WO2008066298A1 - Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same - Google Patents
Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same Download PDFInfo
- Publication number
- WO2008066298A1 WO2008066298A1 PCT/KR2007/006012 KR2007006012W WO2008066298A1 WO 2008066298 A1 WO2008066298 A1 WO 2008066298A1 KR 2007006012 W KR2007006012 W KR 2007006012W WO 2008066298 A1 WO2008066298 A1 WO 2008066298A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- composition according
- formula
- compound
- formulation
- substituted
- Prior art date
Links
- 0 *C1(*)C(C(C(c2c3cccc2)=O)=O)=C3OC1(*)* Chemical compound *C1(*)C(C(C(c2c3cccc2)=O)=O)=C3OC1(*)* 0.000 description 2
- MMMSEHKHTHHENG-UHFFFAOYSA-N CC(C)(C1(C)Cc(cc2OC)cc(C(C(C)(C3)C4=O)OC3=C)c2C4=O)OC(C2C(C3=O)=CC=CC2)=C1C3=O Chemical compound CC(C)(C1(C)Cc(cc2OC)cc(C(C(C)(C3)C4=O)OC3=C)c2C4=O)OC(C2C(C3=O)=CC=CC2)=C1C3=O MMMSEHKHTHHENG-UHFFFAOYSA-N 0.000 description 1
- HYXITZLLTYIPOF-UHFFFAOYSA-N CC1(C)c2ccc(-c([o]cc3C)c3C(C3=O)=O)c3c2CCC1 Chemical compound CC1(C)c2ccc(-c([o]cc3C)c3C(C3=O)=O)c3c2CCC1 HYXITZLLTYIPOF-UHFFFAOYSA-N 0.000 description 1
- BUJFTKPQXSIZFX-UHFFFAOYSA-N CCC(C)(C)NC Chemical compound CCC(C)(C)NC BUJFTKPQXSIZFX-UHFFFAOYSA-N 0.000 description 1
- AIGAZQPHXLWMOJ-UHFFFAOYSA-N Cc1c[o]c(-c(ccc2c(C)cccc22)c2C2=O)c1C2=O Chemical compound Cc1c[o]c(-c(ccc2c(C)cccc22)c2C2=O)c1C2=O AIGAZQPHXLWMOJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
Definitions
- the present invention relates to a naphthoquinone-based compound having therapeutic effect on the treatment and/or prevention of prostate and/or testicle (seminal glands)-related diseases, and to a pharmaceutical composition of intestinal delivery system containing the same.
- Prostate is a walnut-shape organ that surrounds the distal urethra in men and is located just below the urinary bladder where it produces mucous substances.
- the only function of the prostate is to protect semen and produce seminal fluid for breeding.
- the prostate has characteristics in that it grows larger with aging and that most mammals have the urethra for excreting urine from the bladder which passes through the prostate. Due to such anatomical characteristics of the prostate, men may sometimes have urinary excretion-related problems and induce various diseases including prostate-related diseases.
- the prostate-related disease is a collective term for prostatitis and benign prostatic hyperplasia.
- the prostatitis is defined as an infection or inflammation of the prostate. High fever, acheness and stiffness caused by the prostatitis are generally chronic symptoms, although there are acute cases. Thus, despite the standard therapy, the prostatitis is considered one of incurable diseases with a relatively high relapse rate. Statistically, 30% or more of men in 20s to 50s suffer from a prostate-related disease, and it is a frequently occurring disease which occupies 25% or more of the outpatients in urology. However, the full recovery rate of the disease is very low, thereby having problems that 80 to 90% of the above patients suffers recurrence.
- the prostatitis is induced by inflammation of the prostate tissue, and its symptoms include conspicuously frequent micturition, thinning of urine flow, burning pain during urination, indisposed pain in the abdominal and perineal region, and serious testicle or back pain. In addition, these symptoms become intense after drinking or overworking, thereby progressing to other general symptoms of sexual dysfunction, prospermia, and fatigue.
- chronic prostatitis is a disease which frequently appears in adult men. Prostatitis has substantial relation to all prostate cancer. Even with no other peculiar symptoms, inflammation may be found through histologic examination. When suffering from the chronic prostatitis, pain commonly begins at the pelvic area and progresses to symptoms such as urinary hesitancy, impotence and sterility. A typical symptom, 'urinary hesitancy', associated with prostatitis leads to symptoms such as lack of sleep caused by nocturia, weak urinary stream and urinary retention.
- the benign prostatic hyperplasia (BPH) refers to the increase in size of the prostate. The disease occupies the most frequent occurrence among male urinary dysfunction, and its frequency increases as men get older. Men aged 60 or older suffer from this disease relatively more often. As a result, the disease increases in its rate of occurrence in the aging society.
- the benign prostatic hyperplasia as the prostate enlarges, show symptoms including frequency of urination, especially nocturine (waking up to urinate about 2 to 4 times at night time), difficulty starting the urine flow and decreased unrination force.
- nocturine waking up to urinate about 2 to 4 times at night time
- difficulty starting the urine flow and decreased unrination force.
- the irritative symptoms and urination symptoms that have been observed from beginning become intensed as the conditions develops to finally being unable to urinate. Thereafter, the urine amount is reduced even more, and residual urine amount are increased resulting in obstructive effects on the kidney.
- the chronic prostatitis and benign prostatic hyperplasia may not show serious symptoms in many men. However, they are chronic diseases that give serious influence to the quality of life. Moreover, their diagnosis is difficult and complete cure of the diseases is also difficult. In addition, the occurrence of the prostate cancer tends to rise due to the increase in the average lifespan and westernized diet.
- Another object of the present invention is to provide a pharmaceutical composition of an intestinal delivery system including, as an active ingredient, the naphthoquinone-based compound.
- the above and other objects can be accomplished by the provision of one or more selected from the compounds represented by the following Formula 1 and Formula 2, or a pharmaceutically acceptable salt, prodrug, solvate or isomer thereof, having a therapeutic effect on the treatment and prevention of prostate and/or testicle (seminal glands)-related diseases:
- R 1 and R 2 are each independently hydrogen, halogen, hydroxyl, or Ci-C 6 lower alkyl or alkoxy, or R 1 and R 2 may be taken together to form a substituted or unsubstituted cyclic structure which may be saturated or partially or completely unsaturated;
- R 3 , R 4 , R 5 , R 6 , R 7 and Rg are each independently hydrogen, hydroxyl, amino, Q-C 2O alkyl, alkene or alkoxy, C 4 -C 2O cycloalkyl, heterocycloalkyl, aryl or heteroaryl, or two substituents of R 3 to Rg may be taken together to form a cyclic structure which may be saturated or partially or completely unsaturated;
- X is selected from a group consisting of C(R)(R'), N(R"), O and S, preferably O or S, and more preferably O, wherein R, R' and R" are each independently hydrogen or C 1 -C 6 lower alkyl;
- Y is C, S or N, with proviso that when Y is S, R 7 and R 8 are not any substituent, and when Y is N, R 7 is hydrogen or C 1 -C 6 lower alkyl and R 8 are not any substituent;
- n is 0 or 1, with proviso that when n is 0, carbon atoms adjacent to n form a cyclic structure via a direct bond, or a pharmaceutically acceptable salt, prodrug, solvate or isomer thereof.
