WO2008057585A1 - Novel compounds, pharmaceutical compositions containing same, and methods of use for same - Google Patents
Novel compounds, pharmaceutical compositions containing same, and methods of use for same Download PDFInfo
- Publication number
- WO2008057585A1 WO2008057585A1 PCT/US2007/023552 US2007023552W WO2008057585A1 WO 2008057585 A1 WO2008057585 A1 WO 2008057585A1 US 2007023552 W US2007023552 W US 2007023552W WO 2008057585 A1 WO2008057585 A1 WO 2008057585A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- pharmaceutical composition
- subject
- halogen
- Prior art date
Links
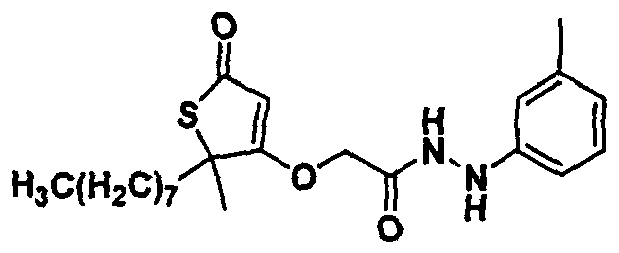
- 0 C*(C)C(C)(C(OCC(NNC(c1ccncc1)=O)=O)=C1)SC1=O Chemical compound C*(C)C(C)(C(OCC(NNC(c1ccncc1)=O)=O)=C1)SC1=O 0.000 description 5
- IEMMBWWQXVXBEU-UHFFFAOYSA-N CC(c1ccc[o]1)=O Chemical compound CC(c1ccc[o]1)=O IEMMBWWQXVXBEU-UHFFFAOYSA-N 0.000 description 1
- VFQXVTODMYMSMJ-UHFFFAOYSA-N NC(c1ccncc1)=O Chemical compound NC(c1ccncc1)=O VFQXVTODMYMSMJ-UHFFFAOYSA-N 0.000 description 1
- AKCRQHGQIJBRMN-UHFFFAOYSA-O [NH3+]c(cccc1)c1Cl Chemical compound [NH3+]c(cccc1)c1Cl AKCRQHGQIJBRMN-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/28—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/30—Oxygen or sulfur atoms
- C07D233/32—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/32—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- Fatty acids have three primary roles in the physiology of cells. First, they are the building bocks of biological membranes. Second, fatty acid derivatives serve as hormones and intracellular messengers. Third, and of particular importance to the present invention, fatty acids are fuel molecules that can be stored in adipose tissue as triacylglycerols, which are also known as neutral fats.
- FAS fatty acid synthase
- ACC acetyl CoA carboxylase
- malic enzyme acetyl CoA carboxylase
- citrate lyase The principal enzyme, FAS, catalyzes the NADPH-dependent condensation of the precursors malonyl-CoA and acetyl CoA to produce fatty acids.
- NADPH is a reducing agent that generally serves as the essential electron donor at two points in the reaction cycle of FAS.
- the other three enzymes i.e., ACC, malic enzyme, and citrate lyase
- Other enzymes for example the enzymes that produce NADPH, are also involved in fatty acid synthesis.
- FAS has an Enzyme Commission (RC.) No. 2.3.1.85 and is also known as fatty acid synthase, fatty acid ligase, as well as its systematic name acyl-CoA:malonyl-CoA C- acyltransferase (decarboxylating, oxoacyl- and enoyl-reducing and thioester-hydrolysing).
- RC. Enzyme Commission
- acetyl transacylase malonyl transacylase
- beta-ketoacyl synthetase condensing enzyme
- beta-ketoacyl reductase beta-hydroxyacyl dehydrase
- enoyl reductase thioesterase
- FAS inhibitors can be identified by the ability of a compound to inhibit the enzymatic activity of purified FAS.
- FAS activity can be assayed by measuring the incorporation of radiolabeled precursor (i.e., acetyl CoA or malonyl-CoA) into fatty acids or by spectrophotometrically measuring the oxidation of NADPH. (DiIs, et al., Methods Enzymol., 35:74-83).
- N-ethylmaleimide oxalyl thiol esters such as S-oxalylglutathione gossypol phenylglyoxal
- TDG 2-tetradecanylglutarate
- the condensing enzyme of the FAS complex is well characterized in terms of structure and function.
