Cyclisch substituierte 3,5-Dicyano-2-thiopyridine und ihre Verwendung
Die vorliegende Anmeldung betrifft neue 4-Cycloalkyl- und 4-Heterocycloalky 1-3,5 -dicyano-2- thiopyridin-Derivate, Verfahren zu ihrer Herstellung, ihre Verwendung zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Be- handlung und/oder Prophylaxe von Krankheiten, vorzugsweise zur Behandlung und/oder Prävention von Hypertonie und anderen kardiovaskulären Erkrankungen.
Adenosin, ein Purin-Nukleosid, ist in allen Zellen vorhanden und wird unter einer Vielzahl von physiologischen und pathophysiologischen Stimuli freigesetzt. Adenosin entsteht intrazellulär beim Abbau von Adenosin-5'-monophosphat (AMP) und S-Adenosylhomocystein als Zwischen- produkt, kann jedoch aus der Zelle freigesetzt werden und übt dann durch Bindung an spezifische Rezeptoren Funktionen als hormonähnliche Substanz oder Neurotransmitter aus.
Unter normoxischen Bedingungen ist die Konzentration des freien Adenosin im Extrazellulärraum sehr niedrig. Die extrazelluläre Konzentration von Adenosin erhöht sich in den betroffenen Organen jedoch dramatisch unter ischämischen bzw. hypoxischen Bedingungen. So ist beispiels- weise bekannt, dass Adenosin die Thrombozyten- Aggregation hemmt und die Durchblutung der Herzkranzgefäße steigert. Weiterhin wirkt es auf den Blutdruck, die Herzfrequenz, auf die Ausschüttung von Neurotransmittern und auf die Lymphozyten-Differenzierung. In Adipozyten ist Adenosin in der Lage, die Lipolyse zu hemmen und somit die Konzentration an freien Fettsäuren und Triglyzeriden im Blut zu senken.
Diese Wirkungen von Adenosin zielen darauf ab, das Sauerstoffangebot der betroffenen Organe zu erhöhen bzw. den Stoffwechsel dieser Organe zu drosseln, um damit unter ischämischen oder hypoxischen Bedingungen eine Anpassung des Organstoffwechsels an die Organdurchblutung zu erreichen.
Die Wirkung von Adenosin wird über spezifische Rezeptoren vermittelt. Bekannt sind bisher die Subtypen Al, A2a, A2b und A3. Als "Adenosinrezeptor-selektive Liganden" werden erfindungsgemäß solche Substanzen bezeichnet, die selektiv an einen oder mehrere Subtypen der Adenosin- rezeptoren binden und dabei entweder die Wirkung des Adenosin nachahmen (Adenosin- Agonisten) oder dessen Wirkung blockieren (Adenosin-Antagonisten) können.
Die Wirkungen dieser Adenosin-Rezeptoren werden intrazellulär durch den Botenstoff cAMP vermittelt. Im Falle der Bindung von Adenosin an die A2a- oder A2b-Rezeptoren kommt es über eine Aktivierung der membranständigen Adenylatzyklase zu einem Anstieg des intrazellulären
cAMP, während die Bindung des Adenosin an die Al- oder A3 -Rezeptoren über eine Hemmung der Adenylatzyklase eine Abnahme des intrazellulären cAMP-Gehalts bewirkt.
Im Herz-Kreislaufsystem sind die Hauptwirkungen der Aktivierung von Adenosin-Rezeptoren: Bradykardie, negative Inotropie und Protektion des Herzens vor Ischämie ("preconditioning") über Al -Rezeptoren, Dilation der Gefäße über A2a- und A2b-Rezeptoren sowie Inhibition der Fibroblasten und Glattmuskelzellproliferation über A2b-Rezeptoren.
Im Falle von Al-Agonisten (Kopplung bevorzugt über G,-Proteine) wird dabei eine Abnahme des intrazellulären cAMP -Gehaltes beobachtet (bevorzugt nach direkter Vorstimulation der Adenylatzyklase durch Forskolin). Entsprechend führen A2a- und A2b-Agonisten (Kopplung bevorzugt über Gs-Proteine) zu einer Zunahme und A2a- und A2b-Antagonisten zu einer Abnahme im cAMP-Gehalt der Zellen. Im Falle der A2-Rezeptoren ist eine direkte Vorstimulation der Adenylatzyklase durch Forskolin nicht hilfreich.
Die Aktivierung von A2b-Rezeptoren durch Adenosin oder spezifische A2b-Agonisten führt über die Erweiterung von Gefäßen zu einer Blutdrucksenkung. Die Blutdrucksenkung ist von einem reflektorischen Herzfrequenzanstieg begleitet. Der Herzfrequenzanstieg kann durch die Aktivierung von Al -Rezeptoren durch spezifische Al-Agonisten reduziert werden.
Die kombinierte Wirkung von selektiven Al/A2b-Agonisten auf das Gefäßsystem und die Herzfrequenz resultiert somit in einer systemischen Blutdrucksenkung ohne relevanten Herzfrequenzanstieg. Mit einem solchen pharmakologischen Profil könnten duale Al/A2b-Agonisten zur Behandlung z.B. der Hypertonie beim Menschen eingesetzt werden.
In Adipozyten bewirkt die Aktivierung von Al- und A2b-Rezeptoren eine Inhibition der Lipolyse. Die kombinierte Wirkung von Al/A2b-Agonisten auf den Lipidstoffwechsel führt somit zu einer Senkung von freien Fettsäuren und Triglyzeriden. Eine Senkung der Lipide wiederum führt bei Patienten mit Metabolischem Syndrom und bei Diabetikern zur Verringerung der Insulinresistenz und zur Verbesserung der Symptomatik.
Die zuvor genannte Rezeptor-Selektivität lässt sich bestimmen durch die Wirkung der Substanzen an Zelllinien, die nach stabiler Transfektion mit der entsprechenden cDNA die jeweiligen Rezeptorsubtypen exprimieren (siehe hierzu die Druckschrift M. E. Olah, H. Ren, J. Ostrowski, K. A. Jacobson, G. L. Stiles, "Cloning, expression, and characterization of the unique bovine Al adenosine receptor. Studies on the ligand binding site by site-directed mutagenesis", J. Biol. Chem. 267 (1992), Seiten 10764-10770, deren Offenbarung hiermit im vollen Umfang durch Bezugnahme eingeschlossen ist).
Die Wirkung der Substanzen an solchen Zelllinien lässt sich erfassen durch biochemische Messung des intrazellulären Botenstoffes cAMP (siehe hierzu die Druckschrift K. N. Klotz, J. Hess- ling, J. Hegler, C. Owman, B. KuIl, B. B. Fredholm, M. J. Lohse, "Comparative pharmacology of human adenosine receptor Subtypes - characterization of stably transfected receptors in CHO cells", Naunyn Schmiedebergs Arch. Pharmacol. 357 (1998), Seiten 1-9, deren Offenbarung hiermit im vollen Umfang durch Bezugnahme eingeschlossen ist).
Bei den aus dem Stand der Technik bekannten, als "Adenosinrezeptor-spezifisch" geltenden Liganden handelt es sich überwiegend um Derivate auf Basis des natürlichen Adenosins [S. -A. Poulsen und R. J. Quinn, "Adenosine receptors: New opportunities for future drugs", Bioorganic and Medicinal Chemistry 6 (1998), Seiten 619-641]. Diese aus dem Stand der Technik bekannten Adenosin-Liganden haben jedoch meistens den Nachteil, dass sie nicht wirklich rezeptorspezifϊsch wirken, schwächer wirksam sind als das natürliche Adenosin oder nach oraler Applikation nur sehr schwach wirksam sind. Deshalb werden sie überwiegend nur für experimentelle Zwecke verwendet. In klinischer Entwicklung befindliche Verbindungen dieser Art eignen sich bislang nur zur intravenösen Applikation.
hi WO 01/25210 und WO 02/070485 werden substituierte 2-Thio-3,5-dicyano-4-aryl-6-amino- pyridine als Adenosinrezeptor-Liganden für die Behandlung von Erkrankungen beschrieben. In WO 03/053441 werden spezifisch substituierte 2-Thio-3,5-dicyano-4-phenyl-6-aminopyridine als selektive Liganden des Adenosin Al -Rezeptors offenbart, und in WO 2006/027142 werden substi- tuierte Phenylaminothiazol-Derivate als duale Adenosin Al/A2b-Agonisten für die Behandlung der Hypertonie und anderer kardiovaskulärer Erkrankungen beansprucht. Allerdings zeigte es sich, dass diese Verbindungen zum Teil hinsichtlich ihrer physikochemischen und/oder pharmakokinetischen Eigenschaften, wie beispielsweise ihrer Löslichkeit in Wasser und anderen physiologischen Medien oder ihres Absorptionsverhaltens im Körper, Nachteile aufweisen.
In WO 01/62233 werden verschiedene Pyridin- und Pyrimidin-Derivate sowie ihre Verwendung als Adenosinrezeptor-Modulatoren offenbart. Die Verwendung von 4-Cycloalkyl- und 4-Hetero- cycloalkyl-substituierten 3,5-Dicyanopyridinen als Calcium-abhängige Kaliumkanalöffner zur Behandlung urologischer Erkrankungen wird in EP 1 302 463-A1 beschrieben. In WO 2004/054505 wird die Verwendung von 2-Amino-3-cyanopyridin-Derivaten als MK-2-Inhibitoren zur Behand- lung unterschiedlicher Erkrankungen beansprucht. Verschiedene heterocyclisch substituierte Pyri- din-Derivate und ihre Verwendung zur Behandlung von Krankheiten werden in WO 99/32117, WO 2005/046603, WO 2005/007647 und WO 2006/034446 beschrieben. In WO 02/50071 werden Aminothiazol-Derivate als Tyrosin-Kinase-Inhibitoren für die Behandlung von Krebs sowie immunologischer und allergischer Erkrankungen offenbart.
Aufgabe der vorliegenden Erfindung ist die Bereitstellung neuer Verbindungen, die als selektive Agonisten des Adenosin Al -Rezeptors oder als selektive duale Agonisten des Adenosin Al- und A2b-Rezeptors wirken, als solche zur Behandlung und/oder Prävention insbesondere von Hypertonie und anderen kardiovaskulären Erkrankungen, des Metabolischen Syndroms, Diabetes und Dyslipidämien sowie zur Organprotektion bei Transplantationen und operativen Eingriffen geeignet sind und darüber hinaus ein verbessertes physikochemisches und/oder pharmakokinetisches Profil gegenüber den aus dem Stand der Technik bekannten Substanzen aufweisen.
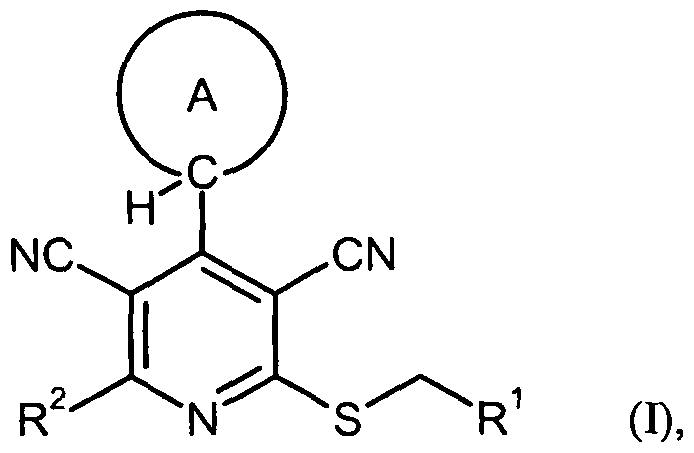
Gegenstand der vorliegenden Erfindung sind Verbindungen der Formel (I)
in welcher
der Ring A für einen (C4-C7)-Cycloalkyl- oder (C4-C7)-Cycloalkenyl-Ring oder für einen 4- bis 7- gliedrigen, C-verknüpften Heterocyclus, der ein oder zwei Ringglieder aus der Reihe N-R3 und/oder O enthält, steht,
worin (C4-C7)-Cycloalkyl und (C4-C7)-Cycloalkenyl ein- oder zweifach, gleich oder ver- schieden, mit einem Rest ausgewählt aus der Reihe (CrC6)-Alkyl, Hydroxy, (C1-C6)-
Alkoxy, Amino, Mono-(Ci-C6)-alkylamino und Di-(Ci -C6)-alkylamino substituiert sein können, wobei die genannten (CrC6)-Alkyl- und (CrC6)-Alkoxy-Reste ihrerseits ein- oder zweifach, gleich oder verschieden, mit Hydroxy, (Ci-C4)-Alkoxy und/oder (C3-C6)-Cyclo- alkyl substituiert sein können,
und worin
R3 Wasserstoff, (Ci-C6)-Alkyl, welches ein- oder zweifach, gleich oder verschieden, mit Hydroxy, (Ci-C4)-Alkoxy, (Ci-C4)-Acyloxy und/oder (C3-C6)-Cycloalkyl substituiert sein kann, oder (CrC6)-Acyl, welches mit Hydroxy oder (CrC4)-Alkoxy substituiert sein kann, bedeutet,
R1 für (C6-Cio)-Aryl oder 5- bis 10-gliedriges Heteroaryl mit bis zu drei Ring-Heteroatomen aus der Reihe N, O und/oder S steht, welche jeweils
(0 ein- oder zweifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe
Halogen, Nitro, Cyano, (CrC6)-Alkyl, Trifluormethyl, Hydroxy, (C1-C6)-Alkoxy)
Amino, Mono-(CrC6)-alkylamino, Di-(C i-C6)-alkylamino, Carboxyl, (C,-C6)-Alkoxy- carbonyl, Carbamoyl, Mono-(Ci-C6)-alkylaminocarbonyl und Di-(CrC6)-alkylamino- carbonyl
und/oder
(H) mit Pyrrolidino, Piperidino, Morpholino, Piperazino, N-(Ci -C4)-Alkylpiperazino, Tetrazolyl oder einer Gruppe der Formel -L-R4, worin
L eine Bindung, NH oder O bedeutet
und
R4 Phenyl oder 5- oder 6-gliedriges Heteroaryl mit bis zu drei Ring-Heteroatomen aus der Reihe N, O und/oder S bedeutet, welche jeweils ein- bis dreifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe Halogen, Nitro, Cyano, (CrC6)-Alkyl, Trifluormethyl, Hydroxy, (CrC6)-Alkoxy, Difluormeth- oxy, Trifluormethoxy, Amino, Mono-(CrC6)-alkylamino, Di-(Ci-C6)-alkyl- amino, (Ci-C6)-Alkoxycarbonyl und Carboxyl substituiert sein können,
substituiert sein können,
und
R2 für Wasserstoff oder für (Ci-C6)-Alkyl oder (CrC6)-Alkoxy, welche jeweils mit Hydroxy, (CrC4)-Alkoxy, Carboxyl, (Ci-C4)-Alkoxycarbonyl oder bis zu dreifach mit Fluor substituiert sein können, steht
oder
R2 für eine Gruppe der Formel -NR5R6 steht, worin
R5 und R6 gleich oder verschieden sind und unabhängig voneinander Wasserstoff oder (C1- C6)-Alkyl, das ein- oder zweifach, gleich oder verschieden, mit Hydroxy, (Ci-C4)-
Alkoxy, Amino, Mono-(Ci-C4)-alkylamino, Di-(CrC4)-alkylamino, Carboxyl, (C1-
C4)-Alkoxycarbonyl und/oder einem 4- bis 7-gliedrigen Heterocyclus substituiert sein kann, bedeuten,
wobei der genannte Heterocyclus ein oder zwei Ring-Heteroatome aus der Reihe N, O und/oder S enthält und seinerseits ein- oder zweifach, gleich oder verschieden, mit (CrC4)-Alkyl, Hydroxy, Oxo und/oder (Ci-C4)-Alkoxy substituiert sein kann,
oder
R5 und R6 zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7- gliedrigen Heterocyclus bilden, der ein weiteres Ring-Heteroatom aus der Reihe N,
O oder S enthalten und ein- oder zweifach, gleich oder verschieden, mit (Ci-C4)-
Alkyl, Hydroxy, Oxo, (Ci-C4)-Alkoxy, Azetidino, Pyrrolidino, Piperidino und/oder Morpholino substituiert sein kann,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze,
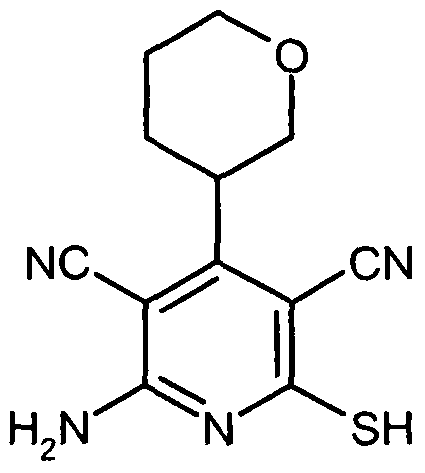
mit Ausnahme der Verbindung 2-Amino-6-(benzylthio)-4-(tetrahydro-2H-pyran-2-yl)-pyridin-3,5- dicarbonitril.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung umfasst deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsaure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbin- düngen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbin- düngen. Der Begriff "Prodrugs" umfaßt Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungs gemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
(Cr-CftVAlkyl und (C
1-Q)-AIkVl stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger oder verzweigter Alkylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, iso-Butyl, sec.-Butyl, tert.-Butyl, 1-Ethyl- propyl, n-Pentyl und n-Hexyl.
stehen im Rahmen der Erfindung für einen monocycli sehen, gesättigten Carbocyclus mit 4 bis 7, 3 bis 6 bzw. 3 bis 5 Ring-Kohlen-
stoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
(Ca-CjVCvcloalkenyl steht im Rahmen der Erfindung für einen monocyclischen Carbocyclus mit 4 bis 7 Ring-Kohlenstoffatomen und einer Doppelbindung. Beispielhaft und vorzugsweise seien ge- nannt: Cyclobut-2-en-l-yl, Cyclopent-2-en-l-yl, Cyclopent-3-en-l-yl, Cyclohex-2-en-l-yl, Cyclo- hex-3-en-l-yl und Cyclohept-3-en-l-yl.
(C1-Cn)-AIkOXV und (C1-Q)-AIkOXV stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein gerad- kettiger oder verzweigter Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugs- weise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy, tert.-Butoxy, n-Pentoxy und n-Hexoxy.
(Cι-Cft)-Alkoxycarbonyl und (G-QVAlkoxycarbonyl stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen, der über eine Carbonylgruppe verknüpft ist. Bevorzugt ist ein geradkettiger oder verzweigter Alkoxy- carbonylrest mit 1 bis 4 Kohlenstoffatomen in der Alkoxy-Gruppe. Beispielhaft und vorzugsweise seien genannt: Methoxycarbonyl, Ethoxycarbonyl, n-Propoxycarbonyl, Isopropoxycarbonyl und tert . -Butoxycarbony 1.
(C
1-C^)-AcVl und (C
1-C^)-AcVl [(C
rC
6)-Alkanoyl und (Ci-C
4)-Alkanoyl] stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoff- atomen, der in der 1 -Position ein doppelt gebundenes Sauerstoffatom trägt und über die 1 -Position verknüpft ist. Bevorzugt ist ein Acylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Formyl, Acetyl, Propionyl, n-Butyryl, wo-Butyryl, n-Pentanoyl, Pivaloyl und n-Hexanoyl.
steht im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkyl- Rest mit 1 bis 4 Kohlenstoffatomen, der in der 1 -Position ein doppelt gebundenes Sauerstoffatom trägt und in der 1 -Position über ein weiteres Sauerstoffatom verknüpft ist. Beispielhaft und vorzugsweise seien genannt: Acetoxy, Propionoxy, rc-Butyroxy und /so-Butyroxy.
Mono-(C1-Cft)-alkylamino und Mono-(Ci-C4)-alkylamino stehen im Rahmen der Erfindung für eine
Amino-Gruppe mit einem geradkettigen oder verzweigten Alkylsubstituenten, der 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweist. Bevorzugt ist ein geradkettiger oder verzweigter Monoalkyl- amino-Rest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl-
amino, Ethylamino, n-Propylamino, Isopropylamino, n-Butylamino, tert.-Butylamino, n-Pentyl- amino und n-Hexylamino.
stehen im Rahmen der Erfindung für eine
Amino-Gruppe mit zwei gleichen oder verschiedenen geradkettigen oder verzweigten Alkyl- substituenten, die jeweils 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweisen. Bevorzugt sind gerad- kettige oder verzweigte Dialkylamino-Reste mit jeweils 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: NN-Dimethylamino, NN-Diethylamino, N-Ethyl-N-methyl- amino, N-Methyl-N-n-propylamino, N-Isopropyl-N-n-propylamino, N,N-Diisopropylamino, N-n-
Butyl-N-methylamino, N-tert.-Butyl-N-methylamino, N-Ethyl-N-n-pentylamino und N-n-Hexyl-N- methylamino.
Mono- bzw. Di-fCi^-Cfil-alkylaminocarbonyl steht im Rahmen der Erfindung für eine Amino- Gruppe, die über eine Carbonylgruppe verknüpft ist und die einen geradkettigen oder verzweigten bzw. zwei gleiche oder verschiedene geradkettige oder verzweigte Alkylsubstituenten mit jeweils 1 bis 6 Kohlenstoffatomen aufweist. Bevorzugt ist ein Mono- bzw. Dialkylaminocarbonyl-Rest mit 1 bis 4 Kohlenstoffatomen in der Alkylgruppe. Beispielhaft und vorzugsweise seien genannt: Methylaminocarbonyl, Ethylaminocarbonyl, n-Propylaminocarbonyl, Isopropylaminocarbonyl, n- Butylaminocarbonyl, tert.-Butylaminocarbonyl, NN-Dimethylaminocarbonyl, N,N-Diethylamino- carbonyl, N-Ethyl-N-methylaminocarbonyl, N-Methyl-N-n-propylaminocarbonyl, N-«-Butyl-N- methylaminocarbonyl und N-tert.-Butyl-N-methylaminocarbonyl.
("CVCuiVAryl steht im Rahmen der Erfindung für einen aromatischen Carbocyclus mit 6 oder 10 Ring-Kohlenstoffatomen. Bevorzugte Arylreste sind Phenyl und Νaphthyl.
Ein 4- bis 7-gliedriger Heterocvclus steht im Rahmen der Erfindung für einen gesättigten Hetero- cyclus mit insgesamt 4 bis 7 Ringatomen, der ein oder zwei Ring-Heteroatome aus der Reihe Ν, O und/oder S enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls ein Ring-Stickstoff- atom verknüpft ist. Bevorzugt ist ein 5- oder 6-gliedriger Heterocvclus mit ein oder zwei Ring- Heteroatomen aus der Reihe Ν und/oder O. Beispielhaft seien genannt: Azetidinyl, Oxetanyl, Pyrrolidinyl, Pyrazolidinyl, Tetrahydrofuranyl, Piperidinyl, Piperazinyl, Tetrahydropyranyl, Mor- pholinyl, Thiomorpholinyl, Hexahydroazepinyl und Hexahydro-l,4-diazepinyl. Bevorzugt sind Pyrrolidinyl, Tetrahydrofuranyl, Piperidinyl, Piperazinyl, Tetrahydropyranyl und Morpholinyl.