- pharmaceutically acceptable salt means a formulation of a compound that does not cause significant irritation to an organism to which it is administered and does not abrogate the biological activity and properties of the compound.
- Examples of the pharmaceutical salt may include acid addition salts of the compound with acids capable of forming a non-toxic acid addition salt containing pharmaceutically acceptable anions, for example, inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, hydrobromic acid and hydroiodic acid; organic carbonic acids such as tartaric acid, formic acid, citric acid, acetic acid, trichloroacetic acid, trifluoroacetic acid, gluconic acid, benzoic acid, lactic acid, fumaric acid, maleic acid and salicylic acid; or sulfonic acids such as methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid and p-toluenesulfonic acid.
- inorganic acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, hydrobromic acid and hydroiodic acid
- organic carbonic acids such as tartaric acid, formic acid
- examples of pharmaceutically acceptable carboxylic acid salts include salts with alkali metals or alkaline earth metals such as lithium, sodium, potassium, calcium and magnesium, salts with amino acids such as arginine, lysine and guanidine, salts with organic bases such as dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)rnethylarnine, diethanolamine, choline and triethylamine.
- the compound 1 or 2 in accordance with the present invention may be converted into salts thereof, by conventional methods well-known in the art.
- prodrug means an agent that is converted into the parent drug in vivo.
- Prodrugs are often useful because, in some situations, they may be easier to administer than the parent drug. They may, for instance, be bioavailable by oral administration, whereas the parent may be not.
- the prodrugs may also have improved solubility in pharmaceutical compositions over the parent drug.
- An example of a prodrug would be a compound of the present invention which is administered as an ester (the "prodrug") to facilitate transport across a cell membrane where water-solubility is detrimental to mobility, but which then is metabolically hydrolyzed to the carboxylic acid, the active entity, once inside the cell where water solubility is beneficial.
- a further example of the prodrug might be a short peptide (polyamino acid) bonded to an acidic group, where the peptide is metabolized to reveal the active moiety.
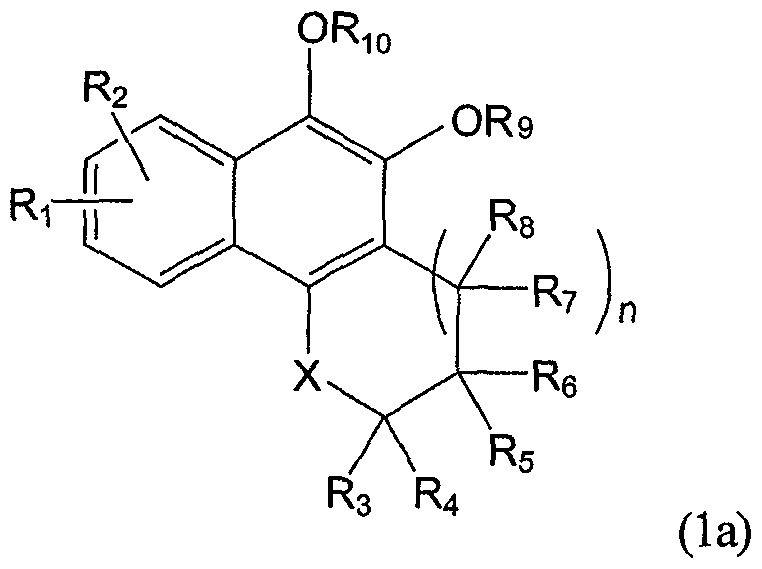
- the pharmaceutical compounds in accordance with the present invention can include a prodrug represented by Formula Ia below:
- R 1 , R. 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , X and n are as defined in Formula 1.
- R 9 and Ri 0 are each independently -SO 3 -Na + or substituent represented by Formula A below or a salt thereof,
- R 11 and R 12 are each independently hydrogen or substituted or unsubstituted Ci-C 20 linear alkyl or C 1 -C 20 branched alkyl
- R 13 is selected from the group consisting of substituents i) to viii) below:
- R, R' and R" are each independently hydrogen or substituted or unsubstituted C 1 -C 20 linear alkyl or C 1 -C 2O branched alkyl, R 14 is selected from the group consisting of hydrogen, substituted or unsubstituted amine, cycloalkyl, heterocycloalkyl, aryl and heteroaryl, 1 is selected from the 1-5;
- k is selected from the 0-20, with proviso that when k is 0, Rn and Ri 2 are not anything, and R ⁇ is directly bond to a carbonyl group.
- solvate means a compound of the present invention or a salt thereof, which further includes a stoichiometric or non-stoichiometric amount of a solvent bound thereto by non-covalent intermolecular forces.
- Preferred solvents are volatile, non-toxic, and/or acceptable for administration to humans. Where the solvent is water, the solvate refers to a hydrate.
- the term "isomer” means a compound of the present invention or a salt thereof, that has the same chemical formula or molecular formula but is optically or sterically different therefrom.
- D type optical isomer and L type optical isomer can be present in the Formula 1 or Formula 2, depending on the R 3 ⁇ R « types of substituents selected.
- compound of Formula 1 or 2 is intended to encompass a compound per se, and a pharmaceutically acceptable salt, prodrug, solvate and isomer thereof.
- alkyl refers to an aliphatic hydrocarbon group.
- the alkyl moiety may be a "saturated alkyl” group, which means that it does not contain any alkene or alkyne moieties.
- the alkyl moiety may also be an "unsaturated alkyl” moiety, which means that it contains at least one alkene or alkyne moiety.
- alkene moiety refers to a group in which at least two carbon atoms form at least one carbon-carbon double bond
- an “alkyne” moiety refers to a group in which at least two carbon atoms form at least one carbon-carbon triple bond.
- heterocycloalkyl means a carbocyclic group in which one or more ring carbon atoms are substituted with oxygen, nitrogen or sulfur and which includes, for example, but is not limited to furan, thiophene, pyrrole, pyrroline, pyrrolidine, oxazole, thiazole, imidazole, imidazoline, imidazolidine, pyrazole, pyrazoline, pyrazolidine, isothiazole, triazole, thiadiazole, pyran, pyridine, piperidine, mo ⁇ holine, thiomorpholine, pyridazine, pyrimidine, pyrazine, piperazine and triazine.
- aryl refers to an aromatic substituent group which has at least one ring having a conjugated pi ( ⁇ ) electron system and includes both carbocyclic aryl (for example, phenyl) and heterocyclic aryl (for example, pyridine) groups. This term includes monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups.