- the active site of the condensing enzyme contains a critical cysteine thiol, which is the target of antilipidemic reagents, such as, for example, the inhibitor cerulenin.
- Preferred inhibitors of the condensing enzyme include a wide range of chemical compounds, including alkylating agents, oxidants, and reagents capable of undergoing disulphide exchange.
- the binding pocket of the enzyme prefers long chain, E, E, dienes.
- Cerulenin [(2S, 3i?)-2,3-epoxy-4-oxo-7,10 dodecadienoyl amide] is an example:
- Cerulenin covalently binds to the critical cysteine thiol group in the active site of the condensing enzyme of fatty acid synthase, inactivating this key enzymatic step (Funabashi, et al., J. Biochem., 105:751-755, 1989). While cerulenin has been noted to possess other activities, these either occur in microorganisms which may not be relevant models of human cells (e. g. , inhibition of cholesterol synthesis in fungi, Omura (1976), Bacteriol. Rev., 40:681-697; or diminished RNA synthesis in viruses, Perez, et al.
- FEBS, 280: 129-133 occur at a substantially higher drug concentrations (inhibition of viral HIV protease at 5 mg/ml, Moelling, et al. (1990), FEBS, 261:373-377) or may be the direct result of the inhibition of endogenous fatty acid synthesis (inhibition of antigen processing in B lymphocytes and macrophages, FaIo, et al. (1987), J. Immunol., 139:3918-3923).
- cerulenin does not specifically inhibit myristoylation of proteins (Simon, et al., J. Biol. Chem., 267:3922-3931, 1992).
- FAS inhibitors are disclosed in U.S. Patent No. 5,614,551 , the disclosure of which is hereby incorporated by reference. Included are inhibitors of fatty acid synthase, citrate lyase, acetyl CoA carboxylase, and malic enzyme.
- Triacsin C (sometimes termed WS- 1228A), a naturally occurring acyl-CoA synthetase inhibitor, which is a product of Streptomyces sp. SK-1894.
- the chemical structure of Triacsin C is l-hydroxy-3-(£, E, E-2 ⁇ T- undecatrienylidine) triazene.
- Triacsin C causes 50% inhibition of rat liver acyl-CoA synthetase at 8.7 uM; a related compound, Triacsin A, inhibits acyl CoA-synthetase by a mechanism which is competitive with long-chain fatty acids. Inhibition of acyl-CoA synthetase is toxic to animal cells.
- Tomoda et al. (Tomoda el. al., J. Biol. Chem. 266:4214-4219, 1991) teaches mat Triacsin C causes growth inhibition in Raji cells at 1.0 ⁇ M, and have also been shown to inhibit growth of Vero and HeIa cells. Tomoda el. al.
- acyl-CoA synthetase is essential in animal cells and that inhibition of the enzyme has lethal effects.
- a family of compounds (gamma-substituted-alpha-methylene-beta-carboxy- gamma-butyrolactones) has been shown inU.S. Patent No. 5,981,575 (the disclosure of which is hereby incorporated by reference) to inhibit fatty acid synthesis, inhibit growth of tumor cells, and induce weight loss.
- the compounds disclosed in the '575 Patent have several advantages over the natural product cerulenin for therapeutic applications: [1] they do not contain the highly reactive epoxide group of cerulenin, [2] they are stable and soluble in aqueous solution, [3] they can be produced by a two-step synthetic reaction and thus easily produced in large quantities, and [4] they are easily tritiated to high specific activity for biochemical and pharmacological analyses.
- the synthesis of this family of compounds, which are fatty acid synthase inhibitors, is described in the '575 Patent, as is their use as a means to treat tumor cells expressing FAS 5 and their use as a means to reduce body weight.
- the '575 Patent also discloses the use of any fatty acid synthase inhibitors to systematically reduce adipocyte mass (adipocyte cell number or size) as a means to reduce body weight.
- adipocyte mass adipocyte cell number or size
- the primary sites for fatty acid synthesis in mice and humans are the liver ⁇ see
- Cerulenin was originally isolated as a potential antifungal antibiotic from the culture broth of Cephalosporium caerulens. Structurally cerulenin has been characterized as (2/?,3S)-epoxy-4-oxo-7,10-trans J trans-dodecanoic acid amide. Its mechanism of action has been shown to be inhibition, through irreversible binding, of beta-ketoacyl-ACP synthase, the condensing enzyme required for the biosynthesis of fatty acids. Cerulenin has been categorized as an antifungal, primarily against Candida and Saccharomyces sp.
