25 ZA ZA2009/04446A patent/ZA200904446B/en unknown
- 2009-07-20 NO NO20092710A patent/NO343748B1/en unknown
- 2009-07-20 NO NO20092711A patent/NO344315B1/en not_active IP Right Cessation
-
2011
- 2011-04-20 US US13/090,694 patent/US20110230509A1/en not_active Abandoned
- 2011-07-27 US US13/191,545 patent/US8227509B2/en active Active
- 2011-12-22 HK HK11113839.5A patent/HK1158989A1/en unknown
-
2012
- 2012-05-18 US US13/475,352 patent/US8895576B2/en active Active
- 2012-05-18 US US13/475,510 patent/US20130108583A1/en not_active Abandoned
-
2013
- 2013-10-15 US US14/054,350 patent/US9145422B2/en active Active
-
2014
- 2014-03-10 US US14/203,297 patent/US9492435B2/en active Active
-
2015
- 2015-08-31 US US14/841,001 patent/US9669011B2/en active Active
-
2016
- 2016-10-10 US US15/289,887 patent/US9951083B2/en active Active
-
2017
- 2017-05-02 US US15/585,038 patent/US10045970B2/en active Active
-
2018
- 2018-03-27 US US15/936,919 patent/US10406139B2/en active Active
- 2018-07-27 US US16/047,008 patent/US10314827B2/en active Active
-
2019
- 2019-04-26 US US16/396,565 patent/US10821102B2/en active Active
- 2019-07-24 US US16/521,447 patent/US11007181B2/en active Active
-
2020
- 2020-09-28 US US17/035,611 patent/US11602527B2/en active Active
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7875628B2 (en) | 2004-08-27 | 2011-01-25 | Infinity Discovery, Inc. | Cyclopamine analogues and methods of use thereof |
| US8236956B2 (en) | 2004-08-27 | 2012-08-07 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogues and methods of use thereof |
| US9145422B2 (en) | 2006-12-28 | 2015-09-29 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US8017648B2 (en) | 2006-12-28 | 2011-09-13 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US10045970B2 (en) | 2006-12-28 | 2018-08-14 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US10314827B2 (en) | 2006-12-28 | 2019-06-11 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US11602527B2 (en) | 2006-12-28 | 2023-03-14 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US9951083B2 (en) | 2006-12-28 | 2018-04-24 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogs |
| US10821102B2 (en) | 2006-12-28 | 2020-11-03 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US7812164B2 (en) | 2006-12-28 | 2010-10-12 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogs |
| US11007181B2 (en) | 2006-12-28 | 2021-05-18 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogs |
| US10406139B2 (en) | 2006-12-28 | 2019-09-10 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogs |
| US8227509B2 (en) | 2006-12-28 | 2012-07-24 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US9669011B2 (en) | 2006-12-28 | 2017-06-06 | Infinity Pharmaceuticals, Inc. | Methods of use of cyclopamine analogs |
| US8785635B2 (en) | 2006-12-28 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogs |
| US8669365B2 (en) | 2006-12-28 | 2014-03-11 | Infinity Pharmaceuticals, Inc. | Cyclopamine analogs |
| US7964590B2 (en) | 2007-03-07 | 2011-06-21 | Infinity Discovery, Inc. | Cyclopamine lactam analogs and methods of use thereof |
| US7648994B2 (en) | 2007-03-07 | 2010-01-19 | Infinity Discovery, Inc. | Heterocyclic cyclopamine analogs and methods of use thereof |
| US8426436B2 (en) | 2007-03-07 | 2013-04-23 | Infinity Discovery, Inc. | Heterocyclic cyclopamine analogs and methods of use thereof |
| US7994191B2 (en) | 2007-03-07 | 2011-08-09 | Infinity Discovery, Inc. | Heterocyclic cyclopamine analogs and methods of use thereof |
| US8431566B2 (en) | 2007-03-07 | 2013-04-30 | Infinity Discovery, Inc. | Cyclopamine lactam analogs and methods of use thereof |
| US8293760B2 (en) | 2007-03-07 | 2012-10-23 | Infinity Discovery, Inc. | Cyclopamine lactam analogs and methods of use thereof |
| US8716479B2 (en) | 2007-12-27 | 2014-05-06 | Infinity Pharmaceuticals, Inc. | Methods for stereoselective reduction |
| CN104059124A (en) * | 2007-12-27 | 2014-09-24 | 无限药品股份有限公司 | Methods For Stereoselective Reduction |