- heteroaryl refers to an aromatic group that contains at least one heterocyclic ring.
- aryl or heteroaryl examples include, but are not limited to, phenyl, furan, pyran, pyridyl, pyrimidyl and triazyl.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R7 and R 8 in Formula 1 in accordance with the present invention may be optionally substituted.
- the substituent group(s) is(are) one or more group(s) individually and independently selected from cycloalkyl, aryl, heteroaryl, heteroalicyclic, hydroxy, alkoxy, aryloxy, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, thiocarbonyl, O-carbamyl, N carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, S-sulfonamido, N-sulfonamido, C-carboxy, 0-carboxy, isocyanato, thiocyanato, isothiocyanato, nitro, silyl, trihalomethanesulfonyl
- Compounds of Formula 3 are compounds wherein n is 0 and adjacent carbon atoms form a cyclic structure (furan ring) via a direct bond therebetween and are often referred to as “furan compounds” or “furano-o-naphthoquinone derivatives” hereinafter.
- Compounds of Formula 4 are compounds wherein n is 1 and are often referred to as “pyran compounds” or “pyrano-o-naphthoquinone” hereinafter.
- furan compounds of Formula 3 preferred are compounds of Formula 3a wherein R 1 , R 2 and R 4 are hydrogen, or compounds of Formula 3b wherein Rj, R 2 and R 6 are hydrogen.
- furan compounds of Formula 3 particularly preferred are compounds below.
- pyran compounds of Formula 4 particularly preferred are compounds of Formula 4a wherein Ri, R 2 , R 5 , R 6 , R 7 and R 8 are respectively hydrogen, or compounds of Formula 4b or Formula 4c wherein Rj and R 2 are taken together to form a cyclic structure which is substituted or unsubstituted.
- Compounds of Formula 2a below are compounds wherein n is 0 and adjacent carbon atoms form a cyclic structure via a direct bond therebetween and Y is C.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 and X are as defined in Formula 2a or 2b.
- active ingredient Effective substance which exerts therapeutic effect on the treatment and/or prevention of prostate and/or testicle (seminal glands)-related diseases in the present invention is often referred to as "active ingredient" hereinafter.
- compounds of Formula 1 or Formula 2 can be prepared by conventional methods known in the art and/or various processes which are based upon the general technologies and practices in the organic chemistry synthesis field.
- the preparation processes described below are only exemplary ones and other processes can also be employed. As such, the scope of the instant invention is not limited to the following processes.
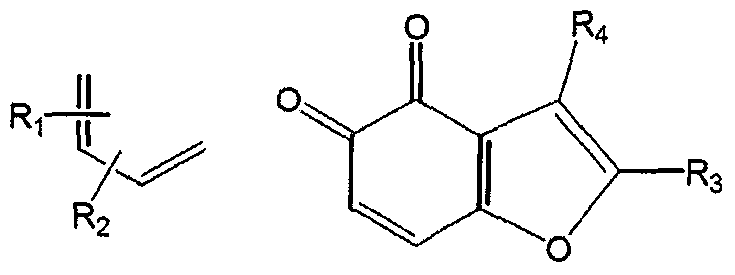
- tricyclic naphthoquinone (pyrano-o-naphthoquinone and furano-o- naphthoquinone) derivatives can be synthesized by two methods mainly.
- One is to derive cyclization reaction using 3-allyl-2-hydroxy-l,4-naphthoquinone in acid catalyst condition, as the following ⁇ -lapachone synthesis scheme.
- 3-allyloxy-l,4-phenanthrenequinone can be obtained by deriving Diels- Alder reaction between 2-allyloxy-l,4-benzoquinone and styrene or 1-vinylcyclohexane derivatives and dehydrating the resulting intermediates using oxygen present in the air or oxidants such as NaIO 4 and DDQ.
- 2-allyl-3-hydroxy-l,4- phenanthrenequinone of Lapachole form can be synthesized via Claisen rearrangement.
- 3-allyloxy-l,4-phenanthrenequinone is hydrolyzed to 3-oxy-l,4- phenanthrenequinone, in the condition of acid (H + ) or alkali (OH " ) catalyst, which is then reacted with various allyl halides to synthesize 2-allyl-3 -hydroxy- 1,4-phenanthrenequinone by C-alkylation.
- the thus obtained 2-allyl-3-hydroxy-l,4-phenanthrenequinone derivatives are subject to cyclization in the condition of acid catalyst to synthesize various 3,4- phenanthrenequinone-based or 5,6,7,8-tetrahydro-3,4-naphthoquinone-based compounds.
- Preparation method 1 is a synthesis of active ingredient by acid-catalyzed cyclization which may be summarized in the general chemical reaction scheme as follows.
- C-alkylated derivatives thus obtained may be subjected to cyclization using sulfuric acid as a catalyst, thereby being capable of synthesizing pyrano-o-naphthoquinone or furano-o-naphthoquinone derivatives among the compounds.
- Preparation method 2 is Diels-Alder reaction using 3 -methylene- 1,2,4- [3UJnaphthalenetrione.
- V. Nair et al Tetrahedron Lett. 42 (2001), 4549-4551, it is reported that a variety of pyrano-o-naphthoquinone derivatives can be relatively easily synthesized by subjecting 3-methylene-l,2,4-[3H]naphthalenetrione, produced upon heating 2- hydroxy-l,4-naphthoquinone and formaldehyde together, to Diels-Alder reaction with various olefin compounds.
- This method is advantageous in that various forms of pyrano-o-naphthoquinone derivatives can be synthesized in a relatively simplified manner, as compared to induction of cyclization using sulfuric acid as a catalyst.
- Preparation method 3 is haloakylation and cyclization by radical reaction.
- the same method used in synthesis of cryptotanshinone and 15,16-dihydro-tanshinone can also be conveniently employed for synthesis of furano-o-naphthoquinone derivatives. That is, as taught by A. C. Baillie et a! (J. Chem. Soc.
- 2-haloethyl or 3-haloethyl radical chemical species derived from 3-halopropanoic acid or 4-halobutanoic acid derivative
- 2-hydroxy-l,4-naphthoquinone can be reacted with 2-hydroxy-l,4-naphthoquinone to thereby synthesize 3-(2-haloethyl or 3- halopropyl)-2-hydroxy-l,4-naphthoquinone, which is then subjected to cyclization under suitable acidic catalyst conditions to synthesize various pyrano-o-naphthoquinone or furano-o- naphthoquinone derivatives.
- Preparation method 4 is cyclization of 4,5-benzofurandione by Diels-Alder reaction.
- Another method used in synthesis of cryptotanshinone and 15,16-dihydro- tanshinone may be a method taught by J. K. Snyder et al (Tetrahedron Letters 28 (1987), 3427-3430).
- furano-o-naphthoquinone derivatives can be synthesized by cycloaddition via Diels-Alder reaction between 4,5-benzofurandione derivatives and various diene derivatives.