- in vitro activity has been shown against some bacteria, actinomycetes, and mycobacteria, although no activity was found against Mycobacterium tuberculosis.
- the activity of fatty acid synthesis inhibitors and cerulenin in particular has not been evaluated against protozoa such as Toxoplasma gondii or other infectious eucaryotic pathogens such as Pneumocystis carinii, Giardia lamblia, Plasmodium sp., Trichomonas vaginalis, Cryptosporidium, Trypanosoma, Leishmania, and Schistosoma.
- Infectious diseases which are particularly susceptible to treatment are diseases which cause lesions in externally accessible surfaces of the infected animal.
- Externally accessible surfaces include all surfaces that may be reached by non-invasive means (without cutting or puncturing the skin), including the skin surface itself, mucus membranes, such as those covering nasal, oral, gastrointestinal, or urogenital surfaces, and pulmonary surfaces, such as the alveolar sacs.
- Susceptible diseases include: (1) cutaneous mycoses or tineas, especially if caused by Microsporum, Trichophyton, Epidermophyton, or Mucocutaneous candidiasis', (2) mucotic keratitis, especially if caused by Aspergillus, Fusarium or Candida; (3) amoebic keratitis, especially if caused by Acanthamoeba; (4) gastrointestinal disease, especially if caused by Giardia lamblia, Entamoeba, Cryptosporidium, Microsporidium, or Candida (most commonly in immunocompromised animals); (5) urogenital infection, especially if caused by Candida albicans or Trichomonas vaginalis; and (6) pulmonary disease, especially if caused by Mycobacterium tuberculosis, Aspergillus, or Pneumocystis carinii.
- Infectious organisms that are susceptible to treatment with fatty acid synthesis inhibitors include Mycobacterium tuberculosis, especially multiply-drug resistant strains, and protozoa such as Toxoplasma. Any compound that inhibits fatty acid synthesis may be used to inhibit microbial cell growth. However, compounds administered to a patient must not be equally toxic to both patient and the target microbial cells. Accordingly, it is beneficial to select inhibitors that only, or predominantly, affect target microbial cells. Eukaryotic microbial cells which are dependent on their own endogenously synthesized fatty acid will express Type I FAS.
- FAS inhibitors are growth inhibitory and by the fact that exogenously added fatty acids can protect normal patient cells but not these microbial cells from FAS inhibitors. Therefore, agents which prevent synthesis of fatty acids by the cell may be used to treat infections.
- fatty acids are synthesized by Type I FAS vising the substrates acetyl CoA, malonyl CoA and NADPH.
- other enzymes which can feed substrates into this pathway may also effect the rate of fatty acid synthesis and thus be important in microbes that depend on endogenously synthesized fatty acid.
- the product of Type I FAS differs in various organisms. For example, in the fungus S. cerevisiae the products are predominately palmitate and stearate esterified to coenzyme-A. In Mycobacterium smegmatis, the products are saturated fatty acid CoA esters ranging in length from 16 to 24 carbons. These lipids are often further processed to fulfill the cells need for various lipid components.
- Inhibition of key steps in down-stream processing or utilization of fatty acids may be expected to inhibit cell function, whether the cell depends on endogenous fatty acid or utilizes fatty acid supplied from outside the cell, and so inhibitors of these down-stream steps may not be sufficiently selective for microbial cells that depend on endogenous fatty acid.
- Type I fatty acid synthesis inhibitor to such microbes makes them more sensitive to inhibition by inhibitors of down-stream fatty acid processing and/or utilization. Because of this synergy, administration of a fatty acid synthesis inhibitor in combination with one or more inhibitors of down-stream steps in lipid biosynthesis and/or utilization will selectively affect microbial cells that depend on endogenously synthesized fatty acid.
- Preferred combinations include an inhibitor of FAS and acetyl CoA carboxylase, or FAS and an inhibitor of MAS.
- the mammal or patient may be treated by administering a fatty acid synthesis inhibitor (Pat No. 5,614,551).