5- bis 10-gliedriges Heteroaryl steht im Rahmen der Erfindung für einen mono- oder gegebenenfalls bicyclischen aromatischen Heterocyclus (Heteroaromaten) mit insgesamt 5 bis 10 Ringatomen, der bis zu drei gleiche oder verschiedene Ring-Heteroatome aus der Reihe Ν, O und/oder S enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls über ein Ring-Stickstoffatom
verknüpft ist. Beispielhaft seien genannt: Furyl, Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thia- zolyl, Oxazolyl, Isoxazolyl, Isothiazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimi- dinyl, Pyridazinyl, Pyrazinyl, Triazinyl, Benzofuranyl, Benzothienyl, Benzimidazolyl, Benzoxa- zolyl, Benzothiazolyl, Benzotriazolyl, Indolyl, Indazolyl, Chinolinyl, Isochinolinyl, Naphthyri- dinyl, Chinazolinyl, Chinoxalinyl, Phthalazinyl, Pyrazolo[3,4-b]pyridinyl. Bevorzugt sind mono- cyclische 5- oder 6-εliedrige Heteroaryl-Reste mit bis zu drei Ring-Heteroatomen aus der Reihe N, O und/oder S wie beispielsweise Furyl, Thienyl, Thiazolyl, Oxazolyl, Isothiazolyl, Isoxazolyl, Pyrazolyl, Imidazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl.
Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und Iod ein. Bevorzugt sind Chlor oder Fluor.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevorzugt. Ganz besonders bevorzugt ist die Substitution mit einem oder zwei gleichen oder verschiedenen Substituenten.
Von besonderer Bedeutung im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 für Phenyl oder 5- oder 6-gliedriges Heteroaryl mit bis zu drei Ring-Heteroatomen aus der Reihe N, O und/oder S steht, welche jeweils
(0 ein- oder zweifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe
Halogen, Nitro, Cyano, (Ci-C6)-Alkyl, Trifluormethyl, Hydroxy, (Ci-C6)-Alkoxy,
Amino, Mono-(CrC6)-alkylamino, Di-(C i-C6)-alkylamino, Carboxyl, (d-C6)-Alkoxy- carbonyl, Carbamoyl, Mono-(Ci-C6)-alkylaminocarbonyl und Di-(C)-C6)-alkylamino- carbonyl
und/oder
(H) mit Pyrrolidino, Piperidino, Morpholino, Piperazino, N'-(Ci-C4)-Alkylpiperazino, Tetrazolyl oder einer Gruppe der Formel -L-R4, worin
L eine Bindung, NH oder O bedeutet
und
R4 Phenyl oder 5- oder 6-gliedriges Heteroaryl mit bis zu drei Ring-Heteroatomen aus der Reihe N, O und/oder S bedeutet, welche jeweils ein- bis dreifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe Halogen, Nitro, Cyano, (C,-C6)-Alkyl, Trifluormethyl, Hydroxy, (C,-C6)-Alkoxy, Difluormeth- oxy, Trifluormethoxy, Amino, Mono-(Ci-C6)-alkylamino, Di-(Ci-C6)-alkyl- amino, (CrC6)-Alkoxycarbonyl und Carboxyl substituiert sein können,
substituiert sind,
oder
R1 für N-Oxidopyridyl steht,
sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
der Ring A für Cyclopentyl, Cyclohexyl, Cyclopent-2-en-l-yl, Cyclopent-3-en-l-yl, Cyclohex-2- en-l-yl oder Cyclohex-3-en-l-yl, für einen 5- oder 6-gliedrigen, C-verknüpften Hetero- cyclus, der ein Ringglied aus der Reihe N-R3 oder O enthält, oder für einen C-verknüpften Heterocyclus der Formel
steht, worin
Cyclopentyl, Cyclohexyl, Cyclopentenyl sowie Cyclohexenyl ein- oder zweifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe (Ci-C4)-Alkyl, Hydroxy, (Ci- C4)-Alkoxy, Amino, Mono-(Ci-C4)-alkylamino und Di-(C rC4)-alkylamino substituiert sein können, wobei die genannten (Ci-C4)-Alkyl- und (CrC4)-Alkoxy-Reste ihrerseits ein- oder zweifach, gleich oder verschieden, mit Hydroxy, (Ci-C4)-Alkoxy und/oder (C3-Cs)-CyCIo- alkyl substituiert sein können,
* die Verknüpfungsstelle mit dem Pyridin-Ring bedeutet
und
R Wasserstoff, (Ci-C4)-Alkyl, welches ein- oder zweifach, gleich oder verschieden, mit Hydroxy, (CrC4)-Alkoxy, (Ci-C4)-Acyloxy und/oder (C3-C5)-Cycloalkyl substituiert sein kann, oder (Ci-C4)-Acyl, welches mit Hydroxy oder (CrC4)-Alkoxy substituiert sein kann, bedeutet,
R für Phenyl oder 5- oder 6-gliedriges Heteroaryl mit bis zu drei Ring-Heteroatomen aus der
Reihe N, O und/oder S steht, welche jeweils
(0 ein- oder zweifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe Fluor, Chlor, Cyano, (CrC4)-Alkyl, Amino, Mono-(Ci-C4)-alkylamino, Di-(Ci-C4)- alkylamino, (C)-C4)-Alkoxycarbonyl, Carboxyl und Carbamoyl
und/oder
(H) mit Morpholino, N'-(Ci-C4)-Alkylpiperazino oder einer Gruppe der Formel -L-R , worin
L eine Bindung oder NH bedeutet
und
R4 Phenyl oder 5- oder 6-gliedriges Heteroaryl mit bis zu drei Ring-Heteroatomen aus der Reihe N, O und/oder S bedeutet, welche jeweils ein- bis dreifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe Fluor, Chlor, Cyano, (C,-C4)-Alkyl, Trifluormethyl, (CrC4)-Alkoxy, Trifluormethoxy, (C,- C4)-Alkoxycarbonyl und Carboxyl substituiert sein können,
substituiert sind,
oder
R1 für N-Oxidopyridyl steht,
und
R2 für Wasserstoff oder für (C]-C4)-Alkoxy, das bis zu dreifach mit Fluor substituiert sein kann, steht
oder
R2 für eine Gruppe der Formel -NR5R6 steht, worin
R5 Wasserstoff oder (CrC4)-Alkyl, das mit Hydroxy, (CrC4)-Alkoxy, Amino, Mono- (CrC4)-alkylamino, Di-(Ci-C4)-alkylamino, Carboxyl, (Ci-C4)-Alkoxycarbonyl oder einem 5- oder 6-gliedrigen Heterocyclus substituiert sein kann, bedeutet,
wobei der genannte Heterocyclus ein oder zwei Ring-Heteroatome aus der Reihe N und/oder O enthält und seinerseits ein- oder zweifach, gleich oder verschieden, mit Methyl, Ethyl, Hydroxy, Methoxy und/oder Ethoxy substituiert sein kann,
Rb Wasserstoff oder Methyl bedeutet
oder
R5 und R6 zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 5- oder 6- gliedrigen Heterocyclus bilden, der ein weiteres Ring-Heteroatom aus der Reihe N oder O enthalten und ein- oder zweifach, gleich oder verschieden, mit Methyl, Ethyl, Hydroxy, Methoxy und/oder Ethoxy substituiert sein kann,
sowie ihre Salze, Solvate und Solvate der Salze.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
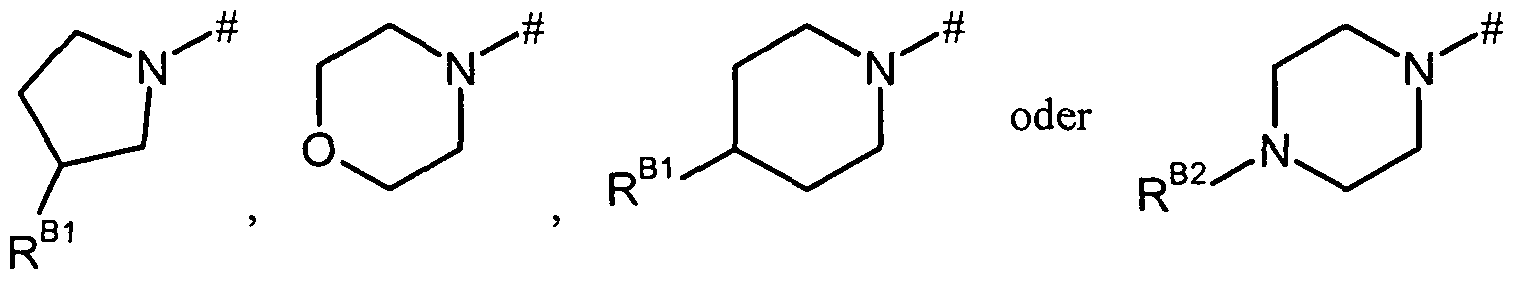
der Ring A für eine Gruppe der Formel
steht, worin
die Verknüpfungsstelle mit dem Pyridin-Ring,
R Wasserstoff, Hydroxy, Methoxy, Ethoxy oder 2-Hydroxyethoxy
und
R3 Methyl, Ethyl, 2-Hydroxyethyl, 2-Acetoxyethyl, 3-Hydroxypropyl, 3-Acetoxy- propyl oder Hydroxyacetyl
bedeuten,
R1 für Phenyl, Oxazolyl, Thiazolyl oder Pyridyl steht, welche jeweils
(/) ein- oder zweifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe Fluor, Chlor, Methyl, Amino, Methoxycarbonyl, Ethoxycarbonyl, Carboxyl und Carbamoyl
oder
00 mit einer Gruppe der Formel -L-R4, worin
L eine Bindung oder NH bedeutet
und
R4 Phenyl oder Pyridyl bedeutet, welche jeweils ein- oder zweifach, gleich oder verschieden, mit einem Rest ausgewählt aus der Reihe Fluor, Chlor, Cyano, Methyl, Methoxy und Carboxyl substituiert sein können,
substituiert sind,
und
R2 für Wasserstoff, Methoxy oder eine Gruppe der Formel -NR5R6 steht, worin
R5 Wasserstoff oder (Ci-C4)-Alkyl, das mit Hydroxy, Amino, Methylamino, Ethyl- amino, Dimethylamino, Diethylamino oder einem Heterocyclus der Formel
oder
substituiert sein kann, bedeutet,
R6 Wasserstoff bedeutet
oder
R5 und R6 zusammen mit dem Stickstoffatom, an das sie gebunden sind, eine Gruppe der Formel
bilden, worin jeweils
** die Verknüpfungsstelle mit dem (Ci-C4)-Alkylrest,
# die Verknüpfungsstelle mit dem Pyridin-Ring,
RB1 Wasserstoff oder Hydroxy
und
RB2 Wasserstoff oder Methyl
bedeuten,
sowie ihre Salze, Solvate und Solvate der Salze.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Verbindungen der Formel (I), in welcher R2 für NH2 steht, dadurch gekennzeichnet, dass man eine Verbindung der Formel (II)
in welcher der Ring A die oben angegebene Bedeutung hat,
in einem inerten Lösungsmittel in Gegenwart einer Base mit einer Verbindung der Formel (HI)
x^^R1 (m),
in welcher R1 die oben angegebene Bedeutung hat und
X für eine geeignete Abgangsgruppe, vorzugsweise für Halogen, insbesondere Chlor, Brom oder Iod, oder für Mesylat, Tosylat oder Triflat steht,
zu einer Verbindung der Formel (I- A)
in welcher R1 und der Ring A die oben angegebenen Bedeutungen haben,
umsetzt
und die Verbindungen der Formel (I-A) gegebenenfalls mit den entsprechenden (/) Lösungsmitteln und/oder (U) Basen oder Säuren in ihre Solvate, Salze und/oder Solvate der Salze überführt.
Das zuvor beschriebene Verfahren kann durch das folgende Reaktionsschema erläutert werden:
Schema 1
Als Lösungsmittel für das erfindungsgemäße Verfahren eignen sich alle organischen Lösungsmittel, die unter den Reaktionsbedingungen inert sind. Hierzu gehören Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol und tert.-Butanol, Ketone wie Aceton und Methyl- ethylketon, acyclische und cyclische Ether wie Diethylether, Methyl-tert.-butylether, 1,2-Dimeth-
oxyethan, Tetrahydrofuran und Dioxan, Ester wie Essigsäureethylester oder Essigsäurebutylester, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan und Cyclohexan, chlorierte Kohlenwasserstoffe wie Dichlormethan, Trichlormethan und Chlorbenzol, oder andere Lösungsmittel wie Di- methylformamid (DMF), Dimethylsulfoxid (DMSO), N-Methylpyrrolidinon (ΝMP), Acetonitril oder Pyridin. Wasser ist als Lösungsmittel ebenfalls geeignet. Ebenso ist es möglich, Gemische der zuvor genannten Lösungsmittel einzusetzen. Bevorzugt wird Dimethylformamid als Lösungsmittel verwendet.
Als Base für die Umsetzung (II) + (UJ) -» (I-A) eignen sich die üblichen anorganischen oder organischen Basen. Hierzu gehören bevorzugt Alkalihydroxide wie beispielsweise Lithium-, Νatrium- oder Kaliumhydroxid, Alkalicarbonate wie Lithium-, Natrium-, Kalium- oder Cäsiumcarbonat, Alkalihydrogencarbonate wie Natrium- oder Kaliumhydrogencarbonat, Alkalialkoholate wie Natrium- oder Kaliummethanolat, Natrium- oder Kaliumethanolat oder Kalium-tert.-butylat, Amide wie Natriumamid, Lithium-, Natrium- oder Kalium-bis-(trimethylsilyl)amid oder Lithium- diisopropylamid, metallorganische Verbindungen wie Butyllithium oder Phenyllithium, oder orga- nische Amine wie Triethylamin, Diisopropylethylamin, Pyridin, l,8-Diazabicyclo[5.4.0]undec-7- en (DBU) oder l,5-Diazabicyclo[4.3.0]non-5-en (DBN). Bevorzugt sind Alkalicarbonate und Alkalihydrogencarbonate.
Die Base kann hierbei in einer Menge von 1 bis 10 Mol, bevorzugt von 1 bis 5 Mol, insbesondere von 1 bis 4 Mol, bezogen auf 1 Mol der Verbindung der Formel (II), eingesetzt werden.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von -78°C bis +140°C, bevorzugt im Bereich von -200C bis +800C, insbesondere bei 00C bis +500C. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck durchgeführt werden (z.B. im Bereich von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die Verbindungen der Formel (U) können in Analogie zu literaturbekannten Methoden beispiels- weise dadurch hergestellt werden, dass man Aldehyde der Formel (IV)
in welcher der Ring A die oben angegebene Bedeutung hat,
in Gegenwart einer Base mit zwei Äquivalenten Cyanothioacetamid umsetzt [siehe Schema 2; vgl. z.B. Dyachenko et al., Russ. J. Chem. 33 (7), 1014-1017 (1997), 34 (4), 557-563 (1998); Dyachenko et al., Chemistry of Heterocyclic Compounds 34 (2), 188-194 (1998); Qintela et al., Ew. J. Med. Chem. 33. 887-897 (1998); Kandeel et al., Z. Naturforsch. 42b, 107-111 (1987); Reddy et al., J. Med. Chem. 49, 607-615 (2006); Evdokimov et al., Org. Lett. 8, 899-902 (2006)].
Schema 2
[EtOH = Ethanol, NMM = N-Methylmorpholin].
Alternativ können Verbindungen der Formel (II) auch ausgehend von Verbindungen der Formel (V)
in welcher der Ring A die oben angegebene Bedeutung hat,
durch Reaktion mit einem Alkalisulfid hergestellt werden. Diese Herstellungsmethode kann durch folgendes Formelschema beispielhaft erläutert werden:
Schema 3
Na
9S
AIs Alkalisulfid wird vorzugsweise Natriumsulfid in einer Menge von 1 bis 10 Mol, bevorzugt von 1 bis 5 Mol, insbesondere von 1 bis 4 Mol, bezogen auf 1 Mol der Verbindung der Formel (V), eingesetzt.
Als Lösungsmittel sind hierfür alle organischen Lösungsmittel geeignet, die unter den Reaktions- bedingungen inert sind. Hierzu gehören insbesondere Dimethylformamid, N-Methylpyrrolidinon, Pyridin und Acetonitril. Ebenso ist es möglich, Gemische der zuvor genannten Lösungsmittel einzusetzen. Bevorzugt wird Dimethylformamid verwendet.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von +200C bis +1400C, bevorzugt im Bereich von +200C bis +1200C, insbesondere bei +600C bis +1000C. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck durchgeführt werden (z.B. im Bereich von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die Verbindungen der Formel (V) können in Analogie zu literaturbeschriebenen Verfahren hergestellt werden [vgl. z.B. Kambe et al., Synthesis, 531-533 (1981); Elnagdi et al., Z. Naturforsch. 47b. 572-578 (1991); Reddy et al., J. Med. Chem. 49, 607-615 (2006); Evdokimov et al., Org. Lett. 8, 899-902 (2006)].
Die Verbindungen der Formel (III) sind kommerziell erhältlich, literaturbekannt oder nach literaturbekannten Methoden herstellbar. So können beispielsweise durch Reaktion von Amiden, Thio- amiden bzw. Thioharnstoff-Derivaten mit einem 1,3-Dihalogenaceton substituierte Oxazol- und Thiazol-Derivate der Formel (III-A), (III-B) bzw. (III-C) erhalten werden (siehe Schema 4):
Schema 4
(ΠI-A)
(III-C)
Im Falle der Verbindungen (HI-C) können diese entweder analog zur Literatur hergestellt und isoliert werden [vgl. z.B. I. Simiti et al., Chem. Ber. 95, 2672-2679 (1962)], oder sie können in situ erzeugt und direkt weiter mit einer Verbindung der Formel (II) umgesetzt werden. Bevorzugt ist die in situ-Eτzeugang unter Verwendung von 1 ,3-Dichloraceton in Dimethylformamid oder Ethanol als Lösungsmittel. Die Darstellung erfolgt im Allgemeinen in einem Temperaturbereich von 00C bis +14O0C, bevorzugt im Bereich von +2O0C bis +12O0C, insbesondere bei +600C bis +1000C.
Die Aldehyde der Formel (FV) sind kommerziell erhältlich, in der Literatur beschrieben oder nach Standardmethoden aus bekannten Ausgangsverbindungen herstellbar.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Verbindungen der Formel (I), in welcher R
2 für die Gruppe -NR
5R
6 steht, worin mindestens einer der beiden Reste R
5 und R
6 nicht Wasserstoff bedeutet, dadurch gekennzeichnet, dass man Verbindungen der Formel (I- A) zunächst mit Kupfer(H)chlorid und Isoamylnitrit in einem geeigneten Lösungsmittel in Verbindungen der Formel (VI)
in welcher R1 und der Ring A die zuvor angegebenen Bedeutungen haben,
überfuhrt und diese anschließend mit einer Verbindung der Formel (VII)
H R5A/N^R6A
in welcher
R5A die oben angegebene Bedeutung von R5 hat,
R6Λ die oben angegebene Bedeutung von R6 hat,
jedoch mindestens einer der beiden Reste R5A und R6A nicht für Wasserstoff steht,
zu Verbindungen der Formel (I-B)
in welcher R1, R5Λ, R6A und der Ring A jeweils die zuvor angegebenen Bedeutungen haben,
umsetzt
und die Verbindungen der Formel (I-B) gegebenenfalls mit den entsprechenden (/) Lösungsmitteln und/oder (H) Basen oder Säuren in ihre Solvate, Salze und/oder Solvate der Salze überführt.
Das zuvor beschriebene Verfahren kann durch das folgende Reaktionsschema erläutert werden:
Schema 5
Die Umsetzung (I- A) — > (VI) erfolgt im Allgemeinen in einem Molverhältnis von 2 bis 12 Mol Kupfer(II)chlorid und 2 bis 12 Mol Isoamylnitrit bezogen auf 1 Mol der Verbindung der Formel (I-A).
Als Lösungsmittel für den Verfahrensschritt (I-A) -> (VI) eignen sich alle organischen Lösungsmittel, die unter den Reaktionsbedingungen inert sind. Hierzu gehören acyclische und cyclische Ether wie Diethylether und Tetrahydrofuran, Ester wie Essigsäureethylester oder Essigsäurebutyl- ester, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan und Cyclohexan, chlorierte Kohlen- Wasserstoffe wie Dichlormethan, 1 ,2-Dichlorethan und Chlorbenzol, oder andere Lösungsmittel wie Dimethylformamid, Acetonitril oder Pyridin. Ebenso ist es möglich, Gemische der zuvor genannten Lösungsmittel einzusetzen. Bevorzugte Lösungsmittel sind Acetonitril und Dimethylformamid.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von -78°C bis +18O0C, bevor- zugt im Bereich von 00C bis +1000C, insbesondere bei +200C bis +800C. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck durchgeführt werden (z.B. im Bereich von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die Umsetzung (VI) + (VE) → (I-B) erfolgt im Allgemeinen in einem Molverhältnis von 1 bis 8 Mol der Verbindung der Formel (VH) bezogen auf 1 Mol der Verbindung der Formel (VI).
Als Lösungsmittel für den Verfahrensschritt (VI) + (VIT) -> (I-B) eignen sich alle organischen Lösungsmittel, die unter den Reaktionsbedingungen inert sind. Hierzu gehören Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol und tert.-Butanol, Ketone wie Aceton und Methylethylketon, acyclische und cyclische Ether wie Diethylether, 1 ,2-Dimethoxyethan, Tetra- hydrofuran und Dioxan, Ester wie Essigsäureethylester oder Essigsäurebutylester, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan und Cyclohexan, chlorierte Kohlenwasserstoffe wie Di- chlormethan, 1 ,2-Dichlorethan und Chlorbenzol, oder andere Lösungsmittel wie Dimethylform- amid, Acetonitril, Pyridin oder Dimethylsulfoxid. Wasser ist als Lösungsmittel ebenfalls geeignet. Ebenso ist es möglich, Gemische der zuvor genannten Lösungsmittel einzusetzen. Bevorzugtes Lösungsmittel ist Dimethylformamid.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von O0C bis +180°C, bevorzugt im Bereich von +200C bis +1500C, insbesondere bei +200C bis +1000C. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck durchgeführt werden (z.B. im Bereich von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die Verbindungen der Formel (VIT) sind entweder kommerziell erhältlich, dem Fachmann bekannt oder nach üblichen Methoden herstellbar.
Die erfindungsgemäßen Verbindungen der Formel (I), in welcher R2 für Wasserstoff steht, können hergestellt werden, indem man Verbindungen der Formel (I-A) in einem geeigneten Lösungsmittel mit Isoamylnitrit in Gegenwart einer katalytischen Menge Kupfer(II)chlorid umsetzt. Diese Methode kann durch das folgende Reaktionsschema erläutert werden:
Schema 6
Die Umsetzung (I-A) → (I-C) erfolgt im Allgemeinen in einem Molverhältnis von 0.01 bis 0.2 Mol Kupfer(π)chlorid und 2 bis 12 Mol Isoamylnitrit bezogen auf 1 Mol der Verbindung der Formel (I-A).