- composition means a mixture of a compound of Formula 1 or Formula 2 with other chemical components, such as diluents or carriers.
- the pharmaceutical composition facilitates administration of the compound to an organism.
- Various techniques of administering a compound are known in the art and include, but are not limited to oral, injection, aerosol, parenteral and topical administrations.
- Pharmaceutical compositions can also be obtained by reacting compounds of interest with acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
- a therapeutically effective amount means an amount of an active ingredient that is effective to relieve or reduce to some extent one or more of the symptoms of the disease in need of treatment, or to retard initiation of clinical markers or symptoms of a disease in need of prevention, when the compound is administered.
- a therapeutically effective amount refers to an amount of the active ingredient which exhibit effects of (i) reversing the rate of progress of a disease; (ii) inhibiting to some extent further progress of the disease; and/or, (iii) relieving to some extent (or, preferably, eliminating) one or more symptoms associated with the disease.
- the therapeutically effective amount may be empirically determined by experimenting with the compounds concerned in known in vivo and in vitro model systems for a disease in need of treatment.
- the pharmaceutical composition of the present invention may be manufactured in a manner that is itself known, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or lyophilizing processes.
- compositions for use in accordance with the present invention may be additionally comprised of a pharmaceutically acceptable carrier, a diluent or an excipient, or any combination thereof. That may be formulated in a conventional manner using one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries which facilitate processing of the active compounds into preparations which can be used pharmaceutically.
- the pharmaceutical composition facilitates administration of the compound to an organism.
- carrier means a chemical compound that facilitates the incorporation of a compound into cells or tissues.
- DMSO dimethyl sulfoxide
- carrier facilitates the uptake of many organic compounds into the cells or tissues of an organism.
- diot defines chemical compounds diluted in water that will dissolve the compound of interest as well as stabilize the biologically active form of the compound. Salts dissolved in buffered solutions are utilized as diluents in the art.
- buffer solution is phosphate buffered saline (PBS) because it mimics the ionic strength conditions of human body fluid. Since buffer salts can control the pH of a solution at low concentrations, a buffer diluent rarely modifies the biological activity of a compound.
- PBS phosphate buffered saline
- the compounds described herein may be administered to a human patient per se, or in the form of pharmaceutical compositions in which they are mixed with other active ingredients, as in combination therapy, or suitable carriers or excipient(s). Proper formulation is dependent upon the route of administration chosen. Techniques for formulation and administration of the compounds may be found in "Remington's Pharmaceutical Sciences,” Mack Publishing Co., Easton, PA, 18th edition, 1990.
- compositions can also be obtained by reacting compounds of interest with acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
- acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
- composition may be formulated in a conventional manner.
- the formulation may be in a form of pharmaceutically acceptable oral, parenteral, topical or membranal administration, or injection and more preferably oral administration.
- the naphthoquinone-based compound is a sparingly soluble substance which dissolves a small amount (about 2 to 4%) only in solvents with excellent solubility, such as CH 2 Cl 2 , CHCl 3 , CH 2 ClCH 2 Cl, CH 3 CCl 3 , monoglyme, or diglyme, whereas it hardly dissolves in other general polar or non-polar solvents. Therefore, despite the excellent pharmaceutical effects of the compound, mere are many difficulties in formulation for the administration to a living body. Due to the characteristics of the naphthoquinone-based compound with sparing solubility, formulation is markedly restricted. Even though the physical activities of the naphthoquinone-based compound have been revealed, the formulation form is limited to intraperitoneal or intravenous injection for administration of the compound into a body.
- the medicine containing the naphthoquinone-based compound as an active ingredient must be absorbed in vivo to some extent with a certain concentration or more to finally establish its pharmaceutical efficacy.
- the absorption rate of the quinine-based compound, i.e., cryptotanshinone, via oral administration is 2.05%, which is very low.
- the pharmaceutical composition according to the present invention for improving sparing solubility problem and bioavailability of the naphthoquinone-based compound, may be preferably an oral pharmaceutical composition which is prepared into intestine-targeted formation.
- an oral pharmaceutical composition passes through the stomach upon oral administration, is largely absorbed by the small intestine and then diffused into all the tissues of the body, thereby exerting therapeutic effects on the target tissues.
- the oral pharmaceutical composition according to the present invention enhances bioabsorption and bioavailability of a compound of Formula 1 or Formula 2 active ingredient via intestine-targeted formulation of the active ingredient. More specifically, when the active ingredient in the pharmaceutical composition according to the present invention is primarily absorbed in the stomach, and upper parts of the small intestine, the active ingredient absorbed into the body directly undergoes liver metabolism which is then accompanied by substantial degradation of the active ingredient, so it is impossible to exert a desired level of therapeutic effects. On the other hand, it is expected that when the active ingredient is largely absorbed around and downstream of the lower small intestine, the absorbed active ingredient migrates via lymph vessels to the target tissues to thereby exert high therapeutic effects.
- the pharmaceutical composition according to the present invention targets up to the intestine which is a final destination of the digestion process, it is possible to increase the in vivo retention time of the drug and it is also possible to minimize decomposition of the drug which may take place due to the body metabolism upon administration of the drug into the body. As a result, it is possible to improve pharmacokinetic properties of the drug, to significantly lower a critical effective dose of the active ingredient necessary for the treatment of the disease, and to obtain desired therapeutic effects even with administration of a trace amount of the active ingredient. Further, in the oral pharmaceutical composition, it is also possible to minimize the absorption variation of the drug by reducing the between- and within-individual variation of the bioavailability which may result from intragastric pH changes and dietary uptake patterns.
- the intestine-targeted formulation according to the present invention is configured such that the active ingredient is largely absorbed in the small and large intestines, more preferably in the jejunum, and the ileum and intestine corresponding to the lower small intestine, particularly preferably in the ileum or colon.
- the intestine-targeted formulation may be designed by taking advantage of numerous physiological parameters of the digestive tract, through a variety of methods.
- the intestine-targeted formulation may be prepared by (1) a formulation method based on a pH-sensitive polymer, (2) a formulation method based on a biodegradable polymer which is decomposable by an intestine-specific bacterial enzyme, (3) a formulation method based on a biodegradable matrix which is decomposable by an intestine- specific bacterial enzyme, or (4) a formulation method which allows release of a drug after a given lag time, and any combination thereof.
- the intestine-targeted formulation (1) using the pH-sensitive polymer is a drug delivery system which is based on pH changes of the digestive tract.
- the pH of the stomach is in a range of 1 to 3, whereas the pH of the small and large intestines has a value of 7 or more, which is higher as compared to that of the stomach.
- the pH- sensitive polymer may be used in order to ensure that the pharmaceutical composition reaches the lower intestinal parts without being affected by pH fluctuations of the digestive tract.