- a new class of compounds has been discovered which has a variety of therapeutically valuable properties, eg. FAS-inhibition, and anti-cancer and anti-microbial properties.
- FIG. 1 show synthentic schemes for making compounds and intermediates pertinent to the invention.
- FIG. 2 shows a synthetic scheme for making a compound under the invention.
- the compounds of the invention can be prepared by conventional means. The synthesis of a number of the compounds is described in the examples. The compounds may be useful for the treatment of obesity, cancer, or microbially-based infections.
- One embodiment of the invention is compounds having the following general formula:
- R 1 H, C 1 -C 2O alkyl, cycloalkyl, alkenyl, aryl, arylalkyl, or alkylaryl, cyanomethyl, -OCH3,
- R 2 -OCH 2 C(O)NHNH-R 5 , where R 5 is
- a heterocycle selected from the group consisting of imidazole, thiazole, benzimidazole, benzoxazole, benzthiazole, tetrazole, triazole, and aminothiazole; or
- R 7 is a Cj-C 2O alkyl, cycloalkyl, alkenyl, aryl, arylalkyl, or alkylaryl, or a heterocycle selected from the group consisting of pyridyl, imidazole, thiazole, benzimidizole, benzoxazole, benzthiazole, tetrazole, triazole, and aminothiazole; and R 3 and R 4 , the same or different from each other, are Ci-C 2O alkyl, cycloalkyl, alkenyl, aryl, arylalkyl, or alkylaryl; with the proviso that when R 1 is -H, -OCH 3 , or -OC(O)CF 3 and R 3 is -(CH 2 ) T CH 3 , then R 2 is not
- R 1 is H.
- R 5 is C 1 -C 1 Q alkyl, cycloalkyl, alkenyl, aryl, arylalkyl, or alkylaryl.
- R 3 is -H or -CH 3 .
- R 4 is n-C ⁇ -Cg alkyl.
- R 6 is C]-C]O alkyl.
- Another embodiment of this invention is a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutical diluent and a compound of formula I.
- compositions of the present invention can be presented for administration to humans and other animals in unit dosage forms, such as tablets, capsules, pills, powders, granules, sterile parenteral solutions or suspensions, oral solutions or suspensions, oil in water and water in oil emulsions containing suitable quantities of the compound, suppositories and in fluid suspensions or solutions.
- unit dosage forms such as tablets, capsules, pills, powders, granules, sterile parenteral solutions or suspensions, oral solutions or suspensions, oil in water and water in oil emulsions containing suitable quantities of the compound, suppositories and in fluid suspensions or solutions.
- pharmaceutical diluent and “pharmaceutical carrier,” have the same meaning.
- solid or fluid unit dosage forms can be prepared.
- the compound can be mixed with conventional ingredients such as talc, magnesium stearate, dicalcium phosphate, magnesium aluminum silicate, calcium sulfate, starch, lactose, acacia, methylcellulose and functionally similar materials as pharmaceutical diluents or carriers.
- Capsules are prepared by mixing the compound with an inert pharmaceutical diluent and rilling the mixture into a hard gelatin capsule of appropriate size.
- Soft gelatin capsules are prepared by machine encapsulation of a slurry of the compound with an acceptable vegetable oil, light liquid petrolatum or other inert oil.
- Fluid unit dosage forms or oral administration such as syrups, elixirs, and suspensions can be prepared.
- the forms can be dissolved in an aqueous vehicle together with sugar or another sweetener, aromatic flavoring agents and preservatives to form a syrup.
- Suspensions can be prepared with an aqueous vehicle with the aid of a suspending agent such as acacia, tragacanth, methylcellulose and the like.
- a suspending agent such as acacia, tragacanth, methylcellulose and the like.
- For parenteral administration fluid unit dosage forms can be prepared utilizing the compound and a sterile vehicle.
- the compound can be dissolved in water for injection and filter sterilized before filling into a suitable vial or ampoule and sealing.
- Adjuvants such as a local anesthetic, preservative and buffering agents can be dissolved in the vehicle.
- the composition can be frozen after filling into a vial and the water removed under vacuum. The lyophilized powder can then be scaled in the vial and reconstituted prior to use.