Als Lösungsmittel für die Reaktion (I-A) — > (I-C) eignen sich alle organischen Lösungsmittel, die unter den Reaktionsbedingungen inert sind. Hierzu gehören acyclische und cyclische Ether wie Di- ethylether und Tetrahydrofuran, Ester wie Essigsäureethylester oder Essigsäurebutylester, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan und Cyclohexan, chlorierte Kohlenwasserstoffe wie Dichlormethan, 1,2-Dichlorethan und Chlorbenzol, oder andere Lösungsmittel wie Dimethyl- formamid, Acetonitril oder Pyridin. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugte Lösungsmittel sind Tetrahydrofuran und Dimethylformamid.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von -78°C bis +1500C, bevorzugt im Bereich von 00C bis +800C, insbesondere bei +100C bis +400C. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck durchgeführt werden (z.B. im Bereich von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die erfindungsgemäßen Verbindungen der Formel (I), in welcher R2 für gegebenenfalls substituiertes (CrC6)-Alkyl oder (Ci-Ce)-Alkoxy steht, sind in Analogie zu literaturbeschriebenen Methoden ausgehend von Verbindungen der Formel (VI) herstellbar [vgl. z.B. D. Mabire et al., J. Med. Chem. 48, 2134-2153 (2005)]. Alternativ können die Verbindungen der Formel (I), in welcher R2 für gegebenenfalls substituiertes (C1-Ce)-AIkOXy steht, auch durch Alkylierung von Verbindungen der Formel (VIII) erhalten werden (siehe Schema 7):
Schema 7
Die Verbindungen der Formel (VITI) ihrerseits sind nach literaturbekannten Methoden aus Verbindungen der Formel (VI) oder (I-A) zugänglich [vgl. z.B. G. Lavecchia et al., Tetrahedron Lett. 45, 6633-6636 (2004)].
Weitere erfindungsgemäße Verbindungen können gegebenenfalls auch hergestellt werden durch Umwandlungen von funktionellen Gruppen einzelner Reste und Substituenten, insbesondere den unter R1, R2 und zum Ring A aufgeführten, ausgehend von den nach obigen Verfahren erhaltenen Verbindungen der Formel (I). Diese Umwandlungen werden nach üblichen, dem Fachmann be- kannten Methoden durchgeführt und umfassen beispielsweise Reaktionen wie nukleophile oder elektrophile Substitution, Oxidation, Reduktion, Hydrierung, Alkylierung, Acylierung, Aminie- rung, Bildung von Carbonsäureamiden und -estern, Esterhydrolyse, Veretherung, Etherspaltung sowie die Einführung und Entfernung temporärer Schutzgruppen.
Überraschenderweise zeigen die erfindungsgemäßen Verbindungen ein nicht vorhersehbares, wert- volles pharmakologisches Wirkspektrum und sind daher insbesondere zur Prophylaxe und/oder Behandlung von Erkrankungen geeignet. Darüber hinaus weisen die erfindungsgemäßen Substanzen gegenüber den Verbindungen aus dem Stand der Technik ein verbessertes Absorptionsverhalten im Körper und/oder eine verbesserte Löslichkeit in Wasser und anderen physiologischen Medien auf, was beispielsweise für ihre galenische Formulierbarkeit und/oder die parenterale An- wendung von Vorteil ist.
Die pharmazeutische Wirksamkeit der erfmdungsgemäßen Verbindungen lässt sich durch ihre Wirkung als potente, selektive Liganden an Adenosin Al- und/oder A2b-Rezeptoren erklären. Sie wirken hierbei als selektive Al- oder als selektive duale Al/A2b-Agonisten.
Als "selektive Liganden an Adenosin Al- und/oder A2b-Rezeptoren" werden im Rahmen der vorliegenden Erfindung solche Adenosin-Rezeptorliganden bezeichnet, bei denen einerseits eine deutliche Wirkung an Al- und/oder A2b-Adenosinrezeptor-Subtypen und andererseits keine oder eine deutliche schwächere Wirkung (Faktor 10 oder höher) an A2a- und A3-Adenosinrezeptor- Subtypen zu beobachten ist, wobei bezüglich der Testmethoden für die Wirk-Selektivität Bezug genommen wird auf die im Abschnitt B-I . beschriebenen Tests.
Die Verbindungen der Formel (I) sind allein oder in Kombination mit einem oder mehreren anderen Wirkstoffen zur Prophylaxe und/oder Behandlung verschiedener Erkrankungen geeignet, so beispielsweise insbesondere bei Hypertonie und anderen Erkrankungen des Herzkreislauf- Systems (kardiovaskulären Erkrankungen) sowie zur Kardioprotektion.
Im Sinne der vorliegenden Erfindung sind unter Erkrankungen des Herzkreislauf-Systems bzw. kardiovaskulären Erkrankungen neben der Hypertonie beispielsweise insbesondere die folgenden
Erkrankungen zu verstehen: periphere und kardiale Gefäßerkrankungen, koronare Herzerkrankung, koronare Restenose wie z.B. Restenose nach Ballondilatation von peripheren Blutgefäßen, akutes
Koronarsyndrom, stabile und instabile Angina pectoris, Herzinsuffizienz, Tachykardien, Arrhythmien, Vorhof- und Kammerflimmern sowie periphere Durchblutungsstörungen.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen insbesondere auch zur Reduktion des von einem Infarkt betroffenen Myokardbereichs sowie zur Prophylaxe von Sekundärinfarkten.
Des weiteren sind die erfindungsgemäßen Verbindungen insbesondere zur Prophylaxe und/oder Behandlung von thromboembolischen Erkrankungen und Ischämien wie Myokardinfarkt, Hirnschlag und transitorischen ischämischen Attacken sowie zur Protektion von Organen bei Transplantationen und operativen Eingriffen, beispielsweise am Herzen, geeignet.
Weitere Indikationsgebiete, für die die erfindungsgemäßen Verbindungen verwendet werden kön- nen, sind beispielsweise insbesondere die Prophylaxe und/oder Behandlung von Erkrankungen des
Urogenitalbereiches, wie z.B. Reizblase, erektile Dysfunktion und weibliche sexuelle Dysfunktion, daneben aber auch die Prophylaxe und/oder Behandlung von inflammatorischen Erkrankungen, wie z.B. Asthma und entzündliche Dermatosen, von neuroinflammatorischen Erkrankungen des
Zentralnervensystems, wie beispielsweise Zustände nach Hirninfarkt, der Alzheimer-Erkrankung, weiterhin auch von neurodegenerativen Erkrankungen sowie von Schmerzzuständen, Krebs und
Übelkeit und Erbrechen in Verbindung mit Krebstherapien.
Ein weiteres Indikationsgebiet sind beispielsweise insbesondere die Prophylaxe und/oder Behandlung von Erkrankungen der Atemwege wie beispielsweise Asthma, chronische Bronchitis, Lungenemphysem, Bronchiektasien, zystische Fibrose (Mukoviszidose) und pulmonale Hypertonie.
Schließlich kommen die erfindungsgemäßen Verbindungen beispielsweise insbesondere auch für die Prophylaxe und/oder Behandlung von Diabetes, insbesondere Diabetes mellitus, diabetischen Folgeerkrankungen wie z.B. Nephropathie und Neuropathie, des Metabolischen Syndroms sowie von Dyslipidämien in Betracht.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Ver- bindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine der erfmdungsgemäßen Verbindungen und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prophylaxe der zuvor genannten Erkrankungen.
Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt: den Fettstoff- Wechsel verändernde Wirkstoffe, Antidiabetika, Blutdruck-Senker, durchblutungsfördernd und/ oder antithrombotisch wirkende Mittel, Antioxidantien, Chemokin-Rezeptor- Antagonisten, p38- Kinase-Inhibitoren, NPY-Agonisten, Orexin-Agonisten, Anorektika, PAF-AH-Inhibitoren, Anti- phlogistika (COX -Inhibitoren, LTB4-Rezeptor-Antagonisten) sowie Analgetika wie beispielsweise Aspirin.
Gegenstand der vorliegenden Erfindung sind insbesondere Kombinationen mindestens einer der erfindungsgemäßen Verbindungen mit mindestens einem den Fettstoffwechsel verändernden Wirkstoff, einem Antidiabetikum, einem blutdrucksenkenden Wirkstoff und/oder einem antithrombotisch wirkenden Mittel.
Die erfindungsgemäßen Verbindungen können vorzugsweise mit einem oder mehreren
• den Fettstoffwechsel verändernden Wirkstoffen, beispielhaft und vorzugsweise aus der Gruppe der HMG-CoA-Reduktase-Inhibitoren, Inhibitoren der HMG-CoA-Reduktase-Expression, Squalensynthese-Inhibitoren, ACAT-Inhibitoren, LDL-Rezeptor-Induktoren, Cholesterin- Absorptionshemmer, polymeren Gallensäureadsorber, Gallensäure-Reabsorptionshemmer, MTP-Inhibitoren, Lipase-Inhibitoren, LpL-Aktivatoren, Fibrate, Niacin, CETP-Inhibitoren, PPAR-α-, PPAR-γ- und/oder PPAR-δ-Agonisten, RXR-Modulatoren, FXR-Modulatoren, LXR-
Modulatoren, Thyroidhormone und/oder Thyroidmimetika, ATP-Citrat-Lyase-Inhibitoren, Lp(a)-Antagonisten, Cannabinoid-Rezeptor 1 -Antagonisten, Leptin-Rezeptor-Agonisten, Bom- besin-Rezeptor-Agonisten, Histamin-Rezeptor-Agonisten sowie der Antioxidantien/Radikal- fänger;
• Antidiabetika, die in der Roten Liste 2004/π, Kapitel 12 genannt sind, sowie beispielhaft und vorzugsweise jenen aus der Gruppe der Sulphonylharnstoffe, Biguanide, Meglitinid-Derivate, Glukosidase-Inhibitoren, Oxadiazolidinone, Thiazolidindione, GLP 1 -Rezeptor- Agonisten, GIu-
kagon-Antagonisten, Insulin-Sensitizer, CCK 1 -Rezeptor- Agonisten, Leptin-Rezeptor- Agonisten, Inhibitoren von Leberenzymen, die an der Stimulation der Glukoneogenese und/ oder Glykogenolyse beteiligt sind, Modulatoren der Glukoseaufnahme sowie der Kaliumkanalöffner, wie z.B. denjenigen, die in WO 97/26265 und WO 99/03861 offenbart sind;
• den Blutdruck senkenden Wirkstoffen, beispielhaft und vorzugsweise aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, Renin-Inhibitoren, beta- Rezeptoren-B locker, alpha-Rezeptoren-Blocker, Diuretika, Phosphodiesterase-Inhibitoren, sGC-Stimulatoren, Verstärker der cGMP-Spiegel, Aldosteron-Antagonisten, Mineralocorticoid- Rezeptor- Antagonisten, ECE-Inhibitoren sowie der Vasopeptidase-Inhibitoren; und/oder
• antithrombotisch wirkenden Mitteln, beispielhaft und vorzugsweise aus der Gruppe der Thrombozytenaggregationshemmer oder der Antikoagulantien
kombiniert werden.
Unter den Fettstoffwechsel verändernden Wirkstoffen werden vorzugsweise Verbindungen aus der Gruppe der HMG-CoA-Reduktase-Inhibitoren, Squalensynthese-Inhibitoren, ACAT-Inhibitoren, Cholesterin-Absorptionshemmer, MTP-Inhibitoren, Lipase-Inhibitoren, Thyroidhormone und/oder Thyroidmimetika, Niacin-Rezeptor-Agonisten, CETP-Inhibitoren, PPAR-α-Agonisten, PPAR-γ- Agonisten, PP AR-δ- Agonisten, polymeren Gallensäureadsorber, Gallensäure-Reabsorptionshem- mer, Antioxidantien/Radikalfänger sowie der Cannabinoid-Rezeptor 1 -Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem HMG-CoA-Reduktase-Inhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin, Cerivastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK-475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Cholesterin-Absorptionshemmer, wie beispielhaft und vorzugsweise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS-201038, R-103757 oder JTT-130, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidhormon und/oder Thyroidmimetikum, wie beispielhaft und vorzugsweise D-Thyroxin oder 3,5,3'-Triiodothyronin (T3), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Agonisten des Niacin-Rezeptors, wie beispielhaft und vorzugsweise Niacin, Acipimox, Acifran oder Radecol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP-Inhibitor, wie beispielhaft und vorzugsweise Torcetrapib, JTT-705, BAY 60-5521 , BAY 78-7499 oder CETP Vaccine (Avant), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-γ-Agonisten, wie beispielhaft und vorzugsweise Piogli- tazone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem PPAR-δ-Agonisten, wie beispielhaft und vorzugsweise GW- 501516 oder BAY 68-5042, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugsweise ASBT (= IBAT)-Inhibitoren wie z.B. AZD-7806, S-8921 , AK-105, BARJ-1741, SC-435 oder SC-635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Antioxidans/Radikalfänger, wie beispielhaft und vorzugsweise Probucol, AGI-1067, BO-653 oder AEOL-10150, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cannabinoid-Rezeptor 1 -Antagonisten, wie beispielhaft und vorzugsweise Rimonabant oder SR-147778, verabreicht.
Unter Antidiabetika werden vorzugsweise Insulin und Insulinderivate sowie oral wirksame hypo- glykämische Wirkstoffe verstanden. Insulin und Insulinderivate umfasst hierbei sowohl Insuline tierischen, menschlichen oder biotechnologischen Ursprungs als auch Gemische hieraus. Die oral wirksamen hypoglykämischen Wirkstoffe umfassen vorzugsweise Sulphonylharnstoffe, Biguanide, Meglitinid-Derivate, Glukosidase-Inhibitoren und PPAR-γ-Agonisten.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit Insulin verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Sulphonylharnstoff, wie beispielhaft und vorzugsweise Tolbutamid, Glibenclamid, Glimepirid, Glipizid oder Gliclazid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Biguanid, wie beispielhaft und vorzugsweise Metformin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Meglitinid-Derivat, wie beispielhaft und vorzugsweise Repagli- nid oder Nateglinid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Glukosidase-Inhibitor, wie beispielhaft und vorzugsweise Mig- litol oder Acarbose, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfmdungsgemäßen Verbindungen in Kombination mit einem PPAR-γ-Agonisten beispielsweise aus der Klasse der Thiazoli- dindione, wie beispielhaft und vorzugsweise Pioglitazone oder Rosiglitazone, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, beta-Rezeptoren-Blocker, alpha-Rezeptoren-B locker und Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Calcium-Antagonisten, wie beispielhaft und vorzugsweise Nifedipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin Aü-Antagonisten, wie beispielhaft und vorzugsweise Losartan, Valsartan, Candesartan, Embusartan, Olmesartan oder Telmisartan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Lisinopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem beta-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Meti- pranolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol, Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucin- dolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem alpha-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise Furosemid, Bumetanid, Torsemid, Bendroflumethiazid, Chlorthiazid, Hydrochlorthiazid, Hydroflumethiazid, Methyclothiazid, Polythiazid, Trichlormethiazid, Chlorthalidon, Indapamid, Metolazon, Quineth- azon, Acetazolamid, Dichlorphenamid, Methazolamid, Glycerin, Isosorbid, Mannitol, Amilorid oder Triamteren, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Antisympathotonika wie Reserpin, Clonidin oder alpha-Methyl-Dopa, mit Kaliumkanal-Agonisten wie Minoxidil, Diazoxid, Dihydralazin oder Hydralazin, oder mit Stickoxid freisetzenden Stoffen wie Glycerinnitrat oder Nitroprussidnatrium verabreicht.
Unter antithrombotisch wirkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer oder der Antikoagulantien verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximela- gatran, Melagatran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem GPÜb/IIIa-Antagonisten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Riva- roxaban (BAY 59-7939), DU-176b, Apixaban, Otamixaban, Fidexaban, Razaxaban, Fondaparinux, Idraparinux, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021 , DX 9065a, DPC 906, JTV 803, SSR-126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nicht- toxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit
magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszuberei- tungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augen- präparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale und die intravenöse Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überfuhrt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige PoIy- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindest- menge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Verwendete Abkürzungen:
Bsp. Beispiel
C Konzentration
DC Dünnschichtchromatographie
DCI direkte chemische Ionisation (bei MS)
DMF NN-Dimethylformamid
DMSO Dimethylsulfoxid d. Th. der Theorie (bei Ausbeute) ee Enantiomerenüberschuss
EI Elektronenstoß-Ionisation (bei MS) ent Enantiomer / enantiomerenrein
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl
Fp. Schmelzpunkt
GC-MS Gaschromatographie-gekoppelte Massenspektrometrie ges. gesättigt h Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie kat. katalytisch konz. konzentriert
LC-MS Flüssigchromatographie-gekoppelte Massenspektrometrie
Lit. Literatur(stelle) min Minute(n)
MS Massenspektrometrie
NMR Kernresonanzspektrometrie rac. racemisch
RP-HPLC reverse phase HPLC
RT Raumtemperatur
R1 Retentionszeit (bei HPLC)
TBME /err.-Butylmethylether
TFA Trifluoressigsäure
THF Tetrahydrofuran verd. verdünnt wässr. wässrig
HPLC-. LC-MS- und GC-MS-Methoden:
Methode 1 (HPLC):
Instrument: Hewlett Packard Series 1050; Säule: Symmetry TM Cl 8 3.9 x 150 mm; Fluss: 1.5 ml/min; Eluent A: Wasser, Eluent B: Acetonitril; Gradient: → 0.6 min 10% B -> 3.8 min 100% B — > 5.0 min 100% B → 5.5 min 10% B; Stopzeit: 6.0 min; Injektionsvolumen: 10 μl; Diodenarray- detektor-Signal: 214 und 254 nm.
Methode 2 (LC-MS):
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith SpeedROD RP-18e 100 mm x 4.6 mm; Eluent A: Wasser + 500 μl 50%-ige Ameisensäure / 1, Eluent B: Acetonitril + 500 μl 50%-ige Ameisensäure / 1; Gradient: 0.0 min 10% B → 7.0 min 95% B → 9.0 min 95% B; Ofen: 35°C; Fluss: 0.0 min 1.0 ml/min → 7.0 min 2.0 ml/min -→ 9.0 min 2.0 ml/min; UV-Detektion: 210 nm.
Methode 3 (LC-MS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Methode 4 (LC-MS):
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A -> 3.0 min 5% A — > 4.5 min 5% A; Fluss: 0.0 min 1 ml/min — > 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 5 (LC-MS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Onyx Monolithic Cl 8, 100 mm x 3 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B:
1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A -> 2 min 65% A → 4.5 min 5% A → 6 min 5% A; Fluss: 2 ml/min; Ofen: 400C; UV-Detektion: 208-400 nm.
Methode 6 (präparative HPLC):
Gerätetyp HPLC: Abimed/Gilson Pump 305/306; Manometric Module 806; UV Knauer Variable Wavelength Monitor; Säule: Gromsil Cl 8, 10 nm, 250 mm x 30 mm; Eluent A: 1 1 Wasser + 0.5 ml 99% TFA, Eluent B: 1 1 Acetonitril; Gradient: 0.0 min 2% B → 10 min 2% B → 50 min 90% B; Fluss: 20 ml/min; Volumen: 628 ml A und 372 ml B.
Methode 7 (LC-MS):
Gerätetyp MS: Waters ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith RPl 8e, 100 mm x 3 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A ^ 2 min 65% A — » 4.5 min 5% A → 6 min 5% A; Fluss: 2 ml/min; Ofen: 400C; UV-Detektion: 210 nm.
Methode 8 TGC-MS):
Instrument: Micromass GCT, GC6890; Säule: Restek RTX-35, 15 m x 200 μm x 0.33 μm; kon- stanter Fluss mit Helium: 0.88 ml/min; Ofen: 700C; Inlet: 25O0C; Gradient: 7O0C, 30°C/min → 3100C (3 min halten).
Methode 9 (GC-MS):
Instrument: Micromass GCT, GC6890; Säule: Restek RTX-35MS, 30 m x 250 μm x 0.25 μm; konstanter Fluss mit Helium: 0.88 ml/min; Ofen: 600C; Inlet: 2500C; Gradient: 600C (0.30 min hal- ten), 50°C/min → 1200C, 16°C/min → 2500C, 30°C/min → 3000C (1.7 min halten).
Methode 10 (chirale HPLC):
Gerätetyp HPLC: HP 1100 mit DAD-Detektion; Säule: Daicel Chiralpak IA, 5 μm, 250 mm x 4.6 mm; Eluent: 50% iso-Hexan, 5% Methanol, 45% tert.-Butylmethylether; Fluss: 15 ml/min.
Methode 11 (chirale HPLC):
Gerätetyp HPLC: HP 1100 mit DAD-Detektion; Säule: Daicel Chiralpak AD-H, 5 μm, 250 mm x 20 mm; Eluent: 50% iso-Hexan, 50% 2-Propanol; Fluss: 15 ml/min.
Methode 12 TLC-MSV
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Syn- ergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A — > 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 13 (LC-MS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1 100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Methode 14 (LC-MSV
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Thermo Hypersil GOLD 3μ 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 100% A → 0.2 min 100% A → 2.9 min 30% A → 3.1 min 10% A → 5.5 min 10% A; Fluss: 0.8 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 15 (LC-MSV
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A — > 2.5 min 30% A → 3.0 min 5% A -> 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 16 (chirale HPLC):
Gerätetyp HPLC: HP 1100 mit DAD-Detektion; Säule: Daicel Chiralpak IA-H, 5 μm, 250 mm x 20 mm; Eluent: 80% TBME, 20% Methanol; Fluss: 15 ml/min.
Methode 17 (chirale HPLO:
Gerätetyp HPLC: HP 1100 mit DAD-Detektion; Säule: Daicel Chiralpak IA, 5 μm, 250 mm x 4.6 mm; Eluent: 80% TBME, 20% Methanol; Fluss: 1.0 ml/min.
Methode 18 (chirale HPLC):
Gerätetyp HPLC: HP 1100 mit DAD-Detektion; Säule: Daicel Chiralpak IA-H, 5 μm, 250 mm x 20 mm; Eluent: 50% iso-Hexan, 45% TBME, 5% Methanol; Fluss: 15 ml/min.
Methode 19 (chirale HPLO:
Gerätetyp HPLC: HP 1 100 mit DAD-Detektion; Säule: Daicel Chiralpak IA, 5 μm, 250 mm x 4.6 mm; Eluent: 40% iso-Hexan, 54% TBME, 6% Methanol; Fluss: 1.0 ml/min.
Methode 20 (HPLC):
Gerätetyp HPLC: HP 1100 mit DAD-Detektion; Säule: Kromasil 100 C 18, 5 μm, 250 mm x 20 mm; Eluent: 25% 0.2%-ige Essigsäure, 75% Acetonitril; Fluss: 25 ml/min.