- pH-sensitive polymer may include, but are not limited to, at least one selected from the group consisting of methacrylic acid-ethyl acrylate copolymer (Eudragit: Registered Trademark of Rohm Pharma GmbH), hydroxypropylmethyl cellulose phthalate (HPMCP) and a mixture thereof.
- the pH-sensitive polymer may be added by a coating process.
- addition of the polymer may be carried out by mixing the polymer in a solvent to form an aqueous coating suspension, spraying the resulting coating suspension to form a film coating, and drying the film coating.
- the intestine-targeted formulation (2) using the biodegradable polymer which is decomposable by the intestine-specific bacterial enzyme is based on the utilization of a degradative ability of a specific enzyme that can be produced by enteric bacteria.
- the specific enzyme may include azoreductase, bacterial hydrolase glycosidase, esterase, polysaccharidase, and the like.
- the biodegradable polymer may be a polymer containing an azoaromatic linkage, for example, a copolymer of styrene and hydroxyethylmethacrylate (HEMA).
- HEMA hydroxyethylmethacrylate
- the active ingredient may be liberated into the intestine by reduction of an azo group of the polymer via the action of the azoreductase which is specifically secreted by enteric bacteria, for example, Bacteroides fragilis and Eubacterium limosum.
- the biodegradable polymer may be a naturally- occurring polysaccharide or a substituted derivative thereof.
- the biodegradable polymer may be at least one selected from the group consisting of dextran ester, pectin, amylose, ethyl cellulose and a pharmaceutically acceptable salt thereof.
- the active ingredient may be liberated into the intestine by hydrolysis of the polymer via the action of each enzyme which is specifically secreted by enteric bacteria, for example, Bifidobacteria and Bacteroides spp. These polymers are natural materials, and have an advantage of low risk of in vivo toxicity.
- the intestine-targeted formulation (3) using the biodegradable matrix which is decomposable by an intestine-specific bacterial enzyme may be a form in which the biodegradable polymers are cross-linked to each other and are added to the active ingredient or the active ingredient-containing formulation.
- the biodegradable polymer may include naturally-occurring polymers such as chondroitin sulfate, guar gum, chitosan, pectin, and the like.
- the degree of drug release may vary depending upon the degree of cross-linking of the matrix-constituting polymer.
- the biodegradable matrix may be a synthetic hydrogel based on N-substituted acrylamide.
- a hydrogel synthesized by cross-linking of N-tert-butylacryl amide with acrylic acid or copolymerization of 2-hydroxyethyl methacrylate and 4-methacryloyloxyazobenzene as the matrix.
- the cross-linking may be, for example an azo linkage as mentioned above, and the formulation may be a form where the density of cross-linking is maintained to provide the optimal conditions for intestinal drug delivery and the linkage is degraded to interact with the intestinal mucous membrane when the drug is delivered to the intestine.
- the intestine-targeted formulation (4) with time-course release of the drug after a lag time is a drug delivery system utilizing a mechanism that is allowed to release the active ingredient after a predetermined time irrespective of pH changes.
- the formulation should be resistant to the gastric pH environment, and should be in a silent phase for 5 to 6 hours corresponding to a time period taken for delivery of the drug from the body to the intestine, prior to release of the active ingredient into the intestine.
- the time-specific delayed-release formulation may be prepared by addition of the hydrogel prepared from copolymerization of polyethylene oxide with polyurethane.
- the delayed-release formulation may have a configuration in which the formulation absorbs water and then swells while it stays within the stomach and the upper digestive tract of the small intestine, upon addition of a hydrogel having the above-mentioned composition after applying the drug to an insoluble polymer, and then migrates to the lower part of the small intestine which is the lower digestive tract and liberates the drug, and the lag time of drug is determined depending upon a length of the hydrogel.
- ethyl cellulose may be used in the delayed- release dosage formulation.
- EC is an insoluble polymer, and may serve as a factor to delay a drug release time, in response to swelling of a swelling medium due to water penetration or changes in the internal pressure of the intestines due to a peristaltic motion.
- the lag time may be controlled by the thickness of EC.
- hydroxypropylmethyl cellulose (PIPMC) may also be used as a retarding agent that allows drug release after a given period of time by thickness control of the polymer, and may have a lag time of 5 to 10 hours.
- the active ingredient may have a crystalline structure with a high degree of crystallinity, or a crystalline structure with a low degree of crystallinity.
- the term "degree of crystallinity" is defined as the weight fraction of the crystalline portion of the total compound and may be determined by a conventional method known in the art. For example, measurement of the degree of crystallinity may be carried out by a density method or precipitation method which calculates the crystallinity degree by previous assumption of a preset value obtained by addition and/or reduction of appropriate values to/from each density of the crystalline portion and the amorphous portion, a method involving measurement of the heat of fusion, an X-ray method in which the crystallinity degree is calculated by separation of the crystalline diffraction fraction and the noncrystalline diffraction fraction from X-ray diffraction intensity distribution upon X-ray diffraction analysis, or an infrared method which calculates the crystallinity degree from a peak of the width between crystalline bands of the infrared absorption spectrum.
- the crystallinity degree of the active ingredient is preferably 50% or less. More preferably, the active ingredient may have an amorphous structure from which the intrinsic crystallinity of the material was completely lost.
- the amorphous naphthoquinone compound exhibits a relatively high solubility, as compared to the crystalline naphthoquinone compound, and can significantly improve a dissolution rate and in vivo absorption rate of the drug.
- the amorphous structure may be formed during preparation of the active ingredient into microparticles or fine particles (micronization of the active ingredient).
- the microparticles may be prepared, for example by spray drying of active ingredients, melting methods involving formation of melts of active ingredients with polymers, co-precipitation involving formation of co-precipitates of active ingredients with polymers after dissolution of active ingredients in solvents, inclusion body formation, solvent volatilization, and the like. Preferred is spray drying.
- the spray drying is a method of making fine particles by dissolving the active ingredient in a certain solvent and the spray-drying the resulting solution. During the spray- drying process, a high percent of the crystallinity of the naphthoquinone compound is lost to thereby result in an amorphous state, and therefore the spray-dried product in the form of a fine powder is obtained.
- the mechanical milling is a method of grinding the active ingredient into fine particles by applying strong physical force to active ingredient particles.
- the mechanical milling may be carried out by using a variety of milling processes such as jet milling, ball milling, vibration milling, hammer milling, and the like. Particularly preferred is jet milling which can be carried out using an air pressure, at a temperature of less than 40 ° C .
- the particle diameter of the active ingredient may be in a range of 5 nm to 500 [M. In this range, the particle agglomeration or aggregation can be maximally inhibited, and the dissolution rate and solubility can be maximized due to a high specific surface area of the particles.
- a surfactant may be additionally added to prevent the particle agglomeration or aggregation which may occur during formation of the fine particles, and/or an antistatic agent may be additionally added to prevent the occurrence of static electricity.
- a moisture-absorbent material may be further added during the milling process.