- the clinical therapeutic indications envisioned for the compounds of the invention include: (1) infections due to invasive micro-organisms such as staphylococci and enter ococci; (2) cancers arising in many tissues whose cells over-express fatty acid synthase, and (3) obesity due to the ingestion of excess calories.
- Dose and duration of therapy will depend on a variety of factors, including (1) the patient's age, body weight, and organ function ( " e.g., liver and kidney function); (2) the nature and extent of the disease process to be treated, as well as any existing significant co-morbidity and concomitant medications being taken, and (3) drug-related parameters such as the route of administration, the frequency and duration of dosing necessary to effect a cure, and the therapeutic index of the drug.
- the dose will be chosen to
- breast cancer cells known to possess high levels of FAS and fatty acid synthesis activity, using
- This compound was prepared according to the scheme shown in FIG. 2. To 26 (83.0 mg, 0.29 mmol) and 4-chlorophenylhydrazine hydrochloride (68.0 mg, 0.38 mmol), following general procedure A compound 21 was obtained (34.0 mg, 30 %).
- Human FAS was purified from cultured ZR-75-1 human breast cancer cells obtained from the American Type Culture Collection. The procedure, adapted from Linn et at., 1981, and Kuhajda et al, 1994, utilizes hypotonic lysis, successive polyethyleneglycol (PEG) precipitations, and anion exchange chromatography. ZR-75-1 cells are cultured at 37 0 C with 5% CO 2 in RPMI culture medium with 10% fetal bovine serum, penicillin and streptomycin.
- Ten Tl 50 flasks of confluent cells are lysed with 1.5 ml lysis buffer (20 mM Tris- HCl, pH 7.5, 1 mM EDTA, 0.1 mM phenylmethanesulfonyl fluoride (PMSF) 9 0.1% Igepal CA- 630) and dounce homogenized on ice for 20 strokes.
- the lysate is centrifuged in JA-20 rotor (Beckman) at 20,000 rpm for 30 minutes at 4 0 C and the supernatant is brought to 42 ml with lysis buffer.
- a solution of 50% PEG 8000 in lysis buffer is added slowly to the supernatant to a final concentration of 7.5%.
- the solution is centrifuged in JA-20 rotor (Beckman) at 15,000 rpm for 30 minutes at 4 0 C.
- Solid PEG 8000 is then added to the supernatant to a final concentration of 15%.
- the pellet is resuspended overnight at 4 0 C in 10 ml of Buffer A (20 mM K 2 HPO4, pH 7.4).
- Buffer A (20 mM K 2 HPO4, pH 7.4
- the protein solution is applied to a Mono Q 5/5 anion exchange column (Pharmacia).
- the assay is performed on a Molecular Devices SpectraMax Plus
- Spectrophotometer The plate containing FAS, buffers, inhibitors, and controls are placed in the spectrophotometer heated to 37 0 C. Using the kinetic protocol, the wells are blanked on duplicate wells containing 100 ⁇ l of 100 mM K 2 HPO 4 , pH 6.5 and the plate is read at OD 3 40 at 10 sec intervals for 5 minutes to measure any malonyl-CoA independent oxidation of NADPH. The plate is removed from the spectrophotometer and malonyl-CoA (67.4 ⁇ M, final concentration per well) and alkynyl-CoA (61.8 ⁇ M, final concentration per well) are added to each well except to the blanks.
- malonyl-CoA 67.4 ⁇ M, final concentration per well
- alkynyl-CoA (61.8 ⁇ M, final concentration per well
- the plate is read again as above with the kinetic protocol to measure the malonyl- CoA dependent NADPH oxidation.
- the difference between the ⁇ OD 340 for the malonyl-CoA dependent and non-malonyl-CoA dependent NADPH oxidation is the specific FAS activity. Because of the purity of the FAS preparation, non-malonyl-CoA dependent NADPH oxidation is negligible.
- the IC 50 for the compounds against FAS is determined by plotting the ⁇ OD 3 40 for each inhibitor concentration tested, performing linear regression and computing the best-fit line, r 2 values, and 95% confidence intervals. The concentration of compound yielding 50% inhibition of FAS is the IC 50 .