Methode 21 (LC-MS):
Instrument: Micromass QuattroPremier mit Waters UPLC Acquity; Säule: Thermo Hypersil GOLD 1.9μ 50 mm x 1 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A -> 0.1 min 90% A → 1.5 min 10% A → 2.2 min 10% A; Fluss: 0.33 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 22 (LC-MS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2.5μ MAX-RP 100A Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 0.1 min 90% A → 3.0 min 5% A → 4.0 min 5% A → 4.1 min 90% A; Fluss: 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Ausgangsverbindungen und Intermediate:
Beispiel IA
4-(2-Hydroxyethoxy)cyclohexancarbonsäure
1.00 g (5.49 mmol) 4-(2-Hydroxyethoxy)benzoesäure werden in 30 ml trockenem THF und 30 ml trockenem Ethanol gelöst und mit 1.13 mg (0.55 mmol) Rhodium auf Aluminiumoxid versetzt. Das Reaktionsgemisch wird 12 h lang bei +50°C mit 50 bar Wasserstoffdruck hydriert. Anschließend wird für weitere 48 h bei +600C mit 80 bar Wasserstoffdruck hydriert. Nach Filtration wird das Filtrat am Rotationsverdampfer eingeengt. Der Rückstand wird ohne weitere Reinigung direkt in der Folgereaktion eingesetzt.
Ausbeute: 1.00 g (84% d. Th., 87% Reinheit)
1H-NMR (400 MHz, DMSO-d6): δ = 12.3 (br. s, IH), 4.52 (br. s, IH), 3.49-3.34 (m, 4H), 3.25-3.14 (m, IH), 2.32-2.22 (m, IH), 2.20-0.90 (m, 8H).
GC-MS (Methode 8): R, = 1.43 min; MS (ESIpos): m/z = 222 [M+H]+.
Beispiel 2A
4-(2-Hydroxyethoxy)cyclohexancarbonsäuremethylester
Das Rohprodukt aus Beispiel IA wird in 40 ml Methanol gelöst und mit 200 mg Dowex 50 WX8- 100-Ionenaustauscher (zuvor 7 x mit je 20 ml 2 M Salzsäure und anschließend 7 x mit je 20 ml
Methanol gewaschen) versetzt. Es wird 20 h bei +64°C gerührt. Nach Abkühlen auf RT wird der Ionenaustauscher abfiltriert und das Filtrat am Rotationsverdampfer eingeengt. Das Rohprodukt wird direkt ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 1.00 g (79% d. Th., 76% Reinheit)
GC-MS (Methode 8): R, = 5.26 min; MS (ESIpos): m/z = 183 [M+H]+.
Beispiel 3A
4-(2- { [tert.-Butyl(diphenyl)silyl]oxy} ethoxy)cyclohexancarbonsäuremethylester
Das Rohprodukt aus Beispiel 2A wird in 20 ml Dichlormethan gelöst und mit 0.83 ml (5.93 mmol) Triethylamin sowie 1.54 ml (5.93 mmol) tert.-Butyldiphenylchlorsilan versetzt. Nach Zugabe von 24.2 mg (0.20 mmol) 4-NN-Dimethylaminopyridin wird 20 h bei RT gerührt. Der Ansatz wird danach mit je 10 ml Ethylacetat und ges. wässriger Νatriumhydrogencarbonat-Lösung versetzt. Nach Trennen der Phasen werden die vereinigten organischen Phasen über Magnesiumsulfat getrocknet. Das Lösungsmittel wird am Rotationsverdampfer entfernt und der Rückstand an Kiesel- gel 60 chromatographisch gereinigt (Laufmittel: Gradient Cyclohexan/Ethylacetat 100:1 — » 20:1).
Ausbeute: 1.90 g (87% d. Th., cis/trans-Gemisch, 87% Reinheit)
GC-MS (Methode 8): R, = 10.36 min und 10.44 min; MS (ESIpos): m/z = 458 [M+NHJ1".
Beispiel 4A
[4-(2-{[terΛ-Butyl(diphenyl)silyl]oxy}ethoxy)cyclohexyl]methanol
1.90 g (3.75 mmol, 87% Reinheit) der Verbindung aus Beispiel 3A werden in 15 ml Diethylether gelöst und zu einer Suspension von 170.9 mg (4.50 mmol) Lithiumaluminiumhydrid in 15 ml Diethylether getropft. Anschließend wird 20 h bei RT gerührt. Es werden weitere 136.7 mg (3.60 mmol) Lithiumaluminiumhydrid zugegeben und die Mischung erneut für 20 h bei RT gerührt. Der Ansatz wird dann mit 257 μl Wasser und 257 μl 15%-iger Kalilauge verrührt. Es werden weitere 10 ml Wasser zugegeben und die Phasen getrennt. Die organische Phase wird über Magnesiumsulfat getrocknet. Das Lösungsmittel wird am Rotationsverdampfer entfernt und der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Cyclohexan/Ethylacetat 50: 1 — > 10:1).
Ausbeute: 1.20 g (72% d. Th., 93% Reinheit, cis/trans-Gemisch)
1H-NMR (400 MHz, DMSO-d6): δ = 7.71-7.63 (m, 4H), 7.49-7.39 (m, 6H), 4.43-4.30 (m, IH), 3.79-3.70 (m, 2H), 3.54-3.46 (m, 2H), 3.21-3.14 (m, 2H), 2.00-1.56 (m, 3H), 1.48-0.78 (m, 6H), 0.98 (s, 9H).
LC-MS (Methode 12): R, = 3.10 min und 3.16 min; MS (ESIpos): m/z = 435 [M+Na]+.
Beispiel 5A
4-(2-{[tert.-Butyl(diphenyl)silyl]oxy}ethoxy)cyclohexancarbaldehyd
553.7 mg (4.36 mmol) Oxalsäuredichlorid werden in 25 ml Dichlormethan gelöst und auf -78°C gekühlt. Es werden langsam 681.6 mg (8.72 mmol) Dimethylsulfoxid zugegeben. Anschließend wird eine Lösung von 1.20 g (2.91 mmol) der Verbindung aus Beispiel 4A in 10 ml Dichlormethan zugetropft. Es wird 1 h bei -78°C nachgerührt. Man versetzt dann mit 2.0 ml (14.54 mmol) Tn- ethylamin und lässt innerhalb von 1 h auf RT erwärmen. Der Ansatz wird danach auf 20 ml eines l :l-Gemisches aus ges. wässriger Natriumhydrogencarbonat-Lösung und Ethylacetat gegeben. Nach Trennen der Phasen wird die wässrige Phase noch zweimal mit je 10 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden am Rotationsverdampfer vom Lösungsmittel befreit. Der Rückstand wird an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Cyclohexan/Ethylacetat 20:1 → 5:1).
Ausbeute: 1.00 g (84% d. Th., cis/trans-Geπήsch)
1H-NMR (400 MHz, DMSO-d6): δ = 9.57, 9.53 (2 s, IH, cis/trans), 7.70-7.62 (m, 4H), 7.49-7.38 (m, 6H), 3.78-3.71 (m, 2H), 3.56-3.42 (m, 3H), 2.37-2.28 (m, IH), 1.85-1.74 (m, IH), 1.74-1.48 (m, 5H), 1.29-1.15 (m, 2H), 0.98 (s, 9H).
GC-MS (Methode 8): R, = 9.95 min und 9.98 min; MS (ESIpos): m/z = 428 [M+NH4]+.
Beispiel 6A
2-Amino-4-[4-(2-{[/err.-butyl(diphenyl)silyl]oxy}ethoxy)cyclohexyl]-6-mercaptopyridin-3,5-di- carbonitril
1.00 g (2.44 mmol) der Verbindung aus Beispiel 5 A werden in 10 ml trockenem Ethanol gelöst und mit 512.1 mg (5.11 mmol) Cyanothioacetamid sowie 517.3 mg (5.11 mmol) 4-Methylmorpho- Hn versetzt. Die Reaktionsmischung wird 6 h unter Rückfluss und anschließend für 20 h bei RT gerührt. Das Lösungsmittel wird danach am Rotationsverdampfer entfernt. 290 mg des erhaltenen Rohprodukts werden direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 — > 95:5). Man erhält dabei die cis/trans-lsoτneτe in reiner Form. Durch wiederholte HPLC-Trennungen kann auch der Rest des Rohprodukts gereinigt werden; es wird jedoch für die folgende Reaktion als Isomerengemisch eingesetzt.
trans-lsomeτ:
Ausbeute: 14 mg (1% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 12.89 (br. s, IH), 8.09-7.60 (br. s, 2H), 7.71-7.62 (m, 4H), 7.50-7.39 (m, 6H), 3.78-3.71 (m, 2H), 3.61-3.53 (m, 2H), 2.93-2.78 (m, IH), 2.08-1.96 (m, 4H), 1.86-1.70 (m, 2H), 1.27-1.10 (m, 2H), 0.99 (s, 9H).
LC-MS (Methode 5): R, = 4.55 min; MS (ESIpos): m/z = 574 [M+NH4]+.
m-Isomer:
Ausbeute: 33 mg (2% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 12.87 (br. s, IH), 7.82-7.56 (br. s, 2H), 7.70-7.62 (m, 4H), 7.48-7.39 (m, 6H), 3.84-3.78 (m, 2H), 3.64-3.60 (m, IH), 3.54-3.48 (m, 2H), 2.93-2.83 (m, IH), 2.38-2.23 (m, 2H), 2.02-1.89 (m, 2H), 1.52-1.36 (m, 4H), 0.98 (s, 9H).
LC-MS (Methode 5): R, = 4.65 min; MS (ESIneg): m/z = 555 [M-H]".
Beispiel 7 A
2-Amino-4-[4-(2-{[fe^.-butyl(diphenyl)silyl]oxy}ethoxy)cyclohexyl]-6-({[2-(4-chlθφhenyl)-l,3- thiazol-4-yl]methyl}thio)pyridin-3,5-dicarbonitril
300 mg (0.32 mmol) der Verbindung aus Beispiel 6A (Isomerengemisch) werden in 2.4 ml trockenem DMF gelöst, mit 95 mg (0.39 mmol) 4-(Chlormethyl)-2-(4-chlorphenyl)-l,3-thiazol sowie 109 mg Natriumhydrogencarbonat versetzt und 8 h bei RT gerührt. Das Gemisch wird danach direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5), wobei die cis/trans-lsomere getrennt werden.
trans-lsomer.
Ausbeute: 35 mg (14% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.21-7.82 (br. s, 2H), 7.95 (d, 2H), 7.89 (s, IH), 7.70-7.62 (m, 4H), 7.56 (d, 2H), 7.50-7.40 (m, 6H), 4.58 (s, 2H), 3.73 (t, 2H), 3.56 (t, 2H), 2.89-2.78 (m, IH), 2.13-2.05 (m, 2H), 2.05-1.92 (m, 2H), 1.80-1.70 (m, 2H), 1.23-1.11 (m, 2H), 0.99 (s, 9H).
LC-MS (Methode 4): R, = 3.76 min; MS (ESIpos): m/z = 764 [M+H]+.
m-Isomer:
Ausbeute: 43 mg (17% d. Th.)
1H-NMR (400 MHz, DMSO-(I6): δ = 8.16-7.22 (br. s, 2H), 7.96-7.90 (m, 2H), 7.88 (s, IH), 7.69- 7.60 (m, 4H), 7.55 (d, 2H), 7.49-7.38 (m, 6H), 4.57 (s, 2H), 3.80 (t, 2H), 3.61 (br. s, IH), 3.50 (t, 2H), 2.94-2.82 (m, IH), 2.37-2.20 (m, 2H), 2.01-1.91 (m, 2H), 1.48-1.38 (m, 4H), 0.99 (s, 9H).
LC-MS (Methode 4): R1 = 3.80 min; MS (ESIpos): m/z = 764 [M+H]+.
Beispiel 8A
4-Hydroxycyclohexancarbonsäuremethylester
5.00 g (34.68 mmol) 4-Hydroxycyclohexancarbonsäure werden in 80 ml Methanol gelöst und langsam mit 2 ml konz. Schwefelsäure versetzt. Es wird 20 h unter Rückfluss gerührt. Nach Abkühlen auf RT wird der Ansatz vorsichtig in ein Gemisch aus 100 ml Ethylacetat und 100 ml ges. wäss- riger Natriumhydrogencarbonat-Lösung gegossen. Die Phasen werden getrennt, und die organische Phase wird einmal mit 20 ml ges. wässriger Arnmoniumchlorid-Lösung gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Rohprodukt wird ohne weitere Reinigung in der Folgeraktion eingesetzt.
Ausbeute: 4.5 g (74% d. Th., 90% Reinheit)
LC-MS (Methode 13): R, = 1.17 min; MS (ESIpos): m/z = 159 [M+H]+.
Beispiel 9A
Methyl 4- { [tert.-butyl(dimethyl)silyl]oxy} cyclohexancarboxylat
2.00 g (11.38 mmol) des Rohprodukts aus Beispiel 8A (90% Reinheit) werden in 50 ml trockenem DMF gelöst und mit 1.47 g (21.62 mmol) Imidazol sowie 2.40 g (15.93 mmol) tert.-Butyldimethyl- silylchlorid versetzt. Das Reaktionsgemisch wird 20 h bei RT gerührt. Der Ansatz wird danach mit je 40 ml Diethylether und ges. wässriger Natriumhydrogencarbonat-Lösung versetzt. Nach Phasen- trennung wird die organische Phase über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Rohprodukt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 3.9 g (100% d. Th., 80% Reinheit)
GC-MS (Methode 9): R, = 7.30 min; MS (ESIpos): m/z = 215 [M-C4H9J+.
Beispiel IQA
(4-{[ter<.-Butyl(dimethyl)silyl]oxy}cyclohexyl)methanol
200 mg (0.59 mmol) des Rohprodukts aus Beispiel 9A (80% Reinheit) werden in 1.5 ml Diethylether gelöst. Diese Lösung wird bei RT zu einer Suspension von 13.4 mg (0.35 mmol) Lithium- aluminiumhydrid in 1.5 ml Diethylether getropft. Es wird 20 h bei RT gerührt. Der Ansatz wird danach mit 12 μl Wasser, 12 μl 15%-iger Kalilauge sowie 3 ml Diethylether versetzt und 30 min bei RT gerührt. Das Reaktionsgemisch wird anschließend über eine Kartusche, die mit 2.4 g Kieselgel und 2.4 g Extrelut beladen ist, filtriert (Laufmittel: Dichlormethan/Ethanol 10:1). Das Lösungsmittel wird am Rotationsverdampfer entfernt und der Rückstand an Kieselgel 60 chroma- tographisch gereinigt (Laufmittel: Gradient Cyclohexan/Ethylacetat 20:1 — > 10:1).
Ausbeute: 88 mg (61% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 4.31 (t, IH), 3.95-3.89 (m, IH), 3.17 (t, 2H), 1.58-1.49 (m, 2H), 1.43-1.20 (m, 7H), 0.83 (s, 9H), 0.01 (s, 6H).
GC-MS (Methode 9): Rt = 6.94 min; MS (ESIpos): m/z = 245 [M+H]+.
Beispiel IIA
4- { [tert.-Butyl(dimethyl)silyl]oxy} cyclohexancarbaldehyd
0.93 g (7.31 mmol) Oxalsäuredichlorid werden in 50 ml Dichlormethan gelöst, auf -78°C gekühlt und langsam mit 1.14 g (14.63 mmol) Dimethylsulfoxid versetzt. Anschließend wird eine Lösung von 1.49 g (4.88 mmol) der Verbindung aus Beispiel 10A (inkl. Nachsynthesen), gelöst in 10 ml Dichlormethan, zugegeben. Das Reaktionsgemisch wird 1 h bei -78°C nachgerührt. Danach wird langsam mit 3.4 ml (24.38 mmol) Triethylamin versetzt und innerhalb von 1 h auf RT erwärmt. Der Ansatz wird dann mit 50 ml Ethylacetat und 30 ml Natriumhydrogencarbonat-Lösung versetzt. Die Phasen werden getrennt, und die wässrige Phase wird zweimal mit je 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden über Magnesiumsulfat getrocknet. Das Lösungsmittel wird am Rotationsverdampfer entfernt und der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Cyclohexan/Ethylacetat 50:1 — > 10:1).
Ausbeute: 690 mg (56% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 9.53 (s, IH), 3.91-3.82 (m, IH), 2.38-2.28 (m, IH), 1.80-1.21 (m, 8H), 0.87 (s, 9H), 0.02 (s, 6H).
GC-MS (Methode 9): R, = 6.59 min; MS (ESIpos): m/z = 185 [M-C4H9J+.
Beispiel 12A
2-Amino-4-(4-{[/er/.-butyl(dimethyl)silyl]oxy}cyclohexyl)-6-mercaptopyridin-3,5-dicarbonitril
690 mg (2.85 mmol) der Verbindung aus Beispiel I IA und 598 mg (5.98 mmol) Cyanothioacet- amid werden in 20 ml Ethanol gelöst und mit 604 mg (5.98 mmol) N-Methylmorpholin versetzt. Das Reaktionsgemisch wird 4 h bei +900C gerührt. Nach Abkühlen auf RT wird für weitere 20 h bei RT nachgerührt. Der ausgefallene Niederschlag wird abgesaugt und mit 3 ml auf O0C gekühltem Ethanol gewaschen. Das gemeinsame Filtrat wird am Rotationsverdampfer eingeengt und der Rückstand über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 — » 95:5), wobei die cis/trans-lsomeτe getrennt werden.
m-Isomer:
Ausbeute: 197 mg (17% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 12.89 (s, IH), 7.99-7.57 (br. s, 2H), 4.10-4.04 (m, IH), 2.91- 2.81 (m, IH), 2.51-2.32 (m, IH), 1.79-1.69 (m, 2H), 1.57-1.39 (m, 6H), 0.90 (s, 9H), 0.08 (s, 6H).
LC-MS (Methode 4): R, = 3.10 min; MS (ESIpos): m/z = 389 [M+H]+.
transAsomeτ:
Ausbeute: 23 mg (2% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 12.99-12.82 (br. s, IH), 8.10-7.57 (br. s, 2H), 3.68-3.54 (m, IH), 2.88-2.77 (m, IH), 2.15-1.92 (m, 4H), 1.83-1.69 (m, 2H), 1.37-1.21 (m, 2H), 0.87 (s, 9H), 0.08 (s, 6H).
LC-MS (Methode 13): R, = 2.88 min; MS (ESIpos): m/z = 389 [M+H]+.
Beispiel 13A
2-Amino-4-(rran5-4-{[rert.-butyl(dimethyl)silyl]oxy}cyclohexyl)-6-({[2-(4-chlorphenyl)-l,3- thiazol-4-yl]methyl}thio)pyridin-3,5-dicarbonitril
23 mg (0.06 mmol) der Verbindung aus Beispiel 12A (trans-lsomef), 17 mg (0.07 mmol) 4-(Chlor- methyl)-2-(4-chlorphenyl)-l,3-thiazol und 20 mg (0.24 mmol) Natriumhydrogencarbonat werden in 2 ml trockenem DMF vorgelegt und 20 h bei RT gerührt. Der Ansatz wird danach direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Aceto- nitril/Wasser 10:90 → 95:5).
Ausbeute: 25 mg (71% d. Th.)
LC-MS (Methode 5): R1 = 5.33 min; MS (ESIpos): m/z = 596 [M+H]+.
Beispiel 14A
2-Amino-4-(cw-4-{[tert.-butyl(dimethyl)silyl]oxy}cyclohexyl)-6-({[2-(4-chloφhenyl)-l,3-thiazol- 4-yl]methyl}thio)pyridin-3,5-dicarbonitril
35 mg (0.08 mmol) der Verbindung aus Beispiel 12A (cw-Isomer), 23 mg (0.09 mmol) 4-(Chlor- methyl)-2-(4-chlθφhenyl)-l,3-thiazol und 26 mg (0.31 mmol) Natriumhydrogencarbonat werden in 2 ml trockenem DMF vorgelegt und 20 h bei RT gerührt. Der Ansatz wird danach direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Aceto- nitril/Wasser 10:90 → 95:5).
Ausbeute: 24 mg (51% d. Th.)
LC-MS (Methode 5): R1 = 5.40 min; MS (ESIpos): m/z = 596 [M+H]+.
Beispiel 15A
2-Amino-4-(cw-4- {[tert.-butyl(dimethyl)silyl]oxy} cyclohexyl)-6-[(pyridin-3-ylmethyl)thio]- pyridin-3 , 5 -dicarbonitril
97 mg (0.23 mmol) der Verbindung aus Beispiel 12A (αs-Isomer), 46 mg (0.28 mmol) 3-Pyridin- methylchlorid-Hydrochlorid und 78 mg (0.93 mmol) Natriumhydrogencarbonat werden in 2 ml trockenem DMF vorgelegt und 20 h bei RT gerührt. Der Ansatz wird danach filtriert und das FiI- trat direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 80 mg (72% d. Th.)
1H-NMR (500 MHz, DMSO-d6): δ = 8.70 (s, IH), 8.38 (d, IH), 8.20-7.72 (br. s, 2H), 7.87 (d, IH), 7.30-7.25 (m, IH), 4.39 (s, 2H), 4.00 (s, IH), 2.85-2.77 (m, IH), 2.36 (dq, 2H), 1.71-1.62 (m, 2H), 1.52-1.43 (m, 2H), 1.41-1.34 (m, 2H), 0.83 (s, 9H), 0.01 (s, 6H).
LC-MS (Methode 12): Rt = 3.03 min; MS (ESIpos): m/z = 480 [M+H]+.
Beispiel 16A
2-Amino-4-(cw-4-{[/erΛ-butyl(dimethyl)silyl]oxy}cyclohexyl)-6-[({2-[(4-fluoφhenyl)amino]-l,3- thiazol-4-yl } methyl)thio]pyridin-3 , 5 -dicarbonitril
62
- 53 -
43 mg (0.26 mmol) 4-Fluorphenylthioharnstoff und 31 mg (0.24 mmol) 1,3-Dichloraceton werden in 2 ml DMF gelöst und 3 h bei +800C gerührt. Nach Abkühlen auf RT wird das Reaktionsgemisch mit 93 mg (0.23 mmol) der Verbindung aus Beispiel 12A (m-Isomer) sowie 78 mg (0.93 mmol) Natriumhydrogencarbonat versetzt und 20 h bei RT gerührt. Der Ansatz wird danach direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Aceto- nitril/Wasser 10:90 → 95:5).
Ausbeute: 66 mg (43% d. Th.)
1H-NMR (500 MHz, DMSO-d6): δ = 10.22 (s, IH), 8.05-7.87 (br. s, 2H), 7.63-7.56 (m, 2H), 7.12 (t, 2H), 6.92 (s, IH), 4.40 (s, 2H), 4.05 (br. s, IH), 2.91-2.82 (m, IH), 2.48-2.71 (m, 2H), 1.76-1.68 (m, 2H), 1.58-1.47 (m, 2H), 1.47-1.39 (m, 2H), 0.89 (s, 9H), 0.06 (s, 6H).
LC-MS (Methode 13): R, = 3.49 min; MS (ESIpos): m/z = 595 [M+H]+.
Beispiel 17A
tert. -Butyl 4-(2-amino-3,5-dicyano-6-mercaptopyridin-4-yl)piperidin-l-carboxylat
3.00 g (14.07 mmol) tert.-Butyl 4-formylpiperidin-l-carboxylat und 2.82 g (28.13 mmol) Cyano- thioacetamid werden in 32 ml Ethanol gelöst und mit 2.85 g (28.13 mmol) 4-Methylmorpholin versetzt. Es wird 4 h unter Rückfluss und anschließend weitere 8 h bei RT gerührt. Das Reaktions- gemisch wird danach am Rotationsverdampfer eingeengt und der Rückstand an Kieselgel 60 chromatographisch aufgereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 50:1 — » 5:1). Das erhaltene Produkt wird ohne weitere Reinigung in den Folgereaktionen eingesetzt.