- the compound of Formula 1 or Formula 2 has a tendency to be crystallized by water, so incorporation of the moisture-absorbent material inhibits recrystallization of the naphthoquinone-based compound over time and enables maintenance of increased solubility of compound particles due to micronization. Further, the moisture-absorbent material serves to suppress coagulation and aggregation of the pharmaceutical composition while not adversely affecting therapeutic effects of the active ingredient.
- the surfactant may include, but are not limited to, anionc surfactants such as docusate sodium and sodium lauryl sulfate; cationic surfactants such as benzalkonium chloride, benzethonium chloride and cetrimide; nonionic surfactants such as glyceryl monooleate, polyoxyethylene sorbitan fatty acid ester, and sorbitan ester; amphiphilic polymers such as polyethylene-polypropylene polymer and polyoxyethylene-polyoxypropylene polymer (Poloxamer), and GelucireTM series (Gattefosse Corporation, USA); propylene glycol monocaprylate, oleoyl macrogol-6-glyceride, linoleoyl macrogol-6-glyceride, caprylocaproyl macrogol-8-glyceride, propylene glycol monolaurate, and polyglyceryl-6-dioleate. These materials may be used alone or in any combination thereof.
- moisture-absorbent material may include, but are not limited to, colloidal silica, light anhydrous silicic acid, heavy anhydrous silicic acid, sodium chloride, calcium silicate, potassium aluminosilicate, calcium aluminosilicate, and the like. These materials may be used alone or in any combination thereof.
- moisture absorbents may also be used as the antistatic agent.
- the surfactant, antistatic agent, and moisture absorbent are added in a certain amount that is capable of achieving the above-mentioned effects, and such an amount may be appropriately adjusted depending upon micronization conditions.
- the additives may be used in a range of 0.05 to 20% by weight, based on the total weight of the active ingredient.
- water-soluble polymers, solubilizers and disintegration-promoting agents may be further added.
- formulation of the composition into a desired dosage form may be made by mixing the additives and the particulate active ingredient in a solvent and spray-drying the mixture.
- the water-soluble polymer is of help to prevent aggregation of the particulate active ingredients, by rendering surroundings of the compound of Formula 1 or Formula 2 molecules or particles hydrophilic to consequently enhance water solubility, and preferably to maintain the amorphous state of the compound of Formula 1 or Formula 2 which is an active ingredient.
- the water-soluble polymer is a pH-independent polymer, and can bring about crystallinity loss and enhanced hydrophilicity of the active ingredient, even under the between- and within-individual variation of the gastrointestinal pH.
- Preferred examples of the water-soluble polymers may include at least one selected from the group consisting of cellulose derivatives such as methyl cellulose, hydroxymethyl cellulose, hydroxyethyl cellulose, ethyl cellulose, hydroxyethylmethyl cellulose, carboxymethyl cellulose, hydroxypropylmethyl cellulose, hydroxypropylmethyl cellulose phthalate, sodium carboxymethyl cellulose, and carboxymethylethyl cellulose; polyvinyl alcohols; polyvinyl acetate, polyvinyl acetate phthalate, polyvinylpyrrolidone (PVP), and polymers containing the same; polyalkene oxide or polyalkene glycol, and polymers containing the same. Preferred is hydroxypropylmethyl cellulose.
- an excessive content of the water-soluble polymer which is higher than a given level provides no further increased solubility, but disadvantageously brings about various problems such as overall increases in the hardness of the formulation, and non-penetration of an eluent into the formulation, by formation of films around the formulation due to excessive swelling of water-soluble polymers upon exposure to the eluent.
- the solubilizer is preferably added to maximize the solubility of the formulation by modifying physical properties of the compound of Formula 1 or Formula 2.
- the solubilizer serves to enhance solubilization and wettability of the sparingly-soluble compound of Formula 1 or Formula 2, and can significantly reduce the bioavailability variation originating from diets and the time difference of drug administration after dietary uptake.
- the solubilizer may be selected from conventionally widely used surfactants or amphiphiles, and specific examples of the solubilizer may refer to the surfactants as defined above.
- the disintegration-promoting agent serves to improve the drug release rate, and enables rapid release of the drug at the target site to thereby increase bioavailability of the drug.
- Preferred examples of the disintegration-promoting agent may include, but are not limited to, at least one selected from the group consisting of croscarmellose sodium, crospovidone, calcium carboxymethylcellulose, starch glycolate sodium and lower substituted hydroxypropyl cellulose. Preferred is croscarmellose sodium.
- the solvent for spray drying is a material exhibiting a high solubility without modification of physical properties thereof and easy volatility during the spray drying process.
- Preferred examples of such a solvent may include, but are not limited to, dichloromethane, chloroform, methanol, and ethanol. These materials may be used alone or in any combination thereof.
- a content of solids in the spray solution is in a range of 5 to 50% by weight, based on the total weight of the spray solution.
- the above-mentioned intestine-targeted formulation process may be preferably carried out for formulation particles prepared as above.
- the oral pharmaceutical composition according to the present invention may be formulated by a process comprising the following steps: (a) adding the compound of Formula 1 or Formula 2 alone or in combination with a surfactant and a moisture-absorbent material, and grinding the compound of Formula 1 or 2 with a jet mill to prepare active ingredient microparticles;
- the surfactant, moisture-absorbent material, water-soluble polymer, solubilizer and disintegration-promoting agent are as defined above.
- the plasticizer is an additive added to prevent hardening of the coating, and may include, for example polymers such as polyethylene glycol.
- formulation of the active ingredient may be carried out by sequential or concurrent spraying of vehicles of Step (b) and intestine-targeted coating materials of Step (c) onto jet-milled active ingredient particles of Step (a) as a seed.
- the oral pharmaceutical composition suitable for use in the present invention contains the active ingredient in an amount effective to achieve its intended purpose, that is therapeutic purpose. More specifically, a therapeutically effective amount refers to an amount of the compound effective to prevent, alleviate or ameliorate symptoms of disease. Determination of the therapeutically effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- the oral pharmaceutical composition according to the present invention is particularly useful for the treatment and/or prevention of prostate and/or testicle (seminal glands)-related diseases, as defined above.
- the prostate and/or testicle (seminal glands)-related diseases preferably include, but are not limited to, prostatitis or benign prostatic hyperplasia.
- treatment of the disease syndromes refers to stopping or delaying of the disease progress, when the drug is used in the subject exhibiting symptoms of disease onset.
- prevention refers to stopping or delaying of symptoms of disease onset, when the drug is used in the subject exhibiting no symptoms of disease onset but having high risk of disease onset.
- FIG. 1 is a graph showing a residual amount of a naphthoquinone-based compound in the jejunum, ileum and large intestine, respectively, when single-pass intestinal perfusion was carried out according to Experimental Example 4;
- FIG. 2 is a graph showing outlet steady-state concentrations of a naphthoquinone- based compound under perfusion in Experimental Example 4; and FIG. 3 and FIG. 4 are respectively a photograph showing prostate tissue staining of SHRs in a control group and an administration group of the compound of Example 3 in Experimental Example 7; and
- FIG. 5 and FIG. 6 are respectively photographs showing prostate tissue staining of WKYs in a control group and an administration group of the compound of Example 3 in Experimental Example 7.