- Crystal Violet Cell Growth Assay The crystal violet assay measure cell growth but not cytotoxicity. This assay employs crystal violet staining of fixed cells in 96-well plates with subsequent solubilization and measurement of OD 490 on a spectrophotometer. The OD 490 corresponds to cell growth per unit time measured. Cells are treated with the compounds of interest or vehicle controls and IC 5 0 for each compound is computed. To measure the cytotoxicity of specific compounds against cancer cells, 5 x 10 4
- MCF-7 human breast cancer cells obtained from the American Type Culture Collection are plated per well in 24 well plates in DMEM medium with 10% fetal bovine serum, penicillin, and streptomycin. Following overnight culture at 37 0 C and 5% CO 2 , the compounds to be tested, dissolved in DMSO, are added to the wells in 1 ⁇ l volume at the following concentrations: 50, 40, 30, 20, and 10 ⁇ g/ml in triplicate. Additional concentrations are tested if required. 1 ⁇ l of DMSO is added to triplicate wells are the vehicle control. C75 is run at 10, and 5 ⁇ g/ml in triplicate as positive controls.
- XTT Cytotoxicity Assay The XTT assay is a non-radioactive alternative for the [ 51 Cr] release cytotoxicity assay.
- XTT is a tetrazolium salt that is reduced to a formazan dye only by metabolically active, viable cells. The reduction of XTT is measured spectrophotometrically as OD4 90 - OD ⁇ so-
- 9 x 10 3 MCF-7 human breast cancer cells obtained from the American Type Culture Collection are plated per well in 96 well plates in DMEM medium with 10% fetal bovine serum, insulin, penicillin, and streptomycin. Following overnight culture at 37 0 C and 5% CO 2 , the compounds to be tested, dissolved in DMSO, are added to the wells in 1 ⁇ l volume at the following concentrations: 80, 40, 20, 10, 5, 2.5, 1.25, and 0.625 ⁇ g/ml in triplicate. Additional concentrations are tested if required. 1 ⁇ l of DMSO is added to triplicate wells are the vehicle control. C75 is run at 40, 20, 10, 15, 12.5, 10, and 5 ⁇ g/ml in triplicate as positive controls.
- XTT Cell Proliferation Kit ⁇ (XTT) Roche). Plates are read at OD4 90 and OD6 50 on a Molecular Devices SpectraMax Plus Spectrophotometer. Three wells containing the XTT reagent without cells serve as the plate blank. XTT data are reported as
- the IC 50 for the compounds is defined as the concentration of drug leading to a
- This assay measures the incorporation of [ l4 C]acetate into total lipids and is a
- MCF, -7 human breast cancer cells cultured as above, are plated at 5 x 10 4 cells per
- DMSO is added to triplicate wells for a vehicle control.
- C75 is run at 5 and 10 ⁇ g/ml
- the lower (organic) phase is transferred into a scintillation vial and dried at 40 0 C under N 2 gas. Once dried, 3 ml of scintillant (APB #NBC5104) is added and vials are counted for 14 C. The Beckman Scintillation counter calculates the average cpm values for triplicates.
- the ICso for the compounds is defined as the concentration of drug leading to a 50% reduction in [ 14 C]acetate incorporation into lipids compared to controls. This is determined by plotting the average cpm for each inhibitor concentration tested, performing linear regression and computing the best-fit line, r 2 values, and 95% confidence intervals. The average cpm values are computed by the Beckman scintillation counter (Model LS6500) for each compound concentration. Computation of linear regression, best-fit line, r 2 , and 95% confidence intervals are calculated using Prism Version 3.0 (Graph Pad Software).
- mice (Jackson Labs) are utilized for the initial weight loss screening. Animals are housed in temperature and 12 hour day/night cycle rooms and fed mouse chow and water ad lib. Three mice are utilized for each compound tested with vehicle controls in triplicate per experiment. For the experiments, mice are housed separately for each compound tested three mice to a cage. Compounds are diluted in DMSO at 10 mg/ml and mice are injected intraperitoneally with 60 mg/kg in approximately 100 ⁇ l of DMSO or with vehicle alone. Mice are observed and weighed daily; average weights and standard errors are computed with Excel (Microsoft). The experiment continues until treated animals reach their pretreatment weights.
- a broth microdilution assay is used to assess the antimicrobial activity of the compounds.
- Microorganisms tested include Staphylococcus aureus (ATCC # 29213), Enterococcus faecalis (ATCC # 29212), Pseudomonas aeruginosa (ATCC # 27853), and Escherichia coli (ATCC # 25922).
- the assay is performed in two growth media, Mueller Hinton Broth and Trypticase Soy Broth.
- a blood (Tsoy/5% sheep blood) agar plate is inoculated from frozen stocks maintained in T soy broth containing 10% glycerol and incubated overnight at 37° C. Colonies are suspended in sterile broth so that the turbidity matches the turbidity of a 0.5 McFarland standard. The inoculum is diluted 1:10 in sterile broth (Mueller Hinton or Trypticase soy) and 195 ul is dispensed per well of a 96-well plate. The compounds to be tested, dissolved in DMSO, are added to the wells in 5 ul volume at the following concentrations: 25, 12.5, 6.25, 3.125, 1.56 and 0.78 ug/ml in duplicate.
- OD ⁇ oo values are computed using SOFTmax Pro Software (Molecular Devices) and MIC values are determined by linear regression analysis using Prism version 3.02 (Graph Pad Software, San Diego). The MIC is defined as the concentration of compound required to produce an ODO O O reading equivalent to 10% of the vehicle control reading.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Child & Adolescent Psychology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
Abstract
Description


































Claims














Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/513,888 US20100168176A1 (en) | 2006-11-08 | 2007-11-08 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| MX2009004950A MX2009004950A (en) | 2006-11-08 | 2007-11-08 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same. |
| CA002668840A CA2668840A1 (en) | 2006-11-08 | 2007-11-08 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| AU2007317794A AU2007317794A1 (en) | 2006-11-08 | 2007-11-08 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| JP2009536300A JP2010509335A (en) | 2006-11-08 | 2007-11-08 | NOVEL COMPOUND, PHARMACEUTICAL COMPOSITION CONTAINING THE SAME, AND METHOD OF USING THE SAME |
| BRPI0719058-1A BRPI0719058A2 (en) | 2006-11-08 | 2007-11-08 | COMPOUNDS, PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME AND METHODS OF USE |
| EP07853099A EP2086943A4 (en) | 2006-11-08 | 2007-11-08 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| EA200970460A EA200970460A1 (en) | 2006-11-08 | 2007-11-08 | NEW COMPOUNDS CONTAINING THEIR PHARMACEUTICAL COMPOSITIONS AND METHODS OF THEIR USE |
| IL198634A IL198634A0 (en) | 2006-11-08 | 2009-05-07 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US85751306P | 2006-11-08 | 2006-11-08 | |
| US60/857,513 | 2006-11-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008057585A1 true WO2008057585A1 (en) | 2008-05-15 |
Family
ID=39364833
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/023552 WO2008057585A1 (en) | 2006-11-08 | 2007-11-08 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US20100168176A1 (en) |
| EP (1) | EP2086943A4 (en) |
| JP (1) | JP2010509335A (en) |
| KR (1) | KR20090098804A (en) |
| CN (1) | CN101611017A (en) |
| AU (1) | AU2007317794A1 (en) |
| BR (1) | BRPI0719058A2 (en) |
| CA (1) | CA2668840A1 (en) |
| EA (1) | EA200970460A1 (en) |
| IL (1) | IL198634A0 (en) |
| MX (1) | MX2009004950A (en) |
| WO (1) | WO2008057585A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2285215A1 (en) * | 2008-06-02 | 2011-02-23 | Fasgen, Inc. | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| US8729239B2 (en) | 2009-04-09 | 2014-05-20 | Nuclea Biotechnologies, Inc. | Antibodies against fatty acid synthase |
| US9346769B2 (en) | 2010-05-05 | 2016-05-24 | Infinity Pharmaceuticals, Inc. | Tetrazolones as inhibitors of fatty acid synthase |
| US11202795B2 (en) | 2014-11-20 | 2021-12-21 | Vib Vzw | Means and methods for treatment of early-onset Parkinson's disease |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001049278A2 (en) * | 2000-01-06 | 2001-07-12 | University College Cardiff Consultants Limited | Compounds and compositions for use in inhibiting endoparasitic fatty acid biosynthesis |
| WO2004005277A1 (en) * | 2002-07-09 | 2004-01-15 | Fasgen, Inc. | Novel compunds, pharmaceutical compositions containing same, and methods of use for same |
-
2007
- 2007-11-08 CA CA002668840A patent/CA2668840A1/en not_active Abandoned
- 2007-11-08 US US12/513,888 patent/US20100168176A1/en not_active Abandoned
- 2007-11-08 CN CNA2007800445821A patent/CN101611017A/en active Pending