| EP2224807A4 (en) * | 2007-12-27 | 2012-12-12 | Infinity Pharmaceuticals Inc | Methods for stereoselective reduction |
| US9238672B2 (en) | 2007-12-27 | 2016-01-19 | Infinity Pharmaceuticals, Inc. | Methods for stereoselective reduction |
| EP2225254A4 (en) * | 2007-12-27 | 2011-03-30 | Infinity Pharmaceuticals Inc | Therapeutic cancer treatments |
| EP2224807A1 (en) * | 2007-12-27 | 2010-09-08 | Infinity Pharmaceuticals, Inc. | Methods for stereoselective reduction |
| EP2225254A1 (en) * | 2007-12-27 | 2010-09-08 | Infinity Pharmaceuticals, Inc. | Therapeutic cancer treatments |
| EP3190121A1 (en) * | 2007-12-27 | 2017-07-12 | Infinity Pharmaceuticals, Inc. | Methods for stereoselective reduction of cyclopamine 4-en-3-one derivative |
| EP2389068A4 (en) * | 2009-01-23 | 2012-07-18 | Cancer Rec Tech Ltd | Hedgehog pathway inhibitors |
| EP2389068A1 (en) * | 2009-01-23 | 2011-11-30 | Cancer Research Technology Limited | Hedgehog pathway inhibitors |
| US9879293B2 (en) | 2009-08-05 | 2018-01-30 | Infinity Pharmaceuticals, Inc. | Enzymatic transamination of cyclopamine analogs |
| US20110135739A1 (en) * | 2009-11-06 | 2011-06-09 | Bennett Carter | Oral Formulations of a Hedgehog Pathway Inhibitor |
| US9879025B2 (en) | 2010-09-14 | 2018-01-30 | Infinity Pharmaceuticals, Inc. | Transfer hydrogenation of cyclopamine analogs |
| US9394313B2 (en) | 2010-09-14 | 2016-07-19 | Infinity Pharmaceuticals, Inc. | Transfer hydrogenation of cyclopamine analogs |
| US9376447B2 (en) | 2010-09-14 | 2016-06-28 | Infinity Pharmaceuticals, Inc. | Transfer hydrogenation of cyclopamine analogs |
| WO2013191144A1 (en) | 2012-06-21 | 2013-12-27 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Novel indanesulfamide derivative |
| WO2015093515A1 (en) | 2013-12-19 | 2015-06-25 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Therapeutic and/or preventive agent comprising 1-indansulfamide derivative for pain |
| US10695344B2 (en) | 2015-06-04 | 2020-06-30 | PellePharm, Inc. | Topical formulations for delivery of hedgehog inhibitor compounds and use thereof |
| US10369147B2 (en) | 2015-06-04 | 2019-08-06 | PellePharm, Inc. | Topical formulations for delivery of hedgehog inhibitor compounds and use thereof |
| US11413283B2 (en) | 2015-06-04 | 2022-08-16 | PellePharm, Inc. | Topical formulations for delivery of hedgehog inhibitor compounds and use thereof |
| WO2017139497A1 (en) * | 2016-02-11 | 2017-08-17 | PellePharm, Inc. | Hedgehog inhibitor for use in relief of and treatment of pruritus or itching |
| US10035816B2 (en) | 2016-02-14 | 2018-07-31 | Shandong University | Compound for treating respiratory syncytial virus infection and preparation method and use thereof |
| WO2017137003A1 (en) * | 2016-02-14 | 2017-08-17 | 山东大学 | Compound for treating respiratory syncytial virus infection and preparation method and use thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11007181B2 (en) | Cyclopamine analogs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780051114.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07870001 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 577916 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007339782 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2673995 Country of ref document: CA Ref document number: 2007870001 Country of ref document: EP Ref document number: 12009501298 Country of ref document: PH Ref document number: MX/A/2009/007077 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2009544276 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2545/KOLNP/2009 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2007339782 Country of ref document: AU Date of ref document: 20071227 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2009128997 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097015913 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0720972 Country of ref document: BR Kind code of ref document: A2 Effective date: 20090629 |