- Octanol and phosphate buffer (pH 7.4) were saturated with a counter-solvent for 24 hours or more.
- a given amount of a naphthoquinone-based compound (compound 1 of Table 1 below) was dissolved in the thus-saturated octanol, mixed with triple-distilled water and stirred using a magnetic stirrer at 200 rpm for 13 hours or more.
- Samples were taken, filtered through a 0.45 [M RC Membrane filter and diluted with methanol. The diluted sample materials were analyzed by HPLC. A partition coefficient versus an amount of the compound 1 was determined. The results thus obtained are given in Table 2.
- the partition coefficient was a value of 2.299, thus representing that the compound 1 is relatively fat-soluble. This result means that the compound 1 has octanol-solubility 100-fold higher than water-solubility, and sufficiently passes through a hydrophobic layer inside the cell membrane, followed by intracellular absorption.
- Example 1 Micronization of active ingredient using Jet mill
- Micronizing of an active ingredient was carried out using a Jet mill (SJ-IOO, Nisshin, Japan). Operation was run at a supply pressure of 0.65 Mpa, and a feed rate of 50 to 100 g/hr. 0.2 g of sodium lauryl sulfate (SLS) and 10 g of a naphthoquinone-based compound (Compound 1 of Table 1) were mixed and ground. Micronized particles were recovered and a particle size was determined by zeta potential measurement. An average particle diameter was 1500 nm.
- Example 2 Preparation of spray-dried product
- the synthesized naphthoquinone-based compound (Compound 1 of Table 1) or the naphthoquinone-based compound of Example 1 (including micronized and non-micronized particles) was added to methylene chloride, and a salt such as sodium chloride, a saccharide such as white sugar or lactose, or a vehicle such as microcrystalline cellulose, monobasic calcium phosphate, starch or mannitol, a lubricant such as magnesium stearate, talc or glyceryl behenate, and a solubilizer such as Poloxamer were added to a given amount of ethanol, followed by homogeneous dispersion to prepare a spray-drying solution which will be used for subsequent spray-drying.
- a salt such as sodium chloride
- a saccharide such as white sugar or lactose
- a vehicle such as microcrystalline cellulose, monobasic calcium phosphate, starch or
- Example 2 To the spray-dried product of Example 2 were added approximately an equal amount of a water-soluble polymer (hydroxypropylmethyl cellulose) relative to an active ingredient, and vehicles such as Croscarmellose sodium and light anhydrous silicic acid, and the mixture was formulated without causing interference of disintegration. A drug dissolution test was carried out in a buffer (pH 6.8). All the compositions exhibited drug dissolution of 90% or higher after 6 hours.
- a water-soluble polymer hydroxypropylmethyl cellulose
- vehicles such as Croscarmellose sodium and light anhydrous silicic acid
- the steady-state intestinal effective permeability (P eff ) can be expressed according to the following equation.
- Pe ff [ -Qin - ln (Cou t /Ci n )] / A
- the radius (r) and length (L) of the jejunum, ileum and large intestine used in experiments are as follows: (r. jejunum, 0.21 cm; ileum, 0.22 cm; large intestine, 0.23 cm, and L: 10 cm)
- the steady-state was confirmed by the ratio of the outlet to inlet concentrations (C out /Cjn) versus time.
- the outlet steady-state concentration of the compound under perfusion was calculated.
- the results thus obtained are given in Table 4 and FIG. 2, respectively.
- the effective permeability was measured at 4 points of each intestinal tissue. As shown in Table 4 and FIG. 2, it can be seen that the highest permeability was observed in the large intestine.
- the spray-dried formulation prepared in Experimental Example 2 was added to an ethanol solution containing about 20% by weight of Eudragit S-100 as a pH-sensitive polymer and about 2% by weight of PEG #6,000 as a plasticizer, and the mixture was then spray-dried to prepare an intestine-targeted formulation.
- the intestine-targeted formulation prepared in Example 3 was exposed to pH 1.2 and pH 6.8, respectively. After 6 hours, the intestine-targeted formulation was removed and washed, and a content of an active ingredient was analyzed by HPLC. An effective amount of the active ingredient was assessed as a measure of the acid resistance. The acid resistance exhibited a very excellent result of 90 to 100%, thus suggesting that the intestine-targeted formulation is chemically stable in the stomach or small intestine.
- SHRs spontaneous hypertensive rats
- WKYs Wistar-Kyoto rats
- Kurea normal blood pressure rats
- Animals were raised in a breeding room maintained at a temperature of 23°C, 55% humidity, illumination of 300 to 500 lux, a 12-h light/dark (L/ D) cycle, and ventilation of 10 to 18 times/hr. Animals were fed ad libitum pellets of Purina Rodent Laboratory Chow 5001 (purchased from Purina Mills Inc., St. Louis, MO, USA) as a solid feed for experimental animals and tap water as drinking water.
- Purina Rodent Laboratory Chow 5001 purchasedd from Purina Mills Inc., St. Louis, MO, USA
- mice were allowed to acclimate to new environment of the breeding room for two weeks and undergo preprocess for measuring blood pressure for two weeks. Then, 13-week-old SHRs and WKYs were divided into two groups, respectively, in which a first group was administered with a formulation for intestinal delivery system prepared in Example 3 according to the present invention for 10 weeks through oral route. A second group was a control group administered with saline excluding all other medicines.
- FIGs. 3 through 6 The results are shown in FIGs. 3 through 6 (FIG. 3: SHR in a control group; FIG. 4: SHR in an administration group of the compound for intestinal delivery system; FIG. 5: WKY in a control group; and FIG. 6: WKY in an administration group of the compound of Example 3).
- the size of the prostate fibroblastic tissue of both SHRs and WKYs significantly reduced in the administration group of the compound for intestinal delivery system compared with the control group. Especially, it can be seen that the compound had greater effect on the treatment of benign prostatic hyperplasia of SHRs. Therefore, the compound of the present invention is believed to be useful for substantial treatment of prostate and/or testicle (seminal glands)-related diseases, such as benign prostatic hyperplasia.
- a compound represented by Formula 1 or 2 according to the present invention is pharmaceutically effective for the treatment of prostate and/or testicle (seminal glands)-related diseases, especially benign prostatic hyperplasia and prostatitis.
- the pharmaceutical composition for oral administration of intestinal delivery system increases absorption amount of the active ingredient and extends the duration time of the its efficacy in vivo, thereby having effects of improving pharmacokinetic properties of the drug. Consequently, by increasing availability of the naphthoquinone-compound in vivo, an excellent effect on the treatment of prostate and/or testicle (seminal glands)-related diseases, especially benign prostatic hyperplasia and prostatitis can be established.