- 2007-11-08 EA EA200970460A patent/EA200970460A1/en unknown
- 2007-11-08 KR KR1020097011742A patent/KR20090098804A/en not_active Application Discontinuation
- 2007-11-08 BR BRPI0719058-1A patent/BRPI0719058A2/en not_active Application Discontinuation
- 2007-11-08 EP EP07853099A patent/EP2086943A4/en not_active Withdrawn
- 2007-11-08 AU AU2007317794A patent/AU2007317794A1/en not_active Abandoned
- 2007-11-08 WO PCT/US2007/023552 patent/WO2008057585A1/en active Search and Examination
- 2007-11-08 JP JP2009536300A patent/JP2010509335A/en active Pending
- 2007-11-08 MX MX2009004950A patent/MX2009004950A/en unknown
-
2009
- 2009-05-07 IL IL198634A patent/IL198634A0/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001049278A2 (en) * | 2000-01-06 | 2001-07-12 | University College Cardiff Consultants Limited | Compounds and compositions for use in inhibiting endoparasitic fatty acid biosynthesis |
| WO2004005277A1 (en) * | 2002-07-09 | 2004-01-15 | Fasgen, Inc. | Novel compunds, pharmaceutical compositions containing same, and methods of use for same |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2086943A4 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2285215A1 (en) * | 2008-06-02 | 2011-02-23 | Fasgen, Inc. | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| CN102111998A (en) * | 2008-06-02 | 2011-06-29 | 法斯根公司 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| JP2011521978A (en) * | 2008-06-02 | 2011-07-28 | ファスジェン, インコーポレイテッド | Novel compounds, pharmaceutical compositions containing them, and methods of use thereof |
| EP2285215A4 (en) * | 2008-06-02 | 2012-04-04 | Fasgen Inc | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
| US8729239B2 (en) | 2009-04-09 | 2014-05-20 | Nuclea Biotechnologies, Inc. | Antibodies against fatty acid synthase |
| US9732158B2 (en) | 2009-04-09 | 2017-08-15 | Nmdx, Llc | Antibodies against fatty acid synthase |
| US9346769B2 (en) | 2010-05-05 | 2016-05-24 | Infinity Pharmaceuticals, Inc. | Tetrazolones as inhibitors of fatty acid synthase |
| US11202795B2 (en) | 2014-11-20 | 2021-12-21 | Vib Vzw | Means and methods for treatment of early-onset Parkinson's disease |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2086943A1 (en) | 2009-08-12 |
| IL198634A0 (en) | 2010-02-17 |
| EA200970460A1 (en) | 2009-12-30 |
| US20100168176A1 (en) | 2010-07-01 |
| BRPI0719058A2 (en) | 2013-11-26 |
| CN101611017A (en) | 2009-12-23 |
| MX2009004950A (en) | 2009-07-27 |
| CA2668840A1 (en) | 2008-05-15 |
| AU2007317794A1 (en) | 2008-05-15 |
| KR20090098804A (en) | 2009-09-17 |
| JP2010509335A (en) | 2010-03-25 |
| EP2086943A4 (en) | 2010-08-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2767092C (en) | Novel compounds, pharmaceutical compositions containing same and methods of use for same | |
| AU2005249437A1 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same | |
| AU2003248810B2 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same | |
| US20110288052A1 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same | |
| JP2011521978A5 (en) | ||
| WO2008057585A1 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same | |
| US20100029752A1 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same | |
| US20100029761A1 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780044582.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07853099 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2668840 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 198634 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2009536300 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007317794 Country of ref document: AU Ref document number: MX/A/2009/004950 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1778/KOLNP/2009 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007853099 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2007317794 Country of ref document: AU Date of ref document: 20071108 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097011742 Country of ref document: KR Ref document number: 200970460 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12513888 Country of ref document: US |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: PI0719058 Country of ref document: BR Kind code of ref document: A2 Effective date: 20090508 |