Ausbeute: 2.59 g (32% d. Th., 63% Reinheit)
LC-MS (Methode 13): R, = 2.02 min; MS (ESIneg): m/z = 358 [M-H]".
Beispiel 18A
tert.-Butyl 4-{2-amino-3,5-dicyano-6-[({2-[(4-fluoφhenyl)amino]-l,3-thiazol-4-yl}methyl)thio]- pyridin-4-yl } piperidin- 1 -carboxylat
100 mg (0.20 mmol) der Verbindung aus Beispiel 17A (70% Reinheit) und 130 mg (0.24 mmol) 4- (Chlormethyl)-N-(4-fluorphenyl)-l,3-thiazol-2-amin (aus 4-Fluorphenylthioharnstoff und 1,3-Di-
chloraceton) werden in 2.5 ml trockenem DMF gelöst und mit 100 mg (0.98 mmol) Natriumhydro- gencarbonat versetzt. Das Reaktionsgemisch wird 8 h bei RT gerührt. Nach Filtration wird das Filtrat im Vakuum eingeengt und der verbleibende Rückstand über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 40 mg (32% d. Th., 88% Reinheit)
LC-MS (Methode 13): R, = 2.89 min; MS (ESIpos): m/z = 566 [M+H]+.
Beispiel 19A
/erf.-Butyl 4-[2-amino-6-({[2-(4-chlorphenyl)-l,3-thiazol-4-yl]methyl}thio)-3,5-dicyanopyridin-4- yl]piperidin-l -carboxylat
Die Titelverbindung wird analog zu Beispiel 18A ausgehend von Beispiel 17A und 4-(Chlor- methyl)-2-(4-chlorphenyl)-l ,3-thiazol erhalten.
Ausbeute: 41% d. Th.
LC-MS (Methode 13): R, = 3.20 min; MS (ESIpos): m/z = 567 [M+H]+.
Beispiel 2OA
4-({[tert.-Butyl(diphenyl)silyl]oxy}methyl)piperidin
5.00 g (43.41 mmol) 4-Piperidinmethanol werden in 400 ml Dichlormethan gelöst und nacheinander mit 9.1 ml (65.11 mmol) Triethylamin, 212 mg (1.74 mmol) 4-NN-Dimethylaminopyridin und 16.9 ml (65.12 mmol) fert.-Butyldiphenylchlorsilan versetzt. Das Reaktionsgemisch wird 8 h bei RT gerührt. Nach Zugabe von 50 ml Dichlormethan wird einmal mit 20 ml Wasser und einmal mit 20 ml ges. wässriger Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet. Das Rohprodukt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 21.16 g (96% d. Th., 70% Reinheit)
LC-MS (Methode 4): R1 = 1.91 min; MS (ESIpos): m/z = 354 [M+H]+.
Beispiel 21A
2-[4-( { [tert.-Butyl(diphenyl)silyl]oxy} methyl)piperidin- 1 -yl]-2-oxoethyl-acetat
5 g des Rohprodukts aus Beispiel 20A werden in 24 ml Dichlormethan suspendiert und auf 00C gekühlt. Es werden 1.88 g (13.79 mmol) Acetoxyessigsäurechlorid und 5.9 ml (42.42 mmol) Tri-
ethylamin zugegeben. Das Reaktionsgemisch wird 8 h bei RT gerührt. Nach Zugabe von 25 ml Di- chlormethan wird einmal mit 10 ml Wasser und einmal mit 10 ml ges. wässriger Natriumchlorid- Lösung gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittel am Rotationsverdampfer wird der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Cyclohexan/Ethylacetat 20: 1 — > 1 : 1).
Ausbeute: 4.04 g (84% d. Th.)
LC-MS (Methode 4): R, = 3.21 min; MS (ESIpos): m/z = 454 [M+H]+.
Beispiel 22A
2-[4-(Hydroxyrnethyl)piperidin-l-yl]-2-oxoethyl-acetat
4.04 g (8.91 mmol) der Verbindung aus Beispiel 21 A werden in 69 ml trockenem THF gelöst und mit 9.8 ml (9.8 mmol) einer 1 M Lösung von Tetrabutylammoniumfluorid in THF versetzt. Das Reaktionsgemisch wird 48 h bei RT gerührt. Das Lösungsmittel wird danach am Rotationsverdampfer entfernt und der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 200: 1 → 1 : 1 ).
Ausbeute: 800 mg (42% d. Th.)
GC-MS (Methode 8): R, = 6.59 min; MS (ESIpos): m/z = 216 [M+H]+.
Beispiel 23A
2-(4-Formylpiperidin- 1 -yl)-2-oxoethyl-acetat
600 mg (2.79 mmol) der Verbindung aus Beispiel 22A werden in 6 ml trockenem Dichlormethan gelöst und mit 1.5 g gepulvertem Molekularsieb (4Ä) und 490 mg (4.18 mmol) N-Methylmorpho- lin-N-oxid versetzt. Anschließend werden 49 mg (0.14 mmol) Tetrapropylammoniumperruthenat zugegeben und der Ansatz 1 h bei RT gerührt. Das Reaktionsgemisch wird danach direkt an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 100:1 — > 20:1).
Ausbeute: 194 mg (33% d. Th.)
GC-MS (Methode 8): R, = 6.78 min; MS (ESIpos): m/z = 214 [M+H]+.
Beispiel 24A
2-[4-(2-Amino-3,5-dicyano-6-mercaptopyridin-4-yl)piperidin-l-yl]-2-oxoethyl-acetat
194 mg (0.91 mmol) der Verbindung aus Beispiel 23A und 182 mg (1.82 mmol) Cyanothioacet- amid werden in 2 ml trockenem Ethanol vorgelegt und mit 184 mg (1.82 mmol) 4-Methylmorpho- lin versetzt. Das Reaktionsgemisch wird 4 h bei +78°C gerührt. Nach Abkühlen auf RT wird weitere 8 h bei dieser Temperatur nachgerührt. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand an Kieselgel 60 chromatographisch aufgereinigt (Laufmittel: Gra-
dient Dichlormethan/Ethanol 20:1 -> 1 :1). Das erhaltene Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 102 mg (17% d. Th., 55% Reinheit)
LC-MS (Methode 12): R, = 1.13 min; MS (ESIpos): m/z = 360 [M+H]+.
Beispiel 25A
2-{4-[2-Amino-6-({[2-(4-chlorphenyl)-l,3-thiazol-4-yl]methyl}thio)-3,5-dicyanopyridin-4-yl]- piperidin- 1 -yl } -2-oxoethyl-acetat
34 mg (0.06 mmol) der Verbindung aus Beispiel 24A werden in 1.2 ml trockenem DMF gelöst und mit 17 mg (0.07 mmol) 4-(Chlormethyl)-2-(4-chlorphenyl)-l,3-thiazol sowie 19 mg (0.23 mmol) Natriumhydrogencarbonat versetzt. Das Reaktionsgemisch wird 8 h bei RT gerührt. Der Ansatz wird danach filtriert und das Filtrat mit ca. 0.5 ml Wasser versetzt. Der ausgefallene Niederschlag wird abgesaugt und im Vakuum bei +5O0C getrocknet.
Ausbeute: 29 mg (74% d. Th., 88% Reinheit)
LC-MS (Methode 3): R1 = 2.58 min; MS (ESIpos): m/z = 567 [M+H]+.
Beispiel 26A
3-[4-({[terΛ-Butyl(diphenyl)silyl]oxy}methyl)piperidin-l-yl]propyl-acetat
5.00 g (12.44 mmol) der Verbindung aus Beispiel 2OA werden in 57 ml Acetonitril gelöst und mit 4.51 g (24.89 mmol) 3-Brompropylacetat sowie 3.44 g (24.89 mmol) Kaliumcarbonat versetzt. Das Reaktionsgemisch wird 8 h bei +800C gerührt. Nach Filtration wird das Lösungsmittel am Rota- tionsverdampfer entfernt. Der Rückstand wird in 50 ml Ethylacetat aufgenommen und jeweils einmal mit 10 ml Wasser und 10 ml ges. wässriger Natriumhydrogencarbonat-Lösung gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Rohprodukt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 6.38 g (93% d. Th., 82% Reinheit)
LC-MS (Methode 2): R, = 4.64 min; MS (ESIpos): m/z = 454 [M+H]+.
Beispiel 27A
3-[4-(Hydroxymethyl)piperidin-l-yl]propyl-acetat
6.38 g (14.06 mmol) des Rohprodukts aus Beispiel 26A werden in 108 ml trockenem THF gelöst und mit 4.04 g (15.47 mmol) Tetra-«-butylammoniumfluorid versetzt. Das Reaktionsgemisch wird 48 h bei RT gerührt. Nach Entfernen des Lösungsmittels wird der Rückstand direkt an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 20:1 -» 1 :1).
Ausbeute: 1.59 g (53% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 4.39 (t, IH), 4.00 (t, 2H), 3.22 (t, 2H), 2.89-2.78 (m, 2H), 2.37-2.24 (m, 2H), 2.00 (s, 3H), 1.90-1.78 (m, 2H), 1.76-1.67 (m, 2H), 1.66-1.53 (m, 2H), 1.39- 1.23 (m, IH), 1.17-1.02 (m, 2H).
MS (ESIpos): m/z = 216 [M+H]+.
Beispiel 28A
3-(4-Formylpiperidin-l-yl)propyl-acetat
1.36 g (6.30 mmol) der Verbindung aus Beispiel 27A werden in 14 ml trockenem Dichlormethan gelöst und nacheinander mit 3.39 g Molekularsieb (4Ä), 1.11 g (9.44 mmol) N-Methylmorpholin- N-oxid sowie 111 mg (0.32 mmol) Tetrapropylammoniumperruthenat versetzt. Das Reaktionsgemisch wird 1 h bei RT gerührt und dann direkt an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 100: 1 → 10:1).
Ausbeute: 634 mg (48% d. Th.)
GC-MS (Methode 8): R, = 5.53 min; MS (ESIpos): m/z = 214 [M+H]+.
Beispiel 29A
3-[4-(2-Amino-3,5-dicyano-6-mercaptopyridin-4-yl)piperidin-l-yl]propyl-acetat
508 mg (2.38 mmol) der Verbindung aus Beispiel 28A und 477 mg (4.77 mmol) Cyanothioacet- amid werden in 5 ml Ethanol vorgelegt und mit 482 mg (4.77 mmol) 4-Methylmorpholin versetzt. Das Reaktionsgemisch wird 4 h bei +78°C gerührt. Nach Abkühlen auf RT wird für weitere 8 h bei dieser Temperatur nachgerührt. Das Lösungsmittel wird am Rotationsverdampfer entfernt, der Rückstand auf Diatomeenerde aufgezogen und dann an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 50: 1 → 3:1).
Ausbeute: 435 mg (48% d. Th.)
LC-MS (Methode 14): R, = 2.09 min; MS (ESIpos): m/z = 360 [M+H]+.
Beispiel 3OA
Tetrahydro-2H-pyran-2-carbaldehyd
1000 mg (8.61 mmol) 2-(Ηydroxymethyl)-tetrahydro-2H-pyran werden in 27 ml trockenem Di- chlormethan gelöst und mit 1.5 g gepulvertem Molekularsieb (4Ä) sowie 1.5 g (12.91 mmol) N- Methylmorpholin-N-oxid versetzt. Anschließend werden 151 mg (0.43 mmol) Tetrapropylammo- niumperruthenat zugegeben und das Reaktionsgemisch 1 h bei RT gerührt. Nach Entfernen des Lösungsmittels wird der Ansatz an Kieselgel 60 chromatographisch vorgereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 200:1 → 20:1). Das erhaltene Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 502 mg (51 % d. Th.)
GC-MS (Methode 8): R1 = 2.07 min; MS (ESIpos): m/z = 114 [M+H]+.
Beispiel 31A
2-Amino-6-mercapto-4-(tetrahydro-2H-pyran-2-yl)pyridin-3,5-dicarbonitril
370 mg (2.66 mmol) des Rohprodukts aus Beispiel 30A und 921 mg (9.20 mmol) Cyanothioacet- amid werden in 8.2 ml Ethanol gelöst und mit 930 mg (9.20 mmol) 4-Methylmorpholin versetzt. Das Reaktionsgemisch wird 3 h bei +78°C gerührt. Nach Abkühlen auf RT wird für weitere 8 h bei dieser Temperatur nachgerührt. Nach Entfernen des Lösungsmittels wird der Ansatz direkt an Kieselgel 60 chromatographisch vorgereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 200:1 — » 20: 1 ). Das erhaltene Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 387 mg (26% d. Th., 76% Reinheit)
LC-MS (Methode 7): R, = 1.93 min; MS (ESIpos): m/z = 261 [M+H]+.
Beispiel 32A
2-Amino-6-mercapto-4-(tetrahydro-2H-pyran-3-yl)pyridin-3,5-dicarbonitril
670 mg (5.87 mmol) Tetrahydro-2H-pyran-3-carbaldehyd [herstellbar gemäß E. J. Corey et al., J. Am. Chem. Soc. 120, 13000-13001 (1998)] und 1.23 g (12.33 mmol) Cyanothioacetamid werden in 11 ml Ethanol gelöst und mit 1.23 g (12.33 mmol) 4-Methylmorpholin versetzt. Das Reaktionsgemisch wird 3 h bei +78°C gerührt. Nach Abkühlen auf RT wird für weitere 8 h bei dieser Tem-
peratur nachgerührt. Dabei fällt ein gelber Niederschlag aus. Dieser wird abfiltriert, auf Diatomeenerde adsorbiert und an Kieselgel 60 chromatographisch vorgereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 200:1 → 1:1). Das erhaltene Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 564 mg (25% d. Th., 69% Reinheit)
LC-MS (Methode 7): R, = 1.62 min; MS (ESIpos): m/z = 261 [M+H]\
Beispiel 33A
2-Amino-4-cyclohexyl-6-mercaptopyridin-3,5-dicarbonitril
4.00 g (35.66 mmol) Cyclohexylcarbaldehyd und 7.14 g (71.32 mmol) Cyanothioacetamid werden in 80 ml Ethanol gelöst und mit 7.21 g (71.32 mmol) 4-Methylmorpholin versetzt. Das Reaktionsgemisch wird 3 h bei +78°C gerührt. Nach Abkühlen auf RT wird für weitere 8 h bei dieser Temperatur nachgerührt. Nach Absaugen des entstandenen Niederschlags wird das Filtrat am Rotationsverdampfer eingeengt und der verbleibende Rückstand an Kieselgel 60 chromatogra- phisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 100:1 — » 20:1).
Ausbeute: 7.78 g (82% d. Th.)
1H-NMR (400 MHz, DMSO-d6): 6 = 7.37 (br. s, 2H), 2.92-2.79 (m, IH), 2.09-1.92 (m, 2H), 1.90- 1.77 (m, 2H), 1.75-1.54 (m, 3H), 1.37-1.08 (m, 3H).
LC-MS (Methode 4): R, = 2.21 min; MS (ESIpos): m/z = 259 [M+H]+.
Beispiel 34A
2-Amino-6-mercapto-4-(tetrahydro-2H-pyran-4-yl)pyridin-3,5-dicarbonitril
600 mg (5.26 mmol) Tetrahydropyran-4-carbaldehyd und 1.05 g (10.50 mmol) Cyanothioacetamid werden in 10 ml Ethanol gelöst und mit 1.06 g (10.50 mmol) 4-Methylmorpholin versetzt. Das Reaktionsgemisch wird 3 h bei +800C gerührt. Der entstandene Niederschlag wird abfϊltriert und ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 720 mg (45% d. Th., 87% Reinheit)
1H-NMR (400 MHz, DMSOd6): δ = 12.94 (br. s, IH), 7.82 (br. s, 2H), 3.99 (dd, 2H), 3.38 (dd, 2H), 3.13 (m, IH), 2.29-2.19 (m, 2H), 1.60 (d, 2H).
LC-MS (Methode 12): R, = 1.15 min; MS (ESIpos): m/z = 261 [M+H]+.
Beispiel 35A
[6-(Pyridin-4-ylamino)pyridin-2-yl]methanol
1.35 g (14.3 mmol) 4-Aminopyridin und 1.34 g (7.1 mmol) (6-Brompyridin-2-yl)-methanol werden für 4 h bei 15O0C gerührt. Nach Abkühlen auf RT wird das Reaktionsgemisch mit 50 ml Aceto- nitril versetzt und 20 min gerührt. Der entstandene Niederschlag wird bei 00C abgesaugt und mit 10 ml Acetonitril gewaschen.
Ausbeute: 1.25 g (39% d. Th., 89% Reinheit)
LC-MS (Methode 14): R, = 1.76 min; MS (ESIpos): m/z = 202 [M+H]+.
Beispiel 36A
6-(Chlormethyl)-N-pyridin-4-yl-pyridin-2-amin
50 mg (0.22 mmol) der Verbindung aus Beispiel 35A und 53 mg (0.44 mmol) Thionylchlorid werden bei 00C in 1.5 ml Dichlormethan vorgelegt und nach Erwärmen auf RT 12 h bei dieser Temperatur gerührt. Das Lösungsmittel wird am Rotationsverdampfer entfernt und das verbleibende Produkt direkt weiter umgesetzt.
Ausbeute: 65 mg (99% d. Th., 74% Reinheit)
LC-MS (Methode 14): Rt = 2.26 min; MS (ESIpos): m/z = 220 [M+H]+.
Beispiel 37A
2-Chlor-6-({[2-(4-chlθφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-cyclohexylpyridm-3,5-dicarbonitril
1.63 g (13.91 mmol) Isopentylnitrit und 1.87 g (13.91 mmol) Kupfer(II)-chlorid werden in 18 ml Acetonitril vorgelegt und mit 1.08 g (2.32 mmol) der Verbindung aus Beispiel 29 versetzt. Das Reaktionsgemisch wird 3 h bei +600C gerührt. Nach Abkühlen auf RT wird das Reaktionsgemisch mit 20 ml 1 N Salzsäure versetzt. Die wässrige Phase wird zweimal mit je 30 ml Ethylacetat extra- hiert. Die vereinigten organischen Phasen werden einmal mit 15 ml ges. wässriger Natriumhydro- gencarbonat-Lösung und einmal mit 15 ml ges. wässriger Natriumchlorid-Lösung gewaschen. Nach Trocknen über Magnesiumsulfat wird das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wird an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Cyclo- hexan/Ethylacetat 100:1 → 5:1).
Ausbeute: 626 mg (56% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 7.94 (d, 2H), 7.72 (s, IH), 7.58 (d, 2H), 4.72 (s, 2H), 3.08- 2.96 (m, IH), 2.04-1.78 (m, 6H), 1.77-1.57 (m, 2H), 1.42-1.11 (m, 2H).
LC-MS (Methode 12): Rt = 3.37 min; MS (ESIpos): m/z = 485 [M+H]+.
Beispiel 38A
2-Amino-4-cyclohex-3-en-l-yl-6-mercaptopyridin-3,5-dicarbonitril
Die Titelverbindung ist analog zu Beispiel 33 A durch Umsetzung von Cyclohex-3-en-l-carbalde- hyd mit 2 Äquivalenten Cyanothioacetamid in Gegenwart von 4-Methylmorpholin erhältlich.
1H-NMR (400 MHz, CDCl3): δ = 12.91 (s, IH), 8.09-7.56 (br. s, 2H), 5.82-5.71 (m, 2H), 3.14-3.02 (m, IH), 2.61-2.52 (m, IH), 2.29-2.03 (m, 4H), 1.78 (d, IH).
LC-MS (Methode 5): R, = 2.82 min; MS (ESIpos): m/z = 257 [M+H]+.
Beispiel 39A
2-Amino-6-methyl-4-(tetrahydro-2H-pyran-2-yl)pyridin-3,5-dicarbonitril
870 mg (7.62 mmol) Tetrahydro-2H-pyran-2-carbaldehyd, 504 mg (7.62 mmol) Malonsäuredinitril und 626 mg (7.62 mmol) 3-Aminocrotonsäurenitril werden in 8.4 ml trockenem Ethanol gelöst. Die Lösung wird auf 00C gekühlt und portionsweise mit 412 mg (7.62 mmol) Natriummethylat
versetzt. Das Reaktionsgemisch wird anschließend auf RT erwärmt und 10 min gerührt. Danach wird 10 h zum Rückfluss erhitzt. Nach dem Abkühlen wird der entstandene braune Niederschlag abgesaugt, dreimal mit insgesamt 100 ml Ethanol gewaschen und anschließend im Vakuum bei 5O0C getrocknet. Das Produkt wird ohne weitere Aufreinigung in der Folgereaktion eingesetzt. Eine Reinigung kann mittels präparativer HPLC erfolgen (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 610 mg (20% d. Th., 61% Reinheit)
LC-MS (Methode 7): R, = 2.45 min; MS (ESIpos): m/z = 243 [M+H]+.
Beispiel 4OA
2-Chlor-6-methyl-4-(tetrahydro-2H-pyran-2-yl)pyridin-3,5-dicarbonitril
610 mg (2.52 mmol) der Verbindung aus Beispiel 39A werden in 44 ml Acetonitril vorgelegt und mit 590 mg (5.04 mmol) Isopentylnitrit sowie 677 mg (5.04 mmol) Kupfer(II)chlorid versetzt. Das Reaktionsgemisch wird 30 min bei 600C gerührt. Der Ansatz wird danach mit 40 ml 1 N Salzsäure versetzt und das Gemisch dreimal mit je 100 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 471 mg (53% d. Th., 75% Reinheit)
LC-MS (Methode 4): R, = 2.50 min; MS (ESIpos): m/z = 262 [M+Η]+.
Beispiel 41A
2-Methyl-6-sulfanyl-4-(tetrahydro-2H-pyran-2-yl)pyridin-3,5-dicarbonitril
471 mg (1.35 mmol) der Verbindung aus Beispiel 4OA werden in 4 ml trockenem DMF vorgelegt und mit 126 mg (1.62 mmol) Natriumsulfid versetzt. Das Reaktionsgemisch wird 12 h bei RT gerührt. Das Lösungsmittel wird anschließend am Rotationsverdampfer entfernt und der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 200:1 → 5: 1).
Ausbeute: 232 mg (64% d. Th.)
LC-MS (Methode 4): R, = 2.01 min; MS (ESIpos): m/z = 260 [M+H]+.
Beispiel 42A
4-(Chlormethyl)-2-(4-chlorphenyl)-l ,3-oxazol
408 mg (3.21 mmol) 1 ,3-Dichloraceton und 500 mg (3.21 mmol) 4-Chlorbenzamid werden vereint und 1 h bei 135°C gerührt. Anschließend wird auf RT abgekühlt, vorsichtig mit 1.1 ml konz. Schwefelsäure versetzt und 5 min nachgerührt. Das Gemisch wird dann vorsichtig auf Eis gegos- sen. Der Niederschlag wird abgesaugt und mit Wasser nachgewaschen. Nach Trocknen wird das Rohprodukt ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 426 mg (49% d. Th., 85% Reinheit)
LC-MS (Methode 14): R1 = 3.78 min; MS (ESIpos): m/z = 228 [M+H]+.
Beispiel 43A
rac-2-Chlor-6-({[2-(4-chlorphenyl)-l,3-oxazol-4-yl]methyl}sulfanyl)-4-(tetrahydro-2H-pyran-3- y l)pyridin-3 , 5 -dicarbonitri 1
244 mg (2.08 mmol) Isopentylnitrit und 280 mg (2.08 mmol) Kupfer(II)chlorid werden in 26 ml Acetonitril vorgelegt und bei RT mit 470 mg (1.04 mmol) der Verbindung aus Beispiel 58 versetzt. Es wird 3 h bei 600C gerührt. Nach Abkühlen auf RT wird die Reaktionslösung mit 20 ml 1 N Salzsäure versetzt. Es wird zweimal mit je 20 ml Ethylacetat extrahiert. Die organischen Phasen werden über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels am Rotationsver- dampfer wird der Rückstand in 20 ml Ethanol suspendiert und der Niederschlag abgesaugt. Der erhaltene Feststoff wird im Vakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung in der Folgereaktion eingesetzt. Eine Reinigung ist mittels präparativer ΗPLC möglich (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 239 mg (40% d. Th., 82% Reinheit)
LC-MS (Methode 2): R1 = 6.45 min; MS (ESIpos): m/z = 471 [M+Η]+.
Beispiel 44A
4-(Chlormethyl)-2-(4-fluor-3-methylphenyl)-l,3-oxazol
2.00 g (12.80 mmol) 4-Fluor-3-methylbenzamid und 1.79 g (14.08 mmol) 1,3-Dichloraceton werden 2 Tage lang bei 130
0C gerührt, wobei sich eine Schmelze bildet. Der Ansatz wird danach auf RT abgekühlt, bei dieser Temperatur vorsichtig mit 3.0 ml konz. Schwefelsäure versetzt und 15 min lang gerührt. Die erhaltene Suspension wird auf 20 ml Eiswasser gegossen und über Nacht bei RT gerührt. Der entstandene Niederschlag wird abfiltriert und über Nacht bei 40
0C im Vaku- umtrockenschrank getrocknet.
Ausbeute: 2.05 g (64% d. Th., 90% Reinheit)
LC-MS (Methode 7): R, = 2.05 min; MS (ESIpos): m/z = 226 [M+H]+.
Die weiteren zur Synthese von Ausführungsbeispielen eingesetzten 4-(Chlormethyl)-2-aryl-l ,3- oxazole werden aus den entsprechenden kommerziell erhältlichen Edukten auf analoge Weise hergestellt.
Beispiel 45A
2-(4-Chlorphenyl)-4,5-dimethyl-l,3-oxazol-3-oxid
1.00 g (9.89 mmol) Diacetylmonoxim und 1.53 g (10.88 mmol) 4-Chlorbenzaldehyd werden in 2 ml (34.94 mmol) Eisessig vorgelegt. Dann wird 30 min lang Chlorwasserstoff-Gas unter Eiskühlung des Reaktionsgemisches eingeleitet. Anschließend wird das Reaktionsgemisch mit 10 ml Diethylether versetzt. Es fällt ein Niederschlag aus, der abgesaugt und zweimal mit je 2 ml Di- ethylether gewaschen wird. Der Niederschlag wird in ca. 5 ml Wasser re-suspendiert und die Sus- pension mit Ammoniak basisch gestellt. Es wird dann viermal mit je 10 ml Dichlormethan extrahiert. Die vereinigten organischen Phasen werden über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 1.85 g (84% d. Th.)
LC-MS (Methode 5): R, = 2.29 min; MS (ESIpos): m/z = 224 [M+H]+.
Beispiel 46A
4-(Chlormethyl)-2-(4-chloφhenyl)-5-methyl-l,3-oxazol
1.00 g (4.47 mmol) der Verbindung aus Beispiel 45 A werden in 15 ml Chloroform vorgelegt und vorsichtig mit 1.5 ml (16.10 mmol) Phosphorylchlorid versetzt. Das Reaktionsgemisch wird 30 min unter Rühren zum Rückfluss erhitzt. Der Ansatz wird anschließend auf 00C abgekühlt und durch Zugabe von Ammoniak schwach basisch gestellt. Das Gemisch wird dreimal mit je 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden zweimal mit je 5 ml Wasser gewaschen und anschließend über Magnesiumsulfat getrocknet. Das Lösungsmittel wird am Rota- tionsverdampfer entfernt. Der Rückstand wird ohne weitere Reinigung in den Folgestufen eingesetzt.
Ausbeute: 1.33 g (96% d. Th., 78% Reinheit)
1H-NMR (400 MHz, DMSO-d6): δ = 7.95 (d, 2H), 7.60 (d, 2H), 4.77 (s, 2H), 2.44 (s, 3H).
LC-MS (Methode 3): R, = 2.80 min; MS (ESIpos): m/z = 242 [M+H]+.
Beispiel 47 A
Methyl-3-cyanobenzoat
100 mg (0.68 mmol) 3-Cyanobenzoesäure werden in 4 ml Toluol und 3.5 ml Methanol vorgelegt und bei RT mit 0.51 ml (1.02 mnmol) einer 2 M Lösung von Trimethylsilyldiazomethan in n- Hexan versetzt. Das Reaktionsgemisch wird 1.5 h bei RT gerührt. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand im Vakuum getrocknet. Das Produkt wird rein erhalten und direkt weiter umgesetzt.
Ausbeute: 1 16 mg (100% d. Th.)
LC-MS (Methode 4): R, = 1.93 min; MS (ESIpos): m/z = 162 [M+H]+.
Beispiel 48A
Methyl-3-(lH-tetrazol-5-yl)benzoat
1.00 g (6.21 mmol) der Verbindung aus Beispiel 47 A werden in 18 ml trockenem DMF vorgelegt. Anschließend werden 2.42 g (37.23 mmol) Natriumazid und 1.99 g (37.23 mmol) Ammoniumchlorid hinzugegeben. Das Reaktionsgemisch wird 2.5 h bei 1200C gerührt. Nach Abkühlen auf RT wird der Ansatz auf ein Gemisch von 30 ml Eiswasser und 10 ml Ethylacetat gegossen. Zu dieser Mischung werden 2.57 g (37.23 mmol) Natriumnitrit gegeben, um überschüssiges Azid zu vernichten. Anschließend wird durch Zugabe von 6 N Salzsäure der pH-Wert auf 1 -2 gestellt und 30 min bei RT gerührt. Die Mischung wird dreimal mit je 20 ml eines 1 : 1 -Gemisches von Ethylacetat und TΗF extrahiert. Der ausgefallene Niederschlag wird abgesaugt und im Vakuum getrocknet. Zur Isolierung weiteren Produkts wird die abgetrennte organische Phase je einmal mit je 10 ml Wasser und gesättigter wässriger Natriumchlorid-Lösung gewaschen. Nach Trocknen über Magnesiumsulfat wird das Lösungsmittel am Rotationsverdampfer entfernt. Beide Feststoffe werden vereint und in der Folgestufe eingesetzt.
Ausbeute: 1.18 g (91% d. Th.)
LC-MS (Methode 5): R, = 1.87 min; MS (ESIpos): m/z = 205 [M+Η]+.
Beispiel 49A
[3 -( 1 H-Tetrazol-5 -y l)phenyl] methanol
438 mg (11.56 mmol) Lithiumaluminiumhydrid werden in 60 ml trockenem TΗF vorgelegt, auf 00C abgekühlt und mit einer Lösung von 1.18 g (5.78 mmol) der Verbindung aus Beispiel 48A in 40 ml trockenem TΗF versetzt. Man lässt die Reaktionsmischung auf RT kommen und rührt 2 h
bei dieser Temperatur. Das Reaktionsgemisch wird dann vorsichtig bei 00C mit 4 M Salzsäure versetzt, bis kein Wasserstoff mehr entwickelt wird. Die Lösung wird danach dreimal mit je 10 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden je einmal mit je 10 ml Wasser und gesättigter wässriger Natriumchlorid-Lösung gewaschen und über Magnesiumsulfat getrock- net. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand im Vakuum getrocknet. Das Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 0.80 g (77% d. Th.)
LC-MS (Methode 5): R1 = 0.90 min; MS (ESIpos): m/z = 177 [M+H]+.
Beispiel 5OA
3-( 1 H-Tetrazol-5 -yl)benzylmethansulfonat
50 mg (0.28 mmol) der Verbindung aus Beispiel 49A werden in 5 ml trockenem Dichlormethan gelöst und mit 33 μl (0.43 mmol) Methansulfonsäurechlorid sowie 0.06 ml (0.43 mmol) Triethyl- amin versetzt. Das Reaktionsgemisch wird 10 h bei RT gerührt. Der Reaktionsansatz wird dann mit 10 ml Dichlormethan verdünnt und je einmal mit je 5 ml Wasser, 1 N Salzsäure und gesättigter wässriger Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels am Rotationsverdampfer erhält man einen Feststoff, der ohne weitere Reinigung in der Folgereaktion eingesetzt wird.
Ausbeute: 65 mg (40% d. Th., 44% Reinheit)
LC-MS (Methode 4): R, = 1.58 min; MS (ESIpos): m/z = 255 [M+Η]+.
Beispiel 51A
Methyl-ci.s-4-hydroxycyclohexancarboxylat
2.95 Liter Methanol und 9.7 ml konz. Schwefelsäure werden vorgelegt und bei RT portionsweise mit 175.0 g (1.21 mol) c/s-4-Hydroxycyclohexancarbonsäure versetzt. Es wird 16 h bei RT gerührt. Das Lösungsmittel wird danach nahezu vollständig am Rotationsverdampfer entfernt und der Rückstand in 2 Liter eines 1 : 1 -Gemisches von gesättigter wässriger Natriumhydrogencarbonat- Lösung und Ethylacetat aufgenommen. Die Phasen werden getrennt, und die organische Phase wird mit 1 Liter 10%-iger wässriger Ammoniumchlorid-Lösung gewaschen und anschließend über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Ethylacetat/ Petrolether 3:7 → 1 : 1).
Ausbeute: 128.4 g (60% d. Th., 89% Reinheit)
1H-NMR (400 MHz, CDCl3): δ = 3.89 (s, IH), 3.68 (s, 3H), 2.44-2.35 (m, IH), 2.07-1.90 (m, 2H), 1.90-1.58 (m, 8H).
GC-MS (Methode 8): R, = 4.10 min; MS (ESIpos): m/z = 140 [M-H2O]+.
Beispiel 52A
Methyl-cjs-4- { [tert.-butyl(dimethyl)silyl]oxy} cyclohexancarboxylat
99.0 g (0.63 mol) der Verbindung aus Beispiel 51 A werden in 2 Liter trockenem DMF gelöst und bei RT mit 132.0 g (0.88 mol) ter/.-Butyldimethylsilylchlorid sowie 80.9 g (1.19 mol) Imidazol versetzt. Es wird 16 h bei RT gerührt. Das Lösungsmittel wird danach am Rotationsverdampfer fast bis zur Trockene entfernt und der Rückstand in 2 Liter eines 1 : 1 -Gemisches von tert.-BuXyϊ- methylether und gesättigter wässriger Natriumhydrogencarbonat-Lösung aufgenommen. Die Phasen werden getrennt, die organische Phase über Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 160.3 g (94% d. Th.)
1H-NMR (400 MHz, CDCl3): δ = 3.89 (s, IH), 3.65 (s, 3H), 2.35-2.25 (m, IH), 1.99-1.88 (m, 2H), 1.70-1.55 (m, 4H), 1.55-1.41 (m, 2H), 0.88 (s, 9H), 0.04 (s, 6H).
GC-MS (Methode 8): R, = 4.96 min; MS (ESIpos): m/z = 273 [M+H]+.
Beispiel 53A
(cis-4- { [terr.-Butyl(dimethyl)silyl]oxy} cyclohexyl)methanol
2.8 g (73.96 mmol) Lithiumaluminiumhydrid werden in 250 ml terf.-Butylmethylether vorgelegt und bei RT tropfenweise mit einer Lösung von 22.9 g (84.05 mmol) der Verbindung aus Beispiel 52A in 250 ml terf.-Butylmethylether versetzt. Es wird 16 h bei 400C gerührt. Anschließend werden weitere 0.7 g (18.44 mmol) Lithiumaluminiumhydrid hinzugegeben und das Reaktionsgemisch 10 h zum Rückfluss erhitzt. Nach Abkühlen auf RT wird vorsichtig mit 20 ml Wasser versetzt. Anschließend werden 20 ml einer 15%-igen Lösung von Kaliumhydroxid in Wasser zugegeben. Die Phasen werden getrennt, die organische Phase über Natriumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Das erhaltene Produkt wird ohne weitere Reinigung in der Folgereaktion eingesetzt.
Ausbeute: 21.1 g (100% d. Th.)
1H-NMR (400 MHz, CDCl3): δ = 3.96-3.91 (br. s, IH), 3.48-3.40 (br. s, 2H), 1.68-1.58 (m, 2H), 1.53-1.35 (m, 8H), 0.87 (s, 9H), O.Ol (s, 6H).
GC-MS (Methode 8): R1 = 4.77 min; MS (ESIpos): m/z = 245 [M+H]+.
Beispiel 54A
ds-4-{[/erΛ-Butyl(dimethyl)silyl]oxy}cyclohexancarbaldehyd
16.4 g (129.47 mmol) Oxalsäuredichlorid werden in 600 ml Dichlormethan gelöst und auf -78°C gekühlt. Dann werden langsam 20.2 g (258.95 mmol) Dimethylsulfoxid zugetropft. Es wird 5 min nachgerührt. Nach Zugabe einer Lösung von 21.1 g (86.32 mmol) der Verbindung aus Beispiel 53A in 200 ml Dichlormethan wird 1 h bei -78°C gerührt. Dann werden langsam 60 ml (431.58 mmol) Triethylamin zugetropft. Anschließend wird innerhalb einer Stunde auf RT erwärmt und das Reaktionsgemisch danach mit 500 ml gesättigter wässriger Natriumhydrogencarbonat-Lösung versetzt. Die Phasen werden getrennt, die organische Phase über Natriumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wird an Kieselgel 60 chromato- graphisch gereinigt (Laufmittel: Gradient Petrolether — > Petrolether/Ethylacetat 9: 1).
Ausbeute: 15.6 g (75% d. Th.)
1H-NMR (400 MHz, CDCl3): δ = 3.90-3.82 (br. s, IH), 2.22-2.12 (m, IH), 1.91-1.77 (m, 2H), 1.67- 1.47 (m, 6H), 0.85 (s, 9H), 0.01 (s, 6H).
Beispiel 55A
2-Amino-4-(cjs-4-hydroxycyclohexyl)-6-sulfanylpyridin-3,5-dicarbonitril
15.6 g (64.35 mmol) der Verbindung aus Beispiel 54A und 13.53 g (135.13 mmol) Cyanothioacet- amid werden in 450 ml Ethanol vorgelegt und mit 9.86 g (135.13 mmol) 4-Methylmorpholin versetzt. Es wird 4 h zum Rückfluss erhitzt und anschließend 16 h bei RT nachgerührt. Es fällt ein Niederschlag aus, der abgesaugt wird (und der ter/.-Butyldimethylsilyl-geschützten Zielverbindung entspricht). Das Filtrat wird am Rotationsverdampfer vom Lösungsmittel befreit und der Rückstand mittels präparativer HPLC (Methode 20) aufgereinigt.
Ausbeute: 2.55 g (14% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 12.95-12.83 (br. s, IH), 8.06-7.52 (br. s, 2H), 4.88-4.46 (br. s, IH), 3.46-3.22 (m, IH), 2.84-2.73 (m, IH), 2.13-1.92 (m, 4H), 1.77-1.67 (m, 3H)1 1.28-1.12 (m, 3H).
LC-MS (Methode 21): Rt = 0.53 min; MS (ESIpos): m/z = 275 [M+H]+.
Ausführungsbeispiele:
Beispiel 1
2-Amino-6-({[2-(4-chloφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-[ira«5-4-(2-hydroxyethoxy)cyclo- hexyl]pyridin-3,5-dicarbonitril
35 mg (0.05 mmol) der Verbindung aus Beispiel 7A (trans-lsomev) werden in 5 ml trockenem THF gelöst, mit 18 mg (0.07 mmol) Tetra-«-butylammoniumfluorid versetzt und 20 h bei RT gerührt. Der Ansatz wird dann mit 15 ml Ethylacetat versetzt. Es wird zweimal mit je 3 ml ges. wässriger Natriumhydrogencarbonat-Lösung gewaschen. Die organische Phase wird über Magne- siumsulfat getrocknet. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird das Rohprodukt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 19 mg (79% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.32-7.77 (br. s, 2H), 7.92 (d, 2H), 7.88 (s, IH), 7.56 (d, 2H), 4.61-4.51 (m, IH), 4.58 (s, 2H), 3.51-3.42 (m, 4H), 3.30-3.20 (m, IH), 2.89-2.78 (m, IH), 2.19- 2.09 (m, 2H), 2.08-1.93 (m, 2H), 1.81-1.71 (m, 2H), 1.26-1.12 (m, 2H).
LC-MS (Methode 5): R, = 3.86 min; MS (ESIpos): m/z = 526 [M+H]+.
Beispiel 2
2-Amino-6-({[2-(4-chlorphenyl)-l,3-thiazol-4-yl]methyl}thio)-4-[c«-4-(2-hydroxyethoxy)cyclo- hexyl]pyridin-3,5-dicarbonitril
43 mg (0.06 mmol) der Verbindung aus Beispiel 7A (ci's-Isomer) werden in 5 ml trockenem THF gelöst, mit 22 mg (0.08 mmol) Tetra-n-butylarnmoniumfluorid versetzt und 20 h bei RT gerührt. Der Ansatz wird dann mit 15 ml Ethylacetat versetzt. Es wird zweimal mit je 3 ml ges. wässriger Natriumhydrogencarbonat-Lösung gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird das Rohprodukt zunächst über präparative HPLC (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 — » 95:5) und dann nochmals an Kieselgel 60 (Laufmittel: Gradient Di- chlormethan/Ethanol 200: 1 — > 10:1) chromatographisch gereinigt.
Ausbeute: 21 mg (68% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.18-7.82 (br. s, 2H), 7.93 (d, 2H), 7.88 (s, IH), 7.57 (d, 2H), 4.58 (s, 2H), 4.31 (t, IH), 3.61 (s, IH), 3.56-3.48 (m, 2H), 3.43-3.36 (m, 2H), 2.95-2.84 (m, IH), 2.36-2.20 (m, 2H), 2.03-1.92 (m, 2H), 1.49-1.37 (m, 4H).
LC-MS (Methode 7): R, = 3.89 min; MS (ESIpos): m/z = 526 [M+H]+.
Beispiel 3
2-Amino-6-({[2-(4-chloφhenyl)-l ,3-thiazol-4-yl]methyl}thio)-4-(ira«5-4-hydroxycyclohexyl)- pyridin-3,5-dicarbonitril
25 mg (0.04 mmol) der Verbindung aus Beispiel 13A (trans-lsomeτ) werden in 1 ml trockenem Acetonitril gelöst und mit 0.1 ml (2.30 mmol) 40%-iger Fluorwasserstoffsäure versetzt. Das Reaktionsgemisch wird 2 h bei RT gerührt. Der Ansatz wird danach direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 20 mg (99% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.21-7.81 (br. s, 2H), 7.94 (d, 2H), 7.88 (s, IH), 7.56 (d, 2H), 4.70 (d, IH), 4.58 (s, 2H), 3.48-3.37 (m, IH), 2.85-2.75 (m, IH), 2.06-1.92 (m, 4H), 1.77-1.68 (m, 2H), 1.28-1.14 (m, 2H).
LC-MS (Methode 7): R, = 3.65 min; MS (ESIpos): m/z = 482 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 3 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 7
2-Amino-6-[({2-[(4-fluoφhenyl)amino]-l,3-thiazol-4-yl}methyl)thio]-4-piperidin-4-ylpyridin-3,5- dicarbonitril
40 mg (0.07 mmol) der Verbindung aus Beispiel 18A werden in 0.9 ml Dioxan gelöst und mit 0.9 ml einer 4 M Lösung von Chlorwasserstoff in Dioxan versetzt. Das Reaktionsgemisch wird 2 h
bei RT gerührt und anschließend direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS- AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 17 mg (52% d. Th.)
1H-NMR (500 MHz, DMSO-d6): δ = 10.27 (s, IH), 8.20-7.72 (br. s, 2H), 7.61 (dd, 2H), 7.12 (dd, 2H), 6.93 (s, IH), 4.40 (s, 2H), 3.74-3.66 (m, IH), 3.53-3.37 (m, 2H), 3.04 (d, 2H), 2.96-2.87 (m, IH), 2.10-1.91 (m, 2H), 1.54 (d, 2H).
LC-MS (Methode 12): Rt = 1.52 min; MS (ESIpos): m/z = 466 [M+H]+.
Beispiel 8
2-Amino-6-({[2-(4-chloφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-piperidin-4-ylpyridin-3,5-dicarbo- nitril
Die Titelverbindung wird auf analoge Weise ausgehend von Beispiel 19A erhalten.
Ausbeute: 57% d. Th.
LC-MS (Methode 12): R, = 1.74 min; MS (ESIpos): m/z = 467 [M+H]+.
Beispiel 9
2- Amino-6-( { [2-(4-chlorphenyl)- 1 ,3-thiazol-4-yl]methyl } thio)-4-( 1 -glycoloylpiperidin-4-yl)- pyridin-3 , 5 -dicarbonitril
29 mg (0.05 mmol) der Verbindung aus Beispiel 25A werden in einem Gemisch aus 1.5 ml Dioxan und 0.8 ml Wasser gelöst und mit 2.4 mg (0.10 mmol) Lithiumhydroxid versetzt. Das Reaktionsgemisch wird zwei Stunden bei RT gerührt. Nach Entfernen des Lösungsmittels am Rotationsver- dampfer wird der Rückstand über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 13 mg (47% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.29-7.83 (br. s, 2H), 7.94 (d, 2H), 7.89 (s, IH), 7.57 (d, 2H), 4.61-4.47 (m, 2H), 4.48 (s, 2H), 4.17-4.03 (m, 2H), 3.89-3.78 (m, IH), 3.19-2.97 (m, 2H), 2.72- 2.61 (m, IH), 2.13-1.90 (m, 2H), 1.80-1.69 (m, 2H).
LC-MS (Methode 3): R, = 2.47 min; MS (ESIpos): m/z = 525 [M+H]+.
Beispiel 10
3-{4-[2-Amino-6-({[2-(4-chloφhenyl)-l,3-thiazol-4-yl]methyl}thio)-3,5-dicyanopyridin-4-yl]- piperidin- 1 -yl } propyl-acetat
60 mg (0.16 mmol) der Verbindung aus Beispiel 29A werden in 3 ml trockenem DMF gelöst und nacheinander mit 46 mg (0.19 mmol) 4-(Chlormethyl)-2-(4-chlorphenyl)-l,3-thiazol und 53 mg (0.63 mmol) Natriumhydrogencarbonat versetzt. Das Reaktionsgemisch wird 8 h bei RT gerührt. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand direkt über prä- parative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/ Wasser 10:90 -> 95:5).
Ausbeute: 65 mg (72% d. Th.)
LC-MS (Methode 3): R, = 1.77 min; MS (ESIpos): m/z = 568 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 10 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 13
2-Amino-6-({[2-(4-chloφhenyl)-l ,3-thiazol-4-yl]methyl}thio)-4-[l-(3-hydroxypropyl)piperidin-4- yl]pyridin-3,5-dicarbonitril
65 mg (0.12 mmol) der Verbindung aus Beispiel 10 werden in einem Gemisch aus 3.5 ml Dioxan und 1.7 ml Wasser vorgelegt und mit 11 mg Lithiumhydroxid versetzt. Es wird zunächst 8 h bei RT gerührt. Danach werden nochmals 22 mg Lithiumhydroxid zugegeben und die Mischung für weitere 16 h bei RT gerührt. Das Lösungsmittel wird dann am Rotationsverdampfer entfernt. Der Rückstand wird in 10 ml Ethylacetat aufgenommen und einmal mit 3 ml ges. wässriger Natrium- hydrogencarbonat-Lösung gewaschen. Es wird über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Dichlormethan/Ethanol 50: 1 — > 10:1).
Ausbeute: 12 mg (19% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.25-7.77 (br. s, 2H), 7.94 (d, 2H), 7.87 (s, IH), 7.56 (d, 2H), 4.59 (s, 2H), 4.41 (br. s, IH), 3.42 (br. s, 2H), 3.00 (d, 2H), 2.88-2.77 (m, IH), 2.40-2.29 (m, 2H), 2.24-2.10 (m, 2H), 1.97-1.85 (m, 2H), 1.70-1.52 (m, 4H).
LC-MS (Methode 5): R, = 2.67 min; MS (ESIpos): m/z = 525 [M+H]+.
Beispiel 14
rac-2-Amino-6-({[2-(4-chloφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-(tetrahydro-2H-pyran-2-yl)- pyridin-3,5-dicarbonitril
33 mg (0.10 mmol) der Verbindung aus Beispiel 31A, 29 mg (0.12 mmol) 4-(Chlormethyl)-2-(4- chloφhenyl)-l,3-thiazol und 33 mg (0.39 mmol) Natriumhydrogencarbonat werden in 2 ml trockenem DMF suspendiert. Das Reaktionsgemisch wird 20 h bei RT gerührt. Nach Filtration des Ansatzes wird direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 30 mg (66% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.27-7.82 (br. s, 2H), 7.93 (d, 2H), 7.87 (s, IH), 7.55 (d, 2H), 4.59 (s, 2H), 4.57-4.50 (m, IH), 4.01 (d, IH), 3.55-3.47 (m, IH), 1.97-1.83 (m, IH), 1.74-1.48 (m, 5H).
LC-MS (Methode 4): R, = 3.15 min; MS (ESIpos): m/z = 468 [M+H]+.
Beispiel 15 und Beispiel 16
e«r-2-Amino-6-({[2-(4-chlorphenyl)-l,3-thiazol-4-yl]methyl}thio)-4-(tetrahydro-2H-pyran-2-yl)- pyridin-3,5-dicarbonitril (Enantiomer 1 und Enantiomer 2)
9962
20 mg der Verbindung aus Beispiel 14 werden in 0.5 ml Methanol sowie 4.5 ml tert.-Butylmethyl- ether gelöst und mittels präparativer HPLC an chiraler Phase (Methode 10) in die Enantiomere getrennt:
Beispiel 15 (Enantiomer 1):
Ausbeute: 8 mg
HPLC (Methode 10): R, = 8.13 min; ee >98%.
Beispiel 16 (Enantiomer 2):
Ausbeute: 9 mg
HPLC (Methode 10): R, = 8.62 min; ee >98%.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 14 aus den entsprechenden Ausgangsverbindungen in racemischer Form hergestellt:
Beispiel 20 und Beispiel 21 ent-Ethy\ 4-[4-( { [6-amino-3 ,5-dicyano-4-(tetrahydro-2H-pyran-2-yl)pyridin-2-yl]thio} methyl)-l ,3- thiazol-2-yl]benzoat (Enantiomer 1 und Enantiomer 2)
500 mg der Verbindung aus Beispiel 19 werden bei ca. 40
0C in 35 ml 2-Propanol gelöst und mittels präparativer HPLC an chiraler Phase (Methode 11) in die Enantiomere getrennt:
Beispiel 20 (Enantiomer 1):
Ausbeute: 231 mg
HPLC (Methode 11 ): R, = 9.70 min; ee >99%
optische Drehung: -0.059° (c = 0.45 g / 100 ml, Chloroform).
Beispiel 21 (Enantiomer 2):
Ausbeute: 209 mg
HPLC (Methode 11): R1 = 11.74 min; ee >98%
optische Drehung: +0.054° (c = 0.49 g / 100 ml, Chloroform).
Beispiel 22
rac-φμ-dfö-Amino-S^-dicyano^^tetrahydro^H-pyran^-yOpyridin^-ylJthiojmethy^-l.S- thiazol-2-yl]benzoesäure
50 mg (0.10 mmol) der Verbindung aus Beispiel 19 und 16 mg (0.40 mmol) Natriumhydroxid werden in einer Mischung aus 4 ml 1 ,2-Dimethoxyethan, 1 ml Ethanol und 4 ml Wasser gelöst. Das Reaktionsgemisch wird 3 h bei RT gerührt. Nach Entfernen des Lösungsmittels wird direkt über
präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Aceto- nitril/Wasser 10:90 → 95:5 unter Zusatz von 0.1% Salzsäure).
Ausbeute: 29 mg (61% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.26-7.85 (br. s, 2H), 7.91 (d, 2H), 7.79 (s, IH), 7.78 (d, 2H), 4.60 (s, 2H), 4.58-4.51 (m, IH), 4.03 (d, IH), 3.55-3.46 (m, IH), 1.96-1.84 (m, IH), 1.76-1.50 (m, 5H).
LC-MS (Methode 5): R, = 3.53 min; MS (ESIpos): m/z = 478 [M+H]+.
Die in der folgenden Tabelle aufgeführten Enantiomere werden auf analoge Weise aus Beispiel 20 bzw. Beispiel 21 erhalten:
Beispiel 25
rac-2-({[2-(4-Chloφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-(tetrahydro-2H-pyran-2-yl)pyridin-3,5- dicarbonitril
100 mg (0.21 mmol) der Verbindung aus Beispiel 14 werden in 5 ml trockenem THF vorgelegt und mit 167 mg (1.42 mmol) Isopentylnitrit und 3 mg (0.02 mmol) Kupfer(II)-chlorid versetzt. Das Reaktionsgemisch wird 8 h bei RT gerührt. Nach erneuter Zugabe von 3 mg (0.02 mmol) Kupfer(II)-chlorid wird das Reaktionsgemisch für weitere 12 h bei RT gerührt. Der Ansatz wird dann mit 6 ml 1 N Salzsäure versetzt. Die wässrige Phase wird zweimal mit je 10 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden einmal mit 5 ml ges. wässriger Natrium- hydrogencarbonat-Lösung und einmal mit 5 ml ges. wässriger Natriumchlorid-Lösung gewaschen. Anschließend wird über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels wird der Rückstand über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittel- gradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 43 mg (44% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 13.19 (s, IH), 9.12 (s, IH), 8.04 (s, 4H), 7.81 (s, IH), 4.79- 4.70 (m, IH), 4.68 (s, 2H), 4.08 (dd, IH), 3.63-3.53 (m, IH), 1.99-1.79 (m, IH), 1.87-1.74 (m, IH), 1.73-1.52 (m, 4H).
LC-MS (Methode 4): Rt = 2.69 min; MS (ESIpos): m/z = 463 [M+H]+.
Beispiel 26
rac-2-Amino-6-({[2-(4-chlθφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-(tetrahydro-2H-pyran-3-yl)- pyridin-3 ,5-dicarbonitril
50 mg (0.13 mmol) der Verbindung aus Beispiel 32A, 39 mg (0.16 mmol) 4-(Chlormethyl)-2-(4- chlorphenyl)-l ,3-thiazol und 45 mg (0.54 mmol) Natriumhydrogencarbonat werden in 3 ml trockenem DMF vorgelegt. Das Reaktionsgemisch wird 12 h bei RT gerührt und dann direkt über präpa-
rative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/ Wasser 10:90 → 95:5).
Ausbeute: 37 mg (58% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.28-7.83 (br. s, 2H), 7.93 (d, 2H), 7.88 (s, IH), 7.57 (d, 2H), 4.59 (s, 2H), 3.97-3.88 (m, IH), 3.86-3.79 (m, 2H), 3.20-3.09 (m, IH), 2.23 (dq, IH), 1.93-1.84 (m, IH), 1.77-1.56 (m, 2H).
LC-MS (Methode 7): R, = 3.98 min; MS (ESIpos): m/z = 468 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 26 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 29
2-Amino-6-({[2-(4-chloφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-cyclohexylpyridin-3,5-dicarbo- nitril
2.00 g (5.42 mmol) der Verbindung aus Beispiel 33A werden in 11 1 ml trockenem DMF vorgelegt und mit 1.46 g (5.96 mmol) 4-(Chlormethyl)-2-(4-chlorphenyl)-l,3-thiazol sowie 1.82 g (21.68 mmol) Natriumhydrogencarbonat versetzt. Das Reaktionsgemisch wird 12 h bei RT gerührt. Der Ansatz wird danach mit 20 ml Wasser verdünnt und zweimal mit je 100 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden einmal mit 15 ml ges. wässriger Natriumhydrogen-
carbonat-Lösung gewaschen und über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels wird der Rückstand an Kieselgel 60 chromatographisch gereinigt (Laufmittel: Gradient Cyclohexan/Ethylacetat 50:1 → 2:1).
Ausbeute: 1.08 g (40% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.20-7.80 (br. s, 2H), 7.93 (d, 2H), 7.88 (s, IH), 7.57 (d, IH), 4.59 (s, 2H), 2.92-2.80 (m, IH), 2.01-1.88 (m, 2H), 1.87-1.77 (m, 2H), 1.76-1.60 (m, 3H), 1.37- 1.10 (m, 3H).
LC-MS (Methode 12): R, = 3.10 min; MS (ESIpos): m/z = 466 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 29 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 33
2-({[2-(4-Chloφhenyl)-l,3-thiazol-4-yl]methyl}thio)-4-cyclohexyl-6-{[3-(diethylamino)propyl]- amino } pyridin-3 ,5-dicarbonitril
50 mg (0.10 mmol) der Verbindung aus Beispiel 37A und 30 mg (0.23 mmol) 3-(Diethylamino)- propylamin werden in 0.7 ml trockenem DMF gelöst. Das Reaktionsgemisch wird 12 h bei RT
gerϋhrt und dann direkt über präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 -> 95:5).
Ausbeute: 35 mg (58% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.58 (t, IH), 7.93 (d, 2H), 7.63 (s, IH), 7.55 (d, 2H), 4.66 (s, 2H)1 3.53-3.45 (m, 2H), 2.92-2.84 (m, IH), 2.43-2.31 (m, 6H), 2.02-1.90 (m, 2H), 1.89-1.81 (m, 2H), 1.76-1.68 (m, 3H), 1.65-1.56 (m, 2H), 1.39-1.14 (m, 3H), 0.89 (t, 6H).
LC-MS (Methode 2): Rt = 4.66 min; MS (ESIpos): m/z = 579 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 33 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 43
2- Amino-6-( { [2-(4-chlorphenyl)- 1 , 3 -thiazol-4-yl]methyl } thio)-4-cyclohex-3-en- 1 -ylpyridin-3 , 5 -di- carbonitril
50 mg (0.20 mmol) der Verbindung aus Beispiel 38 A, 52 mg (0.22 mmol) 4-(Chlormethyl)-2-(4- chlorphenyl)-l ,3-thiazol sowie 49 mg (0.59 mmol) Natriumhydrogencarbonat werden in 2.5 ml trockenem DMF vorgelegt. Das Reaktionsgemisch wird 12 h bei RT gerührt und dann direkt über
präparative HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Aceto- nitril/Wasser 10:90 → 95:5).
Ausbeute: 42 mg (46% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.26-7.86 (br. s, 2H), 7.94 (d, 2H), 7.89 (s, IH), 7.58 (d, 2H), 5.81-5.71 (m, 2H), 4.60 (s, 2H), 3.13-3.03 (m, IH), 2.26-2.08 (m, 4H)1 1.83-1.76 (m, IH).
LC-MS (Methode 13): Rt = 3.20 min; MS (ESIpos): m/z = 464 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 43 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 46
2-Amino-6-({[2-(4-chlorphenyl)-l ,3-thiazol-4-yl]methyl}thio)-4-(tetrahydro-2H-pyran-4-yl)- pyridin-3,5-dicarbonitril
100 mg (0.34 mmol) der Verbindung aus Beispiel 34A, 83 mg (0.34 mmol) 4-(Chlormethyl)-2-(4- chlorphenyl)-l,3-thiazol sowie 57 mg (0.68 mmol) Natriumhydrogencarbonat werden in 5 ml trockenem DMF vorgelegt. Das Reaktionsgemisch wird 16 h bei RT gerührt. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand mit ca. 2 ml Acetonitril ausgerührt. Dabei fällt ein Niederschlag aus, der abgesaugt und getrocknet wird.
Ausbeute: 72 mg (45% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.04 (br. s, 2H), 7.93 (d, 2H), 7.88 (s, IH), 7.56 (d, 2H), 4.59 (s, 2H), 3.98 (dd, 2H), 3.39 (dd, 2H), 3.13 (m, IH), 2.24-2.15 (m, 2H), 1.61 (d, 2H).
LC-MS (Methode 15): R, = 2.96 min; MS (ESIpos): m/z = 468 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 46 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 49
Methyl 3-({[6-amino-3,5-dicyano-4-(tetrahydro-2H-pyran-2-yl)pyridin-2-yl]thio}methyl)benzoat
Eine Lösung von 50 mg (0.19 mmol) der Verbindung aus Beispiel 31 A und 48 mg (0.21 mmol) 3- (Brommethyl)benzoesäuremethylester in 2 ml trockenem DMF werden mit 48 mg (0.58 mmol) Natriumhydrogencarbonat versetzt und 20 h bei RT gerührt. Der Ansatz wird danach direkt mittels präparativer HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5 / 15 μm; Laufmittelgradient: Aceto- nitril/Wasser 10:90 -> 95:5).
Ausbeute: 44 mg (53% d. Th.)
1H-NMR (400 MHz, CDCl3): δ = 8.25-7.77 (br. s, 2H), 8.06 (s, IH), 7.82 (pseudo-dd, 2H), 7.46 (t, IH), 4.58-4.49 (m, IH), 4.53 (s, 2H), 4.02 (dd, IH), 3.85 (s, 3H), 3.55-3.45 (m, IH), 1.97-1.85 (br. s, IH), 1.73-1.51 (m, 5H).
LC-MS (Methode 5): R, = 3.79 min; MS (ESIpos): m/z = 409 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 49 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 52
2-({[2-(4-Chlorphenyl)-l,3-thiazol-4-yl]methyl}sulfanyl)-6-methyl-4-(tetrahydro-2H-pyran-2-yl)- pyridin-3,5-dicarbonitril
Analog zur Herstellung der Verbindung in Beispiel 14 werden 50 mg (0.14 mmol) der Verbindung aus Beispiel 41 A, 40 mg (0.16 mmol) 4-(Chlormethyl)-2-(4-chlorphenyl)-l,3-thiazol und 45 mg (0.54 mmol) Natriumhydrogencarbonat in 2.8 ml trockenem DMF umgesetzt.
Ausbeute: 32 mg (50% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 7.94 (d, 2H), 7.73 (s, IH), 7.57 (d, 2H), 4.73 (s, 2H)1 4.71- 4.69 (m, IH), 4.07 (dd, IH), 3.62-3.53 (m, IH), 2.78 (s, 3H), 2.00-1.89 (br. s, IH), 1.82-1.72 (m, IH), 1.71-1.53 (m, 4H).
LC-MS (Methode 4): R, = 3.36 min; MS (ESIpos): m/z = 467 [M+H]+.
Beispiel 53
rac-Ethyl-4-[4-({[3,5-dicyano-6-methyl-4-(tetrahydro-2H-pyran-2-yl)pyridin-2-yl]sulfanyl}- methyl)- 1 ,3-thiazol-2-yl]benzoat
Die Titelverbindung wird analog zu Beispiel 52 aus den entsprechenden Edukten erhalten.
Ausbeute: 192 mg (37% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.06 (d, 4H), 7.81 (s, IH), 4.75 (s, 2H), 4.70 (d, IH), 4.33 (q, 2H), 4.07 (dd, IH), 3.62-3.53 (m, IH), 2.79 (s, 3H), 1.99-1.89 (m, IH), 1.82-1.73 (m, IH), 1.70- 1.55 (m, 4H), 1.34 (t, 3H).
LC-MS (Methode 4): Rt = 3.32 min; MS (ESIpos): m/z = 505 [M+H]+.
Beispiel 54 und Beispiel 55
e«£-Ethyl-4-[4-( { [3 ,5-dicyano-6-methyl-4-(tetrahydro-2H-pyran-2-yl)pyridin-2-yl] sulfanyl } - methyl)-! ,3-thiazol-2-yl]benzoat (Enantiomer 1 und Enantiomer 2)
190 mg der Verbindung aus Beispiel 53 werden bei ca. 300C in 4 ml Methanol und 10 ml TBME gelöst und mittels präparativer HPLC an chiraler Phase (Methode 16) in die Enantiomere getrennt:
Beispiel 54 (Enantiomer 1):
Ausbeute: 90 mg
HPLC (Methode 17): R, = 5.44 min; ee >99%
optische Drehung: +0.073° (c = 0.50 g / 100 ml, Chloroform).
Beispiel 55 (Enantiomer 2):
Ausbeute: 82 mg
HPLC (Methode 17): R, = 5.83 min; ee >98%.
Beispiel 56
(+)-4-[4-({[3,5-Dicyano-6-methyl-4-(tetrahydro-2H-pyran-2-yl)pyridin-2-yl]sulfanyl}methyl)-l,3- thiazol-2-yl]benzoesäure
Die Titelverbindung wird analog zu Beispiel 22 aus der Verbindung in Beispiel 54 erhalten.
Ausbeute: 17 mg (19% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 13.22-13.13 (br. s, IH), 8.04 (s, 4H), 7.71 (s, IH), 4.76 (s, 2H), 4.74-4.68 (m, IH), 4.06 (dd, IH), 3.62-3.53 (m, IH), 2.79 (s, 3H), 1.98-1.89 (m, IH), 1.82- 1.72 (m, IH), 1.72-1.55 (m, 4H).
LC-MS (Methode 4): Rt = 2.84 min; MS (ESIpos): m/z = 477 [M+H]+
optische Drehung: +0.009° (c = 0.17 g / 100 ml, Methanol).
Beispiel 57
(-)-4-[4-( {[3,5-Dicyano-6-methyl-4-(tetrahydro-2H-pyran-2-yl)pyridin-2-yl]sulfanyl}methyl)-l ,3- thiazol-2-yl]benzoesäure
Die Titelverbindung wird analog zu Beispiel 22 aus der Verbindung in Beispiel 55 erhalten.
Ausbeute: 11 mg (12% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 13.22-13.11 (br. s, IH), 8.04 (s, 4H), 7.70 (s, IH), 4.76 (s, 2H), 4.74-4.68 (m, IH), 4.07 (dd, IH), 3.62-3.52 (m, IH), 2.79 (s, 3H), 1.98-1.89 (m, IH), 1.82- 1.72 (m, IH), 1.71-1.54 (m, 4H).
LC-MS (Methode 22): Rt = 2.39 min; MS (ESIpos): m/z = 477 [M+H]+.
Beispiel 58
2-Amino-6-({[2-(4-chlorphenyl)-l,3-oxazol-4-yl]methyl}sulfanyl)-4-(tetrahydro-2H-pyran-3-yl)- pyridin-3 , 5 -dicarbonitril
Die Titelverbindung wird analog zu Beispiel 14 aus 200 mg (0.67 mmol) der Verbindung in Beispiel 32A und 197 mg (0.74 mmol) der Verbindung in Beispiel 42A erhalten.
Ausbeute: 324 mg (95% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.33 (s, IH), 8.28-7.88 (br. s, 2H), 7.97 (d, 2H), 7.61 (d, 2H), 4.48 (s, 2H), 3.95-3.88 (m, IH), 3.83 (s, 2H), 3.82-3.78 (m, IH), 3.19-3.08 (m, IH), 2.30-2.17 (m, IH), 1.93-1.86 (m, IH), 1.77-1.68 (m, IH), 1.68-1.55 (m, IH).
LC-MS (Methode 4): R, = 2.87 min; MS (ESIpos): m/z = 452 [M+H]+.
Beispiel 59
rac-Ethyl-4-[4-({[6-amino-3,5-dicyano-4-(tetrahydro-2H-pyran-3-yl)pyridin-2-yl]sulfanyl}methyl)- l,3-thiazol-2-yl]benzoat
Die Titelverbindung wird analog zu Beispiel 14 aus 282 mg (0.76 mmol) der Verbindung in Beispiel 32A und 290 mg (0.83 mmol) Ethyl-4-[4-(chlormethyl)-l,3-thiazol-2-yl]benzoat erhalten.
Ausbeute: 181 mg (47% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 8.33-7.89 (br. s, 2H), 8.08 (s, 4H), 7.96 (s, IH), 4.61 (s, 2H), 4.36 (q, 2H), 3.96-3.87 (m, IH), 3.83 (s, 2H), 3.83-3.79 (m, IH), 3.19-3.08 (m, IH), 2.29-2.18 (m, IH), 1.93-1.85 (m, IH), 1.76-1.69 (m, IH), 1.69-1.57 (m, IH), 1.35 (t, 3H).
LC-MS (Methode 4): R, = 2.87 min; MS (ESIpos): m/z = 452 [M+H]+.
Beispiel 60 und Beispiel 61
e«r-Ethyl-4-[4-({[6-amino-3,5-dicyano-4-(tetrahydro-2H-pyran-3-yl)pyridin-2-yl]sulfanyl}methyl)- l,3-thiazol-2-yl]benzoat (Enantiomer 1 und Enantiomer 2)
180 mg der Verbindung aus Beispiel 59 werden bei ca. 3O0C in 5 ml Methanol und 20 ml TBME gelöst und mittels präparativer ΗPLC an chiraler Phase (Methode 18) in die Enantiomere getrennt:
Beispiel 60 (Enantiomer 1):
Ausbeute: 59 mg
ΗPLC (Methode 19): R, = 9.1 1 min; ee >99%
optische Drehung: +0.057° (c = 0.455 g / 100 ml, Chloroform).
Beispiel 61 (Enantiomer 2):
Ausbeute: 77 mg
ΗPLC (Methode 19): R, = 10.29 min; ee >99%.
Beispiel 62
(+)-4-[4-({[6-Amino-3,5-dicyano-4-(tetrahydro-2H-pyran-3-yl)pyridin-2-yl]sulfanyl}methyl)-l,3- thiazol-2-yl]benzoesäure
Die Titelverbindung wird analog zu Beispiel 22 aus der Verbindung in Beispiel 60 erhalten.
Ausbeute: 18 mg (48% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 13.23-13.10 (br. s, IH), 8.35-7.83 (br. s, 2H), 8.05 (s, 4H), 7.96 (s, IH), 4.61 (s, 2H), 3.91 (dd, IH), 3.84-3.78 (m, 2H), 3.19-3.08 (m, IH), 2.30-2.18 (m, IH), 1.93-1.85 (m, IH), 1.77-1.69 (m, IH), 1.69-1.56 (m, IH).
LC-MS (Methode 4): R, = 2.41 min; MS (ESIpos): m/z = 478 [M+H]+.
Beispiel 63
(-)-4-[4-({[6-Amino-3,5-dicyano-4-(tetrahydro-2H-pyran-3-yl)pyridin-2-yl]sulfanyl}methyl)-l,3- thiazol-2-yl]benzoesäure
Die Titelverbindung wird analog zu Beispiel 22 aus der Verbindung in Beispiel 61 erhalten.
Ausbeute: 30 mg (53% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 13.22-13.13 (br. s, IH), 8.29-7.88 (br. s, 2H), 8.04 (s, 4H), 7.95 (s, IH), 4.60 (s, 2H), 3.92 (dd, IH), 3.83-3.78 (m, 2H), 3.19-3.09 (m, IH), 2.31-2.18 (m, IH), 1.95-1.85 (m, IH), 1.77-1.68 (m, IH), 1.68-1.57 (m, IH).
LC-MS (Methode 4): R, = 2.42 min; MS (ESIpos): m/z = 478 [M+H]+
optische Drehung: -0.050° (c = 0.495 g / 100 ml, Methanol/Dichlormethan 1 : 1).
Beispiel 64
rac-4-[4-({[6-Amino-3,5-dicyano-4-(tetrahydro-2H-pyran-3-yl)pyridin-2-yl]sulfanyl}methyl)-l,3- thiazol-2-yl]benzoesäure
Die Titelverbindung wird analog zu Beispiel 22 aus 69 mg (0.14 mmol) der Verbindung aus Beispiel 59 erhalten.
Ausbeute: 51 mg (78% d. Th.)
LC-MS (Methode 5): R, = 3.37 min; MS (ESIpos): m/z = 478 [M+H]+.
Beispiel 65
rac-4-[4-({[3,5-Dicyano-4-(tetrahydro-2H-pyran-3-yl)pyridin-2-yl]sulfanyl}methyl)-l,3-thiazol-2- yl]benzoesäure
51 mg (0.11 mmol) der Verbindung aus Beispiel 64 werden in 2.6 ml trockenem THF vorgelegt und mit 84 mg (0.72 mmol) Isopentylnitrit sowie 1.43 mg (0.01 mmol) Kupfer(II)chlorid versetzt. Das Reaktionsgemisch wird 10 h bei RT gerührt. Der Ansatz wird danach mit 4 ml 1 N Salzsäure versetzt und die wässrige Phase zweimal mit je 10 ml Ethylacetat extrahiert. Die vereinigten orga- nischen Phasen werden einmal mit 5 ml gesättigter wässriger Natriumhydrogencarbonat-Lösung und einmal mit 5 ml gesättigter wässriger Natriumchlorid-Lösung gewaschen und über Magnesiumsulfat getrocknet. Nach Entfernen des Lösungsmittels am Rotationsverdampfer wird der Rückstand mittels präparativer HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 — > 95:5, mit 0.3% Salzsäure). Man erhält einen weißen Fest- stoff.
Ausbeute: 7 mg (14% d. Th.)
1H-NMR (400 MHz, DMSO-d6): δ = 13.18 (s, IH), 9.10 (s, IH), 8.04 (s, 4H), 7.80 (s, IH), 4.78 (s, 2H), 3.99-3.91 (m, 2H), 3.82 (t, IH), 3.42-3.31 (m, 2H), 2.34-2.20 (m, IH), 2.05-1.97 (m, IH), 1.81-1.60 (m, 2H).
LC-MS (Methode 7): R, = 3.45 min; MS (ESIpos): m/z = 463 [M+H]+.
Beispiel 66
rαc-Methyl-N-[6-({[2-(4-chloφhenyl)-l,3-oxazol-4-yl]methyl}sulfanyl)-3,5-dicyano-4-(tetrahydro- 2H-pyran-3-yl)pyridin-2-yl]-N-methylglycinat
239 mg (0.42 mmol) der Verbindung aus Beispiel 43A werden in 6 ml trockenem THF gelöst und nacheinander mit 116 mg (0.83 mmol) Sarcosinmethylester-Hydrochlorid sowie 126 mg (1.25 mmol) Triethylamin versetzt. Das Reaktionsgemisch wird 10 h bei RT gerührt. Das Lösungsmittel
wird danach am Rotationsverdampfer entfernt und der Rückstand mittels präparativer HPLC gereinigt (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 → 95:5).
Ausbeute: 123 mg (55% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.16 (s, IH), 7.97 (d, 2H), 7.61 (d, 2H), 4.56 (s, 2H), 4.36 (s, 2H), 3.97-3.81 (m, 3H), 3.63 (s, 3H), 3.42 (s, 3H), 3.30 (s, 2H), 2.38-2.25 (m, IH), 1.98-1.89 (m, IH), 1.79-1.71 (m, IH), 1.71-1.58 (m, IH).
LC-MS (Methode 2): R1 = 2.47 min; MS (ESIpos): m/z = 538 [M+H]+.
Beispiel 67
rac-3-({[6-Amino-3,5-dicyano-4-(tetrahydro-2H-pyran-2-yl)pyridin-2-yl]sulfanyl}methyl)benz- amid
Analog zur Herstellung der Verbindung in Beispiel 14 werden 50 mg (0.14 mmol) der Verbindung aus Beispiel 31A, 36 mg (0.21 mmol) 3-(Chlormethyl)benzamid und 48 mg (0.58 mmol) Natrium- hydrogencarbonat in 2.0 ml trockenem DMF umgesetzt.
Ausbeute: 23 mg (29% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.20-7.82 (br. s, 2H), 7.99-7.93 (m, 2H), 7.76 (d, IH), 7.65 (d, IH), 7.43-7.34 (m, 2H), 4.57-4.45 (m, IH), 4.49 (s, 2H), 4.02 (dd, IH), 3.56-3.45 (m, IH), 1.95- 1.85 (br. s, IH), 1.73-1.50 (m, 5H).
LC-MS (Methode 5): R, = 3.12 min; MS (ESIpos): m/z = 394 [M+H]+.
Beispiel 68
λ-flc-Z-Amino-ό-IfCZ-amino-l .S-thiazol^-yOmethyπsulfanylJ^-^etrahydro-ZH-pyran^-y^pyridin- 3,5-dicarbonitril
Analog zur Herstellung der Verbindung in Beispiel 14 werden 50 mg (0.14 mmol) der Verbindung aus Beispiel 31 A, 39 mg (0.21 mmol) 4-(Chlormethyl)-l,3-thiazol-2-amin und 48 mg (0.58 mmol) Natriumhydrogencarbonat in 2.0 ml trockenem DMF umgesetzt.
Ausbeute: 35 mg (49% d. Th.)
1H-NMR (400 MHz, DMSO-(I6): δ = 8.20-7.79 (br. s, 2H), 7.08-6.93 (br. s, 2H), 6.60 (s, IH), 4.59- 4.49 (m, IH), 4.27 (s, 2H), 4.02 (dd, IH), 2.55-2.46 (m, IH), 1.96-1.85 (m, IH), 1.74-1.65 (m, 2H), 1.65-1.51 (m, 3H).
LC-MS (Methode 5): R, = 2.31 min; MS (ESIpos): m/z = 373 [M+H]+.
Beispiel 69
Methyl-3-{[(6-amino-3,5-dicyano-4-cyclohexylpyridin-2-yl)sulfanyl]methyl}benzoat
Analog zur Herstellung der Verbindung in Beispiel 29 werden 140 mg (0.54 mmol) der Verbindung aus Beispiel 33A, 137 mg (0.60 mmol) Methyl-3-(brommethyl)benzoat und 182 mg (2.17 mmol) Natriumhydrogencarbonat in 2.0 ml trockenem DMF umgesetzt.
Ausbeute: 30 mg (14% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 8.23-7.78 (br. s, 2H), 8.07 (s, IH), 7.83 (d, 2H), 7.47 (t, IH), 4.53 (s, 2H), 3.87 (s, 3H), 2.93-2.82 (m, IH), 2.02-1.89 (m, 2H), 1.89-1.80 (m, 2H), 1.76-1.65 (m, 3H), 1.39-1.13 (m, 3H).
LC-MS (Methode 7): R, = 4.05 min; MS (ESIpos): m/z = 407 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen werden analog zu Beispiel 69 aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 80
rac-2-Amino-4-(tetrahydro-2H-pyran-2-yl)-6- { [3 -( 1 H-tetrazol-5 -yl)benzyl] sulfanyl } pyridin-3 ,5-di- carbonitril
55 mg (0.16 mmol) der Verbindung aus Beispiel 31 A, 65 mg (0.18 mmol) der Verbindung aus Beispiel 5OA und 41 mg (0.49 mmol) Natriumhydrogencarbonat werden in 1.7 ml trockenem DMF suspendiert und 10 h bei RT gerührt. Der Ansatz wird danach auf 2 ml Wasser gegossen und durch Zugabe von wenig 1 N Salzsäure auf pH 4 gebracht. Es fällt ein brauner Niederschlag aus, der abfiltriert und mittels präparativer HPLC weiter gereinigt wird (Säule: YMC GEL ODS-AQ S-5, 15 μm; Laufmittelgradient: Acetonitril/Wasser 10:90 -> 95:5).
Ausbeute: 5 mg (7% d. Th.)
1H-NMR (400 MHz, DMSOd6): δ = 16.96-16.76 (br. s, IH), 8.18-7.86 (br. s, 2H), 8.14 (s, IH), 7.92 (d, IH), 7.75 (d, IH), 7.54 (t, IH), 4.56 (s, 2H), 4.56-4.49 (m, IH), 4.01 (d, IH), 3.58-3.47 (m, IH), 1.96-1.87 (m, IH), 1.75-1.51 (m, 5H).
LC-MS (Methode 4): R, = 2.50 min; MS (ESIpos): m/z = 419 [M+H]+.
B. Bewertung der pharmakologischen und physiologischen Wirksamkeit
Die pharmakologische und physiologische Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
B-I . Indirekte Bestimmung des Adenosin-Agonismus über Genexpression
Zellen der permanenten Linie CHO (Chinese Hamster Ovary) werden stabil mit der cDNA für die Adenosin-Rezeptor-Subtypen Al, A2a und A2b transfiziert. Die Adenosin-Al -Rezeptoren sind über G,-Proteine und die Adenosin-A2a- und A2b-Rezeptoren über Gs-Proteine an die Adenylat- cyclase gekoppelt. Entsprechend wird die cAMP -Bildung in der Zelle inhibiert bzw. stimuliert. Über einen cAMP-abhängigen Promotor wird danach die Expression der Luziferase moduliert. Der Luziferase-Test wird mit dem Ziel hoher Sensitivität und Reproduzierbarkeit, geringer Varianz und guter Eignung für die Durchführung auf einem Robotersystem optimiert durch Variation mehrerer Testparameter, wie z.B. Zelldichte, Dauer der Anzuchtphase und der Testinkubation, Forskolin- Konzentration und Medium-Zusammensetzung. Zur pharmakologischen Charakterisierung der Zellen und zum Roboter-gestützten Substanz-Screening wird das folgende Testprotokoll verwen- det:
Die Stammkulturen werden in DMEM/F12-Medium mit 10% FCS (fötales Kälberserum) bei 37°C unter 5% CO2 gezüchtet und jeweils nach 2-3 Tagen 1 :10 gesplittet. Testkulturen werden mit 2000 Zellen pro Napf in 384-well-Platten ausgesät und ca. 48 Stunden bei 370C angezogen. Dann wird das Medium durch eine physiologische Kochsalzlösung (130 mM Natriumchlorid, 5 mM Kalium- chlorid, 2 mM Calciumchlorid, 20 mM HEPES, 1 mM Magnesiumchlorid-Hexahydrat, 5 mM Natriumhydrogencarbonat, pH 7.4) ersetzt. Die in DMSO gelösten zu testenden Substanzen werden in einer Verdünnungsreihe von 5 x 10"" M bis 3 x 10"6 M (Endkonzentration) zu den Testkulturen pipettiert (maximale DMSO-Endkonzentration im Testansatz: 0.5%). 10 Minuten später wird Forskolin zu den Al -Zellen zugegeben und anschließend werden alle Kulturen für vier Stunden bei 37°C inkubiert. Danach wird zu den Testkulturen 35 μl einer Lösung, bestehend zu 50% aus Lyse-Reagenz (30 mM Dinatriumhydrogenphosphat, 10% Glycerin, 3% TritonXIOO, 25 mM TrisHCl, 2 mM Dithiotreitol (DTT), pH 7.8) und zu 50% aus Luciferase-Substrat-Lösung (2.5 mM ATP, 0.5 mM Luciferin, 0.1 mM Coenzym A, 10 mM Tricin, 1.35 mM Magnesiumsulfat, 15 mM DTT, pH 7.8) zugegeben, ca. 1 Minute geschüttelt und die Luciferase-Aktivität mit einem Kamerasystem gemessen. Bestimmt werden die EC50-WeIIe, d.h. die Konzentrationen, bei denen bei der Al -Zelle 50% der Luciferase-Antwort inhibiert bzw. bei den A2b- und A2a-Zellen 50% der maximalen Stimulierbarkeit mit der entsprechenden Substanz erreicht sind. Als Referenzverbindung dient in diesen Experimenten die Adenosin-analoge Verbindung NECA (5-N-Ethylcarbox- amido-adenosin), die mit hoher Affinität an alle Adenosin-Rezeptor-Subtypen bindet und eine
agonistische Wirkung besitzt [Klotz, K.N., Hessling, J., Hegler, J., Owman, C, KuIl, B., Fredholm, B.B., Lohse, M.J., "Comparative pharmacology of human adenosine receptor Subtypes - characteri- zation of stably transfected receptors in CHO cells", Naunyn Schmiedebergs Arch. Pharmacol. 357. 1-9 (1998)].
In der folgenden Tabelle 1 sind die EC50-Werte repräsentativer Ausführungsbeispiele für die Rezeptorstimulation an Adenosin Al-, A2a- und A2b-Rezeptor-Subtypen aufgeführt:
Tabelle 1
B-2. Untersuchung an isolierten Gefäßen
Aus narkotisierten Ratten wird die Arteria caudalis präpariert und in eine konventionelle Apparatur zur Messung isolierter Gefäße eingespannt. Die Gefäße werden in einem Wärmebad perfun- diert und mit Phenylephrin kontrahiert. Das Maß der Kontraktion wird über einen Kontraktionsmesser ermittelt. Zu den vorkontrahierten Gefäßen werden Testsubstanzen gegeben und die Abnahme der Kontraktion der Gefäße gemessen. Eine Abnahme der Kontraktion entspricht einer Dilatation der Gefäße. Als EC5o-Wert einer Testsubstanz bzgl. ihrer relaxierenden Eigenschaften wird die Konzentration angegeben, bei der die Kontraktion der Gefäße um 50% verringert ist.
B-3. Blutdruck- und Herzfrequenz-Messungen an wachen Ratten
Wachen SHR (spontaneously hypertensive rats)-Ratten, die einen internen Sender tragen, der dauerhaft sowohl Blutdruck als auch Herzfrequenz messen kann (telemetrische Erfassung von hämodynamischen Parametern), werden Testsubstanzen in verschiedenen Dosierungen oral verabreicht. Anschließend werden über 24 Stunden Blutdruck und Herzfrequenz und deren Veränderun- gen aufgezeichnet.
B-4. Blutdruck- und Herzfrequenz-Messungen an wachen Krallenaffen
Wachen Krallenaffen, die einen internen Sender tragen, der dauerhaft sowohl Blutdruck als auch Herzfrequenz messen kann (telemetrische Erfassung von hämodynamischen Parametern), werden Testsubstanzen in verschiedenen Konzentrationen oral verabreicht. Anschließend werden über 6-24 Stunden Blutdruck und Herzfrequenz und deren Veränderungen aufgezeichnet.
B-5. Bestimmung der Löslichkeit
Benötigte Reagenzien:
• PBS-Puffer pH 7.4: 90.00 g NaCl p.a. (z.B. Fa. Merck, Art.-Nr. 1.06404.1000), 13.61 g KH2PO4 p.a. (z.B. Fa. Merck, Art.-Nr. 1.04873.1000) und 83.35 g 1 N NaOH (z.B. Fa. Bernd Kraft GmbH, Art.-Nr. 01030.4000) in einen 1 Liter-Messkolben einwiegen, mit Wasser auffüllen und ca. 1 Stunde rühren;
• Acetatpuffer pH 4.6: 5.4 g Natriumacetat x 3 H2O p.a. (z.B. Fa. Merck, Art.-Nr. 1.06267.0500) in einen 100 ml-Messkolben einwiegen, in 50 ml Wasser lösen, mit 2.4 g Eisessig versetzen, auf 100 ml mit Wasser auffüllen, pH-Wert überprüfen und falls notwendig auf pH 4.6 ein- stellen;
• Dimethylsulfoxid (z.B. Fa. Baker, Art.-Nr. 7157.2500);
• destilliertes Wasser.
Herstellung der Kalibrierlösungen:
Herstellung der Ausgangslösung für Kalibrierlösungen (Stammlösung): In ein 2 ml Eppendorf- Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 0.5 mg der Testsubstanz genau eingewogen, zu einer Konzentration von 600 μg/ml mit DMSO versetzt (z.B. 0.5 mg Substanz + 833 μl DMSO) und bis zur vollständigen Lösung mittels eines Vortexers geschüttelt.
Kalibrierlösung 1 (20 μg/ml): 34.4 μl der Stammlösung werden mit 1000 μl DMSO versetzt und homogenisiert.
Kalibrierlösung 2 (2.5 μg/ml): 100 μl der Kalibrierlösung 1 werden mit 700 μl DMSO versetzt und homogenisiert.
Herstellung der Probenlösungen:
Probenlösung für Löslichkeit bis 10 g/l in PBS-Puffer pH 7.4: In ein 2 ml Eppendorf-Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 5 mg der Testsubstanz genau eingewogen und zu einer Konzentration von 5 g/l mit PBS-Puffer pH 7.4 versetzt (z.B. 5 mg Substanz + 500 μl PBS-Puffer pH 7.4).
Probenlösung für Löslichkeit bis 10 g/l in Acetatpuffer pH 4.6: In ein 2 ml Eppendorf-Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 5 mg der Testsubstanz genau eingewogen und zu einer Konzentration von 5 g/l mit Acetatpuffer pH 4.6 versetzt (z.B. 5 mg Substanz + 500 μl Acetatpuffer pH 4.6).
Probenlösung für Löslichkeit bis 10 g/l in Wasser: In ein 2 ml Eppendorf-Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 5 mg der Testsubstanz genau eingewogen und zu einer Konzentration von 5 g/l mit Wasser versetzt (z.B. 5 mg Substanz + 500 μl Wasser).
Durchführung:
Die so hergestellten Probenlösungen werden 24 Stunden bei 1400 rpm mittels eines temperierbaren Schüttlers (z.B. Fa. Eppendorf Thermomixer comfort Art.-Nr. 5355 000.011 mit Wechselblock Art.-Nr. 5362.000.019) bei 200C geschüttelt. Von diesen Lösungen werden jeweils 180 μl abgenommen und in Beckman Polyallomer Centrifuge Tubes (Art.-Nr. 343621) überführt. Diese Lösungen werden 1 Stunde mit ca. 223.000 x g zentrifugiert (z.B. Fa. Beckman Optima L-90K
Ultracentrifuge mit Type 42.2 Ti Rotor bei 42.000 rpm). Von jeder Probenlösung werden 100 μl des Überstandes abgenommen und 1 :5, 1 :100 und 1 : 1000 mit dem jeweils verwendeten Lösungsmittel (Wasser, PBS-Puffer 7.4 oder Acetatpuffer pH 4.6) verdünnt. Es wird von jeder Verdünnung eine Abfüllung in ein geeignetes Gefäß für die HPLC-Analytik vorgenommen.
Analytik:
Die Proben werden mittels RP-HPLC analysiert. Quantifiziert wird über eine Zwei-Punkt-Kalibra- tionskurve der Testverbindung in DMSO. Die Löslichkeit wird in mg/1 ausgedrückt. Analysensequenz: 1) Kalibrierlösung 2.5 mg/ml; 2) Kalibrierlösung 20 μg/ml; 3) Probenlösung 1 :5; 4) Probenlösung 1: 100; 5) Probenlösung 1 :1000.
HPLC-Methode für Säuren:
Agilent 1100 mit DAD (Gl 315A), quat. Pumpe (G1311 A), Autosampier CTC HTS PAL, Degaser (G1322A) und Säulenthermostat (G1316A); Säule: Phenomenex Gemini Cl 8, 50 mm x 2 mm, 5 μ; Temperatur: 400C; Eluent A: Wasser/Phosphorsäure pH 2; Eluent B: Acetonitril; Flussrate: 0.7 ml/min; Gradient: 0-0.5 min 85% A, 15% B; Rampe: 0.5-3 min 10% A, 90% B; 3-3.5 min 10% A, 90% B; Rampe: 3.5-4 min 85% A, 15% B; 4-5 min 85% A, 15% B.
HPLC-Methode für Basen:
Agilent 1100 mit DAD (Gl 315A), quat. Pumpe (Gl 31 IA), Autosampier CTC HTS PAL, Degaser (G1322A) und Säulenthermostat (G1316A); Säule: VDSoptilab Kromasil 100 C18, 60 mm x 2.1 mm, 3.5 μ; Temperatur: 3O0C; Eluent A: Wasser + 5 ml Perchlorsäure/l; Eluent B: Acetonitril; Flussrate: 0.75 ml/min; Gradient: 0-0.5 min 98% A, 2% B; Rampe: 0.5-4.5 min 10% A, 90% B; 4.5-6 min 10% A, 90% B; Rampe: 6.5-6.7 min 98% A, 2% B; 6.7-7.5 min 98% A, 2% B.
B-6. Bestimmung pharmakokinetischer Kenngrößen nach intravenöser und oraler Gabe
Die zu untersuchende Substanz wird Tieren (z.B. Maus, Ratte, Hund) intravenös als Lösung appliziert, die orale Applikation erfolgt als Lösung oder Suspension über eine Schlundsonde. Nach Substanzgabe wird den Tieren zu festgelegten Zeitpunkten Blut entnommen. Dieses wird heparini- siert, anschließend wird daraus durch Zentrifugation Plasma gewonnen. Die Substanz wird im Plasma über LC/MS-MS analytisch quantifiziert. Aus den so ermittelten Plasmakonzentration- Zeit-Verläufen werden die pharmakokinetischen Kenngrößen wie AUC, Cmax, T1/2 (Halbwertszeit) und CL (Clearance) mittels eines validierten pharmakokinetischen Rechenprogramms berechnet.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Tablette:
Zusammensetzung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.