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Urology & Nephrology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description












































Claims


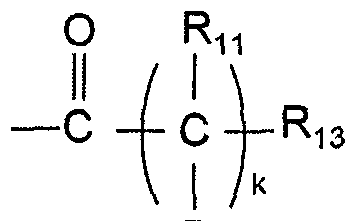








Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009538340A JP2010510983A (en) | 2006-11-27 | 2007-11-26 | Compounds for the treatment and prevention of prostate related diseases and pharmaceutical compositions of colon delivery systems containing them |
| US12/515,014 US20100239685A1 (en) | 2006-11-27 | 2007-11-26 | Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same |
| EP07834307A EP2101757A4 (en) | 2006-11-27 | 2007-11-26 | Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2006-0117685 | 2006-11-27 | ||
| KR20060117685 | 2006-11-27 | ||
| KR1020070111183A KR20080047975A (en) | 2006-11-27 | 2007-11-01 | Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same |
| KR10-2007-0111183 | 2007-11-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008066298A1 true WO2008066298A1 (en) | 2008-06-05 |
Family
ID=39468046
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2007/006012 WO2008066298A1 (en) | 2006-11-27 | 2007-11-26 | Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008066298A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2217225A2 (en) * | 2007-10-11 | 2010-08-18 | Mazence Inc. | Pharmaceutical composition containing micronized particles of naphthoquinone-based compound |
| WO2014138357A1 (en) * | 2013-03-06 | 2014-09-12 | The University Of Akron | Novel tashinone drugs for alzheimer disease |
| US9050267B2 (en) | 2011-02-04 | 2015-06-09 | Novartis Ag | Dry powder formulations of particles that contain two or more active ingredients for treating obstructive or inflammatory airways diseases |
| US9492387B2 (en) | 2010-10-29 | 2016-11-15 | Western University Of Health Sciences | Ternary mixture formulations |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20020092160A (en) * | 2001-01-15 | 2002-12-11 | 서영거 | Novel ortho-naphthopyranoquinone derivatives and using for antimicrobial agent and antifungal agent thereof |
| WO2005063232A1 (en) * | 2003-12-30 | 2005-07-14 | Md Bioalpha Co., Ltd. | Obesity and metabolic syndrome treatment with tanshinone derivatives which increase metabolic activity |
| WO2006088315A1 (en) * | 2005-02-16 | 2006-08-24 | Md Bioalpha Co., Ltd. | Pharmaceutical composition for the treatment or prevention of diseases involving obesity, diabetes, metabolic syndrome, neuro-degenerative diseases and mitochondria dysfunction diseases |
-
2007
- 2007-11-26 WO PCT/KR2007/006012 patent/WO2008066298A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20020092160A (en) * | 2001-01-15 | 2002-12-11 | 서영거 | Novel ortho-naphthopyranoquinone derivatives and using for antimicrobial agent and antifungal agent thereof |
| WO2005063232A1 (en) * | 2003-12-30 | 2005-07-14 | Md Bioalpha Co., Ltd. | Obesity and metabolic syndrome treatment with tanshinone derivatives which increase metabolic activity |
| WO2006088315A1 (en) * | 2005-02-16 | 2006-08-24 | Md Bioalpha Co., Ltd. | Pharmaceutical composition for the treatment or prevention of diseases involving obesity, diabetes, metabolic syndrome, neuro-degenerative diseases and mitochondria dysfunction diseases |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2101757A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2217225A2 (en) * | 2007-10-11 | 2010-08-18 | Mazence Inc. | Pharmaceutical composition containing micronized particles of naphthoquinone-based compound |
| EP2217225A4 (en) * | 2007-10-11 | 2012-12-19 | Mazence Inc | Pharmaceutical composition containing micronized particles of naphthoquinone-based compound |
| US9492387B2 (en) | 2010-10-29 | 2016-11-15 | Western University Of Health Sciences | Ternary mixture formulations |
| US9808426B2 (en) | 2010-10-29 | 2017-11-07 | Western University Of Health Sciences | Ternary mixture formulations |
| US9050267B2 (en) | 2011-02-04 | 2015-06-09 | Novartis Ag | Dry powder formulations of particles that contain two or more active ingredients for treating obstructive or inflammatory airways diseases |
| WO2014138357A1 (en) * | 2013-03-06 | 2014-09-12 | The University Of Akron | Novel tashinone drugs for alzheimer disease |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20100239685A1 (en) | Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same | |
| EP3125872B1 (en) | Amorphous solid dispersion comprising taxane, tablet comprising the same, and method for preparing the same | |
| WO2008136642A1 (en) | Naphthoquinone-based pharmaceutical composition for treatment or prevention of diseases involving obesity, diabetes, metabolic syndrome, neuro-degenerative diseases and mitochondria dysfunction diseases | |
| BRPI0414000B1 (en) | tacrolimus oral sustained release solid pharmaceutical composition in the form of a solid dispersion, dosage form and use of the pharmaceutical composition | |
| WO2008066301A1 (en) | Anticancer composition containing naphthoquinone-based compound for intestine delivery system | |
| TW200302734A (en) | Pharmaceutical compositions for hepatitis c viral protease inhibitors | |
| US20140154319A1 (en) | Pharmaceutical composition for the treatment and prevention of cardiac disease | |
| JP2011500557A (en) | Pharmaceutical composition containing atomized particles of naphthoquinone compound | |
| WO2008066298A1 (en) | Compound for treatment or prevention of prostate-related diseases and pharmaceutical composition of colon delivery system containing the same | |
| US20130302422A1 (en) | Pharmaceutical composition for treatment and prevention of kidney diseases | |
| US20090124680A1 (en) | Use of prodrug composition containing naphthoquinone-based compound for manufacture of medicament for treatment or prevention of diseases involving metabolic syndrome | |
| WO2008066299A1 (en) | Pharmaceutical composition for the treatment and prevention of diseases involving impotence | |
| WO2008066295A1 (en) | Pharmaceutical composition containing naphthoquinone-based compound for intestine delivery system | |
| WO2008066296A1 (en) | Pharmaceutical composition containing phenanthrenequinone-based compound for intestine delivery system | |
| WO2008066300A1 (en) | Naphthoquinone-based pharmaceutical composition for treatment or prevention of diseases involving obesity, diabetes, metabolic syndrome, neuro-degenerative diseases and mitochondria dysfunction diseases | |
| WO2008066297A1 (en) | Pharmaceutical composition for treatment and prevention of restenosis | |
| WO2014140695A1 (en) | Solid oral formulation of a pyrrolidine substituted flavone compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07834307 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12515014 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097010386 Country of ref document: KR Ref document number: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2009538340 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007834307 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |