WO2008064093A2 - Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile - Google Patents
Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile Download PDFInfo
- Publication number
- WO2008064093A2 WO2008064093A2 PCT/US2007/084893 US2007084893W WO2008064093A2 WO 2008064093 A2 WO2008064093 A2 WO 2008064093A2 US 2007084893 W US2007084893 W US 2007084893W WO 2008064093 A2 WO2008064093 A2 WO 2008064093A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- methyl
- quinolin
- crystalline form
- diffraction
- Prior art date
Links
- VXDPOGVDHHJTDY-UHFFFAOYSA-N CC(C)(c(cc1)ccc1N)C#N Chemical compound CC(C)(c(cc1)ccc1N)C#N VXDPOGVDHHJTDY-UHFFFAOYSA-N 0.000 description 1
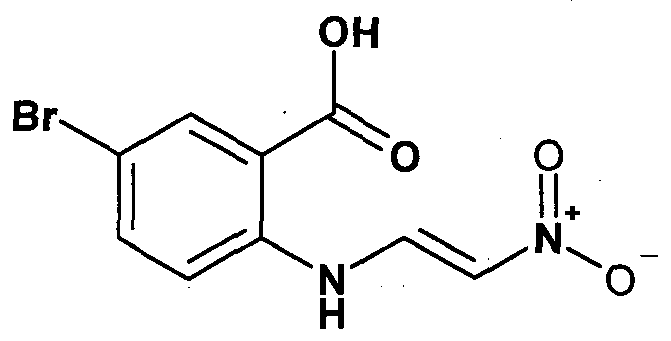
- PCSRUURXNNOYEZ-ONEGZZNKSA-N [O-][NH+](/C=C/Nc(c(C(O)=O)c1)ccc1Br)[O-] Chemical compound [O-][NH+](/C=C/Nc(c(C(O)=O)c1)ccc1Br)[O-] PCSRUURXNNOYEZ-ONEGZZNKSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/29—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of non-condensed six-membered aromatic rings
- C07C309/30—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of non-condensed six-membered aromatic rings of six-membered aromatic rings substituted by alkyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the invention relates to particular solid, preferably crystalline or amorphous, especially crystalline, forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5- c]quinolin-1-yl)-phenyl]-propionitrile (compound I, see below), its hydrates and solvates, its salts and hydrates and solvates of its salts, certain processes for their preparation, pharmaceutical compositions containing these solid forms, and their use in diagnostic methods or, preferably, for the therapeutic treatment of warm-blooded animals, especially humans, and their use as an intermediate or for the preparation of pharmaceutical preparations for use in diagnostic methods or, preferably, for the therapeutic treatment of warm-blooded animals, especially humans.
- 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenylj-propionitrile its inhibition of the activity of the lipid kinases, such as the PI3-kinase and/or members of the PI3-kinase-related protein kinase family (also called PIKK and include DNA-PK, ATM, ATR, hSMG-1 and mTOR), such as the DNA protein-kinase; its preparation; and its use, especially as an anti-tumour agent, are described in WO2006/122806.
- the compound is exemplified therein in free form (see for instance Example 7) and as 4-toluenesulfonic acid salt in a stoichiometric ratio of 1:1.
- the synthesis of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]-propionitrile is also described in the experimental part as Example 1.
- the invention relates especially to essentially pure crystal forms of 2-methyl-2-[4-(3-methyl- 2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile of formula I (compound I),
- the angle of diffraction 2Theta is plotted on the horizontal axis (x-axis) and the intensity (counts) on the vertical (y-axis).
- FIG. 1 Form A of 2-methyl-2-[4-(3-methyl-2-ox ⁇ '8-quinolin-3-yl-2,3-dihydro- imidazo[4, 5-c]quinolin-1-yl)-phenyl]-propionitrile
- FIG. 3 Simulated X-ray powder pattern of form C of 2-methyl-2-[4-(3-methyl-2-oxo- 8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile
- FIG. 4 Form D of2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin'3-yl-2,3-dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionithle
- FIG. 5 Form H A of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3 ⁇ dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monohydrate
- FIG. 6 Form A of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monotosylate
- FIG. 7 Form B of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monotosylate
- FIG. 8 Simulated X-ray powder pattern of form H A of 2-methyl-2-[4-(3-methyl-2- oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monotosylate monohydrate (calculated from the corresponding single crystal structure) Crystalloqraphic data of compound I monotosylate monohydrate:
- FIG. 9 Simulated X-ray powder pattern of form H B of 2-methyl-2-[4-(3-methyl-2- oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenylJ-propionitrile monotosylate dihydrate (calculated from the corresponding single crystal structure)
- FIG. 11 Form A of2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro- imidazo[4, 5-c]quinolin- 1-yl)-phenyl]-propionitrile ditosylate
- FIG. 12 Simulated X-ray powder pattern of form H A of 2-methyl-2-[4-(3-methyl-2- oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionithle ditosylate trihydrate (calculated from the corresponding single crystal structure)
- FIG. 13 Amorphous form of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3- dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monotosylate
- FIG. 14 Raman spectra of amorphous form of 2-methyl-2-[4-(3-methyl-2-oxo-8- quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monotosylate
- FIG. 15 FT-IR spectra of amorphous form of2-methyl-2-[4'(3-methyl-2-oxo-8- quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinoHn-1-yl)-phenyl]-propionitrile monotosylate
- FIG. 16 Simulated X-ray powder pattern of form S 0 of 2-methyl-2-[4-(3-methyl-2- oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monotosylate diformic acid solvate monohydrate (calculated from the corresponding single crystal structure)
- essentially pure is understood in the context of the present invention to mean especially that at least 90, preferably at least 95, and most preferably at least 99 per cent by weight of the crystals of the compound of formula I, its hydrates or solvates, its salts or hydrates or solvates of its salts are present in the specified crystal form according to the invention.
- solid form includes crystalline forms and amorphous forms.
- Preferred solid forms are crystalline forms
- the crystal form of the compound of formula I, its hydrates or solvates, its salts or its hydrates or solvates of its salts exhibits an X-ray diffraction diagram essentially as outlined in one of the Figures.
- solid, preferably crystalline, form of the compound of formula I, its hydrates and solvates, its salts and hydrates or solvates of its salts obtainable as described in the Examples.
- One of the advantages of having access to different crystal forms of the compound of formula I, its hydrates or solvates, its salts or hydrates or solvates of its salts is the fact that distinct crystal forms are prone to incorporate distinct impurities upon crystallization, i.e. an impurity incorporated in crystal form AA is not necessarily also incorporated in the crystal form BB or in the crystal form CC.
- distinct crystal forms display different physical properties such as melting points, hygroscopicities, solubilities, flow properties or thermodynamic stabilities, and, hence, distinct crystal forms allow the choice of the most suitable form for a certain use or aspect, e.g. the use as an intermediate in the process of drug manufacture or in distinct administration forms like tablets, capsules, ointments or solutions.
- the solid, preferably crystalline, forms of the compound of formula I, its hydrates or solvates, its salts and hydrates or solvates of its salts possess valuable pharmacological properties and may, for example, be used in the treatment of conditions which are mediated by the activation of the PI3 kinase enzymes, such as proliferative, inflammatory or allergic conditions, or disorders commonly occurring in connection with transplantation.
- the solid, amorphous or crystalline, preferably crystalline, forms of the compound of formula I, its hydrates or solvates, its salts and hydrates or solvates of its salts may preferably used in the treatment of a proliferative disease selected from a benign or malignant tumor, carcinoma of the brain, kidney, liver, adrenal gland, bladder, breast, stomach, gastric tumors, ovaries, colon, rectum, prostate, pancreas, lung, vagina or thyroid, sarcoma, glioblastomas, multiple myeloma or gastrointestinal cancer, especially colon carcinoma or colorectal adenoma or a tumor of the neck and head, an epidermal hyperproliferation, psoriasis, prostate hyperplasia, a neoplasia, a neoplasia of epithelial character, lymphomas, a mammary carcinoma or a leukemia.
- Other diseases include Cowden syndrome, Lhermitte- Dudos disease
- the present invention relates especially to form A of 2-methyl-2-[4-(3-methyl-2-oxo-8- quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile monotosylate in the treatment of one of the said diseases mentioned herein or in the preparation of a pharmacological agent for the treatment thereof.
- the invention relates also to a method for the treatment of warm-blooded animals suffering from said diseases, wherein a quantity of the solid, preferably crystalline, form of the compound of formula I, its hydrates or solvates, its salts or hydrates or solvates of its salts which is effective against the disease concerned, especially a quantity with antiproliferative efficacy, is administered to warm-blooded animals in need of such treatment.
- the invention relates moreover to the use of solid, preferably crystalline, forms of the compound of formula I, its hydrates or solvates, its salts and hydrates or solvates of its salts for the preparation of pharmaceutical compositions for use in treating the human or animal body, especially for the treatment of proliferative disease, such as benign or malignant tumor, carcinoma of the brain, kidney, liver, adrenal gland, bladder, breast, stomach, gastric tumors, ovaries, colon, rectum, prostate, pancreas, lung, vagina or thyroid, sarcoma, glioblastomas, multiple myeloma or gastrointestinal cancer, especially colon carcinoma or colorectal adenoma or a tumor of the neck and head, an epidermal hyperproliferation, psoriasis, prostate hyperplasia, a neoplasia, a neoplasia of epithelial character, lymphomas, a mammary carcinoma or a leukemia.
- proliferative disease such
- the solid, preferably crystalline, forms of the compound of formula I, its hydrates or solvates, its salts or hydrates or solvates of its salts described herein can be utilized to prepare stable pharmaceutical dosage forms.
- the invention relates also to pharmaceutical preparations which contain an amount, especially an therapeutically effective amount for prevention or treatment of one of the diseases mentioned herein, of the solid, preferably crystalline, form of the compound of formula I 1 its hydrates or solvates, its salts or hydrates or solvates of its salts, together with pharmaceutically acceptable carriers which are suitable for topical, enteral, for example oral or rectal, or parenteral administration and may be inorganic or organic and solid or liquid.
- the present pharmaceutical preparations which, if so desired, may contain further pharmacologically active substances, are prepared in a manner known per se, for example by means of conventional mixing, granulating, coating, dissolving or lyophilising processes, and contain from about 1% to 100%, especially from about 1 % to about 20%, of the active substance or substances.
- the present invention relates also to a process for the preparation of a pharmaceutical composition which comprises mixing a solid, preferably crystalline, form of the compound of formula I, its hydrates or solvates, its salts or hydrates or solvates of its salts of the invention together with at least one pharmaceutically acceptable carrier or diluent.
- composition is intended to encompass a product comprising the active ingredient(s), pharmaceutically acceptable excipients that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- pharmaceutical compositions of the present invention encompass any composition made by admixing the active ingredient, optionally additional active ingredient(s) and pharmaceutically acceptable excipients.
- excipient means a component of a pharmaceutical product that is not the active ingredient, such as filler, diluent and carrier.
- the excipients that are useful in preparing a pharmaceutical composition are preferably generally safe, non-toxic and neither biologically nor otherwise undesirable, and are acceptable for veterinary use, as well as human pharmaceutical use.
- a pharmaceutically acceptable excipient includes both one and more than one such excipient.
- Therapeutically effective amount means the amount of a compound that, when administered for treating or preventing a disease, is sufficient to effect such treatment or prevention for the disease. The “therapeutically effective amount” will vary depending on the compound, the disease and its severity and the age, weight, etc., of the patient to be treated.
- the present invention relates also to a process for the preparation of solid, preferably crystalline, forms of the compound of formula I, its hydrates or solvates, its salts and hydrates or solvates of its salts.
- the precise conditions under which crystals are formed may now be empirically determined and a number of methods are suitable in practice, including the crystallization conditions as described in Examples 3 to 17.
- Crystallization-inducing conditions normally involve the use of an appropriate crystallization- inducing solvent, such as t-butylmethylether (TBME), methanol, ethanol, isopropanol or water or mixtures thereof.
- TBME t-butylmethylether
- the amorphous compound is dissolved in the solvent at a temperature of normally at least 10° C.
- the solution may be produced by dissolving in a solvent any one or more of amorphous forms of the compound, and solvates thereof, such as hydrates, methanolates, ethanolates, isopropanolates, or formiates. Crystals may then be formed by conversion from solution, crystallization taking place at a temperature of between about 0° C. and the boiling point of the solvent.
- amorphous compound may be dissolved in a solvent or a mixture of solvents in which it is readily soluble at elevated temperatures but in which it is only sparingly soluble at lower temperatures.
- Dissolution at elevated temperature is followed by cooling during which the desired crystals crystallize out of solution.
- a cooling and reheating step may be carried out several times, e.g. at least once, at least twice, at least 3x, at least 5x.
- the cooling and reheating temperatures are e.g. at least 5° C, at least 10° C or at least 15° C.
- the low temperature of the cooling/heating cycles may e.g. be less than 15° C, less than 10° C, less than 5° C or less than 0° C, whereas the high temperature may e.g. be at least 15 0 C, at least 20° C, at least 25°C or at least 30° C.
- Mixed solvents comprising a good solvent in which the compound is readily soluble, preferably, in amounts of at least 1% by weight at 30° C, and a poor solvent in which it is more sparingly soluble, preferably in amounts of not more than about 0.01% by weight at 30° C, may also be employed provided that crystallization from the mixture at a reduced temperature, of normally at least about, 0° C, is possible using the selected solvent mixture. Alternatively, the difference in solubility of the crystals in different solvents may be used.
- the amorphous compound may be dissolved in a good solvent in which it is highly soluble such as one in which it is soluble in amounts of at least 1% by weight at about 30° C and the solution subsequently mixed with a poor solvent in which it is more sparingly soluble, such as one in which it is soluble in amounts of not more than about 0.01 % by weight at about 30° C.
- a good solvent in which it is highly soluble such as one in which it is soluble in amounts of at least 1% by weight at about 30° C
- a poor solvent in which it is more sparingly soluble, such as one in which it is soluble in amounts of not more than about 0.01 % by weight at about 30° C.
- the solution of the compound in the good solvent may be added to the poor solvent, while maintaining normally a temperature in excess of about 0° C, or the poor solvent may be added to the solution of the compound in the good solvent, again while normally maintaining a temperature in excess of about 0° C.
- Examples of good solvents may include lower alcohols, such as methanol, ethanol and isopropanol, formic acid acetic acid or acetone.
- An example of a poor solvent is e.g. water.
- crystallization is effected at a temperature in the range of about 0° C to about 40° C.
- solid amorphous compound is suspended at a temperature of normally at least about 0° C in a solvent in which it is incompletely soluble, preferably only sparingly soluble, at that temperature.
- a suspension results in which particles of solid are dispersed, and remain incompletely dissolved in the solvent.
- the solids are maintained in a state of suspension by agitation e.g. by shaking or stirring.
- the suspension is kept at a temperature of normally about 0° C or higher in order to effect a transformation of the starting solids into crystals.
- the amorphous solid compound suspended in a suitable solvent may be a solvate, e.g. hydrate, methanolate, ethanolate, acetate or formiate.
- the amorphous powder may be derived by drying a solvate.
- seeds of crystalline material to the solution in order to induce crystallization.
- the crystalline forms of formula I, its hydrates or solvates, its salts and hydrates or solvates of its salts have a high crystallinity.
- a crystal form is defined herein as having a "high crystallinity" or being “crystallographically pure” when it contains at most about 0.5% (w/w), e.g. at most about 0.1% (w/w) of other form.
- crystalrystallographically pure Form AA contains about 0.5% (w/w) or less, e.g. about 0.1% (w/w) or less of Form BB and/or another crystalline form.
- a "crystallographically pure" form contains less than about 5% of amorphous form or an amount below the limit of detection (i.e. no detactable amount) of amorphous form.
- Form A of compound I can be manufactured in the following way: 241 g of free base are dissolved 2.4 I acetic acid at 50 0 C. The solution is clearfiltered, washed with 250 ml acetic acid and then at 50 0 C 7.2 I of water are added. The free base starts precipitating. The mixture is cooled within 1 h to 25 0 C, is then filtered and washed with 10 I H 2 O. The free base is then dried in vacuo at 50 0 C over night to yield 204 g of free base.
- Compound I changes its polymorphic form after equilibration in different solvents (slurry experiment with approx. 20 mg sample and 0.5 ml solvent at 25°C for 24 hours equilibration time (with agitation)). In methanol, methanol/water, DMF, ethanol, ethylacetate and THF the new form C can be observed.
- compound I is dissolved in formic acid at 60°C, clearfiltered and then methanol is added. After stirring for 2 hours at 65°C, the mixture is cooled to room temperature, the salt is filtered and washed with ice cold methanol to yield form C of compound I.
- Compound I changes its polymorphic form after equilibration in different solvents (slurry experiment with approx. 20 mg sample and 0.5 ml solvent at 25°C for 24 hours equilibration time (with agitation)). In isopropanol as solvent the new form D can be observed.
- This compound is obtained after crystallization by slow solvent evaporation from DMF solution at room temperature.
- a phase transformation of form A prepared according to Example 8 to another crystalline form, further on named form B, can be observed at temperatures above 7O 0 C (this can also be detected in the corresponding DSC.
- the transformation is reversible as found by DSC experiments.
- Form A and Form B have an enantiotropic relationship.
- a saturated solution of compound I ditosylate in ethanol/acetone (1 :1) can be used in a slow solvent evaporation experiment at 25°C.
- the formation of single crystals of compound 1 monotosylate monohydrate (form H A ) has been observed and the single crystal structure could be calculated.
- the ditosylate salt of compound I can be isolated as a second crop from crystallization from the mother liquor after filtering off the monotosylate salt.
- the compound I ditosylate salt has initial loss on drying of 0.4% (up to 140 0 C).
- DSC data showed a melting at approx. 262°C with a melting enthalpy of approx. 93 J/g.
- Amorphous material has been produced by spray drying of compound I monotosylate.
- the glass transition, Tg 1 has been observed by DSC at approx. 128 0 C. After recrystallization at approx. 175°C the substance melted at approx. 279°C with an melting enthalpy of approx. 65 J/g.
- the single crystal structure of compound I monotosylate diacetic acid solvate was determined by X-ray diffraction (modification S c ). Suitable single crystals were obtained by equilibration of compound I monotosylate in methylisobutylketone/acetic acid (1 :1 (v/v)) solvent mixture at 5O 0 C after cooling down to room temperature.
Abstract
Description










Claims

Priority Applications (22)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/514,976 US8436177B2 (en) | 2006-11-20 | 2007-11-16 | Salts and crystall forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| EP07864492.9A EP2094700B1 (en) | 2006-11-20 | 2007-11-16 | Crystalline monotosylate salt of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| EA200900638A EA015677B1 (en) | 2006-11-20 | 2007-11-16 | SALTS AND CRYSTAL FORMS OF 2-METHYL-2-[4-(3-METHYL-2-OXO-8-QUINOLIN-3-YL-2,3-DIHYDROIMIDAZO[4,5-c]QUINOLIN-1-YL)PHENYL]PROPIONITRILE |
| NZ576357A NZ576357A (en) | 2006-11-20 | 2007-11-16 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| UAA200905006A UA98473C2 (en) | 2006-11-20 | 2007-11-16 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| BRPI0719112-0A BRPI0719112A2 (en) | 2006-11-20 | 2007-11-16 | 2-METHYL-2- [4- (3-METHYL-2-OXO-8-QUINOLIN-3-IL-2,3-DI-HYDRO-IMI DAZO [4,5-C] QUINOLIN CRYSTAL SALTS AND FORMS -1-IL) -Phenyl] -PROPIONITRIL |
| AU2007323820A AU2007323820B2 (en) | 2006-11-20 | 2007-11-16 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl] -propionitrile |
| MX2009005360A MX2009005360A (en) | 2006-11-20 | 2007-11-16 | Salts and crystall forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quino lin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propio nitrile. |
| KR1020147029446A KR20140129396A (en) | 2006-11-20 | 2007-11-16 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| RS20140278A RS53335B (en) | 2006-11-20 | 2007-11-16 | Crystalline monotosylate salt of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| KR1020147014039A KR20140091718A (en) | 2006-11-20 | 2007-11-16 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| JP2009537381A JP5562033B2 (en) | 2006-11-20 | 2007-11-16 | 2-Methyl-2- [4- (3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo [4,5-c] quinolin-1-yl) -phenyl]- Salt and crystalline form of propionitrile |
| CA2669199A CA2669199C (en) | 2006-11-20 | 2007-11-16 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| IL198467A IL198467A (en) | 2006-11-20 | 2009-04-30 | Crystal form of monotosylate salt of 2-methyl-2-[4-(3-methyl-2-0xo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-i-yl)-phenyl]-propionitrile |
| TNP2009000191A TN2009000191A1 (en) | 2006-11-20 | 2009-05-15 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| SM200900041T SMP200900041B (en) | 2006-11-20 | 2009-05-28 | Salts and crystalline forms of 2-methyl-2- [4- (3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo [4,5-c] quinolin-1yl) - phenyl] propionitrile |
| NO20092227A NO20092227L (en) | 2006-11-20 | 2009-06-09 | Salts and crystal forms of 2-methyl-2- [4- (3-methyl-2-oxo-8-conolin-3-yl-2,3-dihydro-imidazo [4,5-C] quinolin-1-yl) phenyl] -propionitrile |
| HK10100151.3A HK1132739A1 (en) | 2006-11-20 | 2010-01-08 | Crystalline monotosylate salt of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin- 3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile 2--2-[4-(3--2--8--3--23--[45-c]-1-)- ]- |
| IL219877A IL219877A0 (en) | 2006-11-20 | 2012-05-17 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| US13/856,458 US20130289064A1 (en) | 2006-11-20 | 2013-04-04 | Salts and crystall forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| PH12013502100A PH12013502100A1 (en) | 2006-11-20 | 2013-10-10 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin -3-yl-2,3-dyhydroimidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| HRP20140627TT HRP20140627T1 (en) | 2006-11-20 | 2014-07-01 | Crystalline monotosylate salt of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US86648306P | 2006-11-20 | 2006-11-20 | |
| US60/866,483 | 2006-11-20 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/856,458 Division US20130289064A1 (en) | 2006-11-20 | 2013-04-04 | Salts and crystall forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008064093A2 true WO2008064093A2 (en) | 2008-05-29 |
| WO2008064093A3 WO2008064093A3 (en) | 2008-08-14 |
Family
ID=39345299
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/084893 WO2008064093A2 (en) | 2006-11-20 | 2007-11-16 | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
Country Status (36)
| Country | Link |
|---|---|
| US (2) | US8436177B2 (en) |
| EP (2) | EP2094700B1 (en) |
| JP (2) | JP5562033B2 (en) |
| KR (3) | KR20140091718A (en) |
| CN (5) | CN106045993A (en) |
| AR (1) | AR064256A1 (en) |
| AU (1) | AU2007323820B2 (en) |
| BR (1) | BRPI0719112A2 (en) |
| CA (1) | CA2669199C (en) |
| CL (1) | CL2007003316A1 (en) |
| CO (1) | CO6382134A2 (en) |
| CR (1) | CR10757A (en) |
| DO (1) | DOP2009000116A (en) |
| EA (1) | EA015677B1 (en) |
| EC (1) | ECSP099340A (en) |
| GE (1) | GEP20125436B (en) |
| GT (1) | GT200900133A (en) |
| HK (1) | HK1132739A1 (en) |
| HR (1) | HRP20140627T1 (en) |
| IL (2) | IL198467A (en) |
| JO (1) | JO2903B1 (en) |
| MA (1) | MA30967B1 (en) |
| MX (1) | MX2009005360A (en) |
| MY (1) | MY150216A (en) |
| NI (1) | NI200900091A (en) |
| NO (1) | NO20092227L (en) |
| NZ (1) | NZ576357A (en) |
| PE (2) | PE20081780A1 (en) |
| PH (1) | PH12013502100A1 (en) |
| RS (1) | RS53335B (en) |
| SM (1) | SMP200900041B (en) |
| TN (1) | TN2009000191A1 (en) |
| TW (2) | TWI417292B (en) |
| UA (1) | UA98473C2 (en) |
| UY (1) | UY30728A1 (en) |
| WO (1) | WO2008064093A2 (en) |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009118324A1 (en) * | 2008-03-26 | 2009-10-01 | Novartis Ag | 5imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| WO2013049300A1 (en) * | 2011-09-30 | 2013-04-04 | Dana-Farber Cancer Institute, Inc. | Method of treating mucoepidermoid carcinoma |
| US8476294B2 (en) | 2009-06-04 | 2013-07-02 | Novartis Ag | 1H-imidazo[4,5-c]quinolinone derivatives |
| WO2013152717A1 (en) | 2012-04-10 | 2013-10-17 | 上海昀怡健康管理咨询有限公司 | Fused pyrimidine compound, and preparation method, intermediate, composition, and uses thereof |
| WO2015043398A1 (en) | 2013-09-30 | 2015-04-02 | 上海璎黎药业有限公司 | Fused pyrimidine compound, intermediate, preparation method therefor, and composition and application thereof |
| WO2015055071A1 (en) | 2013-10-16 | 2015-04-23 | 上海璎黎药业有限公司 | Fused heterocyclic compound, preparation method therefor, pharmaceutical composition, and uses thereof |
| US9358232B2 (en) | 2013-04-17 | 2016-06-07 | Signal Pharmaceuticals, Llc | Methods for treating cancer using TOR kinase inhibitor combination therapy |
| US9474757B2 (en) | 2013-04-17 | 2016-10-25 | Signal Pharmaceuticals, Llc | Methods for treating cancer using TOR kinase inhibitor combination therapy |
| US9555033B2 (en) | 2010-02-03 | 2017-01-31 | Signal Pharmaceuticals, Llc | Identification of LKB1 mutation as a predictive biomarker for sensitivity to TOR kinase inhibitors |
| US9557338B2 (en) | 2012-10-18 | 2017-01-31 | Signal Pharmaceuticals, Llc | Inhibition of phosphorylation of PRAS40, GSK3-beta or P70S6K1 as a marker for tor kinase inhibitory activity |
| US9604939B2 (en) | 2013-05-29 | 2017-03-28 | Signal Pharmaceuticals, Llc | Pharmaceutical compositions of 7-(6-(2-hydroxypropan-2-YL)pyridin-3-YL)-1-((trans)-4-methoxycyclohexyl)-3,4-dihydropyrazino [2,3-B]pyrazin-2(1H)-one, a solid form thereof and methods of their use |
| US9630966B2 (en) | 2013-04-17 | 2017-04-25 | Signal Pharmaceuticals, Llc | Treatment of cancer with dihydropyrazino-pyrazines |
| US9782427B2 (en) | 2013-04-17 | 2017-10-10 | Signal Pharmaceuticals, Llc | Methods for treating cancer using TOR kinase inhibitor combination therapy |
| US9827243B2 (en) | 2013-04-17 | 2017-11-28 | Signal Pharmaceuticals, Llc | Pharmaceutical formulations, processes, solid forms and methods of use relating to 1-ethyl-7-(2-methyl-6-(1H-1,2,4-triazol-3-yl)pyridin-3-yl)-3,4-dihydropyrazino[2,3-b]pyrazin-2(1H)-one |
| US9937169B2 (en) | 2013-04-17 | 2018-04-10 | Signal Pharmaceuticals, Llc | Methods for treating cancer using dihydropyrazino-pyrazine compound combination therapy |
| US9975898B2 (en) | 2014-04-16 | 2018-05-22 | Signal Pharmaceuticals, Llc | Solid forms of 1-ethyl-7-(2-methyl-6-(1H-1,2,4-triazol-3-yl)pyridin-3-YL)-3,4-dihydropyrazino [2,3-b]pyrazin-2(1H)-one as tor kinase inhibitors |
| US9980963B2 (en) | 2013-04-17 | 2018-05-29 | Signal Pharmaceuticals, Llc | Treatment of cancer with dihydropyrazino-pyrazines |
| US9981971B2 (en) | 2014-04-16 | 2018-05-29 | Signal Pharmaceuticals, Llc | Solid forms of 1-ethyl-7-(2-methyl-6-(1H-1,2,4-triazol-3-yl)pyridin-3-yl)-3,4-dihydropyrazino[2,3-b]pyrazin-2(1H)-one as TOR kinase inhibitors |
| US10441584B2 (en) | 2016-11-23 | 2019-10-15 | Novartis Ag | Methods of enhancing immune response |
| US10576076B2 (en) | 2015-05-20 | 2020-03-03 | Novartis Ag | Pharmaceutical combination of everolimus with dactolisib |
| US10596165B2 (en) | 2018-02-12 | 2020-03-24 | resTORbio, Inc. | Combination therapies |
| US11096940B2 (en) | 2017-06-22 | 2021-08-24 | Celgene Corporation | Treatment of hepatocellular carcinoma characterized by hepatitis B virus infection |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101965336B (en) | 2008-01-04 | 2015-06-17 | 英特利凯恩有限责任公司 | Certain chemical entities, compositions and methods |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| JP5731978B2 (en) | 2008-09-26 | 2015-06-10 | インテリカイン, エルエルシー | Heterocyclic kinase inhibitor |
| CA2760791C (en) | 2009-05-07 | 2017-06-20 | Intellikine, Inc. | Heterocyclic compounds and uses thereof |
| ES2593256T3 (en) | 2010-05-21 | 2016-12-07 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulations |
| DE102010035744A1 (en) * | 2010-08-28 | 2012-03-01 | Merck Patent Gmbh | Imidazolonylchinoline |
| CA2817577A1 (en) | 2010-11-10 | 2012-05-18 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| DK2663309T3 (en) | 2011-01-10 | 2017-06-19 | Infinity Pharmaceuticals Inc | METHODS FOR PRODUCING ISOQUINOLINONES AND SOLID FORMS OF ISOQUINOLINONES |
| AU2012284088B2 (en) | 2011-07-19 | 2015-10-08 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| MX2014000648A (en) | 2011-07-19 | 2014-09-25 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof. |
| KR20140075693A (en) | 2011-08-29 | 2014-06-19 | 인피니티 파마슈티칼스, 인코포레이티드 | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| SI2976106T1 (en) * | 2013-03-21 | 2021-08-31 | Array Biopharma Inc. | Combination therapy comprising a b-raf inhibitor and a second inhibitor |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| NZ718430A (en) | 2013-10-04 | 2021-12-24 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof |
| EA201691872A1 (en) | 2014-03-19 | 2017-04-28 | Инфинити Фармасьютикалз, Инк. | HETEROCYCLIC COMPOUNDS FOR APPLICATION IN THE TREATMENT OF PI3K-GAMMA-MEDIATED DISORDERS |
| WO2015160975A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| CN114230571A (en) | 2015-09-14 | 2022-03-25 | 无限药品股份有限公司 | Solid forms of isoquinolinones, methods of making, compositions containing, and methods of use thereof |
| WO2017161116A1 (en) | 2016-03-17 | 2017-09-21 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as pi3k kinase inhibitors |
| WO2017214269A1 (en) | 2016-06-08 | 2017-12-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| JP7054681B2 (en) | 2016-06-24 | 2022-04-14 | インフィニティー ファーマシューティカルズ, インコーポレイテッド | Combination therapy |
| CN111187181B (en) * | 2019-11-22 | 2023-05-05 | 吉林大学 | Preparation method of 2- (4-aminophenyl) -2-methylpropanenitrile compound |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005054237A1 (en) | 2003-11-21 | 2005-06-16 | Novartis Ag | 1h-imidazoquinoline derivatives as protein kinase inhibitors |
| WO2006122806A2 (en) | 2005-05-20 | 2006-11-23 | Novartis Ag | 1,3-dihydro-imidazo [4,5-c] quinolin-2-ones as lipid kinase inhibitors |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4929624A (en) | 1989-03-23 | 1990-05-29 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo(4,5-c)quinolin-4-amines |
| US5268376A (en) | 1991-09-04 | 1993-12-07 | Minnesota Mining And Manufacturing Company | 1-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| JP2000119271A (en) | 1998-08-12 | 2000-04-25 | Hokuriku Seiyaku Co Ltd | 1h-imidazopyridine derivative |
| GB0211649D0 (en) * | 2002-05-21 | 2002-07-03 | Novartis Ag | Organic compounds |
| US7563748B2 (en) | 2003-06-23 | 2009-07-21 | Cognis Ip Management Gmbh | Alcohol alkoxylate carriers for pesticide active ingredients |
| AR046845A1 (en) | 2003-11-21 | 2005-12-28 | Novartis Ag | DERIVATIVES OF 1H-IMIDAZO [4,5-C] QUINOLINE FOR THE TREATMENT OF PROTEIN-KINASE DEPENDENT DISEASES |
| EP1769092A4 (en) | 2004-06-29 | 2008-08-06 | Europ Nickel Plc | Improved leaching of base metals |
-
2007
- 2007-11-16 GE GEAP200711260A patent/GEP20125436B/en unknown
- 2007-11-16 BR BRPI0719112-0A patent/BRPI0719112A2/en not_active IP Right Cessation
- 2007-11-16 AU AU2007323820A patent/AU2007323820B2/en not_active Ceased
- 2007-11-16 EA EA200900638A patent/EA015677B1/en not_active IP Right Cessation
- 2007-11-16 EP EP07864492.9A patent/EP2094700B1/en active Active
- 2007-11-16 CA CA2669199A patent/CA2669199C/en not_active Expired - Fee Related
- 2007-11-16 AR ARP070105114A patent/AR064256A1/en unknown
- 2007-11-16 KR KR1020147014039A patent/KR20140091718A/en not_active Application Discontinuation
- 2007-11-16 JP JP2009537381A patent/JP5562033B2/en not_active Expired - Fee Related
- 2007-11-16 CN CN201610405653.7A patent/CN106045993A/en active Pending
- 2007-11-16 KR KR1020147029446A patent/KR20140129396A/en not_active Application Discontinuation
- 2007-11-16 CN CN2012102686473A patent/CN102993202A/en active Pending
- 2007-11-16 RS RS20140278A patent/RS53335B/en unknown
- 2007-11-16 MX MX2009005360A patent/MX2009005360A/en active IP Right Grant
- 2007-11-16 UA UAA200905006A patent/UA98473C2/en unknown
- 2007-11-16 MY MYPI20091920A patent/MY150216A/en unknown
- 2007-11-16 WO PCT/US2007/084893 patent/WO2008064093A2/en active Application Filing
- 2007-11-16 EP EP10176298A patent/EP2364981A1/en not_active Withdrawn
- 2007-11-16 KR KR1020097010203A patent/KR20090080530A/en not_active Application Discontinuation
- 2007-11-16 CN CNA2007800420773A patent/CN101541793A/en active Pending
- 2007-11-16 CN CN201910073048.8A patent/CN109970735A/en active Pending
- 2007-11-16 US US12/514,976 patent/US8436177B2/en active Active
- 2007-11-16 CN CN2011103337901A patent/CN102336752A/en active Pending
- 2007-11-16 NZ NZ576357A patent/NZ576357A/en not_active IP Right Cessation
- 2007-11-19 CL CL200703316A patent/CL2007003316A1/en unknown
- 2007-11-19 TW TW096143742A patent/TWI417292B/en not_active IP Right Cessation
- 2007-11-19 PE PE2007001607A patent/PE20081780A1/en not_active Application Discontinuation
- 2007-11-19 TW TW102134663A patent/TW201402567A/en unknown
- 2007-11-19 PE PE2011001508A patent/PE20120083A1/en not_active Application Discontinuation
- 2007-11-19 JO JO2007482A patent/JO2903B1/en active
- 2007-11-20 UY UY30728A patent/UY30728A1/en not_active Application Discontinuation
-
2009
- 2009-04-29 CR CR10757A patent/CR10757A/en unknown
- 2009-04-30 IL IL198467A patent/IL198467A/en not_active IP Right Cessation
- 2009-05-15 TN TNP2009000191A patent/TN2009000191A1/en unknown
- 2009-05-18 GT GT200900133A patent/GT200900133A/en unknown
- 2009-05-18 EC EC2009009340A patent/ECSP099340A/en unknown
- 2009-05-18 CO CO09050683A patent/CO6382134A2/en active IP Right Grant
- 2009-05-18 NI NI200900091A patent/NI200900091A/en unknown
- 2009-05-19 DO DO2009000116A patent/DOP2009000116A/en unknown
- 2009-05-28 SM SM200900041T patent/SMP200900041B/en unknown
- 2009-06-03 MA MA31947A patent/MA30967B1/en unknown
- 2009-06-09 NO NO20092227A patent/NO20092227L/en not_active Application Discontinuation
-
2010
- 2010-01-08 HK HK10100151.3A patent/HK1132739A1/en not_active IP Right Cessation
-
2012
- 2012-05-17 IL IL219877A patent/IL219877A0/en unknown
-
2013
- 2013-04-04 US US13/856,458 patent/US20130289064A1/en not_active Abandoned
- 2013-05-13 JP JP2013101604A patent/JP2013173788A/en active Pending
- 2013-10-10 PH PH12013502100A patent/PH12013502100A1/en unknown
-
2014
- 2014-07-01 HR HRP20140627TT patent/HRP20140627T1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005054237A1 (en) | 2003-11-21 | 2005-06-16 | Novartis Ag | 1h-imidazoquinoline derivatives as protein kinase inhibitors |
| WO2006122806A2 (en) | 2005-05-20 | 2006-11-23 | Novartis Ag | 1,3-dihydro-imidazo [4,5-c] quinolin-2-ones as lipid kinase inhibitors |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2009228797B2 (en) * | 2008-03-26 | 2012-07-19 | Novartis Ag | 5imidazoquinolines and pyrimidine derivatives as potent modulators of VEGF-driven angiogenic processes |
| EP2474323A3 (en) * | 2008-03-26 | 2012-10-10 | Novartis AG | Imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| WO2009118324A1 (en) * | 2008-03-26 | 2009-10-01 | Novartis Ag | 5imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| US8476294B2 (en) | 2009-06-04 | 2013-07-02 | Novartis Ag | 1H-imidazo[4,5-c]quinolinone derivatives |
| US9555033B2 (en) | 2010-02-03 | 2017-01-31 | Signal Pharmaceuticals, Llc | Identification of LKB1 mutation as a predictive biomarker for sensitivity to TOR kinase inhibitors |
| WO2013049300A1 (en) * | 2011-09-30 | 2013-04-04 | Dana-Farber Cancer Institute, Inc. | Method of treating mucoepidermoid carcinoma |
| US9499561B2 (en) | 2012-04-10 | 2016-11-22 | Shanghai Yingli Pharmaceutical Co., Ltd. | Fused pyrimidine compound, and preparation method, intermediate, composition, and uses thereof |
| WO2013152717A1 (en) | 2012-04-10 | 2013-10-17 | 上海昀怡健康管理咨询有限公司 | Fused pyrimidine compound, and preparation method, intermediate, composition, and uses thereof |
| US9557338B2 (en) | 2012-10-18 | 2017-01-31 | Signal Pharmaceuticals, Llc | Inhibition of phosphorylation of PRAS40, GSK3-beta or P70S6K1 as a marker for tor kinase inhibitory activity |
| US10052322B2 (en) | 2013-04-17 | 2018-08-21 | Signal Pharmaceuticals, Llc | Pharmaceutical formulations, processes, solid forms and methods of use relating to 1-ethyl-7-(2-methyl-6-(1H-1,2,4-triazol-3-yl)pyridin-3-yl)-3,4-dihydropyrazino[2,3-b]pyrazin-2(1H)-one |
| US9827243B2 (en) | 2013-04-17 | 2017-11-28 | Signal Pharmaceuticals, Llc | Pharmaceutical formulations, processes, solid forms and methods of use relating to 1-ethyl-7-(2-methyl-6-(1H-1,2,4-triazol-3-yl)pyridin-3-yl)-3,4-dihydropyrazino[2,3-b]pyrazin-2(1H)-one |
| US10391092B2 (en) | 2013-04-17 | 2019-08-27 | Signal Pharmaceuticals, Llc | Methods for treating cancer using dihydropyrazino-pyrazine compound combination therapy |
| US9474757B2 (en) | 2013-04-17 | 2016-10-25 | Signal Pharmaceuticals, Llc | Methods for treating cancer using TOR kinase inhibitor combination therapy |
| US9937169B2 (en) | 2013-04-17 | 2018-04-10 | Signal Pharmaceuticals, Llc | Methods for treating cancer using dihydropyrazino-pyrazine compound combination therapy |
| US9630966B2 (en) | 2013-04-17 | 2017-04-25 | Signal Pharmaceuticals, Llc | Treatment of cancer with dihydropyrazino-pyrazines |
| US10183019B2 (en) | 2013-04-17 | 2019-01-22 | Signal Pharmaceuticals, Llc | Treatment of cancer with dihydropyrazino-pyrazines |
| US9358232B2 (en) | 2013-04-17 | 2016-06-07 | Signal Pharmaceuticals, Llc | Methods for treating cancer using TOR kinase inhibitor combination therapy |
| US9782427B2 (en) | 2013-04-17 | 2017-10-10 | Signal Pharmaceuticals, Llc | Methods for treating cancer using TOR kinase inhibitor combination therapy |
| US9980963B2 (en) | 2013-04-17 | 2018-05-29 | Signal Pharmaceuticals, Llc | Treatment of cancer with dihydropyrazino-pyrazines |
| US9795603B2 (en) | 2013-05-29 | 2017-10-24 | Signal Pharmaceuticals, Llc | Pharmaceutical compositions of 7-(6-(2-hydroxypropan-2-yl)pyridin-3-yl)-1-((trans)-4-methoxycyclohexyl)-3,4-dihydropyrazino [2,3-B]pyrazin-2(1H)-one, a solid form thereof and methods of their use |
| US9604939B2 (en) | 2013-05-29 | 2017-03-28 | Signal Pharmaceuticals, Llc | Pharmaceutical compositions of 7-(6-(2-hydroxypropan-2-YL)pyridin-3-YL)-1-((trans)-4-methoxycyclohexyl)-3,4-dihydropyrazino [2,3-B]pyrazin-2(1H)-one, a solid form thereof and methods of their use |
| US9974786B2 (en) | 2013-05-29 | 2018-05-22 | Signal Pharmaceuticals, Llc | Pharmaceutical compositions of 7-(6-(2-hydroxypropan-2-yl)pyridin-3-yl)-1-((trans)-4-methoxycyclohexyl)-3,4-dihydropyrazino[2,3- B]pyrazin-2(1H)-one, a solid form there of and methods of their use |
| US10052323B2 (en) | 2013-05-29 | 2018-08-21 | Signal Pharmaceuticals, Llc | Pharmaceutical compositions of 7-(6-(2-hydroxypropan-2-yl)pyridin-3-yl)-1-(trans)-4-methoxycyclohexyl)-3,4-dihydropyrazino [2,3-b]pyrazin-2(1H)-one, a solid form thereof and methods of their use |
| US9745321B2 (en) | 2013-09-30 | 2017-08-29 | Shanghai Yingli Pharmaceutical Co., Ltd | Fused pyrimidine compound, intermediate, preparation method therefor, and composition and application thereof |
| WO2015043398A1 (en) | 2013-09-30 | 2015-04-02 | 上海璎黎药业有限公司 | Fused pyrimidine compound, intermediate, preparation method therefor, and composition and application thereof |
| US9656996B2 (en) | 2013-10-16 | 2017-05-23 | Shanghai Yingli Pharmaceutical Co., Ltd. | Fused heterocyclic compound, preparation method therefor, pharmaceutical composition, and uses thereof |
| WO2015055071A1 (en) | 2013-10-16 | 2015-04-23 | 上海璎黎药业有限公司 | Fused heterocyclic compound, preparation method therefor, pharmaceutical composition, and uses thereof |
| US9981971B2 (en) | 2014-04-16 | 2018-05-29 | Signal Pharmaceuticals, Llc | Solid forms of 1-ethyl-7-(2-methyl-6-(1H-1,2,4-triazol-3-yl)pyridin-3-yl)-3,4-dihydropyrazino[2,3-b]pyrazin-2(1H)-one as TOR kinase inhibitors |
| US9975898B2 (en) | 2014-04-16 | 2018-05-22 | Signal Pharmaceuticals, Llc | Solid forms of 1-ethyl-7-(2-methyl-6-(1H-1,2,4-triazol-3-yl)pyridin-3-YL)-3,4-dihydropyrazino [2,3-b]pyrazin-2(1H)-one as tor kinase inhibitors |
| US10576076B2 (en) | 2015-05-20 | 2020-03-03 | Novartis Ag | Pharmaceutical combination of everolimus with dactolisib |
| US10441584B2 (en) | 2016-11-23 | 2019-10-15 | Novartis Ag | Methods of enhancing immune response |
| US10993940B2 (en) | 2016-11-23 | 2021-05-04 | Novartis Ag | Methods of enhancing immune response |
| US11045463B2 (en) | 2016-11-23 | 2021-06-29 | Novartis Ag | Methods of enhancing immune response |
| US11096940B2 (en) | 2017-06-22 | 2021-08-24 | Celgene Corporation | Treatment of hepatocellular carcinoma characterized by hepatitis B virus infection |
| US10596165B2 (en) | 2018-02-12 | 2020-03-24 | resTORbio, Inc. | Combination therapies |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2094700B1 (en) | Crystalline monotosylate salt of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile | |
| ES2857805T3 (en) | Polycrystalline form of free base or acid salt of EGFR inhibitor, method of preparation and application | |
| KR20180059446A (en) | Pharmaceutically acceptable salts of beta -guanidino propionic acid with improved properties and uses thereof | |
| EP2631234A1 (en) | Solid forms of dabigatran etexilate mesylate and processes for their preparation | |
| WO2023284537A1 (en) | Kras g12d inhibitors and uses thereof | |
| EP4143188A1 (en) | Crystalline forms of gepotidacin | |
| WO2021164793A1 (en) | Compound used as kinase inhibitor and use thereof | |
| WO2017020869A1 (en) | B crystal form of 2-[(2r)-2-methyl-2-pyrrolidinyl]-1h-benzimidazole-7-carboxamide, preparation method and use | |
| AU2012202996A1 (en) | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydroimidazo[4,5-c]quinolin-1-yl)-phenyl] -propionitrile | |
| JP2022525780A (en) | N- (5-((4-ethylpiperazine-1-yl) methyl) pyridin-2-yl) -5-fluoro-4- (3-isopropyl-2-methyl-2H-indazole-5-yl) pyrimidine- Crystalline and amorphous forms of 2-amine and its salts, as well as their production methods and therapeutic uses. | |
| WO2021073494A1 (en) | The salts of a compound and the crystalline forms thereof | |
| JP2018008977A (en) | Novel form of indazolo[5,4-a]pyrrolo[3,4-c]carbazole compound | |
| TW202409016A (en) | Salt and crystal forms of an epidermal growth factor receptor inhibitor | |
| EP4222148A1 (en) | Crystalline form of tegavivint, method of preparation, and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780042077.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07864492 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007864492 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2551/DELNP/2009 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 576357 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007323820 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2009-010757 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 198467 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2669199 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09050683A Country of ref document: CO Ref document number: 12009500980 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2009537381 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097010203 Country of ref document: KR Ref document number: 2009050737 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11260 Country of ref document: GE Ref document number: MX/A/2009/005360 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2007323820 Country of ref document: AU Date of ref document: 20071116 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200900638 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2009000367 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A200905006 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12514976 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 219877 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: PI0719112 Country of ref document: BR Kind code of ref document: A2 Effective date: 20090519 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2014/0278 Country of ref document: RS |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020147029446 Country of ref document: KR |